
Analeptic Amides of d-α-Methylphenethylamine
7
NO\-. 30, 195s *bVIDES OF d-a-xf ETHYLPHENETHYLAMINE 6065 [ COYTRIBCTIOY FROM THE RESEARCH LABORATORIES OF THE u. s. VITAMIN CORPORATIOX] Analeptic Amides of d-a-Methylphenethylamine BY SEYMOUR L. SHAPIRO, IRA M. ROSE AND LOUIS FREEDMAN RECEIVED JUSE 16, 1988 A series of amides of d-a-methylphenethylamine (I) have been synthesized and examined for excitant activity. Such The structural parameters to which activity has been found in the formamide and with selected a-oxyacylating groups. activity is confined are discussed, and a molecular model of the active form of the products proposed. The noted cardinvascular side effects' of d- and dl-a-methylphenethylamine have detracted from the wider clinical application of these compounds in a variety of therapeutic indications. With the premise that relatively small quantities of free d-a-methylphenethylamine (I) are required for pharmacologic r e s p o n ~ e , ~ . ~ we have investigated the effect of acyl derivatives of I in the search for active structures which might be free of the noted cardiovascular side effects of the parent amine. Since activity is associated largely with d-a- methylphenethylamine, our explorations were largely limited to derivatives of this substance. A broad program of synthesis was undertaken and the compounds prepared are described in Tables 1-111. In selected instances] parallel series of dL- and I-, as well as d-derivatives were studied. As the work progressed, it became evident that activity was substantially confined to selected structures containing an a-oxy function on the acylating group and these were explored in more detail. The role of the acylating groups containing an o-oxy function was explored with the sympathomi- metic amines, d-N-methyl-a-methylphenethyl- amine (Table 11, compounds 41, 53) and phenyl- propanolamine (Table III), to establish whether desirable modification of the properties of these substances would also be obtained. The synthesis of the compounds followed es- tablished procedures and the acylating component used, reflected in general, its commercial accessi- bility, and this was varied as esters, acid chlorides, free acids, lactides and lactones. Although a-methylphenethylamine is a struc- turally hindered amine, no complication occurred in the aminolysis of ethyl formate5 though the higher boiling ethylene diformate6 was more con- venient and afforded a cleaner product. By contrast, under parallel conditions, the hin- dered a-methylphenethylamine failed to react with ethyl acetate' although under forcing condi- tions under pressure, excellent yields of the acet- (1) (a) L. S. Goodman and A. Gilman, "The Pharmacological Basis of Therapeutics," 2nd ed., The Macmillan Co., New York, N. Y., 1955, p. 516; (b) K. Soehring and G. Ergenzinger, Arzneimitlel-Forsch., 6, 478 (1955). (2) S. L. Shapiro and L. Freedman, Arch. intern. pharmacodynamie, 112, 419 (1957). (3) J. Axelrod, J. Pharmacal. Exp. Therap., 110, 315 (1954). (4) For specific reaction rate constants for sympathomimetic amines with methyl acetate, see S. L. Jung, J. G. Miller and A. R. Day, THIS JOURNAL, 76, 4604 (1953). (5) W. H. Watanabe and L. R. DeFonso, ibid., 78, 4542 (1956). (0) S. L. Shapiro, V. 8. Parrino, K. Geiger, S. Kobrin and L. Freed- man, ibid., 79, 5064 (19571. (7) Failure of aminolysis of ethyl acetate by t-butylamine is noted: W. P. Ratchford and C. H. Fisher, J. Org. Chew., 15, 317 (1950); see ref. 4. amide were obtainable.8 The compound (Table I, compound 3) also was prepared readily using acetyl chloride. The aminolysis of the a-hydroxy esters such as ethyl glycolate, ethyl lactate and ethyl a-hydroxy- isobutyrate proceeded readily. Here, reactivity was probably increased as a result of forms such as those postulated by Ratchford.g An asymmetric a-carbon atom in the acylating moiety reflected the possibility of isolation of the dd- and dl-forms of the resultant amides. Such separation was effected in selected instances (Table 11, compounds 59, 60 and 66, 67) by fractional crystallization procedures. The aminolysis reaction with benzilic acid pro- ceeded with difficulty and yielded the benzilic acid salt of the amine, and from the breakdown prod- uct] benzophenone,'O a 34y0 yield of N-a-methyl- phenethylbenzhydrylimine. Reaction with methyl benzilate" (as above) afforded only the benzilic acid salt of d- a-methylphenethylamine. The corresponding a-chlorodiphenylacetamide (Table I, compound 34) was prepared readily from a-chlorodiphenylacetyl chloride, but hydrolysis12 and isolation of the desired benzilamide was un- successful. The aminolysis with dibasic esters was effected readily in most instances. With the malonates, the required bisamide was obtained using diethyl malonate and diethyl ethyl malonate. With di- ethyl phenylmalonate, no reaction was noted,13 and upon addition of sodium methoxide as a basic catalyst, the reactant ester suffered decarbethoxyl- ation l4 and N-d-a-methylphenethylphenylacetamide (Table I, compound 31) and bis-N,N'-d-a-methyl- phenethylurea were obtained. Under similar con- ditions, diethyl ethylphenylmalonate afforded the urea and N-d-a-methylphenylethyl-a-phenylbutyr- amide (Table I, compound 32). The noted decarbethoxylation of the substituted malonate esters14 when treated with alkoxide to give the phenyl acetate and ethyl carbonate fol- lowed by reaction with I, would account for the product isolated. The amine I, however, yielded (8) For enhancement of the rate of another sterically unfavorable reaction through the use of forcing conditions, see I. S. Bengelsdorf, THIS JOURNAL, 80, 1442 (1958). (9) W. P. Ratchford, J. Org. Chem., 15, 326 (1950). (10) H. H. Wasserman, H. W. Ackerman, H. H. Wotiz and T. Liu, THIS JOURNAL, 77, 973 (1955). (11) A. Rahman and M. 0. Farooq, Natu*~issenschaJten. 41, 1 (1954). (12) Possible complicating side products may he 2,2'-oxyhis-(n- phenylphenylacetamides) similar to the bisamides reported by A. H. Schlesinger and E. J. Prill, THIS JOURNAL, 78, 6123 (1956). (13) S. B. Speck, ibid., 74, 2876 (1952). (14) V. H. Wallingford, A. H. Homeyer and D. M. Jones, ibid., 63. 2056 (1941).
Transcript of Analeptic Amides of d-α-Methylphenethylamine

NO\-. 30, 195s *bVIDES OF d-a-xf ETHYLPHENETHYLAMINE 6065
[ COYTRIBCTIOY FROM THE RESEARCH LABORATORIES OF THE u. s. VITAMIN CORPORATIOX]
Analeptic Amides of d-a-Methylphenethylamine BY SEYMOUR L. SHAPIRO, IRA M. ROSE AND LOUIS FREEDMAN
RECEIVED JUSE 16, 1988
A series of amides of d-a-methylphenethylamine (I) have been synthesized and examined for excitant activity. Such The structural parameters t o which activity has been found in the formamide and with selected a-oxyacylating groups.
activity is confined are discussed, and a molecular model of the active form of the products proposed.
The noted cardinvascular side effects' of d- and dl-a-methylphenethylamine have detracted from the wider clinical application of these compounds in a variety of therapeutic indications.
With the premise that relatively small quantities of free d-a-methylphenethylamine (I) are required for pharmacologic r e s p o n ~ e , ~ . ~ we have investigated the effect of acyl derivatives of I in the search for active structures which might be free of the noted cardiovascular side effects of the parent amine.
Since activity is associated largely with d-a- methylphenethylamine, our explorations were largely limited to derivatives of this substance.
A broad program of synthesis was undertaken and the compounds prepared are described in Tables 1-111. In selected instances] parallel series of dL- and I - , as well as d-derivatives were studied.
As the work progressed, i t became evident that activity was substantially confined to selected structures containing an a-oxy function on the acylating group and these were explored in more detail.
The role of the acylating groups containing an o-oxy function was explored with the sympathomi- metic amines, d-N-methyl-a-methylphenethyl- amine (Table 11, compounds 41, 5 3 ) and phenyl- propanolamine (Table I I I ) , to establish whether desirable modification of the properties of these substances would also be obtained.
The synthesis of the compounds followed es- tablished procedures and the acylating component used, reflected in general, its commercial accessi- bility, and this was varied as esters, acid chlorides, free acids, lactides and lactones.
Although a-methylphenethylamine is a struc- turally hindered amine, no complication occurred in the aminolysis of ethyl formate5 though the higher boiling ethylene diformate6 was more con- venient and afforded a cleaner product.
