
Priming of Human Resting NK Cells by Autologous M1 ... · gli Investimenti della Ricerca di Base...
12
of February 2, 2019. This information is current as Pathways , and IL-15 β , IFN- β Engagement of IL-1 Autologous M1 Macrophages via the Priming of Human Resting NK Cells by Locati and Domenico Mavilio Molgora, Emanuela Marcenaro, Enrico Lugli, Massimo Irene Mattiola, Matthieu Pesant, Paolo F. Tentorio, Martina http://www.jimmunol.org/content/195/6/2818 doi: 10.4049/jimmunol.1500325 August 2015; 2015; 195:2818-2828; Prepublished online 14 J Immunol Material Supplementary 5.DCSupplemental http://www.jimmunol.org/content/suppl/2015/08/14/jimmunol.150032 References http://www.jimmunol.org/content/195/6/2818.full#ref-list-1 , 19 of which you can access for free at: cites 67 articles This article average * 4 weeks from acceptance to publication Fast Publication! • Every submission reviewed by practicing scientists No Triage! • from submission to initial decision Rapid Reviews! 30 days* • Submit online. ? The JI Why Subscription http://jimmunol.org/subscription is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/About/Publications/JI/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/alerts Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved. Copyright © 2015 by The American Association of 1451 Rockville Pike, Suite 650, Rockville, MD 20852 The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on February 2, 2019 http://www.jimmunol.org/ Downloaded from by guest on February 2, 2019 http://www.jimmunol.org/ Downloaded from
Transcript of Priming of Human Resting NK Cells by Autologous M1 ... · gli Investimenti della Ricerca di Base...

of February 2, 2019.This information is current as
Pathways, and IL-15β, IFN-βEngagement of IL-1
Autologous M1 Macrophages via the Priming of Human Resting NK Cells by
Locati and Domenico MavilioMolgora, Emanuela Marcenaro, Enrico Lugli, Massimo Irene Mattiola, Matthieu Pesant, Paolo F. Tentorio, Martina
http://www.jimmunol.org/content/195/6/2818doi: 10.4049/jimmunol.1500325August 2015;
2015; 195:2818-2828; Prepublished online 14J Immunol
MaterialSupplementary
5.DCSupplementalhttp://www.jimmunol.org/content/suppl/2015/08/14/jimmunol.150032
Referenceshttp://www.jimmunol.org/content/195/6/2818.full#ref-list-1
, 19 of which you can access for free at: cites 67 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2015 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on February 2, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on February 2, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
Priming of Human Resting NK Cells by Autologous M1Macrophages via the Engagement of IL-1b, IFN-b, andIL-15 Pathways
Irene Mattiola,*,†,‡,1 Matthieu Pesant,*,1 Paolo F. Tentorio,† Martina Molgora,*,‡
Emanuela Marcenaro,x Enrico Lugli,† Massimo Locati,*,‡,2 and Domenico Mavilio†,‡,2
The cross talk between NK cells and macrophages is emerging as a major line of defense against microbial infections and tumors.
This study reveals a complex network of soluble mediators and cell-to-cell interactions allowing human classically activated (M1)
macrophages, but not resting (M0) or alternatively activated (M2) macrophages, to prime resting autologous NK cells. In this article,
we show that M1 increase NK cell cytotoxicity by IL-23 and IFN-b–dependent upregulation of NKG2D, IL-1b–dependent upreg-
ulation of NKp44, and trans-presentation of IL-15. Moreover, both IFN-b–dependent cis-presentation of IL-15 on NK cells and
engagement of the 2B4-CD48 pathway are used by M1 to trigger NK cell production of IFN-g. The disclosure of these synergic
cellular mechanisms regulating the M1–NK cell cross talk provides novel insights to better understand the role of innate immune
responses in the physiopathology of tumor biology and microbial infections. The Journal of Immunology, 2015, 195: 2818–2828.
Natural killer cells are important components of the in-nate immune system and play an active role in the clear-ance of tumor-transformed and viral-infected cells either
lacking or down-modulating molecules of the MHC class I withouta prior sensitization (1). Impairments of the interactions between
the heterogeneous family of inhibitory NK cell receptors andMHC class I molecules allow the NK cell–mediated killing via theengagement of a large group of activatingNKRs (aNKRs) that bindspecific ligands expressed on stressed (i.e., infected by pathogens,transplanted, inflamed) or tumor-transformed cells (2–4). Uponactivation, NK cells also secrete a variety of proinflammatorycytokines and chemokines, which play important regulatory func-tions in the context of immune responses, hematopoiesis, and cel-lular homeostasis (1, 5).Macrophages are well-known for being important players in
the physiopathology of microbial infections and cancer. In linewiththe Th1/Th2 T cell polarization, the stimulation of resting macro-phages (M0) with Th1 cytokines (i.e., IFN-g) and the TLR4 ligands(i.e., LPS) induces the classical polarization toward M1 macro-phages (M1), which display strong microbicidal and tumoricidalactivities and preferentially promote inflammatory responses. Incontrast, the alternative polarization toward M2 macrophages (M2)is induced by the Th2 cytokine IL-4 (6, 7). M2 are involved in thecontrol of parasite infections and in tissue remodeling and fi-brosis, and they are endowed with potent proangiogenic featuresthat favor tumor progression by virtue of their immunoregulatoryfunctions (8, 9). The different biological properties of M1 andM2 are also mirrored by distinctive cytokine and chemokine rep-ertoires (10). Tumor-associated macrophages (TAMs), present insolid tumors at high frequency, display both proangiogenic andimmune-regulatory features resembling the M2 phenotype (11). Itis now widely accepted that TAMs sustain cancer growth by en-gaging vicious cellular cross talks with malignant cells and otherimmune effectors within the tumor mass, thus worsening clinicalprognosis and favoring disease progression (12–14).Over the past decade, several lines of evidence clearly demon-
strated that NK cells are able to engage bidirectional interactionswith other members of innate immunity, such as autologous den-dritic cells (DCs), macrophages, and neutrophils (2, 15–20). Thefinal outcome of these synergic interactions mediated by both cell-to-cell contacts and soluble mediators is the coordination andoptimization of both innate and adaptive immune responses. Inparticular, the NK cell–macrophage cross talk is highly relevantin the context of host–pathogen interactions (15, 16, 19) and
*Leukocyte Biology Unit, Humanitas Clinical and Research Center, I-20089 Roz-zano, Milan, Italy; †Unit of Clinical and Experimental Immunology, HumanitasClinical and Research Center, I-20089 Rozzano, Milan, Italy; ‡Department of Med-ical Biotechnologies and Translational Medicine, University of Milan, I-20089 Roz-zano, Milan, Italy; and xDipartimento di Medicina Sperimentale and Centro diEccellenza per le Ricerche Biomediche, Universita degli Studi di Genova, I-16132Genoa, Italy
1I.M. and M.P. contributed equally to this work.
2M.L. and D.M. are joint senior authors on this work.
ORCIDs: 0000-0003-3077-590X (M.L.); 0000-0001-6147-0952 (D.M.).
Received for publication February 11, 2015. Accepted for publication July 19, 2015.
This work was supported by Italian Ministry of Health (Bando Giovani Ricercatori)Grant GR-2008-1135082 (to D.M.), a Ministry of University and Research Fondo pergli Investimenti della Ricerca di Base “Futuro in Ricerca” project (to M.L.), ItalianAssociation for Cancer Research Grants IG 9104 and 14687 (to D.M.) and IG 14685(to M.L.), European Union Marie Curie International Reintegration Grant 322093 (toE.L.) and Collaborative Project TIMER-HEALTH-F4-2011-281608 (to M.L.), theintramural research program of Humanitas Clinical and Research Center (to D.M.),a European School of Molecular Medicine/Structured International PostdoctoralMarie Curie International Mobility Fellowship (to M.P.), and a “Mario e ValeriaRindi” Fellowship from the Italian Foundation for Cancer Research (to I.M.).
I.M., M.P., M.L., and D.M. conceived and designed the experiments; I.M., M.P.,P.F.T., M.M., and E.L. performed the experiments; I.M. and M.P. analyzed the data;E.M. provided reagents; and I.M., M.P., M.L., and D.M. wrote the manuscript.
Address correspondence and reprint requests to Prof. Massimo Locati or Prof. Do-menico Mavilio, Leukocyte Biology Unit, Department of Medical Biotechnologiesand Translational Medicine, University of Milan School of Medicine, HumanitasClinical and Research Center, Via Alessandro Manzoni 113, I-20089 Rozzano, Mi-lan, Italy (M.L.) or Clinical and Experimental Immunology Unit, Department ofMedical Biotechnologies and Translational Medicine, University of Milan Schoolof Medicine, Humanitas Clinical and Research Center, Via Alessandro Manzoni113, I-20089 Rozzano, Milan, Italy (D.M.). E-mail addresses: [email protected] (M.L.) or [email protected] (D.M.)
The online version of this article contains supplemental material.
Abbreviations used in this article: aNKR, activating NKR; DC, dendritic cell; ILC,innate lymphoid cell; IL-15Ra, IL-15R a-chain; rh, recombinant human; RT, roomtemperature; TAM, tumor-associated macrophage.
Copyright� 2015 by TheAmericanAssociation of Immunologists, Inc. 0022-1767/15/$25.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1500325
by guest on February 2, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

during the course of viral infections (20). However, the mechanism(s)adopted by activated human macrophages to regulate autologousresting NK cell functions are largely unknown. A proper activationof NK cells can be achieved with different proinflammatory cyto-kines, such as IL-2, IL-12, IL-15, IL-18, and type I IFNs (1, 21, 22).In particular, IL-15 has been shown to play an important role in thecontext of NK development, homeostasis, and functions. After itsbinding to the high-affinity IL-15R a-chain (IL-15Ra) expressedby DCs and monocytes/macrophages, IL-15 is trans-presented tosurrounding cells bearing the lower-affinity IL-15Rbg-chains (22,23). The membrane-associated IL-15–IL-15Ra complex can bealso cleaved from cell surface and released in biologic fluids, suchas plasma (24, 25). More recently, it emerged that IL-15 can alsobe presented by IL-15Ra to IL-15Rbg-chains expressed on thesame cell, thus allowing a mechanism of IL-15 cis-presentation thattriggers NK cell effector functions (26). Another potent proin-flammatory cytokine produced by M1 is IL-1b, which has beenreported to modulate homeostasis and activation of innate lym-phoid cells (ILCs) in secondary lymphoid tissues (27–29). ILCshave been recently identified as an additional lymphocytic pop-ulation that could be divided in different subsets (ILC1, ILC2,ILC3) (30). Because NK cells differ from ILC1 by the expressionof eomesodermin during their differentiation (30), they are con-sidered as a distinct immune cell population. However, whetherNK cells are responsive to IL-1b is still being debated in bothhuman and murine settings (26–28, 31, 32).This study investigates on the cross talk between human resting
NK cells and autologous polarized macrophages, and discloses theselective ability of M1 to prime NK cells through a complex net-work of interactions that comprises mechanisms associated withboth the production of soluble mediators and cell-to-cell contacts.
