
Micronização Supercrítica do β-Caroteno · “Morre lentamente quem não vira a mesa quando...
90
Micronização Supercrítica do β-Caroteno Sofia Inês de Matos Antunes Dissertação para obtenção do Grau de Mestre em Engenharia Biológica Júri Presidente: Professor Doutor Luís Joaquim Pina da Fonseca Orientador: Professor Doutor António Manuel de Figueiredo Palavra Vogal: Professor Doutor Vítor Manuel Geraldes Fernandes Setembro de 2007
Transcript of Micronização Supercrítica do β-Caroteno · “Morre lentamente quem não vira a mesa quando...

Micronização Supercrítica do β-Caroteno
Sofia Inês de Matos Antunes
Dissertação para obtenção do Grau de Mestre em
Engenharia Biológica
Júri
Presidente: Professor Doutor Luís Joaquim Pina da Fonseca
Orientador: Professor Doutor António Manuel de Figueiredo Palavra
Vogal: Professor Doutor Vítor Manuel Geraldes Fernandes
Setembro de 2007

i
AGRADECIMENTOS
Em primeiro lugar, um especial agradecimento ao Professor António Palavra, pelo convite para a
realização deste trabalho, pela orientação do mesmo, pela confiança demonstrada e pela experiência e
conhecimentos transmitidos.
Agradeço igualmente ao Eng. Miguel Cardoso, por todo o apoio, orientação e conhecimentos que me
forneceu. Pela amizade, companheirismo e espírito de equipa que muito me ensinaram. O trabalho não
teria sido possível de outra forma.
A todo o pessoal da BioTrend por todo o apoio prestado na realização das análises laboratoriais.
A todos aqueles que, directa ou indirectamente, contribuíram para a conclusão deste trabalho.
Às minhas caríssimas amigas Ausenda Pires, pelo esclarecimento de dúvidas fulcrais sobre estatística, e
Ida Griffith, pela ajuda na revisão dos textos em inglês. Um grande Obrigada pela vossa amizade e
disponibilidade, hoje e sempre.
A todos os meus amigos e familiares, pelo apoio, pela amizade e pelos bons momentos que me têm
vindo a proporcionar ao longo do meu percurso académico e da minha vida.
Aos meus pais, por sempre acreditarem e pelo seu apoio e confiança em cada instante.
Ao meu namorado, por tudo.
A todos, a minha mais sincera gratidão.
Obrigada.
“Morre lentamente quem não vira a mesa quando está infeliz com o seu trabalho,
quem não arrisca o certo pelo incerto para ir atrás de um sonho,
quem não se permite, pelo menos uma vez na vida, fugir dos conselhos sensatos.
Morre lentamente quem passa os dias queixando‐se da má sorte ou da chuva que cai incessante.”
(Pablo Neruda)

ii
RESUMO
Neste trabalho foi realizado um estudo de micronização do β‐caroteno, um carotenóide de suma
importância para a indústria, graças às suas propriedades corantes, antioxidantes e como precursor na
síntese da vitamina A. A micronização do β‐caroteno tem por principal objectivo o aumento da sua taxa
de dissolução em água, permitindo, simultaneamente, a preparação de suspensões aquosas contendo
este carotenóide.
O processo foi desenvolvido num aparelho de micronização supercrítica do tipo “SAS”, utilizando como
anti‐solvente o dióxido de carbono supercrítico e como solvente o tetrahidrofurano, tendo sido
estudado o efeito da pressão na dimensão e na forma das partículas obtidas. Os ensaios de
micronização permitiram ainda a realização de um estudo da solubilidade do β‐caroteno na mistura
supercrítica em várias condições de pressão, temperatura e caudal. Foi ainda efectuado um ensaio de
micronização de um extracto de β‐caroteno natural, com o intuito de avaliar o processo em termos de
purificação do produto.
O estudo de solubilidades permitiu estabelecer algumas relações importantes entre as variáveis
estudadas e a solubilidade do β‐caroteno na mistura supercrítica. Concluiu‐se que a solubilidade
depende fortemente da temperatura e da fracção de THF e é pouco dependente da pressão.
Foi possível verificar que a morfologia das partículas é alterada quando se varia a pressão, sendo
também dependente da posição do ponto operatório relativamente ao ponto crítico. Verificou‐se,
ainda, que o tamanho de partícula aumenta com o aumento da pressão, quando se opera acima do
ponto crítico, e estabeleceu‐se uma relação entre o tamanho de partícula e a densidade da mistura
binária.
A micronização com anti‐solvente supercrítico foi bem sucedida no processamento do β‐caroteno,
sendo possível o controlo da morfologia e da dimensão das partículas através da manipulação das
condições experimentais. A micronização do β‐caroteno natural também foi possível, mas a purificação
não foi bem sucedida.
Palavras‐chave: β‐caroteno; fluidos supercríticos; dióxido de carbono; micronização; solubilidade;
tetrahidrofurano

iii
ABSTRACT
The aim of the present work was the micronization of β‐carotene, an important carotenoid, due to its
properties as colorant, antioxidant and as a precursor in the synthesis of vitamin A. The micronization of
β‐carotene has as main goal the increase of its dissolution rate in water, allowing, simultaneously, the
preparation of aqueous suspensions containing this carotenoid.
The process was developed in a supercritical micronization apparatus type SAS, using carbon dioxide as
antisolvent and tetrahydrofuran as solvent. The effect of operating pressure in the particle size and
morphology of the final product has been studied. The runs of micronization have also permitted to
study the solubility of β‐carotene in the supercritical mixture under several conditions of pressure,
temperature and flow rate. The micronization of an extract of natural β‐carotene has also been made, in
order to evaluate the purification ability of the process.
Some relationships between experimental variables studied and solubility of β‐carotene in the
supercritical mixture were recognized. It has been concluded that solubility strongly depends on
temperature and on THF fraction and does not depend much on pressure.
It has been shown that particle morphology is changed when the operating pressure changes and it also
depends on the position of the operating point relatively to the mixture’s critical point. Additionally, it’s
been shown that particle size increases with the operating pressure, when one operates over the critical
point, and a relationship between the particle size and the density of the binary mixture has been
established.
The supercritical antisolvent micronization was well done in the processing of β‐carotene and the
control of particle size and morphology through the manipulation of the operating conditions is
possible. Moreover, this process allows the micronization of the natural extract of β‐carotene, although
purification is not achievable.
Keywords: β‐carotene; supercritical fluids; micronization; carbon dioxide; solubility; tetrahydrofuran

iv
ÍNDICE
Agradecimentos .................................................................................................................................. i
Resumo .............................................................................................................................................. ii
Abstract ............................................................................................................................................. iii
Índice ................................................................................................................................................. iv
Índice de tabelas ............................................................................................................................... vii
Índice de figuras ............................................................................................................................... viii
Lista de abreviaturas .......................................................................................................................... xi
1. Introdução ..................................................................................................................................... 1
1.1. IMPORTÂNCIA DO TAMANHO DE PARTÍCULA ................................................................................................. 1
1.2. MICRONIZAÇÃO – FORMAÇÃO DE MICROPARTÍCULAS ..................................................................................... 4
1.2.1. Métodos tradicionais de moagem .............................................................................................. 4
1.2.2. Engenharia de partículas ............................................................................................................ 5
1.3. OS FLUIDOS SUPERCRÍTICOS E AS SUAS APLICAÇÕES ........................................................................................ 6
1.3.1. Estado supercrítico ..................................................................................................................... 6
1.3.1.1. O dióxido de carbono como fluido supercrítico ................................................................................... 8
1.3.2. Extracção supercrítica ................................................................................................................ 9
1.3.3. Micronização supercrítica (“RESS” e “PGSS”) ............................................................................. 9
1.3.3.1. Expansão rápida de uma solução supercrítica (“RESS”) ..................................................................... 10
1.3.3.2. Precipitação de soluções saturadas em gás (“PGSS”) ........................................................................ 10
1.3.4. Micronização com anti‐solvente supercrítico (“SAS”) ............................................................... 11
1.3.4.1. Fundamentos teóricos ....................................................................................................................... 12
1.3.4.2. O estado da arte da micronização “SAS” – trabalhos elaborados ...................................................... 14
1.3.4.3. Variáveis que influenciam a micronização “SAS” ............................................................................... 16
1.3.5. Atomização assistida por fluidos supercríticos (“SAA”) ............................................................ 17
1.4. CAROTENÓIDES .................................................................................................................................... 18
1.4.1. Características estruturais, químicas e físicas .......................................................................... 18
1.4.2. Distribuição na Natureza .......................................................................................................... 20
1.4.2.1. Plantas superiores .............................................................................................................................. 20
1.4.2.2. Algas ................................................................................................................................................... 21
1.4.2.3. Bactérias ............................................................................................................................................ 21
1.4.2.4. Fungos ................................................................................................................................................ 21
1.4.3. Biossíntese ................................................................................................................................ 21
1.4.4. Fontes alimentares ................................................................................................................... 23

v
1.4.5. Estabilidade .............................................................................................................................. 24
1.4.6. Funções biológicas .................................................................................................................... 24
1.4.6.1. Nas plantas ......................................................................................................................................... 24
1.4.6.2. No ser humano................................................................................................................................... 25
1.5. Β‐CAROTENO ....................................................................................................................................... 27
1.5.1. Química ..................................................................................................................................... 27
1.5.2. Biossíntese ................................................................................................................................ 28
1.5.3. Funções biológicas .................................................................................................................... 29
1.6. OBJECTIVOS DO TRABALHO ..................................................................................................................... 29
2. Parte experimental: Materiais, aparelhos e técnicas ...................................................................... 31
2.1. MATERIAIS .......................................................................................................................................... 31
2.2. MICRONIZAÇÃO SUPERCRÍTICA ................................................................................................................ 31
2.2.1. Descrição geral do aparelho ..................................................................................................... 31
2.2.2. Técnica experimental ................................................................................................................ 34
2.3. ANÁLISE DOS PRODUTOS ........................................................................................................................ 34
2.3.1. Espectrofotometria ................................................................................................................... 34
2.3.2. Cromatografia líquida de alta resolução (HPLC) ...................................................................... 34
2.3.3. “Dynamic light scattering” ....................................................................................................... 35
2.3.4. Microscopia electrónica de varrimento .................................................................................... 35
3. Resultados experimentais e discussão ........................................................................................... 36
3.1. ESTUDO DE SOLUBILIDADES ..................................................................................................................... 36
3.1.1. Estimativa de densidades da mistura binária ........................................................................... 38
3.1.2. Relação entre solubilidade, densidade e variáveis do processo ............................................... 40
3.2. ESTUDO DA PRECIPITAÇÃO DO Β‐CAROTENO SINTÉTICO ................................................................................. 45
3.2.1. Análise macroscópica dos cristais ............................................................................................. 45
3.2.2. Análise da pureza do produto ................................................................................................... 47
3.2.2.1. HPLC ................................................................................................................................................... 47
3.2.2.2. Espectrofotometria ............................................................................................................................ 49
3.2.3. Influência da pressão de trabalho no produto obtido .............................................................. 50
3.2.3.1. Análise do produto não processado .................................................................................................. 51
3.2.3.2. Análise do β‐caroteno processado a 75 bar ....................................................................................... 52
3.2.3.3. Análise do β‐caroteno processado a 100 bar ..................................................................................... 55
3.2.3.4. Análise do β‐caroteno processado a 130 bar ..................................................................................... 57
3.2.3.5. Discussão do efeito da pressão .......................................................................................................... 59
3.3. ESTUDO DA PRECIPITAÇÃO DE UM EXTRACTO NATURAL DE Β‐CAROTENO .......................................................... 62
3.3.1. Análise macroscópica ............................................................................................................... 62
3.3.2. Análise da pureza do produto ................................................................................................... 62
3.3.2.1. HPLC ................................................................................................................................................... 63

vi
3.3.2.2. Espectrofotometria ............................................................................................................................ 65
3.3.3. Conclusão acerca da precipitação do β‐caroteno natural ........................................................ 66
3.4. CONCLUSÕES E PERSPECTIVAS FUTURAS ..................................................................................................... 66
Referências bibliográficas .................................................................................................................. 68
Apêndice A .......................................................................................................................................... I
Apêndice B ......................................................................................................................................... II
Apêndice C ........................................................................................................................................ III
Apêndice D ....................................................................................................................................... IV
Apêndice E ......................................................................................................................................... V
Apêndice F ........................................................................................................................................ VI

vii
ÍNDICE DE TABELAS
Tabela 1.1 – Constantes críticas de alguns fluidos com interesse em extracção supercrítica [40]. ............. 7
Tabela 1.2 – Ordens de grandeza de propriedades termofísicas dos fluidos supercríticos [5]. ................... 8
Tabela 1.3 – Efeito das condições operatórias no tamanho de partícula de vários produtos micronizados
através de técnicas de anti‐solvente supercrítico [70]. .............................................................................. 17
Tabela 1.4 – Quantidades típicas de carotenóides em vegetais [19]. ........................................................ 24
Tabela 3.1 – Dados de pesos moleculares dos compostos envolvidos no trabalho. .................................. 36
Tabela 3.2 – Condições de ensaios, resultados experimentais e fracções molares calculadas. ................. 36
Tabela 3.3 – Parâmetros dos compostos puros [33]. ................................................................................. 39
Tabela 3.4 – Composições da mistura binária nas diferentes condições de trabalho. .............................. 39
Tabela 3.5 – Densidades calculadas pela equação de Peng—Robinson para as várias condições de
trabalho. ..................................................................................................................................................... 40
Tabela 3.6 – Registo de rendimento dos ensaios realizados. ..................................................................... 47
Tabela 3.7 – Resumo dos resultados obtidos para o tamanho de partícula em função da pressão de
processamento. .......................................................................................................................................... 60
Tabela A. 1 – Pontos para o cálculo da recta de calibração do espectrofotómetro. ................................... IV

viii
ÍNDICE DE FIGURAS
Figura 1.1 – Diagramas de fase do dióxido de carbono: pressão vs temperatura (a); densidade vs pressão
(b). ................................................................................................................................................................ 7
Figura 1.2 – Diagrama pressão–composição da mistura etanol–dióxido de carbono a 35°C [15]. ............ 13
Figura 1.3 – Estrutura molecular do isopreno (a) e da sua forma biologicamente activa, difosfato de
isopentenilo (b). ......................................................................................................................................... 19
Figura 1.4 – Estrutura molecular de alguns carotenóides. ......................................................................... 19
Figura 1.5 – Formação do difosfato de isopentenilo [19]. ......................................................................... 22
Figura 1.6 – Biossíntese do fitoeno a partir do IPP e do DMAPP [19]. ....................................................... 23
Figura 1.7 – Estrutura molecular do β‐caroteno. ....................................................................................... 28
Figura 1.8 – Ciclização do licopeno (formação do β‐caroteno) [19]. .......................................................... 29
Figura 2.1 – Aparelho de micronização supercrítica: aspecto geral. .......................................................... 31
Figura 2.2 – Diagrama esquemático do aparelho de micronização supercrítica. B1 e B2, bombas de
recirculação; BP1 e BP2, “backpressure” ou regulador de pressão; BT, banho termostático; C,
condensador; CG, contador de gás; DR, disco de ruptura; G, garrafa de CO2 com tubo prolongador; P,
indicador de pressão; Q, medidor de caudal; S, bureta de solução; T, indicador de temperatura; V,
válvula; VM, válvula micrométrica; VP, vaso de precipitação; VS, vaso de recolha de solvente. .............. 32
Figura 2.3 – Aparelho de micronização supercrítica: banho termostático de água (a); célula de alta
pressão (b). ................................................................................................................................................. 32
Figura 2.4 – Aparelho de micronização supercrítica (secção de bombagem): CO2 (a); solução (b); aspecto
geral (c). ...................................................................................................................................................... 33
Figura 2.5 – Aparelho de micronização supercrítica: recolha de solvente (a); aspecto geral (b). .............. 33
Figura 3.1 – Representação gráfica da variação da solubilidade do β‐caroteno em CO2/THF com a
temperatura. .............................................................................................................................................. 37
Figura 3.2 – Representação gráfica da variação da solubilidade do β‐caroteno em CO2/THF com a fracção
de THF (caudal de líquido). ......................................................................................................................... 37
Figura 3.3 – Representação gráfica da variação da solubilidade do β‐caroteno em CO2/THF com a
pressão. ...................................................................................................................................................... 38
Figura 3.4 – Variação da densidade da mistura binária e da solubilidade do β‐caroteno com a
temperatura. .............................................................................................................................................. 40
Figura 3.5 – Variação da densidade da mistura binária e da solubilidade do β‐caroteno com a pressão. 41
Figura 3.6 – Variação da densidade da mistura binária e da solubilidade do β‐caroteno com a fracção de
THF.............................................................................................................................................................. 41
Figura 3.7 – Solubilidade do β‐caroteno em função da densidade da mistura CO2/THF, quando se varia a
temperatura. .............................................................................................................................................. 42
Figura 3.8 – Solubilidade do β‐caroteno em função da densidade da mistura CO2/THF, quando se varia o
caudal (fracção) de THF. ............................................................................................................................. 42

ix
Figura 3.9 – Solubilidade do β‐caroteno em função da densidade da mistura CO2/THF, quando se varia a
pressão. ...................................................................................................................................................... 43
Figura 3.10 – Representação gráfica da variação da solubilidade do β‐caroteno em CO2/THF com a
fracção de THF (a 100 e a 130 bar). ............................................................................................................ 44
Figura 3.11 – Recolha do β‐caroteno micronizado acima do ponto crítico: copo da célula de alta pressão
(a); tampa da célula de alta pressão (b). .................................................................................................... 45
Figura 3.12 – Recolha do β‐caroteno micronizado abaixo do ponto crítico (75 bar): copo da célula de alta
pressão (a); pormenor do fundo da célula (filtro) (b). ............................................................................... 46
Figura 3.13 – Recolha do β‐caroteno micronizado: aspecto dos cristais. .................................................. 46
Figura 3.14 – Representação gráfica do rendimento da micronização em função da solubilidade (ensaios
T1, P2, Q2 e Q3). ......................................................................................................................................... 47
Figura 3.15 – Cromatograma do β‐caroteno original, micronizado e recolhido no efluente (líquido), em
THF, com detecção a 454 nm. .................................................................................................................... 48
Figura 3.16 – Cromatograma do β‐caroteno original, micronizado e recolhido no efluente (líquido), em
THF, com detecção a 260 nm. .................................................................................................................... 48
Figura 3.17 – Espectros de várias misturas de isómeros do β‐caroteno em ciclohexano [63]. .................. 49
Figura 3.18 – Espectros de absorção do β‐caroteno não processado (original), precipitado e solúvel do
ensaio T1. ................................................................................................................................................... 50
Figura 3.19 – Aspecto do β‐caroteno antes do processamento (imagem de microscopia electrónica de
varrimento com ampliação de 1000 vezes). ............................................................................................... 51
Figura 3.20 – Aspecto do β‐caroteno antes do processamento (imagem de microscopia electrónica de
varrimento com ampliação de 3000 vezes). ............................................................................................... 51
Figura 3.21 – Representação gráfica da distribuição de tamanhos de partícula contabilizada por
contagem e por tratamento estatístico (β‐caroteno não processado). ..................................................... 52
Figura 3.22 – Aspecto do β‐caroteno processado a 75 bar (imagem de microscopia electrónica de
varrimento com ampliação de 50 vezes). ................................................................................................... 53
Figura 3.23 – Aspecto do β‐caroteno processado a 75 bar (imagem de microscopia electrónica de
varrimento com ampliação de 100 vezes). ................................................................................................. 53
Figura 3.24 – Aspecto do β‐caroteno processado a 75 bar (imagens de microscopia electrónica de
varrimento com ampliação de: 500 vezes (a); 3000 vezes (b)). ................................................................. 54
Figura 3.25 – Representação gráfica da distribuição de tamanhos de partícula contabilizada por
contagem e por tratamento estatístico (β‐caroteno processado a 75 bar). .............................................. 55
Figura 3.26 – Aspecto do β‐caroteno processado a 100 bar (imagem de microscopia electrónica de
varrimento com ampliação de 100 vezes). ................................................................................................. 56
Figura 3.27 – Aspecto do β‐caroteno processado a 100 bar (imagem de microscopia electrónica de
varrimento com ampliação de 500 vezes). ................................................................................................. 56
Figura 3.28 – Aspecto do β‐caroteno processado a 100 bar (imagens de microscopia electrónica de
varrimento com ampliação de: 3000 vezes (a); 5000 vezes (b)). ............................................................... 56

x
Figura 3.29 – Representação gráfica da distribuição de tamanhos de partícula contabilizada por
contagem e por tratamento estatístico (β‐caroteno processado a 100 bar). ............................................ 57
Figura 3.30 – Aspecto do β‐caroteno processado a 130 bar (imagem de microscopia electrónica de
varrimento com ampliação de 100 vezes). ................................................................................................. 58
Figura 3.31 – Aspecto do β‐caroteno processado a 130 bar (imagem de microscopia electrónica de
varrimento com ampliação de 200 vezes). ................................................................................................. 58
Figura 3.32 – Aspecto do β‐caroteno processado a 130 bar (imagens de microscopia electrónica de
varrimento com ampliação de: 200 vezes (a); 500 vezes (b)). ................................................................... 58
Figura 3.33 – Representação gráfica da distribuição de tamanhos de partícula contabilizada por
contagem e por tratamento estatístico (β‐caroteno processado a 130 bar). ............................................ 59
Figura 3.34 – Distribuição de tamanhos de partícula obtida para cada um dos ensaios do estudo do
efeito da pressão. ....................................................................................................................................... 60
Figura 3.35 – Relação entre o tamanho de partícula obtido e a densidade estimada para a mistura
CO2/THF em cada uma das condições de pressão...................................................................................... 61
Figura 3.36 – Aspecto do β‐caroteno natural processado. ........................................................................ 62
Figura 3.37 – Cromatograma do β‐caroteno natural micronizado e recolhido no efluente (líquido), em
THF, com detecção a 454 nm. .................................................................................................................... 63
Figura 3.38 – Cromatograma do β‐caroteno natural micronizado e recolhido no efluente (líquido), em
THF, com detecção a 260 nm. .................................................................................................................... 64
Figura 3.39 – Cromatograma do β‐caroteno natural micronizado e sintético, em THF, com detecção a
454 nm. ....................................................................................................................................................... 64
Figura 3.40 – Espectros de absorção do β‐caroteno natural micronizado e recolhido no efluente líquido,
após o processamento. .............................................................................................................................. 65
Figura 3.41 – Espectros de absorção do β‐caroteno sintético (não processado) e no extracto natural
(após o processamento). ............................................................................................................................ 65
Figura A. 1 – Espectro de absorção do β‐caroteno em THF. ....................................................................... III
Figura A. 2 – Representação gráfica da recta de calibração do espectrofotómetro. .................................. IV
Figura A. 3 – Diagrama de fases da mistura CO2/THF à temperatura de 40°C, determinado por Li et al.
[33]. .............................................................................................................................................................. V
Figura A. 4 – Cromatograma do tetrahidrofurano com detecção a 260 nm. .............................................. VI
Figura A. 5 – Cromatograma do tetrahidrofurano com detecção a 454 nm. .............................................. VI

xi
LISTA DE ABREVIATURAS
T – temperatura
P – pressão
ρ – densidade
S – sobressaturação
x – fracção molar
C – concentração
Q – caudal
a, b, κ – parâmetros da equação de Peng–Robinson
k, l – parâmetros das regras de mistura
ω – factor acêntrico
R – constante dos gases perfeitos
v – volume molar
i, j ‐ componentes
c – crítico
0 – inicial
eq – equilíbrio
R – variável reduzida
L – líquido

1
1. INTRODUÇÃO
1.1. Importância do tamanho de partícula
O projecto envolvendo a produção de partículas pequenas com distribuição de tamanho controlada tem
atraído o interesse das comunidades científica e industrial com aplicações nas indústrias farmacêutica,
alimentar e química.
O tamanho de partícula influencia fortemente vários processos, como sejam a combustão de explosivos
sólidos [29] , onde é possível obter uma maior energia de detonação através do uso de partículas mais
pequenas; a eficiência de coloração, que pode ser aumentada quando o material corante é aplicado sob
a forma de partículas muito pequenas; ou a actividade dos catalisadores, que está também associada a
elevadas áreas superficiais, que podem tipicamente ser obtidas através do uso de nanopartículas. As
micropartículas de polímeros podem também ser usadas como fase estacionária em cromatografia, com
a função de adsorventes e suportes de catalisadores, assim como em sistemas de administração de
drogas [51]. Através do uso de nanopartículas, os supercondutores, que têm um grande potencial na
indústria electrotécnica, podem ver ultrapassados alguns dos problemas relacionados com a
manutenção das suas propriedades em diferentes condições operatórias, aquando do aumento de
escala [58]. Em aplicações farmacêuticas, são especialmente importantes a estreita distribuição de
tamanhos de partícula, a uniformidade da morfologia das partículas e a pureza enantiomérica [2].
As partículas de ingredientes farmacêuticos, activos ou não, existem, na maioria dos produtos, sob a
forma de pós secos e de dispersões líquidas e semi‐sólidas, que podem ir de nanocolóides a grânulos à
escala milimétrica, dependendo da dosagem e da via de administração. O tamanho e a forma da
partícula podem influenciar uma vasta gama de propriedades físicas, processos de fabrico e atributos de
qualidade [64]:
• Taxa de dissolução e biodisponibilidade dos princípios activos.
• Velocidade (controlada) de libertação da droga.
• Distribuição e deposição das partículas in vivo, taxa de absorção e tempo de permanência,
especialmente para aerossóis e sistemas de colóides desenhados para atingir um alvo
específico.
• Uniformidade da composição, bem como outras propriedades relacionadas com a estabilidade
físico‐química.
• Comportamento de aerosolização e desempenho das formulações respiratórias.
• Propriedades de fluxo e empacotamento, mistura e segregação de pós, características
reológicas das formulações líquidas e semi‐sólidas.
• Granulosidade das partículas sólidas nas formulações mastigáveis, em cremes para aplicação
tópica e em preparações oftálmicas.

