







γλώσσες
Σελίδες
Νομικός


Peixuan Guo
Structure and Function of φ29 Hexameric RNA That Drives the Viral DNA Packaging Motor: Review PEIXUAN GUO Department of Pathobiology and Purdue Cancer Center, Purdue University, West Lafayette, IN 47907, USA
INTRODUCTION I. APPROACHES AND STRATEGIES IN THE STUDY OF PRNA A. Production of infection virion in vitro with synthetic and purified components – a sensitive system for the assay of pRNA B. Construction of active circularly permuted pRNA (cp-pRNA) C. Determination of the stoichiometry of pRNA 1. Stoichiometry determination by binomial distribution 2. Stoichiometry determination by log/log plot with a serial of dilution 3. Stoichiometry determination by finding the common multiple of 2 and 3 4. Is there a transition from hexamer to pentamer inferred from cry-EM imaging? D. Construction of a pRNA inactive in DNA packaging while competent in procapsid binding II. STUDIES ON pRNA STRUCTURE A. Complementary modification B. Phylogenetic analysis C. Photoaffinity crosslinking with GMPS/Aryl Azide 1. Intra-molecular crosslinking of monomers 2. Intermolecular crosslinking of dimers D. Crosslinking by psoralen E. Crosslinking by phenphi F. Chemical modification. G. Chemical modification interference H. Ribonuclease probing I. Genetic analysis by truncation, insertion, deletion and mutation J. SELEX K. Cryo-AFM (atomic force microscopy) L. Modeling the three-dimensional structure of pRNA M. Conformational change of pRNA
1. Transition from monomer to dimer--- conformational change induced by Mg++
2. Transition from dimer to hexamer 3. Conformational shift in executing the motion task III. RNA/PROTEIN INTERACTION
A. Binding of pRNA to connector gp10 but not to capsid protein B. Foot printing and chemical probing of procapsid-bound pRNA C. Connector domain and sequence for pRNA binding
D. Interaction with DNA packaging protein gp16? IV. EFFECT OF MONO AND DIVALENT CATIONS V. FUNCTION OF pRNA A. pRNA remained associated with procapsids during DNA packaging B. pRNA leaves DNA-filled capsid after the addition of the tail protein gp9 C. Separation of the pRNA into two domains: procapsid binding domain and DNA packaging domain.
D. Dimer as the building block in pRNA formation E. Sequence and size requirement of the loops in dimer formation
F. Minimal sequence requirement for dimer formation
Progress in Nucleic Acid Research Copyright 2002, Elsevier Science (USA). And Molecular Biology, Vol. 72 All Rights reserved.

Peixuan Guo
G.. Minimal sequence requirement for DNA packaging H. Mechanism and role of pRNA in phi29 DNA translocation
1. Sequential action of six pRNA 2. Evidence to support sequential action
a. DNA packaging is block by one pRNA in the chain b. pRNA is very sensitive to mutation c. Complementation cannot rescue inactive pRNA with a single base mutation d. No compensation by wild type e. Math simulation and modeling VI. SIGNIFICANCE AND APPLICATION IN pRNA STUDIES A. A model for the study of RNA dimers and trimers B. A model for the study of viral DNA packaging and design of new antiviral strategies 1. Inhibition of ø29 assembly by antisense DNA targeting pRNA 2.Complete inhibition of phi29 assembly with mutant pRNA C. Similar mechanism between viral DNA translocation and other nucleic acid sliding/riding process D. Insight hints for macromolecular translocation through barrier wall VII. CONCLUDING REMARKS
INTRODUCTION A virus is composed of both protein and nucleic acid, either DNA or RNA. Their genomic DNA or RNA is enclosed in a protein shell or capsid. During replication, the synthesis of viral proteins and genomic DNA or RNA is accomplished by two different sets of machinery. After the synthesis of structural proteins and the viral genome, these components must interact with each other to form a complete virion, a process referred to as virus assembly 1-4. One striking feature in the assembly of many linear double-stranded DNA (dsDNA) viruses, including adenovirus 5;5-9, herpesvirus 10-12, poxvirus 13-15, bacteriophages T116, T317, T418, T519, T7 20;21, P122, P223;24, P2225-27, Mu28, ø2129, ø2930-33, SP0134;35, SPP136;37, λ38-41, and their relatives, is that the viral genome is inserted into a preformed shell called a procapsid during maturation (Fig. 1) 1-3;42-44. Procapsids of dsDNA phages contain a connector (or portal vertex) that serves as part of the DNA translocation machinery 1;36;42. Interestingly, a connector with 12 protein subunits and with a function similar to that of dsDNA phage connectors has been found recently in herpes simplex virus type 145. The translocation of DNA into the procapsid is energetically unfavorable due to the very compact of DNA arrangement within the capsid 43;46;47, therefore ATP hydrolysis is required for packaging 4;44;48-50. Quantification of ATP consumption in DNA packaging with purified components revealed that one ATP is needed for the translocation of two base pairs of DNA into the procapsid 50;51.
Fig. 1: Steps in the production of infectious virion phi29 in vitro using purified and synthetic components (reproduced from 31with permission from J. Virology). An exciting aspect of ø29 DNA packaging is the discovery that a small viral RNA, called pRNA has a novel and essential role in viral DNA packaging 52 (Fig. 2). This pRNA is not a structural component of the mature virion 52, rather, it is a transient factor that

Peixuan Guo
serves as a vital component of the viral motor to drive genomic DNA into the viral procapsid 53. Since the discovery of RNA catalytic activities in 1982 54-56, there has been a revolution in the concept about RNA function, that is that RNA can act like an enzyme, thus an aforesaid term "ribozyme”. Except the newly emerged telomerase, which is the ribonucleoprotein enzyme responsible for the replication of chromosome ends 57, most ribozymes - including RNase P 58; self-splicing introns 54; the hepatitis delta ribozyme 59-61; as well as the hammerhead62-65 and hairpin ribozymes 66;67 - are all involved in catalytic reactions involving RNA substrates. In contrast, the phi29 pRNA provides a model for a ribozyme that acts on DNA.
Fig. 2: Secondary structure, domain and location of pRNA on phi29 viral particle. A. Secondary structure of wild-type pRNA A-b’. The binding domain (shaded area) and the DNA translocation domain are marked with bold lines. The four bases in the right and left loops, which are responsible for inter-RNA interactions, are boxed. B. Power rangers depict the formation of pRNA hexamer by hand-in-hand interaction. C. Diagrams depicting the formation of a pRNA hexameric ring and it’s location in the phi29 DNA packaging machine. Hexamer formation is via right-and-left-loop sequence interactions of pRNA A-b’ and B-a’. The shaded hexagon stands for the connector and the surrounding pentagon stands for the fivefold symmetrical capsid vertex, viewed as end-on with the virion as side-view. The six protrusions stand for six pRNAs with the central region bound to the connector and the 5’/3’ paired region extending outward (adapted from 99with permission from RNA). The phi29 pRNA was first discovered from experiments showing that the packaging of phi29 DNA was sensitive to treatment with RNase A or T1 52. DNA packaging was not inhibited by RNase that was pretreated with RNase inhibitor, indicating that the inhibitory effect on packaging was due specifically to RNase. Electrophoresis of purified procapsids revealed the presence of a 120-base polynucleotide that was resistant to DNase 52. This pRNA can be isolated from purified procapsids by incubation with EDTA, and the purified pRNA can bind to RNA-free procapsids in the presence of Mg++. pRNA-free procapsids are inactive in DNA packaging. However, the addition of pRNA to procapsids reconstitutes DNA packaging activity, and the subsequent DNA filled capsid can be converted into infectious virus. RNA-DNA hybridization analysis was used as a first step to determine the origin of the pRNA 52. The pRNA purified from procapsids was labeled with [32P] and used as a probe in RNA-DNA hybridization with restriction fragments of phi29 DNA. The pRNA hybridized specifically to restriction fragments from the left end of the phi29 DNA, which was the end packaged first during DNA encapsidation. Further analysis confirmed that the pRNA was transcribed from bases 320 to 147 of phi29 genome, driven by the early promotor PE1 (A1)68-70, with transcription being from right to left 48. The pRNA was transcribed primarily as a 174-base molecule in phi29-infected cells. However, nuclease cleavage during procapsid purification eliminated 54 bases from its 3’-end 71. Both the 174- and 120-base RNA molecules have similar activity in DNA packaging in vitro 70-72. This review will focus on the work produced in the author’s laboratory during the last eleven years. Some related works from other labs will also be discussed. The entire article will deal with the structure and function of the pRNA, as well as the methods and approaches that were used to investigate pRNA structure and function. I. APPROACHES AND STRATEGIES IN THE STUDY OF pRNA Four major approaches have been undertaken in this lab to study the structure and function of pRNA. The first one is the development of a highly sensitive in vitro phi29 virion assembly system for the assay of pRNA activity. With this system, up to 5x109 infectious virions per ml can be obtained in the presence of pRNA, yet not a single infectious virion is detected in the absence of the pRNA. Therefore, a system with 9-order of magnitude sensitivity could be used for the analysis of pRNA structure and function. The second approach is the construction of circularly permuted pRNA (cp-pRNA) in which any internal base of the pRNA could be reassigned to serve as new 5’- or 3’-termus. The circular permutation system greatly facilitates the construction of mutant pRNA via PCR and makes it possible to label any specific internal base by radioisotopes or photoaffinity agents. The third approach to analyze
Peixuan Guo
pRNA is to design new methods to study the stoichiometry of pRNA in DNA packaging. A fourth approach is the construction of mutant pRNAs that were inactive in DNA packaging, but competent to compete with wild type pRNA for procapsid binding. A. Production of infectious virions in vitro with synthetic and purified components–--a sensitive system for the assay of pRNA Assembly of infectious phi29 virus requires the following components: a procapsid into which the viral DNA genome will be inserted, a viral DNA genome, DNA-packaging protein gp16, and pRNA. These viral components allow formation of DNA-filled capsids. DNA-filled capsids can be converted into infectious virions with the addition of three additional structural proteins gp9 (tail), gp11 (collar), gp12 (anti-receptor), and a factor gp13 that is required for the assembly of gp11 and gp12 onto the DNA-filled capsid. ATP serve as energy source for DNA translocation and magnesium is required for the appropriate folding of pRNA. The procapsid of the Bacillus subtilis bacteriophage ø29 consists of the major capsid protein gp8 (235 copies), head fiber protein gp8.5 (47 copies), scaffolding protein gp7 (26 copies), and connector protein gp10 (12 copies)73;74 42;75,76. The genome of ø29 is a linear ds-DNA of 19,285 bp. A virus-encoded terminal protein, gp3, is covalently linked to each 5’-end of the genome through the formation of a phosphodiester bond between the -OH group of serine residue 232 of gp3 and the 5’ dAMP of genomic DNA 76 77-79. The terminal protein gp3 functions both as a “primer” in the initiation of DNA replication 77;80;80-82, and as an enhancer for DNA packaging 48;83. Full-length ø29 genomic DNA has been synthesized in vitro 31;80-82. The gp16 protein is part of an ATPase complex 50;84. The genes coding for the structural protein gp7, gp8 and gp10 of phi29 procapsid have been cloned and overexpressed in E.coli, allowing the assembly and purification of procapsids that are competent in in vitro DNA packaging 53;85. Other components, gp16 49, gp9, gp11, gp12, and gp13 31;86 are all purified from cloned and overexpressed genes.
Table 1. (reprinted from 31 with permission from J. Virology). To perform in vitro phi29 assembly, purified procapsids are dialyzed against an EDTA-containing buffer at ambient temperature to unblock the binding sites for pRNA. pRNA is subsequently added, and the mixtures are then dialyzed against magnesium-containing buffer to allow pRNA to attach to the procapsid. The pRNA-enriched procapsids are mixed with gp16, DNA-gp3, and a reaction buffer (containing ATP) to complete the DNA packaging reaction. After DNA packaging, gp9, gp11, gp12, and gp13 are sequentially added to the DNA packaging reactions to convert the DNA-filled capsids into infectious virions (Fig. 1). The yield is then assayed by standard plaque formation. Up to 5 x 109 pfu/ml of the infectious phi29 virions have been assembled in vitro with the exclusive use of ten purified components (Table 1). Omitting the pRNA resulted in no plaque formation. Electron microscopy and genome restriction mapping have confirmed the identity of the infectious ø29 virions synthesized in this system. Due to the absence of any background, the generation of a single infectious virion could be detected. With this system, it has been possible to detect the activity of mutant pRNAs with 105 -fold reductions in DNA packaging efficiency 86, which is too low to be detected by other assay methods available 49;83. The system has been used extensively to study the structure and function of pRNA, including functional domains 87-89 and stoichiometry. B. Constructions of Active Circularly Permuted pRNA (cp-pRNA) Utilizing 5' to 3'-end ligation, Pan et al were able to construct circular tRNA, which was subsequently cleaved with limited alkaline hydrolysis to generate one random break per molecule 90 and generated RNA molecules that were used for structural analysis. Similarly, Nolan et al. synthesized tRNAs with native 5' and 3' ends linked by a synthetic loop and new termini in the interior of the native sequence 91, which they used to map tRNA binding sites on RNase P. The feasibility of constructing circularly permuted RNAs rests on the close proximity of the native RNA 5' and 3' ends. The ø29 pRNA 5'- and 3'-ends are in close proximity 87, hence a series of pRNA molecules with circular permutations have been constructed (Fig. 3). To construct cp-pRNA, two tandem pRNA-coding sequences separated by a 3-base or 17-base loop sequence have been cloned into a plasmid (Fig. 3). PCR primer pairs, such as P6/P5, complementary to various locations within pRNA coding sequences, could be designed to synthesize PCR fragments for transcription of cp-pRNAs. It has been shown that neither a small nor a large linker loop interferes with the biological activity of the pRNA molecule 92. Additionally, most of the bases, especially the nonessential bases, can be used as new termini for constructing active cp-pRNA 93.

