
INTERAÇÃO DE RADIAÇÃO IONIZANTE COM POLÍMERO … · figura 4 - disposiÇÃo dos orbitais Π...
126
HUGO SANTOS SILVA INTERAÇÃO DE RADIAÇÃO IONIZANTE COM POLÍMERO SEMICONDUTOR DERIVADO DE POLI(TIOFENO-FENILENO): ABORDAGEM TEÓRICO- EXPERIMENTAL UNIVERSIDADE FEDERAL DE UBERLÂNDIA INSTITUTO DE FÍSICA 2011
-
Upload
phungduong -
Category
Documents
-
view
214 -
download
0
Transcript of INTERAÇÃO DE RADIAÇÃO IONIZANTE COM POLÍMERO … · figura 4 - disposiÇÃo dos orbitais Π...

HUGO SANTOS SILVA
INTERAÇÃO DE RADIAÇÃO IONIZANTE COM
POLÍMERO SEMICONDUTOR DERIVADO DE
POLI(TIOFENO-FENILENO): ABORDAGEM TEÓRICO-
EXPERIMENTAL
UNIVERSIDADE FEDERAL DE UBERLÂNDIA
INSTITUTO DE FÍSICA
2011

HUGO SANTOS SILVA
INTERAÇÃO DE RADIAÇÃO IONIZANTE COM POLÍMERO
SEMICONDUTOR DERIVADO DE POLI(TIOFENO-FENILENO):
ABORDAGEM TEÓRICO-EXPERIMENTAL
Monografia apresentada à
Coordenação do Curso de Graduação em
Física de Materiais, do Instituto de Física
da Universidade Federal de Uberlândia
como parte dos requisitos para obtenção do
título de BACHAREL em Física com
ênfase em Matéria Condensada.
Orientadora: Profa. Dra. Raigna Augusta
da Silva Zadra Armond
UBERLÂNDIA - MG
2011

HUGO SANTOS SILVA
INTERAÇÃO DE RADIAÇÃO IONIZANTE COM POLÍMERO
SEMICONDUTOR DERIVADO DE POLI(TIOFENO-FENILENO):
ABORDAGEM TEÓRICO-EXPERIMENTAL
Monografia apresentada à Coordenação do Curso de
Graduação em Física de Materiais, do Instituto de Física da
Universidade Federal de Uberlândia como parte dos requisitos
para obtenção do título de BACHAREL em Física com ênfase
em Matéria Condensada, submetida à aprovação da banca
examinadora composta pelos seguintes membros:
_____________________________________________
Profa. Dra. Raigna Augusta da Silva Zadra Armond
(orientadora)
_____________________________________________
Prof. Dr. Augusto Miguel Alcade Milla
_____________________________________________
Prof. Dr. Raimundo Lora Serrano
UBERLÂNDIA - MG
2011

Aos meus pais que, por entre encontros e desencontros,
erros e acertos, sempre ali estavam por mim.

“[...] Será possível? Não seremos mais que um simples
conglomerado vulgar de moléculas sem solidariedade
umas com as outras? Deve desaparecer para sempre nossa
individualidade cheia de amor e, do que foi um homem, não
restará verdadeiramente senão um cadáver destinado
a desagregar-se, lentamente, na fria noite do túmulo? [...]”
Gabriel Delanne
“[...] É a matéria quintessenciada, mas sem analogia
para vós outros, e tão etérea que escapa inteiramente
dos vossos sentidos. [...]”
Allan Kardec

AGRADECIMENTOS
A Deus, pela graça da vida e da evolução dos seres e do Homem. À minha mãe e ao meu pai
que sempre foram meu suporte e minha base, que sempre batalharam tanto por mim. Aos
meus irmãos, por serem a atual razão de viver dos meus pais. A toda a minha família, que
direta ou indiretamente, contribuiu para minha formação como ser humano. Um
agradecimento especial à Jack, Jana e Tereza, por terem me acolhido como parte da família
também, além da Janice, por razões indescritíveis.
Aos meus amigos, que por serem como irmãos, são a família que me escolheram. Obrigado
pelo carinho e amizade.
Em especial, gostaria de agradecer à Isadora por ter me acompanhado por todos esses anos.
Um agradecimento especial à Sandra, que possui um caráter dual: de amiga e de mãe. A todos
que conheci dentro dessa casa, que é o Instituto de Física, em especial ao pessoal do Grupo de
Espectroscopia de Materiais, Marcia, Silésia, Gustavo, Therézio, Paulo, Gilberto, Maurício,
Bebel, Estácio e aos que não estão mais aqui, como o Siri e o Kaká, assim como ao Dominike,
que tanto me ajudou no começo da estrada da simulação computacional.
À professora Raigna, pela orientação, amizade e, principalmente, pela paciência, sem a qual
este trabalho não se realizaria. Pelas tantas oportunidades que me tem proporcionado, por
investir tanto em mim, preocupar-se com meu desenvolvimento acadêmico e pessoal e pela fé.
Enfim, por ser outra 'mãe' que tive e que decididamente me adotou como um filho. MUITO
OBRIGADO.
Aos professores Alexandre e Newton que sempre quiseram meu melhor, e tanto têm investido
no meu crescimento e desenvolvimento, profissional e pessoal. Por tratarem aos alunos do
GEM como filhos.
Ao professor J. E. Manzoli, que passou da figura de colaborador para a figura de AMIGO.
Aos professores do Instituto de Física e aos funcionários deste por serem sempre tão
atenciosos com o crescimento acadêmico de cada um.
Ao pessoal do Laboratório de Irradiação do CTR-IPEN/CNEN pelas irradiações.
À Universidade Federal de Uberlândia, FAPEMIG, CNPq e CAPES pelo apoio financeiro e
de pessoal durante esses anos.

SANTOS SILVA, H. Interação de radiação ionizante com polímero semicondutor
derivado de poli(tiofeno-fenileno): abordagem teórico-experimental. 2011, 110 f.
Monografia (graduação) – Instituto de Física, Universidade Federal de Uberlândia,
Uberlândia 2011.
RESUMO
A influência da radiação gama sobre o polímero conjugado poli(2,5-tiofeno-1,4-
diálcoxifenileno) é investigada usando absorção óptica UV-VIS e fotoluminescência. As
amostras, em solução de solvente orgânico não-halogenado, foram irradiadas com uma fonte
de Cobalto 60 com 20 e 40 kGy de dose. A fim de determinar como essa interação ocorre e
como ela interfere nas propriedades estruturais, eletrônicas e vibracionais do polímero, são
realizados cálculos semi-empíricos sob a parametrização de Hartree-Fock AM1 para
determinação da estrutura conformacional, dos sítios mais reativos, estimativa do grau de
cisão da cadeia e de como ocorre o ataque pela radiação. O comportamento da estrutura
vibrônica em função da dose é estudado dentro do formalismo da Regra de Ouro de Fermi e
da Aproximação de Franck-Condon. Esses dados permitem estimar como os defeitos são
distribuídos, de que forma eles modificam a energia de gap e como modulam os acoplamentos
entre elétrons e fônons. Além disso, com base na estrutura eletrônica do material, é discutido
como esse efeito pode ser utilizado de forma consciente para a manufatura de dispositivos
opto-eletrônicos orgânicos mais eficientes.
Palavras-chave: polímeros conjugados, defeitos, radiação gama, cálculos semi-empíricos.

SANTOS SILVA, H. Interaction of ionizing radiation with semiconductor polymer
derived from poly(tiophene-phenylene): theoretical-experimental approach. 2011, 110 f.
Monografia (graduação) – Instituto de Física, Universidade Federal de Uberlândia,
Uberlândia 2011.
ABSTRACT
The influence of gamma radiation on the conjugated polymer poly(2,5-thiophene-1,4-
dialkoxyphenylene) is investigated using UV-VIS optical absorption and Photoluminescence
measurements. Samples in solution of non-halide organic solvent were irradiated in a Cobalt
60 gamma source with 20 and 40 kGy of total dose. In order to determine how this interaction
occurs and how it changes the structural, electronic and vibrational properties of the polymer,
we carried out semi-empirical calculations under AM1 Hartree-Fock‟s parameterization to
determine conformational structure, most reactive sites, estimative of chain scission degree
and how the radiation attack occurs. The vibronic structure‟s behavior in function of the dose
is studied within the Fermi‟s Golden rule scope with Franck-Condon‟s approximation. The
data allow us to estimate how defects are distributed, how they modify the gap energy and
modulate coupling among electrons and phonons. Moreover, based on the electronic structure
of the material, it is discussed how this effect can be used in a conscientious way to produce
more efficient organic optoelectronic devices.
Keywords: conjugated polymers, defects, gamma radiation, semi-empirical calculations

NOTA DO AUTOR
Os cálculos e a modelagem computacional realizados nesse trabalho são chamados de
„teóricos‟, mesmo que o nível de teoria utilizado não corresponda àquele indicado pela Escola
atuante de Estrutura Eletrônica no Brasil. Podemos então, sem perca de generalidade e
confiança, considerá- los, assim como faz a literatura, como cálculos de Química Quântica,
desde que separações de tal natureza entre a Física e a Química não são mais permitidas a
Grupos que desejem realmente realizar Ciência de ponta em qualquer parte do mundo.
Delimitar uma fronteira entre Química, Física, Matemática e Engenharia não é uma opção ao
Estudo da Matéria Condensada, principalmente quando se trata de Sistemas Complexos, na
qual está englobado o estudo de polímeros e moléculas de interesse, que está desenvolvido
nesse trabalho.

SUMÁRIO
LISTA DE FIGURAS ............................................................................................................... 12
LISTA DE TABELAS............................................................................................................... 15
LISTA DE EQUAÇÕES ........................................................................................................... 16
LISTA DE ABREVIATURAS .................................................................................................. 19
CAPÍTULO I ............................................................................................................................ 22
1 – INTRODUÇÃO, COM JUSTIFICATIVA E SÍNTESE DA BIBLIOGRAFIA
FUNDAMENTAL .................................................................................................................... 22 1.1 - CONTEXTUALIZAÇÃO ............................................................................................ 22
1.2 – PROBLEMÁTICA ...................................................................................................... 25 1.3 – FÍSICA DE MATERIAIS POLIMÉRICOS................................................................. 28
CAPÍTULO II ........................................................................................................................... 38
2 – METODOLOGIA ............................................................................................................... 38
2.1 – MATERIAL E MÉTODO ................................................................................................ 38
2.1.1 – O POLÍMERO CONJUGADO FSE59..................................................................... 38 2.1.2 – A INTERAÇÃO RADIAÇÃO COM MATÉRIA: RADIAÇÃO GAMA DE 60Co. . 45
2.2 – EXPERIMENTAL ......................................................................................................... 47
2.2.1 – ABSORÇÃO ÓPTICA .......................................................................................... 47 2.2.2 – FOTOLUMINESCÊNCIA .................................................................................... 52
2.3 – TEÓRICA ...................................................................................................................... 55 2.3.1 – O ESTADO FUNDAMENTAL: MÉTODO DE HARTREE-FOCK-ROOTHANN E PÓS-HARTREE-FOCK .................................................................................................... 56
2.3.2 – O ESTADO EXCITADO: DESCRIÇÃO SEMI-EMPÍRICA ............................... 69 2.3.3 – FORMALISMO DE TRANSIÇÕES VERTICAIS DE FERMI E ACOPLAMENTOS VIBRÔNICOS DE FRANCK-CONDON........................................... 71
2.3.4 – O FORMALISMO DOS ÍNDICES DE REATIVIDADE DE FUKUI ................. 82 2.3.5 – O SOFTWARE MOPAC2009®.............................................................................. 84
CAPÍTULO III.......................................................................................................................... 85
3 – RESULTADOS .............................................................................................................. 85
3.1 – EXPERIMENTAIS .......................................................................................................... 85
3.1.1 – ABSORÇÃO ÓPTICA ............................................................................................. 86 3.1.2 – FOTOLUMINESCÊNCIA ....................................................................................... 87

3.2 – TEÓRICOS .................................................................................................................... 89
3.2.1 – GEOMETRIAS DE EQUILÍBRIO DO ESTADO FUNDAMENTAL ................. 89 3.2.2 – TRANSIÇÕES ELETRÔNICAS EM FUNÇÃO DO TAMANHO DO
SEGMENTO CONJUGADO ............................................................................................... 92 3.2.3 – AJUSTE VIBRÔNICO DA FOTOLUMINESCÊNCIA ....................................... 96 3.2.4 – ÍNDICES DE REATIVIDADE DE FUKUI .......................................................... 98
CAPÍTULO IV - DISCUSSÃO DOS RESULTADOS........................................................... 105
CAPÍTULO V - CONSIDERAÇÕES FINAIS ...................................................................... 110
CAPÍTULO VI ....................................................................................................................... 112
1 – REFERÊNCIAS BIBLIOGRÁFICAS ............................................................................. 112
ANEXO I ................................................................................................................................ 119

LISTA DE FIGURAS
FIGURA 1 - ORBITAIS MOLECULARES DA MOLÉCULA DE METANO. ..................... 32
FIGURA 2 - SOBREPOSIÇÃO DOS ORBITAIS PZ FORMANDO O ORBITAL π NA
MOLÉCULA DE ETILENO. ........................................................................................... 33
FIGURA 3 - HIDROCARBONETOS AROMÁTICOS CONJUGADOS DE INTERESSE
(DA ESQUERDA PARA A DIREITA, DE CIMA PARA BAIXO): BENZENO,
TIOFENO, PIRROL E ANTRACENO. OBSERVE QUE O ANÉIS DE TIOFENO E DE
PIRROL POSSUEM HETEROÁTOMOS E QUE MESMO ASSIM MANTÊM A
CONJUGAÇÃO. .............................................................................................................. 34
FIGURA 4 - DISPOSIÇÃO DOS ORBITAIS Π ABAIXO E ACIMA DA MOLÉCULA DE
ANTRACENO. EM MOLÉCULAS COM CONJUGAÇÃO MAIOR DO QUE ESSA, A
DELOCALIZAÇÃO ABRANGE TODO O COMPRIMENTO DA MOLÉCULA
DESDE QUE NÃO HAJA INTERRUPÇÕES POR DEFEITOS E/OU SATURAÇÕES
SP3..................................................................................................................................... 34
FIGURA 5 - POLIACETILENO: A) DIMERIZAÇÃO DA CADEIA POLIMÉRICA; (B)
ESTRUTURA DE BANDA ANTES E APÓS A DIMERIZAÇÃO. ................................ 35
FIGURA 6 - A) FORMAÇÃO DE PÓLARONS NA CADEIA DE POLIPIRROL, COM
REPRESENTAÇÃO DOS NOVOS NÍVEIS ENERGÉTICOS CRIADOS NO
INTERIOR DO GAP; B) FORMAÇÃO DE BIPÓLARONS, QUE CRIAM NÍVEIS
CUJA DIFERENÇA DE ENERGIA É AINDA MENOR DO QUE OS PÓLARONS E C)
FORMAÇÃO DE BANDAS DE BIPÓLARONS, QUE ACONTECEM QUANDO
OCORRE SUPEROXIDAÇÃO DA CADEIA, INDUZINDO A FORMAÇÃO DE
VÁRIOS BIPÓLARONS PRÓXIMOS ESPACIALMENTE, QUE FORMAM UM
SEMI-CONTÍNUO DE ESTADOS BIPOLARÔNICOS................................................. 37
FIGURA 7 - PROPOSTA DE HEEGER E COLABORADORES PARA AUMENTAR A
EFICIÊNCIA QUÂNTICA DE EMISSÃO DOS POLITIOFENOS................................ 39
FIGURA 8 - ROTA DE SÍNTESE DO POLÍMERO CONJUGADO FSE59. ........................ 40
FIGURA 9 - CARACTERIZAÇÃO ESTRUTURAL DA ORGANIZAÇÃO DA CADEIA
POLIMÉRICA DO FSE59: D REPRESENTA A DISTÂNCIA INTERCADEIAS E D É
A SEPARAÇÃO INTERDIGITAL ENTRE ELAS. ......................................................... 42
FIGURA 10 - VARREDURA DA REGIÃO CATÓDICA (ESQUERDA) E ANÓDICA
(DIREITA) DO POLÍMERO FSE59. OBSERVE O ALTO POTENCIAL ELÉTRICO

NECESSÁRIO PARA REDUZIR A CADEIA................................................................. 43
FIGURA 11 - A) UV-VIS IN SITU COM APLICAÇÃO DE POTENCIAL, OBSERVANDO
CRÔMICO EM FUNÇÃO DO POTENCIAL E B) SOLUÇÃO ANTES (ESQUERDA)
E APÓS (DIREITA) A APLICAÇÃO DE POTENCIAL ELÉTRICO............................. 44
FIGURA 12 – ESQUEMA DO PROCESSO DE EXCITAÇÃO............................................. 47
FIGURA 13 - NÍVEIS ELETRÔNICOS E TRANSIÇÕES. ................................................... 48
FIGURA 14 - HOMO E LUMO DA MOLÉCULA NO ESPAÇO DAS POSIÇÕES [(A) E
(B), RESPECTIVAMENTE] E (C) COMO ESSES ESTADOS ELETRÔNICOS SÃO
DISTRIBUÍDOS EM FUNÇÃO DA ENERGIA, DADA PELA DENSIDADE DE
ESTADOS (DOS) E PELA DENSIDADE PROJETADA DE ESTADOS (PDOS). EF
REFERE-SE À ENERGIA DE FERMI............................................................................ 51
FIGURA 15 - TRANSFERÊNCIA DE ENERGIA AO LONGO DE UMA CADEIA
POLIMÉRICA OU ENTRE CADEIAS POLIMÉRICAS, INDICANDO QUE O
PROCESSO DE LUMINESCÊNCIA SÓ OCORRE A PARTIR DAS CADEIAS DE
MAIOR SEGMENTO CONJUGADO, OU SEJA, DE MENOR GAP............................ 53
FIGURA 16 - CICLO DE AUTOCONSISTÊNCIA NAS EQUAÇÕES DE HARTREE-
FOCK-ROOTHAAN-HALL. ........................................................................................... 66
FIGURA 17 - ESTRUTURA DAS METODOLOGIAS DE CÁLCULO DA EQUAÇÃO DE
HARTREE-FOCK. ........................................................................................................... 66
FIGURA 18 - PRINCÍPIO DE FRANCK-CONDON. ............................................................ 79
FIGURA 19 - ESPECTROS DE ABSORÇÃO ÓPTICA UV-VIS PARA AS AMOSTRAS DE
FSE59 NÃO IRRADIADA, IRRADIADAS COM 20 E 40 KGY E PARA O
OLIGÔMERO SL128G. ................................................................................................... 87
FIGURA 20 - ESPECTROS DE FOTOLUMINESCÊNCIA PARA AS AMOSTRAS DE
FSE59 NÃO IRRADIADA, IRRADIADAS COM 20 E 40 KGY E PARA O
OLIGÔMERO SL128G. ................................................................................................... 88
FIGURA 21 - GEOMETRIAS DE EQUILÍBRIO PARA (A) 2 ANÉIS, (B) 4 ANÉIS, (C) 5
ANÉIS, (D) 6 ANÉIS E (E) 16 ANÉIS. ........................................................................... 91
FIGURA 22 - ESPECTROS DE ABSORÇÃO SIMULADOS EM FUNÇÃO DO NÚMERO
DE ANÉIS (P) DOS OLIGÔMEROS. ............................................................................. 94
FIGURA 23 - ESPECTRO DE ABSORÇÃO CALCULADO (----) E EXPERIMENTAL (-O-
) DO OLIGÔMERO SL128G, DE 5 ANÉIS, EVIDENCIANDO A DISCREPÂNCIA

TEÓRICA. ........................................................................................................................ 95
FIGURA 24 - ESPECTROS DE PL AJUSTADOS COM O FORMALISMO DE FRANCK-
CONDON. ........................................................................................................................ 97
FIGURA 25 - ESTRUTURA A. .............................................................................................. 98
FIGURA 26 - ESTRUTURA B. .............................................................................................. 99
FIGURA 27 - VARIAÇÃO DA CARGA MEDIDA PELOS ÍNDICES DE FUKUI PARA A
ESTRUTURA A. ............................................................................................................ 100
FIGURA 28 - VARIAÇÃO DA DENSIDADE DE CARGA PARA A ESTRUTURA A. ..... 101
FIGURA 29 - DENSIDADE DE PROBABILIDADE DE OCORRER UMA INTERAÇÃO
DA RADIAÇÃO COM A CADEIA POLIMÉRICA, POR SÍTIO, NA ESTRUTURA A.
........................................................................................................................................ 101
FIGURA 30 - APRESENTAÇÃO GRÁFICA DOS ÍNDICES DE FUKUI PARA ATAQUE
RADICAL NA CADEIA, EM QUE QUANTO MAIS ESCURO O SÍTIO, MAIOR A
PROBABILIDADE DE OCORRER UMA REAÇÃO DE CISÃO. .............................. 102
FIGURA 31 - VARIAÇÃO DA CARGA MEDIDA PELOS ÍNDICES DE FUKUI PARA A
ESTRUTURA B. ............................................................................................................ 102
FIGURA 32 - VARIAÇÃO DA DENSIDADE DE CARGA PARA A ESTRUTURA B. ..... 103
FIGURA 33 - DENSIDADE DE PROBABILIDADE DE OCORRER UMA INTERAÇÃO
DA RADIAÇÃO COM A CADEIA POLIMÉRICA, POR SÍTIO, NA ESTRUTURA B.
........................................................................................................................................ 103
FIGURA 34 - APRESENTAÇÃO GRÁFICA DOS ÍNDICES DE FUKUI PARA ATAQUE
RADICAL NO RAMO SOLUBILIZANTE, EM QUE QUANTO MAIS ESCURO O
SÍTIO, MAIOR A PROBABILIDADE DE OCORRER UMA REAÇÃO DE CISÃO. 104
FIGURA 35 - ESPECTROS DE ABSORÇÃO ÓPTICA UV-VIS DO POLÍMERO FSE59
NÃO-IRRADIADO E IRRADIADO E DO OLIGÔMERO SL128G, EVIDENCIANDO
A SOBREPOSIÇÃO COM A BANDA DE EMISSÃO NO VISÍVEL DO SOL........... 108

LISTA DE TABELAS
TABELA 1 - ENERGIA DE GAP EM FUNÇÃO DO NÚMERO DE ANÉIS DA CADEIA.96
TABELA 2 - ESTIMATIVA DO GRAU DE CISÃO DA CADEIA EM FUNÇÃO DA DOSE
IRRADIADA. ................................................................................................................... 96
TABELA 3 - FATORES DE HUANG-RHYS E PARÂMETROS DE AJUSTE OBTIDOS A PARTIR DO FORMALISMO DE FRANCK-CONDON UTILIZADO PARA AJUSTAR OS ESPECTROS DE PL. ................................................................................................. 97
TABELA 4 - ÍNDICE DE SOBREPOSIÇÃO DO ESPECTRO DE ABSORÇÃO DAS
ESPÉCIES ESTUDADAS COM O ESPECTRO DE EMISSÃO SOLAR, DADO PELA ÁREA INTEGRADA DA CURVA-PRODUTO DOS DOIS ESPECTROS (AMOSTRA E SOL) NORMALIZADOS. .......................................................................................... 108

LISTA DE EQUAÇÕES
EQUAÇÃO 1 - LEI DE BEER-LAMBERT. ........................................................................... 52
EQUAÇÃO 2 - EQUAÇÃO DE SCHRÖDINGER INDEPENDENTE DO TEMPO (ESIT).
.......................................................................................................................................... 56
EQUAÇÃO 3 - OPERADOR HAMILTONIANO ELETRÔNICO ESCRITO EM
COORDENADAS ATÔMICAS. ...................................................................................... 57
EQUAÇÃO 4 - APROXIMAÇÃO DE HARTREE-FOCK. ................................................... 58
EQUAÇÃO 5 - FORMA DIDÁTICA DA APROXIMAÇÃO DE HF. ................................... 58
EQUAÇÃO 6 - FUNÇÃO DE ONDA DE HF NA FORMA DO DETERMINANTE DE
SLATER............................................................................................................................ 58
EQUAÇÃO 7 - EQUAÇÃO DE AUTOVALORES DE HARTREE-FOCK. ......................... 59
EQUAÇÃO 8 - OPERADOR DE FOCK. ............................................................................... 59
EQUAÇÃO 9 - OPERADOR DE FOCK EXPLÍCITO. ......................................................... 59
EQUAÇÃO 10 - OPERADOR DE FOCK RESTRITO. ......................................................... 60
EQUAÇÃO 11 - EQUAÇÃO DE AUTOVALORES DE HF EXPLÍCITA 1.......................... 60
EQUAÇÃO 12 - EQUAÇÃO DE AUTOVALORES DE HF EXPLÍCITA 2. ........................ 61
EQUAÇÃO 13 - EXPANSÃO DAS FUNÇÕES DE ONDA MOLECULAR EM FUNÇÕES
DE BASE ATÔMICAS..................................................................................................... 62
EQUAÇÃO 14 - EQUAÇÃO DE HF COM LCAO - 1. ......................................................... 62
EQUAÇÃO 15 - EQUAÇÃO DE HF COM LCAO - 2. ......................................................... 62
EQUAÇÃO 16 - MATRIZ DE FOCK..................................................................................... 63
EQUAÇÃO 17 - MATRIZ DE SOBREPOSIÇÃO. ................................................................ 63
EQUAÇÃO 18 - MATRIZ DE FOCK E DE SOBREPOSIÇÃO NA NOTAÇÃO DE DIRAC.
.......................................................................................................................................... 63
EQUAÇÃO 19 - EQUAÇÃO DE HARTREE-FOCK-ROOTHAAN-HALL......................... 63
EQUAÇÃO 20 - DECOMPOSIÇÃO DO OPERADOR DE FOCK....................................... 64
EQUAÇÃO 21 - HAMILTONIANO DE CAROÇO............................................................... 64
EQUAÇÃO 22 - HAMILTONIANO DE CAROÇO EM NOTAÇÃO DE DIRAC................ 64
EQUAÇÃO 23 – DEFINIÇÃO DE EM TERMOS DO OPERADOR DENSIDADE. ....... 64
EQUAÇÃO 24 - ELEMENTOS DA MATRIZ DENSIDADE................................................ 65
EQUAÇÃO 25 - INTEGRAIS DE DOIS ELÉTRONS. ......................................................... 65
EQUAÇÃO 26 - EQUAÇÃO GLOBAL EXPLÍCITA DE HARTREE-FOCK-ROOTHAAN-

HALL................................................................................................................................ 65
EQUAÇÃO 27 - RELAÇÃO DE MATAGA-NISHIMOTO. .................................................. 70
EQUAÇÃO 28 - HAMILTONIANO DE UM SISTEMA QUÂNTICO. ................................ 71
EQUAÇÃO 29 - AUTOESTADOS DO SISTEMA QUÂNTICO NO ESTADO N................ 72
EQUAÇÃO 30 - HAMILTONIANO SEMI-CLÁSSICO DE INTERAÇÃO DA
RADIAÇÃO-MATÉRIA. ................................................................................................. 72
EQUAÇÃO 31 - EVOLUÇÃO TEMPORAL DO HAMILTONIANO................................... 72
EQUAÇÃO 32 - COEFICIENTES DO ESTADO EXCITADO - 1. ....................................... 73
EQUAÇÃO 33 - COEFICIENTES DO ESTADO EXCITADO - 2. ....................................... 73
EQUAÇÃO 34 - PROBABILIDADE DE SE ENCONTRAR O SISTEMA NO ESTADO .
.......................................................................................................................................... 73
EQUAÇÃO 35 - PROBABILIDADE DE SE ENCONTRAR O SISTEMA NO ESTADO
CORRELACIONADO COM O ESTADO POR UMA FUNÇÃO DE DENSIDADE
DE ESTADOS . ....................................................................................................... 73
EQUAÇÃO 36 - REGRA DE OURO DE FERMI. ................................................................. 74
EQUAÇÃO 37 - HAMILTONIANO SSH. ............................................................................. 74
EQUAÇÃO 38 - FUNÇÃO DE ONDA TOTAL PARA UM HAMILTONIANO DO TIPO
SSH. .................................................................................................................................. 75
EQUAÇÃO 39 - 1ª CONSEQÜÊNCIA DA APROXIMAÇÃO DE BO -1. ........................... 75
EQUAÇÃO 40 - 1ª CONSEQÜÊNCIA DA APROXIMAÇÃO DE BO -2. ........................... 76
EQUAÇÃO 41 - EQUAÇÃO DE SCHRÖDINGER PARA O HAMILTONIANO SSH COM
APROXIMAÇÃO DE BO................................................................................................ 76
EQUAÇÃO 42 - PARTE ESTÁTICA DA EQUAÇÃO DE SCHRÖDINGER NO MODELO
SSH. .................................................................................................................................. 76
EQUAÇÃO 43 - PARTE DINÂMICA DA EQUAÇÃO DE SCHRÖDINGER NO MODELO
SSH. .................................................................................................................................. 76
EQUAÇÃO 44 - FUNÇÃO DE ONDA VIBRÔNICA EM TERMOS DAS FUNÇÕES DE
ONDA DO OSCILADOR HARMÔNICO QUÂNTICO UNIDIMENSIONAL. ............ 77
EQUAÇÃO 45 - ENERGIA VIBRÔNICA EM FUNÇÃO DA PARTE ELETRÔNICA E
VIBRACIONAL. .............................................................................................................. 77
EQUAÇÃO 46 - TAXA DE PROBABILIDADE DE TRANSIÇÃO ENTRE DOIS NÍVEIS
VIBRÔNICOS. ................................................................................................................. 78
EQUAÇÃO 47 - FATOR DE BOLTZMANN DE OCUPAÇÃO TÉRMICA (OBSERVE QUE

NADA MAIS É DO UMA FUNÇÃO DE PARTIÇÃO CANÔNICA). ........................... 78
EQUAÇÃO 48 - APROXIMAÇÃO DE FRANCK-CONDON. ............................................. 79
EQUAÇÃO 49 - TAXA DE PROBABILIDADE DE TRANSIÇÃO ENTRE DOIS NÍVEIS
ELETRÔNICOS ACOPLADOS COM NÍVEIS VIBRACIONAIS. ............................... 80
EQUAÇÃO 50 - INTEGRAL DE FRANCK-CONDON........................................................ 80
EQUAÇÃO 51 - FATOR DE HUANG-RHYS. ...................................................................... 80
EQUAÇÃO 52 - COEFICIENTE DE EMISSÃO COM ACOPLAMENTO ELÉTRON-
FÔNON............................................................................................................................. 81
EQUAÇÃO 53 - FUNÇÃO DE FUKUI.................................................................................. 82
EQUAÇÃO 54 - FUNÇÕES DE FUKUI PARA (A) ATAQUE NUCLEOFÍLICO, (B)
ELETROFÍLICO E (C) ATAQUE DE RADICAL. .......................................................... 83
EQUAÇÃO 55 - ÍNDICES CONDENSADOS DE FUKUI. .................................................. 83
EQUAÇÃO 56 - EQUAÇÃO DE AJUSTE HIPERBÓLICO PELO MODELO DO
ELÉTRON CONFINADO EM UMA CAIXA. ................................................................ 92
EQUAÇÃO 57 - AJUSTE HIPERBÓLICO COM INCLUSÃO DO PARÂMETRO
CORRETIVO. .................................................................................................................. 93

LISTA DE ABREVIATURAS
UFU – Universidade Federal de Uberlândia
INFIS – Insituto de Física da UFU
GEM – Grupo de Espectroscopia de Materiais (INFIS/UFU)
USP – Universidade de São Paulo
CNEN – Conselho Nacional de Energia Nuclear
IPEN – Instituto de Pesquisas Energéticas e Nucleares (CNEN/USP)
FSE59 – Nome não-oficial do polímero poli(2,5-tiofeno-1,4-dialcóxi- fenileno)
SL128G – Nome não-oficial do oligômero de 5 anéis do FSE59
PPV – Poli(p- fenileno-vinileno)
MEH-PPV – Poli(2-metóxi-5-(2-etil-hexilóxi)-p-fenileno-vinileno)
PT – Politiofeno
P3AT – Poli(3-alquil- tiofeno)
P3HT – Poli(3-hexil-tiofeno)
AO – Optical Abosorption (Absorção óptica)
UV-VIS – Ultravioleta-Visível
PL – Photoluminescence (Fotoluminescência)
K – Kelvin (referente à escala de temperatura)
ES – Equação de Schrödinger
ESIT – Equação de Schrödinger Independente do Tempo
ESDT – Equação de Schrödinger Dependente do Tempo
DFT – Density Functional Theory (Teoria do Funcional da Densidade)
HF – Hartee-Fock
AO – Atomic Orbitals (Orbitais Atômicos)
MO – Molecular Orbitals (Orbitais Moleculares)

NDO – Neglect of Differential Overlap (Método de negligência da sobreposição diferencial)
CNDO – Complete Neglect of Differential Overlap (Método de negligência completa da
sobreposição diferencial)
MNDO – Modified Neglect of Differential Overlap (Método de negligência da sobreposição
diferencial modificado)
INDO – Intermediate Neglect of Differential Overlap (Método de negligência intermediária
da sobreposição diferencial)
ZINDO – Zerner‟s Intermediate Neglect of Differential Overlap (Método de negligência
intermediária da sobreposição diferencial na parametrização de Zerner)
ZINDO-S – Zerner‟s Intermediate Neglect of Differential Overlap - Spectroscopy (Método de
negligência intermediária da sobreposição diferencial na parametrização de Zerner
espectroscópica)
CI – Configuration of Interactions (Interações de Configuração)
CIS – Single-electron Configuration of Interactions (Interações de Configuração de um único
elétron)
MOPAC2009 – Molecular Orbital Package (Pacote de Orbitais Moleculares)
AM1 – Austin Model 1
PM3 – Parametric Method 3
PM5 – Parametric Method 5
PM6 – Parametric Method 6
RHF – Restrict Hartree-Fock (Método de Hartree-Fock restrito)
UHF – Unrestrict Hartree-Fock (Método de Hartree-Fock não-restrito)
ROHF – Restrict Open-Shell Hartree-Fock (Método de Hartree-Fock restrito de camada
aberta)
BO – Aproximação de Born-Oppenheimer
FC – Aproximação de Franck-Condon
f 0 – Função de Fukui para ataque de radical

f + – Função de Fukui para ataque nucleofílico
f - – Função de Fukui para ataque eletrofílico
OLED – Organic Light-Emitting Diode (Diodo emissor de luz orgânico)
OLEC – Organic Light Electrochromic Cell (Célula eletroquímica orgânica emissora de luz)
OPV – Organic Photovoltaic Cell (Célula fotovoltaica orgânica)
OFET – Organic Field-Effect Transistor (Transistor de Efeito de Campo)
p - Número de anéis conjugados da cadeia
pH – Potencial Hidrogeniônico
BV – Banda de Valência
BC – Banda de Condução

22
CAPÍTULO I
Nesse capítulo será realizada uma contextualização do conhecimento acerca da Física de
Materiais Poliméricos assim como da Física de Polímeros Semicondutores. Serão
apresentados os elementos básicos necessários à sua compreensão. Os elementos básicos de
Mecânica Quântica e Física do Estado Sólido não são apresentados e é aconselhável, caso
necessário, que o leitor acompanhe esse capítulo com o auxílio da literatura indicada 1. A fim
de situar o leitor do cenário desse trabalho, é realizado uma breve contextualização da
aplicação de materiais poliméricos semicondutores em dispositivos eletrônicos como também
apresenta-se a problemática à qual se propõe esse texto.
1 – INTRODUÇÃO, COM JUSTIFICATIVA E SÍNTESE DA BIBLIOGRAFIA
FUNDAMENTAL
1.1 - CONTEXTUALIZAÇÃO
Para acompanhar o progresso tecnológico e industrial, novas soluções de produção,
estocagem e utilização de energia precisaram ser desenvolvidas a fim de se evitar um colapso
que seria capaz de afetar a economia mundial. Além disso, a necessidade de se red uzir as
emissões de gases estufa e tóxicos, assim como de rejeitos prejudiciais ao meio ambiente,
induziu o surgimento do conceito de energia limpa assim como consumo limpo. Esses
conceitos que até pouco tempo atrás pareciam futuristas já podem ser vistos na utilização de
1 David Griffiths – Introduction to Quantum Mechanics, 2
nd Edit ion, Benjamin Cummings, 2004; e Ivan S.
Oliveira – Introdução à Física do Estado Sólido , 1ª Edição, Ed itora Livraria da Física, 2005.

23
fontes de energia eólica, solar, de células a hidrogênio, etc., e têm atraído investimentos
bilionários.
Dentre esses projetos de novas fontes renováveis e limpas de energia, a solar talvez é a
que mais chame atenção pela abundância e pela relativa facilidade de implantação.
Aquecedores a energia solar já têm sido usados comumente, no entanto as células de
conversão solar ainda têm participação modesta no mercado energético.
Isso advém do fato de ainda ser alto o custo/benefício apresentado pelas atuais células
feitas de Silício cristalino e/ou amorfo. Outras desvantagens que repelem os investimentos
para popularização dessas células é o seu peso, rigidez, baixo rendimento de conversão e
custo de instalação.
Nas últimas décadas, a partir da descoberta dos polímeros condutores de corrente
elétrica por Heeger e colaboradores, uma nova solução energética nesse âmbito foi
visualizada: a utilização de polímeros semicondutores como matrizes de células fotovoltaicas
leves, flexíveis, baratas, de fácil implantação e cujo rendimento pode ser controlado pelas
técnicas de processamento, síntese e produção. Assim, um novo leque de possibilidades foi
aberto para a utilização de moléculas orgânicas como componentes ativos em dispositivos
emissores (Brédas, Beljonne, Cornil, dos Santos, & Shuai, 1997; Burroughes et al., 1990;
Ding, Pei, Lai, & Huang, 2001; Friend, 2001; Friend et al., 1999; Inganäs et al., 1998;
Sirringhaus, Tessler, & Friend, 1998; Zempo & et al., 2008) e captadores de luz (Coakley &
McGehee, 2004; Friend, 2001) (LEDs e células fotovoltaicas), sensores de temperatura
(Friend, 2001), radiação (Ferreira et al., 2009; Graham, Friend, Fung, & Moratti, 1997; E. A.
B. Silva et al., 2005; Vasconcelos, 2010), pH, etc., supercapacitores, transistores e tantos
outros.
Dentre essas moléculas orgânicas, os polímeros conjugados (ou seja, semicondutores)
se mostraram altamente eficazes para a aplicação tecnológica. A sua composição carbônica
permite, pela estrutura eletrônica do carbono, uma variação imensa das propriedades opto-
eletrônicas (Birgerson et al., 2001; Zempo & et al., 2008) desses materiais por conta de
pequenas alterações provocadas na síntese, purificação, processamento e/ou armazenamento.
Isso fornece uma gama quase infinita de propriedades distintas, cada uma voltada
especificadamente para uma aplicação. Além disso, os o ligômeros, que são moléculas com
estrutura idêntica aos polímeros, porém com unidade repetida menor, têm sido indicados
também para aplicações em que o nível de refinamento das propriedades requerido seja maior,
já que apresentam propriedades similares a dos polímeros iniciadores, com uma maior

24
solubilidade e processabilidade.
O comportamento eletrônico desses materiais é devido à alternância de ligações
simples e duplas ao longo da cadeia que, por dimerização, provoca uma distorção da
densidade de estados, induzindo o surgimento de duas bandas de energias, conhecidas como
HOMO (Highest Occupied Molecular Orbital) e LUMO (Lowest Unoccupied Molecular
Orbital) (Pope, 1999). Esses orbitais, também chamados de π e π*, respectivamente, possuem,
na maior parte desses materiais, uma diferença de energia entre eles de aproximadamente 2
eV, o que justifica seu uso na faixa de freqüências do UV-Vis. Mais sobre as propriedades
eletrônicas desse tipo de material será apresentado na seção 1.4 desse mesmo capítulo.
Essa aplicabilidade dos polímeros conjugados e outras moléculas com propriedades
semelhantes só é possível graças a dois fenômenos de dissociação e recombinação de
portadores de carga: o efeito fotovoltaico (Coakley & McGehee, 2004) e a
eletroluminescência (Friend et al., 1999), respectivamente. O primeiro caracteriza-se pela
captura por eletrodos dos portadores de carga associados que foram formados durante a
absorção de luz, enquanto que o segundo é justamente o efeito inverso: a injeção de
portadores permite uma recombinação entre eles e a conseqüente emissão de luz. Estudos
recentes têm mostrado que o rendimento quântico desses processos é altamente dependente
não só da estrutura eletrônica do polímero, mas também de sua estrutura conformacional
(Salaneck, Friend, & Brédas, 1999), efeitos supramoleculares (Brunsveld, Folmer, Meijer, &
Sijbesma, 2001) como interações inter- e intra-cadeias, presença de substituintes e,
principalmente, de defeitos estruturais(D. Wang, 2008; Gregg, 2009b; Liang, Nardes, Wang,
Berry, & Gregg, 2009; Wong et al., 2001; Yan, Rothberg, Papadimitrakopoulos, Galvin, &
Miller, 1994; Yurtsever & Yurtsever, 1999). Esses, em especial os foto- induzidos por radiação
gama em matriz polimérica conjugada, serão estudados nesse trabalho por caracterização
óptica usando uma abordagem teórica complementar, que propõe modelos de interação da
radiação com a matéria. Assim, é possível determinar como a interação ocorre, como os
defeitos são foto-criados e de que forma eles determinam as novas propriedades básicas do
material estudado.