2
É, contudo, a biodisponibilidade que mais desafia a investigação farmacêutica. A lipofilicidade de muitas
drogas obriga à suspensão de muitos testes farmacológicos, devido à sua baixa biodisponibilidade.
Também muitas das novas drogas altamente eficientes, desenvolvidas na base da medicina molecular
moderna, e projectadas para atingir locais de reconhecimento da superfície das células, possuem
solubilidade em água, e consequente biodisponibilidade, muito baixas. Apesar de a generalidade das
drogas com elevada lipofilicidade poder permear rapidamente as biomembranas, a solubilidade nos
fluidos gastrointestinais e a taxa de dissolução são factores limitantes, na maioria dos casos. É, assim, de
extrema importância o desenvolvimento de estratégias que permitam o aumento da taxa de dissolução
deste tipo de drogas.
As diferentes vias de administração dos ingredientes farmacêuticos exigem diferentes formulações e
dosagens, o que implica também diferentes tamanhos de partícula. Os avanços recentes na terapia da
inalação têm despontado um interesse considerável no desenvolvimento de novas tecnologias da
formulação de drogas. A preparação de pós adequados para a inalação e carregados com biomoléculas é
também de particular interesse para a terapia génica e para a vacinação [9].
A investigação na área do tamanho de partícula tem mostrado que o diâmetro óptimo das partículas
para formulações orais de aerossóis que penetram efectivamente nos pulmões se situa na gama de 1 a 5
µm, sendo o mecanismo predominante a sedimentação nas regiões brônquica e alveolar [64]. As
partículas de tamanho superior a 5 µm colidem com as paredes nas vias respiratórias superiores, sendo
depois transportadas por fluxo ciliar até à boca e atingindo o sistema, primeiramente, por ingestão. As
partículas de tamanho inferior a 1 µm podem permanecer suspensas no ar inspirado e expirado, não
chegando a atingir os pulmões [56].
A produção de micropartículas pode ser importante no envio de drogas directamente para os pulmões.
A deposição selectiva dessas drogas no tracto respiratório humano pode ser optimizada através do
controlo do tamanho das partículas, maximizando a sua efectividade contra doenças que não se
manifestam uniformemente no pulmão (por exemplo, a bronquite) e minimizando os efeitos laterais
adversos [56]. Por exemplo, os antibióticos, quando administrados sob forma de aerossóis, têm a
vantagem de fornecer elevadas concentrações directamente ao local da infecção, no caso de infecções
pulmonares. Por outro lado, os pulmões podem ainda ser considerados um local de recepção de drogas
que permite ultrapassar problemas de absorção e processos metabólicos, que são limitantes da
eficiência da droga por outras vias de administração [55].
As nanopartículas constituem também uma promessa na deposição pulmonar de drogas, devido à
homogeneidade, e consequente eficiência acrescida, das nanosuspensões. As nanopartículas de drogas
pouco solúveis em água têm uma taxa global de dissolução mais elevada e podem ter uma via de
interacção específica com os epitélios traqueo‐brônquico e alveolar. As nanopartículas ultrafinas (<150
nm) têm uma libertação mais lenta do pulmão, maior interacção com certas proteínas e maior
translocação do epitélio para a circulação e, subsequentemente, para os órgãos alvo [64].

3
A dimensão de partícula adequada para comprimidos tende a situar‐se no intervalo de 100 a 200 µm,
devido ao comportamento de compactação exigido e às propriedades de transporte do pó. Contudo,
partículas mais pequenas, de 20 a 50 µm, são passíveis de ter boas características para o tipo de
formulações mastigáveis ou de rápida desintegração, onde a dissolução controlada e a trituração são
importantes. Por outro lado, o tamanho de partícula tem grande influência em praticamente todas as
etapas da produção de comprimidos, incluindo a mistura, granulação, compressão e revestimento [64].
O aumento da biodisponibilidade das drogas insolúveis em água e administradas por via oral pode
também ser atingido por nanopartículas, através da homogeneidade que estas podem conferir a uma
suspensão [64]. A camada de difusão à volta das partículas pequenas é mais fina, especialmente para
diâmetros inferiores a 5 µm, o que resulta numa mais rápida distribuição das moléculas dissolvidas,
devido à diminuição da resistência à transferência de massa [49].
O tamanho de partícula de formulações oftálmicas de libertação controlada é muito importante no
balanço entre a velocidade de libertação da droga, o aumento da biodisponibilidade e a facilidade de
aplicação. Quando formuladas apropriadamente para esta via de administração, as partículas são
retidas no canto do olho, sendo a droga libertada a uma velocidade que não é nem muito rápida nem
muito lenta para permitir a penetração adequada da droga no tecido ocular. As nanopartículas
(tipicamente de cerca de 300 nm) sem bioadesão podem ser eliminadas quase tão rapidamente como as
soluções aquosas, mas a presença de uma fracção de partículas grosseiras acima dos 25 µm pode tornar
o medicamento irritante para o olho. Assim, os maiores desafios do desenvolvimento deste tipo de
sistemas de partículas residem, precisamente, na complexidade do seu fabrico e no controlo do
tamanho de partícula na produção em grande escala [64].
O tamanho de partícula também exerce uma influência significativa nas vias de penetração cutâneas:
partículas com tamanho superior a 10 µm permanecem à superfície da pele; partículas entre 3 a 10 µm
concentram‐se nos folículos capilares; partículas inferiores a 3 µm podem penetrar os folículos e a
epiderme. Assim, a escolha ponderada do tamanho de partícula para formulações tópicas pode não só
maximizar a eficácia local, como também minimizar reacções adversas [64].
As formulações intravenosas são essencialmente produtos baseados em soluções, cujo controlo tem de
ser apertado ao nível da contaminação por parte de partículas, que, em caso de precipitação ou má
formulação, podem provocar a oclusão vascular ou embolia pulmonar do paciente. Mesmo as
micropartículas de tamanho mais elevado podem ainda diminuir a injectabilidade do produto, bem
como o seu manuseamento em seringas [64].
Na área já bastante explorada da administração direccionada a alvos específicos, os fagócitos
mononucleares, células dendríticas, endoteliais, e tumores (células e novos vasos sanguíneos) são alvos
importantes para as nanopartículas com tamanhos médios muitas vezes inferiores a 100 nm. As
nanopartículas com diâmetro inferior a 150 nm têm um tempo de meia vida prolongado na circulação,
devido à sua baixa taxa de aquisição por parte dos micrófagos [64].
Tendo já sido provado que micelas em forma de bastonete, com alguns mícrons de comprimento e
secção transversal de 10 a 100 nm, são capazes de se manter em circulação durante vários dias, pode‐se

4
afirmar que o tamanho não é o único factor preponderante. Diferentes estudos in vivo mostram que
estes bastonetes se comportam como verdadeiras nanopartículas, nas células e no corpo, o que indica
que todas as propriedades das partículas, como o tamanho, forma e características de superfície,
necessitam de ser investigadas neste tipo de formulações [64].
1.2. Micronização – formação de micropartículas
Micronização é o termo usado para designar, duma forma generalizada, o processo de redução de
tamanho de partícula, resultando da ordem de grandeza do tamanho obtido através dos processos de
moagem tradicionais. Actualmente, as exigências da indústria, essencialmente na área farmacêutica, já
se centram num tamanho de partícula ainda mais diminuto, que atinge a escala dos nanómetros, mas o
termo micronização mantém‐se em utilização.
Foram já referidas as inúmeras vantagens da redução do tamanho de partícula de ingredientes
farmacêuticos. A engenharia das pequenas partículas tem, a este nível, dois objectivos principais. Por
um lado, a modificação do tamanho de partícula, porosidade e densidade de um princípio activo. Por
outro, permite ainda a incorporação de um princípio activo numa formulação para administração
dirigida a um alvo específico (mistura com excipientes, por exemplo) [60].
Dos mais tradicionais aos mais recentes, existem vários processos de micronização, baseados em
diferentes fundamentos. A sensibilidade do produto às condições físico‐químicas operacionais, a pureza
e tamanho de partícula pretendidos, podem ditar o tipo de processo a implementar.
A técnica mais comum é a tradicional trituração de partículas maiores, através de processos de moagem
mecânica, em moinhos de jacto, de bolas, ou homogeneizadores de alta pressão. A redução de tamanho
das partículas dá‐se, portanto, através de forças físicas de fricção e atrito, impacto, corte, ou da pressão.
No extremo oposto estão as técnicas de produção de partículas pequenas de tamanho controlado, das
quais são exemplo o “spray drying” ou a precipitação com fluidos supercríticos.
1.2.1. Métodos tradicionais de moagem
A produção de partículas à escala industrial é, normalmente, realizada num processo de várias etapas,
que inclui uma cristalização descontínua, filtração e secagem, seguidas da micronização [9]. No entanto,
apesar de comercializadas e bem estabelecidas, as técnicas tradicionais de moagem mecânica trazem as
desvantagens associadas ao processamento físico de produtos biológicos sensíveis.
Os moinhos de jacto são os aparelhos mais usados. Aplicam uma pressão na câmara de 3 a 10 bar, o que
faz com que o ar injectado, ao sofrer uma expansão que o coloca a alta velocidade, provoque a colisão
das partículas do produto a micronizar. As partículas mais finas são então descarregadas numa câmara
de classificação. Apesar dos gastos energéticos, o stresse térmico a que o produto é sujeito é baixo. Os
moinhos de bolas consistem num tanque rotativo que está parcialmente preenchido com bolas. A

5
rotação deste sistema provoca a redução de tamanho da droga, através das forças de atrito e de
impacto provocadas.
A homogeneização de alta pressão é também muito usada na indústria farmacêutica para a preparação
de nanosuspensões de drogas pouco solúveis em água. Devido às forças de corte e cavitação
provocadas através de uma expansão rápida, ocorre a divisão das partículas e criam‐se estruturas
amorfas na sua superfície, o que as torna termodinamicamente activadas. A solubilidade da droga é
então aumentada através de dois mecanismos: redução do tamanho da partícula e activação da
superfície. Esta técnica aplica pressões da ordem dos 1000 bar às suspensões a processar, durante
vários ciclos, provocando uma elevada tensão de corte no produto [49].
A limitação de todas as técnicas tradicionais de moagem está principalmente na dificuldade de controlo
de características importantes do produto, como o tamanho, a forma, a morfologia, as propriedades da
superfície e a carga electrostática. A micronização mecânica resulta numa larga banda na distribuição de
tamanho e na heterogeneidade de formas das partículas. As superfícies amorfas termodinamicamente
activadas podem converter‐se de novo em material cristalino durante o armazenamento, o que leva a
uma natureza dinâmica do produto, através de modificações das propriedades físico‐químicas, e a
recristalização pode ainda levar à aglomeração. Esta instabilidade torna o produto mais susceptível à
decomposição química e à absorção de água, sendo o conjunto de todas estas alterações altamente
indesejável, podendo prejudicar o desempenho in vitro e in vivo das formulações. Por outro lado, os
custos associados ao processo de micronização mecânica, ainda bastante elevados, devido à grande
quantidade de energia em jogo, e a natureza lábil de muitos produtos farmacêuticos podem limitar a
aplicação deste tipo de micronização.
Devido às desvantagens enumeradas dos processos de moagem, torna‐se necessária a procura de
alternativas mais eficientes e económicas, através da engenharia de partículas, que permitam a
manipulação e o controlo das propriedades desejadas na produção de drogas.
1.2.2. Engenharia de partículas
Dadas as limitações dos processos de moagem para micronização de produtos biológicos, têm vindo a
ser estudadas e desenvolvidas técnicas alternativas, baseadas essencialmente em processos de
separação de drogas em solução.
A produção controlada de partículas de um determinado produto (que pode ser uma droga ou a sua
mistura com moléculas transportadoras, para administração dirigida) cujas características físicas
(tamanho, morfologia e estrutura) estão optimizadas é designada por “engenharia de partículas”. O
objectivo principal desta disciplina é a incorporação de atributos desejáveis nas partículas, tais como
uma estreita distribuição de tamanhos, elevada dispersibilidade, estabilidade físico‐química, maior
biodisponibilidade, distribuição controlada e administração dirigida [9].
As técnicas desenvolvidas mais recentemente no domínio da micronização envolvem o uso de líquidos
convencionais, gases comprimidos, líquidos em condições próximas do ponto crítico, ou fluidos

6
supercríticos, que funcionam como solvente ou antisolvente, ou meios criogénicos que permitem a
congelação ultra rápida. Estas técnicas envolvem a separação de fases entre o solvente e a droga, por
evaporação, expansão rápida, mudança na composição do solvente, ou solidificação por congelação. A
configuração do atomizador em muitos destes processos permite a produção de gotas com elevada área
superficial, o que resulta na separação de fases e rápida nucleação, levando a partículas muito pequenas
[60].
Para além do controlo das propriedades das partículas, estes novos processos trazem vantagens
relativamente aos métodos tradicionais, pois requerem menor manuseamento, o que permite um
aumento do rendimento e simplifica os procedimentos de limpeza e esterilização, e estão mais aptos
para o aumento de escala. Estes processos podem também operar em modo contínuo, ao contrário dos
processos de moagem, que operam em descontínuo [60].
O início da aplicação do “spray drying” remota aos anos 40 do século XX, no processamento de
alimentos e de produtos bioquímicos e farmacêuticos na indústria. O facto de ser uma técnica
relativamente simples, de fácil operação, com disponibilidade de equipamento em grande escala e
capacidade de produção de materiais compostos, levou a uma popularidade generalizada deste
processo [9].
As drogas processadas por esta técnica atingem tamanhos de partícula reduzidos e distribuições
homogéneas. O limite de tamanho de partícula obtido por “spray drying” situa‐se no intervalo de 1 a 5
µm, o que ainda restringe a sua aplicabilidade em alguns produtos farmacêuticos com exigências de
tamanho de partícula mais reduzido. Contudo, embora bem desenvolvida e versátil, esta técnica é
bastante limitada no que diz respeito à aplicação a produtos biológicos. Apesar de reduzido pelo efeito
do termómetro húmido, o stresse provocado pelas elevadas temperaturas pode causar alterações
conformacionais, ou mesmo a desnaturação das biomoléculas. Também a rápida solidificação resulta na
formação de partículas amorfas que podem sofrer recristalização, instabilizando o produto [9]. O uso de
solventes orgânicos também deve ser evitado, devido a problemas toxicológicos e ambientais, pelo que
esta técnica só deve ser aplicada a drogas solúveis em água [49].
Mais recentemente, nomeadamente a partir da década de 1990, têm vindo a ser desenvolvidas várias
técnicas que empregam os fluidos supercríticos nos processos de micronização, as quais serão
averiguadas com maior detalhe no decorrer deste texto.
1.3. Os fluidos supercríticos e as suas aplicações
1.3.1. Estado supercrítico
Os fluidos supercríticos caracterizam‐se por a sua temperatura e pressão serem superiores aos
correspondentes valores críticos (Figura 1.1). Acima do ponto crítico, deixa de haver tensão superficial e
separação entre as fases líquida e gasosa em equilíbrio, formando‐se uma única fase supercrítica, cujas
7
propriedades são intermédias daqueles dois estados. A Tabela 1.1 regista a pressão e temperatura
críticas de alguns fluidos com interesse em processos de extracção supercrítica.
Tabela 1.1 – Constantes críticas de alguns fluidos com interesse em extracção supercrítica [40].
Fluido Tc (K) Pc (bar) Etileno 282,4 50,4 Dióxido de carbono 304,1 73,8 Etano 305,4 48,8 Óxido nitroso 309,6 72,4 Propano 369,8 42,5 Etanol 513,9 61,4 Benzeno 562,1 48,9 Tolueno 591,8 41,0 Água 674,3 221,2
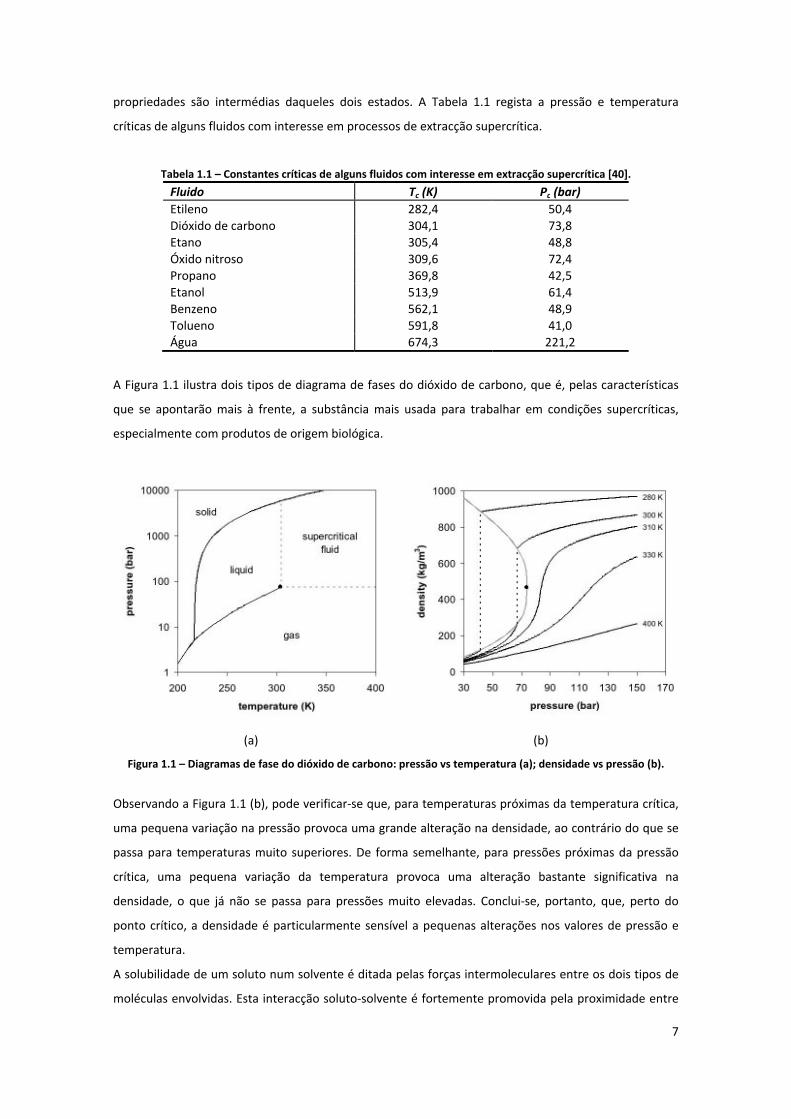
A Figura 1.1 ilustra dois tipos de diagrama de fases do dióxido de carbono, que é, pelas características
que se apontarão mais à frente, a substância mais usada para trabalhar em condições supercríticas,
especialmente com produtos de origem biológica.
(a) (b)
Figura 1.1 – Diagramas de fase do dióxido de carbono: pressão vs temperatura (a); densidade vs pressão (b).
Observando a Figura 1.1 (b), pode verificar‐se que, para temperaturas próximas da temperatura crítica,
uma pequena variação na pressão provoca uma grande alteração na densidade, ao contrário do que se
passa para temperaturas muito superiores. De forma semelhante, para pressões próximas da pressão
crítica, uma pequena variação da temperatura provoca uma alteração bastante significativa na
densidade, o que já não se passa para pressões muito elevadas. Conclui‐se, portanto, que, perto do
ponto crítico, a densidade é particularmente sensível a pequenas alterações nos valores de pressão e
temperatura.
A solubilidade de um soluto num solvente é ditada pelas forças intermoleculares entre os dois tipos de
moléculas envolvidas. Esta interacção soluto‐solvente é fortemente promovida pela proximidade entre
8
as moléculas e, portanto, pela densidade da fase fluida. Consequentemente, deve‐se esperar que um
fluido supercrítico tenha um poder de dissolução elevado em estados de elevada densidade, sendo esse
poder menor para densidades mais baixas [5]. Tendo em conta as considerações anteriores, é fácil
aceitar que o controlo do poder solvente de um fluido supercrítico é possível, através da manipulação
da sua pressão, a temperatura constante.
Os fluidos supercríticos possuem também propriedades de transporte que os tornam únicos.
Parâmetros como a viscosidade, condutividade térmica e difusibilidade contribuem significativamente
para o comportamento do fluido supercrítico, pois são essas propriedades que determinam a força
motriz para a transferência de calor e de massa.
Tal como evidencia a Tabela 1.2, a densidade do fluido supercrítico pode aproximar‐se à do estado
líquido, tornando o poder solvente dos fluidos supercríticos semelhante ao dos líquidos. Por outro lado,
as propriedades de transporte são intermédias entre as dos gases e dos líquidos, o que faz com que um
fluido supercrítico se comporte como uma fase extremamente móvel, capaz de se misturar rápida e
profundamente com outras substâncias, reduzindo ainda os custos de transporte através de bombas,
devido à sua baixa viscosidade.
Tabela 1.2 – Ordens de grandeza de propriedades termofísicas dos fluidos supercríticos [5].
Estado Densidade (kg m‐3)
Viscosidade (N s m‐2)
Coeficiente de difusão (m2 s‐1)
Condutividade térmica
(W m‐1 K‐1) Gasoso 1 – 100 10‐5 – 10‐4 10‐5 – 10‐4 2×10‐5 – 5×10‐4
Fluido Supercrítico 250 – 800 10‐4 – 10‐3 10‐8 – 10‐7 5×10‐2 – 10‐1
Líquido 800 – 1200 10‐3 – 10‐2 10‐9 – 10‐8 ≈ 10‐1
1.3.1.1. O dióxido de carbono como fluido supercrítico
No que diz respeito a técnicas que envolvem o uso de fluidos supercríticos, o dióxido de carbono é,
quase impreterivelmente, a substância escolhida. A sua pressão e temperatura críticas,
respectivamente, 73,8 bar e 31°C, são facilmente atingíveis e permitem o processamento, em condições
moderadas e inofensivas, de produtos sensíveis à temperatura, como é o caso da maioria dos produtos
de origem e aplicação biológica. Adicionalmente, o dióxido de carbono tem ainda as vantagens da sua
natureza inerte, não tóxica, e do seu baixo custo.
Em termos de micronização supercrítica, o dióxido de carbono permite a extracção e a separação
eficiente de solventes orgânicos, o que possibilita a produção de partículas secas, ou sob a forma de
suspensão aquosa, livres de contaminação por parte dos solventes. Este facto, aliado à facilidade da
realização de uma precipitação limpa e reciclável, constitui, claramente, uma vantagem, nomeadamente
no que diz respeito ao processamento de produtos farmacêuticos e alimentares.
É importante referir, ainda, as vantagens do dióxido de carbono supercrítico na precipitação selectiva,
na separação de impurezas e no controlo das formas cristalinas. Em certas aplicações, tais como a
engenharia de partículas de drogas de elevado valor e sensibilidade, o dióxido de carbono supercrítico

9
pode reduzir a complexidade de fabrico, as necessidades de energia e de solventes, e geralmente
constitui um processo mais benigno e amigo do ambiente do que as técnicas convencionais [9].
Devido ao seu comportamento não polar, o dióxido de carbono tem pouca afinidade para compostos
polares. No entanto, a sua mistura com uma pequena quantidade de uma outra substância, designada
de co‐solvente ou “entrainer”, como a água, o etanol ou o metanol, pode aumentar consideravelmente
o poder solvente do dióxido de carbono [40].
1.3.2. Extracção supercrítica
A extracção supercrítica é uma das mais conhecidas e estudadas aplicações dos fluidos supercríticos,
que se baseia no poder de dissolução deste tipo de fluidos. Devido às suas características, estes fluidos
têm uma boa capacidade para dissolver solutos e, portanto, um grande potencial para os separar de
outros compostos, sendo relativamente fácil a manipulação da selectividade do fluido supercrítico por
alteração das condições experimentais de pressão e/ou temperatura.
As grandes vantagens da extracção supercrítica em relação às técnicas convencionais são a possibilidade
de processamento a condições moderadas de temperatura e o uso de fluidos não tóxicos e baratos, o
que torna este processo bastante atractivo para a separação de produtos biológicos. O uso de co‐
solventes neste processo é também particularmente atractivo, na medida em que permite a extracção
de compostos polares.
A extracção supercrítica tem aplicações nas indústrias farmacêutica e alimentar, na extracção de
compostos de produtos naturais (como a cafeína, a baunilha, óleos vegetais ou carotenóides) ou na
remoção de extractos indesejáveis (pesticidas ou químicos perigosos). Ao nível da indústria biológica,
este tipo de extracção tem também algumas aplicações interessantes, como a remoção de agentes
biostáticos de caldos de fermentação, a extracção de solutos orgânicos de soluções aquosas, a ruptura
celular, a destruição de desperdícios industriais, o tratamento de materiais linhocelulósicos, a
recuperação e purificação de produtos biológicos, o fraccionamento de óleos ou a preparação de
lipossomas [5]. Um exemplo de aplicação da extracção supercrítica de produtos biológicos é na
extracção de compostos com importância farmacêutica a partir de microalgas [40].
1.3.3. Micronização supercrítica (“RESS” e “PGSS”)
O papel dos fluidos supercríticos no processo de micronização pode ser de solvente ou anti‐solvente.
Dentro destas duas possibilidades, existem quatro técnicas principais de micronização que envolvem
este tipo de fluidos, sendo as duas primeiras abordadas nesta secção e as outras nas secções seguintes:
• Expansão rápida de uma solução supercrítica (“Rapid expansion of supercritical solutions”) –
“RESS”;
• Precipitação a partir de soluções saturadas em gás (“Precipitation from gas‐saturated
solutions”) – “PGSS”;

