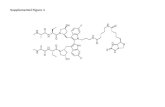
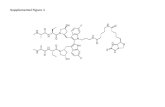
Supplemental Figures and Tables Supplemental Figures and Tables Figure S1. Phosphoamidate Analogues...
34
S1 Supplemental Figures and Tables Figure S1. Phosphoamidate Analogues Figure S2. Single Incorporation of modified nucleotides. (A) compound 11b, HIV-1 RT (0.025 U/μL), P1T2 (125 nM); (B) compound 11a,Therminator polymerase (0.05 U/μL), P1T2 (125 nM) ; (C) compound 11b, Therminator polymerase (0.05 U/μL), P1T2 (125 nM); (D) compound 11c, Therminator polymerase (0.05 U/μL), P1T1 (125 nM); (E) compound 18, Therminator polymerase (0.05 U/μL), P1T2 (125 nM)). dTTP (10 μM) and dATP (10 μM) were used as reference.
Transcript of Supplemental Figures and Tables Supplemental Figures and Tables Figure S1. Phosphoamidate Analogues...

S1
Supplemental Figures and Tables
Figure S1. Phosphoamidate Analogues
Figure S2. Single Incorporation of modified nucleotides. (A) compound 11b, HIV-1 RT (0.025 U/μL), P1T2 (125 nM); (B) compound 11a,Therminator polymerase (0.05 U/μL), P1T2 (125 nM) ; (C) compound 11b, Therminator polymerase (0.05 U/μL), P1T2 (125 nM); (D) compound 11c, Therminator polymerase (0.05 U/μL), P1T1 (125 nM); (E) compound 18, Therminator polymerase (0.05 U/μL), P1T2 (125 nM)). dTTP (10 µM) and dATP (10 µM) were used as reference.

S2

S3
Figure S3. Incorporation of dTTP into P1T2 (125 nM) by Therminator polymerase (0.00005 U µL-1).
Figure S4. Mass spectra of a single nucleotide incorporation experiment of compound 11a by Therminator
polymerase; (A) P1T2, compound 11a, Therminator polymerase. (B) P1T2, Therminator polymerase.
Five different oligonucleotides were detected in the reaction mixture: a primer elongated with two
10a (Mw 5860, peak 2); a primer elongated with one 10a and one dTMP (Mw 5840, peak 1); a
template degraded by two nucleotides (G and T) at the 3’ end and elongated with one 10a (Mw 6383,
peak 3); the degraded template elongated with two 10a (Mw 6707, peak 5); the degraded template
elongated with one 10a and one dTMP (Mw 6687, peak 4).

S4
Figure S5. Schemes for Polymerization and Pyrophosphorolysis by Therminator Polymerases using
Compound 11a and P1T2. (A) Chain Elongation of Primer; (B) Formation of dTTP and dGTP by
Template Degradation and Chain Elongation of Template. Symbol for Compound 10a (5-chloro-2’-
deoxyuridine 5’-monophosphonate) is X.

S5
Figure S6. Template Degradation by Pyrophosphorolysis. Compound 11b (1 mM), P1T2 (125 nM),
Therminator polymerase (0.05 U/μL), inorganic PPase (0.125 U/μL); Similar results are obtained
using 11c as substrate (data not shown)

S6
Figure S7. Elongation into P1T4 (125 nM, A) and P1T3 (125 nM, B) by Therminator polymerase (0.05 U/μL).
Time points: 15, 30, 60, 90 and 120 min. dTTP (50µM) and dATP (50µM) were used as reference.

S7
Figure S8. T-ROESY spectra of Compound 10b

S8
Figure S9. RMS deviation from normal B-DNA duplex for 20 ns simulations.

S9
Figure S10. Rdf, radial distribution function of solvent around O5’ and O4’’ in the dsDNA (A) and the
XNA:DNA duplex (B).
A)
B)

S10
Table S1. Elongation results of modified nucleotide analogues. Condition: P1T3 (125 nM) or P1T4
(125 nM), [Therminator polymerase] = 0.05 U/μL, at 500 μM substrate concentration in 120 min
incubation time; percentage of total radio-emitting oligonucleotides in the mixture.
11a 11b 11c 15a 15b 15c 16a 16b 16c 17a 17b 17c 18
P + 7 0 0 58 0 0 12.3 0 0 0 0 0 0 0 P + 6 0 21.4 27.8 0 0 37.6 5.3 0 0 0 0 0 0
P + 5 7.1 59.7 6.5 0 0 47.4 18.9 0 0 4.9 0 0 0
P + 4 84.3 18.9 0 0 0 2.7 20.8 0 0 9.8 0 0 0
P + 3 4.9 0 0 1.8 0 0 29.2 0 6.4 27.1 9 2.8 17.5
P + 2 3.7 0 0 21.7 0 0 20.6 17.7 48.4 35.1 33 28.2 72.6
P + 1 0 0 0 42 17.7 0 5.1 33.2 27.4 16 35.7 37.8 7.4
p 0 0 0 34.5 72.8 0 0 49 17.9 7.1 22.3 31.2 2.5
Primer P1 5'-CAGGAAACAGCTATGAC-3' Template T3 3'-GTCCTTTGTCGATACTGTTTTTTTGGAC-5' Template T4 3'-GTCCTTTGTCGATACTGAAAAACTGC-5'

S11
Table S2. Results from a Curves 5.3 calculation (Lavery and Skllenar, 1988) on 10 snapshot
structures from 15 to 20 ns. All values are averages over all nucleotide pairs in the helix, except for
the groove parameters that were taken from a central nucleotide pair in the helix (pair 6 A/T). * are
global parameters, relative to the helix axis, ** are local parameters calculated from one base pair to
the next one. Mgr is major groove, mgr = minor groove.
DNA:DNA
dx* -2.02 -2.63 -2.17 -0.88 -1.18 -1.94 -1.93 -2.14 -1.58 -1.77 dy* 0.25 0.10 0.23 -0.70 0.00 -0.36 0.61 -0.03 0.16 0.22
Inclin* 9.00 14.31 9.16 4.71 5.92 7.92 12.36 10.65 3.68 8.36
Tip* -4.23 -7.03 -3.34 2.89 -3.45 2.09 -8.42 -1.67 -2.76 0.60
Shift* 0.03 -0.05 0.13 -0.01 0.09 0.08 -0.03 0.04 0.17 0.14
Slide** -0.10 -0.16 -0.14 -0.13 -0.15 -0.16 -0.01 -0.12 -0.06 -0.13
Rise** 3.31 3.16 3.37 3.46 3.35 3.24 3.38 3.21 3.45 3.34
Tilt** -1.08 -1.43 0.24 1.01 -0.24 1.91 -2.49 0.07 -0.04 0.82
Roll** 0.87 1.58 2.88 0.51 2.59 1.27 -1.68 2.10 1.55 0.88
Twist** 32.88 32.48 31.75 33.09 32.35 33.90 35.13 32.48 32.95 33.40
Propeller -15.70 -14.69 -7.99 -11.75 -15.15 -16.82 -12.42 -17.80 -12.46 -15.49
mgr width 5 8 7 4 4 6 7 4 5 5
mgr depth 5 4 4 5 5 4 4 5 5 5
Mgr width 11 13 15 14 12 13 12 11 14 12
Mgr depth 6 8 9 1 6 7 7 4 5 6
helix diam 20 21 21 19 19 20 20 21 21 20
Sugar C2’endo + C1’exo
XNA:DNA
dx* -1.83 -2.07 -1.95 -2.92 -3.05 -2.25 -2.69 -2.75 -1.91 -1.34 dy* 0.63 0.37 0.07 0.05 0.02 0.93 0.63 0.84 -0.16 -0.03
Inclin* 13.19 17.66 18.85 15.58 7.08 7.41 10.28 10.78 8.91 10.93
Tip* -8.52 -2.0 1.99 -1.64 1.07 -6.69 -7.52 -4.51 -0.69 1.22
Shift* -0.03 0.12 0.19 0.10 0.16 0.12 0.08 0.16 0.14 0.16
Slide** 0.04 -0.16 -0.21 -0.15 -0.10 -0.92 -0.29 -0.11 -0.11 -0.28
Rise** 3.46 3.56 3.29 3.18 3.46 3.40 3.19 3.37 3.40 3.47
Tilt** -1.77 -1.16 1.47 -0.60 -1.47 0.89 -2.55 -4.12 -0.10 0.48
Roll** 2.58 0.01 -0.13 2.69 4.75 5.09 7.20 6.07 1.29 3.20
Twist** 27.07 31.38 31.82 29.66 25.51 27.66 27.15 25.35 31.38 29.99
Propeller -13.46 -11.48 -16.99 -6.62 1.09 -14.65 -12.04 -3.13 -12.76 -13.91
mgr width 4 7 7 9 8 5 7 7 7 9
mgr depth 5 4 4 2 3 6 4 3 4 3
Mgr width 17 17 15 14 17 15 10 12 14 13
Mgr depth 6 8 2 11 6 9 10 12 9 9
helix diam 20 21 21 21 22 21 22 21 20 20
Sugar See Table S3

S12
Table S3. Average ribose Sugar Phase angles (Altona and Sundaralingam, 1972) in the different
residues in the antiparallel XNA:DNA duplex during the last 5 ns of the trajectory. The circular
variance is close to 1 when the spread in the angles is high.
XNA DNA
Phase Circular Phase Circular
132.20 0.06 142.98 0.07 175.98 0.04 199.44 0.12
87.53 0.29 155.60 0.44
50.90 0.25 16.05 0.78
69.92 0.46 142.33 0.21
58.77 0.32 174.72 0.22
78.98 0.38 152.63 0.16
58.93 0.18 143.64 0.45
74.39 0.27 146.12 0.08
75.87 0.53 198.55 0.28
92.32 0.40 132.43 0.26
117.34 0.40 125.69 0.14

S13
Table S4. Atomic charges of XNA nucleotides used in the Amber MD simulations.
Ade Gua Thy Cyt
P 1.1694 P 1.1694 P 1.1694 P 1.1694 O1P -0.8028 O1P -0.8028 O1P -0.8028 O1P -0.8028
O2P -0.8028 O2P -0.8028 O2P -0.8028 O2P -0.8028
C5’ 0.2191 /-0.0775
C5’ 0.2191 /-0.0775
C5’ 0.2191 /-0.0775
C5’ 0.2191 /-0.0775
H5’1 -0.0746 /0.0879
H5’1 -0.0746 /0.0879
H5’1 -0.0746 /0.0879
H5’1 -0.0746 /0.0879
H5’2 -0.0746 /0.0879
H5’2 -0.0746 /0.0879
H5’2 -0.0746 /0.0879
H5’2 -0.0746 /0.0879
H5’3 /0.0879 H5T /0.0879 H5T /0.0879 H5T /0.0879
O5’ -0.3755 /-0.3008
O5’ -0.3755 /-0.3008
O5’ -0.3755 /-0.3008
O5’ -0.3755 /-0.3008
C4’ 0.0043 C4’ 0.0043 C4’ 0.0043 C4’ 0.0043
H4’ 0.1350 H4’ 0.1350 H4’ 0.1350 H4’ 0.1350
O4’ -0.2988 O4’ -0.2988 O4’ -0.2988 O4’ -0.2988
C1’ 0.0431 C1’ 0.0358 C1’ 0.0680 C1’ -0.0116
H1’ 0.1838 H1’ 0.1746 H1’ 0.1804 H1’ 0.1963
N9 -0.0268 N9 0.0577 N1 -0.0239 N1 -0.0339
C8 0.1607 C8 0.0736 C6 -0.2209 C6 -0.0183
H8 0.1877 H8 0.1997 H6 0.2607 H6 0.2293
N7 -0.6175 N7 -0.5725 C5 0.0025 C5 -0.5222
C5 0.0725 C5 0.1991 C7 -0.2269 H5 0.1863
C6 0.6897 C6 0.4918 H71 0.0770 C4 0.8439
N6 -0.9123 O6 -0.5699 H72 0.0770 N4 -0.9773
H61 0.4167 N1 -0.5053 H73 0.0770 H41 0.4314
H62 0.4167 H1 0.3520 C4 0.5194 H42 0.4314
N1 -0.7624 C2 0.7432 O4 -0.5563 N3 -0.7748
C2 0.5716 N2 -0.9230 N3 -0.4340 C2 0.7959
H2 0.0598 H21 0.4235 H3 0.3420 O2 -0.6548
N3 -0.7417 H22 0.4235 C2 0.5677
C4 0.3800 N3 -0.6636 O2 -0.5881
C4 0.1814
C3’ 0.3337 C3’ 0.3337 C3’ 0.3337 C3’ 0.3337
H3’ 0.0659 H3’ 0.0659 H3’ 0.0659 H3’ 0.0659
C2’ -0.0859 C2’ -0.0859 C2’ -0.0859 C2’ -0.0859
H2’1 0.0368 H2’1 0.0368 H2’1 0.0368 H2’1 0.0368
H2’2 0.0368 H2’2 0.0368 H2’2 0.0368 H2’2 0.0368
O3’ -0.6076 /0.6689
O3’ -0.6076 /0.6689
O3’ -0.6076 /0.6689
O3’ -0.6076 /0.6689
H3T /0.4340 H3T /0.4340 H3T /0.4340 H3T /0.4340