By contrast, under parallel conditions, the hin- dered a-methylphenethylamine failed to react with ethyl acetate' although under forcing condi- tions under pressure, excellent yields of the acet-
(1) (a) L. S. Goodman and A. Gilman, "The Pharmacological Basis of Therapeutics," 2nd ed., T h e Macmillan Co., New York, N. Y., 1955, p. 516; (b) K. Soehring and G. Ergenzinger, Arzneimit le l -Forsch. , 6 , 478 (1955).
( 2 ) S. L. Shapiro and L. Freedman, A r c h . i n t e r n . p h a r m a c o d y n a m i e , 112, 419 (1957).
( 3 ) J. Axelrod, J. Pharmacal. E x p . Therap. , 110, 315 (1954). (4) For specific reaction ra te constants for sympathomimetic amines
with methyl acetate, see S. L. Jung, J. G. Miller and A. R. Day, THIS JOURNAL, 76, 4604 (1953).
( 5 ) W. H. Watanabe and L. R. DeFonso, ibid. , 78, 4542 (1956). (0) S. L. Shapiro, V. 8. Parrino, K. Geiger, S. Kobrin and L. Freed-
man, ibid. , 79, 5064 (19571. (7) Failure of aminolysis of ethyl acetate b y t-butylamine is noted:
W. P. Ratchford and C. H. Fisher, J . Org. C h e w . , 15, 317 (1950); see ref. 4.
amide were obtainable.8 The compound (Table I, compound 3) also was prepared readily using acetyl chloride.
The aminolysis of the a-hydroxy esters such as ethyl glycolate, ethyl lactate and ethyl a-hydroxy- isobutyrate proceeded readily. Here, reactivity was probably increased as a result of forms such as those postulated by Ratchford.g
An asymmetric a-carbon atom in the acylating moiety reflected the possibility of isolation of the dd- and dl-forms of the resultant amides. Such separation was effected in selected instances (Table 11, compounds 59, 60 and 66, 67) by fractional crystallization procedures.
The aminolysis reaction with benzilic acid pro- ceeded with difficulty and yielded the benzilic acid salt of the amine, and from the breakdown prod- uct] benzophenone,'O a 34y0 yield of N-a-methyl- phenethylbenzhydrylimine. Reaction with methyl benzilate" (as above) afforded only the benzilic acid salt of d- a-methylphenethylamine.
The corresponding a-chlorodiphenylacetamide (Table I, compound 34) was prepared readily from a-chlorodiphenylacetyl chloride, but hydrolysis12 and isolation of the desired benzilamide was un- successful.
The aminolysis with dibasic esters was effected readily in most instances. With the malonates, the required bisamide was obtained using diethyl malonate and diethyl ethyl malonate. With di- ethyl phenylmalonate, no reaction was noted,13 and upon addition of sodium methoxide as a basic catalyst, the reactant ester suffered decarbethoxyl- ation l4 and N-d-a-methylphenethylphenylacetamide (Table I, compound 31) and bis-N,N'-d-a-methyl- phenethylurea were obtained. Under similar con- ditions, diethyl ethylphenylmalonate afforded the urea and N-d-a-methylphenylethyl-a-phenylbutyr- amide (Table I, compound 32) .
The noted decarbethoxylation of the substituted malonate esters14 when treated with alkoxide to give the phenyl acetate and ethyl carbonate fol- lowed by reaction with I, would account for the product isolated. The amine I, however, yielded (8) For enhancement of the ra te of another sterically unfavorable
reaction through the use of forcing conditions, see I. S. Bengelsdorf, THIS JOURNAL, 80, 1442 (1958).
(9) W. P. Ratchford, J. Org. Chem., 15, 326 (1950). (10) H. H. Wasserman, H. W. Ackerman, H. H. Wotiz and T. Liu,
THIS JOURNAL, 77, 973 (1955). (11) A. Rahman and M. 0. Farooq, N a t u * ~ i s s e n s c h a J t e n . 41, 1
(1954). (12) Possible complicating side products may he 2,2'-oxyhis-(n-
phenylphenylacetamides) similar t o the bisamides reported b y A. H. Schlesinger and E. J. Prill, THIS JOURNAL, 78, 6123 (1956).
(13) S. B. Speck, ibid., 74, 2876 (1952). (14) V. H. Wallingford, A. H. Homeyer and D. M. Jones, ibid. , 63.
2056 (1941).
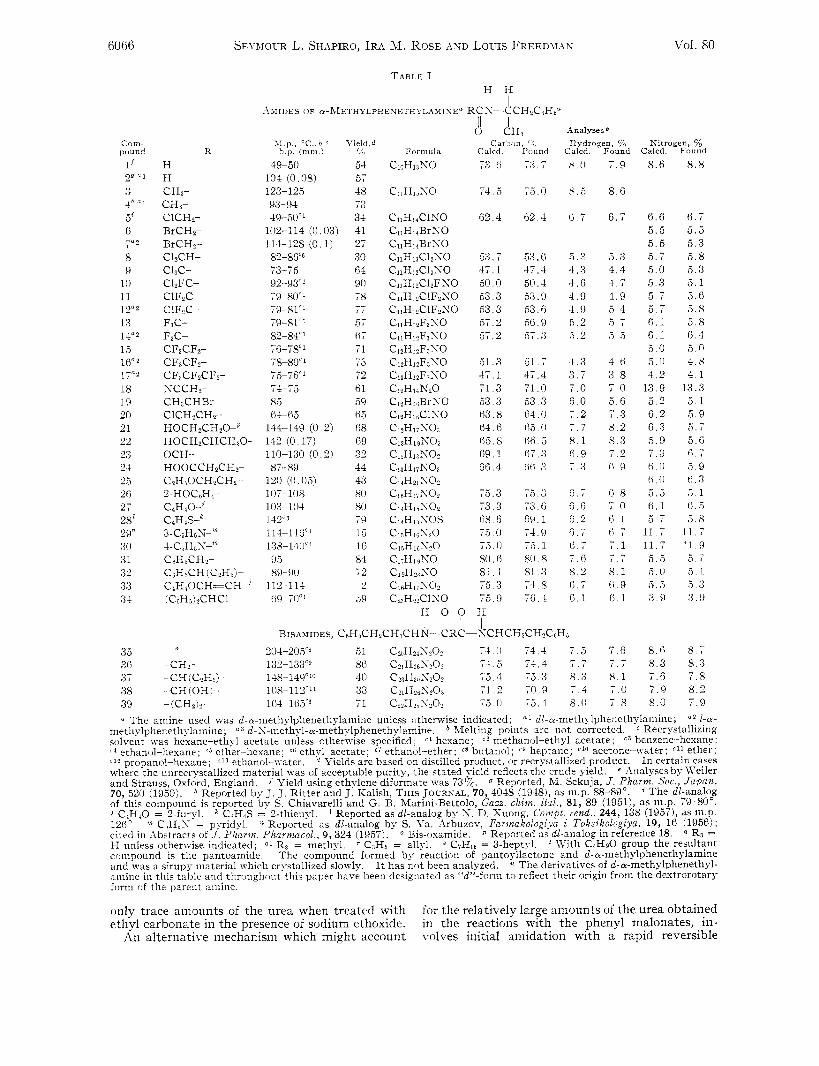
SEI-BIOUR L. SHAPIRO, IRA hl. ROSE hiYD LOUIS FREEDhrah Vol. 80
Com- pound
1' 2Q "1
3 1 h (1 1
5% 6 7" 2
8 9
10 11 12"2 1 3 14"* 1 5 16"2 1 7" 2
18 19 20 21 22 23 24 2 5 26 27 28l 29" 30 3 1 32 3 3 8 1
35 3fi 3 7 3 8 39
K
H H C H r CH3- C l C H r BrCH2- BrCHl- C12CH- C&c- ClZFC- ClFZC ClFJC- F3C- FdC- CF3CF2- CFaCF2- C F ~ C F ~ C F I - SCCH2- CH,CHRr- ClCHaCHr H O C H ~ C H Z O - ~ HOCHzCHCHtO- OCH- HOOCCHzCHz- C?HdOCH2CH?- 2-H OCoH4- ~ ~ ~ ~ 0 - 7 C4H4S--h 3-C0H 6 S-'" I-C,H,S-'" CoHbCHa- C6H5CH (CzH,)- CIHaOCH=CH- (CeHdLCHCl-
