
Enfermedad de pompe
41
Enfermedad de Pompe Carmen Díaz Diego Romo Integración Avanzada en AP
-
Upload
carmen-perez -
Category
Health & Medicine
-
view
61 -
download
4
Transcript of Enfermedad de pompe

Enfermedad de Pompe
Carmen DíazDiego Romo
Integración Avanzada en AP

Concepto de la enfermedad
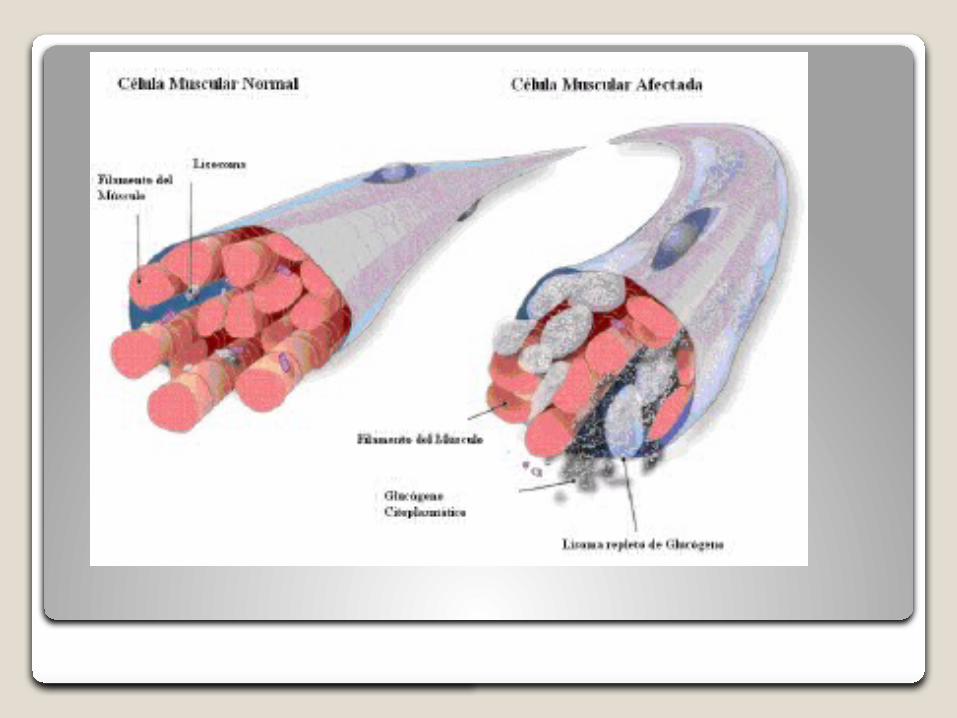
La enfermedad de Pompe, también llamada glucogenosis tipo II o deficiencia de maltasa ácidas es una miopatía metabólica rara, progresiva y letal es un tipo de trastornos de depósito lisosomal, provocada por una deficiencia de hidrolisis lisosómica de α-glucosidasa ácida (GAA) que degrada el glucógeno lisosomal a glucosa.
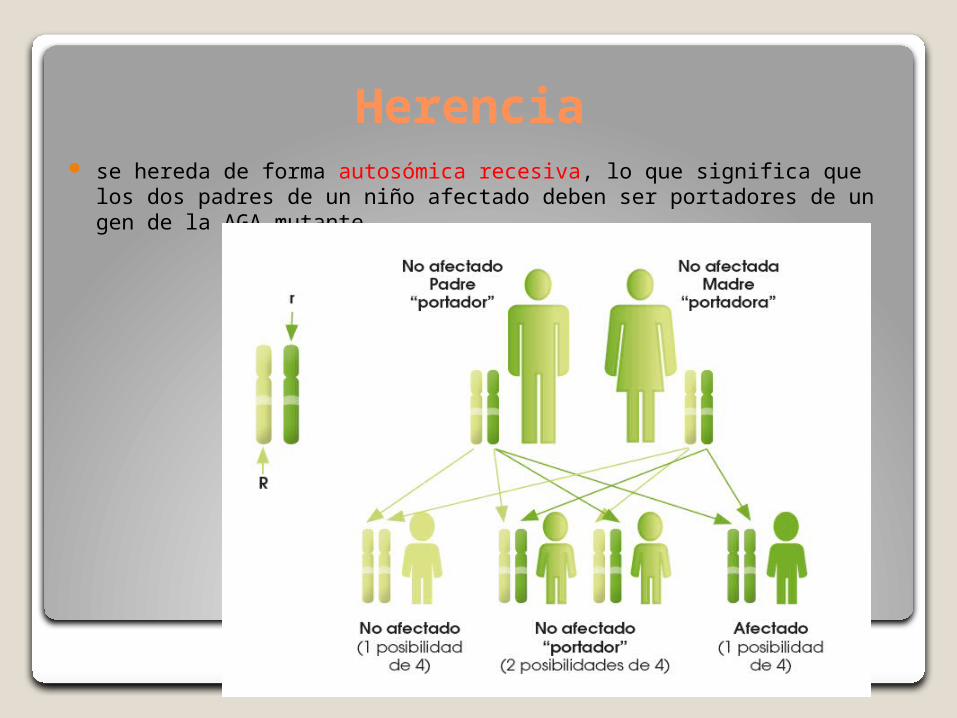
Herencia se hereda de forma autosómica recesiva, lo que significa que los dos
padres de un niño afectado deben ser portadores de un gen de la AGA mutante.