10
• Anti‐solvente supercrítico (“Supercritical antisolvent”) – “SAS”;
• Atomização assistida por fluidos supercríticos (“Supercritical assisted atomization”) – “SAA”.
1.3.3.1. Expansão rápida de uma solução supercrítica (“RESS”)
A expansão rápida de uma solução supercrítica é uma técnica de recristalização, redução de tamanho de
partícula, e mistura de componentes [18]. Esta técnica foi já aplicada na produção de pós de produtos
cerâmicos, polímeros e esteróides farmacêuticos [60][25].
O processo de micronização por “RESS” baseia‐se na precipitação de partículas através da rápida
expansão da solução em cujo solvente se encontram dissolvidas. Nesta técnica, o soluto é dissolvido
directamente no dióxido de carbono supercrítico, sendo a solução depois atomizada através de um
restritor para uma câmara de recolha, que está, tipicamente, em condições atmosféricas. Esta expansão,
que faz com que o dióxido de carbono evapore rapidamente, provoca uma redução brusca na
densidade, e, logo, no seu poder solvente, incitando um elevado grau de sobressaturação na solução e
promovendo a rápida nucleação das partículas de soluto, o que produz o pó micronizado.
Quando um composto é solúvel no dióxido de carbono supercrítico em fracções molares de
aproximadamente 10‐4, esta técnica é a escolhida, dado que permite a produção contínua, simples,
directa, e livre de solventes, de um pó seco. A “RESS” pode ser optimizada para atingir distribuições de
tamanho de partícula relativamente estreitas, podendo ser usada para o revestimento de
micropartículas com polímeros solúveis no dióxido de carbono na microencapsulação de drogas, por
exemplo [9].
A grande desvantagem deste processo é que a substância a precipitar não pode ser pouco solúvel no
dióxido de carbono, o que limita a aplicabilidade desta técnica no processamento de produtos
biológicos e farmacêuticos, na sua maioria polares e insolúveis neste solvente. A aplicação da “RESS” em
grande escala também sofre de alguns problemas, nomeadamente, ao nível da expansão do solvente,
que provoca um arrefecimento muito acentuado no sistema. Esta queda de temperatura, que ocorre na
zona do restritor, pode provocar o entupimento do mesmo, ou ainda promover a formação de gotas,
com a consequente redissolução e perda das partículas formadas. No processamento de produtos
termolábeis, o pré‐aquecimento do sistema pode não ser possível, o que exige a adopção de outros
meios, como a administração de calor à câmara de recolha com um fluido quente, o que implica,
também, gastos adicionais [18].
1.3.3.2. Precipitação de soluções saturadas em gás (“PGSS”)
A precipitação de soluções saturadas em gás faz uso da elevada solubilidade do dióxido de carbono em
certos materiais, como os polímeros, o que resulta na sua plasticização, redução da viscosidade, e
abaixamento dos pontos de fusão e vitrificação [9]. Estas propriedades têm sido exploradas na mistura,

11
revestimento e encapsulação, de alguns produtos, desde pós inorgânicos a formulações farmacêuticas,
sem que seja necessário o uso de solventes orgânicos [25].
A técnica e o fundamento da “PGSS” são muito similares à “RESS”. No entanto, embora sejam poucos os
compostos orgânicos solúveis em dióxido de carbono, existe um número considerável de polímeros que
podem ser embebidos em dióxido de carbono supercrítico e, portanto, processados por “PGSS” [60].
Como as solubilidades dos gases comprimidos em líquidos e sólidos como os polímeros são usualmente
elevadas, e muito mais elevadas do que as solubilidades desses sólidos e líquidos na fase gasosa
comprimida, o processo consiste na dissolução de dióxido de carbono em substâncias fundidas ou
suspensas em líquidos, conduzindo à formação de uma solução ou suspensão saturada em gás [25].
Assim, o dióxido de carbono supercrítico é disperso para dentro de um composto fundido, para criar
uma suspensão gás‐líquido, cujo ponto de fusão e viscosidade diminuem com a concentração do fluido
supercrítico. Esta suspensão é então atomizada, através de um restritor, para dentro de uma câmara de
recolha, que se encontra em condições atmosféricas. Ao expandir, o dióxido de carbono arrefece e
promove também o arrefecimento e a solidificação do material fundido, formando‐se micropartículas
porosas, através de um mecanismo de sobressaturação semelhante ao já descrito para a “RESS”.
As vantagens da técnica de “PGSS” são também idênticas às da “RESS”, no que diz respeito à
simplicidade do processo, aos seus custos, e à possibilidade de ter um produto final livre de solventes
orgânicos. Adicionalmente, a “PGSS” requer, geralmente, pressões mais reduzidas e menores consumos
de gás do que a “RESS” [18].
Como desvantagem da “PGSS”, tem‐se que esta só permite o processamento de compostos que se
encontram fundidos abaixo das condições do fluido supercrítico, limitando grandemente a aplicação
desta técnica com formulações biológicas.
1.3.4. Micronização com anti‐solvente supercrítico (“SAS”)
Os anti‐solventes líquidos são amplamente conhecidos e aplicados na indústria. São baseados no uso de
dois solventes, completamente miscíveis entre si, sendo o soluto a precipitar apenas solúvel num
daqueles (o primeiro). A mistura do solvente com o anti‐solvente provoca a sobressaturação da solução
e a consequente precipitação do soluto. No entanto, o uso de solventes líquidos tem a grande
desvantagem do complexo pós‐processamento, necessário para a completa eliminação dos resíduos de
solvente.
Devido às características de transporte dos fluidos supercríticos e à possibilidade da sua recuperação (e
do solvente orgânico) sem processamento posterior, este tipo de fluidos tem sido proposto, nas últimas
décadas, como alternativa aos anti‐solventes líquidos [51]. A micronização baseada neste conceito foi
apresentada, pela primeira vez, numa proposta de patente, em 1988 [25].
Do ponto de vista termodinâmico, o processo de micronização com anti‐solvente supercrítico (“SAS”)
tem de obedecer às seguintes especificações [52]:

12
• O soluto tem de ser solúvel no solvente orgânico à temperatura de trabalho e insolúvel no
fluido supercrítico;
• O solvente e o fluido supercrítico têm de ser miscíveis.
O dióxido de carbono supercrítico, como anti‐solvente, traz várias vantagens relativamente aos anti‐
solventes líquidos. Por um lado, a completa remoção do anti‐solvente é conseguida com uma simples
redução de pressão, que provoca a sua passagem ao estado gasoso. Por outro, o anti‐solvente
supercrítico é caracterizado pela sua elevada difusibilidade, que pode ser, como foi já referido (secção
1.3.1), até duas ordens de grandeza superior à dos líquidos. Assim, a sua rápida difusão e mistura no
solvente líquido produz a rápida sobressaturação da solução e a precipitação do soluto em
micropartículas.
Apesar de ainda pouco difundidos na indústria, os processos de precipitação que envolvem fluidos
supercríticos têm vindo a ser vistos como excelentes alternativas aos processos tradicionais, pela
possibilidade que oferecem de garantir a elevada qualidade de certos produtos farmacêuticos, facto que
se sobrepõe aos problemas técnicos associados às altas pressões praticadas [65]. Devido à baixa
solubilidade de muitos compostos farmacêuticos em dióxido de carbono, o processo “SAS” torna‐se o
mais apelativo, relativamente aos outros processos de micronização com fluidos supercríticos já
abordados (secção 1.3.3).
1.3.4.1. Fundamentos teóricos
Na técnica de micronização “SAS”, a precipitação ocorre de acordo com a selecção adequada das
substâncias e das condições operatórias, de forma a que o solvente e o anti‐solvente sejam parcial ou
totalmente miscíveis e o soluto tenha baixa solubilidade no anti‐solvente [15].
A micronização “SAS” é um processo complexo que envolve a interacção de vários mecanismos de
transferência de massa, hidrodinâmica, equilíbrio de fases, nucleação e/ou crescimento dos cristais.
Uma descrição completa deste processo teria de ter em conta todos estes mecanismos, o que, até aos
dias de hoje, ainda não foi possível, apesar dos esforços por parte de muitos autores na modelação
deste processo [36].
Quando as condições operatórias se encontram abaixo do ponto crítico da mistura, tira‐se partido da
capacidade do dióxido de carbono supercrítico para dissolver e expandir solventes orgânicos. Quando a
expansão é suficientemente acentuada, o poder de solvatação do solvente torna‐se tão pequeno que
provoca a sobressaturação da solução, permitindo a precipitação do soluto em pequenas partículas [51].
Em condições supercríticas da mistura, o solvente orgânico difunde‐se rapidamente no dióxido de
carbono e o este no solvente, formando uma única fase. Ambas as taxas de difusão são bastante mais
rápidas do que em anti‐solventes líquidos convencionais. Assim, as elevadas taxas de transferência de
massa resultam numa nucleação muito mais rápida e uniforme, permitindo atingir tamanhos de
partícula muito mais pequenos e distribuições de tamanho mais estreitas [60].
13
Termodinâmica de misturas binárias
As condições operatórias do processo de micronização “SAS” devem ser seleccionadas com base na
análise do diagrama de fases do sistema em estudo. Na carência de diagramas ternários para o sistema
soluto, solvente e anti‐solvente, o comportamento do sistema pode ser previsto, embora não sem
algumas reservas, com base no diagrama binário da mistura solvente/anti‐solvente. Este diagrama
permite saber qual o ponto crítico desta mistura, bem como as fracções molares dos dois componentes
em cada uma das fases em equilíbrio, quando em condições subcríticas.
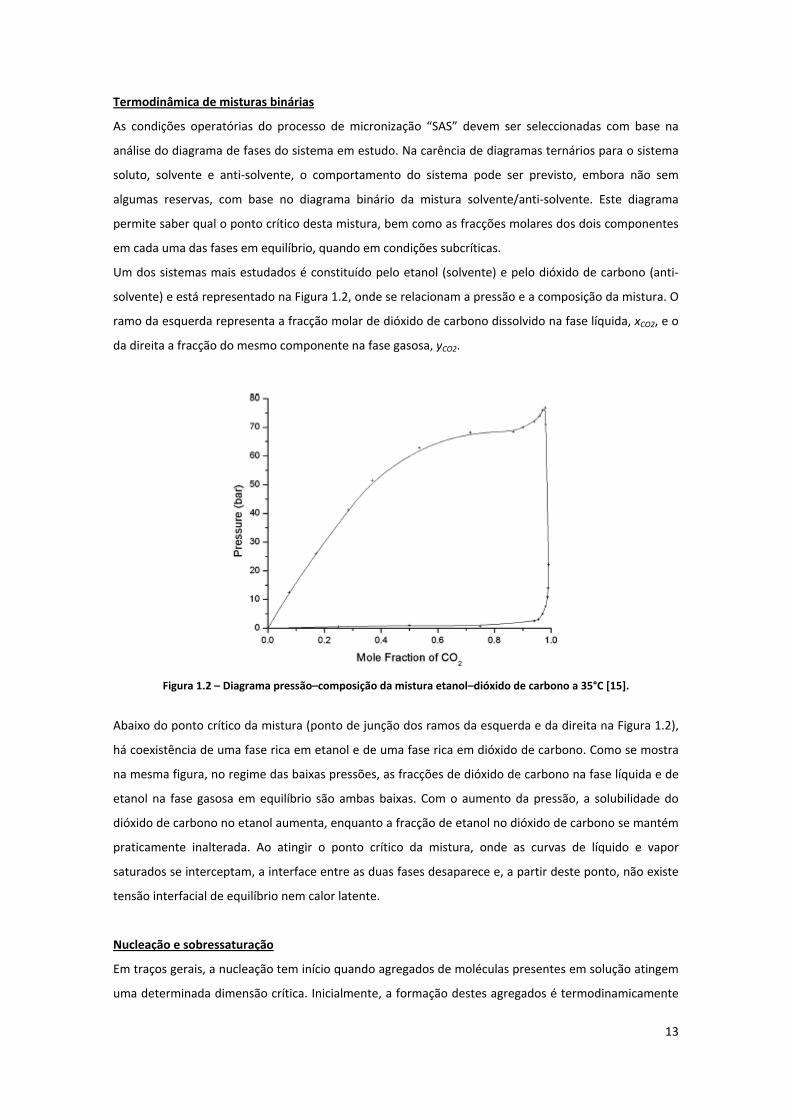
Um dos sistemas mais estudados é constituído pelo etanol (solvente) e pelo dióxido de carbono (anti‐
solvente) e está representado na Figura 1.2, onde se relacionam a pressão e a composição da mistura. O
ramo da esquerda representa a fracção molar de dióxido de carbono dissolvido na fase líquida, xCO2, e o
da direita a fracção do mesmo componente na fase gasosa, yCO2.
Figura 1.2 – Diagrama pressão–composição da mistura etanol–dióxido de carbono a 35°C [15].
Abaixo do ponto crítico da mistura (ponto de junção dos ramos da esquerda e da direita na Figura 1.2),
há coexistência de uma fase rica em etanol e de uma fase rica em dióxido de carbono. Como se mostra
na mesma figura, no regime das baixas pressões, as fracções de dióxido de carbono na fase líquida e de
etanol na fase gasosa em equilíbrio são ambas baixas. Com o aumento da pressão, a solubilidade do
dióxido de carbono no etanol aumenta, enquanto a fracção de etanol no dióxido de carbono se mantém
praticamente inalterada. Ao atingir o ponto crítico da mistura, onde as curvas de líquido e vapor
saturados se interceptam, a interface entre as duas fases desaparece e, a partir deste ponto, não existe
tensão interfacial de equilíbrio nem calor latente.
Nucleação e sobressaturação
Em traços gerais, a nucleação tem início quando agregados de moléculas presentes em solução atingem
uma determinada dimensão crítica. Inicialmente, a formação destes agregados é termodinamicamente

14
desfavorável, mas ocorre, devido ao movimento Browniano, em solução homogénea, ou à presença de
substratos que facilitam a nucleação1, em solução heterogénea. Contudo, quando os agregados atingem
a dimensão crítica, a absorção de qualquer molécula suplementar já é termodinamicamente favorável,
pelo que a partícula cresce regular e irreversivelmente.
À partícula de dimensão crítica dá‐se o nome de núcleo e a etapa de formação destes núcleos, a
primeira do processo de precipitação, é, portanto, designada de nucleação. O número de núcleos que se
formam por unidade de tempo e de volume é a velocidade de nucleação. A segunda etapa deste
processo é o crescimento, durante o qual não se formam mais núcleos e o que ocorre é o crescimento
dos que se formaram na etapa anterior [62].
A força motriz para que se dê a precipitação do soluto é a sobressaturação, S, definida pela equação
(1.1), onde x0 é a concentração inicial do soluto na solução e xeq a correspondente concentração de
equilíbrio.
eqx
xS 0= (1.1)
A sobressaturação ocorre devido à difusão do anti‐solvente nas gotas atomizadas do solvente contendo
o soluto. Assim que a sobressaturação se torna suficientemente elevada num determinado elemento de
volume, a nucleação tem início e a sua velocidade aumenta grandemente com o aumento de S. Quando
o processo de difusão é rápido, o equilíbrio entre o solvente e o anti‐solvente é estabelecido antes que o
primeiro núcleo se forme, o que faz com que a sobressaturação seja uniforme em todo o volume da
mistura, bem como a respectiva velocidade de nucleação. Assim, para maior grau de sobressaturação,
são mais e mais pequenas as partículas que se formam por unidade de tempo e de volume.
Tomando novamente como exemplo o sistema etanol–dióxido de carbono (Figura 1.2 da secção 0),
verifica‐se que, perto do ponto crítico da mistura, o valor de xCO2 aumenta até 0,95. Nestas condições, a
correspondente concentração de equilíbrio do soluto, xeq, decresce várias vezes relativamente à sua
concentração inicial, x0, o que faz com que a sobressaturação seja máxima perto do ponto crítico. Acima
deste ponto, há miscibilidade total do solvente com o anti‐solvente, qualquer que seja a composição da
mistura, pelo que a maior a fracção de dióxido de carbono no sistema torna menor a concentração de
equilíbrio, xeq, o que permite tornar a sobressaturação máxima.
1.3.4.2. O estado da arte da micronização “SAS” – trabalhos elaborados
Ao longo, essencialmente, da última década, têm sido realizados vários estudos que incidem sobre o
processo de micronização com anti‐solvente supercrítico. A aplicação desta técnica estende‐se, hoje em
dia, a vários tipos de produto, desde explosivos a fármacos, e a investigação que sobre ela recai diz 1 Na presença de uma superfície heterogénea na solução sobressaturada, a energia interfacial aparente decresce, porque a criação de uma interface cristal–partícula é energeticamente favorável, relativamente à criação de uma interface cristal–solução. [59]

15
respeito, principalmente, à influência das variáveis operatórias sobre as características do produto final
e aos solventes utilizados.
No início dos anos noventa, um trabalho de Randolph et al. [8] mostrou que a precipitação de sólidos a
partir de solventes líquidos, com a sobressaturação induzida pela dissolução de dióxido de carbono a
alta pressão, pode ser um processo de cristalização atractivo. Os autores utilizaram como sistema
modelo o β‐caroteno, utilizando como solvente a ciclohexanona e uma mistura de tolueno/butanol, e
verificaram ainda que é possível separar o β‐caroteno dos seus produtos de degradação (óxidos), bem
como purificar o isómero geométrico trans do β‐caroteno.
Recentemente, Kikic et al. [1] estudaram a influência das condições operatórias na precipitação e co‐
precipitação do β‐caroteno com um polímero biodegradável (PVP K30), utilizando como solvente o
clorofórmio.
Cocero & Ferrero [10] estudaram também a micronização de carotenóides pela técnica “SAS”. O β‐
caroteno foi micronizado com sucesso em condições subcríticas, utilizando como solventes o acetato de
etilo e o dicloromentano. Neste trabalho, foi também estudada a solubilidade deste carotenóide na
mistura dióxido de carbono/acetato de etilo.
Martín et al. [38] estudaram a aplicação e a influência das variáveis operatórias da micronização “SAS”
na co‐precipitação do β‐caroteno e da luteína com polietileno glicol (PEG). Este é um polímero solúvel
em água e amplamente utilizado na indústria, devido à sua compatibilidade fisiológica. As vantagens da
sua mistura com carotenóides são o aumento da estabilidade e da taxa de dissolução destes em água. A
micronização dessa mistura permite obter um produto final livre de solventes e com degradação
térmica e oxidação dos carotenóides reduzidas.
Os mesmos autores analisaram a precipitação do ácido mandélico em etil acetato por “SAS” [37], com
vista ao aumento de escala deste processo para aplicação nas indústrias farmacêutica e cosmética. Este
produto pode ser utilizado como anti‐séptico urinário e na síntese orgânica de drogas, corantes e
pesticidas. A influência das variáveis operatórias (pressão, temperatura, concentração inicial e caudais)
nas características do produto final foi avaliada, assim como foi realizada a modelação do processo.
Um outro trabalho de Miguel et al. [42] reporta a micronização do licopeno e a influência das variáveis
operatórias nas características do produto final.
Majerik et al. [34] investigaram a co‐precipitação (“SAS”) do oxeglizatar (uma droga recente, fracamente
solúvel em água, utilizada no combate à diabetes tipo II) com diferentes excipientes e solventes. O seu
estudo incidiu sobre o efeito dos solventes e dos excipientes nas propriedades das formulações sólidas
(grau de cristalinidade, morfologia, pureza polimórfica, conteúdo em solvente residual, cinética de
dissolução e rendimento da precipitação), sendo que se concluiu ser possível a preparação de partículas
de transportadores de drogas através da técnica “SAS”.
Costa et al. [11] estudaram a precipitação de micropartículas de PHBV (co‐precipitados de poli(3‐
hidroxibutirato) com 3‐hidroxivalerato), um co‐polímero que tem vindo a ver aumentadas as suas
aplicações no campo da biomedicina, devido às suas potencialidades na administração controlada de
drogas. A micronização do PHBV foi possível, sendo ainda estudada a influência dos parâmetros

16
operacionais (temperatura, pressão e caudal) sobre a morfologia das partículas, o seu tamanho, e
distribuição de tamanhos.
O grupo de trabalho de Reverchon et al. tem vindo a desenvolver vários estudos nesta área. A
micronização de precursores de supercondutores [58][52], antibióticos [55][54][53] e biopolímeros [57].
A influência dos vários parâmetros do processo e dos solventes tem sido avaliada na morfologia e
tamanho de partícula dos produtos finais.
Tenorio et al. investigaram a influência dos solventes e da pressão na micronização da ampicilina
[71][70]. O trabalho permitiu concluir que a variação do solvente líquido provoca uma grande variação
no tamanho e distribuição de tamanho de partícula, bem como na morfologia do pó obtido.
Adicionalmente, verificou‐se que a variação da pressão operatória permite controlar a forma das
partículas e a sua coalescência.
Hwang et al. [47] realizaram recentemente um estudo no qual avalia a influência da pressão,
temperatura, e solvente utilizado, nas características morfológicas do fluconazol (um agente anti‐
fúngico) recristalizado pelo processo “SAS”. Este grupo de trabalho observou diferenças nas formas
polimórficas do produto causadas pelas alterações das condições experimentais do processo,
concluindo que as características do estado sólido do fluconazol podem ser controladas pela técnica
“SAS”.
1.3.4.3. Variáveis que influenciam a micronização “SAS”
A influência das variáveis operatórias sobre o tamanho de partícula obtido utilizando técnicas de
micronização “SAS” tem sido estudada por vários autores. Entre essas variáveis, a pressão (P) e a
temperatura (T) de trabalho, a concentração da solução inicial (C) e os caudais de CO2 (QCO2) e de
solução (QL) têm um papel fundamental nos mecanismos de precipitação, actuando quer na
hidrodinâmica do sistema, quer na termodinâmica da mistura ternária e, portanto, na força motriz para
a precipitação – a sobressaturação.
Reverchon [51] e Tenorio et al. [70] registaram alguns resultados importantes a este nível, fazendo uma
revisão sobre alguns trabalhos já elaborados e o efeito das variáveis estudadas sobre o produto final. A
Tabela 1.3 resume alguns desses resultados, evidenciando que não existe uma regra geral para a
influência de cada uma das variáveis no processo, o que permite concluir que a micronização com anti‐
solvente supercrítico está ainda longe de ser explicada à luz de uma só disciplina.
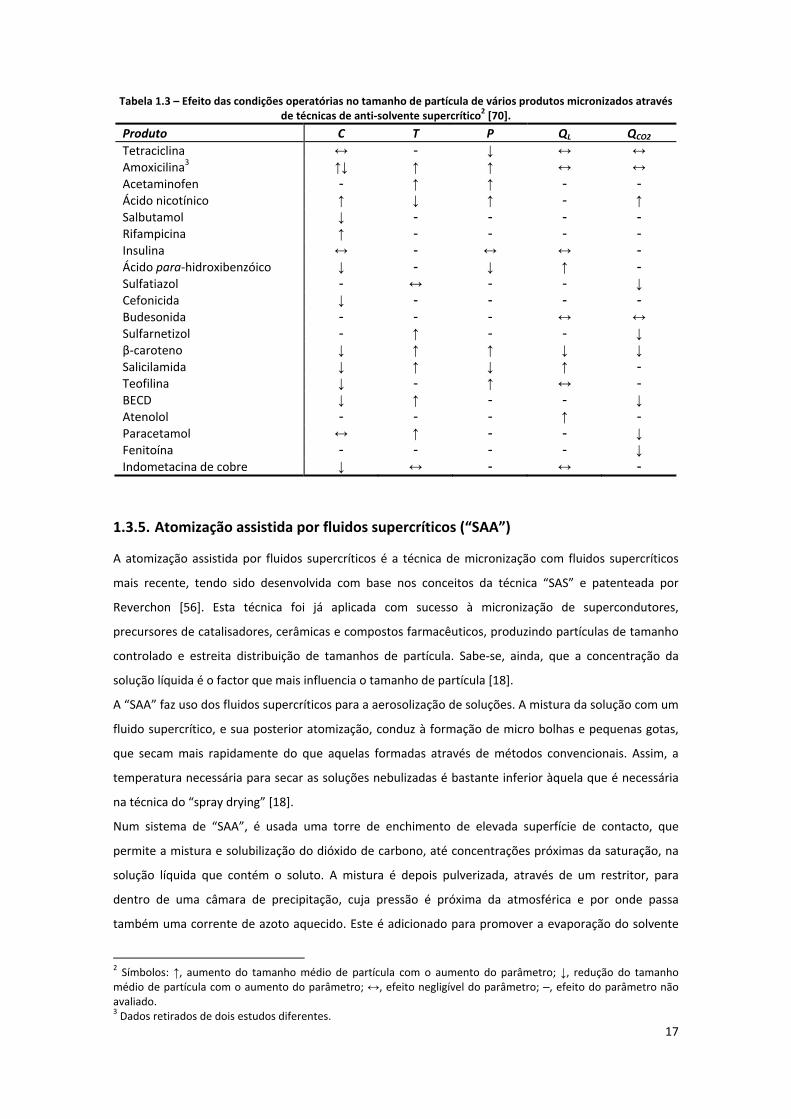
17
Tabela 1.3 – Efeito das condições operatórias no tamanho de partícula de vários produtos micronizados através de técnicas de anti‐solvente supercrítico2 [70].
Produto C T P QL QCO2 Tetraciclina ↔ - ↓ ↔ ↔ Amoxicilina3 ↑↓ ↑ ↑ ↔ ↔ Acetaminofen - ↑ ↑ - - Ácido nicotínico ↑ ↓ ↑ - ↑ Salbutamol ↓ - - - - Rifampicina ↑ - - - - Insulina ↔ - ↔ ↔ - Ácido para‐hidroxibenzóico ↓ - ↓ ↑ - Sulfatiazol - ↔ - - ↓ Cefonicida ↓ - - - - Budesonida - - - ↔ ↔ Sulfarnetizol - ↑ - - ↓ β‐caroteno ↓ ↑ ↑ ↓ ↓ Salicilamida ↓ ↑ ↓ ↑ - Teofilina ↓ - ↑ ↔ - BECD ↓ ↑ - - ↓ Atenolol - - - ↑ - Paracetamol ↔ ↑ - - ↓ Fenitoína - - - - ↓ Indometacina de cobre ↓ ↔ - ↔ -
1.3.5. Atomização assistida por fluidos supercríticos (“SAA”)
A atomização assistida por fluidos supercríticos é a técnica de micronização com fluidos supercríticos
mais recente, tendo sido desenvolvida com base nos conceitos da técnica “SAS” e patenteada por
Reverchon [56]. Esta técnica foi já aplicada com sucesso à micronização de supercondutores,
precursores de catalisadores, cerâmicas e compostos farmacêuticos, produzindo partículas de tamanho
controlado e estreita distribuição de tamanhos de partícula. Sabe‐se, ainda, que a concentração da
solução líquida é o factor que mais influencia o tamanho de partícula [18].
A “SAA” faz uso dos fluidos supercríticos para a aerosolização de soluções. A mistura da solução com um
fluido supercrítico, e sua posterior atomização, conduz à formação de micro bolhas e pequenas gotas,
que secam mais rapidamente do que aquelas formadas através de métodos convencionais. Assim, a
temperatura necessária para secar as soluções nebulizadas é bastante inferior àquela que é necessária
na técnica do “spray drying” [18].
Num sistema de “SAA”, é usada uma torre de enchimento de elevada superfície de contacto, que
permite a mistura e solubilização do dióxido de carbono, até concentrações próximas da saturação, na
solução líquida que contém o soluto. A mistura é depois pulverizada, através de um restritor, para
dentro de uma câmara de precipitação, cuja pressão é próxima da atmosférica e por onde passa
também uma corrente de azoto aquecido. Este é adicionado para promover a evaporação do solvente
2 Símbolos: ↑, aumento do tamanho médio de partícula com o aumento do parâmetro; ↓, redução do tamanho médio de partícula com o aumento do parâmetro; ↔, efeito negligível do parâmetro; –, efeito do parâmetro não avaliado. 3 Dados retirados de dois estudos diferentes.