-CH?- -CH (C?Hj) -CH(OH)- -(CHz)z-
\ I 11 , "C , bsc h P ( m m ) c
49-50 54 104 (0 08) 57 123-125 48 93-94 73 49-50" 34
102-114 (0 03) 41
Y i:ld, d
114-128 (0 1) 82-8GC6 73-75 92-93c' 79-80'' 79-8 1' I
79-81C1 82-84<1 76-X' 78-80'' 75-76'' 74-75 85 64-65
144-149 (0 2) 142 (0 17) 110-130 (0 2) 87-89
107-108 103-104
114-116<1 138-140L' 95 89-UO
112-114 h9-70L'
120 (0 05)
142~3
27 30 64 90 78
57 67 71
72 61 59 (i 5 68 69 32 44 43 80 80 79 1 5 16 84 72 2
29
-- I /
-r I 5
d d H , C a r h n , ,,
Calcd. Found
7 3 . A
74,5
62.4
53 .7 47 .1 50.0 53 .3 53 .3 57.2 57.2
<5l. 3 17.1 71.3 5 3 . 3 63.8 6 4 . 6 6 5 . 8 G9.1 66 .4
75.3 73.3 68 ,6 i o . 0 l S . 0
80.6 8 1 . 1 1 3 . 3 ( 0 . 9
H I
- - - -
-- r -
73.7
, 3 . 0
0 2 . 4
--
53.6 47 .4 50.4 53.9 53 , fj 3:. 9 57 .3
51 .7 37 .4 71.0 5 3 . 3 64.0 ri5 , 0 66 5 67 .3 ri6 . 3
, , I . 3 73 . 0 69.1 74 .9 i 0 . l
80.8 8 1 . 3 74.8 76 .4
- -
--
Analyses Hydrogen, $& Sitrogen, YO
Calcd. Found Calcd. Found
x . 0
x. .5
ci , 7
5 . 2 4 . 3 4 .6 4 .9 4 .9 5 . 2 r, 2
4 . 3 3 . 7 7 . 0 6 0 7 . 2
8 . 1 6 . 9 7 . 3
(i. 7 6.6 6. 2 6,7 6 . 7 7 . 6 8 . 2 6 . 7 6 . 1
- - , . I
! I , I BISAMIDES, C~H~CH~CH~CHN--CRC-SCHCH~CH?C~H:,
204-205cn ,5l C~IIH~IS,C'~ 74 0 74,4 7 . 5 132-133'' 86 C?iH26S?0? 74 .5 74.4 7 . 7 148-149c1n 40 c ~ ~ H ~ ~ s ~ o ~ 75 .4 7 j . 3 8 .3 1u8-112c" 33 C2,HP6S?Oi 7 1 . 2 70.9 7 . 4 -- 1G4-165'6 71 C,pHy<N,O:, i ~ 1 0 7 5 , 1 8 .0
7 . 9 8 .6
8.6
6.7 6 . 6 5 . 5 5 .5
5.3 5 . 7 4 . 4 5 . 0 4 . 7 5 . 3 4 . 9 5 . 7 5 . 4 5 .7 ; I , , 6.1 5 . 5 6 . 1
5 . 0 4 . 6 5.0 3 . 8 4 . 2 7 . 0 13 .9 5 . 6 5 . 2 7 . 3 6 . 2 8 . 2 6 . 3 8 . 3 5 . 9 7 . 2 7 . 3 6 . 9 6 . 0
6 . 0 0 . 8 5 . 5 7 . 0 6 . 1 0 . 1 j . 7 r i . 7 1 1 . 7 7 . 1 11 .7 / . I 5 . 5 8 . 1 5 . 0 6 . 9 5 . 3 6 . 1 3 . 9
- -
- -
7 . 6 8 .6 7 . 7 8 .3 8.1 7 .6 7 . 0 7 . 9 7 . 8 8 .0
8 . 8
6 . 7 5 . 5 5.3 5.8 5 , 3 5.1 5 . 6 5 . 8 5 . 8 f i . 4 5 , 0 4 . 8 4 . 1
13.3 5 . 1 5 . 9 ti . 7 5 . 6 6 . 7 5 9 6 . 3 5 . 1 ii 5 5 . 8
1 1 . 7 11 .9 5 . 7 5 . 1 5 . 3 :3 . 9
8 . 7 8 . 3 7 . 8 8 . 2 7 . 9
The amine used was d-a-methylphenethylamine unless otherwise indicated; dl-a-methylphenethylamine; u z 1-e- niethylphenethylamine; d-S-methyl-a-methylphenethylamine. Melting points are not corrected. Recrystallizing solvent was hexane-ethyl acetate unless otherwise specified; c 1 hexane; c 2 methanol-ethyl acetate; c 3 benzene-hexane ; r d ethanol-hexane; ej ether-hexane; c6 ethyl acetate; c7 ethanol-ether; c8 butanol; c9 heptane; clo acetone-water: cll ether; < 1 2 propanol-hexane; ethanol-water. Uields are based on distilled product, or recrystallized product. I n certain cases where the unrecrystallized material was of acceptable purity, the stated yield reflects the crude yield. e -4nalyses by n'eiler and Strauss, Oxford, England. Reported, M. Sekuja, J . Pharm. Soc., Jupan, 70, 520 (1950). * Reported by J. J. Ritter and J. Kalish, THIS JOURNAL, 70, 4038 (1948), as m.p. 88-89". The dl-analog of this compound is reported by S. Chiavarelli and G. B. Marini-Bettolo, Gaze. chim. itul., 81, 89 (1951), as m.p. 79-80", j C 4 H 4 0 = 2-furyl. Reported as dl-analog by S. D. Xuong, Comfit. rend., 244, 138 (19571, dS 1ii.P. 126" * Reported as dl-analog by S. \'a. Airbuzov, Farinakologiya i Toksikologiya, 19, 16 (1956); cited in Abstracts of J . Pharni. Pharnzecol., 9, 324 (1957). * R3 = H unless otherwise indicated; Q' Rg = methyl. C3H5 = allyl. 6 C7H16 = 3-heptyl. IVith C4Hg0 group the resultant compound is the pantoamide. The compound formed by reaction of pantoyllactone and d-wmethylphenethylamine and was a sirupy material which crl-stalked slowly. The derivatives of d-a-methylphenethyl- amine in this table and throughout this paper have been designated as "8'-form to reflect their origin from the dextrorotary form of the parent amine.
only trace amounts of the urea when treated with ethyl carbonate in the presence of sodium ethoxide.
An alternative mechanism which might account
f Yield using ethylene diformate was i3Yc,
CjHjS = 2-thienyl. 'n C:HiS = pyridyl.
* Bis-oxamide. p Reported as dl-analog in reference 18.
It has not been analyzed.