Manifestaciones clínicas
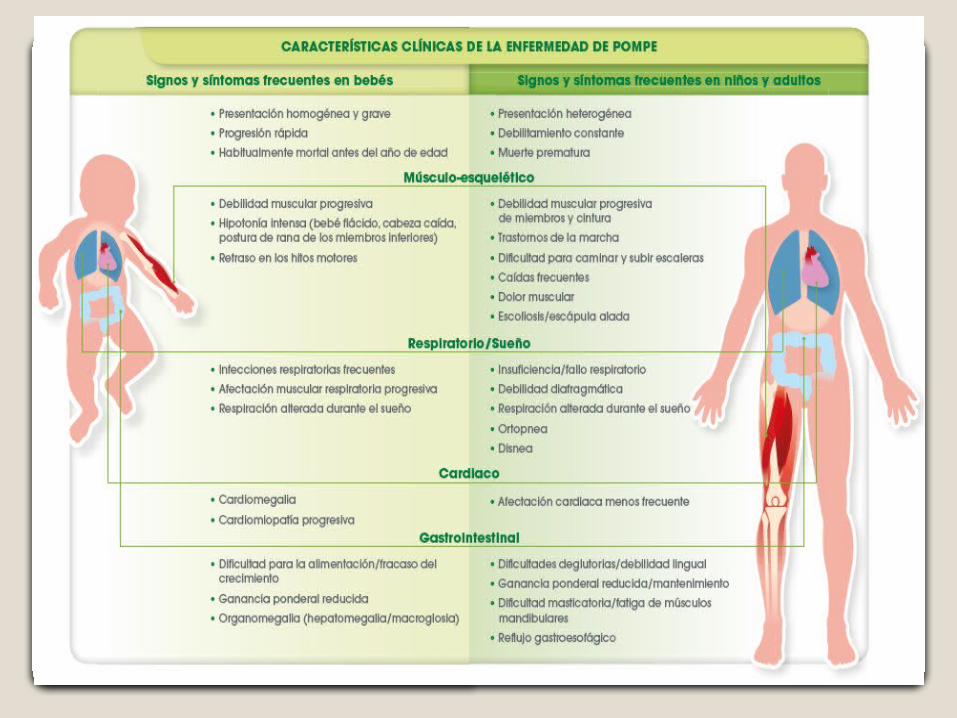
Es una enfermedad neuromuscular que se presenta con un espectro variable de edad de inicio, implicación orgánica y grado de miopatía (afectación muscular).
Forma infantil: alrededor de los 2 meses de vida Forma tardía: después del primer año de vida y se distinguen tres
formas,infantil,juvenil y del adulto (<1% en la forma infantil, hasta el 10% en la juvenil y < 40% pero más del 10% en la forma del adulto).

