
Malattia di Parkinson -...
26
Malattia di Parkinson
Transcript of Malattia di Parkinson -...

Malattia
di Parkinson
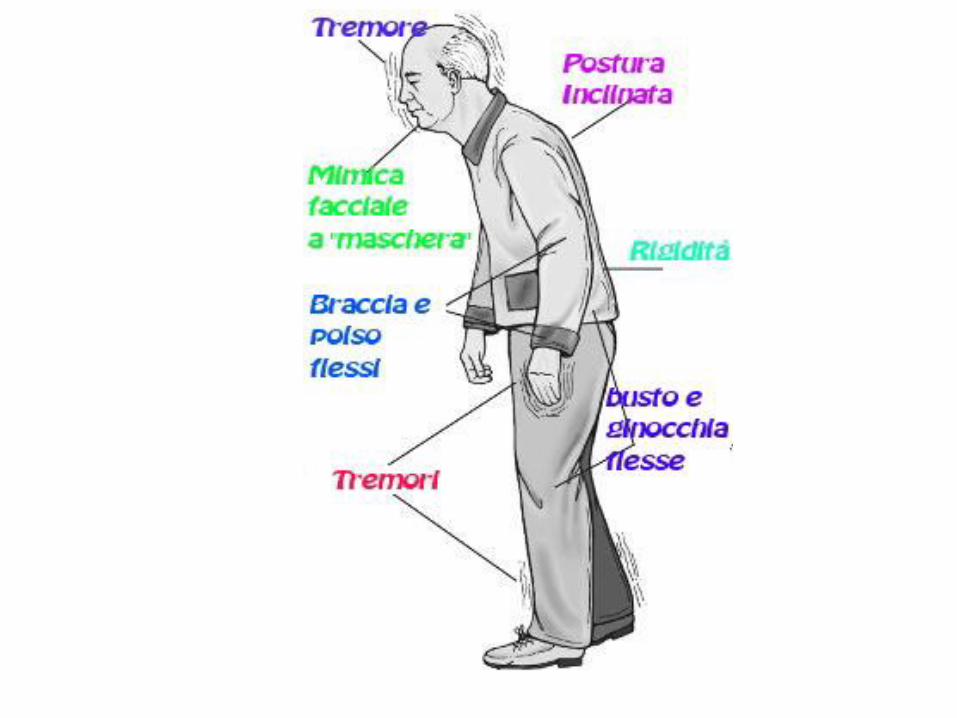


Sintomi cardinali della MP:bradicinesia, ipo/acinesia, rigidità muscolare, tremore a riposo
(insorgenza asimmetrica), instabilità posturale, disturbi nella parola e nella
scrittura, postura flessa in avanti, andatura a piccoli passi e freezing
(arresti improvvisi nell’andatura)
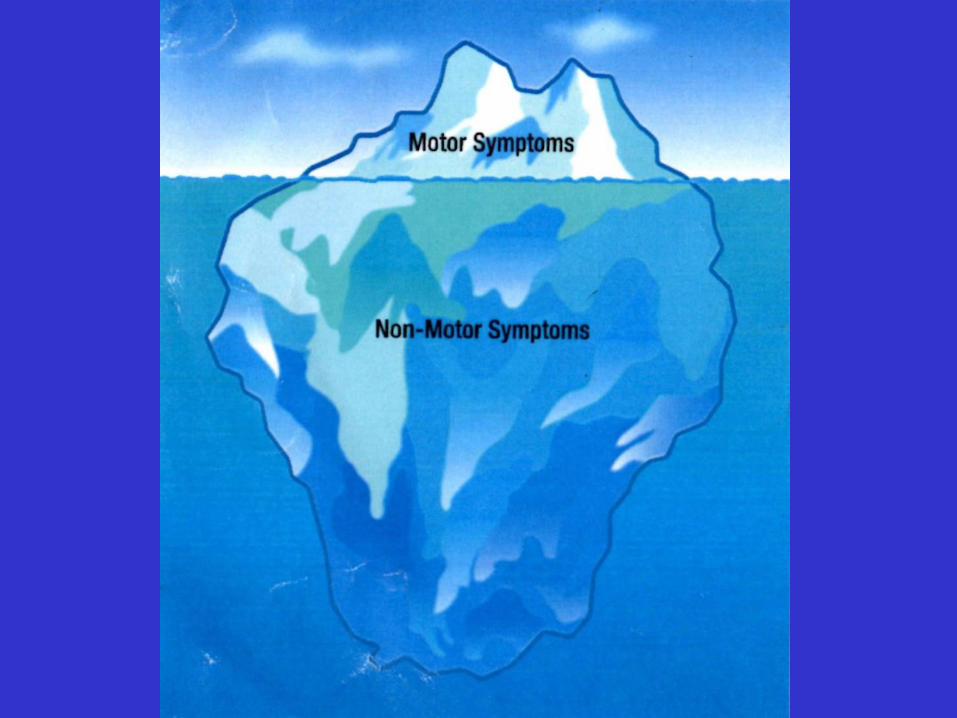
Sintomi non motori:ipotensione, stipsi, disfunzioni della vescica e della termoregolazione
insieme a disturbi del sonno, fatica e perdita di peso
Depressione, ansia, deficit cognitivi.
