
Protein Families (Relating Protein Sequence, Structure, and Function) || The Nucleophilic Attack...
33
SECTION II IN-DEPTH REVIEWS OF PROTEIN FAMILIES
Transcript of Protein Families (Relating Protein Sequence, Structure, and Function) || The Nucleophilic Attack...

SECTION II
IN-DEPTH REVIEWS OF PROTEINFAMILIES

6THE NUCLEOPHILIC ATTACKSIX-BLADED β-PROPELLER (N6P)SUPERFAMILY
Michael A. Hicks, Alan E. Barber II,and Patricia C. BabbittDepartment of Bioengineering and Therapeutic Sciences, UCSF Mission Bay, SanFrancisco, CA,USA
CHAPTER SUMMARY
Functionally diverse enzyme superfamilies (SFs) are defined in this chapteras groups of evolutionarily related proteins that conserve constellations offunctionally important active site residues but whose substrates, products,and even overall reactions can be substantially different. Several of theseSFs are classified in a publically available resource, the Structure–FunctionLinkage Database (SFLD), which links protein sequences and structures withtheir chemical reactions. The classification of some protein families into SFs,however, can be especially difficult when complex variations in sequence,combined with an incomplete knowledge of the determinants of function,prevent a full understanding of the sequence–structure–function relationship.The nucleophilic attack six-bladed β-propeller (N6P) SF provides an interest-ing model for evaluating these complexities based both on the limited numberof functionally characterized proteins in the SF and on the great diversity ofits sequences and functions. This chapter describes the SF and its classifi-cation in the SFLD and other protein databases. Additionally, we describethe N6P SF in the context of other proteins sharing the β-propeller fold and
Protein Families: Relating Protein Sequence, Structure, and Function, First Edition.Edited by Christine Orengo and Alex Bateman.© 2014 John Wiley & Sons, Inc. Published 2014 by John Wiley & Sons, Inc.
127

128 THE NUCLEOPHILIC ATTACK SIX-BLADED β-PROPELLER (N6P) SUPERFAMILY
highlight the challenges in defining accurate functional boundaries betweenstructurally similar groups. Finally, we discuss how the SF context providesa strategy for selecting protein targets for experimental characterization thatcan be especially informative for functional inference.
6.1 INTRODUCTION
By August 2013, the UniProtKB/TrEMBL protein database contained more than41 million sequences and was rapidly increasing in size, with the rate of genomesequencing continuing to increase as technological advances drive down sequenc-ing costs. Metagenomics and other new sequencing efforts also continue to rampup, promising to multiply the number of available protein sequences many timesover. As these databases grow, a decreasing proportion of these proteins canbe experimentally characterized, necessitating assignment of molecular functionalmost exclusively using computational methods. To meet these challenges, anno-tation strategies have become more sophisticated, benefiting especially from theuse of multiple orthogonal methods to improve the accuracy of functional pre-diction (reviewed in Frishman, 2007; Loewenstein et al., 2009; Rentzsch andOrengo, 2009). Accompanying these, new resources have been developed to clas-sify a large proportion of the known protein universe of evolutionarily relatedproteins into groupings so that what is known about their sequence as well asstructural and functional relationships can be used as a starting point for inferenceof their functional properties (see Chapters 2, 4, and 5).
Additional resources target specific classes of proteins for exploration and clas-sification of structure–function relationships. This chapter introduces an impor-tant subset of enzyme SFs that we describe as “functionally diverse” and forwhich data and information is accessible using one of these specialized resources,the SFLD (Pegg et al., 2006) (http://sfld.rbvi.ucsf.edu/). The SFLD defines prop-erties of enzyme SFs with a focus on mapping information about their molecularfunctions and mechanisms to similarity-based classification of their sequences,structures, and active site features. The SFLD also provides tools for investigat-ing the evolution of the many different reaction families these SFs contain andpredicting the reaction and substrate specificities of new members discovered ingenome and other sequencing projects.
The next section of this chapter provides some basic background importantfor understanding our analysis of one SFLD SF, namely the N6P SF. First, wediscuss the concept of functionally diverse enzyme SFs using as an examplethe well-curated enolase SF (Babbitt et al., 1996; Gerlt et al., 2012; Gerlt et al.,2005) and show how the study of these types of SFs can be used to provide cluesabout how new enzyme reactions evolve from a conserved structural scaffold andactive site features. This is followed by a brief summary of the organization of theSFLD and the tools and information it contains, accompanied by a description ofhow the SF context can be used to improve our ability to predict the molecular

BACKGROUND, RESOURCES, AND TOOLS 129
functions of enzymes, annotate new members of an SF as they are added tothe public databases, and identify those that have been misannotated. Finally,to describe our large-scale analysis of structure–function relationships acrossthe 2500 proteins of the N6P SF, we introduce protein similarity networks, arelatively new application of network technology to examine and illustrate broadfunctional trends in protein SFs from the context of sequence and structuralsimilarity (Atkinson et al., 2009).
We focus in the body of this chapter on the N6P SF. This SF is much lesswell understood than the enolase SF and illustrates how large-scale computationalanalysis to define structure–function relationships can aid in predicting the reac-tion specificity for enzymes of unknown function (called “unknowns” in the restof this chapter). The N6P SF currently contains approximately 2500 sequences(Hicks et al., 2011) and represents a subset of greater than 17,000 proteins weanalyzed within an even larger six-bladed β-propeller fold class. (These >17,000proteins represent the closest matches outside of the N6P SF but do not includesome SFs considered by other resources to belong to the fold class, such as theneuraminidases.) In this chapter, we describe in some detail how the SF contextallows us to make inferences about how differences in the sequence and structuralproperties of the N6P SF map to differences in their functional properties. ThisSF was chosen because of the importance of its known reactions in many typesof biological processes, including the synthesis of natural products that are usedas precursors for the development of drugs targeting a broad group of humandiseases (Ma et al., 2006). Because of the complexity of their structure–functionrelationships and the relative lack of experimentally or structurally characterizedmembers, this SF also exemplifies especially well some of the challenges asso-ciated with classification and annotation of new enzymes as they are discoveredin genome projects.
An online classification of the N6P SF as described in this chapter is availableat the SFLD, along with interactive versions of sequence similarity networkssuch as those shown in this chapter. Additional tools provided by the SFLDenable users to classify their sequences of interest into subgroups and familieswithin the SF.
6.2 BACKGROUND, RESOURCES, AND TOOLS IMPORTANTFOR UNDERSTANDING THIS CHAPTER
6.2.1 Functionally Diverse Enzyme Superfamilies
Functionally diverse enzyme SFs are sets of evolutionarily related proteins (alsocalled divergent proteins) that conserve constellations of functionally importantactive site residues but whose substrates, products, and even overall reactionscan be substantially different (Babbitt and Gerlt, 1997; Gerlt and Babbitt, 2001).Within each SF, we define as a family the set of sequences, typically frommultiple organisms, that catalyze the same reaction in the same way. Betweenthese two levels, subgroups are defined as needed to describe subsets of sequences

130 THE NUCLEOPHILIC ATTACK SIX-BLADED β-PROPELLER (N6P) SUPERFAMILY
that cluster together on the basis of sequence and structural features. Using thishierarchy, the SFLD can link sequence, structure, and functional properties in anespecially useful way: that is, it allows us to associate these properties to enzymesat both the SF and family levels. Figure 6.1 illustrates the SFLD hierarchy for theenolase SF. This classification hierarchy also allows us to address problems of“over-annotation” to a specific function that cannot be validated from availableinformation, thereby avoiding misannotation of reaction specificity. For example,enzymes that can only be assigned to an SF on the basis of structural and activesite motifs conserved among all of its members but not to a specific family areannotated only as belonging to that SF. This allows us to associate functionalproperties of an enzyme only with the specific aspects of its active site that enablethose properties. As a result, this classification scheme provides a blueprint for theinference of functional properties that is more reliable for functional annotationthan simple approaches based on overall sequence similarity (Schnoes et al.,2009). Many other approaches use related principles, some of which are describedin other chapters in this book (Chapters 7 and 8).
In functionally diverse enzyme SFs, divergent evolution of such broadly dif-ferent chemical reactions can be described by the “chemistry-constrained” modelof enzyme evolution. In this model, the ancestral structural scaffold can be “re-engineered” by nature for a variety of functions by reusing a conserved activesite that mediates a fundamental chemical capability such as a partial reaction.
Figure 6.1 Classification hierarchy for some representative subgroups and families of theenolase SF. At the SF level, all members share a subset of active site residues required toinitiate the partial reaction they all have in common. Subgroups designate subsets of the SFfor each of which an additional set of active site residues are conserved, distinguishing thesubgroups from each other. The family level describes an isofunctional group of proteinsthat catalyze the same reaction in the same way. Typically, each is distinguished by afamily-specific set of residues associated with reaction and substrate specificity. Althoughdifferent families can evolve at different rates, the sequence identity within families isgenerally greater than between families.
BACKGROUND, RESOURCES, AND TOOLS 131
Built on this foundation, variations in substrate binding and overall chemistryevolve (Babbitt and Gerlt, 1997; Gerlt and Babbitt, 2001; Glasner et al., 2007).Ultimately, the new overall reactions that result must be consonant with both theconstraints imposed by the SF’s common chemical capability and the nature of thereactions that a new substrate can undergo. While this model has been useful fordeveloping a hierarchical classification of linked structure–function properties inenzymes, we note that evolutionary relationships within an SF, especially for verydivergent members, can be difficult to define, as we show below for the N6P SF.
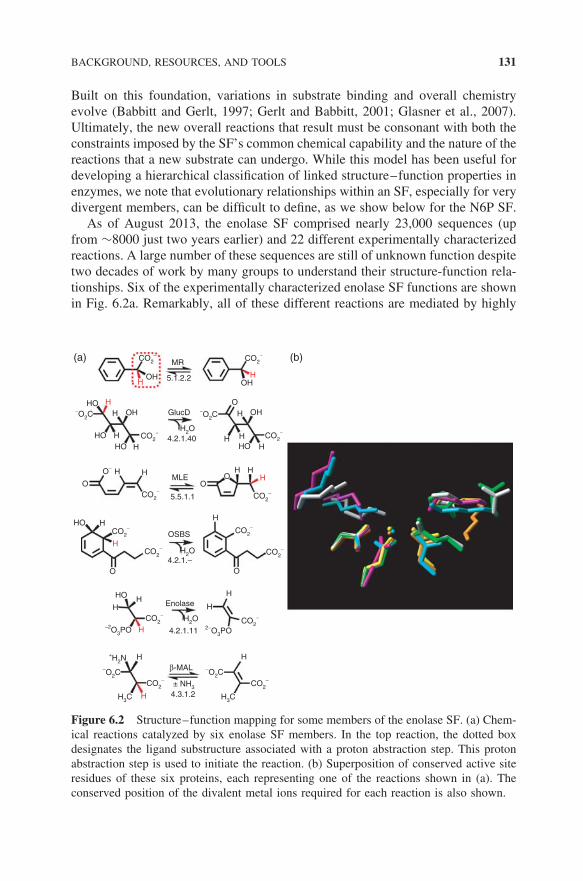
As of August 2013, the enolase SF comprised nearly 23,000 sequences (upfrom ∼8000 just two years earlier) and 22 different experimentally characterizedreactions. A large number of these sequences are still of unknown function despitetwo decades of work by many groups to understand their structure-function rela-tionships. Six of the experimentally characterized enolase SF functions are shownin Fig. 6.2a. Remarkably, all of these different reactions are mediated by highly
HO
HOHO
HO
HO
HO
OH
OHOH
OH
H HH
OHH
H
H
H
H
HH
H
H
H H
H
H
H H
H
H
H
H
H H
O
O O
OO
H GlucD−O2C−O2C
H2O
H2O
H2O
CO2−
O−
CO2− CO2
−
CO2−
CO2−CO2
−
CO2−
CO2−
CO2−
−2O3PO
H3C H3C
± NH3
2−O3PO
−O2C−O2C
+H2N
CO2−
CO2−
CO2−
CO2−
CO2−
4.2.1.40
MR
5.1.2.2
MLE
5.5.1.1
OSBS
Enolase
4.2.1.−
4.2.1.11
β-MAL
4.3.1.2
(a) (b)
Figure 6.2 Structure–function mapping for some members of the enolase SF. (a) Chem-ical reactions catalyzed by six enolase SF members. In the top reaction, the dotted boxdesignates the ligand substructure associated with a proton abstraction step. This protonabstraction step is used to initiate the reaction. (b) Superposition of conserved active siteresidues of these six proteins, each representing one of the reactions shown in (a). Theconserved position of the divalent metal ions required for each reaction is also shown.

