
FABULOUS 2016 - Preliminary Program in Full is found that torsion angle, τ, defined by the...
15
This event is endorsed and organized by FABULOUS 2016 - Preliminary Program in Full http://fabulous-conf.org/2016/show/program-preliminary 1 of 5 18/12/2017, 16:46
Transcript of FABULOUS 2016 - Preliminary Program in Full is found that torsion angle, τ, defined by the...

This event is endorsedand organized by
FABULOUS 2016 - Preliminary Program in Full http://fabulous-conf.org/2016/show/program-preliminary
1 of 5 18/12/2017, 16:46

FABULOUS 2016 - Preliminary Program in Full http://fabulous-conf.org/2016/show/program-preliminary
2 of 5 18/12/2017, 16:46

FABULOUS 2016 - Preliminary Program in Full http://fabulous-conf.org/2016/show/program-preliminary
3 of 5 18/12/2017, 16:46
This event is endorsedand organized by
FABULOUS 2016 - Organizing Committee http://fabulous-conf.org/2016/show/org-com
1 of 4 18/12/2017, 16:48

FABULOUS 2016 - Organizing Committee http://fabulous-conf.org/2016/show/org-com
2 of 4 18/12/2017, 16:48

EAI Institutional Members
FABULOUS 2016 - Organizing Committee http://fabulous-conf.org/2016/show/org-com
3 of 4 18/12/2017, 16:48
This event is endorsedand organized by
FABULOUS 2016 - Technical Program Committee http://fabulous-conf.org/2016/show/prog-com
1 of 3 18/12/2017, 16:49
EAI Institutional Members
FABULOUS 2016 - Technical Program Committee http://fabulous-conf.org/2016/show/prog-com
2 of 3 18/12/2017, 16:49
This event is endorsedand organized by
FABULOUS 2016 - Steering Committee http://fabulous-conf.org/2016/show/steering-com
1 of 3 18/12/2017, 16:48

Experimental and theoretical study of the UV-Vis
spectrum of a new coumarine-derived ligand
Dejan Milenković1*, Srećko Trifunović2, Edina Avdović2, Nenad Vuković2, Milena
Vukić2, Jasmina Dimitrić-Marković3, and Zoran Marković1,4
1Bioengineering Research and Development Center, Prvoslava Stojanovića 6, 34000
Kragujevac, Serbia 2Faculty of Science, University of Kragujevac, 12 Radoja Domanovića, P.O. Box 60, 34000
Kragujevac, Serbia 3Faculty of Physical Chemistry, University of Belgrade, Studentski trg 12-16,11000
Belgrade, Republic of Serbia 4Department of Chemical-Technological Sciences, State University of Novi Pazar, Vuka
Karadžića bb, 36300 Novi Pazar, Serbia
Abstract. The UV-Vis properties of new coumarine-derived ligand (3-(1-(3-
hydroxypropylamino)propylidene)-chroman-2,4-dione) were investigated. The
time-dependent density functional theory (TDDFT) approach in combination
with the B3LYP functional was used for simulation of UV-Vis spectra of
examined compound. The agreement between the observed wavelengths and
intensities in the UV spectrum of ligand 1 and those predicted with B3LYP
functional, is satisfactory.
Keywords: UV-Vis spectra, TDDFT, B3LYP, coumarine-derived ligand
(ligand 1), oscillator strengths
1 Introduction
Coumarins (2H-1-benzopyran-2-one) consist of a large class of phenolic substances
found in plants and are made of fused benzene and 𝛼-pyrone rings [1]. Coumarine and
its derivatives are natural compounds with high and significant bilogical activities like
spasmolitic, antiarrhytmic, cardiotonic and photodynamic properties [2]. Аlso,
coumarine and its derivatives were tested against several tumor cell lines [3]. These
compounds can be found in different food sources such as fruits, herbs and
vegetables [4]. Coumarin and its derivatives have important role in the fields of
biology, medicine, industry, botany and chemistry. Some metal complexes with
coumarine derivatives showed significant anticoagulant [5] and antitumor activity
[2,6].
* Dejan Milenković, Bioengineering Research and Development Center, Prvoslava Stojanovića
6, 34000 Kragujevac, Serbia ([email protected])