Materials and MethodsNK cell and macrophage isolation and culture
Human PBMCs were isolated from buffy coats of healthy donors obtainedin accordance with clinical protocols approved by the Institutional ReviewBoard of Desio Hospital, Milan, Italy. After centrifugation on a Ficoll den-sity gradient (GE Healthcare Biosciences), NK cells and monocytes wereisolated by negative magnetic cell purification (STEMCELLTechnologies)(33, 34). The purity of enriched resting NK cells was $95%, and the fre-quency of other contaminating cells was #3% for NKT/T cells, #2%for CD56neg/CD127pos ILCs,#0.5% for CD19pos B cells,#0.2 for CD14pos
monocytes, and #0.2% for M-DC8pos slan-DCs (Supplemental Fig. 1A,1B). Freshly purified and enriched NK cells were frozen in FBS supple-mented with 10% DMSO (Sigma) immediately after their isolation, thawedwhen autologous resting or polarized macrophages where ready, and cul-tured in absence of any stimuli for 24 h at 33 106 cells/ml. The viability ofNK cells before the coculture was always$90%, as assessed by trypan blueexclusion (data not shown). Resting NK cells did not undergo a signifi-cant cellular activation after being thawed and cultured (SupplementalFig. 1C). To generate resting macrophage (M0), we cultured CD14pos
monocytes (cell purity $ 95%) for 7 d with RPMI 1640 supplemented with10% FBS, 1% L-glutamine, 1% penicillin/streptomycin (Lonza) and 100 ng/mlrecombinant human (rh) M-CSF (R&D Systems). M0 macrophages werethen incubated for 24 h with 100 ng/ml LPS purified from Escherichia coli055:B5 (Sigma) and 20 ng/ml rhIFN-g (Peprotech) to induce M1 polarization(evaluated by CD80 expression) or 20 ng/ml rhIL-4 (Peprotech) to induceM2 polarization (evaluated by CD206 and CD209 expression; SupplementalFig. 2A–C). The role of inflammasome was investigated treating restingmacrophages for 1 h with 50 mg/ml glibenclamide (Labogen) or its vehicle(DMSO) before stimulation with 100 ng/ml LPS and 20 ng/ml rhIFN-g.
Cell lines
K562 (erythroleukemia), HEK-293T (embryonic kidney), JA3 (Jurkat leu-kemia), Raji (Burkitt’s lymphoma), and 221 (B lymphoblastoid) cell lineswere cultured in DMEM supplemented with 10% FBS, 1% L-glutamine, 1%pen/strept. TheMOLT-4 (acute lymphoblastic leukemia) cell linewas grownin RPMI 1640 medium supplemented with 10% FBS, 1% L-glutamine, 1%pen/strept, and 1% HEPES (Lonza).
Flow cytometry and cell sorting
For direct multicolor flow-cytometry analysis (FACSCanto II; BD Bio-sciences) or cell sorting (FACSAria II; BD Biosciences), NK cells, mono-cytes, and polarized macrophages were washed with HBSS (Lonza)supplemented with 2% FBS and incubated for 20 min at 4˚C with thefollowing directly conjugated Abs labeled with indicated fluorochromes:CD3/CD56-FITC/PC5 (Beckman Coulter), CD3-FITC (BD Pharmingen),CD3-Brilliant Violet 650 (Biolegend), CD14-FITC (BD Pharmingen), CD14-Brilliant Violet 570 (Biolegend), CD16-PE-Cy7 (BD Pharmingen), CD16-PerCP-Cy5.5 (BDPharmingen), CD19-allophycocyanin-Cy7 (BDPharmingen),CD48-PE (BDPharmingen), CD56-Brilliant Violet 421 (Biolegend),CD69-Pe-Cy7 (BD Pharmingen), CD80-PE (BD Pharmingen), CD117-PE-Cy7(Biolegend), CD127-PE-Cy5 (eBioscience), CD206-FITC (BD Pharmingen),CD209-PE (BD Pharmingen), M-DC8-allophycocyanin (Miltenyi), NKp46-PE (Beckman Coulter), NKp44-PE (Beckman Coulter), NKp30-PE (BeckmanCoulter), NKG2D-PE (Beckman Coulter), DNAM-1–PE (Beckman Coulter),2B4-PE (R&D Systems), IL-15Ra–PE (Biolegend), IL-1RII–PE (R&D Sys-tems), and their appropriate isotype controls. For IL-1RI indirect staining,NK cells were incubated for 45 min at room temperature (RT) with purifiedgoat anti-human IL-1RI (R&D Systems) or its polyclonal IgG control andfor 30 min at RT with mouse anti-goat Alexa Fluor 647–labeled sec-ondary Ab (Invitrogen). For sorting experiments, thawed NK cells werestained for 15 min at RTwith LIVE/DEADAqua (Invitrogen), followedby incubation for 20 min at RT with fluorochrome-conjugated Abs andthen sorted. FACS-sorted NK cells showed a purity of $98%, and thefrequencies of cellular contaminants were #0.4% for CD19pos B cellsand CD3pos T/NKT cells,#0.8% for CD56neg/CD127pos ILCs, and#0.2%for CD14pos monocytes andM-DC8pos slan-DCs (Supplemental Fig. 1D, 1E).Data were analyzed with FACSDiva (BD) and FlowJo (Tree Star) software.
Incubation of NK cells with macrophage-conditioned media orautologous macrophages
NK cells were incubated for 24 h at 1.25–2.5 3 105 cells/ml with30% conditioned media taken from macrophage (M0/M1/M2) cultures,200 U/ml rhIL-2, 20 ng/ml rhIL-12, 10 ng/ml rhIL-15 (Peprotech), 1 ng/mlrhIL-1b (Miltenyi), 100 ng/ml rhIL-18, or 200 U/ml rhIFN-b (R&D Sys-tems). For direct NK cell–macrophage coculture, 1 3 105 NK cells weregrown for 24 h at a 1:1 cell ratio with resting or polarized macrophages,which were extensively washed to eliminate all soluble factors accumu-lated during the polarization period before analysis.
Fluorescence microscopy
NK cells treated with macrophage-conditioned media or cytokines wereattached to poly-L-lysin solution (Sigma)–coated cover glasses. NK cellswere fixed and nonspecific staining were prevented by incubation for1 h with PBS (Biosera) supplemented with 2% BSA (Sigma) and 5%donkey serum (Sigma). NK cells were then incubated for 1 h at RTwith 2.5mg/ml purified goat anti-human IL-1RI (R&D Systems), 5 mg/ml purifiedgoat anti-human IL-15Ra (R&D Systems), or their corresponding isotypecontrols, then washed and incubated for 1 h at RT with a donkey anti-goat Alexa Fluor 488–labeled secondary Ab (Invitrogen). After repeatedwashes, NK cells were incubated 10 min at RT with DAPI (Invitrogen).CD14pos monocytes were plated directly on cover glasses, differentiatedinto resting macrophages, and then polarized, as described earlier. Theywere then fixed, incubated for 1 h with PBS supplemented with 2% BSAand 5% donkey serum, and stained for IL-15Ra or IL-1RI, following thesame protocol used for NK cells. NK cells and macrophages were notpermeabilized. Images were acquired in oil with an Olympus FV1000(UPL SAPO 603ONA: 1.35) and analyzed with FV1000 using the Imarissoftware.
Real-time PCR
Cells were lysed with QIAzol Reagent (Qiagen), and total RNAwas ex-tracted using miRNeasy mini kit (Qiagen). RNAwas converted in cDNAusing the High-Capacity cDNA Reverse Transcription Kit (Applied Bio-systems), and quantification of the following transcripts was performedfollowing the recommended protocols for SYBR Green Master Mix(Applied Biosystems) or TaqMan Fast Advanced Master Mix (Applied Bio-systems): IFN-g (forward: 59-CTCTTGGCTGTTACTGCCAGG-39; re-verse: 59-CTCCACACTCTTTTGGATGCT-39), IL-1RI (TaqMan assayHs00991002_m1), IL-15 (TaqMan assay Hs01003716_m1), and IL-15Ra(TaqMan assay Hs00542604_m1). Results were normalized on s18 (for-ward: 59-CCGCAGCTAGGAATAATGGAATA-39; reverse: 59-CGAAA-ACCAACAAAATAGAACCG-39; or TaqMan assay Hs99999901_s1) forNK cell analysis or GAPDH (TaqMan assay Hs99999905_m1) for mac-
The Journal of Immunology 2819
by guest on February 2, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
rophage analysis. Data were analyzed by SDS 2.4 software with a 7900HTFast Real Time PCR System (Applied Biosystems) and Opticon Monitor 3with a PTC-200 PCR (MJ Research).
Masking experiments
Saturating concentrations of the following neutralizing Abs were added tothe NK cell culture: NKG2D (1 mg/ml; Biolegend), IL-1b (5 mg/ml; BDPharmingen), IL-15 (0.5 mg/ml; R&D Systems), IL-18 (5 mg/ml; MBLInternational Corporation), IL-23p19 (0.8 mg/ml; R&D Systems), IFN-b(0.2 mg/ml; R&D Systems), IL-1RI (2 mg/ml; R&D Systems), and IL-15Ra(5 mg/ml; R&D Systems). Anti-NKp30 (clone F252), anti-NKp44 (cloneKS38), anti-NKp46 (clone KL247), anti-2B4 (clone C054), and anti-DNAM-1 (clone DX11) mAbs were kindly provided by Prof. AlessandroMoretta (University of Genova, Italy). Macrophage-conditioned mediawere incubated for 45 min at RTwith blocking Abs against cytokines be-fore NK cell stimulation. IL-1RI on NK cells was blocked at RT for 45 minbefore macrophage-conditioned media treatment and left until harvestof the cells. Anti–IL-15Ra was given at the beginning of the treatmentwith macrophage-conditioned media or NK cell–macrophage cocultureand left until the conclusion of the experiment. Blocking Abs to activatingreceptors were incubated for 45 min at RT with macrophage-primedNK cells before they were incubated with target cells and left until har-vest of the cells and on resting NK cells before they were cocultured withpolarized macrophages and left for all the period of the coculture. Eachneutralizing condition was compared with a matched isotype control.
CD107a degranulation assay and cytokine secretion
We used CD107a degranulation assay as indicative of NK cell cytotox-icity, as previously demonstrated (35). NK cells treated with macrophage-conditioned media were incubated with K562 at 2:1 E:T ratio orHEK-293T at 5:1 E:T ratio for 4 h in the presence of PE-labeled CD107aAb (BD Pharmingen) or its isotype control. K562, JA3, MOLT-4, Raji, or221 (E:T ratio 2:1) were incubated for 4 h directly into NK cell–macro-phage cocultures, together with PE-labeled CD107a Ab or its isotypecontrol. Thereafter, cells were washed, stained with CD56 and CD3 Abs toallow gating on NK cells, and analyzed by flow cytometry. IFN-g secretionbyNKcellswas dosed by commercialELISAkit (Duoset; R&DSystems).NKcells were washed extensively after treatment with autologous macrophage-conditioned media to eliminate any cytokine carryover. NK cells were
stimulated with K562 cells (E:T ratio 2:1) for 4 h in both experimentalapproaches (indirect or direct culture). Macrophage secretion of IL-1b,IL-12p70, IL-15, and IL-15/IL-15Rawere dosed by commercial ELISAkit (Duoset; R&D Systems). Data were analyzed by SoftMaxPro 5.3software.
