
Adição nucleofílica diastereosseletiva a iminas e...
256
1 UNIVERSIDADE FEDERAL DO RIO DE JANEIRO NÚCLEO DE PESQUISAS DE PRODUTOS NATURAIS Adição nucleofílica diastereosseletiva a iminas e nitroolefinas quirais oriundas de α-aminoácidos naturais: Síntese do Ácido-(2R,3S)-2- nitroisopropil-3-N,N-dibenzilamino-4-fenilbutanóico. A N D R É L U I Z D A S I L V A M O U R A Rio de Janeiro 2007
Transcript of Adição nucleofílica diastereosseletiva a iminas e...

1
UNIVERSIDADE FEDERAL DO RIO DE JANEIRO NÚCLEO DE PESQUISAS DE PRODUTOS NATURAIS
Adição nucleofílica diastereosseletiva a iminas e nitroolefinas quirais oriundas de α-aminoácidos naturais: Síntese do Ácido-(2R,3S)-2-
nitroisopropil-3-N,N-dibenzilamino-4-fenilbutanóico.
A N D R É L U I Z D A S I L V A M O U R A
Rio de Janeiro
2007

Livros Grátis
http://www.livrosgratis.com.br
Milhares de livros grátis para download.

2
André Luiz da Silva Moura
ADIÇÃO NUCLEOFÍLICA DIASTEREOSSELETIVA A IMINAS E NITROOLEFINAS QUIRAIS ORIUNDAS DE α-AMINOÁCIDOS NATURAIS: SÍNTESE DO ÁCIDO-(2R,3S)-2-NITROISOPROPIL-3-N,N-DIBENZILAMINO-4-FENILBUTANÓICO.
Tese de Doutorado apresentada ao Programa de Pós-Graduação em Química de Produtos Naturais, junto ao Núcleo de Pesquisas de Produtos Naturais (NPPN) da Universidade Federal do Rio de Janeiro, como parte dos requisitos necessários à obtenção do título de Doutor em Ciências.
Orientadora: Profª. Drª. Vera Lúcia Patrocinio Pereira.
Rio de Janeiro
2007

3
Ficha Catalográfica
Moura, André Luiz da Silva Adição nucleofílica diastereosseletiva a iminas e nitroolefinas quirais oriundas de α-aminoácidos naturais. Síntese do Ácido-(2R,3S)-2-nitroisopropil-3-N,N-dibenzilamino-4-fenilbutanóico. Rio de Janeiro, UFRJ, NPPN, 2007, xvi, 231 f. 1. Adição de Michael 4. Síntese de β-aminoácidos-α-substituídos 2. Nitroolefinas deficientes de elétrons 5. Nitrodiaminas quirais 3. Aldiminas quirais 6. Reação de Nef
Tese: Doutor em Ciências I – Universidade Federal do Rio de Janeiro Núcleo de Pesquisas de Produtos Naturais II – Título

4
A todos aqueles que me apoiaram e, de alguma forma, contribuíram para que este
trabalho fosse possível.

5
Agradecimentos
• À Professora Dra. Vera Lúcia Patrocinio Pereira, por todos estes dez anos de
convívio. • Ao amigo Leandro Lara de Carvalho, pelo apoio inestimável durante estes anos de
LASESB. • Aos companheiros do LASESB pela convivência. • À Central Analítica do NPPN e demais funcionários. • Ao CNPq pelo apoio financeiro. • A Daniel Macedo.

6
“La creación sigue incesantemente por medio del hombre. Pero el hombre no crea: descubre.”
Antoni Gaudí.
“Le hasard ne favorise que les esprits préparés.”
Louis Pasteur.
"Der erste Trunk aus dem Becher der Naturwissenschaft macht atheistisch; aber auf dem Grund des Bechers wartet Gott."
“Natural science does not simply describe and explain nature; it is part of the interplay between nature and ourselves; it describes nature as exposed to our method of questioning.”
Werner Heisenberg.
"Não quero ter a terrível limitação de quem vive apenas do que é passível de fazer sentido. Eu não: quero é uma verdade inventada."
Clarice Lispector.

i
SUMÁRIO
Lista de abreviaturas e fórmulas químicas...................................................................v
Índice de Esquemas e/ou Esquemas/Tabela................................................................vii
Índice de Esquemas retrossintéticos.............................................................................ix Índice de Quadros............................................................................................................x Índice de Figuras ............................................................................................................x
Índice de Espectros e Cromatogramas.........................................................................xi
RESUMO...........................................................................................................................xv
ABSTRACT .....................................................................................................................xvi
1. INTRODUÇÃO.
1.1 Nitroalcanos: Características gerais, ocorrência natural e atividade biológica, métodos de preparação e versatilidade sintética
1.1.1 - Características gerais...........................................................................................1 1.1.2 - Ocorrência natural e atividade biológica.............................................................2 1.1.3 - Métodos de preparação........................................................................................4 1.1.4 - Versatilidade sintética.........................................................................................5 1.2 Iminas: Características gerais e reatividade, métodos de preparação e adição
nucleofílica de nitroalcanos a iminas 1.2.1 - Características gerais e reatividade.....................................................................6 1.2.2 - Métodos de preparação......................................................................................10 1.2.3 - Adição nucleofílica de nitroalcanos a iminas – Reação de aza-Henry..............11 1.2.3.1 - Versão estereosseletiva da reação de aza-Henry...............................................14 1.2.3.1.1 - Utilização de catalisadores organometálicos..................................................14 1.2.3.1.2 - Utilização de catalisador orgânico.................................................................17 1.2.3.1.3 - Utilização da metodologia chiron approach..................................................20 1.3 Nitroolefinas: Características gerais, métodos de preparação e adições conjugadas
estereosseletivas a nitroolefinas 1.3.1 - Características gerais.........................................................................................24 1.3.2 - Métodos de preparação......................................................................................26 1.3.3 - Adições conjugadas estereosseletivas a nitroolefinas.......................................29 1.3.3.1- Adição de Michael enatiosseletiva empregando aditivos ou catalisadores enantiopuros.....................................................................................................................29 1.3.3.1.1 - Utilização de organocatalisador.....................................................................29 1.3.3.1.2 - Utilização de catalisador organometálico......................................................32 1.3.3.2- Adição de Michael enantiosseletiva empregando nucleófilos quirais...............35

ii
1.3.3.4- Adição de Michael diastereosseletiva controlada pelo substrato (metodologia chiron approach).............................................................................................................37 1.4 β-aminoácidos: Ocorrência natural e atividades biológicas, principais métodos de
obtenção 1.4.1 - Ocorrência natural e atividades biológicas........................................................39 2. OBJETIVOS .................................................................................................................42 3. ESTRATÉGIA SINTÉTICA..............................................................................................43 4. RESULTADOS E DISCUSSÃO.........................................................................................45 5. CONCLUSÃO................................................................................................................72 6. MATERIAIS E MÉTODOS..............................................................................................74 7. PARTE EXPERIMENTAL...............................................................................................76 7.1) Síntese dos ésteres 85a-d.........................................................................................76 7.1.1) Síntese do (2S)-N,N-dibenzilamino-4-metilpentanoato de benzila (85c)..............76 7.1.2) Síntese do (2S)-N,N-dibenzilamino-3-metilbutanoato de benzila (85d)...............79 7.1.3) Síntese do (2S)-N,N-dibenzilamino-3-fenilpropanoato de benzila (85b)..............84 7.1.4) Síntese do (2S)-N,N-dibenzilaminopropanoato de benzila (85a)..........................87 7.2) Síntese dos álcoois 86a-d ........................................................................................91 7.2.1) Síntese do (2S)-N,N-dibenzilamino-4-metil-1-pentanol (86c)..............................90 7.2.2) Síntese do (2S)-N,N-dibenzilamino-3-metil-1-butanol (86d)...............................93 7.2.3) Síntese do (2S)-N,N-dibenzilaminopropanol (86a)...............................................98 7.2.4) Síntese do (2S)-N,N-dibenzilamino-3-fenil-1-propanol (86b)............................103 7.3) Síntese dos aldeídos 87a-d ....................................................................................107 7.3.1) Síntese do (2S)-N,N-dibenzilamino-4-metilpentanal (87c).................................107 7.3.2) Síntese do (2S)-N,N-dibenzilaminopropanal (87a).............................................109 7.4) Síntese dos nitroálcoois 92a-d...............................................................................111 7.4.1) Síntese do (2R,3S)-1-nitro-3-N,N-dibenzilamino-4-fenil-2-butanol (92b).........111 7.4.2) Síntese do (2R,3S)-1-nitro-3-N,N-dibenzilamino-2-butanol (92a).....................116 7.4.3) Síntese do (3R,4S)-2-nitro-4-N,N-dibenzilamino-3-pentanol (92c)....................120 7.4.4) Síntese do (3R,4S)-2-nitro-4-N,N-dibenzilamino-6-metil-3-eptanol (92d).........124 7.5) Síntese das iminas 88a-d........................................................................................129 7.5.1) Síntese da (2S)-N,N-dibenzil-4-metil-1-N-benzilpentanaldimina (88c)..............129 7.5.2) Síntese da (2S)-N,N-dibenzil-3-fenil-1-N-benzilpropanaldimina (88b)..............131

iii
7.6) Síntese das nitroolefinas 93a,b...............................................................................135 7.6.1) Síntese do (1E,3S)-3-N,N-dibenzilamino-1-nitrobuteno (93a)...........................135 7.6.2) Síntese do (1E,3S)-3-N,N-dibenzilamino-4-fenil-1-nitrobuteno (93b)...............142 7.7) Síntese do 2-hidroxi-2-metilnitropropano (90) .....................................................148 7.8) Síntese do nitrobutano............................................................................................152 7.9) Procedimento geral para as reações de adição de nitroalcanos às iminas 88a-d...156 7.9.1) Síntese do (2R,3S)-2-N-benzilamino-3-N,N-dibenzilamino-5-metil-1-nitroexano (89a)...............................................................................................................................157 7.9.2) Síntese do (3R,4S)-3-N-benzilamino-4-N,N-dibenzilamino-2-nitroeptano (89b)..............................................................................................................................162 7.9.3) Síntese do (4S,5S)-3-nitro-4-N-benzilamino-5-N,N-dibenzilamino-7-metiloctanoato de metila (89c)......................................................................................166 7.9.4) Síntese do (4R,5S)-2,7-dimetil-3-nitro-4-N-benzilamino-5-N,N-dibenzilamino-2-octanol (89d)..................................................................................................................171 7.9.5) Síntese do (2R,3S)-2-N-benzilamino-3-N,N-dibenzilamino-1-nitrobutano (89e)...............................................................................................................................176 7.9.6) Síntese do (2R,3S)-2-N-benzilamino-3-N,N-dibenzilamino-4-fenil-1-nitrobutano (89f)...............................................................................................................................179 7.9.7) Síntese do (2S,3R)-1-fenil-2-N,N-dibenzilamino-3-N-benzilamino-4-nitroeptano (89g)...............................................................................................................................184 7.10) Procedimento geral para a síntese das nitroazidas 94b e 94e..............................188 7.10.1) Síntese do (2R,3S)-2-azido-3-N,N-dibenzilamino-4-fenil-1-nitrobutano (94e)...............................................................................................................................188 7.10.2) Síntese do (2R,3S)-2-azido-3-N,N-dibenzilamino-1-nitrobutano (94b)............194 7.11) Procedimento geral para a síntese dos nitroéteres 94a e 94d...............................197 7.11.1) Síntese do (2R,3S)-2-metoxi-3-N,N-dibenzilamino-1-nitrobutano (94a).........199 7.11.2) Síntese do (2R,3S)-2-metoxi-3-N,N-dibenzilamino-4-fenil-1-nitrobutano (94d)..............................................................................................................................199 7.12) Síntese das dinitroaminas 94c e 94f (Procedimento I).........................................203 7.12.1) Síntese do (2S,3S)-1-fenil-2-N,N-dibenzilamino-3-nitrometil-4-metil-4-nitropentano (94f)..........................................................................................................203

iv
7.12.2) Síntese do (3S,4S)-2-metil-3-nitrometil-4-N,N-dibenzilamino-2-nitropentano (94c)...............................................................................................................................208 7.13) Síntese da dinitroamina 94f (Procedimento II)....................................................215 7.13.1) Síntese do (2S,3S)1-fenil-2-N,N-dibenzilamino-3-nitrometil-4-metil-4-nitropentano (94f)..........................................................................................................215 7.14) Síntese do (3S)-dibenzilamino-4-fenil-2-metilen-butanonitrila (94g).................216 7.15) Síntese do β-aminoácido 95f: Ácido-(2R,3S)-2-nitroisopropil-3-N,N-dibenzilamino-4-fenilbutanóico.....................................................................................220 7.16) Síntese do β-aminoácido 95d: Ácido-(2R,3S)-2-metoxi-3,3-N,N-bibenzilamino-4-fenilbutanóico................................................................................................................225 REFERÊNCIAS................................................................................................................227
v
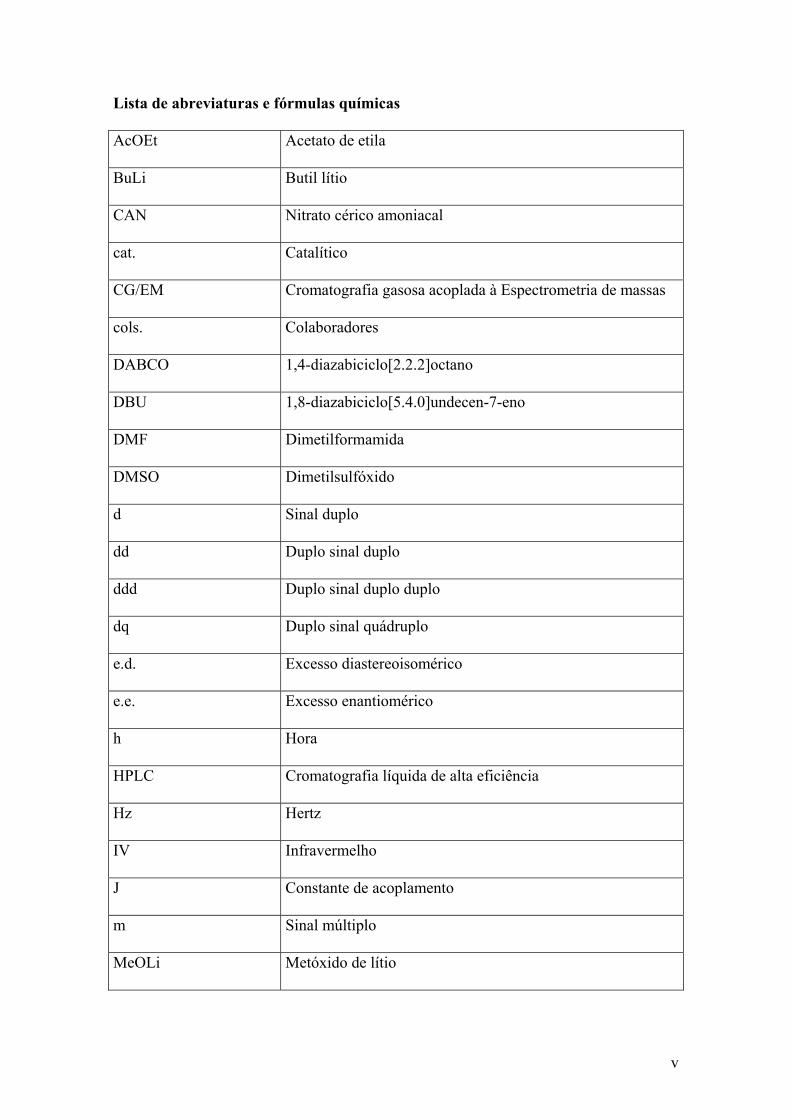
Lista de abreviaturas e fórmulas químicas
AcOEt Acetato de etila
BuLi Butil lítio
CAN Nitrato cérico amoniacal
cat. Catalítico
CG/EM Cromatografia gasosa acoplada à Espectrometria de massas
cols. Colaboradores
DABCO 1,4-diazabiciclo[2.2.2]octano
DBU 1,8-diazabiciclo[5.4.0]undecen-7-eno
DMF Dimetilformamida
DMSO Dimetilsulfóxido
d Sinal duplo
dd Duplo sinal duplo
ddd Duplo sinal duplo duplo
dq Duplo sinal quádruplo
e.d. Excesso diastereoisomérico
e.e. Excesso enantiomérico
h Hora
HPLC Cromatografia líquida de alta eficiência
Hz Hertz
IV Infravermelho
J Constante de acoplamento
m Sinal múltiplo
MeOLi Metóxido de lítio

vi
min Minuto
μ Momento de dipolo
Nu Nucleófilo
PMP para-metoxifenila
pTsOH Ácido para-toluenossulfônico
q Sinal quádruplo
r.d. Razão diastereoisomérica
rend. Rendimento
RMN 13C Ressonância Magnética Nuclear de Carbono 13
RMN 1H Ressonância Magnética Nuclear de Hidrogênio
[α]D25 Rotação óptica específica medida na raia D do sódio a 25oC.
sl Sinal largo
solv. Solvente
t Sinal triplo
t.a. Temperatura ambiente
TBAF Fluoreto de tetrabutilamônio
TBDMS t-butildimetilsilano
TEA Trietilamina
THF Tetraidrofurano
UV Ultravioleta

vii
Índice de Esquemas e/ou Esquemas/Tabela
Esquema 1: Formação do ânion 1-nitropropano estabilizado por estruturas de
ressonância.........................................................................................................................1
Esquema/Tabela 2: Reações de obtenção de nitroalcanos..............................................4
Esquema 3: Reação secundária de formação de azaenolatos promovida pelo uso de
reagentes organometálicos.................................................................................................8
Esquema 4: Formação da imina 2 através da adição de benzilamina ao aldeído quiral
1.......................................................................................................................................10
Esquema 5: Formação de iminas cíclicas 6 a partir da reação de aza-Wittig................10
Esquema/Tabela 6: Síntese de iminas com o emprego de microondas e do catalisador
sólido Envirocat EPZG®..................................................................................................11
Esquema 7: Adição de ânions nitronatos a N-(p-metoxibenzil)-iminas. Síntese de 1,2-
diaminas 9a-d..................................................................................................................13
Esquema/Tabela 8: Adição de nitrometano às N-fosfinoiliminas 12a-d na presença de
catalisador heterobimetálico 13.......................................................................................14
Esquema/Tabela 9: Adição de nitroalcanos ao α-iminoéster N-protegido 15 na
presença do catalisador organometálico 17.....................................................................15
Esquema/Tabela 10: Síntese das nitroaminas 20a-d através de catálise
organometálica.................................................................................................................17
Esquema/Tabela 11: Adição de nitrometano às N-fosfonoiliminas 21a,b e 22a-c.......18
Esquema/Tabela 12: Síntese das nitroaminas 27a-e através da reação de aza-Henry
catalisada pela tiouréia 26................................................................................................19
Esquema 13: Adição do sililnitronato 29 à N-PMP-N,N-dibenzilfenilalaninaldimina
28.....................................................................................................................................20
Esquema 14: Redução da nitrodiamina 30 às triaminas 31a,b.......................................21
Esquema 15: Obtenção das α-amidoalquilfenilsulfonas 33a,b a partir do (R)-
gliceraldeídoacetonídeo...................................................................................................22
Esquema 16: Obtenção das 1,2 nitroaminas N-protegidas 35a,b...................................22
Esquema/Tabela 17: Síntese das 3-nitropirrolidinas-2-substituídas 40a-d...................23
Esquema 18: Adição conjugada de espécieis nucleofílicas a nitroolefinas...................24
Esquema 19: Reação de Diels-Alder empregando o nitroestireno como dienófilo........25
Esquema 20: Reação de Diels-Alder empregando nitroolefina 43 como heterodieno...26
Esquema 21: Formação de nitroolefinas a partir de nitroálcoois...................................27

viii
Esquema/Tabela 22: Síntese das nitroolefinas 47a-e a partir da desidratação dos
nitroálcoois 46a-e............................................................................................................27
Esquema/Tabela 23: Síntese de nitroolefinas 49a-e empregando zeólitas....................28
Esquema 24: Adição de Michael estereosseletiva utilizando β-cetoésteres-γ,δ-
insaturados 50a-d na presença da tiouréia 51 como catalisador.....................................29
Esquema 25: Análise retrossintética da síntese total da (-)-Epibatidina........................30
Esquema/Tabela 26: Adição conjugada de 2-nitropropano a derivados de nitroestirenos
59a – k mediada por catalisador quiral 60 derivado de alcalóides da Cinchona sp........31
Esquema 27: Obtenção de substâncias 1,3-dinitradas 64 e 65a-e utilizando o catalisador
organometálico 62.........................................................................................33
Esquema 28: Obtenção da tiouréia 67 a partir da substância 1,3-dinitrada 65a...........33
Esquema 29: Síntese do (+)-Ibuprofen...........................................................................34
Esquema 30: Adição de enolato do (S)-ácido mandélico 71 ao nitroestireno................35
Esquema 31: Obtenção da lactama 73 através da redução do grupamento nitro seguida
de aminólise do acetal 72a...............................................................................................35
Esquema 32: Síntese de α-aminoácidos 77a-d a partir da adição da (R)-4-fenil-2-
oxazolidinona a nitroolefinas aquirais 74a-d..................................................................36
Esquema/Tabela 33: Adição conjugada de reagentes organometálicos às nitroolefinas
78 e 80.............................................................................................................................37
Esquema/Tabela 34: Adição conjugada de reagentes organometálicos funcionalizados
à nitroolefina 82...............................................................................................................38
Esquema 35: Síntese dos ésteres N,N-dibenzilbenzílicos 85a-d oriundos dos
aminoácidos naturais 84a-d.............................................................................................45
Esquema 36: Redução dos ésteres perbenzilados 85a-d................................................46
Esquema 37: Oxidação de Swern de 86a-d....................................................................46
Esquema 38: Síntese das N,N-dibenzil-N-benzilaldiminas 88a-d..................................47
Esquema/Tabela 39: Diastereosseletividade da adição de ânions nitronatos às
aldiminas quirais enantiopuras 88a-d..............................................................................48
Esquema 40: Tentativas de N-proteção da amina secundária 89a.................................51
Esquema 41: Tentativa de acetilaçao da nitrodiamina 89e e geração da nitroolefina 93a
por eliminação.................................................................................................................52
Esquema 42: Tentativa de desnitração da nitrodiamina 89g..........................................52
Esquema 43: Possibilidade de cristalização através de derivatização da amina
secundária 89a.................................................................................................................53

ix
Esquema/Tabela 44: Síntese diastereosseletiva dos nitroálcoois 92a-d........................54
Esquema 45: Obtenção dos nitroálcoois 92a-c via metodologia de Hanessian.............55
Esquema 46: Síntese das nitroolefinas quirais 94a-b.....................................................56
Esquema/Tabela 47: Adição conjugada diastereosseletiva de variados nucleófilos às
nitroolefinas quirais 94a-b...............................................................................................58
Esquema 48: Adição conjugada do ânion cianeto a 93b, seguida de eliminação de
ácido nitroso....................................................................................................................61
Esquema 49: Reatividade de ânions nitronatos como espécies 1,3-dipolares................62
Esquema 50: Adição de 2-nitropropano à nitroolefina 93a: Obtenção majoritária de um
provável produto de cicloadição [3 + 2] (100) segundo análise de CG/EM, RMN de 1H
e IV..................................................................................................................................63
Esquema 51: Justificativa de quebra para a formação do fragmento m/z = 224 no
Espectro 101....................................................................................................................64
Esquema 52: Formação do produto de cicloadição 1,3-dipolar 100 através da
aproximação exo e ataque pela face Re-Re do dipolarófilo 93a......................................65
Esquema/Tabela 53: Síntese do β-aminoácido 95f por vários metodologias da reação
de Nef..............................................................................................................................67
Esquema 54: Mecanismo proposto por Criegee da reação de ozonólise frente a olefinas
substituídas......................................................................................................................68
Esquema 55: Proposta mecanística para a ozonólise do sal de nitronato 105 proveniente
da dinitroamina 94f..........................................................................................................69
Esquema 56: Ozonólise de olefinas mono-substituídas gerando ésteres metílicos........70
Esquema 57: Obtenção do β-aminoácido 95d................................................................71
Índice de Esquemas retrossintéticos
Esquema retrossintético 1: Obtenção diastereosseletiva das nitroaminas 89a-g a partir
da adição de nitroalcanos às iminas 88a-c oriundas de L-aminoácidos naturais............43
Esquema retrossintético 2: Obtenção diastereosseletiva do β-aminoácido-α-
substituído 95a-g.............................................................................................................44

x
Índice de Quadros
Quadro 1: Algumas substâncias naturais portadoras do grupamento nitro e sua
atividade biológica.............................................................................................................2
Quadro 2: Algumas drogas sintéticas que portam o grupo nitro em sua estrutura..........3
Quadro 3: Alguns nitroalcanos comerciais e seus respectivos preços.............................4
Quadro 4: Eletrofilicidade de iminas comparada a aldeídos e cetonas............................7
Quadro 5: Exemplos de iminas N-ativadas......................................................................9
Quadro 6: Incorporação da β-Alanina na estrutura do (R)-(+)-Ácido Pantotênico........39
Quadro 7: Alguns produtos naturais com atividade biológica que exibem em sua
estrutura um β-aminoácido..............................................................................................41
Índice de Figuras
Figura 1: Formação do ânion nitronato a partir do nitroetano tratado com base.............1
Figura 2: Conversão do grupo nitro para diversas funcionalidades.................................5
Figura 3: Obtenção de substâncias aminadas a partir da adição nucleofílica a iminas....6
Figura 4: Reatividade das iminas frente a uma adição nucleofílica na presença de
reagente organometálico....................................................................................................8
Figura 5: Obtenção de 1,2-diaminas e α-aminoácidos a partir de iminas N-
protegidas.........................................................................................................................12
Figura 6: Modelo de estado de transição em cadeira para a catálise enantiosseletiva
empregando 17................................................................................................................16
Figura 7: Abstração do hidrogênio α-nitro pelo catalisador tiouréia 25.......................19
Figura 8: Energias das estruturas de transição otimizadas através do método semi-
empírico PM3 levando aos nitroderivados A [(S,S)-35a] e B [(S,R)-35a]......................23
Figura 9: Possibilidades estruturais de β-aminoácidos acíclicos e cíclicos....................40
Figura 10: Classificação de β-aminoácidos segundo Seebach.......................................40
Figura 11: Modelo de estado de transição de Felkin-Ahn para o ataque nucleofílico de
ânions nitronatos à leucinaldimina 88d...........................................................................50
Figura 12: Projeções de Newman para o ataque nucleofílico de nitronatos aos aldeídos
87a-b................................................................................................................................56
Figura 13: a) Representação da formação de espécie 1,3-dipolo; b) Faces
diastereotópicas da nitroolefina 93a (dipolarófilo).........................................................63

xi
Figura 14: Modelo de estado de transição de Felkin-Ahn para o ataque nucleofílico de
variados nucleófilos à nitroolefina 93b...........................................................................66
Índice de Espectros e Cromatogramas
Espectro 1: RMN de 1H de 85c......................................................................................77
Espectro 2: RMN de 13C de 85c.....................................................................................78
Espectro 3: RMN de 1H de 85d......................................................................................80
Espectro 4: RMN de 13C de 85d.....................................................................................81
Espectro 5: APT de 85d.................................................................................................82
Espectro 6: Infravermelho de 85d..................................................................................83
Espectro 7: RMN de 1H de 85b......................................................................................85
Espectro 8: RMN de 13C de 85b.....................................................................................86
Espectro 9: RMN de 1H de 85a......................................................................................88
Espectro 10: RMN de 13C de 85a...................................................................................89
Espectro 11: RMN de 1H de 86c....................................................................................91
Espectro 12: RMN de 13C de 86c...................................................................................92
Espectro 13: RMN de 1H de 86d....................................................................................94
Espectro 14: RMN de 13C de 86d...................................................................................95
Espectro 15: APT de 86d...............................................................................................96
Espectro 16: Infravermelho de 86d................................................................................97
Espectro 17: RMN de 1H de 86a....................................................................................99
Espectro 18: RMN de 13C de 86a.................................................................................100
Espectro 19: APT de 86a.............................................................................................101
Espectro 20: Infravermelho de 86a..............................................................................102
Espectro 21: RMN de 1H de 86b..................................................................................104
Espectro 22: RMN de 13C de 86b.................................................................................105
Espectro 23: APT de 86b.............................................................................................106
Espectro 24: RMN de 1H de 87c..................................................................................108
Espectro 25: RMN de 1H de 87a..................................................................................110
Espectro 26: RMN de 1H de 92b..................................................................................112
Espectro 27: RMN de 13C de 92b.................................................................................113
Espectro 28: APT de 92b.............................................................................................114
Espectro 29: Infravermelho de 92b..............................................................................115

xii
Espectro 30: RMN de 1H de 92a..................................................................................117
Espectro 31: RMN de 13C de 92a.................................................................................118
Espectro 32: APT de 92a..............................................................................................119
Espectro 33: RMN de 1H de 92c..................................................................................121
Espectro 34: RMN de 13C de 92c.................................................................................122
Espectro 35: APT de 92c..............................................................................................123
Espectro 36: RMN de 1H de 92d..................................................................................125
Espectro 37: RMN de 13C de 92d.................................................................................126
Espectro 38: APT de 92d.............................................................................................127
Espectro 39: Infravermelho de 92d..............................................................................128
Espectro 40: RMN de 1H de 88c..................................................................................130
Espectro 41: RMN de 1H de 88b..................................................................................132
Espectro 42: RMN de 13C de 88b.................................................................................133
Espectro 43: APT de 88b.............................................................................................134
Espectro 44: RMN de 1H de 93a..................................................................................136
Espectro 45: RMN de 13C de 93a.................................................................................137
Espectro 46: APT de 93a..............................................................................................138
Espectro 47: Fragmentograma de 93a..........................................................................140
Espectro 48: Infravermelho de 93a..............................................................................141
Espectro 49: RMN de 1H de 93b..................................................................................143
Espectro 50: RMN de 13C de 93b.................................................................................144
Espectro 51: APT de 93b.............................................................................................145
Espectro 52: Fragmentograma de 93b..........................................................................146
Espectro 53: Infravermelho de 93b..............................................................................147
Espectro 54: RMN de 1H de 90....................................................................................149
Espectro 55: RMN de 13C de 90...................................................................................150
Espectro 56: Infravermelho de 90................................................................................151
Espectro 57: RMN de 1H do nitrobutano.....................................................................153
Espectro 58: Fragmentograma do nitrobutano.............................................................154
Espectro 59: Infravermelho do nitrobutano.................................................................155
Espectro 60: RMN de 1H de 89a..................................................................................158
Espectro 61: RMN de 13C de 89a.................................................................................159
Espectro 62: APT de 89a..............................................................................................160
Espectro 63: Infravermelho de 89a..............................................................................161

xiii
Espectro 64: RMN de 1H de 89b..................................................................................163
Espectro 65: RMN de 13C de 89b.................................................................................164
Espectro 66: Expansão do espectro 65.........................................................................165
Espectro 67: RMN de 1H de 89c..................................................................................167
Espectro 68: RMN de 13C de 89c.................................................................................168
Espectro 69: APT de 89c..............................................................................................169
Espectro 70: Infravermelho de 89c..............................................................................170
Espectro 71: RMN de 1H de 89d..................................................................................172
Espectro 72: RMN de 13C de 89d.................................................................................173
Espectro 73: APT de 89d.............................................................................................174
Espectro 74: Infravermelho de 89d..............................................................................175
Espectro 75: RMN de 1H de 89e..................................................................................177
Espectro 76: RMN de 13C de 89e.................................................................................178
Espectro 77: RMN de 1H de 89f...................................................................................180
Espectro 78: RMN de 13C de 89f..................................................................................181
Espectro 79: APT de 89f..............................................................................................182
Espectro 80: Infravermelho de 89f...............................................................................183
Espectro 81: RMN de 1H de 89g..................................................................................185
Espectro 82: RMN de 13C de 89g.................................................................................186
Espectro 83: APT de 89g..............................................................................................187
Espectro 84: RMN de 1H de 94e..................................................................................189
Espectro 85: APT de 94e..............................................................................................190
Espectro 86: Infravermelho de 94e..............................................................................191
Espectro 87: Fragmentograma do diastereoisômero majoritário de 94e......................193
Espectro 88: RMN de 1H de 94b..................................................................................195
Espectro 89: Infravermelho de 94b..............................................................................196
Espectro 90: RMN de 1H de 94a..................................................................................198
Espectro 91: RMN de 1H de 94d..................................................................................200
Espectro 92: RMN de 13C de 94d.................................................................................201
Espectro 93: Infravermelho de 94d..............................................................................202
Espectro 94: RMN de 1H de 94f...................................................................................204
Espectro 95: RMN de 13C de 94f..................................................................................205
Espectro 96: APT de 94f..............................................................................................206
Espectro 97: Infravermelho de 94f...............................................................................207

xiv
Espectro 98: RMN de 1H de 95c e provável produto de cicloadição 100....................209
Espectro 99: Expansão do espectro 98.........................................................................210
Espectro 100: Fragmentograma do possível produto de cicloadição1,3-dipolar.........212
Espectro 101: Fragmentograma de 94c........................................................................213
Espectro 102: Infravermelho de 94c e produto de cicloadição....................................214
Espectro 103: RMN de 1H de 94g................................................................................217
Espectro 104: RMN de 13C de 94g...............................................................................218
Espectro 105: Infravermelho de 94g............................................................................219
Espectro 106: APT de 95f............................................................................................223
Espectro 107: Infravermelho de 95f.............................................................................224
Espectro 108: APT de 95d...........................................................................................226
Cromatograma 1: Nitroolefina 93a.............................................................................133
Cromatograma 2: Identificação dos dois diastereoisômeros de 94e...........................192
Cromatograma 3: : Indicação de produto majoritário diferente de 94c......................211

xv
RESUMO
Moura, André Luiz da Silva. Adição nucleofílica diastereosseletiva a iminas e nitroolefinas quirais oriundas de α-aminoácidos naturais. Síntese do Ácido-(2R,3S)-2-nitroisopropil-3-N,N-dibenzilamino-4-fenilbutanóico. Rio de Janeiro, 2007. Tese (Doutorado em Química de Produtos Naturais) – Núcleo de Pesquisas de Produtos Naturais, Universidade Federal do Rio de Janeiro, Rio de Janeiro, 2007. A diastereosseletividade da reação de adição nucleofílica de diversos ânions nitronatos às aldiminas quirais enatiomericamente enriquecidas sintetizadas a partir de α-aminoácidos naturais como L-valina, L-leucina, L-alanina e L-fenilalanina foi investigada. Os ânions nitronatos foram gerados a partir dos nitroalcanos correspondentes na presença da resina básica Amberlist A-21 sem solvente.
Um outro parâmetro investigado por nós consisitiu na utilização de nitroalcanos volumosos, sintetizados em nosso grupo de pesquisa, e sua influência na diastereosseletividade desta reação. As correspondentes nitrodiaminas oriundas da reação de aza-Henry foram obtidas com rendimentos químicos que variaram na faixa de moderados (55-81%) e os excessos diastereoisoméricos obtidos variaram de moderados a bons (66-80%). Adicionalmente, não foi observado sinal de racemização ou de outras reações secundárias. Aumentando o escopo do estudo da diastereosseletividade de adições nucleofílicas a eletrófilos quirais oriundos de α-aminoácidos naturais, foram sintetizadas nitroolefinas quirais deficientes de elétrons, até então inéditas na literatura, a partir da L-alanina e L-fenilalanina, onde foram empregadas no estudo da adição de conjugada de variados nucleófilos. Diversas condições reacionais como base, tempo, solvente e temperatura foram estudadas. Foram obtidos os adutos em uma faixa de rendimento químico moderado (58 a 77%) e os excessos diastereoisoméricos variaram de baixos a bons (38 a 86%). Não foi observado sinal de racemização nestas reações, embora em um único caso, nós constatamos a ocorrência de uma reação de cicloadição [3 + 2] que ocorrera em concomitância com adição conjugada. Adicionalmente, estendemos a aplicação desta nova metodologia para a síntese de β-aminoácidos, onde variados métodos para a reação de Nef também foram investigados.

xvi
ABSTRACT Moura, André Luiz da Silva. Diastereoselective nucleophilic addition to chiral imines and nitroolefins from natural α-amino acids. Synthesis of (2R,3S)-2-nitroisopropyl-3-N,N-dibenzylamino-4-phenylbutanoic acid. Rio de Janeiro, 2007. Tese (Doutorado em Química de Produtos Naturais) – Núcleo de Pesquisas de Produtos Naturais, Universidade Federal do Rio de Janeiro, Rio de Janeiro, 2007.
The nucleophilic addition of several nitronate anions to chiral enantiomeric enriched aldimines synthetized from natural α-amino acids such as L-valine, L-leucine, L-alanine and L-phenylalanine had its diastereoselectivity investigated. The nitronate anions were generated from corresponding nitroalkanes in presence of basic resin Amberlyst A-21 neat.
Another analyzed parameter has consisited on the use of bulky nitroalkanes, produced by our research group, and its influence on the diastereoselectivity of this reaction. The corresponding nitrodiamines afforded by aza-Henry reaction were synthetized with moderated yields (55-81%) and diastereomeric excess have ranged from moderated to good (66-88%). Additionally, it was not observed any sign of racemization or side reactions. Aiming to increase the scope of the study on diastereoselective nucleophilic additions to chiral eletrophiles furnished from natural α-amino acids, chiral electron-deficient nitroolefins were synthetized, hitherto unknown in literature, from L-alanine e L-phenylalanine, where they were employed as Michael acceptors in the study of conjugate additions of several nucleophiles. Several reactional conditions, namely base, time, solvent and temperature were studied. The Michael adduts were obtained with moderated to good yields (58-77%) and diastereomeric excess have ranged from low to good (38-86%). Once it was not observed any sign of racemization in these reactions, although, in a single case, we have noticed the occurence of [3 + 2] cycloaddition reaction, which had carried out concurrently with desired conjugate addition.
Furthermore, we have extended the application of this novel methodology to the synthesis of β-amino acids, where we have been investigating various methods to accomplish the Nef reaction.

1
1. INTRODUÇÃO.
1.5 Nitroalcanos: Características gerais, ocorrência natural e atividade biológica,
métodos de preparação e versatilidade sintética.
1.5.1 - Características gerais.
O interesse em nitroalcanos vem aumentando significativamente durante os
últimos anos devido à melhoria de métodos de preparação e interconversão do
grupamento nitro em outras funcionalidades, tais como: aminas, álcoois, substâncias
carboniladas entre outras.1a,b Assim sendo, nitroalcanos são importantes e versáteis
intermediários sintéticos, extremamente úteis no processo de formação de ligação C-C.2
A propriedade físico-química mais importante dos nitroalcanos consiste na alta
acidez dos hidrogênios na posição α ao grupamento nitro. O pKa destes hidrogênios
apresenta-se na faixa de 7,8 a 10 em água. Tal acidez é justificada pelo fato de que o
carbânion gerado é muito estabilizado devido à deslocalização de carga pela estrutura,
através das formas canônicas de ressonância. Veja Figura 1 a seguir:
H C
H
H
C
H
H
N
O-
O
H C
H
H
C
H
N
O-
O
:BBase
H C
H
H
C
H
N
O-
O-
Formas canônicas de ressonância do ânion nitronatoNitroetanopΚa = 8,5Ácido forte Base cojugada estável
Facilidade de formação Estabilidade alta
+ + +
Figura 2: Formação do ânion nitronato a partir do nitroetano tratado com base.
O 1-nitropropano, por exemplo, exibe relativa acidez (pKa = 7,8), visto que, em
água, apresenta-se parcialmente ionizado. Veja Esquema 1 a seguir:
+N
H H
H2O
O
O- +N_
H
O
O-
+N_
H
O-
O+NO-
O-
1-nitropropano
H3O+
ânion nitronato(1-propilnitronato)
+
Esquema 1: Formação do ânion 1-propilnitronato estabilizado por estruturas de ressonância.

2
Devido à sua alta acidez, nitroalcanos primários ou secundários, quando tratados
com uma espécie fracamente básica são transformados nos seus respectivos ânions.
Estes têm sido empregados como nucleófilos seletivos em variadas reações de adição
nucleofílica, por exemplo, reação de Henry (ou nitroaldólica), reação de Michael,
reação de imino-Henry. Exemplos destas reações serão abordados posteriormente.
1.5.2 - Ocorrência natural e atividade biológica.
Nitrocompostos, tais como nitroalcanos, nitroalcenos e nitroaromáticos, fazem
parte de uma classe de substâncias que é escassamente distribuída na natureza. Os
poucos exemplos que ocorrem naturalmente exibem uma variada atividade biológica.
Alguns destes exemplos estão mostrados no Quadro 1 a seguir:
O2N
O2N
OH
OH
NHCOCHCl2
Cloranfenicol - isolado de Streptomyces venezuelae -utilizado em infecções causadas por H. influenza (conjuntivite bacteriana e febre tifóide).3
(1E)-1-nitro-1-pentadeceno - Substância de defesade cupins.3
HO NO2
O
Ácido 3-nitropropanóico - veneno isoladode plantas e fungos.5
OHO
HO
HOH2C
OH
O NO2
Miserotoxina - presente emforraginosas e causa envenenamento de animais.3
NO2
(1E, 1Z)-nitroetenilbenzeno - isolado de Eucondylodesmus elegans - exibe atividade antipredatória.4
Quadro 1: Algumas substâncias naturais portadoras do grupamento nitro e suas atividades biológicas. Por outro lado, existem várias substâncias sintéticas com atividade biológica que
também portam o grupamento nitro em sua estrutura, veja Quadro 2 a seguir:
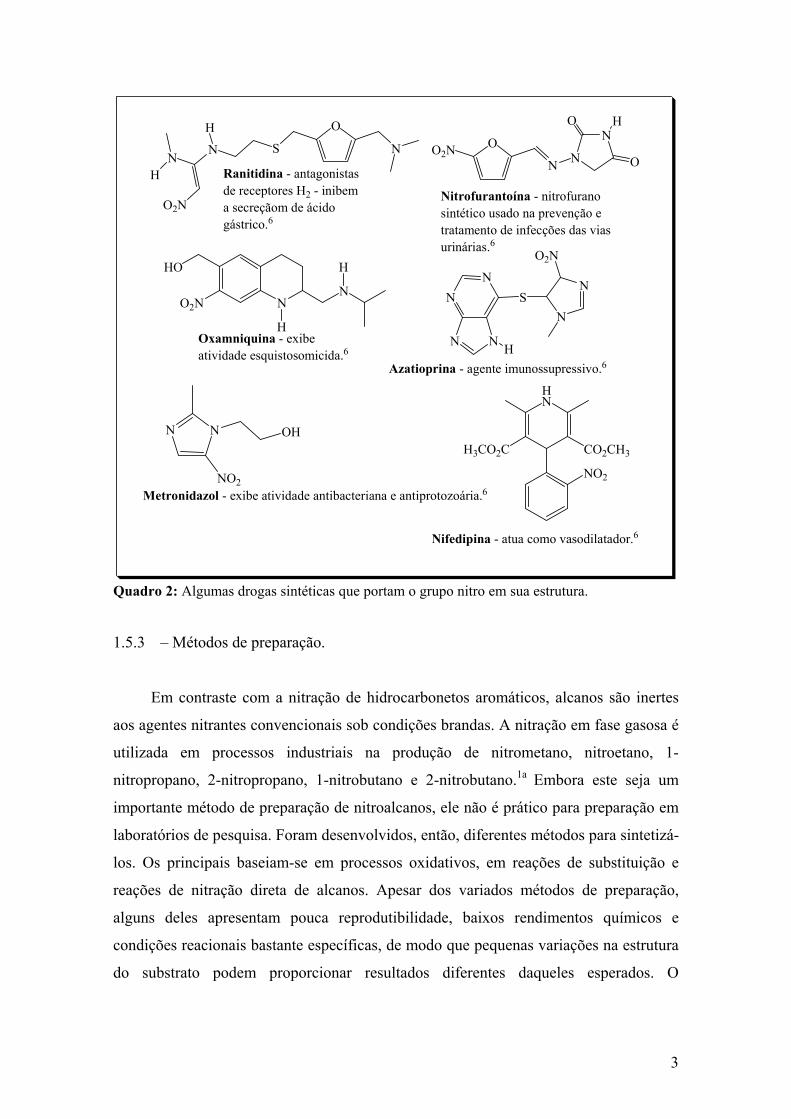
3
O
NSNN
O2N
OO2NN N
N
O
O HH
H Ranitidina - antagonistas de receptores H2 - inibem a secreçãom de ácido gástrico.6
Nitrofurantoína - nitrofurano sintético usado na prevenção e tratamento de infecções das vias urinárias.6
O2N
HO
N
H
N
H
Oxamniquina - exibe atividade esquistosomicida.6
N N OH
NO2Metronidazol - exibe atividade antibacteriana e antiprotozoária.6
NN
N N
SN
N
O2N
HAzatioprina - agente imunossupressivo.6
NO2
HN
CO2CH3H3CO2C
Nifedipina - atua como vasodilatador.6
Quadro 2: Algumas drogas sintéticas que portam o grupo nitro em sua estrutura. 1.5.3 – Métodos de preparação.
Em contraste com a nitração de hidrocarbonetos aromáticos, alcanos são inertes
aos agentes nitrantes convencionais sob condições brandas. A nitração em fase gasosa é
utilizada em processos industriais na produção de nitrometano, nitroetano, 1-
nitropropano, 2-nitropropano, 1-nitrobutano e 2-nitrobutano.1a Embora este seja um
importante método de preparação de nitroalcanos, ele não é prático para preparação em
laboratórios de pesquisa. Foram desenvolvidos, então, diferentes métodos para sintetizá-
los. Os principais baseiam-se em processos oxidativos, em reações de substituição e
reações de nitração direta de alcanos. Apesar dos variados métodos de preparação,
alguns deles apresentam pouca reprodutibilidade, baixos rendimentos químicos e
condições reacionais bastante específicas, de modo que pequenas variações na estrutura
do substrato podem proporcionar resultados diferentes daqueles esperados. O
4
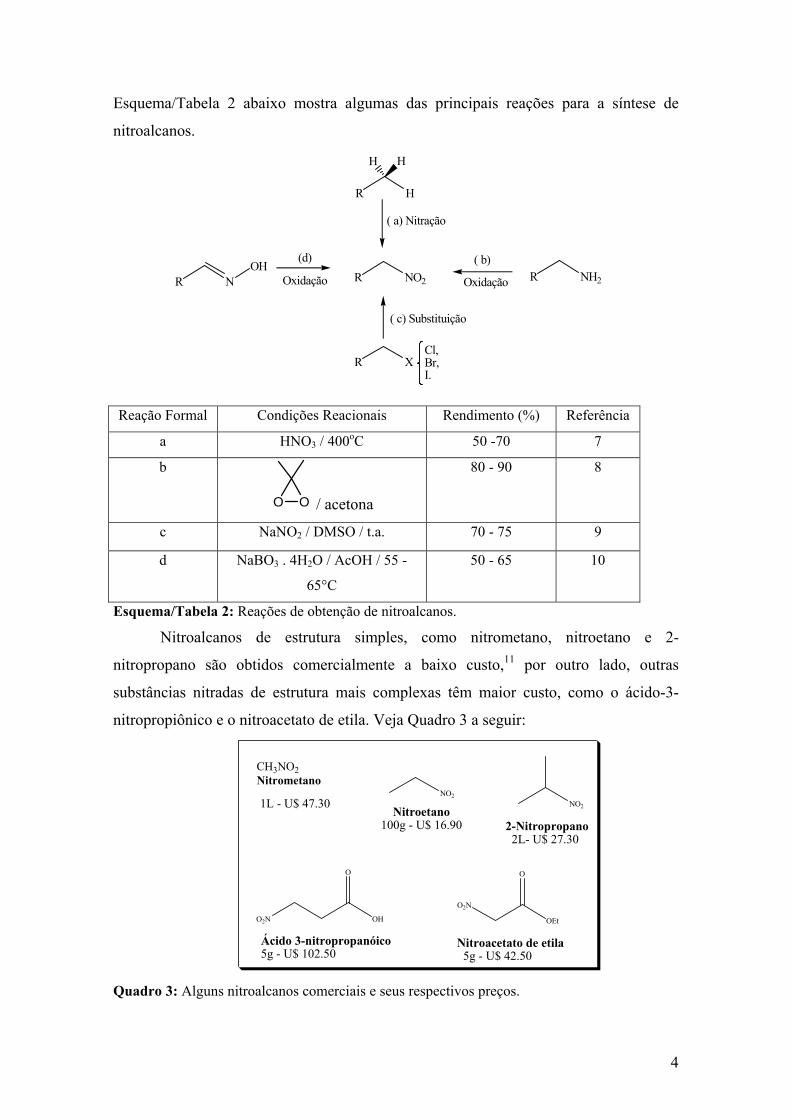
Esquema/Tabela 2 abaixo mostra algumas das principais reações para a síntese de
nitroalcanos.
Reação Formal Condições Reacionais Rendimento (%) Referência
a HNO3 / 400oC 50 -70 7
b
O O / acetona
80 - 90 8
c NaNO2 / DMSO / t.a. 70 - 75 9
d NaBO3 . 4H2O / AcOH / 55 -
65°C
50 - 65 10
Esquema/Tabela 2: Reações de obtenção de nitroalcanos.
Nitroalcanos de estrutura simples, como nitrometano, nitroetano e 2-
nitropropano são obtidos comercialmente a baixo custo,11 por outro lado, outras
substâncias nitradas de estrutura mais complexas têm maior custo, como o ácido-3-
nitropropiônico e o nitroacetato de etila. Veja Quadro 3 a seguir:
CH3NO2 Nitrometano
NO2NO2
O2N OH
O
O2N
OEt
O
1L - U$ 47.30Nitroetano
100g - U$ 16.90 2-Nitropropano 2L- U$ 27.30
Ácido 3-nitropropanóico5g - U$ 102.50
Nitroacetato de etila 5g - U$ 42.50
Quadro 3: Alguns nitroalcanos comerciais e seus respectivos preços.
R NO2 R NH2
R X
R NOH
( a) Nitração
( b)
( c) Substituição
(d)
OxidaçãoOxidação
R H
H H
Cl, Br, I.

5
1.5.4 - Versatilidade sintética.
O grupo nitro é particularmente versátil em síntese orgânica, portanto, os
derivados de nitroalcanos podem ser transformados em uma grande variedade de
moléculas com distintas funcionalidades.3 Veja Figura 2 abaixo:
R1R3
R2
O
R1 R3
NH2R2
R1 R3
HR2
CN
R1
R2
R1R3
R2
NOH
R1OH
R2
O
R1H
R2
O
R1 R3
NO2R2
Figura 2: Conversão do grupo nitro para diversas funcionalidades.
Assim, podemos ter um nitro primário sendo transformado em nitrila,12
adicionalmente, pode-se ter o carbono α-nitro oxidado à carbonila de aldeído, cetona
ou ácido carboxílico (reação de Nef),13a,b ainda ter o grupo nitro reduzido para produzir
aminas e oximas14a,b e também pode haver a remoção de grupamento nitro secundário
e/ou terciário (desnitração).15 Algumas outras transformações são bastante utilizadas e
exploradas nas mais diversas rotas sintéticas, tornando os nitroalcanos extremamente
úteis em síntese orgânica.

6
1.6 Iminas: Características gerais e reatividade, métodos de preparação e adição nucleofílica de nitroalcanos a iminas.
1.6.1 - Características gerais e reatividade.
Iminas são intermediários sintéticos fundamentais para a obtenção de uma
grande variedade de substâncias nitrogenadas.16 Mais especificamente, substâncias que
exibem em sua estrutura a funcionalidade diamina vicinal têm atraído considerável
atenção de grupos de pesquisa devido ao importante uso como ligantes quirais em
síntese enantiosseletiva,17a,b a elevada atividade biológica em química medicinal18 e por
estarem presentes em produtos naturais biologicamente ativos.19
A adição de nucleófilos ao carbono imínico (C=N) permite um rápido acesso a
substâncias aminadas como mostrada na Figura 3 abaixo:
R R´
NR"
RR´
HNR"
Ar
RR´
HNR"
R M
R´R
HNR"
R
R-M
RR´
HNR"
M
RR´
HNR"
CN
HCN
R
HNR"
R´
O
OR
Ar-M R
RR´
HNR"
H
RR´
HNR"
P
O
OROR
P
O
OROR
H
[H]
44
45
46
47
48
49
50
51
52
Figura 3: Obtenção de substâncias aminadas a partir da adição nucleofílica a iminas.
Quando comparamos aos estudos feitos sobre a ligação dupla C=O, os estudos
sobre a ligação C=N da imina é uma área de estudo pouco explorada. Embora haja
muita similaridade entre a reatividade de ambas, algumas diferenças podem ser
enumeradas:

7
(1) A reatividade de iminas frente a adições nucleofílicas é menor devido à
menor eletrofilicidade do carbono imínico (μ C=N < μ C=O). Veja Quadro 4 abaixo:
R1 R2R1 R2
ON
δC=N << δC=OμC=N<< μC=O
R3
R1 = H, alquil e arilR2 = H, alquilR3 = alquil e aril.
Quadro 4: Eletrofilicidade de iminas comparada a aldeídos e cetonas.
(2) Iminas não são sempre facilmente preparadas, especialmente cetiminas.
Adicionalmente, muitas aldiminas são instáveis.
(3) Aldeídos ou cetonas não têm isômeros geométricos, todavia, em iminas, há a
possibilidade da ocorrência de dois isômeros geométricos, sendo o isômero trans,
geralmente preferido, devido a considerações estereoquímicas.
(4) No caso de iminas alifáticas, que apresentam um hidrogênio na posição α ao
grupo imino, pode haver desprotonação pelo uso de nucleófilos organometálicos (bases
fortes) e reações laterais, como acoplamentos, podem ocorrer. Conseqüentemente, essas
iminas podem ser tautomerizadas a enaminas. Veja Figura 4 a seguir:
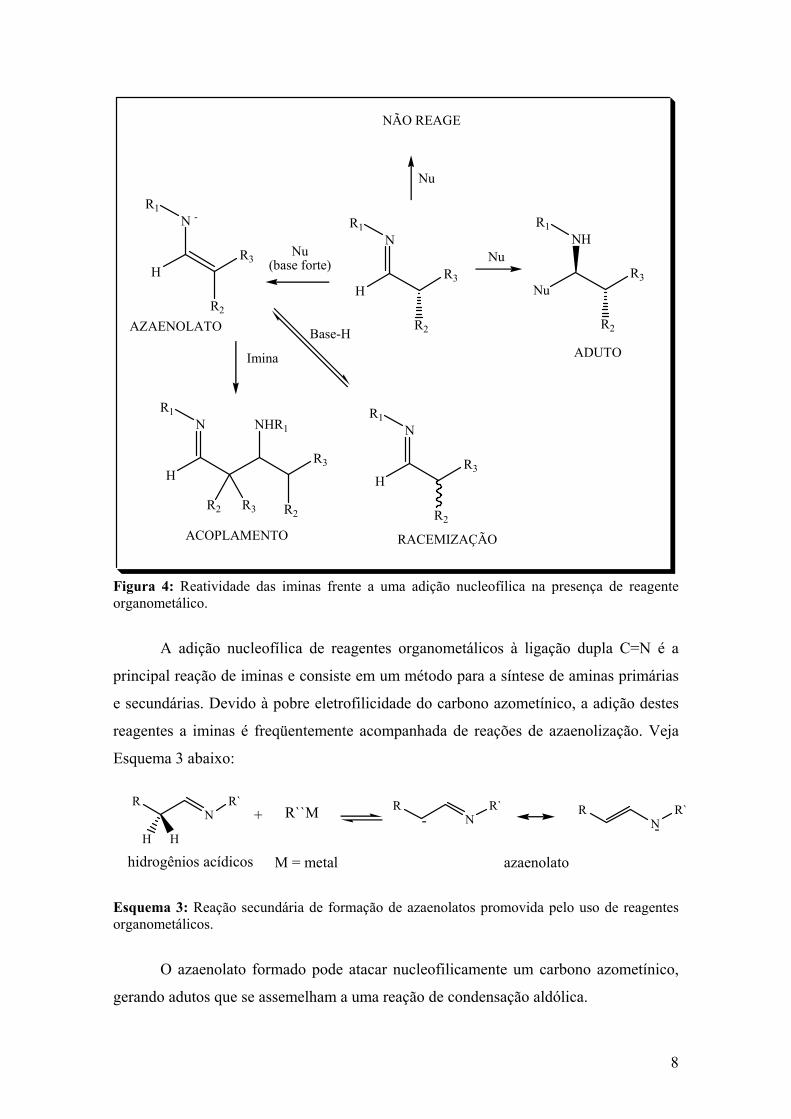
8
HR3
N
R2
R1
Nu
NuNu (base forte)
NÃO REAGE
NuR3
NH
R2
R1
HR3
N -
R2
R1
HR3
NR1
AZAENOLATO
HR3
N
R2 R3
NHR1
R2
R1
ACOPLAMENTOR2
RACEMIZAÇÃO
ADUTOImina
Base-H
Figura 4: Reatividade das iminas frente a uma adição nucleofílica na presença de reagente organometálico.
A adição nucleofílica de reagentes organometálicos à ligação dupla C=N é a
principal reação de iminas e consiste em um método para a síntese de aminas primárias
e secundárias. Devido à pobre eletrofilicidade do carbono azometínico, a adição destes
reagentes a iminas é freqüentemente acompanhada de reações de azaenolização. Veja
Esquema 3 abaixo:
Esquema 3: Reação secundária de formação de azaenolatos promovida pelo uso de reagentes organometálicos.
O azaenolato formado pode atacar nucleofilicamente um carbono azometínico,
gerando adutos que se assemelham a uma reação de condensação aldólica.
NR`R
HHN
R`RN
R`R
hidrogênios acídicos
+ R``M
M = metal
- -
azaenolato

9
Para aumentar a utilidade das adições de organometálicos a iminas, muitos
métodos para contornar os problemas de reatividade têm sido explorados, como: (a)
aumento de eletrofilicidade da ligação C=N por N-inserção de grupamentos retiradores
de elétrons20a,b,c ou por N-coordenação com ácidos de Lewis;21 (b) uso de reagentes
organometálicos menos básicos para minimizar a reação de azaenolização.22
Desta forma, com o intuito de aumentar a eletrofilicidade da ligação C=N e,
portanto, minimizar os problemas de baixa reatividade e azaenolização frente às adições
nucleofílicas, a síntese de iminas onde há substituintes retiradores de elétrons acoplados
ao átomo de nitrogênio é freqüentemente relatada na literatura. Veja alguns exemplos de
iminas N-ativadas mostradas no Quadro 5 abaixo.
R1 R2
NP
O
Ph
Ph
R1 R2
N
O
R2
R1 R2
N
Ts
R1 = alquil, ArR2 = H, aquil R1 R2
NSi(Me)3
R1 R2
NBH2
Quadro 5: Exemplos de iminas N-ativadas.
Dentre as iminas N-ativadas, podemos citar como exemplos as N-sililiminas, N-
boriliminas, N-fosfinoiliminas, N-aciliminas e N-tosiliminas. Grande parte destes
métodos, entretanto, é restrita a aldiminas não-azaenolizáveis e, apesar de aumentar
significativamente o rendimento químico nas adições nucleofílicas, requer condições
drásticas para a remoção dos grupos ativantes, o que pode inviabilizar a metodologia de
ativação, dependendo do substrato em questão.23

10
1.6.2 - Métodos de preparação.
Geralmente, iminas são preparadas a partir de uma substância carbonilada, que
pode ser um aldeído ou uma cetona, e uma amina primária na presença de um agente
dessecante. Por exemplo, Cativiela e cols.24 sintetizaram a N-benzilimina 2, em 93% de
rendimento, por tratamento do aldeído 1 com benzilamina, em éter etílico como
solvente e sulfato de magnésio como agente dessecante. Veja Esquema 4 abaixo:
(R)
OBn
BnO
O
BnNH2
MgSO4 / Et2O 3h, 0°C 93%
(S) NBnO
OBn
H
Bn
1 2
Esquema 4: Formação da imina 2 através da adição de benzilamina ao aldeído quiral 1.
Outros métodos de preparação de iminas foram relatados, embora sejam bem menos
utilizados, como, por exemplo, o método desenvolvido por Vaultier e cols.25 que leva à
formação eficiente de iminas cíclicas, intermediários importantes utilizados para a
síntese de alcalóides de estruturas simples ou complexas. Este método, que conduz à
formação destas iminas cíclicas, é conhecido como aza-Wittig. Veja Esquema 5 abaixo:
XR
R`` R`
O
n N3R
R`` R`
O
n Ph3P=NR
R`` R`
O
n
NR
R`
R``
n
NaN3
PPh3- N2
- P(O)Ph3
R= Me, Pr, Ph, ciclopropilaR'= Me, Et, HR''=H, MeX =Br, Cln = 1,2,3,4
78 - 96%3 4 5
6
70 - 83%
Esquema 5: Formação de iminas cíclicas 6 a partir da reação de aza-Wittig.
Inicialmente, a ω-halocetona 3, com variados substituintes, é tratada com azida
de sódio, o que leva à formação da azida correspondente 4. O tratamento desta azida
com um equivalente de trifenilfosfina forneceu um intermediário sintético 5,
equivalente a uma fosforana, onde este é transformado, através de uma reação de aza-
Wittig intramolecular, a uma imina cíclica 6, com rendimento químico na faixa de 70 a
83%.

11
Posteriormente, Varma e cols.26 sintetizaram várias iminas aromáticas 8 sem o uso
de solventes, utilizando Envirocat EPZGR (semelhante à argila montmorilonita) como
catalisador. Veja Esquema/Tabela 6:
R
CHONH2
R
NPh
+ Envirocat EPZGR
microondas7 Anilina 8
R Tempo (min) Rend. (%)
H 1,0 97
o-OH 1,0 92
p-OH 3,0 91
p-Me 2,5 90
p-NMe2 3,0 96
p-OMe 2,5 95
Esquema/Tabela 6: Síntese de iminas com o emprego de microondas e do catalisador sólido Envirocat EPZG®.
Este método faz parte da chamada Química Verde, onde há a preocupação com o
meio ambiente, já que esta reação não utiliza solventes orgânicos, principalmente os
solventes aromáticos, que são geralmente usados para produzir um azeótropo com a
água formada na reação e sua posterior remoção. Adicionalmente, a catálise com esta
fase sólida Envirocat EPZG® não mostrou necessidade de quaisquer agentes
dessecantes.
1.2.3 - Adição nucleofílica de nitroalcanos a iminas - Reação de aza-Henry.
Escolhemos, exclusivamente, relatar os trabalhos onde é feita a adição de ânions
nitronatos a iminas, pois este é um dos pontos estudados desta tese. Todavia, é
imprescindível relatarmos que a grande maioria dos estudos feitos sobre adição
nucleofílica a iminas concentra-se em empregá-las com grupamentos ativadores
apropriados, seja ele acoplado ou coordenado ao átomo de nitrogênio da ligação C=N,
visando, como mostrado anteriormente, aumentar a eletrofilicidade desta referida
ligação frente ao ataque nucleofílico e, também, o emprego de reagentes

12
organometálicos, como nucleófilos mais potentes, o que sugere em contrapartida, o uso
de iminas não propensas à azaenolização.
A reação de aza-Henry, também conhecida como reação de nitro-Mannich,
consiste na adição nucleofílica de ânions nitronatos a iminas, fornecendo substâncias
1,2-nitroaminadas. A versatilidade sintética desta reação reside na formação de ligação
carbono-carbono, bem como na obtenção de importantes intermediários sintéticos,
como, por exemplo, 1,2-nitroaminas e α-aminoácidos. Veja Figura 5 abaixo:
R
N
GP
+NO2
R' R
HN
GP
R'
NO2
R
NH2
R'
NH2
R
NH2
R'=H
O
OH
Espéciebásica
GP = grupo de proteção
1,2-diaminas
α-aminoácidos
Figura 5: Obtenção de 1,2-diaminas e α-aminoácidos a partir de iminas N-protegidas.
A redução do grupo nitro seguida da desproteção do grupo amino fornece
substâncias com a porção 1,2-diamina. Tais substâncias diaminadas estão presentes em
produtos naturais, são utilizadas em química medicinal e também são ligantes quirais
em catálise enantiosseletiva. Já a oxidação do carbono α-nitro, por intermédio da reação
de Nef, seguida da desproteção do grupo amino, leva à formação de α-aminoácidos, que
desempenham um importante papel como blocos de construção de moléculas
biologicamente ativas.
Existem dois antigos relatos da reação de aza-Henry na literatura, datados de 1946,27a,b
onde 1,2-nitroaminas foram produzidas a partir de aminas primárias e secundárias,
formaldeído e nitroalcanos. Nestes trabalhos, as nitroaminas foram obtidas em baixo
rendimento químico e nada foi dito sobre a diastereosseletividade da reação.

13
Após essa data, de acordo com os nossos conhecimentos, somente em 1998,
Anderson e cols.28 estudaram essa reação na sua versão diastereosseletiva, porém
racêmica. Veja Esquema 7 a seguir:
O2N
R1
1) n-BuLi, THF, -78oC2) PMBN=CHR2
3) THF, AcOH,-78o - 0oC(74-95%)
R2
R1
NH
NO2
PMB
SmI2
THF / MeOH(60-77%)
R2
R1
NH
NH2
PMB
CAN
MeCN / H2O(86-100%) R2
R1
NH2
NH2
9a-d
11a,b,d10a,b,d
20-82 % de e.d.Produto majoritário anti
R1= Et, PhR2 = Ph, CH2OBn, n-PnPMB = p-MeOBn 9a, R1= Et, R2 = Ph
9b, R1= Et, R2 = n-Pn9c, R1= Ph, R2 = Ph9d, R1= Et, R2 = CH2OBn
Esquema 7: Adição de ânions nitronatos a N-(p-metoxibenzil)-iminas. Síntese de 1,2-diaminas 11a,b,d.
Os autores relataram a adição de nitronatos de lítio, gerados a partir de
nitroalcanos e n-BuLi, a N-(p-metoxibenzil)-iminas aquirais na presença de ácido de
Brønsted. As nitroaminas 9 a-d foram obtidas em uma faixa de 20 a 82% de excesso
diastereoisomérico e rendimentos químicos que variaram de 74 a 95%. Os
diastereoisômeros anti 9a,b,d foram obtidos majoritariamente e convertidos nas 1,2-
diaminas racêmicas 11a,b,d diastereoisomericamente enriquecidas.
Um crescente interesse pela reação de aza-Henry tem sido observado nos
últimos anos,16,29 em grande parte devido ao crescente emprego de metodologias que
envolvem catálise enantiosseletiva, veja Gráficos 1 e 2 abaixo. Desta forma, um
considerável esforço tem sido direcionado, até o presente momento, para o
desenvolvimento de novas metodologias visando à versão estereosseletiva da reação de
aza-Henry. A seguir, serão demonstrados alguns dos exemplos mais representativos
descritos na literatura.

14
Gráfico 1 Palavra chave: aza-Henry
Gráfico 2
Palavra chave: nitro-Mannich
Gráficos 1 e 2: Interesse pela reação de aza-Henry (ou nitro-Mannich) nos últimos anos.
1.2.3.1 - Versão estereosseletiva da reação de aza-Henry.
1.2.3.1.1 - Utilização de catalisadores organometálicos.
No ano de 1999, Shibasaki e cols.30 realizaram a primeira catálise
enantiosseletiva da reação de aza-Henry. Os autores relatam um estudo sobre a adição
estereosseletiva de nitrometano a N-fosfinoiliminas aromáticas 12a-d, utilizando o
catalisador heterobimetálico 13, veja Esquema /Tabela 8 a seguir:
Ar NPPh2
O
+ CH3NO2
20 % mol cat. 13
PhMe, THF (7:1)-40 0C Ar
NO212a-d14a-d
HNP
Ph
O
Ph
Ar Rend. (%) Produto e.e. (%)
12a, Ph 79 14a 91
12b, 4-Cl-C6H4 93 14b 87
12c, p-toluil 85 14c 89
12d, 2-furil 57 14d 83
Esquema/Tabela 8: Adição de nitrometano às N-fosfinoiliminas 12a-d na presença de catalisador heterobimetálico 13.
Após algumas tentativas preliminares, o catalisador 13, preparado a partir da
combinação de Yb(OiPr)3, KOtBu e binaftol na proporção de (1:1:3), respectivamente,
foi escolhido para a reação. A proposta desta catálise heterobimetálica pode ser
13

15
classificada como multifuncional por reunir em um único catalisador, ambas as
funcionalidades de ácido de Lewis e base de Brønsted. Isto permite que haja uma
ativação simultânea da imina (via coordenação da ligação dupla P=O com o átomo de
itérbio) e do nitrometano (via desprotonação do carbono alfa nitro) para a reação de
adição. Os produtos 14a-d foram obtidos em bons rendimentos químicos e
enantiocontrole utilizando as quatro iminas aromáticas 12a-d, contudo, foram
necessárias grandes quantidades de catalisador, a baixas temperaturas, além de longos
tempos de reação (mínimo de 60 horas).
Baseados nos resultados de Shibasaki, Jøgensen e cols.31a,b em 2001, relataram a
adição de diferentes nitroalcanos ao α-iminoéster N-protegido 15, catalisada por
complexos de cobre, na síntese de β-nitro-α-aminoésteres 16a-e, veja Esquema/Tabela
9 abaixo:
R
NO2+
EtOOC
NPMP
Catalisador 17 ( 20 % mol )
Et3N (20 % mol)CH2Cl2, t.a NO2(R)
(R)
R
EtO2C
HNPMP
15
16a-ePMP = p-metoxifenil
N N
OO
Ph PhCu
TfO OTf
cat. 17 =
Entrada R Produto-Rendimento
(%) r.d. (sin:anti) e.e (%)a
1 H 16a, (38) - 87
2 Me 16b, (61) 70:30 97
3 Et 16c, (81) 95:5 97
4 Ph 16d, (59) 55:45 74
5 PhCH2 16e, (80) 95:5 95
a) Excesso enantiomérico para o produto sin.
Esquema/Tabela 9: Adição de nitroalcanos ao α-iminoéster N-protegido 15 na presença do catalisador organometálico 17.
Embora um grande número de ligantes quirais da bisoxazolina e sais de cobre
tenha sido examinado, o resultado mais promissor foi obtido usando 17 e quantidade
16
catalítica de triflato de cobre (II), à temperatura ambiente. A adição de trietilamina
como base foi necessária para desprotonação dos nitroalcanos a serem adicionados.
De acordo com as entradas 3 e 5, os produtos 16c e 16e foram obtidos em
excelentes rendimentos químicos e altas estereosseletividades. Nos demais casos, os
produtos foram obtidos em rendimentos químicos moderados com pobre
diastereosseletividade, porém com moderada a excelente enantiosseletividade. Um
modelo de estado de transição em cadeira foi proposto para racionalizar a
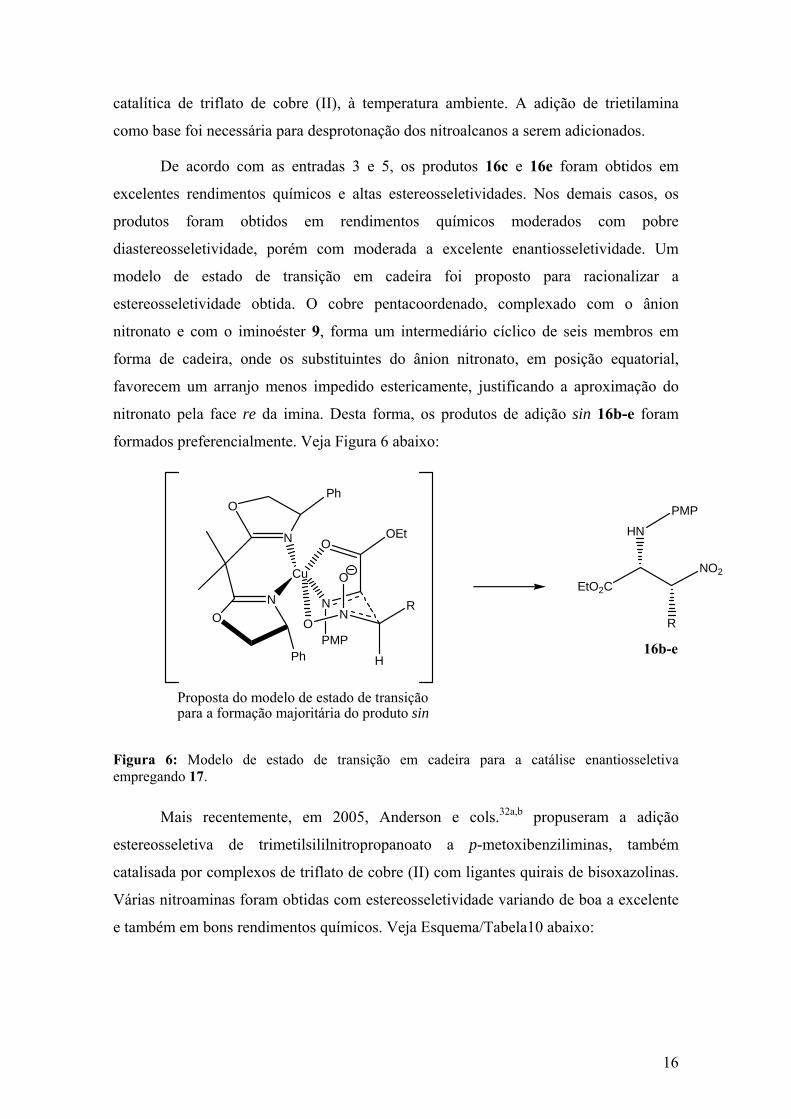
estereosseletividade obtida. O cobre pentacoordenado, complexado com o ânion
nitronato e com o iminoéster 9, forma um intermediário cíclico de seis membros em
forma de cadeira, onde os substituintes do ânion nitronato, em posição equatorial,
favorecem um arranjo menos impedido estericamente, justificando a aproximação do
nitronato pela face re da imina. Desta forma, os produtos de adição sin 16b-e foram
formados preferencialmente. Veja Figura 6 abaixo:
N
O
Cu
PMP
N
N
OPh
O NO
O
R
H
NO2
EtO2C
R
HNOEt
PMP
Ph 16b-e
Proposta do modelo de estado de transiçãopara a formação majoritária do produto sin
Figura 6: Modelo de estado de transição em cadeira para a catálise enantiosseletiva empregando 17.
Mais recentemente, em 2005, Anderson e cols.32a,b propuseram a adição
estereosseletiva de trimetilsililnitropropanoato a p-metoxibenziliminas, também
catalisada por complexos de triflato de cobre (II) com ligantes quirais de bisoxazolinas.
Várias nitroaminas foram obtidas com estereosseletividade variando de boa a excelente
e também em bons rendimentos químicos. Veja Esquema/Tabela10 abaixo:

17
R1 R2
NPMP
+
Et H
NOTMSO
Cu(OTf)2 (10% molar)(S)-18 (11% molar)
THF, -30°C , 40h R1
HN R2
NO2
PMP
19a-d 20a-d
N
O
N
O18 =
Entrada Imina R1 R2 Rend. (%)a anti/sinb e.e. (anti)c
1 19a Ph H 84 >15:1 94%
2 19b 4-Cl-C6H4 H 84 >15:1 92%
3 19c 2-furil H 89 11:1 94%
4 19d c-hex H 88 >15:1 87% a) Rendimento isolado e purificado de mistura diastereoisomérica inseparável; b) Diastereosseletividade medida via RMN de 1H; c) Medição realizada por HPLC quiral. Esquema/Tabela 10: Síntese das nitroaminas 20a-d através de catálise organometálica.
Em ensaios preliminares, Anderson e cols. selecionaram o catalisador mais
adequado a ser empregado na síntese das nitroaminas 20a-d, como a otimização das
condições reacionais empregadas, a porcentagem molar do catalisador e do sal de cobre
e o solvente. Foi verificado que a bisoxazolina 18, com os dois grupamentos t-butila,
fornece uma melhor estereosseleção nessas adições, onde N-PMP-iminas aromáticas e
alifáticas 19a-d foram empregadas.
Os resultados mostrados no Esquema/Tabela 10 acima mostram que este
protocolo de reação de aza-Henry enantiosseletiva é particularmente eficiente para a
conversão de simples aldiminas às nitroaminas correspondentes com excelentes níveis
de diastereo- e enantiosseletividade.
1.2.3.1.2 - Utilização de catalisador orgânico.
A utilização de catalisadores orgânicos quirais, que contenham a porção tiouréia
em sua estrutura, vem recebendo bastante atenção nos últimos anos33 em versões
estereosseletivas da reação de aza-Henry. Takemoto e cols.34 em 2004, descreveram a

18
utilização de uma tiouréia quiral na primeira organocatálise enantiosseletiva envolvendo
uma reação de aza-Henry. Veja Esquema/Tabela 11 abaixo:
Ar NPPh2
O
+ 10 eq. CH3NO2
Catalisador 25(10 % mol)
CH2Cl2, t.a. Ar
HN
HN
HN
S
N(CH3)2F3C
CF3
Cat. 25 =
21a,b 22a-c 23a,b
24a-c
P(O)Ph2
NO2
Ar Nitroamina / rendimento (%) e.e. (%)
21a, Ph 23a / (87) 67
21b, 2-furil 23b / (85) 76
22a, 2-naftil 24a / (78) 70
22b, 2-piridil 24b / (91) 68
22c, cinamil 24c / (68) 65
Esquema/Tabela 11: Adição de nitrometano às N-fosfonoiliminas 21a,b e 22a-c.
O catalisador 25 utilizado reúne a porção tiouréia e o grupamento básico N,N-
dimetilamina em sua estrutura. Este foi originalmente desenhado para adição
enantiosseletiva de dietilmalonatos a β-nitroestirenos. A sua utilização na reação de aza-
Henry, entretanto, forneceu moderada enantiosseletividade e bons rendimentos
químicos na obtenção dos produtos 23a,b e 24a-c, a partir da adição de nitrometano às
N-fosfonoiliminas 21a,b e 22a-c.
Embora o mecanismo com o qual o catalisador 25 promove a reação não seja
inteiramente conhecido, foi proposta uma coordenação da porção tiouréia com os dois
átomos de oxigênio do grupo nitro, enquanto a amina terciária facilita a enolização
(abstração do hidrogênio α-nitro), veja Figura 7 a seguir:
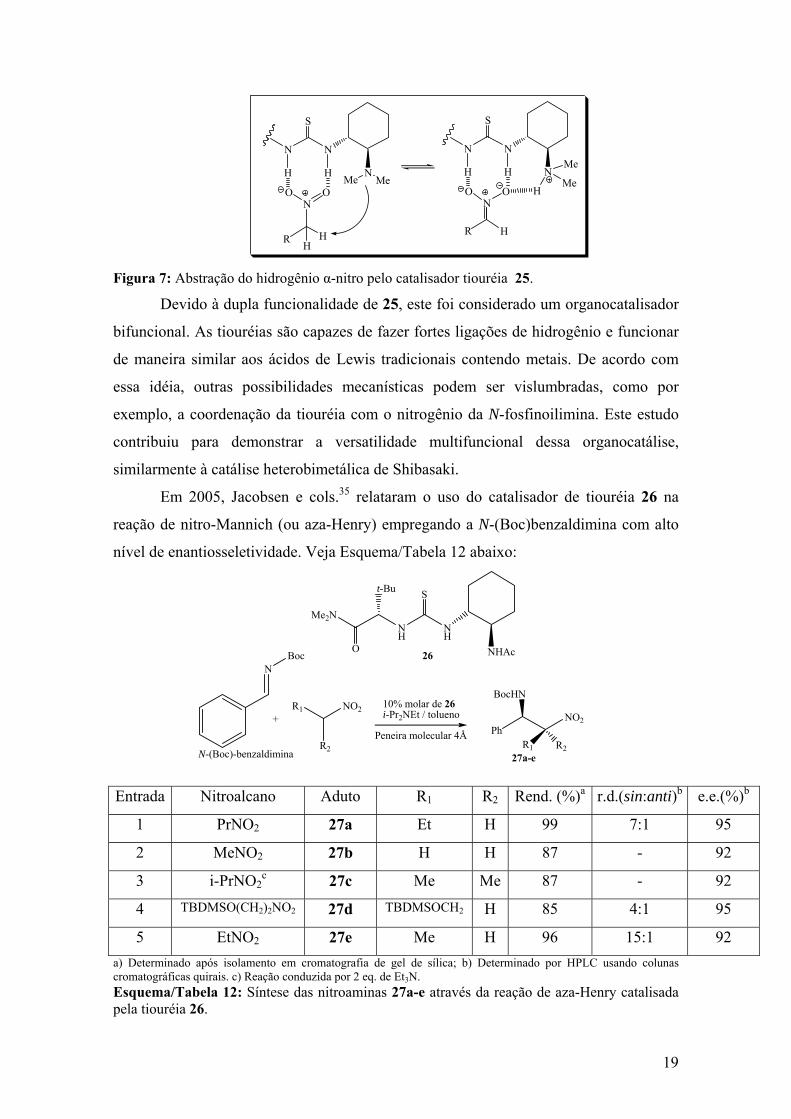
19
N N
S
H H NMeMe
NO O
R HH
N N
S
H H NMe
Me
NO O
R H
H
Figura 7: Abstração do hidrogênio α-nitro pelo catalisador tiouréia 25.
Devido à dupla funcionalidade de 25, este foi considerado um organocatalisador
bifuncional. As tiouréias são capazes de fazer fortes ligações de hidrogênio e funcionar
de maneira similar aos ácidos de Lewis tradicionais contendo metais. De acordo com
essa idéia, outras possibilidades mecanísticas podem ser vislumbradas, como por
exemplo, a coordenação da tiouréia com o nitrogênio da N-fosfinoilimina. Este estudo
contribuiu para demonstrar a versatilidade multifuncional dessa organocatálise,
similarmente à catálise heterobimetálica de Shibasaki.
Em 2005, Jacobsen e cols.35 relataram o uso do catalisador de tiouréia 26 na
reação de nitro-Mannich (ou aza-Henry) empregando a N-(Boc)benzaldimina com alto
nível de enantiosseletividade. Veja Esquema/Tabela 12 abaixo:
NH
NH
Me2N
O
t-Bu S
NHAcN
Boc
10% molar de 26i-Pr2NEt / tolueno
Peneira molecular 4Å+
R1
R2
NO2
PhNO2
BocHN
R2R1
26
27a-eN-(Boc)-benzaldimina
Entrada Nitroalcano Aduto R1 R2 Rend. (%)a r.d.(sin:anti)b e.e.(%)b
1 PrNO2 27a Et H 99 7:1 95
2 MeNO2 27b H H 87 - 92
3 i-PrNO2c 27c Me Me 87 - 92
4 TBDMSO(CH2)2NO2 27d TBDMSOCH2 H 85 4:1 95
5 EtNO2 27e Me H 96 15:1 92 a) Determinado após isolamento em cromatografia de gel de sílica; b) Determinado por HPLC usando colunas cromatográficas quirais. c) Reação conduzida por 2 eq. de Et3N. Esquema/Tabela 12: Síntese das nitroaminas 27a-e através da reação de aza-Henry catalisada pela tiouréia 26.

