
Prussian blue nanoparticles protected by the water · PDF filePrussian blue nanoparticles...
9
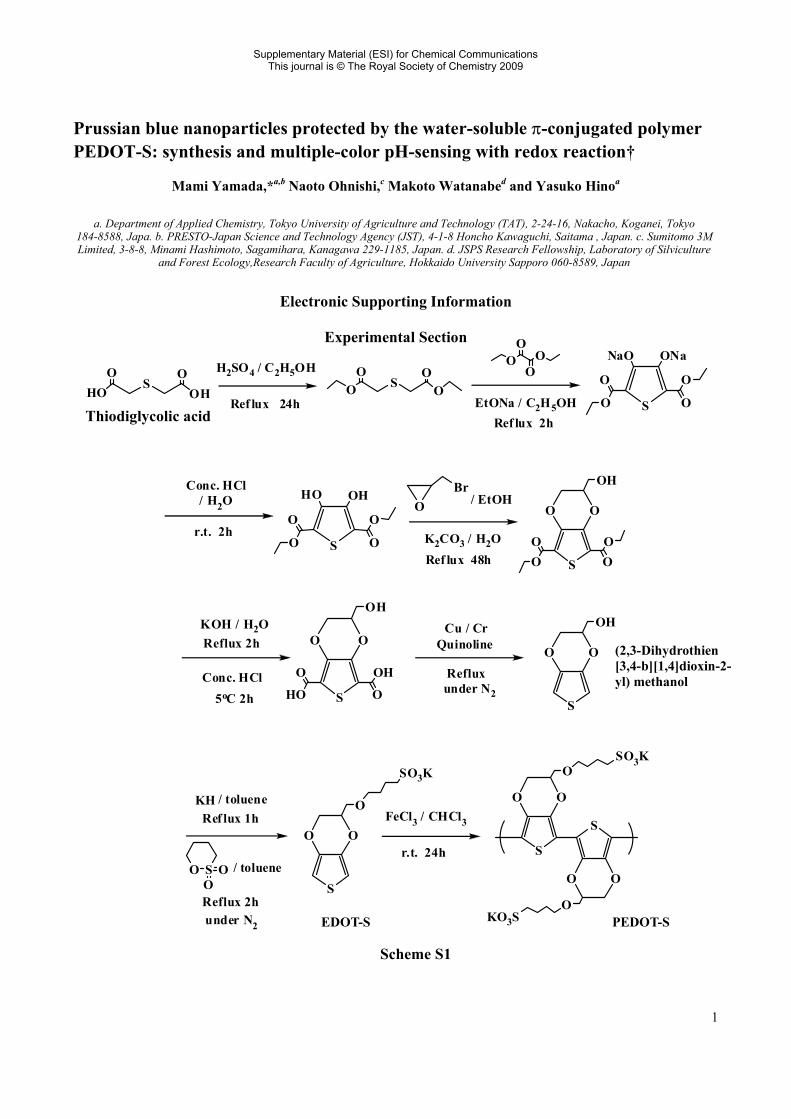
1 Prussian blue nanoparticles protected by the water-soluble π-conjugated polymer PEDOT-S: synthesis and multiple-color pH-sensing with redox reaction† Mami Yamada,* a,b Naoto Ohnishi, c Makoto Watanabe d and Yasuko Hino a a. Department of Applied Chemistry, Tokyo University of Agriculture and Technology (TAT), 2-24-16, Nakacho, Koganei, Tokyo 184-8588, Japa. b. PRESTO-Japan Science and Technology Agency (JST), 4-1-8 Honcho Kawaguchi, Saitama , Japan. c. Sumitomo 3M Limited, 3-8-8, Minami Hashimoto, Sagamihara, Kanagawa 229-1185, Japan. d. JSPS Research Fellowship, Laboratory of Silviculture and Forest Ecology,Research Faculty of Agriculture, Hokkaido University Sapporo 060-8589, Japan Electronic Supporting Information Experimental Section HO S OH O O O S O O O Ref lux 24h EtONa / C 2 H 5 OH O O O O Reflux 2h H 2 SO 4 /C 2 H 5 OH S ONa NaO O O O O Conc. HCl /H 2 O r.t. 2h S OH HO O O O O Br O / EtOH K 2 CO 3 /H 2 O Reflux 48h S O O O SO 3 K S O O O KO 3 S S O O O O O O OH KOH / H 2 O Reflux 2h Conc. HCl 5 o C 2h S O O OH HO O O OH S O O OH Cu / Cr Quinoline under N 2 Reflux S O O O SO 3 K KH / toluene Reflux 1h S O O O / toluene Reflux 2h under N 2 FeCl 3 / CHCl 3 r.t. 24h PEDOT-S (2,3-Dihydrothien [3,4-b][1,4]dioxin-2- yl) methanol EDOT-S Thiodiglycolic acid Scheme S1 Supplementary Material (ESI) for Chemical Communications This journal is © The Royal Society of Chemistry 2009
Transcript of Prussian blue nanoparticles protected by the water · PDF filePrussian blue nanoparticles...
