
Revised M2:11282 Red cell ICAM-4 is a novel ligand for platelet ...
29
1 Revised M2:11282 Red cell ICAM-4 is a novel ligand for platelet activated α IIb β 3 -integrin Patricia Hermand # , Pierre Gane # , Martine Huet # , Vincent Jallu + , Cécile Kaplan + , HH Sonneborn°, Jean-Pierre Cartron #§ and Pascal Bailly # From # INSERM U76 and + Unité d’Immunologie plaquettaire, Institut National de la Transfusion Sanguine, 6 rue Alexandre Cabanel, 75015 Paris, France, °Biotest AG, 63276 Dreieich, Germany *This investigation was supported in part by the Institut National de la Transfusion Sanguine (INTS), the Institut National de la Santé et de la Recherche Médicale (INSERM. This article must therefore be hereby marked "advertisement" in accordance with 18 U.S.C. section 1734 solely to indicate this fact. § correspondence : Dr Jean-Pierre Cartron INSERM U76 Institut National de la Transfusion Sanguine 6 rue Alexandre Cabanel, 75015 Paris, France. Phone : (33) 1.44.49.30.00 Fax : (33) 1.43.06.50.19 e-mail : [email protected] Copyright 2002 by The American Society for Biochemistry and Molecular Biology, Inc. JBC Papers in Press. Published on December 10, 2002 as Manuscript M211282200 by guest on March 22, 2018 http://www.jbc.org/ Downloaded from
Transcript of Revised M2:11282 Red cell ICAM-4 is a novel ligand for platelet ...

1
Revised M2:11282
Red cell ICAM-4 is a novel ligand for platelet activated αIIbβ3-integrin
Patricia Hermand#, Pierre Gane#, Martine Huet#, Vincent Jallu+, Cécile Kaplan+, HH
Sonneborn°, Jean-Pierre Cartron#§ and Pascal Bailly#
From #INSERM U76 and +Unité d’Immunologie plaquettaire, Institut National de la
Transfusion Sanguine, 6 rue Alexandre Cabanel, 75015 Paris, France, °Biotest AG,
63276 Dreieich, Germany
*This investigation was supported in part by the Institut National de la Transfusion Sanguine (INTS),
the Institut National de la Santé et de la Recherche Médicale (INSERM. This article must therefore be
hereby marked "advertisement" in accordance with 18 U.S.C. section 1734 solely to indicate this fact.
§ correspondence : Dr Jean-Pierre Cartron
INSERM U76
Institut National de la Transfusion Sanguine
6 rue Alexandre Cabanel, 75015 Paris, France.
Phone : (33) 1.44.49.30.00
Fax : (33) 1.43.06.50.19
e-mail : [email protected]
Copyright 2002 by The American Society for Biochemistry and Molecular Biology, Inc.
JBC Papers in Press. Published on December 10, 2002 as Manuscript M211282200 by guest on M
arch 22, 2018http://w
ww
.jbc.org/D
ownloaded from

2
Running title : ICAM4/αIIbβ3-integrin interaction, RBC, platelet GPIIb-IIIa, CHO
transfectants, hemostasis, thrombosis.
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

3
ABSTRACT
ICAM-4 (LW blood group glycoprotein) is an erythroid specific membrane
component that belongs to the family of intercellular adhesion molecules and
interacts in vitro with different members of the integrin family, suggesting a potential
role in adhesion or cell interaction events, including hemostasis and thrombosis. To
evaluate the capacity of ICAM-4 to interact with platelets, we have immobilized
RBCs, platelets and ICAM-Fc fusion proteins to a plastic surface and analyzed their
interaction in cell adhesion assays with RBCs and platelets from normal individuals
and patients, as well as with cell transfectants expressing the αIIbβ3 integrin. The
platelet fibrinogen receptor αIIbβ3 (platelet GPIIb-IIIa) in high affinity state following
GRGDSP peptide activation was identified for the first time as the receptor for RBC
ICAM-4. The specificity of the interaction was demonstrated by showing that (i)
activated platelets adhered less efficiently to immobilized ICAM-4-negative than to
ICAM-4-positive RBCs, (ii) monoclonal antibodies specific for the β3-chain alone and
for a complex-specific epitope of the αIIbβ3 integrin, and specific for ICAM-4 to a
lesser extent, inhibited platelet adhesion, whereas monoclonal antibodies to GPIb,
CD36, and CD47 did not, (iii) activated platelets from two unrelated type-I
glanzmann’s thrombasthenia patients did not bind to coated ICAM-4. Further
support to RBC-platelet interaction was provided by showing that DTT-activated
αIIbβ3-CHO transfectants strongly adhere to coated ICAM-4-Fc protein but not to
ICAM-1-Fc, and was inhibitable by specific antibodies. Deletion of individual Ig-
domains of ICAM-4 and inhibition by synthetic peptides showed that the αIIbβ3
integrin binding site encompassed the first and second Ig-domains, and that the G65-
V74 sequence of domain D1 might play a role in this interaction. Although normal
RBCs are considered non adhesive during coagulation and are reported passively
entrapped in fibrin polymers, these studies identify ICAM-4 as the first RBC protein
ligand of platelets that may have relevant physiological significance.
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

4
INTRODUCTION
The main physiological function of red blood cells (RBCs)1 which encapsulate
hemoglobin is to ensure the respiratory gases transport throughout of the human
body. However, the recent demonstration that mature RBCs express a growing
number of adhesion molecules, many of which exhibit blood group specificities (1-3),
reinforces the necessity to revisit the functional interaction of RBCs with leukocytes,
platelets and vascular endothelium in normal and pathological conditions.
It is interesting that many RBC adhesion molecules contain protein domains
characteristic of the immunoglobulin superfamily, suggesting some recognition
function. These molecules might participate to the normal RBC physiology, by
playing a role during erythropoiesis (differentation, maturation, enucleation, release),
self recognition mechanisms, red cell turnover and cell aging through cellular
interactions with counter receptors present on macrophages from bone marrow or
reticuloendothelial system in spleen and liver (1,4-9). Along this process, some
adhesion molecules are rapidly down-regulated and others are expressed at different
stages and remain on RBCs (10,11, and references herein). Finally, mature RBCs still
express adhesion molecules which are usually associated to leukocytes (CD44, CD47,
CD58) and others which have potential adhesion properties such as LW/ICAM-4
(CD242), Lu (CD239), Oka (CD147), CD99/Xg, JMH (CD108 ) and DO (1-3).
Nevertheless, normal RBCs do not adhere to circulating cells and vessel walls under
normal circumstances, suggesting that the RBC adhesion molecules are inaccessible
to their ligands. In contrast, the conversion of non adherent RBCs to adherent state
arise in several diseases. In such circumstances, adhesion molecules might be
involved in the pathophysiology of malaria (12,13), sickle cell disease (14-17) and
diabetes (18,19), mainly through an abnormal adhesion to the vascular endothelium
(1,20). Additionally, both phosphatidylserine exposure at the RBC surface and
adhesion molecules on these cells might also play a role in hemostasis and
thrombosis, for instance through interaction with cells expressing integrins, like
activated leukocytes, monocytes, platelets and endothelial cells (21,22). Interestingly
also, RBCs have the necessary signal transduction pathways to mediate these
functions (23).
Among RBC adhesion molecules, ICAM-4 (LW blood group glycoprotein, CD242)
emerges from the others by its structure similarities with ICAM-family and its
interaction characterized in vitro with different members of the β-integrin subfamilies
(αLβ2 (LFA-1), αMβ2 (Mac-1), α4β1 (VLA-4) (24-26), αV-integrins (αVβ1 and αVβ5) (27)).
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

5
These two families of proteins are well known to play crucial role in cell-cell
interactions and to be involved in large range of biological functions (28-31). For
instance, ICAM-4/integrin interaction might play a role during erythroid maturation
in bone-marrow or in the red cell turnover by spleen macrophages that express the
αdβ2 integrin (25,27,32). Additionally, ICAM-4 as well as the Lu blood group protein
might be involved in adhesion of sickle RBCs to TNF-α activated endothelial cells
(HUVEC) (7) and to laminin (33,34), respectively. It is suspected that abnormal
adhesion of sickle RBCs to endothelial cells and extracellular matrix proteins might
be responsible for the painful crisis of the disease that result from vaso-occlusive
episodes (35).
The purpose of this report was to examine the potential role of ICAM-4 in RBC-
platelet interaction and to demonstrate that this protein interacts in vitro with the
high affinity state of activated platelet αIIbβ3-integrin.
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