132 THE NUCLEOPHILIC ATTACK SIX-BLADED β-PROPELLER (N6P) SUPERFAMILY
similar overall structures and active sites (Fig. 6.2b). This finding can be ratio-nalized by the observation that these active site similarities are associated witha partial reaction common to all members of the SF, for example, abstractionof a proton on a carbon alpha to a carboxylate group (as shown in the commonsubstructure box in Fig. 6.2a), leading to the formation of a common type ofenolate anion intermediate. Although some of their pairwise sequence identitiesare less than 10%, all sequences assigned to the SF show conservation of thisproton abstraction machinery and metal-binding ligands.
Formation of the intermediate in the enolase and other functionally diverse SFsis typically followed by an additional partial reaction(s) that differ in each enzymefamily, leading to a range of different products. Thus, even for the thousandsof enolase SF sequences for which we cannot assign a reaction specificity, wecan reasonably infer that all will catalyze the common partial reaction and thattheir substrates will contain the substructure moiety associated with the protonabstraction step (Fig. 6.2a). Thus, the SF context provides a rules-based approachfor inference of the SF’s common functional capability of all of its members.Further, the nature of this step restricts the search space for functional inferenceof their reaction and substrate specificities.
Members of functionally diverse SFs are difficult to annotate and easy tomisannotate because they all look alike because of the conserved sequence andstructural and functional characteristics they share (Fig. 6.2). Since they havediverged to catalyze different reactions, families within these SFs can be distin-guished by additional sequence and structural features that define their reactionand substrate specificities. But because the different families in an SF often evolveat different rates and exhibit low sequence similarity both within and betweenfamilies, their specificity-conferring features are often challenging to identify(see, e.g., Glasner et al., 2006; Nguyen et al., 2008). As a result, misannotationlevels for these families can be alarmingly high (Schnoes et al., 2009).
An estimate of the number of functionally diverse enzyme SFs in nature canbe made using PDBSprotEC (Martin, 2004) and SCOP (Andreeva et al., 2008)to map the enzyme nomenclature (EC) (International Union of Biochemistry andMolecular Biology: Nomenclature Committee and Webb, 1992) subsubclasses(reaction types) to structural SFs. Defining as functionally diverse those SFscontaining two or more families different at the third digit of the EC class(representing reaction specificity), over 250 SFs are predicted (Almonacid andBabbitt, 2011). Although currently comprising about a third of known enzymeSFs, this count is likely an under-representation, as it only takes into accountthose enzymes for which EC numbers have been identified and for which thereaction specificity is known. Thus, not only are structure–function relationshipsin functionally diverse enzyme SFs complex, making them especially challengingfor understanding the roles of these proteins in many different applications, butthe significant proportion of the enzyme universe they represent also underscoresthe importance of addressing these challenges.

BACKGROUND, RESOURCES, AND TOOLS 133
6.2.2 Structure–Function Linkage Database (SFLD)
The SFLD is a publicly available resource that links evolutionarily relatedsequences and structures from functionally diverse SFs to their chemicalreactions. It is unique in linking the overall and partial reactions (or otherchemical capabilities) common to all members of a SF, subgroup, or familywith conserved sequence and structural elements that enable them. The SFLDontology and schema were designed to represent enzyme structure–functionlinkage at these multiple levels of granularity to allow annotation of a functioncommon to SFs even if family-level reaction and substrate specificity could notbe predicted (Pegg et al., 2005). This schema also allows us to address problemsof “over-annotation” to a specific function that cannot be validated from availableinformation. The SFLD principally targets SFs that are believed to have evolvedaccording to the chemistry-constrained model of evolution described above,as these types of SFs present special challenges for inference of reaction andsubstrate specificity. In tying together a theoretical model that uniquely linksstructure and function in each of the SFs it contains, the SFLD provides a levelof organization and highly curated information that allows users to leveragethe data for biological insights (Pegg et al., 2006). A wide range of browse andsearch tools are available to aid users in accessing SFLD information, along withvisualization tools providing interactive access to such data as curated structures,chemical reactions, multiple alignments, and similarity networks. The SFLDalso includes orthogonal information such as operon context and links to severalother information resources devoted to classification and analysis of enzymes.
6.2.3 Introduction to Protein Similarity Networks
The sequences in the SFs targeted by the SFLD typically number in the thou-sands, so that even using redundance filtering it is not possible to create a globalpicture of their sequence/structure relationships using phylogenetic trees or mul-tiple alignments, as these are slow to generate and realistically cannot be usedwith thousands of diverse sequences (or hundreds of structures). Moreover, fortheir structure–function mappings, many different visualizations of each of theirconstituent subgroups are often needed, requiring a method for querying the datathat is very fast to construct, easy to update on a regular basis, and able to capturelarge amounts of information in a single view. To meet these requirements, wehave begun to use sequence- and structure-similarity networks.
For a particular SF, many networks can be generated in a few days usingseveral types of similarity metrics, for example, based on protein sequence orstructure, or ligand or reaction similarity, and viewed using various clusteringalgorithms, layouts, and thresholds. In capturing all connections among proteinsbetter than a user-specified threshold, networks provide information not availablefrom alignments or trees (Atkinson et al., 2009). Especially useful, similaritynetworks can be linked to functional properties by overlaying the networks withdifferent types of biological and chemical information, providing a simple and fast

134 THE NUCLEOPHILIC ATTACK SIX-BLADED β-PROPELLER (N6P) SUPERFAMILY
way to investigate functional trends from the context of sequence or structuralsimilarity. For example, by painting these networks with the known reactionspecificities of previously characterized families, it is easy to identify clusters ofknown function, sometimes allowing us to hypothesize functional properties ofunknowns that cluster nearby.
Although networks have previously been used extensively to capture manytypes of biological relationships such as genetic or protein interactions, applica-tion of network methods to structure–function mapping in proteins is relativelynew (see Adai et al., 2004; Enright et al., 2002; Finn et al., 2006; Frickey andLupas, 2004; Medini et al., 2006 for some examples). In our work on functionallydiverse enzyme SFs, we have now established the utility of similarity networks formany applications. Examples include choosing targets for experimental analyses(Pieper et al., 2009), showing the relative outlier status of specific families withina large SF (Nguyen et al., 2008), and illustrating the correlation of active siteconservation patterns with lineage (Hall et al., 2007). Additionally, as describedhere, similarity networks currently offer the only practical way to summarizestructure–function relationships in large and complex SFs in a single, intuitivelyaccessible representation (see Atkinson and Babbitt, 2009, for example).
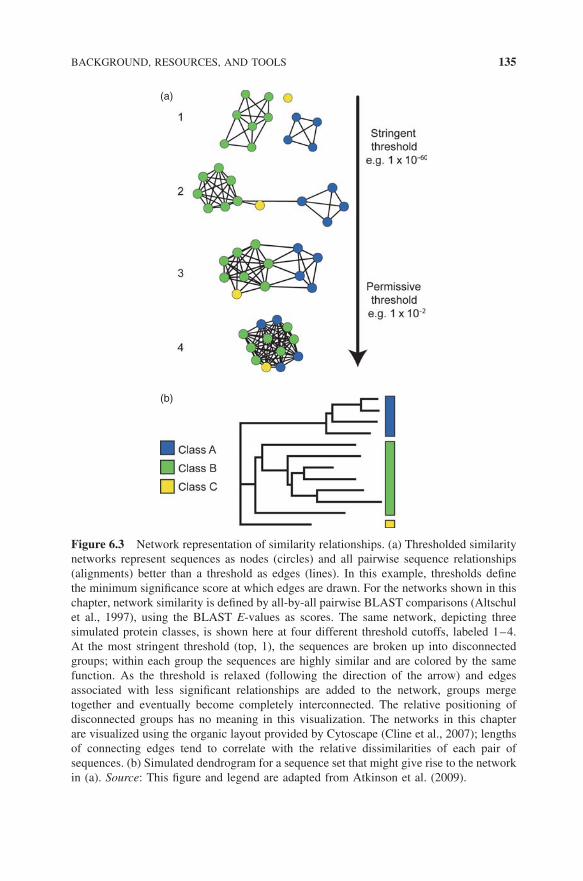
Figure 6.3 illustrates some basic principles in the creation and application ofthresholded similarity networks such as those provided in this chapter. Althoughthese network figures are static images, the downloadable versions available fromthe SFLD are fully interactive so that clicking on a node pulls up accessioncodes to several data resources, species, and other metadata. Such networks canbe easily painted with these and other types of information such as the presenceof conserved motifs, common genome context, cellular location, or similarity ofsubstrates, making them useful for interpreting structure–function relationshipsin individual proteins or across entire SFs. Using such orthogonal information tohelp reveal trends across similarity clusters from these types of functional cluesis a major strength of the approach.
Validation of similarity networks using several well-characterized SFs hasidentified many advantages of network representations, showing that, in general,sequence networks generated from all-by-all pairwise comparisons of a largenumber of divergent queries correlate well with distances from phylogenetictrees and recapitulate much of the information in them (Atkinson et al., 2009).They also provide new information not available from trees, as they can capturemultiple similarity connections between nodes, rather than showing only thetwo best connections for any one leaf, as is provided in visualizations of trees.Network topologies are generally robust to missing data and agree with knownsequence and structural relationships. In addition, structure-based and sequence-based networks largely agree, providing a useful check especially on sequence-based networks that depict very divergent relationships.
A practical advantage of networks is that they can easily handle many moresequences than can be generally evaluated using trees or multiple alignments.However, networks still have size limitations, especially using laptops or desktop
BACKGROUND, RESOURCES, AND TOOLS 135
(a)
(b)
Figure 6.3 Network representation of similarity relationships. (a) Thresholded similaritynetworks represent sequences as nodes (circles) and all pairwise sequence relationships(alignments) better than a threshold as edges (lines). In this example, thresholds definethe minimum significance score at which edges are drawn. For the networks shown in thischapter, network similarity is defined by all-by-all pairwise BLAST comparisons (Altschulet al., 1997), using the BLAST E-values as scores. The same network, depicting threesimulated protein classes, is shown here at four different threshold cutoffs, labeled 1–4.At the most stringent threshold (top, 1), the sequences are broken up into disconnectedgroups; within each group the sequences are highly similar and are colored by the samefunction. As the threshold is relaxed (following the direction of the arrow) and edgesassociated with less significant relationships are added to the network, groups mergetogether and eventually become completely interconnected. The relative positioning ofdisconnected groups has no meaning in this visualization. The networks in this chapterare visualized using the organic layout provided by Cytoscape (Cline et al., 2007); lengthsof connecting edges tend to correlate with the relative dissimilarities of each pair ofsequences. (b) Simulated dendrogram for a sequence set that might give rise to the networkin (a). Source: This figure and legend are adapted from Atkinson et al. (2009).

