A Novel Assay for Measurement of Membrane-Protein Surface Expression using a β-lactamase Reporter
12
This article is protected by copyright. All rights reserved Development of a new assay for quantitative measurement of surface expression of membrane proteins Vincent M Lam 1* , Pieter Beerepoot 1* , Stephane Angers 2 , and Ali Salahpour 1# 1 Department of Pharmacology and Toxicology, University of Toronto, Toronto, Ontario, Canada 2 Leslie Dan Faculty of Pharmacy, University of Toronto, Toronto, Ontario, Canada *Two authors contributed equally. #Corresponding Author: Ali Salahpour, Ph.D. [email protected] Synopsis The trafficking of membrane proteins is dynamic and contributes to the homeostatic control of their cell surface localization and their function in signal transduction. Here, we describe a simple, low cost, and potentially high-throughput assay, for measurement of surface expression of proteins. The assay uses β-lactamase enzyme (βlac) as a reporter, and its cell-impermeable substrate, nitrocefin. The utility of this assay is demonstrated using well-established paradigms of internalization and molecular chaperoning, applied to two GPCRs and a monoamine transporter. Abstract: The trafficking of membrane proteins is dynamic and contributes to the homeostatic control of their cell surface localization and their function in signal transduction. It is therefore important to have sensitive techniques that allow measurement of surface expression. The current assays for such measurement are time consuming and low throughput. Here, we describe a quantitative, one step, and potentially high- throughput assay, using the β-lactamase enzyme (βlac) as a reporter, for measurement of surface expression of proteins. In this assay, the βlac is fused to the extracellular portion of the plasma membrane protein of interest. To selectively measure surface expression, a cell-impermeable substrate of βlac, nitrocefin, is used. We demonstrate the utility of the βlac assay using well-established paradigms of internalization and molecular chaperoning, applied to two GPCRs and a monoamine transporter. Considering its simplicity and low cost, this assay could become a standard technique in the measurement of protein surface expression Key words: β2AR, DAT, βlac, GPCR, surface expression, molecular chaperone, internalization This article has been accepted for publication and undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process, which may lead to differences between this version and the Version of Record. Please cite this article as doi: 10.1111/tra.12073 © 2013 John Wiley & Sons A/S
Transcript of A Novel Assay for Measurement of Membrane-Protein Surface Expression using a β-lactamase Reporter

This article is protected by copyright. All rights reserved
Development of a new assay for quantitative measurement of surface expression of membrane proteins
Vincent M Lam1*, Pieter Beerepoot1*, Stephane Angers2, and Ali Salahpour1#
1 Department of Pharmacology and Toxicology, University of Toronto, Toronto, Ontario, Canada 2 Leslie Dan Faculty of Pharmacy, University of Toronto, Toronto, Ontario, Canada *Two authors contributed equally. #Corresponding Author: Ali Salahpour, Ph.D. [email protected]
Synopsis The trafficking of membrane proteins is dynamic and contributes to the homeostatic control of their cell surface localization and their function in signal transduction. Here, we describe a simple, low cost, and potentially high-throughput assay, for measurement of surface expression of proteins. The assay uses β-lactamase enzyme (βlac) as a reporter, and its cell-impermeable substrate, nitrocefin. The utility of this assay is demonstrated using well-established paradigms of internalization and molecular chaperoning, applied to two GPCRs and a monoamine transporter.