25
1.2 – PROBLEMÁTICA
Defeitos em materiais semicondutores têm sido assunto de discussão e estudo durante
as últimas duas décadas quando, da necessidade de se miniaturizar ainda mais os dispositivos,
impurezas e distorções da cristalização prejudicavam drasticamente o rendimento do ma terial.
Primeiramente estudado em semicondutores inorgânicos, como o GaAs, InAs, InP e GaP, Si e
Ge, os defeitos também se mostraram como ferramentas para alterar a estrutura eletrônica
desses semicondutores pela injeção de elétrons / buracos na estrutura eletrônica deles, por um
processo de dopagem, favorecendo o caráter do tipo p ou n desses materiais. Além disso,
essas imperfeições da estrutura cristalina foram encaradas como forma de melhorar
características quando o factível parecia ser o extremo oposto. Esse é o caso recente, a título
de exemplificação, do Óxido de Háfnio (HfO) (Milgrom, 2007), material que mesmo não
sendo semicondutor, é utilizada na forma cristalina como camada isolante de transistores em
microprocessadores. Utilizado em processadores recém-lançados pela Intel®, a linha i3, i5 e
i7, essa camada de óxido cristalino tem sua constante dielétrica aumentada muitas vezes
quando defeitos pontuais de Silício são inseridos na estrutura, o que provoca uma amorfização
da estrutura, com perca da „memória de cristal‟ pelo bulk de HfO, aumentando ainda mais o
desempenho dessas estruturas, apresentando uma alta constante dielétrica, sem formação de
fronteiras de grão e sem canais de fuga de corrente.
Já os defeitos em semicondutores orgânicos também têm sido estudados pela literatura
(Gregg, 2009b; Liang et al., 2009; Yan et al., 1994), mas, por serem bem mais comuns de
ocorrerem do que nos semicondutores inorgânicos, tendo em vista os processos de síntese,
manipulação, device manufacturing e caracterização, o foco dos estudos têm sido em como
eles podem alterar e definir o desempenho desses materiais, principalmente os poliméricos.
Alguns autores acreditam (Liang et al., 2009) que determinados defeitos estruturais não-
carregados, quando induzidos de forma controlada, são capazes de, ao contrário do que é
esperado, aumentar a eficiência das propriedades elétr icas de polímeros π-conjugados. Um
exemplo relatado recentemente é o de filmes poliméricos de P3HT, ricos em defeitos
estruturais induzidos por Me2SO4 e LiH e, por conta disso, apresentam um photobleaching
menor que aqueles filmes de P3HT pristine. Além disso, a presença desses defeitos não-
carregados na estrutura polimérica é capaz de aumentar a concentração e a mobilidade de
portadores de carga, como mostrado por Liang et al. (2009). Esse efeito se deve ao fato de
que a indução de deformações da estrutura planar e trigonal sp2 da cadeia polimérica do

26
polímero pode gerar estados eletrônicos no gap que nem sempre são prejudiciais (Gregg,
2009a). Além disso, a inclusão de alguns desses defeitos pode acomodar o stress inevitável
sofrido pelo filme polimérico na produção dos dispositivos opto-eletrônicos (Liang et al.,
2009).
Entre os métodos reportados pela literatura para criação e estudo de defeitos, podem
ser citados a dopagem iônica, foto e termo-degradação, saturação e isomerização (Liang et al.,
2009). No entanto, ultimamente tem sido relatado o uso de radiação ionizante como forma de
induzir alterações estruturais e eletrônicas em macromoléculas conjugadas, como é o caso de
nanotubos de carbono (Cataldo, 2000; Chen, Wu, Liu, & Long, 2005; Crespi, Chopra, Cohen,
Zettl, & Louie, 1996; Lee & Kim, 2008; Smith & Luzzi, 2001), e outros materiais orgânicos.
A interação da radiação ionizante com materiais poliméricos tem sido uma área muito
estudada em diversos trabalhos (Chen et al., 2005; Lee & Kim, 2008; Xu, Wang, Zhang, &
Liu, 2006) envolvendo, principalmente, aplicação mecânica. No entanto, é recente a utilização
desse tipo de radiação para aplicação em materiais com potencial de aplicação eletrônica
(Graham et al., 1997). A forma de interação é relativamente conhecida pela literatura e
envolve três processos que são basicamente: efeito fotoelétrico, espalhamento Compton e
formação de pares elétrons-pósitrons pela interação de Feynmann (Reilly, 1991). Para
materiais carbonosos interagindo com radiação gama com energia inferior a 2 MeV, a
formação de pares é um processo que ocorre com uma freqüência menor que 2%. O
espalhamento elástico e inelástico, portanto, são os fenômenos predominantes e são
responsáveis por (Davenas et al., 2002) ionização, quebras de ligação,
hidrogenação/saturação, crosslinks, degradação, entre outros processos que são ainda
discutidos. Em especial, os processos de quebra de ligação serão estudados nesse trabalho
utilizando 60Co como fonte de radiação gama, cuja emissão ocorre em duas raias com energia
média de 1.25 MeV. Na faixa de dose utilizada, a radiação provoca praticamente somente
cisões na cadeia e induz crosslinkings graças à ionização de átomos reativos da cadeia. A
controlabilidade dos defeitos pode ser garantida pelo fato de que nessa faixa de dose, a taxa de
criação de defeitos é aproximadamente linear com a dose, como pode ser visualizado no
decorrer do trabalho.
Os primeiros relatos da interação de radiação ionizante, em especial a gama, com
polímeros conjugados datam da década de 80 (Kudoh, Sasuga, Seguchi, & Katsumura, 1996;
Yoshino, Hayashi, & Inuishi, 1982), com a tentativa de produzir dosímetros poliméricos de
baixo custo com parâmetro de medida mais fácil do que os até então muito usados

27
termoluminescentes inorgânicos. Os pioneiros nessa área são Burroughes e colaboradores,
estudando a interação de radiação gama com o PPV (Bernius, Inbasekaran, O'Brien, & Wu,
2000; Burroughes et al., 1990; M. D. R. Silva, Gançalves Jr, Silva, & Marletta, 2010), Graeff
e colaboradores com o MEH-PPV em soluções de solvente halogenados (gama) (E. A. B.
Silva et al., 2005) e Bianchi e colaboradores que focaram no desenvolvimento de sensores de
raios-x e ultravioleta para terapia neonatal (Ferreira et al., 2009; Vasconcelos, 2010). São
muito poucos os relatos, no entanto, do uso consciente da radiação ionizante como forma de
alterar as propriedades eletrônicas de materiais poliméricos conjugados para aplicação direta
em dispositivos (Graham et al., 1997).
Os mecanismos de interação da radiação ionizante com esse tipo de material ainda são
desconhecidos em sua totalidade e envolvem, na sua quase totalidade, a presença de radicais
foto-gerados do solvente, como tem sido relatado por Graeff et al. (2005). Em solução de
solventes orgânicos halogenados, como tetracloreto de carbono, di-bromo-metano, di- iodo-
metano e outros, o MEH-PPV tem sua estrutura atacada pelos radicais halogenados e, por
conta disso, sofre quebra da cadeia e, conseqüentemente, da conjugação, apresentando os
deslocamentos hipsocrômicos dos espectros de absorção e fotoluminescência, assim como
hipocromia. Quando em solução de benzeno, esses autores não notaram alterações nas
propriedades eletrônicas desse material, tendo, então, assumindo que a interação da radiação
com a cadeia ocorre unicamente quando intermediada por radicais. Bianchi et al., estudando
(Ferreira et al., 2009; Vasconcelos, 2010) também o MEH-PPV com UV percebeu que a
proporção dos gases O2/N2 era um parâmetro chave na mediação da interação
radiação/matéria, atribuindo, assim, os efeitos deletérios à estrutura e letrônica do polímero
conjugado unicamente à degradação foto- induzida pela presença de O2.
Nesses estudos, as alterações nas propriedades eletrônicas são devidas unicamente à
diminuição do segmento conjugado por conta dos defeitos foto- induzidos e de cisão de cadeia
e de cisão dos ramos laterais solubilizantes. Semelhante abordagem é capaz de abrir um leque
de novas possibilidades para aplicação de semicondutores orgânicos na indústria eletrônica,
criando dispositivos mais eficientes, além de unicamente o desenvolvimento de dosimetria.
Nesse trabalho, estudamos a influência da radiação gama sobre as propriedades
eletrônicas, estruturais e vibracionais do polímero conjugado derivado de tiofeno-fenileno
poli(2,5-tiofeno-1,4-dialcóxifenileno) (Bouachrine et al., 2002; Ding et al., 2001; Lois et al.,
2007; S. Bouzakraoui, 2010; R. A. Silva et al., 2006; R. A. Silva et al., 2005; Raigna A. Silva,
Serein-Spirau, Bouachrine, Lere-Porte, & Moreau, 2004) através de absorção óptica e

28
fotoluminescência. O ajuste gaussiânico dos espectros de emissão dentro do formalismo
(Marletta, Guimarães, & Faria, 2002) de transições verticais de Franck-Condon é realizado a
fim de entender como o acoplamento vibrônico é influenciado pela dose, já que um
comportamento não esperado é verificado nesses espectros. De forma a elucidar como a
interação da radiação ionizante com a matéria ocorre, apela-se para modelagem molecular por
métodos semi-empíricos AM1 (Dewar & Thiel, 1977; Dewar, Zoebisch, Healy, & Stewart,
1985) da estrutura conformacional e geometria de equilíbrio de oligômeros da cadeia
polimérica conjugada, visando entender as alterações e defeitos induzidos em função da dose.
A partir das geometrias de equilíbrio, as transições eletrônicas são simuladas também por
parametrização semi-empírica ZINDO/S-CI (Anderson, Edwards, Zerner, & Canuto, 1982;
Bacon & Zerner, 1979; Edwards & Zerner, 1987; Head & Zerner, 1985, 1986; J. Ridley &
Zerner, 1973; J. E. Ridley & Zerner, 1976) para entender como a radiação induz transições
eletrônicas.
Finalmente, com os resultados desse trabalho é possível propor uma nova rota de
tratamento de dados de polímeros irradiados, assim como uma contribuição para a literatura
de defeitos.
1.3 – FÍSICA DE MATERIAIS POLIMÉRICOS
Tendo pontuado essa síntese do panorama da aplicação de semicondutores poliméricos
no mercado tecnológico, resta-nos pontuar os elementos químico-físicos que definem o
comportamento eletrônico desses materiais. A Física de Materiais Poliméricos que será
apresentada aqui pode ser encontrada em uma literatura mais especializada 2 de forma que
satisfaça desde leigos a profissionais no assunto. É interessante ressaltar que a notação e a
nomenclatura utilizada doravante pode ser a corrente em literatura química e cabe ao leitor
realizar as „transformações‟ necessárias.
Uma cadeia polimérica é uma macromolécula, formada a partir de unidades de
repetição (meros) unidas por ligações primárias fortes, que podem ser iônicas, coordenadas,
metálicas ou covalentes, sendo este último tipo o mais comum. Estas ligações são chamadas
2 – Sebastião V. Canevarolo Jr. – Ciência dos Polímeros, 2
a Ed ição, Artliber Ed ., 2006; Elo ísa B. Mano –
Introdução a Polímeros, 2ª Ed ição, Ed igard Blücher, 1999; e Len i Akcelrud – Fundamentos da Ciência dos
Polímeros, 1ª Edição, Manole, 2006.

29
de intramoleculares, em oposição às intermoleculares, que ocorrem entre cadeias diferentes,
em função de forças secundárias fracas. Essas ligações intermoleculares podem ser: interação
de van der Waals (dipolo-dipolo e dipolo-dipolo induzido), forças de dispersão e pontes de
hidrogênio/sulfeto.
As forças intramoleculares covalentes determinam, com o arranjo das unidades de
repetição, a estrutura química e o tipo de cadeia polimérica, incluindo o t ipo de configuração.
Estas também vão influenciar na rigidez/flexibilidade da cadeia polimérica e,
conseqüentemente, do polímero, assim como na sua estabilidade térmica, química,
fotoquímica, etc. As forças intermoleculares, por sua vez, determinam decisivamente a
maioria das propriedades físicas do polímero: temperatura de fusão cristalina, solubilidade,
cristalinidade, difusão, permeabilidade a gases e vapores, deformação, etc. Quanto mais fortes
forem estas forças, maior a atração entre as cadeias, tornando-se mais difícil todo e qualquer
evento que envolva a separação e/ou fluxo de uma cadeia sobre a outra.
Outro ponto a destacar na estrutura molecular dos polímeros é a funcionalidade das
moléculas formadoras. Funcionalidade de uma molécula é o número de pontos reativos
(passíveis de reação em condições favoráveis) presentes nessa molécula. Para que uma
molécula de baixo peso molecular produza um polímero, é necessário que sua funcionalidade
seja pelo menos igual a dois. Isso significa dizer que existem no mínimo dois sítios reativos
na molécula onde a reação pode se propagar nos dois sentidos, formando uma
macromolécula. A reação de duas moléculas monofuncionais produz apenas uma ligação, com
a conseqüente formação de outra molécula, também pequena. Moléculas polifuncionais
produzem redes poliméricas tridimensionais, provocando uma rigidez da matriz polimérica.
Os polímeros formados dessa forma são conhecidos como termorrígidos e são bastante
utilizados para aplicações mecânicas.
Partindo dessa definição, as cadeias poliméricas formadas podem se apresentar de
várias formas e arquiteturas, a saber: a) cadeias lineares, cuja cadeia polimérica é constituída
apenas de uma cadeia principal e é formada pela polimerização de monômeros bifuncionais;
b) cadeias ramificadas, que a partir da cadeia principal partem prolongamentos formado pelo
mesmo mero formador da cadeia principal ou por um outro mero diferente, formando
diferentes arquiteturas; c) cadeias com ligações cruzadas, que são cadeias poliméricas que
estão ligadas entre si através de segmentos de cadeia unidos por forças primárias covalentes
fortes.

30
Do ponto de vista de aplicação tecnológica em eletrônica molecular, somente os
polímeros da primeira classe são interessantes, visto que é justamente o confinamento
eletrônico unidimensional que promove as propriedades semicondutoras dos polímeros
conjugados, conforme será explorado mais profundamente na próxima seção (1.4). Polímeros
da classe b) e c) são visados do ponto de vista de isoladores de eletricidade e para aplicações
em que a rigidez mecânica é a propriedade de interesse.
Quanto à composição da cadeia polimérica, os polímeros podem ser divididos em
homopolímeros ou copolímeros. Homopolímeros são aqueles cuja unidade de repetição é
sempre a mesma em todo o comprimento da cadeia, ao passo que copolímeros são aqueles
polímeros que possuem mais de um tipo de mero diferente formando a cadeia polimérica,
podendo eles serem distribuídos de forma aleatória (em que não há uma seqüência definida de
disposição dos meros), alternada (meros dispostos de maneira alternada), em bloco (formação
de grandes seqüências de uma dado mero alternado com outras grandes seqüências de outro
mero) e grafitizados ou enxertados (em que sobre a cadeia de um homopolímero liga-se
covalentemente outra cadeia polimérica diferente).
Antes de discutirmos como determinada estrutura química dos polímeros pode induzir
a formação de uma estrutura eletrônica semicondutora, é interessante discutirmos também a
Estrutura Molecular do Estado Sólido desse tipo de materiais, tentando entender,
previamente, como é possível realmente aplicar esses materiais em dispositivos e como suas
propriedades se conservam e/ou se alteram em estado sólido. A estrutura do estado sólido em
polímeros consiste no modo como as cadeias moleculares estão empacotadas formando a
massa sólida. Este pode ser desordenado, formando a fase amorfa; ou ordenado, regular e
repetitivo, definindo a fase cristalina. Assim, a cristalinidade em polímeros consiste no
alinhamento de segmentos de cadeias em um arranjo tridimensionalmente perfeito.
O processo de cristalização de polímeros difere dos sólidos cristalinos convencionais
devido à natureza peculiar deste se apresentar na forma de longas cadeias poliméricas. Os
domínios cristalinos, chamados “cristalitos”, são muito menores do que os cristais normais,
contêm mais imperfeições e estão interconectados com as regiões amorfas, não havendo uma
divisão clara entre regiões cristalinas e amorfas. Além disso, uma completa transformação
para o estado cristalino é impossível porque normalmente apenas uma parte da molécula
adota a conformação ordenada necessária.
A facilidade com que a cristalização ocorre depende da estrutura química, presença de

31
impurezas e condições de cristalização do polímero. Polímeros cristalizáveis típicos são os
que possuem cadeias lineares. Se tiverem ramificações ou grupos laterais, estes devem ser
suficientemente pequenos ou dispostos regularmente e simetricamente ao longo das cadeias.
Esta disposição regular, dita estéreo-regularidade (também conhecida como régio-
regularidade), é essencial para o desenvolvimento da cristalinidade. A cristalização pode ser
favorecida também pela existência de grupos que promovam fortes ligações intermoleculares
secundárias, tais como grupos polares, ou que permitam a formação de pontes de hidrogênio
entre as moléculas.
Esses detalhes sobre cristalização de estruturas poliméricas foram apresentados tendo
em vista que são evitados do ponto de vista de produção de semicondutores orgânicos.
Conforme será elucidado no próximo tópico, a presença de cristalinidade polimérica cria
canais onde os portadores de carga excitados, chamados de éxcitons, podem se difundir sem
que ocorra, de maneira eficiente, recombinação entre eles com emissão de luz. O polímero
conjugado que será utilizado nesse trabalho, no entanto, é um copolímero de tiofeno-fenileno,
alternado, com braços alcóxicos grafitizados lateralmente no anel fenilênico, com alta régio-
regularidade e que apresenta uma fase cristalográfica que ainda assim não interfere nas
propriedades eletrônicas de estado sólido da cadeia. Esse fato habilita-o ao uso em
dispositivos opto-eletrônicos orgânicos, mesmo tendo em vista o empacotamento de estado
sólido, com constância das propriedades eletrônicas. Melhores detalhes são discutidos na
seção 1.4 deste Capítulo e na seção 2.1 do Capítulo seguinte.
1.4 – FÍSICA DE POLÍMEROS SEMICONDUTORES
A fim de não tornar a leitura desse texto extensa e cansativa, será assumido que o
leitor já tenha conhecimento básico prévio de Física de Materiais Semicondutores. Não serão
apresentados aqui os elementos de Mecânica Quântica e Física de Estado Sólido que levaram
ao desenvolvimento dessa área da Física, apenas será mostrado como é possível a existência
de estados semicondutores em moléculas orgânicas a partir da Teoria do Orbital Molecular. A
teoria desenvolvida nessa seção pode facilmente ser encontrada em (Pope, 1999).
A configuração eletrônica do estado fundamental do carbono, pelo diagrama de Linus

32
Pauling, é 1s2, 2s2, 2p2. Há quatro elétrons no nível eletrônico mais externo; os dois elétrons
no orbital s são emparelhados enquanto os dois elétrons p são desemparelhados. Outra
configuração eletrônica possível para o carbono, mas menos estável, pode ser conseguida pela
„fusão‟ dos orbitais 2s e 2p, criando um conjunto de quatro orbitais equivalentes degenerados,
referidos como orbitais híbridos sp3. A energia adicional requerida para localizar os quatro
elétrons de valência em quatro orbitais sp3 de arquitetura tetraédrica é provida pelo ganho de
energia em fazer quatro ligações químicas com ligantes em torno do átomo de carbono. A
molécula de metano (Figura 1)3 exemplifica esse tipo de ligação, com a ilustração das
respectivas geometrias dos orbitais sp3 (observe que enquanto a geometria do orbital s é
esférica, a geometria do orbital p é de halteres, e a geometria do orbital híbrido é o resultado
da intersecção entre essas duas geometrias):
Figura 1 - Orbitais Moleculares da Molécula de Metano.
Também é possível que os orbitais s e p se combinem para formar três orbitais sp2
(processo conhecido como hibridização sp2) que são referidos como orbitais planares
trigonais. Nesse caso, um dos três orbitais atômicos p originais, o orbital pz, que é
mutuamente ortogonal aos outros dois orbitais, px e py, permanece inalterado enquanto os
orbitais 2s se fundem com os orbitais px e py. Esses três orbitais híbridos sp2 são todos
coplanares e direcionados de 120° um do outro. As ligações formadas a partir desses três
orbitais são chamadas de ligações σ enquanto que o orbital pz mantém-se perpendicular ao
plano formado pela hibridização sp2. O esquema de orbital híbrido provê um modelo
conveniente para entender a formação de estruturas carbonosas de dupla ligação, como é o
3 Créditos a Mart in Pope e Charles E. Swenberg – Electronic Process in Organic Crystals and Polymers, pág. 3,
2nd Edition, Oxford University Press, 1999.

33
caso do etileno (CH2= CH2) (Figura 2).
z
C
H
C
H
H
H
z
Figura 2 - Sobreposição dos orbitais pz formando o orbital π na molécula de etileno.
No entanto, o mais importante para esse trabalho é entender como ocorre a formação
de estruturas conjugadas, ou seja, de que forma a alternância entre hibridizações sp2 e sp3 na
cadeia polimérica (leia-se alternância de ligações simples e duplas) induz a formações de
bandas de energia nesse tipo de material. Para isso, consideremos, a título de simplicidade e
ilustração, a molécula de benzeno. Os orbitais pz de cada átomo de carbono são
perpendiculares ao plano que contém todos os átomos de carbono, e o espaçamento entre os
átomos de carbono é tal que existe uma sobreposição entre orbitais pz vizinhos. Essa
sobreposição, do inglês overlap, entre orbitais vizinhos cria uma ligação chamada de ligação
π. A ligação π estabelece uma delocalização (leia-se „movimentação‟) da densidade eletrônica
acima e abaixo do plano dos átomos de carbono, com nenhuma densidade eletrônica no plano
nodal coincidindo com o plano da molécula. Em contraste, os orbitais sp2 geram densidades
eletrônicas altamente localizadas no plano do anel e entre os átomos de carbono e hidrogênio.
As ligações π-delocalizadas aumentam a estabilidade da molécula de benzeno em comparação
com o cenário em que existe uma alternância fixa entre ligações simples e duplas.
O grau de delocalização e como o sistema π-eletrônico de uma molécula interage com
o de outra molécula vizinha é a chave para a compreensão de como ocorrem as excitações
coletivas nesse tipo de arquitetura molecular. Os compostos ilustrados na (Figura 3) são
exemplos do que podemos chamar como hidrocarbonetos aromáticos conjugados. O termo
conjugado refere-se à alternância da seqüência de ligações simples e duplas na molécula. O
termo aromático advém do odor característico dos compostos naturais nos quais esses

34
compostos foram primeiramente encontrados. A (Figura 4) ilustra de que forma o orbital π se
localiza nos planos perpendiculares acima e abaixo de uma molécula de antraceno (formada
de três anéis aromáticos acoplados). Os átomos de hidrogênio não são mostrados.
Figura 3 - Hidrocarbonetos aromáticos conjugados de interesse (da esquerda para a direita, de cima para baixo) :
benzeno, tiofeno, pirro l e antraceno. Observe que o anéis de tiofeno e de pirrol possuem heteroátomos e que
mes mo assim mantêm a conjugação.
Figura 4 - Disposição dos orbitais π abaixo e acima da molécu la de antraceno. Em molécu las com conjugação
maior do que essa, a delocalização abrange todo o comprimento da molécula desde que não haja interrupções por
defeitos e/ou saturações sp3.
As propriedades semicondutoras ou condutoras dos polímeros conjugados estão
relacionadas à presença da extensão do orbital , formado pela combinação das hibridizações
sp2 + pz. Sabe-se que a ligação entre os átomos de carbono é do tipo , porém, a fraca
interação van der Waals entre os planos dos orbitais pz e a extensão dos orbitais gera as
características condutoras e semicondutoras dos polímeros, dando a origem ao orbital
molecular -ligante ( ) e ao orbital molecular -antiligante ( *). Estes orbitais moleculares
também são chamados de banda de valência e banda de condução *.
A delocalização dos orbitais acima e abaixo da cadeia polimérica permite que haja

35
uma mobilidade de carga na extensão desta cadeia. Porém, para um polímero conjugado, há
diferenças na distância entre as ligações dos átomos de carbono, ou seja, para as duplas
ligações entre os carbonos da cadeia polimérica, a distância é menor do que entre os carbonos
com ligações simples, variando conforme a Figura 5.a para o poliacetileno. Esta variação entre
as distâncias promove a distorção de Peierls4 (Figura 5.b) e a abertura na estrutura de banda
após a dimerização.
a) b)
Figura 5 - Poliacetileno: a) dimerização da cadeia polimérica; (b) estrutura de banda antes e após a dimerização.
A distorção de Peierls faz com que os orbitais ocupados e os orbitais vazios * se
expandam originando orbitais moleculares análogos às conhecidas bandas de condução e de
valência (BC e BV) dos semicondutores. A largura da banda proibida ou a energia de gap
(EGAP) entre BC e BV é quem determina as propriedades elétricas intrínsecas do polímero.
No estudo de propriedades eletrônicas de polímeros conjugados, é suficiente,
conforme ficará claro no próximo Capítulo, focarmos nas propriedades dos elétrons ,
4 Distorção de Peierls ou Transição de Peierls (Rudolf Peierls, 1930), também conhecida como dimerização, é
uma distorção da rede periódica de um cristal unidimensional devido às oscilações em torno da posição de
equilíbrio das posições atômicas. O Teorema de Peierls estabelece que uma cadeia unidimensional igualmente
espaçada com um elétron por íon é instável. Isso pode ser provado usando um modelo simples do potencial para
um elétron em um cristal unidimensional de espaçamento da rede a. A periodicidade do cristal geral gaps de
energia na estrutura de bandas (diagrama E versus k ) nos múlt iplos do vetor de onda k tal que k=π/a, de maneira
similar ao modelo de Kronig-Penney. Se cada um dos íons contribui com um elétron, então a banda será meio -
preenchida, até valores de k=± π/2a no estado fundamental. Em uma distorção da rede em que cada íon se move
mais perto de um vizinho e mais distante do outro, o período da rede dobrou de a para 2a. De fato, a prova do
teorema está no fato de que dobrando o período introduz-se novos band gaps localizados em múltip los de
k=π/2a. Isso iria provocar uma pequena diminuição da energia total da distribuição eletrôn ica, baseada na
distorção das bandas na vizinhança dos novos gaps. Aproximando-se de k=π/2a pela esquerda, a distorção
devido a introdução de um novo band gap fará com que os elétrons encontrem-se em uma energia mais baixa do
que eles estariam em um cristal perfeito. Portanto, essa distorção da rede torna-se energeticamente mais
favorável quando a diminuição da energia devido a essa nova organização supera a energia elástica de rearranjo
dos íons.
- /2 a /2 a - /a /a k
k
g a p

36
excluindo a rede de ligações dos elétrons σ. Os elétrons estão nos orbitais moleculares de
mais alta energia ocupados e são, portanto, os mais facilmente excitáveis. Desde que os
orbitais desocupados de mais baixa energia também são orbitais , as transições eletrônicas
entre eles são referidas como transições *. Diversos modelos podem ser empregados
para explicar como essas transições podem ocorrer na forma de absorção óptica,
fotoluminescência e por condução de corrente elétrica. Sendo assim, as propriedades ópticas
podem ser determinadas apenas pelo estudo das bandas e * do polímero conjugado e este
assunto será melhor discutido no Capítulo II, seção 2.2.1.
De forma geral, quando um fóton de energia , onde é a constante reduzida de
Planck e é a freqüência angular do fóton, incide sobre um segmento conjugado, caso a
condição em que a energia do fóton seja maior ou igual à energia de gap entre as bandas - *,
, uma transição eletrônica entre as bandas ocorrerá 5. Isso significa dizer que um
elétron da banda de valência foi promovido para a banda de condução * e, pelo Princípio
de Conservação da Carga, uma „lacuna‟ foi deixada para trás na banda de valência. A essa
lacuna de carga dá-se o nome de buraco (hole, do Inglês), h+, e ela possui mobilidade na
banda de valência assim como o elétron possui na banda de condução. No entanto, a massa
específica deles é diferente e essa diferença define se o polímero semicondutor é
preferencialmente um condutor de elétrons ou de buracos. A maioria dos polímeros
conjugados que são estudados pela literatura são condutores preferenciais de buracos, que
possuem maior mobilidade do que os elétrons, os quais sentem um potencial da rede cristalina
maior do que os portadores de carga positiva. Quando gerado, o par elétron-buraco mantém-se
ligado devido à atração eletrostática entre eles, formando uma quasipartícula chamada de
éxciton. Sendo formados, os éxcitons possuem mobilidade pelo material e podem ser
dissociados na presença de potenciais elétricos capazes de vencer a energia coulombiana
atrativa. Tanto o efeito de fotoluminescência quanto o de eletroluminescência consistem-se na
dissociação de éxcitons: na fotoluminescência, o par elétron-buraco é gerado pela absorção de
um fóton, que quando dissociado, recombina-se e emite luz e na eletroluminescência, a
injeção de portadores gera o par, que quando dissociado e recombinado, também emite
5 Quando essa condição não é satisfeita, ainda assim o processo de absorção pode ocorrer, por meio de um
fenômeno conhecido como absorção de múltip los fótons. Quando dois ou mais fótons com energia menor do que
o gap interagem com o material semicondutor, separados temporalmente por um intervalo de tempo muito curto
(da ordem de 10-15
s), o primeiro fóton pode levar o elétron para um nível virtual no interior do gap e o segundo
fóton excita-o para a banda de condução. Existe um estudo sobre absorção de dois fótons para o polímero FSE59
e pode ser encontrado em M. G. Vivas, S. L. Nogueira, H. Santos Silva et al. The Journal of Physical Chemistry
A – submetido em 06/04/2011.

37
radiação luz.
No entanto, outro processo ocorre quando um elétron é excitado da banda de valência
para a de condução: que é o processo de ionização da cadeia polimérica. O buraco deixado
para trás na banda de valência possui delocalização e por ser um portador de carga ele
provoca uma deformação estrutural da rede, que sofre alteração conformacional para
comportar energeticamente esse „defeito‟. A essa quasipartícula, que existe concomitante ao
éxciton, damos o nome de pólaron. Quando, em um sítio próximo de onde ocorreu a primeira
excitação, ocorre uma segunda, o par de pólarons forma um estado ligado entre eles,
provocando uma forte distorção do retículo local cuja energia é superior à repulsão
coulombiana entre as quasipartículas. À esse novo estado de oxidação, dá-se o nome de
bipólarons.
A formação de pólarons e bipólarons cria novos estados de energia permitidos dentro
do gap, permitindo a ocorrência de novas transições eletrônicas de menor energia que antes
eram proibidas. Note que, além do fato dessas transições serem de menor energia, como
veremos no Capítulo seguinte, elas são intensificadas ainda por uma „quebra‟ temporária de
algumas regras de seleção para o processo de absorção, devido à distorção sentida pelo
retículo (spin-flip). A Figura 6 ilustra como a formação de pólarons, bipólarons e bandas de
bipólarons podem ser formadas no polipirrol, como ilustrado por Foschini, 2009.
Figura 6 - a) fo rmação de pólarons na cadeia de polipirrol, com representação dos novos níveis energéticos
criados no interior do gap; b) formação de bipólarons, que criam níveis cuja diferença de energia é ainda menor
do que os pólarons e c) formação de bandas de bipólarons, que acontecem quando ocorre superoxidação da
cadeia, induzindo a formação de vários bipólarons próximos espacialmente, que fo rmam um semi-contínuo de
estados bipolarônicos.

38
CAPÍTULO II
Nesse capítulo será apresentada a abordagem teórica utilizada na parte experimental e
teórica desse trabalho. Serão também apresentados os detalhes de síntese, caracterização
teórica e experimental do polímero, assim como de seu oligômero. Em caso de dúvida do
leitor, é indicado uma literatura auxiliar para a compreensão. A abordagem dos métodos
teóricos aqui apresentados pode ser encontrada também em uma referência especializada 6.
2 – METODOLOGIA
2.1 – MATERIAL E MÉTODO
2.1.1 – O POLÍMERO CONJUGADO FSE59
Os politiofenos têm sido uma classe de polímeros conjugados estudados desde a
década de 90, visto que devido à alta delocalização eletrônica na cadeia, eles apresentam
propriedades de interesse em condução, luminescência e cromismo. Dentre a classe de
polímeros derivados de tiofeno, podemos destacar o politiofeno (P T) em si, o poli(tiofeno-
vinileno) (PTV) e, principalmente, os poli(3-alquil- tiofenos) (P3AT), que têm se mostrado
bastante apropriados para aplicação em dispositivos opto-eletrônicos orgânicos, tendo em
vista sua maior solubilidade em solventes orgânicos providenciada pela grafitização da cadeia
principal pelos ramos alquílicos. Uma das características mais interessantes desses derivados
é justamente a capacidade de cromismo, ou seja, alteram drasticamente sua cor dependendo
das condições ambientais em que se encontram, devido à temperatura (termocromismo),
solvente (solvatocromismo) e potencial aplicado (eletrocromismo), o que os habilita
principalmente para aplicações em sensores, como tem sido extensivamente estudado pela
6 José David M. Vianna, Adalberto Fazzio e Sy lvio Canuto – Teoria Quântica de Moléculas e Sólidos, 1
a Ed ição,
Livraria da Física Ed., 2004; Efthimios Kaxiras – Atomic and Electronic Structure of Solids, 1ª Edição,
Cambridge, 2003; e Andrew R. Leach – Molecular Modelling – Principles and Applications, 2ª Ed ição, Prentice
Hall, 2001.

39
literatura, inclusive pela Profa. Débora Balogh, do Instituto de Física de São Carlos, USP, e
pelo nosso próprio grupo.
No entanto, os politiofenos apresentam um rendimento quântico de fotoluminescência
(Pei, Yu, Huang, & Heeger, 2000) e, conseqüentemente7, de eletroluminescência muito baixo
(~1-3%) e, portanto, não é considerado viável para aplicação em dispositivos eletrônicos de
emissão de luz. Esse fato é devido ao processo de difusão tridimensional de éxciton entre as
cadeias formadoras do bulk, impedindo que pares elétrons-buracos se recombinem e decaiam
radiativamente e, além disso, é atribuído também à alta taxa de cruzamento intersis temas de
níveis de estado singleto para estado tripleto (spin-flip).
Algumas alternativas têm sido propostas para contornar essa baixa eficiência quântica
em processos luminescentes dos politiofenos e uma delas é a copolimerização deles com
monômeros fenilênicos (Pei et al., 2000), o que produziria polímeros conjugados baseados no
politiofeno, mantendo suas propriedades elétricas e de estabilidade ambiental tanto em estado
neutro como dopado, ao passo que as suas propriedades ópticas seriam aumentadas
consideravelmente. Essa abordagem acoplada à grafitização do anel tiofênico é capaz ainda de
fornecer boa solubilidade aos polímeros resultantes, o que é essencial para as propriedades
emissivas desse tipo de material. Essa abordagem foi primeiramente proposta por Heeger e
colaboradores (Pei et al., 2000) e gera polímeros cuja cadeia principal tem a estrutura
apresentada na Figura 7 :
Figura 7 - Proposta de Heeger e colaboradores para aumentar a eficiência quântica de emissão dos politiofenos.
No entanto, percebeu-se que essa abordagem, além de ainda assim produzir polímeros
com baixa eficiência quântica de emissão, os grupamentos conectados ao anel tiofênico
induziam fortes interações estéricas8 entre as cadeias, propiciando a cristalização. Uma leve
7 Isso é devido ao fato de que aproximadamente apenas 1/4 dos pares elétrons -buracos formados durante a
absorção de um fóton ou pela injeção de portadores possuem mesmo momento e, conseqüentemente, podem se
recombinar emitindo luz. Por conta disso, é costume d izer que o rendimento quântico de eletroluminescência,
, é menor ou igual a 25% do rendimento quântico de fotoluminescência, . 8 Os efeitos estéricos advêm do fato de que cada átomo dentro de uma molécula ocupa uma determinada

40
modificação da estrutura apresenta Figura 7 permitiu que, retirando os grupos R3 e R4, e
deixando somente grupos alcóxicos conectados ao anel fenilênico, o rendimento de emissão
pode chega a 74% (de fotoluminescência) (R. A. Silva et al., 2006). Partindo dessa estrutura
então que a classe de copolímeros conjugados derivados de poli(tiofeno-fenileno) foi pela
primeira vez sintetizada no início de 1998.
O copolímero conjugado FSE59 é sintetizado pela rota organometálica de
acoplamento cruzado catalisada por Paládio, desenvolvida pelo químico norte-americano
Richard F. Heck (Delaware University, Estados Unidos) e pelos químicos japoneses Ei- ichi
Negishi (Purdue University, Estados Unidos) e Akira Suzuki (Universidade de Hokkaido,
Japão), trabalho esse iniciado por Heck na década de 1970. Essa rota, ganhadora do prêmio
Nobel de Química de 2010 (Foundation, 2010), é responsável pela síntese orgânica de
moléculas conjugadas utilizadas na agro- indústria e na síntese de fármacos e, recentemente,
começou a ser utilizada na produção de moléculas conjugadas complexas emissoras de luz.
A rota de síntese consiste na reação entre 2,5-bis-(tributil-estanil)-tiofeno com 1,4-
dibromo-2,5-bis-(dióxi-3,6-decilóxi)-benzeno, na presença de uma mistura 1:1 (v/v) de
tetrahidrofurano (THF) com dimetilformamida (DMF), destilados e peneirados
molecularmente utilizando como catalisador o tetra(trifenil- fosfato) de paládio (Pd(PPh3)4). A
reação apresenta rendimento variando de 84 a 90%. Esta rota está simplificada na Figura 8.
Figura 8 - Rota de síntese do polímero conjugado FSE59.
Após sintetizado, o polímero foi caracterizado9 por Ressonância Magnética Nuclear de
Hidrogênio e de Carbono (1H e 13C RMN), cromatografia de permeação em gel (GPC) para
quantidade de espaço que associa uma energia de sobreposição das nuvens eletrônicas de outros átomos e são
capazes de alterar propriedades conformacionais da cadeia, assim como aumentar ou diminuir sua reatividade. 9 As técnicas de caracterização aqui apresentadas não são discutidas por não fazerem parte da proposta deste
trabalho. São utilizados dados fornecidos pelo grupo autor da síntese e semelhantes medidas não foram
realizadas pelo autor deste texto. Mais informações sobre essas técnicas de caracterização podem ser encontradas
em T. W. Graham Solomons – Química Orgânica, 2ª Edição, Ed itora LTC, 2009.