Patogenia
La deficiencia de la enzima glucosidasa ácida, provoca la acumulación de glucógeno en diversos tejidos, especialmente el músculo cardíaco, respiratorio y esquelético, lo que provoca el desarrollo de una cardiomiopatía hipertrófica y una debilidad muscular progresiva, incluyendo una alteración de la función respiratoria.
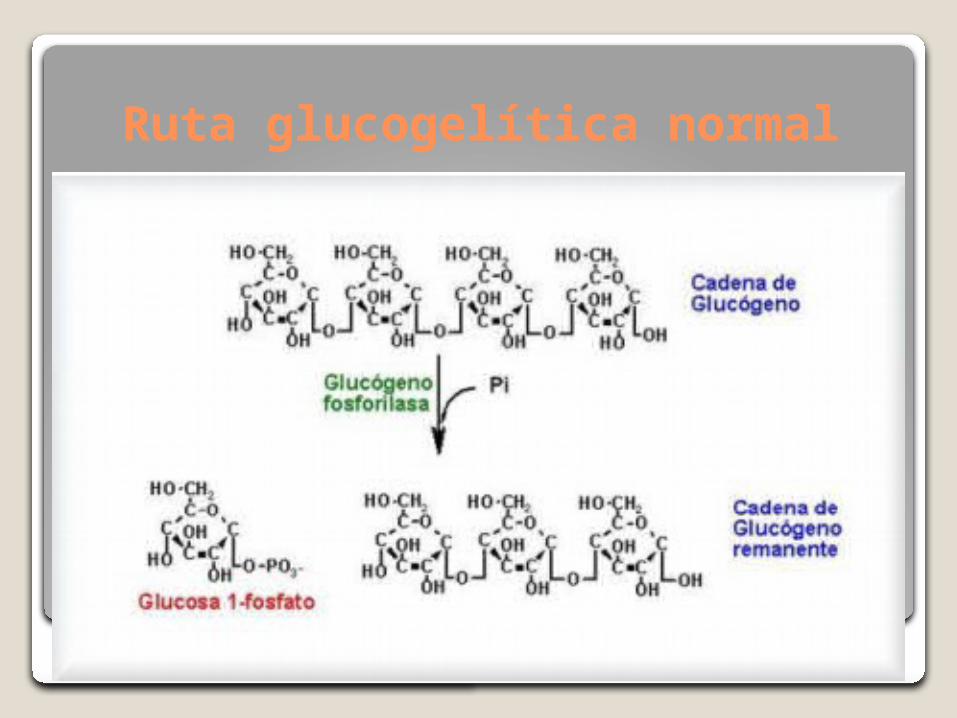
Ruta glucogelítica normal
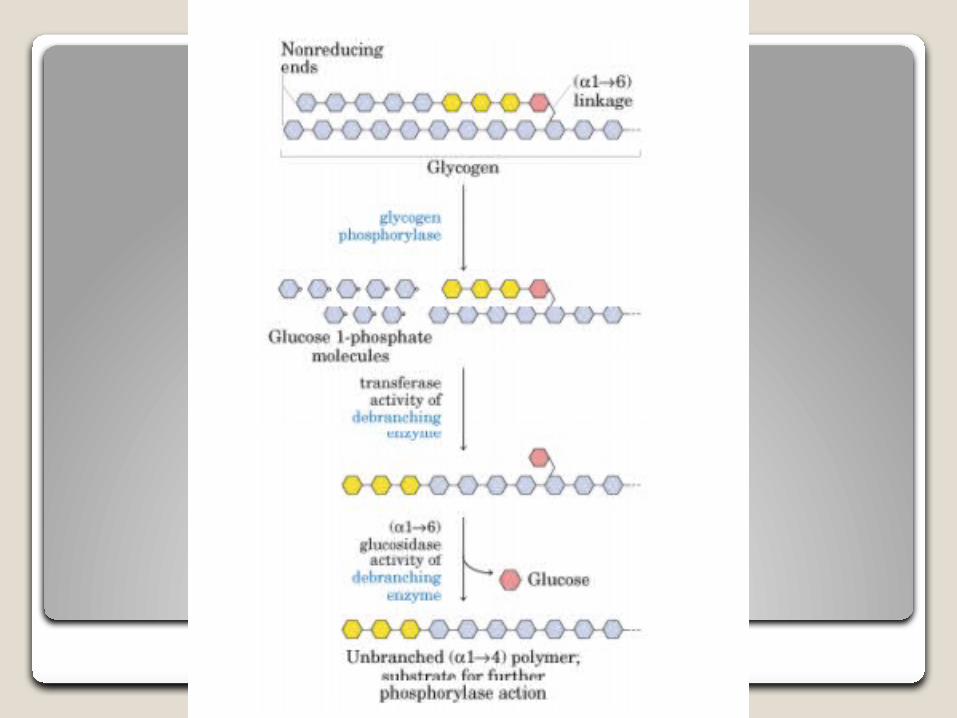
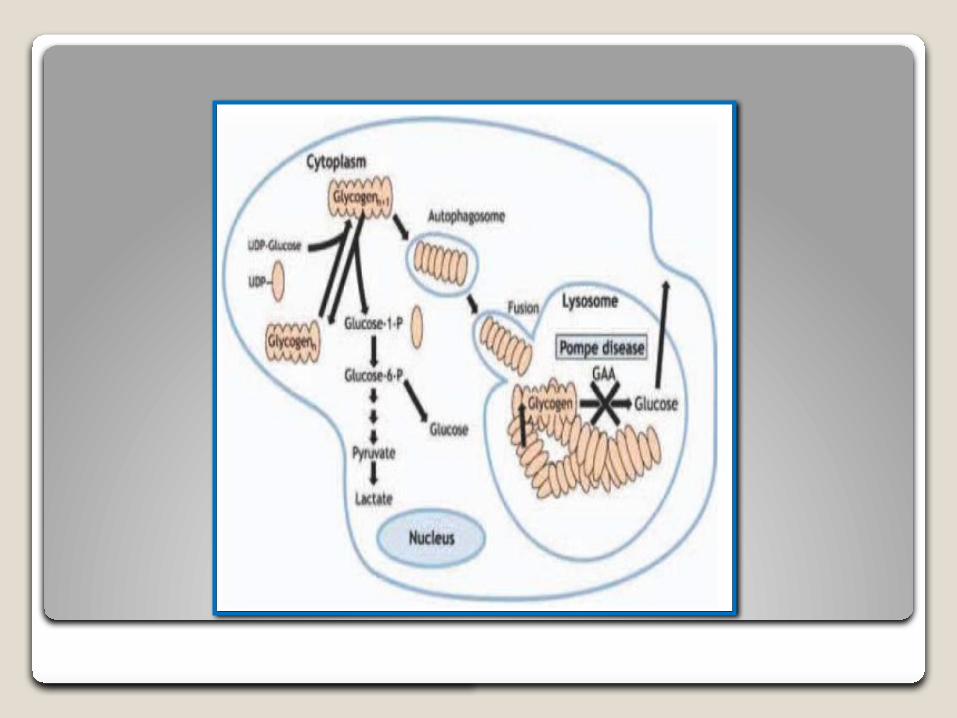
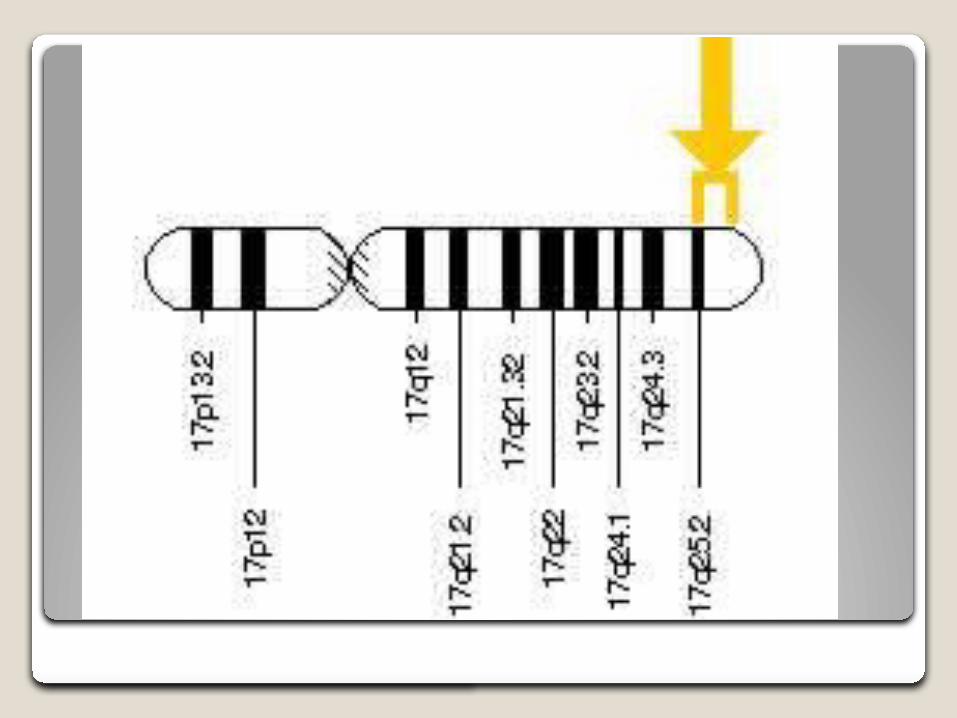
Características moleculares El gen de la α-glucosidasa se encuentra en el brazo largo del
cromosoma 17 (17q25.2-q25.3), y se han identificado más de 350 mutaciones
La mutación más frecuente en pacientes caucásicos es IVS 1 (–13 T>G), que está en uno de los alelo en más del 50% de estos afectados.
Dependiendo del tipo de mutación en el gen, existirá una deficiencia total o parcial de la actividad de la enzima en todas las células del organismo.