18
líquido, que pode ser água ou um solvente orgânico, e a consequente formação de pequenas partículas
de soluto.
Neste processo, a solubilização do dióxido de carbono na solução líquida é um dos parâmetros chave
que controlam a eficiência da micronização. A quantidade máxima de dióxido de carbono que pode ser
dissolvida depende do solvente líquido e da temperatura e pressão dentro do dispositivo onde ocorre a
mistura.
A “SAA” já provou ser uma técnica vantajosa na produção de farmacêuticos micronizados, sem que
ocorra degradação química, com distribuições de tamanhos de partícula mais estreitas do que as
obtidas através de métodos convencionais. Contudo, a grande vantagem da “SAA” relativamente às
outras técnicas de micronização que envolvem fluidos supercríticos é a versatilidade do solvente líquido
usado. A possibilidade de utilizar soluções aquosas estende a aplicabilidade das técnicas de
micronização com fluidos supercríticos ao processamento de produtos biológicos como as proteínas.
1.4. Carotenóides
Os carotenóides pertencem a uma família de compostos químicos lipofílicos, os pigmentos, que
absorvem luz na região visível do espectro. A cor produzida por estes compostos deve‐se à presença de
uma estrutura específica de cada molécula, o cromóforo, que capta a energia. A luz que não é absorvida
por esta estrutura é reflectida, o que lhe confere a sua cor.
Os pigmentos estão amplamente distribuídos pelos mais variados domínios da vida, mas são as plantas
os principais produtores, onde se encontram nas folhas, frutos, caules e flores. Os pigmentos existem
também em algumas estruturas animais, como os olhos e a pele, em bactérias e em fungos. A
importância deste tipo de compostos é relevante, na medida em que, sem eles, seriam impossíveis
funções vitais como a fotossíntese, onde têm o seu papel as clorofilas e os carotenóides, ou o transporte
de oxigénio e dióxido de carbono, pela hemoglobina e a mioglobina.
As aplicações dos pigmentos naturais e sintéticos na indústria são vastas e conhecidas em
medicamentos, alimentos, vestuário, mobiliário, cosméticos, entre outros produtos.
O metabolismo dos carotenóides e potenciais funções biológicas e relacionadas com a saúde têm sido
alvo de um interesse generalizado, devido às fortes correlações encontradas entre a ingestão de frutos e
vegetais e a diminuição do risco de doenças crónicas como o cancro e a aterosclerose.
1.4.1. Características estruturais, químicas e físicas
Graças às suas características estruturais, os carotenóides pertencem à classe dos pigmentos que deriva
dos isoprenóides (também designados de terpenóides). Os derivados dos isoprenóides contam com
mais de 23000 compostos e são constantemente identificadas mais moléculas. Dada a sua abundância e
estrutura, têm sido considerados três subgrupos principais: as quinonas, os carotenóides e os iridóides.

19
Em geral, os carotenóides são constituídos por oito unidades isoprenóides (Figura 1.3 a), cuja orientação
é invertida no centro da molécula. No entanto, apesar de derivarem da molécula de isopreno, este não é
o precursor directo dos isoprenóides. Na biossíntese, esse papel é levado a cabo pela sua forma
biologicamente activa, o difosfato de isopentenilo (Figura 1.3 b), que é formado do ácido mevalónico,
via acetato [30].
(a) (b)Figura 1.3 – Estrutura molecular do isopreno (a) e da sua forma biologicamente activa, difosfato de isopentenilo
(b).
A característica mais óbvia das moléculas de carotenóides é a sua longa cadeia poliénica, que pode
conter entre três a quinze ligações duplas conjugadas (cromóforo), e cujo comprimento determina o seu
espectro de absorção e, portanto, a sua cor. Alguns exemplos estão representados na Figura 1.4.
Figura 1.4 – Estrutura molecular de alguns carotenóides.
Nos compostos com uma única ligação dupla conjugada, os comprimentos de onda de absorção estão
na gama do ultravioleta, o que faz com que não sejam corados. Contudo, se uma molécula tem várias
ligações duplas conjugadas, o seu comprimento de onda de máxima absorção desloca‐se para a região
visível do espectro (≈400‐600 nm). Para o olho humano, os carotenóides com sete a quinze ligações

20
duplas conjugadas aparentam uma cor que vai de amarelo a vermelho. Este sistema de electrões π é
ainda influenciado por ligações duplas adicionais, grupos funcionais e diferentes conformações [30].
Todos os carotenóides podem ser considerados derivados do licopeno (C40H56), adquirindo a sua
estrutura própria através de reacções envolvendo hidrogenação, desidrogenação, ciclização, inserção de
oxigénio, migração de ligações duplas, migração de grupos metilo, alongamento ou encurtamento de
cadeias.
Os carotenóides são classificados segundo a sua estrutura química em carotenos (constituídos por
carbono e hidrogénio) e oxicarotenóides ou xantofilas (constituídos por carbono, hidrogénio e oxigénio).
Podem ainda ser classificados, segundo a sua função, como carotenóides primários ou secundários. Os
primeiros agrupam aqueles que são essenciais para a fotossíntese (β‐caroteno, violaxantina e
neoxantina), enquanto os secundários são aqueles que estão localizados nos frutos e nas flores (α‐
caroteno, β‐criptoxantina, zeaxantina, anteraxantina, capsantina, capsorubina).
1.4.2. Distribuição na Natureza
Os carotenóides são o grupo de pigmentos mais amplamente distribuído. A sua presença foi já
identificada em organismos fotossintéticos e não‐fotossintéticos: em plantas superiores, algas, fungos,
bactérias e, pelo menos, em uma espécie de cada forma de vida animal. São responsáveis pela coloração
vermelha, cor‐de‐laranja ou amarela dos frutos, vegetais, fungos, flores e também pássaros, insectos,
crustáceos e trutas. Contudo, apenas os microrganismos e as plantas são capazes de sintetizar
carotenóides de novo. Os carotenóides que existem nos animais são provenientes de uma das duas
fontes anteriores, apesar de poderem ser modificados ao longo do seu metabolismo, de forma a
acumularem‐se nos tecidos.
1.4.2.1. Plantas superiores
Os carotenóides acumulam‐se nos cloroplastos de todas as plantas verdes, numa mistura de α‐ e β‐
caroteno, β‐criptoxantina, luteína, zeaxantina, violaxantina e neoxantina. Estes pigmentos encontram‐se
sob a forma de complexos formados por uma ligação não covalente com proteínas. Nas folhas verdes,
os carotenóides estão livres, não esterificados, e a sua composição depende da planta e das suas
condições de desenvolvimento. Algumas gimnospérmicas acumulam carotenóides pouco vulgares, como
a rodoxantina ou a β‐carotenona, em pequenas gotas de óleo que se encontram fora dos plastos. Nos
tecidos reprodutivos, foram já encontrados carotenóides como a liliaxantina e a crocetina. Também as
flores podem sintetizar carotenóides altamente oxigenados (frequentemente 5,8‐epóxidos), β‐
carotenos, ou carotenóides que são específicos da espécie. Os frutos são considerados ainda mais
surpreendentes do que as flores na síntese de carotenóides, tendo sido descritos mais de setenta
carotenóides que lhes são característicos.

21
1.4.2.2. Algas
Nas algas, os carotenóides encontram‐se nos cloroplastos, em misturas complexas características de
cada classe. A excepção vai para as algas verdes (filo Chlorophyta), que têm tendência a acumular os
pigmentos característicos das plantas superiores.
As algas vermelhas (filo Rhodophyta) possuem α‐ e β‐caroteno e os seus derivados hidroxilados. Nos
dinoflagelados (filo Pyrrophyta), os pigmentos principais são a peridininda, a dinoxantina e a
fucoxantina. As crisófitas (filo Chrysophyta) acumulam epoxi‐carotenóides, e carotenóides alénicos e
acetilénicos, e ainda fucoxantina e diadinoxantina. A eutreptielanona foi identificada em euglenófitas
(filo Euglenophyta). O filo Chloromonadophyta tem como principais carotenóides a diadinoxantina, a
heteroxantina e a vaucheriaxantina. As Chryptophyta são caracterizadas pelos seus carotenóides
acetilénicos, como a aloxantina, a monadoxantina e a crocoxantina. As Phaeophyta são caracterizadas
pelo seu pigmento principal, a fucoxantina.
1.4.2.3. Bactérias
São cerca de oitenta os diferentes carotenóides sintetizados por bactérias fotossintéticas. Estes
carotenóides são caracterizados por ser, na sua maioria, alifáticos, embora nas Chlorobiaceae e
Chloroflexaceae alguns carotenóides tenham anéis aromáticos ou β. Cada espécie possui várias classes
de carotenóides, que se encontram, na sua totalidade, ligados aos complexos de captação de luz ou aos
centros de reacção nos sistemas membranares das células bacterianas.
Nas bactérias não fotossintéticas, os carotenóides podem aparecer esporadicamente e, quando
presentes, têm características únicas. Por exemplo, algumas Staphylococcus acumulam carotenóides C30,
as flavobactérias C45 e C50, enquanto algumas micobactérias acumulam glicosídeos de carotenóides C40.
1.4.2.4. Fungos
A distribuição dos carotenóides pelos fungos, organismos não fotossintéticos, é aparentemente
caprichosa. No entanto, estas espécies acumulam geralmente carotenos e carotenóides mono‐ e bi‐
cíclicos.
1.4.3. Biossíntese
A biossíntese dos carotenóides inicia‐se com a síntese do difosfato de isopentenilo (IPP) envolve a
condensação da hidroxietil tiamina, derivada do piruvato, com o carbono C1 do grupo aldeído do D‐
gliceraldeído‐3‐fosfato (G3P), reacção que dá origem à 1‐desoxi‐D‐xilulose 5‐fosfato (DXP). Esta reacção
é catalisada pela 1‐desoxi‐D‐xilulose 5‐fosfato sintase (DXPS). De seguida, um rearranjo molecular e
subsequente redução da DXP, catalisada pela DXP redutoisomerase, que requer NADPH e Mn2+ como
cofactores, dá origem ao 2‐C‐metil‐D‐eritriol 4‐fosfato (MEP). Este é convertido a IPP e dimetil‐alil

22
pirofosfato (DMAPP) via 4‐(citidina 5’‐difosfo)‐2‐C‐metil‐D‐eritritol (CDP‐ME), 2‐fosfo‐4‐(citidina 5’‐
difosfo)‐2‐C‐metil‐D‐eritritol (CDP‐MEP), 2‐C‐metil‐D‐eritritol‐2,4‐ciclodifosfato e 1‐hidroxi‐2‐metil‐2‐(E)‐
butenil 4‐fosfato (HMBPP). As enzimas envolvidas nestas reacções são a 2‐C‐metil‐D‐eritritol 4‐fosfato
citidil transferase (MCT), 4‐(citidina 5’‐difosfo)‐2‐C‐metil‐D‐eritritol cinase (CMK), 2‐C‐metil‐D‐eritritol
2,4‐ciclodifosfato (MCS) e 1‐hidroxi‐2‐metil‐2‐(E)‐butenil 4‐fosfato sintase (HDS).
A enzima responsável pela conversão de HMBPP numa mistura de IPP e DMAPP é a IPP/DMAPP sintase.
Um esquema da via metabólica que leva à formação do IPP está representado na Figura 1.5.
Figura 1.5 – Formação do difosfato de isopentenilo [19].
O IPP é a unidade básica para a construção dos terpenóides de cadeia mais longa. Contudo, o IPP não é
suficientemente reactivo para iniciar as reacções de condensação, pelo que o primeiro passo é a sua
isomerização a dimetil‐alil pirofosfato (DMAPP), reacção que é catalisada pela IPP isomerase, que utiliza
como cofactor um ião metálico divalente. O passo seguinte é a condensação do IPP e do DMAPP,
formando geranil pirofosfato (GPP). De seguida, duas moléculas de IPP são adicionadas ao GPP,
obtendo‐se o geranilgeranil pirofosfato (GGPP), por catálise com a GGPP sintase, enzima que necessita
de dois iões, Mg2+ e Mn2+, por centro catalítico. O intermediário destas reacções é o farnesil difosfato
(FPP).
O primeiro passo específico para a síntese dos carotenóides é a condensação de duas moléculas de
GGPP, para obter o cis‐fitoeno (C40), tendo a reacção o prefitoeno pirofosfato (PPPP) como
intermediário. No tomate e na pimenta, foi verificado que uma enzima (fitoeno sintase) catalisa ambos
os passos. A biossíntese do fitoeno está representada na Figura 1.6.

23
Figura 1.6 – Biossíntese do fitoeno a partir do IPP e do DMAPP [19].
O fitoeno sofre quatro reacções de dessaturação, para se obter fitoflueno, ζ‐caroteno, neurosporeno e,
finalmente, licopeno, nas quais se sabe que o ferro tem um papel fundamental na cadeia de transporte
electrónico. A biossíntese dos outros carotenóides dá‐se a partir deste ponto, através de reacções cuja
molécula de partida é o licopeno. A enzima responsável pela dessaturação do fitoeno a ζ‐caroteno é a
fitoeno dessaturase (PDS), sendo a ζ‐caroteno dessaturase (ZDS) responsável pela dessaturação do ζ‐
caroteno a licopeno, via neurosporeno.
1.4.4. Fontes alimentares
Os carotenóides que se encontram na dieta humana derivam essencialmente de plantas cultivadas,
onde os carotenóides estão localizados nas raízes, folhas, galhos, sementes, frutos e flores. Cerca de
sessenta carotenóides diferentes foram já identificados em frutos e vegetais consumidos pelo Homem
[19]. Em menor proporção, os carotenóides podem também ser encontrados em ovos, aves domésticas
ou peixe, animais que se alimentam tipicamente de plantas ou algas. As quantidades típicas de
carotenóides em plantas cultivadas são mostradas na Tabela 1.4.
No sangue humano em circulação, os sete carotenóides predominantes são o β‐caroteno, o licopeno, a
luteína, o α‐caroteno, a α‐criptoxantina, a β‐criptoxantina e a zeaxantina. Cerca de 25 a 75% do β‐
caroteno percorre o intestino e é expelido com as fezes, mas o restante é absorvido inalterado, circula
através do corpo e é armazenado no tecido adiposo, devido à sua lipossolubilidade, o que dá às
gorduras a sua cor amarela. A eliminação ocorre depois, lentamente, através da via fecal. Carotenóides
diferentes são acumulados em diferentes órgãos: o licopeno e o β‐caroteno na próstata, e a luteína e a
zeaxantina na mácula do olho, por exemplo [45].
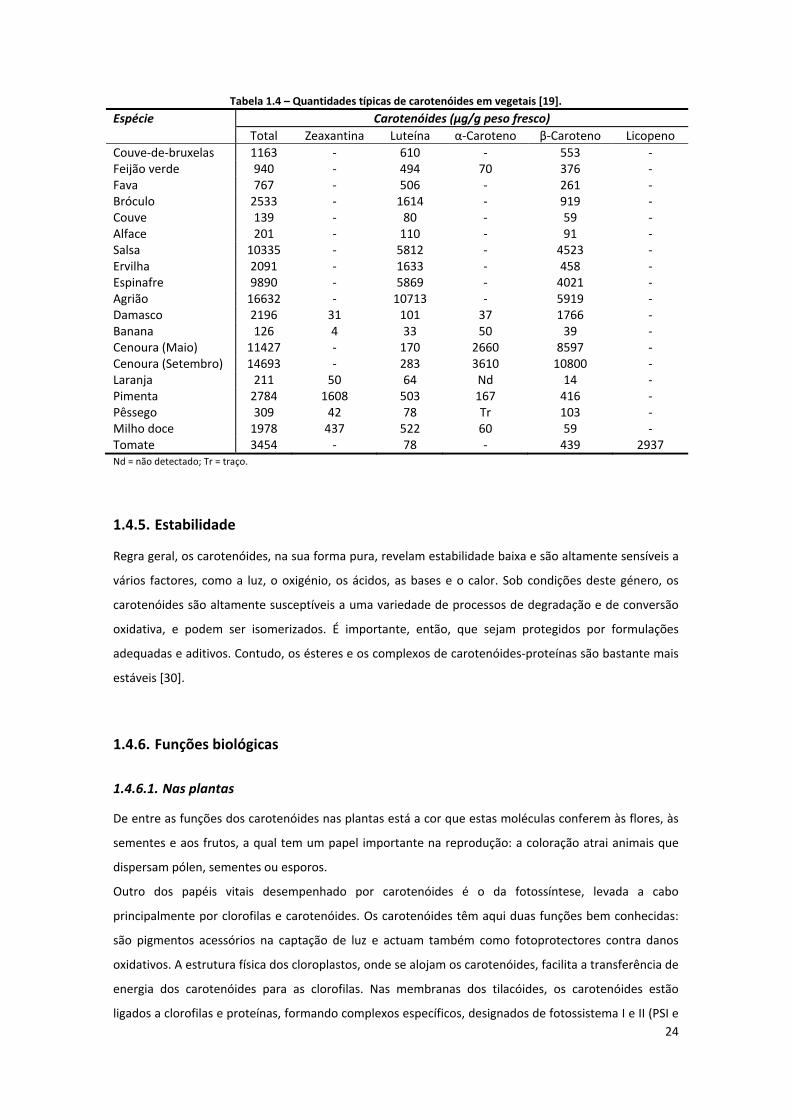
24
Tabela 1.4 – Quantidades típicas de carotenóides em vegetais [19].
Espécie Carotenóides (µg/g peso fresco)Total Zeaxantina Luteína α‐Caroteno β‐Caroteno Licopeno
Couve‐de‐bruxelas 1163 ‐ 610 ‐ 553 ‐ Feijão verde 940 ‐ 494 70 376 ‐ Fava 767 ‐ 506 ‐ 261 ‐ Bróculo 2533 ‐ 1614 ‐ 919 ‐ Couve 139 ‐ 80 ‐ 59 ‐ Alface 201 ‐ 110 ‐ 91 ‐ Salsa 10335 ‐ 5812 ‐ 4523 ‐ Ervilha 2091 ‐ 1633 ‐ 458 ‐ Espinafre 9890 ‐ 5869 ‐ 4021 ‐ Agrião 16632 ‐ 10713 ‐ 5919 ‐ Damasco 2196 31 101 37 1766 ‐ Banana 126 4 33 50 39 ‐ Cenoura (Maio) 11427 ‐ 170 2660 8597 ‐ Cenoura (Setembro) 14693 ‐ 283 3610 10800 ‐ Laranja 211 50 64 Nd 14 ‐ Pimenta 2784 1608 503 167 416 ‐ Pêssego 309 42 78 Tr 103 ‐ Milho doce 1978 437 522 60 59 ‐ Tomate 3454 ‐ 78 ‐ 439 2937Nd = não detectado; Tr = traço.
1.4.5. Estabilidade
Regra geral, os carotenóides, na sua forma pura, revelam estabilidade baixa e são altamente sensíveis a
vários factores, como a luz, o oxigénio, os ácidos, as bases e o calor. Sob condições deste género, os
carotenóides são altamente susceptíveis a uma variedade de processos de degradação e de conversão
oxidativa, e podem ser isomerizados. É importante, então, que sejam protegidos por formulações
adequadas e aditivos. Contudo, os ésteres e os complexos de carotenóides‐proteínas são bastante mais
estáveis [30].
1.4.6. Funções biológicas
1.4.6.1. Nas plantas
De entre as funções dos carotenóides nas plantas está a cor que estas moléculas conferem às flores, às
sementes e aos frutos, a qual tem um papel importante na reprodução: a coloração atrai animais que
dispersam pólen, sementes ou esporos.
Outro dos papéis vitais desempenhado por carotenóides é o da fotossíntese, levada a cabo
principalmente por clorofilas e carotenóides. Os carotenóides têm aqui duas funções bem conhecidas:
são pigmentos acessórios na captação de luz e actuam também como fotoprotectores contra danos
oxidativos. A estrutura física dos cloroplastos, onde se alojam os carotenóides, facilita a transferência de
energia dos carotenóides para as clorofilas. Nas membranas dos tilacóides, os carotenóides estão
ligados a clorofilas e proteínas, formando complexos específicos, designados de fotossistema I e II (PSI e

25
PSII, respectivamente), que desempenham funções primárias na fotossíntese. O carotenóide mais
abundante no PSI é o β‐caroteno, sendo a luteína mais abundante no PSII.
O ciclo da xantofila desempenha também um papel importante no mecanismo de protecção das plantas
contra os danos provocados pela luz. Quando as folhas estão expostas a uma iluminação muito forte, os
grupos epoxi‐xantofila são removidos da violaxantina, para inicialmente formar a anteraxantina e a
zeaxantina. O nível de carotenóides do ciclo da xantofila (violaxantina, anteraxantina e zeaxantina) é
aumentado nas folhas expostas ao sol, fenómeno que é particularmente importante na dissipação de
parte da energia absorvida, para protecção do aparelho fotossintético.
1.4.6.2. No ser humano
As primeiras correlações entre o elevado consumo de carotenóides e benefícios para a saúde surgiram
na literatura na década de 1970. Dietas ricas em frutos e vegetais eram associadas a reduzidas
incidências de cancro e doenças coronárias, verificando‐se também o inverso.
Ao longo dos anos, têm sido realizados inúmeros estudos que evidenciam os benefícios dos
carotenóides para o ser humano. Foi já observado, entre outras propriedades, que os carotenóides
modulam o metabolismo do citocromo P450 (β‐caroteno, criptoxantina, luteína); inibem o metabolismo
do ácido araquidónico (β‐caroteno); actuam como antioxidantes e aniquiladores de radicais livres e
espécies reactivas (astaxantina, cantaxantina, α‐caroteno, β‐caroteno, crocetina, criptoxantina, luteína,
licopeno, zeaxantina); modulam funções imunitárias (astaxantina, cantaxantina, β‐caroteno); induzem a
diferenciação e a comunicação intercelular (astaxantina, cantaxantina, α‐caroteno, β‐caroteno, luteína,
licopeno); inibem a instabilidade cromossómica (cantaxantina, β‐caroteno); e inibem alterações
bioquímicas associadas à proliferação celular (α‐caroteno, β‐caroteno). O β‐caroteno pode ainda
influenciar a apoptose, levando à morte celular, ao invés da sua proliferação [45].
Pro‐vitamina A
A mais bem estabelecida função dos carotenóides é a de pro‐vitamina A. Esta está restringida a cerca de
cinquenta e três carotenóides com anéis β nas suas extremidades, tais como o β‐caroteno, a zeaxantina
e a β‐criptoxantina. Quando ingeridos, estes carotenóides são convertidos em retinal por uma 15‐15’‐
desoxigenase intestinal. A vitamina A na dieta pode provir de fontes animais, sob a forma de ésteres de
retinil, retinol e retinal, ou de fontes vegetais, sob a forma de carotenóides. Os carotenóides pro‐
vitamina A nos vegetais e nos frutos atingem a actividade de vitamina A quando são convertidos, no
corpo, em retinol. Este pode inibir a proliferação e induzir a diferenciação de células epiteliais, através
de ligações a receptores nucleares e subsequente modificação da actividade dos genes [45].
Os carotenóides pro‐vitamina A na dieta podem ainda prevenir doenças como a xeroftalmia [19],
doença que se refere em conjunto a todas as manifestações oculares da deficiência de vitamina A. O
primeiro sinal da carência neste nutriente é a cegueira nocturna, que pode progredir a danos estruturais

26
no olho, como a secura das membranas e da córnea, podendo eventualmente resultar no
enfraquecimento da visão ou na cegueira irreversível.
A vitamina A é indispensável para o normal funcionamento do sistema imunitário, da visão, para a
manutenção epitelial, segregação mucosa, resistência a infecções e reprodução. No processo da
degeneração macular, o retinol está relacionado com a indução de uma cascata de genes que permite a
fagocitose da célula danificada na retina, processo que é crítico para a sobrevivência do fotoreceptor.
Adicionalmente, está estabelecido que os retinóides afectam muitos processos biológicos, como a
proliferação celular, a diferenciação e a morfogénese [14].
Antioxidante
Os carotenóides são também bem conhecidos pela sua actividade antioxidante e está estabelecido que
a sua estrutura tem aqui uma forte influência. Os carotenóides têm assim a capacidade de actuar como
aniquiladores do oxigénio molecular singleto, funcionando como protectores das células e dos
organismos contra a foto‐oxidação, evitando a presença de radicais livres e de espécies excitadas que
podem causar danos irreversíveis ao nível do DNA e dos lípidos. Estas propriedades dos carotenóides
fazem deles uma potencial arma contra o cancro e outras doenças que se acredita serem despontadas
por radicais livres, como a aterosclerose, as cataratas, a degeneração macular relacionada com a idade e
a esclerose múltipla [19]. Estudos epidemiológicos que examinam o risco de doença e a aquisição de
carotenóides específicos têm sugerido uma correlação inversa entre os níveis de α‐tocoferol e de
carotenóides e a incidência de doenças coronárias [61].
O stresse oxidativo tem sido implicado na patofisiologia de inflamações crónicas e no cancro, assim
como em doenças como a de Alzheimer, Parkinson, esclerose lateral amiotrófica, ou arteriosclerose. Foi
já demonstrado que o licopeno e o β‐caroteno intracelulares são capazes de proteger as células contra
danos no DNA causados por peroxinitritos formados a partir da geração simultânea de óxido nítrico e do
radical superóxido. Estes carotenóides são também inibidores potentes de quebras na cadeia simples de
DNA plasmídico [44].
O transporte de carotenóides até aos tecidos dá‐se através de lipoproteínas, essencialmente
lipoproteínas de baixa densidade. Devido às suas características antioxidantes, é possível que a função
dos carotenóides seja, em parte, proteger as lipoproteínas da oxidação. Esta protecção poderá ser
importante no desenvolvimento da aterosclerose, após ter sido posta a hipótese de que o processo
desta doença envolve a oxidação das lipoproteínas de baixa densidade [61].
Este tipo de descobertas permitem explicar a razão pela qual os frutos e os vegetais estão muitas vezes
ligados a baixos riscos de desenvolvimento de processos patológicos como o cancro, a inflamação
crónica e as doenças cardiovasculares e neurológicas. Em combinação com vários outros mecanismos,
as propriedades antioxidantes dos carotenóides podem proteger as células contra os efeitos adversos
do stresse oxidativo [44].
Os carotenóides reagem contra os radicais livres através de mais do que um mecanismo. Podem
fornecer electrões em falta aos radicais livres de outras moléculas ou formar aductos com esses radicais,