Peixuan Guo
Fig. 3: Construction of tandem DNA templates for the synthesis of circularly permuted pRNA (cp-pRNA). Two pRNA-coding sequences are cloned into a plasmid with head-to-tail linkage via three nucleotides AAA. Each pairs of PCR primers, such as P6/P5, containing SP6 or T7 promoter, are used to generate a DNA fragment for RNA transcription. Arrows indicate positions of new 5’/3’ termini that have been used (Adapted from 88;93 with permission). With the synthesis of biologically active cp-pRNA, the analysis of specific interactions of unique internal bases with components of the phi29 DNA packaging complex is now possible. By labeling the new 5’-or 3’-terminus with photoaffinity cross-linking agents, specific interactions of the modified bases with proteins, DNA, or RNA can be elucidated 91. C. Determination of the stoichiometry of pRNA To elucidate the role of the pRNA in DNA packaging, it is crucial to know how many copies of the pRNA are involved in each DNA packaging event. Three approaches have been developed to determine the stoichiometry of the pRNA that have led to the conclusion that six pRNAs are present in each DNA translocating motor. The quantification of pRNA has been investigated by several approaches 71;94-96. This section will focus only on innovative methods developed in this reviewer’s lab. 1. Determination of the stoichiometry by binomial distribution The first approach of pRNA stoichiometry determination involves the use of binomial distribution 95;97. pRNAs with mutations in the 5’/3’ paired region retain their procapsid binding capacity but lose their DNA packaging function (Section I.D & V.C). When mutant pRNA and wild-type pRNA are mixed at various ratios in in vitro assembly assays (i.e. 10% mutant and 90% wild-type etc.), the probability of procapsids possessing a certain amount of mutant and a certain amount of wild-type can be determined by the
expansion of a binomial: (p+q)z=
z0
p
z+
z1
p
z-1q+
z2
p
z-2q2+...
zz − 1
pq
z-1+
zz
q
z= Z
M
M =0
z
∑ pz-M
qM
where
is equal to:
(
ZM
Z!M!(Z − M)!
), and Z represents the total number of pRNA per procapsid, while p and q represent the % of mutant and wild type
pRNA, respectively, which are used in the reaction mixture. For example, if the total number of pRNA per procapsid required for DNA packaging (Z) is 3, then the probability of all combinations of mutant (M) and wild type (N) pRNAs on a given procapsid can be determined by the expansion of the binomial: (p + q)3 = p3 + 3p2q +3pq2 +q3=100%. That is, in the procapsid population, there is the probability of a procapsid possessing either three copies of mutant pRNA, two copies of mutant and one copy of wild type, one
copy of mutant and two copies of wild type, or three copies of wild type, which are p3, 3p2q, +3pq2, or q3, respectively. Suppose that there are 70% mutant and 30% wild type pRNA in a reaction mixture, then, the percentage of procapsids that possess one copy of
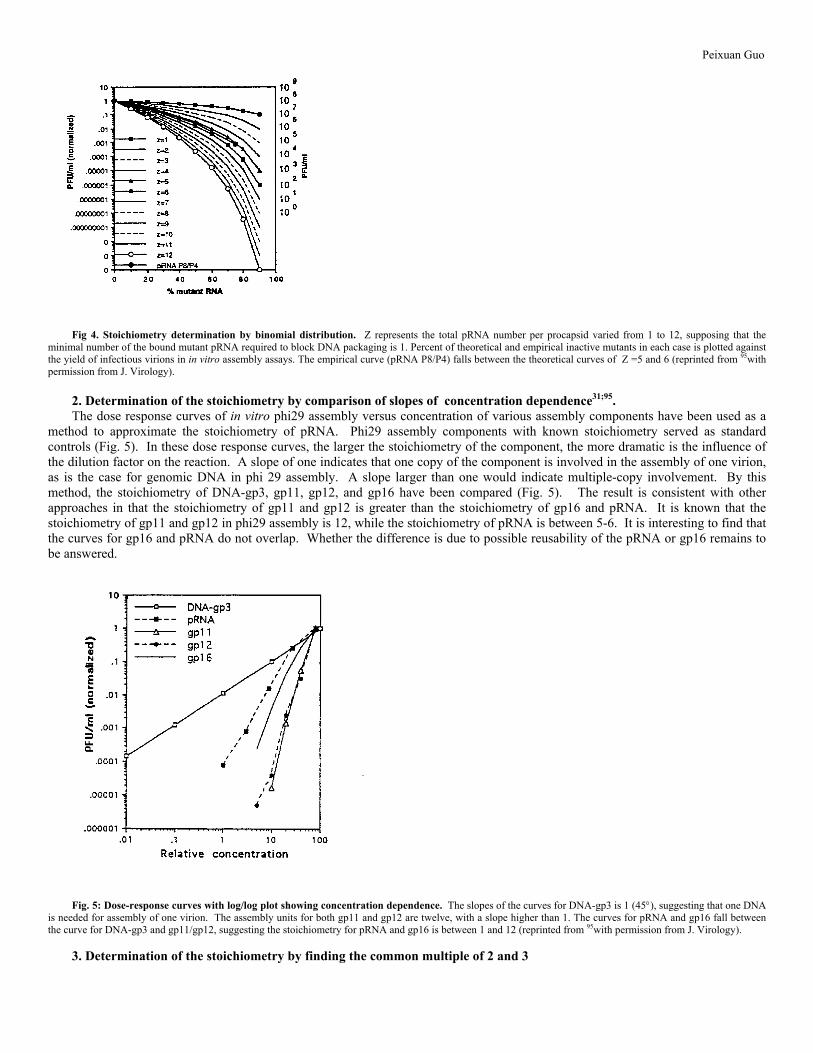
mutant and two copies of wild type is 3pq2, that is, 3(0.7)(0.3)2= 19%. If only one mutant pRNA per procapsid is sufficient to render the procapsid unable to package DNA, then only those procapsids with 3 wild type pRNAs bound (0 mutant pRNA, q3) would package DNA. The yield of virions from the empirical data is plotted and compared to a series of predicted curves to find a best fit (Fig. 4). It was found that the experimental inhibition curve most closely resembled, in both slope and magnitude, the predicted curves where Z=5 or 695.
Peixuan Guo
Fig 4. Stoichiometry determination by binomial distribution. Z represents the total pRNA number per procapsid varied from 1 to 12, supposing that the minimal number of the bound mutant pRNA required to block DNA packaging is 1. Percent of theoretical and empirical inactive mutants in each case is plotted against the yield of infectious virions in in vitro assembly assays. The empirical curve (pRNA P8/P4) falls between the theoretical curves of Z =5 and 6 (reprinted from 95with permission from J. Virology). 2. Determination of the stoichiometry by comparison of slopes of concentration dependence31;95. The dose response curves of in vitro phi29 assembly versus concentration of various assembly components have been used as a method to approximate the stoichiometry of pRNA. Phi29 assembly components with known stoichiometry served as standard controls (Fig. 5). In these dose response curves, the larger the stoichiometry of the component, the more dramatic is the influence of the dilution factor on the reaction. A slope of one indicates that one copy of the component is involved in the assembly of one virion, as is the case for genomic DNA in phi 29 assembly. A slope larger than one would indicate multiple-copy involvement. By this method, the stoichiometry of DNA-gp3, gp11, gp12, and gp16 have been compared (Fig. 5). The result is consistent with other approaches in that the stoichiometry of gp11 and gp12 is greater than the stoichiometry of gp16 and pRNA. It is known that the stoichiometry of gp11 and gp12 in phi29 assembly is 12, while the stoichiometry of pRNA is between 5-6. It is interesting to find that the curves for gp16 and pRNA do not overlap. Whether the difference is due to possible reusability of the pRNA or gp16 remains to be answered.
Fig. 5: Dose-response curves with log/log plot showing concentration dependence. The slopes of the curves for DNA-gp3 is 1 (45°), suggesting that one DNA is needed for assembly of one virion. The assembly units for both gp11 and gp12 are twelve, with a slope higher than 1. The curves for pRNA and gp16 fall between the curve for DNA-gp3 and gp11/gp12, suggesting the stoichiometry for pRNA and gp16 is between 1 and 12 (reprinted from 95with permission from J. Virology).
3. Determination of the stoichiometry by finding the common multiple of 2 and 3

Peixuan Guo
The stoichiometry of pRNA has also been investigated by the mixing of two inactive mutant pRNAs with loops complementary intermolecularly, and then assaying the activity of such mixtures in DNA packaging (Fig. 6) 96;98. The predicted secondary structure of the pRNA (Fig. 2) reveals two loops, called left and right hand loops 99. Sequences of these two loops (bases 45-48 of right hand loop and bases 85-82 of left-hand loop) are complementary and were originally proposed to form a pseudoknot 100. However, extensive studies have revealed that these two sequences interact intermolecularly, allowing the formation of pRNA oligomers. Several pRNAs with mutated left and right hand loop sequences were constructed. To simplify the description of these mutants, uppercase and lowercase letters are used to represent the right and left-hand loop sequences of the pRNA, respectively (Fig. 2). The same letter in upper and lower cases symbolizes a pair of complementary sequences. For example, in pRNA A-a’, the right loop A
(5’GGAC48) and the left loop a’ 3’CCUG82) are complementary, while in pRNA A-b’, the four bases of the right loop A are not complementary to the sequence of the left loop b’ (3’UGCG82). Mutant pRNAs with complementary loop sequences (such as pRNA A/a') are active in phi29 DNA packaging, while mutants with non-complementary loops (such as pRNA A/b') are inactive (Table 2-4).
Table 2. (Adapted from 96 with permission).
Table 3. (Adapted from 96 with permission).

Peixuan Guo
a. A set of two interlocking pRNAs96 A key finding in pRNA research was that the mixing of two mutant pRNAs with trans-complementary loops restored DNA packaging activity. For example, pRNAs A/b' and B/a' were inactive in DNA packaging alone, but when mixed together restored DNA packaging activity. This result can be explained by the trans-complementarily of pRNA loops, i.e. the right hand loop A of pRNA A/b' could pair with the left hand loop a' of pRNA B/a'. Since mixing two inactive pRNAs with interlocking loops, such as when pRNA I-j’ and J-i’ are mixed in a 1:1 molar ratio, resulted in production of infectious virions, the stoichiometry of the pRNA is predicted to be a multiple of two (Table 2-4). Together with the results from binomial distribution and serial dilution analyses, it has been confirmed that the stoichiometry of pRNA is six.
Table 4. (Adapted from 96 with permission). b. A set of three interlocking pRNAs96 Another set of mutants is composed of three pRNAs, J-p’, P-k’ and K-j’. This set is expected geometrically to be able to form a 3-, 6-, 9-, or 12-mer ring that carries each of the three mutants. When tested alone, each individual pRNA shows little or no activity (Table 2-4). When any two of the three mutants are mixed, again little or no activity is detected. However, when all three pRNAs are mixed in a 1:1:1 ratio, DNA packaging activity is restored. The lack of activity in mixtures of only two mutant pRNAs and the restored activity in mixtures of three mutant pRNAs is expected since the mutations in each RNA are engineered in such a way that only the presence of all three RNAs will produce a closed ring. The fact that the three inactive pRNAs are fully active when mixed together suggests that the number of pRNAs in the DNA packaging complex is a multiple of 3, in addition to being a multiple of two (Table 2-4). Thus the number of pRNAs required for DNA packaging is a common multiple of 2 and 3, which is 6 (or 12 but this number has been excluded by the approach of binomial distribution (Section I.C.1). c. A set of six inter-locking pRNAs96 DNA packaging activity is also achieved by mixing six mutant pRNAs each being inactive when used alone (Table 2-4). Thus, an interlocking hexameric ring can be predicted to form by the base pairing of the interlocking loops. 4. Is there a transition from hexamer to pentamer inferred from cryo-EM imaging? Two laboratories using cryo-electronic microscopy have also reported the stoichiometry of the pRNA on procapsids. Both labs compared the images of pRNA-free procapsids to images of pRNA-bound procapsids. After extracting the image of the pRNA-free procapsid from the procapsid with pRNA bound, Carrascosa and coworkers revealed a pRNA hexamer bound to procapsids (Fig. 6-A) 101. In contrast, using the same techniques and approaches, Baker, Rossmann and coworkers demonstrated a pRNA pentamer on phi29 procapsids (Fig.6-B)102. One explanation by Baker, Rossmann and coworkers is that pRNA hexamers are formed initially, but after binding to procapsids, one of the pRNAs is dissociated from the procapsids leaving five pRNAs still bound. A resolution to this controversy has not come about, however, it is worth noting that due to the low electron density of RNA molecules and the resolution limit of EM along with the interference of the fivefold symmetrical environment of the procapsid shell around the pRNA complex, EM image reconstruction for such a molecule remains difficult. Other biochemical and biophysical approaches are needed to clarify the discrepancy. It would be very interesting to see how the five-fold symmetry pRNA pentamer could interact with a six-fold symmetrical connector. The other explanation for the appearance of a pentamer by Baker, Rosen and coworkers is that, the size prediction by computer suggests that the pRNA hexamer with the predicted computer model 98 could not fit into the structural environment of the connector embedded in the procapsid 102. With reference to the pentamer, they predicted that the best explanation for the pentameric pRNA with smaller size is that the five copy of the pRNA binds to the capsid protein gp8 with a five-fold symmetry, rather than binding to the 12 or 6-fold symmetrical connector. However, extensive investigation by filter binding assay, sedimentation analysis, and genetic studies concludes that pRNA binds to the connector (Section III.A) If such a shift in binding target does occur, it might represent anther novel mechanism in pRNA function. Another possibility might be that six pRNAs bind to procapsid in such a way that two of them lay on top of each other to get ready to gear DNA translocation motor, resulting in an appearance of pRNA pentamer.

Peixuan Guo
Fig. 6: Cryo-EM images showing that the pRNA forms hexamer A 99 and pentamer (B) 100 from two different laboratories using the similar approaches (Reprinted by permission from J. Mol. Biol99 copy right 2000 Academic Press LTD, and by Nature 100 copy right 2000 Macmillan Magazines Led). D. Construction of a pRNA inactive in DNA packaging while competent in procapsid binding Extensive investigation has confirmed that pRNA contains two functional domains. The procapsid binding domain is located at the middle part of the molecule, and the DNA translocating domain is located at the 5’-/3’-paired ends. Deletion or mutation of the DNA translocating domain generates a mutant pRNA inactive in DNA packaging while having a procapsid binding efficiency equivalent to wild type pRNA95;103. This special feature of mutant pRNA has greatly facilitated the study of pRNA structure and function. As described in the previous and following sections, this mutant has been used for competitive inhibition in procapsid binding103, for stoichiometry determination using binomial distribution95, for design of antiviral approach103;104, for model simulation leading to the finding of the mechanism of sequential action105, and for the design of chemical modification interference106. II. STUDIES ON pRNA STRUCTURE The astonishing diversity in RNA function is attributed to the flexibility in RNA structure. The question of how some RNA molecules perform their versatile and novel functions is largely unknown. To elucidate this question, it is crucial to understand the principles and rules that regulate RNA structure building. Due to its complexity and versatility, the mechanisms of RNA folding remain to be determined. NMR and X-ray crystallography 107-109 have been utilized to obtain a physical tertiary structure of RNAs. The very limited number of reported RNA crystallographic structures 109-113 compels the use of alternative approaches to obtain information on RNA structures. A. Complementary modification Computer analysis of pRNA secondary structure 114 shows that the 5’ and 3’ ends are paired. Complementary modification was used to confirm the pRNA secondary structure in this and other areas of the molecule 93;100;115. An extensive series of helix disruptions by base substitutions almost always resulted in the loss of DNA packaging activity. Additional compensatory mutations that restored the predicted base pairings rescued the activity of pRNA. Such complementary modification has led to the conclusion that bases 1-3 are paired with bases 117-115; bases 7-9 are paired with bases 112-110; bases 14-16 are paired with bases 103-101; and bases 76-78 are base paired with 90-88 (Fig. 2). This second site suppression confirmed the existence of a helical structure and that this helical structure are essential for pRNA function. Complementary modification has also been applied to the study of loop/loop interaction in dimer formation 96;98;99. A series of mutant pRNAs carrying mutated right and/or left-hand loop sequences have been constructed to determine the intermolecular interaction by the mixing of inactive mutant pRNAs, each having interactive complementary loops, with each other. All mutant pRNAs that had unpaired right and left loops, such as pRNA J-i’, were inactive in in vitro phi29 assembly when used alone. However, when two inactive pRNAs that were trans-complementary in their right and left loops, for example pRNA I-j’ and J-i’, were mixed in a 1:1 molar ratio, full activity was restored. The observed activity of a mixture of two inactive mutants (Table 2-4) confirmed that the right loop interacted with the left loop of a different RNA, and was involved in dimer formation. B. Phylogenetic analysis Phylogenetic analysis of pRNAs from Bacillus subtilis phages SF5, B103 116, phi29, PZA, M2, NF, and GA1 117, shows very low sequence identity and few conserved bases, yet, the family of pRNAs appears to have similar predicted secondary structures99;117 (Fig. 7). The requirement for pRNA in ø29 assembly appears to be very specific in that pRNAs from other phages cannot replace the ø29 pRNA in in vitro packaging assays 117, and that a single base mutation can render the pRNA completely inactive 115.

Peixuan Guo
All seven pRNAs of these phages contain both the right and left hand loops. Complementary sequences within the two loops are found in each of these pRNAs. The numbers of paired bases are from five (5’-GUUUU/CAAAA-5’) for SF5 to four (5’-AACC/UUGG-5’) for phi29/PZA and B103, to three (5’-AUC/UAG-5’) for M2/NF, and two (5’-CC/GG-5’) for GA1. Phylogenetic analysis led to the conclusion that there must be at least one G/C pair in the pairing of the right and left hand loops. This conclusion is based on the observation that all predicted paired sequences in the right and left-hand loops contain at least one G/C pair. Two G/C pairs are found in phi29 PZA and GA1 pRNAs, and only one G/C pair is present in pRNAs of B103, M2/NF and SF5. The finding that the paired loop sequences in GA1 consist of only two G/C pairs supports the conclusion that two G/C pairs are sufficient to mediate pRNA interactions 96;98. C. Photoaffinity Crosslinking with GMPS/Aryl Azide Aryl azides can be incorporated into RNA molecules to obtain three-dimensional structural information 91. These reagents contain groups that are chemically inert in the absence of light, but can be converted to a reactive nitrene by UV irradiation at long wavelengths 118;119. Aaryl azides have been specifically attached to the 5’ end of the pRNA or to cpRNAs. To accomplish this 5’-end labeling, 5'-thiophosphate pRNA or cpRNA is synthesized by in vitro transcription in the presence of excess GMPS (guanine-monophosphorothioate) over GTP 119. GMPS is an efficient primer of RNA synthesis by T7 RNA polymerase but cannot be used for chain elongation. The 5'-thio-pRNA and the 5'-thio-cpRNAs are then treated with azidophenacyl bromide to produce the 5'-azido-pRNA and 5'-azido-cpRNAs, respectively, by the nucleophilic displacement of bromine 119. The azido group is converted to a reactive nitrene by long wavelength UV irradiation, which then inserts into nearby bonds resulting in covalent crosslinks 118. Our previous work showed that it is possible to generate active cpRNAs by assigning certain internal sites of the pRNA as new 5’- and 3’-termini (Section I.B) 88;93. Therefore, with the use of cpRNA, specific internal bases of the pRNA were uniquely labeled with photoaffinity crosslinking agents to analyze inter/intra-molecular interactions. When necessary, the 5’-end of the RNA can also be labeled with [32P]. Crosslinked RNAs can be separated from uncrosslinked RNAs by denaturing gel electrophoresis, and crosslink sites are determined by primer extension106;120. Bases identified as crosslink sites by primer extension indicate bases that are in close proximity to the photoagent labeled base. The use of cpRNAs allows the identification of intra-molecular contacts throughout the pRNA molecule. Crosslinking data have been used as distance constraints in molecular modeling studies 88;93;106;120(Section II.L).
Fig. 7: Phlyogenetic analysis and predicted secondary structures of pRNAs in phages SF5’, B103, phi29, PZA, M2, NF, and GA1. The bases in the right and left loops responsible for inter-RNA interaction are boxed (reprinted from 99 with permission from RNA).