1
Prussian blue nanoparticles protected by the water-soluble π-conjugated polymer PEDOT-S: synthesis and multiple-color pH-sensing with redox reaction†
Mami Yamada,*a,b Naoto Ohnishi,c Makoto Watanabed and Yasuko Hinoa
a. Department of Applied Chemistry, Tokyo University of Agriculture and Technology (TAT), 2-24-16, Nakacho, Koganei, Tokyo 184-8588, Japa. b. PRESTO-Japan Science and Technology Agency (JST), 4-1-8 Honcho Kawaguchi, Saitama , Japan. c. Sumitomo 3M Limited, 3-8-8, Minami Hashimoto, Sagamihara, Kanagawa 229-1185, Japan. d. JSPS Research Fellowship, Laboratory of Silviculture
and Forest Ecology,Research Faculty of Agriculture, Hokkaido University Sapporo 060-8589, Japan
Electronic Supporting Information
Experimental Section
HOS
OHO O
O S OO O
Ref lux 24h EtONa / C2H5OH
O OO
O
Ref lux 2h
H2SO4 / C2H5OH
S
ONaNaO
O
O O
O
Conc. HCl/ H2O
r.t. 2hS
OHHO
O
O O
O
BrO / EtOH
K2CO3 / H2ORef lux 48h
S
OO
OSO3K
S
O O
OKO3S
S OO OO
OO
OH
KOH / H2OReflux 2h
Conc. HCl5oC 2h S O
O OH
HO
OO
OH
S
OO
OHCu / CrQuinoline
under N2
Reflux
S
OO
O
SO3K
KH / tolueneRef lux 1h
SO OO
/ toluene
Reflux 2hunder N2
FeCl3 / CHCl3
r.t. 24h
PEDOT-S
(2,3-Dihydrothien [3,4-b][1,4]dioxin-2-yl) methanol
EDOT-S
Thiodiglycolic acid
Scheme S1
Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2009

2
Chemicals: All starting chemicals were of the purest commercially available grade. Thiodiglycolic acid, sodium carbonate, anhydrous magnesium sulfate, diethyl oxalate, sodium ethoxide, potassium chloride, potassium hydroxide, iron(III) chloride, sulfuric acid, ethanol, concentrated hydrochloric acid, chloroform, cyclohexane, toluene, acetone, methanol, hydrazine monohydrate, iron(II) chloride tetrahydrate, potassium hexacyanoferrate(III), hydrochloride, poly(vinylpyrrolidone) (PVP, Mw = 30,000) and KBr for FT-IR were purchased from Wako Chemicals. Epibromohydrin was purchased from Tokyo Chemical Industry. Potassium hydrade (30 wt.% in mineral oil), poly(diallyldimethylammonium chloride) (PDDA) (30 wt% in water, Mw = 200,000-350,000), and D2O for NMR were purchased from Aldrich. Deionized water was used for all syntheses, and was obtained with a Millipore Milli-Q SP. The resistance of the water was confirmed to be over 18.3 MΩ. Synthesis of potassium 4-(2,3-dihydrothieno[3,4-b][1,4]dioxin-2-yl)methoxybutane-1-sulfonate (EDOT-S) (Scehme S1): EDOT-S was synthesized in seven steps based on the previous literature (ref. 10a in the text), replacing sodium hydrate with potassium hydrate. Briefly, potassium hydrate (0.070 g, 0.180 mmol) and (2,3-dihydrothien [3,4-b][1,4]dioxin-2-yl) methanol (0.26 g, 1.51 mmol) were mixed in 18 mL of toluene and refluxed for 1h under a N2 atmosphere. After the mixture was cooled at rt, 6 mL of butane sulfone (0.210 g, 1.51 mmol) in toluene was added through a drop funnel, and the solution was refluxed for 2h. After cooling, the mixture was added to methanol (100 mL). The resulting precipitate was washed with acetone and then filtered to give an off-brown powder (0.340 g, 1.03 mmol) after drying in vacuo at rt at 65.4 % yield. The total yield from the first step (starting material: thiodiglycolic acid) was 3.4 %.
