
ΤΟΜΟΣ 3 - ΤΕΥΧΟΣ 1 ΙΑΝΟΥΑΡΙΟΣ - ΜΑΡΤΙΟΣ 2012 · μάζα στην...
113
ΕΛΛΗΝΙΚΗ ΑΙΜΑΤΟΛΟΓΙΚΗ ΕΤΑΙΡΕΙΑ HELLENIC SOCIETY OF HAEMATOLOGY ISSN: 1792-7110 Διευθυντής Σύνταξης: Καθηγητής Φώτης Ν. Μπερής Συνεκδότες: Καθηγήτρια Ελένη A. Παπαδάκη, Καθηγητής Κωνσταντίνος Τσαταλάς Aναπληρωτής Εκδότης: Επίκουρος Καθηγητής Θεόδωρος Π. Βασιλακόπουλος Μυελοϋπερπλαστικά σύνδρομα Παναγιώτης Παναγιωτίδης Guest Editor ΤΟΜΟΣ 3 - ΤΕΥΧΟΣ 1 ΙΑΝΟΥΑΡΙΟΣ - ΜΑΡΤΙΟΣ 2012 Αίμα 3 (1) Μυελοϋπερπλαστικά σύνδρομα ΙΑΝΟΥΑΡΙΟΣ - ΜΑΡΤΙΟΣ 2012 Β Α C
Transcript of ΤΟΜΟΣ 3 - ΤΕΥΧΟΣ 1 ΙΑΝΟΥΑΡΙΟΣ - ΜΑΡΤΙΟΣ 2012 · μάζα στην...

ΕΛΛΗΝΙΚΗ ΑΙΜΑΤΟΛΟΓΙΚΗ ΕΤΑΙΡΕΙΑ HELLENIC SOCIETY OF HAEMATOLOGY
ISSN: 1792-7110
Διευθυντής Σύνταξης: Καθηγητής Φώτης Ν. Μπερής
Συνεκδότες: Καθηγήτρια Ελένη A. Παπαδάκη, Καθηγητής Κωνσταντίνος ΤσαταλάςAναπληρωτής Εκδότης: Επίκουρος Καθηγητής Θεόδωρος Π. Βασιλακόπουλος
Μυελοϋπερπλαστικά σύνδρομα
Παναγιώτης ΠαναγιωτίδηςGuest Editor
ΤΟΜΟΣ 3 - ΤΕΥΧΟΣ 1 ΙΑΝΟΥΑΡΙΟΣ - ΜΑΡΤΙΟΣ 2012
Αίμ
α 3
(1) Μ
υελοϋπερπ
λαστικά σύνδρομα ΙΑΝ
ΟΥΑ
ΡΙΟ
Σ - ΜΑ
ΡΤΙΟ
Σ 2012
ΒΑ C

ix
ΕΛΛΗΝΙΚΗ ΑΙΜΑΤΟΛΟΓΙΚΗ ΕΤΑΙΡΕΙΑ
The Journalof the Hellenic Society of
HEMATOLOGY
Περιοδική Έκδοση της Ελληνικής ΑιμΑτολογικηςΕταιρείας
HELLENIC SOCIETY OF HAEMATOLOGY
BOARD OF THE HELLENICSOCIETY OF HAEMATOLOGY
President: Nikolaos Charchalakis Vice Presidents: Dimitrios Karakassis Ioanna Sakellari General Secretary: Elissavet Grouzi Executive Secretary: Ioannis Kakkas Treasurer: Panagiotis Panagiotidis Members: Georgios Vassilopoulos Konstantinos Tsatalas Panagiotis Tsaftaridis Panagiotis Tsirigotis
ΔΙΟΙΚΗΤΙΚΟ ΣΥΜΒΟΥΛΙΟ ΤΗΣ ΕΛΛΗΝΙΚΗΣ ΑΙΜΑΤΟΛΟΓΙΚΗΣ ΕΤΑΙΡΕΙΑΣ
Πρόεδρος: Νικόλαος Χαρχαλάκης Αντιπρόεδροι: Δημήτριος Καρακάσης Ιωάννα Σακελλάρη Γεν. Γραμματέας: Ελισάβετ Γρουζή Ειδ. Γραμματέας: Ιωάννης Κάκκας Ταμίας: Παναγιώτης Παναγιωτίδης Μέλη: Γεώργιος Βασιλόπουλος Κωνσταντίνος Τσαταλάς Παναγιώτης Τσαφταρίδης Παναγιώτης Τσιριγώτης
ΕΚΔΟΤΗΣ
Φώτης Ν. ΜπερήςΚαθηγητής Αιματολογίας
Ιατρικής Σχολής Πανεπιστημίου ΓενεύηςΚινητό: +41 79 957 5461
e-mail: [email protected]
EDITOR
Photis N. BerisProfessor of Haematology
Medical School Geneva University, Switzerland Modile: +41 79 957 5461
e-mail: [email protected]
ΣΥΝ-ΕΚΔOΤΕΣ
Κωνσταντίνος ΤσαταλάςΚαθηγητής Αιματολογίας
Πανεπιστημιακή Αιματολογική ΚλινικήΠανεπιστημιακό Γενικό Νοσοκομείο Αλεξανδρούπολης
E-mail: [email protected]
CO-EDITORS
Konstantinos TsatalasProfessor of Haematology
University Haematology ClinicUniversity General Hospital of Alexandroupolis
E-mail: [email protected]
Ελένη A. ΠαπαδάκηΚαθηγήτρια Αιματολογίας
Επικεφαλής Αιματολογικού ΤμήματοςΠανεπιστημιακού Νοσοκομείου Ηρακλείου Κρήτης
Ιατρικής Σχολής Πανεπιστημίου ΚρήτηςΤηλ.: 2810 394629Fax.: 2810 394632
e-mail: [email protected]
Helen A. PapadakiProfessor of Haematology
University of Crete School of MedicineHead of Dept of Haematology
University Hospital of Heraklion, CreteΤel.: +30 2810 394629Fax.: +30 2810 394632
e-mail: [email protected]
ΙΔΙΟΚΤΗΤΗΣ: Ελληνική Αιματολογική Εταιρεία, Κηφισίας 27, 115 23 Αθήνα, Τηλ.: 210 7211806ΓΡΑΦΕΙΟ ΕΚΔΟΣΗΣ - ΔΙΑΦΗΜΙΣΤΙΚΕΣ ΚΑΤΑΧΩΡΗΣΕΙΣ: Vita Congress - Β. Βουραζέρης, Παπαδιαμαντοπούλου 4 & Βασ. Σοφίας, 11528 Αθήνα, Τηλ.: 210.72.54.360, Fax: 210.72.54.363, E-mail: [email protected], http://www.vitacongress.grΕΚΤΥπΩΣΗ: ΤΕΧΝΟΓΡΑΜΜΑmed, Λ. Μεσογείων 380, 153 41 Αγία Παρασκευή, Τηλ.: +30210.6000643, Fax: +30 210 6002295, E-mail: [email protected]
πΡΟΗΓΟΥΜΕΝΟΙ ΣΥΝ-ΕΚΔOΤΕΣΝικόλαος Κ. Ζούμπος
PAST CO-EDITORSNicolaos C. Zoumbos

Vol. 3, No 1
January - March 2012
CONTENTS Intoduction ........................................................................................................................................................................................1
Panayiotis Panayiotidis 1. Historical Background in CML ......................................................................................................................................................2
Georgios Georgiou, Photis Beris 2. Prognostic factors in CML and treatment choices ............................................................................................................. 10
Maria Ν. Pagoni, Ifigeneia A. Tzannou 3. First Line Treatment for Chronic Phase Chronic Myelogenous Leukemia
(CP-CML): Treatment Options .................................................................................................................................................... 23Constantinos Tsatalas, Emmanouil Spanoudakis, Ioannis Kotsianidis
4. Criteria for treatment switch from imatinib to second generation TKIs..................................................................... 37Nora-Athina Viniou, Panagiotis Diamantopoulos, Vassiliki Papadopoulou
5. The role of Stem Cell Transplantation in CML: For which patients and when .......................................................... 45Ifigeneia Tzannou and Ioannis Baltadakis
6. Guidelines for the molecular monitoring of patients with CML treaτed with tyrosine kinase inhibitors ...... 57Tassoula Touloumenidou, Apostolia Papalexandri, Kostas Stamatopoulos
7. CML stem cells : the pathway to CML cure? ......................................................................................................................... 65Panagiotis Panagiotidis
8. Classification and current prognostication of Myeloproliferative Neoplasms ........................................................ 78Theodoros Marinakis, George Kanavos, Nikolaos I. Anagnostopoulos
9. Genetic aberrations in Ph negative myeloproliferative neoplasms ............................................................................ 89Georgios Georgiou, Vasiliki Mpartzi
10. Therapeutic management of Ph negative Myeloproliferative Neoplasms ............................................................... 99Despoina Pantelidou, Maria Papaioannou
ΜΥΕLOPROLIFERATIVE SYNDROMES Guest editor: P. Panagiotidis
Cover page: A. Nowell PC and Hungerford DA. Ph+ chromosome in CML. Β. ΒCR-Abl fusion gene in CML. C. Point mutation JAK2 V617F in Ph- myeloproliferative syndromes.

Τόμος 3, Τεύχος 1
Iανουάριος - Μάρτιος 2012
ΠΕΡΙΕΧΟΜΕΝΑ Εισαγωγή ..............................................................................................................................................................................................1
Παναγιώτης Παναγιωτίδης 1. Ιστορική αναδρομή στη ΧΜΛ .......................................................................................................................................................2
Γεώργιος Γεωργίου, Φώτης Μπερής 2. Προγνωστικοί παράγοντες στη ΧΜΛ και θεραπευτικές επιλογές ................................................................................ 10
Μαρία Ν. Παγώνη, Ιφιγένεια Α. Τζάννου 3. Θεραπεία 1ης γραμμής σε ασθενείς με χρόνια μυελογενή λευχαιμία χρόνιας φάσης:
Θεραπευτικές επιλογές ................................................................................................................................................................ 23Κωνσταντίνος Τσαταλάς, Εμμανουήλ Σπανουδάκης, Ιωάννης Κοτσιανίδης
4. Κριτήρια αλλαγής από imatinib σε ΤΚΙs δεύτερης γενιάς ................................................................................................ 37Νόρα-Αθηνά Βύνιου, Παναγιώτης Διαμαντόπουλος, Βασιλική Παπαδοπούλου
5. Ο ρόλος της μεταμόσχευσης στη ΧΜΛ: Σε ποιους ασθενείς και πότε ........................................................................ 45Ιφιγένεια Τζάννου, Ιωάννης Μπαλταδάκης
6. Εργαστηριακή παρακολούθηση ασθενών με ΧΜΛ και κλινική αξιολόγηση ............................................................ 57Τασούλα Τουλουμενίδου, Αποστολία Παπαλεξανδρή, Κώστας Σταματόπουλος
7. Βλαστικά κύτταρα της χρόνιας μυελογενούς λευχαιμίας (CML stem cells) - Η οδός προς την ίαση; .................................................................................................................................................................. 65
Παναγιώτης Παναγιωτίδης 8. Tαξινόμηση και προγνωστικοί παράγοντες στα Philadelphia αρνητικά Μυελοϋπερπλαστικά
Νεοπλάσματα .................................................................................................................................................................................. 78Θεόδωρος Μαρινάκης, Γεώργιος Καναβός, Νικόλαος Ι. Αναγνωστόπουλος
9. Γενετικές βλάβες στα Ph- μυελοϋπερπλαστικά νοσήματα ............................................................................................... 89Γιώργος Γεωργίου, Βασιλική Μπάρτζη
10. Θεραπευτική αντιμετώπιση των Ph- Μυελοϋπερπλαστικών Νεοπλασμάτων (Ph-ΜΥΝ) .................................... 99Δέσποινα Παντελίδου, Μαρία Παπαϊωάννου
ΜΥΕΛΟϋΠΕΡΠΛΑΣΤΙΚΑ ΣΥΝΔΡΟΜΑGuest editor: Π. Παναγιωτίδης
Εικόνα εξωφύλλου: A. Nowell PC and Hungerford DA. Περιγραφή του χρωμοσώματος Ph+ στη ΧΜΛ. Β. BCR-Abl υβριδικό γονίδιο στη ΧΜΛ. C. Σημει-ακή μετάλλαξη JAK2 V617F στα Ph- μυελοϋπερπλαστικά σύνδρομα.

ΜΕΛΛΟΝΤΙΚΑ ΤΕΥΧΗ Έτος 2012 Ιούλιος Θέμα Μυελοδυσπλαστικά σύνδρομα – 2ο μέρος Guest editor Ν.Α. Βύνιου, Α. Συμεωνίδης
Σεπτέμβριος Θέμα Νόσος Hodgkin Guest editor Θεόδωρος Βασιλακόπουλος
Νοέμβριος Θέμα Περιλήψεις 23ου Ετήσιου Πανελληνίου Αιματολογικού Συνεδρίου
Έτος 2013 Μάρτιος Θέμα Αναιμία Guest editor Ελένη Παπαδάκη, Χαράλαμπος Ποντίκογλου
Ιούλιος Θέμα Χρόνιες β λεμφοϋπερπλαστικές παθήσεις Guest editor Κώστας Σταματόπουλος
Σεπτέμβριος Θέμα Αυτόλογη Μεταμόσχευση Guest editor Μαρία K. Αγγελοπούλου

Αγαπητοί Συνάδελφοι,
Στα μυελοϋπερπλαστικά σύνδρομα περιλαμβάνεται ένα σύνολο νοσημάτων κάποια από τα οποία έχουν μικρή επίπτωση στο προσδόκιμο και την ποιότητα ζωής και κάποια μέχρι πρόσφατα ανήκαν στην κατηγορία των δυνητικά θανατηφόρων νοσημάτων. Τα τελευταία χρόνια υπήρξε μία έκρηξη, τόσο στις γνώσεις για τη μοριακή βιολογία των νοσημάτων αυτών, όσο και στη θεραπευτική αντι-μετώπισή τους.
Χαρακτηριστικό παράδειγμα αποτελεί η Χρόνια Μυελογενής Λευχαιμία που από το 1960 με την περιγραφή του χρωμοσώματος Φιλαδέλφεια και την περιγραφή της υβριδικής πρωτεΐνης BCR-Abl το 1990 βρεθήκαμε στη θεραπευτική εφαρμογή των αναστολέων τυροσινικής κινάσης (TKIs) το 2001. Σήμερα με τη δυνατότητα επιλογής τριών διαφορετικών TKIs για τη θεραπεία της Χρόνιας Μυελογενούς Λευχαιμίας, όχι απλώς έχει εξασφαλιστεί η μακρόχρονη επιβίωση των ασθενών αυ-τών με άριστη ποιότητα ζωής, αλλά έχει ήδη ανοίξει το κεφάλαιο της πιθανότητας διακοπής της χορήγησης ΤΚΙs σε επιλεγμένους ασθενείς με ΧΜΛ που έχουν επιτύχει «λειτουργική ίαση» της νό-σου. Φυσικά η αλλογενής μεταμόσχευση μυελού των οστών διατηρεί το ρόλο της στη θεραπεία της ΧΜΛ αλλά προφανώς με διαφορετικά κριτήρια σήμερα στην εποχή των ΤΚΙs.
Κλινικά διλήμματα που αφορούν τη «σωστή» επιλογή των ΤΚΙs στη θεραπεία της ΧΜΛ, τον ρό-λο της μεταμόσχευσης και την εργαστηριακή παρακολούθηση των ασθενών είναι υπαρκτά και ορι-σμένες φορές φλέγοντα.
Στην οικογένεια των Φιλαδέλφεια αρνητικών μυελοϋπερπλαστικών νοσημάτων υπάρχει πρόσφατα (2005) η περιγραφή της σημειακής μετάλλαξης JAK V617F που προσέφερε τόσο στην προσπάθεια κατανόησης της μοριακής βάσης των νόσων αυτών, αλλά κυρίως στη διευκόλυνση της γρήγορης και ασφαλούς διαγνωστικής προσπέλασης. Στην οικογένεια αυτή των νοσημάτων πρόσφατα εμφα-νίστηκε θεραπευτικά ένας νέος TKI με θεραπευτική ένδειξη τη μυελοίνωση.
Οι πρόοδοι στη βιολογία και τη θεραπευτική των νόσων αυτών αφήνει στους αιματολόγους την ανάγκη σωστής/λελογισμένης ενσωμάτωσης τους στην καθημερινή κλινική πράξη, σύμφωνα με τις διεθνείς οδηγίες (όπου υπάρχουν!).
Ευχαριστώ όλους τους συγγραφείς για τη συγγραφή των αντίστοιχων κεφαλαίων του τεύχους αυτού και πιστεύω ότι θα αποτελέσουν ένα χρήσιμο βοήθημα τόσο στην κλινική, όσο και στη βιο-λογική τους σκέψη.
Αθήνα Ιούνιος 2012
ΠαναγιώτηςΠαναγιωτίδης Αναπληρωτής Καθηγητής Αιματολογίας Ιατρική Σχολή Πανεπιστημίου Αθηνών
Haema 2012; 3(1): 1
Εισαγωγή

Ιστορική αναδρομή στη ΧΜΛ
Γεώργιος Γεωργίου1, Φώτης Μπερής1,2
ΠΕΡΊληΨη: η Xρονία Mυελογενής λευχαιμία (Χμλ) κατά πάσα πιθανότητα περιεγράφει για πρώτη φορά το 1845. η καθιέρωση όμως των μυελουπερπλαστικών συνδρόμων ως ξεχωριστή οντότητα, οφεί-λεται στον William Dameshek το 1951, ο οποίος και θεωρείται επισήμως ο 1ος που προσπάθησε να την ταξινομήσει στα αιματολογικά νεοπλάσματα εκτός των οξειών λευχαιμιών. το 1960 για 1η φορά παρα-τηρήθηκε μία χρωμοσωματική ανωμαλία στον καρυότυπο, στην οποία δόθηκε το όνομα Philadelphia από την πόλη στην οποία ανακαλύφθηκε. η αντιμετάθεση όμως t(9;22) περιγράφη για 1η φορά από την Janet Rowley στις ηΠΑ το 1972. Δέκα χρόνια αργότερα χαρτογραφήθηκε το ανθρώπινο ομόλογο του v-abl και σε μικρό σχετικά διάστημα (1984) οι ερευνητές έδειξαν ότι το σημείο τομής στο χρωμόσωμα 22 βρίσκεται πάντοτε σε μία περιοχή σχετικώς περιορισμένη γνωστή ως bcr. τέλος περί τις αρχές του 1990 απoδείχθηκε η ογκογόνος δραστηριότης του bcr - abl και η δυνατότητά του να προκαλεί το νόση-μα σε ποντίκια. η θεραπεία της Χμλ ήταν στην αρχή παρηγορητική και το νόσημα εθεωρείτο ανίατο. η βουσουλφάνη ήταν το 1ο κυτταροστατικό που πιο εύκολα σε σχέση με την ακτινοβολία μπορούσε να ελέγξει τη λευκοκυττάρωση που απέβαινε μοιραία, χωρίς θεραπευτική επέμβαση. η υδροξυουρία εισή-χθη στη θεραπευτική το 1960, ήτο και είναι το κατ’εξοχήν φάρμακο για τον έλεγχο της λευκοκυττάρω-σης και δεν επιταχύνει την εξέλιξη της νόσου στην οξεία της μορφή. Δεν μπορεί όμως να επηρεάσει τη φυσική εξέλιξη της νόσου. η εισαγωγή της αυτόλογης μεταμόσχευσης στην ιατρική, υπήρξε η πρώτη ελπίδα «διαιώνισης» της νόσου στη χρόνια φάση, γεγονός όμως το οποίο δεν μπόρεσε ποτέ κανείς εκ των ερευνητών να επιτύχει. η 1η ελπίδα μακρού ελέγχου της νόσου ήταν η ιντερφερόνη, η οποία επέτυχε επιμήκυνση του ολικού χρόνου επιβίωσης των ασθενών κατά 1 - 2 χρόνια. ο συνδυασμός της με αρα-συτίνη εθεωρείτο η ιδανική θεραπεία πριν από την έλευση των αναστολέων της κινάσης της τυροσίνης.
Haema 2012; 3(1): 2-9 Copyright EAE
1Unilabs, Département d’Hématologie, Coppet Switzerland;2Département de Médecine Interne, Faculté de Médecine, Université de
Genève, Genève SuisseΔιεύθυνση αλληλογραφίας: Ερευνητικό Αιματολογικό Εργαστήριο Α΄
ΠΠ Κλινικής, ΕΚΠΑ, Μιχαλακοπούλου 176, 115 27 Αθήνα, e-mail: [email protected]
Professeur Photis Beris, Unilabs, Laboratoire central de Suisse romande, Chemin des Perrières 2, 1296 Coppet – CH [email protected]
Aνασκόπηση
ΕΙΣΑΓΩΓΗη ιστορία της αιματολογίας αποτελεί για τους νεω-
τέρους συναδέλφους μια κληρονομιά που τους άφησαν οι «πατέρες» της επιστήμης μας, αυτοί που χωρίς τα εκ-πληκτικά μέσα του εργαστηρίου και της πληροφορικής που διαθέτουμε σήμερα, μπόρεσαν με όπλο την κλινική παρατήρηση και εξέταση να ταξινομήσουν τα νοσήμα-τα, να μελετήσουν τη φυσική τους εξέληξη και να διατυ-πώσουν τις πρώτες θεωρίες που ωδήγησαν στη μετέπειτα συστηματική μελέτη στο εργαστήριο. η Χμλ αποτελεί
ένα κλασσικό παράδειγμα αυτής της επίπονης και μα-κράς μελέτης που κατέληξε τα τελεύταία 20 χρόνια στη ριζική αλλαγή στην αντιμετώπιση ενός νοσήματος που εθεωρείτο θανατηφόρο για τους πάσχοντες. η συντακτι-κή επιτροπή του παροντος τεύχους του «ΑιμΑ» θεώρη-σε απαραίτητη αυτή την ιστορική αναδρομή την οποία ελπίζουμε επιτυχώς να προσφέρουμε στους αναγνώστες του περιοδικού μας.
ΚΛΙΝΙΚΗ ΕΙΚΟΝΑ, πΑθΟΦΥΣΙΟΛΟΓΙΑτο 1845, ο παθολογοανατόμος John Hughes Bennett
από το Εδιμβούργο (33 ετών τότε) περιέγραψε μια “Case of Hypertrophy of the Spleen and Liver in which Death Took Place from Suppuration of the Blood” στο Edinburgh Medical Journal. O ασθενής αυτός ονομαζόταν John Menteith, και ήταν ένας 28χρονος επισκευαστής σκεπών (slater) από το Εδιμβούργο, ο οποίος είχε αντιληφθεί μια

Ιστορική αναδρομή στη ΧΜΛ 3
μάζα στην αριστερή πλευρά της κοιλιάς του επί 8 μήνες πριν το θάνατό του. ςτη νεκροψία που έγινε, βρέθηκε μα-ζική διόγκωση του ήπατος, του σπληνός και των λεμφαδέ-νων. η εξέταση του αίματός του αποκάλυψε την ύπαρξη: «πραγματικού πύου, που είναι σχηματισμένο σε όλο το αγγειακό σύστημα, ανεξάρτητα από οποιαδήποτε τοπική πυώδη συλλογή από την οποία μπορεί να προερχόταν».
Έξι εβδομάδες αργότερα ο Rudolph Virchow (εικό-να 1), στο Βερολίνο (24 ετών τότε) περιέγραψε τη νόσο μιας ασθενούς ονόματι Marie Straide. η ασθενής ήταν μια 50χρονη μαγείρισσα που πέθανε με έναν τεράστιο σπλήνα 6 μήνες μετά την πρώτη της επίσκεψη στο για-τρό της. ςτο αίμα της η αναλογία των «αχρωμάτιστων προς τα χρωματισμένα σωματίδια ήταν ανεστραμμένη».
Αν και κανένας δε μπορεί να γνωρίζει με ακρίβεια, αυτές οι δύο κλινικές περιπτώσεις είναι πολύ πιθανό να είναι οι πρώτες περιγραφές της Χρόνιας μυελογενούς λευχαιμίας. Ενώ ο Bennet νόμισε ότι ο ασθενής του εί-χε λοίμωξη, ο Virchow υποπτεύθηκε ότι πρόκειται για «νεοπλασματική νόσο» που γρήγορα ονόμασε λευχαιμία.
ο Ernst Neumann ήταν ο πρώτος που αναγνώρισε τον κεντρικό ρόλο του μυελού των οστών στη λευχαιμία (Neumann, 1870). ο Neumann, ήταν μόνιμος κάτοικος στο Königsberg (πρωτεύουσα της Ανατολικής Πρωσσί-ας) και αναγνώρισε δύο τύπους διήθησης του μυελού των οστών από τη λευχαιμία 1. την ‘πυώδη υπερπλα-
σία’ που χαρακτηριζόταν από υπερκοκκιωμένα κύτταρα και 2. τη ‘λεμφική υπερπλασία’ όπου τα κύτταρα είχαν ομοιογενή πυρήνα και σχεδόν καθόλου πρωτόπλασμα (Neumann, 1878).1
ςτα χρόνια που ακολούθησαν, έγινε η περιγραφή και άλλων νοσημάτων με υπερπλασία της μυελικής σειράς (ερυθροκυττάρωση, μυελοΐνωση, θρομβοκυττάρωση) και η πρώτη προσπάθεια ταξινόμησης έγινε από τον Dameshek.
O William Dameshek (1900-1969, εικόνα 2) γεννή-θηκε στις 22 μαΐου 1900 στη Ρωσία και μετακόμισε στις ηνωμένες Πολιτείες με τους γονείς του το 1903. το 1946, μαζί με τον Henry μ Stratton, ίδρυσε το BLOOD-το ση-μερινό περιοδικό of the American Society of Hematology και έγινε ο πρώτος editor-in-chief.2 O Dameshek έπαιξε σημαντικό ρόλο στη δημιουργία τόσο της Διεθνούς Αι-ματολογικής Εταιρείας το 1946 όσο και της Αμερικανι-κής Εταιρείας Αιματολογίας (ASH) το 1958. Αν και ο Dameshek είχε ευρέα κλινικά και εργαστηριακά ενδιαφέ-ροντα, έγινε ιδιαίτερα γνωστός περιγράφοντας την ιδέα των μΥΝ (μυελοϋπερπλαστικών νοσημάτων) το 1951. Θα πρέπει να σημειωθεί, ωστόσο, ότι και άλλοι πριν τον Dameshek είχαν αναγνωρίσει την υπερπλασία και των τριών σειρών στα μυελοϋπερπλαστικά νοσήματα. την πρώτη δεκαετία του εικοστού αιώνα οι Turk3 Weber4 και Watson5 είχαν παρατηρήσει υπερπλασία της ερυθράς και
Εικόνα 1. Rudolf Virchow (πηγή: http://www.nlm.nih.gov/hmd/ihm/
Εικόνα 2. William Dameshek (πηγή: http://www.nlm.nih.gov/hmd/ihm/

Γ. Γεωργίου και Φ. Μπερής4
λευκής σειράς στην πολυκυτταραιμία, ενώ χαρακτηρι-στική είναι και η περιγραφή των Vaughan και Harrison: «φαίνεται ότι η πολυκυτταραιμία, η μεγακαρυοκυτταρική λευχαιμία και η μυελοσκλήρυνση με λευκο-ερυθροβλα-στική αναιμία, σχηματίζουν μια ομάδα στενά σχετιζόμε-νων οντοτήτων». 6 Δύο χρόνια μετά την περιγραφή και κατάταξη των μΥΝ από το Dameshek, (1953) βρέθηκε ότι το γενετικό υλικό των κυττάρων, το DNA, σχημάτι-ζε διπλή έλικα7 ενώ το 1956 οι Tjio και Levon δημοσί-ευσαν μια εργασία όπου έδειχναν ότι ο άνθρωπος έχει 46 και όχι 48 χρωμοσώματα όπως μέχρι τότε πιστευόταν. 8
Χρωμοσωμικές μελέτες στους καρκίνους είχαν αρχίσει λίγα χρόνια νωρίτερα και είχαν δείξει ποικίλες ιδιότυπες χρωμοσωμικές βλάβες.9 Πολλοί επιστήμονες ερευνούσαν ανώμαλες πρωτεΐνες και εξωτερικά αίτια του καρκίνου, όπως χημικά (βενζένιο) και ιονίζουσα ακτινοβολία. Αυτά ήταν γνωστό ότι προκαλούσαν χρωμοσωμικές θράυσεις και αναδιατάξεις αλλά θεωρήθηκαν τότε ότι ήταν τυχαί-ες και δεν προκαλούσαν από μόνες τους εξαλλαγή. με τον καιρό, η χρήση υποτονικών διαλυμάτων και διαλύ-ματος οξεικού οξέος και αλκοόλης ως μονιμοποιητικού μέσου, βελτίωσαν το διαχωρισμό των χρωμοσωμάτων στα κύτταρα και η καταμέτρησή τους ήταν πιο αξιόπι-στη και αναπαραγώγιμη.
το 1960, ο Peter Nowell, ένας Αμερικανός βιολόγος, ανακάλυψε τη φυτοαιματογλουτινίνη (PHA) η οποία διε-γείρει τα τ λεμφοκύτταρα και τα προκαλεί να πολλαπλα-σιαστούν, με αποτέλεσμα να μην ήταν πλέον αναγκαία η προσφυγή σε βιοψία δέρματος για συλλογή ινοβλαστών προκειμένου να τεθούν σε καλλιέργειες για τη δημιουρ-γία μεταφάσεων.10 ο ίδιος, εργαζόταν στο Πανεπιστήμιο της Πενσυλβάνια των ηΠΑ, όταν απροσδόκητα απεικό-νισε μεταφασικά χρωμοσώματα σε λευχαιμικά κύττα-ρα, πάνω σε πλακίδια τα οποία είχαν πρώτα (από λάθος) πλυθεί σε νερό της βρύσης πριν βαφτούν με χρώση Giemsa.11 ο Nowell αμέσως μετά συνεργάστηκε με τον David Hungerford (1927-1993), ο οποίος την εποχή εκεί-νη δούλευε ως μεταπτυχιακός φοιτητής σε μελέτες πάνω στα ανθρώπινα χρωμοσώματα, στο Fox Chase Institute for Cancer Research στις ηΠΑ. μαζί ανακάλυψαν ένα αφύσικα μικρό χρωμόσωμα που έμοιαζε με το χρωμόσω-μα Υ, σε δύο άντρες ασθενείς με Χμλ και έκαναν την πρώτη ανακοίνωση της παρατήρησής τους το 1960. οι δυό τους στη συνέχεια περιέγραψαν τη συστηματική πα-ρουσία του ανώμαλου αυτού χρωμοσώματος σε επτά άλ-λες τυπικές περιπτώσεις Χμλ, συμπεριλαμβανομένων δύο γυναικών. λόγω της συνύπαρξης και κυττάρων με φυσιολογικό καρυότυπο οι Nowell και Hungerford συ-μπέραναν ότι το μικρό χρωμόσωμα στη Χμλ δεν είναι ιδιοσυστασιακό και ότι μπορεί να συνδέεται αιτιολογι-κά με την ασθένεια.12
ςτην Πρώτη Διεθνή Διάσκεψη για την ονοματολογία των χρωμοσωμάτων το 1960 στο Denver των ηΠΑ, το ανώμαλο χρωμόσωμα στη Χμλ ονομάστηκε Φιλαδέλ-
φεια, από την πόλη στην οποία ανακαλύφθηκε (Ph1). ο εκθέτης ‘1’ χρησιμοποιήθηκε εν αναμονή και άλλων πα-ρόμοιων ανακαλύψεων από την ομάδα των ερευνητών στην πόλη αυτή. το χρωμόσωμα Φιλαδέλφεια (Ph) είναι η πρώτη χρωμοσωμική ανωμαλία ειδική για συγκεκριμέ-νο νόσημα που περιγράφηκε και επιβεβαίωσε τη θεωρία του Theodore Boveri, ενός γερμανού βιολόγου, ο οποί-ος το 1914 υποστήριξε ότι ο καρκίνος είναι αποτέλεσμα επίκτητων κυτταρικών αλλαγών.13
το 1961 η Mary Frances Lyon (Αγγλίδα γενετιστής) και το 1962 ο Ernest Beutler (Αμερικάνος αιματολόγος) έδειξαν τη λειτουργική αδρανοποίηση των ετεροχρωμα-τικών χρωμοσωμάτων Χ, όπως επίσης και το ότι η αδρα-νοποίηση αυτή αφορούσε και το πατρικό και το μητρικό χρωμόσωμα Χ κατά έναν τυχαίο τρόπο.14,15
με βάση αυτή τη λογική, το 1965, πολυμορφισμοί του γονιδίου του εδραζώμενου στο χρωμόσωμα Χ, glucose-6-phosphate dehydrogenase (G-6-PD), χρησιμοποιήθηκαν για να αποδειχτεί η κλωνική προέλευση του λειομειώμα-τος.16 ςτη συνέχεια οι Fialkow και συνεργάτες εφήρμοσαν παρόμοια τεχνική για να επιβεβαιώσουν την κλωνική φύ-ση της Χμλ (1967), της πολυκυτταραιμίας (1976), της μυελοϊνωσης (1978) και της πρωτοπαθούς θρομβοκυτ-τάρωσης (1981).17-20
μέχρι το 1970 τα χρωμοσώματα βαφόταν μόνο με Giemsa και ήταν δύσκολη η αναγνώρισή τους. το 1970 και 1971 εισήχθησαν δύο τεχνικές χρώσεις των χρωμο-σωμάτων, μια παραλλαγή της Giemsa (G-banding) και η quinacrine mustard (Q-banding).21
η εισαγωγή των νέων τεχνικών χρώσης επέτρεψε στον Caspersson και συνεργάτες να αναγνωρίσουν μορφολογι-κά το χρωμόσωμα Φιλαδέλφεια, το 1970, σαν ένα χρωμό-σωμα 22 με έλλειψη χρωμοσωμικού υλικού.22 το 1972, η Janet Rowley (Αμερικανίδα γενετίστρια), γυρίζοντας από το ηνωμένο Βασίλειο όπου είχε εκπαιδευτεί στην τεχνική Q-banding, βρήκε την αντιμετάθεση t(8;21)(q22;q22) σε δύο ασθενείς με λευχαιμία και ουσιαστικά ήταν η πρώτη αντιμετάθεση που περιγράφτηκε σε λευχαιμία (εικόνα 3). ςτη συνέχεια επιβεβαίωσε την παρατήρηση της ύπαρξης του χρωμοσώματος Φιλαδέλφεια και επιπλέον χρησιμο-ποιώντας την τεχνική Q-banding αποκάλυψε τη σύστασή του, ως το αποτέλεσμα αμοιβαίας μετάθεσης μεταξύ των χρωμοσωμάτων 9 και 22 t(9;22)(q34;q11).24
λίγο πριν την ανακάλυψη της αντιμετάθεσης t(9;22)(q34;q11), το 1969 είχε ανκαλύφθεί ο ιός Abelson (Abel-son murine leukemia virus), που περιέχει την Ρ160 gag/v-abl ογκοπρωτεΐνη.25
το 1982, μια ομάδα Αμερικανών επιστημόνων (Heisterkamp και συνεργάτες) χαρτογράφησαν το αν-θρώπινο ομόλογο του v-abl (ABL) και στη συνέχεια σε συνεργασία με τους de Klein και συνεργάτες στο Πανε-πιστήμιο Erasmus στο Ρότερνταμ της ολλανδίας κατέδει-ξαν τη συμμετοχή αυτού του γονιδίου στην αντιμετάθεση t(9;22)(q34;q11).26,27

Ιστορική αναδρομή στη ΧΜΛ 5
Εικόνα 3. Τμήματα καρυοτύπων που δείχνουν τίς κοινές με-ταθέσεις που βρήκε η Rowley. Οι μεταθέσεις εμφανίζονται με την σειρα με την οποίαν ανευρέθησαν. (πηγή: Rowley JD, Chromosomal translocations: revisited yet again. Blood, 2008; 112:2183-9).
το 1984 η ίδια συνεργατική ομάδα έδειξε το σημείο τομής στο χρωμόσωμα 22 σε μια περιοχή 5.8 kb που την ονόμασαν ‘breakpoint cluster region (bcr)’, που αργότε-ρα έδειξαν ότι είναι μέρος του γονιδίου BCR.28
το 1984 επίσης, περιγράφηκε ένα άλλο μετάγρα-φο του φυσιολογικού γονιδίου ABL στους ασθενείς με Χμλ που στη συνέχεια αναγνωρίστηκε ως το χιμαιρικό μετάγραφο BCR–ABL που μεταφραζόταν στη χιμαιρι-κή πρωτεΐνη P210.29
το 1984 δείχθηκε ότι η πρωτεΐνη ABL στη Χμλ έχει αυξημένη δραστηριότητα τυροσινικής κινάσης. το 1987 και το 1988 δείχθηκε η ογκογόνος δραστηριότητα της P210 BCR–ABL σε μυελό των οστών ποντικών και σε κυτταρικές σειρές (Ba/F3). το 1989 και το 1990 το χιμαι-ρικό γονίδιο BCR-ABL αποδείχθηκε ότι προκαλεί λέμφω-μα και οξεία λευχαιμία σε ποντίκια. το 1990 επιμόλυνση αιμοποιητικών stem cells με το χιμαιρικό γονίδιο BCR-ABL με τη βοήθεια ρετροϊών έδειξε ότι προκαλεί νόσο προσομοιάζουσα στη Χμλ σε ποντίκια. Έτσι το χιμαι-ρικό γονίδιο καθιερώθηκε ως η μετάλλαξη που προκαλεί την εμφάνιση Χμλ.30
θΕΡΑπΕΙΑη θεραπεία στη λευχαιμία εξελίχθηκε αργά. τον 18ο
αιώνα ο, Thomas Fowler παρασκεύασε ένα διάλυμα τρι-οξειδίου του αρσενικού (As2O3) με διττανθρακικό κά-λιο που έφερε το όνομά του. το 1865 ο Heinrich Lissauer περιέγραψε τη χρήση του διαλύματος Fowler’s solution σε ασθενή με χρόνια μυελογενή λευχαιμία και ανέφερε κλινική ύφεση (τα οξείδια του αρσενικού έχουν ξαναέρ-θει στο προσκήνιο ως πολύ αποτελεσματική θεραπεία
ως πρώτη γραμμή θεραπείας ή σαν θεραπεία διάσωσης σε ασθενείς με οξεία προμυελοκυτταρική λευχαιμία).31
Παράγωγα του αρσενικού βρισκόταν σε χρήση ήδη πάνω από 2.000 χρόνια, λόγω των θεραπευτικών τους ιδιοτήτων σε διάφορες ασθένειες.32
τα παράγωγα του αρσενικού έγιναν στη συνέχεια η θεραπεία εκλογής στις μυελογενείς λευχαιμίες, αν και οι υφέσεις ήταν απρόβλεπτες και συνήθως παροδικές. το 1892, ο Osler στο βιβλίο του έγραφε: «Υπάρχουν διά-φορες θεραπείες που έχουν επίδραση στη νόσο (λευχαι-μία). Από αυτές το αρσενικό, όταν δίνεται σε μεγάλες δόσεις, είναι το καλύτερο. Προσωπικά έχω επανειλλη-μένα δει βελτιώσεις μετά από τη χρήση του.». οι ακτί-νες Röntgen ανακαλύφθηκαν το 1895 και η χρήση τους υποσκέλισε το αρσενικό στη θεραπεία της χρόνιας μυε-λογενούς λευχαιμίας τα πρώτα χρόνια του 20ου αιώνα.33
Ωστόσο, η χρήση του αρσενικού στην Αιματολογία γνώρισε νέα δημοτικότητα τη δεκαετία του 1930, όταν αναφέρθηκε η αποτελεσματικότητά του σε ασθενείς με χρόνια μυελογενή λευχαιμία.34
ςτη δεκαετία του 30 χρησιμοποιήθηκαν ακόμα ο αντιλευκοκυτταρικός ορός, το βενζένιο και ο ραδιενερ-γός φώσφορος.35-37
το ουρεθάνιο (Ethylcarbamate-urethane) βρισκόταν σε χρήση πριν το 1950 για αρκετά χρόνια σαν αναισθη-τικό για πειραματόζωα. Είχε επίσης δειχθεί ότι καταστέλ-λει τις μιτώσεις των κυττάρων του κερατοειδούς τους και μειώνει τον αριθμό των λευκών τους.38
Αυτές του οι δράσεις όπως και η καταστολή της ανά-πτυξης μικροοργανισμών καθώς και η μεταλλαξιογόνος του δράση στη μύγα των φρούτων (fruit-fly), οδήγησαν στην υπόθεση ότι μπορεί να έχει ανάλογη θεραπευτι-κή δράση με την ιονίζουσα ακτινοβολία και οδήγησε το 1946 στη διερεύνηση της επίδρασης του ουρεθανίου σε όγκους ζώων.39
κλινικές μελέτες [WATKINS, C., et al. (1948), Blood, 3, 892., CRESKOFF, A.J., et al. (1948), Ibid., 3, 896, HIRSCHBECK, J.S. (1948), Y.A.M.A., 136, 90] έδει-ξαν ότι η επίδραση του ουρεθανίου στη λευχαιμία είναι ανάλογη της ιονίζουσας ακτινοβολίας αλλά με μικρότε-ρης διάρκειας υφέσεις.40
τη δεκαετία του 50 χρησιμοποιήθηκαν και άλλες θε-ραπείες στη Χμλ με άλλοτε άλλη αποτελεσματικότητα, όπως η thio-Triethylenethiophosphoramide (thio-TEPA).41
τη δεκαετία του 1950 επίσης, εισήχθη στη θεραπευτι-κή της Χρόνιας μυελογενούς λευχαιμίας η βουσουλφάνη (Myleran). το «Myleran» (1: 4-dimethanesulphonyloxy-butane) παρασκευάστηκε από τους Haddow και Timmis το 1953 με την ελπίδα ότι η βιολογική του δράση μπορεί να έμοιαζε εκείνη της μουστάρδας (nitrogen mustard) λό-γω της αλκυλιωτικής ικανότητας που είχε. 42
η χρήση του Myleran ήταν πιο εύκολη σε σχέση με την ακτινοβολία, δεν είχε σοβαρές παρενέργειες (όταν

Γ. Γεωργίου και Φ. Μπερής6
χορηγούνταν σε μικρές δόσεις), αλλά δεν έδειξε πλεονέ-κτημα όσον αφορά την επιβίωση των ασθενών και επι-πλέον παρατηρήθηκαν και πολλές οξείες υποτροπές σε ασθενείς που ελάμβαναν το φάρμακο συνεχώς (ανάπτυ-ξη αντοχής). ςήμερα η βουσουλφάνη δε χρησιμοποιείται παρά μόνο σε μεταμοσχεύσεις μυελού των οστών γιατί βρέθηκε ότι είναι πιο αποτελεσματική στο να «σκοτώ-νει» τα φυσιολογικά stem cells από τα λευχαιμικά stem cells στη Χμλ. 43
τη δεκαετία του 1960 εισήχθη στη θεραπευτική η υδροξυουρία η οποία χρησιμοποιήθηκε σε μεγάλο φάσμα κακόηθων νεοπλασμάτων (λιποσάρκωμα, νευροβλάστω-μα, μελάνωμα κλπ) αλλά εκεί που έδειξε τη μεγαλύτερη αποτελεσματικότητα ήταν στη Χμλ: μείωση της ηπατο-σπληνομεγαλίας, ευκολότερος έλεγχος των λευκών αιμο-σφαιρίων, άνοδος της αιμοσφαιρίνης κ.λπ. 44
η υδροξυουρία αναστέλλει τη ριβονουκλεοτιδική ρε-δουκτάση και in vivo φαίνεται ότι μεταβολίζεται σε οξεί-δια του αζώτου. Παρέμεινε και παραμένει σε χρήση σε ασθενείς με μυελικές κακοήθειες κυρίως ως παρηγορητική θεραπεία για τον έλεγχο του αριθμού των λευκοκυττάρων.
Άλλες θεραπείες που κατά καιρούς χρησιμοποιήθη-καν στη Χμλ είναι η λευκαφαίρεση, η σπληνεκτομή, η εντατικοποιημένη χημειοθεραπεία, ακόμα και η ανοσο-θεραπεία με Bacillus Calmette-Guérin σε συνδυασμό με αλλογενείς μυελοβλάστες και ήπια χημειοθεραπεία (βου-σουλφάνη per os).45-50
Ωστόσο καμία από αυτές τις θεραπευτικές προσεγ-γίσεις δεν επηρέασε τη φυσική εξέλιξη της νόσου, που ήταν η βλαστική εκτροπή και ο σύντομος θάνατος με-τά από αυτή.
Αυτόλογη μεταμόσχευσηκατά τα τέλη του 1970 αρκετοί ερευνητές προσπά-
θησαν να σταθεροποιήσουν οξείες λευχαιμίες οι οποί-ες είχαν εισέλθει σε ύφεση με τη χορήγηση αφανιστικής χημειοθεραπείας ακολουθούμενης με διάσωση από αυ-τόλογη μεταμόσχευση αιμοποιητικών κυττάρων. ςε γενι-κές γραμμές τα αποτελέσματα αυτών των μελετών ήταν απογοητευτικά και στην καλύτερη των περιπτώσεων η ύφεση η οποία επιτυγχάνοταν ήταν μικρής διάρκειας. ο ρόλος όμως της αυτόλογης μεταμόσχευσης στη χρόνια μυελογενή λευχαιμία υπήρξε αντικείμενο μακρόχρονης μελέτης κατά τη δεκαετία του 1980 με κύριο κέντρο το νοσοκομείο Hammersmith στο λονδίνο και κύριο ερευ-νητή τον John Goldman. ςε μία σειρά 21 ασθενών όπου τα κύτταρα είχαν συλλεγεί είτε στη διάγνωση είτε αργό-τερα στη χρόνια φάση51,52, 10 από αυτούς επέζησαν κατά μέσο όρο 84 μήνες μετά την αυτομεταμόσχευση ( εύρος 12 με 118 μήνες). Από τους 11 που πέθαναν, 7 κατέληξαν λόγω μετατροπής της νόσου σε οξεία μορφή. Από τους επιζήσαντες πλείστοι απέκτησαν φυσιολογική αιμοποί-
ηση με μερική παροδική εξάλειψη του Ph1 χρωματοσώ-ματος. Αυτά τα αποτελέσματα έδωσαν ελπίδες κατά τα τέλη του 1980 αρχές 1990, ότι καλλιέργειες μακράς δι-αρκείας κυττάρων ασθενών με Χμλ θα ευνοούσαν την επικράτηση Φιλαδέλφεια αρνητικών προγόνων αιμοποιη-τικών κυττάρων. Εκτός από τις καλλιέργειες ιn vitro των προγονικών κυττάρων, έγιναν προσπάθειες κάθαρσης μυ-ελού των οστών από τα λευχαιμικά κύτταρα με τη χρησι-μοποίηση antisense ολιγονουκλεοτιδίων53.
πΡΩΤΕΣ ΕΛπΙΔΕΣ ΜΑΚΡΟΥ ΕΛΕΓΧΟΥ ΤΗΣ ΝΟΣΟΥ ΜΕ ΤΗΝ ΙΝΤΕΡΦΕΡΟΝΗ.
η στρατηγική της αυτόλογης μεταμόσχευσης διεκόπη αφενός μεν, με την εφαρμογή της αλλογενούς μεταμό-σχευσης, είτε από συγγενή είτε από ηLΑ εθελοντή συμ-βατό δότη, και αυτό χάρις στην πρόοδο για την αποφυγή και πρόληψη επιπλοκών στην αλλογενή μεταμόσχευση, αφετέρου δε με τον ερχομό της ιντερφερόνης η οποία χά-ρις στις νέες μεθόδους παραγωγής της βασιζόμενες στην ανασχεδιαζόμενη τεχνολογία DNA (recombinant DNA technology), επέτρεψε τη χρήση αυτής σε μεγάλες δό-σεις σε πλείστα κακοήθη αιματολογικά νοσήματα μετα-ξύ των οποίων και στη χρόνια μυελογενή λευχαιμία54,55. ηδη από τα μέσα της δεκαετίας του 1980 παρατηρήθη-κε ότι 70% των ασθενών τύχαιναν απόλυτης αιματολο-γικής ύφεσης με περίπου στα 15 με 20% από αυτούς να εμφανίζουν μία μείζονα κυτταρογεννετική απάντηση. τέ-λος, στα μέσα της δεκαετίας του 1990, όταν η αλυσιδωτή αντίδραση της πολυμεράσης για τα μετάγραφα ΒCR-ABL κατέστη δυνατή, αποδείχτηκε ότι 1 με 2 % των ασθενών που ελάμβαναν ιντερφερόνη-α καθίσταντο απολύτως αρνητικοί σε μοριακό επίπεδο για το χρωμόσωμα Φιλα-δέλφειας56-58. το σπουδαιότερο όμως ήταν ότι, για πρώτη φορά ασθενείς που ελάμβαναν ιντερφερόνη αλφα είχαν μέσο χρόνο επιβίωσης 1 με 2 χρόνια μεγαλύτερο σε σύ-γκριση με αυτούς που ελάμβαναν υδροξυουρία. οι ιτα-λοί αιματολόγοι για πρώτη φορά απέδειξαν σε προοπτική τυχιοποιημένη μελέτη ότι η ιντερφερόνη αλφα οδηγεί σε μεγαλύτερες κυτταρογεννετικές απαντήσεις, επιβραδύνει την εξέλιξη σε πιό προχωρημένα στάδια της νόσου και το κυριώτερο αυξάνει την ολική επιβίωση57. Βελτίωση των αποτελεσμάτων της ιντερφερόνης άλφα υπήρξε ο συνδυασμός με χαμηλές δόσεις ARA-C, συνδυασμός ο οποίος αποτελούσε το «gold standard » για ασθενείς με χρόνια μυελογεννή και χωρίς συμβατό δότη ή σε ασθε-νείς άνω των 60 ετών. Βρισκόμαστε ήδη στα τέλη του 20 ου αιώνα και η θερaπευτική αντιμετώπιση της χρό-νιας μυελογενούς λευχαιμίας θα αλλάξει ριζικά με την πλήρη κατανόηση του μοριακού μηχανισμού της νόσου και με την επιτυχή εφαρμογή της πρώτης στοχευμένης θεραπείας ογκολογικού νοσήματος με τους αναστολείς των κινασών της τυροσίνης. γι’αυτήν όμως τη θεραπευ-

Ιστορική αναδρομή στη ΧΜΛ 7
Historical Background in CMLby Georgios Georgiou1, Photis Beris1,2
1Unilabs, Département d’Hématologie, Coppet Switzerland; 2Département de Médecine Interne, Faculté de Médecine, Université de Genève, Genève Suisse
ABSTRACT: Chronic myelogenous leukemia was most probably described for the fist time in 1845. The establishment however of myeloproliferative disorders as a separate entity, belongs to William Dameshek in 1951 who is considered officially the haematologist that classified CML in haematologic malignan-cies outside acute leukemias. In 1960 a chromosomal abnormality was observed for the first time and the name Philadelphia chromosome was coined due to the city where it was described. Janet Rowley, from the USA, in 1972 described for the first time the translocation t(9;22). Ten years later the human homolog of v-abl was isolated and in relatively short period (1984) researchers showed that the break point in chromosome 22 was always occurred in relatively small region named bcr. Finally at the begin-ning of 1990 the oncogenic effect of bcr - abl was demonstrated by the induction of the disease in mouse. At the beginning, treatment of CML was palliative and the disease was considered incurable. Busulfan was the first cytostatic which relatively easier than radiation, controls extreme leucocytosis which with-out any therapeutic intervention was always fatal. Hydroxyurea was introduced in therapeutics in 1960 and was and still is, the best drug to control leucocytosis without acceleration of the disease towards its acute phase. However hydroxyurea can not modify the physical evolution of the disease. The introduc-tion of autologous bone marrow transplantation in medicine was the first hope to perpetuate the disease in its chronic phase, fact that it was never achieved by any investigator. The first real hope to prolong the chronic phase of the disease was interferon – α, which achieved a significant increase of overall surviv-al of patients for 1 – 2 years as compared to hydroxyurea. Association of interferon – α with AraC was considered the goal standard of treatment of CML before the era of tyrosine kinase inhibitors.
Βιβλιογραφία 1. Νeumann E. Ueber die Bedeutung des Knochenmarkes fur
die Blutbildung. Ein Beitrag zur Entwicklungsgeschichte der Blutkorperchen. Archives Heilkunde.1869; 10:68–102.
2. Stratton HM. William Dameshek, a personal remem-brance. Blood. 1970; 35:4–5.
3. Turk W. Beitrage zur kenntnis des symptomenbildes poly-zythamie mit milztumor und zyanose. Wiener Medizinische Wochenschrift. 1904; 17:153–160, 189–193.
4. Weber FP, Watson JH. Chronic polycythemia with enlarged spleen, probably a disease of the bone marrow. Trans Clin Soc. 1904; 37:115.
5. Parkes-Weber F. Polycythemia, erythrocytosis and eryth-raemia. QJM. 1908; 2:85–134.
6. Vaughan JM, Harrison CV. Leuco-erythrobalstic anaemia and myelosclerosis. J Pathol Bacteriol. 1939; 48:339–352.
7. Watson JD, Crick FH. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature. 1953; 171:737–738
τική εξέλιξη έχουν αφιερωθεί άλλα κεφάλαια του παρό-ντος τεύχους του «ΑιμΑ».
ΕπΙΛΟΓΟΣη ιστορική αναδρομή της Χμλ αποτελεί παράδειγ-
μα της μεγάλης σημασίας που έχει η κλινική παρατήρη-ση και εξέταση, η οποία επέτρεψε εδώ και 150 χρόνια να ξεχωρίσει ως ιδιαίτερο νόσημα αυτή η μορφή της λευχαι-μίας. Ακολούθως η συστηματική μελέτη μεγάλου αριθ-μού ασθενών που διεγιγνώσκοντο αποκλειστικά και μόνο με κλινικά κριτήρια, οδήγησε την ανεύρεση του πρώτου ογκολογικού δείκτη, του χρωματοσώματος Φιλαδέλφεια
[t(9;22)]. Αυτή η ανακάλυψη αποτέλεσε την αφετηρία για την κατανόηση της κλονικής παθολογικής αιμοποί-ησης σε αντιδιαστολή με την πολυκλονική φυσιολογική αιμοποίηση. τούτο επέτρεψε τη δοκιμή διαφόρων θερα-πευτικών σχημάτων και την ανεύρεση της θεραπευτικης δράσης της ιντερφερόνης Α, η περαιτέρω δε εξέλιξη της μοριακής Βιολογίας και η εφαρμογή της στο μοντέλο της Χμλ, επέτρεψαν την εξεύρεση της πρώτης στοχευμένης θεραπείας η οποία όπως θα αναφερθεί λεπτομερειακά στα επόμενα κεφάλαια άλλαξαν ριζικά τη φυσική εξέλιξη της νόσου και έθεσε τις προυποθέσεις για την εξεύρεση, ελ-πίζουμε σε βραχύ χρονικό διάστημα, νέων φαρμάκων που αποβλέπoυν όχι πλέον στη μακρά ύφεση, αλλά την ίαση του κακκοήθους αυτού αιματολογικού νοσήματος.

Γ. Γεωργίου και Φ. Μπερής8
8. Tjio JH, Levan A The chromosome number of man. He-reditas. 1956; 42:1–6.
9. Yoshida T. The Yoshida sarcoma, an ascites tumor. Jpn J Cancer Res. 1949; 40:1–21.
10. Nowell PC. Phytohemagglutinin: an initiator of mitosis in cultures of normal human leukocytes. Cancer Res. 1960; 20:462–466.
11. Nowell PC. Progress with chronic myelogenous leuke-mia: a personal perspective over four decades. Annu Rev Med. 2002; 53:1–13.
12. Nowell PC, Hungerford DA. A minute chromosome in human granulocytic leukemia. Science. 1960; 132:1497.
13. Boveri T. Zur Frage der Entstehung maligner Tumoren. Gus-tav Fischer, Jena 1914.
14. Beutler E, Yeh M, Fairbanks VF. The normal human female as a mosaic of X-chromosome activity: studies using the gene for C-6-PD-deficiency as a marker. Proc Natl Acad Sci USA. 1962; 48:9–16
15. Lyon MF. Gene action in the X-chromosome of the mouse (Mus musculusL.). Nature. 1961; 190:372–373.
16. Linder D, Gartler SM. Glucose-6-phosphate dehydroge-nase mosaicism: utilization as a cell marker in the study of leiomyomas. Science. 1965; 150:67–69.
17. Fialkow PJ, Gartler SM, Yoshida A. Clonal origin of chronic myelocytic leukemia in man. Proc Natl Acad Sci USA. 1967; 58:1468–1471.
18. Adamson JW, Fialkow PJ, Murphy S, Prchal JF, Steinmann L. Polycythemia vera: stem-cell and probable clonal origin of the disease. N Engl J Med. 1976; 295:913–916.
19. Jacobson RJ, Salo A, Fialkow PJ. Agnogenic myeloid meta-plasia: a clonal proliferation of hematopoietic stem cells with secondary myelofibrosis. Blood. 1978; 51:189–194.
20. Fialkow PJ, Faguet GB, Jacobson RJ, Vaidya K, Murphy S. Evidence that essential thrombocythemia is a clonal disorder with origin in a multipotent stem cell. Blood. 1981; 58:916–919.
21. Drets ME, Shaw MW. Specific banding patterns of human chromosomes.Proc Natl Acad Sci USA. 1971;68:2073-2077.
22. Caspersson T, Gahrton G, Lindsten J, Zech L. Identifi-cation of the Philadelphia chromosome as a number 22 by quinacrine mustard fluorescence analysis. Exp Cell Res. 1970; 63:238–240
23. Rowley JD. Letter: a new consistent chromosomal ab-normality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Na-ture. 1973; 243:290–293
25. Abelson HT, Rabstein LS. Lymphosarcoma: virus-in-duced thymic-independent disease in mice. Cancer Res. 1970; 30:2213–2222.
26. Heisterkamp N, Groffen J, Stephenson JR, et al. Chromo-somal localization of human cellular homologues of two viral oncogenes. Nature. 1982; 299:747–749.
27. de Klein A, van Kessel AG, Grosveld G, et al. A cellular oncogene is translocated to the Philadelphia chromosome in chronic myelocytic leukaemia. Nature. 1982; 300:765–767.
28. Heisterkamp N, Stam K, Groffen J, de Klein A, Grosveld
G. Structural organization of the bcr gene and its role in the Ph’ translocation. Nature. 1985; 315:758–761.
29. Ben-Neriah Y, Daley GQ, Mes-Masson AM, Witte ON, Baltimore D. The chronic myelogenous leukemia-specific P210 protein is the product of the bcr/abl hybrid gene. Sci-ence. 1986; 233:212–214.
30. Elefanty AG, Hariharan IK, Cory S. bcr–abl, the hallmark of chronic myeloid leukaemia in man, induces multiple hae-mopoietic neoplasms in mice. EMBO J. 1990; 9:1069–1078
31. Lissauer H. Zwei Falle von Leucaemie. Berl Klin Wochen-schr. 1865; 2:403–404.
32. Klaassen CD. Heavy metals and heavy-metal antagonists. In: Hardman JG, Gilman AG, Limbird LE, eds. Goodman & Gilman›s The Pharmacological Basis of Therapeutics. New York: McGraw-Hill, 1996:1649-1672.
33. Senn N. Case of splenomedullary leukaemia successfully treated by the use of the Rontgen ray. Med Rec. 1903; 64:281–282.
34. Forkner CE, Scott TFM. Arsenic as a therapeutic agent in chronic myelogenous leukemia. JAMA. 1931; 97:3–5.
35. Hueper WC, Russell M. Some immunological aspects of leukemia. Arch Intern Med. 1932; 49:113–122.
36. Kalapos I. Die Wirkung des Benzols bei der Leukamie. Klin Wochenschr. 1935; 14:864–867.
37. Lawrence JH. Nuclear physics and therapy: preliminary report on a new method for the treatment of leukemia and polycythemia vera. Radiology. 1940; 35:51–60.
38. Hawkins JA, Murphy JB. Exp Med. 1925; 7:42, 609. 39. Haddow A, Sexton WA. Nature. 1946; 157:500. 40. Cooper T, Watkins CH. Ethyl carbamate (urethane) in the
treatment of chronic myelocytic leukemia; results of a three-year study. Med Clin North Am. 1950; 34:1205–1215.
41. Smith NJ, Rosellos S, Shay H. Effect of triethylene thiophos-phoramide in treatment of leukemia and certain lymphomas in infants and children. J Pediat. 1955; 46:493. 505.
42. Galton DA. Myleran in chronic myeloid leukaemia; results of treatment. Lancet. 1953; 264:208–213.
43. Blackburn EK, King GM, Swan HT. Myleran in treatment of chronic myeloid leukaemia. Br J Med. 1956; 14:835-837.
44. Kennedy BJ, Yarbro JW. Metabolic and therapeutic effects of hydroxyurea in chronic myelogenous leukemia. Trans Assoc Am Physicians. 1965; 78:391–399.
45. Buckner D, Graw RG Jr, Eisel RJ, Henderson ES, Per-ry S. Leukapheresis by continuous flow centrifugation (CFC) in patients with chronic myelocytic leukemia (CML). Blood. 1969; 33:353–369.
46. Cutting HO. The effect of splenectomy in chronic granulo-cytic leukemia. Report of a case. Arch Intern Med. 1967; 120: 356–360.
47. Tura S, Baccarani M, Mandelli F, et al. Splenectomy in early chronic myeloid leukaemia: preliminary report on 37 cases. Haematologica. 1974; 59:428–439.
48. Baker M, Taub R, Carter W, et al, and the Toronto Leukemia Study Group. Immunotherapy for Chronic Myelogenous Leukemia: Survival is not Affected by Treatment in the Stable Phase1 Cancer Research. 1984; 44:383-385.

Ιστορική αναδρομή στη ΧΜΛ 9
49. Cunningham I, Gee T, Dowling M, Chaganti R, Bailey R, Hopfan S et al. Results of treatment of Ph’+ chronic my-elogenous leukemia with an intensive treatment regimen (L-5 protocol). Blood. 1979; 53:375–395.
50. Kantarjian HM, Vellekoop L, McCredie KB, et al. Intensive combination chemotherapy (ROAP 10) and splenectomy in the management of chronic myelogenous leukemia. J Clin Oncol. 1985; 3:192–200.
51. Mughal T, Hoyle C, Goldman JM. Autografting for patients with chronic myeloid leukemia – The Hammersmith experi-ence. Stem cells. 1993; 11(Suppl 3):20–22.
52. Hoyle C, Gray R, Goldman J on behalf of the Hammersmith BMT team. Autografting for patients with CML in chronic phase – an update. Brit J Haematol. 1994; 86:76–81.
53. de Fabritiis P, Amadori S, Calabretta B, Mandelli F: Elimina-tion of clonogenic Philadelphia-positive cells using BCR-ABL antisense oligonucleotides. Bone Marrow Transplant. 1993; 12:261–265.
54. Talpaz M, McCredie KB, Mavligit GM, Gutterman J.
Leukocyte interferon-induced myeloid cytoreduction in chronic myelogenous leukemia. Blood. 1983; 62:689–692.
55. Talpaz M, Kantarjian HM, McCredie K et al. Hematologic remission and cytogenetic improvement induced by recom-binant human interferon alpha in chronic myelogenous leukaemia. N Eng J Med. 1986; 314:1065–1069.
56. Talpaz M, Kantarjian HM, Kurzrock R et al. Interferon-alpha produces sustained cytogenetics responses in chronic myelogenous leukemia. Ann Intern Med. 1991; 114:532–538.
57. The Italian Cooperative study group on Chronic Myeloid Leukemia. Interferon alfa-2a as compared with conven-tional chemotherapy for the treatment of chronic myeloid leukaemia. N Engl J Med. 1994; 330:820–825.
58. Ozer H, George SL, Schiffer CA, et al. Prolonged subcutane-ous administration of recombinant 2b interferon in patients with previously untreated Philadelphia chromosome-positive chronic-phase chronic myelogenous leukaemia: effect on remission duration and survival: Cancer and Leukemia Group B Study 8583. Blood. 1993; 82:2975–2984.

Προγνωστικοί παράγοντες στη ΧΜΛ και θεραπευτικές επιλογές
Μαρία Ν. Παγώνη,1 Ιφιγένεια Α. Τζάννου1
ΠΕΡΊληΨη: η αναγνώριση των προγνωστικών δεικτών θεωρείται σημαντική στην κλινική πρακτι-κή για την επιλογή της βέλτιστης θεραπείας και την έγκαιρη αλλαγή της θεραπευτικής στρατηγικής. Υπάρχουν δύο κατηγορίες προγνωστικών παραγόντων στη Χμλ, ήτοι οι παράγοντες που αναγνωρίζο-νται στη διάγνωση (baseline) και εκείνοι που εκτιμώνται κατά τη διάρκεια της θεραπείας και ως επί το πλείστον σχετίζονται με την ανταπόκριση. τα πιο συχνά χρησιμοποιούμενα προγνωστικά συστήματα είναι το Sokal και το Hasford score. Παρόλο που αυτά τα δύο μοντέλα δημιουργήθηκαν και αξιολογή-θηκαν στην προ TKIs εποχή, η προγνωστική τους αξία έχει τεκμηριωθεί και στην εποχή των TKIs. το EUTOS score δημιουργήθηκε για τους ασθενείς που λαμβάνουν imatinib, όμως φαίνεται ότι χρήζει πε-ραιτέρω αξιολόγησης σε προοπτικές μελέτες. για την έγκαιρη αναγνώριση των ασθενών που δε θα αντα-ποκριθούν ικανοποιητικά στη θεραπεία με TKIs, προσδιορίστηκαν βιοδείκτες που παίζουν σημαντικό ρόλο στην πρόγνωση των ασθενών με Χμλ. τέτοιοι δείκτες είναι οι επιπρόσθετες κυτταρογενετικές ανωμαλίες στο λευχαιμικό κλώνο και ειδικότερα η παρουσία των major-route ACAs και η γονοδιακή υπογραφή στη διάγνωση. Βιολογικοί δείκτες, όπως είναι η έκφραση της αντλίας οργανικών κατιόντων OCT-1 και της Ρ-γλυκοπρωτεΐνης, η φωσφορυλίωση από τους TKIs του υποστρώματος της bcr-abl κι-νάσης Crkl έχουν αποδειχθεί χρήσιμοι για την πρόβλεψη της ανταπόκρισης στο imatinib, αλλά η ευρεία εφαρμογή τους δεν έχει καθιερωθεί. ςτην καθημερινή κλινική πράξη, η ανταπόκριση στη θεραπεία σε συγκεκριμένες χρονικές στιγμές από την έναρξη της αγωγής, αποτελεί το σημαντικότερο προγνωστικό παράγοντα, με την ισχυρότερη προγνωστική αξία για την έκβαση των ασθενών να έχει η επίτευξη πλή-ρους κυτταρογενετικής ύφεσης (CCgR). η σημασία του χρόνου επίτευξης της μοριακής ανταπόκρισης και του βάθους της έχουν επίσης αξιολογηθεί. ςε πρόσφατη μελέτη, η μέτρηση των μεταγράφων BCR-ABL στους 3 μήνες αποτελεί τον καλύτερο τρόπο ταυτοποίησης των ασθενών με δυσμενή πρόγνωση και άρα επιτρέπει την πρώιμη κλινική παρέμβαση. ςτις περισσότερες περιπτώσεις, η ύπαρξη μεταλλά-ξεων στην abl κινάση συνεπάγεται μη ικανοποιητική ανταπόκριση ή αποτυχία στο imatinib και αποτε-λεί δυσμενή προγνωστικό παράγοντα για την επιβίωση χωρίς πρόοδο νόσου και τη συνολική επιβίωση. η αναζήτηση των μεταλλάξεων είναι απαραίτητη για το σχεδιασμό της θεραπευτικής στρατηγικής με-τά την αποτυχία της θεραπείας 1ης γραμμής. τα δεδομένα για τους προγωστικούς παράγοντες που προ-βλέπουν την ανταπόκριση στους αναστολείς 2ης γενιάς είναι λιγότερα, όμως φαίνεται ότι σοβαρό ρόλο παίζει η παρουσία και το είδος μεταλλάξεων, ενώ η επίτευξη CCgR αποτελεί και σε αυτήν την περί-πτωση τη σημαντικότερη προγνωστική παράμετρο. με την εισαγωγή των αναστολέων δεύτερης γενιάς στη θεραπεία 1ης γραμμής, φαίνεται ότι η έκβαση των ασθενών βελτιώνεται, οι δε παράγοντες που δια-τηρούν την προγνωστική τους αξία είναι η επίτευξη CCgR και το επίπεδο της μοριακής ανταπόκρισης.
Haema 2012; 3(1): 10-22 Copyright EAE
1Αιματολογική κλινική και μονάδα μεταμόσχευσης μυελού οστών, γΝΑ «Ευαγγελισμός», Αθήνα.
Διεύθυνση αλληλογραφίας: Μαρία Ν. Παγώνη, Αιματολογική Κλινική και Μονάδα Μεταμόσχευσης Μυελού Οστών, ΓΝΑ «Ευαγγελισμός», Αθήνα. E-mail: [email protected]
Aνασκόπηση
ΕΙΣΑΓΩΓΗη φυσική πορεία της Χρόνιας μυελογενούς λευχαι-
μίας (Χμλ) περιλαμβάνει τρεις φάσεις, τη χρόνια φάση, την επιταχυνόμενη και τη βλαστική κρίση. ςτην πλειοψη-

Προγνωστικοί παράγοντες στη ΧΜΛ και θεραπευτικές επιλογές 11
φία των ασθενών, το νόσημα διαγιγνώσκεται σε χρόνια φάση. Πριν την εισαγωγή ειδικών θεραπειών, η εξέλιξη του νοσήματος οδηγούσε στο θάνατο σε διάμεσο χρό-νο 3 ετών από τη διάγνωση. ςτις αρχές του 20ου αιώνα το αρσενικό και η ακτινοβόληση του σπληνός χρησιμο-ποιήθηκαν για την ανακούφιση από τα συμπτώματα. με τη χρήση της βουσουλφάνης επετεύχθη επιμήκυνση του προσδόκιμου επιβίωσης σε 4-5 έτη. Όμως, το φάρμακο ενοχοποιήθηκε για ταχύτερη πρόοδο νόσου και έδωσε τη θέση του στην υδροξυκαρβαζίνη, η οποία επίσης δεν οδήγησε σε καθυστέρηση της εξέλιξης της νόσου, ούτε σε βελτίωση της επιβίωσης.1,2 ςτις αρχές της δεκαετίας του 1980 η χορήγηση ανασυνδυασμένης ιντερφερόνης-α (IFN-α), είχε ως αποτέλεσμα την επίτευξη μείζονος κυτ-ταρογενετικής ανταπόκρισης (MCgR) σε ένα ποσοστό περίπου 15-25% των ασθενών και βελτίωση του προσδό-κιμου επιβίωσης κατά 1-2 έτη επιπλέον. μετά το 1980, η αλλογενής μεταμόσχευση αιμοποιητικών κυττάρων (αλ-λο-μΑκ) απετέλεσε τη θεραπεία εκλογής για τους ασθε-νείς με διαθέσιμο ιστοσυμβατό δότη, παρά την αυξημένη νοσηρότητα και θνητότητα, ως η μοναδική θεραπευτική προσέγγιση που δυνητικά οδηγεί σε πλήρη ίαση του νο-σήματος.1,2 η εμφάνιση του πρώτου αναστολέα τυροσι-νικής κινάσης (TKI), του imatinib mesylate, στις αρχές του 21ου αιώνα, άλλαξε εντυπωσιακά την πορεία και την έκβαση των ασθενών με Χμλ. το νέο φάρμακο είχε ικα-νοποιητικό προφίλ ασφάλειας και ήταν εξαιρετικά αποτε-λεσματικό, επιτυγχάνοντας συνολική επιβίωση 86% στα 8 έτη.3 ςήμερα είναι διαθέσιμοι και άλλοι αναστολείς 2ης και 3ης γενιάς. οι αναστολείς 2ης γενιάς αρχικά πήραν έγκριση ως θεραπεία 2ης γραμμής, μετά από δυσανεξία ή αποτυχία στο imatinib. το 2010 έλαβαν έγκριση από τον FDA και τον ΕμΕΑ ως 1ης γραμμής θεραπεία στη Χμλ.
Παρότι στους περισσότερους ασθενείς που λαμβά-νουν τκι ως 1ης γραμμής θεραπεία παρατηρούνται ικανο-ποιητικές κυτταρογενετικές και μοριακές ανταποκρίσεις, 20-30% των ασθενών δεν επιτυγχάνουν ποτέ μείζονα μο-ριακή ανταπόκριση, ενώ περίπου 10% θα παρουσιάσουν πρόοδο νόσου. Είναι, λοιπόν, αναγκαίο να μπορούμε να διακρίνουμε, όσο το δυνατόν νωρίτερα, τους ασθενείς που δε θα ανταποκριθούν ικανοποιητικά στη θεραπεία. η αναγνώριση των προγνωστικών παραγόντων θεωρεί-ται σημαντική στην κλινική πρακτική για την επιλογή της βέλτιστης θεραπείας και την έγκαιρη αλλαγή της θεραπευτικής στρατηγικής (π.χ. προγραμματισμός για αλλο-μΑκ), όταν ενδείκνυται. Επιπλέον, ο ορισμός ευ-ρέως χρησιμοποιούμενων προγνωστικών παραγόντων, μπορεί να βελτιώσει τα αποτελέσματα των κλινικών δο-κιμών, καθώς επιτρέπει καλύτερη διαστρωμάτωση των ασθενών στις ομάδες της μελέτης, ενώ καθίσταται δυνα-τή η σύγκριση αποτελεσμάτων από διαφορετικές μελέτες. ςε γενικές γραμμές, υπάρχουν δύο κατηγορίες προγνω-στικών παραγόντων στη Χμλ, ήτοι οι παράγοντες που αναγνωρίζονται στη διάγνωση (baseline) και εκείνοι που
εκτιμώνται κατά τη διάρκεια της θεραπείας και ως επί το πλείστον σχετίζονται με την ανταπόκριση.4,5
πΡΟΓΝΩΣΤΙΚΟΙ πΑΡΑΓΟΝΤΕΣ ΣΤΗ ΔΙΑΓΝΩΣΗ ΚΑΙ πΡΟΓΝΩΣΤΙΚΑ ΣΥΣΤΗΜΑΤΑ
η φάση του νοσήματος κατά τη διάγνωση της Χμλ είναι αναμφισβήτητα ο κυριότερος προγνωστικός παρά-γοντας για την ανταπόκριση, τη διάρκειά της και τη συ-νολική επιβίωση.6
ςύμφωνα με τους ορισμούς της WHO 2008, η χρό-νια φάση χαρακτηρίζεται από ποικίλου βαθμού λευκο-κυττάρωση στο περιφερικό αίμα, λόγω αύξησης τόσο των ουδετερόφιλων όσο και αωρότερων μορφών της κοκκιώδους σειράς, χωρίς εμφανή δυσπλασία. το ποσο-στό των βλαστικών κυττάρων δεν ξεπερνά το 2%, ενώ συνυπάρχει αύξηση των βασεόφιλων και ηωζινόφιλων κυττάρων. η επιταχυνόμενη φάση καθορίζεται από την παρουσία τουλάχιστον ενός από τα παρακάτω κριτήρια: εμμένουσα λευκοκυττάρωση ή αύξηση λευκοκυττάρων >10x109/L ή αυξανόμενη σπληνομεγαλία παρά τη θερα-πεία, εμμένουσα θρομβοκυττάρωση >1000x109/L που δεν έλέγχεται από τη θεραπεία, εμμένουσα θρομβοπενία <100x109/L ανεξάρτητη από τη θεραπεία, κυτταρογενε-τική εξέλιξη, παρουσία ≥20% βασεόφιλων στο αίμα και 10-19% βλάστες στο αίμα ή στο μυελό. η βλαστική κρί-ση, τέλος, χαρακτηρίζεται από παρουσία βλαστών σε πο-σοστό ≥20% στο αίμα ή στο μυελό ή εξωμυελική νόσο.
οι ασθενείς που διαγιγνώσκονται σε επιταχυνόμενη φάση ή βλαστική κρίση έχουν σαφώς χειρότερη έκβαση από τους ασθενείς σε χρόνια φάση. ςύμφωνα με τις τρέ-χουσες οδηγίες, οι ασθενείς σε χρόνια φάση αντιμέτωπί-ζονται με τκιs (imatinib ή δεύτερης γενιάς αναστολέα). οι ασθενείς σε βλαστική κρίση, που είναι κατάλληλοι για μεταμόσχευση, προγραμματίζονται για αλλο-μΑκ, αφού προηγηθεί χορήγηση τκι με ή χωρίς χημειοθερα-πεία με σκοπό την επίτευξη δεύτερης χρόνιας φάσης. η ομάδα των ασθενών σε επιταχυνόμενη φάση είναι ετε-ρογενής, καθώς περιλαμβάνει ασθενείς με νόσο ελαφρώς πιο προχωρημένη από τη χρόνια φάση έως και κοντά στη βλαστική εκτροπή. για τους ασθενείς αυτούς, συνιστά-ται διενέργεια αλλο-μΑκ μετά από θεραπεία με imatinib (600mg ή 800mg) ή με δεύτερης γενιάς αναστολέα.7
ςτη μελέτη των Jiang και συν, συγκρίθηκαν τα αποτε-λέσματα για ασθενείς που διαγνώστηκαν σε επιταχυνόμενη φάση και έλαβαν imatinib ή αλλο-μΑκ. Αναγνωρίστη-καν τρεις ομάδες κινδύνου βάσει των εξής προγνωστικών παραγόντων: διάρκεια νόσου >12 μήνες, αιμοσφαιρίνη <10 g/dL, βλάστες περιφερικού αίματος >5%. οι ασθε-νείς υψηλού κινδύνου είχαν στατιστικά σημαντικά καλύ-τερα αποτελέσματα για 5ετή ΕFS, OS και PFS στο σκέλος της αλλο-μΑκ. για τους ασθενείς ενδιάμεσου κινδύνου

Μ.Ν. Παγώνη και Ι.Α. Τζάννου12
δεν υπήρχε διαφορά στην ΕFS και OS ανάλογα με τη θεραπεία, όμως οι ασθενείς που υποβλήθηκαν σε αλλο-μΑκ είχαν σημαντικά καλύτερη 6ετή PFS. τέλος, για τους ασθενείς χαμηλού κινδύνου τα αποτελέσματα ήταν εξίσου καλά και με τις δύο θεραπευτικές προεσεγγίσεις.8
η πρώτη προσπάθεια δημιουργίας προγνωστικού συστήματος, που έτυχε ευρείας εφαρμογής στην κλινι-κή πρακτική, δημοσιεύτηκε το 1984 από τους Sokal και συν, μετά από μελέτη σε ασθενείς με Χμλ, οι οποίοι έλα-βαν αγωγή με βουσουλφάνη. Χρησιμοποιήθηκαν τέσσε-ρις κλινικές και εργαστηριακές παράμετροι, η ηλικία, το μέγεθος του σπληνός, ο αριθμός των αιμοπεταλίων και το ποσοστό των βλαστών στο περιφερικό αίμα. με βάση τα κριτήρια αυτά, αναγνωρίστηκαν τρεις προγνωστικές ομάδες. οι ασθενείς χαμηλού κινδύνου είχαν 4ετή επιβί-ωση 63%, ενώ για τους ασθενείς ενδιάμεσου και υψηλού κινδύνου η 4ετής επιβίωση ήταν 43% και 33% αντίστοι-χα.9 η προγνωστική αξία του Sokal score στην εποχή της θεραπείας με IFN-α, διατηρήθηκε για την ομάδα υψηλού κινδύνου. Όμως δεν ήταν δυνατόν, βάσει του προγνωστι-κού αυτού μοντέλου, να γίνει σαφής διαχωρισμός μετά-ξυ των ομάδων ενδιάμεσου και χαμηλού κινδύνου.5 οι Hasford και συν σε μετα-ανάλυση δεδομένων από προ-οπτικές μελέτες, αξιολόγησαν ένα νέο προγνωστικό σύ-στημα, προσθέτοντας δύο ακόμα παραμέτρους σε αυτές που είχαν χρησιμοποιηθεί στο Sokal score, τα ποσοστά των βασεόφιλων και των ηωζινόφιλων κυττάρων στο αί-μα. Προέκυψαν τρεις προγνωστικές ομάδες και η συνολι-κή επιβίωση βρέθηκε 76%, 55% και 25% για την ομάδα χαμηλού, ενδιάμεσου και υψηλού κινδύνου αντίστοιχα.10
Παρά το γεγονός ότι αυτά τα δύο μοντέλα δημιουρ-γήθηκαν και αξιολογήθηκαν στην προ TKIs εποχή, η προγνωστική τους αξία έχει τεκμηριωθεί και στην επο-χή των τκιs, καθώς φαίνεται ότι μπορούν να διαχωρί-σουν τους ασθενείς που θα ανταποκριθούν ικανοποιητικά στο imaitinib από αυτούς που δε θα έχουν το επιθυμητό αποτέλεσμα. Χρησιμοποιώντας τόσο το Sokal όσο και το Hasford score μπορεί να εκτιμηθεί η πιθανότητα επίτευξης πλήρους κυτταρογενετικής (CCgR) και μείζονος μοριακής ανταπόκρισης (MMolR) για τους ασθενείς που λαμβά-νουν imatinib. ςτις αρχικές μελέτες η συνολική επιβίω-ση στους 54 μήνες ήταν 94%, 88% και 81% αντίστοιχα για τους ασθενείς χαμηλού, ενδιάμεσου και υψηλού κιν-δύνου σύμφωνα με το Sokal score. Όμως, από τα αποτε-λέσματα της μελέτης IRIS προκύπτει ότι το Sokal score χάνει την προγνωστική του αξία στους ασθενείς υψηλού κινδύνου που επιτυγχάνουν CCgR με το imatinib.11-13 με βάση, λοιπόν, την αξιοσημείωτη βελτίωση της έκβασης των ασθενών στην εποχή των TKIs, κανένας ασθενής, ακόμα και αν ανήκει σε ομάδα υψηλού κινδύνου σύμ-φωνα με τα δύο μοντέλα, δε δικαιολογείται να μη λάβει την καθιερωμένη θεραπεία 1ης γραμμής.14
το πιο πρόσφατο προγνωστικό μοντέλο είναι το EUTOS και δημιουργήθηκε μετά από αξιολόγηση 2060
ασθενών που έλαβαν imatinib ως μονοθεραπεία ή σε συνδυασμό με άλλους παράγοντες. Χρησιμοποιώντας μόνο το ποσοστό των βασεόφιλων κυττάρων στο αίμα και το μέγεθος του σπληνός, διαχωρίζει τους ασθενείς σε δύο ομάδες κινδύνου. η 5ετής επιβίωση χωρίς νόσο στην ομάδα υψηλού κινδύνου ήταν 82%, ενώ στην ομά-δα χαμηλού κινδύνου 90%.15 Ωστόσο οι Marin και συν, ελέγχοντας την εγκυρότητα του EUTOS συγκριτικά με το Sokal score, σε μία αναδρομική μελέτη ασθενών που είχαν λάβει imatinib, διαπίστωσαν ότι η προγνωστική του αξία, σε αντίθεση με αυτή του Sokal, δεν ήταν στατιστικά σημαντική. ςυνεπώς, χρήζει περαιτέρω αξιολόγησης σε προοπτικές μελέτες, πριν ενταχθεί στην κλινική πράξη.16
πΡΟΓΝΩΣΤΙΚΟΙ ΒΙΟΔΕΙΚΤΕΣ ΣΤΗ ΔΙΑΓΝΩΣΗ ΣΧΕΤΙζΟΜΕΝΟΙ ΜΕ ΤΗΝ πΟΡΕΙΑ ΤΗΣ ΝΟΣΟΥ
η αδυναμία των κλασικών κλινικών και εργαστη-ριακών παραμέτρων να αναγνωρίσουν επακριβώς τους ασθενείς που δε θα ανταποκριθούν ικανοποιητικά στις νέες θεραπείες στόχευσης, οδήγησε στην αναζήτηση και-νούργιων προγνωστικών βιοδεικτών.
Κυτταρογενετικοί δείκτεςη κυτταρογενετική ανάλυση του μυελού, εκτός από
την παρουσία του χρωμοσώματος Philadelphia, είναι δυ-νατόν να αποκαλύψει κι άλλες κυτταρογενετικές ανωμα-λίες στο λευχαιμικό κλώνο. ςε 10-15% των ασθενών με Χμλ μπορεί να παρατηρηθεί μεγάλου μεγέθους απώλεια γενετικού υλικού πέριξ του σημείου τομής στο χιμαιρι-κό χρωμόσωμα. οι απώλειες στο χιμαιρικό χρωμόσωμα συμβαίνουν κατά τη διαδικασία της αντιμετάθεσης, ποι-κίλουν σε μέγεθος, περιλαμβάνουν ακολουθίες τόσο από το χρωμόσωμα 9 όσο και από το 22 και συχνά συμμετέ-χουν περισσότερα από 300 γονίδια.
η απώλεια στο der(9) χρωμόσωμα δεν αντιστοιχεί σε συγκεκριμένα κλινικά χαρακτηριστικά ή εργαστηριακά ευρήματα, ούτε σχετίζεται με τη φάση του νοσήματος και τα γνωστά προγνωστικά σκορ. Παρόλο που αποτελεί δυ-σμενή προγνωστικό παράγοντα στους ασθενείς που λαμ-βάνουν θεραπεία με IFN-α ή υδροξυουρία, τα δεδομένα για την προγνωστική της σημασία στους ασθενείς που αντιμετωπίζονται με imatinib είναι ακόμη ασαφή.17-23 η μελέτη των Huntly και συν έδειξε ότι το imatinib βελ-τιώνει την έκβαση των ασθενών με απώλεια γενετικού υλικού στο der(9), χωρίς όμως να εξαλείφει τη δυσμενή προγνωστική αξία της συγκεκριμένης βλάβης.19 Αντίθε-τα, άλλες μελέτες, όπως αυτή των Quintas-Cardama και συν24 και της ιταλικής ομάδας GIMEMA22, έδειξαν ότι η εν λόγω κυτταρογενετική ανωμαλία δεν επηρεάζει την ανταπόκριση και τη διάρκειά της ή τη συνολική επιβίω-