6
MATERIALS AND METHODS
Blood samples, reagents and antibodies
RBC from donors with common and rare phenotypes (Donull, Lunull of the Lu(a-b-)
type, LWnull, JMHnull) came from the frozen RBC collection of the Centre National de
Référence pour les Groupes Sanguins (Paris, France). Fresh blood samples from two
unrelated type-I glanzmann’s thrombasthenia patients were obtained after informed
consent. Apyrase, prostaglandin-E1 (PGE1), thrombin from human origin and anti-
glycophorin-A mAb (clone E4) and the peptide Arg-Gly-Glu (RGE) were purchased
from Sigma (St. Louis, MO). Peptides Gly-Arg-Gly-Asp-Ser-Pro (GRGDSP), Arg-Gly-
Asp (RGD) and the Fibrinogen Binding Inhibitor (FBI) peptide (residues 400-411 of
the fibrinogen γ chain) were from Bachem (Budendorf, Switzerland). Other peptides
Gly-Trp-Val-Ser-Tyr-Gln-Leu-Leu-Asp-Val (G-V, residues 65-74 of ICAM-4), Cys-
His-Ala-Arg-Leu-Asn-Leu-Asp-Gly-Leu-Val-Val-Arg (C-R, residues 180-192 of
ICAM-4) and corresponding random (rd) peptides used were synthesized and
purified by Neosystem (Strasbourg, France). Specific mAbs used in this study include
clones P2 and SZ22 recognizing the αIIb-chain (CD41) in the presence and the absence
of the β3-chain, respectively, clones SZ21 and SZ2 specific for the β3-chain (CD61) and
GpIb protein (CD42b), respectively, clone FA6.152 specific for CD36 and clone
AICD58 specific for CD58, which were purchased from Coulter/Immunotech
(Marseille, France). The mAb AP-2 specific for a complex-specific epitope of the αIIbβ3
integrin came from GTI (Brookfield, WI). PAC-1 and AK-4 mAbs specific for
activated αIIbβ 3 complex and P-selectin (CD62P), respectively, came from BD-
Pharmingen (SanDiego, CA). The mAb 3E12 to CD47 was from BioAtlantique
(Nantes, France). The murine mAb BS56 to ICAM-4/LWab was previously described
(36). ImmunoPure mouse IgG from Pierce (Rockford, IL, USA) was used as negative
control IgG. Chimeric ICAM-pIgI constructs derived from intact ICAM-4 (LWa allele)
carrying the two Ig-like domains D1 and D2 (residues 1-208), or deletion mutants D1-
ICAM-4 (residues 1-101) or D2-ICAM-4/ (residues 102-208) were used to produce
soluble Fc-fusion proteins as described (26). ICAM-1- and ICAM-2-pIgI constructs
(kindly provided by Dr. D. Simmons and E. Ferguson, Oxford, UK) were used to
produce ICAM-Fc soluble fusion proteins as above.
GRGDSP-activated platelets
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

7
Human platelets were obtained from fresh ACD-anticoagulated blood from
volunteers not taking any medication and were washed three times in modified
Tyrode’s albumin buffer (5 mM Hepes, 150 mM NaCl, 2.5 mM KCl, 12 mM NaHCO3,
5.5 mM glucose, 0.1% (w/v) bovine serum albumin (pH 6.5), 250 ng/ml PGE1, 25
mg/ml apyrase) by centrifugation at 1,200 x g for 10 min. Platelets were activated as
previously described (37,38). Briefly, 1x108 washed platelets resuspended in 0.1 ml
Tyrode’s-albumin buffer (pH 7.4) containing 2 mM CaCl2 and 1 mM MgCl2, were
incubated at 22°C for 5 min with 1 mM GRGDSP peptide. Then, an equal volume of
phosphate buffered saline (PBS, 10 mM phosphate buffer in 0.15 M NaCl, pH 7.2)
containing PGE1 (250 ng/ml), apyrase (25 mg/ml), 2 mM CaCl2, 1 mM MgCl2 and
1% (w/v) paraformaldehyde, was added and the mixture was incubated for 1 hour at
22°C . Then, 0.2 ml of 500 mM NH4Cl was added to stop the reaction in PBS. Fixed
activated platelets were washed several times to remove the activating peptide prior
to assays and resuspended in modified Tyrode’s buffer pH 7.4 containing divalent
cations. Fixed unactivated platelets used as control, were prepared by omitting
divalent cations and the activating peptide in the different buffers.
Platelet adhesion assays to immobilized RBCs
RBCs were immobilized on microtiter plates through binding to coated anti-
glycophorin A. Briefly, mAb E4 at 20 µg/ml (50 µl/well) in 25 mM Tris pH 8, 150 mM
NaCl, was adsorbed overnight at 4°C on flat-bottom 96-well microtiter plates (Nunc
A/S, Roskilde, Denmark). After two washes of wells with the same buffer, RBCs
(2.0x106/well in a final volume of 300 µl) resuspended in modified Tyrode’s buffer
pH 7.4 with or without cations (2 mM MgCl2 and 2 mM CaCl2) were added. After 1
hour incubation at 22°C, fixed GRGDSP-activated or unactivated platelets
(5.0x106/well in a final volume of 100 µl) in modified Tyrode’s buffer pH 7.4 with or
without divalent cations, respectively, were added to RBC coated wells. After 90 min
at 22°C , non adherent cells were removed by filling the wells with binding buffer,
and the microplates were put to float upside down in a PBS solution. Cells that
sticked to the plastic wells were recovered by vigorous shaking in 400 µl of PBS and
were counted by flow cytometric analysis using a FACSCalibur (Becton Dickinson,
San Diego, CA). Platelets and RBCs were distinguished by forward scatters and
platelet staining with the FITC-anti-human CD61 mAb (clone VI-PL2, BD
Biosciences).
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

8
RBCs adhesion to adherent platelets
Following isolation, unactivated platelets (1x107/well in a final volume of 100 µl)
resuspended in RPMI 1640, 10 mM Hepes containing PGE1 and apyrase were added
to wells to adhere overnight at 37°C. After washing, adherent platelets were
stimulated with thrombin (0.5 unit in 100 µl/well) diluted in Hank’s Balanced Salts
(HBSS) containing 2 mM CaCl2 during 20 min at room temperature. After another
washing, RBCs (3.3x106/well in a final volume of 300 µl) resuspended in HBSS with 2
mM CaCl2, 1 mM MgCl2, were added to each well. After 90 min at 22°C , non
adherent RBCs were removed by filling the wells with binding buffer, and the
microplates were put to float upside down in a PBS solution. Then RBCs numeration
was done using a Nikon Eclipse TE300 microscope (Nikon, Paris, France) (10 x
objective) coupled to a Biocom informatic system of images integration (Biocom, les
Ulis, France). For blocking experiments, RBCs and adherent platelets stimulated by
thrombin were pretreated with specific mAbs (2.5 µg/well) and ICAM-Fc protein (2.5
µg/well), respectively, for 30 min at 22°C.
αIIbβ3-CHO transfectants and DTT activation
Chinese hamster ovary cell line (CHO) was grown in Iscove’s modified Dulbecco
medium with Glutamax-1 (Life Technologies, Inc.) supplemented with amphotericin-
B-penicillin-streptamycin and 10% fetal calf serum (FCS). CD41 (αIIb chain) and CD61
(β 3 chain) cDNAs subcloned into pcDNA3.1 vector (Invitrogen, Leek, The
Netherlands), kindly provided by Dr. P.J. Newman (Blood Center of Southeastern
Wisconsin, Milwaukee, WI), were cotransfected into CHO cells using the Lipofectin-
Reagent according to the manufacturer’s instructions (Life Technologies,
Gaithersburg, MD). Stable transformants resistant to G418 (0.6 mg/ml of geneticin)
were selected for CD41 and CD61 expression by immuno-magnetic separation using
mAb AP-2 and magnetic beads coated with anti-mouse IgG (Dynabeads-M-450,
DYNAL, Oslo, Norway). CD41 and CD61 expression of stable clones was analyzed
and quantified by flow cytometric analysis with Qifikit calibration beads, used
according to manufacturer’s instructions (Dako, Denmark). One clone with the
strongest expression of αIIbβ3-integrin was selected. For adhesion assays, αIIbβ3-CHO
transfectant and wild-type (parental) CHO cells were treated with or without 10 mM
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