1H-NMR (300 MHz): solvent D2O (4.63 ppm): 6.93 (2H, s), 4.34 (1H, m), 4.19 (1H, d, J = 5.5 Hz), 3.99 (1H, m), 3.65 (2H, m), 3.48 (2H, m), 2.80 (2H, t), 1.61 (4H, m).
13C-NMR (500 MHz): solvent D2O; δ ppm: 141.3 (h), 141.1 (j), 101.1 (i), 100.9 (k), 73.5 (f), 71.6 (e), 69.2 (d), 66.8 (g), 51.3 (a), 30.9 (c), 21.5 (b).
Synthesis of poly(4-(2,3-dihydrothieno[3,4-b][1,4]dioxin-2yl-methoxy-1-butanesulfonic acid, potassium salt) (PEDOT-S) (Scheme S1): PEDOT-S was synthesized by the chemical oxidation of PEDOT-S with iron(III) chloride according to the previous literature (ref. 10b in the text). The prepared EDOT-S monomer (0.10 g, 0.30 mmol) and iron(III) chloride (0.14 g, 0.99 mmol) were vigorously stirred in 15 mL of chloroform under a N2 atmosphere for 24h. The resulting solution was then added to 100 mL of methanol containing ~ 0.5 mL of hydrazine hydrate. The filtered precipitate was dispersed in 200 mL of 1M potassium hydroxide in methanol for 48h in order to exchange the Fe ions with K ions electrostatically bound to pendant sulfonate groups of the obtained polymer. The filtered black precipitate was then redissolved in 200 mL of deionized water with stirring for 24 h. The filtrate was evaporated, and the obtained residue was washed with 300 mL of acetone. The resulting solid was dissolved in deionized water, and was dialyzed for a week using a 3500 g/mol cutoff membrane to remove oligomers. The dialyzed solution was evaporated and dried in vacuo to give a black PEDOT-S powder.
S
OO
O
SO3K
a b
c d
e f
g
h i
j
k
Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2009