Προγνωστικοί παράγοντες στη ΧΜΛ και θεραπευτικές επιλογές 13
ση στους ασθενείς που λαμβάνουν imatinib.H γενετική αστάθεια που προκύπτει, λόγω της αντιμε-
τάθεσης t(9;22), έχει ως αποτέλεσμα τη συνεχή εμφάνι-ση επιπρόσθετων χρωμοσωμικών ανωμαλιών (additional chromosomal aberrations - ACAs) και μεταλλάξεων που οδηγούν στην εξέλιξη του νοσήματος σε επιταχυνόμενη φάση και βλαστική εκτροπή. τέτοιου είδους αλλοιώσεις διαπιστώνονται στις προχωρημένες φάσεις σε ποσοστό 80% αλλά μπορεί να είναι παρούσες και κατά τη διάγνω-ση σε 10-12% των περιπτώσεων. Περιλαμβάνουν δε τις παραλλαγές του χρωμοσώματος Philadelphia [(variant αντιμεταθέσεις), t(v;22)], απώλεια του χρωμοσώματος Υ (-Υ) και ACAs. οι ΑCAs διακρίνονται σε 2 κατηγορί-ες: minor-route [όπως t(3;12), t(4;6), t(2;16), t(1;21)] και major-route [+8, i(17)(q10), +der(22)t(9;22)(q34;q11), ider(22)(q10) t(9;22)(q34;q11)]. οι τελευταίες έχει φανεί ότι είναι οι σημαντικότερες, όσον αφορά στην κυτταρο-γενετική εξέλιξη της νόσου, δεδομένου ότι απαντώνται με μεγάλη συχνότητα στη βλαστική εκτροπή.25 Έχει ήδη παρατηρηθεί από παλαιότερες μελέτες ότι οι ασθενείς με επιπρόσθετες χρωμοσωμικές ανωμαλίες και κυρίως major-route ACAs ή σύνθετο καρυότυπο, επιτυγχάνουν μικρότερου βαθμού κυτταρογενετική ανταπόκριση μετά από θεραπεία με imatinib. ο τύπος των ACAs δε φάνη-κε να επηρεάζεται από την αγωγή με τκιs και δε μετα-βάλλεται καθ΄ όλη τη διάρκεια της θεραπείας και την πορεία της Χμλ.26-29 με βάση αυτές τις ενδείξεις, σύμ-φωνα με τις συστάσεις του European LeukemiaNet (ELN), η παρουσία ACAs στη διάγνωση θεωρείται προειδοποι-ητικό σημείο (warning) για αποτυχία στη θεραπεία με imatinib7,27. ςε πρόσφατη μελέτη των Fabarius και συν, σε 1151 ασθενείς, που συμπεριελήφθησαν στην προοπτι-κή τυχαιοποιημένη κλινική δοκιμή CML IV, επιβεβαιώ-θηκε η παρατήρηση ότι η παρουσία major-route ACAs στη διάγνωση αποτελεί δυσμενή προγνωστικό παράγο-ντα για την επιβίωση και συνεπάγεται ταχύτερη πρόοδο νόσου. ςτη συγκεκριμένη μελέτη το ποσοστό των ασθε-νών με παρουσία major-route ACAs στη διάγνωση ήταν 1,4%. με διάμεσο χρόνο παρακολούθησης 5,3 έτη, το διάμεσο διάστημα για CCgR και MMolR ήταν μεγαλύ-τερο, ενώ η 5ετής PFS και η OS στατιστικά σημαντικά μικρότερες για τους ασθενείς που έφεραν major-route ΑCAs.25 Ειδικότερα, η 5ετής PFS σε ασθενείς με χρω-μόσωμα Philadelphia ήταν 90%, με t(v;22) 81%, με -Υ 88%, με minor-route ΑCAs 96% και major-route ΑCAs 50% (p<0.001), ενώ η OS ήταν 92%, 87%, 91%, 96% και 53% αντίστοιχα (p<0.001). Όπως προτείνουν οι συγ-γραφείς, η μικρή ομάδα ασθενών με Χμλ που φέρουν major-route ΑCAs στην πρωτοδιάγνωση, θα μπορούσαν να είναι υποψήφιοι για εντατικοποιημένη θεραπεία πρώ-της γραμμής με αλλο-μΑκ ή νεότερους παράγοντες.
Παραλλαγές της αντιμετάθεσης [t(v;22)] προκύπτουν όταν ένα ή περισσότερα χρωμοσώματα, εκτός των 9 και 22, εμπλέκονται στην αντιμετάθεση. τέτοιου είδους ανω-
μαλίες διαπιστώνονται σε 5-10% των νεοδιαγνωσθέντων ασθενών. Παλαιότερες μελέτες με μικρό αριθμό ασθενών έδειξαν δυσμενέστερη πρόγνωση σε εκείνους που έφεραν κάποια παραλλαγή της αντιμετάθεσης. Παρ΄ όλα αυτά, η πρόσφατη μελέτη της GIMEMA, σε ασθενείς που ελάμ-βαναν imatinib ως θεραπεία 1ης γραμμής, καταλήγει ότι τα κλινικά χαρακτηριστικά και η έκβαση των ασθενών στους οποίους ανιχνεύονται παραλλαγές της αντιμετάθε-σης Ph στη διάγνωση δε διαφέρουν από εκείνων στους οποίους δεν ανιχνεύονται.30,31
Γονιδιακοί δείκτεςςτην προσπάθεια να προβλεφθεί η ανταπόκριση στη
θεραπεία των ασθενών με Χμλ, έγινε μελέτη του γονι-διώματος των CD34+ κυττάρων με μικροσυστοιχίες. Από διάφορες μελέτες αναγνωρίστηκαν γονιδιακοί πολυμορ-φισμοί που φαίνεται να επηρεάζουν την εξέλιξη της νό-σου και την ανταπόκριση στο imatinib. μάλιστα, φάνηκε ότι σε πολλές περιπτώσεις υπάρχει ταύτιση μεταξύ των πολυμορφισμών που ανιχνεύονται σε πρόοδο νόσου και αυτών που προδιαθέτουν σε μη ικανοποιητική ανταπό-κριση στη θεραπεία. η μελέτη των πολυμορφισμών έφερε στο προσκήνιο αρκετά γονίδια προς εξέταση σε προοπτι-κές μελέτες προκειμένου να αξιολογηθεί η προγνωστι-κή τους σημασία.32,33 Πράγματι, πρόσφατα οι Oehler και συν αναγνώρισαν έξι γονίδια (NOB1, DDX47, IGSF2, LTB4R, SCARB1 και SLC25A3) η έκφραση των οποίων διαφέρει ανάλογα με τη φάση της νόσου. με βάση τα ίδια αυτά γονίδια, οι ερευνητές αναφέρουν, ότι σε αντίθεση με τα χρησιμοποιούμενα συστήματα ταξινόμησης, μπο-ρεί να γίνει αξιόπιστη διάκριση μετάξυ πρώιμης (early) και όψιμης (late) χρόνιας φάσης. Εφόσον η ανταπόκριση στη θεραπεία είναι άμεσα συνδεδεμένη με τη φάση της νόσου, μια ακριβέστερη μέθοδος εκτίμησης της φάσης της Χμλ στηριζόμενη σε ένα τέτοιο γονιδιακό μοντέ-λο, μπορεί να επιτρέπει την εφαρμογή αποτελεσματικό-τερης θεραπείας ανάλογα με την ομάδα κινδύνου, όπως καθορίζεται από το γονιδιακό προφίλ.34
πΡΟΓΝΩΣΤΙΚΟΙ ΒΙΟΔΕΙΚΤΕΣ ΣΤΗ ΔΙΑΓΝΩΣΗ ΣΧΕΤΙζΟΜΕΝΟΙ ΜΕ ΤΗΝ ΑΝΤΑπΟΚΡΙΣΗ ΣΤΗ θΕΡΑπΕΙΑΦαρμακοκινητική
η ευαισθησία του λευχαιμικού κλώνου στο imatinib είναι δυνατόν να προσδιοριστεί πριν από την έναρξη της θεραπείας, μετρώντας in vitro τη συγκέντρωση του φαρ-μάκου που απαιτείται για να προκαλέσει μείωση της δρα-στικότητας της bcr-abl κινάσης κατά 50% (IC50imatinib). οι White και συν εφάρμοσαν αυτήν τη μέθοδο και διεπίστω-σαν ότι ασθενείς με χαμηλή IC50imatinib στη διάγνωση, εί-

Μ.Ν. Παγώνη και Ι.Α. Τζάννου14
χαν καλύτερες μοριακές ανταποκρίσεις μετά τη θεραπεία με imatinib. Επιπλέον, έδειξαν ότι δεν υπάρχει συσχέτι-ση μεταξύ της IC50imatinib και του Sokal score. Δεν υπήρ-ξε στατιστικά σημαντική διαφορά ανάμεσα στις ομάδες κινδύνου κατά Sokal όσον αφορά στη μείωση του bcr-abl κατά 2 λογαρίθμους στους 3 μήνες. Ειδικά για την ομάδα χαμηλού κινδύνου, φάνηκε ότι η προγνωστική αξία της IC50imatinib για την επίτευξη μοριακής ύφεσης αυξάνεται. Αξίζει να σημειωθεί ότι μεταξύ των ασθενών χαμηλού κιδύνου κατά Sokal, περισσότεροι από 50% αυτών που είχαν χαμηλή IC50imatinib πέτυχαν μείωση του bcr-abl κα-τά 2 λογαρίθμους στους 3 μήνες, ενώ κανένας από τους ασθενείς με υψηλή IC50imatinib δεν παρουσίασε αντίστοι-χη ανταπόκριση.35
η ετερογένεια των λευχαιμικών κλώνων όσον αφορά στην ευαισθησία τους στο imatinib, σχετίζεται με την ικα-νότητα του φαρμάκου να εισέρχεται στο κύτταρο και την ταχύτητα απομάκρυνσής του από αυτό. το imatinib εισέρ-χεται στο κύτταρο με ενεργητική εισροή μέσω της αντλί-ας οργανικών κατιόντων OCT-1. η έκφραση της OCT-1 έχει μελετηθεί εκτενώς. ςτη μελέτη της ομάδας από την Αυστραλία, διαπιστώθηκε ότι η διαφορετική in vitro ευ-αισθησία διαφόρων κυτταρικών σειρών στο imatinib είχε άμεση συσχέτιση με την έκφραση της OCT-1 στα κύττα-ρα της κάθε σειράς.36 Επίσης, οι Crossman και συν παρα-τήρησαν ελαττωμένη έκφραση OCT-1 στα μονοπύρηνα κύτταρα του μυελού ασθενών που δεν κατόρθωσαν να επιτύχουν τουλάχιστον ελάσσονα μοριακή ανταπόκριση μετά από 12 μήνες θεραπείας με imatinib.37 τα αποτελέ-σματα επιβεβαιώθηκαν και από μία μελέτη 70 ασθενών που έδειξε στατιστικά σημαντική διαφορά στην επίτευξη CCgR στους 6 μήνες, καθώς επίσης στην επιβίωση χωρίς πρόοδο νόσου και τη συνολική επιβίωση, ανάλογα με τα επίπεδα OCT-1 πριν από την έναρξη θεραπείας.38 ςυνη-γορητικά είναι και τα αποτελέσματα της μελέτης TOPS, στην οποία όσοι από τους ασθενείς με χαμηλή έκφραση OCT-1 έλαβαν αγωγή με υψηλή δόση imatinib είχαν κα-λύτερα ποσοστά μμolR έναντι αυτών που έλαβαν την καθιερωμένη δόση.39
ςτην απομάκρυνση του imatinib από το λευχαιμικό κύτ-ταρο συμμετέχει η πρωτεΐνη/μεταφορέας Ρ-γλυκοπρωτεΐνη (P-gp). η πρωτεΐνη αυτή είναι ενεργητικός μεταφορέας του imatinib προς τον εξωκυττάριο χώρο και η υπερέκ-φρασή της θεωρήθηκε ότι συμβάλλει στην αντοχή στο imatinib. Φάνηκε, όμως, ότι η μέτρηση της έκφρασης της P-gp δεν αντιπροσωπεύει και τη δραστικότητά της. οι Dulucq και συν μελέτησαν τους τρεις συχνότερους πο-λυμορφισμούς στην περιοχή MDR (Multidrug resistance) και παρατήρησαν ότι το ποσοστό επίτευξης μμolR ήταν 85% στους ασθενείς που ήταν ομόζυγοι για το αλλήλιο 1236τ έναντι ποσοστού 47,7% που βρέθηκε στους ασθε-νείς με τους άλλους γονότυπους.40
Μεταλλάξεις της bcr-abl κινάσηςςτους μισούς περίπου ασθενείς που δεν ανταποκρίνο-
νται στο imatinib, η ανθεκτικότητα οφείλεται σε μεταλλά-ξεις της bcr-abl κινάσης. οι Gorre και συν αναγνώρισαν πρώτοι τη μετάλλαξη τ315ι, η οποία αργότερα φάνηκε ότι αποτελεί δυσμενή προγνωστικό παράγονται για την επιβίωση χωρίς πρόοδο νόσου και τη συνολική επιβίωση των ασθενών με Χμλ.41 ο αριθμός των αναφερόμενων μεταλλάξεων σε όλο το μήκος της abl ακολουθίας συνε-χώς αυξάνεται, όμως η κλινική τους σημασία ποικίλει. μεταλλάξεις ανιχνεύονται σε μεγαλύτερη συχνότητα σε προχωρημένες φάσεις της νόσου. Εκτός από την τ315ι, οι μεταλλάξεις που βρίσκονται στην Ρ-αγκύλη (P-loop), φάνηκε ότι οδηγούν συχνότερα σε αποτυχία θεραπείας με imatinib και εξέλιξη νόσου.42,43 Παρ΄όλα αυτά η αναζήτη-ση μεταλλάξεων στην πρωτοδιάγνωση δε συνιστάται, πα-ρά μόνο σε ασθενείς που βρίσκονται σε επιταχυνόμενη ή βλαστική φάση. ςτην περίπτωση αυτήν, είναι σημαντικό ο θεράπων να γνωρίζει την ευαισθησία της μετάλλαξης στους διαθέσιμους αναστολείς, ώστε να γίνει ο βέλτιστος θεραπευτικός σχεδιασμός με τον κατάλληλο TKI πριν τη διενέργεια αλλο-μΑκ στις περιπτώσεις που ενδείκνυται.
Εκτός από τα προγνωστικά μοντέλα Sokal, Hasford και EUTOS, οι υπόλοιποι προγνωστικοί παράγοντες που αναλύθηκαν αφενός μεν απαιτούν ιδιαίτερα εξειδικευμέ-να εργαστήρια, αφετέρου δε χρειάζονται περαιτέρω αξι-ολόγηση της προγνωστικής τους αξίας και μέχρι σήμερα τουλάχιστον δεν έχουν τύχει ευρείας εφαρμογής. Έτσι, στην κλινική πρακτική οι θεραπευτικές αποφάσεις βασί-ζονται, κατά κύριο λόγο, στα δεδομένα που σχετίζονται με την ανταπόκριση στη θεραπεία.
προγνωστικοί δείκτες στη διάρκεια της θεραπείας σχετιζόμενοι με την ανταπόκρισηΑιματολογική ανταπόκριση
Είναι προφανές ότι η μη επίτευξη αιματολογικής ύφε-σης στους πρώτους 3 μήνες της θεραπείας με imatinib απο-τελεί όχι μόνο δυσμενή προγνωστικό παράγοντα, αλλά θεραπευτική αποτυχία. Όσον αφορά στις κυτταροπενίες κατά τη διάρκεια της θεραπείας, δεν έχουν συσχετιστεί μεν με δυσμενή πρόγνωση, υποδηλώνουν όμως αυξημέ-νη τοξικότητα που μπορεί να απαιτήσει μείωση δόσεων ή αναβολή της θεραπείας με αποτέλεσμα, σε ορισμένες περιπτώσεις, την πλημμελή ανταπόκριση.44
Κυτταρογενετική ανταπόκρισηη πρώιμη κυτταρογενετική απάντηση θεωρείται ο ση-
μαντικότερος από τους προγνωστικούς παράγοντες που

Προγνωστικοί παράγοντες στη ΧΜΛ και θεραπευτικές επιλογές 15
σχετίζονται με την ανταπόκριση στη θεραπεία. ςύμφω-να με τις συστάσεις του ELN κυτταρογενετικός έλεγχος πραγματοποιείται στους 3, 6, 12 και 18 μήνες μετά την έναρξη θεραπείας 1ης γραμμής.7 Έχει διαπιστωθεί ότι ση-μείο αναφοράς για την εκτίμηση της κυτταρογενετικής απάντησης και την προγνωστική της αξία αποτελούν οι 6 μήνες. ςε πολλές μελέτες, συμπεριλαμβανομένης της IRIS, απουσία κυτταρογενετικής ανταπόκρισης στους 6 μήνες σχετίζεται με μειωμένη πιθανότητα επίτευξης CCgR και μειωμένη επιβίωση χωρίς πρόοδο νόσου.45-48 ςτη μελέτη IRIS επισημαίνεται η προγνωστική σημασία της επίτευξης CCgR για την επιβίωση. Ασθενείς που πέ-τυχαν CCgR μετά από 12, 18 ή 24 μήνες θεραπείας με imatinib είχαν καλύτερη επιβίωση ελεύθερη νόσου, συ-γκριτικά με εκείνους, οι οποίοι στις ίδιες χρονικές στιγ-μές είχαν μερική κυτταρογενετική ανταπόκριση.48 Όσον αφορά στο βέλτιστο χρόνο επίτευξης CCgR οι Quintas-Cardama και συν καταλήγουν ότι η πιθανότητα MMolR και η επιβίωση χωρίς πρόοδο νόσου είναι σημαντικά χα-μηλότερες για τους ασθενείς εκείνους που στους 12 πρώ-τους μήνες θεραπείας με imatinib δεν καταφέρνουν να επιτύχουν CCgR.45 Παρά το γεγονός, ότι καλές ανταπο-κρίσεις μπορούν να επιτευχθούν καθυστερημένα και να οδηγήσουν σε εξίσου ευνοϊκή έκβαση, οι ασθενείς που δεν ανταποκρίνονται εγκαίρως (εντός των 12 πρώτων μη-νών), έχουν αυξημένο κίνδυνο εξέλιξης νόσου. οι Marin και συν, σε μια προσπάθεια να ελέγξουν την εγκυρότητα των κριτηρίων του ELN αναφορικά με την ανταπόκριση, βρήκαν ότι οι ασθενείς με CCgR στους 12 μήνες είχαν PFS 96,1% έναντι των ασθενών με MCgR για τους οποί-ους η PFS βρέθηκε 73,4%, διαφορά που ήταν στατιστικά σημαντική. το τελευταίο ποσοστό, όμως, ήταν συγκρί-σιμο με τo αντίστοιχο για τoυς ασθενείς που, σύμφωνα με τα ELN κριτήρια, είχαν θεραπευτική αποτυχία στο imatinib.47 με βάση τις παρατηρήσεις αυτές και τη δια-θεσιμότητα των τκιs 2ης γενιάς, θα μπορούσε κανείς να υποστηρίξει ότι η μη βέλτιστη (suboptimal) ανταπόκριση διακρίνει πρώιμα την ομάδα των ασθενών που μπορούν να ωφεληθούν από αλλαγή της θεραπείας με χορήγηση ενός από τους νεότερους παράγοντες.
Μοριακή ανταπόκρισηΌσον αφορά στην επίτευξη MMolR, η σημασία της
έχει ήδη τεκμηριωθεί από τη μελέτη IRIS. Ασθενείς με MMolR στους 18 μήνες από την έναρξη της αγωγής, έχουν PFS που αγγίζει το 95% και πιθανότητα απώλειας της CCgR 3% στα 7 έτη, έναντι πιθανότητας 26% για τους ασθενείς οι οποίοι έχουν επιτύχει CCgR χωρίς MMolR. Επιπλέον, η μελέτη έδειξε ότι ασθενείς, στο περιφερικό αίμα των οποίων ανιχνεύθηκαν μετάγραφα BCR-ABL >10% στους 6 μήνες και >1% στους 12 μήνες, είχαν υπο-δεέστερη επιβίωση ελεύθερη συμβαμάτων (EFS) και με-
γαλύτερη συχνότητα εξέλιξης σε επιταχυνόμενη φάση ή βλαστική κρίση.49 Άλλες μελέτες έδειξαν ότι το βάθος της μοριακής ύφεσης 3 μήνες μετά την έναρξη θεραπείας δί-νει αξιόπιστες προγνωστικές πληροφορίες. ςυγκεκριμέ-να, στους ασθενείς με μείωση των μεταγράφων κατά 1-2 και >2 λογαρίθμους, η συχνότητα MMolR στους 24 μή-νες ήταν 69% και 100% αντίστοιχα, ενώ σε ασθενείς που δεν εμφάνισαν μείωση κατά 1 λογάριθμο τουλάχιστον, η συχνότητα MMolR στους 24 μήνες ήταν μόλις 13%. οι Quintas-Cardama και συν έδειξαν ότι ασθενείς με λό-γο BCR-ABL1/ABL1 >1% και <10% στους 3 μήνες θε-ραπείας με imatinib είχαν 92% πιθανότητα να πετύχουν CCgR, ποσοστό συγκρίσιμο με το 98% που αναφέρεται σε ασθενείς με λόγο <1%. Αντίθετα, ασθενείς με BCR-ABL1/ABL1 >10% στους 3 μήνες είχαν πιθανότητα να πετύχουν CCgR μόνο 67%. Όμως, αξίζει να σημειωθεί ότι ο κίνδυνος εξέλιξης νόσου ήταν 11% για τους ασθε-νείς με λόγο BCR-ABL1/ABL1 μεταξύ 1% και 10%, ποσοστό τριπλάσιο από το αντίστοιχο για τους ασθενείς με λόγο <1%.45
μια πρόσφατη δημοσίευση των Marin και συν ανα-φέρει ότι ασθενείς με επίπεδα μεταγράφων BCR-ABL1 >9,84% στους 3 μήνες θεραπείας με imatinib έχουν ση-μαντικά μικρότερη 8ετή επιβίωση έναντι των ασθενών με χαμηλότερα επίπεδα μεταγράφων (56,9% vs 93.3%). Αντίστοιχα είναι και τα αποτελέσματα για την επιβίωση χωρίς νόσο, την αθροιστική επίπτωση CCgR και MMolR. οι συγγραφείς καταλήγουν ότι η μοριακή ανταπόκριση στους 3 μήνες αποτελεί τη μοναδική παράμετρο για την εκτίμηση της έκβασης των ασθενών με Χμλ που λαμ-βάνουν θεραπεία με imatinib, παρέχοντας τη δυνατότητα πρώιμης αναγνώρισης αυτών που χρήζουν θεραπείας 2ης γραμμής και άρα θα ωφεληθούν από την έγκαιρη χορή-γηση των νεότερων αναστολέων.50 η μελέτη CML IV της γερμανικής ομάδας οδηγεί σε ανάλογα συμπεράσματα, τα οποία ενισχύουν την επιλογή της πρώιμης αλλαγής θερα-πείας, καθώς για τους ασθενείς με επίπεδα μεταγράφων BCR-ABL1 >10% στους 3 μήνες θεραπείας με imatinib η 5ετής OS ήταν 87%, ενώ για τους ασθενείς με BCR-ABL1 <1% ήταν 97%.51
ΑΛΛΟΙ πΡΟΓΝΩΣΤΙΚΟΙ ΔΕΙΚΤΕΣ ΣΤΗ ΔΙΑΡΚΕΙΑ ΤΗΣ θΕΡΑπΕΙΑΣ
H ετερογένεια των ανταποκρίσεων μεταξύ των ασθε-νών που λαμβάνουν αγωγή με imatinib οφείλεται, ως ένα βαθμό, στις διακυμάνσεις που παρατηρούνται στα επί-πεδα του φαρμάκου στο πλάσμα. ςτη μελέτη των Picard και συν μετρήθηκαν τα επίπεδα του imatinib στο πλά-σμα μετά από έναν τουλάχιστον χρόνο συνεχούς θερα-πείας. Διαπιστώθηκε ότι τα επίπεδα του φαρμάκου ήταν σημαντικά υψηλότερα στην ομάδα των ασθενών που εί-χαν πετύχει MMolR. οι ερευνητές θεωρούν ότι με τον προσδιορισμό των επιπέδων του imatinib στο πλάσμα

Μ.Ν. Παγώνη και Ι.Α. Τζάννου16
μπορεί να προβλεφθεί η πιθανότητα επίτευξης μοριακής ανταπόκρισης. Ασθενείς με επίπεδα φαρμάκου >1002 ng/ml έχουν σημαντικά μεγαλύτερη πιθανότητα επίτευξης MMolR.52 ςε μια υπο-ανάλυση της μελέτης IRIS φάνη-κε επίσης ότι τα επίπεδα του imatinib στο πλάσμα απο-τελούν προγνωστικό παράγοντα για την επίτευξη CCgR, ανεξάρτητα από την ομάδα κινδύνου κατά Sokal.53 Πα-ρά την προγνωστική της αξία, η συστηματική μέτρηση των επιπέδων του φαρμάκου στο αίμα προς το παρόν δε συνιστάται. μπορεί, όμως, να εφαρμοστεί σε ασθενείς με μη ικανοποιητική ανταπόκριση ή αποτυχία στη θερα-πεία. Πάντως, σε μια μελέτη από την τσεχία, που εφαρ-μόστηκε η μέτρηση επιπέδων imatinib σε 151 ασθενείς με Χμλ, εκτός θεραπευτικού πρωτοκόλλου, δε διαπι-στώθηκε συσχέτιση μεταξύ των επιπέδων του φαρμάκου και της ανταπόκρισης.54
κατ’αναλογία με την in vitro μέτρηση της IC50imatinib, οι White και συν εκτίμησαν την in vivo αναστολή της δράσης της bcr-abl κινάσης σε ασθενείς υπό θεραπεία με imatinib, μετρώντας το φωσφορυλιωμένο Crkl (CT10 regulator of kinase like), υπόστρωμα της bcr-abl κινάσης. Παρατηρήθηκαν μεγάλες διακυμάνσεις μεταξύ των ασθε-νών και από την περαιτέρω ανάλυση των αποτελεσμά-των διαπιστώθηκε ότι η αναστολή της δράσης της bcr-abl κινάσης είχε μεγάλη προγνωστική αξία για την επίτευξη κυτταρογενετικής και μοριακής ανταπόκρισης.55 Ανάλογα συμπεράσματα για την προγνωστική αξία της μέτρησης εξάγονται και από τη μελέτη TIDΕL.56 η μέθοδος, ωστό-σο, δεν έχει ενταχθεί στην καθ΄ ημέρα κλινική πράξη.
πΡΟΓΝΩΣΤΙΚΟΙ ΔΕΙΚΤΕΣ ΣΤΗΝ ΕπΟΧΗ ΤΩΝ ΑΝΑΣΤΟΛΕΩΝ ΔΕΥΤΕΡΗΣ ΓΕΝΙΑΣ
ςαφώς λιγότερα δεδομένα υπάρχουν αναφορικά με τους προγνωστικούς παράγοντες για την έκβαση των ασθενών που λαμβάνουν 2ης γενιάς TKIs και οι πλείστες μελέτες αναφέρονται στη χορήγησή τους ως 2ης γραμμής θεραπεία μετά από αποτυχία στο imatinib.
Στη δεύτερης γραμμής θεραπείατο 2011 στο συνέδριο της ASH ανακοινώθηκαν απο-
τελέσματα της γερμανικής μελέτης CML IV για 120 ασθε-νείς σε 1η χρόνια φάση, που έλαβαν dasatinib ή nilotinib μετά από αστοχία της θεραπείας με imatinib. Παρατηρή-θηκε ότι οι ασθενείς που άλλαξαν θεραπεία λόγω δυσανε-ξίας στο imatinib είχαν καλύτερη 3ετή PFS σε σύγκριση με αυτούς που έλαβαν 2ης γενιάς TKI λόγω αποτυχίας (96% vs 76%), ενώ η OS ήταν 96% και 80% αντίστοι-χα για τις δύο ομάδες. Δεδομένων των αποτελεσμάτων, προτείνεται να αναζητηθούν εναλλακτικές θεραπείες για τους ασθενείς της δεύτερης ομάδας.57
οι Tam και συν, με βάση την αποδεδειγμένη σημα-σία της κυτταρογενετικής ανταπόκρισης, νωρίς μετά την έναρξη θεραπείας με imatinib, για την επίτευξη CCgR και MMolR, εξέτασαν αν η προγνωστική της αξία δια-τηρείται και στη θεραπεία με τους 2ης γενιάς TKIs. με-λέτησαν 113 ασθενείς που έλαβαν dasatinib ή nilotinib και παρατήρησαν ότι όσοι, μετά από 12 μήνες θεραπεί-ας, πέτυχαν τουλάχιστον MCgR είχαν καλύτερη επιβίωση (επιβίωση ενός έτους 97% vs 84%, p=0.02) και εξέλιξη νόσου σε ένα έτος 3% vs 17% (p=0.003). η πρώιμη CgR φάνηκε να έχει προγνωστική σημασία, καθώς από τους ασθενείς που δεν πέτυχαν καμία κυτταρογενετική αντα-πόκριση στους 3-6 μήνες θεραπείας, λιγότεροι από 10% κατάφεραν τελικά να αποκτήσουν MCgR.58 Επομένως, ασθενείς χωρίς CgR στους 3-6 μήνες, πρέπει να θεωρεί-ται ότι δεν ανταποκρίνονται στη θεραπεία με 2ης γενιάς TKIs και χρήζουν αλλαγής της θεραπευτικής προσέγγι-σης, με διενέργεια αλλο-μΑκ ή ένταξη σε ερευνητικά πρωτόκολλα.
οι Branford και συν παρατήρησαν ότι τα επίπεδα BCR-ABL στους 3 μήνες μετά την έναρξη θεραπείας 2ης γραμμής με νεότερο αναστολέα, είχαν προγνωστική σημασία για τη μετέπειτα επίτευξη MMolR και MCgR. ςυγκεκριμένα, ασθενείς με BCR-ABL ≤1% IS στους 3 μήνες πέτυχαν MMolR σε ποσοστό 80% και MCgR 91% μετά από 24 μήνες θεραπείας έναντι αντίστοιχων ποσο-στών μόνο 4% και 11% για εκείνους που είχαν BCR-ABL >10% IS στους 3 μήνες.59
οι μεταλλάξεις που ανευρίσκονται στο γονίδιο abl, πριν από την έναρξη θεραπείας με dasatinib ή nilotinib μετά από αποτυχία του imatinib, αναφέρονται ως baseline μεταλλάξεις και έχει φανεί ότι επηρεάζουν την ανταπό-κριση στη θεραπεία με 2ης γραμμής τκιs και την εξέλιξη του νοσήματος. Είναι γνωστό ότι η παρουσία της μετάλ-λαξης τ315ι συνεπάγεται αντίσταση τόσο στο dasatinib όσο και στο nilotinib και άρα θα ήταν λογική μια διαφο-ρετική θεραπευτική προσέγγιση, όπως η αλλο-μΑκ ή η χορήγηση 3ης γενιάς τκι. Όσον αφορά σε άλλες μεταλ-λάξεις, πρώιμες μελέτες έδειξαν ότι η ανταπόκριση στους αναστολείς 2ης γενιάς μπορεί να προβλεφθεί με το score μετάλλαξης, δηλαδή τον in vitro προσδιορισμό της IC50 του εν λόγω φαρμάκου, που απαιτείται για να αναστείλει κατά 50% τη δράση της bcr-abl κινάσης με τη συγκεκρι-μένη μετάλλαξη. Από μια μελέτη των Hughes και συν σχε-τικά με την αποτελεσματικότητα του nilotinib σε ασθενείς με baseline μεταλλάξεις (εξαιρουμένης της τ315ι), βρέ-θηκαν τρεις μεταλλάξεις με δυσμενή προγνωστική ση-μασία. οι ασθενείς που έφεραν τις μεταλλάξεις Υ253H, Ε255V/K, F359V/C είχαν MCgR 19%, MMolR 0% και CCgR 4% στους 12 μήνες. για τους ασθενείς με άλλες μεταλλάξεις τα αντίστοιχα ποσοστά ήταν 59%, 43% και 29%. ςτατιστικά σημαντική διαφορά παρατηρήθηκε και στο ποσοστό προόδου νόσου.60 Ανάλογη μελέτη για το dasatinib, αναγνώρισε ως προγνωστικής σημασίας τις με-

Προγνωστικοί παράγοντες στη ΧΜΛ και θεραπευτικές επιλογές 17
ταλλάξεις F317L/I/V, T315A, V299L.61 Έτσι μετά από την αναζήτηση και ανίχνευση των μεταλλάξεων, είναι εφικτή η επιλογή του αποτελεσματικότερου αναστολέα, ενώ η παρουσία της T315I αποτελεί απόλυτη ένδειξη για αλλο-μΑκ.62
Επίσης, η εμφάνιση νέας μετάλλαξης σε οποιαδήπο-τε φάση της θεραπείας αποτελεί δυσμενή προγνωστικό παράγοντα, ως σημείο γενετικής αστάθειας και ανθεκτι-κότητας στη χορηγούμενη αγωγή.7
Πρόσφατα έγινε προσπάθεια δημιουργίας προγνω-στικών μοντέλων για την εκτίμηση της αποτελεσματικό-τητας των νεότερων τκιs, ως θεραπεία 2ης γραμμής. οι Milojkovic και συν αναγνώρισαν ως ανεξάρτητους προ-γνωστικούς παράγοντες την κυτταρογενετική ανταπόκρι-ση στην προηγηθείσα θεραπεία με imatinib, την ομάδα κινδύνου κατά Sokal, την εμφάνιση ουδετεροπενίας κα-τά τη διάρκεια της θεραπείας με imatinib, με αποτέλεσμα μείωση ή παράλειψη δόσης φαρμάκου και το μεσοδιά-στημα μεταξύ της διακοπής του imatinib και της έναρξης της θεραπείας 2ης γραμμής. οι ασθενείς ταξινομήθηκαν σε 3 ομάδες κινδύνου με βάση τις τρεις πρώτες παραμέ-τρους και η αθροιστική επίπτωση CCgR σε 2,5 έτη βρέ-θηκε 100% για την ομάδα χαμηλού κινδύνου, 52,2% για την ομάδα ενδιάμεσου κινδύνου και 13,8% για την ομά-δα υψηλού κινδύνου.63
ςτη μελέτη των Jabbour και συν διαπιστώθηκε κα-λύτερη επιβίωση μετά από θεραπεία με δεύτερης γενιάς αναστολείς σε ασθενείς με κυτταρογενετική υποτροπή ή δυσανεξία στο imatinib σε σχέση με ασθενείς που εί-χαν αιματολογική υποτροπή ή πρωτογενή αντίσταση στο imatinib. Ανεξάρτητοι δυσμενείς προγνωστικοί παράγο-ντες για την EFS ήταν η μη επίτευξη οποιασδήποτε κυτ-ταρογενετικής ανταπόκρισης στην προηγηθείσα θεραπεία με imatinib και η κατάσταση ικανότητας κατά ECOG (PS ≥1). Ασθενείς με 0, 1 ή 2 δυσμενείς προγνωστικούς παράγοντες είχαν 2ετή EFS 78%, 49% και 20%, ενώ η 2ετής OS βρέθηκε αντίστοιχα 95%, 85% και 40%.64 τα δύο παραπάνω προγνωστικά συστήματα δεν έχουν αξιο-λογηθεί ακόμα επαρκώς σε μεγάλο αριθμό ασθενών και ως εκ τούτου δεν έχουν ενταχθεί στην κλινική πρακτι-κή. Θα μπορούσαν, όμως, να χρησιμοποιηθούν για να αναγνωρίσουν τους ασθενείς που δεν θα ανταποκριθούν ικανοποιητικά στη θεραπεία 2ης γραμμής με dasatinib ή nilotinib, ώστε να οδηγηθούν σε αλλο-μΑκ εφ΄όσον υπάρχει διαθέσιμος δότης.
Στην πρώτης γραμμής θεραπείαςχετικά πρόσφατα, οι αναστολείς δεύτερης γενιάς πή-
ραν έγκριση ως θεραπεία 1ης γραμμής. Προκειμένου να λάβουν την έγκριση, έπρεπε να απαντηθούν ερωτήματα σχετικά με την αποτελεσματικότητα, την ασφάλεια, αλ-λά και την αξιολόγηση της ανταπόκρισης και ποιοί δεί-
κτες πρόγνωσης έχουν αξία.Από την ομάδα του MDACC ανακοινώθηκαν απο-
τελέσματα από διαδοχικές μελέτες φάσης 2 για νεοδια-γνωσθέντες ασθενείς με Χμλ, που έλαβαν imatinib 400 mg, imatinib 800 mg, αναστολέα 2ης γενιάς (dasatinib ή nilotinib). Αναλύθηκαν τα δεδομένα από τις μελέτες αυτές για να εκτιμηθεί η συσχέτιση της επίτευξης CCgR, πρώ-ιμα κατά τη διάρκεια της θεραπείας, με την έκβαση. η συνολική CCgR ήταν 87% για το imatinib 400 mg, 91% για το imatinib 800 mg και 96% με τους τκιs δεύτερης γενιάς και η MMolR ήταν 77%, 87% και 89% αντίστοι-χα. η 3ετής EFS βρέθηκε 85%, 92% και 97%, ενώ η OS 93%, 97% και 100% αντίστοιχα. με landmark ανάλυση οι ασθενείς με CCgR σε 3, 6, 12 μήνες είχαν στατιστι-κά σημαντικά καλύτερη έκβαση με 3ετή ΕFS 98%, 97%, 98% και OS 99%, 99% και 99% αντίστοιχα, σε σύγκρι-ση με ΕFS 83%, 72%, 67% και OS 95%, 90% και 94% αντίστοιχα για όσους δεν είχαν πετύχει CCgR. η επίτευ-ξη CCgR διαπιστώθηκε σε διάμεσο χρόνο 6 μηνών υπό imatinib 400 mg και 3 μηνών υπό imatinib 800mg και dasatinib ή nilotinib. Δεδομένου ότι η επίτευξη MMolR υπό imatinib βρέθηκε να σχετίζεται με καλύτερη πιθανό-τητα διατήρησης της κυτταρογενετικής ανταπόκρισης και της EFS, οι ερευνητές μελέτησαν τα μακροχρόνια αποτε-λέσματα των ασθενών με CCgR στους 12 μήνες σε σχέ-ση με την MMolR κατά την ίδια χρονική στιγμή. μεταξύ των ασθενών με CCgR στους 12 μήνες δεν υπήρχαν δι-αφορές στην OS ή την ΕFS, ανεξάρτητα αν οι ασθενείς είχαν πετύχει ή όχι MMolR. Παρά το γεγονός, λοιπόν, ότι η ανταπόκριση είναι ταχύτερη με τους αναστολείς 2ης γενιάς, φάνηκε ότι η πρόγνωση είναι η ίδια για όλους τους ασθενείς με CCgR σε μια δεδομένη χρονική στιγ-μή, ανεξαρτήτως θεραπείας. τελικά, η επίτευξη πρώιμης CCgR παραμένει σημαντικός προγνωστικός παράγοντας για την έκβαση των ασθενών, ανεξάρτητα από την επί-τευξη ή όχι MMolR.65
το 2011 στο Αιματολογικό συνέδριο των ηΠΑ, ανα-κοινώθηκαν δεδομένα σχετικά με προγνωστικούς παρά-γοντες για την ανταπόκριση στους νεότερους αναστολείς όταν χορηγούνται ως θεραπεία 1ης γραμμής. η ανάλυση των αποτελεσμάτων της μοριακής ανταπόκρισης μεταξύ τω ασθενών που έλαβαν dasatinib vs imatinib στην τυχαι-οποιημένη μελέτη DASISION έδειξε ότι η μοριακή αντα-πόκριση ήταν ταχύτερη και βαθύτερη στους ασθενείς που έλαβαν dasatinib, με ποσοστά αθροιστικής ανταπόκρισης MMolR 65% vs 47%, και επίπεδα BCR-ABL ≤0,01% σε 29% vs 19% και BCR-ABL ≤0,0032% σε 17% vs 9% αντί-στοιχα. ο διάμεσος χρόνος επίτευξης της MMolR ήταν κατά 20 μήνες βραχύτερος για την ομάδα του dasatinib (15 vs 35 μήνες). Ανεξάρτητα του σκέλους θεραπείας, η μείωση των μεταγράφων, ώστε BCR-ABL ≤10% στους 3 μήνες σχετίζεται με αυξημένη πιθανότητα επίτευξης CCgR στους 12 μήνες (96% vs 27%) και MMolR στους 24 (76% vs 16%), μικρότερη πιθανότητα εξέλιξης νόσου

Μ.Ν. Παγώνη και Ι.Α. Τζάννου18
(2% vs 8%) και μεγαλύτερη PFS στους 24 μήνες (97% vs 83%).66 ςτην τυχαιοποιημένη μελέτη ΕNESTnd αξι-ολογήθηκαν τα αποτελέσματα των ασθενών που έλαβαν nilotinib (600 mg ή 800 mg ημερησίως) έναντι imatinib σαν θεραπεία 1ης γραμμής. ο διάμεσος χρόνος για την επίτευξη MMolR ήταν 8,3 μήνες για τους ασθενείς που έλαβαν nilotinib έναντι 11,1 μηνών για εκείνους που έλα-βαν imatinib. η MMolR στους 24 μήνες ήταν στατιστι-κά σημαντικά καλύτερη στην ομάδα του nilotinib (71% vs 67% vs 44%, p<0.0001), όπως επίσης και το ποσοστό των ασθενών που πέτυχαν επίπεδα BCR-ABL ≤0,0032% (26% vs 21% vs 10%). Επιπροσθέτως, οι ασθενείς υπό nilotinib είχαν σημαντικά μικρότερη πιθανότητα εξέλι-ξης νόσου.67 Από την ανάλυση των αποτελεσμάτων της ENESTnd φάνηκε ότι η πιθανότητα εμφάνισης μεταλλά-ξεων στην abl κινάση και η πρόοδος νόσου ήταν μικρό-τερη μεταξύ των ασθενών που έλαβαν nilotinib, γεγονός που υποδηλώνει ότι οι βαθύτερες μοριακές ανταποκρί-σεις προφυλάσσουν από την ανάπτυξη μεταλλάξεων και την εξέλιξη νόσου.68
Προγνωστικοί παράγοντες, που αναγνωρίστηκαν στην εποχή του imatinib, χάνουν την αξία τους στην επο-χή των νεότερων αναστολέων. Παράδειγμα αποτελεί η OCT-1, η οποία δεν επηρεάζει τη δράση των τκιs 2ης γε-νιάς, αφού δεν αποτελούν υπόστρωμα αυτής, σε αντίθε-ση με το imatinib.
ςυνεπώς, η επιλογή των νεότερων αναστολέων ως θεραπεία 1ης γραμμής φαίνεται ότι βελτιώνει τα ήδη πο-λύ καλά αποτελέσματα του imatinib. Όμως απαιτείται αυστηρή παρακολούθηση της ανταπόκρισης, ώστε να αναγνωρίζεται έγκαιρα η θεραπευτική αστοχία και να λαμβάνονται θεραπευτικές αποφάσεις, δεδομένου ότι ο βέλτιστος χρόνος αλλαγής θεραπείας και η καταλληλό-τερη προσέγγιση, επί αποτυχίας στους νεότερους ανα-στολείς, όταν χορηγούνται ως θεραπεία 1ης γραμμής, δεν είναι ακριβώς καθορισμένα με τα σημερινά δεδομένα.
ΣΥΜπΕΡΑΣΜΑΤΑη εισαγωγή των αναστολέων τυροσινικής κινάσης στη
θεραπευτική αντιμετώπιση της Χμλ απετέλεσε πραγμα-τική επανάσταση και άλλαξε τα δεδομένα για τους ασθε-νείς αλλά και για τους θεράποντες. μόλις μισό αιώνα πριν, η διάγνωση της Χμλ σήμαινε βέβαιο θάνατο σε σύντο-μο χρονικό διάστημα. ςήμερα η Χμλ είναι ένα χρόνιο νόσημα και οι ασθενείς μπορούν να έχουν προσδόκιμο επιβίωσης ανάλογο ακόμα και με αυτό υγιών ατόμων της ίδιας ηλικίας. Ωστόσο, ριζική ίαση του νοσήματος δεν επιτυγχάνεται, παρά μόνο μέσω της αλλο-μΑκ. τα αποτελέσματα συνεχώς βελτιώνονται και οι προσδοκίες των ασθενών και των γιατρών αυξάνονται. το τελικό ζη-τούμενο μπορεί να είναι κάθε φορά διαφορετικό, μακρά επιβίωση, η επιβίωση χωρίς συμβάματα, η ίαση. Όσο οι
Πίνακας 1. Προγνωστικά συστήματα – Εκτίμηση κινδύνου
Sokal score - Στην εποχή της βουσουλφάνης
Παράμετροι Ηλικία (έτη), Μέγεθος Σπληνός (σε cm, η μέ-γιστη απόσταση από το πλευρικό τόξο), Αριθμός Αιμοπεταλίων (x109/l), Ποσοστό Βλαστών στο αίμα
Υπολογισμός Exp 0.0116 x (age - 43.4) + 0.0345 x (spleen - 7.51) + 0.188 x {(platelet count ÷ 700)2 - 0.563} + 0.0887 x (blast cells - 2.10)
Προγνωστικές Ομάδες
Χαμηλού κινδύνου: < 0.8Eνδιάμεσου κινδύνου: 0.8 – 1.2Υψηλού κινδύνου: >1.2
Hasford score - Στην εποχή της ιντερφερόνης
Παράμετροι Ηλικία (έτη), Μέγεθος Σπληνός (σε cm, η μέ-γιστη απόσταση από το πλευρικό τόξο), Αριθμός Αιμοπεταλίων (x109/l), Ποσοστό Βλαστών στο αίμα, Ποσοστό Βασεόφιλων στο αίμα, Ποσοστό Ηωζινόφιλων στο αίμα
Υπολογισμός 0.0666 when age ≥50 years + (0.042 x spleen) + 1.0956 when platelet count >1500 x x109/l + (0.0584 x blast cells) + 0.20399 when basophils >3% + (0.0413 x eosinophils) x 100
Προγνωστικές Ομάδες
Χαμηλού κινδύνου: ≤ 780Eνδιάμεσου κινδύνου: 781 – 1480Υψηλού κινδύνου: >1480
EUTOS score - Στην εποχή των αναστολέων τυροσινικής κινάσης
Παράμετροι Μέγεθος Σπληνός (σε cm, η μέγιστη από-σταση από το πλευρικό τόξο), Ποσοστό Βασεόφιλων στο αίμα
Υπολογισμός (7 x basophils) + (4 x spleen)
Προγνωστικές Ομάδες
Χαμηλού κινδύνου: ≤ 87Υψηλού κινδύνου: >87
Hammersmith score - Για την ανταπόκριση στους αναστολείς 2ης γενιάς
Παράμετροι Η καλύτερη κυτταρογενετική ανταπόκριση με το imatinib, ομάδα κινδύνου κατά So-kal, ουδετεροπενία υπό imatinib
Υπολογισμός Κυτταρογενετική ανταπόκριση: 1 – 94% Ph+ μεταφάσεις = 0 και ≥95% = 1
Ομάδα κινδύνου κατά Sokal: χαμηλού = 0, ενδιάμεσου ή υψηλού = 0.5
Ουδετεροπενία: απουσία = 0, βαθμού 3 και 4 = 1
Προγνωστικές Ομάδες
Χαμηλού κινδύνου: < 1.5Ενδιάμεσου κινδύνου: 1.5 – 2.5Υψηλού κινδύνου: >2.5

Προγνωστικοί παράγοντες στη ΧΜΛ και θεραπευτικές επιλογές 19
Prognostic factors in CML and treatment choicesby Maria Ν. Pagoni and Ifigeneia A. Tzannou
Department: Department of Hematology and Stem Cell Transplantation Unit, Evaggelismos General Hospital, Athens, Greece
ABSTRACT: The definition of prognostic factors is considered to be important for the selection of the optimal treatment modality and the prompt modification of treatment strategy. Two categories of prog-nostic factors can be established in CML. Those, that can be identified at the time of diagnosis (baseline) and those that can be assessed during treatment, and are usually relative to response. The most wide-ly used prognostic scoring systems are Sokal and Hasford scores. Even though both of these systems were established and validated before the introduction of TKIs, their predictive power has been proven in the TKI era. The EUTOS score has been recently developed for patients treated with imatinib, how-ever it still needs further evaluation in prospective clinical trials. Biological markers may be helpful in the timely recognision of those patients that are not going to respond to TKI therapy. Additional chromo-somal aberrations in the malignant clone, most importantly major-route ACAs, and gene profiling sig-
θεραπευτικές επιλογές αυξάνονται και τα αποτελέσματα βελτιώνονται τόσο αυξάνεται η δυσκολία λήψης θεραπευ-τικών αποφάσεων. ςε αυτό ακριβώς το σημείο η γνώση και αναγνώριση προγνωστικών παραγόντων για τον κα-θορισμό της βέλτιστης θεραπείας για τον κάθε ασθενή είναι όχι απλώς χρήσιμη αλλά αναγκαία (Πίνακες 1, 2).
Ήδη από τη δεκαετία του 1980 έγινε προσπάθεια ανα-γνώρισης προγνωστικών παραγόντων στη Χμλ. ορισμέ-νοι από αυτούς έχουν τεκμηριωμένη προγνωστική αξία,
ενώ για άλλους η αξία αυτή αμφισβητείται και χρήζει πε-ραιτέρω αξιολόγησης. κάποιοι είναι δυνατόν να προσ-διορισθούν εύκολα, ενώ άλλοι απαιτούν εξειδικευμένη τεχνολογία στην οποία δεν μπορούν να έχουν πρόσβαση όλοι. τελικά, αυτό που έχει σημασία είναι η βέλτιστη αξι-οποίηση όλων των διαθέσιμων προγνωστικών δεικτών, ώστε να προσφέρεται στον κάθε ασθενή η καλύτερη δι-αθέσιμη θεραπεία ή να άλλαζει η θεραπευτική στρατη-γική τη σωστή στιγμή.
Πίνακας 2. ΧΜΛ – Προγνωστικοί Παράγοντες
Πριν από την έναρξη θεραπείας
Βιοδείκτες σχετιζόμενοι με την ανταπόκριση στο imatinib
Σχετιζόμενοι με το επίπεδο ανταπόκρισης στο imatinib
Σχετιζόμενοι με την ανταπόκριση στους 2ης γενιάς TKIs
Σχετιζόμενοι με την ανταπόκριση σε 2ης γενιάς TKIs, ως 1ης γραμμής θεραπεία
Φάση νόσου Sokal score Hasford score EUTOS score Επιπρόσθετες
κυτταρογενετικές ανωμαλίες (ACAs)
Γονιδιακό προφίλ
Έκφραση OCT-1 Έκφραση P-gp
Φωσφορυλίωση Crkl
Μεταλλάξεις bcr-abl κινάσης
Επίπεδα του φαρμάκου στο πλάσμα
Αιματολογική ανταπόκριση στους 3 μήνες
Κυτταρογενετική ανταπόκριση στους 3, 6 και 12 μήνες
Μοριακή ανταπόκριση στους 3, 6, 12 και 18 μήνες
Δυσανεξία ή αντίσταση στο imatinib
Hammersmith score
Μεταλλάξεις bcr-abl κινάσης
PS κατά ECOG Κυτταρογενετική ανταπόκριση στους 3, 6 και 12 μήνες
Μοριακή ανταπόκριση στους 3, 6 και 12 μήνες
Κυτταρογενετική ανταπόκριση στους 3, 6 και 12 μήνες
Μοριακή ανταπόκριση στους 3, 6 και 12 μήνες

Μ.Ν. Παγώνη και Ι.Α. Τζάννου20
Βιβλιογραφία1. Goldman JM. How I treat chronic myeloid leukemia in the
imatinib era. Blood. 2007; 110:2828-2837.2. Goldman JM. Initial treatment for patients with CML.
Hematology. 2009; 453-460.3. Hochhaus A, O’Brien SG, Guilhot F, et al. Six year follow-up
of patients receiving imatinib for the first-line treatment of chronic myeloid leukemia. Leukemia. 2009; 213:1054-1061.
4. Hochhaus A. Prognostic factors in Chronic Myeloid Leukemia (CML) Onkologie. 2008; 31:576–578.
5. Hernandez-Boluda JC and Cervantes F. Prognostic factors in chronic myeloid leukemia. Best Pract Res Clin Haem. 2009; 22:343-353.
6. Baccarani μ, Saglio G, Goldman J, et al. Evolving concepts in the management of chronic myeloid leukemia: recom-mendations from an expert panel on behalf of the European LeukemiaNet. Blood. 2006; 108:1809-1820.
7. Baccarani M, Cortes J, Pane F, et al. Chronic myeloid leukemia: an update of concepts and management recom-mendations of European LeukemiaNet. J Clin Oncol. 2009; 27:6041-6051.
8. Jiang Q, Xu L-P, Liu D-H, et al. Imatinib mesylate versus allogeneic hematopoietic stem cell transplantation for patients with chronic myelogenous leukemia in the accelerated phase. Blood. 2011; 117:3032-3040.
9. Sokal JE, Cox EB, Baccarani M, et al. Prognostic discrimina-tion in “good-risk” chronic granulocytic leukemia. Blood. 1984; 63:789-799.
10. Hasford J, Pfirrmann M, Hehlmann R, et al. A new prog-nostic score for survival of patients with chronic myeloid leukemia treated with interferon alfa. Writing Committee for the Collaborative CML. Prognostic Factors Project Group. J Natl Cancer Inst. 1998; 90:850-858.
11. Hughes TP, Kaeda J, Branford S, et al. Frequency of major molecular responses to imatinib or interferon alpha plus cytarabine in newly diagnosed chronic myeloid leukemia. New Eng J Med. 2003; 349:142101432.
12. Rosti G, Trabacchi E, Bassi E, et al. Risk and early cytoge-netic response to imatinib and interferon in chronic myeloid leukemia. Hematologica. 2003; 88:256-259.
13. Simonsson B, on behalf of the IRIS study group. Beneficial effects of cytogenetic and molecular response on long term outcome in patients with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP) treated with imatinib (IM): update from the IRIS study [abstract] Blood. 2005; 106:52a.
14. Hughes T. ABL kinase inhibitor therapy for CML: Baseline assessments and response monitoring. Hematology. 2006; 221-218.
15. Hasford J, Baccarani M, Hoffmann V, et al. Predicting com-plete cytogenetic response and subsequent progression-free survival in 2060 patients with CML on imatinib treatment: the EUTOS score. Blood. 2011;118:686-692.
16. Marin D, Ibrahim AR, Goldman JM. European Treatment and Outcome Study (EUTOS) Score for Chronic Myeloid Leukemia Still Requires More Confirmation. J Clin Onc. 2011; 29:944-3945.
17. Kolomietz E, Al-Maghrabi J, Brennan S, et al. Primary chromosomal rearrangements of leukemia are frequently accompanied by extensive submicroscopic deletions and may lead to altered prognosis. Blood. 2001; 97:3581-3588.
18. Huntly BJ, Reid AG, Bench AJ et al. Deletions of the derivative chromosome 9 occur at the time of the Philadel-phia translocation and provide a powerful and independent prognostic indicator in chronic myeloid leukemia. Blood. 2001; 98:1732-1738.
19. Huntly BJ, Guilhot F, Reid AG, et al. Imatinib improves but may not fully reverse the poor prognosis of patients with CML with derivative chromosome 9 deletions. Blood. 2003; 102:2205-2212.
20. Dewald GW, Wyatt WA, Silver RT. Atypical BCR and ABL D-FISH patterns in chronic myeloid leukemia and their pos-sible role in therapy. Leuk Lymphoma. 1999; 34:481-491.
21. Huntly BJ, Bench A, Green AR. Double jeopardy from a
nature at the time of diagnosis may be useful in this way. Furthermore, the organic cation pump OCT-1 and P-glycoprotein expression, as well as phosphorylisation of Crkl may be predictive of the patient’s response to imatinib. However, they have not been incorporated in the clinical practice. Response to treatment in certain time milestones is the most important prognostic factor, and, among all prognostic factors, is the most powerful in predicting complete cytogenetic response (CCgR). The importance of the time and deapth of molecular response have also been evaluated. In a recent study, a single measurement of BCR-ABL transcript levels was found to be the best tool to identify patients with dismal prognosis, therefore allowing early intervention. BCR-ABL kinase domain mutations may result in suboptimal re-sponses or failure to imatinib, and are associated with poor prognosis regarding event free and overall survival. Screening for mutations is mandatory upon first line treatment failure, in order to make the op-timal therapeutic choice. In regard with second generation TKIs, the data concerning factors predictive of responce are limited. BCR-ABL kinase domain mutations and maintenance of CCgR are concidered to be the most important prognostic parameters. The introduction of second generation TKIs in first line treatment of CML has resulted in better outcomes. As far as prognostic factors are concerned, achieve-ment of CCgR and BCR-ABL transcript levels in certain timepoints remain crucial.

Προγνωστικοί παράγοντες στη ΧΜΛ και θεραπευτικές επιλογές 21
single translocation: Deletions of the derivative chromosome 9 in chronic myeloid leukemia. Blood. 2003; 102:1160-1168.
22. Castagnetti F, Testoni N, Luatti S, et al. Deletions of the De-rivative chromosome 9 do not influence the response and the outcome of chronic myeloid leukemia in early chronic phase treated with imatinib mesylate: GIMEMA CML Working Party analysis J Clin Onc. 2010; 28:2748-2754.
23. Kreil S, Pfirrmann M, Haferlach C, et al. Heterogeneous prognostic impact of derivative chromosome 9 deletions in chronic myelogenous leukemia. Blood. 2007; 110:1283-1290.
24. Quintas-Cardama A, Kantarjian H, Talpaz M, et al. Ima-tinib mesylate therapy may overcome the poor prognostic significance of deletions of derivative chromosome 9 in patients with chronic myelogenous leukemia. Blood. 2005; 105:2281-2286.
25. Fabarius A, Leitner A, Hochhaus A, et al. Impact of additional cytogenetic aberrations at diagnosis on prognosis of CML: long-term observation of 1151 patients from the randomized CML Study IV. Blood. 2011; 26:6760-6768.
26. Cortes JE, Talpaz M, Giles F, et al. Prognostic significance of cytogenetic clonal evolution in patients with chronic myelogenous leukemia on imatinib mesylate therapy. Blood. 2003; 101:3794-3800.
27. Schoch C, Haferlach T, Kern W, et al. Occurrence of addi-tional chromosome aberrations in chronic myeloid leukemia patients treated with imatinib mesylate. Leukemia. 2003; 17:461-463.
28. Verma D, Kantarjian H, Shan J, et al. Survival outcomes for clonal evolution in chronic myeloid leukemia patients on second generation tyrosine kinase inhibitor therapy. Cancer. 2010; 116:2673-2681.
29. Haferlach C, Bacher U, Schnittger S, et al. Similar patterns of chromosome abnormalities in CML occur in addition to the Philadelphia chromosome with or without tyosine kinase inhibitor treatment. Leukemia. 2010; 24:638-640.
30. El-Zimaity MMT, Kantarjian H, Talpaz M, et al. Results of imatinib mesylate therapy in chronic myelogenous leukae-mia with variant Philadelphia chromosome. Br J Haematol. 2004; 125:187-195.
31. Marzocchi G, Castagnetti F, Luatti S, et al. Variant Philadel-phia translocations: molecular-cytogenetic characterization and prognostic influence on frontline imatinib therapy, a GIMEMA Working Party on CML analysis. Blood. 2011; 117:6793-6800.
32. Radich JP, Dai H, Mao M, et al. Gene expression changes associated with progression and response in chronic myeloid leukemia. Proc Natl Acad Sci USA. 2006; 103:2794-2799.
33. Mc Weeney SK, Falinski R, Nowicki MO, et al. A gene expression signature of CD34+ cells to predict major cytoge-netic response in chronic phase chronic myeloid leukemia patients treated with imatinib. Blood. 2010; 115:315-325.
34. Oehler VG, Yeung KY, Choi YE, et al. The derivation of diagnostic markers of chronic myeloid leukemia progres-sion from microarray data. Blood. 2009; 114:3292-3298.
35. White D, Saunders V, Lyons B, et al. In vitro sensitivity to imatinib-induced inhibition of ABL kinase activity is
predictive of molecular response in patients with de novo CML Blood. 2005; 106:2520-2526.
36. Thomas J, Wang L, Clark RE, et al. Active transport of imatinib into and out of cells: implications for drug resist-ance. Blood. 2004; 104:3739-3745.
37. Crossman LC, Druker BJ, Deninger MW, et al. hOCT1 and resistance to imatinib. Blood. 2005; 106:1133-1134.
38. Wang L, Giannoudis A, Lane S, et al. Expression of the uptake drug transporter hOCT1 is an important clinical determinant of the response to imatinib in chronic myeloid leukemia. Clin Pharmacol Ther. 2008; 83:258-264.
39. White DL, Saunders VA, Dang P, et al. CML patients with low OCT-1 activity achieve better molecular responses on high dose imatinib than on standard dose. Those with high OCT-1 activity have excellent responses on either dose: a TOPS correlative study. Blood. 2008;112:Abstract 3187.
40. Dulucq S, Bouchet S, Turcq B, et al. Multidrug resistance gene (MDR1) polymorphisms are associated with major molecular responses ti standaed-dose imatinib in chronoc myeloid leukemia. Blood. 2008; 112:2024-2027.
41. Gorre ME, Mohammed M, Ellwood K, et al. Clinical resist-ance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001; 293:876-880.
42. Nicolini FE, Corm S, Le QH, et al. Mutation status and clinical outcome of 89 imatinib mesylate-resistant chronic myelogenous leukemia patients: a retrospective analysis from the French intergroup of CML. Leukemia. 2006; 20:1061-1066.
43. Branford S, Rudzki Z, Walsh S et al. Detection of BCR-ABL mutations in patients with CML treated with imatinib is virtually always accompanied by clinical resistance, and mutations in the ATP phoesphate binding loop (P-loop) are associated with a poor prognosis. Blood. 2003; 102:276-283.
44. Hughes TP, Branford S, Reynolds J, et al. Maintenance of imatinib dose intensity in the first six months of therapy for newly diagnosed patients with CML is predictive of molecular response, independent of the ability to increase dose at a later point. Blood. 2005; 106:Abstract 164.
45. Quintas-Cardama A, Kantarjian H, Jones D, et al. Delayed achievement of cytogenetic and molecular response is associ-ated with increased risk of progression among patients with chronic myeloid leukemia in early chronic phase receiving high-dose or standard-dose imatinib therapy. Blood. 2009; 113:6315-6321.
46. Kantarjian H, Talpaz M, O’Brien S, et al. Survival benefit with imatinib mesylate versus interferon-alpha-based regimens in newly diagnosed chronic phase chronic myelogenous leukemia. Blood. 2006; 108:1835-1840.
47. Marin D, Milojkovic D, Olavarria E, et al. European Leu-kemiaNet criteria for failure or suboptimal response reliably identify patients with CML in early chronic phase treated with imatinib whose eventual outcome is poor. Blood. 2008; 112:4437-4444.
48. Baccarani M, Guilhot F, Larson RA, et al. Outcomes by cytogenetic and molecular response at 12 and 18 months of imatinib in patients with newly diagnosed chronic myeloid

Μ.Ν. Παγώνη και Ι.Α. Τζάννου22
leukemia (CML) in Chronic Phase (CP) in the IRIS Trial. Blood. 2006; 108:Abstract 2138.
49. Hughes TP, Hochhaus A, Branford S, et al. Long-term prog-nostic significance of early molecular response to imatinib in newly diagnosed chronic myeloid leukemia: an analysis from the International Randomized Study of Interferon and STI571 (IRIS). Blood. 2010; 116:3758-3765.
50. Marin D, Ibrahim AR, Lucas C, et al. Assessment of BCR-ABL1 transcript levels at 3 months is the only requirement for predicting outcome for patients with chronic myeloid leukemia treated with tyrosine kinase inhibitors. J Clin Oncol. 2012; 30:232-238.
51. Hanfstein B, Muller MC, Erben P, et al. Molecular and cytogenetic response after 3 months of imatinib treatment is predictive of the risk of disease progression and death in newly diagnosed chronic myeloid leukemia patients: a follow-up analysis of the German CML study IV. Blood. 2011; 118: Abstract 783.
52. Picard S, Titier K, Etienne G, et al. Trough imatinib plasma levels are associated with both cytogenetic and molecular responses to standard-dose imatinibin chronic myeloid leukemia. Blood. 2007; 109:3496-3499.
53. Larson GA, Druker BJ, Guilhot F, et al. Imatinib pharma-cokinetics and its correlation with response and safety in chronic-phase chronic myeloid leukemia: a subanalysis of the IRIS study. Blood. 2008; 111:4022-4028.
54. Faber E, Friedecky D, Micova K, et al. Imatinib trough plasma levels do not correlate with the response to therapy in patients with chronic myeloid leukemia in routine clinical setting. Ann Haematol. Epub Jan 2012.
55. White D, Saunders V, Grigg A, et al. Measurement of in vivo BCR-ABL kinase inhibition to monitor imatinib-induced target blockade and predict response in chronic myeloid leukemia. J Clin Oncol. 2007; 25:4445-4451.
56. White DL, Saunders VA, Branford S, et al. The in-vivo level of imatinib induced kinase inhibition achieved in de-novo CML patients during the first 28 days of therapy is a powerful predictor of molecular outcome. Blood. 2005; 106: Abstract 436.
57. Saussele S, Lauseker M, Proetel U, et al. Second line therap;y with second generation TKI after intolerance to imatinib based treatments showed high overall survival in contrast to second line therapy after resistance; results of the randomized CML study IV. Blood. 2011; 118: Abstract 781.
58. Tam CS, Kantarjian H, Garcia-Manero G, et al. Failure to achieve a major cytogenetic response by 12 months defines inadequate response in patients receiving nilotinib or dasatinib as second or subsequent line therapy for chronic myeloid leukemia. Blood. 2008; 112:516-518.
59. Branford S, Lawrence R, Fletcher L, et al. The initial mo-
lecular response of chronic phase CML patients treated with second generation ABL inhibitor therapy after imatinib Failure can predict inadequate response and provide indications for rational mutation screening. Blood. 2008; 112: Abstract 331.
60. Hughes T, Saglio G, Branford S, et al. Impact of baseline BCR-ABL mutations on response to nilotinib in chronic myelogenous meukemia patients in chronic phase. J Clin Oncol. 2009; 27:4204-4210.
61. Muller M, Cortes J, Kim DW, et al. Dasatinib treatment of chronic-phase chronic myeloid leukemia: analysis of responses according to preexisting BCR-ABL mutations. Blood. 2009; 114:4944-4953.
62. Branford S, Melo JV and Hughes TP. Selecting optimal second-line tyrosine kinase inhibitor therapy for chronic myeloid leukemia patients after imatinib failure: does the BCR-ABL mutation status really matter? Blood. 2009; 114: 5426-5435.
63. Milojkovic D, Nicholson E, Apperley JF, et al. Early predic-tion of success or failure of treatment with second-generation tyrosine kinase inhibitors in patients with chronic myeloid leukemia. Hematologica. 2005; 92:224-231.
64. Jabbour E, Kantarjian H, O’Brien S, et al. Predictive factors for outcome and response in patients treated with second-generation tyrosine kinase inhibitorsforchronicmyeloidleu-kemiainchronic phase after imatinib failure. Blood. 2011; 117:1822-1827.
65. Jabbour E, Kantarjian H, O’Brien S, et al. The achievement of an early complete cytogenetic response is a major determinant for outcome in patients with early chronic phase chronic myeloid leukemia treated with tyrosine kinase inhibitors. Blood. 2011; 118:4541-4546.
66. Hochhaus A, Saglio G, Chuah C, et al. Dasatinib and imatinib-induced reductions in BCR-ABL transcript levels below 10% at 3 months are associated with improved responses in patients with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP): analysis of molecular response kinetics in the DASISION trial. Blood. 2011; 118: Abstract 2767.
67. Kantarjian H, Hochhaus A, Saglio G, et al. Nilotinib versus imatinib for the treatment of patients with newly diagnosed chronic phase . Philadelphia chromosome-positive chronic myeloid leukemia: a 24 month minimum follow-up of the phase 3 randomised ENESTnd trial. Lancet Oncol. 2011; 12:841-851.
68. Hughes TP, Kim DW, Etienne G, et al. The incidence of BCR-ABL mutations and their impact on outcome of pa-tients with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP) treated with nilotinib or imatinib in ENESTnd trial: a 36-month follow-up. Blood. 2011; 118:Abstract 2755.

Θεραπεία 1ης γραμμής σε ασθενείς με χρόνια μυελογενή λευχαιμία χρόνιας φάσης: Θεραπευτικές επιλογές
Κωνσταντίνος Τσαταλάς, Εμμανουήλ Σπανουδάκης, Ιωάννης Κοτσιανίδης
ΠΕΡΊληΨη: η χρήση του ιμ στην καθημερινή πράξη την τελευταία δεκαεπενταετία έχει επιφέρει επανάσταση στην αντιμετώπιση των ασθενών με χρόνια μυελογενή λευχαιμία (Χμλ). η πρόοδος αυ-τή οφείλεται στην ανακάλυψη των αναστολέων της κινάσης της τυροσίνης (Tyrosine Kinase Inhibi-tors-TKIs), που ο ρόλος τους είναι να αποσιωπήσουν τη λειτουργία της ογκοπρωτεΐνης BCR-ABL. το imatinib (IM) αποτέλεσε τον πρώτο τκι με τον οποίο επιτεύχθηκαν πλήρεις κυτταρογενετικές ανταπο-κρίσεις και βελτιωμένη συνολική επιβίωση στους ασθενείς με Χμλ. Παρ’ όλα αυτά, περίπου το 1/3 των ασθενών δεν είχαν την επιθυμητή έκβαση, εξαιτίας αντίστασης ή δυσανεξίας στο φάρμακο. οι ασθενείς αυτοί ενδεχομένως να ωφεληθούν από άλλες εναλλακτικές θεραπείες 1ης γραμμής. οι ασθενείς που εμ-φανίζουν καθυστερημένη ανταπόκριση στο ιμ διατρέχουν τον κίνδυνο εξέλιξης της νόσου. ο κίνδυ-νος αυτός ελαττώνεται εφόσον το χιμαιρικό γονίδιο BCR-ABL, που αποτελεί την αιτία της γενετικής αστάθειας, υποστεί ταχεία και ισχυρή αναστολή. Πρόσφατες μελέτες έδειξαν ότι με τη χρήση του da-satinib και nilotinib στη θεραπεία 1ης γραμμής επιτυγχάνονται ταχύτερες και βαθύτερες κυτταρογενε-τικές και μοριακές ανταποκρίσεις σε σχέση με το IM, και επιπρόσθετα μειώνεται ο κίνδυνος εξέλιξης της νόσου. με βάση τα αποτελέσματα αυτά, οι τκιs 2ης γενεάς αποτελούν πλέον εγκεκριμένες επιλο-γές θεραπείας 1ης γραμμής με την προοπτική ότι σύντομα η παρουσία τους θα μεταβάλλει τη θεραπευ-τική στρατηγική στους νεοδιαγνωσμένους ασθενείς με Χμλ. η απουσία δεδομένων σύγκρισης μεταξύ των δεν διευκολύνει την επιλογή του καταλληλότερου αναστολέα, η οποία θα εξατομικευθεί με βάση το προφίλ υγείας του ασθενούς, τις υπάρχουσες μεταλλάξεις και την αναμενόμενη τοξικότητα του κά-θε φαρμάκου. Ωστόσο, σημαντικά ζητήματα συζήτησης είναι: α) κατά πόσον οι πρώιμες κυτταρογενε-τικές και μοριακές ανταποκρίσεις θα μεταφρασθούν σε μακροχρόνιο όφελος επιβίωσης β) η έλλειψη μακροχρόνιων δεδομένων ασφάλειας και γ) το φαρμακευτικό κόστος, ιδιαίτερα ενόψει της επικείμενης κυκλοφορίας γενόσημης μορφής IM.
Haema 2012; 3(1): 23-36 Copyright EAE
Πανεπιστημιακή Αιματολογική κλινική Δημοκριτείου Παν/μίου ΘράκηςΔιεύθυνση Αλληλογραφίας: κωνσταντίνος τσαταλάς, καθηγητής Αι-
ματολογίας ιατρικού τμήματος Δημοκριτείου Πανεπιστημίου Θρά-κης, Πανεπιστημιακή Αιματολογική κλινική, Πανεπιστημιακό γενι-κό Νοσοκομείο Αλεξανδρούπολης, Δραγάνα, 68100 Αλεξανδρούπολη, τηλ.: 25510 76149 / Fax: 25510 76154, e-mail: [email protected]
Aνασκόπηση
ΕΙΣΑΓΩΓΗμέχρι τα μέσα του προηγούμενου αιώνα, η θεραπεία
της Χμλ περιορίζονταν στην παρηγορητική αντιμετώ-πιση της νόσου με σπληνική ακτινοβολία και λίγο αργό-τερα στη χορήγηση busulphan και hydroxyurea.1 με τα φάρμακα αυτά οι επιδημιολογικές μελέτες έδειχναν πο-σοστά πενταετούς επιβίωσης περίπου 24%. η εισαγω-γή στη θεραπεία της νόσου της interferon alpha (IFN-α) και της αλλογενούς μεταμόσχευσης στελεχιαίων αιμο-
ποιητικών κυττάρων (Αλλο-μΑκ) βελτίωσε την έκβα-ση της νόσου με ποσοστά 5ετούς επιβίωσης στο χρονικό διάστημα 1996-2004 περίπου 50%.2 την τελευταία δε-καετία η χορήγηση του εκλεκτικού αναστολέα της κινά-σης της τυροσίνης (τκι) imatinib (ιμ) (Glivec Novartis Pharmaceuticals), αρχικά σε ασθενείς με Χμλ και αντοχή στην IFN-α και ακολούθως ως θεραπείας πρώτης γραμ-μής, βελτίωσε δραματικά τη σχετιζόμενη με τη νόσο θνη-τότητα κατά 74%.2
IMATINIB (IM)To imatinib mesylate (Gleevec, STI-571), είναι μία
2-φαινυλαμινοπυριμιδίνη, η οποία αποτελεί εκλεκτικό και ισχυρό αναστολέα του BCR-ABL και μερικών άλλων

Κ. Τσαταλάς και συν24
κινασών της τυροσίνης στις οποίες περιλαμβάνονται ο c-kit, PDGF-R α και β και το ABL-σχετιζόμενο γονίδιο.3 Χορηγούμενο από το στόμα εμφανίζει βιοδιαθεσιμότητα 98% και χρόνο ημίσειας ζωής 13-16 ώρες.
η αποτελεσματικότητα του ιμ αξιολογήθηκε αρχικά σε ασθενείς με Χμλ που ήσαν ανθεκτικοί σε προηγη-θείσα θεραπεία με IFN-α. ςτη μελέτη αυτή πλήρη αιμα-τολογική ανταπόκριση (complete hematologic response, CHR) πέτυχε το 95% των ασθενών και μείζονα κυττα-ρογενετική ανταπόκριση (major cytogenetic response, MCyR), δηλαδή, συνδυασμό πλήρους και μερικής κυτ-ταρογενετικής ανταπόκρισης το 60%, με διάμεσο χρόνο παρακολούθησης 18 μηνών.4 ς’ αυτό το χρονικό διάστη-μα η νόσος δεν εξελίχθηκε σε επιταχυνόμενη φάση (ac-celerated phase, ΑΡ) ή και βλαστική μετατροπή (blastic phase, BP) στο 89% των ασθενών, ενώ το 95% αυτών παρέμειναν ζώντες. με βάση τα αποτελέσματα αυτά το ιμ εγκρίθηκε ως φάρμακο εκλογής στη θεραπεία 2ης γραμμής ασθενών με Χμλ σε δόση 400mg ημερησίως.
η κλινική ωστόσο αποτελεσματικότητα του ιμ εκτι-μήθηκε κυρίως στη διεθνή μελέτη φάσεως ιιι, την IRIS study (International Randomized Study of Interferon and STI571), στην οποία 1106 μη προθεραπευθέντες ασθε-νείς με Χμλ τυχαιοποιήθηκαν μεταξύ του ιμ σε δόση 400mg/ημέρα και του συνδυασμού IFN-α και cytarabine.5
μετά από διάμεσο χρόνο παρακολούθησης 12 μηνών, η συχνότητα πλήρους κυτταρογενετικής ανταπόκρισης (complete cytogenetic response, CCyR) στους ασθενείς που έλαβαν IM και συνδυασμό IFN-α+cytarabine ήταν 94% και 85%, αντίστοιχα. Έτσι, το IM έλαβε ένδειξη στη θεραπεία 1ης γραμμής της Χμλ. Πρόσφατα, ανακοινώ-θηκαν τα μακροχρόνια αποτελέσματα της μελέτης IRIS.6 μετά από 8 έτη παρακολούθησης, η συσσωρευτική CCyR ήταν 83% (18% των ασθενών απώλεσαν την CCyR και 3% εξελίχθηκαν σε επιταχυνόμενη ή βλαστική φάση), η ελεύθερη συμβαμάτων επιβίωση (event free survival, EFS) 81% και η ελεύθερη εξέλιξης της νόσου επιβίωση (progression free survival, PFS) 92%. ςτους ασθενείς που πέτυχαν μείζονα μοριακή ανταπόκριση (major molecu-lar response, MMR) στους 12 μήνες η PFS ήταν 100% στα 8 χρόνια. η συχνότητα εξέλιξης σε επιταχυνόμενη ή βλαστική φάση της νόσου ελαττώνονταν με την πάροδο του χρόνου και ήταν 1,5% (πρώτο έτος), 2,8% (δεύτερο έτος), 1,6% (τρίτο έτος), 0,9% (τέταρτο έτος), 0,5% (πέ-μπτο έτος), 0% (έκτο και έβδομο έτος) και 0,4% (όγδοο έτος). ςτα 8 έτη, η συνολική επιβίωση (overall survival, OS) ήταν 85% (93% λαμβάνοντας υπόψη μόνο τους θα-νάτους από Χμλ).
ΟΙ ΤΚΙs ΑπΟΤΕΛΟΥΝ θΕΡΑπΕΙΑ ΕΚΛΟΓΗΣ ΣΤΗ ΧΜΛ ΧΡΟΝΙΑΣ ΦΑΣΗΣ
η αλλογενής μεταμόσχευση στελεχιαίων αιμοποι-
ητικών κυττάρων (Αλλο-μΑκ) αποτελεί τη μοναδική θεραπευτική προσέγγιση που δυνητικά εξασφαλίζει ία-ση στη Χμλ με ποσοστά δεκαπενταετούς επιβίωσης 53% (φάση plateau μετά τα 2,5 έτη).7 Ωστόσο, αφενός η πλειονότητα των ασθενών δεν διαθέτει συμβατό δότη και αφετέρου η σχετιζόμενη με τη μεταμόσχευση νοση-ρότητα και θνητότητα, ακόμη και επί παρουσίας πλήρους συμβατού δότη, παραμένει υψηλή (θνητότητα των αρχι-κών 100 ημερών περίπου 15%).7,8 μία τυχαιοποιημένη μελέτη σύγκρισης της Αλλο-μΑκ με την καλύτερη δι-αθέσιμη φαρμακευτική αγωγή, βασισμένη στην ύπαρξη ή μη διαθέσιμου ιστοσυμβατού δότη, έδειξε υπεροχή της συμβατικής προσέγγισης.9 Ωστόσο, θα πρέπει να αναφερ-θεί ότι στο σκέλος της φαρμακευτικής αγωγής μόνο το 20% των ασθενών έλαβε ιμ, γεγονός που καταδεικνύει ότι η αποτελεσματικότητα αυτής της θεραπείας υποεκτι-μήθηκε. Από τη μελέτη IRIS δεν ήταν δυνατόν να δει-χθεί πλεονέκτημα επιβίωσης για το ιμ, δεδομένου ότι οι ασθενείς που δεν ανταποκρίνονταν στην IFN-α (90% των ασθενών μετά 9 μήνες) ελάμβαναν ιμ με αποτέλε-σμα να έχουν εξίσου καλή έκβαση με τους ασθενείς του σκέλους του ιμ.5,6 Έτσι, μέχρι πρόσφατα η υπεροχή του ιμ έναντι των θεραπειών με βάση την IFN-α (ιντερφερό-νη) προέκυπτε από αναδρομικές μελέτες σύγκρισης της επιβίωσης.10,11 τελευταία, δύο μελέτες αξιολόγησης της μακροχρόνιας έκβασης ασθενών με Χμλ που ελάμβα-ναν ιμ έδειξαν ότι η επιβίωσή τους ήταν παρόμοια με αυτή του γενικού πληθυσμού.12,13
ςυμπερασματικά, ευρέως αποδεκτή είναι σήμερα η άποψη ότι το ιμ (ή ένας από τους νεότερους τκιs που πρόσφατα εγκρίθηκαν στη θεραπεία 1ης γραμμής) αποτελεί τη θεραπεία εκλογής για τη χρόνια φάση της Χμλ, με μοναδική εξαίρεση τους πολύ νεαρούς ασθε-νείς (ηλικίας<20 ετών) με HLA-συμβατό αδελφό και χα-μηλό ΕΒμτ score μεταμοσχευτικού κινδύνου (European Group for Blood and Marrow Transplantation Risk As-sessment Score of 0).14
θΕΡΑπΕΥΤΙΚΟΙ ΣΤΟΧΟΙ ΣΤΗ θΕΡΑπΕΙΑ 1ης ΓΡΑΜΜΗΣ ΜΕ ΤΚΙs
η αναδρομική ανάλυση δεδομένων ασθενών με Χμλ που αντιμετωπίστηκαν τόσο με παλαιότερες συμβατικές θεραπείες, όπως η IFN, όσο και με ιμ έδειξε ότι η επί-τευξη CCyR εξασφαλίζει βελτίωση της συνολικής επιβί-ωσης (overal survival, OS).15,16 Έτσι, η CCyR αποτέλεσε τον ιδανικό θεραπευτικό στόχο. Επιπρόσθετα, η παρατή-ρηση ότι μεταξύ των ασθενών με CCyR εκείνοι που εμ-φάνιζαν την ελάχιστη μοριακά ανιχνεύσιμη νόσο είχαν μεγαλύτερη πιθανότητα διατήρησης της ανταπόκρισης στη θεραπεία, έφερε στο προσκήνιο την έννοια της μο-ριακής ανταπόκρισης. ςήμερα, θεωρούμε ως μείζονα μο-ριακή ανταπόκριση (major molecular response, MMR)

Θεραπεία 1ης γραμμής σε ασθενείς με ΧΜΛ χρόνιας φάσης: Θεραπευτικές επιλογές 25
την ελάττωση του “φορτίου” των BCR-ABL μεταγρά-φων κατά τρεις λογαρίθμους σε σχέση με το διαγνωστικό δείγμα (έκφραση των αποτελεσμάτων αναφορικά με το διεθνές πρότυπο, IRIS scale).17 Δεδομένης της μεταβλη-τότητας των μετρήσεων μεταξύ των διαφόρων διαγνω-στικών κέντρων τα αποτελέσματα εκφράζονται καλύτερα στα πλαίσια ενός διεθνούς προτύπου (International scale, IS), ώστε η MMR να αντιστοιχεί σε ένα λόγο του BCR-ABL προς το γονίδιο αναφοράς που είναι ≤0,1% (εκα-τοστιαία αναλογία έκφρασης BCR-ABL/ABLX100).18 Ως πλήρης μοριακή ανταπόκριση (complete molecular response, CMR) ορίζεται η μη ανίχνευση χιμαιρικών μεταγράφων BCR-ABL. Επειδή σε πολύ χαμηλά φορ-τία μεταγράφων προκύπτουν δυσκολίες στην ποσοτική τους έκφραση, συμβατικά ως CMR ορίζεται η ελάττωση του φορτίου κατά 4 τουλάχιστον λογαρίθμους (CMR4), ή κατ’ άλλους 4,5 (CMR4,5) ή και 5 (CMR5), σε σχέση με το διαγνωστικό δείγμα.18-20 Πρόσφατα, στην κλινική πράξη αντί της CMR προτείνεται η έννοια της μοριακής ανταπόκρισης (molecular response, MM) με τον προσ-διορισμό μμ4, μμ4,5, μμ5 ανάλογα με τους λογαρίθ-μους ελάττωσης του φορτίου των BCR-ABL μεταγράφων σε σχέση με το διαγνωστικό δείγμα (ενδελεχής ανάλυση του θέματος γίνεται στην ανασκόπηση “Εργαστηριακή παρακολούθηση ασθενών με Χμλ και κλινική αξιολό-γηση” του παρόντος τεύχους).
η προγνωστική σημασία της επίτευξης MMR στους ασθενείς που ήδη βρίσκονται σε CCyR αξιολογήθηκε σε 4 μεγάλης έκτασης μελέτες (CCyR έναντι CCyR+MMR σε Χμλ):21-24 α) η μακροχρόνια παρακολούθηση 476 ασθενών της μελέτης IRIS έδειξε ότι η πέραν της CCyR η επίτευξη μμR παρατείνει την EFS και ελαττώνει την πιθανότητα εξέλιξης της νόσου, χωρίς ωστόσο να έχει καμιά επίπτω-ση στη συνολική επιβίωση.21 με βάση τις παρατηρήσεις αυτές η τρέχουσα σύσταση του ELN (European Leuke-mia Net) για τη Χμλ είναι ότι η μη επίτευξη ή απώλεια της MMR δεν αποτελεί κριτήριο αποτυχίας της θεραπείας και συνεπώς καμία θεραπευτική παρέμβαση (λ.χ. αύξη-ση δόσης χορηγουμένου φαρμάκου ή αλλαγή σε νεότε-ρης γενεάς τκι) δεν είναι δικαιολογημένη.20,25 Ωστόσο, πολλοί ειδικοί στη Χμλ θεωρούν ότι η παρουσία CCyR χωρίς MMR μετά από 18 μήνες θεραπείας, που σύμφωνα με τις αναθεωρημένες οδηγίες του ELN χαρακτηρίζεται ως υποβέλτιστη ανταπόκριση (Suboptimal Response), θα πρέπει να προβληματίζει το θεράποντα ιατρό και να απο-τελεί ένδειξη στενότερης παρακολούθησης (λ.χ. Q-PCR κάθε 3 μήνες αντί 6μήνου).26 Επίσης, συνιστούν έλεγχο μεταλλάξεων στους ασθενείς που εμφανίζουν απώλεια MMR ή αύξηση “φορτίου” των BCR-ABL μεταγράφων (διπλασιασμό του κλάσματος BCR-ABL προς γονίδιο αναφοράς ή αύξηση κατά ένα λογάριθμο).23,27-29 Επιση-μαίνεται, ότι οι ιατροί της κοινότητας εκλαμβάνουν συ-χνά την παραπάνω σύσταση ως ένδειξη αλλαγής του τκι που χορηγείται στον ασθενή, μολονότι καμία μελέτη μέ-
χρι σήμερα δεν έδειξε ότι η αλλαγή θεραπείας σε ασθε-νείς με υποβέλτιστη ανταπόκριση (CCyR χωρίς MMR) θα μπορούσε να βελτιώσει την οS, PFS ή και EFS26 β) στη δεύτερη μελέτη οι de Lavallade και συν.22 (ομάδα Νοσοκομείου Hammersmith του λονδίνου) ανέλυσαν την έκβαση 204 ασθενών με νεοδιαγνωσμένη Χμλ που ελάμβαναν ιμ και διαπίστωσαν ότι ενώ η επίτευξη της CCyR στους 12 μήνες εξασφάλιζε καλύτερη PFS και OS, η παρουσία MMR δεν προσέθετε κανένα πλεονέκτημα γ) στη μελέτη του MD Anderson Cancer Center σε 276 ασθενείς23 με νεοδιαγνωσμένη Χμλ που έλαβαν θερα-πεία με ιμ παρατηρήθηκε ότι η επίτευξη MCyR στους 6 και 12 μήνες συσχετίζονταν στενά με την οS και PFS, ενώ η επίτευξη MMR στους ασθενείς που ήταν ήδη σε CCyR επηρέαζε την PFS αλλά όχι τη συνολική επιβίω-ση δ) παρόμοια αποτελέσματα αναφέρθηκαν από τη με-λέτη των Hehlmann και συν.24 (γερμανική ομάδα Χμλ): η επίτευξη MMR σε ασθενείς με CCyR δεν εξασφάλιζε κανένα επιπρόσθετο θεραπευτικό όφελος απ’ ότι η πα-ρουσία CCyR χωρίς μμR.
ςυμπερασματικά, από τα παραπάνω δεδομένα προ-κύπτει ότι: 1) η CCyR αποτελεί τον ιδανικό θεραπευτικό στόχο στη θεραπεία της Χμλ, δεδομένου ότι εξασφαλί-ζει πλεονέκτημα επιβίωσης 2) η επιβίωση των ασθενών με CCyR δεν εξαρτάται από το εάν αυτή ακολουθείται ή μη από την παρουσία μμR και 3) η επιπρόσθετη πα-ρουσία μμR ενδεχομένως ελαττώνει την πιθανότητα εμφάνισης συμβαμάτων (events) και, δευτερευόντως, με-τατροπής σε επιταχυνόμενη ή βλαστική φάση της νόσου.
η επίτευξη CMR σε οποιαδήποτε χρονική στιγμή κα-τά τη διάρκεια της θεραπείας, μολονότι δεν περιλαμβά-νεται στα χρονικά ορόσημα των συστάσεων του ELN για την αντιμετώπιση της Χμλ, φαίνεται ότι μειώνει ακόμη περισσότερο την πιθανότητα υποτροπής30,31 και κυρίως αποτελεί τη βασική προϋπόθεση διακοπής της θεραπεί-ας.32 Όπως θα αναφερθεί παρακάτω, η διακοπή της θερα-πείας με ιμ σε ασθενείς με σταθερή CMR, στα πλαίσια της μελέτης STIM (Stop Imatinib), έδειξε ενθαρρυντικά αποτελέσματα.33
ΒΕΛΤΙΩΣΗ θΕΡΑπΕΙΑΣ 1ης ΓΡΑΜΜΗΣ: ΕΙΝΑΙ ΕΦΙΚΤΗ ΚΑΙ ΑΝΑΓΚΑΙΑ;
μολονότι η αντιμετώπιση της χρόνιας φάσης της Χμλ με την καθιερωμένη δόση του ιμ άλλαξε ριζικά τη φυσική ιστορία του νοσήματος, αιτία προβληματισμού αποτέλεσε η διαπίστωση στις διαδοχικές αναλύσεις των αποτελεσμάτων της μελέτης IRIS (η τελευταία μετά 8 έτη παρακολούθησης) ότι το 1/3 των ασθενών δεν είχε την επιθυμητή έκβαση.6 Ειδικότερα, η απώλεια της CHR και CCyR αφορούσε στο 8% και 18% των ασθενών αντίστοι-χα, εξέλιξη σε επιταχυνόμενη ή βλαστική φάση στο 8% και διακοπή της θεραπείας ή μετακίνηση στο άλλο σκέ-