for the relatively large amounts of the urea obtained in the reactions with the phenyl malonates, in- volves initial amidation with a rapid reversible
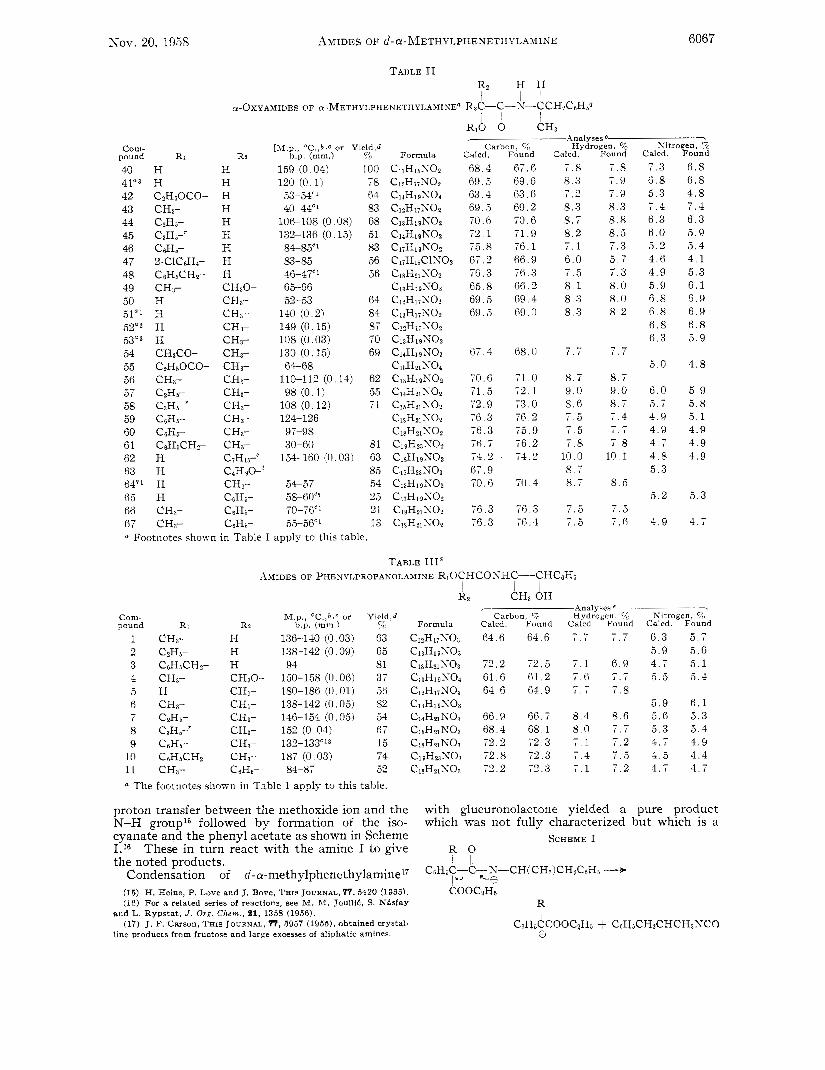
Nov. 20, 1958 AMIDES OF ~ - C Y - ~ ~ E T H Y L P H E X E T H Y L A M I N E 6067
Com- pound Ri
Analyses e-
[hi.p., "C.,b.C or Yield,d Carbon, 5% Hydrogen, % R2 b.p. (mm.) V0 Formula Calcd. Found Calcd. Found
40 H H 159 (0.04) 100 41a3 H H 120 (0.1) 78 42 C2HjOCO- H 53-5.iC' 64 43 CH3- H 40-44'' 83 44 C2Hj- H 106-108 (0.08) 68 45 CIHE-' H 132-136 (0.15) 51 46 C6Hs- H 84-85'' 83 47 2-C1CcjHa- H 83-85 56 48 CsHjCHz- H 46-47'' 56
50 H CH3- 52-53 64 51a' H CH3- 140 (0.2) 84 52a2 H CH3- 149 (0.15) 87 53"3 H CH3- 108 (0.03) TO 54 CH3CO- CH3- 130 (0.15) 69
49 CH3- CHaO- 65-66
55 C2HrOCO- CH3- 64-68 56 CH3- CH3- 110-112 (0.14) 62 57 C2Hs- CHa- 98 (0.1) 5 $5 58 C~HB-' CHI- 108 (0.12) 71 59 CtjH6- CH3- 124-126 60 CsHj- CHI- 97-98 61 CsHjCH2- CHz- 30-60 81
63 H CaHgO-' 85 64"' H CH3- 54-57 54 65 H C6H5- 58-6OC5 25 66 CH3- CsHj- 70-76" 21 67 CHI- C ~ H B - 55-56" 13 a Footnotes shown in Table I apply to this table.
62 H CiHij-' 154-160 (0,03) 63
CllH1jS02 68 .4 67.6 7 . 8 7 . 8
ClaHlgSOI 63 .4 63 .6 7 .2 7 . 9
C13H1gN02 70.6: 70.6 8 . 7 8.8 Cl4HIgK02 72 .1 71.9 8 . 2 8 . 5 CliH1&02 75.8 76 .1 7 . 1 7 . 3 CIjH18ClS02 67 .2 66 .9 6 . 0 5 . 7 C ~ S H ~ ~ S O ~ 76 .3 76 .3 7 . 5 7 . 3
CleHljNO? 69 .5 69.6 8.3 7 . 9
C12H17SOz 69 .5 69.2 8.3 8 . 3
C13H1gxo3 65 .8 6 6 . 2 8 . 1 8.0 Cl?HiiSO? 69 .5 69 .4 8 . 3 8 . 0 C12H17S02 N9.j 69.0 8 . 3 8 . 2 CizHijS02 CnH19S02 ClaH1gx03 67 .4 68.0 7 . 7 - - CisHziSOi C13HlgN02 70.6 71.0 8 . 7 8 . 7 Cl4Hz1KO2 71 .5 72 .1 9 . 0 9 . 0 C1jH21S02 72.9 73.0 8 . 6 8 . 7 C18H21S02 76 .3 76.2 7 . 5 7 . 4 C18H21S02 76.3 75.9 7 . 5 7 . 7 C ~ ~ H ~ ~ S O P 76.7 76.2 7 . 8 7 . 8 ClaHl~SOz 74.2 74.2 10 .0 1 0 . 1 CljH23N03 67. 9 8 . 7 C13H1gS02 70.6 70.4 8 . 7 8 . 5 C17HigS02 C18H21S02 76.3 76.3 7 . 5 7 . 5 C I ~ H ~ I S O Z 76 .3 76.4 7 . 5 7.6
1 . 1
TABLE 111" AMIDES OF PHENYLPROPANOLAMINE R10CHCOSHC-CHC6Hj
I I 1
Com- hl.p., 'C.,b.c or Yield,d pound Ri R? b.p. (mm.) /c
1 CH3- H 136-140 (0.03) 63 2 C2Hj- H 138-142 (0.09) 65 3 CsHjCH2- H 94 81 4 CHa- C&O- 150-158 (0.06) 37 5 H CH3- 180-186 (0.01) 56 6 CH3- CHI- 138-142 (0.05) 82 7 CzHs- CH3- 146-154 (0.05) 54
C'
8 C3Hj-' CH3- 152 (0.04) 67 9 C6Hj- CH3- 132-133"' 15
10 C6HjCH2- CH3- 187 (0.03) 74 11 CH3- CcHj- 84-87 52 a The footnotes shown in Table I apply to this table.
CH3 OH 7 Analyses*-
Calcd Found Calcd Found Carbon, 4 Hydrogen R
64.6 64 6 7 7 7 7
72.2 72 .5 7 . 1 6 . 9 61.6 61 .2 7 . 6 7 . 7 64 .6 64 .9 7 . 7 7 . 8
66 .9 6 6 . i 8 . 4 8 . 6 68 .4 68 .1 8 . 0 7 . 7 72.2 72.3 7 . 1 7 . 2 72 .8 72.3 7 . 4 7 . 5 72.2 72.3 7 . 1 7 . 2
Kitrogen, "c Calcd. Found
7 . 3 6 . 8 6 . 8 6 . 8 5 . 3 4 . 8 7 . 4 7 . 4 6 . 3 6 . 3 6 . 0 5 . 9 5 . 2 5 . 4 4 . 6 4 . 1 4 . 9 5 . 3 5 . 9 6 . 1 6 . 8 6 .9 6 . 8 6 .9 6 . 8 6 . 8 6 . 3 5 . 9
5.0 4 . 8
6 .0 5 . 9 5 .7 5 . 8 4 . 9 5 . 1 4 . 9 4 .9 4 . 7 4 . 9 4 . 8 4 . 9 5 . 3
5 . 2 5 . 3
4 . 9 4 . 7
Nitrogen, 70 Calcd. Found
6 . 3 5 . 7 5 . 9 5 . 6 4 . 7 5 .1 5 . 5 5 4
5 . 9 6 . 1 5 . 6 5 . 3 5 . 3 5 . 4 4 . 7 4 . 9 4 .5 4 . 4 4 . 7 4 . 7
proton transfer between the methoxide ion and the with glucuronolactone yielded a pure product N-H group15 followed by formation of the iso- which was not fully characterized but which is a
SCHEME I cyanate and the phenyl acetate as shown in Scheme I.I6 These in turn react with the amine I to give R O -
the noted products. I I 1 Condensation of d-~~methylphenethylaminel~ CsH5c-c-sS-cH~CH3)CH~C6H5 I1J =4 - -
(15) H. Heine, P. Love and J. Bove, THIS JOURNAL, ' IT , 5420 (1955). COOCaHj R (16) For a related series of reactions, see M. M. Joullif, S. Ndsfay
(17) J. F. Carson, THIS JOURNAL, 77, 5957 ( 1 9 5 3 , obtained crystal- I and L. Ryps ta t , J . O r g . Chem., 21, 1358 (1956).
line products from fructose and large excesses of aliphatic amines. CGH~CCOOC!H~ f C G Y ~ C H ~ C H C H ~ S C O
6

SEYMOUR L. SHAPIRO, IRA 31. ROSE AND LOCIS FREEDMAX Vol. 80
TABLE 11’ SUMMARY OF PHARMACOLOGICAL F I S D I N G S “ ~
Excitant response with compoundsc of Tables I and I I d 4 + 3 + 2 + 1 4 S e g .