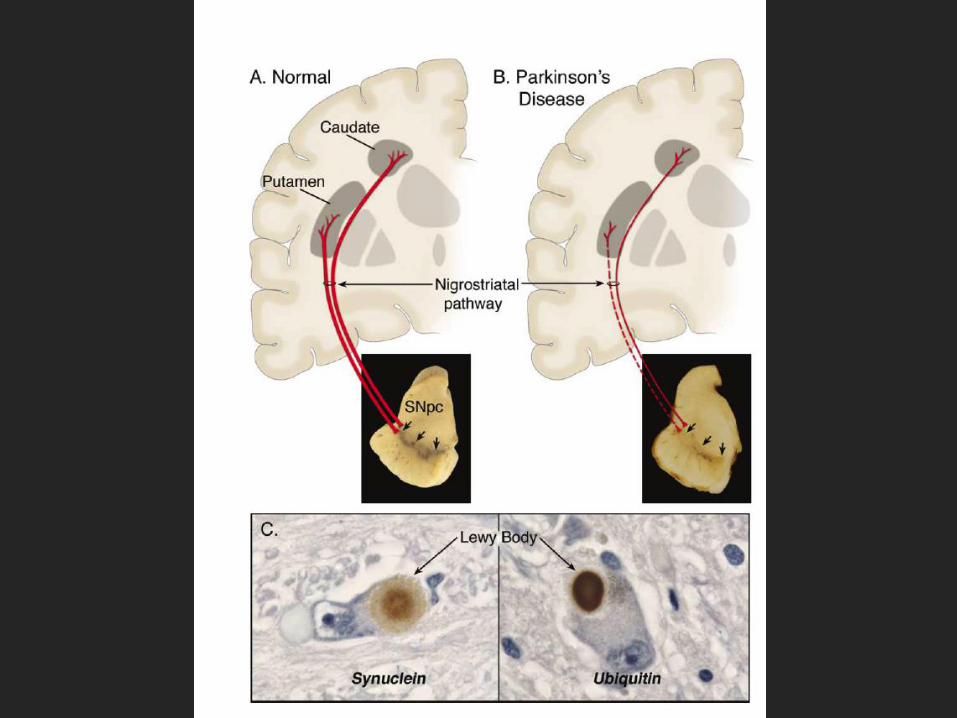
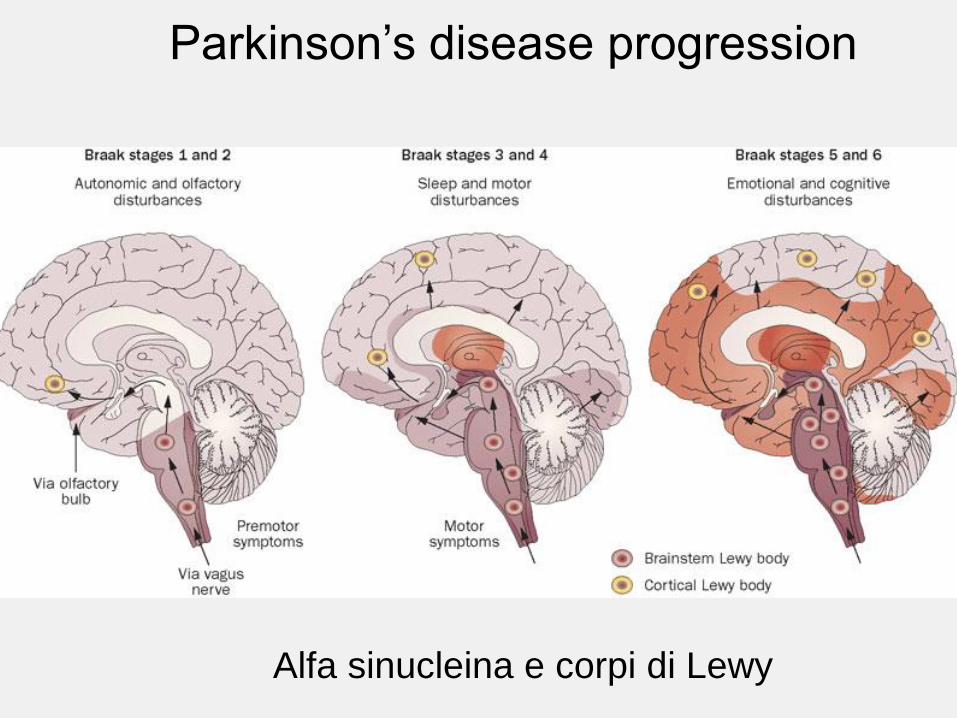
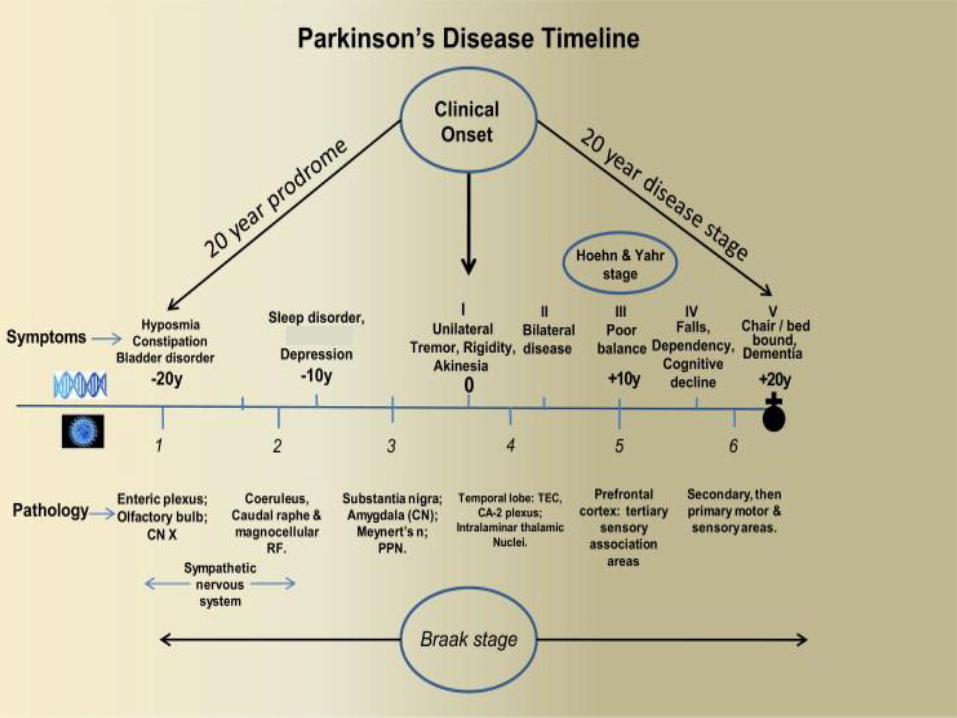
Parkinson’s disease progression
Alfa sinucleina e corpi di Lewy
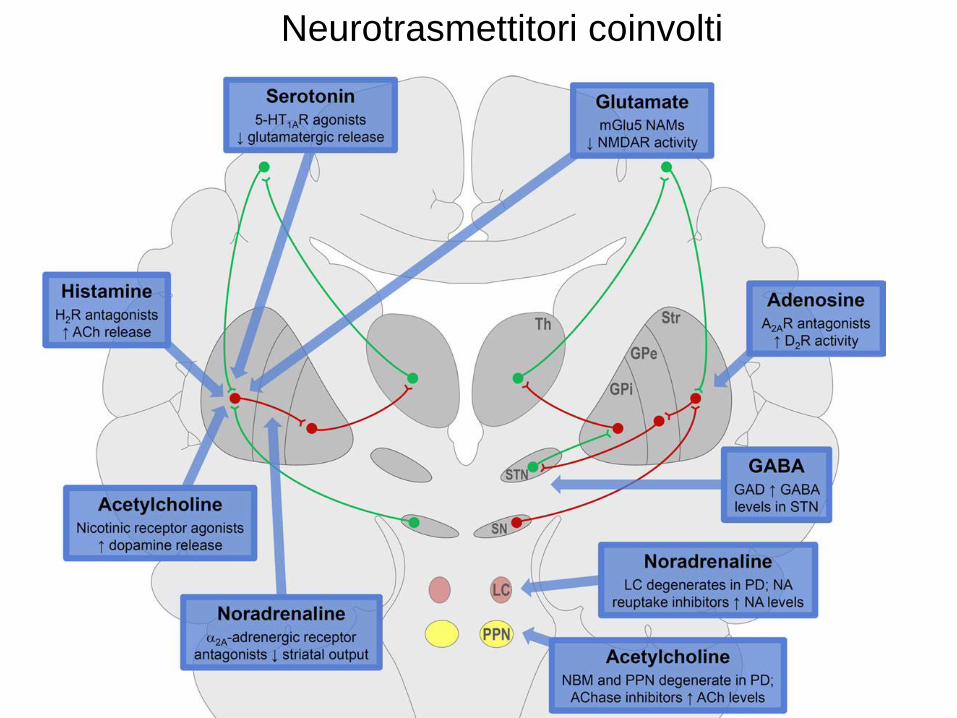
Neurotrasmettitori coinvolti
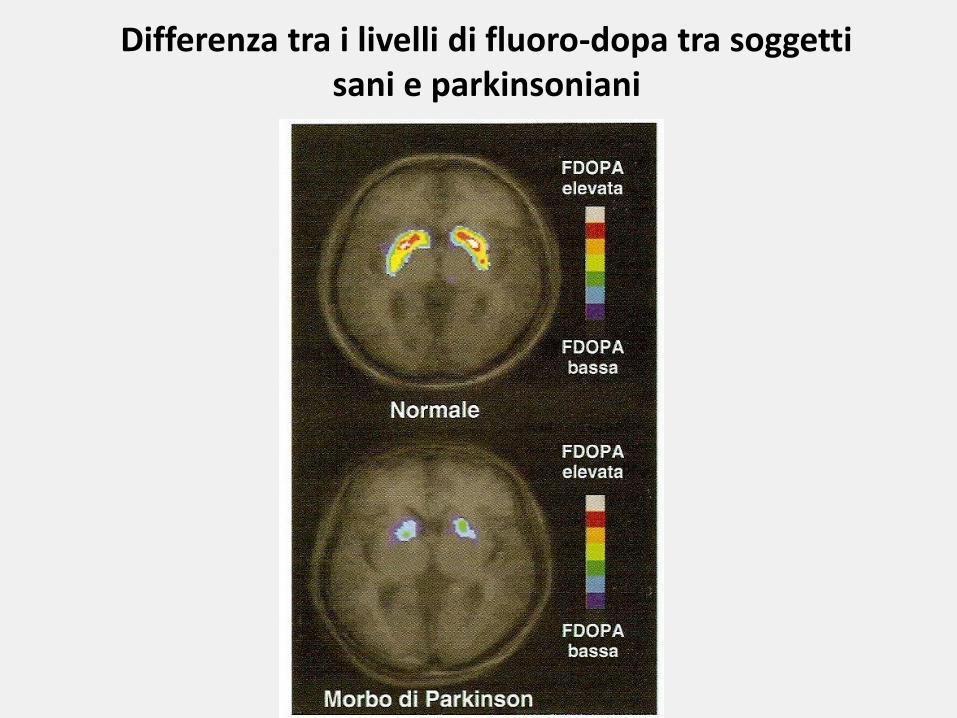
Differenza tra i livelli di fluoro-dopa tra soggetti sani e parkinsoniani
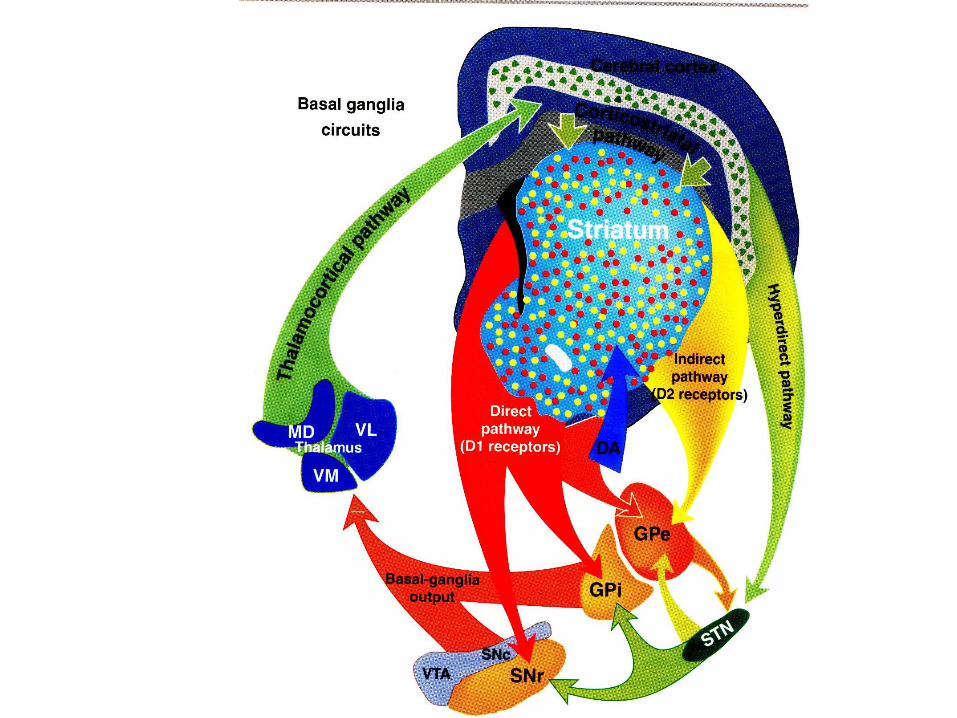
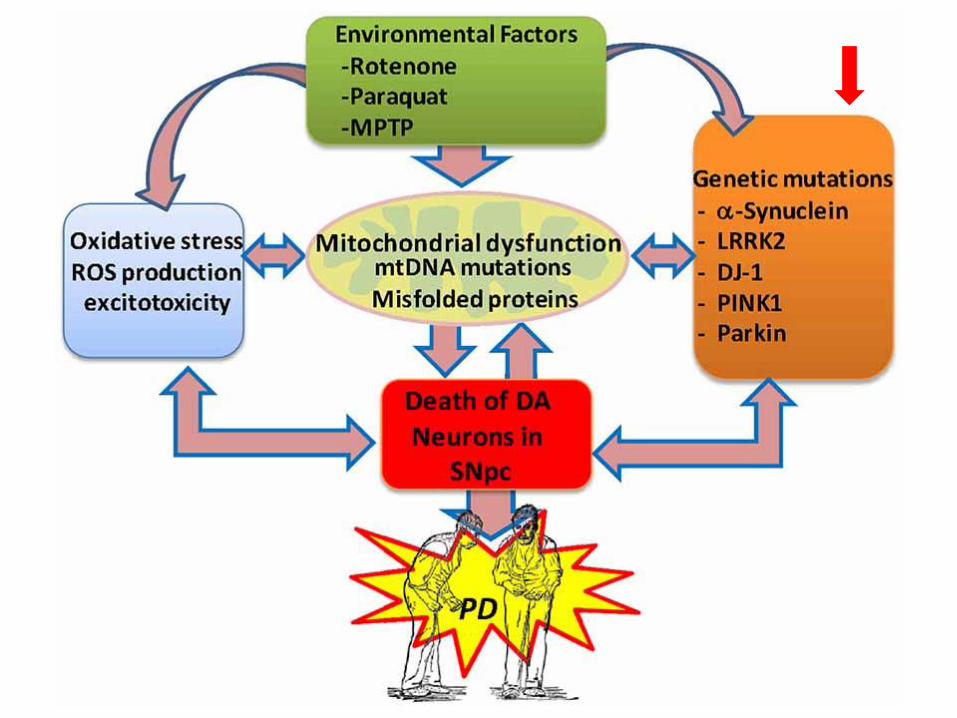