3
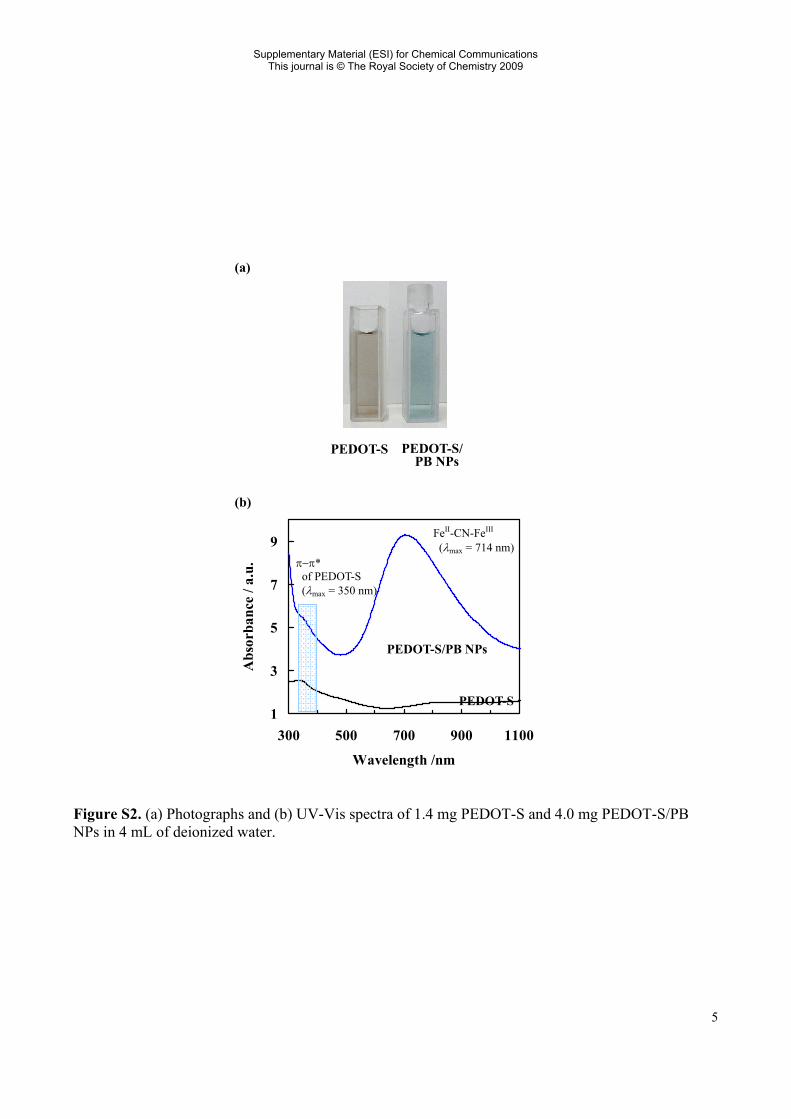
The polymerization was confirmed as follows. In the FT-IR spectrum of the obtained compound, the peak at 3112 cm−1 due to α C-H in the thiophene unit of the EDOT-S monomer completely disappeared. The symmetric (1047 cm−1) and asymmetric (1203 cm−1) S=O stretching modes did not change after polymerization, suggesting that the pendant sulfonate groups are very stable. In the UV-Vis spectrum, the absorption peak due to the π−π* transition of EDOT-S at 263 nm was red-shifted after the polymerization, with significant broadening of the peak in the range of 350 ~ 600 nm (Figure S2). The new broad peak at ca 800 nm is characteristic of p-doped polythiophenes, indicating that the as-prepared polymer had already self-doped with pendant sulfonate groups (ref. 12 in the text). The molecular weight was determined as Mn = 91,700, Mw = 104,000, Mw/Mn = 1.14 (see below). Layer-by-Layer Film Formation of PEDOT-S/PB NPs/PDDA: A glass slide (1 × 6 cm2) pre-cleaned in 5 % HCl, acetone, and deionized water with ultrasonic waves for 5 min each was used as a substrate. A multilayer film of PEDOT-S/PB NPs and PDDA can be prepared by alternately dipping the substrate into 10 mL of each aqueous solution containing 1 wt% PDDA or 19.5 mg PEDOT-S/ PB NPs. Between each dipping step, the substrate was rinsed with deionized water and dried by blowing with N2 for 10 s. The positively charged polyelectrolyte of PDDA was adsorbed first, enabling the sequential deposition of PEDOT-S/PB NPs possessing a negative polarity on a glass substrate.