Abstract: The trafficking of membrane proteins is dynamic and contributes to the homeostatic control of their cell surface localization and their function in signal transduction. It is therefore important to have sensitive techniques that allow measurement of surface expression. The current assays for such measurement are time consuming and low throughput. Here, we describe a quantitative, one step, and potentially high-throughput assay, using the β-lactamase enzyme (βlac) as a reporter, for measurement of surface expression of proteins. In this assay, the βlac is fused to the extracellular portion of the plasma membrane protein of interest. To selectively measure surface expression, a cell-impermeable substrate of βlac, nitrocefin, is used. We demonstrate the utility of the βlac assay using well-established paradigms of internalization and molecular chaperoning, applied to two GPCRs and a monoamine transporter. Considering its simplicity and low cost, this assay could become a standard technique in the measurement of protein surface expression Key words: β2AR, DAT, βlac, GPCR, surface expression, molecular chaperone, internalization
This article has been accepted for publication and undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process, which may lead to differences between this version and the Version of Record. Please cite this article as doi: 10.1111/tra.12073
© 2013 John Wiley & Sons A/S

This article is protected by copyright. All rights reserved
The trafficking of membrane proteins is a tightly-regulated, dynamic process that is linked to the pathophysiology of many diseases such as cystic fibrosis (see reviews[1][2]). For most membrane proteins, the homeostatic control of their cell surface localization is regulated by a number of different mechanisms including rate of synthesis, internalization, recycling, and degradation. Furthermore, small molecules can affect the levels of surface expression for many membrane proteins, a property that can be exploited as a therapeutic approach. For example, ligands for G-Protein Coupled Receptors (GPCRs) induce internalization of receptors as a mechanism of desensitization/ resensitization [3]. For monoamine transporters, both substrates and inhibitors have been shown to affect cell surface expression[4]. Additionally some membrane proteins require the presence of specific molecular chaperones required for cell surface expression[5]. Considering all the regulatory mechanisms affecting surface expression, and the functional impact of altered trafficking, there is a clear need for sensitive and quantitative techniques to study membrane protein surface expression. The assays that are classically used to measure membrane protein surface expression work by extracellular labeling of proteins using biotinylation or antibodies. Whole-cell ELISA and flow cytometry depend on the use of a primary antibody towards an extracellular epitope and a secondary antibody that is either enzyme-linked (ELISA) or fluorescent (flow cytometry). In biotinylation assays, cell surface proteins are labeled with biotin, purified using streptavidin affinity, and subsequently visualized using western blotting. Both antibody-dependent and biotinylation assays, require long incubation and multiple washing steps, which is impractical for drug screening purposes Furthermore these assays typically exhibit considerable variability that makes their use challenging for reliable and quantitative determination of cell surface protein expression. The insertion of fluorescent protein tags, such as green fluorescent protein (GFP), is often used to track surface expression. Surface expression of such recombinant proteins can be determined by examining distribution of GFP providing useful information about cellular distribution, but limiting the throughput [6]. Assays more suitable for high throughput screening to measure surface expression have also been developed. For example, fluorogen activated proteins (FAP) have been used to tag extracellular portions of membrane proteins[7]. The resulting FAP fusion protein can then be labeled by incubating cells with fluorogens. Another approach that has been used is the labeling of proteins with small enzyme reporters, notably the SNAP and CLIP tags, which use mutant forms of an alkylguanine-DNA alkyltransferase and
modified substrates for intra- and extracellular labelling[8]. Although these assays are promising, the equipment and reagent costs are relatively high. Other enzymes, such as β-galactosidase and β-lactamase (βlac), have also been used to tag membrane proteins [9,10]. In particular βlac has been shown to be amenable to creation of fusion proteins as a sensitive reporter for cytosolic, membrane-associated, and excreted proteins. Furthermore, βlac is a versatile reporter with many chromogenic and fluorescent commercially available substrates. We have taken advantage of these properties of βlac to develop a novel, simple, one-step assay for the quantitative measurement of surface expression of plasma membrane proteins. Results and Discussion The assay uses βlac as a reporter that is genetically fused to a flexible extracellular portion of the plasma membrane protein of interest. The βlac clone (Gi9910661) is used for these studies since it can be expressed