300:; and ovcr 200-300‘i; 100-299‘:: 20-09c’, 0- 20 ‘1 Inhibits 10 mg. 10 mg . 10 mg. 1 0 mg, 10 m g .
I , 23, 40, 50 32 (100 mg.) 42, 54, 55 2, 25. 51, 05 3, 4, 5, 19, 20, 22, 24, 32, 34, 37, 62 36, 38,39, 41, 52, 53, 03 64 (40 mg.)
27 (50 tng.) 2 0 mg. 20 mg. 20 mg. 20 nib’.
13, 44, 45, 48, 56, 58 The excitor!: action is
registered on a counter and kymograph responsive to motion and was established over a 16-hour test period. The coni- pounds were administered at the indicated dosage levels in mg./kg. subcutaneously aiid evaluated in six rats, with si.; additional rats serving as controls during intervals from 5 P.M. to 9 A.M. the next day. The activity (excitant response) is expressed by the formula: A = % increase in activity = (activityincrease of test group/activitycontrolgroup) X 100. Four activity ranges were arbitrarily set up as shown in the table and expressed as 4+ through 0 (little or no activity), Several com- pounds showed depressant effects and these are grouped under the “Inhibits” column. Cardiovascular effects were investigated in the isolated guinea pig heart and in the anesthetized dog. While toxicity of individual compounds is not listed, the range for the active structures indicated an LD,i,. in mice of 153-500 mg./kg. As a control, benzedrine (dZ-~~-methylphenethyl- ainine) hydrochloride a t 10 mg./kg. showed a 4+ response. Table I11 compounds 1 [1+ ( l O O ) ] , 2, 3, 5, 7 and 9 were nega-
61, 18 (100 mg). 29, 30, 46, 47, 49, 66 26, 28, 33, G7 a The individual compounds Tvere evaluated for their excitant effect in rats using activity cages.
tive
condensation of two moles of d-a-methylphenethyl- amine and one mole of glucuronolactone with elimination of one mole of water.
Additional syntheses of interest concerned the preparation of glycol carbonates from reaction of d-a-methylphenethylamine’s with ethylene car- bonate and propylene carbonate (Table I, com-
PhaPmaco1ogy.-The compounds herein de- scribed were assessed for their effects on increasing the motor activity of rats, and results have been noted in Table IV. The data obtained reflect the results of screening tests as established upon sub- cutaneous administration of the compounds. The more active derivatives were subsequently evalu- ated per os, and for indication of cardiovascular side effects.
Inspection of Table IT’ reflects a definite struc- ture us. activity relationship in that all compounds which retained the excitant effects of the parent :Imine upon acylation, with the exception of com- pound 1, contained an a-oxy function in the acylat- ing function.
m’ithin the active compounds a number of structural features were required for retention of activity. Thus, variation of the aniine as dl-a- methylphenethylamine (compound 1 LIS. 2, 50 2’s.
I ) , or I-a-methylphenethylamine (compound 50 7 ’ 5 . 3 2 ) , or d-X-methyl-a-methyiphenethylamine (compound 40 tis. 41, 30 LIS. 53), was associated with sharp diminution or disappearance of activity. IVhile the presence of a hydroxy group, an alkoxy group or carbonyl oxygen on the a-carbon atom of the acylating group yielded compounds with high activity (compounds 40, ,io; -1.3, 44, 45, 48, 56, 58, 01, and 23) , this effect could be modified by other substituents on the a-carbon atom or on the a-oxy function. Thus, two methyl groups on the a-car- bon (compound 64) or one phenyl group (compound (Lj) resulted in depressant effect and slight excitant effect, respectively, as did a larger alkyl group (compound 62). LVhen derivatives of the active structures had substituents capable of withdrawing clectrons from the a-oxy atom such as acetyl (com- pound 50 vs. X), or carbethoxy (compound 50 vs.
[’OUllds 2 1, 22).
(IH) K Hazard, T Cheymol, P Chabrier, Y Gay and 31 P Muller, A I I o l i u i v i l v n n ( , 9 , 390 (1951).
55, 40 vs. 42) or phenyl (compound 40 LIS . 46, 4 i ) , activity was reduced. An alkoxy group on the P- carbon atom yielded but slight activity (compound 25). Other compounds containing an a-oxy func- tion which showed little or no response were the bisamide derived from tartronic acid (compound 3 8 ) ) the furoamide (compound 27) and the panto- amide (compound 63).
An indication of criticality dependent on the con figuration a t the a-carbon of the acyl group was, re- flected in the noted activity of compound 06 vs. 67. This factor is being explored further with optically active lactate as a reactant and will be reported at a later date.1g
The body of data thus far presented is not con- sistent with a mechanism involving in vielo hydrol- ysis of the acylated amide to free d-a-methylphen-- ethylamine. Although systematic studies of the hydrolysis of amides are scant, i t is indicatec120s2L that the hydrolysis of amides parallels observa- tions with esters. The use of the ester data cited by HineZ3 translatcd per sc to the amides of this series would indicate that the order of increasing difficulty of hydrolysis would be compound 3, 1 , 40, S , which is clearly inconsistent with the pharma- cdogical observations.
A structural parameter which is more conclusive is one associ3ted with a hydrogen bonded cyclic structure.
It has been quite clearly established that in the simple amides a trans configuration is found be- tween the carbonyl oxygen and the amide hydro- geii.23 X hydrogen bonded structure of the type 11 is proposed for the actire structures noted herein.
(19) S L. Shapiro, I. If. Rose and I*. Freedman, in preparation. (20) J. Hine, “Physical 0r::anic Chemistry,” McGraw-Hill Boob
(21) 12. L. Bender and 12. D. Ginger, THIS J U U R K A L , 77, 343
1.22) Reference 90, p. 27.7. (23 ) 1 . 1;. \\‘orsham. J r . , and .\I. E Hobbs. THIS IouKx, t i , 76, 201;
Co., Inc.. N e w York, N. Y . , 195l;, pi?. 298-299.
(1955).
(1954).

Nov. 20, 1958 AMIDES OF d-(Y-h~ETHYLPHENETHYLL4M1XE 6069
TABLE V a - 0 ~ ~ ACIDS ASD DERIVATIVES, RIOCHR~COR,
Yield, C' 112% pound Ri R2 R4 O C . hIm. /o
1" CzHc- H -0c211; 56 17 74 2" C3H5-b H -OH 104-128 9 3i 1.4538 3 d C6HbCHn- H -OH 120-122 0 . 1 77 1.5280 4" CHI- -0CH3 62 16 19 1.4056 5f CH3- CH3- -OH 84 10 64 6' CH3- CHa- -c1 38-58 90 95
Si C3Hs- CH3- -OH 104-106 10 72
Com- B.p.
C&- CHI- -0CzHs 56 17 68 1.4014 1.4384
9j C6HjCH2- CH3- -OH 118-120 0 .15 80 1.5140 1 0 k C6HjCH2- CH3- -c1 68 0 .12 81 1.5104
-h
S . M. McElvain and W, R. Davie, THIS JOURNAL, 73, 1400 (1951), report b.p. 155-158". * C3Hs is allyl. R . M. Evans and L. N. Owen, J . Chem. Soc., 244 (1949), report b.p. 120" a t 13 mm. B. Rothstein, Bull. soc. chim., 51, 691 (1932), report b.p. 180-152" a t 15 mm., n 1 9 ~ 1.5250. e E. E. Royals and A. G. Robinson 111, THIS JOURNAL, 78, 4163 (1986), re- port b.p. 54" a t 5 mm., f C. Niemann, A. A. Benson and J. F. Mead, J . Org. Chem., 8, 397 (1943), repor: b.p. 87-89' a t 10 mm. a t 755 mm., ~ Z O D 1.4013. A n d . Calcd. for C7HI4O3: neut. equiv., 130.1. Found: neut. equiv., 135.4. 1 P. A. Levene and M. Kuara, J . Bid. Chem., 113, 153 (1936), report b.p. 123-128" a t 0.2 mm. Referencej, report b.p. 79-81' a t 0.5
1.4047. Reference f, report b.p. 46-49" a t 52 mm. * "Beilstein" Vol. 111, p. 109, rep0rts.b.p. 153-155
mm.
This structure is consistent with the effects of electron-attracting and electron-donating substitu- ents on the hydroxy groups of compounds 40 and 50 discussed above. The compounds which could assume this structure, but had no excitant effect (compounds 64, 65)) could conceivably exist in an alternate conformationz4 unfavorable for formation of the structure 11.
While a similar cyclic structure could be written for the pharmacologically inactive a-halo amides (compounds 3, 19), it is of interest that Katayamaz5 i n a crystal study of chloroacetamide, could find no evidence of hydrogen bond formation of the type shown in 11.
