
Inhibition of p38 mitogen-activated protein kinase attenuates interleukin-1β-induced thermal...
11
Inhibition of p38 mitogen-activated protein kinase attenuates interleukin-1b-induced thermal hyperalgesia and inducible nitric oxide synthase expression in the spinal cord Chun-Sung Sung,* , Zhi-Hong Wen,à , § Wen-Kuei Chang, Kwok-Hon Chan, Shung-Tai Ho,à Shen-Kou Tsai, Yi-Chen Chang,à , § and Chih-Shung Wongà *Graduate Institute of Medical Sciences, National Defense Medical Center, Taipei, Taiwan Department of Anesthesiology, Veterans General Hospital-Taipei and School of Medicine, National Yang-Ming University, Taipei, Taiwan àDepartment of Anesthesiology, National Defense Medical Center, and Tri-Service General Hospital, Taipei, Taiwan §Shin Kong Wu Ho-Su Memorial Hospital, Department of Education and Research, Taipei, Taiwan Abstract We have reported recently that intrathecal (i.t.) injection of interleukin-1b (IL-1b), at a dose of 100 ng, induces inducible nitric oxide synthase (iNOS) expression and nitric oxide (NO) production in the spinal cord and results in thermal hyper- algesia in rats. This study further examines the role of mi- togen-activated protein kinase (MAPK) in i.t. IL-1b-mediated iNOS–NO cascade in spinal nociceptive signal transduction. All rats were implanted with an i.t. catheter either with or without an additional microdialysis probe. Paw withdrawal latency to radiant heat is used to assess thermal hyperal- gesia. The iNOS and MAPK protein expression in the spinal cord dorsal horn were examined by western blot. The [NO] in CSF dialysates were also measured. Intrathecal IL-1b leads to a time-dependent up-regulation of phosphorylated p38 (p-p38) MAPK protein expression in the spinal cord 30– 240 min following IL-1b injection (i.t.). However, neither the phosphorylated extracellular signal-regulated kinase (p-ERK) nor phosphorylated c-Jun NH2-terminal kinase (p-JNK) was affected. The total amount of p38, ERK, and JNK MAPK proteins were not affected following IL-1b injection. Intrathecal administration of either selective p38 MAPK, or JNK, or ERK inhibitor alone did not affect the thermal noci- ceptive threshold or iNOS protein expression in the spinal cord. However, pretreatment with a p38 MAPK inhibitor significantly reduced the IL-1b-induced p-p38 MAPK expression by 38–49%, and nearly completely blocked the subsequent iNOS expression (reduction by 86.6%), NO production, and thermal hyperalgesia. In contrast, both ERK and JNK inhibitor pretreatments only partially ( 50%) inhibited the IL-1b-induced iNOS expression in the spinal cord. Our results suggest that p38 MAPK plays a pivotal role in i.t. IL-1b-induced spinal sensitization and nociceptive sig- nal transduction. Keywords: interleukin-1, mitogen-activated protein kinase; nitric oxide synthase, nitric oxide, spinal cord, thermal hyper- algesia. J. Neurochem. (2005) 94, 742–752. Received November 8, 2004; revised manuscript received March 10, 2005; accepted April 4, 2005. Address correspondence and reprint requests to Chih-Shung Wong, Department of Anesthesiology, Tri-Service General Hospital and National Defense Medical Center, #325, Sec. 2, Chenggung Road, Neihu 114, Taipei, Taiwan. E-mail: [email protected] Abbreviations used: aCSF, artificial cerebrospinal fluid; CSF, cere- brospinal fluid; ERK, extracellular signal-regulated kinase; ERKI, extracellular signal-regulated kinase inhibitor; IL-lb, interleukin-lb; IL- lra, interleukin-1 receptor antagonist; iNOS, inducible nitric oxide syn- thase; i.t., intrathecal; JNK, c-Jun NH 2 -terminaI kinase; JNKI, c-Jun NH 2 -terminal kinase inhibitor; MAPK, mitogen-activated protein kinase; NO, nitric oxide; p-ERK, phosphorylated extracellular signal- regulated kinase; p-JNK, phosphorylated c-Jun NH 2 -terminal kinase; p-p38, phosphorylated p38; PWL, paw Withdrawal latency; SB, SB203580. Journal of Neurochemistry , 2005, 94, 742–752 doi:10.1111/j.1471-4159.2005.03226.x 742 Ó 2005 International Society for Neurochemistry, J. Neurochem. (2005) 94, 742–752
-
Upload
chun-sung-sung -
Category
Documents
-
view
213 -
download
1
Transcript of Inhibition of p38 mitogen-activated protein kinase attenuates interleukin-1β-induced thermal...

Inhibition of p38 mitogen-activated protein kinase attenuatesinterleukin-1b-induced thermal hyperalgesia and inducible nitricoxide synthase expression in the spinal cord
Chun-Sung Sung,*,� Zhi-Hong Wen,�,§ Wen-Kuei Chang,� Kwok-Hon Chan,� Shung-Tai Ho,�Shen-Kou Tsai,� Yi-Chen Chang,�,§ and Chih-Shung Wong�
*Graduate Institute of Medical Sciences, National Defense Medical Center, Taipei, Taiwan
�Department of Anesthesiology, Veterans General Hospital-Taipei and School of Medicine, National Yang-Ming University, Taipei,
Taiwan
�Department of Anesthesiology, National Defense Medical Center, and Tri-Service General Hospital, Taipei, Taiwan
§Shin Kong Wu Ho-Su Memorial Hospital, Department of Education and Research, Taipei, Taiwan
Abstract
We have reported recently that intrathecal (i.t.) injection of
interleukin-1b (IL-1b), at a dose of 100 ng, induces inducible
nitric oxide synthase (iNOS) expression and nitric oxide (NO)
production in the spinal cord and results in thermal hyper-
algesia in rats. This study further examines the role of mi-
togen-activated protein kinase (MAPK) in i.t. IL-1b-mediated
iNOS–NO cascade in spinal nociceptive signal transduction.
All rats were implanted with an i.t. catheter either with or
without an additional microdialysis probe. Paw withdrawal
latency to radiant heat is used to assess thermal hyperal-
gesia. The iNOS and MAPK protein expression in the spinal
cord dorsal horn were examined by western blot. The [NO] in
CSF dialysates were also measured. Intrathecal IL-1b leads
to a time-dependent up-regulation of phosphorylated p38
(p-p38) MAPK protein expression in the spinal cord 30–
240 min following IL-1b injection (i.t.). However, neither the
phosphorylated extracellular signal-regulated kinase (p-ERK)
nor phosphorylated c-Jun NH2-terminal kinase (p-JNK) was
affected. The total amount of p38, ERK, and JNK MAPK
proteins were not affected following IL-1b injection.
Intrathecal administration of either selective p38 MAPK, or
JNK, or ERK inhibitor alone did not affect the thermal noci-
ceptive threshold or iNOS protein expression in the spinal
cord. However, pretreatment with a p38 MAPK inhibitor
significantly reduced the IL-1b-induced p-p38 MAPK
expression by 38–49%, and nearly completely blocked the
subsequent iNOS expression (reduction by 86.6%), NO
production, and thermal hyperalgesia. In contrast, both ERK
and JNK inhibitor pretreatments only partially (� 50%)
inhibited the IL-1b-induced iNOS expression in the spinal
cord. Our results suggest that p38 MAPK plays a pivotal role
in i.t. IL-1b-induced spinal sensitization and nociceptive sig-
nal transduction.
Keywords: interleukin-1, mitogen-activated protein kinase;
nitric oxide synthase, nitric oxide, spinal cord, thermal hyper-
algesia.
J. Neurochem. (2005) 94, 742–752.
Received November 8, 2004; revised manuscript received March 10,2005; accepted April 4, 2005.Address correspondence and reprint requests to Chih-Shung Wong,
Department of Anesthesiology, Tri-Service General Hospital andNational Defense Medical Center, #325, Sec. 2, Chenggung Road, Neihu114, Taipei, Taiwan. E-mail: [email protected] used: aCSF, artificial cerebrospinal fluid; CSF, cere-
brospinal fluid; ERK, extracellular signal-regulated kinase; ERKI,
extracellular signal-regulated kinase inhibitor; IL-lb, interleukin-lb; IL-lra, interleukin-1 receptor antagonist; iNOS, inducible nitric oxide syn-thase; i.t., intrathecal; JNK, c-Jun NH2-terminaI kinase; JNKI,c-Jun NH2-terminal kinase inhibitor; MAPK, mitogen-activated proteinkinase; NO, nitric oxide; p-ERK, phosphorylated extracellular signal-regulated kinase; p-JNK, phosphorylated c-Jun NH2-terminal kinase;p-p38, phosphorylated p38; PWL, paw Withdrawal latency; SB,SB203580.
Journal of Neurochemistry, 2005, 94, 742–752 doi:10.1111/j.1471-4159.2005.03226.x
742 � 2005 International Society for Neurochemistry, J. Neurochem. (2005) 94, 742–752

IL-1b has been demonstrated to modulate pain transmission.It is upregualted in the spinal cord in both inflammatory andneuropathic pain animal models (DeLeo et al. 1997; Wanget al. 1997; Sweitzer et al. 1999; Samad et al. 2001).Intrathecal (i.t.) administration of IL-1b alone or in combi-nation with cytokines produces pain in animals (Meller et al.1994; Falchi et al. 2001), and i.t. administration of eitherIL-1 receptor antagonist (IL-1ra) or monoclonal anti-IL1 Igblocks this pain (Sommer et al. 1999; Samad et al. 2001;Chacur et al. 2004). Nitric oxide (NO) has also beendemonstrated to participate in nociceptive signaling afterinflammation, neuropathy, and trauma (Maihofner et al.2000; Omote et al. 2001). Up-regulation of nitric oxidesynthase (NOS) and increased NO production in the spinalcord lead to hyperalgesia and allodynia in rats (Wu et al.2001; Tao and Johns 2002), and blocking of NOS activityreduces the nociceptive responses in rats (Roche et al. 1996;Wu et al. 2001). In our previous study, we demonstrated thati.t. administration of IL-1b, at doses of 100, 200, and 500 ng,produced thermal hyperalgesia in rats in a dose-dependentmanner. The inducible NOS (iNOS) protein expression andNO production induced by IL-1b occurred in a time-dependent manner in the spinal cord, while i.t. pretreatmentwith iNOS inhibitor blocked the IL-1b-induced NO produc-tion and thermal hyperalgesia in the rats. It was suggestedthat the i.t. IL-1b-induced thermal hyperalgesia might bethrough activation of the iNOS–NO cascade that inducedspinal nociceptive sensitization (Sung et al. 2004).