Statistical analysis
Results were expressed as mean 6 SEM from multiple independent ex-periments. Two-tailed Student t test or one-sample two-tailed t tests forcomparison against fixed values (% of max, set to 100%) were performedusing Prism (GraphPad 4) and/or Excel (Microsoft) software: n.s., not sig-nificant; *p, 0.05, **p, 0.01, ***p, 0.001, ****p, 0.0001.
ResultsPriming of resting NK cells requires M1 polarization
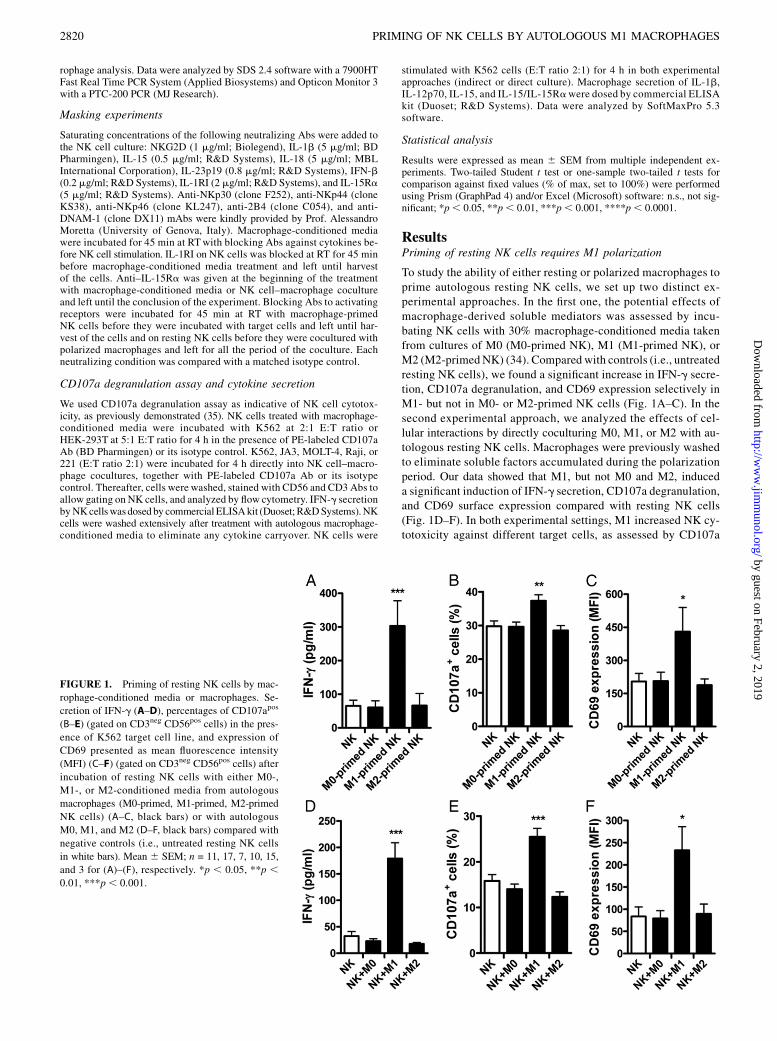
To study the ability of either resting or polarized macrophages toprime autologous resting NK cells, we set up two distinct ex-perimental approaches. In the first one, the potential effects ofmacrophage-derived soluble mediators was assessed by incu-bating NK cells with 30% macrophage-conditioned media takenfrom cultures of M0 (M0-primed NK), M1 (M1-primed NK), orM2 (M2-primedNK) (34). Compared with controls (i.e., untreatedresting NK cells), we found a significant increase in IFN-g secre-tion, CD107a degranulation, and CD69 expression selectively inM1- but not in M0- or M2-primed NK cells (Fig. 1A–C). In thesecond experimental approach, we analyzed the effects of cel-lular interactions by directly coculturing M0, M1, or M2 with au-tologous resting NK cells. Macrophages were previously washedto eliminate soluble factors accumulated during the polarizationperiod. Our data showed that M1, but not M0 and M2, induceda significant induction of IFN-g secretion, CD107a degranulation,and CD69 surface expression compared with resting NK cells(Fig. 1D–F). In both experimental settings, M1 increased NK cy-totoxicity against different target cells, as assessed by CD107a
FIGURE 1. Priming of resting NK cells by mac-
rophage-conditioned media or macrophages. Se-
cretion of IFN-g (A–D), percentages of CD107apos
(B–E) (gated on CD3neg CD56pos cells) in the pres-
ence of K562 target cell line, and expression of
CD69 presented as mean fluorescence intensity
(MFI) (C–F) (gated on CD3neg CD56pos cells) after
incubation of resting NK cells with either M0-,
M1-, or M2-conditioned media from autologous
macrophages (M0-primed, M1-primed, M2-primed
NK cells) (A–C, black bars) or with autologous
M0, M1, and M2 (D–F, black bars) compared with
negative controls (i.e., untreated resting NK cells
in white bars). Mean 6 SEM; n = 11, 17, 7, 10, 15,
and 3 for (A)–(F), respectively. *p , 0.05, **p ,0.01, ***p, 0.001.
2820 PRIMING OF NK CELLS BY AUTOLOGOUS M1 MACROPHAGES
by guest on February 2, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
degranulation assay (35, 36) (Supplemental Fig. 3A, 3C). Takentogether, these results demonstrate that proinflammatory M1prime resting NK cells by both secreting soluble mediators andestablishing cell-to-cell interactions. In contrast, neither M0 norM2 influence NK cell activation and effector functions underthese experimental settings.
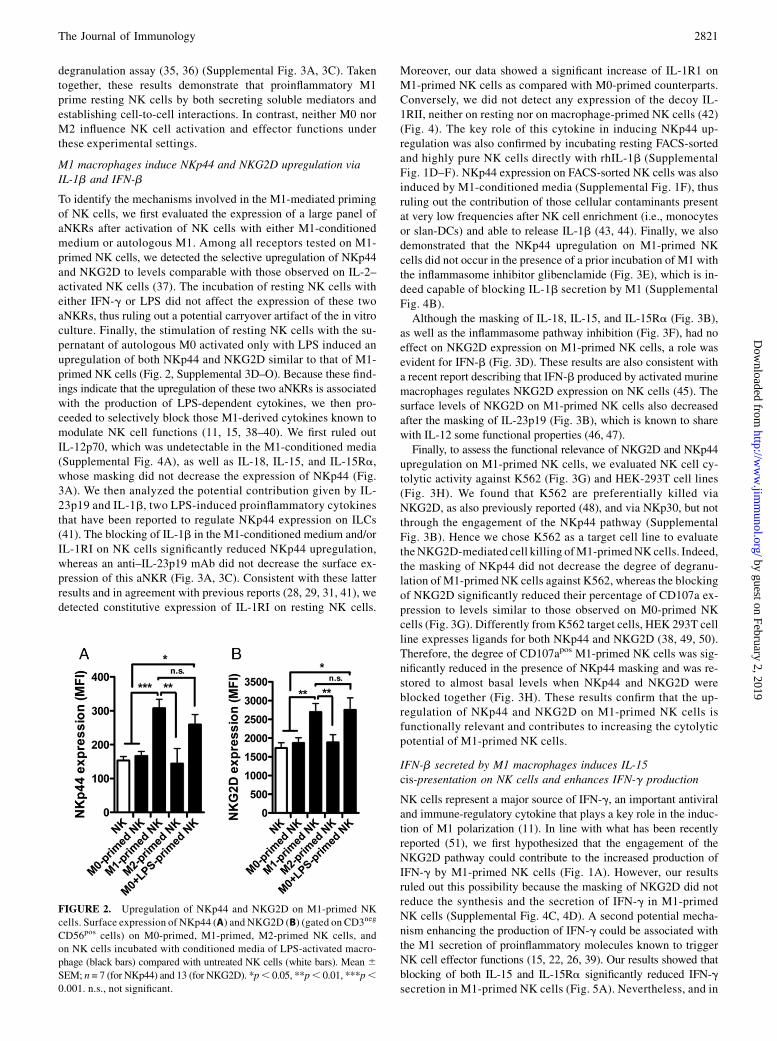
M1 macrophages induce NKp44 and NKG2D upregulation viaIL-1b and IFN-b
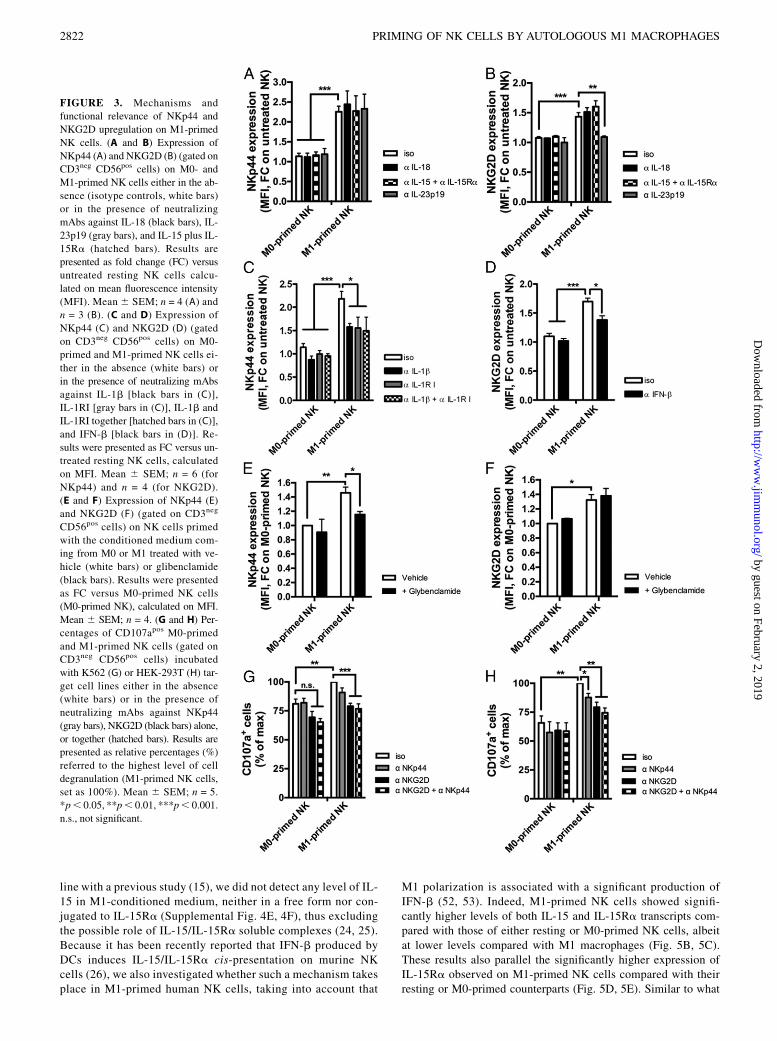
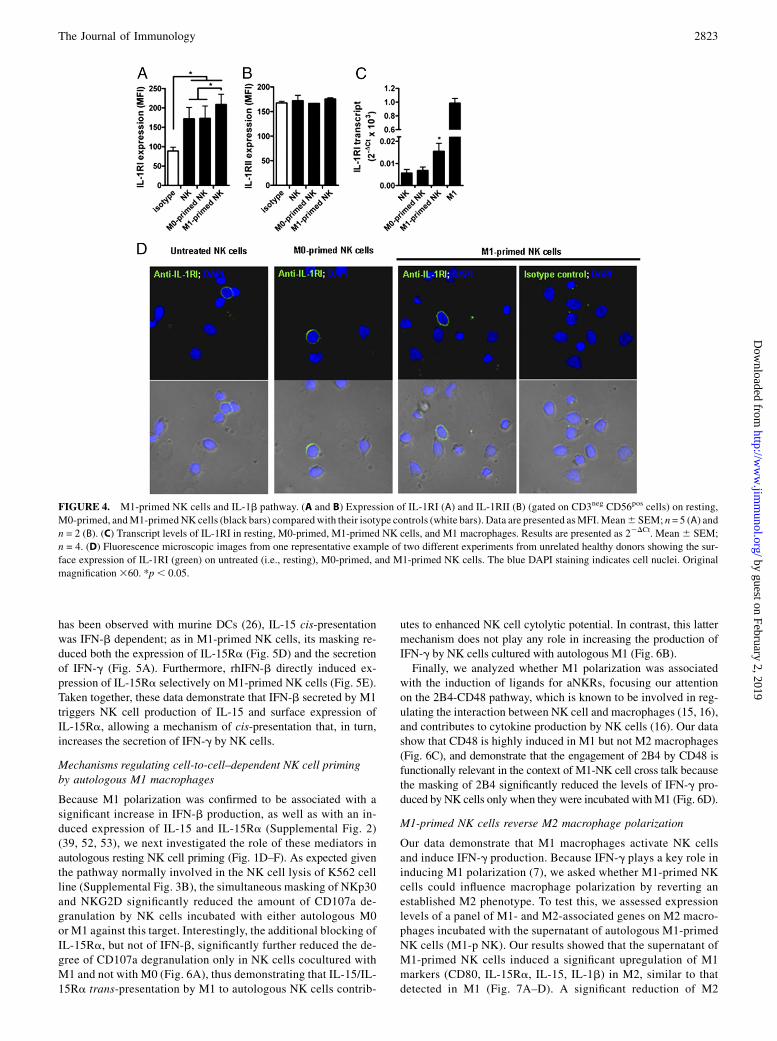
To identify the mechanisms involved in the M1-mediated primingof NK cells, we first evaluated the expression of a large panel ofaNKRs after activation of NK cells with either M1-conditionedmedium or autologous M1. Among all receptors tested on M1-primed NK cells, we detected the selective upregulation of NKp44and NKG2D to levels comparable with those observed on IL-2–activated NK cells (37). The incubation of resting NK cells witheither IFN-g or LPS did not affect the expression of these twoaNKRs, thus ruling out a potential carryover artifact of the in vitroculture. Finally, the stimulation of resting NK cells with the su-pernatant of autologous M0 activated only with LPS induced anupregulation of both NKp44 and NKG2D similar to that of M1-primed NK cells (Fig. 2, Supplemental 3D–O). Because these find-ings indicate that the upregulation of these two aNKRs is associatedwith the production of LPS-dependent cytokines, we then pro-ceeded to selectively block those M1-derived cytokines known tomodulate NK cell functions (11, 15, 38–40). We first ruled outIL-12p70, which was undetectable in the M1-conditioned media(Supplemental Fig. 4A), as well as IL-18, IL-15, and IL-15Ra,whose masking did not decrease the expression of NKp44 (Fig.3A). We then analyzed the potential contribution given by IL-23p19 and IL-1b, two LPS-induced proinflammatory cytokinesthat have been reported to regulate NKp44 expression on ILCs(41). The blocking of IL-1b in the M1-conditioned medium and/orIL-1RI on NK cells significantly reduced NKp44 upregulation,whereas an anti–IL-23p19 mAb did not decrease the surface ex-pression of this aNKR (Fig. 3A, 3C). Consistent with these latterresults and in agreement with previous reports (28, 29, 31, 41), wedetected constitutive expression of IL-1RI on resting NK cells.
Moreover, our data showed a significant increase of IL-1R1 onM1-primed NK cells as compared with M0-primed counterparts.Conversely, we did not detect any expression of the decoy IL-1RII, neither on resting nor on macrophage-primed NK cells (42)(Fig. 4). The key role of this cytokine in inducing NKp44 up-regulation was also confirmed by incubating resting FACS-sortedand highly pure NK cells directly with rhIL-1b (SupplementalFig. 1D–F). NKp44 expression on FACS-sorted NK cells was alsoinduced by M1-conditioned media (Supplemental Fig. 1F), thusruling out the contribution of those cellular contaminants presentat very low frequencies after NK cell enrichment (i.e., monocytesor slan-DCs) and able to release IL-1b (43, 44). Finally, we alsodemonstrated that the NKp44 upregulation on M1-primed NKcells did not occur in the presence of a prior incubation of M1 withthe inflammasome inhibitor glibenclamide (Fig. 3E), which is in-deed capable of blocking IL-1b secretion by M1 (SupplementalFig. 4B).Although the masking of IL-18, IL-15, and IL-15Ra (Fig. 3B),