Κ. Τσαταλάς και συν26
λος της μελέτης εξαιτίας ανεπιθύμητων ενεργειών ή εργα-στηριακή τοξικότητα στο 6%. Επιπλέον, τα αποτελέσματα από τη χρήση του φαρμάκου εκτός κλινικών πρωτοκόλ-λων στην καθημερινή ιατρική πράξη –όπως η μελέτη του Νοσοκομείου Hammersmith με EFS 63% στα 5 έτη22 και μελέτη Northwestern Britain CCyR 50% στα 2 έτη–34 υπο-λείπονταν αυτών της μελέτης IRIS. με βάση τα παραπά-νω δεδομένα, η ανάγκη βελτίωσης των αποτελεσμάτων της θεραπείας 1ης γραμμής φάνηκε ότι είναι επιτακτική. Προς την κατεύθυνση αυτή υπάρχουν οι εξής επιλογές:
Χορήγηση υψηλών δόσεων ΙΜμετά τις αρχικές μελέτες φάσης ιι ενός σκέλους,35-38
που έδειξαν ότι με τη χορήγηση ιμ σε δόσεις 600-800mg ημερησίως επιτυγχάνονταν CCyR και μμR σε συντο-μότερο χρονικό διάστημα και υψηλότερα ποσοστά, ακο-λούθησαν δύο μεγάλες τυχαιοποιημένες μελέτες φάσεως ιιι.39,40 ςτη μελέτη TOPS (Tyrosine Kinase Inhibitor Op-timization and Selectively Study)39 συγκρίθηκε η αποτε-λεσματικότητα της υψηλής δόσης ιμ (800mg, ασθενείς: 319) με την καθιερωμένη δόση (400mg, ασθενείς: 157) σαν θεραπεία 1ης γραμμής σε ασθενείς με χρόνια φάση Χμλ. ςτους 12 μήνες, δεν παρατηρήθηκε καμία διαφο-ρά μεταξύ του σκέλους υψηλής δόσης και συμβατικής δόσης τόσο στη συχνότητα της CCyR (70% έναντι 66%) όσο και στην MMR (46% έναντι 40%), η οποία αποτε-λούσε και τον αρχικό καταληκτικό σημείο της μελέτης. Ωστόσο, οι ανταποκρίσεις ήταν ταχύτερες στους ασθε-νείς που έλαβαν υψηλή δόση ιμ με υψηλότερα ποσο-στά MMR στους 3 και 6 μήνες και CCyR στους 6 μήνες (57% έναντι 45%). ςημειώνεται, ότι η πιθανότητα αντα-πόκρισης ήταν συγκρίσιμη μεταξύ των διαφόρων υποομά-δων κινδύνου κατά Sokal score. μια πρόσφατη ανάλυση των αποτελεσμάτων της μελέτης έδειξε ότι στους 24 μή-νες τα ποσοστά της MMR δεν διέφεραν μεταξύ των δύο σκελών (54% έναντι 51%), όπως και η EFS (95% έναντι 95%), PFS (97% έναντι 98%) και OS (97% έναντι 98%). Επιπλέον, οι ανεπιθύμητες ενέργειες βαθμού ιιι/ιV ήταν συχνότερες στους ασθενείς που έλαβαν την υψηλότερη δόση του φαρμάκου (63,6% έναντι 33%). Παρόμοια τυ-χαιοποιημένη μελέτη φάσεως ιιι του ELN σύγκρινε την αποτελεσματικότητα 400mg και 800mg σαν θεραπεία 1ης γραμμής σε 216 ασθενείς υψηλού κινδύνου κατά So-kal score.40 τα ποσοστά επίτευξης CCyR στο 1ο έτος της θεραπείας, που αποτελούσε και τον αρχικό καταληκτικό στόχο της μελέτης, ήταν 64% και 58% για την υψηλή δό-ση (800mg) και συμβατική (400mg) δόση του ιμ αντί-στοιχα. Επίσης, δεν παρατηρήθηκε καμία διαφορά στην MMR (49% έναντι 41%), καθώς και στα ποσοστά των άλλων παραμέτρων όπως της OS στα 3 έτη (91% έναντι 84%), PFS (88% έναντι 86%) και EFS (62% έναντι 66%).
ςτη γερμανική μελέτη CML IV24 η σύγκριση των δύο
δοσολογικών σχημάτων ιμ (800mg και 400mg) έδειξε ότι η μμR (αρχικό καταληκτικό σημείο) επιτυγχάνο-νταν ταχύτερα και σε υψηλότερα ποσοστά στους ασθενείς που ελάμβαναν 800mg ιμ (59% έναντι 44%, p=0,0003). Ωστόσο, η ταχύτερη επίτευξη ανταπόκρισης δεν συνεπά-γονταν πλεονέκτημα στην οS και PFS με ποσοστά 95% και 94% αντίστοιχα.
Πρόσφατα δημοσιεύθηκε μία συστηματική ανασκό-πηση και μετα-ανάλυση των τυχαιοποιημένων μελετών σύγκρισης της καθιερωμένης ημερήσιας δόσης των 400 mg ιμ με τις υψηλότερες δόσεις σε ασθενείς με νεοδια-γνωσθείσα Χμλ σε χρόνια φάση.41 η ανάλυση βασίσθη-κε στις τυχαιοποιημένες μελέτες τοPS,39 γερμανική IV,24 γαλλική SPIRIT42 και του ELN.40 οι 3 πρώτες μελέτες συμπεριελάμβαναν ασθενείς όλων των ομάδων κινδύνου, ενώ η μελέτη του ELN μόνο ασθενείς υψηλού κινδύνου. η μετα-ανάλυση έδειξε ότι στους 12 μήνες το υψηλότε-ρο δοσολογικό σχήμα βελτίωσε τα ποσοστά της CCyR και μμR. Ωστόσο, κατά τη διάρκεια της συνολικής πα-ρακολούθησης των ασθενών δεν παρατηρήθηκε καμία διαφορά στη θνητότητα από κάθε αιτία ή την πιθανότη-τα εξέλιξης της νόσου. ςημειώνεται, ότι οι ανεπιθύμητες ενέργειες που επέβαλαν τη διακοπή της θεραπείας ήταν συχνότερες στο σκέλος των υψηλών δόσεων. Tο βασικό συμπέρασμα της μετα-ανάλυσης ήταν ότι προς το παρόν δεν υπάρχουν επαρκείς ενδείξεις που να δικαιολογούν τη χρήση των υψηλών δόσεων ιμ στη θεραπεία 1ης γραμ-μής ασθενών με Χμλ χρόνιας φάσης.
Συνδυασμός ΙΜ και IFNτα λευχαιμικά στελεχιαία κύτταρα, που εμφανίζουν
ενδογενή αντίσταση στους τκιs, θα μπορούσαν να στο-χοποιηθούν πιο αποτελεσματικά από το συνδυασμό IFN και ενός τκι. ςτην κατεύθυνση αυτή τα πλέον υποσχό-μενα αποτελέσματα προέρχονται από τη γαλλική μελέτη SPIRIT (STI571 Prospective Randomized Trial),42 στην οποία συγκρίθηκαν 400mg IM (159 ασθενείς) με 400mg IM+pegylated IFN (157 ασθενείς). ςτους 12 μήνες τα πο-σοστά επίτευξης μμR και CMR4 ήταν υψηλότερα στην ομάδα του συνδυασμού ιμ + pegylated IFN (μμR 57% έναντι 38%, CMR4 30% έναντι 14%). Επισημαίνεται, ότι το 45% των ασθενών στην ομάδα του συνδυασμού διέ-κοψε τη θεραπεία εξαιτίας τοξικότητας. Παρόμοια απο-τελέσματα προέκυψαν με τη χρησιμοποίηση αναλόγου συνδυασμού IM + pegylated IFN στη μελέτη της ςκανδι-ναβικής ομάδας (Νordic CML Study Group).43 Αντίθετα, στη γερμανική CML IV μελέτη η σύγκριση των 400mg IM με το συνδυασμό ιμ και συμβατικής IFN δεν έδειξε καμία διαφορά.24 οι διαφορές που παρατηρήθηκαν στα αποτελέσματα των παραπάνω μελετών ενδεχομένως να ερμηνεύονται από τη διαφορετική δραστικότητα της pe-gylated έναντι της συμβατικής IFN. Ωστόσο, επειδή η

Θεραπεία 1ης γραμμής σε ασθενείς με ΧΜΛ χρόνιας φάσης: Θεραπευτικές επιλογές 27
τοξικότητα προκαλεί συχνές διακοπές της θεραπείας δεν προέκυψε σημαντική διαφορά στην ΕFS, PFS και οS με-ταξύ των δύο σκελών θεραπείας (IM έναντι συνδυασμού IM + IFN), ακόμη και στις περιπτώσεις που παρατηρή-θηκε διαφορά στην MMR και CMR.
ςυμπερασματικά, προς το παρόν, δεν υπάρχει ομο-φωνία για τη χρησιμότητα των συνδυασμών τκι και IFN στη θεραπεία 1ης γραμμής της Χμλ χρόνιας φάσης. το πλεονέκτημα της πρώϊμης ανταπόκρισης από το συν-δυασμό ιμ+ιFN δεν είναι βέβαιο ότι βελτιώνει μακρο-πρόθεσμα την έκβαση της νόσου, ενώ η τοξικότητα του συνδυασμού είναι αξιοσημείωτη. οι παραπάνω θεραπευ-τικές προσεγγίσεις πρέπει να συγκριθούν με τους TKIs 2ης γενεάς στη θεραπεία 1ης γραμμής, στα πλαίσια τυχαι-οποιημένων μελετών.
Νεότερης γενεάς ΤΚΙsοι πρώτοι δύο νεότεροι αναστολείς της κινάσης της
τυροσίνης που χρησιμοποιήθηκαν στη θεραπεία της Χμλ και φέρονται με την ονομασία τκιs 2ης γενεάς είναι το dasatinib και nilotinib.
το dasatinib (BMS-354825, Sprycel, Bristol-Myers Squibb) είναι δραστικός αναστολέας των κινασών bcr-abl, SFK, ephrin receptor kinases, PDGFR και kit. Επι-πλέον, το dasatinib συνδέεται και με άλλες κινάσες της τυροσίνης, σερίνης και θρεονίνης, όπως κινάσες της οι-κογένειας TEC, της mitogen-activated protein και του υποδοχέα της κινάσης της τυροσίνης, discoid in domain receptor 1.44 το dasatinib, σε αντίθεση με το IM, μπορεί να προσδεθεί στο ενεργό κέντρο της ABL κινάσης είτε βρίσκεται σε ενεργοποιημένη είτε σε αδρανή διαμόρφω-ση, καθώς επίσης και όταν υπάρχουν σημειακές μεταλ-λάξεις, εκτός της T315I, που επιφέρουν αντίσταση στο IM. ςε in vitro μελέτες BCR-ABL+ κυτταρικών σειρών παρουσιάζει 325 φορές μεγαλύτερη δραστικότητα σε σύ-γκριση με το IM.45,46
To nilotinib (Tasigna, AMN107, Novartis Pharma), το από του στόματος χορηγούμενο παράγωγο της αμινοπυ-ριμιδίνης, αποτελεί νεότερο, δεύτερης γενεάς εκλεκτικό αναστολέα της κινάσης της τυροσίνης, με δραστικότητα 10-50 φορές ισχυρότερη του IM.47 η ανακάλυψη του ni-lotinib προέκυψε στην προσπάθεια βελτίωσης των φαρ-μακοκινητικών ιδιοτήτων του IM, με την αντικατάσταση μίας ομάδας μεθυλπιπεραζίνης. το nilotinib, εκτός της ABL κινάσης αναστέλλει τη δραστικότητα των Αrg, Kit και υποδοχέων των PDGF α και β, αλλά καθόλου των κι-νασών της Src οικογένειας.44 με εξαίρεση τη μετάλλαξη T315I, είναι δραστικό έναντι των περισσοτέρων ανθεκτι-κών στο IM μεταλλάξεων.
η χορήγηση των TKIs δεύτερης γενεάς στη θεραπεία 1ης γραμμής της Χμλ χρόνιας φάσης βασίσθηκε α) στην ισχυρότερη σε σχέση με το ιμ in vitro δραστικότητα τους
β) τη μικρότερη μεταλλαξιογόνο δράση τους και γ) στην αποτελεσματική και ασφαλή δράση τους στη θεραπεία 2ης γραμμής σε ασθενείς με αντίσταση ή δυσανεξία στο ιμ. ςτην κατεύθυνση αυτή έχουν πραγματοποιηθεί 3 με-λέτες φάσεως ιι με ένα σκέλος θεραπείας που περιελάμ-βαναν nilotinib ή dasatinib.48-50 και οι 3 μελέτες έδειξαν ταχεία επίτευξη κυτταρογενετικών και μοριακών αντα-ποκρίσεων με ποσοστά CCyR στους 6 μήνες μεγαλύτε-ρα των 90%. τα ποσοστά της MMR στους 12 μήνες ήταν 71% για τους ασθενείς που έλαβαν dasatinib48 και 81% και 85% για τους ασθενείς υπό θεραπεία με nilotinib στις μελέτες του MD Anderson49 και GIMEMA50 αντίστοιχα.
με σκοπό την επιβεβαίωση των παραπάνω αποτε-λεσμάτων τα δύο φάρμακα συγκρίθηκαν με το ιμ στα πλαίσια τυχαιοποιημένων μελετών φάσεως ιιι. ςτην Da-satinib versus Imatinib Study IΝ Treatment-Naïve CML (DASISION) μελέτη51 το dasatinib σε δόση 100mg ημε-ρησίως συγκρίθηκε με το ιμ σε δόση 400mg ημερησί-ως, ενώ στην Evaluating Nilotinib Efficacy and Safety in Clinical τrials-Newly Diagnosed Patients (ENESTnd) μελέτη52 συγκρίθηκαν δύο δοσολογικά σχήματα του ni-lotinib (400mg δις ημερησίως και 300mg δις ημερησίως) με ιμ 400 mg ημερησίως. και στις δύο μελέτες παρατη-ρήθηκε υπεροχή του πειραματικού σκέλους ως προς το αρχικό καταληκτικό σημείο της μελέτης, το οποίο στη DA-SISION ήταν επιβεβαιωμένη CCyR στους 12 μήνες και στην ENESTnd MMR στους 12 μήνες. η υπεροχή αυτή επιβεβαιώθηκε και με τα αποτελέσματα της πρόσφατης ανάλυσης της ENESTnd μετά 36 μήνες53 και της DASI-SION μετά 24 μήνες παρακολούθησης54 (Πίνακας 1). οι ασθενείς που έλαβαν nilotinib εμφάνισαν σημαντικά ελατ-τωμένο κίνδυνο εξέλιξης της νόσου, ενώ παρόμοια, στα-τιστικώς σημαντική, διαφορά δεν προέκυψε στη μελέτη DASISION. Αναφορικά με την τοξικότητα, στους 36 μή-νες παρακολούθησης της ENESTnd μελέτης επιβεβαιώ-θηκε ότι η δόση του nilotinib 300mg δις ημερησίως ήταν καλώς ανεκτή με χαμηλότερα ποσοστά αιματολογικής το-ξικότητας και εργαστηριακών ανωμαλιών σε σχέση με το nilotinib 400mg δις ημερησίως και την καθιερωμένη δό-ση του ιμ 400mg ημερησίως.53,55 οι αιματολογικές ανε-πιθύμητες ενέργειες που προκαλούνται από το dasatinib ήταν συχνότερες (22% βαθμού 3/4 θρομβοπενία έναντι 10% από ιμ), καθώς και οι πλευριτικές συλλογές που εγ-γίζουν το 16% (κυρίως βαθμού 1-2 και <1% βαθμού 3)54,55 (Πίνακας 2). με βάση τα αποτελέσματα των παραπάνω μελετών το nilotinib και dasatinib εγκρίθηκαν ως φάρ-μακα 1ης γραμμής για την αντιμετώπιση των ασθενών με Χμλ, τόσο στις ηΠΑ όσο και στις Ευρωπαϊκές χώρες.
ςυμπερασματικά, το nilotinib και dasatinib προκαλούν ταχεία ελάττωση του λευχαιμικού φορτίου της νόσου σε ασθενείς με Χμλ, με αποτέλεσμα αυξημένα ποσοστά επίτευξης CCyR μετά 3 μήνες θεραπείας και ελάττωση του ποσοστού ασθενών με αποτυχία ή υποβέλτιστη αντα-πόκριση στους 12 μήνες, σύμφωνα με τις συστάσεις του
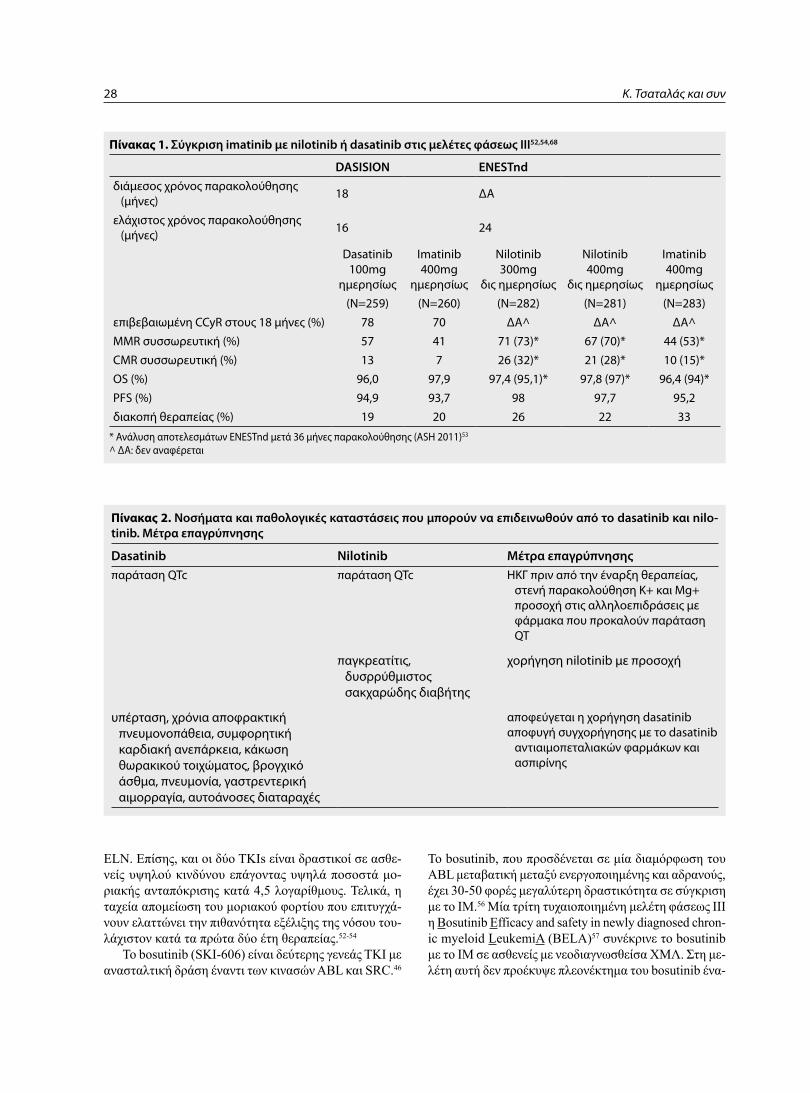
Κ. Τσαταλάς και συν28
ELN. Επίσης, και οι δύο τκιs είναι δραστικοί σε ασθε-νείς υψηλού κινδύνου επάγοντας υψηλά ποσοστά μο-ριακής ανταπόκρισης κατά 4,5 λογαρίθμους. τελικά, η ταχεία απομείωση του μοριακού φορτίου που επιτυγχά-νουν ελαττώνει την πιθανότητα εξέλιξης της νόσου του-λάχιστον κατά τα πρώτα δύο έτη θεραπείας.52-54
το bosutinib (SKI-606) είναι δεύτερης γενεάς τκι με ανασταλτική δράση έναντι των κινασών ABL και SRC.46
το bosutinib, που προσδένεται σε μία διαμόρφωση του ABL μεταβατική μεταξύ ενεργοποιημένης και αδρανούς, έχει 30-50 φορές μεγαλύτερη δραστικότητα σε σύγκριση με το ιμ.56 μία τρίτη τυχαιοποιημένη μελέτη φάσεως ιιι η Bosutinib Efficacy and safety in newly diagnosed chron-ic myeloid LeukemiA (BELA)57 συνέκρινε το bosutinib με το ιμ σε ασθενείς με νεοδιαγνωσθείσα Χμλ. ςτη με-λέτη αυτή δεν προέκυψε πλεονέκτημα του bosutinib ένα-
πίνακας 1. Σύγκριση imatinib με nilotinib ή dasatinib στις μελέτες φάσεως ΙΙΙ52,54,68
DASISION ENESTndδιάμεσος χρόνος παρακολούθησης
(μήνες) 18 ΔΑ
ελάχιστος χρόνος παρακολούθησης (μήνες) 16 24
Dasatinib 100mg
ημερησίως
Imatinib 400mg
ημερησίως
Nilotinib 300mg
δις ημερησίως
Nilotinib 400mg
δις ημερησίως
Imatinib 400mg
ημερησίως(N=259) (N=260) (N=282) (N=281) (N=283)
επιβεβαιωμένη CCyR στους 18 μήνες (%) 78 70 ΔΑ^ ΔΑ^ ΔΑ^MMR συσσωρευτική (%) 57 41 71 (73)* 67 (70)* 44 (53)*CMR συσσωρευτική (%) 13 7 26 (32)* 21 (28)* 10 (15)*OS (%) 96,0 97,9 97,4 (95,1)* 97,8 (97)* 96,4 (94)*PFS (%) 94,9 93,7 98 97,7 95,2διακοπή θεραπείας (%) 19 20 26 22 33
* Ανάλυση αποτελεσμάτων ENESTnd μετά 36 μήνες παρακολούθησης (ASH 2011)53
^ ΔΑ: δεν αναφέρεται
πίνακας 2. Νοσήματα και παθολογικές καταστάσεις που μπορούν να επιδεινωθούν από το dasatinib και nilo-tinib. Μέτρα επαγρύπνησης
Dasatinib Nilotinib Μέτρα επαγρύπνησηςπαράταση QTc παράταση QTc ΗΚΓ πριν από την έναρξη θεραπείας,
στενή παρακολούθηση K+ και Mg+ προσοχή στις αλληλοεπιδράσεις με φάρμακα που προκαλούν παράταση QT
παγκρεατίτις, δυσρρύθμιστος σακχαρώδης διαβήτης
χορήγηση nilotinib με προσοχή
υπέρταση, χρόνια αποφρακτική πνευμονοπάθεια, συμφορητική καρδιακή ανεπάρκεια, κάκωση θωρακικού τοιχώματος, βρογχικό άσθμα, πνευμονία, γαστρεντερική αιμορραγία, αυτοάνοσες διαταραχές
αποφεύγεται η χορήγηση dasatinib αποφυγή συγχορήγησης με το dasatinib
αντιαιμοπεταλιακών φαρμάκων και ασπιρίνης

Θεραπεία 1ης γραμμής σε ασθενείς με ΧΜΛ χρόνιας φάσης: Θεραπευτικές επιλογές 29
ντι του ιμ ως προς το αρχικό καταληκτικό σημείο που ήταν η επίτευξη CCyR στους 12 μήνες, με αποτέλεσμα το bosutinib να μην εγκριθεί από τον FDA ως φάρμακο 1ης γραμμής για τη Χμλ. ςημειώνεται, ότι τα απογοη-τευτικά αποτελέσματα ενδεχομένως να οφείλονται στις συχνές διακοπές του φαρμάκου εξαιτίας διαρροιών, οι οποίες αποτελούν συνήθη ανεπιθύμητη ενέργεια του bo-sutinib και θα μπορούσαν να αντιμετωπιστούν με καταλ-ληλότερη υποστηρικτική αγωγή.58
ΑΝΑζΗΤΗΣΗ ΤΗΣ ΚΑΛΥΤΕΡΗΣ θΕΡΑπΕΥΤΙΚΗΣ ΣΤΡΑΤΗΓΙΚΗΣ
τα εντυπωσιακά αποτελέσματα από τις μελέτες φά-σεως ιι και ιιι, στις οποίες οι TKIs 2ης γενεάς χρησιμο-ποιήθηκαν ως θεραπεία 1ης γραμμής στη χρόνια φάση της Χμλ, θέτουν επιτακτικά το ερώτημα εάν επιβάλ-λεται η αλλαγή στη στρατηγική της αντιμετώπισης των ασθενών αυτών. ςυγκεκριμένα, ο προβληματισμός ανα-φέρεται στις παρακάτω δύο επιλογές α) παραμένει το ιμ ως αρχική θεραπεία, με τη δυνατότητα αλλαγής σε TKIs 2ης γενεάς επί αποτυχίας ή δυσανεξίας του φαρμάκου και β) όλοι οι ασθενείς λαμβάνουν εξ αρχής TKIs 2ης γενε-άς. Αρκετά σημεία σχετικά με το παραπάνω ερώτημα θα πρέπει να συζητηθούν:
1) τα αποτελέσματα της θεραπείας με το ιμ μετά από 8 έτη παρακολούθησης είναι εξαιρετικά, γεγονός που επι-βεβαιώνει τη σταθερότητα των ανταποκρίσεων, την άρι-στη ανοχή του φαρμάκου στην πλειονότητα των ασθενών και την απουσία απρόβλεπτων ανεπιθύμητων ενεργειών μετά από μακροχρόνια χρήση.6 Αποτελεί η καθιέρωση των TKIs 2ης γενεάς ως θεραπείας 1ης γραμμής συνετή πράξη χωρίς να υπάρχουν διαθέσιμα μακροχρόνια απο-τελέσματα ασφάλειας;
2) με βάση την εμπειρία από τη μελέτη IRIS, 30-35% των ασθενών που λαμβάνουν ιμ θα χρειαστούν σε κά-ποια χρονική στιγμή αλλαγή θεραπείας.6,59 η χορήγηση ενός TKI 2ης γενεάς στους ασθενείς που ανέπτυξαν αντί-σταση στο ιμ θα έχει ως αποτέλεσμα την επίτευξη CCyR στο 50% και διετή PFS στο 64-82%.60,61 Επομένως, με-τά την αποτυχία της θεραπείας με ιμ θα διασωθεί τελι-κά το 30-40% των ασθενών.32 Αναφορικά δε με την EFS των ασθενών που αντιμετωπίζονται μία διαδοχική χορή-γηση TKIs, προκύπτει η έννοια της πραγματικής (actual) ή τρέχουσας EFS (current event-free survival, CEFS) κα-θώς ο συμβατικός ορισμός της EFS θεωρεί ότι ένα σύμ-βαμα (event) (λ.χ. μετατροπή της νόσου) συντελείται αμετάκλητα, χωρίς ωστόσο να λαμβάνει υπ’ όψη το γε-γονός ότι ο ασθενής, μετά από αποτυχία στο ιμ, μπορεί να αντιμετωπιστεί επιτυχώς με ένα διαφορετικό TKI.62,63 Έτσι, η EFS μετά θεραπεία με ιμ είναι 81% στα 7 έτη,32 ενώ για το ίδιο χρονικό διάστημα η CEFS είναι 88%.63
3) τις παραπάνω απόψεις ενισχύουν τα αποτελέσματα
πρόσφατης μελέτης παρατήρησης πέντε Βρετανικών κέ-ντρων που έδειξαν ότι η επιβίωση των ασθενών που έλα-βαν TKIs 2ης γενεάς, μετά αποτυχία της θεραπείας με ιμ και επέτυχαν CCyR ήταν παρόμοια με αυτή των ασθενών που ανταποκρίθηκαν εξαρχής στο ιμ.64
4) μία εναλλακτική προσέγγιση αποτελεί η Αυστρα-λιανή μελέτη TIDEL II65 στην οποία νεοδιαγνωσθέντες ασθενείς σε χρόνια φάση Χμλ λαμβάνουν αρχικά ιμ 600mg ημερησίως και ακολούθως ιμ 800mg ημερησί-ως ή nilotinib 400mg δις ημερησίως εάν αποτύχουν δια-δοχικά στην επίτευξη πολύ φιλόδοξων μοριακών στόχων που περιλαμβάνουν επίπεδα BCR-ABL μεταγράφων 10% στους 3 μήνες, ≤1% (ισοδύναμο της CCyR) στους 6 μή-νες και ≤0,1% (ισοδύναμο της MMR) στους 12 μήνες θεραπείας. Επίσης, αλλαγή της θεραπείας σε nilotinib ακολουθεί την εμφάνιση μη αιματολογικής τοξικότητας βαθμού III/IV ή απώλεια της ανταπόκρισης στο ιμ. τα αποτελέσματα της μελέτης με διάμεσο χρόνο παρακο-λούθησης 30 μήνες έδειξαν ότι 35 από τους 105 ασθενείς (32%) άλλαξαν θεραπεία σε nilotinib με ποσοστά MMR 66% και 81% και CMR 12% και 24% στους 12 και 24 μήνες αντίστοιχα. ςημειώνεται ότι τα παραπάνω αποτε-λέσματα είναι συγκρίσιμα με αυτά που παρατηρήθηκαν με τη χορήγηση nilotinib και dasatinib στις μελέτες EN-ESTnd και DASISION.
5) οι μελέτες φάσεως ιιι (DASISION και ΕΝΕSτnd) έδειξαν ότι το dasatinib και nilotinib εξασφαλίζουν υψη-λότερα ποσοστά CCyR και MMR και σε συντομότερο χρονικό διάστημα από ότι το ιμ. Ωστόσο, για να δειχθεί ότι το πλεονέκτημα στα ποσοστά ανταποκρίσεων διατη-ρείται με την πάροδο του χρόνου απαιτείται πιο μακρο-χρόνια παρακολούθηση. η στατιστικά σημαντική όμως ελάττωση στη συχνότητα μετατροπής της νόσου σε επι-ταχυνόμενη ή βλαστική φάση που παρατηρήθηκε στη μελέτη ENESTnd53 αποτελεί μία πρώτη ισχυρή ένδειξη βελτίωσης της μακροχρόνιας έκβασης της νόσου με τους πιο ισχυρούς νεότερης γενιάς TKIs. ςημειώνεται, ότι η μετατροπή της νόσου στη Χμλ προκαλείται από δευτε-ρογενή τυχαία (stochastic) γενετικά συμβάματα σε γε-νωμικώς ασταθή BCR-ABL θετικά κύτταρα.46,66,67 κατά συνέπεια, η αποτελεσματική πρόληψη της εξέλιξης της νόσου απαιτεί έντονη ελάττωση του αριθμού αυτών των κυττάρων (βάθος ανταπόκρισης), που συντελείται ταχέ-ως (ταχύτητα ανταπόκρισης) και είναι διατηρήσιμη (δι-άρκεια ανταπόκρισης).46,67
6) Επειδή η Χμλ αποτελεί νόσημα με σχετικά μακρά κλινική πορεία απαιτείται μακροχρόνια παρακολούθηση για να καταδειχθεί ότι οι νεότεροι TKIs εξασφαλίζουν πλεονέκτημα συνολικής επιβίωσης. Έτσι, μολονότι τα πρόσφατα αποτελέσματα της μελέτης ENESTnd μετά 36 μήνες παρακολούθησης δεν έδειξαν διαφορά στη OS, φαίνεται ότι η θεραπεία με nilotinib συγκρινόμενη με το ιμ συνοδεύεται από μικρότερο αριθμό προκαλουμένων από τη νόσο θανάτων.53,68

Κ. Τσαταλάς και συν30
7) Αντικείμενο προβληματισμού και στις δύο μελέ-τες, ENESTnd και DASISION, αποτελεί το γεγονός ότι 25% των ασθενών από το πειραματικό σκέλος διέκοψαν τη θεραπεία.69 η ανοχή των τριών τκιs φαίνεται ότι είναι συγκρίσιμη και η συχνότητα διακοπής λόγω ανεπιθύμη-των ενεργειών παρόμοια. Ωστόσο, αρκετοί ασθενείς δι-έκοψαν το χορηγούμενο φάρμακο εξαιτίας των ορισμών αποτυχίας της θεραπείας και υποβέλτιστης ανταπόκρισης που υιοθετήθηκαν στις μελέτες αυτές με βάση την εμπε-ρία της μελέτης IRIS. Ανοιχτό παραμένει το ερώτημα αν οι ορισμοί αυτοί έχουν την ίδια σημασία σε ασθενείς που λαμβάνουν τκιs 2ης γενεάς σαν θεραπεία 1ης γραμμής.
8) ςτις ίδιες μελέτες52-54, η EFS των ασθενών χαμη-λού κινδύνου κατά Sokal και Hasford score ήταν εξαιρε-τική, εύρημα το οποίο είναι υποδηλωτικό ότι οι ασθενείς αυτοί θα μπορούσαν με ασφάλεια να λάβουν αγωγή με φθηνότερα φάρμακα. Άλλωστε το κόστος της θεραπείας με τκιs, η οποία χορηγείται εφόρου ζωής, θα αποτελέσει σύντομα σημαντικό ζήτημα για τις υπηρεσίες υγείας διε-θνώς, δεδομένου ότι το γενόσημο σκεύασμα ιμ θα δια-τίθεται στη φαρμακευτική αγορά από το 2015, σε τιμές 10-50 φορές χαμηλότερες.70 Ωστόσο, με δεδομένο ότι οι ασθενείς που λαμβάνουν TKIs 2ης γενεάς επιτυγχάνουν υψηλότερα ποσοστά πλήρους μοριακής ανταπόκρισης, το ενδεχόμενο διακοπής της θεραπείας, όπως υποδηλώνεται και από τη μελέτη STIM (Stop-ιmatinib),33 θα συμβάλλει στην ελάττωση του θεραπευτικού κόστους. Επιπρόσθετα, η θεραπευτική στρατηγική της εφόδου με TKI 2ης γενεάς που θα ακολουθείται από συντήρηση με ιμ, αποτελεί μία προοπτική προς τη κατεύθυνση της περιστολής της φαρ-μακευτικής δαπάνης η οποία εξετάζεται ήδη σε μία υπό εξέλιξη κλινική μελέτη (NCTO1316250).67
9) οι αρχικές επιφυλάξεις, ότι ενδεχομένως η χρήση των TKIs 2ης γενεάς στη θεραπεία 1ης γραμμής θα οδη-γήσει στην εμφάνιση περισσότερων τκι-ανθεκτικών με-ταλλάξεων συγκριτικά με αυτές που προκύπτουν από το ιμ, μετριάσθηκαν από τα πρόσφατα αποτελέσματα της μελέτης ENESTnd στους 36 μήνες, η οποία έδειξε μικρό-τερο αριθμό μεταλλάξεων στο σκέλος της θεραπείας με nilotinib.53 ςημειώνεται, ότι η εμφάνιση της πολυανθε-κτικής μετάλλαξης τ315I ήταν παρόμοια στα δύο σκέλη.
10) Ως μειονέκτημα σχεδιασμού στις μελέτες ENESTnd και DASISION καταγράφεται ότι και στις δύο δεν προ-βλέφθηκε διασταύρωση (crossover) μεταξύ των σκελών της θεραπείας, ούτε και σύγκριση των TKIs 2ης γενεάς με την τρέχουσα αποδεκτή κλινική πρακτική, δηλαδή την αρχική θεραπεία με ιμ και τη συνέχιση με TKI 2ης γενε-άς επί αποτυχίας ή δυσανεξίας του πρώτου φαρμάκου.71
ςυμπερασματικά, το ερώτημα κατά πόσον η στρατη-γική της αρχικής θεραπείας με ιμ και αλλαγή σε TKI 2ης γενεάς με βάση τα “ορόσημα” (milestones) που θέτει το ΕLN είναι εξίσου ή πλέον αποτελεσματική της θεραπεί-ας με TKI 2ης γενεάς χορηγούμενους από την αρχή, θα πρέπει να απαντηθεί σε προοπτική τυχαιοποιημένη κλι-
νική μελέτη, της οποίας το ένα σκέλος θα περιλαμβάνει ιμ± TKI 2ης γενεάς και το άλλο TKI 2ης γενεάς. τέλος, διαφωτιστική θα ήταν μια μελέτη απ’ ευθείας σύγκρι-σης nilotinib με dasatinib στη θεραπεία της 1ης γραμμής, η οποία σύμφωνα με την εκτίμηση των συγγραφέων της παρούσας ανασκόπησης, αποτελεί μια απομακρυσμένη προοπτική (ςχήμα 1).
ΔΙΑΚΟπΗ ΤΗΣ θΕΡΑπΕΙΑΣΌπως προκύπτει από τα παραπάνω η πλειονότητα των
ασθενών σε χρόνια φάση Χμλ εμφανίζει ευνοϊκή εξέλι-ξη με προσδόκιμο επιβίωσης που εγγίζει αυτό του γενικού πληθυσμού.12,13,72 Έτσι, συζητούμε για “λειτουργική ίαση” των περισσοτέρων ασθενών, οι οποίοι ενόσω συνεχίζουν τη φαρμακευτική αγωγή με τκι προστατεύονται από την εμφάνιση αντίστασης στη θεραπεία και εξέλιξης της νό-σου σε επιταχυνόμενη ή βλαστική φάση.73 Ωστόσο, καθώς οι τκιs δεν εξαφανίζουν τα μετάγραφα BCR-ABL στον πληθυσμό των αρχέγονων ηSCs (Hemopoietic Stem Cell, HSC),74 υπάρχει μεγάλη πιθανότητα υποτροπής της Χμλ και ως εκ τούτου η εφόρου ζωής συνέχιση της αγωγής αποτελεί την τρέχουσα θεραπευτική οδηγία.20 Έτσι ανα-φύονται προβλήματα που σχετίζονται με το ενδεχόμενο της υποτροπής ενώ συνεχίζεται η θεραπεία, τη μακροχρό-νια ανοχή των τκιs, τη συμμόρφωση των ασθενών στη θεραπεία και το κόστος της φαρμακευτικής δαπάνης για τις υγειονομικές υπηρεσίες υγείας των διαφόρων χωρών. ςημειώνεται, ότι η εξαιρετική αποτελεσματικότητα της θεραπείας με τκιs, που έχει μετατρέψει τη Χμλ σε μία νόσο πραγματικά χρόνια (λ.χ. όπως ο σακχαρώδης δια-βήτης) οδηγεί στην πρόβλεψη ότι ο συνολικός αριθμός των ασθενών στις ηΠΑ πριν από το 2040 θα υπερβαί-νει τις 250.000.75 η έρευνα λοιπόν στρέφεται προς νέες κατευθύνσεις με στόχο την ανεύρεση θεραπευτικών μο-νοπατιών που θα οδηγήσουν στην πραγματική ίαση της νόσου. Έτσι τα ερωτήματα: α) πότε και σε ποιους ασθε-νείς μπορεί να επιχειρηθεί η διακοπή της θεραπείας με τκι και β) ποια είναι η στρατηγική που θα μειώσει μετά τη διακοπή το ενδεχόμενο υποτροπής, αποτελούν σήμε-ρα σημαντική ερευνητική πρόκληση.
κατά τη διάρκεια της θεραπείας 1ης γραμμής με ιμ παρατηρείται συνήθως μια συνεχής μείωση του φορτίου των BCR-ABL μεταγράφων.21 τελικά, σε μερικούς ασθε-νείς η νόσος σταθερά δεν είναι μοριακά ανιχνεύσιμη, με αποτέλεσμα να ομιλούμε περί “πλήρους μοριακής αντα-πόκρισης” (CMR). Αξιοσημείωτο είναι ότι η μη ανίχνευση του BCR-ABL θα πρέπει να θεωρείται απλά ως απουσία μετρήσιμης υπολειμματικής νόσου με τη χρήση των δι-αθέσιμων μεθόδων RQ-PCR και όχι ως βέβαια απουσία υπολειμματικών λευχαιμικών κυττάρων. Έτσι, η καλού-μενη “πλήρης μοριακή ανταπόκριση” μπορεί να συνοδεύ-εται από υπολειμματικό φορτίο της νόσου που ποικίλει

Θεραπεία 1ης γραμμής σε ασθενείς με ΧΜΛ χρόνιας φάσης: Θεραπευτικές επιλογές 31
μεταξύ των ασθενών. Επιπρόσθετα, οι υπάρχοντες περι-ορισμοί στην ανίχνευση της υπολειμματικής νόσου εν-δεχομένως μελλοντικά να μεταβληθούν με τη βελτίωση της ευαισθησίας των τεχνικών της RQ-PCR, με αποτέ-λεσμα το ποσοστό των ασθενών με μη ανιχνεύσιμη μο-ριακή νόσο να μειωθεί ακόμη περισσότερο. Εύλογο είναι λοιπόν το ερώτημα εάν σε ασθενείς με μη ανιχνεύσιμη μοριακά νόσο μπορεί να θεωρηθεί ότι η Χμλ έχει ιαθεί και ως εκ τούτου να διακοπεί η χορηγουμένη θεραπευτι-κή αγωγή. η αρχική εμπειρία από μικρό αριθμό ασθενών με μη ανιχνεύσιμα BCR-ABL μετάγραφα που διέκοψαν τη θεραπεία κατόπιν επιθυμίας τους, εξαιτίας εγκυμο-σύνης ή σοβαρής τοξικότητας, ήταν μάλλον αποθαρρυ-ντική, δεδομένου ότι η νόσος σύντομα υποτροπίασε και χρειάσθηκε να επαναχορηγηθεί το ιμ.73,76 η διακοπή του ιμ εξετάσθηκε συστηματικά σε δύο προοπτικές κλινικές μελέτες. η μελέτη STIM (Stop Imatinib) από τη γαλλι-κή ομάδα Fi-LMC33 αφορούσε 100 ασθενείς σε χρόνια φάση Χμλ που ελάμβαναν ιμ για τουλάχιστον 36 μή-νες και εμφάνιζαν σταθερά μη-ανιχνεύσιμα BCR-ABL μετάγραφα για 2 ή περισσότερα έτη (ελάχιστος αποδε-κτός αριθμός μεταγράφων του γονιδίου αναφοράς ABL ήταν 50.000, στην εφαρμοζόμενη τεχνική RQ-PCR). με
διάμεση παρακολούθηση 17 μηνών, 54 ασθενείς εμφάνι-σαν υποτροπή, στην πλειονότητα των περιπτώσεων στη διάρκεια των πρώτων 6 μηνών. η συνολική πιθανότητα διατήρηση της CMR στους 12 μήνες ήταν 43%, ενώ για τους 69 ασθενείς που παρακολουθούνταν για περισσό-τερο από 12 μήνες η ελεύθερη υποτροπής επιβίωση ήταν 41% και 38% στους 12 και 24 μήνες αντίστοιχα. το γυ-ναικείο φύλο, το υψηλό Sokal risk score και η συντομότε-ρη διάρκεια θεραπείας με τκι προδιέθεταν σε συχνότερη εμφάνιση υποτροπής, ενώ η προηγούμενη θεραπεία με ιFN-α δεν προφύλασσε από την επανεμφάνιση της νό-σου.33 ςημειώνεται, ότι οι ασθενείς που υποτροπίασαν ανταποκρίθηκαν ευνοϊκά στην εκ νέου χορήγηση του ιμ. Παρόμοια αποτελέσματα με τα παραπάνω παρατη-ρήθηκαν στη μελέτη CML-8 της Αυστραλιανής ομάδας λευχαιμιών.77 Από τους 18 ασθενείς με μη-ανιχνεύσιμα BCR-ABL μετάγραφα (RQ-PCR με ευαισθησία τουλά-χιστον 4,5 λογαρίθμους) μετά από 2 έτη τουλάχιστον θε-ραπείας με ιμ, 8 παρέμειναν σε CMR με διάμεσο χρόνο παρακολούθησης 2 ετών χωρίς θεραπεία.
η εμπειρία από τη διακοπή της θεραπείας με dasatinib ή nilotinib είναι προς το παρόν περιορισμένη και προέρχε-ται κυρίως από τη συνεχιζόμενη γαλλική μελέτη διακοπής
Σχήμα 1. Αλγόριθμος θεραπείας ΧΜΛ χρόνιας φάσης.

Κ. Τσαταλάς και συν32
των φαρμάκων σε ασθενείς με μη-ανιχνεύσιμα BCR-ABL μετάγραφα για 2 τουλάχιστον έτη (ελάχιστος αποδεκτός αριθμός μεταγράφων του γονιδίου αναφοράς ABL ήταν 40.000, στην εφαρμοζόμενη τεχνική RQ-PCR).78 Πρό-σφατη ανάλυση των αποτελεσμάτων της μελέτης, στην οποία 33 ασθενείς διέκοψαν το dasatinib ή nilotinib έδει-ξε ότι μετά διάμεσο χρόνο παρακολούθησης 9 μηνών, η συχνότητα ελεύθερης από θεραπεία MMR ήταν 72,8%.
οι παραπάνω μελέτες έστω και αν στερούνται μακρο-χρόνιας παρακολούθησης, θέτουν σε αμφισβήτηση τις τρέχουσες οδηγίες που συνιστούν ότι η θεραπεία με TKIs δεν πρέπει ουδέποτε να διακόπτεται. Ωστόσο, υπάρχουν πολλαπλές ενδείξεις που συνηγορούν ότι στους ασθενείς που διακόπτουν τον τκι και δεν ξαναρχίζουν θεραπεία η δεξαμενή των λευχαιμικών κυττάρων παραμένει, όπως: α) διαδοχικές εκτιμήσεις με RQ-PCR αποκαλύπτουν τη σταθερή ή περιστασιακή παρουσία στο περιφερικό αίμα BCR-ABL μεταγράφων, έστω και σε χαμηλές ποσότη-τες,33,77 β) η εφαρμογή τεχνικής βασιζόμενης σε γενωμι-κό DNA ανιχνεύει αναδιατάξεις του γονιδίου BCR-ABL οι οποίες δεν είναι αναγκαίο να εκφράζονται77 και γ) η παρουσία BCR-ABL μεταγράφων σε in-vitro σύντομης και μακράς διάρκειας καλλιέργειες CD34+ κυττάρων από ασθενείς εκτός θεραπείας, που παραμένουν για μα-κρό χρονικό διάστημα χωρίς ανιχνεύσιμα BCR-ABL με-τάγραφα στο περιφερικό αίμα.79
ςυμπερασματικά: α) η παρουσία ανιχνεύσιμου με μοριακές τεχνικές γενετικού υλικού στο περιφερικό αί-μα ασθενών, που διέκοψαν τη θεραπεία με τκι, δεν συ-νεπάγεται υποχρεωτικά υποτροπή της νόσου, β) προς το παρόν δεν υπάρχει ομοφωνία ποιος είναι στην κλινική πράξη ο βαθμός ανταπόκρισης που επιτρέπει την ασφα-λή διακοπή της θεραπείας, γ) μένει να καθορισθεί το όριο της παρουσίας ανιχνεύσιμης υπολειμματικής νόσου πέ-ρα από το οποίο θα επιβάλλεται η θεραπευτική παρέμ-βαση (επαναχορήγηση του φαρμάκου) μετά τη διακοπή του τκι και δ) τα κλινικά και βιολογικά χαρακτηριστι-κά των υποψηφίων για διακοπή του φαρμάκου ασθενών θα πρέπει να καθορισθούν σε μεγάλες κλινικές μελέτες.
θΕΡΑπΕΙΑ ΑΣθΕΝΩΝ ΑΝθΕΚΤΙΚΩΝ ΣΤΗΝ 1ης ΓΡΑΜΜΗΣ θΕΡΑπΕΙΑ ΜΕ ΤΚΙs 2ης ΓΕΝΕΑΣ
Προς το παρόν δεν υπάρχουν δεδομένα από μελέ-τες αντιμετώπισης ασθενών που ανέπτυξαν αντίσταση ή δυσανεξία στο dasatinib ή nilotinib ενώ αυτά χορηγού-νταν ως θεραπεία 1ης γραμμής. μία λογική θεραπευτική προσέγγιση θα μπορούσε να βασίζεται στο προφίλ με-ταλλάξεων και το φάσμα ανεπιθύμητων ενεργειών που συνοδεύει τον κάθε τκι ξεχωριστά.69 με εξαίρεση την BCR-ABL/T315I μετάλλαξη, όλες οι άλλες μεταλλάξεις που προκαλούν αντίσταση στο dasatinib ή nilotinib μπο-
ρεί να είναι ευαίσθητες στον άλλο τκι. Επιπλέον, το ιμ ενδεχομένως να είναι δραστικό σε επιλεγμένες περιπτώ-σεις απώλειας της ανταπόκρισης στο dasatinib.69 μελλο-ντική προοπτική χρήσης νεότερων τκιs στη θεραπεία 1ης ή 2ης γραμμής αποτελούν οι TKIs bosutinib (2ης γενεάς) και ponatinib (3ης γενεάς), Επιπλέον, η Aλλο-μΑκ θα πρέπει να αποτελεί θεραπευτική επιλογή σε επιλεγμένους ασθενείς, ιδιαίτερα μετά προηγούμενη αποτυχία σε δύο τκιs.32 (ενδελεχής ανάπτυξη του θέματος στην ανασκό-πηση «ο ρόλος της μεταμόσχευσης στη Χμλ: σε ποιους ασθενείς και πότε» του παρόντος τεύχους).
θΕΡΑπΕΙΑ ΑΣθΕΝΩΝ ΜΕ Τ315Ι ΜΕΤΑΛΛΑξΗ
η BCR-ABL/T315I μετάλλαξη είναι πλήρως ανθε-κτική και στους τρεις διαθέσιμους TKIs (IM, nilotinib, dasatinib) επειδή η πλάγια άλυσος ισολευκίνης του με-ταλλαγμένου χιμαιρικού γονιδίου BCR-ABL δεν σχημα-τίζει δεσμό υδρογόνου με τους TKIs με αποτέλεσμα να παρεμποδίζεται η σύνδεση του φαρμάκου με το στόχο. μολονότι η Αλλο-μΑκ έχει απόλυτη ένδειξη για τους ασθενείς με Χμλ που φέρουν τη μετάλλαξη και είναι επιλέξιμοι γι’ αυτή τη θεραπευτική επιλογή, υπάρχουν διάφορα πειραματικά φάρμακα τα οποία δοκιμάζονται σε κλινικές μελέτες. ιδιαίτερο ενδιαφέρον παρουσιάζει το ponatinib (γνωστό και ως ΑΡ24534, Ariad Pharmaceuti-cals, Cambridge, MA) που είναι ο πρώτος 3ης γενιάς τκι με ισχυρή και σταθερή δραστικότητα έναντι της μετάλλαξης τ315ι.80 η αποτελεσματικότητα του ponatinib βασίζεται στο ότι παρακάμπτει το δεσμό υδρογόνου και προσδένε-ται καταλυτικά σε περιοχή ενεργού κινάσης με τη βοή-θεια εύκαμπτου τριακαρβονικού αιθυλικού συνδέτη.80 το ponatinib αναστέλλει τόσο το αμετάλλακτο (IC50 0,37nm) όσο και το μεταλλαγμένο (IC50 2,0nm) BCR-ABL/T315I γονίδιο. Επίσης έχει δραστικότητα έναντι διαφόρων κοι-νών μεταλλάξεων του BCR-ABL όπως Ε255K, V253H και G250Ε και άλλων κινασών της τυροσίνης όπως SFK, PDGFa και κιτ.80
τα αποτελέσματα από τη μελέτη φάσεως ι του pona-tinib σε ασθενείς με προχωρημένες αιματολογικές κα-κοήθειες παρουσιάσθηκαν πρόσφατα.81 το φάρμακο χορηγήθηκε σε δόσεις 2mg έως 60mg ημερησίως και οι συνηθέστερες ανεπιθύμητες ενέργειες ήταν θρομβοπενία (23%), εξάνθημα (22%), αρθραλγίες (15%) και παγκρεα-τίτις, η οποία οδήγησε στον καθορισμό της μέγιστης ανε-κτής δόσης στα 45mg. Από τους 38 ασθενείς με Χμλ σε χρόνια φάση CηR πέτυχε το 95%, MCyR το 66% και CCyR το 53%. Από τους 9 ασθενείς με μετάλλαξη τ315ι που αξιολογήθηκαν το 100% πέτυχε CηR και CMyR και το 89% CCyR. ςτο ASH meeting του 2011 ανακοινώθη-καν τα αποτελέσματα της μελέτης φάσεως ιι PACE που αξιολογεί περαιτέρω το ponatinib σε Ph+ λευχαιμίες.82

Θεραπεία 1ης γραμμής σε ασθενείς με ΧΜΛ χρόνιας φάσης: Θεραπευτικές επιλογές 33
First Line Treatment for Chronic Phase Chronic Myelogenous Leukemia (CP-CML): Treatment Options
by Constantinos Tsatalas, Emmanouil Spanoudakis, Ioannis Kotsianidis Department of Haematology, Democritous University of Thrace Medical School, Alexandroupolis
ABSTRACT: The clinical outcome for patients with CP-CML has changed dramatically in the past 15 years. This has been due to the development of tyrosine kinase inhibitors (TKIs), compounds that inhib-it the activity of the oncogenic BCR-ABL protein. Imatinib (IM) was the first TKI developed for CML, and it led to high rates of complete cytogenetic responses and improved survival for patients with this disease. However, approximately one-third of patients experience IM resistance or intolerance. These patients may benefit from alternative more effective treatment options. Patients who have delayed re-sponses to IM are at increased risk of disease progression. As BCR-ABL activity has a critical role in disease progression by promoting genetic instability, more effective BCR-ABL inhibition may decrease the risk of disease progression. Randomized phase III studies have demonstrated that first-line treatment with dasatinib or nilotinib results in higher rates of CCyR and MMR by 12-24 months compared with IM, in addition to a lower rate of disease progression. Based on these data, it is reasonable to use sec-ond-generation TKIs in the treatment of newly diagnosed patients with CP-CML. As there are no data comparing both agents, one agent cannot be recommended over the other. Therefore, physicians should consider the BCR-ABL mutation profile and the patient’s history to make an educated decision on the best choice. The obvious debating points regarding the use of second-generation TKIs up front are: a) whether or not the benefit in early cytogenetic and molecular response will translate to a long-lasting survival benefit b) the wisdom of switching to second-generation drugs without long-term safety data and c) cost issues, especially with the prospect of generic IM in the foreseeable future.
Άλλα πειραματικά φάρμακα που χρησιμοποιούνται στη θεραπεία της τ315ι μετάλλαξης είναι η omacetax-ine (homoharringtonine), DCC2036, PHA739358 και XL228 η λεπτομερής περιγραφή των οποίων ξεφεύγει από το σκοπό της παρούσης ανασκόπησης.
θΕΡΑπΕΙΑ 1ης ΓΡΑΜΜΗΣ ΧΜΛ ΧΡΟΝΙΑΣ ΦΑΣΗΣ. ΑΝΟΙΧΤΑ ζΗΤΗΜΑΤΑ
Παρά τη σημαντική πρόοδο που συντελέσθηκε στη Χμλ ανοιχτά παραμένουν τα παρακάτω ζητήματα: α) σε ασθενείς υψηλού κινδύνου, χορηγούμε αρχικά ιμ και αλλάζουμε σε τκιs 2ης γενεάς εάν δεν επιτευχθούν σε σύντομο χρονικό διάστημα πολύ φιλόδοξα ορόσημα ή εναλλακτικά, αρχίζουμε με τκιs 2ης γενεάς και αλλά-ζουμε τους ασθενείς που πέτυχαν βέλτιστη ανταπόκρι-ση σε θεραπεία συντήρησης με το μικροτέρου κόστους ιμ; β) η ταχύτερη και συχνότερη επίτευξη κυτταρογε-νετικών και μοριακών ανταποκρίσεων με τους TKIs 2ης γενεάς ενδεχομένως επιβάλλει την αναθεώρηση των κα-
τευθυντήριων οδηγιών του ELN, καθώς οι ορισμοί της βέλτιστης και υποβέλτιστης απάντησης και αποτυχίας της θεραπείας δεν διευκολύνουν πλέον τους θεραπευτικούς χειρισμούς των ασθενών που λαμβάνουν νεότερους TKIs γ) μία περιοχή σημαντικού προβληματισμού αποτελεί η ποιότητα των μεταλλάξεων του χιμαιρικού γονιδίου BCR-ABL που θα προκύψουν από τη χρήση των τκιs 2ης γε-νεάς στη θεραπεία 1ης γραμμής δ) προκαλεί επιφυλάξεις η απουσία μακροχρόνιων δεδομένων που σχετίζονται με την ασφάλεια μιας πολύ ισχυρής αναστολής προκαλού-μενης από τους τκιs 2ης γενεάς ε) απαιτείται βελτίωση της κατανόησης των μηχανισμών αντίστασης που είναι ανεξάρτητοι του BCR-ABL και της βιολογίας του αρχέ-γονου προγονικού Χμλ κυττάρου στ) αναζήτηση πιο αξιόπιστων μεθόδων εκτίμησης της CMR με σκοπό την αναγνώριση των ασθενών που αγγίζουν την «ίαση» και ως εκ τούτου μπορούν να ενταχθούν σε προοπτικές με-λέτες οριστικής διακοπής του τκι και ζ) η αποτελεσμα-τική θεραπεία της μετάλλαξης τ315ι.
Βιβλιογραφία1. Hehlmann R, Hochhaus A, Baccarani M. Chronoc myeloid
leukemia. Lancet. 2007; 370:342-350.2. Baccarani M. New directions in the treatment of patients with
chronic myeloid leukemia: introduction. Semin Hematol. 2009; 46(Suppl3):S1-S4.
3. Jabbour E, Parikh SA, Kantarjian H, Cortes J. Chronic myeloid leukemia: mechanisms of resistance and treatment.

Κ. Τσαταλάς και συν34
Hematol Oncol Clin N Am. 2011; 25:981-995.4. Kantarjian H, Sawyers C, Hochhaus A, Guilhot F, et al.
Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukaemia. N Eng J Med. 2002; 346:645-652.
5. O’Brien SG, Guilhot F, Larson RA, et al. Imatinib compared with interferon and low dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukaemia. N Engl J Med. 2003; 348:994-1004.
6. Deininger MW, O’ Brien SG, Guilhot F, et al. International randomized study of interferon vs STI571 (IRIS) 8-year follow-up: sustained survival and low risk for progression or events in patients with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP) treated with imatinib. Blood. 2009; 114:1126.
7. Robin M, Guardiola P, Devergie A, et al. A 10-year median follow-up study after allogeneic stem cell transplantation for chronic myeloid leukemia in chronic phase from HLA-identical sibling donors. Leukemia. 2005; 19:1613-1620.
8. Fausel C. Targeted chronic myeloid leukaemia therapy: seeking a cure. Am J Health Syst Pharm. 2007; 64(Suppl 15):S9-15.
9. Hehlmann R, Berger U, Pfirmann M, et al. Drug treatment is superior to allografting as first-line therapy in chronic myeloid lekemia. Blood. 2007; 109:4686-4692.
10. Roy L, Guilhot J, Krahnke T, et al. Survival advantage from imatinib compared with the combination interferon-alpha plus cytarabine in chronic-phase chronic myelogenous leu-kemia : historical comparison between two phase 3 trials. Blood. 2006; 108:1478-1484.
11. Kantarjian HM, Talpaz M, O’Brien S, et al. Survival benefit with imatinib mesylate versus interferon-alpha-based regi-mens in newly diagnosed chronic-phase chronic myelogenous leukaemia. Blood. 2006; 108:1835-1840.
12. Gambacorti-Passerini C, Antolini L, Mahon FX, et al. Multicenter independent assessment of outcomes in chronic myeloid leukemia patients treated with imatinib. J Natl Cancer Inst. 2011; 103(7):553-561.
13. Björkholm M, Ohm L, Eloranta S, et al. Success story of targeted therapy in chronic myeloid leukemia: a population-based study of patients diagnosed in Sweden from 1973 to 2008. J Clin Oncol. 2011; 29:2514-2520.
14. Goldman JM. Initial treatment for patients with CML. He-matology Am Soc Hematol Educ Program. 2009:453-460.
15. Kantarjian H, O’Brien S, Cortes J, et al. Complete cytogenetic and molecular responses to interferon-alpha-based therapy for chronic myelogenous leukemia are associated with ex-cellent long-term prognosis. Cancer. 2003; 97:1033-1041.
16. Druker B, Guilhot F, O’Brien S, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006; 355:2408-2417.
17. Hughes TP, Kaeda J, Branford S, et al. for the International Randomised Study of Interferon versus STI571 (IRIS) Study Group. Frequency of major molecular responses to imatinib or interferon alpha plus cytarabine in newly chronic myeloid leukemia. N Engl J Med. 2003; 349:1423-1432.
18. Branford S, Hughes TP. Practical considerations for monitor-ing patients with chronic myeloid leukemia. Semin Hematol. 2010; 47(4):327-334.
19. Baccarani M, Saglio G, Goldman J, et al. European Leuke-mia Net. Evolving concepts in the management of chronic meyloid leukemia: recommendations from an expert panel on behalf of the European Leukemia Net. Blood. 2006; 108:1809-1820.
20. Baccarani M, Cortes J, Pane F, et al. Chronic myeloid leukemia: an update of concepts and management recom-mendations of European Leukemia Net. J Clin Oncol. 2009; 27:6041-6051.
21. Hughes TP, Hochhaus A, Branford S, et al. Long-term prog-nostic significance of early molecular response to imatinib in newly diagnosed chronic myeloid leukaemia: an analysis from the international randomized study of interferon and STI571 (IRIS). Blood. 2010; 116:3758-3765.
22. de Lavallade H, Apperley JF, Khorashad JS, et al. Imatinib for newly diagnosed patients with chronic myeloid leukemia: incidence of sustained responses in an intention-to-treat analysis. J Clin Oncol. 2008; 26:3358-3363.
23. Kantarjian H, O’Brien S, Cortes J, et al. Significance of increasing levels of minimal residual disease in patients with Philadelphia chromosome-positive chronic myelogenous leukemia in complete cytogenetic response. J Clin Oncol. 2009; 27:3659-3663.
24. Hehlmann R, Lauseker S, Jung-Munkwitz S, et al. Tolera-bility-adapted imatinib 800 mg/d versus 400 mg/d versus 400 mg/d plus interferon-alpha in newly diagnosed chronic myeloid leukemia. J Clin Oncol. 2011; 29:1634-1642.
25. Santos FP, Kantarjian H, Quintas-Cardama A, Cortes J. Evolution of therapies for chronic myelogenous leukemia. Cancer J. 2011; 17:465-476.
26. Kantarjian H, Cortes J. Considerations in the management of patients with Philadelphia chromosome-positive chronic myeloid leukemia receiving tyrosine kinase inhibitor therapy. J Clin Oncol. 2011; 29:1512-1516.
27. Press R, Love Z, Tronnes A, et al. BCR-ABL mRNA levels at and after the time of a complete cytogenetic response (CCR) predict the duration of CCR in imatinib mesylate-treated patients with CML. Blood. 2006; 107:4250-4256.
28. Radich JP. Chronic myeloid loukemia 2010: where are we now and where can we go? Hematology Am Soc Hematol Educ Program. 2010:122-128.
29. Kantarjian H, Schiffer C, Jones D, et al. Monitoring the response and course of chronic myeloid leukaemia in the modern era of BCR-ABL tyrosine kinase inhibitors: practical advice on the use and interpretation of monitoring methods. Blood. 2008; 111:1774-1780.
30. Press RD, Galderisi C, Yang E, et al. A half-log increase in BCR-ABL RNA predicts a higher risk of relapse in patients with chronic myeloid leukemia with an imatinib-induced complete cytogenetic response. Clin Cancer Res. 2007; 13:6136-6143.
31. Verma D, Kantarjian H, Shan J, et al. Sustained complete molecular response to imatinib in chronic myeloid leukemia:

Θεραπεία 1ης γραμμής σε ασθενείς με ΧΜΛ χρόνιας φάσης: Θεραπευτικές επιλογές 35
a target worth aiming and achieving? Blood. 2009; 114:505.32. Cortes J, Hochhaus A, Hughes T, Kantarjian H. Front-line
and salvage therapies with tyrosine kinase inhibitors and other treatments in chronic myeloid leukemia. J Clin Oncol. 2011; 29:524-531.
33. Mahon FX, Rea D, Guilhot J, et al. Discontinuation of imatinib in patients with chronic myeloid leukemia who have maintained complete molecular remission for at least 2 years: the prospective , multicentre, Stop Imatinib (STIM) trial. Lancet Oncol. 2010; 11:1029-1035.
34. Lucas CM, Wang L, Austin GM, et al. A population study of imatinib in chronic myeloid leukaemia demonstrates lower efficacy than in clinical trials. Leukemia. 2008; 22:1963-1966.
35. Castagnetti F, Palandri F, Amabile M, et al. Results of high-dose imatinib mesylate in intermediate Sokal risk chronic myeloid leukemia patients in early chronic phase: a phase 2 trial of the GIMEMA CML Working Party. Blood. 2009; 113:3428-3434.
36. Cortes JE, Kantarjian HM, Goldberg SL, et al. High-dose imatinib in newly diagnosed chronic-phase chronic myeloid leukemia: high rates of rapid cytogenetic and molecular responses. J Clin Oncol. 2009; 27:4754-4759.
37. Hughes TP, Branford S, White DL, et al. Impact of early dose intensity on cytogenetic and molecular responses in chronic-phase CML patients receiving 600 mg/day of imatinib as initial therapy. Blood. 2008; 112:3965-3973.
38. Kantarjian H, Talpaz M, O’Brien S, et al. High-dose imatinib mesylate therapy in newly diagnosed Philadelphia chromo-some-positive phase chronic myeloid leukaemia. Blood. 2004; 103:2873-2878.
39. Cortes JE, Baccarani F, Guilhot F, et al. Phase III, rand-omized, open-label study of daily imatinib mesylate 400 mg versus 800 mg in patients with newly diagnosed, previously untreated chronic myeloid leukaemia in chronic phase using molecular end points: tyrosine kinase inhibitor optimization and selectivity study. J Clin Oncol. 2010; 28:424-430.
40. Baccarani M, Rosti G, Castagnetti F, et al. Comparison of imatinib 400 mg and 800 mg daily in the front-line treat-ment of high risk, Philadelphia-positive chronic myeloid leukaemia; a European Leukemia Net Study. Blood. 2009; 113:4497-4504.
41. Gafter-Gvili A, Leader A, Gurion R, et al. High-dose imatinib for newly diagnosed chronic phase chronic myeloid leuke-mia patients - Systematic review and meta-analysis. Am J Haematol. 2011; 86:657-662.
42. Preudhomme C, Guilhot J, Nicolini FE, et al. SPIRIT In-vestigators, France Intergroup des Leucémies Myéloïdes Chroniques (Fi-LMC). Imatinib plus peginterferon alfa-2α in chronic myeloid leukemia. N Engl J Med. 2010; 363:2511-2521.
43. Simonsson B, Gedde-Dahl T, Markevärn B, et al. Nordic CML Study Group. Combination of pegylated IFN-aRb with imatinib increases molecular response rates in patients with low or intermediate-risk chronic myeloid leukaemia. Blood. 2011; 118:3228-3335.
44. Melo JV, Chuah C. Novel agents in CML therapy: tyrosine
kinase inhibitors and beyond. Hematology Am Soc Hematol Educ Program. 2008:427-435.
45. O’Hare T, Walters DK, Stoffregen EP, et al. In vitro activity of Bcr-Abl inhibitors AMN107 and BMS- 354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res. 2005; 65:4500-4505.
46. Shami PJ, Deininger M. Evolving treatment strategies for patients newly diagnosed with chronic myeloid leukaemia: the role of second-generation BCR-ABL inhibitors as first-line therapy. Leukemia. 2011; 26:1-11.
47. Weisberg E, Manley PW, Breitenstein W, et al. Characteriza-tion of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005; 7:129-141.
48. Cortes JE, Jones D, O’Brien S, et al. Results of dasatinib therapy in patients with early chronic-phase chronic myeloid leukaemia. J Clin Oncol. 2010; 28:398-404.
49. Cortes JE, Jones D O’Brien S, et al. Nilotinib as front-line treatment for patients with chronic myeloid leukemia in early chronic phase. J Clin Oncol. 2010; 28:392-397.
50. Rosti G, Palandri F, Castagnetti F, et al. Nilotinib for the frontline treatment of Ph(+) chronic myeloid leukemia. Blood. 2009; 114:4933-4938.
51. Kantarjian H, Shah NP, Hochhaus A, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukaemia. N Engl Med. 2010; 362:2260-2270.
52. Saglio G, Kim DW, Issaragrisil S, et al. ENESTnd Investiga-tors: Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med. 2010; 362:2251-2259.
53. Saglio G, le Coutre P, Pasquini R, et al. Nilotinib versus imatinib in patients with newly diagnosed Philadelphia chromosome-positive chronic myeloid leukaemia in chronic phase: ENESTnd 36-month follow-up. Blood. 2011; 118:452.
54. Kantarjian HM, Shah NP, Cortes JE, et al. Dasatinib or imatinib in newly diagnosed chronic phase chronic myeloid leukemia: 2-year follow-up from a randomized phase 3 trial (DASISION). Blood. 2012; 119:1123-1129.
55. Hochhaus A. Managing chronic myeloid leukaemia as a chronic disease. Hematology Am Soc Hematol Educ Pro-gram 2011:128-135.
56. Puttini M, Coluccia AM, Boschelli F, et al. In vitro and in vivo activity of SKI-606, a novel Src-Abl inhibitor, against imatinib-resistant Bcr-Abl+ neoplastic cells. Cancer Res. 2006; 66:11314-11322.
57. Gambacorti-Passerini C, Kim DW, Kantarjian HM, et al. An ongoing phase 3 study of Bosutinib (SKI-606) versus imatinib in patients with newly diagnosed chronic phase chronic myeloid leukemia. Blood 2010. 116:208.
58. Eiring AM, Khorashad J, Morley K, Deininger MW. Ad-vances in the treatment of chronic myeloid leukemia. BMC Medicine. 2011; 26:9:99.
59. Druker BJ, Guilhot F, O’Brien SG, et al. IRIS Investigators: Five-years follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl Med J. 2006; 355:2409-2417.
60. Hochhaus A, Baccarani M, Deininger M, et al. Dasatinib induces durable cytogenetic responses in patients with chronic myelogenous leukemia in chronic phase with resistance or

Κ. Τσαταλάς και συν36
intolerance to imatinib. Leukemia. 2008; 22:1200-1206.61. Kantarjian H, Giles F, Bhalla K, et al. Update on imatinib
resistant chronic myeloid leukaemia patients in chronic phase (CML-CP) on nilotinib therapy at 24 months. Clini-cal response, safety and long term outcomes. Blood. 2009; 114:1129 .
62. Okimoto RA, Van Etten RA. Navigating the road toward optimal initial therapy for chronic myeloid leukemia. Curr Opin Hematol. 2011; 18:89-97.
63. Al-Kali A, Kantarjian H, Shan J, et al. Current event-free survival after sequential tyrosine kinase inhibitor therapy for chronic myeloid leukemia. Cancer. 2011; 117:327-335.
64. Ibrahim AR, Clark RE, Holyoake TL, et al. Second generation tyrosine kinase inhibitors improve the survival of patients with chronic myeloid leukaemia in whom imatinib therapy has failed. Haematologica. 2011; 96:1779-1782.
65. Yeung DT, Osborn M, White DL, et al. Upfront imatinib therapy in CML patients with rapid switching to nilotinib for failure to achieve molecular targets or intolerance achieves high overall rates of molecular response and a low risk progression-an update of the TIDEL-II trial. Blood. 2011; 118:451.
66. Calabretta B, Perrotti D. The biology of CML blast crisis. Blood. 2004; 103:4010-4022.
67. Davies J. First-line therapy for CML: nilotinib comes of age. Lancet Oncol. 2011; 12:826-827.
68. Kantarjian HM, Hochhaus A, Saglio G, et al. Nilotinib versus imatinib for the treatment of patients with newly diagnosed chronic phase, Philadelphia chromosome-positive, chronic myeloid leukaemia: 24-month minimum follow-up of the phase 3 randomised ENESTnd trial. Lancet Oncol. 2011; 12:841-851.
69. Smith CC, Shah NP. Tyrosine kinase inhibitor therapy for chronic myeloid leukemia: approach to patients with treatment-naïve or refractory chronic-phase disease. Hemato-lolgy Am Soc Hematol Educ. Program 2011. 2010:121-127.
70. Mueller JM. Taking TRIPS to India: Novartis, patent law, and access to medicines. N Engl J Med. 2007; 356:541-543.
71. Tefferi A. Second-generation tyrosine kinase inhibitors in chronic myelogenous leukemia. Cancer. 2011; 117:234-237.
72. Pinilla-Ibarz J, Flinn I. The expanding options for front-line treatment in patients with newly diagnosed CML. Crit Rev Oncol Hematol. 2012 April 2007 [Epub ahead of print].
73. Rea D, Rousselot P, Guilhot J, Guilhot F, Mahon FX. Curing chronic myeloid leukemia. Curr Hematol Malig Rep. 2012 Mar 13. [Epub ahead of print].
74. Bhatia R, Holtz M, Niu N, et al. Persistence of malignant hematopoietic progenitors in chronic myelogenous leuke-mia patients in complete cytogenetic remission following imatinib mesylate treatment. Blood. 2003; 101:4701-4707.
75. Mughal TI, Radich JP, Van Etten R, et al. Chronic myeloid leukemia 2011: successes, challenges and strategies- Proceed-ings of the 5th Annual BCR-ABL1 positive and BCR-ABL1 negative myeloproliferative neoplasms workshop. Am J Hematol. 2011; 86:811-819.
76. Cortes J, O’ Brien S, Katarjian H. Discontinuation of ima-tinib therapy after achieving a molecular response. Blood. 2004; 104:2204-2205.
77. Ross DM, Branford S, Seymour JF, et al. Patients with chronic myeloid leukemia who maintain a complete mo-lecular response after stopping imatinib treatment have evidence of persistent leukemia by DNA PCR. Leukemia. 2010; 24:1719-1724.
78. Rea D, Rousselot P, Nicolini FE, et al. Discontinuation of dasatinib or nilotinib in chronic myeloid leukemia patients with stable undetectable Bcr-Abl transcripts: results from the French CML group (FILMC). Blood. 2011; 118:604.
79. Chomel JC, Bonnet ML, Sorel N, et al. Leukemic stem cell persistence in chronic myeloid leukemia patients with sustained undetectable molecular residual disease. Blood. 2011; 118:3657-3660.
80. O’Hare T, Shakespeare WC, Zhu X, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukaemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009; 16:401-412.
81. Cortes J, Talpaz M, Bixby D, et al. A phase 1 trial of oral ponatinib (AP24534) in patients with refractory chronic myelogenous leukaemia (CML) and other hematologic malignancies: emerging safety and clinical response find-ings. Blood. 2010; 116:210.
82. Cortes JE, Kim DW, Pinilla-Ibarz J, et al. Initial findings from the PACE trial: a pivotal phase 2 study of ponatinib in patients with CML and Ph+ ALL resistant or intolerant to dasatinib or nilotinib, or with the T315I mutation. Blood. 2011; 118:452.

Κριτήρια αλλαγής από imatinib σε ΤΚΙs δεύτερης γενιάς
Νόρα-Αθηνά Βύνιου, Παναγιώτης Διαμαντόπουλος, Βασιλική Παπαδοπούλου
ΠΕΡΊληΨη: η ανταπόκριση των ασθενών με χρόνια μυελογενή λευχαιμία στη θεραπεία με imatinib δεν είναι δεδομένη. Ως σήμερα δεν υπάρχει ασφαλής τρόπος ανίχνευσης των ασθενών που θα εμφανίσουν πρωτοπαθή ή δευτεροπαθή (απώλεια της ανταπόκρισης) αντοχή στο φάρμακο. ο έλεγχος της ανταπό-κρισης στηρίζεται σε κλινικά, αιματολογικά, κυτταρογενετικά και μοριακά κριτήρια και πρέπει να δι-ενεργείται σε προκαθορισμένα χρονικά διαστήματα, με βάση τις συστάσεις του Ευρωπαϊκού Δικτύου λευχαιμίας (ELN) και του ςυνεργατικού Δικτύου κατά του καρκίνου (NCCN). ςτην εποχή των ανα-στολέων τυροσινικής κινάσης 2ης γενιάς, οι ασθενείς που δεν επιτυγχάνουν το επιθυμητό αποτέλεσμα της θεραπείας με imatinib, όπως και οι ασθενείς που εμφανίζουν δυσανεξία στο φάρμακο είναι υποψή-φιοι για αλλαγή θεραπείας σε dasatinib ή nilotinib. ςημαντικό μέρος της θεραπευτικής αποτυχίας οφεί-λεται στην ανάπτυξη ανθεκτικών στο φάρμακο μεταλλάξεων της BCR/ABL κινάσης. μεταξύ αυτών, η T315I είναι ανθεκτική και στους τρεις ως τώρα διαθέσιμους αναστολείς. για τις υπόλοιπες περιπτώσεις, η επιλογή του καταλληλότερου αναστολέα 2ης γενιάς γίνεται κυρίως με βάση τον έλεγχο μεταλλάξεων, το προφίλ ανεπιθυμήτων ενεργειών του κάθε αναστολέα και παράγοντες που αφορούν στη φαρμακο-κινητική τους, όπως οι διαιτητικοί περιορισμοί που συνοδεύουν τη λήψη τους. Παράγοντες που συμ-μετέχουν στην αποτυχία της θεραπείας με imatinib είναι και η εμφάνιση πρόσθετων χρωμοσωμικών ανωμαλιών, όπως επίσης και η χαμηλή ενδοκυττάρια συγκέντρωση του ανθρώπινου μεταφορέα οργανι-κών κατιόντων – 1 (hOCT-1), η οποία όμως δεν επηρεάζει τη δραστικότητα των αναστολέων 2ης γενιάς. Νεώτερα δεδομένα προκύπτουν συνεχώς από συγκριτικές μελέτες των τριών φαρμάκων που αφορούν στην ορθολογική τους χρήση στη θεραπεία πρώτης και δεύτερης γραμμής.
Haema 2012; 3(1): 37-44 Copyright EAE
Α΄ Παθολογική κλινική ΕκΠΑ, Νοσοκομείο «λαϊκό»Διεύθυνση Αλληλογραφίας: Νόρα-Αθηνά Βύνιου, Α΄Παθολογική κλινι-
κή ΕκΠΑ, γΝΑ «λαϊκό», email : [email protected]
Aνασκόπηση
ΕΙΣΑΓΩΓΗτο imatinib θεωρείται ο θεμέλιος λίθος της θεραπεί-
ας της χρονίας φάσης της χρόνιας μυελογενούς λευχα-μίας (Χμλ) κατά το μεγαλύτερο μέρος της τελευταίας δεκαετίας. το Ευρωπαϊκό Δίκτυο λευχαιμίας (ELN) και το ςυνεργατικό Δίκτυο κατά του καρκίνου (NCCN) θέ-σπισαν μεθόδους παρακολούθησης και κριτήρια ανταπό-κρισης που διαχωρίζουν τους ασθενείς που είναι πιθανό να ανταποκριθούν καλά στη θεραπεία με imatinib από αυτούς που έχουν λιγότερες πιθανότητες επίτευξης κα-λού θεραπευτικού αποτελέσματος.1,2 ςτους πίνακες 1 και 2 αναγράφονται τα κριτήρια ανταπόκρισης και τα αντί-στοιχα χρονοδιαγράμματα για ασθενείς που βρίσκονται σε χρόνια φάση Χμλ σύμφωνα με τις κατευθυντήριες οδηγίες του ELN και του NCCN.
το ELN έχει προσδιορίσει την ανταπόκριση κατά την χρόνια φάση της Χμλ ως «επιθυμητή», «κατώτερη της επιθυμητής» και «αποτυχία» (Πίνακας 1).
η επιθυμητή ανταπόκριση καταδεικνύει ότι δεν χρειά-ζεται μεταβολή στη θεραπεία και η επιβίωση αναμένεται πλησίον του 100% στα επόμενα 6-7 χρόνια. με ανταπό-κριση κατώτερη της επιθυμητής ο ασθενής εξακολου-θεί να έχει σημαντικό όφελος από την θεραπεία αλλά δυνητικά έχει πιο επιβαρυμένη πρόγνωση από ασθενείς με επιθυμητή ανταπόκριση. Αυτή η ομάδα ασθενών εί-ναι υποψήφια για αλλαγή θεραπείας. Επιπλέον, ασθενείς που αποτυγχάνουν να έχουν πλήρη αιματολογική ύφεση στους 3 μήνες, οποιαδήποτε κυτταρογενετική ανταπόκριση στους 12 μήνες και τέλος πλήρη κυτταρογενετική αντα-πόκριση στους 18 μήνες από την έναρξη της θεραπείας, θεωρείται ότι υπάγονται στην κατηγορία της θεραπευτι-κής αποτυχίας και πρέπει να τους χορηγείται θεραπεία 2ης γραμμής. η αποτυχία επίτευξης συγκεκριμένης ελάττω-σης του λευχαιμικού φορτίου σε συγκεκριμένο χρονικό

Ν.-Α. Βύνιου και συν38
Πίνακας 1. Κριτήρια ανταπόκρισης ασθενών σε χρόνια φάση ΧΜΛ στη θεραπεία με imatinib (όπως προσδιο-ρίζονται από το ELN)
Χρόνος Εκτίμησης (Μήνες)
ΑνταπόκρισηΕπιθυμητή Κατώτερη της επιθυμητής Αποτυχία
0 0 0 0
3 Πλήρης αιματολογική ύφεση, ≤65% Ph+
Πλήρης Αιματολογική ύφεση, >65% Ph+
Απουσία αιματολογικής ύφεσης
6 ≤35% Ph+ 35-95% Ph+ >95% Ph+
12 Πλήρης κυτταρογενετική ύφεση (0% Ph+ )
1-35% Ph+ >35% Ph+
18 Μέγιστη μοριακή ύφεση < μέγιστης μοριακής ύφεσης >0% Ph+
Όποτεδήποτε Σταθερή ή βελτιούμενη μέγιστη μοριακή ύφεση
Απώλεια μέγιστης μοριακής ύφεσης, εμφάνιση μεταλλάξεων με ευαισθησία στο imatinib
Απώλεια πλήρους αιματολογικής ύφεσης, απώλεια πλήρους κυτταρογενετικής ύφεσης, νέες επιπλέον κυτταρογενετικές ανωμαλίες, μεταλλάξεις χωρίς ευαισθησία στο imatinib
διάστημα σε ασθενή που ακόμη ανέχεται καλά τη θερα-πεία υποδεικνύει ανθεκτική νόσο κατά την οποία ο ασθε-νής έχει στατιστικά μεγαλύτερες πιθανότητες επιδείνωσης εφόσον συνεχίσει την ίδια θεραπεία. η προαναφερθείσα κατάσταση περιγράφεται με τον όρο πρωτοπαθής αντο-χή. Ως δευτεροπαθής αντοχή ορίζεται η επιβεβαιωμένη απώλεια οποιουδήποτε βαθμού κυτταρογενετικής ή μο-ριακής ύφεσης είχε επιτευχθεί.
με μια πρώτη ματιά τα αποτελέσματα στην επιβίωση μετά από θεραπεία με imatinib φαίνονται εξαιρετικά. Όμως με τα κριτήρια του ELN περίπου 25% των περιπτώσεων ασθενών σε χρόνια φάση της νόσου θα αποδειχθεί ότι εμ-φανίζουν «αποτυχία» ή ανταπόκριση «κατώτερη της επι-θυμητής».4 Επίσης ένα άλλο ποσοστό ασθενών εμφανίζει «δυσανεξία» στο φάρμακο. ςτα 8 χρόνια παρακολούθη-σης των ασθενών στη μελέτη IRIS, η ολική επιβίωση στο σκέλος του imatinib ήταν 85%. Όταν στην αξιολόγηση συμπεριλαμβάνονται μόνο οι θάνατοι ασθενών που είχαν σχέση με την υποκείμενη Χμλ, το ποσοστό της ολικής επιβίωσης φτάνει το 93%.5 οι προαναφερθείσες 3 ομάδες ασθενών με την ένδειξη «αποτυχία», ανταπόκριση «κα-τώτερη της επιθυμητής» και δυσανεξία στο imatinib εί-ναι υποψήφιες για θεραπεία 2ης γραμμής. οι μηχανισμοί ανάπτυξης αντοχής στη θεραπεία με imatinib έχουν γίνει αντικείμενο εκτενούς μελέτης. ςημαντικό ποσοστό αν-θεκτικών περιπτώσεων οφείλεται στην αδυναμία ελέγ-χου της δραστικότητας της BCR/ABL κινάσης. με τη
δημιουργία εναλλακτικών ισχυροτέρων BCR/ABL ανα-σταλτών με δραστικότητα σε περιπτώσεις αντοχής στο imatinib λόγω μεταλλάξεων στο BCR/ABL τμήμα της κινάσης, προέκυψε αισιοδοξία για την περαιτέρω αντι-μετώπισης της νόσου.
οι μεταλλάξεις μπορούν να διακριθούν στις ακόλου-θες βασικές κατηγορίες:6-10
1. μεταλλάξειες οι οποίες προκαλούν ελάττωση ή παύ-ση της αναστολής της BCR/ABL δραστηριότητας. οι μεταλλάξεις αυτές εντοπίζονται εσωτερικά στο τμή-μα της BCR/ABL κινάσης και εμποδίζουν την σύν-δεση του imatinib.
2. Πολλαπλασιασμός του γενετικού υλικού ή απόκτηση επιπλέον χρωμοσωμάτων Ph με αποτέλεσμα υπερπα-ραγωγή BCR/ABL πρωτεϊνης.
3. Ανεξάρτητοι μηχανισμοί απώλειας ανταπόκρισης, λόγω βλαβών στο BCR/ABL τμήμα. Ευθύνονται για απώλεια της ανταπόκρισης σε ορισμένες περιπτώσεις Χμλ.ςτη μελέτη START-C 46% των ασθενών με Χμλ
σε χρόνια φάση και ανάπτυξη αντοχής στη θεραπεία με imatinib, δεν είχαν ανιχνεύσιμες μεταλλάξεις στο τμήμα BCR/ABL. τα στοιχεία αυτά επιβεβαίωσαν ότι η δευ-τερογενής αντοχή στο imatinib είναι πολυπαραγοντική. Φαίνεται ότι εκτός των ήδη αναφερθέντων μηχανισμών ενίσχυσης της δραστηριότητας της BCR/ABL τυροσινι-κής κινάσης, εμπλέκονται οι ενεργοποιημένες κινάσες της
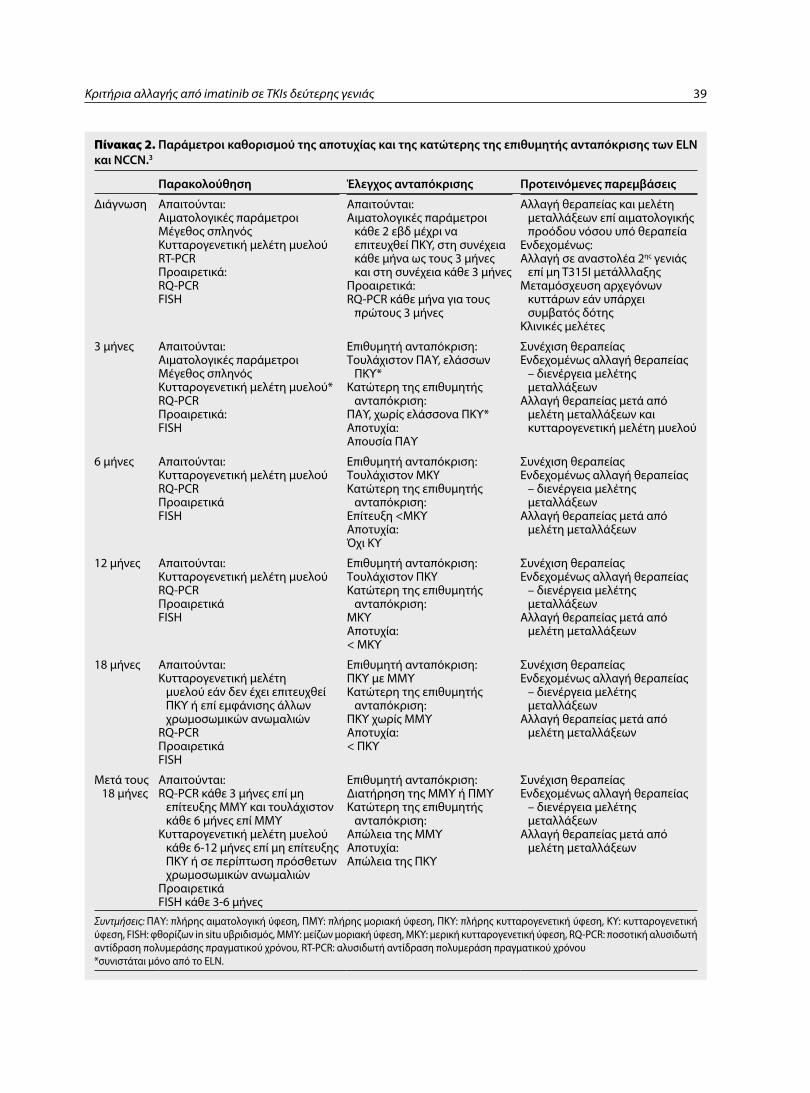
Κριτήρια αλλαγής από imatinib σε ΤΚΙs δεύτερης γενιάς 39
Πίνακας 2. Παράμετροι καθορισμού της αποτυχίας και της κατώτερης της επιθυμητής ανταπόκρισης των ELN και NCCN.3
Παρακολούθηση Έλεγχος ανταπόκρισης Προτεινόμενες παρεμβάσεις
Διάγνωση Απαιτούνται:Αιματολογικές παράμετροιΜέγεθος σπληνόςΚυτταρογενετική μελέτη μυελούRΤ-PCRΠροαιρετικά:RQ-PCRFISH
Απαιτούνται:Αιματολογικές παράμετροι
κάθε 2 εβδ μέχρι να επιτευχθεί ΠΚΥ, στη συνέχεια κάθε μήνα ως τους 3 μήνες και στη συνέχεια κάθε 3 μήνες
Προαιρετικά:RQ-PCR κάθε μήνα για τους
πρώτους 3 μήνες
Αλλαγή θεραπείας και μελέτη μεταλλάξεων επί αιματολογικής προόδου νόσου υπό θεραπεία
Ενδεχομένως:Αλλαγή σε αναστολέα 2ης γενιάς
επί μη T315I μετάλλλαξηςΜεταμόσχευση αρχεγόνων
κυττάρων εάν υπάρχει συμβατός δότης
Κλινικές μελέτες
3 μήνες Απαιτούνται:Αιματολογικές παράμετροιΜέγεθος σπληνόςΚυτταρογενετική μελέτη μυελού*RQ-PCRΠροαιρετικά:FISH
Επιθυμητή ανταπόκριση:Τουλάχιστον ΠΑΥ, ελάσσων
ΠΚΥ*Κατώτερη της επιθυμητής
ανταπόκριση:ΠΑΥ, χωρίς ελάσσονα ΠΚΥ*Αποτυχία:Απουσία ΠΑΥ
Συνέχιση θεραπείαςΕνδεχομένως αλλαγή θεραπείας
– διενέργεια μελέτης μεταλλάξεων
Αλλαγή θεραπείας μετά από μελέτη μεταλλάξεων και κυτταρογενετική μελέτη μυελού
6 μήνες Απαιτούνται:Κυτταρογενετική μελέτη μυελούRQ-PCRΠροαιρετικάFISH
Επιθυμητή ανταπόκριση:Τουλάχιστον MKYΚατώτερη της επιθυμητής
ανταπόκριση:Επίτευξη <ΜΚΥΑποτυχία:Όχι ΚΥ
Συνέχιση θεραπείαςΕνδεχομένως αλλαγή θεραπείας
– διενέργεια μελέτης μεταλλάξεων
Αλλαγή θεραπείας μετά από μελέτη μεταλλάξεων
12 μήνες Απαιτούνται:Κυτταρογενετική μελέτη μυελούRQ-PCRΠροαιρετικάFISH
Επιθυμητή ανταπόκριση:Τουλάχιστον ΠKYΚατώτερη της επιθυμητής
ανταπόκριση:ΜΚΥΑποτυχία:< ΜΚΥ
Συνέχιση θεραπείαςΕνδεχομένως αλλαγή θεραπείας
– διενέργεια μελέτης μεταλλάξεων
Αλλαγή θεραπείας μετά από μελέτη μεταλλάξεων
18 μήνες Απαιτούνται:Κυτταρογενετική μελέτη
μυελού εάν δεν έχει επιτευχθεί ΠΚΥ ή επί εμφάνισης άλλων χρωμοσωμικών ανωμαλιών
RQ-PCRΠροαιρετικάFISH
Επιθυμητή ανταπόκριση:ΠΚΥ με ΜΜΥΚατώτερη της επιθυμητής
ανταπόκριση:ΠΚΥ χωρίς ΜΜΥΑποτυχία:< ΠΚΥ
Συνέχιση θεραπείαςΕνδεχομένως αλλαγή θεραπείας
– διενέργεια μελέτης μεταλλάξεων
Αλλαγή θεραπείας μετά από μελέτη μεταλλάξεων
Μετά τους 18 μήνες
Απαιτούνται:RQ-PCR κάθε 3 μήνες επί μη
επίτευξης ΜΜΥ και τουλάχιστον κάθε 6 μήνες επί ΜΜΥ
Κυτταρογενετική μελέτη μυελού κάθε 6-12 μήνες επί μη επίτευξης ΠΚΥ ή σε περίπτωση πρόσθετων χρωμοσωμικών ανωμαλιών
ΠροαιρετικάFISH κάθε 3-6 μήνες
Επιθυμητή ανταπόκριση:Διατήρηση της ΜΜΥ ή ΠΜΥΚατώτερη της επιθυμητής
ανταπόκριση:Απώλεια της ΜΜΥΑποτυχία:Απώλεια της ΠΚΥ
Συνέχιση θεραπείαςΕνδεχομένως αλλαγή θεραπείας
– διενέργεια μελέτης μεταλλάξεων
Αλλαγή θεραπείας μετά από μελέτη μεταλλάξεων
Συντμήσεις: ΠΑΥ: πλήρης αιματολογική ύφεση, ΠΜΥ: πλήρης μοριακή ύφεση, ΠΚΥ: πλήρης κυτταρογενετική ύφεση, ΚΥ: κυτταρογενετική ύφεση, FISH: φθορίζων in situ υβριδισμός, ΜΜΥ: μείζων μοριακή ύφεση, ΜΚΥ: μερική κυτταρογενετική ύφεση, RQ-PCR: ποσοτική αλυσιδωτή αντίδραση πολυμεράσης πραγματικού χρόνου, RT-PCR: αλυσιδωτή αντίδραση πολυμεράση πραγματικού χρόνου*συνιστάται μόνο από το ELN.