Epidemiología
Basado en los datos disponibles, los investigadores han calculado que la enfermedad ocurre en aproximadamente 1 en 40.000 nacimientos vivos mundialmente.
Prevalencia de menos de 200.000 personas en los Estados Unidos de Norteamérica y no más de 5 en 10.000 personas en Europa.
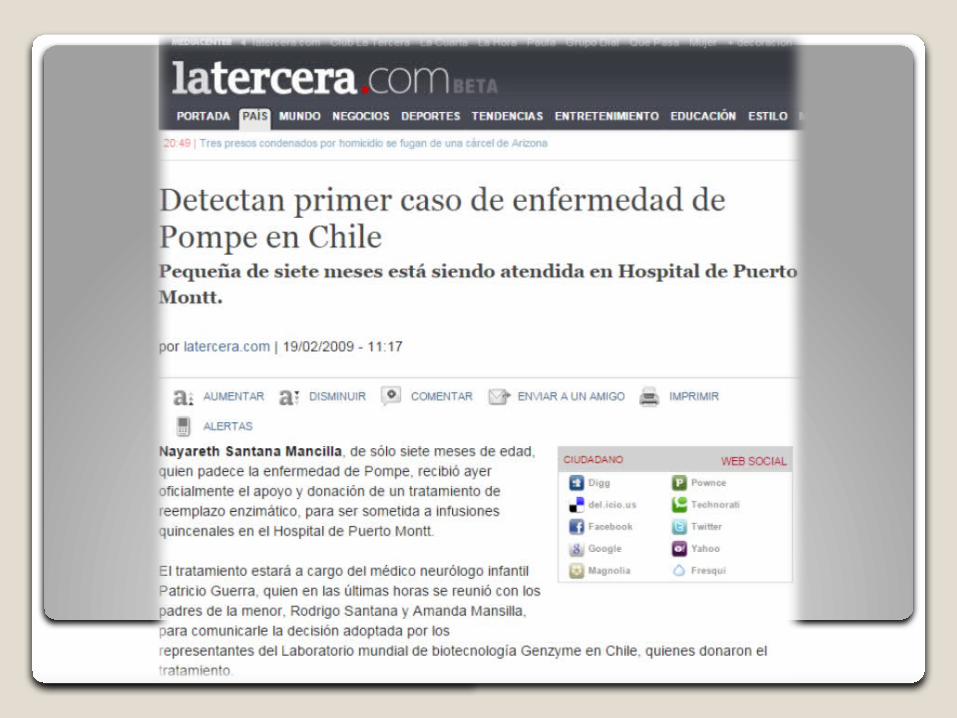

Claudia Saladriga (23), una de las cuatro personas en Chile diagnosticadas con el mal de Pompe. Revista Paula 2013
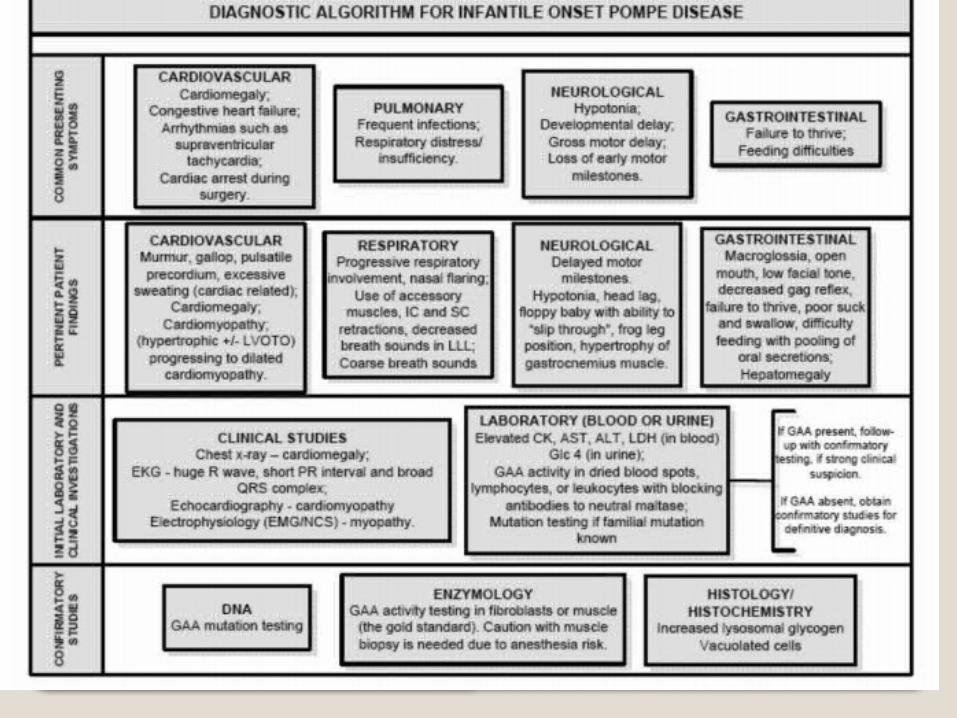

Exámenes diagnósticos CPK (creatina-fosfocinasa)ALT (Alanina aminotransferasa)ASTDeshidrogenasa lácticaLactatoElectromiografía y conducción nerviosaBiopsia

Examen de creatina-fosfocinasavalor normal:10 a 120 microgramos por litro (mcg/L)
Examen de sangreCuando el nivel total de creatina-
fosfocinasa es muy alto, generalmente significa que ha habido lesión o estrés en el corazón, el cerebro o el tejido muscular.
cuando se presenta un daño en el músculo, la creatina-fosfocinasa se filtra al torrente sanguíneo.

Alanina aminotransferasa rango normal es de 10 a 40 unidades internacionales por litro (UI/L).
Muestra de sangreEs una enzima que se encuentra en
mayores cantidades en el hígado. La lesión a este órgano ocasiona la liberación de la sustancia dentro de la sangre.
Este examen se usa para determinar si un paciente tiene daño hepático

Aspartato aminotransferasa El rango normal es de 10 a 34 UI/L.
Muestra de sangre Es una enzima que se encuentra en altas
cantidades en las células del hígado, el corazón y los músculos
El aumento en los niveles de AST generalmente es un signo de una enfermedad hepática.
Aumento fisiológico en embarazo y después del ejercicio

Examen deshidrogenasa láctica valores normales es de 105 a 333 UI/L
Muestra de sangrese mide para verificar daño tisular. La proteína
deshidrogenasa láctica se encuentra en muchos tejidos del cuerpo, especialmente el corazón, el hígado, el riñón, los músculos, el cerebro, las células sanguíneas y los pulmones.