20
Jacobsen e cols., após testarem variadas condições reacionais e também
catalisadores com pequenas variações estruturais, estabeleceram que o emprego da
tiouréia quiral 26 deu-se de maneira significantemente mais reativa nas condições
reacionais otimizadas.
Analisando o Esquema/Tabela 12 anteriormente observado, observa-se nas
entradas 2-4 rendimentos químicos menores em relação àqueles observados nas entradas
1 e 5, devido a uma menor reatividade, principalmente do nitrometano, entrada 2, que
requereu um tempo maior de reação (40 horas). Na entrada 3, observa-se a reação com o
nitroalcano mais estericamente impedido, 2-nitropropano, que só se procedeu quando a
amina empregada foi substituída pela amina terciária Et3N. As nitroaminas 27a-e foram
obtidas com diastereosseletividades que variaram de moderadas a boas, embora com um
alto nível de enantiosseletividade.
1.2.3.1.3 - Utilização da metodologia chiron approach.
De acordo com os nossos conhecimentos, o emprego da metodologia chiron
approach na reação de Aza-Henry só foi relatado mais recentemente por Bernardi e
cols.36 em 2002, por Petrini e cols.37 em 2003, Benetti38 em 2004 e por nós em 2002,39
na obtenção diastereosseletiva de 1,2-nitroaminas enantiopuras.
Em 2003, Bernardi e cols.36 relataram a síntese de triaminas pseudo-C2
simétricas utilizadas em inibidores de HIV-protease, empregando a adição do ânion
sililnitronato 29 à N-PMP-N,N-dibenzilfenilalaninaldimina 28, como mostrado no
Esquema 13 abaixo:
Ph
N
NBn2
PMP Ph
NOMe3SiO
+ 5% molar de Sc(OTf)3
MeCN / 0ºCPh
HN
NBn2
PMP
NO2
Ph
2930
28 68%
2 diastereoisômeros(60:40)
Esquema 13: Adição do sililnitronato 29 à N-PMP-N,N-dibenzilfenilalaninaldimina 28.

21
De acordo com procedimentos realizados por Jørgensen31b e Anderson,32b
Benetti e cols., após várias tentativas, verificaram que o uso de porção catalítica de
triflato de escândio mostrou ser a melhor rota para o acesso à síntese das nitrodiaminas
diastereoisoméricas 30. Embora este ácido de Lewis tenha se mostrado eficiente na
adição catalítica de sililnitronatos à aldimina 28, a razão diastereoisomérica foi pobre.
As nitroaminas 30 foram obtidas em 68 % de rendimento químico - após purificação em
coluna cromatográfica - como uma mistura de dois diastereoisômeros na razão
distereoisomérica de 60:40 verificada por RMN de 1H (Esquema 13).
Posteriormente, com o intuito de evitar a já relatada reação de retro-aza-Henry28
que ocorre parcialmente em sílica gel, os adutos 30 foram submetidos à redução sem
quaisquer purificações. Após a etapa de redução, as triaminas 31a,b foram separadas em
sílica desativada e puderam ser isoladas como únicos diastereoisômeros em 66% de
rendimento total a partir da imina 28. Veja Esquema 14 abaixo:
Ph
HN
NBn2
PMP
NO2
Ph
30
NaBH4 / NiCl2 6H2O / MeOH0ºC / 30 min Ph
HN
NBn2
PMP
NH2
Ph
31a,b
cromatografia
Ph
HN
NBn2
PMP
NH2
Ph Ph
HN
NBn2
PMP
NH2
Ph+
31a 31b
Esquema 14: Redução da nitrodiamina 30 às triaminas 31a,b.
No estudo realizado por Petrini e cols.,37 os autores relataram o uso de α-
amidoalquilfenilsulfonas 33a,b como precursores para gerar in situ as N-
carbamoiliminas 34a,b e, em seguida, sofrerem adição de nitrometano. Veja Esquemas
15 e 16 abaixo:

22
(R)
OO (R) SO2Ph
NHCO2R
(R)
OO
O
RO NH2
O
+
PhSO2H
MgSO4, CH2Cl2
32a, R = t-Bu 32b, R = Bn 33a, 70%, r.d. > 95:5
33b, 65%, r.d. > 95:5(R)-gliceraldeído-acetonídeo
Esquema 15: Obtenção das α-amidoalquilfenilsulfonas 33a,b a partir do (R)-gliceraldeídoacetonídeo.
O (R)-gliceraldeídoacetonídeo reagiu com os carbamatos 32a,b e ácido
benzenossulfínico para dar origem às α-amidoalquilfenilsulfonas 33a,b em bons
rendimentos químicos e alta diastereosseletividade para o produto sin (Esquema 15). A
reação das sulfonas 33a,b com nitrometanonato de sódio forneceu as correspondentes
1,2 nitroaminas N-protegidas 35a,b também em bons rendimentos químicos e alta
diastereosseletividade para o produto anti (Esquema 16).
(R)
OO (R) SO2Ph
NHCO2R
(S)
OO (S)
NHCO2R
CH3NO2, NaH[CH2NO2
-]
-PhSO2- NO2
33a, R = t-Bu 33b, Bn
35a, = 75%, r.d. > 95:535b, = 74%, r.d. > 95:5
(S)
OO
NCO2Rin situ
34a,b
Esquema 16: Obtenção das 1,2 nitroaminas N-protegidas 35a,b.
Nesta etapa, os autores relatam que as iminas 34a,b são formadas in situ e uma
adição posterior do ânion nitronato foi efetuada. As energias das estruturas de transição,
otimizadas através do método semi-empírico PM3, revelaram que o ataque do nitronato
de sódio pela face Si da imina 34a (confôrmero A) é favorecido por 3,92 kcal.mol-1, em
relação à adição pela face Re (confôrmero B) levando à formação do produto anti (S,S)-
35a, como exemplificado na Figura 8 mostrada a seguir:
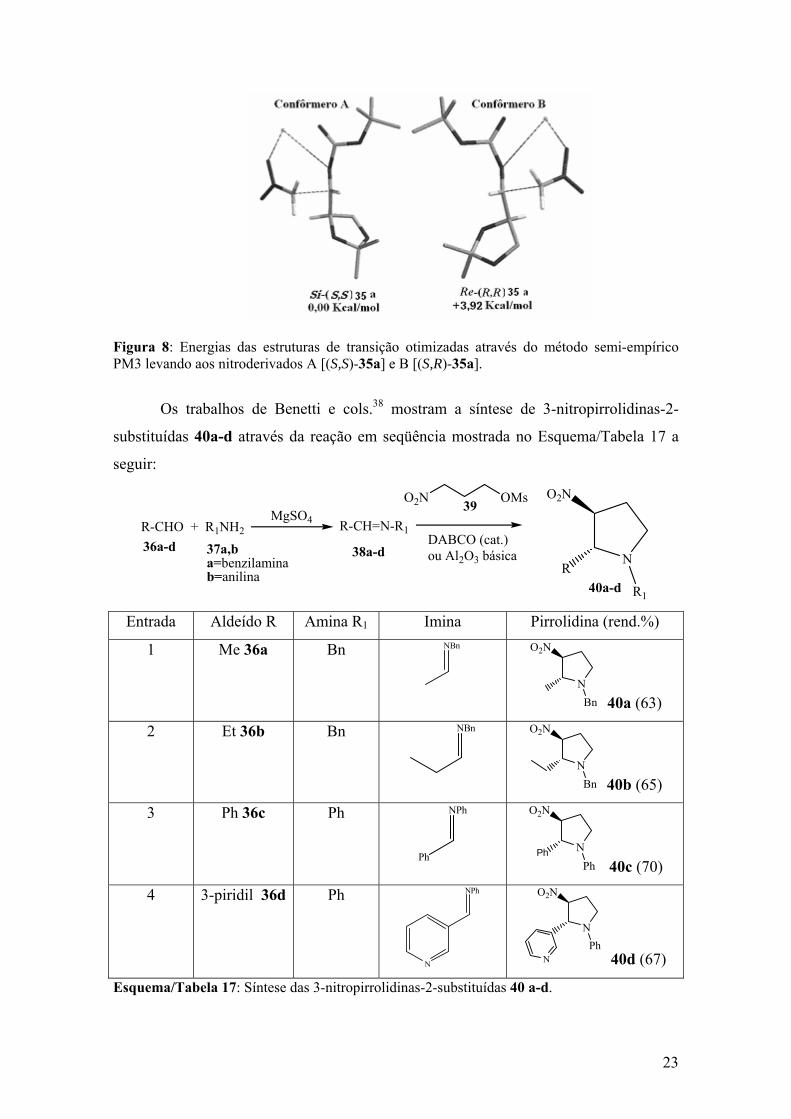
23
Figura 8: Energias das estruturas de transição otimizadas através do método semi-empírico PM3 levando aos nitroderivados A [(S,S)-35a] e B [(S,R)-35a].
Os trabalhos de Benetti e cols.38 mostram a síntese de 3-nitropirrolidinas-2-
substituídas 40a-d através da reação em seqüência mostrada no Esquema/Tabela 17 a
seguir:
R-CHO + R1NH2MgSO4 R-CH=N-R1
O2N OMs
DABCO (cat.)ou Al2O3 básica N
R
O2N
R1
36a-d 37a,ba=benzilaminab=anilina
38a-d
39
40a-d Entrada Aldeído R Amina R1 Imina Pirrolidina (rend.%)
1 Me 36a Bn NBn
N
O2N
Bn 40a (63)
2 Et 36b Bn NBn
N
O2N
Bn 40b (65)
3 Ph 36c Ph
Ph
NPh
NPh
O2N
Ph 40c (70)
4 3-piridil 36d Ph NPh
N
N
O2N
PhN 40d (67)
Esquema/Tabela 17: Síntese das 3-nitropirrolidinas-2-substituídas 40 a-d.

24
Inicialmente, Benetti e cols. relataram que as primeiras tentativas de realizar a
seqüência de reações mostradas anteriormente no Esquema/Tabela 17, misturando
apenas quantidades equimolares dos três componentes (aldeído, amina e 3-
nitropropanolmetanossulfonato 39 em diclorometano e à temperatura ambiente)
produziram apenas e em baixos rendimentos, o produto esperado 40a-d. Os
pesquisadores, entretanto, otimizaram as condições reacionais para a obtenção das
desejadas pirrolidinas 40a-d, através da formação das aldiminas 38a-d, seguida da
reação com 39 e quantidades catalíticas de DABCO ou alumina básica.
Desta forma, as pirrolidinas 40a-d foram sintetizadas com rendimentos químicos
satisfatórios e, na maioria dos casos, como um único diastereoisômero, o qual possui
estereoquímica trans entre o grupamento nitro na posição 3 e o substituinte na posição
2. O isômero cis esteve em alguns casos presente sob forma de traços.
As estruturas dos compostos 40a-d puderam ser assinaladas inequivocamente
através de análises de RMN de 1H das constantes de acoplamento entre H-2 e H-3 (J= 0
–2,5 Hz), que é consistente com um ângulo diedro de 90º.
*
Os resultados por nós obtidos, sobre a adição nucleofílica diastereosseletiva de
nitroalcanos a iminas quirais azaenolizáveis serão abordados posteriormente na seção de
Resultados e discussão.
1.7 Nitroolefinas: Características gerais, métodos de preparação e adições conjugadas
estereosseletivas a nitroolefinas.
1.7.1 – Características gerais.
Nitroolefinas são alcenos cuja ligação dupla está em conjugação com o grupo
nitro (~NO2) - fortemente retirador de elétrons – o que as tornam excelentes aceptores
de Michael, ou seja, ótimos eletrófilos para reações do tipo adição conjugada 1,4.1b,c
Veja Esquema 18 abaixo:
R1 R2
NO2
NuR1 R2
NO2NuAceptores de Michael
Adutos de Michael
Adição de Michael
Nu = Espécie nucleofílica
Esquema 18: Adição conjugada de espécieis nucleofílicas a nitroolefinas.

25
A adição conjugada de nucleófilos a nitroolefinas é considerada uma valiosa
ferramenta na formação de ligações carbono-carbono e carbono-heteroátomo,
dependendo do nucleófilo empregado em questão. Uma grande variedade de derivados
de nitroalcanos pode ser obtida como produto desta reação. Além disso, o grupo nitro,
como já visto anteriormente, é particularmente versátil em síntese orgânica, portanto, os
adutos obtidos podem ser transformados em uma grande variedade de moléculas com
distintas funcionalidades (veja Figura 2). Posteriormente, serão abordados os mais
significativos exemplos retirados da literatura de adições conjugadas a nitroolefinas.
Outras reações nas quais nitroolefinas podem estar envolvidas são as reações de
cicloadição,40 como a reação de Diels-Alder, onde são empregadas como potentes
dienófilos41 e também heterodienos42. Veja Esquemas 19 e 20 a seguir:
TBDMSO
N
O
41
+
Ph
NO2
Nitroestireno
i) MeOH, -80ºC a t.a.ii) AcOH / NaOAc, pH=4,6iii) recristalização
O
NO2
Ph
OTBDMS
42e.e.> 95%
Esquema 19: Reação de Diels-Alder empregando o nitroestireno como dienófilo.
A reação de Diels-Alder, mostrada no Esquema 19, emprega o nitroestireno
como dienófilo e o dieno quiral 41. Inicialmente, o nitroestireno e o dieno 41 foram
submetidos à reação de cicloadição do tipo [4+2] em metanol, à baixa temperatura.
Após deixar-se o sistema, gradualmente, atingir a temperatura ambiente promoveu-se,
em meio ácido tamponado, a conversão da enamina intermediária formada à cetona 42.
Dessa forma, houve a remoção da porção quiral que foi responsável pela alta
enantiosseletividade da cicloexanona tetra-substituída 42.

26
N
OMe
OMe
OBu
+
Tolueno, 0°C80%
MAD
43 OMe
OMe
NOO OBu
44
O
O
Esquema 20: Reação de Diels-Alder empregando nitroolefina 43 como heterodieno.
Alquilenol éteres são extensamente empregados como dienófilos em
cicloadições do tipo [4+2] com nitroolefinas. As reações podem ser promovidas pelo
uso de variados ácidos de Lewis para fornecer nitronatos cíclicos em bons rendimentos
químicos. No Esquema 20 mostrado acima, um exemplo representativo é a cicloadição
do derivado do nitroestireno 43 com o n-butil vinil éter promovido pelo volumoso ácido
de Lewis bis(2,6-di-t-butil-4-metilfenóxido)-metilalumínio (MAD) em tolueno a 0ºC
para fornecer o nitronato de alquila 44 em excelente rendimento químico (80%).
1.7.2 - Métodos de preparação.
O método mais prático e usual de preparação de nitroolefinas envolve a reação
de Henry ou nitroaldólica, sendo esta reconhecida como uma das mais clássicas reações
para a formação de ligação C-C. Essencialmente, trata-se da condensação entre uma
substância carbonilada (aldeído ou cetona) e um nitroalcano que contenha um
hidrogênio alfa ao grupamento nitro, na presença de uma base. O produto formado desta
reação é um nitroálcool que, após sofrer eliminação, fornece a nitroolefina desejada.
McMurry e cols.43 descreveram a reação onde nitroálcoois são submetidos à formação,
in situ, de um mesilato e sua posterior eliminação através do tratamento com base para
fornecer as nitroolefinas 45 em bons rendimentos químicos. Veja Esquema 21 abaixo:

27
R1
OH
NO2
R2
R1 e R2 = grupos alquila
R1NO2
R2
MsCl, TEA
15 min., 0 0C(67-78 %) 45
Esquema 21: Formação de nitroolefinas a partir de nitroálcoois.
Diferentemente do método de McMurry para a síntese de nitroolefinas
conjugadas, onde são empregados cloreto de metanossulfonila (MsCl) e trietilamina
como base, outros métodos também estão relatados na literatura. Como por exemplo,
Barua e cols.44 desenvolveram a síntese de nitroolefinas conjugadas a partir da
desidratação de um nitroálcool, oriundo de uma reação nitroaldólica, através do
emprego de trifenilfosfina e trietilamina conduzida em tetracloreto de carbono sob
refluxo, obtendo o produto desejado em uma faixa de rendimento químico que variou
de 80 a 95% . Veja Esquema/Tabela 22 abaixo:
R1
R2
OH
NO2
R1
NO2
R2
PPh3 / CCl4 / Et3N / refluxo
1 - 4 h80 - 95 %46 47
46, 47 R1 R2
a Ph H
b Ph Me
c 4-MeOC6H4 Me
d 4-Cl-C6H4 Me
e Pr Me
Esquema/Tabela 22: Síntese das nitroolefinas 47 a-e a partir da desidratação dos nitroálcoois 46a-e.
Os autores relataram que os nitroálcoois 46a-e são transformados nos haletos
correspondentes pelo tratamento com trifenilfosfina e o tetra-haleto de carbono, no caso,
CCl4, e simultaneamente com o emprego de uma base adequada gera, in situ, a
eliminação do haleto de hidrogênio para fornecer as nitroolefinas conjugadas em ótimos
rendimentos químicos e exclusivamente com geometria E.

28
Em 1998, Sreekumar e cols.45 descreveram a síntese de nitroolefinas conjugadas
usando zeólitas (peneira molecular – aluminossilicatos hidratados) como catalisadores.
Veja Esquema/Tabela 23 abaixo:
NO (1atm)CCl4, 2h
HY-Zeólita, 75°CAlceno48a-e
Nitroolefina49a-e
Entrada Alceno Nitroolefina Rendimento
1
48a
NO2
49a
78
2 n-C8H17 48b n-C8H17NO2
49b 71
3
48c
NO2
49c
79
4
48d
NO2
49d
81
5
48e
NO2
49e
76
Esquema 23 /Tabela: Síntese de nitroolefinas 49a-e empregando zeólitas.
A síntese de nitroolefinas conjugadas baseia-se, comumente, em uma inicial
reação de nitroaldólica com posterior eliminação de H2O. Diferentemente, no estudo
realizado por Sreekumar e cols. mostrado acima, foram sintetizadas as desejadas
nitroolefinas a partir de alcenos, empregando o gás óxido nítrico (NO) e zeólitas.
As nitroolefinas sintetizadas apresentaram rendimentos químicos que variaram de
71 a 81% e os autores relataram que a facilidade deste método catalítico heterogêneo
para a conversão de olefinas em nitroolefinas resulta em um uso de zeólitas de baixo
custo, fácil manipulação experimental e regeneração, permitindo o reúso do catalisador.
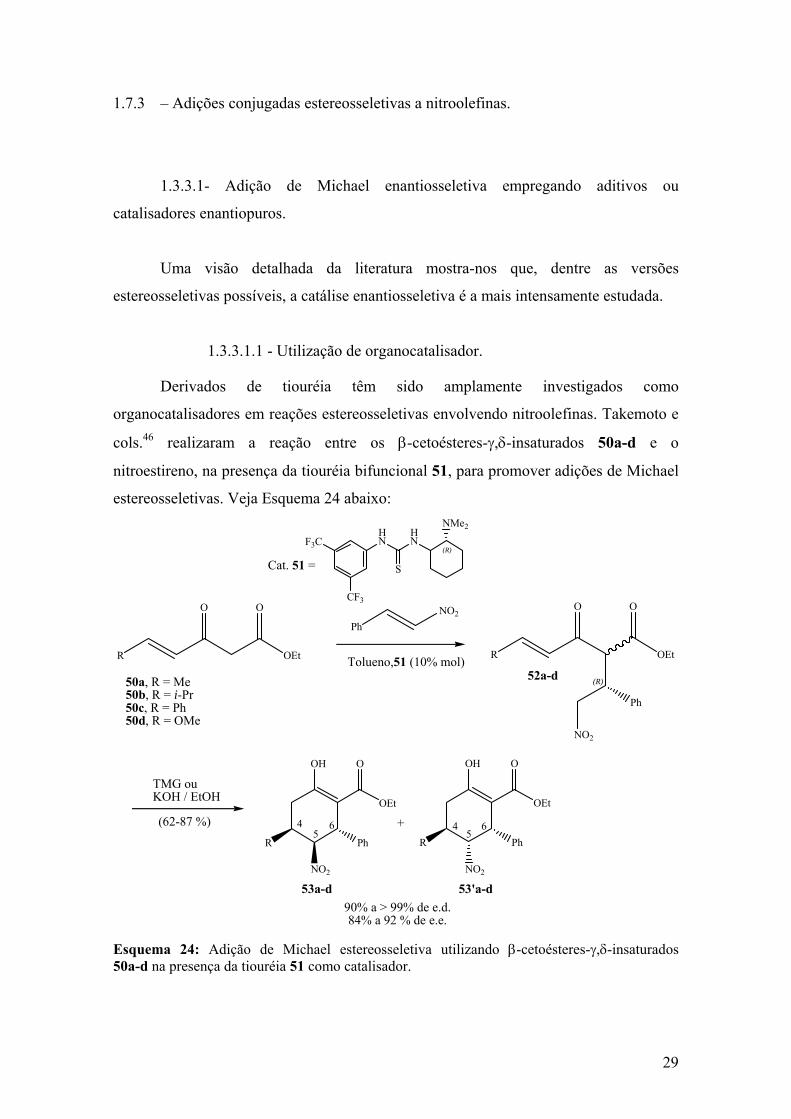
29
1.7.3 – Adições conjugadas estereosseletivas a nitroolefinas.
1.3.3.1- Adição de Michael enantiosseletiva empregando aditivos ou
catalisadores enantiopuros.
Uma visão detalhada da literatura mostra-nos que, dentre as versões
estereosseletivas possíveis, a catálise enantiosseletiva é a mais intensamente estudada.
1.3.3.1.1 - Utilização de organocatalisador.
Derivados de tiouréia têm sido amplamente investigados como
organocatalisadores em reações estereosseletivas envolvendo nitroolefinas. Takemoto e
cols.46 realizaram a reação entre os β-cetoésteres-γ,δ-insaturados 50a-d e o
nitroestireno, na presença da tiouréia bifuncional 51, para promover adições de Michael
estereosseletivas. Veja Esquema 24 abaixo:
R OEt
O O
R OEt
O O
(R)
NO2
Ph
PhNO2
HN
HN
(R)
S
F3C
CF3
NMe2
50a, R = Me50b, R = i-Pr50c, R = Ph50d, R = OMe
Tolueno,51 (10% mol)
TMG ou KOH / EtOH
52a-d
(62-87 %)
OH O
OEt
Ph
NO2
R
OH O
OEt
Ph
NO2
R
45
6 45
6+
53a-d
Cat. 51 =
90% a > 99% de e.d.84% a 92 % de e.e.
53'a-d
Esquema 24: Adição de Michael estereosseletiva utilizando β-cetoésteres-γ,δ-insaturados 50a-d na presença da tiouréia 51 como catalisador.
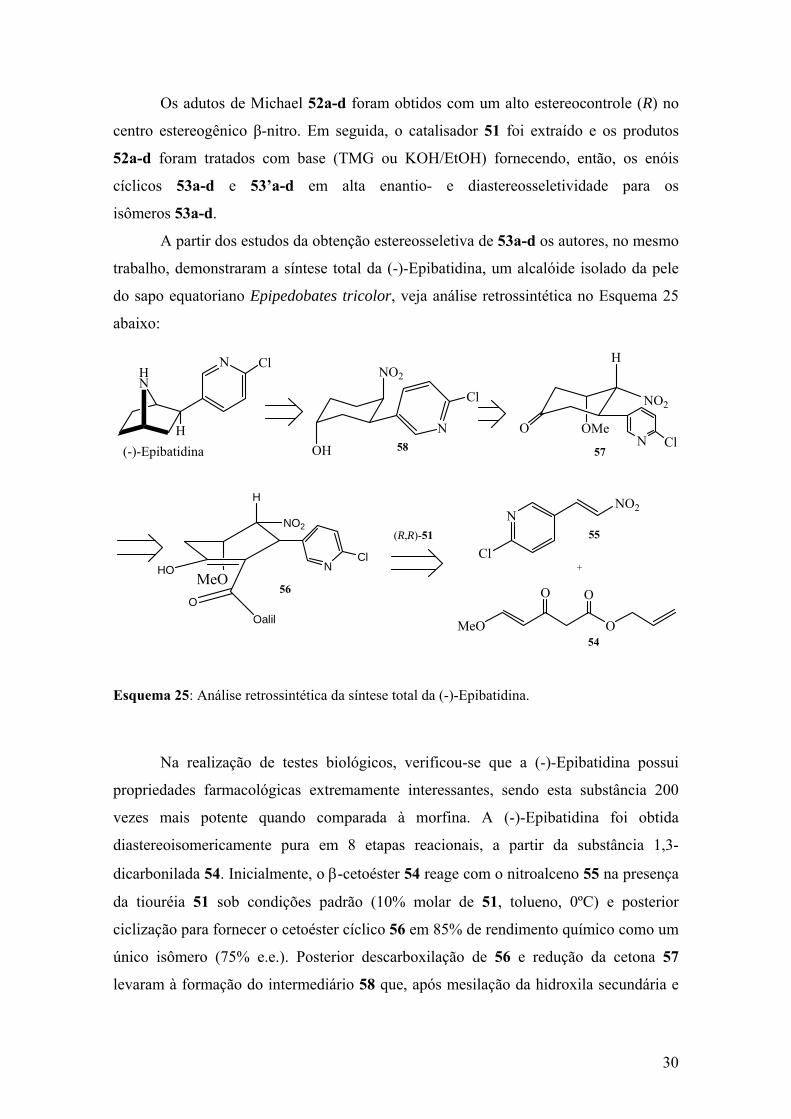
30
Os adutos de Michael 52a-d foram obtidos com um alto estereocontrole (R) no
centro estereogênico β-nitro. Em seguida, o catalisador 51 foi extraído e os produtos
52a-d foram tratados com base (TMG ou KOH/EtOH) fornecendo, então, os enóis
cíclicos 53a-d e 53’a-d em alta enantio- e diastereosseletividade para os
isômeros 53a-d.
A partir dos estudos da obtenção estereosseletiva de 53a-d os autores, no mesmo
trabalho, demonstraram a síntese total da (-)-Epibatidina, um alcalóide isolado da pele
do sapo equatoriano Epipedobates tricolor, veja análise retrossintética no Esquema 25
abaixo:
HO
OalilO
H
NO2
NCl
MeO
HN
H
N Cl
(-)-Epibatidina OH
NO2
N
Cl
O OMe
H
NO2
N Cl
(R,R)-51N
Cl
NO2
MeO O
55
54
+
58 57
O O56
Esquema 25: Análise retrossintética da síntese total da (-)-Epibatidina.
Na realização de testes biológicos, verificou-se que a (-)-Epibatidina possui
propriedades farmacológicas extremamente interessantes, sendo esta substância 200
vezes mais potente quando comparada à morfina. A (-)-Epibatidina foi obtida
diastereoisomericamente pura em 8 etapas reacionais, a partir da substância 1,3-
dicarbonilada 54. Inicialmente, o β-cetoéster 54 reage com o nitroalceno 55 na presença
da tiouréia 51 sob condições padrão (10% molar de 51, tolueno, 0ºC) e posterior
ciclização para fornecer o cetoéster cíclico 56 em 85% de rendimento químico como um
único isômero (75% e.e.). Posterior descarboxilação de 56 e redução da cetona 57
levaram à formação do intermediário 58 que, após mesilação da hidroxila secundária e
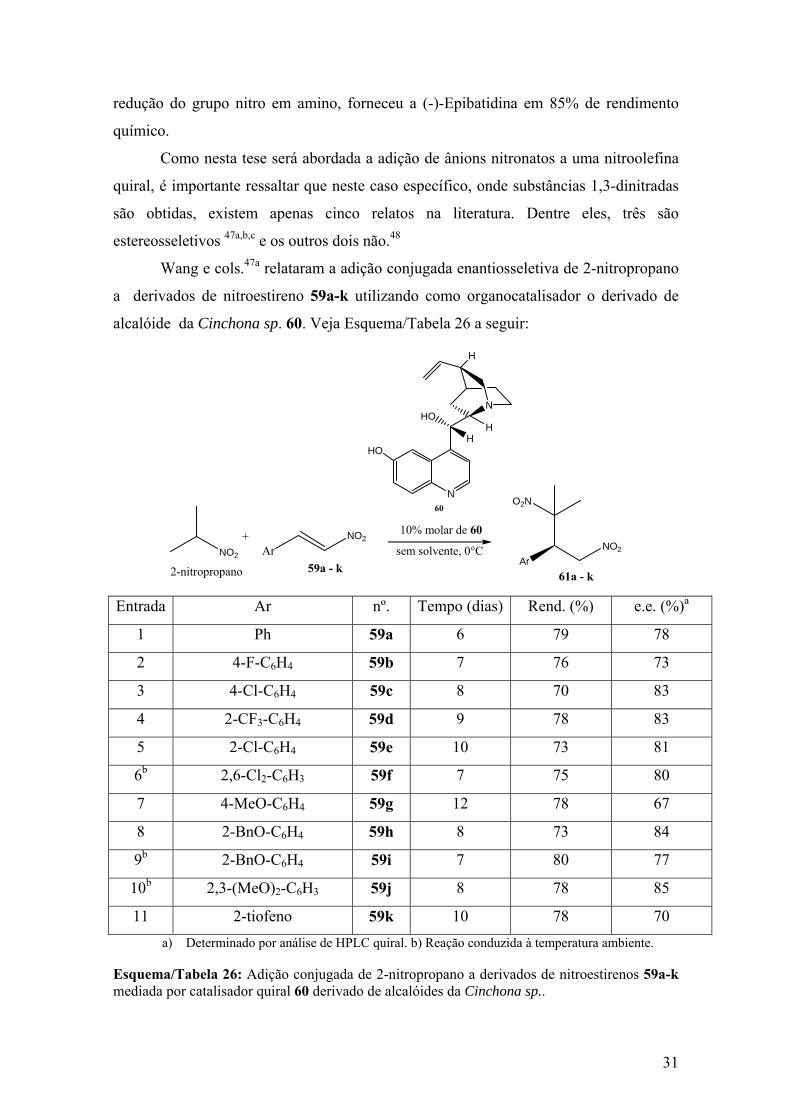
31
redução do grupo nitro em amino, forneceu a (-)-Epibatidina em 85% de rendimento
químico.
Como nesta tese será abordada a adição de ânions nitronatos a uma nitroolefina
quiral, é importante ressaltar que neste caso específico, onde substâncias 1,3-dinitradas
são obtidas, existem apenas cinco relatos na literatura. Dentre eles, três são
estereosseletivos 47a,b,c e os outros dois não.48
Wang e cols.47a relataram a adição conjugada enantiosseletiva de 2-nitropropano
a derivados de nitroestireno 59a-k utilizando como organocatalisador o derivado de
alcalóide da Cinchona sp. 60. Veja Esquema/Tabela 26 a seguir:
N
HO
H
N
H
H
HO
60
NO2
+Ar
NO210% molar de 60
sem solvente, 0°C NO2
O2N
Ar59a - k61a - k2-nitropropano
Entrada Ar nº. Tempo (dias) Rend. (%) e.e. (%)a
1 Ph 59a 6 79 78
2 4-F-C6H4 59b 7 76 73
3 4-Cl-C6H4 59c 8 70 83
4 2-CF3-C6H4 59d 9 78 83
5 2-Cl-C6H4 59e 10 73 81
6b 2,6-Cl2-C6H3 59f 7 75 80
7 4-MeO-C6H4 59g 12 78 67
8 2-BnO-C6H4 59h 8 73 84
9b 2-BnO-C6H4 59i 7 80 77
10b 2,3-(MeO)2-C6H3 59j 8 78 85
11 2-tiofeno 59k 10 78 70 a) Determinado por análise de HPLC quiral. b) Reação conduzida à temperatura ambiente.
Esquema/Tabela 26: Adição conjugada de 2-nitropropano a derivados de nitroestirenos 59a-k mediada por catalisador quiral 60 derivado de alcalóides da Cinchona sp..

32
Estudos prévios realizados por Wang e cols. demonstraram que o
organocatalisador derivado de alcalóide da Cinchona sp. 60 promoveu o acesso às
substâncias 1,3-dinitradas em satisfatórios rendimentos químicos (70 – 82%) e níveis de
enantiosseletividade (67 – 85% e.e.).
Analisando o Esquema/Tabela 26 mostrado anteriormente, verificou-se que os
anéis aromáticos das nitroolefinas que apresentam substituintes neutros (entrada 1),
retiradores de elétrons (entradas 2 – 6) e doadores de elétrons (entradas 7 – 10) reagiram
semelhantemente, indicando que características eletrônicas têm pouco efeito sobre o
processo, bem como efeitos estéricos (entradas 4, 5, e 8-10) parecem governar
minimamente a reatividade e enantiosseletividade das adições conjugadas. Uma
nitroolefina heteroaromática foi empregada (entrada 11) e também foi verificado o
mesmo padrão de reatividade e estereosseletividade das demais nitroolefinas estudadas.
1.3.3.1.2 - Utilização de catalisador organometálico:
Du e cols.47b utilizaram um complexo tridentado de zinco e bis(oxazolina) 62 na
adição estereosseletiva de nitroetano ao nitroestireno e derivados (63a-e). Os produtos
64 e 65a-e foram obtidos em bons rendimentos químicos com alta enantiosseletividade
(e.e. 88 - 91 %) e de moderada a boa diastereosseletividade (3,8 - 11,7 : 1). Os adutos de
Michael com configuração sin foram obtidos majoritariamente, conforme visto no
Esquema 27:
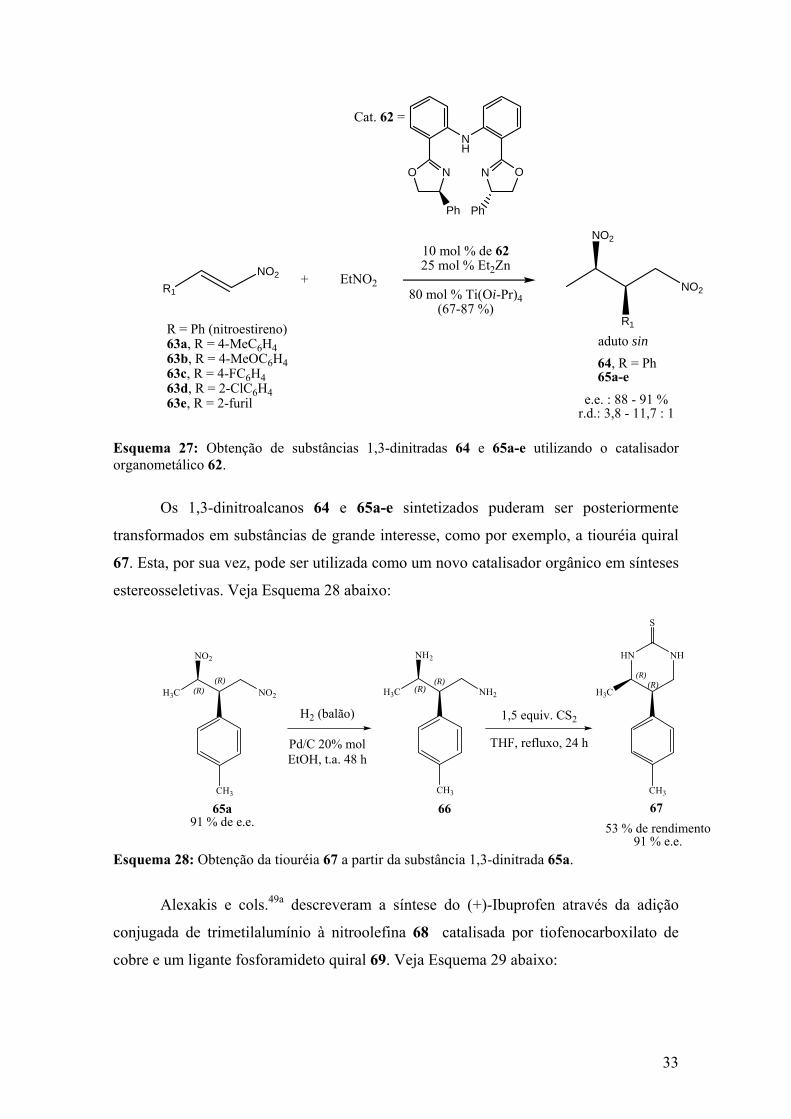
33
R1
NO2 + NO2
R1
NO2
R = Ph (nitroestireno)63a, R = 4-MeC6H463b, R = 4-MeOC6H463c, R = 4-FC6H463d, R = 2-ClC6H463e, R = 2-furil
10 mol % de 6225 mol % Et2Zn
80 mol % Ti(Oi-Pr)4(67-87 %)
aduto sin
e.e. : 88 - 91 % r.d.: 3,8 - 11,7 : 1
NH
O N N O
PhPh
Cat. 62 =
64, R = Ph65a-e
EtNO2
Esquema 27: Obtenção de substâncias 1,3-dinitradas 64 e 65a-e utilizando o catalisador organometálico 62.
Os 1,3-dinitroalcanos 64 e 65a-e sintetizados puderam ser posteriormente
transformados em substâncias de grande interesse, como por exemplo, a tiouréia quiral
67. Esta, por sua vez, pode ser utilizada como um novo catalisador orgânico em sínteses
estereosseletivas. Veja Esquema 28 abaixo:
H3C (R)(R)
NO2
NO2
CH3
H3C (R)(R)
NH2
NH2
CH3
H3C
(R)(R)
NHHN
CH3
S
H2 (balão)
Pd/C 20% molEtOH, t.a. 48 h
1,5 equiv. CS2
THF, refluxo, 24 h
65a 91 % de e.e.
66 6753 % de rendimento
91 % e.e.Esquema 28: Obtenção da tiouréia 67 a partir da substância 1,3-dinitrada 65a.
Alexakis e cols.49a descreveram a síntese do (+)-Ibuprofen através da adição
conjugada de trimetilalumínio à nitroolefina 68 catalisada por tiofenocarboxilato de
cobre e um ligante fosforamideto quiral 69. Veja Esquema 29 abaixo:
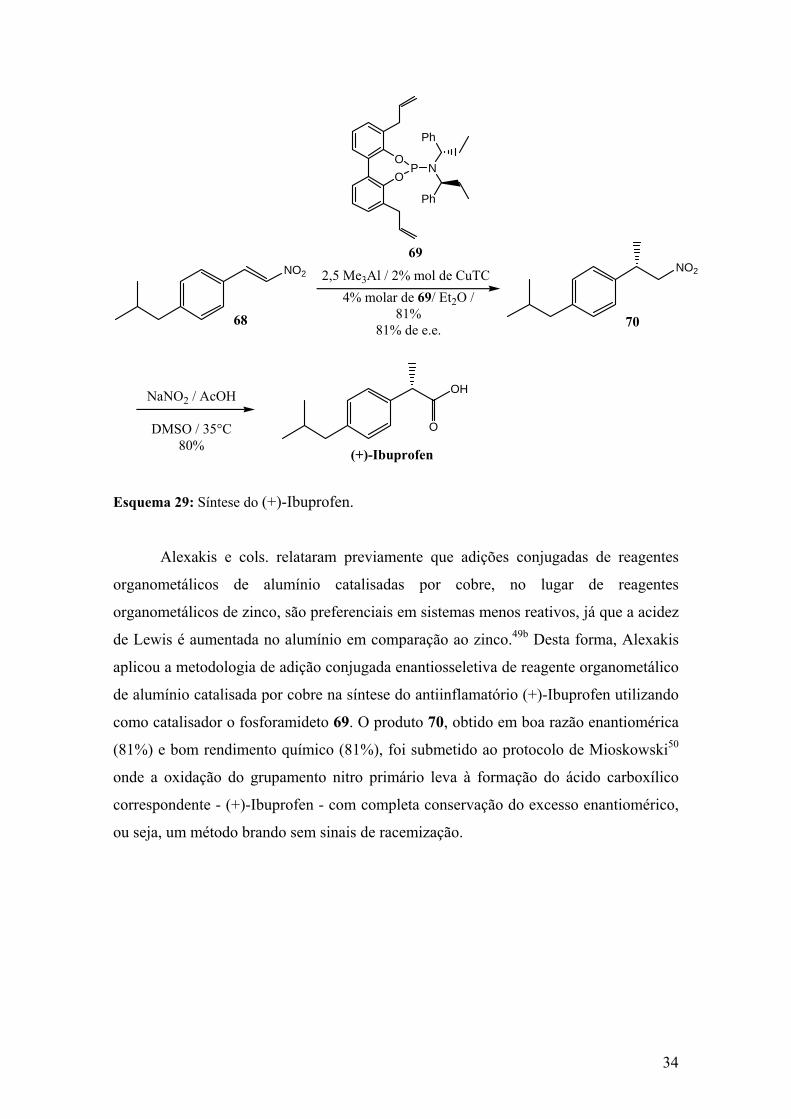
34
NO2 2,5 Me3Al / 2% mol de CuTC4% molar de 69/ Et2O /
81%81% de e.e.
NO2
68 70
OO
P N
Ph
Ph
69
OH
(+)-Ibuprofen
O
NaNO2 / AcOH
DMSO / 35°C80%
Esquema 29: Síntese do (+)-Ibuprofen.
Alexakis e cols. relataram previamente que adições conjugadas de reagentes
organometálicos de alumínio catalisadas por cobre, no lugar de reagentes
organometálicos de zinco, são preferenciais em sistemas menos reativos, já que a acidez
de Lewis é aumentada no alumínio em comparação ao zinco.49b Desta forma, Alexakis
aplicou a metodologia de adição conjugada enantiosseletiva de reagente organometálico
de alumínio catalisada por cobre na síntese do antiinflamatório (+)-Ibuprofen utilizando
como catalisador o fosforamideto 69. O produto 70, obtido em boa razão enantiomérica
(81%) e bom rendimento químico (81%), foi submetido ao protocolo de Mioskowski50
onde a oxidação do grupamento nitro primário leva à formação do ácido carboxílico
correspondente - (+)-Ibuprofen - com completa conservação do excesso enantiomérico,
ou seja, um método brando sem sinais de racemização.
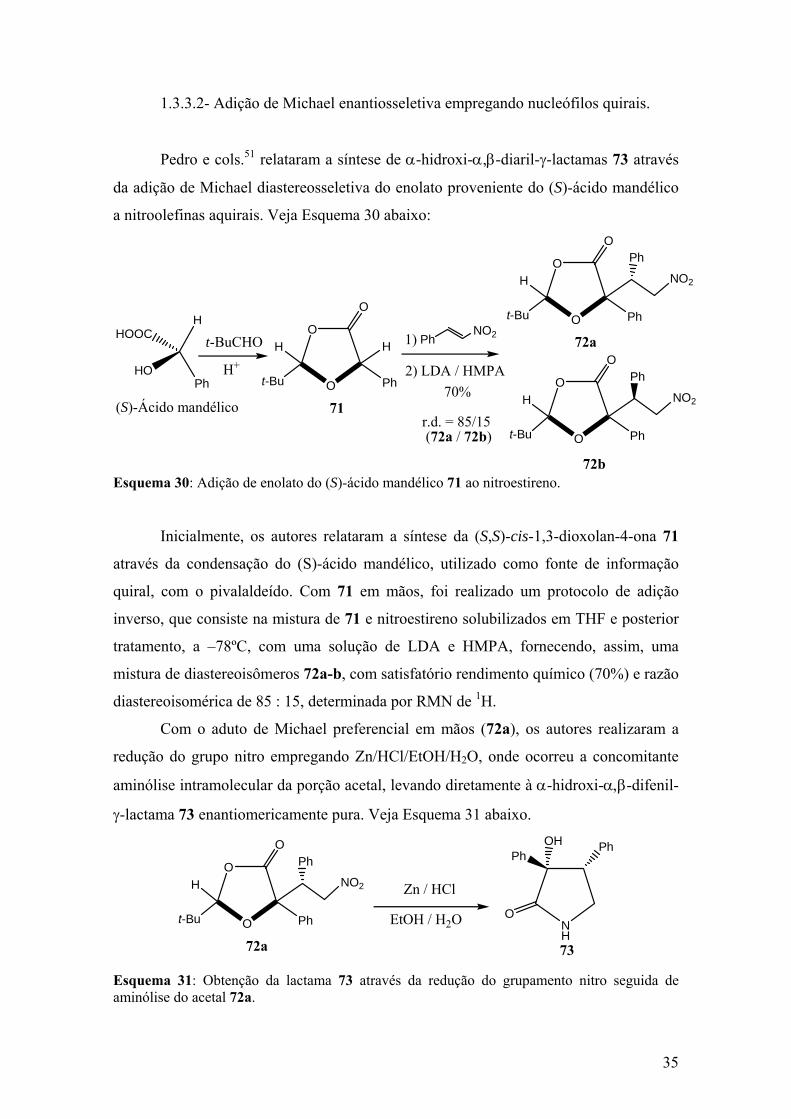
35
1.3.3.2- Adição de Michael enantiosseletiva empregando nucleófilos quirais.
Pedro e cols.51 relataram a síntese de α-hidroxi-α,β-diaril-γ-lactamas 73 através
da adição de Michael diastereosseletiva do enolato proveniente do (S)-ácido mandélico
a nitroolefinas aquirais. Veja Esquema 30 abaixo:
Ph
HHOOC
HO
(S)-Ácido mandélico
t-BuCHO
H+
O
O
O
H
Ph
H
t-Bu
71
1)
2) LDA / HMPA
O
O
O
Ph
H
t-Bu
72aPhNO2
NO2
O
O
O
Ph
H
t-Bu
NO2
72b
Ph
Ph 70%
r.d. = 85/15 (72a / 72b)
Esquema 30: Adição de enolato do (S)-ácido mandélico 71 ao nitroestireno.
Inicialmente, os autores relataram a síntese da (S,S)-cis-1,3-dioxolan-4-ona 71
através da condensação do (S)-ácido mandélico, utilizado como fonte de informação
quiral, com o pivalaldeído. Com 71 em mãos, foi realizado um protocolo de adição
inverso, que consiste na mistura de 71 e nitroestireno solubilizados em THF e posterior
tratamento, a –78ºC, com uma solução de LDA e HMPA, fornecendo, assim, uma
mistura de diastereoisômeros 72a-b, com satisfatório rendimento químico (70%) e razão
diastereoisomérica de 85 : 15, determinada por RMN de 1H.
Com o aduto de Michael preferencial em mãos (72a), os autores realizaram a
redução do grupo nitro empregando Zn/HCl/EtOH/H2O, onde ocorreu a concomitante
aminólise intramolecular da porção acetal, levando diretamente à α-hidroxi-α,β-difenil-
γ-lactama 73 enantiomericamente pura. Veja Esquema 31 abaixo.
O
O
O
Ph
H
t-Bu
72a
NO2
Ph
Zn / HCl
EtOH / H2O NH
O
PhPh
OH
73 Esquema 31: Obtenção da lactama 73 através da redução do grupamento nitro seguida de aminólise do acetal 72a.

36
Um outro exemplo de emprego de nucleófilos quirais na adição conjugada a
nitroolefinas aquirais pode ser visto no estudo de Mioskowski e cols.,50 onde foi
relatada a síntese de α-aminoácidos 77a-d a partir de nitroolefinas 74a-d. Veja
Esquema 32 abaixo:
1) t-BuOK, THF
2) NO2
R
O
NH
OPh
O
NO
Ph
NO2
R(R)-4-fenil-2-oxazolidinona
R
R
75a-d
NaNO2 / AcOH
DMSO / 40°C
O
NO
Ph
OHR
R
R
76a-dO
1) Li / NH3THF, t-BuOH, -78°C
2) DOWEX 50WX4-400H2O / NH4OH aq.
NH2
OHR
77a-dO
a, R= Me (25%)b, R= n-Pr (48%)c, R= t-Bu (89%)d, R= cicloexil (88%)
(62 - 99%)
74a-d
(43 - 87%)e.d. > 98%
a, R= Meb, R= n-Prc, R= t-Bud, R= cicloexil
Esquema 32: Síntese de α-aminoácidos 77a-d a partir da adição da (R)-4-fenil-2-oxazolidinona a nitroolefinas aquirais 74a-d.
O tratamento da (R)-4-fenil-2-oxazolidinona com t-butóxido de potássio gera o
amideto quiral correspondente que é, então, adicionado às nitroolefinas
monossubstituídas 74a –d para produzir os nitro-derivados 75a-d em uma faixa de 43 a
87% de rendimento químico, com excessos diastereoisoméricos superiores a 98%. Os
nitro-derivados obtidos são levados aos ácidos carboxílicos correspondentes, 76a-d,
através de condições brandas, com rendimentos químicos que variaram de 43 a 87 %.
Observou-se que os produtos com substituintes alquílicos de maior massa (76c,d) foram
obtidos em maior rendimento químico em comparação àqueles com grupamento alquila
de pouca massa (76a-b), o que sugere a perda de produto devido a sua solubilidade em
fase aquosa durante os procedimentos de isolamento. Para a obtenção dos α-
aminoácidos 77a-d, o grupamento amino pode ser obtido através da reação de Birch,
que consiste no tratamento das oxazolidinonas 76a-d com lítio, amônia em THF e
tBuOH a – 78ºC por 30 minutos. Desta forma, os correspondentes α-aminoácidos foram
isolados após purificação em resina de troca iônica e obtidos em 62 – 99% de
rendimento químico. A pureza enantiomérica dos α-aminoácidos 77a-c obtidos foi
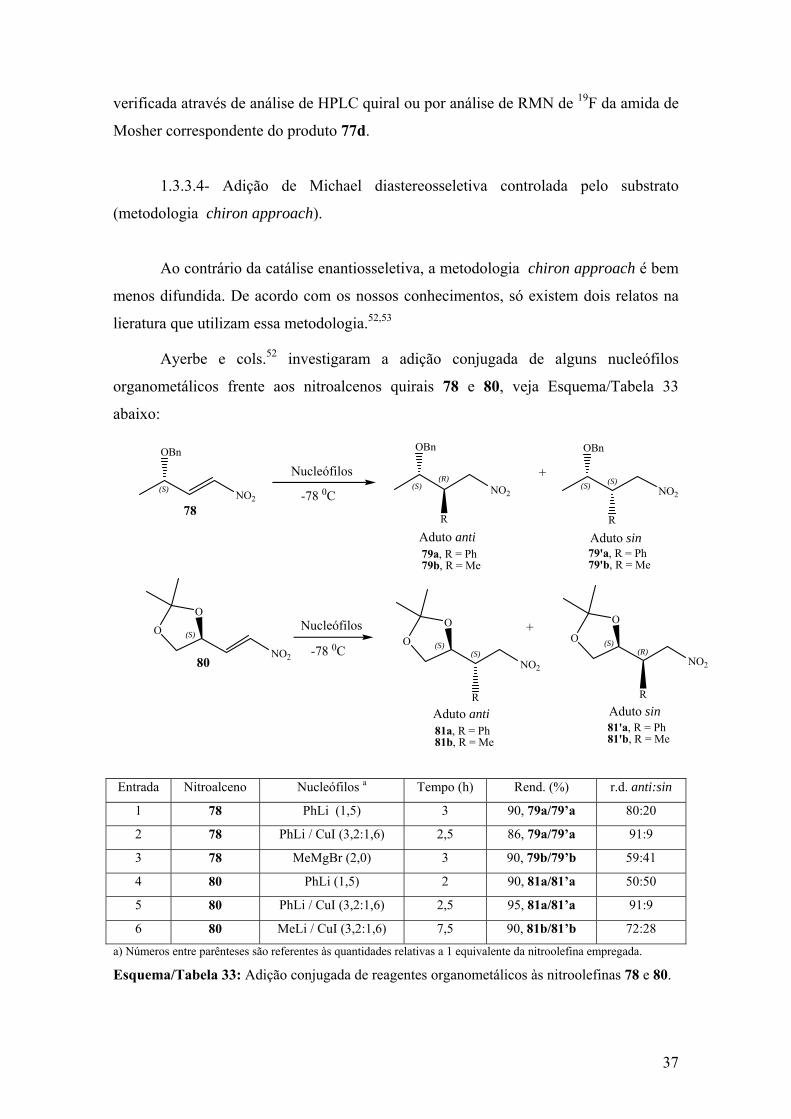
37
verificada através de análise de HPLC quiral ou por análise de RMN de 19F da amida de
Mosher correspondente do produto 77d.
1.3.3.4- Adição de Michael diastereosseletiva controlada pelo substrato
(metodologia chiron approach).
Ao contrário da catálise enantiosseletiva, a metodologia chiron approach é bem
menos difundida. De acordo com os nossos conhecimentos, só existem dois relatos na
lieratura que utilizam essa metodologia.52,53
Ayerbe e cols.52 investigaram a adição conjugada de alguns nucleófilos
organometálicos frente aos nitroalcenos quirais 78 e 80, veja Esquema/Tabela 33
abaixo:
NO2(S)
OBn
NO2
(R)(S)
OBn
R
NO2(S)(S)
OBn
R
+
79a, R = Ph79b, R = Me
79'a, R = Ph79'b, R = Me
Nucleófilos
(S)
O
O
NO2(S)
O
O(S)
NO2
(S)
O
O(R)
NO2
Nucleófilos +
78
80
R R
-78 0C
-78 0C
Aduto anti Aduto sin
81a, R = Ph81b, R = Me
81'a, R = Ph81'b, R = Me
Aduto anti Aduto sin
Entrada Nitroalceno Nucleófilos a Tempo (h) Rend. (%) r.d. anti:sin
1 78 PhLi (1,5) 3 90, 79a/79’a 80:20
2 78 PhLi / CuI (3,2:1,6) 2,5 86, 79a/79’a 91:9
3 78 MeMgBr (2,0) 3 90, 79b/79’b 59:41
4 80 PhLi (1,5) 2 90, 81a/81’a 50:50
5 80 PhLi / CuI (3,2:1,6) 2,5 95, 81a/81’a 91:9
6 80 MeLi / CuI (3,2:1,6) 7,5 90, 81b/81’b 72:28
a) Números entre parênteses são referentes às quantidades relativas a 1 equivalente da nitroolefina empregada.
Esquema/Tabela 33: Adição conjugada de reagentes organometálicos às nitroolefinas 78 e 80.

38
Analisando os dados acima, as nitroolefinas 78 e 80 induziram, em geral, de um
baixo a moderado estereocontrole na obtenção dos seus respectivos adutos de Michael
79/79’ e 81/81’. Os melhores resultados foram obtidos utilizando transmetalação com
CuI (entradas 2 e 5). Sendo assim, os nucleófilos mais promissores para essas adições
diastereosseletivas foram os derivados de lítio organocupratos do tipo LiR2Cu. Os
autores relataram que os produtos de configuração anti 79a,b e 81a,b foram obtidos
majoritariamente.
Um outro estudo foi realizado por Pätzel e cols.,53 onde foram adicionados
reagentes organometálicos funcionalizados ao nitroalceno 82, e é mostrado no
Esquema/Tabela 34 a seguir:
(S)
O
O
NO2(S)
O
O(S)
NO2
(S)
O
O(R)
NO2
Nucleófilos +
Aduto anti83a-d
Aduto sin83'a-d
82
R R
Entrada Nucleófilos Condições
reacionais Produtos
Rend.
(%) r.d. (anti:sin)
1 CH2=CH-MgBr/ZnI2/CuCN - 78°C → t.a, THF 83a/83’a 57 91:9
2 CH2=CH-MgBr - 78°C → t.a, THF 83b/83’b 68 10:90
3 PhC≡CLi - 78°C, THF 83c/83’c 82 73:27
4 Me3SiC≡CLi - 78°C, THF 83d/83’d 71 60:40
Esquema/Tabela 34: Adição conjugada de reagentes organometálicos funcionalizados à nitroolefina 82.
Nos exemplos demonstrados acima, os adutos de Michael foram obtidos em
bons rendimentos químicos seguidos de uma pobre diastereosseletividade nas entradas 3
e 4, e boa diastereosseletividade nas entradas 1 e 2. Nestes estudos, os autores também
relatam que os produtos de configuração anti 83a,c,d são formados majoritariamente,
exceto na entrada 2, pois a estereoquímica inversa para o aduto-sin 83’b, poderia ser
explicada pela formação de um intermediário coordenado.

39
1.8 β-Aminoácidos: Ocorrência natural e atividades biológicas.
1.8.1 - Ocorrência natural e atividades biológicas.
Contrariamente aos α-aminoácidos proteinogênicos, que são constituintes de
todas as enzimas que controlam o metabolismo em seres vivos, a grande parte dos β-
aminoácidos ocorre somente como constituintes de produtos naturais distintos, como
peptídeos, ciclopeptídeos, depsipeptídeos, glicopeptídeos, alcalóides ou terpenóides.54
Aparentemente, bactérias, cianobactérias, fungos e plantas freqüentemente
incorporam β-aminoácidos em metabólitos secundários, que servem como ferramentas
para garantir a sua sobrevivência em competição com outros organismos. Desta
maneira, estes compostos são freqüentemente caracterizados pelas suas potentes
atividades biológicas e fisiológicas – baseadas, crucialmente, sobre suas subestruturas
de β-aminoácidos. Como conseqüência, muitos produtos naturais com uma porção de β-
aminoácido fornecem um manancial de estruturas para o desenvolvimento de novas
drogas.55
O β-aminoácido mais simples é o ácido-β-aminopropanóico (β-Alanina), que
não é um aminoácido proteinogênico, embora seja um componente essencial de muitas
substâncias biologicamente ativas relevantes, como a vitamina B3 [(R)-(+)-Ácido
Pantotênico]. Veja Quadro 6 a seguir:
H2N OH
O
β-Alanina
NH
OH
OO
OH
HO
(R)-(+)-Ácido Pantotênico Quadro 6: Incorporação da β-Alanina na estrutura do (R)-(+)-Ácido Pantotênico.
Existem três tipos gerais de β-aminoácidos quirais de cadeia aberta, dependendo
de onde se situa a substituição: ou no carbono que comporta o grupamento carboxila
(posição α), ou no carbono que comporta o grupamento amino (posição β) ou em ambas
as posições (α,β-dissubstituição) Adicionalmente, β-aminoácidos cíclicos podem
apresentar os grupamentos carboxila e amino como substituintes em um anel
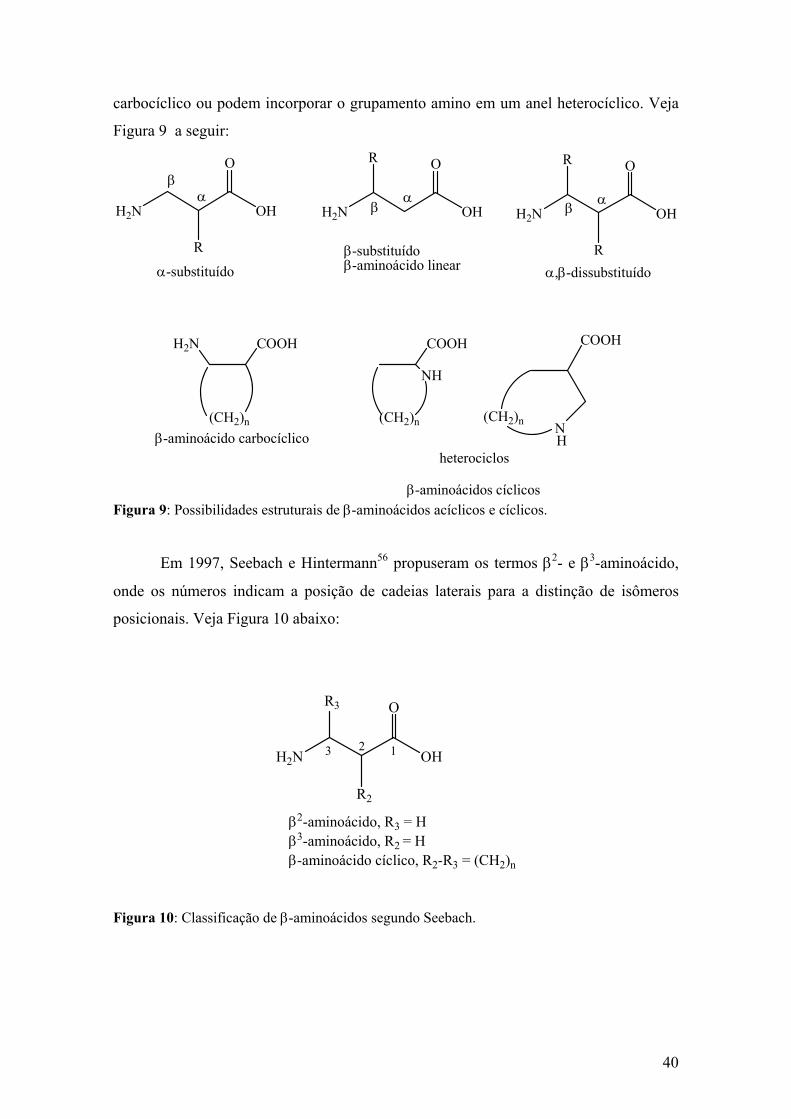
40
carbocíclico ou podem incorporar o grupamento amino em um anel heterocíclico. Veja
Figura 9 a seguir:
H2N OH
O
R
αβ
H2N OH
O
αβ
R
H2N OH
O
R
αβ
R
α-substituídoβ-substituídoβ-aminoácido linear α,β-dissubstituído
H2N COOH
(CH2)n
COOH
(CH2)n
NH
COOH
N(CH2)n
Hβ-aminoácido carbocíclico
β-aminoácidos cíclicos
heterociclos
Figura 9: Possibilidades estruturais de β-aminoácidos acíclicos e cíclicos.
Em 1997, Seebach e Hintermann56 propuseram os termos β2- e β3-aminoácido,
onde os números indicam a posição de cadeias laterais para a distinção de isômeros
posicionais. Veja Figura 10 abaixo:
H2N OH
O
R2
R3
123
β2-aminoácido, R3 = Hβ3-aminoácido, R2 = Hβ-aminoácido cíclico, R2-R3 = (CH2)n
Figura 10: Classificação de β-aminoácidos segundo Seebach.
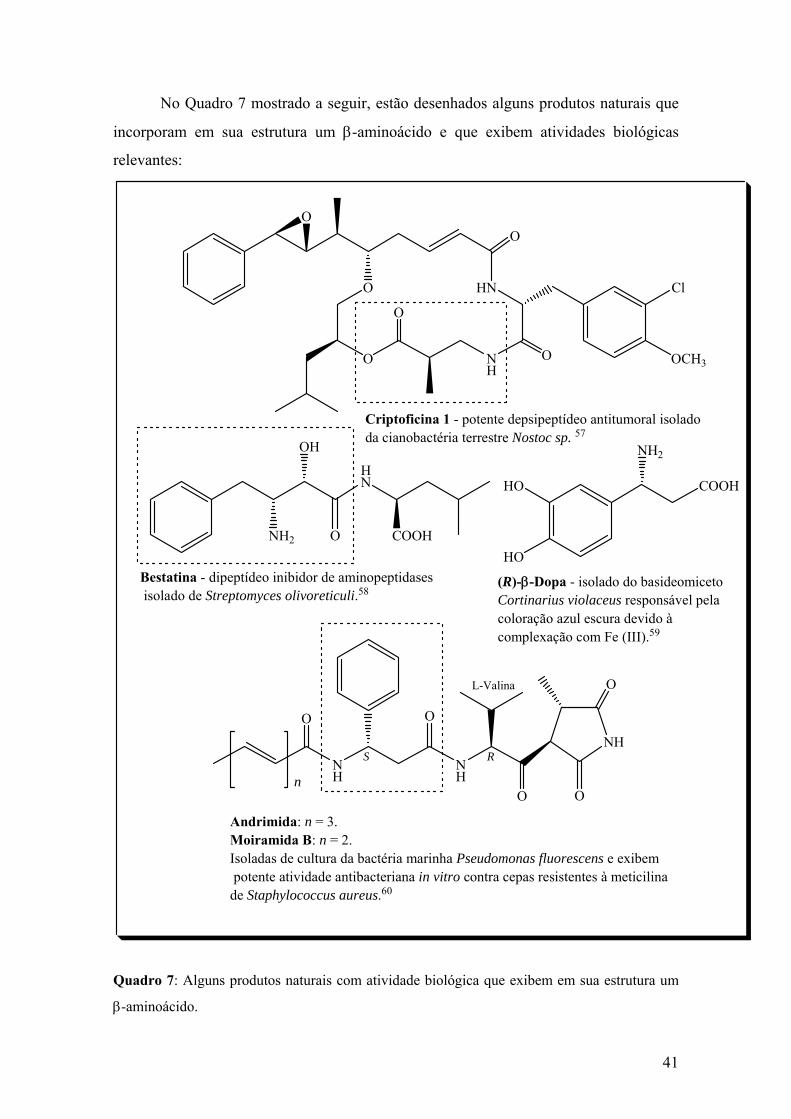
41
No Quadro 7 mostrado a seguir, estão desenhados alguns produtos naturais que
incorporam em sua estrutura um β-aminoácido e que exibem atividades biológicas
relevantes:
OO
O
O
O
NH
O
HN Cl
OCH3
Criptoficina 1 - potente depsipeptídeo antitumoral isolado da cianobactéria terrestre Nostoc sp. 57
HN
ONH2
OH
COOH
Bestatina - dipeptídeo inibidor de aminopeptidases isolado de Streptomyces olivoreticuli.58
COOH
NH2
HO
HO
(R)-β-Dopa - isolado do basideomiceto Cortinarius violaceus responsável pela coloração azul escura devido à complexação com Fe (III).59
NHn
O
NH
O
O
NH
O
O
S R
L-Valina
Andrimida: n = 3. Moiramida B: n = 2.Isoladas de cultura da bactéria marinha Pseudomonas fluorescens e exibem potente atividade antibacteriana in vitro contra cepas resistentes à meticilina de Staphylococcus aureus.60
Quadro 7: Alguns produtos naturais com atividade biológica que exibem em sua estrutura um
β-aminoácido.

42
2. OBJETIVOS.
Devido à inexistência, até o momento, de relatos na literatura em que ânions
nitronatos não metálicos sejam adicionados diastereosseletivamente a eletrófilos como
iminas quirais azaenolizáveis enantiopuras, nós objetivamos investigar, primeiramente,
a reatividade e a diastereosseletividade da adição de vários ânions nitronatos a aldiminas
quirais enantiopuras oriundas de L-aminoácidos naturais tais como L-leucina, L-valina,
L-alanina e L-fenilalanina, empregando as condições reacionais otimizadas por nós em
trabalhos anteriores. Em um segundo momento, objetivou-se estender o escopo deste
estudo à adição conjugada de variados nucleófilos a nitroolefinas, até então inéditas na
literatura, também derivadas dos mesmos L-aminoácidos naturais empregados
anteriormente. Adicionalmente, vislumbramos a aplicação desta nova metodologia
proposta na síntese diastereosseletiva de β-aminoácidos-α-substituídos, intermediários
sintéticos de considerável importância em Química de Produtos Naturais.
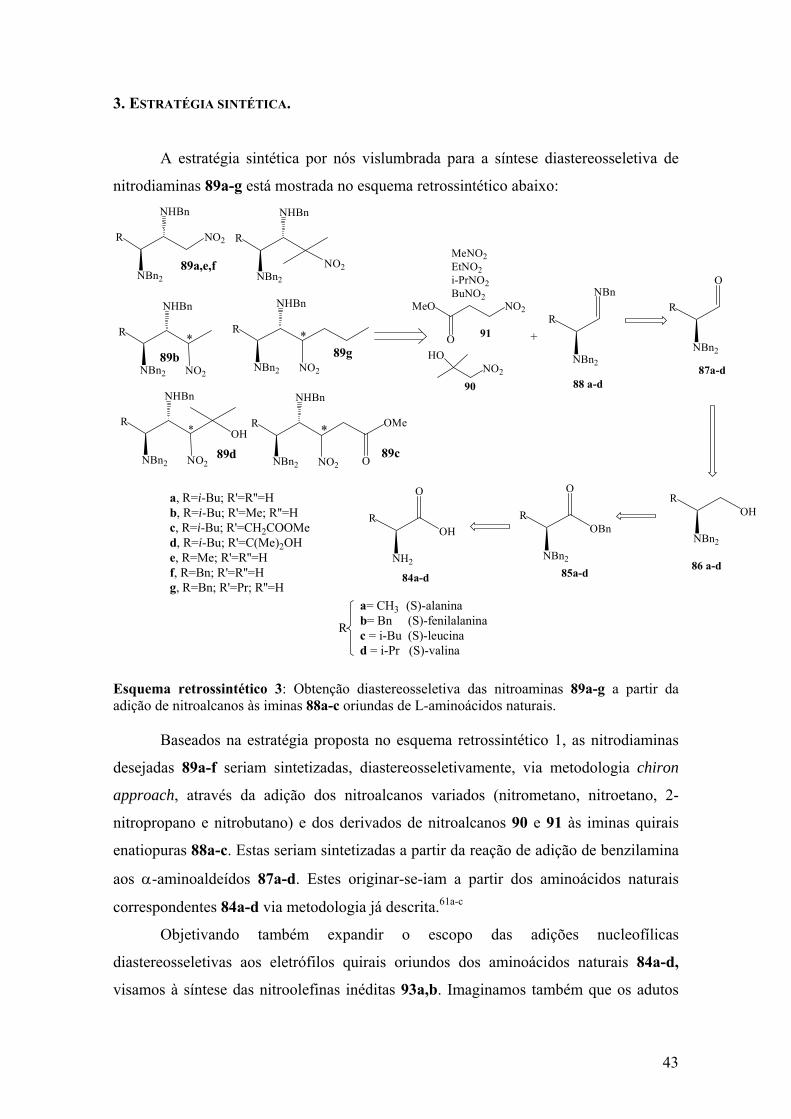
43
3. ESTRATÉGIA SINTÉTICA.
A estratégia sintética por nós vislumbrada para a síntese diastereosseletiva de
nitrodiaminas 89a-g está mostrada no esquema retrossintético abaixo:
a= CH3 (S)-alanina b= Bn (S)-fenilalaninac = i-Bu (S)-leucina d = i-Pr (S)-valina
R
NBn2
NBnR
NBn2
O
R
NH2
O
OH
+
88 a-d87a-d
R
84a-d
R
NBn2
O
OBn
R
NBn2
OH
MeNO2EtNO2i-PrNO2BuNO2
NO2MeO
O
NO2
HO
86 a-d85a-d
90
91
a, R=i-Bu; R'=R''=Hb, R=i-Bu; R'=Me; R''=Hc, R=i-Bu; R'=CH2COOMed, R=i-Bu; R'=C(Me)2OHe, R=Me; R'=R''=Hf, R=Bn; R'=R''=Hg, R=Bn; R'=Pr; R''=H
R
NBn2
NHBn
NO2
R
NBn2
NHBn
∗
NO2
R
NBn2
NHBn
∗
NO2
OHR
NBn2
NHBn
∗
NO2
OMe
O
R
NBn2
NHBn
∗
NO2
R
NBn2
NHBn
NO289a,e,f
89b 89g
89d 89c
Esquema retrossintético 3: Obtenção diastereosseletiva das nitroaminas 89a-g a partir da adição de nitroalcanos às iminas 88a-c oriundas de L-aminoácidos naturais.
Baseados na estratégia proposta no esquema retrossintético 1, as nitrodiaminas
desejadas 89a-f seriam sintetizadas, diastereosseletivamente, via metodologia chiron
approach, através da adição dos nitroalcanos variados (nitrometano, nitroetano, 2-
nitropropano e nitrobutano) e dos derivados de nitroalcanos 90 e 91 às iminas quirais
enatiopuras 88a-c. Estas seriam sintetizadas a partir da reação de adição de benzilamina
aos α-aminoaldeídos 87a-d. Estes originar-se-iam a partir dos aminoácidos naturais
correspondentes 84a-d via metodologia já descrita.61a-c
Objetivando também expandir o escopo das adições nucleofílicas
diastereosseletivas aos eletrófilos quirais oriundos dos aminoácidos naturais 84a-d,
visamos à síntese das nitroolefinas inéditas 93a,b. Imaginamos também que os adutos

44
originados a partir dessas adições conjugadas pudessem ser empregados na síntese dos
β-aminoácidos-α-substituídos 95a-g conforme mostrado no esquema retrossintético 2.
R
NBn2
NO2
a= CH3 (S)-alanina b= Bn (S)-fenilalanina
R
NBn2
OR
NH2
O
OH
87a-b R
84a-b
R
NBn2
OH
NO2 + MeNO2
R
NBn2
NO2
R'
R
NBn2
OH
O
Reação de Nef
Reação de Henry
+ i-PrNO2MeOLi
(Me)3SiN3(Me)3SiCN
R'
92a-b
93a-b94a-g
95a-g
PerbenzilaçãoReduçãoOxidação
a, R=Me; R'=OMeb, R=Me; R'=N3c, R=Me; R'=C(Me)2(NO2)d, R=Bn; R'=OMee, R=Bn; R'=N3f, R=Bn; R'=C(Me)2(NO2)g, R=Bn; R'=CN
Esquema retrossintético 4: Obtenção diastereosseletiva dos β-aminoácidos-α-substituídos 95a-g.
Baseados na estratégia proposta no esquema retrossintético 2, os β-aminoácidos-
α-substituídos 95a-g seriam sintetizados através da reação de Nef a partir dos adutos
94a-g. Estes poderiam ser sintetizados, diastereosseletivamente, a partir da adição
conjugada de nucleófilos variados às nitroolefinas quirais inéditas enatiopuras 93a e
93b. Estas, por sua vez, seriam sintetizadas por desidratação dos nitroálcoois 92a-b.
Estes nitroálcoois são obtidos diastereosseletivamente através da reação de Henry entre
nitrometano e os α-aminoaldeídos 87a-b. Estes últimos são oriundos dos aminoácidos
naturais correspondentes 84a-b através da mesma metodologia empregada
anteriormente (veja Esquema retrossintético 1).
Embora o novo centro estereogênico formado nos nitroálcoois será perdido na
etapa posterior de eliminação que leva à formação das nitroolefinas 93a e 93b, também
é valioso relatar que estudaremos as condições de realização da reação de Henry,
visando uma melhora na eficiência da reação em relação às metodologias já descritas na
literatura.62

45
4. RESULTADOS E DISCUSSÃO.
Objetivando dar continuidade ao estudo sobre a reatividade e
diastereosseletividade das adições de ânions nitronatos não metálicos às iminas quirais
enantiopuras e azaenolizáveis, oriundas de L-aminoácidos, estudo este já iniciado em
nosso grupo de pesquisa,63 foi mister a síntese das iminas 88a-d (veja esquema
retrossintético 1, mostrado na página 43).
Imaginamos, então, que as iminas 88a-d, obtidas facilmente a partir de α-
aminoácidos naturais correspondentes (84a-d), matéria-prima de baixo custo, disponível
em mercado nacional, pudessem ser mais estáveis e, portanto, adequadas ao nosso
estudo de reatividade e diastereosseletividade frente a adições de diferentes ânions
nitronatos.
Iniciamos, então, a síntese das iminas necessárias 88a-d através de rota já
descrita na literatura.61a-d Primeiramente, realizamos uma reação de esterificação e
proteção do grupamento amino simultaneamente de quatro aminoácidos naturais,
conforme mostrado no Esquema 35 abaixo:
R
NH2
O
OH BnBrK2CO3
EtOH : H2O (5:1)
R
NBn2
O
OBn
a R= CH3 (S)-alanina b R = Bn (S)-fenilalanina c R= i-Pr (S)-valina d R= i-Bu (S)-leucina
84a-d 85a-d(85 - 93%)
Esquema 35: Síntese dos ésteres N,N-dibenzilbenzílicos 85a-d oriundos dos aminoácidos naturais 84a-d.
Esta reação foi utilizada por Beaulieu61c e por Warren61d e teve a sua execução
melhorada por nós através da utilização de uma proporção ótima de etanol / água com o
intuito de melhorar a solubilidade dos aminoácidos empregados, bem como reduzir o
tempo reacional através do emprego de refluxo, uma vez que Beaulieu propusera 5 dias
de reação à temperatura abiente. O procedimento modificado, então, consiste em refluxo
do aminoácido na presença de carbonato de potássio, brometo de benzila em uma
mistura de etanol/ água na proporção de 5:1. Após o tempo reacional de 16 horas, um

46
produto perbenzilado foi, então, submetido a uma purificação em coluna cromatográfica
de gel de sílica, fornecendo os aminoácidos perbenzilados 85a-d na faixa de 85 a 93%
de rendimento.
Tendo os ésteres benzílicos 85a-d já sintetizados, a próxima etapa consistiu em
uma redução com hidreto de lítio e alumínio conduzido em THF seco, em refluxo por 4
horas. Após purificação por destilação sob pressão reduzida, os aminoálcoois desejados
são obtidos em uma faixa de 85 a 93% de rendimento purificado. Veja Esquema 36 a
seguir:
R
NBn2
O
OBnR
NBn2
OH
LiAlH4THF,
refluxo
a R= CH3 b R= Bn c R= i-Pr d R= i-Bu
85a-d 86a-d(85 - 93%)
Esquema 36: Redução dos ésteres perbenzilados 85a-d.
Para dar prosseguimento ao trabalho, uma reação de oxidação foi necessária,
convertendo os álcoois primários 86a-d em seus respectivos aminoaldeídos 87a-d. Para
isso, o método de oxidação de escolha foi aquele desenvolvido por Swern e cols.64,61a
Veja Esquema 37 abaixo:
R
NBn2
OH
R
NBn2
O(CO)2Cl2, DMSO
CH2Cl2, TEA,- 78°C a 0°C
86a-d 87a-da R= CH3 b R= Bn c R= i-Pr d R= i-Bu
(96-98%)
Esquema 37: Oxidação de Swern de 86a-d.
Os aminoálcoois foram, então, submetidos à oxidação de Swern, sendo obtidos
os α-amionoaldeídos correspondentes em uma faixa de 96 a 98% de rendimento bruto.