in eukaryotic cells[11]. To selectively detect plasma membrane-localized proteins, the assay takes advantage of nitrocefin, a well-characterized, commercially-available chromogenic substrate of βlac (Fig1)[11–13]. βlac enzymatic activity can be easily quantified by measuring the change in absorbance of nitrocefin from λ=390nm (pH 7.0) to λ=486nm (pH 7.0), which is produced by hydrolysis of the beta-lactam ring within nitrocefin. Cell impermeability of nitrocefin was established by examining nitrocefin conversion in cells expressing the cytosolic protein β-arrestin tagged with βlac. There was no substrate hydrolysis when intact β-arrestin-βlac cells were incubated with nitrocefin. Substrate hydrolysis only occurred when these cells were lysed/permeabilised by hypotonic shock/ and mechanical lysis, indicating that nitrocefin is indeed cell impermeable (Supplemental Figure 1). Therefore measurements of βlac enzymatic activity using nitrocefin reflect the surface expression of the plasma membrane protein of interest, as the intracellular βlac is unable to metabolize the hydrophilic nitrocefin (Fig 1). Importantly, the rate of nitrocefin hydrolysis by βlac is directly proportional to the amount of surface expression of the membrane protein of interest, as there is no amplification of the signal. This is in contrast to antibody-mediated assays like ELISA or flow cytometry. In this study we demonstrate the utility of this assay to monitor the surface expression of two types of membrane proteins: two GPCRs, the β2-adrenergic receptor (β2AR) and the metabotropic GABAB receptor 1 (GBR1), and one monoamine transporter (the dopamine transporter DAT). Utilizing well-established principles such as internalization and

This article is protected by copyright. All rights reserved
molecular chaperoning, we show that the βlac assay detects surface protein levels with the same sensitivity as classical ELISA and flow cytometry, while having the major advantages of being less expensive, more rapid, and one-step, requiring no incubations. For the GPCR studies, the βlac was cloned in frame at the N-terminus of the receptor preceded by a signal sequence for proper targeting to the secretory pathway[14]. For DAT, βlac was inserted in the second extracellular loop between amino acid residues 193-203. It has previously been shown that an insertion of an HA epitope at this same position preserved DAT activity[15]. In addition to the βlac, an HA epitope was also added 5’ to the βlac in all constructs, allowing the direct comparison of the βlac assay in parallel with classical ELISAs and flow cytometry. Importantly all βlac constructs were properly expressed and retained functional activity similar to untagged proteins (Supplemental figure 2). As a first demonstration of the βlac assay’s utility, we studied the dynamics of β2AR internalization, a prototypical family A GPCR [16]. Stimulation of β2AR with 10µM of the full agonist isoproterenol led to a time-dependent internalization of the receptor reaching a maximum at 30 minutes post-stimulation (Fig 2A). The half-life of internalization is 6.54 min, in accordance to what has previously been reported for β2AR[17]. The results obtained through the βlac assay (Fig 2A) were qualitatively replicated in parallel using classical ELISA and flow cytometry with half lives of 6.39 and 4.52 min respectively. (Fig 2B-C). This result shows that all assays report a similar time course for β2AR internalization (compare Fig 2A to Fig 2B and 2C). We next compared these assays by measuring the dose-dependent internalization of β2AR with isoproterenol. As assessed using the βlac assay, the EC50 of β2AR internalization with isoproterenol is 21.8 ±34.7 nM at 30 minutes post-stimulation (Fig 2D). Whereas the βlac assay demonstrated a classical sigmoidal dose response, the dose response using ELISA yielded a linear relationship (Fid 2E) indicating that for this type of experimentation, the βlac assay has a better resolution than classical ELISA (compare Fig 2D to Fig 2E). For flow cytometry the dose response yielded an EC50 of 24.54 ± 12.66 nM (Fig 2F), similar to what is obtained with the βlac assay (Compare Fig 2D to 2F). It is important to note that the maximum internalization of these assays are also similar, 40% for βlac and ELISA and 50% for flow cytometry. The marginal difference in maximum internalisation between βlac/ELISA and flow cytometry could potentially be due to the innate differences of the assays. Having established that the βlac assay can be used to study ligand-mediated internalization of a GPCR, we next investigated whether the assay could be used to assess the effects of molecular chaperones on surface expression. It is well documented that