as well as the inflammasome pathway inhibition (Fig. 3F), had noeffect on NKG2D expression on M1-primed NK cells, a role wasevident for IFN-b (Fig. 3D). These results are also consistent witha recent report describing that IFN-b produced by activated murinemacrophages regulates NKG2D expression on NK cells (45). Thesurface levels of NKG2D on M1-primed NK cells also decreasedafter the masking of IL-23p19 (Fig. 3B), which is known to sharewith IL-12 some functional properties (46, 47).Finally, to assess the functional relevance of NKG2D and NKp44
upregulation on M1-primed NK cells, we evaluated NK cell cy-tolytic activity against K562 (Fig. 3G) and HEK-293T cell lines(Fig. 3H). We found that K562 are preferentially killed viaNKG2D, as also previously reported (48), and via NKp30, but notthrough the engagement of the NKp44 pathway (SupplementalFig. 3B). Hence we chose K562 as a target cell line to evaluatetheNKG2D-mediated cell killing ofM1-primedNK cells. Indeed,the masking of NKp44 did not decrease the degree of degranu-lation of M1-primed NK cells against K562, whereas the blockingof NKG2D significantly reduced their percentage of CD107a ex-pression to levels similar to those observed on M0-primed NKcells (Fig. 3G). Differently fromK562 target cells, HEK 293T cellline expresses ligands for both NKp44 and NKG2D (38, 49, 50).Therefore, the degree of CD107apos M1-primed NK cells was sig-nificantly reduced in the presence of NKp44 masking and was re-stored to almost basal levels when NKp44 and NKG2D wereblocked together (Fig. 3H). These results confirm that the up-regulation of NKp44 and NKG2D on M1-primed NK cells isfunctionally relevant and contributes to increasing the cytolyticpotential of M1-primed NK cells.
IFN-b secreted by M1 macrophages induces IL-15cis-presentation on NK cells and enhances IFN-g production
NK cells represent a major source of IFN-g, an important antiviraland immune-regulatory cytokine that plays a key role in the induc-tion of M1 polarization (11). In line with what has been recentlyreported (51), we first hypothesized that the engagement of theNKG2D pathway could contribute to the increased production ofIFN-g by M1-primed NK cells (Fig. 1A). However, our resultsruled out this possibility because the masking of NKG2D did notreduce the synthesis and the secretion of IFN-g in M1-primedNK cells (Supplemental Fig. 4C, 4D). A second potential mecha-nism enhancing the production of IFN-g could be associated withthe M1 secretion of proinflammatory molecules known to triggerNK cell effector functions (15, 22, 26, 39). Our results showed thatblocking of both IL-15 and IL-15Ra significantly reduced IFN-gsecretion in M1-primed NK cells (Fig. 5A). Nevertheless, and in
FIGURE 2. Upregulation of NKp44 and NKG2D on M1-primed NK
cells. Surface expression of NKp44 (A) and NKG2D (B) (gated on CD3neg
CD56pos cells) on M0-primed, M1-primed, M2-primed NK cells, and
on NK cells incubated with conditioned media of LPS-activated macro-
phage (black bars) compared with untreated NK cells (white bars). Mean 6SEM; n = 7 (for NKp44) and 13 (for NKG2D). *p, 0.05, **p, 0.01, ***p,0.001. n.s., not significant.
The Journal of Immunology 2821
by guest on February 2, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
line with a previous study (15), we did not detect any level of IL-15 in M1-conditioned medium, neither in a free form nor con-jugated to IL-15Ra (Supplemental Fig. 4E, 4F), thus excludingthe possible role of IL-15/IL-15Ra soluble complexes (24, 25).Because it has been recently reported that IFN-b produced byDCs induces IL-15/IL-15Ra cis-presentation on murine NKcells (26), we also investigated whether such a mechanism takesplace in M1-primed human NK cells, taking into account that
M1 polarization is associated with a significant production ofIFN-b (52, 53). Indeed, M1-primed NK cells showed signifi-cantly higher levels of both IL-15 and IL-15Ra transcripts com-pared with those of either resting or M0-primed NK cells, albeitat lower levels compared with M1 macrophages (Fig. 5B, 5C).These results also parallel the significantly higher expression ofIL-15Ra observed on M1-primed NK cells compared with theirresting or M0-primed counterparts (Fig. 5D, 5E). Similar to what
FIGURE 3. Mechanisms and
functional relevance of NKp44 and
NKG2D upregulation on M1-primed
NK cells. (A and B) Expression of
NKp44 (A) and NKG2D (B) (gated on
CD3neg CD56pos cells) on M0- and
M1-primed NK cells either in the ab-
sence (isotype controls, white bars)
or in the presence of neutralizing
mAbs against IL-18 (black bars), IL-
23p19 (gray bars), and IL-15 plus IL-
15Ra (hatched bars). Results are
presented as fold change (FC) versus
untreated resting NK cells calcu-
lated on mean fluorescence intensity
(MFI). Mean6 SEM; n = 4 (A) and
n = 3 (B). (C and D) Expression of
NKp44 (C) and NKG2D (D) (gated
on CD3neg CD56pos cells) on M0-
primed and M1-primed NK cells ei-
ther in the absence (white bars) or
in the presence of neutralizing mAbs
against IL-1b [black bars in (C)],
IL-1RI [gray bars in (C)], IL-1b and
IL-1RI together [hatched bars in (C)],
and IFN-b [black bars in (D)]. Re-
sults were presented as FC versus un-
treated resting NK cells, calculated
on MFI. Mean 6 SEM; n = 6 (for
NKp44) and n = 4 (for NKG2D).