Ν.-Α. Βύνιου και συν40
οικογένειας SrC (SFKs) καθώς και η ανάπτυξη παθολογι-κών χρωμοσωμικών ανωμαλιών στα Ph θετικά κύτταρα.9,11
οι σημειακές μεταλλάξεις στο τμήμα της κινάσης ABL φαίνεται να είναι ο πλέον συχνός μηχανισμός αντί-στασης. ςτη μεγάλη μελέτη 319 ασθενών σε χρόνια φάση Χμλ των Khorashad και συν. βρέθηκε οτι οι μεταλλά-ξεις στο τμήμα της κινάσης ήταν ο μόνος ανεξάρτητος προγνωστικός παράγων για απώλεια της πλήρους κυττα-ρογενετικής ύφεσης και υψηλότερο κίνδυνο μετατροπής της νόσου (κίνδυνος x3,8 και x3,7 αντιστοίχως) σε σύ-γκριση με την ομάδα ασθενών χωρίς μεταλλάξεις.12 οι ασθενείς που είχαν μεταλλάξεις στην P-loop εμφάνιζαν υψηλό κίνδυνο εξέλιξης της νόσου.13 το ίδιο έχει επιβε-βαιωθεί και από άλλες μελέτες σε ασθενείς που λάμβαναν imatinib. Όμως οι Jabbour και συνεργάτες δεν επιβεβαί-ωσαν τα παραπάνω ευρήματα στη μελέτη τους14 ενώ οι Branford και συνεργάτες έδειξαν μεγαλύτερη συχνότητα μεταλλάξεων στην προχωρημένη χρονία φάση και στην επιταχυνόμενη φάση της νόσου.15
τόσο το dasatinib όσο και το nibotinib είναι ισχυροί αναστολείς με δραστηριότητα έναντι ενός ευρέως φά-σματος μεταλλάξεων με αντοχή στο imatinib.16,17 και τα δύο φάρμακα αρχικά πήραν έγκριση από το FDA για την θεραπεία ασθενών με Χμλ με αντοχή ή δυσανεξία στη θεραπεία με imatinib.
Από όλες τις μεταλλάξεις στο τμήμα της ABL κινάσης η μετάλλαξη T315I παρουσίαζει τη μεγαλύτερη αντίσταση στη θεραπεία με imatinib, dasatinib, nibotinib. ορισμένες μελέτες βρίσκουν ότι η μετάλλαξη T315I συσχετίζεται με εξέλιξη της νόσου και πτωχή επιβίωση.18,19 οι Jabbour και συνεργάτες ανέφεραν ότι η επιβίωση των ασθενών με τ315ι εξαρτάται από την φάση της νόσου στην οποία αναπτύσσεται η μετάλλαξη. Ασθενείς με την τ315ι σε χρονία φάση της νόσου είχαν καλοήθη πορεία, δηλαδή 2ετή επιβίωση 87%.17 το dasatinib και το nibotinib έχουν δραστηριότητα έναντι των περισσοτέρων μεταλλάξεων που συνδέονται με αντοχή στο imatinib με εξαίρεση την μετάλλαξη τ315ι που εμφανίζει υψηλή αντοχή και στα τρία φάρμακα.20,21
Ειδικά το dasatinib δεν είναι το καταλληλότερο και για την μετάλλαξη F317L που συσχετίζεται με μέτρια αντοχή στο φάρμακο. η F317L συχνά συνοδεύεται από αιματο-λογική ανταπόκριση, σπάνια όμως από κυτταρογενετική ανταπόκριση και τελικά από ανάπτυξη αντοχής κατά την θεραπεία με dasatinib.22 οι μεταλλάξεις G250E, E255ν/κ και V299L εμφανίζουν μέτρια αντοχή στο dasatinib, κα-θώς έχουν αναφερθεί ορισμένες περιπτώσεις ασθενών με μικρή κυτταρογενετική ανταπόκριση.19
το nibotinib είναι ένα δομικό παραγωγό του imatinib με κλινική δραστικότητα έναντι των περισσοτερων με-ταλλάξεων με αντοχή στο imatinib όπως και στις σχετι-κά ανθεκτικές Υ253η, Ε255ν/κ και F359V.16-20
η επιλογή της θεραπείας 2ης γραμμής μεταξύ του
dasatinib και του nibotinib σε περιπτώσεις ανάπτυξης αντο-χής ή εμφάνισης δυσανεξίας στη θεραπεία με imatinib, μπορεί να γίνει με βάση την αναγνώριση συγκεκριμένων μεταλλάξεων στο ABL τμήμα και την ειδική τους απο-δεδειγμένη ευαισθησία στους αναστολείς τυροσίνης κι-νάσης 2ης γενιάς.
η επιλογή της αγωγής πρέπει να λαμβάνει υπόψιν συ-νοδές παθήσεις του ασθενούς οι οποίες επηρεάζουν την λήψη του συγκεκριμένου αναστολέα. ςαν παραδείγματα παρενεργειών αναφέρουμε την πλευριτική συλλογή με το dasatinib και τον αυξημένο κίνδυνο επιμήκυνσης του QT, την αύξηση των επιπέδων της γλυκόζης και της λιπάσης του ορού και την αύξηση των ηπατικών ενζύμων με το nibotinib. ο κάθε αναστολέας έχει συγκεκριμένο προφίλ παρενεργειών που θα πρέπει να ληφθούν υπόψιν κατά την εξατομίκευση της θεραπείας των ασθενών με Χμλ.
ςε ασθενείς που χάνουν την ανταπόκριση τους και εμφανίζουν αντοχή κατά την θεραπεία με dasatinib ή nibotinib, είναι λογικό να δοθεί ο εναλλακτικός αναστο-λέας 2ης γενιάς εκτός από την περίπτωση ανάπτυξης της τ315ι μετάλλαξης. η πλειοψηφία των εμφανιζόμενων μεταλλάξεων συνήθως δεν έχει διασταυρούμενη αντοχή με τον άλλο αναστολέα.
ιδιαίτερο ενδιαφέρον παρουσίαζει το γεγονός ότι συ-γκεκριμένες μεταλλάξεις κατά τη θεραπεία με dasatinib όπως η V299L, F317I και τ315ι διατηρούν ευαισθησία στο imatinib in vitro. Αντίθετα οι μεταλλάξεις που προ-καλούν αντοχή κατά την λήψη nibotinib δεν έχουν καμία ευαισθησία στο imatinib.23 Ένας άλλος παράγοντας που πρέπει να ληφθεί υπόψιν στην επιλογή του αναστολέα 2ης γενιάς, είναι η δυνατότητα του ασθενούς να συμμορ-φωθεί με τον τρόπο λήψης του φαρμάκου. το dasatinib χορηγείται μια φορά την ημέρα χωρίς διαιτητικό περιο-ρισμό. το nibotinib λαμβάνεται 2 φορές την ημέρα και χρειάζεται διαιτητικός περιορισμός.
ςτο τμήμα του κεφαλαίου που αναφέρεται στην δευ-τεροπαθή αντίσταση μετά από θεραπεία με imatinib θα αναφερθούμε στην εμφάνιση επιπλέον κλωνικών χρω-μοσωματικών ανωμαλιών. ςε ασθενείς σε επιταχυνόμε-νη φάση, η ανάπτυξη νέου κλώνου είχε σαν αποτέλεσμα μικρότερα ποσοστά ανταπόκρισης και σε σύντομο χρο-νικό διάστημα αποτυχία της θεραπείας.24 Ενδιαφέρον πα-ρουσιάζουν μελέτες, όπως αυτή που περιλαμβάνει 498 ασθενείς με Χμλ σε επιταχυνόμενη ή χρόνια φάση. η ανάπτυξη κλωνικής ανωμαλίας δεν ήταν σημαντικός πα-ράγων που επηρέαζε αρνητικά την μέγιστη ή πλήρη κυτ-ταρογενετική ανταπόκριση κατά την θεραπεία με imatinib. Αναδείχθηκε όμως ως ανεξάρτητος προγνωστικός παρά-γων που συνδέεται με χαμηλή επιβίωση στη χρόνια και επιταχυνόμενη φάση της Χμλ.25 ςε ασθενείς σε χρόνια φάση Χμλ στους οποίους είχε αποτυχία η θεραπεία με imatinib και ελάμβαναν αναστολείς 2ης γενιάς, η αιμα-τολογική ανταπόκριση, τα ποσοστά κυτταρογενετικής

Κριτήρια αλλαγής από imatinib σε ΤΚΙs δεύτερης γενιάς 41
ανταπόκρισης, η ολική επιβίωση και η ελευθερη νόσου επιβίωση δεν διέφεραν στην χρόνια φάση μεταξύ αυτών που είχαν επιπλέον χρωμοσωμική ανωμαλία και αυτών που δεν εμφάνιζαν νέο κλώνο.26 Πρέπει όμως να σημειω-θεί ότι η ανάπτυξη νέας κλωνικής ανωμαλίας είχε σημα-ντικά δυσμενή επίπτωση όταν συσχετιζόταν και με άλλα χαρακτηριστικά της επιταχυνόμενης φάσης. οι ασθενείς που εμφάνιζαν τρισωμία 8, ανωμαλία στο χρωμόσωμα 17 και σύμπλοκες κυτταρογενετικές βλάβες είχαν τη χειρό-τερη πρόγνωση, ανεξάρτητα του αριθμού των μεταφά-σεων που ανιχνεύονταν.27
ςε τελική ανάλυση, οι μελέτες αναδεικνύουν την χρη-σιμότητα της ανάλυσης των μεταλλάξεων γιατί αναγνω-ρίζουν μια ομάδα ασθενών οι οποίοι χρήζουν στενότερης παρακολούθησης γιατί εμφανίζουν μεγαλύτερο κίνδυνο επιδείνωσης της νόσου. ςτην ομάδα αυτή των ασθενών περιλαμβάνονται εκείνοι για τους οποίους η αλλογενής μεταμόσχευση μυελού είναι η ενδεδειγμένη θεραπεία.
ςτα κριτήρια αλλαγής θεραπείας από imatinib σε αναστολείς τυροσινικής κινάσης 2ης γενιάς, οφείλουμε να παραθέσουμε την εμπειρία από την χορήγηση υψη-λότερων δόσεων imatinib συγκριτικά με την καθιερομέ-νη δοσολογία των 400mg ημερησίως στην προσπάθεια προσπέλασης της πρωτογενούς αντοχής. Ένας από τους λόγους πρωτογενούς αντοχής είναι η χαμηλή ενδοκυτ-τάρια συγκέντρωση του imatinib.26,28 ο ανθρώπινος με-ταφορέας οργανικών κατιόντων (human organic cation transporter 1 – hOCT-1 ) θεωρείται ως ο πλέον ισχυρός δείκτης ανταπόκρισης στη θεραπεία με imatinib.29 Πρό-σφατα αναφέρθηκε ότι οι περισσότεροι ασθενείς με μη ικανοποιητική ανταπόκριση στο imatinib, είχαν χαμηλή δραστηριότητα hOCT1.30 ςτην πρόσφατη μετανάλυση ασθενών της μελέτης TIDEL, η μεγίστη μοριακή αντα-πόκριση στους 60 μήνες ήταν μεγαλύτερη στους ασθε-νείς με υψηλή δραστηριότητα hOCT1 συγκρινόμενη με αυτήν ασθενών με χαμηλή δραστηριότητα OCT1 (89% έναντι 55% αντίστοιχα). η χαμηλή δραστηριότητα OCT1 συσχετίστηκε με στατιστικά σημαντικά χαμηλότερη ολι-κή επιβίωση (87% έναντι 96%). Ενδιαφέρον παρουσιάζει η συσχέτιση ανάλυσης μεταλλάξεων με την δραστηριό-τητα της OCT1. Αναφέρεται ότι η χαμηλή δραστηριότη-τα OCT1 συσχετίζεται με υψηλό ποσοστό μεταλλάξεων του ABL τμήματος (21% έναντι 4%). οι διαφορές αυτές ήταν στατιστικά σημαντικότατες στους ασθενείς της μελέ-της TIDEL που ελάμβαναν λιγότερο από 600 mg imatinib την ημέρα.31 Εδώ πρέπει να αναφερθεί ότι η δραστικότητα του nibotinib και του dasatinib δεν εξαρτάται από την έκ-φραση του hOCT1 επομένως, όπως αναφέρεται και στις οδηγίες του NCCN του 2012 για την Χμλ, οι ασθενείς με χαμηλή έκφραση OCT1 είναι πιθανόν να οφελούνται από την χορήγηση nibotinib ή dasatinib.32
Παρά το γεγονός ότι υπάρχουν αποτελέσματα μελε-τών που δείχνουν ότι οι ασθενείς που μπορούν να ανε-
χτούν υψηλότερες δόσεις imatinib, έχουν κλινικό όφελος, η μεγάλη συχνότητα τοξικότητας με τις υψηλότερες δό-σεις imatinib, καθώς το μεγάλο κόστος της θεραπείας μας καθοδηγούν να χορηγούμε σε ασθενείς υψηλότερες δό-σεις imatinib από τα 400 mg ημερησίως μόνο αν υπάρ-χει συγκεκριμένο κλινικό πρωτόκολλο.22
ιδιαίτερο ενδιαφέρον παρουσίαζει η μελέτη φάσης ιι START-R που εκτίμησε την αποτελεσματικότητα των ψηλών δόσεων imatinib συγκριτικά με το dasatinib.33 Αξιολογήθηκαν 150 ασθενείς σε χρονία φάση Χμλ αν-θεκτικοί στο imatinib, οι οποίοι τυχαιοποίηθηκαν μεταξύ 800 mg imatinib και 140 mg (70mg δυο φορές την ημέ-ρα) dasatinib. To dasatinib υπερείχε σημαντικά των 800 mg imatinib εφόσον είχε αποτύχει η θεραπεία με 600 mg imatinib. Αντίθετα υπήρχαν ισοδύναμα αποτελέσματα με-ταξύ dasatinib και imatinib στους ασθενείς με αποτυχία στη δόση των 400 mg imatinib. Όμως στα 2 χρόνια παρα-κολούθησης, το dasatinib υπερείχε των 800 mg imatinib στους ασθενείς που ήταν ανθεκτικοί στα 400 mg και 600 mg ημερήσιας δόσης imatinib. ςτα 2 χρόνια το dasatinib εμφάνιζε υπεροχή στην αιματολογική ύφεση (93% έναντι 82%) στη μείζονα κυτταρογενετική ύφεση (53% έναντι 33%) και στην πλήρη κυτταρογενετική ύφεση (29% ένα-ντι 12%). Επίσης το dasatinib υπερείχε ως προς την επιβί-ωση ελεύθερης νόσου, καταδεικνύοντας ότι το dasatinib είναι αποτελεσματικό στους ασθενείς με Χμλ σε χρόνια φάση, ανθεκτικούς τόσο στα 400 mg όσο και σε υψηλό-τερες δόσεις imatinib.34
Iδιαίτερη αναφορά πρέπει να γίνει και στη θεραπευ-τική αντιμετώπιση της ομάδας ασθενών με κατώτερη της επιθυμητής κυτταρογενετική ανταπόκριση. η στρατηγι-κή της παρακολούθησης και αναμονής (wait and watch), η αύξηση της δόσης του Imatinib στα 800 mg, εφόσον είναι ανεκτή και τέλος η χορήγηση αναστολέων 2ης γε-νιάς είναι πιθανοί εναλλακτικοί χειρισμοί. ςτην παρού-σα φάση δεν υπάρχουν δεδομένα που να αποδεικνύουν τη σαφή υπεροχή συγκεκριμένης θεραπευτικής επιλογής ως προς την ολική επιβίωση ή την επιβίωση ελευθέρης νόσου,35 όμως μελέτες αναφέρουν ότι οι κυτταρογενετι-κές ανταποκρίσεις που είναι κατώτερες των επιθυμητών ίσως τελικά δεν διαφέρουν από τις «αποτυχίες» κυτταρο-γενετικής ανταπόκρισης ως προς την εξέλιξη της νόσου. τα καλά αποτελέσματα και η καλή ανοχή της θεραπείας με αναστολείς 2ης γενιάς προοδευτικά κερδίζουν έδαφος στη θεραπευτική αντιμετώπιση αυτής της ομάδας ασθε-νών.36 Όσον αφορά τη μεγίστη μοριακή ανταπόκριση δεν επιβεβαιώθηκαν από άλλες μελέτες τα ευρήματα της με-λέτης IRIS.37 ςτη μελέτη αυτή είχε δειχθεί ότι στους 72 μήνες παρακολούθησης, η ομάδα ασθενών που επιτυγχά-νει πλήρη κυτταρογενετική και μεγίστη μοριακή ύφεση στους 18 μήνες συγκρινόμενη με αυτή με πλήρη κυττα-ρογενετική αλλά όχι μεγίστη μοριακή ύφεση στο ίδιο χρονικό διάστημα είχε στατιστικά σημαντικά αυξημένη

Ν.-Α. Βύνιου και συν42
πιθανότητα επιβίωσης ελεύθερης νόσου,38 όμως έχει επι-βεβαιωθεί από μελέτες ότι η επίτευξη μεγίστης μοριακής ύφεσης συμπληρωματικά στην πλήρη κυτταρογενετική ύφεση ελαχιστοποιεί τον κίνδυνο απώλειας της πλήρους κυτταρογενετικής ύφεσης3 και της προόδου της νόσου και συσχετίζεται με σταθερή ανταπόκριση.39 Επίσης έχει δειχθεί ότι η επίτευξη πλήρους μοριακής ύφεσης με διάρ-κεια 2 ετών δίνει τη δυνατότητα διακοπής της θεραπεί-ας με imatinib.40 οι πιο πρόσφατες συστάσεις του ELN βασίζονται στα προαναφερθέντα και ορίζουν τον επιθυ-μητό χρόνο επίτευξης μεγίστης μοριακής ανταπόκρισης στους 18 μήνες. Δεν θεωρούν ότι η μη επίτευξη μεγίστης μοριακής ανταπόκρισης ή η απώλεια της μεγίστης μορι-ακής ανταπόκρισης χωρίς να συνοδεύεται από απώλεια της πλήρους κυτταρογενετικής ύφεσης αποτελεί «αποτυ-χία» της θεραπείας, αλλά εμπίπτει στην κατηγορία της «κατώτερης της επιθυμητής» ανταπόκρισης. τα υπάρχο-ντα κλινικά στοιχεία από μελέτες δεν αποδεικνύουν ότι η αλλαγή θεραπείας σε αναστολείς 2ης γενιάς θα βελτί-ωνε την απώτερη εξέλιξη της νόσου σε αυτή την ομάδα ασθενών.1,41 η μόνη εξαίρεση είναι όταν η μη επίτευξη ή η απώλεια της μεγίστης μοριακής ανταπόκρισης συ-σχετίζεται με μεταλλάξεις ανθεκτικές στο imatinib όπως η T315I ή Ε255κ/ν και η Υ253η. οι περιπτώσεις αυτές θεωρούνται «αποτυχίες» και πρέπει να αντιμετωπίζονται
με άμεση τροποποίηση στη θεραπευτική αντιμετώπιση.1
μελέτη μεταλλάξεων συστείνεται από το ELN τό-σο κατά την «αποτυχία» όσο και για την «κατώτερη της επιθυμητής» ανταπόκριση. ο λόγος είναι η πιθανή προ-σαρμογή ή αλλαγή της θεραπείας και η επιλογή του κα-ταλλήλου αναστολέα 2ης γενιάς για την αντιμετώπιση του υπολειμματικού κλώνου που φέρει την ειδική μετάλλαξη. τέλος, η μελέτη των μεταλλάξεων μπορεί να προσδιορίσει εάν πρέπει να γίνει αλλογενής μεταμόσχευση μυελού.4243
Δυστυχώς εκτός από τις παραμέτρους που μας επι-τρέπουν την αναγνώριση της πρωτοπαθούς αντοχής, δεν υπάρχουν οδηγά σημεία για τη διάκριση των ασθενών που θα εμφανίσουν δευτεροπαθή αντοχή ή επιδείνωση νό-σου. τόσο η πρωτοπαθής όσο και η δευτεροπαθής αντο-χή μοιάζει να εμφανίζονται πιο συχνά στους ασθενείς οι οποίοι ταξινομούνται σε ενδιαμέσου και υψηλού κινδύνου ομάδες με τις αιματολογικές και κλινικές παραμέτρους που χρησιμοποιούν τα διεθνή προγνωστικά συστήματα (π.χ. SOKAL, EURO, EUTOS).44,45 ςε αυτή τη φάση δεν υπάρχουν ειδικές θεραπευτικές οδηγίες για αυτούς τους ασθενείς. Είναι πιθανό να προκύψουν από τις μελέτες σύ-γκρισης των αποτελεσμάτων του imatinib στα 400 mg με τα nilotinib και dasatinib ως θεραπείες 1ης γραμμής και να τροποποιήσουν τις απόψεις μας στο μέλλον.
Criteria for treatment switch from imatinib to second generation TKIsby Nora-Athina Viniou, Panagiotis Diamantopoulos, Vassiliki Papadopoulou
1st Department of Internal Medicine, School of Medicine, National and Kapodistrian University of Athens, “Laiko” General Hospital, Athens
ABSTRACT: Patients with chronic myelogenous leukemia do not invariably respond to treatment with imatinib. To date, there is no safe way to predict which patient will show primary or secondary resist-ance to the drug. Clinical, hematologic, cytogenetic and molecular criteria are used to assess treatment response. Treatment response assessment should be made in predetermined intervals according to the European Leukemia Net (ELN) and the National Comprehensive Cancer Network (NCCN) recommen-dations. In the era of second generation tyrosine kinase inhibitors (TKIs), patients not achieving a robust response to treatment with imatinib, as well as those with treatment intolerance are candidates for treat-ment switch to dasatinib or nilotinib. Treatment failure is largely caused by the emergence of mutations of the BCR/ABL kinase that are resistant to the drug. Among them, T315I is a mutation resistant to all currently available TKIs. For all the rest, the choice of the more suitable 2nd generation TKI should be made based on mutation analysis, the adverse event profile of the drugs as well as their kinetics, which determine the dietary restrictions accompanying their use. Other factors, such as the emergence of addi-tional karyotypic aberrations and the low intracellular concentration of the human organic cation trans-porter – 1 (hOCT-1) may be responsible for imatinib treatment failure, although hOCT-1 intracellular concentration does not affect the effectiveness of 2nd generation TKIs. Comparative studies keep on pro-viding data about the rational use of all three drugs, both as first and second line treatment.

Κριτήρια αλλαγής από imatinib σε ΤΚΙs δεύτερης γενιάς 43
Βιβλιογραφία 1. Barani M, Cortes J, Pane F, et al. Chronic myeloid leukemia:
an update of concepts and management recommendations of European LeukemiaNet. J Clin Oncol. 2009; 27(35):6041-6051.
2. O’Brien S, Berman E, Borghaei H, et al; National Compre-hensive Cancer Network. NCCN clinical practice guidelines in oncology: chronic myelogenous leukemia. J Natl Compr Canc Netw. 2009; 7(9):984-1023.
3. Saglio G, Fava C. Practical monitoring of chronic myelog-enous leukemia: when to change treatment. Journal of the National Comprehensive Cancer Network 2012; 10:121-129
4. Marin D, Milojkovic D, Olavarria E, et al. European Leu-kemiaNet criteria for failure or suboptimal response reliably identify patients with CML in early chronic phase treated with imatinib whose eventual outcome is poor. Blood. 2008; 112(12):4437-4444. Epub 2008 Aug 20.
5. Dreininger M, O’brien SG, Guilhaut F, et al. International randomized study of interferon versus STI571 (IRIS) 8-year follow up: sustained survival and low risk for progression or event in patients with newly diagnosed chronic myeloid leukemia in chronic phase with imatinib. Abstract Blood. 2009; 114:1126.
6. Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, Sawyers CL. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001; 293(5531):876-880. Epub 2001 Jun 21.
7. Shah NP, Nicoll JM, Nagar B, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002; 2(2):117-125.
8. Branford S, Rudzki Z, Walsh S, et al. High frequency of point mutations clustered within the adenosine triphosphate-binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood. 2002; 99(9):3472-3475.
9. von Bubnoff N, Schneller F, Peschel C, Duyster J. BCR-ABL gene mutations in relation to clinical resistance of Philadelphia-chromosome-positive leukaemia to STI571: a prospective study. Lancet. 2002; 359(9305):487-491.
10. Hochhaus A, Kreil S, Corbin AS, et al. Molecular and chro-mosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia. 2002; 16(11):2190-2196.
11. Apperley JF. Part I: mechanisms of resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol. 2007; 8(11):1018-1029.
12. Khorashad JS, de Lavallade H, Apperley JF, et al. Finding of kinase domain mutations in patients with chronic phase chronic myeloid leukemia responding to imatinib may identify those at high risk of disease progression. J Clin Oncol. 2008; 26(29):4806-4813. Epub 2008 Jul 21.
13. Soverini S, Martinelli G, Rosti G, et al. ABL mutations in
late chronic phase chronic myeloid leukemia patients with up-front cytogenetic resistance to imatinib are associated with a greater likelihood of progression to blast crisis and shorter survival: a study by the GIMEMA Working Party on Chronic Myeloid Leukemia. J Clin Oncol. 2005; 23(18):4100-4109. Epub 2005 May 2.
14. Jabbour E, Kantarjian H, Jones D, et al. Frequency and clinical significance of BCR-ABL mutations in patients with chronic myeloid leukemia treated with imatinib mesylate. Leukemia. 2006; 20(10):1767-1773. Epub 2006 Jul 20.
15. Branford S, Rudzki Z, Walsh S, et al. Detection of BCR-ABL mutations in patients with CML treated with imatinib is virtually always accompanied by clinical resistance, and mutations in the ATP phosphate-binding loop (P-loop) are associated with a poor prognosis. Blood. 2003; 102(1):276-283. Epub 2003 Mar 6.
16. Talpaz M, Shah NP, Kantarjian H, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006; 354(24):2531-2541.
17. Kantarjian H, Giles F, Wunderle L, et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med. 2006; 354(24):2542-2551.
18. Jabbour E, Kantarjian H, Jones D, et al. Characteristics and outcomes of patients with chronic myeloid leukemia and T315I mutation following failure of imatinib mesylate therapy. Blood. 2008; 112(1):53-55. Epub 2008 Apr 10.
19. Nicolini FE, Hayette S, Corm S, et al. Clinical outcome of 27 imatinib mesylate-resistant chronic myelogenous leukemia patients harboring a T315I BCR-ABL mutation. Haematologica. 2007; 92(9):1238-1241.
20. Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004; 305(5682):399-401.
21. O’Hare T, Walters DK, Stoffregen EP, et al. In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res. 2005; 65(11):4500-4505.
22. Shah NP, Skaggs BJ, Branford S, et al. Sequential ABL kinase inhibitor therapy selects for compound drug-resistant BCR-ABL mutations with altered oncogenic potency. J Clin Invest. 2007;117(9):2562-9.
23. Smith CC, Shah NP. Tyrosine kinase inhibitor therapy for chronic myeloid leukemia: approach to patients with treatment-naïve or refractory chronic phase disease. Educa-tion program book, American Society of Hematology, San Diego, California, December 10-13, 2011.
24. O’Dwyer ME, Mauro MJ, Kurilik G, et al. The impact of clonal evolution on response to imatinib mesylate (STI571) in accelerated phase CML. Blood. 2002;100(5):1628-1633.
25. Cortes JE, Talpaz M, Giles F, et al. Prognostic significance of cytogenetic clonal evolution in patients with chronic myelogenous leukemia on imatinib mesylate therapy. Blood. 2003 May 15;101(10):3794-3800. Epub 2003 Jan 30.
26. Verma D, Kantarjian H, Shan J, et al. Survival outcomes for clonal evolution in chronic myeloid leukemia patients on second generation tyrosine kinase inhibitor therapy.

Ν.-Α. Βύνιου και συν44
Cancer. 2010 Jun 1;116(11):2673-81. 27. Larson RA, Druker BJ, Guilhot F, et al; IRIS (International
Randomized Interferon vs STI571) Study Group. Imatinib pharmacokinetics and its correlation with response and safety in chronic-phase chronic myeloid leukemia: a subanalysis of the IRIS study. Blood. 2008;111(8):4022-4028. Epub 2008 Feb 6.
28. Picard S, Titier K, Etienne G, et al. Trough imatinib plasma levels are associated with both cytogenetic and molecular responses to standard-dose imatinib in chronic myeloid leukemia. Blood. 2007;109(8):3496-3499. Epub 2006 Dec 27.
29. Wang L, Giannoudis A, Lane S, Williamson P, Pirmohamed M, Clark RE. Expression of the uptake drug transporter hOCT1 is an important clinical determinant of the response to imatinib in chronic myeloid leukemia. Clin Pharmacol Ther. 2008;83(2):258-264. Epub 2007 Jun 13.
30. White DL, Saunders VA, Dang P, et al. Most CML patients who have a suboptimal response to imatinib have low OCT-1 activity: higher doses of imatinib may overcome the negative impact of low OCT-1 activity. Blood. 2007;110(12):4064-4072. Epub 2007 Aug 30.
31. White DL, Dang P, Engler J, et al. Functional activity of the OCT-1 protein is predictive of long-term outcome in patients with chronic-phase chronic myeloid leukemia treated with imatinib. J Clin Oncol. 2010;28(16):2761-2767. Epub 2010 Apr 26.
32. Giannoudis A, Davies A, Lucas CM, Harris RJ, Pirmohamed M, Clark RE. Effective dasatinib uptake may occur without human organic cation transporter 1 (hOCT1): implications for the treatment of imatinib-resistant chronic myeloid leukemia. Blood. 2008; 112(8):3348-3354. Epub 2008 Jul 31.
33. Kantarjian H, Pasquini R, Hamerschlak N, et al. Dasatinib or high-dose imatinib for chronic-phase chronic myeloid leukemia after failure of first-line imatinib: a randomized phase 2 trial. Blood. 2007; 109(12):5143-5150. Epub 2007 Feb 22.
34. Kantarjian H, Pasquini R, Lévy V, et al. Dasatinib or high-dose imatinib for chronic-phase chronic myeloid leukemia resistant to imatinib at a dose of 400 to 600 milligrams daily: two-year follow-up of a randomized phase 2 study (START-R). Cancer. 2009;115(18):4136-47.
35. Jabbour E, Cortes JE, Kantarjian HM. Suboptimal response to or failure of imatinib treatment for chronic myeloid leukemia: what is the optimal strategy? Mayo Clin Proc.
2009;84(2):161-169. 36. Saglio G, Kim DW, Issaragrisil S, et al; ENESTnd Investiga-
tors. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med. 2010;362(24):2251-2259. Epub 2010 Jun 5.
37. Hehlmann R, Lauseker M, Jung-Munkwitz S, et al. Toler-ability-adapted imatinib 800 mg/d versus 400 mg/d versus 400 mg/d plus interferon-α in newly diagnosed chronic myeloid leukemia. J Clin Oncol. 2011;29(12):1634-16342. Epub 2011 Mar 21.
38. Druker BJ, Guilhot F, O’Brien SG, et al; IRIS Investigators. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355(23):2408-17.
39. Kantarjian H, O’Brien S, Shan J, et al. Cytogenetic and molecular responses and outcome in chronic myelogenous leukemia: need for new response definitions? Cancer. 2008;112(4):837-45.
40. Mahon FX, Réa D, Guilhot J, et al; Intergroupe Français des Leucémies Myéloïdes Chroniques. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010;11(11):1029-1035. Epub 2010 Oct 19.
41. Cortes J, Hochhaus A, Hughes T, Kantarjian H. Front-line and salvage therapies with tyrosine kinase inhibitors and other treatments in chronic myeloid leukemia. J Clin Oncol. 2011;29(5):524-531. Epub 2011 Jan 10.
42. Soverini S, Hochhaus A, Nicolini FE, et al. BCR-ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: recom-mendations from an expert panel on behalf of European LeukemiaNet. Blood. 2011;118(5):1208-1215. Epub 2011 May 11.
43. Branford S, Hughes TP. Mutational analysis in chronic my-eloid leukemia: when and what to do? Curr Opin Hematol. 2011;18(2):111-116.
44. Sokal JE, Cox EB, Baccarani M, et al. Prognostic discrimina-tion in «good-risk» chronic granulocytic leukemia. Blood. 1984;63(4):789-799.
45. Hasford J, Baccarani M, Hoffmann V, et al. Predicting com-plete cytogenetic response and subsequent progression-free survival in 2060 patients with CML on imatinib treatment: the EUTOS score. Blood. 2011; 118(3):686-692. Epub 2011 May 2.

Ο ρόλος της μεταμόσχευσης στη ΧΜΛ: Σε ποιους ασθενείς και πότε
Ιφιγένεια Τζάννου, Ιωάννης Μπαλταδάκης
ΠΕΡΊληΨη: η αλλογενής μεταμόσχευση αιμοποιητικών κυττάρων (αλλο-μΑκ) υπήρξε η πρώτη θερα-πευτική μέθοδος που οδήγησε στην ίαση της χρόνιας μυελογενούς λευχαιμίας (Χμλ) μέσω εκρίζωσης του Philadelphia θετικού λευχαιμικού κλώνου. μετά την εισαγωγή του πρώτου αναστολέα τυροσινικής κινάσης (imatinib) και τα εξαιρετικά αποτελέσματα με τη θεραπεία στόχευσης, η αλλο-μΑκ δεν χρη-σιμοποιείται πλέον ως 1ης γραμμής θεραπεία στην πλειοψηφία των περιπτώσεων Χμλ. Ωστόσο, η αλ-λο-μΑκ αποτελεί σήμερα επιλογή για αξιόλογο ποσοστό ασθενών (40%) που παρουσιάζουν αποτυχία στην αρχική θεραπεία με imatinib. η απόφαση για διενέργεια αλλο-μΑκ βασίζεται στην ακριβή στάθ-μιση του οφέλους και των κινδύνων από τη μεταμόσχευση. ο κίνδυνος από τη μεταμόσχευση μπορεί να εκτιμηθεί με το σύστημα βαθμολόγησης του European Group for Blood and Marrow Transplantation (ΕΒμτ score) που διατηρεί ακόμα και σήμερα την αξία του, παρότι την τελευταία δεκαετία παρατηρεί-ται βελτίωση της επιβίωσης κατά 10-15% σε όλες τις ομάδες κινδύνου. η συνεκτίμηση του ΕΒμτ score με τον ειδικό για τη μεταμόσχευση δείκτη συννοσηρότητας (hematopoietic cell transplantation-specific comorbidity index, HCT-CI), μπορεί να οδηγήσει σε ακριβέστερη και εξατομικευμένη πρόβλεψη της έκ-βασης της αλλο-μΑκ. Διευκολύνεται, έτσι, η επιλογή των πλέον κατάλληλων για μεταμόσχευση ασθε-νών και καθίσταται δυνατή η προσαρμογή της μεθόδου ανάλογα με το βαθμό κινδύνου. Αυτό μπορεί να επιτευχθεί με την επιλογή του ενδεικνυόμενου σχήματος προετοιμασίας (μυελοαφανιστικού ή μειωμέ-νης έντασης), σε συνδυασμό με τη χορήγηση τκι μετά την εγκατάσταση του μοσχεύματος και την αξι-οποίηση της δράσης του μοσχεύματος έναντι της λευχαιμίας (GVL) με εγχύσεις λεμφοκυττάρων δότη (DLI). για ασθενείς με προχωρημένη νόσο, η θεραπεία με τκι πριν τη μεταμόσχευση, με σκοπό την επίτευξη δεύτερης χρόνιας φάσης, μπορεί να βελτιώσει τα αποτελέσματα της αλλο-μΑκ. Επιπλέον, η έκβαση της αλλο-μΑκ από μη συγγενείς δότες, είναι πλέον συγκρίσιμη με αυτήν της μεταμόσχευσης από συμβατό αδελφό, λόγω επιλογής περισσότερο συμβατών δοτών με μοριακή HLA τυποποίηση υψη-λής ευκρίνειας. ςύμφωνα με τις τρέχουσες κατευθυντήριες οδηγίες του European Leukemia Net (2009), η αλλο-μΑκ συνιστάται για ασθενείς με προχωρημένη νόσο στη διάγνωση ή εξέλιξη νόσου υπό θερα-πεία με imatinib, καθώς και σε αποτυχία στους τκι δεύτερης γενιάς (2G-TKI). Όμως, οι ενδείξεις και ο κατάλληλος χρόνος για τη διενέργεια αλλο-μΑκ στην εποχή των τκι αναπροσαρμόζονται συνεχώς, με βάση δεδομένα που προκύπτουν από πρόσφατες κλινικές μελέτες. οι ασθενείς με πτωχή ανταπόκρι-ση στη θεραπεία 2ης γραμμής με τκι μπορούν να αναγνωριστούν εκ των προτέρων με βάση το Sokal score στη διάγνωση, την κυτταρογενετική ανταπόκριση στην προηγηθείσα θεραπεία με imatinib και την ουδετεροπενία κατά τη διάρκεια της αγωγής με imatinib (Hammersmith score), καθώς και από την απουσία κυτταρογενετικής ανταπόκρισης στους 3 μήνες θεραπείας με 2G-TKI. Ακόμη, το επίπεδο της μοριακής ανταπόκρισης στους 3 μήνες μπορεί να καθορίσει πρώιμα την υποομάδα των ασθενών (πε-ρίπου 10%) που δεν θα ανταποκριθούν σε 1ης ή 2ης γραμμής θεραπεία με 2G-TKI. ςτο προσεχές μέλ-λον, η μεταμόσχευση θα αποτελεί έγκαιρη θεραπεία 2ης γραμμής της Χμλ για κατάλληλους ασθενείς, με βάση τα επίπεδα μεταγράφων BCR-ABL στους 3 μήνες αγωγής με τκι. η προσέγγιση αυτή επιτρέ-πει ορθολογικό σχεδιασμό της μεθόδου και βελτίωση των αποτελέσμάτων στη Χμλ, που εξαρτώνται κυρίως από τη φάση της νόσου και το χρόνο από τη διάγνωση έως τη μεταμόσχευση.
Haema 2012; 3(1): 45-56 Copyright EAE
Αιματολογική κλινική και μονάδα μεταμόσχευσης μυελού οστών, γ.Ν.Α. «ο Ευαγγελισμός», Αθήνα
Διεύθυνση Αλληλογραφίας: ιωάννης μπαλταδάκης, Αιματολογική κλι-νική και μμμο, γΝΑ «ο Ευαγγελισμος», Αθήνα. E-mail: [email protected]
Aνασκόπηση

Ι. Τζάννου και Ι. Μπαλταδάκης46
ΕΙΣΑΓΩΓΗη αλλογενής μεταμόσχευση αρχέγονων αιμοποιητι-
κών κυττάρων (αλλο-μΑκ) ήταν η πρώτη θεραπευτική μέθοδος που αποδείχθηκε ότι οδηγεί σε εκρίζωση του Philadelphia-θετικού (Ph+) κλώνου και τελικά στην ίαση της Χμλ.1 για περισσότερα από 20 χρόνια, η αλλο-μΑκ αποτελούσε τη θεραπεία εκλογής για ασθενείς με Χμλ ηλικίας κάτω των 50 ετών, που διέθεταν ιστοσυμβατό αδελφό δότη.2 μάλιστα μέχρι το 2000, η Χμλ παρέμε-νε η συχνότερη ένδειξη για αλλο-μΑκ τόσο από αδελφό όσο και από μη συγγενή ιστοσυμβατό δότη. Αυτό υποδή-λωνε αφενός τον πλημμελή έλεγχο της νόσου με τα έως τότε διαθέσιμα φάρμακα (βουσουλφάνη, υδροξυουρία, ιντερφερόνη-α), αφετέρου δε την αποτελεσματικότητα της αλλο-μΑκ που οφείλεται στην ισχυρή ανοσολογι-κή δράση του μοσχεύματος έναντι της λευχαιμίας (Graft-versus-Leukemia, GvL) στη Χμλ.3
το 2001 εισήχθη στην κλινική πράξη ο πρώτος ανα-στολέας τυροσινικής κινάσης (τκι), το imatinib, που αντικατέστησε την αλλο-μΑκ ως πρώτης γραμμής θε-ραπεία, με αποτέλεσμα ο αριθμός των μεταμοσχεύσεων στη Χμλ να μειωθεί σημαντικά κατά την τελευταία δε-καετία.4 με την πάροδο του χρόνου, έγινε αντιληπτό ότι περίπου 40% των ασθενών που λαμβάνουν imatinib δεν ανταποκρίνονται στη θεραπεία, εμφανίζουν δυσανεξία ή αναπτύσσουν δευτεροπαθή αντοχή.5 ςε αυτές τις περι-πτώσεις η αλλο-μΑκ συνεχίζει να αποτελεί θεραπευτι-κή επιλογή, αλλά με την εισαγωγή στην κλινική πράξη των τκι δεύτερης γενιάς (2G-TKI), οι ενδείξεις για αλ-λο-μΑκ αναθεωρούνται. Παρά το γεγονός, όμως, ότι ο αριθμός των υποψηφίων προς μεταμόσχευση έχει περι-οριστεί, η αλλο-μΑκ έχει ακόμα θέση στη θεραπεία της Χμλ.6 η απόφαση για μεταμόσχευση σε ένα συγκεκρι-
μένο ασθενή πρέπει να λαμβάνεται αφού σταθμιστούν με ακρίβεια οι κίνδυνοι και τα οφέλη από την αλλο-μΑκ συγκριτικά προς τις εναλλακτικές θεραπείες.
ΕΚΤΙΜΗΣΗ ΚΙΝΔΥΝΟΥ ΣΧΕΤΙζΟΜΕΝΟΥ ΜΕ ΤΗ ΜΕΤΑΜΟΣΧΕΥΣΗΠρογνωστικά συστήματα βαθμολόγησης του κινδύνου
η αλλο-μΑκ προσφέρει μεν τη δυνατότητα οριστικής ίασης στους ασθενείς που πάσχουν από Χμλ, συνοδεύε-ται όμως από αξιοσημείωτη νοσηρότητα και θνητότητα. η προσεκτική επιλογή των ασθενών που έχουν σημαντι-κή πιθανότητα να ωφεληθούν από την αλλο-μΑκ έχει μεγάλη σημασία.7 για το λόγο αυτόν, το European Group for Blood and Marrow Transplantation (ΕΒμτ) δημοσί-ευσε το 1998 ένα προγνωστικό σύστημα βαθμολόγησης (ΕΒμτ score), με βάση πέντε παράγοντες κινδύνου, ειδι-κά για τη Χμλ που ήταν τότε η κύρια ένδειξη για αλλο-μΑκ. οι προγνωστικοί αυτοί παράγοντες είναι το είδος του δότη, η φάση της νόσου κατά τη μεταμόσχευση, η ηλικία του ασθενούς, ο συνδυασμός φύλου δότη/λήπτη και το χρονικό διάστημα από τη διάγνωση μέχρι τη με-ταμόσχευση. με βάση το μοντέλο αυτό, η σχετιζόμενη με τη μεταμόσχευση θνητότητα (TRM) και η επιβίωση διαφοροποιούνται ανάλογα με τη συνολική βαθμολογία του ασθενούς για τους παραπάνω παράγοντες (Πίνακες 1,2).8 Πρέπει να σημειωθεί, ότι οι ασθενείς που εντάχθη-καν στην εν λόγω ανάλυση είχαν μεταμοσχευθεί πριν από το 2000. με την πρόοδο που έχει συντελεστεί την τελευ-ταία δεκαετία στη μεταμόσχευση (βελτίωση υποστηρι-κτικής αγωγής, υψηλής ευκρίνειας HLA-τυποποίηση και μεγαλύτερη δυνατότητα ανεύρεσης συμβατού εθελοντή
Πίνακας 1. Προγνωστικές παράμετροι κινδύνου σχετιζόμενου με τη μεταμόσχευση κατά ΕΒΜΤΠρογνωστικός παράγοντας ΒαθμολογίαΗλικία <20 ετών 0
20-40 ετών 1>40 ετών 2
Διάστημα από τη διάγνωση μέχρι τη μεταμόσχευση ≤1 έτος 0>1 έτος 1
Φάση νόσου κατά τη μεταμόσχευση 1η χρόνια φάση 0Επιταχυνόμενη ή 2η χρόνια φάση 1Βλαστική κρίση 2
Συνδυασμός φύλου δότη-λήπτη Γυναίκα δότρια και άνδρας λήπτης 1Όλοι οι υπόλοιποι συνδυασμοί 0
Είδος δότη HLA ιστοσυμβατός αδελφός 0Όλοι οι υπόλοιποι δότες 1
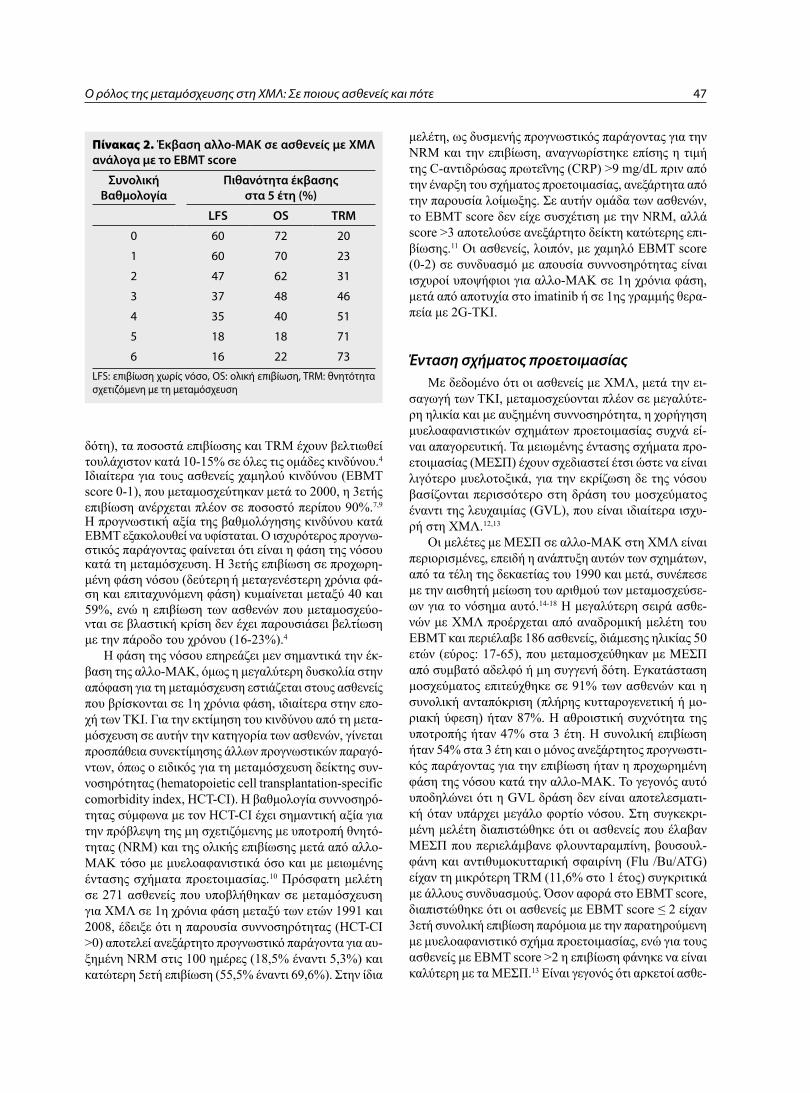
Ο ρόλος της μεταμόσχευσης στη ΧΜΛ: Σε ποιους ασθενείς και πότε 47
δότη), τα ποσοστά επιβίωσης και TRM έχουν βελτιωθεί τουλάχιστον κατά 10-15% σε όλες τις ομάδες κινδύνου.4 ιδιαίτερα για τους ασθενείς χαμηλού κινδύνου (ΕΒμτ score 0-1), που μεταμοσχεύτηκαν μετά το 2000, η 3ετής επιβίωση ανέρχεται πλέον σε ποσοστό περίπου 90%.7,9 η προγνωστική αξία της βαθμολόγησης κινδύνου κατά ΕΒμτ εξακολουθεί να υφίσταται. ο ισχυρότερος προγνω-στικός παράγοντας φαίνεται ότι είναι η φάση της νόσου κατά τη μεταμόσχευση. η 3ετής επιβίωση σε προχωρη-μένη φάση νόσου (δεύτερη ή μεταγενέστερη χρόνια φά-ση και επιταχυνόμενη φάση) κυμαίνεται μεταξύ 40 και 59%, ενώ η επιβίωση των ασθενών που μεταμοσχεύο-νται σε βλαστική κρίση δεν έχει παρουσιάσει βελτίωση με την πάροδο του χρόνου (16-23%).4
η φάση της νόσου επηρεάζει μεν σημαντικά την έκ-βαση της αλλο-μΑκ, όμως η μεγαλύτερη δυσκολία στην απόφαση για τη μεταμόσχευση εστιάζεται στους ασθενείς που βρίσκονται σε 1η χρόνια φάση, ιδιαίτερα στην επο-χή των τκι. για την εκτίμηση του κινδύνου από τη μετα-μόσχευση σε αυτήν την κατηγορία των ασθενών, γίνεται προσπάθεια συνεκτίμησης άλλων προγνωστικών παραγό-ντων, όπως ο ειδικός για τη μεταμόσχευση δείκτης συν-νοσηρότητας (hematopoietic cell transplantation-specific comorbidity index, HCT-CI). η βαθμολογία συννοσηρό-τητας σύμφωνα με τον HCT-CI έχει σημαντική αξία για την πρόβλεψη της μη σχετιζόμενης με υποτροπή θνητό-τητας (ΝRM) και της ολικής επιβίωσης μετά από αλλο-μΑκ τόσο με μυελοαφανιστικά όσο και με μειωμένης έντασης σχήματα προετοιμασίας.10 Πρόσφατη μελέτη σε 271 ασθενείς που υποβλήθηκαν σε μεταμόσχευση για Χμλ σε 1η χρόνια φάση μεταξύ των ετών 1991 και 2008, έδειξε ότι η παρουσία συννοσηρότητας (HCT-CI >0) αποτελεί ανεξάρτητο προγνωστικό παράγοντα για αυ-ξημένη ΝRM στις 100 ημέρες (18,5% έναντι 5,3%) και κατώτερη 5ετή επιβίωση (55,5% έναντι 69,6%). ςτην ίδια
μελέτη, ως δυσμενής προγνωστικός παράγοντας για την ΝRM και την επιβίωση, αναγνωρίστηκε επίσης η τιμή της C-αντιδρώσας πρωτεΐνης (CRP) >9 mg/dL πριν από την έναρξη του σχήματος προετοιμασίας, ανεξάρτητα από την παρουσία λοίμωξης. ςε αυτήν ομάδα των ασθενών, το ΕΒμτ score δεν είχε συσχέτιση με την ΝRM, αλλά score >3 αποτελούσε ανεξάρτητο δείκτη κατώτερης επι-βίωσης.11 οι ασθενείς, λοιπόν, με χαμηλό ΕΒμτ score (0-2) σε συνδυασμό με απουσία συννοσηρότητας είναι ισχυροί υποψήφιοι για αλλο-μΑκ σε 1η χρόνια φάση, μετά από αποτυχία στο imatinib ή σε 1ης γραμμής θερα-πεία με 2G-TKI.
Ένταση σχήματος προετοιμασίαςμε δεδομένο ότι οι ασθενείς με Χμλ, μετά την ει-
σαγωγή των τκι, μεταμοσχεύονται πλέον σε μεγαλύτε-ρη ηλικία και με αυξημένη συννοσηρότητα, η χορήγηση μυελοαφανιστικών σχημάτων προετοιμασίας συχνά εί-ναι απαγορευτική. τα μειωμένης έντασης σχήματα προ-ετοιμασίας (MEςΠ) έχουν σχεδιαστεί έτσι ώστε να είναι λιγότερο μυελοτοξικά, για την εκρίζωση δε της νόσου βασίζονται περισσότερο στη δράση του μοσχεύματος έναντι της λευχαιμίας (GVL), που είναι ιδιαίτερα ισχυ-ρή στη Χμλ.12,13
οι μελέτες με μΕςΠ σε αλλο-μΑκ στη Χμλ είναι περιορισμένες, επειδή η ανάπτυξη αυτών των σχημάτων, από τα τέλη της δεκαετίας του 1990 και μετά, συνέπεσε με την αισθητή μείωση του αριθμού των μεταμοσχεύσε-ων για το νόσημα αυτό.14-18 η μεγαλύτερη σειρά ασθε-νών με Χμλ προέρχεται από αναδρομική μελέτη του ΕΒμτ και περιέλαβε 186 ασθενείς, διάμεσης ηλικίας 50 ετών (εύρος: 17-65), που μεταμοσχεύθηκαν με μΕςΠ από συμβατό αδελφό ή μη συγγενή δότη. Εγκατάσταση μοσχεύματος επιτεύχθηκε σε 91% των ασθενών και η συνολική ανταπόκριση (πλήρης κυτταρογενετική ή μο-ριακή ύφεση) ήταν 87%. η αθροιστική συχνότητα της υποτροπής ήταν 47% στα 3 έτη. η συνολική επιβίωση ήταν 54% στα 3 έτη και ο μόνος ανεξάρτητος προγνωστι-κός παράγοντας για την επιβίωση ήταν η προχωρημένη φάση της νόσου κατά την αλλο-μΑκ. το γεγονός αυτό υποδηλώνει ότι η GVL δράση δεν είναι αποτελεσματι-κή όταν υπάρχει μεγάλο φορτίο νόσου. ςτη συγκεκρι-μένη μελέτη διαπιστώθηκε ότι οι ασθενείς που έλαβαν μΕςΠ που περιελάμβανε φλουνταραμπίνη, βουσουλ-φάνη και αντιθυμοκυτταρική σφαιρίνη (Flu /Bu/ATG) είχαν τη μικρότερη TRM (11,6% στο 1 έτος) συγκριτικά με άλλους συνδυασμούς. Όσον αφορά στο ΕΒμτ score, διαπιστώθηκε ότι οι ασθενείς με ΕΒμτ score ≤ 2 είχαν 3ετή συνολική επιβίωση παρόμοια με την παρατηρούμενη με μυελοαφανιστικό σχήμα προετοιμασίας, ενώ για τους ασθενείς με ΕΒμτ score >2 η επιβίωση φάνηκε να είναι καλύτερη με τα μΕςΠ.13 Είναι γεγονός ότι αρκετοί ασθε-
Πίνακας 2. Έκβαση αλλο-ΜΑΚ σε ασθενείς με ΧΜΛ ανάλογα με το ΕΒΜΤ score
Συνολική Βαθμολογία
Πιθανότητα έκβασης στα 5 έτη (%)
LFS OS TRM
0 60 72 20
1 60 70 23
2 47 62 31
3 37 48 46
4 35 40 51
5 18 18 71
6 16 22 73
LFS: επιβίωση χωρίς νόσο, OS: ολική επιβίωση, TRM: θνητότητα σχετιζόμενη με τη μεταμόσχευση

Ι. Τζάννου και Ι. Μπαλταδάκης48
νείς που υποβάλλονται σήμερα σε αλλο-μΑκ για Χμλ έχουν ΕΒμτ score >2, αφού η μεταμόσχευση καθυστε-ρεί σημαντικά. ςε γενικές γραμμές, τα μΕςΠ φαίνεται ότι έχουν θέση στην αντιμετώπιση ασθενών με Χμλ ηλι-κίας άνω των 50 ετών με συννοσηρότητα. Δεν υπάρχουν επαρκή δεδομένα για νεότερους ασθενείς, παρόλο που τα μΕςΠ θα μπορούσαν να αποτελέσουν θεραπευτική επιλογή σε περιπτώσεις ασθενών που επιθυμούν να δια-τηρήσουν ικανότητα τεκνοποίησης. η αυξημένη, όμως, συχνότητα υποτροπής αποτελεί τον κυριότερο προβλη-ματισμό για τη χρήση μΕςΠ, ιδιαίτερα στους νεότερους ασθενείς. μια εναλλακτική προσέγγιση είναι ο συνδυα-σμός μΕςΠ με προληπτική χορήγηση imatinib από την εγκατάσταση του μοσχεύματος και για 1 έτος περίπου με-τά την αλλο-μΑκ, με σκοπό τη μείωση της συχνότητας της υποτροπής. Όπως φάνηκε από μια μικρή μελέτη, η οποία περιελάμβανε αφαίρεση τ-λεμφοκυττάρων in vivo με τη χορήγηση alemtuzumab στο σχήμα προετοιμασίας, η χορήγηση του imatinib καθυστερεί την υποτροπή και την ανάγκη έγχυσης λεμφοκυττάρων του δότη (DLI). η πλειοψηφία, όμως, των ασθενών υποτροπιάζουν αμέσως μετά τη διακοπή του imatinib και πρέπει να υποβληθούν όψιμα σε DLI για την επίτευξη μοριακής ύφεσης.19 Άρα, η διατήρηση και η ενίσχυση της GVL δράσης αποτελεί προϋπόθεση για την ίαση της Χμλ μετά από μεταμό-σχευση με μΕςΠ.
Είδος δότη και πηγή μοσχεύματος με τον έλεγχο της HLA συμβατότητας με μοριακές
τεχνικές σε επίπεδο αλληλίου, τα αποτελέσματα των αλ-λο-μΑκ από συμβατούς μη συγγενείς δότες είναι συ-γκρίσιμα με αυτά των μεταμοσχεύσεων από συμβατό αδελφό.20 Έτσι, η μεταμόσχευση από πλήρως συμβατό μη συγγενή δότη (συμβατότητα 8/8 στους τόπους HLA-A, -B, -C και -DRB1) είναι λογική επιλογή σε ασθενείς που παρουσιάζουν πρόοδο νόσου υπό imatinib ή μη ικανοποι-ητική ανταπόκριση (suboptimal response) σε 2G-TKI.21
Δεν υπάρχουν σαφή δεδομένα ότι το περιφερικό αίμα υπερτερεί έναντι του μυελού των οστών σε αλλο-μΑκ για Χμλ ή για το αντίθετο. γενικά, η μεταμόσχευση αρ-χέγονων αιμοποιητικών κυττάρων περιφερικού αίματος (PBSC) προσφέρει ταχύτερη αποκατάσταση της αιμο-ποιίας συγκριτικά με το μυελό των οστών, αλλά συνο-δεύεται από μεγαλύτερη συχνότητα οξείας και κυρίως εκτεταμένης χρόνιας νόσου μοσχεύματος έναντι ξενιστή (GVHD).22 Αναφορικά με τη Χμλ, μια αναδρομική με-λέτη βασισμένη σε δεδομένα από τα αρχεία του EBMT και του IΒMTR έδειξε ότι η έκβαση των ασθενών σε χρό-νια φάση, που υποβλήθηκαν σε αλλο-μΑκ από συγγενή ιστοσυμβατό δότη, ήταν ανεξάτητη της πηγής του μοσχεύ-ματος.23 ςε μεταγενέστερη ανάλυση της ίδιας μελέτης με μεγαλύτερο χρόνο παρακολούθησης (6 έτη), φάνηκε
ότι η επιβίωση χωρίς νόσο ήταν σημαντικά υψηλότερη με τα PBSC συγκριτικά με το μυελό των οστών για τους ασθενείς που μεταμοσχεύθηκαν σε προχωρημένη φάση νόσου (33% έναντι 25%), αλλά χαμηλότερη σε αυτούς που μεταμοσχεύθηκαν σε πρώτη χρόνια φάση (41% ένα-ντι 61%), λόγω μεγαλύτερης όψιμης TRM.24
ςτην περίπτωση αλλο-μΑκ από εθελοντές μη συγ-γενείς δότες, δεν υπάρχουν ειδικά για τη Χμλ δεδομέ-να σχετικά με τη σύγκριση των δύο πηγών μοσχεύματος. Πάντως, πρόσφατα ανακοινώθηκαν τα αποτελέσματα μιας πολυκεντρικής μελέτης φάσης ιιι από τις η.Π.Α., στην οποία 551 ασθενείς με διάφορες αιματολογικές κα-κοήθειες, συμπεριλαμβανομένης της Χμλ, τυχαιοποιή-θηκαν να υποβληθούν σε μεταμόσχευση με PBSC ή με μυελό των οστών από μη συγγενή δότη. Δεν διαπιστώθη-κε στατιστικά σημαντική διαφορά μεταξύ των δύο πηγών μοσχεύματος ως προς την ολική επιβίωση, την επιβίωση χωρίς νόσο και τη συχνότητα της υποτροπής. Παρατηρή-θηκε, όμως, σημαντικά μεγαλύτερη συχνότητα χρόνιας GVHD στην ομάδα των PBSC συγκριτικά με το μυελό (53% έναντι 40%). η δε αθροιστική επίπτωση της εκτε-ταμένης χρόνιας GVHD ήταν 46% με τα PBSC και 31% με το μυελό των οστών. η συχνότητα ανεπάρκειας του μοσχεύματος ήταν μεγαλύτερη στην ομάδα του μυελού (8% έναντι 0%).25 Αν και από τα υπάρχοντα δεδομένα δεν είναι δυνατό να καθοριστεί ποια είναι η προτιμητέα πηγή μοσχεύματος, η χρήση των PBSC έχει επικρατήσει ένα-ντι του μυελού στην κλινική πράξη. η αυξημένη, όμως, συχνότητα της χρόνιας GVHD και η νοσηρότητα που αυτή συνεπάγεται πρέπει να λαμβάνεται υπόψη, ιδιαίτε-ρα στις περιπτώσεις ασθενών με Χμλ στους οποίους η GVHD αναμένεται να είναι βαριά ή δεν είναι επιθυμητή (μεταμόσχευση σε 1η χρόνια φάση, ασθενείς μεγαλύτε-ρης ηλικίας, γυναίκα δότρια και άνδρας λήπτης, ασυμβα-τότητα HLA μεταξύ δότη και λήπτη). ςτις περιπτώσεις αυτές, η χρήση μυελού των οστών ως πηγής μοσχεύμα-τος είναι προτιμητέα.
η εμπειρία από τη μεταμόσχευση μη συγγενικού ομ-φαλοπλακουντιακού αίματος (οΠΑ) στη Χμλ είναι πολύ περιορισμένη, ιδιαίτερα σε ενήλικες ασθενείς.26,27 λόγω της υψηλής πρώιμης TRM (20-40%), το οΠΑ επί του παρόντος αποτελεί εναλλακτική πηγή μοσχεύματος μό-νο για τους ασθενείς που έχουν απόλυτη ανάγκη αλλογε-νούς μεταμόσχευσης και δεν διαθέτουν ενήλικα δότη με αποδεκτή συμβατότητα (³7/8 αλλήλια HLA).
Επίδραση προηγηθείσας θεραπείας με ΤΚΙ στην έκβαση της αλλο-ΜΑΚ
Όλοι οι ασθενείς που υποβάλλονται σε αλλο-μΑκ για Χμλ έχουν προηγουμένως λάβει τουλάχιστον μία γραμμή θεραπείας με τκι. ςε αρκετές περιπτώσεις η δι-άρκεια της θεραπείας αυτής ξεπερνάει τα 2 έτη. Υπήρξε

Ο ρόλος της μεταμόσχευσης στη ΧΜΛ: Σε ποιους ασθενείς και πότε 49
ανησυχία ότι η μακροχρόνια χρήση τκι μπορεί να έχει αρνητική επίδραση στην αλλο-μΑκ. ςυγκεκριμένα, η άμεση τοξικότητα του φαρμάκου, η διαταραχή της λει-τουργίας ζωτικών οργάνων, η ανοσοκαταστολή, η επι-λογή περισσότερο ανθεκτικών και επιθετικών κλώνων και η καθυστέρηση της μεταμόσχευσης για περισσότε-ρο από 1 έτος από τη διάγνωση θα μπορούσαν να αυξή-σουν τη συχνότητα και τη βαρύτητα των επιπλοκών από την αλλο-μΑκ.28
Από το 2003 που δημοσιεύτηκαν οι πρώτες μελέτες αναφορικά με την επίδραση του imatinib στην αλλο-μΑκ, έγινε κατανοητό ότι οι παραπάνω υποθέσεις κατά κανόνα δεν ισχύουν. οι Shimoni και συν έδειξαν πως λόγω των αποτελεσμάτων της θεραπείας με τκι, οι περισσότεροι ασθενείς ήταν σε λιγότερο προχωρημένη φάση της νό-σου, επομένως και σε καλύτερη κατάσταση ικανότητας, σε σχέση με τους ασθενείς που δεν είχαν προηγουμένως λάβει τκι.28 H προηγηθείσα χρήση imatinib δεν φάνηκε να επηρεάζει δυσμενώς την εγκατάσταση του μοσχεύματος ούτε την τRμ, παρόλο που σημειώθηκε αυξημένη ηπατι-κή τοξικότητα και επίπτωση φλεβοαποφρακτικής νόσου του ήπατος. Επίσης, το imatinib δεν ενοχοποιήθηκε για μεγαλύτερη συχνότητα GVHD.29-31 η γερμανική ομάδα μελέτης της Χμλ δημοσίευσε το 2010 τα αποτελέσμα-τα της αλλο-μΑκ σε 84 ασθενείς που είχαν αποτύχει στη θεραπεία με imatinib. η TRM ήταν 8% στο σύνολο των ασθενών, η δε 3ετής ολική επιβίωση ήταν 91% για τους ασθενείς που μεταμοσχεύθηκαν σε χρόνια φάση και 59% για τους ασθενείς με προχωρημένη νόσο κατά την αλλο-μΑκ.9 Φαίνεται ότι η ελάττωση του φορτίου της νόσου μετά από αγωγή με imatinib, τελικά έχει ευεργετικά απο-τελέσματα για την έκβαση της αλλο-μΑκ, ιδιαίτερα στη χρόνια φάση της Χμλ, ενώ δεν έχει διαπίστωθεί παρό-μοια θετική επίδραση του imatinib όταν η φάση της νό-σου κατά τη μεταμόσχευση είναι προχωρημένη.
Αν και ο αριθμός των μελετών και των ασθενών είναι περιορισμένος, ούτε το dasatinib ή το nilotinib φαίνεται να έχουν αρνητική επίπτωση στα αποτελέσματα της αλ-λο-μΑκ, όσον αφορά στην τοξικότητα, την TRM και την ολική επιβίωση. η συχνότερη ανεπιθύμητη ενέργεια των φαρμάκων αυτών είναι η μυελοτοξικότητα, η οποία μπορεί να επηρεάσει δυσμενώς την αλλο-μΑκ μόνον αν είναι σοβαρού βαθμού, συνοδευόμενη από λοίμω-ξη. η ηπατική τοξικότητα της μεταμόσχευσης μετά από θεραπεία με δεύτερης γενιάς τκι δεν διαφέρει από την αναμενόμενη.32,33 Πρέπει να σημειωθεί ότι στις υπάρχου-σες μελέτες η διάμεση διάρκεια προηγηθείσας θεραπεί-ας με τκι ήταν μικρότερη από 2 έτη και μόνο ένα μικρό ποσοστό των ασθενών (περίπου 10%) είχε εκτεθεί στο imatinib για περισσότερο από 3 έτη.9,29 Παραμένει ανα-πάντητο, λοιπόν, το ερώτημα εάν η μακροχρόνια αγω-γή με τκι μπορεί τελικά να επιδεινώσει την έκβαση της μεταμόσχευσης.
Η ΑΛΛΟ-ΜΑΚ ΩΣ πΡΩΤΗΣ ΓΡΑΜΜΗΣ θΕΡΑπΕΙΑΠρώτη χρόνια φάση
ο ρόλος της αλλο-μΑκ στη χρόνια φάση της Χμλ έχει διαφοροποιηθεί σημαντικά στην εποχή των τκι.34-36 οι Hehlmann και συν, σε τυχαιοποιημένη μελέτη, συ-νέκριναν την αλλο-μΑκ με τους τκι ως θεραπεία 1ης γραμμής στη Χμλ σε χρόνια φάση. Διαπιστώθηκε πως παρά το γεγονός ότι μετά από αλλο-μΑκ επιτυγχάνο-νται μεγαλύτερα ποσοστά πλήρους κυτταρογενετικής (CCgR) και μείζονας μοριακής ύφεσης (MMR), η συ-νολική επιβίωση ελαττώνεται συνεχώς σε βάθος χρό-νου, λόγω όψιμης υποτροπής και κυρίως λόγω TRM.35 Αντίθετα, η θεραπεία με τκι υπερέχει σαφώς της αλλο-μΑκ από άποψη επιβίωσης στα 8 έτη παρακολούθη-σης, ιδιαίτερα σε ασθενείς χαμηλού κινδύνου, σύμφωνα με το Hasford score.37,38 Ασθενείς υψηλού κινδύνου κατα Sokal39 ή Hasford40, αλλά χαμηλού κινδύνου για μεταμό-σχευση, σύμφωνα με τα κριτήρια του ΕΒμτ, θεωρητικά θα μπορούσαν να υποβληθούν σε αλλο-μΑκ εξαρχής. Όμως, επειδή η TRM παραμένει υπολογίσιμη ακόμα και σε αυτήν την ομάδα των ασθενών (10-15%), η θεραπεία 1ης γραμμής με τκι προτιμάται έναντι της αλλο-μΑκ, ιδιαίτερα μετά την εισαγωγή των 2G-TKI. Εξαίρεση αποτελούν οι ασθενείς ηλικίας <20 ετών που διαθέτουν ιστοσυμβατό αδελφό δότη.21
Επιπρόσθετες κυτταρογενετικές ανωμαλίες κατά τη διάγνωση
Είναι γνωστό ότι 10-12% των ασθενών με Χμλ φέ-ρουν επιπρόσθετες χρωμοσωμικές ανωμαλίες (ΑCA) κατά τη διάγνωση, που περιλαμβάνουν παραλλαγές της αντι-μετάθεσης Philadelphia [t(v;22)], απώλεια του χρωμο-σώματος Υ (-Υ), καθώς και «γνήσιες» ACA σε ποσοστό <5%. κατά την εξέλιξη της νόσου, το ποσοστό των ACA ανέρχεται στο 80% των περιπτώσεων.41,42 οι ACA που εμφανίζονται σταθερά και σε υψηλή συχνότητα (>80%) σε προχωρημένη φάση Χμλ είναι η τρισωμία 8, ο δι-πλασιασμός του χρωμοσώματος Ph και το ισοχρωμόσωμα 17(q10). Αυτές χαρακτηρίζονται ως η «κύρια οδός» κυτ-ταρογενετικής εξέλιξης (major-route ACA), σε αντιδια-στολή με τη «δευτερεύουσα οδό» (minor-route ACA) που περιλαμβάνει ACA που παρατηρούνται σπάνια (<15%) σε επιταχυνόμενη ή βλαστική φάση της νόσου, όπως είναι οι αντιμεταθέσεις t(3;12), t(4;6), t(2;16) και t(1;21).43 η επίδραση της ύπαρξης ΑCA κατά τη διάγνωση στην πρό-γνωση της Χμλ, μελετήθηκε πρόσφατα σε 1151 ασθε-νείς από την τυχαιοποιημένη γερμανική μελέτη CML IV. οι ερευνητές εξέτασαν την επίδραση στην έκβαση της νόσου των t(v;22) και των major- και minor-route ΑCA σε ασθενείς που έλαβαν imatinib ως μονοθεραπεία ή σε

Ι. Τζάννου και Ι. Μπαλταδάκης50
συνδυασμό με ιντερφερόνη-α ή αρασυτίνη. Από τη με-λέτη διαπιστώθηκε ότι οι ασθενείς με major-route ACA έχουν σαφώς χαμηλότερη επιβίωση χωρίς εξέλιξη νόσου (PFS) και ολική επιβίωση (50% και 53% στα 5 έτη αντί-στοιχα). Ακόμη, το διάμεσο διάστημα για την επίτευξη CCgR και MMR ήταν σημαντικά μεγαλύτερο στους ασθε-νείς με major-route ACA. Αυτή η μικρή ομάδα ασθενών (1,4% στη συγκεκριμένη μελέτη) έχει δυσμενή πρόγνω-ση και πρέπει να αντιμετωπίζεται πρώιμα με αλλο-μΑκ ή εναλλακτική θεραπεία.44
Βλαστική ή επιταχυνόμενη φάση κατά τη διάγνωση
ο ρόλος της αλλο-μΑκ ως 1ης γραμμής θεραπείας ασθενών σε βλαστική κρίση Χμλ κατά τη διάγνωση εί-ναι σαφής. οι ασθενείς που διαγιγνώσκονται σε βλαστική φάση παρουσιάζουν χαμηλή συχνότητα κυτταρογενετικής ανταπόκρισης (16%) στο imatinib. οι ανταποκρίσεις εί-ναι βραχύβιες και η διάμεση επιβίωση δεν ξεπερνά τους 7 μήνες.45 Όπως προαναφέρθηκε, η έκβαση της αλλο-μΑκ εξαρτάται από το στάδιο της νόσου κατά τη μεταμόσχευ-ση. η επιβίωση των ασθενών που μεταμοσχεύονται σε έκδηλη βλαστική φάση είναι κάτω από 20%. Προκειμέ-νου, λοιπόν, να βελτιωθούν τα αποτελέσματα για τους ασθενείς με προχωρημένη νόσο, θεωρείται απαραίτητη η επίτευξη δεύτερης χρόνιας φάσης με χορήγηση ενός τκι, χημειοθεραπείας ή συνδυασμού αυτών πριν τη δι-ενέργεια της αλλο-μΑκ.
ςτην επιταχυνόμενη φάση, όπου η 6-ετής επιβίωση υπό imatinib φτάνει το 50%, η θέση της αλλο-μΑκ δεν έχει διευκρινιστεί πλήρως. οι περιπτώσεις Χμλ που χα-ρακτηρίζονται ως επιταχυνόμενη φάση κατά τη διάγνω-ση αποτελούν μια ιδιαίτερα ετερογενή ομάδα ασθενών. η κατάσταση της νόσου μπορεί να ποικίλλει από ελα-φρά πιο προχωρημένη νόσο σε σχέση με τη χρόνια φά-ση μέχρι σχεδόν βλαστική κρίση. Ασθενείς με λιγότερο προχωρημένη νόσο μπορούν να έχουν καλή ανταπόκρι-ση στους TKI και πρέπει να αντιμετωπίζονται παρόμοια με τους ασθενείς που διαγιγνώσκονται σε χρόνια φάση. Αντίθετα, οι ασθενείς που είναι στα πρόθυρα βλαστικής κρίσης πρέπει να υποβάλλονται σε επιθετική θεραπεία και έγκαιρα σε αλλο-μΑκ. Δεν υπάρχουν, προς το παρόν, αξιόπιστοι βιολογικοί δείκτες για τη διάκριση προγνωστι-κών όμαδων μεταξύ των ασθενών που διαγιγνώσκονται σε επιταχυνόμενη φάση. μια πρόσφατη προοπτική, αλ-λά όχι τυχαιοποιημένη, μελέτη από την κίνα συνέκρινε τα αποτελέσματα της χορήγησης imatinib έναντι της αλ-λο-μΑκ στην αντιμετώπιση ασθενών με Χμλ σε επι-ταχυνόμενη φάση. Αναγνωρίστηκαν τρεις ανεξάρτητοι δυσμενείς προγνωστικοί παράγοντες για την επιβίωση και την PFS: διάρκεια Χμλ ≥12 μήνες, αιμοσφαιρίνη
<10 g/dL και ποσοστό βλαστών στο περιφερικό αίμα ≥5%. οι ασθενείς ταξινομήθηκαν σε τρεις προγνωστικές ομάδες, δηλαδή χαμηλού, ενδιάμεσου ή υψηλού κινδύ-νου, ανάλογα με την παρουσία κανενός, ενός ή δύο από τους ανωτέρω παράγοντες αντίστοιχα. Παρατήθηκε ότι στους ασθενείς χαμηλού κινδύνου η επιλογή της θερα-πείας δεν επηρέασε την έκβαση. τα αποτελέσματα ήταν εξίσου καλά με το imatinib και την αλλο-μΑκ, με 6ετή PFS και ολική επιβίωση της τάξης του 80%. Αντίθετα, στις ομάδες ενδιάμεσου και υψηλού κινδύνου, τα απο-τελέσματα της αλλο-μΑκ υπερείχαν. ιδιαίτερα για τους ασθενείς υψηλού κινδύνου, η 5ετής επιβίωση ήταν 100% με την αλλο-μΑκ έναντι 18,8% με το imatinib. Επομέ-νως, η αλλο-μΑκ αποτελεί θεραπεία εκλογής για τους ασθενείς με επιταχυνόμενη φάση Χμλ υψηλού και εν-διάμεσου κινδύνου. Από την άλλη, στους ασθενείς χαμη-λού κινδύνου προτείνεται έναρξη θεραπείας με imatinib, στενή παρακολούθηση της ανταπόκρισης και επί αποτυ-χίας, αλλο-μΑκ.46
Η ΑΛΛΟ-ΜΑΚ ΩΣ ΔΕΥΤΕΡΗΣ ΚΑΙ ΤΡΙΤΗΣ ΓΡΑΜΜΗΣ θΕΡΑπΕΙΑ
οι ασθενείς που εμφανίζουν αποτυχία στην 1ης γραμ-μής θεραπεία με imatinib, μπορούν να αντιμετωπιστούν με 2G-TKI ή αλλο-μΑκ. η αντίσταση στο imatinib, σύμ-φωνα με το European Leukemia Net, ορίζεται ως απο-τυχία επίτευξης πλήρους αιματολογικής ύφεσης στους 3 μήνες, οποιασδήποτε κυτταρογενετικής ύφεσης στους 6 μήνες, πλήρους κυτταρογενετικής ύφεσης στους 18 μή-νες ή απώλεια αιματολογικής ή πλήρους κυτταρογενε-τικής ύφεσης ή εμφάνιση νέων μεταλλάξεων.21 Αξίζει να σημειωθεί ότι κάποιοι από τους ασθενείς που εμφα-νίζουν αντίσταση στο imatinib παραμένουν σε χρόνια φάση, ενώ άλλοι εμφανίζουν εξέλιξη νόσου σε επιταχυ-νόμενη φάση ή βλαστική κρίση. Πιθανολογείται ότι οι διαφορετικές μορφές έκφρασης της αντίστασης οφείλο-νται σε διαφορετικούς μηχανισμούς, με αποτέλεσμα να διαφοροποιείται και η πρόγνωση σε κάθε περίπτωση. οι ασθενείς με αντίσταση στο imatinib αποτελούν μια ετε-ρογενή ομάδα και δεν είναι δυνατό να αντιμετωπιστούν με τον ίδιο τρόπο.12
Εξέλιξη σε επιταχυνόμενη ή βλαστική φάση υπό θεραπεία με imatinib
η εξέλιξη σε προχωρημένη φάση της νόσου υπό imatinib έχει δυσμενέστατη πρόγνωση και επιβάλλεται προγραμματισμός για αλλο-μΑκ το ταχύτερο δυνατό.45 ςτο μεσοδιάστημα μέχρι τη μεταμόσχευση, είναι σκόπιμη η προσπάθεια επίτευξης δεύτερης χρόνιας φάσης με χο-ρήγηση 2G-TKI, χημειοθεραπείας ή συνδυασμού αυτών.

Ο ρόλος της μεταμόσχευσης στη ΧΜΛ: Σε ποιους ασθενείς και πότε 51
Μεταλλάξεις στην περιοχή κινάσης (KD) του χιμαιρικού γονιδίου BCR-ABL
ςε ασθενείς σε χρόνια φάση που λαμβάνουν θεραπεία 1ης γραμμής με imatinib, έλεγχος μεταλλάξεων BCR/ABL KD ενδείκνυται μόνο σε αποτυχία ή μη ικανοποι-ητική ανταπόκριση σύμφωνα με τα κριτήρια του ELN.21 η ανίχνευση τέτοιων μεταλλάξεων μπορεί να καθοδηγή-σει στην επιλογή του κατάλληλου 2G-TKI (dasatinib ή nilotinib) και στην περίπτωση της μετάλλαξης T315I, που δεν ανταποκρίνεται σε κανένα από τα παραπάνω φάρμα-κα, να υποδείξει την ανάγκη για αλλο-μΑκ ή πειραματι-κή θεραπεία.47,48 ςε ασθενείς με αντοχή στο imatinib που λαμβάνουν 2ης γραμμής θεραπεία με 2G-TKI, έλεγχος μεταλλάξεων απαιτείται στην περίπτωση αποτυχίας αι-ματολογικής ή κυτταρογενετικής ανταπόκρισης.49
Πρέπει να σημειωθεί ότι η εμφάνιση μεταλλάξεων υπό imatinib, ανεξάρτητα από την ευαισθησία τους στους 2G-TKI, σηματοδοτεί κίνδυνο εξέλιξης της νόσου και απαιτεί εγρήγορση για το ενδεχόμενο διενέργειας αλλο-μΑκ. Αυτό υποδεινύει πρόσφατη μελέτη από το M.D. Anderson, που εξέτασε την επίδραση της παρουσίας με-ταλλάξεων στην έκβαση της αλλο-μΑκ σε ασθενείς με προηγούμενη αποτυχία σε τκι. ςτην υποομάδα των ασθε-νών που παρουσίαζαν μεταλλάξεις, η ολική επιβίωση στα 2 έτη μετά την αλλο-μΑκ ήταν σημαντικά κατώτερη συ-γκριτικά με τους ασθενείς με αμετάλλακτη BCR-ABL KD (44% έναντι 76% αντίστοιχα).50 Πρέπει να ληφθεί υπόψη ότι, οι 15 από τους 19 ασθενείς που είχαν μεταλ-λάξεις μεταμοσχεύθηκαν σε επιταχυνόμενη φάση ή βλα-στική κρίση. με δεδομένο τον κίνδυνο για εξέλιξη του νοσήματος και τη δυσμενή έκβαση της μεταμόσχευσης, η αναζήτηση δότη για αλλο-μΑκ είναι σκόπιμο να γί-νεται έγκαιρα στους ασθενείς που φέρουν μεταλλάξεις, ιδιαίτερα δε σε όσους έχουν χαμηλή πιθανότητα ανταπό-κρισης στους 2G-TKI.
Πιθανότητα ανταπόκρισης σε δεύτερης γενιάς ΤΚΙ
ςήμερα, οι περισσότεροι ασθενείς σε χρόνια φάση με αποτυχία στο imatinib τίθενται σε αγωγή με 2G-τκι. η πιθανότητα πλήρους κυτταρογενετικής ανταπόκρισης στη θεραπεία με dasatinib ή nilotinib είναι της τάξης του 40-45% και συνεπώς οι μισοί περίπου ασθενείς με αντο-χή στο imatinib θα χρειαστούν θεραπεία τρίτης γραμμής. Επομένως, είναι απαραίτητο να γίνεται HLA τυποποίη-ση και αναζήτηση συμβατού δότη στους νεότερους ασθε-νείς από τη φάση της αποτυχίας στο imatinib, ώστε να είναι εφικτή η διενέργεια αλλο-μΑκ έγκαιρα σε όσους δεν παρουσιάσουν ικανοποιητική ανταπόκριση στη θε-ραπεία 2ης γραμμής.
ςύμφωνα με τις τρέχουσες συστάσεις του ELN, η απο-
τυχία της θεραπείας με dasatinib ή nilotinib ορίζεται με προσωρινά (provisional) κριτήρια, που αφορούν κυρίως στην κυτταρογενετική ανταπόκριση: απουσία κυτταρογε-νετικής ανταπόκρισης (CgR) μετά από 3 μήνες θεραπεί-ας, ελάχιστη CgR στους 6 μήνες, απουσία μερικής CgR μετά από 1 έτος. Επίσης, αποτυχία θεωρείται η εμφάνι-ση νέων μεταλλάξεων της ΒCR-ABL KD με ανεπαρκή ευαισθησία στους TKI 2ης γενιάς σε οποιαδήποτε φάση της θεραπείας με αυτούς.21
Ένα προγνωστικό σύστημα για την πρώιμη πρό-βλεψη της ανταπόκρισης στη θεραπεία 2ης γραμμής με dasatinib ή nilotinib δημιουργήθηκε από την ομάδα του Hammersmith (Hammersmith score). η βαθμολο-γία προκύπτει από τη συνεκτίμηση τριών παραμέτρων, που αποτελούν ανεξάρτητους προγνωστικούς παράγο-ντες για την ανταπόκριση στη θεραπεία 2ης γραμμής με TKI: 1) μέγιστη κυτταρογενετική ανταπόκριση στη θε-ραπεία με imatinib (πλήρης: 0, Ph-θετικές μεταφάσεις 1-94%: 1 βαθμός, Ph-θετικές μεταφάσεις ≥95%: 3 βαθ-μοί), 2) Sokal score στη διάγνωση (χαμηλό: 0, ενδιάμε-σο ή υψηλό: 0,5 βαθμός) και 3) ουδετεροπενία κατά τη διάρκεια της θεραπείας με imatinib (απουσία ουδετερο-πενίας: 0, επανειλημμένα επεισόδια ουδετεροπενίας που απαίτησαν μείωση της δόσης του imatinib κάτω από 400 mg/ημέρα: 1 βαθμός). με βάση το Hammersmith score, οι ασθενείς με αποτυχία στο imatinib διακρίνονται σε τρεις προγνωστικές ομάδες, δηλαδή σε ομάδα καλής (βαθμολο-γία: <1,5), ενδιάμεσης (βαθμολογία: 1,5-2,5) και πτωχής πρόγνωσης (βαθμολογία: >2,5). η αθροιστική συχνότη-τα πλήρους κυτταρογενετικής ύφεσης με 2G-TKI στην ομάδα καλής πρόγνωσης είναι 100%, ενώ στις ομάδες εν-διάμεσης και πτωχής πρόγνωσης ήταν 52,2% και 13,8% αντίστοιχα. ςτην παραπάνω μελέτη διαπιστώθηκε ακό-μη, ότι το επίπεδο της κυτταρογενετικής ανταπόκρισης στους 3 μήνες από την έναρξη dasatinib ή nilotinib μπο-ρεί να προβλέψει την πιθανότητα επίτευξης πλήρους κυτταρογενετικής ύφεσης στους 12 μήνες. οι ασθενείς που παρουσιάζουν τουλάχιστον ελάχιστη κυτταρογενε-τική ανταπόκριση (Ph+ μεταφάσεις <95%) στους 3 μή-νες έχουν σημαντικά μεγαλύτερη πιθανότητα επίτευξης πλήρους κυτταρογενετικής ύφεσης στο έτος, συγκριτικά με τους ασθενείς που δεν έχουν καμιά κυτταρογενετι-κή ανταπόκριση (100% έναντι 0% αντίστοιχα).51 Παρό-μοια συμπεράσματα προκύπτουν και από μελέτη του MD Anderson, στην οποία διαπιστώθηκε ότι η πρώιμη κυτ-ταρογενετική ανταπόκριση στην αγωγή με 2G-TKI είναι ισχυρός καθοριστικός παράγοντας για την επίτευξη μείζο-νας κυτταρογενετικής ύφεσης στους 12 μήνες. Από τους ασθενείς που δεν παρουσίασαν καμιά κυτταρογενετική ανταπόκριση στους 3-6 μήνες, μόνο το 10% πέτυχε το στόχο της μείζονας κυτταρογενετικής ύφεσης στο έτος.52 οι ασθενείς, λοιπόν, με χαμηλό Hammersmith score, ανα-μένεται ότι θα ωφεληθούν πρωτίστως από τη θεραπεία με 2G-TKI. Αντίθετα, οι ασθενείς με υψηλό Hammersmith

Ι. Τζάννου και Ι. Μπαλταδάκης52
score πρέπει να θεωρηθούν υποψήφιοι για αλλο-μΑκ, ιδιαίτερα εαν ταξινομούνται ως χαμηλού κινδύνου κατά ΕΒμτ. οι ασθενείς με ενδιάμεσο Hammersmith score, καθώς και οι ασθενείς με υψηλό score που ταξινομού-νται ως υψηλού κινδύνου για μεταμόσχευση κατά ΕΒμτ, μπορούν να θεραπευθούν αρχικά με 2G-TKI και η ένδει-ξη για αλλο-μΑκ να εκτιμηθεί με βάση την κυτταρογε-νετική ανταπόκριση στους 3 μήνες.51
Πολύ πρόσφατες μελέτες δείχνουν ότι, η πρόβλε-ψη της ανταπόκρισης στους τκι είναι εφικτή ακόμη πιο πρώιμα και συγκεκριμένα στους πρώτους 3 μήνες από την έναρξη θεραπείας 1ης γραμμής. Από την ομάδα του Hammersmith, σε 282 ασθενείς με Χμλ σε χρόνια φά-ση που έλαβαν imatinib ως 1ης γραμμής θεραπεία και επί αποτυχίας dasatinib ή nilotinib, μελετήθηκαν τα επίπεδα των μεταγράφων BCR-ABL στους 3, 6 και 12 μήνες από την έναρξη του imatinib. Διαπιστώθηκε ότι τα επίπεδα των μεταγράφων στους 3 μήνες ήταν ο ισχυρότερος πα-ράγοντας πρόβλεψης της ανταπόκρισης στη θεραπεία με TKI και της επιβίωσης. οι ασθενείς που είχαν BCR-ABL >9,84% στους 3 μήνες, είχαν σημαντικά χαμηλότερη 8ετή επιβίωση (56,9% έναντι 93,3%, p <0,001) και PFS (57% έναντι 92,8%, p <0,001), καθώς και μικρότερη αθροιστι-κή συχνότητα πλήρους κυταρογενετικής (CCgR) και πλή-ρους μοριακής ύφεσης (CMR), συγκριτικά με αυτούς που είχαν επίπεδα BCR-ABL <9,84%. Ακόμη, η επακόλουθη θεραπεία με 2G-TKI δεν διαφοροποιούσε σημαντικά την πρόγνωση, που βασιζόταν στην πρώιμη μέτρηση των με-ταγράφων BCR-ABL υπό την αγωγή με imatinib.53 Από την τυχαιοποιημένη μελέτη DASISION, σε ασθενείς που έλαβαν dasatinib ή imatinib ως θεραπεία 1ης γραμμής, ανακοινώθηκε τελευταία ότι η μείωση των μεταγράφων BCR-ABL σε επίπεδα ≤10% στους 3 μήνες συσχετίζε-ται με αυξημένη πιθανότητα επίτευξης CCgR στους 12 μήνες και μείζονας μοριακής ύφεσης (μμR) στους 24 μήνες, καθώς και μειωμένη συχνότητα εξέλιξης σε επι-ταχυνόμενη/βλαστική φάση. ςτο σκέλος του dasatinib, η PFS στους 24 μήνες ήταν 97% για τους ασθενείς που είχαν BCR-ABL ≤10% στους 3 μήνες έναντι 83% στους ασθενείς με BCR-ABL >10%. Παρόμοια αποτελέσματα προέκυψαν και στο σκέλος του imatinib (PFS 96% ένα-ντι 85% αντίστοιχα).54
Επίσης, σε 150 νεοδιαγνωσθέντες ασθενείς που έλα-βαν dasatinib στo πλαίσιo της μελέτης UK SPIRIT 2, η πιθανότητα επίτευξης πλήρους CgR ήταν 89% όταν τα επίπεδα των μεταγράφων BCR-ABL ήταν <10% στους 3 μήνες από την έναρξη της θεραπείας, ενώ ήταν μόλις 50,2% με υψηλότερα επίπεδα μεταγράφων. τα προκα-ταρκτικά αυτά δεδομένα παρουσιάζουν μεγάλο ενδιαφέ-ρον, επειδή καθιστούν δυνατή την πρώιμη αναγνώριση της υποομάδας των ασθενών (περίπου 10%) που δεν πρό-κειται ανταποκριθούν σε 1ης ή 2ης γραμμής θεραπεία με 2G-TKI. Έτσι στο εξής, η αλλο-μΑκ έχει δυνητικά έν-δειξη ως έγκαιρη θεραπεία διάσωσης στους ασθενείς που
δεν παρουσιάζουν ικανοποιητική πρώιμη μοριακή αντα-πόκριση στην αγωγή με τκι.55
ΣΥΜπΕΡΑΣΜΑΤΑ ΚΑΙ πΡΟΟπΤΙΚΕΣη αλλο-μΑκ εξακολουθεί να έχει θέση στην αντι-
μετώπιση της Χμλ, ακόμα και μετά την εισαγωγή των τκι στην κλινική πράξη και για ορισμένους ασθενείς αποτελεί τη μοναδική θεραπεία που προσφέρει δυνα-τότητα ίασης. ςύμφωνα με τις πλέον πρόσφατες κατευ-θυντήριες οδηγίες του European Leukemia Net (2009), υποψήφιοι για αλλο-μΑκ είναι ασθενείς σε χρόνια φά-ση με αποτυχία στη θεραπεία με imatinib που φέρουν τη μετάλλαξη τ315ι, ασθενείς χωρίς ανταπόκριση στη θε-ραπεία με τκι δεύτερης γενιάς ή ασθενείς που παρουσι-άζουν επιταχυνόμενη/βλαστική φάση κατά τη διάγνωση ή εξέλιξη νόσου υπό αγωγή με τκι (Πίνακας 3). Παρά την αισθητή μείωση του αριθμού των μεταμοσχεύσεων στη Χμλ την τελευταία δεκαετία, η αλλο-μΑκ παρα-
Πίνακας 4. Πρώιμες ενδείξεις για αλλο-ΜΑΚ στη ΧΜΛ στο άμεσο μέλλον
Θεραπεία 1ης γραμμής
Παρουσία “major-route” επιπρόσθετων χρωμοσωμικών ανωμαλιών κατά τη διάγνωση
Θεραπεία 2ης γραμμής
Χαμηλή πιθανότητα ανταπόκρισης στην αγωγή με 2G-TKI μετά από αποτυχία στο imatinib (Hammersmith score >2,5)
Απουσία κυτταρογενετικής ανταπόκρισης (Ph+ μεταφάσεις >95%) στους 3 μήνες αγωγής με 2G-TKI
Επίπεδα μεταγράφων BCR-ABL >10% στους 3 μήνες αγωγής με 2G-TKI
Πίνακας 3. Ενδείξεις αλλο-ΜΑΚ στη ΧΜΛ σύμφωνα με τις οδηγίες του European Leukemia Net (2009)Θεραπεία 1ης
γραμμήςΔιάγωση σε επιταχυνόμενη ή
βλαστική φάση
Θεραπεία 2ης γραμμής
Εξέλιξη σε επιταχυνόμενη ή βλαστική φάση υπό αγωγή με ΤΚΙ
Ασθενείς που φέρουν την μετάλλαξη Τ315Ι
Θεραπεία 3ης γραμμής
Όλοι οι ασθενείς με αποτυχία σε 2ης γενιάς ΤΚΙ

Ο ρόλος της μεταμόσχευσης στη ΧΜΛ: Σε ποιους ασθενείς και πότε 53
μένει επίκαιρη, γιατί ο ρόλος της αναπροσαρμόζεται συ-νεχώς υπό το φως των νεότερων δεδομένων (Πίνακας 4). με βάση, λοιπόν, τα τελευταία ευρήματα από μεγάλες προοπτικές μελέτες, η εκτίμηση της μοριακής ανταπό-κρισης πρώιμα κατά τη θεραπεία με τκι, ιδιαίτερα 2ης γενιάς, ενδέχεται να οδηγήσει στην πιο έγκαιρη επιλογή των ασθενών που χρειάζονται μεταμόσχευση. η προoπτι-κή αυτή αφενός παρέχει τη δυνατότητα ανεύρεσης πλέ-ον κατάλληλου δότη, αφετέρου ενδέχεται να βελτιώσει σημαντικά την έκβαση της μεταμόσχευσης, που εξαρτά-ται κατά κύριο λόγο από τη φάση της νόσου και το χρό-
νο από τη διάγνωση, ακόμη και στην εποχή των τκι.29 ςτο προσεχές μέλλον, λοιπόν, η μεταμόσχευση μπορεί να αποτελεί θεραπεία 2ης γραμμής της Χμλ, με βάση τα επίπεδα των μεταγράφων BCR-ABL σε καθορισμέ-να χρονικά ορόσημα κατά την πρώιμη φάση της αγωγής με τκι. ςτο πλαίσιο αυτό, καθίσται δυνατός ο ορθολο-γικός σχεδιασμός της αλλο-μΑκ (στάθμιση κινδύνου/οφέλους, επιλογή σχήματος προετοιμασίας, είδους δότη και πηγής μοσχεύματος), που ενδέχεται να αποτελέσει υπόδειγμα για τη μεταμόσχευση και σε άλλες αιματολο-γικές κακοήθειες.
The role of Stem Cell Transplantation in CML: For which patients and whenby Ifigeneia Tzannou and Ioannis Baltadakis
Department of Hematology and Stem Cell Transplantation Unit, Evaggelismos General Hospital, Athens, Greece
ABSTRACT: Allogeneic stem cell transplantation (allo-SCT) was the first modality proven to be cura-tive for chronic myeloid leukemia (CML) by eradication of the malignant Philadelphia-positive clone. Since the introduction of the first tyrosine kinase inhibitor (TKI), imatinib, and the outstanding results obtained by targeted therapy, allo-SCT is no longer a first-line treatment for the majority of CML cases. However, allo-SCT is currently a therapeutic option for the considerable proportion of patients (40%) who fail up front treatment with imatinib. The decision to proceed to allo-SCT depends on carefully weighting the expected benefits against the estimated risks of the procedure. The risk of transplantation is assessed by the European Group for Blood and Marrow Transplantation (ΕΒμτ) score which main-tains its validity nowadays,with an improvement of survival by 10-15% for all risk categories over the last decade. The ΕΒμτ risk scorecombined with the recently developed hematopoietic cell transplan-tation-specific comorbidity index (HCT-CI) could result in a more accurate prediction of the outcome of allo-SCT on individual patient basis, thereby facilitating better selection of suitable transplant can-didates and allowing for customization of the procedure according to risk. The latter can be pursued by the choice of the appropriate conditioning regimen (myeloablative or reduced-intensity) in concert with the preemptive use of TKI and exploitation of the graft-versus-leukemia (GVL) effect by donor lym-phocyte infusions (DLI) post transplant. For patients with advanced disease, improvement of the results of allo-SCT can been accomplished by pre-transplant use of TKI in order to achieve a second chronic phase. Moreover, the outcomes of unrelated donor SCT, have become comparable to those of matched sibling transplants, due to selection of more compatible donors by DNA-based HLA typing techniques. According to current European Leukemia Net guidelines (2009), allo-SCT is mainly recommended for patients with advanced phase CML at presentation or with disease progression under imatinib treatment, as well as upon failure of therapy with second generation TKI (2G-TKI). However, the indications and the timing of allo-SCT in the era of TKI are subject to continuous revision in the light of emerging data from recent clinical studies. Patients with unfavorable outcomes with second-line TKI can be identified in advance, based on Sokal score at diagnosis, prior cytogenetic response (CgR) to imatinib, and occur-rence of neutropenia during imatinib treatment (Hammersmith score), as well as by the absence of any CgRat 3 months of therapy with 2G-TKI. Furthermore, the level of molecular response at 3 months of treatment may define a subgroup of about 10% of patients, who are not going to respond to first- or sec-ond-line therapy with 2G-TKI. Therefore, in the immediate future, transplantation may be considered as a timely, second-line therapy for suitable patients according to the molecular response at the first 3 months of treatment with TKI. This approach will allow for a more rational design of the procedure, and may improve further the outcomes of allo-SCT in CML, which are largely dependent on disease phase and on the interval from diagnosis to transplant.