S14
Experimental Procedures
Procedures of chemical synthesis
General Experimental; For all reactions, analytical grade solvents were used. All moisture-sensitive
reactions were carried out in oven-dried glass-ware (135 °C). 1H and 13C NMR spectra were recorded
with a Bruker Advance 300 (1H NMR: 300 MHz, 13C NMR: 75 MHz) or 500 (1H NMR: 500 MHz, 13C
NMR: 125 MHz), using tetramethylsilane as internal standard for 1H NMR spectra and DMSO-d6 (39.5
ppm) or CDCl3 (77.2 ppm) for 13C NMR spectra. Abbreviations used are: s = singlet, d = doublet, t =
triplet, q = quartet, m = multiplet, br s = broad signal. Coupling constants are expressed in Hertz.
Mass spectra are obtained with a a quadrupole orthogonal acceleration time-of-flight mass
spectrometer (Synapt G2 HDMS, Waters, Milford, MA). Samples were infused at 3µL/min and spectra
were obtained in positive (or: negative) ionization mode with a resolution of 15000 (FWHM) using
leucine enkephalin as lock mass. Precoated aluminum sheets (Fluka Silica gel/TLC-cards, 254 nm)
were used for TLC. Column chromatography was performed on ICN silica gel 63-200, 60 Å.
Purification by HPLC was performed on a reverse phase C18 column by gradient elution (acetonitrile
and 100 mM triethylammonium bicarbonate (TEAB) buffer).
General method to synthesize compound 3
To a stirred solution of 2-deoxy-D-ribose (10.0 g, 74.6 mmol) in methanol (250 ml) was added HCl
(3.0 ml, 1.25 M in MeOH). The reaction mixture was stirred for 1 h at room temperature under an
Argon atmosphere then sodium bicarbonate (5.0 g) was added and the mixture was stirred for further
30 min. The solids were filtered and the solvent removed in vacuo to give as orange oil. The crude
residue 1 (11.0 g) was dissolved in pyridine (250 ml) and cooled to 0 °C and tert-butyldimethylsilyl
chloride (13.4 g, 89.1 mmol) was added. The mixture was stirred at room temperature until the
starting material 1 was completely consumed as monitored by TLC (~ 3 h). After conversion, benzoyl
chloride (9.48 ml, 81.7 mmol) and DMAP (2.27 g, 18.6 mmol) was added to the mixture and stirred for
2 h. The volatile was removed under reduced pressure and the crude residue was diluted with ethyl
acetate and washed with water, NaHCO3, brine, dried over Na2SO4 and concentrated under reduced
pressure. The crude residue 2 was dissolved in THF (250 ml) and cooled to 0 °C, acetic acid (1 ml)
and TBAF (96 ml, 1 M in THF, 96.5 mmol) were added. The mixture was stirred at room temperature

S15
for 2 h and quenched with MeOH (5 ml). The volatile was removed under reduced pressure, the crude
residue was diluted with ethyl acetate and washed with water, NaHCO3, brine, dried over Na2SO4 and
concentrated under reduced pressure. The crude residue was purified by column chromatography on
silica gel (EtOAc: Hexane = 1: 2) to afford compound 3 (13.0 g, 69.4 % for 4 steps) as a colorless
liquid.
(2R,3S,5R)-2-((Tert-butyldimethylsilyloxy)methyl)-5-methoxytetrahydrofuran-3-ol (1):
1H NMR (300 MHz, CDCl3, 25 °C): δ 5.04 (dd, J= 5.3 Hz, J= 1.9 Hz, 1H, H1), 4.40 (q, J= 4.4 Hz, 1H,
H3), 4.88-4.82 (m, 1H, H4), 3.76 (dd, J= 9.8 Hz, J= 5.2 Hz, 1H, H5), 3.53 (dd, J= 9.5 Hz, J= 8.1 Hz,
1H, H5), 3.30 (s, 3H, OCH3), 2.23-2.16 (m, 2H, H2, OH), 2.06 (dd, J= 12.0 Hz, J= 6.1 Hz, 1H, H2),
0.89 (s, 9H, C(CH3)3), 0.066 (s, 6H, SiCH3) ppm. 13 C NMR (75 MHz, CDCl3, 25 °C): δ 105.21, 85.89,
73.68, 65.13, 55.17, 41.11, 26.06, 18.46, -5.22, -5.27 ppm. HRMS: calcd for C12H26NaO4Si [M+Na]+
285.1493, found 285.1480.
(2R,3S,5S)-2-((Tert-butyldimethylsilyloxy)methyl)-5-methoxytetrahydrofuran-3-ol (1):
1H NMR (300 MHz, CDCl3, 25 °C): δ 5.09 (d, J= 4.5 Hz, 1H, H1), 4.18 (dd, J= 10.6 Hz, J= 5.6 Hz, 1H,
H3), 4.12 (t, J= 3.2 Hz, 1H, H4), 3.73 (dd, J= 10.9 Hz, J= 3.6 Hz, 1H, H5), 3.56 (dd, J= 10.9 Hz, J= 4.8
Hz, 1H, H5), 3.38 (s, 3H, OCH3), 2.81 (d, J= 10.6 Hz, 1H, OH), 2.17-2.09 (m, 1H, H2), 1.98 (d, J=
13.6 Hz, 1H, H2), 0.88 (s, 9H, C(CH3)3), 0.056 (s, 3H, SiCH3), 0.046 (s, 3H, SiCH3) ppm. 13 C NMR
(75 MHz, CDCl3, 25 °C): δ 105.83, 88.06, 63.95, 54.94, 41.25, 26.04, 18.46, -5.22, -5.32 ppm.
(2R,3S,5R)-2-((Tert-butyldimethylsilyloxy)methyl)-5-methoxytetrahydrofuran-3-yl benzoate (2)
1H NMR (300 MHz, CDCl3, 25 °C): δ 8.03 (d, J= 7.2 Hz, 2H, Ar-H), 7.59-7.54 (m, 1H, Ar-H), 7.43 (t, J=
7.7 Hz, 2H, Ar-H), 5.50 (quint, J= 3.9 Hz, 1H, H3), 5.20 (dd, J= 5.5 Hz, J= 3.2 Hz, 1H, H1), 4.28-4.23
(m, 1H, H4), 3.78 (dd, J= 10.4, Hz, J= 5.6 Hz, 1H, H5), 3.71 (dd, J= 10.5 Hz, J= 6.7 Hz, 1H, H5), 3.38
(s, 3H, OCH3), 2.45 (ddd, J= 14.2 Hz, J= 6.8 Hz, J= 3.1 Hz, 1H, H2), 2.25 (dt, J= 14.3 Hz, J= 5.3 Hz,
1H, H2), 0.88 (s, 9H, C(CH3)3), 0.079 (s, 6H, SiCH3) ppm. 13 C NMR (75 MHz, CDCl3, 25 °C): δ
166.12, 133.24, 130.16, 129.79, 128.51, 105.79, 84.67, 76.13, 64.56, 55.46, 39.42, 26.00, 18.43, -
5.22, -5.25 ppm. HRMS: calcd for C19H30NaO5Si [M+Na]+ 389.1760, found 389.1792.
(2R,3S,5S)-2-((Tert-butyldimethylsilyloxy)methyl)-5-methoxytetrahydrofuran-3-yl benzoate (2)
1H NMR (300 MHz, CDCl3, 25 °C): δ 8.05 (d, J= 7.1 Hz, 2H, Ar-H), 7.58-7.72 (m, 1H, Ar-H), 7.43 (t, J=
7.7 Hz, 2H, Ar-H), 5.38 (ddd, J= 7.6 Hz, J= 2.9 Hz, J= 1.7 Hz, 1H, H3), 5.16 (d, J= 5.2 Hz, 1H, H1),
4.28 (q, J= 3.1 Hz, 1H, H4), 3.91 (dd, J= 11.1, Hz, J= 3.3 Hz, 1H, H5), 3.80 (dd, J= 11.0 Hz, J= 3.2 Hz,