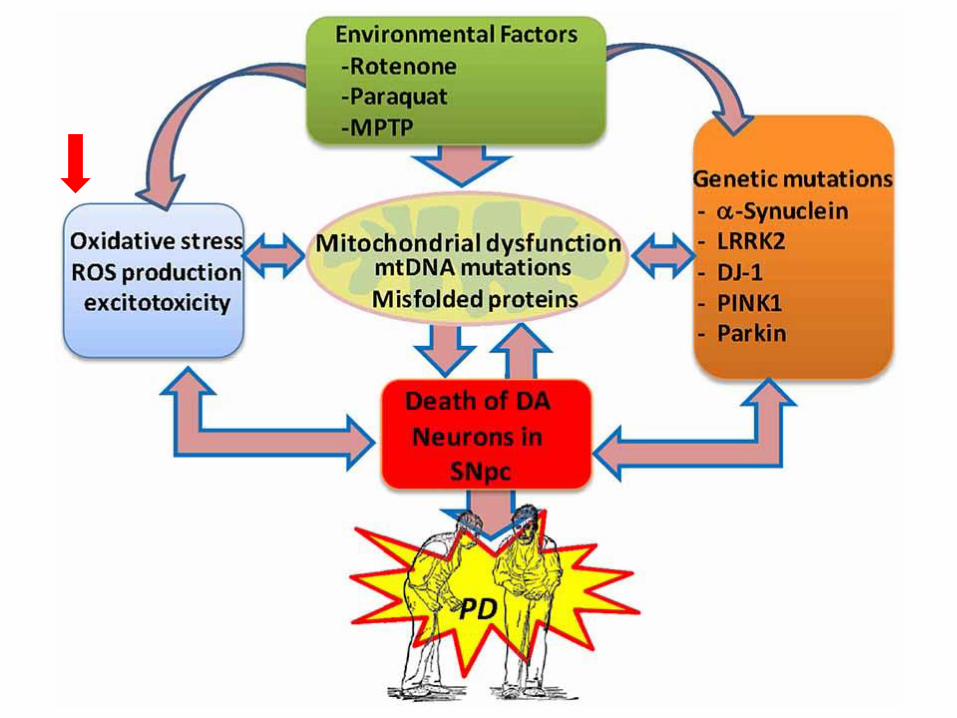
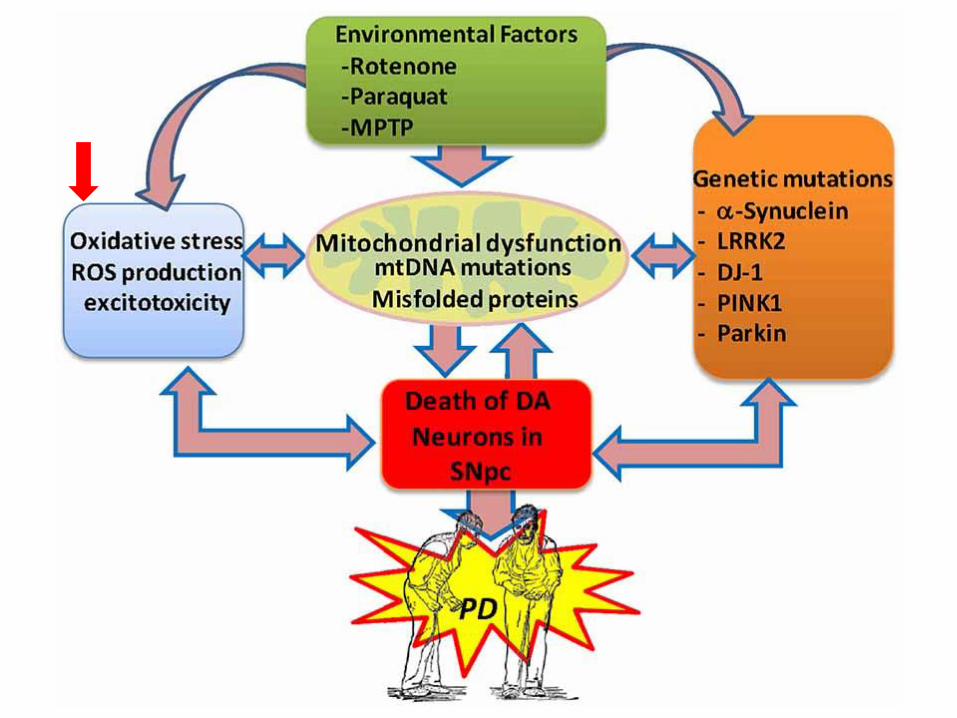
Meccanismi genetici coinvolti nella malattia di
Parkinson
α-sinucleina nel locus 4q21-23, (insorgenza intorno ai 45 anni e con decorso
rapido dei sintomi)
leucine-rich repeat kinase 2 (LRRK2), (interagisce con il c terminale della
parkina), la LRRK2 mutata causa apoptosi. Presente in citoplasma e mitocondri
DJ-1, nel locus 1p36, provoca modificazioni dell’ α-sinucleina