Ι. Τζάννου και Ι. Μπαλταδάκης54
Βιβλιογραφία1. Fefer A, Cheever MA, Thomas ED, et al. Disappearance
of Ph1-positive cells in four patients with chronic granulo-cytic leukemia after chemotherapy, irradiation and marrow transplantation from an identical twin. N Engl J Med 1979; 300:333-337.
2. Goldman JM. Allogeneic stem cell transplantation for chronic myeloid leukemia-status in 2007. Bone Mar Transpl 2008; 42:S11-S13.
3. Boehm A, Walcherberger B, Sperr WR, et al. Improved outcome in patients with chronic myelogenous leukemia after allogeneic hematopoietic stem cell transplantation over the past 25 years: a single center experience. Biol Blood Marrow Transplant 2011; 17:133-140.
4. Gratwohl A, Brand R, Apperley J, et al. Allogeneic he-matopoietic stem cell transplantation for chronic myeloid leukemia in Europe 2006: transplant activity, long-term data and current results. An analysis by the Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation (EBMT). Haematologica 2006; 91:513-521.
5. O’ Brien SG, Guilhot F, Goldman JM, et al. International Randomized Study of Interferon Versus STI571 (IRIS) 7-year follow-up: sustained survival, low rate of transformation and increased rate of major molecular response (MMR) in patients (pts) with newly diagnosed chronic myeloid leukemia in chronic phase (CMLCP) treated with imatinib (IM). Blood 2008; 112:Abstract 186.
6. Venepalli N, Rezvani K, Mielke S, Savani BN. Role of allo-SCT for CML in 2010. Bone Marrow Transplant 2010; 45:1579-1586.
7. Pavlu J, Szydlo RM, Goldman JM, Apperley J. Three decades of transplantation in chronic myeloid leukemia: what have we learned? Blood 2011; 117:755-763.
8. Gratwohl A, Hermans J, Goldman JM, et al. Risk assessment of patients with chronic myeloid leukemia before allogeneic blood and marrow transplantation: Chronic Leukemia Work-ing Party of the European Group for Blood and Marrow Transplantation. Lancet 1998; 352:1087-1092.
9. Saussele S, Lauseker M, Gratwohl A, et al. Allogeneic hematopoietic cell transplantation (allo-SCT) for chronic myeloid leukemia in the imatinib era: evaluation of its impact within a subgroup of the randomized German CML study IV. Blood 2010; 115:1880-1885.
10. Sorror ML, Maris MB, Storb R, et al. Hematopoietic cell transplantation (HCT)-specific comorbidity index: a new tool for risk assessment before allogeneic HCT. Blood 2005; 106:2912-2919.
11. Pavlu J, Kew A, Taylor-Roberts B, et al. Optimizing patient selection for allogeneic hematopoietic cell transplantation in chronic myeloid leukemia in chronic phase. Blood 2010; 115:4018- 4020.
12. Apperley JF. Managing the patient with chronic myeloid leukemia through and after allogeneic stem cell transplanta-tion. Hematology 2006:226-232.
13. Crawley C, Szydlo R, Lalancette M et al. Outcomes of re-
duced-intensity transplantation for chronic myeloid leukemia: an analysis of prognostic factors from the Chronic Leukemia Working Party of the EBMT. Blood 2005; 106:2969-2976.
14. Bornhauser M, Kiehl M, Siegert W et al. Dose-reduced conditioning for allografting in 44 patients with chronic myeloid leukaemia: a retrospective analysis. Br J Haematol 2001; 115:119-124.
15. Das M, Saikia TK, Advani SH, et al. Use of a reduced-intensity conditioning regimen for allogeneic transplantation in patients with chronic myeloid leukemia. Bone Marrow Transplant 2003; 32:125-129.
16. Sloand E, Childs RW, Solomon S et al. The graft-versus- leukemia effect of nonmyeloablative stem cell allografts may not be sufficient to cure chronic myelogenous leukemia .Bone Marrow Transplant 2003; 32:897-901.
17. Weisser M, Schleuning M, Ledderose G et al. Reduced- intensity conditioning using TBI (8 Gy), fludarabine, cy-clophosphamide and ATG in elderly CML patients provides excellent results especially when performed in the early course of the disease. Bone Marrow Transplant 2004; 34:1083-1088.
18. Kubriaei P, Derty MA, Giralt S, et al. Long-term follow-up of allogeneic stem cell transplantation with reduced-intensity conditioning for patients with chronic myeloid leukemia. Blood 2007; 110:3456-3462.
19. Olavarria E, Siddique S, Griffiths MJ, et al. Posttransplan-tation imatinib as a strategy to postpone the requirement for immunotherapy in patients undergoing reduced inten-sity allografts for chronic myeloid leukemia. Blood 2007; 110:4614-4617.
20. Davies SM, DeFor TE, McGlave PB, et al. Equivalent outcomes in patients with chronic myelogenous leukemia after early transplantation of phenotypically matched bone marrow from related or unrelated donors. Am J Med 2001; 110:339-346.
21. Baccarani M, Cortes J, Pane F, et al. Chronic myeloid leukemia: an update of concepts and management recom-mendations of European Leukemia Net. J Clin Oncol 2009; 27:6041-6051.
22. Stem Cell Trialists’ Collaborative Group. Allogeneic Pe-ripheral Blood Stem-Cell Compared With Bone Marrow Transplantation in the Management of Hematologic Ma-lignancies: An Individual Patient Data Meta-Analysis of Nine Randomized Trials. J Clin Oncol 2005; 23:5074-5087.
23. Champlin RE, Schmitz N, Horowitz MM et al. Blood stem cells compared with bone marrow as a source of hematopoietic cells for allogeneic transplantation. IBMTR Histocompat-ibility and Stem Cell Sources Working Committee and the European Group for Blood and Marrow Transplantation (EBMT). Blood 2000; 95:3702-370.
24. Schmitz N, Eapen M, Horowitz MM, et al. Long-term outcome of patients given transplants of mobilized blood or bone marrow: a report from the International Bone Marrow Transplantation Registry and the European Group for Blood and Marrow Transplantation. Βlood 2006; 108:4288-4290.
25. Anasetti C, Brent LR, Lee SJ, et al. Increased incidence of chronic chronic graft-versus-host disease (GVHD) and no

Ο ρόλος της μεταμόσχευσης στη ΧΜΛ: Σε ποιους ασθενείς και πότε 55
survival advantage with filgrastim-mobilized peripheral blood stem cells (PBSC) compared to bone marrow (BM) transplants from unrelated donors: results of Blood and Marrow Transplant Clinical Trials Network (BMT CTN) protocol 0201, a phase III, prospective, randomized trial. Blood 2011; 118:Abstract 1.
26. Nagamura-Inoue T, Kai S, Azuma H, et al. Unrelated cord blood transplantation in CML: Japan Cord Blood Bank Net-work analysis. Bone Marrow Transplant 2008; 42:241–251.
27. Sanz GF, Saavedra S, Jimenez C, et al. Unrelated donor cord blood transplantation in adults with chronic myelogenous leukemia: results in nine patients from a single institution. Bone Marrow Transplant 2001; 27:693–701.
28. Shimoni A, Kroger N, Zander AR, et al. Imatinib mesylate (STI571) in preparation for allogeneic hematopoietic stem cell transplantation and donor lymphocyte infusions in pa-tients with Philadelphia positive acute leukemias. Leukemia 2003; 17:290-297.
29. Lee SJ, Kukreja M, Wang T, et al. Impact of prior imatinib mesylate on the outcome of hemotopoietic cell transplantation for chronic myeloid leukemia. Blood 2008; 112:3500-3507.
30. Oehler VG, Gooley T, Snyder DS, et al. The effects of ima-tinib mesylate treatment before allogeneic transplantation for chronic myeloid leukemia. Blood 2007; 109:1782-1789.
31. Deininger M, Schleuning M, Greinix H, et al. The effect of prior exposure to imatinib on transplant- related mortality. Hematologica 2006; 91:452-459.
32. Jabbour E, Cortes J, Kartarjian H, et al. Novel tyrosine kinase inhibitor therapy before allogeneic stem cell transplantation in patients with chronic myeloid leukemia. Cancer 2007; 110:340-344.
33. Shimoni A, Leiba M, Schleuning M. et al. Prior treatment with the tyrosine kinase inhibitors dasatinib and nilotinib allows stem cell transplantation (SCT) in a less advanced disease phase and does not increase SCT toxicity in patients with chronic myelogenous leukemia and Philadelphia positive acute lymphoblastic leukemia. Leukemia 2009; 23:190-194.
34. Bittencourt H, Funke V, Fogliatto L, et al. Imatinib mesylate versus allogeneic BMT for patients with chronic myeloid leukemia in first chronic phase. Bone Marrow Transplant 2008; 42:597-600.
35. Hehlmann R, Berger U, Pfirrmann M, et al. Drug treatment is superior to allografting as first line therapy in chronic myeloid leukemia. Blood 2007; 109:4686-4692.
36. Goldman JM, Navneet SM, Klein JP, et al. Relapse and late mortality in 5-year survivors of myeloablative allogeneic stem cell transplantation for chronic myeloid leukemia in first chronic phase. J Clin Oncol 2010; 28:1888-1895.
37. Copelan EA, Crilley PA, Szer J, et al: Late mortality and relapse following BuCy2 and HLA-identical sibling marrow transplantation for chronic myelogenous leukemia. Biol Blood Marrow Transplant 2009; 15:851-855.
38. Robin M, Guardiola P, Devergie A, et al: A 10-year median follow-up study after allogeneic stem cell transplantation for chronic myeloid leukemia in chronic phase from HLA-identical sibling donors. Leukemia 2005; 19:1613-1620.
39. Sokal JE, Cox EB, Baccarani M, et al. Prognostic discrimi-nation in “good risk” chronic granulocytic leukemia. Blood 1984; 63:789-799.
40. Hasford J, Pfirmann M, Hehlmann R, et al. A new prog-nostic score for survival of patients with chronic myeloid leukemia treated with interferon alfa. J Natl Cancer Inst 1998; 90:850-858.
41. Skorski T. BCR/ABL, DNA damage and DNA repair: Implications for new treatment concepts. Leuk Lymphoma 2008; 49:610-614.
42. Hehlmann R, Hochhaus A, Baccarani M. Chronic myeloid leukaemia. Lancet. 2007; 370:342-350.
43. Mitelman F. The Cytogenetic Scenario of Chronic Myeloid Leukemia. Leuk Lymphoma 1993; 11(s1):11-15.
44. Fabarius A, Leitner A, Hochhaus A, et al. Impact of additional cytogenetic aberrations at diagnosis on prognosis of CML: long-term observation of 1151 patients from the randomized CML Study IV. Blood 2011; 26:6760-6768.
45. Kartarjian H, Cortes J, O’Brien S, et al. Imatinib mesylate (STI571) therapy for Philadelphia chromosome-positive chronic myelogenous leukemia in blast phase. Blood 2002; 99:3547-3553.
46. Jiang Q, Xu L-P, Liu D-H, et al. Imatinib mesylate versus allogeneic hematopoietic stem cell transplantation for patients with chronic myelogenous leukemia in the accelerated phase. Blood 2011; 117:3032-3040.
47. Jabbour E, Jones D, Kantarjian HM, et al. Long-term out-come of patients with chronic myeloid leukemia treated with second-generation tyrosine kinase inhibitors after imatinib failure is predicted by the in vitro sensitivity of BCR-ABL kinase domain mutations. Blood 2009; 114:2037-2043.
48. Muller MC, Cortes JE, Kim DW, et al. Dasatinib treatment of chronic-phase chronic myeloid leukemia: analysis of responses according to preexisting BCR-ABL mutations. Blood 2009; 114:4944-4953.
49. Soverini S, Hochhaus A, Nicolini FE, et al. BCR-ABL kinase domain mutation analysis in CML patients treated with TKIs: recommendations from ELN expert panel. Blood 2011; 118:1208-1215.
50. Jabbour E, Cortes J, Santos FPS, et al. Results of allogeneic hematopoietic stem cell transplantation for chronic myelog-enous leukemia patients who failed tyrosine kinase inhibitors after developing BCR-ABL kinase domain mutations. Blood 2011; 117:3641-3647.
51. Milojkovic D, Nicholson E, Apperley JF, et al. Early predic-tion of success or failure using second generation tyrosine kinase inhibitors for chronic myeloid leukemia. Haemato-logica 2010; 95:224-231.
52. Tam CS, Kantarjian H, Garcia-Manero G, et al. Failure to achieve a major cytogenetic response by 12 months defines inadequate response in patients receiving nilotinib or dasatinib as second or subsequent line therapy for chronic myeloid leukemia. Blood 2008; 112:516-518.
53. Marin D, Ibrahim AR, Lucas C, et al. Assessment of BCR-ABL1 transcript levels at 3 months is the only requirement for predicting outcome for patients with chronic myeloid

Ι. Τζάννου και Ι. Μπαλταδάκης56
leukemia treated with tyrosine kinase inhibitors. J Clin Oncol 2012; 30:232-238.
54. Hochhaus A, Saglio G, Chuah C, et al. Dasatinib and imatinib-induced reductions in BCR-ABL transcript levels below 10% at 3 months are associated with improved responses in patients with newly diagnosed chronic myeloid leukemia in chronic
phase (CML-CP): analysis of molecular response kinetics in the DASISION trial. Blood 2011; 118:Abstract 2767.
55. Marin D, Hedgley C, Clark RE, et al. The predictive value of early molecular response in chronic phase CML pa-tients treated with dasatinib first line therapy. Blood 2011; 118:Abstract 785.

Εργαστηριακή παρακολούθηση ασθενών με ΧΜΛ και κλινική αξιολόγηση
Τασούλα Τουλουμενίδου, Αποστολία Παπαλεξανδρή, Κώστας Σταματόπουλος
ΠΕΡΊληΨη: οι γενετικές βλάβες που συμβαίνουν στα πλαίσια της καρκινογένεσης είναι ειδικοί δεί-κτες των καρκινικών κυττάρων, βοηθούν στη διάκρισή τους από τα φυσιολογικά κύτταρα και χρησιμο-ποιούνται ευρέως τόσο για την ταξινόμηση των νεοπλασιών, ιδίως του αιμοποιητικού ιστού, όσο και για την επιλογή της κατάλληλης θεραπευτικής αγωγής. η αλυσιδωτή αντίδραση πολυμεράσης (polymerase chain reaction, PCR) είναι ευαίσθητη και ειδική μέθοδος για την αναζήτηση χιμαιρικών μεταγράφων που προκύπτουν από χρωμοσωμικές μεταθέσεις σε ασθενείς με αιματολογικές κακοήθειες, μεταξύ άλ-λων και χρόνια μυελογενή λευχαιμία (Χμλ). ςήμερα, η μοριακή παρακολούθηση της Χμλ ουσιαστικά ταυτίζεται με την ποσοτική μέτρηση των χιμαιρικών μεταγράφων BCR-ABL με τεχνική real-time PCR (RQ-PCR). με το συγκεκριμένο ζήτημα έχουν ασχοληθεί συστηματικά κι εντατικά ερευνητικές ομάδες από την Ευρώπη, τις ηΠΑ και την Αυστραλία, καταλήγοντας στη διατύπωση κατευθυντήριων οδηγι-ών ώστε να διασφαλίζεται η αξιοπιστία των αποτελεσμάτων του μοριακού ελέγχου. οι συγκεκριμένες οδηγίες καλύπτουν κάθε παράμετρο, από τη συλλογή του δείγματος έως την έκφραση του αποτελέσμα-τος, και θέτουν τους ορισμούς για την αξιολόγηση της ανταπόκρισης (μείζων μοριακή ανταπόκριση, πλήρης μοριακή ανταπόκριση). Επίσης, προτείνουν το ενδεδειγμένο χρονοδιάγραμμα παρακολούθησης και υποδεικνύουν τις περιστάσεις κατά τις οποίες πρέπει να διενεργείται έλεγχος για ανίχνευση μεταλ-λάξεων στην περιοχή κινάσης της τυροσίνης της χιμαιρικής πρωτεΐνης BCR-ABL, όπως επίσης και τη μεθοδολογία ανίχνευσης.
Haema 2012; 3(1): 57-64 Copyright EAE
Αιματολογική κλινική και μμμο, γΝ γ. Παπανικολάου, ΘεσσαλονίκηΔιεύθυνση Αλληλογραφίας: κώστας ςταματόπουλος, Αιματολογική κλι-
νική και μμμο, γΝ «γ. Παπανικολάου», Εξοχή, 57010 Θεσσαλονίκη, τηλ.: 2313.307992 / Fax: 2313.307579, E-mail: [email protected]
Aνασκόπηση
ΕΙΣΑΓΩΓΗη χρόνια μυελογενής λευχαιμία (Χμλ) είναι η πρώ-
τη ανθρώπινη κακοήθεια η οποία συσχετίσθηκε με μια συγκεκριμένη χρωμοσωμική ανωμαλία. ςυγκεκριμένα, το 1960 στη Φιλαδέλφεια των ηνωμένων Πολιτειών της Αμερικής οι Peter Nowell και David Hungerford ανέφε-ραν την παρουσία ενός βραχύτερου χρωμοσώματος 22 μετά από παρατήρηση στο μικροσκόπιο λευχαιμικών κυτ-τάρων μυελού των οστών ασθενών με Χμλ. γι’ αυτό το λόγο, η συγκεκριμένη χρωμοσωμική ανωμαλία ονομά-στηκε χρωμόσωμα Φιλαδέλφειας (Ph).
το 1973, η Janet Rowley ανακάλυψε ότι η δημιουρ-γία του χρωμοσώματος Ph είναι αποτέλεσμα αμοιβαίας μετάθεσης γενετικού υλικού ανάμεσα στα χρωμοσώμα-
τα 9 και 22 και, συγκεκριμένα, ανάμεσα στις ζώνες q34 και q11. ςε μοριακό επίπεδο συμβαίνει θραύση δύο γο-νιδίων που βρίσκονται σε αυτές τις ζώνες (c-ABL και c-BCR, αντιστοίχως) και δημιουργία του χιμαιρικού γο-νιδίου BCR-ABL.
Ανάλογα με τη θέση θραύσης του γονιδίου BCR, η χιμαιρική πρωτεΐνη BCR/ABL μπορεί έχει μέγεθος από 190 έως 230 KDa. το σημείο θραύσης στο γονίδιο BCR εντοπίζεται σχεδόν αποκλειστικά στην περιοχή M-bcr, μεταξύ των εξωνίων 13 και 14, ή αλλιώς b2 και b4. η σύνδεση γίνεται σταθερά με τo ιντρόνιο μεταξύ των εξω-νίων 1b και 2 του ABL. το αποτέλεσμα είναι ένα χιμαιρι-κό μετάγραφο που περιλαμβάνει αλληλουχίες του BCR και το a2 εξώνιο του ABL και κωδικοποιεί τη χιμαιρική πρωτεΐνη p210.
οι περισσότεροι ασθενείς με Χμλ έχουν χιμαιρικά μετάγραφα με τις συμβολές b3-a2 (55%) ή b2-a2 (40%), ενώ σε ποσοστό 5% μπορεί ν’ ανιχνευθούν και τα δύο μετάγραφα. ο τύπος του χιμαιρικού μεταγράφου δε συ-σχετίζεται με την κλινική πορεία της νόσου. ςπανιότερα,

Τ. Τουλουμενίδου και συν58
το σημείο θραύσης στο γονίδιο BCR μπορεί να βρίσκεται στην περιοχή m-bcr, οδηγώντας στην παραγωγή της χι-μαιρικής πρωτεΐνης p190 (συχνότερη στην ολλ). ςπο-ραδικά αναφέρεται η δημιουργία μεγαλύτερης πρωτεΐνης p230, από τη θραύση του γονιδίου BCR στο εξώνιο 191-3.
το γονίδιο ABL κωδικοποιεί την πρωτεΐνη p145abl, μια κυτταροπλασματική κινάση της τυροσίνης η οποία περιέχει κρίσιμες λειτουργικές αλληλουχίες γι’ αλληλε-πίδραση με άλλες πρωτεΐνες που συμμετέχουν σ’ ενδοκυτ-τάρια μονοπάτια σηματοδότησης, για την ενδοκυττάρια εντόπισή της και για την αλληλεπίδραση με τον κυττα-ροσκελετό. η λειτουργία της πρωτεΐνης p145 βρίσκεται υπό αυστηρό έλεγχο ώστε ν’ αποφεύγεται το ενδεχόμενο απρόσφορης ή υπέρμετρης ενεργοποίησης των κυττάρων.
η πρωτεΐνη p210 που κωδικοποιείται από το χιμαιρι-κό γονίδιο BCR-ABL έχει ιδιοσύστατα ενεργοποιημένη δραστικότητα κινάσης τυροσίνης και μάλιστα πολύ με-γαλύτερη σε σχέση με την αντίστοιχη της p145. Επίσης, αντιθέτα από την p145, η p210 εμφανίζει ικανότητα αυ-τοφωσφορυλίωσης με συνέπεια να παραμένει συνεχώς ενεργός, διαταράσσοντας πολλές θεμελιώδεις λειτουρ-γίες του κυττάρου (μεταξύ άλλων, πολλαπλασιασμός, απόπτωση, διακυττάρια επικοινωνία, εγκατάσταση στο σωστό μικροπεριβάλλον, προσκόλληση στο στρώμα)4,5.
ΒΑΣΙΚΕΣ ΑΡΧΕΣ ΜΟΡΙΑΚΗΣ ΑΝΙΧΝΕΥΣΗΣ ΧΙΜΑΙΡΙΚΩΝ ΓΟΝΙΔΙΩΝ ΚΑΙ ΜΕΤΑΓΡΑΦΩΝ
οι διάφορες μορφές καρκίνου προκύπτουν από απορ-ρύθμιση της λειτουργίας γονιδίων που εμπλέκονται στον έλεγχο της αύξησης και διαφοροποίησης όπως επίσης και του πολλαπλασιασμού και της απόπτωσης των κυτ-τάρων. η απορρύθμιση μπορεί να επέλθει από ποικίλες δομικές μεταβολές του DNA, π.χ. σημειακές μεταλλάξεις και αμοιβαίες χρωμοσωμικές μεταθέσεις. οι συγκεκρι-μένες δομικές μεταβολές είναι ειδικοί δείκτες των καρ-κινικών κυττάρων, βοηθούν στη διάκρισή τους από τα φυσιολογικά κύτταρα και χρησιμοποιούνται ευρέως τό-σο για την ταξινόμηση των νεοπλασιών, ιδίως του αιμο-ποιητικού ιστού, όσο και για την επιλογή της κατάλληλης θεραπευτικής αγωγής.
η αλυσιδωτή αντίδραση πολυμεράσης (polymerase chain reaction, PCR) είναι ευαίσθητη και ειδική μέθοδος για την αναζήτηση χιμαιρικών μεταγράφων που προκύ-πτουν από χρωμοσωμικές μεταθέσεις σε ασθενείς με αιμα-τολογικές κακοήθειες. τα χιμαιρικά μετάγραφα παρέχουν έναν ειδικό δείκτη για την αναγνώριση του λευχαιμικού κυττάρου, τόσο κατά τη διάγνωση και τυποποίηση της νόσου, όσο και κατά τη αναζήτηση της ελάχιστης υπο-λειμματικής νόσου (ΕΥΝ).
η αναζήτηση των χιμαιρικών μεταγράφων με τη μέ-
θοδο PCR στηρίζεται στο σχεδιασμό ειδικών εκκινητών (primers) που υβριδίζονται με αλληλουχίες των δύο δι-αφορετικών γονιδίων που συμμετέχουν στη δημιουργία του χιμαιρικού γονιδίου, ώστε το προιόν της PCR να πε-ριλαμβάνει τη χιμαιρική αλληλουχία. Επειδή τα σημεία θραύσης των χιμαιρικών γονιδίων μπορεί να ποικίλουν και να κατανέμονται σε μεγάλη έκταση, δημιουργώντας προβλήματα στην επιλογή της περιοχής-στόχου του DNA με βάση την οποία θα σχεδιαστούν οι εκκινητές, ακο-λουθείται μια εναλλακτική στρατηγική. ςυγκεκριμένα, με υπόστρωμα ολικό κυτταρικό RNA (περιέχει το σύνο-λο των μεταγράφων, κανονικών και χιμαιρικών) συντί-θεται συμπληρωματικό (complementary DNA, cDNA) με τη δράση του ενζύμου αντίστροφη μεταγραφάση (re-verse transcriptase, RT). το cDNA είναι απαλλαγμένο από ιντρόνια, οπότε η ενίσχυση των χιμαιρικών αλληλουχιών είναι ευχερέστερη. Έτσι, σε πολλές εφαρμογές, αντί για PCR σε γενωμικό DNA χρησιμοποιείται RT-PCR, οπότε αντί για χιμαιρικά γονίδια στην ουσία ανιχνεύονται χι-μαιρικά μετάγραφα.
ΜΟΡΙΑΚΗ ΑΝΙΧΝΕΥΣΗ ΧΙΜΑΙΡΙΚΩΝ ΜΕΤΑΓΡΑΦΩΝ BCR-ABL
η RT-PCR απαιτεί μικρή ποσότητα αρχικού υλικού, ολοκληρώνεται σε σχετικά σύντομο χρονικό διάστημα κι έχει τη μεγαλύτερη ευαισθησία από όλες τις χρησιμο-ποιούμενες τεχνικές, βασικό πλεονέκτημα στο πλαίσιο ανίχνευσης ΕΥΝ μετά από θεραπεία. η ευαισθησία της φθάνει στην ανίχνευση ενός νεοπλασματικού κυττάρου μέσα σε 105-106 φυσιολογικά κύτταρα. Ωστόσο, βασικό μειονέκτημα της “συμβατικής” (ποιοτικής) PCR είναι η αδυναμία ποσοτικής εκτίμησης του φορτίου της νόσου. με δεδομένη αυτή την αδυναμία, έχουν αναπτυχθεί τε-χνικές ποσοτικής σε πραγματικό χρόνο PCR (Real time Quantitative PCR, RQ-RCR) που επιτρέπουν τον ποσο-τικό προσδιορισμό της ΕΥΝ και χρησιμοποιούνται ευρέ-ως στην παρακολούθηση των ασθενών με αιματολογικές κακοήθειες γενικότερα και Χμλ ειδικότερα6-10.
ςήμερα, η μοριακή παρακολούθηση της Χμλ ου-σιαστικά ταυτίζεται με την ποσοτική μέτρηση των χι-μαιρικών μεταγράφων BCR-ABL με RQ-PCR. με το συγκεκριμένο ζήτημα έχουν ασχοληθεί συστηματικά κι εντατικά ερευνητικές ομάδες από την Ευρώπη, τις ηΠΑ και την Αυστραλία, έχοντας ως βασική επιδίωξη τη δια-τύπωση κατευθυντήριων οδηγιών ώστε να διασφαλίζεται η αξιοπιστία των αποτελεσμάτων του μοριακού ελέγχου. ςτις ενότητες που ακολουθούν θα παρουσιαστούν οι σχε-τικές δραστηριότητες και κατευθυντήριες οδηγίες όπως επίσης και η «στρατηγική» μοριακής παρακολούθησης στην εποχή των αναστολέων της κινάσης της τυροσίνης BCR-ABL6-11.

Εργαστηριακή παρακολούθηση ασθενών με ΧΜΛ και κλινική αξιολόγηση 59
RQ-PCR ΣΤΗΝ πΑΡΑΚΟΛΟΥθΗΣΗ ΑΣθΕΝΩΝ ΜΕ ΧΜΛ: ΔΥΝΑΤΟΤΗΤΕΣ, πΕΡΙΟΡΙΣΜΟΙ ΚΑΙ ζΗΤΗΜΑΤΑ ΕΝΑΡΜΟΝΙΣΗΣ ΑπΟΤΕΛΕΣΜΑΤΩΝ ΜΕΤΑξΥ ΕΡΓΑΣΤΗΡΙΩΝ
η RQ-RCR πραγματοποιείται σε ειδικούς θερμοκυ-κλοποιητές που φέρουν ανιχνευτή φθορισμού και κατα-γράφουν την ποσότητα του προϊόντος PCR σε κάθε κύκλο της αντίδρασης. ςήμερα υπάρχουν ποικίλα όργανα για RQ-PCR με διαφορετική μεθοδολογία, όπως το σύστη-μα TaqMan που βασίζεται στην υδρόλυση διπλά σημα-σμένων ανιχνευτών, και η τεχνολογία LightCycler, που χρησιμοποιεί ένα ζεύγος γειτονικών ανιχνευτών υβριδι-σμού. το αποτέλεσμα μπορεί να δοθεί ως απόλυτη τιμή ή να εκφραστεί σε σχέση μ’ ένα γονίδιο αναφοράς.
η ετερογένεια ως προς τις τεχνικές, τα γονίδια ανα-φοράς και τον τρόπο έκφρασης του αποτελέσματος στις διάφορες μελέτες δημιουργούν προβλήματα στη συγκρι-τική αξιολόγηση δεδομένων από διαφορετικές ερευνη-τικές ομάδες. ςτα πλαίσια αυτά, εκπονήθηκαν διεθνή συνεργατικά προγράμματα με σκοπό την προτυποποίη-ση της RQ-PCR8,9. τα συμπεράσματα των προγραμμάτων και οι βασικές κατευθυντήριες οδηγίες έχουν ως εξής:11,12
1. Είδος δείγματος: για την ανίχνευση ΕΥΝ μπορεί να χρησιμοποιηθεί αίμα ή αναρρόφημα μυελού των οστών. Ωστόσο, ο ταυτόχρονος προσδιορισμός μεταγράφων BCR-ABL σε αίμα και μυελό έδειξε αποκλίσεις σε ορι-σμένους ασθενείς (διαφορά αριθμού μεταγράφων έως και ένα λογάριθμο μεταξύ των δύο δειγμάτων). ςυνεπώς, για τη διαχρονική παρακολούθηση του ασθενούς συνιστάται η χρησιμοποίηση αποκλειστικά του ίδιου είδους δείγμα-τος (αίμα ή αναρρόφημα μυελού των οστών).
2. Κυτταροβρίθεια δείγματος: προϋπόθεση για την αξιοπιστία των αποτελεσμάτων είναι η ικανοποιητική κυτταροβρίθεια του δείγματος (τουλάχιστον 1-2Χ107 λευ-κοκύτταρα, που αντιστοιχούν σε 5-10 ml αίμα).
3. Επεξεργασία δείγματος: η απομόνωση του RNA πραγματοποιείται σε λευκοκύτταρα μετά από λύση των ερυθροκυττάρων και όχι σε στοιβάδα μονοπυρήνων.
4. Πρωτόκολλο RQ-PCR: συνήθως χρησιμοποιούνται ανιχνευτές υδρόλυσης ή υβριδισμού.
5. Εσωτερικός ποιοτικός έλεγχος – Γονίδιο αναφοράς: ένας σημαντικός εσωτερικός έλεγχος για την ποιότητα του δείγματος συνίσταται σε ποσοτικοποίηση των μετα-γράφων ενός γονιδίου αναφοράς. το γονίδιο αναφοράς πρέπει να έχει ορισμένα χαρακτηριστικά:
• να μη διαθέτει σχετιζόμενα ψευδογονίδια• να εκφράζεται σε συγκρίσιμο επίπεδο με το γονίδιο-
στόχο • να εκφράζεται σε όλες τις κυτταρικές σειρές • να μην επηρεάζεται από την υποκείμενη νόσο• να μην εμφανίζει διακυμάνσεις έκφρασης ανάλογα
με τη φάση της νόσου.
για τη μοριακή παρακολούθηση ΕΥΝ σε ασθενείς με Χμλ, ως γονίδια αναφοράς χρησιμοποιούνται τα γονί-δια ABL, BCR13 ή BGUS, με πιο διαδεδομένο το ABL14. ο ελάχιστος αποδεκτός αριθμός μεταγράφων του γονι-δίου αναφοράς ABL είναι 10.000. Ωστόσο, προκειμένου να επιτευχθεί υψηλή ευαισθησία και να εξασφαλιστεί η αξιοπιστία του αποτελέσματος σε πολύ χαμηλά φορτία μεταγράφων BCR-ABL, ο απαιτούμενος αριθμός μετα-γράφων του γονιδίου ABL είναι τουλάχιστον 30.00011. Πρόκειται για κρίσιμο ζήτημα στο πλαίσιο αξιολόγησης της πλήρους μοριακής ύφεσης, τόσο υπό αναστολείς της κινάσης BCR-ABL όσο και μετά μεταμόσχευση αλλογε-νών αιμοποιητικών κυττάρων.
6. Ποσοτικός προσδιορισμός των χιμαιρικών μεταγρά-φων: δημιουργείται καμπύλη αναφοράς με πλασμίδια που περιέχουν γνωστές συγκεντρώσεις του γονιδίου-στόχου και του γονιδίου αναφοράς, αντιστοίχως. Ως ευαισθησία της μεθόδου ορίζεται η κατώτερη αραίωση πλασμιδίου στην οποία προκύπτει θετικό αποτέλεσμα (5-10 αντίγρα-φα). το αποτέλεσμα εκφράζεται ως εκατοστιαία αναλο-γία έκφρασης του BCR-ABL προς το γονίδιο αναφοράς ABL (BCR-ABL/ABL X100).
7. Ομαλοποίηση των αποτελεσμάτων: τ’ αποτελέσματα κάθε ανάλυσης ομαλοποιούνται με τη χρήση συντελεστή μετατροπής (conversion factor, CF)11,12, ειδικού για κάθε εργαστήριο, που προσδιορίζεται με μια προτυποποιημέ-νη κι επίπονη διαδικασία ανταλλαγής δειγμάτων μεταξύ εργαστηρίου(-ων) αναφοράς και κατά τόπους εργαστη-ρίων. η ομαλοποίηση επιτρέπει την έκφραση των απο-τελεσμάτων σε «Διεθνή κλίμακα», δηλαδή υπό μορφή που μπορεί ν’ αξιολογηθεί για την εξαγωγή συμπερασμά-των ανεξάρτητα από τις τυχόν μεθοδολογικές διαφορές μεταξύ εργαστηρίων.
«ΜΟΡΙΑΚΗ ΑΝΤΑπΟΚΡΙΣΗ» ΣΤΗ ΧΜΛ: ΟΡΙΣΜΟΙ ΚΑΙ ΑΝΟΙΧΤΑ ζΗΤΗΜΑΤΑ
ςύμφωνα με τις διεθνείς κατευθυντήριες οδηγίες (Eu-ropean LeukemiaNet), η ανταπόκριση όπως αξιολογείται με βάση την RQ-PCR προσδιορίζεται με βάση δύο «ορό-σημα». ςυγκεκριμένα:
ως μείζων μοριακή ανταπόκριση (major molecular re-sponse, MMR) ορίζεται η ελάττωση του «φορτίου» των BCR-ABL μεταγράφων κατά τρεις λογάριθμους σε σχέση με το διαγνωστικό δείγμα (έκφραση των αποτελεσμάτων αναφορικά με το διεθνές πρότυπο, IRIS scale)15 .
ως πλήρης μοριακή ανταπόκριση (complete molecu-lar response, CMR) θα έπρεπε να ορίζεται η μη ανίχνευση χιμαιρικών μεταγράφων BCR-ABL. Ωστόσο, το ζήτημα δεν είναι απλό και, προς το παρόν, δεν υπάρχει ομοφωνία ως προς την ακριβή σημασία της CMR επειδή σε πολύ χαμηλά φορτία μεταγράφων η ποσοτικοποίηση παρουσι-άζει εγγενείς δυσχέρειες, ιδίως όταν δεν υπάρχει ικανο-

Τ. Τουλουμενίδου και συν60
ποιητικός αριθμός μεταγράφων του γονιδίου αναφοράς. κατά σύμβαση, ως CMR ορίζεται η ελάττωση του φορτί-ου των BCR-ABL μεταγράφων κατά τουλάχιστον 4 (κατ’ άλλους 4.5, 5 ή και περισσότερο) λογάριθμους σε σχέση με το διαγνωστικό δείγμα11,16,17.
ΣΤΡΑΤΗΓΙΚΗ πΑΡΑΚΟΛΟΥθΗΣΗΣ ΕΛΑΧΙΣΤΗΣ ΥπΟΛΕΙΜΜΑΤΙΚΗΣ ΝΟΣΟΥ ΣΤΗ ΧΜΛ: ΑΣθΕΝΕΙΣ ΣΕ ΧΡΟΝΙΑ ΦΑΣΗ
ςύμφωνα με τις αναθεωρημένες οδηγίες του Europe-an LeukemiaNet16, η παρακολούθηση ασθενών με Χμλ υπό αναστολείς των κινασών της τυροσίνης BCR-ABL (Tyrosine Kinase Inhibitors. TKI) βασίζεται στις εξής εξετάσεις: αιμοδιάγραμμα, κυτταρογενετικός έλεγχος, μοριακή ανίχνευση μεταγράφων BCR-ABL με RQ-PCR και ανίχνευση μεταλλάξεων BCR-ABL σε ειδικές περι-πτώσεις (Πίνακας 1).
κατά τη διάγνωση είναι απαραίτητος ο προσδιορι-σμός του τύπου του μεταγράφου που φέρει ο ασθενής, ο οποίος πραγματοποιείται με ποιοτική RT-PCR18, συνήθως σύμφωνα με προτυποποιημένα πρωτόκολλα του Euro-pean LeukemiaNet14. ςε σπάνιες περιπτώσεις με κλινική και αιματολογική εικόνα Χμλ, η RT-PCR για χιμαιρι-κά μετάγραφα BCR-ABL που κωδικοποιούν την πρωτε-ΐνη p210 είναι αρνητική. ςε τέτοιες περιπτώσεις πρέπει ν’ αποκλειστεί το ενδεχόμενο της παρουσίας εναλλακτι-
κών μεταγράφων BCR-ABL, όπως τα μετάγραφα που κω-δικοποιούν τις χιμαιρικές πρωτεΐνες p190 ή p230 (<1% του συνόλου των ασθενών), για τα οποία δεν υπάρχουν πρωτυποποιημένα πρωτόκολλα ποσοτικοποίησης με RQ-PCR (ιδίως για τα p230).
Απαραίτητη διαγνωστική ενέργεια κατά τη διάγνωση είναι και η ποσοτικοποίηση του φορτίου των χιμαιρικών μεταγράφων με RQ-PCR, ώστε να υπάρχει σημείο ανα-φοράς για τη μετέπειτα παρακολούθηση της ανταπόκρι-σης στη θεραπεία.
Ανίχνευση ελάχιστης υπολειμματικής νόσου σε ασθενείς υπό Imatinib mesylate
ςύμφωνα με τις διεθνείς συστάσεις16, η ανίχνευση ελά-χιστης υπολειμματικής νόσου με τεχνική RQ-PCR μετά θεραπεία με Imatinib Mesylate (Glivec) πρέπει να διενερ-γείται κάθε 3 μήνες έως την επίτευξη MMR. Έπειτα, ο μο-ριακός έλεγχος μπορεί να διενεργείται τουλάχιστον κάθε 6 μήνες, με βέλτιστη συχνότητα τους 3-4 μήνες. τακτικό-τερη παρακολούθηση συνιστάται σε ασθενείς με υποβέλ-τιστη απάντηση (με βάση τα κριτήρια ELN, Πίνακας 2) και ασθενείς με χαρακτηριστικά υψηλού κινδύνου (υψη-λό Sokal score, επιπρόσθετες χρωμοσωμικές ανωμαλίες).
Ανίχνευση ελάχιστης υπολειμματικής νόσου σε ασθενείς υπό 2ης γενιάς TKI ως θεραπεία 1ης γραμμής
οι ασθενείς που λαμβάνουν 2ης γενιάς αναστολείς κι-νασών της τυροσίνης (Nilotinib, Dasatinib) ως θεραπεία 1ης γραμμής επιτυγχάνουν βαθύτερες υφέσεις και ταχύ-τερη CCyR. ςε αυτό το πλαίσιο τίθεται ο προβληματι-σμός αν οι συγκεκριμένοι ασθενείς πρέπει να ελέγχονται με RQ-PCR με μεγαλύτερη συχνότητα μέχρι την επίτευ-ξη MMR16, άποψη την οποία συμμερίζονται και οι συγ-γραφείς του παρόντος άρθρου.
Ανίχνευση ελάχιστης υπολειμματικής νόσου σε ασθενείς μετά από αλλογενή μεταμόσχευση
οι ασθενείς με Χμλ που υποβάλλονται σε αλλογενή μεταμόσχευση πρέπει να παρακολουθούνται επ’αόριστον σε διαστήματα 3- ή 6- μηνών. η ανίχνευση μεταγράφων BCR-ABL σε χαμηλό φορτίο (<0.02%) σ’ ένα μόνο στιγ-μιότυπο δεν σηματοδοτεί μοριακή υποτροπή. οι ασθε-νείς με περιοδική θετικότητα για μετάγραφα BCR-ABL σε χαμηλό επίπεδο μπορεί να παραμείνουν σε μακρόχρο-νη ύφεση. οι ασθενείς με σταθερά αρνητικό φορτίο (ή ένα στιγμιότυπο χαμηλού φορτίου) BCR-ABL μεταγρά-
Πίνακας 1. Κατευθυντήριες οδηγίες για την παρα-κολούθηση ασθενών με ΧΜΛ υπό αναστολείς κινα-σών της τυροσίνης.
Αιμοδιάγραμμα Στη διάγνωση, ανά 15 μέρες μέχρι την πλήρη αιματολογική ύφεση και στη συνέχεια τουλάχιστον ανά 3 μήνες.
Κυτταρογενετικός έλεγχος
Στη διάγνωση, στους 3 και 6 μήνες. Στη συνέχεια, ανά 6 μήνες μέχρι την επίτευξη CCyR και, έπειτα, ανά 12 μήνες. Σε κάθε περίπτωση αποτυχίας ή ανεξήγητων κυτταροπενιών.
RQ-PCR Ανά 3 μήνες μέχρι την επίτευξη MMR. Έπειτα, τουλάχιστον ανά 6 μήνες.
Ανίχνευση μεταλλάξεων BCR-ABL
Σε πρωτογενή αντίσταση ή υποβέλτιστη απάντηση. Σε αλλαγή της θεραπείας.
CCyR: πλήρης κυτταρογενετική ανταπόκριση. MMR: μείζων μοριακή ανταπόκριση.
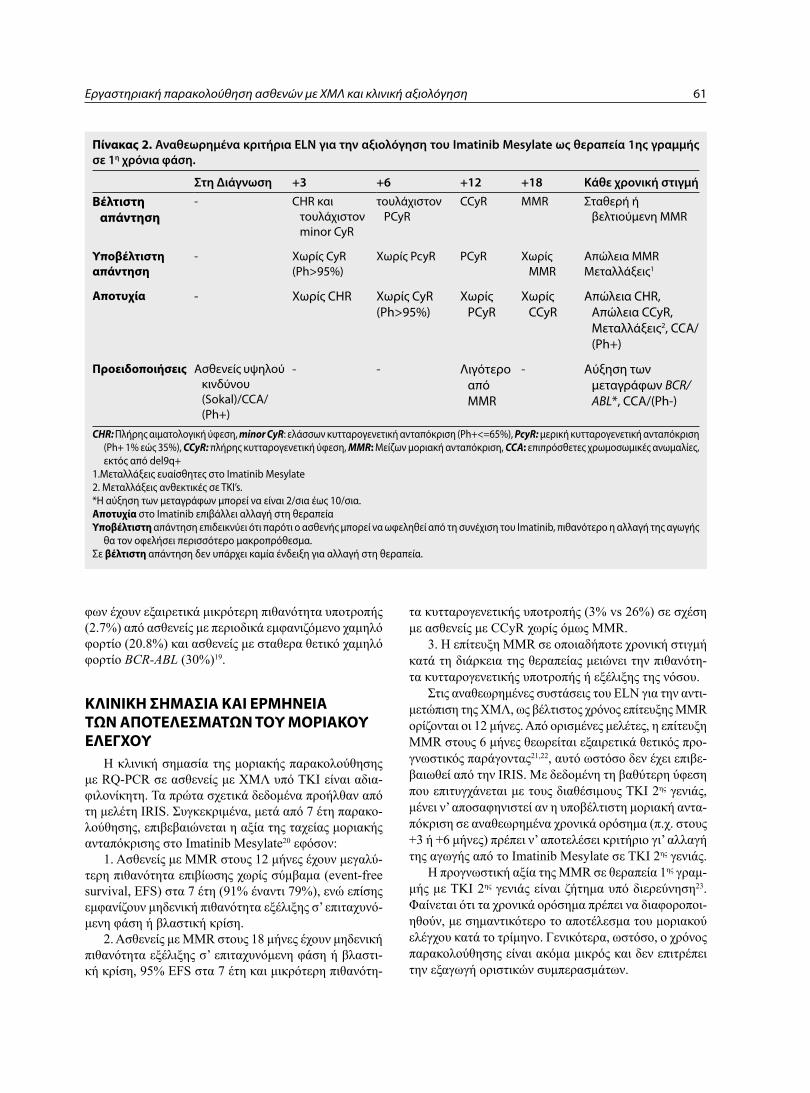
Εργαστηριακή παρακολούθηση ασθενών με ΧΜΛ και κλινική αξιολόγηση 61
φων έχουν εξαιρετικά μικρότερη πιθανότητα υποτροπής (2.7%) από ασθενείς με περιοδικά εμφανιζόμενο χαμηλό φορτίο (20.8%) και ασθενείς με σταθερα θετικό χαμηλό φορτίο BCR-ABL (30%)19.
ΚΛΙΝΙΚΗ ΣΗΜΑΣΙΑ ΚΑΙ ΕΡΜΗΝΕΙΑ ΤΩΝ ΑπΟΤΕΛΕΣΜΑΤΩΝ ΤΟΥ ΜΟΡΙΑΚΟΥ ΕΛΕΓΧΟΥ
η κλινική σημασία της μοριακής παρακολούθησης με RQ-PCR σε ασθενείς με Χμλ υπό TKI είναι αδια-φιλονίκητη. τα πρώτα σχετικά δεδομένα προήλθαν από τη μελέτη IRIS. ςυγκεκριμένα, μετά από 7 έτη παρακο-λούθησης, επιβεβαιώνεται η αξία της ταχείας μοριακής ανταπόκρισης στο Imatinib Mesylate20 εφόσον:
1. Ασθενείς με MMR στους 12 μήνες έχουν μεγαλύ-τερη πιθανότητα επιβίωσης χωρίς σύμβαμα (event-free survival, EFS) στα 7 έτη (91% έναντι 79%), ενώ επίσης εμφανίζουν μηδενική πιθανότητα εξέλιξης σ’ επιταχυνό-μενη φάση ή βλαστική κρίση.
2. Ασθενείς με MMR στους 18 μήνες έχουν μηδενική πιθανότητα εξέλιξης σ’ επιταχυνόμενη φάση ή βλαστι-κή κρίση, 95% EFS στα 7 έτη και μικρότερη πιθανότη-
τα κυτταρογενετικής υποτροπής (3% vs 26%) σε σχέση με ασθενείς με CCyR χωρίς όμως μμR.
3. η επίτευξη MMR σε οποιαδήποτε χρονική στιγμή κατά τη διάρκεια της θεραπείας μειώνει την πιθανότη-τα κυτταρογενετικής υποτροπής ή εξέλιξης της νόσου.
ςτις αναθεωρημένες συστάσεις του ELN για την αντι-μετώπιση της Χμλ, ως βέλτιστος χρόνος επίτευξης MMR ορίζονται οι 12 μήνες. Από ορισμένες μελέτες, η επίτευξη MMR στους 6 μήνες θεωρείται εξαιρετικά θετικός προ-γνωστικός παράγοντας21,22, αυτό ωστόσο δεν έχει επιβε-βαιωθεί από την IRIS. με δεδομένη τη βαθύτερη ύφεση που επιτυγχάνεται με τους διαθέσιμους TKI 2ης γενιάς, μένει ν’ αποσαφηνιστεί αν η υποβέλτιστη μοριακή αντα-πόκριση σε αναθεωρημένα χρονικά ορόσημα (π.χ. στους +3 ή +6 μήνες) πρέπει ν’ αποτελέσει κριτήριο γι’ αλλαγή της αγωγής από το Imatinib Mesylate σε τκι 2ης γενιάς.
η προγνωστική αξία της MMR σε θεραπεία 1ης γραμ-μής με TKI 2ης γενιάς είναι ζήτημα υπό διερεύνηση23. Φαίνεται ότι τα χρονικά ορόσημα πρέπει να διαφοροποι-ηθούν, με σημαντικότερο το αποτέλεσμα του μοριακού ελέγχου κατά το τρίμηνο. γενικότερα, ωστόσο, ο χρόνος παρακολούθησης είναι ακόμα μικρός και δεν επιτρέπει την εξαγωγή οριστικών συμπερασμάτων.
πίνακας 2. Αναθεωρημένα κριτήρια ELN για την αξιολόγηση του Imatinib Mesylate ως θεραπεία 1ης γραμμής σε 1η χρόνια φάση.
Στη Διάγνωση +3 +6 +12 +18 Κάθε χρονική στιγμήΒέλτιστη
απάντηση- CHR και
τουλάχιστον minor CyR
τουλάχιστον PCyR
CCyR MMR Σταθερή ή βελτιούμενη MMR
Υποβέλτιστηαπάντηση
- Χωρίς CyR(Ph>95%)
Χωρίς PcyR PCyR Χωρίς MMR
Απώλεια MMRΜεταλλάξεις1
Αποτυχία - Χωρίς CHR Χωρίς CyR(Ph>95%)
Χωρίς PCyR
Χωρίς CCyR
Απώλεια CHR, Απώλεια CCyR, Μεταλλάξεις2, CCA/(Ph+)
προειδοποιήσεις Ασθενείς υψηλού κινδύνου (Sokal)/CCA/(Ph+)
- - Λιγότερο από MMR
- Αύξηση των μεταγράφων BCR/ABL*, CCA/(Ph-)
CHR: Πλήρης αιματολογική ύφεση, minor CyR: ελάσσων κυτταρογενετική ανταπόκριση (Ph+<=65%), PcyR: μερική κυτταρογενετική ανταπόκριση (Ph+ 1% εώς 35%), CCyR: πλήρης κυτταρογενετική ύφεση, MMR: Μείζων μοριακή ανταπόκριση, CCA: επιπρόσθετες χρωμοσωμικές ανωμαλίες, εκτός από del9q+
1.Μεταλλάξεις ευαίσθητες στο Imatinib Mesylate2. Μεταλλάξεις ανθεκτικές σε ΤΚΙ’s.*Η αύξηση των μεταγράφων μπορεί να είναι 2/σια έως 10/σια.Αποτυχία στο Imatinib επιβάλλει αλλαγή στη θεραπείαΥποβέλτιστη απάντηση επιδεικνύει ότι παρότι ο ασθενής μπορεί να ωφεληθεί από τη συνέχιση του Imatinib, πιθανότερο η αλλαγή της αγωγής
θα τον οφελήσει περισσότερο μακροπρόθεσμα.Σε βέλτιστη απάντηση δεν υπάρχει καμία ένδειξη για αλλαγή στη θεραπεία.

Τ. Τουλουμενίδου και συν62
ΑΝΤΟΧΗ ΣΤΗ θΕΡΑπΕΙΑ ΜΕ ΑΝΑΣΤΟΛΕΙΣ ΚΙΝΑΣΩΝ ΤΗΣ ΤΥΡΟΣΙΝΗΣ ΚΑΙ ΜΕΤΑΛΛΑξΕΙΣ ΤΟΥ ΧΙΜΑΙΡΙΚΟΥ ΓΟΝΙΔΙΟΥ BCR-ABL
Παρότι οι αναστολείς των κινασών της τυροσίνης έχουν βελτιώσει εντυπωσιακά την κλινική πορεία των ασθενών με Χμλ, μερικοί ασθενείς χάνουν την ανταπόκριση ή εμφα-νίζουν αντοχή. ςτα υποκείμενα αίτια συμπεριλαμβάνονται, μεταξύ άλλων, τροποποίησεις σε μεταφορείς φαρμάκων, επιπρόσθετες κυτταρογενετικές ανωμαλίες, επαύξηση (am-plification) του γονιδίου BCR-ABL, ενεργοποιήση άλλων κινασών τυροσίνης κ.λπ.24,25. Ωστόσο, ίσως το σημαντι-κότερο ρόλο (ιδίως από την άποψη των περαιτέρω θερα-πευτικών χειρισμών) διαδραματίζουν οι μεταλλάξεις που ανιχνεύονται στην περιοχή κινάσης του γονιδίου BCR-ABL.
ςήμερα γνωρίζουμε ότι 45-50% των ασθενών που εμφανίζουν αντοχή στο Imatinib Mesylate έχουν μεταλ-λάξεις του γονιδίου BCR-ABL. Όλες οι μεταλλάξεις δεν έχουν την ίδια συχνότητα ούτε τον ίδιο κλινικό αντίκτυ-πο26,27, καθώς κάποιες μεταλλάξεις φαίνεται να μην παί-ζουν ρόλο στην εξέλιξη της νόσου. Ωστόσο, υπάρχει ομοφωνία για το γεγονός ότι τη δυσμενέστερη πρόγνω-ση μεταξύ όλων των μεταλλάξεων έχει η T315I. Εφόσον ανιχνευθεί η συγκεκριμένη μετάλλαξη, προς το παρόν, η μοναδική αποτελεσματική επιλογή για τους ασθενείς εί-ναι η μεταμόσχευση αλλογενών αιμοποιητικών κυττάρων. Επίσης, πρόσφατα αναφέρθηκε ότι η παρουσία πολλα-πλών μεταλλάξεων επηρεάζει αρνητικά την πρόγνωση28.
ΜΕθΟΔΟΙ ΑΝΙΧΝΕΥΣΗΣ ΤΩΝ ΜΕΤΑΛΛΑξΕΩΝ ΤΟΥ ΧΙΜΑΙΡΙΚΟΥ ΓΟΝΙΔΙΟΥ BCR-ABL
Υπάρχουν αρκετές διαθέσιμες τεχνικές για την ανί-χνευση μεταλλάξεων του γονιδίου BCR-ABL που ποικί-λουν ως προς την ευαισθησία, το κόστος και το βαθμό δυσκολίας. ορισμένες (π.χ. pyrosequencing, ligation-de-pendent competitive PCR, φασματοσκοπία μαζών, muta-tion-specific PCR, polymerase colony assay) έχουν μεν εξαιρετική ευαισθησία (0.0003% έως 0.1%) αλλά είναι τεχνικά δύσκολες και (προς το παρόν) ιδιαίτερα δαπα-νηρές. Έτσι, στην εργαστηριακή πρακτική χρησιμοποι-ούνται η αποδιατακτική υγρή χρωματογραφία υψηλής απόδοσης (D-HPLC) και η αλληλούχηση του DNA29,30. η πρώτη τεχνική υπερτερεί από την άποψη της ευαισθη-σίας (1% έναντι 10-15%), αλλά είναι ακριβή και δεν τυ-ποποιεί τη μετάλλαξη, γι’ αυτό και κυρίως εφαρμόζεται η αλληλούχηση του DNA. ςτη συγκεκριμένη προσέγγιση αναλύονται οι αλληλουχίες των εξωνίων 4-10 του γονιδί-ου ABL που συμμετέχει στη δημιουργία του χιμαιρικού γονιδίου, στις οποίες συμπεριλαμβάνεται και η περιοχή με ενεργότητα κινάσης της τυροσίνης.
Πρόσφατα ολοκληρώθηκε η διεθνής συνεργατική προσπάθεια για τη διατύπωση κατευθυντήριων οδηγιών σχετικά με τη μεθοδολογία ανίχνευσης μεταλλάξεων και το χρόνο διενέργειας της εξέτασης (Πίνακας 3)31. ςύμ-φωνα με αυτές τις οδηγίες, ο έλεγχος κατά τη διάγνωση πρέπει να γίνεται μόνο όταν η νόσος εκδηλώνεται σ’ επι-ταχυνόμενη φάση (ΕΦ) ή βλαστική κρίση (Βκ) επειδή σε χρόνια φάση πολύ σπάνια ανιχνεύονται μεταλλάξεις, των οποίων μάλιστα η κλινική σημασία είναι αδιευκρί-νιστη. Αντίθετα, σε ΕΦ/Βκ οι μεταλλαξεις είναι συχνές, πιθανότατα στα πλαίσια γενετικής αστάθειας του λευχαι-μικού κλώνου, και η ανίχνευσή τους μπορεί να καθορί-σει την επιλογή της θεραπείας.
κατά τη θεραπεία 1ης γραμμής με Imatinib Mesylate, ο έλεγχος για μεταλλάξεις δεν πρέπει να πραγματοποιεί-ται ως εξέταση ρουτίνας. Αντίθετα, 29% των ασθενών με αποτυχία στο Imatinib έχουν μεταλλάξεις και η ανίχνευ-σή τους μπορεί να καθοδηγήσει την επιλογή της θερα-πείας 2ης γραμμής. μεταξύ των ασθενών με υποβέλτιστη απάντηση, 16% φέρουν μεταλλάξεις, ιδίως πρόκειται για υποβέλτιστη κυτταρογενετική παρά μοριακή απάντηση. τέλος, σε αύξηση των μεταγράφων BCR-ABL που οδηγεί σε απώλεια της MMR κρίνεται απαραίτητη η ανίχνευση μεταλλάξεων ως αίτιο αντίστασης του κλώνου στη θερα-πεία. Ασθενείς που αυξάνουν τον αριθμό των μεταγράφων BCR-ABL αλλά παραμένουν σε MMR πρέπει να παρακο-λουθούνται προσεκτικά ώστε να διαπιστωθεί αν το φορ-τίο των μεταγράφων έχει αυξητική τάση. γενικότερα, δεν έχει τεκμηριωθεί συσχέτιση τέτοιων μικρών διακυμάν-σεων (στα όρια του τεχνικού λάθους) με την παρουσία μεταλλάξεων στην περιοχή της κινάσης της τυροσίνης.
τέλος, σε θεραπεία 2ης γραμμής με Dasatinib/Nilotinib, μεταλλάξεις αναζητούνται σε περίπτωση αιματολογικής ή κυτταρογενετικής αποτυχίας (καμία κυτταρογενετική απάντηση σε 3 μήνες, ελάχιστη σε 6 μήνες ή μικρότερη από μερική σε 12 μήνες). Προς το παρόν, τα διαθέσιμα δεδομένα από τη χορήγηση αυτών των φαρμάκων ως θε-ραπεία 1ης γραμμής δεν επαρκούν για την εξαγωγή συ-μπερασμάτων και κατευθυντήριων γραμμών.
Πίνακας 3. Κατευθυντήριες οδηγίες του ELN για το χρόνο ανίχνευσης μεταλλάξεων.
Στη διάγνωση Μόνο σε επιταχυνόμενη φάση ή βλαστική κρίση
Θεραπεία 1ης γραμμής με Imatinib Mesylate
Αποτυχία θεραπείαςΑπώλεια MMRΥποβέλτιστη απάντηση
Θεραπεία 2ης γραμμής με Dasatinib/Nilotinib
Σε περίπτωση αιματολογικής ή κυτταρογενετικής αποτυχίας

Εργαστηριακή παρακολούθηση ασθενών με ΧΜΛ και κλινική αξιολόγηση 63
Guidelines for the molecular monitoring of patients with CML treaτed with tyrosine kinase inhibitors
by Tassoula Touloumenidou, Apostolia Papalexandri, Kostas Stamatopoulos Hematology Department and HCT Unit, G. Papanicolaou Hospital, Thessaloniki, Greece
ABSTRACT: The advent of therapies with a curative potential for patients with hematological malig-nancies created the need for better assessment of response. Hence, sensitive and accurate detection of minimal residual disease (MRD) is considered as a pre-requisite for therapeutic success. MRD assess-ment in chronic myeloid leukemia (CML) is based on the detection of the chimeric BCR-ABL mRNA transcripts which serve as a “molecular signature” which can be used for the discrimination of residu-al malignant cells from normal hematopoietic cells. Modern PCR protocols for the detection of BCR-ABL transcripts fulfil some important criteria: in particular, (i) they provide specificity for malignancy and, (ii) they are characterized by satisfactory sensitivity. However, limitations persist: thus, standardi-zation is suboptimal or not fully implemented, while the precise quantification of MRD levels is still an open issue. Today, the molecular monitoring of BCR-ABL transcripts in CML is performed by real-time quantitative PCR (RQ-PCR) protocols. Thanks to international multi-institutional initiatives, recom-mendations for harmonizing the differing RQ-PCR methodologies with the objective of maximizing re-liability of analysis for clinical decision making now exist: all aspects of the analysis are covered, from source and number of cells to validation of PCR amplification efficiencies, ensuring the comparability of the results from different trials and/or medical centers. A cornerstone of the follow-up of CML pa-tients treated with tyrosine kinase inhibitors relates to the accurate detection of somatic mutations with-in the BCR-ABL kinase domain, which represent the major mechanism of acquired resistance. Several approaches are available for mutation detection; however, presently the most widely used method is di-rect sequencing. More sensitive approaches may be beneficial, but are yet to prove their clinical utility.
Βιβλιογραφία1. Vardiman JW. Chronic myelogenous leukemia, BCR-ABL1+.
Am J Clin Pathol. 2009; 132(2):250-60. 2. Jabbour E, Fava C, Kantarjian H. Advances in the biology
and therapy of patients with chronic myeloid leukaemia. Best Pract Res Clin Haematol. 2009; 22(3):395-407.
3. Burke BA, Carroll M. BCR-ABL: a multi-faceted promoter of DNA mutation in chronic myelogeneous leukemia. Leu-kemia. 2010; 24(6):1105-12.
4. Sloma I, Jiang X, Eaves AC, Eaves CJ. Insights into the stem cells of chronic myeloid leukemia. Leukemia. 2010; 24(11):1823-33.
5. Carter BZ, Mak DH, Cortes J, Andreeff M. The elusive chronic myeloid leukemia stem cell: does it matter and how do we eliminate it? Semin Hematol. 2010; 47(4):362-70.
6. Gabert J, Beillard E, van der Velden VH, et al. Standardization and quality control studies of ‘real-time’ quantitative reverse transcriptase polymerase chain reaction of fusion gene tran-scripts for residual disease detection in leukemia - a Europe Against Cancer program. Leukemia. 2003; 1 2:2318-57.
7. van der Velden VH, Hochhaus A, Cazzaniga G, et al. Detec-tion of minimal residual disease in hematologic malignancies by real-time quantitative PCR: principles, approaches, and laboratory aspects. Leukemia. 2003; 6:1013-34.
8. Branford S, Cross N, Hochhaus A, et al. Rationale for the recommendations for harmonizing current methodology
for detecting BCR-ABL transcripts in patients with chronic myeloid leukaemia. Leukemia. 2006; 20:1925–1930.
9. Hughes T, Deininger M, Hochhaus A, et al. Monitoring CML patients responding to treatment with tyrosine kinase inhibitors: review and recommendations for harmonizing current methodology for detecting BCR-ABL transcripts and kinase domain mutations and for expressing results. Blood. 2006; 108:28-37.
10. Melo JV, Ross DM. Minimal residual disease and discon-tinuation of therapy in chronic myeloid leukemia: can we aim at a cure? Hematology Am Soc Hematol Educ Program. 2011; 2011:136-42.
11. Branford S, Hughes TP. Practical considerations for monitor-ing patients with chronic myeloid leukemia. Semin Hematol. 2010; 47(4):327-34.
12. Müller MC, Cross NC, Erben P, et al. Harmonization of molecular monitoring of CML therapy in Europe. Leukemia. 2009; 23:1957–1963.
13. Ross DM, Branford S, Seymoyr JF, et al. Patients with chronic myeloid leukemia who maintain a complete mo-lecular response after stopping imatinib treatment have evidence of persistent leukemia by DNA PCR. Leukemia. 2010; 24:1719-1724.
14. van Dongen JJ, Macintyre EA, Gabert JA, et al. Standardized RT-PCR analysis of fusion gene transcripts from chromo-some aberrations in acute leukemia for detection of minimal

Τ. Τουλουμενίδου και συν64
residual disease. Report of the BIOMED-1 Concerted Action: investigation of minimal residual disease in acute leukemia. Leukemia. 1999; 13(12):1901-1928.
15. Hughes TP, Kaeda J, Branford S, et al, for the International Randomised Study of Interferon versus STI571 (IRIS) Study Group. Frequency of Major Molecular Responses to Imatinib or Interferon Alfa plus Cytarabine in Newly Chronic Myeloid Leukemia N Engl J Med. 2003; 349(15):1423-1432.
16. Baccarani M, Cortes J, Pane F, et al. Chronic myeloid leukemia: an update of concepts and management recom-mendations of European LeukemiaNet., J Clin Oncol. 2009; 27(35):6041-6051.
17. Baccarani M, Saglio G, Goldman J, et al; European Leuke-mia Net.Evolving concepts in the management of chronic myeloid leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood. 2006; 108(6):1809-1820.
18. Hughes T, Branford S. Molecular monitoring of BCR-ABL as a guide to clinical management in chronic myeloid leu-kaemia. Blood Rev. 2006; 20(1):29- 41.
19. Kaeda J, O’Shea D, Szydlo RM, et al. Serial measurement of BCR-ABL transcripts in the peripheral blood after allogeneic stem cell transplantation for chronic myeloid leukemia: an attempt to define patients who may not require further therapy. Blood. 2006; 107(10):4171-6.
20. Hughes TP, Hochhaus A, Branford S, et al, IRIS investiga-tors. Long-term prognostic significance of early molecular response to imatinib in newly diagnosed chronic myeloid leukemia: an analysis from the International Randomized Study of Interferon and STI571 (IRIS) Blood. 2010; 116: 3758-3765.
21. Branford S, Lawrence R, Grigg A, et al. Long term follow up of patients with CML in chronic phase treated with first-line imatinib suggests that earlier achievement of a major molecular response leads to greater stability of response. Blood. 2008; 112:2113.
22. Quintas-Cardama A, Kantarjian H, Jones D, et al. Delayed achievement of cytogenetic and molecular response is associ-
ated with increased risk of progression among patients with chronic myeloid leukemia in early chronic phase receiving high-dose or standard-dose imatinib therapy. Blood. 2009; 113(25):6315-21.
23. Jabbour E, Kantarjian HM, O’Brien S, et al. Front-line therapy with second-generation tyrosine kinase inhibitors in patients with early chronic phase chronic myeloid leukemia: what is the optimal response? J Clin Oncol. 2011; 29(32):4260-4265.
24. Apperley JF, Part I. Mechanisms of resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol. 2007; 8:1018-29.
25. Mathisen MS, Kantararjian HM, Cortes J, Jabbour E. Mutant BCR-ABL clones in chronic myeloid leukemia Haemato-logica. 2011; 96(3):347-349.
26. O’ Hare T, Eide CA, Deininger MWN. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia Blood. 2007; 110:2242-2249.
27. Branford S, Melo JV, Hughes TP. Selecting optimal second-line tyrosine kinase inhibitor therapy for chronic myeloid leukemia patients after imatinb failure: does the BCR-ABL mutation status really matter? Blood. 2009; 114:5426-5435.
28. Parker WT, Ho M, Scott HS, Hughes TP, Branford S. Poor response to second-line kinase inhibitors in CML patients with multiple low level mutations, irrespective of their resistance profile Blood 2011 (Epub a head of print), PMID 22210874.
29. Ernst T, Erben P, Mueller MC, et al. Dynamics of BCR-ABL mutated clones prior to hematologic or cytogenetic resistance to imatinib. Haematologica. 2008; 93(2):187-192.
30. Ernst T, Hoffmann J, Erben P, et al. ABL single nucleotide polymorphisms may masquerade as BCR-ABL mutations associated with resistance to tyrosine kinase inhibitors in patients with chronic myeloid leukemia. Haematologica. 2008; 93(9):1389-1393.
31. Soverini S, Hochhaus A, Nicolini FE, et al. BCR-ABL ki-nase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: recom-mendations from an expert panel on behalf of European LeukemiaNet. Blood. 2011; 118(5):1208-1215.

Βλαστικά κύτταρα της Χρόνιας Μυελογενούς Λευχαιμίας (CML Stem Cells) - Η οδός προς την ίαση;
Mαρία Δήμου, Παναγιώτης Παναγιωτίδης
ΠΕΡΊληΨη: η Χρόνια μυελογενής λευχαιμία είναι ένα μυελοϋπερπλαστικό νόσημα, το οποίο φέρει την χρωμοσωμική μετάθεση t(9;22). η παθογένεση της νόσου είναι άμεσα συνδεδεμένη με την παρα-γωγή της υβριδικής πρωτεϊνης BCR-ABL που έχει δραστηριότητα τυροσινικής κινάσης. η θεραπεία της Χμλ με αναστολείς των τυροσινικών κινασών (Tyrosin Kinase Inhibitors-TKIs) έχει οδηγήσει σε θεαματικά αποτελέσματα όσον αφορά την επίτευξη πλήρους κυτταρογεννετικής ανταπόκρισης και την μακρά επιβίωση ασθενών με Χμλ. ςε ορισμένους ασθενείς έχει επιτευχθεί μακροχρόνια «πλήρης μο-ριακή ύφεση» της νόσου ; σε ορισμένους διεκόπη η χορήγηση TKIs αλλά στους περισσότερους επανεμ-φανίστηκε μοριακά η νόσος. το γεγονός αυτό υποδεικνύει ότι τα αρχέγονα κύτταρα της Χμλ επιβίωσαν της μακροχρόνιας αγωγής με Imatinib και ευθύνονται για την «μοριακή επανεμφάνιση» της Χμλ. τα αρχέγονα κύτταρα της Χμλ χρησιμοποιούν πλειάδα βιοχημικών μονοπατιών για να εξασφαλίσουν την επιβίωση, την αυτοανανέωσή τους, πολλαπλασιασμό, αναστολή απόπτωσης κ.λπ. το Imatinib mesylate και σε μεγαλύτερο βαθμό οι νεώτεροι τκιs (nilotinib, dasatinib κ.λπ.) οδηγούν σε μοριακές υφέσεις, αλ-λά in vitro μελέτες έχουν δείξει ότι δεν καταφέρνουν να εξαφανίσουν τον πληθυσμό των Χμλ-HSCs. οι κυτταρικές οδοί που χρησιμοποιούν τα αρχέγονα κύτταρα της Χμλ αποτελούν σήμερα αντικείμε-νο εντατικής έρευνας, προκειμένου να σχεδιαστούν θεραπείες που στοχεύουν στην εξαφάνισή τους σε συνδυασμό με χορήγηση τκιs. μια τέτοια συνδυαστική θεραπεία αναστολέων μπορεί να οδηγήσει σε μακροχρόνια «λειτουργική ίαση» σε ασθενείς με Χμλ.
Haema 2012; 3(1): 65-77 Copyright EAE
Α΄ Προπαιδευτική Παθολογική κλινική ΕκΠΑ, Νοσοκομείο «λαϊκό», Αθήνα
Διεύθυνση Αλληλογραφίας: Π. Παναγιωτίδης, Α΄ ΠΠκ ΕκΠΑ, γΝΑ «λαϊκό», e-mail: [email protected]
Aνασκόπηση
ΕΙΣΑΓΩΓΗη Χρόνια μυελογενής λευχαιμία (Χμλ) είναι μία
κλωνική μυελοϋπερπλαστική νόσος που ξεκινά από το επίπεδο του αιμοποιητικού βλαστικού κυττάρου (Hemo-poietic Stem Cell-HSC) και χαρακτηρίζεται από την πα-ρουσία του ογκογονιδίου σύντηξης, BCR-ABL1. το γονίδιο αυτό κωδικοποιεί μία καινοτόμα πρωτεΐνη με λειτουργία τυροσινικής κινάσης (τκ), η οποία προκαλεί αυξημένη παραγωγή και πρώιμη απελευθέρωση μυελοειδών κυτ-τάρων στο περιφερικό αίμα. το ογκογονίδιο BCR-ABL μπορεί να στοχευθεί με αναστολείς τυροσινικών κινασών (Tyrosine Kinase Inhibitors-TKIs) των οποίων o ρόλος είναι να αποσιωπήσουν την λειτουργία της ογκοπρωτε-ϊνης. τκιs όπως το Imatinib mesylate (Glivec®) χρησι-μοποιούνται πια ως καθιερωμένη θεραπεία σε ασθενείς
με πρωτοδιαγνωσμένη Χμλ. Αν και οι τκιs αποτέλε-σαν μια εξαιρετικά σημαντική πρόοδο στη θεραπεία της Χμλ, δεν καταφέρνουν να εξαφανίσουν τελείως τη νό-σο. οι TKIs οδηγούν σε ταχύτατες μοριακές υφέσεις στην πλειονότητα των ασθενών με Χμλ σε χρόνια φάση, αλλά ως επί τω πλείστον στοχεύουν σε διαιρούμενα κύτταρα και συνήθως δεν εξαφανίζουν τα μετάγραφα BCR-ABL στον πληθυσμό των αρχέγονων HSCs2.
Προς επιβεβαίωση των ανωτέρω οι Su Chu και συνερ-γάτες στο ASH meeting τo 20083, έδειξαν ότι οι ασθενείς με Χμλ που λαμβάνουν συνεχώς Imatinib και έχουν επι-τύχει σταθερή πλήρη κυτταρογενετική ύφεση για πάνω από 5 έτη, εξακολουθούν να έχουν μετρήσιμη έκφραση BCR-ABL στον πληθυσμό των αρχέγονων HSCs.
ςε μία πιο πρόσφατη μελέτη, την STIM trial (Stop IMatinib) από την γαλλική ομάδα των Mahon και συνερ-γατών4, μετά τη διακοπή του Imatinib σε ασθενείς που βρίσκονταν σε πλήρη μοριακή ύφεση για τουλάχιστον 2 έτη, παρατηρήθηκαν 61% υποτροπές, η οποίες όμως αντα-ποκρίθηκαν ασφαλώς στην επαναχορήγηση του ιmatinib.

M. Δήμου και Π. Παναγιωτίδης66
Ανάλογα ευρήματα έχουμε και από τη χρήση των νεό-τερων TKIs – Nilotinib, Dasatinib & Bosutinib5-8. τα μό-ρια αυτά αν και έχουν εξαιρετικά αντιπολλαπλασιαστικά αποτελέσματα, η ικανότητά τους να οδηγούν τα Χμλ HSCs και τα πρόδρομα Χμλ κύτταρα σε απόπτωση εί-ναι πολύ περιορισμένη.
καθώς λοιπόν υπάρχει μεγάλη πιθανότητα υποτρο-πής της Χμλ μετά τη διακοπή των τκιs, η σύσταση επί του παρόντος είναι να χορηγούνται εφόρου ζωής. Παρό-λα αυτά οι ανησυχίες που υπάρχουν αναφορικά με την πιθανότητα υποτροπής ενώ συνεχίζεται η θεραπεία με TKIs (αφού δεν υπάρχει πραγματική ίαση), οι ανεπιθύ-μητες ενέργειες από τη θεραπεία, το τεράστιο οικονομικό φορτίο και στοιχεία που αφορούν την έλλειψη συμμόρ-φωσης των ασθενών σε μία δια βίου θεραπεία (ιδίως αν αναφερόμαστε σε νέους ασθενείς αναπαραγωγικής ηλι-κίας), δημιουργούν ένα εξαιρετικά ισχυρό κίνητρο για την αναζήτηση της απόλυτης ίασης. Προφανώς αυτή θα είχε ως στόχο τον πληθυσμό των Χμλ HSCs που όπως είδαμε ανιχνεύεται ακόμα και μετά από πολλά χρόνια επιτυχούς θεραπείας με TKIs και που είναι η αιτία της υποτροπής μετά από τη διακοπή τους.
οι μηχανισμοί με τους οποίους δημιουργείται αυτή η έλλειψη ευαισθησίας των Χμλ HSCs στους TKIs παρα-μένουν σχετικά αδιευκρίνιστοι . Παράγοντες όπως η κυτ-ταρική ηρεμία (quiescence), επίκτητες μεταλλάξεις στο ογκογονίδιο BCR-ABL, υψηλά επίπεδα έκφρασής του και υπερέκφραση διαμεμβανικών πρωτεϊνικών μεταφο-ρέων στα HSCs ενδέχεται να παίζουν ρόλο.
Εν τούτοις, πολύ πρόσφατα η Αmie Corbin και οι συ-νεργάτες της από την ομάδα του καθηγητή Druker στο Oregon9, έδειξαν ότι: το Imatinib in vitro ανέστειλε την δραστικότητα της BCR-ABL TK στον ίδιο βαθμό σε όλα τα HSCs, καθώς και στα πρόδρομα Χμλ κύτταρα από νεοδιαγνωσμένους ασθενείς με Χμλ, τα οποία είτε βρίσκονταν σε φάση ηρεμίας είτε διαιρούνταν. Αν και η σύντομης διάρκειας in vitro θεραπεία με Imatinib ελάτ-τωνε τον πολλαπλασιασμό των Χμλ HSCs και προδρό-μων, τα κύτταρα αυτά μπορούσαν παρουσία κυτοκινών και αυξητικών παραγόντων να πολλαπλασιάζονται όπως και τα φυσιολογικά ομόλογά τους HSCs και πρόδρομα αιμοποιητικά κύτταρα, παρά την απουσία της BCR-ABL δραστικότητας. τα ευρήματα αυτά αποδεικνύουν ότι τα Χμλ HSCs δεν είναι εξαρτώμενα από την παρουσία του ογκογονιδίου για την επιβίωσή τους και ότι μπορούν να επιβιώνουν και να πολλαπλασιάζονται ανεξάρτητα από την μακροχρόνια καταστολή του BCR-ABL ογκογονιδί-ου με τους διάφορους τκιs.
Παρόμοια αποτελέσματα έδειξε και η ακόμα πιο πρό-σφατη μελέτη των Ashley Hamilton και συνεργατών στο Blood10. οι ερευνητές εδώ προχώρησαν σε πειράματα με διαγονιδιακά ποντίκια με Χμλ με τη χρήση του da-satinib. H εργασία αυτή απέδειξε ότι λειτουργικά Χμλ HSCs και πρόδρομα Χμλ κύτταρα μπορούσαν να επι-
βιώνουν in vitro για μεγάλα χρονικά διαστήματα, παρά την πλήρη καταστολή του ογκογονιδίου και παρά την έλ-λειψη κυτοκινών. τα Χμλ HSCs που επιβίωσαν είχαν πλήρως αδρανοποιημένη τη λειτουργία της BCR-ABL TK. Επιπλέον φάνηκε ότι αυτός ο πληθυσμός των αν-θεκτικών στο dasatinib HSCs ανήκε όντως στον κλώνο και ήταν πλούσιος σε αρχέγονα κύτταρα, τα οποία ούτε πολλαπλασιασμό των BCR-ABL μεταγράφων είχαν, ούτε μεταλλάξεις στο τμήμα της τυροσινικής κινάσης παρου-σίαζαν (στοιχεία δηλαδή που θα μπορούσαν να εξηγή-σουν την αντοχή στον TKI ).
Όλα τα ανωτέρω ευρήματα υποδεικνύουν ότι πρέπει να στραφούμε σε στόχους ανεξάρτητους από το BCR-ABL, προκειμένου να εξαφανίσουμε τα Χμλ HSCs και κατά συνέπεια να επιτύχουμε την ίαση για τη νόσο.
ςτην προσπάθειά μας να ανιχνεύσουμε πιθανούς θε-ραπευτικούς στόχους για την εξόντωση των Χμλ HSCs, είναι απαραίτητο να κατανοήσουμε βασικά στοιχεία της βιολογίας τους, που όπως πλέον γνωρίζουμε δεν διαφέ-ρει από την βιολογία των φυσιολογικών HSCs. η παρού-σα ανασκόπηση σκοπεύει να παρουσιάσει με συντομία τα κυριότερα στοιχεία της βιολογίας των Χμλ HSCs, τα οποία ενδέχεται να αποδειχθούν στο μέλλον άριστοι θεραπευτικοί στόχοι προς επίτευξη της απόλυτης ίασης.
ΟΡΙΣΜΟΣ ΚΑΙ ΙΔΙΟΤΗΤΕΣ ΤΩΝ XMΛ HSCsΩς φυσιολογικά HSCs ορίζονται μακράς διάρκειας
κύτταρα που μπορούν να δημιουργήσουν αποικίες (Long Term Culture Initiating Cells-LTCIC), ικανές να εμβο-λιαστούν σε ανοσοκατασταλμένο ποντίκι. μετά από τη διαμεταφορά σε NOD/SCID ποντικό μπορούν να επανα-δημιουργήσουν τα διάφορα όργανα και να σχηματίσουν λειτουργικά κύτταρα του αίματος στο δότη. ομοίως τα Χμλ HSCs όταν διαμεταφέρονται σε υποθανατηφό-ρα ακτινοβολημένα NOD/SCID ποντίκια, καταφέρνουν να εμβολιαστούν και να δημιουργήσουν λευχαιμία. τα Χμλ HSCs είναι επίσης LTCIC και κινητικά ήρεμα (qui-escent). H κατάσταση ηρεμίας είναι παροδική και ανα-στρέψιμη. κάτω από συγκεκριμένες συνθήκες τα ήρεμα Χμλ HSCs μπορούν να δημιουργήσουν λευχαιμία. τα Χμλ HSCs όπως και τα φυσιολογικά HSCs βρίσκονται στο μυελό των οστών in vivo, ο ποίος αποτελεί καταφύ-γιο και προστασία από ποικίλους κινδύνους, μεταξύ των οποίων και οι θεραπείες11.
ΦΑΙΝΟΤΥπΟΣ ΤΩΝ ΧΜΛ HSCsTα Χμλ HSCs είναι ανοσοφαινοτυπικά όμοια με τα
φυσιολογικά HSCs. και τα δύο στη κυτταρομετρία ροής χαρακτηρίζονται από χαμηλές πρόσθιες και πλάγιες δια-χύσεις, που αντιπροσωπεύουν ένα μικρού μεγέθους χω-ρίς κοκκίωση πρωτόπλασμα.