136 THE NUCLEOPHILIC ATTACK SIX-BLADED β-PROPELLER (N6P) SUPERFAMILY
computers. Currently, the practical limit for opening and exploring networksusing Cytoscape is approximately 106 edges, which typically correlates to a fewthousand fairly divergent sequences.
Similarity networks are not without issues, however, and several significantlimitations can affect their use. For example, it is important to confirm thatthe similarity signal captured by BLAST (Altschul et al., 1997) (or even morecomplex similarity metrics such as hidden Markov models (Eddy, 1996), or someof those used in structure comparison algorithms) is comparable across a target setof highly diverse proteins. For interpreting similarity networks, we also cautionthat coming to a final conclusion based on networks, especially those generatedby pairwise comparisons using algorithms such as BLAST, should usually beaccompanied by additional analyses. Unlike phylogenetic trees, BLAST-based orstructure-based networks are not based on an explicit evolutionary model andtherefore cannot be used as a substitute for a phylogenetic tree. As with othercomparisons of very divergent proteins, variations between sequence or structuralfamilies associated with domain organization, the presence or absence of multipledomains, or inserts that may differ widely across a divergent set of proteins canalso complicate interpretation of relationships. Such issues can be mitigated ifcare is used in interpreting the results.
6.3 SEQUENCE/STRUCTURE/FUNCTION RELATIONSHIPSIN THE NUCLEOPHILIC ATTACK 6-BLADED β-PROPELLER (N6P)SUPERFAMILY
6.3.1 Classification of Proteins in the 6-Bladed β-Propeller Fold Classand the N6P Superfamily
As discussed in other chapters of this book (see Chapter 3 and 7), CATH (class,architecture, topology, homology) (Cuff et al., 2011) and SCOP (structural clas-sification of proteins) (Andreeva et al., 2008) provide frameworks for identifyingsimilarities in structure across the protein universe. In this section, we use theseresources to provide a context for our definition of the N6P SF within the largerscope of the 6-bladed β-propeller fold.
Examination of similarity relationships between a set of 41 nonredundant crys-tal structures of six-bladed β-propeller fold proteins (Fig. 6.4) identified onemajor cluster and several smaller clusters of related proteins. Mapping of func-tional information designating major functional classes (defined as Pfam families(Finn et al., 2010)) onto the network shows that structures of known functionalgroups cluster together. Additionally, this view allows us to begin to see con-nections between proteins of different functions. In the major cluster of 18 pro-tein structures, eight families are represented: glucose-sorbosone dehydrogenase(GSDH), senescence marker protein-30/gluconolactonase/luciferin-regeneratingenzyme-like region (SGL), arylesterase, strictosidine synthase (SS), major royaljelly protein (MRJP), NHL, PD40, and low-density lipoprotein repeat class B(Ldl_recept_b).

RELATIONSHIPS IN THE N6P SF 137
Figure 6.4 Structure-based similarity network of crystal structures sharing the six-bladedβ-propeller fold. Forty-one nonredundant structures were compared pairwise using thealgorithm TM-align (Zhang and Skolnick, 2005). The structural representatives of theN6P SF are boxed. Structures are colored by Pfam family as shown. Each node representsa structure and is labeled with its Protein Data Bank (PDB) ID. Edges are drawn betweennodes only if the alignment score is greater than 0.7, a score considered statisticallysignificant by the authors of TM-align. For this network, the median alignment length forthose edges displayed is 233 residues and the median root-mean-square deviation (RMSD)is 2.75 A. (See insert for color representation of the figure.)
Classification of this fold class and of the proteins of the N6P SF are generallysimilar among the three comprehensive structure and sequence resources (SCOP,CATH, and Pfam) and are generally consistent with those shown in Fig. 6.4.Of the structures represented in this figure, 61% are classified in SCOP v1.75and 78% in CATH v3.4.0. Among these, several belong to an identical homolo-gous SF in CATH: the TolB, C-terminal domain SF (2.120.10.30). These includeproteins from the SGL (1pjx.pdb, 2ghs.pdb, 2p4o.pdb), arylesterase (1v04.pdb),MRJP (2qe8.pdb), NHL (1rwi.pdb, 1q7f.pdb), PD40 (2ojh.pdb), and Ldl_recep-t_b (1npe.pdb, 1ijq.pdb) Pfam families. CATH also includes proteins not in this

138 THE NUCLEOPHILIC ATTACK SIX-BLADED β-PROPELLER (N6P) SUPERFAMILY
central cluster as belonging to the same homologous SF, notably two additionalPD40 proteins (1crz.pdb, 2hqs.pdb), an additional Ldl_recept_b (1n7d.pdb), anda GSDH protein (1cru.pdb) that does not connect into our central cluster at thisthreshold (though three other pdb structures that are not currently classified inCATH do connect). Other proteins in the network are classified into differenthomologous SFs in CATH: the neuraminidases and sialidases (2.120.10.10) andthe phytases (2.120.10.20). SS (str_synth) and the six-bladed β-propeller domainof the folate receptor (Folate_rec) are not yet classified in CATH.
The classification of SFs by SCOP is mostly in agreement with CATH, thoughthere are key differences. Like CATH, SCOP classifies the SGL (2dg1.pdb,1pjx.pdb, 2ghs.pdb, 2p4o.pdb) and arylesterase (1v04.pdb) structures in thesame SF (b.68.6). However, SCOP differs from CATH in that NHL (1rwi.pdb,1q7f.pdb) and Ldl_recept_b (1npe.pdb, 1ijq.pdb) proteins belong to two distinctSFs (b.68.9 and b.68.5, respectively).
The definition of the N6P SF that is used in the SFLD differs from the SFdefinition used by SCOP, CATH, and Pfam principally because the SFLD addscriteria associated with conserved catalytic strategies to refine further definitionsat the SF level. In some SFs, such strategies can be associated with specificfeatures conserved in members’ active sites as well. Our definition of the N6PSF is most similar to the SCOP definition, but includes SS (neither CATH norSCOP has yet classified the SS structures (2fp8.pdb in the network)) and thesubgroup we describe as the strictosidine synthase-like (SSL) proteins. Consistentwith our definition, in this chapter we evaluate the boundaries of the N6P SFwith respect to its nearest neighbors in sequence, structure, and features of thechemical strategy by which catalysis is enabled. Below, the N6P SF and itsconstituent subgroups are defined as they are classified in the SFLD.
6.3.2 Definition of the N6P Superfamily
The 2500 members of the N6P SF can be distinguished from the other proteinsof the six-bladed β-propeller fold class (∼36,000 total members for the familieslisted in Fig. 6.4 from Pfam 25.0) by sequence and structure similarity and bysimilarity in the fundamental chemical strategy by which catalysis is enabled. Asmall number of proteins within the SF, that is, members of the SFLD-definedSS family, differ in additional features that distinguish them from the rest of theSF as well. These are described in detail below.
Three criteria distinguish the N6P SF from the rest of the fold class. First,proteins in the three subgroups that comprise the N6P SF share higher overallsequence similarity with each other than they do with any other protein sharing thesame fold. Second, despite the quite different overall reactions represented by thefew members of the SF that have been biochemically characterized, all membersof the N6P SF can be united by a common catalytic step, that is, nucleophilicattack on an electrophilic substrate (Fig. 6.5). Finally, with the exception ofthe small number of enzymes that include those biochemically characterized (orpredicted) to catalyze the SS reaction, nearly all members of the SF appear to be

WHAT WE KNOW AND DO NOT KNOW ABOUT THE N6P SF 139
Figure 6.5 Examples of chemical reactions catalyzed by the N6P SF. Asterisk indicateselectrophilic atom that is attacked. (a) SS reaction. (b) Examples of lactonase, esterase,and phosphotriesterase reactions catalyzed by members of the senescence marker protein-30/gluconolactonase/luciferin-regenerating enzyme-like region (SGL) and arylesterase-like subgroups. Source: Figure and legend adapted from Hicks et al. (2011).
metal dependent, sharing in their active site architectures four highly conservedmetal-binding residues that coordinate to a divalent metal. These conserved metal-binding ligands can be identified in multiple sequence alignments representing thehuge majority of the 2500 members of the N6P SF (not shown). Although othermembers of the fold class are metal dependent, the particular metal-dependentbinding modality found in the N6P SF distinguishes it from all of these otherproteins.
The members of the N6P SF are represented by three highly diverse subgroups,designated by the different colors within the rounded rectangle shown in Fig. 6.4.Each of these subgroups was previously identified as part of the same SF bysequence similarity and by one of the known reactions its members catalyze(Kobayashi et al., 1998), some of which are depicted in Fig. 6.5. A more detaileddescription of structure–function relationships among these subgroups is givenbelow.
6.4 WHAT WE KNOW AND DO NOT KNOW ABOUT THE ENZYMESOF THE N6P SUPERFAMILY
6.4.1 The Senescence Marker Protein-30/Gluconolactonase/Luciferin-Regenerating Enzyme-Like (SGL) Subgroup
The SGL subgroup includes about 1500 members (Fig. 6.6), several of whichare known to catalyze lactonase, esterase, and organophosphatase activities

140 THE NUCLEOPHILIC ATTACK SIX-BLADED β-PROPELLER (N6P) SUPERFAMILY
Figure 6.6 Sequence similarity network for the SGL subgroup downloaded from the cur-rent SFLD. The 1449 proteins of the SGL subgroup of the N6P SF are shown. Sequencesimilarity was computed from pairwise BLAST comparisons of all of the members of thesubgroup using the SFLD as a background model. Nodes are colored by the type of lifeas designated in the key. Small circular nodes represent biochemically uncharacterizedproteins; large diamond-shaped nodes with white outlines indicate biochemically charac-terized proteins. Edges are drawn if the pairwise similarity score between two nodes isless than 1 × 10−60. An arrow indicates the Burholderia sp. H160 discussed in the text.(See insert for color representation of the figure.)
(Fig. 6.5b). Senescence marker protein-30 (SMP-30), also known as regucalcin,functions as a lactonase in the biosynthesis of l-ascorbic acid in nonprimatemammals (Kondo et al., 2006) and has been shown to hydrolyze toxicorganophosphates in mouse liver (Billecke et al., 1999; Kondo et al., 2004).Luciferin-regenerating enzyme (LRE), as the name suggests, catalyzes theregeneration of luciferin in fireflies through hydrolysis of oxyluciferin to 2-cyano-6-hydroxybenzothiazole, which then is condensed with d-cysteine (Gomiand Kajiyama, 2001). Additional biochemically characterized proteins from this