27
por exemplo. Em ambos os casos, é a riqueza em electrões nativa dos carotenóides que os torna
atractivos para os radicais, permitindo a protecção de lípidos, proteínas e DNA, dos danos provocados
pelos radicais. Como consequência, há um interesse crescente no uso e na medida da capacidade
antioxidante em preparações alimentares e farmacêuticas e em estudos clínicos. O interesse é
principalmente devido ao papel das espécies de oxigénio reactivas no processo de envelhecimento e na
patogénese de muitas doenças nas quais aquelas espécies estão envolvidas. Muitos estudos têm
mostrado que estas espécies de oxigénio reactivas, incluindo radicais livres de oxigénio, são factores
importantes na etiologia de doenças degenerativas, incluindo algumas hepatopatias e danos
importantes noutros orgãos. Estes radicais livres são também responsáveis pelo ataque aos ácidos
gordos insaturados das membranas biológicas, o que resulta na peroxidação lipídica e na desnaturação
de proteínas e do DNA, causando uma série de alterações deteriorativas nos sistemas biológicos,
levando à inactivação celular [43].
Anti‐cancerígeno
Culturas de células in vitro mostram que os carotenóides inibem a proliferação celular, a transformação,
a formação de micronúcleos, e a expressão de certos genes. Estas propriedades são consistentes com o
efeito protector contra a carcinogénese. Vários estudos têm sido realizados, sugerindo relações entre o
consumo elevado de carotenóides e o menor risco de cancro do pulmão, da mama, da próstata e do
recto [19]. Os carotenóides de maior relevância neste campo são o β‐caroteno e o licopeno, o primeiro
associado ao pulmão e à mama e o segundo à próstata e ao recto. Contudo, nem todos os estudos nesta
área são consistentes entre si, revelando um misto de mecanismos ainda desconhecido e uma dúvida
permanente acerca da verdadeira actividade anticancerígena dos carotenóides [6].
1.5. β‐caroteno
De todos os carotenóides, o β‐caroteno é o de actividade biológica mais relevante, sendo a sua função
mais conhecida a de pro‐vitamina A. O β‐caroteno possui uma coloração que vai desde o amarelo até ao
vermelho forte, dependendo do seu grau de pureza, fonte e localização, ocorre naturalmente em muitas
plantas, nomeadamente vegetais e frutos. Contudo, as concentrações mais elevadas de β‐caroteno são,
de longe, as encontradas na microalga halotolerante Dunaliella salina, atingindo até 100 g/kg de peso
seco [31]. O β‐caroteno é também o carotenóide mais importante em termos de biodisponibilidade e
ocorrência na natureza. Aparentemente, todas as amostras de plantas carotenogénicas comestíveis
analisadas até à data contêm o β‐caroteno como constituinte, principal ou não [17].
1.5.1. Química
Se o aldeído retinal (C20, H18, O) for representado por R=O, então o retinol será H‐R‐OH, o ácido
retinóico será H‐R‐OOH, e o β‐caroteno com todas as ligações trans será R=R. Como os outros

28
carotenóides, o β‐caroteno é um isoprenóide biossintetisado através da ligação das extremidades finais
(cadeia linear) de duas moléculas C20. Assim, o β‐caroteno é baseado num esqueleto de quarenta
átomos de carbono, com onze ligações duplas conjugadas C=C. Este sistema de ligações C‐C duplas e
simples alternadas forma o cromóforo, com electrões π deslocalizados ao longo da cadeia poliénica,
gerando a sua distinta forma molecular, reactividade química e propriedades de absorção de luz. Esta
ocorre na gama visível de baixa energia do espectro, que confere aos frutos e plantas as suas cores
amarela, laranja, vermelha e verde brilhantes. As extremidades da molécula do β‐caroteno são anéis
monoinsaturados de seis átomos de carbono (Figura 1.7).
Figura 1.7 – Estrutura molecular do β‐caroteno.
O β‐caroteno pode ocorrer sob a forma de vários estereoisómeros, sendo o mais estável a forma em
que todas as ligações são trans. Contudo, as formas cis são significantemente mais lipossolúveis,
podendo ser absorvidas de forma melhor pelo corpo humano. Estima‐se que as formas cis apresentem
uma actividade de pro‐vitamina A de menos de 50% da forma completamente trans. No entanto,
estudos recentes levados a cabo com o isómero nomeado 9‐cis‐β‐caroteno revelaram que este pode ser
isomerisado quase completamente ao β‐caroteno com todas as ligações trans no intestino delgado do
homem. Isto implica que o 9‐cis‐β‐caroteno é tão bom como o β‐caroteno de configuração trans, em
termos de fonte de vitamina A.
1.5.2. Biossíntese
A biossíntese do β‐caroteno dá‐se por ciclização do licopeno, cuja biossíntese foi já descrita (secção
1.4.3), por acção da β‐ciclase (LCY‐b). A ciclização é limitada à formação de um anel de seis membros nas
duas extremidades do precursor acíclico (licopeno). A formação dos grupos cíclicos nas extremidades é
iniciada por ataque no carbono C2 da ligação dupla do terminal C‐1,2, que está isolada da cadeia
poliénica. Podem ocorrer dois tipos de anéis, α e β, os quais diferem da posição da ligação dupla no anel
ciclohexeno e dependem da natureza da enzima ciclase (Figura 1.8). Estudos sugerem que o substrato
para a ciclização é o todo‐trans‐licopeno e o cofactor necessário pelas ciclases é o NADPH.

29
Figura 1.8 – Ciclização do licopeno (formação do β‐caroteno) [19].
1.5.3. Funções biológicas
Para o ser humano, a função mais importante do β‐caroteno é a de precursor da vitamina A.
Teoricamente, a clivagem enzimática de uma molécula de β‐caroteno pode render duas de retinol,
enquanto a clivagem de outros dois carotenóides pro‐vitamina A importantes, o α‐caroteno e a β‐
criptoxantina, rendem apenas uma molécula de retinol cada. Um dos benefícios da administração de β‐
caroteno é que a sua conversão a vitamina A só se dá quando esta é requerida pelo corpo, o que se
opõe à potencial toxicidade associada com uma overdose de vitamina A [17].
Para lá da sua função como precursor da síntese de vitamina A pelo corpo, a actividade antioxidante do
β‐caroteno é também bem conhecida. O β‐caroteno é um tipo de antioxidante pouco usual, que actua
por aniquilação dos radicais, sendo a sua actividade óptima à pressão de oxigénio nos tecidos.
Evidências epidemiológicas têm mostrado que o β‐caroteno é capaz de prevenir o cancro em vários
órgãos, como o pulmão, o estômago, o colo do útero, o pâncreas, o cólon, o recto, a mama, a próstata e
o ovário, através da sua actividade antioxidante. Outras funções deste carotenóide são a sua influência
na comunicação intracelular, resposta imunitária, transformação neoplástica e controlo do crescimento
[43]. Os resultados de um estudo in vitro revelam ainda evidências para a morte de uma linha celular
leucémica de linfoblasto T provocada pelo β‐caroteno [6].
1.6. Objectivos do trabalho
As propriedades do β‐caroteno tornam‐no numa substância de grande potencial de aplicação nas
indústrias farmacêutica e alimentar, como corante, antioxidante ou precursor da vitamina A, entre
outros potenciais efeitos benéficos. Contudo, a elevada lipofilicidade deste carotenóide faz com que

30
seja altamente insolúvel em preparações aquosas, pelo que é importante a procura de uma solução que
ultrapasse esta limitação.
A redução de tamanho de partícula é, como foi já referido, uma das formas de aumentar a taxa de
dissolução de substâncias de baixa solubilidade perante a preparação de suspensões dessas mesmas
substâncias.
O presente trabalho tem por objectivo principal a micronização do β‐caroteno sintético, utilizando a
técnica de micronização por anti‐solvente supercrítico. O solvente escolhido é o tetrahidrofurano (THF),
devido à solubilidade relativamente elevada do β‐caroteno neste solvente (10 mg/mL).
A recolha de resultados de solubilidade é importante para vários processos da tecnologia supercrítica,
como a cromatografia, a extracção e a precipitação. Assim, simultaneamente, medidas de solubilidade
do β‐caroteno na mistura supercrítica CO2/THF serão possíveis e estudar‐se‐á a variação da solubilidade
com as variáveis do processo – temperatura, pressão e composição da mistura.
O estudo da morfologia e da dimensão das partículas obtidas aquando do processamento a diferentes
pressões será realizado, de forma a tentar estabelecer uma relação entre o tamanho de partícula e a
pressão de trabalho. Serão realizados ensaios acima e abaixo da pressão crítica, para determinar as
diferenças existentes entre uma e outra condição.
Por fim, será efectuado um ensaio de precipitação de um extracto de β‐caroteno produzido por via
biológica. A pureza do produto final será analisada e a micronização “SAS” como processo de separação
e purificação será avaliada.

31
2. PARTE EXPERIMENTAL: MATERIAIS, APARELHOS E TÉCNICAS
2.1. Materiais
Todos estudos de micronização foram realizados com β‐caroteno sintético (isómero trans) Tipo I da
Sigma (grau de pureza ≈95% UV), à excepção de um ensaio, em que se utilizou um extracto natural β‐
caroteno produzido e fornecido pela BioTrend.
O solvente utilizado nestes estudos, bem como em todas as análises necessárias, foi o tetrahidrofurano
(THF) pró‐análise da Merck, com grau de pureza ≥99,8% e estabilizado com BHT (hidroxitolueno
butilado).
O dióxido de carbono (pureza de 99,998%) foi fornecido pela Air Liquide.
2.2. Micronização supercrítica
2.2.1. Descrição geral do aparelho
Os ensaios de micronização supercrítica foram realizados num aparelho construído para esse fim, no
âmbito do trabalho de doutoramento do Eng. Miguel Cardoso, sob a orientação do Professor António
Palavra, no Instituto Superior Técnico. O aparelho, representado na Figura 2.1, está esquematizado na
Figura 2.2 e está descrito na literatura [69]. Mostra‐se também em maior pormenor algumas partes
deste aparelho (Figura 2.3, Figura 2.4 e Figura 2.5).
Figura 2.1 – Aparelho de micronização supercrítica: aspecto geral.

32
Figura 2.2 – Diagrama esquemático do aparelho de micronização supercrítica. B1 e B2, bombas de recirculação; BP1 e BP2, “backpressure” ou regulador de pressão; BT, banho termostático; C, condensador; CG, contador de gás; DR, disco de ruptura; G, garrafa de CO2 com tubo prolongador; P, indicador de pressão; Q, medidor de caudal; S, bureta de solução; T, indicador de temperatura; V, válvula; VM, válvula micrométrica; VP, vaso de
precipitação; VS, vaso de recolha de solvente.
O aparelho é constituído por uma célula (vaso de precipitação) de alta pressão de aço inoxidável e
volume igual a 300 mL (VP), imersa num banho termostático de água (BT), à qual é alimentada uma
corrente de gás (CO2) e outra de solução líquida com a substância a micronizar. O fundo da célula está
equipado com um filtro de aço inoxidável, que permite a recolha do pó micronizado, enquanto a mistura
orgânica passa através daquele. Ambas as correntes são também mantidas à temperatura da célula,
através da passagem pelo banho. A temperatura deste é mantida usando um termóstato (Tectron Bio,
modelo Selecta) e verificada por um termómetro. A pressão e temperatura na célula são medidas,
respectivamente, por um manómetro e por um termopar, ambos digitais (Omega Engineering, Inc.).
(a) (b)Figura 2.3 – Aparelho de micronização supercrítica: banho termostático de água (a); célula de alta pressão (b).

33
A corrente de gás provém de uma garrafa de dióxido de carbono (G) e é refrigerada num banho de gelo
(C), para que a alimentação à respectiva bomba (Gilson) (B1) se mantenha no estado líquido. A pressão
a jusante desta bomba, medida por um manómetro analógico (Omega Engineering, Inc.) é controlada
por um sistema de regulação de pressão (Tescom) (BP1), que permite manter a pressão constante,
através da “recirculação” do fluido para a bomba, caso a pressão na célula seja superior à pretendida.
A corrente de líquido, por sua vez, provém de uma bureta (S), onde é injectada a solução. À saída da
bureta, a bomba de líquido (Gilson) (B2) pressuriza a solução para a célula, sendo a pressão à saída da
bomba medida por um manómetro analógico (Omega Engineering, Inc.). A solução é injectada também
no topo da célula, mas num local distinto do CO2, através de um restritor de aço inoxidável (125 µm de
diâmetro e 1 cm de comprimento).
(a) (b) (c)Figura 2.4 – Aparelho de micronização supercrítica (secção de bombagem): CO2 (a); solução (b); aspecto geral (c).
A corrente de saída da célula passa por uma válvula micrométrica (Hoke) (VM) aquecida através de uma
fita de aquecimento. Esta válvula permite controlar o caudal de saída da célula. A expansão da mistura
supercrítica e separação do solvente e do CO2 dá‐se no vaso de recolha do solvente (VS), cuja pressão,
medida por um manómetro analógico (Omega Engineering, Inc.), é regulada por um segundo sistema de
controlo de pressão (Tescom) (BP2).
(a) (b)Figura 2.5 – Aparelho de micronização supercrítica: recolha de solvente (a); aspecto geral (b).

34
A corrente gasosa que abandona o vaso de recolha de solvente passa por um medidor de caudal
(Omega Engineering, Inc.) (Q) e por um contador de gás (AMC) (CG). No final de cada ensaio, o solvente
é recolhido através da abertura da válvula do vaso (V1).
2.2.2. Técnica experimental
Todos os ensaios foram conduzidos obedecendo a um procedimento semelhante.
O primeiro passo consiste no bombeamento de CO2 (caudal na bomba de 25 mL/min) até se atingir a
pressão desejada, para a qual é regulada a válvula BP1. Simultaneamente, é estabelecida a pressão (30
bar) no vaso de expansão da mistura solvente/CO2, por regulação da válvula BP2.
A injecção de uma quantidade previamente calculada de solvente orgânico (exemplo deste cálculo no
Apêndice A) é efectuada após se estabilizar a pressão, de forma a garantir o estado estacionário.
Quando o solvente orgânico dentro da célula atinge a concentração pretendida, estabelece‐se o caudal
de saída (leitura no medidor de caudal igual a 354), por regulação da válvula micrométrica (VM).
Após a injecção da solução e do solvente, a passagem de CO2 pela célula durante cerca de 75 minutos
(cálculo no Apêndice B) permite que praticamente todo o solvente abandone a célula, por
arrastamento, restando apenas o pó seco.
No final da secagem, o sistema é despressurizado, a célula aberta para recolha do pó micronizado e o
solvente obtido é retirado do vaso de expansão.
2.3. Análise dos produtos
2.3.1. Espectrofotometria
Os espectros de varrimento e a medição da absorvância do solvente recolhido no final de cada ensaio
foram realizados com recurso a um espectrofotómetro da marca Shimadzu, modelo UV PharmaSpec –
1700, utilizando o programa UV Probe Versão 2.10, da Shimadzu.
Foi previamente realizado um espectro de varrimento, para o qual foi seleccionado o comprimento de
onda de máxima absorção, sendo feita de seguida uma recta de calibração (lei de Lambert‐Beer) a esse
mesmo comprimento de onda (458 nm), de forma a relacionar a absorvância com a concentração das
soluções medidas (espectro de varrimento e recta de calibração nos Apêndices C e D, respectivamente).
2.3.2. Cromatografia líquida de alta resolução (HPLC)
A pureza dos produtos obtidos foi analisada com recurso a um cromatógrafo de líquido da Shimadzu,
modelo LC.2010CHT, com detector UV/visível e uma coluna de fase reversa Chromolith Performance RP‐
4 correspondente a cerca de 11,704 g/min de CO2.

35
18 encapped (diâmetro 3 mm e comprimento 100 mm). Os cromatogramas foram obtidos com o
programa LC Real Time Analysis (Shimadzu).
2.3.3. “Dynamic light scattering”
O tamanho de partícula e distribuição de tamanho de partícula foram analisados por “dynamic light
scattering” (“DLS”) usando um equipamento da Brookhaven Instruments (goniómetro BI‐200SM e
correlacionador BI‐9000AT), com laser de He‐Ne (632,8 nm, 35 mW) da Spectra Physics (modelo 127)
como fonte de luz. Os resultados foram analisados usando o software BI‐ZP da Brookhaven.
2.3.4. Microscopia electrónica de varrimento
As amostras de pó analisadas foram observadas por um microscópio electrónico de varrimento da
Philips (XL 30 FEG). As amostras foram cobertas com 250 Å de ouro, usando um equipamento de
revestimento por pulverização da Jeol (modelo JFC‐1100).
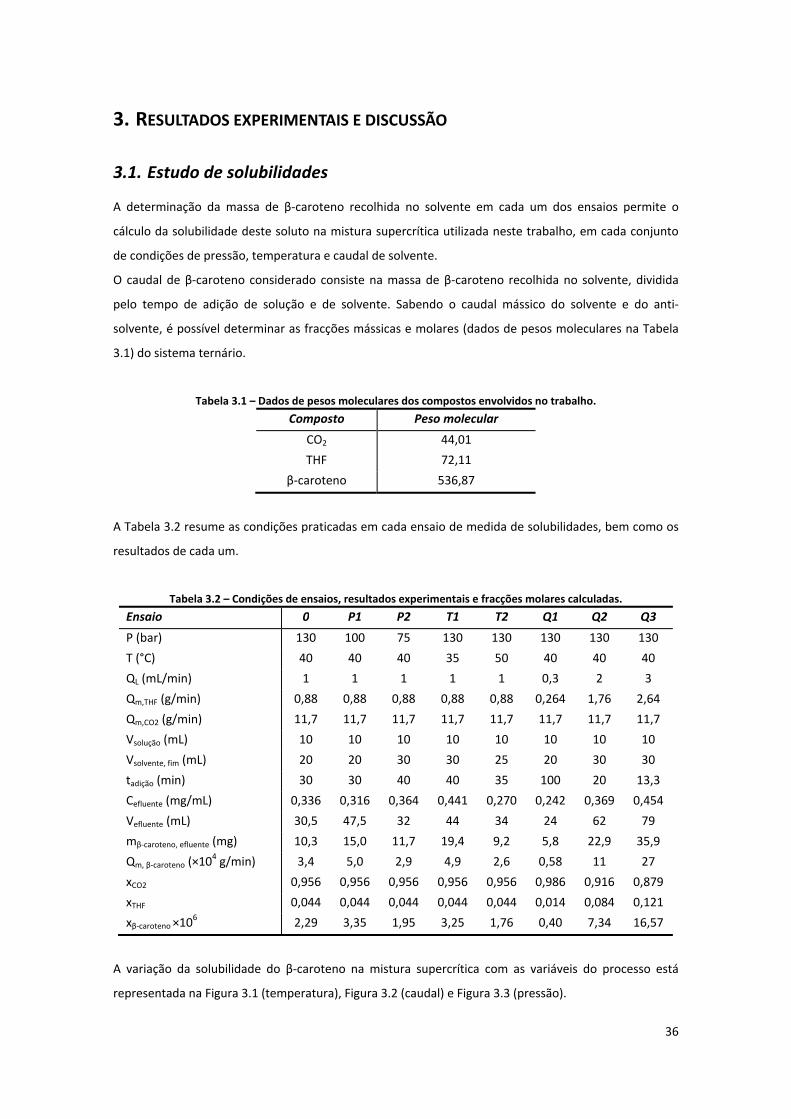
36
3. RESULTADOS EXPERIMENTAIS E DISCUSSÃO
3.1. Estudo de solubilidades
A determinação da massa de β‐caroteno recolhida no solvente em cada um dos ensaios permite o
cálculo da solubilidade deste soluto na mistura supercrítica utilizada neste trabalho, em cada conjunto
de condições de pressão, temperatura e caudal de solvente.
O caudal de β‐caroteno considerado consiste na massa de β‐caroteno recolhida no solvente, dividida
pelo tempo de adição de solução e de solvente. Sabendo o caudal mássico do solvente e do anti‐
solvente, é possível determinar as fracções mássicas e molares (dados de pesos moleculares na Tabela
3.1) do sistema ternário.
Tabela 3.1 – Dados de pesos moleculares dos compostos envolvidos no trabalho.
Composto Peso molecular
CO2 44,01
THF 72,11
β‐caroteno 536,87
A Tabela 3.2 resume as condições praticadas em cada ensaio de medida de solubilidades, bem como os
resultados de cada um.
Tabela 3.2 – Condições de ensaios, resultados experimentais e fracções molares calculadas.
Ensaio 0 P1 P2 T1 T2 Q1 Q2 Q3
P (bar) 130 100 75 130 130 130 130 130
T (°C) 40 40 40 35 50 40 40 40
QL (mL/min) 1 1 1 1 1 0,3 2 3
Qm,THF (g/min) 0,88 0,88 0,88 0,88 0,88 0,264 1,76 2,64
Qm,CO2 (g/min) 11,7 11,7 11,7 11,7 11,7 11,7 11,7 11,7
Vsolução (mL) 10 10 10 10 10 10 10 10
Vsolvente, fim (mL) 20 20 30 30 25 20 30 30
tadição (min) 30 30 40 40 35 100 20 13,3
Cefluente (mg/mL) 0,336 0,316 0,364 0,441 0,270 0,242 0,369 0,454
Vefluente (mL) 30,5 47,5 32 44 34 24 62 79
mβ‐caroteno, efluente (mg) 10,3 15,0 11,7 19,4 9,2 5,8 22,9 35,9
Qm, β‐caroteno (×104 g/min) 3,4 5,0 2,9 4,9 2,6 0,58 11 27
xCO2 0,956 0,956 0,956 0,956 0,956 0,986 0,916 0,879
xTHF 0,044 0,044 0,044 0,044 0,044 0,014 0,084 0,121
xβ‐caroteno ×106 2,29 3,35 1,95 3,25 1,76 0,40 7,34 16,57
A variação da solubilidade do β‐caroteno na mistura supercrítica com as variáveis do processo está
representada na Figura 3.1 (temperatura), Figura 3.2 (caudal) e Figura 3.3 (pressão).
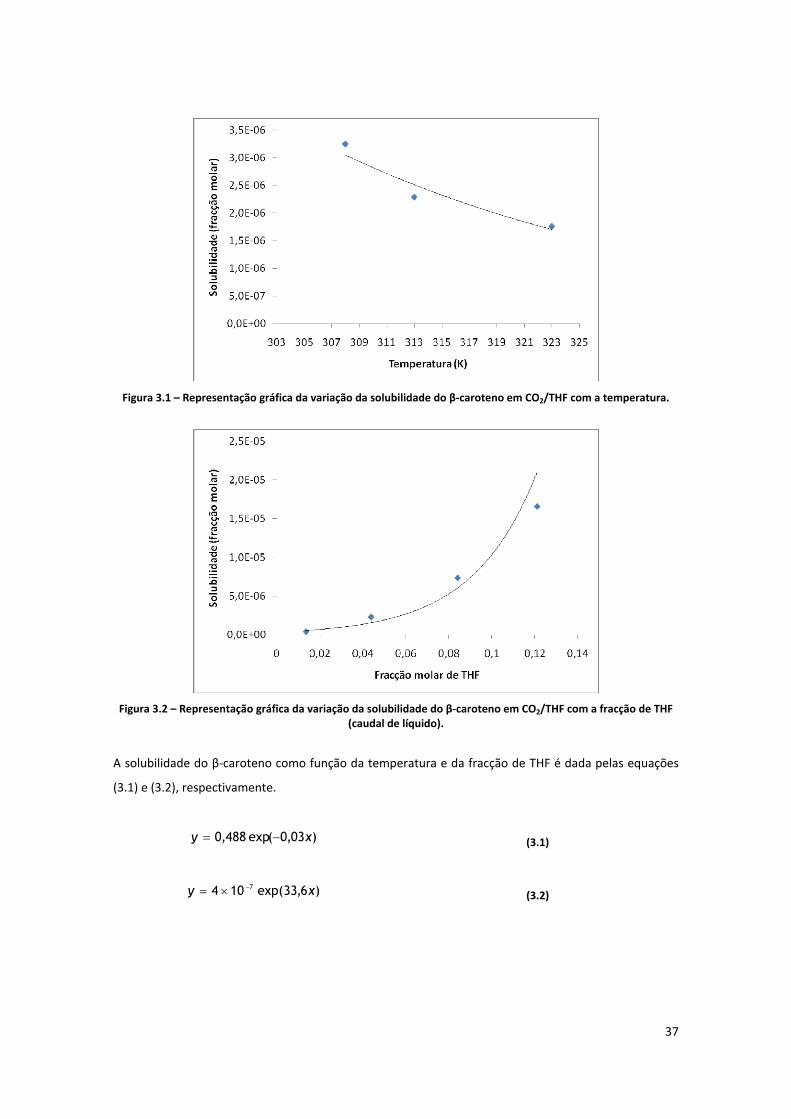
37
Figura 3.1 – Representação gráfica da variação da solubilidade do β‐caroteno em CO2/THF com a temperatura.
Figura 3.2 – Representação gráfica da variação da solubilidade do β‐caroteno em CO2/THF com a fracção de THF (caudal de líquido).
A solubilidade do β‐caroteno como função da temperatura e da fracção de THF é dada pelas equações
(3.1) e (3.2), respectivamente.
)03,0(exp488,0 xy −= (3.1)
)6,33exp(104 7 xy −×= (3.2)
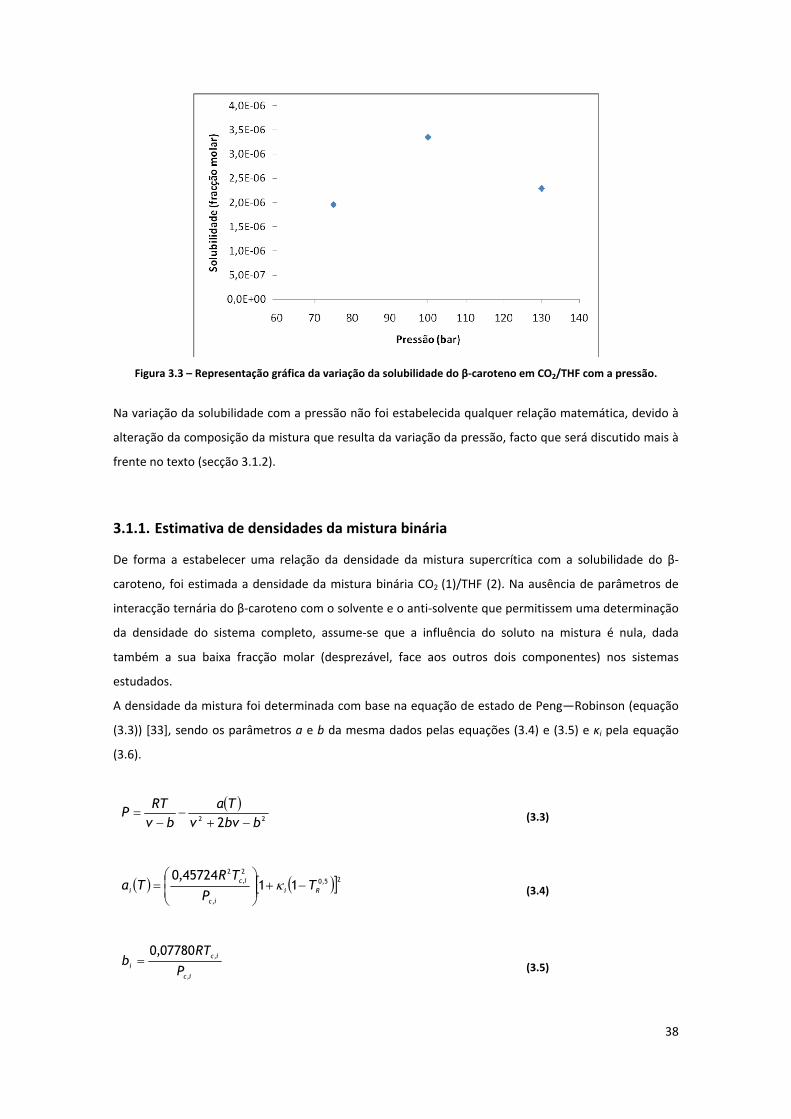
38
Figura 3.3 – Representação gráfica da variação da solubilidade do β‐caroteno em CO2/THF com a pressão.
Na variação da solubilidade com a pressão não foi estabelecida qualquer relação matemática, devido à
alteração da composição da mistura que resulta da variação da pressão, facto que será discutido mais à
frente no texto (secção 3.1.2).
3.1.1. Estimativa de densidades da mistura binária
De forma a estabelecer uma relação da densidade da mistura supercrítica com a solubilidade do β‐
caroteno, foi estimada a densidade da mistura binária CO2 (1)/THF (2). Na ausência de parâmetros de
interacção ternária do β‐caroteno com o solvente e o anti‐solvente que permitissem uma determinação
da densidade do sistema completo, assume‐se que a influência do soluto na mistura é nula, dada
também a sua baixa fracção molar (desprezável, face aos outros dois componentes) nos sistemas
estudados.
A densidade da mistura foi determinada com base na equação de estado de Peng—Robinson (equação
(3.3)) [33], sendo os parâmetros a e b da mesma dados pelas equações (3.4) e (3.5) e κi pela equação
(3.6).
( )22 2 bbvv
Tabv
RTP
−+−
−= (3.3)
( ) ( )[ ]25,0
,
2,
2
1145724,0
Ri
ic
ici T
P
TRTa −+⎟
⎟⎠
⎞⎜⎜⎝
⎛= κ (3.4)
ic
ic
i P
RTb
,
,07780,0= (3.5)