Examen lactato o acido láctico 4.5 a 19.8 mg/dL (0.5 - 2.2 mmol/L)
Muestra de sangreEl ácido láctico se produce principalmente en las
células musculares y en los glóbulos rojos. Dicho ácido se forma cuando el cuerpo descompone carbohidratos para utilizarlos como energía durante momentos de niveles bajos de oxígeno
Los resultados anormales quieren decir que los tejidos corporales no están obteniendo suficiente oxígeno.
No realizar ejercicio físico previo a toma de examen*
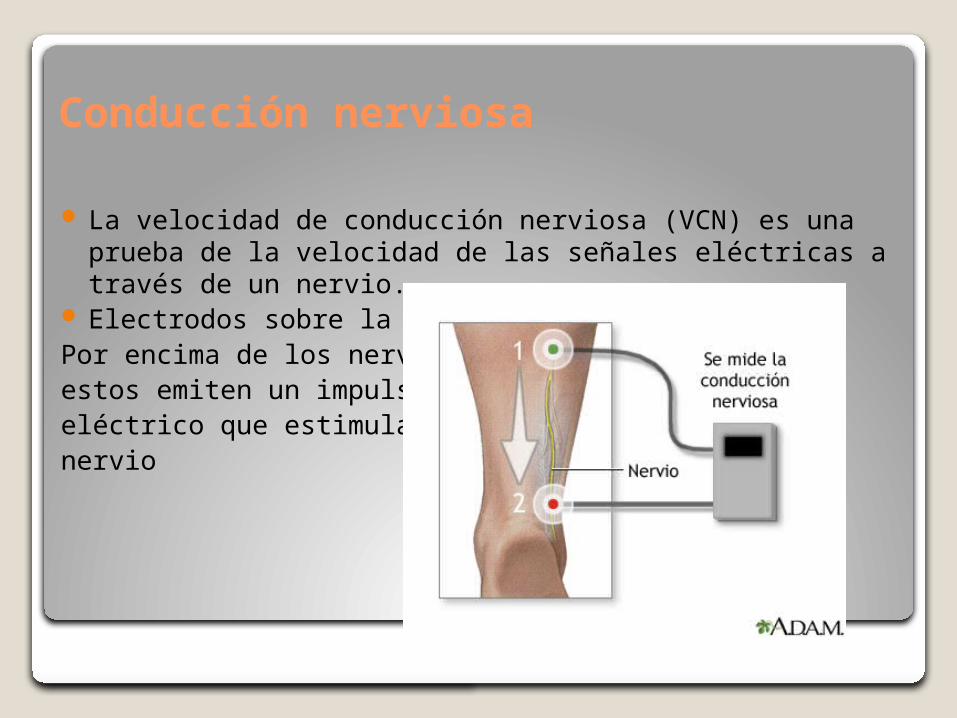
Conducción nerviosa
La velocidad de conducción nerviosa (VCN) es una prueba de la velocidad de las señales eléctricas a través de un nervio.
Electrodos sobre la pielPor encima de los nerviosestos emiten un impulsoeléctrico que estimula alnervio

Algunas de las causas posibles de un resultado anómalo son:axonopatía (daño en la célula nerviosa)bloqueo de la conducción (obstáculo al impulso
en el propio nervio)desmielinización (daño en la vaina de mielina)
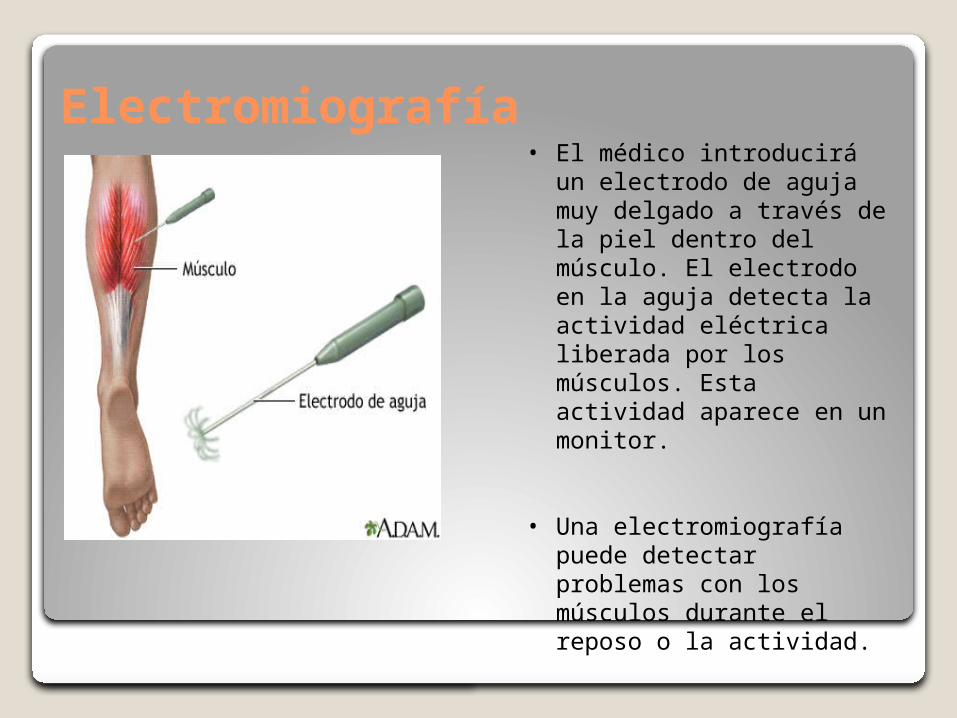
Electromiografía • El médico introducirá un
electrodo de aguja muy delgado a través de la piel dentro del músculo. El electrodo en la aguja detecta la actividad eléctrica liberada por los músculos. Esta actividad aparece en un monitor.
• Una electromiografía puede detectar problemas con los músculos durante el reposo o la actividad.
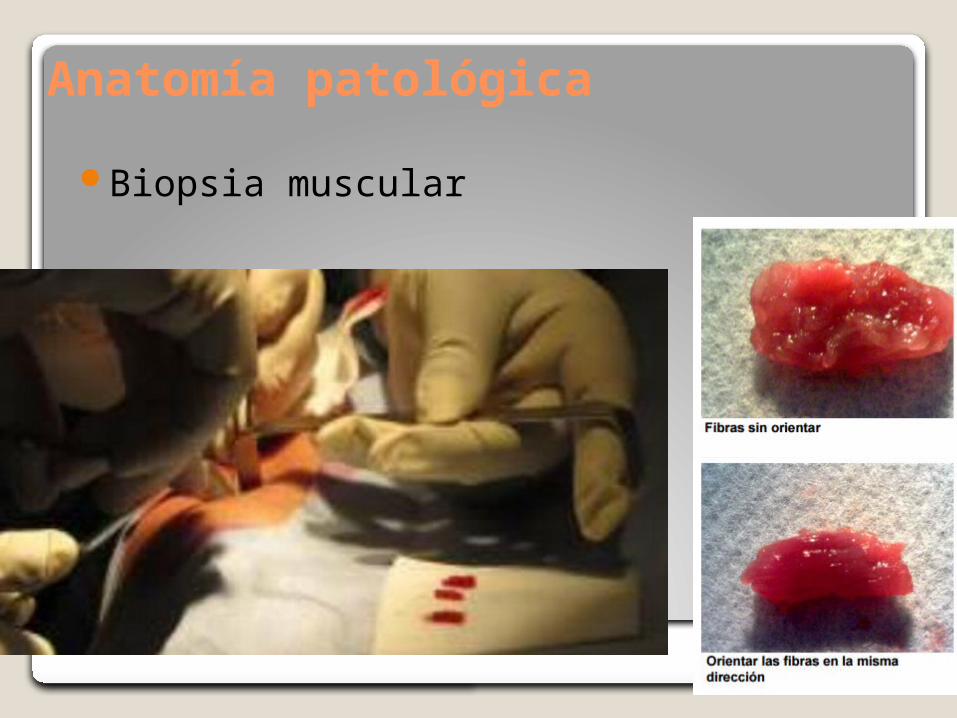
Anatomía patológica
Biopsia muscular