idrolasi dell’ubiquitina C-terminale L-1 (UHC-L1), nel locus 4p-14, (le proteine
destinate ad essere degradate dal sistema dell’ubiquitina vengono
preventivamente marcate con catene di poli-ubiquitina e successivamente
degradate dal proteasoma)
PINK 1 (PTEN-induced kinase 1) localizzata nei mitocondri
parkina, nel locus 6q25.2-27, (forme giovanili, con insorgenza intorno ai 32 anni,
caratterizzate dall’assenza di corpi di Lewy nel cervello) è una ubiquitina ligasi
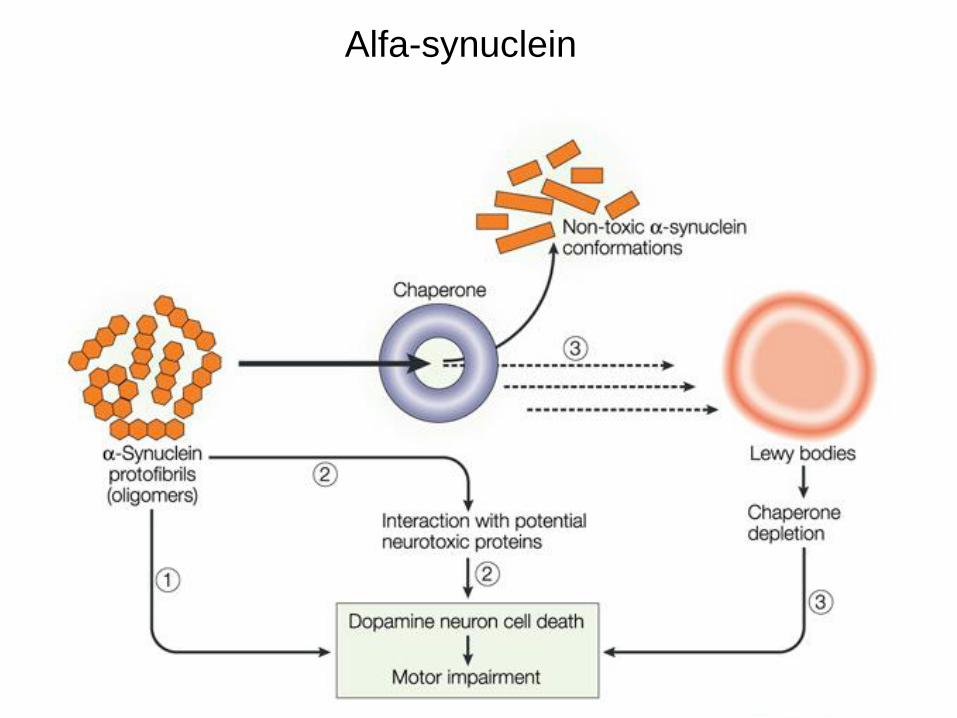
Alfa-synuclein
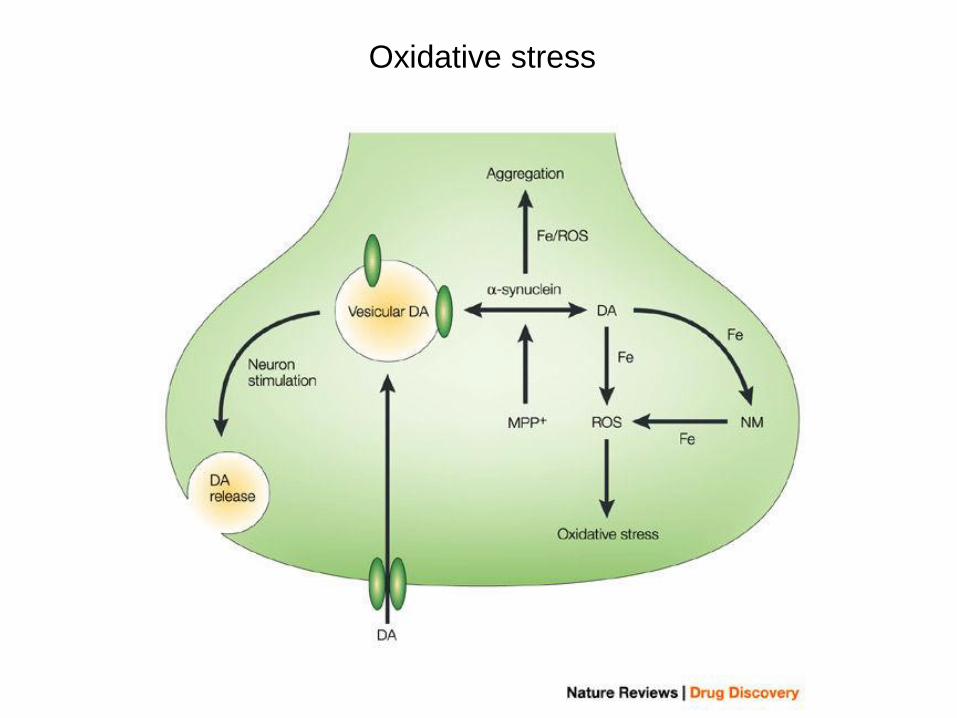
Oxidative stress
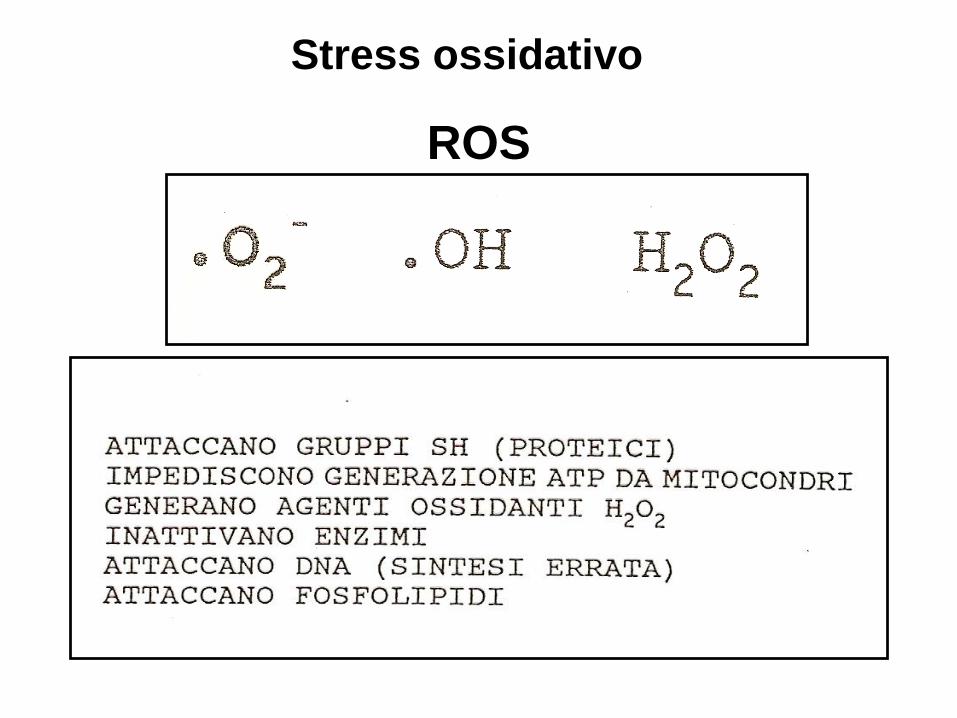
Stress ossidativo
ROS

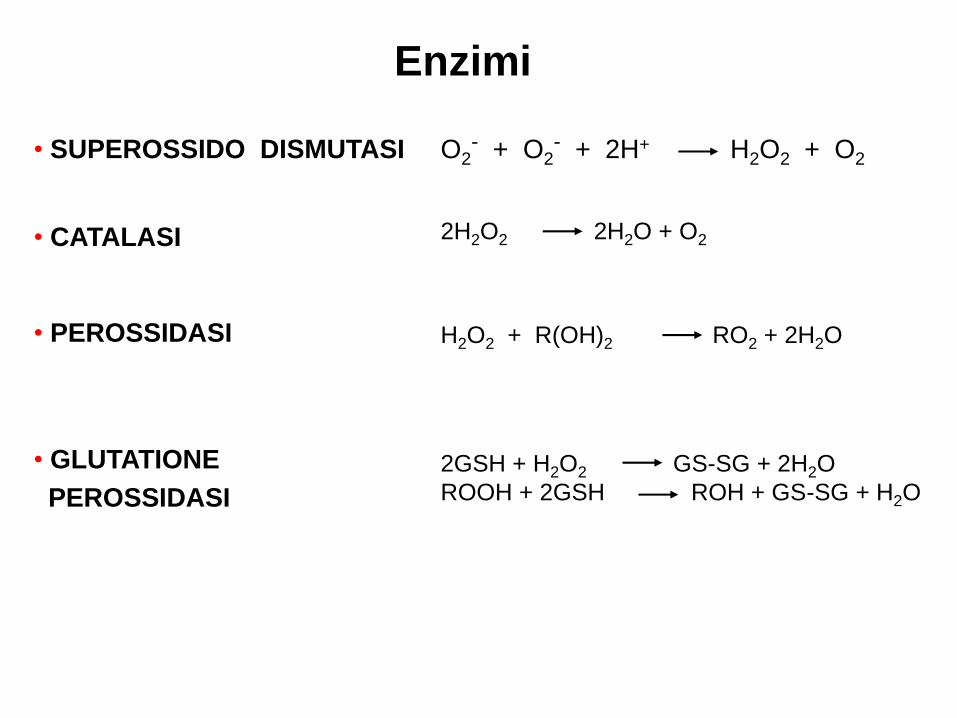
Enzimi
• SUPEROSSIDO DISMUTASI
• CATALASI
• PEROSSIDASI
• GLUTATIONE
PEROSSIDASI
O2- + O2
- + 2H+ H2O2 + O2
2H2O2 2H2O + O2
H2O2 + R(OH)2 RO2 + 2H2O
2GSH + H2O2 GS-SG + 2H2O
ROOH + 2GSH ROH + GS-SG + H2O