Peixuan Guo
1. Intra-molecular crosslinking of monomers106 Three nucleotides have been selected as new 5’ terminus for labeling with Aryl azides (Table 5). One of the new 5’ termini, G108, is located within the 5'/3'-end helix necessary for DNA packaging, while two of the other sites, G75 and G78, are located within the interior sequences involved in procapsid binding. The photosensitive group was coupled to the 5’-thiophosphate of each cpRNA. Each photoagent-coupled cpRNA is fully functional in DNA packaging. After irradiation, the crosslinked pRNAs are isolated from denaturing gels. Crosslinked pRNA migrates slower than uncrosslinked one. Bands representing intra-molecular and intermolecular crosslinks have been identified and purified. Crosslinked sites are mapped by primer extension. Reverse transcription stops occur one base prior to the crosslinked bases. For example, a stop at base 32 would mean that base 31 is crosslinked. G108 is crosslinked to C10 and G11; G75 is cross-linked to A26, U27, G28, U29 and G30; while G78 is cross-linked to U31. The azidophenacyl group is only 9 Å, but experimental data has demonstrated that the cross-linking group can reach distances of 12 Å (Norman Pace, personal communications). The data suggest that G108 is proximate to C10 and G11; G75 is proximate to bases 26-30; while G78 is proximate to U31. These distances have been used as constraints in computer modeling of pRNA structure (Section II.L) (Fig. 10). 2. Intermolecular crosslinking of dimers120 The methods used for crosslinking of dimers are basically the same as those used in monomer crosslinking work. The cpRNA B-a' was labeled with aryl azides on G82 and incubated with an equimolar amount of unlabeled pRNA A-b’. The left and right loop sequences of these two pRNAs are complementary, and therefore they are able to form dimers. After UV-crosslinking, dimers were isolated from the gel, and the crosslinking site was identified by reverse transcriptase primer extension using a pRNA A-b'-specific primer.. G82 was found cross-linked to G39, G40, A41, C49, G62, C63, and C64 120, indicating that all these eight bases are in close proximity as dimers, as seen in computer models (Fig. 10). D. Crosslinking by Psoralen Psoralen has been used as a photoactive probe for nucleic acid structure because it can intercalates into RNA or DNA helices and, upon irradiation with 320-400 nm light, freezes the uridine of RNA or the thymidine of DNA that are in close proximity (helix or pseudoknot) by covalent attachment 121-123. The sites of crosslinks can be determined by primer extension 124 and/or Mung bean nuclease treatment 125. The psoralen derivative, AMT (4'-aminomethyl-4,5',8-trimethyl psoralen), has been selected due to its solubility 124. One advantage of psoralen crosslinking is that it crosslinks RNA or DNA but not protein. This is different from the azido group described above which crosslinks non-specifically to both protein or nucleic acids. Psoralen, however, can induce intra-molecular crosslinks within the pRNA even in the presence of other proteins, such as procapsid or gp16. Thus, pRNA conformations in different environments can be detected. Psoralen crosslinks can also be reversed by 254 nm irradiation. With the use of 2-dimensional gel 121;123;126 and 5’-end radiolabeled cpRNAs, pRNA conformational change in the presence of different packaging components can be investigated. The result reveals that pRNA had at least two conformations, one which was able to bind procapsid
and the other which did not (Section II.M). It has been found that in the absence of Mg++, the region of C67 to A70 is proximate to U31 to U36, since these two areas can be crosslinked by Psoralen 124.
Table 5. (Adapted from 139 with permission).

Peixuan Guo
E. Crosslinking by phenphi Unlike psoralen, phenphi [(cis-Rh(phen)(phi)Cl2
+ (phen = 1,10-phenanthroline; and phi = 9,10-phenanthrenequinone diimine)] induces covalent bonds between guanosine bases upon UV activation. Phenphi has also been shown to crosslink pRNA and revealed the proximity of G75 with G28, and G30 of pRNA 127. F. Chemical Modification Various chemicals alter specific reactive groups of RNA and thus provide information regarding base pairing, base stacking, and the tertiary interactions of specific bases of RNA. The modifying agents include dimethyl sulfate (DMS), which methylates A at N1, G at N7 and C at N3 128;129; kethoxal, which modifies G at N1 and N2 130; and 1-cyclohexyl-3-(2-morpholinoehtyl)-carbodiimide metho-p-toluene sulfonate (CMCT), which attacks U at N3 and G at N1 128-130. Locations of modified bases can be identified by primer extension with reverse transcriptase 128;131. Reverse transcriptase does not stop at methylated N7 of G. Thus, aniline-induced chain scission is required in this case prior to extension 128;130. It is common to find that some nonspecific stops occur in primer extension of unmodified RNA; thus, it is critical to use unmodified RNA as negative controls in primer extension. When pRNA/protein mixtures are being analyzed, reactions are phenol-extracted, and the RNA is precipitated prior to the extension reaction. Chemical modification of a base is a good indication that the base is unpaired. Lack of modification will most likely be due to base pairing, especially in helical regions. Lack of modification may also be the result of tertiary interactions or non-canonical base-base, base-sugar, or base-phosphate interactions 130. Such reactions often occur in phylogenetically predicted stem loop or bulge regions of RNA. Chemical modification data can provide information on base accessibility, which can be used to assess predicted secondary structures and evaluate molecular models of the tertiary structure. Phi29 pRNAs have been modified with DMS, CMCT, and kethoxal 128;130;132. Reaction conditions were obtained empirically to produce, on the average, one modification per molecule by titrating the concentration of the modifying reagents needed. Chemical modification showed that the sequence C18C19A20 is accessible to chemicals in monomers and dimers as well as procapsid-bound pRNA 132;133. The results indicate that CCA, though not involved in procapsid binding 93;134, is present on the surface of the pRNA as a bulge, perhaps to interact with other DNA packaging components 133. This conclusion is supported by studies of pRNAs with mutations in the CCA bulge (Section II-I).
Bases U85U84G83G82 and A45A46C47C48 of monomer are accessible to chemicals, but in dimer they are not accessible to modification. These bases compose the right and left loop. Results confirm that these bases are involved in inter-pRNA interaction 96;98. Bases G57, A56, and G55 are also protected from chemical modification in dimer, but can be modified in monomer. Comparison of the modification pattern of monomer and dimer revealed that the major loops- the right, left and head loops- are all involved in pRNA/pRNA contact to form dimer, since all of them are strongly modified in monomer but protected in dimers. All five single base bulges in wild type pRNA are modified fairly strongly. Each of the five single base bulges is dispensable for pRNA activity 93;100. In fact, only base U5 has mild negative effect on pRNA activity when deleted, and the detrimental effect is marginal (a 10-fold reduction) 115. The three large loops of the pRNA (right hand, left hand, and head loops) were each extensively modified by chemicals, supporting the pRNA secondary structural prediction. G. Chemical modification interference Chemical modification interference has been performed to determine which pRNA bases are involved in dimer formation. Normally, RNA B-a’ would be able to interact with A/b’ to form dimers. The monomer RNA B/a’ was treated with either DMS or CMCT and then mixed with unmodified monomer A/b’ in order to test their competency in dimer formation. If the modified base is involved in dimer formation, pRNA B-a’ carrying this modified base would not be able to form a dimer with pRNA A-b’, and thus this pRNA will be present in the fast migrating band representing monomers. After being isolated from the gel, both monomers and dimers were subjected to primer extension to identify the modified bases. The concentration of the chemicals was pretitrated to ensure that on the average only one base per RNAs was modified 106. The results of chemical modification interference analysis revealed that bases U54, G55, U59, C65, A66, A68, U69, A70, C71, C84, C85, C88, A89, A90 and C92 were involved in dimerization, while bases 72-81 were not106.

Peixuan Guo
Fig. 8: Summary of chemical modification (A and B) and Chemical modification interference (C). The black short arrow, double-lined arrow, and empty-square indicate a strong, moderate, and weak modification, respectively. A and B summarized the chemical modification of monomer and dimer, respectively, as a comparison. D is the depicting of RNA/RNA interaction in dimer formation bases on the data from A-C (adapted from 132 and 106 with permission from RNA and J. Biol Chem, respectively). H. Ribonuclease probing Some ribonucleases are sensitive to RNA secondary structure. For example, RNases T1 (specific for GpN linkages), U2 (specific for ApN linkages), and S1 prefer to cleave to single-stranded RNA. Nuclease V1 is specific for double stranded RNA. End-labeled pRNA and cpRNA in various solutions containing Mg++, procapsid, or gp16, individually or in combination, have been probed by T1, U2 or V1 117;124. T1 and V1 are used to distinguish the loops and helices of four RNA with similar functions 117. Footprinting has also been conducted using ribonuclease to detect the sequence that binds procapsid 94. T1 has been used to study the change of pRNA conformations 124. Since the activity of RNase T1 is Mg++ independent, this enzyme can be used to investigate the conformational change of pRNA induced by ATP in the presence or absence of Mg++.
Mg++-induced pRNA conformational change has been verified by T1 ribonuclease probing124. The pattern of partial digestion of pRNA by T1 provided strong evidence for the presence of two conformations, dependent on the presence or absence of Mg++. Without Mg++, strong cleavages by T1 were seen in G28, G30, and G34. While with Mg++, these three bases became more resistant to T1 attack, indicating a conformation change or refolding of pRNA caused by Mg++. I. Genetic analysis by truncation, insertion, deletion and mutation The availability of the highly sensitive pRNA assay procedure using the in vitro phi29 assembly system (Section I-A) greatly facilitated the genetics analysis. Taking advantage of the circularly permuted pRNA system (Section I-B) and the technique of two step PCR, mutant pRNA can be easily constructed with truncation, deletion, insertion and mutation targeting at any defined position. Plasmids with pRNA coding sequence were used as templates to generate PCR DNA fragments with primer pairs containing either the

Peixuan Guo
T7 or SP6 promoter and mutations to pRNA. The PCR fragments were used to transcribe mutant or circularly permuted pRNAs in vitro with either T7 or SP6 RNA polymerase. By the use of this procedure in combination with the aforementioned in vitro assembly assay system, tens of mutant pRNAs can be obtained and tested in one or two weeks. Complementary modification 100;135 is one of the mutation studies that has been used to identify helix regions and intermolecular loop/loop interaction in dimer formation 93;99;115(Section II-A). Nucleotides U72U73U74 are predicted to be a bulge located at a three-helix junction (Fig 2). Mutation studies have shown that the deletion of these three nucleotides abolishes the activity of the mutant pRNA F5, which has native 5’/3’ ends 100. However, a circularly permuted cpRNA 75/71 that has had deletion of these three nucleotides but then had new 5’ and 3’ ends located at bases 75 and 71, respectively, was fully active in in vitro phi29 assembly 93. This suggested that the UUU bulge in this area provided flexibility to the pRNA. In pRNA F5 with normal 5’ and 3’ ends, deletion of the UUU bulge eliminated the flexibility in folding of the three-way junction, thereby creating an inactive mutant. In cpRNA 75/71, this flexibility was compensated for by the discontinuity of the phosphodiester bond in this area. The result suggested that the UUU presents as a bulge at the three-way junction to provide flexibility in folding and to serve as a hinge for the twisting of the lower stem-loop. Deletion 100 and insertion93 have been used to study the function of the C18C19A20 bulge. A pRNA with three bases, 3’-GGU-5' inserted between A99 and A100 to pair with the bases C18C19A20 in the bulge, generates the pRNA 7/GGU 93. This pRNA was fully competent to form dimers and bind procapsids, however its activity in DNA packaging and virion assembly was completely lost 93. A pRNA with a deletion of all three bases of the CCA bulge 134 exhibited the same biological activity as pRNA 7/GGU concerning procapsid binding, DNA packaging and virion assembly, indicating that the CCA bulge, though not involved in procapsid binding, likely interacts with other DNA packaging components 133. J. SELEX In vitro evolution is a powerful tool to study consensus elements of RNA structure and function. Starting with a library containing pRNA sequences with random mutations within a defined region, in vitro evolution techniques allow the selection of pRNA variants that are able to bind a specific ligand. Such selection for interacting species is based on different primary structures that would adopt the same structural feature as wild type RNA. SELEX (Systematic Evolution of Ligands by Exponential Enrichment) allows screening for co-variation of several nucleotides and can be used to reveal noncanonical interactions that are difficult to prove by classic genetic and biochemical approaches 136;137. SELEX has been used for the selection of pRNA sequences that bind procapsids 138. Using a DNA template pool with random mutations covering residues 30-91,they found that after five rounds of selection, most clones selected were wild type. Co-variation is found in predicted helix region, supporting the prediction of helical structures in these regions138. A second pool of pRNA sequences was also constructed to test the loop/loop interaction in pRNA dimer formation 138. Using randomized mutation from bases 45 to 62 that cover the right hand loop and bases 81 to 84 that cover the left hand loop, it was found that after five rounds of selection, the pRNA pool can compete with wild-type pRNA for procapsid binding. It was concluded that the wild type pRNA sequence was in all probability the most suitable sequence for procapsid binding. It is important to point out that special care must be taken when considering dimer formation with regard to the data in this experiment. A finding of unpaired right and left loops from clones selected from the pool that can also bind procapsid is, by no means, an indication that paring is not absolutely necessary for procapsid binding. For example, if mutant pRNA A-b’ and B-a’ are present in the pool, both of them could pass the screening in binding selection, since A-b’ will interact with B-a’ to form dimer and will bind procapsids, and be selected. However, the isolation of A-b’ from the pool does not mean that A-b’ by itself could bind procapsids. It is probable that A-b' was selected based on its ability to bind procapsids in combination with a trans-complementary pRNA, such as B-a'. K. Cryo-AFM (atomic force microscopy) Atomic force microscopy has been used by several investigators to detect images of RNA (Fig. 9) in a denatured conformation. As the first attempt to test whether this technique can be used to detect the 3D structure of RNA in native conformation, cryo-AFM has been performed on phi29 pRNA 106;132;139. 1. Cryo-AFM images of monomer132;139 Cryo-AFM imaging reveals that the pRNA monomer folded into a “√” (check mark-shaped) structure. The color in Fig. 9 indicates the thickness or height of the image but does not reflect the atom density observed end on. The brighter or whiter the color, the thicker the surface; the darker the color, the thinner the surface. The color and contrast of the image clearly indicate that the area around the head loop (the elbow of the " ") is the thickest/tallest (Fig. 9), as seen in the computer model (Fig. 10). 2. Cryo-AFM images of dimers106;132;139. Cryo-AFM has been used to directly visualize purified pRNA dimers. The native dimers have an elongated shape (Fig. 9) 132. It appears that head to head contact is involved in dimer formation, resulting in a complex almost twice as long as a monomer. The conclusion of head to head contact agrees with chemical modification results showing that nucleotides of the head loop in dimers were protected from chemical attack. Cryo-AFM images of the fused dimer, a pRNA construct consisting of two tandemly linked pRNAs (Table 5), exhibit a similar structure to the non-covalently linked pRNA dimer106. The dimensions of the covalently linked fused dimer are comparable to native dimer 132;139. L. Modeling the three-dimensional structure of pRNA The goal of modeling pRNA is to organize collections of structural data from crosslinking, chemical or ribonuclease probing, cryo-AFM and other genetic data into a three-dimensional form. Since a large number of structural constraints are available,