9
DTT in RPMI 1640, 10 mM Hepes, at 22°C for 20 min to activate the αIIbβ3 complex
receptor (39).
Cell adhesion assays to immobilized proteins
Purified ICAM-Fc proteins diluted in 25 mM Tris pH 8.0, 150 mM NaCl, 2 mM MgCl2
and 2 mM CaCl2, were absorbed to flat-bottom 96-well microtiter plates overnight at
4°C, at 2.5-20 µg/ml (50 µl/well in triplicate). The wells were then blocked for 2 hrs at
22°C with 1% non-fat milk in the same buffer. For adhesion assays, either fixed
GRGDSP-activated or unactivated platelets (5x106/well in a final volume of 100 µl) in
modified Tyrode’s buffer pH 7.4 with or without divalent cations, respectively, wild-
type CHO cells, DTT-activated or unactivated αIIbβ3-CHO transfectants (1x105/well
in a final volume of 100 µl) resuspended in RPMI 1640, 10 mM Hepes containing 2
mM MgCl2 and 2 mM CaCl2, were added to the coated wells and incubated for 90
mins at 22°C. Non adherent cells were removed by washings before microscopic
observation and CHO cell numeration was done as indicated above. Platelets were
counted by flow cytometric analysis as above. For blocking experiments, the cells
were pretreated with specific peptides and their corresponding random counterpart
(125 µM final concentration) or with different mAbs (5 µg for 5x106 platelets or 1x105
CHO cells/100 µl) for 30 min at 22°C prior addition to protein-coated wells.
RESULTS
RBCs interact with activated platelets
To analyze molecular events occurring during RBC-platelet interaction, in vitro
cell adhesion assays were developed using RBCs from donors of common and rare
phenotypes immobilized to plastic surface via anti-GPA binding and platelets from
normal healthy donors, pretreated or not with the synthetic GRGDSP peptide in the
presence of inhibitors of platelet activation, thus resulting in specific αIIbβ3-integrin
activation and the acquisition of high affinity Fg-binding state without addition of a
cellular agonist (37). Accordingly, in addition to bind Fg, GRGDSP-treated platelets
reacted strongly with the mAb PAC-1, which binds to the activated αIIbβ3 complex
(40), but no reactivity with the mAb AK-4 (41), which binds to P-selectin normally
contained in intracellular α-granules (not shown). As shown in Fig. 1, GRGDSP-
activated platelets adhered more efficiently than unactivated platelets to immobilized
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

10
ICAM-4 positive RBCs from control donors. The 100% relative binding was
equivalent to 220 ± 100 GRGDSP-activated platelets adhered to 1.0 x 103 immobilized
RBCs. When unactivated platelets were used as control, a 69% reduced adhesion was
noted that corresponds to a mean background of 31±12%. As preliminary assays
showed that similar results were obtained with fresh and unfrozen RBCs (not
shown), the following studies were performed with unthawed RBCs since rare RBC
variants lacking different membrane proteins were available from our frozen
collection.
Activated platelets bind to coated RBCs lacking the blood group proteins Lu (CD239,
laminin receptor of 78-85 kDa), JMH (CD108, 80 kDa) and DO (ADP-
ribosyltransferase 4 of 47-67 kDa) but expressing normal levels of ICAM-4, as
efficiently as would normal ICAM-4 positive RBCs. Interestingly, when ICAM-4
negative (LWnull) RBCs lacking of the ICAM-4/LW glycoprotein (42 kDa) from three
unrelated donors were coated to plastic wells, a 40 % decrease binding of GRGDSP-
activated platelets was observed after deduction of the mean background
corresponding to the unactivated platelet adhesion to all types of RBCs (p<0.001 vs
unactivated platelets and p<0.05 vs controls).
To confirm that ICAM-4 plays a role in RBC-platelet interactions, RBC adhesion on
adherent platelets stimulated by thrombin, a more physiologically relevant platelet
activator than the RGDS peptide, was also analyzed although in this assay platelets
are more activated with α-granule release than GRGDSP-activated platelets (see
above). Although ICAM-4-positive RBCs did not bind to unstimulated adherent
platelets in the presence of PGE1 and apyrase (not shown), they bind strongly to
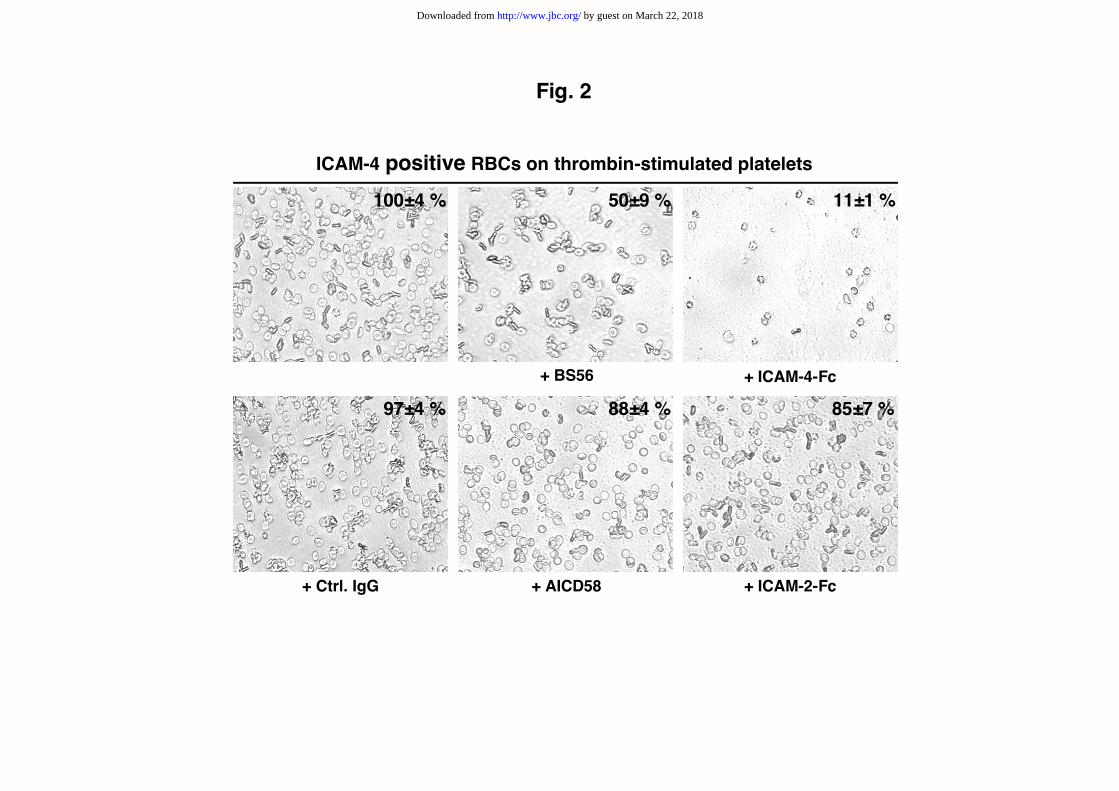
thrombin-stimulated platelets (Fig. 2). This binding was efficiently decrease to 50 ±
9% and 11 ± 1% by mAb BS56 and soluble ICAM-4-Fc protein, respectively, whereas
the mAb AICD58 reacting with the erythroid membrane CD58 protein and the
soluble ICAM-2-Fc protein had only a minor inhibitory effect (88 ± 4 and 85 ± 7%,
respectively). Similarly, mAbs anti-RhD (LOR-15C9), anti-Fy6 (BAM9917) and anti-
MER2 (1D12 or 2F7) directed against various RBC surface membrane proteins did not
exhibit any effect (not shown). Unfortunately, the non specific adherence of frozen
RBCs in this assay made impossible the comparative analysis between the ICAM-4
positive and negative RBCs. Altogether, these data suggests that ICAM-4 might take
a significant part (about 50%) in the adhesion of RBCs to activated platelets. As the
GRGDSP peptide is a trigger of a high affinity state of αIIbβ3-integrin which mediates
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

11
Fg binding and platelet aggregation (37), our data suggested that ICAM-4 might
interact with αIIbβ3-integrin but also with other adhesive molecules.
RBC-platelet interaction is mediated via ICAM-4
To obtain further evidence that ICAM-4 might interact with a high affinity
state of α IIbβ3-integrin, type-I glanzmann's thrombastenia platelets from two
unrelated patients who both exhibit a 6 bp deletion in exon 7 of the β3 gene (42), were
used for cell adhesion assay to coated ICAM-4-Fc protein. Fig. 3A shows that
unactivated platelets from normal control donors did not bind to immobilized ICAM-
4-Fc, as expected from above data, whereas the same platelets activated by the
GRGDSP peptide bound readily to coated ICAM-4-Fc, but not to immobilized ICAM-
1. The 100% relative binding of GRGDSP-activated platelets to ICAM-4-Fc was
equivalent to 12.5±3.0% of the total added platelets. Conversely, platelets from the
thrombasthenic patients type 1 with a severe defect of αIIbβ3-integrin surface
expression, either unactivated (not shown) or GRGDSP-activated, failed to bind to
coated ICAM-4-Fc (Fig. 3A).
In order to determine the specificity of these interactions, the effect of different mAbs
and synthetic peptides on the platelet adhesion to immobilized ICAM-4-Fc protein
was investigated (Fig. 3B). Adhesion of activated platelets from normal control
donors was efficiently blocked (approximately, 70% and 60%, respectively) by P2 and
AP2 mAbs specific for the αIIb-chain in the presence of the β3-chain and the complex-
specific epitope of the αIIbβ3 integrin, respectively. SZ21 and SZ22 mAbs which
recognize the β3- and αIIb-chains alone, respectively, and the BS56 mAb specific for
ICAM-4, partially but significantly inhibited the interaction between ICAM-4 and
activated platelets, whereas the SZ2 mAb directed against the GPIb platelet
glycoprotein and the control mouse IgG had no significant effect (Fig. 3B). In
addition, mAbs FA6 and 3E12 directed against CD36 and CD47, respectively, did not
inhibit the platelet-ICAM-4 interaction. Blocking experiments by synthetic peptides
revealed that the RGD peptide that binds to αIIbβ3-integrin and inhibits Fg binding,
strongly reduced by 75 % the adhesion of activated platelets to ICAM-4, whereas the
RGE peptide had no effect.
RBC-platelet interaction is mediated via ICAM-4 /αIIbβ3 –integrin
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