41
determinação do peso molecular, difração de raios-X (XRD), voltametria cíclica (CV),
absorção óptica (OA), fotoluminescência (PL), Espalhamento Raman e absorção de
infravermelho por transformada de Fourier (FT-IR). Detalhes da caracterização podem ser
encontrados na referência (Raigna A. Silva et al., 2004).
Por hora, no entanto, convém-nos10 ater à cristalinidade do polímero estudada por
medidas de difração de raios-X. Sabe-se que as propriedades físicas e de aplicação de
polímeros conjugados é altamente dependente do grau de cristalinidade e observa-se que altas
condutividades com dopagem são obtidas para materiais a ltamente organizados. Ao contrário,
altos rendimentos quânticos de fotoluminescência e, conseqüentemente, de
eletroluminescência exigem uma baixa cristalinidade, a fim de se evitar a difusão
tridimensional de éxcitons e confinar unidimensionalmente a mobilidade dos portadores.
A ordem estrutural de P3AT tem sido estudada por difração de raios-X (A. Bolognesi
et al., 2004; Alberto Bolognesi, Botta, & Porzio, 2001). Para amostras régio-regulares que
contém apenas ligações cabeça-cauda, os padrões de raios-X são consistentes com estruturas
lamelares11 bem organizadas que distanciam uma cadeia principal da outra apenas pelo ramo
alquílico da cadeia. A distância de espaçamento D entre as cadeias de P3AT depende do
tamanho da cadeia alquílica e da sua interdigitalização. Os copolímeros sintetizados pela rota
descrita acima apresentam alta régio-regularidade, assim como os derivados de PT e
apresentam padrões de raios-X similares a esses. No caso especial desse polímero estudado
nesse trabalho, o FSE59, um pico de difração intenso e bem-definido é observado para
ângulos pequenos (2θ=4,0º), significando uma distância D de aproximadamente 22 Å, e um
pico largo próximo de 2θ=23º, o que indica uma distância para o estaqueamento12,13 π de 3,8
Å com uma larga dispersão em torno de um valor médio, que está consistente com resultados
de previsão teórica (3,75 Å) realizados pelo autor desse trabalho e que não são apresentados
aqui. Para essa distância medida D, é interessante ressaltar que ela é justamente o
comprimento da cadeia alquílica lateral, que, assumindo a coplanaridade das ligações C-O e
10
A discussão realizada doravante pelos próximos capítu los pode parecer ao leitor um pouco cansativa e sem
sentido, mas ela será pertinente à análise dos resultados, no Capítulo IV. 11
Só lidos lamelares são sólidos com estrutura em folhas. Isso quer dizer que seus átomos estão ligados
fortemente em duas direções formando lâminas contínuas e fracamente ligadas por interação de van der Waals na
direção perpendicular a estas lâminas. 12
A palavra estaqueamento pode ser considerada um neologis mo. 13
Estaqueamento π (π-stacking) é o empilhamento sofrido por moléculas π-conjugadas tendo em vista a
interação por fo rças dispersivas entre esses elétrons. O comportamento da energia de interação é do t ipo
Lennard-Jones com correção de dipolo induzido (Potencial de Stockmayer) e as forças responsáveis são do tipo
de dispersão e de van der Waals.

42
C-C é de 13 Å. Esse valor para essa distância implica que as cadeias alquílicas são
interdigitadas por uma distância de 4 Å, que corresponde ao tamanho do grupo butílico final.
Com a variação do braço alcóxico lateral conectado ao anel fenilênico, assim como foi
também sintetizado pelos responsáveis pela síntese do FSE59, nenhum padrão de cristalização
é notado, nem como arranjo lamelar das cadeias. A Figura 9 ilustra essa interação intercadeias.
Figura 9 - Caracterização estrutural da organização da cadeia polimérica do FSE59: D representa a distância
intercadeias e d é a separação interdigital entre elas.
Como visto, a estrutura de Estado Sólido do polímero estudado é semi-cristalina, com
fases lamelares definidas pelas interações entre os grupos butílicos terminais do ramo
dialcóxico. Percebe-se que a distância intercadeias é considerada alta o que mantém o
confinamento eletrônico unidimensional, não afetando a taxa de recombinação excitônica.
Será discutido no Capítulo IV de que forma a interação da radiação ionizante com esse
polímero pode diminuir esse grau de cristalização lamelar, o que indica que possíveis
rendimentos quânticos superiores de fotoluminescência podem ser atingidos.
As propriedades elétricas foram estudadas pela técnica de voltametria cíclica 14, em
filmes depositados sobre eletrodos de platina a partir da solução polimérica em clorofórmio
(CHCl3), usando como solução suporte acetonitrila (CH3CN) e como eletrólito
hexaflúorfosfato de tetrabutilamônio (Bu4NPF6). O potencial de oxidação encontrado na
14
Técnica que consiste-se na aplicação de um potencial elétrico por um eletrodo conhecido como trabalho, cuja
corrente é aquisicionada por um eletrodo chamado de contra-eletrodo e as flutuações das curvas eqüipotenciais
na solução eletrolítica são corrigidas pelo eletrodo de trabalho a partir da diferença de potencial entre ele e a
própria solução que é medida por um eletrodo conhecido como de referência. Essa técnica fornece informações
acerca do comportamento de oxi-redução do material, assim como informações de potencial de ionização,
eletroafin idade, energia de HOMO/LUMO e gap.
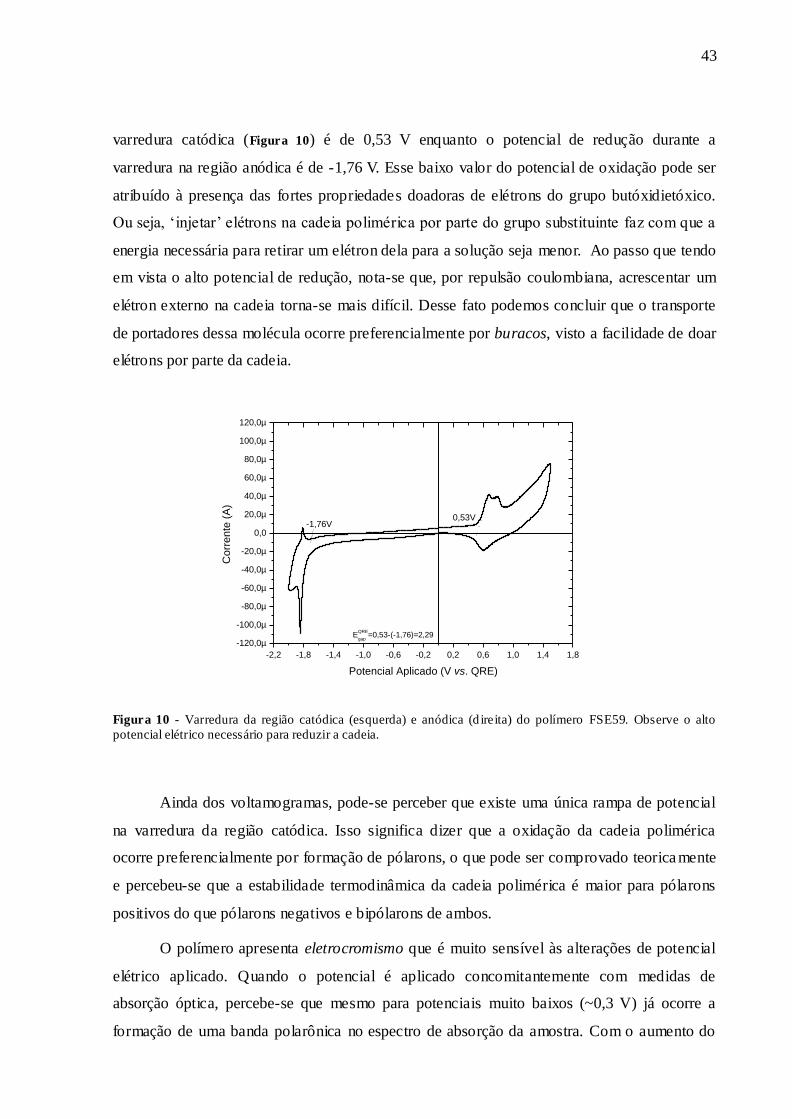
43
varredura catódica (Figura 10) é de 0,53 V enquanto o potencial de redução durante a
varredura na região anódica é de -1,76 V. Esse baixo valor do potencial de oxidação pode ser
atribuído à presença das fortes propriedades doadoras de elétrons do grupo butóxidietóxico.
Ou seja, „injetar‟ elétrons na cadeia polimérica por parte do grupo substituinte faz com que a
energia necessária para retirar um elétron dela para a solução seja menor. Ao passo que tendo
em vista o alto potencial de redução, nota-se que, por repulsão coulombiana, acrescentar um
elétron externo na cadeia torna-se mais difícil. Desse fato podemos concluir que o transporte
de portadores dessa molécula ocorre preferencialmente por buracos, visto a facilidade de doar
elétrons por parte da cadeia.
-2,2 -1,8 -1,4 -1,0 -0,6 -0,2 0,2 0,6 1,0 1,4 1,8
-120,0µ
-100,0µ
-80,0µ
-60,0µ
-40,0µ
-20,0µ
0,0
20,0µ
40,0µ
60,0µ
80,0µ
100,0µ
120,0µ
-1,76V
Co
rre
nte
(A
)
Potencial Aplicado (V vs. QRE)
0,53V
EQRE
gap=0,53-(-1,76)=2,29
Figura 10 - Varredura da região catódica (esquerda) e anódica (d ireita) do polímero FSE59. Observe o alto
potencial elétrico necessário para reduzir a cadeia.
Ainda dos voltamogramas, pode-se perceber que existe uma única rampa de potencial
na varredura da região catódica. Isso significa dizer que a oxidação da cadeia polimérica
ocorre preferencialmente por formação de pólarons, o que pode ser comprovado teorica mente
e percebeu-se que a estabilidade termodinâmica da cadeia polimérica é maior para pólarons
positivos do que pólarons negativos e bipólarons de ambos.
O polímero apresenta eletrocromismo que é muito sensível às alterações de potencial
elétrico aplicado. Quando o potencial é aplicado concomitantemente com medidas de
absorção óptica, percebe-se que mesmo para potenciais muito baixos (~0,3 V) já ocorre a
formação de uma banda polarônica no espectro de absorção da amostra. Com o aumento do

44
potencial, ocorre uma mudança brusca do espectro de absorção que pode ser entendido de
maneira análoga a uma „transição de fase de primeira ordem‟. Ou seja, a banda polarônica se
torna responsável pela atividade óptica do polímero, ocorrendo justamente em menores
energias do que as bandas puramente eletrônicas, o que representa de fato a criação de níveis
permitidos no meio do gap. A Figura 11.a) (extraída de Silva, 2004) mostra as medidas de
absorção UV-VIS in situ enquanto a Figura 11.b) mostra a diferença visual da amostra
polimérica que pode ser notada quando essa „transição de fase‟ ocorre. Essa técnica será
apresentada com mais detalhes na seção 2.2.1. Observe, também, nessa mesma medida, a
presença de um ponto fixo nos espectros de absorção em função do potencial, ponto este
conhecido como isobéstico. Este ponto fixo é capaz de separar o comportamento do espectro
em duas bandas muito bem definidas, que podem ser tratadas como bandas polarônicas,
decorrentes de transições entre níveis induzidos por pólarons no gap de energia (à direita do
ponto) e como bandas puramente eletrônicas, que são fruto de transições entre orbitais
eletrônicos somente.
a) b)
Figura 11 - a) UV-VIS in situ com aplicação de potencial, observando crômico em função do potencial e b)
solução antes (esquerda) e após (direita) a aplicação de potencial elétrico.

45
2.1.2 – A INTERAÇÃO RADIAÇÃO COM MATÉRIA: RADIAÇÃO GAMA DE 60Co.
Ondas eletromagnéticas podem interagir com a matéria por diversas formas já
relatadas pela literatura. Como o objetivo desse trabalho é contribuir para o entendimento de
que forma essa interação se processa com meio moleculares, podemos citar alguns efeitos
resultantes dessa interação, com a respectiva faixa de freqüência da radiação: transições entre
níveis rotacionais em moléculas assim como ressonância paramagnética em elétrons
desemparelhados (microondas), ressonância magnética nuclear (rádio), transições eletrônicas
de elétrons de valência (ultravioleta e visível), transições eletrônicas de elétrons de caroço
(raios-X), transições entre níveis vibracionais (infravermelho), efeito fotoelétrico (ultravioleta
e raios-X), efeito Mössbauer (para complexos organometálicos), efeito Compton (raios-X e
gama), quebra de ligações químicas (ultravioleta, raios-X e radiação gama), isomerização
(visível e infravermelho), etc.
Em especial, para a geração de radiação gama, que é o foco desse trabalho, é
necessário que qualquer que sejam os processos de decaimento radiativo que os geram, estes
devem ser altamente energéticos, já que fótons gama possuem energia da ordem de keV a
MeV. Para conseguir acessar tais energias, os níveis de energia devem ser suficientemente
afastados e nem ao menos correspondem às diferenças de energia em níveis eletrônicos. Tais
níveis energéticos só são alcançados por núcleos e assim a radiação gama nada mais é do que
os fótons emitidos no processo de decaimento do estado excitado de um núcleo.
Esses decaimentos radiativos (que emitem radiação) costumam ocorrer em geral
acompanhados de decaimentos radioativos (que apresentam atividade comparada com
decaimentos do elemento Rádio, ou seja, que apresentem radioatividade). Entre esses
processos, podemos citar como os mais eficientes o decaimento beta tanto do Césio-137 como
o do Cobalto-60. O decaimento do 137Cs por emissão beta tem tempo de meia-vida de 1,17
anos e converte-se em 137Ba que libera a energia restante acumulada no núcleo pela emissão
de um fóton gama. Enquanto que o decaimento do 60Co, que é sintético, se dá também pela
emissão beta, decaindo em um núcleo de 60Ni, com meia-vida de 5,27 anos, que também
relaxa radiativamente com emissão de um fóton gama com energia média de 1,25 MeV. Diz-
se energia média porque, de fato, o decaimento ocorre via dois processos monocromáticos
com fótons com energias de 1,173 MeV e 1,332 MeV. É interessante ressaltar dois fatos
envolvendo esses radioisótopos: 1) ambos são utilizados para aplicação em radioterapia,
sendo o Cobalto preferido tendo em vista que o seu tempo de meia-vida é maior do que o do

46
Césio e 2) o Cobalto-60 é um dos maiores causadores de estragos em detonações atômicas de
fissão nuclear, como as bombas de Urânio, Neptúnio e Plutônio: os nêutrons liberados nesses
processos de fissão, quando interagem com Ferro-58 (abundância natural de 0,28%, mas que
estão presentes nas hemácias de humanos) convertem-no em 60Co, prolongando ainda mais os
efeitos da radiação, processo que é conhecido como dirty bomb (bomba suja).
Nesse trabalho, tendo em vista a disponibilidade de uma planta de irradiação no
Instituto Nacional de Pesquisas Energéticas e Nucleares, optou-se por trabalhar com a
interação da radiação gama com fótons emitidos por Cobalto, ao passo que são extremamente
monocromáticos e assim os efeitos da radiação podem ser estudados com o parâmetro energia
dos fótons mantido constante.
As amostras foram irradiadas com duas doses15 bem definidas de 20 e 40 kGy, a uma
taxa de dose de 1,98 kGy.h-1, assim escolhidas tendo em vista um objetivo inicial desse
trabalho que era o estudo concomitante de compósitos de FSE59 com nanotubos de carbono
irradiados. No entanto, tal linha de pesquisa não se mostrou mais atraente tendo em vista a
completa perda de definição das propriedades eletrônicas dos nanotubos após a irradiação e
assim se preferiu estudar a influência dela somente na cadeia polimérica.
5 mL de solução com concentração de 0,04 mg/mL foram irradiadas com 20 e 40 kGy
de dose durante esse trabalho. Disso, a massa de polímero em cada amostra foi de 0,2 mg e,
considerando que o peso molecular do monômero seja de aproximadamente 478,65 g/mol,
tem-se uma quantidade total de monômeros de aproximadamente 4,17.10-7 moles. Irradiando
as amostras com 20 e 40 kGy, obtém-se que a quantidade de energia absorvida pelas amostras
irradiadas com 20 kGy é de 2,5.1016 eV e de 5,0.1016 eV para as de 40 kGy. Considerando a
quantidade de moles de monômeros, tem-se que a densidade aproximada da distribuição de
energia é de aproximadamente 0,10 eV/monômero para a primeira dose e 0,20 eV/monômero
para a segunda. De forma melhor apresentada, temos uma distribuição da densidade de
energia absorvida pelo sistema de 2,31 kcal/mol e 4,62 kcal/mol para as duas doses. Veremos
posteriormente que essas energias absorvidas, descontando os efeitos de solvente, são
justamente as energias necessárias para que ocorram determinadas cisões na cadeia
polimérica.
15
Pode-se definir dose absorvida de radiação pela unidade Gray (Gy) como sendo a quantidade de energia em
Joules (J) absorvida por 1 kilograma (kg) de matéria. Sendo assim, 1 Gy = 1 J/1 kg.

47
2.2 – EXPERIMENTAL
2.2.1 – ABSORÇÃO ÓPTICA
Quando uma radiação contínua passa por um material transparente, uma porção da
radiação pode ser absorvida. Se isso ocorre, a radiação residual, quando passa por um prisma
ou por uma grade de difração, por exemplo, resulta em um espectro com hiatos de freqüência
chamado de espectro de absorção. Como um resultado da absorção de energia, átomos ou
moléculas passam de um estado de baixa energia (o estado inicial ou chamado de estado
fundamental) para um estado de maior energia (o estado excitado). Figura 12 mostra como esse
processo de excitação, que é quantizado, ocorre entre dois níveis.
Figura 12 – Esquema do processo de excitação.
No caso da espectroscopia de absorção no ultravioleta e visível, as transições que
resultam na absorção da radiação eletromagnética nessa região do espectro são transições
entre níveis eletrônicos de energia. Conforme a molécula absorve energia, um elétron é
promovido de um orbital ocupado para um orbital desocupado de maior energia potencial.
Geralmente, a transição mais provável de ocorrer é do orbital molecular mais alto ocupado
(Highest Occupied Molecular Orbital – HOMO) para o orbital molecular mais baixo
desocupado (Lowest Unoccupied Molecular Orbital – LUMO).
Para a maioria das moléculas, os orbitais ocupados de menor energia são os orbitais σ,
que correspondem às ligações σ. Os orbitais π encontram-se em níveis de energia ligeiramente
superiores, enquanto que os orbitais ocupados por elétrons não compartilhados, os orbitais
não-ligantes (n) encontram-se ainda em maiores energias. Os orbitais desocupados, também
conhecidos de orbitais anti-ligantes (π*e σ*) são os orbitais de mais alta energia. A Figura 13
mostra uma típica progressão que pode ocorrer nesse tipo de moléculas, como também as
diferentes possibilidades de transições em função da energia e do tipo de composto que dá

48
origem às bandas de absorção. Observe que nos preocuparemos nesse trabalho apenas com as
transições π π*, que ocorrem em alquenos.
Figura 13 - Níveis eletrônicos e transições.
É importante fazer duas ressalvas: a primeira é que apenas as transições de mais baixa
energia são importantes, ou seja, para o caso de compostos conjugados, apenas as transições
π π* o são (e não π π*+1, que é notado por HL+1, por exemplo) e que nem todas as
transições apresentadas no diagrama de níveis eletrônicos são observadas. Certas restrições,
chamadas de regras de seleção devem ser consideradas. Uma das mais importantes dessas
regras afirma que transições que envolvam uma troca do número quântico de spin durante
uma transição eletrônica não são permitidas; a essas transições damos o nome de transições
proibidas. Outra regra de seleção estabelece o número de elétrons que pode ser excitado de
uma única vez, com propriedades de simetria da molécula e de estados eletrônicos e com
outros fatores16. Transições que são formalmente proibidas pelas regras de seleção são
freqüentemente não observadas nos espectros de absorção. No entanto, como o tratamento
teórico é muito aproximado, em certos casos essas transições são observadas, embora a
intensidade da absorção tende a ser muito menor do que para transições permitidas pelas
regras de seleção.
Para um átomo que absorve luz na região UV-VIS, o espectro de absorção consiste de
linhas muito finas, como seria esperado para processos quantizados que ocorrem entre dois
níveis discretos de energia. Para moléculas, no entanto, a absorção óptica ocorre sobre uma
larga faixa de comprimentos de onda devido ao fato de que moléculas possuem muitos modos
vibracionais e rotacionais excitados à temperatura ambiente. De fato, as vibrações das
16
É vasta a literatura de espectroscopia disponível e exemplos podem ser citados, tal como: Manfred Hesse et al.
– Spectroscopic Methods in Organic Chemistry, 2ª Ed., Thieme Medical Publishers, 2008.

49
moléculas não podem ser totalmente congeladas mesmo a zero absoluto. Os níveis de energia
desses estados são muito próximos um dos outros e correspondem a diferenças de energias
bem menores do que aquelas de transições eletrônicas. Os níveis rotacionais e vibracionais
são então superpostos os níveis eletrônicos. Uma molécula pode então sofrer transições
eletrônicas e vibracionais-rotacionais ao mesmo tempo. Para polímeros, no entanto, tendo em
vista o grande comprimento linear da cadeia, considera-se que as barreiras de energia
potencial para uma rotação da molécula são suficientemente grandes para que esses
compostos não apresentem essas transições sob condições normais. Os níveis vibracionais, no
entanto, são mais facilmente atingidos e as transições eletrônicas, na maioria das vezes,
apresentam resolução suficiente para que se detecte pelo espectro de absorção as transições
vibracionais. Conforme será visto no tópico seguinte, essas transições vibracionais são ainda
mais perceptíveis no processo inverso ao de absorção, que é o de luminescência.
Pelo fato de haver muitas transições possíveis, separadas por diferenças de energias
pequenas, cada transição eletrônica consiste de um vasto número de linhas espaçadas que os
espectrofotômetros não são capazes de resolvê- las. Assim, o instrumento traça um „envelope‟
sobre o padrão de absorção inteiro. O que é observado para esse tipo de transições
combinadas é que o espectro de absorção de uma molécula usualmente consiste de bandas
largas de absorção centradas próximo do comprimento de onda da principal transição naquela
região.
Por se tratar de um processo ressonante entre dois níveis discretos de energia, ou, no
caso de moléculas com bandas eletrônicas dispersas, entre bandas de energias, o processo de
absorção de um fóton por um cromóforo17 pode ser modelado por um oscilador harmônico,
cujo poço de potencial parabólico é corrigido para anarmonicidade, resultando então em um
poço com potencial do estilo Morse. Por essa aproximação, fica entendido que a absorção de
energia de um fóton faz o papel da força externa em um sistema massa-mola, cuja solução das
equações diferenciais acopladas do problema é uma curva chamada de lorentziana. Para o
caso da absorção óptica, sabemos que os níveis energéticos são discretos e o modelo pode
ainda ser entendido como o oscilador harmônico quântico, enquanto que a solução lorentziana
17
Por mais que a absorção seja o resultado da excitação de elétrons do estado fundamental para o estado
excitado, os núcleos dos átomos nas ligações químicas que formam a molécu la desempenham um papel
importante determinando a região onde a absorção ocorrerá. Os núcleos determinam a força com a que os
elétrons são presos e, portanto, no espaçamento de energia entre o estado fundamental e o estado excitado. Logo,
a energia característica de uma transição eletrônica e o comprimento de onda da radiação absorvid a são
propriedades de um grupo de átomos ao invés de apenas dos elétrons sozinhos. A esse grupo de átomos que
produz tal absorção, damos o nome de cromóforo.

50
continua válida. Por esse modelo, entende-se que tanto a convolução como a deconvolução de
um espectro de absorção devem ser feitas por meio de funções lorentzianas, já que são elas
que explicam as transições eletrônicas por um modelo físico que melhor descreve o processo.
Alguns autores, em contrapartida, afirmam que os espectros de absorção devem ser
convoluídos/deconvoluídos por funções do tipo gaussianas, que são soluções para o modelo
físico em que tratam uma distribuição estatística gaussiana dos segmentos absorvedores de
radiação, assim como a distribuição de carga na molécula também possui uma forma
gaussiana. De forma geral, convencionou-se utilizar nesse trabalho uma deconvolução
loretziana dos espectros de absorção experimentais, assim como uma convolução também
lorentziana das transições eletrônicas calculadas teoricamente para a formação do espectro de
absorção teórico. Entende-se que essa forma de análise dos dados fornece as energias
aproximadas dos níveis entre os quais as transições ópticas ocorrem, ao passo que um
tratamento gaussiano do espectro de absorção é capaz de fornecer, somente, a forma da
polidispersão de segmentos conjugados de moléculas absorvedoras que constituem o bulk,
mesmo que o(s) máximo(s) da(s) gaussiana(s) corresponda(m) ao(s) máximo(s) da(s)
lorentziana(s) utilizada(s) no ajuste espectral ou no envelopamento das transições.
Antes de finalizar a apresentação desse tópico, é interessante destacar que, do ponto de
vista didático, o processo de absorção óptica, entendido como a transição eletrônica entre dois
níveis intermediada por um fóton, pode ser reinterpretado, no espaço das posições, como
sendo a alteração da „posição‟ da nuvem eletrônica em determinada região da molécula. Ou
seja, tomando por base o monômero do polímero conjugado estudado nesse trabalho, o seu
orbital de fronteira do estado fundamental, o HOMO, possui uma distribuição espacial da
densidade eletrônica π conforme a mostrada pela Figura 14(a), enquanto que o orbital de
fronteira de mais baixa energia do estado excitado, já possui a distribuição conforme a Figura
14(b). Observe que o processo de absorção óptica nada mais é do que o fato de ceder uma
quantidade de energia bem-definida (quantizada) para que a nuvem eletrônica „altere sua
posição‟ na molécula, passando da situação da (a) para a da (b) (ou, como uma transição entre
dois „níveis‟ apresentados na distribuição da densidade de estados eletrônicos (c)).
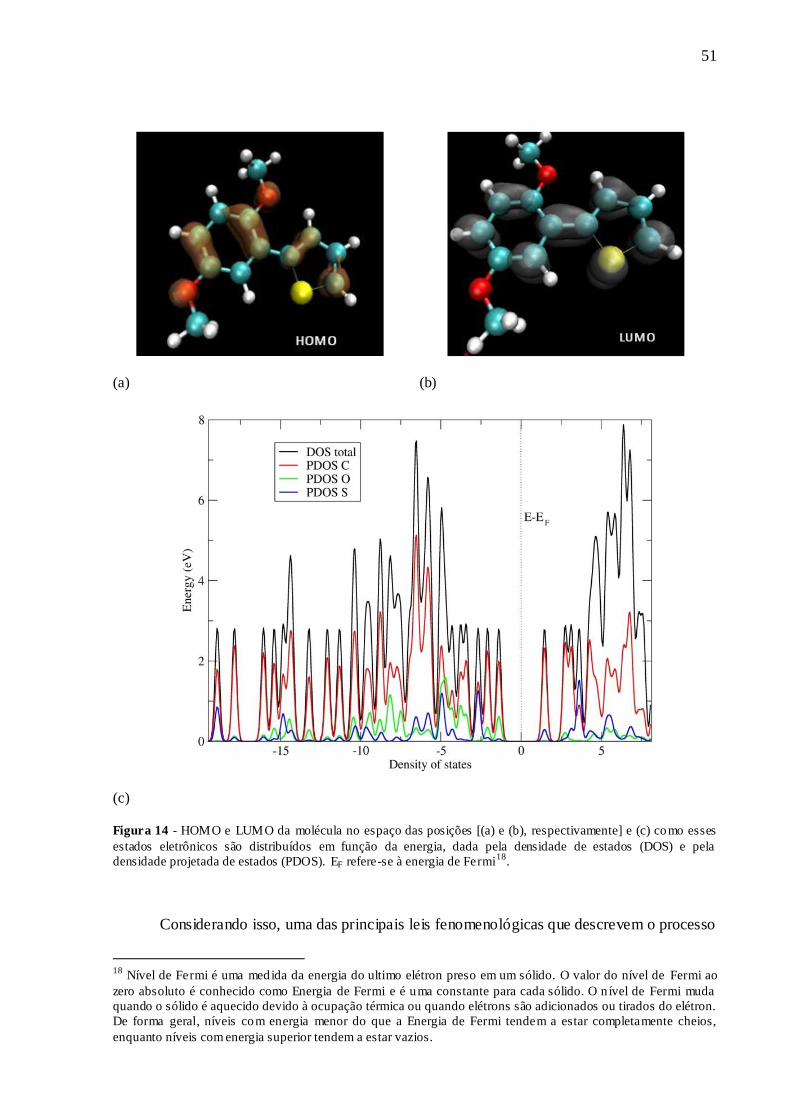
51
(a) (b)
(c)
Figura 14 - HOMO e LUMO da molécula no espaço das posições [(a) e (b), respectivamente] e (c) como esses
estados eletrônicos são distribuídos em função da energia, dada pela densidade de estados (DOS) e pela
densidade projetada de estados (PDOS). EF refere-se à energia de Fermi18
.
Considerando isso, uma das principais leis fenomenológicas que descrevem o processo
18
Nível de Fermi é uma medida da energia do ultimo elétron preso em um sólido. O valor do nível de Fermi ao
zero absoluto é conhecido como Energia de Fermi e é uma constante para cada sólido. O n ível de Fermi muda
quando o sólido é aquecido devido à ocupação térmica ou quando elétrons são adicionados ou tirados do elétron.
De forma geral, níveis com energia menor do que a Energia de Fermi tendem a estar completamente cheios,
enquanto níveis com energia superior tendem a estar vazios.

52
de absorção óptica é conhecida como Lei de Beer-Lambert. Essa lei pode ser encontrada a
partir das equações de Maxwell utilizando o coeficiente de absorção como sendo a parte
complexa do índice de refração (que resulta em função trigonométrica hiperbólica) e também
por uma análise muito mais trivial que é apresentada a seguir: a absorbância de um
determinado material em solução deve ser proporcional à sua concentração (c), assim como o
passo da cubeta (l) e a constante de proporcionalidade deve ser uma constante intrínseca de
cada material e chamada de absortividade molar ( ), que é a única variável dentro da Lei de
Beer-Labert que depende do tipo de material estudado. A absorbância, de fato, é um número
dado pelo logaritmo natural da razão entre a intensidade da luz transmitida pela amostra e a
intensidade do feixe de referência, que passa por nenhuma amostra. Assim, essa lei
fenomenológica (que se assemelha bastante à Lei de Decaimento Radioativo, já que as duas
são leis de decaimento: enquanto a de decaimento radioativo considera a diminuição da
quantidade de núcleos de um elemento-pai no tempo, a Lei de Beer-Lambert considera a
diminuição da intensidade do feixe-pai no espaço) pode ser escrita como:
(Equação 1)
Equação 1 - Lei de Beer-Lambert.
Do ponto de vista de instrumentação, um típico espectrofotômetro UV-VIS consiste-se
de uma fonte de luz, geralmente uma lâmpada de Deutério (UV) e de Tungstênio (VIS) que
conseguem varrer todo o espectro com intensidade aproximadamente constante, na região de
190 a 1100 nm, um monocromador e um detector. O monocromador é uma rede de difração
cujo papel é separar o feixe em seus comprimentos de ondas constituintes enquanto que o
detector é geralmente uma fotomultiplicadora ou fotodiodos.
2.2.2 – FOTOLUMINESCÊNCIA
Quando excitados para a banda de condução, os elétrons podem sofrer transições
radiativas de volta para a banda de valência, liberando sua energia potencial adquirida devido
ao processo de absorção pela emissão de um fóton com energia igual à diferença de energia
entre o fundo da banda de condução (orbital LUMO) e o topo da banda de valência (orbital
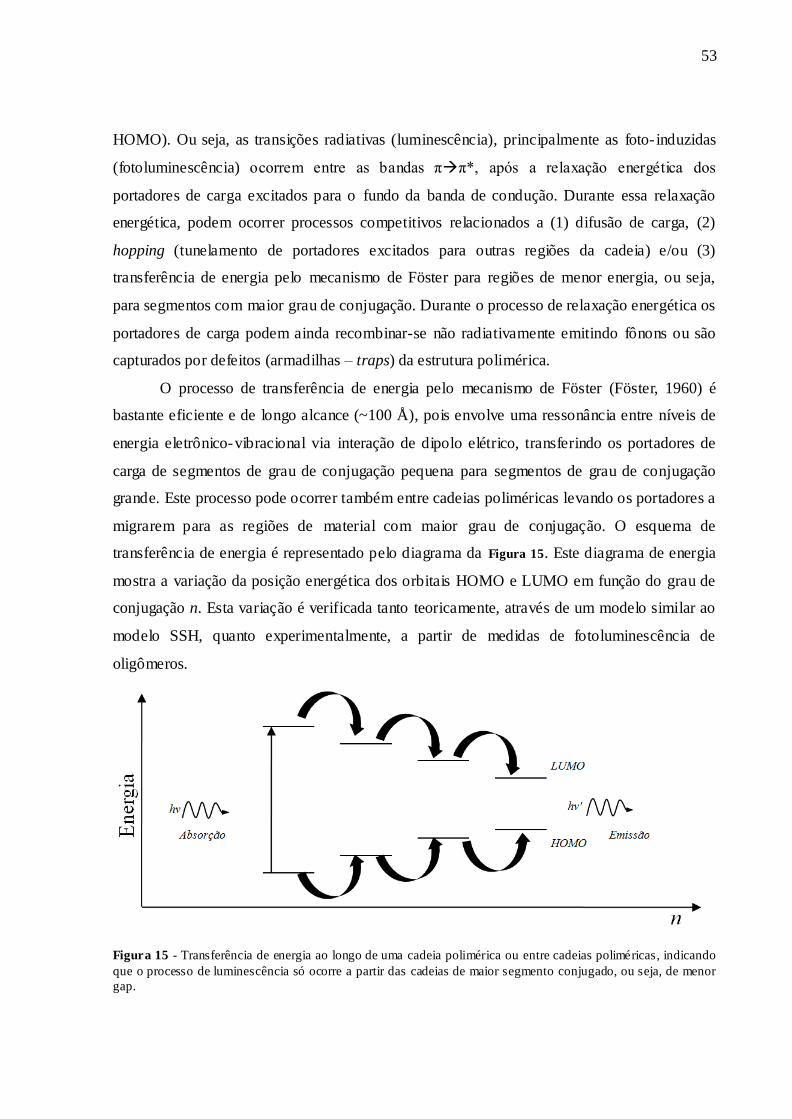
53
HOMO). Ou seja, as transições radiativas (luminescência), principalmente as foto- induzidas
(fotoluminescência) ocorrem entre as bandas ππ*, após a relaxação energética dos
portadores de carga excitados para o fundo da banda de condução. Durante essa relaxação
energética, podem ocorrer processos competitivos relacionados a (1) difusão de carga, (2)
hopping (tunelamento de portadores excitados para outras regiões da cadeia) e/ou (3)
transferência de energia pelo mecanismo de Föster para regiões de menor energia, ou seja,
para segmentos com maior grau de conjugação. Durante o processo de relaxação energética os
portadores de carga podem ainda recombinar-se não radiativamente emitindo fônons ou são
capturados por defeitos (armadilhas – traps) da estrutura polimérica.
O processo de transferência de energia pelo mecanismo de Föster (Föster, 1960) é
bastante eficiente e de longo alcance (~100 Å), pois envolve uma ressonância entre níveis de
energia eletrônico-vibracional via interação de dipolo elétrico, transferindo os portadores de
carga de segmentos de grau de conjugação pequena para segmentos de grau de conjugação
grande. Este processo pode ocorrer também entre cadeias poliméricas levando os portadores a
migrarem para as regiões de material com maior grau de conjugação. O esquema de
transferência de energia é representado pelo diagrama da Figura 15. Este diagrama de energia
mostra a variação da posição energética dos orbitais HOMO e LUMO em função do grau de
conjugação n. Esta variação é verificada tanto teoricamente, através de um modelo similar ao
modelo SSH, quanto experimentalmente, a partir de medidas de fotoluminescência de
oligômeros.
Figura 15 - Transferência de energia ao longo de uma cadeia polimérica ou entre cadeias poliméricas, indicando
que o processo de luminescência só ocorre a partir das cadeias de maior segmento conjugado, ou seja, de menor
gap.