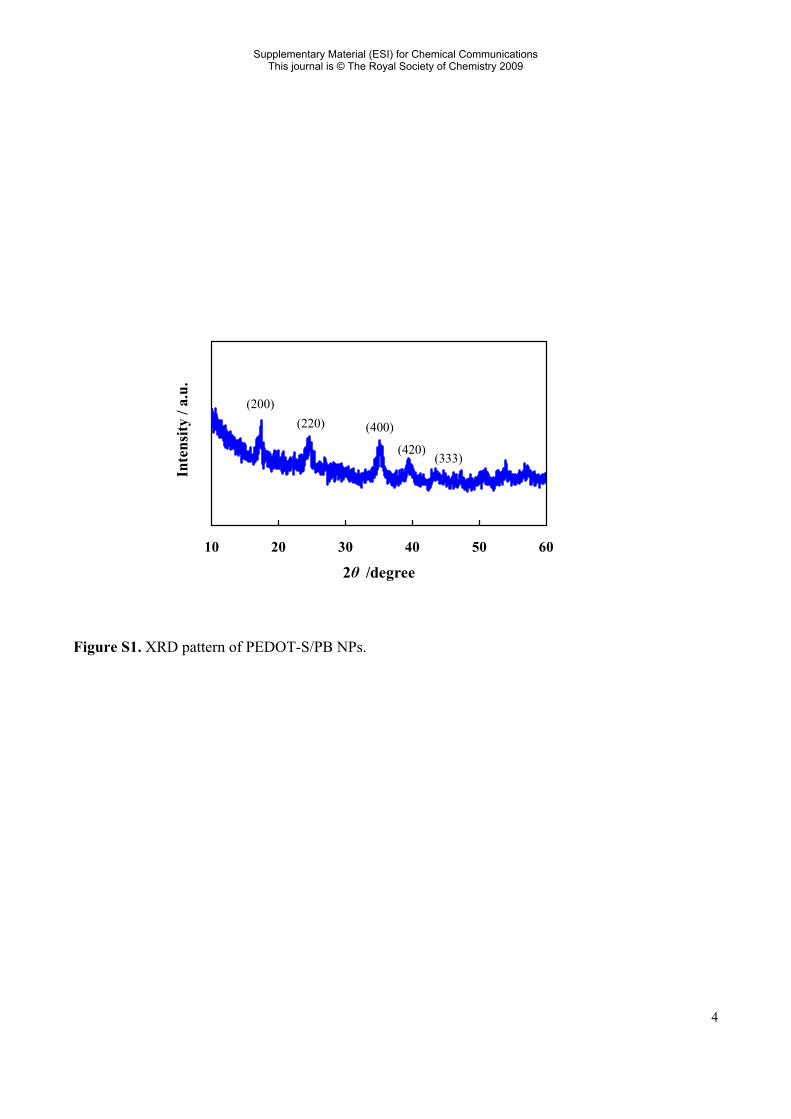
Characterization: 1H-NMR and 13C-NMR spectra of EDOT-S in D2O were obtained with a Varian Gemini2000 (300 mHz) and a Varian UNITY 500Inova (500 MHz), respectively. The number-average (Mn) and weight-average molecular weight (Mw) of PEDOT-S was determined by gel permeation chromatography (GPC) with a Jasco RI-2031Plus using pullulan as a standard polymer material. TEM images of PEDOT-S/PB NPs were taken by a Hitachi H-7100 with an operating voltage of 100 kV. The sample for TEM was prepared by casting the aqueous solution of PEDOT-S/PB NPs over standard carbon-coated copper grids (Okenshoji, 200 mesh). UV-Vis-NIR spectra of the compounds were taken with a JASCO V-670. XRD patterns of PEDOT-S/PB NPs mounted on an amorphous silicon plate were measured with a Rigaku RINT 2000 diffractometer with Cu Kα (40 mV, 100 mA) radiation from 10 to 60 degrees (2θ value) at a scan rate of 1.000°/min. FT-IR spectra of the samples confined to a KBr disk were recorded on a Horiba FT-720 spectrophotometer. The value of pH in solution was monitored by a Thermo Orion 4 Star after making a two point calibration using standard buffer solutions of pH 4 and pH 7. The cyclic voltammograms of the compounds were measured with a Hokuto HZ-5000.
Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2009
4
Figure S1. XRD pattern of PEDOT-S/PB NPs.
10 20 30 40 50 60
2θ /degree
(200) (220) (400)
(420) (333)
Inte
nsity
/ a.
u.
Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2009
5
Figure S2. (a) Photographs and (b) UV-Vis spectra of 1.4 mg PEDOT-S and 4.0 mg PEDOT-S/PB NPs in 4 mL of deionized water.
1
3
5
7
9
300 500 700 900 1100
Wavelength /nm
Abs
orba
nce
/ a.u
.