47
Estes aldeídos foram utilizados sem qualquer purificação, já que são conhecidos
os mecanismos de degradação dos mesmos na presença de sílica gel.65 Adicionalmente,
eles são conhecidos como substratos relativamente estáveis e menos propensos à
epimerização devido às dibenzilas do grupamento amino.61b
Nós demos início, então, às sínteses das iminas 88a-d. Estas puderam ser obtidas
por tratamento dos α-amionoaldeídos 87a-d com benzilamina na presença de sulfato de
sódio, em diclorometano24. Após três horas de agitação magnética, à temperatura
ambiente, as iminas 87a-d foram obtidas praticamente em rendimento químico
quantitativo. Fez-se necessário um rápido processo de purificação, das iminas recém-
formadas, em coluna cromatográfica de gel de sílica, fornecendo as iminas puras na
faixa de 88 a 95% de rendimento. Veja Esquema 38 abaixo:
R
NBn2
O
R
NBn2
NBn
BnNH2
Na2SO4 / CH2Cl2 / t.a.2h
87a-d 88a-da R= CH3 b R= Bn c R= i-Pr d R= i-Bu
R= Bn - [α]D= -63,3° (0,1; CHCl3)R= i-Bu - [α]D= -29,5° (0,1; CHCl3)
(88 - 95%)
Esquema 38: Síntese das N,N-dibenzil-N-benzilaldiminas 88a-d.
Com as iminas 88a-d em mãos, demos prosseguimento à rota I, realizando as
adições de vários ânions nitronatos conforme condição reacional mostrada no
Esquema/Tabela 39 abaixo:
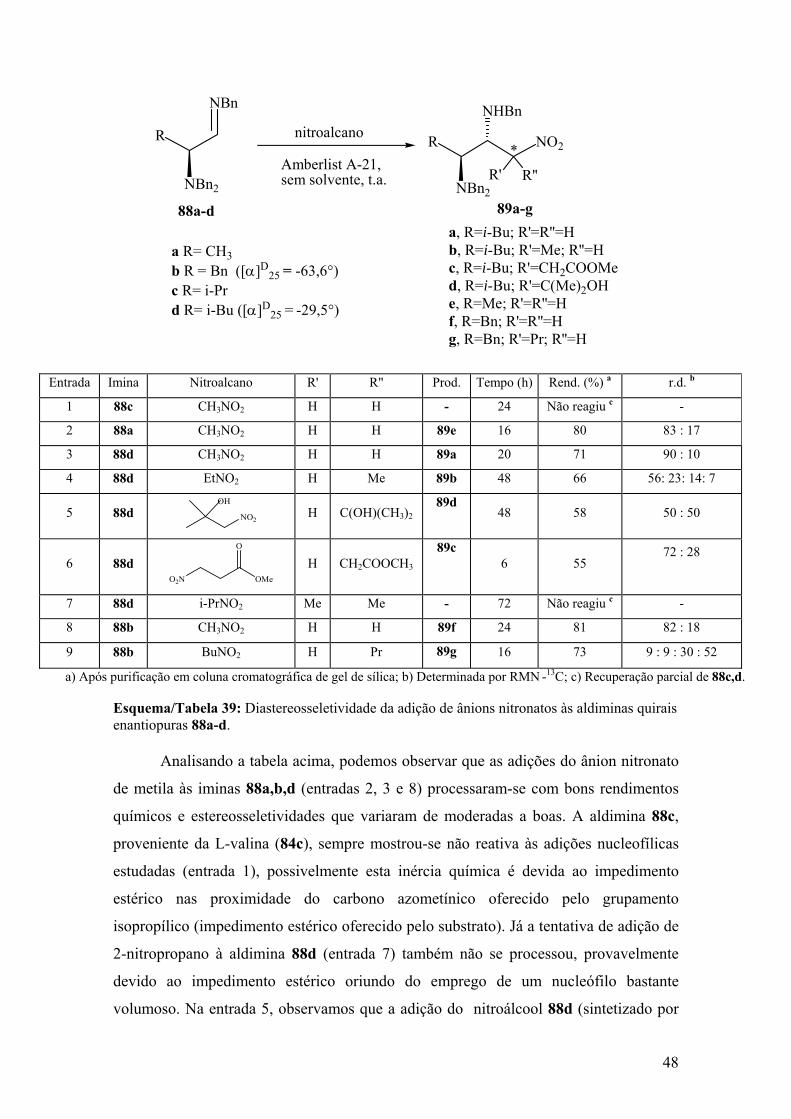
48
a R= CH3 b R = Bn ([α]D
25 = -63,6°) c R= i-Pr d R= i-Bu ([α]D
25 = -29,5°)
R
NBn2
NBn
nitroalcano
Amberlist A-21, sem solvente, t.a.
R
NBn2
NHBn
* NO2
R' R''
88a-d 89a-ga, R=i-Bu; R'=R''=Hb, R=i-Bu; R'=Me; R''=Hc, R=i-Bu; R'=CH2COOMed, R=i-Bu; R'=C(Me)2OHe, R=Me; R'=R''=Hf, R=Bn; R'=R''=Hg, R=Bn; R'=Pr; R''=H
Entrada Imina Nitroalcano R' R" Prod. Tempo (h) Rend. (%) a r.d. b
1 88c CH3NO2 H H - 24 Não reagiu c -
2 88a CH3NO2 H H 89e 16 80 83 : 17
3 88d CH3NO2 H H 89a 20 71 90 : 10
4 88d EtNO2 H Me 89b 48 66 56: 23: 14: 7
5 88d NO2
OH
H C(OH)(CH3)2
89d 48 58 50 : 50
6 88d O2N OMe
O
H CH2COOCH3
89c 6 55
72 : 28
7 88d i-PrNO2 Me Me - 72 Não reagiu c -
8 88b CH3NO2 H H 89f 24 81 82 : 18
9 88b BuNO2 H Pr 89g 16 73 9 : 9 : 30 : 52
a) Após purificação em coluna cromatográfica de gel de sílica; b) Determinada por RMN -13C; c) Recuperação parcial de 88c,d.
Esquema/Tabela 39: Diastereosseletividade da adição de ânions nitronatos às aldiminas quirais enantiopuras 88a-d.
Analisando a tabela acima, podemos observar que as adições do ânion nitronato
de metila às iminas 88a,b,d (entradas 2, 3 e 8) processaram-se com bons rendimentos
químicos e estereosseletividades que variaram de moderadas a boas. A aldimina 88c,
proveniente da L-valina (84c), sempre mostrou-se não reativa às adições nucleofílicas
estudadas (entrada 1), possivelmente esta inércia química é devida ao impedimento
estérico nas proximidade do carbono azometínico oferecido pelo grupamento
isopropílico (impedimento estérico oferecido pelo substrato). Já a tentativa de adição de
2-nitropropano à aldimina 88d (entrada 7) também não se processou, provavelmente
devido ao impedimento estérico oriundo do emprego de um nucleófilo bastante
volumoso. Na entrada 5, observamos que a adição do nitroálcool 88d (sintetizado por

49
adição de nitrometano à propanona, usando Amberlist A-21 como base, em 80% de
rendimento), que apresenta acentuado impedimento estérico por ser neopentílico, é
totalmente diastereosseletiva na indução 1,2 (C2-C3). Tal adição levaria à formação de
quatro possíveis diastereoisômeros e, entretanto, apenas dois foram formados. Veja
Espectro 71 de RMN-1H. A adição do nitroéster 88d (entrada 6), sintetizado segundo
metodologia desenvolvida pelo nosso grupo,66 mostrou-se igualmente diastereosseletiva
em C2-C3. Neste caso, o volume do nucleófilo não explica a excelente
diastereosseletividade (entrada 6). Ao adicionarmos nitroetano (entrada 4) e nitrobutano
(entrada 9) às iminas 88d e 88b, respectivamente, observou-se a formação de quatro
diastereoisômeros conforme o esperado. Podemos propor, baseados em modelos de
estados de transição, que serão discutidos à frente, que a estereoquímica relativa em C2-
C3 seja anti, e que o centro estreogênico nitrometínico (CHNO2) seja também formado
estereosseletivamente, mas que devido à alta acidez do próton nitrometínico e às
condições reacionais básicas, este estereocontrole seja perdido (epimerização), o que
justifica a formação dos dois estereoisômeros nas entradas 5 e 6 e os quatro nas demais
entradas. É importante mencionar que estas adições nucleofílicas não apresentaram
quaisquer indícios de reação de retro-aza-Henry, contrariamente ao comportamento da
gliceraldimina investigada em estudos prévios.63
Como se pôde observar, os rendimentos químicos decresceram de acordo com o
aumento de volume do nitroalcano (entradas 5 e 6). Em relação às adições à
valinaldimina 88c (entrada 1) e à fenilalaninaldimina 88d (entrada 7), devido à não
obtenção dos adutos esperados, houve, então, a recuperação parcial destas iminas 88c,d
após o tempo reacional estipulado, sem observação de decomposição. As iminas
derivadas dos (L)-aminoácidos, 88a-d, mostraram-se bem mais estáveis às condições
reacionais empregadas se comparadas com a gliceraldimina, que mostrou uma rápida
degradação.
É importante ressaltar a ausência de racemização por abstração do hidrogênio α-
imínico de 88d, fato este corroborado pela conservação do valor de [α]D25 de 88d antes
da submissão à reação nucleofílica e sua posterior recuperação do meio reacional
([α]D25 = - 29,5º).

50
Vale lembrar que reações de adição a iminas são menos estudadas em
comparação àquelas onde se empregam substâncias carboniladas, devido à baixa
eletrofilicidade e facilidade de α-desprotonação, como já discutido anteriormente. Desta
forma, verifica-se que na maioria dos relatos vistos na literatura, empregam-se iminas
com um ativador apropriado, o qual pode estar acoplado ou mesmo coordenado ao
átomo de nitrogênio, desta forma, aumentando a eletrofilicidade e uma maior variedade
de nucleófilos pode ser empregada.
No estudo relatado por nós, não empregamos quaisquer ativantes da ligação
C=N, via acoplamento ou coordenação ao átomo de nitrogênio, tampouco o uso de
nucleófilos metálicos fortes e/ou catalisadores metálicos, como ácidos de Lewis.
A formação do diastereoisômero majoritário, que nós inferimos possuir a
configuração relativa anti, pode ser racionalizada baseada no modelo de estado de
transição do tipo Felkin-Ahn,67 pois é assumido que a conformação no estado
fundamental manter-se-á no estado de transição, uma vez que não temos a intervenção
de metais, o que descarta a possibilidade de atuação de um estado de transição quelado
(Cram quelado).68 Veja Figura 11 abaixo:
Figura 11: Modelo de estado de transição de Felkin-Ahn para o ataque nucleofílico de ânions nitronatos à leucinaldimina 88d.
NBn2
NHBn
ANTI
NBn2
H
BnN
face Re
Nu
Nu
H
NBn2
NBn
NBn2
H
NBn
H
>.
Projeção de Newman
Modelo de estado detransição de Felkin-Ahn H
109o
86d

51
O modelo de estado de transição de Felkin-Ahn apresenta a conformação semi-
estrelada que evita a tensão torcional que surgiria em um arranjo eclipsado. O ataque do
nucleófilo dar-se-á pelo lado oposto (anti) ao grupo mais volumoso (Bn2N~) pela face
Re, segundo a trajetória de Bürgi-Dunitz (109o com o plano da ligação C=N),69 levando
ao diastereoisômero anti.
Com o intuito de estabelecer a estereoquímica relativa e, conseqüentemente, a
absoluta dos adutos 89a-g sintetizados, fazia-se imperioso a proteção do grupamento
amino secundário formado pela adição nucleofílica, a fim de proporcionar uma melhor
separação cromatográfica das nitrodiaminas obtidas como uma mistura de
diastereoisômeros - já que foi verificado por nós que o grupamento amino livre
prejudicava a análise através de cromatografia gasosa e confere instabilidade química
com posterior degradação das nitrodiaminas formadas - e um possível meio de obtê-las
em seu estado sólido para uma análise do resultado estereoquímico da reação estudada
através da técnica de cristalografia de raios-X. Infelizmente, exaustivas tentativas de
proteção do grupamento amino com Boc foram feitas sem, contudo, obter qualquer
êxito. Veja Esquema 40 abaixo:
NBn2
NBn
88d
MeNO2
Amberlist A-2120h/71% NBn2
NHBn
89a
NO2t-BuO O
O
Et3N ou ImidazolaCH2Cl2
NBn2
NBn
NO2
Boc
Ot-Bu
O
Esquema 40: Tentativas de N-proteção da amina secundária 89a.
Outras tentativas de proteção do grupamento amino através de acetilação foram
feitas sem, contudo, obter o produto desejado, e sim, ocorrer uma reação de eliminação
e formação da nitroolefina 93a. Veja Esquema 41 abaixo:

52
NBn2
NHBn
NO2
89e
(Ac)2O
HCl cat. / 0ºC30 min NBn2
NBn
NO2
Ac
NBn2
NO2
93a
PRODUTO ACETILADONÃO FORMADO
68%
Esquema 41: Tentativa de acetilação da nitrodiamina 89e e geração da nitroolefina 93a por eliminação.
O produto obtido nesta reação, em rendimento de 68% foi a inesperada
nitroolefina 93a. Esta, em um próximo momento, deverá ser sintetizada por nós em um
maior rendimento químico e terá a sua reatividade e diastereosseletividade investigada
frente a diversos nucleófilos.
Uma outra possibilidade de desvendar a natureza estereoquímica da reação, seria
a síntese do nitrobutano e posterior adição à aldimina 88b; isto levaria a um
intermediário sintético para a realização de uma correlação química à idêntica diamina
quiral sintetizada por Reetz e cols.22 Veja Esquema 42 abaixo:
NBn2
NBn
88b
BuNO2
Amberlist A-2116h/76% NBn2
NHBn
*
NO2
89g
AIBN / benzeno80ºC
n-Bu3SnH
NBn2
NHBn
Bn
NH2
NH2
Bu
Diamina quiralsintetizada por Reetz22
Esquema 42: Tentativa de desnitração da nitrodiamina 89g.
Desafortunadamente, o nitro secundário presente na substância 89g não foi
passível de remoção através de protocolo já descrito na literatura por Ono.15 Desta
forma, a realização de uma filiação química da diamina que seria obtida por nós e
relatada por Reetz via adição de reagentes organometálicos a uma aldimina ativada (N-
tosilaldimina) não pôde ser alcançada.

53
Como perspectiva de solução para a comprovação da estereoquímica anti
aventada por nós e respaldada pela análise do modelo de estado de transição de Felkin-
Ahn, parece-nos viável a tentativa de cristalização das nitrodiaminas 89a-g via reação
de amidação com ácido p-bromobenzóico e posterior análise cristalográfica de raios-X.
Veja Esquema 43 abaixo:
NBn2
NHBn
89a
NO2
COOH
Br
NBn2
NBn
O
NO2
Br
Esquema 43: Possibilidade de cristalização através de derivatização da amina secundária 89a.
***
Com o estudo da reatividade e da diastereosseletividade das iminas 88a-d frente
às adições de nitroalcanos estando em processo de confirmação da
diastereosseletividade anti inferida e, por conseguinte, a estereoquímica absoluta das
nitrodiaminas 89a-g determinada, pensamos em realizar um novo estudo onde outros
eletrófilos quirais pudessem ser também sintetizados a partir dos α-aminoácidos já
estudados 84a-d.
Inspirados, então, na síntese inesperada da nitroolefina 93a, veja esquema 41,
demos início à uma nova rota para a síntese eficiente das nitroolefinas quirais 93a-b, até
hoje inéditas na literatura, a partir dos α-aminoácidos naturais 84a-b. Utilizaremos para
isso, a reação de eliminação de McMurry43 dos nitroálcoois correspondentes 92a-b
obtidos, através da reação de Henry,2,70a,b entre os aminoaldeídos 87a-b e nitrometano.
Veja Esquema/Tabela 44 a seguir:

54
RO
NBn2
R
NBn2
OH
NO2
92a,R=Me92b,R=Bn
MeNO2
TBAF 20%THF seco
87a R=Me (L-alanina)87b R=Bn (L-fenilalanina)
87a
EtNO2
TBAF 20% THF seco
∗
NBn2
OH
NO2
92c
NBn2
∗ NO2
OH
92d
87d R=i-Bu (L-leucina)
Entrada α-aminoaldeído Tempo (h) Produto, Rendimentoa
r.d.b
1 87a 1,5 92a,(95%) 90 : 10
2 87b 2,0 92b,(91%) 93 : 7
3 87a 1,5 92c,(84%) 90 : 10c
4 87d 1,5 92d,(82%) 23 : 13 : 53 : 11
a) Após purificação de mistura dos diastereoisômeros em coluna cromatográfica de gel de sílica; b) Determinado por RMN de 13C; c) Apenas dois diastereoisômeros formados. Esquema/Tabela 44: Síntese diastereosseletiva dos nitroálcoois 92a-d.
De acordo com os resultados mostrados no Esquema/Tabela 44 acima, podemos
verificar nas entradas 1-3 excelentes resultados obtidos quando se empregou o N,N-
dibenzilamino-alaninaldeído (87a) como substrato para a reação nitroaldólica,
utilizando, respectivamente, nitrometano e nitroetano e também o N,N-dibenzilamino-
fenilalaninaldeído (87b) como substrato e nitrometano. Na entrada 1, a reação se
processou rapidamente, com observação de conversão química total no tempo relatado,
mostrando excelente rendimento químico e boa razão diastereoisomérica anti do
nitroálcool 92a obtido. Surpreendentemente, na entrada 3, ainda tendo como substrato
o aldeído 87a, a adição de nitroetano geraria dois estereocentros no nitroálcool 92c, fato
este não observado em análises espectroscópicas, o que nos sugere alta
diastereosseleção na formação do novo centro α-amino. Na entrada 2, quando foi

55
empregado o aldeído 87b, observamos uma maior diastereosseletividade da reação de
Henry em comparação à entrada 1, embora um menor rendimento químico. Ao
adicionarmos nitroetano ao aldeído 87d (entrada 4), então, houve a formação dos
quatro possíveis diastereoisômeros previstos para esta reação. É provável que,
analisando atentamente a razão diastereoisomérica obtida, que o segundo centro
estereogênico formado sendo no carbono α-nitro, seja parcial ou totalmente
epimerizado ao longo do percurso reacional.
A atribuição da estereosseletividade anti para 92a-d foi possível baseados nos
estudos realizados por Hanessian e cols.,62 no qual foram obtidos, dentre outros, os
mesmos nitroálcoois 92a-c, assim uma filiação química pôde ser realizada. Hanessian
conduziu a reação nitroaldólica usando de 1 e/ou 2 equivalentes de TBAF. Veja alguns
resultados sumarizados no Esquema/Tabela 45 a seguir:
R
NBn2
O
+
1-2 eq. deTBAF hidratado
0°C, 10 -30 minR'R
NBn2
OH
NO2
R'
NO2
87 a,b92a-c
Entrada α-aminoaldeído R’ Rend. (%) r.d.a Métodob
1 87a H 88 80 : 20 A
2 87b H 97 83 : 17 B
3 87a Me 68 80 : 12 : 8 A a) Determinado por RMN de 1H a partir da integração do próton nitrometínico; b) A: Uso 1 eq. de TBAF. B= Uso 2 eq. de TBAF.
Esquema/Tabela 45: Obtenção dos nitroálcoois 92a-c via metodologia de Hanessian.
Analisando os resultados obtidos por Hanessian e cols., verificamos que o
emprego de quantidades equimolares, ou mesmo de dois equivalentes, de TBAF apenas
diminui significativamente o tempo reacional em comparação aos nossos resultados. Na
entrada 1, Hanessian obteve o nitroálccol 92a em 88% de rendimento químico e com
excesso diastereoisomérico de 60%; em comparação aos nossos resultados, o mesmo
nitroálcool foi obtido em 95% de rendimento químico e com excesso diastereoisomérico
(e.d.) igual a 76%, empregando quantidade catalítica de TBAF (20% molar). Seguindo a
análise comparativa, na entrada 2, Hanessian sintetizou 92b em excelente rendimento
químico (empregou 2 equivalentes de TBAF) com e.d. igual a 66%; no nosso caso, 92b
foi obtido em 87% de rendimento químico com e.d. apenas de 42%. Já na entrada 3, dos
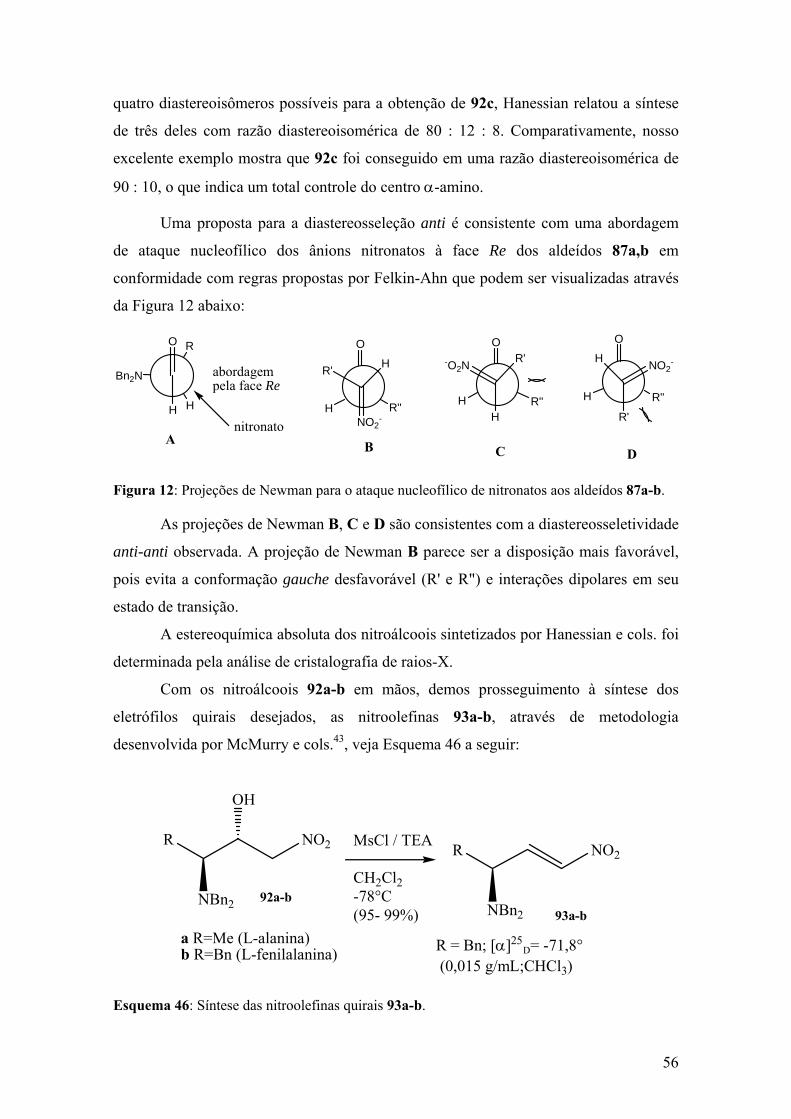
56
quatro diastereoisômeros possíveis para a obtenção de 92c, Hanessian relatou a síntese
de três deles com razão diastereoisomérica de 80 : 12 : 8. Comparativamente, nosso
excelente exemplo mostra que 92c foi conseguido em uma razão diastereoisomérica de
90 : 10, o que indica um total controle do centro α-amino.
Uma proposta para a diastereosseleção anti é consistente com uma abordagem
de ataque nucleofílico dos ânions nitronatos à face Re dos aldeídos 87a,b em
conformidade com regras propostas por Felkin-Ahn que podem ser visualizadas através
da Figura 12 abaixo:
R
H
Bn2N
O
H
abordagem pela face Re
nitronatoA B C D
O
NO2-
O O
-O2NR' H
H
R' H
R'
NO2-
H R'' H R'' H R''
Figura 12: Projeções de Newman para o ataque nucleofílico de nitronatos aos aldeídos 87a-b.
As projeções de Newman B, C e D são consistentes com a diastereosseletividade
anti-anti observada. A projeção de Newman B parece ser a disposição mais favorável,
pois evita a conformação gauche desfavorável (R' e R") e interações dipolares em seu
estado de transição.
A estereoquímica absoluta dos nitroálcoois sintetizados por Hanessian e cols. foi
determinada pela análise de cristalografia de raios-X.
Com os nitroálcoois 92a-b em mãos, demos prosseguimento à síntese dos
eletrófilos quirais desejados, as nitroolefinas 93a-b, através de metodologia
desenvolvida por McMurry e cols.43, veja Esquema 46 a seguir:
R
NBn2
OH
NO2 R
NBn2
NO2
92a-b93a-b
MsCl / TEA
CH2Cl2-78°C(95- 99%)
a R=Me (L-alanina)b R=Bn (L-fenilalanina) R = Bn; [α]25
D= -71,8° (0,015 g/mL;CHCl3)
Esquema 46: Síntese das nitroolefinas quirais 93a-b.

57
Apesar de McMurry ter realizado a reação de eliminação a 0ºC, o nosso grupo
de pesquisa verificou a necessidade de temperaturas mais baixas para que a reação se
processe de forma mais eficaz e sem a formação de produtos colaterais.71
As nitroolefinas 93a,b foram obtidas em excelente rendimento químico, quase
quantitativo, e com ótimo grau de pureza, o que nos permitiu empregá-las nas reações
posteriores sem qualquer necessidade de purificação prévia. Para a aferição da atividade
óptica, fez-se necessário uma rápida purificação em coluna cromatográfica de gel de
sílica. Estas nitroolefinas mostraram-se instáveis ao contato com sílica gel, sendo
perdido muito material ao final do processo de purificação. A caracterização estrutural
da nitroolefina 93b, por exemplo, pôde ser evidenciada pela análise do espectro de
RMN de H1 (veja Espectro 49) que apresentou um conjunto de sinais na faixa de 6,78 a
6,96 ppm referente aos hidrogênios olefínicos; em relação aos carbonos olefínicos, estes
puderam ser evidenciados pelos deslocamentos a 140,57 e 140,03 ppm no espectro de
RMN de 13C (veja Espectro 50). Adicionalmente, as bandas de absorção a 1526 e
1350 cm-1, no espectro de infravermelho (veja Espectro 53) de forte intensidade
referentes à deformação axial assimétrica e simétrica, respectivamente, do grupo nitro
conjugado confirma a estrutura proposta para 93b. A caracterização estrutural da
nitroolefina 93a deu-se de forma similar à relatada anteriormente, tendo como fator
contundente, a evidência do pico do íon molecular - m/z = 296 - (veja Espectro 47),
confirmando, assim, a obtenção de 93a.
Como as nitroolefinas 93a,b são substâncias, até o nosso conhecimento, inéditas
na literatura, foi necessária a síntese de uma rota racêmica para que pudesse ser
confirmada a pureza óptica das nitroolefinas em questão, ou seja, verificar se a reação
de obtenção das mesmas processa-se sem racemização e/ou perda parcial de atividade
óptica.
Desta forma, a síntese da rota racêmica deu-se a partir do uso da (±)-
fenilalanina, e todas as etapas de perbenzilação, redução, oxidação, reação de Henry, até
a reação de formação da nitroolefina 93b’ (racêmica) foi alcançada. Similarmente, veja
Esquema retrossintético 2. Tendo, agora, em mãos 93b e 93b’, realizamos experimentos
através de cromatografia gasosa com coluna quiral (ciclodextrina, 30 metros; D. I. 0,25;
filme 0,25 μm), com o intuito de verificar a pureza óptica de 93b. Contudo, as análises
obtidas através do experimento de CG quiral nas condições utilizadas por nós, não
puderam ser concludentes, pois não houve a resolução do racemato 93b’.

58
Dando prosseguimento ao trabalho, iniciamos o estudo da reatividade e
diastereosseletividade das nitroolefinas 93a,b recém-sintetizadas, frente a variados tipos
de nucleófilos. O átomo nucleofílico pode ser oxigênio (adição de metóxido de lítio),
nitrogênio (empregando-se trimetilsililazida como reagente) e carbono (empregando-se
o 2-nitropropano como reagente e também trimetilsililcianeto). Veja Esquema/Tabela
47 abaixo:
R
NBn2
NO2
93a-b
a R=Me (L-alanina)b R=Bn (L-fenilalanina)
Condições reacionais R
NBn2
NO2
Nu
a, R=Me; R'=OMeb, R=Me; R'=N3c, R=Me; R'=C(Me)2(NO2)d, R=Bn; R'=OMee, R=Bn; R'=N3f, R=Bn; R'=C(Me)2(NO2)g, R=Bn; R'=CN
94a-g
Veja Tabela
Entrada Nitroolefina Nucleófilo Condições Reacionais Produto, Rend.a
r.d.b
1 93a MeOLi MeOH / t.a. / 16h 94a, 77% 69 : 31
2 93a (Me)3SiN3 TBAF / THF / t.a./ 20h 94b, 74% 74 : 26
3 93a i-PrNO2 TBAF 20% /THF / t.a./20h 94c, 75% c
4 93b MeOLi MeOH / t.a. / 20h 94d, 70% 57 : 43
5 93b (Me)3SiN3 TBAF / THF / t.a./ 24h 94e, 67% 86 : 14d
6 93b i-PrNO2 TBAF 20% /THF / t.a./24h 94f, 65% 93 : 7
7 93b i-PrNO2 DBU20%/MeCN / t.a./12h 94f, 58% 85 : 15
8 93b (Me)3SiCN TBAF / MeCN / t.a./ 24h 94g, 63% e
a) Rendimento químico obtido após purificação em coluna cromatográfica de gel de sílica; b) Razão diastereoisomérica analisada através de RMN de 1H e/ou 13C; c) Provável produto de cicloadição [3 + 2] e substrato 93a; d) Razão diastereoisomérica determinada através de CG; e) Isolado um produto oriundo de eliminação de HNO2. Esquema/Tabela 47: Adição conjugada diastereosseletiva de variados nucleófilos às nitroolefinas quirais 94a-b.

59
Ao analisarmos os resultados obtidos sumarizados no Esquema/Tabela 47
mostrado anteriormente, podemos verificar que, no geral, os rendimentos químicos
foram sempre alcançados em uma faixa que variou de moderada (58 – 67%) a boa (70 –
77%). Interessantemente, ao compararmos as estereosseletividades obtidas para cada
nitroolefina empregada neste estudo, repararemos que ao utilizarmos a nitroolefina 93a,
entradas 1 e 2 (momentaneamente excetuaremos a entrada 3 para comentá-la a
posteriori) as razões diastereoisoméricas obtidas não foram satisfatórias. Na entrada 1,
empregamos uma solução a 1M de metóxido de lítio em metanol para adicionarmos o
nucleófilo metóxido à nitroolefina 93a. Observamos que o produto formado compartilha
do mesmo Rf (razão em relação ao front – cromatografia em camada fina) do substrato,
dificultando, assim, num primeiro momento, a monitoração do tempo reacional. Foi
obtido o produto 94a em bom rendimento químico e baixa diastereosseletividade.
O assinalamento estrutural para o nitroéter formado (94a) foi realizado pela
ausência dos sinais olefínicos em 7,0 ppm referentes ao substrato (nitroolefina 93a) e
aparecimento do sinal característico de hidrogênios metilênicos α-nitro (4,7 – 5,0 ppm)
e dos hidrogênios da metoxila adicionada (3,4 ppm) – veja Espectro 90.
Ao adicionarmos trimetilsililazida (entrada 2), onde faz-se necessário a adição
de um ácido de Lewis (BF3 - Et2O) ou um sal de fluoreto (TBAF) para liberar o
nucleófilo, o comportamento reacional não variou muito em relação ao anterior e a
nitroazida obtida (94b) em 74 % de rendimento químico e exibiu uma moderada
diastereosseleção. O seu assinalamento estrutural deu-se analogamente ao relatado
anteriormente para 94a (desaparecimento do sinal olefínico do substrato e aparecimento
de grupamento ~CH2NO2), sendo a característica mais marcante da adição de azida a
sua intensa absorção no infravermelho em 2131 e 2100 cm-1 (veja Espectro 89).
Resolvemos, então, estudar o perfil reacional da nitroolefina 93b, adicionando os
mesmos nucleófilos já empregados. Na entrada 4, o mesmo padrão foi repetido em
relação à entrada 1, o nitroéter 94d possui o mesmo Rf do substrato 93b e o excesso
diastereoisomérico obtido foi praticamente nulo (veja Espectro 91). O assinalamento
estrutural de 94d deu-se através da observação do desaparecimento dos prótons
olefínicos do substrato 93b (6,78 a 6,96 ppm – espectro 49) e aparecimento dos sinais
característicos de ~CH2NO2 (4,7 – 4,84 ppm) e ~OCH3 (3,3 ppm) – veja Espectro 91.

60
A entrada 5, entretanto, mostra um resultado bem diferente daquele
anteriormente obtido na entrada 2, uma vez que a nitroazida 94e foi formada com uma
melhor diastereosseleção (86:14), como revelado pela análise em cromatografia gasosa
(veja Cromatograma 2), embora houve um ligeiro decréscimo no rendimento químico
(67%). O assinalamento estrutural de 94e foi realizado através da confirmação de uma
absorção a 2132 e 2100 cm-1, referente ao grupamento azido e deformações assimétrica
e simétrica do grupamento nitro a 1556 e 1374 cm-1 (veja Espectro 86), fato este que
confirma o aparecimento de hidrogênios α-nitro na faixa de 4,78 a 4,87 ppm (veja
Espectro de Infravermelho 84).
Encorajados pelo melhor resultado estereosseletivo, a adição de 2-nitropropano,
na entrada 6, forneceu o aduto de Michael 94f - substância 1,3-dinitrada - em moderado
rendimento químico (65%) e ótima razão diastereoisomérica (93:7), determinada através
de análises dos Espectros 94 (RMN de 1H) e 95 (RMN de 13C). O seu assinalamento
químico procedeu através da obvservação não usual de duplos sinais de estiramento
assimétrico dos diferentes grupamentos nitro - primário e terciário - em 1556 e
1544 cm-1 e simétrico em 1377 e 1345 cm-1 (veja Espectro 97), bem como o
desaparecimento dos prótons olefínicos do substrato 93b e o aparecimento dos prótons
α-nitro em 4,6 a 4,98 ppm (veja Espectro 94). Este produto (94f), único obtido na forma
de um sólido após purificação em coluna de gel de sílica, foi enviado ao grupo de
pesquisa do professor Carlos Alberto de Simone da Universidade Federal de Alagoas
(UFAL) para ser submetido a experimentos de cristalografia de raios-X com intuito de
comprovar a estereoquímica anti das reações de adição conjugada às nitroolefinas. Esta
anti-diastereosseletividade, foi proposta por nós a partir de análise de modelo de estado
de transição de Felkin-Ahn, que será discustido posteriormente.
Adicionalmene, uma variante da condição reacional para a obtenção de 94f
também foi paralelamente investigada em nosso laboratório,72 que consiste no emprego
catalítico de DBU como base. Apesar da reação ter se processado em um menor tempo
reacional, a razão diastereoisomérica e o rendimento químico observados foram
consideravelmente menores em relação àqueles onde foi empregado TBAF catalítico
como base.
Como relatado anteriormente, a melhor diastereosseleção obtida na adição
conjugada de variados nucleófilos às nitroolefinas 94a,b mostrou ser a adição de 2-
nitropropano à nitroolefina 94b (entrada 6), onde permitiu-nos o acesso a uma

61
substância 1,3-dinitrada. A despeito do uso potencial de 94f para a síntese de 1,3-
diaminas opticamente ativas, dentre outras substâncias de interesse, este acoplamento
C-C entre nitroalcanos e nitroolefinas é extremamente raro na literatura.47c É importante
mencionar que, ao nosso conhecimento, não existe na literatura corrente qualquer
exemplo da adição de um nitroalcano a uma nitroolefina quiral enantiopura.
Finalmente, na entrada 8, um outro nucleófilo centrado em carbono foi testado.
O ânion cianeto foi liberado, no meio reacional, através da adição de um ácido de Lewis
(BF3 - Et2O) ou de um sal de fluoreto (TBAF) ao reagente trimetilsililcianeto.
Interessantemente, foi determinado por análises espectroscópicas que o produto 94g,
originalmente oriundo de uma simples adição de Michael de cianeto à nitoolefina 93b,
sofreu eliminação de ácido nitroso, dando origem a uma nitrila α,β-insaturada - outro
aceptor de Michael - obtida em 63% de rendimento químico. Veja Esquema 48 abaixo:
NBn2
NO2Si(Me)3CN
TBAF/CH3CN/t.a.NBn2
CN
HNO2
94g93b63%
Esquema 48: Adição conjugada do ânion cianeto a 93b, seguida de eliminação de ácido nitroso.
O assinalamento estrutural foi realizado pela presença de sinais olefínicos a 6,41
– 6,04 ppm (veja Espectro 103) e a não-observação dos sinais esperados de ~CH2NO2,
que ocorrem de 4,5 a 5,0 ppm. Fato este corroborado pela análise do Espectro 105 de
infravermelho, onde observamos as ausências das bandas de absorção de grupamento
nitro (1557 e 1375 cm-1, aproximadamente), bem como a banda de absorção em
2218 cm-1, comprobatória de grupamento nitrila sob influência de um grupamento
puxador de elétrons (~NO2 ligado ao carbono α). Adicionalmente, observa-se o
aparecimento, no Espectro 104, de sinal referente a carbonos olefínicos em 147,7 e
114,0 ppm, sinal em 117,3 do carbono do grupamento nitrila em conjugação;
concomitantemente, observou-se a ausência do sinal referente ao carbono metilênico
ligado ao grupamento nitro (80 a 90 ppm, aproximadamente).
Analisando o resultado obtido na entrada 3 do Esquema/Tabela 47, vemos que
uma possível reação de cicloadição se processou. Desse modo, o ânion nitronato de
isopropila pode ter atuado como uma espécie 1,3-dipolar.73 Este resultado evidencia
uma diferente e rara reatividade para os ânions nitronatos. Veja esquema 49 abaixo:

62
R1
R2
N
O
O
R1
R2
N
OM
O
X O
N
X
OM
R1
R2
N
OM
O
R1
R2
R1
R2
N
OM
O
i) ou ii)
X = Haleto, CO2R, NO2 e etc.
base
99a, M = R3Si99b, M = alquil
i) R3SiClii) cloreto de alquila
97a, M = R3Si97b, M = alquil
9896 97
espécie 1,3-dipolar
Esquema 49: Reatividade de ânions nitronatos como espécies 1,3-dipolares.
Segundo o Esquema 49, os ânions nitronatos podem ser derivatizados a silil-
nitronatos 97a ou alquil-nitronatos 97b, gerando, assim, espécies 1,3-dipolares e serem
capazes de reagir com olefinas elétron-deficientes 98, em cicloadições 1,3-dipolar para
fornecer N-sililoxiisoxazolidinas 99a ou N-alcoxiisoxazolidinas 99b como produtos. É
importante mencionar que alquilnitronatos e sililnitronatos são substâncias
rotineiramente utilizadas como 1,3-dipolos em reações de cicloadições, entretanto, não
conseguimos selecionar um exemplo sequer da literatura onde um ânion nitronato
estivesse agindo como um 1,3-dipolo, muito embora, Jørgensen e Gothelf74, numa
rápida menção, tivessem considerado essa possibilidade.
O resultado obtido na entrada 3 mostra que não apenas os nitronatos de silila e
alquila, gerados a partir do tratamento dos ânions nitronatos com cloretos de silila ou
alquila, respectivamente geram espécies 1,3-dipolares. O ânion nitronato de isopropila,
gerado da reação entre 2-nitropropano e uma base, através de sua estrutura de
ressonância, pode agir como uma espécie 1,3-dipolar. Veja Figura 13 a seguir:
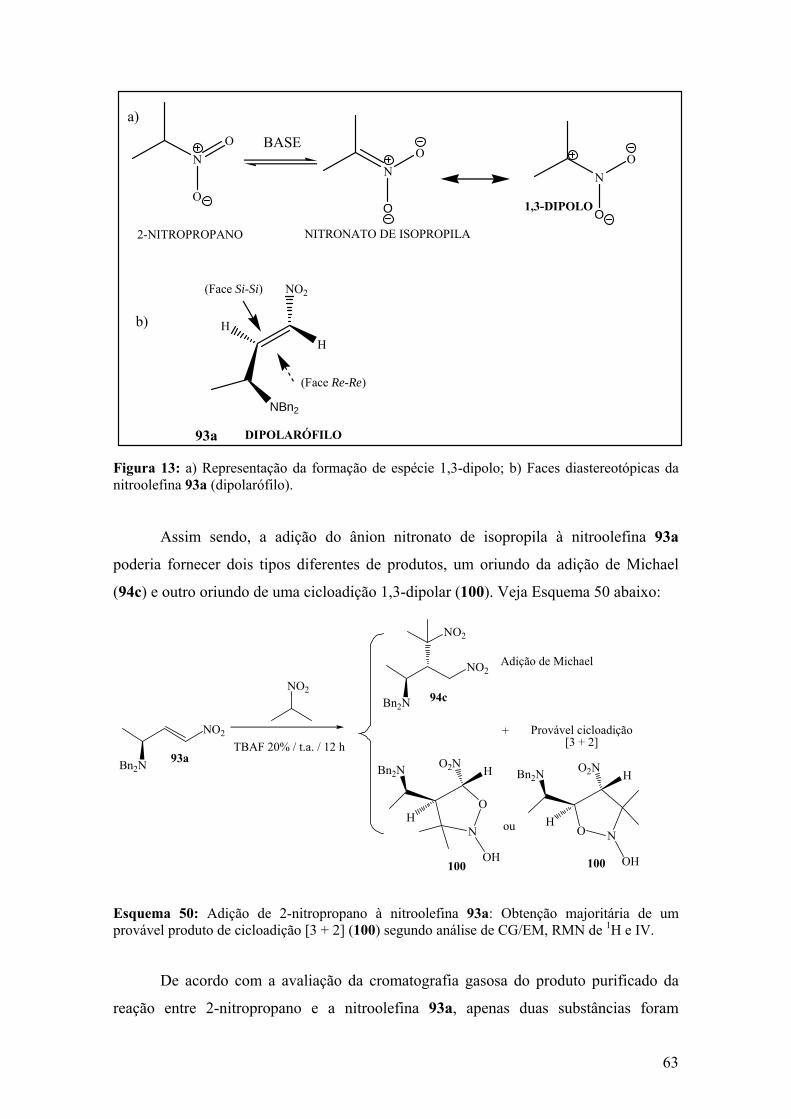
63
NO
O
2-NITROPROPANO
NO
NO
O1,3-DIPOLO
H
NO2
(Face Re-Re)
(Face Si-Si)
DIPOLARÓFILO
a)
b)
93a
O
H
BASE
NITRONATO DE ISOPROPILA
NBn2
Figura 13: a) Representação da formação de espécie 1,3-dipolo; b) Faces diastereotópicas da nitroolefina 93a (dipolarófilo).
Assim sendo, a adição do ânion nitronato de isopropila à nitroolefina 93a
poderia fornecer dois tipos diferentes de produtos, um oriundo da adição de Michael
(94c) e outro oriundo de uma cicloadição 1,3-dipolar (100). Veja Esquema 50 abaixo:
NO2
Adição de Michael
+
HN
O
OH
O2N
Provável cicloadição[3 + 2]
Bn2N
NO2
Bn2N
NO2
94c
93a
NO2
TBAF 20% / t.a. / 12 h
100
Bn2N H
ou HO N
OH
O2NBn2N H
100
Esquema 50: Adição de 2-nitropropano à nitroolefina 93a: Obtenção majoritária de um provável produto de cicloadição [3 + 2] (100) segundo análise de CG/EM, RMN de 1H e IV.
De acordo com a avaliação da cromatografia gasosa do produto purificado da
reação entre 2-nitropropano e a nitroolefina 93a, apenas duas substâncias foram

64
identificadas (veja Cromatograma 3). Inicialmente, esperamos encontrar os dois
possíveis diastereoisômeros provenientes da reação conjugada do nitroalcano em
questão ao aceptor de Michael 93a. Entretanto, o produto majoritário formado (100)
indica ser proveninte de uma reação de cicloadição, enquanto a outra substância
identificada parece-nos ser o aduto 94c, evidenciada através de sinais referentes a
hidrogênios α-nitro em 4,4 ppm (veja Espectro 98), bem como análise do fragmento de
massa 224, que indica perda de 161 (C2H2NO2) a partir do íon molecular (296),
característico desta nitroolefina (veja Espectro 101). Veja Esquema 51 abaixo:
NBn2
NO2
NO2
-NO2
NO2
massa = 161
PM=296
NBn2
m/z = 224
Esquema 51: Justificativa de quebra para a formação do fragmento m/z = 224 no Espectro 101.
Surpreendentemente, através de análise espectroscópica na região de
infravermelho (veja Espectro 102), observamos sinais referentes a ligações químicas
não justificadas para o aduto de Michael então esperado (94c), como absorções inferidas
para ligações do tipo C-O (1126 e 1073 cm-1) e O-H (3380 cm-1), por exemplo. E,
finalmente, sob análise espectroscópica de RMN de 1H, confirmamos sinais referentes à
mistura dos dois produtos, oriundos de reações químicas distintas (94c e 100).
Uma análise do modelo de estado de transição que justifica o único produto
formado a partir de cicloadição 1,3-dipolar (100) entre o ânion nitronato de isopropila e
a nitroolefina 93a está mostrada no Esquema 52 a seguir:

65
NO2
O
N
OH
H
NO2
HH
Me
N
O
O
1,3-DIPOLOMe
H
H+
NBn2
Bn2N
100
NO2
HH
NBn2O
N
O
Me
Me
H+
ou
NO2
O N
OH
HH
Bn2N
100
Esquema 52: Formação do produto de cicloadição 1,3-dipolar 100 através da aproximação exo ou endo e ataque pela face Re-Re do dipolarófilo 93a.
Como observado pelo Esquema 52 acima, o ataque da espécie 1,3-dipolar dar-
se-á, principalmente, pela face menos impedida estericamente da nitroolefina 93a (face
Re-Re), fato observado pela formação de um único produto de cicloadição. Em relação
à regiosseletividade da cicloadição, estudos mais detalhados de espectroscopia serão
realizados com o objetivo de determinar se a espécie 1,3-dipolar aproximar-se-á de
forma exo (sendo aquela onde o oxigênio da espécie dipolar, ânion 2-nitropropanoato,
ligar-se-á ao carbono α-nitro) ou endo (o oxigênio ligar-se-á ao carbono eletrofílico da
nitroolefina).
E com a finalidade de explicar uma possível diastereosseleção anti para os
adutos formados nas entradas 1,2 e 4-7, retomamos o modelo de estado de transição
proposto por Felkin-Ahn como visto anteriormente, e consideramos a nitroolefina 93b
sendo atacada preferencialmente pela face Re-Re, menos impedida estericamente.
Observe Figura 14 a seguir:
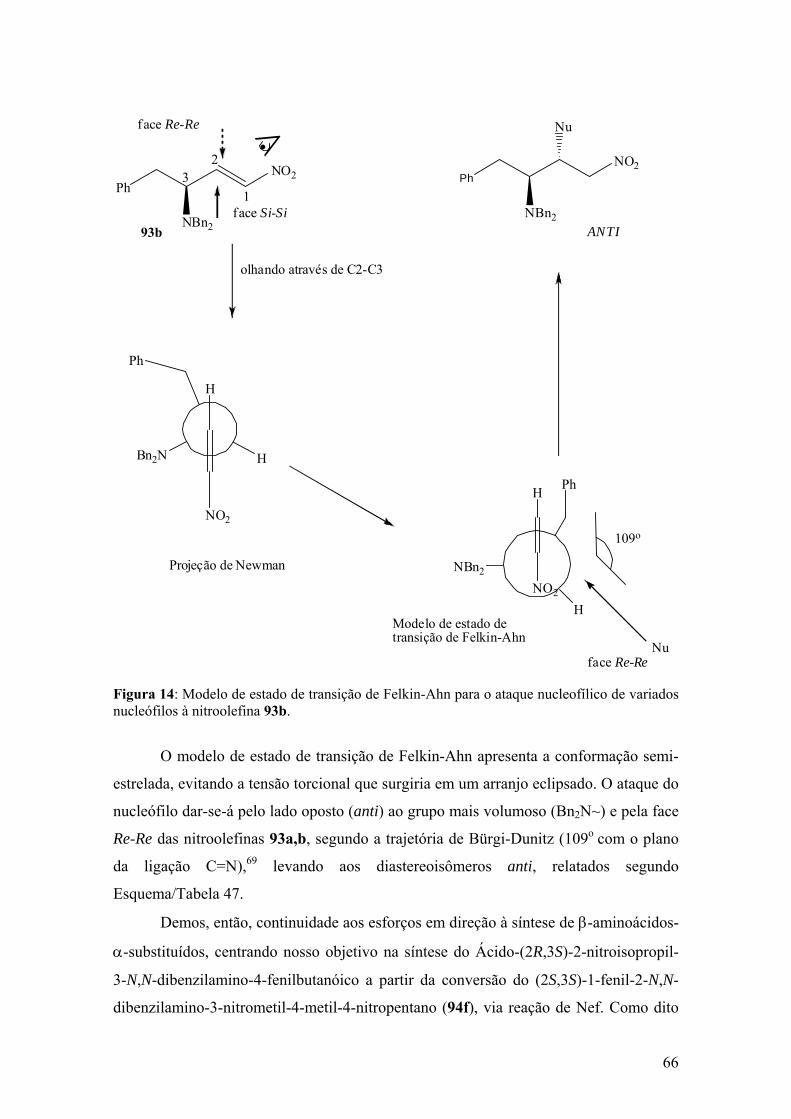
66
Ph
NBn2ANTI
NBn2
face Re-ReNu
Ph
NBn2
Bn2N H
Ph
>.
Projeção de Newman
Modelo de estado detransição de Felkin-Ahn
H
109o
93b
NO2
H
NO2
face Re-Re
face Si-Si
olhando através de C2-C3
Ph
NO2
Nu
1
23
NO2
H
Figura 14: Modelo de estado de transição de Felkin-Ahn para o ataque nucleofílico de variados nucleófilos à nitroolefina 93b.
O modelo de estado de transição de Felkin-Ahn apresenta a conformação semi-
estrelada, evitando a tensão torcional que surgiria em um arranjo eclipsado. O ataque do
nucleófilo dar-se-á pelo lado oposto (anti) ao grupo mais volumoso (Bn2N~) e pela face
Re-Re das nitroolefinas 93a,b, segundo a trajetória de Bürgi-Dunitz (109o com o plano
da ligação C=N),69 levando aos diastereoisômeros anti, relatados segundo
Esquema/Tabela 47.
Demos, então, continuidade aos esforços em direção à síntese de β-aminoácidos-
α-substituídos, centrando nosso objetivo na síntese do Ácido-(2R,3S)-2-nitroisopropil-
3-N,N-dibenzilamino-4-fenilbutanóico a partir da conversão do (2S,3S)-1-fenil-2-N,N-
dibenzilamino-3-nitrometil-4-metil-4-nitropentano (94f), via reação de Nef. Como dito

67
anteriormente, a reação de Nef consiste na conversão do grupo nitro primário ou
secundário em funções carboniladas, como aldeídos (nitro primário), cetonas (nitro
secundário) e até mesmo ácidos carboxílicos (nitro primário). Sendo assim, com o
objetivo de aplicar esta metodologia por nós estudada na síntese de β-aminoácidos-α-
substituídos, realizamos o estudo da reação de Nef tendo como substrato o aduto de
Michael oriundo da adição do ânion nitronato de isopropila à nitroolefina 93b,
conforme exemplificado no Esquema/Tabela 53 abaixo:
NBn2
NO2
Condiçõesreacionais
NO2
Reação de Nef
NBn2
OH
NO2
O94f 95f
Entrada Condições Reacionais Rendimento (%)a Referência
1 KMnO4 / KOH / K2HPO4 / t-BuOH / t.a. / 1h 55 75
2 Oxone® traços 76
3 NaNO2 / DMSO / AcOH / t.a. / 7h traços 77
4 MeOLi / MeOH / O3 / -78°C / 0,5h 79 78a,b
a) Determinado após purificação em coluna cromatográfica de gel de sílica. Esquema 53/Tabela: Síntese do β-aminoácido 95f por várias metodologias da reação de Nef. Foram selecionados três procedimentos mais utilizados na literatura para essa
reação. Um baseia-se na oxidação da ligação dupla do ânion nitronato por uma solução
de permanganato de potássio (entrada 1), e o outro pela oxidação mediada por Oxone®
(2 KHSO5.KHSO4.K2SO4) (entrada 2). O terceiro utiliza nitrito de sódio e ácido acético
para a transformação de nitros primários em ácido carboxílico, empregando condições
ácidas moderadas (entrada 3).
Embora vários relatos na literatura apontam os métodos de oxidação descritos
nas entradas 2 e 3 como os mais reprodutivos e com ótimos rendimentos químicos, para
o nosso sistema, apenas os métodos de Nef oxidativa utilizando KMnO4 em tampão de
sais de fosfato (entrada 1), onde forneceu o produto desejado em 55% de rendimento

68
químico. O método relatado por McMurry e cols., que em geral mostra-se eficaz na
conversão de nitros primários e secundários a carbonilas de aldeídos e cetonas,
respectivamente, via a ozonólise de sais de nitronatos, em nosso caso, providenciou-nos
a obtenção de um produto cabonilado, em 79% de rendimento químico, que acreditamos
se tratar de um ácido (entrada 4). Há poucos relatos na literatura, onde este protocolo de
ozonólise de nitronatos leva a tal função.79
Observamos, através de cromatografia em camada fina (CCF), apenas uma
conversão parcial do substrato 94f quando utilizado o método visto na entrada 1; ao
passo que houve conversão total do mesmo substrato (observado em CCF) quando o
método de ozonólise foi utilizado (entrada 4), embora o rendimento químico após
isolamento não fora quantitativo, o que indica possível perda de produto no processo de
extração do meio reacional.
Uma explicação para a formação do ácido 95f através do procedimento de
ozonólise pode ser analogamente racionalizada em duas etapas: Primeiramente, de
forma análoga ao mecanismo proposto por Criegee79, veja Esquema 54 a seguir. O
ozônio reage com a olefina 101, formando o intermediário de cinco membros 102. Este,
por sua vez, sofre um rearranjo para gerar as substâncias carboniladas 103a,b e outros
derivados de carbonila 104a,b. Na presença de uma substância redutora, como por
exemplo, dimetilsulfeto, 104a,b são convertidos a compostos carbonilados.
R2R1
R3 R4
OO
R4
R3R1
R2
O
R1 R2R3 R4
O O
O
R3 R4 R1 R2
OO
Oa b
O3
Ozonídeo
a
a
b
b++
133
134 135a 136a135b136b
Esquema 54: Mecanismo proposto por Criegee da reação de ozonólise frente a olefinas substituídas.

69
Analogamente,
N
OO
O
O
OH
H
OO
+ NO3-
Ozonídeo
H
NOO Li+
Nitronato de lítio
O3 LiOMe (1,0M)MeOH -78 0C(R)
NO2
94f 105 106Óxido decarbonila
107
NO2
NBn2
Ph
Esquema 55: Proposta mecanística para a ozonólise do sal de nitronato 105 proveniente da dinitroamina 94f.
Sugerimos que, analogamente ao mecanismo visualizado no Esquema 54, ao
passar um fluxo de ozônio através de uma solução que contém o sal do nitronato 105, o
ozonídeo 106 tenha sido gerado com um átomo de nitrogênio presente no anel de cinco
membros. Após a possível formação de 106, este sofre um rearranjo gerando o
fragmento 107 e o íon nitrato, veja Esquema 55.
Objetivando justificar a formação do ácido 95f sob este protocolo, podemos
fazer uma analogia com os relatos mecanísticos de Marshall e Garofalo78b. Os autores
realizaram um estudo sobre a clivagem oxidativa de olefinas, na obtenção de ésteres
metílicos, através de ozonólise em metanol e hidróxido de sódio, utilizando
diclorometano como co-solvente, veja Esquema 56 a seguir:
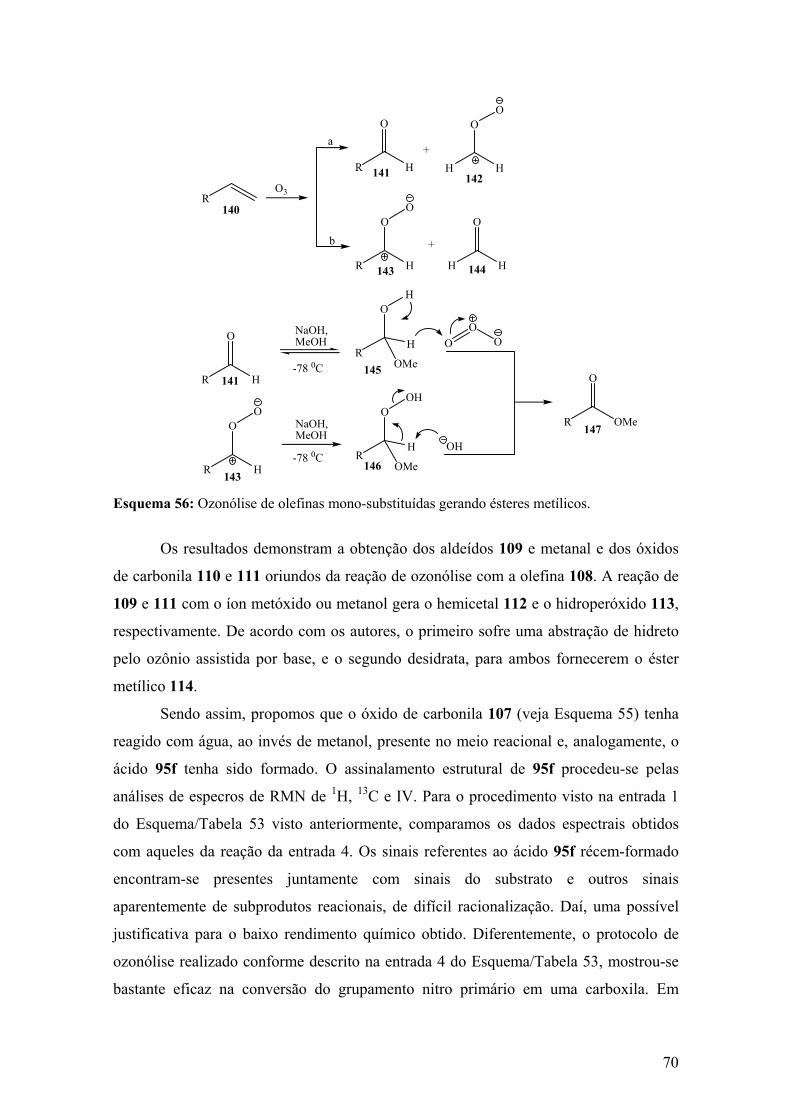
70
R
R H
O
R H
OO
R H
O
R H
OO
NaOH,MeOH
NaOH,MeOH
RH
OH
OMe
RH
OOH
OMe
OO
O
OH
R OMe
O
+
+
a
b
O3
O
HH
O
HH
O
140
141 142
143 144
141
143
145
146
147
-78 0C
-78 0C
Esquema 56: Ozonólise de olefinas mono-substituídas gerando ésteres metílicos.
Os resultados demonstram a obtenção dos aldeídos 109 e metanal e dos óxidos
de carbonila 110 e 111 oriundos da reação de ozonólise com a olefina 108. A reação de
109 e 111 com o íon metóxido ou metanol gera o hemicetal 112 e o hidroperóxido 113,
respectivamente. De acordo com os autores, o primeiro sofre uma abstração de hidreto
pelo ozônio assistida por base, e o segundo desidrata, para ambos fornecerem o éster
metílico 114.
Sendo assim, propomos que o óxido de carbonila 107 (veja Esquema 55) tenha
reagido com água, ao invés de metanol, presente no meio reacional e, analogamente, o
ácido 95f tenha sido formado. O assinalamento estrutural de 95f procedeu-se pelas
análises de especros de RMN de 1H, 13C e IV. Para o procedimento visto na entrada 1
do Esquema/Tabela 53 visto anteriormente, comparamos os dados espectrais obtidos
com aqueles da reação da entrada 4. Os sinais referentes ao ácido 95f récem-formado
encontram-se presentes juntamente com sinais do substrato e outros sinais
aparentemente de subprodutos reacionais, de difícil racionalização. Daí, uma possível
justificativa para o baixo rendimento químico obtido. Diferentemente, o protocolo de
ozonólise realizado conforme descrito na entrada 4 do Esquema/Tabela 53, mostrou-se
bastante eficaz na conversão do grupamento nitro primário em uma carboxila. Em

71
trabalhos prévios realizados em nosso laboratório,71 verificou-se que este protocolo leva
à formação de éster metílico, conforme exemplificado no Esquema 56 visto
anteriormente. Contudo, devido às condições dos reagentes empregados, solução 1,0 M
de metóxido de lítio e etanol seco, parece-nos provável que os mesmos não foram
suficientes para isentar o meio reacional da presença de água, que, por sua vez ataca
preferencialmente a espécie 107 (óxido de carbonila – veja Esquema 55), levando à
formação de 95f, que teve seu assinalmento estrutural através da observação de uma
banda intensa de absorção de carboxila de ácido carboxílico a 1716 cm-1 (veja Espectro
107) e sinal em 169,6 ppm, referentes ao mesmo carbono (veja Espectro 106).
Um segundo exemplo de reação de Nef nos adutos de Michael obtidos (94a-g)
foi realizado empregando o aduto 94d, oriundo da adição de metóxido à nitroolefina
93b, de acordo com o procedimenmto relatado na entrada 1 do Esquema/Tabela 51.
Veja Esquema 57 abaixo:
NO2
NBn2
OMe
94d
KMnO4
KOH / K2HPO4t-BuOH / t.a. / 1h
OH
NBn2
OMe
O
95d60% Esquema 57: Obtenção do β-aminoácido 95d.
De acordo com o Esquema 56 acima, a reação de Nef oxidativa mostrou-se
eficaz na transformação de um grupamento nitro primário em 94d a uma carboxila de
ácido em 95d. O β-aminoácido 95d foi obtido em um rendimento químico moderado de
60%, sendo um análogo metilado e um epímero da Bestatina (veja Quadro 6, página
41).

72
5. CONCLUSÃO. As aldiminas quirais 88a-d sintetizadas a partir dos α-aminoácidos L-valina, L-
leucina, L-alanina e L-fenilalanina, foram submetidas a adições nucleofílicas de ânions
nitronatos oriundos de nitroalcanos comerciais como nitrometano, nitroetano e 2-
nitropropano, bem como de derivados de nitroalcanos sintetizados por nós como 1-
nitrobutano, 90 (nitroálcool) e 91 (nitroéster) tendo como base a resina Amberlist A-21
e reação conduzida sem solvente. No caso da aldimina 88d, oriunda da L-valina, não
houve sinal da conversão química desejada, pois o ataque nucleofílico torna-se
impedido devido a considerações estéricas provenientes do grupamento isopropil nas
adjacências do carbono azometínico. Para as demais aldiminas sintetizadas, foram
obtidas as nitrodiaminas correspondentes em rendimentos químicos que variaram de
moderados (55 - 66%) a bons (73 - 82%), com excessos diastereoisoméricos obtidos na
faixa de baixos (48%) a bons (80%). No caso do emprego de nitroalcanos volumosos,
90 e 91, foi observado um aumento na diastereosseletividade, pois foram formados
apenas dois dos quatro diastereoisômeros possíveis. Em todas as reações realizadas, não
foram constatados sinais de reações laterais e também de racemização, já que, em
alguns casos onde houve baixo rendimento químico, o substrato empregado foi
recuperado após todo o tempo reacional e teve a sua atividade óptica conferida,
mantendo-se igualmente àquela antes de ser submetido à reação em questão,
corroborando o fato de que nas condições reacionais empregadas, a base empregada
para a geração dos ânions nitronatos e eles mesmos não apresentam basicidade
suficiente para promover a abstração do hidrogênio α-imínico.
O sucesso deste método consiste em uma rota rápida para a síntese
estereosseletiva de nitroaminas, cujo grupo nitro pode ser facilmente hidrogenado em
bons rendimentos químicos ou transformado em diversas outras funcionalidades.
Adicionalmente, as nitroaminas podem ser transformadas em diaminas quirais, que vêm
sendo utilizadas como ligantes quirais em síntese enantiosseletiva.
O estudo da adição conjugada de variados nucleófilos às nitroolefinas quirais
sintetizadas a partir dos α-aminoácidos L-alanina e L-fenilalanina mostrou-se bastante
promissor para uma simples rota de acesso à síntese de β-aminoácidos com variados
padrões de substituição na posição α. As nitroolefinas empregadas 93a-b, até então
inéditas na literatura, mostraram-se bastante estáveis quimicamente, e, novamente após

73
inúmeras condições reacionais empregadas, como, principalmente, base, tempo,
temperatura e solvente, não foram observados indícios de racemização do substrato,
tendo sua atividade óptica conferida após todo o tempo reacional em casos onde não
houve conversão total, evidenciado por cromatografia em camada fina. Em apenas um
caso, onde se empregou a nitroolefina 93a e foi adicionado o ânion nitronato de
isopropila, verificou-se a competição entre a reação de adição conjugada versus uma
reação de cicloadição do tipo [3 + 2].
A variedade de nucleófilos empregados permitiu a obtenção das 1,3-nitroaminas
94a-g com diferentes padrões de substituição na posição 2. Os produtos obtidos foram
sintetizados em uma faixa de rendimento químico que variou de moderada (58 – 67%) a
boa (70 - 77%) após purificação em coluna cromatográfica de gel de sílica e,
comparativamente aos resultados obtidos no estudo realizado com as aldiminas, os
excessos diastereoisoméricos alcançados foram mais satisfatórios; no caso da
nitroolefina 93b, tendo o melhor exemplo, a adição de nitronato de isopropila à
nitroolefina 93b, oriunda da L-fenilalanina, com uma razão diastereoisomérica anti:sin
de 93 : 7 (94f).
Com este ótimo resultado obtido, esta nova metodologia estudada pôde ser
empregada na síntese de β-aminoácidos-α-substituídos. Foi, então, realizado diversos
estudos sobre condições reacionais da reação de Nef para a síntese do β-aminoácido
95f, que teve sua síntese realizada por nós com 79% de rendimento químico.

74
6. MATERIAIS E MÉTODOS.
As substâncias D-manitol, L-leucina, L-valina, L-alanina, L-fenilalanina,
propanona, brometo de butila, ácido p-toluenossulfônico, periodato de sódio,
bicarbonato de sódio, hidróxido de potássio, carbonato de potássio, permanganato de
potássio, hidrogenofosfato de potássio, hidróxido de sódio, cloreto de sódio, sulfato de
sódio anidro, nitrito de sódio, florisil, ácido clorídrico, ácido acético foram adquiridas
através da VETEC QUÍMICA FINA Ltda. O 2,2-dimetoxipropano e metóxido de lítio
em metanol (solução a 1,0 M) foram adquiridos pela ALDRICH®. As substâncias
trimetilsililazida e trimetilsililcianeto foram adquiridas através da ACROS ORGANIC. Os
nitroalcanos (nitrometano, nitroetano e 2-nitropropano), tetraidrofurano, benzilamina,
brometo de benzila, hidreto de alumínio e lítio, cloreto de oxalila, fluoreto de
tetrabutilamônio, cloreto de mesila, Amberlist A-21, alumina básica, DBU e
dimetilsulfeto foram adquiridos através da MERCK®. Estas substâncias não foram
previamente purificadas. Já os solventes utilizados para fins sintéticos e analíticos, tais
como acetato de etila, diclorometano, hexano, éter dietílico, metanol, dimetilsulfóxido,
dimetilformamida, acetonitrila, trietilamina passaram por uma destilação simples.
A secagem do tetraidrofurano foi efetuada por refluxo com sódio metálico, na
presença de benzofenona, seguida de destilação. O metanol foi seco somente com sódio
metálico seguido de destilação. Já o diclorometano, 2,2-dimetoxipropano,
dimetilsulfóxido e dimetilformamida foram secos por refluxo com hidreto de cálcio
seguido de destilação.
Para a técnica de separação cromatográfica em coluna foram utilizadas colunas
de vidro (2 x 20cm e 3 x 50cm) com fase estacionária composta por gel de sílica de
granulometria 0,063mm – 0,20mm 60 Å (MERCK).
O acompanhamento das separações cromatográficas e dos processos de
transformações químicas foram realizados sobre cromatofolhas de alumínio recobertas
com gel de sílica 60 F254 de dimensões 2,5 x 6,0 cm, irradiadas com luz ultravioleta de
comprimento de onde de 254 nm, sendo, então, esta cromatografia em camada fina
(CCF) revelada através da imersão em uma solução etanólica a 7 % de ácido
fosfomolibdênico, submetendo-a a posterior aquecimento.

75
Os espectros de RMN de ¹H foram obtidos em um aparelho Varian Gemini 200
(200MHz) e em um aparelho Bruker-DRX (400MHz), utilizando-se o tetrametilsilano
(TMS) como referência interna dos valores de deslocamento químico (δ), que foram
referidos em ppm, em relação ao TMS e os das constantes de acoplamento (J) em Hertz.
As multiplicidades referentes a cada absorção são expressas da seguinte forma: sinal
simples (s), sinal duplo (d), sinal triplo (t), sinal quádruplo (q), duplo sinal duplo (dd),
sinal múltiplo (m) e sinal simples largo (sl). Os espectros de RMN de ¹³C foram obtidos
em aparelho Varian Gemini 200 (50MHz) e Brücker (100MHz), utilizando-se o TMS
como referência interna. As multiplicidades dos sinais dos carbonos foram observadas
pelo uso da técnica APT (Attached Proton Test). Os espectros de IV foram obtidos em
um aparelho Nicolet Magna-IR-760 e somente as principais bandas foram relatadas. Os
espectros de massa de baixa resolução foram obtidos por impacto de elétrons a 70 eV,
em um aparelho CGMS-QP5000 da Shimadzu utilizando a coluna ZB-5 ms (30m –
0,25mm Ø) com uma rampa de temperatura de 60-290 0C, injetor a 270 0C e interface a
230 0C.
As medidas de rotação óptica específica foram efetuadas em um polarímetro
Perkin-Elmer modelo 243-B.

76
7. PARTE EXPERIMENTAL.
7.1) Síntese dos ésteres 85a-d:
Procedimento geral:
7.1.1) Síntese do (2S)-N,N-dibenzilamino-4-metilpentanoato de benzila (85c).
A uma mistura de 5 g de (S)-leucina (38,2 mmoles) e 15,8 g de carbonato de
potássio (114,6 mmoles) em 40,0 mL de EtOH e 8,0 mL de H2O, respectivamente, sob
agitação magnética e refluxo, foram adicionados 15,9 mL de brometo de benzila (133,7
mmoles) após os sólidos se dissolverem parcialmente. A mistura reacional permaneceu
sob refluxo por 16 horas. Em seguida, o meio reacional foi filtrado, os sólidos foram
lavados com EtOAc e a solução foi concentrada sob pressão reduzida. O líquido pouco
viscoso e ligeiramente amarelado obtido foi submetido à purificação em gel de sílica,
utilizando apenas hexano como eluente. O produto desejado foi obtido em 86% de
rendimento químico.
- Dados espectrais:
RMN 1H (200 MHz, CDCl3): δ (200MHz, CDCl3): δ 7,5 – 7,2 (m, 15 H), 5,3 (d, 2H,
J=12,18), 5,16 (d, 2H, J=12,18), 3,9 (d, 2H, J=12,2), 3,4 (d, 2H, J=12,2), 3,2 (m, 1H),
1,7 (m, 1H), 1,57 – 1,2 (m, 1H), 0,8 (d, 3H), 0,6 (d, 3H).
RMN 13C (50,4 MHz, CDCl3): δ 173,0 (C), 139,5 (C), 136,0 (C), 128,7 – 126,8 (CH –
aromático), 65,7 (CH2), 58,6 (CH), 54,3 (CH2), 38,5 (CH2), 24,2 (CH), 23,0 (CH3),
21,4 (CH3).
OBn
NBn2
O
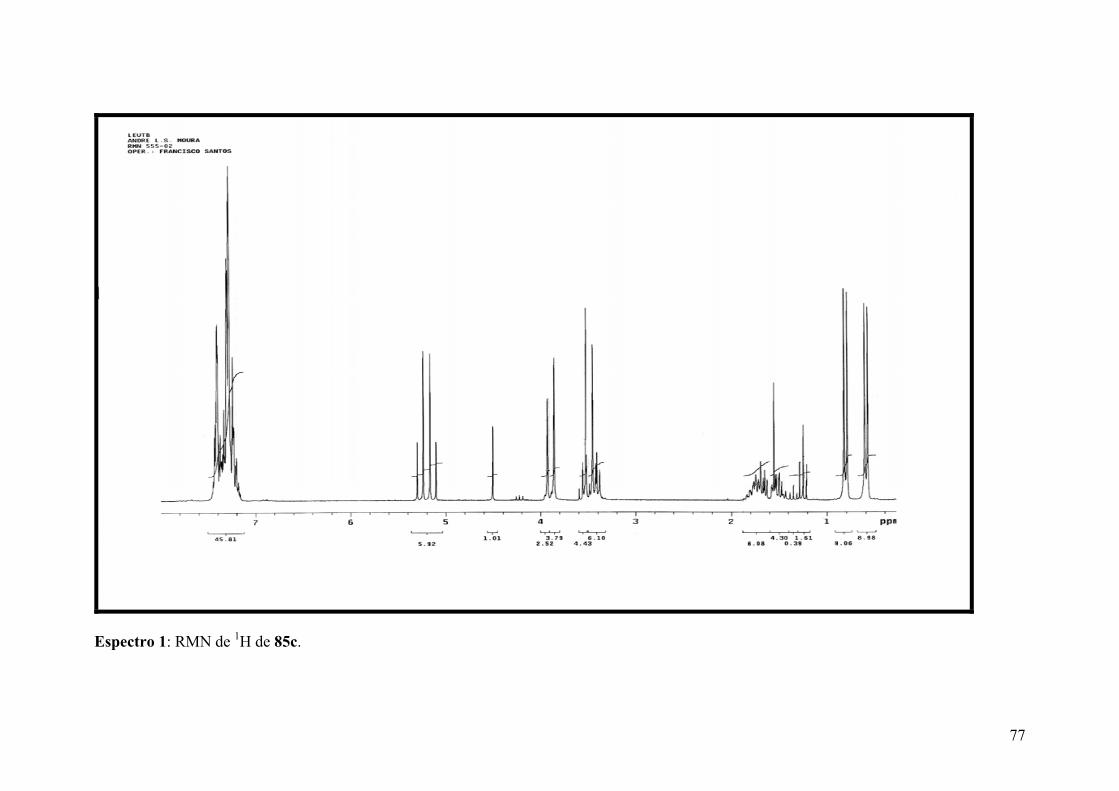
77
Espectro 1: RMN de 1H de 85c.
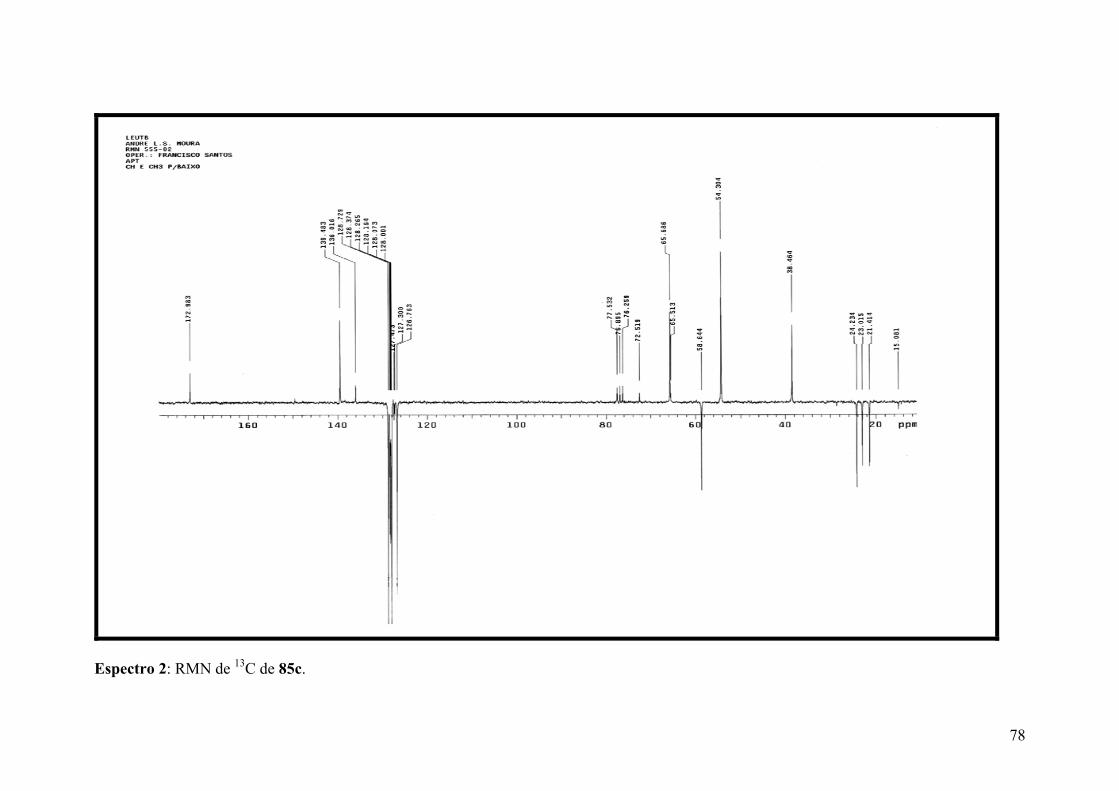
78
Espectro 2: RMN de 13C de 85c.