WHAT WE KNOW AND DO NOT KNOW ABOUT THE N6P SF 141
subgroup include drug-responsive protein 35 (Drp35), which functions in theresistance to antibiotics by Staphylococcus aureus (Murakami et al., 1999; Tanakaet al., 2007), and diisopropylfluorophosphatase (DFPase) (Scharff et al., 2001),whose native activity is unknown but which can break down organophosphates,including nerve toxins such as soman (Hoskin and Roush, 1982).
A conserved set of metal-coordinating ligands is present in the active site ofall structurally characterized proteins of the SGL subgroup. Detailed mechanisticstudies done on the DFPase enzyme have implicated these residues as critical forits organophosphatase activity (Blum and Chen, 2010; Blum et al., 2006; Blumet al., 2008; Melzer et al., 2009). In the proposed DFPase mechanism, an asparticacid coordinated to a divalent metal performs a direct nucleophilic attack on thephosphorous atom of the phosphoryl group of diisopropyl fluorophosphate (DFP),forming a phosphoenzyme intermediate. Water then completes the hydrolyticreaction through regeneration of the aspartic acid, as demonstrated by H2
18Oincorporation experiments (Blum et al., 2006).
Mutation studies of Drp35 suggest that the structurally equivalent metal-coordinating ligands are necessary to catalyze the lactonase reaction, althoughthe identity of one of the ligands differs from that of DFPase (Asn120 in DFPase)and is in a position structurally equivalent to Asp138 in Drp35. In this case, it isproposed that Asp138 activates a water molecule, leading to a nucleophilic attackon the carbon atom of the carbonyl group of the lactone. The oxygen atom nextto the scissile bond is proposed to be protonated by the other metal-coordinatedaspartate (Tanaka et al., 2007).
For other enzymes in this subgroup, the details of their reaction mechanismsare unclear. Though the metal-coordinating ligands likely play a role, additionalresidues may act as specificity determinants. Furthermore, the vast majority ofproteins in the SGL subgroup are biochemically uncharacterized. Gene contextanalysis of SGL members from bacteria suggests that several proteins likely playroles in sugar metabolism and transport (Hicks M. et al., Unpublished), thoughtheir reaction specificities are also currently unknown.
6.4.2 The Arylesterase-Like Subgroup
The arylesterase-like subgroup has one defined family in the SFLD: the paraox-onases (which include PON1, PON2, and PON3), with the reaction specificitiesof the rest of the subgroup classified as unknowns (Fig. 6.7). Sequences fromthis family share about 60% identity and conserve a set of seven residues thoughtto be important in catalysis. Five coordinate to the catalytic calcium ion (fourof which are conserved at the SF level) and the other two, a His–His dyad,appear to be critical for their lactonase and esterase activities. These proteins dif-fer both in their expression patterns and the degree to which they catalyze theirrespective reactions. In humans, PON1 expression is found in the liver, PON3 isfound in both the liver and kidney, and PON2 is found in most tissues, includ-ing the heart, liver, kidney, lung, small intestine, spleen, stomach, placenta, andtestis (for a review, see Ng et al., 2005). PON1 catalyzes lactonase, esterase, and

142 THE NUCLEOPHILIC ATTACK SIX-BLADED β-PROPELLER (N6P) SUPERFAMILY
Figure 6.7 Sequence similarity network for the arylesterase-like subgroup downloadedfrom the SFLD. The 123 proteins of the arylesterase-like subgroup of the N6P SF areshown. Sequence similarity was computed from pairwise BLAST comparisons of allof the members of the subgroup using the SFLD as a background model. Small circu-lar nodes represent biochemically uncharacterized proteins; large diamond-shaped nodeswith white outlines indicate biochemically characterized proteins. Edges are drawn if thepairwise similarity score between two nodes is less than 1 × 10−30. (See insert for colorrepresentation of the figure.)
organophosphatase reactions, though lactonase appears to be its native activity(Billecke et al., 2000; Draganov et al., 2005; Khersonsky and Tawfik, 2005).PON2 and PON3 also appear to be lactonases, yet differ from PON1 in that theyhave very limited arylesterase activity and almost no organophosphatase activity(Draganov, 2010). Additionally, high density lipoprotein-associated PON1, thebest studied member, exerts its antiatherogenic properties through the inhibitionof cell-mediated oxidation of low-density lipoproteins and stimulation of choles-terol efflux, likely via its lactonase activity (Rosenblat et al., 2006). Furthermore,paraoxonases play a role in acylhomoserine lactone-dependent quorum-sensingsystems, helping to prevent microbial infection through their lactonase activity(Draganov et al., 2005). For example, in Pseudomonas aeruginosa, PON1 inserum has been shown to prevent the formation of bacterial biofilm in vitro.Interestingly, PON1-deficient mice show enhanced resistance to P. aeruginosa

FUNCTIONAL PREDICTIONS AND PREDICTION OF MISANNOTATION 143
infection, likely due to increased expression of PON2 and PON3 (Ozer et al.,2005).
Structural and mutational analyses indicate that the lactonase and esterasereactions catalyzed by the PON enzymes are likely due to a proton-shuttle mech-anism involving the activation of a metal-coordinated water by a His–His dyad,leading to a nucleophilic attack on the carbon atom of a carbonyl group (Harelet al., 2004). It is believed that the organophosphatase reaction catalyzed by PON1may be performed in a manner analogous to DFPase of the SGL subgroup, whichinvolves a direct nucleophilic attack by a metal-coordinated aspartic acid (Blumet al., 2006).
As the vast majority of proteins in the arylesterase-like subgroup remainuncharacterized, both biochemically and structurally, other functions likelyremain to be discovered.
6.4.3 The Strictosidine Synthase-like (SSL) Subgroup
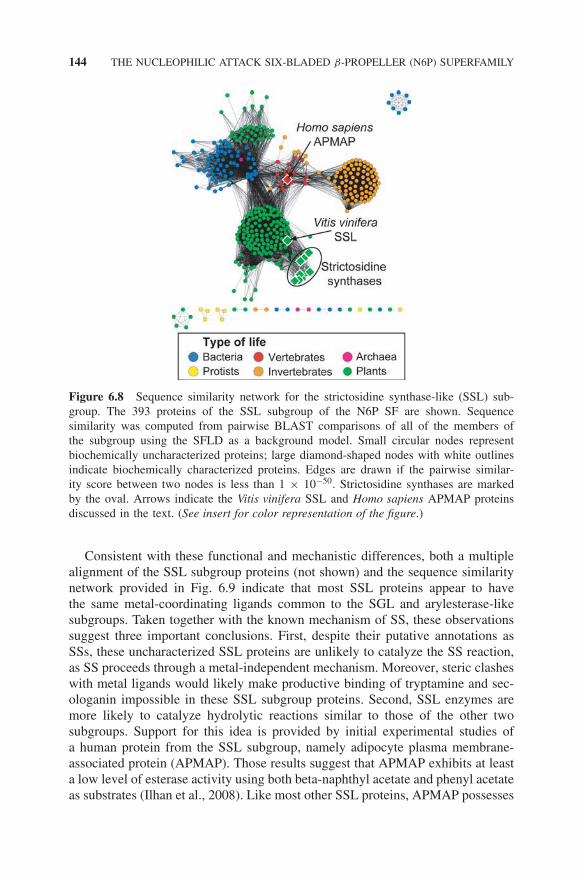
The SSL subgroup represents a group of nearly 400 proteins and is namedfor the small number of proteins within the subgroup that have been shown tocatalyze the SS reaction (Fig. 6.8). Strictosidine is the precursor to the monoter-penoid indole alkaloid biosynthesis pathway, which produces about 2000 knowncompounds, several of which are used in the treatment of cancer, malaria, hyper-tension, schizophrenia, and arrhythmic heart disorders (Ma et al., 2006). The hugemajority of the rest of the subgroup, comprising approximately 400 proteins, areuncharacterized.
6.5 FUNCTIONAL PREDICTIONS AND PREDICTIONOF MISANNOTATION OF SSL SUBGROUP ENZYMES
SS catalyzes a Pictet–Spengler reaction, resulting in the condensation oftryptamine and secologanin to form the strictosidine product (Maresh et al.,2008). This bisubstrate condensation reaction is quite different from theother known hydrolytic reactions catalyzed by the SGL and arylesterase-likesubgroups of the SF (Fig. 6.5). And although the condensation reactioncatalyzed by SS requires a nucleophilic attack on an electrophilic substrate,which is the fundamental chemistry common to the N6P SF, it differs frommost of the other SF members in that it is metal independent and lacks allfour metal-coordinating ligands common to both the SGL and arylesterase-likesubgroups. Thus, the mechanism by which SS catalyzes this fundamental stepis quite distinct from the reactions of the SGL and arylesterase-like subgroups.Instead of metal-assisted catalysis, SS uses a functionally required glutamate,Glu309, which activates tryptamine by abstracting a proton, allowing for anucleophilic attack on a carbon atom of the aldehyde of a second substrate,secologanin, forming an iminium species. The indole then attacks the iminiummoiety to form the strictosidine product (Maresh et al., 2008).
144 THE NUCLEOPHILIC ATTACK SIX-BLADED β-PROPELLER (N6P) SUPERFAMILY
Figure 6.8 Sequence similarity network for the strictosidine synthase-like (SSL) sub-group. The 393 proteins of the SSL subgroup of the N6P SF are shown. Sequencesimilarity was computed from pairwise BLAST comparisons of all of the members ofthe subgroup using the SFLD as a background model. Small circular nodes representbiochemically uncharacterized proteins; large diamond-shaped nodes with white outlinesindicate biochemically characterized proteins. Edges are drawn if the pairwise similar-ity score between two nodes is less than 1 × 10−50. Strictosidine synthases are markedby the oval. Arrows indicate the Vitis vinifera SSL and Homo sapiens APMAP proteinsdiscussed in the text. (See insert for color representation of the figure.)
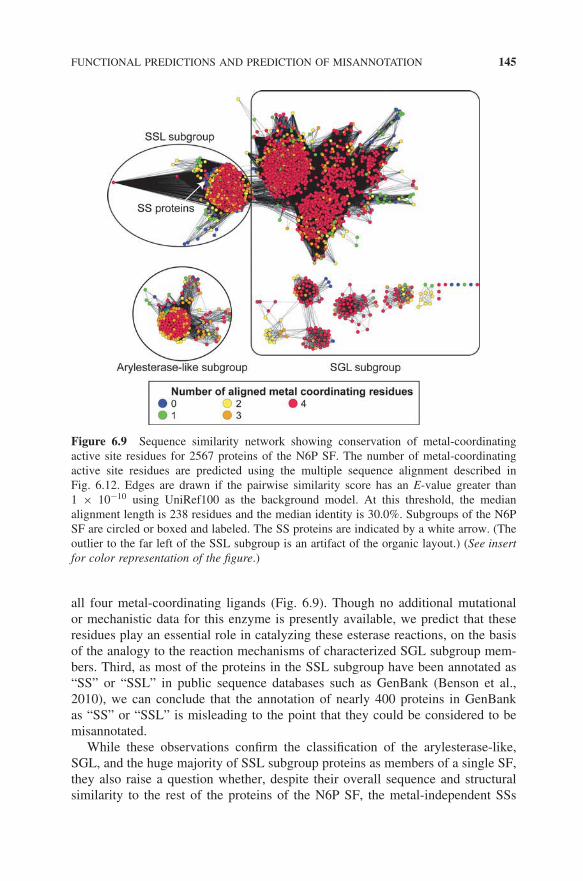
Consistent with these functional and mechanistic differences, both a multiplealignment of the SSL subgroup proteins (not shown) and the sequence similaritynetwork provided in Fig. 6.9 indicate that most SSL proteins appear to havethe same metal-coordinating ligands common to the SGL and arylesterase-likesubgroups. Taken together with the known mechanism of SS, these observationssuggest three important conclusions. First, despite their putative annotations asSSs, these uncharacterized SSL proteins are unlikely to catalyze the SS reaction,as SS proceeds through a metal-independent mechanism. Moreover, steric clasheswith metal ligands would likely make productive binding of tryptamine and sec-ologanin impossible in these SSL subgroup proteins. Second, SSL enzymes aremore likely to catalyze hydrolytic reactions similar to those of the other twosubgroups. Support for this idea is provided by initial experimental studies ofa human protein from the SSL subgroup, namely adipocyte plasma membrane-associated protein (APMAP). Those results suggest that APMAP exhibits at leasta low level of esterase activity using both beta-naphthyl acetate and phenyl acetateas substrates (Ilhan et al., 2008). Like most other SSL proteins, APMAP possesses
FUNCTIONAL PREDICTIONS AND PREDICTION OF MISANNOTATION 145
Figure 6.9 Sequence similarity network showing conservation of metal-coordinatingactive site residues for 2567 proteins of the N6P SF. The number of metal-coordinatingactive site residues are predicted using the multiple sequence alignment described inFig. 6.12. Edges are drawn if the pairwise similarity score has an E-value greater than1 × 10−10 using UniRef100 as the background model. At this threshold, the medianalignment length is 238 residues and the median identity is 30.0%. Subgroups of the N6PSF are circled or boxed and labeled. The SS proteins are indicated by a white arrow. (Theoutlier to the far left of the SSL subgroup is an artifact of the organic layout.) (See insertfor color representation of the figure.)
all four metal-coordinating ligands (Fig. 6.9). Though no additional mutationalor mechanistic data for this enzyme is presently available, we predict that theseresidues play an essential role in catalyzing these esterase reactions, on the basisof the analogy to the reaction mechanisms of characterized SGL subgroup mem-bers. Third, as most of the proteins in the SSL subgroup have been annotated as“SS” or “SSL” in public sequence databases such as GenBank (Benson et al.,2010), we can conclude that the annotation of nearly 400 proteins in GenBankas “SS” or “SSL” is misleading to the point that they could be considered to bemisannotated.
While these observations confirm the classification of the arylesterase-like,SGL, and the huge majority of SSL subgroup proteins as members of a single SF,they also raise a question whether, despite their overall sequence and structuralsimilarity to the rest of the proteins of the N6P SF, the metal-independent SSs