(E and F) Expression of NKp44 (E)
and NKG2D (F) (gated on CD3neg
CD56pos cells) on NK cells primed
with the conditioned medium com-
ing from M0 or M1 treated with ve-
hicle (white bars) or glibenclamide
(black bars). Results were presented
as FC versus M0-primed NK cells
(M0-primed NK), calculated on MFI.
Mean 6 SEM; n = 4. (G and H) Per-
centages of CD107apos M0-primed
and M1-primed NK cells (gated on
CD3neg CD56pos cells) incubated
with K562 (G) or HEK-293T (H) tar-
get cell lines either in the absence
(white bars) or in the presence of
neutralizing mAbs against NKp44
(gray bars), NKG2D (black bars) alone,
or together (hatched bars). Results are
presented as relative percentages (%)
referred to the highest level of cell
degranulation (M1-primed NK cells,
set as 100%). Mean 6 SEM; n = 5.
*p, 0.05, **p, 0.01, ***p, 0.001.
n.s., not significant.
2822 PRIMING OF NK CELLS BY AUTOLOGOUS M1 MACROPHAGES
by guest on February 2, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
has been observed with murine DCs (26), IL-15 cis-presentationwas IFN-b dependent; as in M1-primed NK cells, its masking re-duced both the expression of IL-15Ra (Fig. 5D) and the secretionof IFN-g (Fig. 5A). Furthermore, rhIFN-b directly induced ex-pression of IL-15Ra selectively on M1-primed NK cells (Fig. 5E).Taken together, these data demonstrate that IFN-b secreted by M1triggers NK cell production of IL-15 and surface expression ofIL-15Ra, allowing a mechanism of cis-presentation that, in turn,increases the secretion of IFN-g by NK cells.
Mechanisms regulating cell-to-cell–dependent NK cell primingby autologous M1 macrophages
Because M1 polarization was confirmed to be associated with asignificant increase in IFN-b production, as well as with an in-duced expression of IL-15 and IL-15Ra (Supplemental Fig. 2)(39, 52, 53), we next investigated the role of these mediators inautologous resting NK cell priming (Fig. 1D–F). As expected giventhe pathway normally involved in the NK cell lysis of K562 cellline (Supplemental Fig. 3B), the simultaneous masking of NKp30and NKG2D significantly reduced the amount of CD107a de-granulation by NK cells incubated with either autologous M0or M1 against this target. Interestingly, the additional blocking ofIL-15Ra, but not of IFN-b, significantly further reduced the de-gree of CD107a degranulation only in NK cells cocultured withM1 and not with M0 (Fig. 6A), thus demonstrating that IL-15/IL-15Ra trans-presentation by M1 to autologous NK cells contrib-
utes to enhanced NK cell cytolytic potential. In contrast, this lattermechanism does not play any role in increasing the production ofIFN-g by NK cells cultured with autologous M1 (Fig. 6B).Finally, we analyzed whether M1 polarization was associated
with the induction of ligands for aNKRs, focusing our attentionon the 2B4-CD48 pathway, which is known to be involved in reg-ulating the interaction between NK cell and macrophages (15, 16),and contributes to cytokine production by NK cells (16). Our datashow that CD48 is highly induced in M1 but not M2 macrophages(Fig. 6C), and demonstrate that the engagement of 2B4 by CD48 isfunctionally relevant in the context of M1-NK cell cross talk becausethe masking of 2B4 significantly reduced the levels of IFN-g pro-duced by NK cells only when they were incubated withM1 (Fig. 6D).
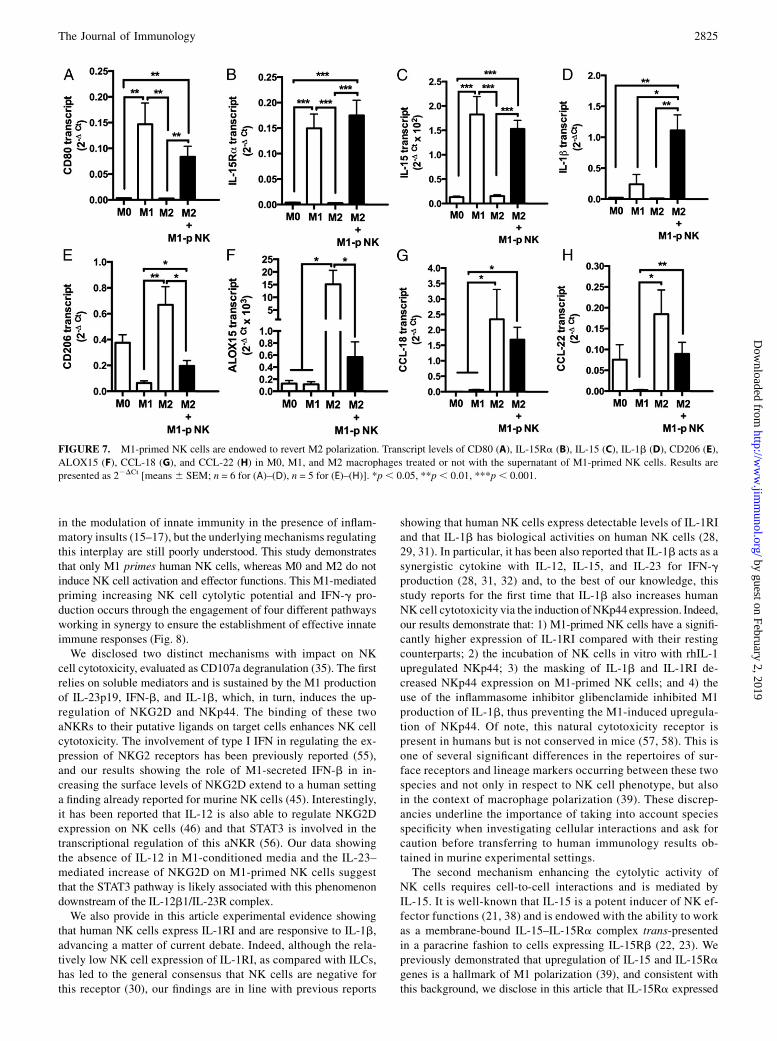
M1-primed NK cells reverse M2 macrophage polarization
Our data demonstrate that M1 macrophages activate NK cellsand induce IFN-g production. Because IFN-g plays a key role ininducing M1 polarization (7), we asked whether M1-primed NKcells could influence macrophage polarization by reverting anestablished M2 phenotype. To test this, we assessed expressionlevels of a panel of M1- and M2-associated genes on M2 macro-phages incubated with the supernatant of autologous M1-primedNK cells (M1-p NK). Our results showed that the supernatant ofM1-primed NK cells induced a significant upregulation of M1markers (CD80, IL-15Ra, IL-15, IL-1b) in M2, similar to thatdetected in M1 (Fig. 7A–D). A significant reduction of M2
FIGURE 4. M1-primed NK cells and IL-1b pathway. (A and B) Expression of IL-1RI (A) and IL-1RII (B) (gated on CD3neg CD56pos cells) on resting,
M0-primed, andM1-primedNK cells (black bars) comparedwith their isotype controls (white bars). Data are presented asMFI.Mean6 SEM; n= 5 (A) and
n = 2 (B). (C) Transcript levels of IL-1RI in resting, M0-primed, M1-primed NK cells, and M1 macrophages. Results are presented as 22DCt. Mean6 SEM;
n = 4. (D) Fluorescence microscopic images from one representative example of two different experiments from unrelated healthy donors showing the sur-
face expression of IL-1RI (green) on untreated (i.e., resting), M0-primed, and M1-primed NK cells. The blue DAPI staining indicates cell nuclei. Original
magnification360. *p, 0.05.
The Journal of Immunology 2823
by guest on February 2, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
markers in already polarized M2 was also observed, althoughthe M2 phenotype was fully reverted for some markers (CD206,
ALOX15) and only partially for others (CCL18, CCL22; Fig. 7E–
H). Taken together, these results reveal a number of different
pathways sustaining the ability of M1 to promote NK cell anti-
tumoral properties (Fig. 8) and suggest that conversely soluble
mediators released by M1-primed NK cells have the potential
to re-educate the protumoral M2 phenotype.
DiscussionBeyond being effector cells able to eliminate virally infected ortumor-transformed target cells, NK cells are also endowed withimmune-regulatory functions. Indeed, their ability to interact withother innate immunity cells, such as DCs or neutrophils, is rele-vant for the development of optimal immune responses againstpathogens and tumors (54). The cross talk between NK cells andmacrophages has also been recently reported to play a key role
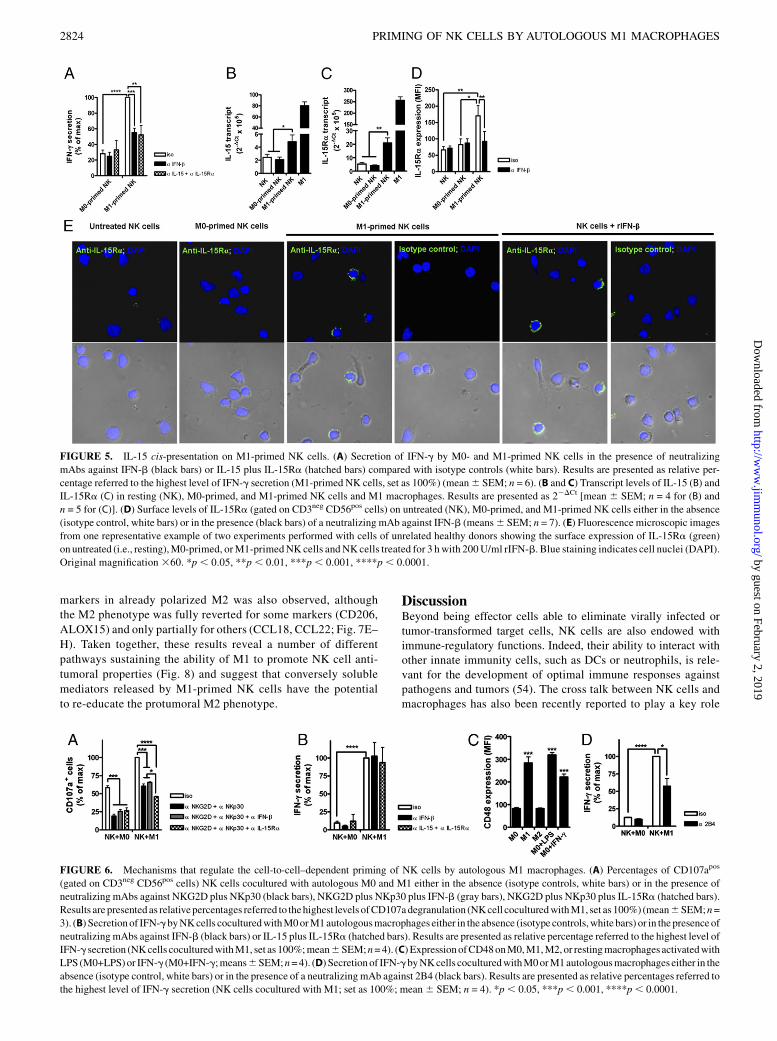
FIGURE 5. IL-15 cis-presentation on M1-primed NK cells. (A) Secretion of IFN-g by M0- and M1-primed NK cells in the presence of neutralizing
mAbs against IFN-b (black bars) or IL-15 plus IL-15Ra (hatched bars) compared with isotype controls (white bars). Results are presented as relative per-
centage referred to the highest level of IFN-g secretion (M1-primed NK cells, set as 100%) (mean6 SEM; n = 6). (B and C) Transcript levels of IL-15 (B) and
IL-15Ra (C) in resting (NK), M0-primed, and M1-primed NK cells and M1 macrophages. Results are presented as 22DCt [mean 6 SEM; n = 4 for (B) and
n = 5 for (C)]. (D) Surface levels of IL-15Ra (gated on CD3neg CD56pos cells) on untreated (NK), M0-primed, and M1-primed NK cells either in the absence
(isotype control, white bars) or in the presence (black bars) of a neutralizing mAb against IFN-b (means6 SEM; n = 7). (E) Fluorescence microscopic images
from one representative example of two experiments performed with cells of unrelated healthy donors showing the surface expression of IL-15Ra (green)
on untreated (i.e., resting),M0-primed, orM1-primedNK cells andNKcells treated for 3 hwith 200U/ml rIFN-b. Blue staining indicates cell nuclei (DAPI).