54
No entanto, um espectro de emissão experimental possui uma forma de linha espectral
não homogênea, ou seja, a forma espectral é dependente de outros fatores além de
simplesmente da posição da transição e da intensidade (Marletta et al., 2002). O momento de
dipolo elétrico de transição, por exemplo, assim como o acoplamento entre as transições
eletrônicas e as transições vibracionais modificam a forma de linha e induzem o aparecimento
de picos menos intensos nos espectros. A esses acoplamentos entre as transições de níveis
eletrônicos e as transições de níveis vibracionais, damos o nome de acoplamento vibrônico e
devido ao fato de ser o fônon o quantum de energia vibracional definido no espaço recíproco,
eles também são conhecidos como acoplamentos elétron-fônon.
Partindo disso, o processo de emissão de uma molécula pode ser simplificado a partir
do entendimento de que o elétron, após excitado a partir do topo da banda de valência, por
exemplo, atinge um nível vibracional que é acoplado a um nível eletrônico dentro da banda de
condução. Esse elétron sofre decaimentos não-radiativos, ativando outros fônons conforme
ele „passa‟ pelos níveis vibrônicos de menor energia e com isso, ele relaxa até o fundo da
banda. Só então ocorre o processo de transição vertical (radiativo) com a emissão de um
fóton. Embora ocorra esse processo de decaimento não-radiativo antes da emissão de ocorrer
o processo radiativo, pode o elétron experimentar uma transição conhecida como puramente
eletrônica, também conhecida como transição 0-0. Ou seja, o elétron é excitado do topo da
banda de valência para um nível vibrônico da banda de condução, porém não ocorre processos
de decaimento por ativação de fônons nos níveis vibracionais e o elétron transiciona
diretamente para a banda de valência, sendo a transição de maior intensidade do espectro.
O acoplamento elétron-fônon pode ainda ser entendido por um modelo simplificado e
bem ilustrativo19 que entende o elétron de um cromóforo da cadeia polimérica conjugada
como sendo um elétron preso em um poço de potencial unidimensional, que em um momento
t≠0 recebe uma quantidade de energia e por conta disso acessa um estado excitado. Esse
ganho de energia por parte do elétron faz com que ele „acione‟ alguns graus de liberdade desse
poço de potencial e que quanto maior for a energia que esse elétron recebeu, mais „graus de
liberdade‟ ele aciona da cadeia. A analogia pode ser estendida para a realidade física entendo
que quanto maior a energia do fóton absorvido, mais níveis vibrônicos são acionados na
banda de condução.
O aparato experimental para medidas de fotoluminescência é bem parecido com
aquele utilizado para a medida de absorção UV-VIS, porém ao invés de usar lâmpadas como
19
Enfatiza-se que esse modelo apresentado é didático e não corresponde à realidade física do processo.

55
fonte de excitação, costuma-se utilizar um laser com spot monocromático cuja energia dos
fótons seja ligeiramente maior do que o a energia de gap aproximada da cadeia polimérica.
Comumente são utilizados lasers de íons de Argônio (Ar+) com emissão em 455 ou 487 nm e
fotodiodos como detector que consigam ver a resposta de emissão para um dado comprimento
de onda.
2.3 – TEÓRICA
O problema de se descrever a estrutura de moléculas e sólidos para previsão de
propriedades estruturais, eletrônicas, vibracionais, etc., consiste-se basicamente em, com base
na teoria quântica, resolver a equação de Schröndiger (ES) para um conjunto de partículas,
que são os elétrons e os núcleos que constituem a molécula. A solução dessa equação
fornecerá as funções de onda dos átomos assim como suas autoenergias e distribuição espacial
na molécula. No entanto, a solução exata da ES para átomos com mais de um elétron torna-se
impossível, ainda mais quando temos sistemas moleculares, formados por centenas e milhares
de elétrons. Assim, encontrar as autoenergias permitidas do sistema, assim como sua posição
translacional de equilíbrio, torna-se uma tarefa que exige o uso de métodos aproximativos que
permitam, com ajustável requinte de confiabilidade, descrever e prever propriedades desses
materiais.
Não é a proposta deste texto revisar as várias abordagens que já foram criadas para a
resolução da ES para sistemas moleculares assim como para descrever o comportamento
temporal delas, no entanto, o nível de aproximação no qual, em prática, modelos de estrutura
eletrônica moderna são baseados, é definido sem elaboração de detalhes, e o leitor é
referenciado a uma leitura mais aprofundada na literatura especializada (ver nota 21).

56
2.3.1 – O ESTADO FUNDAMENTAL: MÉTODO DE HARTREE-FOCK-
ROOTHANN E PÓS-HARTREE-FOCK
A primeira consideração a ser realizada para que possam ser introduzidos os métodos
de descrição tanto do estado fundamental como do estado excitado de moléculas e sólidos é
que como os núcleos são muito mais pesados do que os elétrons (razão massa do
elétron/massa do núcleo é da ordem de 10-4 a 10-5), eles se movem bem mais lentamente que
os próprios elétrons. Assim, para a análise das propriedades eletrônicas de uma molécula,
costuma-se assumir o núcleo com uma posição fixa no espaço. Além disso, a posição dos
núcleos é mais localizada na molécula e as vibrações nucleares interagem fracamente com os
níveis eletrônicos, por uma aproximação que chamamos de adiabática (também conhecida
como Born-Oppenheimer (BO) e que será mais bem discutida em seguida).
Intuitivamente entende-se que tanto a geometria de equilíbrio como as propriedades
físicas de moléculas no estado fundamental sejam função apenas da energia total do sistema,
valendo a minimização do funcional de energia no equilíbrio. A energia total de sistemas
como esse podem ser descritos em termos unicamente da energia eletrônica, energia
vibracional e rotacional da molécula, desprezando a energia cinética translacional. É fácil
mostrar, por argumentos semi-clássicos, que a ordem de grandeza da energia eletrônica é
maior do que a vibracional que é maior que a rotacional. Assim, a energia eletrônica é muito
maior que a energia vibracional na maioria das vezes, e podemos desacoplar o operador
hamiltoniano eletrônico ( ) do hamiltoniano vibracional (ou, hamiltoniano nuclear), para
que a equação de Schrödinger seja resolvida somente para os elétrons20. Além disso, para
potenciais não-dependentes do tempo, a equação de Schrödinger pode ser desacoplada em um
termo espacial e um termo temporal que descreve a evolução da função de onda no tempo.
Partindo disso e de forma geral, a equação de Schrödinger independente do tempo para os
elétrons pode ser escrita da forma:
(Equação 2)
Equação 2 - Equação de Schrödinger independente do tempo (ESIT).
Em que, é a função de onda eletrônica do sistema e o operador hamiltoniano eletrônico
20
O que é uma conseqüência direta da aproximação de Born -Oppenheimer.

57
para uma molécula poliatômica pode ser escrito como21 (em unidades atômicas):
(Equação 3)
Equação 3 - Operador hamiltoniano eletrônico escrito em coordenadas atômicas22
.
onde o índice {i,j} corresponde aos elétrons, o índice {A,B} aos núcleos de massa MA e carga
ZA, enquanto é a distância entre os elétrons {i,j} e é a distância entre o núcleo A e o i-
ésimo elétron. Os termos da equação acima representam, em ordem, a energia cinética dos
elétrons escrita na forma quântica, o potencial de Coulomb clássico dos núcleos que é
sentido pelos elétrons, assim como as repulsões coulombianas entre elétrons-elétrons.
Note que termos correspondentes à energia cinética dos núcleos e repulsão coulombiana
núcleo-núcleo não aparecem nesse hamiltoniano, devido justamente ao fato de que na
aproximação de Born-Oppenheimer, o termo de energia cinética do núcleo é negligenciado e
o termo de repulsão núcleo-núcleo é estabelecido como uma constante, permitindo a completa
separação do operador energia entre as partes eletrônicas e nucleares. Dessa forma, a função
de onda eletrônica e sua energia eletrônica dependem dos núcleos apenas de forma
paramétrica e podemos tratar os elétrons como partículas que se movem em um campo de
núcleos fixos.
O problema de se resolver a ES para muitos-corpos, que são os N elétrons se movendo
no campo de M núcleos, ainda é de difícil solução, desde que se trata de uma equação
diferencial parcial de muitas variáveis acopladas. Assim, uma aproximação bastante útil
inserida em 1928, foi a aproximação de Hartree-Fock (HF) (Szabo, 1996), que substitui os N
elétrons interagentes se movendo no campo nuclear por N problemas de um elétron se
movendo no mesmo campo nuclear acrescido do campo dos (N-1) elétrons restantes. Esse
modelo, chamado de campo efetivo, permite ainda, caso seja necessário, a inserção das
vibrações nucleares na solução do problema através de métodos perturbativos. Em termos do
potencial eletrostático fixo, as soluções estacionárias para um elétron da ES, ou seja, as
funções de onda, são chamadas de orbitais atômicos (AO – atomic orbitals) para átomos e
orbitais moleculares (molecular orbitals), no caso de moléculas. Assim, a aproximação HF,
21
Veja, por exemplo, (Szabo, 1996) 22
Observe que a condição imposta no somatório evita que haja uma singularidade na função hamiltoniana.

58
de maneira geral, diz que:
(Equação 4)
Equação 4 - Aproximação de Hartree-Fock.
Que pode ainda ser simplificada em uma forma didática (sem rigor matemático),
dizendo que:
(Equação 5)
Equação 5 - Forma didática da aproximação de HF.
Nesse contexto, desde que as moléculas em questão não contêm elementos pesados, os
cálculos são não-relativísticos23. As funções de onda de HF total para os N elétrons é então
construída como um produto antissimétrico24 dos orbitais moleculares ocupados por um
elétron. Tal função de onda, escrita na forma de um determinante (conhecido como
determinante de Slater) é chamada de função de onda HF restrita e o uso dessa forma
determinante permite que a antissimetria da função de onda de um elétron seja explícita 25.
Dessa forma, tem-se:
(Equação 6)
Equação 6 - Função de onda de HF na forma do determinante de Slater.
Onde o fator (N!)-1/2 é uma constante de normalização. Os são as funções de onda
individuais j das variáveis espaciais e de spin do elétron i, ou seja, são os orbitais j do elétron
i.
23
Ou seja, não incorporam efeito de acoplamento spin-órbita, por exemplo . Esse efeito pode ser interpretado de
uma forma simples (embora incorreta) que considera que para um elétron orbitando em torno de um núcleo, no
referencial do elétron, é o núcleo que gira em torno dele. Este movimento orb ital da carga positiva do núcleo dá
origem a um campo magnético que age sobre o elétron e, por efeito Zeeman, quebra a degenerescência entre
duas possíveis orientações do spin eletrônico. 24
Uma matriz A é antissimétrica se A é uma matriz quadrada de forma que sua transposta At seja tal que A
t=-A.
25 A antissimetria da função de onda deve ser sempre estabelecida em um sistema de vários férmions tendo em
vista a indistinguibilidade entre eles. Ou seja, a função de onda não pode manter-se inalterada mediante a
permutação entre dois elétrons.

59
Tendo em mãos essa função de onda, com ajuda do cálculo variacional, que assegura
que a função de onda do sistema será aquela que minimiza a energia eletrônica e com
manipulações algébricas usando as propriedades dos operadores hermitianos, chega-se a uma
equação de autovalores, parecida com ES, chamada de Equação de Hartree-Fock:
(Equação 7)
Equação 7 - Equação de autovalores de Hartree-Fock.
Em que o termo é o operador de Fock para o elétron 1 aplicado no orbital i do
mesmo elétron, resultando no próprio orbital multiplicado por um autovalor de energia
característico do orbital i, ou seja, é uma equação de autovalores. O operador de Fock é uma
aproximação físico-matemática do operador hamiltoniano, porém com o acréscimo de dois
termos cuja origem é tratada no Apêndice II, por se tratar de um formalismo mais avançado.
De forma geral, ele pode ser escrito como:
(Equação 8)
Equação 8 - Operador de Fock.
Em que pode ser encarado como um pseudo-hamiltoniano do i-ésimo elétron,
é o operador de Coulomb que define a força de repulsão entre os elétrons j e i no sistema
e é conhecido como operador de troca (Stolarczyk, Jeziorska, & Monkhorst, 1988), que
define o efeito quântico produzido pela troca de orbitais entre os elétrons i e j. Explicitamente,
tem-se:
(Equação 9)
Equação 9 - Operador de Fock Explícito.
Onde o primeiro termo refere-se ao operador “pseudo-hamiltoniano”, o segundo é o

60
operador de Coulomb e o terceiro é o operador de troca. No entanto, pelo Princípio de
Exclusão de Pauli, sabemos que dois elétrons não podem ter o mesmo conjunto de números
quânticos (espaciais e de spin). Mas como para cada conjunto de números quânticos espaciais
existem dois números quânticos de spin possíveis (up e down ), a ocupação dos orbitais
cai de N para N/2 orbitais ocupados. Assim, o método de Hartree-Fock fica conhecido como
Hartree-Fock Restrito (RHF – Restricted Hartree-Fock) e o operador de Fock para o elétron 1
pode ser reescrito como:
(Equação 10)
Equação 10 - Operador de Fock Restrito.
Observe que o índice i para os operadores de Coulomb e de troca foram trocados por
índices a, que não se refere mais ao elétron i isolado e sim ao conjunto de números quânticos
espaciais degenerados. Observe também que, enquanto o operador de Coulomb teve que ser
compensado pela troca dos índices (fator 2 multiplicativo) o operador de troca não. Isso é
devido ao fato de que o operador de Coulomb é insensível ao spin eletrônico (é um operador
clássico) enquanto que o operador de troca, por ser quântico, já consegue discernir entre dois
elétrons de spin contrários que são trocados.
Dessa forma, a equação de HF que permite encontrar os orbitais (leia-se funções de
onda) que minimizem a energia eletrônica do sistema (leia-se autoenergia) é explicitamente
escrita da forma, para o elétron 1 com conjunto de números quânticos espaciais a:
(Equação 11)
Equação 11 - Equação de autovalores de HF exp lícita 1.
Ou:

61
(Equação 12)
Equação 12 - Equação de autovalores de HF exp lícita 2.
Que substitui a ES para encontrar as autoenergias do sistema. O primeiro termo
entre colchetes se refere ao operador pseudo-hamiltoniano, o segundo ao operador de
Coulomb e o terceiro ao operador de troca. Além disso, note também que a integração dos
operadores de Coulomb e de troca do elétron 1 são efetuadas no volume ( ) orbital do
elétron 2, indicando justamente essa interação.
Dois pontos devem ser ressaltados nesse contexto: 1) observe que a equação de HF
não incorpora um termo de correlação eletrônica, ou seja, trata os elétrons como partículas
interagentes unicamente a partir do operador de Coulomb, desconsiderando o fato de que o
movimento do i-ésimo elétron depende do movimento dos outros (N – 1) elétrons e a esse
fato, chamamos de correlação eletrônica26,27 (Löwdin, 1955); e 2) por mais que
simplificações físico-matemáticas tenham sido realizadas, a Equação 12 ainda é uma equação
integro-diferencial complexa que deve ser resolvida de forma auto-consistente28.
Para resolver esse problema, que é encontrar os autovalores de energia da função
desconhecida , é realizada uma expansão dela em funções conhecidas (que podem ser
ondas planas ou funções gaussianas), que são também chamadas de funções de base. Esse
método é conhecido como Combinação Linear de Orbitais Atômicos (LCAO – Linear
26
Esse fato não contradiz a aproximação de Fock, que diz que o i-ésimo elétron se move em um campo atrativo
nuclear e um campo repulsivo dos outros (N-1) elét rons. Observe que a correlação eletrônica é um fator
adicional não incluído pela descrição monodeterminantal da função de onda eletrônica. Maiores informações
podem ser obtidas no paper original de Löwdin e Per-Olov. 27
Um cálculo autoconsistente supostamente é capaz de corrig ir tal característica (e não, „deficiência‟) do método
de HF (vide próxima nota). 28
Uma equação é dita de ser resolvida de forma autoconsistente quando é impossível, a partir apenas de
constantes fundamentais e dados do problema, chegar a um resultado. Ou seja, a solução da equação depende de
uma solução anterio r, de forma que o ciclo p recisa ser in iciado por uma solução tentativa, e a partir dela é
encontrada uma nova solução, e no caso de equações de autovalores, os autovalo res para essas duas autofunções
(a tentativa e a primeira solução) são encontrados e a diferença entre eles é avaliada. O processo continua até que
a diferença entre os autovalores do ciclo k e do ciclo k-1 seja menor ou igual do que um critério de convergência
estabelecido pelo usuário do método.

62
Combinations of Atomic Orbitals), desde que as funções de onda da molécula (orbital
molecular) é expandido como uma combinação linear das funções de onda dos orbitais dos
átomos formadores da molécula (orbitais atômicos), através de funções simples que sejam
capazes de descrever de forma aproximada a distribuição espacial de carga do átomo. Assim,
temos:
(Equação 13)
Equação 13 - Expansão das funções de onda molecular em funções de base atômicas.
Em que , ou seja, quanto maior a expansão, melhor a aproximação. Além disso,
os coeficientes indicam o quanto de cada função de onda do orbital atômico está
presente na função de onda do orbital molecular a. Inserindo essa expansão na Equação 7
(Equação de HF):
(Equação 14)
Equação 14 - Equação de HF com LCAO - 1.
Usando a propriedade da completude da base (multiplicando pelo complexo conjugado
da função de onda ) e integrando, tem-se:
(Equação 15)
Equação 15 - Equação de HF com LCAO - 2.
Perceba que o termo integral do lado esquerdo é uma matriz, chamada de matriz de
Fock [F], cujo conjunto de elementos é dado por:

63
(Equação 16)
Equação 16 - Matriz de Fock.
E que o termo integral do lado direito também é uma matriz, chamada de matriz de
Sobreposição [S], cujo conjunto de elementos é:
(Equação 17)
Equação 17 - Matriz de Sobreposição.
Na notação de Dirac, elas ficam determinadas como:
(Equação 18)
Equação 18 - Matriz de Fock e de Sobreposição na notação de Dirac.
Ou seja, a matriz sobreposição quantifica o tanto de um orbital atômico que se
sobrepõe ao outro (overlapping) e os elementos da matriz de Fock fornece os autovalores de
energia quando um orbital interage com um .
Sendo assim, a Equação 15 pode ser rescrita na forma matricial:
(Equação 19)
Equação 19 - Equação de Hartree-Fock-Roothaan-Hall.
Onde é a matriz dos coeficientes. A equação de HF escrita dessa forma é
conhecida como Equação de Hatree-Fock-Roothaan-Hall.
Ainda assim, pela definição do operador de Fock, podemos separá-lo em uma parte
com caráter hamiltoniano para o caroço29 dos átomos mais um termo que reúna o caráter do
29
Entende-se por caroço (mais precisamente caroço iônico) como sendo a união entre o núcleo e os elétrons
internos do átomo, ou seja, aqueles que não interagem com outros átomos e por conta disso não participam d e

64
operador de Coulomb e do operador de troca. Dessa forma:
(Equação 20)
Equação 20 - Decomposição do Operador de Fock.
Em que o hamiltoniano de caroço nada mais é do que:
(Equação 21)
Equação 21 - Hamiltoniano de Caroço.
Ou, em notação de Dirac:
(Equação 22)
Equação 22 - Hamiltoniano de Caroço em notação de Dirac.
E o operador pode ser escrito em função do operado que atua nos orbitais
atômicos e , de forma que:
(Equação 23)
Equação 23 – Defin ição de em termos do operador densidade.
E a matriz resultante é conhecida como matriz densidade e fornece a probabilidade
de se localizar elétrons em determinadas regiões dos orbitais moleculares, ou seja, indica a
probabilidade de se encontrar um elétron i de um orbital atômico em algum outro orbital
atômico pertencente à descrição do orbital molecular. Os elementos da matriz densidade, a
partir de álgebra linear, são escritos a partir dos coeficientes da expansão LCAO dos orbitais
e no conjunto de números quânticos espaciais degenerados a:
ligações químicas nem interferem nas propriedades do átomo.

65
(Equação 24)
Equação 24 - Elementos da matriz densidade.
E ainda, as integrais definidas pelos termos são chamadas de integrais de
dois centros ou também integrais de dois elétrons, desde que correspondem ao cálculo do
tanto que um orbital atômico do elétron 1 interage com os orbitais atômicos do elétron 2 na
formação dos orbitais moleculares. Essas integrais serão de extrema importância na definição
das parametrizações semi-empíricas, definidas em seguida, e por conta disso são definidas
explicitamente:
(Equação 25)
Equação 25 - Integrais de dois elétrons.
A equação de Hartree-Fock-Roothaan-Hall é então definida globalmente:
(Equação 26)
Equação 26 - Equação global explícita de Hartree-Fock-Roothaan-Hall.
Que é a base de todo cálculo de estrutura eletrônica dentro desse formalismo. Essa
equação deve ser resolvida de forma autoconsistente (SCF – Self-Consistent Field), visto que
somente a partir de é possível determinar os coeficientes da expansão LCAO . O chute
inicial para dar início à autoconsistência é fazer o operador de Fock ser igual ao operador
hamiltoniano de caroço e a partir disso definir uma matriz densidade inicial, que determina
um conjunto de coeficientes da expansão. As autoenergias dos orbitais são calculadas e
armazenadas e um novo ciclo é iniciado a partir dessa função de onda orbital encontrada no

66
primeiro ciclo. Um esquema que descreve o processo é apresentado na Figura 16 :
Figura 16 - Ciclo de autoconsistência nas equações de Hartree-Fock-Roothaan-Hall.
A Equação de HF (Equação 26) possui um grande número de integrais para serem
resolvidas numericamente. Para sua solução existem ainda duas maneiras de fazê- lo: por ab
initio, ou seja, que só incluiu até então aproximações físico-matemáticas, ou de forma semi-
empírica (SE), em que parâmetros experimentais são inseridos no cálculo das integrais de
forma que o processo SCF seja acelerado. Dentro da forma SE de resolução da Equação 26,
existem ainda duas abordagens distintas: 1) que intenta reproduzir o cálculo ab initio com o
menor número possível de funções de base e (conhecidas como parametrizações CNDO,
INDO e NDDO, a serem explicadas em seguida), 2) que além de reproduzir o cálculo ab
initio tenta ainda obter dados que concordem com dados experimentais (parametrizações
MINDO, MNDO, AM1, PM3 e posteriores). Esses métodos, conhecidos como Pós-Hartree-
Fock, foram propostos inicialmente por Pople, 1960 para a abordagem 1) e por Dewar para a
abordagem 2). O esquema da (Figura 17) explica bem como essa divisão ocorre.
Figura 17 - Estrutura das metodologias de cálculo da Equação de Hartree-Fock.
Hartree-Fock
ab initio
Semi-empírico
CNDO, INDO, NDDO.
MINDO, MNDO, AM1, PM3...

67
As parametrizações decorrentes da metodologia 2) são conseqüência direta daquelas
da metodologia 1), com a inserção de dados adicionais que são apresentados a seguir. De
forma pouco abrangente, essas parametrizações incluem as seguintes modificações da Equação
26:
1. CNDO: Complete Neglect of Differential Overlap
Despreza todas as integrais que envolvem funções de base diferentes,
ou seja, a sobreposição de orbitais não é considerada.
A matriz sobreposição é dita como sendo igual à matriz identidade, o
que é conseqüência direta da primeira parametrização ([S]=[I]).
No hamiltoniano de caroço, ao invés de tratar as massas atômicas no
termo de atração Coulombiana núcleo-elétron, é introduzido o conceito
de carga efetiva de caroço, que é a carga do íon.
As integrais de dois centros (dois elétrons) são iguais a um parâmetro
que depende somente do átomo A, do átomo B e da distância entre
eles.
2. INDO: Intermediate Neglect of Differential Overlap
A principal introdução paramétrica é a consideração das integrais de
troca entre orbitais diferentes ( ) que estejam centradas no mesmo
átomo, ou seja, orbitais atômicos delocalizados sobre átomos diferentes
não são considerados.
3. NDDO: Neglect of Diatomic Differential Overlap
Nessa parametrização as integrais de dois centros (dois elétrons) cujos
orbitais estejam em átomos diferentes são desprezadas.
A partir dessas parametrizações, propostas por Pople, as de Dewar seguem:

68
4. MNDO: Modified Neglect of Differential Overlap
Inclui uma forma peculiar de análise do hamiltoniano de caroço, muito
baseado no método CNDO.
As integrais de três e quatro centros são anuladas.
As integrais de um centro não são calculadas numericamente, mas sim
tem seus valores substituídos por parâmetros obtidos a partir de
espectroscopia atômica.
As integrais de dois centros também não são avaliadas numericamente,
mas sim avaliadas a partir das integrais de um centro, utilizando-se de
expansões de multipolos das distribuições de carga.
A repulsão entre os caroços iônicos é do tipo Morse.
Por mais que a repulsão iônica preveja um termo atrativo e repulsivo ao
mesmo tempo, ele não é suficiente na descrição de pontes de
hidrogênio, por exemplo.
5. AM1: Austin Method 1
A principal modificação do método MNDO é a inserção de um termo
de correção dependente do tipo de átomo na repulsão iônica, cujos
parâmetros são grandezas a serem otimizadas.
Por conta disso, a interação de van der Waals é mais bem descrita do
que no método MNDO, visto que a correção do hamiltoniano de caroço
inclui uma ou mais gaussianas atrativas que compensam a repulsão
excessiva diretamente. Gaussianas repulsivas foram também inseridas
centradas em uma separação internuclear menor, levando a uma
redução global do termo na expressão de repulsão de caroço e então
reduzindo a repulsão em distâncias internucleares maiores. Para
elementos altamente eletronegativos (como o Oxigênio), apenas
gaussianas atrativas foram incluídas, enquanto que para aqueles de
eletronegatividade moderada (Carbono, Hidrogênio, Nitrogênio e
Enxofre), ambos os tipos foram inseridos.

69
Nesse trabalho, os cálculos foram realizados utilizando a forma semi-empírica, pela
parametrização AM1 (Austin Model 1). Essa parametrização foi escolhida tendo em vista a
sua vasta aplicação na literatura de moléculas conjugadas, com boa descrição de propriedades
estruturais e eletrônicas desses compostos, principalmente aqueles formados de carbono,
hidrogênio, oxigênio, nitrogênio, enxofre, flúor, cloro e iodo. Além disso, sua boa descrição
de ligações não-covalentes, como pontes de hidrogênio e sulfeto, faz dessa parametrização
apropriada para a determinação da estrutura de equilíbrio do polímero estudado, o FSE59.
Parametrizações mais recentes, como a PM3 e PM5 (Parametric Method 3,5) também foram
testadas, mas os resultados obtidos não são condizentes com dados experimentais do FSE59,
portanto tal análise não foi incluída nesse trabalho e a parte teórica dessas parametrizações
nem foi apresentada, assim como é o caso da parametrização PM6, que por ser muito recente,
não possui muita literatura disponível atestando a confiabilidade do método.
2.3.2 – O ESTADO EXCITADO: DESCRIÇÃO SEMI-EMPÍRICA
Para a descrição do estado excitado de moléculas, existem vários metodologias
disponíveis na literatura que conseguem boa concordância com dados experimentais, sendo a
discrepância devida unicamente a efeitos de bulk que interferem nos resultados medidos.
Entre esses métodos, podem ser destacados: Teoria da Perturbação de Muitos-Corpos (MBPT
– Many-Body Perturbation Theory), Teoria do Funcional da Densidade Dependente do Tempo
(TDDFT – Time-Dependent Density Functional Theory), Hartree-Fock Dependente do Tempo
(TDHF – Time-Dependent Hartree-Fock) e parametrizações semi-empíricas de Hartree-Fock
com Interações de Configurações (CI – Configurantion Interactions) e Interações de
Configurações de Único Elétron (CIS – Configuration Interactions of Single-Electron). Esse
trabalho é baseado no formalismo CI da reparametrização de Zerner do método INDO com
parâmetros espectroscópicos (ZINDO/S-CI). A seguir são apresentadas as bases teóricas
desses dois métodos, tanto o CI como o ZINDO/S.
Como dito anteriormente, o método INDO considera nos cálculos as integrais de troca
entre orbitais diferentes centrados no mesmo átomo, mas mesmo assim incorpora a
característica do CNDO de que integrais centradas em dois átomos são iguais a um parâmetro

70
que depende do átomo A, do átomo B e da distância entre eles. Em 1991, M. Zerner
introduziu uma reparametrização do método INDO, em que o parâmetro é dado por uma
relação conhecida como Mataga-Nishimoto (Stolarczyk et al., 1988):
(Equação 27)
Equação 27 - Relação de Mataga-Nishimoto.
Em que é proporcional à diferença entre o potencial de ionização e a afinidade
eletrônica do átomo I, e é obtido por ajuste de dados experimentais e costuma ser tomado
como igual a 1,2. Essa reparametrização, também conhecida como ZINDO/S ou apenas
INDO/S, é realizada com um método de Interações de Configurações (CI), que permite que as
funções de onda do método INDO sejam avaliadas em um estado excitado.
A função de onda do estado fundamental para o método de HF é construída com os
orbitais de mais baixa energia até a=N/2. Esses orbitais são chamados de orbitais ocupados,
denotados por . Se, no entanto, usarmos os orbitais desocupados na construção da
função de onda, tem-se então uma configuração excitada do sistema, dada pela função de
onda , que pode ser construída tanto para estado singleto (em não há alteração da
multiplicidade30 do estado fundamental e o momento angular de spin total continua sendo
nulo) ou para estado tripleto (em que ocorre alteração da multiplicidade e o momento angular
de spin total é aumentado, pelo desemparelhamento de spin).
O método de interações de configurações permite que a partir da permutação dos
orbitais ocupados e virtuais, como uma nova base para descrever o estado fundamental, seja
escrita uma função de onda que é entendida como de estado fundamental. Mas, de fato, por
possuir mais contribuições de orbitais desocupados do que ocupados, acaba sendo uma função
de onda do estado excitado. A descrição das propriedades eletrônicas, como espectro de
absorção óptica, que nada mais é do que o envelopamento de transições eletrônicas calculadas
por funções do tipo gaussianas ou lorentzianas, de compostos orgânicos e lantanídeos é
30
Esse conceito é exp licado na seção 2.3.4.

71
bastante condizente com dados experimentais e tem validado o uso desse método como uma
ferramenta eficiente para a previsão teórica de energia de gap, afinidade eletrônica, entre
outras propriedades de estado excitado.
Nesse trabalho, o método ZINDO/S-CI foi utilizado pela implementação que existe no
software ArgusLab® (Thompson, 2010), que foi escolhido pela sua gratuidade e facilidade de
uso.
2.3.3 – FORMALISMO DE TRANSIÇÕES VERTICAIS DE FERMI E
ACOPLAMENTOS VIBRÔNICOS DE FRANCK-CONDON
Para entender o comportamento dos espectros de emissão em função da dose irradiada,
faz-se necessário um estudo mais aprofundado dos fatores que interferem nas taxas de
decaimento radiativos e não-radiativos do estado excitado do polímero conjugado estudado.
Para isso, será desenvolvido superficialmente aqui um formalismo que pode se encontrado
facilmente em livros de Mecânica Quântica de nível médio 31. Particularmente, a abordagem
tratada aqui é devida à Marletta, Guimarães e Faria, 2002, e tal formalismo pode ser
encontrado ou na referência (Marletta et al., 2002), visto que boa parte será suprimida por
fugir do escopo desse trabalho.
Considere inicialmente um sistema quântico em um estado não-degenerado n que
sofre uma transição para outro estado também não degenerado m. A taxa de probabilidade de
transição em função do tempo deste sistema pode ser calculada considerando a teoria da
perturbação dependente do tempo e o seguinte hamiltoniano:
(Equação 28)
Equação 28 - Hamiltoniano de um sistema quântico.
Onde é o hamiltoniano não-perturbado e é o hamiltoniano dependente do tempo. Os
autoestados desse hamiltoniano podem ser escrito por:
31 J. J. Sakurai – Modern Quantum Mechanics, Ed. Revisada, Addison-Wesley Publishing Company, Los
Angeles, 1994.

72
(Equação 29)
Equação 29 - Autoestados do sistema quântico no estado n.
Onde são os coeficientes da expansão da função de onda não-dependente do
tempo e é o termo de evolução temporal do sistema, assim como são as
autoenergias do hamiltoniano não-perturbado. O hamiltoniano de interação da radiação com a
matéria , dada pela formulação semi-clássica da Teoria Eletromagnética32:
(Equação 30)
Equação 30 - Hamiltoniano semi-clássico de interação da radiação-matéria.
Onde é a i-ésima carga sob ação do campo de radiação quantizado , V é o campo
eletrostático e é o momento generalizado da partícula, que é utilizado para a montagem do
hamiltoniano perturbado.
Sabe-se, da teoria das perturbações, que as transições são calculadas (em primeira
ordem) pelo elemento de matriz entre os estados iniciais (n) e finais (m). Assim, a evolução
temporal do operador hamiltoniano perturbado pode ser escrita pela dependência temporal
explícita:
(Equação 31)
Equação 31 - Evolução temporal do hamiltoniano.
E os coeficientes do nível m é dado por:
32
Kleber D. Machado – Teoria do Eletromagnetismo, 1ª Ed., Vol. III, pag. 20, Ed. UEPG, Ponta Grossa, PR,
2006.

73
(Equação 32)
Equação 32 - Coeficientes do estado excitado - 1.
De toda forma, assumindo que em t=0 somente o estado n seja populado e que em t≠0
o estado m seja populado, então:
(Equação 33)
Equação 33 - Coeficientes do estado excitado - 2.
E a probabilidade de se encontrar o sistema em um estado para um t qualquer
é dada pelo módulo quadrático:
(Equação 34)
Equação 34 - Probabilidade de se encontrar o sistema no estado .
Porém, os estados e estão correlacionados por uma densidade de estados
considerada aproximadamente constante de forma que:
(Equação 35)
Equação 35 - Probabilidade de se encontrar o sistema no estado correlacionado com o estado por uma
função de densidade de estados .
De forma que a probabilidade por unidade de tempo de que uma transição entre os
estados e ocorra é dada por:

74
(Equação 36)
Equação 36 - Regra de Ouro de Fermi.
Que é conhecida como Regra de Ouro de Fermi e permite o cálculo da taxa de
transição de um autoestado de uma determinada energia (estado excitado) de um sistema
quântico para um contínuo de autoestados (estado fundamental) de energia devido a uma
perturbação dependente do tempo. Este resultado será utilizado logo à frente no cálculo das
transições verticais acopladas por fônons.
Esses acoplamentos em que há transferência de parte da energia eletrônica para
ativação de modos vibracionais na banda de condução foram primeiramente estudados por
S.H. Lin (S. Lin, 1968; Sheng Hsien Lin, 1966; S. H. Lin, 1973, 1989) e A.D. Brailsford
(Brailsford & Chang, 1970), na década de 60/70 e a forma de linha de absorção e emissão
com acoplamento elétron-fônon de macromoléculas, como os polímeros, por J. Cornil
(Cornil, Beljonne, Friend, & Brédas, 1994) e R. Chang (Chang et al., 2000; Yu et al., 1996) na
década de 90. Para a descrição do acoplamento elétron-fônon nessas estruturas, parte-se do
hamiltoniano modelo utilizado para a descrição da estrutura eletrônica de polímeros
conjugados, que foi primeiramente proposto por Su, Schrieffer e Heeger (Su, Schrieffer, &
Heeger, 1979) (SSH) (1979). Esse modelo é essencialmente um tratamento de Hückel
aplicado a uma molécula conjugada linear em que as interações entre os elétrons σ e π são
ignoradas. Apenas os elétrons π, um de cada átomo de carbono são considerados móveis e a
interação dominante é tomada como sendo o acoplamento elétron-rede (elétron-fônon). Além
disso, todos os efeitos de correlação eletrônica (interação de muitos-corpos) são
negligenciados e uma conseqüência direta desse fato é a exclusão do tratamento de éxcitons
no modelo, entendendo que a teoria de um-elétron, aplicada a semicondutores inorgânicos, é
apropriada a semicondutores orgânicos.
O hamiltoniano SSH é, então:
(Equação 37)
Equação 37 - Hamiltoniano SSH.

75
Onde , e são, respectivamente, o hamiltoniano eletrônico não-
relativístico com os núcleos fixos, o de interação elétron-fônon escrito nas coordenadas
eletrônicas (r) e nucleares (R) e o vibracional, escrito nas coordenadas nucleares.
Pela aproximação de Born-Oppenheimer, a função de onda do sistema, nas
coordenadas eletrônicas, nucleares e eletrônico-nucleares acopladas, pode ser
desacoplada de forma que:
(Equação 38)
Equação 38 - Função de onda total para um hamiltoniano do tipo SSH.
Em que é a função de onda eletrônica para o a-ésimo elétron, a
função de onda vibrônica (resultante do acoplamento eletrônico e vibracional) do a-ésimo
elétron acoplado com o -ésimo fônon e a função de onda vibracional do -ésimo
modo vibracional. O argumento da aproximação de Born-Oppenheimer será usado por duas
vezes agora e será justificado logo em seguida:
1ª Aproximação: Qualquer propriedade eletrônica de interesse, como
condutividade elétrica ou atividade óptica, pode ser considerada apenas como uma
função da estrutura eletrônica e da estrutura vibrônica, sendo possível desprezar
os efeitos vibracionais puros, visto que a energia associada a esses modos são
aproximadamente 3 a 4 ordens de grandeza menor do que a energia eletrônica.
Isso é equivalente a dizer que é muito menor do que os outros termos do
hamiltoniano SSH e pode ser desprezado. A conseqüência dessa aproximação é
considerar que a função de onda total pode ser aproximada a:
(Equação 39)
Equação 39 - 1ª Conseqüência da aproximação de BO -1.