Mitogen-activated protein kinase (MAPK) is an import-ant intracellular signaling molecule that controls cellfunction and survival in eukaryotes (Robinson and Cobb1997; Widmann et al. 1999; Johnson and Lapadat 2002).Three subfamilies of MAPKs have been identified inmammals: extracellular signal-regulated kinase (ERK),c-Jun NH2-terminal kinase (JNK), and p38 MAPK. BothJNK and p38 MAPKs are activated by inflammatoryagents and environmental stress, whereas ERK is stimu-lated by growth factors, virus infection and carcinogens(Bogoyevitch et al. 1995; Bhat et al. 1998; Aggeli et al.2001). Peripheral inflammatory, neuropathic and noxiousstimuli induce MAPK protein expression which in turnresults in nociceptive responses in rodents; these effectsare attenuated by inhibition of either ERK (Ji et al. 1999;Dai et al. 2002; Galan et al. 2003), JNK (Ma and Quirion2002), or p38 MAPK (Kim et al. 2002; Jin et al. 2003;Svensson et al. 2003a; b; Tsuda et al. 2004). These resultssuggest that ERK, p38, and JNK may play important rolesin pain signaling in the spinal cord. IL-1b has beendemonstrated to up-regulate MAPK which, in turn,activates the NOS–NO cascade, while pharmacologicalinhibition of MAPK activity or transfecting with anegative-mutant MAPK construct inhibits the IL-1b-induced iNOS–NO activation. IL-1b alone or in combina-tion with other cytokines activates p38 MAPK, which
increases iNOS expression in various cells (Da Silva et al.1997; Larsen et al. 1998; Guan et al. 1999; Xu andMalave 2000; Bhat et al. 2002; Hua et al. 2002; Jeohnet al. 2002). In addition, IL-1b was shown to activateERK and induced iNOS expression in rat ventricularmyocytes and cardiac microvascular endothelial cells, andthese effects were blocked by an ERK inhibitor (Singhet al. 1996). In contrast, p38 MAPK inhibited the IL-1b-mediated iNOS expression in rat pulmonary artery smoothmuscle and glomerular mesangial cells (Guan et al. 1997;Finder et al. 2001).
To date, there is little information about the role of MAPKactivation in i.t. IL-1b-activated spinal iNOS–NO signalingcascade and the thermal hyperalgesia development in rats.The aim of our present study was to examine the role ofspinal MAPK in the i.t. IL-1b-induced spinal sensitizationand thermal hyperalgesia.
Materials and methods
Animal preparation and intrathecal drug delivery
The treatment and use of the animals conformed to the guidelines of
the International Association for the Study of Pain (Zimmermann
1983), and were approved by the Animal Care and Use Committee
of our institute. Animal preparation and methods of construction and
implantation of the intrathecal catheter and microdialysis probe were
according to the procedure described previously (Sung et al. 2004).Briefly, Male Wistar rats (350–400 g) under chloral hydrate
anesthesia (350 mg/kg i.p.) were implanted with an i.t. catheter.
For the microdialysis study, a microdialysis probe was also
implanted. After implantation, all rats were allowed a 5-day
recovery period before use. All rats were housed individually with
ad libitum access to food and water, and maintained under a
standard 12-h light : 12-h dark cycle at room temperature (25�C).Rats with any neurological deficits were excluded from the study.
The effect of MAPKs on IL-1b-induced iNOS–NO activation and
thermal hyperalgesia was examined by i.t. injection of SB203580 (a
highly selective p38 inhibitor), (L)-JNK inhibitor 1 (a highly
selective cell-permeable JNK inhibitor), or ERK activation inhibitor
peptide II (a highly selective cell-permeable ERK inhibitor). The
dose-dependent effect of these MAPK inhibitors on the nociceptive
response in the rats were examined in our preliminary study, and
5 lg was chosen for all three MAPK inhibitors for the following
experiments.
On the sixth day after i.t. catheter implantation, rats were
assigned to one of 10 groups: (i) control group, rats received 25 lLof artificial cerebrospinal fluid (aCSF; 151.1 mM Na+, 2.6 mM K+,
122.7 mM Cl–, 21.0 mM HCO3–, 0.9 mM Mg2+, 1.3 mM Ca2+,
2.5 mM HPO42–, 3.5 mM dextrose, bubbled with 5% CO2 in 95%
O2, adjusted to pH 7.3); (ii) IL group, rats received 100 ng (2 lL) ofIL-1b, followed by 23 lL of aCSF to flush the catheter; (iii) SB
group, rats received 5 lg (5 lL) of SB203580 (a p38 MAPK
inhibitor), followed by 20 lL of aCSF to flush the catheter; (iv)
SB + IL group, rats received 5 lg (5 lL) of SB203580 and flushed
with 10 lL of aCSF 1 h before i.t. injection of IL-1b (100 ng, 2 lL)flushed with 8 lL of aCSF; (v) JNKI group, rats received 5 lg
Spinal MAPK and IL-1b-induced thermal hyperalgesia 743
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 94, 742–752

(2.5 lL) of (L)-JNK inhibitor 1 (a JNK inhibitor), followed by
22.5 lL of aCSF to flush the catheter; (vi) JNKI + IL group, rats
received 5 lg (2.5 lL) of (L)-JNKI 1 and flushed with 10.5 lL of
aCSF 1 h before i.t. injection of IL-1b (100 ng, 2 lL) flushed with
10 lL of aCSF; (vii) ERKI group, rats received 5 lg (5 lL) of ERKactivation inhibitor peptide II (an ERK inhibitor), followed by
20 lL of aCSF to flush the catheter; (viii) ERKI + IL group, rats
received 5 lg (5 lL) of ERK activation inhibitor peptide II and
flushed with 10 lL of aCSF 1 h before i.t. injection of IL-1b(100 ng, 2 lL) flushed with 8 lL of aCSF; (ix) IL-1ra group, rats
received 20 lg (39.2 lL) of IL-1 receptor antagonist (IL-1ra),
followed by 10 lL of aCSF to flush the catheter and (x) IL-1ra + IL
group, rats received 20 lg (39.2 lL) of IL-1ra and flushed with
10 lL of aCSF 1 h before IL-1b (100 ng/2 lL, i.t.) injection and
flushed with 8 lL of aCSF.
Behavioral assessment
Paw withdrawal latency (PWL) to radiant heat was used to assess
thermal hyperalgesia (Biological Research Apparatus type 7370,
Plantar Test; UGOBasile, Comerio, Italy). Rats were placed in plastic
cages on a glass platform and the heat source positioned directly
beneath the right hind paw. The infra-red heat intensity 20 at UGO
Basile plantar test apparatuswas adjusted to obtain an average PWLof
17.6 ± 0.4 s, and the cut-off time was set at 22 ± 0.4 s to prevent
tissue damage. The PWL was assessed at baseline (before i.t.
injection) and at 1, 2, 3, 4, 5, 6, 8, 10, 12, and 24 h after drug delivered.
Western blot analysis
Rats were killed at 30, 45, 60, 75, 90 min or 2, 4, or 6 h after i.t.
injection, and spinal cords were removed. The dorsal part of the
lumbar spinal cord was dissected and immediately frozen and stored
at )80�C until used. The lumbar spinal cord preparation and the
western blot analysis procedure were modified from our previous
study (Sung et al. 2004). Briefly, the spinal cord samples were
homogenized in cold lysis buffer (50 mM Tris, pH 7.5, 150 mM
NaCl, 2% Triton X-100, 0.1 mM EDTA, 0.1 mM EGTA, 100 lg/mL
of phenylmethylsulfonyl fluoride, 1 lg/mL of aprotinin, 20 mM
NaF, 0.2 mM Na3VO4). The tissue extracts were electrophoresed and
transferred to a polyvinylidene difluoride (PVDF) membrane
(Immobilon�-p, Millipore, Bedford, MA. USA). The membrane
was blocked with 5% non-fat dry milk in Tris-buffered saline
(TBST; 0.1% Tween-20, 20 mM Tris-HCl, 137 mM NaCl, pH 7.4)
for 30 min at room temperature followed by incubation with
primary antibodies for overnight at 4�C. The membrane was
washed, blocked with 5% non-fat dry milk in TTBS, and then
incubated with appropriate horseradish peroxidase-conjugated sec-
ondary antibody for 1 h at room temperature (25�C). Bound
antibody was detected by chemiluminescence (ECL; Perkin-Elmer,
Boston, MA, USA) using X-ray film (KODAK X-OMAT LS,
Kodak, Rochester, NY, USA). The PVDF membranes were stripped
(Restore� western blot stripping buffer, Pierce, Rockford, IL, USA)
and reblotted with different antibodies. The antibodies used in this
study were mouse monoclonal iNOS antibody (1 : 1000 dilution;
Transduction Laboratories, Lexington, KY, USA); rabbit polyclonal
p38 or phospho-p38 MAPK antibodies (PhosphoPlus� p38 MAP
Kinase Kit, New England BioLab, Beverly, MA, USA); rabbit
polyclonal JNK or phospho-JNK antibodies (PhosphoPlus� SAPK/
JNK Antibody Kit, New England BioLab), or rabbit polyclonal
ERK or phospho-ERK antibodies (PhosphoPlus� p42/44 MAP
Kinase Antibody Kit, New England BioLab), respectively. The
relative densities of the p-p38 MAPK bands were calculated and
normalized relative to the total p38 MAPK, and p-JNK bands were
also normalized relative to the total JNK in each sample. Similarly,
p-ERK bands were normalized relative to the total ERK in each
sample. The spinal cord b-actin blot was used as the internal control
of protein content. Densitometry was used to express the density of
the band as a relative density compared to the background.
CSF dialysates collection and NO analysis
All i.t. catheters and microdialysis probes were perfused with aCSF
once a day to keep catheter patent. On day 6, CSF dialysate
collection and NO analysis were performed as described (Sung et al.2004). In brief, one end of the microdialysis probe was connected to
a microdialysis pump (CMA102, Acton, MA, USA) for continuous
aCSF infusion, and the other end used to collect the CSF dialysate.
The microdialysis probe was perfused with aCSF (5 lL/min) and
dialysates were collected prior to each drug administration (baseline)
and at 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, 5, 5.5, 6, 8, 10, 12, and 24 h after
i.t. drugs administration. The dialysate samples were collected and
frozen to )80�C until assayed.