Histoprocesamiento: Criofijación


Tinciones
Rutina(H-E)Ácido peryódico-Schiff (PAS)Coloración histoenzimática:- Fosfatasa ácidaIHQMET
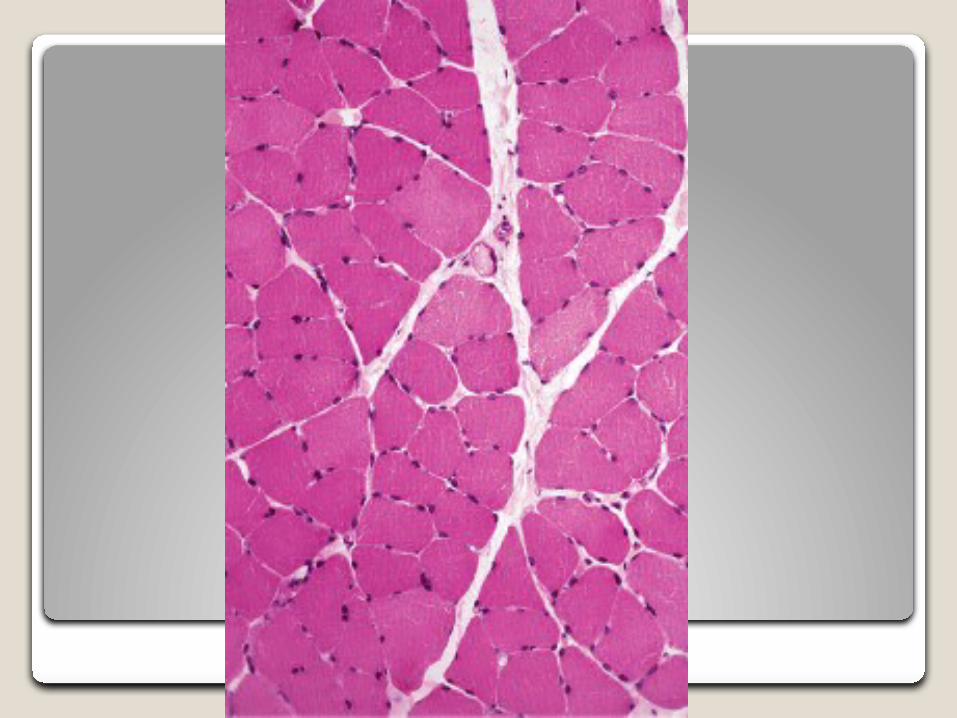
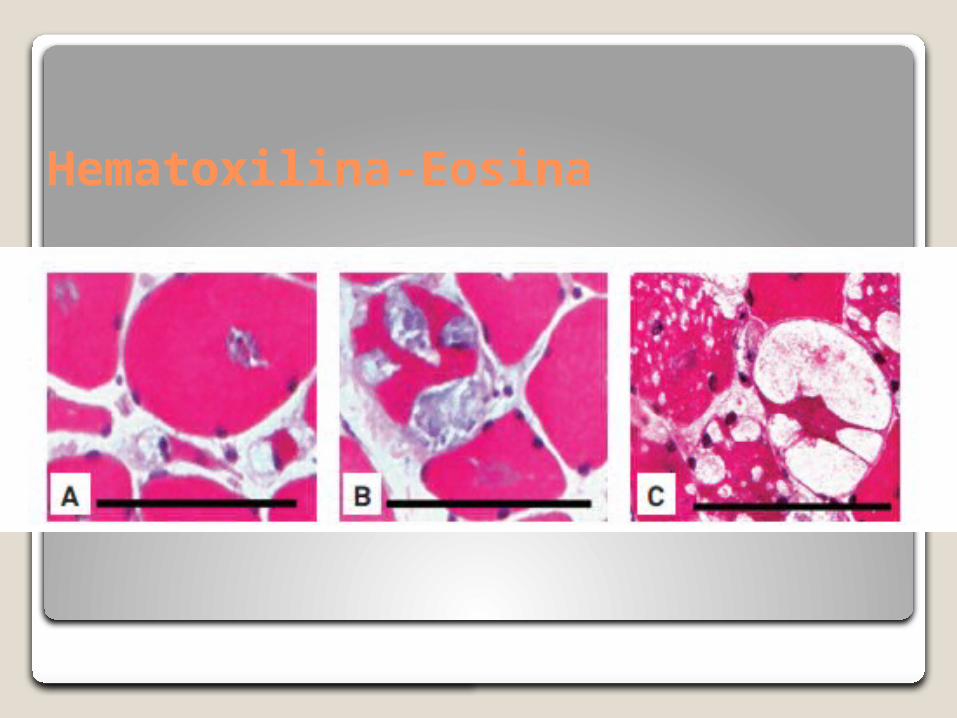
Hematoxilina-Eosina
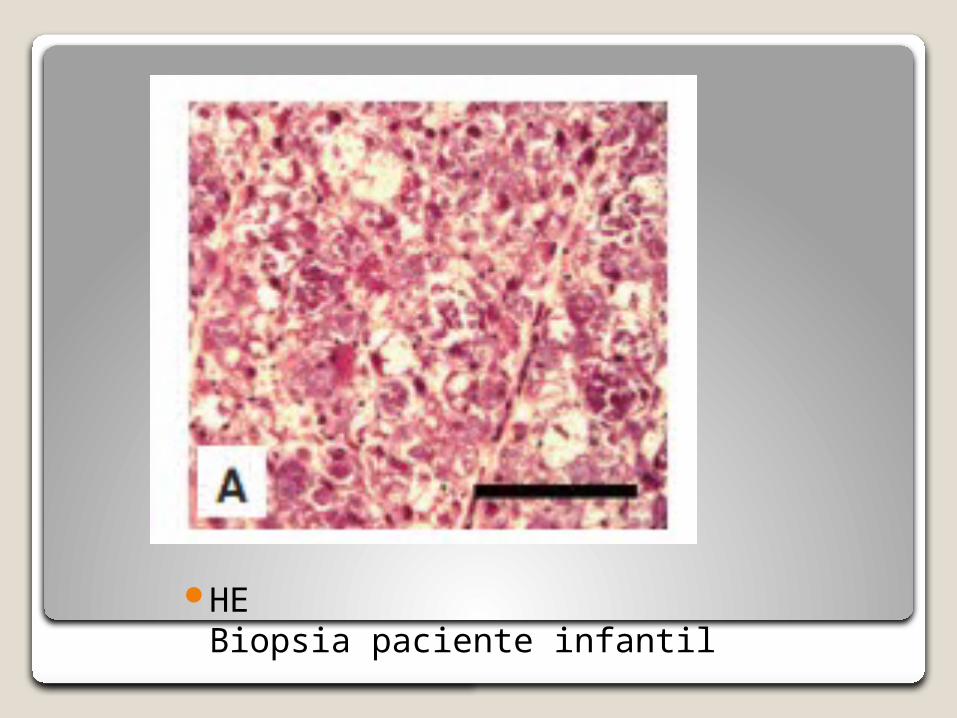