76
O que faz com que a função de onda seja escrita como:
(Equação 40)
Equação 40 - 1ª Conseqüência da aproximação de BO -2.
Assim, a equação de Schrödinger pode ser escrita como:
(Equação 41)
Equação 41 - Equação de Schrödinger para o hamiltoniano SSH com aproximação de BO.
Que pode ainda ser separada em uma parte estática (em relação aos núcleos) e uma
parte dinâmica, devido à dependência em R. Assim:
(Equação 42)
Equação 42 - Parte estática da equação de Schrödinger no modelo SSH.
Observe que o operador hamiltoniano eletrônico , como é escrito somente nas
coordenadas eletrônicas r não atua na função de onda vibrônica. Pela regra da cadeia, tem-se
também:
(Equação 43)
Equação 43 - Parte d inâmica da equação de Schrödinger no modelo SSH.
Que é nada mais do que a definição do operador hamiltoniano em função do operador
laplaciano nas coordenadas nucleares, e que a soma é feita sobre todos os b-ésimos núcleos.
Aqui, a 2ª Aproximação baseada na de BO é inserida e estabelece que:
2ª Aproximação: depende fracamente de R e por conta disso o primeiro e
segundo termos do laplaciano podem ser desprezados frente ao último termo, que

77
é predominante. Isto significa dizer que os estados eletrônicos são inalterados com
a variação de R e que se ajustam gradualmente ou adiabaticamente, uma vez que o
movimento eletrônico é mais rápido (~10-15 s) do que o movimento nuclear (~10-12
s), sendo tal aproximação válida para estados não-degenerados e que em casos que
isso não seja verdade, basta modificar a função de onda para um produto de
funções de onda eletrônicas e vibracionais separadamente.
O potencial vibracional para pequenas oscilações pode ser aproximado por um
potencial parabólico e assim a função de onda vibrônica pode ser escrita como um produtório
das funções de onda de osciladores harmônicos quânticos unidimensionais de forma que:
(Equação 44)
Equação 44 - Função de onda vibrônica em termos das funções de onda do oscilador harmônico quântico
unidimensional.
Em que são as coordenadas normais e o produtório é feito sobre os p-ésimos modos
vibracionais com energia . Assim, a energia para uma coordenada nuclear R é
dada como:
(Equação 45)
Equação 45 - Energia v ibrônica em função da parte eletrônica e v ibracional.
Considerando a ocupação térmica dos estados vibracionais, a situação física descrita
pela Equação 45 pode ser visualidade através da Figura 18 para o estado fundamental a e
excitado b. Nesta figura estão representados os níveis de energia a e b e o potencial vibrônico,
assim como a probabilidade de ocupação térmica destes níveis (o que pode ser mais bem
entendido logo adiante).
As taxas de probabilidade de transição eletrônica intermediada por níveis vibracionais

78
entre os estados inicial ( ) e final ( ) são calculadas utilizando a Regra de Ouro de Fermi
(Equação 36). Assim, a probabilidade de transição , na aproximação de dipolo
elétrico, com a absorção de um fóton de energia é (S. Lin, 1968; Sheng Hsien Lin, 1966;
S. H. Lin, 1973, 1989):
(Equação 46)
Equação 46 - Taxa de probabilidade de transição entre dois níveis vibrônicos.
Onde é o operador momento de dipolo elétrico, é a densidade de radiação por
unidade de freqüência, é a função densidade de estados e é o fator de
Boltzmann da ocupação térmica dos estados vibracionais de energia dado por:
(Equação 47)
Equação 47 - Fator de Boltzmann de ocupação térmica (observe que nada mais é do uma função de partição
canônica).
Com k sendo a constante de Boltzmann e T a temperatura absoluta do sistema.
Detalhes sobre o grau de ocupação térmica podem ser encontrados a partir do Ensemble
(Grande-) Canônico em literatura especializada de Mecânica Estatística, tratando os fônons
como bósons.
Considerando que os núcleos variem lentamente nas coordenadas normais de vibração
durante a transição eletrônica, a aproximação adiabática permite que os elementos de matriz
da equação Equação 46 possam ser calculados usando a chamada aproximação de
Franck-Condon. Esta aproximação estabelece que durante as transições eletrônicas, uma
mudança de um nível de energia vibracional para outro é mais provável de ocorrer se as duas
funções de onda vibracionais se sobrepõem. Ou seja, esta é um complemento da aproximação
de Born-Oppenheimer, porém com o adendo de que se a molécula sofre uma transição
vibracional junto com a transição eletrônica, o novo nível vibracional deve ser
instantaneamente compatível com as posições nucleares e o momento do nível vibracional da
molécula no estado original. Ainda assim, classicamente, o princípio de Franck-Condon é
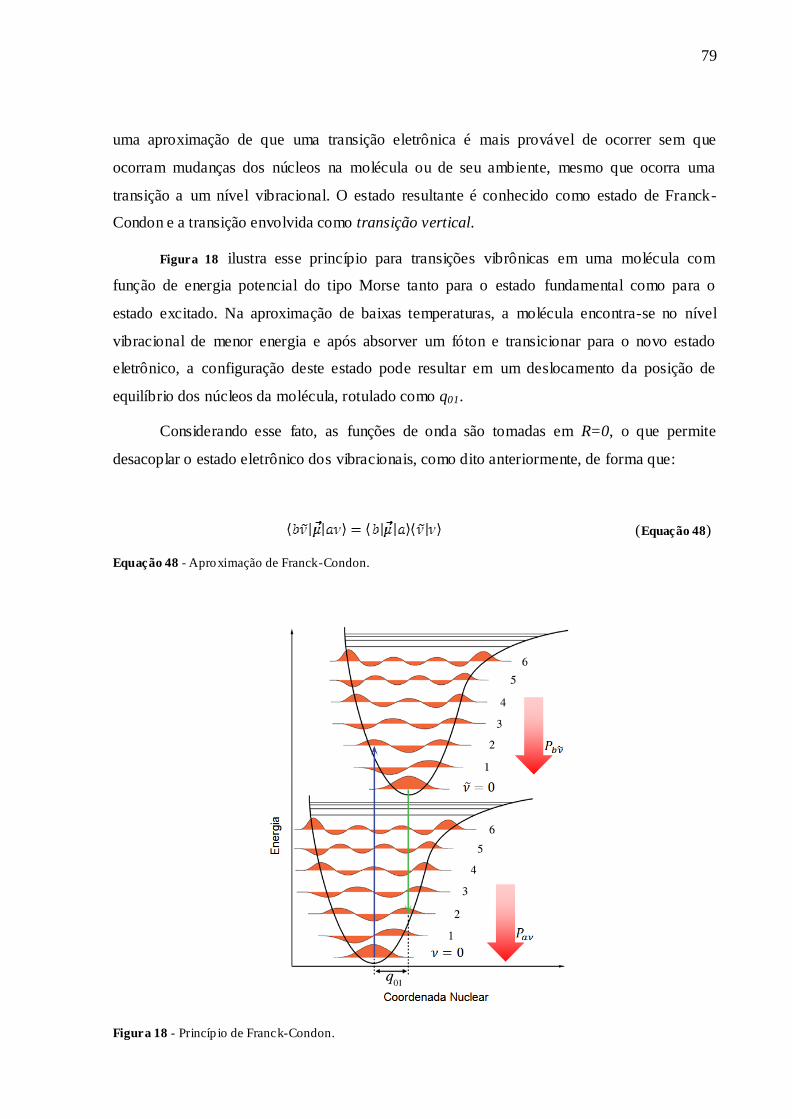
79
uma aproximação de que uma transição eletrônica é mais provável de ocorrer sem que
ocorram mudanças dos núcleos na molécula ou de seu ambiente, mesmo que ocorra uma
transição a um nível vibracional. O estado resultante é conhecido como estado de Franck-
Condon e a transição envolvida como transição vertical.
Figura 18 ilustra esse princípio para transições vibrônicas em uma molécula com
função de energia potencial do tipo Morse tanto para o estado fundamental como para o
estado excitado. Na aproximação de baixas temperaturas, a molécula encontra-se no nível
vibracional de menor energia e após absorver um fóton e transicionar para o novo estado
eletrônico, a configuração deste estado pode resultar em um deslocamento da posição de
equilíbrio dos núcleos da molécula, rotulado como q01.
Considerando esse fato, as funções de onda são tomadas em R=0, o que permite
desacoplar o estado eletrônico dos vibracionais, como dito anteriormente, de forma que:
(Equação 48)
Equação 48 - Aproximação de Franck-Condon.
Figura 18 - Princíp io de Franck-Condon.

80
E se considerarmos também a função densidade de estados na sua
forma integral, utilizando da aproximação de que os potenciais vibracionais do estado
fundamental e excitado são iguais, porém com os mínimos deslocados (Figura 18), obtém-se a
seguinte taxa de probabilidade de transição (ref. 66 tese do marletta):
(Equação 49)
Equação 49 - Taxa de probabilidade de transição entre dois níveis eletrônicos acoplados com níveis
vibracionais.
Onde é o elemento de matriz do dipolo elétrico e a integral que contém é
conhecida como integral de Franck-Condon e fornece de que forma os níveis eletrônicos e
vibracionais estão acoplados, definidas por:
(Equação 50)
Equação 50 - Integral de Franck-Condon.
Em que é a distribuição estatística de Bose-Einstein, aplicável para bósons
(fônons), e é conhecido como fator de Huang-Rhys para o -ésimo modo vibracional,
responsável pelo acoplamento e que expressa o quanto de energia é perd ida em forma de
ativação de modos vibrônicos (fônons) quando um elétron sofre uma transição vertical de um
estado excitado para um estado fundamental. Qualitativamente, o fator de Huang-Rhys (HR) é
definido como:
(Equação 51)
Equação 51 - Fator de Huang-Rhys.
Que também pode ser entendido como a relaxação de energia por unidade de energia
do fônon ativado na rede. Com uma matemática mais elaborada, o coeficiente de e missão
pode ser encontrado, a partir da Equação 49, e é dado por:

81
(Equação 52)
Equação 52 - Coeficiente de emissão com acoplamento elétron-fônon.
Em que descreve o efeito do meio, c é a velocidade da luz e é a
diferença de energia entre os estados eletrônicos localizados LUMO ( ) e HOMO ( ) do
segmento copolimérico com o maior comprimento de conjugação (máximo do espectro de
emissão) e d é um parâmetro que descreve a forma de linha gaussiana inomogênea do
espectro (ou seja, que considera a distribuição gaussiana de segmentos).
Os espectros ajustados por esse formalismo fornecem dados acerca dos acoplamentos
electrons-fônons dados pelo fator Huang-Rhys para o -ésimo modo vibracional acoplado
com o a-ésimo nível eletrônico. Assim, é possível mapear de forma bastante completa de que
forma níveis vibracionais estão sendo ativados em cada transição eletrônica, por uma
metodologia complementar a métodos mais caros e mais dispendiosos, como espectroscopia
Raman e Absorção de Infravermelho.
Após a apresentação de todo esse formalismo, o leitor que seja versado minimamente
em Física do Estado Sólido pode estar pensando de que forma é possível fazer considerações
sobre estrutura vibrônica intermediada por fônons em materiais amorfos, como os polímeros,
visto que essas quasipartículas são definidas só mediante a alta simetria da rede recíproca.
Além do que, o tratamento apresentado visa uma aplicação das matrizes em solução, sistema
que possui nenhuma cristalinidade e preenche muito mal as propriedades exigidas pelo Estado
Sólido para a definição de tais entes. Embora seja nebulosa semelhante abordagem e de difícil
explicação por um rigor matemático, similar descrição dos modos vibrônicos só é realizada
com a condição imposta de que a cadeia polimérica pode ser aproximada por uma cadeia
linear e infinita, com repetição periódica, em que estruturas complexas, como anéis
aromáticos, tiofênicos, pirrólicos, etc., são aproximados por um conceito de átomo efetivo da
rede, assumindo que seu comportamento possa ser ainda aproximado ao de outro átomo
qualquer que substitua essa estrutura, porém com o mesmo efeito sobre a rede. Sendo assim, é
pedido ao leitor a abstração para que visualize as vibrações da rede como quantizadas e ainda
assim, em uma estrutura ideal, como a cadeia linear infinita.

82
2.3.4 – O FORMALISMO DOS ÍNDICES DE REATIVIDADE DE FUKUI
A fim de se determinar em quais sítios a cadeia polimérica de FSE59 possui a maior
possibilidade de interagir com a radiação gama, realizou-se nesse trabalho um estudo dos
Índices de Reatividade de Fukui (Misra & Sannigrahi, 1996; Roy, Hirao, Krishnamurty, &
Pal, 2001) em duas configurações moleculares que serão apresentadas em detalhes na seção
seguinte. Dessa forma, tanto a reatividade da cadeia principal como a da cadeia lateral pode
ser determinada.
A Função de Fukui (Korchowiec, 2000; Méndez & Gázquez, 1994) descreve de
que forma a densidade eletrônica de uma determinada molécula varia em função da variação
do seu número de elétrons. Ou seja, matematicamente, por se tratar de uma taxa de variação,
fica determinada como uma derivada da densidade em relação ao número N de elétrons
dado um potencial externo constante:
(Equação 53)
Equação 53 - Função de Fukui.
A função de Fukui indica como a densidade eletrônica varia devido à retirada
(dopagem p) ou inclusão (dopagem n) de um elétron no sistema (com resultado „extrapolável‟
para maiores níveis de dopagem) para uma dada geometria molecular. Uma interpretação
complementar da função de Fukui entende o processo a variação do número de elétrons está
diretamente relacionada a uma alteração da densidade eletrônica dos orbitais HOMO e
LUMO da molécula, de forma que é capaz de informar de que forma esses orbitais são
modificados com um processo de dopagem, dentro de uma aproximação adiabática (do tipo
Born-Oppenheimer). Contudo, a interpretação mais prática dessa função se dá entendendo
como um indicativo da reatividade de um determinado átomo da molécula, ou seja, um
índice local, capaz de dizer onde na molécula é mais provável que um ataque eletrônico
ocorra. Além disso, a função de Fukui difere de outras grandezas como dureza (hardness) e
moleza (softness), muito utilizadas na literatura química, desde que estas são grandezas que
descrevem a reatividade global da molécula e não são capazes de fornecer informações sobre
a reatividade local, conforme aquela o é.

83
Em um sistema físico em que o número de elétrons é finito, como em uma molécula, a
derivada apresenta na Equação 54 é descontínua e assim existe uma dependência da função de
Fukui com a inserção ou retirada de elétrons. Para solucionar tal problema, são definidas três
funções distintas que descrevem o aumento, a redução ou a não alteração do número de
elétrons do sistema. Tais funções assim definidas são chamadas de Índices de Reatividade de
Fukui Condensados e são definidos, rigorosamente, por:
(Equação 54a)
(Equação 54b)
(Equação 54c)
Equação 54 - Funções de Fukui para (a) ataque nucleofílico, (b) eletrofílico e (c) ataque de radical.
Em que descreve o ataque nucleofílico (ataque por ânions, provocando
dopagem n), ataque eletrofílico (ataque por cátions, provocando dopagem p) e o
ataque de radicais livres. Uma forma mais prática para calcular esses Índices, proposta por
Yang e Mortier, considera a molécula de N elétrons cuja reatividade deseja-se determinar
como a espécie M, quando com excesso de elétrons (N+1) como espécie M- e quando com
falta (N-1) como espécie M+. Assim, os índices ficam determinados:
(Equação 55a)
(Equação 55b)
(Equação 55c)
Equação 55 - Índices Condensados de Fukui.
Sendo que corresponde a e assim sucessivamente; e ,
e representam a população eletrônica sobre o k-ésimo átomo da molécula M-, M+ e M,

84
calculada a partir da partição de cargas parciais de Mulliken ou de Merz-Singh-Kollman
(MSK), sendo a primeira a mais comumente utilizada. Além disso, mesmo sendo os cálculos
realizados para as configurações M-, M+ e M em uma mesma geometria (aproximação de
BO), a informação contida nos índices de Fukui dizem respeito à espécie M, cuja estrutura
otimizada pelo método AM1, nesse caso, deve ser levada em questão. Outros pontos
interessantes a se ressaltar é que, 1) quando um cálculo conformacional é realizado para
espécies carregadas, como é o caso de M- e M+, a função de onda molecular deve ainda ser
restrita, como é o caso do método Hartree-Fock Restrito (RHF), mesmo que a espécie tenha
camada aberta, a fim de se evitar o fenômeno de contaminação de spin caso o método não
restrito fosse utilizado (o que é bem discutido na referência XX); e 2) para o cálculo dessas
estruturas, a multiplicidade de spin é alterada seguindo a relação , onde S é a
projeção do número quântico de spin sobre o eixo Z das coordenadas eletrônicas, ou seja, a
multiplicidade da molécula passa de para durante o cálculo das espécies
carregadas.
2.3.5 – O SOFTWARE MOPAC2009®
O software utilizado para previsão das estruturas de equilíbrio deste trabalho foi o
MOPAC2009®, desenvolvido por James Stewart no grupo de Michael Dewar (Stewart,
2008). Os algoritmos implementados no código incluem praticamente todas as
parametrizações semi-empíricas, desde as mais antigas, como a MINDO, MNDO, AM1, PM3
até as mais recentes, como PM5 e PM6, cujos resultados têm sido citados pela literatura como
os mais próximos possíveis, a partir desse nível de teoria, dos cálculos ab initio. A licença
acadêmica do software é gratuita e existe vasta literatura de como ele pode ser utilizado.

85
CAPÍTULO III
Neste capítulo serão apresentados os resultados tanto experimentais como teóricos obtidos ao
longo desse trabalho. De forma a explicar os resultados experimentais, a modelagem
computacional realizada é apresentada, suprimindo detalhes que podem facilmente serem
encontrados em literatura especializada de Química Quântica.
3 – RESULTADOS
3.1 – EXPERIMENTAIS
As soluções poliméricas do FSE59 foram preparadas usando como solvente o
tetrahidrofurano (THF), em uma concentração de 0,04 mg/mL. As soluções foram
hermeticamente lacradas e irradiadas com gama em uma fonte de Cobalto-60 com doses de 20
e 40 kGy, na ausência de iluminação e oxigênio, para evitar fotodegradação. Um ponto
interessante a se ressaltar é a utilização do oligômero do polímero FSE59, conhecido como
SL128G. Essa molécula possui a mesma estrutura química do polímero, no entanto, com um
tamanho da cadeia muito bem definido, tendo apenas cinco anéis conjugados (fenileno-
tiofeno-fenileno-tiofeno-fenileno). A grafitização lateral do fenileno é similar àquela que
existe no FSE59 e a síntese desse material é obtida por alterações no processo de
polimerização que foi discutido na seção 2.1.1.

86
3.1.1 – ABSORÇÃO ÓPTICA
As medidas de absorção óptica foram realizadas em solução, usando uma cubeta com
passo de 2 mm, em um espectrofotômetro FEMTO 800XI UV-VIS-NIR, com range de 190 a
1100 nm, com resolução de 1 nm, usando como fonte de excitação uma lâmpada de Deutério-
Tungstênio.
A Figura 19 apresenta os espectros de absorção das amostras irradiadas com 20 e 40
kGy em comparação com aquela não irradiada, com ênfase na banda principal. Além disso, é
apresentado o espectro de absorção da amostra de SL128G preparada sob as mesmas
condições, não irradiada, a título de comparação, já que a estrutura do oligômero SL128G é
muito bem definida pela síntese. Percebe-se um deslocamento hipsocrômico (blue-shift) nos
espectros, o que pode ser atribuído à diminuição do tamanho do segmento conjugado,
responsável pela absorção. Observa-se também um alargamento espectral da banda principal.
Estudos anteriores não-publicados para esse tipo de polímero com o tamanho do braço lateral
(segmento R1/R2 da Figura 7) variável mostram que o espectro de absorção daqueles com os
braços mais curtos é mais largo do que aqueles com braços mais longos. Esse fato é um
indício de que o polímero pode estar sofrendo alterações nos braços laterais solubilizantes, o
que vai ser discutido na seção seguinte. Outro fator que também pode justificar o alargamento
da banda principal do espectro de absorção é a cisão heterogênea dos segmentos conjugados,
provocando o surgimento de diversos segmentos conjugados de tamanhos diferentes, cada um
absorvendo em uma região preferencial. O efeito disso no bulk provoca, pela propriedade
aditiva da absorção óptica, um alargamento espectral.
Em corroboração a essas hipóteses, nota-se que os espectros das amostras irradiadas
tendem, em energia, ao espectro do oligômero, enquanto que seguem um comportamento
oposto quanto à largura. Por ser o oligômero uma estrutura bem definida, sua banda principal
de absorção pode facilmente ser ajustada por uma única lorentziana e nesse caso não existe
nenhum sentido em um ajuste gaussiânico, conforme discutido no capítulo anterior, desde que
não existe uma polidispersão gaussiana dos segmentos em torno de um valor médio. Essa
tendência em energia ao oligômero em face do polímero pelas amostras irradiadas pode
explicar a cisão dos segmentos absorvedores enquanto que o comportamento da largura de
linha pode explicar a polidispersão dos segmentos conjugados e uma cisão dos segmentos
laterais. Esses fatos serão analisados com mais detalhes no capítulo seguinte.
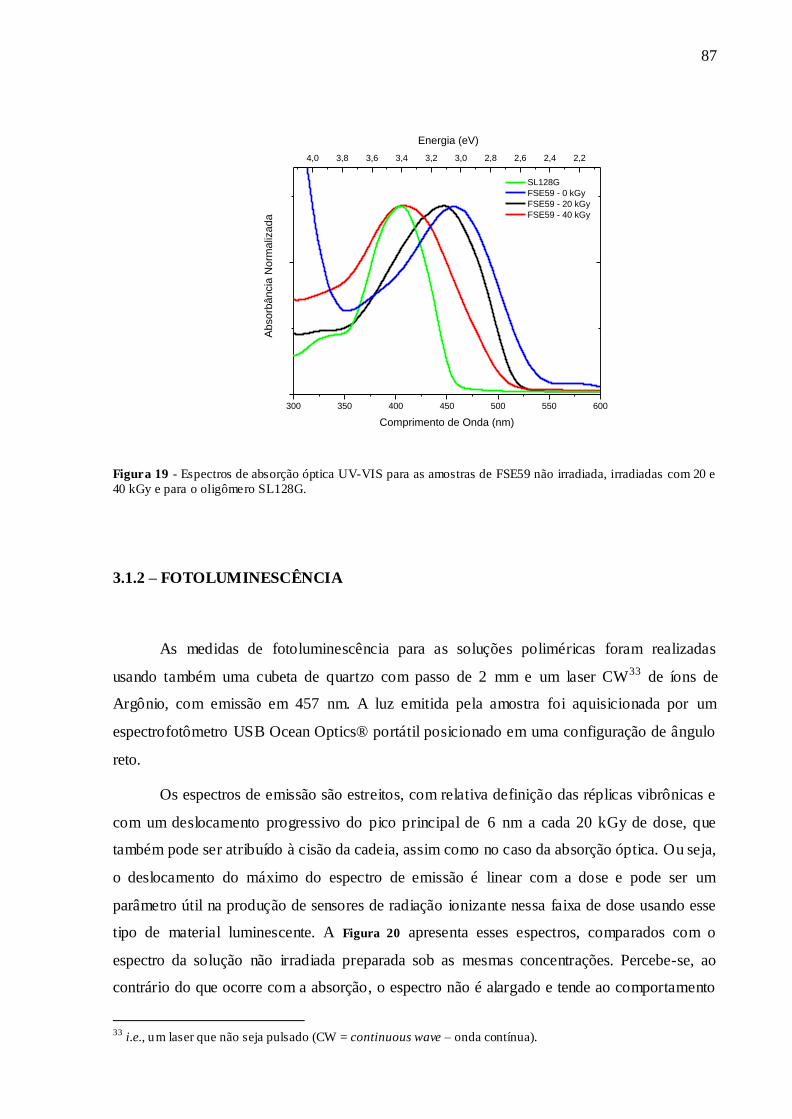
87
300 350 400 450 500 550 600
4,0 3,8 3,6 3,4 3,2 3,0 2,8 2,6 2,4 2,2
SL128G
FSE59 - 0 kGy
FSE59 - 20 kGy
FSE59 - 40 kGy
Ab
so
rbâ
ncia
No
rma
liza
da
Comprimento de Onda (nm)
Energia (eV)
Figura 19 - Espectros de absorção óptica UV-VIS para as amostras de FSE59 não irradiada, irradiadas com 20 e
40 kGy e para o oligômero SL128G.
3.1.2 – FOTOLUMINESCÊNCIA
As medidas de fotoluminescência para as soluções poliméricas foram realizadas
usando também uma cubeta de quartzo com passo de 2 mm e um laser CW33 de íons de
Argônio, com emissão em 457 nm. A luz emitida pela amostra foi aquisicionada por um
espectrofotômetro USB Ocean Optics® portátil posicionado em uma configuração de ângulo
reto.
Os espectros de emissão são estreitos, com relativa definição das réplicas vibrônicas e
com um deslocamento progressivo do pico principal de 6 nm a cada 20 kGy de dose, que
também pode ser atribuído à cisão da cadeia, assim como no caso da absorção óptica. Ou seja,
o deslocamento do máximo do espectro de emissão é linear com a dose e pode ser um
parâmetro útil na produção de sensores de radiação ionizante nessa faixa de dose usando esse
tipo de material luminescente. A Figura 20 apresenta esses espectros, comparados com o
espectro da solução não irradiada preparada sob as mesmas concentrações. Percebe-se, ao
contrário do que ocorre com a absorção, o espectro não é alargado e tende ao comportamento
33
i.e., um laser que não seja pulsado (CW = continuous wave – onda contínua).
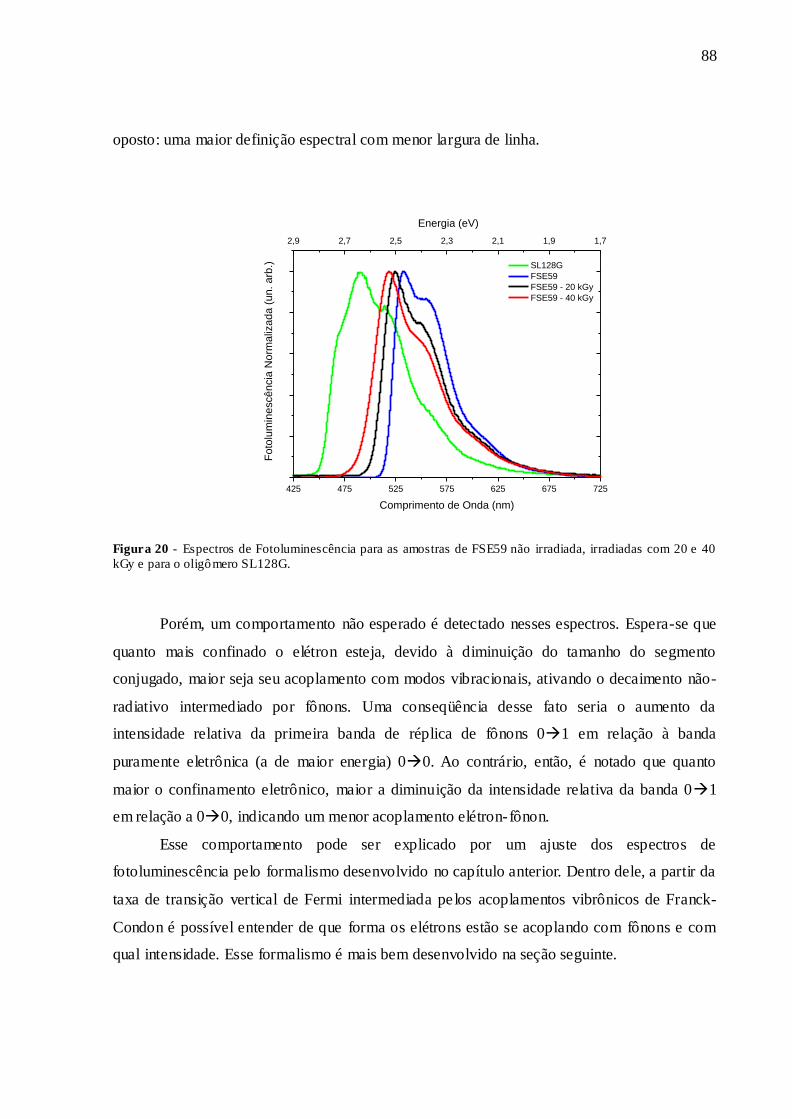
88
oposto: uma maior definição espectral com menor largura de linha.
425 475 525 575 625 675 725
2,9 2,7 2,5 2,3 2,1 1,9 1,7
SL128G
FSE59
FSE59 - 20 kGy
FSE59 - 40 kGy
Fo
tolu
min
escê
ncia
No
rma
liza
da
(u
n. a
rb.)
Comprimento de Onda (nm)
Energia (eV)
Figura 20 - Espectros de Fotoluminescência para as amostras de FSE59 não irradiada, irradiadas com 20 e 40
kGy e para o oligômero SL128G.
Porém, um comportamento não esperado é detectado nesses espectros. Espera-se que
quanto mais confinado o elétron esteja, devido à diminuição do tamanho do segmento
conjugado, maior seja seu acoplamento com modos vibracionais, ativando o decaimento não-
radiativo intermediado por fônons. Uma conseqüência desse fato seria o aumento da
intensidade relativa da primeira banda de réplica de fônons 01 em relação à banda
puramente eletrônica (a de maior energia) 00. Ao contrário, então, é notado que quanto
maior o confinamento eletrônico, maior a diminuição da intensidade relativa da banda 01
em relação a 00, indicando um menor acoplamento elétron-fônon.
Esse comportamento pode ser explicado por um ajuste dos espectros de
fotoluminescência pelo formalismo desenvolvido no capítulo anterior. Dentro dele, a partir da
taxa de transição vertical de Fermi intermediada pelos acoplamentos vibrônicos de Franck-
Condon é possível entender de que forma os elétrons estão se acoplando com fônons e com
qual intensidade. Esse formalismo é mais bem desenvolvido na seção seguinte.

89
3.2 – TEÓRICOS
Tendo isso em vista, para compreender melhor como a interação induz defeitos na
estrutura polimérica, mesmo com um solvente orgânico que não produz deliberadamente
radicais livres (não-halogenado), faz-se necessário o apelo para uma modelagem teórica que
seja capaz de explicar como as propriedades estruturais e eletrônicas do polímero fazem com
que a interação ocorra preferencialmente em determinados sítios. Além disso, faz-se
necessário identificar os elementos estruturais e eletrônicos alterados pela interação com a
radiação e como isso pode alterar substancialmente o rendimento de dispositivos constituídos
a partir de materiais tratados dessa maneira.
3.2.1 – GEOMETRIAS DE EQUILÍBRIO DO ESTADO FUNDAMENTAL
A fim de entender como esses parâmetros interferem nas propriedades eletrônicas do
polímero e de que forma ele interage com a radiação gama, primeiramente as geometrias de
oligômeros com número crescente de monômeros foram otimizadas usando o formalismo
semi-empírico de Hartree-Fock, sob a parametrização AM1. Esse formalismo foi escolhido
tendo em vista a boa concordância dos resultados com aqueles obtidos por cálculos com nível
de teoria mais rigoroso e que demandam um maior custo computacional, como é o caso da
teoria do funcional da densidade (DFT). As geometrias foram otimizadas a partir de uma
geometria inicial planar, isotática, com um critério de convergência criterioso.
Depois de otimizadas, as geometrias apresentam uma torção helicoidal da cadeia,
tendo em vista a atração das pontes de sulfeto entre o oxigênio do grupo solubilizante e o
enxofre do anel tiofênico. Além disso, essas interações induzem ângulos diedros da cadeia
que variam de 23 a 63º, que tende a um valor constante a partir de 14 anéis, o que não é capaz
de provocar quebra de conjugação com um tamanho maior dos oligômeros. Sendo assim,
espera-se que os espectros de absorção simulados para os oligômeros em função do número
de segmentos conjugados para essas estruturas apresentem um comportamento batocrômico,
ou seja, as energias das transições eletrônicas devem diminuir conforme a cadeia aumente,
tendo em vista a maior delocalização eletrônica.

90
No entanto, antes de apresentar as geometrias otimizadas dos oligômeros com número
crescente de anéis, é interessante destacar o procedimento teórico utilizado para o cálculo
dessas estruturas: como os braços solubilizantes grafitizados aos anéis fenilênicos possuem
vários graus de liberdade, a convergência do cálculo a uma estrutura cuja energia seja um
mínimo global torna-se computacionalmente cara e duvidosa, desde que existem vários
mínimos locais na hipersuperfície de energia potencial. Sendo assim, os braços com três
oxigênios e terminação butílica, como aqueles indicados na Figura 8, são substituídos por um
grupo alcóxico composto de um único oxigênio e um metil, o que reduz o custo
computacional e não altera as propriedades eletrônicas da cadeia conjugada, desde que
estudos mostram que o efeito doador de elétrons pelo grupo alcóxico é intermediado pelo
primeiro oxigênio somente, sendo que o grupo que vem depois dele é de interesse quando se
trata de bulk, pelas razões já apresentadas no Capítulo II, seção 2.1.1 (solubilidade e formação
de filmes finos em substratos).
Considerando isso, abaixo (Figura 21) são apresentadas as geometrias de equilíbrio do
estado fundamental para as estruturas oligoméricas com número crescente de anéis
conjugados, aos pares, para 2, 4, 6 e 16 anéis. É apresentada, especialmente, a estrutura
reduzida34 do oligômero SL128G, com 5 anéis conjugados.
É interessante ressaltar que a otimização de estruturas com mais de 20 anéis
conjugados de comprimento se torna dispendiosa temporal e computacionalmente, desde que
o tempo de processamento em métodos derivados de Hartree-Fock possui custo
computacional proporcional a M3, onde M é o número de átomos da simulação. Além disso,
entende-se que, para os objetivos dessa simulação, esse conjunto de geometrias já seja
suficiente para criar uma base de transições eletrônicas que seja capaz de, a partir de uma
equação geral que as descreva segundo um modelo apropriado, estimar a quantidade de anéis
conjugados das amostras poliméricas após a irradiação. Essa metodologia é desenvolvida na
seção seguinte.
34
Entende-se como estrutura reduzida das geometrias como sendo aquelas que possuem o braço lateral reduzido
para as otimizações teóricas.
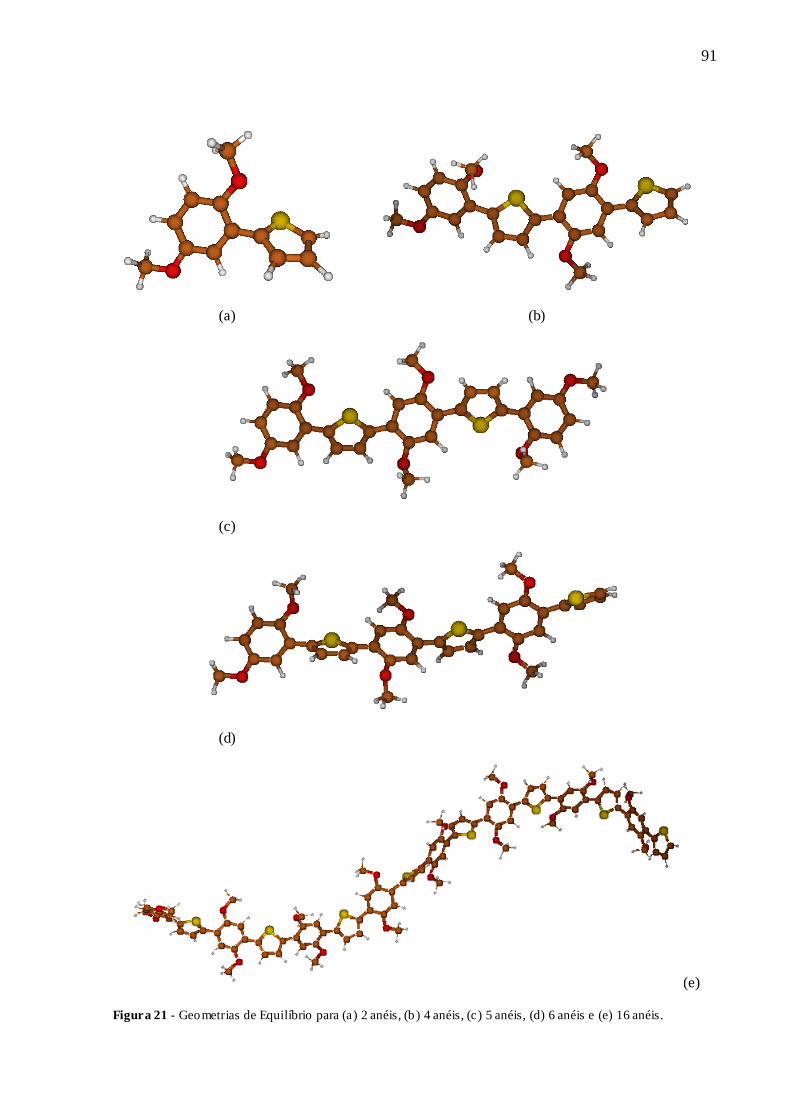
91
(a) (b)
(c)
(d)
(e)
Figura 21 - Geometrias de Equilíbrio para (a) 2 anéis, (b ) 4 anéis, (c) 5 anéis, (d) 6 anéis e (e) 16 anéis.

92
3.2.2 – TRANSIÇÕES ELETRÔNICAS EM FUNÇÃO DO TAMANHO DO
SEGMENTO CONJUGADO
A fim de estimar o grau de cisão da cadeia polimérica em função da dose irradiada, é
traçada uma metodologia que pretende estimar o tamanho da cadeia polimérica do FSE59 a
partir dos seus espectros de absorção das soluções irradiadas. O caminho traçado é
esquematizado da seguinte maneira: primeiramente, a partir das geometrias de equilíbrio
calculadas pela metodologia apresentada, 10 transições eletrônicas UV-VIS são calculadas ao
nível de teoria ZINDO/S-CI, conforme apresentado no Capítulo II; essas transições
eletrônicas, são envelopadas com lorentzianas de 35 nm de largura à meia altura35 (FWHM –
Full-Width at Half Maximum) e a soma desses envelopes lorentzianos assume-se ser um
espectro de absorção óptica UV-VIS teórico; tendo esses espectros de absorção em função do
número de anéis conjugados da cadeia oligomérica, ajusta-se o máximo dos espectros em
função do tamanho da cadeia por uma função hiperbólica36 (Equação 56). Tendo esse ajuste, o
espectro de absorção da estrutura SL128G é calculado e comparado com o seu espectro
experimental (Figura 23), e então, a partir disso, a discrepância teórico-experimental é
estabelecida, para que os outros espectros sejam corrigidos para o número de anéis
conjugados. Nessa passagem é assumido que a discrepância entre os resultados teóricos e
experimentais inserida pelo método mantém-se constante e independente do tamanho da
cadeia, o que é plausível tendo em vista que nenhum novo parâmetro além desse foi inserido.
A equação usada para o ajuste dos máximos do espectro de absorção teóricos (leia-se
gap óptico) é:
(Equação 56)
Equação 56 - Equação de ajuste hiperbólico pelo modelo do elétron confinado em uma caixa.
35
Escolheu-se essa FWHM desde que, quando ajustado, o espectro de absorção experimental do FSE59
apresenta três transições cujas lorentzianas que as descrevem possuem justamente esse valor de largura à meia
altura. Assim, os espectros ditos teóricos podem ser comparados a pé de igualdade com os espectros
experimentais. 36
A função de ajuste hiperbólica foi escolhida partindo do modelo quântico do elétron preso em um poço de
potencial unidimensional: as autoenergias que são solução da equação de Schrondiger são proporcionais a L-2
,
onde L é a largura do poço (leia-se número de anéis conjugados). Mas, como uma função L-2
possui o mesmo
comportamento do que uma função com dependência L-1
, além desta possuir menor grau de liberdade, preferiu-
se utilizá-la como ajuste, assumindo este modelo como uma boa aproximação para o confinamento eletrônico
unidimensional que ocorre em moléculas lineares conjugadas.