79
7.1.2) Síntese do (2S)-N,N-dibenzilamino-3-metilbutanoato de benzila (85d).
Líquido pouco viscoso e pálido obtido em 85% de rendimento após purificação
em coluna cromatográfica de gel de sílica, tendo como eluente hexano.
- Dados espectrais:
IV (KBr): ν 3068-3030, 2961-2808, 1728, 1181, 1140, 1108, 746, 698 cm-1.
RMN 1H (200MHz, CDCl3): δ 7,5 – 7,2 (m, 15 H), 5,3 (d, 2H, J=12,18), 5,16 (d, 2H,
J=12,18), 3,97 (d, 2H), 3,28 (d, 2H), 2,9 (d, 1H), 2,16 (m, 1H), 1,02 (d, 3H), 0,78 (d,
3H).
RMN 13C (50,4 MHz, CDCl3): δ 171,7 (C), 139,3 (C), 136,0 (C), 128,6 – 126,7 (CH),
72,5 (CH2), 67,9 (CH), 65,5 (CH2), 54,4 (CH2), 27,1 (CH), 19,8 (CH3), 19,4 (CH3).
OBn
NBn2
O
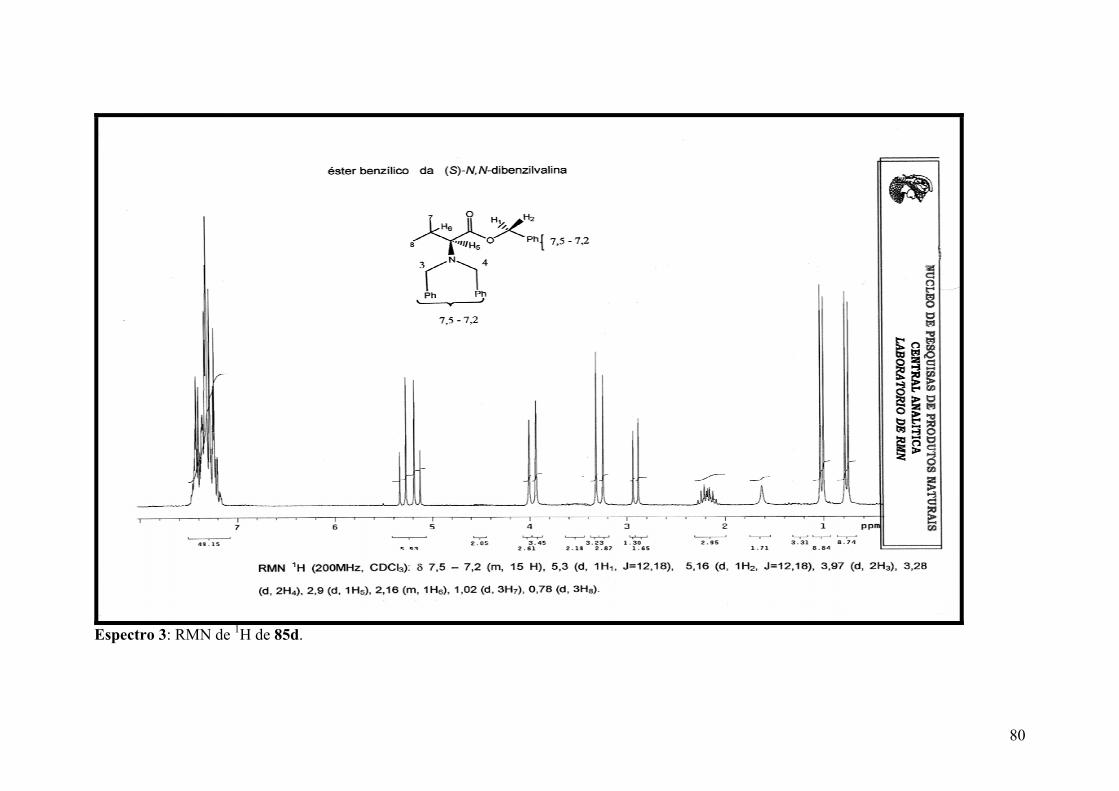
80
Espectro 3: RMN de 1H de 85d.
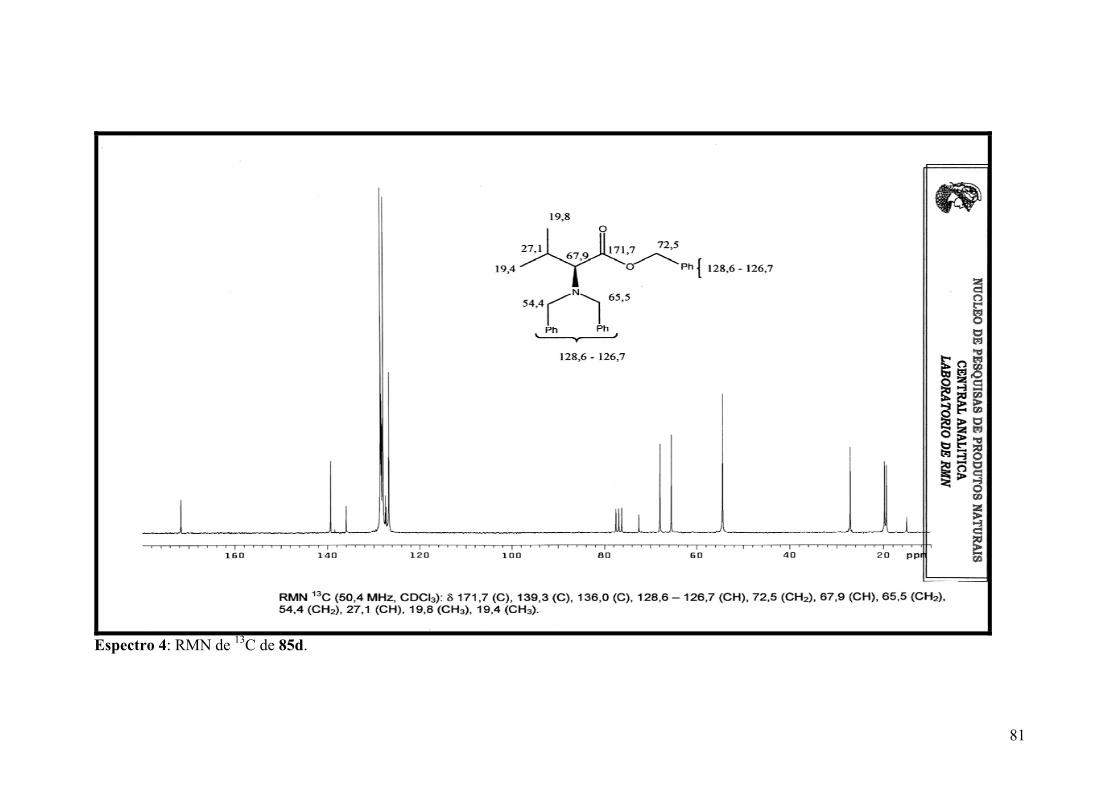
81
Espectro 4: RMN de 13C de 85d.
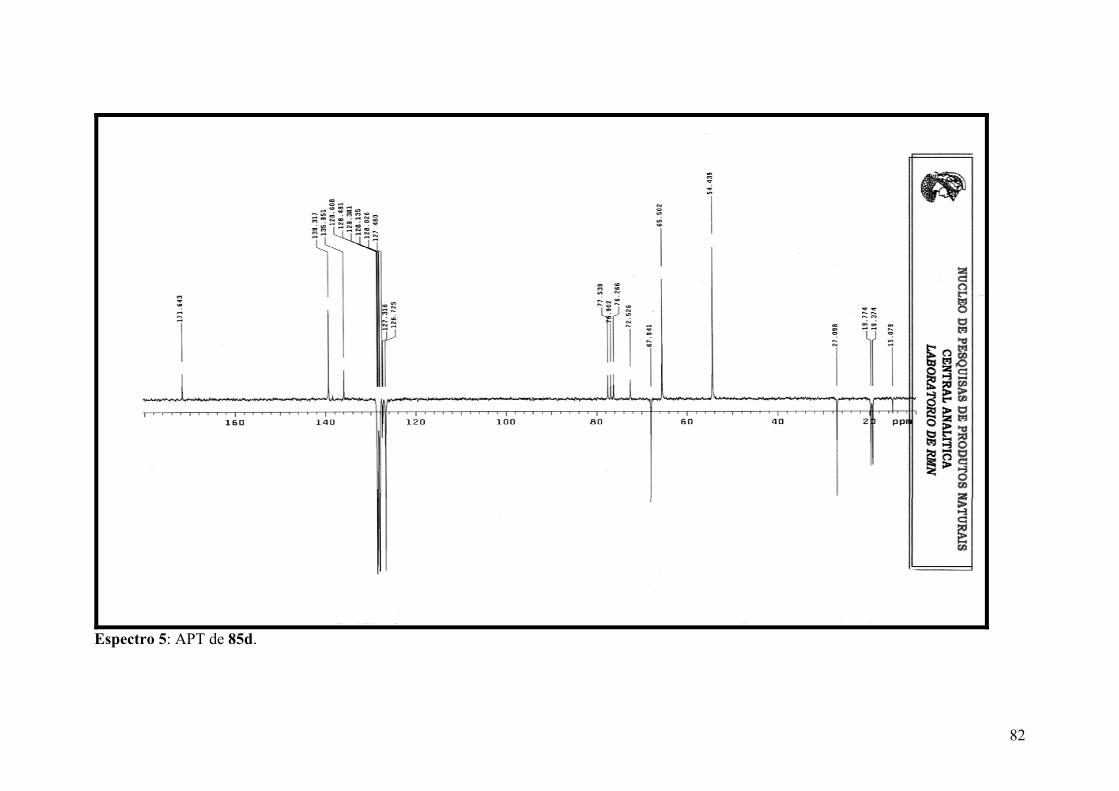
82
Espectro 5: APT de 85d.
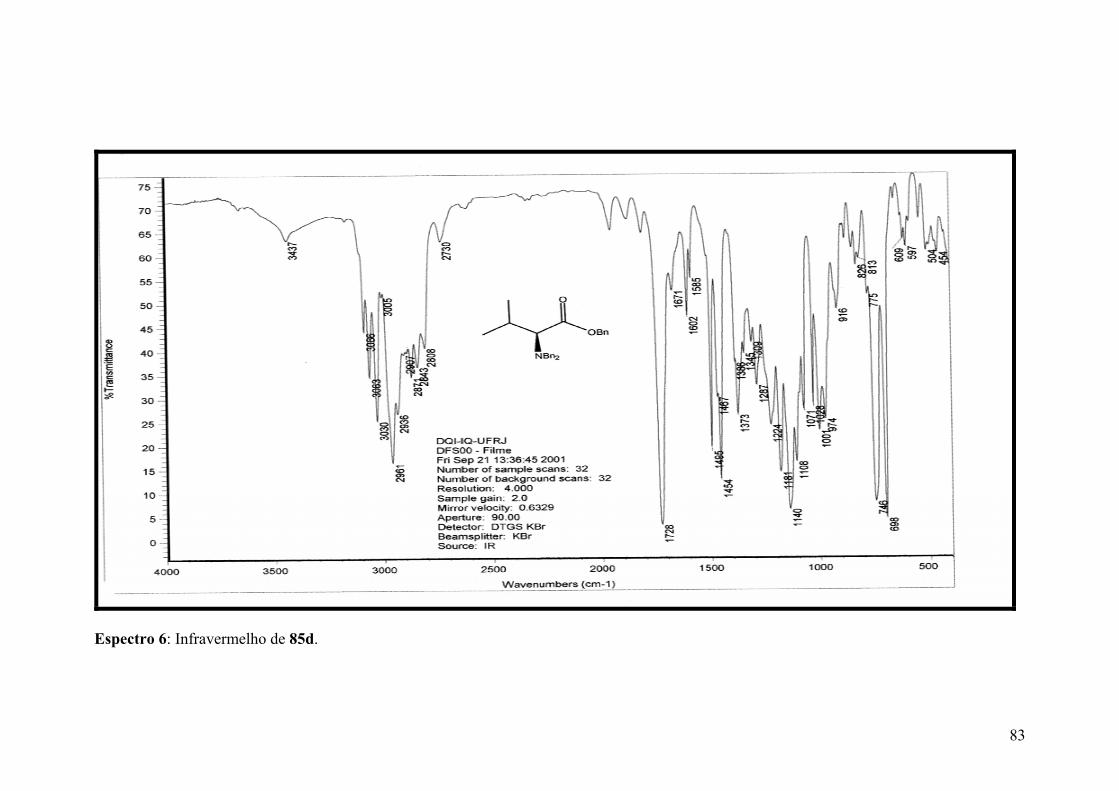
83
Espectro 6: Infravermelho de 85d.

84
7.1.3) Síntese do (2S)-N,N-dibenzilamino-3-fenilpropanoato de benzila (85b).
OBn
O
NBn2
Líquido viscoso e amarelado obtido em 93% de rendimento químico após
purificação em coluna cromatográfica de gel de sílica, tendo como eluente hexano.
- Dados espectrais:
RMN 1H (200 MHz, CDCl3): δ 7,2-7,0 (m, 20H), 5,2 (d, 1H, J=12,3), 5,15 (d, 1H,
J=12,3), 3,9 (d, 1H, J=12,2), 3,7 (t, 1H), 3,5 (d, 1H, J=12,3), 3,1 (ddd, 2H).
RMN 13C (50,4 MHz, CDCl3): δ 172,0 (C), 139,0 – 135,8 (C), 129,3-126,1 (CH-
aromático), 65,9 (CH2), 62,2 (CH), 54,3 (CH2), 35,5 (CH2).
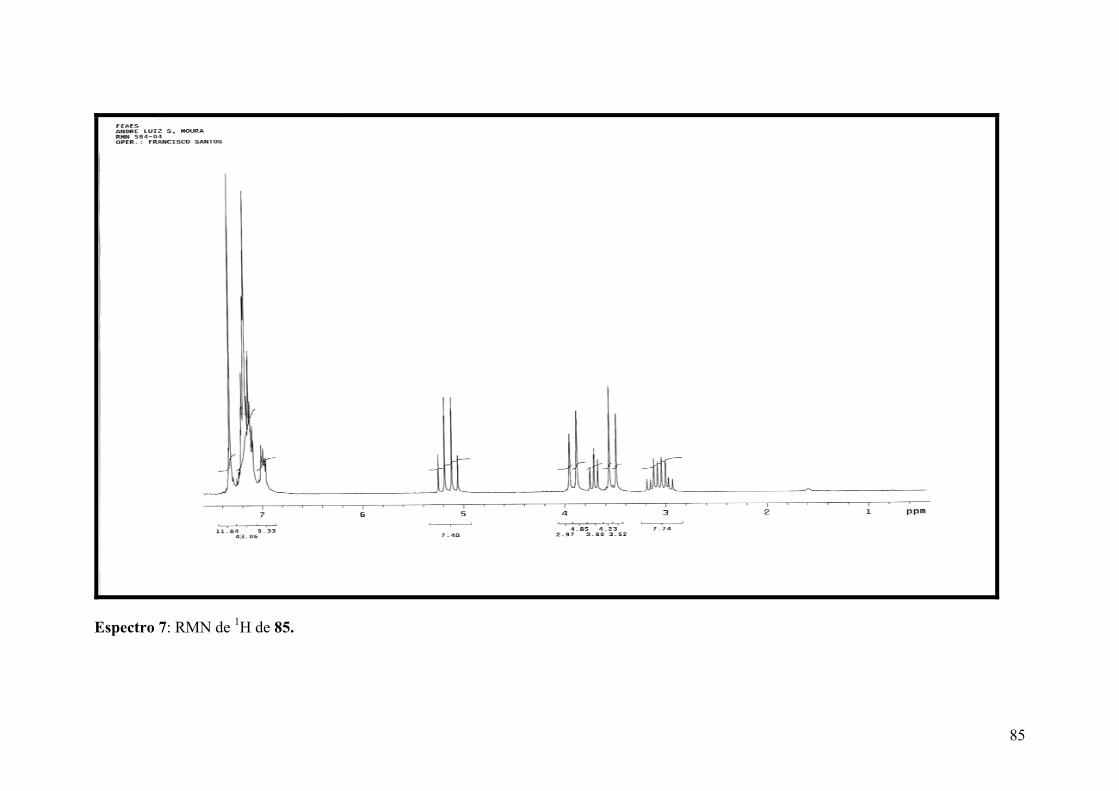
85
Espectro 7: RMN de 1H de 85.
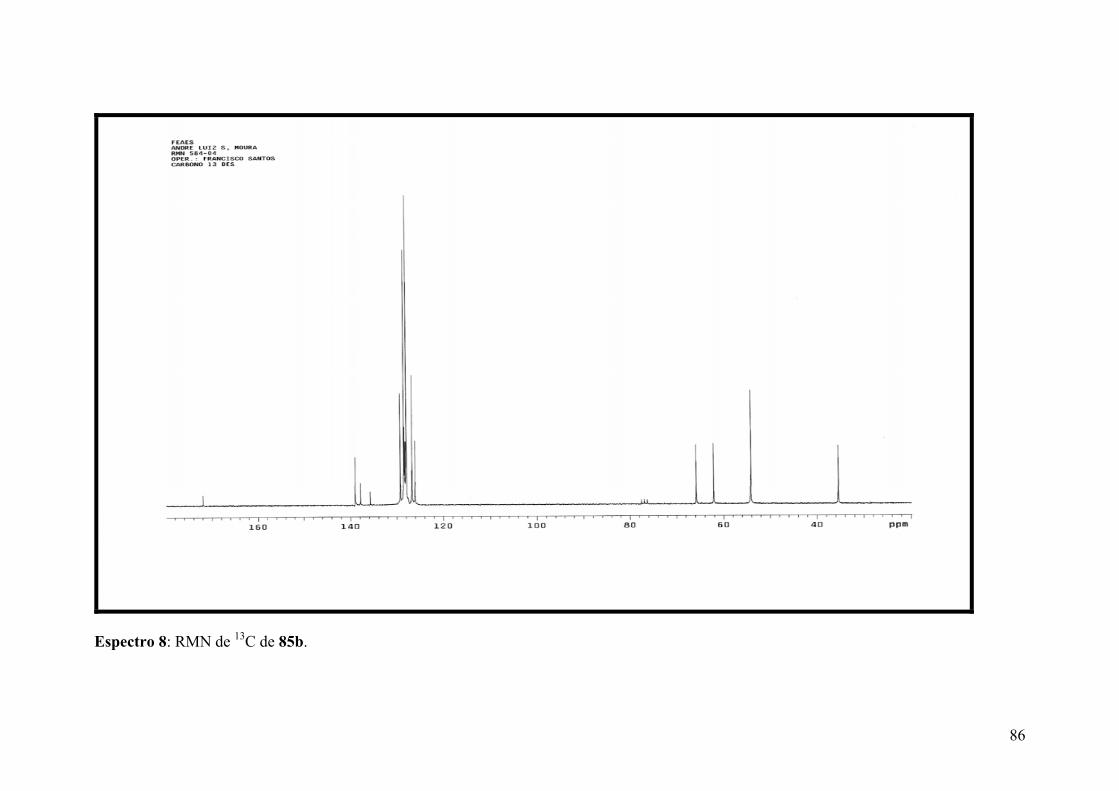
86
Espectro 8: RMN de 13C de 85b.
87
7.1.4) Síntese do (2S)-N,N-dibenzilaminopropanoato de benzila (85a).
OBn
O
NBn2
Líquido viscoso e pálido obtido em 90% de rendimento químico após purificação
em coluna cromatográfica de gel de sílica, tendo como eluente hexano.
- Dados espectrais:
RMN 1H (200 MHz, CDCl3): δ 7,4-7,2 (m, 15H), 5,23 (d, 1H), 5,17 (d, 1H), 3,81 (d,
1H), 3,6 (d, 1H), 3,5 (m, 1H), 1,28 (d, 3H).
RMN 13C (50,4 MHz, CDCl3): δ 174,1 (C), 140,4 e 136,7 (C), 129,6-127,5 (CH-
aromático), 66,6 (CH2), 56,8 (CH), 55,0 (CH2), 15,5 (CH3).
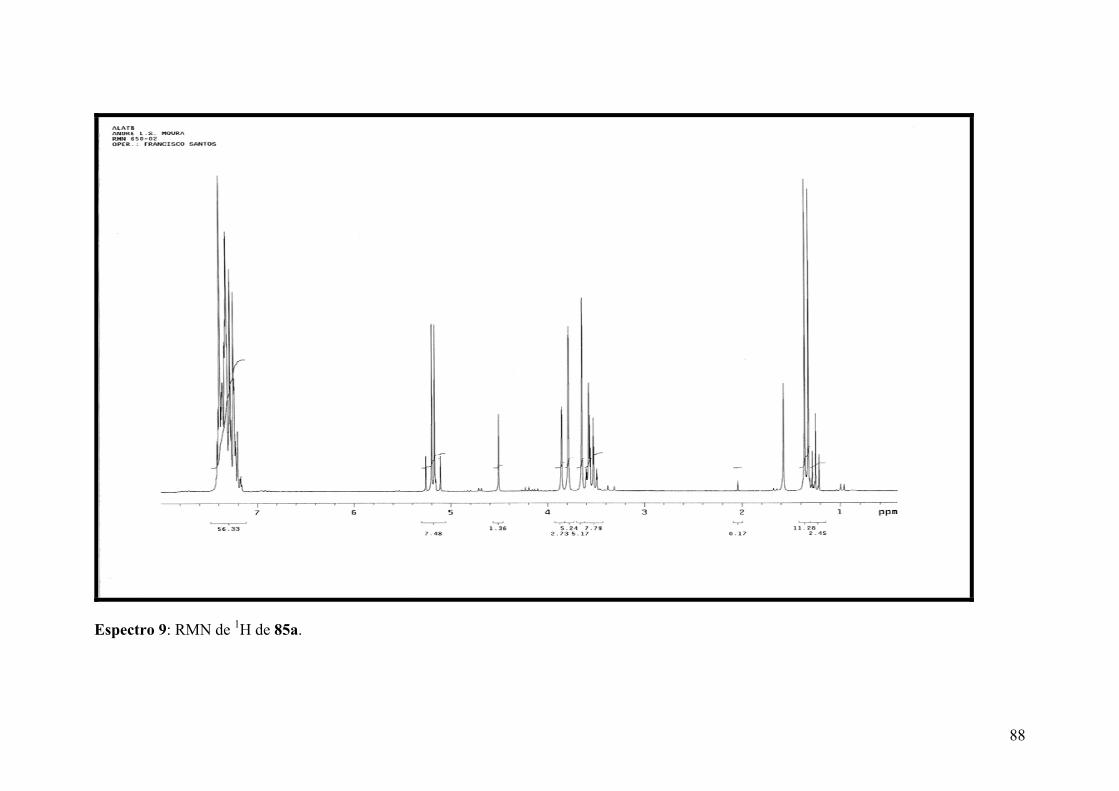
88
Espectro 9: RMN de 1H de 85a.
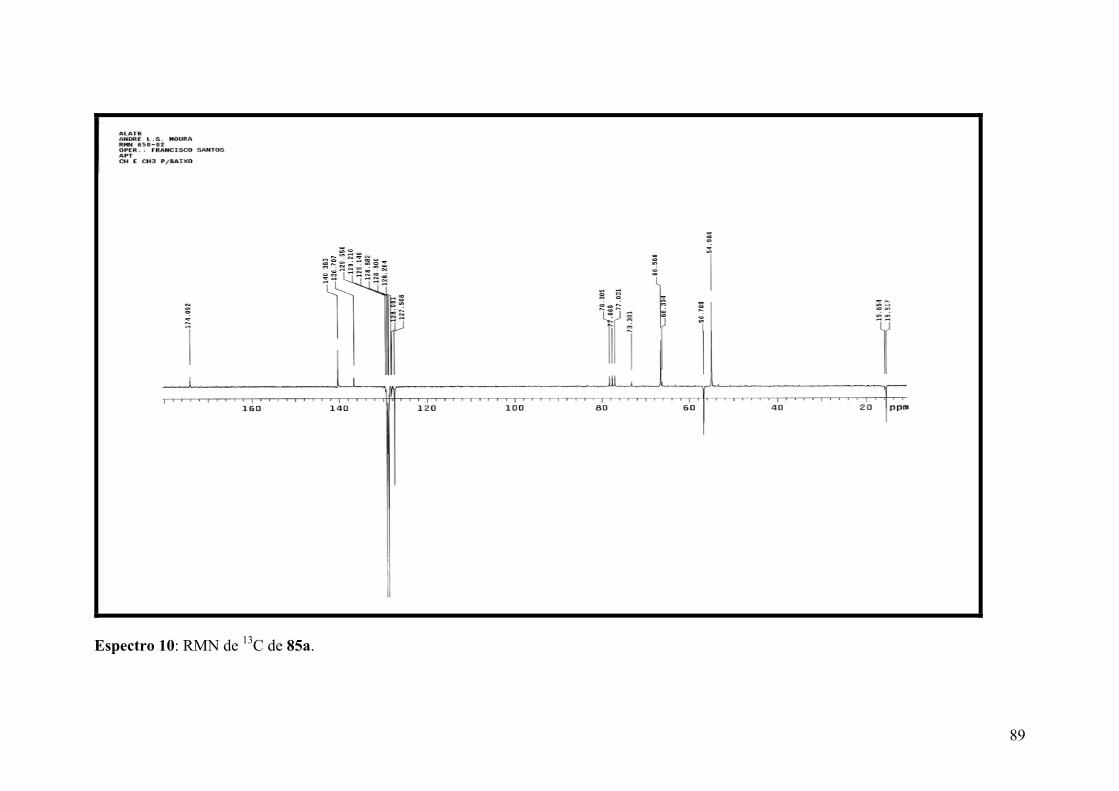
89
Espectro 10: RMN de 13C de 85a.

90
7.2) Síntese dos álcoois 86a-d:
Procedimento geral:
7.2.1) Síntese do (2S)-N,N-dibenzilamino-4-metil-1-pentanol (86c).
Foram adicionados a uma suspensão de 0,9 g de LiAlH4 (22,5 mmoles) em 20,0
mL de THF seco, a 0°C, 3,0 g (7,5 mmoles) do éster 85c, solubilizados em 10,0 mL de
THF seco. O sistema permaneceu sob agitação e refluxo por 4 horas. Em seguida,
adicionou-se uma solução de NaOH 10 % até a completa formação de um precipitado
branco. O produto esperado foi extraído por duas lavagens com EtOAc. O aminoálcool,
um líquido muito viscoso de cor ligeiramente amarelada, foi obtido em 85 % de
rendimento químico após purificação por destilação sob pressão reduzida em um
aparelho de Kugelrhor (p.e. = 170°C, 1,0 mmHg).
- Dados espectrais:
RMN 1H (200MHz, CDCl3): δ 7,19 - 7,89 (m, 10H), 3,81 (d, 2H, J=13,27), 3,34 - 3,54
(m, 2H), 3,36 (d, 2H, J=13,27), 3,19 (sl, 1H), 2,84 (m, 1H), 1,50 (m, 2H), 1,16 (m, 1H),
0,92 (d, 3H, J=6,13), 0,86 (d, 3H, J=6,04).
RMN 13C (50,4 MHz, CDCl3): δ 139,7 (C), 129,0-127,1 (CH-aromático), 61,5 (CH2),
56,7 (CH), 53,1 (CH2), 34,0 (CH2), 25,8 (CH), 24,1 (CH3), 22,2 (CH3).
OH
NBn2
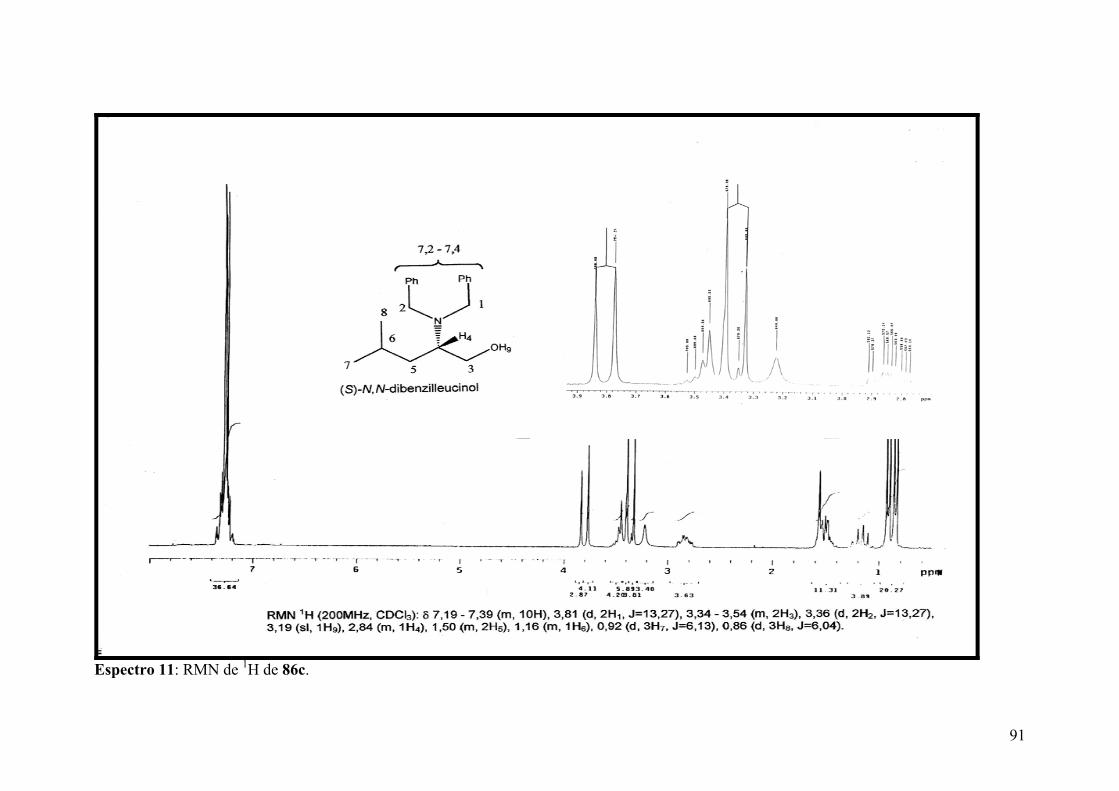
91
Espectro 11: RMN de 1H de 86c.
92
Espectro 12: RMN de 13C de 86c.

93
7.2.2) Síntese do (2S)-N,N-dibenzilamino-3-metil-1-butanol (86d).
Líquido muito viscoso de cor ligeiramente amarelada obtido em 85 % de rendimento
químico após purificação por destilação sob pressão reduzida em um aparelho de
Kugelrhor (p.e. = 170°C, 1,0 mmHg).
- Dados espectrais:
IV (KBr): ν 3424, 3104-3027, 1072, 1065, 1028, 748, 699 cm-1.
RMN 1H (200 MHz, CDCl3): δ 7,35 – 7,2 (m, 10H), 3,84 (d, 2H, J=13,2), 3,66 (d, 2H,
J=13,2), 3,56 (dd, 1H), 3,42 (t, 1H, J=10,0), 2,7 (sl, 1H), 2,52 (ddd, 1H), 2,02 (m, 1H),
1,16 (d, 3H, J=6,7), 0,87 (d, 3H, J=6,7).
RMN 13C (50,4MHz, CDCl3): δ 139,6 (C), 129,0 – 127,0 (CH), 64,5 (CH), 59,1 (CH2),
54,1 (CH2), 27,5 (CH), 22,6 (CH3), 20,0 (CH3).
OH
NBn2
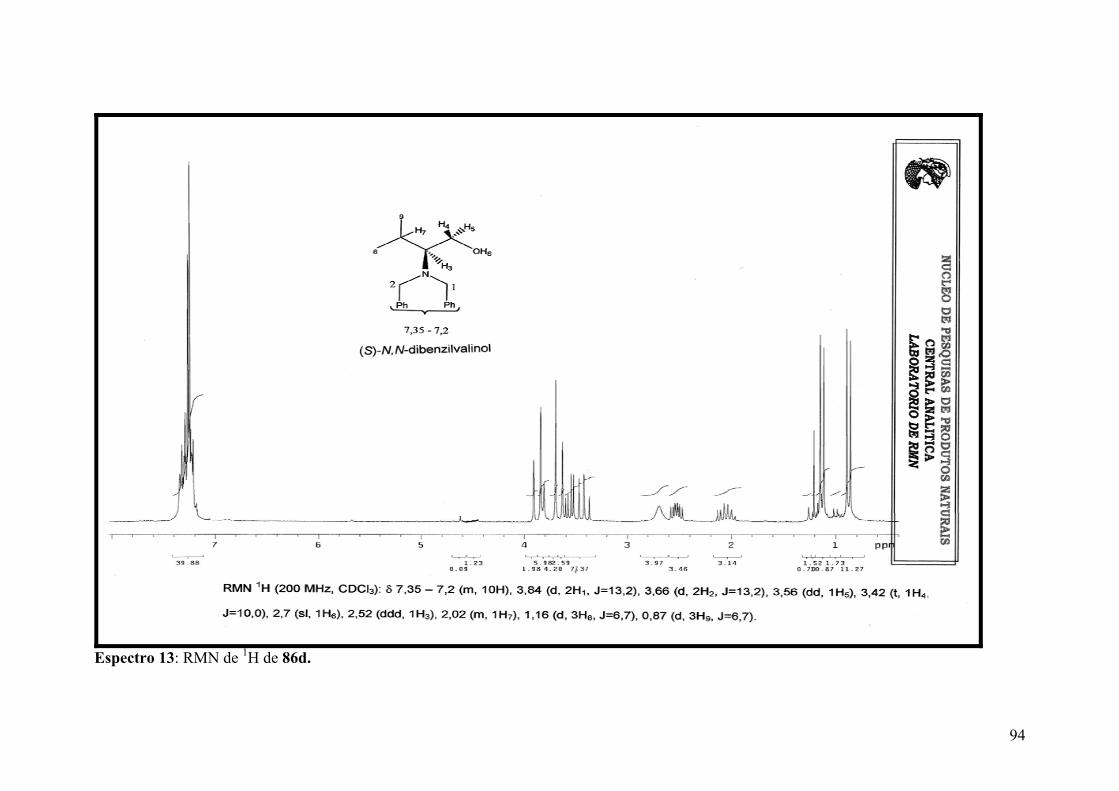
94
Espectro 13: RMN de 1H de 86d.
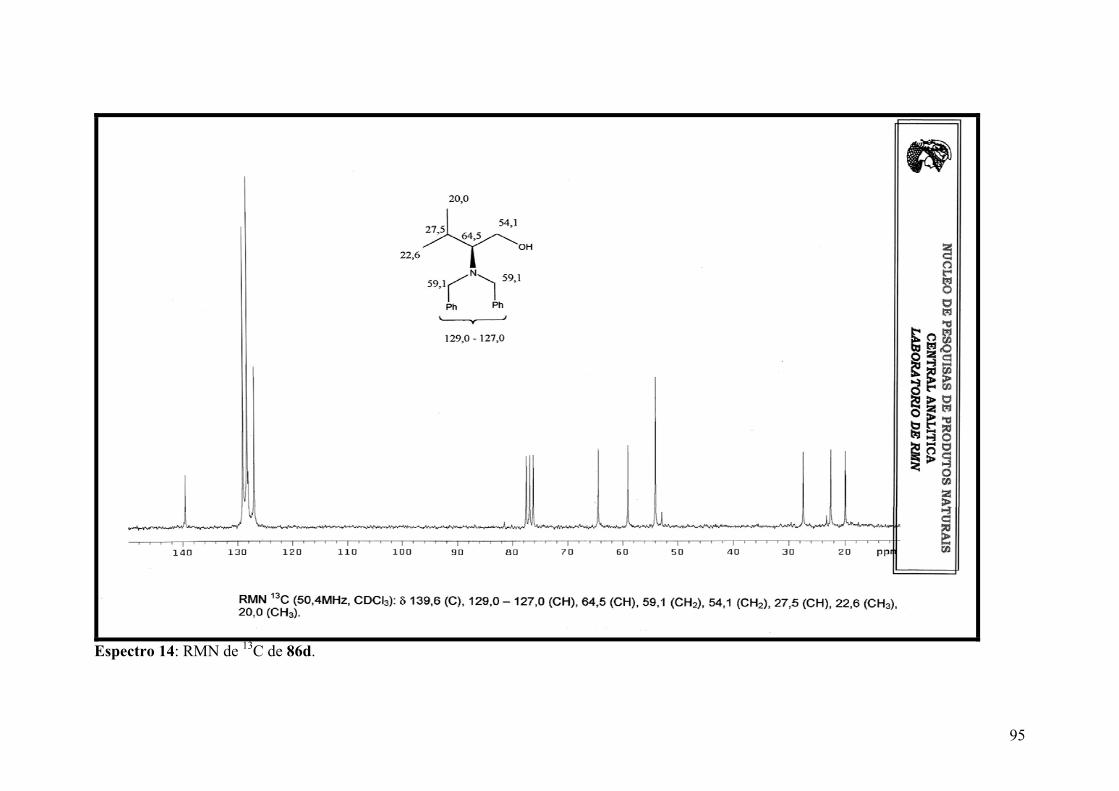
95
Espectro 14: RMN de 13C de 86d.

96
Espectro 15: APT de 86d.
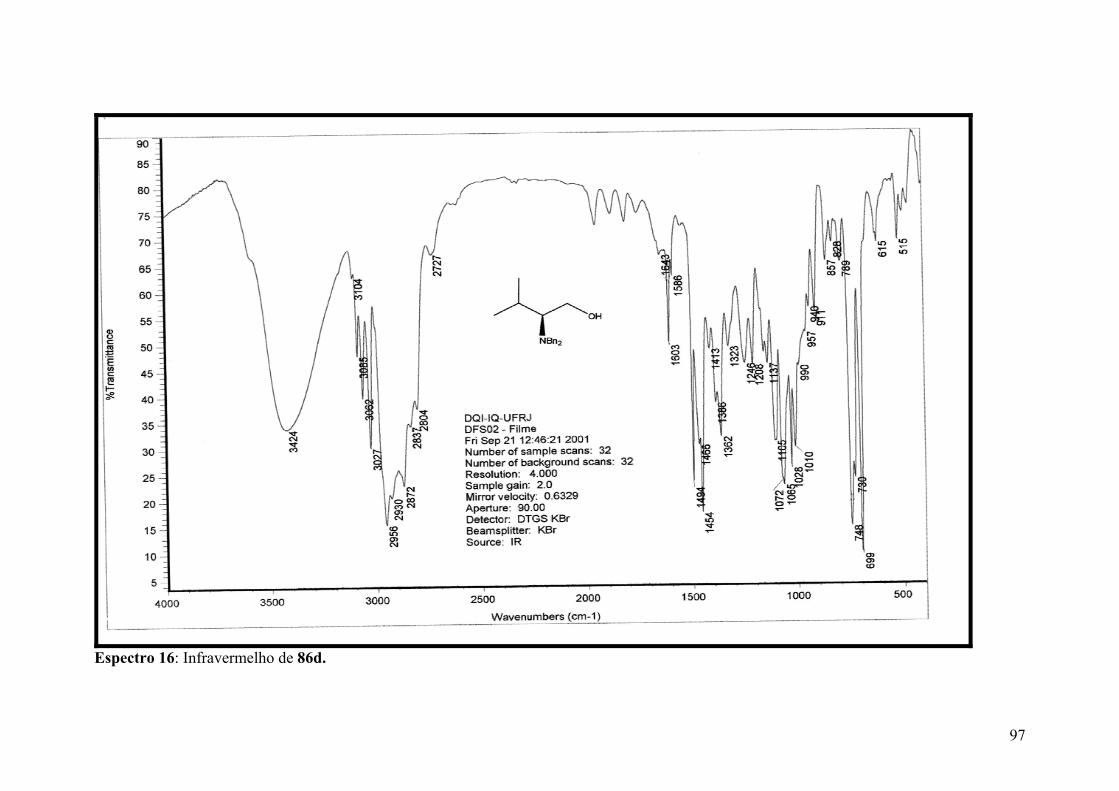
97
Espectro 16: Infravermelho de 86d.

98
7.2.3) Síntese do (2S)-N,N-dibenzilaminopropanol (86a).
Líquido muito viscoso de cor pálida obtido em 87 % de rendimento químico após
purificação por destilação sob pressão reduzida em um aparelho de Kugelrhor (p.e. =
170°C, 1,0 mmHg).
- Dados espectrais:
IV (KBr): ν 3451, 3082-3026, 2963-2848, 1018, 742, 694 cm-1.
RMN 1H (200 MHz, CDCl3): δ 7,4 - 7,2 (m, 10H), 4,0 (m, 1H), 3,82 (d, 2H, J=13,4),
3,58 (d, 1H, J=13,4), 2,7 (ddd, 1H), 1,4 (d, 3H, J=6,7).
RMN 13C (50,4 MHz, CDCl3): δ 139,0 (C), 128,7 - 126,9 (CH), 61,9 (CH2), 58,9(CH2)
55,3 (CH), 22,9 (CH3).
NBn2
OH
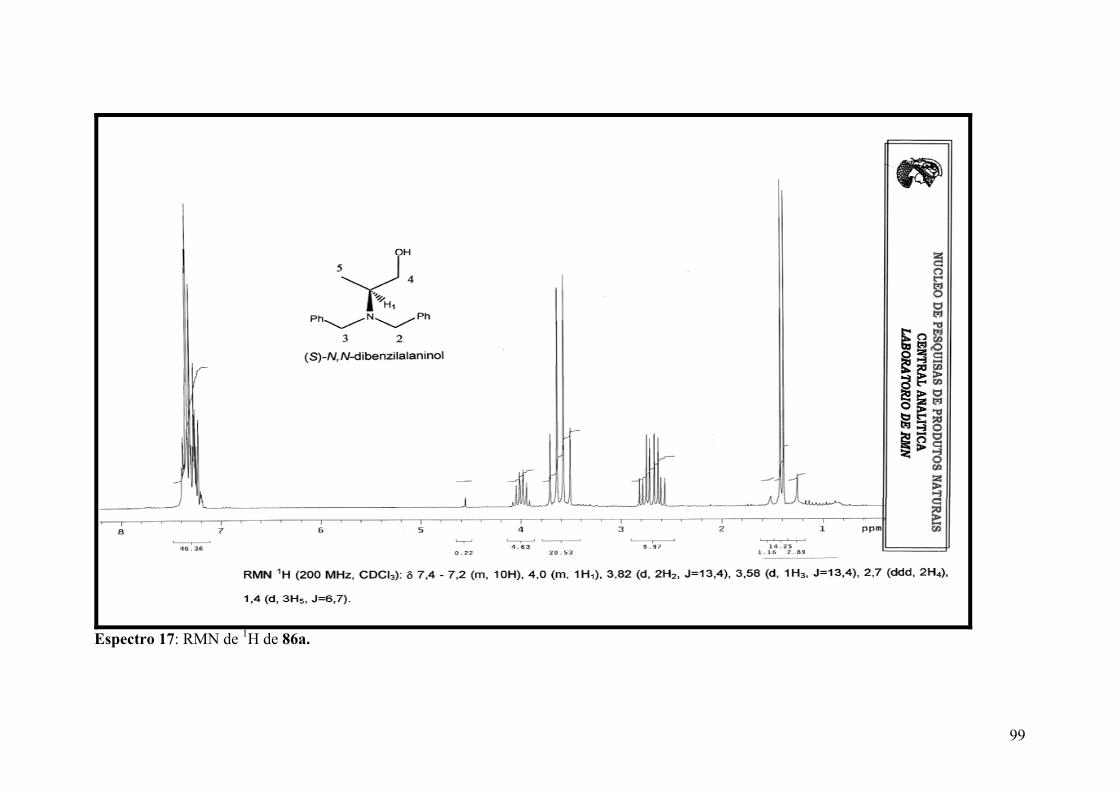
99
Espectro 17: RMN de 1H de 86a.
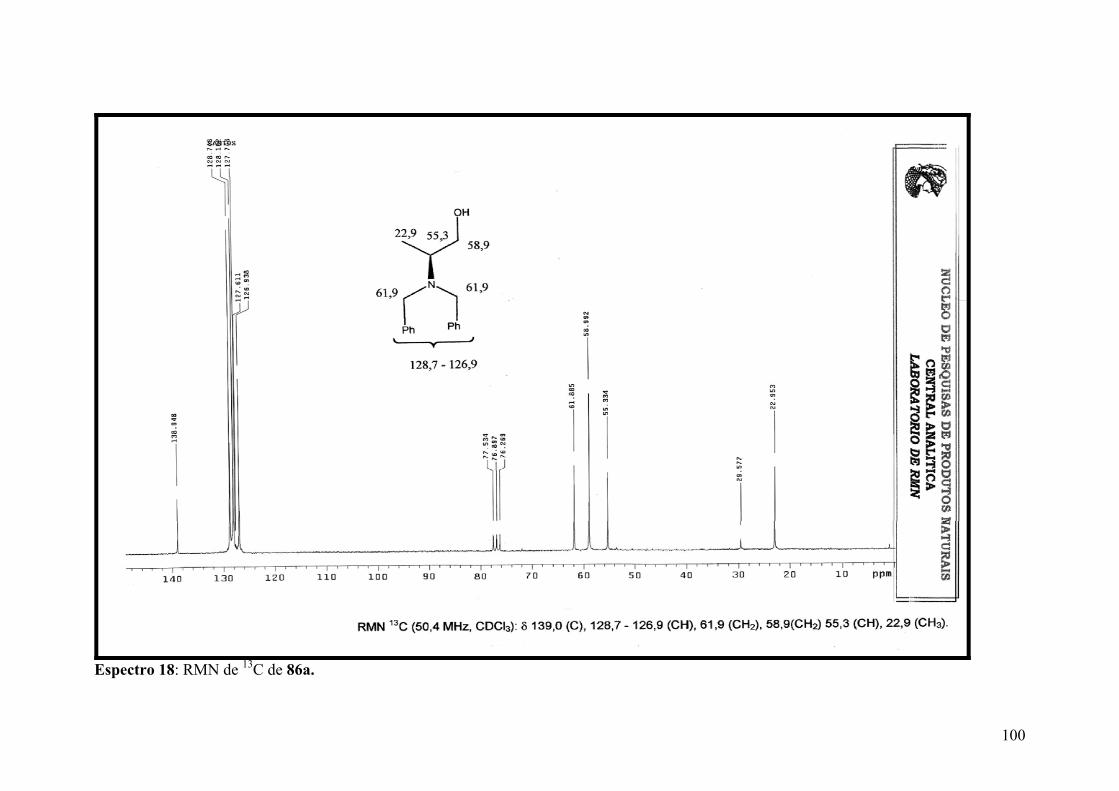
100
Espectro 18: RMN de 13C de 86a.
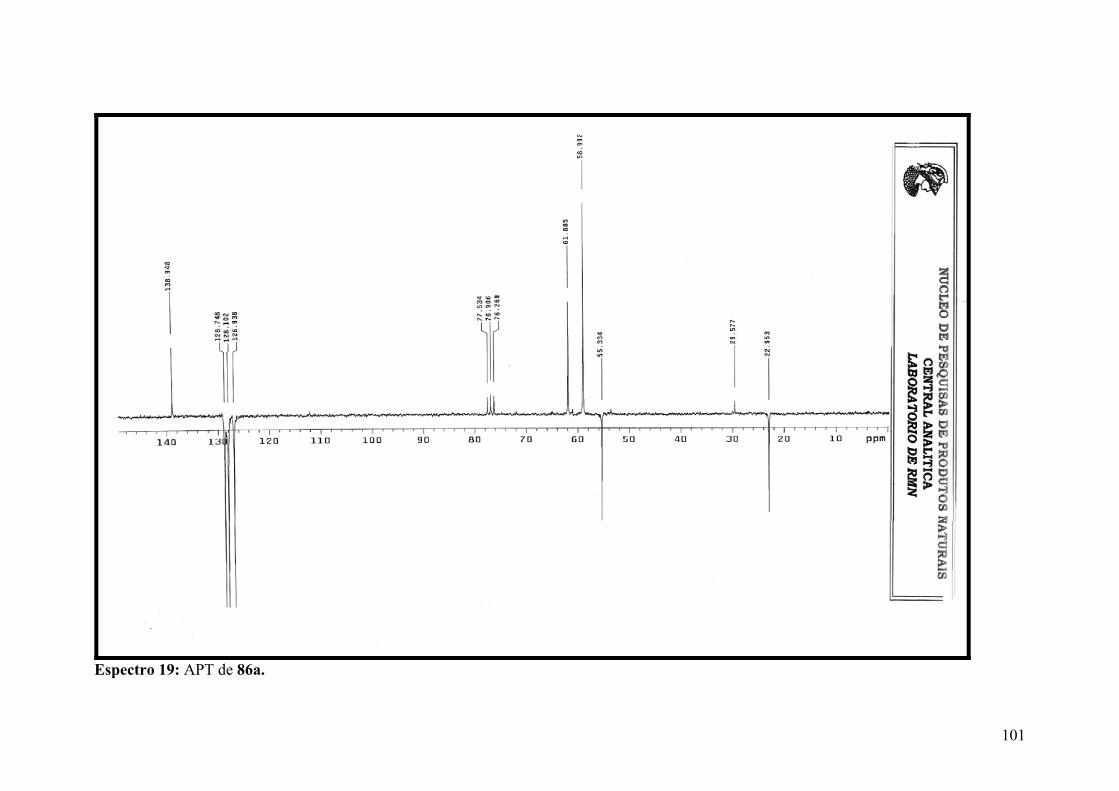
101
Espectro 19: APT de 86a.
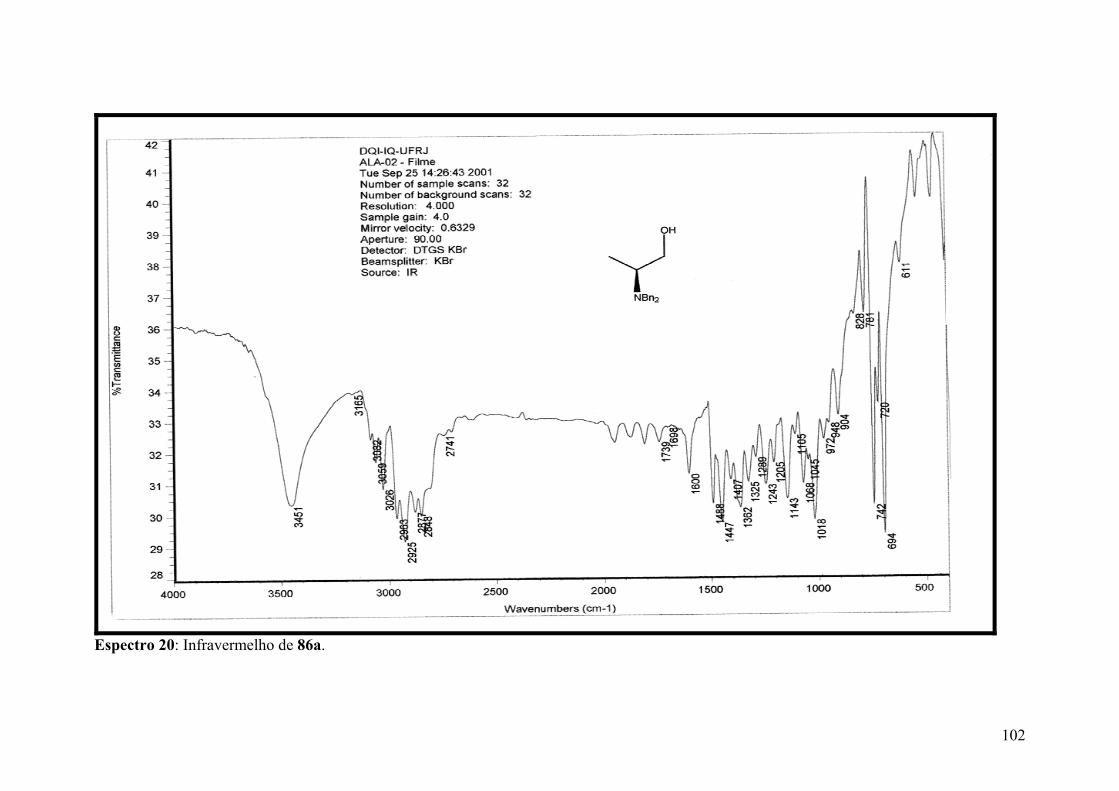
102
Espectro 20: Infravermelho de 86a.
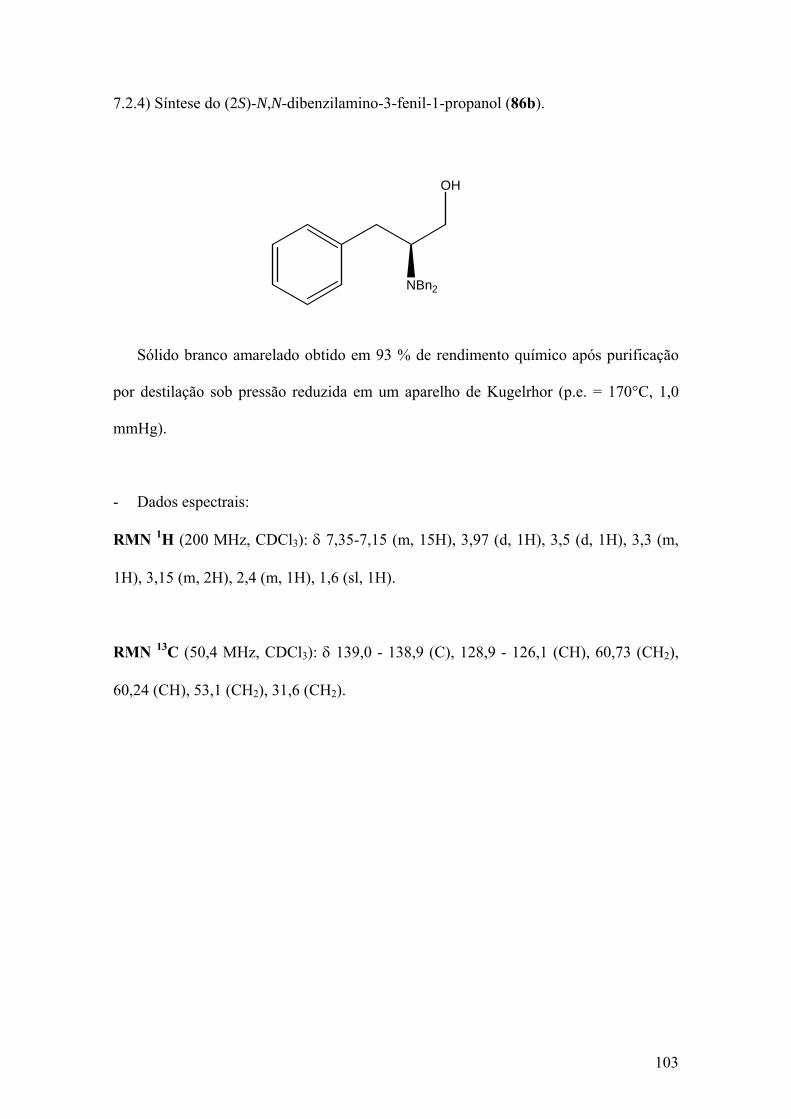
103
7.2.4) Síntese do (2S)-N,N-dibenzilamino-3-fenil-1-propanol (86b).
Sólido branco amarelado obtido em 93 % de rendimento químico após purificação
por destilação sob pressão reduzida em um aparelho de Kugelrhor (p.e. = 170°C, 1,0
mmHg).
- Dados espectrais:
RMN 1H (200 MHz, CDCl3): δ 7,35-7,15 (m, 15H), 3,97 (d, 1H), 3,5 (d, 1H), 3,3 (m,
1H), 3,15 (m, 2H), 2,4 (m, 1H), 1,6 (sl, 1H).
RMN 13C (50,4 MHz, CDCl3): δ 139,0 - 138,9 (C), 128,9 - 126,1 (CH), 60,73 (CH2),
60,24 (CH), 53,1 (CH2), 31,6 (CH2).
NBn2
OH
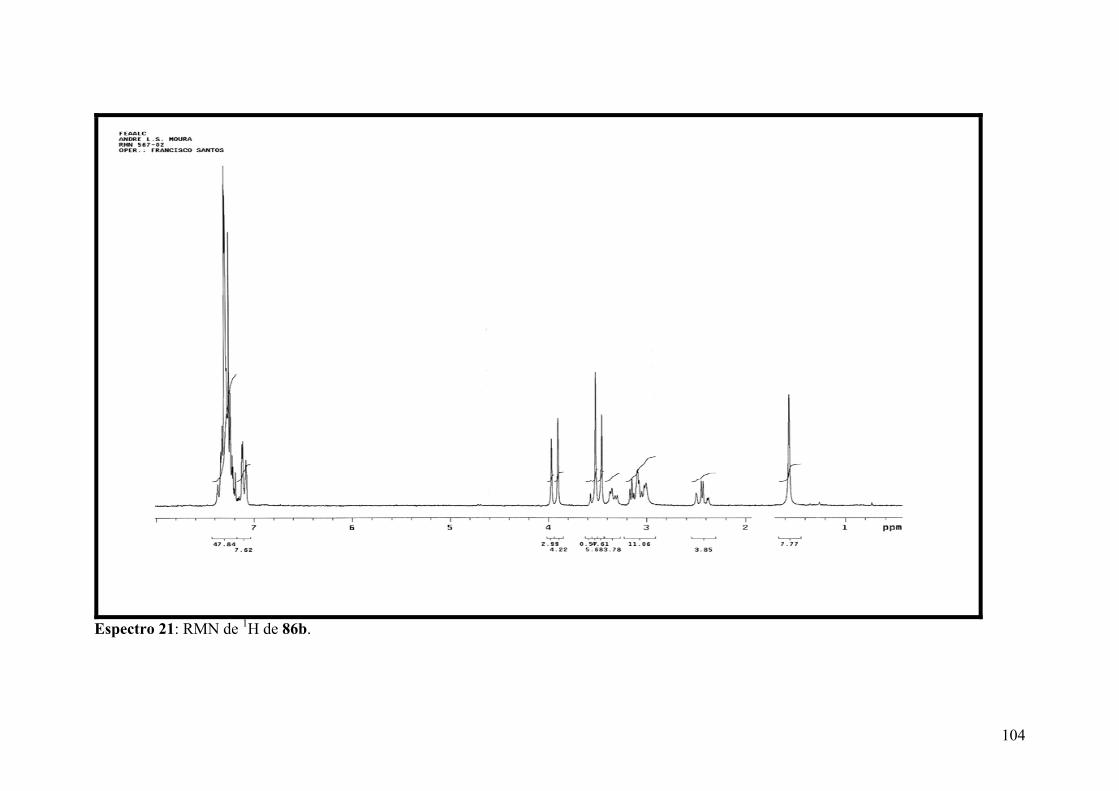
104
Espectro 21: RMN de 1H de 86b.
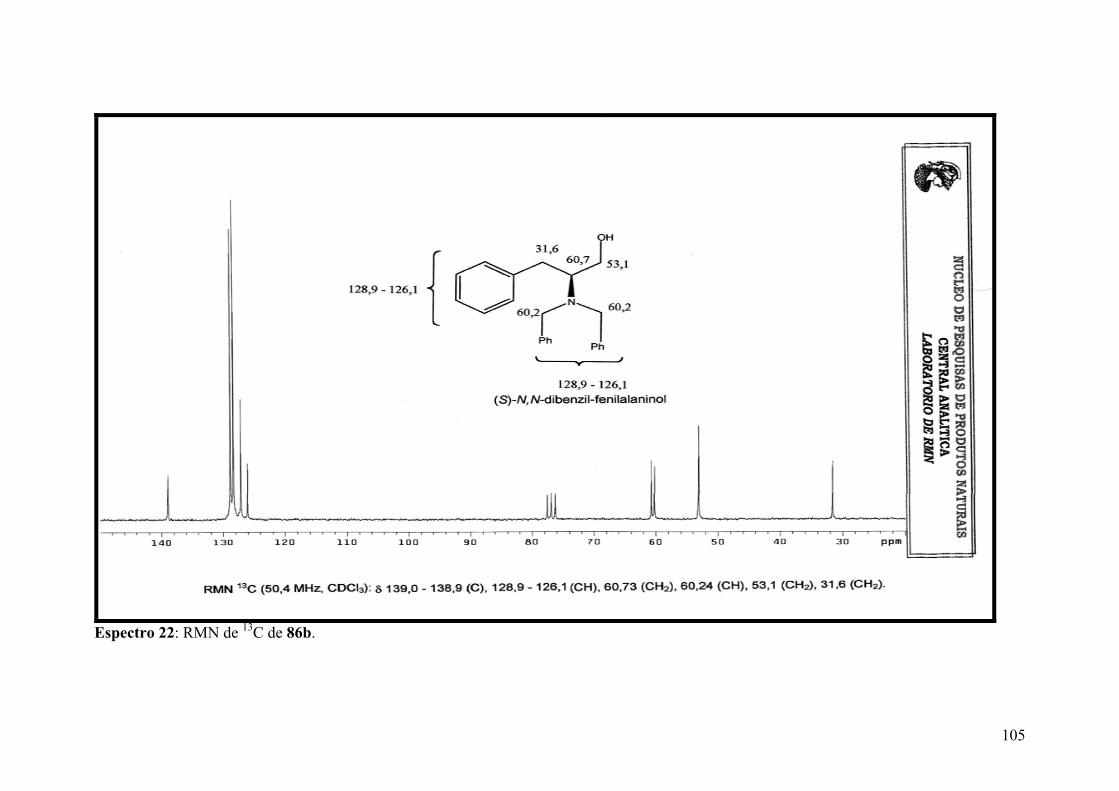
105
Espectro 22: RMN de 13C de 86b.
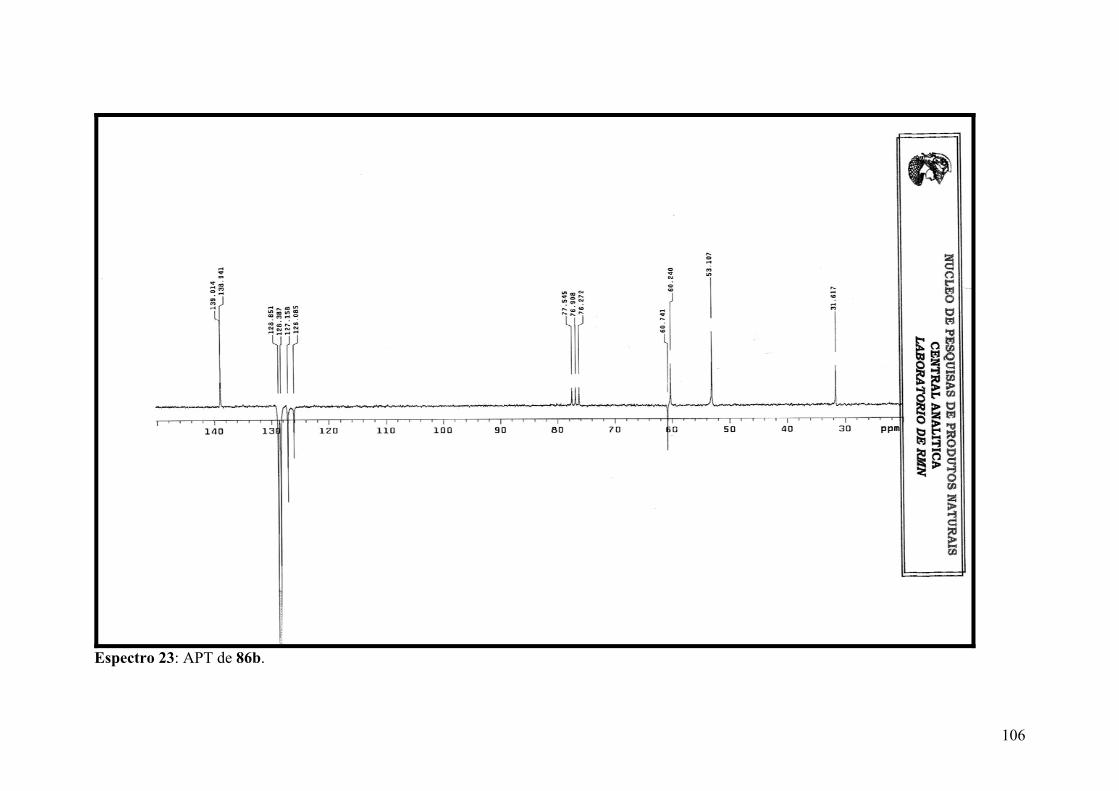
106
Espectro 23: APT de 86b.

107
7.3) Síntese dos aldeídos 87a-d:
Procedimento geral:
7.3.1) Síntese do (2S)-N,N-dibenzilamino-4-metilpentanal (87c).
A uma solução de 0,8 mL (9,4 mmoles) de cloreto de oxalila em 5,0 mL de
diclorometano seco, a – 78°C, foram adicionados 1,3 mL (18,8 mmoles) de DMSO
seco. Após 15 minutos sob agitação, a esta solução foram adicionados, gota à gota, 2,5 g
(8,4 mmoles) do aminoálcool 86c diluídos em 15,0 mL de diclorometano seco. A
solução foi agitada por 30 minutos a - 78°C. Findo este tempo, adicionou-se, à mistura
reacional, 6,0 mL de trietilamina seca e a solução foi lavada com água (1 x 15,0 mL) e
salmoura (1 x 10,0 mL). A fase orgânica foi seca sob sulfato de sódio e evaporada sob
pressão reduzida, fornecendo o desejado aminoaldeído - líquido amarelado - em
rendimento próximo ao quantitativo e utilizado posteriormente sem qualquer
purificação.
- Dados espectrais:
RMN 1H (200 MHz, CDCl3): δ 9,8 (s, 1H), 7,2-7,1 (m, 10H), 3,8 (d, 1H), 3,62 (d, 1H),
3,2 (m, 1H), 1,6 (m, 1H), 1,4 (t, 2H), 0,82 (d, 3H), 0,79 (d, 3H).
(dados referentes a um espectro de produto não purificado).
NBn2
O
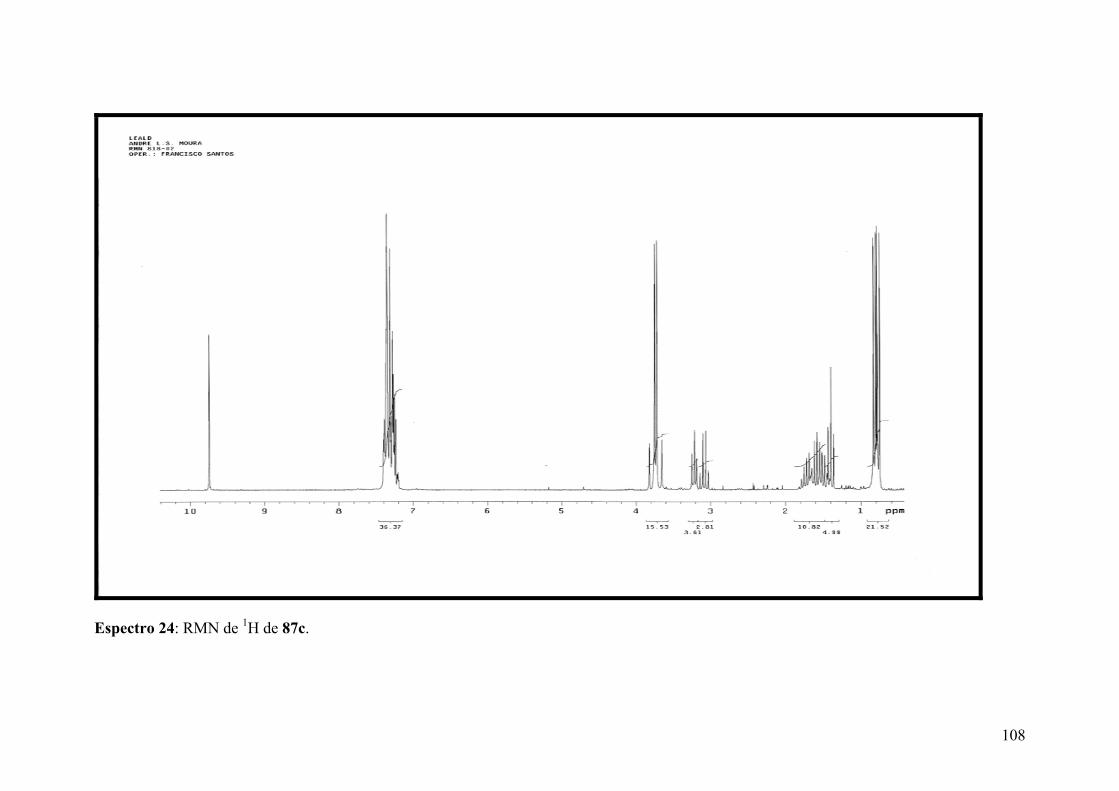
108
Espectro 24: RMN de 1H de 87c.
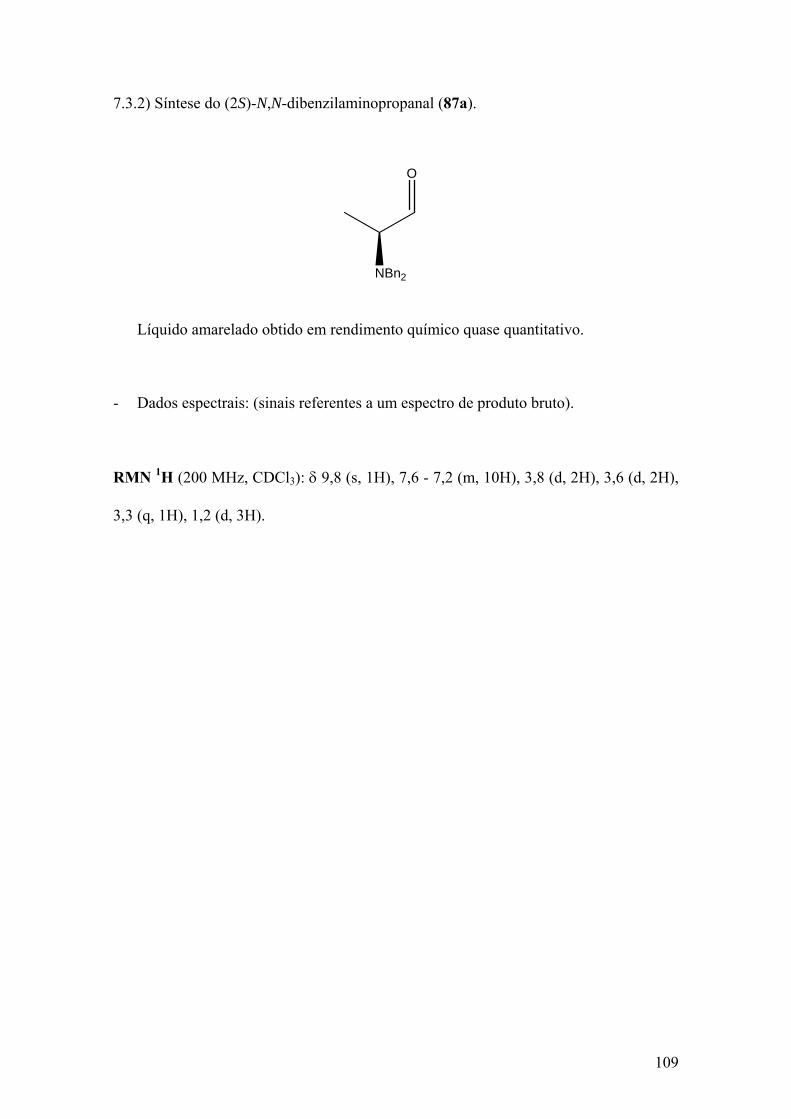
109
7.3.2) Síntese do (2S)-N,N-dibenzilaminopropanal (87a).
Líquido amarelado obtido em rendimento químico quase quantitativo.
- Dados espectrais: (sinais referentes a um espectro de produto bruto).
RMN 1H (200 MHz, CDCl3): δ 9,8 (s, 1H), 7,6 - 7,2 (m, 10H), 3,8 (d, 2H), 3,6 (d, 2H),
3,3 (q, 1H), 1,2 (d, 3H).
NBn2
O
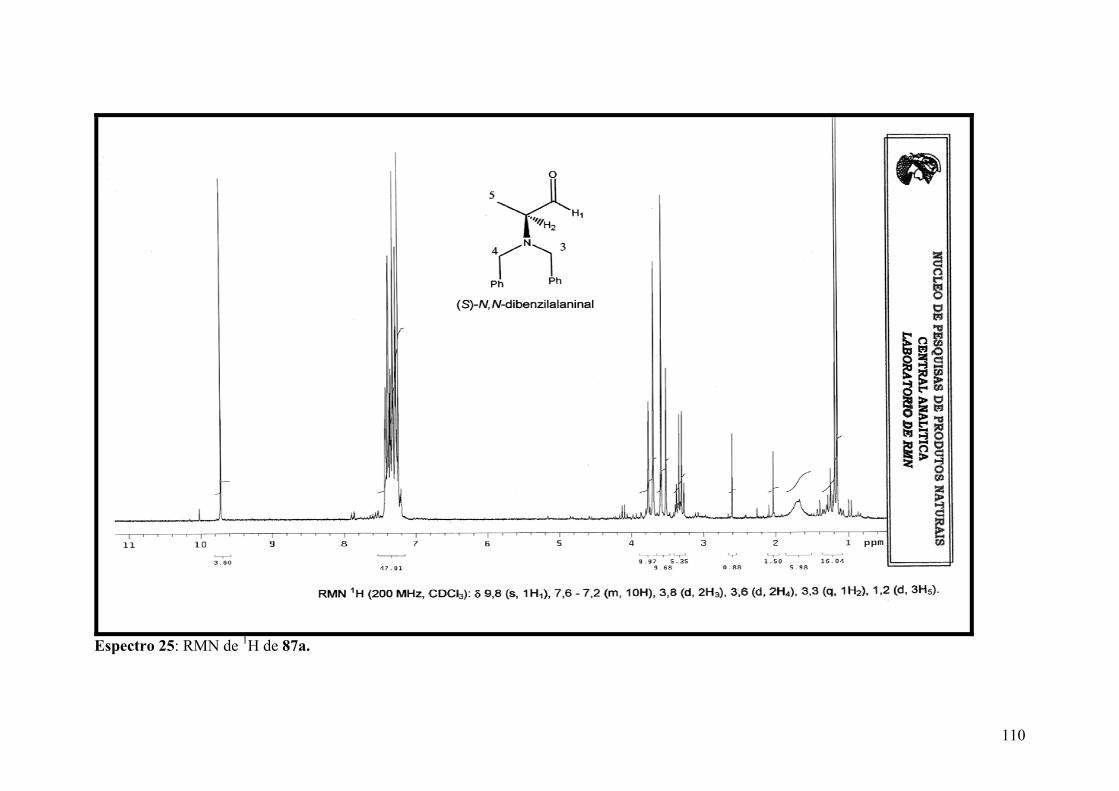
110
Espectro 25: RMN de 1H de 87a.