Original magnification 360. *p , 0.05, **p , 0.01, ***p , 0.001, ****p , 0.0001.
FIGURE 6. Mechanisms that regulate the cell-to-cell–dependent priming of NK cells by autologous M1 macrophages. (A) Percentages of CD107apos
(gated on CD3neg CD56pos cells) NK cells cocultured with autologous M0 and M1 either in the absence (isotype controls, white bars) or in the presence of
neutralizing mAbs against NKG2D plus NKp30 (black bars), NKG2D plus NKp30 plus IFN-b (gray bars), NKG2D plus NKp30 plus IL-15Ra (hatched bars).
Results are presented as relativepercentages referred to thehighest levels ofCD107adegranulation (NKcell coculturedwithM1, set as 100%) (mean6SEM;n=
3). (B) Secretionof IFN-gbyNKcells coculturedwithM0orM1autologousmacrophages either in the absence (isotype controls,white bars) or in thepresence of
neutralizingmAbs against IFN-b (black bars) or IL-15 plus IL-15Ra (hatched bars). Results are presented as relative percentage referred to the highest level of
IFN-g secretion (NKcells coculturedwithM1, set as 100%;mean6SEM;n=4). (C) ExpressionofCD48onM0,M1,M2,or restingmacrophages activatedwith
LPS (M0+LPS)or IFN-g (M0+IFN-g;means6SEM;n=4). (D) Secretionof IFN-gbyNKcells coculturedwithM0orM1autologousmacrophages either in the
absence (isotype control, white bars) or in the presence of a neutralizingmAb against 2B4 (black bars). Results are presented as relative percentages referred to
the highest level of IFN-g secretion (NK cells cocultured with M1; set as 100%; mean6 SEM; n = 4). *p, 0.05, ***p, 0.001, ****p, 0.0001.
2824 PRIMING OF NK CELLS BY AUTOLOGOUS M1 MACROPHAGES
by guest on February 2, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
in the modulation of innate immunity in the presence of inflam-matory insults (15–17), but the underlying mechanisms regulatingthis interplay are still poorly understood. This study demonstratesthat only M1 primes human NK cells, whereas M0 and M2 do notinduce NK cell activation and effector functions. This M1-mediatedpriming increasing NK cell cytolytic potential and IFN-g pro-duction occurs through the engagement of four different pathwaysworking in synergy to ensure the establishment of effective innateimmune responses (Fig. 8).We disclosed two distinct mechanisms with impact on NK
cell cytotoxicity, evaluated as CD107a degranulation (35). The firstrelies on soluble mediators and is sustained by the M1 productionof IL-23p19, IFN-b, and IL-1b, which, in turn, induces the up-regulation of NKG2D and NKp44. The binding of these twoaNKRs to their putative ligands on target cells enhances NK cellcytotoxicity. The involvement of type I IFN in regulating the ex-pression of NKG2 receptors has been previously reported (55),and our results showing the role of M1-secreted IFN-b in in-creasing the surface levels of NKG2D extend to a human settinga finding already reported for murine NK cells (45). Interestingly,it has been reported that IL-12 is also able to regulate NKG2Dexpression on NK cells (46) and that STAT3 is involved in thetranscriptional regulation of this aNKR (56). Our data showingthe absence of IL-12 in M1-conditioned media and the IL-23–mediated increase of NKG2D on M1-primed NK cells suggestthat the STAT3 pathway is likely associated with this phenomenondownstream of the IL-12b1/IL-23R complex.We also provide in this article experimental evidence showing
that human NK cells express IL-1RI and are responsive to IL-1b,advancing a matter of current debate. Indeed, although the rela-tively low NK cell expression of IL-1RI, as compared with ILCs,has led to the general consensus that NK cells are negative forthis receptor (30), our findings are in line with previous reports
showing that human NK cells express detectable levels of IL-1RIand that IL-1b has biological activities on human NK cells (28,29, 31). In particular, it has been also reported that IL-1b acts as asynergistic cytokine with IL-12, IL-15, and IL-23 for IFN-gproduction (28, 31, 32) and, to the best of our knowledge, thisstudy reports for the first time that IL-1b also increases humanNK cell cytotoxicity via the induction ofNKp44 expression. Indeed,our results demonstrate that: 1) M1-primed NK cells have a signifi-cantly higher expression of IL-1RI compared with their restingcounterparts; 2) the incubation of NK cells in vitro with rhIL-1upregulated NKp44; 3) the masking of IL-1b and IL-1RI de-creased NKp44 expression on M1-primed NK cells; and 4) theuse of the inflammasome inhibitor glibenclamide inhibited M1production of IL-1b, thus preventing the M1-induced upregula-tion of NKp44. Of note, this natural cytotoxicity receptor ispresent in humans but is not conserved in mice (57, 58). This isone of several significant differences in the repertoires of sur-face receptors and lineage markers occurring between these twospecies and not only in respect to NK cell phenotype, but alsoin the context of macrophage polarization (39). These discrep-ancies underline the importance of taking into account speciesspecificity when investigating cellular interactions and ask forcaution before transferring to human immunology results ob-tained in murine experimental settings.The second mechanism enhancing the cytolytic activity of
NK cells requires cell-to-cell interactions and is mediated byIL-15. It is well-known that IL-15 is a potent inducer of NK ef-fector functions (21, 38) and is endowed with the ability to workas a membrane-bound IL-15–IL-15Ra complex trans-presentedin a paracrine fashion to cells expressing IL-15Rb (22, 23). Wepreviously demonstrated that upregulation of IL-15 and IL-15Ragenes is a hallmark of M1 polarization (39), and consistent withthis background, we disclose in this article that IL-15Ra expressed
FIGURE 7. M1-primed NK cells are endowed to revert M2 polarization. Transcript levels of CD80 (A), IL-15Ra (B), IL-15 (C), IL-1b (D), CD206 (E),
ALOX15 (F), CCL-18 (G), and CCL-22 (H) in M0, M1, and M2 macrophages treated or not with the supernatant of M1-primed NK cells. Results are
presented as 22DCt [means 6 SEM; n = 6 for (A)–(D), n = 5 for (E)–(H)]. *p, 0.05, **p, 0.01, ***p, 0.001.
The Journal of Immunology 2825
by guest on February 2, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

by M1 trans-presents IL-15 to surrounding NK cells, thus en-hancing their cytotoxicity properties. Different from IFN-b andIL-1b, which selectively upregulate specific aNKRs, this IL-15trans-presentation pathway might favor NK cell cytotoxicityby increasing intracellular levels of granzyme B (59).Our study also provides experimental evidence of two addi-
tional pathways involved in the M1-induced NK cell secretion ofIFN-g. The first mechanism relies on the ability of M1-polarizedmacrophages to synthetize IFN-b (45). We demonstrate thatNK cells respond to IFN-b by increasing their surface expressionof IL-15Ra and their synthesis of IL-15. This allows autocrineIL-15 cis-presentation on NK cells, which, in turn, enhances theirproduction of IFN-g. These results are in line with a recent reportinvestigating the DC–NK cell cross talk in the murine setting(26). The second cell contact–dependent mechanism enhancingIFN-g production by NK cells relies on the activation of theCD48-2B4 pathway (15, 16, 20). We show in this article that thisbinding occurs only in the presence of M1 polarization. In fact,although NK cells constitutively express high levels of 2B4,M0 and M2 are negative for the expression of its natural ligandCD48. It has been reported that the engagement of the CD48-2B4pathway induces to the activation of FYNT, which, in turn, inducesthe NK cell production of IFN-g (60), thus further supporting ourfindings showing the importance of the CD48–2B4–IFN-g axisin the context of NK cell–macrophage cross talk.The M1-induced NK cell production of IFN-g likely plays a role
in the context of rescuing M2 toward a proinflammatory M1 profileto boost and optimize innate immune responses against infections.Indeed, a defective or aberrant NK cell–M1 cross talk might favorviral or bacterial infections, as reported in HIV-1–infected patients.Indeed, the reported NK cell impairment in the clearance of autol-ogous HIV-1–infected CD4pos T cells, in the killing of cell targets
either tumor-transformed or infected by opportunistic pathogens,in the production of antiviral cytokines/chemokines (includingIFN-g), and in the interaction with autologous DCs explain, atleast in part, HIV-1 escape from innate immune responses of thehost (5). These pathologic NK cell effector functions are directlyassociatedwith the expansion of a highly anergic CD56neg/CD16pos
(CD56neg) NK cell subset, whose frequency is very low in healthyindividuals (35, 37). Moreover, the presence of these aberrantCD56neg NK cells is also paralleled by a preferential expansion ofM2 in AIDS patients (61). In contrast, macrophages are suscep-tible to HIV-1 infection and represent an important reservoir forthe virus (61, 62). Hence it is conceivable to hypothesize that thephysiopathology of NK cells and macrophages in HIV-1 infectionis associated with a dysfunctional M1–NK cell cross talk. Thisstudy sheds new light on the mechanisms required for an effectiveNK cell–M1 interplay, and therefore paves the groundwork tobetter understanding and breaks this vicious cycle to overcomethe defective host immune responses against HIV-1.Both NK cells and macrophages are also present in tumor
stroma and are naturally endowed with antitumoral activities(1, 9). However, under the effect of tumor-derived signals, macro-phages acquire a protumoral M2-like functional profile, whichnot only develops protumoral functions (63) but also, as shown inthis study, compromises their priming effects on NK cells. Thus,acting on TAMs, the tumor microenvironment interferes with theantitumoral potential of both cell types. Several approaches havebeen proposed to target TAMs therapeutically, including blockingtheir homing to tumors (64) or depleting them in situ (65). Asrecently demonstrated with CD40 agonists in pancreatic cancer(66), TAM reprogramming to M1-like phenotypes represents apromising approach that not only detracts the tumor from its pro-tumoral activities but also converts them into efficient antitumor
FIGURE 8. Macrophage–NK cell interactions. Diagram summarizes the cellular pathways that regulate the cross talk between human NK cells
and M1 macrophages. First, polarization to M1 induces CD48 expression on macrophages, whose binding to 2B4 triggers NK cell production of IFN-g
(cellular interaction, upper left). Second, M1 secretion of IFN-b induces NK cell production and surface expression of IL-15 and IL-15Ra, respectively.