2 Methodology section
The equilibrium geometry of coumarine-derived ligand (1) was optimized by density
functional theory (DFT) using B3LYP exchange correlation functional first proposed
by Becke [7], in combination with the 6–311+G(d,p) basis set. The optimization was
carried out using Gaussian 09 package [8]. The structure was optimized at 298 K
without any geometrical restrictions. The nature of the stationary points was
determined by performing frequency analysis: equilibrium geometries have no
imaginary vibrations. The effect of methanol as solvent was taken into account in
geometry optimization and energy calculation by using the SMD model [9].
To simulate the UV spectrum of investigated compound TDDFT (Time Dependent
Density Functional Theory) approach [10] was employed for predicting the electronic
transitions. TD calculation was performed in methanol. All transitions were
considered. All parameters important for the simulation of UV-VIS spectra such as
excited state energies, oscillator strengths (f), and a list of the transitions that gave rise
to each excited state were calculated. Natural Bond orbital (NBO) analysis [11] was
performed for the explanation of the interactions inside of molecule and visualization
of orbitals involved in electronic transitions. The B3LYP ground state geometry was
used to perform the NBO.
3 Results and discussion
3.1 Structural analysis
The structure of most stable conformation of ligand 1 defined by using B3LYP
local density functional method in conjunction with 6–311+G(d,p) basis set, in
methanol as solvent. From the optimized molecular structure of the ligand 1 (Fig. 1),
it is found that torsion angle, τ, defined by the C4–C3–C1'–N1 atoms is 172°, which
indicated nonplanarity of molecule. The investigated molecule has one internal
hydrogen bond (IHB) which additionally stabilize corresponding compound. For this
IHB the NBO analysis revealed that electron density is donated from the p and sp2
orbitals of the oxygen atoms into the proximate σ* antibonding H–N1 bond. These
donor-acceptor interactions are responsible for hydrogen bond formation.

Figure 1. Optimized geometry of the ligand 1 obtained at the B3LYP/6-311+G(d,p) level of
theory in methanol.
3.2 UV spectrum
The experimental and computed UV spectra are presented in Fig. 2, whereas
calculated electronic transitions, absorption wavelengths, and oscillator strength, as
well as experimental absorption wavelengths are presented in Table 1.
Table 1. UV spectral data for compound 1.
Expt B3LYP/6-311+G(d,p)
λmax (nm) λmax f Orbital description
319 294 0.52 H → L (66%)
243 248 0.18 H → L+1 (48%)
234 238 0.28 H-1→ L+1 (58%)
The experimental spectrum of 1 in of methanol shows two wide peaks at 319 and 243
nm. The theoretical investigation of electronic transition was also performed in
methanol. The UV spectrum of the compound 1 was simulated using the TDDFT
approach. In Table 1 are given data for the UV spectrum. The two absorption peaks
are prominent, i.e., at 294 nm which is absorption maximum and the second peak has
been observed at 248 nm which are in good agreement with the experimental
evidences (Table 1, Fig. 2). As mentioned above, electronic transitions have been
investigated. The first transition state at 294 nm dominated with maximum absorption
which is caused by HOMO →LUMO with 66% contribution. The second transition
state caused by HOMO → LUMO+1 at 248 nm with 48% contribution. The third
state has 58% contribution from HOMO-1 → LUMO+1. The orbitals cooresponding
for these electronic transitions were presenred in Fig. 3.
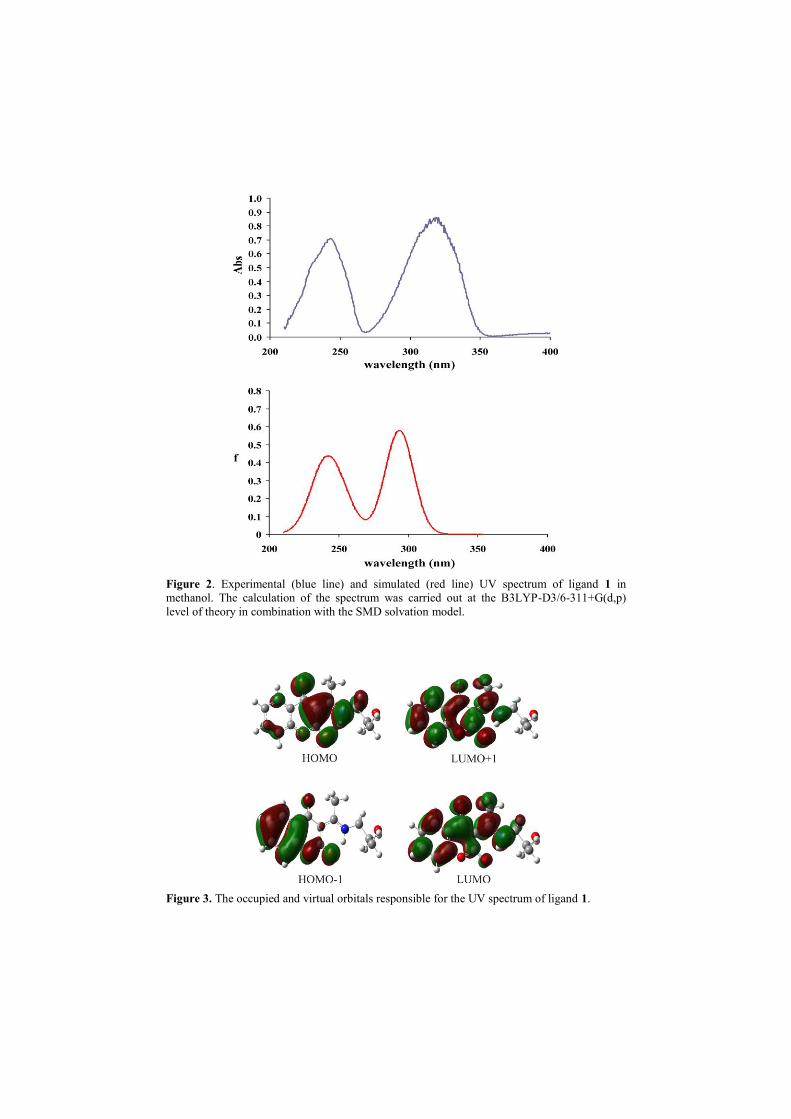
Figure 2. Experimental (blue line) and simulated (red line) UV spectrum of ligand 1 in
methanol. The calculation of the spectrum was carried out at the B3LYP-D3/6-311+G(d,p)
level of theory in combination with the SMD solvation model.
Figure 3. The occupied and virtual orbitals responsible for the UV spectrum of ligand 1.