HE Biopsia paciente infantil
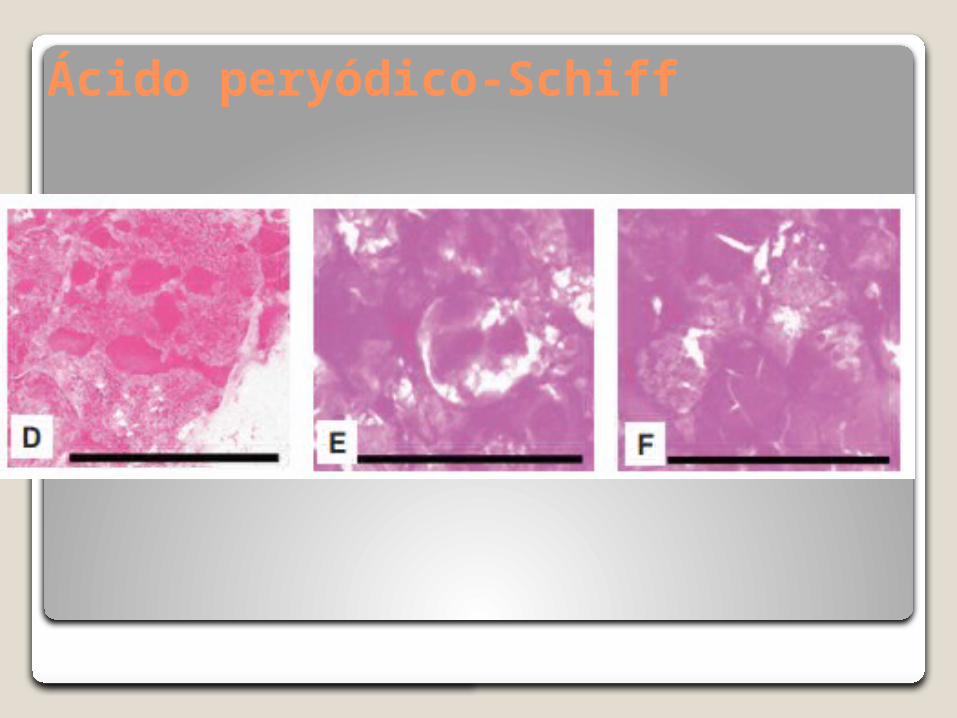
Ácido peryódico-Schiff
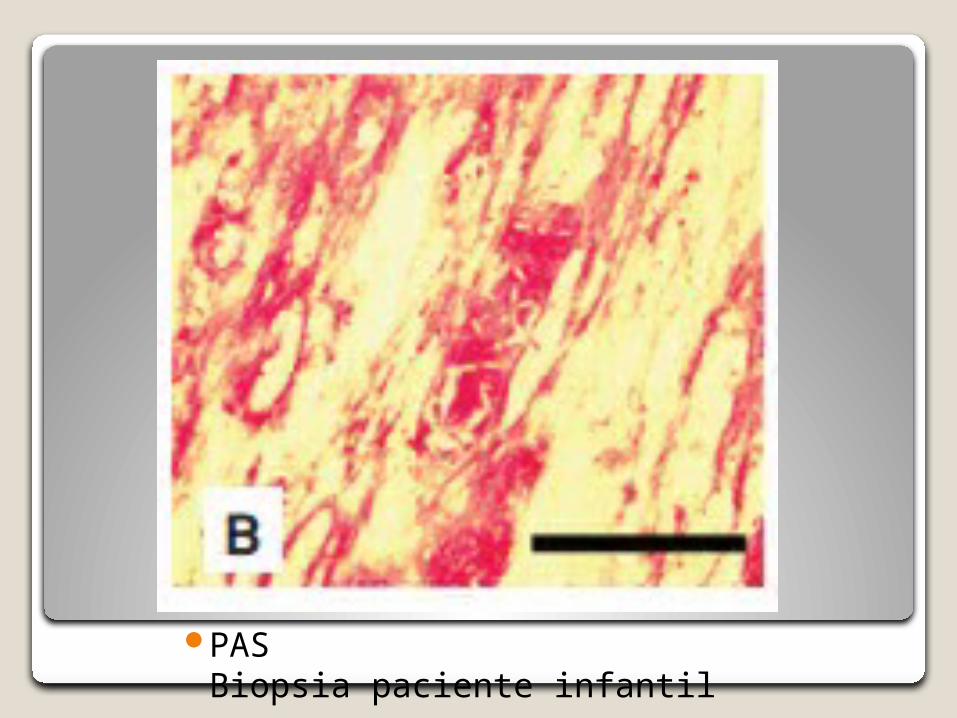
PASBiopsia paciente infantil
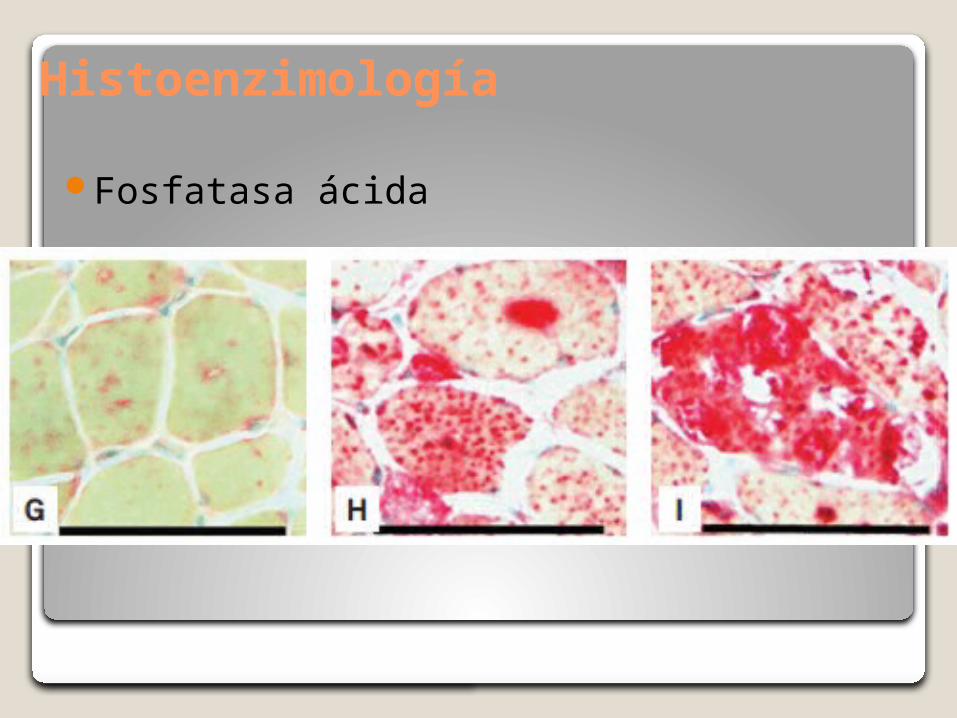
Histoenzimología
Fosfatasa ácida
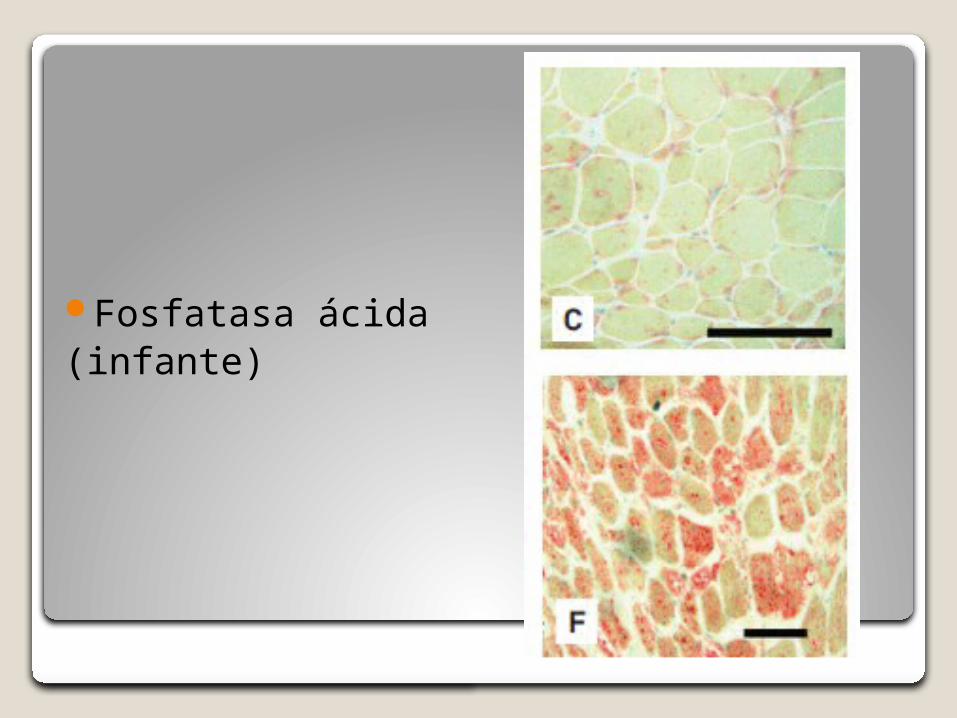
Fosfatasa ácida(infante)