The total amount of NOx (NO + NO2– + NO3–) in the samples
was used to determine the NO concentration in the CSF. NOx in the
dialysates was analyzed by using a chemiluminescence detector
(NOA 280, Sievers Instruments Inc., Boulder, CO, USA). The
method was modified from those described in previous studies
(Braman and Hendrix 1989; Bush et al. 1992). The standard curve
was generated by using sodium nitrate and 0.1 M vanadium (III)
chloride, heated to 90�C in a water-jacketed purge vessel (NOA 280
component, Sievers Instruments Inc., Boulder, CO, USA) constantly
bubbled with oxygen-free nitrogen. The NOx signals from the
chemiluminescence detector were displayed and analyzed by using a
computer-based data recording and analyzing system (Sievers
Instruments Inc., Boulder, CO, USA). Background concentrations
of NOx with different agents in aCSF were measured and subtracted.
All standards and samples were analyzed in duplicate.
Chemicals
p38 inhibitor SB203580 [4-(4-fluorophenyl)-2-(4-methylsulfonyl-
phenyl)-5-(4-pyridynyl) imidazole]; (L)-JNK inhibitor 1 [(L)-HIV-
TAT48-57-PP-JBD20], and ERK activation inhibitor peptide II
(MTPTAT-G-MEK113) (Calbiochem, La Jolla, CA, USA) were
dissolved at 1 lg/lL, 2 lg/lL and 1 lg/lL, respectively, in aCSF.
Recombinant rat IL-1b (R&DSystems,Minneapolis,MN,USA)was
dissolved in aCSF (50 ng/lL). Recombinant rat IL-1ra (lot number
IHS02502A) was purchased from R&DSystems (Minneapolis, MN,
USA) and supplied at a concentration of 0.512 lg/lL.
Data analysis
Eight to 10 rats were included in each group for the thermal
hyperalgesia measurement, and eight animals per group were used
for the assessment of NO release. For western blot analysis, four
animals were used at each time point in each group. All data are
presented as mean ± SEM after normalization to the baseline values.
Differences between groups were analyzed using one-way analysis
of variance (ANOVA) followed by Tukey post hoc test. A p-value£ 0.05 was considered statistically significant.
744 C.-S. Sung et al.
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 94, 742–752
Results
Number of rats used in the experiments
In our study design, all rats were intrathecally implanted witheither an i.t. catheter only or in combination with amicrodialysis probe before the following experiments. Afterimplantation, all rats were allowed a 5-day recovery periodbefore use; meanwhile, the i.t. catheter and microdialysisprobe were perfused with aCSF once a day to keep thecatheter patent. By visual observation of daily activity andfood intake/stool passage, the intrathecally implanted ratswhich developed any abnormal behaviors (such as poorappetite, decreased stool passage, decreased daily activity,frequent vocalizations and flaccidity) and motor deficitduring the 5 days’ observation were excluded fromexperiments. The numbers of rats used in the followingexperiments were listed in Table 1. Four rats did not receiveany drug or vehicle (aCSF) treatment and were used as shamcontrols to exclude the possibility of mechanical stress-induced protein change after implantation.
The dose–behavior effect of these MAPK inhibitors onnociception in the rat was examined initially. Eight rats wereassigned to each MAPK inhibitor group at a dose of 10 lg.Eight rats were intrathecally injected with 10 lg of SB203580did not develop any abnormal behavior or motor deficit.
However, three rats developed spontaneous nociceptivebehaviors (e.g. decreased activity and flaccidity) and one ofthe three rats died 6 h after i.t. administration of 10 lg of (L)-JNK inhibitor 1 (n ¼ 8) (Table 1). Similarly, toxicity occurredin three rats and one of them died 4 h after i.t. injection with10 lg ERK activation inhibitor peptide II (n ¼ 8) (Table 1).Therefore, the dose of each MAPK inhibitor was reduced to5 lg due to potentially toxic effects at the higher dose.Fortunately, no grossly abnormal behavior or toxic effectswere observed at the dose 5 lg. The dose of 5 lg for eachselective MAPK inhibitor was chosen for all followingexperiments. The other rats were assigned randomly to eachgroup and none developed abnormal behaviors or motordeficits after i.t. injections with either aCSF or drug (Table 1).
IL-1b-activated p38 MAPK and p38 MAPK inhibition
attenuated the IL-1b-up-regulated p-p38 MAPK protein
expression in spinal cord
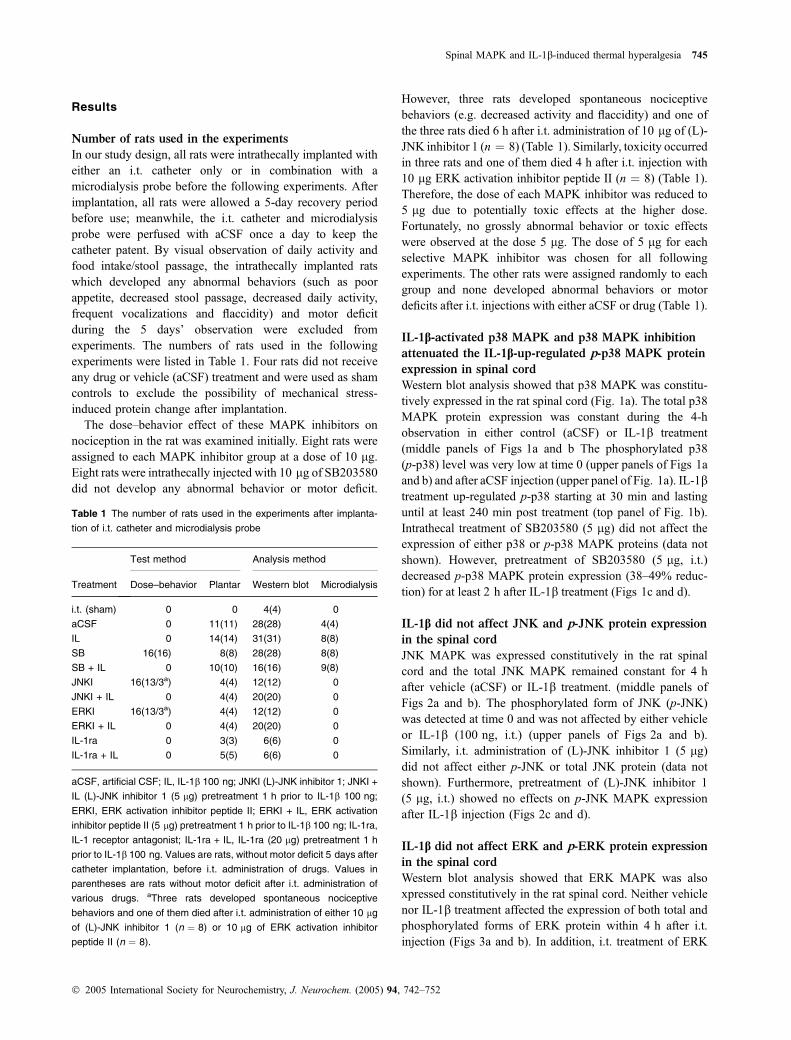
Western blot analysis showed that p38 MAPK was constitu-tively expressed in the rat spinal cord (Fig. 1a). The total p38MAPK protein expression was constant during the 4-hobservation in either control (aCSF) or IL-1b treatment(middle panels of Figs 1a and b The phosphorylated p38(p-p38) level was very low at time 0 (upper panels of Figs 1aand b) and after aCSF injection (upper panel of Fig. 1a). IL-1btreatment up-regulated p-p38 starting at 30 min and lastinguntil at least 240 min post treatment (top panel of Fig. 1b).Intrathecal treatment of SB203580 (5 lg) did not affect theexpression of either p38 or p-p38 MAPK proteins (data notshown). However, pretreatment of SB203580 (5 lg, i.t.)decreased p-p38 MAPK protein expression (38–49% reduc-tion) for at least 2 h after IL-1b treatment (Figs 1c and d).
IL-1b did not affect JNK and p-JNK protein expression
in the spinal cord
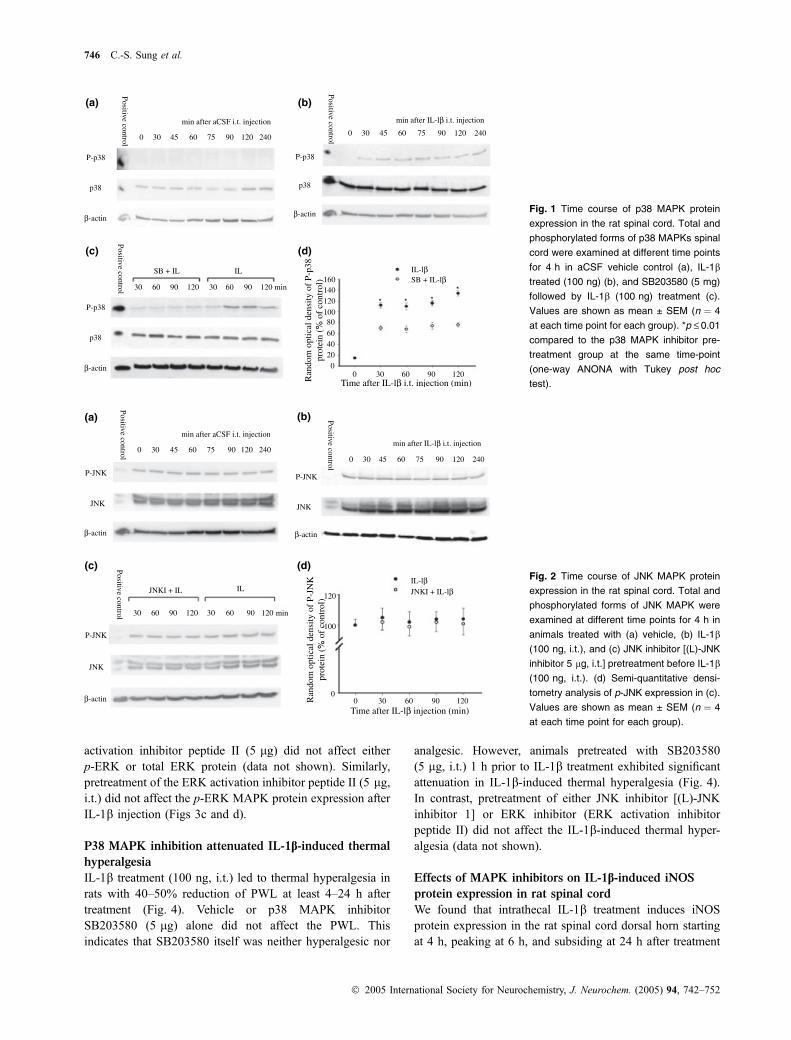
JNK MAPK was expressed constitutively in the rat spinalcord and the total JNK MAPK remained constant for 4 hafter vehicle (aCSF) or IL-1b treatment. (middle panels ofFigs 2a and b). The phosphorylated form of JNK (p-JNK)was detected at time 0 and was not affected by either vehicleor IL-1b (100 ng, i.t.) (upper panels of Figs 2a and b).Similarly, i.t. administration of (L)-JNK inhibitor 1 (5 lg)did not affect either p-JNK or total JNK protein (data notshown). Furthermore, pretreatment of (L)-JNK inhibitor 1(5 lg, i.t.) showed no effects on p-JNK MAPK expressionafter IL-1b injection (Figs 2c and d).