Peixuan Guo
computer programs can successfully construct three-dimensional structures 140;141. The first attempt for construct the 3D structure of the pRNA monomer and hexamer is based only on the information available for pRNA secondary structure 98;142 and the finding of the intermolecular interaction of two loops with four bases. In considering the construction of a RNA 3D structure without 3-D structure data, these models are plausable. Due to the subsequent extensive investigation on pRNA 3-D structure, new models of 3D structure on pRNA monomer and hexamer have also been constructed 143. A computer model of pRNA dimer has also been constructed based on the constraints obtained from chemical modification, chemical modification interference, crosslinking, and cryo-AFM (Fig. 10) 143.
Fig. 9: Cryo-AFM (atomic force microscopy) showing monomers (left column) and dimers (right column) of pRNA. The magnification in A and B is x5,000,000. C and D are enlarged images of monomers and dimers, respectively. E and F are the illustrations of monomers and dimers, respectively (reproduced from 132 with permission from RNA). New models of pRNA monomer, dimer and hexamer (Fig. 10) are produced on Silicon Graphics Octane and Indigo2 computers running IRIX 6.2 or 6.5, using the programs NAHELIX, MANIP, PRENUC, NUCLIN, and NUCMULT 144,113.The modeling was performed based on the following assumptions. 1). All helices are modeled as a regular A-form double helix. 2) Internal loops and mismatched bases are constructed by maintaining the integrity of the double helix while optimizing base pairing and stacking inside the loop, as suggested by most structural data from X-ray and NMR analysis. 3) A general rule for the modeling of RNA hairpin loop has been proposed 145, which involves maximal stacking on the 3' side of the stem and enough nucleotides stacked on the 5' side to allow loop closure, as found in the anticodon loop of tRNA. 4) Bulges less than four bases are modeled either sticking out from stems to ovoid helical distortion, loops are constructed either protruding from the stems, or within the helical domain, causing the helical axis to bend. Parameters for stacking energy are considered to decide whether bulges should be protruding or within the helical stems 146. 5) Helix untwisting or twisting, helix-helix interactions, triple base interactions 147, pseudoknots, or other higher order structures have been built into the model at constant geometrical distances but allowing certain torsion angle variation. The program regarding RNA flexibility, a consequence of rotational freedom about seven intrastrand bonds, has been applied to the construction of the UUU bulge at the three-way junction. These three-base bulges have been found to provide flexibility for the appropriate folding of the three-way junction. Conventional computer algorithms involving the minimization of empirical energy functions have been considered. 12 angstroms has been considered as a maximum distance constraint when bases are crosslinked by GMPS/Aryl Azide. Modified distance geometry and molecular mechanics algorithms, using simplified "pseudo atom" representations, have been considered to generate structures consistent with data from crosslinking, chemical modification and chemical modification interference. A constraint satisfaction algorithm is combined with discrete representations of nucleotide conformations to take care of the disturbed area in order to ensure the normal representation of all atoms. Crystal structure reveals that the connector contains three sections, a narrower end, a central section, and a wider end with a diameter of 6.6nm, 9.4nm, 13.8nm, respectively (Fig. 10D) 102;148. The hexamer model by Hoeprich and Guo 143 contains a central channel with a diameter of 7.6nm, that perhaps can sheath onto the narrowed end of the connector and anchored by the connector central section, which is wider than the central channel of the pRNA hexamer (Fig. 2E & F).
Peixuan Guo
Fig. 10: Computer modeling of phi29 pRNA. A, monomer in Ball & Stick format; B. dimer in Ribbon format; C. hexaer in Spacefill format; D. Crystal structure of connector 102 with the sequences predicted for pRNA binding149 in blue; E. pRNA hexamer and connector complex. In C, the six protrusions represent the DNA packaging domain composed of the 5’ (in blue) and 3’ (in red) end (adapted from 143with permission). As noted earlier (Section V.C), pRNA contains two functional domains (Fig. 2), one is for connector binding and the other is for DNA translocation. The connector binding domain is located at the middle, bases 23-97, and the DNA translocation domain is located in the 5’/3’ paired ends. It has been predicted that the connector protein (gp10) contains a conserved RNA recognition motif (RRM), residues 148-214, located at the narrow end of the connector that protrudes from the procapsid 149;150. The hexamer model by Hoeprich and Guo 143 complies with the aforementioned data showing that the bases 23-97 (colored green in Fig. 10C, E & F), which is the connector binding domain, interact with the predicted RRM of connector (Fig. 2E and F in blue), while the 5’/3’ paired region (Fig. 2E in red and cyan), which is the DNA translocation domain, extends away from the connector. M. Conformational change of pRNA The ability of RNA to perform diverse biological functions despite being comprised of only four different building blocks (A,C, G, and U) can be attributed to the flexibility in RNA folding. In two instances of pRNA action, the tasks performed could not be accomplished without considering a conformational change. The first instance is the transition from pRNA dimers to hexamers. The other is the execution of the physical motion required to translocate phi29 genomic DNA into empty procapsids. It has also been demonstrated that pRNA monomers possess two conformations. Conformational changes in pRNA have been demonstrated by assessing the structure of pRNA in the presence or absence of Mg++
and other DNA-packaging components. The conformation of pRNA in the presence and absence of Mg++ has been investigated with psoralen crosslinking 124, nuclease probing 124, and chemical modification 132. The conformation of the pRNA bound to procapsids has been analyzed by RNase foot printing 94 and chemical probing 142.
1. Transition from monomer to dimer--- conformational change induced by Mg++ Free pRNA in solution can adapt two conformations. Mg++ induces a pRNA conformational change as revealed by pRNA migration rate changes in EDTA and Mg++ gels, by T1 ribonuclease probing, and by psoralen crosslinking 124. In the presence of 1 mM Mg++ and absence of EDTA, pRNA migrates faster in gel, indicating a conformational change. Indeed, an Mg++ concentration of 1 mM was found to be sufficient to induce this conformational change, and this concentration is the same as that required for pRNA/procapsid binding and DNA packaging. Moreover, this concentration was also close to the Mg++ concentration in normal host cells. With Mg++, pRNA seems to adopt a more compact conformation that may facilitate its attachment to procapsid. Ribonuclease T1 digestion confirms the presence of two conformations. In the absence of Mg++, strong cleavages by T1 are seen in pRNA G28, G30, and G34. With Mg++, these three bases became more resistant to T1 attack. A conformational change in pRNA induced by Mg++ has been further investigated by psoralen crosslinking (Section II.D) 124. In
the absence of Mg++, pRNA is crosslinked and shows slower migration rate in gel. When 1 mM Mg++ is present, the crosslinked band is undetectable. It is known that Mg++ does not affect the crosslinking efficiency of RNA with psoralen 121. The induced conformational change is reversible by additions of EDTA. Nucleotide U69 has been confirmed to crosslink to U31, U33 and U35.
Mn++, Ca++, and Co++ , at concentrations of 5 mM, can also inhibit the crosslinking by psoralen. This is in agreement with later experiments showing that Mn++ and Ca++could replace Mg++ in pRNA dimer formation and procapsid binding 139. Chemical modification of the pRNA in the absence of Mg++ has also been performed. Both the right hand loop and the head loop show a relative lack of modification in the absence of Mg++ compared to when Mg++ is present. A similar reduction in modification occurs in the stem of the head stem loop. The presence of drastically altered chemical modification patterns within this procapsid

Peixuan Guo
binding domain suggests that Mg++ plays a crucial role in pRNA folding to generate the appropriate right and head loop structures, and
explains why Mg++ is needed for dimer formation, procapsid binding, DNA packaging, and phi29 assembly.
Other evidence points to a conformational shift from monomer to dimer. For example, in pRNA monomers, base A56 in the head loop is accessible to chemicals, while in dimers, A56 in the head loop is not modified by DMS132. This is an indication of a conformational change in the head loop area during the formation of dimers - the building blocks of the hexamer motor complex 139. A conformational change in the pRNA has also been observed upon procapsid binding. It was found that the cleavage of four guanines, G37G38G39G40, by ribonucleases was enhanced in procapsid-bound pRNA, compared to cleavage of free pRNA. Such a result implies that pRNA undergoes a conformational change upon binding to procapsid 94. 2. Transition from dimer to hexamer It is concluded that dimers are the building blocks in hexamer assembly with a pathway of dimer → tetramer → hexamer 139. Closed dimers, two molecules linked together by the holding of two pairs of hands, are active in procapsid binding and DNA packaging, while open dimers formed by the holding of only one pair of hands, are unstable in solution 139. (Section V.D). Both tandem and fused pRNA dimers (Table 5) with complementary loops designed to form even-numbered rings are active in DNA packaging, while those without complementary loops are inactive 139. These results imply that the true pRNA intermediary in hexamer assembly is the closed dimer with the holding of two pairs of hands (i.e. right and left hand loops of each pRNA paired with those of the other), and that the two interacting loops of one dimer play a key role in recruiting an incoming dimer 96;99. In dimers, each pRNA holds hands of only one additional pRNA. However, in hexamers, each pRNA holds hands of two additional pRNAs. It seems paradoxical then that a dimer with no unpaired loops would be the precursor to hexamers where each pRNA interacts with two others. Thus, how is the conclusion that dimer is the building block for pRNA hexamer interpreted? Recent results reveal that the pRNA has a strong tendency to form a circular ring by hand-in-hand contact, regardless of whether the pRNA is in dimer, trimer or hexamer form (to be published), suggesting that the structure of pRNA is bendable. Therefore, a conformational shift is expected during the transition from dimer to hexamer. It is reasonable to believe that dimer formation is a prerequisite to generate an appropriate 3D interface for procapsid binding. In such a scenario, one of the hands of the dimer that is already paired with its complementary loop sequence on the other pRNA would release after binding to the procapsid. The dimer with a released hand is similar to the open (linear) dimer that has been demonstrated to be unstable in solution yet still active in procapsid binding and DNA packaging 99. The open hand may serve as a welcoming hand to recruit an incoming dimer to the procapsid (Fig. 12). Indeed, pRNA conformational changes before and after binding to procapsid have been documented by nuclease probing94 and chemical modification 142 (Section II.F). 3. Conformational shift in executing the motion task Many macromolecules undergo a conformational change in executing tasks involving motion, rotation, or migration. In the phi29 system, procapsids contain a six-fold symmetrical connector embedded in a protein shell with a five-fold rotational symmetry. The relative motion of two rings could allow the motion of DNA translocation to occur 105;151. The finding that a hexameric pRNA complex is bound to the connector and that 6 pRNAs worked sequentially is potentially relevant to this model (Section V.H and Fig. 13). It has been shown that pRNA contains two domains: one for connector binding and the other for DNA translocation (Section V.C). The pRNA binds to the connector leaving the 5’/3' domain free to interact with other DNA packaging components. It is proposed that the pRNA is part of an ATPase and possesses at least two conformations, a relaxed form and a contracted form. Alternating between contraction and relaxation, each member of the hexameric RNA complex, driven by ATP hydrolysis, helps rotate the DNA translocation machine105. III. pRNA PROTEIN INTERACTIONS
A. Strong binding of pRNA to connector but undetectable biding to capsid proteins One important aspect in investigating the role of an enzyme is to find out to where it binds. Co-sedimentation of the pRNA with purified connector (portal vertex) in sucrose gradients, and retention of radioactive-labeled pRNA with this protein in nitrocellulose binding assays, indicate that the pRNA is attached to the connector and is part of the translocating machinery 70. Moreover, the isometric particle, a defective procapsid lacking the portal vertex protein, does not contain pRNA 70;152. It would be interesting to know whether pRNA binds to the connector gp10 only, or whether it binds to the capsid protein gp8 to bridge the connector and capsid. UV crosslinking has been performed to answer these questions 153. Briefly, pRNA was made in the
presence of alpha-32P[UTP] and 5’-iodo-UTP. After binding, the RNA/procapsid complex was irradiated with short wavelength UV light. When [32P]-pRNA was crosslinked with procapsid, the band in SDS gel corresponding to gp10 was specifically labeled, while the gp8 was not, indicating that pRNA bound selectively to gp10 (Fig. 11)153. 5S rRNA did not compete with radioactive-labeled pRNA for gp10 binding, but unlabelled pRNA did, indicating the binding is specific. Later on the same group who claim that 5S RNA was able to produce some DNA packaging in the in vitro assay 154;155 also publish another report to show that the specific pRNA was able to discriminate and select phi29 DNA for packaging from a pool containing both bacterial and phage DNAs 156. Our first reports showed that the presence of 5S RNA was able to produce some DNA packaging in the in vitro assay. Later, we showed that the specific pRNA was able to select fhi29 DNA to be packaged from a mixture containing bacterial and phage DNAs. These reports are not at all contradictory. The nonappearance of pRNA/gp8 binding in cross-linking experiment does not completely exclude the possibility of transient

Peixuan Guo
interaction of pRNA with capsid protein. The data can only conclude that the affinity of pRNA to connector is much stronger than to capsid, and connector is the foothold for pRNA.
Fig. 11: Specific binding of radiolabeled pRNA to connector demonstrated by crosslinking. Panel I and II are Coomassie blue stain and x-ray film of the same SDS ployacrylamide gel. Lanes a,b,c,d corresponds to A,B,C,D, respectively. In addition to the components noted on the bottom of the figures, all lanes contain [32P]pRNA. Reaction mixtures were treated with RNase A before loading to the gel. The smears in lane a’ suggests that the RNase A could not digest pRNA completely due to the presence of high concentration of procapsid in the reaction (adapted from 153 with permission from RNA). B. Foot printing and chemical probing of prohead-bound pRNA Foot printing of the pRNA/procapsid complex with nuclease A, T1 and V1 reveals that bases 22-84 are protected from enzyme digestion 94. This is an indication that the region from bases 22-84 contacts with connector. Chemical probing of procapsid-bound pRNA has also been performed to investigate the pRNA/procapsid interaction 142. The chemicals, DMS, CMCT, Kethoxal, used in this experiment are much smaller than ribonucleases; thus, a finer mapping of procapsid-protected bases can be achieved than by RNase probing. The most striking feature of the modification of procapsid-bound pRNA was the relative lack of modification of the RNA compared to wild type pRNA alone. The entire left stem loop was completely unreactive. Additionally, only base A56 of the head loop was modified. In the right loop, which was modified strongly and completely in wild type pRNA, only bases 42, 43, and 44 were significantly modified and base 45 was only moderately modified. The helical regions throughout the molecule were completely uncreative, with the exception of base U36, which was strongly modified in both bound and free pRNAs. The bases C18, C19, A20, and U35 were all strongly modified in both procapsid bound and free pRNAs, suggesting that this 3-base bulge is extended from the procapsid. The bases U5 and U73 were moderately modified in free pRNA and strongly modified in procapsid bound pRNA. Also, bases U17 and U21 were moderately modified in procapsid bound pRNA, but not in free pRNA, suggesting that the binding of pRNA to procapsids changed the conformation of pRNA.
C. Connector domain and sequence for pRNA binding By sequence homologous comparison, it was predicted that the connector protein gp10 contains three conserved RNA recognition motifs (RRM). The first motif was predicted to be residues 7-57 149, and the second one is residues 22-95. However, by comparison with the crystal structure of the purified connector 33;102;157;158, these two motifs fall into the upper half part of the wider connector end that is embedded in the procapsid. Since this region is not exposed, it is impossible for pRNA to bind if the crystal structure of the purified connector possesses an identical conformation as the one embedded in the procapsid. The third motif, residues 148-214, is located at the narrow end of the connector that protrudes from the procapsid. It is possible that this might represent the true RRM for pRNA binding based on the assumption that the conformation of the purified connector is identical to the one embedded in the procapsid. Site directed mutation of several charged and aromatic residues of connector protein gp10 render the procapsid unable to package DNA 159. The RNA binding sequence of gp10 is predicted to be the amino acid residues 21 to 94. Specific antibodies against polypeptides covering different stretches of the gp10 sequence have been generated to investigate the RNA binding region of the connector160. In combination with immuno-electron microscopy, the RNA binding domain has been mapped to be at the end of the narrow side of the connector 101. Again, the crystallography structure of a purified connector reveals that residues 21 to 94 are located at the wider end that is embedded in the procapsid. It would not be possible for pRNA to bind residues 21-94. Several domains of the connector have been mapped by antibody 160;161. The domains and sequence mapped by antibody 160 do not agree with the structural data from X-ray crystallography 162. But the immunolabelling detection of the pRNA site, other than sequence, at the narrow end of the connector is fully consistent with the crystallographic data. The global connector structure obtained from EM studies 150;161;163 164were very similar to the structure solved by crystallography 102. D. Can pRNA interact with gp16? Filter binding assay has been performed to study protein binding of pRNA70. Glass filter could bind proteins nonspecifically, but do not bind RNA. When radioactive-label pRNA binds to protein, pRNA will attach to the filter due to the retention of the protein on the filter. Both connector and gp16 have been tested for pRNA binding. gp16 does not bind to purified pRNA, but the connector does.