Of the more active structures, compound 50, the N- (d- a-methylphenethy1)-lactamide, was shown to be orally effective without significant cardiovascular effects, and is under clinical trial.
Inspection of the proposed structure for com- pound 60 (111) along with some of the newly de- scribed analeptic drugsz6 all bearing the important
I11 27 IV CtiHj I
(24) S. Mizushima, e l ai., THIS JOI:RNAL, 78, 2038 (1956). (25) M. Katayama, Acta Cryst., 9, 986 (195G). (26) (a) Structure IV, 3-methyl-2-phenylmorpholine hydrochloride,
E. P. Galvin, T. H. McGavack and S. Kenigsberg, A m . J . Dig. Dis., 1, 155 (1956); (b) structure V, or-(Z-piperidyl)-benzhydrol hydrochlo- ride, B. B. Brown and H. W. Werner, J . Pharmacol. Ex# . Theualh., 110, 180 (1954) : (c) structure VI , methyl a-(2-piperidyl)-phenylacetate hy- drochloride, E. Sury and K. Hoffman, Hela. Chim. Acta, 37, 2133 (1954); (d) F. Leuschner, A. Leuschner and F. Heim, Avch. ex$. Pathol. Pharmakol., Naunyn-Schmiedeberp's 227, 129 (1955).
(27) An interpretation of t h e infrared spectrum of this compound, received through the courtesy of Mr. E. S. Todero, Perkin-Elmer Cor- poration, indicates hydrogen bonding as shown.
phenylisopropylamine group26d indicates an inter- esting parallelism.
Experimental Materials.-The d-, I - and dl-a-methylphenethylamines
were purchased from True Synthetics, Long Island City, N. Y., and were of U.S.P. standard. Phenylpropanolamine hydrochloride was obtained from Hexagon Laboratories, S. Y. , X. Y . The majority of the acylating agents were commercial materials; dimethyl tartronate was prepared as previously described,29 and the a-oxy acids synthesized as intermediates have been gathered and described in Table v. N-(d-a-Methylphenethy1)-lactamide (Table 11, Compound
50 (from Ethyl Lactate).--.$ mixture of 135.2 g. (1.0 mole) of d-a-methylphenethylamine and 120 ml. (1.04 moles) of ethyl lactate were heated under reflux. After 15 hr. the initial internal reflux temperature of 159' had decreased to 115' by the formed ethandl of reaction. The condenser was replaced by a l-igreux column and after 41 ml. of ethanol had distilled, the internal temperature had increased to 18.1'. The column was replaced by a condenser and the re- action continued under reflux for 7 hr. The reaction mixture was then subjected to distillation. About 10 g. of unreacted amine was initially obtained and the product, the lacta- mide of d-a-methylphenethylamine, distilled 146-183' a t 0.20-0.28 mm., there being obtai;ed 186 g. (90%) which crystallized on seeding, m.p. 47-48 .
"hen lactide was heated with the amine for 4 hr. a t 140_0, the same product was obtained in 937, yield, m.p. 4,- 4 9 O .
Unless specified below, the products of Tables 1-111 were obtained using the requisite ester by the procedure de- scribed above.
N-(d-a-Methylphenethy1)-a-allyloxypropionamide (Table 11, Compound 58).-Over 16 hr. , 1.4 ml. (theoretical amount) of water was removed by azeotropic distillation from a refluxing solution of 9.1 g. (0.07 mole) of dl-a-allyl- oxypropionic acid, 9.46 g. (0.07 mole) of d-a-methylpheneth- ylamine and 100 ml. of xylene. The cooled xylene solution was washed with dilute hydrochloric acid, water, dilute sodium hydroxide solution, water and then filtered. The xylene was removed a t 45' a t 30 mm. and the residue was distilled in vacuo. After removal of low boiling materials and a small forerun, the product distilled a t 108" a t 0.12 mm. yielding 12.3 g. ( i ly,) .
The following compounds were also prepared from the acid and d-a-methylphenethylamine: 45, 48, 61, 62. N-(d-a-Methylphenethy1)-salicylamide (Table I , Com-
pound 26).--A solution of 6.6 g. (0.042 mole) of salicyloyl chloride in 25 ml. of benzene was added dropwise over 30 minutes with stirring, to a solution of 22 g. of d-a-methyl- phenethylamine in 125 ml. of benzene with the reaction
(28) Descriptive data shown in t h e tables are not herein reproduced (29) S. L. Shapiro, K. Geiger and H. Solowa?, 1Zleeting-in-~inia-
ture, Westchester Branch, American Chemical Society, April. 19,jfi.

maintained a t about 10" by external cooling. The benzene phase was separated and then washed with dilute hydro- ehloric acid, water, dilute sodium hydroxide and finally, water. After removal of the benzene, the residue crystal- lized yielding 8.45 g. (79.571) of amide product, 111.p. 89- 9 i o . 111 1 rp -30
moved on the stearn-bath, the residue dissolved iii benzene, the benzene removed on the steam-bath to dry the product and the residue distilled in ' ~ " c u o , yielding 0.60 g. (31.4:;) of an oil, b.p. 110-130' a t 0.20 mm. The analyses intli- cated that the resultant compound was not analytically
The following products were similarly prepared from the acid chloride and d-a-methl-lphenethylamine: compounds 5-9, 19,20,25,27,28,31,32,34,35,39, 46, 47, 56 and 65 of Tables I and 11.
N-(d-a-Methylphenethylj-acetamide from Ethyl Acetate under Pressure.--.4 mixture of 13.5 g. (0.1 mole) of d-a- inethylphenethylamine and 11 ml. of ethyl acetate in a sealed steel bomb was immersed in an oil-bath a t a tempera- ture of 200'. hfter 12 hr. , the bomb \vas allowed to cool to room temperature and the contents removed (methanol wash). The solvents were hoiled off under vacuuni and the crystalline solid obtained was washed with hexane. Xfter trituration with 50 ml. of water containing 10 ml. of 3 LV hydrochloric acid, and filtration, the crystals were washed with water, air-dried and finally dried in vaczio. The product 10.57 g: (60',2), melted at 124-125". -1 mixed m.p. with the acetamide prepared from acetyl chloride (Table I, com- pound 3) was 125126'.
\I.'heii ethyl acetate and d-a-iiietli!-lphenethylaniine were allowed to react under reflux a t atmospheric pressure for 8 hr., no product was isolated.
Ethyl Carbonate Ester of N-(d-a-Methylphenethy1)-gly- colamide (Table 11, Compound 42).-S-(d-a-Methylplien- ethyl)-glycolamide (Table I, compound 40), 10.0 g., 0.05 mole) was dissolved in a mixture of 10 ml. of pyridine and 15 inl. of acetonitrile, the solution cooled in an ice-bath, and 7 nil. (excess) of ethyl chloroformate added dropwise over 10 minutes with stirring and cooling. .After standing 1 hr. a t 2O0, the solvents were removed a t reduced pressure aiid the residue dissolved in a mixture of 100 ml. rif benzene and 100 nil. of 0.5 N hydrochloric acid. The benzene layer T W ~
washed with water and the benzene removed on the steain- bath. The residue of white needles was triturated irith cold hexane, there being obtained 8.5 g.
Compound 55, the ethyl carbonate ester of N-(d-a-metli- ylp1ienethyl)-lactamide was similarly prepared.