146 THE NUCLEOPHILIC ATTACK SIX-BLADED β-PROPELLER (N6P) SUPERFAMILY
should be included in the SF as well. We argue that the preponderance of evidencesupports the inclusion of the SSs in the N6P SF, as discussed below.
6.6 DO THE STRICTOSIDINE SYNTHASES REALLY BELONGTO THE N6P SUPERFAMILY?
As is clear from our analysis of the SSL subgroup, complex variations in sequenceamong seemingly related proteins can complicate function annotation transferfrom biochemically characterized to uncharacterized proteins, resulting in misan-notation of function for many members of this subgroup. Moreover, defining whatto include in a functionally diverse SF is challenging, as exemplified especiallyfor the SS enzymes.
With respect to the first criterion defined by the SFLD for inclusion of proteinsin the N6P SF (see Section 6.3.2 “Definition of the N6P Superfamily”), theSS proteins can be classified as members of the N6P SF on the basis of theiroverall similarities in sequence and structure to other SF members. For the secondsimilarity in the fundamental catalytic strategy by which catalysis is enabled,the SSs also fit as members of the N6P SF. As shown in Fig. 6.5, like thereactions of other functionally characterized members of the SF, the SS reactioninvolves a nucleophilic attack on an electrophilic substrate, albeit using a differentmechanism than the other known SF members. At first pass, the SSs appear tofail the last required criterion for inclusion in the SF, however: that is, similarityin active site architecture or functionally important residues. Figure 6.10, whichcompares the active site of SS with those of metal-dependent enzymes from theSGL and arylesterase-like subgroups, illustrates how very different the activesites of the SSs are relative to the rest of the SF. It is these differences inthe active sites, taken together with the dissimilarity between the condensationreaction catalyzed by SS and the hydrolytic reactions of the rest of the SF, thatmake it difficult to rationalize the inclusion of the SSs as members of the N6PSF.
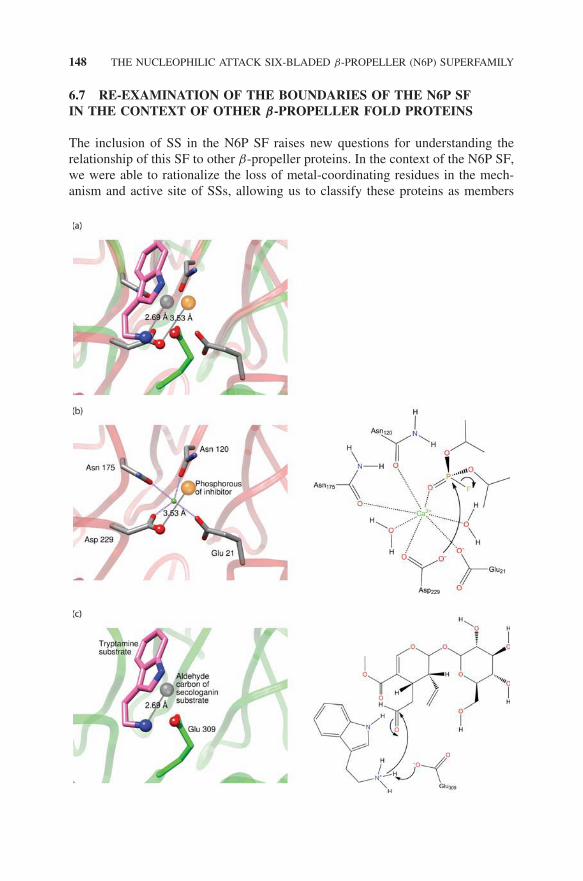
Further comparison of the SSs with other N6P SF members moderates thisview, however, leading us to classify the SSs as members of the N6P SF afterall. First, a phylogenetic analysis of the SSL subgroup (not shown) suggeststhat SS likely arose from a metal-dependent ancestor (Hicks et al., 2011). Sec-ond, as shown in Fig. 6.11a, comparison of a liganded structure from the SGLsubgroup (DFPase) with liganded SS proteins shows that both enzymes initiatenucleophilic attack from the same position in the active site. These similari-ties in active sites allow us to speculate that SS evolved to bind tryptaminein the region of the active site occupied by two of the four metal binding-ligands present in other SF members such as DFPase and leads to a hypoth-esis for a mechanistic role of tryptamine that is analogous to the roles of themetals in DFPase. The mechanistic diagrams shown on the right of Fig. 6.11describe this mechanistic hypothesis. In DFPase (Fig. 6.11b), the reaction isinitiated by a metal-assisted nucleophilic attack of the oxygen of Asp229 of

DO THE STRICTOSIDINE SYNTHASES REALLY BELONG TO THE N6P 147
Figure 6.10 Active site superposition of (a) SGL (Drp35, pdb_id: 2dg1; red) andarylesterase-like (PON1, pdb_id: 1v04; cyan) subgroup proteins. Conserved metal-coordinating residues are labeled and colored by element; gray: carbon, blue: nitrogen,red: oxygen. (b) The same superposition as shown in (a), but with the metals removed forclarity and SS (pdb_id: 2fpb; green) added. Four SS residues that superimpose with alphacarbon positions for metal-coordinating residues in Drp35 and PON1 are labeled black.The glutamate (Glu309) required for SS activity in 2fpb is also labeled black. These fiveSS residues are colored by element; green: carbon, blue: nitrogen, red: oxygen. Source:Figure and legend are adapted from Hicks et al. (2011). (See insert for color representationof the figure.)
DFPase on the phosphorus of the substrate, DFP. We speculate that the analogyin SS might involve substrate-assisted catalysis in which the amine group oftryptamine is deprotonated by Glu309. This step is then followed by the subse-quent attack of the amine on the aldehyde moiety of secologanin (Fig. 6.11c).Mutagenesis of Glu309 has previously established that it is required for SSactivity.
148 THE NUCLEOPHILIC ATTACK SIX-BLADED β-PROPELLER (N6P) SUPERFAMILY
6.7 RE-EXAMINATION OF THE BOUNDARIES OF THE N6P SFIN THE CONTEXT OF OTHER β-PROPELLER FOLD PROTEINS
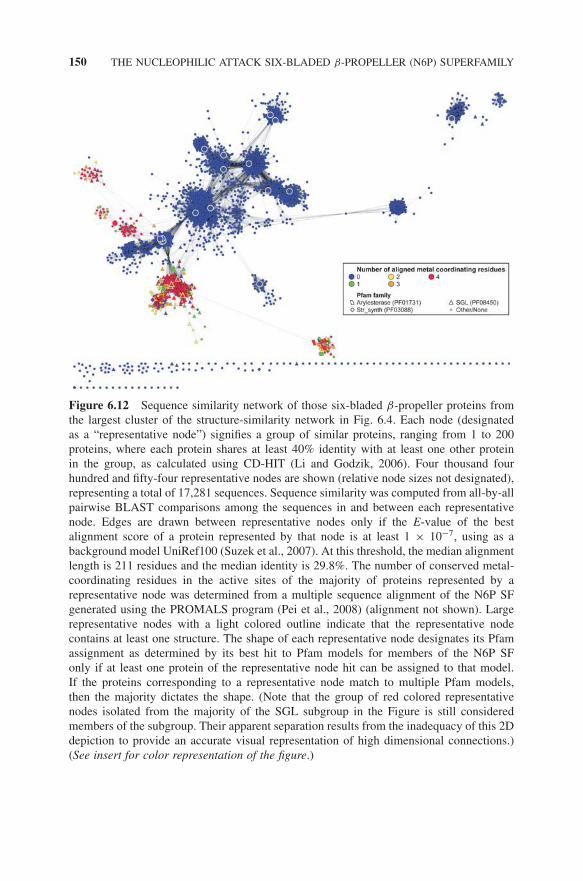
The inclusion of SS in the N6P SF raises new questions for understanding therelationship of this SF to other β-propeller proteins. In the context of the N6P SF,we were able to rationalize the loss of metal-coordinating residues in the mech-anism and active site of SSs, allowing us to classify these proteins as members