39
226992,054226,137464,0 iii ωωκ −+= (3.6)
Para a mistura binária, foram adoptadas as regras de mistura clássicas de van der Waals (equações (3.7)
e (3.8)).
( ) ( )iji j
jiji kaaxxa −= ∑∑ 121
(3.7)
( )iji j
ji
ji lbb
xxb −⎟⎟⎠
⎞⎜⎜⎝
⎛ += ∑∑ 1
2 (3.8)
Os parâmetros dos compostos puros são dados pela Tabela 3.3 e os parâmetros de interacção,
provenientes da literatura [33], tomam o valor de k12 = 0,01678 e l12 = ‐0,02395.
Tabela 3.3 – Parâmetros dos compostos puros [33].
Composto Tc (K) Pc (bar) ω
CO2 (1) 304,21 73,8 0,225
THF (2) 540,1 51,9 0,226
A composição da mistura binária foi determinada com base na pressão e na quantidade de THF
adicionada à célula antes da micronização, tendo em conta a densidade do dióxido de carbono em cada
conjunto de condições de trabalho, o volume da célula (300 mL) e a densidade do THF (0,88 g/mL).
Nos ensaios em que QL = 1 mL/min, tomou‐se um volume médio (20 mL) de THF a adicionar à célula
para se atingir o estado estacionário antes de se dar início à injecção da solução. Nos ensaios em que se
variou o caudal, o volume de THF adicionado foi recalculado.
Tabela 3.4 – Composições da mistura binária nas diferentes condições de trabalho.
T (K) P (bar) ρCO2 (g/mL) QL (mL/min) xTHF xCO2
308 130 0,78570 1 0,044 0,956
313 75 0,23153 1 0,134 0,866
313 100 0,62861 1 0,054 0,946
313 130 0,74304 0,3 0,014 0,986
313 130 0,74304 1 0,044 0,956
313 130 0,74304 2 0,084 0,916
313 130 0,74304 3 0,121 0,879
323 130 0,63612 1 0,053 0,947
Com os valores da composição da mistura binária registados na Tabela 3.4, é possível calcular os
parâmetros da equação de Peng—Robinson, cuja única incógnita passa a ser o volume molar, v.
Determinado este valor, e tomando um peso molecular médio da mistura (ponderado pelas fracções
molares dos seus dois constituintes), calcula‐se a respectiva densidade (Tabela 3.5).
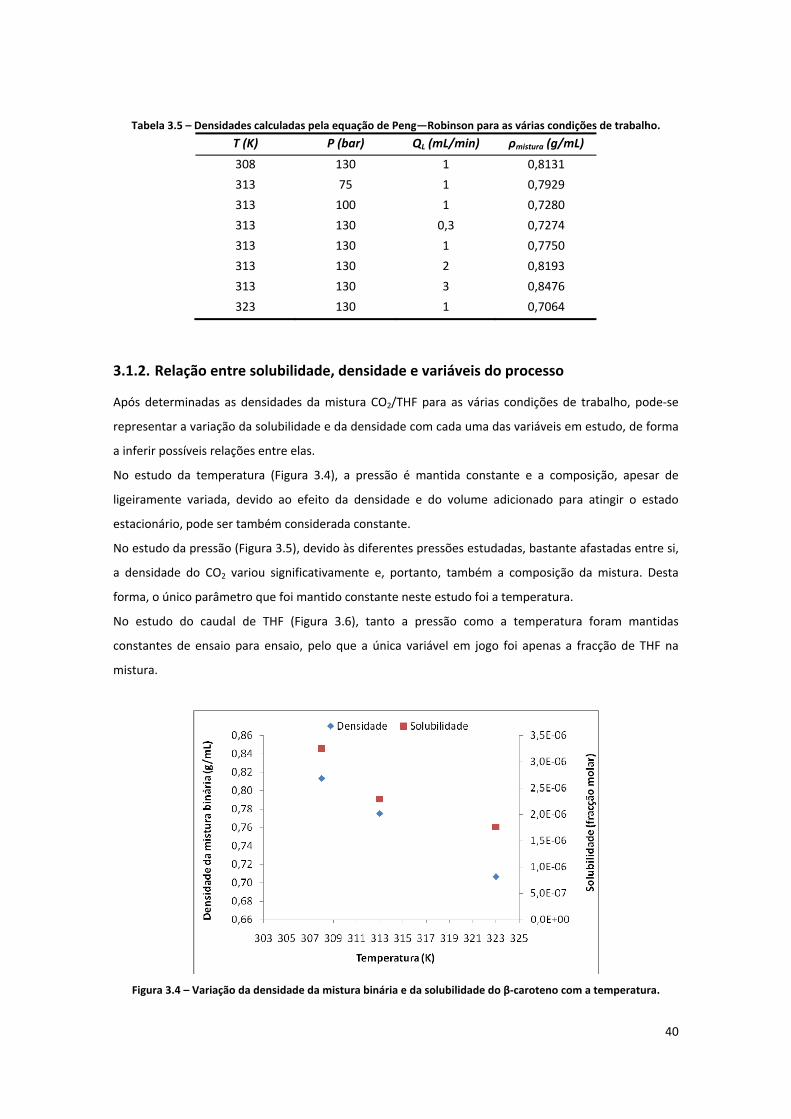
40
Tabela 3.5 – Densidades calculadas pela equação de Peng—Robinson para as várias condições de trabalho.
T (K) P (bar) QL (mL/min) ρmistura (g/mL)
308 130 1 0,8131
313 75 1 0,7929
313 100 1 0,7280
313 130 0,3 0,7274
313 130 1 0,7750
313 130 2 0,8193
313 130 3 0,8476
323 130 1 0,7064
3.1.2. Relação entre solubilidade, densidade e variáveis do processo
Após determinadas as densidades da mistura CO2/THF para as várias condições de trabalho, pode‐se
representar a variação da solubilidade e da densidade com cada uma das variáveis em estudo, de forma
a inferir possíveis relações entre elas.
No estudo da temperatura (Figura 3.4), a pressão é mantida constante e a composição, apesar de
ligeiramente variada, devido ao efeito da densidade e do volume adicionado para atingir o estado
estacionário, pode ser também considerada constante.
No estudo da pressão (Figura 3.5), devido às diferentes pressões estudadas, bastante afastadas entre si,
a densidade do CO2 variou significativamente e, portanto, também a composição da mistura. Desta
forma, o único parâmetro que foi mantido constante neste estudo foi a temperatura.
No estudo do caudal de THF (Figura 3.6), tanto a pressão como a temperatura foram mantidas
constantes de ensaio para ensaio, pelo que a única variável em jogo foi apenas a fracção de THF na
mistura.
Figura 3.4 – Variação da densidade da mistura binária e da solubilidade do β‐caroteno com a temperatura.
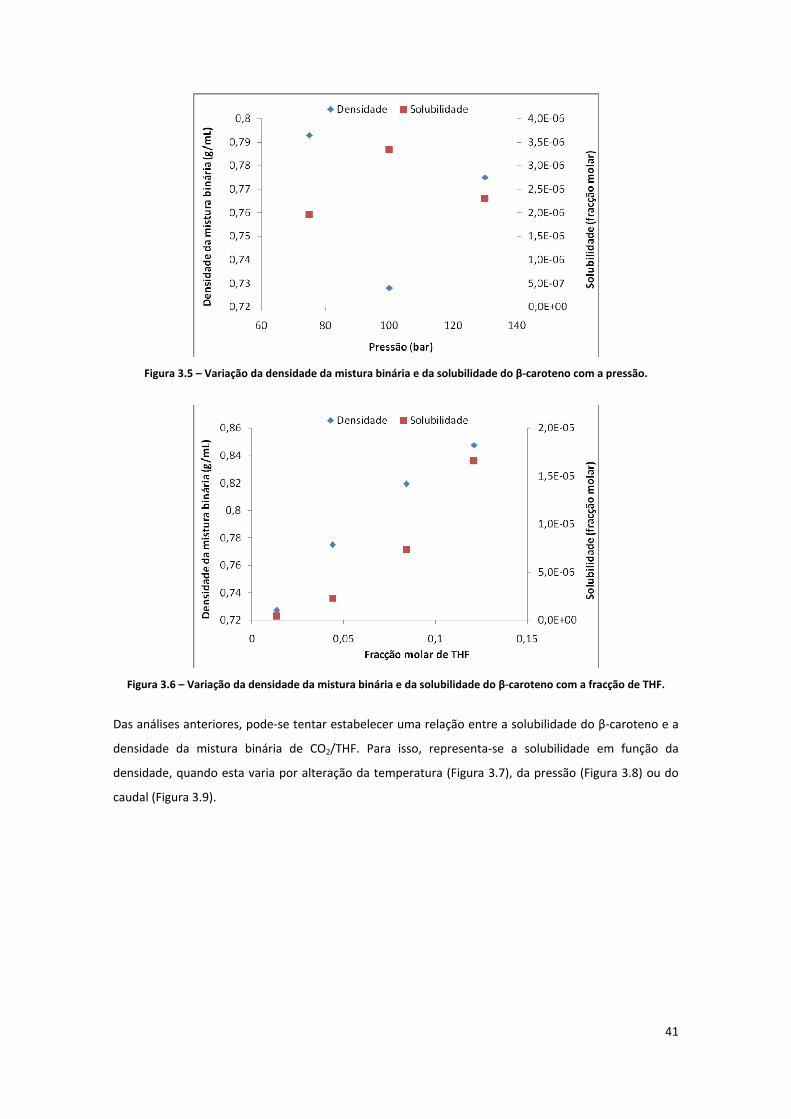
41
Figura 3.5 – Variação da densidade da mistura binária e da solubilidade do β‐caroteno com a pressão.
Figura 3.6 – Variação da densidade da mistura binária e da solubilidade do β‐caroteno com a fracção de THF.
Das análises anteriores, pode‐se tentar estabelecer uma relação entre a solubilidade do β‐caroteno e a
densidade da mistura binária de CO2/THF. Para isso, representa‐se a solubilidade em função da
densidade, quando esta varia por alteração da temperatura (Figura 3.7), da pressão (Figura 3.8) ou do
caudal (Figura 3.9).
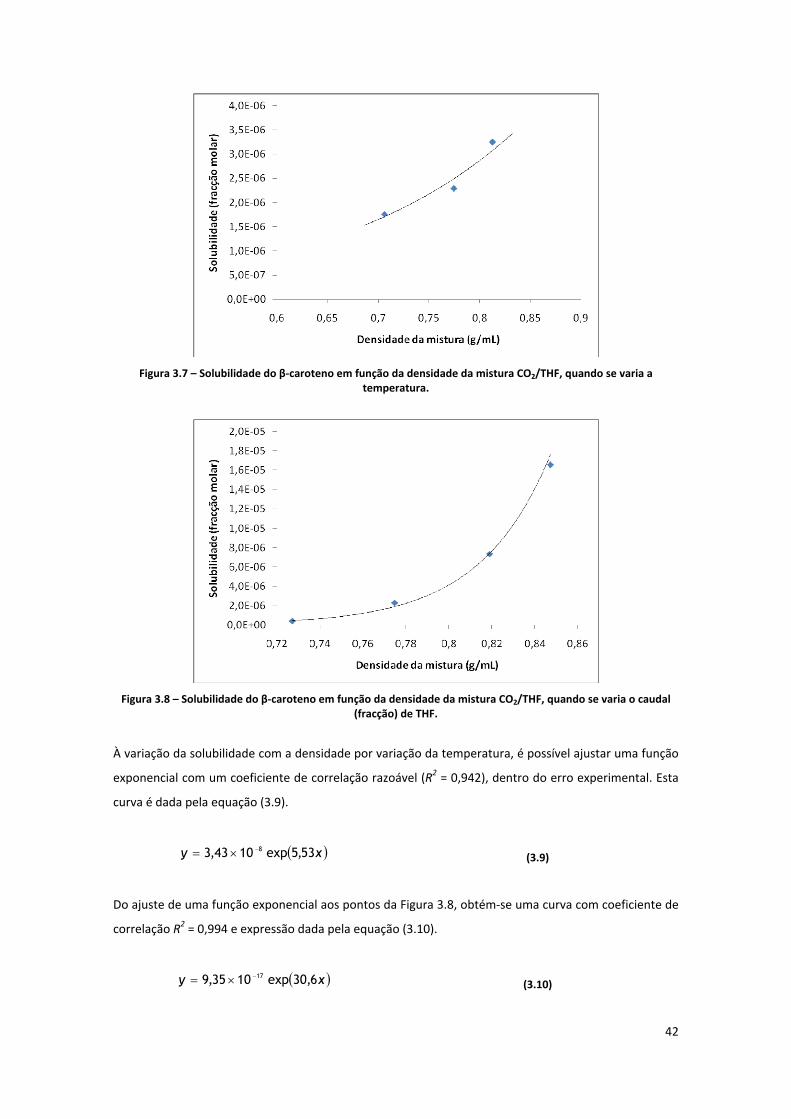
42
Figura 3.7 – Solubilidade do β‐caroteno em função da densidade da mistura CO2/THF, quando se varia a temperatura.
Figura 3.8 – Solubilidade do β‐caroteno em função da densidade da mistura CO2/THF, quando se varia o caudal (fracção) de THF.
À variação da solubilidade com a densidade por variação da temperatura, é possível ajustar uma função
exponencial com um coeficiente de correlação razoável (R2 = 0,942), dentro do erro experimental. Esta
curva é dada pela equação (3.9).
( )xy 53,5exp1043,3 8−×= (3.9)
Do ajuste de uma função exponencial aos pontos da Figura 3.8, obtém‐se uma curva com coeficiente de
correlação R2 = 0,994 e expressão dada pela equação (3.10).
( )xy 6,30exp1035,9 17−×= (3.10)
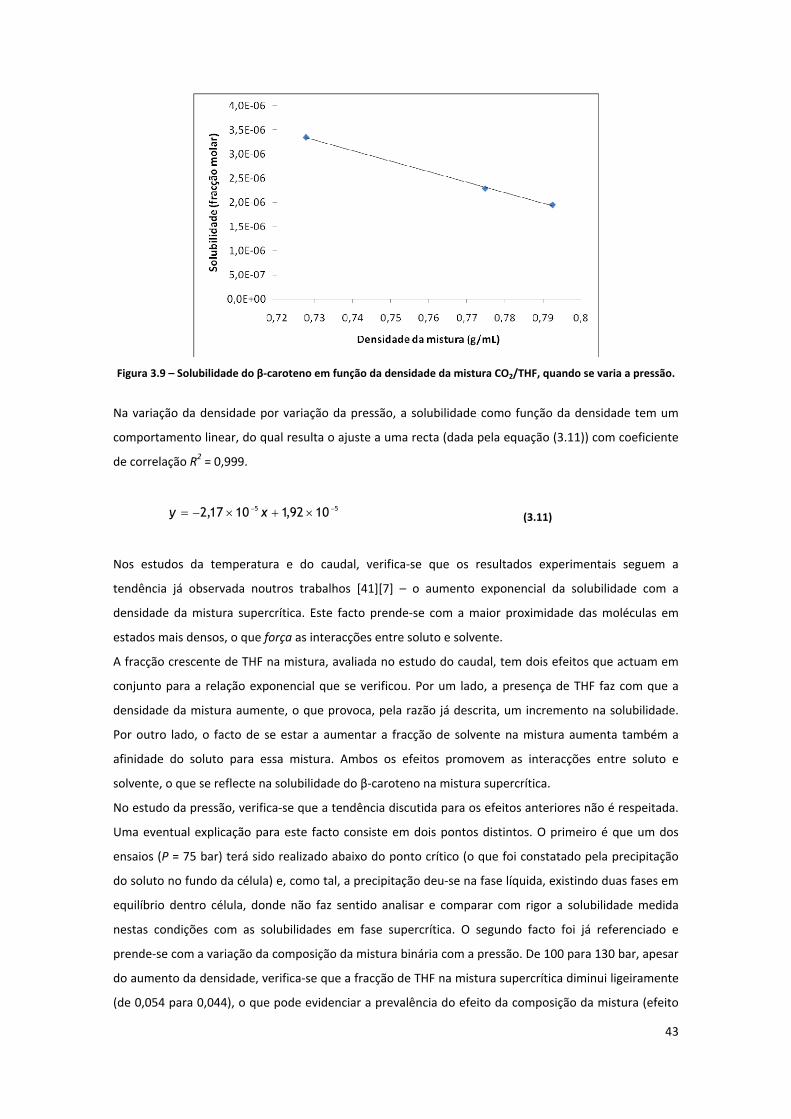
43
Figura 3.9 – Solubilidade do β‐caroteno em função da densidade da mistura CO2/THF, quando se varia a pressão.
Na variação da densidade por variação da pressão, a solubilidade como função da densidade tem um
comportamento linear, do qual resulta o ajuste a uma recta (dada pela equação (3.11)) com coeficiente
de correlação R2 = 0,999.
55 1092,11017,2 −− ×+×−= xy (3.11)
Nos estudos da temperatura e do caudal, verifica‐se que os resultados experimentais seguem a
tendência já observada noutros trabalhos [41][7] – o aumento exponencial da solubilidade com a
densidade da mistura supercrítica. Este facto prende‐se com a maior proximidade das moléculas em
estados mais densos, o que força as interacções entre soluto e solvente.
A fracção crescente de THF na mistura, avaliada no estudo do caudal, tem dois efeitos que actuam em
conjunto para a relação exponencial que se verificou. Por um lado, a presença de THF faz com que a
densidade da mistura aumente, o que provoca, pela razão já descrita, um incremento na solubilidade.
Por outro lado, o facto de se estar a aumentar a fracção de solvente na mistura aumenta também a
afinidade do soluto para essa mistura. Ambos os efeitos promovem as interacções entre soluto e
solvente, o que se reflecte na solubilidade do β‐caroteno na mistura supercrítica.
No estudo da pressão, verifica‐se que a tendência discutida para os efeitos anteriores não é respeitada.
Uma eventual explicação para este facto consiste em dois pontos distintos. O primeiro é que um dos
ensaios (P = 75 bar) terá sido realizado abaixo do ponto crítico (o que foi constatado pela precipitação
do soluto no fundo da célula) e, como tal, a precipitação deu‐se na fase líquida, existindo duas fases em
equilíbrio dentro célula, donde não faz sentido analisar e comparar com rigor a solubilidade medida
nestas condições com as solubilidades em fase supercrítica. O segundo facto foi já referenciado e
prende‐se com a variação da composição da mistura binária com a pressão. De 100 para 130 bar, apesar
do aumento da densidade, verifica‐se que a fracção de THF na mistura supercrítica diminui ligeiramente
(de 0,054 para 0,044), o que pode evidenciar a prevalência do efeito da composição da mistura (efeito
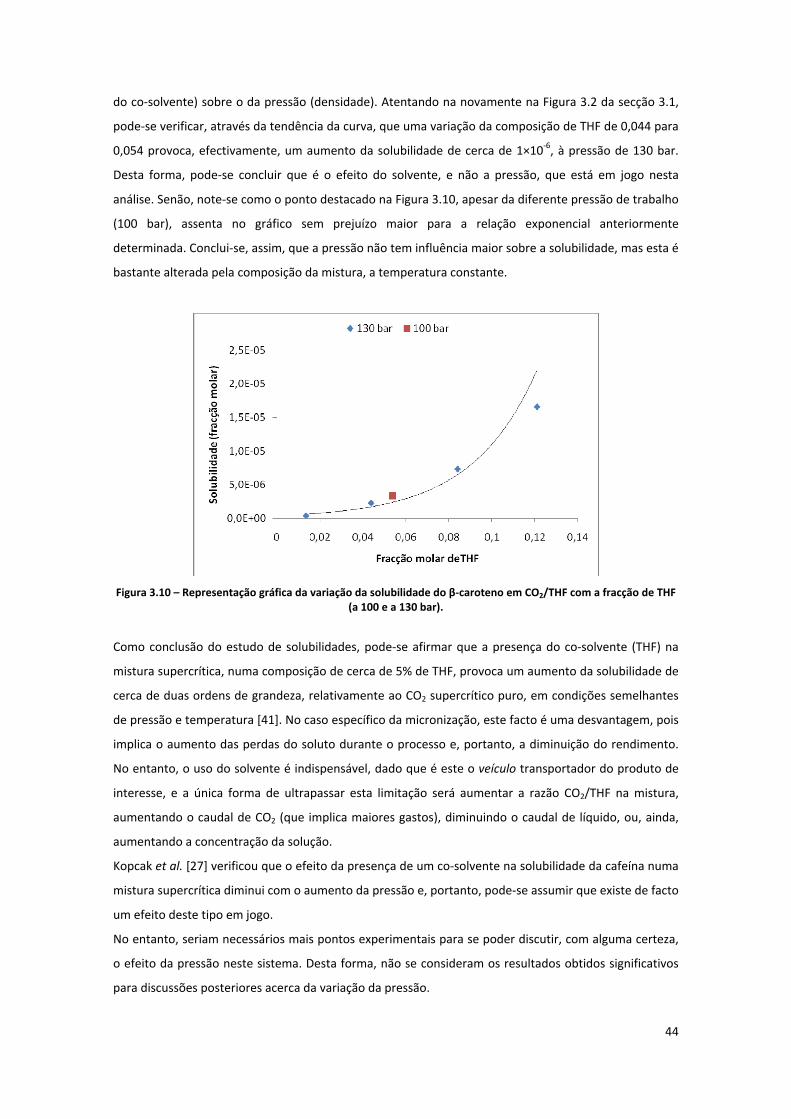
44
do co‐solvente) sobre o da pressão (densidade). Atentando na novamente na Figura 3.2 da secção 3.1,
pode‐se verificar, através da tendência da curva, que uma variação da composição de THF de 0,044 para
0,054 provoca, efectivamente, um aumento da solubilidade de cerca de 1×10‐6, à pressão de 130 bar.
Desta forma, pode‐se concluir que é o efeito do solvente, e não a pressão, que está em jogo nesta
análise. Senão, note‐se como o ponto destacado na Figura 3.10, apesar da diferente pressão de trabalho
(100 bar), assenta no gráfico sem prejuízo maior para a relação exponencial anteriormente
determinada. Conclui‐se, assim, que a pressão não tem influência maior sobre a solubilidade, mas esta é
bastante alterada pela composição da mistura, a temperatura constante.
Figura 3.10 – Representação gráfica da variação da solubilidade do β‐caroteno em CO2/THF com a fracção de THF (a 100 e a 130 bar).
Como conclusão do estudo de solubilidades, pode‐se afirmar que a presença do co‐solvente (THF) na
mistura supercrítica, numa composição de cerca de 5% de THF, provoca um aumento da solubilidade de
cerca de duas ordens de grandeza, relativamente ao CO2 supercrítico puro, em condições semelhantes
de pressão e temperatura [41]. No caso específico da micronização, este facto é uma desvantagem, pois
implica o aumento das perdas do soluto durante o processo e, portanto, a diminuição do rendimento.
No entanto, o uso do solvente é indispensável, dado que é este o veículo transportador do produto de
interesse, e a única forma de ultrapassar esta limitação será aumentar a razão CO2/THF na mistura,
aumentando o caudal de CO2 (que implica maiores gastos), diminuindo o caudal de líquido, ou, ainda,
aumentando a concentração da solução.
Kopcak et al. [27] verificou que o efeito da presença de um co‐solvente na solubilidade da cafeína numa
mistura supercrítica diminui com o aumento da pressão e, portanto, pode‐se assumir que existe de facto
um efeito deste tipo em jogo.
No entanto, seriam necessários mais pontos experimentais para se poder discutir, com alguma certeza,
o efeito da pressão neste sistema. Desta forma, não se consideram os resultados obtidos significativos
para discussões posteriores acerca da variação da pressão.