IL-1b did not affect ERK and p-ERK protein expression
in the spinal cord
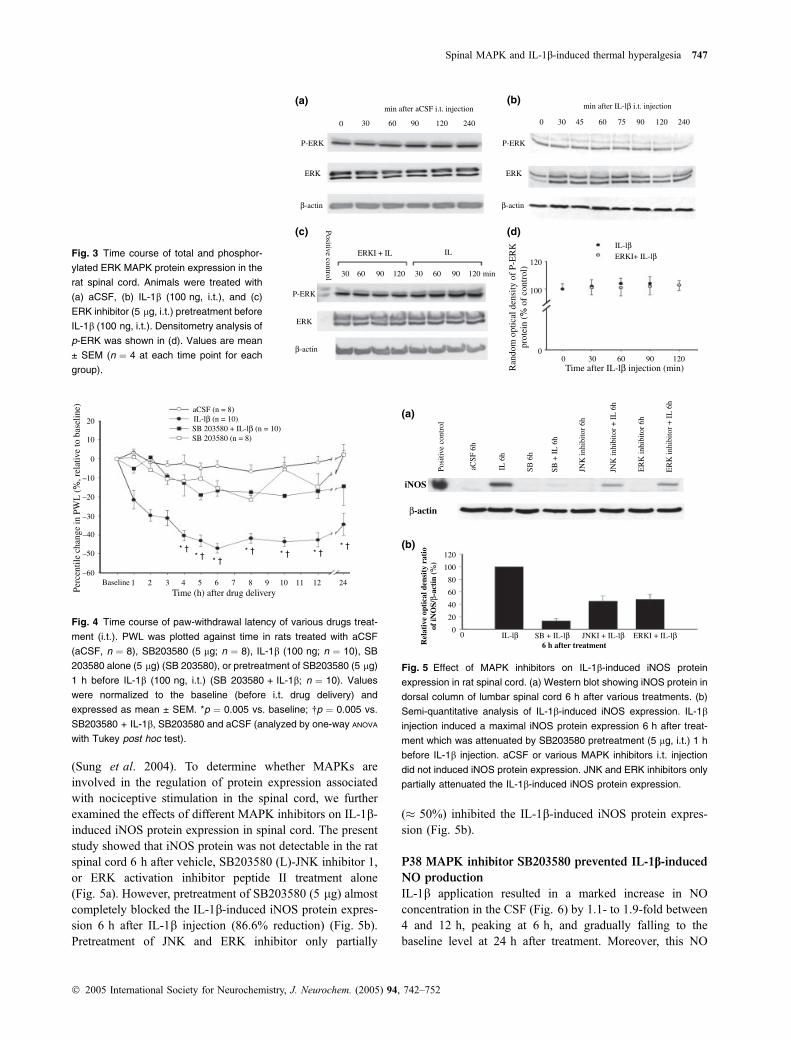
Western blot analysis showed that ERK MAPK was alsoxpressed constitutively in the rat spinal cord. Neither vehiclenor IL-1b treatment affected the expression of both total andphosphorylated forms of ERK protein within 4 h after i.t.injection (Figs 3a and b). In addition, i.t. treatment of ERK
Table 1 The number of rats used in the experiments after implanta-
tion of i.t. catheter and microdialysis probe
Treatment
Test method Analysis method
Dose–behavior Plantar Western blot Microdialysis
i.t. (sham) 0 0 4(4) 0
aCSF 0 11(11) 28(28) 4(4)
IL 0 14(14) 31(31) 8(8)
SB 16(16) 8(8) 28(28) 8(8)
SB + IL 0 10(10) 16(16) 9(8)
JNKI 16(13/3a) 4(4) 12(12) 0
JNKI + IL 0 4(4) 20(20) 0
ERKI 16(13/3a) 4(4) 12(12) 0
ERKI + IL 0 4(4) 20(20) 0
IL-1ra 0 3(3) 6(6) 0
IL-1ra + IL 0 5(5) 6(6) 0
aCSF, artificial CSF; IL, IL-1b 100 ng; JNKI (L)-JNK inhibitor 1; JNKI +
IL (L)-JNK inhibitor 1 (5 lg) pretreatment 1 h prior to IL-1b 100 ng;
ERKI, ERK activation inhibitor peptide II; ERKI + IL, ERK activation
inhibitor peptide II (5 lg) pretreatment 1 h prior to IL-1b 100 ng; IL-1ra,
IL-1 receptor antagonist; IL-1ra + IL, IL-1ra (20 lg) pretreatment 1 h
prior to IL-1b 100 ng. Values are rats, without motor deficit 5 days after
catheter implantation, before i.t. administration of drugs. Values in
parentheses are rats without motor deficit after i.t. administration of
various drugs. aThree rats developed spontaneous nociceptive
behaviors and one of them died after i.t. administration of either 10 lg
of (L)-JNK inhibitor 1 (n ¼ 8) or 10 lg of ERK activation inhibitor
peptide II (n ¼ 8).
Spinal MAPK and IL-1b-induced thermal hyperalgesia 745
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 94, 742–752
activation inhibitor peptide II (5 lg) did not affect eitherp-ERK or total ERK protein (data not shown). Similarly,pretreatment of the ERK activation inhibitor peptide II (5 lg,i.t.) did not affect the p-ERK MAPK protein expression afterIL-1b injection (Figs 3c and d).
P38 MAPK inhibition attenuated IL-1b-induced thermal
hyperalgesia
IL-1b treatment (100 ng, i.t.) led to thermal hyperalgesia inrats with 40–50% reduction of PWL at least 4–24 h aftertreatment (Fig. 4). Vehicle or p38 MAPK inhibitorSB203580 (5 lg) alone did not affect the PWL. Thisindicates that SB203580 itself was neither hyperalgesic nor
analgesic. However, animals pretreated with SB203580(5 lg, i.t.) 1 h prior to IL-1b treatment exhibited significantattenuation in IL-1b-induced thermal hyperalgesia (Fig. 4).In contrast, pretreatment of either JNK inhibitor [(L)-JNKinhibitor 1] or ERK inhibitor (ERK activation inhibitorpeptide II) did not affect the IL-1b-induced thermal hyper-algesia (data not shown).
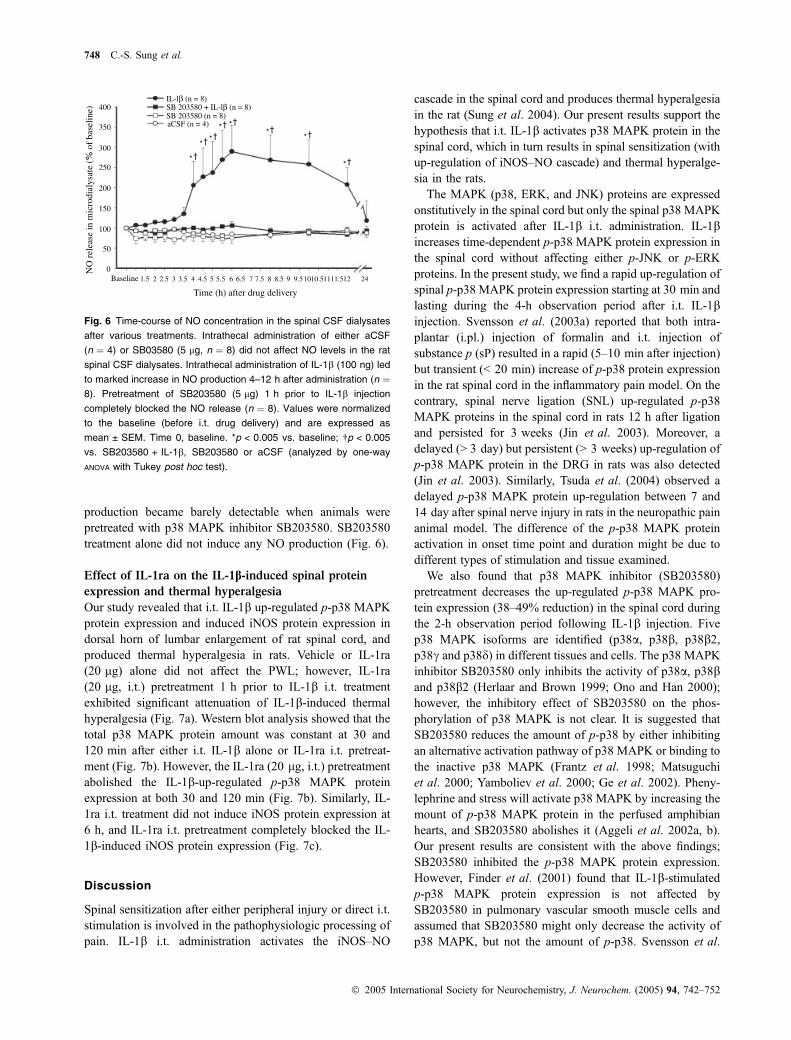
Effects of MAPK inhibitors on IL-1b-induced iNOS
protein expression in rat spinal cord
We found that intrathecal IL-1b treatment induces iNOSprotein expression in the rat spinal cord dorsal horn startingat 4 h, peaking at 6 h, and subsiding at 24 h after treatment
Positive control
0
P-JNK
JNK
JNKI + IL ILIL-lβJNKI + IL-lβ
30 45 60
min after aCSF i.t. injection
75 90 120 240min after IL-lβ i.t. injection
Positive control0 30 45 60 75 90 120 240
β-actin
P-JNK
JNK
β-actin
P-JNK
JNK
β-actin
Positive control 30 60 90 120 30 60 90 120 min
00
30Time after IL-lβ injection (min)
60 90 120
120
100
Ran
dom
opt
ical
den
sity
of
P-JN
Kpr
otei
n (%
of
cont
rol)
(a)
(c)
(b)
(d)Fig. 2 Time course of JNK MAPK protein
expression in the rat spinal cord. Total and
phosphorylated forms of JNK MAPK were
examined at different time points for 4 h in
animals treated with (a) vehicle, (b) IL-1b
(100 ng, i.t.), and (c) JNK inhibitor [(L)-JNK
inhibitor 5 lg, i.t.] pretreatment before IL-1b
(100 ng, i.t.). (d) Semi-quantitative densi-
tometry analysis of p-JNK expression in (c).