S16
1H, H5), 3.41 (s, 3H, OCH3), 2.44 (ddd, J= 14.4 Hz, J= 7.7 Hz, J= 5.4 Hz, 1H, H2), 2.13 (d, J= 14.4
Hz, 1H, H2), 0.89 (s, 9H, C(CH3)3), 0.089 (s, 3H, SiCH3), 0.074 (s, 3H, SiCH3) ppm. 13 C NMR (75
MHz, CDCl3, 25 °C): δ 166.70, 133.17, 130.34, 129.89, 128.49, 105.58, 84.52, 75.36, 63.73, 55.09,
39.61, 26.04, 18.46, -5.17, -5.29 ppm.
(2R,3S,5R)-2-(Hydroxymethyl)-5-methoxytetrahydrofuran-3-yl benzoate (3)
1H NMR (300 MHz, CDCl3, 25 °C): δ 8.02 (d, J= 7.1 Hz, 2H, Ar-H), 7.60-7.55 (m, 1H, Ar-H), 7.44 (t, J=
6.9 Hz, 2H, Ar-H), 5.58-5.55 (m, 1H, H3), 5.25 (dd, J= 5.7 Hz, J= 2.6 Hz, 1H, H1), 4.38 (q, J= 2.3 Hz,
1H, H4), 3.80-3.84 (m, 2H, H5), 3.45 (s, 3H, OCH3), 2.97 (t, J= 6.4 Hz, 1H, OH), 2.55-2.37 (m, 2H, H2)
ppm. 13 C NMR (75 MHz, CDCl3, 25 °C): δ 166.50, 133.42, 129.85, 129.79, 128.57, 105.87, 86.37,
76.18, 64.12, 55.72, 40.16 ppm. HRMS: calcd for C13H16NaO5 [M+Na]+ 275.0895, found 275.0891.
(2R,3S,5S)-2-(Hydroxymethyl)-5-methoxytetrahydrofuran-3-yl benzoate (3)
1H NMR (300 MHz, CDCl3, 25 °C): δ 8.05 (d, J= 7.1 Hz, 2H, Ar-H), 7.59-7.54 (m, 1H, Ar-H), 7.44 (t, J=
7.8 Hz, 2H, Ar-H), 5.33 (ddd, J= 8.3 Hz, J= 3.9 Hz, J= 2.3 Hz, 1H, H3), 5.16 (d, J= 4.7 Hz, 1H, H1),
4.26 (q, J= 3.8 Hz, 1H, H4), 3.88-3.85 (m, 2H, H5), 3.41 (s, 3H, OCH3), 2.47 (ddd, J= 14.4 Hz, J= 8.3
Hz, J= 5.3 Hz, 1H, H2), 2.22 (br s, 1H, OH), 2.20 (d, J= 14.4 Hz, 1H, H2) ppm. 13 C NMR (75 MHz,
CDCl3, 25 °C): δ 166.88, 133.35, 129.96, 129.89, 128.52, 105.12, 83.74, 74.88, 62.81, 55.12, 39.46
ppm.
(2S,3S)-3-(Benzoyloxy)-5-methoxytetrahydrofuran-2-carboxylic acid (4)
The compound 3 (4.7 g, 18.6 mmol) was dissolved in acetonitrile (50 ml). PhI(OAc)2 (13.2 g, 41.0
mmol) was added to the solution followed by water (50 ml). The mixture was then treated with
TEMPO (0.58 g, 3.73 mmol) and stirred for 16 h. The mixture was then concentrated under reduced
pressure and the crude product was purified by column chromatography on silica gel (CH2Cl2: MeOH
= 50: 1) to afford the compound 4 (4.5 g, 90.7 %) as colorless liquid.
(2S,3S,5R)-3-(Benzoyloxy)-5-methoxytetrahydrofuran-2-carboxylic acid (4: C1-beta)
1H NMR (300 MHz, CDCl3, 25 °C): δ 11.47 (br s, 1H, CO2H), 8.04 (d, J= 7.1 Hz, 2H, Ar-H), 7.61-7.56
(m, 1H, Ar-H), 7.45 (t, J= 7.8 Hz, 2H, Ar-H), 5.92-5.89 (m, 1H, H3), 5.32 (dd, J= 5.2 Hz, J= 2.1 Hz,1H,
H1), 4.79 (d, J= 2.7 Hz, 1H, H4), 3.48 (s, 3H, OCH3), 2.57 (ddd, J= 14.2 Hz, J= 7.1 Hz, J= 2.1 Hz, 1H,
H2), 2.29 (dt, J= 14.1 Hz, J= 2.2 Hz, 1H, H2) ppm. 13 C NMR (75 MHz, CDCl3, 25 °C): δ 174.49,

S17
166.15, 133.57, 129.83, 129.35, 128.55, 106.69, 81.39, 55.89, 39.47 ppm. HRMS: calcd for C13H13O6
[M-H]- 265.0712, found 265. 0700.
(2S,3S,5S)-3-(Benzoyloxy)-5-methoxytetrahydrofuran-2-carboxylic acid (4: C1-alpha)
1H NMR (300 MHz, CDCl3, 25 °C): δ 8.08 (d, J= 7.8 Hz, 2H, Ar-H), 7.63-7.58 (m, 1H, Ar-H), 7.47 (t, J=
7.8 Hz, 2H, Ar-H), 5.55-5.51 (m, 1H, H3), 5.35 (d, J= 5.2 Hz, 1H, H1), 4.80 (d, J= 3.0 Hz, 1H, H4),
3.45 (s, 3H, OCH3), 2.48 (ddd, J= 14.5 Hz, J= 7.5 Hz, J= 5.3 Hz, 1H, H2), 2.28 (d, J= 14.5 Hz, 1H, H2)
ppm. 13 C NMR (75 MHz, CDCl3, 25 °C): δ 173.59, 166.74, 133.66, 130.06, 129.47, 128.61, 106.17,
81.38, 76.36, 55.70, 38.66 ppm.
(2R,3S)-2-Acetoxy-5-methoxytetrahydrofuran-3-yl benzoate (5)
The compound 4 (4.5 g, 16.9 mmol) was dissolved in dry THF (85 ml) and treated with pyridine (3.0
ml, 37.2 mmol). The reaction mixture was flushed with argon and then treated with lead tetraacetate
(8.25 g, 18.6 mmol). The reaction mixture was protected from light and stirred for 16 h. The solid was
filtered through celite and the filtrate was concentrated under reduced pressure. The residue was
diluted with ethyl acetate and washed with saturated NaHCO3, brine, dried over Na2SO4 and
concentrated under reduced pressure. The crude product was purified by column chromatography on
silica gel (EtOAc : Hexane = 1/ 6) to afford the compound 5 (3.4 g, 71 %) as colorless liquid.
(2R,3S,5R)-2-Acetoxy-5-methoxytetrahydrofuran-3-yl benzoate (5: C1 beta)
1H NMR (300 MHz, CDCl3, 25 °C): δ 8.01 (dd, J= 7.1 Hz, J= 1.4 Hz, 2H, Ar-H), 7.59-7.53 (m, 1H, Ar-
H), 7.43 (t, J= 7.3 Hz, 2H, Ar-H), 6.34 (s, 1H, H4), 5.53-5.50 (m, 1H, H1), 5.40-5.37 (m, 1H, H3), 3.42
(s, 3H, OCH3), 2.53-2.45 (m, 1H, H2), 2.39-2.32 (m, 1H, H2), 2.11 (s, 3H, COCH3) ppm. 13 C NMR (75
MHz, CDCl3, 25 °C): δ 169.63, 165.52, 133.44, 129.71, 129.32, 128.46, 107.44, 99.73, 77.77, 55.96,
37.49, 21.08 ppm. HRMS: calcd for C14H16NaO6 [M+Na]+ 303.0845, found 303.0844.
(2R,3S,5S)-2-Acetoxy-5-methoxytetrahydrofuran-3-yl benzoate (5: C1-alpha)
1H NMR (300 MHz, CDCl3, 25 °C): δ 8.05 (dd, J= 8.3 Hz, J= 1.2 Hz, 2H, Ar-H), 7.60-7.55 (m, 1H, Ar-
H), 7.45 (t, J= 7.8 Hz, 2H, Ar-H), 6.42 (s, 1H, H4), 5.36 (t, J= 1.4 Hz, 1H, H1), 5.34 (t, J= 1.3 Hz, 1H,
H3), 3.47 (s, 3H, OCH3), 2.59 (dt, J= 14.9 Hz, J= 6.1 Hz, 1H, H2), 2.17 (d, J= 14.7 Hz, 1H, H2), 2.11
(s, 3H, COCH3) ppm. 13C NMR (75 MHz, CDCl3, 25 °C): δ 169.48, 165.91, 133.48, 130.01, 129.68,
128.56, 107.08, 100.01, 76.36, 56.19, 36.37, 21.22 ppm.

S18
(2R,3S)-2-((Diisopropoxyphosphoryl)methoxy)-5-methoxytetrahydrofuran-3-yl benzoate (6)
The compound 5 (3.7 g, 13.2 mmol) was dissolved in dichloromethane (70 ml) and then cooled to -
20 °C. The cooled solution was then treated with phosphonate (3.89 g, 19.8 mmol) followed by
TMSOTf (4.78 ml, 26.4 mmol). The mixture was stirred for 16 h at -20 °C and then quenched with
triethylamine (4 ml). The mixture was washed with saturated NaHCO3, brine, dried over Na2SO4 and
concentrated under reduced pressure. The crude product was purified by column chromatography on
silica gel (EtOAc: Hexane = 1/ 1) to afford the compound 6 (4.0 g, 73 %) as colorless liquid.
1H NMR (300 MHz, CDCl3, 25 °C): δ 8.01 (d, J= 7.4 Hz, 2H, Ar-H), 7.60-7.55 (m, 1H, Ar-H), 7.44 (t, J=
7.5 Hz, 2H, Ar-H), 5.46 (dd, J= 5.7 Hz, J= 1.9 Hz, 1H, H1), 5.40-5.31 (m, 1H, H3), 5.10 (s, 1H, H4),
4.77 (septet, J= 4.7 Hz, 2H, CH), 3.99 (dd, J= 13.8 Hz, J= 8.5 Hz, 1H, PCH2), 3.78 (dd, J= 13.8 Hz, J=
8.8 Hz, 1H, PCH2), 3.47 (s, 3H, OCH3), 2.42-2.33 (m, 2H, H2), 1.34 (d, J= 4.7 Hz, 12H, CH3) ppm. 13
C NMR (75 MHz, CDCl3, 25 °C): δ 165.67, 133.47, 130.03, 129.83, 128.57, 107.35, 106.59 (t, J= 10.8
Hz), 78.28, 71.54 (d, J= 6.4 Hz), 71.35 (d, J= 6.7 Hz), 61.16 (d, J= 167.5 Hz), 56.40, 37.44, 24.24,
24.20, 24.12, 24.06 ppm. HRMS: calcd for C19H29NaO8P [M+Na]+ 439.1498, found 439.1494.
(2R,3S)-5-Acetoxy-2-((diisopropoxyphosphoryl)methoxy)tetrahydrofuran-3-yl benzoate (7)
A cold mixture of acetic anhydride, acetic acid, and conc. sulfuric acid was added to a solution of
compound 6 (4.0 g, 9.6 mmol) in dry CH2Cl2 at 0 °C and stirred for 15 min. The reaction mixture was
slowly poured into cold saturated NaHCO3 solution, and stirring was maintained for 3 h. The mixture
was extracted with CH2Cl2 from the water phase and dried over Na2SO4 and concentrated under
reduced pressure. The crude product was purified by column chromatography on silica gel (EtOAc:
Hexane = 1/ 1) to afford the compound 7 (4.0 g, 73 %) as colorless liquid.
1H NMR (300 MHz, CDCl3, 25 °C): δ 8.07-7.99 (m, 4H, Ar-H), 7.62-7.55 (m, 2H, Ar-H), 7.48-7.42 (m,
4H, Ar-H), 6.56 (dd, J= 6.0 Hz, J= 4.3 Hz, 1H, H1), 6.46 (d, J= 5.7 Hz, 1H, H1), 5.50 (dd, J= 5.8 Hz,
J= 1.8 Hz, 1H, H3), 5.44 (s, 1H, H4), 5.38 (d, J= 5.1 Hz, 1H, H3), 5.34 (s, 1H, H4), 4.77 (septet, J= 6.1
Hz, 4H, CH), 4.00-3.69 (m, 4H, PCH2), 2.71-2.43 (m, 3H, H2), 2.24 (d, J= 15.1 Hz, 1H, H2), 2.11 (s,
3H, OAc), 2.07 (s, 3H, OAc), 1.36-1.33 (m, 24H, CH3) ppm. 13 C NMR (75 MHz, CDCl3, 25 °C): δ
170.35, 169.94, 165.60, 133.62, 133.56, 130.00, 129.86, 129.38, 128.61, 128.58, 128.50, 107.93 (d,
J= 12.1 Hz), 107.49 (d, J= 12.3 Hz), 77.45, 75.70, 71.45 (d, J= 6.2 Hz), 71.36 (d, J= 6.3 Hz), 61.94 (d,