111
7.4) Síntese dos nitroálcoois 92a-d:
Procedimento geral:
7.4.1) Síntese do (2R,3S)-1-nitro-3-N,N-dibenzilamino-4-fenil-2-butanol (92b).
NBn2
NO2
OH
Foram pesados, em um balão de vidro com atmosfera inerte de N2, 0,1928 g de
TBAF (0,2 eq. ; 0,61 mmol) e, logo em seguida, foram adicionados 10,0 mL de THF
seco. Após a solubilização do TBAF, 1,0 g do aminoaldeído 87b (3,04 mmoles)
solubilizados em pequena quantidade de THF seco foram vertidos à solução de TBAF.
A mistura reacional permaneceu sob agitação magnética, à temperatura ambiente, por 1
hora. Depois de transcorrido o tempo reacional, a mistura reacional foi evaporada sob
pressão reduzida e o resíduo resultante foi purificado em uma coluna de gel de sílica,
tendo como eluente uma mistura de Hexano / AcOEt a 20%, fornecendo o desejado
nitroálcool em um rendimento químico de 91% como um líquido pouco viscoso de cor
amarelada.
- Razão diastereoisomérica de 93 : 7 (medida no CH em 54,6 ppm – veja Espectro 27).
- Dados espectrais:
IV (KBr): ν 3354, 3085-3004, 2928-2806, 1553, 1350, 1121, 1028, 745, 699 cm-1.
RMN 1H (200 MHz, CDCl3): δ 7,4-7,2 (m, 15H), 4,8 (dd, 1H, J=2,2, J=13,15), 4,46 (m,
1H, J=2,2), 4,0 (m, 1H), 3,8 (d, 1H), 3,5 (d, 1H),3,2 (m, 1H), 3,0 (m, 1H), 2,0 (s, 1H).
RMN 13C (50,4 MHz, CDCl3): δ 140,1-138,7 (C), 129,2-126,1 (CH-aromático), 79,8
(CH2), 70,6 (CH), 61,5 (CH), 54,6 (CH2), 32,3 (CH2).
Nota: Os dados abaixo referentes aos espectros de RMN 1H e RMN 13C de 92a-d são de misturas de diastereoisômeros.
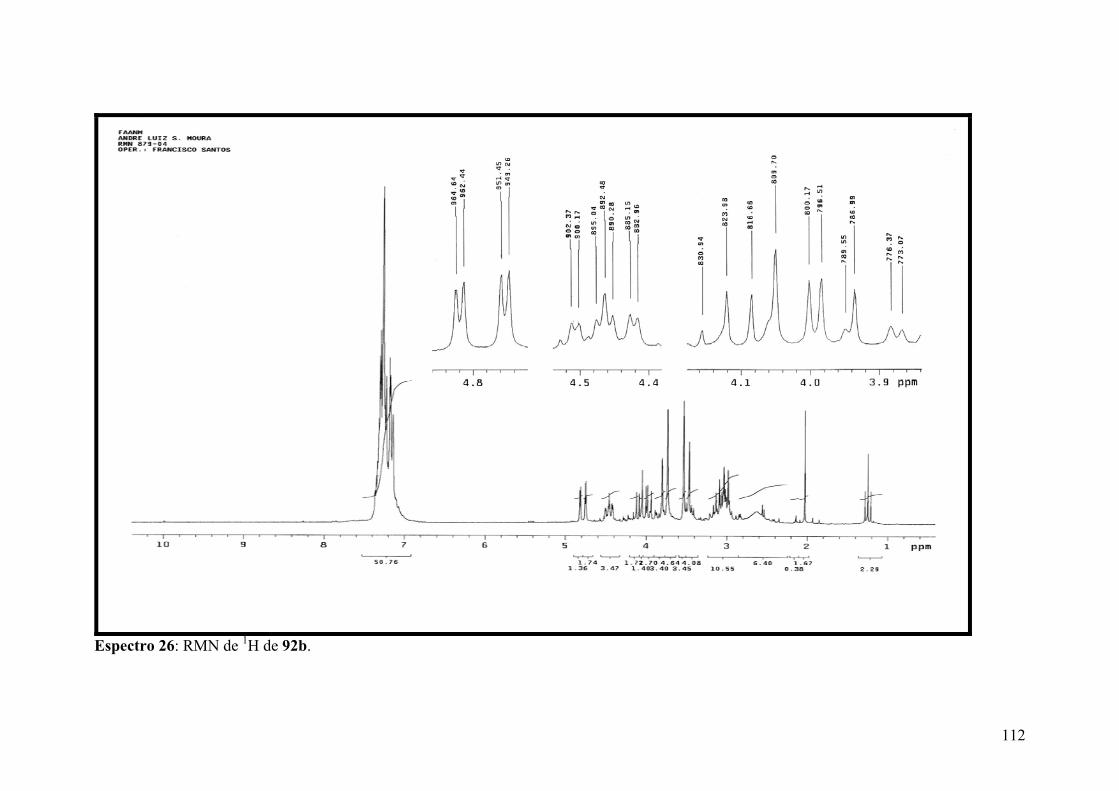
112
Espectro 26: RMN de 1H de 92b.
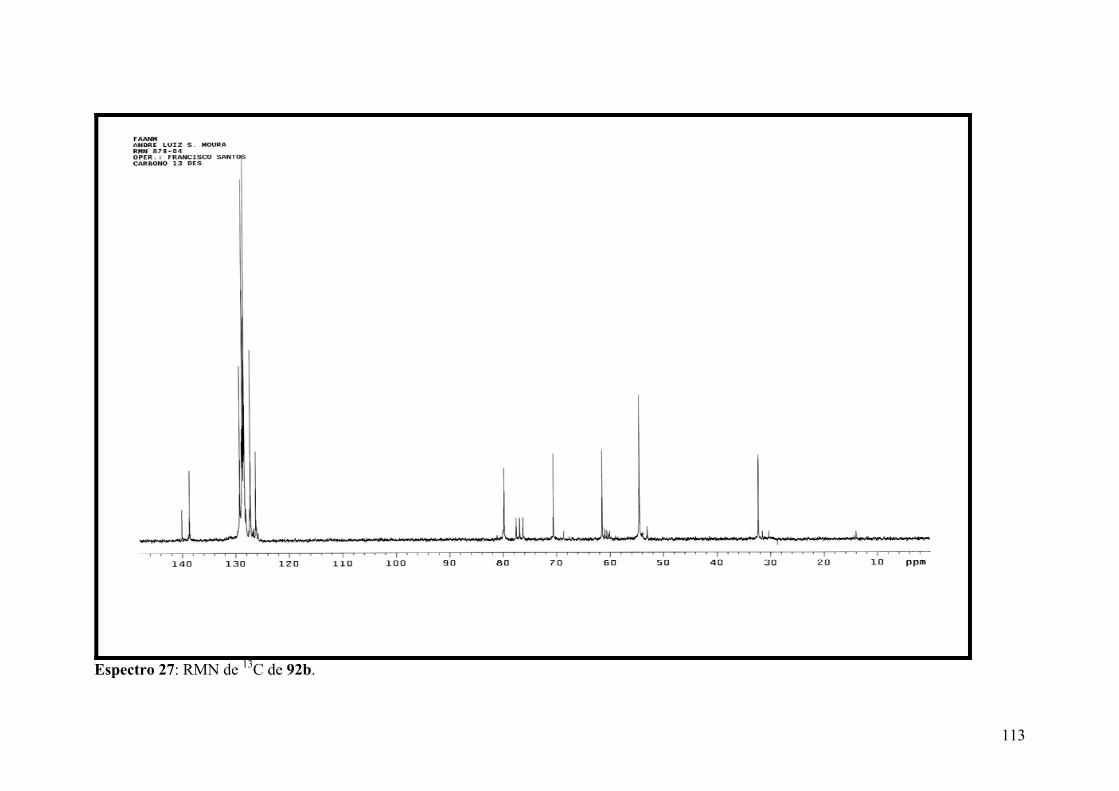
113
Espectro 27: RMN de 13C de 92b.
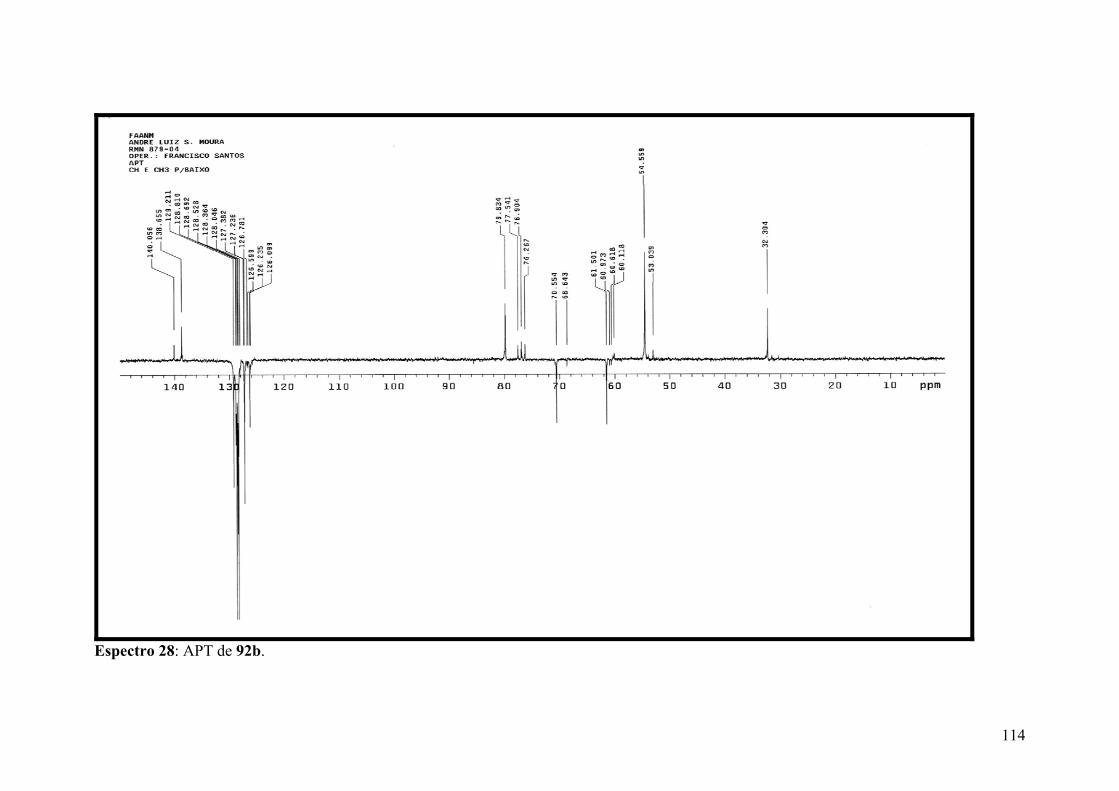
114
Espectro 28: APT de 92b.
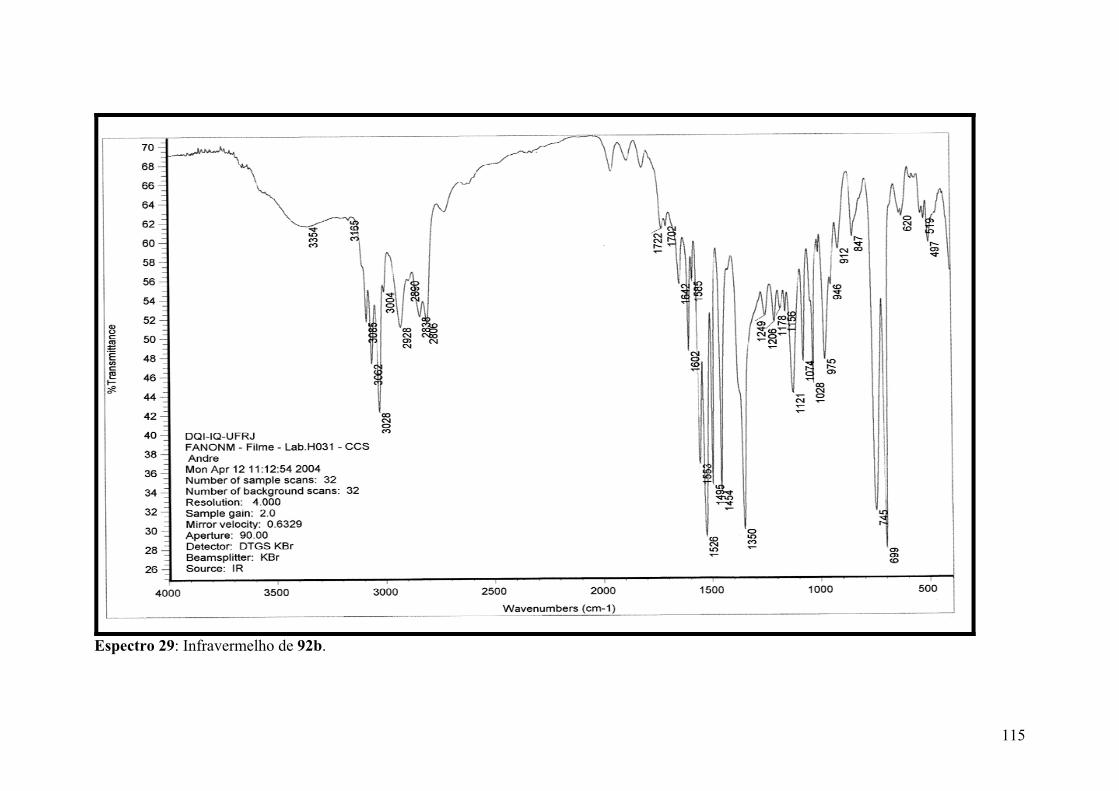
115
Espectro 29: Infravermelho de 92b.
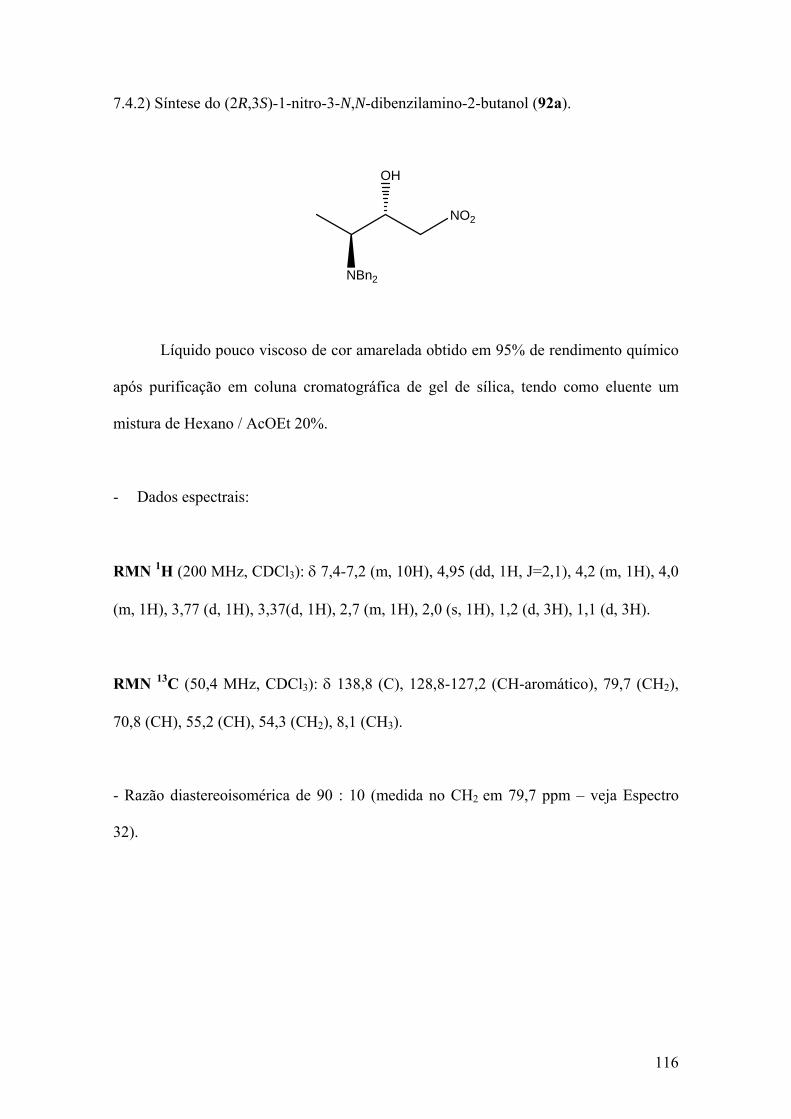
116
7.4.2) Síntese do (2R,3S)-1-nitro-3-N,N-dibenzilamino-2-butanol (92a).
NO2
NBn2
OH
Líquido pouco viscoso de cor amarelada obtido em 95% de rendimento químico
após purificação em coluna cromatográfica de gel de sílica, tendo como eluente um
mistura de Hexano / AcOEt 20%.
- Dados espectrais:
RMN 1H (200 MHz, CDCl3): δ 7,4-7,2 (m, 10H), 4,95 (dd, 1H, J=2,1), 4,2 (m, 1H), 4,0
(m, 1H), 3,77 (d, 1H), 3,37(d, 1H), 2,7 (m, 1H), 2,0 (s, 1H), 1,2 (d, 3H), 1,1 (d, 3H).
RMN 13C (50,4 MHz, CDCl3): δ 138,8 (C), 128,8-127,2 (CH-aromático), 79,7 (CH2),
70,8 (CH), 55,2 (CH), 54,3 (CH2), 8,1 (CH3).
- Razão diastereoisomérica de 90 : 10 (medida no CH2 em 79,7 ppm – veja Espectro
32).
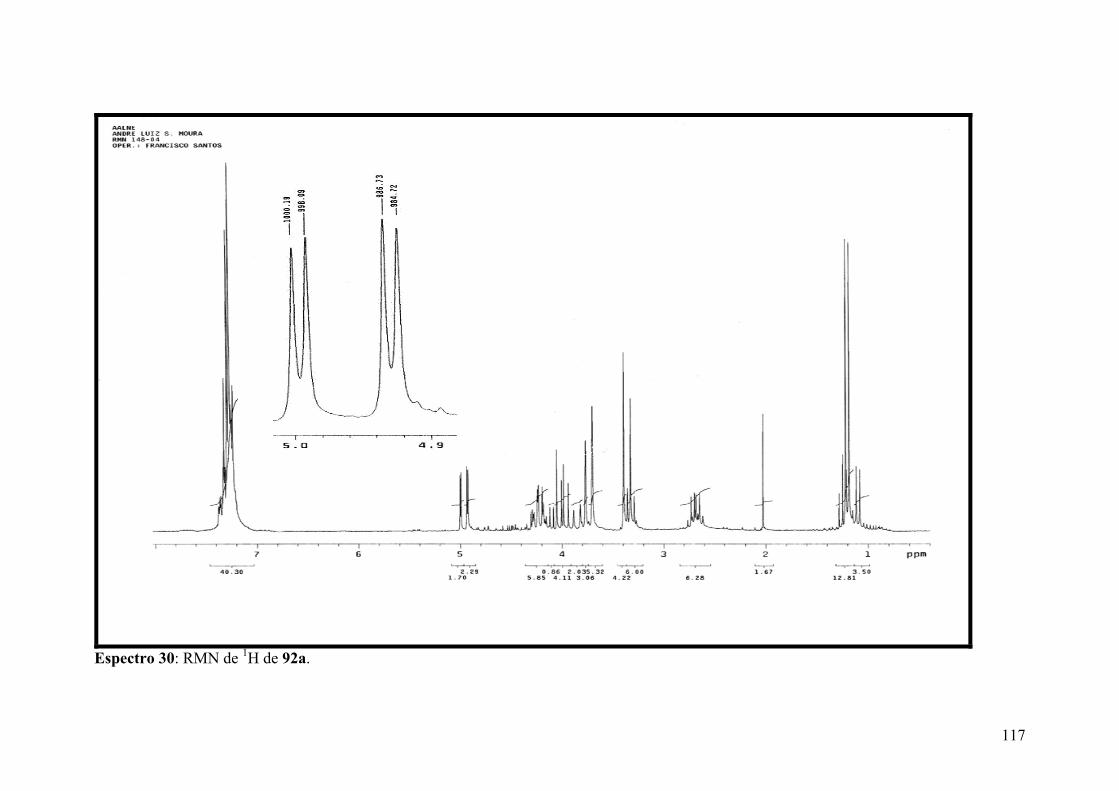
117
Espectro 30: RMN de 1H de 92a.
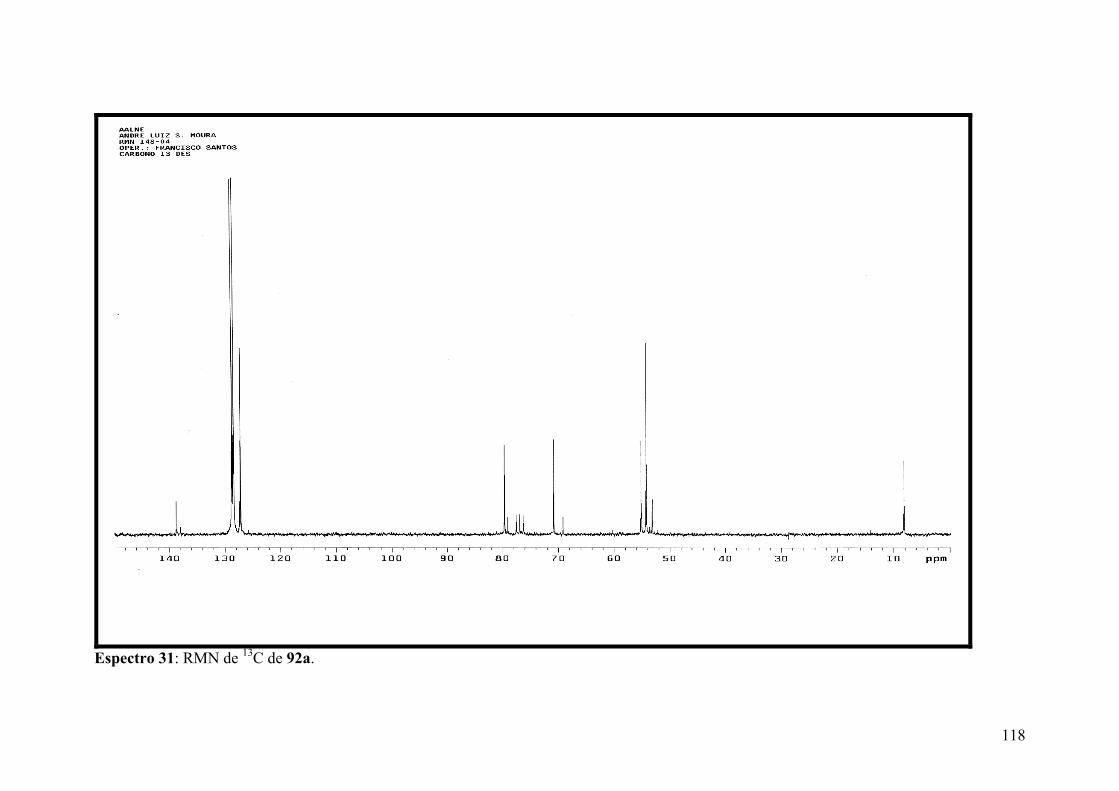
118
Espectro 31: RMN de 13C de 92a.

119
Espectro 32: APT de 92a.

120
7.4.3) Síntese do (3R,4S)-2-nitro-4-N,N-dibenzilamino-3-pentanol (92c).
NBn2
OH
NO2∗
Líquido pouco viscoso de cor amarelada obtido em 84% de rendimento químico
após purificação em coluna cromatográfica de gel de sílica, tendo como eluente um
mistura de Hexano / AcOEt 20%.
- Dados espectrais:
RMN 1H (200 MHz, CDCl3): δ 7,4-7,2 (m, 10H), 5,16 (dq, 1H, J=1,4), 4,2 (m, 1H),
3,77 (d, 1H), 3,3 (d, 1H), 2,6 (m, 1H), 2,0 (s, 1H), 1,2 (d, 3H), 0,8 (d, 3H).
RMN 13C (50,4 MHz, CDCl3): δ 139,1-138,1 (C), 129,1-127,2 (CH-aromático), 84,0
(CH), 73,6 (CH), 54,0 (CH2), 53,3 (CH), 9,9 (CH3), 8,3 (CH3).
- Razão diastereoisomérica de 90 : 10 (medida no CH em 84,0 ppm – veja Espectro 35).
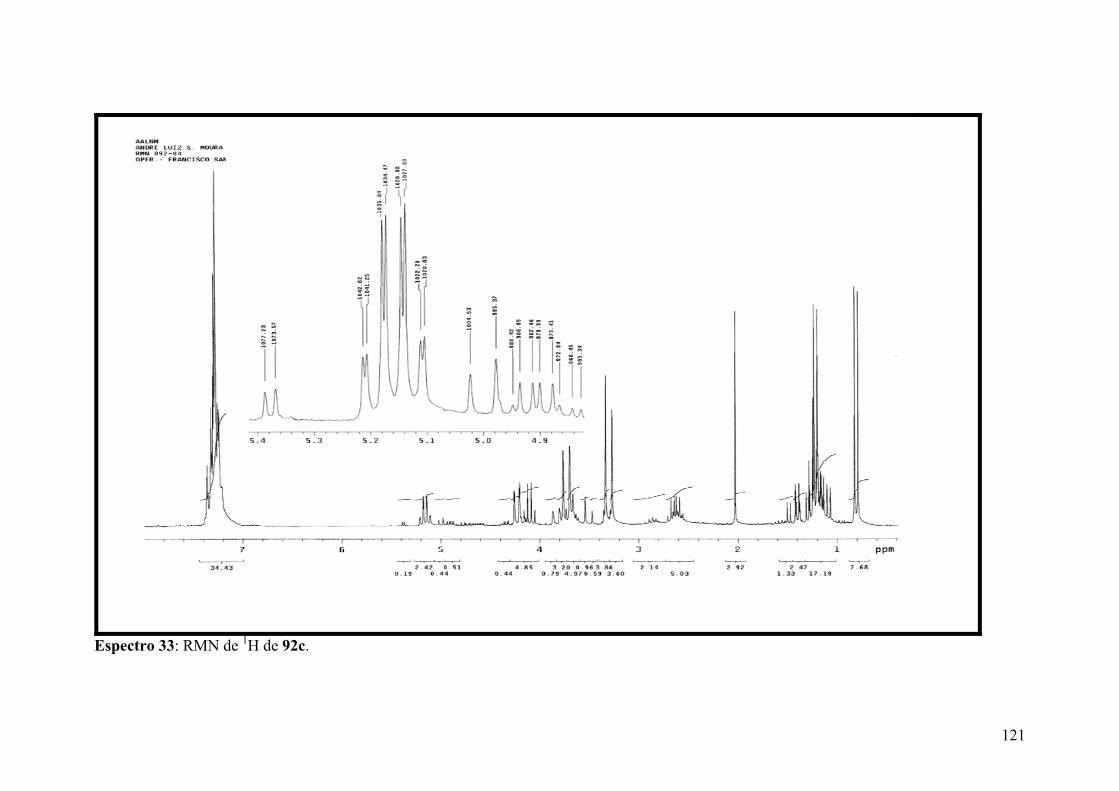
121
Espectro 33: RMN de 1H de 92c.
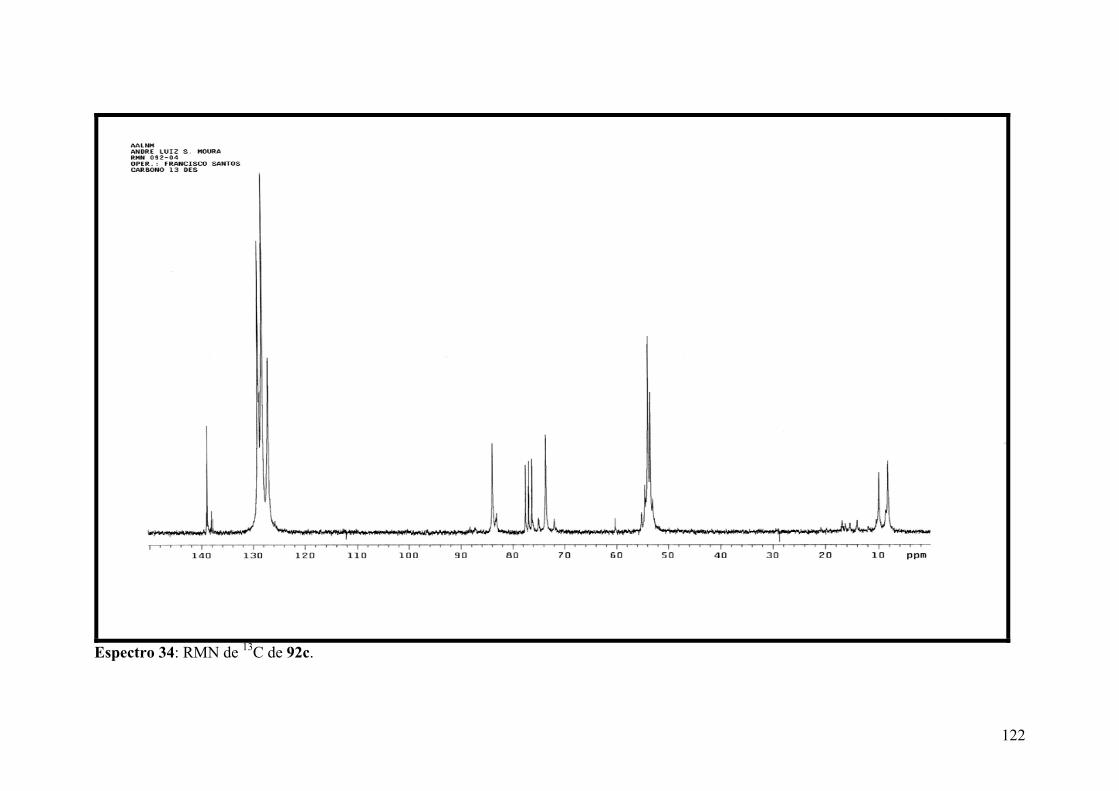
122
Espectro 34: RMN de 13C de 92c.
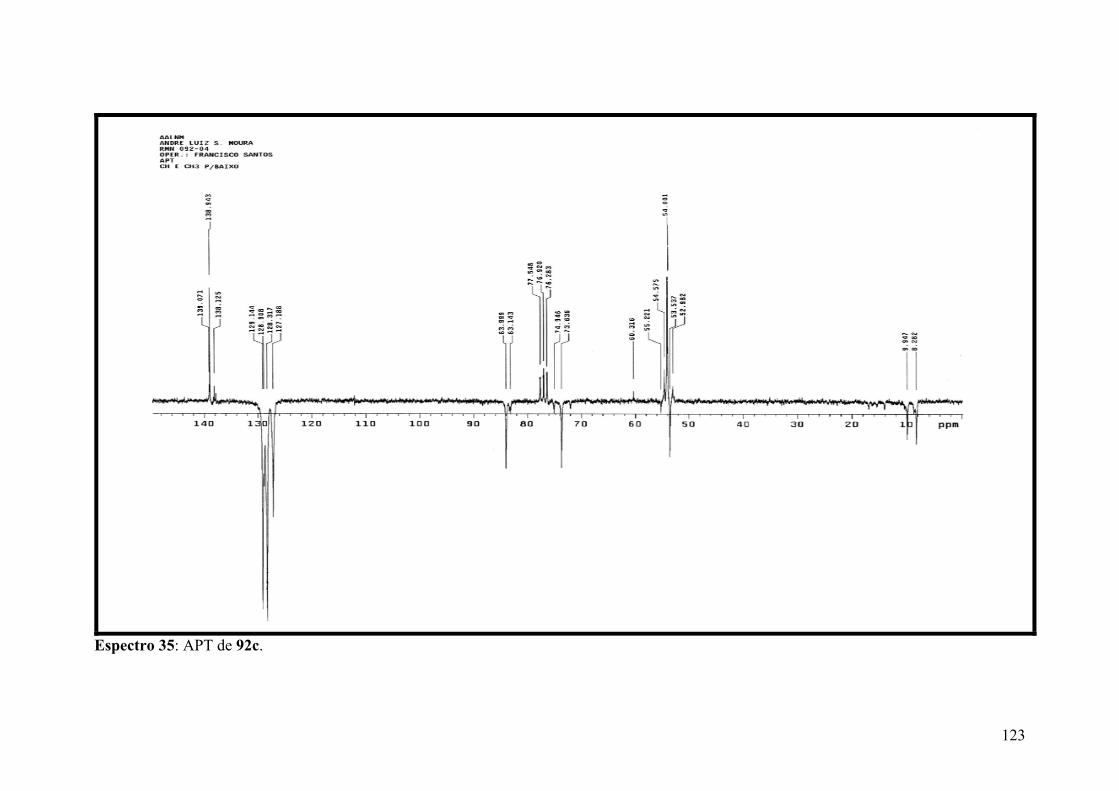
123
Espectro 35: APT de 92c.
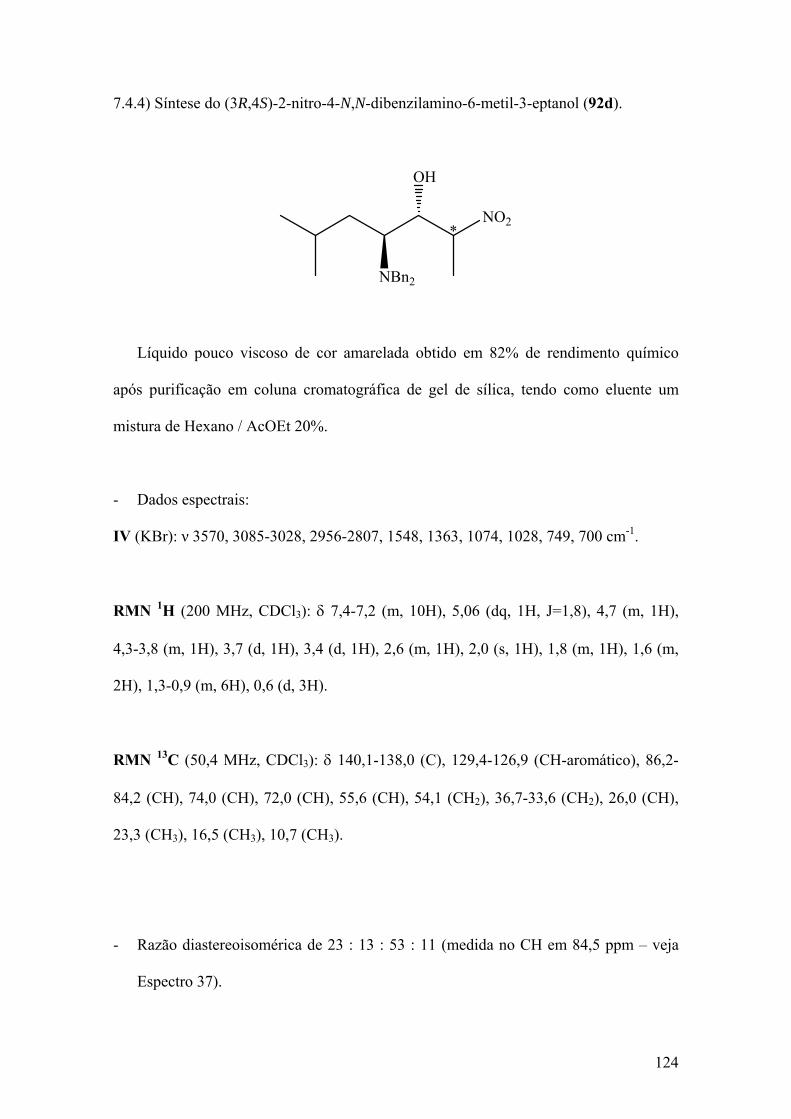
124
7.4.4) Síntese do (3R,4S)-2-nitro-4-N,N-dibenzilamino-6-metil-3-eptanol (92d).
*NO2
NBn2
OH
Líquido pouco viscoso de cor amarelada obtido em 82% de rendimento químico
após purificação em coluna cromatográfica de gel de sílica, tendo como eluente um
mistura de Hexano / AcOEt 20%.
- Dados espectrais:
IV (KBr): ν 3570, 3085-3028, 2956-2807, 1548, 1363, 1074, 1028, 749, 700 cm-1.
RMN 1H (200 MHz, CDCl3): δ 7,4-7,2 (m, 10H), 5,06 (dq, 1H, J=1,8), 4,7 (m, 1H),
4,3-3,8 (m, 1H), 3,7 (d, 1H), 3,4 (d, 1H), 2,6 (m, 1H), 2,0 (s, 1H), 1,8 (m, 1H), 1,6 (m,
2H), 1,3-0,9 (m, 6H), 0,6 (d, 3H).
RMN 13C (50,4 MHz, CDCl3): δ 140,1-138,0 (C), 129,4-126,9 (CH-aromático), 86,2-
84,2 (CH), 74,0 (CH), 72,0 (CH), 55,6 (CH), 54,1 (CH2), 36,7-33,6 (CH2), 26,0 (CH),
23,3 (CH3), 16,5 (CH3), 10,7 (CH3).
- Razão diastereoisomérica de 23 : 13 : 53 : 11 (medida no CH em 84,5 ppm – veja
Espectro 37).
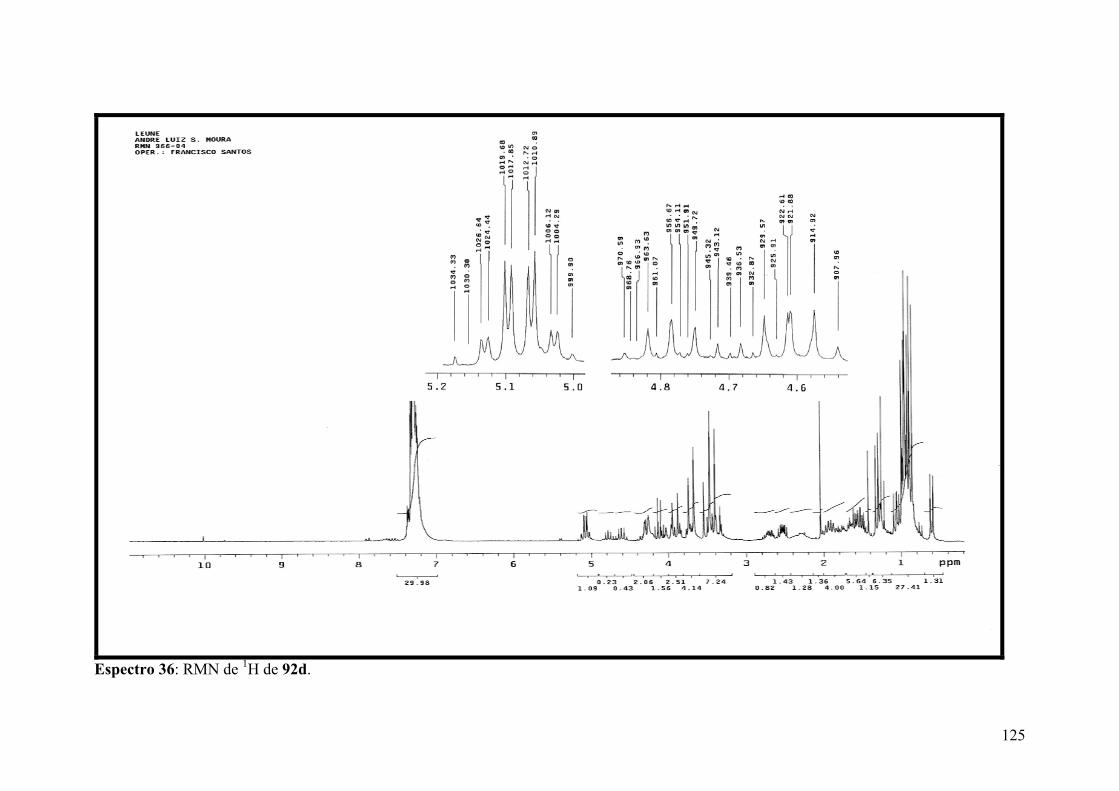
125
Espectro 36: RMN de 1H de 92d.
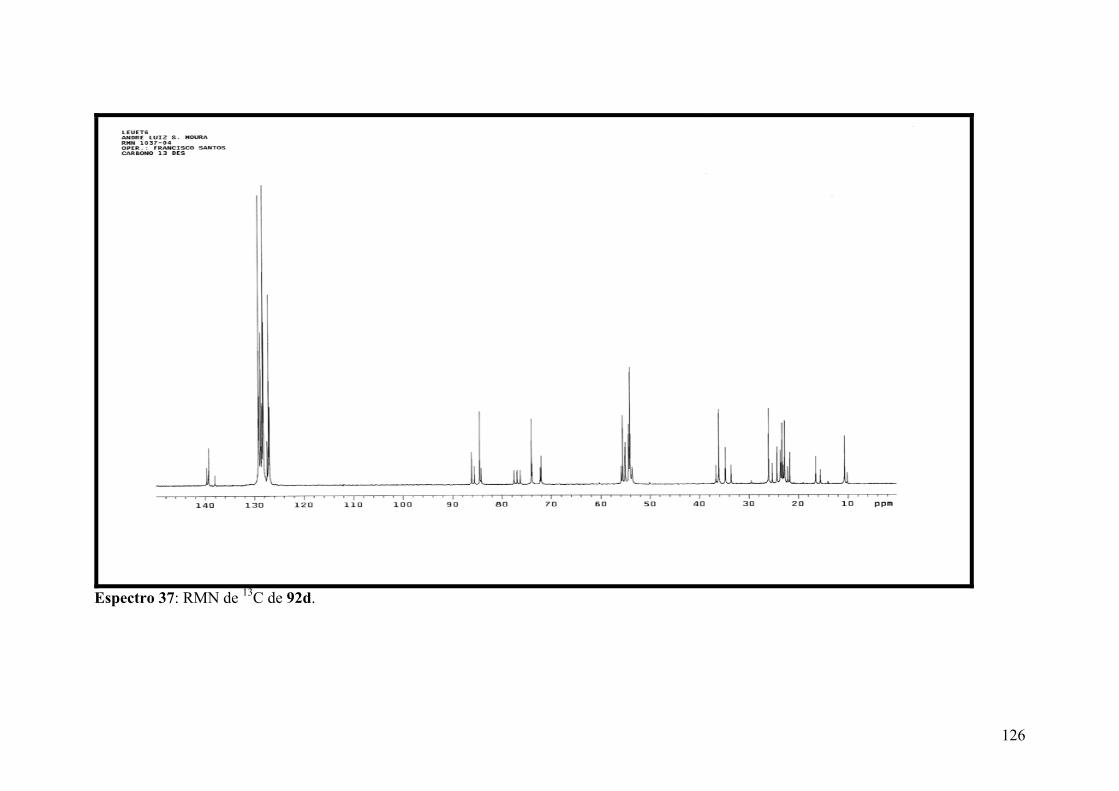
126
Espectro 37: RMN de 13C de 92d.
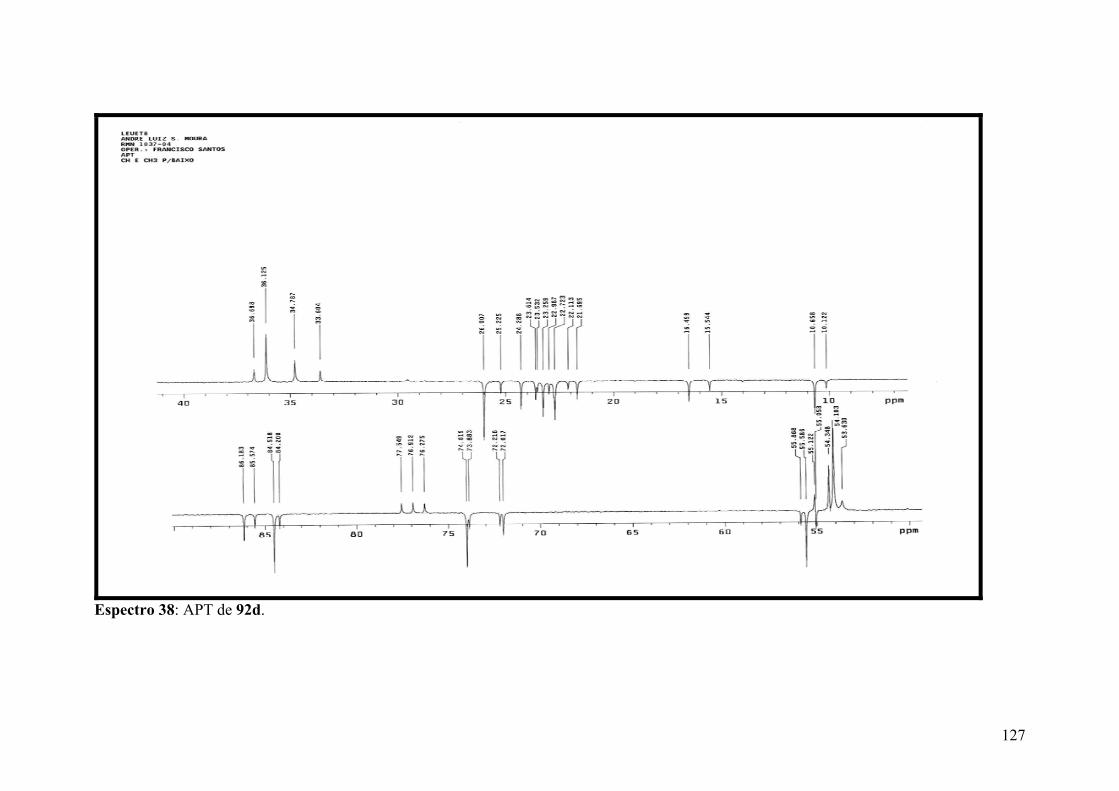
127
Espectro 38: APT de 92d.
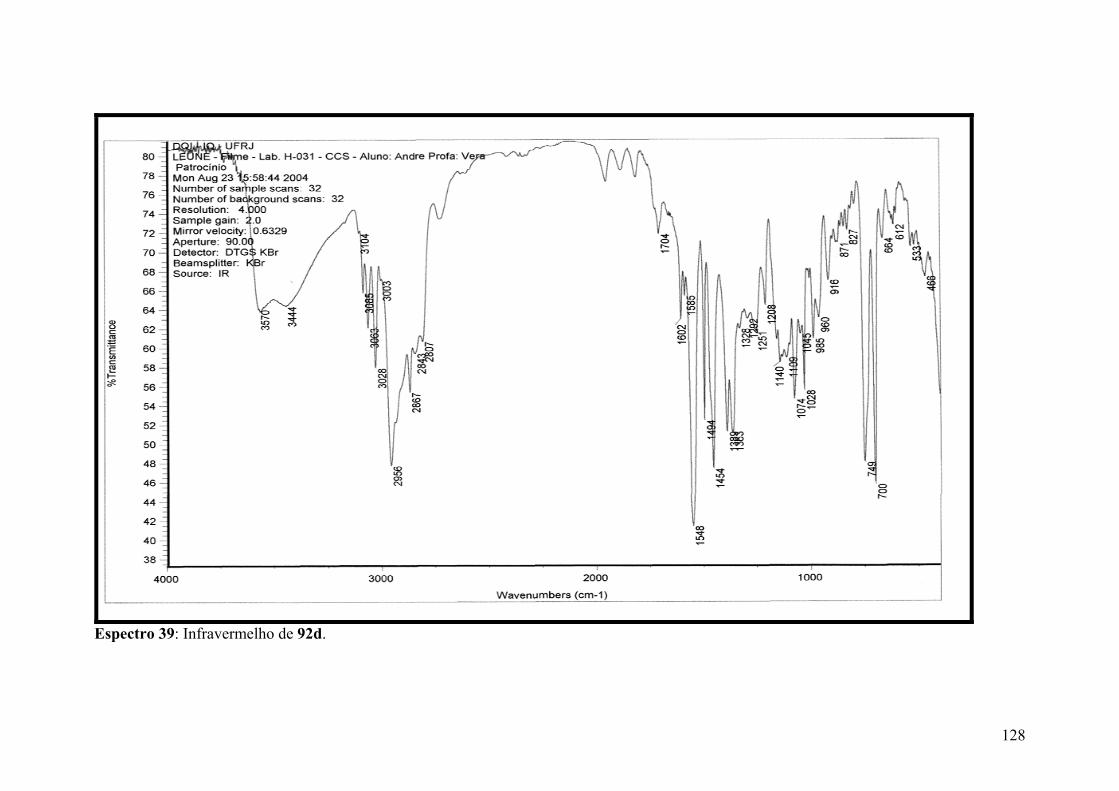
128
Espectro 39: Infravermelho de 92d.

129
7.5) Síntese das iminas 88a-d:
Procedimento geral:
7.5.1) Síntese da (2S)-N,N-dibenzil-4-metil-1-N-benzilpentanaldimina (88c).
Adicionou-se 0,8 ml de benzilamina (7,5 mmoles) a 2,2 g do aminoaldeído 87c
(7,5 mmoles), solubilizados em 5,0 mL de diclorometano seco em 1,4 g de Na2SO4
anidro. O meio reacional permaneceu sob agitação por três horas. Isolou-se, então, o
produto por uma filtração simples. A imina bruta foi purificada em coluna
cromatográfica de gel de sílica eluída com hexano / acetato de etila 20%, fornecendo
2,7 g da imina desejada, como um líquido pouco viscoso de cor amarelada, em 93% de
rendimento químico.
- Dados espectrais:
RMN 1H (200MHz, CDCl3): δ 7,88 (m, 1H), 7,39 - 7,19 (m, 15H), 4,60 (d, 2H, J=2,75),
3,80 (d, 2H, J=13,73), 3,63 (d, 2H, J=13,74), 3,42 - 3,30 (m, 1H), 1,88- 1,46 (m, 3H),
0,80 (d, 3H, J=6,51), 0,70 (d, 3H, J=6,50).
NBn2
NBn
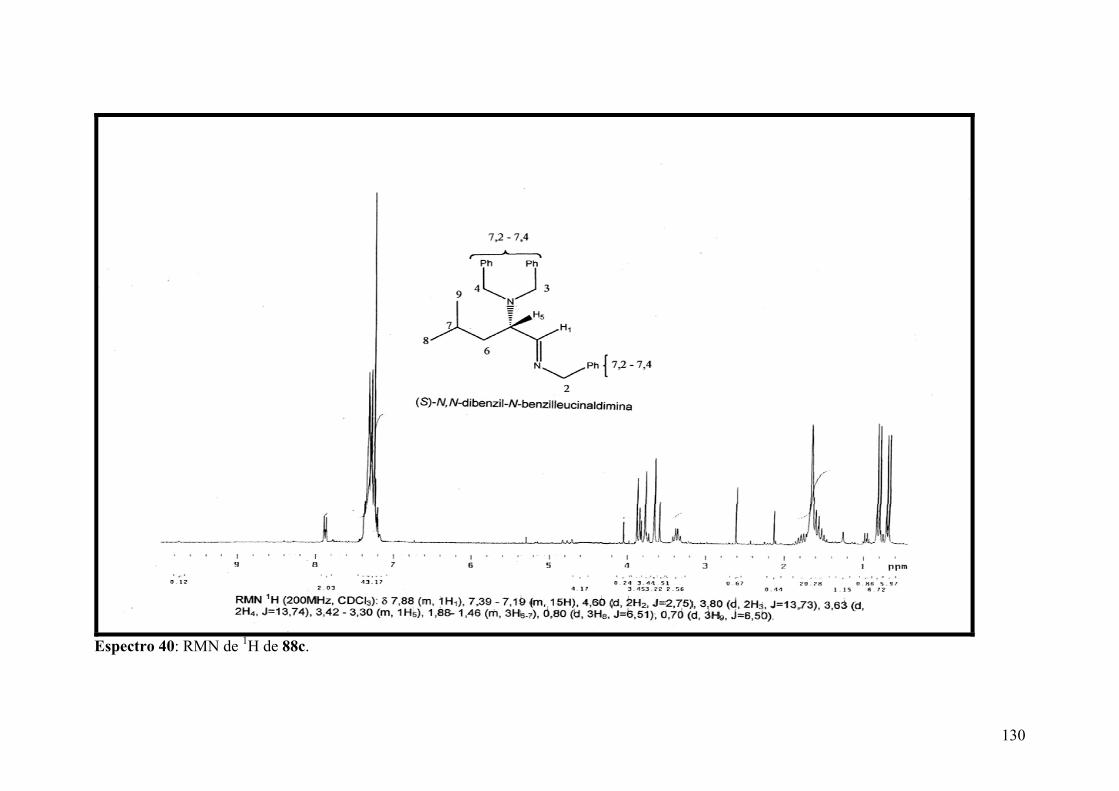
130
Espectro 40: RMN de 1H de 88c.

131
7.5.2) Síntese da (2S)-N,N-dibenzil-3-fenil-1-N-benzilpropanaldimina (88b).
NBn2
NBn
NBn2
NBn
Líquido pouco viscoso de coloração amarelo-acastanhada obtido em 95% de
rendimento químico após isolamento e rápida purificação em coluna cromatográfica de
gel de sílica, tendo como eluente uma mistura de hexano / acetato de etila 20%.
- Dados espectrais:
RMN 1H (200 MHz, CDCl3): δ 7,9 (m, 1H), 7,6-7,0 (m, 20H), 3,9 (d, 1H), 3,7 (m, 2H),
3,5 (d, 1H), 3,05 (m, 1H), 1,3 (m, 2H).
RMN 13C (50,4 MHz, CDCl3): δ 172,0 (C), 139,5-136,0 (C), 129,7-126,0 (CH-
aromático), 65,8 (CH2), 62,1 (CH), 54,2 (CH2), 35,4 (CH2).
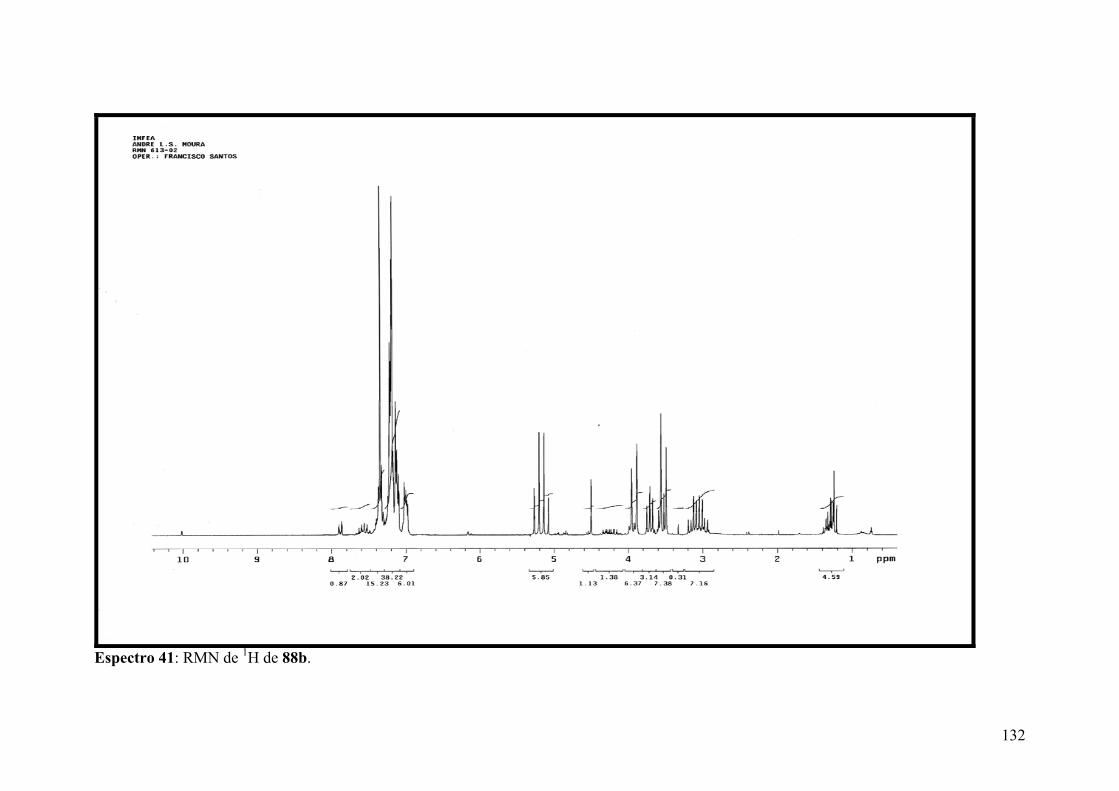
132
Espectro 41: RMN de 1H de 88b.
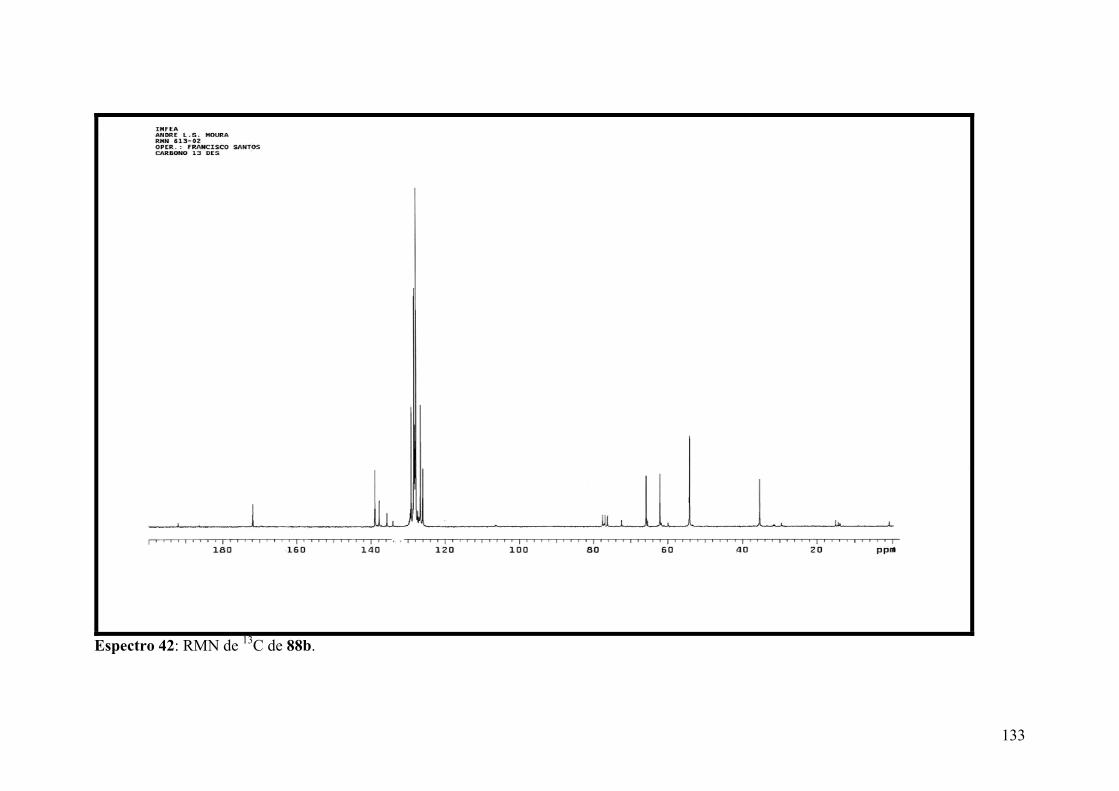
133
Espectro 42: RMN de 13C de 88b.
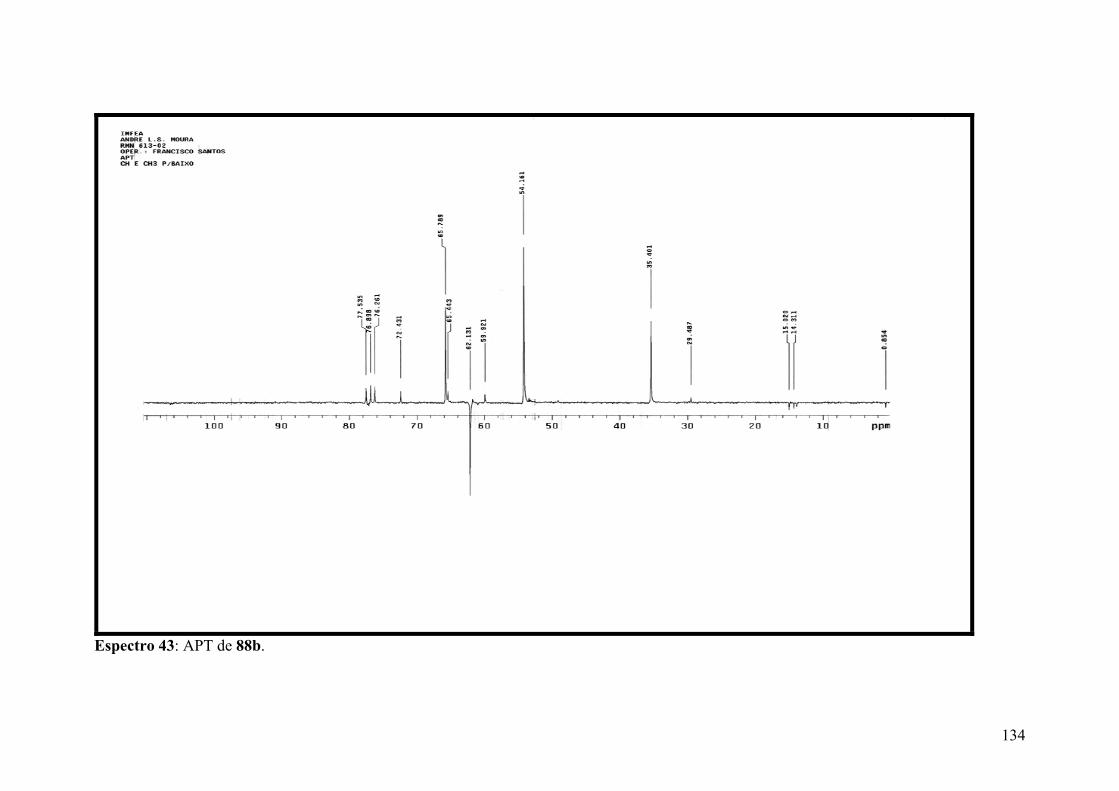
134
Espectro 43: APT de 88b.