Peixuan Guo
Later on, it was found that gp16 could protect the procapsid bound pRNA from nuclease digestion149 by the addition of a gp16-containing cell extract to the reaction, but not or little protection was observed by the addition of purified gp16. The difference in these two gp16s is that the cell extract contains a large amount of unspecific proteins. The RNA recognition motif of gp16 has also been predicted, though not proven, to be residues 47 to 119. Whether gp16, after binding to the procapsid, interacts with pRNA remains an interesting unsolved problem. It is worth noting that due to the hydrophobicity, gp16 has a strong intention of self-aggregation. This special feature of gp16 must be dealt with care to avoid nonspecific binding due to the strong self-aggregation. IV. EFFECT OF MONO AND DIVALENT CATIONS ON pRNA DIMER FORMATION, PROCAPSID BINDING AND VIRAL ASSEMBLY124;139 Mg++ is required for dimer formation. For circularly permuted cpRNAs, the Mg++ concentration required for 50% dimer formation is 4 mM; while for pRNAs with wild type 5’/3’ ends it is 0.4 mM139. However, the mechanism behind this difference is unknown. Effects of other cations on dimer formation, procapsid binding, and viral assembly have also been investigated. At least a 1M concentration of monovalent ions is needed for pRNA dimerization, while as low as 5 mM of divalent ions is sufficient. Spermidine, a positively charged compound, can also stimulate dimer formation at a concentration of 5mM, indicating that dimerization is a result of a cation effect. CoCl2 or NiCl2 could not promote dimer formation, while FeCl2, ZnCl2 or CdCl2 caused the precipitation of pRNA.. These data suggest that pRNAs form dimers in the presence of positively charged cations, including mono- or divalent cations, as well as spermidine. Dimer formation is an intrinsic feature of pRNAs, and cations play a facilitating role. Contrary to the lack of ion specificity for dimer formation, the binding of dimers to procapsid showed a specific requirement for divalent cations. Neither monovalent cations nor spermidine could stimulate the binding of dimers to procapsids. Only Mg++ and Ca++ could, while Mn++ showed a reduced efficiency. These data indicate that dimer formation is not sufficient for procapsid binding, DNA packaging and viral assembly. Divalent cations, Mg++, Ca++, and Mn++, play other roles in addition to promoting dimer formation. V. FUNCTIONS OF pRNA A. Separation of the pRNA into two domains: procapsid binding domain and DNA packaging domain Extensive investigation has provided strong evidence that the pRNA molecule contains two functional domains (Fig. 2). This conclusion comes from the results of different approaches, including a) base deletion and mutation 87;93;100;115;165; b) ribonuclease probing 94;124; c) oligo targeting89;104; d) competition assays to inhibit phage assembly95;103;104; e) crosslinking to portal protein by UV153; and f) psoralen crosslinking and primer extension166. Mutations of more than two bases at the 5’ or 3’ end of the pRNA to non-complementarity within the 5'/3' end terminal helix resulted in complete loss of DNA packaging activity. However, these mutants were able to bind procapsids with an affinity similar to wild-type pRNA 87;95;104;153. Chemical and ribonuclease probing revealed that the binding pattern of these mutant pRNAs couldn’t be distinguished from that of wild type pRNA132;142. These mutants were able to compete with wild-type pRNA for procapsid binding and also strongly inhibited phage assembly in vitro. Antisense oligos targeting the procapsid-binding domain blocked the binding of pRNA to procapsid, and therefore, completely inhibited viral assembly in vitro 89;89. Antisense oligos targeting the 5’/3’ DNA translocating domain (Fig. 2) completely inhibited viral assembly, but did not block procapsid binding 89. A truncated pRNA, comprised of bases 28-91, can still be UV-crosslinked to the phi29 connector protein gp10 specifically153. A 75-base RNA segment, comprised of bases 23-97, was able to form dimers, interlock into hexamers, compete with full-length pRNA for procapsid binding, and therefore inhibit phi29 assembly in vitro139. All of the above data support a conclusion that pRNA contains two functional domains, one for procapsid binding and the other one for a yet undefined role in DNA translocation. The procapsid binding domain is located at the central part of the molecule94;139;153, bases 23-97, and the DNA translocation domain is located in the 5’/3’ paired ends87. B. pRNA remained associated with DNA-filled capsids105 One question that remains to be answered is whether the pRNA is needed only for the initiation of DNA packaging, or whether it participates in the DNA translocation process. [3H]-pRNA was used to package unlabeled genomic DNA-gp3 in vitro. Following sucrose sedimentation to separate DNA-filled capsid from empty procapsids, it was found that [3H]-pRNA was present in the radioactive peak corresponding to the location of DNA-filled capsids. These results suggest that the pRNA is associated with the DNA packaging machinery during the DNA translocation. C. pRNA leaves DNA-filled capsid after the addition of the tail protein gp9105 Although the evidence suggests the pRNA participates in the DNA packaging process of phage phi29, it is not known when the pRNA leaves the procapsid. Sucrose gradient sedimentation has been used to detect assembly intermediates. After the addition of purified gp9 following DNA packaging, the pRNA is no longer associated with the DNA-filled capsids. D. Dimer as the building block in pRNA formation It has been shown that the hexamer is built from dimer98;139. The pathway of building a hexamer is: dimer → tetramer → hexamer (Fig. 12)139. This conclusion comes from the following evidence. 1. Homogenous molecules with two complementary loops such as pRNA I-i’ can form dimers 96;98;120;139. Two heterogeneous molecular such A-b’ plus B-a’ can also form stable pRNA dimers in solution. Both dimers have been purified from native gel and/or sucrose gradient. The purified dimers are active in procapsid binding and phi29 DNA packaging 120;139. 2. Homologous pRNAs with non-complementary right and left loops, such as A’-b’ appear predominantly as monomers. Such monomeric pRNAs can not bind procapsid nor package DNA 96;98;139. Monomeric pRNAs are unable to compete with dimers formed by homologous pRNA containing complementary loops, e.g. I-i’, for procapsid binding, while . dimer containing two heterogeneous pRNA are potent competitors. Monomeric pRNA is also unable to inhibit dimeric pRNA for DNA packaging 96;139.

Peixuan Guo
3. Cryo-AFM also directly confirms the existence of dimers 106;132;139. The detection of dimeric pRNAs in solution suggests that it might be a stable intermediate during assembly of pRNA hexamer.
Fig. 12: A diagram depicts the pathway of pRNA hexamer formation. The pentagon represents the procapsid shell with five-fold symmetry. The dodecagon represents the connector with a six-fold central hole for DNA entry 33;164;203. The black elliptical particles represent the pRNA (reproduced from 143 with permission). 4. The fact that the minimum size of pRNA for procapsid binding is the same as the minimum size for dimer formation 99;153also supports the conclusion that dimers are the binding unit for hexamer assembly. 5. Covalently linked dimeric pRNAs are able to package DNA in vitro. The strongest evident to support the conclusion that dimers are the building blocks of RNA hexamer comes from the finding that covalently linked dimers are active in phi29 DNA packaging. Three kinds of dimers including cross-linked dimer 120, fused dimer and tandem dimer 139 have been constructed and all of them are active in phi29 DNA packaging (Table 5). Cross-linked dimers are generated by linking pRNA A-b’ to a pRNA with a photosensitive agent attached at G82 of pRNA B-a’ and then linking it to A-b’ by UV activation and purified by gel 120 (Tabel 2). . Fused pRNA dimers are generated by merging the right loop from one pRNA with the left loop sequences of the other pRNA. The merging site is located at the crosslinking sites (Table 5). Tandem pRNAs dimers are generated by joining two pRNA together via head-to-tail linkage (Table 5). If dimers are the building blocks, there are two possible pathways to assemble a hexamer. The first one is the independent addition of individual dimers (2 x 3 = 6). The second way is the cooperative addition of dimers through hand-in-hand interaction to recruit the in-coming dimers (2 + 2 + 2 = 6). pRNA binding analysis reveals that the Hill coefficient167 is 2.5, strongly suggesting that on the connector there are three binding sites for dimer binding and that the binding process is cooperative. Therefore, the sequence of hexamer formation is (2 + 2 + 2 = 6)139. E. Sequence and size requirement of the loops in dimer formation As described earlier, four bases in each of the two wild type pRNA loops are involved in RNA/RNA interactions to form a hexagonal complex(Section I.C.3). It has been suggested that two GC pairs are sufficient for loop/loop interaction98. The size and sequence requirement for such loop/loop interactions in dimer formation has been investigated thoroughly 99. In some cases only two G/C pairs are sufficient for mediating dimer formation. When all four bases are paired, at least one G/C pair is needed. The maximum number of base pairings between the two loops to give full activity is five, while the minimum loop length is five bases for one loop and three bases for the other. Phylogenetic analysis of seven pRNAs from phages SF5, B103, phi29 , PZA, M2, NF, and GA1 support these conclusions 139. Truncation, deletion, mutation and phylogenetic analysis have been performed to determine the sequence requirement for loop/loop interaction in dimer formation. Without considering the tertiary interaction, in some cases only two G/C pairs between the interacting loops could provide certain pRNAs with activity98. When all four bases are paired, at least one G/C pair is required99. The maximum number of base pairings between the two loops to allow pRNA to retain wild-type activity is five, while the minimum number is five for one loop and three for the other. These findings are supported by phylogenetic analysis of seven pRNAs from different phages99. F Minimal RNA size requirement for dimer formation. A 75-base RNA segment, bases 23-97, is able to form dimer, to interlock into the hexamer, to compete with full-length pRNA for procapsid binding, and, therefore, to inhibit phi29 assembly in vitro. It suggests that segments 23-97 are a self-folded independent domain involved in procapsid binding and RNA/RNA interaction in dimer and hexamer formation, while bases 1-22 and 98-120 are involved in DNA translocation but dispensable for RNA/RNA interaction 99. Therefore, this 75-base RNA could be a model for structural studies in RNA dimerization. G. Minimal sequence requirement for procapsid binding and DNA packaging It has also been reported153 that a 55-bases RNA (bases 37-91) is able to bind procapsid specifically, but the efficiency is much lower. . bases 6-113 (105 nucleotides with the additional deletion of two nonessential bases C109 and A106) are the minimum sequence required for full procapsid binding activity as wild type. Bases 1-117 are the minimum sequence needed for full DNA packaging activity. H. Mechanism and role of pRNA in phi29 DNA translocation 1. Sequential action of six pRNA

Peixuan Guo
In 1997, a comprehensive paper was published to describe the role of phi29 pRNA in DNA packaging 105. This was the first paper to describe the specific role of pRNA in phi29 DNA packaging and to provide strong evidence for concluding that six pRNAs work sequentially in driving the DNA translocation motor. This paper also provides an elegant model for elucidating the mechanism of rotation and the quantification of energy (ATP) usage was also provided 105. It advocates that phi29 DNA packaging is accomplished by a mechanism similar to driving a bolt with a hex nut, which consists of six DNA-packaging pRNAs (Fig. 13). Recently, phi29 DNA packaging has become a popular subject. Publications102 168;169 in this area have affirmed that the aforementioned inference concerning the consecutive action of six pRNA in driving the DNA rotation machine is convincing. All icosahedral viruses contain a five-fold symmetrical capsid face. Six copies of pRNA bind to the connector (portal vertex) that is embedded in such a five fold symmetrical area 105;151. Rotation of the hexameric pRNA within a 5-fold symmetrical environment could constitute a mechanical motor in which the relative motion of the two rings could produce a driving force to translocate the DNA into the procapsid. Analogous to the sequential action of six cylinders in a car engine, sequential action of six pRNAs is one way to achieve the turning of this DNA packaging motor. If the six cylinders fired synchronously, the engine would not run continuously (Fig. 13). The pRNA may collaborate with gp16 to perform this rotation job. By sequential action, it is meant that the multiple pRNAs involved in DNA packaging appear to act in a step-by-step process, with each pRNA exerting its individual effect alternatively (Fig.13). The pRNA contains two functional domains: one for connector binding and the other for DNA translocation(Section V.C). pRNA binds to the connector leaving the 5’/3' essential domain free for interaction with other components, such as gp16 or DNA-gp3. It has been reported that the C18C19A20 bulge is located on the surface of a pRNA bound procapsid132;142 and is critical for DNA translocation, while being dispensable for procapsid binding 93;99;105;134;142. The C18C19A20 bulge might be directly involved in interacting with ATP, gp16 or DNA-gp3. It is predicted that pRNA is part of an ATPase and possesses at least two conformations (Section II. M), a relaxed and a contracted one. Alternating between contraction and relaxation and driven by ATP hydrolysis, each member of the hexameric RNA complex helps rotate the translocation machine. The requirement of loop/loop interaction in hexamer formation 96 98 supports the pRNA sequential action model. The pRNAs may need to communicate with each other to ensure that the motion progress is consecutive. Inter-pRNA interactions via loops might serve as a link to pass a signal to adjacent pRNAs. Thus base pairing between the right and left-hand loops might be necessary to transfer a conformational change from one pRNA to an adjacent one.
Fig. 13: A model depicting the sequential action of pRNA in phi29 DNA packaging. The hexagon represents the connector (portal vertex) and the surrounding pentagon represents the head membrane. Six protrusions represent six pRNAs. The variable shapes and patterns portray the pRNA (alone or with other components) in serial energetic states. Some, such as pRNA 4 panel A, are in a contracted conformation, and others, such as pRNA 1 in panel A, are in extended conformation. Arrows point to the different energetic states of pRNA 1. A to G represent six steps of rotation. Each step rotates 12°, since a five to six-fold symmetry mismatch generates 30 equivalent orientations (360°/30 = 12°). The portal vertex turns 72° after six steps. For example, pRNA 1 moves from vertex a (A) to vertex b (G) and rotates 72°. Each step requires one ATP to initiate one pRNA conformation change, and six ATPs are needed for the transition from one vertex to another vertex. Therefore, 30 ATPs are needed for one 360° rotation. The drawing is the view from inside the procapsid (reprinted from 105 with permission from J. Virology). 2. Evidence to support sequential action The conclusion of sequential consecutive action of pRNA comes from the following evidence 105. a. DNA packaging is blocked by only one inactive pRNA in the chain When any one of six pRNAs was replaced with an inactive pRNA, DNA packaging was completely blocked95;103;104. This is an indication that six pRNAs work in a pattern similar to a serial, not parallel, circuit. It strongly supports the speculation that individual pRNAs take turns mediating successive steps of DNA packaging. b. pRNA is very sensitive to mutation A single base change can render the pRNA completely inactive in DNA packaging 115. c. Complementation cannot rescue inactive pRNA with a single base mutation

Peixuan Guo
Extensive investigation shows that not a single plaque can be produced in any complementation test with any two inactive pRNAs with mutations at two different locations 105. Moreover, the yield of a complementation test with two partially active pRNAs has never been higher than the yield produced with one of the more active pRNA alone. See comment The result suggests that the action of pRNA is cooperative and synergetic. d. pRNA mutation cannot be compensated by wild type pRNA Compensation test showed that the wild type phenotype pRNA could not compensate for the defect of any one of these mutants 105. The failure of wild type pRNA to compensate for the defect in mutant pRNAs is clearly documented by the results of inhibition assays in which the activity of wild type pRNA is strongly inhibited by mutant pRNA 95;103;105;139. The failure of wild type pRNA to compensate for mutant pRNAs and the success of mutant pRNAs in inhibiting wild type pRNA is also demonstrated by an almost perfect match in comparing the empirical with the theoretical yield predicted based on sequential model (Fig.14)105.
Fig. 14: Overlapping of empirical curve with the computer simulation curve representing the model of sequential action of six pRNA. The semilog plot of the empirical data is compared with the plots predicted by integrated, sequential and random models of pRNA action, simulated by binomial distribution. Curves represent the yield of virion production versus percentage (molar ratio of mutant to wild type) of mutant pRNA in the assembly mixture. Wild-type pRNA was mixed with various amounts of mutant pRNA 26A/27 and assayed with the in vitro phi29 assembly system e. Math simulation and modeling The strongest support for sequential action comes from the extensive quantification and math simulation as described below. Fig. 16. Models to simulate the mechanism of pRNA action. Three models, Integrated, Sequential Individual, and Random Individual, have been used to simulate the mechanism of pRNA action.(Fig. 16.) Compensation and complementation studies did not support the integrated model, but favored individual models105. Concerning two individual models, competitive inhibition excluded the Random Individual model. Therefore, only the sequential model is favored.
To investigate the role of pRNA in DNA packaging, it is crucial to determine whether the DNA packaging machine is an entirety and should be studied as an intact assemblage (an Integrated model), or whether it can be dissected into the
functions of each individual pRNA molecule. If six pRNA molecules work individually, then do the six pRNAs work consecutively (Sequential Individual model), or does each pRNA contribute one step of each reaction cycle consisting of six steps but in random order (Random Individual model)? These three models have been proposed to distinguish these possibilities. The principle behind the model discrimination and the mathematical formula for quantification has been described in detail 105.
Mechanism of
pRNA actionIntegrated modelIndividual model
Random model
Sequential mod
The models that have been simulated using binomial distribution ( Section I.C.1) are compared empirical curves. The Our results agreed with the Sequential Individual model, since the empirical curve overlapped with the curve predicted with this model (Fig. 14). IV SIGNIFICANCE AND APPLICATION OF THE STUDY ON pRNA A. Model for the study of RNA dimers and trimers Stable dimers and trimers of the pRNA in the absence of protein can be isolated 139. The dimerization/trimerization assay provides a simple and stable system to study the solution of the structure of pRNA complexes and to investigate the mechanism of RNA/RNA interactions. RNA dimerization or oligomerization has been found to play a vital role in certain living systems. Dimerization of retroviral genomic RNA is critical for retroviral life cycle, including translation, reverse transcription, RNA