S19
J= 169.3 Hz), 61.62 (d, J= 169.4 Hz), 36.85, 35.87, 24.22, 24.16, 24.09, 21.23, 21.04 ppm. HRMS:
calcd for C20H29NaO9P [M+Na]+ 467.1447, found 467.1431.
(2R,3S,5R)-5-(5-Chloro-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-
((diisopropoxyphosphoryl)methoxy)tetrahydrofuran-3-yl benzoate (8a)
To a solution of 5-chlorouracil (0.33 g, 2.25 mmol) in CH3CN (5 ml), N,O-bis(trimethylsilyl)acetamide
(1.65 ml, 6.75 mmol) was added and the mixture stirred for 20 min at room temperature. To the
mixture a solution of the compound 7 (0.50 g, 1.13 mmol) in CH3CN (2 ml) was added. After cooling at
0 °C TMSOTf (0.41 ml, 2.25 mmol) was added dropwise and the reaction mixture was stirred
overnight at 4 °C. The mixture was poured into a mixture of CH2Cl2 and saturated NaHCO3 solution
(5/1). The CH2Cl2 layer was removed and the water layer was extracted with CH2Cl2. The combined
CH2Cl2 extracts were washed with brine and dried over Na2SO4. After the solvent was removed the
residue was purified by column chromatography (CH2Cl2/MeOH = 50:1) on silica gel to give the
compound 8a (beta isomer 0.15 g, 25 %, alpha isomer 0.25 g, 42 %) as a white solid.
1H NMR (300 MHz, CDCl3, 25 °C): δ 9.24 (br s, 1H, NH), 8.04 (d, J = 7.2 Hz, 2H, Ar-H), 7.81 (s, 1H,
H6), 7.64-7.59 (m, 1H, Ar-H), 7.47 (t, J = 7.5 Hz, 2H, Ar-H), 6.71 (t, J = 7.8 Hz, 1H, H1’), 5.53 (t, J =
4.6 Hz, 1H, H3’), 5.37 (s, 1H, H4’), 4.82 (septet, J = 6.2 Hz, 2H, OCH(CH3)2), 4.03-3.85 (m, 2H, PCH2),
2.70 (dd, J1= 14.8 Hz, J2= 6.4 Hz, 1H, H2’), 2.40-2.37 (m, 1H, H2’), 1.37 (d, J = 6.2 Hz, 12H,
OCH(CH3)2) ppm. 31P NMR (121.5 MHz, CDCl3, 25 °C): 17.43 ppm. 13 C NMR (75 MHz, CDCl3, 25
°C): δ 165.53, 158.93, 149.97, 136.46, 133.85, 129.95, 128.95, 128.69, 110.68, 107.30 (d, Jc-p= 11.0
Hz), 86.64, 76.99, 71.89 (d, Jc-p= 6.4 Hz), 71.81 (d, Jc-p= 6.4 Hz), 62.64 (d, Jc-p= 169.5 Hz), 35.26,
24.17, 24.12 ppm. HRMS: calcd for C22H27ClN2O9P [M-H]- 529.1143, found 529.1134.
Diisopropyl ((2R,3S,5R)-5-(5-chloro-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3-
hydroxytetrahydrofuran-2-yloxy)methylphosphonate (9a)
The compound 8a (0.25 g, 0.47 mmol) was dissolved in 7 N ammonia in methanol (5 ml) and the
reaction mixture was stirred overnight at room temperature. The reaction mixture was concentrated
under reduced pressure and the crude residue was purified by column chromatography
(CH2Cl2/MeOH = 20:1) on silica gel to give the compound 9a (0.19 g, 94 %) as a white solid.
1H NMR (300 MHz, CDCl3, 25 °C): δ 9.93 (br s, 1H, NH), 7.71 (s, 1H, H6), 6.67 (t, J = 7.4 Hz, 1H, H1’),

S20
5.23 (s, 1H, H4’), 4.78 (septet, J = 6.5 Hz, 2H, OCH(CH3)2), 4.43 (br s, 1H, H3’), 4.29 (br s, 1H, OH),
3.94 (dd, J1= 14.1 Hz, J2= 9.1 Hz, 1H, PCH2), 3.83 (dd, J1= 14.1 Hz, J2= 7.6 Hz, 1H, PCH2), 2.51 (dd,
J1= 13.9 Hz, J2= 6.2 Hz, 1H, H2’), 2.13-2.04 (m, 1H, H2’), 1.35 (br s, 12H, OCH(CH3)2) ppm. 31P NMR
(121.5 MHz, CDCl3, 25 °C): 18.09 ppm. 13 C NMR (75 MHz, CDCl3, 25 °C): δ 159.25, 150.10, 137.05,
110.36, 110.31 (d, Jc-p= 7.8 Hz), 87.17, 74.97, 72.19 (d Jc-p= 6.7 Hz), 72.04 (d Jc-p= 6.7 Hz), 62.57
(d, Jc-p= 170.3 Hz), 37.73, 24.19 ppm. HRMS: calcd for C15H23ClN2O8P [M-H]- 425.0881, found
425.0875.
((2R,3S,5R)-5-(5-Chloro-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3-hydroxytetrahydrofuran-2-
yloxy)methylphosphonic acid (10a)
The compound 9a (0.26 g, 0.61 mmol) in dry DCM (5 ml) were treated with HMDS (1.9 ml, 9.14 mmol),
and then TMSI (0.52 ml, 3.66 mmol) was added dropwise under stirring at 0 °C. The mixture was
stirred for 1h at 0 °C. The reaction was quenched with 1.0 M TEAB solution (2 ml). The mixture was
concentrated, and the residue was purified by RP-HPLC running a gradient of CH3CN in 0.1 M TEAB
buffer solution to afford the compound 10a (0.11 g, 53 %) as a white solid.
1H NMR (300 MHz, D2O, 25 °C): δ 7.94 (s, 1H, H6), 6.51 (t, J = 7.7 Hz, 1H, H1’), 5.21 (s, 1H, H4’),
4.47(d, J = 3.5 Hz, 1H, H3’), 3.70 (dd, J1= 13.0 Hz, J2= 8.0 Hz, 1H, PCH2), 3.49 (dd, J1= 13.0 Hz, J2=
8.5 Hz, 1H, PCH2), 2.41-2.26 (m, 2H, H2’) ppm. 31P NMR (121.5 MHz, D2O, 25 °C): 12.65 ppm. 13 C
NMR (75 MHz, D2O, 25 °C): δ 161.79, 151.05, 138.32, 109.67 (d, Jc-p= 10.5 Hz), 109.32, 86.19,
73.87, 64.42 (d, Jc-p= 154.2 Hz), 36.02 ppm. HRMS: calcd for C9H11ClN2O8P [M-H]- 340.9942, found
340.9944.
((2R,3S,5R)-5-(5-Chloro-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3-hydroxytetrahydrofuran-2-
yloxy)methylphosphonic diphosphoric anhydride (11a)
The compound 10a (40 mg, 0.07 mmol) was dissolved in anhydrous DMF (1 ml) and Bu3N (53 µl,
0.22 mmol) was added. The mixture was stirred at room temperature for 30 min. and concentrated in
vacuo and then redissolved in anhydrous DMF (1 ml) and treated with N,N-carbonyldiimidazole (0.12
g, 0.73 mmol). The mixture was stirred for 30 min and tris(tetrabutylammonium) hydrogen
pyrophosphate (0.53 g, 0.59 mmol) in DMF (2 ml) was added. The mixture was stirred overnight.
Excess aqueous ammonia (1 ml) was added and the mixture concentrated in vacuo. The residue was

S21
purified by chromatography on DEAE-cellulose (gradient: water/ 1M TEAB (v/v); 1/0, 1/0.2, 1/0.5, 1/1).
Further purification was performed by RP-HPLC by gradient elution to afford the compound 11a (12
mg, ~23 %) as a white solid.
1H NMR (300 MHz, D2O, 25 °C): δ 8.03 (s, 1H, H6), 6.58 (t, J = 7.2 Hz, 1H, H1’), 5.27 (s, 1H, H4’),
4.56 (d, J = 4.0 Hz, 1H, H3’), 3.96 (d, J= 8.8 Hz, 2H, PCH2), 2.43-2.38 (m, 2H, H2’) ppm. 31P NMR
(121.5 MHz, D2O, 25 °C): δ 7.53 (d, J= 26.2 Hz, Pα), -10.67 (d, J= 20.1 Hz, Pγ), -23.39 (dd, J= 26.2 Hz,
J= 20.1 Hz, Pβ) ppm. 13 C NMR (75 MHz, D2O, 25 °C): δ 161.48, 150.79, 138.43, 109.67 (d, Jc-p=
10.2 Hz), 109.36, 86.32, 73.82, 63.49 (d, Jc-p= 163.1 Hz), 35.77 ppm. HRMS: calcd for
C9H13ClN2O14P3 [M-H]- 500.9268, found 500.9284.
P-(((2R,3S,5R)-5-(5-Chloro-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3-hydroxytetrahydrofuran-2-
yloxy)methyl)-N,N-bis(3-methoxy-3-oxopropyl)phosphonamidic acid (12a)
In a two-neck flask, the compound 10a (40 mg, 0.07 mmol) and dimethyl iminodipropionate
hydrochloride (66 mg, 0.29 mmol) were suspended in a mixture of water (0.5 ml) and t-BuOH (1.5 ml).
One drop of Et3N was added to the solution and then the mixture was heated at 80 °C. A solution of
DCC (0.106 g, 0.51 mmol) in t-BuOH (1 ml) was added and the reaction mixture was stirred for 1h at
80 °C. After cooling, the solvent was removed under reduced pressure. The resulting crude residue
was purified by chromatography on silica gel (gradient : CHCl3/ MeOH/ H2O (v/v/v) 5:1:0, 5:2:0.25,
5:3: 0.25) to yield the compound 12a (16 mg, 42 %) as a white solid.
1H NMR (300 MHz, MeOD, 25 °C): δ 7.93 (s, 1H, H6), 6.50 (t, J = 6.5 Hz, 1H, H1’), 5.07 (s, 1H, H4’),
4.35 (d, J = 3.9 Hz, 1H, H3’), 3.77 (dd, J = 13.0 Hz, J = 6.9 Hz, 1H, PCH2), 3.64 (s, 6H, OCH3), 3.54
(dd, J = 13.0 Hz, J = 9.1 Hz, 1H, PCH2), 3.32-3.30 (m, 4H, NCH2), 2.60 (t, J= 7.5 Hz, 4H, CH2CO),
2.31-2.24 (m, 2H, H2’) ppm. 31P NMR (121.5 MHz, MeOD, 25 °C): δ 16.11 ppm. 13 C NMR (125 MHz,
MeOD, 25 °C): δ 174.63, 161.82, 151.85, 139.08, 112.16 (d, Jc-p= 9.7 Hz), 110.19, 88.15, 75.62,
66.56 (d, Jc-p= 145.4 Hz), 51.94, 43.50 (d, Jc-p= 4.7 Hz), 38.52, 36.03 ppm. HRMS: calcd for
C17H24ClN3O11P [M-H]- 512.0837, found 512.0848.
3,3'-((((2R,3S,5R)-5-(5-Chloro-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3-hydroxytetrahydrofuran-2-
yloxy)methyl)(hydroxy)phosphorylazanediyl)dipropanoic acid (16a)
The compound 10a (16 mg, 0.03 mmol) was dissolved in 0.4 M NaOH/ MeOH (1/1, 1 ml) and stirred