12
To provide further evidence that ICAM-4 may interact with the αIIbβ3 integrin,
stable CHO transfectants expressing recombinant human αIIbβ3 were generated and
used in cell adhesion assays (Fig. 4). Several αIIbβ3-CHO transfectants were obtained
and one clone expressing a high level of αIIbβ3-integrin (αIIb, 18,600 molecules/cell
and β 3 , 67,000 molecules/cell, as estimated by by flow cytometric analysis with
specific mAbs) was chosen for further studies. The αIIbβ3 integrin of these cells was
activated by DTT treatment and the adhesion of DTT-activated and unactivated
αIIbβ3-CHO transfectants to immobilized ICAM-4-Fc was examined (Fig. 4). In a
preliminary experiment we found that these cells also reacted with PCA-1 Mab that
recognized the activated αIIbβ3 integrin complex (not shown). DTT-activated αIIbβ3-
CHO transfectants dose-dependently bind to coated ICAM-4-Fc protein, whereas
untreated αIIbβ3-CHO transfectants as well as parental CHO cells, either or not
treated with DTT, did not bind at all. About 32% of the total added DTT-activated
αIIbβ3-CHO transfectants adhered to coated ICAM-4-Fc, but there was no binding to
immobilized ICAM-1-Fc protein used as control (Fig. 4B). Identical results were
obtained when the αIIbβ3-CHO transfectants were activated by the GRGDSP-peptide
instead of DTT (not shown). The binding of DTT-activated αIIbβ3-CHO transfectants
to immobilized ICAM-4-Fc could be blocked by approximately 50 % by mAbs
specific for ICAM-4 (BS56) or for the complex-specific epitope of the αIIbβ3 integrin
(AP2), but not with mAbs to the β3-chain (SZ21) and αIIb-chain (SZ22 and P2) of the
αIIbβ3 integrin, as shown on Fig. 4B. The absence of inhibition noted with the mAb P2,
which efficiently blocked activated platelet adhesion (see Fig. 3) might result from
conformational changes and/or glycosylation variations of αIIbβ3 integrin in platelets
and the CHO-transfectants independently of the mode of integrin activation. As
expected, control mouse IgG had no effect.
Putative domains on ICAM-4 that interact with the αIIbβ3 integrin
As an attempt to localize the αIIbβ3 integrin binding site on ICAM-4, domain deletion
mutants lacking either extracellular Ig-like domain D1 or domain D2 were produced
and used in cell adhesion assays to chimeric Fc-proteins. Fig. 5A showed that the
binding of DTT-activated αIIbβ3-CHO transfectants via the αIIbβ3 integrin required the
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

13
presence of both domains D1 and D2, since a 50% decrease binding was observed in
the absence of either domain D1 or D2. Similar effects with less amplitude were
observed using GRGDSP-activated platelets (see Fig. 5B).
Further blocking experiments with synthetic peptides were performed. Adhesion of
activated-platelet was efficiently inhibited to 14% and 58% by the Fibrinogen Binding
Inhibitor (FBI, Fg γ chain residues 400-411) peptide and the ICAM-4 peptide G-V
(residues 65-74), respectively, two peptides exhibiting a QxxDV motif involved in the
fibrinogen/αIIbβ3-integrin interaction (Fig. 5B). When DTT-activated α IIbβ3-CHO
transfectants were used, a 78% decrease binding was observed in the presence of the
ICAM-4 derived peptide G-V whereas the peptide FBI failed to inhibit (Fig. 5A). The
lack of inhibition by the peptide FBI, might result from some changes of αIIbβ3
integrin when expressed in CHO-transfectants, as suspected for the reactivity of mAb
P2 (see above). Of not, neither the random peptides of FBI and G65-V74 nor a control
ICAM-4 peptide (C-R, residues 180-192) had any inhibitory effect. Altogether, these
results demonstrate that the αIIbβ3 binding site on ICAM-4 encompassed domains D1
and D2, and that it seems to reside at the tip of the E strand of domain D1 which is in
contact with the loop C’-E of domain D2.
DISCUSSION
In this report in vitro cell adhesion assays have been developed to evaluate the
capacity of red cell ICAM-4 to interact with platelets and to identify the molecular
basis of the interaction. The αIIbβ3 integrin (platelet fibrinogen receptor GPIIb-IIIa)
was identified as the receptor for RBC ICAM-4. However, we found that the αIIbβ3
integrin had to be in its high affinity state to bind ICAM-4, as the interaction occurred
only after synthetic GRGDSP peptide activation, but not with untreated resting
platelets. This was based on the following evidence: (i) activated platelets adhered
less efficiently to immobilized ICAM-4-negative (LWnull) than to ICAM-4-positive
RBCs, (ii) monoclonal antibodies specific for a complex-specific epitope of the αIIbβ3
integrin or to the β3-chain alone and specific for ICAM-4 to a lesser extent, inhibited
platelet adhesion, (iii) activated platelets from two unrelated type-I glanzmann’s
thrombasthenia patients that are deficient for the αIIbβ3 integrin (and vitronectin
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

14
receptor αvβ3) did not bind to coated ICAM-4-Fc protein, and (iv) DTT-activated
αIIbβ3-CHO transfectants strongly adhere to coated ICAM-4-Fc protein but not to
coated ICAM-1-Fc, and this was inhibitable by specific antibodies. It should be
mentioned that αIIbβ3 integrin activation occurred in the absence of any signaling or
secretion (37,39) and that antibodies specific for GPIb, the von Willebrand receptor of
platelets, for CD36 (platelet GPIV) and CD47 (IAP, integrin-associated protein), two
multifunctional membrane proteins acting as thrombospondin receptors (43,44), did
not inhibit the platelet adhesion to immobilized ICAM4-Fc protein. As
thrombasthenic platelets that expressed normal levels of other platelet receptors like
the α2β1 integrin (collagen receptor), and the fibronectin and laminin receptors
(GPIa-IIa and GPIc’-IIa, respectively), did not adhere to ICAM4-Fc protein, it is
assumed that these proteins do not play a significant role in RBC-platelet interaction
under the experimental conditions used.
Further analysis with platelets, αIIbβ3–CHO transfectants and ICAM-4 Fc mutant
proteins have shown that the two Ig-like domains of ICAM-4 are required for αIIbβ3
integrin interaction, since domain deletion mutants lacking either the first (D1) or
second (D2) Ig-domain exhibited significant reduced binding (see Fig. 5A and B). A
similar effect has been observed when ICAM-4 mutant proteins interact with the
leukocyte αMβ2 (Mac-1) integrin, whereas interaction with the αLβ2 (LFA-1) integrin
requires predominantly the first Ig domain D1 (26). The binding of adhesive proteins
to αIIbβ3 integrin is predominantly mediated by the RGD peptide motif present on the
respective adhesive ligands (45), but this peptide which is absent from ICAM-4,
blocks the ICAM-4/αIIbβ3 interaction. The platelet αIIbβ3 integrin also binds to the
carboxy-terminal end of the Fg γ chain via a dodecapeptide sequence (peptide FBI,
residues 400-411) containing the motif QAGDV (46). Interestingly, ICAM-4 contains a
similar motif at position 70-74 (QLLDV) located in the first ICAM-4 Ig-like domain
(26) and the ICAM-4 peptide G-V (residues 65-74) including this motif is a potent
inhibitor of the ICAM-4/αIIbβ3 interaction. Moreover, the peptide FBI also inhibited
platelet binding to ICAM-4-Fc. These findings suggest that the platelet αIIbβ3 integrin
might interact with the G65-V74 sequence of ICAM-4 that includes a QxxDV motif
known to be involved in the fibrinogen/αIIbβ3-integrin interaction. The G65-V74
sequence motif which forms the tip of the E strand of domain D1 and is in contact
with the loop C’-E of domain D2 (26), most probably constitutes a major part of the
αIIbβ3 integrin binding site on ICAM-4 Therefore, binding inhibitions by RGD and FBI
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