Peixuan Guo
encapsidation and virion assembly 170-172. An RNA-RNA intermolecular interaction is required for the formation of a specific ribonucleoprotein particle, bicoid mRNA 3’ UTR-STAUFEN, that determines the formation of the anterior pattern of the Drosophila embryo 173. RNA-RNA interaction, is one essential step in the cleavage reaction of RNase P on tRNA 55;174;175. Replications of the plasmid ColE1 are regulated by plasmid-specified small RNAs (RNA I and RNA II) that form complexes by complementary RNA stem-loop interactions 176. The system, which we have developed, is a very simple and stable system that could be used as a model for the study of RNA/RNA intermolecular interactions. B. A model for the study of viral DNA packaging and for the design of new antiviral strategies Extensive investigation on DNA packaging of the dsDNA viruses documents certain common features in this step of the viral life cycle. The commonalties include the use of a pair of non-capsid enzymes to translocate the viral DNA into a procapsid coupled with the hydrolysis of ATP 38;44;50;51;105;177-182. Similarities in DNA packaging between dsDNA bacteriophages and herpes viruses, adenovirus, pavovirus and poxviruses (see Introduction) justify the use of phi29 as model system for the design of new antiviral strategies. Using phi29 pRNA as a target, several methods have been modeled 103 853104 for the inhibition of viral assembly. 1. Inhibition of ø29 assembly by antisense DNA targeting pRNA in vitro Antisense oligos include antisense RNA and antisense DNA. They are small regulatory single-stranded polynucleotides that bind to complementary regions on a specific target in order to control their biological function. Antisense DNA could bind to the pRNA and cause a change in the pRNA electrophoretic mobility 89. Antisense DNA oligos were shown to hybridize to phi29 pRNA by gel shift assays 89. Phi 29 pRNA contains two functional domain (Section V.C). One domain that is located at the central region is required for procapsid binding. The other domain, consisting of the paired 5'- and 3'-ends, is needed for DNA translocation into procapsids, but it is not required for procapsid binding. Oligo P6 targeting the left-hand loop of the procapsid binding domain could block the binding of pRNA to procapsids, resulting in the inhibition of ø29 assembly in vitro. Oligos P11 and P15 targeting either the 5'- or the 3'-end, resistively, of the pRNA did not prevent pRNA from binding to procapsid, but strongly inhibited DNA packaging 87;89. 2. Complete inhibition of phi29 assembly with mutant pRNA in viro Phi29 is used as a model to explore new avenues in antiviral research beyond the in vitro inhibition with antisense oligos targeting pRNA. As already noted, pRNA contains two functional domains (Section V.C). Sequence alterations that disrupt the base pairing at the 5'/3' ends of the DNA translocating domain result in mutant pRNAs that are completely inactive in phage assembly in vitro, but retain wild type procapsid binding affinity 87;95;103;153. These mutant pRNAs are able to compete with wild type pRNAs for procapsid binding and efficiently inhibit viral assembly in vitro and in vivo 103. For in vivo studies, , a plasmid expressing a pRNA with a 4 base mutation at the 3’ end was constructed and transformed into host cells. Cells harboring this plasmid were completely resistant to plaque formation by wild type ø29. The novelty here is the result of “complete” inhibition. It indicates that factors involved in viral assembly can be a targeted for efficient and specific antiviral treatment. The high efficiency was due to two pivotal features. First, the pRNA contains two domains, one for procapsid binding and the other for DNA-packaging. Mutation of the DNA-packaging domain resulted in a pRNA with no DNA-packaging activity, but intact procapsid binding competence. Second, six pRNAs were involved in the packaging of one genome. This higher order dependence of pRNA concomitantly resulted in a higher order of inhibitory effect due to the fact that blocking only one of the six positions occupied by the mutant pRNA could result in the complete block of virion assembly 95. The principle of using molecules containing two functional domains and requiring multiple copy involvement as inhibitors could be applied to gene therapy, intracellular immunization, antiviral drug design or construction of transgenic plants resistant to viral infection, using certain viral structural proteins, enzymes, and other RNAs involved in the viral life cycle. C. Similar mechanism between viral DNA translocation and other nucleic acid sliding/riding process There is a group of nucleic acid-binding proteins that plays a similar role in DNA or RNA riding or sliding related to DNA replication, translocation, recombination, and RNA transcription96;183;184. They can be categorized into two subsets, proteins that bind nucleic acid and proteins that act on nucleic acid. The common feature of these two subsets is that they interact with RNA or DNA in a polymer conformation with a ring-shape morphology. Most members of this group are hexamers. The subset that acts on nucleic acid includes helicase 185;186, the E. coli transcription termination protein Rho187;188, the yeast DNA polymerase processivity factor 189, bovine papillomavirus E1 replication initiator 190, and the E. coli DNA polymerase III holoenzyme191;192, which exist as hexamers 185;193-196. Though the mechanisms of this kind of DNA-protein interaction remain to be elucidated, the finding that six copies of pRNA are attached to a six-fold symmetrical connector during DNA translocation indicates it might have something in common. The process of both viral DNA packaging and DNA replication (or RNA transcription) involves the relative motion of two components, one of which is nucleic acid. It would be intriguing to show how the pRNA may play a role similar to that which protein enzymes, such as DNAS-helicases or the termination factor Rho, play.
Peixuan Guo
A
B Fig. 15: Comparison of phi29 DNA packaging machine (A) with the DNA replication apparatus (B) (A is reprinted from 184 with permission from Elsevier Science. B is reprinted from the newspaper Indianapolis Star with permission). D. Insight hints for macromolecular across the barrier of cell membrane translocation through barrier wall Migration or transportation of macromolecules, such as proteins or nucleic acids, through a barrier or cell membrane is a common process in life systems. After transcription in the nucleus, mRNA and tRNA must pass the nuclear membrane to reach the translation machinery in the cytoplasm. Similarly, nuclear proteins must pass from the cytoplasm, where they are synthesized, to the nucleus where they function. After infection or transfection, most viral or plasmid DNA must pass the nuclear membrane to serve as templates for gene expression (reviewed in 197). The Rev protein of HIV helps in the translocation of viral mRNA from the nucleus to the cytoplasm through a nuclear pore 198;199. Varieties of proteins and other elements migrate into and out of the cell and nucleus to perform their respective functions. Molecular migration or translocation also is manifest in the tracking and rail riding of enhancers or transcription factors along DNA 195, the translocation of the transcription termination protein Rho along RNA 196, and the migration of helicases along single stranded DNA during DNA replication 185;200-202. One of the most complex and intricate translocation processes is viral DNA encapsidation. Our study on DNA encapsidation could provide hints for macromolecular translocation through cell membrane. V. CONCLUDING REMARKS Phi29 DNA packaging system exhibits an extraordinary number of unique features. The in vitro DNA packaging efficiency is the highest in comparison with all in vitro systems available. All the components required for the assembly of the infectious virion and to

Peixuan Guo
interact with pRNA have been cloned in and purified from E. coli. Infectious virion can assemble in vitro with the exclusive use of all purified components and synthetic pRNA with zero background. It is composed of a highly sensitive system with a nine-order of magnitude for the assay of pRNA function. The DNA packaging motor is the strongest nanometer-motor with a highest stalling force. The crystal structure of one of the important motor components, the connector to where the pRNA binds, has been solved. This motor with 1/1027 cubic inch can be made with other purified chimeric components artificially designed (unpublished data). Therefore, this is an unparalleled system and model for the studies on viral DNA packaging, for function analysis of molecular motors, for design of imitating of nanomotor and other machines, for the characterization of biochemical energy transformation into motion, for the deciphering of the process of DNA or RNA riding or sliding enzyme such as helicase and bio-clamp, for the understanding of the mechanism of macromolecule crossing cell membranes and other barrier, and for the development of antiviral therapy to block DNA packaging, a unique process that is not used by eukaryotic cells. The illustration of the role of pRNA in this fascinating biological process would certainly provide new insight in modern biology. This pRNA is a novel and intriguing molecule. It is unusually stable with a very compact and tightly folded structure. It interacts with ions and proteins to adapt numerous conformations depending on the environment and solution condition. It can be manipulated freewheeling to form homologous monomers, dimers, trimers and hexamers; all of the oligomers have a strong intention to form a circle by hand-in-hand interaction intra – or inter-molecularly. Therefore, many unknown properties of RNA can be unfolded by the investigation of this unusual molecule.
Acknowledgement I thank Drs. Mark Trottier, Chaoping Chen for critical comments and review of this manuscript, Jane Kovach, Dan Shu and Steve Hoeprich for the manuscript preparation. Dr. Jose Carrascosa for information on his research data. Dr. Michael Rossmann for his support by using the crystal structure data of the connector. The work in the author’s laboratory is supported by NIH grants GM59944, GM60529, GM48159 and GM46490, as well as NSF grant MCB9723923.

Peixuan Guo
Reference List 1. C.Bazinet and King,J.: The DNA translocation vertex of dsDNA bacteriophages. Ann.Rev.Microbiol. 39:109-129, 1985. 2. Casjens,S. and Hendrix,R.: Control mechanisms in dsDNA bacteriophage assembly, In: The Bacteriophages Vol.1. Ed.
R.Calendar. Plenum Pubishing Corp., New York, 1988, pp. 15-92. 3. P.Guo: Introduction: Principles, perspectives, and potential applications in viral assembly. Seminars in Virology (Editor's
Introduction) 5(1):1-3, 1994. 4. W.C.Earnshaw and Casjens,S.R.: DNA packaging by the double-stranded DNA bacteriophages. Cell 21:319-331, 1980. 5. K.E.Gustin and Imperiale,M.J.: Encapsidation of viral DNA requires the adenovirus L1 52/55-kilodalton protein. J Virol.
72:7860-7870, 1998. 6. S.I.Schmid and Hearing,P.: Bipartite structure and functional independence of adenovirus type 5 packaging elements. J Virol.
71:3375-3384, 1997. 7. J.C.D'Halluin, Milleville,M., Boulanger,P.A., and Martin,G.R.: Temperature-sensitive mutants of adenovirus type 2 blocked in
virion assembly: accumulation of light intermediate particles. J.Virol. 26:344-356, 1978. 8. C.L.Cepko and Sharp,P.A.: Assembly of adenovirus major capsid protein is mediated by a nonvirion protein. Cell 31:407-415,
1982. 9. B.Edvardsson, Everitt,E., Joernvall,E., Prage,L., and Philipson,L.: Intermediates in adenovirus assembly. J.Virol. 19:533-547,
1976. 10. F.J.Rixon and McNab,D.: Packaging-competent capsids of a herpes simplex virus temperature- sensitive mutant have properties
similar to those of in vitro-assembled procapsids. J Virol. 73:5714-5721, 1999. 11. J.Y.Lee, Irmiere,A., and Gibson,W.: Primate cytomegalovirus assembly: evidence that DNA packaging occurs subsequent to B
capsid assembly. Virology 167:87-96, 1988. 12. A.Dasgupta and Wilson D.W.: ATP Depletion Blocks Herpes Simplex Virus DNA Packaging and Capsid Maturation. J
Virology 73(3):2006-2015, 1999. 13. Moss,B.: Replication of Poxviruses, In: Virology. Eds. B.N.Fields and et al. Raven Press, New York, 1985, pp. 685-703. 14. A.M.DeLange, Reddy,M., Scraba,D., Upton,C., and McFadden,G.: Replication and resolution of cloned poxvirus telomeres in
vivo generates linear minichromosomes with intact viral hairpin termini. J.Virol. 59:249-259, 1986. 15. B.L.Parsons and Pickup,D.J.: Transcription of orthopoxvirus telomeres at late times during infection. Virology 175:69-80,
1990. 16. H.Drexler: Initiation by bacteriophage T1 of DNA packaging at a site between the P and Q genes of bacteriophage l. J.Virol.
49:754-759, 1984. 17. H.Shibata, Fujisawa,H., and Minagawa,T.: Early events in a defined in vitro system for packaging of bacteriophage T3 DNA.
Virology 159:250-258, 1987. 18. V.B.Rao and Black,L.W.: Evidence that a phage T4 DNA packaging enzyme is a processed form of the major capsid gene
product. Cell 42:967-977, 1985. 19. R.D.Everett: DNA replication of bacteriophage T5. 3. Studies on the structure of concatemeric T5 DNA. J.gen.virol. 52:25-38,
1981. 20. M.E.Cerritelli and Studier,F.W.: Purification and characterization of T7 head-tail connectors expressed from the cloned gene. J
Mol Biol 285:299-307, 1996. 21. M.Sun, Louie,D., and Serwer,P.: Single-event analysis of the packaging of bacteriophage T7 DNA concatemers in vitro.
Biophys J 77(3):1627-1637, 1999. 22. K.Skorupski, Sauer,B., and Sternberg,N.: Faithful cleavage of the P1 packaging site (pac) requires two phage proteins, PacA
and PacB, and two Escherichia coli proteins, IHF and HU. J Mol Biol 243:268-282, 1994. 23. G.Pruss and Calendar,R.: Maturation of bacteriophage P2 DNA. Virology 86:454-467, 1978. 24. S.Rishovd, Holzenburg,A., Johansen,B.V., and Lindqvist,B.H.: Bacteriophage P2 and P4 morphogenesis: structure and function
of the connector. Virology 245:11-17, 1998. 25. B.Greene and King,J.: In vitro unfolding/refolding of wild type phage P22 scaffolding protein reveals capsid-binding domain. J
Biol Chem 274:16135-16140, 1999. 26. S.D.Moore and Prevelige,P.E., Jr.: Structural transformations accompanying the assembly of bacteriophage P22 portal protein
rings in vitro. J.Biol.Chem. 276:6779-6788, 2001. 27. K.Eppler, Wyckoff,E., Goates,J., Parr,R., and Casjens,S.: Nucleotide sequence of the bacteriophage P22 genes required for
DNA packaging. Virology 183:519-538, 1991. 28. C.M.Burns, Chan,H.L.B., and DuBow,M.S.: In vitro maturation and encapsidation of the DNA of transposable µ-like phage
D108. Proc.Natl.Acad.Sci.USA 87:6092-6096, 1990. 29. M.P.Smith and Feiss,M.: Sequence analysis of the phage 21 genes for prohead assembly and head completion. Gene 126:1-7,
1993. 30. S.Grimes and Anderson,D.: The bacteriophage phi29 packaging proteins supercoil the DNA ends. J Mol Biol 266:901-914,
1997. 31. C.S.Lee and Guo,P.: In vitro assembly of infectious virions of ds-DNA phage φ29 from cloned gene products and synthetic
nucleic acids. J.Virol. 69:5018-5023, 1995.

Peixuan Guo
32. C.Gutierrez, Freire,R., Salas,M., and Hermoso,J.M.: Assembly of phage phi29 genome with viral protein p6 into a compact complex. EMBO 13(1):269-276, 2001.
33. J.M.Valpuesta, Fernandez,J.J., Carazo,J.M., and Carrascosa,J.L.: The three-dimensional structure of a DNA translocating machine at 10 A resolution. Structure.Fold.Des 7:289-296, 1999.
34. M.H.Levner and Cozzarelli,N.R.: Replication of viral DNA in SPO1-infected Bacillus subtilis. I. Replicative intermediates. Virology 48:402-416, 1972.
35. L.P.Gage and Geiduschek,E.P.: RNA synthesis during bacteriophage SPO1 development: six classes of SPO1 RNA. J.Mol.Biol. 57:279-297, 1971.
36. P.Dubé, Tavares,P., Lurz,R., and van Heel,M.: The portal protein of bacteriophage SPP1: a DNA pump with 13-fold symmetry. EMBO J. 12:1303-1309, 1993.
37. A.Gual, Camacho,A.G., and Alonso,J.C.: Functional analysis of the terminase large subunit, G2P, of Bacillus subtilis bacteriophage SPP1. J.Biol.Chem. 275:35311-35319, 2000.
38. A.Becker and Gold,M.: Prediction of an ATP reactive center in the small subunit, gpNul of phage lambda terminase enzyme. J Mol Biol 199:219-222, 1988.
39. T.Dokland and Murialdo,H.: Structural transitions during maturation of bacteriophage lambda capsids. J Mol.Biol 233:682-694, 1993.
40. L.Woods and Catalano,C.E.: Kinetic characterization of the GTPase activity of phage lambda terminase: evidence for communication between the two "NTPase" catalytic sites of the enzyme. Biochemistry Nov 2;38(44):14624-14630, 1999.
41. J.Q.Hang, Catalano,C.E., and Feiss,M.: The Functional Asymmetry of cosN, the Nicking Site for Bacteriophage lambda DNA Packaging, Is Dependent on the Terminase Binding Site, cosB. Biochemistry 40:13370-13377, 2001.
42. Anderson,D.L. and Reilly,B.: Morphogenesis of bacteriophage φ29, In: Bacillus subtilis and other gram-positive bacteria: Biochemistry, physiology, and molecular genetics. Eds. A.L.Sonenshein, J.A.Hoch, and R.Losick. American Society for Microbiology, Washington, D.C., 1993, pp. 859-867.
43. L.W.Black: DNA Packaging in dsDNA bacteriophages. Ann Rev Microbiol 43:267-292, 1989. 44. P.Guo and Trottier,M.: Biological and biochemical properties of the small viral RNA (pRNA) essential for the packaging of the
double-stranded DNA of phage φ29. Seminars in Virology 5:27-37, 1994. 45. W.W.Newcomb, Juhas,R.M., Thomsen,D.R., Homa,F.L., Burch,A.D., Weller,S.K., and Brown,J.C.: The UL6 Gene Product
Forms the Portal for Entry of DNA into the Herpes Simplex Virus Capsid. J.Virol. 75:10923-10932, 2001. 46. T.Hohn, Wurtz,M., and Hohn,B.: Capsid transformation during packaging of bacteriophage λ DNA. Philos Trans R Soc
London Ser B 276:51-61, 1976. 47. T.Hohn: Packaging of genomes in bacteriophages: a comparison of ssRNA bacteriophages and dsDNA bacteriophages. Philos
Trans R Soc London Ser B 276:143-150, 1976. 48. M.A.Bjornsti, Reilly,B.E., and Anderson,D.L.: Morphogenesis of bacteriophage φ29 of Bacillus subtilis: oriented and quantized
in vitro packaging of DNA protein gp3. J.Virol. 45:383-396, 1983. 49. P.Guo, Grimes,S., and Anderson,D.: A defined system for in vitro packaging of DNA-gp3 of the Bacillus subtilis bacteriophage
φ29. Proc Natl Acad Sci USA 83:3505-3509, 1986. 50. P.Guo, Peterson,C., and Anderson,D.: Prohead and DNA-gp3-dependent ATPase activity of the DNA packaging protein gp16
of bacteriophage φ29. J Mol Biol 197:229-236, 1987. 51. M.Morita, Tasaka,M., and Fujisawa,H.: DNA packaging ATPase of bacteriophage T3. Virology 193:748-752, 1993. 52. P.Guo, Erickson,S., and Anderson,D.: A small viral RNA is required for in vitro packaging of bacteriophage φ29 DNA. Science
236:690-694, 1987. 53. P.Guo, Rajogopal,B., Anderson,D., Erickson,S., and Lee,C.-S.: sRNA of bacteriophage φ29 of B.subtilis mediates DNA
packaging of φ29 proheads assembled in E. coli. Virology 185:395-400, 1991. 54. K.Kruger, Grabowski,P.J., Zaug,A.J., Sands,J., Gottschling,D.E., and Cech,T.R.: Self-splicing RNA: autoexcision and
autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell 31:147-157, 1982. 55. C.Guerrier-Takada, Gardiner,K., Marsh,T., Pace,N., and Altman,S.: The RNA moiety of ribonuclease P is the catalytic subunit
of the enzyme. Cell 35:849-857, 1983. 56. T.R.Cech: The chemistry of self-splicing RNA and RNA enzymes. Science 236:1532-1539, 1987. 57. S.Aigner, Lingner,J., Goodrich,K.J., Grosshans,C.A., Shevchenko,A., Mann,M., and Cech,T.R.: Euplotes telomerase contains
an La motif protein produced by apparent translational frameshifting. EMBO J. 19:6230-6239, 2000. 58. D.J.Lane, Pace,B., Olsen,G.L., Stahl,D.A., Sogin,M.L., and Pace,N.R.: Rapid-determination of 16s ribosomal-RNA sequences
for phylogenetic analysis. Proc.Natl.Acad.Sci.USA 82:6955-6959, 1985. 59. D.W.Lazinski and Taylor,J.M.: Intracellular Cleavage and Lifation of Hepatitis Delta Virus Genomic RNA: Regulation of
Ribozyme Activity by cis-Acting Sequences and Host Factors. J.Virol. 69:1190-1120, 1995. 60. T.B.Macnaughton, Wang,Y.J., and Lai,M.M.: Replication of hepatitis delta virus RNA: effect of mutations of the autocatalytic
cleavage sites. J.Virol. 67:2228-2234, 1993. 61. M.M.Lai: The molecular biology of hepatitis delta virus. Annu.Rev.Biochem. 64:259-286, 1995. 62. N.A.Sarver, Cantin,E.M., Chang,P.S., Zaia,J.A., Ladne,P.A., Stephens,D.A., and Rossi,J.J.: Ribozymes as potential anti-HIV-1
therapeutic agents. Science 247:1222-1225, 1990.