45
3.2. Estudo da precipitação do β‐caroteno sintético
Os ensaios realizados para o estudo da micronização do β‐caroteno sintético foram conduzidos de forma
a avaliar o tipo de precipitação obtida acima e abaixo do ponto crítico, bem como analisar o tamanho de
partícula obtido quando se varia a pressão de trabalho. O comprimento do restritor através do qual é
injectada a solução também foi variado, com o objectivo de averiguar a sua influência no local de
precipitação do produto dentro da célula.
As condições dos ensaios são as descritas na Tabela 3.2 da secção 3.1. Foi ainda realizado um ensaio a
65 bar (P3) e um outro com as condições do ensaio 0 e com o restritor de 10 cm (N1).
3.2.1. Análise macroscópica dos cristais
Aquando da recolha do pó micronizado da célula de alta pressão, verificou‐se que, à excepção dos
ensaios P2 e N1, o produto se encontrava espalhado por toda a célula. Nestes ensaios acima do ponto
crítico da mistura, como sugere o diagrama de fases da mistura binária (Apêndice E), existe dentro da
célula uma única fase (supercrítica), em que a sobressaturação da solução ocorre em todos os
elementos de volume da célula, dado que a adição da solução e do CO2 se dão no topo da mesma. A
cristalização deu‐se a partir das paredes da célula, que devem ter servido como centro de nucleação
e/ou de crescimento dos cristais (Figura 3.11).
No ensaio N1, a precipitação ocorreu a meio da célula, formando um anel à volta do local onde se
situava a extremidade do restritor.
(a) (b)Figura 3.11 – Recolha do β‐caroteno micronizado acima do ponto crítico: copo da célula de alta pressão (a);
tampa da célula de alta pressão (b).
Segundo o diagrama de fase da mistura binária CO2/THF, os ensaios P2 (75 bar) e P3 (65 bar) foram
realizados abaixo do ponto crítico da mistura. Este facto pôde ainda ser constatado através da forma
como ocorreu a micronização do β‐caroteno no ensaio P2. Evidenciando a existência de duas fases em
equilíbrio, e a sua afinidade para a mais densa das duas, o produto surge acumulado no fundo da célula
(Figura 3.12). A sua precipitação ocorre na fase líquida, segundo um mecanismo de expansão

46
volumétrica do solvente e subsequente sobressaturação da solução, por dissolução de uma grande
quantidade de CO2.
No ensaio P3 não ocorreu a precipitação do β‐caroteno dentro da célula, mas foi observada uma massa
muito reduzida de cristais no lado exterior do filtro, os quais deverão ter sido formados por flutuações
de concentração locais. Assume‐se, portanto, que a 65 bar não ocorreu a precipitação do β‐caroteno,
sendo o limite mínimo de pressão para que se dê a micronização a praticada no ensaio P2 (75 bar).
(a) (b)Figura 3.12 – Recolha do β‐caroteno micronizado abaixo do ponto crítico (75 bar): copo da célula de alta pressão
(a); pormenor do fundo da célula (filtro) (b).
Macroscopicamente, o β‐caroteno processado apresenta‐se sob a forma de cristais planos de dimensão
considerável, de cor vermelha escura e com reflexos prateados, ao contrário do produto original, que
apresenta uma coloração completamente uniforme. O β‐caroteno original tem também uma tendência
para aderir mais às superfícies do que o processado. A Figura 3.13 mostra o aspecto dos cristais após o
processamento e a recolha.
Figura 3.13 – Recolha do β‐caroteno micronizado: aspecto dos cristais.
O rendimento de β‐caroteno micronizado em cada um dos ensaios está registado na Tabela 3.6. É
também mostrado o volume de solvente que foi adicionado no final de cada ensaio, de forma a arrastar
o β‐caroteno acumulado no volume morto do aparelho, dado que este influencia a quantidade de
produto obtido no final. Em média, a quantidade de β‐caroteno perdido em cada ensaio, que

47
corresponde à massa que é retida nas tubagens e às perdas que ocorrem na recolha do pó, é de cerca
de 20% da massa inicial.
Tabela 3.6 – Registo de rendimento dos ensaios realizados.
Ensaio 0 P1* P2 P3** T1 T2 Q1 Q2 Q3 N1
Solvente (mL) 20 20 30 30 30 25 20 30 30 30
Rendimento 41% ‐ 79% ‐ 62% 61% 37% 48% 51% 57%
*, massa não contabilizada; **, micronização não teve lugar.
O rendimento médio, que depende tanto das perdas no aparelho, como da solubilidade nas condições
dos ensaios, é de aproximadamente 60%. A título ilustrativo, mostra‐se na Figura 3.14 a relação entre o
rendimento e a solubilidade, para condições semelhantes de volume de solvente adicionado e perdas no
sistema (ensaios T1, P2, Q2 e Q3). Esta figura prova que se tem todo o interesse em trabalhar em
condições de baixa solubilidade do β‐caroteno, de forma a maximizar o rendimento.
Figura 3.14 – Representação gráfica do rendimento da micronização em função da solubilidade (ensaios T1, P2, Q2 e Q3).
3.2.2. Análise da pureza do produto
3.2.2.1. HPLC
A pureza do β‐caroteno micronizado e da solução recolhida no efluente de um dos ensaios (Q2) foram
analisadas e comparadas com o β‐caroteno de partida por HPLC.
A análise do trans‐β‐caroteno e do cis‐β‐caroteno foi realizada com detecção a 454 nm. A presença do
isómero cis em qualquer das soluções analisadas implica o aparecimento de dois picos, em tempos de
retenção ligeiramente diferentes, no cromatograma. A presença de eventuais produtos de degradação
(óxidos) foi avaliada com detecção a 260 nm, tal como sugere Randolph et al. [8]. Os cromatogramas
detectados a 454 e a 260 nm estão patentes na Figura 3.15 e na Figura 3.16, respectivamente.
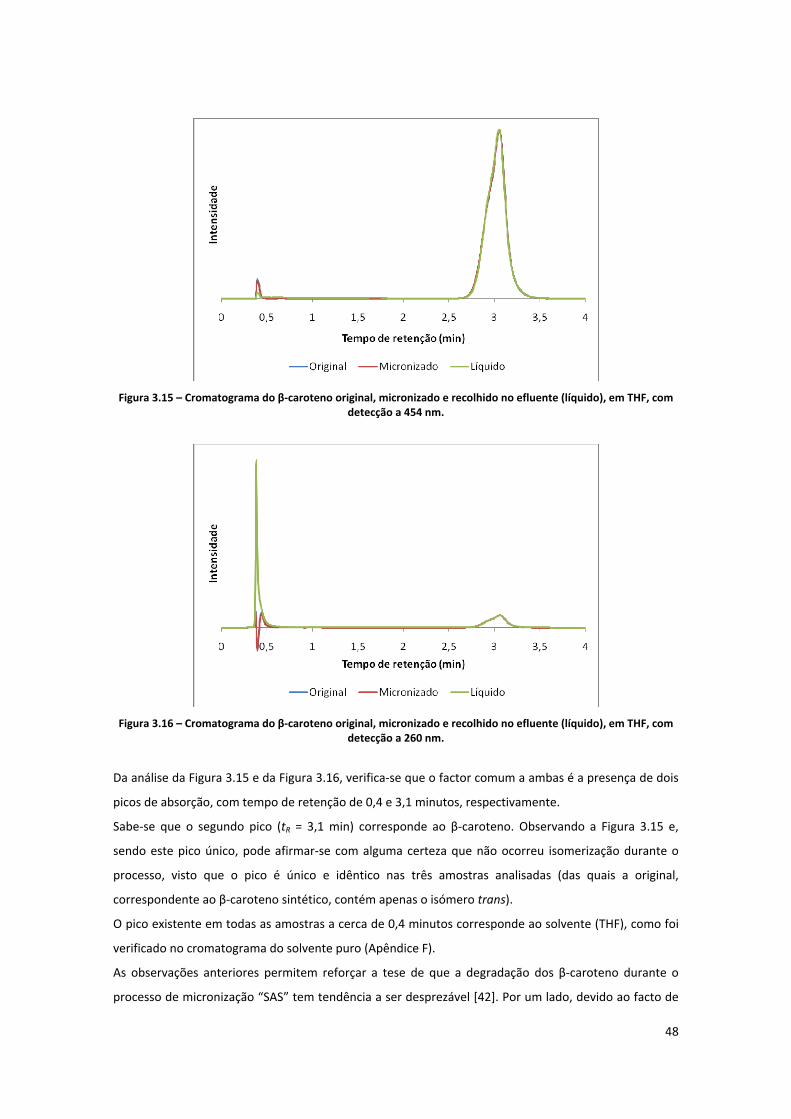
48
Figura 3.15 – Cromatograma do β‐caroteno original, micronizado e recolhido no efluente (líquido), em THF, com detecção a 454 nm.
Figura 3.16 – Cromatograma do β‐caroteno original, micronizado e recolhido no efluente (líquido), em THF, com detecção a 260 nm.
Da análise da Figura 3.15 e da Figura 3.16, verifica‐se que o factor comum a ambas é a presença de dois
picos de absorção, com tempo de retenção de 0,4 e 3,1 minutos, respectivamente.
Sabe‐se que o segundo pico (tR = 3,1 min) corresponde ao β‐caroteno. Observando a Figura 3.15 e,
sendo este pico único, pode afirmar‐se com alguma certeza que não ocorreu isomerização durante o
processo, visto que o pico é único e idêntico nas três amostras analisadas (das quais a original,
correspondente ao β‐caroteno sintético, contém apenas o isómero trans).
O pico existente em todas as amostras a cerca de 0,4 minutos corresponde ao solvente (THF), como foi
verificado no cromatograma do solvente puro (Apêndice F).
As observações anteriores permitem reforçar a tese de que a degradação dos β‐caroteno durante o
processo de micronização “SAS” tem tendência a ser desprezável [42]. Por um lado, devido ao facto de
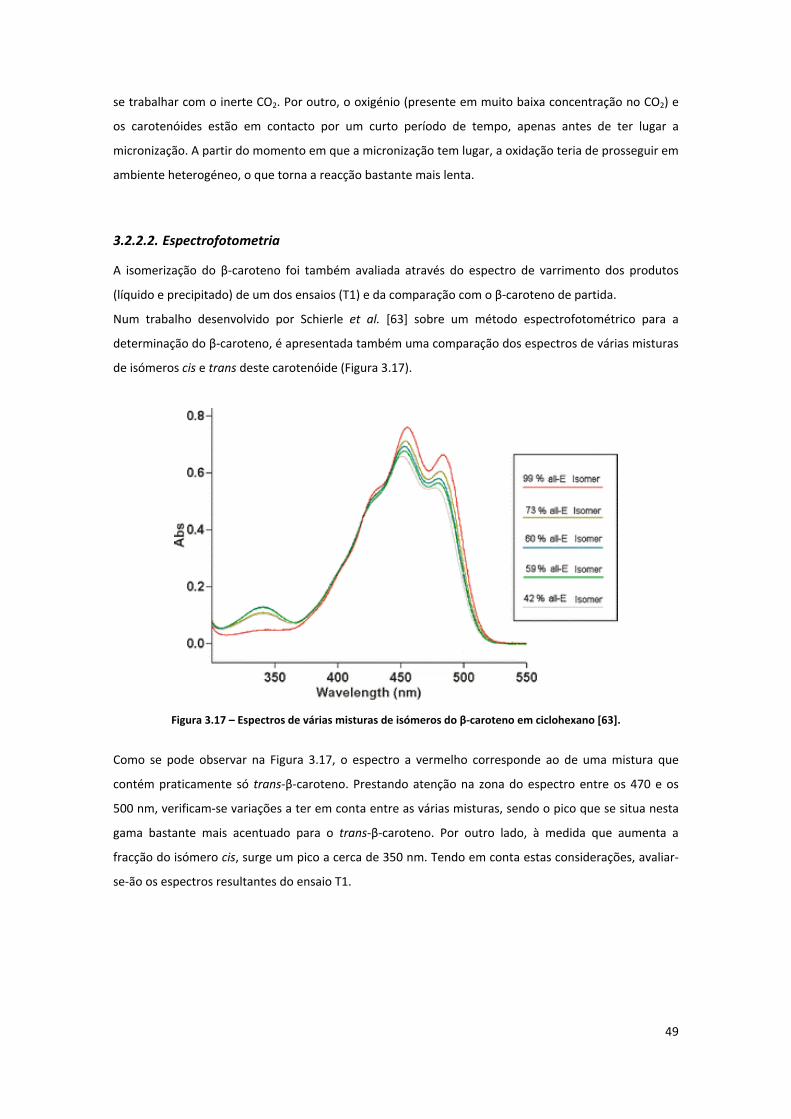
49
se trabalhar com o inerte CO2. Por outro, o oxigénio (presente em muito baixa concentração no CO2) e
os carotenóides estão em contacto por um curto período de tempo, apenas antes de ter lugar a
micronização. A partir do momento em que a micronização tem lugar, a oxidação teria de prosseguir em
ambiente heterogéneo, o que torna a reacção bastante mais lenta.
3.2.2.2. Espectrofotometria
A isomerização do β‐caroteno foi também avaliada através do espectro de varrimento dos produtos
(líquido e precipitado) de um dos ensaios (T1) e da comparação com o β‐caroteno de partida.
Num trabalho desenvolvido por Schierle et al. [63] sobre um método espectrofotométrico para a
determinação do β‐caroteno, é apresentada também uma comparação dos espectros de várias misturas
de isómeros cis e trans deste carotenóide (Figura 3.17).
Figura 3.17 – Espectros de várias misturas de isómeros do β‐caroteno em ciclohexano [63].
Como se pode observar na Figura 3.17, o espectro a vermelho corresponde ao de uma mistura que
contém praticamente só trans‐β‐caroteno. Prestando atenção na zona do espectro entre os 470 e os
500 nm, verificam‐se variações a ter em conta entre as várias misturas, sendo o pico que se situa nesta
gama bastante mais acentuado para o trans‐β‐caroteno. Por outro lado, à medida que aumenta a
fracção do isómero cis, surge um pico a cerca de 350 nm. Tendo em conta estas considerações, avaliar‐
se‐ão os espectros resultantes do ensaio T1.
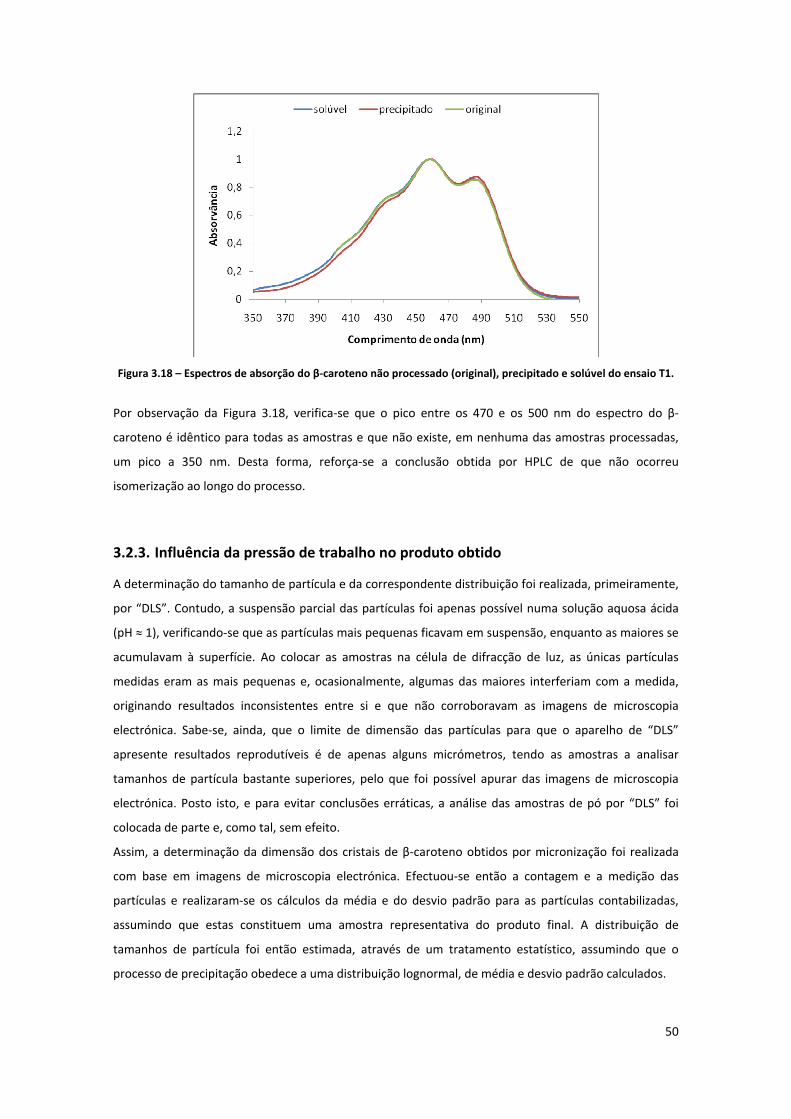
50
Figura 3.18 – Espectros de absorção do β‐caroteno não processado (original), precipitado e solúvel do ensaio T1.
Por observação da Figura 3.18, verifica‐se que o pico entre os 470 e os 500 nm do espectro do β‐
caroteno é idêntico para todas as amostras e que não existe, em nenhuma das amostras processadas,
um pico a 350 nm. Desta forma, reforça‐se a conclusão obtida por HPLC de que não ocorreu
isomerização ao longo do processo.
3.2.3. Influência da pressão de trabalho no produto obtido
A determinação do tamanho de partícula e da correspondente distribuição foi realizada, primeiramente,
por “DLS”. Contudo, a suspensão parcial das partículas foi apenas possível numa solução aquosa ácida
(pH ≈ 1), verificando‐se que as partículas mais pequenas ficavam em suspensão, enquanto as maiores se
acumulavam à superfície. Ao colocar as amostras na célula de difracção de luz, as únicas partículas
medidas eram as mais pequenas e, ocasionalmente, algumas das maiores interferiam com a medida,
originando resultados inconsistentes entre si e que não corroboravam as imagens de microscopia
electrónica. Sabe‐se, ainda, que o limite de dimensão das partículas para que o aparelho de “DLS”
apresente resultados reprodutíveis é de apenas alguns micrómetros, tendo as amostras a analisar
tamanhos de partícula bastante superiores, pelo que foi possível apurar das imagens de microscopia
electrónica. Posto isto, e para evitar conclusões erráticas, a análise das amostras de pó por “DLS” foi
colocada de parte e, como tal, sem efeito.
Assim, a determinação da dimensão dos cristais de β‐caroteno obtidos por micronização foi realizada
com base em imagens de microscopia electrónica. Efectuou‐se então a contagem e a medição das
partículas e realizaram‐se os cálculos da média e do desvio padrão para as partículas contabilizadas,
assumindo que estas constituem uma amostra representativa do produto final. A distribuição de
tamanhos de partícula foi então estimada, através de um tratamento estatístico, assumindo que o
processo de precipitação obedece a uma distribuição lognormal, de média e desvio padrão calculados.

51
A influência da pressão sobre o tamanho das partículas formadas foi estudada com base em dois ensaios
acima do ponto crítico da mistura (100 e 130 bar) e um abaixo deste ponto (75 bar). Nestes ensaios, a
temperatura foi de 40°C, o caudal de 1 mL/min e a concentração inicial de β‐caroteno em THF igual a 6
mg/mL.
3.2.3.1. Análise do produto não processado
De forma a possibilitar a comparação do produto processado com o produto inicial, foi realizada a
análise deste. A Figura 3.19 e a Figura 3.20 representam duas imagens do β‐caroteno antes do
processamento e a Figura 3.21 a distribuição de tamanhos de partícula obtida na contagem e a
distribuição estimada pelo tratamento estatístico.
Figura 3.19 – Aspecto do β‐caroteno antes do processamento (imagem de microscopia electrónica de varrimento com ampliação de 1000 vezes).
Figura 3.20 – Aspecto do β‐caroteno antes do processamento (imagem de microscopia electrónica de varrimento com ampliação de 3000 vezes).
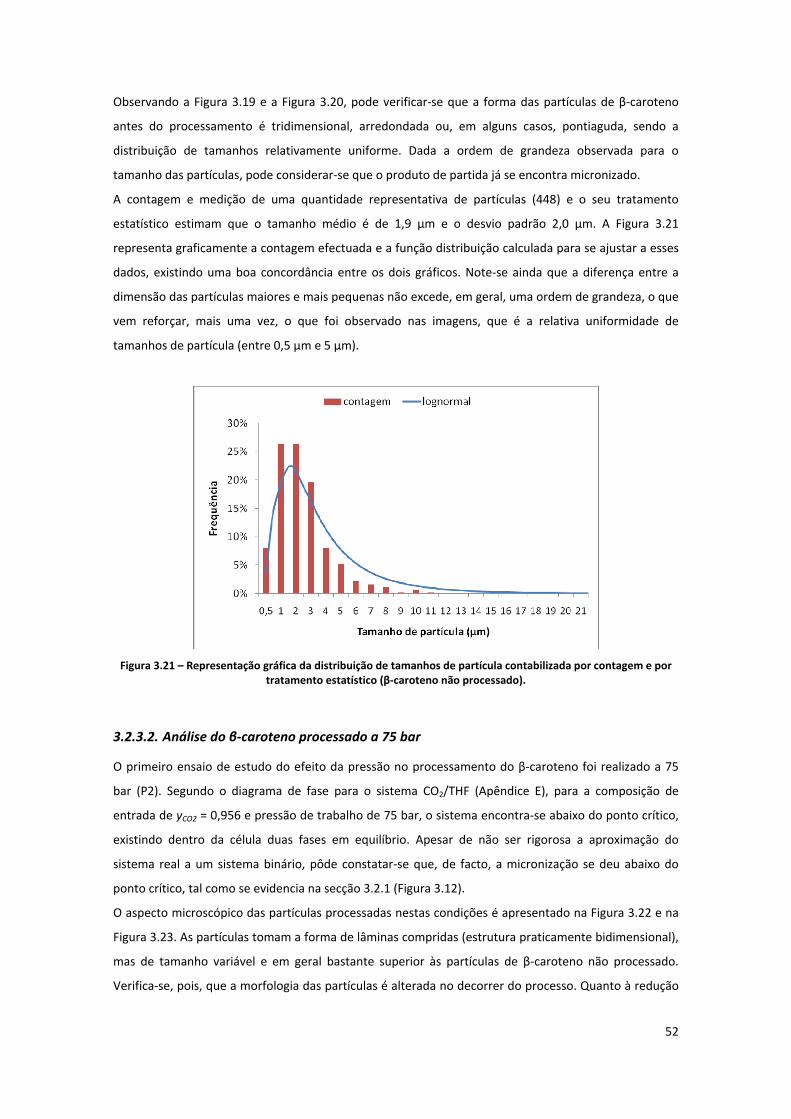
52
Observando a Figura 3.19 e a Figura 3.20, pode verificar‐se que a forma das partículas de β‐caroteno
antes do processamento é tridimensional, arredondada ou, em alguns casos, pontiaguda, sendo a
distribuição de tamanhos relativamente uniforme. Dada a ordem de grandeza observada para o
tamanho das partículas, pode considerar‐se que o produto de partida já se encontra micronizado.
A contagem e medição de uma quantidade representativa de partículas (448) e o seu tratamento
estatístico estimam que o tamanho médio é de 1,9 µm e o desvio padrão 2,0 µm. A Figura 3.21
representa graficamente a contagem efectuada e a função distribuição calculada para se ajustar a esses
dados, existindo uma boa concordância entre os dois gráficos. Note‐se ainda que a diferença entre a
dimensão das partículas maiores e mais pequenas não excede, em geral, uma ordem de grandeza, o que
vem reforçar, mais uma vez, o que foi observado nas imagens, que é a relativa uniformidade de
tamanhos de partícula (entre 0,5 µm e 5 µm).
Figura 3.21 – Representação gráfica da distribuição de tamanhos de partícula contabilizada por contagem e por tratamento estatístico (β‐caroteno não processado).
3.2.3.2. Análise do β‐caroteno processado a 75 bar
O primeiro ensaio de estudo do efeito da pressão no processamento do β‐caroteno foi realizado a 75
bar (P2). Segundo o diagrama de fase para o sistema CO2/THF (Apêndice E), para a composição de
entrada de yCO2 = 0,956 e pressão de trabalho de 75 bar, o sistema encontra‐se abaixo do ponto crítico,
existindo dentro da célula duas fases em equilíbrio. Apesar de não ser rigorosa a aproximação do
sistema real a um sistema binário, pôde constatar‐se que, de facto, a micronização se deu abaixo do
ponto crítico, tal como se evidencia na secção 3.2.1 (Figura 3.12).
O aspecto microscópico das partículas processadas nestas condições é apresentado na Figura 3.22 e na
Figura 3.23. As partículas tomam a forma de lâminas compridas (estrutura praticamente bidimensional),
mas de tamanho variável e em geral bastante superior às partículas de β‐caroteno não processado.
Verifica‐se, pois, que a morfologia das partículas é alterada no decorrer do processo. Quanto à redução

53
do tamanho das partículas, esta não acontece, quando comparada com o produto de partida (já
micronizado), mas pode considerar‐se que o objectivo da formação de micropartículas foi atingido.
Figura 3.22 – Aspecto do β‐caroteno processado a 75 bar (imagem de microscopia electrónica de varrimento com ampliação de 50 vezes).
Figura 3.23 – Aspecto do β‐caroteno processado a 75 bar (imagem de microscopia electrónica de varrimento com ampliação de 100 vezes).
Na Figura 3.24 (a) mostra‐se em maior detalhe a forma dos cristais de β‐caroteno processados a 75 bar.
Nesta figura, é particularmente evidente a sua estrutura praticamente bidimensional, bem como a
forma irregular das arestas dos cristais, que podem evidenciar que as partículas maiores são produto da
agregação de partículas mais pequenas em fases mais adiantadas do crescimento dos cristais, tal como
foi proposto por Bristow et al. [4]. Segundo este autor, as partículas formadas abaixo da pressão crítica
da mistura estão muito mais agregadas do que as que são formadas a pressões mais elevadas. Este
mecanismo de aglomeração pode ser explicado pela precipitação que ocorre dentro das gotas ricas em
THF, onde as partículas submicrométricas começam a nuclear, coalescem e fundem durante a fase de
crescimento que prossegue a nucleação.