15
peptides, which binds to the β3 chain (GPIIIa) and αIIb chain (GPIIb) respectively (47),
suggest that ICAM-4 binds to the same or overlapping site(s) on the αIIbβ3 complex. It
should be noticed that the G70R substitution responsible for the blood group LWa-
>LWb polymorphism (48) which corresponds to the first position of the QxxDV
motif, had no effect on RBC-platelet adhesion reported here (unpublished
observation), suggesting that this polymorphism is neutral with regards to ICAM-
4/αIIbβ3 integrin interaction.
Our studies therefore indicate that adhesion of normal RBCs to activated platelets
occur through a specific ligand/receptor interaction. Whether or not signaling events
across the platelet and/or RBC membranes are triggered by the interaction of the
αIIbβ3 integrin receptor with its RBC ICAM-4 ligand is currently unknown.
As LWnull RBCs still adhere to activated platelets and RBCs adhere to adherent
platelets stimulated by thrombin, which have release their α-granule contents, it is
assumed that other factors critical for interaction may exist. αIIbβ3 integrin activation
with either RGD-containing peptide or DTT induces Fg binding (37,39), suggesting
that indirect RBC-platelet contacts via this adhesive macro molecule might occur.
However, although ICAM-1 binds to Fg (49), interaction of the structurally related
ICAM-4 protein with Fg has not yet been documented. If such interaction exists, Fg
could form RBC-platelet and RBC-endothelium cross-bridges via RBC ICAM-4 and
αIIbβ3 integrin on activated-platelet and ICAM-1 and/or αvβ3-integrin present on the
stimulated vascular endothelium (50,51). Still other adhesion pathways mediating
cross-bridges of RBCs with platelets and/or endothelial cells with a variety of
adhesive proteins might also be operating, but this need further investigations.
Additionally, erythroid receptors for adhesive molecules, like the Lutheran (CD239,
laminin receptor) (33,34) and sulfated glycolipids (receptors of laminin, TSP, and
vWF) (52,53) are present on the RBCs and might also take part in the RBC-platelet
and endothelial cell interactions. Moreover, direct RBC-endothelial cell interaction
might also occur as ICAM-4 has been reported to bind to α4β1 (VLA-4), and to αvβ1-
and αvβ5-integrins present on hematopoietic cells and might also account for the
binding of sickle RBCs to vascular endothelium (27).
All these findings indicate that ICAM4 is an unusual adhesion molecule which has a
broad ligand binding specificity, including at least some β1, β2, β3 (this report), and β5
integrins, but the binding affinity for these ligands may vary widely. Another
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

16
example of receptor with a promiscuous specificity is the DARC protein (Duffy
Antigen receptor for Chemokines) which binds to CC and CXC families of
chemokines (54). Therefore, it is anticipated that ICAM-4 may have a potential role in
a number of physiological processes, including hemostasis and thrombosis (21,22).
Although further investigation of RBC interaction with blood cells and vascular cells
under various flow conditions should delineate more precisely the physiological
relevance of these interactions, RBC interaction with activated platelets, is supported
by several observations : (i) an active role in hemostasis and thrombosis (21,22) as
interaction between metabolically active RBCs and platelets is known to enhance
platelet reactivity (21), including the enhancement of αIIbβ3 integrin activation and P-
selectin expression (55), (ii) the presence of RBCs as well as of leukocytes in a
developing thrombus (56-58), in which platelets are activated and may interact with
RBCs, (iii) the presence of platelet-erythrocyte aggregates in patients with sickle cell
anemia (59,60) and end-stage renal disease (61). However, the molecular target(s)
responsible for RBCs-platelet interaction have not been characterized. Our results
provide the first direct characterization of a molecular interaction between normal
RBCs and platelets and together with the findings discussed above they strongly
suggest that RBCs may play an active role in hemostasis and thrombosis.
After this paper was submitted, adhesion of normal RBCs to fMLP (formyl-Met-Leu-
Phe peptide)-activated neutrophils and collagen-activated platelets, as well as to
fibrin, was shown under low shear rates conditions (62). Interestingly, the data
suggested that adhesion of RBCs to neutrophils might be mediated through Mac-1
(CD11b/CD18) and ICAM-4, supporting recent findings indicating that β2 integrins
and ICAM-4 interact with each other (26). Additionally, RBC-platelet interaction was
strongly reduced by soluble fibrinogen and EDTA and was partially inhibited by
antibodies to CD36 and GPIb, but no inhibition was noted with a single antibody
against the α IIb chain (CD41) of the α IIbβ3 integrin (62). Although no effect of
monoclonal antibodies to GPIb and CD36 was found in static conditions of assay, our
results indicate that the interaction of thrombin-activated platelets with intact RBCs
(Fig. 2) and of GRGDSP-activated platelets with immobilized ICAM-4 (Fig. 3B) could
be inhibited by soluble ICAM-4 (by 89%) and to at least 50% by the monoclonal
antibody P2 recognizing the αIIb-chain in the presence of the β3-chain, or by the
monoclonal antibody AP-2 specific for a complex-specific epitope of the αIIbβ3
integrin, respectively. Monoclonal antibodies SZ22 and SZ21 to the αIIb-chain and β3-
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

17
chain alone, respectively, were weak inhibitors in the latter condition (Fig. 3B).
Consistent with the above results, our studies have shown that ICAM-4 interaction
with β -integrins is calcium-dependent (26). Obviously, distinct experimental
conditions (platelet activation, flow conditions) and monoclonal antibodies used may
explain the reported differences.
In conclusion, although passive entrapment of RBCs during coagulation or
thrombosis is commonly accepted, these data provide independent evidence
indicating that a physiological interaction between RBCs and activated platelets (and
neutrophils) mediated by specific receptor/ligand interactions can occur in a variety
of biological process, notably during normal hemostatic conditions (clot formation),
pathological occlusion conditions (deep vein thrombosis, sickle cell disease) and
possibly inflammation, particularly under low blood flow conditions, close to static,
which may facilitate RBC adhesion events. Although ICAM-4 may play a significant
role, clearly other receptor/ligand interactions are likely to occur which deserves
further analysis.
Acknowledgements.
We would like to thank Dr David Simmons and Dr Elaine Ferguson for the supply of
the ICAM-pIgI constructs, and Dr P.J Newman for the CD41 and CD61 cDNAs in
pcDNA3.1 vector.
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

18
FOOTNOTES
1the abbreviations used are : RBCs, red blood cells; FBI, Fibrinogen Binding Inhibitor; TSP, thrombospondin ; vWF, von Willebrand Factor ; mAb, monoclonal antibody ;PGE1, prostaglandin-E1 ; TNF-α, Tumor Necrosis Factor-α ; HUVEC, HumanUmbilical Vein Endothelium Cells.
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

19
REFERENCES
1. Telen, M. J. (2000) Semin Hematol 37, 130-1422. Spring, F. A., and Parsons, S. F. (2000) Transfus Med Rev 14, 351-3633. Cartron, J. P., and Colin, Y. (2001) Transfus Clin Biol 8, 163-1994. Verfaillie, C., Hurley, R., Bhatia, R., and McCarthy, J. B. (1994) Crit Rev Oncol
Hematol 16, 201-2245. Hanspal, M. (1997) Curr Opin Hematol 4, 142-1476. Bratosin, D., Mazurier, J., Tissier, J. P., Estaquier, J., Huart, J. J., Ameisen, J. C.,
Aminoff, D., and Montreuil, J. (1998) Biochimie 80, 173-1957. Parsons, S. F., Spring, F. A., Chasis, J. A., and Anstee, D. J. (1999) Baillieres Best
Pract Res Clin Haematol 12, 729-7458. Oldenborg, P. A., Zheleznyak, A., Fang, Y. F., Lagenaur, C. F., Gresham, H. D., and
Lindberg, F. P. (2000) Science 288, 2051-20549. Chan, J. Y., and Watt, S. M. (2001) Br J Haematol 112, 541-55710. Bony, V., Gane, P., Bailly, P., and Cartron, J. P. (1999) Br J Haematol 107, 263-27411. Southcott, M. J., Tanner, M. J., and Anstee, D. J. (1999) Blood 93, 4425-443512. Ward, T. M., Chitnis, C. E., and Miller, L. H. (1994) Baillère’s Clinical Infectious
Diseases 1, 155-19013. Oh, S. S., Chishti, A. H., Palek, J., and Liu, S. C. (1997) Curr Opin Hematol 4, 148-
15414. Wick, T. M., and Eckman, J. R. (1996) Curr Opin Hematol 3, 118-12415. Hebbel, R. P. (1997) J Clin Invest 99, 2561-256416. Serjeant, G. R. (1997) Lancet 350, 725-73017. Bunn, H. F. (1997) N Engl J Med 337, 762-76918. Wautier, J. L., Wautier, M. P., Schmidt, A. M., Anderson, G. M., Hori, O.,
Zoukourian, C., Capron, L., Chappey, O., Yan, S. D., Brett, J., and et al. (1994) ProcNatl Acad Sci U S A 91, 7742-7746
19. Schmidt, A. M., Hofmann, M., Taguchi, A., Yan, S. D., and Stern, D. M. (2000)Semin Thromb Hemost 26, 485-493
20. Cines, D. B., Pollak, E. S., Buck, C. A., Loscalzo, J., Zimmerman, G. A., McEver, R.P., Pober, J. S., Wick, T. M., Konkle, B. A., Schwartz, B. S., Barnathan, E. S.,McCrae, K. R., Hug, B. A., Schmidt, A. M., and Stern, D. M. (1998) Blood 91, 3527-3561
21. Marcus, A. J., and Safier, L. B. (1993) Faseb J 7, 516-52222. Andrews, D. A., and Low, P. S. (1999) Curr Opin Hematol 6, 76-8223. Minetti, G., and Low, P. S. (1997) Curr Opin Hematol 4, 116-12124. Bailly, P., Hermand, P., Callebaut, I., Sonneborn, H. H., Khamlichi, S., Mornon, J. P.,
and Cartron, J. P. (1994) Proc Natl Acad Sci U S A 91, 5306-531025. Bailly, P., Tontti, E., Hermand, P., Cartron, J. P., and Gahmberg, C. G. (1995) Eur J
Immunol 25, 3316-332026. Hermand, P., Huet, M., Callebaut, I., Gane, P., Ihanus, E., Gahmberg, C. G., Cartron,
J. P., and Bailly, P. (2000) J Biol Chem 275, 26002-2601027. Spring, F. A., Parsons, S. F., Ortlepp, S., Olsson, M. L., Sessions, R., Brady, R. L.,
and Anstee, D. J. (2001) Blood 98, 458-46628. Springer, T. A. (1990) Nature 346, 425-43429. Luscinskas, F. W., and Lawler, J. (1994) Faseb J 8, 929-93830. Gahmberg, C. G., Tolvanen, M., and Kotovuori, P. (1997) Eur J Biochem 245, 215-
23231. Hayflick, J. S., Kilgannon, P., and Gallatin, W. M. (1998) Immunol Res 17, 313-327
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