Peixuan Guo
63. A.Woisard, Fourrey,J.L., and Favre,A.: Multiple folded conformations of a hammerhead ribozyme domain under cleavage conditions. J Mol Biol 239:366-370, 1994.
64. J.B.Murray, Adams,C.J., Arnold,J.R.P., and Stockley,P.G.: The roles of the conserved pyrimidine bases in hammerhead ribozyme catalysis: evidence for a magnesium ion-binding site. Biochem.J. 311:487-494, 1995.
65. A.L.Feig, Scott,W.G., and Uhlenbeck,O.C.: Inhibition of the Hammerhead Ribozyme Cleavage Reaction by Site-Specific Binding of Tb(III). Science 279:81-84, 1998.
66. B.M.Chowrira, Berzal-Herranz,A., and Burke,J.M.: Novel guanosine requirement for catalysis by the hairpin ribozyme. Nature 354:320-322, 1991.
67. S.A.Strobel and Ryder,S.P.: Biological catalysis. The hairpin's turn. Nature 410:761-763, 2001. 68. F.Kawamura and Ito,J.: Transcription of the genome of bacteriophage φ29: Isolation and mapping of the major early mRNA
synthesized in vivo and in vitro. J.Virol. 23:562-577, 1977. 69. J.M.Sogo, Inciarte,M.R., Corral,J., Vinuela,E., and Salas,M.: RNA polymerase binding sites and transcription map of the DNA
of a Bacillus subtilis phage φ29. J Mol Biol 127:411-436, 1979. 70. P.Guo, Bailey,S., Bodley,J.W., and Anderson,D.: Characterization of the small RNA of the bacteriophage φ29 DNA packaging
machine. Nucleic Acids Res. 15:7081-7090, 1987. 71. J.Wichitwechkarn, Bailey,S., Bodley,J.W., and Anderson,D.: Prohead RNA of bacteriophage φ29: size, stoichiometry and
biological activity. Nucelic Acids Research 17(9):3459-3468, 1989. 72. A.McGregor, Rao,M.V., Duckworth,G., Stockley,P.G., and Connolly,B.A.: Preparation of oligoribonucleotides containing 4-
thiouridine using Fpmp chemistry. Photo-crosslinking to RNA binding proteins using 350 nm irradiation. Nucleic Acids Res. 24(16):3173-3180, 1996.
73. M.E.Tosi, Reilly,B.E., and Anderson,D.L.: Morphogenesis of bacteriophage φ29 of Bacillus subtilis: cleavage and assembly of the neck appendage protein. J.Virol. 16:1282-1295, 1975.
74. E.W.Hagen, Reilly,B.E., Tosi,M.E., and Anderson,D.L.: Analysis of gene function of bacteriophage φ29 of Bacillus subtilis: identification of cistrons essential for viral assembly. J.Virol. 19(2):501-517, 1976.
75. E.Vinuela, Camacho,A., Jimenez,F., Carrascosa,J.L., Ramirez, G, and Salas,M.: Structure and assembly of phage phi29. Philosophical Transactions of the Royal Society of London - Series B: Biological Sciences 276:29-35, 1976.
76. A.Camacho, Jimenez,F., De La,T.J., Carrascosa,J.L., Mellado,R.P., Vasquez,C., Vinuela,E., and Salas,M.: Assembly of Bacillus subtilis phage phi29. 1. Mutants in the cistrons coding for the structural proteins. Eur.J Biochem. 73:39-55, 1977.
77. M.Salas: Protein-Priming of DNA Replication. Ann.Rev.Biochem. 60:39-71, 1991. 78. D.L.Anderson, Hickman,H.H., and Reilly,B.E.: Structure of Bacillus subtilis bacteriophage φ 29 and the length of φ29
deoxyribonucleic acid. J Bact 91:2081-2089, 1966. 79. J.Ortín, Viñuela,E., Salas,M., and Vásquez,C.: DNA-protein complex in circular DNA from phage φ29. Nature New Biol
234:275-277, 1971. 80. J.Méndez, Blanco,L., Esteban,J.A., Bernad,A., and Salas,M.: Initiation of φ29 DNA replications occurs at the second 3+
nucleotide of the linear template: A sliding-back mechanism for protein-primed DNA replication. Proc Natl Acad Sci USA 89:9579-9583, 1992.
81. L.Blanco, Bernad,A., Lázaro,J.M., Martin,G., Garmendia,C., and Salas,M.: Highly efficient DNA synthesis by the phage φ29 DNA polymerase symmetrical mode of DNA replication. J.Biol.Chem. 264:8935-8940, 1989.
82. L.Blanco, Lazaro,J.M., de Vega,M., Bonnin,A., and Salas,M.: Terminal protein-primed DNA amplification. Proc.Natl.Acad.Sci.USA 91:12198-12202, 1994.
83. S.Grimes and Anderson,D.: In Vitro Packaging of Bacteriophage φ29 DNA Restriction Fragments and the Role of the Terminal Protein gp3. J.Mol.Biol. 209:91-100, 1989.
84. M.A.Bjornsti, Reilly,B.E., and Anderson,D.L.: In vitro assembly of the Bacillus subtilis bacteriophage φ29. Proc.Natl.Acad.Sci.USA 78:5861-5865, 1981.
85. P.Guo, Erickson,S., Xu,W., Olson,N., Baker,T.S., and Anderson,D.: Regulation of the phage φ29 prohead shape and size by the portal vertex. Virology 183:366-373, 1991.
86. C.S.Lee and Guo,P.: A highly sensitive system for the in vitro assembly of bacteriophage φ 29 of Bacillus subtilis. Virology 202:1039-1042, 1994.
87. C.L.Zhang, Lee,C.-S., and Guo,P.: The proximate 5' and 3' ends of the 120-base viral RNA (pRNA) are crucial for the packaging of bacteriophage φ29 DNA. Virology 201:77-85, 1994.
88. C.L.Zhang, Trottier,M., and Guo,P.X.: Circularly permuted viral pRNA active and specific in the packaging of bacteriophage φ29 DNA. Virology 207:442-451, 1995.
89. C.L.Zhang, Garver,K., and Guo,P.: Inhibition of phage φ29 assembly by antisense oligonucleotides targeting viral pRNA essential for DNA packaging. Virology 211:568-576, 1995.
90. T.Pan, Gutell,R.R., and Uhlenbeck,O.C.: Folding of Circularly Permuted Transfer RNAs. Science 254:1361-1364, 1991. 91. J.M.Nolan, Burke,D.H., and Pace,N.R.: Circularly Permuted tRNAs as Specific Photoaffinity Probes of Ribonuclease P RNA
Structure. Science 261:762-765, 1993. 92. J.B.Katz, Henderson,L.M., and Erickson,G.A.: Recombination in vivo of pseudorabies vaccine strains to produce new virus
strains. Vaccine 8:286-288, 1990.

Peixuan Guo
93. C.L.Zhang, Tellinghuisen,T., and Guo,P.: Use of circular permutation to assess six bulges and four loops of DNA-Packaging pRNA of bacteriophage φ29. RNA 3:315-322, 1997.
94. R.J.D.Reid, Bodley,J.W., and Anderson,D.: Characterization of the prohead-pRNA interaction of bacteriophage φ29. J Biol Chem 269:5157-5162, 1994.
95. M.Trottier and Guo,P.: Approaches to determine stoichiometry of viral assembly components. J.Virol. 71:487-494, 1997. 96. P.Guo, Zhang,C., Chen,C., Trottier,M., and Garver,K.: Inter-RNA interaction of phage phi29 pRNA to form a hexameric
complex for viral DNA transportation. Mol.Cell. 2:149-155, 1998. 97. C.Chen, Trottier,M., and Guo,P.: New approaches to stoichiometry determination and mechanism investigation on RNA
involved in intermediate reactions. Nucleic Acids Symposium Series 36:190-193, 1997. 98. F.Zhang, Lemieux,S., Wu,X., St.-Arnaud,S., McMurray,C.T., Major,F., and Anderson,D.: Function of hexameric RNA in
packaging of bacteriophage phi29 DNA in vitro. Mol.Cell. 2:141-147, 1998. 99. C.Chen, Zhang,C., and Guo,P.: Sequence requirement for hand-in-hand interaction in formation of pRNA dimers and
hexamers to gear phi29 DNA translocation motor. RNA 5:805-818, 1999. 100. R.J.D.Reid, Zhang,F., Benson,S., and Anderson,D.: Probing the structure of bacteriophage φ29 prohead RNA with specific
mutations. J Biol Chem 269:18656-18661, 1994. 101. Borja Ibarra, Jose R.Caston, Oscar Llorca, Mikel Valle, Jose M.Valpuesta, and Jose L.Carrascosa: Topology of the components
of the DNA packaging machinery in the phage phi29 prohead. J.Mol.Biol. 298:807-815, 2000. 102. A.A.Simpson, Tao,Y., Leiman,P.G., Badasso,M.O., He,Y., Jardine,P.J., Olson,N.H., Morais,M.C., Grimes,S., Anderson,D.L.,
Baker,T.S., and Rossmann,M.G.: Structure of the bacteriophage phi29 DNA packaging motor. Nature 408:745-750, 2000. 103. M.Trottier, Zhang,C.L., and Guo,P.: Complete inhibition of virion assembly in vivo with mutant pRNA essential for phage φ29
DNA packaging. J.Virol. 70:55-61, 1996. 104. M.Trottier, Garver,K., Zhang,C., and Guo,P.: DNA-packaging pRNA as target for complete inhibition of viral assembly in vitro
and in vivo. Nucleic Acids Symposium Series 36:187-189, 1997. 105. C.Chen and Guo,P.: Sequential action of six virus-encoded DNA-packaging RNAs during phage phi29 genomic DNA
translocation. J.Virol. 71:3864-3871, 1997. 106. Y.Mat-Arip, Garver,K., Chen,C., Sheng,S., Shao,Z., and and Guo,P.: Three-dimensional Interaction of Phi29 pRNA Dimer
Probed by Chemical Modification Interference, Cryo-AFM, and Cross-linking. J Biol Chem 276:32575-32584, 2001. 107. P.Ayback, Sandstrom,A., Yamakage,S., Sund,C., Glemarec,C., and Chattopadtryaya,J.: Solution structure of lariat RNA by
500MHz NMR spectroscopy and molecular dynamics studies in water. J Biochem Biophys Meth 27:229-259, 1993. 108. R.D.Peterson, Bartel,D.P., Szostak,J.W., and Feigon,J.: 1H NMR studies of the high-affinity Rev binding site of the Rec
responsive element of HIV-1 mRNA base pairing in the core binding element. Biochemistry 33:5357-5366, 1994. 109. J.H.Cate, Gooding,A.R., Podell,E., Zhou,K., Golden,B.L., Kundrot,C.E., Cech,T.R., and Doudna,J.A.: Crystal structure of a
group I ribozyme domain: primciples of RNA packaging. Science 273:1678-1685, 1996. 110. H.W.Pley, Flaherty,K.M., and McKay,D.B.: Three-dimensional structure of a hammerhead ribozym. Nature 372:68-74, 1994. 111. J.L.Sussman, Holbrook,S.R., Warrant,W.R., Church,G.M., and Kim,S.H.: Crystal structure of yeast phenylalanine transfer
RNA. I. Crystallograhpic refinement. J.Mol.Biol. 123:607-630, 1978. 112. B.Hingerty, Brown,R.S., and Jack,A.: Further refinement of the structure of yeast tRNAphe. J.Mol.Biol. 124:523, 1987. 113. E.Westhof, Dumas,P., and Moras,D.: Crystallographic refinement of yerst aspartic acid transfer RNA. J.Mol.Biol. 184:119,
1985. 114. M.Zuker: On finding all suboptimal foldings of an RNA molecule. Science 244:48-52, 1989. 115. C.L.Zhang, Tellinghuisen,T., and Guo,P.: Conformation of the helical struture of the 5'/3' termini of the essential DNA
packaging pRNA of phage φ29. RNA 1:1041-1050, 1995. 116. T.Pecenkova, Benes,V., Paces,J., Vlcek,C., and Paces,V.: Bacteriophage B103: complete DNA sequence of its genome and
relationship to other Bacillus phages. Gene 199:157-163, 1997. 117. S.Bailey, Wichitwechkarn,J., Johnson,D., Reilly,B., Anderson,D., and Bodley,J.W.: Phylogenetic analysis and secondary
structure of the Bacillus subtilis bacteriophage RNA required for DNA packaging. J Biol Chem 265:22365-22370, 1990. 118. S.H.Hixson and Hixson,S.S.: p-Azidophenacyl bromide, a versatile photolabile bifunctional reagent. Reaction with
glyceraldehyde-3-phosphate dehydrogenase. Biochemistry 14:4251-4254, 1975. 119. A.B.Burgin and Pace,N.R.: Mapping the active site of ribonuclease P RNA using a substrate containing a photoaffinity agent.
EMBO J 9:4111-4118, 1990. 120. K.Garver and Guo,P.: Mapping the inter-RNA interaction of phage phi29 by site-specific photoaffinity crosslinking. J Biol
Chem 275(4):2817-2824, 2000. 121. D.A.Wassarman: Psoralen crosslinking of small RNAs in vitro. Molecular Biology Reports 17:143-151, 1993. 122. K.Tyc and Steitz,J.A.: A new interaction between the mouse 5' external transcribed spacer of pre-rRNA and U3 snRNA
detected by psoralen crosslinking. Nucleic Acids Res. 20:5375-5382, 1992. 123. G.D.Cimino, Gamper,H.B., Isaacs,S.T., and Hearst,J.E.: Psoralens as photoactive probes of nucleic acid structure and function:
organic chemistry, photochemistry, and biochemistry. Ann.Rev.Biochem. 54:1151-1193, 1985. 124. C.Chen and Guo,P.: Magnesium-induced conformational change of packaging RNA for procapsid recognition and binding
during phage phi29 DNA encapsidation. J.Virol. 71:495-500, 1997.