Values are shown as mean ± SEM (n ¼ 4
at each time point for each group).
(a)
(c)
(b)
(d)
Positive control 0
P-p38
p38
30 45 60
min after aCSF i.t. injection min after IL-lβ i.t. injection
IL-lβ
* * **
SB + IL-lβ
β-actin
P-p38
p38
β-actin
P-p38
p38
β-actin
75 90 120 240
Positive control 30 60
SB + IL IL
90 120 30 60 90 120160140120
10080
6040200
0 30Time after IL-lβ i.t. injection (min) R
ando
m o
ptic
al d
ensi
ty o
f P-
p38
prot
ein
(% o
f co
ntro
l)
60 90 120
min
Positive control
0 30 45 60 75 90 120 240
Fig. 1 Time course of p38 MAPK protein
expression in the rat spinal cord. Total and
phosphorylated forms of p38 MAPKs spinal
cord were examined at different time points
for 4 h in aCSF vehicle control (a), IL-1b
treated (100 ng) (b), and SB203580 (5 mg)
followed by IL-1b (100 ng) treatment (c).
Values are shown as mean ± SEM (n ¼ 4
at each time point for each group). *p £ 0.01
compared to the p38 MAPK inhibitor pre-
treatment group at the same time-point
(one-way ANONA with Tukey post hoc
test).
746 C.-S. Sung et al.
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 94, 742–752
(Sung et al. 2004). To determine whether MAPKs areinvolved in the regulation of protein expression associatedwith nociceptive stimulation in the spinal cord, we furtherexamined the effects of different MAPK inhibitors on IL-1b-induced iNOS protein expression in spinal cord. The presentstudy showed that iNOS protein was not detectable in the ratspinal cord 6 h after vehicle, SB203580 (L)-JNK inhibitor 1,or ERK activation inhibitor peptide II treatment alone(Fig. 5a). However, pretreatment of SB203580 (5 lg) almostcompletely blocked the IL-1b-induced iNOS protein expres-sion 6 h after IL-1b injection (86.6% reduction) (Fig. 5b).Pretreatment of JNK and ERK inhibitor only partially
(� 50%) inhibited the IL-1b-induced iNOS protein expres-sion (Fig. 5b).
P38 MAPK inhibitor SB203580 prevented IL-1b-inducedNO production
IL-1b application resulted in a marked increase in NOconcentration in the CSF (Fig. 6) by 1.1- to 1.9-fold between4 and 12 h, peaking at 6 h, and gradually falling to thebaseline level at 24 h after treatment. Moreover, this NO
0
P-ERK
ERK
30 60
min after aCSF i.t. injection
90 120 240
min after IL-lβ i.t. injection
0 30 45 60 75 90 120 240
β-actin
P-ERK
ERK
β-actin
P-ERK
ERK
β-actin 0
120
100
Ran
dom
opt
ical
den
sity
of
P-E
RK
prot
ein
(% o
f co
ntro
l)
IL-lβERKI+ IL-lβ
0 30Time after IL-lβ injection (min)
60 90 120
Positive control 30 60 90 120 30 60 90 120 min
ERKI + IL IL
(a)
(c)
(b)
(d)
Fig. 3 Time course of total and phosphor-
ylated ERK MAPK protein expression in the
rat spinal cord. Animals were treated with
(a) aCSF, (b) IL-1b (100 ng, i.t.), and (c)
ERK inhibitor (5 lg, i.t.) pretreatment before
IL-1b (100 ng, i.t.). Densitometry analysis of
p-ERK was shown in (d). Values are mean
± SEM (n ¼ 4 at each time point for each
group).
aCSF (n = 8)
20
10
0
–10
–20
–30
–40
–50
–60Baseline
SB 203580 + IL-lβ (n = 10) SB 203580 (n = 8)
IL-lβ (n = 10)
1 2
* †* †
* †* † * † * †
* †
3Time (h) after drug deliveryPe
rcen
tile
chan
ge in
PW
L (
%, r
elat
ive
to b
asel
ine)
4 5 6 7 8 9 10 11 12 24
Fig. 4 Time course of paw-withdrawal latency of various drugs treat-
ment (i.t.). PWL was plotted against time in rats treated with aCSF
(aCSF, n ¼ 8), SB203580 (5 lg; n ¼ 8), IL-1b (100 ng; n ¼ 10), SB
203580 alone (5 lg) (SB 203580), or pretreatment of SB203580 (5 lg)
1 h before IL-1b (100 ng, i.t.) (SB 203580 + IL-1b; n ¼ 10). Values
were normalized to the baseline (before i.t. drug delivery) and
expressed as mean ± SEM. *p ¼ 0.005 vs. baseline; �p ¼ 0.005 vs.
SB203580 + IL-1b, SB203580 and aCSF (analyzed by one-way ANOVA
with Tukey post hoc test).
Posi
tive
cont
rol
aCSF
6h
iNOS
β-actin
(a)
(b)
IL 6
h
SB 6
h
SB +
IL
6h
JNK
inhi
bito
r 6h
ER
K in
hibi
tor
6h
JNK
inhi
bito
r +
IL
6h
ER
K in
hibi
tor
+ I
L 6
h
120
100
80
60
40
20
00 IL-lβ SB + IL-lβ
6 h after treatmentRel
ativ
e op
tica
l den
sity
rat
ioof
iNO
S/β-
acti
n (%
)
JNKI + IL-lβ ERKI + IL-lβ
Fig. 5 Effect of MAPK inhibitors on IL-1b-induced iNOS protein
expression in rat spinal cord. (a) Western blot showing iNOS protein in
dorsal column of lumbar spinal cord 6 h after various treatments. (b)
Semi-quantitative analysis of IL-1b-induced iNOS expression. IL-1b
injection induced a maximal iNOS protein expression 6 h after treat-
ment which was attenuated by SB203580 pretreatment (5 lg, i.t.) 1 h
before IL-1b injection. aCSF or various MAPK inhibitors i.t. injection
did not induced iNOS protein expression. JNK and ERK inhibitors only
partially attenuated the IL-1b-induced iNOS protein expression.
Spinal MAPK and IL-1b-induced thermal hyperalgesia 747
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 94, 742–752
production became barely detectable when animals werepretreated with p38 MAPK inhibitor SB203580. SB203580treatment alone did not induce any NO production (Fig. 6).
Effect of IL-1ra on the IL-1b-induced spinal protein
expression and thermal hyperalgesia
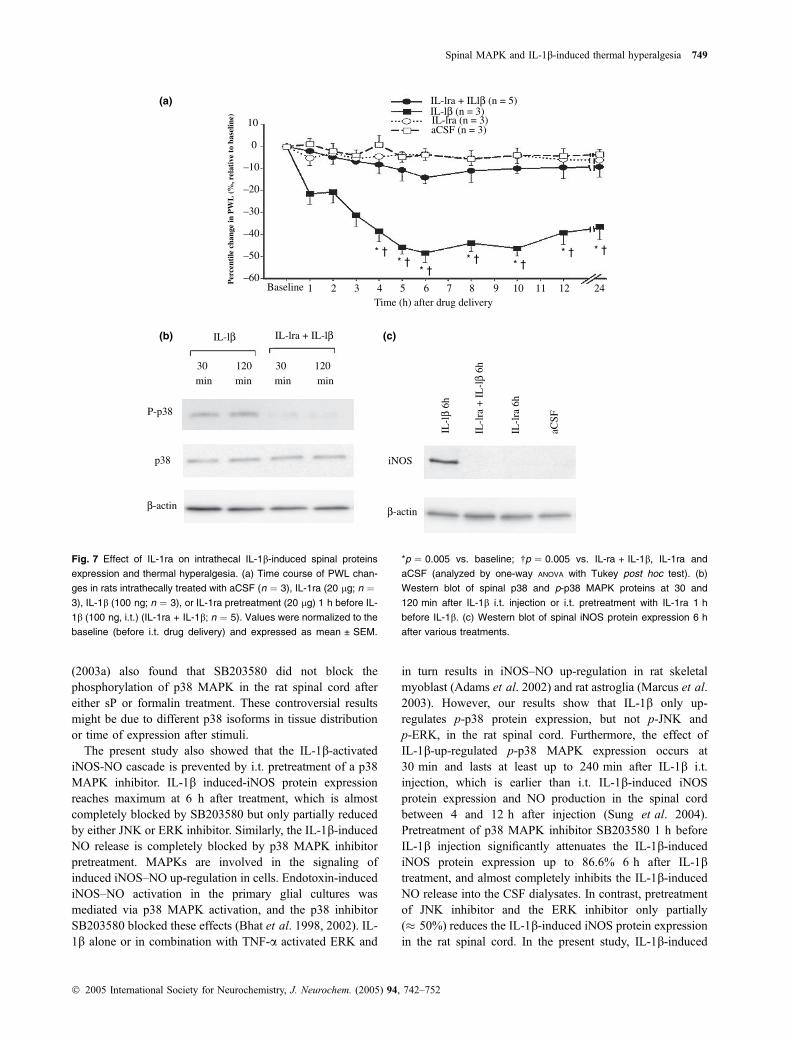
Our study revealed that i.t. IL-1b up-regulated p-p38 MAPKprotein expression and induced iNOS protein expression indorsal horn of lumbar enlargement of rat spinal cord, andproduced thermal hyperalgesia in rats. Vehicle or IL-1ra(20 lg) alone did not affect the PWL; however, IL-1ra(20 lg, i.t.) pretreatment 1 h prior to IL-1b i.t. treatmentexhibited significant attenuation of IL-1b-induced thermalhyperalgesia (Fig. 7a). Western blot analysis showed that thetotal p38 MAPK protein amount was constant at 30 and120 min after either i.t. IL-1b alone or IL-1ra i.t. pretreat-ment (Fig. 7b). However, the IL-1ra (20 lg, i.t.) pretreatmentabolished the IL-1b-up-regulated p-p38 MAPK proteinexpression at both 30 and 120 min (Fig. 7b). Similarly, IL-1ra i.t. treatment did not induce iNOS protein expression at6 h, and IL-1ra i.t. pretreatment completely blocked the IL-1b-induced iNOS protein expression (Fig. 7c).