S22
at room temperature for 3 h. The reaction solution was cooled at 0 °C and 1 N HCl (0.2 ml) was added.
The mixture was purified by RP-HPLC by gradient elution to the compound 16a (12 mg, ~56 %) as a
white solid.
1H NMR (500 MHz, DMSO-d6, 25 °C): δ 7.77 (s, 1H, H6), 6.36 (t, J = 6.6 Hz, 1H, H1’), 5.07 (s, 1H,
H4’), 4.14 (d, J = 4.4 Hz, 1H, H3’), 3.51 (dd, J = 13.0 Hz, J = 7.7 Hz, 1H, PCH2), 3.41 (dd, J = 13.0 Hz,
J = 7.9 Hz, 1H, PCH2), 3.17-3.13 (m, 4H, NCH2), 2.39 (t, J = 6.8 Hz, 4H, CH2CO), 2.17 (dd, J = 13.9
Hz, J = 6.5 Hz, 1H, H2’), 2.08-2.04 (m, 1H, H2’) ppm. 31P NMR (202.5 MHz, DMSO-d6, 25 °C): δ
14.64 ppm. 13 C NMR (125 MHz, DMSO-d6, 25 °C): δ 173.76, 158.99, 149.78, 137.31, 109.56 (d, Jc-
p= 7.5 Hz), 108.16, 86.00, 73.61, 64.56 (d, Jc-p= 142.5 Hz), 42.63, 37.56, 36.23 ppm. HRMS: calcd
for C15H20ClN3O11P [M-H]- 484.0524, found 484.0537.
P-(((2R,3S,5R)-5-(5-Chloro-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3-hydroxytetrahydrofuran-2-
yloxy)methyl)-N-((S)-1,4-dimethoxy-1,4-dioxobutan-2-yl)phosphonamidic acid (13a)
This compound was synthesized according to the procedure for the synthesis of compound 12a,
using L-aspartic acid hydrochloride in a yield of 67 % (24 mg).
1H NMR (300 MHz, MeOD, 25 °C): δ 7.89 (s, 1H, H6), 6.54 (t, J = 7.7 Hz, 1H, H1’), 5.06 (s, 1H, H4’),
4.30 (d, J = 4.2 Hz, 1H, H3’), 4.29-4.22 (m, 1H, CH), 3.79 (dd, J = 12.7 Hz, J = 7.8 Hz, 1H, PCH2),
3.72 (s, 3H, OCH3), 3.64 (s, 3H, OCH3), 3.58 (dd, J = 12.8 Hz, J = 9.1 Hz, 1H, PCH2), 2.83 (t, J= 5.4
Hz, 2H, CH2CO), 2.35-2.20 (m, 2H, H2’) ppm. 31P NMR (121.5 MHz, MeOD, 25 °C): δ 14.10 ppm. 13 C
NMR (125 MHz, MeOD, 25 °C): δ 175.46 (d, Jc-p= 4.3 Hz), 173.16, 164.93, 154.40, 138.78, 111.68 (d,
Jc-p= 9.9 Hz), 110.58, 88.19, 75.79, 67.45 (d, Jc-p= 147.9 Hz), 52.79, 52.19, 40.82 (d, Jc-p= 3.4 Hz),
38.67 ppm. HRMS: calcd for C15H26ClN3O11P [M-H]- 484.0524, found 484.0517.
(2S)-2-((((2R,3S,5R)-5-(5-Chloro-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3-hydroxytetrahydrofuran-
2-yloxy)methyl)(hydroxy)phosphorylamino)succinic acid (17a)
This compound was synthesized according to the procedure for the synthesis of compound 16a, from
the compound 13a in a yield of 49 % (16 mg).
1H NMR (500 MHz, DMSO-d6, 25 °C): δ 7.76 (s, 1H, H6), 6.37 (t, J = 6.7 Hz, 1H, H1’), 5.08 (s, 1H,
H4’), 4.15 (d, J = 4.5 Hz, 1H, H3’), 3.79-3.76 (m, 1H, CH), 3.53 (dd, J = 12.9 Hz, J = 7.9 Hz, 1H,
PCH2), 3.41 (dd, J = 12.9 Hz, J = 8.0 Hz, 1H, PCH2), 2.49-2.47 (m, 2H, CH2CO), 2.18 (dd, J = 14.0 Hz,

S23
J = 6.6 Hz, 1H, H2’), 2.08-2.04 (m, 1H, H2’) ppm. 31P NMR (202.5 MHz, DMSO-d6, 25 °C): δ 13.57
ppm. 13 C NMR (125 MHz, DMSO-d6, 25 °C): δ 175.54, 173.50, 159.01, 149.82, 137.43, 109.50 (d,
Jc-p= 8.1 Hz), 108.22, 86.00, 73.73, 65.46 (d, Jc-p= 146.0 Hz), 51.43, 37.60 ppm. HRMS: calcd for
C13H16ClN3O11P [M-H]- 456.0211, found 456.0215.
Bis(((2R,3S,5R)-5-(5-chloro-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3-hydroxytetrahydrofuran-2-
yloxy)methyl)diphosphonic acid (15a)
1H NMR (300 MHz, D2O, 25 °C): δ 7.96 (s, 1H, H6), 6.51 (t, J = 6.8 Hz, 1H, H1’), 5.15 (s, 1H, H4’),
4.49 (d, J = 4.0 Hz, 1H, H3’), 4.03-3.96 (m, 1H, PCH2), 3.86-3.79 (m, 1H, PCH2), 2.45-2.32 (m, 2H,
H2’) ppm. 31P NMR (121.5 MHz, D2O, 25 °C): δ 6.86 ppm. 13 C NMR (75 MHz, D2O, 25 °C): δ 162.03,
151.14, 138.26, 109.59, 109.54 (d, Jc-p= 7.7 Hz), 109.36, 86.32, 73.82, 63.4986.20, 73.83, 63.78 (dd,
Jc-p= 168.4, Jc-p= 4.8 Hz), 35.86 ppm. HRMS: calcd for C18H21Cl2N4O15P2 [M-H]- 664.9856, found
664.9850.
((2R,3S,5R)-5-(6-Amino-9H-purin-9-yl)-3-hydroxytetrahydrofuran-2-yloxy)methylphosphonic acid (10c)
To a solution of 6-chloropurine (0.27 g, 1.76 mmol) in CH3CN (5 ml), N,O-bis(trimethylsilyl)acetamide
(0.66 ml, 2.70 mmol) was added and the mixture stirred for 20 min at room temperature. To the
mixture a solution of the compound 7 (0.60 g, 1.35 mmol) in CH3CN (2 ml) was added. After cooling at
0 °C TMSOTf (0.27 ml, 1.49 mmol) was added dropwise and the reaction mixture was stirred for 3 h
at room temperature. The mixture was poured into a mixture of CH2Cl2 and saturated NaHCO3
solution (5/1). The CH2Cl2 layer was removed and the water layer was extracted with CH2Cl2. The
combined CH2Cl2 extracts were washed with brine and dried over Na2SO4. After the solvent was
removed the residue was dissolved in 7 N ammonia in MeOH (10 ml) and stirred at room temperature
for overnight. After removing the solvent under reduced pressure, the crude residue was dissolved in
dry DCM (5 ml). HMDS (1.9 ml, 9.14 mmol), and then TMSI (0.52 ml, 3.66 mmol) were added
dropwise under stirring at 0 °C. The mixture was stirred for 1h at 0 °C. The reaction was quenched
with 1.0 M TEAB solution (2 ml). The mixture was concentrated, and the residue was purified by RP-
HPLC running a gradient of CH3CN in 0.1 M TEAB buffer solution to afford the compound 10c (0.08 g,
10 %) as a white solid.

S24
1H NMR (500 MHz, D2O, 25 °C): δ 8.28 (s, 1H, H2), 8.10 (s, 1H, H8), 6.55 (t, J = 7.1 Hz, 1H, H1’),
4.59 (d, J= 1H, H4’), 4.47(d, J = 4.7 Hz, 1H, H3’), 3.54 (dd, J1= 13.0 Hz, J2= 8.8 Hz, 1H, PCH2), 3.47
(dd, J1= 13.0 Hz, J2= 9.8 Hz, 1H, PCH2), 2.80 (ddd, J= 14.9 Hz, J= 7.3 Hz, J= 5.2 Hz, 1H, H2’), 2.61
(dd, J= 14.8 Hz, J= 7.1 Hz, 1H, H2’) ppm. 31P NMR (121.5 MHz, D2O, 25 °C): 14.57 ppm. 13 C NMR
(125 MHz, D2O, 25 °C): δ 155.27, 152.53, 148.67, 140.12, 118.15, 109.77 (d, Jc-p= 10.0 Hz), 83.54,
74.39, 63.75 (d, Jc-p= 130.0 Hz), 37.21 ppm. HRMS: calcd for C10H13N5O6P [M-H]- 330.0603, found
330.0603.
((2R,3S,5R)-5-(6-Amino-9H-purin-9-yl)-3-hydroxytetrahydrofuran-2-yloxy)methylphosphonic
diphosphoric anhydride (11c)
This compound was synthesized according to the procedure for the synthesis of compound 11a, from
the compound 10c in a yield of 22 % (10 mg).
1H NMR (300 MHz, D2O, 25 °C): δ 8.48 (s, 1H, H2), 8.30 (s, 1H, H8), 6.70 (t, J = 7.1 Hz, 1H, H1’),
5.40 (s, 1H, H4’), 4.74 (d, J = 5.1 Hz, 1H, H3’), 3.90-3.74 (m, 2H, PCH2), 2.99-2.87 (m, 1H, H2’), 2.66
(dd, J1= 15.0 Hz, J2= 7.1 Hz, 1H, H2’) ppm. 31P NMR (121.5 MHz, D2O, 25 °C): δ 7.72 (d, J= 26.2 Hz,
Pα), -10.06 (d, J= 20.2 Hz, Pγ), -23.23 (dd, J= 25.7 Hz, J= 20.3 Hz, Pβ) ppm. 13 C NMR (125 MHz,
D2O, 25 °C): δ 155.33, 152.46, 148.78, 140.14, 118.11, 109.62 (d, Jc-p= 11.3 Hz), 83.37, 74.11,
62.89 (d, Jc-p= 163.7 Hz), 36.68 ppm. HRMS: calcd for C10H15N5O12P3 [M-H]- 489.9930, found
489.9943.
P-(((2R,3S,5R)-5-(6-amino-9H-purin-9-yl)-3-hydroxytetrahydrofuran-2-yloxy)methyl)-N,N-bis(3-
methoxy-3-oxopropyl)phosphonamidic acid (12c)
This compound was synthesized according to the procedure for the synthesis of compound 12a, from
the compound 10c in a yield of 35 % (20 mg).
1H NMR (500 MHz, MeOD, 25 °C): δ 8.42 (s, 1H, H2), 8.20 (s, 1H, H8), 6.66 (t, J = 7.1 Hz, 1H, H1’),
5.09 (s, 1H, H4’), 4.48 (d, J = 4.2 Hz, 1H, H3’), 3.64 (dd, J1= 12.9 Hz, J2= 7.4 Hz, 1H, PCH2), 3.63 (s,
6H, OCH3), 3.44 (dd, J1= 12.8 Hz, J2= 9.3 Hz, 1H, PCH2), 3.31-3.29 (m, 4H, NCH2), 2.78-2.73 (m, 1H,
H2’), 2.57 (t, J= 7.6 Hz, 4H, CH2CO), 2.51 (ddd, J1= 14.1 Hz, J2= 6.8 Hz, J3= 0.9 Hz) ppm. 31P NMR
(121.5 MHz, MeOD, 25 °C): δ 16.40 ppm. 13 C NMR (75 MHz, MeOD, 25 °C): δ 174.64, 157.34,
153.91, 150.71, 141.04, 120.03, 112.03 (d, Jc-p= 11.3 Hz), 85.31, 76.00, 66.14 (d, Jc-p= 146.3 Hz),