PEDOT-S/PB NPs
PEDOT-S
FeII-CN-FeIII
(λmax = 714 nm) π−π* of PEDOT-S (λmax = 350 nm)
(a)
(b)
PEDOT-S PEDOT-S/PB NPs
Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2009

6
Figure S3. Photograph of the synthetic mixture prepared using the same procedure as for PEDOT-S/PB NPs, replacing PEDOT-S with PVP. In this synthetic condition, with a monomer to iron ratio of 1, stable PB NPs could not be obtained, and the PB instead precipitated.
Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2009

7
Figure S4. Photographs of 1.0 mg PEDOT-S in 4 mL of deionized water at various pH values of 9.2, 7.04, 6.00, 5.06, 3.52, 2.53 and 1.70 (from left to right)adjusted by the addition of 4 µL of hydrazine followed by sequential additions of conc. HCl.
Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2009
8
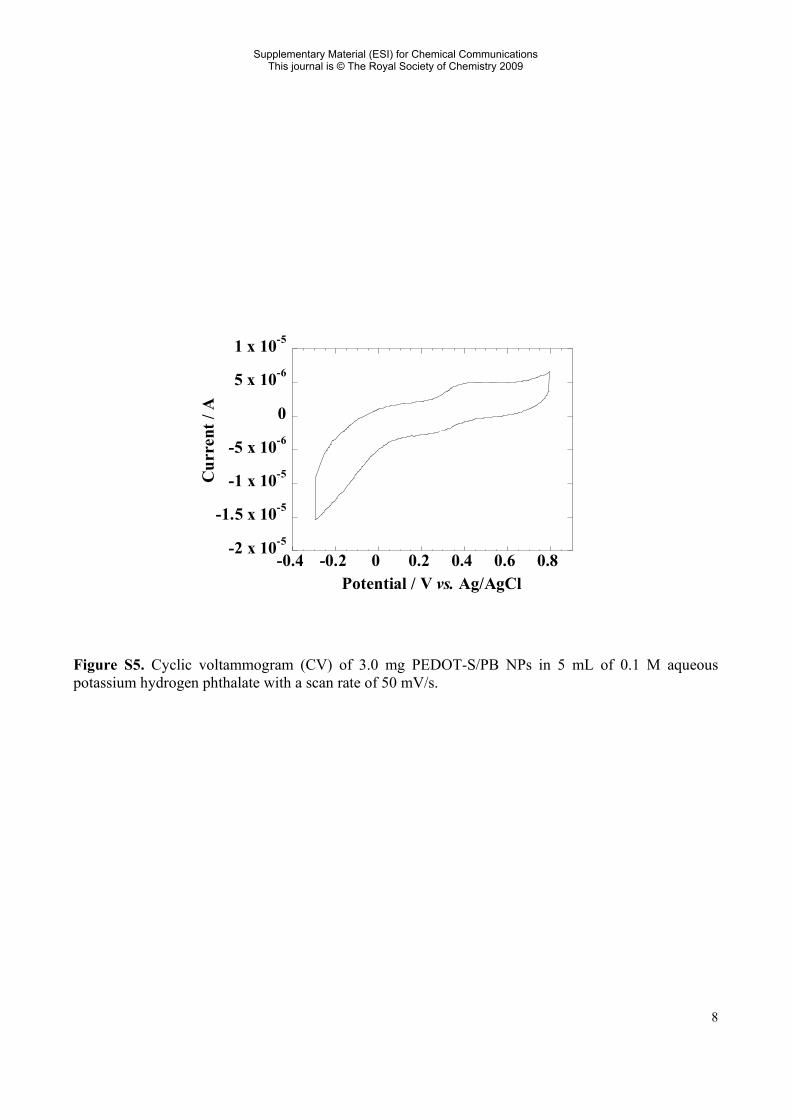
Figure S5. Cyclic voltammogram (CV) of 3.0 mg PEDOT-S/PB NPs in 5 mL of 0.1 M aqueous potassium hydrogen phthalate with a scan rate of 50 mV/s.