Βλαστικά κύτταρα της χρόνιας μυελογενούς λευχαιμίας (CML stem cells) - Η οδός προς την ίαση 67
τα Χμλ HSCs εκφράζουν τους δείκτες CD34, Thy1/CD90 και CD133 , ενώ δεν εκφράζουν το CD38, CD45RA, CD71 και πολλούς δείκτες των ώριμων αιμοποιητικών κυττάρων11.
το CD123 (η α αλυσίδα του υποδοχέα της IL3) και το CD33 που ως γνωστόν αποτελούν δείκτες των οξει-ών μυελογενών λευχαιμιών, έχει φανεί ότι συνεκφράζο-νται στην πλειονότητα των CD34+CD38- Χμλ HSCs στη διάγνωση12.
ςε μία πολύ πρόσφατη μελέτη στο Hematologica από τους Harald Herrmann και συνεργάτες13, φάνηκε ότι τα Χμλ HSCs εκφράζουν σημαντικά υψηλότερα επίπεδα CD33 από τα φυσιολογικά HSCs. ςτην ίδια μελέτη HSCs από ασθενείς με χρόνια φάση Χμλ καλλιεργήθηκαν in vitro με διάφορες δόσεις από το μονοκλωνικό anti-CD33 αντίσωμα gemtuzumab/ozogamycin (GO) και παρατηρή-θηκε σε όλα αναστολή της αύξησής τους . η δράση ήταν δοσοεξαρτώμενη και ακολουθούνταν από απόπτωση.
οι συγγραφείς υπονοούν ότι το μονοκλωνικό GO θα μπορούσε να δοκιμαστεί και in vivo με στόχο την εξα-φάνιση των Χμλ HSCs και κατά συνέπεια την ίαση.
ΤΑ ΒΑΣΙΚΑ ΣΗΜΑΤΟΔΟΤΙΚΑ «ΜΟΝΟπΑΤΙΑ» πΟΥ ΕΝΕΡΓΟπΟΙΟΥΝΤΑΙ ΑπΟ ΤΟ BCR-ABL (εικόνα 1)
Το PI3-K/AKT «μονοπάτι»η συγκεκριμένη οδός έχει φανεί ότι παίζει σημαντικό
ρόλο σε πολλές μορφές καρκίνων και στη Χμλ. Είναι γνωστό ότι ενεργοποιείται με τη σύνδεση υποδοχέων με δράση TK με αυξητικούς παράγοντες ή από την έκφραση του BCR-ABL. Πολλά μόρια κατωφερώς του Ακτ έχει φανεί ότι παίζουν σημαντικό ρόλο στη λευχαιμογένεση από το BCR-ABL, είτε μέσω ενεργοποίησης είτε μέσω απενεργοποίησης.
τέτοια μόρια είναι οι πρωτεϊνες FOXO (FOrkhead Box O)(FOXO1, FOXO3a & FOXO4), οι οποίες αποτε-λούν μεταγραφικούς παράγοντες. Απενεργοποίηση των FOXO πρωτεϊνών οδηγεί σε αυξημένο πολλαπλασια-σμό των Χμλ κυττάρων, αναστολή της απόπτωσης ή και στα δύο15. Επιπλέον στα φυσιολογικά HSCs ποντι-κού, οι παράγοντες FOXO1, 3a & 4 τα προστατεύουν από το οξειδωτικό stress. Απώλειά τους οδηγεί σε παραγωγή ελεύθερων ριζών οξυγόνου, επιτάχυνση του κυτταρικού κύκλου και τελικά εξάντληση των HSCs16.
ςχετικά πρόσφατα οι Naka και συνεργάτες παρουσί-ασαν στο Nature μία πολύ σημαντική εργασία. Χρησιμο-ποίησαν ένα συνγονιδιακό σύστημα μεταμόσχευσης σε ποντίκια, το οποίο ήταν ικανό να δημιουργήσει μυελοϋ-περπλαστική νόσο που θύμιζε Χμλ. ςτο μοντέλο αυτό απέδειξαν ότι ο FOXO3a έχει βασικότατο ρόλο στην επι-βίωση των Χμλ HSCs17. οι πληθυσμοί των κυττάρων
που ήταν ικανοί να δημιουργήσουν λευχαιμία (Leukemia Initiating Cells-LIC) απέδειξαν ιδιαίτερα αυξημένη δρα-στικότητα του FOXO3a. Επιπλέον η ικανότητά τους να δημιουργούν νόσο ελαττωνόταν σημαντικά με την ανα-στολή του FOXο3a. Εξαιρετικά ενδιαφέρον στην ίδια με-λέτη ήταν το γεγονός ότι ο TGFβ (transforming growth factor β) μπορούσε να καταστείλει το «μονοπάτι» Ακτ, οδηγώντας έτσι σε ενεργοποίηση του FOXO3a. ςυνδυ-άζοντας το Imatinib με έλλειψη FOXO3a και αναστολή του TGFβ, η XMλ μπορούσε πλήρως να εξαφανιστεί από αυτό το συνγονιδιακό μοντέλο του ποντικού.
Έχουμε ήδη αναφέρει ότι οι TKIs ασκούν μία ισχυρή αντιπολλαπλασιαστική δράση στα Χμλ HSCs in vitro, και ως εκ τούτου δυνητικά συμβάλουν στην κυτταρική ηρεμία τους και την παραμονή της νόσου9. Αυτό ενδέχε-ται να εξηγείται από το γεγονός ότι οι τκιs ενεργοποι-ούν κυτταρικά μονοπάτια, όπως τις πρωτεΐνες FOXO, οι οποίες με τη σειρά τους οδηγούν σε έξοδο από τον κυττα-ρικό κύκλο και σταμάτημα στη G1 φάση. κάποια πρώι-μα στοιχεία δείχνουν ότι το σταμάτημα στη φάση G1 του κυτταρικού κύκλου των Χμλ HSCs, ενδέχεται να προ-καλείται από επανενεργοποίηση των πρωτεϊνών FOXO18.
Είναι πολύ πιθανό η πολύ καλή κατανόηση της αντι-πολλαπλασιαστικής δράσης των TKIs στα Χμλ HSCs, να μπορέσει στο μέλλον να βελτιστοποιήσει τη θεραπευ-τική τους στόχευση. Αυτό φαίνεται ότι μπορεί να επι-τευχθεί και μέσω αναστολής της επίκτητης κατάστασης κυτταρικής ηρεμίας που προκαλείται με την ενεργοποί-ηση των FOXO πρωτεϊνών, η οποία θα υποχρεώσει τα HSCs της νόσου να εισέλθουν στον κυτταρικό κύκλο και να υποστούν απόπτωση.
H τυροσινική κινάση JAK2H Janus Kinase (JAK) είναι μία οικογένεια από εν-
δοκυττάριες τκs που δεν αποτελούν υποδοχείς, οι οποί-ες μεταδίδουν κυτοκινο-εξαρτώμενα μηνύματα μέσω της οδού JAK/STAT και έχει φανεί ότι κατέχει καίριο ρόλο σε μία ποικιλία μυελοϋπερπλαστικών νοσημάτων19. ο σχη-ματισμός της πρωτεΐνης BCR-ABL ενεργοποιεί διάφο-ρα σηματοδοτικά μονοπάτια, μεταξύ αυτών και του JAK/STAT20,21. Είναι πλέον επιβεβαιωμένο ότι η JAK2 είναι ενεργοποιημένη στα ανεξάρτητα από κυτοκίνες BCR-ABL θετικά κύτταρα22. H BCR-ABL πρωτεΐνη σχηματίζει ένα σύμπλοκο με τη JAK2 προκειμένου να γίνει η φω-σφωρυλίωση της τελευταίας. η ενεργοποίηση της JAK2 γνωρίζουμε πια ότι παίζει βασικό ρόλο στην ογκογενητι-κή ικανότητα του BCR-ABL23. ςυνέπεια αυτού είναι ότι κινάσες όπως η JAK2 αποτελούν πολλά υποσχόμενους στόχους για τη θεραπεία της Χμλ.
ςε μία σχετικά πρόσφατη εργασία24 το μόριο ΑG490, ένας εξειδικευμένος JAK2 αναστολέας, φάνηκε ότι προκα-λεί σημαντικό βαθμό απόπτωσης σε κύτταρα μυελού των
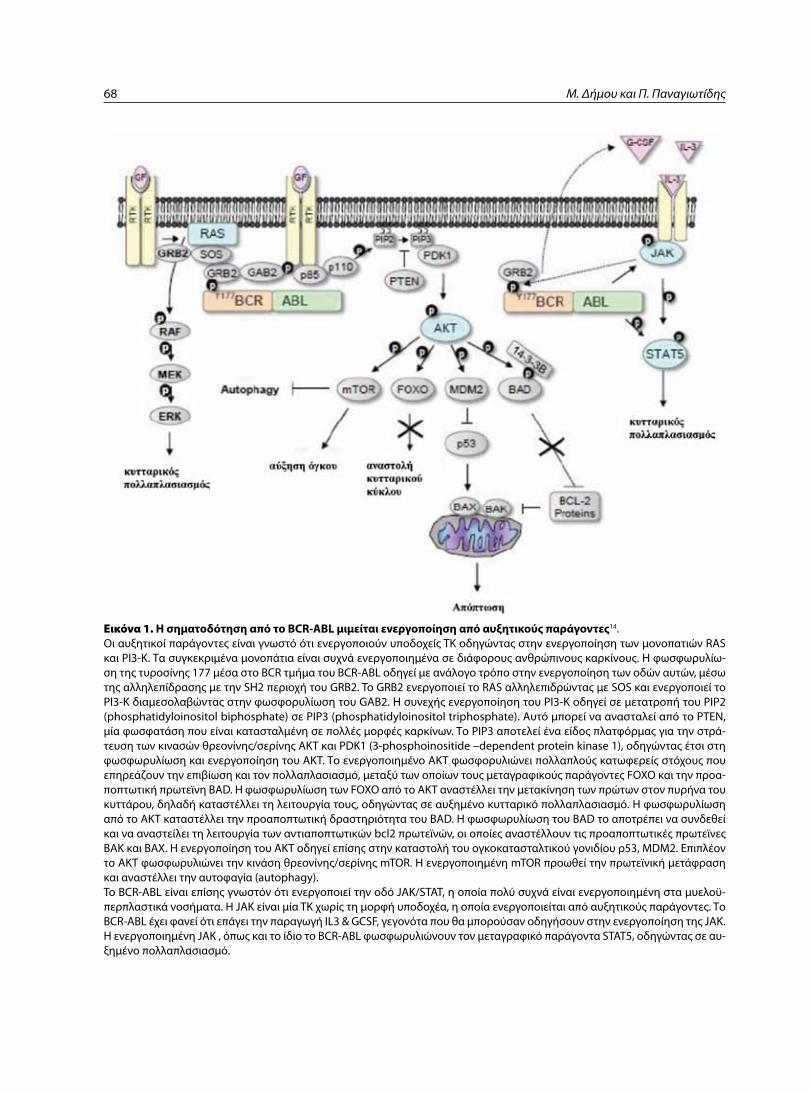
M. Δήμου και Π. Παναγιωτίδης68
Εικόνα 1. Η σηματοδότηση από το BCR-ABL μιμείται ενεργοποίηση από αυξητικούς παράγοντες14.Οι αυξητικοί παράγοντες είναι γνωστό ότι ενεργοποιούν υποδοχείς ΤΚ οδηγώντας στην ενεργοποίηση των μονοπατιών RAS και PI3-K. Τα συγκεκριμένα μονοπάτια είναι συχνά ενεργοποιημένα σε διάφορους ανθρώπινους καρκίνους. Η φωσφωρυλίω-ση της τυροσίνης 177 μέσα στο BCR τμήμα του BCR-ABL οδηγεί με ανάλογο τρόπο στην ενεργοποίηση των οδών αυτών, μέσω της αλληλεπίδρασης με την SH2 περιοχή του GRB2. To GRB2 ενεργοποιεί το RAS αλληλεπιδρώντας με SOS και ενεργοποιεί το PI3-K διαμεσολαβώντας στην φωσφορυλίωση του GAB2. H συνεχής ενεργοποίηση του PI3-K οδηγεί σε μετατροπή του PIP2 (phosphatidyloinositol biphosphate) σε ΡΙΡ3 (phosphatidyloinositol triphosphate). Αυτό μπορεί να ανασταλεί από το ΡΤΕΝ, μία φωσφατάση που είναι κατασταλμένη σε πολλές μορφές καρκίνων. Το ΡΙΡ3 αποτελεί ένα είδος πλατφόρμας για την στρά-τευση των κινασών θρεονίνης/σερίνης ΑΚΤ και PDK1 (3-phosphoinositide –dependent protein kinase 1), οδηγώντας έτσι στη φωσφωρυλίωση και ενεργοποίηση του ΑΚΤ. Το ενεργοποιημένο ΑΚΤ φωσφορυλιώνει πολλαπλούς κατωφερείς στόχους που επηρεάζουν την επιβίωση και τον πολλαπλασιασμό, μεταξύ των οποίων τους μεταγραφικούς παράγοντες FOXO και την προα-ποπτωτική πρωτεϊνη BAD. H φωσφωρυλίωση των FOXO από το ΑΚΤ αναστέλλει την μετακίνηση των πρώτων στον πυρήνα του κυττάρου, δηλαδή καταστέλλει τη λειτουργία τους, οδηγώντας σε αυξημένο κυτταρικό πολλαπλασιασμό. Η φωσφωρυλίωση από το ΑΚΤ καταστέλλει την προαποπτωτική δραστηριότητα του BAD. H φωσφωρυλίωση του BAD το αποτρέπει να συνδεθεί και να αναστείλει τη λειτουργία των αντιαποπτωτικών bcl2 πρωτεϊνών, οι οποίες αναστέλλουν τις προαποπτωτικές πρωτεϊνες ΒΑΚ και ΒΑΧ. Η ενεργοποίηση του ΑΚΤ οδηγεί επίσης στην καταστολή του ογκοκατασταλτικού γονιδίου ρ53, MDM2. Επιπλέον το ΑΚΤ φωσφωρυλιώνει την κινάση θρεονίνης/σερίνης mTOR. Η ενεργοποιημένη mTOR προωθεί την πρωτεϊνική μετάφραση και αναστέλλει την αυτοφαγία (autophagy).To ΒCR-ABL είναι επίσης γνωστόν ότι ενεργοποιεί την οδό JAK/STAT, η οποία πολύ συχνά είναι ενεργοποιημένη στα μυελοϋ-περπλαστικά νοσήματα. Η JAK είναι μία ΤΚ χωρίς τη μορφή υποδοχέα, η οποία ενεργοποιείται από αυξητικούς παράγοντες. Το BCR-ABL έχει φανεί ότι επάγει την παραγωγή IL3 & GCSF, γεγονότα που θα μπορούσαν οδηγήσουν στην ενεργοποίηση της JAK. H ενεργοποιημένη JAK , όπως και το ίδιο το BCR-ABL φωσφωρυλιώνουν τον μεταγραφικό παράγοντα STAT5, οδηγώντας σε αυ-ξημένο πολλαπλασιασμό.

Βλαστικά κύτταρα της χρόνιας μυελογενούς λευχαιμίας (CML stem cells) - Η οδός προς την ίαση 69
οστών από ασθενείς με Χμλ σε χρόνια, επιταχυνόμενη ή βλαστική φάση24. το μόριο ήταν αποτελεσματικό ακόμα και σε κύτταρα από ασθενείς ανθεκτικούς στο Imatinib. ςτην ίδια εργασία άλλοι JAK2 αναστολείς (τG101209, HBC) οδήγησαν σε απόπτωση, Imatinib ευαίσθητες και μη Χμλ κυτταρικές σειρές.
μία πιο πρόσφατη μελέτη έδειξε ότι η διπλή ανα-στολή της JAK2 και των ΑΒL κινασών με το μόριο οΝ44580, στοχεύει αποτελεσματικά ιmatinib ευαίσθη-τα και μη κύτταρα από ασθενείς με Χμλ ακόμα και σε βλαστική κρίση25.
Είναι λογικό λοιπόν ποικίλοι αναστολείς της JAK2 να βρίσκονται σε διάφορα προκλινικά και κλινικά στάδια έρευνας, καθώς αντιπροσωπεύουν ελκυστικούς εναλλα-κτικούς τρόπους στόχευσης των Χμλ HSCs (Πίνακας 1).
ΣΗΜΑΤΟΔΟΤΙΚΑ ΜΟΝΟπΑΤΙΑ πΟΥ ΕΧΟΥΝ ΣΧΕΣΗ ΜΕ ΤΗΝ ΑΥΤΟ-ΑΝΑΝΕΩΣΗ ΤΩΝ ΧΜΛ HSCs
Αν και η ικανότητα για αυτό-ανανέωση θεωρείται κε-φαλαιώδους σημασίας για την διατήρηση των Χμλ HSCs, οι μηχανισμοί που τη διέπουν και τα σηματοδοτικά μο-νοπάτια που εμπλέκονται, κατά βάση δεν είναι γνωστά.
HοδόςWnt (εικόνα 2)26 προάγει την αυτό-ανανέω-ση, πολλαπλασιασμό και παροδική διαφοροποίηση των φυσιολογικών HSCs και εμπλέκεται στην ανάπτυξη και αναγέννηση των κυττάρων των σπονδυλωτών. ςτα θη-λαστικά διατηρεί τoν πληθυσμό των HSCs που βρίσκο-νται σε ιστούς με ταχύ πολλαπλασιασμό (όπως το δέρμα
και οι ενδοκρινείς αδένες), ενώ πολύ συχνά ανευρίσκε-ται μονίμως ενεργοποιημένη σε διάφορες μορφές καρκί-νου και στη Χμλ.
Πρόσφατη μελέτη από τους Zhao και συνεργάτες27 έδωσε ισχυρές ενδείξεις ότι η σηματοδότηση μέσω Wnt/β-catenin είναι απαραίτητη για την επιβίωση των φυσι-ολογικών και Χμλ HSCs. Αυτό το εύρημα μαζί με μία παλαιότερη ανακοίνωση που απέδειξε ότι η ενεργοποίηση της β-catenin σε πρόδρομα της μυελικής σειράς κύτταρα, μπορεί να ευοδώσει την πρόοδο από τη χρόνια φάση στη βλαστική κρίση28, αποδεικνύουν ότι η σηματοδοτική οδός Wnt/β-catenin μπορεί να αποτελέσει πιθανό μελλοντικό θεραπευτικό στόχο, με σκοπό την πλήρη ίαση από την Χμλ. Βεβαίως μεγάλη προσοχή πρέπει να δοθεί στο γε-γονός ότι από αυτή την οδό εξαρτώνται και τα φυσιολο-γικά HSCs. To θεραπευτικό παράθυρο πιθανώς υπάρχει γιατί στα Χμλ HSCs η Wnt/ β-catenin ενεργοποιείται με παρεκκλίνοντα τρόπο.
Ενθαρρυντικά είναι τα αποτελέσματα που υπάρχουν από μελέτες με διάφορους αναστολείς της β-catenin (π.χ. ΑV65, BC2059) σε κυτταρικές σειρές από ασθενείς με Χμλ ακόμα και από Imatinib ανθεκτικούς ή με τη με-τάλλαξη τ315ι29,30.
HοδόςHedgehog εν συντομία θα λέγαμε ότι προά-γει τον κυτταρικό πολλαπλασιασμό και αυτο-ανανέωση των HSCs, μέσω αύξησης της έκφρασης των γονιδίων cyclin D1, myc & snail (εικόνα 3)26. Yπάρχουν 3 γνω-στοί Hedgehog (Hh) συνδέτες. ο καλύτερα μελετημένος είναι ο Sonic Hedgehog (SHH) συνδέτης.
H SMO (smoothened) είναι μία διαμεμβρανική πρω-τεΐνη η οποία αναμεταδίδει το σήμα της SHH. Χρησιμο-
Εικόνα 2. Η οδός Wnt26.

M. Δήμου και Π. Παναγιωτίδης70
ποιώντας ποντίκια που είχαν έλλειψη της SMO ή άλλα που έλαβαν ένα αναστολέα της SMO, την κυκλοπαμί-δη, ο Zhao και οι συνεργάτες του31 απέδειξαν ελάττωση στον αριθμό των Χμλ HSCs και παράταση επιβίωσης σε ακτινοβολημένα ποντίκια που εξέφραζαν BCR-ABL HSCs. Αντίθετα φάνηκε ότι η διαγονιδιακή έκφραση μί-ας ενεργοποιημένης μορφή SMO οδήγησε σε αυξημέ-νο αριθμό Χμλ HSCs και επιτάχυνση της αύξησης του όγκου. με αυτόν τον τρόπο οι συγγραφείς υπονοούν ότι η διαταραχή στην οδό Hedgehog μπλοκάρει την πρόοδο της Χμλ με το να επηρεάζει τα Χμλ HSCs. Εν τούτοις στη συγκεκριμένη μελέτη υπήρχε ελάττωση της επιβίω-σης και των φυσιολογικών HCSs, γεγονός που αποθάρ-ρυνε την κλινική εφαρμογή της μεθόδου.
τρείς όμως διαφορετικές μελέτες έδειξαν ότι τα φυ-σιολογικά HCSs δεν εξαρτώνται από την οδό Hedgehog για την επιβίωσή τους32-34.
ςε μία σχετικά πρόσφατη εργασία στο ASH Annual Meeting το 2009 οι Irvine και οι συνεργάτες έδειξαν ότι ο αναστολέας SMO, LDE225, σε συνδυασμό με το nilo-tinib, εξαφανίζει τα Χμλ HSCs και τα προγονικά κύτταρα.
Επί του παρόντος είναι ενεργείς διάφορες κλινικές μελέτες φάσης ι/ιι που μελετούν τη δράση SMO ανα-στολέων σε συνδυασμό τκιs (πίνακας 1). μία εξ’αυ-τών είναι η μελέτη του συνδυασμού του dasatinib με τον SMO αναστολέα BMS-833923 για ασθενείς με χρόνια φάση Χμλ που εμφάνισαν πρόοδο νόσου ή αποτυχία ενώ βρίσκονταν υπό θεραπεία με TKIs (NCT01218477). Άλλοι αναστολείς όπως τα μόρια LDE225 & LEQ506 χρησιμοποιούνται σε φάσεις ι μελέτες για συμπαγείς
όγκους (NCT00880308, NCT00961896, NCT01033019, NCT01125800, NCT01106508).
O ΡΟΛΟΣ ΤΗΣ ΟΓΚΟΚΑΤΑΣΤΑΛΤΙΚΗΣ πΡΩΤΕϊΝΗΣ PML (PROMYELOCYTIC LEUKEMIA)
Προσφάτως φάνηκε ότι η ογκοκατασταλτική πρωτεϊνη PML επιτρέπει στα Χμλ HSCs να διατηρούν την ηρεμία τους. η PML είναι μία πρωτεΐνη που κατέχει βασικότα-το ρόλο στην απόπτωση, την κυτταρική διαφοροποίη-ση και την κυτταρική γήρανση. η PML υπερεκφράζεται σε βλάστες από ασθενείς με Χμλ, ενώ χαμηλά επίπεδα PML αντιστοιχούν με καλύτερη ανταπόκριση στη θερα-πεία και βελτίωση επιβίωσης35. Όπως φάνηκε από την εργασία των Ito και συνεργατών35, τα Χμλ HSCs που δεν εκφράζουν την PML, μπαίνουν στον κυτταρικό κύ-κλο και οδηγούνται σε εξάντληση και στη συνέχεια δεν έχουν την ικανότητα να προκαλέσουν λευχαιμία σε πει-ραματόζωα όταν μεταμοσχεύονται.
το τριοξείδιο του αρσενικού (As2O3), παραδοσιακό κινέζικο φάρμακο, γνωρίζουμε ότι οδηγεί σε αποδόμηση της PML και έχει ήδη χρησιμοποιηθεί σε κλινικές μελέ-τες, αποδεικνύοντας δραστικότητα κατά των Χμλ HSCs , χωρίς να είναι τοξικό. το ενδιαφέρον είναι ότι τα φυσιο-λογικά HSCs που δεν εκφράζουν PML είναι λιγότερο ευ-άλωτα στη χημειοθεραπεία από τα λευχαιμικά αντίστοιχά τους, γεγονός που αποτελεί ένα είδος «θεραπευτικού πα-ράθυρου» στη στοχευμένη θεραπεία της Χμλ.
Εικόνα 3. Η οδός Hedgehog26.

Βλαστικά κύτταρα της χρόνιας μυελογενούς λευχαιμίας (CML stem cells) - Η οδός προς την ίαση 71
μία φάσης ι/ιι μελέτη για την ασφάλεια ,την ανεκτι-κότητα και την αντιλευχαιμική δράση του As2O3 σε συν-δυασμό με Imatinib σε ασθενείς με ανθεκτική Χμλ σε χρόνια φάση, ολοκληρώθηκε πρόσφατα (NCT00053248) και τα αποτελέσματά της αναμένονται. Επιπλέον μία άλλη ανάλογη φάσης ιι μελέτη είναι ακόμα ανοιχτή για έντα-ξη ασθενών (πίνακας 1).
H ΒΟΗθΗΤΙΚΗ πΡΩΤΕϊΝΗ ΤΟΥ ΥπΟΔΟΧΕΑ ΤΗΣ ΙΝΤΕΡΛΕΥΚΙΝΗΣ 1- (INTERLEUKIN-1 RECEPTOR ACCESSORY PROTEIN-IL1RAP)
H IL1RAP είναι ένας Toll-like υποδοχέας που με-σολαβεί στη λειτουργία της IL-1 και στη μετάδοση του μηνύματος της IL-33. H IL-33 είναι μία κυτοκίνη που ενεργοποιεί τα τ λεμφοκύτταρα και τα μαστοκύτταρα, μέσω της σύνδεσης με τον υποδοχέα της, ST2 και στη συνέχεια διμερίζεται με την ιL1RAP. η IL1RAP έχει φα-νεί ότι υπερεκφράζεται στα Χμλ HSCs36,37. τα τελευταία χρόνια αποκαλύφθηκε ότι IL1RAP αποτελεί επιφανεια-κό δείκτη των Χμλ HSCs και αυτή η ιδιότητα επέτρε-ψε την επαγωγή κυτταροτοξικότητας εξαρτώμενης από το αντίσωμα (ADCC).
In vitro η υπερέκφραση του BCR-ABL σε ομφαλο-πλακουντιακά κύτταρα, οδηγεί σε υπερέκφραση και του IL1RAP, επιβεβαιώνοντας τη σχέση εξάρτησης με το BCR-ABL ογκογονίδιο1.
Χρησιμοποιώντας ένα καινοτόμο πρωτόκολλο FISH (fluorescence in situ hybridization), το οποίο ονομάζεται Flow-drop-FISH σε LTCIC καλλιέργειες, οι Jaras και συ-νεργάτες έδειξαν ότι τα CD34+CD38-,IL1RAP+ κύτταρα είναι κυρίως λευχαιμικά, ενώ τα CD34+CD38-, IL1RAP- είναι βασικά Ph αρνητικά36.
Ένα πολυκλωνικό anti-IL1RAP1 αντίσωμα έχει κα-τασκευαστεί και έχει φανεί ότι μπορεί να στοχεύσει τα λευχαιμικά και όχι τα φυσιολογικά HSCs. Εν τούτοις η επιλεκτική κυτταροτοξική δραστικότητα του νέου αυτού αντισώματος κατά των λευχαιμικών έναντι των φυσιολο-γικών HSCs δεν έχει ακόμα επιβεβαιωθεί σε αυστηρώς καθορισμένες σειρές από HSCs ή με μεταμοσχευτικές με-θόδους σε ανοσοκατασταλμένα ποντίκια. Παρόλα αυτά το αντίσωμα anti-IL1RAP είναι πολύ πιθανό να χρησι-μοποιηθεί στο μέλλον μόνο του ή σε συνδυασμό με άλ-λα φάρμακα για τη θεραπεία της Χμλ38.
πΑΡΑΓΟΝΤΕΣ πΟΥ ΕΧΟΥΝ ΣΧΕΣΗ ΜΕ ΤΗΝ ΑπΟπΤΩΣΗ ΣΤΑ ΧΜΛ HSCs
η απόπτωση, δηλαδή ο προγραμματισμένος κυτ-ταρικός θάνατος, ελέγχεται από μία εξωγενή οδό μέσω υποδοχέων που μεσολαβούν στον κυτταρικό θάνατο και από την ενδογενή οδό στην οποία βασικό ρόλο παίζουν τα μιτοχόνδρια (εικόνα 4). η απόπτωση με διάφορους τρόπους διαδραματίζει βασικό ρόλο στη φυσιολογία των ζωικών ειδών.
Πίνακας 1.
Συνδυασμός φαρμάκων NCT-identifier Φάση- Αs2O3+ Imatinib για ΧΜΛ σε ΧΦ- Αs2O3+ TKIs για ΧΜΛ σε ΧΦ
0005324801397734
II/ ολοκληρώθηκε η ένταξη ασθενών
ΙΙ/ανοιχτή
- Nilotinib +Peg-IFN-2α (ΝΙLOPEG) για πρωτοδιαγνωσμένες ΧΜΛ σε ΧΦ
- Imatinib + Peg-IFN-α2α σε ΧΜΛ σε ΧΦ- Nilotinib + IFN-a σε δυσανεξία ή ανθεκτικότητα στο Ιmatinib σε
ΧΜΛ/ΧΦ (NICOLI)
012946180057337801220648
ΙΙ/ανοιχτήΙΙ/ανοιχτήII/ανοιχτή
Ιmatinib+/- Hydrochloroquine (CHOICES) σε ΧΜΛ/ΧΦ 01227135 ΙΙ/ανοιχτή
- Dasatinib + SMO antagonist (BMS-833923) σε ΧΜΛ/ΧΦ μετά από αντοχή στο Imatinib
- Nilotinib+ SMO antagonist (LDE225) σε ΧΜΛ/ΧΦ σε ανθεκτικότητα σε άλλους ΤΚΙs
01218477001456676
ΙΙ/ανοιχτή II/ανοιχτή
JAK2 inhibitor (SB1518) σε ανθεκτικές ή σε υποτροπή αιματολογικές κακοήθειες (και ΧΜΛ)
00719836 Ι/ΙΙ/ συνεχίζεται-ολοκληρώθηκε η ένταξη ασθενών

M. Δήμου και Π. Παναγιωτίδης72
η απορρύθμιση της απόπτωσης πιθανότατα παί-ζει σημαντικό ρόλο στην προστασία των Χμλ-HSCs από τον κυτταρικό θάνατο. ςτην πραγματικότητα HSCs που εκφράζουν το BCR-ABL, έχει φανεί ότι εκφράζουν υψηλά επίπεδα από τις αντιαποπτωτικές πρωτεϊνες Bcl-xL & Mcl-1 και χαμηλά επίπεδα από την προαποπτωτι-κή πρωτεΐνη Bim. H αναστολή της τυροσινικής κινάσης BCR-ABL σε αυτά τα κύτταρα οδήγησε σε αύξηση των επιπέδων της Mcl-1 και Bim39,40.
To ABT-737, ένας εκλεκτικός αναστολέας Bcl-2, Bcl-xL& Bcl-w (εικόνα 4), έχει δείξει ισχυρή αντιλευχαι-μική δράση, μεταξύ άλλων σε προγονικά κύτταρα από ομλ41. η αναστολή των Bcl-2/BclxL από το ΑΒτ-737, φάνηκε ότι ενισχύει τη δράση του Imatinib στα Χμλ κύτταρα42. Επιπλέον ο συνδυασμός του ΑΒτ-737 με τον δεύτερης γενιάς τκι, ιΝΝο-406, έχει φανεί ότι αυξάνει την απόπτωση, ακόμα και στα ιΝΝο-ανθεκτικά κύττα-ρα με σημειακές μεταλλάξεις του BCR-ABL, εκτός από τη μετάλλαξη τ315ι43.
το Bcl-2 antisence ολιγονουκλεοτίδιο-Genasence- έδειξε δραστικότητα εναντίον Imatinib-ανθεκτικών BCR-ABL+ κυττάρων44.
Επιπλέον η surviving, μία πρωτεϊνη που ανήκει στην οικογένεια ιΑΡ (inhibitors of apoptosis proteins), ελέγχε-ται από την κινάση BCR-ABL και υπερεκφράζεται στα XMλ κύτταρα, ενώ η στόχευση της surviving ξεπερνά την αντίσταση στο Imatinib45.
η πρωτεϊνη ΧιΑΡ, ο πιο ισχυρός αναστολέας κα-σπάσης της οικογένειας ιΑΡ, υπερεκφράζεται στα Χμλ κύτταρα και το triptolide (εικόνα 4), ένας παράγων που απομονώθηκε από κινέζικα βότανα, ελαττώνει τα επί-πεδα του ΧιΑΡ, του Mcl1 και του BCR-ABL και προά-γει την ανεξάρτητη του BCR-ABL απόπτωση των Χμλ
κυττάρων. Χρησιμοποιώντας αυτή ακριβώς την ιδιότη-τα του triptolide, οι μak και συνεργάτες μελέτησαν τη δραστικότητα του στη Χμλ και έδειξαν ότι το triptolide προκαλεί κυτταρικό θάνατο, ανεξάρτητα από τις ανταπο-κρίσεις στο Imatinib σε Χμλ HSCs46, είτε μόνος του είτε σε συνδυασμό με τον αναστολέα ABT-73747.
Επαγωγή απόπτωσης ώστε εκλεκτικά να θανατώνο-νται τα Χμλ HSCs, έχει φανεί ότι μπορεί να προκληθεί από τον αναστολέα της φαρνεσυλτρανσφεράσης BMS-214662, εν μέρει μέσω ενεργοποίησης του Bax. H κυτ-ταροτοξικότητα αφορούσε κυτταρικές σειρές HSCs που είχαν μεταλλάξεις ανθεκτικές στο Imatinib ακόμα και την τ315ι48.
Η Ιντερφερόνη άλφα (ΙFN-α) πέρα από τις γνωστές έμ-μεσες επιδράσεις στις ανοσιακές και αντιοαγγειογενετικές απαντήσεις, άμεσα μπορεί να τροποποιήσει την έκφραση πληθώρας γονιδίων (όπως TRAIL, Fas/FasL, caspase 8, XIAP associated factor -1 ή XAF-1), τα οποία προάγουν τον κυτταρικό θάνατο. Mε άλλα λόγια δηλαδή η IFN-α επάγει την διαδικασία της απόπτωσης11. Πολύ ενδιαφέ-ρουσα είναι η σχετικά πρόσφατη ανακοίνωση από τους Essers και συνεργάτες στο Nature49, η οποία έδειξε ότι η IFN-α ενεργοποιεί τα αδρανή HSCs και προάγει τον πολλαπλασιασμό τους με το να αυξάνει τη φωσφορυλί-ωση των STAT1 και Αkt και την έκφραση των γονιδίων στόχων, μέσω της δραστηριοποίησης του αντιγόνου των βλαστικών κυττάρων-1, σε in vivo πειραματικά μοντέλα ποντικών. Αυτή η παρατήρηση σημαίνει ότι κατά πάσα πιθανότητα ότι η IFN-α στοχεύει τα Χμλ HSCs με το να ωθεί ήρεμα αρχέγονα CD34+ XMλ πρόδρομα κύτταρα μέσα στον κυτταρικό κύκλο. ο συνδυασμός της εξόδου των Χμλ HSCs από την κατάσταση ηρεμίας με την αυ-ξημένη απόπτωση, που προκαλείται από την IFN-α, πι-θανότατα είναι η αιτία πρόσφατων παρατηρήσεων ότι η η IFN-α μπορεί να οδηγήσει σε μακροχρόνιες υφέσεις σε ασθενείς με Χμλ50.
Υπάρχουν επίσης μελέτες που δείχνουν ότι ο συνδυ-ασμός IFN-α και Imatinib μπορεί να οδηγήσει σε υψη-λότερα ποσοστά υφέσων. η πιο πρόσφατη από αυτές τις μελέτες, η SPITIT trial51 έδειξε ότι ο συνδυασμός PEG-IFN-α και Imatinib οδηγεί σε σημαντικά υψηλότερα πο-σοστά μείζονος και πλήρους μοριακής ύφεσης. Επί του παρόντος είναι ενεργείς αρκετές μελέτες φάσης ιι που μελετούν τον συνδυασμό IFN-α και TKIs σε νεοδιαγνω-σθένετες ασθενείς με Χμλ (πίνακας 1)
Η ΑΥΤΟΦΑΓΙΑ (AUTOPHAGY)H αυτοφαγία είναι μία διαδικασία ανακύκλωσης, η
οποία οδηγεί σε εγκλωβισμό και αποδόμηση κατεστραμ-μένων πρωτεϊνών και άλλων ενδοκυττάριων πρωτεϊνών μέσα στα λυσοσώματα (εικόνα 5). η ανακύκλωση αυτών των ενδοκυττάριων υλικών μπορεί να χρησιμοποιηθεί ως
Εικόνα 411. Η εξωγενής αποπτωτική οδός μέσω υποδοχέων που μεσολαβούν στον κυτταρικό θάνατο και η ενδογενής απο-πτωτική οδός μέσω μιτοχονδρίων. Η ΑΒΤ-737 προάγει την από-πτωση με το να ανταγωνίζεται την Bcl-2 και την Bcl-xL, ενώ το triptolide προκαλεί απόπτωση εν μέρει με το να ελαττώνει την ΧΙΑΡ και την Μcl-1.
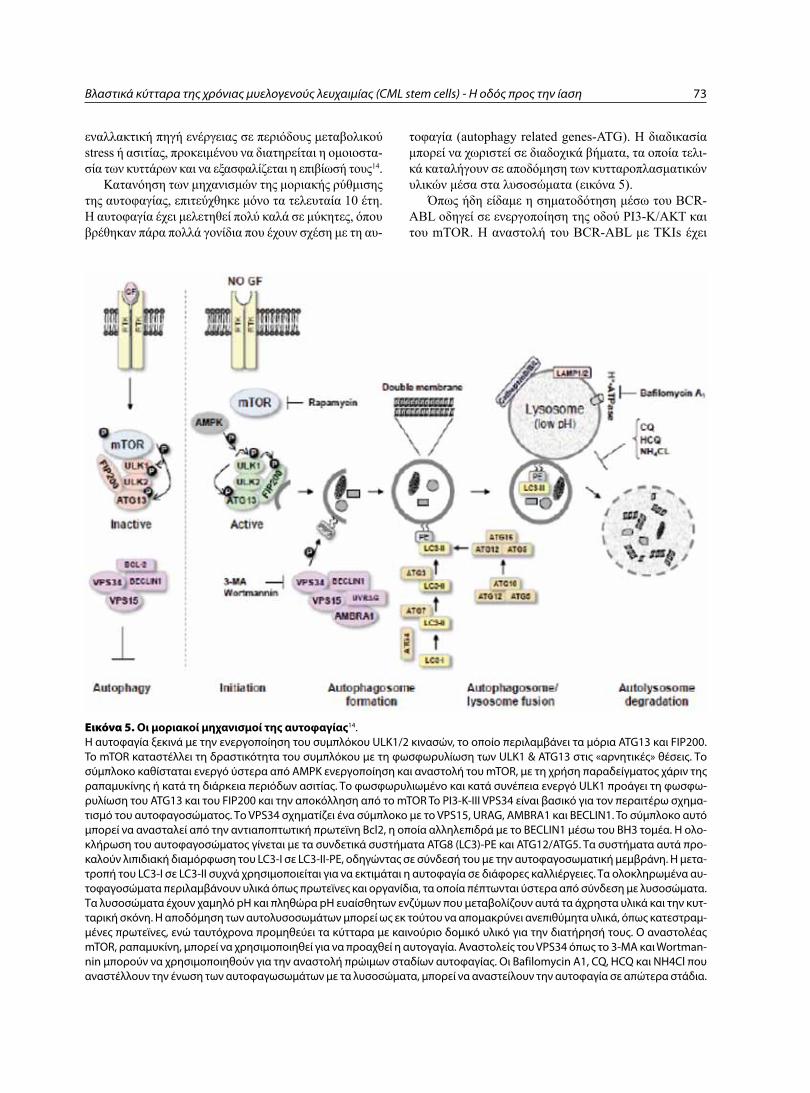
Βλαστικά κύτταρα της χρόνιας μυελογενούς λευχαιμίας (CML stem cells) - Η οδός προς την ίαση 73
εναλλακτική πηγή ενέργειας σε περιόδους μεταβολικού stress ή ασιτίας, προκειμένου να διατηρείται η ομοιοστα-σία των κυττάρων και να εξασφαλίζεται η επιβίωσή τους14.
κατανόηση των μηχανισμών της μοριακής ρύθμισης της αυτοφαγίας, επιτεύχθηκε μόνο τα τελευταία 10 έτη. η αυτοφαγία έχει μελετηθεί πολύ καλά σε μύκητες, όπου βρέθηκαν πάρα πολλά γονίδια που έχουν σχέση με τη αυ-
Εικόνα 5. Οι μοριακοί μηχανισμοί της αυτοφαγίας14.Η αυτοφαγία ξεκινά με την ενεργοποίηση του συμπλόκου ULK1/2 κινασών, το οποίο περιλαμβάνει τα μόρια ΑΤG13 και FIP200. To mTOR καταστέλλει τη δραστικότητα του συμπλόκου με τη φωσφωρυλίωση των ULK1 & ATG13 στις «αρνητικές» θέσεις. Το σύμπλοκο καθίσταται ενεργό ύστερα από ΑΜΡΚ ενεργοποίηση και αναστολή του mTOR, με τη χρήση παραδείγματος χάριν της ραπαμυκίνης ή κατά τη διάρκεια περιόδων ασιτίας. Το φωσφωρυλιωμένο και κατά συνέπεια ενεργό ULK1 προάγει τη φωσφω-ρυλίωση του ATG13 και του FIP200 και την αποκόλληση από το mTOR To PI3-K-III VPS34 είναι βασικό για τον περαιτέρω σχημα-τισμό του αυτοφαγοσώματος. Το VPS34 σχηματίζει ένα σύμπλοκο με το VPS15, URAG, AMBRA1 και BECLIN1. To σύμπλοκο αυτό μπορεί να ανασταλεί από την αντιαποπτωτική πρωτεϊνη Bcl2, η οποία αλληλεπιδρά με το BECLIN1 μέσω του BH3 τομέα. Η ολο-κλήρωση του αυτοφαγοσώματος γίνεται με τα συνδετικά συστήματα ATG8 (LC3)-PE και ATG12/ATG5. Τα συστήματα αυτά προ-καλούν λιπιδιακή διαμόρφωση του LC3-I σε LC3-II-PE, οδηγώντας σε σύνδεσή του με την αυτοφαγοσωματική μεμβράνη. Η μετα-τροπή του LC3-I σε LC3-II συχνά χρησιμοποιείται για να εκτιμάται η αυτοφαγία σε διάφορες καλλιέργειες. Τα ολοκληρωμένα αυ-τοφαγοσώματα περιλαμβάνουν υλικά όπως πρωτεϊνες και οργανίδια, τα οποία πέπτωνται ύστερα από σύνδεση με λυσοσώματα.Τα λυσοσώματα έχουν χαμηλό pH και πληθώρα pH ευαίσθητων ενζύμων που μεταβολίζουν αυτά τα άχρηστα υλικά και την κυτ-ταρική σκόνη. Η αποδόμηση των αυτολυσοσωμάτων μπορεί ως εκ τούτου να απομακρύνει ανεπιθύμητα υλικά, όπως κατεστραμ-μένες πρωτεϊνες, ενώ ταυτόχρονα προμηθεύει τα κύτταρα με καινούριο δομικό υλικό για την διατήρησή τους. Ο αναστολέας mTOR, ραπαμυκίνη, μπορεί να χρησιμοποιηθεί για να προαχθεί η αυτογαγία. Αναστολείς του VPS34 όπως το 3-MA και Wortman-nin μπορούν να χρησιμοποιηθούν για την αναστολή πρώιμων σταδίων αυτοφαγίας. Οι Bafilomycin A1, CQ, HCQ και NH4Cl που αναστέλλουν την ένωση των αυτοφαγωσωμάτων με τα λυσοσώματα, μπορεί να αναστείλουν την αυτοφαγία σε απώτερα στάδια.
τοφαγία (autophagy related genes-ATG). H διαδικασία μπορεί να χωριστεί σε διαδοχικά βήματα, τα οποία τελι-κά καταλήγουν σε αποδόμηση των κυτταροπλασματικών υλικών μέσα στα λυσοσώματα (εικόνα 5).
Όπως ήδη είδαμε η σηματοδότηση μέσω του BCR-ABL οδηγεί σε ενεργοποίηση της οδού Ρι3-κ/Ακτ και του mTOR. H αναστολή του BCR-ABL με τκιs έχει

M. Δήμου και Π. Παναγιωτίδης74
φανεί ότι όχι μόνο προάγει την απόπτωση, αλλά και την αυτοφαγία52.
το 200653 οι Yan και συνεργάτες έδειξαν ότι η νευ-ροτοξίνη crotoxin επάγει την αυτοφαγία στην κυτταρι-κή σειρά κ562 και ότι το σταμάτημα της αυτοφαγίας με την 3-μΑ (3-methyladenine) και το Νη4Cl προάγει την κυτταροτοξικότητα από τη νευροτοξίνη.
το 2007 οι Carew και συνεργάτες54 έδειξαν ότι η ανα-στολή της αυτοφαγίας με CQ (chloroquine) ενισχύει την αντινεοπλασματική δράση του αναστολέα αποακετυλί-ωσης ιστονών (SAHA), σε πρωτοπαθώς ανθεκτικά στο Imatinib Χμλ κύτταρα. Αυτές οι δύο μελέτες ήταν η πρώτη απόδειξη ότι τα Χμλ κύτταρα επάγουν την αυ-τοφαγία ως μηχανισμό κυτταρικής επιβίωσης ύστερα από θεραπεία με αντινεοπλασματικά φάρμακα, υπονο-ώντας ότι η αναστολή της αυτοφαγίας θα μπορούσε να είναι μία καινοτόμος θεραπευτική οδός σε περιπτώσεις αντοχής στο Imatinib.
η πιο ισχυρή εν τούτοις ένδειξη ότι η αναστολή της αυτοφαγίας μπορεί να ενισχύει την αποτελεσματικότητα του Imatinib στη θεραπεία της Χμλ ήρθε με τις εργασίες των Βellodi και συνεργατών το 200955 και στη συνέχεια το 2011 από την εργασία των Crowely και συνεργατών56 . οι δύο αυτές εργασίες έδειξαν ότι η αναστολή της αυτο-φαγίας σε συνδυασμό με τη θεραπεία με τκιs (imatinib, dasatinib ή nilotinib) , προκαλεί σχεδόν πλήρη εξαφάνιση των φαινοτυπικά και λειτουργικά καθορισμένων Χμλ ηSCs. To πιο πιθανό είναι ότι η θεραπεία με τκιs προ-άγει την αυτοφαγία με το να αναστέλλει τον mTOR, τον αρνητικό ρυθμιστή της αυτοφαγίας και οι ανωτέρω ερ-γασίες έδειξαν ότι πιθανότατα συμμετέχει το stress του ενδοπλασματικού δικτύου. Παρόλα αυτά ο ακριβής μη-χανισμός δεν έχει ακόμα ξεκαθαριστεί.
Όλα τα προαναφερθέντα ενθαρρυντικά αποτελέσμα-τα σε κυτταρικές σειρές με Χμλ ηSCs οδήγησαν στη δημιουργία μίας κλινικής μελέτης φάσης ιι, την CHOIC-ES (CHlOroquine and Imatinib Combination to Eliminate Stem cells-ΝCT01227135)-πίνακας 1. Πρόκειται για μία τυχαιοποιημένη μελέτη του Imatinib έναντι Imatinib και χλωροκίνη για ασθενείς με Χμλ σε μείζονα κυτταρο-γενετική ύφεση (MCyR), με ανιχνεύσιμη νόσο με την ποσοτική PCR και είναι η πρώτη κλινική μελέτη που χρησιμοποιεί την αναστολή της αυτοφαγίας στη Χμλ.
Ο ΜΕΤΑΓΡΑΦΙΚΟΣ πΑΡΑΓΩΝ BMI1 (Β CELL SPECIFIC MOLONEY MURINE LEUKEMIA vIRUS INTEGRATION SITE 1)
O BMI1 είναι μέλος της ομάδας γονιδίων polycomb, και λειτουργεί ως κατασταλτής μεταγραφής, διατηρώντας βασικό ρόλο στην γονιδιακή έκφραση κατά τη διάρκεια της ανάπτυξης. κατέχει σημαντική θέση τόσο στα φυσι-ολογικά όσο και στα λευχαιμικά HSCs54. H έκφραση του
BMI1 είναι εντονότερη σε πιο αρχέγονους πληθυσμούς HSCs και ελαττώνεται κατά τη διάρκεια της ωρίμανσης58,59.
οι Rizo και συνεργάτες έδειξαν ότι εισάγοντας το Βμι1 σε βλαστικά και πρόδρομα αιμοποιητικά ομφαλοπλακου-ντιακά κύτταρα ανθρώπου, παρατείνεται η διατήρηση της δεξαμενής τους μέσω αυτοανανέωσης των HSCs60.
To BMI1 έχει φανεί ότι έχει εντονότερη έκφραση στα στα Χμλ CD34+ από τα φυσιολογικά ομόλογά τους. Επιπλέον η επαγωγή έκφρασης Βμι1 με ρετροϊούς σε Χμλ CD34+ κύτταρα από ασθενείς με πρωτοδιάγνωση Χμλ σε χρόνια φάση, οδήγησε σε αύξηση της πολλα-πλασιαστικής τους ικανότητας και των ιδιοτήτων αυτο-ανανέωσης61.
Αν και δεν υπάρχουν ακόμα επαρκή στοιχεία ότι το Βμι1 από μόνο του μπορεί να επάγει λευχαιμία, η συνέκ-φραση του Βμι και του BCR-ABL σε ανθρώπινα CD34+ κύτταρα, είναι ικανή να οδηγήσει σε μεταμοσχεύσιμη λευχαιμία σε NOD/SCID ποντίκια60,61.
Εν κατακλείδι το Βμι1 είναι βασικό στοιχείο της φυ-σιολογικής και λευχαιμικής αιμοποίησης και ως εκ τούτου αντιπροσωπεύει ιδανικό στόχο για τη θεραπεία της Χμλ.
H ΦΩΣΦΑΤΑΣΗ PP2A (PROTEIN POSPHATASE 2A)
H ΡΡΑ2 είναι βασική κυτταρική φωσφατάση σερί-νης/θρεονίνης και εμπλέκεται σε μία πλειάδα κυτταρι-κών λειτουργιών. Έχει αναφερθεί ότι παίζει σημαντικό ρόλο στην ογκογένεση ως ογκοκατασταλτικός παράγων και υπάρχουν ενδείξεις ότι μπορεί η επανενεργοποίησή της να βοηθήσει στη θεραπεία της Χμλ62.
οι Neviani και συνεργάτες έδειξαν με την πολύ ση-μαντική εργασία τους63 ότι η ΡΡΑ2 αδρανοποιείται στα CD34+ Χμλ προγονικά κύτταρα. η αδρανοποίηση αυτή είναι αποτέλεσμα της δράσης του αναστολέα της ΡΡΑ2, της πρωτεϊνης SET. To BCR-ABL επάγει την έκφραση της πρωτεϊνης SET με έναν δοσοεξαρτώμενο και κινα-σο-εξαρτώμενο τρόπο. Επιπλέον πρόσφατες μελέτες έδει-ξαν ότι η JAK2 κατέχει επίσης ρόλο στην ενεργοποίηση του SET κατωφερώς του BCR-ABL24. τόσο σε Imatinib ευαίσθητα όσο και σε Imatinib ανθεκτικά κύτταρα αλλά και σε κύτταρα από ασθενείς με Χμλ σε βλαστική κρί-ση, η επαγωγή της δραστικότητας της ΡΡΑ2, προάγει την φωσφωρυλίωση του BCR-ABL, οδηγώντας σε αναστολή της αύξησης του όγκου, αυξημένη απόπτωση και ελατ-τωμένη in vivo ικανότητα λευχαιμογένεσης63.
καθώς με βάση τα ανωτέρω η επαγωγή της δρά-σης της φωσφατάσης ΡΡΑ2 είναι πιθανός θεραπευτικός στόχος για τη Χμλ, η Novartis δοκιμάζει έναν ισχυρό ενεργοποιητή της, το μόριο FTY720 (Gilenia®) με πολύ εντυπωσιακά αποτελέσματα σε προκλινικό στάδιο προς το παρόν, ακόμα και για τις κυτταρικές σειρές με την με-τάλλαξη τ315ι64.

Βλαστικά κύτταρα της χρόνιας μυελογενούς λευχαιμίας (CML stem cells) - Η οδός προς την ίαση 75
ςτον πίνακα 1 παρουσιάζονται οι τρέχουσες κλινικές μελέτες φαρμάκων που στοχεύουν τα Χμλ-HSCs με ή χωρίς συνδυασμό με τκιs.
ΣΥΜπΕΡΑΣΜΑΤΙΚΑγια πολλά χρόνια η Χμλ αντιπροσώπευε ένα μοντέλο
και ένα παράδειγμα για τα καρκινικά βλαστικά κύτταρα, την εξάρτηση από το ογκογονίδιο και τις στοχευμένες θε-ραπείες. με την είσοδο των τκιs στη θεραπεία της Χμλ «ξεκίνησε ένα ταξίδι» με στόχο την ίαση, χρησιμοποιώ-ντας φάρμακα μη τοξικά από του στόματος. το βασικό
σημερινό εμπόδιο για την πλήρη ίαση των ασθενών σε χρόνια φάση βρίσκεται στον πληθυσμό των βλαστικών κυττάρων της Χμλ,ο οποίος ούτε ογκοεξαρτώμενος ούτε τκι ευαίσθητος είναι. η παρούσα ανασκόπηση με στοι-χεία από τη βασική φυσιολογία των Χμλ HSCs έδειξε ότι αυτά έχουν πολλαπλές ιδιαιτερότητες, οι οποίες επά-γονται εν μέρει από το BCR-ABL, αλλά μπορεί να είναι και ανεξάρτητες από αυτό και οι οποίες ενδέχεται να απο-τελέσουν ιδανικούς θεραπευτικούς στόχους. οι νέοι αυ-τοί θεραπευτικοί στόχοι σε συνδυασμό με τους τκιs θα μπορέσουν στο μέλλον να οδηγήσουν σε αυτό που ονομά-ζουμε απόλυτη ίαση στη Χρόνια μυελογενή λευχαιμία.
CML stem cells: the pathway to CML cure?by Maria Dimou, Panayiotis Panayiotidis
1st Department of Propaedeutic Medicine, School of Medicine, National and Kapodistrian University of Athens, Greece
ABSTRACT: Chronic Myelogenous Leukemia (CML) is a myeloproliferative disorder which originates from Hemopoietic Stem Cells-HSC carrying the chromosomal translocation t( 9;22). The hallmark of the disease is the production of the fusion gene/protein BCR-ABL which is responsible for the patho-genesis of CML. Tyrosin Kinase Inhibitors-TKIs, target BCR-ABL and have revolutionized the therapy of CML, leading to complete cytogenetic responses and long term survival of patients. In some patients “complete molecular responses” have been obtained ; studies in CML patients who were in “complete molecular response” and discontinued treatment with Imatinib have shown that in the majority of pa-tients a molecular relapse of CML has occurred. This proves that the stem cell population of CML is not eliminated by long term Imatinib therapy. CML stem cells use a variety of biochemical pathways for their survival, autorenewal, inhibition of apoptosis etc. Both Imatinib mesylate and mainly newer TKIs (nilotinib, dastinib etc) lead to deep molecular responses but in vitro studies have shown that they are not able to eliminate the CML HSCs population. Combination studies in CML patients using inhibitors targeting both BCR-ABL and other biochemical pathways present in CML stem cells may lead to “func-tional cure” in a substantial proportion of CML patients.
Βιβλιογραφία1. Ren R. Mechanisms of BCR-ABL in the pathogenesis of
chronic myelogenous leukaemia. Nat Rev Cancer. 2005; 5:172-83.
2. Βhatia R, Holtz M, Niu N, et al. Persistence of malignant hematopoietic progenitors in chronc myelogenous leukemia patients in complete cytogenetic remission following imatinib mesylate treatment. Blood. 2003;101(12):4701-7.
3. Su Chu AL, Mc Donald T, Snyder DS, et al. Persistence of leukemia stem cells in chronic myelogenous leukemia patients in complete cytognetic remission on imatinib treatment for 5 years (abstract) Blood (ASH Annual Meeting Abstracts). 2008; 112:194.
4. Mahon FX, Rea D, Guillot J, et al. Discontinuation of Imatinib in patients with Chronic Myelogenous Leukemia who have maintained complete molecular remission for at
least 2 years: The prospective multicenter Stop IMatinib trial. Lamcet Oncol. 2010; 11(11):1029-35.
5. Jorgensen HG, Allan EK, Jordanides NE, Mountford JC, Holyoake TL. Nilotinib exerts equipotent antiproliferative effects to imatinib and does not induce apoptosis in VD34+ CML cells. Blood 2007; 109(9):4016-4019.
6. Copland M, Hamilton A, Elrick LJ, et al. Dasatinib (BMS 354825) targets an earlier progenitor population than Imatinib in primary CML but does not eliminate the quiescence frac-tion. Blood. 2006; 107(11):4532-4539.
7. Konig H, Copland M, Chu S, Jove R, Holyoake TL, Bhatia R. Effects of dastinib on SRC kinase activity and down-stream intracellular signaling in primitive chronic myelog-enous leukemia hematopoietic cells. Cancer Res. 2008; 68(23):9624-9633.
8. Kοnig H, Holyoake TL, Bahtia R. Effective and selective inhibition of chronic myeloid leukemia primitive hemat-

M. Δήμου και Π. Παναγιωτίδης76
opoietic progenitors by the dual Src/ Abl kinase inhibitor SKI-606. Blood. 2008; 111 (4): 2329-2338.
9. Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, Druker BJ. Human Cronic Myeloid Leukemia Stem Cells are insensitive to Imatinib despite inhibition of BCR-ABL activity. J Clin Invest. 2011; 121:396-409.
10. Hamilton A, Hegalson V, Schemionek M, et al. Chronic myeloid leukemia stem cells are not dependent on BCR-ABL kinase activity for their survival. Blood. 2012; 119 (6):1501-1510.
11. Carter B, Mac D, Cortes J, Andreeff M. The elusive CML stem cell: does it matter and how do we eliminate it? Semin Hematol. 2010; 47(4): 362-370.
12. Florian S, Sonneck K, Hauswirth AW, et al. Detection of molecular targets on the surface of CD34+/CD38- stem cells in various myeloid malignancies. Leuk Lymphoma 2006; 47:207-222.
13. Herrmann H, Cerny-Reiterer C, Gleixner CV, et al. CD34+/CD38- stem cells in chronic myeloid leukemia express Siglec-3 (CD33) and are responsive to the CD33-target-ing drug gemtuzumab/ozogamycin. Hematologica. 2012; 97(2):219-226.
14. Helgason VG, Karvela M, Holyoake TL. Kill one bird with two stones: potential efficacy of BCR-ABL and autophagy inhibition in CML. Blood (prepublished on line 21 June 2011. doi: 10. 1182/blood-2011-01-330621)
15. Skorski T, Kanakaraj P, Nieborowska-Skorska M, et al. Phosphatidylinositol-3 kinase activity is regulated by BCR/ABL and is required for the growth of Philadelphia chromo-somepositive cells. Blood. 1995; 86(2):726–36.
16. Tothova Z, Gilliland DG. FOXO transcription factors and stem cell homeostasis: insights from the hematopoietic system. Cell Stem Cell. 2007; 1(2):140–52.
17. Naka K, Hoshii T, Muraguchi T, et al. TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature. 2010; 463(7281):676–80.
18. Pellicano F, Helgason GV, Allan E, et al. FOXO transcrip-tion factor activity is partially retained in quiescent CML stem cells and induced by tyrosine kinase inhibitors in CML progenitor cells [abstract]. Haematologica (EHA Annual Meeting Abstract). 2010; 95(Suppl 1):48.
19. Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005; 352(17):1779–90.
20. Sattler M, Salgia R. Activation of hematopoietic growth factor signal transduction pathways by the human oncogene BCR/ABL. Cytokine Growth Factor Rev 1997; 8(1):63–79.
21. Jiang X, Lopez A, Holyoake T, et al. Autocrine production and action of IL-3 and granulocyte colony-stimulating fac-tor in chronic myeloid leukemia. Proc Natl Acad Sci USA. 1999; 96(22):12804–9.
22. Xie S, Wang Y, Liu J, et al. Involvement of Jak2 tyrosine phosphorylation in Bcr-Abl transformation. Oncogene. 2001; 20(43):6188–95.
23. Xie S, Lin H, Sun T, Arlinghaus RB. Jak2 is involved in c-Myc induction by Bcr-Abl. Oncogene 2002; 21(47):7137–46.
24. Samanta AK, Chakraborty SN, Wang Y, et al. Jak2 inhibi-tion deactivates Lyn kinase through the SET-PP2A-SHP1 pathway, causing apoptosis in drug-resistant cells from chronic myelogenous leukemia patients. Oncogene 2009; 28(14):1669–81.
25. Samanta AK, Chakraborty SN, Wang Y, et al. Destabiliza-tion of Bcr-Abl/Jak2 network by a Jak2/Abl kinase inhibitor ON044580 overcomes drug resistance in blast crisis chronic myelogenous leukemia (CML). Genes Cancer. 2010;19; 1(4):346–59.
26. Caligur V. The stem cell hypothesis. BioFiles 2008, 3.5,427. Zhao C, Blum J, Chen A, et al. Loss of beta-catenin impairs
the renewal of normal and CML stem cells in vivo. Cancer Cell 2007; 12:528–541.
28. Jamieson CH, Ailles LE, Dylla SJ, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Eng J Med 2004; 351:657–667.
29. Nagao R, Kimura S, Takeuchi M, et al. A novel β-catenin inhibitor, AV65, suppresses the growth of CML cell lines which aquire Imatinib resistance because of ABL kinase domain mutations including T315I and Hypoxia-Adaptation. ASH Annual Meeting Abstracts, 2010 No 1081.
30. Warren F, Smith J, Mudunuru U, et al. Treatment with β-catenin antagonist BC2059 exhibits single agent efficacy and exerts superior activity with Tyrosine Kinase Inhibitors (TKI) or histone deacetylase (HDAC) inhibitor against hu-man AML, CML and myeloproliferative neoplasms (MPN) progenitor cells. Molec Cancer Therapeutics. 2011 (10) suppl 1, abstract C144.
31. Zhao C, Chen A, Jamieson CH, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 2009; 458:776–779.
32. Dierks C, Beigi R, Guo GR, et al. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell 2008; 14:238–249.
33. Gao J, Graves S, Koch U, et al. Hedgehog signaling is dispensable for adult hematopoietic stem cell function. Cell Stem Cell 2009; 4:548–558.
34. Hofmann I, Stover EH, Cullen DE, et al. Hedgehog signal-ing is dispensable for adult murine hematopoietic stem cell function and hematopoiesis. Cell Stem Cell 2009; 4:559–567.
35. Ito K, Bernardi R, Morotti A, et al. PML targeting eradi-cates quiescent leukaemia-initiating cells. Nature 2008; 19(453):1072–1078.
36. Jaras M, Johnels P, Hansen N, et al. Isolation and killing of candidate chronic myeloid leukemia stem cells by antibody targeting of IL-1 receptor accessory protein. Proc Natl Acad Sci USA. 2010; 107(37):16280–5.
37. Subramaniam S, Stansberg C, Cunningham C. The interleukin 1 receptor family. Dev Comp Immunol. 2004;28(5):415–28.
38. Pellicano F, Sinclair A, Holyake TL. In Search of CML stem cells’ deadly weakness. Curr Hematol Malig Rep. 2011; 6:82-87.
39. Aichberger KJ, Mayerhofer M, Krauth MT, et al. Identifica-tion of mcl-1 as a BCR/ABL-dependent target in chronic myeloid leukemia (CML): evidence for cooperative antileuke-

Βλαστικά κύτταρα της χρόνιας μυελογενούς λευχαιμίας (CML stem cells) - Η οδός προς την ίαση 77
mic effects of imatinib and mcl-1 antisense oligonucleotides. Blood 2005; 105:3303–3311
40. Aichberger KJ, Mayerhofer M, Krauth MT, et al. Low-level expression of proapoptotic Bcl-2- interacting mediator in leukemic cells in patients with chronic myeloid leukemia: role of BCR/ABL, characterization of underlying signal-ing pathways, and reexpression by novel pharmacologic compounds. Cancer Res 2005; 65:9436–9444.
41. Konopleva M, Contractor R, Tsao T, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell, 2006
42. Kuroda J, Kimura S, Andreeff M, et al. ABT-737 is a useful component of combinatory chemotherapies for chronic my-eloid leukaemias with diverse drug-resistance mechanisms. Br J Haematol 2007; 140:181–190.
43. Kuroda J, Kimura S, Strasser A, et al. Apoptosis-based dual molecular targeting by INNO-406, a second-generation Bcr-Abl inhibitor, and ABT-737, an inhibitor of antiapoptotic Bcl-2 proteins, against Bcr-Abl-positive leukemia. Cell Death Differ 2007; 14:1667–1677.
44. Tauchi T, Sumi M, Nakajima A, et al. BCL-2 antisense oli-gonucleotide genasense is active against imatinib-resistant BCR-ABL-positive cells. Clin Cancer Res 2003;9:4267–4273.
45. Carter BZ, Mak D, Schober WD, et al. Regulation of sur-vivin expression through bcr-abl/ MAPK cascade: target-ing survivin overcomes Imatinib resistance and increases Imatinib sensitivity in Imatinib responsive CML cells. Blood 2006;107:1555–1563.
46. Mak DH, Schober WD, Chen W, et al. Triptolide induces cell death independent of cellular responses to imatinib in blast crisis chronic myelogenous leukemia cells including quiescent CD34+ primitive progenitor cells. Mol Cancer Ther 2009;8:2509–2516.
47. Mak DH, Wang RY, Schober WD, et al. Activation of apopto-sis signaling eliminates CD34+ progenitor cells in blast crisis CML independent of response to tyrosine kinase inhibitors. Leukemia (28 October 2011) | doi:10.1038/leu.2011.285.
48. Copland M, Pellicano F, Richmond L, et al. BMS-214662 potently induces apoptosis of chronic myeloid leukemia stem and progenitor cells and synergizes with tyrosine kinase inhibitors. Blood 2008;111:2843–2853.
49. Essers MA, Offner S, Blanco-Bose WE, et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature 2009;458:904–908
50. Kantarjian HM, O’Brien S, Cortes JE, et al. Complete cy-togenetic and molecular responses to interferon-alpha-based therapy for chronic myelogenous leukemia are associated with excellent longterm prognosis. Cancer 2003;97:1033–1041.
51. Guilhot F, Preudhomme C, Guilhot J, et al. Significant higher rates of undetectable molecular residual disease and molecular responses with pegylated form of interferon a2a in combination with Imatinib (IM) for the treatment of newly
diagnosed chronic phase (CP) chronic myeloid leukaemia (CML) patients (pts): confirmatory results at 18 months of part 1 of the spirit phase III randomized trial of the french CML group (FI LMC) [abstract]. ASH 2009. 2009
52. Bellodi C, Lidonnici MR, Hamilton A, et al. Targeting au-tophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J Clin Invest 2009;119(5):1109-1123.
53. Yan CH, Liang ZQ, Gu ZL, Yang YP, Reid P, Qin ZH. Con-tributions of autophagic and apoptotic mechanisms to CrTX-induced death of K562 cells. Toxicon 2006;47(5):521-530.
54. Carew JS, Nawrocki ST, Kahue CN, et al. Targeting au-tophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl mediated drug resistance. Blood 2007;110(1):313-322.
55. Bellodi C, Lidonnici MR, Hamilton A, et al. Targeting au-tophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J Clin Invest 2009;119(5):1109-1123.
56. Crowley LC, Elzinga BM, O’Sullivan GC, McKenna SL. Autophagy induction by Bcr-Abl-expressing cells facilitates their recovery from a targeted or nontargeted treatment. Am J Hematol 2011;86(1):38-47.
57. Schuringa JJ, Vellenga E. Role of the polycomb group gene BMI1 in normal and leukemic hematopoietic stem and progenitor cells. Curr Opin Hematol 2010;17(4):294–299.
58. Lessard J, Baban S, Sauvageau G. Stage-specific expres-sion of polycomb group genes in human bone marrow cells. Blood 1998;91(4):1216–1224
59. Lessard J, Schumacher A, Thorsteinsdottir U, et al. Func-tional antagonism of the Polycomb-Group genes eed and Bmi1 in hemopoietic cell proliferation. Genes Dev 1999;13(20):2691–2703.
60. Rizo A, Dontje B, Vellenga E, et al. Long-term maintenance of human hematopoietic stem/progenitor cells by expression of BMI1. Blood 2008;111(5):2621–2630.
61. Rizo A, Horton SJ, Olthof S, et al. BMI1 collaborates with BCRABL in leukemic transformation of human CD34+ cells. Blood 2010;116(22):4621–4630.
62. Westermarck J, Hahn WC. Multiple pathways regulated by the tumor suppressor PP2A in transformation. Trends Mol Med 2008;14(4):152–160.
63. Neviani P, Santhanam R, Trotta R, et al. The tumor sup-pressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell 2005;8(5):355–368.
64. Neviani P, Santhanam R, Oaks JJ, et al. FTY720, a new alternative for treating blast crisis chronic myelogenous leukemia and Philadelphia chromosome-positive acute lymphocytic leukemia. J Clin Invest 2007;117(9):2408–2421.

Tαξινόμηση και προγνωστικοί παράγοντες στα Philadelphia αρνητικά Μυελοϋπερπλαστικά Νεοπλάσματα
Θεόδωρος Μαρινάκης, Γεώργιος Καναβός, Νικόλαος Ι. Αναγνωστόπουλος
ΠΕΡΊληΨη: το 2008 ο Παγκόσμιος οργανισμός Υγείας βασίστηκε και σε νεότερες πληροφορίες για να κατατάξει τα Χρόνια μυελοϋπερπλαστικά Νοσήματα. Έτσι στο διαγνωστικό αλγόριθμο προστέθηκε η κατάδειξη του παθολογικού κλώνου, που τεκμηριώνεται πλέον και με τη παρουσία της JAK2V617F μετάλλαξης ή άλλων λιγότερο συχνών μοριακών δεικτών καθώς και τη παρουσία ιστολογικών ευρημά-των από τη βιοψία μυελού. ςτα «κλασικά» Ph αρνητικά μυελοϋπερπλαστικά Νεοπλάσματα (Ph- MYN) περιλαμβάνονται η Αληθής Πολυκυτταραιμία (AΠ), η ιδιοπαθής Θρομβοκυττάρωση (ιΘ) και η Πρω-τοπαθής μυελοΐνωση (Πμι). λιγότερο συχνά Ph- μΥΝ είναι η Χρόνια ουδετεροφιλική λευχαιμία, η Χρόνια ηωσινοφιλική λευχαιμία μη άλλως ταξινομούμενη, η μαστοκυττάρωση και άλλες νοσολογι-κές οντότητες με χαρακτηριστικά μυελοϋπερπλαστικών νοσημάτων, που αναφέρονται ως μυελοϋπερ-πλαστικά Νεοπλάσματα, αταξινόμητα. Ως παράγοντες κινδύνου για την ΑΠ και την ιΘ αναγνωρίζονται σήμερα η ηλικία >60 ετών και το ιστορικό προηγούμενης θρόμβωσης. Ως ασθενείς χαμηλού κινδύνου χαρακτηρίζονται αυτοί, που δεν διαθέτουν κανέναν από τους παραπάνω παράγοντες, ενώ αντίθετα αυ-τοί, που έχουν και έναν παράγοντα κινδύνου χαρακτηρίζονται ως ασθενείς υψηλού κινδύνου και πρέπει να λαμβάνουν κυτταροστατική αγωγή. οι ασθενείς χαμηλού κινδύνου με επιβαρυντικούς καρδιαγγεια-κούς παράγοντες (υπέρταση, σακχαρώδη διαβήτη, κάπνισμα, υπερλιπιδαιμία) θα πρέπει να λαμβάνουν αγωγή μόνο έναντι των καρδιαγγειακών παραγόντων. ςτην Πμι βαθμολογώντας παράγοντες με ανε-ξάρτητη αρνητική προγνωστική αξία (μεγάλη ηλικία, αναιμία, συστηματικά συμπτώματα, λευκοκυττά-ρωση, παρουσία βλαστών στο περιφερικό αίμα) διάφορα προγνωστικά συστήματα έχουν σχεδιαστεί σε σχέση με την επιβίωση, όπως το Διεθνές Προγνωστικό ςύστημα (IPSS) και το δυναμικό Διεθνές Προ-γνωστικό ςύστημα (DIPSS), που ανέδειξαν 4 ομάδες κινδύνου με σημαντικές διαφορές στη διάμεση επιβίωση. Πρόσφατα ενσωματώθηκαν στο τελευταίο και άλλοι παράγοντες (καρυότυπος, αριθμός αι-μοπεταλίων, εξάρτηση από μεταγγίσεις ερυθρών) για την καλύτερη πρόγνωση της επιβίωσης σε όλη τη διάρκεια της νόσου. τα παραπάνω συστήματα συμβάλλουν σημαντικά στην εξατομικευμένη θεραπευ-τική αγωγή σε ασθενείς με Πμι.
Haema 2012; 3(1): 78-88 Copyright EAE
Αιματολογική κλινική γ.Ν.Α. «γ. γεννηματάς»Διεύθυνση Αλληλογραφίας: Θεόδωρος μαρινάκης, Αιματολογική κλινι-
κή γ.Ν.Α. «γ. γεννηματάς», μεσογείων 154, 11527 Αθήνα, τηλ./Fax: 210 7470473, E- mail: [email protected]
Aνασκόπηση
1. ΙΣΤΟΡΙΚΗ ΑΝΑΔΡΟΜΗ. ΤΑξΙΝΟΜΗΣΗO William Dameshek χρησιμοποίησε το 1951 για
πρώτη φορά τον όρο «Mυελοϋπερπλαστικές διαταρα-χές- μΥΔ» (Myeloproliferative Disorders–MPDs) για να ομαδοποιήσει τέσσερα νοσήματα με μεγάλη συνά-φεια μεταξύ τους, τόσο από κλινικής, όσο και από παθο-φυσιολογικής άποψης. η ομάδα αυτή περιελάμβανε τη Χρόνια μυελογενή λευχαιμία, την Αληθή Πολυκυττα-
ραιμία (ΑΠ), την ιδιοπαθή Θρομβοκυττάρωση (ιΘ) και την Πρωτοπαθή μυελοΐνωση (Πμι), που αποτέλεσαν τις «κλασικές μΥΔ»1. μεταγενέστερες μελέτες ξεχώρι-σαν τη Χμλ από την ομάδα αυτή καθώς διαλευκάνθηκε η παθογένειά της, η χαρακτηριζόμενη από την παρουσία του χρωμοσώματος Philadelphia (Ph) και του χιμαιρικού γονιδίου BCR ABL.
το 2001, ο Παγκόσμιος οργανισμός Υγείας (World Health Organization–WHO) κατέταξε τις κλασικές μΥΔ στην ευρύτερη κατηγορία των «Χρόνιων μυελοϋπερπλα-στικών Νοσημάτων» (Chronic Myeloproliferative Dis-eases–CMPDs), που συμπεριελάμβανε επίσης τη Χρόνια ουδετεροφιλική λευχαιμία, τη Χρόνια ηωσινοφιλική

Tαξινόμηση και προγνωστικοί παράγοντες στα Philadelphia αρνητικά Μυελοϋπερπλαστικά Νεοπλάσματα 79
λευχαιμία/Υπερηωσινοφιλικό ςύνδρομο και τα αταξι-νόμητα Χρόνια μυελοϋπερπλαστικά Νοσήματα2. τα νοσήματα αυτά αποτελούσαν μιά ετερογενή ομάδα με κοινό χαρακτηριστικό τους τη κλωνική υπερπλασία της μυελικής σειράς, που αντιστοιχούσε στο περιφερικό αί-μα σε αύξηση των κοκκιοκυττάρων, των αιμοπεταλίων ή/και των ερυθροκυττάρων χωρίς να συνοδεύεται από δυσερυθροποίηση, δυσπλασία της κοκκιώδους σειράς ή μονοκυττάρωση.
Επειδή συχνά υπάρχουν ομοιότητες των Χρόνιων μυ-ελοϋπερπλαστικών Νοσημάτων με αντιδραστικές μορφές, που μπορεί να συμπεριφέρονται ανάλογα, τα επόμενα έτη ακολούθησε προσπάθεια κατάδειξης κλωνικότητας σε αυτά τα νοσήματα. ςτην καθημερινή πρακτική αυτό δεν είναι συχνά εφικτό, εάν η κυτταρογενετική μελέτη δεν είναι δυνατή ή εάν ο καρυότυπος αποδειχθεί φυσιο-λογικός. Από το 2005 παρατηρήθηκε σημαντική εξέλιξη με τη ταυτοποίηση στο γονίδιο JAK2 στο εξώνιο 14 της επίκτητης μετάλλαξης JAK2V617F3,4. η μετάλλαξη αυ-τή, που θέτει τη κινάση JAK2 σε κατάσταση συνεχούς δραστηριοποίησης και οδηγεί σε συνεχή πολλαπλασια-σμό των αιμοποιητικών κυττάρων, ανευρίσκεται στο 95% τουλάχιστον των περιπτώσεων με ΑΠ και στο 50% των περιπτώσεων με ιΘ και Πμι5. τα επόμενα έτη αναδεί-χθηκαν νέες μεταλλάξεις, όπως της MPLW515K/L στο γονίδιο του υποδοχέα της θρομβοποιητίνης MPL, που ανευρίσκεται στην Πμι σε ποσοστό 5-10% (και σπανι-ότερα στην ιΘ), καθώς και αυτές στο γονίδιο JAK2 στο εξώνιο 12, που ήταν πιό ειδικές στην ΑΠ σε μικρότερα ποσοστά5. Είναι σαφές ότι τα παραπάνω ευρήματα μπο-ρούν να κάνουν τη διάγνωση αυτών των νοσημάτων πιο ευχερή από την «προ-JAK2» εποχή.
το 2008 η WHO βασίστηκε σε νεότερες και προϋπάρ-χουσες πληροφορίες σε κλινικό, φαινοτυπικό και γενετι-κό επίπεδο για να κατατάξει τα αιματολογικά νοσήματα6.
Έτσι στην αναθεωρημένη έκδοση της WHO (2008)7 για τις Χρόνιες μυελικές Νεοπλασίες ο όρος «νόσημα» έχει αντικατασταθεί από τον όρο «νεόπλασμα», τόσο για τα Χρόνια μυελοϋπερπλαστικά Νοσήματα, που μετονο-μάζονται πλέον σε μυελοϋπερπλαστικά Νεοπλάσματα (μΥΝ), όσο και για τα μυελοδυσπλαστικά ςύνδρομα/μυελοϋπερπλαστικά Νοσήματα.
- Έγινε δυνατή η ενσωμάτωση στα διαγνωστικά κρι-τήρια και μοριακών δεικτών πιο ειδικών για τα μΥΝ και στο διαγνωστικό αλγόριθμο προστέθηκε η κατάδειξη πα-θολογικού κλώνου, που τεκμηριώνεται με τη παρουσία της JAK2V617F μετάλλαξης ή άλλων λιγότερο συχνών μοριακών δεικτών καθώς και ιστολογικών ευρημάτων από τη βιοψία μυελού.
- για τη διάγνωση της ιΘ μειώθηκε ο απαιτούμενος αριθμός ΑμΠ από 600x109/L σε 450x109/L.
- οι περιπτώσεις της Χρόνιας ηωσινοφιλικής λευχαι-μίας/Υπερηωσινοφιλικού ςυνδρόμου εντάχθηκαν πλέον στα μυελικά και λεμφικά Νεοπλάσματα με ηωσινοφι-
λία και αναδιατάξεις των γονιδίων PDGFRA, PDGFRB, FGFR1, ενώ εάν δεν ανιχνεύονται οι παραπάνω αναδι-ατάξεις καθώς και το χιμαιρικό γονίδιο BCR ABL, αυ-τές κατατάσσονται πλέον στην κατηγορία της Χρόνιας ηωσινοφιλικής λευχαιμίας μη άλλως ταξινομούμενης.
- τέλος, η μαστοκυττάρωση περιλαμβάνεται πλέον στην κατηγορία των μΥΝ.
Έτσι στα «κλασικά» Ph αρνητικά μυελοϋπερπλαστι-κά Νεοπλάσματα (Ph- MYN) περιλαμβάνονται η Αλη-θής Πολυκυτταραιμία, η ιδιοπαθής Θρομβοκυττάρωση και η Πρωτοπαθής μυελοΐνωση. λιγότερο συχνά Ph- μΥΝ -αναφερόμενα και ως «μη κλασικά ή παραλλαγές Ph- MYN»- είναι η Χρόνια ουδετεροφιλική λευχαιμία, η Χρόνια ηωσινοφιλική λευχαιμία μη άλλως ταξινομού-μενη, η μαστοκυττάρωση και άλλες νοσολογικές οντότη-τες με χαρακτηριστικά μυελοϋπερπλαστικών νοσημάτων χωρίς κάποια ιδιαίτερη κλινική, φαινοτυπική ή μοριακή σφραγίδα (μυελοϋπερπλαστικά Νεοπλάσματα, αταξινό-μητα) 7,8 (Πίνακας 1).
πίνακας 1. Ταξινόμηση των Μυελικών Νεοπλασιών και Μυελοϋπερπλαστικών Νεοπλασμάτων (WHO 2008)7.
1. Οξεία Μυελογενής Λευχαιμία
2.Μυελοδυσπλαστικά Σύνδρομα
3. Μυελοϋπερπλαστικά Νεοπλάσματα
3.1. Χρόνια Μυελογενής Λευχαιμία, BCR-ABL θετική
3.2. Αληθής Πολυκυτταραιμία
3.3. Ιδιοπαθής Θρομβοκυττάρωση
3.4. Πρωτοπαθής Μυελοΐνωση
3.5. Χρόνια Ουδετεροφιλική Λευχαιμία
3.6. Χρόνια Ηωσινοφιλική Λευχαιμία, μη άλλως ταξινομούμενη
3.7. Μαστοκυττάρωση3.8. Μυελοϋπερπλαστικά Νεοπλάσματα,
αταξινόμητα4. Μυελοδυσπλαστικά/Μυελοϋπερπλαστικά
Νεοπλάσματα5. Μυελικά και Λεμφικά Νεοπλάσματα με Ηωσινοφιλία
και αναδιατάξεις των γονιδίων PDGFRA, PDGFRB ή FGFR1
2. ΚΛΑΣΙΚΑ PHILADELPHIA ΑΡΝΗΤΙΚΑ ΜΥΕΛΟϋπΕΡπΛΑΣΤΙΚΑ ΝΕΟπΛΑΣΜΑΤΑ 2.1. Διαγνωστική προσέγγιση
οι τρεις κλινικές οντότητες Αληθής Πολυκυτταραιμία