20
32. Van der Vieren, M., Le Trong, H., Wood, C. L., Moore, P. F., St John, T., Staunton,D. E., and Gallatin, W. M. (1995) Immunity 3, 683-690
33. Udani, M., Zen, Q., Cottman, M., Leonard, N., Jefferson, S., Daymont, C., Truskey,G., and Telen, M. J. (1998) J Clin Invest 101, 2550-2558
34. El Nemer, W., Gane, P., Colin, Y., Bony, V., Rahuel, C., Galacteros, F., Cartron, J. P.,and Le Van Kim, C. (1998) J Biol Chem 273, 16686-16693
35. Hebbel, R. P., and Mohandas, N. (1994) Sickle Cell Diease : Basic Principles andClinical Pracrice, 217-230
36. Sonneborn, H. H., Ernst, M., and Voak, D. (1994) Vox Sanguinis 67, 11437. Du, X. P., Plow, E. F., Frelinger, A. L., 3rd, O'Toole, T. E., Loftus, J. C., and
Ginsberg, M. H. (1991) Cell 65, 409-41638. Xia, Z., and Frojmovic, M. M. (1994) Biophys J 66, 2190-220139. Zucker, M. B., and Masiello, N. C. (1984) Thromb Haemost 51, 119-12440. Abrams, C. S., Ellison, N., Budzynski, A. Z., and Shattil, S. J. (1990) Blood 75, 128-
13841. Lasky, L. A. (1992) Science 258, 964-96942. Morel-Kopp, M. C., Kaplan, C., Proulle, V., Jallu, V., Melchior, C., Peyruchaud, O.,
Aurousseau, M. H., and Kieffer, N. (1997) Blood 90, 669-67743. Greenwalt, D. E., Lipsky, R. H., Ockenhouse, C. F., Ikeda, H., Tandon, N. N., and
Jamieson, G. A. (1992) Blood 80, 1105-111544. Brown, E. J., and Frazier, W. A. (2001) Trends Cell Biol 11, 130-13545. Pytela, R., Pierschbacher, M. D., Ginsberg, M. H., Plow, E. F., and Ruoslahti, E.
(1986) Science 231, 1559-156246. Kloczewiak, M., Timmons, S., Lukas, T. J., and Hawiger, J. (1984) Biochemistry 23,
1767-177447. Plow, E. F., D'Souza, S. E., and Ginsberg, M. H. (1992) Semin Thromb Hemost 18,
324-33248. Hermand, P., Gane, P., Mattei, M. G., Sistonen, P., Cartron, J. P., and Bailly, P. (1995)
Blood 86, 1590-159449. Tsakadze, N. L., Zhao, Z., and D'Souza, S. E. (2002) Trends Cardiovasc Med 12, 101-
10850. Altieri, D. C. (1999) Thromb Haemost 82, 781-78651. Byzova, T. V., Rabbani, R., D'Souza, S. E., and Plow, E. F. (1998) Thromb Haemost
80, 726-73452. Hillery, C. A., Du, M. C., Montgomery, R. R., and Scott, J. P. (1996) Blood 87, 4879-
488653. Roberts, D. D., Williams, S. B., Gralnick, H. R., and Ginsburg, V. (1986) J Biol Chem
261, 3306-330954. Neote, K., Darbonne, W., Ogez, J., Horuk, R., and Schall, T. J. (1993) J Biol Chem
268, 12247-1224955. Valles, J., Santos, M. T., Aznar, J., Martinez, M., Moscardo, A., Pinon, M.,
Broekman, M. J., and Marcus, A. J. (2002) Blood 99, 3978-398456. Saniabadi, A. R., Lowe, G. D., Barbenel, J. C., and Forbes, C. D. (1985) Thromb Res
38, 225-23257. Joseph, R., Welch, K. M., D'Andrea, G., and Riddle, J. M. (1989) Thromb Res 53,
485-49158. Bernstein, E., and Kairinen, E. (1971) Science 173, 76559. Wun, T., Paglieroni, T., Tablin, F., Welborn, J., Nelson, K., and Cheung, A. (1997) J
Lab Clin Med 129, 507-516
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

21
60. Wun, T., Paglieroni, T., Field, C. L., Welborn, J., Cheung, A., Walker, N. J., andTablin, F. (1999) J Investig Med 47, 121-127
61. Sirolli, V., Strizzi, L., Di Stante, S., Robuffo, I., Procopio, A., and Bonomini, M.(2001) Thromb Haemost 86, 834-839
62. Goel, M. S., and Diamond, S. L. (2002) Blood 100, 3797-3803
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

22
FIGURE LEGENDS
Fig. 1. Adhesion of platelets to immobilized RBCs.
Adhesion of GRGDSP-activated platelets (gray bars) from normal donors to ICAM-4
positive (ctrl, Lunull, JMHnull, Donull) and ICAM-4 negative (LWnull) frozen RBCs
immobilized onto plastic wells. Unactivated platelets (hatched bars) were used as
controls and the horizontal line visualized the mean background (31±12% )
corresponding to the unactivated platelet adhesion to all types of RBCs. The results
are expressed as the relative percentage of bound platelets, where 100% is calculated
from the total number of normal GRGDSP-activated platelets bound to immobilized
RBCs. The mean ± SEM from at least six experiments is shown. By Student's t test
analysis, ***p<0.001 versus unactivated platelet and ∆p<0.05 or less versus control.
Fig. 2. Adhesion of RBCs to adherent platelets.
Adhesion of ICAM-4 positive RBCs to adherent platelets stimulated by thrombin (0.5
unit/well). RBCs and stimulated adherent platelets were pretreated or not with
saturating concentration of mAbs to ICAM-4 (BS56) and ICAM-4-Fc protein,
respectively. The percentage of bound RBCs is indicated on the top right of each field
of view. The 100% corresponds to the total number of RBCs bound to adherent
platelets. Controls include mAb anti-CD58 (AICD58) which binds to RBCs, unrelated
mouse IgG antibody (ctrl IgG) and ICAM-2-Fc protein.
Fig. 3. Adhesion of platelets to immobilized ICAM-4-Fc protein.
A, Microphotographs showing the comparative adhesion of GRGDSP-activated
platelets from normal or type-I glanzmann’s thrombasthenia (GT type-I) patients to
ICAM-Fc proteins coated to flat-bottom 96-well microtiter plates (1 µg/well). ICAM-
1-Fc protein and unactivated normal platelets were used as negative controls. B,
Adhesion of normal GRGDSP-activated platelets to coated ICAM-Fc proteins (1
µg/well) and pretreated or not with saturating concentrations of mAbs specific for
ICAM-4 (BS56), β3-chain (SZ21), αIIb-chain (SZ22 and P2), αIIbβ3 complex (AP2), CD47
(3E12) and CD36 (FA6.152), or pretreated with 125 µM (final concentration) of RGE
and RGD peptides. The results are expressed as the relative percentage of activated
platelets bound to coated ICAM-Fc proteins. The 100% value is calculated from the
total number of activated platelets bound in the absence of peptides or mAbs.
Negative controls include mAb SZ2 specific for platelet gpIb, unrelated mouse IgG
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