N-(d-a-Methylphenethy1)-a-acetoxypropionamide (Table 1 I , Compound 54) .-?;-(d-a-SlethS-lphenethyl)-lactamide (Table 11, compound 50, 10.35 g. , 0.05 mole), was dissolved in a mixture of 20 ml. of acetonitrile and 6 nil. of pyridine. This solution was cooled to 10" and 3 mi. (excess) of acetyl chloride was added dropwise over 15 minutes with contin- ued cooling. Xfter 2 hr. a t 20" the solvents were removed. The residue was t r e a t d with 50 ml. of benzene and 15 ml. of ; j S hydrochloric acld, the benzene solution washed with water, filtered, and evaporated to a small volume on the steam-bath. Distillation of the residue gave 8.55 g. (69';) o f the product, b.p. 128-130' a t 0.15 mm. N-(d-a-Methylphenethy1)-succinamic Acid (Table I , Coni-
~ ) I I L I I I ~ 21).-To a refluxing suspension of 4.0 g . (0.01 mole) ( i f .;uceinic anhydride in 40 ml. of cliloroforrn, a solution of ,541 g. (0.04 mole) of ci-a-itietliylphenetliylamiiie in 10 inl. of chloroform was added dropwise over 1 lir. The anhydride tlissolved slowly during addition, and heating was continued f:ir 15 minutes after complete solution. The product vas extracted into 55, sodium hydroxide, washed with ether, acidified and re-extracted into chloroform. Upon removal of the solvent, the oily residue obtained (7.65 g . ) solidified when scratched and cooled under hexane. N,N'-Di-(d-a-Methylphenethylj-malonamide (Table I ,
C~inipound 36).-.\ mixture of 10.0 g. (0.0624 mole) of di- ethyl malonate arid 20 ml. (excess) of d-a-methylphenethyl- ;linine was run through two cycles of reflux to ccmstant in- ternal temperature and remov:tl of the formed ethanol. \\'hen cool, the reaction mixture crystallized. After tritu- I-atioti with dilute hydrochloric acid, there was obtained 3).0 g. (94(jL,) of crude product, m.p. 124-126'. N-(d-a-Methylphenethy1)-glyoxalamide (Table I , Coin-
~ , ~ ~ u n d 2,3).-:\ suspension of 2.:37 g., (0.01 mole) of s-(&- inethylphenethyl)-dinieth etatnldc (Table 11, com- pound 49) in 15 nil. of 5 furic acid was treated under reflux for :{ hr. The solution 5rl obtained was saturated with bodiutn chloride and the product extrxted with seven E?- 1111. 11~1rtions of ether. Tlic ci)nibirierl ether extracts werc I\ ;tsllctl with aqueous sodiuni bicurbonate. The bicarlx)ti;ite
ash was extracted with two 25-1111. prirti(ins c;f ether \vhich \verc cmibinetl with the m i i i i estrcict. Tlic ether IWS re-
.4nal. Calcd. for CllH13SOL: C, 69.1; H, 6.9; S, 7.3. Found: C,67.3; H,7.2; S, 6.7.
Attempted Preparation of N-(d-a-Methylphenethy1)-ben- zi1amide.-\Yhen 22.8 g. (0.1 mole) of benzilic acid and 13 A8 g. (0.1 mole) of d-a-methylphenethylamine in xylene were heated as described above, 1.2 ml. of water was col- lected in 30 hr. The cooled xylene deposited a large quan- tit>- of crystals which were filtered, washed with benzene and dried; 15.35 g. (42%,), 1n.p. 179-180°, on recrystallization (ethyl acetate) n1.p. 181-182'.
Anal. Calcd. for C23HZ5S03: C, 76.0; H, 6.9; S, 3.9. Found: C, 76.3; H,6.4; L-,3.8.
The compound proved to be the benzilic acid salt of d-a- niethylphenethylamine, aiid did not depress the melting point of the authentic salt described below.
The d-a-methylphenethylamine salt of benzilic acid \vas prepared by mixing ethyl acetate solutions of the acid and base, n1.p. 180-182".
The xylene filtrate afforded a11 oily residue which yielded 10.12 g. (34'30) of the S-(d-a-methylphenethy1)-benzhydryli- mine, b.p. 148' a t 0.08 mm.
.lnal. Calcd. for C21HLlS: C , 88.3; H, 7.1; S, 4.7. Found: C, 88.1; H , 7.5, S, 4.6. N-(d-a-Methylphenethylj-benzhydry1imine.-A solutiou
of 9.1 g. (0.05 mole) of benzophenone and 6.8 g. (0.05 mole) of d-a-methylphenethylamine in 75 ml. of xylene was heated under reflux in a Dean-Stark apparatus for 24 hr. Only 0.5 inl . of mater was obtained. Upon removal of the xylene, distillation of the residue after separation of unchanged re- actants gave 2.53 g. (17%) of product, b.p. 134-144' at 0.04
..lnaZ. Calcd. for Cs2Hg1S: C, 88.3; H, 7.1; S, 4.7. Found: C, 88.6; H, 7.7; S,4.9.
Reaction of Methyl Benzilate and d-a-Methylphenethyl- amine.--.A mixture of 9.0 g. (0.067 mole) of d-a-methyl- phenethylamine arid 8.1 g. (0.033 mole) of methyl benzilatc under an air-cooled condenser was heated in an oil-bath a t lCj7-l62" for 7.5 hr. Shortly after initiation, the internal temperature of the melt rose to 170' and then fell to 160". On cooling, a semi-solid mass was produced which on tri- turation with hexane gave 10.95 g, (90%;) of d-n-methyl- phenethylamine salt of benzilic acid, m.p. 181-182" alone, or mixed with authentic material prepared from d-n-meth>-l- phenethylamine and benzilic acid in ethyl acetate.
Reaction of d-a-Methylphenethylamine with Glucurono- la~tone.3~--Xtidition of 14.85 g. (0.110 mole) of d-a-methyl- phenethylaniine to a suspension of 9.68 g. (0.055 mole) of glucuronolactone in -55 tnl. of methanol resulted in complete
Xfter 2 hr., the solution had -1ddition of 375 ml. of
olution which after standing yielded hich after trituration with benze!ie ), m.1,. 115-1 18". T~ecrystalliz~ition
(ethyl acetate) raised the m . p . to 121-123'. Anal. Calcd. for C21H3g>03: C, 67.3; H , 7.5; S,
6.5. Found: C, 66.5; H , i .3; S, 6.8. The analytical figure indicates two moles of d-a-methyl-
phenethj-lamine reacting with one mole c?f glucuronolactone with elimination of one mole of water. The compound was not further investigated.
Hydroxyethyl Urethan of d-a-Methylphenethylamine (Table I, Compound 21).--.1 mixture of 6.75 g. (0.05 mole) o f d-a-ineth?-lphenethylaniine and 4.8 g. (1070 excess) o f ethylene carbonate was heated at 60' for 24 hr. and then a t i).2 inin. and 100" for 1.5 hr. On distillation of the residue there wvas obtained 68''; yield of product, b.p. 144-149' a t 11.2 111171.
Innl.
lution within a few ininutes. )lored to an intense cherry red.
Coinp~iuritl 22, Table 1 \v:ti siinihrly prepared.
(30) The compound has been described herein in view of the yharma- cologicd acti\.ity i t s iml , r ted
(31) T h e authors apprcclrtc t h c hainl,Ic of gluciirt,tiiililct~,oc lira \ i< l ed b y - 13r 11 \I . I iathmann. Ccxn Products Refining Co., S e a
Y(irk. ?-. T.
-. ~~~ .

Nov. 20, 1958 AMIDES OF d-(U-METHYLPHENETHYLAMII\'E 607 1
The Diastereoisomers of N-(d-a-Methylphenethy1)-a- methoxyphenylacetamide (Table 11, Compounds 66 and 67) .-When 8.3 g: (0.05 mole) of dl-a-methoxyphenylacetic acid and 6.76 g. (0.05 mole) of d-a-methylphenethylamine in 100 ml. of xylene were treated as above, 0.9 ml. of water was collected in 15 hr. After the removal of the xylene, the resi- due of 12.5 g. crystallized on cooling. The solid was dis- solved in 100 ml. of boiling hexane and five 20-1111. portions of hexane added as the solution cooled. On standing for 4 hr., 5.9 g. (m.p. 50-68") separated, called fraction I. The fil- trate stored a t 10" for 72 hr. afforded 4.0 g. (m.p. 58-60'), called fraction 11. Fraction I was dissolved in 150 ml. of boiling hexane and after standing 4 hr., 2.3 g. (compound 66), m.p. 70-76", was obtained, CYD (CHCla) -24.6". Frac- tion I1 was dissolved in 100 ml. of hexane. On standing for several hours, the hard needles of compound 66 formed. These mere separated and on standing for 20 hr., 1.8 g. of fluffv needles of comDound 67 was obtained, m.D. 55-56", LYD (CHC13) -55.1O.-
The Diastereoisomers of N-id-a-MethvlDhenethvli-a- phenoxypropionamide (Table 11, Compound; 59 a n d 60). A solution of 9.23 g. (0.05 mole) of dl-a-phenoxypropionyl chloride in 25 ml. of benzene was added to a stirred cooled solution of 13.85 g. (0.015 mole) of d-a-methylphenethyl- amine in 75 ml. of benzene a t 15". The stirred suspension was allowed to warm to room temperature over 30 minutes. The amine hydrochloride was separated and the benzene solution was washed with dilute hydrochloric acid, water, dilute sodium hydroxide and finally water. Upon removal of the benzene, there was obtained 13.1 g. (92Yc) of the mix- ture of isomers, m.p. 80-110'.