the metabotropic GABAB R1 receptor subunit requires the co-expression of its molecular chaperone GABAB R2 (GBR2) to traffic to the plasma membrane[5]. We therefore evaluated surface expression of GBR1 with increasing levels of GBR2 using the βlac assay. As shown in Figure 3A and B, GBR2 expression dose-dependently increased surface expression of GBR1. The results obtained with the βlac assay (Fig 3A) are similar to what can be measured using ELISA (Fig 3B), once again demonstrating the utility and reliability of the βlac for quantitative measurement of surface expression of GPCRs and its similarities to well established techniques. In order to demonstrate that the βlac assay can be applied to other plasma membrane proteins besides GPCRs, we studied the internalization of the twelve transmembrane domain protein DAT. In HEK cells DAT internalizes in response to phorbol myristate ester (PMA) stimulation, and this internalization can be blocked with GBR12909, a selective DAT blocker[18]. As shown in Fig 3C and 3D, treatment of cells expressing HA-βlac-DAT with PMA for 1 hour results in 23% ± 4 and 24% ± 3 internalization of the transporter as measured by the βlac and ELISA assays respectively. Pre-incubation with 1μM of GBR12909 blocks PMA-induced internalization as previously described using cell surface biotinylation experiments[18]. These results further demonstrate the utility of the βlac assay for measuring changes in DAT surface expression and once again show that the βlac assay performs similarly to the well established ELISA assay (compare Fig 3C to 3D). Finally the suitability of the βlac assay for High Throughput Screening (HTS) was evaluated. For this, the Z’ Factor of βlac assay for isoproterenol induced β2AR internalization was measured. The Z’ factor is a measure of the quality of an assay that takes into account both the signal dynamic range and variation of experimental data of the assay [19]. A Z’ of 0.5 and above is considered excellent for HTS applications. As shown in Supplemental Figure 3, the Z’ Factor of the βlac assay is 0.52 when measuring agonist induced internalization of β2AR. This indicates that the βlac assay is very robust and potentially an excellent assay for HTS screening of compounds affecting surface expression of membrane proteins. In this study, we describe a, simple one-step assay based on measurement of βlac activity to quantitatively measure the surface expression of plasma membrane proteins. As shown in figures 2 and 3, the results obtained from the βlac assay are qualitatively and quantitatively similar to the well established assays of ELISA and flow cytometry. However, the major advantage of the current assay is that it is 4-5 times faster to execute compared to ELISA or flow cytometry and it is 7-12 times less expensive depending on the cost of antibody used (See Table 1). Using β2AR, a prototypical GPCR, we demonstrated that time and dose dependent internalization of this receptor can be measured using the βlac assay. Using the GABAB receptor as

This article is protected by copyright. All rights reserved
a model, we showed that the βlac assay can be utilised for the study of molecular chaperones of GPCRs. We also demonstrated that the assay can be applied to study the surface expression of the dopamine transporter, indicating the potential for this assay to be generalised to all plasma membrane proteins. Finally, measuring the Z’ factor of the assay for β2AR internalization, we showed that the βlac assay is suitable for HTS screening of compounds affecting surface expression of membrane proteins. To employ the βlac assay, recombinant proteins must be generated by inserting the βlac in an extracellular portion of the protein which could interfere with protein function. However, we observed no effect on function on any of the proteins examined in this study (Supp Figure 2). This is in line with other reports indicating that N-terminal fusion of GFP or SNAP tag to various receptors does not interfere with their activity [6,8]. However, because the assay requires a recombinant protein it can not be utilised for measurement of endogenous native proteins. Nevertheless, considering all the advantages of the βlac assay (commercially available reagents, one step, fast, cheap, robust (Z’ factor)), we anticipate that the βlac assay can become an important tool for measurement of surface expression of membrane proteins Materials and Methods Reagents Nitrocefin (BD biosciences) was dissolved in DMSO at a concentration of 10mM. Isoproterenol (Sigma) was dissolved in PBS containing 170 μM ascorbic acid. GBR12909 (Tocris) was dissolved in distilled water with 5% DMSO. PMA (Bioshop) was dissolved in DMSO. Mouse anti-HA primary antibody (12CA5 hybridoma) and anti-mouse HRP conjugated (Cell Signaling Technology) secondary antibodies were diluted in PBS containing 1% BSA. Plasmid Construction The cDNA expression vectors for human GBR1 and β2AR were provided by Dr. Michel Bouvier [20,21]. The cDNA expression vector for human GBR2 was obtained from Missouri S&T cDNA Resource Center. A peYFP-c1 vector with a human DAT with a YFP on the n-terminus and an HA sequence replacing residues 193-203 was provided by Dr. Alexander Sorkin [10]. The β-lactamase sequence was cloned from the ampicillin resistance gene within the pcDNA3.1 plasmid, with the restriction sites HindIII and AscI at 5’ and 3’ of the βlac respectively. The βlac was further modified by the additions of a His-tag, chicken α7 nicotinic receptor signal sequence (SS) [9], and an HA epitope at the N-terminus, yielding the following cDNA: His-SS-HA-βlac. The βlac construct was subsequently cloned into the multiple cloning site of the pcDNA3.1 plasmid. A 5’ Asc I and 3’ Not I restriction site were added to the GBR1 and β2AR. These cDNA constructs were then ligated into the plasmid in frame with the βlac. For the DAT