RE-EXAMINATION OF THE BOUNDARIES OF THE N6P SF 149
of the SF. But this interpretation is complicated in the context of the larger foldclass, which shows that other potentially homologous members of the fold classalso lack conservation of metal-binding ligands typical of the N6P SF. Thus, thislarger view forces us to re-evaluate our initial criteria for excluding from the SFother proteins in the fold class on the basis of their lack of metal-binding ligands(Fig. 6.12). We also note that different databases classify relationships amongespecially highly divergent groups of proteins differently. For example, we notedearlier that CATH classifies protein domains from the SGL subgroup of the N6PSF together with members of the Pfam families MRJP, NHL, PD40, and Ldl_re-cept_b, and a GSDH protein in a single structural SF they designate as the TolBC-terminal domain SF. This differs from the N6P SF definition currently usedby the SFLD. We argue, however, that neither definition should be considered asmore “correct” than the other. Rather, each merely reflects the different criteriaeach resource has chosen to use in the classification of sequence, structural, andfunctional relationships.
From the perspective of the SFLD classification, these relationships becomemore complicated than classifications based primarily on sequence and structuralsimilarity, as the SFLD attempts to address how functional features are bestincluded in the formal classification of functionally diverse enzyme SFs. Here,we examine further whether other proteins sharing the six-bladed β-propeller foldthat also catalyze hydrolytic reactions should be considered as members in the SF.These include phytases, which catalyze the hydrolysis of phytic acid to phosphate,and neuraminidases, which hydrolyze glycosidic linkages of neuraminic acids,
←−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−Figure 6.11 A related catalytic strategy unites the SS enzymes with the rest of theN6P SF. Left panels: active site representations; right panels: diagrams depicting stepsin the catalytic mechanisms as described in the text. (a) Superposition of the active siteof DFPase (pdb_id: 2gvv; SGL subgroup) with dicyclopentyl phosphoramidate inhibitorbound (only the phosphorous of the inhibitor is depicted) (as seen in (b)) and of SS(green backbone) with tryptamine bound (pdb_id: 2fpb) and with secologanin bound (pdbid: 2fpc; only the aldehyde carbon is depicted) (as seen in (c)). Atoms and ligands aredepicted as in (b) and (c), with the metal from (b) removed for clarity. (b) The left panelshows the active site of DFPase as in (a) except that the coordination of the metal to themetal binding ligands is shown. The nucleophilic oxygen of the catalytic Asp229, andthe electrophilic phosphorous atom of the inhibitor are depicted as ball and stick. Twowater molecules and the phosphoryl oxygen coordinated to the metal have been removedfor clarity. The right panel depicts the nucleophilic attack of the oxygen of Asp229 onthe phosphorous of the substrate DFP. (c) The left panel shows the superposition of SSwith tryptamine bound and with secologanin as in (a). The acidic oxygen atom of thecatalytic Glu309, the reactive nitrogen atom of tryptamine, and the electrophilic carbonof secologanin are shown as ball and stick. The right panel shows the deprotonation ofthe amine group of tryptamine by Glu309 and the subsequent nucleophilic attack of theamine on the aldehyde of secologanin. Source: Figure and legend are adapted from Hickset al. (2011).
150 THE NUCLEOPHILIC ATTACK SIX-BLADED β-PROPELLER (N6P) SUPERFAMILY
Figure 6.12 Sequence similarity network of those six-bladed β-propeller proteins fromthe largest cluster of the structure-similarity network in Fig. 6.4. Each node (designatedas a “representative node”) signifies a group of similar proteins, ranging from 1 to 200proteins, where each protein shares at least 40% identity with at least one other proteinin the group, as calculated using CD-HIT (Li and Godzik, 2006). Four thousand fourhundred and fifty-four representative nodes are shown (relative node sizes not designated),representing a total of 17,281 sequences. Sequence similarity was computed from all-by-allpairwise BLAST comparisons among the sequences in and between each representativenode. Edges are drawn between representative nodes only if the E-value of the bestalignment score of a protein represented by that node is at least 1 × 10−7, using as abackground model UniRef100 (Suzek et al., 2007). At this threshold, the median alignmentlength is 211 residues and the median identity is 29.8%. The number of conserved metal-coordinating residues in the active sites of the majority of proteins represented by arepresentative node was determined from a multiple sequence alignment of the N6P SFgenerated using the PROMALS program (Pei et al., 2008) (alignment not shown). Largerepresentative nodes with a light colored outline indicate that the representative nodecontains at least one structure. The shape of each representative node designates its Pfamassignment as determined by its best hit to Pfam models for members of the N6P SFonly if at least one protein of the representative node hit can be assigned to that model.If the proteins corresponding to a representative node match to multiple Pfam models,then the majority dictates the shape. (Note that the group of red colored representativenodes isolated from the majority of the SGL subgroup in the Figure is still consideredmembers of the subgroup. Their apparent separation results from the inadequacy of this 2Ddepiction to provide an accurate visual representation of high dimensional connections.)(See insert for color representation of the figure.)

USING THE SUPERFAMILY CONTEXT TO SELECT PROTEIN TARGETS 151
neither of which connects to the main cluster of proteins in the structure-basedsimilarity network (Fig. 6.4).
To evaluate whether phytases are good candidates for inclusion in the N6P SF,a superposition of phytase with DFPase was examined (not shown). Although thissuperposition revealed a couple of calcium-coordinating ligands in the phytaseactive site that are somewhat similar in position to the metal-binding ligands ofDFPase, these side chains align poorly relative to alignments between structuresclassified as members of the SF. Overall, this alignment suggests a different bind-ing modality relative to the N6P SF and suggests a distinct structure–functionmapping in phytases. Superposition of this phytase structure with that of SS is notany better (also not shown), further arguing against the inclusion of the phytasesas members of the N6P SF. Structural alignments between neuraminidases andN6P SF members are also poor and suggest that these proteins are too dissim-ilar to include them in the SF. Outside of the families that catalyze hydrolyticreactions, including the NHL, PD40, GSDH, and MRJP Pfam families, a furtherexamination of those structures most similar to the N6P SF in Fig. 6.4 suggeststhat these structural relationships are too weak to justify inclusion in the N6P SF(not shown).
Interestingly, we find strong conservation patterns between SGL subgroupproteins and seven-bladed β-propeller proteins (virginiamycin B lyases), whichmay indicate that our initial search for SF members, limited to using the canoni-cally defined 6-bladed β-propeller fold, was insufficient. In β-propeller proteins,blade count may not be highly relevant in defining characteristics of an SF, assequence similarity between proteins of different blade counts can be more sig-nificant than that within proteins sharing the same blade count (Chaudhuri et al.,2008). For example, sequences of the sialidases, which share the six-bladed β-propeller fold with the N6P SF, are more distant from the SGL subgroup proteinsthan the SGL subgroup is to the seven-bladed virginiamycin B lyases. How-ever, given the current lack of functional information for the SGL subgroup,a strong link between the N6P SF and the virginiamycin B lyases cannot beestablished.
6.8 USING THE SUPERFAMILY CONTEXT TO SELECT PROTEINTARGETS FOR EXPERIMENTAL CHARACTERIZATION
Sequence similarity networks that map characterized proteins together withunknowns allow identification of clusters of unknowns that have not yet beencharacterized, enabling a strategy for choosing proteins for characterization thatcould be especially informative for functional inference (Pieper et al., 2009). Forexample, sequence similarity networks can help identify “functional boundaries”within a given SF and provide an estimation of how many functions remain tobe discovered. Addressing this problem is a significant challenge for functionalinference, as clustering by sequence and structure alone frequently does not trackwith functional properties (see Glasner et al., 2006 and Schnoes et al., 2009 for