135
7.6) Síntese das nitroolefinas 93a,b.
Procedimento geral:
7.6.1) Síntese do (1E,3S)-3-N,N-dibenzilamino-1-nitrobuteno (93a).
NBn2
NO2
O nitroálcool 92a (0,8g; 3,14 mmoles) foi dissolvido em 5,0 mL de
diclorometano a uma temperatura de -78 0C sob atmosfera de argônio. Em seguida, 0,24
mL de cloreto de mesila (3,14 mmoles) foi adicionado à solução. Após 5 minutos, sob
agitação magnética, 1,75 mL de trietilamina (4 eq. ; 12,6 mmoles) foi adicionado ao
meio reacional gota à gota por um período de 15 minutos. A mistura reacional
permaneceu sob agitação magnética por uma hora. Após esse tempo, a mistura reacional
foi transferida para um funil de separação onde foram adicionados 20,0 mL de
diclorometano, e então, lavada com água, HCl 5% e salmoura. Obteve-se um líquido
amarelo, que, após purificação em coluna de gel de sílica, tendo como eluente uma
mistura de hexano / AcOEt 4:1, forneceu o produto em 95% de rendimento químico.
- Dados espectrais:
IV (KBr): ν 3085-3029, 2973-2807, 1644, 1602, 1525, 1351, 1028, 747, 699 cm-1.
RMN 1H (200 MHz, CDCl3): δ 7,6-7,2 (m, 10H), 7,0 (dd, 2H, J= 13,55, J=1,46), 3,6
(m, 5H), 1,3 (d, 3H).
RMN 13C (50,4 MHz, CDCl3): δ 144,0 –140,0 (CH-olefina), 129,5-127,0 (CH-
aromático), 54,0 (CH2), 51,8 (CH), 13,9 (CH3).
CG/EM (70 eV): único pico em 31,3min; m/z: 296 (íon molecular), 281 (perda de
metila), 91 (pico base – íon tropílio).
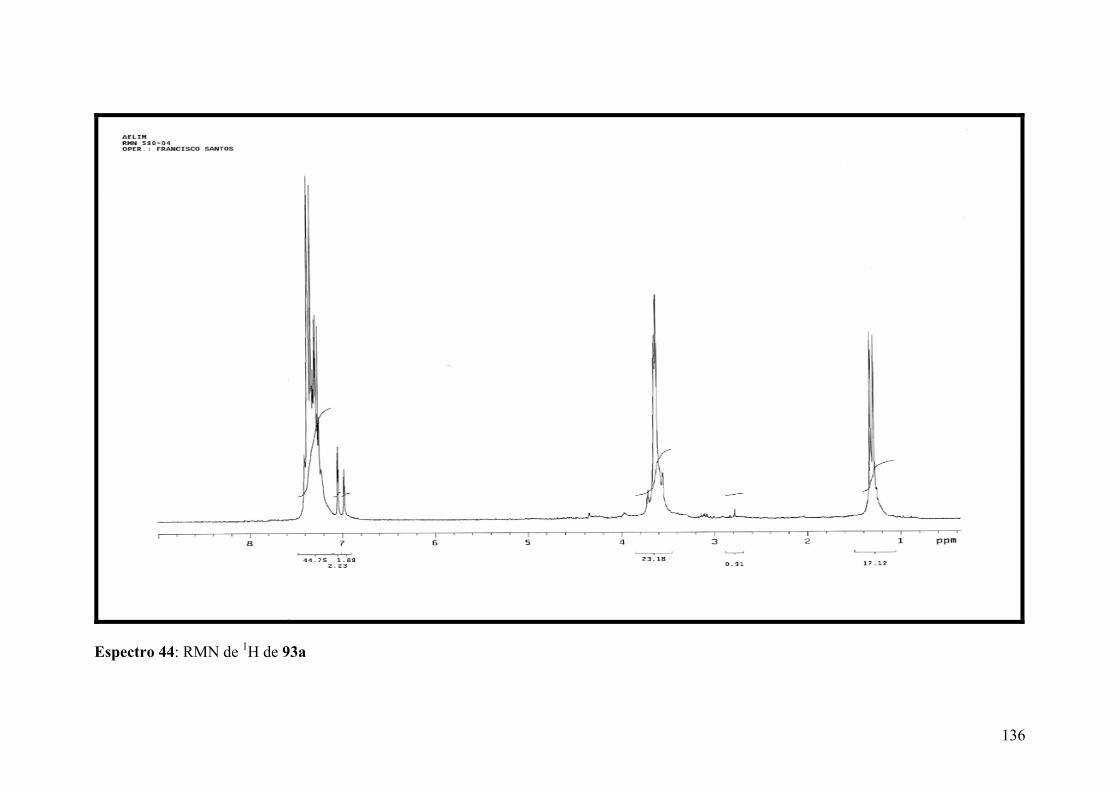
136
Espectro 44: RMN de 1H de 93a
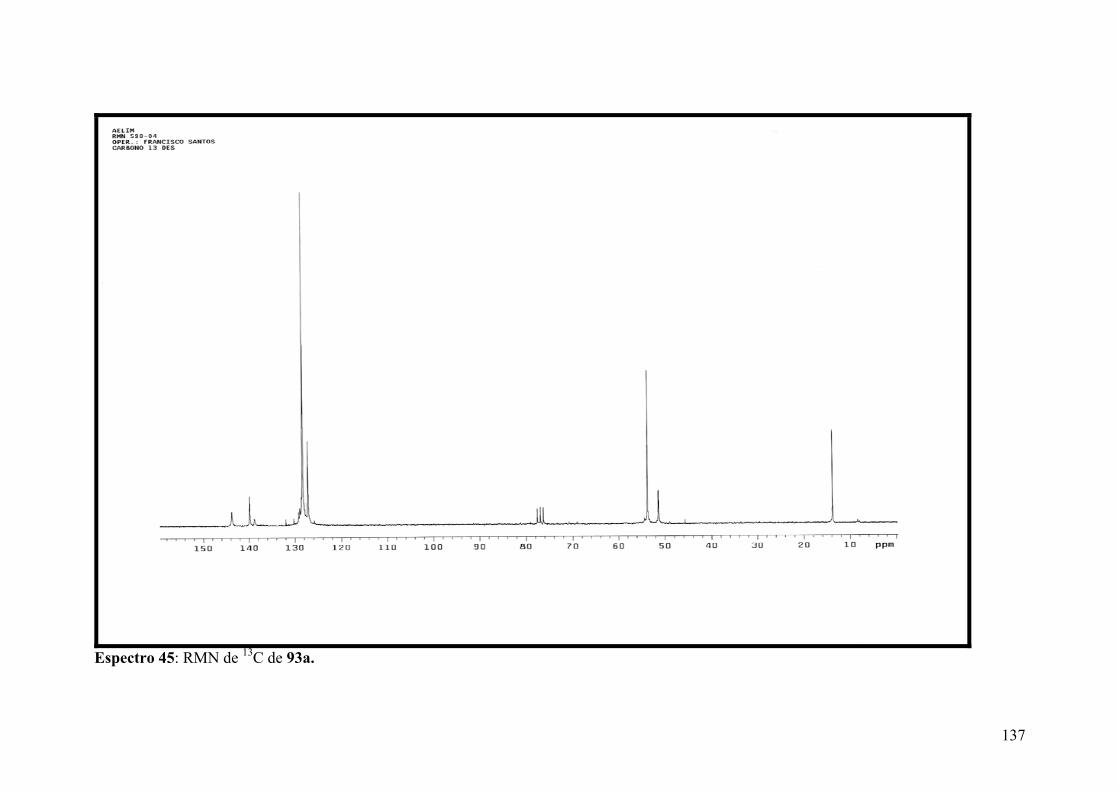
137
Espectro 45: RMN de 13C de 93a.
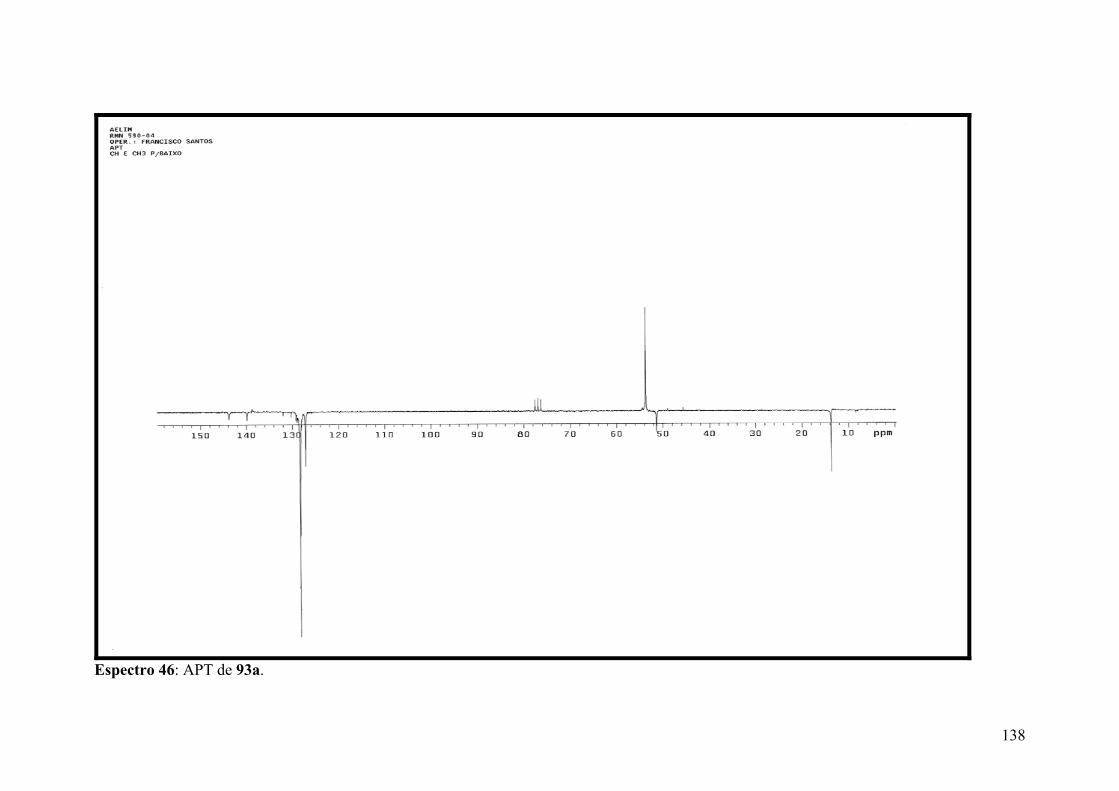
138
Espectro 46: APT de 93a.
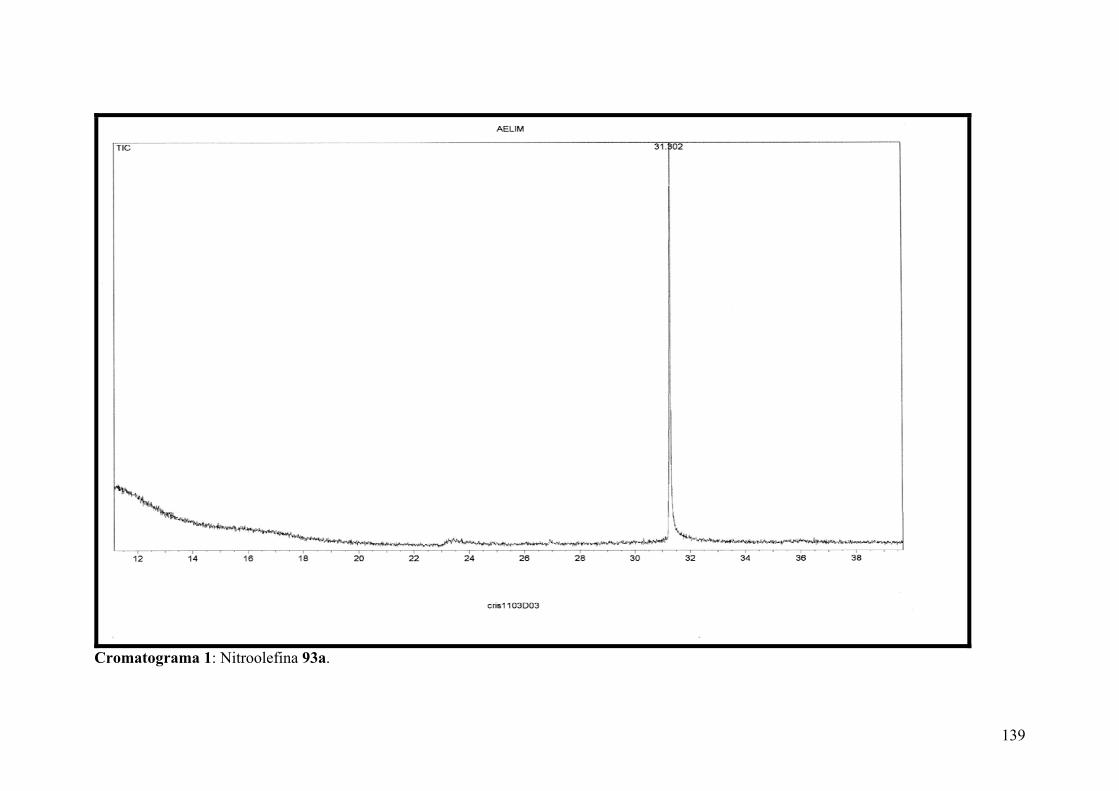
139
Cromatograma 1: Nitroolefina 93a.
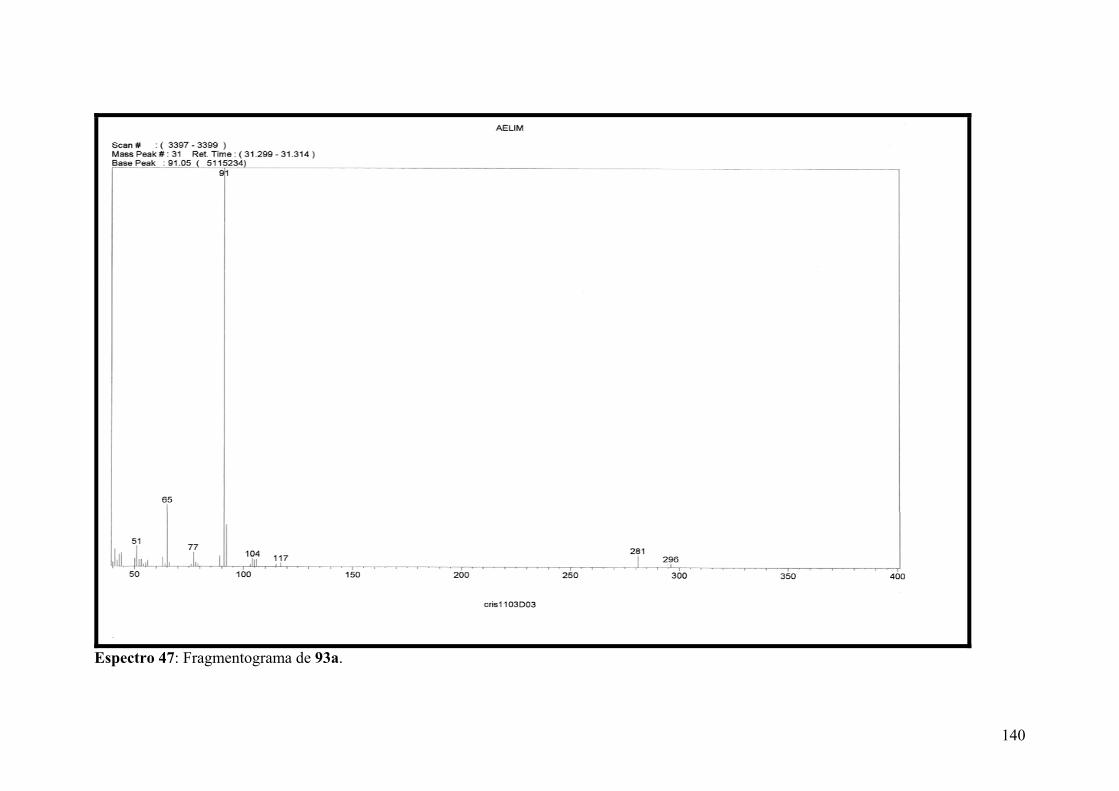
140
Espectro 47: Fragmentograma de 93a.
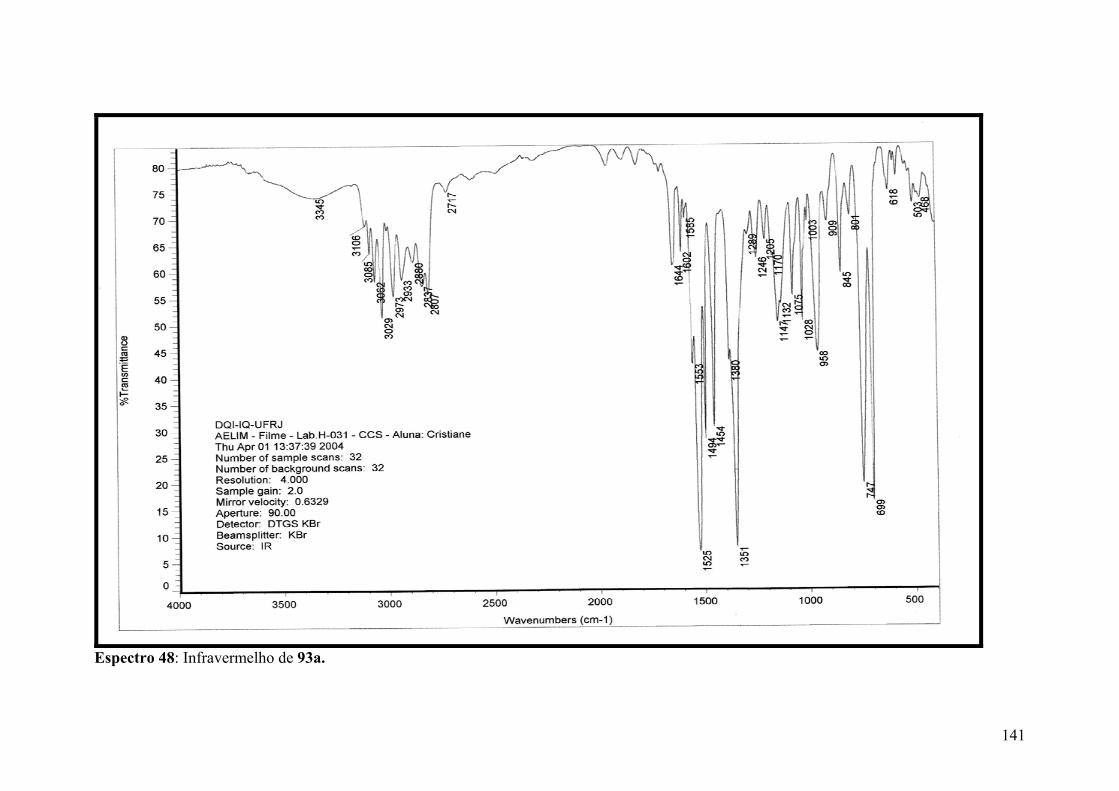
141
Espectro 48: Infravermelho de 93a.
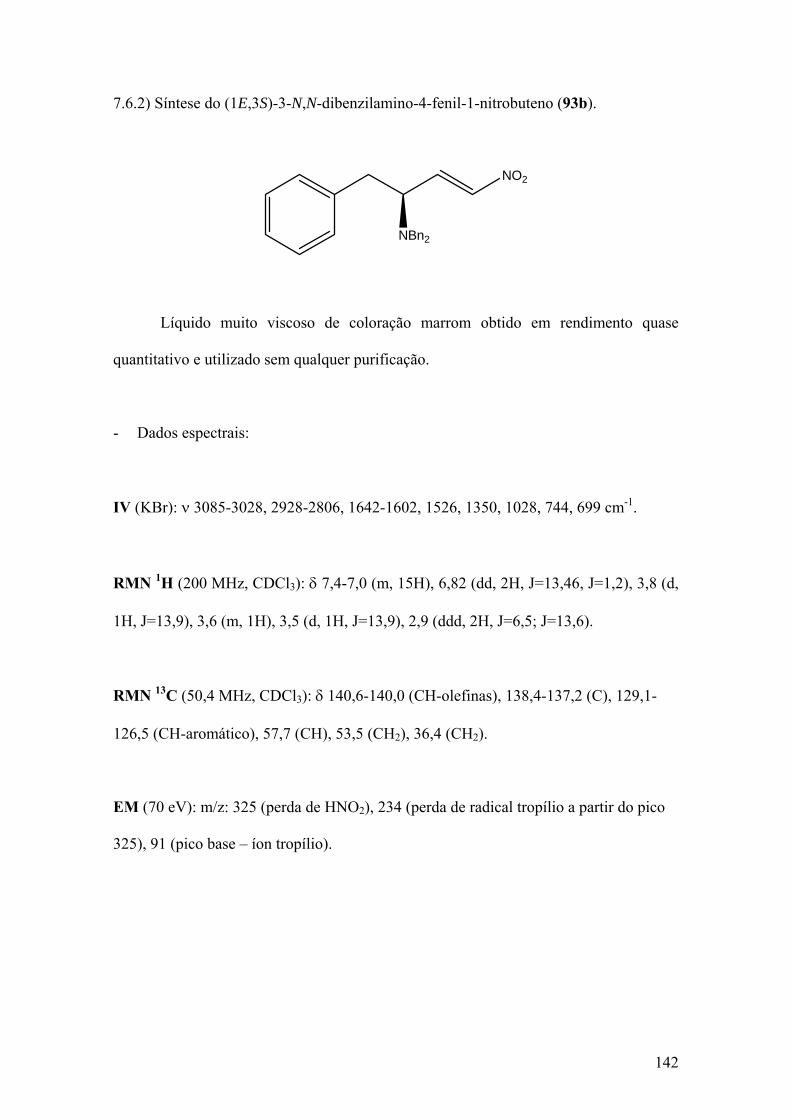
142
7.6.2) Síntese do (1E,3S)-3-N,N-dibenzilamino-4-fenil-1-nitrobuteno (93b).
NBn2
NO2
Líquido muito viscoso de coloração marrom obtido em rendimento quase
quantitativo e utilizado sem qualquer purificação.
- Dados espectrais:
IV (KBr): ν 3085-3028, 2928-2806, 1642-1602, 1526, 1350, 1028, 744, 699 cm-1.
RMN 1H (200 MHz, CDCl3): δ 7,4-7,0 (m, 15H), 6,82 (dd, 2H, J=13,46, J=1,2), 3,8 (d,
1H, J=13,9), 3,6 (m, 1H), 3,5 (d, 1H, J=13,9), 2,9 (ddd, 2H, J=6,5; J=13,6).
RMN 13C (50,4 MHz, CDCl3): δ 140,6-140,0 (CH-olefinas), 138,4-137,2 (C), 129,1-
126,5 (CH-aromático), 57,7 (CH), 53,5 (CH2), 36,4 (CH2).
EM (70 eV): m/z: 325 (perda de HNO2), 234 (perda de radical tropílio a partir do pico
325), 91 (pico base – íon tropílio).
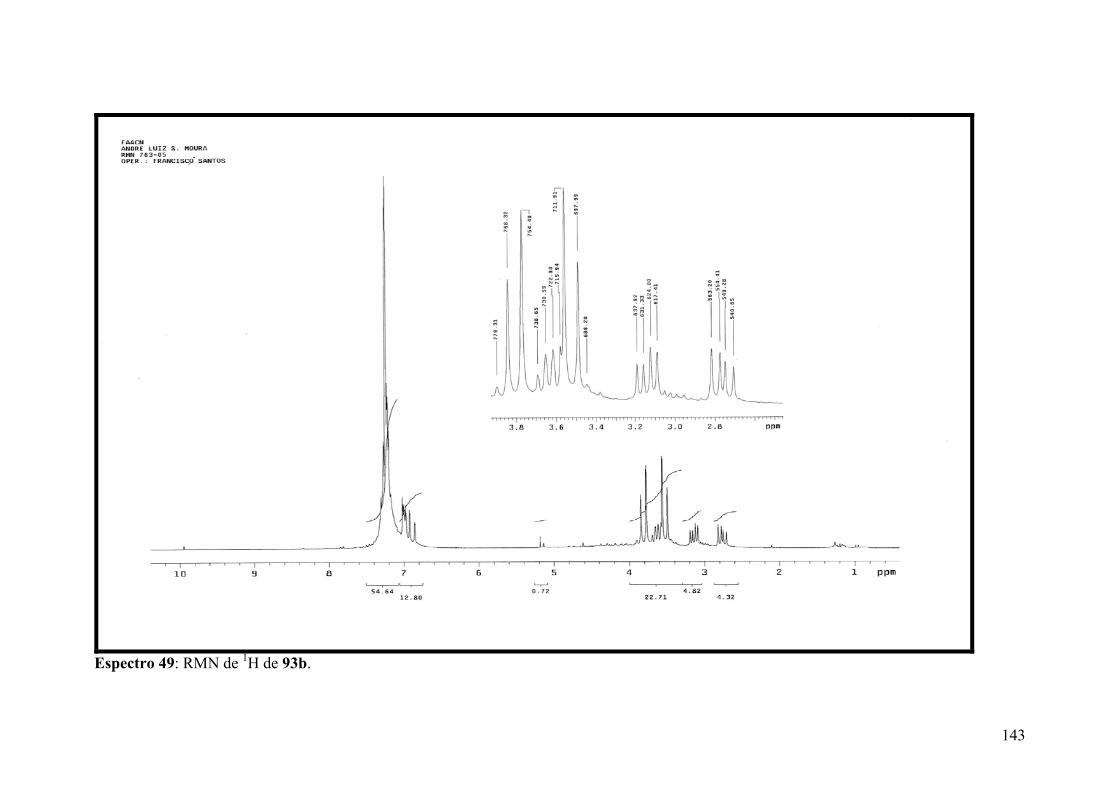
143
Espectro 49: RMN de 1H de 93b.
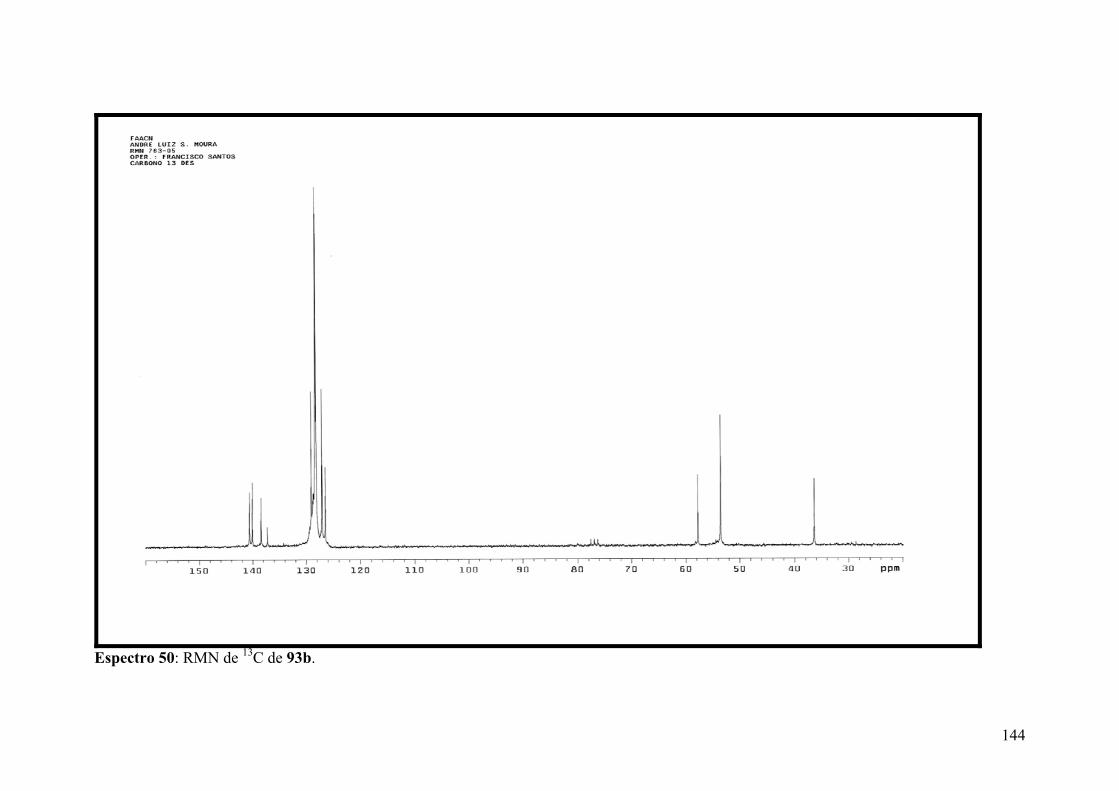
144
Espectro 50: RMN de 13C de 93b.
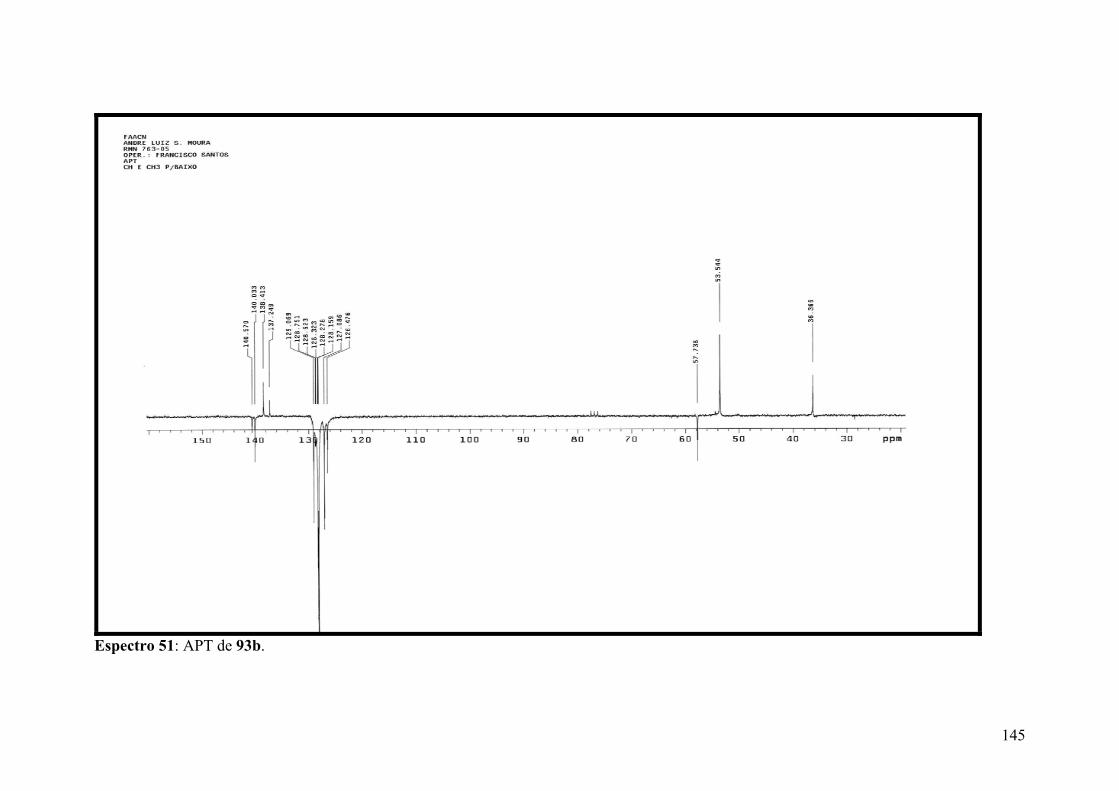
145
Espectro 51: APT de 93b.
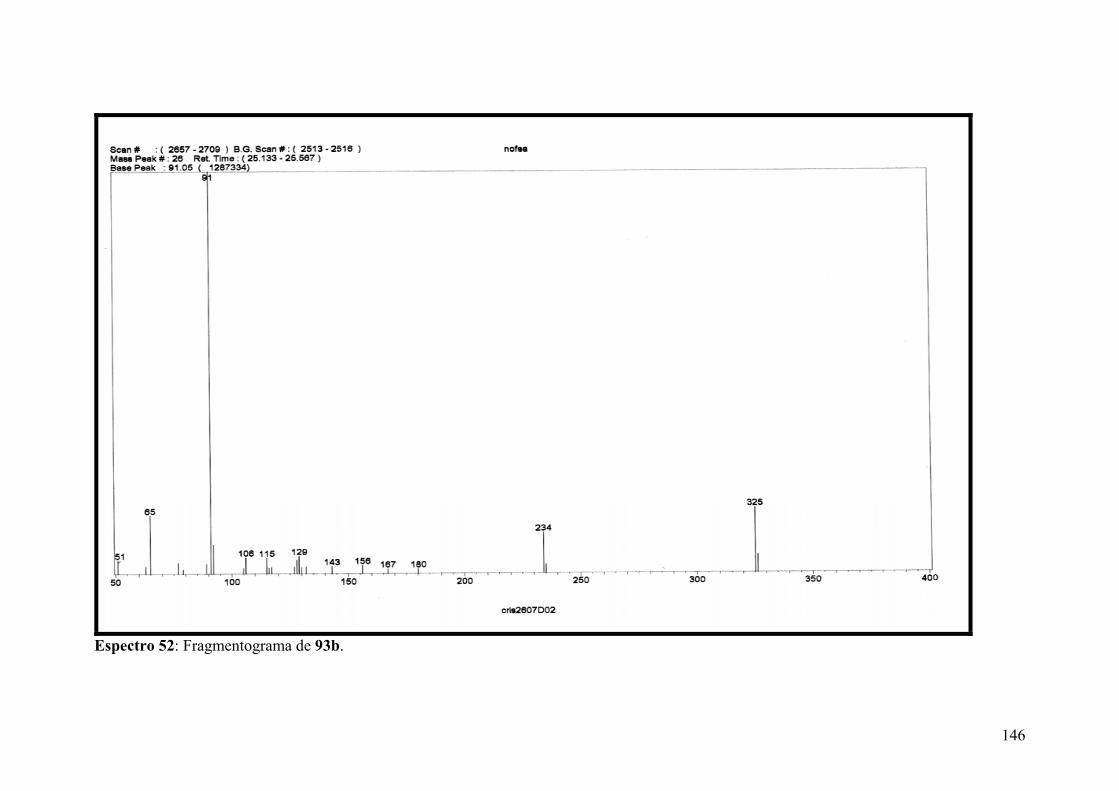
146
Espectro 52: Fragmentograma de 93b.
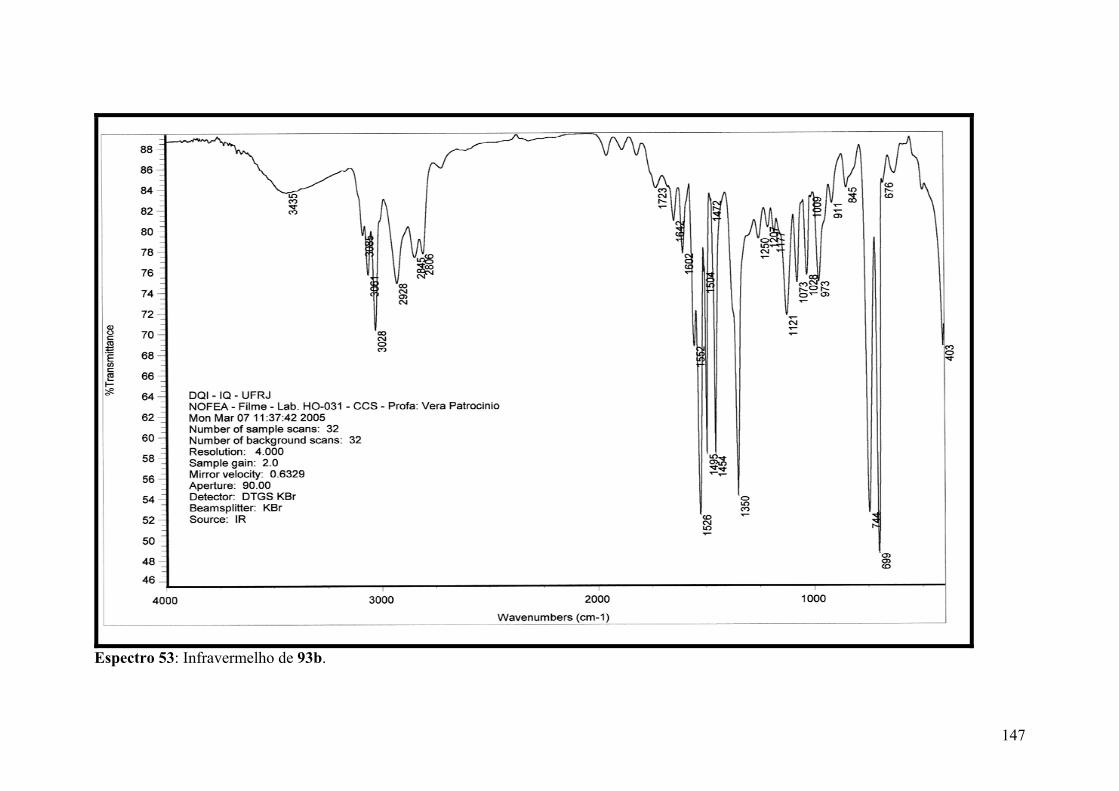
147
Espectro 53: Infravermelho de 93b.

148
7.7) Síntese do 2-hidroxi-2-metilnitropropano (90).
NO2
HO
Foram adicionados 2,7 ml de propanona (37,0 mmoles) a uma mistura de 2,0 ml
de nitrometano (37,0 mmoles) em 11,4 g de Amberlist A-21 ou 14,5 g de Al2O3 básica.
A mistura reacional permaneceu por 20 horas sob temperatura ambiente. Após este
período, o meio reacional foi filtrado e lavado com diclorometano (2 x 10,0 ml). A
solução resultante foi evaporada sob pressão reduzida, fornecendo um líquido claro em
rendimento químico de 83%.
- Dados espectrais:
IV (KBr): ν 3530, 3423, 2982, 1552, 1378, 1158.
RMN 1H (200MHz, CDCl3): δ 4,4 (s, 2H), 3,1 (sl, 1H), 1,4 (s, 2 CH3).
RMN 13C (50,4 MHz, CDCl3): δ 85,0 (CH2), 69,5 (C), 26,6 (2 CH3).
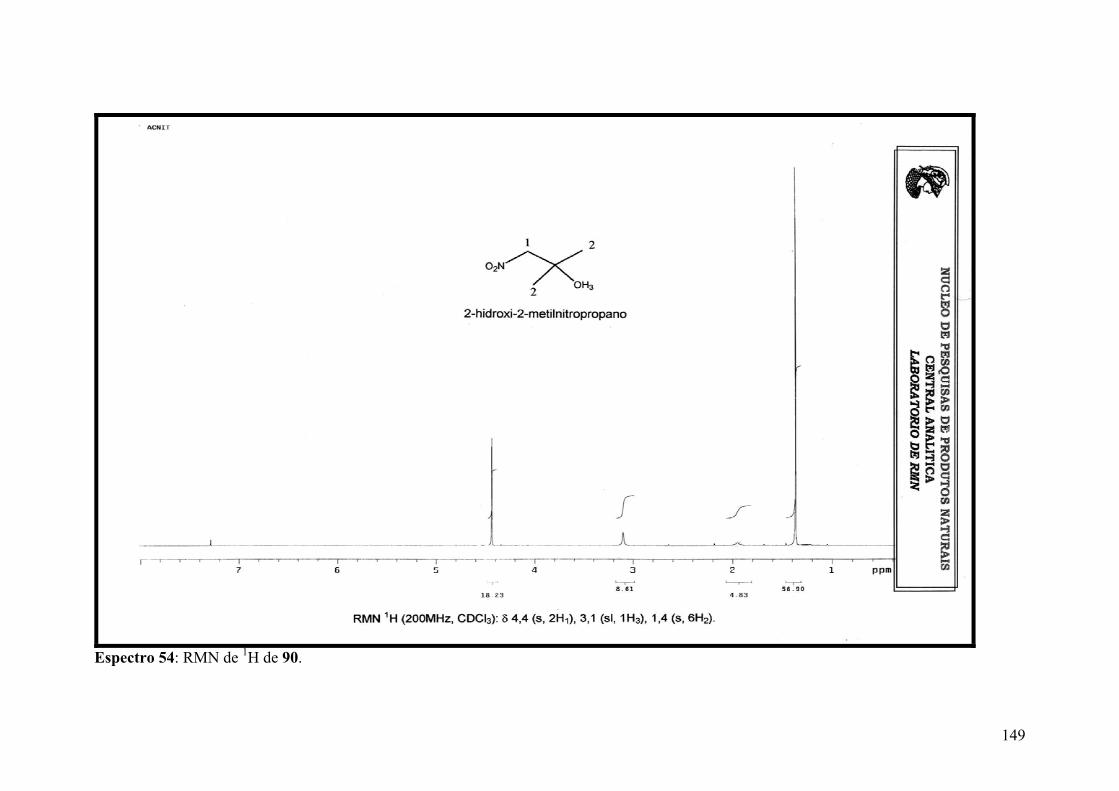
149
Espectro 54: RMN de 1H de 90.
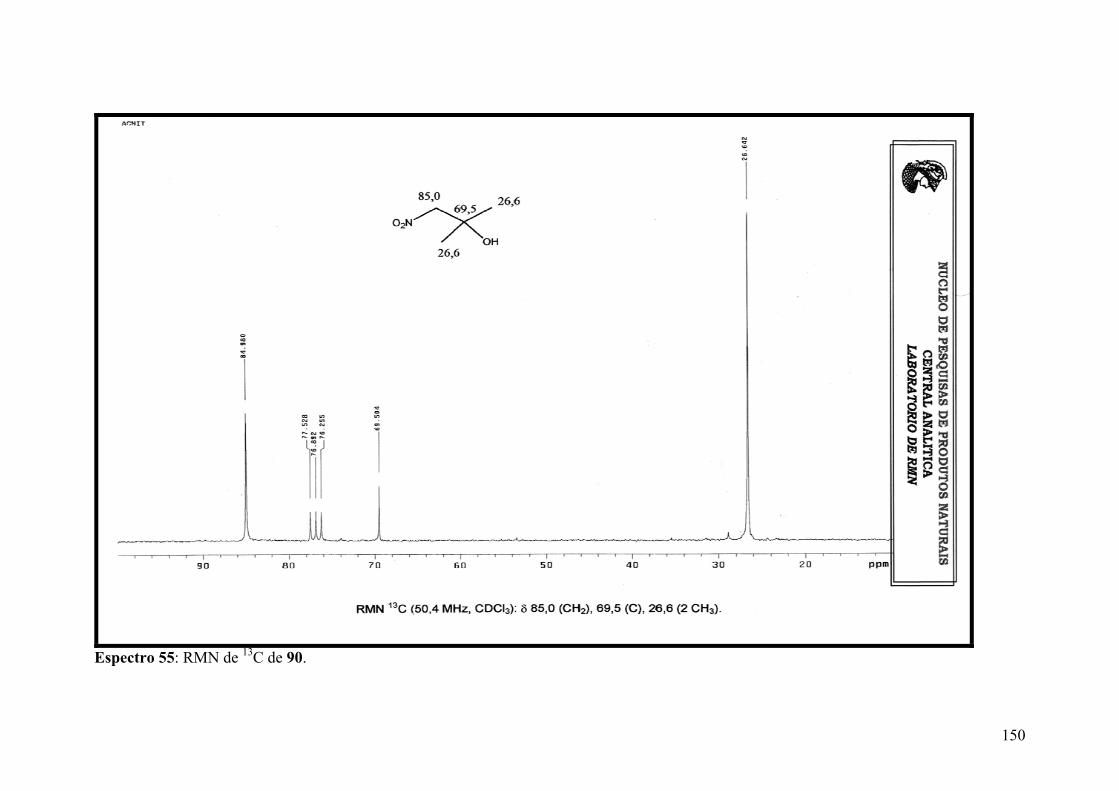
150
Espectro 55: RMN de 13C de 90.
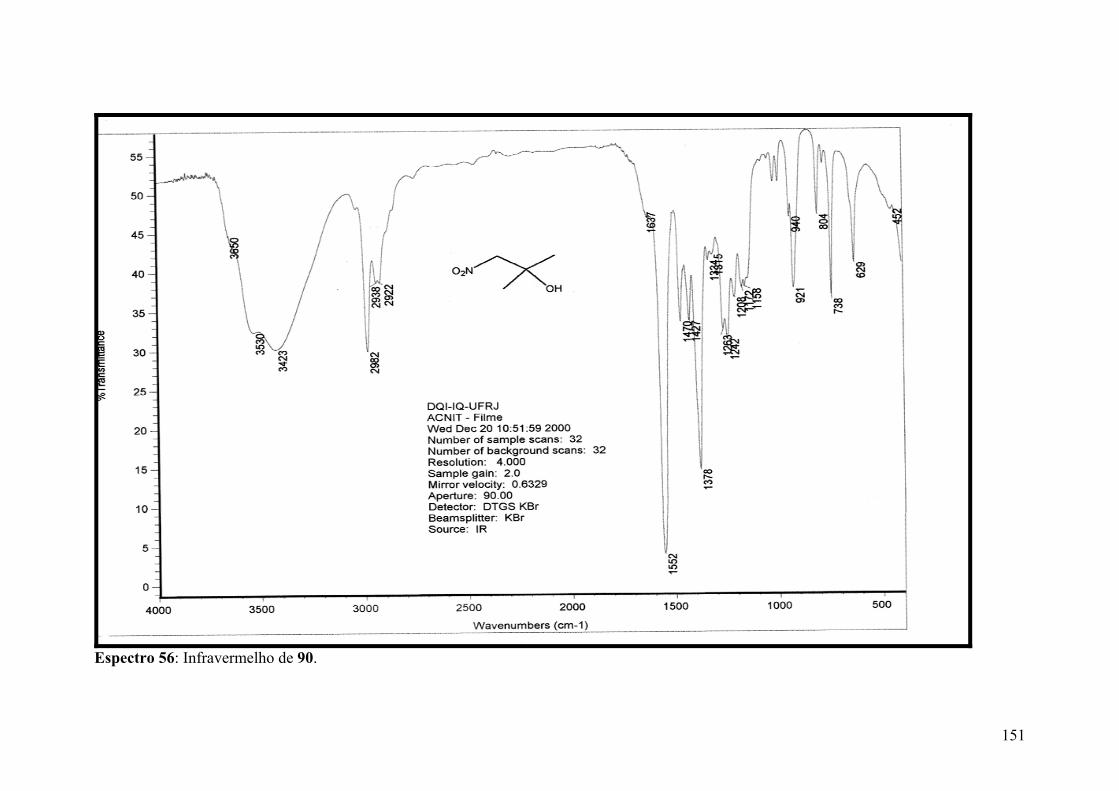
151
Espectro 56: Infravermelho de 90.

152
7.8) Síntese do nitrobutano.
NO2
Foram adicionados 4,11 g de brometo de butila (30 mmoles) gota à gota a uma
solução de NaNO2 (3,73g ; 54 mmoles) em 60,0 mL de DMF. A mistura reacional foi
agitada por 7 horas à temperatura ambiente. Após o término desse tempo, 100 mL de
água gelada foram adicionados a essa mistura reacional e, logo em seguida, foi extraída
com éter dietílico (3 x 50,0mL). Os extratos etéreos foram combinados e secos com
Na2SO4, passados por uma camada de Florisil® e concentrados sob pressão reduzida
para fornecer o nitroalcano em 61% de rendimento químico que pôde ser utilizado
posteriormente sem qualquer purificação.
- Dados espectrais (referentes a um produto bruto).
IV (KBr): ν 1552, 1386, 1094.
RMN 1H (200 MHz, CDCl3): δ 3,4 (t, 2H), 1,83 (q, 2H), 1,5 (sex, 2H), 0,9 (t, 2H).
EM (70 eV): m/z 57 (perda de NO2), 41 (pico base).
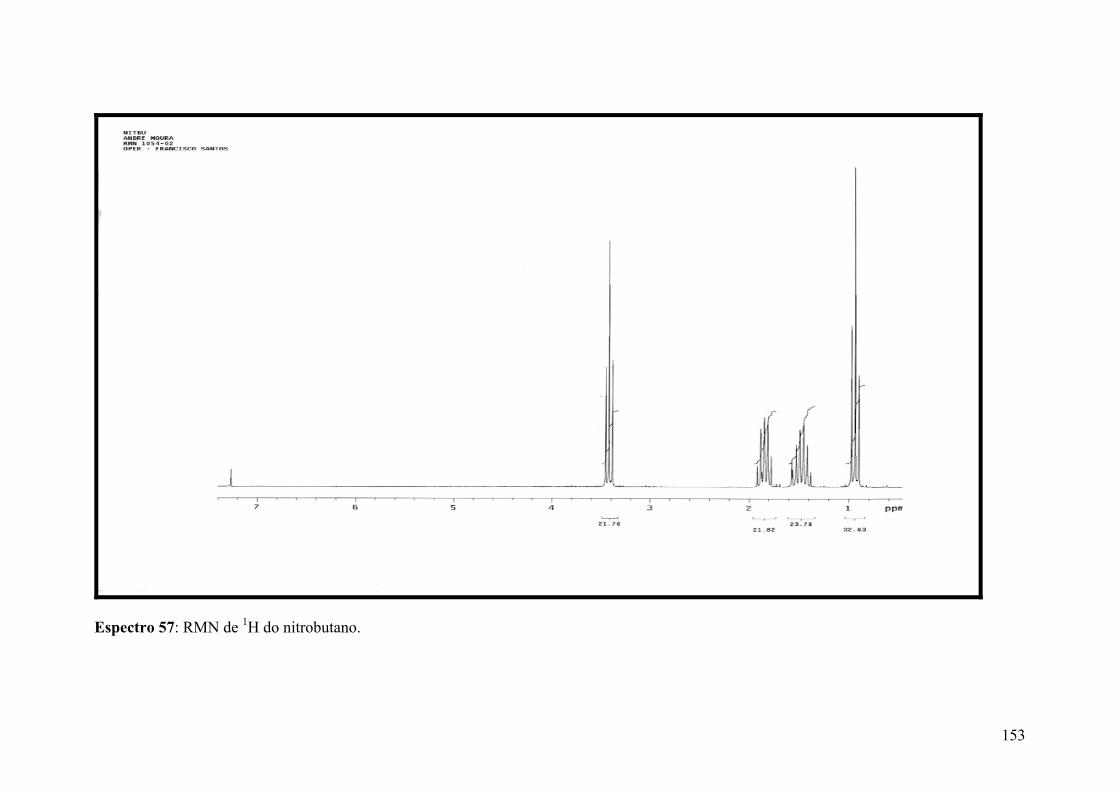
153
Espectro 57: RMN de 1H do nitrobutano.
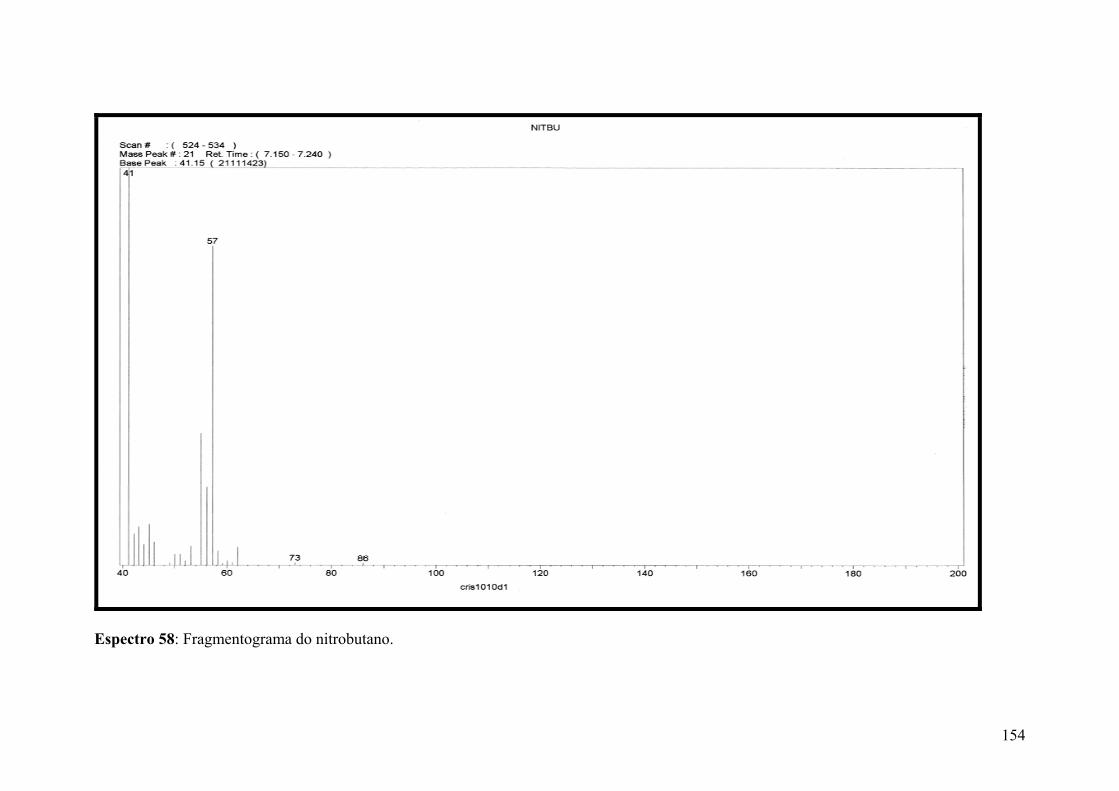
154
Espectro 58: Fragmentograma do nitrobutano.
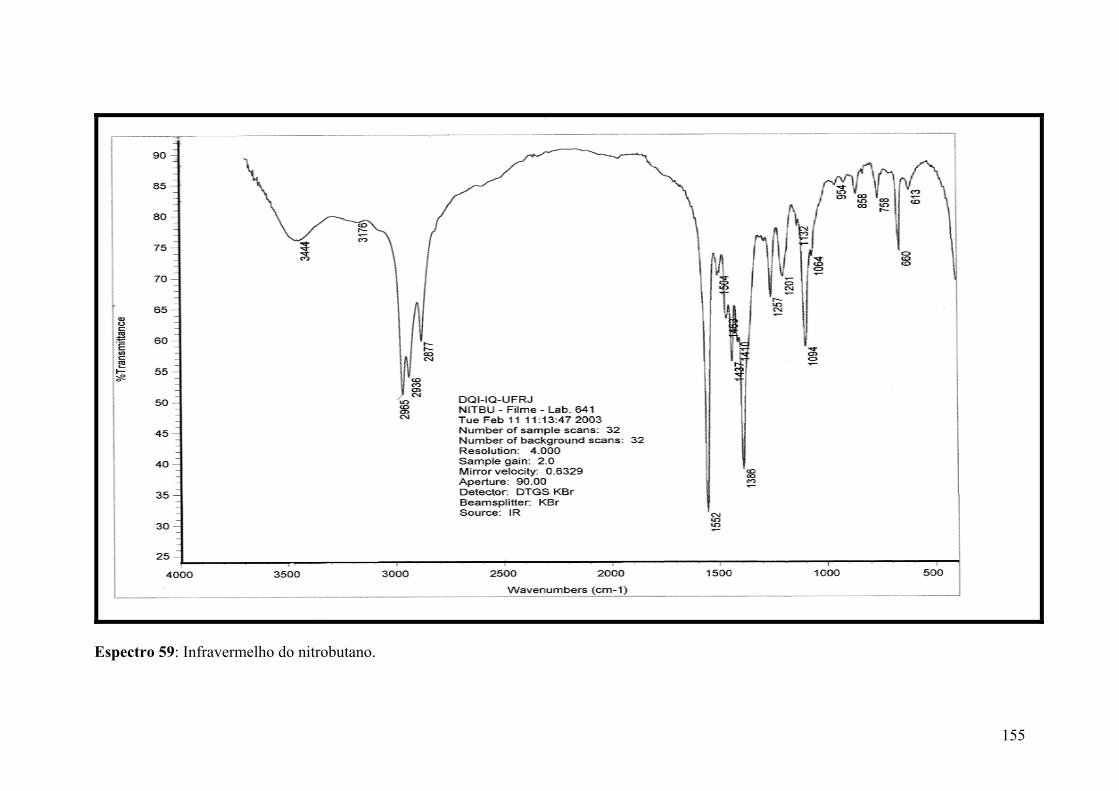
155
Espectro 59: Infravermelho do nitrobutano.

156
7.9) Procedimento geral para as reações de adição de nitroalcanos às iminas 88a-d.
Uso de Amberlist A-21 como base:
Foram adicionados 5,3 mmoles da imina (88a, b, c ou d) a 5,3 mmoles do
nitroalcano em 1,7 g de Amberlist A-21. O tempo reacional foi avaliado de acordo com
a evolução da reação acompanhada através de cromatografia em camada fina. Após o
tempo decorrido, a suspensão foi lavada com CH2Cl2 (2 x 15,0 mL), seguida por uma
filtração simples e concentração do filtrado sob pressão reduzida.
O produto bruto obtido é purificado em coluna cromatográfica de gel de sílica.
As nitrodiaminas obtidas (89a-g) têm um aspecto de líquido viscoso e de
coloração amarelo-acastanhada.
Nota: Os dados abaixo referentes aos espectros de RMN 1H e RMN 13C são de misturas
de diastereoisômeros.
157
7.9.1) Síntese do (2R,3S)-2-N-benzilamino-3-N,N-dibenzilamino-5-metil-1-nitroexano (89a).
- Dados espectrais:
IV (KBr): ν 3568, 3400, 2958, 1563, 1557, 1378, 1250, 750, 701.
RMN 1H (200 MHz, CDCl3): δ 7,3 (m, 15H), 4,74 (d, 2H, J=3,57), 4,67 (d, 2H,
J=3,76), 4,56(d, 2H, J=5,22), 4,48(d, 2H, J=5,31), 4,7 - 3,9 (m, 2H), 3,7 - 3,4 (m, 6H),
3,0 (m, 2H), 1,7 - 1,2 (m, 3H), 1,0 - 0,9 (t, 6H).
RMN 13C (50,4 MHZ, CDCl3): δ 138,4 (C), 128,7 - 127,1 (CH), 74,1 (CH2), 73,9
(CH2), 54,8 (CH), 54,4 (CH2), 38,5 (CH), 35,9 (CH2), 25,4 (CH), 22,8 (CH3), 22,3
(CH3).
- Razão diastereoisomérica: 90 : 10 ( medida no CH em 54,8 ppm – veja Espectro 62).
NO2
NBn2
NHBn
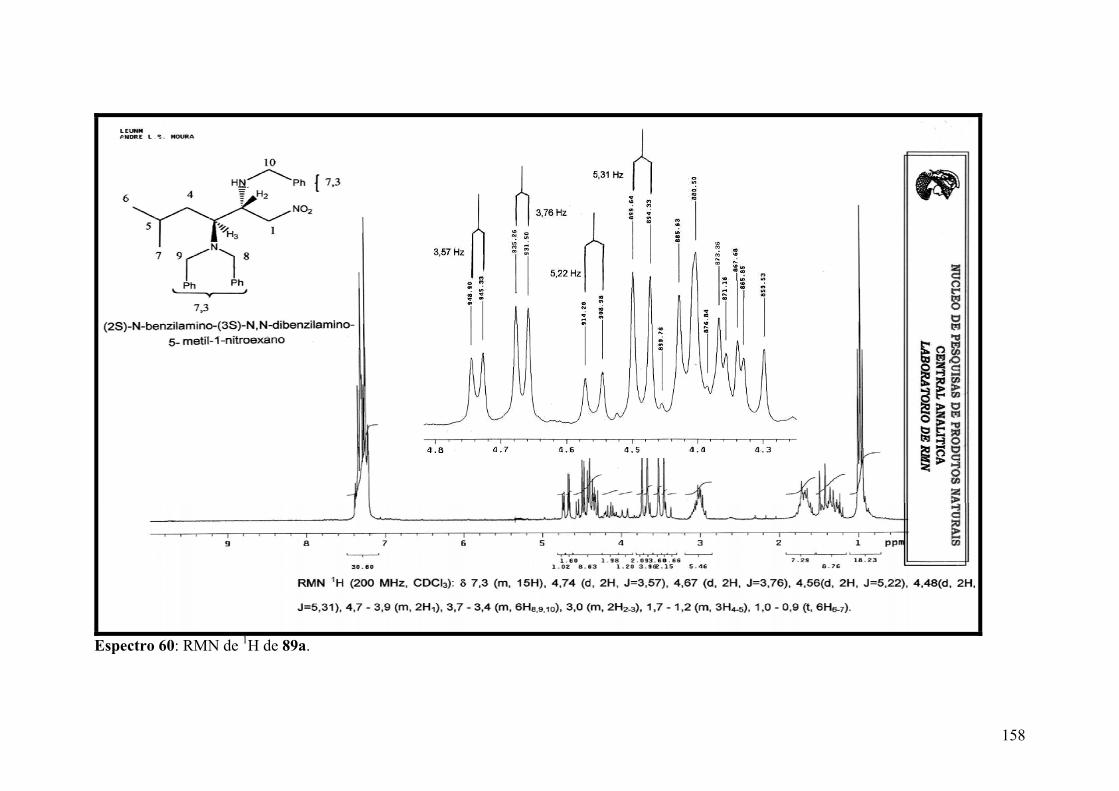
158
Espectro 60: RMN de 1H de 89a.
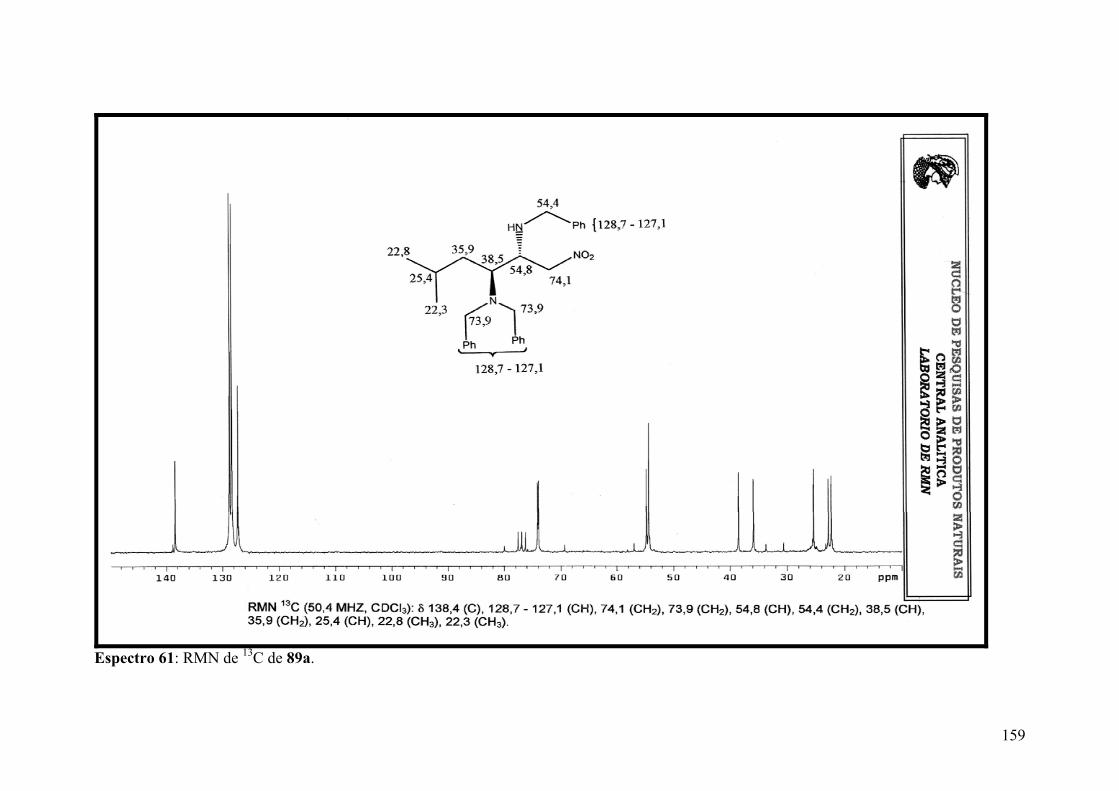
159
Espectro 61: RMN de 13C de 89a.
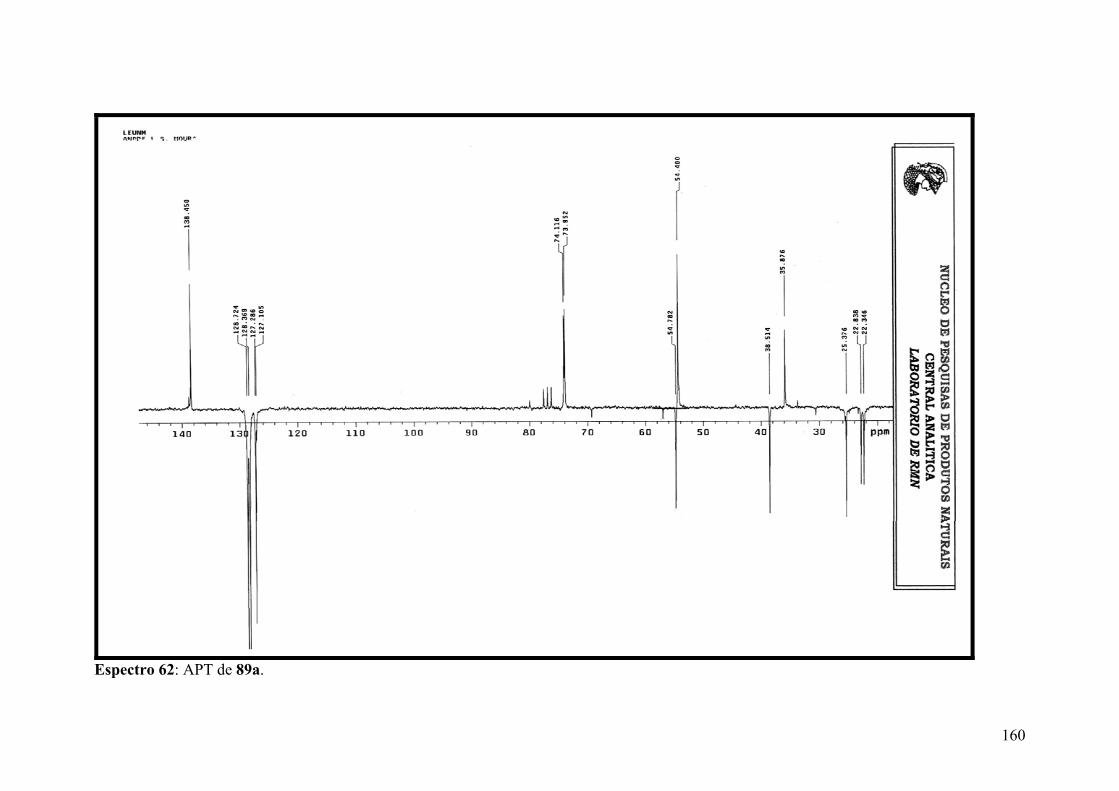
160
Espectro 62: APT de 89a.
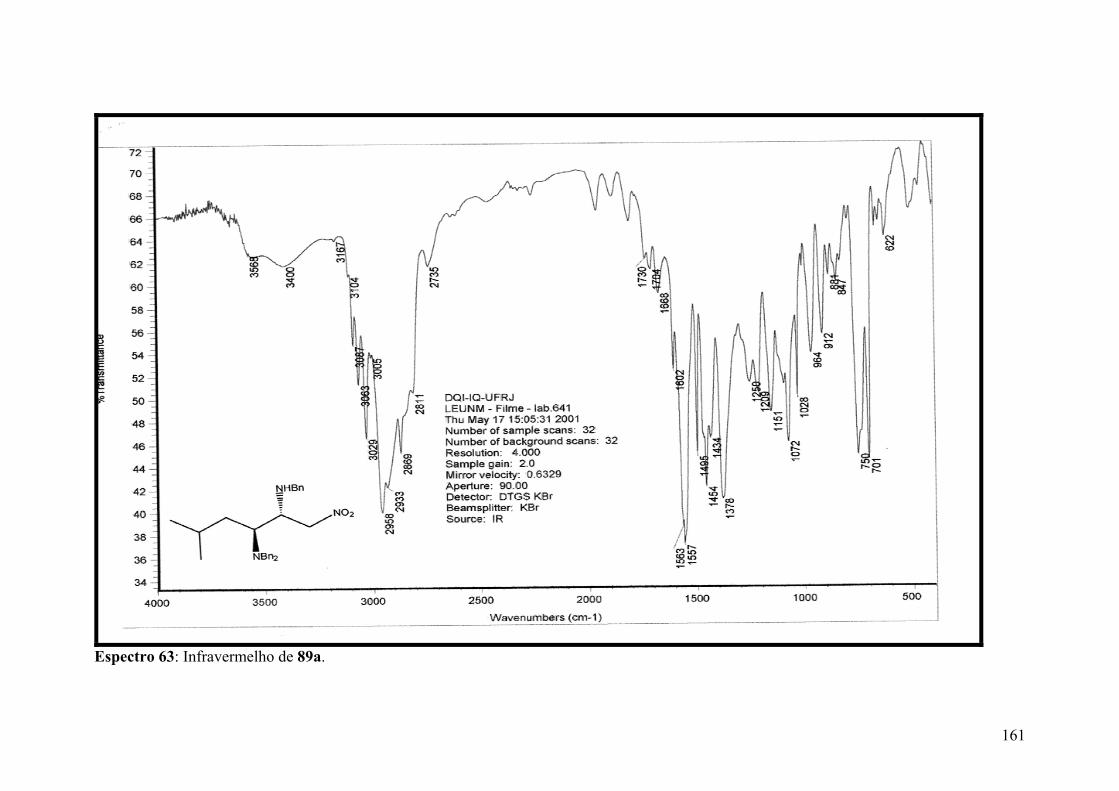
161
Espectro 63: Infravermelho de 89a.
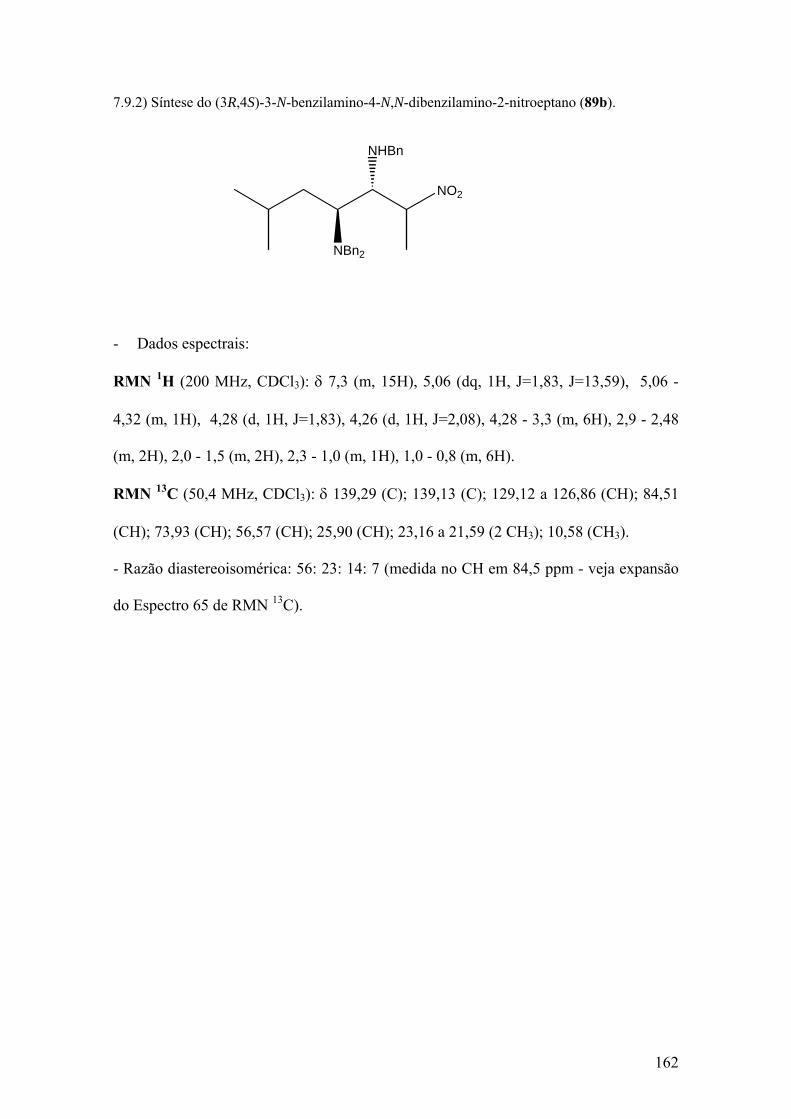
162
7.9.2) Síntese do (3R,4S)-3-N-benzilamino-4-N,N-dibenzilamino-2-nitroeptano (89b).
- Dados espectrais:
RMN 1H (200 MHz, CDCl3): δ 7,3 (m, 15H), 5,06 (dq, 1H, J=1,83, J=13,59), 5,06 -
4,32 (m, 1H), 4,28 (d, 1H, J=1,83), 4,26 (d, 1H, J=2,08), 4,28 - 3,3 (m, 6H), 2,9 - 2,48
(m, 2H), 2,0 - 1,5 (m, 2H), 2,3 - 1,0 (m, 1H), 1,0 - 0,8 (m, 6H).
RMN 13C (50,4 MHz, CDCl3): δ 139,29 (C); 139,13 (C); 129,12 a 126,86 (CH); 84,51
(CH); 73,93 (CH); 56,57 (CH); 25,90 (CH); 23,16 a 21,59 (2 CH3); 10,58 (CH3).
- Razão diastereoisomérica: 56: 23: 14: 7 (medida no CH em 84,5 ppm - veja expansão
do Espectro 65 de RMN 13C).
NO2
NBn2
NHBn
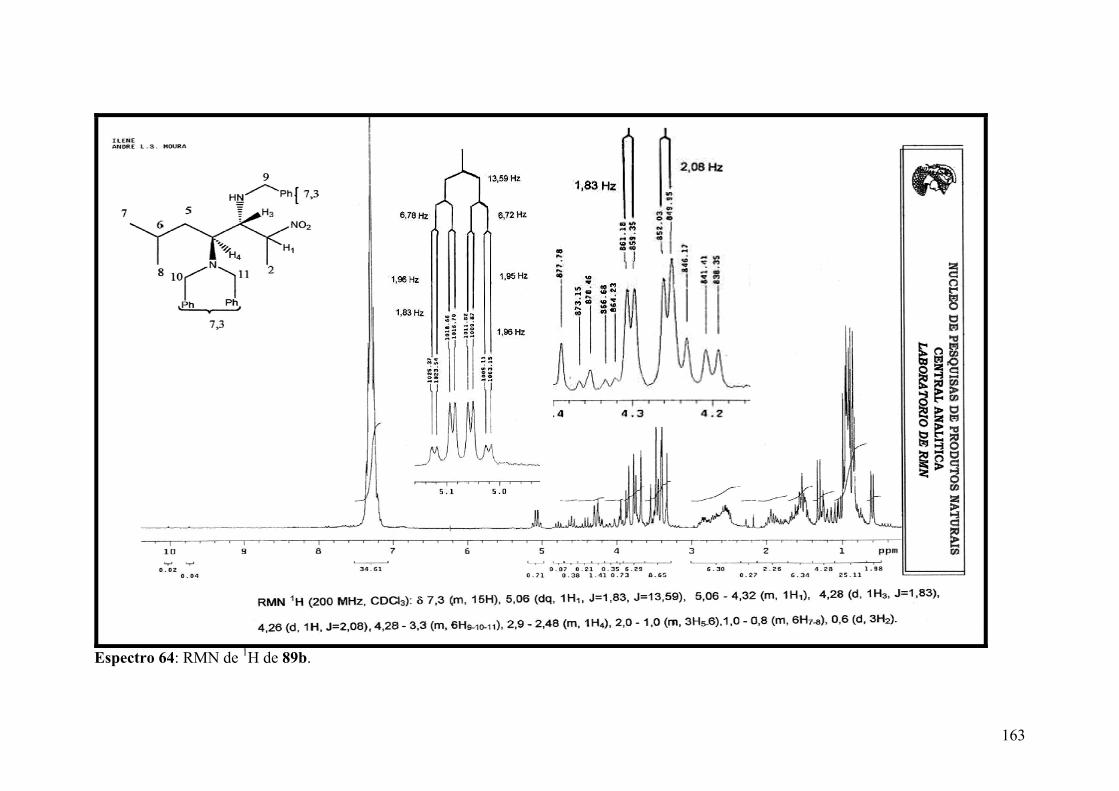
163
Espectro 64: RMN de 1H de 89b.
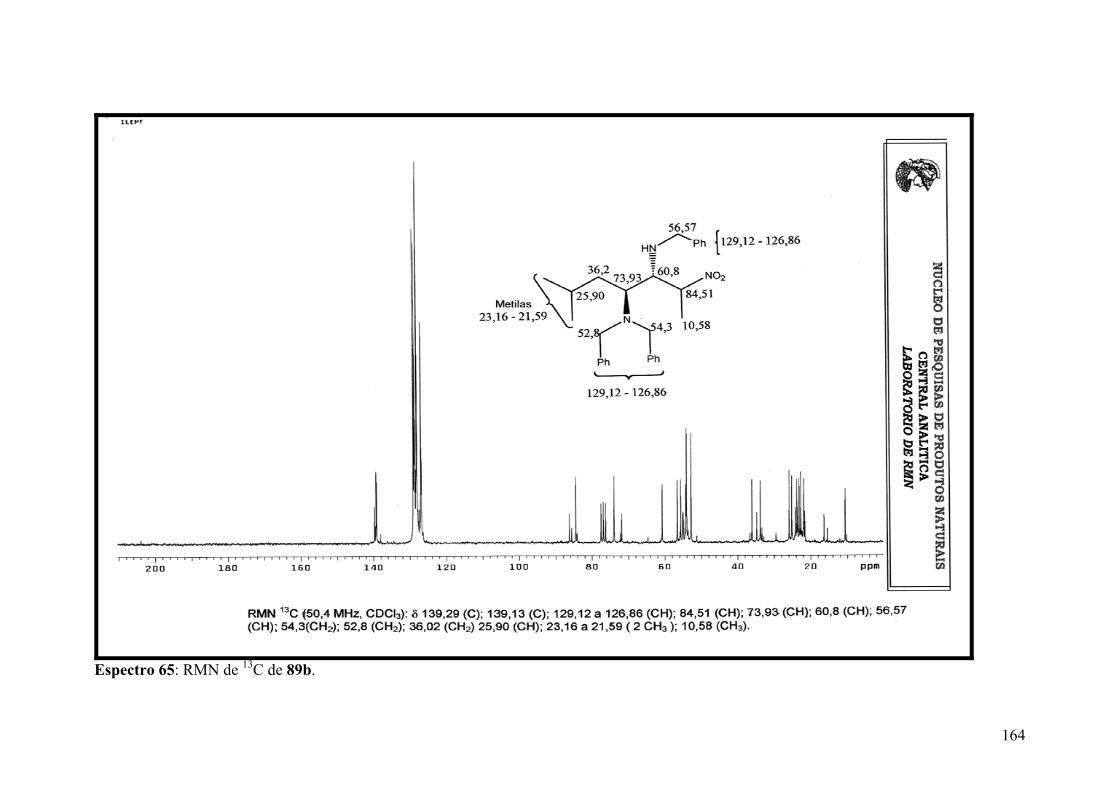
164
Espectro 65: RMN de 13C de 89b.
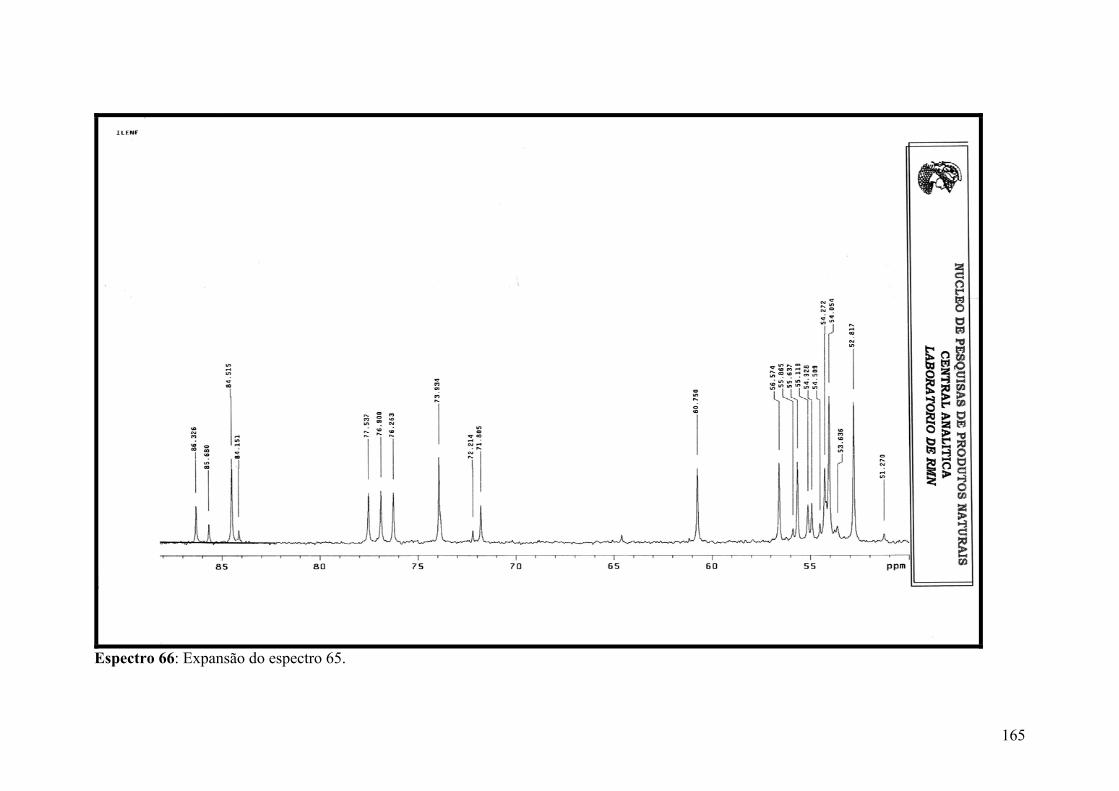
165
Espectro 66: Expansão do espectro 65.
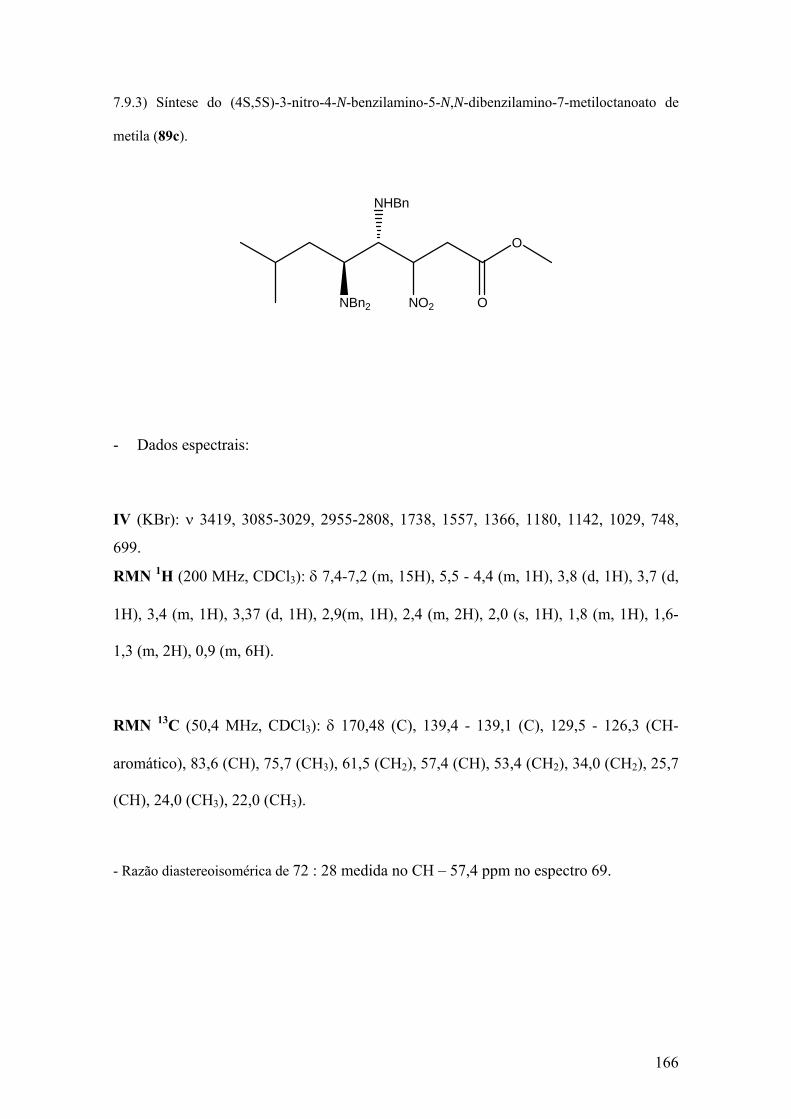
166
7.9.3) Síntese do (4S,5S)-3-nitro-4-N-benzilamino-5-N,N-dibenzilamino-7-metiloctanoato de
metila (89c).
NBn2
NHBn
NO2
O
O
- Dados espectrais:
IV (KBr): ν 3419, 3085-3029, 2955-2808, 1738, 1557, 1366, 1180, 1142, 1029, 748,
699.
RMN 1H (200 MHz, CDCl3): δ 7,4-7,2 (m, 15H), 5,5 - 4,4 (m, 1H), 3,8 (d, 1H), 3,7 (d,
1H), 3,4 (m, 1H), 3,37 (d, 1H), 2,9(m, 1H), 2,4 (m, 2H), 2,0 (s, 1H), 1,8 (m, 1H), 1,6-
1,3 (m, 2H), 0,9 (m, 6H).
RMN 13C (50,4 MHz, CDCl3): δ 170,48 (C), 139,4 - 139,1 (C), 129,5 - 126,3 (CH-
aromático), 83,6 (CH), 75,7 (CH3), 61,5 (CH2), 57,4 (CH), 53,4 (CH2), 34,0 (CH2), 25,7
(CH), 24,0 (CH3), 22,0 (CH3).
- Razão diastereoisomérica de 72 : 28 medida no CH – 57,4 ppm no espectro 69.
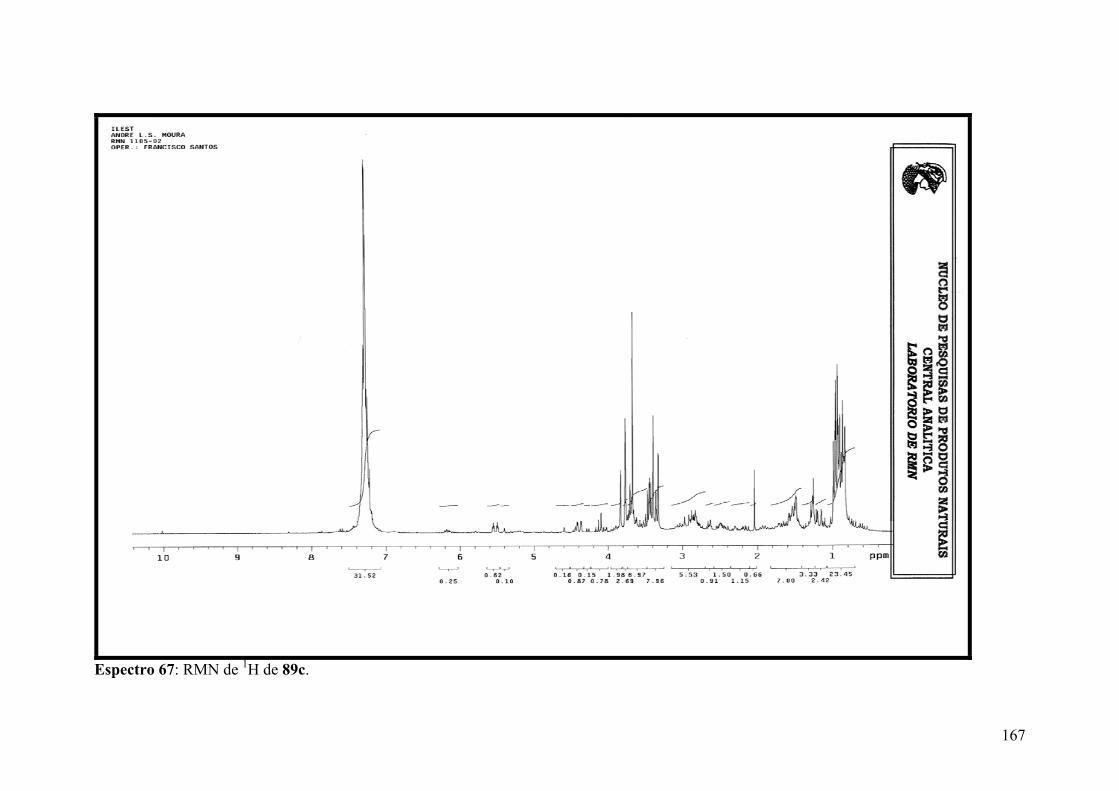
167
Espectro 67: RMN de 1H de 89c.
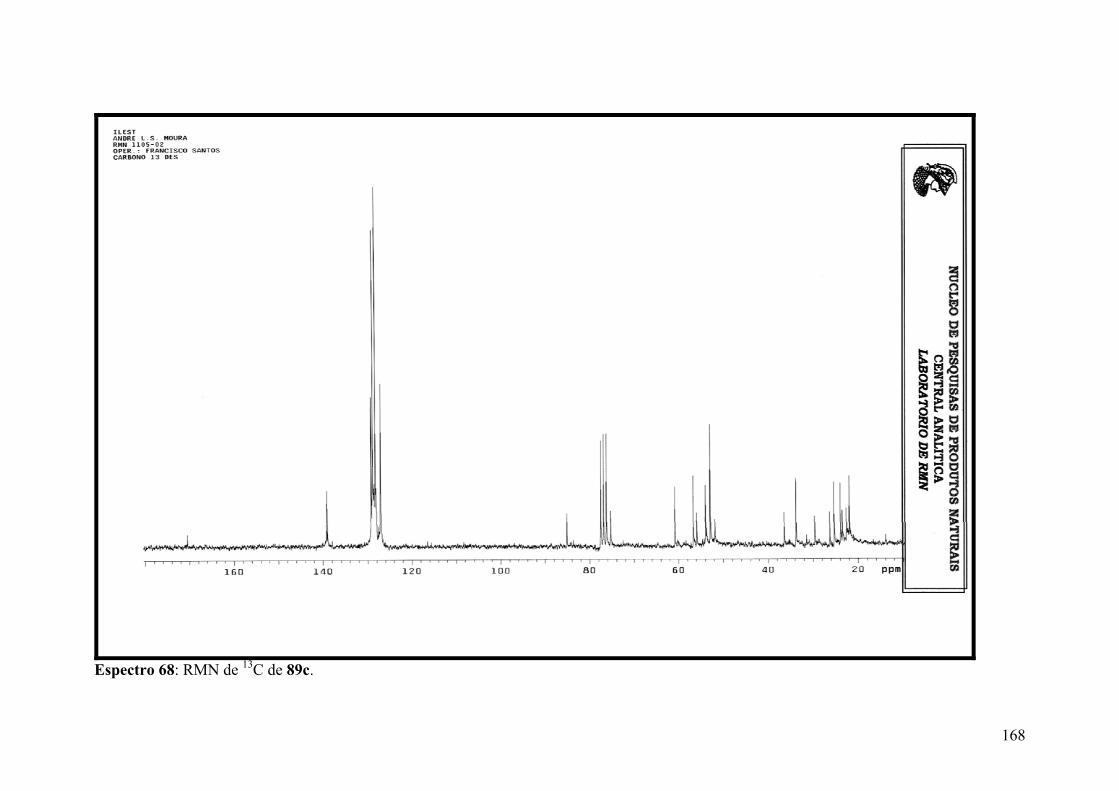
168
Espectro 68: RMN de 13C de 89c.
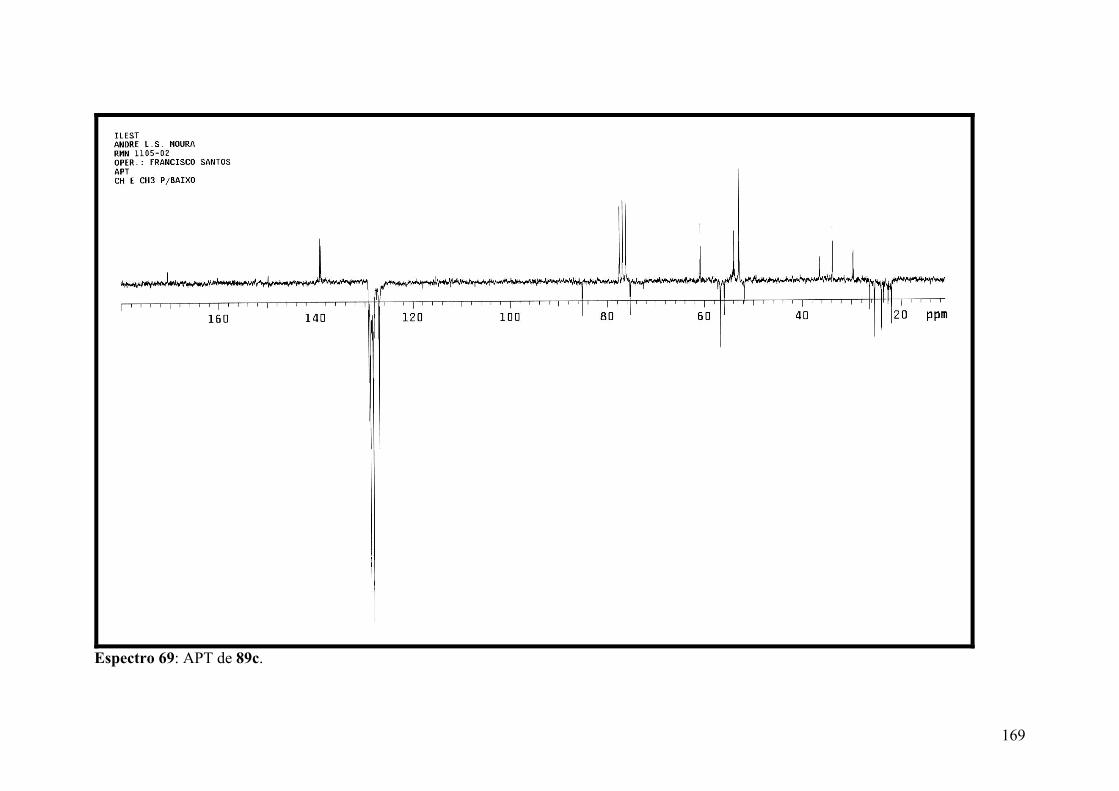
169
Espectro 69: APT de 89c.
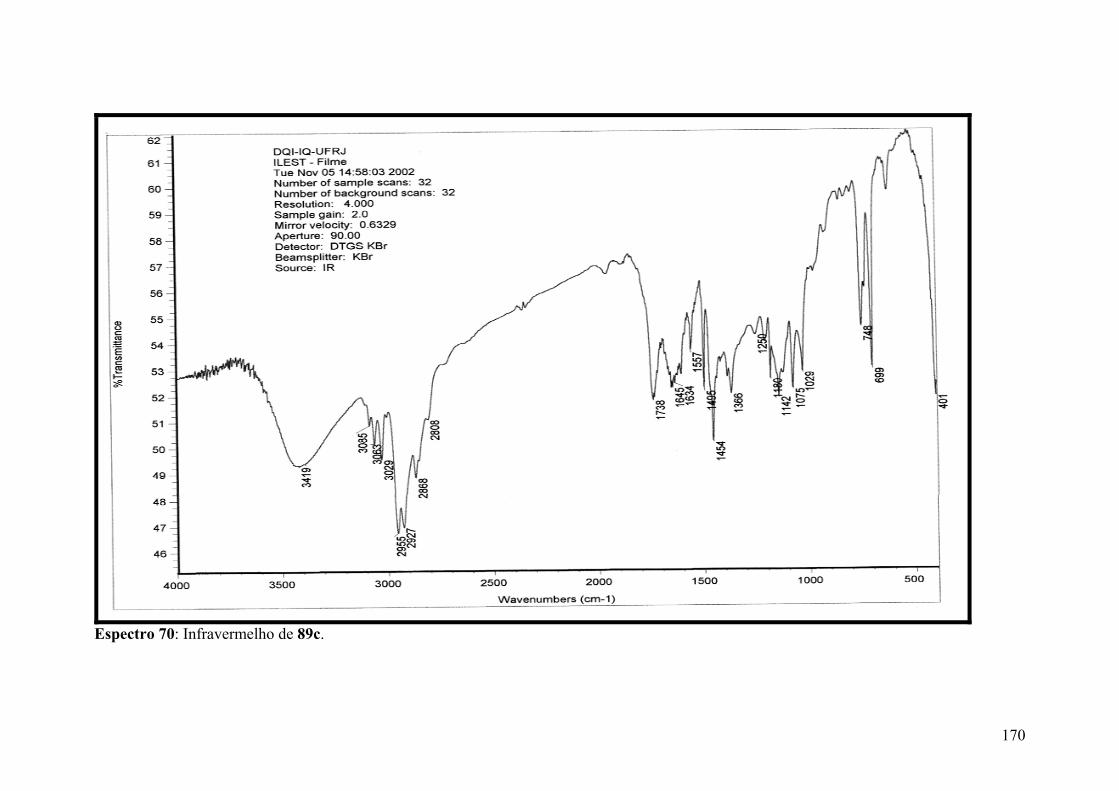
170
Espectro 70: Infravermelho de 89c.