Compound 59, Table 11.-The crude mixture was stirred with 1 liter of hexane a t room teniperature and filtered. The insoluble material, 4 g. melting about 123", was recrystal- lized from a mixture of 110 ml. of hexane and 20 ml. of eth) 1 acetate, yielding 3.2 g. j22yLL) of needles, m.p. 124-125.5".
Compound 60, Table 11.-To the filtrate above, hexane was added to a total volume of 1.5 liters and a small addi- tional crude crop of compound 59 was separated and dis- carded. On cooling a t 10" for 48 hr., an additional 0.6 g . of mixed product, m.p. 87-94', was obtained and discarded. The filtrate was concentrated to 250 ml., carbon added and the solution filtered. After several hours, the formed crys- tals of compound 60 were separated, washed with hexane and dried; yield 4.0 g. (28761, m.p. 97-98'.
Reaction of d-a-Methylphenethylamine with Diethyl Phenylmalonate in the Presence of Sodium Methoxide .--A mixture of 11.8 g. (0.05 mole) of diethyl phenylmalonate, 20 ml. (excess) of d-a-methylphenethylamine and 1.0 g. of sodium methoxide was heated under reflux. After 1.25 hr. the formed ethanol was removed (4.0 ml.) . \Vhen cool, the solid residue was triturated successively with 1 -V hydro- chloric acid, water and hexane, yielding 20.2 g. of a mixture of products, m.p. 8G90".
Isolation of N,N'-Bis-(d-a-methylphenethy1)-urea.--A so- lution of 2.0 E. of the above material in 15 ml. of hot ethanol (charcoal) was filtered, diluted with water (5.0 ml.) till just cloudy, and seeded. After standing overnight, the crkstals which had formed were filtered, washed with 5 0 7 ethanol and dried over phosphoru2 pentoxide a t 80" giving 0.53 g. of the urea, m.p. 147-151 . h mixed m.p. with an authentic sample of the urea (m.p. 149-151') was 148-151'.
Anal. Calcd. for C~SHZ~S?O: C, 77.0; H, 8.2; N, 9.5. Found: C, 77.6; H , 8.0; N, 9.7.
Isolation of N- (d-a-Methylphenethy1)-phenylacetamide .- The product mixture, 2.0 g., was extracted with 100 ml. of boiling hexane. On coooling, the filtrate deposited 0.42 g . of a mixture, m.p. 88-91 . l f t e r several recrystallizations from hexane, most of the urea wau removed and the m.p. of the needles (0.12 9.) was raised to 89-96". The mixed melting point with authentic amide (Table I , compound 31) wa?
N,N'-Bis-(d-a-methylphenethy1)-urea.--B solution of 8.0 g. (0.059 mole) of d-a-methylphenethylamine, 2.0 g. (0.033 mole) of urea and 10.0 g. of glacial acetic acid was heated in an oil-bath until 4.0 ml. of acetic acid distilled 0~ t .32 The cool, still liquid residue was diluted with 70 ml. of methanol and 30 ml. of water was added to the cloud Doint. The
92-98'.
product Crystallized and was separated, 0.76' g. (8.5%), m.p. 149-151'.
(32) R. H. Wiley, P. Beasley and L. H. Knabeschuh, THIS JOUR-
XAL. 76, 311 (1954).
Reaction of d-0-Methylphexiethylamine with Diethyl Ethyl- phenylmalonate in the Presence of Sodium Methoxide.-A solution of 13.2 g. (0.05 mole) nf diethyl ethylphenylmalo- nate and 20 ml. (excess) of d-a-methylphenethylamine heated under reflux showed no reaction, with the internal tempera- ture remaining a t 214' for 2 hr. Upon addition of 1.1 g. of sodium methoxide, the internal temperature fell to 116' after 2 hr. under reflux. Distillation a t atmospheric pres- sure up to 190" yielded 3.7 ml. of formed alcohol. Upon additional heating under reflux, the internal temperature fell from 185 t o 173" after 3.5 hr. IVhen cool, thesolid resi- due was washed successively with 1 N hydrochloric acid, water and hexane to give 19.8 g. of a mixture of products which was dissolved (charcoal) in a mixture of 700 ml. of heptane and 65 ml. of ethyl acetate, and filtered. On stand- ing 5 hr., 6.1 g. (41%) of N,I\;'-bis-(d-a-methyl-@-pheneth- y1)-urea, m.p. 148-149", was obtained. Upon recrystalli- zation (ethyl acetate), the m.p. was 151-152'. A mixed melting point with a n authentic sample of the urea (m.p. 149-151') was 150-151".
Anal. Calcd. for C I ~ H ? ~ K ; ~ O : C, 77.0; H, 8.2; N, 9.5. Found: C, 77.0; H,7.9; hT,9.1.
The heptane mother liquor, stored a t lo", gave 6.62 g. (4770) of material, m.p. 80-85", still containing some of the urea. Recrystallization (hexane-ethyl acetate) gave 4.3 g. of N-(d-a-methylphenethy1)-a-phenylbutyramide, m.p. 89- 90". A mixed melting point with authentic material (Table I, Compound 32) was 89-90'.
Anal. Calcd. for C I ~ H ~ ~ N O : C, 81.1; H , 8.2; N, 5.0. Found: C, 81.5; H , 8 . 1 ; iY,4.8. N-(dl-a-Methyl-p-hydroxyphenethy1)-lactamide (Table
111, Compound 5).-.4 mixture of 7.2 g. (0.05 mole) of lac- tide and 17.5 g. (0.16 mole) of phenylpropanolamine was heated a t 140' for 4 hr. IVhen cool, 50 ml. of water was added, the solution acidified with hydrochloric acid and the product extracted with nine 100-ml. portions of ether. After removal of the ether, the residue was distilled in vacuo yielding 12.5 g. (56%) of amide, b.p. 180-186" a t 0.1 mm.
N- (dl-a-Methyl-@-hydroxyphenethyl)-a-ethoxypropion- amide (Table 111, Compound 7).--A solution of 8.0 g. (0.053 mole) of phenylpropanolamine and 7.32 g. (0.050 mole) of ethyl a-ethoxypropionate was heated under reflux to a constant internal temperature and the formed ethanol then was distilled out until the internal reaction temperature reached 180'. The reflux step was repeated. The residue, after cooling, was dissolved in ether and the solution washed with dilute hydrochloric acid. On removal of the ether, the residue was dried by boiling with benzene, the benzene re- moved on the steam-bath and the residue distilled. There was obtained 6.8 g. (547,) of amide, b.p. 146-154" a t 0.05 mm .
Compounds 1, 2 and 4 in Table I11 were prepared in a s'milar manner.
N-(dZ-a-Methyl-p-hydroxyphenethyl)-a-benzyloxypro- iionamide (Table 111, Compound lO).--iZ solution of 7.8 g. (1.0393 mole) of dZ-a-benzyloxypropionyl chloride in 25 ml. : f acetonitrile was added dropwise over 30 minutes with :irring to a solution of 12.48 g. (0.0825 mole) of phenylpro-
I molamine in 500 ml. of acetonitrile while maintaining the .eaction temperature below 25'. Stirring was continued 'or 30 minutes after addition was complete, the phenylpro- lanolamine hydrochloride was separated and the acetoni- trile removed from the filtrate in vacuo. The residue was dissolved in 300 ml. of benzene and 100 ml. of 0.5 N hydro- chloric acid. The benzene layer was washed with water, dilute sodium hydroxide solution, water, and then the ben- zene was evaporated on the steam-bath. The residue on dis- tillation yielded 9.17 g. (74%) of product boiling a t 187- 189' a t 0.03 mm.
Compounds 3, 6, 8, 9 and 11 in Table 111 were prepared in a similar manner.
Acknowledgment.-The authors are indebted to Dr. G. Ungar and his staff for the pharmacological results herein reported, to V. Parrino and E. Rogow for the synthesis of compounds 6-18 and K. Geiger for the synthesis of compound 38 of Table I, and especially to E. Roskin for his technical assistance. YONKERS 1, S. Y .














![Primul cuvânt D · Primul cuvânt 342 D d, D, s.m. "litera d/D "; "sunetul [d]" "litera §/» "; "sunetul [§]" "grupul de litere dh/DH " "sunetul [dh/ δ]" d, D , s.f. invar.: cu](https://static.fdocument.org/doc/165x107/5e4b02b8ccbf8f281c58ecc6/primul-cuvnt-d-primul-cuvnt-342-d-d-d-sm-litera-dd-sunetul.jpg)