93
Em que os parâmetros u, v e k foram mantidos livres para variar. Após o ajuste, o
parâmetro aditivo u foi desconsiderado e a equação, agora em função de u e p foi re-ajustada
para a situação em que p=5 (estrutura do SL128G) e então o parâmetro u foi determinado.
Tendo isso em vista, a equação encontrada que descreve o comportamento semi-empírico do
gap em função do número de anéis conjugados de oligômeros de FSE59 é:
(Equação 57)
Equação 57 - A juste hiperbólico com inclusão do parâmetro corret ivo.
Onde o parâmetro u assume claramente o papel da energia de gap para a molécula
infinita, ou seja, quando , o que está condizente com resultados experimentais obtidos
anteriormente em medidas de baixas temperaturas, por Silva e colaboradores, 2010. Além
disso, o parâmetro k é muitas vezes desprezado na literatura que tenta descrever esse tipo de
comportamento, tendo em vista que se entende que o modelo do elétron confinado em uma
caixa não apresenta um termo aditivo corretivo como esse. Preferiu-se usar tal termo nesse
ajuste semi-empírico a fim de que ele corrija levemente efeitos de desordem e
inomogeneidade da amostra submetida ao processo de radiação, não sendo então justificável
pelo modelo e sim como uma ferramenta através de qual seja possível incluir efeitos de
desordem não-controlada. Desde que os valores de p são números inteiros maiores que a
unidade, um parâmetro corretivo da ordem do valor encontrado não inviabiliza o ajuste, pelo
contrário, corrobora o fato de que a cisão heterogênea induzida pela radiação é de certa forma
„controlada‟, por razões que se verão brevemente.
Os espectros de absorção calculados são apresentados na Figura 22 em função do
número p de anéis. Na Figura 23 a comparação entre os espectros é apresentada, mostrando a
discrepância teórico-experimental introduzida pelo cálculo ZINDO/S-CI.
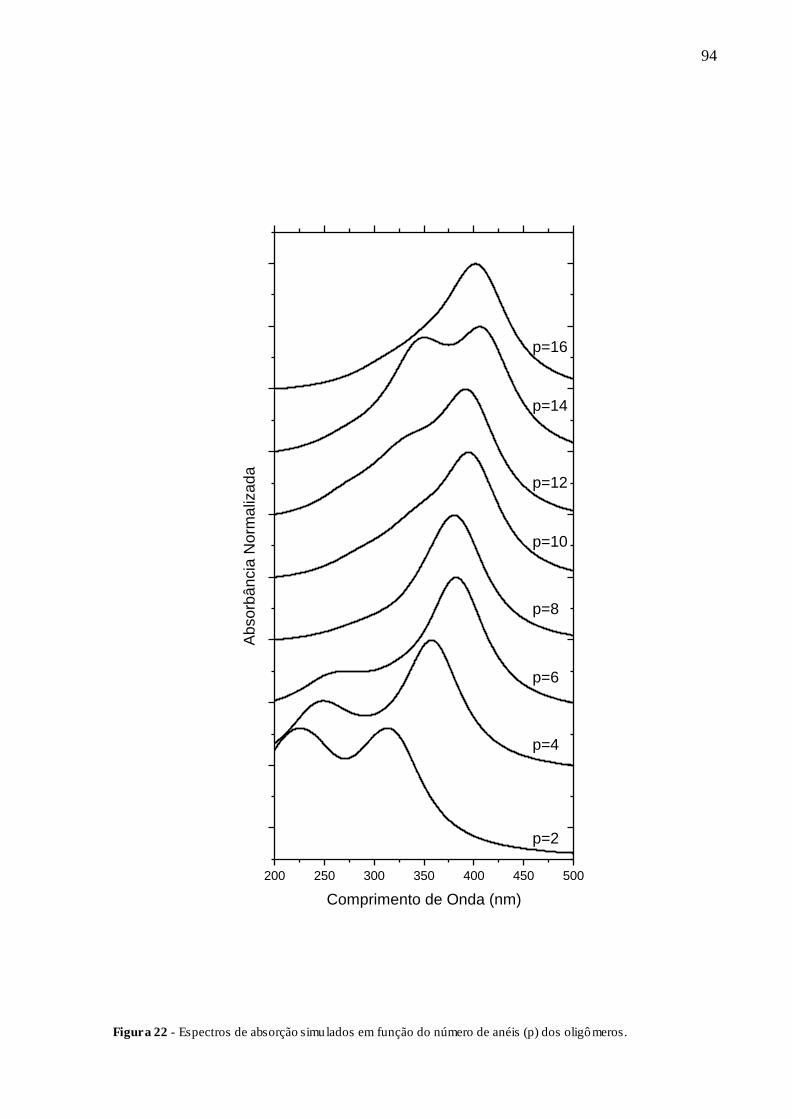
94
200 250 300 350 400 450 500
p=2
Comprimento de Onda (nm)
Ab
so
rbâ
ncia
No
rma
liza
da
p=4
p=6
p=8
p=10
p=12
p=16
p=14
Figura 22 - Espectros de absorção simulados em função do número de anéis (p) dos oligômeros.
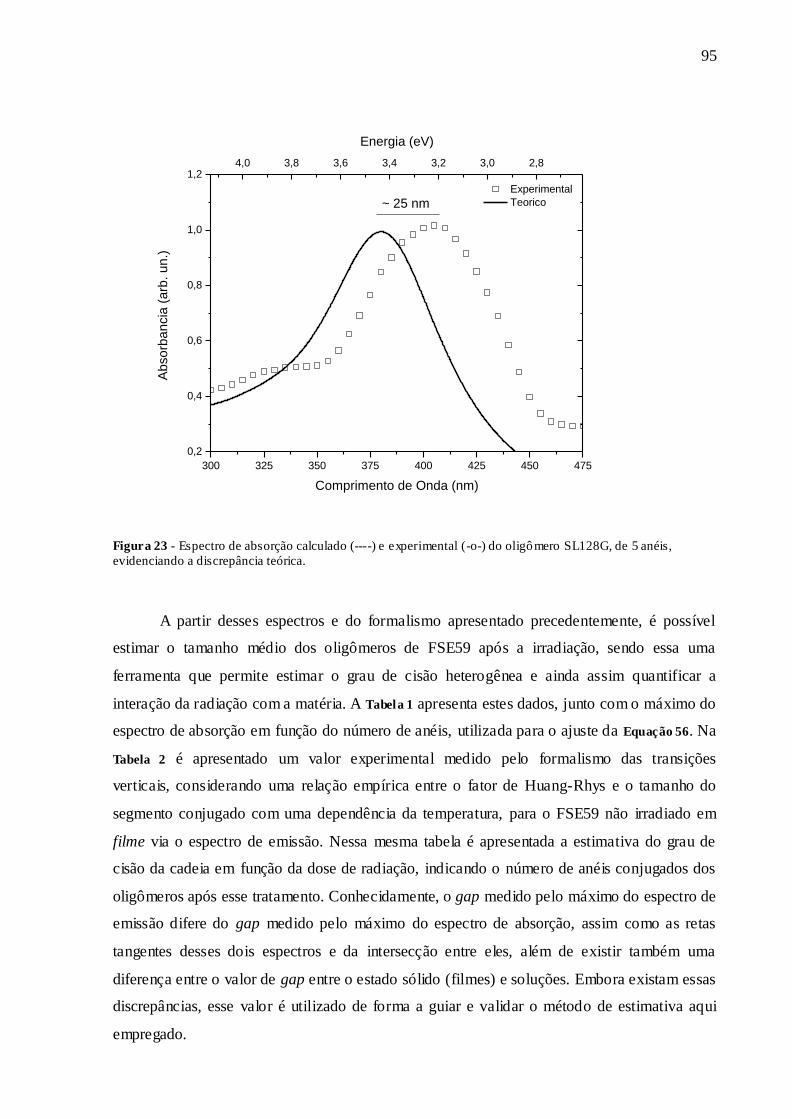
95
300 325 350 375 400 425 450 475
0,2
0,4
0,6
0,8
1,0
1,2
A
bso
rba
ncia
(a
rb. u
n.)
Comprimento de Onda (nm)
Experimental
Teorico
4,0 3,8 3,6 3,4 3,2 3,0 2,8
Energia (eV)
~ 25 nm
Figura 23 - Espectro de absorção calculado (----) e experimental (-o-) do oligômero SL128G, de 5 anéis,
evidenciando a discrepância teórica.
A partir desses espectros e do formalismo apresentado precedentemente, é possível
estimar o tamanho médio dos oligômeros de FSE59 após a irradiação, sendo essa uma
ferramenta que permite estimar o grau de cisão heterogênea e ainda assim quantificar a
interação da radiação com a matéria. A Tabela 1 apresenta estes dados, junto com o máximo do
espectro de absorção em função do número de anéis, utilizada para o ajuste da Equação 56. Na
Tabela 2 é apresentado um valor experimental medido pelo formalismo das transições
verticais, considerando uma relação empírica entre o fator de Huang-Rhys e o tamanho do
segmento conjugado com uma dependência da temperatura, para o FSE59 não irradiado em
filme via o espectro de emissão. Nessa mesma tabela é apresentada a estimativa do grau de
cisão da cadeia em função da dose de radiação, indicando o número de anéis conjugados dos
oligômeros após esse tratamento. Conhecidamente, o gap medido pelo máximo do espectro de
emissão difere do gap medido pelo máximo do espectro de absorção, assim como as retas
tangentes desses dois espectros e da intersecção entre eles, além de existir também uma
diferença entre o valor de gap entre o estado sólido (filmes) e soluções. Embora existam essas
discrepâncias, esse valor é utilizado de forma a guiar e validar o método de estimativa aqui
empregado.
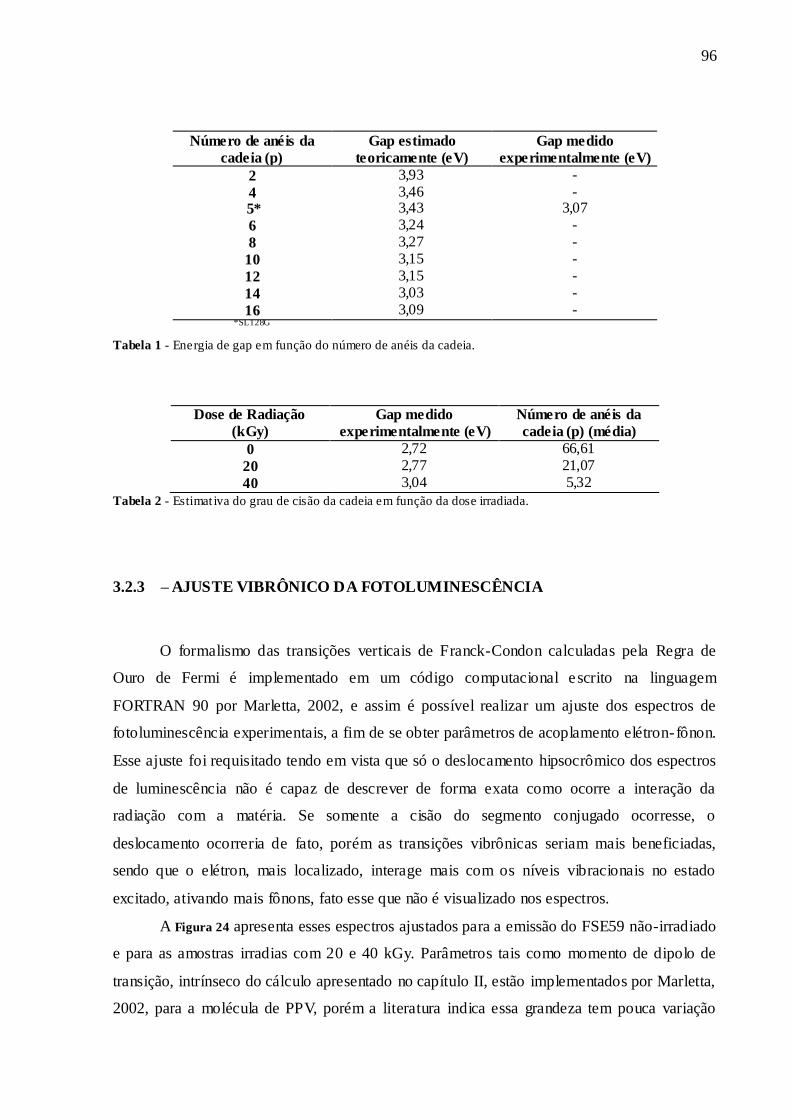
96
Número de anéis da
cadeia (p)
Gap estimado
teoricamente (eV)
Gap medido
experimentalmente (eV)
2 3,93 -
4 3,46 - 5* 3,43 3,07
6 3,24 -
8 3,27 -
10 3,15 -
12 3,15 -
14 3,03 -
16 3,09 - *SL128G
Tabela 1 - Energia de gap em função do número de anéis da cadeia.
Dose de Radiação
(kGy)
Gap medido
experimentalmente (eV)
Número de anéis da
cadeia (p) (média)
0 2,72 66,61
20 2,77 21,07
40 3,04 5,32
Tabela 2 - Estimat iva do grau de cisão da cadeia em função da dose irradiada.
3.2.3 – AJUSTE VIBRÔNICO DA FOTOLUMINESCÊNCIA
O formalismo das transições verticais de Franck-Condon calculadas pela Regra de
Ouro de Fermi é implementado em um código computacional escrito na linguagem
FORTRAN 90 por Marletta, 2002, e assim é possível realizar um ajuste dos espectros de
fotoluminescência experimentais, a fim de se obter parâmetros de acoplamento elétron-fônon.
Esse ajuste foi requisitado tendo em vista que só o deslocamento hipsocrômico dos espectros
de luminescência não é capaz de descrever de forma exata como ocorre a interação da
radiação com a matéria. Se somente a cisão do segmento conjugado ocorresse, o
deslocamento ocorreria de fato, porém as transições vibrônicas seriam mais beneficiadas,
sendo que o elétron, mais localizado, interage mais com os níveis vibracionais no estado
excitado, ativando mais fônons, fato esse que não é visualizado nos espectros.
A Figura 24 apresenta esses espectros ajustados para a emissão do FSE59 não-irradiado
e para as amostras irradias com 20 e 40 kGy. Parâmetros tais como momento de dipolo de
transição, intrínseco do cálculo apresentado no capítulo II, estão implementados por Marletta,
2002, para a molécula de PPV, porém a literatura indica essa grandeza tem pouca variação
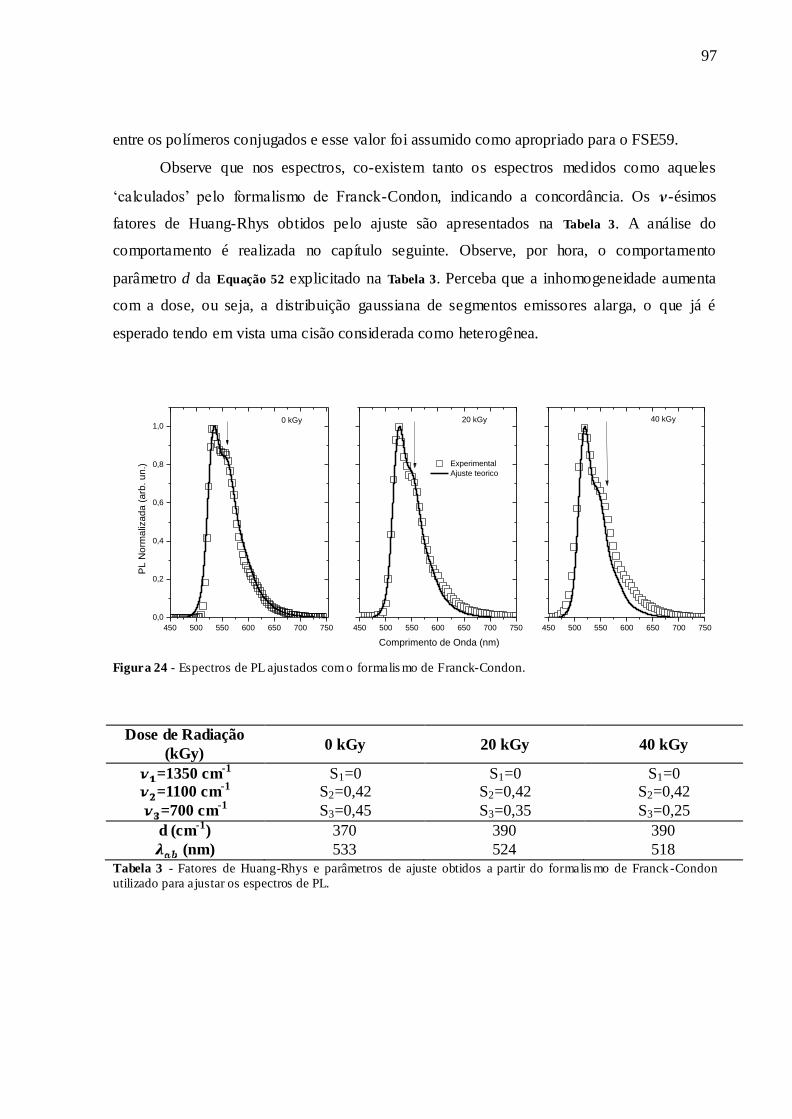
97
entre os polímeros conjugados e esse valor foi assumido como apropriado para o FSE59.
Observe que nos espectros, co-existem tanto os espectros medidos como aqueles
„calculados‟ pelo formalismo de Franck-Condon, indicando a concordância. Os -ésimos
fatores de Huang-Rhys obtidos pelo ajuste são apresentados na Tabela 3. A análise do
comportamento é realizada no capítulo seguinte. Observe, por hora, o comportamento
parâmetro d da Equação 52 explicitado na Tabela 3. Perceba que a inhomogeneidade aumenta
com a dose, ou seja, a distribuição gaussiana de segmentos emissores alarga, o que já é
esperado tendo em vista uma cisão considerada como heterogênea.
450 500 550 600 650 700 750 450 500 550 600 650 700 750450 500 550 600 650 700 750
0,0
0,2
0,4
0,6
0,8
1,00 kGy
Comprimento de Onda (nm)
20 kGy
40 kGy
Experimental
Ajuste teorico
PL
No
rma
liza
da
(a
rb. u
n.)
Figura 24 - Espectros de PL ajustados com o formalis mo de Franck-Condon.
Dose de Radiação
(kGy) 0 kGy 20 kGy 40 kGy
=1350 cm-1 S1=0 S1=0 S1=0 =1100 cm-1 S2=0,42 S2=0,42 S2=0,42
=700 cm-1 S3=0,45 S3=0,35 S3=0,25
d (cm-1) 370 390 390
(nm) 533 524 518 Tabela 3 - Fatores de Huang-Rhys e parâmetros de ajuste obtidos a partir do formalis mo de Franck-Condon
utilizado para ajustar os espectros de PL.

98
3.2.4 – ÍNDICES DE REATIVIDADE DE FUKUI
De forma a elucidar o tipo de cisão fotoinduzida que ocorre e em quais sítios isso
acontece, os índices de reatividade de Fukui foram calculados. Eles, como dito anteriormente,
permitem avaliar como a densidade de carga varia em cada sítio atômico da cadeia, indicando
assim quais são as regiões mais propensas a sofrer um ataque de radical ou sofrer uma
alteração físico-química. Para tanto, consideramos duas estruturas para avaliar esse índice
para a cadeia infinita idealizada: a primeira, chamada de estrutura A (Figura 25), que é o
trímero (6 anéis) do FSE59 com a estrutura reduzida, ou seja, com os braços laterais alcóxico-
metílicos; e a segunda, chamada de estrutura B (Figura 26), que é um monômero (2 anéis) com
a estrutura total, não-reduzida, com os braços laterais como o experimental.
Para a estrutura A, o comportamento da reatividade da cadeia principal conjugada
pode ser monitorado, enquanto que para a estrutura B esse comportamento já foi monitorado
para a cadeia grafitizada à principal, saturada (ramo solubilizante). Essa metodologia foi
utilizada tendo em vista as razões indiciadas pelos espectros de fotoluminescência em função
da dose de radiação, que não podem ser explicados somente pela cisão da cadeia principal e
conseqüente diminuição do segmento conjugado emissor. Além disso, as mesmas estruturas
não puderam ser otimizadas e ter os índices de Fukui calculados devido ao alto custo
computacional envolvido em otimizar geometrias radicalares. Além disso, a presença dos
braços solubilizantes em estruturas extensas, como já é o caso do trímero, por possuírem
vários graus de liberdade com geometrias isoenergéticas, as estruturas obtidas não são
totalmente confiáveis.
Figura 25 - Estrutura A.

99
Figura 26 - Estrutura B.
A apresentação dos índices de Fukui será feita separadamente para a estrutura A e
estrutura B.
Estrutura A: A Figura 27 apresenta a variação da carga medida pelos índices de
Fukui quando um ataque de radical ocorre e a carga é inalterada (situação Δq=0) ( ),
situação em que há um ataque nucleofílico ( ) e a situação em que ocorre um ataque
eletrofílico ( ). O ataque nucleofílico ocorre por uma espécie que possui um par de elétrons
para efetuar ligações e se liga a moléculas que sejam capazes de comportar esses elétrons, ou
seja, é um ataque por conta de um ânion, que cede elétrons para a estrutura polimérica, no
caso, caracterizando uma situação de n-doping (dopagem do tipo n). Por outro lado, o ataque
eletrofílico se dá por uma espécie com alta afinidade eletrônica e se liga a estruturas capazes
de doar elétrons, caracterizando assim uma situação de p-doping.
Já a Figura 28 apresenta a variação da densidade de carga na cadeia, tomada como a
derivada primeira do polinômio interpolador da variação de carga, dados pelo índice de Fukui
de ataque radical. Observe que as maiores variações ocorrem para os átomos de carbono 1, 4,
11, 14, 21 e 24 e para os átomos de enxofre 38, 39 e 40. Para os átomos de enxofre, tal
variação era esperada, tendo em vista sua alta eletronegatividade. É interessante, no entanto,
notar que os átomos de carbono onde essa variação é maior são aqueles do anel tiofênico que
se ligam ao anel fenilênico e, portanto, essa região da cadeia é a mais susceptível de interação.
Observe que não há indícios de quebra dos anéis tiofênicos e fenilênicos, já que a reatividade
dos outros átomos dos anéis é baixa. A conservação do comportamento eletrônico do polímero
também corrobora esse fato.
Assumindo que qualquer reação de cisão que ocorra na cadeia polimérica ocorra nos
sítios mais reativos e que a reatividade do sítio é diretamente proporcional à sua densidade de
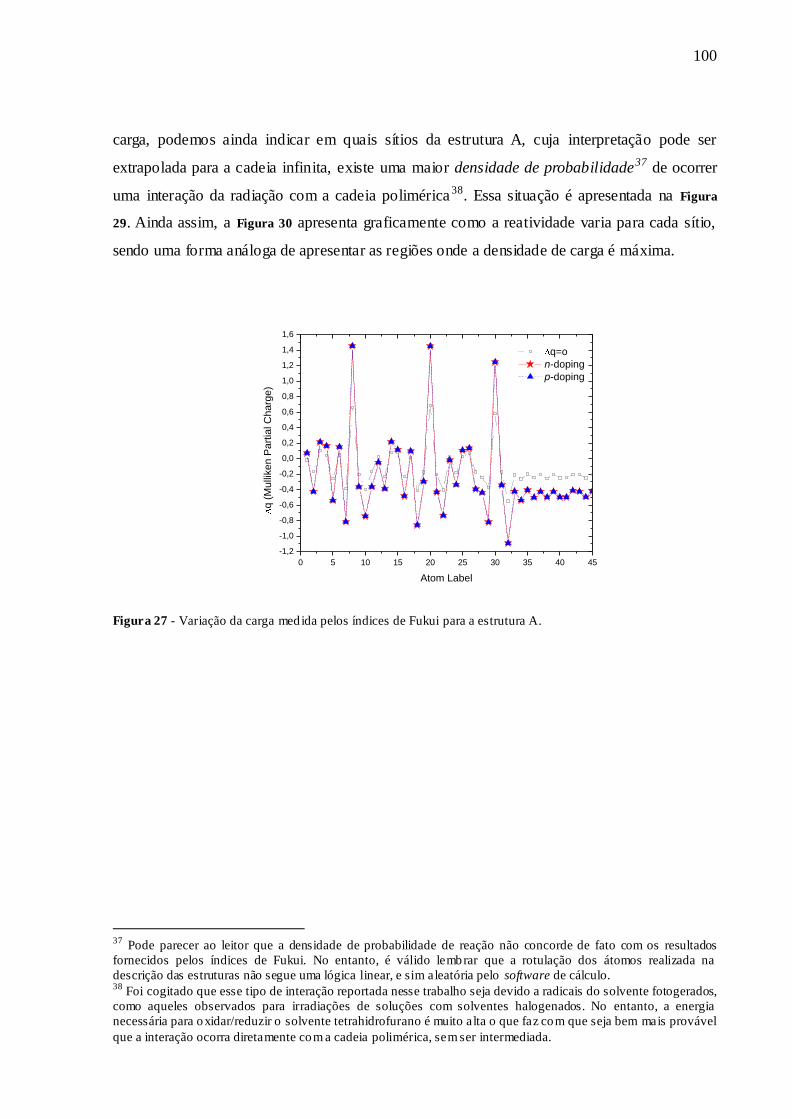
100
carga, podemos ainda indicar em quais sítios da estrutura A, cuja interpretação pode ser
extrapolada para a cadeia infinita, existe uma maior densidade de probabilidade37 de ocorrer
uma interação da radiação com a cadeia polimérica38. Essa situação é apresentada na Figura
29. Ainda assim, a Figura 30 apresenta graficamente como a reatividade varia para cada sítio,
sendo uma forma análoga de apresentar as regiões onde a densidade de carga é máxima.
0 5 10 15 20 25 30 35 40 45
-1,2
-1,0
-0,8
-0,6
-0,4
-0,2
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
q (
Mu
llike
n P
art
ial C
ha
rge
)
Atom Label
q=o
n-doping
p-doping
Figura 27 - Variação da carga medida pelos índices de Fukui para a estrutura A.
37
Pode parecer ao leitor que a densidade de probabilidade de reação não concorde de fato com os resultados
fornecidos pelos índices de Fukui. No entanto, é válido lembrar que a rotulação dos átomos realizada na
descrição das estruturas não segue uma lógica linear, e sim aleatória pelo software de cálculo. 38
Foi cogitado que esse tipo de interação reportada nesse trabalho seja devido a radicais do solvente fotogerados,
como aqueles observados para irradiações de soluções com solventes halogenados. No entanto, a energia
necessária para oxidar/reduzir o solvente tetrahidrofurano é muito alta o que faz com que seja bem mais provável
que a interação ocorra diretamente com a cadeia polimérica, sem ser intermediada.
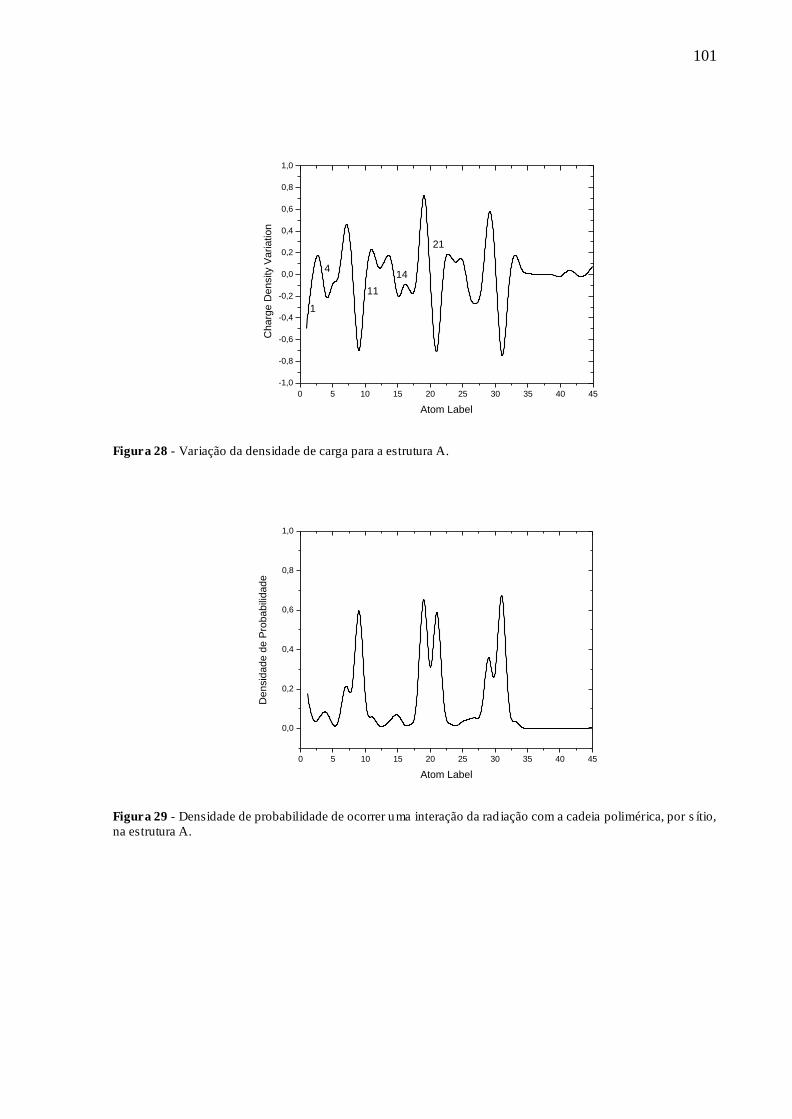
101
0 5 10 15 20 25 30 35 40 45
-1,0
-0,8
-0,6
-0,4
-0,2
0,0
0,2
0,4
0,6
0,8
1,0
Ch
arg
e D
en
sity V
ari
atio
n
Atom Label
1
4
11
14
21
Figura 28 - Variação da densidade de carga para a estrutura A.
0 5 10 15 20 25 30 35 40 45
0,0
0,2
0,4
0,6
0,8
1,0
De
nsid
ad
e d
e P
rob
ab
ilid
ad
e
Atom Label
Figura 29 - Densidade de probabilidade de ocorrer uma interação da rad iação com a cadeia polimérica, por s ítio,
na estrutura A.
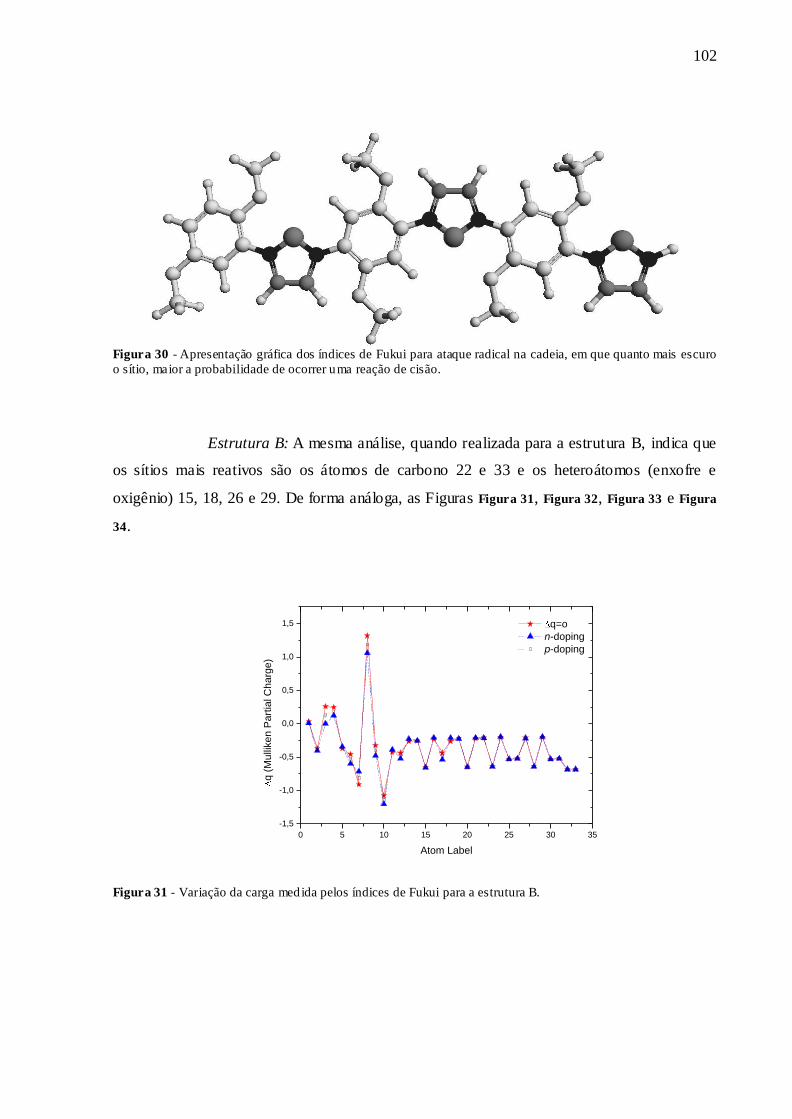
102
Figura 30 - Apresentação gráfica dos índices de Fukui para ataque radical na cadeia, em que quanto mais escuro
o sítio, maior a probabilidade de ocorrer uma reação de cisão.
Estrutura B: A mesma análise, quando realizada para a estrutura B, indica que
os sítios mais reativos são os átomos de carbono 22 e 33 e os heteroátomos (enxofre e
oxigênio) 15, 18, 26 e 29. De forma análoga, as Figuras Figura 31, Figura 32, Figura 33 e Figura
34.
0 5 10 15 20 25 30 35
-1,5
-1,0
-0,5
0,0
0,5
1,0
1,5 q=o
n-doping
p-doping
Atom Label
q (
Mu
llike
n P
art
ial C
ha
rge
)
Figura 31 - Variação da carga medida pelos índices de Fukui para a estrutura B.
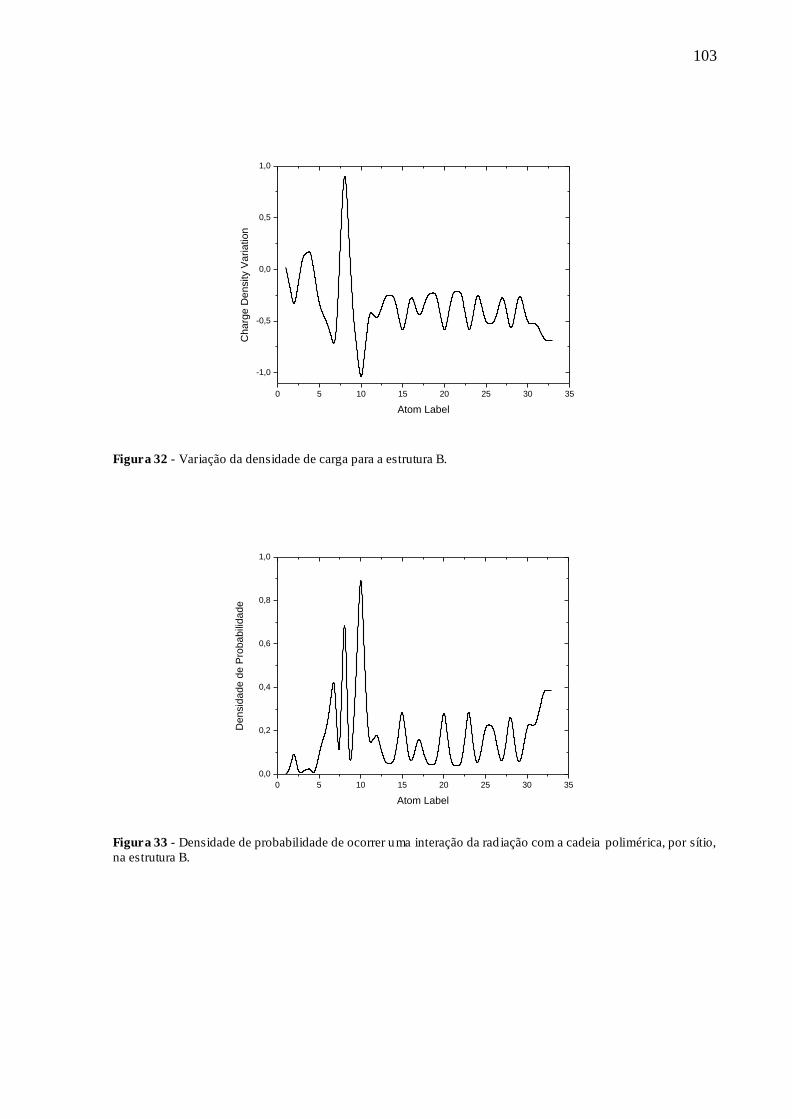
103
0 5 10 15 20 25 30 35
-1,0
-0,5
0,0
0,5
1,0
C
ha
rge
De
nsity V
ari
atio
n
Atom Label
Figura 32 - Variação da densidade de carga para a estrutura B.
0 5 10 15 20 25 30 35
0,0
0,2
0,4
0,6
0,8
1,0
D
en
sid
ad
e d
e P
rob
ab
ilid
ad
e
Atom Label
Figura 33 - Densidade de probabilidade de ocorrer uma interação da rad iação com a cadeia polimérica, por sítio,
na estrutura B.

104
Figura 34 - Apresentação gráfica dos índices de Fukui para ataque radical no ramo solubilizante, em que quanto
mais escuro o sítio, maior a probabilidade de ocorrer uma reação de cisão.
Assumindo o mesmo fato que para a estrutura A, a cisão ocorre, então, nas regiões do
segundo e terceiro átomo de oxigênio da cadeia dialcóxica. Observe que mediante tal evento,
a estrutura eletrônica da cadeia principal mantém-se inalterada desde que o segmento saturado
após o primeiro oxigênio já não influencia nas propriedades eletrônicas da cadeia principal,
sendo apenas artefatos físico-químicos utilizados para promoção de solubilidade e adesão em
substratos vítreos.