-2 x 10-5
-1.5 x 10-5
-1 x 10-5
-5 x 10-6
0
5 x 10-6
1 x 10-5
-0.4 -0.2 0 0.2 0.4 0.6 0.8
Cur
rent
/ A
Potential / V vs. Ag/AgCl
Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2009
9
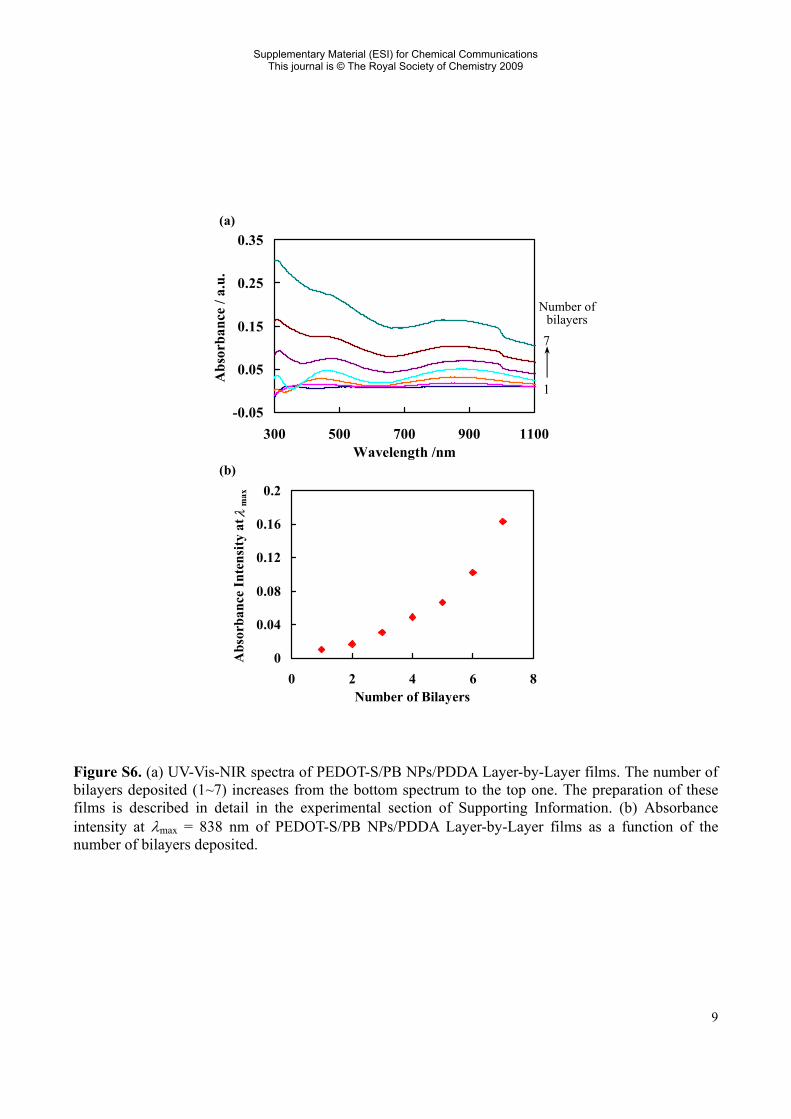
Figure S6. (a) UV-Vis-NIR spectra of PEDOT-S/PB NPs/PDDA Layer-by-Layer films. The number of bilayers deposited (1~7) increases from the bottom spectrum to the top one. The preparation of these films is described in detail in the experimental section of Supporting Information. (b) Absorbance intensity at λmax = 838 nm of PEDOT-S/PB NPs/PDDA Layer-by-Layer films as a function of the number of bilayers deposited.
-0.05
0.05
0.15
0.25
0.35
300 500 700 900 1100Wavelength /nm
Abs
orba
nce
/ a.u
.
1
7
Number of bilayers
0
0.04
0.08
0.12
0.16
0.2
0 2 4 6 8Number of Bilayers
Abs
orba
nce
Inte
nsity
at λ
max
(a)
(b)
Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2009

![Conference Poster - [email protected]](https://static.fdocument.org/doc/165x107/6203b130da24ad121e4c5b7c/conference-poster-emailprotected.jpg)
