In turn, the IL-15–IL-15Ramembrane-bound complex is cis-presented in an autocrine loop to the same NK cell expressing the IL-15Rb–IL-15Rg receptor
complex, thus enhancing NK cell production of IFN-g (soluble interaction, upper right). Third, M1 secretion of IL-1b and IFN-b or IL-23 induces the
upregulation of NKp44 and NKG2D, respectively, that, in turn, increases NK cell cytotoxicity against target cells expressing their putative ligands (soluble
interaction, lower right). Fourth, the IL-15–IL-15Ra membrane-bound complex expressed by M1 is trans-presented in a paracrine loop to surrounding
NK cells expressing the IL-15Rb–IL-15Rg receptor complex and induces NK cell activation and cytotoxicity (cellular interaction, lower left).
2826 PRIMING OF NK CELLS BY AUTOLOGOUS M1 MACROPHAGES
by guest on February 2, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

cell mediators (67). Our results indicate that the ability of M1 toengage a positive cross talk with NK cells improves NK cytolyticpotential and increases IFN-g production, which would feedbackon macrophages and potentiate their antitumoral activity (Fig. 8).On this important matter, we also demonstrate in this article thatM1-primed NK cells are able to downmodulate the expression ofprotumoral genes (M2 markers) and to increase the expression ofantitumoral genes (M1 markers) in M2. Interestingly, the condi-tioned media from M1-primed NK cells induced in M2 the ex-pression of inflammatory genes (IL-15, IL-15Ra, IL-1b), which,in turn, sustain NK cell activation. This suggests that therapeuticapproaches able to re-educate TAM to M1-like macrophages mayalso take advantage of this positive feed-forward loop with NKcells, which should, in principle, result is setting in motion a pow-erful antitumoral immune mechanism.In summary, this study describes a complex network of cellu-
lar pathways that regulate, in the context of a human autologoussetting, cellular and soluble pathways operated by macrophages toefficiently prime resting NK cells. In particular, we disclose inthis article cytokine (IL-15, IL-1b, IFN-b) and membrane receptorpathways (CD48-2B4) working in synergy to ensure appropriatesupport from M1-polarized macrophages to NK cell–mediatedimmune responses, highly relevant with respect to tumor biologyand microbial infections. Our findings also open new insights withregard to the potential ability of M1-activated NK cells in revertingM2 polarization and boosting a preferential proinflammatory mac-rophage response.
DisclosuresThe authors have no financial conflicts of interest.
References1. Vivier, E., E. Tomasello, M. Baratin, T. Walzer, and S. Ugolini. 2008. Functions
of natural killer cells. Nat. Immunol. 9: 503–510.2. Moretta, A. 2002. Natural killer cells and dendritic cells: rendezvous in abused
tissues. Nat. Rev. Immunol. 2: 957–964.3. Moretta, A., C. Bottino, M. Vitale, D. Pende, R. Biassoni, M. C. Mingari, and
L. Moretta. 1996. Receptors for HLA class-I molecules in human natural killercells. Annu. Rev. Immunol. 14: 619–648.
4. Lanier, L. L. 2005. NK cell recognition. Annu. Rev. Immunol. 23: 225–274.5. Brunetta, E., K. L. Hudspeth, and D. Mavilio. 2010. Pathologic natural killer cell
subset redistribution in HIV-1 infection: new insights in pathophysiology andclinical outcomes. J. Leukoc. Biol. 88: 1119–1130.
6. Mills, C. D., K. Kincaid, J. M. Alt, M. J. Heilman, and A. M. Hill. 2000. M-1/M-2macrophages and the Th1/Th2 paradigm. J. Immunol. 164: 6166–6173.
7. Murray, P. J., J. E. Allen, S. K. Biswas, E. A. Fisher, D. W. Gilroy, S. Goerdt,S. Gordon, J. A. Hamilton, L. B. Ivashkiv, T. Lawrence, et al. 2014. Macrophageactivation and polarization: nomenclature and experimental guidelines. [Pub-lished erratum appears in 2014 Immunity. 41: 339–340.] Immunity 41: 14–20.
8. Mosser, D. M., and J. P. Edwards. 2008. Exploring the full spectrum of mac-rophage activation. Nat. Rev. Immunol. 8: 958–969.
9. Murray, P. J., and T. A. Wynn. 2011. Protective and pathogenic functions ofmacrophage subsets. Nat. Rev. Immunol. 11: 723–737.
10. Mantovani, A., A. Sica, S. Sozzani, P. Allavena, A. Vecchi, and M. Locati. 2004.The chemokine system in diverse forms of macrophage activation and polari-zation. Trends Immunol. 25: 677–686.
11. Biswas, S. K., and A. Mantovani. 2010. Macrophage plasticity and interactionwith lymphocyte subsets: cancer as a paradigm. Nat. Immunol. 11: 889–896.
12. Steidl, C., T. Lee, S. P. Shah, P. Farinha, G. Han, T. Nayar, A. Delaney,S. J. Jones, J. Iqbal, D. D. Weisenburger, et al. 2010. Tumor-associated mac-rophages and survival in classic Hodgkin’s lymphoma. N. Engl. J. Med. 362:875–885.
13. Mantovani, A., P. Allavena, A. Sica, and F. Balkwill. 2008. Cancer-related in-flammation. Nature 454: 436–444.
14. Noy, R., and J. W. Pollard. 2014. Tumor-associated macrophages: from mech-anisms to therapy. [Published erratum appears in 2014 Immunity. 41: 866.] Im-munity 41: 49–61.
15. Bellora, F., R. Castriconi, A. Dondero, G. Reggiardo, L. Moretta, A. Mantovani,A. Moretta, and C. Bottino. 2010. The interaction of human natural killer cellswith either unpolarized or polarized macrophages results in different functionaloutcomes. Proc. Natl. Acad. Sci. USA 107: 21659–21664.
16. Nedvetzki, S., S. Sowinski, R. A. Eagle, J. Harris, F. Vely, D. Pende,J. Trowsdale, E. Vivier, S. Gordon, and D. M. Davis. 2007. Reciprocal regulationof human natural killer cells and macrophages associated with distinct immunesynapses. Blood 109: 3776–3785.
17. Michel, T., F. Hentges, and J. Zimmer. 2012. Consequences of the crosstalkbetween monocytes/macrophages and natural killer cells. Front. Immunol. 3: 403.
18. Costantini, C., F. Calzetti, O. Perbellini, A. Micheletti, C. Scarponi, S. Lonardi,M. Pelletier, K. Schakel, G. Pizzolo, F. Facchetti, et al. 2011. Human neutrophilsinteract with both 6-sulfo LacNAc+ DC and NK cells to amplify NK-derivedIFNgamma: role of CD18, ICAM-1, and ICAM-3. Blood 117: 1677–1686.
19. Lapaque, N., T. Walzer, S. Meresse, E. Vivier, and J. Trowsdale. 2009. Inter-actions between human NK cells and macrophages in response to Salmonellainfection. J. Immunol. 182: 4339–4348.
20. Romo, N., G. Magri, A. Muntasell, G. Heredia, D. Baıa, A. Angulo, M. Guma,and M. Lopez-Botet. 2011. Natural killer cell-mediated response to humancytomegalovirus-infected macrophages is modulated by their functional polari-zation. J. Leukoc. Biol. 90: 717–726.
21. Cooper, M. A., T. A. Fehniger, and M. A. Caligiuri. 2001. The biology of humannatural killer-cell subsets. Trends Immunol. 22: 633–640.
22. Fehniger, T. A., and M. A. Caligiuri. 2001. Interleukin 15: biology and relevanceto human disease. Blood 97: 14–32.
23. Jakobisiak, M., J. Golab, and W. Lasek. 2011. Interleukin 15 as a promisingcandidate for tumor immunotherapy. Cytokine Growth Factor Rev. 22: 99–108.
24. Steel, J. C., T. A. Waldmann, and J. C. Morris. 2012. Interleukin-15 biology andits therapeutic implications in cancer. Trends Pharmacol. Sci. 33: 35–41.
25. Bergamaschi, C., J. Bear, M. Rosati, R. K. Beach, C. Alicea, R. Sowder,E. Chertova, S. A. Rosenberg, B. K. Felber, and G. N. Pavlakis. 2012. Circu-lating IL-15 exists as heterodimeric complex with soluble IL-15Ra in humanand mouse serum. Blood 120: e1–e8.
26. Zanoni, I., R. Spreafico, C. Bodio, M. Di Gioia, C. Cigni, A. Broggi, T. Gorletta,M. Caccia, G. Chirico, L. Sironi, et al. 2013. IL-15 cis presentation is requiredfor optimal NK cell activation in lipopolysaccharide-mediated inflammatoryconditions. Cell Reports 4: 1235–1249.
27. Cella, M., K. Otero, and M. Colonna. 2010. Expansion of human NK-22 cellswith IL-7, IL-2, and IL-1beta reveals intrinsic functional plasticity. Proc. Natl.Acad. Sci. USA 107: 10961–10966.
28. Hughes, T., B. Becknell, A. G. Freud, S. McClory, E. Briercheck, J. Yu, C. Mao,C. Giovenzana, G. Nuovo, L. Wei, et al. 2010. Interleukin-1beta selectivelyexpands and sustains interleukin-22+ immature human natural killer cells insecondary lymphoid tissue. Immunity 32: 803–814.
29. Glatzer, T., M. Killig, J. Meisig, I. Ommert, M. Luetke-Eversloh, M. Babic,D. Paclik, N. Bl€uthgen, R. Seidl, C. Seifarth, et al. 2013. RORgt⁺ innate lym-phoid cells acquire a proinflammatory program upon engagement of the acti-vating receptor NKp44. Immunity 38: 1223–1235.
30. Spits, H., D. Artis, M. Colonna, A. Diefenbach, J. P. Di Santo, G. Eberl,S. Koyasu, R. M. Locksley, A. N. McKenzie, R. E. Mebius, et al. 2013. Innatelymphoid cells–a proposal for uniform nomenclature. Nat. Rev. Immunol. 13:145–149.
31. Cooper, M. A., T. A. Fehniger, A. Ponnappan, V. Mehta, M. D. Wewers, andM. A. Caligiuri. 2001. Interleukin-1beta costimulates interferon-gamma pro-duction by human natural killer cells. Eur. J. Immunol. 31: 792–801.