S25
51.94, 43.52 (d, Jc-p= 4.8 Hz), 39.70, 36.00 ppm. HRMS: calcd for C18H26N6O9P [M-H]- 501.1499,
found 501.1502.
3,3'-((((2R,3S,5R)-5-(6-Amino-9H-purin-9-yl)-3-hydroxytetrahydrofuran-2-
yloxy)methyl)(hydroxy)phosphorylazanediyl)dipropanoic acid (16c)
This compound was synthesized according to the procedure for the synthesis of compound 16a, from
the compound 12c in a yield of 45 % (12 mg).
1H NMR (300 MHz, DMSO-d6, 25 °C): δ 8.34 (s, 1H, H2), 8.15 (s, 1H, H8), 7.27 (br s, 2H, CO2H), 6.54
(t, J = 6.9 Hz, 1H, H1’), 5.02 (s, 1H, H4’), 4.26 (d, J = 4.1 Hz, 1H, H3’), 3.45 (dd, J1= 12.5 Hz, J2= 8.8
Hz, 1H, PCH2), 3.32 (dd, J1= 12.5 Hz, J2= 8.2 Hz, 1H, PCH2), 3.18-3.11 (m, 4H, NCH2), 2.41 (t, J= 6.6
Hz, 4H, CH2CO), 2.50-2.39 (m, 2H, H2’) ppm. 31P NMR (121.5 MHz, DMSO-d6, 25 °C): δ 14.98 ppm.
13 C NMR (75 MHz, DMSO-d6, 25 °C): δ 173.68, 155.91, 152.64, 149.31, 138.91, 118.35, 109.22 (d,
Jc-p= 10.8 Hz), 82.78, 73.99, 63.95 (d, Jc-p= 143.5 Hz), 42.56, 36.12 ppm. HRMS: calcd for
C16H22N6O9P [M-H]- 473.1186, found 473.1191.
P-(((2R,3S,5R)-5-(6-amino-9H-purin-9-yl)-3-hydroxytetrahydrofuran-2-yloxy)methyl)-N-((S)-1,4-
dimethoxy-1,4-dioxobutan-2-yl)phosphonamidic acid (13c)
This compound was synthesized according to the procedure for the synthesis of compound 12a, from
the compound 10c, using L-aspartic acid hydrochloride in a yield of 83 % (20 mg).
1H NMR (300 MHz, D2O, 25 °C): δ 8.39 (s, 1H, H2), 8.26 (s, 1H, H8), 6.66 (t, J = 6.9 Hz, 1H, H1’),
5.20 (s, 1H, H4’), 4.61 (d, J = 4.8 Hz, 1H, H3’), 4.19-4.13 (m, 1H, CH), 3.69 (s, 3H, OCH3), 3.66 (s, 3H,
OCH3), 3.62-3.51 (m, 2H, PCH2), 2.93-2.86 (m, 1H, H2’), 2.83-2.80 (m, 2H, CH2CO), 2.70 (dd, J1=
14.7 Hz, J2= 7.1 Hz, 1H, H2’) ppm. 31P NMR (202.5 MHz, D2O, 25 °C): δ 15.70 ppm. 13 C NMR (125
MHz, D2O, 25 °C): δ 175.09 (d, Jc-p= 3.9 Hz), 173.15, 155.30, 152.52, 148.68, 139.86, 118.22,
109.78 (d, Jc-p= 12.2 Hz), 83.55, 74.08, 65.15 (d, Jc-p= 146.7 Hz), 52.52, 52.02, 50.95, 38.98 (d, Jc-
p= 4.1 Hz), 36.89 ppm. HRMS: calcd for C16H22N6O9P [M-H]- 473.1186, found 473.1202.
(2S)-2-((((2R,3S,5R)-5-(6-Amino-9H-purin-9-yl)-3-hydroxytetrahydrofuran-2-
yloxy)methyl)(hydroxy)phosphorylamino)succinic acid (17c)
This compound was synthesized according to the procedure for the synthesis of compound 16a, from

S26
the compound 13c in a yield of 44 % (12 mg).
1H NMR (300 MHz, D2O, 25 °C): δ 8.46 (s, 1H, H2), 8.28 (s, 1H, H8), 6.68 (t, J = 7.1 Hz, 1H, H1’),
5.19 (s, 1H, H4’), 4.65 (d, J = 4.7 Hz, 1H, H3’), 3.92-3.85 (m, 1H, CH), 3.61-3.46 (m, 2H, PCH2), 2.95-
2.85 (m, 1H, H2’), 2.69-2.57 (m, 2H, CH2CO), 2.46 (dd, J1= 14.5 Hz, J2= 8.0 Hz, 1H, H2’) ppm. 31P
NMR (202.5 MHz, DMSO, 25 °C): δ 16.29 ppm. 13 C NMR (125 MHz, DMSO, 25 °C): δ 175.25,
173.11, 155.92, 152.66, 149.31, 118.32, 109.25 (d, Jc-p= 9.9 Hz), 82.74, 73.99, 64.79 (d, Jc-p= 144.3
Hz), 51.38, two peaks are hidden under solvent signals. ppm. HRMS: calcd for C14H18N6O9P [M-H]-
445.0873, found 445.0882.
((2R,3S,5R)-5-(6-Amino-9H-purin-9-yl)-3-hydroxytetrahydrofuran-2-yloxy)methyl((5-(6-amino-9H-
purin-9-yl)-3-hydroxytetrahydrofuran-2-yloxy)methyl)diphosphonic acid (15c)
1H NMR (300 MHz, D2O, 25 °C): δ 8.14 (s, 1H, H2), 8.06 (s, 1H, H8), 6.56 (t, J = 7.1 Hz, 1H, H1’),
5.27 (s, 1H, H4’), 4.63 (d, J = 4.4 Hz, 1H, H3’), 4.20-4.13 (m, 1H, PCH2), 3.73-3.66 (m, 1H, PCH2),
2.77-2.68 (m, 1H, H2’), 2.58 (dd, J1= 14.6 Hz, J2= 7.1 Hz, 1H, H2’) ppm. 31P NMR (121.5 MHz, D2O,
25 °C): δ 8.06 ppm. 13 C NMR (125 MHz, D2O, 25 °C): δ 154.79, 152.32, 148.28, 139.85, 117.66,
109.74 (d, Jc-p= 13.7 Hz), 109.739, 83.41, 74.37, 63.70 (d, Jc-p= 171.3 Hz), 37.68 ppm. HRMS:
calcd for C20H25N10O11P2 [M-H]- 643.1179, found 643.1160.
(2R,3S,5R)-2-((Diisopropoxyphosphoryl)methoxy)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-
yl)tetrahydrofuran-3-yl benzoate (8b)
This compound was synthesized according to the procedure for the synthesis of compound 8a, using
thymine in a yield of 87 % (α/β = 1:1).
1H NMR (300 MHz, CDCl3, 25 °C): δ 8.98 (br s, 1H, NH), 8.045 (d, J = 7.1 Hz, 2H, Ar-H), 7.65-7.59 (m,
1H, Ar-H), 7.51-7.46 (m, 3H, Ar-H, H6), 6.83 (dd, J = 8.4 Hz, J = 6.4 Hz, 1H, H1’), 5.53 (d, J = 4.6 Hz,
1H, H3’), 5.26 (s, 1H, H4’), 4.81 (septet, J = 6.2 Hz, 2H, OCH(CH3)2), 3.98 (dd, J = 13.2. Hz, J = 9.5
Hz, 1H, PCH2), 3.79 (dd, J = 13.2. Hz, J = 10.4 Hz, 1H, PCH2), 2.63 (dd, J1= 14.7 Hz, J2= 6.4 Hz, 1H,
H2’), 2.46-2.41 (m, 1H, H2’), 2.05 (d, J = 0.8 Hz, 3H, CH3), 1.40-1.34 (m, 12H, OCH(CH3)2) ppm. 31P
NMR (121.5 MHz, CDCl3, 25 °C): 17.67 ppm. 13 C NMR (75 MHz, CDCl3, 25 °C): δ 165.66, 163.64,
150.95, 135.24, 133.91, 130.01, 129.07, 128.75, 113.22, 106.93 (d, Jc-p= 14.3 Hz), 85.94, 71.81 (d,

S27
Jc-p= 7.2 Hz), 71.68 (d, Jc-p= 6.4 Hz), 62.49 (d, Jc-p= 171.7 Hz), 34.73, 24.23, 12.55 ppm. HRMS:
calcd for C23H32N2O9P [M+H]+ 511.1845, found 511.1854.
Diisopropyl ((2R,3S,5R)-3-hydroxy-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-
yl)tetrahydrofuran-2-yloxy)methylphosphonate (9b)
This compound was synthesized according to the procedure for the synthesis of compound 9a, from
the compound 8b in a yield of 90 % .
1H NMR (300 MHz, CDCl3, 25 °C): δ 10.05 (br s, 1H, NH), 7.36 (s, 1H, H6), 6.79 (t, J = 6.7 Hz, 1H,
H1’), 5.11 (s, 1H, H4’), 4.80-4.74 (m, 2H, OCH(CH3)2), 4.69 (br s, 1H, H3’), 4.43 (br s, 1H, OH), 3.92
(dd, J = 13.6. Hz, J = 9.3 Hz, 1H, PCH2), 3.75 (dd, J = 13.5. Hz, J = 9 Hz, 1H, PCH2), 2.44 (dd, J1=
13.9 Hz, J2= 6.3 Hz, 1H, H2’), 2.17-2.08 (m, 1H, H2’), 1.98 (s, 3H, CH3), 1.36-1.32 (m, 12H,
OCH(CH3)2) ppm. 31P NMR (121.5 MHz, CDCl3, 25 °C): 18.61 ppm. 13 C NMR (75 MHz, CDCl3, 25
°C): δ 164.21, 151.31, 135.70, 112.54, 109.85 (d, Jc-p= 11.6 Hz), 86.30, 75.03, 71.84 (d, Jc-p= 6.4
Hz), 71.78 (d, Jc-p= 6.5 Hz), 62.16 (d, Jc-p= 171.2 Hz), 37.19, 24.14, 12.56 ppm. HRMS: calcd for
C16H28N2O8P [M+H]+ 407.1583, found 407.1566.
((2R,3S,5R)-3-Hydroxy-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-2-
yloxy)methylphosphonic acid (10b)
This compound was synthesized according to the procedure for the synthesis of compound 10a, from
the compound 9b in a yield of 42 % (0.15 g).
1H NMR (500 MHz, D2O, 25 °C): δ 7.58 (d, J = 1 Hz, 1H, H6), 6.57 (t, J = 7.5 Hz, 1H, H1’), 5.51 (s, 1H,
H4’), 4.48 (t, J = 3.0 Hz, 1H, H3’), 3.73 (dd, J1 = 12.5 Hz, J2 = 8.5 Hz, 1H, PCH2), 3.52 (dd, J1= 12.5
Hz, J2= 8.5 Hz, 1H, PCH2), 2.36-2.34 (m, 2H, H2’), 1.90 (d, J = 1 Hz, 3H, CH3) ppm. 31P NMR (121.5
MHz, D2O, 25 °C): 13.68 ppm. 13 C NMR (75 MHz, D2O, 25 °C): δ 167.10, 152.71, 138.13, 113.07,
110.39 (d, Jc-p= 10.6 Hz), 86.35, 74.96, 65.56 (d, Jc-p= 153.2 Hz), 36.59, 12.27 ppm. HRMS: calcd
for C10H14N2O8P [M-H]- 321.0488, found 321.0486.
Diphosphoric ((2R,3S,5R)-3-hydroxy-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-
yl)tetrahydrofuran-2-yloxy)methylphosphonic anhydride (11b)
This compound was synthesized according to the procedure for the synthesis of compound 11a, from