171
7.9.4) Síntese do (4R,5S)-2,7-dimetil-3-nitro-4-N-benzilamino-5-N,N-dibenzilamino-2-octanol (89d).
∗
NBn2
NHBn
NO2
HO
- Dados espectrais:
IV (KBr): ν 3392, 3086-3028, 2956-2810, 1553, 1376, 1073, 1029, 749, 700.
RMN 1H (200 MHz, CDCl3): δ 7,4-7,2 (m, 15H), 4,74 (d, 1H, J=2,29), 4,68 (d, 1H,
J=2,29), 4,1 (d, 1H), 4,05 (d, 1H), 3,8 (d, 1H), 3,6 (d, 1H), 3,5-3,2 (m, 2H), 1,95-1,35
(m, 3H), 1,3 (s, 3H), 1,2 (s, 3H), 0,95 (d, 3H), 0,81 (d, 3H).
RMN 13C (50,4 MHz, CDCl3): δ 139,86 (3C), 129,57-127,06 (CH-aromático), 80,69
(C), 70,74 (CH), 61,47 (CH2), 57,64 (CH), 57,30 (CH), 55,18 (CH2), 53,53 (CH2), 36,35
(CH2), 34,48 (CH2), 26,1-21,53 (4 CH3 e CH).
- Razão diastereoisomérica de 50 : 50 medida no CH2 em 36,35 ppm no espectro 73.
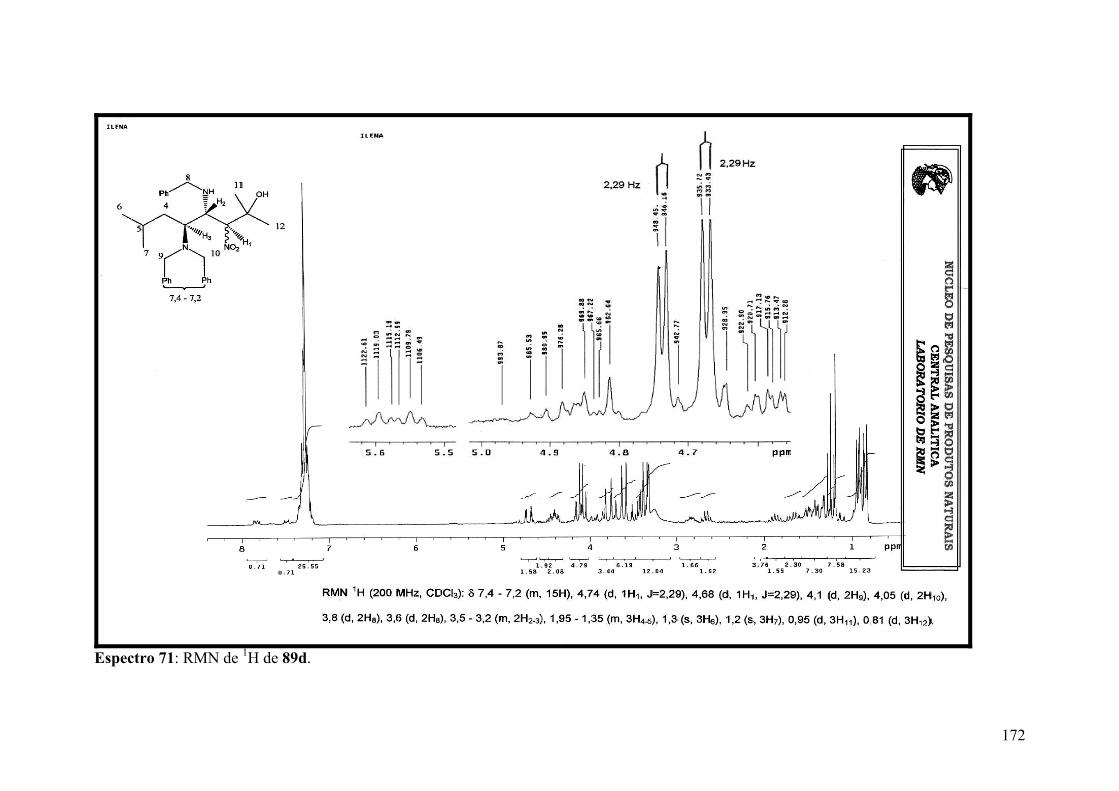
172
Espectro 71: RMN de 1H de 89d.
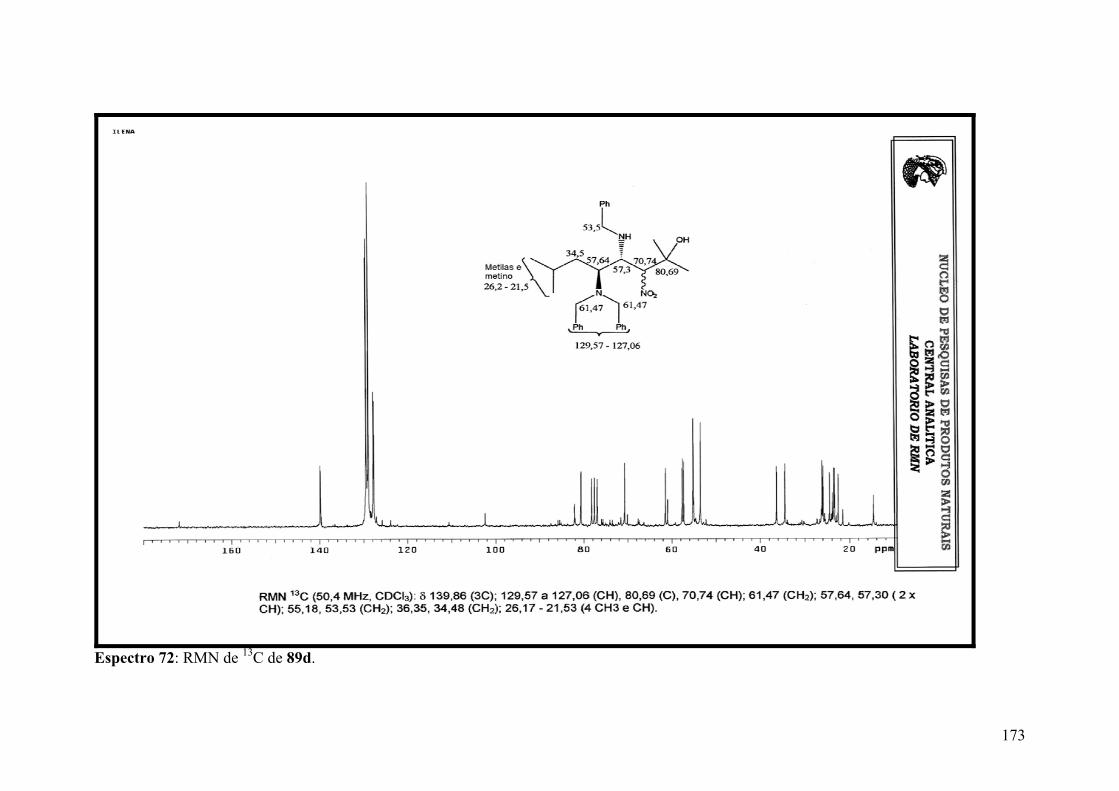
173
Espectro 72: RMN de 13C de 89d.
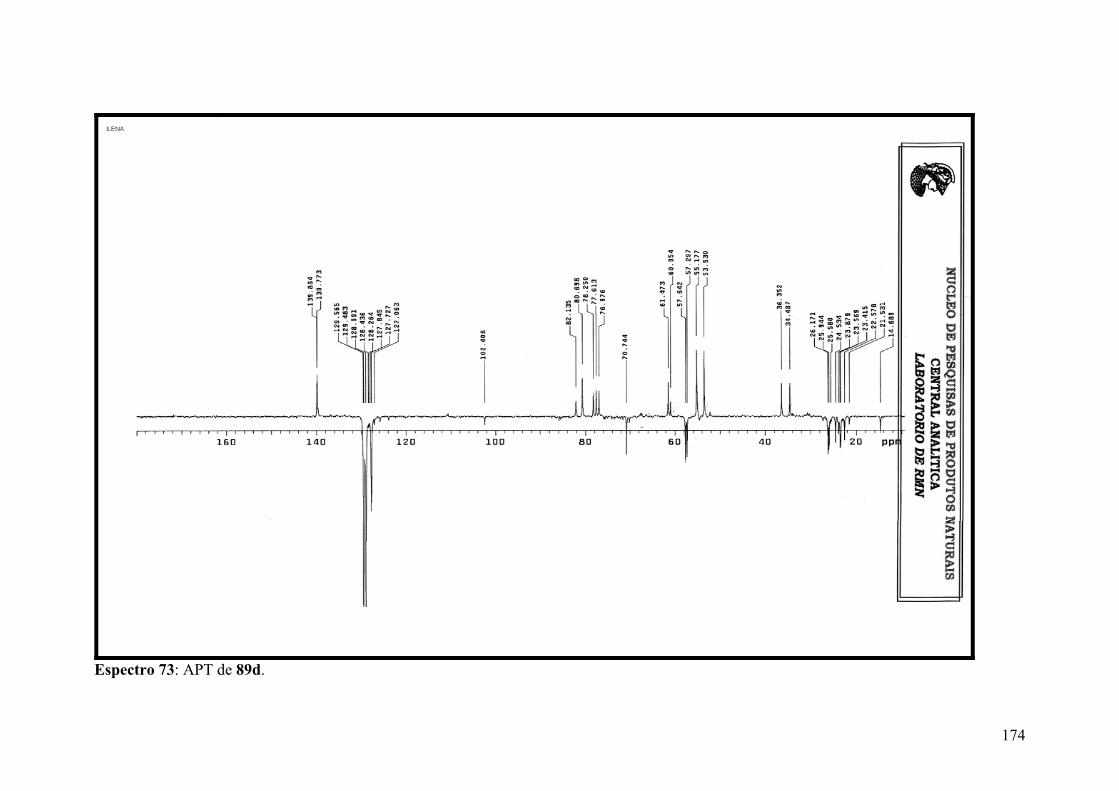
174
Espectro 73: APT de 89d.
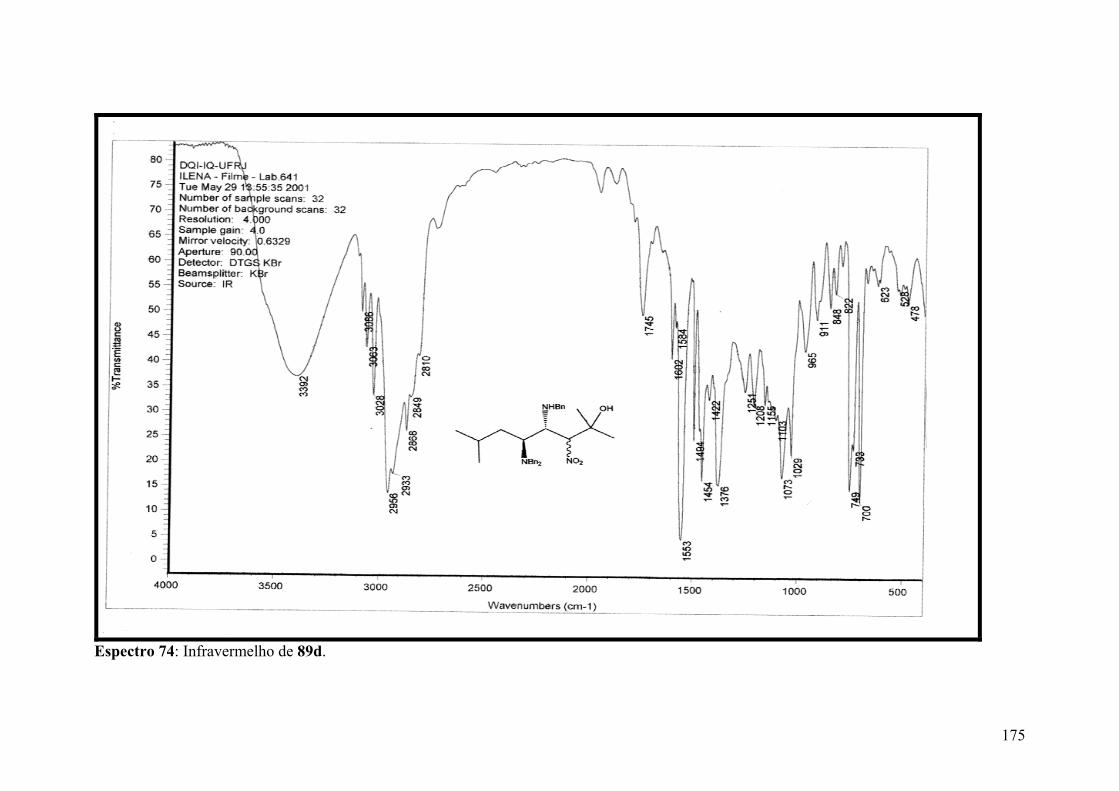
175
Espectro 74: Infravermelho de 89d.
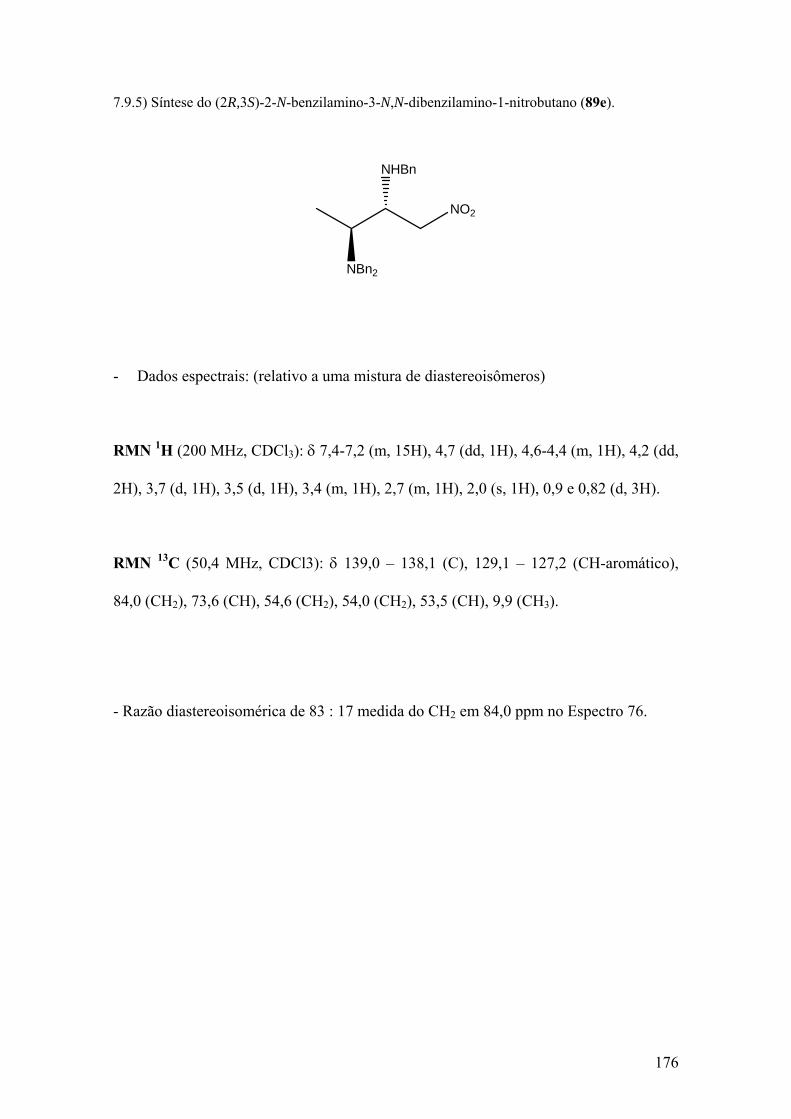
176
7.9.5) Síntese do (2R,3S)-2-N-benzilamino-3-N,N-dibenzilamino-1-nitrobutano (89e).
NBn2
NHBn
NO2
- Dados espectrais: (relativo a uma mistura de diastereoisômeros)
RMN 1H (200 MHz, CDCl3): δ 7,4-7,2 (m, 15H), 4,7 (dd, 1H), 4,6-4,4 (m, 1H), 4,2 (dd,
2H), 3,7 (d, 1H), 3,5 (d, 1H), 3,4 (m, 1H), 2,7 (m, 1H), 2,0 (s, 1H), 0,9 e 0,82 (d, 3H).
RMN 13C (50,4 MHz, CDCl3): δ 139,0 – 138,1 (C), 129,1 – 127,2 (CH-aromático),
84,0 (CH2), 73,6 (CH), 54,6 (CH2), 54,0 (CH2), 53,5 (CH), 9,9 (CH3).
- Razão diastereoisomérica de 83 : 17 medida do CH2 em 84,0 ppm no Espectro 76.
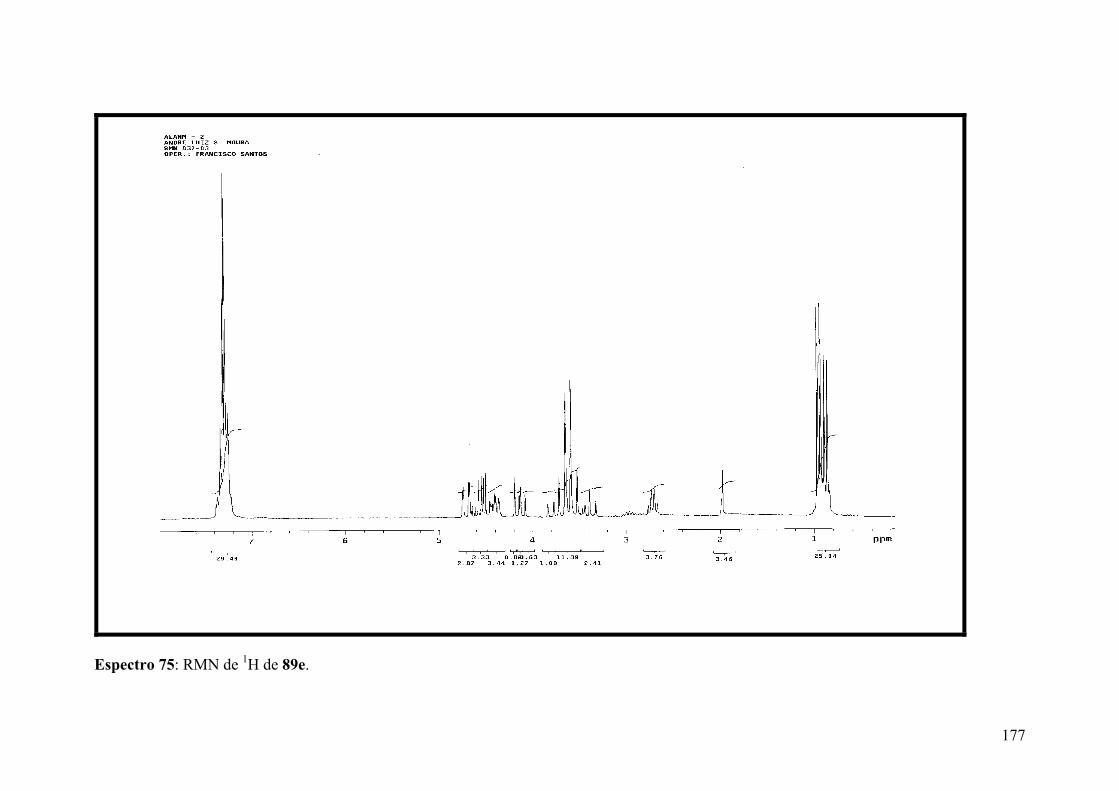
177
Espectro 75: RMN de 1H de 89e.
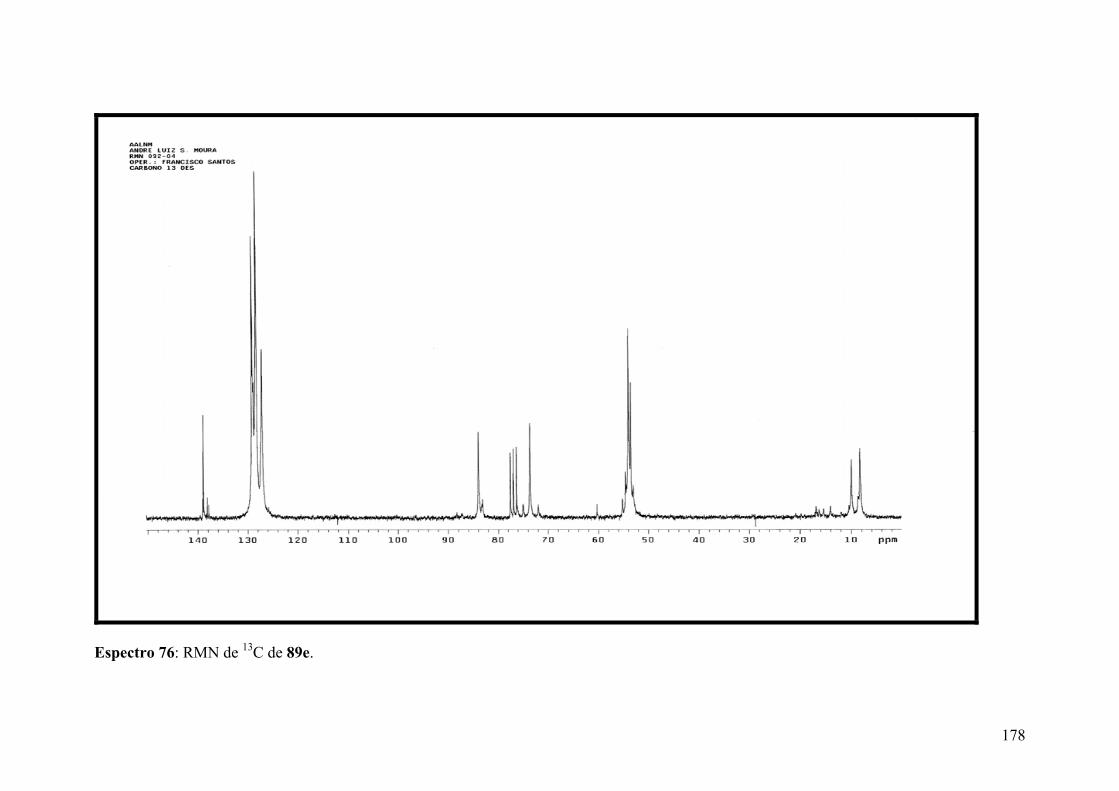
178
Espectro 76: RMN de 13C de 89e.
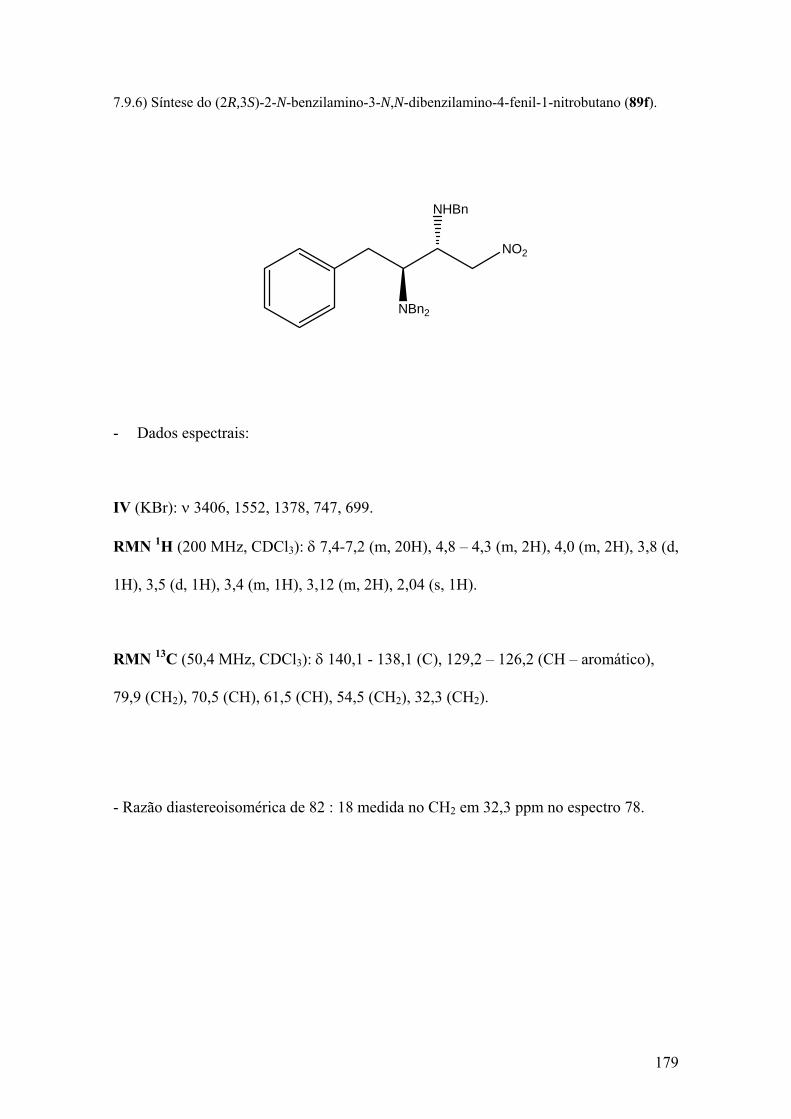
179
7.9.6) Síntese do (2R,3S)-2-N-benzilamino-3-N,N-dibenzilamino-4-fenil-1-nitrobutano (89f).
NBn2
NHBn
NO2
- Dados espectrais:
IV (KBr): ν 3406, 1552, 1378, 747, 699.
RMN 1H (200 MHz, CDCl3): δ 7,4-7,2 (m, 20H), 4,8 – 4,3 (m, 2H), 4,0 (m, 2H), 3,8 (d,
1H), 3,5 (d, 1H), 3,4 (m, 1H), 3,12 (m, 2H), 2,04 (s, 1H).
RMN 13C (50,4 MHz, CDCl3): δ 140,1 - 138,1 (C), 129,2 – 126,2 (CH – aromático),
79,9 (CH2), 70,5 (CH), 61,5 (CH), 54,5 (CH2), 32,3 (CH2).
- Razão diastereoisomérica de 82 : 18 medida no CH2 em 32,3 ppm no espectro 78.
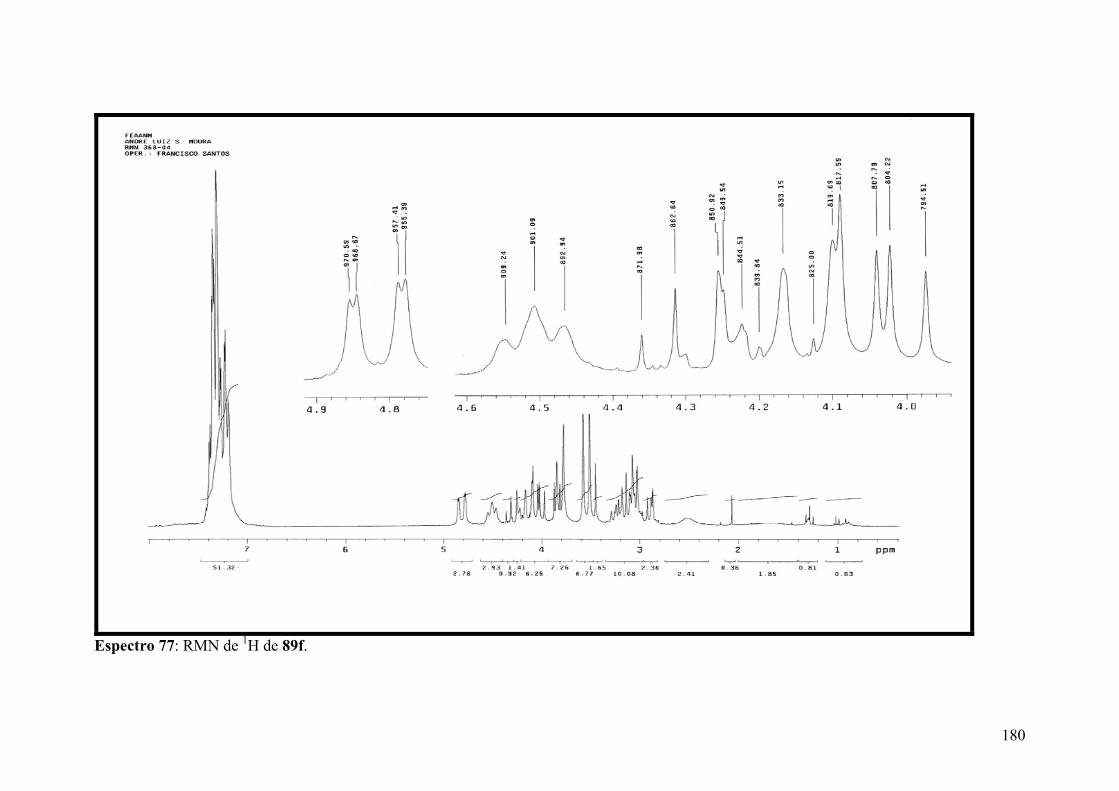
180
Espectro 77: RMN de 1H de 89f.
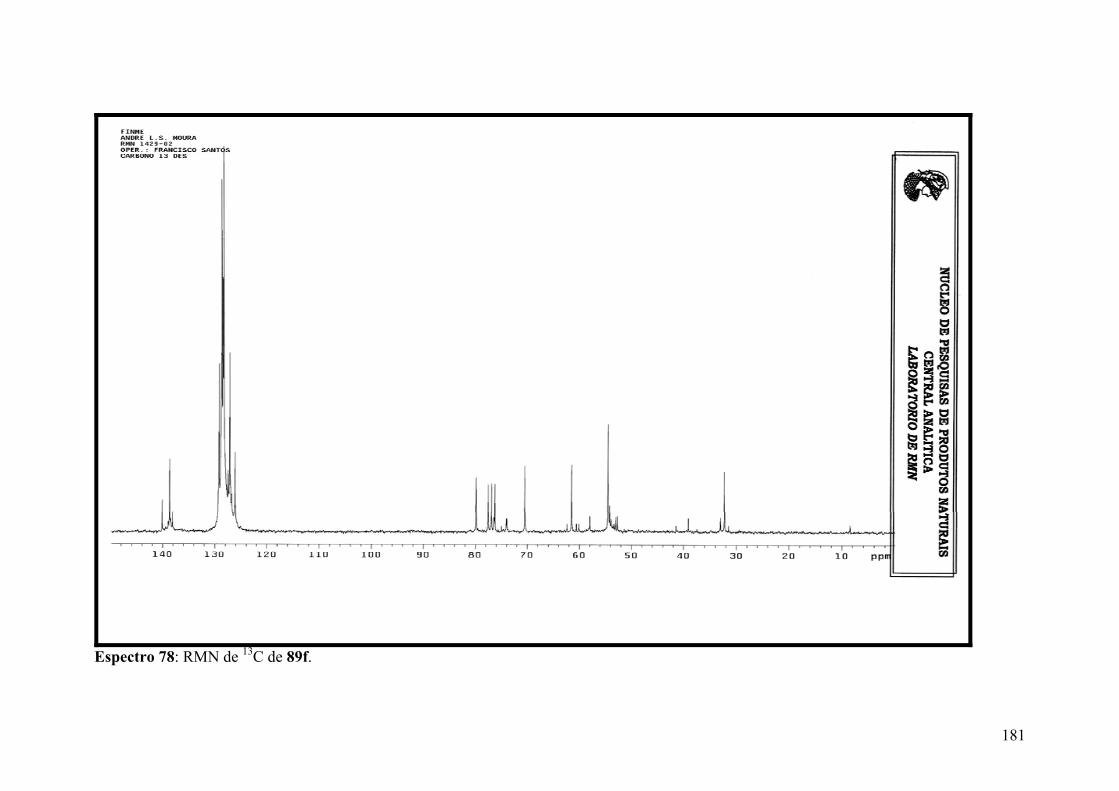
181
Espectro 78: RMN de 13C de 89f.
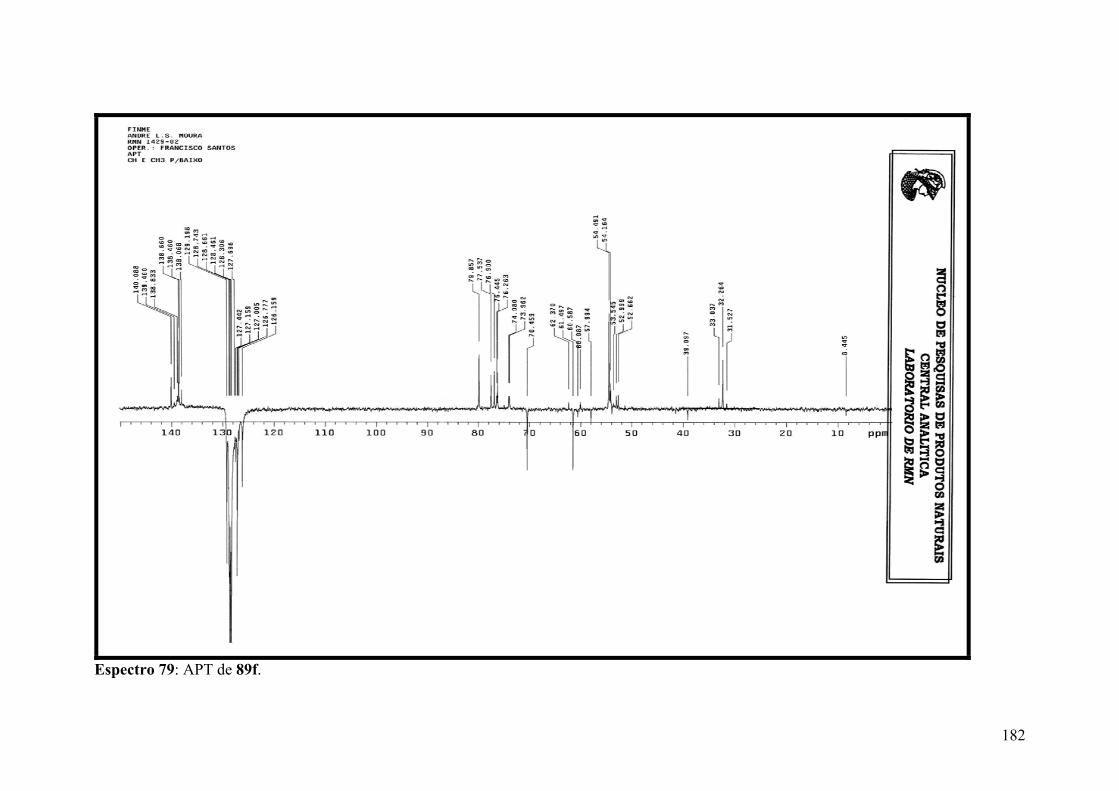
182
Espectro 79: APT de 89f.
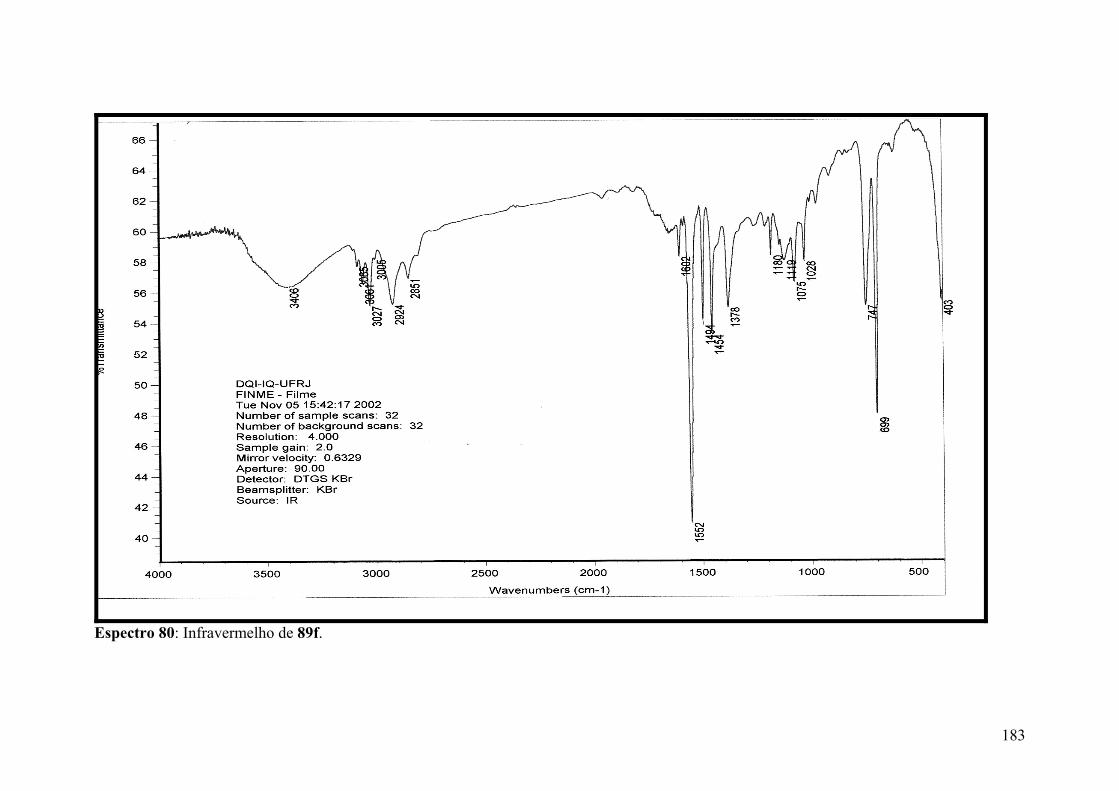
183
Espectro 80: Infravermelho de 89f.
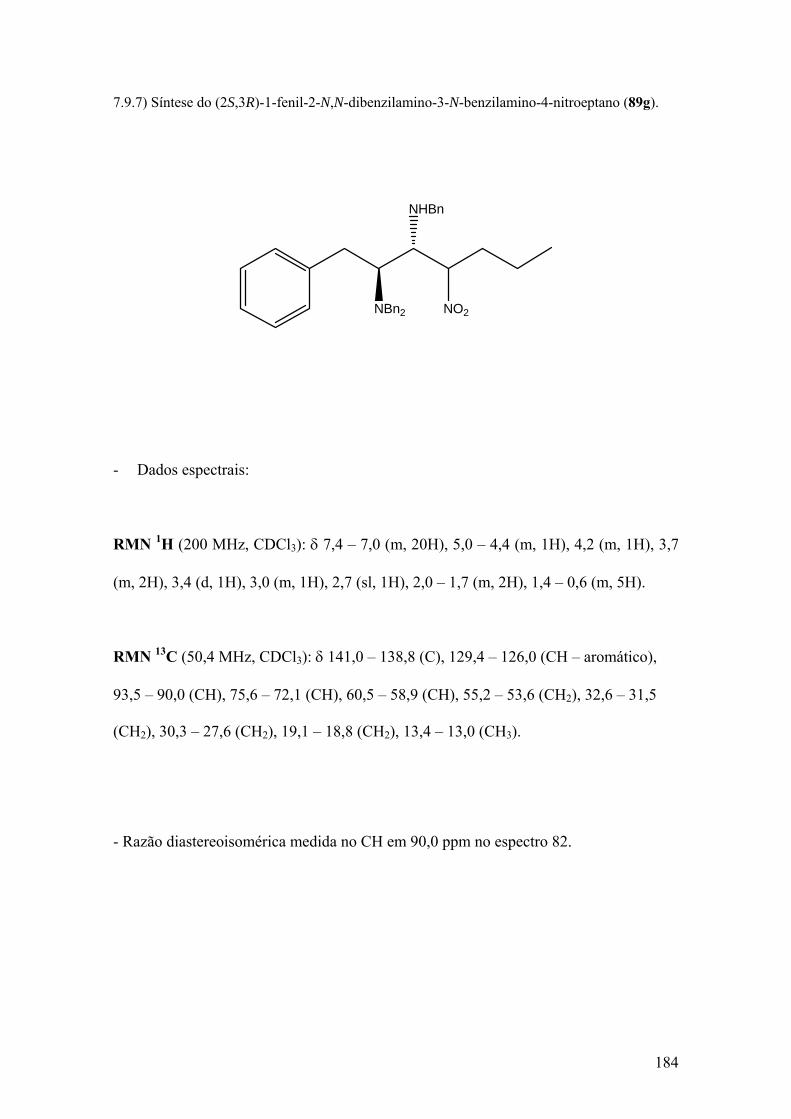
184
7.9.7) Síntese do (2S,3R)-1-fenil-2-N,N-dibenzilamino-3-N-benzilamino-4-nitroeptano (89g).
NBn2
NHBn
NO2
- Dados espectrais:
RMN 1H (200 MHz, CDCl3): δ 7,4 – 7,0 (m, 20H), 5,0 – 4,4 (m, 1H), 4,2 (m, 1H), 3,7
(m, 2H), 3,4 (d, 1H), 3,0 (m, 1H), 2,7 (sl, 1H), 2,0 – 1,7 (m, 2H), 1,4 – 0,6 (m, 5H).
RMN 13C (50,4 MHz, CDCl3): δ 141,0 – 138,8 (C), 129,4 – 126,0 (CH – aromático),
93,5 – 90,0 (CH), 75,6 – 72,1 (CH), 60,5 – 58,9 (CH), 55,2 – 53,6 (CH2), 32,6 – 31,5
(CH2), 30,3 – 27,6 (CH2), 19,1 – 18,8 (CH2), 13,4 – 13,0 (CH3).
- Razão diastereoisomérica medida no CH em 90,0 ppm no espectro 82.

185
Espectro 81: RMN de 1H de 89g.
186
Espectro 82: RMN de 13C de 89g.
187
Espectro 83: APT de 89g.

188
7.10) Procedimento geral para a síntese das nitroazidas 94b e 94e.
7.10.1) Síntese do (2R,3S)-2-azido-3-N,N-dibenzilamino-4-fenil-1-nitrobutano (94e).
NBn2
NO2
N3
A 0,1896g de TBAF (0,6 mmol) solubilizado em 5,0 mL de THF seco, foi
adicionado 0,08 mL de trimetilsililazida (0,6 mmol). A esta solução foi adicionado
0,20 g da nitroolefina 93b (0,54 mmol) solubilizada em 5,0 mL de THF seco. A mistura
reacional foi mantida sob agitação magnética por 20 h à temperatura ambiente. Após o
término da reação, a mistura reacional foi lavada com água e então extraída com AcOEt
(3 x 10,0 mL) e os extratos orgânicos foram secos com Na2SO4 e evaporados sob
pressão reduzida, fornecendo a nitroaminoazida 94e, como um líquido viscoso amarelo-
alaranjado, em 67 % de rendimento químico após purificação em coluna de gel de
sílica tendo como eluente uma mistura de Hexano / AcOEt 20 %.
- Dados espectrais:
IV (KBr): ν 2132, 2100, 1555, 1377, 1075, 1024, 747, 700.
RMN 1H (200 MHz, CDCl3): δ 7,4 – 7,2 (m, 15H), 4,75 (m, 2H,), 4,16 (m, 1H),
3,8 (m, 4H), 3,4 (d, 1H), 3,0 (m, 2H).
RMN 13C (50,4 MHz, CDCl3): δ 140,1 (C), 129,1 – 126,5 (CH – aromático), 77,8
(CH2), 61,0 (CH), 60,6 (CH), 55,3 (CH2), 30,2 (CH2).
CG/EM (70 eV): m/z: 195, 91 (pico base – íon tropílio).
- Razão diastereoisomérica medida através de proporção em cromatografia gasosa (cromatograma 2).
189
Espectro 84: RMN de 1H de 94e.
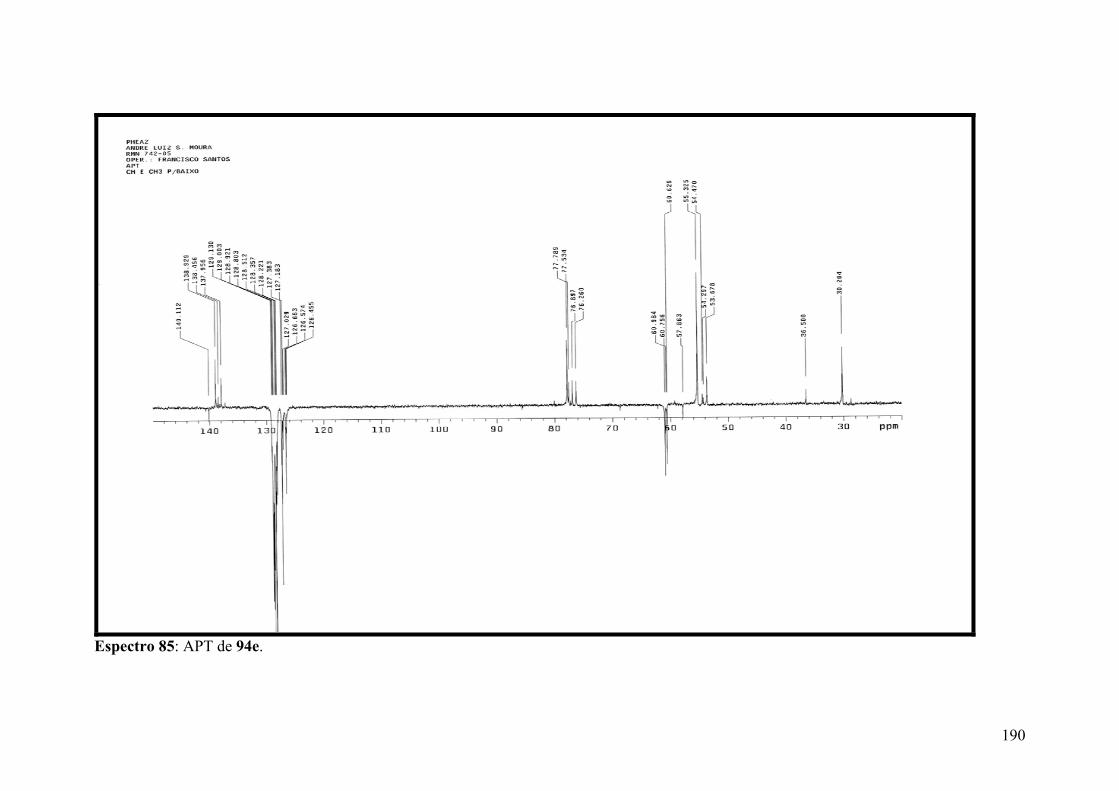
190
Espectro 85: APT de 94e.
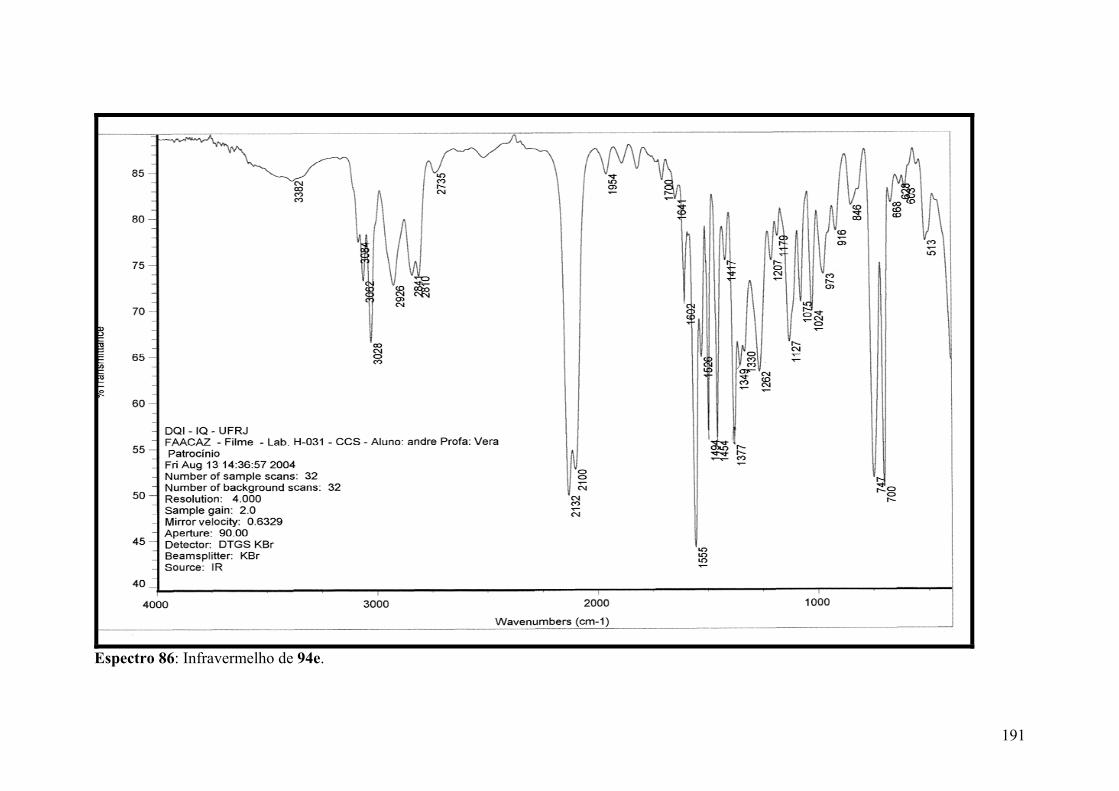
191
Espectro 86: Infravermelho de 94e.
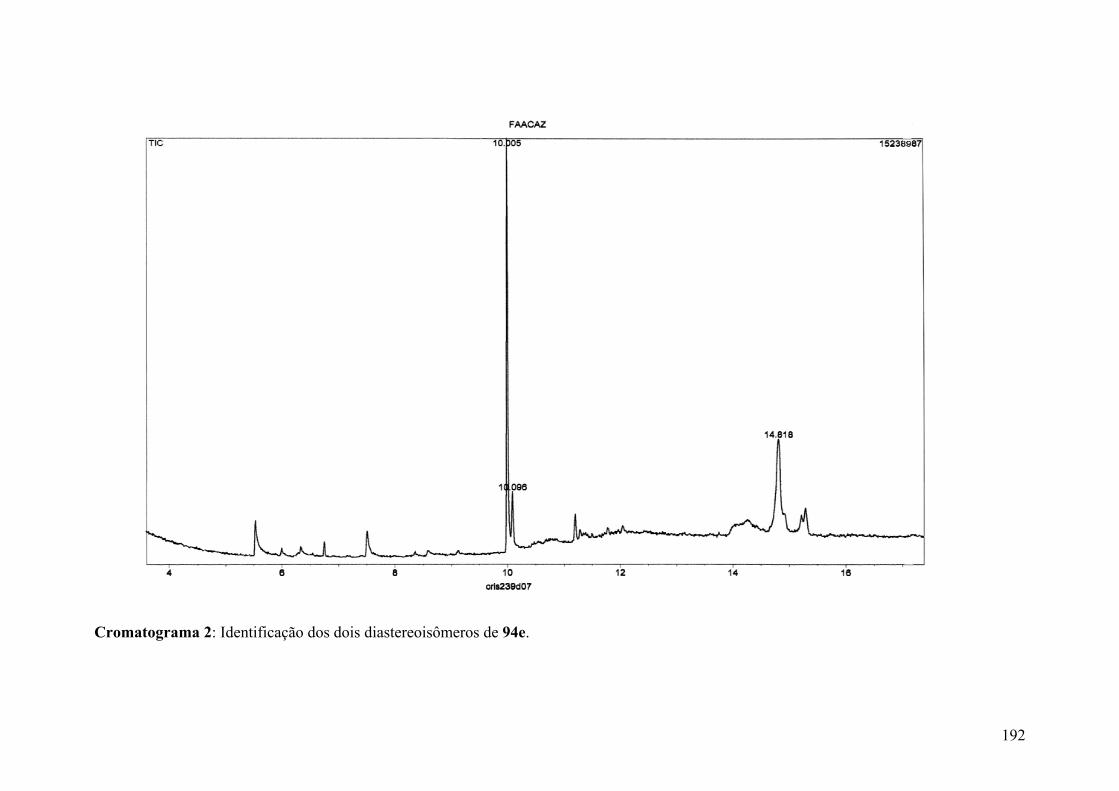
192
Cromatograma 2: Identificação dos dois diastereoisômeros de 94e.
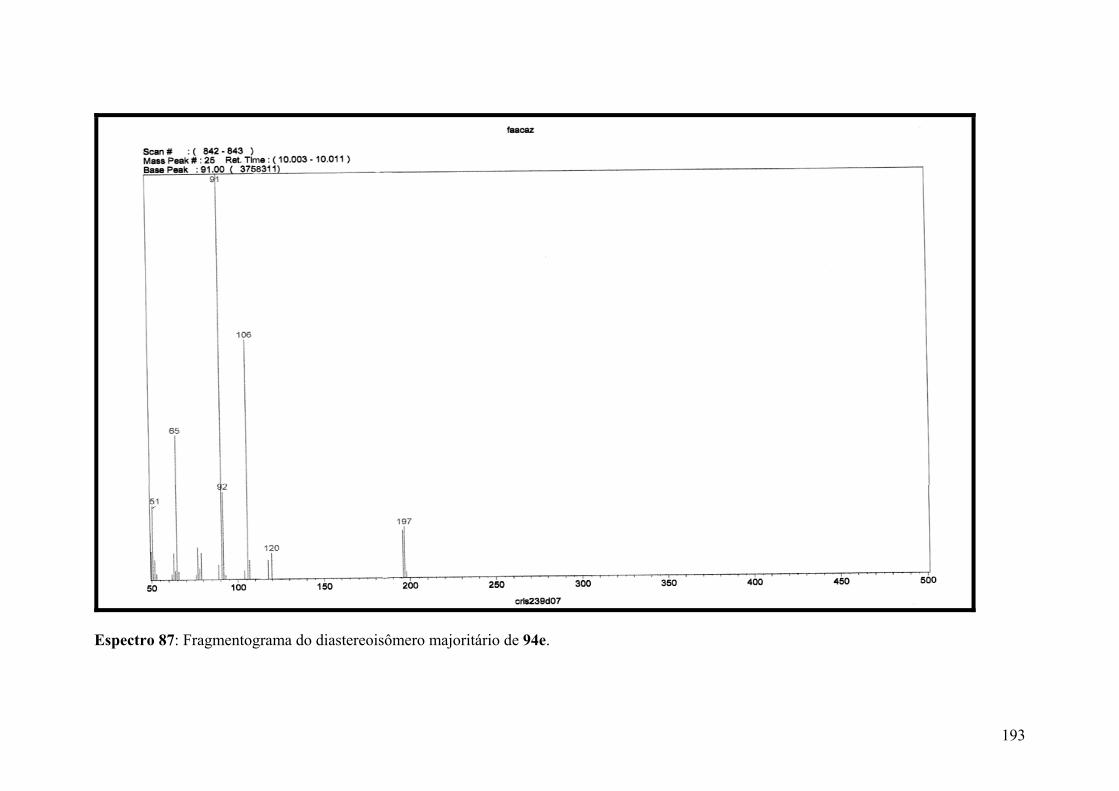
193
Espectro 87: Fragmentograma do diastereoisômero majoritário de 94e.
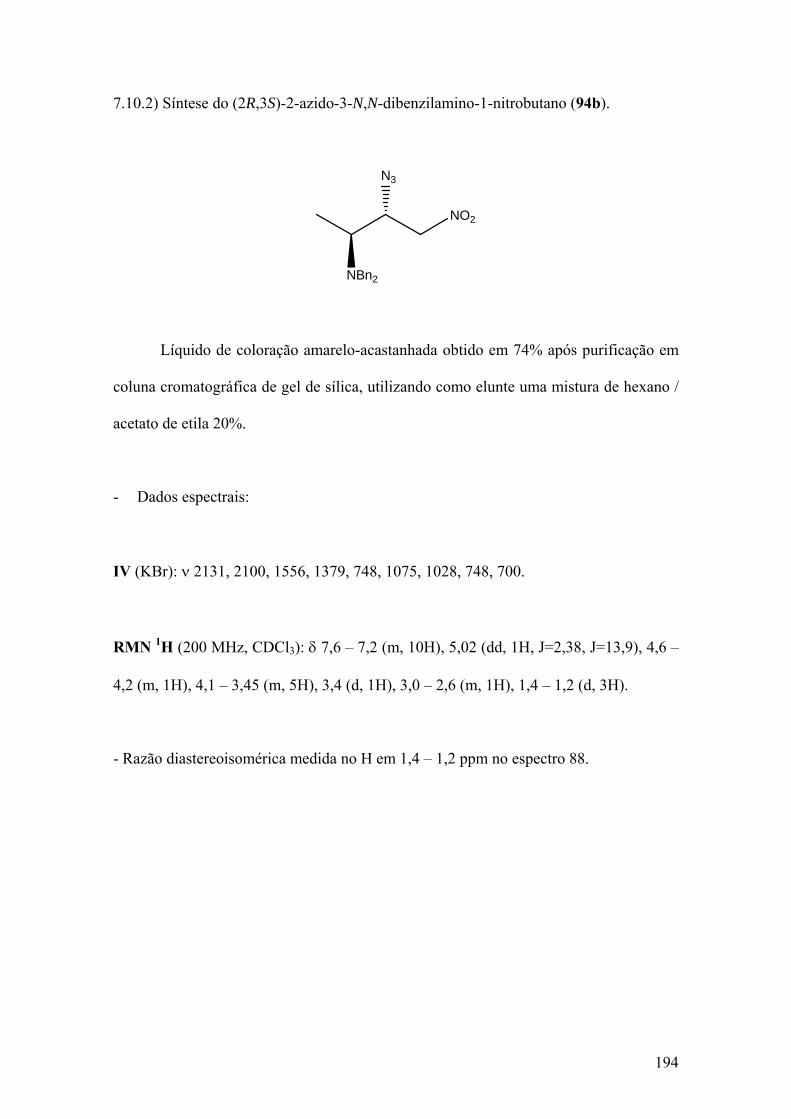
194
7.10.2) Síntese do (2R,3S)-2-azido-3-N,N-dibenzilamino-1-nitrobutano (94b).
NBn2
NO2
N3
Líquido de coloração amarelo-acastanhada obtido em 74% após purificação em
coluna cromatográfica de gel de sílica, utilizando como elunte uma mistura de hexano /
acetato de etila 20%.
- Dados espectrais:
IV (KBr): ν 2131, 2100, 1556, 1379, 748, 1075, 1028, 748, 700.
RMN 1H (200 MHz, CDCl3): δ 7,6 – 7,2 (m, 10H), 5,02 (dd, 1H, J=2,38, J=13,9), 4,6 –
4,2 (m, 1H), 4,1 – 3,45 (m, 5H), 3,4 (d, 1H), 3,0 – 2,6 (m, 1H), 1,4 – 1,2 (d, 3H).
- Razão diastereoisomérica medida no H em 1,4 – 1,2 ppm no espectro 88.
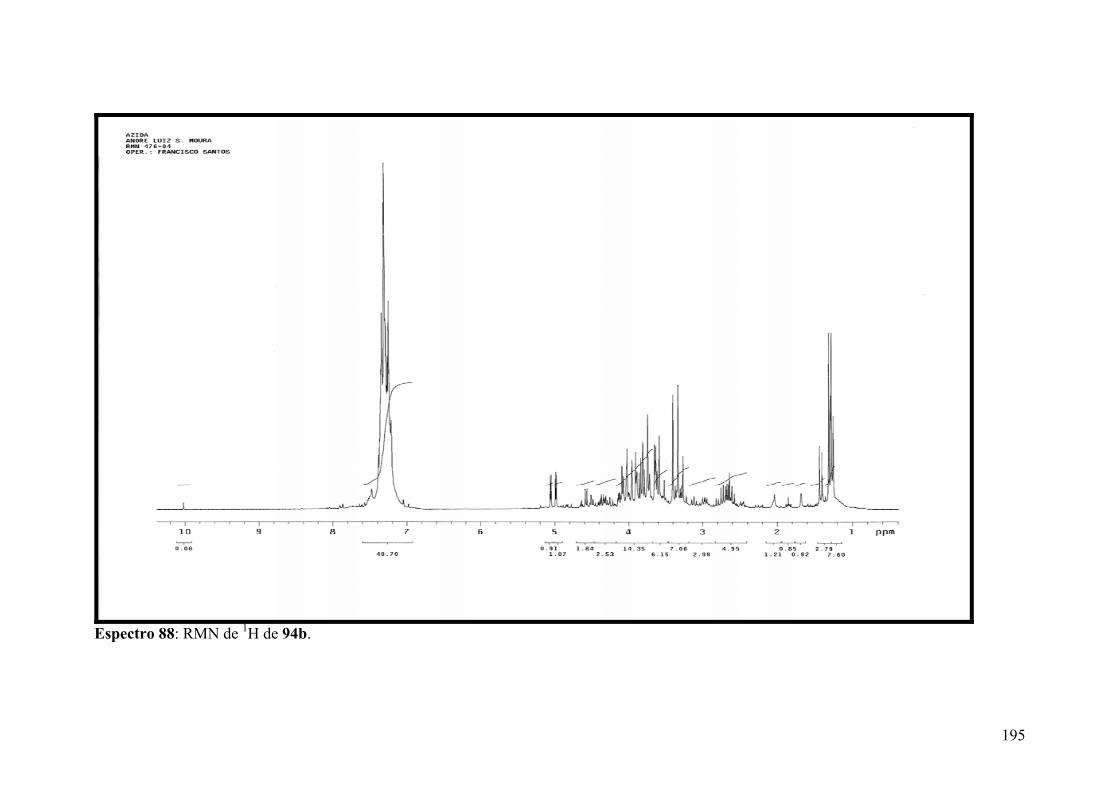
195
Espectro 88: RMN de 1H de 94b.
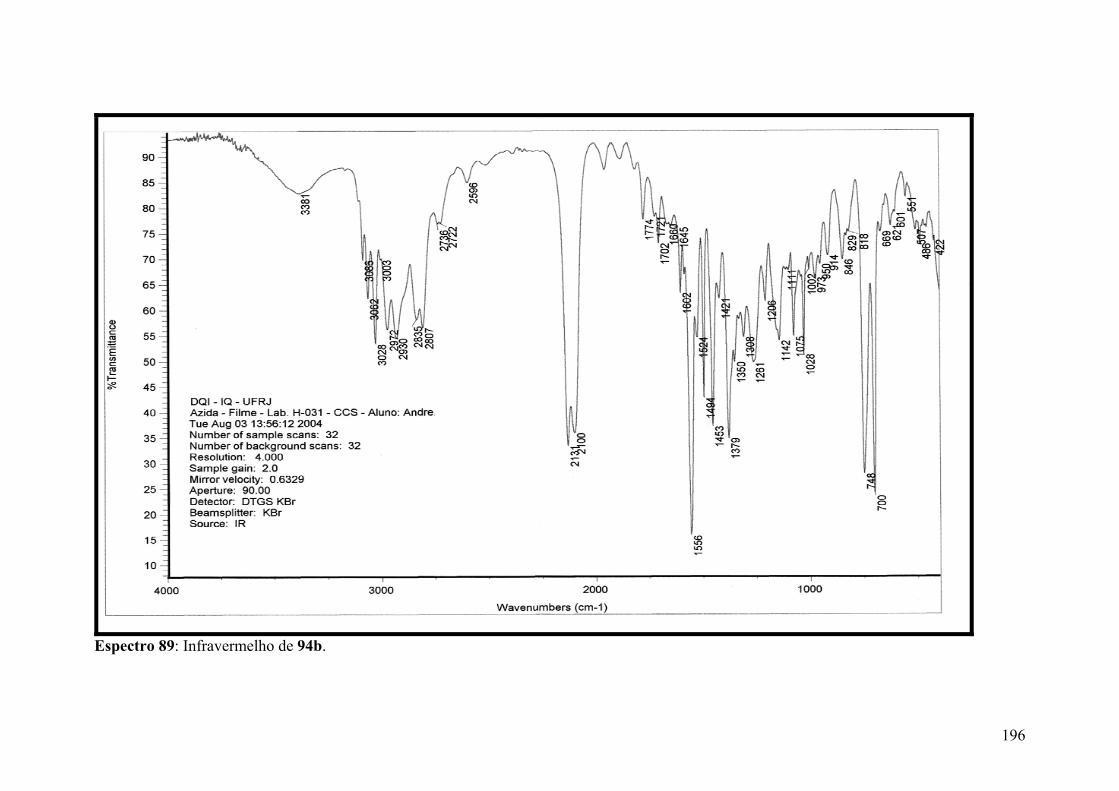
196
Espectro 89: Infravermelho de 94b.

197
7.11) Procedimento geral para a síntese dos nitroéteres 94a e 94d.
7.11.1) Síntese do (2R,3S)-2-metoxi-3-N,N-dibenzilamino-1-nitrobutano (94a).
NBn2
NO2
O
A uma solução de nitroolefina 93a (0,150g ; 0,51 mmol) solubilizada em 5,0 mL
de metanol foram adicionados 0,51 mL de uma solução de metóxido de lítio / metanol
1,0 M gota à gota no intervalo de 3 horas à temperatura ambiente. Findo esse tempo, o
metanol foi evaporado sob pressão reduzida. O produto 94a, líquido amarelado pouco
viscoso, foi purificado através de cromatografia em coluna de gel de sílica eluído com
de 20% hexano / AcOEt. Obteve-se um óleo amarelo em 77% de rendimento químico.
- Dados espectrais:
RMN 1H (200 MHz, CDCl3): δ 7,4 – 7,2 (m, 10H), 4,96 – 4,76 (ddd, 2H, J= 2,2), 4,1
(dd, 1H, J=2,9), 4,0 (d, 1H, J=12,5), 3,9 (m, 1H), 3,8 (d, 1H, J=13,0), 3,4 (s, 3H), 3,3
(m, 1H), 1,2 (m, 3H).
- Razão diastereoisomérica de 69 : 31 medida no H (CH3O~) em 3,4 ppm – espectro 90.
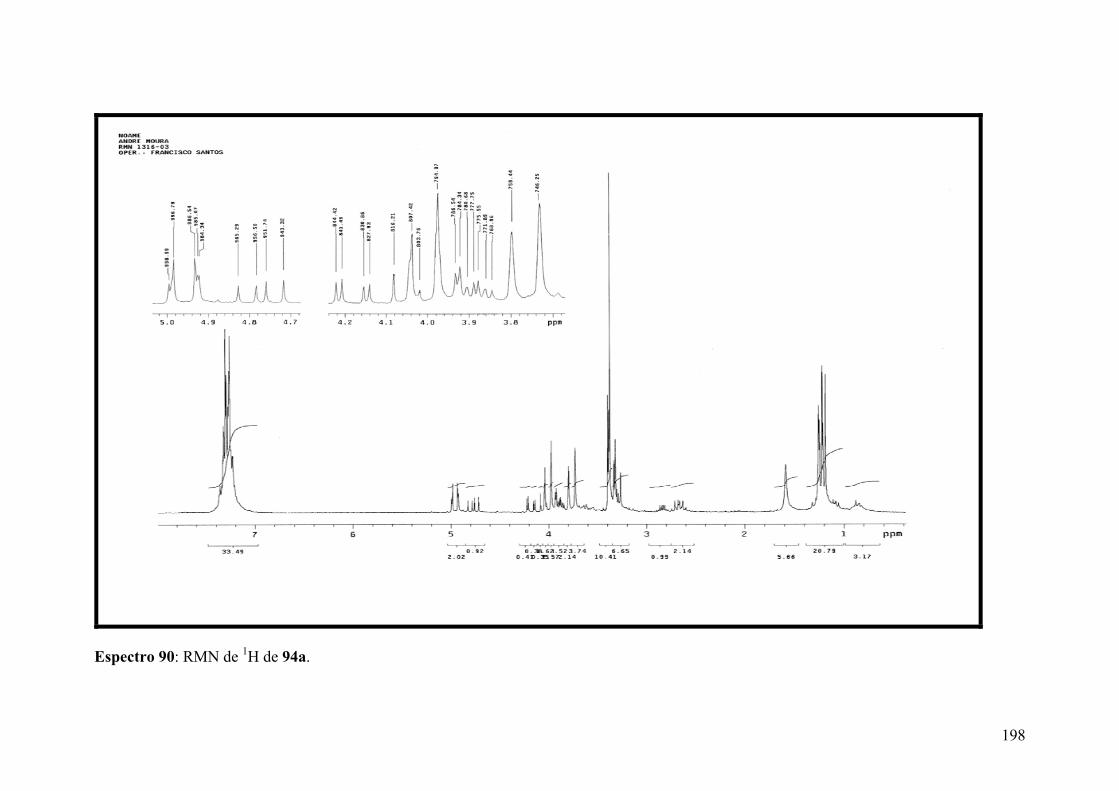
198
Espectro 90: RMN de 1H de 94a.