32. van de Wetering, D., R. A. de Paus, J. T. van Dissel, and E. van de Vosse. 2009.Salmonella induced IL-23 and IL-1beta allow for IL-12 production by mono-cytes and Mphi1 through induction of IFN-gamma in CD56 NK/NK-like T cells.PLoS One 4: e8396.
33. Gupta, N., J. Arthos, P. Khazanie, T. D. Steenbeke, N. M. Censoplano,E. A. Chung, C. C. Cruz, M. A. Chaikin, M. Daucher, S. Kottilil, et al. 2005.Targeted lysis of HIV-infected cells by natural killer cells armed and triggered bya recombinant immunoglobulin fusion protein: implications for immunotherapy.Virology 332: 491–497.
34. Solinas, G., S. Schiarea, M. Liguori, M. Fabbri, S. Pesce, L. Zammataro,F. Pasqualini, M. Nebuloni, C. Chiabrando, A. Mantovani, and P. Allavena. 2010.Tumor-conditioned macrophages secrete migration-stimulating factor: a newmarker for M2-polarization, influencing tumor cell motility. J. Immunol. 185:642–652.
35. Brunetta, E., M. Fogli, S. Varchetta, L. Bozzo, K. L. Hudspeth, E. Marcenaro,A. Moretta, and D. Mavilio. 2009. The decreased expression of Siglec-7 rep-resents an early marker of dysfunctional natural killer-cell subsets associatedwith high levels of HIV-1 viremia. Blood 114: 3822–3830.
36. Alter, G., J. M. Malenfant, and M. Altfed. 2004. CD107a as a functional marker forthe identification of natural killer cell activity. J. Immunol. Methods 294: 15–22.
37. Mavilio, D., J. Benjamin, M. Daucher, G. Lombardo, S. Kottilil, M. A. Planta,E. Marcenaro, C. Bottino, L. Moretta, A. Moretta, and A. S. Fauci. 2003. Naturalkiller cells in HIV-1 infection: dichotomous effects of viremia on inhibitory andactivating receptors and their functional correlates. Proc. Natl. Acad. Sci. USA100: 15011–15016.
38. Varchetta, S., B. Oliviero, D. Mavilio, and M. U. Mondelli. 2013. Differentcombinations of cytokines and activating receptor stimuli are required for humannatural killer cell functional diversity. Cytokine 62: 58–63.
39. Martinez, F. O., S. Gordon, M. Locati, and A. Mantovani. 2006. Transcriptionalprofiling of the human monocyte-to-macrophage differentiation and polarization:new molecules and patterns of gene expression. J. Immunol. 177: 7303–7311.
40. Biswas, S. K., P. Allavena, and A. Mantovani. 2013. Tumor-associated macro-phages: functional diversity, clinical significance, and open questions. Semin.Immunopathol. 35: 585–600.
41. Bernink, J. H., C. P. Peters, M. Munneke, A. A. te Velde, S. L. Meijer, K. Weijer,H. S. Hreggvidsdottir, S. E. Heinsbroek, N. Legrand, C. J. Buskens, et al. 2013.Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues.Nat. Immunol. 14: 221–229.
42. Garlanda, C., F. Riva, E. Bonavita, and A. Mantovani. 2013. Negative regulatoryreceptors of the IL-1 family. Semin. Immunol. 25: 408–415.
The Journal of Immunology 2827
by guest on February 2, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

43. Scotton, C. J., F. O. Martinez, M. J. Smelt, M. Sironi, M. Locati, A. Mantovani,and S. Sozzani. 2005. Transcriptional profiling reveals complex regulation of themonocyte IL-1 beta system by IL-13. J. Immunol. 174: 834–845.
44. Wehner, R., A. Bitterlich, N. Meyer, A. Kloss, K. Schakel, M. Bachmann, andM. Schmitz. 2013. Impact of chemotherapeutic agents on the immunostimula-tory properties of human 6-sulfo LacNAc+ (slan) dendritic cells. Int. J. Cancer132: 1351–1359.
45. Zhou, Z., C. Zhang, J. Zhang, and Z. Tian. 2012. Macrophages help NK cells toattack tumor cells by stimulatory NKG2D ligand but protect themselves fromNK killing by inhibitory ligand Qa-1. PLoS One 7: e36928.
46. Zhang, C., J. Zhang, J. Niu, Z. Zhou, J. Zhang, and Z. Tian. 2008. Interleukin-12improves cytotoxicity of natural killer cells via upregulated expression ofNKG2D. Hum. Immunol. 69: 490–500.
47. Zhu, S., P. V. Phatarpekar, C. J. Denman, V. V. Senyukov, S. S. Somanchi,H. T. Nguyen-Jackson, E. M. Mace, A. F. Freeman, S. S. Watowich, J. S. Orange,et al. 2014. Transcription of the activating receptor NKG2D in natural killer cellsis regulated by STAT3 tyrosine phosphorylation. Blood 124: 403–411.
48. Bae, D. S., Y. K. Hwang, and J. K. Lee. 2012. Importance of NKG2D-NKG2Dligands interaction for cytolytic activity of natural killer cell. Cell. Immunol. 276:122–127.
49. Baychelier, F., A. Sennepin, M. Ermonval, K. Dorgham, P. Debre, andV. Vieillard. 2013. Identification of a cellular ligand for the natural cytotoxicityreceptor NKp44. Blood 122: 2935–2942.
50. Byrd, A., S. C. Hoffmann, M. Jarahian, F. Momburg, and C. Watzl. 2007. Ex-pression analysis of the ligands for the Natural Killer cell receptors NKp30 andNKp44. PLoS One 2: e1339.
51. Horng, T., J. S. Bezbradica, and R. Medzhitov. 2007. NKG2D signaling is coupledto the interleukin 15 receptor signaling pathway. Nat. Immunol. 8: 1345–1352.
52. Tamassia, N., V. Le Moigne, F. Calzetti, M. Donini, S. Gasperini, T. Ear,A. Cloutier, F. O. Martinez, M. Fabbri, M. Locati, et al. 2007. The MyD88-independent pathway is not mobilized in human neutrophils stimulated viaTLR4. J. Immunol. 178: 7344–7356.
53. Yang, H. T., Y. Wang, X. Zhao, E. Demissie, S. Papoutsopoulou, A. Mambole,A. O’Garra, M. F. Tomczak, S. E. Erdman, J. G. Fox, et al. 2011. NF-kB1inhibits TLR-induced IFN-b production in macrophages through TPL-2-dependent ERK activation. J. Immunol. 186: 1989–1996.
54. Hudspeth, K., E. Pontarini, P. Tentorio, M. Cimino, M. Donadon, G. Torzilli,E. Lugli, S. Della Bella, M. E. Gershwin, and D. Mavilio. 2013. The role ofnatural killer cells in autoimmune liver disease: a comprehensive review. J.Autoimmun. 46: 55–65.
55. Zhang, C., J. Zhang, R. Sun, J. Feng, H. Wei, and Z. Tian. 2005. Opposing effectof IFNgamma and IFNalpha on expression of NKG2 receptors: negative regu-lation of IFNgamma on NK cells. Int. Immunopharmacol. 5: 1057–1067.
56. Goriely, S., M. F. Neurath, and M. Goldman. 2008. How microorganisms tip thebalance between interleukin-12 family members. Nat. Rev. Immunol. 8: 81–86.
57. Hayakawa, Y., N. D. Huntington, S. L. Nutt, and M. J. Smyth. 2006. Functionalsubsets of mouse natural killer cells. Immunol. Rev. 214: 47–55.
58. Moretta, A., C. Bottino, M. Vitale, D. Pende, C. Cantoni, M. C. Mingari,R. Biassoni, and L. Moretta. 2001. Activating receptors and coreceptors involvedin human natural killer cell-mediated cytolysis. Annu. Rev. Immunol. 19: 197–223.
59. Mortier, E., R. Advincula, L. Kim, S. Chmura, J. Barrera, B. Reizis,B. A. Malynn, and A. Ma. 2009. Macrophage- and dendritic-cell-derivedinterleukin-15 receptor alpha supports homeostasis of distinct CD8+ T cellsubsets. Immunity 31: 811–822.
60. Veillette, A. 2006. Immune regulation by SLAM family receptors and SAP-related adaptors. Nat. Rev. Immunol. 6: 56–66.
61. Herbein, G., and A. Varin. 2010. The macrophage in HIV-1 infection: fromactivation to deactivation? Retrovirology 7: 33.
62. Varchetta, S., P. Lusso, K. Hudspeth, J. Mikulak, D. Mele, S. Paolucci,R. Cimbro, M. Malnati, A. Riva, R. Maserati, et al. 2013. Sialic acid-binding Ig-like lectin-7 interacts with HIV-1 gp120 and facilitates infection of CD4posT cells and macrophages. Retrovirology 10: 154.
63. Hanahan, D., and L. M. Coussens. 2012. Accessories to the crime: functions ofcells recruited to the tumor microenvironment. Cancer Cell 21: 309–322.
64. Qian, B. Z., J. Li, H. Zhang, T. Kitamura, J. Zhang, L. R. Campion, E. A. Kaiser,L. A. Snyder, and J. W. Pollard. 2011. CCL2 recruits inflammatory monocytes tofacilitate breast-tumour metastasis. Nature 475: 222–225.
65. Ries, C. H., M. A. Cannarile, S. Hoves, J. Benz, K. Wartha, V. Runza, F. Rey-Giraud,L. P. Pradel, F. Feuerhake, I. Klaman, et al. 2014. Targeting tumor-associatedmacrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy.Cancer Cell 25: 846–859.
66. Beatty, G. L., E. G. Chiorean, M. P. Fishman, B. Saboury, U. R. Teitelbaum,W. Sun, R. D. Huhn, W. Song, D. Li, L. L. Sharp, et al. 2011. CD40 agonistsalter tumor stroma and show efficacy against pancreatic carcinoma in mice andhumans. Science 331: 1612–1616.
67. Hagemann, T., T. Lawrence, I. McNeish, K. A. Charles, H. Kulbe,R. G. Thompson, S. C. Robinson, and F. R. Balkwill. 2008. “Re-educating”tumor-associated macrophages by targeting NF-kappaB. J. Exp. Med. 205:1261–1268.
2828 PRIMING OF NK CELLS BY AUTOLOGOUS M1 MACROPHAGES
by guest on February 2, 2019http://w
ww
.jimm
unol.org/D
ownloaded from








![Ricerca del decadimento X(3872) J= ! nell’esperimento LHCb al CERN 1 Exotic charmonia: ... (3872) !J= ˇ+ˇ rules out all the JPC assignments except for 1++ and2 +,ascanbeseeninFigure1.3[16].](https://static.fdocument.org/doc/165x107/611830c7783de947ee64efaa/ricerca-del-decadimento-x3872-j-nellaesperimento-lhcb-al-1-exotic-charmonia.jpg)