The shapes of the corresponding orbitals confirm that these transitions were
associated with significant charge transfer between the coumarine moiety and side
alkyl chain.
4 Conclusion
All results were obtained using the B3LYP/6-311+G(d,p) level of theory. On the basis
of conformational analysis, it was found that 1 is the most stable structure. Re-
optimized structure of 1 in methanol was used to simulate the UV–vis spectra. Both
methods, experimental and theoretical showed as two major peaks. The results
showed that there is good agreement between experimental and calculated values for
λmax. On the basis of new facts, it is clear that this functional (B3LYP) can be used
successfully in the study of this class of compounds. According to the analysis of
frontier orbitals it was found that for these transitions was responsible charge transfer
between the coumarine moiety and side alkyl chain.
Acknowledgments. This work was supported by the Ministry of Science of the
Republic of Serbia (Projects Nos. 172015, 174028 and 172016).
References
1. Aoyama, Y., Katayama, T., Yamamoto, M., Tanaka, H., Kon, K.: A new antitumor
antibiotic product, demethylchartreusin. Isolation and biological activities. J. Antibiot. 45,
875-888 (1992)
2. Manolov, I., Kostova, I., Netzeva, T., Konstantinov, S., Karaivanova, M.: Cytotoxic
activity of cerium complexes with coumarin derivatives. Molecular modeling of the ligands.
Arch. Pharm. (Weinheim) 333, 93–98 (2000).
3. Weber, U.S., Steffen, B., Siegers, C.: Antitumor-activities of coumarin, 7- hydroxy-coumarin
and its glucuronide in several human tumor cell lines. Res. Commun. Mol. Pathol.
Pharmacol. 99, 193-206 (1998)
4. Kleiner, H.E., Vulimiri, S.V., Miller, L., Johnson, W.H. Whitman, C.P., DiGiovanni, J.: Oral
administration of naturally occurring coumarins leads to altered phase I and II enzyme
activities and reduced DNA adduct formation by polycyclic aromatic hydrocarbons in
various tissues of SENCAR mice. Carcinogenesis. 22, 73-82 (2001).
5. Jiang, D., Deng, R., Wu, J.: Synthesis and properties of lanthanide compounds of 3-sulfo-4-
hy-droxycoumarin. Wuji Huaxue. 5, 21–28 (1989)
6. Kostova, I., Manolov, I., Konstantinov, S., Karaivanova, M.: Synthesis, physicochemical
characterisation and cytotoxic screening of new complexes of cerium, lanthanum and
neodymium with Warfarin and Coumachlor sodium salts. Eur. J. Med. Chem. 34, 63–68
(1999)
7. Becke, A.D.: Density-functional exchange-energy approximation with correct asymptotic
behavior. Phys. Rev. A Gen. Phys. 38, 3098-3100 (1988)
8. Frisch, M. J., Trucks, G. W., Schlegel, H. B., et al. Gaussian 09, revision A.1-
SMP.Wallingford, CT: Gaussian, Inc. (2009).

9. Marenich, A.V., Cramer, C.J. Truhlar, D.G.: Universal solvation model based on solute
electron density and on a continuum model of the solvent defined by the bulk dielectric
constant and atomic surface tensions. J. Phys. Chem. B 113, 6378-6396 (2009)
10. Bauernschmitt, R., Ahlrichs, R.: Treatment of electronic excitations within the adiabatic
approximation of time dependentdensity functional theory. Chem. Phys. Lett. 256, 454-464
(1996)
11. Foster, J.P., Weinhold, F.: Natural hybrid orbitals. J. Am. Chem. Soc. 102, 7211–7218
(1980)


