23
antibody (ctrl IgG), ICAM-1-Fc and wells without coated protein. The mean ± SEM
from three experiments is shown. By Student’s t test analysis, **p<0.01, and *p<0.05.
Fig. 4. Adhesion of αIIbβ3-CHO transfectants to immobilized ICAM-4-Fc.
A, Dose-dependent cell adhesion of DTT-activated αIIbβ3-CHO transfectants (�) and
of control including parental CHO cells (DTT-treated or not) and unactivated αIIbβ3-
CHO transfectants (����) to ICAM-4-Fc protein coated to plastic wells at varying
concentration. The results are expressed as mean percentage of bound cells ± SEM of
three experiments. B, Effect of different mAbs on the adhesion of DTT-activated
αIIbβ3 CHO-transfectant to ICAM-4-Fc coated (500 ng/well). Cells were pretreated or
not with saturating concentrations of indicated mAbs specific for ICAM-4 (BS56), β3-
chain (SZ21), αIIb-chain (SZ22 and P2) and αIIbβ3 complex (AP2). The results are
expressed as the relative percentage of activated αIIbβ3-CHO transfectants bound to
coated ICAM-Fc proteins as in Fig. 3. Controls included unrelated mouse IgG
antibody (ctrl IgG) and wells coated with either ICAM-1-Fc (500 ng/well) or no
protein at all. The mean ± SEM from three experiments is shown. By Student's t test
analysis, ***p<0.001.
Fig. 5. ICAM-4 domains interacting with platelet aIIbb3 integrin .
Adhesion of DTT-activated αIIbβ3-CHO transfectants (A) and GRGDSP-activated
platelets (B) to intact ICAM-4-Fc (+) and to ICAM-4-Fc domain deletion mutants (D1
or D2) coated to plastic surface (500 ng/well) and effects of Fg γ chain and ICAM-4
peptides on cells binding. Cells were pretreated with the Fibrinogen Binding
Inhibitor peptide (FBI, residues 400-411) and peptides G-V (residues 65-74) and C-R
(residues 180-192) derived from ICAM-4 at the final concentration of 125 µM. After 30
mins incubation, the cells were tested for binding to coated ICAM-4-Fc (+). For each
peptide tested, the corresponding random (rd) peptide was used as control. C-R
peptide was used as a control ICAM-4 sequence. The results are expressed as
indicated in Fig. 4. Negative adhesion controls include wells without coated protein.
The mean ± SEM from three experiments is shown. By Student’s t test analysis,
***p<0.001, and **p<0.01.
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from
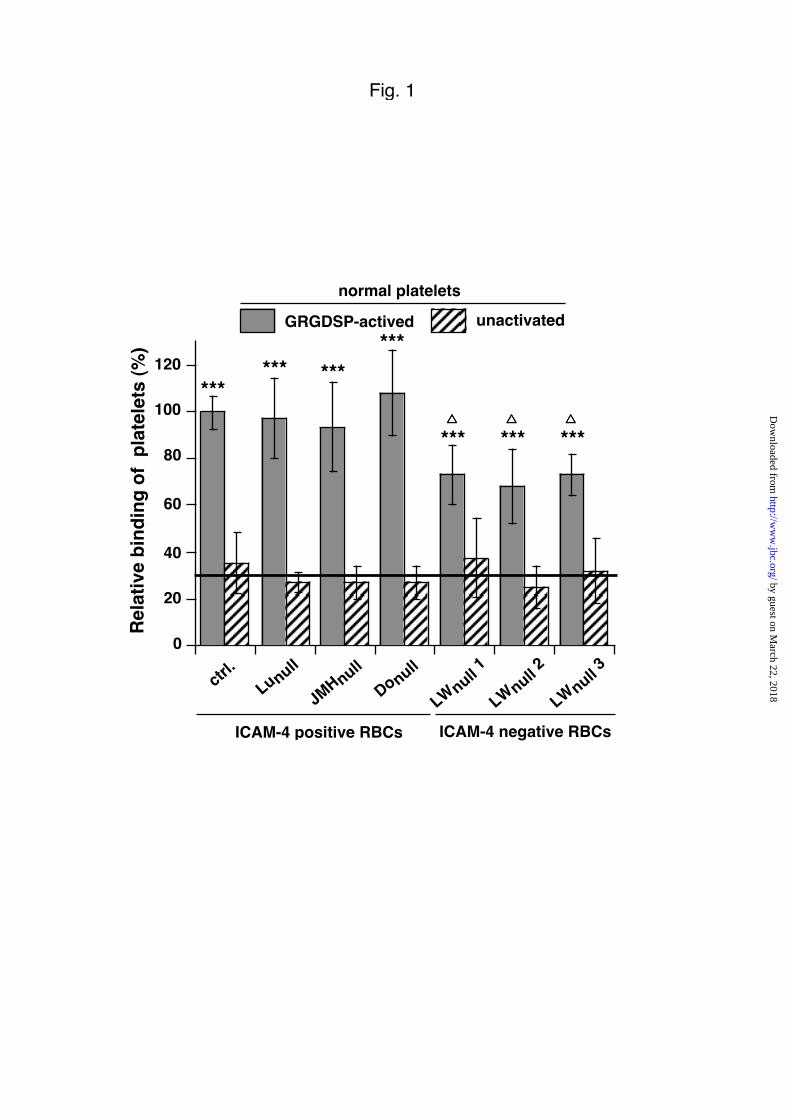
Fig. 1