S28
the compound 10b in a yield of 15 % (10 mg).
1H NMR (500 MHz, D2O, 25 °C): δ 7.60 (s, 1H, H6), 6.61 (t, J = 7.0 Hz, 1H, H1’), 5.19 (s, 1H, H4’),
4.55 (br s, 1H, H3’), 3.94 (dd, J1 = 13.5 Hz, J2 = 8.5 Hz, 1H, PCH2), 3.88 (dd, J1= 13.5 Hz, J2= 8.5 Hz,
1H, PCH2), 2.39-2.37 (m, 2H, H2’), 1.94 (s, 3H, CH3) ppm. 31P NMR (121.5 MHz, D2O, 25 °C): δ 7.72
(d, J= 26.4 Hz, Pα), -11.03 (d, J= 19.9 Hz, Pγ), -23.23 (t, J= 33.7 Hz, Pβ) ppm. 13 C NMR (75 MHz,
D2O, 25 °C): δ 166.14, 151.78, 137.10, 112.20, 109.32 (d, Jc-p= 11.1 Hz), 85.56, 73.95, 63.22 (d, Jc-
p= 162.9 Hz), 35.36, 11.35 ppm. HRMS: calcd for C10H16N2O14P3 [M-H]- 480.9814, found 480.9839,
calcd for C10H15N2O11P2 [M-PO3H]- 401.0151, found 401.0165,.
P-(((2R,3S,5R)-3-hydroxy-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-2-
yloxy)methyl)-N,N-bis(3-methoxy-3-oxopropyl)phosphonamidic acid (12b)
This compound was synthesized according to the procedure for the synthesis of compound 12a, from
the compound 10b in a yield of 45 % (20 mg).
1H NMR (300 MHz, MeOD, 25 °C): δ 7.60 (s, 1H, H6), 6.58 (t, J = 7.2 Hz, 1H, H1’), 4.98 (s, 1H, H4’),
4.34 (br s, 1H, H3’), 3.74 (dd, J= 12.1 Hz, J= 8.3 Hz, 1H, PCH2), 3.64 (s, 6H, OCH3), 3.48 (dd, J= 11.2
Hz, J= 9.1 Hz, 1H, PCH2), 3.34 (t, J = 7.4 Hz, 4H, NCH2), 2.59 (t, J = 7.4 Hz, 4H, C(O)CH2), 2.26-2.24
(m, 2H, H2’), 1.96 (s, 3H, CH3) ppm. 31P NMR (121.5 MHz, D2O, 25 °C): δ 16.36 ppm. 13 C NMR (75
MHz, D2O, 25 °C): δ 174.69, 166.37, 152.71, 138.11, 112.78, 111.60 (d, Jc-p= 11.5 Hz), 87.21, 75.87,
66.2 (d, Jc-p= 146.6 Hz), 51.95, 43.42 (d, Jc-p= 4.6 Hz), 38.00, 35.93, 12.64 ppm. HRMS: calcd for
C18H27N3O11P [M-H]- 492.1383, found 492.1381.
3,3'-(Hydroxy(((2R,3S,5R)-3-hydroxy-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-
yl)tetrahydrofuran-2-yloxy)methyl)phosphorylazanediyl)dipropanoic acid (16b)
This compound was synthesized according to the procedure for the synthesis of compound 16a, from
the compound 12b in a yield of 67 % (12 mg).
1H NMR (500 MHz, D2O, 25 °C): δ 7.61 (s, 1H, H6), 6.57 (t, J = 7.5 Hz, 1H, H1’), 5.11 (s, 1H, H4’),
4.49 (br s, 1H, H3’), 3.76 dd, J= 13.0 Hz, J= 7.6 Hz, 1H, PCH2), 3.58 (dd, J= 12.8 Hz, J= (.1 Hz, 1H,
PCH2), 3.26 (t, J = 8.1 Hz, 4H, NCH2), 2.50 (t, J = 8.1 Hz, 4H, C(O)CH2), 2.40-2.38 (m, 2H, H2’), 1.95
(s, 3H, CH3) ppm. 31P NMR (202.5 MHz, DMSO, 25 °C): δ 14.78 ppm. 13 C NMR (125 MHz, DMSO,
25 °C): δ 173.82, 163.81, 150.76, 135.91, 111.00, 109.04 (d, Jc-p= 11.3 Hz), 84.91, 73.93, 64.28 (d,

S29
Jc-p= 143.8 Hz), 42.57, 37.04, 36.20, 11.94 ppm. HRMS: calcd for C16H23N3O11P [M-H]- 464.1070,
found 464.1076.
(2S)-2-(Hydroxy(((2R,3S,5R)-3-hydroxy-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-
yl)tetrahydrofuran-2-yloxy)methyl)phosphorylamino)succinic acid (17b)
This compound was synthesized according to the procedure for the synthesis of compound 17a, from
the compound 10b in a yield of 65 % (25 mg).
1H NMR (300 MHz, D2O, 25 °C): δ 7.62 (s, 1H, H6), 6.59 (t, J = 7.1 Hz, 1H, H1’), 5.06 (s, 1H, H4’),
4.49 (br s, 1H, H3’), 3.89-3.82 (m, 1H, CH), 3.76 (dd, J1= 12.5 Hz, J2= 8.3 Hz, 1H, PCH2O), 3.55 (dd,
J1 = 12.5 Hz, J2 = 9.6 Hz, 1H, PCH2O), 2.57 (dd, J= 14.2 Hz, J= 4.6 Hz, 1H, PCH2), 2.44- 2.36 (m, 3H,
H2’, PCH2), 1.96 (s, 3H, CH3) ppm. 31P NMR (202.5 MHz, DMSO, 25 °C): δ 14.16 ppm. 13 C NMR
(125 MHz, DMSO, 25 °C): δ 175.32, 173.21, 163.79, 150.75, 135.90, 111.01, 109.21 (d, Jc-p= 11.3
Hz), 84.96, 73.93, 65.09 (d, Jc-p= 144.6 Hz), 51.52, one peak is hidden under solvent signals, 37.13,
12.03 ppm. HRMS: calcd for C14H19N3O11P [M-H]- 436.0763, found 436.0765.
Bis(((2R,3S,5R)-3-hydroxy-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-2-
yloxy)methyl)diphosphonic acid (15b)
1H NMR (500 MHz, D2O, 25 °C): δ 7.53 (s, 1H, H6), 6.53 (t, J = 7.5 Hz, 1H, H1’), 5.10 (s, 1H, H4’),
4.48 (d, J = 4.5 Hz, 1H, H3’), 3.94 (dt, J1 = 13.0 Hz, J2 = 4.0 Hz, 1H, PCH2), 3.77 (dt, J1= 13.0 Hz, J2=
4.0 Hz, 1H, PCH2), 2.35 (dd, J1 = 14.5 Hz, J2 = 6.5 Hz, 1H, H2’), 2.31-2.25 (m, 1H, H2’), 1.92 (s, 3H,
CH3) ppm. 31P NMR (121.5 MHz, D2O, 25 °C): δ 6.78 ppm. 13 C NMR (75 MHz, D2O, 25 °C): δ 166.29,
151.78, 136.92, 112.19, 109.26 (t, Jc-p= 5.7 Hz), 85.55, 63.76 (dd, Jc-p= 168.3 Hz, Jc-p= 4.6 Hz),
35.65, 11.39 ppm. HRMS: calcd for C20H27N4O15P2 [M-H]- 625.0948, found 625.0938.
N-(3-(Dimethoxyphosphoryl)-1-methoxy-1-oxopropan-2-yl)-P-(((2R,3S,5R)-3-hydroxy-5-(5-methyl-2,4-
dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-2-yloxy)methyl)phosphonamidic acid (14)
1H NMR (300 MHz, MeOD, 25 °C): δ 7.60 (s, 1H, H6), 6.59 (t, J = 7.3 Hz, 1H, H1’), 4.96 (s, 1H, H4’),
4.24 (br s, 1H, H3’), 4.31-4.21 (m, 1H, CH), 3.78 (dd, J1 = 12.5 Hz, J2 = 8.3 Hz, 1H, PCH2O), 3.75 (d,
J= 0.9 Hz, 3H, OCH3), 3.74 (s, 3H, OCH3), 3.71 (d, J= 0.9 Hz, OCH3), 3.51 (dd, , J1 = 12.5 Hz, J2 =
9.3 Hz, 1H, PCH2O), 2.38 (dd, J= 17.6 Hz, J= 6.1 Hz, 2H, PCH2), 2.25-2.22 (m, 2H, H2’), 1.97 (s, 3H,

S30
CH3) ppm. 31P NMR (121.5 MHz, D2O, 25 °C): δ 30.91, 13.47 ppm. 13 C NMR (75 MHz, MeOD, 25
°C): δ 174.88, 166.35, 152.71, 138.16, 112.83, 111.38 (d, Jc-p= 11.3 Hz), 87.15, 75.90, 67.41 (d, Jc-
p= 146.8 Hz), 53.34 (d, J= 4.5 Hz), 53.26 (d, J= 4.7 Hz), 52.81, 51.66 (d, J= 3.2 Hz), 38.07, 31.48 (d,
Jc-p= 134.3 Hz), 12.62 ppm. HRMS: calcd for C16H27N3NaO12P2 [M+Na]+ 538.0968, found 538.0875.
P-(((2R,3S,5R)-3-hydroxy-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-2-
yloxy)methyl)-N-(1-methoxy-1-oxo-3-phosphonopropan-2-yl)phosphonamidic acid (14-a)
1H NMR (300 MHz, D2O, 25 °C): δ 7.61 (s, 1H, H6), 6.59 (t, J = 7.3 Hz, 1H, H1’), 5.16 (s, 1H, H4’),
4.48 (br s, 1H, H3’), 4.49-3.99 (m, 1H, CH), 3.78 (dd, , J1 = 13.1 Hz, J2 = 8.0 Hz, 1H, PCH2O), 3.73 (s,
3H, OCH3), 3.62 (dd, , J1 = 12.8 Hz, J2 = 8.7 Hz, 1H, PCH2O), 2.39-2.37 (m, 2H, H2’), 1.96 (s, 3H,
CH3), 1.96-1.83 (m, 2H, PCH2) ppm. 31P NMR (121.5 MHz, D2O, 25 °C): δ 16.37, 16.23 ppm. 13 C
NMR (75 MHz, D2O, 25 °C): δ 177.10, 166.51, 152.00, 137.14, 112.09, 109.48 (d, Jc-p= 10.7 Hz),
85.56, 73.94, 64.88 (d, Jc-p= 145.2 Hz), 52.19, 51.52, 35.66, 33.73 (d, Jc-p= 132.1 Hz), 11.44 ppm.
HRMS: calcd for C14H22N3O12P2 [M-H]- 486.0679, found 486.0673.
2-(Hydroxy(((2R,3S,5R)-3-hydroxy-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-
yl)tetrahydrofuran-2-yloxy)methyl)phosphorylamino)-3-phosphonopropanoic acid (18)
1H NMR (600 MHz, DMSO, 25 °C): δ 7.47 (s, 1H, H6), 6.45 (t, J = 7.2 Hz, 1H, H1’), 4.97 (s, 1H, H4’),
4.15 (d, J = 4.8 Hz, 1H, H3’), two peaks are hidden under water signal (CHCOOH, PCH2O), 2.12-1.98
(m, 4H, PCH2O, PCH2CH), 1.83 (s, 3H, CH3) ppm. 31P NMR (121.5 MHz, D2O, 25 °C): δ 20.56, 16.69
ppm. 13 C NMR (150 MHz, DMSO, 25 °C): δ 171.24, 163.85, 150.81, 136.00, 110.99, 109.12, 84.97,
73.94, 64.56 (d, Jc-p= 151.5 Hz), 50.24, 39.19, 30.09 (d, Jc-p= 123 Hz), 12.10 ppm. HRMS: calcd for
C13H20N3O12P2 [M-H]- 472.0522, found 472.0525.
DNA polymerase reactions
Oligonucleotides P1, T1-T4 were purchased from Sigma Genosys. The concentrations were deter
mined with a Varian Cary-300-Bio UV Spectrophotometer. The primer was 5’-33P-labeled with 5’-
[γ33P]-ATP (Perkin Elmer) using T4 polynucleotide kinase (New England Biolabs) according to the
procedure provided by the supplier. The labeled oligonucleotide was further purified using Illustra
TM Microspin TM G-25 columns (GE Healthcare).