construct, a Quikchange II site-directed mutagenesis kit (Agilent) was used to insert Spe I and an Asc I sites on either side of the HA-sequence. The HA-sequence was then removed, and the HA-βlac sequence flanked by the same restriction sites was ligated to the DAT. Site-directed mutagenesis was then used to remove both restriction sites, resulting in a YFP-HA-βlac–DAT. Cell Culture HEK 293 and HEK 293T cells were maintained in Dulbecco’s modified Eagle medium (DMEM, Wisent) and supplemented with 10% FBS (Wisent), 100 U/ml penicillin and 100µg/ml streptomycin. HEK 293 cells stably expressing β2AR or GBR1 were further supplemented with 1µg/ml puromycin. Cell lines stably transfected with DAT were maintained with 500µg/mL G418 (Bioshop). All cells were kept at 37˚C and 5% atmospheric CO2. Generation of Stable Cell Lines and Transient Transfections Cells (2x106 cells) were seeded into 10cm tissue culture plates. The following day, cells were transfected with 3 µL of polyethylenimine (1mg/mL) (Polyscience Inc) per µg of plasmid DNA. For transient transfections, cells (HEK 293T) were seeded 6h post-transfection for experiments. For stable cell line creation (HEK 293), media was replaced with the proper selection antibiotic 24 hours post transfection. Clonal cell lines were generated by picking individual colonies and expression was confirmed by western blot. For ELISA and βlac experiments, 1x105 cells were plated into individual wells of a poly-D-lysine coated 48 well plate. βlac Assay For the βlac assay, 1x105 cells were plated into individual wells of a poly-D-lysine coated 48 well plate. The assay was performed 24 hours after plating. Nitrocefin was first diluted to a final concentration of 100 μM in PBS. After drug treatment. the cells were washed once with PBS and, after removal of the PBS wash, 200 μL of the nitrocefin solution was added to the each well. Immediately after the addition of the nitrocefin solution, absorbance for each well was read once every minute for 30 minutes at 486 nm using the EPOCH microplate spectrophotometer (Biotek). The rate of reaction (slope of the curve in the linear range) was taken as the readout for this assay (See Supplemental Figure 4 for an example). ELISA All ELISAs were performed as described previously[22]. Briefly, 24 hours after plating the cells and after drug treatment, plates were washed with PBS and blocked with PBS containing 1%BSA and kept on ice. Primary antibody (1:1000) was then incubated for one hour on ice. The cells were then fixed with 4% PFA for 15 minutes and subsequently blocked at room temperature with PBS with 1% BSA for 30 minutes. Secondary

This article is protected by copyright. All rights reserved
antibody (1:1000) was added and incubated for 30 minutes. The cells were washed 3 times with PBS and 1% BSA with a final wash of PBS where the HRP substrate Sigmafast OPD (Sigma) was subsequently added. After 30-45 minutes of substrate incubation, the reaction was stopped with the addition of 3M HCl. The supernatant was then transferred to a 96 well plate and absorbance was read using the EPOCH microplate spectrophotometer (Biotek) at 492 nm. Flow Cytometry Cells were plated in 6-well plates and incubated for 24 hours before drug treatment. Immediately after completion of drug treatment cells were put on ice and washed with cold PBS. The cells were lifted off by incubation for 10 min with PBS 0.02%EDTA.The EDTA was neutralized by adding cell culture media (DMEM 10% FBS). The suspensions were then spun down in a centrifuge with a swinging bucket rotor at 1500 rpm for 5 minutes and washed twice with PBS 2% FBS. Cells were spun down and resuspended in PBS 2%FBS with 1:250 primary antibody and incubated for 30 minutes. Subsequently, cells were spun down and washed twice in PBS 2%FBS. After centrifugation cells were incubated in 1:100 Alexa Fluor 647 Donkey anti-mouse IgG antibody (Invitrogen) in 100 μL PBS 2% FBS for 15 minutes. After 2 more PBS 2% FBS washes, the cells were fixed in PBS 2% PFA for 30 minutes, spun down, and resuspended in PBS 2%FBS. Subsequently, the cell suspensions were strained through cell strainer caps (BD Falcon) into round bottom tubes. Fluoresence was then acquired on a BD LSR Fortessa flow cytometer with an excitation wavelength of 640 nm, and a 670/14 bandpass filter, collecting 10000 events per sample. All steps were performed on ice and samples were protected from light after addition of secondary antibody. Data Analyses Data analyses were performed with Graphpad Prism 5.01 (Graphpadsoftware inc). Linear regression was performed on βlac data to determine the slope of the curve for comparisons. Two-tailed Student’s t-test was used for two-sample comparisons and ANOVA was used for multiple comparisons. Data acquisition and analysis for flow cytometry was performed with BD FACS Diva 6.1.2. Median fluorescence intensity was used to determine % internalization. Acknowledgement We thank Dr A. Ramsey for critical reading of the manuscript and helpful discussion. This work was partially supported by the CIHR and NSERC operating grant to AS. VL was partially funded by OGS. PB was partially funded by OGS and CIHR. We thank Dr. Alexander Sorkin for the YFP-HA-DAT cDNA and Dr. Michel Bouvier for the GBR1 and B2AR cDNA.