105
CAPÍTULO IV
Nesse capítulo, tanto os resultados experimentais como teóricos serão analisados sob uma
abordagem „complementarista‟, em que estes servem como argumentos que corroborem as
hipóteses levantadas por aqueles. Não haverá uma separação evidente entre os resultados
obtidos por cada técnica, visando à unificação do conhecimento adquirido sobre a forma de
interação da radiação com a matéria.
A cisão dos segmentos conjugados absorvedores na região UV-VIS influencia
diretamente em deslocamentos espectrais observados para os espectros de absorção
experimental, segundo o modelo do elétron confinado em uma caixa. Semelhante processo
também é capaz de justificar os deslocamentos observados no espectro de emissão
experimental. De forma geral, a cisão do segmento conjugado é ocasionada pela cisão da
própria cadeia em sítios determinados, como foi determinado pelos índices de reatividade de
Fukui para a estrutura A.
Por mais que a cisão ocorra em locais determinados, ainda existe uma polidispersão
dos novos segmentos formados que pode ser decomposta em duas causas: a própria
polidispersão inicial do polímero e a forma heterogênea com que a radiação interage com a
cadeia. Pelos índices de Fukui percebe-se que os sítios mais reativos são os mais internos da
cadeia, devido a ser essa região a de maior probabilidade de se encontrar um elétron π-
delocalizado. Esse fato pode ainda ser acompanhado pelo parâmetro d apresentado na Tabela 3
obtido do ajuste de Franck-Condon da emissão para as amostras irradiadas. Como dito no
Capítulo II, Seção 2.2.3, a interpretação desse parâmetro é a dispersão gaussiana dos
segmentos emissores e o seu aumento pode ser atribuído justamente a essas considerações
realizadas até agora. Observe, contudo, a sua aparente constância para as duas amostras
irradiadas, que indica que o simples fato de interação com radiação ionizante já é capaz de
alargar a distribuição de segmentos de forma não-dependente com a intensidade. A constância
desse parâmetro corrobora também a hipótese de que os efeitos da radiação gama em

106
materiais orgânicos são lineares para essa faixa e taxa de dose.
Ainda, o ajuste dos espectros de emissão é capaz de explicar não somente a
diminuição do segmento conjugado como também explicar o comportamento dos espectros de
fotoluminescência em função da dose. Ao contrário do que é esperado, conforme os elétrons
fossem mais confinados na estrutura polimérica, maior deveria ser a localização da função de
onda eletrônica sobre ligações específicas, o que é uma conseqüência da menor delocalização.
Assim, maiores acoplamentos entre elétrons e fônons seriam esperados e os espectros de
fotoluminescência apresentariam bandas vibrônicas melhor resolvidas, com intensidades das
bandas 0 1 crescente em relação à transição puramente eletrônica 00. Porém, o que se
nota pelos espectros de absorção é uma perda da resolução vibrônica, com a diminuição da
intensidade relativa da primeira banda em relação à banda puramente eletrônica.
A partir do ajuste, é possível perceber que o modo vibrônico alterado sofre justamente
um ganho de liberdade vibracional com uma menor localização da função de onda nessa
região, mediante o argumento apresentado recentemente. O fônon cuja expressão é alterada é
o de número de onda 700 cm-1, que corresponde ao modo vibracional do grupo -O-CH2- e O-
CH3, correspondente ao grupo dialcóxico grafitizado na cadeia. Conforme indicado pelos
índices de Fukui para a estrutura B, a liberdade desse grupo aumenta já que o segmento
pendente a partir do segundo e terceiro átomo de oxigênio é quebrado. Assim, um novo
caminho de relaxação vibracional que não compete com a relaxação eletrônica, conforme os
espectros de emissão, é criado. Esse novo modo vibracional não-competitivo com os modos
de decaimento radiativo pode permitir que a cadeia polimérica libere parte do stress sofrido na
formação de filmes devido à rigidez induzida pela conjugação π.
Percebe-se também, que a irradiação, sob esse ponto de vista, não induz uma
desordem prejudicial às propriedades eletrônicas do material, o que poderia ser monitorado
pelo alargamento do espectro de emissão, por exemplo, ou seja, a interação da radiação com a
estrutura vibrônica é bastante seletiva e não induz defeitos de forma aleatória. Essa
seletividade dos modos pode ser utilizada para um controle preciso das propriedades de
emissão eletro-óptica, por exemplo, em dispositivos produzidos a partir desse tipo de
material. Além disso, a indução de diminuição do segmento conjugado, com a conseqüente
modulação de gap, é um artifício interessante do ponto de vista prático, tendo em vista a
precisão que é possível na escolha do comprimento de onda de emissão, parâmetro que não é
tão bem controlado pela síntese do material.
Essa quebra lateral do ramo solubilizante corrobora também a segunda causa do

107
alargamento dos espectros de absorção. A primeira, já definida no início dessa seção, é devida
à polidispersividade induzida e a segunda, que só pode ser explicada por essa cisão, devido a
efeitos de empacotamento das cadeias. Cadeias com segmentos laterais menores empacotam
mais e esse processo induz a formação de regiões cristalinas altamente organizadas, que
possuem um comprimento de conjugação efetivo maior e por isso absorvem em energias
menores (comprimento de onda maior), ao passo que semelhantes regiões também induzem
desordem estrutural na região vizinha, diminuindo o comprimento efetivo de conjugação e
deslocando hipsocromicamente a absorção. Esses efeitos, somados, induzem um alargamento
desses espectros.
Com a diminuição do braço lateral, a cristalinidade do polímero é alterada, induzindo
que ocorram maiores interação cadeia-cadeia. Conhece-se da literatura que, cadeias
empacotadas apresentam um rendimento quântico de fotoluminescência e, conseqüentemente,
de eletroluminescência menor do naquelas matrizes em que a confo rmação molecular tende à
single-molecule-like (comportamento dos cromóforos que, por não interagirem com outros
cromóforos, emitem como se estivessem isolados), devido à difusão bi/tridimensional de
éxcitons, diminuindo a taxa de recombinação e transição radiativa. Isso leva a pensar que a
irradiação pode diminuir esses rendimentos quânticos da matriz polimérica, diminuindo assim
a eficiência de dispositivos desenvolvidos com materiais tratados dessa maneira. Embora isso
de fato possa ocorrer, o aumento da largura do espectro de absorção, induzida pela irradiação,
é interessante do ponto de vista de aplicação em células fotovoltaicas, desde que uma maior
sobreposição com o espectro de emissão solar pode ser obtido, conforme a Figura 35 mostra. A
sobreposição do espectro de absorção dessas espécies com o espectro de emissão do Sol é
dada pela Tabela 4. Observe que a sobreposição do polímero não- irradiado é maior do que
aqueles irradiados, no entanto a razão da sobreposição entre o não-irradiado e o irradiado com
20 kGy é maior do que a razão do não- irradiado e do oligômero, o que indica a possibilidade
de se construir células fotovoltaicas com moléculas de cadeia pequena e ainda assim com o
espectro de absorção em energias pequenas.
Por outro lado, o estreitamento do espectro de emissão é interessante para aplicação
desse material em dispositivos emissores de luz que apresentem um pico de emissão muito
estreito para aplicações que assim o requeiram. Semelhante comportamento não é obtido caso
fosse usado o oligômero SL128G no lugar da amostra de FSE59 irradiada com 40 kGy
(FSE59-40), por exemplo. Ambos apresentam aproximadamente o mesmo número de anéis,
porém o espectro de emissão do SL128G é bem mais largo do que o do FSE59-40 enquanto o
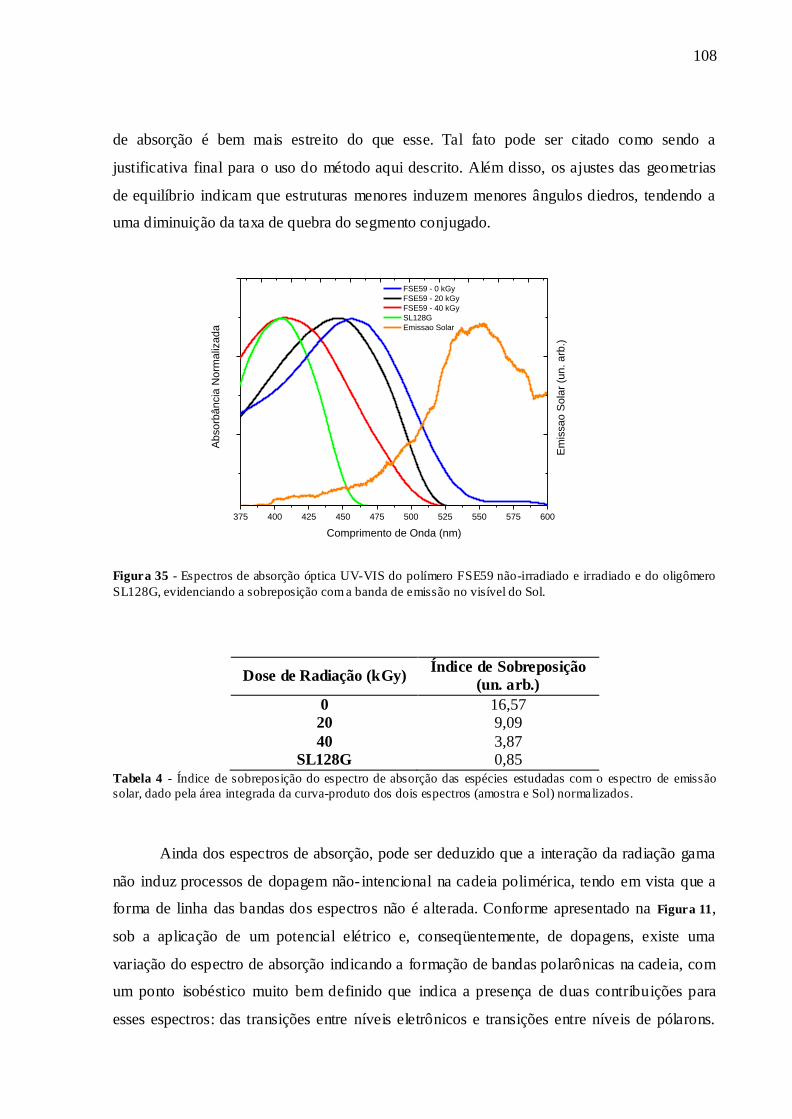
108
de absorção é bem mais estreito do que esse. Tal fato pode ser citado como sendo a
justificativa final para o uso do método aqui descrito. Além disso, os ajustes das geometrias
de equilíbrio indicam que estruturas menores induzem menores ângulos diedros, tendendo a
uma diminuição da taxa de quebra do segmento conjugado.
375 400 425 450 475 500 525 550 575 600
Em
issa
o S
ola
r (u
n. a
rb.)
FSE59 - 0 kGy
FSE59 - 20 kGy
FSE59 - 40 kGy
SL128G
Emissao Solar
Ab
so
rbâ
ncia
No
rma
liza
da
Comprimento de Onda (nm)
Figura 35 - Espectros de absorção óptica UV-VIS do polímero FSE59 não-irradiado e irradiado e do oligômero
SL128G, evidenciando a sobreposição com a banda de emissão no visível do Sol.
Dose de Radiação (kGy) Índice de Sobreposição
(un. arb.)
0 16,57 20 9,09
40 3,87 SL128G 0,85
Tabela 4 - Índice de sobreposição do espectro de absorção das espécies estudadas com o espectro de emissão
solar, dado pela área integrada da curva-produto dos dois espectros (amostra e Sol) normalizados.
Ainda dos espectros de absorção, pode ser deduzido que a interação da radiação gama
não induz processos de dopagem não- intencional na cadeia polimérica, tendo em vista que a
forma de linha das bandas dos espectros não é alterada. Conforme apresentado na Figura 11,
sob a aplicação de um potencial elétrico e, conseqüentemente, de dopagens, existe uma
variação do espectro de absorção indicando a formação de bandas polarônicas na cadeia, com
um ponto isobéstico muito bem definido que indica a presença de duas contribuições para
esses espectros: das transições entre níveis eletrônicos e transições entre níveis de pólarons.

109
Esse mesmo comportamento não é observado nos espectros de absorção das amostras
irradiadas, indicando que mediante irradiação, o FSE59 não sofre dopagem, o que também
corrobora para a não formação de radicais fotogerados do solvente.
Dos espectros de emissão, por extrapolação, é possível notar que uma dose
aproximada de aproximadamente 90 kGy é capaz de aniquilar completamente o modo de
fônon 700 cm-1. Com essa dose, o acoplamento vibrônico seria intermediado por um único
fônon com energia de 1100 cm-1 devido à não-alteração da estrutura correspondente a esse
modo vibrônico, que é o estiramento simétrico φ alcóxico.

110
CAPÍTULO V
Serão apresentadas aqui as considerações finais do trabalho, assim como as conclusões
obtidas, estruturadas na forma de um apanhado do capítulo anterior.
1 – CONSIDERAÇÕES FINAIS
De acordo com os dados apresentados neste trabalho, a irradiação gama é considerada
como uma forma viável e rápida de introduzir defeitos em matrizes poliméricas emissoras de
luz, devido á interação com fótons altamente energéticos. A forma como esses defeitos são
induzidos e como eles modulam o gap e os acoplamentos vibrônicos, ou seja, a estrutura
vibracional e estrutural da molécula é estudada por uma abordagem experimental com
comprovação teórica não conhecida na literatura.
A abordagem experimental aliada à modelagem por métodos semi-empíricos de
simulação computacional tanto do estado fundamental como do estado excitado permitem
entender de que forma a interação radiação-matéria ocorre de uma forma mais precisa. A
energia de gap se mostrou facilmente alterável pela dose, assim como a estrutura vibracional
acoplada com os modos eletrônicos. Ainda assim, essas alterações puderam ser monitoradas
por esse formalismo e por um formalismo mecânico-quântico a partir da taxa de transição
vertical de Fermi aplicada às transições entre estados de Franck-Condon.
Para esse tipo especial de matriz polimérica, um derivado de poli(2,5-tiofeno-1,4-
dialcóxifenileno), os fótons gama induzem cisão da cadeia nos ramos solubilizantes laterais e
na cadeia principal, desativando um modo vibracional de uma forma bem específica e
modulando o gap com um controle preciso. Finalmente, a abordagem apresentada aqui se
mostrou como sendo eficiente para entender, simular e melhorar as propriedades de polímeros
conjugados emissores de luz que, quando submetidos a tratamentos como esse, possam gerar
dispositivos mais eficientes com aumento de rendimento quântico, já que induz novos canais

111
de decaimento não radiativo que não são competitivos com o decaimento eletrônico radiativo.
Além disso, esses canais permitem liberar o filme polimérico do stress sofrido pelas
conformações quando em estado sólido.
2 – CONCLUSÕES
A influência da radiação gama sobre o polímero conjugado poli(2,5-tiofeno-1,4-
diálcoxifenileno) foi investigada usando absorção óptica UV-VIS e fotoluminescência. As
amostras, em solução tetrahidrofurano (THF), foram irradiadas com uma fonte de 60Co com
20 e 40 kGy de dose e 1,98 kGy.h-1. A fim de determinar como essa interação ocorre e como
ela interfere nas propriedades estruturais, eletrônicas e vibracionais do polímero, foram
realizados cálculos semi-empíricos AM1 para determinação da estrutura conformacional, dos
sítios mais reativos, estimativa do grau de cisão da cadeia e de como ocorre o ataque pela
radiação. O comportamento da estrutura vibrônica em função da dose é estudado dentro do
formalismo da Regra de Ouro de Fermi e da Aproximação de Franck-Condon. Observou-se a
diminuição do acoplamento eletrônico com o fônon de 700 cm-1, indicando que o braço lateral
ligado ao anel fenilênico tenha ganhado novos graus de liberdade com a irradiação. Esse fato
foi comprovado pelos índices de reatividade de Fukui, que indicam que a densidade de carga
é maior no segundo e terceiro átomo de oxigênio da cadeia lateral solubilizante e, portanto,
são os sítios mais propensos à interação com a radiação. Além disso, esses índices também
permitiram comprovar os sítios onde ocorre cisão da cadeia principal, o que explica o
comportamento dos espectros de absorção e parcialmente os de emissão. Esses dados
permitem estimar como os defeitos são distribuídos, de que forma eles modificam a energia
de gap e como modulam os acoplamentos entre elétrons e fônons.

112
CAPÍTULO VI
1 – REFERÊNCIAS BIBLIOGRÁFICAS
Anderson, W. P., Edwards, W. D., Zerner, M. C., & Canuto, S. (1982). A comparison of
theoretical models for interpreting the photoelectron spectrum of borazine. Chemical
Physics Letters, 88(2), 185-192.
Bacon, A. D., & Zerner, M. C. (1979). An intermediate neglect of differential overlap theory
for transition metal complexes: Fe, Co and Cu chlorides. Theoretical Chemistry
Accounts: Theory, Computation, and Modeling (Theoretica Chimica Acta), 53(1), 21-
54.
Bernius, M. T., Inbasekaran, M., O'Brien, J., & Wu, W. (2000). Progress with Light-Emitting
Polymers. Advanced Materials, 12(23), 1737-1750.
Birgerson, J., Johansson, N., Pohl, A., Lögdlund, M., Tsukahara, Y., Kaeriyama, K., et al.
(2001). Electronic structure of some conjugated polymers for electron transport.
Synthetic Metals, 122(1), 67-72.
Bolognesi, A., Botta, C., Mercogliano, C., Porzio, W., Jukes, P. C., Geoghegan, M., et al.
(2004). Structural features in aligned poly(3-alkylthiophene) films revealed by grazing
incidence X-ray diffraction. Polymer, 45(12), 4133-4138.
Bolognesi, A., Botta, C., & Porzio, W. (2001). Highly Regioregular Poly-3-alkylthiophenes:
Influence of the Structure on Photoluminescence. Monatshefte für Chemie / Chemical
Monthly, 132(1), 121-128.
Bouachrine, M., Lère-Porte, J.-P., Moreau, J. J. E., Spirau, F. S., da Silva, R. A., Lmimouni,
K., et al. (2002). A thienylene-phenylene copolymer with di(ethylene oxide) side
chains and its use in light emitting diodes. Synthetic Metals, 126(2-3), 241-244.
Brailsford, A. D., & Chang, T. Y. (1970). Nonradiative Decay of Individual Vibronic Levels in
Large Molecules. The Journal of Chemical Physics, 53(8), 3108-3113.

113
Brédas, J. L., Beljonne, D., Cornil, J., dos Santos, D. A., & Shuai, Z. (1997).
Electroluminescence in semiconducting conjugated polymers and oligomers: a
quantum–chemical approach. Philosophical Transactions of the Royal Society of
London. Series A: Mathematical, Physical and Engineering Sciences, 355(1725), 735-
747.
Brunsveld, L., Folmer, B. J. B., Meijer, E. W., & Sijbesma, R. P. (2001). Supramolecular
Polymers. Chemical Reviews, 101(12), 4071-4098.
Burroughes, J. H., Bradley, D. D. C., Brown, A. R., Marks, R. N., Mackay, K., Friend, R. H.,
et al. (1990). Light-emitting diodes based on conjugated polymers. Nature, 347(6293),
539-541.
Cataldo, F. (2000). A Raman study on radiation-damaged graphite by [gamma]-rays. Carbon,
38(4), 634-636.
Chang, R., Hsu, J. H., Fann, W. S., Liang, K. K., Chang, C. H., Hayashi, M., et al. (2000).
Experimental and theoretical investigations of absorption and emission spectra of the
light-emitting polymer MEH-PPV in solution. Chemical Physics Letters, 317(1-2),
142-152.
Chen, S., Wu, G., Liu, Y., & Long, D. (2005). Preparation of Poly(acrylic acid) Grafted
Multiwalled Carbon Nanotubes by a Two-Step Irradiation Technique.
Macromolecules, 39(1), 330-334.
Coakley, K. M., & McGehee, M. D. (2004). Conjugated Polymer Photovoltaic Cells.
Chemistry of Materials, 16(23), 4533-4542.
Cornil, J., Beljonne, D., Friend, R. H., & Brédas, J. L. (1994). Optical absorptions in
poly(paraphenylene vinylene) and poly(2,5-dimethoxy-1,4-paraphenylene vinylene)
oligomers. Chemical Physics Letters, 223(1-2), 82-88.
Crespi, V. H., Chopra, N. G., Cohen, M. L., Zettl, A., & Louie, S. G. (1996). Anisotropic
electron-beam damage and the collapse of carbon nanotubes. Physical Review B,
54(8), 5927.
D. Wang, M. R., N. Kopidakis, B.A. Gregg. (2008). Do the Defects Make it Work? Defect
Engineering in Pi-Conjugated Polymers and Their Solar Cells, 33rd IEEE Photovoltaic
Specialists Conference. San Diego, California: National Renewable Energy
Laboratory.
Davenas, J., Stevenson, I., Celette, N., Cambon, S., Gardette, J. L., Rivaton, A., et al. (2002).
Stability of polymers under ionising radiation: The many faces of radiation

114
interactions with polymers. Nuclear Instruments and Methods in Physics Research
Section B: Beam Interactions with Materials and Atoms, 191(1-4), 653-661.
Dewar, M. J. S., & Thiel, W. (1977). Ground states of molecules. 38. The MNDO method.
Approximations and parameters. Journal of the American Chemical Society, 99(15),
4899-4907.
Dewar, M. J. S., Zoebisch, E. G., Healy, E. F., & Stewart, J. J. P. (1985). Development and use
of quantum mechanical molecular models. 76. AM1: a new general purpose quantum
mechanical molecular model. Journal of the American Chemical Society, 107(13),
3902-3909.
Ding, A.-L., Pei, J., Lai, Y.-H., & Huang, W. (2001). Phenylene-functionalized polythiophene
derivatives for light-emitting diodes: their synthesis, characterization and properties.
Journal of Materials Chemistry, 11(12), 3082-3086.
Edwards, W. D., & Zerner, M. C. (1987). A generalized restricted open-shell Fock operator.
Theoretical Chemistry Accounts: Theory, Computation, and Modeling (Theoretica
Chimica Acta), 72(5), 347-361.
Ferreira, G. R., Vasconcelos, C. K. B. d., Silva, M. M., Santos, F. A. d., Pires, J. G., Duarte, A.
S., et al. (2009). A Novel and Low-Cost Disposable Device for Phototherapy of
Neonatal Jaundice. MRS Online Proceedings Library, 1209, null-null.
Föster, T. (1960). Transfer Mechanism of Electronic Excitation Energy. Radiation Research
Supplement, 2, 326-339.
Foundation, T. N. (2010). The Nobel Prize in Chemistry 2010. Retrieved 24 de Março, 2011
Friend, R. H. (2001). Conjugated polymers. New materials for optoelectronic devices. Pure
and Applied Chemistry, 73(3), 425-430.
Friend, R. H., Gymer, R. W., Holmes, A. B., Burroughes, J. H., Marks, R. N., Taliani, C., et al.
(1999). Electroluminescence in conjugated polymers. Nature, 397(6715), 121-128.
Graham, S. C., Friend, R. H., Fung, S., & Moratti, S. C. (1997). The effect of X-ray
irradiation on poly(p-phenylene vinylene) and derivatives. Synthetic Metals, 84(1-3),
903-904.
Gregg, B. A. (2009a). Charged defects in soft semiconductors and their influence on organic
photovoltaics. Soft Matter, 5(16), 2985-2989.
Gregg, B. A. (2009b). Transport in Charged Defect-Rich π-Conjugated Polymers. The Journal
of Physical Chemistry C, 113(15), 5899-5901.
Head, J. D., & Zerner, M. C. (1985). A Broyden--Fletcher--Goldfarb--Shanno optimization

115
procedure for molecular geometries. Chemical Physics Letters, 122(3), 264-270.
Head, J. D., & Zerner, M. C. (1986). An approximate Hessian for molecular geometry
optimization. Chemical Physics Letters, 131(4-5), 359-366.
Inganäs, O., Granlund, T., Theander, M., Berggren, M., Andersson, M. R., Ruseekas, A., et al.
(1998). Optical emission from confined poly(thiophene) chains. Optical Materials,
9(1-4), 104-108.
Korchowiec, J. (2000). Recognition of the electrophilic and nucleophilic centers in molecules
via the radical charge transfer Fukui function. Computers & Chemistry, 24(3-4), 259-
262.
Kudoh, H., Sasuga, T., Seguchi, T., & Katsumura, Y. (1996). High-energy- ion- irradiation
effects on polymer materials: 3. The sensitivity of cellulose triacetate and poly(methyl
methacrylate). Polymer, 37(14), 2903-2908.
Lee, K.-Y., & Kim, K.-Y. (2008). 60Co [gamma]-ray irradiation effect and degradation
behaviors of a carbon nanotube and poly(ethylene-co-vinyl acetate) nanocomposites.
Polymer Degradation and Stability, 93(7), 1290-1299.
Liang, Z., Nardes, A., Wang, D., Berry, J. J., & Gregg, B. A. (2009). Defect Engineering in π-
Conjugated Polymers. Chemistry of Materials, 21(20), 4914-4919.
Lin, S. (1968). Spectral band shape of absorption and emission of molecules in dense media.
Theoretical Chemistry Accounts: Theory, Computation, and Modeling (Theoretica
Chimica Acta), 10(4), 301-310.
Lin, S. H. (1966). Rate of Interconversion of Electronic and Vibrational Energy. The Journal
of Chemical Physics, 44(10), 3759-3767.
Lin, S. H. (1973). Effect of high pressures on molecular electronic spectra and electronic
relaxation. The Journal of Chemical Physics, 59(8), 4458-4467.
Lin, S. H. (1989). Theory of photoinduced intramolecular electron transfer in condensed
media. The Journal of Chemical Physics, 90(12), 7103-7113.
Lois, S., Florès, J.-C., Lère-Porte, J.-P., Serein-Spirau, F., Moreau, J. J. E., Miqueu, K., et al.
(2007). How to Build Fully π-Conjugated Architectures with Thienylene and
Phenylene Fragments. European Journal of Organic Chemistry, 2007(24), 4019-4031.
Löwdin, P.-O. (1955). Quantum Theory of Many-Particle Systems. III. Extension of the
Hartree-Fock Scheme to Include Degenerate Systems and Correlation Effects.
Physical Review, 97(6), 1509.
Marletta, A., Guimarães, F. E. G., & Faria, R. M. (2002). Line shape of emission spectra of

116
the luminescent polymer poly(p-phenylene vinylene).
Méndez, F., & Gázquez, J. (1994). The Fukui function of an atom in a molecule: A criterion to
characterize the reactive sites of chemical species. Journal of Chemical Sciences,
106(2), 183-193.
Milgrom, L. (2007). Hafnium oxide helps make chips smaller and faster. Retrieved 23 de
Março, 2011
Misra, G. P., & Sannigrahi, A. B. (1996). A comparison of condensed fukui function, free
valency and unpaired spin population as reactivity indices for open-shell molecules.
Journal of Molecular Structure: THEOCHEM, 361(1-3), 63-68.
Pei, J., Yu, W.-L., Huang, W., & Heeger, A. J. (2000). A Novel Series of Efficient Thiophene-
Based Light-Emitting Conjugated Polymers and Application in Polymer Light-
Emitting Diodes. Macromolecules, 33(7), 2462-2471.
Pope, M. a. S., Charles E. (1999). Electronic Processes in Organic Crystals and Polymers (2
ed. Vol. 1). New York: Oxford University Press.
Reilly, D. (1991). Passive Nondestructive Assay of Nuclear Materials. Washington, D.C.:
Nuclear Regulatory Commission.
Ridley, J., & Zerner, M. (1973). An intermediate neglect of differential overlap technique for
spectroscopy: Pyrrole and the azines. Theoretical Chemistry Accounts: Theory,
Computation, and Modeling (Theoretica Chimica Acta), 32(2), 111-134.
Ridley, J. E., & Zerner, M. C. (1976). Triplet states via intermediate neglect of differential
overlap: Benzene, pyridine and the diazines. Theoretical Chemistry Accounts: Theory,
Computation, and Modeling (Theoretica Chimica Acta), 42(3), 223-236.
Roy, R. K., Hirao, K., Krishnamurty, S., & Pal, S. (2001). Mulliken population analysis based
evaluation of condensed Fukui function indices using fractional molecular charge. The
Journal of Chemical Physics, 115(7), 2901-2907.
S. Bouzakraoui, H. Z., S.M.Bouzzine, R.Augusta Dasilva, J-P Lère-Porte, F. Serein-Spirau, K.
Alimi, M. Hamidi, M. Bouachrine. (2010). Synthesis and theoretical study of a new
conjugated copolymer based on thiophene and phenylene. Phys. Chem. News, 51,
111-121.
Salaneck, W. R., Friend, R. H., & Brédas, J. L. (1999). Electronic structure of conjugated
polymers: consequences of electron- lattice coupling. Physics Reports, 319(6), 231-
251.
Silva, E. A. B., Borin, J. F., Nicolucci, P., Graeff, C. F. O., Netto, T. G., & Bianchi, R. F.

117
(2005). Low dose ionizing radiation detection using conjugated polymers. Applied
Physics Letters, 86(13), 131902-131903.
Silva, M. D. R., Gançalves Jr, A. A., Silva, R. A., & Marletta, A. (2010). Gamma radiation
effects on absorbance and emission properties of layer-by-layer PPV/DBS films.
Journal of Non-Crystalline Solids, 356(44-49), 2429-2432.
Silva, R. A., Cury, L. A., Marletta, A., Guimarães, P. S. S., Bouachrine, M., Lère-Porte, J. P.,
et al. (2006). Absorption and photoluminescence of a new thienylene-phenylene
copolymer. Journal of Non-Crystalline Solids, 352(32-35), 3685-3688.
Silva, R. A., Cury, L. A., Mazzoni, M. S., Soares, E., Guimarães, P. S. S., Serein-Spirau, F., et
al. (2005). Study of [Thienylene-dialkoxy phenylene] Conjugated Materials.
Macromolecular Symposia, 229(1), 194-196.
Silva, R. A., Serein-Spirau, F., Bouachrine, M., Lere-Porte, J.-P., & Moreau, J. J. E. (2004).
Synthesis and characterization of thienylene-phenylene copolymers with
oligo(ethylene oxide) side chains. Journal of Materials Chemistry, 14(20), 3043-3050.
Sirringhaus, H., Tessler, N., & Friend, R. H. (1998). Integrated Optoelectronic Devices Based
on Conjugated Polymers. Science, 280(5370), 1741-1744.
Smith, B. W., & Luzzi, D. E. (2001). Electron irradiation effects in single wall carbon
nanotubes. Journal of Applied Physics, 90(7), 3509-3515.
Stewart, J. J. P. (2008). MOPAC2009. Colorado Springs, CO: Stewart Computational
Chemistry.
Stolarczyk, L. Z., Jeziorska, M. g., & Monkhorst, H. J. (1988). Exact Hartree-Fock exchange
in one-dimensional metals. Physical Review B, 37(18), 10646.
Su, W. P., Schrieffer, J. R., & Heeger, A. J. (1979). Solitons in Polyacetylene. Physical Review
Letters, 42(25), 1698.
Szabo, A. e. Ö., Neil S. (1996). Modern Quantum Chemistry: Introduction to Advanced
Electronic Structure Theory. Mineola, New York: Dover Publications, Inc. .
Thompson, M. A. (2010). ArgusLab (Version 4.0). Seattle, WA: Planaria Software LLC.
Vasconcelos, C. K. B. d. F., Giovana R. and Bianchi, Rodrigo F. (2010). Desenvolvimento e
caracterização de sensor de acúmulo de dose de radiação azul polimérico. Polímeros
[online], 20(1), 14-18.
Wong, K. F., Skaf, M. S., Yang, C.-Y., Rossky, P. J., Bagchi, B., Hu, D., et al. (2001).
Structural and Electronic Characterization of Chemical and Conformational Defects in
Conjugated Polymers. The Journal of Physical Chemistry B, 105(26), 6103-6107.

118
Xu, H., Wang, X., Zhang, Y., & Liu, S. (2006). Single-Step in Situ Preparation of Polymer-
Grafted Multi-Walled Carbon Nanotube Composites under 60Co γ-Ray Irradiation.
Chemistry of Materials, 18(13), 2929-2934.
Yan, M., Rothberg, L. J., Papadimitrakopoulos, F., Galvin, M. E., & Miller, T. M. (1994).
Defect Quenching of Conjugated Polymer Luminescence. Physical Review Letters,
73(5), 744.
Yoshino, K., Hayashi, S., & Inuishi, Y. (1982). Effects of Electron Irradiation on Electrical
Conductivity of Polyacetylene. Japanese Journal of Applied Physics, 21(Copyright (C)
1982 Publication Board, Japanese Journal of Applied Physics), L569.
Yu, J., Hayashi, M., Lin, S. H., Liang, K. K., Hsu, J. H., Fann, W. S., et al. (1996).
Temperature effect on the electronic spectra of poly(p-phenylenevinylene). Synthetic
Metals, 82(2), 159-166.
Yurtsever, E., & Yurtsever, M. (1999). A theoretical study of structural defects in conjugated
polymers. Synthetic Metals, 101(1-3), 335-336.
Zempo, Y., & et al. (2008). Optical properties in conjugated polymers. Journal of Physics:
Condensed Matter, 20(6), 064231.

119
ANEXO I
PUBLICAÇÕES CORRELACIONADAS COM ESSE TRABALHO
Esse trabalhou gerou as seguintes produções científicas:
10 (dez) apresentações de pôsteres em eventos e congressos nacionais e
internacionais;
1 (uma) publicação em Livro de Proceedings;
1 (um) prêmio de Student Travel Grant Award, concedido para viagem
ao exterior para apresentação;
1 (uma) apresentação oral em congresso internacional;
1 (um) artigo científico publicado no The Journal of Physical
Chemistry A (segue em anexo).
Outras produções indiretas não listadas.

rXXXX American Chemical Society A dx.doi.org/10.1021/jp203244z | J. Phys. Chem. A XXXX, XXX, 000–000
ARTICLE
pubs.acs.org/JPCA
Controlling Bandgap Energy and Multivibronic Modes of aPoly(2,5-thiophene-1,4-dialkoxyphenylene) Derivative byGamma PhotonsH. Santos Silva,*,† S. L. Nogueira,† J. E. Manzoli,‡ N. M. Barbosa Neto,† A. Marletta,† F. Serein-Spirau,§
J.-P L�ere-Porte,§ Sandrine Lois,§ and R. A. Silva†,||
†Instituto de F�isica, Universidade Federal de Uberl̂andia, CP 593, 38400-902, Uberl̂andia - MG, Brazil‡Instituto de Pesquisas Energ�eticas e Nucleares, IPEN/CNEN-SP, Av. Prof. Lineu Prestes 2242, Cidade Universit�aria, 05508-000,S~ao Paulo - SP, Brazil§AM2N-Architectures Mol�eculaires et Mat�eriaux Nanostructur�es, Institut Charles Gerhardt-Ecole Nationale Superieure de Chimie deMontpellier (ENSCM) 34296, Montpellier, Cedex 5, France
)Divis~ao de Metrologia de Materiais, Instituto Nacional de Metrologia, Normalizac)~ao e Qualidade Industrial, 25250-020,Duque de Caxias - RJ, Brazil
1. INTRODUCTION
The applicability of conjugated polymers is only possible dueto two charge carriers process of dissociation and recombination:the photovoltaic effect1 and electroluminescent effect,2,3 respec-tively. The former is characterized by the electrode capture ofbound charge carriers produced during light absorption, whilethe last is the opposite effect, that is, an injection of chargecarriers that allows a recombination among them, followed bylight emission. Recent studies have shown that the quantum yieldof these processes is highly dependent not only on the electronicstructure of the polymer, but also on its conformational structure,4
supramolecular5 effects like inter- and intrachain interactions,substituents presence, and mainly, structural defects.6�11
Defects have also been studied in the literature, attempting toelucidate how the material synthesis, manipulation, characteriza-tion, and device manufacturing6,7 can be crucial and, in most ofthe times, harmful to polymericmaterial yields through undesirabledoping processes,11 electronic structure degradation, or altera-tion. However, some authors believe that some noncharged, well-controlled structural defects are able to, instead of what isexpected, increase electrical and maybe optical properties ofπ-conjugated polymers.6 For instance, we can cite Liang et al.6
who have shown that poly(3-hexyl-thiophene) (P3HT) filmsrich in structural defects induced by Me2SO4 and LiAlH4
present a smaller photobleaching than pristine P3HT films.In addition, these defects can increase charge carriers concen-tration and mobility. This can be attributed to the inducteddeformations on the trigonal planar sp2 polymeric chainstructure, a fact that can create electronic states inside the bandgap that not always can hinder other electronic processes.11,12
Moreover, the insertion of some defects can relieve the chainfrom inevitable stress suffered by film formation in optoelec-tronic devices’ production.
Among the methods used to create and study defects reportedin literature, we can highlight ionic doping, photo- and thermo-degradation, saturation, and isomerization.6 However, the use ofγ radiation has been lately reported as an efficient way to inducestructural and electronic modifications in conjugated macromo-lecules, such as carbon nanotubes,13�17 quantum dots�conjugatedpolymers composites,18 porphyrins,19,20 and so on.
Received: April 7, 2011Revised: May 31, 2011
ABSTRACT: In this work, the influence of γ radiation on electronic,structural, and vibrational properties of a poly(2,5-thiophene-1,4-dia-lkoxyphenylene) derivative is studied by optical absorption and photo-luminescence. A Gaussian fit of emission spectra within Franck�Condonvertical transitions formalism was carried out in order to understand howvibronic coupling is affected by the dose, because an unexpected lumines-cence behavior was observed. Aiming to understand the ionizing radiation�matter interaction processes, we employed a molecular modeling procedure,through the use of a semiempirical method (AM1) applied to conjugatedoligomers’ conformational structure and equilibrium geometries, to clarifythe defects induction for the used doses. From AM1 optimized structures,electronic transitions were calculated by ZINDO/S�CI semiempiricalmethod to measure the chain scission degree. Moreover, with the resultspresented in this work, it is possible to comeupwith a newphysical�chemical route to treat and increase conjugated polymers’ efficiency.Finally, we believe that the present paper contributes to the literature about defects on conjugated polymers.