Microscopía electrónica de transmisión

TratamientoTerapias físicas y ocupacionales puede ser
beneficiosa para algunos pacientes. Las alteraciones en la dieta pueden proporcionar mejoras temporales, pero no alteraran el curso de la enfermedad.
El asesoramiento genético puede proveer a las familias la información con respecto al riesgo en los embarazos futuros.

Sustitución enzimática (Por excelencia)
Consiste en la administración intravenosa de una forma precursora de la enzima α-1, 4 glucosidasa, que es capaz de penetrar en los lisosomas.

GRACIAS
Bibliografía http://
www.guiametabolica.org/ecm/enfermedad-pompe/info/causas-manifestacion-clinica-enfermedad-pompe
http://www.fundaciongenzyme.es/Enfermedades/EnfermedadesLisosomales/EnfermedaddePompe.aspx
http://www.scielo.org.co/pdf/anco/v27n4/v27n4a06.pdf http://www.latercera.com/contenido/654_102917_9.shtml http://www.genzyme.cl/thera/pompe/gzla_p_tp_thera_pompe.asp

![MANUAL DE INSTRUC}IUNI - alphapompe.ro · Pompe centrifuge verticale tip SV SV 2, 4, 8, 16 SV 33, 46, 66, 92 Produc\tori: LOWARA - Italia . Manual de instruc]iuni 2 1. GENERALIT|}I](https://static.fdocument.org/doc/165x107/5b9c27a009d3f2194e8bcbbb/manual-de-instruciuni-pompe-centrifuge-verticale-tip-sv-sv-2-4-8-16-sv.jpg)
