0
20
40
60
80
100
120
Rel
ativ
e b
ind
ing
of
pla
tele
ts (
%)
ICAM-4 negative RBCs
ctrl
.
Donull
Lunull
JMHnull
***
*********
***����
***����
***����
LWnull 1
LWnull 2
LWnull 3
ICAM-4 positive RBCs
GRGDSP-actived
normal platelets
unactivated
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from
+ BS56
+ Ctrl. IgG
+ ICAM-4-Fc
+ ICAM-2-Fc
50±9 % 11±1 %
97±4 %
100±4 %
88±4 % 85±7 %
+ AICD58
ICAM-4 positive RBCs on thrombin-stimulated platelets
Fig. 2
by guest on March 22, 2018http://www.jbc.org/Downloaded from
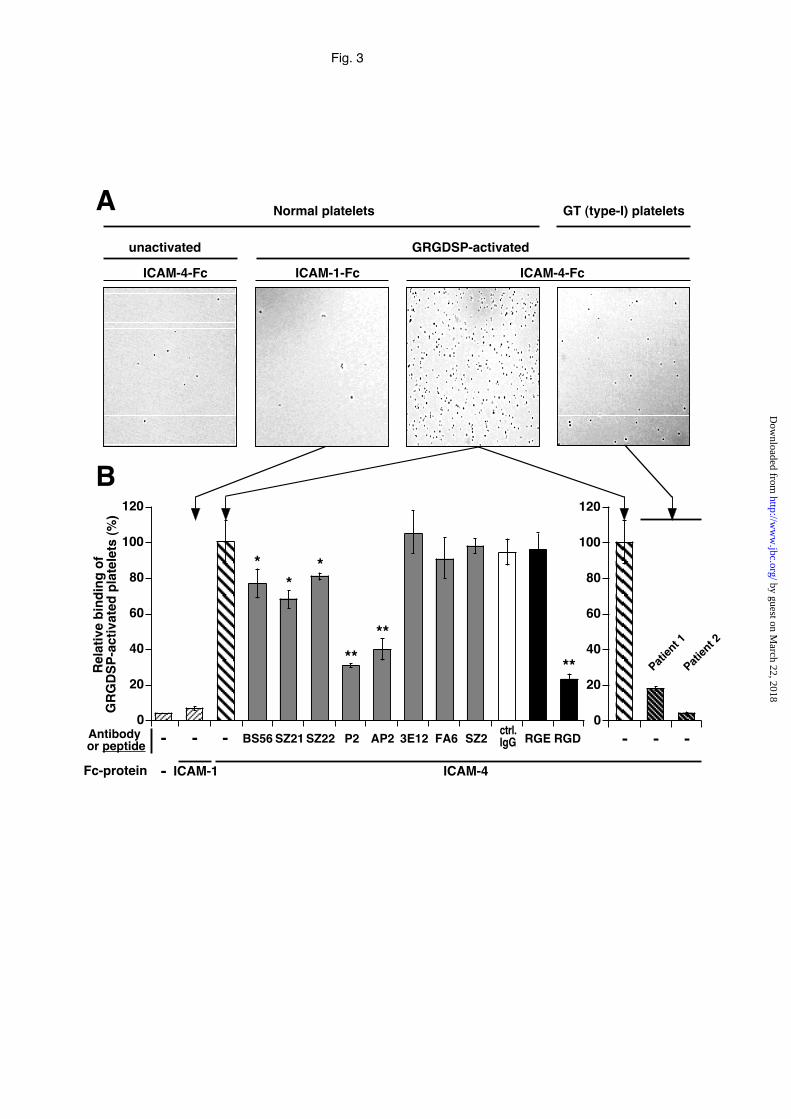
Fig. 3
ICAM-1-Fc ICAM-4-Fc
Normal platelets GT (type-I) plateletsA
GRGDSP-activated
ICAM-4-Fc
unactivated
ICAM-4
- -
B
-
20
40
60
80
100
120
0
R
elat
ive
bin
din
g o
fG
RG
DS
P-a
ctiv
ated
pla
tele
ts (
%)
ICAM-1
Antibody or peptide
-
- - -
Fc-protein
20
40
60
80
100
120
0
Patien
t 1
Patien
t 2
RGE RGD
**
SZ21 SZ22 P2 AP2BS56 3E12ctrl.IgGSZ2FA6
**
*
****
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from
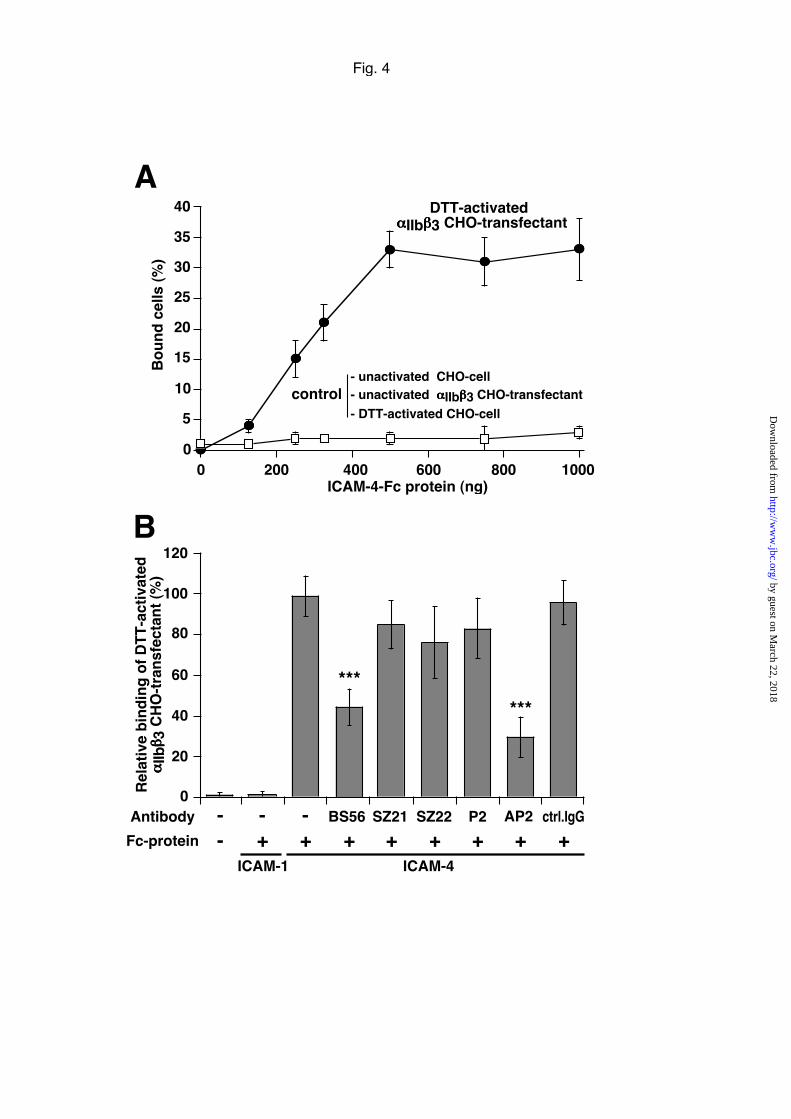
Fig. 4
- BS56 AP2SZ21 SZ22 ctrl.IgGP2
+ + + + + + +--
0
20
40
60
80
100
120
Rel
ativ
e b
ind
ing
of
DT
T-a
ctiv
ated
αααα
IIbββββ 3
CH
O-t
ran
sfec
tan
t (%
)
Antibody
Fc-protein
B
-+
ICAM-1 ICAM-4
***
***
DTT-activatedααααIIbββββ3 CHO-transfectant
Bo
un
d c
ells
(%
)
ICAM-4-Fc protein (ng)
10
15
20
25
30
35
40
0 200 400 600 800 1000
5
0
A
- unactivated CHO-cell- unactivated ααααIIbββββ3 CHO-transfectant
- DTT-activated CHO-cell
control
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from
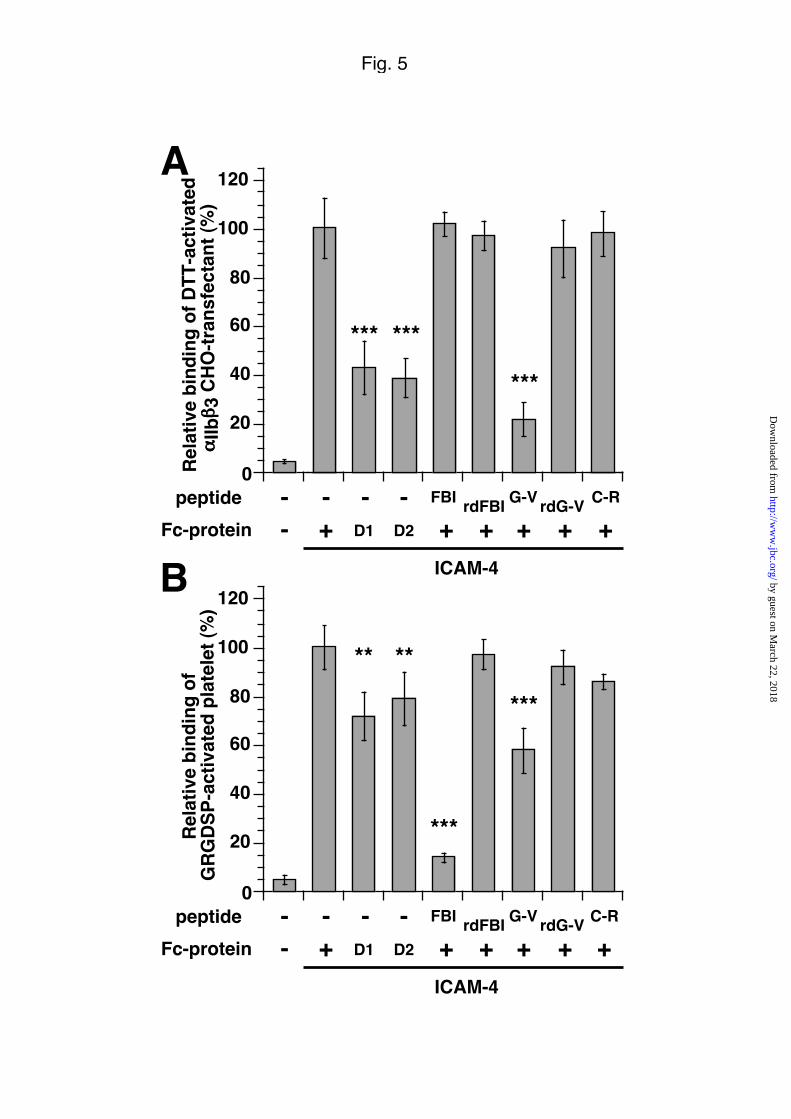
Fig. 5
Rel
ativ
e b
ind
ing
of
DT
T-a
ctiv
ated
αααα
IIbββββ
3 C
HO
-tra
nsf
ecta
nt
(%)
-D1 D2 + +
--
peptide
Fc-protein
-
ICAM-4
0
20
40
60
80
100
120
+FBI rdFBI G-V rdG-V C-R-
+ + +
A
*** ***
***
Rel
ativ
e b
ind
ing
of
GR
GD
SP
-act
ivat
ed p
late
let
(%)
-D1 D2 + +
--
peptide
Fc-protein
-
ICAM-4
0
20
40
60
80
100
120
+FBI rdFBI G-V rdG-V C-R-
+ + +
B
***
***
** **
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Sonneborn, Jean-Pierre Cartron and Pascal BaillyPatricia Hermand, Pierre Gane, Martine Huet, Vincent Jallu, Cécile Kaplan, Hans H
(sub3}-integrinβIIbαRed cell ICAM-4 is a novel ligand for platelet activated
published online December 10, 2002J. Biol. Chem.
10.1074/jbc.M211282200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from


