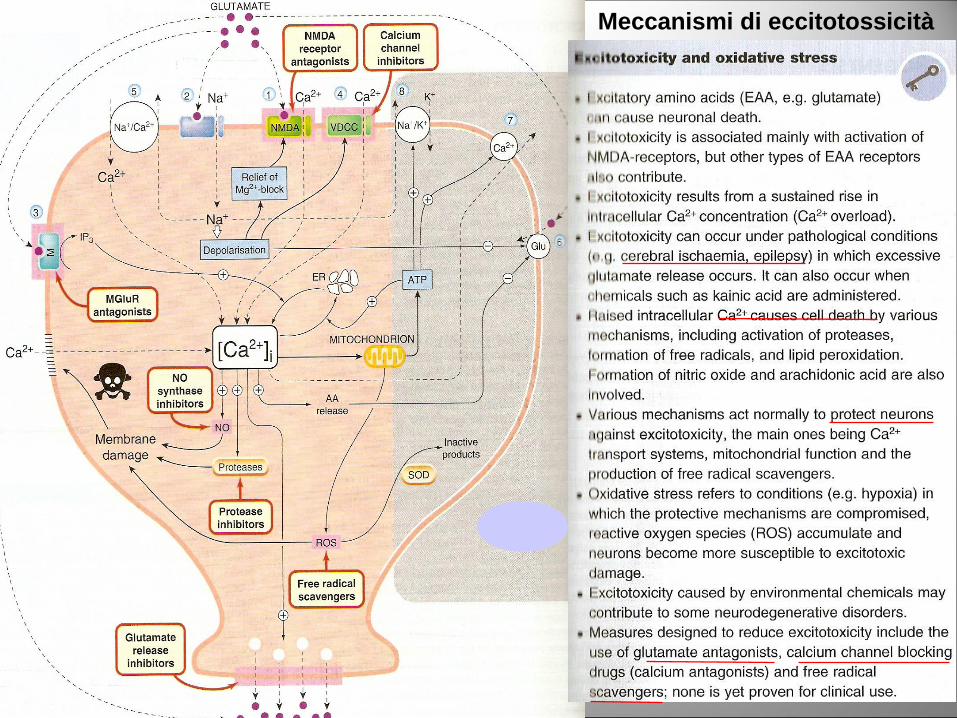
Meccanismi di eccitotossicità

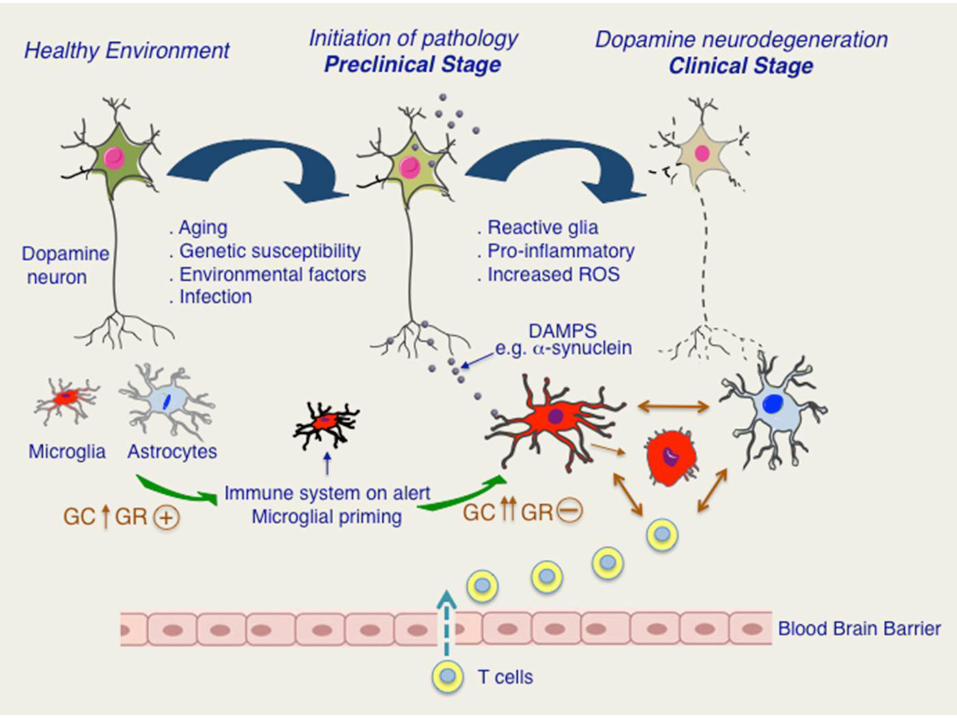
Neuroinfiammazione

Carenza di Glucocerebrosidasi
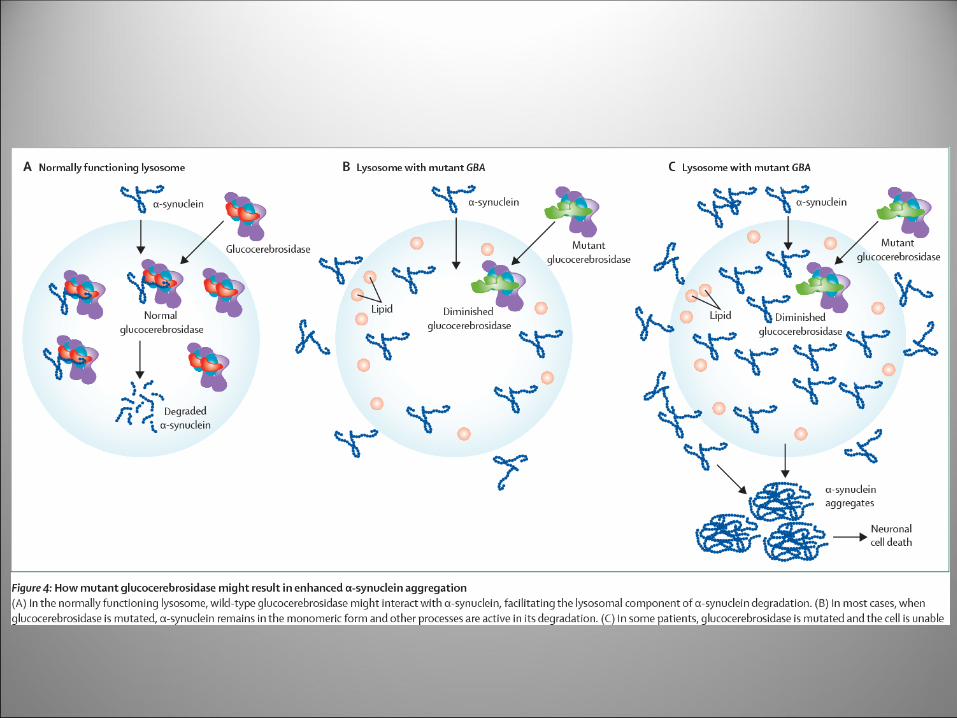

Mutations in the glucosylceramidase beta (GBA) gene are
associated with neurodegenerative diseases marked by
protein aggregation
GBA encodes the lysosomal enzyme glucocerebrosidase,
which breaks down glucosylceramide
The link between GBA mutations and protein aggregation is
that lysosomal accumulation of glucosylceramide causes
impaired autophagy
Changes in the turnover and abundance of proteins is
associated with extracellular vesicles (EVs), which are
vehicles for the spread of protein aggregates in
neurodegenerative disease
Gba1b mutants had six times as many EVs as controls
EV abundance contributed to the accumulation of protein
aggregates.
Glucocerebrosidase deficiency causes pathogenic changes
in EV metabolism and may promote the spread of protein
aggregates through extracellular vesicles
![Einzelmolekülanalyse von eiseninduzierten, porenbildenden ... · sich der Morbus Parkinson häufiger in Europa und Nordamerika als in Asien [160]. Hinsichtlich der jährlichen Neuerkrankungen](https://static.fdocument.org/doc/165x107/5e226280e00ea56e94608285/einzelmoleklanalyse-von-eiseninduzierten-porenbildenden-sich-der-morbus-parkinson.jpg)

