Peixuan Guo
125. C.F.Hui and Cantor,C.R.: Mapping the location of psoralen crosslinks on RNA by mung bean nuclease sensitivity of RNA-DNA hybrids. Proc.Natl.Acad.Sci USA 82:1381-1385, 1985.
126. D.A.Wassarman and Steitz,J.A.: Interactions of small nuclear RNA's with precursor messenger RNA during in vitro splicing. Science 257:1918-1925, 1992.
127. T.Mohammad, Chen,C., Guo,P., and Morrison,H.: Photoinduced cross-linking of RNA by cis-Rh(phen)2Cl2+ and cis- Rh(phen)(phi)Cl2+: a new family of light activatable nucleic acid cross-linking agents. Bioorg.Med.Chem Lett. 9:1703-1708, 1999.
128. U.Moazed, Stern,S., and Noller,H.F.: Rapid chemical probing of conformation in 16S ribosomal RNA and 30S ribosomal subunits using primer extension. J Mol Biol 187:399-416, 1986.
129. L.P.Yap and Musier-Forsyth,K.: Transfer RNA aminoacylation: Identification of a critical ribose 2'-hydroxyl-base interaction. RNA 1:418-424, 1995.
130. C.Ehresmann, Baudin,F., Mougel,M., Romby,P., Ebel,J.-P., and Ehresmann,B.: Probing the structure of RNAs in solution. Nucleic Acids Res. 15:9109-9128, 1987.
131. S.Stern, Moazed,D., and Noller,H.F.: Structural analysis of RNA using chemical and enzymatic probing monitored by primer extension. Meth.Enzymol. 164:481-489, 1988.
132. M.Trottier, Mat-Arip,Y., Zhang,C., Chen,C., Sheng,S., Shao,Z., and Guo,P.: Probing the structure of monomers and dimers of the bacterial virus phi29 hexamer RNA complex by chemical modification. RNA 6:1-10:1257-1266, 2000.
133. C.Zhang, Trottier,M., Chen,C., and Guo,P.: Chemical modification patterns of active and inactive as well as procapsid-bound and unbound DNA-packaging RNAof bacterial virus Phi29. Virology 281:281-293, 2001.
134. R.J.D.Reid, Bodley,J.W., and Anderson,D.: Identification of bacteriophage φ29 prohead RNA (pRNA) domains necessary for in vitro DNA-gp3 packaging. J.Biol.Chem. 269:9084-9089, 1994.
135. D.G.Knorre and Vlassov,V.V.: Complementary-addressed (sequence-specific) modification of nucleic acids. Prog Nucleic Acid Res Mol.Biol. 32:291-320, 1985.
136. A.D.Ellington and Szostak,J.W.: In vitro selection of RNA molecules that bind specific ligands. Nature 346:818-822, 1990. 137. G.Tuerk and Gold,L.: Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA
ploymerase. Science 249:505-510, 1990. 138. F.Zhang and Anderson,D.: In vitro selection of Bacteriophage phi29 prohead RNA aptamers for prohead binding. J Biol Chem
273:2947-2953, 1998. 139. C.Chen, Sheng,S., Shao,Z., and Guo,P.: A Dimer as a Building Block in Assembling RNA. A hexamer that gears bacterial virus
phi29 DNA-translocating machinery. J Biol Chem 275(23):17510-17516, 2000. 140. M.S.Babcock, Pednault,E.P.D., and Olson,W.K.: Nucleic Acid Structure Analysis. Mathematics for local cartesian and helical
structure parameters that are truly comparable between structures. J.Mol.Biol. 237:125-156, 1994. 141. D.Gautheret and Cedergren,R.: Modeling the three-dimensional structure of RNA. FASEB-J 7 :97-105, 1993. 142. C.Zhang, Trottier,M., Chen,C., and Guo,P.: Chemical Modification Patterns of Active and inactive as Well as Procapsid-Bound
and Unbound DNA-Packaging RNA of Bacterial Virus Phi29. Virology 281:281-293, 2001. 143. S.Hoeprich and Guo,P.: Computer modeling of 3-D structure of pRNA monomer, dimer and hexamer of phi29 DNA packaging
motor. J Biol.Chem 2001. 144. C.Massire and Westhof,E.: MANIP: an interactive tool for modelling RNA. J Mol.Graph.Model. 16:197, 1998. 145. C.A.G.Haasnoot, Hilbers,C.W., van der Marel,G.A., van Broom,J.H., Singh,U.C., Pattabiraman,N., and Kollman,P.A.: On loop
folding in nucleic acid hairpin-type structures. J.Biomol.Struct.& Dyn 3:843-857, 1986. 146. D.H.Turner, Sugimoto,N., and Freier,S.M.: RNA structure prediction. Annu.Rev.Biophys.Chem. 17:167-192, 1988. 147. F.Michel, Ellington,A.D., Couture,S., and Szostak,J.W.: Phylogenetic and genetic evidence for base-triples in the catalytic
domain of group I introns. Nature 34:578-580, 1990. 148. A.A.Simpson, Leiman,P.G., Tao,Y., He,Y., Badasso,M.O., Jardine,P.J., Anderson,D.L., and Rossmann,M.G.: Structure
determination of the head-tail connector of bacteriophage phi29. Acta Crystallogr.D.Biol.Crystallogr. 57:1260-1269, 2001. 149. S.Grimes and Anderson,D.: RNA Dependence of the Bateriophage phi29 DNA Packaging ATPase. Mol.Biol. 215:559-566,
1990. 150. J.M.Carazo, Donate,L.E., Herranz,L., Secilla,J.P., and Carrascosa,J.L.: Three-dimensional reconstruction of the connector of
bacteriophage φ29 at 1.8 nm resolution. J Mol Biol 192:853-867, 1986. 151. R.W.Hendrix: Summetry mismatch and DNA packaging in large bacteriophages. Proc.Natl.Acad.Sci.USA 75:4779-4783, 1978. 152. Y.Tao, Olson,N.H., Xu,W., Anderson,D.L., Rossmann,M.G., and Baker,T.S.: Assembly of a Tailed Bacterial Virus and Its
Genome Release Studied in Three Dimensions. Cell 95 :431-437, 1998. 153. K.Garver and Guo,P.: Boundary of pRNA functional domains and minimum pRNA sequence requirement for specific
connector binding and DNA packaging of phage phi29. RNA. 3:1068-1079, 1997. 154. L.E.Donate and Carrascosa,J.L.: Characterization of a versatile in vitro DNA packaging system based on hybrid λ/φ29
proheads. Virology 182:534-544, 1991. 155. L.E.Donate, Murialdo,H., and Carrascosa,J.L.: Production of λ-phi29 Chimeras. Virology 179:936-940, 1990. 156. J.M.Valpuesta, Donate,L.E., Mier,C., Herranz,L., and Carrascosa,J.L.: RNA-mediated specificity of DNA packaging into
hybrid λ/φ29 proheads. The EMBO Journal 12 (11):4453-4459, 1993.

Peixuan Guo
157. M.O.Badasso, Leiman,P.G., Tao,Y., He,Y., Ohlendorf,D.H., Rossmann,M.G., and Anderson,D.: Purification, crystallization and initial X-ray analysis of the head- tail connector of bacteriophage phi29. Acta Crystallogr D Biol Crystallogr 56 ( Pt 9):1187-1190, 2000.
158. A.Guasch, Parraga,A., Pous,J., Valpuesta,J.M., Carrascosa,J.L., and Coll,M.: Purification, crystallization and preliminary X-ray diffraction stydies of the bacteriophage phi 29 connector particle. FEBS 430(1998):283-287, 1998.
159. L.E.Donate, Valpuesta,J.M., Mier,C., Rojo,F., and Carrascosa,J.L.: Characterization of an RNA-binding domain in the bacteriophage phi29 connector. J.Biol.Chem. 268(27):20198-20204, 1993.
160. M.Valle, Kremer,L., Martinez,A., Roncal,F., Valpuesta,J.M., Albar,J.P., and Carrascosa,J.L.: Domain architecture of the bacteriophage phi29 connector protein. J Mol.Biol 288:899-909, 1999.
161. J.L.Carrascosa and Valpuesta,J.M.: Bacteriophage connectors: Structural features of a DNA translocating motor. Recent Res.Devel.Virol. 1:449-465, 1999.
162. M.C.Morais, Tao Y, Olsen,N.H., Grimes,S., Jardine,P.J., Anderson,D., Baker TS, and Rossmann,M.G.: Cryoelectron-Microscopy Image Reconstruction of Symmetry Mismatches in Bacteriophage phi29. J Struct Biol 135:38-46, 2001.
163. J.M.Valpuesta and Carrascosa,J.: Structure of viral connectors and their funciton in bacteriophage assembly and DNA packaging. Quart.Rev.Biophys. 27:107-155, 1994.
164. J.Jimenez, Santisteban,A., Carazo,J.M., and Carrascosa,J.L.: Computer graphic display method for visualizing three-dimensional biological structures. Science 232:1113-1115, 1986.
165. J.Wichitwechkarn, Johnson,D., and Anderson,D.: Mutant prohead RNAs in the in vitro packaging of bacteriophage phi 29 DNA-gp3. J Mol.Biol 223:991-998, 1992.
166. M.L.Perdue, Cohen,J.C., Kemp,M.C., Randall,C.C., and O'Callaghan,D.J.: Characterization of three species of nucleocapsids of equine herpesvirus type-1 (EHV-1). Virology 64:187-204, 1975.
167. J.N.Weiss: The Hill equation revisited: uses and misuses. The FASEB Journal 11:835-841, 1997. 168. Davenport R.J.: Crossover Research Yield Scents and Sensitivity---Watching a virus get stuffed. Science 291:2071-2072, 2001. 169. D.E.Smith, Tans,S.J., Smith,S.B., Grimes,S., Anderson,D.L., and Bustamante,C.: The bacteriophage phi29 portal motor can
package DNA against a large internal force. Nature 413:748-752, 2001. 170. W.I.Sundquist and Heaphy,S.: Evidence for interstand quadruplex formation in the dimerization of human immunodeficiency
virus 1 genomic RNA. Proc.Natl.Acad.Sci.USA 90:3393-3397, 1993. 171. E.Skripkin, Paillart,J.C., Marquet,R., Ehresmann,B., and Ehresmann,C.: Identification of the primary site of the human
immunodeficiency virus type 1 RNA dimerization in vitro. Proc.Natl.Acad.Sci USA 91:4945-4949, 1994. 172. J.C.Paillart, Skripkin,E., Ehresmann,B., Ehresmann,C., and Marquet,R.: A loop-loop "kissing" complex is the essential part of
the dimer linkage of genomic HIV-1 RNA. Proc Natl Acad Sci U.S.A 93:5572-5577, 1996. 173. D.Ferrandon, Koch,I., Westhof,E., and Nusslein-Volhard,C.: RNA-RNA interaction is required for the formation of specific
bicoid mRNA 3' UTR-STAUFEN ribonucleoprotein particles. EMBO J 16:1751-1758, 1997. 174. B.K.Oh and Pace,N.R.: Interaction of the 3'-end of tRNA with ribonuclease P RNA. Nucleic Acids Res. 22(20):4087-4094,
1994. 175. M.F.Baer, Reilly,R.M., McCorkle,G.M., Hai,T.Y., Altman,S., and RajBhandary,U.L.: The recognition by RNase P of precursor
tRNAs. J.Biol.Chem. 263:2344-2351, 1988. 176. Y.Eguchi and Tomizawa,J.: Complex formed by complementary RNA stem-loops and its stabilization by a protein: function of
CoIE1 Rom protein. Cell 60:199-209, 1990. 177. M.Feiss, Sippy,J., and Miller,G.: Processive action of terminase during sequential packaging of bacteriophage λ chromosomes.
J Mol Biol 186:759-771, 1985. 178. R.R.Higgins, Lucko,H.J., and Becker,A.: Mechanism of cos DNA cleavage by bacteriophage lambda terminase-multiple roles
of ATP. Cell 54:765-775, 1988. 179. J.R.Panuska and Goldthwait,D.A.: A DNA dependent ATPase from T4-infected E. coli. Purification and properties of a
63,000-dalton enzyme and its conversion to a 22,000-dalton form. J.Biol.Chem. 255:5208-5214, 1980. 180. V.S.Manne, Rao,V.B., and Black,L.W.: A bacteriophage T4 DNA packaging related DNA-dependent ATPase-endonuclease. J
Biol Chem 257:13223-13232, 1982. 181. K.Hamada, Fujisawa,H., and Minagawa,T.: Characterization of ATPase activity of a defined in vitro system for packaging of
bacteriophage T3 DNA. Virology 159:244-249, 1987. 182. P.Serwer: The Source of Energy for Bacteriophage DNA Packaging: An Osmotic Pump Explains the Data. Biopolymers
27:165-169, 1988. 183. V.Ellison and Stillman,B.: Opening of the clamp: an intimate view of an ATP-driven biological machine. Cell 106:655-660,
2001. 184. M.M.Hingorani and O'Donnell,M.: Sliding Clamps: a (tail)ored fit. Curr.Biol. 10:25-29, 2000. 185. S.C.West: DNA helicases: New breeds of translocating motors and molecular pumps. Cell 86 :177-180, 1996. 186. T.Niedenzu, Roleke,D., Bains,G., Scherzinger,E., and Saenger,W.: Crystal structure of the hexameric replicative helicase RepA
of plasmid RSF1010. J.Mol.Biol. 306:479-487, 2001. 187. E.P.Gogol, Seifried,S.E., and von Hippel,P.H.: Structure and assembly of the Escherichia coli transcription termination factor
rho and its interaction with RNA. I. Cryoelectron microscopic studies. J Mol Biol 221:1127-1138, 1991.

Peixuan Guo
188. B.R.Burgess and Richardson,J.P.: RNA passes through the hole of the protein hexamer in the complex with the Escherichia coli Rho factor. J.Biol.Chem. 276:4182-4189, 2001.
189. J.Bowers, Tran,P.T., Joshi,A., Liskay,R.M., and Alani,E.: MSH-MLH complexes formed at a DNA mismatch are disrupted by the PCNA sliding clamp. J.Mol.Biol. 306:957-968, 2001.
190. J.Sedman and Stenlund,A.: The papillomavirus E1 protein forms a DNA-dependent hexameric complex with ATPase and DNA helicase activities. J Virol. 72:6893-6897, 1998.
191. F.P.Leu and O'Donnell,M.: Interplay of a clamp loader subunits in opening the (beta) sliding clamp of E. coli DNA polymerase III holoenzyme. J.Biol.Chem. 2001.
192. M.S.Song, Dallmann,H.G., and McHenry,C.S.: Carboxyl-terminal Domain III of the delta ' Subunit of the DNA Polymerase III Holoenzyme Binds delta. J.Biol.Chem. 276:40668-40679, 2001.
193. E.P.Geiduschek: Riding the (mono)rails: the structure of catenated DNA-tracking proteins. Chem.& Biol. 2:123-125, 1997. 194. M.C.Young, Schultz,D.E., Ring,D., and von Hippel,P.H.: Kinetic Parameters of the Translocation of Bacteriophage T4 Gene 41
Protein Helicase on Single-stranded DNA. J Mol Biol 235:1447-1458, 1994. 195. D.R.Herendeen, Kassavetis,G.A., and Geiduschek,E.P.: A transcriptional enhancer whose function imposes a requirement that
proteins track along DNA. Science 256:1298-1303, 1992. 196. J.Geiselmann, Wang,Y., Seifried,S.E., and von Hippel,P.H.: A physical model for the translocation and helicase activities of
Escherichia coli transcription termination protein rho. Proc Natl Acad Sci USA 90:7754-7758, 1993. 197. L.I.Davis: The nuclear pore complex. Ann.Rev.Biochem. 64:865-896, 1995. 198. R.M.Krug: The regulation of export of mRNA from nucleus to cytoplasm. Current Opinion in Cell Biology 5:944-949, 1993. 199. K.Pfeifer, Weiler,B.E., Ugarkovic,D., Bachmann,M., Schroder,H.C., and Muller,W.E.: Evidence for a direct interaction of Rev
protein with nuclear envelope mRNA-translocation system. Eur.J.Biochem. 199:53-64, 1991. 200. M.Young, Kuhl,S., and von Hippel,P.: Kinetic theory of ATP-driven translocases on one-dimensional polymer lattices. J Mol
Biol 235:1436-1446, 1994. 201. E.H.Egelman: Homomorphous hexameric helicases: tales from the ring cycle. Structure 4:759-762, 1996. 202. M.C.San Martin, Gruss,C., and Carazo,J.M.: Six molecules of SV40 large T antigen assemble in a propeller-shaped particle
around a channel. J Mol Biol 268:15-20, 1997. 203. A.Guasch, Pous,J., Parraga,A., Valpuesta,J.M., Carrascosa,J.L., and Coll,M.: Crystallographic analysis reveals the 12-fold
symmetry of the bacteriophage phi29 connector particle. J Mol.Biol 281:219-225, 1998.
Top Related