Θ. Μαρινάκης και συν80
(ΑΠ), ιδιοπαθής Θρομβοκυττάρωση (ιΘ) και Πρωτοπα-θής μυελοΐνωση (Πμι) μοιράζονται κοινά χαρακτηρι-στικά: το κύτταρο που έχει υποστεί εξαλλαγή είναι το πολυδύναμο αρχέγονο αιμοποητικό κύτταρο, η κυτταρι-κή ωρίμανση είναι φυσιολογική ή σχεδόν φυσιολογική, εμφανίζουν αλληλοεπικάλυψη στην κλινική εικόνα και περιπτώσεις ΑΠ και ιΘ μπορεί να καταλήξουν με ινωτι-κές αλλοιώσεις στο μυελό των οστών. ςτις περιπτώσεις αυτές η τελική κατάληξη μπορεί να είναι η δευτεροπα-θής μυελοΐνωση (μετα-πολυκυτταραιμική/μετα-θρομβο-κυτταραιμική μυελοΐνωση).
η ανεύρεση μιας από τις μεταλλάξεις που προανα-φέρθηκαν πιστοποιεί την ύπαρξη κλωνικού μΥΝ και αποκλείει την αντιδραστική ερυθροκυττάρωση, θρομ-βοκυττάρωση ή μυελοΐνωση. Όμως η παρουσία τους δεν διαχωρίζει τον υπότυπο μΥΝ- εκτός ίσως από τις με-ταλλάξεις στο εξώνιο 12 του JAK2, που δεν έχουν ανα-φερθεί σε άλλα μΥΝ πλην της ΑΠ και τις μεταλλάξεις του MPL, που δεν έχουν ανευρεθεί σε ασθενείς με ΑΠ.
ςε ασθενείς με αυξημένη αιμοσφαιρίνη η ανάδειξη της μετάλλαξης JAK2V617F επιτρέπει τη διάγνωση της ΑΠ σε περισσότερες από 95% των περιπτώσεων, ενώ σε 2% περίπου των ασθενών με ΑΠ ανευρίσκονται μεταλλάξεις στο εξώνιο 12 του JAK2. για τη διάγνωση της ιΘ απαι-τείται αριθμός αιμοπεταλίων ≥450x109/L, αποκλεισμός της αντιδραστικής θρομβοκυττάρωσης και άλλων μΥΝ, που παρουσιάζονται με θρομβοκυττάρωση και κυρίως της Χμλ, όπου ο αποκλεισμός της με τεχνικές PCR ή FISH είναι υποχρεωτικός. Θετικότητα για JAK2V617F ή MPL μετάλλαξη καλύπτει μόνο το 60-70% των περιπτώσεων ιΘ και επομένως η εκτίμηση της οστεομυελικής βιοψίας είναι αναγκαία για τον καθορισμό της διάγνωσης. ςτην Πμι επίσης η ιστολογική εξέταση παίζει σημαντικό ρόλο καθώς ανευρίσκεται εκτεταμένη εναπόθεση κολλαγόνου και ρετικουλίνης, χωρίς βεβαίως αυτή η εναπόθεση από μόνη της να είναι διαγνωστική, αφού σε μικρή σχετικά έκταση μπορεί να ανευρεθεί και σε ΑΠ και ιΘ. και στην περίπτωση της Πμι πρέπει να αποκλείεται η Χμλ, ενώ η ανεύρεση μεταλλάξεων, όπως της JAK2V617F ή της MPL αποκλείει την αντιδραστική μυελοΐνωση.
τα διαγνωστικά κριτήρια παρατίθενται στο Πίνακα 2.
2.2. Προγνωστικοί παράγοντες στην Αληθή Πολυκυτταραιμία και Ιδιοπαθή Θρομβοκυτταραιμία
2.2.1. Θρομβωτικά επεισόδιαΕπειδή οι εφαρμοζόμενες θεραπείες στα κλασικά
Ph- μΥΝ αποβλέπουν στην ελάττωση του κινδύνου για θρόμβωση και αιμορραγία, τόσο στην ΑΠ, όσο και στην ιΘ, η κατάταξη σε ομάδες κινδύνου σε αυτά τα νοσήμα-τα έχει σχεδιαστεί με βάση την πιθανότητα που έχουν οι
ασθενείς αυτοί να εμφανίσουν θρομβωτικό επεισόδιο. Έτσι, ως παράγοντες κινδύνου αναγνωρίζονται σή-
μερα η ηλικία >60 ετών και το ιστορικό προηγούμενης θρόμβωσης9,10.
Ως ασθενείς χαμηλού κινδύνου χαρακτηρίζονται αυ-τοί, που δεν διαθέτουν κανέναν από τους παραπάνω πα-ράγοντες, ενώ αντίθετα αυτοί, που έχουν έστω και έναν παράγοντα κινδύνου, χαρακτηρίζονται ως ασθενείς υψη-λού κινδύνου και θα πρέπει να λαμβάνουν κυτταροστα-τική αγωγή9.
ςτη μελέτη ECLAP δείχθηκε ότι από τους ασθενείς με ΑΠ, τόσο αυτοί ηλικίας >65 ετών, όσο και αυτοί με ιστο-ρικό προηγούμενης θρόμβωσης παρουσίαζαν μεγαλύτερο κίνδυνο εμφάνισης καρδιαγγειακών επιπλοκών και μάλι-στα αυτοί που είχαν και τους δύο παράγοντες εμφάνιζαν πολύ μεγαλύτερο11. Ανάλογα ευρήματα σε σχέση με τη μεγάλη ηλικία και με ιστορικό προηγούμενης θρόμβω-σης παρατηρήθηκαν και σε ασθενείς με ιΘ12.
Υπάρχει επιπλέον και μια ενδιάμεση ομάδα, που απο-τελείται από ασθενείς χαμηλού κινδύνου, που έχουν όμως επιβαρυντικούς καρδιαγγειακούς παράγοντες (υπέρταση, σακχαρώδη διαβήτη, κάπνισμα, υπερλιπιδαιμία). Αυτοί οι ασθενείς θα πρέπει να λαμβάνουν αγωγή έναντι των καρδιαγγειακών επιβαρυντικών παραγόντων, ενώ δεν συ-στήνεται κυτταροστατική αγωγή9,13.
ςύμφωνα με το Βρετανικό Medical Research Council (MRC)-PT1 τα κριτήρια για τους ασθενείς ενδιάμεσου κινδύνου για ιΘ περιλαμβάνουν ηλικίες 40-60 έτη χωρίς προηγούμενο επεισόδιο θρόμβωσης, και απουσία υπέρ-τασης, σακχαρώδη διαβήτη και ΑμΠ <1.500x109/L. Πα-ρουσία των τριών τελευταίων παραγόντων σε νεότερους ασθενείς <60 έτη χωρίς προηγούμενο επεισόδιο θρόμβω-σης ή αιμορραγίας μπορεί να τους κατατάξει σε κατηγο-ρία υψηλού κινδύνου, ενώ μόνο οι ασθενείς ηλικίας <40 έτη χωρίς τα παραπάνω προβλήματα χαρακτηρίζονται ως χαμηλού κινδύνου14.
κατ’ εξαίρεση, ασθενείς χαμηλού ή ενδιαμέσου κινδύ-νου μπορούν να λάβουν κυτταροστατική θεραπεία, όταν υπάρχει: α) μη ανοχή στις αφαιμάξεις, β) προοδευτική αύ-ξηση των λευκών, γ) προοδευτική συμπτωματική σπλη-νομεγαλία, δ) μη ελεγχόμενα σοβαρά συμπτώματα της νόσου και ε) αύξηση των αιμοπεταλίων >1.500x109/L9.
Ο αριθμός των αιμοπεταλίων δεν έχει συμπεριληφθεί ακόμη στους παράγοντες κινδύνου καθώς δεν έχει αποδει-χθεί συσχέτιση μεταξύ του αριθμού τους και της εμφάνι-σης θρομβωτικών ή αιμορραγικών επεισοδίων, αλλά είναι γεγονός ότι θρομβοκυττάρωση με ΑμΠ >1.500x109/L σχετίζεται με επίκτητη νόσο von Willebrand και αιμορ-ραγική διάθεση15.
Όσον αφορά τον αριθμό των ερυθρών αιμοσφαιρίων, από τη μελέτη ECLAP δεν αποδείχθηκε κάποια συσχέτι-ση μεταξύ υψηλού αιματοκρίτη έως 50% και αυξημένου κινδύνου για θρόμβωση16.
Αντικρουόμενα είναι τα αποτελέσματα μελετών που
Tαξινόμηση και προγνωστικοί παράγοντες στα Philadelphia αρνητικά Μυελοϋπερπλαστικά Νεοπλάσματα 81
προσπαθούν να αποδείξουν συσχέτιση μεταξύ λευκοκυτ-τάρωσης και θρομβώσεων σε ασθενείς με μΥΝ:
ςτη μελέτη ECLAP διαπιστώθηκε ότι οι ασθενείς με ΑΠ και αριθμό λευκών >15x109/L παρουσίαζαν σημαντι-κά αυξημένο κίνδυνο οξέος εμφράγματος του μυοκαρδίου συγκριτικά με αυτούς με αριθμό λευκών <10x109/L16,17. οι Wolanskyj και συν σε μια σειρά 332 ασθενών με ιΘ παρατήρησαν ότι αριθμός λευκών >15x109/L σχετιζόταν με μεγαλύτερο κίνδυνο θρομβωτικού επεισοδίου στους ασθενείς αυτούς σε σύγκριση με άλλους με χαμηλότερο αριθμό18. Ανάλογα ήταν τα αποτελέσματα από μια σειρά 439 ασθενών με ιΘ από τους Carobbio και συν19. ςε άλ-λη μελέτη σε 605 ασθενείς με ιΘ και λευκοκυττάρωση δεν επιβεβαιώθηκε η παραπάνω συσχέτιση, πιθανώς λόγω της εφαρμοζόμενης θεραπείας20. Άλλες αναφορές επιβε-
βαίωσαν ότι μόνο η ηλικία >60 έτη και προηγηθέν θρομ-βωτικό επεισόδιο αποτελούν ανεξάρτητους παράγοντες κινδύνου για θρόμβωση και όχι η παρουσία λευκοκυττά-ρωσης21. ςε άλλη μελέτη σε 407 ασθενείς με ΑΠ και ιΘ αναφέρθηκε ότι η παρουσία λευκοκυττάρωσης στη διά-γνωση με τιμές ΑμΠ <1.000x109/L δεν προαναγγέλλει αρτηριακή ή φλεβική θρόμβωση. Αντίθετα η μεγάλη ηλι-κία σχετιζόταν με αρτηριακή θρόμβωση σε ασθενείς με ΑΠ και η υψηλή τιμή αιμοσφαιρίνης με φλεβικές θρομ-βώσεις σε ασθενείς με ιΘ22.
Έχει μελετηθεί και η πρόγνωση της παρουσίας της μετάλλαξης JAK2V617F στην ΑΠ και ιΘ:
το γεγονός ότι ασθενείς με ιΘ θετικοί στη μετάλλα-ξη εμφανίζονται με υψηλότερες τιμές αιμοσφαιρίνης και λευκών, με μικρότερο αριθμό αιμοπεταλίων και με υπερ-
πίνακας 2. Διαγνωστικά κριτήρια κατά WHO 2008 για τα κλασικά Ph- Μυελοϋπερπλαστικά Νεοπλάσματα8.
Κριτήρια Αληθής Πολυκυτταραιμία (ΑΠ)
Ιδιοπαθής Θρομβοκυτταραιμία (ΙΘ)
ΠρωτοπαθήςΜυελοΐνωση (ΠΜΙ)
Μείζονα 1. Hb >18,5g/dl (άνδρες) και >16,5g/dl (γυναίκες) ή Hb ή Hct> 99η εκατοστιαία θέση των ορίων αναφοράς για την ηλικία, το φύλο ή το υψόμετρο διαβίωσης
ή Hb >17g/dl (άνδρες)και >15g/dl (γυναίκες), αν συνδέεται με σταθερή
αύξηση ≥2g/dl από τη βασική τιμή του ασθενούς και η αύξηση δεν σχετίζεται με διόρθωση σιδηροπενικής αναιμίας
ήΑυξημένη μάζα ερυθρών
>25% της μέσης φυσιολογικά προβλεπόμενης τιμής
2. Παρουσία της μετάλλαξης JAK2V617F ή άλλου κλωνικού δείκτη
1. Αιμοπετάλια ≥450x109/L2. Μυελός με αυξημένο αριθμό
μεγακαρυοκυττάρων χωρίς σημαντική αύξηση της κοκκιώδους ή της ερυθράς σειράς
3. Δεν πληρούνται τα κριτήρια για ΑΠ, ΠΜΙ, ΧΜΛ, ΜΔΣ, ή άλλο μυελικό νεόπλασμα
4. Παρουσία της μετάλλαξης JAK2V617F ή άλλου κλωνικού δείκτη ή αποκλεισμός αντιδραστικής θρομβοκυττάρωσης
1. Αυξημένος αριθμός μεγακαρυοκυττάρων με ατυπία, σε συνδυασμό με ίνωση από αυξημένη εναπόθεση ρετικουλίνης ή/και κολλαγόνου
ή- σε απουσία ίνωσης-
ο αυξημένος αριθμός μεγακαρυοκυττάρων πρέπει να συνοδεύεται από αυξημένη κυτταροβρίθεια, με αύξηση της κοκκιώδους σειράς και συχνά μείωση της ερυθράς σειράς (προϊνωτική κυτταρική φάση της νόσου)
2. Δεν πληρούνται τα κριτήρια για ΑΠ, ΧΜΛ, ΜΔΣ, ή άλλο μυελικό νεόπλασμα
3. Παρουσία της JAK2V617F ή άλλου κλωνικού δείκτη ή αποκλεισμός αντιδραστικής ίνωσης
Ελάσσονα 1.Μυελός με υπερπλασία των τριών κυτταρικών σειρών
2.Ελαττωμένα επίπεδα EPO στον ορό
3.Ανάπτυξη ενδογενών αποικιών ερυθροβλαστών
1. Λευκοερυθροβλαστική αντίδραση
2. Αυξημένη LDH3. Αναιμία4. Ψηλαφητή σπληνομεγαλία
Διαγνωστικοί συνδυασμοί
2 μείζονα + 1 έλασσονήτο πρώτο μείζον + 2 ελάσσονα
Απαραίτητα και τα 4 Και τα 3 μείζονα + 2 ελάσσονα

Θ. Μαρινάκης και συν82
κυτταρικό μυελό με έντονη αιμοποίηση, έχει οδηγήσει κάποιους ερευνητές στην υπόθεση ότι αυτοί οι ασθενείς με ιΘ θα μπορούσαν να πάσχουν από κάποια «άτυπη» μορφή ΑΠ23. οι Vannucchi και συν παρατήρησαν ότι οι ομοζυγώτες JAK2V617F ασθενείς με ΑΠ και ιΘ ήταν με-γαλύτερης ηλικίας, παρουσίαζαν υψηλότερες τιμές αιμα-τοκρίτη και λευκών και σημαντικότερη σπληνομεγαλία σε σχέση με τους ετεροζυγώτες. Ειδικότερα οι ασθενείς με ΑΠ εμφάνιζαν πιο συχνά κνησμό24.
Έχει αναφερθεί επίσης ότι ασθενείς με ιΘ θετικοί στη μετάλλαξη JAK2V617F παρουσιάζουν αυξημένα ποσο-στά φλεβικών θρομβώσεων σε σχέση με τους ασθενείς με αμετάλλακτο το γονίδιο. η τελευταία παρατήρηση επι-βεβαιώθηκε σε αρκετές αναφορές25,26,27,29, αλλά υπήρξαν και άλλες, που δεν διαπίστωσαν συσχέτιση μεταξύ της παρουσίας της JAK2V617F μετάλλαξης και του κινδύ-νου για θρομβωτικά επεισόδια23,28,30.
Εκτός από την παρουσία ή όχι της μετάλλαξης JAK2V617F έχει μελετηθεί και ο ρόλος του φορτίου της, όσον αφορά την πρόγνωση της ΑΠ και ιΘ:
μελέτη της GIMEMA μεταξύ 323 ασθενών με ΑΠ (67,8% ετερόζυγοι, 32,2% ομόζυγοι) και 639 ασθενών με ιΘ (40,2% αμετάλλακτοι, 57,6% ετεροζυγώτες, 2,2% ομο-ζυγώτες) δεν ανέδειξε σημαντική διαφορά στη συχνότητα και στο τύπο των θρομβώσεων μεταξύ ετεροζυγωτών και ομοζυγωτών στη ΑΠ, ενώ οι ομοζυγώτες με ιΘ εμφάνι-ζαν υψηλότερο ποσοστό θρομβώσεων σε σχέση με αυτούς που ήταν αρνητικοί στη μετάλλαξη24. ςε άλλη προοπτική μελέτη σε 173 ασθενείς με ΑΠ που μετρήθηκε το αλληλι-κό φορτίο JAK2V617F στη διάγνωση, διαπιστώθηκε ότι οι ασθενείς με υψηλά ποσοστά αλληλίου στα κοκκιοκύτ-ταρα παρουσίασαν κατά 3,5 φορές μεγαλύτερο κίνδυνο θρομβώσεων σε σχέση με αυτούς με χαμηλότερο φορτίο αλληλίου31. Αντίθετα οι Tefferi και συν μελέτησαν το φορ-τίο JAK2V617F στο μυελό των οστών σε 77 ασθενείς με ΑΠ σε διάφορες στιγμές μετά τη διάγνωση και διαπίστω-σαν ότι το φορτίο της μετάλλαξης δεν σχετιζόταν με ση-μαντικά καρδιαγγειακά συμβάντα10. Αντικρουόμενες είναι και οι αναφορές για συσχέτιση μεταξύ φορτίου του αλλη-λίου και θρομβώσεων σε ασθενείς με ιΘ29,30,32.
Απομένει ερώτημα στο μέλλον, εάν και κατά πόσο και άλλοι παράγοντες, όπως π.χ. η λευκοκυττάρωση, η παρουσία της μετάλλαξης JAK2V617F, κ.ά. μπορεί να αποτελέσουν σε ΑΠ και ιΘ ένα νέο πρόσθετο ανεξάρτη-το παράγοντα κινδύνου για θρομβωτικό επεισόδιο.
2.2.2. Λευχαιμική εκτροπή - Εξέλιξη σε δευτεροπαθή μυελοΐνωση- Επιβίωση
η διάμεση επιβίωση στην ΑΠ και στην ιΘ υπερβαίνει τη 15ετία και ο κίνδυνος να αναπτυχθεί στη 10ετία μυ-ελοΐνωση (<4% και 10% αντίστοιχα) ή λευχαιμία (<2% και 6% αντίστοιχα) είναι σχετικά μικρός33.
ςτη μελέτη ECLAP, 22 από σύνολο 1638 ασθενών
με ΑΠ παρουσίασαν εκτροπή σε οξεία λευχαιμία/μυε-λοδυσπλαστικά σύνδρομα (ολ/μΔς) μετά από 8,4 έτη από τη διάγνωση. η μεγάλη ηλικία ήταν η σημαντικότε-ρη ανεξάρτητη μεταβλητή που σχετιζόταν με τον κίνδυνο εκτροπής της ΑΠ προς ολ/μΔς34. Επίσης είναι γνωστό ότι τα κυτταροστατικά αποτελούν τεκμηριωμένους πα-ράγοντες κινδύνου για την εκτροπή της ΑΠ σε ολ/μΔς. και ενώ ο σχετικός κίνδυνος εκτροπής των ασθενών με ΑΠ μετά από θεραπεία με υδροξυουρία ήταν μικρός, αυτός των ασθενών που ελάμβαναν βουσουλφάνη, πι-ποβρομάνη ή ραδιενεργό φωσφόρο (μόνα ή σε συνδυα-σμό) ήταν πολύ μεγαλύτερος σε σχέση με τους ασθενείς με ΑΠ, που υποβάλλονταν σε αφαιμάξεις ή ελάμβαναν ιντερφερόνη34. ςε άλλη μελέτη σε 459 ασθενείς με ΑΠ δείχθηκε ότι εκτός από την ηλικία (>60 έτη) και η λευ-κοκυττάρωση (>15x109/L) και η αρτηριακή θρόμβωση στη διάγνωση αποτελούν ανεξάρτητους προγνωστικούς παράγοντες για μειωμένη επιβίωση σε ΑΠ35.
τέλος η λευκοκυττάρωση (>15x109/L) στη διάγνω-ση της ΑΠ θεωρήθηκε επίσης παράγοντας κινδύνου για την εξέλιξη σε μετα-πολυκυτταραιμική μυελοΐνωση36.
οι Gangat και συν (2007) μετά από παρακολούθηση 605 ασθενών με ιΘ για 84 μήνες διαπίστωσαν λευχαιμική εκτροπή σε ποσοστό 3,3%. Δείχθηκε ότι η αναιμία απο-τελεί ανεξάρτητο παράγοντα κινδύνου τόσο για τη μει-ωμένη επιβίωση, όσο και για τη λευχαιμική εκτροπή32. ςυμπληρωματικά άλλοι παράγοντες με σημαντικό ρόλο στην επιβίωση ήταν η μεγάλη ηλικία (>60 έτη), η λευ-κοκυττάρωση (>15x109/L), το κάπνισμα, ο σακχαρώδης διαβήτης και ιστορικό προηγούμενης θρόμβωσης. για τη λευχαιμική εκτροπή σε ιΘ ενοχοποιήθηκε επιπλέον και η θρομβοκυττάρωση (ΑμΠ>1000x109/L), αλλά όχι η κυτταροστατική αγωγή. Επίσης δεν δείχθηκε ότι η πα-ρουσία της μετάλλαξης JAK2V617F επηρεάζει τη λευ-χαιμική εκτροπή ή την επιβίωση32.
ςε ανάλογη μελέτη 605 ασθενών με ιΘ οι Passamonti και συν (2008) διαπίστωσαν εξέλιξη σε μυελοΐνωση σε ποσοστό 2,8% (κίνδυνος 3,9% στη 10ετία) και για την εξέλιξη αυτή δείχθηκε να σχετίζεται η αναιμία. λευχαιμία διαπιστώθηκε σε 14 ασθενείς (2,3%) σε διάμεσο χρόνο 11 έτη μετά τη διάγνωση (κίνδυνος 2,6% στη 10ετία). η μεγάλη ηλικία (>60 έτη) στη διάγνωση εμφάνιζε σημα-ντική συσχέτιση με την εμφάνιση της λευχαιμίας και όχι η προηγηθείσα κυτταροστατική αγωγή. Παρατηρήθηκε διάμεση επιβίωση 22,3 έτη, ενώ παράγοντες όπως η ηλι-κία >60 έτη στη διάγνωση και το ιστορικό προηγούμενης θρόμβωσης θεωρήθηκαν ανεξάρτητοι παράγοντες κινδύ-νου για την επιβίωση21.
2.3. Προγνωστικοί παράγοντες στην Πρωτοπαθή Μυελοΐνωση
Αντίθετα από την ΑΠ και την ιΘ, όπου οι θεραπευτι-

Tαξινόμηση και προγνωστικοί παράγοντες στα Philadelphia αρνητικά Μυελοϋπερπλαστικά Νεοπλάσματα 83
κές στρατηγικές αποβλέπουν κυρίως στην ελάττωση του κινδύνου για θρομβωτικό επεισόδιο, στην Πμι η θερα-πεία αποσκοπεί στην παράταση του προσδόκιμου επιβίω-σης και εάν είναι δυνατόν στην ίαση μετά από αλλογενή μεταμόσχευση αρχέγονων αιμοποιητικών κυττάρων. Εάν αυτοί οι δύο στόχοι δεν είναι εφικτοί, τότε η θεραπεία εί-ναι απλά παρηγορητική και αποβλέπει στην ανακούφι-ση των ασθενών από τα συμπτώματα της νόσου και στη βελτίωση της ποιότητας ζωής τους9. για τους παραπάνω λόγους τα προγνωστικά συστήματα για την Πμι έχουν σχεδιαστεί με στόχο τον προσδιορισμό της επιβίωσης και όχι την πιθανότητα θρομβωτικού επεισοδίου κατά την πορεία της νόσου.
Από ιστορικής άποψης, ευρήματα στο περιφερικό αίμα κατά τη διάγνωση, όπως αναιμία (Hb <10g/dl) και λευκοπενία (WBC <4x109/L) ή λευκοκυττάρωση (WBC >30x109/L), ήταν οι πιο σημαντικές μεταβλητές αναφορι-κά με την πρόγνωση. για το λόγο αυτό και οι μεταβλητές αυτές αποτέλεσαν τα κριτήρια του προγνωστικού συ-στήματος της Lille (Dupriez score, 1996)37 (Πίνακας 3).
το 2009 η Διεθνής ομάδα Εργασίας για την Έρευνα και Θεραπεία της μυελοΐνωσης (International Working Group for Myelofibrosis Research and Treatment– IWG-MRT) ανέλυσε αναδρομικά περισσότερες από χίλιες περι-πτώσεις Πμι κατά τη διάγνωση και ανέπτυξε ένα Διεθνές Προγνωστικό ςύστημα (International Prognostic Scor-ing System – IPSS), το οποίο περιελάμβανε 5 παράγο-ντες με ανεξάρτητη αρνητική προγνωστική αξία: ηλικία >65 ετών, αναιμία (Hb<10g/dl ), συστηματικά συμπτώ-ματα, λευκοκυττάρωση (WBC >25x109/L) και παρουσία βλαστών (≥1%) στο περιφερικό αίμα38. Βαθμολογώντας την παρουσία 0 (χαμηλού κινδύνου), 1 (ενδιαμέσου κιν-δύνου-1), 2 (ενδιαμέσου κινδύνου-2) ή περισσότερων παραγόντων (υψηλού κινδύνου) αναδείχθηκαν 4 ομάδες κινδύνου με σημαντικές διαφορές στη διάμεση επιβίω-ση με 135, 95, 48, και 27 μήνες αντίστοιχα38 (Πίνακας 3).
Ακολούθως έγιναν προσπάθειες, ώστε να είναι δυνα-τό να εφαρμοστεί το IPSS σε όλη τη διάρκεια της νόσου, αλλά και σε άλλες ηλικίες, με αποτέλεσμα οι ερευνητές να καταλήξουν σε ένα «δυναμικό» IPSS (dynamic IPSS–DIPSS)39, σύμφωνα με το οποίο η εμφάνιση οποιουδή-ποτε από τους παραπάνω 5 παράγοντες κατά την πορεία της νόσου αποτελεί γεγονός επιβαρυντικό της πρόγνωσης. Από τους 5 αυτούς παράγοντες, η αναιμία (Hb <10g/dl) είχε τη μεγαλύτερη αρνητική προγνωστική αξία. η αξιο-λόγηση προέβλεπε 2 βαθμούς για την αναιμία και 1 βαθ-μό για τις υπόλοιπες μεταβλητές. Έτσι διαμορφώθηκαν 4 ομάδες κινδύνου: χαμηλού κινδύνου (0 βαθμοί), ενδι-άμεσου-1 (1 ή 2 βαθμοί), ενδιάμεσου-2 (3 ή 4 βαθμοί) και υψηλού κινδύνου (5 ή 6 βαθμοί). η διάμεση επιβίω-ση στις παραπάνω ομάδες αντιστοιχούν σε απροσδιόρι-στο χρόνο για την πρώτη ομάδα και 14,2, 4 και 1,5 έτη στις υπόλοιπες.
το 2011 το DIPSS εξελίχθηκε ακόμη περισσότερο κα-θώς προστέθηκαν σε αυτό ακόμη 3 παράγοντες (καρυότυ-πος, αριθμός αιμοπεταλίων και εξάρτηση από μεταγγίσεις ερυθρών), που αποδείχτηκαν ανεξάρτητοι προγνωστικοί παράγοντες μειωμένης επιβίωσης, και μετονομάστηκε σε DIPSS Plus40. ςε αυτό ο καρυότυπος θεωρείται δυσμενής, εάν περιέχει 1-2 κυτταρογενετικές ατυπίες με οποιαδήπο-τε από τις βλάβες +8, -7/7q-, i(17q), -5/5q-, 12p-, inv(3), 11q23 καθώς και ο σύνθετος καρυότυπος (≥3 κυτταρογε-νετικές ατυπίες). η διάμεση επιβίωση στις 4 παραπάνω ομάδες αντιστοιχεί σε 185 μήνες στην ομάδα χαμηλού κινδύνου και 78, 35 και 16 μήνες στις ομάδες ενδιάμε-σου-1, ενδιάμεσου-2 και υψηλού κινδύνου.
Άλλες νεότερες προσπάθειες πρόγνωσης της εξέλιξης σε Πμι στηρίχθηκαν σε ευρήματα από τον καρυότυπο: διακρίθηκαν 4 ομάδες κινδύνου με βάση τον καρυότυ-πο: ευνοϊκός καρυότυπος (+9, 20q-, 13q), φυσιολογικός, δυσμενής (σύνθετος καρυότυπος, +8) και καρυότυπος με άλλες ανωμαλίες. (International Prognostic Scoring system-independent cytogenetic risk categorization) 41.
Όπως φαίνεται στα παραπάνω προγνωστικά συστήμα-τα δεν έχει θέση η παρουσία της JAK2V617F μετάλλα-ξης καθώς η παρουσία της δεν σχετίζεται με την επιβίωση των ασθενών. η έλλειψη προγνωστικής αξίας της παρα-πάνω μετάλλαξης αναδείχθηκε σε 2 μελέτες, που κατέ-δειξαν μειωμένη συνολική επιβίωση σε ασθενείς με Πμι με ελαττωμένο φορτίο JAK2 V617F σε σχέση με ασθε-νείς με αυξημένο φορτίο του μεταλλαγμένου γονιδίου42,43.
Πρόσφατα μελετήθηκε και ο ρόλος του παράγοντα TNFα στην παθοφυσιολογία των Ph- μΥΝ44. οι ερευ-νητές έδειξαν ότι ο TNFα, ένας κατεξοχήν αρνητικός ρυθμιστικός παράγοντας της αιμοποίησης, αποτελεί κε-ντρικό μεσολαβητή της κλωνικής ανάπτυξης των μΥΝ. ςυγκεκριμένα, η έκφραση του μεταλλαγμένου γονιδίου JAK2V617F οδηγεί σε αυξημένα επίπεδα TNFα, καταστέλ-λοντας έτσι τη φυσιολογική αιμοποίηση, ενώ παραδόξως παρατηρήθηκε βιολογική αντίσταση των JAK2V617F-θετικών προγονικών κυττάρων στη δράση του TNFα και αυξημένη ικανότητα πολλαπλασιασμού των πιο ώριμων νεοπλασματικών κυττάρων μυελικής σειράς υπό την επί-δραση του TNFα. Aξιοσημείωτο είναι το γεγονός ότι αυ-ξημένες συγκεντρώσεις TNFα παρατηρήθηκαν και στο πλάσμα ασθενών με μΥΝ JAK2V617F-αρνητικά, γεγο-νός που οδηγεί στην υπόθεση ότι σε αυτούς τους ασθενείς υπάρχουν γενετικές βλάβες, που οδηγούν σε ενεργοποίη-ση της οδού JAK-STAT και υπερπαραγωγή TNFα. γνω-ρίζοντας ήδη ότι ο TNFα κατέχει σημαντικό ρόλο στην έκλυση προφλεγμονωδών κυτταροκινών και στην εμφάνι-ση συστηματικών συμπτωμάτων, πιθανώς ο παράγοντας αυτός να αποτελέσει μία ακόμη προγνωστική μεταβλητή και να συμβάλλει με τον τρόπο αυτό στην ορθότερη δια-στρωμάτωση των ασθενών σε ομάδες κινδύνου.
Θ. Μαρινάκης και συν84
3. ΜΗ ΚΛΑΣΙΚΑ PHILADELPHIA-ΑΡΝΗΤΙΚΑ ΜΥΕΛΟϋπΕΡπΛΑΣΤΙΚΑ ΝΕΟπΛΑΣΜΑΤΑ
3.1. Χρόνια Ουδετεροφιλική Λευχαιμία
Είναι σπάνιο νόσημα, καθώς μόνο περί τις 150 περι-πτώσεις έχουν περιγραφεί συνολικά. Απαντάται συνήθως σε ηλικιωμένα άτομα και χαρακτηρίζεται από λευκοκυτ-τάρωση (λευκά >25x109/L με περισσότερα από 80% ου-
δετερόφιλα και ραβδοπύρηνα), ηπατοσπληνομεγαλία και απουσία του χιμαιρικού γονιδίου BCR ABL. ο μυελός των οστών εμφανίζει υπερπλασία μόνο της κοκκιώδους σειράς χωρίς ίνωση ή δυσπλαστικές αλλοιώσεις. για τη διάγνω-ση της νόσου, απαιτείται αποκλεισμός της αντιδραστικής λευκοκυττάρωσης, της παρουσίας BCR ABL και αναδια-τάξεων των γονιδίων PDGFRA, PDGFRB και FGFR1.
λόγω της σπανιότητας της νόσου, δεν έχει καταστεί δυνατή η αναγνώριση προγνωστικών παραγόντων. η επι-βίωση των ασθενών ποικίλλει από 6 μήνες έως και περισ-
πίνακας 3. Προγνωστικά συστήματα για την πρωτοπαθή μυελοΐνωση (ΠΜΙ)
Παράγοντες κινδύνου (ΠΚ)
Lille (1996) 37 (στη διάγνωση της ΠΜΙ)
IPSS (2009) 38 (στη διάγνωση της ΠΜΙ)
DIPSS (2011)39
(ισχύει σε όλη τη διάρκεια της ΠΜΙ)
DIPSS Plus (2011)40
Αναιμία (Ηb<10g/dl)
X X X X
Αριθμός Λευκών X(<4 ή >30x109/L)
X(>25x109/L)
X X
Βλάστες στο περιφερικό αίμα
Χ Χ Χ
Συστηματικά συμπτώματα*
Χ Χ Χ
Ηλικία >65 ετών Χ Και για ηλικίες <65 έτη Χ
Δυσμενής καρυότυπος#
Χ
Αιμοπετάλια <100 x109/L
Χ
Εξάρτηση απόμεταγγίσεις ερυθρών
Χ
Ομάδα Χαμηλού Κινδύνου
χωρίς ΠΚΜέση επιβίωση93m
χωρίς ΠΚΜέση επιβίωση135m
Λευκά >25x109/LΣχετικός κίνδυνος:1,7
0 βαθμοίΜέση επιβίωση185m
Ομάδα Ενδιαμέσου Κινδύνου-1 1 ΠΚ
Μέση επιβίωση26m
1 ΠΚΜέση επιβίωση95m
Hb <10g/dlΣχετικός κίνδυνος:3,7
1 βαθμόςΜέση επιβίωση78m
Ομάδα Ενδιαμέσου Κινδύνου- 2
2 ΠΚΜέση επιβίωση48m
Βλάστες στο περιφερικό αίμα≥1%Σχετικός κίνδυνος:2,59
2-3 βαθμοίΜέση επιβίωση35m
Ομάδα Υψηλού Κινδύνου
2 ΠΚΜέση επιβίωση13m
≥3 ΠΚΜέση επιβίωση27m
Συστηματικά συμπτώματαΣχετικός κίνδυνος: 3,04
4-6 βαθμοίΜέση επιβίωση16m
* απώλεια βάρους >10%, νυκτερινοί ιδρώτες, πυρετός, # 1-2 κυτταρογενετικές ατυπίες με οποιαδήποτε από τις ακόλουθες βλάβες: +8, -7/7q-, i(17q), -5/5q-, 12p-, inv(3), 11q23, ή σύνθετος καρυότυπος (≥3 κυτταρογενετικές ατυπίες), m: μήνες

Tαξινόμηση και προγνωστικοί παράγοντες στα Philadelphia αρνητικά Μυελοϋπερπλαστικά Νεοπλάσματα 85
σότερο από 20 έτη. μόνο η εμφάνιση μυελοδυσπλαστικών χαρακτηριστικών κατά την πορεία της νόσου σηματοδο-τεί πιθανά την εξέλιξή της σε ομλ45.
3.2. Χρόνια Ηωσινοφιλική Λευχαιμία μη άλλως ταξινομούμενη
Αποτελεί ένα μΥΝ, στο οποίο ανευρίσκεταιι αυτό-νομος, κλωνικός πολλαπλασιασμός ηωσινοφίλων στο μυελό, στο περιφερικό αίμα και στους ιστούς, με κύριο χα-ρακτηριστικό την ηωσινοφιλία στο περιφερικό αίμα (ηω-σινόφιλα >1,5x109/L). για τη διάγνωση της νόσου είναι απαραίτητη η απόδειξη της κλωνικότητας των ηωσινο-φίλων (κυτταρογενετικός κλώνος ή HUMARA X-linked ανάλυση πολυμορφισμού), η απουσία BCR ABL και ανα-διατάξεων των γονιδίων PDGFRA, PDGFRB και FGFR1, η απουσία inv(16)(p13.1q22) ή t(16;16)(p13.1;q22) και αριθμός βλαστών >5% στο μυελό ή >2% στο περιφερικό αίμα. Εάν δεν είναι δυνατή η κατάδειξη της κλωνικότη-τας, τίθεται η διάγνωση του ιδιοπαθούς υπερηωσινοφι-λικού συνδρόμου ή της ιδιοπαθούς υπερηωσινοφιλίας.
Ως δυσμενείς προγνωστικοί παράγοντες έχουν ανα-γνωριστεί η παρουσία εκσεσημασμένης σπληνομεγαλίας, η παρουσία βλαστών στο περιφερικό αίμα, ο αυξημένος αριθμός (αλλά <20%) βλαστών στο μυελό, οι κυτταρο-γενετικές ατυπίες και οι δυσπλαστικές αλλοιώσεις στις υπόλοιπες σειρές του μυελού46.
3.3. Μαστοκυττάρωσηη μαστοκυττάρωση αποτελεί ομάδα νοσημάτων, που
χαρακτηρίζονται από εκτεταμένη συσσώρευση μαστοκυτ-τάρων σε έναν ή περισσότερους ιστούς. Διακρίνεται σε 2 μεγάλες κατηγορίες: τη δερματική και τη συστηματική. Πρόκειται για σπάνια νόσο με άγνωστη επίπτωση. ςτα παιδιά, το 80% των περιπτώσεων εμφανίζεται στο πρώ-το έτος της ζωής και συνήθως πρόκειται για δερματικές μορφές της νόσου, που βελτιώνονται ή υποχωρούν πλή-ρως στην εφηβεία. Αντίθετα, οι ενήλικες με μαστοκυττά-ρωση έχουν συνήθως τη συστηματική μορφή της νόσου. ςτον Πίνακα 4 περιγράφονται τα κριτήρια διάγνωσης της συστηματικής μαστοκυττάρωσης.
Δυσμενείς προγνωστικοί παράγοντες είναι η καθυ-στερημένη εμφάνιση των συμπτωμάτων, η απουσία δερ-ματικής μαστοκυττάρωσης, η θρομβοπενία, η αυξημένη LDH, η αναιμία, η μεγάλη κυτταροβρίθεια του μυελού των οστών, η παρουσία δυσπλαστικών στοιχείων στη γενική αίματος, η αυξημένη αλκαλική φωσφατάση και η ηπα-τοσπληνομεγαλία. Επίσης το ποσοστό και η μορφολογία των μαστοκυττάρων στο μυελό έχουν αναγνωριστεί ως σημαντικοί επιπρόσθετοι και ανεξάρτητοι προγνωστικοί δείκτες. τέλος, σημαντικό ρόλο στην πρόγνωση έχει και η μορφή της νόσου, καθώς πρόκειται για μια ετερογενή
νοσολογική οντότητα, που περιλαμβάνει από απλές δερ-ματικές βλάβες μέχρι νεοπλάσματα υψηλής κακοήθειας. Ασθενείς με επιθετική νόσο, όπως η μαστοκυτταρική λευ-χαιμία, έχουν πολύ μικρή επιβίωση, ενώ αυτοί με ήπια, συστηματική μαστοκυττάρωση έχουν φυσιολογικό προσ-δόκιμο επιβίωσης. η μαστοκυττάρωση που εντοπίζεται αποκλειστικά στο μυελό των οστών έχει άριστη πρόγνω-ση, ενώ εάν συνδέεται με άλλη κλωνική αιματολογική κακοήθεια, η κλινική πορεία και η πρόγνωση συνήθως καθορίζονται από τη συνυπάρχουσα αιματολογική νόσο47.
3.4. Μυελοϋπερπλαστικά Νεοπλάσματα, αταξινόμητα
ςτην ομάδα αυτή περιλαμβάνονται νοσολογικές οντό-τητες με χαρακτηριστικά κλινικά, μορφολογικά και εργα-στηριακά μΥΝ χωρίς όμως να εμφανίζουν τα κριτήρια των μΥΝ. Περιλαμβάνουν αρχικά στάδια ΑΠ, ιΘ ή Πμι, που δεν έχουν παρουσιάσει ακόμη τα υπόλοιπα χαρακτη-ριστικά στοιχεία, προχωρημένα στάδια μΥΝ, όπου η προέχουσα ίνωση επηρεάζει τα υπόλοιπα ή περιπτώσεις με σαφή ευρήματα μΥΝ, όπου συνυπάρχουσα νεοπλα-σία ή φλεγμονή επηρεάζει τα υπόλοιπα. η πρόγνωση των παραπάνω ασθενών ακολουθεί αυτή της ανάλογης ομά-δας που τα χαρακτηριστικά της ομοιάζουν περισσότερο48.
πίνακας 4. Διαγνωστικά κριτήρια κατά WHO 2008 για τη συστηματική μαστοκυττάρωση47
ΚριτήριαΜείζονα 1. Παρουσία στή βιοψία μυελού
ή/και σε άλλο εξωλεμφαδενικό όργανο, πολυεστιακών πυκνών αθροίσεων μαστοκυττάρων, όπως αναγνωρίζονται με τη χρώση για τρυπτάση ή άλλες ειδικές χρώσεις
Ελάσσονα 1. Άτυπη μορφολογία σε >25% των μαστοκυττάρων σε τομές οστεομυελικής βιοψίας, μυελικά επιχρίσματα ή άλλους εξωμυελικούς ιστούς
2. Ανίχνευση της μετάλλαξης KITD816V στο μυελό, στο περιφερικό αίμα ή σε άλλο εξωλεμφαδενικό όργανο
3. Έκφραση των δεικτών CD2 ή/και CD25 στα μαστοκύτταρα του μυελού ή άλλων εξωδερματικών θέσεων
4. Επίπεδα τρυπτάσης ορού >20ng/ml
Διαγνωστικοί συνδυασμοί
1 μείζον + 1 έλασσον ή τουλάχιστον 3 ελάσσονα

Θ. Μαρινάκης και συν86
Classification and current prognostication of Myeloproliferative Neoplasmsby Theodoros Marinakis, George Kanavos, Nikolaos I. Anagnostopoulos
Department of Clinical Hematology, General Hospital of Athens “G. Gennimatas”
ABSTRACT: In the revised World Health Organization classification system for Chronic Myeloid Neo-plasms (2008) the diagnostic algorithms for Polycythemia Vera, Essential Thrombocythemia and Pri-mary Myelofibrosis have been substantially changed to include informations regarding JAK2V617F and similar activating mutations, as well as pertinent histologic features of the bone marrow biopsy as di-agnostic criteria. The Philadelphia negative Myeloproliferative Neoplasms category includes the three classic MPN: Polycythemia Vera, (PV), Essential Thrombocythemia (ET) and Primary Myelofibrosis (PMF), as well as Chronic Neutrophilic Leukemia, Chronic Eosinophilic leukemia not otherwise speci-fied, Mastocytosis and Myeloproliferative neoplasm, unclassifiable. Patients with PV and ET should be defined as high risk if age is greater than 60 years or there is a history of previous thrombosis. High risk patients with PV should be managed with phlebotomy, low dose aspirin and cytoreduction, while high risk patients with ET should be managed with cytoreduction. For PMF several prognostic scoring sys-tems have been proposed based on some adverse prognostic factors for survival including advanced age, marked anemia, leukocytosis, constitutional symptoms and presence of circulating blasts. Risk stratifi-cation in PMF should start with the International Prognostic Scoring System- IPSS for newly diagnosed patients and dynamic IPSS for patients during the disease course with the addition of cytogenetics eval-uation and transfusion status. This prognostic assessment of patients with PMF may be useful for treat-ment decision-making.
Βιβλιογραφία 1. Dameshek W. Some speculations on the myeloproliferative
syndromes. Blood. 1951; 6:372-375. 2. Vardiman JW, Brunning RD, Harris NL. WHO histological
classification of Chronic Myeloproliferative Diseases. In: Jaffe ES, Harris NL, Stein H, Vardiman JW (eds). World Health Organization Classification of Tumors: Tumours of the Haematopoietic and Lymphoid Tissues. International Agency for Research on Cancer (IARC) Press: Lyon, France, 2001:17-44.
3. James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signaling causes polycythemia vera. Nature. 2005; 434:1144-1148
4. Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005; 365:1054–61
5. Levine RL, Gilliland GD. Myeloproliferative disorders. Blood. 2008; 112:2190-2198
6. Tefferi A, Thiele J, Orazi A, et al. Proposals and rationale for revision of the Wηο diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibro-sis: Recommendations from an ad hoc international expert panel. Blood. 2007; 110:1092-1097.
7. Vardiman JW, Brunning RD,Arber Da, LeBeau MM. Introduction and overview of the classification of the my-eloid neoplasms. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW (eds). WHO Classification of Tumours of the Haematopoietic and Lymphoid Tissues. International Agency for Research on
Cancer (IARC) Press: Lyon, France, 2008: 18-30. 8. Tefferi A and Vardiman JW. Classification and diagnosis
of myeloproliferative neoplasms: The 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia. 2008; 22:14-22
9. Barbui T, Barosi G, Birgegard G, et al. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European Leu-kemiaNet. J Clin Oncol. 2011; 29:761-770.
10. Vannucchi AM, Barbui T. Stratified management of essential thrombocythemia and polycythemia vera. In: Hematology Education ; the Education Program for the Congress of the EHA 2008; 2:201-208
11. μarchioli R, Finazzi G, Landolfi R, et al. Vascular and neo-plastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol. 2005; 23:2224-32
12. Cortelazzo S,Viero P, Finazzi G, et al Incidence and risk factors for thrombotic complications in a historical cohort of 100 patients with essential thrombocythemia. J Clin Oncol. 1990; 8:556-562
13. McMullin MF, Bareford D, Campbell P, et al. Guide-lines for the diagnosis, investigation and management of pοlycythemia/erythrocytosis. Br J Haematol. 2005; 130: 174-195.
14. Harrison CN. Essential thrombocythaemia: challenges and evidence-based management. Br J Haematol. 2005; 130:153-165
15. Budde U, van Genderen PJ: Acquired von Willebrand dis-ease in patients with high platelets counts. Semin Thromb

Tαξινόμηση και προγνωστικοί παράγοντες στα Philadelphia αρνητικά Μυελοϋπερπλαστικά Νεοπλάσματα 87
Hemost. 1997; 23:425-431. 16. Landolfi R, Marchioli R, Kutti J, et al. Efficacy and safety
of low-dose aspirin in polycythemia vera. N Eng J Med. 2004. 350:114-124.
17. Landolfi R, Di Gennaro L, Barbui T, et al. Leukocytosis as a major thrombotic risk factor in patients with polycythemia vera. Blood. 2007; 109:2446-2452.
18. Wolanskyj AP, Schwager SM, McClure RF, et al. Essential thrombocythemia beyond the first decade: life expectancy, long-term complication rates, and prognostic factors. Mayo Clin Proc. 2006; 81:159-166.
19. Carobbio A, Finazzi G, Guerini V, et al. Leukocytosis is a risk factor for thrombosis in essential thrombocythemia: interaction with treatment, standard risk factors, and JAK2 mutation status. Blood. 2007; 109:2310-2313.
20. Tefferi A, Gangat N. Wolanskyj A. The interaction between leukocytosis and other risk factors for thrombosis in essential thrombocythemia. Blood. 2007; 109:4105.
21. Passamonti F, Rumi E, Arcaini L, et al. Prognostic factors for thrombosis, myelofibrosis and leukemia in essential thrombocythemia: a study of 605 patients. Haematologica. 2008; 93:1645-1651.
22. Gangat N, Wolansyj AP, Schwager SM, et al. Leukocytosis at diagnosis and the risk of subsequent thrombosis in patients with low-risk essential thrombocythemia and polycythemia vera. Cancer. 2009; 115:5740-5745.
23. Campbell PJ, Scott LM, Buck G, et al. Definition of subtypes of essential thrombocythemia and relation to polycythemia vera based on JAK2V617F mutation status: a prospective study. Lancet. 2005; 366:1945-1953.
24. Vannucchi AM, Antonioli E, Guglielmelli P, et al. Clinical profile of homozygous JAK2V617F mutation in patients with polycythemia vera or essential thrombocythemia. Blood. 2007; 110:840-846.
25. Finazzi G, Rambaldi A, Guerini V, et al. Risk of thrombosis in patients with essential thrombocythemia and polycy-themia vera according to JAK2V617F mutation status. Haematologica. 2007; 92:135-136.
26. Heller PG, Lev PR, Salim JP, et al. JAK2V617F muta-tion in platelets from essential thrombocythemia patiets: correlation with clinical features and analysis of STAT5 phosphorylation status. Eur J Haematol. 2006; 77:210-216.
27. Cheung B, Radia D, Pantelidis P, et al. The presence of the JAK2V617F mutation is associated with a higher haemoglobin and increased risk of thrombosis in essential thrombocythemia. Br J Haematol. 2006; 132:244-245.
28. Antonioli E, Guglielmelli P, Pancrazzi A, et al. Clinical implications of the JAK2V617F mutation in essential thrombocythemia. Leukemia. 2005; 19:1847-1849.
29. Kittur J, Knudson RA, Lasho TL, et al. Clinical correlates of JAK2V617F allele burden in essential thrombocythemia. Cancer. 2007; 109:2279-2284.
30. Pemmaraju N, Moliterno AR, Williams DM, et al. The quantitative JAK2V617F neutrophil allele burden does not correlate with thrombotic risk in essential thrombocytosis. Leukemia. 2007; 21:2210-2212.
31. Vannucchi AM, Antonioli E, Guglielmelli P, et al. Prospec-tive identification of high risk polycythemia vera patients based on JAK2V617F allele burden. Leukemia 2007; 21:1952-1959
32. Gangat N. Wolansyj AP, McClure RF, et al. Risk stratifica-tion for survival and leukemic transformation in essential thrombocythemia: a single institutional study of 605 patients. Leukemia. 2007; 21:270-276.
33. Tefferi A. Essential thrombocythemia, polycythemia vera and myelofibrosis: current management and prospect of targeted therapy. Am J Hematol 2008; 83:491-497
34. Finazzi G, Caruso V, Marchiali R, et al. Acute leukemia in polycythemia vera: an analysis of 1638 patients enrolled in a prospective observational study. Blood. 2005; 105:2664-2670.
35. Gangat N, Strand J, Li CY, et al. Leukocytosis in polycy-themia vera predicts both inferior survival and leukaemic transformation. Br J Haematol. 2007; 138:354-358
36. Passamonti F, Rumi E, Caramella M, et al. A dynamic prognostic model to predict survival in post–polycythemia vera myelofibrosis. Blood 2008; 111:3383-3387.
37. Dupriez B, Morel P, Demory JL, et al. Prognostic factors in agnogenic myeloid metaplasia: a report on 195 cases with a new scoring system. Blood. 1996; 88:1013-1018.
38. Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009; 113:2895-2901.
39. Passamonti F, Cervantes F, Vannucchi AM, et al. A dynamic prognostic model to predict survival in primary myelofi-brosis: a study by the IWG-MRT (International Working Group for Myelofibrosis Research and Treatment). Blood. 2010; 115:1703-1708.
40. Gangat N, Caramazza D, Vaidya R, et al. DIPSS Plus: A Refined Dynamic International Prognostic Scoring System for Primary Myelofibrosis That Incorporates Prognostic Information From Karyotype, Platelet Count, and Transfu-sion Status. J Clin Oncol. 2011; 29:392-397.
41. Hussein K, Pardanani AD, Van Dyke DL, et al. International Prognostic Scoring System independent cytogenetic risk categorization in primary myelofibrosis. Blood. 2010; 115:496-499.
42. Guglielmelli P, Barosi G, Specchia G, et al. Identification of patients with poorer survival in primary myelofibrosis based on the burden of JAK2V617F mutated allele. Blood. 2009; 114:1477-1483.
43. Tefferi A, Lasho TL, Huang J, et al. Low JAK2V617F al-lele burden in primary myelofibrosis, compared to either a higher allele burden or unmutated status, is associated with inferior overall and leukemia-free survival. Leukemia. 2008; 22:756-761.
44. Fleischman AG, Aichberger KJ, Luty SB, et al. TNFα facilitates clonal expansion of JAK2V617F positive cells in myeloproliferative neoplasms. Blood. 2011; 118:6392-6398.
45. Bain BJ, Brunning RD,Vardiman JW, et al. Chronic neutro-

Θ. Μαρινάκης και συν88
philic leukaemia. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW (eds). WHO Classification of Tumours of the Haematopoietic and Lymphoid Tissues. International Agency for Research on Cancer (IARC) Press: Lyon, France, 2008: 38-39.
46. Bain BJ, Gilliland DG, Vardiman JW, et al. Chronic eosino-philic leukaemia, not otherwise specified. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW (eds). WHO Classification of Tumours of the Haematopoietic and Lymphoid Tissues. International Agency for Research on Cancer (IARC) Press: Lyon, France, 2008: 51-53.
47. Horny H-P, Metcalfe DD, Bennett JM, et al. Mastocytosis. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW (eds). WHO Classification of Tumours of the Haematopoietic and Lymphoid Tissues. International Agency for Research on Cancer (IARC) Press: Lyon, France, 2008:54-63.
48. Kvasnicka HM, Bain BJ, Thiele J, et al. Myeloproliferative neoplasm, unclassifiable. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW (eds). WHO Classification of Tumours of the Haematopoietic and Lymphoid Tissues. International Agency for Research on Cancer (IARC) Press: Lyon, France, 2008:64-65.

Γενετικές βλάβες στα Ph- μυελοϋπερπλαστικά νοσήματα
Γιώργος Γεωργίου, Βασιλική Μπάρτζη
ΠΕΡΊληΨη: τα μυελοϋπερπλαστικά νεοπλάσματα είναι κλωνικά νοσήματα που χαρακτηρίζονται από την υπερπαραγωγή ώριμων ως επί των πλείστων κυττάρων. το 2008 ο Παγκόσμιος οργανισμός Υγείας (WHO) κατέταξε τα μυελοϋπερπαλστικά νεοπλάσματα (μΥΝ) σε συγκεκριμένες κατηγορίες. η καινούρ-για αυτή κατάταξη ήταν το αποτέλεσμα της ανακάλυψης συγκεκριμένων γενετικών βλαβών που βρίσκο-νται στα μΥΝ: μεταλλάξεις των γονιδίων JAK2 και MPL. η ανίχνευση αυτών των βλαβών έχει εφαρμογή στην καθημερινή κλινική πράξη αφού αποτελεί αναπόσπαστο στοιχείο της διαγνωστικής προσέγγισης αυ-τών των νοσημάτων. μετά την ανακάλυψη αυτών των μεταλλάξεων, άρχισε να γίνεται αντιληπτό ότι η παθογένεση των μΥΝ είναι αρκετά πιο πολύπλοκη. Όλα τα μΥΝ είναι κλωνικά νοσήματα με την αρχι-κή βλάβη να επισυμβαίνει στο αρχέγονο αιμοποιητικό κύτταρο, προκαλώντας κυτταρική υπερπαραγω-γή είτε λόγω αυξημένης ευαισθησίας ή πλήρους ανεξαρτητοποίησης από την επίδραση κυτταροκινών. τα τελευταία χρόνια έχουν ανιχνευθεί επιπλέον μεταλλάξεις διαφόρων γονιδίων στα μυελοϋπερπλαστικά σύνδρομα: μεταλλάξεις των γονιδίων TET2, ASXL1, CBL, IDH κλπ. To ποσοστό συμμετοχής αυτών των μεταλλάξεων στην αρχική παθογένεση των μΥΝ δεν είναι γνωστή, όπως δεν είναι γνωστό το εάν συνδυ-ασμός διαφορετικών μεταλλάξεων οδηγεί σε διαφορετική φαινοτυπική έκφραση (πχ άλλος συνδυασμός μεταλλάξεων οδηγεί σε αληθή πολυκυτταραιμία, άλλος σε ιδιοπαθή θρομβοκυττάρωση κλπ.). μένει να δειχθεί κατά πόσο αυτή η πολυπλοκότητα γενετικών ανωμαλιών στα μΥΝ μπορεί να φανεί χρήσιμη στην κλινική πράξη όσον αφορά στη διαφορική τους διάγνωση, αλλά ακόμα και στη στοχευμένη θεραπεία τους.
Haema 2012; 3(1): 89-98 Copyright EAE
Ερευνητικό Αιματολογικό Εργαστήριο Α΄Προπαιδευτικής Παθολογικής κλινικής ΕκΠΑ.
Διεύθυνση Αλληλογραφίας: γεώργιος γεωργίου, Ερευνητικό Αιματολο-γικό Εργαστήριο Α΄ΠΠκ [email protected]
Aνασκόπηση
Ι. ΕΙΣΑΓΩΓΗ - ΜΥΕΛΙΚΕΣ ΝΕΟπΛΑΣΙΕΣ το 2008 ο Παγκόσμιος οργανισμός Υγείας (WHO)
χρησιμοποιώντας δεδομένα από την κλινική εικόνα, την κυτταρική μορφολογία, την κυτταρομετρίας ροής, την ανοσοϊστοχημείας και τις γενετικές βλάβες κατέταξε τα αιματολογικά νεοπλάσματα σε συγκεκριμένες κατηγο-ρίες.1 ςύμφωνα με την κατάταξη στην κατηγορία των μυελικών Νεοπλασιών περιλαμβάνονται πέντε νοσο-λογικές οντότητες: 1. οξεία μυελογενής λευχαιμία (ομλ) 2. μυελοδυσπλαστικά ςύνδρομα (μΔς) 3. μυελοϋπερπλαστικά Νεοπλάσματα (μΥΝ) 4. μυελοδυσπλαστικά-μυελοϋπερπλαστικά Νεοπλά-
σματα 5. μυελικά και λεμφικά Νεοπλάσματα με ηωσινοφιλία
και αναδιατάξεις των γονιδίων PDGFRA, PDGFRB, ή FGFR1
Εξαιρώντας την οξεία μυελογενή λευχαιμία και τα μυελοδυσπλαστικά ςύνδρομα, στις άλλες κατηγορίες μυελικών Νεοπλασιών περιλαμβάνονται Νοσολογικές οντότητες που χαρακτηρίζονται από υπερπαραγωγή ώρι-μων ως επί των πλείστων κυττάρων (με ή χωρίς δυσπλα-στικές αλλοιώσεις) της μυελικής σειράς.
ΙΙ. ΜΥΕΛΟϋπΕΡπΛΑΣΤΙΚΑ ΝΕΟπΛΑΣΜΑΤΑ (ΜΥΝ)
ςτα κλασσικά Ph αρνητικά (Ph-) μΥΝ περιλαμβά-νονται η Αληθής Πολυκυτταραιμία (ΑΠ), η ιδιοπαθής Θρομβοκυττάρωση (ιΘ) και η Πρωτοπαθής μυελοΐνωση (Πμ). λιγότερο συχνά Ph αρνητικά μυελοϋπερπλαστικά Νεοπλάσματα είναι η Χρόνια ουδετεροφιλική λευχαι-μία, Χρόνια ηωσινοφιλική λευχαιμία, η μαστοκυττάρω-ση και άλλες νοσολογικές οντότητες με χαρακτηριστικά μυελοϋπερπλαστικών νοσημάτων χωρίς κάποια ιδιαίτερη κλινική, φαινοτυπική ή μοριακή «σφραγίδα» (μυελοϋ-περπλαστικά Νεοπλάσματα, αταξινόμητα).

Γ. Γεωργίου και Β. Μπάρτζη90
Μυελοϋπερπλαστικά Νεοπλάσματα (ΜΥΝ) και γενετικές ανωμαλίες
οι παθογενετικοί μηχανισμοί στα Ph αρνητικά μΥΝ είναι αρκετά σύνθετοι, με συχνό εύρημα τη διαταρα-χή λειτουργίας μιας τυροσινικής κινάσης (αποτέλεσμα μετάλλαξης ή χιμαιρισμού) που μιμείται τον κατταρά-κτη γεγονότων που επισυμβαίνουν μετά από τη διέγερ-ση υποδοχέων κυτταροκινών. τα μΥΝ είναι ετερογενή όσον αφορά τα κλινικά χαρακτηριστικά, τους παθογενε-τικούς μηχανισμούς και την κλινική πορεία. η ανακά-λυψη της σημειακής μετάλλαξης V617F του γονιδίου JAK2 στις περισσότερες περιπτώσεις ασθενών με Αλη-θή Πολυκυτταραιμία οδήγησε σε δραστική αλλαγή της κατάταξης και των κριτηρίων διάγνωσης των μυελοϋ-περπλαστικών ςυνδρόμων.2
α. Κλασσικά ΜΥΝ: Αληθής Πολυκυτταραιμία (ΑΠ), Ιδιοπαθής Θρομβοκυττάρωση (ΙΘ), Πρωτοπαθής Μυελοΐνωση (ΠΜ)
οι τρεις κλινικές οντότητες μοιράζονται κοινά χαρα-κτηριστικά: το κύτταρο που έχει υποστεί εξαλλαγή είναι το πολυδύναμο αρχέγονο αιμοποητικό κύτταρο, η κυτ-ταρική ωρίμανση είναι φυσιολογική ή σχεδόν φυσιολο-γική, εμφανίζουν αλληλοεπικάλυψη στην κλινική εικόνα και στην περίπτωση της ΑΠ και ιΘ μπορεί να καταλή-ξουν σε ινωτικές αλλοιώσεις του μυελού.
Χρωμοσωμικές ανωμαλίες στα κλασσικά ΜΥΝΧρωμοσωμικές ανωμαλίες στα μΥΝ βρίσκονται στο
10-20% των περιπτώσεων ασθνεών με ΑΠ και ιΘ και στο 30-40% ασθενών με Πμ. οι πιο συχνές κυττραογενετι-κές ανωμαλίες στην ΑΠ είναι μερική τρισωμία 1q, +8, +9, del(13q), del(20q), στην ιΘ tr8, tr9, del(13q), del(20q) και στην Πμ μερική τρισωμία 1q(1q21-1q32), der(6)t(1;6)(q23-25;p21-22), del(13q)-η πιο συχνή ανωμαλία στην Πμ, που βρίσκεται στο 25% των περιπτώσεων με παθολογικό καρυότυπο-, del(20q)-η δεύτερη πιο συχνή κυτταρογενετική ανωμαλία στην Πμ.3 ςε μια μελέτη των Vaidya et al., μονοσωματικός καρυότυπος (mono-somal karyotype) βρέθηκε στο 2% των ασθενών με Πμ. οι ασθενείς αυτοί είχαν εξαιρετικά αυξημένη θνητότητα και πολύ υψηλότερα ποσοστά λευχαιμικής εκτροπής σε σύγκριση με όλους τους άλλους ασθενείς. 4
Μοριακές βλάβες στα ΜΥΝΕπειδή συχνά υπάρχουν ομοιότητες των μΥΝ με
αντιδραστικές μορφές που μπορεί να συμπεριφέρονται ως μυελοϋπερπλαστικά νοσήματα, διαμορφώθηκαν δι-αγνωστικά κριτήρια που εμπεριέχουν δείκτες που είναι ειδικοί αυτών των νεοπλασιών. Έτσι, στα διαγνωστικά
κριτήρια κατά WHO 2008 των μΥΝ περιλαμβάνονται και οι μεταλλάξεις των γονιδίων JAK2 και MPL. 1 ςτην πραγματικότητα η ανεύρεση μιας από τις μεταλλάξεις αυτές πιστοποιεί την ύπαρξη κλωνικού μΥΝ και απο-κλείει την αντιδραστική ερυθροκυττάρωση, θρομβο-κυττάρωση ή μυελοΐνωση. Από την άλλη, η ανίχνευσή τους δε διαχωρίζει τον υπο-τύπο μΥΝ εκτός ίσως από τις μεταλλάξεις στο εξώνιο 12 του JAK2 που δεν έχουν αναφερθεί σε άλλα μΥΝ εκτός της ΑΠ και τις μεταλλά-ξεις του MPL που δεν έχουν βρεθεί σε ασθενείς με ΑΠ. ςε ασθενείς με αυξημένη αιμοσφαιρίνη, η ανάδειξη της μετάλλαξης JAK2V617F επιτρέπει τη διάγνωση ΑΠ σε πάνω από 95% των περιπτώσεων, αφού σε λιγότερο από το 2% των ασθενών με ΑΠ ανευρίσκονται μεταλλάξεις στο εξώνιο 12 του JAK2.5 για τη διάγνωση της ιΘ απαι-τείται αριθμός αιμοπεταλίων >450.000/μl, αποκλεισμός αντιδραστικής θρομβοκυττάρωσης και άλλων μΥΝ που παρουσιάζονται με θρομβοκυττάρωση και κυρίως της Χμλ όπου ο αποκλεισμός της με PCR ή FISH είναι υποχρεωτικός. Θετικότητα για JAK2V617F ή MPL με-τάλλαξη καλύπτει μόνο το 60-70% των περιπτώσεων ιΘ και επομένως ιστολογική εκτίμηση οστεομυελικής βιο-ψίας είναι αναγκαία για τον καθορισμό της διάγνωσης.6 ςτην Πμ επίσης η ιστολογική εξέταση παίζει σημαντικό ρόλο, καθώς ανευρίσκεται εκτεταμένη εναπόθεση κολ-λαγόνου και ρετικουλίνης, αν και από μόνη της αυτή η εναπόθεση δεν είναι διαγνωστική καθώς σε μικρή έκτα-ση μπορεί να βρεθεί και στην ΑΠ και ιΘ. και στην περί-πτωση της Πμ πρέπει να αποκλείεται η Χμλ, ενώ και εδώ η ανεύρεση μεταλλάξεων JAK2V617F ή MPL απο-κλείει την αντιδραστική μυελοΐνωση.
Μετάλλαξη JAK2V617F (εξώνιο14)το γονίδιο JAK2 εδράζεται στο χρωμόσωμα 9p24 και
περιλαμβάνει 25 εξώνια και η πρωτεΐνη JAK2 αποτελεί-ται από 1132 αμινοξέα. η οικογένεια των Janus κινασών περιλαμβάνει τις JAK1, JAK2 και TYK2 κινάσες που εκ-φράζονται σε όλα τα κύτταρα των θηλαστικών και την JAK3 η έκφραση της οποίας περιορίζεται στα αιμοποι-ητικά κύτταρα. η ενεργοποίηση υποδοχέων κυτταροκι-νών (αυξητικής ορμόνης, ερυθροποιητίνης, προλακτίνης, λεπτίνης, GM-CSF, θρομβοποιητίνης, πολλών ιντερλευ-κινών) οδηγεί στην ενεργοποίηση της οδού JAK-STAT για τη μετάδοση-μεταφορά του ενδοκυττάριου σήματος. H οδός JAK-STAT είναι σημαντική σε σημαντικές κυτ-ταρικές λειτουργίες όπως κυτταρικός πολλαπλασιασμός, επιβίωση και φυσιολογική λειτουργία αιμοποιητικών, καρ-διακών και άλλων κυττάρων.7-9 μεταλλάξεις των JAK1, JAK2 και JAK3 έχουν συσχετιστεί με μυελικές και λεμ-φικές νεοπλασίες.10 η σημειακή μετάλλαξη, JAK2V617F, σχετίζεται με τα μυελοϋπερπλαστικά νεοπλάσματα και οι πρώτες αναφορές για την ύπαρξή της εμφανίστηκαν το 2005.2 η JAK2V617F προέρχεται από την αντικατάστα-

Γενετικές βλάβες στα Ph- μυελοϋπερπλαστικά νοσήματα 91
ση γουανίνης από Θυμίνη, στη θέση 1849, στο εξώνιο 14 του γονιδίου και η οποία έχει σαν αποτέλεσμα την αντι-κατάσταση της Βαλίνης από Φαινυλαλανίνη στο κωδικό-νιο 617 της ώριμης πρωτεΐνης (εικόνα 1). η μετάλλαξη επηρεάζει τη μη-καταλυτική περιοχή ψευδο-κινάσης της πρωτεΐνης, εκτρέπει τον ρυθμιστικό ρόλο που έχει στη δραστηριότητα της JAK2 κινάσης και οδηγεί σε συνεχή ενεργοποίηση της οδού STAT3/5.10
η JAK2V617F είναι η πιο συχνή μετάλλαξη στα Ph αρνητικά μυελοϋπερπλαστικά Νεοπλάσματα καθώς ανευρίσκεται σε ~95% των ασθενών με Αληθή Πολυ-κυτταραιμία, σε ~60% των ασθενών με ιδιοπαθή Θρομ-βοκυττάρωση και σε ~60% των ασθενών με Πρωτοπαθή μυελοΐνωση. Έχει όμως βρεθεί και σε μερικούς ασθενείς με μυελοδυσπλαστικό/μυελοϋπερπλαστι κό ςύνδρομο (στην Ανθεκτική Αναιμία με δακτυλιοειδείς σιδηροβλά-στες και θρομβοκυττάρωση όπου ανευρίσκεται σε πο-σοστό~50%)11 και σπάνια σε de novo οξεία μυελογενή λευχαιμία, μυελοδυσπλαστικό ςύνδρομο ή Χμλ.12-14
η ανεύρεση της μετάλλαξης και σε άλλα νοσήματα δεν υποβαθμίζει την ευρεία ειδικότητά της σε ασθενείς με μυελικά νεοπλάσματα αφού δεν ανευρίσκεται σε ασθε-
νείς με λεμφοκυτταρικά νεοπλάσματα, αντιδραστικά μυ-ελοϋπερπλαστικά ή υγιείς εθελοντές.15,16 μεταμόσχευση JAK2V617F μεταλλαγμένων κυττάρων σε ποντίκια προκα-λεί φαινότυπο ΑΠ, με συνοδό λευκοκυττάρωση και τελικά ίνωση του μυελού.17 Πρόσφατα, επιδρώντας στα επίπεδα έκφρασης του αλληλίου V617F (χαμηλά επίπεδα έκφρα-σης) αναπτύχθηκαν και ποντίκια με φαινότυπο ιΘ.18 τα μοντέλα αυτά υποδεικνύουν ότι η μετάλλαξη JAK2V617F είναι ικανή να προκαλέσει φαινότυπο μΥΝ σε ποντίκια και ότι τα επίπεδα της έκφρασης μπορεί να σχετίζονται με το φαινότυπο. Παρά την ύπαρξη των ανωτέρω πειρα-ματικών δεδομένων, η JAK2V617F δε φαίνεται να είναι το αρχικό γενετικό γεγονός πρόκλησης νόσου και πιθα-νώς αντιπροσωπεύει έναν υπο-κλώνο που δεν σχετίζεται συχνά με τη λευχαιμική μετατροπή.19 ςτις περισσότερες περιπτώσεις ασθενών με ΑΠ ή Πμ και αντίθετα σε ένα μόνο μικρό ποσοστό ασθενών με ιΘ, η μετάλλαξη βρί-σκεται σε ομόζυγη κατάσταση μέσω μιτωτικής αναδιά-ταξης (uniparental disomy-εικόνα 2).
το υψηλότερο φορτίο μεταλλαγμένου αλληλίου (η αναλογία δηλαδή μεταλλαγμένου προς αμετάλλακτο γο-νίδιο JAK2) βρίσκεται κατά φθίνουσα σειρά σε ασθενείς με ΑΠ, με Πμ και τέλος σε ιΘ.20 η διαφορά αυτή ωστό-σο στο φορτίο του μεταλλαγμένου γονιδίου δεν μπορεί από μόνη της να αποτελέσει κριτήριο διάκρισης μεταξύ των νοσολογικών οντοτήτων, ούτε μπορεί να εξηγήσει το παράδοξο μία μετάλλαξη να προκαλεί διαφορετι-κούς φαινοτύπους. Επίσης η απλή παρουσία μετάλλαξης JAK2V617F ή αυξημένο φορτίο μεταλλαγμένου αλληλίου δε φαίνεται να επηρεάζει την επιβίωση ή τη λευχαιμική μετατροπή.21,22 Αντίθετα χαμηλό φορτίο μεταλλαγμένου αλληλίου έχει συσχετιστεί με μειωμένη επιβίωση στην Πμ. 23 το φορτίο του JAK2V617F αλληλίου αυξάνει με το χρόνο στην ΑΠ και Πμ αλλά όχι στην ιΘ.
Εικόνα 1. Πρωτεΐνη JAK2 και θέση σημειακής μετάλλαξης JAK2V617F.
Εικόνα 2. Μιτωτικός ανασυνδυασμός και ομοζυγωτία σημειακής μετάλλαξης JAK2V617F.
FERM SH2 Ψευδοκινάση
JAK2V617FΕξώνιο 14
Kινάση

Γ. Γεωργίου και Β. Μπάρτζη92
Μεταλλάξεις εξωνίου 12 του γονιδίου JAK2 οι μεταλλάξεις του εξωνίου 12 του JAK2 γονιδίου εί-
ναι σχετικά ειδικές στην Αληθή Πολυκυτταραιμία αρνητι-κή στη μετάλλαξη JAK2V617F και πρωτοπεριγράφηκε το 2007 (εικόνα 3). η πιο συχνή ανάμεσα στις πάνω από 10 μεταλλάξεις του εξωνίου 12 που έχουν περιγραφεί μέχρι τώρα, είναι η μετάλλαξη N542-E543del. οι μεταλλάξεις στο εξώνιο 12 περιλαμβάνουν απαλείψεις, διπλασιασμούς και σημεικακές μεταλλάξεις που δε διασπούν το πλαίσιο ανάγνωσης (in-frame) και κυρίως αφορούν σε μια περι-οχή 7 αμινοξέων (F537–E543).
οι μεταλλάξεις βρίσκονται συνήθως σε ετερόζυγη μορφή και προκαλούν αποκλειστικά ερυθροκυττάρωση με μειωμένα επίπεδα ερυθροποιητίνης ορού ενώ βρίσκο-νται συχνότερα σε νεαρότερες ηλικίες ασθενών κατά τη διάγνωση. η κλινική πορεία των ασθενών φαίνεται να εί-ναι η ίδια με αυτή των JAK2V617F-θετικών ασθενών.24
Μεταλλάξεις MPLτο γονίδιο MPL (myeloproliferative leukemia virus
oncogene homolog), εδράζεται στο χρωμόσωμα 1p34, πε-ριλαμβάνει 12 εξώνια και κωδικοποιεί τον υποδοχέα θρομ-βοποιητίνης (635–680 αμινοξέα). μεταλλάξεις σε γενετικό επίπεδο που προκαλούν ενεργοποίηση του υποδοχέα συ-σχετίζονται με οικογενή θρομβοκυττάρωση (S505N) και παρουσιάζουν φαινότυπο μυελοϋπερπλαστικού νοσήματος όπως σπληνομεγαλία, μυελοΐνωση και αυξημένο κίνδυνο θρομβώσεων.25 Ένας πολυμορφισμός του MPL (G1238T) που έχει σαν αποτέλεσμα την αντικατάσταση K39N βρί-σκεται στο 7% περίπου των Αφρο-Αμερικανών και σχε-τίζεται με υψηλότερο αριθμό αιμοπεταλίων.26 ςωματικές μεταλλάξεις του MPL είναι σπάνιες και ανευρίσκονται σχε-δόν αποκλειστικά στους ασθενείς με μυελοϋπερπλαστι-κά ςύνδρομα (έχουν ανευρεθεί και σε ασθενείς με οξεία μεγακαρυοβλαστική λευχαιμία).27 η σημειακή μετάλλαξη MPLW515L προκαλείται από μια αλλαγή γουανίνης σε Θυμίνη στη θέση 1544 στο εξώνιο 10, με αποτέλεσμα την αντικατάσταση της τρυπτοφάνης σε λευκίνη στο κωδικό-νιο 515 της πρωτεΐνης MPL. η σημειακή αυτή μετάλλα-ξη πρωτοπεριγράφηκε το 2006 σε ασθενείς με Πρωτοπαθή μυελοΐνωση JAK2V617F-αρνητικούς και in vivo προκα-
λεί μυελοϋπερπλαστικό νόσημα στα ποντίκια με ίνωση μυ-ελού θρομβοκυττάρωση. ςτη συνέχεια ανευρέθηκαν και άλλες σημειακές μεταλλάξεις στο εξώνιο 10 του γονιδίου: MPLW515K, MPLW515S και MPLS505N σε ασθενείς με ιδιοπαθή Θρομβοκυττάρωση και Πρωτοπαθή μυελοΐνω-ση με συχνότητα που κυμαίνεται από 3-15%.28 η σημειακή μετάλλαξη MPL515 ανευρίσκεται στα αρχέγονα κύτταρα και προκαλεί επίσης ενεργοποίηση της οδού JAK-STAT. κάποιοι ασθενείς με ιδιοπαθή Θρομβοκυττάρωση ή Πρω-τοπαθή μυελοΐνωση έχουν πάνω από μια μεταλλάξεις στο γονίδιο MPL ενώ άλλοι έχουν μικρό φορτίο JAK2V617F θετικού κλώνου και ταυτόχρονα μεγαλύτερο φορτίο με-ταλλαγμένου MPL κλώνου. ομοζυγωτία μετάλλαξης MPL οφείλεται σε uniparental disomy όπως συμβαίνει και με την JAK2V617F.29 ιδιοπαθής θρομβοκυττάρωση με μεταλ-λαγμένο MPL σχετίζεται με μεγαλύτερη ηλικία, μικρότε-ρη τιμή αιμοσφαιρίνης, υψηλότερο αριθμό αιμοπεταλίων και υψηλότερο κίνδυνο αρτηριακών θρομβώσεων.30 η πα-ρουσία της δε φαίνεται να επηρεάζει τη επιβίωση ή την εξέλιξη σε οξεία λευχαιμία. Πρωτοπαθής μυελοΐνωση με μεταλλαγμένο MPL σχετίζεται με γυναικείο φύλο, μεγα-λύτερη ηλικία και μικρότερη τιμή αιμοσφαιρίνης.31 ο φαι-νότυπος των ασθενών με μετάλλαξη MPL είναι επομένως διαφορετικός από αυτόν με μεταλλάξεις JAK2.
β. Μαστοκυττάρωσηη μαστοκυττάρωση χαρακτηρίζεται από συσσώρευση
αυξημένου αριθμού μαστοκυττάρων σε ένα ή περισσότερα όργανα με συχνότερη εντόπιση το δέρμα, τον μυελό των οστών και το γαστρεντερικό σύστημα. ςύμφωνα με την τα-ξινόμηση κατά WHO 2008, η μαστοκυττάρωση διακρίνε-ται σε: Δερματική μαστοκυττάρωση (CM), λανθάνουσα συστηματική μαστοκυττάρωση (ISM), (η πιο συχνή μορ-φή ςυστηματικής μαστοκυττάρωσης), ςυστηματική μα-στοκυττάρωση με συνοδό αιματολογικό νόσημα κλωνικού τύπου μη μαστοκυτταρικής σειράς (SM-AHNMD), Επιθε-τική ςυστηματική μαστοκυττάρωση (ASM), λευχαιμία εκ μαστοκυττάρων (MCL), ςάρκωμα εκ μαστοκυττάρων (MCS), Εξωδερματικό μαστοκύττωμα. η δερματική μα-στοκυττάρωση είναι αποκλειστικά δερματολογική πάθη-ση και η συσσώρευση των μαστοκυττάρων περιορίζεται στο δέρμα. η συστηματική μαστοκυττάρωση είναι μυελο-ϋπερπλαστικο νεόπλασμα. ο πολλαπλασιασμός των μαστο-κυττάρων βρίσκεται σε ένα τουλάχιστον όργανο (συνήθως στο μυελό των οστών) και θα πρέπει να επιβεβαιωθεί με βιοψία. Δερματική συμμετοχή μπορεί να συνυπάρχει αλ-λά δεν είναι απαραίτητα παρούσα. η διάγνωση της συ-στηματικής μαστοκυττάρωσης απαιτεί την παρουσία ενός μείζονος και ενός ελάσσονος κριτηρίου ή τουλάχιστον τρι-ών ελασσόνων κριτηρίων: μείζον κριτήριο: Πολυεστια-κή πυκνή μαστοκυτταρική διήθηση (>15 μαστοκύτταρα ανά άθροιση) Ελάσσονα κριτήρια: 1. περισσότερο από
Εικόνα 3. Πρωτεΐνη JAK2 και θέση των μεταλλάξεων στο εξώ-νιο 12.
FERM SH2 Ψευδοκινάση
JAK2V617FΕξώνιο 14
JAK2Εξώνιο 12
Kινάση

Γενετικές βλάβες στα Ph- μυελοϋπερπλαστικά νοσήματα 93
το 25% των μαστοκυττάρων να έχουν άτυπη μορφολογία (ατρακτοειδή ή επιμήκη), 2. Ύπαρξη σημειακής μετάλλα-ξης του γονιδίου c-ΚΙΤ στο κωδικόνιο 816, 3. τα μαστο-κύτταρα να εκφράζουν CD2 και/ή CD25, 4. η τρυπτάση ορού >20 ng/ml. η μετάλλαξη D816V του γονιδίου c-KIT ανιχνεύεται στο 95% των ενηλίκων με συστηματική μα-στοκυττάρωση στα παθολογικά μαστοκύτταρα, ενώ στη δερματική μαστοκυττάρωση μόνο το 1/3 παρουσιάζει τη συγκεκριμένη μετάλλαξη στα μαστοκύτταρα. η μετάλλα-ξη αυτή χαρακτηρίζεται από την αντικατάσταση βαλίνης από ασπαρτικό οξύ (Asp816Val) στο κωδικόνιο 816 του εξωνίου 17 και προκαλεί την ενεργοποίηση και φωσφο-ρυλίωση της πρωτεΐνης KIT με αποτέλεσμα τον ανεξέλε-γκτο πολλαπλασιασμό των μαστοκυττάρων. ςπανιότερα, ανευρίσκονται άλλες σημειακές μεταλλάξεις στο εξώνιο 17 όπως D816Y, D816H, D816F.
γ. Χρόνια Ουδετεροφιλική Λευχαιμία, Χρόνια Ηωσινοφιλική Λευχαιμία μη άλλως ταξινομούμενη
Χρόνια Ουδετεροφιλική ΛευχαιμίαΕίναι σπάνια νόσος (έχουν περιγραφεί ~150 περιπτώ-
σεις) που απαντάται κυρίως σε ηλικιωμένα άτομα και χα-ρακτηρίζται από ουδετεροφιλία (>25.000/μl λευκά, 80% ώριμα ουδετερόφιλα), σπληνομεγαλία και απουσία χιμαι-ρικού γονιδίου BCR-ABL. οστεομυελική βιοψία αποκα-λύπτει υπερπλασία μόνο της κοκκιώδους σειράς, χωρίς ίνωση ή μυελοδυσπλαστικές αλλοιώσεις. για τη διάγνω-σή της απαιτείται αποκλεισμός παρουσίας BCR-ABL όσο και αποκλεισμός παρουσίας αναδιατάξεων των γονιδίων PDGFRΑ, PDGFRB και FGFR1. Δεν υπάρχει κάποιος μο-ριακός δείκτης που να κατευθύνει προς τη διάγνωσή της.32
Χρόνια Ηωσινοφιλική Λευχαιμία μη άλλως ταξινομούμενη
Πρόκειται για ένα μΥΝ στο οποίο υπάρχει αυτόνο-μος, κλωνικός πολλαπλασιασμός ηωσινοφίλων στο αίμα, μυελό και στους ιστούς, με την ηωσινοφιλία να είναι το κύριο χαρακτηριστικό στο αίμα (>1.500/μl ηωσινόφιλα). για τη διάγνωσή της πρέπει να υπάρχει απόδειξη κλωνι-κότητας των ηωσινοφίλων (πχ κυτταρογενετικός κλώνος ή HUMARA X-linked ανάλυση πολυμορφισμού), μυελο-βλάστες στο μυελό >5% και <20%, αποκλεισμός παρου-σίας BCR-ABL και αναδιατάξεων των γονιδίων PDGFRΑ, PDGFRB και FGFR1, απουσία inv(16) (p13.1q22) ή t(16;16)(p13.1;q22). Αν δεν μπορεί να αποδειχθεί κλωνι-κότητα τότε τίθεται η διάγνωση του ιδιοπαθούς υπερηω-σινοφιλικού συνδρόμου. και στην περίπτωση της Χρόνια ηωσινοφιλικής λευχαιμίας μη άλλως ταξινομούμενης, δεν υπάρχει κάποιος ειδικός μοριακός δείκτης.
ΙΙΙ. ΜΥΕΛΟΔΥΣπΛΑΣΤΙΚΑ/ΜΥΕΛΟϋπΕΡπΛΑΣΤΙΚΑ ΝΕΟπΛΑΣΜΑΤΑ (ΜΔΣ/ΜΥπ)
Χαρακτηρίζονται από την ταυτόχρονη παρουσία μυ-ελοδυσπλαστικών και μυελοϋπερπλαστικών χαρακτη-ριστικών.
Χρόνια Μυελομονοκυτταρική Λευχαιμία (ΧΜΜΛ)
Χαρακτηρίζεται από την παρουσία μονοκυττάρων στο περιφερικό αίμα (>1.000/μl), απουσία BCR-ABL και απου-σία αναδιατάξεων των γονιδίων PDGFRΑ, PDGFRB και FGFR1, <20% βλάστες στο μυελό και δυσπλασία σε μια ή περισότερες σειρές (ή σε απουσία δυσπλασίας, κλωνι-κός κυτταρογενετικός ή μοριακός δείκτης ή αποκλεισμός άλλης αιτίας μονοκυττάρωσης). το 40% των ασθενών με Χμμλ φέρουν μεταλλάξεις στο γονίδιο RAS.33 ενώ ασθενείς με ETV6-PDGFRB χιμαιρικό γονίδιο [t(5;12)(q31;p12)] κατατάσσονται πλέον στην κατηγορία μυ-ελικά Νεοπλάσματα με ηωσινοφιλία και αναδιατάξεις του PDGFRB.
Άτυπη Χρόνια Μυελογενής Λευχαιμία, BCR-ABL1 αρνητική
Φέρει όλα τα χαρακτηριστικά της Χμλ αλλά χωρίς να υπάρχει BCR-ABL χιμαιρικό γονίδιο και χωρίς να υπάρχουν αναδιατάξεις των γονιδίων PDGFRΑ, PDG-FRB και FGFR1. Περίπου 30% των περιπτώσεων φέρει μεταλλάξεις των γονιδίων KRAS ή NRAS.
Παιδική Χρόνια Μυελομονοκυτταρική Λευχαιμία (JMML)
η συχνότητά της είναι 1,3 περιπτώσεις ανά εκατομύ-ριο ασθενών ηλικίας 0-14 ετών κατά έτος. Χαρακτηρίζεται από μονοκυττάρωση, απουσία BCR-ABL και αναδιατά-ξεων των γονιδίων PDGFRΑ, PDGFRB και FGFR1 και <20% βλάστες στο μυελό. ςωματικές μεταλλάξεις στο γονίδιο PTPN11 βρίσκονται στο 35% των περιπτώσεων, καθώς και στα γονίδια NRAS, KRAS και NF1 σε ποσο-στό 20% για το κάθε γονίδιο.34
Ανθεκτική Αναιμία με δακτυλιοειδείς σιδηροβλάστες και θρομβοκυττάρωση
Είναι σπάνια νόσος και χαρακτηρίζεται από δυσερυ-θροποίηση με δακτυλιοειδείς ερυθροβλάστες στο μυε-λό, θρομβοκυττάρωση και παρουσία αυξημένου αριθμού

Γ. Γεωργίου και Β. Μπάρτζη94
μεγάλων μεγακαρυοκυττάρων. Περίπου 50% των ασθε-νών φέρουν τη μετάλλαξη JAK2V617F και μικρός αριθ-μός ασθενών φέρει μεταλλάξεις του MPL. Περιπτώσεις ασθενών με μεμονωμένη έλλειψη 5q ή inv(3)(q21q26) δεν περιλαμβάνονται σε αυτή την κατηγορία.11,35
Ιv. ΜΥΕΛΙΚΑ ΚΑΙ ΛΕΜΦΙΚΑ ΝΕΟπΛΑΣΜΑΤΑ ΜΕ ΗΩΣΙΝΟΦΙΛΙΑ ΚΑΙ ΑΝΑΔΙΑΤΑξΕΙΣ ΤΩΝ ΓΟΝΙΔΙΩΝ PDGFRA, PDGFRB, Η FGFR1
Πρόκειται για σπάνια νοσήματα στα οποία συχνά έχουν ηωσινοφιλία. Νοσήματα με αναδιατάξεις του PDG-FRA και συγκεκριμένα FIP1L1-PDGFRA παρουσιάζο-νται κυρίως σαν χρόνια ηωσινοφιλική λευχαιμία με συχνή αύξηση των μαστοκυττάρων. λιγότερο συχνά παρουσι-άζονται ως ομλ ή τ-ολλ και στις δύο όμως περιπτώ-σεις πάντα με ηωσινοφιλία. Νοσήματα με αναδιατάξεις του PDGFRΒ παρουσιάζονται κυρίως αλλά όχι αποκλει-στικά σαν Χμμλ με ηωσινοφιλία. Νοσήματα με ανα-διατάξεις του FGFR1 παρουσιάζονται σαν τ-ολλ με ηωσινοφιλία, χρόνια ηωσινοφιλική λευχαιμία, Β-ολλ ή ομλ. Αξίζει να τονιστεί ότι σε περιπτώσεις με κλινι-κή υποψία νοσήματος με αναδιάταξη FIP1L1-PDGFRA απαιτείται η ανάδειξή του με RT-PCR ή FISH καθώς δεν αναγνωρίζεται στην κλασσική κυτταρογενετική μελέτη.
v. ΜΕΤΑΛΛΑξΕΙΣ ΓΟΝΙΔΙΩΝ ΣΤΑ ΜΥΝ ΕΚΤΟΣ ΤΩΝ JAK2 ΚΑΙ MPL
η ανακάλυψη καινούργιων γενετικών ανωμαλιών στα μΥΝ συνεχώς διευρύνεται αλλά η παθογενετική τους αξία περιορίζεται προς το παρόν, από το γεγονός ότι ανευρίσκονται και σε άλλα νοσήματα εκτός από τα μυελικά νεοπλάσματα.
α. Μεταλλάξεις TET2 (tet ongogene family member 2)
η πρωτεΐνη TET2 ανήκει σε μια οικογένεια πρωτεϊνών με τρία μέλη (TET1, TET2 και TET3), η λειτουργία των οποίων μπορεί να περιλαμβάνει τη μετατροπή της 5-με-θυλκυτοσίνης σε 5-υδροξυμεθυλκυτοσίνη και με αυτόν τον τρόπο να επηρεάζει πιθανώς την επιγενετική ρύθμιση της μεταγραφής. το γονίδιο ΤET2 εδράζεται στο χρωμό-σωμα 4q24 σε μια θέση τομής που ανευρίσκεται σε αντι-μεταθέσεις στην ομλ: t(3;4)(q26;q24), t(4;5)(q24;p16), t(4;7)(q24;q21) και del(4)(q23q24). μεταλλάξεις του TET2 πρωτοπεριγράφηκαν το 2008 και περιλαμβάνουν μεταλλάξεις που μετατοπίζουν το πλαίσιο ανάγνωσης ή δημιουργούν κωδικόνιο λήξης ή είναι σημειακές μεταλ-λάξεις με αλλαγή αμινοξέως και βρίσκονται διασκορπι-
σμένες στα 12 εξώνια του γονιδίου. Βρίσκονται και στα JAK2V617F-θετικά (17%) και στα JAK2V617F-αρνητικά (7%) μυελοϋπερπλαστικά Νεοπλάσματα. μεταλλάξεις TET2 συνυπάρχουν επίσης με άλλες μεταλλάξεις ή ανα-διατάξεις γονιδίων στην ομλ: RARA, FLT3, RAS, MLL, CEBPA και NPM1. μεταλλάξεις TET2 σε μυελοϋπερπλα-στικά Νεοπλάσματα μπορεί να προϋπάρχουν ή να ακο-λουθούν την απόκτηση μετάλλαξης JAK2 στο εξώνιο 12 ή 14 ή να υπάρχουν ανεξάρτητα προκαλώντας δικλωνι-κή νόσο. η παρουσία μεταλλάξεων TET2 σε πολλά νο-σήματα κάνει αμφίβολο τον παθογενετικό τους ρόλο στα μυελοϋπερπλαστικά Νεοπλάσματα και επιπλέον, με τα υπάρχοντα στοιχεία, δε φαίνεται να επηρεάζει την επι-βίωση, τη εξέλιξη σε ομλ ή τον κίνδυνο θρομβώσεων σε ασθενείς με Αληθή Πολυκυτταραιμία ή Πρωτοπαθή μυελοΐνωση. Αντίθετα φαίνεται ότι σχετίζονται με αυ-ξημένη επιβίωση στα μΔς και μειωμένη επιβίωση στην ομλ και Χμμλ.36
β. Μεταλλάξεις ASXL1 (additional sex combs like 1 (Drosophila))
το γονίδιο ASXL1 εδράζεται στο χρωμόσωμα 20q11.1 και πιστεύεται ότι προκαλεί αναστολή μεταγραφής που προκαλείται από τον υποδοχέα του ρετινοϊκού οξέος. Εκ-φράζεται σε όλα τα αιμοποιητικά κύτταρα. μεταλλάξεις του γονιδίου βρίσκονται στη χρόνια και στη βλαστική φάση μυελοϋπερπλαστικών Νεοπλασμάτων σε ποσο-στό από 8-19%. οι μεταλλάξεις του ASXL1 μπορεί ή όχι να συνυπάρχουν με μεταλλάξεις JAK2 ή TET2. Επειδή ο αριθμός των ασθενών που εξετάστηκαν σε πρόσφατες μελέτες είναι μικρός, χρειάζονται περαιτέρω, μεγαλύτε-ρες μελέτες για να καθορίσουν την ακριβή επίπτωση στα μυελοϋπερπλαστικά Νεοπλάσματα και να προσδιορί-σουν την προγνωστική αξία αυτών των μεταλλάξεων.37
γ. Μεταλλάξεις CBL (Cas-Br-M (murine) ecotropic retroviral transforming sequence)
το γονίδιο CBL αποτελείται από 16 εξώνια και εδρά-ζεται στο χρωμόσωμα 11q23.3. η πρωτεΐνη CBL (906 αμινοξέα) βρίσκεται στο κυτταρόπλασμα και εμπλέκεται στη μετάδοση ενδοκυτταρίων σημάτων μέσω κινασών αλλά επίσης συμμετέχει και στην αποδόμηση πρωτεϊ-νών (ubiquitination). μεταλλάξεις του CBL λειτουργούν ογκογενετικά σε αρκετές κυτταρικές σειρές και προκαλεί πολλαπλασιασμό ανεξαρτήτως αυξητικών παραγόντων. Έκφραση μεταλλαγμένου CBL με τη βοήθεια ρετροϊών προκάλεσε διήθηση πολλών οργάνων από μαστοκύτταρα και μυελοϋπερπλαστικό φαινότυπο και οξεία λευχαιμία σε ορισμένες περιπτώσεις.38 ςε αντίθεση, σε μια μελέτη δεν ανευρέθησαν μεταλλάξεις του γονιδίου σε κανέναν από τους 60 ασθενείς με συστηματική μαστοκυτάρωση.39

Γενετικές βλάβες στα Ph- μυελοϋπερπλαστικά νοσήματα 95
μεταλλάξεις του CBL πρωτοπεριγράφηκαν σε ομλ με σύντηξη των γονιδίων MLL–CBL ως αποτέλεσμα μερι-κής απάλειψης του CBL. μεταλλάξεις του είναι πιο συ-χνές σε Παιδική Χρόνια μυελομονοκυτταρική λευχαμία (~17%) και στη Χμμλ (~11%), εντοπίζονται κυρίως στο εξώνιο 8 και μπορούν να συνυπάρχουν με μεταλλάξεις άλλων γονιδίων όπως RUNX1, FLT3, JAK2 και TP53.40
μεταλλάξεις του CBL δεν ανευρίσκονται συχνά σε άλλα μυελικά νεοπλάσματα: 6% σε Πρωτοπαθή μυελοΐνωση, 1% σε Χρόνια ηωσινοφιλική λευχαμία, <1% σε de novo ομλ, μΔς, μαστοκυττάρωση, Χρόνια ουδετεροφιλι-κή λευχαιμία, βλαστική κρίση Χμλ.41
δ. Μεταλλάξεις IDH (IDH1=isocitrate dehydrogenase 1 (NADP+), soluble, IDH2=isocitrate dehydrogenase 2 (NADP+), mitochondrial)
τα γονίδια IDH1 (στο 2q33.3; 10 εξώνια) και IDH2 (στο 15q26.1; 11 εξώνια) κωδικοποιούν τις ισοκιτρικές δεϋδρογενάσες 1 και 2 αντίστοιχα. H IDH1 (414 αμινο-ξέα) βρίσκεται στο κυτταρόπλασμα και στα περοξισώματα ενώ η IDH2 (452 αμινοξέα) βρίσκεται στα μιτοχόνδρια. μεταλλάξεις των IDH1 και IDH2 πρωτοπεριγράφηκαν σε γλιώματα και στη συνέχεια σε ομλ και πιο σπάνια σε άλλα νεοπλάσματα.42 ςε μια μελέτη 200 ασθενών σε χρό-νια ή βλαστική φάση μυελοϋπερπλαστικού Νοσήματος, βρέθηκαν 9 μεταλλάξεις στα IDH (5 στο IDH1- R132C και R132S- και 4 στο IDH2- R140Q και R140W) αλλά καμία από αυτές δε βρέθηκε σε ασθενή με Αληθή Πολυ-κυτταραιμία ή ιδιοπαθή Θρομβοκυττάρωση. οι μεταλ-λάξεις αυτές συνυπήρχαν με την JAK2V617F.43
ε. Μεταλλάξεις του IKZF1(IKAROS family zinc finger 1)
το γονίδιο IKZF1 στο χρωμόσωμα 7p12 (επτά εξώ-νια) κωδικοποιεί τους μεταγραφικούς παράγοντες Ikaros που είναι ρυθμιστές διαφοροποίησης λεμφοκυττάρων. μεταλλάξεις του IKZF1 υπάρχουν συχνά στην ολλ και στη βλαστική φάση της Χμλ, υποδηλώνοντας παθογε-νετικό ρόλο στη λευχαιμιογένεση. μια μελέτη έδειξε ότι μεταλλάξεις του γονιδίου είναι σπάνιες στη χρόνια φάση των μυελοϋπερπλαστικών Νεοπλασμάτων αλλά ανευρί-σκονται σε ένα ποσοστό ~19% σε ασθενείς σε βλαστι-κή εκτροπή.44
στ. Mεταλλάξεις LNK (SH2B3)η πρωτεΐνη LNK προσδένεται στην πρωτεΐνη JAK2
μετά από διέγερση του υποδοχέα της θρομβοποιητίνης (τΡο) με αποτέλεσμα να αναστέλλει την ενεργοποίηση
της οδού JAK-STAT, αποτελώντας έτσι έναν εσωτερι-κό μηχανισμό ρύθμισης της οδού. Έχουν βρεθεί μεταλ-λάξεις στο γονίδιο LNK σε ασθενείς με μΥΝ, οι οποίες προκαλούν αναστολή λειτουργίας της πρωτεΐνης (μέσω δημιουργίας πρόωρου κωδικονίου λήξης). με τις μεταλ-λάξεις αυτές φαίνεται ότι χάνεται ο ρυθμιστικός ρόλος της πρωτεΐνης στην οδό JAK-STAT, επιτρέποντας τη συνεχή ενεργοποίησή της και προκαλώντας κλωνική αιμοποίη-ση και ανάπτυξη μυελοϋπερπλαστικού νεοπλάσματος.45
vI. ΓΕΝΕΤΙΚΗ πΡΟΔΙΑθΕΣΗ ΓΙΑ ΑΝΑπΤΥξΗ ΜΥΝ
Επιδημιολογικές μελέτες έδειξαν 5 με 7 πολλαπλάσιο κίνδυνο ανάπτυξης μΥΝ σε συγγενείς πρώτου βαθμού ασθενών με μΥΝ.46 Ένα μεγάλο ποσοστό των ασθενών και των συγγενών τους με μΥΝ είχαν στο χρωμόσωμα 9 στην περιοχή που εδράζεται το γονίδιο JAK2 έναν συγκε-κριμένο απλότυπο, που αναφέρεται ως 46/1 ή GGCC.47 ο τρόπος με τον οποίο ο συγκεκριμένος απλότυπος προδια-θέτει στην απόκτηση μεταλλάξεων του γονιδίου JAK2 και την ανάπτυξη μΥΝ, δεν είναι γνωστός, αλλά μια υπόθε-ση είναι ότι προδιαθέτει σε αυξημένο ρυθμό απόκτησης μεταλλάξεων στην περιοχή ή ότι οδηγεί σε πλεονέκτη-μα επιβίωσης του κυττάρου σε περίπτωση απόκτησης της σημειακής μετάλλαξης JAK2V617F.
vII. ΣΥΝΥπΑΡξΗ JAK2v617F ΜΕΤΑΛΛΑξΗΣ ΚΑΙ BCR-ABL ΧΙΜΑΙΡΙΚΟΥ ΓΟΝΙΔΙΟΥ
τα τελευταία χρόνια έχουν περιγραφεί περιπτώσεις ασθενών με μυελοϋπερπλαστικό νόσημα και ταυτόχρονη παρουσία μετάλλαξης JAK2V617F και χιμαιρικού γονιδί-ου BCR-ABL.48-50 παρά τις αρχικές εκτιμήσεις ότι οι δύο μεταλλάξεις δεν συνυπάρχουν.51 η συνύπαρξη των δύο μεταλλάξεων έχει περιγραφεί σε ασθενείς με Χμλ υπό θεραπεία με αναστολείς τυροσινικής κινάσης (τκιs) και σε ασθενείς με αρχική διάγνωση μΥΝ. ςτην πρώτη περί-πτωση φαίνεται ότι ή ο JAK2V617F κλώνος προϋπήρχε είτε δημιουργήθηκε κατά τη διάρκεια της θεραπείας με τκιs.52,53 Από την άλλη πλευρά κλινική εκδήλωση BCR-ABL+ Χρόνιας μυελογενούς λευχαιμίας σε ασθενή με JAK2V617F μΥΝ έχει επίσης περιγραφεί και φαίνεται ότι δεν εξαρτάται από την προηγούμενη λήψη κυτταρο-στατικής θεραπείας, καθώς τουλάχιστον σε μια περίπτω-ση ο ασθενής ελάμβανε μόνο ασπιρίνη.48,54
η συνύπαρξη των δύο μεταλλάξεων φαίνεται ότι είναι σπάνια. η αναζήτησή τους ταυτόχρονα δεν μπορεί ακόμα προς το παρόν να δικαιολογηθεί παρά μόνο σε ειδικές πε-ριπτώσεις όπως για παράδειγμα ανάπτυξη εικόνας μΥΝ σε ασθενή με Χμλ που λαμβάνει τκι και βρίσκεται σε

Γ. Γεωργίου και Β. Μπάρτζη96
Genetic aberrations in Ph negative myeloproliferative neoplasmsby Georgios Georgiou, Vasiliki Mpartzi
Research Laboratory of APPK
ABSTRACT: The myeloproliferative neoplasms are clonal diseases characterized by overproduction of mainly mature cells. In 2008 the World Health Organization (WHO) classified myeloproliferative neo-plasms (MPN) in specific categories. This new classification system was the result of the discovery of specific genetic lesions found in MPNs: JAK2 and MPL gene mutations. The detection of these lesions is applicable to everyday clinical practice as an integral component of the diagnostic approach to these diseases. After the discovery of these mutations, it became apparent that the pathogenesis of the various MPNs is much more complex. All MPNs are clonal diseases with the initial damage occurring to the primitive hematopoietic cell, causing cell overproduction, due to increased sensitivity or complete in-dependence from the influence of cytokines. The last few years several mutations in various genes have been detected in MPNs: mutations in TET2, ASXL1, CBL, IDH, etc. The involvement of these mutations in the initial pathogenesis of MPN is not known; unknown is also whether the combination of different mutations can lead to different phenotypic expression (eg a particular combination of mutations leads to polycythemia vera, another combination leads to essential thrombocythaemia etc). It remains to be shown whether this complex genetic abnormalities in MPNs can be useful in clinical practice with re-gard to the differential diagnosis, but also in targeted therapy.
Βιβλιογραφία 1. Swerdlow SH, Campo E, Harris NL, et al. WHO Classifica-
tion of Tumours of Haematopoietic and Lymphoid Tissues, Fourth Edition. WHO Classification of Tumours, Volume 2. IARC WHO Classification of Tumours, No 2.
2. Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005; 365:1054–1061.
3. Campbell LJ. Cytogenetics of myeloproliferative neoplasms. Methods Mol Biol. 2011; 730:89-98.
4. Vaidya R, CaramazzaD, Begna KH, et al, Monosomal karyotype in primary myelofibrosis is detrimental to both overall and leukemia-free survival. Blood. 2011 May 26; 117(21):5612-5615.
5. Spivak JL, Silver RT. The revised World Health Organi-zation diagnostic criteria for polycythemia vera, essential thrombocytosis and primary myelofibrosis: an alternative proposal. Blood. 2008; 112:231–239 .
6. Thiele J, Kvasnicka HM, Vardiman J. Bone marrow histo-pathology in the diagnosis of chronic myeloproliferative disorders: a forgotten pearl. Best Pract Res Clin Haematol. 2006; 19:413–437.
7. Kurdi M, Booz GW. JAK redux: a second look at the regula-tion and role of JAKs in the heart. Am J Physiol Heart Circ Physiol. 2009; 297: H1545–H1556.
8. Ghoreschi K, Laurence A, O’Shea JJ. Janus kinases in im-mune cell signaling. Immunol Rev. 2009; 228: 273–287.
9. Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol. 2007; 178:2623–2629.
10. Funakoshi-Tago M, Tago K, Abe M, et al. STAT5 activation is critical for the transformation mediated by myeloprolif-erative disorder-associated JAK2 V617F mutant. J Biol Chem. 2010; 285:5296–5307.
11. Schmitt-Graeff AH, Teo SS, Olschewski M, et al. JAK2V617F mutation status identifies subtypes of refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Haematologica. 2008; 93:34–40.
12. Steensma DP, McClure RF, Karp JE, et al. JAK2 V617F is a rare finding in de novo acute myeloid leukemia, but STAT3 activation is common and remains unexplained. Leukemia. 2006; 20:971–978.
13. Steensma DP, Dewald GW, Lasho TL, et al. The JAK2 V617F activating tyrosine kinase mutation is an infrequent event in both ‘atypical’ myeloproliferative disorders and myelodysplastic syndromes. Blood. 2005; 106:1207–1209.
14. Inami M, Yamaguchi H, Hasegawa S, et al. Analysis of the exon 12 and 14 mutations of the JAK2 gene in Philadelphia chromosome-positive leukemia. Leukemia. 2008; 22:216.
15. Melzner I, Weniger MA, Menz CK, et al. Absence of the JAK2 V617F activating mutation in classical Hodgkin lymphoma and primary mediastinal B-cell lymphoma. Leukemia. 2006; 20:157–158.
16. Passamonti F, Rumi E, Pietra D, et al. JAK2 (V617F) mutation in healthy individuals. Br J Haematol. 2007;
πλήρη κυτταρογενετική και μείζονα/πλήρη μοριακή ύφε-ση ή σε ασθενή με μΥΝ JAK2V617+ που αναπτύσσει
αιματολογική εικόνα Χρόνιας μυελογεμούς λευχαιμίας.