199
7.11.2) Síntese do (2R,3S)-2-metoxi-3-N,N-dibenzilamino-4-fenil-1-nitrobutano (94d).
NBn2
NO2
O
Líquido amarelado claro pouco viscoso obtido em 70% de rendimento químico
após purificação em coluna cromatográfica de gel de sílica, tendo como eluente uma
mistura de hexano/acetato de etila 20%.
- Dados espectrais:
IV (KBr): ν 1552, 1380, 1106, 1075, 746, 699.
RMN 1H (200 MHz, CDCl3): δ 7,4 – 7,2 (m, 15H), 4,76 (m, 2H), 4,1 (m, 1H), 3,7 (d,
1H), 3,5 (d, 1H), 3,3 (s, 3H), 3,0 (m, 2H).
RMN 13C (50,4 MHz, CDCl3): δ 140,5 – 138,9 (C), 129,4 – 126,4 (CH – aromático),
80,62 3 79,88 (CH), 79,00 e 78,71 (CH2), 61,62 e 61,63 (CH), 59,82 e 59,29 (CH3),
55,51 e 54,44 (CH2), 32,65 e 29,23 (CH3).
- Razão diastereoisomérica de 57 : 43 medida no H em 3,3 ppm – espectro 91.
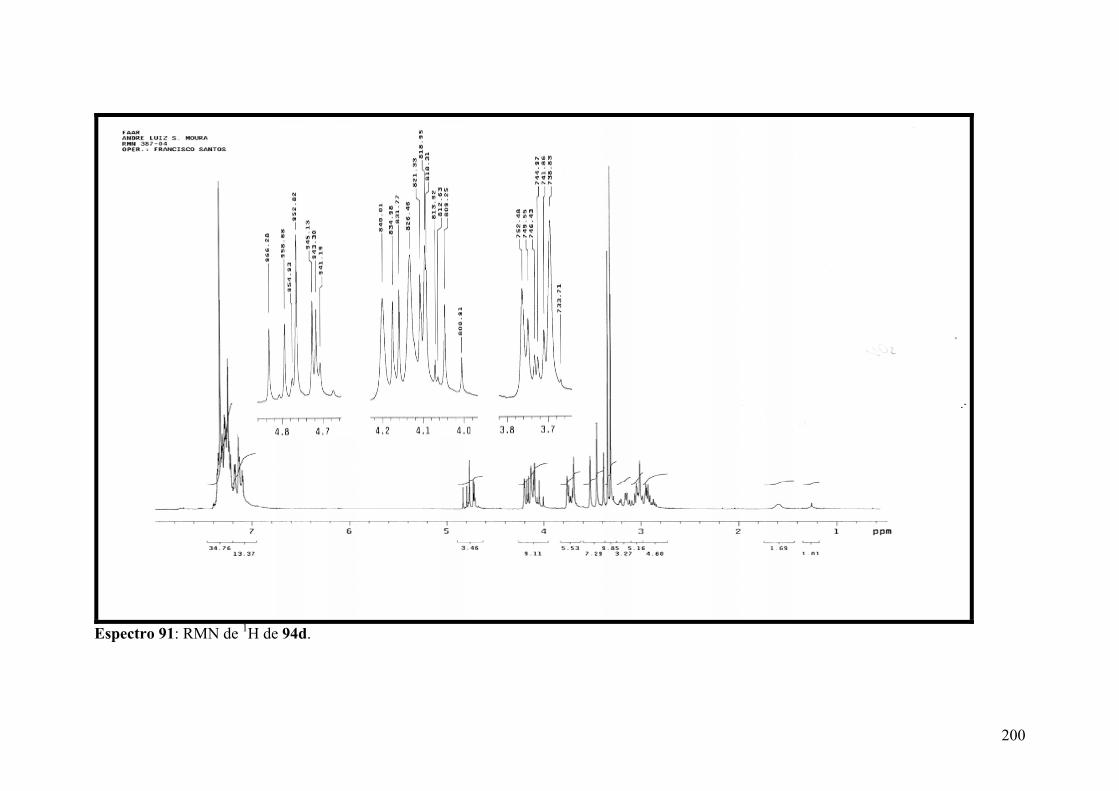
200
Espectro 91: RMN de 1H de 94d.
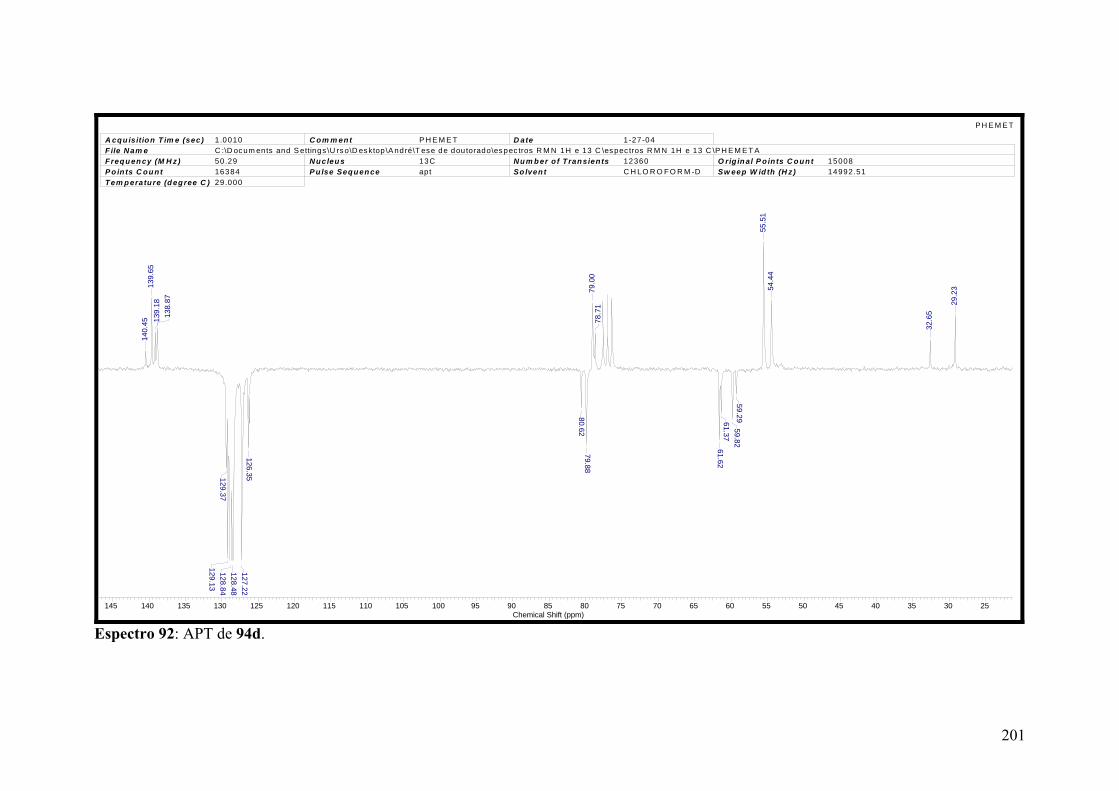
201
P H E M E T
A cqu isition T im e (sec) 1.0010 C om m en t P H E M E T D ate 1-27-04F ile Nam e C :\D ocum ents and S ettings \U rso\D esktop\A ndré\T ese de dou torado\espec tros R M N 1H e 13 C \espec tros R M N 1H e 13 C \P H E M E T AFrequ en cy (M H z ) 50.29 Nucleu s 13C Nu m b er o f T ransien ts 12360 O rig in al P o in ts C o un t 15008P o in ts C ou nt 16384 P u lse Sequ en ce apt So lven t C H LO R O F O R M -D Sw eep W id th (H z ) 14992.51Tem p erature (d eg ree C ) 29.000
145 140 135 130 125 120 115 110 105 100 95 90 85 80 75 70 65 60 55 50 45 40 35 30 25Chemical Shift (ppm)
140.
4513
9.65
139.
1813
8.87
129.37129.13
128.84128.48127.22
126.35
80.6279.88
79.0
078
.71
61.6261.37
59.8259.29
55.5
154
.44
32.6
5
29.2
3
Espectro 92: APT de 94d.
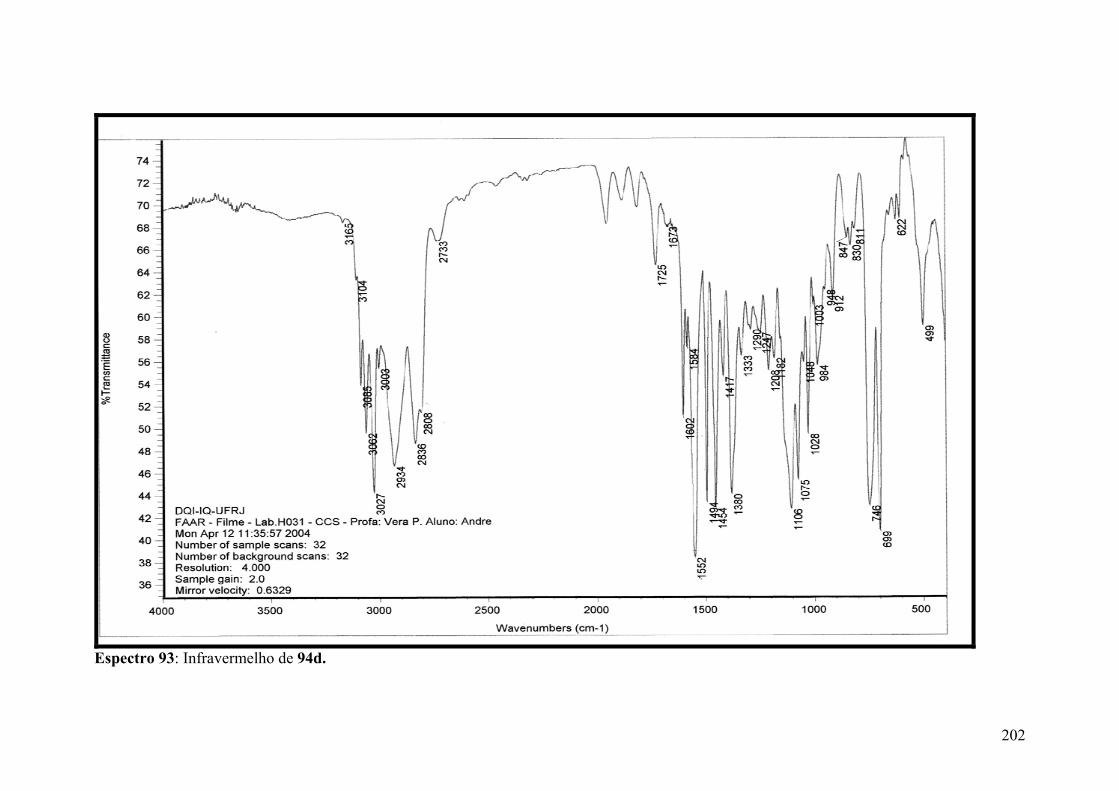
202
Espectro 93: Infravermelho de 94d.
203
7.12) Síntese das dinitroaminas 94c e 94f:
Procedimento I:
7.12.1) Síntese do (2S,3S)-1-fenil-2-N,N-dibenzilamino-3-nitrometil-4-metil-4-
nitropentano (94f).
NBn2
O2N
NO2
A uma solução de 0,0348g de TBAF (0,2 eq. ; 0,11 mmol) solubilizado em
5,0 mL de THF seco e 60,0 μL de 2-nitropropano (1,2 eq. ; 0,65 mmol), foram
adicionados 0,20g da nitroolefina 93b (0,54 mmol) solubilizada em 5,0 mL de THF
seco e mantido sob agitação magnética à temperatura ambiente. A mistura reagiu por 24
horas sendo, então, concentrada sob pressão reduzida. A resíduo remanescente foi
submetido a uma cromatografia em coluna de gel de sílica e eluída com hexano / AcOEt
20%. Obteve-se um líquido marrom muito viscoso em 65% de rendimento químico.
- Dados espectrais:
IV (KBr): ν 3086-3028, 2928-2803, 1556, 1544, 1377, 1345, 745, 700 cm-1.
RMN 1H (200 MHz, CDCl3): δ 7,4 – 6,9 (m, 15H), 4,9 – 4,66 (ddd, 2H, J= 8,0, J= 2,2),
3,9 (d, 1H), 3,6 (m, 1H), 3,4 (d, 1H), 2,9 – 2,3 (m, 2H), 2,2 (m, 1H), 1,6 – 1,4 (s, 6H).
RMN 13C (50,4 MHz, CDCl3): δ 139,1 – 138,3 (C), 130,1 – 126,8 (CH – aromático),
90,1 (C), 73,9 (CH2), 58,2 (CH2), 52,3 (CH2), 44,1 (CH), 35,0 (CH2), 25,8 (CH3), 24,8
(CH3).
- Razão diastereoisomérica de 93 : 7 medida no C em 90,1 ppm – espectro 95.
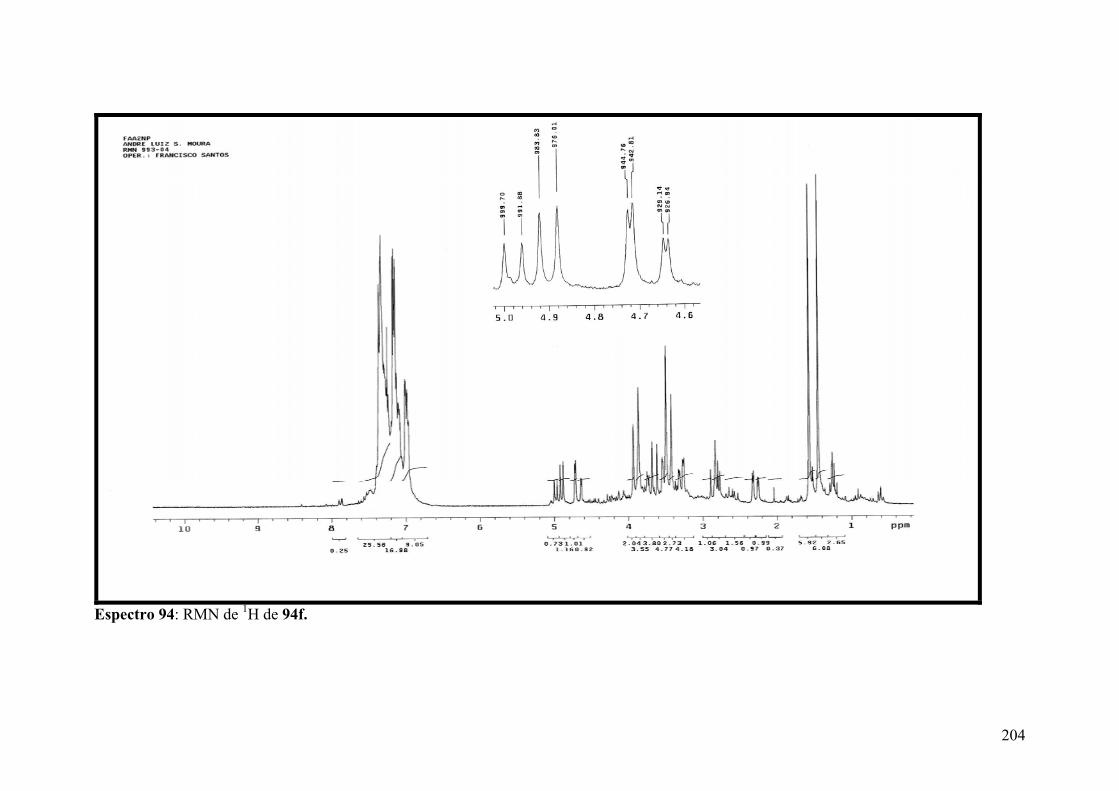
204
Espectro 94: RMN de 1H de 94f.
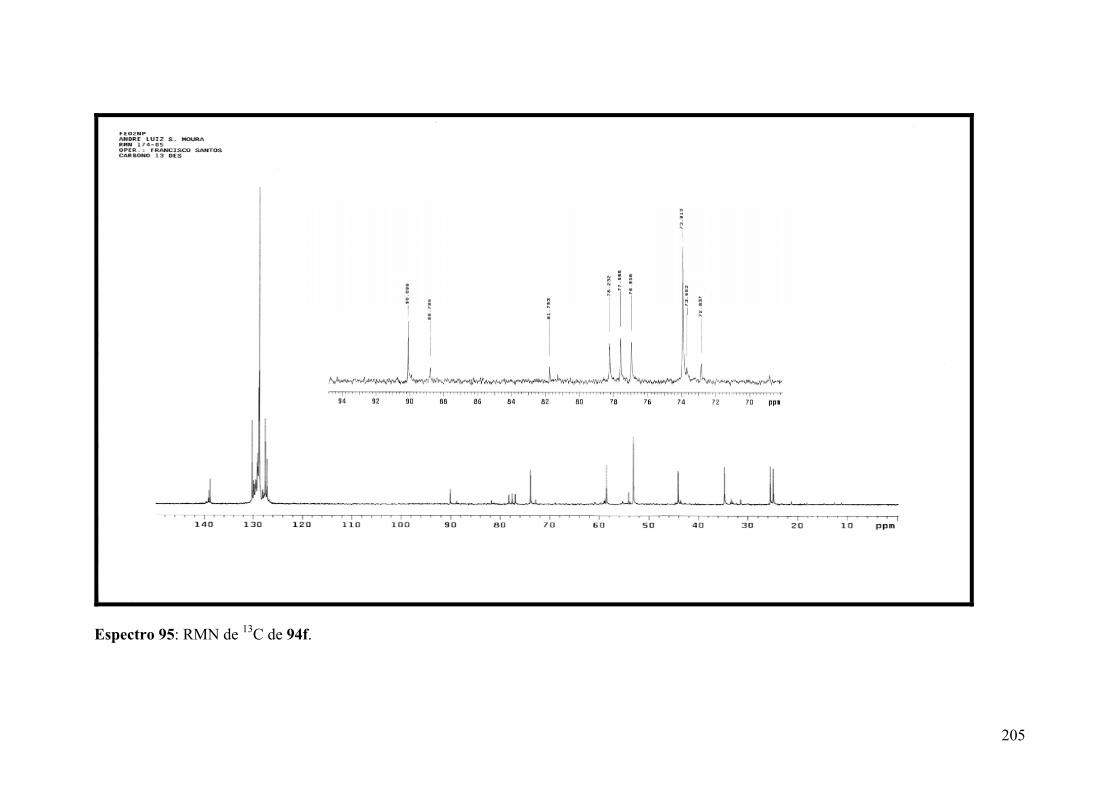
205
Espectro 95: RMN de 13C de 94f.
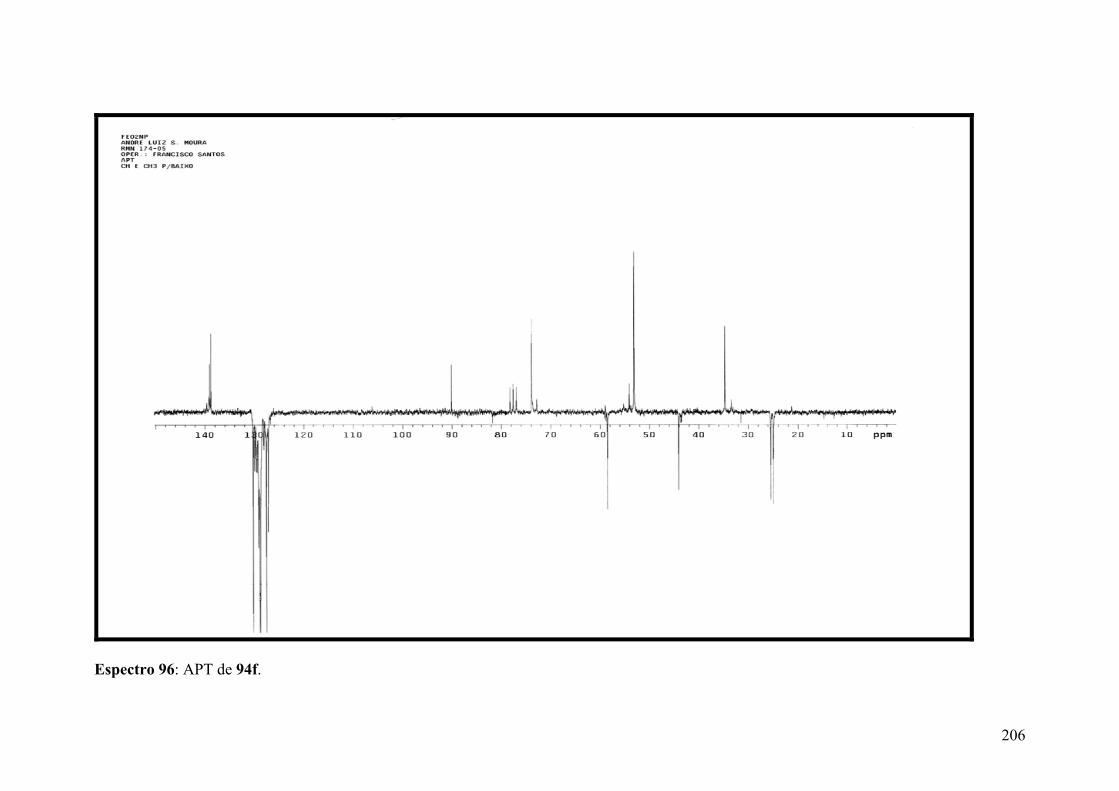
206
Espectro 96: APT de 94f.
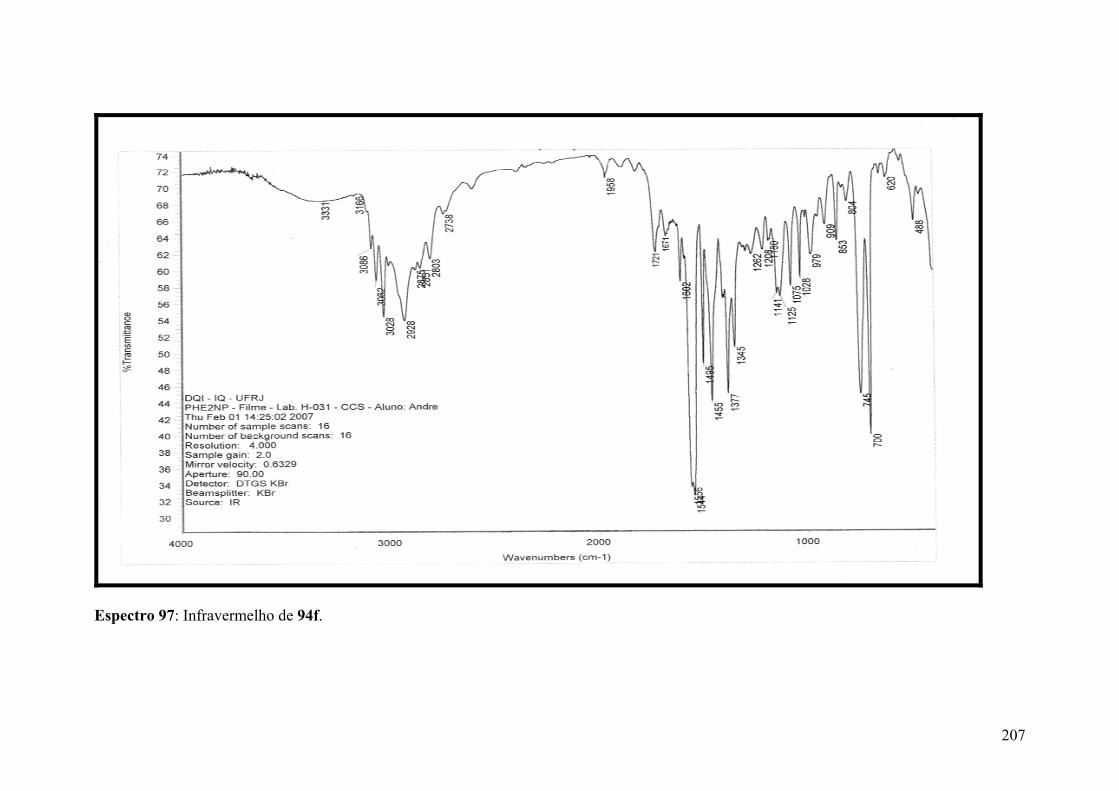
207
Espectro 97: Infravermelho de 94f.
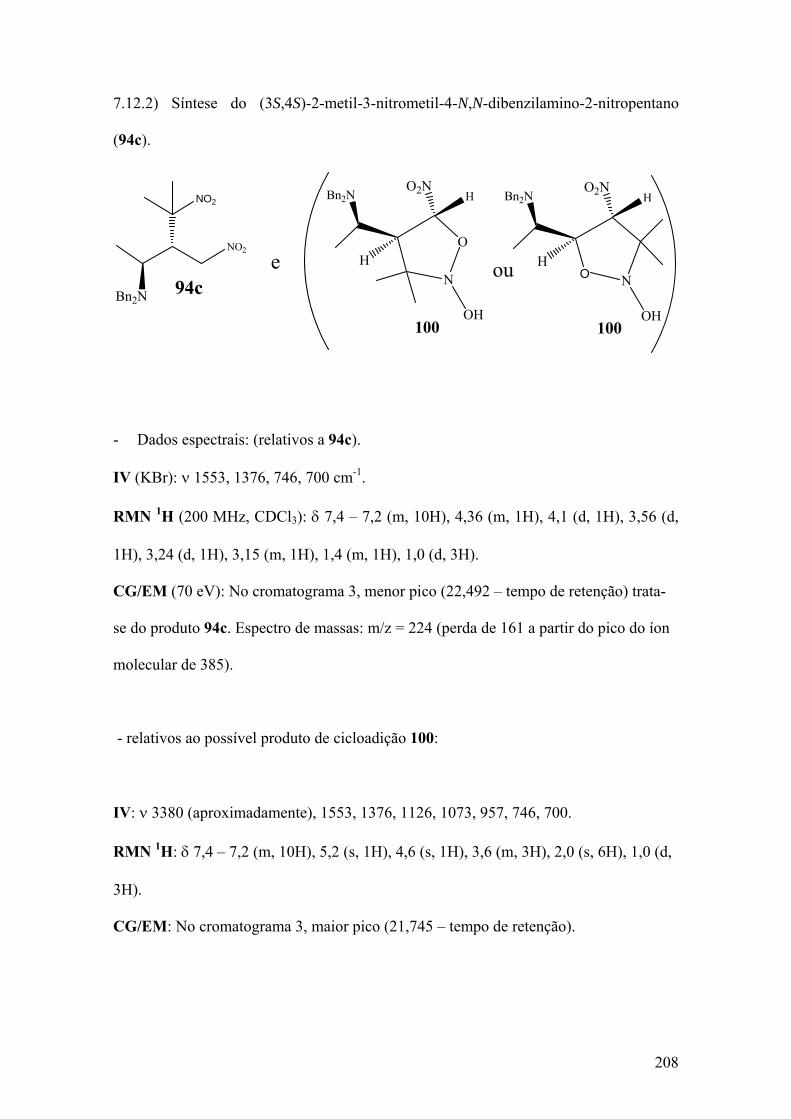
208
7.12.2) Síntese do (3S,4S)-2-metil-3-nitrometil-4-N,N-dibenzilamino-2-nitropentano
(94c).
HN
O
OH
O2N
NO2
Bn2N
NO2
94c
100
Bn2N H
ou HO N
OH
O2NBn2N H
e
100
- Dados espectrais: (relativos a 94c).
IV (KBr): ν 1553, 1376, 746, 700 cm-1.
RMN 1H (200 MHz, CDCl3): δ 7,4 – 7,2 (m, 10H), 4,36 (m, 1H), 4,1 (d, 1H), 3,56 (d,
1H), 3,24 (d, 1H), 3,15 (m, 1H), 1,4 (m, 1H), 1,0 (d, 3H).
CG/EM (70 eV): No cromatograma 3, menor pico (22,492 – tempo de retenção) trata-
se do produto 94c. Espectro de massas: m/z = 224 (perda de 161 a partir do pico do íon
molecular de 385).
- relativos ao possível produto de cicloadição 100:
IV: ν 3380 (aproximadamente), 1553, 1376, 1126, 1073, 957, 746, 700.
RMN 1H: δ 7,4 – 7,2 (m, 10H), 5,2 (s, 1H), 4,6 (s, 1H), 3,6 (m, 3H), 2,0 (s, 6H), 1,0 (d,
3H).
CG/EM: No cromatograma 3, maior pico (21,745 – tempo de retenção).
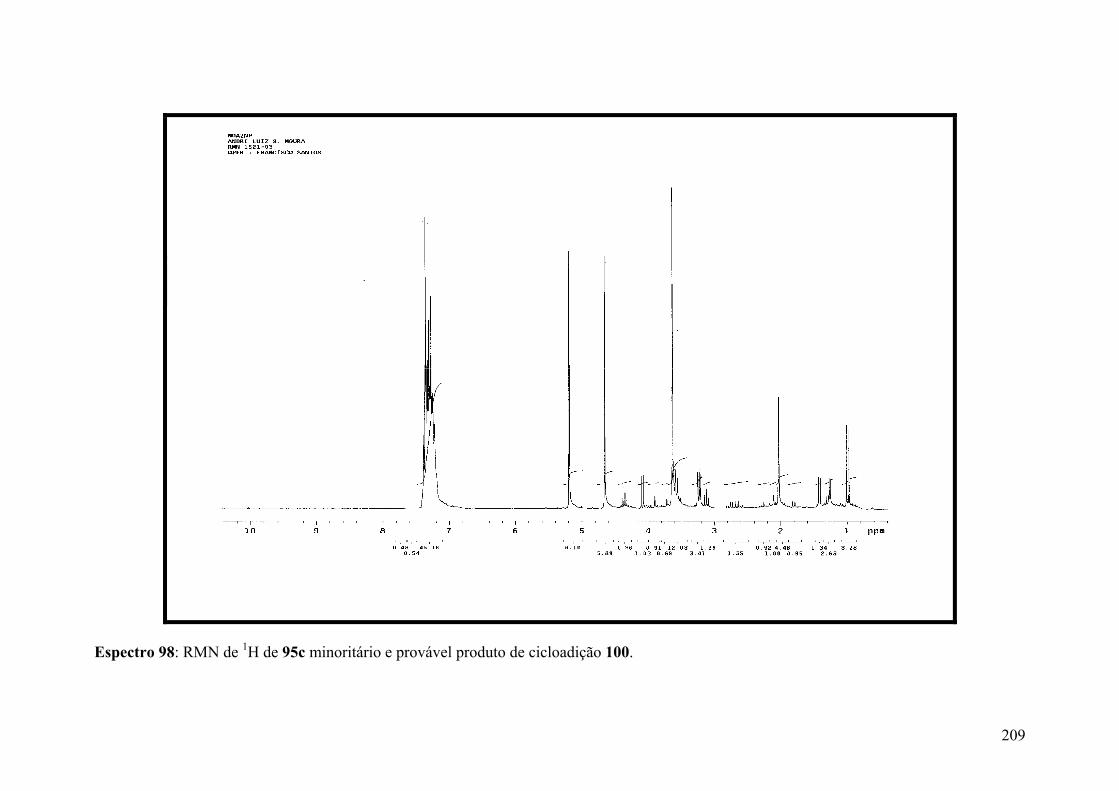
209
Espectro 98: RMN de 1H de 95c minoritário e provável produto de cicloadição 100.
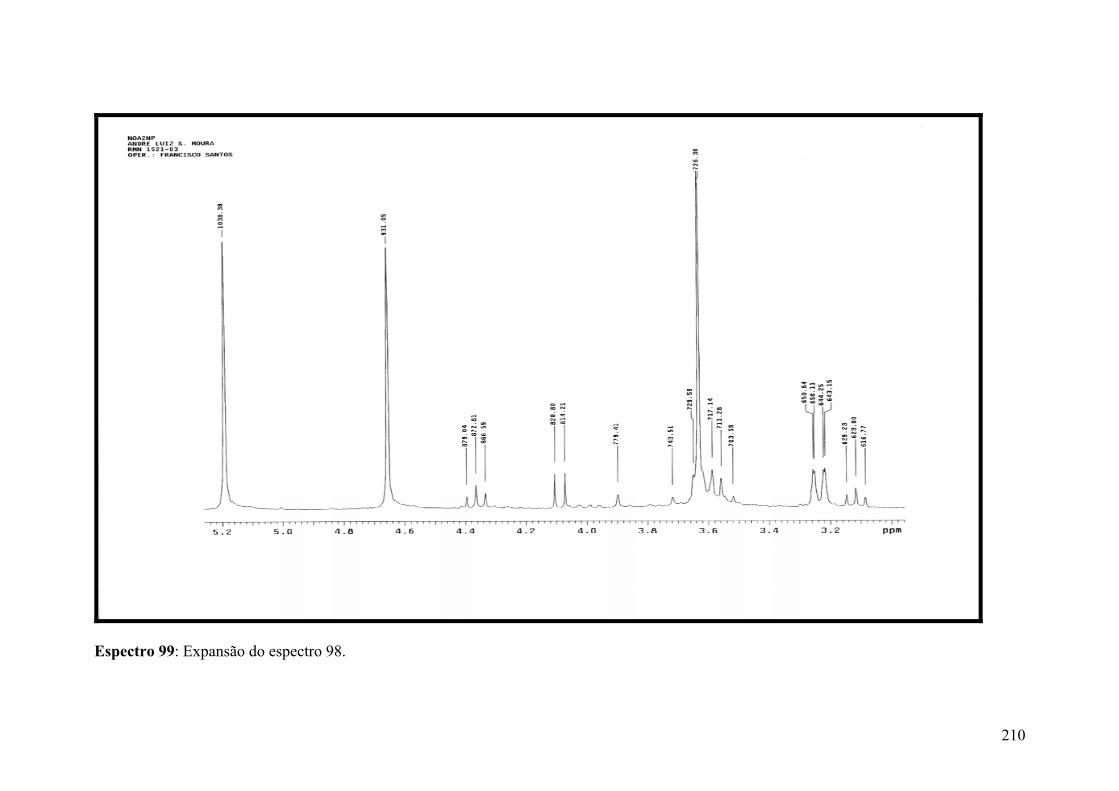
210
Espectro 99: Expansão do espectro 98.
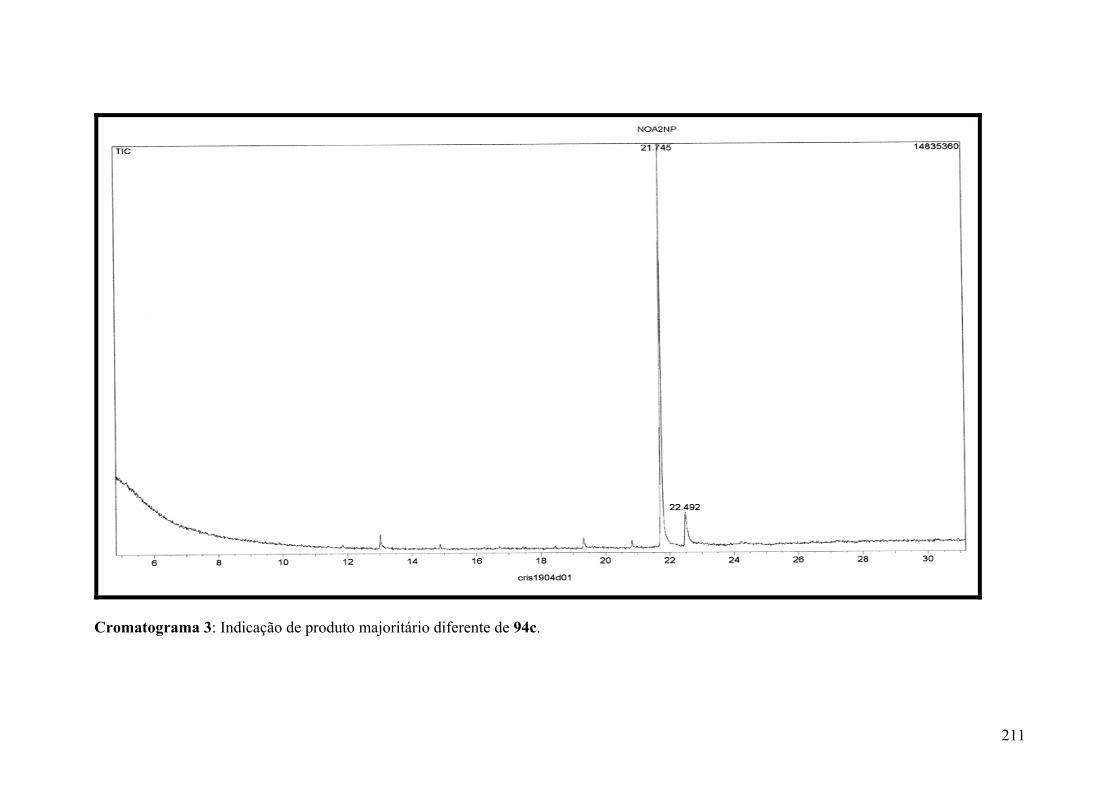
211
Cromatograma 3: Indicação de produto majoritário diferente de 94c.
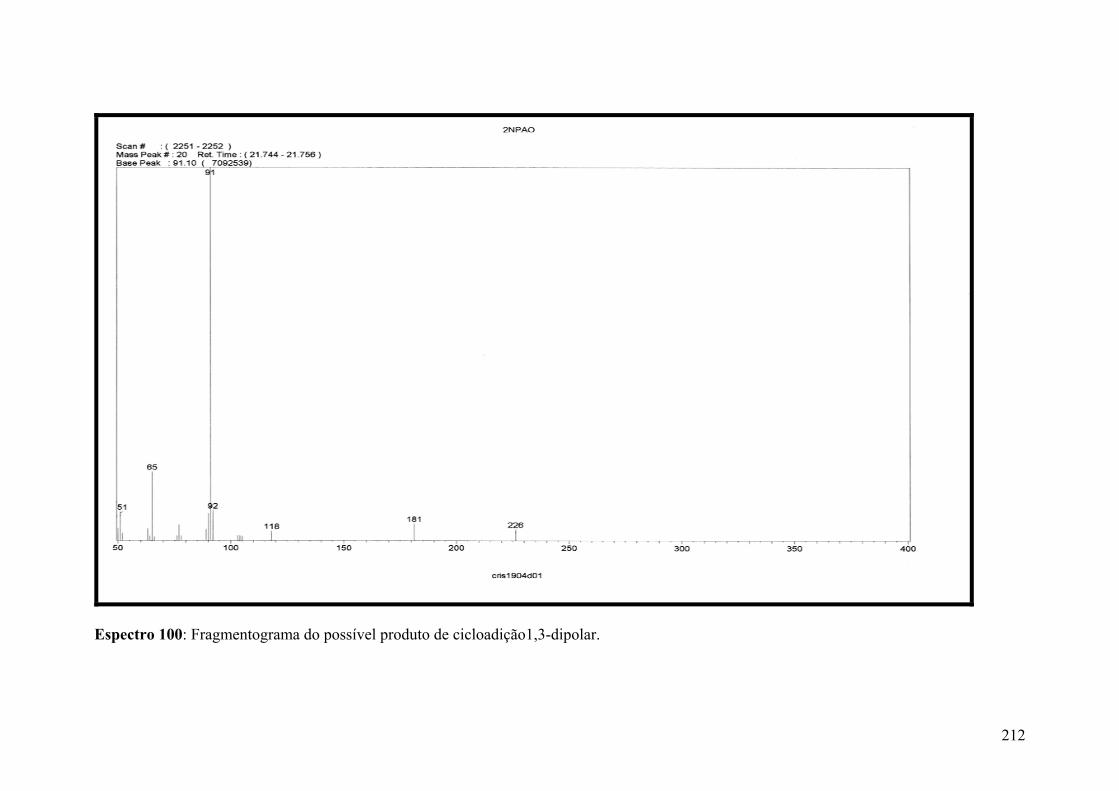
212
Espectro 100: Fragmentograma do possível produto de cicloadição1,3-dipolar.
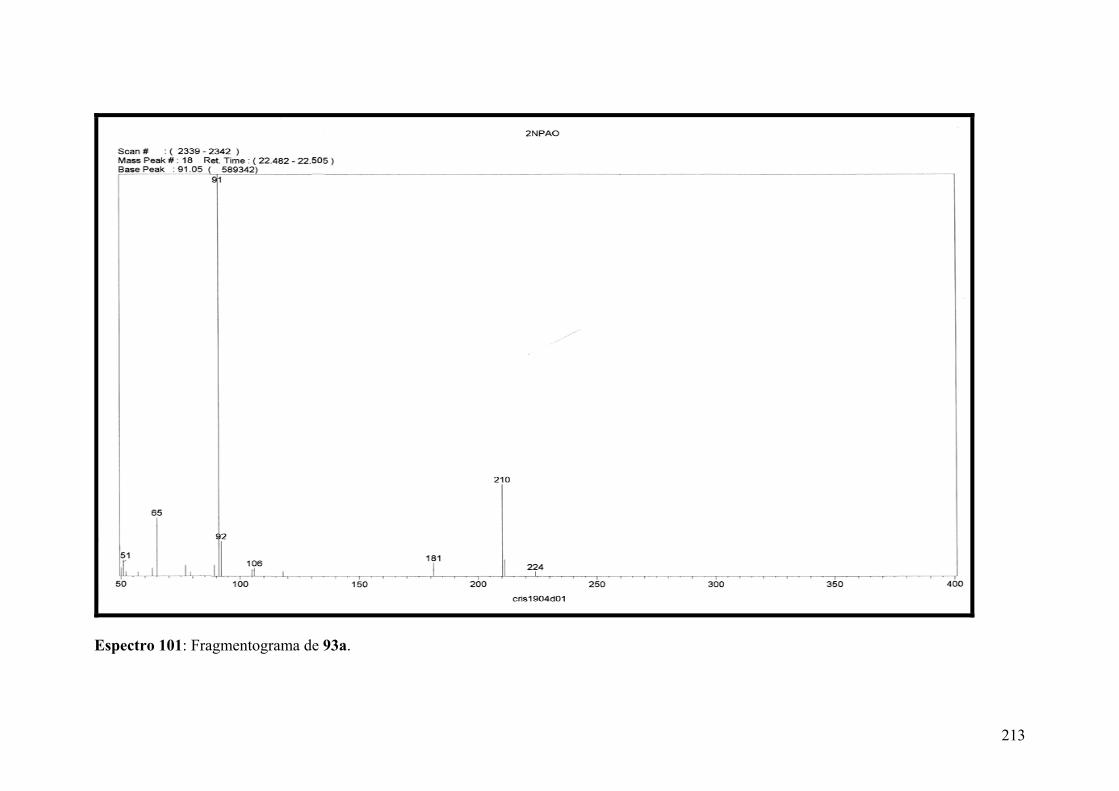
213
Espectro 101: Fragmentograma de 93a.
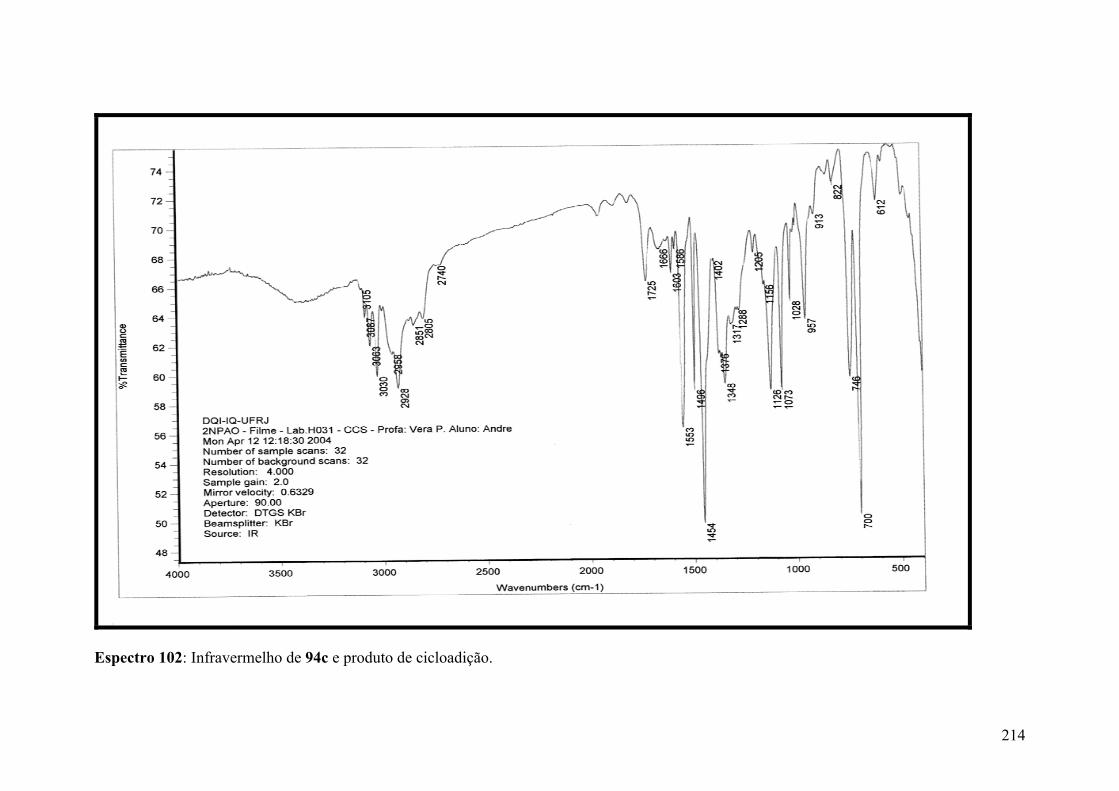
214
Espectro 102: Infravermelho de 94c e produto de cicloadição.

215
7.13) Síntese da dinitroamina 94f:
Procedimento II:
7.13.1) Síntese do (2S,3S)-1-fenil-2-N,N-dibenzilamino-3-nitrometil-4-metil-4-
nitropentano (94f).
NBn2
O2N
NO2
A uma solução de 27 μL de DBU (0,5 eq. ; 0,182 mmol) solubilizado em 5,0 mL
de acetonitrila e 98 μL de 2-nitropropano (1,2 eq. ; 1,09 mmol), foi adicionado 0,3394g
da nitroolefina 93b (0,91 mmol) solubilizada em 5,0 mL de acetonitrila e mantida sob
agitação magnética à temperatura ambiente. A mistura reagiu por 24 horas sendo, então,
concentrada sob pressão reduzida. A resíduo remanescente foi submetido a uma
cromatografia em coluna de gel de sílica e eluída com hexano / AcOEt 20%. Obteve-se
um líquido marrom muito viscoso em 58 % de rendimento químico.

216
7.14) Síntese do (3S)-dibenzilamino-4-fenil-2-metilen-butanonitrila (94g).
NBn2
CN
A 0,2528 g de TBAF (1,1 eq. ; 0,80 mmol) solubilizado em 5,0 mL de
acetonitrila, foi adicionado 0,11 mL de trimetilsililcianeto (0,80 mmol). A esta solução
foi adicionado 0,27 g da nitroolefina 93b (0,73 mmol) solubilizada em 5,0 mL de
acetonitrila. A mistura reacional foi mantida sob agitação magnética por 20 h à
temperatura ambiente. Após o término da reação, adicionou-se 1,0 mL de solução
aquosa de NaHCO3. A mistura reacional foi então extraída com AcOEt (3 x 10,0 mL) e
os extratos orgânicos foram secos com Na2SO4 e evaporados sob pressão reduzida,
fornecendo a aminometilenonitrila 94g em 63 % de rendimento químico após
purificação em coluna de gel de sílica tendo como eluente uma mistura de Hexano /
AcOEt 30 %.
- Dados espectrais:
IV (KBr): ν 3085-3028, 2929-2806, 2218, 1495, 743, 699.
RMN 1H (200 MHz, CDCl3): δ 7,5 – 7,0 (m, 10H), 6,4 (m, 1H), 6,04 (s, 1H), 3,94 (d,
1H), 3,69 (d, 1H), 3,5 (d, 1H), 3,0 (m, 2H).
RMN 13C (50,4 MHz, CDCl3): δ 147,73 (C), 138,8 – 133,7 (C), 129,25 – 126,5 (CH –
aromático), 117,28 (C), 114,0 (CH2), 62,3 (CH), 57,59 (CH2), 37,54 (CH2).
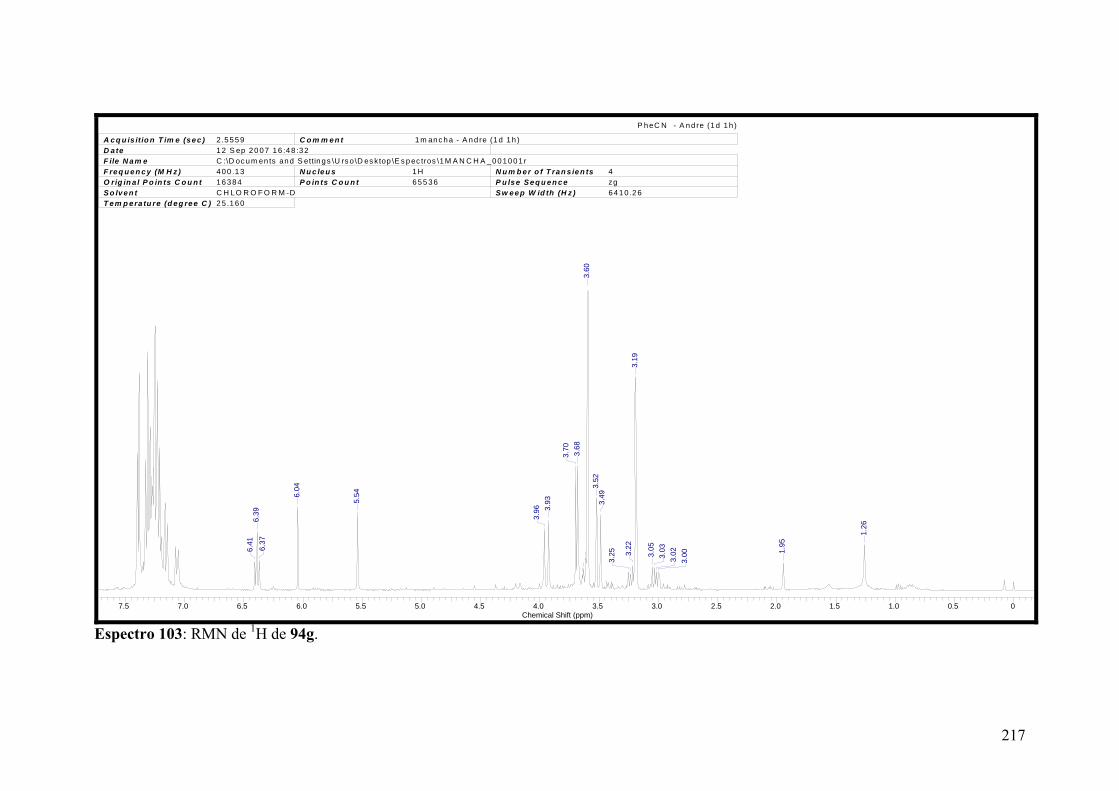
217
P heC N - A nd re (1d 1h )
A cq u is it io n T im e (sec ) 2 .5559 C o m m en t 1m anc ha - A nd re (1d 1h )D ate 12 S ep 20 07 16 :48 :32F ile N am e C :\D oc um en ts and S ettings \U rs o\D es k top \E s p ec tros \1M A N C H A _001001 rF req u en cy (M H z ) 400 .13 N u c leu s 1H N u m b er o f T ran s ien ts 4O rig in a l P o in ts C o u n t 16384 P o in ts C o u n t 655 36 P u lse S e q u e n c e zgS o lv e n t C H LO R O F O R M -D S w e e p W id th (H z ) 6410 .26T em p era tu re (d eg ree C ) 25 .160
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0Chemical Shift (ppm)
6.41
6.39
6.37
6.04
5.54
3.96 3.
93
3.70 3.68
3.60
3.52
3.49
3.25 3.
223.
19
3.05
3.03
3.02
3.00 1.
95
1.26
Espectro 103: RMN de 1H de 94g.
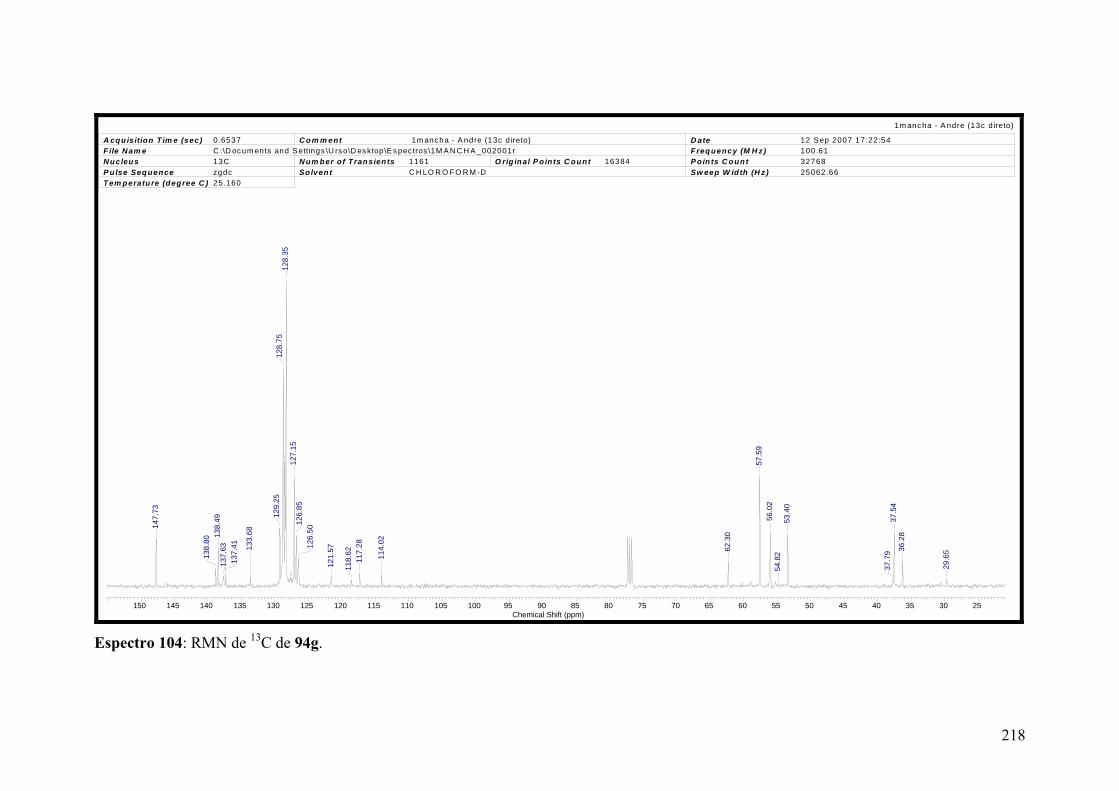
218
1m ancha - A ndre (13c d ireto)
A cqu isition T im e (sec) 0.6537 C om m ent 1m ancha - A ndre (13c d ireto) D ate 12 S ep 2007 17:22:54F ile Nam e C :\D ocum ents and S ettings \U rso\D esktop\E spec tros \1M A N C H A _002001r Frequency (M H z) 100.61N ucleus 13C Num ber o f Transien ts 1161 O rig inal P o ints C ount 16384 P oin ts C ount 32768P ulse Sequence zgdc Solven t C H LO R O FO R M -D Sw eep W id th (H z ) 25062.66Tem perature (degree C ) 25.160
150 145 140 135 130 125 120 115 110 105 100 95 90 85 80 75 70 65 60 55 50 45 40 35 30 25Chemical Shift (ppm)
147.
73
138.
8013
8.49
137.
6313
7.41 13
3.68
129.
2512
8.75
128.
3512
7.15
126.
8512
6.50
121.
57
118.
6211
7.28
114.
02
62.3
0
57.5
956
.02
54.8
253
.40
37.7
937
.54
36.2
8
29.6
5
Espectro 104: RMN de 13C de 94g.
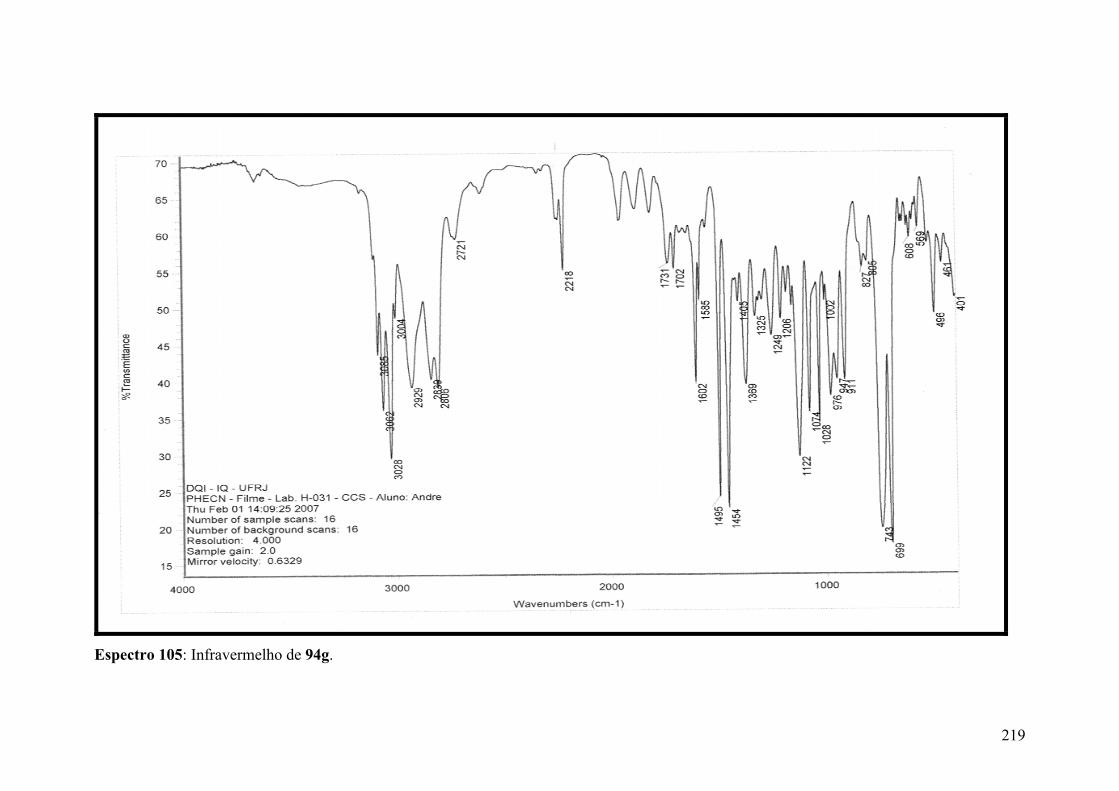
219
Espectro 105: Infravermelho de 94g.

220
7.15) Síntese do β-aminoácido 95f: Ácido-(2R,3S)-2-nitroisopropil-3-N,N-
dibenzilamino-4-fenilbutanóico.
NBn2
OH
NO2
O
Procedimento I:
A dinitroamina 94f (0,0425 g ; 0,092 mmol) solubilizada em 1,0 mL de t-BuOH
à temperatura ambiente foi tratada com 0,55 mL de um solução-tampão de KOH (KOH
0,5M em K2HPO4 1,25 M). A mistura reacional foi mantida sob agitação magnética e
em atmosfera inerte de N2 por 2 minutos. Então, foi adicionado 0,72 mL de KMnO4 0,5
M (4,0 eq. – 0,368 mmol) de modo a manter a temperatura ambiente. Depois de
transcorrido o tempo de 1 hora, adicionou-se, com banho de gelo, 10,0 mL de uma
solução saturada de Na2SO3. A mistura reacional teve o seu pH ajustado para 3 com
adição de HCl 2M e a clara solução resultante foi extraída com AcOEt (5 x 10,0 mL).
Os extratos orgânicos foram secos com Na2SO4 e evaporados sob pressão reduzida,
fornecendo o β-aminoácido 95f em um rendimento químico de 55% como um líquido
de coloração clara ligeiramente amarelado.

221
Procedimento II:
Uma solução de 0,151 g da dinitroamina 94f (0,33 mmol), 0,069 g de NaNO2
(1,0 mmol) e 0,19 mL de AcOH (3,3 mmoles) em 2,0 mL de DMSO foi agitada por 6
horas à temperatura ambiente. Após esse tempo, a mistura reacional foi acidificada com
uma solução de HCl 10% e extraída com éter dietílico (3 x 10,0 mL). As fases orgânicas
foram combinadas, secas com Na2SO4 e concentradas sob pressão reduzida, fornecendo
um óleo amarelo pálido fino, que foi purificado através de coluna cromatográfica de gel
de sílica, tendo como eluente uma mistura de Hexano / AcOEt 50 %.
Procedimento III:
A dinitroamina 94f (0,1295 g ; 0,28 mmol) foi dissolvida em 4,0 mL metanol
seco sob atmosfera de argônio. A -78 0C, adicionou-se 0,28 mL de uma solução de
MeOLi 1,0 M deixando a mistura reagir por 10 minutos. Ao término desse tempo,
um fluxo de O2/O3 foi passado através da mistura reacional por 30 minutos. Terminado
esse tempo, a reação foi purgada com oxigênio para remover o excesso de ozônio, e
então foi tratada com 0,07 mL de dimetilsulfeto até que se atingisse temperatura
ambiente. Após 16 horas, os solventes foram removidos sob pressão reduzida e o
resíduo resultante foi vertido em salmoura (10,0 mL) e extraído com diclorometano (2 x
15,0 mL). Após a separação das fases, a fase aquosa foi extraída mais uma vez com
acetato de etila (1 x 15,0 mL). A fase contendo o acetato de etila foi lavada com água
destilada (1 x 10,0 mL) para só então ser somada as demais fases orgânicas. As fases
orgânicas somadas foram secas com Na2SO4, concentradas para gerar um óleo amarelo
claro fino. O material foi purificado em coluna de sílica gel utilizando uma mistura de
hexano / acetato de etila 60% como eluente para fornecer o produto 95f em 79 % de
rendimento químico.

222
- Dados espectrais (relativos ao Experimento I e III):
Experimento III:
IV (KBr): ν 1716, 1543, 1345, 1016 cm-1.
RMN 13C (50,4 MHz, CDCl3): δ 169,78 (C), 135,9 – 133,6 (C), 133,1 – 128,2 (CH –
aromático), 88,49 (C), 62,53 (CH), 54,87 (CH2), 40,09 (CH), 29,54 (CH2), 25,95 e
22,88 (2 CH3).
Experimento I:
Os dados obtidos no experimento I (protocolo usando KMnO4 e tampão)
confirmam a transformação do nitro primário em carboxila de ácido. Devido ao baixo
rendimento químico da reação e interferência do substrato remanescente, os espectros
de RMN de 1H e 13C mostraram-se complexos. Contudo, os sinais referentes à
interconversão funcional puderam ser visualizados.
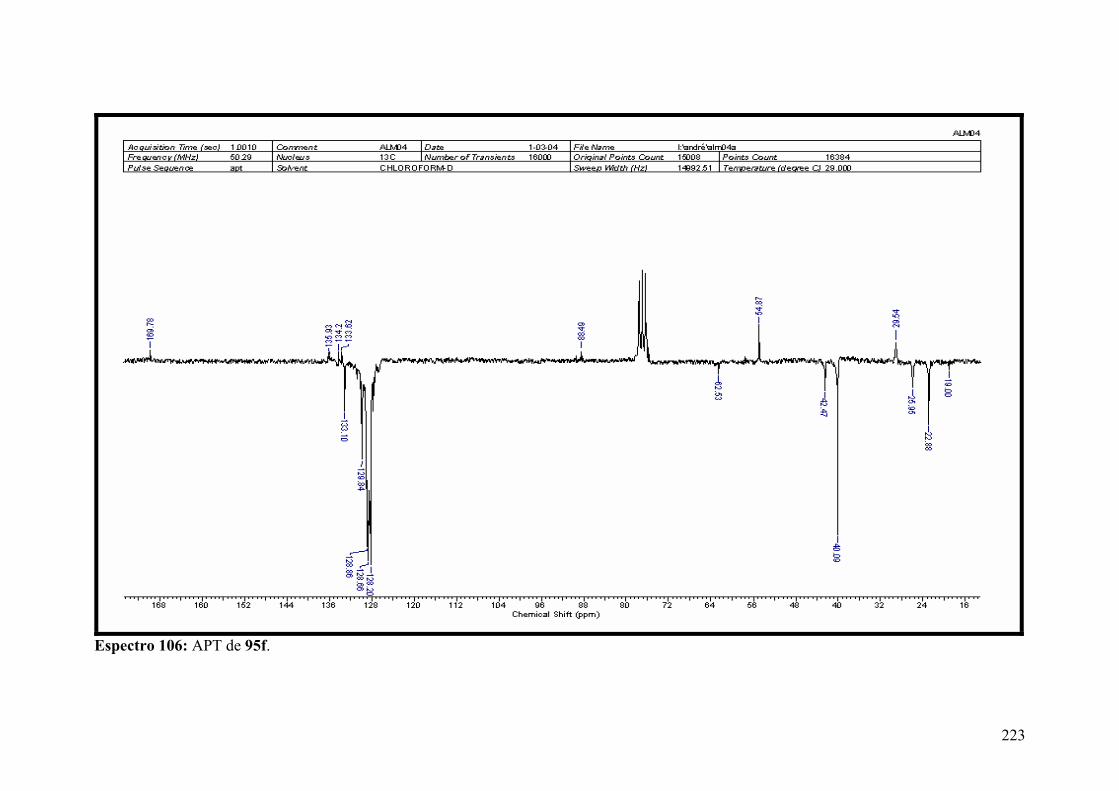
223
Espectro 106: APT de 95f.
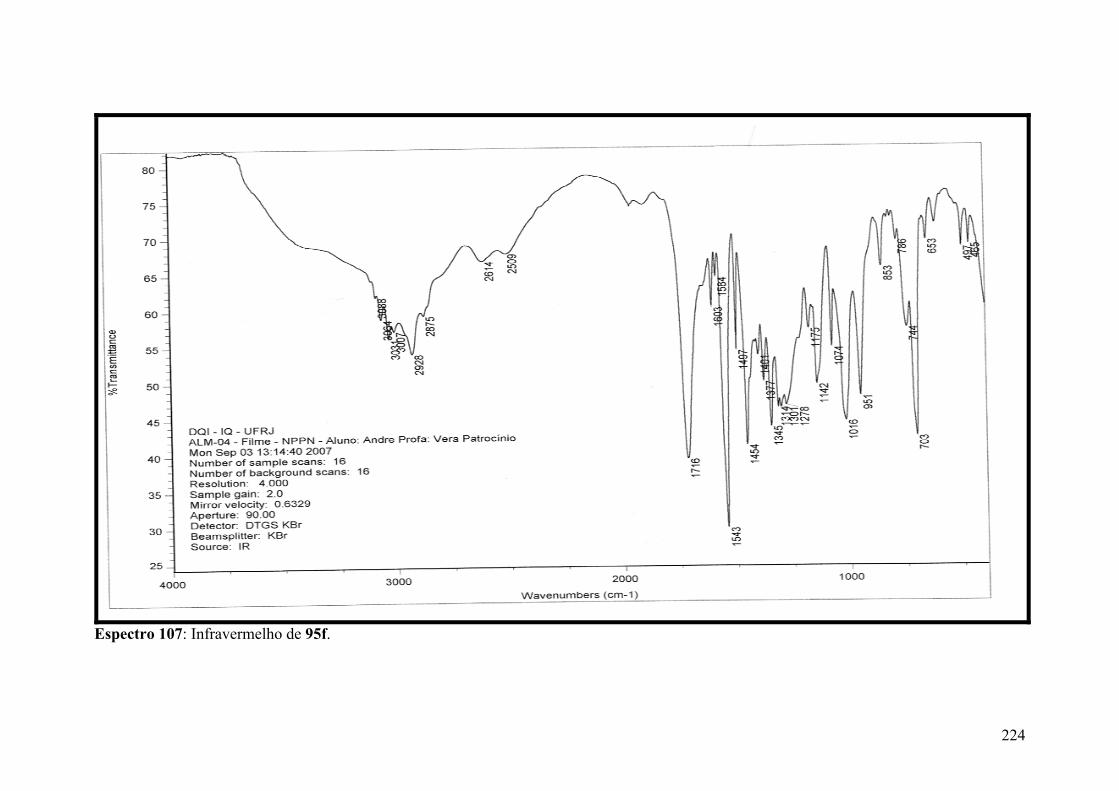
224
Espectro 107: Infravermelho de 95f.

225
7.16) Síntese do β-aminoácido 95d: Ácido-(2R,3S)-2-metoxi-3,3-N,N-bibenzilamino-4-fenilbutanóico.
OH
NBn2
OMe
O
Procedimento Experimental idêntico ao Procedimento I – página 271. - Dados espectrais:
RMN 13C (50,4 MHz, CDCl3): δ 171,0 (C), 135,9 (C), 131,9 – 125,2 (CH-aromático),
105,9 (CH), 80,2 (CH3), 58,3 (CH), 54,1 (CH2), 42,0 (CH2).
Observação: Dados espectrais referentes a um espectro de produto bruto (outros sinais
não-relatados são pertencentes ao substrato).
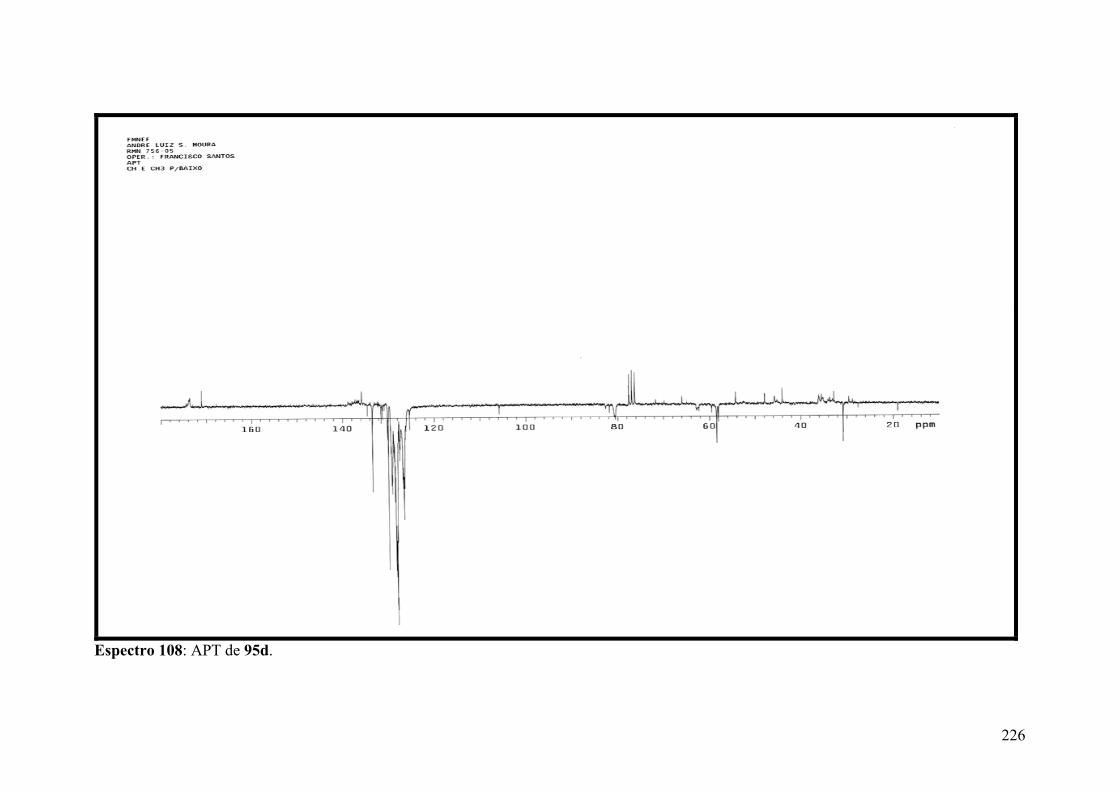
226
Espectro 108: APT de 95d.

227
REFERÊNCIAS. 1. a) Ono, N. The Nitro Group in Organic Synthesis, Wiley-VCH, New York, 2001. b) Berner, O.M.; Tedeschi, L.; Enders, D. Eur. J. Org. Chem., 2002, 1877. c) Sibi, M.P.; Manyem, S. Tetrahedron, 2000, 56, 8033. 2. Luzzio, F. A. Tetrahedron, 2001, 57, 915. 3. Seebach, D. ; Colvin, E. W. ; Lehr, F. ; Weller, T. Chimia, 1979, 33, 1. 4. Kuwahara, Y.; Ômura, H.; Tanabe, T. Naturwiss., 2002, 89, 308. 5. Silva, P. C. ; Costa, J. S. ; Pereira, V. L. P. Synthetic Comm. 2000, 31, 595. 6. Goodman, L.S.; Gilman, A. The Pharmacological Basis of Therapeutics, McGraw-Hill, New York, 1996. 7. March, J. Advanced Organic Chemistry, Wiley-Interscience, New York, 3rd edition, 1985. 8. Murray, R.W.; Jeyaraman, R.; Mohan, L. Tetrahedron Lett., 1986, 27, 2335. 9. Kornblum, N.; Powers, J.W. J. Org. Chem. 1957, 22, 455. 10. Olah, G.A.; Ramaiah, P.; Lee, C.S.; Prakash, G.K.S. Synlett, 1992, 377. 11. Catalog Handbook of Fine Chemicals Aldrich 2006-2007. 12. El Kaim, L.; Gacon, A. Tetrahedron Lett., 1997, 38, 3391. 13. a) Ballini, R.; Petrini, M. Tetrahedron, 2004, 60, 1017. b) Pinnick, H.W.; Org. React. 1990, 38, 655. 14. a) redução do grupamento nitro a aminas: Andruszkiewicz, R.; Silverman, R.B. Synthesis, 1989, 953. b) redução do grupamento nitro a oximas: Ho, T.L.; Wong, T.L. Synthesis, 1974, 196. 15. Ono, N.; Miyake, H.; Kaji, A. J. Org. Chem., 1984, 49, 4997. 16. Vilaivan, T; Bhanthumnavin, W; Anant, Y.S-., Current Organic Chemistry, 2005, 9, 1315. 17. a) Corey, E. J. Pure Appl. Chem. 1990, 62, 1209. b) Volante, R. P. ; Reider, P. J. Tetrahedron Lett., 1994, 35, 673. 18. Uchida, I. ; Shigematsu, N. ; Ezaki, M. ; Hashimoto, M. Chem. Pharm. Bull. 1985, 33, 3053. 19. Li, G.; Kotti, S.R.S.S.; Cody, T. Chem. Biol. Drug Des., 2006; 67, 101.

228
20. a) Sisko, J. ; Weinreb, S. M. J. Org. Chem., 1990, 55, 393. b) Hirao, A. ; Hattori, I. ; Yamaguchi, K. ; Nakahamas, S. ; Yamazaki, N. Synthesis, 1982, 461. 21. Wada, M. A. ; Sakurai, Y. ; Akiba, K. Tetrahedron Lett., 1984, 25, 1079. 22. Reetz, M.T.; Jäger, R.; Drewlies, R.; Hübel, M. Angew. Chem., Int. Ed. Engl. 1991, 30, 103. 23. Weinreb, S. M.; Orr, R. K. Synthesis, 2005, 1, 1–23. 24. Badorrey, R. ; Cativiela, C. ; Díaz-de-Villegas, M. D. ; Gálvez, J. A. Tetrahedron, 1997, 53, 1411. 25. Vaultier, M. ; Lambert, P. H. ; Carrie, R. Bull. Soc. Chim. Fr., 1986, 83. 26. Varma, R. S. ; Dahiya, R. Synlett, 1997, 1245. 27. a) Johnson, H. G. J. Am. Chem. Soc. 1946, 68, 12. b) Senkus, M. J. Am. Chem. Soc. 1946, 68, 10. 28. Adams, H. ; Anderson, J. C. ; Peace, S. ; Pennell, A. M. K. J. Org. Chem. 1998, 63, 9932. 29.Web of Science: http://portal.isiknowledge.com (29/08/2007). 30. Yamada, K. ; Harwood, S. J. ; Groger, H. ; Shibasaki, M. Angew. Chem., Int. Ed. Engl. 1999, 38, 3504. 31. a) Nishiwaki, N, Knudsen, K. R.; Gothelf, K. V.; Jørgensen, K. A. Angew. Chem. Int. Ed. 2001, 40, 2992. b) Knudsen, K. R.; Risgaard, T.; Nishiwaki, N.; Gothelf, K. V.; Jørgensen, K. A. J. Am. Chem. Soc. 2001, 123, 5843. 32. a) Anderson, J.C.; Howell, G.P.; Lawrence, R.M.; Wilson, C.S. J. Org. Chem. 2005, 70, 5665. b) Anderson, J. C.; Peace, S.; Pih, S. Synlett, 2000, 850. 33. Jacobsen, E.N.; Taylor, M.S. Angew. Chem., Int. Ed. Engl. 2006, 45, 2. 34. Okino, T.; Nakamura, S.; Furukawa, T.; Takemoto, Y. Org. Lett. 2004, 6, 625. 35. Yoon, T.P.; Jacobsen, E.N. Angew. Chem. Int. Ed., 2005, 44, 466. 36. Bernardi, L.; Bonini, B.F.; Dessole, G.; Fochi, M.; Franchini, M.C.-; Gavioli, S.; Ricci, A.;Varchi, G. J. Org. Chem., 2003, 68,1418. 37. Petrini, M.; Foresti, E.; Palmieri. G.; Profeta, R., Org. Biomol. Chem., 2003, 1, 4275. 38. Benetti, S.; Baricordi, N.; Biondini, G.; Risi, C.; Pollini, G.P., Tetrahedron Lett., 2004, 45, 1373.

229
39. Neto, J. C. Tese de Mestrado, 2000, NPPN/UFRJ, Rio de Janeiro. 40. Denmark S. E.; Thorarensen, A. Chem. Rev., 1996, 96, 137. 41. Barluenga, J.; Aznar, F.; Ribas, C.; Valdes, C. J. Org. Chem., 1998, 63, 10052. 42. Denmark, S. E.; Marcin, L. R. J. Org. Chem. 1993, 58, 3857. 43. McMurry, J.E.; Melton, J. J. Org. Chem. 1975, 40, 2138. 44. Barua, N.C.; Saikia, A.K.; Sharma, R.P.; Gosh, A.C. Synthesis, 1994, 685. 45. Sreekumar, R.; Padmakumar, R.; Rugmini, P. Tetrahedron Lett., 1998, 39, 2695. 46. Hoashi,Y.; Yabuta, T.; Yuan, P.; Miyabe, H.; Takemoto, Y. Tetrahedron, 2006, 62, 365 e referências nele citadas. 47. a)Wang, J.; Li, H.; Zu, L.; Jiang, W.; Wang, W. Adv. Synth. Catal. 2006, 348, 2047. b) Lu, S.-F.; Du, D.-M.; Xu, J.; Zhang, S.-W. J. Am. Chem. Soc. 2006, 128, 7418. c) Ooi, T.; Takada, S.; Doda, K.; Maruoka, K. Angew. Chem. Int. Ed. 2006, 45, 7606. 48. a) Alcântara, M.-P. D.; Escribano, F. B.; Sánchez, A. G.-; Diánez, M. J.; Estrada, M. D.; Castro, A. L.-; Garrido, S.P.- Synthesis, 1996, 64. b) Ballini, R.; Bosica, G.; Fiorini, D.; Palmieri, A.; Synthesis, 2004, 1938. 49. Alexakis, A.; Polet, D. Tetrahedron Lett., 2005, 46, 1529. 50. Sabelle, S.; Lucet, D.; Le Gall, T.; Mioskowski, C. Tetrahedron Lett., 1998, 39, 2111. 51. Pedro, J.R.; Monje, B.; Fernández, I.; Blay, G. Tetrahedron, 2004, 60, 165. 52. Ayerbe, A., Morao, I., Arrieta, A., Linden, A. Cossío, F, P. Tetrahedron Lett., 1996, 37, 3055. 53. Galley, G. Hübner, J., Anklam, S., Jones, P. G., Pätzel, M. Tetrahedron Lett., 1996, 35, 6307. 54. Juaristi, E.; Soloshonok, V.A. Enantioselective Synthesis of β-Amino Acids, Wiley-Interscience, New Jersey, 2005. 55. Steer, D.L.; Lew, R.A.; Perlmutter, P.; Smith, A.I.; Aguillar, M.-I. Curr. Med. Chem., 2002, 9, 811. 56. Hintermann, T.; Seebach, D. Synlett, 1997, 437. 57. Barrow, R.A.; Hemscheidt, T.; Liang, J.; Paik, S.; Moore, R. E.; Tius, M. J. Am. Chem Soc. 1995, 117, 2479.

230
58. Umezawa, H.; Aoyagi, T.; Suda, H.; Hamada, M.; Takeuchi, T. J. Antibiot. 1976, 29, 97. 59. Nussbaum, F. v.; Spiteller, P.; Rüth, M.; Steglich, W.; Wanner, G.; Gamblin, B.; Stievano, L.; Wagner, F. E. Angew. Chem. Int. Ed., 1998, 37, 3292. 60. Andrimida: Fredenhagen, A.; Tamura, S.Y.; Kenny, P.T.M.; Komura, H.; Naya, Y.; Nakanishi, K. J. Am. Chem. Soc., 1987, 109, 4409. Moiramida B: Needham, J.; Kelly, M. T.; Ishige, M.; Andersen, R.J. J. Org. Chem., 1994, 59, 2058. 61. a) Reetz, M. T.; Drewes, M.W.; Schmitz, A. Angew. Chem., Int. Ed. Engl., 1987, 26, 1141. b) Reetz, M. T. Chem. Rev. 1999, 99, 1021. c) Beaulieu, P. L.; Wernic, D. J. Org. Chem., 1996, 61, 3635. d) O'Brien, P.; Warren, S. Tetrahedron Lett., 1996, 24, 4271. 62. Hanessian, S.; Devasthale, P.V. Tetrahedron Lett., 1996, 37, 987. 63. Moura, A.L.S.Tese de Mestrado, 2002, NPPN/UFRJ, Rio de Janeiro. 64. Swern, D.; Omura, K. Tetrahedron, 1978, 34, 1651. 65. Adia, M. ; Hénaff, N. ; Whiting, A. Tetrahedron Lett., 1997, 38, 3101. 66. Silva, P.C.; Costa, J.S.; Pereira, V.L.P. Synthetic Comm. 2000, 31, 595. 67. Ahn, N. T. ; Eisenstein, O. Tetrahedron Lett., 1976, 155. 68. Cram, D. J. ; Kopecty, K. R. J. Am. Chem. Soc., 1959, 81, 2923. 69. Bürgi, H. B. ; Dunitz, J. D. ; Lehn, J. M. ; Wipff, G. Tetrahedron, 1974, 30, 1563. 70. a) Boruwa, N.; Gogoi, P.P.; Saikia, A.K.; Barua, N.C. Tetrahedron: Asymm., 2006, 17, 3315. b) Palomo, C.; Oiarbide, M.; Laso, A. Eur. J. Org. Chem., 2007, 16, 2561. 71. Carvalho, L.L. Tese de Mestrado, 2007, NPPN/UFRJ, Rio de Janeiro. 72. Vieira, D.P.P.30ª Reunião Anual da Sociedade Brasileira de Química, 2007, Águas de Lindóia, São Paulo, Brasil. 73. Smirnov, V.O.; Ioffe, S.L.; Tishkov, A.A.; Khomutova, Y.A.; Nesterov, I.D.; Antipin, M.Y.; Smit, W.A.; Tartakovsky, V.A. J. Org. Chem. 2004, 69, 8485. 74. Gothelf, K. V.; Jørgensen A. Chem. Rev. 1998, 98, 863. 75. Lindell, S.D.; Stones, E.A.S-. Synlett, 1991, 591. 76. Ceccherelli, P.; Curini, M.; Marcotullio, M.C.; Epifano, F.; Rosati, O. Synth. Commun., 1998, 28, 3054. 77. Mioskowski, C.; Wagner, A.; Matt, C. J. Org. Chem., 1997, 62, 234.

231
78. a) McMurry, J.E.; Melton, J.; Padgett, H. J. Org. Chem., 1974, 39, 259. b) Marshall, J.A.; Garofalo, A.W. J. Org. Chem., 1993, 58, 3675. 79. Bailey, S.P.; Chem. Rev. 1958, 58, 925.
Livros Grátis( http://www.livrosgratis.com.br )
Milhares de Livros para Download: Baixar livros de AdministraçãoBaixar livros de AgronomiaBaixar livros de ArquiteturaBaixar livros de ArtesBaixar livros de AstronomiaBaixar livros de Biologia GeralBaixar livros de Ciência da ComputaçãoBaixar livros de Ciência da InformaçãoBaixar livros de Ciência PolíticaBaixar livros de Ciências da SaúdeBaixar livros de ComunicaçãoBaixar livros do Conselho Nacional de Educação - CNEBaixar livros de Defesa civilBaixar livros de DireitoBaixar livros de Direitos humanosBaixar livros de EconomiaBaixar livros de Economia DomésticaBaixar livros de EducaçãoBaixar livros de Educação - TrânsitoBaixar livros de Educação FísicaBaixar livros de Engenharia AeroespacialBaixar livros de FarmáciaBaixar livros de FilosofiaBaixar livros de FísicaBaixar livros de GeociênciasBaixar livros de GeografiaBaixar livros de HistóriaBaixar livros de Línguas
Baixar livros de LiteraturaBaixar livros de Literatura de CordelBaixar livros de Literatura InfantilBaixar livros de MatemáticaBaixar livros de MedicinaBaixar livros de Medicina VeterináriaBaixar livros de Meio AmbienteBaixar livros de MeteorologiaBaixar Monografias e TCCBaixar livros MultidisciplinarBaixar livros de MúsicaBaixar livros de PsicologiaBaixar livros de QuímicaBaixar livros de Saúde ColetivaBaixar livros de Serviço SocialBaixar livros de SociologiaBaixar livros de TeologiaBaixar livros de TrabalhoBaixar livros de Turismo







![PPC T7 QO Aula6 Aromaticos-1.ppt [Somente leitura] [Modo ... · Etapa determinante (adição do eletrófilo) Adição do Eletrófilo Perda do Próton ... Substituintes Ativadores](https://static.fdocument.org/doc/165x107/5c4e571893f3c34aee57883a/ppc-t7-qo-aula6-aromaticos-1ppt-somente-leitura-modo-etapa-determinante.jpg)



![[14] Ligas ferrosas 2.ppt [Modo de Compatibilidade] · ¾Microestrutura do ferro fundido nodular G formação de nódulos de grafita F adição de P 150 μm adição de “nodulizadores”](https://static.fdocument.org/doc/165x107/5c5ec02209d3f28e758c8934/14-ligas-ferrosas-2ppt-modo-de-compatibilidade-microestrutura-do-ferro.jpg)