References
1. Kim Chiaw P, Eckford PDW, Bear CE. Insights into the mechanisms underlying CFTR channel activity, the molecular basis for cystic fibrosis and strategies for therapy. Essays Biochem 2011;50:233–48.
2. Kerem E. Mutation specific therapy in CF. Paediatr Respir Rev 2006;7 Suppl 1:S166–9.
3. Hanyaloglu AC, Von Zastrow M. Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu Rev Pharmacol Toxicol 2008;48:537–68.
4. Kahlig KM, Galli A. Regulation of dopamine transporter function and plasma membrane expression by dopamine, amphetamine, and cocaine. Eur J Pharmacol 2003;479:153–8.
5. Bouvier M. Oligomerization of G-protein-coupled transmitter receptors. Nat Rev Neurosci 2001;2:274–86.
6. Tsien RY. The green fluorescent protein. Annu Rev Biochem 1998;67:509–44.
7. Fisher GW, Adler SA, Fuhrman MH, Waggoner AS, Bruchez MP, Jarvik JW. Detection and quantification of beta2AR internalization in living cells using FAP-based biosensor technology. J Biomol Screen 2010;15:703–9.
8. Maurel D, Comps-Agrar L, Brock C, Rives M, Bourrier E, Ayoub MA, Bazin H, Tinel N, Durroux T, Prézeau L, Trinquet E, Pin J. Cell-surface protein-protein interaction analysis with time-resolved FRET and snap-tag technologies: application to GPCR oligomerization. Nat Methods 2008;5:561–7.
9. Qureshi SA. Beta-lactamase: an ideal reporter system for monitoring gene expression in live eukaryotic cells. Biotechniques 2007;42:91–6.
10. Watanabe S, Mizukami S, Akimoto Y, Hori Y, Kikuchi K. Intracellular protein labeling with prodrug-like probes using a mutant β-lactamase tag. Chemistry 2011;17:8342–9.

This article is protected by copyright. All rights reserved
11. Moore JT, Davis ST, Dev IK. The development of beta-lactamase as a highly versatile genetic reporter for eukaryotic cells. Anal Biochem 1997;247:203–9.
12. Bebrone C, Moali C, Mahy F, Rival S, Docquier JD, Rossolini GM, Fastrez J, Pratt RF, Frère JM, Galleni M. CENTA as a chromogenic substrate for studying beta-lactamases. Antimicrob Agents Chemother (Bethesda) 2001;45:1868–71.
13. O’Callaghan CH, Morris A, Kirby SM, Shingler AH. Novel method for detection of beta-lactamases by using a chromogenic cephalosporin substrate. Antimicrob Agents Chemother (Bethesda) 1972;1:283–8.
14. Weill C, Ilien B, Goeldner M, Galzi JL. Fluorescent muscarinic EGFP-hM1 chimeric receptors: design, ligand binding and functional properties. J Recep Signal Transduction Res 1999;19:423–36.
15. Sorkina T, Miranda M, Dionne KR, Hoover BR, Zahniser NR, Sorkin A. RNA interference screen reveals an essential role of Nedd4-2 in dopamine transporter ubiquitination and endocytosis. J Neurosci 2006;26:8195–205.
16. Dohlman HG, Thorner J, Caron MG, Lefkowitz RJ. Model systems for the study of seven-transmembrane-segment receptors. Annu Rev Biochem 1991;60:653–88.
17. Moore RH, Sadovnikoff N, Hoffenberg S, Liu S, Woodford P, Angelides K, Trial J a, Carsrud ND, Dickey BF, Knoll BJ. Ligand-stimulated beta 2-adrenergic receptor
internalization via the constitutive endocytic pathway into rab5-containing endosomes. J Cell Sci 1995;108:2983–91.