152 THE NUCLEOPHILIC ATTACK SIX-BLADED β-PROPELLER (N6P) SUPERFAMILY
some examples). Additionally, knowledge gained from the SF perspective cansuggest fundamental catalytic capabilities and constraints that, along with genecontext and other orthogonal information, can be used to improve predictions ofthe functions of unknowns.
An example in the N6P SF is provided by the SSL subgroup protein from Vitisvinifera. The sequence similarity network for this subgroup (Fig. 6.8) shows thatthis uncharacterized protein shares a high degree of overall sequence similaritywith known SSs. As reported previously (Hicks et al., 2011), a structure-guidedsequence alignment representing structures from all three N6P subgroups, severalother sequences that sample the diversity of the SSL subgroup, and the V. viniferaprotein revealed active site properties in the latter protein that appeared “tran-sitional” between the true SSs and majority of the SSL subgroup proteins thatconserve all four of the canonical metal-binding ligands. The V. vinifera proteinconserves three of the four metal-coordinating ligands, with the missing ligandsubstituted by a glycine, a residue it shares in common with all known SSs. Aspredicted by the presence of these metal-binding ligands especially, subsequentbiochemical characterization of this protein demonstrated that it does not catalyzethe SS reaction at a measurable level but instead exhibits a low level of esteraseactivity using p-nitrophenyl acetate as a substrate. This result suggests propertiesof a functional boundary between SSs and SSL proteins and provides insightabout possible paths by which SSs may have evolved within the background ofthe hydrolytic and metal-dependent SSL proteins.
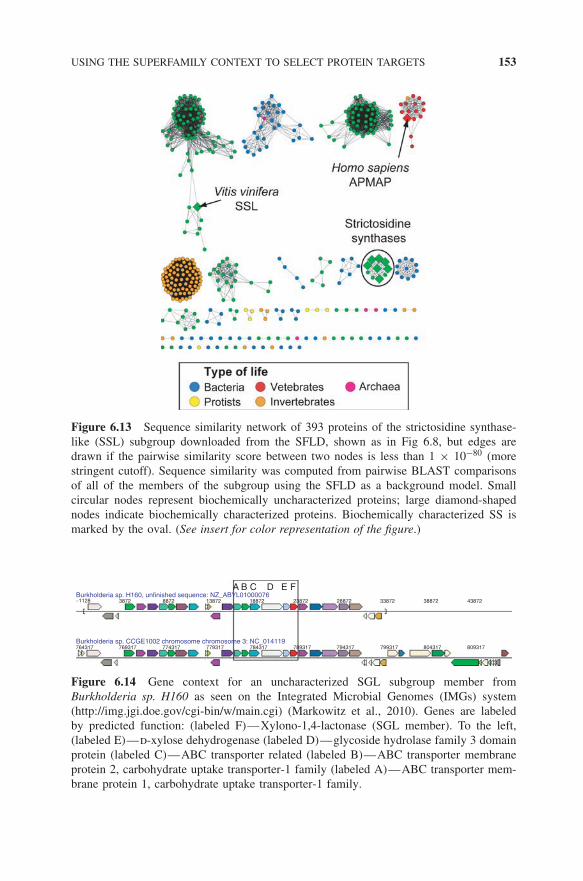
Using a more stringent E-value cutoff than the one used for the SSL subgroupnetwork presented in Fig. 6.8, the similarity network in Fig. 6.13 shows multiplediscrete clusters, one of which cleanly separates all of the SSs into a single clus-ter. Although insufficient information currently exists to infer how many of theother clusters shown in this network are isofunctional, we hypothesize from thisnetwork view that at least several different functional families may remain to beidentified in the subgroup. Further comparison of sequence profiles representingthese clusters may be useful for hypothesizing additional functional boundarieswithin the subgroup and for choosing sequences for structural characterizationthat would likely be useful in answering these questions.
Like the SSL subgroup, the SGL subgroup, which comprises the vast majorityof the N6P SF, is also poorly defined in terms of functional families and identifica-tion of the reactions they catalyze. However, knowing some types of reactions thecharacterized members of this subgroup catalyze (lactonase, esterase, organophos-phatase) provides hints about specific reactions that may be represented. Whenevaluated together with orthogonal information such as well-defined gene con-text, new targets for biochemical and structural characterization can be identified.For example, a potentially attractive target for characterization is the predictedSGL protein encoded in the genome of Burkholderia sp. H160 (indicated by thearrow in Fig. 6.6), for which the gene context suggests a role in a d-xylose uptakeand metabolism pathway (Fig. 6.14). Putting this together with its likely functionas a hydrolase, we speculate that this protein may catalyze the breakdown ofxylono-1,4 lactone to produce d-xylonate.
USING THE SUPERFAMILY CONTEXT TO SELECT PROTEIN TARGETS 153
Figure 6.13 Sequence similarity network of 393 proteins of the strictosidine synthase-like (SSL) subgroup downloaded from the SFLD, shown as in Fig 6.8, but edges aredrawn if the pairwise similarity score between two nodes is less than 1 × 10−80 (morestringent cutoff). Sequence similarity was computed from pairwise BLAST comparisonsof all of the members of the subgroup using the SFLD as a background model. Smallcircular nodes represent biochemically uncharacterized proteins; large diamond-shapednodes indicate biochemically characterized proteins. Biochemically characterized SS ismarked by the oval. (See insert for color representation of the figure.)
A B C D E FBurkholderia sp. H160, unfinished sequence: NZ_ABYL01000076
Burkholderia sp. CCGE1002 chromosome chromosome 3: NC_014119
−1128 3872
764317 769317
8872 13872 18872 23872 28872 33872 38872 43872
774317 779317 784317 789317 794317 799317 804317 809317
Figure 6.14 Gene context for an uncharacterized SGL subgroup member fromBurkholderia sp. H160 as seen on the Integrated Microbial Genomes (IMGs) system(http://img.jgi.doe.gov/cgi-bin/w/main.cgi) (Markowitz et al., 2010). Genes are labeledby predicted function: (labeled F)—Xylono-1,4-lactonase (SGL member). To the left,(labeled E)—d-xylose dehydrogenase (labeled D)—glycoside hydrolase family 3 domainprotein (labeled C)—ABC transporter related (labeled B)—ABC transporter membraneprotein 2, carbohydrate uptake transporter-1 family (labeled A)—ABC transporter mem-brane protein 1, carbohydrate uptake transporter-1 family.

154 THE NUCLEOPHILIC ATTACK SIX-BLADED β-PROPELLER (N6P) SUPERFAMILY
6.9 CONCLUSION
This chapter described the links between structural and functional properties ofthe proteins of the N6P SF and placed their relationships in the larger contextof the six-bladed β-propeller fold proteins. Further dissection of differencesamong the proteins of the three subgroups of the SF, namely the arylesterase-like,SGL, and SSL subgroups, revealed clues about the functions of uncharacterizedproteins in each, allowing the classification of all of the SF members in theSFLD at least at the subgroup level. The limits described here for the predictionand annotation of reaction specificity families also addressed the difficulty ofpredicting functional boundaries and illustrated some important challenges fordiscriminating functions based on sequence or structural similarities, especiallyfor functionally diverse enzyme SFs. These issues complicate our understandingof sequence, structure, and function relationships in such systems and illustratesome of the significant challenges for achieving high-quality annotations in theabsence of experimental validation.
ACCESS TO DATA FROM THIS WORK
The N6P SF has been added to the SFLD and curated into subgroups and familieswhere possible, based on available high-quality experimental information. SFs areautomatically updated on a regular basis, and new sequences and structures areclassified according to the SFLD schema using automated protocols to the extentpossible, which are then further checked by human curators. These sequenceand structure data are freely available at http://sfld.rbvi.ucsf.edu, along with full-length alignments of representative sequences identifying key conserved aminoacids. Interactive versions of SF, subgroup, and family networks are also availablefor download and can be viewed using the freely available Cytoscape programwhich can be downloaded from http://www.cytoscape.org/.
ACKNOWLEDGMENTS
This work was supported by NIH R01 GM60595 to PCB. AEB was supportedby the ARCS Foundation and the PhRMA Foundation Predoctoral Informaticsfellowship. Molecular graphics images were produced using the UCSF Chimerapackage from the Resource for Biocomputing, Visualization, and Informatics atthe University of California, San Francisco (supported by NIH P41 RR001081).The authors wish to thank Jimin Pei and Nick Grishin for their help in generatingthe PROMALS3D alignment.
REFERENCES
Adai, A.T., Date, S.V., Wieland, S., and Marcotte, E.M. (2004) LGL: creating a map ofprotein function with an algorithm for visualizing very large biological networks. JMol Biol, 340, 179–190.

REFERENCES 155
Almonacid, D.E. and Babbitt, P.C. (2011) Toward mechanistic classification of enzymefunctions. Curr Opin Chem Biol, 15, 435–442.
Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein databasesearch programs. Nucleic Acids Res, 25, 3389–3402.
Andreeva, A., Howorth, D., Chandonia, J.M., Brenner, S.E., Hubbard, T.J., Chothia, C.,and Murzin, A.G. (2008) Data growth and its impact on the SCOP database: newdevelopments. Nucleic Acids Res, 36, D419–D425.
Atkinson, H.J. and Babbitt, P.C. (2009) An atlas of the thioredoxin fold class reveals thecomplexity of function-enabling adaptations. PLoS Comput Biol, 5, e1000541.
Atkinson, H.J., Morris, J.H., Ferrin, T.E., and Babbitt, P.C. (2009) Using sequence simi-larity networks for visualization of relationships across diverse protein superfamilies.PLoS ONE, 4, e4345.
Babbitt, P.C. and Gerlt, J.A. (1997) Understanding enzyme superfamilies chemistry asthe fundamental determinant in the evolution of new catalytic activities. J Biol Chem,272, 30591–30594.
Babbitt, P.C., Hasson, M.S., Wedekind, J.E., Palmer, D.R., Barrett, W.C., Reed, G.H.,Rayment, I., Ringe, D., Kenyon, G.L., and Gerlt, J.A. (1996) The enolase superfamily:a general strategy for enzyme-catalyzed abstraction of the alpha-protons of carboxylicacids. Biochemistry, 35, 16489–16501.
Benson, D.A., Karsch-Mizrachi, I., Lipman, D.J., Ostell, J., and Sayers, E.W. (2010)GenBank. Nucleic Acids Res, 38, D46–D51.
Billecke, S., Draganov, D., Counsell, R., Stetson, P., Watson, C., Hsu, C., and Du, B.N.L.(2000) Human serum paraoxonase (pon1) isozymes Q and R hydrolyze lactones andcyclic carbonate esters. Drug Metabolism and Disposition, 28, 1335–1342.
Billecke, S.S., Primo-Parmo, S.L., Dunlop, C.S., Doorn, J.A., La Du, B.N., and Broom-field, C.A. (1999) Characterization of a soluble mouse liver enzyme capable ofhydrolyzing diisopropyl phosphorofluoridate. Chem Biol Interact, 119–120, 251–256.
Blum, M.-M. and Chen, J.C.H. (2010) Structural characterization of the catalytic calcium-binding site in diisopropyl fluorophosphatase (DFPase)–Comparison with related[beta]-propeller enzymes. Chem Biol Interact, 187, 373–379.
Blum, M.-M., Lohr, F., Richardt, A., Ruterjans, H., and Chen, J.C.H. (2006) Binding ofa designed substrate analogue to diisopropyl fluorophosphatase: implications for thephosphotriesterase mechanism. J Am Chem Soc, 128, 12750–12757.
Blum, M.M., Timperley, C.M., Williams, G.R., Thiermann, H., and Worek, F. (2008)Inhibitory potency against human acetylcholinesterase and enzymatic hydrolysis offluorogenic nerve agent mimics by human paraoxonase 1 and squid diisopropyl fluo-rophosphatase. Biochemistry, 47, 5216–5224.
Chaudhuri, I., Soding, J., and Lupas, A.N. (2008) Evolution of the β-propeller fold.Proteins Struct Funct Bioinformatics, 71, 795–803.
Cline, M.S., Smoot, M., Cerami, E., Kuchinsky, A., Landys, N., Workman, C., Christmas,R., Avila-Campilo, I., Creech, M., Gross, B. et al. (2007) Integration of biologicalnetworks and gene expression data using Cytoscape. Nat Protoc, 2, 2366–2382.
Cuff, A.L., Sillitoe, I., Lewis, T., Clegg, A.B., Rentzsch, R., Furnham, N., Pellegrini-Calace, M., Jones, D., Thornton, J., and Orengo, C.A. (2011) Extending CATH:increasing coverage of the protein structure universe and linking structure with func-tion. Nucleic Acids Res, 39, D420–D426.