Discussion
Spinal sensitization after either peripheral injury or direct i.t.stimulation is involved in the pathophysiologic processing ofpain. IL-1b i.t. administration activates the iNOS–NO
cascade in the spinal cord and produces thermal hyperalgesiain the rat (Sung et al. 2004). Our present results support thehypothesis that i.t. IL-1b activates p38 MAPK protein in thespinal cord, which in turn results in spinal sensitization (withup-regulation of iNOS–NO cascade) and thermal hyperalge-sia in the rats.
The MAPK (p38, ERK, and JNK) proteins are expressedonstitutively in the spinal cord but only the spinal p38 MAPKprotein is activated after IL-1b i.t. administration. IL-1bincreases time-dependent p-p38 MAPK protein expression inthe spinal cord without affecting either p-JNK or p-ERKproteins. In the present study, we find a rapid up-regulation ofspinal p-p38 MAPK protein expression starting at 30 min andlasting during the 4-h observation period after i.t. IL-1binjection. Svensson et al. (2003a) reported that both intra-plantar (i.pl.) injection of formalin and i.t. injection ofsubstance p (sP) resulted in a rapid (5–10 min after injection)but transient (< 20 min) increase of p-p38 protein expressionin the rat spinal cord in the inflammatory pain model. On thecontrary, spinal nerve ligation (SNL) up-regulated p-p38MAPK proteins in the spinal cord in rats 12 h after ligationand persisted for 3 weeks (Jin et al. 2003). Moreover, adelayed (> 3 day) but persistent (> 3 weeks) up-regulation ofp-p38 MAPK protein in the DRG in rats was also detected(Jin et al. 2003). Similarly, Tsuda et al. (2004) observed adelayed p-p38 MAPK protein up-regulation between 7 and14 day after spinal nerve injury in rats in the neuropathic painanimal model. The difference of the p-p38 MAPK proteinactivation in onset time point and duration might be due todifferent types of stimulation and tissue examined.
We also found that p38 MAPK inhibitor (SB203580)pretreatment decreases the up-regulated p-p38 MAPK pro-tein expression (38–49% reduction) in the spinal cord duringthe 2-h observation period following IL-1b injection. Fivep38 MAPK isoforms are identified (p38a, p38b, p38b2,p38c and p38d) in different tissues and cells. The p38 MAPKinhibitor SB203580 only inhibits the activity of p38a, p38band p38b2 (Herlaar and Brown 1999; Ono and Han 2000);however, the inhibitory effect of SB203580 on the phos-phorylation of p38 MAPK is not clear. It is suggested thatSB203580 reduces the amount of p-p38 by either inhibitingan alternative activation pathway of p38 MAPK or binding tothe inactive p38 MAPK (Frantz et al. 1998; Matsuguchiet al. 2000; Yamboliev et al. 2000; Ge et al. 2002). Pheny-lephrine and stress will activate p38 MAPK by increasing themount of p-p38 MAPK protein in the perfused amphibianhearts, and SB203580 abolishes it (Aggeli et al. 2002a, b).Our present results are consistent with the above findings;SB203580 inhibited the p-p38 MAPK protein expression.However, Finder et al. (2001) found that IL-1b-stimulatedp-p38 MAPK protein expression is not affected bySB203580 in pulmonary vascular smooth muscle cells andassumed that SB203580 might only decrease the activity ofp38 MAPK, but not the amount of p-p38. Svensson et al.
IL-lβ (n = 8)
aCSF (n = 4)
SB 203580 + IL-lβ (n = 8) SB 203580 (n = 8)
* †
* †* †
* † * †* †
* †
* †
400
350
300
200
100
250
150
50
0Baseline 1.5 2.5 3.5 4.5 5.5 6.5 7.5 8.5 9.51010.51111.512 242 3 4 5 6 7 8 9
Time (h) after drug delivery
NO
rel
ease
in m
icro
dial
ysat
e (%
of
base
line)
Fig. 6 Time-course of NO concentration in the spinal CSF dialysates
after various treatments. Intrathecal administration of either aCSF
(n ¼ 4) or SB03580 (5 lg, n ¼ 8) did not affect NO levels in the rat
spinal CSF dialysates. Intrathecal administration of IL-1b (100 ng) led
to marked increase in NO production 4–12 h after administration (n ¼8). Pretreatment of SB203580 (5 lg) 1 h prior to IL-1b injection
completely blocked the NO release (n ¼ 8). Values were normalized
to the baseline (before i.t. drug delivery) and are expressed as
mean ± SEM. Time 0, baseline. *p < 0.005 vs. baseline; �p < 0.005
vs. SB203580 + IL-1b, SB203580 or aCSF (analyzed by one-way
ANOVA with Tukey post hoc test).
748 C.-S. Sung et al.
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 94, 742–752
(2003a) also found that SB203580 did not block thephosphorylation of p38 MAPK in the rat spinal cord aftereither sP or formalin treatment. These controversial resultsmight be due to different p38 isoforms in tissue distributionor time of expression after stimuli.
The present study also showed that the IL-1b-activatediNOS-NO cascade is prevented by i.t. pretreatment of a p38MAPK inhibitor. IL-1b induced-iNOS protein expressionreaches maximum at 6 h after treatment, which is almostcompletely blocked by SB203580 but only partially reducedby either JNK or ERK inhibitor. Similarly, the IL-1b-inducedNO release is completely blocked by p38 MAPK inhibitorpretreatment. MAPKs are involved in the signaling ofinduced iNOS–NO up-regulation in cells. Endotoxin-inducediNOS–NO activation in the primary glial cultures wasmediated via p38 MAPK activation, and the p38 inhibitorSB203580 blocked these effects (Bhat et al. 1998, 2002). IL-1b alone or in combination with TNF-a activated ERK and
in turn results in iNOS–NO up-regulation in rat skeletalmyoblast (Adams et al. 2002) and rat astroglia (Marcus et al.2003). However, our results show that IL-1b only up-regulates p-p38 protein expression, but not p-JNK andp-ERK, in the rat spinal cord. Furthermore, the effect ofIL-1b-up-regulated p-p38 MAPK expression occurs at30 min and lasts at least up to 240 min after IL-1b i.t.injection, which is earlier than i.t. IL-1b-induced iNOSprotein expression and NO production in the spinal cordbetween 4 and 12 h after injection (Sung et al. 2004).Pretreatment of p38 MAPK inhibitor SB203580 1 h beforeIL-1b injection significantly attenuates the IL-1b-inducediNOS protein expression up to 86.6% 6 h after IL-1btreatment, and almost completely inhibits the IL-1b-inducedNO release into the CSF dialysates. In contrast, pretreatmentof JNK inhibitor and the ERK inhibitor only partially(� 50%) reduces the IL-1b-induced iNOS protein expressionin the rat spinal cord. In the present study, IL-1b-induced
IL-lβ
IL-l
β 6h
β-actin β-actin
P-p38
30
min min min min
120 30 120
iNOSp38
IL-lra + IL-lβ
Baseline 1 2 3 4 5 6 7 8 9 10 11 12 24
IL-l
ra +
IL
-lβ
6h
IL-lra + ILlβ (n = 5)IL-lβ (n = 3)IL-lra (n = 3)aCSF (n = 3)
IL-l
ra 6
h
aCSF
Time (h) after drug delivery
10
(a)
(b) (c)
0
–10
–20
–30
–40
–50
–60Per
cent
ile c
hang
e in
PW
L (
%, r
elat
ive
to b
asel
ine)
* †* †
* †* † * †
* † * †
Fig. 7 Effect of IL-1ra on intrathecal IL-1b-induced spinal proteins
expression and thermal hyperalgesia. (a) Time course of PWL chan-
ges in rats intrathecally treated with aCSF (n ¼ 3), IL-1ra (20 lg; n ¼3), IL-1b (100 ng; n ¼ 3), or IL-1ra pretreatment (20 lg) 1 h before IL-
1b (100 ng, i.t.) (IL-1ra + IL-1b; n ¼ 5). Values were normalized to the
baseline (before i.t. drug delivery) and expressed as mean ± SEM.
*p ¼ 0.005 vs. baseline; �p ¼ 0.005 vs. IL-ra + IL-1b, IL-1ra and
aCSF (analyzed by one-way ANOVA with Tukey post hoc test). (b)
Western blot of spinal p38 and p-p38 MAPK proteins at 30 and
120 min after IL-1b i.t. injection or i.t. pretreatment with IL-1ra 1 h
before IL-1b. (c) Western blot of spinal iNOS protein expression 6 h
after various treatments.
Spinal MAPK and IL-1b-induced thermal hyperalgesia 749
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 94, 742–752

iNOS–NO cascade is reduced by pretreatment with p38MAPK inhibitor, indicating an involvement of the p38MAPK signaling pathway in the spinal cord. However, thepartial inhibitory effect of JNK and ERK inhibitor pretreat-ment on the IL-1b-induced iNOS expression is not clear. AsMAPK can activate transcription factors such as AP-1,activating transcription factor-2 (ATF-2), c-Jun, nucleartranscription factor-jB (NFjB), and Elk-1, and initiatecomplex transcription regulation in response to extracellularstimuli (Meyer et al. 1996; Janssen-Heininger et al. 1999;Yang et al. 2003). It is possible that these transcriptionfactors might be inhibited by either JNK or ERK inhibitors.