18. Gorentla BK, Vaughan RA. Differential effects of dopamine and psychoactive drugs on dopamine transporter phosphorylation and regulation. Neuropharmacology 2005;49:759–68.
19. Zhang J-H. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen 1999;4:67–73.
20. Salahpour A, Angers S, Mercier J-F, Lagacé M, Marullo S, Bouvier M. Homodimerization of the beta2-adrenergic receptor as a prerequisite for cell surface targeting. J Biol Chem 2004;279:33390–7.
21. Villemure J-F, Adam L, Bevan NJ, Gearing K, Chénier S, Bouvier M. Subcellular distribution of GABA(B) receptor homo- and hetero-dimers. J Biochem 2005;388:47–55.
22. Lavoie C, Mercier J-F, Salahpour A, Umapathy D, Breit A, Villeneuve L-R, Zhu W-Z, Xiao R-P, Lakatta EG, Bouvier M, Hébert TE. Beta 1/beta 2-adrenergic receptor heterodimerization regulates beta 2-adrenergic receptor internalization and ERK signaling efficacy. J Biol Chem 2002;277:35402–10.
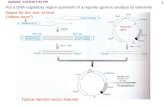
Figure 1 Schematic representation of the β-lac assay for quantitative measurement of membrane protein surface expression. In the absence of membrane protein surface expression, the cell-impermeable substrate nitrocefin is not cleaved (left panel). When a β-lac tagged-membrane protein is present at the surface (right panel), nitrocefin is cleaved resulting in a change of absorbance from (λ=390nm, pH 7.0-yellow) to (λ=486nm, pH 7.0-red).

This article is protected by copyright. All rights reserved

This article is protected by copyright. All rights reserved
Figure 2 Comparison between β-lac assay, ELISA, and Flow Cytometry using the β2AR. Time course of isoproterenol (10 µM) mediated internalization of β2AR as measured using the βlac (A), ELISA (B), and flow cytometry (C) assays (n=4). Dose response of isoproterenol mediated internalization (30 min) of β2AR as measured using the βlac (D), ELISA (E), and flow cytometry (F) assays (n=3). One-way ANOVA with Bonferroni corrected t-test post-hoc were used to determine differences between data sets * P<0.05, ** P<0.01, *** P<0.001. All data are represented as mean % of vehicle treated or mock transfected cells ± S.E.M.

This article is protected by copyright. All rights reserved
Figure 3 Comparison between β-lac assay and ELISA using GBR1, and DAT. Surface expression of GBR1 with increasing amount of GBR2 DNA as measured by the βlac (A) and ELISA (B) assays (n=3). Surface expression of DAT after stimulation with PMA (1μM-60 min) alone or with pre-treatment with GBR12909 (100uM-15 min) as measured using the βlac (C) and ELISA (D) assays (n=4). One-way ANOVA with Bonferroni corrected t-test post-hoc were used to determine differences between data sets * P<0.05, ** P<0.01, *** P<0.001. All data are represented as mean % of vehicle treated or mock transfected cells ± S.E.M.