B dx.doi.org/10.1021/jp203244z |J. Phys. Chem. A XXXX, XXX, 000–000
The Journal of Physical Chemistry A ARTICLE
The interaction of γ rays with polymeric materials has beenwidely studied, focused mainly on mechanical applications.13,16,21
However, such pacific use of ionizing radiation in potentialmaterials for electronics, to intentionally tune their properties,22
is quite recent. The interaction mechanism is well-known by theliterature and involves three basic processes: photoelectric effect,Compton scatter, and pair formation.23 For carbon materialsinteracting with γ radiation of energy lower than 2 MeV, the pairformation effect occurs with less than 2% frequency, even thoughelastic and inelastic scatter are the predominant processes and areresponsible for24 ionization, bond break, hydrogenation, cross-links, degradation, and so on.
Bond break processes in a poly(2,5-thiophene-1,4-dialkoxy-phenylene) derivative25�31 were specially studied in this work,using 60Co as the gamma source, whose emission happens in twospecific energy lines (avg. 1.25 MeV). In the dose range used inthis work, the radiation induces basically only chain scissions andcross-links due to the ionization of the reactive atoms.24 Defectscontrollability can be guaranteed by the fact that, in this doserange and dose rate, the interaction of radiation with matter isapproximately linear, as it can be deduced throughout this work.
Such polythiophene derivatives, poly(3-alkylthiophene) (P3AT)and poly(thiophene-co-phenylene), for instance, are a represen-tative class of conjugated polymers that form environmentallyand thermally stable materials.32,33 These compounds exhibit arange of interesting features associated with the redox activity, areversible transition between the doped and the neutral states,and this results in conductivity, electronic, and electrochemicalproperties.34�36 Copolymers containing both phenylene andthiophene units have also proved to be of interest in combiningthe properties associated with the two different conjugatedrings.31,37 The presence of di(ethyleneoxide) polar side-chainswas shown to give good solubility, excellent adhesion properties,and promote ion solvation and mobility to phenylene�vinylenecopolymers.38 This approach has proved to be efficient inincreasing emission quantum yield of such structures by confin-ing unidimensionally charged carriers, avoiding its diffusion and,consequently, nonradiative decays.
Ionizing radiation interaction, especially the gamma withconjugated polymers, was first reported in the early 80s,39,40 inan attempt to develop low-cost polymeric dosimeters withmeasurement patterns more easily controllable than the thermo-luminescent inorganics used until then. The pioneers in this fieldare Burroughes et al., studying gamma interaction in poly(p-phenylene-vinylene) (PPV)41�43 and poly(2-methoxy-5-[ethyl]-hexyloxy-p-phenylene-vinylene (MEH-PPV)44 by Graeff et al.,and UV/X-rays interaction by Bianchi et al.,45,46 aiming todevelop neonatal therapy dosimeters. There are few examplesusing wittingly ionizing radiation as a way to alter conjugatedpolymeric material electronic properties for direct application indevices.22
The interaction mechanism in such material is yet not totallydescribed and most authors suggest that they are caused andintermediated by photogenerated solvent radicals.44 In halideorganic solvent solutions, such as carbon tetrachloride, dibromo-methane, and diiodomethane, for instance, the MEH-PPV isseverely damaged by halide radicals and suffers both a chain and aconjugation break, with hypsochromic shifts in optical absorp-tion and emission spectra, which presents a strongly reducedintensity after irradiation. When in benzene solution, no changein electronic properties is observed, leading to the assumptionthat the radiation�matter interaction takes place only when
intermediated by the solvent. Bianchi et al. showed that harmfuleffects on MEH-PPV irradiated with UV45,46 are dependent onO2/N2 proportion and attributes the decrease of optical absorp-tion and emission efficiency to the induced photodegradation bythe O2 presence. In these studies, electronic modifications areonly caused by conjugated length diminishing by the photo-induced defects and chain scission; however, how other mod-ifications on structure can change electronic/vibrationalstructure has not been explored by literature. Such an approachis able to open up a range of new possible applications of organicsemiconductors in electronic industry, developing more efficientdevices beyond, solely, developing the dosimetry field.
In the work reported here, an anomalous photoluminescencebehavior for γ-irradiated samples of a poly(2,5-thiophene-1,4-dialkoxyphenylene) (Figure 1) conjugated polymer derivative isobserved and explained by an experimental�theoretical ap-proach. Optical absorption and photoluminescence for increas-ing doses of γ radiation in THF solutions were studied andsemiempirical AM147,48 calculations were performed to deter-mine the most reactive sites in the chain, as well as where theinteraction is most likely to happen. Furthermore, ZINDO/S-CI,49�55 an electronic transitions prevision, was carried out forthe increasing number of rings in the conjugated chain. Such dataallow us to estimate how the defects are distributed and how theymodulate the gap energy and multivibronic couplings, using theFranck�Condon transitions within the Fermi’s Golden rulescope.56 It is also discussed how these defects can be intentionallyused to develop more efficient organics-based opto-electronicdevices.
2. METHODOLOGY
2.1. Experimental Section. The conjugated polymer 1 de-picted in Figure 1 was synthesized by the organometallicpalladium route.29 The other structure (2) in the same figurewas also synthesized, but it was only used as a theoreticalprocedure to reduce computational costs because, for alkoxycompounds, the length of the lateral chain bonded to oxygendoes not interfere in the electronic properties of the conjugatedbackbone, as reported elsewhere,57 and was confirmed by opticalabsorption (OA) measurements.58 But this length does interferewith the semicrystalline solid state structure given by the lamellarphases induced by the interdigitation of the lateral branch chains.By varying this chain, only the polymer with m = 2 and Z = butyl
Figure 1. Conjugated polymers used for experimental (1) and theore-tical (2) analysis.
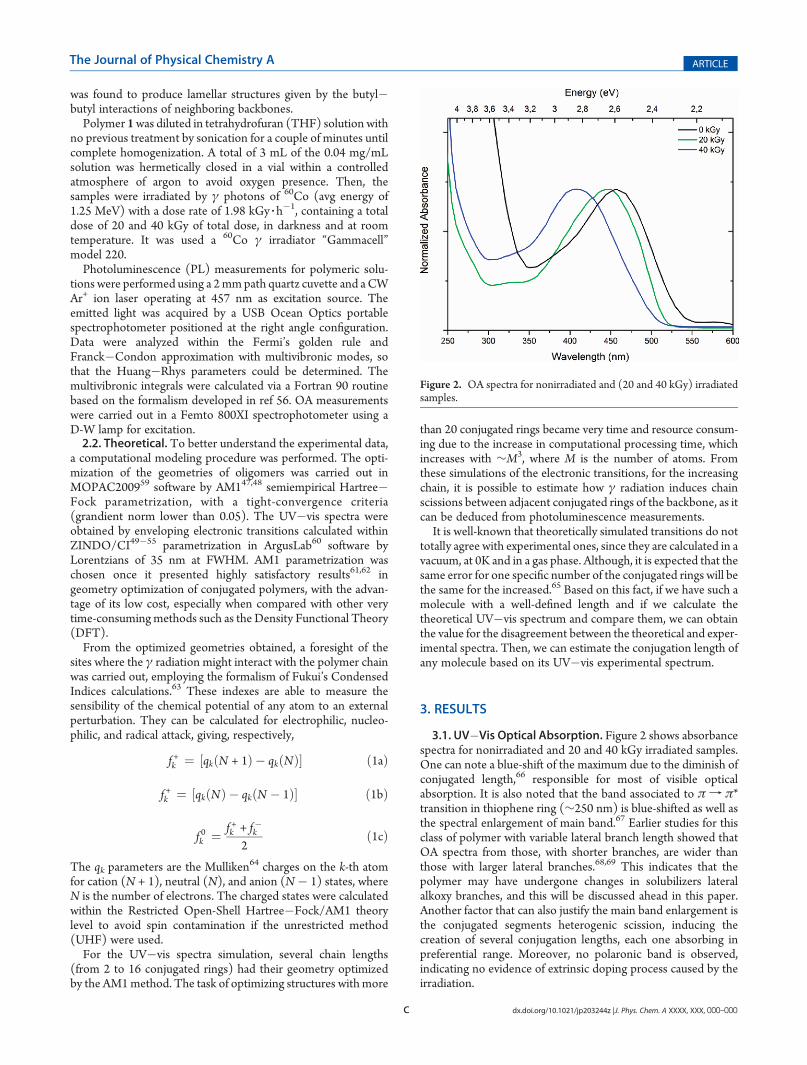
C dx.doi.org/10.1021/jp203244z |J. Phys. Chem. A XXXX, XXX, 000–000
The Journal of Physical Chemistry A ARTICLE
was found to produce lamellar structures given by the butyl�butyl interactions of neighboring backbones.Polymer 1was diluted in tetrahydrofuran (THF) solution with
no previous treatment by sonication for a couple of minutes untilcomplete homogenization. A total of 3 mL of the 0.04 mg/mLsolution was hermetically closed in a vial within a controlledatmosphere of argon to avoid oxygen presence. Then, thesamples were irradiated by γ photons of 60Co (avg energy of1.25 MeV) with a dose rate of 1.98 kGy 3 h
�1, containing a totaldose of 20 and 40 kGy of total dose, in darkness and at roomtemperature. It was used a 60Co γ irradiator “Gammacell”model 220.Photoluminescence (PL) measurements for polymeric solu-
tions were performed using a 2mmpath quartz cuvette and aCWAr+ ion laser operating at 457 nm as excitation source. Theemitted light was acquired by a USB Ocean Optics portablespectrophotometer positioned at the right angle configuration.Data were analyzed within the Fermi’s golden rule andFranck�Condon approximation with multivibronic modes, sothat the Huang�Rhys parameters could be determined. Themultivibronic integrals were calculated via a Fortran 90 routinebased on the formalism developed in ref 56. OA measurementswere carried out in a Femto 800XI spectrophotometer using aD-W lamp for excitation.2.2. Theoretical. To better understand the experimental data,
a computational modeling procedure was performed. The opti-mization of the geometries of oligomers was carried out inMOPAC200959 software by AM147,48 semiempirical Hartree�Fock parametrization, with a tight-convergence criteria(grandient norm lower than 0.05). The UV�vis spectra wereobtained by enveloping electronic transitions calculated withinZINDO/CI49�55 parametrization in ArgusLab60 software byLorentzians of 35 nm at FWHM. AM1 parametrization waschosen once it presented highly satisfactory results61,62 ingeometry optimization of conjugated polymers, with the advan-tage of its low cost, especially when compared with other verytime-consumingmethods such as the Density Functional Theory(DFT).From the optimized geometries obtained, a foresight of the
sites where the γ radiation might interact with the polymer chainwas carried out, employing the formalism of Fukui’s CondensedIndices calculations.63 These indexes are able to measure thesensibility of the chemical potential of any atom to an externalperturbation. They can be calculated for electrophilic, nucleo-philic, and radical attack, giving, respectively,
f +k ¼ ½qkðN + 1Þ � qkðNÞ� ð1aÞ
f +k ¼ ½qkðNÞ � qkðN � 1Þ� ð1bÞ
f 0k ¼ f +k + f�k2
ð1cÞ
The qk parameters are the Mulliken64 charges on the k-th atomfor cation (N + 1), neutral (N), and anion (N� 1) states, whereN is the number of electrons. The charged states were calculatedwithin the Restricted Open-Shell Hartree�Fock/AM1 theorylevel to avoid spin contamination if the unrestricted method(UHF) were used.For the UV�vis spectra simulation, several chain lengths
(from 2 to 16 conjugated rings) had their geometry optimizedby the AM1method. The task of optimizing structures withmore
than 20 conjugated rings became very time and resource consum-ing due to the increase in computational processing time, whichincreases with ∼M3, where M is the number of atoms. Fromthese simulations of the electronic transitions, for the increasingchain, it is possible to estimate how γ radiation induces chainscissions between adjacent conjugated rings of the backbone, as itcan be deduced from photoluminescence measurements.It is well-known that theoretically simulated transitions do not
totally agree with experimental ones, since they are calculated in avacuum, at 0K and in a gas phase. Although, it is expected that thesame error for one specific number of the conjugated rings will bethe same for the increased.65 Based on this fact, if we have such amolecule with a well-defined length and if we calculate thetheoretical UV�vis spectrum and compare them, we can obtainthe value for the disagreement between the theoretical and exper-imental spectra. Then, we can estimate the conjugation length ofany molecule based on its UV�vis experimental spectrum.
3. RESULTS
3.1. UV�Vis Optical Absorption. Figure 2 shows absorbancespectra for nonirradiated and 20 and 40 kGy irradiated samples.One can note a blue-shift of the maximum due to the diminish ofconjugated length,66 responsible for most of visible opticalabsorption. It is also noted that the band associated to π f π*transition in thiophene ring (∼250 nm) is blue-shifted as well asthe spectral enlargement of main band.67 Earlier studies for thisclass of polymer with variable lateral branch length showed thatOA spectra from those, with shorter branches, are wider thanthose with larger lateral branches.68,69 This indicates that thepolymer may have undergone changes in solubilizers lateralalkoxy branches, and this will be discussed ahead in this paper.Another factor that can also justify the main band enlargement isthe conjugated segments heterogenic scission, inducing thecreation of several conjugation lengths, each one absorbing inpreferential range. Moreover, no polaronic band is observed,indicating no evidence of extrinsic doping process caused by theirradiation.
Figure 2. OA spectra for nonirradiated and (20 and 40 kGy) irradiatedsamples.
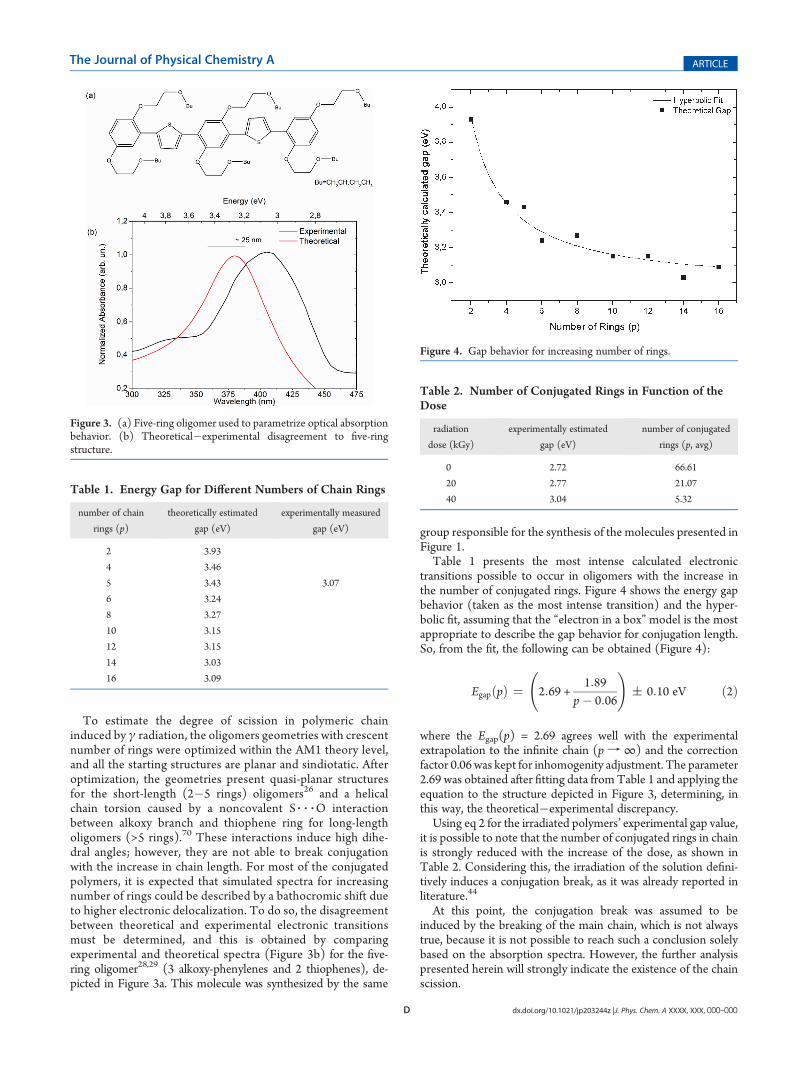
D dx.doi.org/10.1021/jp203244z |J. Phys. Chem. A XXXX, XXX, 000–000
The Journal of Physical Chemistry A ARTICLE
To estimate the degree of scission in polymeric chaininduced by γ radiation, the oligomers geometries with crescentnumber of rings were optimized within the AM1 theory level,and all the starting structures are planar and sindiotatic. Afteroptimization, the geometries present quasi-planar structuresfor the short-length (2�5 rings) oligomers26 and a helicalchain torsion caused by a noncovalent S 3 3 3O interactionbetween alkoxy branch and thiophene ring for long-lengtholigomers (>5 rings).70 These interactions induce high dihe-dral angles; however, they are not able to break conjugationwith the increase in chain length. For most of the conjugatedpolymers, it is expected that simulated spectra for increasingnumber of rings could be described by a bathocromic shift dueto higher electronic delocalization. To do so, the disagreementbetween theoretical and experimental electronic transitionsmust be determined, and this is obtained by comparingexperimental and theoretical spectra (Figure 3b) for the five-ring oligomer28,29 (3 alkoxy-phenylenes and 2 thiophenes), de-picted in Figure 3a. This molecule was synthesized by the same
group responsible for the synthesis of the molecules presented inFigure 1.Table 1 presents the most intense calculated electronic
transitions possible to occur in oligomers with the increase inthe number of conjugated rings. Figure 4 shows the energy gapbehavior (taken as the most intense transition) and the hyper-bolic fit, assuming that the “electron in a box” model is the mostappropriate to describe the gap behavior for conjugation length.So, from the fit, the following can be obtained (Figure 4):
EgapðpÞ ¼ 2:69 +1:89
p� 0:06
!( 0:10 eV ð2Þ
where the Egap(p) = 2.69 agrees well with the experimentalextrapolation to the infinite chain (p f ∞) and the correctionfactor 0.06 was kept for inhomogenity adjustment. The parameter2.69 was obtained after fitting data from Table 1 and applying theequation to the structure depicted in Figure 3, determining, inthis way, the theoretical�experimental discrepancy.Using eq 2 for the irradiated polymers’ experimental gap value,
it is possible to note that the number of conjugated rings in chainis strongly reduced with the increase of the dose, as shown inTable 2. Considering this, the irradiation of the solution defini-tively induces a conjugation break, as it was already reported inliterature.44
At this point, the conjugation break was assumed to beinduced by the breaking of the main chain, which is not alwaystrue, because it is not possible to reach such a conclusion solelybased on the absorption spectra. However, the further analysispresented herein will strongly indicate the existence of the chainscission.
Figure 3. (a) Five-ring oligomer used to parametrize optical absorptionbehavior. (b) Theoretical�experimental disagreement to five-ringstructure.
Table 1. Energy Gap for Different Numbers of Chain Rings
number of chain
rings (p)
theoretically estimated
gap (eV)
experimentally measured
gap (eV)
2 3.93
4 3.46
5 3.43 3.07
6 3.24
8 3.27
10 3.15
12 3.15
14 3.03
16 3.09
Figure 4. Gap behavior for increasing number of rings.
Table 2. Number of Conjugated Rings in Function of theDose
radiation
dose (kGy)
experimentally estimated
gap (eV)
number of conjugated
rings (p, avg)
0 2.72 66.61
20 2.77 21.07
40 3.04 5.32
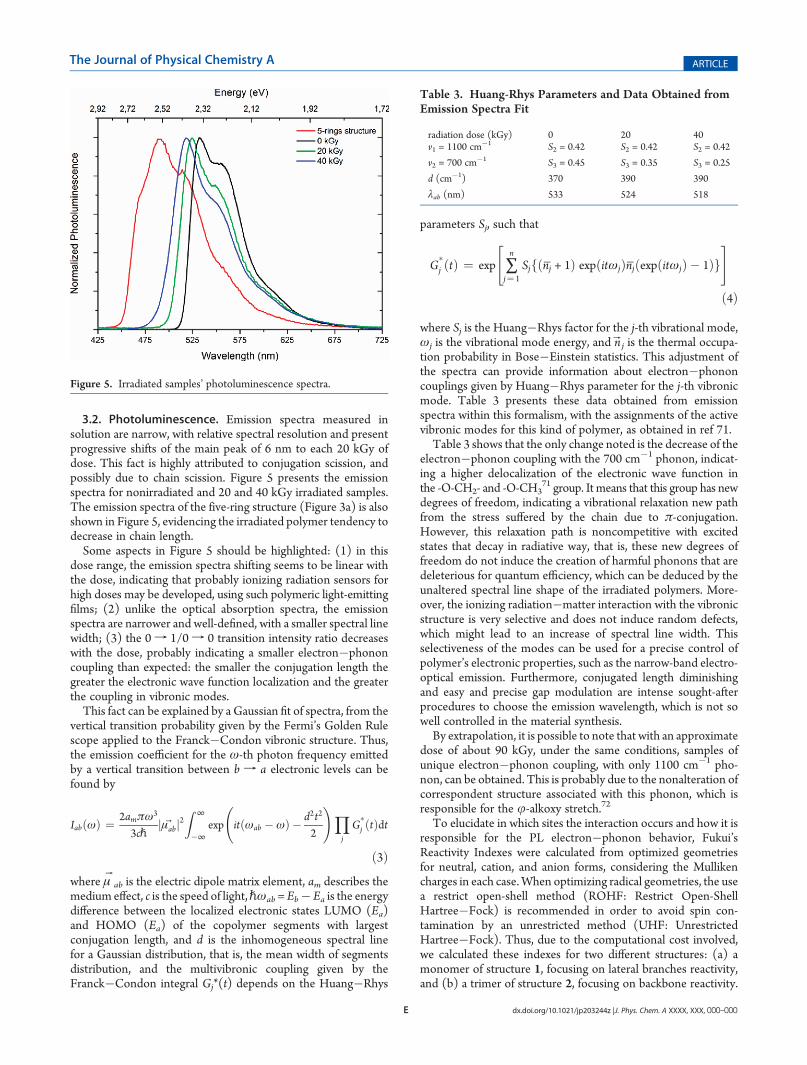
E dx.doi.org/10.1021/jp203244z |J. Phys. Chem. A XXXX, XXX, 000–000
The Journal of Physical Chemistry A ARTICLE
3.2. Photoluminescence. Emission spectra measured insolution are narrow, with relative spectral resolution and presentprogressive shifts of the main peak of 6 nm to each 20 kGy ofdose. This fact is highly attributed to conjugation scission, andpossibly due to chain scission. Figure 5 presents the emissionspectra for nonirradiated and 20 and 40 kGy irradiated samples.The emission spectra of the five-ring structure (Figure 3a) is alsoshown in Figure 5, evidencing the irradiated polymer tendency todecrease in chain length.Some aspects in Figure 5 should be highlighted: (1) in this
dose range, the emission spectra shifting seems to be linear withthe dose, indicating that probably ionizing radiation sensors forhigh doses may be developed, using such polymeric light-emittingfilms; (2) unlike the optical absorption spectra, the emissionspectra are narrower and well-defined, with a smaller spectral linewidth; (3) the 0 f 1/0 f 0 transition intensity ratio decreaseswith the dose, probably indicating a smaller electron�phononcoupling than expected: the smaller the conjugation length thegreater the electronic wave function localization and the greaterthe coupling in vibronic modes.This fact can be explained by a Gaussian fit of spectra, from the
vertical transition probability given by the Fermi’s Golden Rulescope applied to the Franck�Condon vibronic structure. Thus,the emission coefficient for the ω-th photon frequency emittedby a vertical transition between b f a electronic levels can befound by
IabðωÞ ¼ 2amπω3
3cpjμ~abj2
Z ∞
�∞exp itðωab �ωÞ � d2t2
2
!Yj
G�j ðtÞdt
ð3Þwhere μBab is the electric dipole matrix element, am describes themedium effect, c is the speed of light,pωab = Eb� Ea is the energydifference between the localized electronic states LUMO (Ea)and HOMO (Ea) of the copolymer segments with largestconjugation length, and d is the inhomogeneous spectral linefor a Gaussian distribution, that is, the mean width of segmentsdistribution, and the multivibronic coupling given by theFranck�Condon integral Gj*(t) depends on the Huang�Rhys
parameters Sj, such that
G�j ðtÞ ¼ exp ∑
n
j¼ 1Sjfðn̅j + 1Þ expðitωjÞn̅jðexpðitωjÞ � 1Þg
" #
ð4Þ
where Sj is the Huang�Rhys factor for the j-th vibrational mode,ωj is the vibrational mode energy, and nBj is the thermal occupa-tion probability in Bose�Einstein statistics. This adjustment ofthe spectra can provide information about electron�phononcouplings given by Huang�Rhys parameter for the j-th vibronicmode. Table 3 presents these data obtained from emissionspectra within this formalism, with the assignments of the activevibronic modes for this kind of polymer, as obtained in ref 71.Table 3 shows that the only change noted is the decrease of the
electron�phonon coupling with the 700 cm�1 phonon, indicat-ing a higher delocalization of the electronic wave function inthe -O-CH2- and -O-CH3
71 group. It means that this group has newdegrees of freedom, indicating a vibrational relaxation new pathfrom the stress suffered by the chain due to π-conjugation.However, this relaxation path is noncompetitive with excitedstates that decay in radiative way, that is, these new degrees offreedom do not induce the creation of harmful phonons that aredeleterious for quantum efficiency, which can be deduced by theunaltered spectral line shape of the irradiated polymers. More-over, the ionizing radiation�matter interaction with the vibronicstructure is very selective and does not induce random defects,which might lead to an increase of spectral line width. Thisselectiveness of the modes can be used for a precise control ofpolymer’s electronic properties, such as the narrow-band electro-optical emission. Furthermore, conjugated length diminishingand easy and precise gap modulation are intense sought-afterprocedures to choose the emission wavelength, which is not sowell controlled in the material synthesis.By extrapolation, it is possible to note that with an approximate
dose of about 90 kGy, under the same conditions, samples ofunique electron�phonon coupling, with only 1100 cm�1 pho-non, can be obtained. This is probably due to the nonalteration ofcorrespondent structure associated with this phonon, which isresponsible for the j-alkoxy stretch.72
To elucidate in which sites the interaction occurs and how it isresponsible for the PL electron�phonon behavior, Fukui’sReactivity Indexes were calculated from optimized geometriesfor neutral, cation, and anion forms, considering the Mullikencharges in each case.When optimizing radical geometries, the usea restrict open-shell method (ROHF: Restrict Open-ShellHartree�Fock) is recommended in order to avoid spin con-tamination by an unrestricted method (UHF: UnrestrictedHartree�Fock). Thus, due to the computational cost involved,we calculated these indexes for two different structures: (a) amonomer of structure 1, focusing on lateral branches reactivity,and (b) a trimer of structure 2, focusing on backbone reactivity.
Figure 5. Irradiated samples’ photoluminescence spectra.
Table 3. Huang-Rhys Parameters and Data Obtained fromEmission Spectra Fit
radiation dose (kGy) 0 20 40v1 = 1100 cm�1 S2 = 0.42 S2 = 0.42 S2 = 0.42
v2 = 700 cm�1 S3 = 0.45 S3 = 0.35 S3 = 0.25
d (cm�1) 370 390 390
λab (nm) 533 524 518

F dx.doi.org/10.1021/jp203244z |J. Phys. Chem. A XXXX, XXX, 000–000
The Journal of Physical Chemistry A ARTICLE
This was needed because the presence of expanded lateralbranches induces isoenergetic degrees of freedom, making thetrimer structure with expanded branches highly time-consumingand unreliable calculations. Figure 6 shows graphically theabsolute Fukui indexes f0, indicating that the most reactiveinteraction sites are the bonding carbon atoms of the thiophenering, for the main backbone, and the second and third oxygenatoms in the dialkoxy group. This approach allows us to predictthat the radiation induces effectively the main backbone scissionin the thiophene ring (what corroborates to OA results), and thesame occurs to the lateral branch at the second and third oxygenatom, which keeps intact the main conjugated backbone electro-nic structure, agreeing with UV�vis optical absorption andphotoluminescence results. In addition, the high reactivity ofthese sites, mainly in the trimer structure, strongly indicates theoccurrence of a chain scission in these sites, which leads to aconjugation break. The highly energetic gamma photons mayinduce variations in charge density over these sites, whatdestabilizes the structure, turning the broken structure morestable than the bonded. Still, the lateral branch scission partiallyexplains the enlargement of the UV�vis spectra and the narrow-ing of PL spectra.These results can be used for the development of more
efficient conjugated-polymer based devices, such as photovoltaiccells and light-emitting diodes. The absorption spectra over-lapping in irradiated solutions with solar emission is greater thanin nonirradiated ones, resulting in a larger amount of absorbedenergy that can be efficiently used. Moreover, the emissionspectra in OLEDs can be obtained for very-well-specified de-signed devices. Still, such defects can relieve the polymeric chainfrom the inevitable stress that it undergoes during film formationinducing,6 thus, bringing higher yields.
4. DISCUSSION AND CONCLUSION
In this work, a new methodology for understanding a defectinducing process, the gap energy modulation and vibroniccouplings in a conjugated polymer by γ radiation was reported.It was noted that the semiempirical simulations approach candeduct how γ radiation interacts with the light-emitting polymer.Results indicate that electronic properties of conjugated matriceswith such defects can probably provide devices with higherquantum yields, based on themultivibronic coupling. In addition,the gap energy can be modulated by a mere variation of the dose.
According to the data presented in this work, γ irradiation isconsidered a fast and viable method to introduce defects in light-emitting conjugated polymeric matrices, due to their highlyenergetic photons. Moreover, in a special conjugated matrix suchas poly(2,5-dialkoxy-p-phenylene-thienylene), γ photons inducethe chain scission in the alkoxy branch and among conjugatedrings, hindering some vibronic levels in a very specific way. Finally,the approach presented here proved to be an efficient method tounderstand, simulate, and improve the properties of light-emittingconjugated polymers.
’AUTHOR INFORMATION
Corresponding Author*E-mail: [email protected]; [email protected].
’ACKNOWLEDGMENT
The authors would like to thank the Brazilian fundingagencies: INCT/nanomateriais de carbono, INCT/INEO,INCT/INFO, CNPq, CAPES, FAPEMIG, and FAPESP, forthe financial support and the CTR-IPEN Irradiation Laboratorystaff. H.S.S. is deeply thankful to D. P. Andrade for thediscussions.
’REFERENCES
(1) Coakley, K. M.; McGehee, M. D. Chem. Mater. 2004, 16, 4533.(2) Friend, R. H.; Gymer, R. W.; Holmes, A. B.; Burroughes, J. H.;
Marks, R. N.; Taliani, C.; Bradley, D. D. C.; Santos, D. A. D.; Bredas,J. L.; Logdlund, M.; Salaneck, W. R. Nature 1999, 397, 121.
(3) Br�edas, J. L.; Beljonne, D.; Cornil, J.; dos Santos, D. A.; Shuai, Z.Philos. Trans. R. Soc., A 1997, 355, 735.
(4) Salaneck, W. R.; Friend, R. H.; Br�edas, J. L. Phys. Rep. 1999,319, 231.
(5) Brunsveld, L.; Folmer, B. J. B.; Meijer, E. W.; Sijbesma, R. P.Chem. Rev. 2001, 101, 4071.
(6) Liang, Z.; Nardes, A.; Wang, D.; Berry, J. J.; Gregg, B. A. Chem.Mater. 2009, 21, 4914.
(7) Yan, M.; Rothberg, L. J.; Papadimitrakopoulos, F.; Galvin, M. E.;Miller, T. M. Phys. Rev. Lett. 1994, 73, 744.
(8) Wang, D.; M. R., Kopidakis, N.; Gregg, B.A. In 33rd IEEEPhotovoltaic Specialists Conference; National Renewable Energy Laboratory:San Diego, CA, 2008.
(9) Wong, K. F.; Skaf, M. S.; Yang, C.-Y.; Rossky, P. J.; Bagchi, B.;Hu, D.; Yu, J.; Barbara, P. F. J. Phys. Chem. B 2001, 105, 6103.
(10) Yurtsever, E.; Yurtsever, M. Synth. Met. 1999, 101, 335.(11) Gregg, B. A. J. Phys. Chem. C 2009, 113, 5899.(12) Gregg, B. A. Soft Matter 2009, 5, 2985.(13) Lee, K.-Y.; Kim, K.-Y. Polym. Degrad. Stab. 2008, 93, 1290.(14) Crespi, V. H.; Chopra, N. G.; Cohen, M. L.; Zettl, A.; Louie,
S. G. Phys. Rev. B 1996, 54, 5927.(15) Smith, B. W.; Luzzi, D. E. J. Appl. Phys. 2001, 90, 3509.(16) Chen, S.; Wu, G.; Liu, Y.; Long, D. Macromolecules 2005,
39, 330.
Figure 6. Graphical representation of the Fukui indices for each site forboth structures (reduced trimer and expanded monomer). The darkerthe site indicates the more reactive the site is. Geometries do notcorrespond to the optimized ones and are forced to be planar forillustrative reasons.

G dx.doi.org/10.1021/jp203244z |J. Phys. Chem. A XXXX, XXX, 000–000
The Journal of Physical Chemistry A ARTICLE
(17) Cataldo, F. Carbon 2000, 38, 634.(18) Campbell, I. H.; Crone, B. K. Adv. Mater. 2006, 18, 77.(19) Dunning, H. N.; Moore, J. W. Ind. Eng. Chem. 1959, 51, 161.(20) Myers, L. S., Jr.; Rothschild, M.-L.; Kersten, M.; Cosi, L. Radiat.
Res. 1959, 11, 761.(21) Xu, H.; Wang, X.; Zhang, Y.; Liu, S. Chem. Mater. 2006,
18, 2929.(22) Graham, S. C.; Friend, R. H.; Fung, S.; Moratti, S. C. Synth. Met.
1997, 84, 903.(23) Doug Reilly, N. E., Smith, H., Jr.; Kreiner, S. Passive Nondes-
tructive Assay of Nuclear Materials; National Technical InformationService: Washington, DC, 1991.(24) Davenas, J.; Stevenson, I.; Celette, N.; Cambon, S.; Gardette,
J. L.; Rivaton, A.; Vignoud, L. Nucl. Instrum. Methods Phys. Res., Sect. B2002, 191, 653.(25) Silva, R. A.; Cury, L. A.; Marletta, A.; Guimar~aes, P. S. S.;
Bouachrine, M.; L�ere-Porte, J. P.; Moreau, J. E.; Serein-Spirau, F. J. Non-Cryst. Solids 2006, 352, 3685.(26) Lois, S.; Flor�es, J.-C.; L�ere-Porte, J.-P.; Serein-Spirau, F.;
Moreau, J. J. E.; Miqueu, K.; Sotiropoulos, J.-M.; Bayl�ere, P.; Tillard,M.; Belin, C. Eur. J. Org. Chem. 2007, 2007, 4019.(27) Ding, A.-L.; Pei, J.; Lai, Y.-H.; Huang, W. J. Mater. Chem. 2001,
11, 3082.(28) Silva, R. A.; Cury, L. A.; Mazzoni, M. S.; Soares, E.; Guimar~aes,
P. S. S.; Serein-Spirau, F.; L€ois, S.; Moreau, J.; L�ere-Porte, J. P.Macromol.Symp. 2005, 229, 194.(29) Silva, R. A.; Serein-Spirau, F.; Bouachrine, M.; Lere-Porte, J.-P.;
Moreau, J. J. E. J. Mater. Chem. 2004, 14, 3043.(30) Bouzakraoui, S.; Z., H.; Bouzzine, S. M.; Augusta Dasilva, R.;
L�ere-Porte, J-P; Serein-Spirau, F.; Alimi, K.; Hamidi, M.; Bouachrine, M.Phys. Chem. News 2010, 51, 111.(31) Bouachrine, M.; L�ere-Porte, J.-P.; Moreau, J. J. E.; Spirau, F. S.;
da Silva, R. A.; Lmimouni, K.; Ouchani, L.; Dufour, C. Synth. Met. 2002,126, 241.(32) Roncali, J. Chem. Rev. 1992, 92, 711.(33) Roncali, J. Chem. Rev. 1997, 97, 173.(34) McCullough, R. D. Adv. Mater. 1998, 10, 93.(35) Pei, J.; Yu, W.-L.; Huang, W.; Heeger, A. J. Macromolecules
2000, 33, 2462.(36) Schopf, G.; Kossmehl, G. In Polythiophenes - Electrically Con-
ductive Polymers; Springer: Berlin/Heidelberg: 1997; Vol. 129, p 51.(37) Lere-Porte, J.-P.; Moreau, J. J. E.; Serein-Spirau, F.; Torreilles,
C.; Righi, A.; Sauvajol, J.-L.; Brunet, M. J. Mater. Chem. 2000, 10, 927.(38) Pei, Q.; Yang J. Am. Chem. Soc. 1996, 118, 7416.(39) Kudoh, H.; Sasuga, T.; Seguchi, T.; Katsumura, Y. Polymer
1996, 37, 2903.(40) Yoshino, K.; Hayashi, S.; Inuishi, Y. Jpn. J. Appl. Phys. 1982,
21, L569.(41) Bernius,M. T.; Inbasekaran,M.; O’Brien, J.;Wu,W.Adv.Mater.
2000, 12, 1737.(42) Burroughes, J. H.; Bradley, D. D. C.; Brown, A. R.; Marks, R. N.;
Mackay, K.; Friend, R. H.; Burns, P. L.; Holmes, A. B. Nature 1990,347, 539.(43) Silva, M. D. R.; Ganc) alves, A. A., Jr; Silva, R. A.; Marletta, A.
J. Non-Cryst. Solids 2010, 356, 2429.(44) Silva, E. A. B.; Borin, J. F.; Nicolucci, P.; Graeff, C. F. O.; Netto,
T. G.; Bianchi, R. F. Appl. Phys. Lett. 2005, 86, 131902.(45) Ferreira, G. R.; Vasconcelos, C. K. B. d.; Silva, M. M.; Santos,
F. A. d.; Pires, J. G.; Duarte, A. S.; Bianchi, A. G. C.; Bianchi, R. F.MRSOnline Proceedings Library 2009, 1209.(46) Vasconcelos, C. K. B. d. F.; Giovana, R.; Bianchi, R. F. Pol�imeros
[online] 2010, 20, 14.(47) Dewar, M. J. S.; Thiel, W. J. Am. Chem. Soc. 1977, 99, 4899.(48) Dewar, M. J. S.; Zoebisch, E. G.; Healy, E. F.; Stewart, J. J. P.
J. Am. Chem. Soc. 1985, 107, 3902.(49) Anderson, W. P.; Edwards, W. D.; Zerner, M. C.; Canuto, S.
Chem. Phys. Lett. 1982, 88, 185.(50) Bacon, A. D.; Zerner, M. C. Theor. Chim. Acta 1979, 53, 21.
(51) Edwards, W. D.; Zerner, M. C. Theor. Chim. Acta 1987, 72, 347.(52) Head, J. D.; Zerner, M. C. Chem. Phys. Lett. 1985, 122, 264.(53) Head, J. D.; Zerner, M. C. Chem. Phys. Lett. 1986, 131, 359.(54) Ridley, J.; Zerner, M. Theor. Chim. Acta 1973, 32, 111.(55) Ridley, J. E.; Zerner, M. C. Theor. Chim. Acta 1976, 42, 223.(56) Marletta, A.; Guimar~aes, F. E. G.; Faria, R. M. Braz. J. Phys.
2002, 32, 2b.(57) Bowden, K.; M. F. Fabian, W.; Kollenz, G. J. Chem. Soci., Perkin
Trans. 2 1997, 547.(58) Marra, P. H. S.; Silva, R. A. 2006; unpublished work.(59) Stewart, J. J. P. MOPAC2009, version 10.341W; Stewart
Computational Chemistry; web: HTTP://OpenMOPAC.net.(60) Thompson, M. A. ArgusLab 4.0; Planaria Software LLC:
Seattle, WA; web: http://www.arguslab.com.(61) Del Nero, J.; Galv~ao, D. S.; Laks, B. Opt. Mater. 2003, 21, 461.(62) Giro, R.; Galv~ao, D. S. Int. J. Quantum Chem. 2003, 95, 252.(63) Misra, G. P.; Sannigrahi, A. B. THEOCHEM 1996, 361, 63.(64) Roy, R. K.; Hirao, K.; Krishnamurty, S.; Pal, S. J. Chem. Phys.
2001, 115, 2901.(65) Johnson, Kurt A.; Ashcroft, N. W. Phys. Rev. B 1998, 58, 15548.(66) Gartstein, Y. N.; Rice, M. J.; Conwell, E. M. Phys. Rev. B 1995,
51, 5546.(67) Nguyen, T.-Q.; Martini, I. B.; Liu, J.; Schwartz, B. J. J. Phys.
Chem. B 1999, 104, 237.(68) Qi, Z.; Wei, B.; Sun, Y.; Wang, X.; Kang, F.; Hong, M.; Tang, L.
Polym. Bull. 2011, 66, 905.(69) Sundberg, M.; Ingan€as, O.; Stafstr€om, S.; Gustafsson, G.;
Sj€ogren, B. Solid State Commun. 1989, 71, 435.(70) It is interesting to note that the solid state and gas phase
structures have a few differences in S 3 3 3O interactions: when in the gasphase, this interaction is dominant on the Hamiltonian’s potentialenergy terms; in the solid state, there are other concomitant interactions,that are not taken into account in the gas phase calculation, which canpartially hinder the S 3 3 3O interaction. For instance, we can men-tion intermolecular interdigitation, film formation stress, aggregation,pi-stacking, defects, and so on.
(71) Silva, R. A.; Cury, L. A.; Guimar~aes, P. S. S.; Marletta, A.;Serein-Spirau, F.; Bouachrine, M.; Moreau, J. J. E.; L�ere-Porte, J. P.J. Polym. Sci., Part B: Polym. Phys. 2010, 48, 964.
(72) Brito, W. H.; Silva, R. A.; Miwa, R. H. J. Chem. Phys. 2010,133, 204703.


