Intrathecal pretreatment of p38 MAPK inhibitorSB203580 reverses IL-1b-induced thermal hyperalgesia.Neither JNK inhibitor nor ERK inhibitor pretreatmentsignificantly reduces the IL-1b-induced thermal hyperalgesia.Ample evidence suggests that MAPKs are involved innociception. Peripheral inflammation, neuropathy and sPactivate p38 MAPK protein in spinal cord and the resultingnociceptive behaviors are blocked by p38 MAPK inhibition(Kim et al. 2002; Jin et al. 2003; Svensson et al. 2003a;Tsuda et al. 2004). Pretreatment of p38 MAPK inhibitorsprevent the acute hyperalgesia resulting from i.pl. formalininjection, i.t. administration of sP or NMDA and attenuatedthe up-regulated p-p38 MAPK protein in the spinal cord(Svensson et al. 2003a, b). In addition, Ganju et al. (2001)reported that i.pl. injection of either complete Freund’sadjuvant or IL-1b led to mechanical hyperalgesia in rats; thiswas also reversed by premedication of p38 MAPK inhibitor.SNL up-regulates p-p38 MAPK protein in both spinal cordand DRG, and resulted in mechanical allodynia in rats (Jinet al. 2003); this was attenuated by treatment of a p38MAPK inhibitor (Jin et al. 2003). These results indicate thatp38 MAPK is involved in the signaling transduction pathwayin both inflammatory and neuropathic pain. Although bothERK and JNK activation have been reported to be involvedin peripheral inflammation, noxious heat and electric stimu-lation, and the corresponding nociceptive behaviors wereblocked by pretreatment of ERK inhibitors (Ji et al. 1999;Dai et al. 2002; Galan et al. 2002; Ma and Quirion 2002;Galan et al. 2003). Our present findings are consistent withthe above reports that p38 MAPK protein activation in spinalcord occurs earlier than the expression of thermal hyperal-gesia after stimulation, and further indicated that IL-1b-induced thermal hyperalgesia is mediated by p38 MAPK inthe spinal cord.
In the present study, we found that IL-1ra i.t. pretreatmentinhibits the i.t. IL-1b-induced spinal protein activation andthermal hyperalgesia, it further confirmed the specific effectof IL-1b-induced thermal hyperalgesia is via activation ofp38 MAPK. Recombinant human IL-1ra (rhIL-1ra; AmgenInc., Thousand Oaks, CA, USA) i.t. treatment, at the dose of50 or 100 lg/lL, had been demonstrated to effectivelyinhibit the hyperalgesia and allodynia induced by peripheral
inflammation, neuropathy and i.t. injection of human immu-nodeficiency virus envelope glycoprotein (Watkins et al.1997; Milligan et al. 2001; Sweitzer et al. 2001); however,i.t. treatment with 10 lg rhIL-1ra failed to reverse thehyperalgesia produced by intraplantar. formalin injection(Watkins et al. 1997). In our present study, we choose thedose of 20 lg (39.2 lL) recombinant rat IL-1ra (rrIL-1ra; R& D Systems, Minneapolis, MN, USA) for i.t. treatmentrather than 100 lg (195.3 lL) or 50 lg (97.7 lL) under theconsideration of possible volume effect. Intrathecal treatmentwith rrIL-ra (20 lg) alone neither affected the PWL in rats,nor up-regulated p-p38 MAPK protein, nor induced iNOSprotein expression in the rat spinal cord. However, i.t.pretreatment with rrIL-1ra effectively inhibited the i.t. IL-1b-induced spinal nociceptive sensitization. Therefore, theenhancement of nociceptive signalings in spinal cord isspecific for the pathologic effect induced by IL-1b i.t.treatment.
In conclusion, we show that i.t. injection of IL-1b resultsin activation of the iNOS–NO signal transduction cascade inthe rat spinal cord which leads to thermal hyperalgesia. In thepresent study, i.t. IL-1b-induced-spinal iNOS–NO cascadeand consequent thermal hyperalgesia are reduced by i.t.pretreatment with p38 MAPK inhibitor, but neither ERK norJNK inhibitor, indicating an involvement of spinal p38MAPK in mediating thermal hyperalgesia. On the basis ofthe present study, we suggest that p38 MAPK plays a crucialrole in the IL-1b-induced nociceptive signal transduction,and i.t. administration of p38 MAPK inhibitors may havepotential for reducing the proinflammatory cytokine-inducedcentral nociceptive sensitization.
Acknowledgements
We would like to thank Dr Tsai Shih-Yen and Farrer R. G.
(Neurology and Research Services, Hines Veterans Affair Hospital,
Hines, IL) for the critical reading of this manuscript. This work was
supported by the National Science Council (NSC 92–2314-B-075–
094), Taipei Veterans General Hospital (VGH93-330), and Defense
of Ministry (DOD-93-2-08) of Taiwan.
References
Adams V., Nehrhoff B., Spate U., Linke A., Schulze P. C., Baur A.,Gielen S., Hambrecht R. and Schuler G. (2002) Induction of iNOSexpression in skeletal muscle by IL-1b and NFjB activation: anin vitro and in vivo study. Cardiovasc. Res. 54, 95–104.
Aggeli I.-K. S., Gaitanaki C., Lazou A. and Beis I. (2001) Stimulation ofmultiple MAPK pathways by mechanical overload in the perfusedamphibian heart. Am. J. Physiol. Regul. Integr. Comp. Physiol.281, R1689–R1698.
Aggeli I.-K. S., Gaitanaki C., Lazou A. and Beis I. (2002a) Hyperos-motic and thermal stresses activated p38-MAPK in the perfusedamphibian heart. J. Exp. Biol. 205, 443–454.
Aggeli I.-K. S., Gaitanaki C., Lazou A. and Beis I. (2002b) a1- and b-adrenoceptor stimulation differentially activate p38-MAPK and
750 C.-S. Sung et al.
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 94, 742–752

atrial natriuretic peptide production in the perfused amphibianheart. J. Exp. Biol. 205, 2387–2397.
Bhat N. R., Feinstein D. L., Shen Q. and Bhat A. N. (2002) p38 MAPK-mediated transcriptional activation of inducible nitric-oxide syn-thase in glial cells. Roles of nuclear factors, nuclear factor jB,cAMP-response element-binding protein, CCAAT/enhancer-bind-ing protein-b, and activating transcription factor-2. J. Biol. Chem.277, 29 584–29 592.
Bhat N. R., Zhang P., Lee J. C. and Hogan E. L. (1998) Extracellularsignal-regulated kinase and p38 subgroups of mitogen-activatedprotein kinases regulated inducible nitric oxide synthase and tumornecrosis factor-a gene expression in endotoxin-stimulated primaryglial cultures. J. Neurosci. 18, 1633–1641.
Bogoyevitch M. A., Ketterman A. J. and Sugden P. H. (1995) Cellularstresses differentially activate c-Jun N-terminal protein kinases andextracellular signal-regulated protein kinases in cultured ventricularmyocytes. J. Biol. Chem. 270, 29 710–29 719.
Braman R. S. and Hendrix S. A. (1989) Nanogram nitrite and nitratedetermination in environmental and biological materials by vana-dium (III) reduction with chemiluminescence detection. Anal.Chem. 61, 2715–2718.
Bush P. A., Gonzalez N. E., Griscavage J. M. and Ignarro L. J. (1992)Nitric oxide synthase from cerebellum catalyzes the formation ofequimolar quantities of nitric oxide and citrulline from l-arginine.Biochem. Biophys. Res. Commun. 185, 960–966.
Chacur M., GutierreZ. J. M., Milligan E. D., Wieseler-Frank J., Britto L.R., Maier S. F., Watkins L. R. and Cury Y. (2004) Snake venomcomponents enhance pain upon subcutaneous injection: an initialexamination of spinal cord mediators. Pain 111, 65–76.
Da Silva J., Pierrat B., Mary J. L. and Lesslauer W. (1997) Blockade ofp38 mitogen-activated protein kinase pathway inhibits induciblenitric-oxide synthase expression in mouse astrocytes. J. Biol.Chem. 272, 28 373–28 380.
Dai Y., Iwata K., Fukuoka T., Kondo E., Tokunaga A., Yamanaka H.,Tachibana T., Liu Y. and Noguchi K. (2002) Phosphorylation ofextracellular signal-regulated kinase in primary afferent neurons bynoxious stimuli and its involvement in peripheral sensitization.J. Neurosci. 22, 7737–7745.
DeLeo J. A., Colburn R. W. and Ricknian A. J. (1997) Cytokine andgrowth factor immunohistochemical spinal profiles in two animalmodels of mononeuropathy. Brain Res. 759, 50–57.
Falchi M., Ferrara F., Gharib C. and Dib B. (2001) Hyperalgesic effect ofintrathecally administered interleukin-1 in rats. Drugs Exp. Clin.Res. 27, 97–101.
Finder J. D., Petrus J. L., Hamilton A., Villavicencio R. T., Pitt B. R. andSebti S. M. (2001) Signal transduction pathways of IL-1b-medi-ated iNOS in pulmonary vascular smooth muscle cells. Am. J.Physiol. Lung Cell Mol. Physiol. 281, L816–L823.
Frantz. B., Klatt T., Pang M., Parsons J., Rolando A., Williams H., TocciM. J., O’Keefe S. J. and O’Neill E. A. (1998) The activation stateof p38 mitogen-activated protein kinase determines the efficiencyof ATP competition for pyridinylimidazole inhibitor binding.Biochemistry 37, 13 846–13 853.
Galan A., Cervero F. and Laird J. M. (2003) Extracellular signaling-regulated kinase-1 and – 2 (ERK 1/2) mediate referred hyperal-gesia in a murine model of visceral pain. Brain Res. Mol. BrainRes. 116, 126–134.
Galan A., Lopez-Garcia J. A., Cervero F. and Laird J. M. (2002) Acti-vation of spinal extracellular signaling-regulated kinase-1 and -2by intraplantar carrageenan in rodents. Neurosci. Lett. 322, 37–40.
Ganju P., Davis A., Patel S., Nunez X. and Fox A. (2001) p38 Stress-activated protein kinase inhibitor reverses bradykinin B1 receptor-mediated component of inflammatory hyperalgesia. Eur. J.Pharmacol. 421, 191–199.
Ge B., Gram H., Di Padova F., Huang B., New L., Ulevitch R. J., Luo Y.and Han J. (2002) MAPKK-independent activation of p38amediated by TAB1-dependent autophosphorylation of p38a. Sci-ence 295, 1291–1294.
Guan Z., Baier L. D. and Morrison A. R. (1997) p38 mitogen-activatedprotein kinase down-regulates nitric oxide and upregulates pros-taglandin E2 biosynthesis stimulated by interleukin-1b. J. Biol.Chem. 272, 8083–8089.
Guan Z., Buckman S. Y., Springer L. D., and Morrison A. R. (1999)Both p38a MAPK and JNK/SAPK pathways are important forinduction of nitric-oxide synthase by interleukin-1b in rat glo-merular masangial cells. J. Biol. Chem. 274, 36 200–36 206.
Herlaar E. and Brown Z. (1999) p38 MAPK signalling cascades ininflammatory disease. Mol. Med. Today 5, 439–447.