S31
End-labelled primer was annealed to its template by combining primer and template in a molar ratio of
1:2 and heating the mixture to 75 °C for 10 min followed by slow cooling to room temperature over a
period of 1.5 h. For the incorporation of compound 11a-c, 15a-c, 16a-c, 17a-c and 18, a series of 20
µL-batch reactions were performed with the enzyme (HIV-1 RT, pol III, Taq, Vent (exo-), Therminator
DNA polymerase). The final mixture contained 125 nM primer template complex, reaction buffer (50
mM Tris.HCl, 50 mM KCl, 10 mM MgCl2, 0.5 mM spermidine, 10 mM dithiothreitol (DTT); pH 8.3 for
HIV-1 RT; 20 mM Tris.HCl, 10 mM KCl, 2 mM MgSO4, 0.1 % Triton X-100, pH 8.8 for thermostable
polymerase; 20 mM Tris.HCl, 10 mM MgCl2, 10 mM MDTT, 20 μg/mL BSA, 4% glycerol, pH 7.5 for
polymerase III), appropriate concentration of enzyme, and different concentrations of modified
nucleotide building blocks (1 mM, 500 µM, 200 µM and 100 µM). In the control reaction a 10 µM
dATP or dTTP were used as reference. The mixture was incubated at 37 °C or 75 °C respectively,
aliquots were quenched after 10, 20, 30, 60 and 120 min. For elongation experiments, the same
mixture with appropriate primer:template hybrid was incubated at 37 °C or 75 °C and aliquots were
quenched after 15, 30, 60, 90, 120 min. In the reaction with the natural nucleotides, dATP (50 µM) and
dTTP (50 µM) were used as positive control.
For mass analysis, unlabelled primer P1 is annealed to template T2 by adding 10.2 μL of a 90 μM
primer solution to 10 μL of a 110 μM template solution, in a final volume of 20.2 μL. The mixture is
heated at 75 °C for 10 min and cooled down slowly to room temperature during 2 hours. The final
concentration of P1T2 hybrid is 45.4 μM. Compound 11a (2 mM solution) is added to an equal volume
of mixture containing H2O, Thermopol buffer, 4 units of Therminator DNA polymerase and 13.6 μM of
P1T2, in a final volume of 20 μL, the reaction is performed at 75 °C and is quenched after 1.5 hours
by putting the mixture at 95 °C for 5 min. Furthermore, the sample desalted using IllustraTM
MicrospinTM G-25 columns.
For template degradation reaction, template was 5’-33P-labeled with 5’-[γ33P]-ATP using the same
procedure as labeling primer. End labeled template annealed to the corresponding primer by
combining primer and template in a molar radio of 1:2, and heated the mixture at 75 °C for 5 min
followed by slowly cooling to room template over a period of 1.5 h.
Steady-state kinetics of single nucleotide incorporation
The steady-state kinetics of single nucleotide incorporation of compound 16a and the natural
nucleoside triphosphate (dTTP) were determined by a gel-based polymerase assay. In all the

S32
experiments, the template T2 and the primer P1 were used. The primer and template in a 1:2 molar
ration were hybridized in a buffer containing 20 mM Tris.HCl, 10 mM KCl, 2 mM MgSO4, 0.1 % Triton
X-100, pH 8.8 and used in an amount to provide 125 nM concentration of the primer in each 20 µL
reaction. A range of building block concentrations between 10 µM to 1 mM of compound 16a and
between 0.1 µM to 10 µM for the natural building block were used. The final concentration of
Therminator DNA polymerase for the single incorporation of compound 16a is 0.0005 U/µL and for
dTTP is 0.00005 U/µL. Reaction mixtures were incubated at 75 °C and aliquots were drawn at 6
different time intervals. Reactions were quenched by addition of a buffer containing 80 % formamide,
2 mM EDTA and 1X TBE buffer, and putting in 95 °C for 5 min. Analysis of polymerase reaction was
performed by polyacrylamide gel electrophoresis. The incorporation rates (V) were calculated based
on the percentage of the extended oligonucleotide in the mixture (P+1 band). The kinetic parameters
(Vmax and KM) were determined by plotting V (nM/min) versus substrate concentration (µM) and fitting
the data to a non-linear Michaelis-Menten regression using GraphPad Prism Software 5.0.
NMR conformation determination
A Bruker Avance II 600 spectrometer equipped with 5 mm TCI HCN Z gradient cryoprobe was used to
measure 3JHH coupling constants and NOE interactions used to determine a predominant
conformation of the five membered ring. Spectra were processed using Topspin (version 2.1, Bruker
Biospin). The 3JHH coupling constants were measured from 1D proton spectra of a spinning sample at
5, 10, 15 and 20 °C, with 64K points over a spectral width of 12000 Hz. Coupling constant data were
used to determine the ring conformation using the MATLAB pseudorotation GUI (Hendrickx and
Martins, 2008). Two hundred milliseconds of T-ROESY (Hwang and Shaka,1992) spectrum with
200ms mixing time was collected with a spectral width of 5400 Hz in F1 and F2 with 2048 complex
points in F2 and 512 increments in F1. Shifted sinebell function was used in both dimensions. Zero
filling was applied to end up with 2048 · 1024 points.
Molecular modelling of a DNA pyrophosphate-phosphonate nucleotide analogues hybrid
duplex
Amber 11 was used in all simulations (Case et al, 2005).
Electrostatic charges. Atomic electrostatic charges of the p5 molecules, to be used in the amber
software package (Case et al, 2005) were calculated from the electrostatic potential at the 6-31G*
level using the package Gamess (Schmidt et al, 1993) and a RESP fitting procedure (Bayly et al,

S33
1993). charges on the bases were restrained to keep the same atomic charges for the nucleosides as
in the amber 94 library (Table S4).
Amber parameters. The force field parameters for amino acids and nucleic acids used in the amber
simulations are those from the ff99bsc0 dataset (Pérez et al, 2007). The phosphonate angle and
bond parameters were taken from Baaden 2001 (Baaden et al, 2001) (see http://ambermd.org/,
contributed parameters).
Model building. Pdb structure 1BNA, a DNA 12-mer duplex with sequence
CGCGAATTCGCG:CGCGAATTCGCG (Drew et al, 1981) was used as reference structure
(DNA:DNA) as well as a template to construct the antiparallel DNA pyrophosphate-phosphonate
nucleotide analogues (pN) duplex pN:DNA, with sequence
pCpGpCpGpApApTpTpCpGpCpG:CGCGAATTCGCG. In the latter, the C5’ and O5’ were switched
In one chain to get a phosphonate:dna duplex.
Molecular dynamics simulations. Solvated molecular dynamics was used to verify the stability of the
hybrid p5:dna duplex as well as a normal DNA:DNA 12-mer. The duplexes pN:DNA and DNA:DNA
were solvated in a truncated octahedron TIP3P water box (Jorgensen et al, 1983). 22 Na+ counter-
ions were added to get an electrostatic neutral system. The water molecules and counter-ions were
then allowed to relax their positions while keeping the solute fixed. Initially, for 20 ps, the systems
were heated up to 300K with constant-T, constant-V conditions while constraining the position of the
solute and using a Langevin temperature equilibration scheme. The molecular dynamics was then
continued for 200 ps at constant T and constant pressure to equilibrate the system further. The
simulation temperature was 300 K. Molecular dynamics simulations were initiated with no restraints,
with periodic boundary conditions, using a cut-off distance of 10 Å for the non-bonded interactions and
the particle-mesh-Ewald method for the summation of the coulombic interactions(Cheatham et al,
1995), MD time step = 0.002 ps. Total simulation times were 20 ns.
Supplemental References
Altona, C. and Sundaralingam, M. (1972) Conformational analysis of the sugar ring in nucleosides
and nucleotides. New description using the concept of pseudorotation. J. Am. Chem. Soc. 94, 8205-
8212.

S34
Baaden, M.; Burgard, M.; Boehme, C. and Wipff, G. (2001) Lanthanide cation binding to a phosphoryl-
calix[4]arene: the importance of solvent and counterions investigated by molecular dynamics and
quantum mechanical simulations. Phys. Chem. Chem. Phys. 3, 1317-1325.
Bayly, C.I.;Cieplak, P.; Cornell, W. and Kollman, P.A. (1993) A Well-Behaved Electrostatic Potential
Based Method Using Charge Restraints For Determining Atom-Centered Charges: The RESP Model.
J. Phys. Chem. 97, 10269-10280.
Case, D.A.; Cheatham T.E. 3rd; Darden T.; Gohlke H.; Luo R.; Merz K.M. Jr.; Onufriev A.; Simmerling
C.; Wang B.and Woods R.J. (2005) The AMBER biomolecular simulation programs. J. Comput. Chem.
26, 1668-1688.
Cheatham, T.E. III; Miller, J.L.; Fox, T.; Darden, T.A.; Kollman, P.A. (1995) Molecular dynamics
simulations on solvated biomolecular systems–The particle mesh Ewald method leads to stable
trajectories of DNA, RNA and proteins. J. Am. Chem. Soc. 117, 4193-4194.
Drew, H.R.; Wing R.M.; Takano T.; Broka C.; Tanaka S.; Itakura K. and Dickerson R.E. (1981)
Structure of a B-DNA dodecamer: conformation and dynamics. Pro. Natl. Acad. Sci. U S A. 78, 2179-
2183.
Hendrickx, P.M. and Martins, J.C. (2008) A user-friendly Matlab program and GUI for the
pseudorotation analysis of saturated five-membered ring systems based on scalar coupling constants.
Chem. Cent. J. Oct 24, 2:20.
Hwang, T.-L. and Shaka, A.J. (1992) Cross relaxation without TOCSY: transverse rotating-frame
Overhauser effect spectroscopy. J. Am. Chem. Soc. 114, 3157-3159.
Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.; Impey, R.W.; Klein, M.L. (1983) Comparison of
simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926-935.
Lavery, R. and Sklenar, H. (1988). The definition of generalized helicoidal parameters and of axis
curvature for irregular nucleic acids. J. Biomol. Struct. Dyn. 6, 63-91.
Pérez, A.; Marchán I.; Svozil D.; Sponer J.; Cheatham T.E. 3rd; Laughton C.A. and Orozco M. (2007)
Refinement of the AMBER force field for nucleic acids. Improving the description of alpha/gamma
conformers. Biophys. J. 92, 3817-3829.
Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.;
Matsunaga, N.; Nguyen, K.A.; Su, S.; Windus, T.L.; Dupuis, M. and Montgomery, J.A. (1993) General
Atomic and Molecular Electronic Structure System. J. Comput. Chem. 14, 1347-1363.