Γενετικές βλάβες στα Ph- μυελοϋπερπλαστικά νοσήματα 97
136:678–679. 17. Wernig G, Mercher T, Okabe R, et al. Expression of
Jak2V617F causes a polycythemia vera-like disease with associated myelofibrosis in a murine bone marrow transplant model. Blood. 2006; 107:4274–4281.
18. Tiedt R, Hao-Shen H, Sobas MA, et al. Ratio of mutant JAK2–V617F to wild-type Jak2 determines the MPD phe-notypes in transgenic mice. Blood. 2008; 111:3931–3940.
19. Jamal R, Belisle C, Lessard MC, et al. Evidence suggesting the presence of a stem cell clone anteceding the acquisition of the JAK2-V617F mutation. Leukemia. 2008; 22:1472–1474.
20. Antonioli E, Guglielmelli P, Poli G, et al. Influence of JAK2V617F allele burden on phenotype in essential throm-bocythemia. Haematologica. 2008; 93:41–48.
21. Kittur J, Knudson RA, Lasho TL, et al. Clinical correlates of JAK2V617F allele burden in essential thrombocythemia. Cancer. 2007; 109:2279–2284.
22. Campbell PJ, Scott LM, Buck G, et al. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: a prospective study. Lancet. 2005; 366:1945–1953.
23. Barosi G, Bergamaschi G, Marchetti M, et al. JAK2 V617F mutational status predicts progression to large splenomegaly and leukemic transformation in primary myelofibrosis. Blood. 2007; 110:4030–4036.
24. Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 muta-tions in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007; 356:459–468.
25. Teofili L, Giona F, Torti L, Cenci T, Ricerca BM, Rumi C, et al.Hereditary thrombocytosis caused by MPLSer505Asn is associated with ahigh thrombotic risk, splenomegaly and progression to bone marrow fibrosis. Haematologica. 2009;95(1):57-64.
26. Moliterno AR, Williams DM, Gutierrez-Alamillo LI, et al. Mpl Baltimore: a thrombopoietin receptor polymorphism associated with thrombocytosis. Proc Natl Acad Sci USA. 2004; 101:11444–11447.
27. Hussein K, Bock O, Theophile K, et al. MPLW515L muta-tion in acute megakaryoblastic leukaemia. Leukemia. 2009; 23:852–855.
28. Pardanani AD, Levine RL, Lasho T, et al. MPL515 muta-tions in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006; 108:3472–3476.
29. Lasho TL, Pardanani A, McClure RF, et al. Concurrent MPL515 and JAK2V617F mutations in myelofibrosis: chronology of clonal emergence and changes in mutant allele burden over time. Br J Haematol. 2006; 135:683–687.
30. Vannucchi AM, Antonioli E, Guglielmelli P, et al. Characte-ristics and clinical correlates of MPL 515W>L/K mutation in essential thrombocythemia. Blood. 2008; 112:844–847.
31. Guglielmelli P, Pancrazzi A, Bergamaschi G, et al. Anaemia characterises patients with myelofibrosis harbouring Mpl mutation. Br J Haematol. 2007; 137:244–247.
32. Elliott MA. Chronic neutrophilic leukemia and chronic myelomonocytic leukemia: WHO defined. Best Pract Res Clin Haematol. 2006; 19:571–593.
33. Hirsch-Ginsberg C, LeMaistre AC, Kantarjian H, et al. RAS mutations are rare events in Philadelphia chromosome-neg-ative/bcr gene rearrangement-negative chronic myelogenous leukemia, but are prevalent in chronic myelomonocytic leukemia. Blood. 1990; 76:1214–..............
34. Tartaglia M, Niemeyer CM, Fragale A, et al. Somatic mu-tations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003; 34:148–150.
35. Steensma DP, Caudill JS, Pardanani A, et al. MPL W515 and JAK2 V617 mutation analysis in patients with refractory anemia with ringed sideroblasts and an elevated platelet count. Haematologica. 2006; 91:ECR57.
36. Abdel-Wahab O, Mullally A, Hedvat C, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood. 2009; 114:144–147.
37. Abdel-Wahab O, Manshouri T, Patel J, et al. Genetic analysis of transforming events that convert chronic myeloprolifera-tive neoplasms to leukemias. Cancer Res. 2010; 70:447–452.
38. Bandi SR, Brandts C, Rensinghoff M, et al. E3 ligase-defective Cbl mutants lead to a generalized mastocytosis and myeloproliferative disease. Blood. 2009; 114:4197–4208.
39. Grand FH, Hidalgo-Curtis CE, Ernst T, et al. Frequent CBL mutations associated with 11q acquired uniparental disomy in myeloproliferative neoplasms. Blood. 2009; 113:6182–6192.
40. Loh ML, Sakai DS, Flotho C, et al. Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood. 2009; 114:1859–1863.
41. Makishima H, Cazzolli H, Szpurka H, et al. Mutations of e3 ubiquitin ligase cbl family members constitute a novel common pathogenic lesion in myeloid malignancies. J Clin Oncol. 2009; 27:6109–6116.
42. Ward PS, Patel J, Wise DR, et al. The common feature of leukemia-associated idh1 and idh2 mutations is a neo-morphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010; 17:225–234.
43. Pardanani A, Lasho T, Finke C, et al. IDH1 and IDH2 muta-tion analysis in chronic and blast phase myeloproliferative neoplasms. Leukemia 2010.
44. Jager R, Gisslinger H, Berg T, et al. Deletions of the Tran-scription Factor Ikaros in Myeloproliferative Neoplasms at Transformation to Acute Myeloid Leukemia. ASH Annual Meeting Abstracts. 2009; 114:435.
45. Oh ST, Simonds EF, Jones C, et al., Novel mutations in the inhibitory adaptor protein LNK drive JAK-STAT signaling in patients with myeloproliferative neoplasms. Blood. 2010 Aug 12; 116(6):988-92.
46. Landgren O, Goldin LR, Kristinsson SY, et al. Increased risks of polycythemia vera, essential thrombocythemia, and myelofibrosis among 24,577 first-degree relatives of 11,039 patients with myeloproliferative neoplasms in Sweden. Blood. 2008;112(6):2199-2204.
47. Olcaydu D, Skoda RC, Looser R, et al. The ‘GGCC’ haplotype of JAK2 confers susceptibility to JAK2 exon 12 mutationpositive polycythemia vera. Leukemia. 2009;

Γ. Γεωργίου και Β. Μπάρτζη98
23(10):1924-1926. 48. Jallades L, Hayette S, Tigaud I, et al. Emergence of Therapy-
unrelated CML on a Background of BCR-ABL-negative JAK2 V617F-positive Chronic Idiopathicmyelofibrosis. Leuk Res. 2008; 32:1608–1610.
49. Bornhäuser M, Mohr B, Oelschlaegel U, et al. Concurrent JAK2 (V617F) Mutation and BCR-ABL Translocation within Committed Myeloid Progenitors in Myelofibrosis. Leukemia. 2007; 21:1824–1826.
50. Mello Conchon M, Costa JL, Novaes M , et al. Simultaneous detection of JAK2 V617F Mutation and Bcr-Abl Transloca-tion in a Patient with Chronic Myelogenous Leukemia. Int J Hematol. 2008; 88:243–245.
51. Jelinek J, Oki Y, Gharibyan V, et al. JAK2 Mutation 1849G>T
is Rare in Acute Leukemias but Can be Found in CMML, Philadelphia Chromosome-negative CML, and Megakaryo-cytic Leukemia. Blood. 2005; 106:3370–3373.
52. Kramer A, Reiter A, Kruth J, et al. JAK2-V617F Mutation in a Patient with Philadelphia-chromosomepositive Chronic Myeloid Leukaemia. Lancet Oncol. 2007; 8:658–660.
53. Inami M, Inokuchi K, Okabe M, et al. Polycythemia As-sociated with the JAK2V617F Mutation Emerged During Treatment of Chronic Myelogenous Leukemia. Leukemia, 2007; 21:1103–1104.
54. Scott LM, Campbell PJ, Baxter EJ, et al. The V617F JAK2 Mutation is Uncommon in Cancers and in Myeloid Malig-nancies other than the Classic Myeloproliferative Disorders. Blood. 2005; 106:2920–2921.

Θεραπευτική αντιμετώπιση των Ph- Μυελοϋπερπλαστικών Νεοπλασμάτων (Ph-ΜΥΝ)
Δέσποινα Παντελίδου1, Μαρία Παπαϊωάννου1
ΠΕΡΊληΨη: την τελευταία δεκαετία η περιγραφή γενετικών βλαβών που παρατηρούνται στα Ph αρνη-τικά μυελοϋπερπλαστικά Νεοπλάσματα (Ph-μΥΝ), με πλέον αντιπροσωπευτική τη μετάλλαξη V617F στο εξώνιο 14 της JAK2 κινάσης, συνέβαλε σημαντικά στην κατανόηση της παθοφυσιολογίας των νο-σημάτων αυτών, χωρίς ωστόσο να επηρεάσει την θεραπευτική τους αντιμετώπιση. Ενόψει των νέων γε-νετικών και βιολογικών δεδομένων, το Ευρωπαϊκό Δίκτυο ELN (European LeukemiaNet), στα πλαίσια της βελτιστοποίησης και εναρμόνισης των θεραπευτικών χειρισμών, των κριτηρίων ανταπόκρισης και παρακολούθησης των ασθενών με Ph-μΥΝ, πρότεινε αντίστοιχες κατευθυντήριες οδηγίες που συνο-ψίζονται παρακάτω. οι ασθενείς με Αληθή Πολυκυτταραιμία (ΑΠ) και ιδιοπαθή Θρομβοκυτταραιμία (ιΘ) διακρίνονται σε υψηλού κινδύνου εάν η ηλικία τους είναι μεγαλύτερη των 65 ετών ή εάν αναφέ-ρουν ιστορικό θρομβωτικού η αιμορραγικού επεισοδίου. Ασθενείς υψηλού κινδύνου με ΑΠ αντιμετω-πίζονται αρχικά με αφαιμάξεις, χαμηλή δόση ασπιρίνης και κυτταροξική θεραπεία με υδροξυουρία η με ιντερφερόνη σε κάθε ηλικία. Ασθενείς υψηλού κινδύνου με ιΘ αντιμετωπίζονται με κυτταροτοξική θε-ραπεία όπως υδροξυουρία. η παρακολούθηση των ασθενών συνιστάται να γίνει σύμφωνα με τα κριτή-ρια του ELN. η θεραπευτική προσέγγιση των ασθενών με Πρωτοπαθή μυελοΐνωση (Πμι), βασίζεται στη συμπτωματολογία, στα κλινικά και εργαστηριακά ευρήματα των ασθενών, που καθορίζουν ομά-δες κινδύνου. τα κορτικοστεροειδή, τα ανδρογόνα, οι παράγοντες διέγερσης της ερυθροποίησης και τα ανοσοτροποιητικά φάρμακα συνιστώνται σαν θεραπευτικές επιλογές για την αντιμετώπιση των ασθε-νών με βαριά αναιμία ενώ η υδροξυουρία αποτελεί θεραπεία πρώτης γραμμής για την αντιμετώπιση της σπληνομεγαλίας. Ενδείξεις σπληνεκτομής είναι η συμπτωματική πυλαία υπέρταση, η ανθεκτική και επώδυνη σπληνομεγαλία και οι συχνές μεταγγίσεις ερυθρών. η αλλογενής μεταμόσχευση αιμοποιητι-κών κυττάρων λόγω των σοβαρών επιπλοκών της αποτελεί δυνητική επιλογή σε ασθενείς που πληρούν τα κριτήρια υποψηφιότητας για μεταμόσχευση ενώ η διάμεση επιβίωσή τους προβλέπεται να είναι μι-κρότερη από 5 έτη. Ειδικές ομάδες ασθενών με μΥΝ που χρήζουν εξατομικευμένου χειρισμού αποτε-λούν τα παιδιά και οι εγκυμονούσες γυναίκες.
Haema 2012; 3(1): 99-108 Copyright EAE
1Αιματολογικό τμήμα, Α΄ Παθολογική Πανεπιστημιακή κλινική Α.Π.Θ., ΠγΝ ΑΧΕΠΑ, Θεσσαλονίκη
Διεύθυνση Αλληλογραφίας: Δ. Παντελίδου, e-mail: [email protected] μ. Παπαϊωάννου, e-mail: [email protected]Αιματολογικό τμήμα, Α΄ Πανεπιστημιακή Παθολογική κλινική, ΠΠγΝΑΧΕΠΑ, ς. κυριακίδη 1, 54636, Θεσσαλονίκη
Aνασκόπηση
Εισαγωγήη ανακάλυψη ότι η πρωτεϊνική τυροσινική κινάση
JAK2 είναι μεταλλαγμένη (V617F) σε περισσότερους από 90% των ασθενών με αληθή πολυκυτταραιμία και περίπου στο 60% των ασθενών με ιδιοπαθή θρομβοκυτ-ταραιμία και πρωτοπαθή μυελοΐνωση, διαμόρφωσε νέα μοντέλα προσέγγισης στην κατανόηση των κλινικών και
βιολογικών χαρακτηριστικών των Ph- κλασικών μυελο-ϋπερπλαστικών νεοπλασμάτων.1 ο σημαντικός βιολο-γικός ρόλος της μεταλλαγμένης JAK2 υπαγόρευσε την ανάπτυξη αναστολέων της,2 με προοπτική την κλινική εφαρμογή τους. Ωστόσο, στην τρέχουσα χρονική περί-οδο η θεραπευτική αντιμετώπιση των Ph-μΥΝ συνεχί-ζει να αποτελεί πρόκληση. Ενόψει των νέων γενετικών και βιολογικών δεδομένων αναφορικά με τα Ph-μΥΝ, το Ευρωπαϊκό Δίκτυο ELN (European LeukemiaNet), στα πλαίσια της βελτιστοποίησης και εναρμόνισης των θεραπευτικών χειρισμών, των κριτηρίων ανταπόκρισης και παρακολούθησης των ασθενών με Ph-μΥΝ, πρότεινε αντίστοιχες κατευθυντήριες οδηγίες που δημοσιεύτηκαν

Δ. Παντελίδου και Μ. Παπαϊωάννου100
πρόσφατα. ςτις προτάσεις του ELN θα στηριχθεί κατά κύριο λόγο η παρούσα ανασκόπηση.
Ι. θεραπευτική αντιμετώπιση αληθούς πολυκυτταραιμίας (Απ)
ςτόχοι της θεραπείας των ασθενών με ΑΠ αποτελούν: (1) η αποφυγή εμφάνισης θρομβωτικού επεισοδίου και αιμορραγικών επιπλοκών και (2) η ελαχιστοποίηση του κινδύνου εξέλιξης σε μεταπολυκυτταραιμική μυελοΐνω-ση και οξεία λευχαιμία. Επιπλέον, με τη θεραπεία επι-διώκεται ο έλεγχος των συστηματικών εκδηλώσεων της νόσου και η πρόληψη επιπλοκών (θρομβωτικές, αιμορ-ραγικές) σε καταστάσεις υψηλού κινδύνου όπως η κύη-ση και οι χειρουργικές επεμβάσεις.3,4
ο ορθός σχεδιασμός του θεραπευτικού χειρισμού ασθενών με ΑΠ, προϋποθέτει την κατάταξή τους σε ομά-δες κινδύνου. Πολυκεντρικές μελέτες έδειξαν ότι η μεγά-λη ηλικία και το θετικό ιστορικό θρόμβωσης αποτελούν ανεξάρτητους παράγοντες για θρόμβωση.5-7 Ως εκ τού-του οι ασθενείς μεγάλης ηλικίας (>60 ετών) ή με ιστο-ρικό θρομβωτικού επεισοδίου κατατάσσονται σε ομάδα υψηλού κινδύνου. οι υπόλοιποι συνιστούν την ομάδα χαμηλού κινδύνου.
Ανεξάρτητα από την ομάδα κινδύνου , σε όλους τους ασθενείς που πάσχουν από ΑΠ συστήνεται η διακοπή του καπνίσματος και η ρύθμιση άλλων παραγόντων κινδύνου για θρόμβωση όπως: μεταβολικό σύνδρομο, σακχαρώδης διαβήτης, αρτηριακή υπέρταση, υπερχοληστεριναιμία.
Ia. Θεραπεία πρώτης γραμμής. Όλοι οι ασθενείς με ΑΠ πρέπει να αντιμετωπίζονται αρχικά με αφαιμάξεις και στόχο την επίτευξη αιματοκρίτη <45%. η πολυκε-ντρική μελέτη CYTO-PV, που δεν έχει ολοκληρωθεί ακό-μη, προσδοκά να απαντήσει στο ερώτημα εάν η επίτευξη αιματοκρίτη <45% πλεονεκτεί από αιματοκρίτη μεταξύ 45%-50%.8 οι αφαιμάξεις δε συνδέονται με αυξημένο κίνδυνο θρομβώσεων6,9 ενώ, χαμηλές δόσεις ασπιρίνης ελαττώνουν τα θρομβοεμβολικά επεισόδια και τους θα-νάτους που σχετίζονται με αυτά.10 Έτσι, ασπιρίνη σε δό-σεις 75-100mg/ημέρα, συνιστάται σε όλους τους ασθενείς με ΑΠ που δεν έχουν ιστορικό σοβαρής αιμορραγίας ή γαστρικής δυσανεξίας στο φάρμακο.4,11 μετά την αρχι-κή εφαρμογή των αφαιμάξεων, ο περαιτέρω χειρισμός εξαρτάται από την ομάδα κινδύνου. οι ασθενείς χαμη-λού κινδύνου παραμένουν σε αφαιμάξεις και χαμηλές δόσεις ασπιρίνης ενώ οι ασθενείς υψηλού κινδύνου υπο-βάλλονται σε αγωγή με κυτταροστατικά φάρμακα. Επι-πλέον ενδείξεις για έναρξη κυτταροστατικής θεραπείας είναι οι συχνές ανάγκες αφαιμάξεων ή η μη ανοχή τους, η συμπτωματική ή επιδεινούμενη σπληνομεγαλία, ο υψη-λός αριθμός αιμοπεταλίων (>1500 × 109/L), η επιδεινού-μενη λευκοκυττάρωση και τα σοβαρά συμπτώματα που σχετίζονται με τη νόσο.1 η υδροξυουρία και επί δυσανε-
ξίας η ιντερφερόνη αποτελούν πρώτης γραμμής μυελο-κατασταλτικές θεραπείες.12 η λευχαιμογόνος δράση της υδροξυουρίας σε ασθενείς με ΑΠ αποτελεί πεδίο αντι-παράθεσης13-15 και η χορήγησή της χρειάζεται προσοχή σε ασθενείς νεαρής ηλικίας, σε άτομα στα οποία προη-γήθηκε μυελοκατασταλτική αγωγή ή έχουν κυτταρογε-νετικές ανωμαλίες.12 η μπουσουλφάνη θα μπορούσε να χορηγηθεί σαν κυτταροστατική θεραπεία υπό ορισμένες συνθήκες, όπως σε υπερήλικες ασθενείς.
Ιβ. Έλεγχος ανταπόκρισης. η ανταπόκριση στην κυτ-ταροστατική θεραπεία προσδιορίζεται από τα κριτήρια που έχουν προταθεί από το ELN για την επίτευξη φυσι-ολογικών αιματολογικών παραμέτρων και την εξαφά-νιση συμπτωμάτων και κλινικών σημείων της νόσου.16 (Πίνακας 1).
Ιγ. Αλλαγή θεραπείας και δεύτερης γραμμής θεραπεία. ο αυξημένος κίνδυνος εμφάνισης οξείας λευχαιμίας, αν μετά την υδροξυουρία ακολουθήσει αγωγή με άλλο μυ-ελοκατασταλτικό φάρμακο, υπαγορεύει την επιλογή μη λευχαιμογόνων ουσιών, όπως ιντερφερόνη, για θεραπεία δεύτερης γραμμής.17 Έτσι, σε ασθενείς που ελάμβαναν υδροξυουρία και εμφανίζουν δυσανεξία ή ανεπαρκή αντα-πόκριση 18 η ιντερφερόνη θα αποτελέσει θεραπεία δεύτε-ρης γραμμής και το αντίθετο.
πίνακας 1. Κριτήρια κλινικής και αιματολογικής ανταπόκρισης στην κυτταροστατική θεραπεία της Αληθούς πολυκυτταραιμίας (ELN)16
Πλήρης ανταπόκριση
Αιματοκρίτης <45% χωρίς αφαιμάξεις ΚΑΙ
Αριθμός αιμοπεταλίων <400 x 109/L ΚΑΙ
WBC ≤10 x 109/LΚΑΙΦυσιολογικό μέγεθος σπληνός σε
απεικονιστική μέθοδοΚΑΙΑπουσία συμπτωμάτων
σχετιζόμενων με τη νόσο*
Μερική ανταπόκριση
Σε ασθενείς που δεν πληρούν τα κριτήρια για πλήρη ανταπόκριση
Αιματοκρίτης <45% χωρίς αφαιμάξεις ή
Απάντηση σε ≥3 από τα άλλα κριτήρια
Χωρίς ανταπόκριση
Οποιαδήποτε ανταπόκριση που δεν πληρεί τα κριτήρια της μερικής
* Στα συμπτώματα που σχετίζονται με τη νόσο συμπεριλαμβάνονται οι μικροαγγειακές διαταραχές, ο κνησμός και η κεφαλαλγία.

Θεραπευτική αντιμετώπιση των Ph- Μυελοϋπερπλαστικών Νεοπλασμάτων (Ph-ΜΥΝ) 101
Κριτήρια ανθεκτικότητας ή/και δυσανεξίας στην υδροξυουρία σε ασθενείς με ΑΠ18
• Ανάγκη αφαιμάξεων για διατήρηση του αιματοκρίτη σε επίπεδα <45% μετά από 3 μήνες αγωγής με υδρο-ξυουρία, σε δόση τουλάχιστον 2gr/ημέρα ή
• μη ελεγχόμενη υπερπλασία μυελού (π.χ. αριθμός αι-μοπεταλίων >400 × 109/L και αριθμός WBC >10 × 109/L) μετά από 3 μήνες αγωγής με υδροξυουρία, σε δόση τουλάχιστον 2gr/ημέρα ή
• Αποτυχία περιορισμού σπληνομεγαλίας (ψηλαφητός σπλήνας >10 εκ. από το πλευρικό τόξο) κατά 50%, καταγεγραμένης με ψηλάφηση ή αποτυχία πλήρους εξαφάνισης συμπτωμάτων σχετιζόμενων με τη σπλη-νομεγαλία μετά από 3 μήνες αγωγής με υδροξυουρία σε δόση τουλάχιστον 2gr/ημέρα ή
• Απόλυτος αριθμός ουδετεροφίλων <1,0 × 109/L ή αριθμός αιμοπεταλίων <100 × 109/L ή αιμοσφαιρίνη <10gr/dL υπο αγωγή με τη χαμηλότερη δόση υδρο-ξυουρίας που απαιτείται για την επίτευξη πλήρους ή μερικής κλινικής και αιματολογικής ανταπόκρισης ή
• Παρουσία ελκών στα πόδια ή άλλης σημαντικής μη αιματολογικής τοξικότητας σχετιζόμενης με την υδρο-ξυουρία, όπως βλεννογονοδερματικές εκδηλώσεις, συμπτώματα απο το πεπτικό σύστημα, πνευμονίτιδα ή πυρετός σε οποιαδήποτε δόση υδροξυουρίας.με τη χορήγηση ιντερφερόνης, η συνολική ανταπό-
κριση με επίτευξη αιματοκρίτη <45%, χωρίς αφαιμάξεις, διαπιστώνεται σε 50% των ασθενών, η μείωση του μεγέ-θους του σπληνός σε 77% και η ελάττωση του κνησμού σε 75%.19 η μακράς διάρκειας (μέση διάρκεια 13 έτη) θεραπεία με ιντερφερόνη σε 55 ασθενείς με ΑΠ έδειξε επίτευξη του θεραπευτικού στόχου μετά από 1-2 έτη θε-ραπείας, δυνατότητα ελάττωσης της δόσης για συντήρη-ση και απουσία θρομβοαιμορραγικών επεισοδίων κατά τη διάρκεια παρακολούθησης.20 οι πεγκυλιωμένες μορφές ιντερφερόνης-α (Peg-IFNα) επιτρέπουν την εβδομαδιαία χορήγηση και δυνητικά βελτιώνουν τη συμμόρφωση των ασθενών. οι Kiladjian και συν. παρουσίασαν σε πολυκε-ντρική μελέτη μια μικρή ομάδα ασθενών με ΑΠ, στους οποίους χορηγήθηκε η Peg-IFN-α2α και παρατηρήθηκε ελάττωση της έκφρασης της μεταλλαγμένης μορφής του JAK2 σε 24 από 27 ασθενείς (89%) και εξαφάνισή της σε έναν ασθενή, μετά από θεραπεία διάρκειας ενός έτους.21,22 Αντίθετα, σε άλλη μελέτη με ασθενείς πάσχοντες από ΑΠ και ιΘ τα αποτελέσματα που αφορούσαν στην ελάττω-ση της έκφρασης της μεταλλαγμένης μορφής του JAK2 ήταν ιδιαίτερα περιορισμένα.23
οι ασθενείς χαμηλού κινδύνου στην ηλικία των 60 ετών και πάνω ή που εκδήλωσαν ένα σοβαρό θρομβω-τικό ή αιμορραγικό επεισόδιο υποβάλλονται σε κυττα-ροτοξική θεραπεία. η ασπιρίνη θα πρέπει να αποσυρθεί από τη θεραπεία εάν εκδηλωθεί ένα σοβαρό αιμορραγι-κό επεισόδιο, κυρίως από το γαστρεντερικό σύστημα. η
μπουσουλφάνη και ο 32Ρ αποτελούν θεραπευτικές επι-λογές τελευταίας γραμμής σε ασθενείς με ελαττωμένο προσδόκιμο επιβίωσης.24-26
ΙΙ. θεραπευτική αντιμετώπιση της Ιδιοπαθούς θρομβοκυτταραιμίας (Ιθ)
η ιΘ, το συχνότερο από τα Ph- μυελοϋπερπλαστικά νεοπλάσματα, χαρακτηρίζεται από θρομβοκυττάρωση, υπερπλασία των μεγακαρυοκυττάρων στο μυελό και τάση για εμφάνιση αγγειακών επιπλοκών, όπως: θρομβώσεις, αιμορραγίες και μικροαγγειακές διαταραχές. για το λό-γο αυτό η ιΘ επηρεάζει περισσότερο την ποιότητα ζωής των ασθενών από ότι την επιβίωσή τους.27 Έτσι, η θερα-πευτική στρατηγική της αντιμετώπισης της νόσου καθο-ρίζεται από τον κίνδυνο του ασθενούς να αναπτύξει τις παραπάνω επιπλοκές και ο στόχος της θεραπείας θα πρέ-πει να είναι η πρόληψη θρομβωτικών και αιμορραγικών επεισοδίων, χωρίς αύξηση του κινδύνου για εξέλιξη της νόσου.28 Προοπτικές και αναδρομικές μελέτες έδειξαν ότι ηλικία μεγαλύτερη από 60 έτη ή προηγούμενο θρομ-βωτικό επεισόδιο αποτελούν κυρίαρχους παράγοντες που προβλέπουν αγγειακές επιπλοκές29-31 και καθορίζουν ομά-δα ασθενών υψηλού κινδύνου. ςαφής συσχέτιση μεταξύ βαθμού θρομβοκυττάρωσης και θρομβωτικών επιπλοκών δεν έχει βρεθεί, αν και σε πολύ υψηλές τιμές αιμοπεταλί-ων (>1500 × 109/L) μπορεί να εμφανιστεί επίκτητη νόσος von Willebrand και αιμορραγική διάθεση.32
ΙΙα. Θεραπεία πρώτης γραμμής η εμφάνιση θρομβώσε-ων σε ασθενείς χαμηλού κινδύνου (κάτω των 60 ετών, χω-ρίς ιστορικό θρομβωτικού επεισοδίου) είναι παρόμοια με αυτή που παρατηρείται στον υγιή πληθυσμό.33,34 Εντούτοις, οι Alvarez-Larran και συν. σε μια πρόσφατη αναδρομι-κή μελέτη αναλύοντας τα αποτελέσματα της αντιαιμο-πεταλιακής θεραπείας (κυρίως χαμηλή δόση ασπιρίνης) σε 300 ασθενείς με χαμηλού κινδύνου ιΘ, έδειξαν ότι η θεραπεία αυτή μείωσε τον κίνδυνο φλεβικής θρόμβωσης σε JAK2V617F θετικούς ασθενείς και τον κίνδυνο αρτη-ριακής θρόμβωσης στους ασθενείς με καρδιαγγειακούς παράγοντες κινδύνου, ενώ συνδεόταν με αιμορραγικά επεισόδια σε ασθενείς με αριθμό αιμοπεταλίων >1000 x 109/L.35 καλή ρύθμιση των γενικών καρδιαγγειακών πα-ραγόντων κινδύνου (αρτηριακή υπέρταση, σακχαρώδης διαβήτης, υπερχοληστεριναιμία), συμπεριλαμβανομένης της διακοπής του καπνίσματος, επιβάλλεται σε κάθε ασθε-νή με ιΘ, ανεξαρτήτως ομάδας κινδύνου.36
η υδροξυουρία αποτελεί πρώτη επιλογή κυτταροστα-τικής θεραπείας σε ασθενείς υψηλού κινδύνου μετά την τυχαιοποιημένη μελέτη των Cortelazzo και συν.37 η με-λέτη έδειξε ότι η ομάδα των ασθενών υπο υδροξυουρία εμφανίζει μειωμένο αριθμό θρομβώσεων σε σύγκριση με την ομάδα των ασθενών χωρίς θεραπεία (παρακολούθη-ση 27 μηνών, συχνότητα θρομβώσεων 3,6% έναντι 24%

Δ. Παντελίδου και Μ. Παπαϊωάννου102
αντίστοιχα).37 τα ευρήματα αυτά επιβεβαιώθηκαν από τα αποτελέσματα της μελέτης MRCPT-1 (τυχαιοποιη-μένη μελέτη που συνέκρινε την υδροξυουρία σε συνδυ-ασμό με ασπιρίνη έναντι της αναγρελίδης σε συνδυασμό με ασπιρίνη, σε 809 ασθενείς με υψηλού κινδύνου ιΘ).38 ο συνδυασμός υδροξυουρίας με ασπιρίνη αποδείχθηκε ότι πλεονεκτεί του συνδυασμού αναγρελίδης με ασπιρί-νη, στην ελάττωση των συνολικών αγγειακών θρομβω-τικών επεισοδίων και στη μετάπτωση σε μυελοΐνωση. η αναγρελίδη έδειξε ισοδύναμη δραστικότητα με την υδρο-ξυουρία στη μείωση του αριθμού των αιμοπεταλίων. Ως εκ τούτου η υπεροχή της υδροξυουρίας θα μπορούσε να εξηγηθεί απο την ικανότητά της να μειώνει όχι μόνο τα αιμοπετάλια αλλά και τα ερυθρά και λευκά αιμοσφαίρια.39 η ανησυχία για πιθανή μεταλλαξιγόνο δράση της υδρο-ξυουρίας δεν έχει σαφώς αποδειχθεί. ουσιαστικά, πολλές από τις περιπτώσεις μετάπτωσης σε μυελοδυσπλαστικό σύνδρομο ή οξεία λευχαιμία εμφανίστηκαν σε ασθενείς που είχαν εκτεθεί επιπλέον και σε άλλους κυτταροστα-τικούς παράγοντες.40 Εντούτοις, η χρήση της υδροξυου-ρίας ως θεραπεία πρώτης γραμμής σε νεαρά άτομα (<40 έτη) θα πρέπει να εφαρμοστεί μετά από σοβαρή σκέψη. Επιπλέον, επειδή η σοβαρή θρομβοκυττάρωση (>1500 x 109/L) αποτελεί παράγοντα κινδύνου για αιμορραγία, θα μπορούσε να συζητηθεί η χορήγηση κυτταροστατικής θε-ραπείας και στην ομάδα αυτών των ασθενών.41
Όλοι οι ασθενείς με ιΘ και μικροαγγειακές διαταρα-χές όπως ερυθρομελαλγία, ακροπαραισθησία, ισχαιμία δακτύλων, νευρολογικά συμπτώματα (παροδικά ισχαι-μικά επεισόδια, κεφαλαλγία τύπου ημικρανίας, ζάλη), διαταραχές της όρασης (παροδική τύφλωση, αμαύρωση της όρασης, φωταψίες) πρέπει να λαμβάνουν χαμηλές δόσεις ασπιρίνης.42,43
ΙΙβ. Έλεγχος ανταπόκρισης στη θεραπεία. η ανταπό-κριση στη θεραπεία προσδιορίζεται από την επίτευξη φυσιολογικών αιματολογικών παραμέτρων και την εξα-φάνιση συμπτωμάτων και κλινικών σημείων της νόσου (Πίνακας 2). το μυελικό αναρρόφημα και η οστεομυελι-κή βιοψία είναι χρήσιμα όχι για την κλινική παρακολού-θηση της νόσου αλλά για την εκτίμηση μετάπτωσης σε μυελοΐνωση ή οξεία λευχαιμία.28
ΙΙγ. Θεραπεία δεύτερης γραμμής. Ασθενείς με ιΘ που λαμβάνουν περισσότερους από έναν κυτταροτοξικούς πα-ράγοντες βρίσκονται σε σημαντικά υψηλότερο κίνδυνο εμφάνισης μυελοδυσπλαστικού συνδρόμου ή οξείας λευ-χαιμίας.44 ςε ασθενείς λοιπόν που εμφανίζουν αντίσταση ή δυσανεξία στην υδροξυουρία45 συνιστάται η χορήγηση μη λευχαιμογόνων παραγόντων όπως αναγρελίδη. η ανα-γρελίδη μειώνει τον αριθμό των αιμοπεταλίων αναστέλ-λοντας τη διαφοροποίηση των μεγακαρυοκυττάρων. η μικρή επίδρασή της στη συσσώρευση των αιμοπεταλίων είναι η πιθανότερη εξήγηση για τα αυξημένα αιμορρα-γικά επεισόδια που καταγράφηκαν στο συνδυασμό ανα-γρελίδης-ασπιρίνης στη μελέτη MRCPT-1.38 Πρόδρομα
δεδομένα της προοπτικής μελέτης ANAHYDRET που συ-γκρίνει την αναγρελίδη με την υδροξυουρία στην αντιμε-τώπιση της ιΘ, έδειξαν παρόμοια δραστικότητα των δυο ουσιών αν και η διάρκεια παρακολούθησης των ασθενών ήταν μόλις 12 μήνες.46 τα αυξημένα ποσοστά δευτεροπα-θούς μυελοΐνωσης που παρατηρήθηκαν στο συνδυασμό αναγρελίδης με ασπιρίνη, στη μελέτη MRCPT-1, εμφα-νίστηκαν μετά από παρακολούθηση μέσης διάρκειας 39 μηνών.38 ςτις ανεπιθύμητες εκδηλώσεις του φαρμάκου συμπεριλαμβάνονται αίσθημα παλμών (27%), ταχυκαρ-δία και άλλες αρρυθμίες (<10%), συμφορητική καρδιακή ανεπάρκεια (2%), κατακράτηση υγρών και ήπια αναιμία.47 η ιντερφερόνη άλφα (IFN-α) αποτελεί πειραματική θε-ραπεία και πρέπει να χορηγείται σε ειδικές κατηγορίες ασθενών, όπως νεαρές γυναίκες σε αναπαραγωγική ηλι-κία και ασθενείς σε κύηση, στις οποίες είναι αναγκαία η μείωση του αριθμού των αιμοπεταλίων.48 Ωστόσο, οι ανεπιθύμητες εκδηλώσεις της IFN-α (πυρετός, γριππώ-δης συνδρομή, κατάθλιψη, τριχόπτωση) οδηγούν σε δια-κοπή της στο 1/3 των ασθενών.49 η μπουσουλφάνη και ο 32Ρ αποτελούν θεραπευτικές επιλογές τελευταίας γραμμής σε ασθενείς με ελαττωμένο προσδόκιμο επιβίωσης.50-52
Κριτήρια Ανθεκτικότητας ή/και Δυσανεξίας στην υδροξυουρία σε ασθενείς με ΙΘ45
• Αριθμός αιμοπεταλίων >600 x 109/L μετά από 3 μή-νες αγωγής με υδροξυουρία, σε δόση 2gr/ημέρα του-
Πίνακας 2. Κριτήρια κλινικής και αιματολογικής ανταπόκρισης στην κυτταροστατική θεραπεία της Ιδιοπαθούς Θρομβοκυτταραιμίας (ELN)16
Πλήρης ανταπόκριση
Αριθμός αιμοπεταλίων ≤400 x 109/L ΚΑΙΑπουσία συμπτωμάτων
σχετιζόμενων με τη νόσο* ΚΑΙΦυσιολογικό μέγεθος σπληνός σε
απεικονιστική μέθοδο ΚΑΙΑριθμός WBC ≤10 x 109/L
Μερική ανταπόκριση
Σε ασθενείς που δεν πληρούν τα κριτήρια για πλήρη ανταπόκριση αριθμός αιμοπεταλίων ≤600 x 109/L ή ελάττωση >50% από τον αρχικό αριθμό
Χωρίς ανταπόκριση
Οποιαδήποτε απάντηση που δεν πληρεί τα κριτήρια της μερικής ανταπόκρισης
* Στα συμπτώματα που σχετίζονται με τη νόσο συμπεριλαμβάνονται οι μικροαγγειακές διαταραχές, ο κνησμός και η κεφαλαλγία.

Θεραπευτική αντιμετώπιση των Ph- Μυελοϋπερπλαστικών Νεοπλασμάτων (Ph-ΜΥΝ) 103
λάχιστον (2,5 gr/ημέρα υδροξυουρίας σε ασθενείς με βάρος σώματος >85 kg) ή
• Αριθμός αιμοπεταλίων >400 x 109/L και αριθμός WBC < 2,5 x 109/L σε οποιαδήποτε δόση υδροξυουρίας ή
• Αριθμός αιμοπεταλίων >400 x 109/L και αιμοσφαιρί-νη <10gr/dL σε οποιαδήποτε δόση υδροξυουρίας ή
• Παρουσία ελκών στα πόδια ή άλλων σημαντικών βλεννογονοδερματικών βλαβών σε οποιαδήποτε δό-ση υδροξυουρίας ή
• Πυρετός προκαλούμενος από την υδροξυουρία.
ΙΙΙ. θεραπευτική αντιμετώπιση της πρωτοπαθούς μυελοΐνωσης (πΜΙ)
τα συχνότερα κλινικά προβλήματα που αντιμετω-πίζουν οι ασθενείς με Πμι είναι: (1) η συμπτωματική αναιμία, (2) η εκσεσημασμένη ηπατοσπληνομεγαλία, (3) οι εκδηλώσεις απο εξωμυελικές εστίες αιμοποίησης (πιεστικά φαινόμενα στο νωτιαίο μυελό, ασκίτης, πνευ-μονική υπέρταση), (4) τα θρομβοαιμορραγικά αγγειακά συμβάματα, (5) οι επαναλαμβανόμενες κρίσεις ουρικής αρθρίτιδας και (6) τα έντονα γενικά συμπτώματα (καχε-ξία, απώλεια βάρους, οστικά άλγη, πυρετός, νυκτερινές εφιδρώσεις).3 οι συμβατικές θεραπευτικές παρεμβάσεις δεν επιτυγχάνουν ίαση της νόσου. η εφαρμογή της αλ-λογενούς μεταμόσχευσης αρχέγονων αιμοποιητικών κυτ-τάρων υπόσχεται ίαση με αντιστάθμισμα τον κίνδυνο θανάτου ή χρόνιας νοσηρότητας από νόσο μοσχεύματος εναντίον του ξενιστή, έχοντας έτσι αμφισβητούμενο κα-θαρό αποτέλεσμα.53 Πρωταρχικής λοιπόν σημασίας στο σχεδιασμό της θεραπευτικής αντιμετώπισης ενός ασθε-νούς με Πμι, αποτελούν η γενική του κατάσταση, τα προβλήματα που εμφανίζει λόγω νόσου, τα οφέλη και οι κίνδυνοι της προτεινόμενης θεραπείας, η διαθεσιμότητα νέων θεραπευτικών επιλογών και οι επιθυμίες του ιδίου του ασθενούς.54
ςε ασθενείς χωρίς ή με ελάχιστα συμπτώματα συνι-στάται η τακτική «παρακολούθηση και αναμονή» μέχρις εμφάνισης συγκεκριμένων προβλημάτων που θα επιζη-τούν επίλυση.
ΙΙΙα. Αντιμετώπιση της αναιμίας. η συμπτωματική αναιμία, με τιμές αιμοσφαιρίνης συνήθως χαμηλότερες από 10gr/dl, απαιτεί θεραπεία. Δραστικοί παράγοντες εί-ναι τα κορτικοστεροειδή, τα ανδρογόνα, οι παράγοντες διέγερσης ερυθροποίησης και τα ανοσοτροποποιητικά. Πρώτη επιλογή θα μπορούσαν να είναι οι παράγοντες διέγερσης της ερυθροποίησης (ΠΔΕ), όπως ανασυνδυ-ασμένη ερυθροποιητίνη ή νταρμποποιητίνη, χωρίς δια-φορά στην αποτελεσματικότητα,55 σε ασθενείς χωρίς ή με ήπια σπληνομεγαλία (ψηλαφητός σπλήνας <5 εκ από το πλευρικό τόξο), επειδή υπάρχει κίνδυνος η χορήγηση των παραγόντων αυτών να επιδεινώσει τη σπληνομεγα-λία.53 Φαίνεται ότι, οι ασθενείς που εξαρτώνται από με-
ταγγίσεις και έχουν τιμές ενδογενούς ερυθροποιητίνης >125 IU/L δεν ωφελούνται από τους ΠΔΕ.56 Απάντηση διάρκειας ενός περίπου έτους αναμένεται σε ≤56% των ασθενών που δεν εξαρτώνται από μεταγγίσεις και έχουν τιμή αιμοσφαιρίνης <10gr/dl.57 ςε αναιμικούς ασθενείς που δεν πληρούν τα παραπάνω κριτήρια ή που έχασαν την ανταπόκριση στους ΠΔΕ, μπορούν να χορηγηθούν κορτι-κοστεροειδή (προσοχή σε άτομα με ιστορικό σακχαρώδη διαβήτη ή οστεοπόρωση), όπως πρεδνιζολόνη 0,5mg/kg την ημέρα, ανδρογόνα (προσοχή σε ασθενείς με αυξη-μένο προστατικό αντιγόνο), όπως νταναζόλη 600mg την ημέρα,58 ή θαλιδομίδη 50mg την ημέρα με ή χωρίς πρεδ-νιζολόνη,59,60 με απαντήσεις διάρκειας ενός έτους περί-που, σε 20% των ασθενών. η λεναλιδομίδη συνιστάται ως θεραπεία πρώτης γραμμής, σε παρουσία έλλειψης 5q με καλά αποτελέσματα στη βελτίωση της αναιμίας και της σπληνομεγαλίας.61 για τον περιορισμό των θρομβω-τικών επεισοδίων σε ασθενείς που λαμβάνουν θαλιδομί-δη ή λεναλιδομίδη και έχουν αριθμό αιμοπεταλίων >50 x 109/l προτείνεται η προφυλακτική χορήγηση ασπιρίνης.
ΙΙΙβ. Αντιμετώπιση της σπληνομεγαλίας. Θεραπεία επι-λογής για τη συμπτωματική σπληνομεγαλία αποτελεί η υδροξυουρία, με την οποία επιτυγχάνεται και έλεγχος της θρομβοκυττάρωσης και της λευκοκυττάρωσης.62 Ελάττωση του μεγέθους του σπληνός κατά 25% ή 50% επιτυγχάνεται σε ποσοστά 35% και 17% των ασθενών, αντίστοιχα.62,63 οι απαντήσεις αυτές είναι παροδικές, εμφανίζονται κυρίως σε ασθενείς με μετρίου βαθμού σπληνομεγαλία (<10 εκ) και σε ασθενείς που φέρουν τη μετάλλαξη JAK2V617F.53 μπουσουλφάνη ή μελφαλάνη χρησιμοποιείται σε άτομα μεγαλύτερης ηλικίας που δεν ανέχονται ή δεν απαντούν στη θεραπεία με υδροξυουρία, με συχνότερη επιπλοκή τη μυελοκαταστολή. Χαμηλές δόσεις θαλιδομίδης ελατ-τώνουν το μέγεθος του σπληνός και βελτιώνουν τη θρομ-βοπενία σε μικρό ποσοστό ασθενών, περίπου <20%.60,64 για την ογκώδη ανθεκτική σπληνομεγαλία θα μπορού-σε να χορηγηθεί μηνιαίως ενδοφλέβια κλαντριμπίνη με ανταπόκριση μέχρι 50% και κύρια τοξικότητα τις βαριές αλλά αναστρέψιμες κυτταροπενίες.65
η ακτινοβόληση του σπληνός (με 0,1-0,5 Gy σε 5-10 συνεδρίες) επιτυγχάνει συμπτωματική ανακούφιση από τη μηχανική πίεση, παροδικά (μέση διάρκεια 3-6 μήνες) και συνδέεται με θνητότητα >10%, που οφείλεται σε επι-πλοκές των προκαλούμενων κυτταροπενιών.66
Σπληνεκτομή. Ενδείξεις σπληνεκτομής αποτελούν: 1) η συμπτωματική ογκώδης σπληνομεγαλία, 2) η συμπτω-ματική πυλαία υπέρταση (κιρσοί οισοφάγου με/ή χωρίς αιμορραγία), 3) η έκδηλη καχεξία και 4) η βαριά αναι-μία που εξαρτάται από μεταγγίσεις.54 Αντίθετα, η βαριά θρομβοπενία αποτελεί δείκτη επικείμενης λευχαιμικής μεταμόρφωσης και η σπληνεκτομή δεν φαίνεται να επη-ρεάζει τη συνολική έκβαση της κατάστασης.3 η πλειο-ψηφία των ασθενών που υποβάλλονται σε σπληνεκτομή ωφελούνται έτσι ώστε οι περισσότεροι από τους μισούς

Δ. Παντελίδου και Μ. Παπαϊωάννου104
ασθενείς με αναιμία να μη χρειάζονται μεταγγίσεις, να βελτιώνεται η καχεξία καθώς και τα γενικά και μηχανι-κά/πιεστικά συμπτώματά τους. η μέση διάρκεια ανταπό-κρισης στη σπληνεκτομή είναι περίπου 1 έτος. Ωστόσο η σπληνεκτομή σχετίζεται με περιεγχειρητική θνητότητα 5-10% και θνησιμότητα ~25%.67 οι κυριότερες επιπλο-κές της σπληνεκτομής είναι η αιμορραγία, η θρόμβω-ση, οι λοιμώξεις και η αντιδραστική θρομβοκυττάρωση και λευκοκυττάρωση που συνήθως ελέγχονται με υδρο-ξυουρία. για τον περιορισμό του κινδύνου των παραπά-νω επιπλοκών συνιστάται ο αριθμός των αιμοπεταλίων να είναι <200 x 109/L πριν από τη σπληνεκτομή.67 η δε προφυλακτική αντιπηκτική αγωγή συνιστάται να ξεκινά από την 5η-8η μετεγχειρητική ημέρα και να διαρκεί για 4-6 εβδομάδες.53 μετά τη σπληνεκτομή μπορεί να εμφα-νιστεί ηπατομεγαλία (σε 20% των ασθενών), στα πλαί-σια εξωμυελικής αιμοποίησης. Ως εκ τούτου η έκδηλη ηπατομεγαλία αποτελεί αντένδειξη για σπληνεκτομή.54
ΙΙΙγ. Αντιμετώπιση εξωμυελικής μη ηπατοσπληνικής αιμοποίησης. οι συμπτωματικές βλάβες εξωμυελικής αι-μοποίησης συνιστάται να αντιμετωπίζονται με ακτινοβο-λία σε χαμηλές δόσεις (0,1 έως 1,0 Gy σε πέντε έως δέκα συνεδρίες). Δύσπνοια, βήχας και οιδήματα κάτω άκρων εγείρουν την υποψία διάχυτης παρεγχυματικής προσβο-λής των πνευμόνων, που τεκμηριώνεται με καταγραφή αυξημένων συστολικών πιέσεων της πνευμονικής αρτη-ρίας και/ή θετικό σπινθηρογράφημα με τεχνήτιο 99m. Ύφεση των συμπτωμάτων επιτυγχάνεται με ακτινοβό-ληση ολόκληρου του πνεύμονα με 100 cGy.54
ΙΙΙδ. Αλλογενής μεταμόσχευση αρχέγονων αιμοποιη-τικών κυττάρων. η αλλογενής μεταμόσχευση αποτελεί τη μοναδική θεραπευτική παρέμβαση στη μυελοΐνωση, που δυνητικά υπόσχεται την ίαση, αλλά που σχετίζεται και με υψηλή θνητότητα και θνησιμότητα. τα τελευταία χρόνια η εισαγωγή μη μυελοτοξικών σχημάτων προετοι-μασίας στο σχεδιασμό των αλλογενών μεταμοσχεύσεων, αν και επέτρεψε την ηλικιακή διεύρυνση στην επιλογή των υποψηφίων ασθενών για μεταμόσχευση, δεν φαίνε-ται να βελτίωσε τα αποτελέσματα ή την τοξικότητα. με την κλασική αλλογενή μεταμόσχευση η συνολική πεντα-ετής επιβίωση ήταν 37% και 30% (για συγγενή και μη συγγενή δότη αντίστοιχα) ενώ η θνητότητα η σχετιζόμε-νη με τη μεταμόσχευση ήταν 27% στο 1 έτος και 35% στα 5 έτη. με τα σχήματα ελαττωμένης τοξικότητας η τριετής επιβίωση ήταν 39% και 17% (για συγγενή και μη συγγενή δότη αντίστοιχα) χωρίς μείωση των υποτρο-πών και της θνητότητας.68 ςυγκριτικά, σε μια πρόσφατη μελέτη ασθενών με μυελοΐνωση, που πληρούσαν τα κρι-τήρια για μεταμόσχευση (ενδιάμεσου ή υψηλού κινδύ-νου DIPSS69, ηλικίας <60 ετών) αλλά δεν υποβλήθηκαν σε αυτή, η ετήσια και η τριετής επιβίωσή τους κυμάνθη-κε από 71%-95% και 55%-77% αντίστοιχα.70 Δυσμενείς προγνωστικοί παράγοντες για την έκβαση της αλλο-γενούς μεταμόσχευσης αποτελούν:1) η εξάρτηση των
ασθενών από μεταγγίσεις, 2) η παρουσία κυτταρογενε-τικών ανωμαλιών κακής πρόγνωσης, 3) η μεγάλη ηλικία των ασθενών, 4) η λήψη μοσχεύματος από συγγενή δό-τη μη ταυτόσημο HLA ή από μη συγγενή δότη μη πλή-ρως HLA συμβατό.3 Παρά το γεγονός ότι δεν υπάρχουν ισχυρές ενδείξεις για το αν η προηγηθείσα σπληνεκτομή επηρεάζει την έκβαση της μεταμόσχευσης, ο Tefferi προ-τείνει την ελάττωση του μεγέθους του σπλήνα με φαρ-μακευτική αγωγή (υδροξυουρία, αναστολείς JAK) πριν από τη μεταμόσχευση.53,68 τέλος, αξίζει να τονιστεί, ότι η επιλογή της μεταμόσχευσης θα πρέπει να βασιστεί σε μια εκ βαθέων συζήτηση με τον ασθενή , σχετικά με τα υπέρ και τα κατά της μεθόδου, ώστε η τελική απόφαση να τους ικανοποιεί όλους.54
ΙΙΙε. Νέες θεραπείες:Πομαλιδομίδη. η πομαλιδομίδη αποτελεί δεύτερης
γενιάς ανάλογο της θαλιδομίδης και ανήκει στην κα-τηγορία των ανοσορρυθμιστικών παραγόντων. μελέ-τες φάσεως ιι, στις οποίες χορηγήθηκε μόνο του ή σε συνδυασμό με πρεδνιζολόνη, έδειξαν ανταπόκριση της αναιμίας σε 25% των ασθενών, αλλά μόνο σε εκείνους που έφεραν τη μετάλλαξη JAK2V617F και δεν εμφάνι-ζαν ογκώδη σπληνομεγαλία. η απάντηση στη θεραπεία μπορούσε να προβλεφθεί από την εμφάνιση βασεοφιλί-ας σχετιζόμενης με την πομαλιδομίδη.71,72
Αναστολείς JAK2. οι JAK2 αναστολείς σχεδιάστη-καν έτσι ώστε να αποτελέσουν ένα ακόμη παράδειγμα στοχευμένης θεραπείας, όπως συνέβη στη χρόνια μυελο-γενή λευχαιμία. Ωστόσο, ο σχεδιασμός δεν απέδωσε τα αναμενόμενα επειδή δεν έχει βρεθεί παράγοντας ειδικός για τη μεταλλαγμένη πρωτεϊνη JAK2V617F. Έτσι, όλοι οι JAK αναστολείς επιδρούν και στη φυσιολογική πρωτεΐ-νη επηρεάζοντας τη σηματοδότηση της ερυθροποίησης και της θρομβοποίησης στα φυσιολογικά κύτταρα, μέσω της οδού JAK-STAT. η ανταπόκριση που διαπιστώνεται με τη χορήγηση των JAK2 αναστολέων φαίνεται ότι εί-ναι ανεξάρτητη από την παρουσία ή όχι του μεταλλαγ-μένου κλώνου και σχετίζεται όχι τόσο με την καταστολή του παθολογικού κλώνου αλλά με μια πιο συνολική ρύθ-μιση που ασκούν στην οδό μετάδοσης του σήματος των προφλεγμονωδών κυτταροκινών.73
το ruxolitinib (INCB018424) είναι ένας JAK1/JAK2 αναστολέας, που χορηγήθηκε σε 153 ασθενείς με μυε-λοΐνωση, σε μελέτη φάσης ι/ιι και φάνηκε ότι, σε δόση 15mg δυο φορές την ημέρα, ήταν καλά ανεκτό και οδή-γησε σε κλινική ανταπόκριση με ελάττωση της σπληνο-μεγαλίας κατά ≥50% και ταχεία βελτίωση της καχεξίας και των γενικών συμπτωμάτων (αδυναμία, καχεξία, νυ-κτερινοί ιδρώτες, κνησμός) ανεξαρτήτως της παρουσίας ή όχι της μετάλλξης JAK2V617F.74 με βάση τα παραπάνω αποτελέσματα ο Αμερικανικός οργανισμός Φαρμάκων (FDA) έδωσε την έγκριση κυκλοφορίας του παράγο-ντα στις ηνωμένες Πολιτείες το Νοέμβριο του 2011. το ruxolitinib (JAKAFI) μπορεί να χορηγηθεί σε ασθενείς

Θεραπευτική αντιμετώπιση των Ph- Μυελοϋπερπλαστικών Νεοπλασμάτων (Ph-ΜΥΝ) 105
με ενδιάμεσου ή υψηλού κινδύνου μυελοΐνωση (πρωτο-παθή, μετα-πολυκυτταραιμική ή μεταθρομβοκυτταραι-μική). η ημερήσια δόση είναι 40 mg διαιρεμένη σε δυο δόσεις για ασθενείς με αριθμό αιμοπεταλίων υψηλότερο από 200 x109/L και 30 mg διαιρεμένη σε δυο δόσεις για ασθενείς με αριθμό αιμοπεταλίων από 100-200 x109/L. οι κυριότερες επιπλοκές είναι αιματολογικές όπως αναι-μία (βαθμού 3 και 4, σε 45% των ασθενών) και θρομβο-πενία (βαθμού 3 και 4, σε 13% των ασθενών) καθώς και ήπιες μη αιματολογικές (κεφαλαλγία, ζάλη, μώλωπες).75 ςτην επιστημονική συνάντηση της Αμερικανικής Αιμα-τολογικής Εταιρείας (ASH) του 2010, ανακοινώθηκαν τα αποτελέσματα από την χορήγηση ενός άλλου αναστολέα, του CYT387, σε μελέτη φάσεως I/II, που έδειξαν αξιο-σημείωτη βελτίωση της αναιμίας σε 40% των ασθενών, της σπληνομεγαλίας και των γενικών συμπτωμάτων.76 η συνδυαστική χορήγηση των JAK2 αναστολέων με άλλους παράγοντες καθώς και η χρησιμοποίησή τους στη μετα-μόσχευση, αποτελούν αντικείμενα μελλοντικών μελετών.
Ιv. Ειδικές περιπτώσεις μυελοϋπερπλαστικών νεοπλασιών
Παιδιά. τα παιδιά προσβάλλονται σπάνια από μΥΝ με συχνότερα εμφανιζόμενο νόσημα την ιΘ. Εξ ορισμού, τα παιδιά με μΥΝ ανήκουν στην ομάδα χαμηλού κινδύ-νου εκτός αν έχει ήδη εκδηλωθεί αιμορραγικό ή θρομβω-τικό επεισόδιο. η θεραπεία τους εξατομικεύεται λόγω: (1) του θεωρητικού κινδύνου λευχαιμογένεσης μετά μακρά θεραπεία με υδροξυουρία, (2) της αυξημένης συχνότητας εμφάνισης αναιμίας απο αναγρελίδη, (3) των παρενερ-γειών της ιντερφερόνης (νευροψυχιατρικές, αυτοάνοσες εκδηλώσεις) που μπορεί να γίνουν επικίνδυνες στα παι-διά και (4) του κινδύνου συνδρόμου Reye απο την ασπι-ρίνη σε παιδιά ηλικίας <12 ετών.3,77
Κύηση. Πολυκεντρικές μελέτες έχουν δείξει ότι η κύ-ηση γυναικών με ιΘ θεωρείται υψηλού κινδύνου τόσο για το έμβρυο όσο και για τη μητέρα. ο κίνδυνος αποβολής
είναι 3,4 φορές υψηλότερος από εκείνον σε γυναίκες μάρ-τυρες ίδιας ηλικίας. τα ποσοστά των επιτυχών γεννήσε-ων ζωντανών εμβρύων κυμαίνονται από 57% έως 75,4%, οι αποβολές πρώτου τριμήνου από 15,6% έως 35% και οι επιπλοκές στην έγκυο από 8% έως 11%. τα καλύτερα αποτελέσματα έχουν καταγραφεί σε πρόσφατες μελέτες στις οποίες η ιντερφερόνη-α αποτελούσε τη θεραπεία επι-λογής.78 Ασθενείς χωρίς συμπτώματα ή κύηση χαμηλού κινδύνου μπορεί να αντιμετωπιστούν με διατήρηση του αιματοκρίτη σε χαμηλά επίπεδα (<45%) και χαμηλή δό-ση ασπιρίνης (100mg/ημέρα) ή με ηπαρίνη χαμηλού μο-ριακού βάρους σε όλη τη διάρκεια της κύησης και για 6 εβδομάδες μετά τον τοκετό.12 οι ασθενείς με κύηση υψη-λού κινδύνου συνιστάται να αντιμετωπίζονται με ηπαρίνη χαμηλού μοριακού βάρους σε όλη τη διάρκεια της κύησης και για 6 εβδομάδες μετά τον τοκετό.79 Υψηλού κινδύ-νου θεωρείται η κύηση όταν στο ιστορικό της ασθενούς αναφέρονται: (1) θρομβωτικό ή αιμορραγικό επεισόδιο κατά τη διάρκεια προηγούμενης κύησης ή εκτός αυτής, (2) ανεξήγητες επαναλαμβανόμενες αυτόματες αποβο-λές πρώτου τριμήνου ή μικρό για την ηλικία του έμβρυο ή ενδομήτριος θάνατος εμβρύου ή βαριά προεκλαμψία ή αποκόλληση πλακούντα, δηλαδή επιπλοκές προηγούμε-νων κυήσεων που θα μπορούσαν να θεωρηθούν σχετιζό-μενες με μΥΝ. ςε περιπτώσεις κυήσεων που απαιτείται κυτταροτοξική θεραπεία, η IFN-α αποτελεί θεραπεία επι-λογής. Επίσης σε περιπτώσεις κυήσεων με αιμορραγικές επιπλοκές ή πολύ υψηλό αριθμό αιμοπεταλίων συνιστά-ται αποφυγή της ασπιρίνης και έναρξη IFN-α.41
Κνησμός. O ανυπόφορος κνησμός, που επάγεται από το νερό, αποτελεί ένα δυσεπίλυτο πρόβλημα σε μερικούς ασθενείς που πάσχουν κυρίως από ΑΠ. τα αντιϊσταμινι-κά, όπως η κυπροεπταδίνη, αποτελούν θεραπεία πρώτης γραμμής. η χρήση ιντερφερόνης στην κλασική ή την πε-γκυλιωμένη μορφή της έχει δώσει ικανοποιητικά απο-τελέσματα. Άλλες θεραπευτικές επιλογές αποτελούν η παροξετίνη (εκλεκτικός ανταγωνιστής επαναπροόσλη-ψης της σεροτονίνης), η φωτοχημειοθεραπεία με ψωρα-λένιο και η υπεριώδης Α ακτινοβολία.78
Therapeutic management of Ph negative Myeloproliferative Neoplasmsby Despoina Pantelidou1, Maria Papaioannou2
Haematology Unit of 1st Department of Internal Medicine, AHEPA University Hospital, Thessaloniki
ABSTRACT: Despite the dramatic improvement on the understanding of the pathophysiology of Phil-adelphia negative classical Myeloproliferative Neoplasms (Ph-MPN) following the description, in the last years, of recurrent molecular abnormalities represented mainly by the V617F mutation in JAK2 ex-on 14, the treatment of MPN remains a challenge. In practice, therapeutic recommendations are largely based on consensus of experts appointed by the European LeukemiaNet (ELN). Two consensus confer-ences were convened to produce recommendations on the management of Ph(-) MPN, including moni-toring, response definition, first- and second-line therapy, and therapy for special issues. The first step

Δ. Παντελίδου και Μ. Παπαϊωάννου106
for the management of polycythemia vera (PV) and essential thrombocythemia (ET) is to identify the potential risk to develop major thrombotic or hemorrhagic complications and should be defined as high risk if age is greater than 60 years or there is a history of previous thrombosis. High-risk patients with PV should be managed with phlebotomy, low-dose aspirin, and cytoreduction, with either hydroxyurea or interferon at any age. High-risk patients with ET should be managed with cytoreduction, using hy-droxyurea at any age. Monitoring response in PV and ET should use the ELN clinicohematologic crite-ria. The treatment strategy of patients with primary myelofibrosis (PMF) starts with the risk stratification and depends on the clinical symptoms and signs. Corticosteroids, androgens, erythropoiesis-stimulating agents, and immunomodulators are recommended to treat anemia of PMF, whereas hydroxyurea is the first-line treatment of PMF-associated splenomegaly. Indications for splenectomy include symptomatic portal hypertension, drug-refractory painful splenomegaly, and frequent red blood cell transfusions. The risk of allogeneic stem-cell transplantation–related complications is justified in transplantation-eligible patients whose median survival time is expected to be less than 5 years.
Βιβλιογραφία1. Vannucchi AM, Guglielmelli P, Tefferi A. Advances in under-
standing and management of myeloproliferative neoplasms. CA Cancer J Clin. 2009; 59:171-191.
2. Verstovsek S. Therapeutic potential of JAK2 inhibitors. Hematology Am Soc Hematol Educ Program. 2009; 636-642.
3. Barbui T, Barosi G, Birgegard G, et al. Philadelphia-negative classical myeloproliferative neoplasms: Critical concepts and management recommendations from European Leukemia Net. J Clin Oncol. 2011; 29:761-770.
4. McMullin MF, Bareford D, Campbell P, et al. Guidelines for the diagnosis, investigation and management of polycythemia /erythrocytosis. Br J Haematol. 2005; 130:174-195.
5. Berk PD, Goldberg JD, Donovan PB, et al. Therapeutic recommendations in polycythemia vera based on Poly-cythemia Vera Study Group protocols. Semin Hematol. 1986; 23:132-143.
6. Marchioli R, Finazzi G, Landolfi R, et al. Vascular and neo-plastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol. 2005; 23:2224-2232.
7. Finazzi G, Barbui T. Evidence and expertise in the manage-ment of polycythemia vera and essential thrombocythemia. Leukemia. 2008; 22:1494-1502.
8. Marchioli R, Finazzi G, Specchia G, et al. The CYTO-PV: A large-scale trial testing the intensity of cytoreductive therapy to prevent cardiovascular events in patients with polycythemia vera. Thrombosis. 2011; 2011:794240 Epub 2011 May 17.
9. Di Nisio M, Barbui T, Di Gennaro L, et al. The hematocrit and platelet target in polycythemia vera. Br J Haematol. 2006; 136:249-259.
10. Landolphi R, Marchioli R, Kutti J, et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med. 2004; 350:114-124.
11. Barbui T, Finazzi G. Evidence-based management of polycythemia vera. Best Pract Res Clin Haematol. 2006; 19:483-493.
12. Finazzi G, Barbui T. How I treat patients with polycythemia vera. Blood. 2007; 109:5104-5111.
13. Spivak JL, Hasselbalch H. Hydroxykarbamide: a user’s guide for chronic myeloproliferative disorders. Exper Rev Antiancer Ther. 2011; 11:403-414.
14. Najean Y, Rain JD. Treatment of polycythemia vera: The use of hydroxyurea and pipobroman in 292 under the age of 65 years. Blood. 1997; 90:3370-3377.
15. Campbell PJ, Baxter EJ, Beer PA, et al. Mutation of JAK2 in the myeloproliferative disorders: timing, clonality studies, cytogenetic associations and role in leukemic transformation. Blood. 2006; 108:3548-3555.
16. Barosi G, Birgegard G, Finazzi G, et al. Response criteria for essential thrombocythemia and polycythemia vera. Results of a European Leukemia-Net consensus conference. Blood. 2009; 113:4829-4833.
17. Finazzi G, Caruso V, Marchioli R, et al. Acute leukemia in polycythemia vera. An analysis of 1638 patients enrolled in a prospective observational study. Blood. 2005; 105:2664-2670.
18. Barosi G, Birgegard G, Finazzi G, et al. A unified definition of clinical resistance and intolerance to hydroxycarbamide in polycythemia vera and primary myelofibrosis: Results of a European LeukemiaNet (ELN) consecus process. Br J Haematol. 2010; 148:961-963.
19. Lengfelder E, Berger U, Hehlmann R. Interferon alpha in the treatment of polycythemia vera. Ann Hematol. 2000; 79:103-109.
20. Silver RT. Long-term effects of the treatment of poly-cythemia vera with recombinant interferon alpha. Cancer. 2006; 107:451-458.
21. Kiladjian JJ, Cassinat B, Turlure P, et al. High molecular response rate of polycythemia vera patients treated with pegylated interferon alpha-2a. Blood. 2006; 108:2037-2040.
22. Quintas-Cardama A, Kantarjian H, Manshouri T, et al. Pegylated interferon-alpha-2alpha yields high rates of he-matologic and molecular response in patients with advanced essential thrombocythemia and polycythemia vera. J Clin Oncol. 2009; 27:5418-5424.
23. Samuelsson J, Mutschler M, Birgegard G, Gram-Hansen P, Bjorkholm M, Pahl HL. Limited effects on JAK2 mutational status after pegylated interferon-2b therapy in polycythemia

Θεραπευτική αντιμετώπιση των Ph- Μυελοϋπερπλαστικών Νεοπλασμάτων (Ph-ΜΥΝ) 107
vera and essential thrombocythemia. Haematologica. 2006; 91:1281-1282.
24. Najean Y, Rain JD, Dresch C, et al. Risk of leukemia, car-cinoma and myelofibrosis in 32P or chemotherapy-treated patients with polycythemia vera: a prospective analysis of 682 cases. “The French Cooperative Group for the study of Polycythemias” Leuk Lymphoma. 1996; 22(supp1):111-119.
25. Björkholm M, Derolf AR, Hultcrantz M, et al. Treatment-related risk factors for transformation to acute myeloid leu-kaemia and myelodysplastic syndromes in myeloproliferative neoplasms. J Clin Oncol. 2011; 29:2410-2415.
26. Tefferi A. Polycythemia vera and essential thrombocythemia: 2012 update on diagnosis, risk stratification, and manage-ment. Am J Hematol. 2012; 87:284-293.
27. Cervantes F, Passamonti F, Barosi G. Life expectancy and prognostic factors in the classic BCR/ABL- negative my-eloproliferative disorders. Leukemia. 2008; 22:905-914.
28. Beer PA, Erber WN, Campbell PJ, et al. How I treat essential thrombocythemia. Blood. 2011; 117:1472-1482.
29. Wolanskyj AP, Schwager SM, McClure RF, et al. Essential thrombocythemia deyond the first decade: Life expectancy, long-term complication rates and prognostic factors. Mayo Clin Proc. 2006; 81:159-166.
30. Bazzan M, Tamponi G, Schinco P, et al. Thrombosis-free survival and life expectancy in 187 consecutive patients with essential thrombocythemia. Ann Hematol. 1999; 78:539-543.
31. Chim CS, Kwong YL, lie AK, et al. Long-term outcome of 231 pateints with essential thrombocythemia: prognostic fac-tors for thrombosis, bleeding, myelofibrosis and leukaemia. Arch Intern Med. 2005; 165:2651-2658.
32. Budde U, van Genderen PJ. Acquired von Willebrand disease in patients with high platelet counts. Semin Thromb Hemost. 1997; 23:425-431.
33. Tefferi A, Gangat N, Wolanskyj AP. Management of extreme thrombocytosisin otherwise low-riskessential thrombo-cythemia: Does number matter? Blood. 2006; 108:2493-2494.
34. Ruggeri M, FinazziG, Tosetto A, et al. No treatment for low-risk thrombocythemia: Results from a prospective study. Br J Haematol. 1998; 103:772-777.
35. Alvarez-Larran A, Cervantes F, Pereira A, et al. Observa-tion versus antiplatelet therapy as primary prophylaxis for thrombosis in low-risk essential thrombocythemia. Blood. 2010; 116:1205-1210.
36. Cervantes F. Management of essential thrombocythemia. Hematology Am Soc Hematol Educ Program. 2011; 215-221.
37. Cortelazzo S, Finnazi G, Ruggeri M, et al. Hydroxyurea for patients with essential thrombocythemia and a high risk for thrombosis. N Engl J Med. 1995; 332:1132-1136.
38. Harrison CN, Campbell PJ, Buck G, et al. Hydroxyurea compared with anagrelide in high-risk essential thrombo-cythemia. N Engl J Med. 2005; 353:33-45.
39. Falanga A, Marchetti M, Evangelista V, et al. Polymorpho-nuclear leukocyte activation and hemostasis in patients with essential thrombocythemia and polycythemia vera. Blood. 2000; 96:4261-4266.
40. Sterkers Y, Preudhomme C, Lai JL, et al. Acute myeloid
leukaemia and myelodysplastic syndromes following essential thrombocythemia treated with hydroxyurea: high propor-tion of cases with 17p deletion. Blood. 1998; 91:616-622.
41. Pleyer L, Faber V, Neureiter D, Greil R. Essential Throm-bocythemia in Greil R, Pleyer L, Neureiter D, Faber V. (Editors). Chronic Myeloid Neoplasias and Clonal Overlap Syndromes. Wien New York: Springer. 2010; 14-50.
42. van Genderen PJ, Michiels JJ, Van Strik R, et al. Platelet consumption in thrombocythemia complicated by eryth-romelalgia: reversal by aspirin. Thromb Haemost. 1995; 73:210-214.
43. Birgegard G. Long-term management of thrombocytosis in essential thrombocythaemia. Ann Hematol. 2009; 88:1-10.
44. Finnazi G, Ruggeri M, Rodeghiero F, et al. Second malig-nancies in patients with essential thrombocythemia treated with busulphan and hydroxyurea. Long-term follow up of a randomized clinical trial. Br J Haematol. 2000; 110:577-583.
45. Barosi G, Besses C, Birgegard G, et al. A unified definition of clinical resistance/intolerance to hydroxyurea in essential thrombocythemia: results of a consensus process by an international working group. Leukemia. 2007; 21:277-280.
46. Gisslinger H, Gotic M, Holowiecki J, et al. Final results of the ANAHYDRET study: non-inferiority of anagrelide com-pared with hydroxyurea in newly diagnosed WHO-essential thrombocythemia patients [abstract] Blood. 2008; 112:661.
47. Storen EC, Tefferi A. Long-term use of anagrelidein young patients with essential thrombocythemia. Blood. 2001; 97:863-866.
48. Martinelli P, Martinelli V, Agangi A, et al. Interferon alpha treatment for pregnant women affected by essential throm-bocythemia: case reports and a review. Am J Obstet Gynecol 2004; 191:2016-2020.
49. Pogliani EM, Rossini F, Moccolis I, et al. Alpha interferon as initial treatment of essential thrombocythemia . Analysis after two years of follow-up. Tumori. 1995; 81:245-248.
50. Van de Pette JE, Prochazka AV, Pearson TC, et al. Primary thrombocythemia treated with bususlphan. Br J Haematol 1986; 62:229-237.
51. Shvidel L, Sigler E, Haran M, et al. Busulphan is safe and efficient treatment in elderly patients with essential throm-bocythemia. Leukemia. 2007; 21:2071-2072.
52. Brandt L, Anderson H. Survival and risk of leukaemia in polycythemia vera and essential thrombocythemia treated with oral radiophosphorus: are safer drugs available? Eur J Hematol. 1995; 54:21-26.
53. Tefferi A. How I treat myelofibrosis. Blood. 2011; 117:3494-3504.
54. Vannuchi AM. Management of myelofibrosis. Hematology Am Soc Hematol Educ Program. 2011; 222-230.
55. Cervantes F, Alvarez-Larran A, Hernadez-Boluda JC, et al. Darbopoetin-alpha for the anaemia of myelofibrosis with myeloid metaplasia. Br J Haematol. 2006; 134:184-186.
56. Cervantes F, Alvarez-Larran A, Hernandez-Boluda JC, et al. Erythropoietin treatment of the anaemia of myelofibrosis with myeloid metaplasia: results in 20 patients and review of the literature. Br J Haematol. 2004; 127:399-403.

Δ. Παντελίδου και Μ. Παπαϊωάννου108
57. Huang J, Tefferi A. Erythropoiesis stimulating agents have limited therapeutic activity in transfusion-dependent patients with primary myelofibrosis regardless of serum erythropoietin level. Eur J Haematol. 2009; 83:154-155.
58. Cervantes F, Alvarez-Larran A, Domingo A, et al. Efficacy and tolerability of danazol as a treatment for the anaemia of myelofibrosis with myeloid metaplasia: long-term results in 30 patients. Br J Haematol. 2005; 129:771-775.
59. Thapaliya P, Tefferi A, Pardanani A, et al. International work-ing group for myelofibrosis research and treatment response assessment and lond-term follow-up of 50 myelofibrosis patients treated with thalidomide-prednisone based regimens. Am J Hematol. 2011; 86:96-98.
60. Thomas DA, Giles FJ, Verstovsek S, et al. Thalidomide therapy for myelofibrosis with myeloid metaplasia. Cancer. 2006; 106:1974-1984.
61. Tefferi A, Lasho TL, Mesa RA, et al. Lenalidomide ther-apy in del(5)(q31)-associated myelofibrosis: cytogenetic and JAK2V617F molecular remissions. Leukemia. 2007; 21:1827-1828.
62. Martinez-Trillos A, Gaya A, Maffioli M, et al. Efficacy and tolerability of hydroxyurea in the treatment of hyperprolifera-tive manifestations of myelofibrosis: Results in 40 patients. Ann Hematol. 2010; 89:1233-1237.
63. Mesa RA. How I treat symptomatic splenomegaly in patients with myelifibrosis. Blood. 2009; 113:5394-5400.
64. Marchetti M, Barosi G, Balestri F, et al. Low-dose thalidomide ameliorates cytopenias and splenomegaly in myelofibrosis with myeloid metaplasia: a phase II trial. J Clin Oncol. 2004; 22:424-431.
65. Faoro LN, Tefferi A, Mesa RA. Long-term analysis of the pal-liative benefit of 2-chlorodeoxyadenosine for myelofibrosis with myeloid metaplasia. Eur J Haematol. 2005; 74:117-120.
66. Elliot MA, Chen MG, Silverstein MN, et al. Splenic ir-radiation for symptomatic splenomegaly associated with myelofibrosis with myeloid metaplsia. Br J Haematol. 1998; 103:505-511.
67. Mesa RA, Nagomey DS, Schwager S, et al. Palliative goals, patient selection, and perioperative platelet management: outcomes and lessons from 3 dacades of splenectomy for
myelofibrosis with myeloid metaplasia at the Mayo clinic. Cancer. 2006; 107:361-370.
68. Ballen KK, Shrestha S, Sobocinski KA, et al. Outcome of transplantation for myelofibrosis Biol Blood Marrow Transplant. 2010; 16:358-367.
69. Passamonti F, Cervantes F, Vannuchi AM, et al. A dynamic prognostic model to predict survival in primary myelofibrosis: a study dy the IWG-MPT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood. 2010; 115:1703-1708.
70. Siragusa S, Passamonti F, Cervantes F, et al. Survival in young patients with intermediate-/high risk myelofibrosis: Estimates derived from data-basesfor nontransplant patients. Am J Hematol. 2009; 84:140-143.
71. Tefferi a, Verstovsek S, Barosi G, et al. Pomalidomide is active in the treatment of anemia associated with myelofi-brosis. J Clin Oncol. 2009; 27:4563-4569.
72. Begna KH, Mesa RA, Pardanani A, et al. A phase-2 trial of low-dose pomalidomide in myelofibrosis with anemia. Leukemia. 2011; 25:301-304.
73. Cervantes F, Pereira A. Advances in the understandind and management of primary myelofibrosis. Curr Opin Oncol. 2011; 23:665-671.
74. Verstovsek S, Kantarjian H, Mesa RA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010; 363:1117-1127.
75. Full prescribing information for JAKAFI (Ruxolitinib) www.accessdata.fda.gov/drugsatfda_docs/label/2011/202192lbl.pdf
76. Pardanani A, George G, Lasho T, et al. A phase I/II study of CYT387, an oral JAK-1/2 inhibitor, in myelofibrosis: significant response rates in anemia, splenomegaly and con-stitutional symptoms [abstract]. Blood. 2010; 116:abst 460.
77. Barbui T, Finazzi G. Special issues in myeloproliferative neoplasms. Cur Hematol Malig Rep. 2011; 6:28-35.
78. Valera MC, Parant O, Vayssiere C, et al. Essential Thrombo-cythemia and pregnancy. Eur J Obst Gynecol Reprod Biol. 2011; 158:141-147.
79. Tefferi A, Passamonti F. Essential thrombocythemia and pregnancy. Observations from recent studies and manage-ment recommendations. Am J Hematol. 2009; 84:629-630.


