Hua L. L., Zhao M. L., Cosenza M., Kim M. O., Huang H., Tanowitz H.B., Brosnan C. F. and Lee S. C. (2002) Role of mitogen-activatedprotein kinases in inducible nitric oxide synthase and TNFaexpression in human fetal astrocytes. J. Neuroimmunol. 126, 180–189.
Janssen-Heininger Y. M. W., Macara I. and Mossman B. T. (1999)Cooperativity between oxidants and tumor necrosis factor in theactivation of nuclear factor (NF)-jB: requirement of Ras/mitogen-activated protein kinases in the activation of NF-jB by oxidants.Am. J. Respir. Cell Mol. Biol. 20, 942–952.
Jeohn G. H., Cooper C. L., Wilson B., Chang R. C., Jang K. J., Kim H.C., Liu B. and Hong J. S. (2002) p38 MAP kinase is involved inlipopolysaccharide-induced dopaminergic neuronal cell death inrat mesencephalic neuron-glia cultures. Ann. NY Acad. Sci. 962,332–346.
Ji R. R., Baba H., Brenner G. J. and Woolf C. J. (1999) Nociceptive-specific activation of ERK in spinal neurons contributes to painhypersensitivity. Nat. Neurosci. 2, 1114–1119.
Jin S. X., Zhuang Z. Y., Woolf C. J. and Ji R. R. (2003) p38 Mitogen-activated protein kinase is activated after a spinal nerve ligation inspinal cord microglia and dorsal root ganglion neurons and con-tributes to the generation of neuropathic pain. J. Neurosci. 23,4017–4022.
Johnson G. L. and Lapadat R. (2002) Mitogen-activated protein kinasepathways mediated by ERK, JNK, and p38 protein kinases. Sci-ence 298, 1911–1912.
Kim S. Y., Bae J. C., Kim J. Y., Lee H. L., Lee K. M., Kim D. S. and ChoH. J. (2002) Activation of p38 MAP kinase in the rat dorsal rootganglia and spinal cord following peripheral inflammation andnerve injury. Neuroreport 13, 2483–2486.
Larsen C. M., Wadt K. A., Juhl L. F., Andersen H. U., Karlsen A. E., SuM. S., Seedorf K., Shapiro L., Dinarello C. A. and Mandrup-Poulsen T. (1998) Interleukin-1b-induced rat pancreatic islet nitricoxide synthesis requires both the p38 and extracellular signal-regulated kinase 1/2 mitogen-activated protein kinases. J. Biol.Chem. 273, 15 294–15 300.
Ma W. and Quirion R. (2002) Partial sciatic nerve ligation inducesincrease in the phosphorylation of extracellular signal-regulatedkinase (ERK) and c-Jun N-terminal kinase (JNK) in astrocytes inthe lumbar spinal dorsal horn and the gracile nucleus. Pain 99,175–184.
Maihofner C., Euchenhofer C., Tegeder I., Beck K. F., Pfeilschifter J.and Geisslinger G. (2000) Regulation and immunohistochemicallocalization of nitric oxide synthases and soluble guanylyl cyclasein mouse spinal cord following nociceptive stimulation. Neurosci.Lett. 290, 71–75.
Marcus J. S., Karackattu S. L., Fleegal M. A. and Sumners C. (2003)Cytokine-stimulated inducible nitric oxide synthase expression inastroglia: role of Erk mitogen-activated protein kinase and NF-jB.Glia 41, 152–160.
Spinal MAPK and IL-1b-induced thermal hyperalgesia 751
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 94, 742–752

Matsuguchi T., Musikacharoen T., Ogawa T. and Yoshikai Y. (2000)Gene expressions of Toll-like receptor 2, but not Toll-like receptor4, is induced by LPS and inflammatory cytokines in mousemacrophages. J. Immunol. 165, 5767–5772.
Meller S. T., Dykstra C., Grzybycki D., Murphy S. and Gebhart G. F.(1994) The possible role of glia in nociceptive processing andhyperalgesia in the spinal cord of the rat. Neuropharmacology 33,1471–1478.
Meyer C. F., Wang X., Chang C., Templeton D. and Tan T. H. (1996)Interaction between c-Rel and the mitogen-activated protein kinasekinase kinase 1 signaling cascade in mediating jB enhancer acti-vation. J. Biol. Chem. 271, 8971–8976.
Milligan E. D., O’Connor K. A., Nguyen K. T., Armstrong C. B.,Twining C., Gaykema R. P., Holguin A., Martin D., Maier S. F. andWatkins L. R. (2001) Intrathecal HIV-1 envelope glycoproteingp120 induces enhanced pain states mediated by spinal cord pro-inflammatory cytokines. J. Neurosci. 21, 2808–2819.
Omote K., Hazama K., Kawamata T., Kawamata M., Nakayaka Y.,Toriyabe M. and Namiki A. (2001) Peripheral nitric oxide in car-rageenan-induced inflammation. Brain Res. 912, 171–175.
Ono K. and Han J. (2000) The p38 signal transduction pathway: acti-vation and function. Cell Signal 12, 1–13.
Robinson M. J. and Cobb M. H. (1997) Mitogen-activated protein kinasepathways. Curr. Opin. Cell Biol. 9, 180–186.
Roche A. K., Cook M., Wilcox G. L. and Kajander K. C. (1996) A nitricoxide synthesis inhibitor (L-NAME) reduces licking behavior andFos-labeling in the spinal cord of rats during formalin-inducedinflammation. Pain 66, 331–341.
Samad T. A., Moore K. A., Sapirstein A., Billet S., Allchorne A., PooleS., Bonventre J. V. and Woolf C. J. (2001) Interleukin-1b-mediatedinduction of COX-2 in the CNS contributes to inflammatory painhypersensitivity. Nature 410, 471–475.
Singh K., Balligand J. L., Fischer T. A., Smith T. W. and Kelly R. A.(1996) Regulation of cytokine-inducible nitric oxide synthase incardiac myocytes and microvascular endothelial cells. Role ofextracellular signal-regulated kinases 1 and 2 (ERK1/ERK2) andSTAT1a. J. Biol. Chem. 271, 1111–1117.
Sommer C., Petrausch S., Lindenlaub T. and Toyka K. V. (1999)Neutralizing antibodies to interleukin 1-receptor reduce painassociated behavior in mice with experimental neuropathy. Neur-sci. Lett. 270, 25–28.
Sung C. S., Wen Z. H., Chang W. K., Ho S. T., Tsai S. K., Chang Y. C.and Wong C. S. (2004) Intrathecal interleukin-1b administrationinduces thermal hyperalgesia by activating inducible nitric oxidesynthase expression in the rat spinal cord. Brain Res. 1015,1445–1453.
Svensson C. I., Hua X. Y., Protter A. A., Powell H. C. and Yaksh T. L.(2003a) Spinal p38 MAP kinase is necessary for NMDA-induced
spinal PGE2 release and thermal hyperalgesia. Neuroreport 14,1153–1157.
Svensson C. I., Marsala M., Westerlund A., Calcutt N. A., Campana W.M., Freshwater J. D., Catalano R., Feng Y., Protter A. A., Scott B.and Yaksh T. L. (2003b) Activation of p38 mitogen-activatedprotein kinase in spinal microglia is a critical link in inflammation-induced spinal pain processing. J. Neurochem. 86, 1534–1544.
Sweitzer S. M., Colburn R. W., Rutkowski M. and DeLeo J. A. (1999)Acute peripheral inflammation induces moderate glial activationand spinal IL-1b expression that correlates with pain behavior inthe rat. Brain Res. 829, 209–221.
Sweitzer S., Martin D. and DeLeo J. A. (2001) Intrathecal interleukin-1receptor antagonist in combination with soluble tumor necrosisfactor receptor exhibits an anti-allodynic action in a rat model ofneuropathic pain. Neuroscience 103, 529–539.
Tao Y. X. and Johns R. A. (2002) Activation and up-regulation of spinalcord nitric oxide receptor, soluble guanylate cyclase, after formalininjection into the rat hind paw. Neuroscience 112, 439–446.
Tsuda M., Mizokoshi A., Shigemot-Mogami Y., Koizumi S. and InoueK. (2004) Activation of p38 mitogen-activated protein kinase inspinal hyperactivity microglia contributes to pain hypersensitivityfollowing peripheral nerve injury. Glia 45, 89–95.
Wang C. X., Olschowka J. A. and Wrathall J. R. (1997) Increase ofinterleukin-1b mRNA and protein in the spinal cord followingexperimental traumatic injury in the rat. Brain Res. 759,190–196.
Watkins L. R., Martin D., Ulrich P., Tracey K. J. and Maier S. F. (1997)Evidence for the involvement of spinal cord glia in subcutaneousformalin induced hyperalgesia in the rat. Pain 71, 225–235.
Widmann C., Gibson S., Jarpe M. B. and Johnson G. L. (1999) Mitogen-activated protein kinase: conservation of a three-kinase modulefrom yeast to human. Physiol. Rev. 79, 143–180.
Wu J., Fang L., Lin Q. and Willis W. D. (2001) Nitric oxide synthase inspinal cord central sensitization following intradermal injection ofcapsaicin. Pain 94, 47–58.
Xu X. and Malave A. (2000) p38 MAPK, but not p42/p44 MAPKmediated inducible nitric oxide synthase expression in C6 gliomacells. Life Sci. 67, 3221–3230.
Yamboliev I. A., Hedges J. C., Mutnick J. L. M., Adam L. P. andGerthoffer W. T. (2000) Evidence for modulation of smooth muscleforce by p38 MAP kinase/HSP pathway. Am. J. Physiol. HeartCirc. Physiol. 278, H1899–H1907.
Yang S. H., Sharrocks A. D. and Whitmarsh A. J. (2003) Transcrip-tional regulation by the MAP kinase signaling cascades. Gene320, 3–21.
Zimmermann M. (1983) Ethical guidelines for investigations ofexperimental pain in conscious animals. Pain 16, 109–110.
752 C.-S. Sung et al.
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 94, 742–752









![Mechanisms and functions of p38 MAPK signalling and functions of p38 MAPK signalling 405 Both MKK3 and MKK6 are highly specific for p38 MAPKs [14,23].Inaddition,p38αcanbealsophophorylatedbyMKK4,an](https://static.fdocument.org/doc/165x107/5ae2800d7f8b9a097a8d0b79/mechanisms-and-functions-of-p38-mapk-signalling-and-functions-of-p38-mapk-signalling.jpg)