This article is protected by copyright. All rights reserved
Table 1. Comparison of time and cost of ELISA and βlac assays steps ELISA βlac
1 Wash (1 time) Wash (1 time) 2 Block Add nitrocefin 3 Add 1° antibody Read (Abs 486nm) 4 Wash (3 times) 5 Fix (4% PFA) 6 Wash (3 times) 7 Block 8 Add 2° antibody 9 Wash (3 times)
10 Add substrate 11 Stop reaction 12 Read (Abs 496nm)
time 4- 6 hours 15-60 minutes cost 30-50 cents/well 4 cents/well

This article is protected by copyright. All rights reserved
Supplemental information
Supp. Figure 1 Cell permeability of the βlac substrate nitrocefin. A rat β-arrestin2-βlac construct was created by replacing the YFP in the β-arrestin-YFP construct with βlac [1]. Cells were transfected with 0, 0.5, 1, and 2 μg of βarrestin-βlac DNA, and were plated in a 48-well plate 24 hours post-transfection. At 48 hours post-transfection, cells were washed and incubated for 15 minutes in either PBS or ddH20. After incubation, cell solutions in the ddH20 treated wells were mechanically lysed by pipetting up and down. PBS was aspirated from the PBS-treated wells and nitrocefin in PBS was added. To the ddH2O treated wells, a solutioin of 2X nitrocefin in 2X PBS was then added , and nitrocefin hydrolysis was monitored. Unlysed cells showed no increase in signal compared to mock transfected cells, regardless of the amount of βarrestin-βlac DNA that was transfected. Cell lysis resulted in increases in signal with increasing amounts of βarrestin-βlac DNA reaching 33±6.8.fold compared to 2±0.9 fold for lysed mock cells. Mean ± S.E.M, n=3. Supp. Figure 2. Functionality of βlac constructs compared to untagged proteins. A) EPAC stable cells were transfected with 50ng of βlac- β2AR (red), HA-β2AR (blue), or mock transfected (black) and seeded into a 96 well plate 24 hours post transfection. The cells were incubated with increasing doses of isoproterenol and the signalling was quantified through BRET[2]. The EC50’s for isoproterenol induced cAMP production was 3.3 and 1.5 nM for the βlac and HA-β2AR respectively. B) WT-GBR1 or βlac-GBR1 were transfected with GBR2 (0.5-5μg). Treatment with forskolin results in formation of cAMP and therefore a change in BRET. This forskolin signal is partially blocked by -4M, but not –9M baclofen treatment in the wildtype and βlac tagged GBR1. t-tests were performed to compare the effect of -4M baclofen to forskolin only treated cells n=4, * P<0.05, ** P<0.01 C) Uptake of 3H [DA] in cells stably expressing HA-DAT (blue) or βlac-DAT (red) was measured using liquid scintillation counting. Non-linear regression of the data shows similar curves, with Km values of 3.2±0.7 μM and 4.2±1.6 μM for βlac -DAT and HA-DAT respectively. Graph is representative of two experiments, each data point represents the average of three wells. One-way ANOVA with Bonferroni corrected t-test post-hoc were used to determine differences between data sets * P<0.05, ** P<0.01. All data are represented as mean % of vehicle treated or mock transfected cells ± S.E.M.
Supp. Figure 3 Z’ of β2AR internalization. Evaluation of Z’of β2AR internalization after stimulation with isoproterenol. Cells were treated with either vehicle (blue) or 10µM isoproterenol (red) for 30 minutes and internalization was assessed using the using the βlac assay. The Z; was found using the following equation[3]:
Z’= 1 - (3σc+ + 3σc-) / (|µc+ - µc-|) Where µc represents the mean and σc the standard deviations of the controls, where the (+) and (-) denote positive (vehicle) and negative (10µM isoproterenol) controls respectively. The mean is represented by the solid black lines (µc+ = 0.18 µc- = 0.144) and the 3σ (σc+ = 0.006 σc- = 0.006) by the dotted lines for each control. The calculated value is Z’=0.52.
Supp. Figure 4 Representative absorbance/time curves of cells expressing βlac constructs upon addition of nitrocefin. All data analyses in previous figures were performed by taking the slope of the nitrocefin hydrolysis. A) -β2AR cells treated with vehicle (red) or 10μM isoproterenol (blue) with slopes of 0.0033 and 0.0021 units/min respectively. B) βlac-DAT cells treated with vehicle (red) or 1μM PMA (blue) with slopes of 0.0015 and 0.001 units/min respectively. C) GBR1 cells transfected with 0μg GBR2 (red) or 2 μg of GBR2 (Blue) with slopes of 0.0013 and 0.0025 units/min respectively. All curves were fitted with linear regression yielding r2>0.999.

This article is protected by copyright. All rights reserved
References
1. Angers S, Salahpour A, Joly E, Hilairet S, Chelsky D, Dennis M, Bouvier M. Detection of beta 2-adrenergic receptor dimerization in living cells using bioluminescence resonance energy transfer (BRET). Proc Natl Acad Sci U S A 2000;97:3684–9.
2. Barak LS, Salahpour A, Zhang X, Masri B, Sotnikova TD, Ramsey AJ, Violin JD, Lefkowitz RJ, Caron MG, Gainetdinov RR. Pharmacological Characterization of Membrane-Expressed Human Trace Amine-Associated Receptor 1 ( TAAR1 ) by a Bioluminescence Resonance Energy Transfer cAMP Biosensor. Mol Pharmacol 2008;74:585–594.
3. Zhang J-H. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen 1999;4:67–73.