156 THE NUCLEOPHILIC ATTACK SIX-BLADED β-PROPELLER (N6P) SUPERFAMILY
Draganov, D.I. (2010) Lactonases with organophosphatase activity: structural and evolu-tionary perspectives. Chem Biol Interact, 187, 370–372.
Draganov, D.I., Teiber, J.F., Speelman, A., Osawa, Y., Sunahara, R., and La Du, B.N.(2005) Human paraoxonases (PON1, PON2, and PON3) are lactonases with overlap-ping and distinct substrate specificities. J Lipid Res, 46, 1239–1247.
Eddy, S.R. (1996) Hidden Markov models. Current Opin Struct Biol, 6, 361–365.
Enright, A.J., Van Dongen, S., and Ouzounis, C.A. (2002) An efficient algorithm forlarge-scale detection of protein families. Nucleic Acids Res, 30, 1575–1584.
Finn, R.D., Mistry, J., Schuster-Bockler, B., Griffiths-Jones, S., Hollich, V., Lassmann,T., Moxon, S., Marshall, M., Khanna, A., Durbin, R. et al. (2006) Pfam: clans, webtools and services. Nucleic Acids Res, 34, D247–D251.
Finn, R.D., Mistry, J., Tate, J., Coggill, P., Heger, A., Pollington, J.E., Gavin, O.L.,Gunasekaran, P., Ceric, G., Forslund, K. et al. (2010) The Pfam protein familiesdatabase. Nucleic Acids Res, 38, D211–D222.
Frickey, T. and Lupas, A. (2004) CLANS: a Java application for visualizing proteinfamilies based on pairwise similarity. Bioinformatics, 20, 3702–3704.
Frishman, D. (2007) Protein annotation at genomic scale: the current status. Chem Rev,107, 3448–3466.
Gerlt, J.A. and Babbitt, P.C. (2001) Divergent evolution of enzymatic function: Mech-anistically diverse superfamilies and functionally distinct suprafamilies. Annu RevBiochem, 70, 209–246.
Gerlt, J.A., Babbitt, P.C., Jacobson, M.P., and Almo, S.C. (2012) Divergent evolution inthe enolase superfamily: strategies for assigning functions. J Biol Chem, 287, 29–34.
Gerlt, J.A., Babbitt, P.C., and Rayment, I. (2005) Divergent evolution in the enolasesuperfamily: the interplay of mechanism and specificity. Arch Biochem Biophys, 433,59–70.
Glasner, M.E., Fayazmanesh, N., Chiang, R.A., Sakai, A., Jacobson, M.P., Gerlt, J.A.,and Babbitt, P.C. (2006) Evolution of structure and function in the o-succinylbenzoatesynthase/N-acylamino acid racemase family of the enolase superfamily. J Mol Biol,360, 228–250.
Glasner, M.E., Gerlt, J.A., and Babbitt, P.C. (2007) Mechanisms of protein evolutionand their application to protein engineering. Adv Enzymol Relat Areas Mol Biol, 75,193–239 xii–xiii.
Gomi, K. and Kajiyama, N. (2001) Oxyluciferin, a luminescence product of fireflyluciferase, is enzymatically regenerated into luciferin. J Biol Chem, 276, 36508–36513.
Hall, R.S., Brown, S., Fedorov, A.A., Fedorov, E.V., Xu, C., Babbitt, P.C., Almo, S.C.,and Raushel, F.M. (2007) Structural diversity within the mononuclear and binuclearactive sites of N-acetyl-D-glucosamine-6-phosphate deacetylase. Biochemistry, 46,7953–7962.
Harel, M., Aharoni, A., Gaidukov, L., Brumshtein, B., Khersonsky, O., Meged, R., Dvir,H., Ravelli, R.B., McCarthy, A., Toker, L. et al. (2004) Structure and evolution of theserum paraoxonase family of detoxifying and anti-atherosclerotic enzymes. Nat StructMol Biol, 11, 412–419.
Hicks, M.A., Barber, A.E., Giddings, L.-A., Caldwell, J., O’Connor, S.E., and Babbitt,P.C. (2011) The evolution of function in strictosidine synthase-like proteins. ProteinsStruct Funct Bioinformatics, 79, 3082–3098.

REFERENCES 157
Hoskin, F.C.G. and Roush, A.H. (1982) Hydrolysis of nerve gas by squid-type diisopropylphosphorofluoridate hydrolyzing enzyme on agarose resin. Science, 215, 1255–1257.
Ilhan, A., Gartner, W., Nabokikh, A., Daneva, T., Majdic, O., Cohen, G., Bohmig, G.A.,Base, W., Horl, W.H., and Wagner, L. (2008) Localization and characterization of thenovel protein encoded by C20orf3. Biochem J, 414, 485–495.
International Union of Biochemistry and Molecular Biology: Nomenclature Committee,and Webb. (1992). Enzyme nomenclature 1992: recommendations of the NomenclatureCommittee of the International Union of Biochemistry and Molecular Biology on thenomenclature and classification of enzymes. (San Diego: Academic Press).
Khersonsky, O. and Tawfik, D.S. (2005) Structure-reactivity studies of serum paraoxonasePON1 suggest that its native activity is lactonase. Biochemistry, 44, 6371–6382.
Kobayashi, M., Shinohara, M., Sakoh, C., Kataoka, M., and Shimizu, S. (1998) Lactone-ring-cleaving enzyme: Genetic analysis, novel RNA editing, and evolutionary impli-cations. Proc Natl Acad Sci USA, 95, 12787–12792.
Kondo, Y., Inai, Y., Sato, Y., Handa, S., Kubo, S., Shimokado, K., Goto, S., Nishikimi,M., Maruyama, N., and Ishigami, A. (2006) Senescence marker protein 30 functionsas gluconolactonase in L-ascorbic acid biosynthesis, and its knockout mice are proneto scurvy. Proc Natl Acad Sci USA, 103, 5723–5728.
Kondo, Y., Ishigami, A., Kubo, S., Handa, S., Gomi, K., Hirokawa, K., Kajiyama, N.,Chiba, T., Shimokado, K., and Maruyama, N. (2004) Senescence marker protein-30 isa unique enzyme that hydrolyzes diisopropyl phosphorofluoridate in the liver. FEBSLett, 570, 57–62.
Li, W. and Godzik, A. (2006) Cd-hit: a fast program for clustering and comparing largesets of protein or nucleotide sequences. Bioinformatics, 22, 1658–1659.
Loewenstein, Y., Raimondo, D., Redfern, O.C., Watson, J., Frishman, D., Linial, M.,Orengo, C., Thornton, J., and Tramontano, A. (2009) Protein function annotation byhomology-based inference. Genome Biol, 10, 207.
Ma, X., Panjikar, S., Koepke, J., Loris, E., and Stockigt, J. (2006) The structure ofRauvolfia serpentina strictosidine synthase is a novel six-bladed beta-propeller fold inplant proteins. Plant Cell, 18, 907–920.
Maresh, J.J., Giddings, L.A., Friedrich, A., Loris, E.A., Panjikar, S., Trout, B.L., Stockigt,J., Peters, B., and O’Connor, S.E. (2008) Strictosidine synthase: mechanism of a Pictet-Spengler catalyzing enzyme. J Am Chem Soc, 130, 710–723.
Markowitz, V.M., Chen, I.M.A., Palaniappan, K., Chu, K., Szeto, E., Grechkin, Y., Ratner,A., Anderson, I., Lykidis, A., Mavromatis, K. et al. (2010) The integrated microbialgenomes system: an expanding comparative analysis resource. Nucleic Acids Res, 38,D382–D390.
Martin, A.C. (2004) PDBSprotEC: a web-accessible database linking PDB chains to ECnumbers via SwissProt. Bioinformatics, 20, 986–988.
Medini, D., Covacci, A., and Donati, C. (2006) Protein homology network families revealstep-wise diversification of Type III and Type IV secretion systems. PLoS Comput Biol,2, e173.
Melzer, M., Chen, J.C.H., Heidenreich, A., Gab, J.r., Koller, M., Kehe, K., andBlum, M.-M. (2009) Reversed enantioselectivity of diisopropyl fluorophosphataseagainst organophosphorus nerve agents by rational design. J Am Chem Soc, 131,17226–17232.

158 THE NUCLEOPHILIC ATTACK SIX-BLADED β-PROPELLER (N6P) SUPERFAMILY
Murakami, H., Matsumaru, H., Kanamori, M., Hayashi, H., and Ohta, T. (1999) Cellwall-affecting antibiotics induce expression of a novel gene, drp35, in Staphylococcusaureus. Biochem Biophys Res Comm, 264, 348–351.
Ng, C.J., Shih, D.M., Hama, S.Y., Villa, N., Navab, M., and Reddy, S.T. (2005) Theparaoxonase gene family and atherosclerosis. Free Radical Biol Med, 38, 153–163.
Nguyen, T.T., Brown, S., Fedorov, A.A., Fedorov, E.V., Babbitt, P.C., Almo, S.C.,and Raushel, F.M. (2008) At the periphery of the amidohydrolase superfamily:Bh0493 from Bacillus halodurans catalyzes the isomerization of D-galacturonate toD-tagaturonate. Biochemistry, 47, 1194–1206.
Ozer, E.A., Pezzulo, A., Shih, D.M., Chun, C., Furlong, C., Lusis, A.J., Greenberg, E.P.,and Zabner, J. (2005) Human and murine paraoxonase 1 are host modulators of Pseu-domonas aeruginosa quorum-sensing. FEMS Microbiol Lett, 253, 29–37.
Pegg, S.C., Brown, S., Ojha, S., Huang, C.C., Ferrin, T.E., and Babbitt, P.C. (2005)Representing structure-function relationships in mechanistically diverse enzyme super-families. Pac Symp Biocomput, 358–369.
Pegg, S.C., Brown, S.D., Ojha, S., Seffernick, J., Meng, E.C., Morris, J.H., Chang, P.J.,Huang, C.C., Ferrin, T.E., and Babbitt, P.C. (2006) Leveraging enzyme structure-function relationships for functional inference and experimental design: the structure-function linkage database. Biochemistry, 45, 2545–2555.
Pei, J., Kim, B.-H., and Grishin, N.V. (2008) PROMALS3D: a tool for multiple proteinsequence and structure alignments. Nucleic Acids Res, 36, 2295–2300.
Pieper, U., Chiang, R., Seffernick, J.J., Brown, S.D., Glasner, M.E., Kelly, L., Eswar,N., Sauder, J.M., Bonanno, J.B., Swaminathan, S. et al. (2009) Target selection andannotation for the structural genomics of the amidohydrolase and enolase superfamilies.J Struct Funct Genomics, 10, 107–125.
Rentzsch, R. and Orengo, C.A. (2009) Protein function prediction - the power of multi-plicity. Trends Biotechnol, 27(4), 210–219.
Rosenblat, M., Gaidukov, L., Khersonsky, O., Vaya, J., Oren, R., Tawfik, D.S., and Avi-ram, M. (2006) The catalytic histidine dyad of high density lipoprotein-associatedserum paraoxonase-1 (PON1) is essential for PON1-mediated Inhibition of low den-sity lipoprotein oxidation and stimulation of macrophage cholesterol efflux. J BiolChem, 281, 7657–7665.
Scharff, E.I., Koepke, J., Fritzsch, G., Lucke, C., and Ruterjans, H. (2001) Crystal structureof diisopropylfluorophosphatase from Loligo vulgaris. Structure, 9, 493–502.
Schnoes, A.M., Brown, S.D., Dodevski, I., and Babbitt, P.C. (2009) Annotation error inpublic databases: misannotation of molecular function in enzyme superfamilies. PLoSComput Biol, 5, e1000605.
Suzek, B.E., Huang, H., McGarvey, P., Mazumder, R., and Wu, C.H. (2007) UniRef:comprehensive and non-redundant UniProt reference clusters. Bioinformatics, 23,1282–1288.
Tanaka, Y., Morikawa, K., Ohki, Y., Yao, M., Tsumoto, K., Watanabe, N., Ohta, T., andTanaka, I. (2007) Structural and mutational analyses of Drp35 from Staphylococcusaureus: a possible mechanism for its lactonase activity. J Biol Chem, 282, 5770–5780.
Zhang, Y. and Skolnick, J. (2005) TM-align: a protein structure alignment algorithm basedon the TM-score. Nucleic Acids Res, 33, 2302–2309.


