54
Paralelamente à alteração da morfologia das partículas, verificou‐se ainda, numa imagem de maior
ampliação, que os cristais procedentes deste ensaio apresentam uma superfície porosa, característica
que é realçada na Figura 3.24 (b). Este facto demonstra que, apesar do aumento do tamanho das
partículas resultante do processamento a 75 bar, é possível alterar completamente a sua morfologia, o
que pode potenciar igualmente interessantes aplicações, dado o aumento da área superficial que é
possível obter.
(a) (b)Figura 3.24 – Aspecto do β‐caroteno processado a 75 bar (imagens de microscopia electrónica de varrimento com
ampliação de: 500 vezes (a); 3000 vezes (b)).
A distribuição de tamanhos de partícula obtida no processamento do β‐caroteno a 75 bar foi também
estimada com base na contagem e medição das partículas, através de imagens obtidas por microscopia
electrónica.
A medição de uma quantidade considerada representativa da amostra (290 partículas) permitiu o
cálculo do tamanho médio de partícula e do desvio padrão, respectivamente 100,7 µm e 1,9 µm. As
representações gráficas das partículas contadas e da função distribuição (lognormal) estimada
encontram‐se na Figura 3.25, revelando uma razoável concordância entre as duas.
Para uma análise acerca do efeito da pressão sobre o tipo de partículas obtidas em condições de
pressão abaixo do ponto crítico, foi realizado um ensaio à pressão de 65 bar. Contudo, o que se verificou
foi que a precipitação não ocorreu, o que levou a concluir que o limite mínimo de pressão (mantendo
constantes as restantes condições) para que se dê a micronização do β‐caroteno é 75 bar. A realização
de um ensaio intermédio (70 bar, por exemplo) foi descartada, dado que a diferença de pressão seria
provavelmente demasiado pequena para que se observassem diferenças significativas no produto, tal
como foi constatado por Cocero & Ferrero [10].
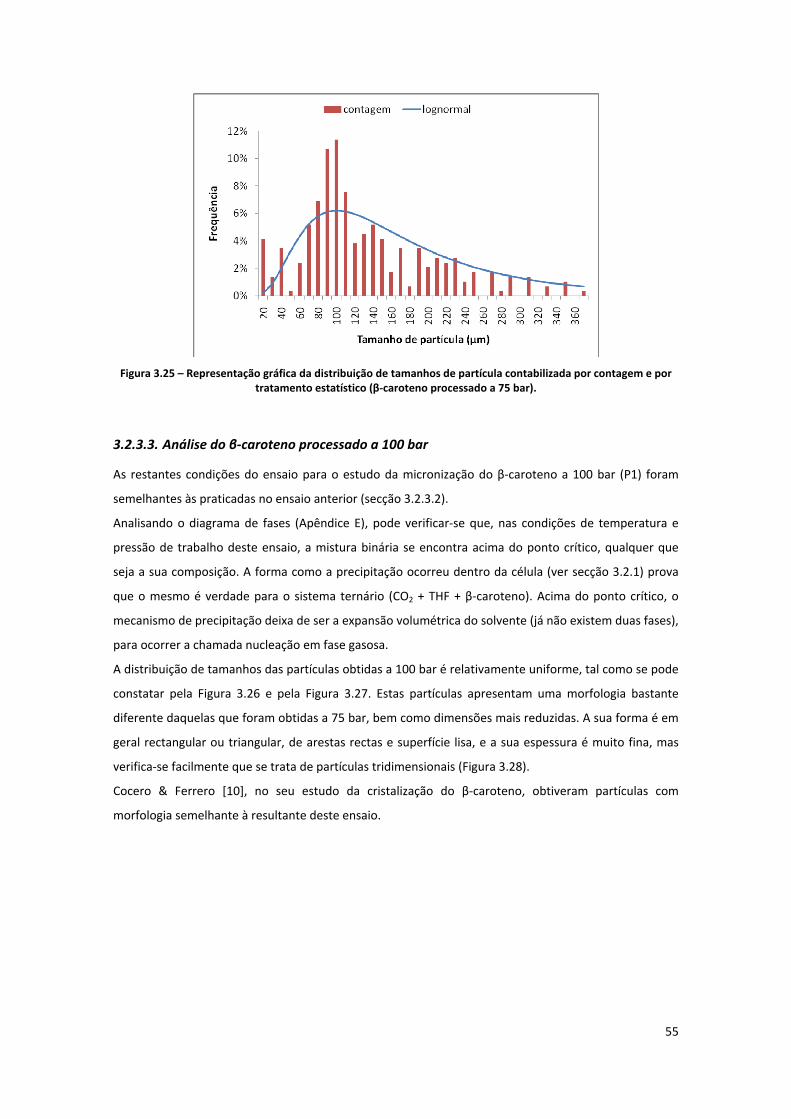
55
Figura 3.25 – Representação gráfica da distribuição de tamanhos de partícula contabilizada por contagem e por tratamento estatístico (β‐caroteno processado a 75 bar).
3.2.3.3. Análise do β‐caroteno processado a 100 bar
As restantes condições do ensaio para o estudo da micronização do β‐caroteno a 100 bar (P1) foram
semelhantes às praticadas no ensaio anterior (secção 3.2.3.2).
Analisando o diagrama de fases (Apêndice E), pode verificar‐se que, nas condições de temperatura e
pressão de trabalho deste ensaio, a mistura binária se encontra acima do ponto crítico, qualquer que
seja a sua composição. A forma como a precipitação ocorreu dentro da célula (ver secção 3.2.1) prova
que o mesmo é verdade para o sistema ternário (CO2 + THF + β‐caroteno). Acima do ponto crítico, o
mecanismo de precipitação deixa de ser a expansão volumétrica do solvente (já não existem duas fases),
para ocorrer a chamada nucleação em fase gasosa.
A distribuição de tamanhos das partículas obtidas a 100 bar é relativamente uniforme, tal como se pode
constatar pela Figura 3.26 e pela Figura 3.27. Estas partículas apresentam uma morfologia bastante
diferente daquelas que foram obtidas a 75 bar, bem como dimensões mais reduzidas. A sua forma é em
geral rectangular ou triangular, de arestas rectas e superfície lisa, e a sua espessura é muito fina, mas
verifica‐se facilmente que se trata de partículas tridimensionais (Figura 3.28).
Cocero & Ferrero [10], no seu estudo da cristalização do β‐caroteno, obtiveram partículas com
morfologia semelhante à resultante deste ensaio.

56
Figura 3.26 – Aspecto do β‐caroteno processado a 100 bar (imagem de microscopia electrónica de varrimento com ampliação de 100 vezes).
Figura 3.27 – Aspecto do β‐caroteno processado a 100 bar (imagem de microscopia electrónica de varrimento com ampliação de 500 vezes).
(a) (b)Figura 3.28 – Aspecto do β‐caroteno processado a 100 bar (imagens de microscopia electrónica de varrimento
com ampliação de: 3000 vezes (a); 5000 vezes (b)).
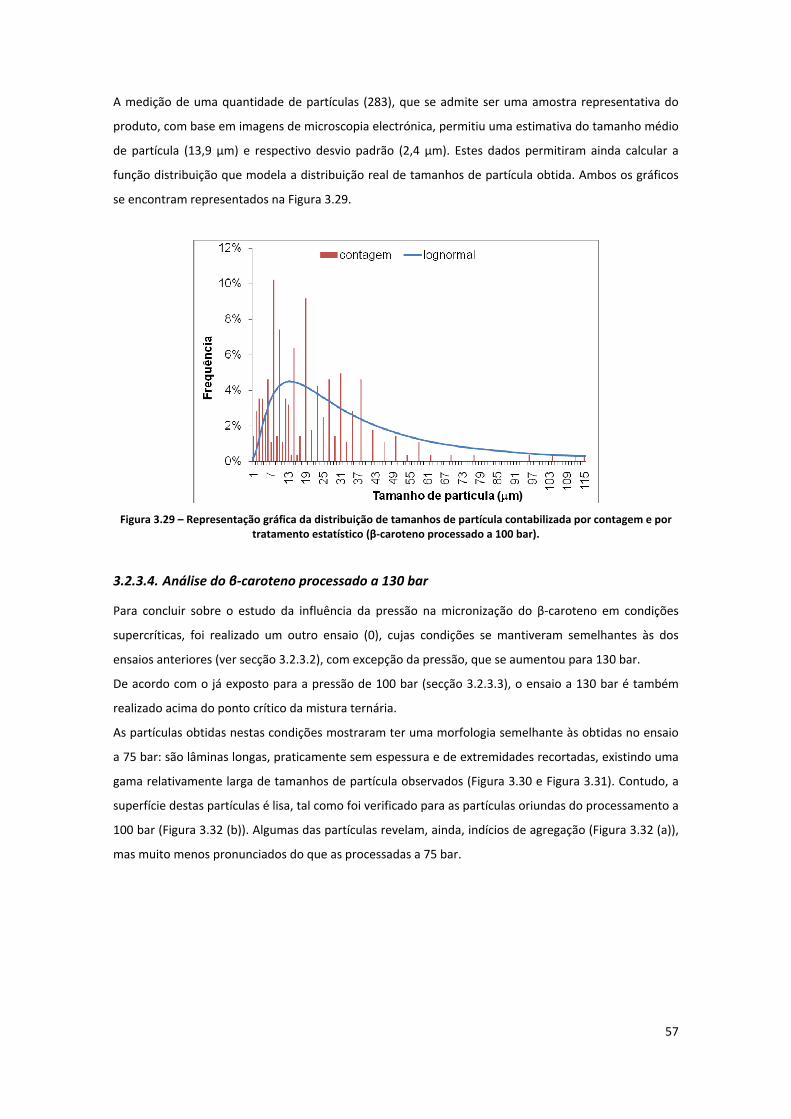
57
A medição de uma quantidade de partículas (283), que se admite ser uma amostra representativa do
produto, com base em imagens de microscopia electrónica, permitiu uma estimativa do tamanho médio
de partícula (13,9 µm) e respectivo desvio padrão (2,4 µm). Estes dados permitiram ainda calcular a
função distribuição que modela a distribuição real de tamanhos de partícula obtida. Ambos os gráficos
se encontram representados na Figura 3.29.
Figura 3.29 – Representação gráfica da distribuição de tamanhos de partícula contabilizada por contagem e por tratamento estatístico (β‐caroteno processado a 100 bar).
3.2.3.4. Análise do β‐caroteno processado a 130 bar
Para concluir sobre o estudo da influência da pressão na micronização do β‐caroteno em condições
supercríticas, foi realizado um outro ensaio (0), cujas condições se mantiveram semelhantes às dos
ensaios anteriores (ver secção 3.2.3.2), com excepção da pressão, que se aumentou para 130 bar.
De acordo com o já exposto para a pressão de 100 bar (secção 3.2.3.3), o ensaio a 130 bar é também
realizado acima do ponto crítico da mistura ternária.
As partículas obtidas nestas condições mostraram ter uma morfologia semelhante às obtidas no ensaio
a 75 bar: são lâminas longas, praticamente sem espessura e de extremidades recortadas, existindo uma
gama relativamente larga de tamanhos de partícula observados (Figura 3.30 e Figura 3.31). Contudo, a
superfície destas partículas é lisa, tal como foi verificado para as partículas oriundas do processamento a
100 bar (Figura 3.32 (b)). Algumas das partículas revelam, ainda, indícios de agregação (Figura 3.32 (a)),
mas muito menos pronunciados do que as processadas a 75 bar.
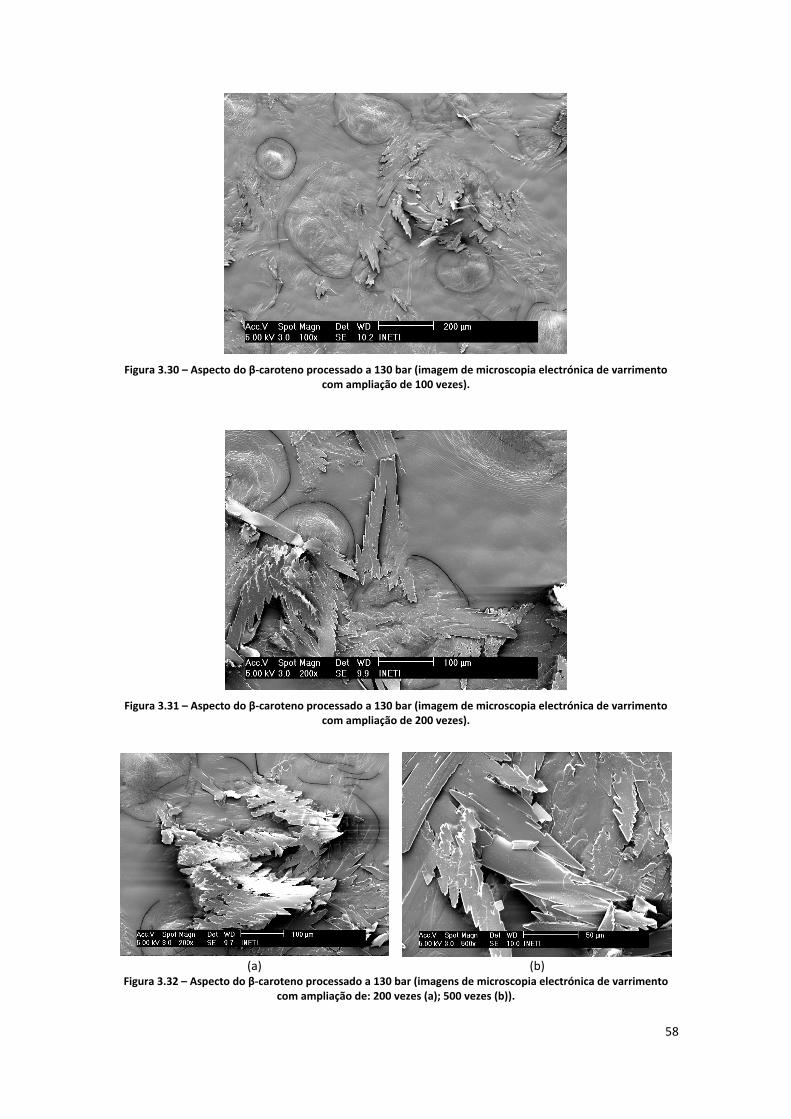
58
Figura 3.30 – Aspecto do β‐caroteno processado a 130 bar (imagem de microscopia electrónica de varrimento com ampliação de 100 vezes).
Figura 3.31 – Aspecto do β‐caroteno processado a 130 bar (imagem de microscopia electrónica de varrimento com ampliação de 200 vezes).
(a) (b)Figura 3.32 – Aspecto do β‐caroteno processado a 130 bar (imagens de microscopia electrónica de varrimento
com ampliação de: 200 vezes (a); 500 vezes (b)).
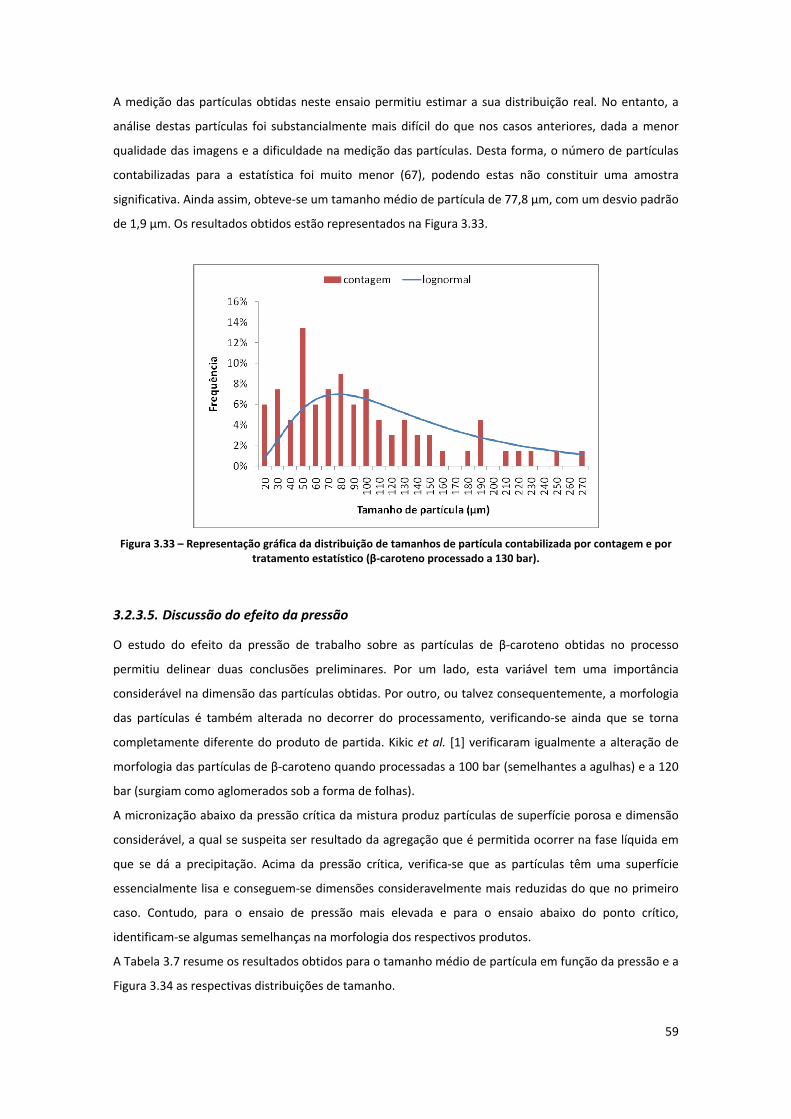
59
A medição das partículas obtidas neste ensaio permitiu estimar a sua distribuição real. No entanto, a
análise destas partículas foi substancialmente mais difícil do que nos casos anteriores, dada a menor
qualidade das imagens e a dificuldade na medição das partículas. Desta forma, o número de partículas
contabilizadas para a estatística foi muito menor (67), podendo estas não constituir uma amostra
significativa. Ainda assim, obteve‐se um tamanho médio de partícula de 77,8 µm, com um desvio padrão
de 1,9 µm. Os resultados obtidos estão representados na Figura 3.33.
Figura 3.33 – Representação gráfica da distribuição de tamanhos de partícula contabilizada por contagem e por tratamento estatístico (β‐caroteno processado a 130 bar).
3.2.3.5. Discussão do efeito da pressão
O estudo do efeito da pressão de trabalho sobre as partículas de β‐caroteno obtidas no processo
permitiu delinear duas conclusões preliminares. Por um lado, esta variável tem uma importância
considerável na dimensão das partículas obtidas. Por outro, ou talvez consequentemente, a morfologia
das partículas é também alterada no decorrer do processamento, verificando‐se ainda que se torna
completamente diferente do produto de partida. Kikic et al. [1] verificaram igualmente a alteração de
morfologia das partículas de β‐caroteno quando processadas a 100 bar (semelhantes a agulhas) e a 120
bar (surgiam como aglomerados sob a forma de folhas).
A micronização abaixo da pressão crítica da mistura produz partículas de superfície porosa e dimensão
considerável, a qual se suspeita ser resultado da agregação que é permitida ocorrer na fase líquida em
que se dá a precipitação. Acima da pressão crítica, verifica‐se que as partículas têm uma superfície
essencialmente lisa e conseguem‐se dimensões consideravelmente mais reduzidas do que no primeiro
caso. Contudo, para o ensaio de pressão mais elevada e para o ensaio abaixo do ponto crítico,
identificam‐se algumas semelhanças na morfologia dos respectivos produtos.
A Tabela 3.7 resume os resultados obtidos para o tamanho médio de partícula em função da pressão e a
Figura 3.34 as respectivas distribuições de tamanho.
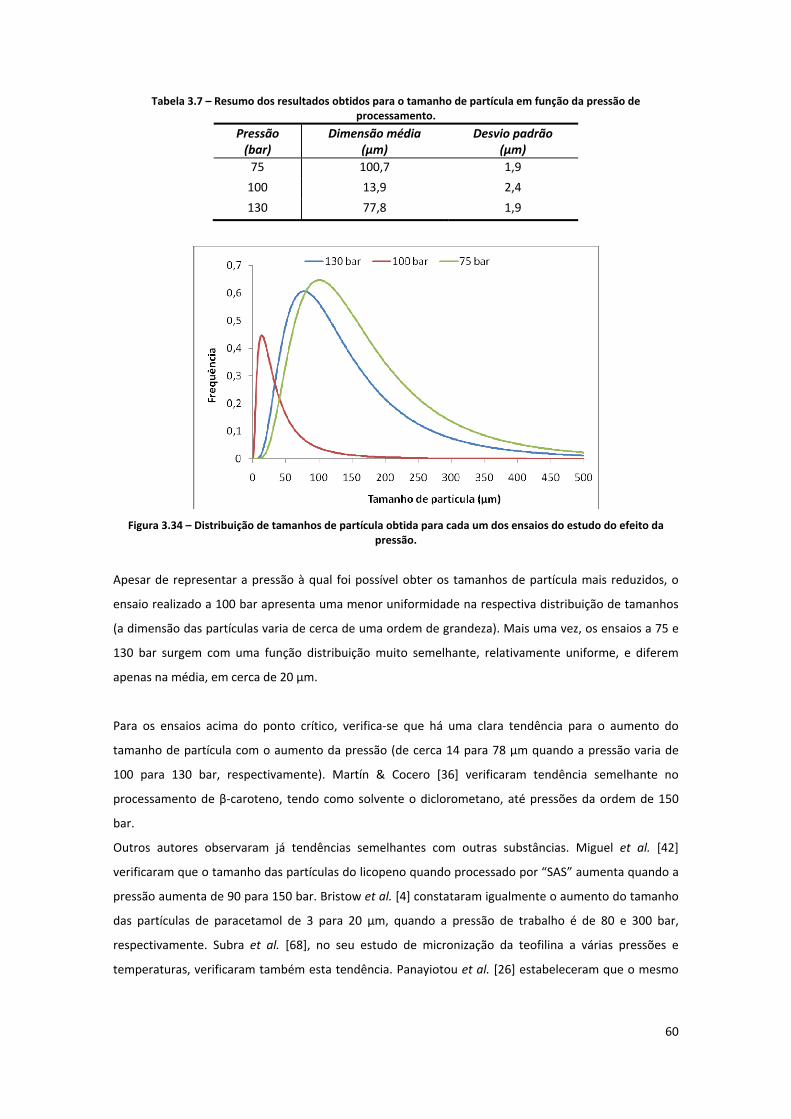
60
Tabela 3.7 – Resumo dos resultados obtidos para o tamanho de partícula em função da pressão de processamento.
Pressão (bar)
Dimensão média(µm)
Desvio padrão(µm)
75 100,7 1,9
100 13,9 2,4
130 77,8 1,9
Figura 3.34 – Distribuição de tamanhos de partícula obtida para cada um dos ensaios do estudo do efeito da pressão.
Apesar de representar a pressão à qual foi possível obter os tamanhos de partícula mais reduzidos, o
ensaio realizado a 100 bar apresenta uma menor uniformidade na respectiva distribuição de tamanhos
(a dimensão das partículas varia de cerca de uma ordem de grandeza). Mais uma vez, os ensaios a 75 e
130 bar surgem com uma função distribuição muito semelhante, relativamente uniforme, e diferem
apenas na média, em cerca de 20 µm.
Para os ensaios acima do ponto crítico, verifica‐se que há uma clara tendência para o aumento do
tamanho de partícula com o aumento da pressão (de cerca 14 para 78 µm quando a pressão varia de
100 para 130 bar, respectivamente). Martín & Cocero [36] verificaram tendência semelhante no
processamento de β‐caroteno, tendo como solvente o diclorometano, até pressões da ordem de 150
bar.
Outros autores observaram já tendências semelhantes com outras substâncias. Miguel et al. [42]
verificaram que o tamanho das partículas do licopeno quando processado por “SAS” aumenta quando a
pressão aumenta de 90 para 150 bar. Bristow et al. [4] constataram igualmente o aumento do tamanho
das partículas de paracetamol de 3 para 20 µm, quando a pressão de trabalho é de 80 e 300 bar,
respectivamente. Subra et al. [68], no seu estudo de micronização da teofilina a várias pressões e
temperaturas, verificaram também esta tendência. Panayiotou et al. [26] estabeleceram que o mesmo
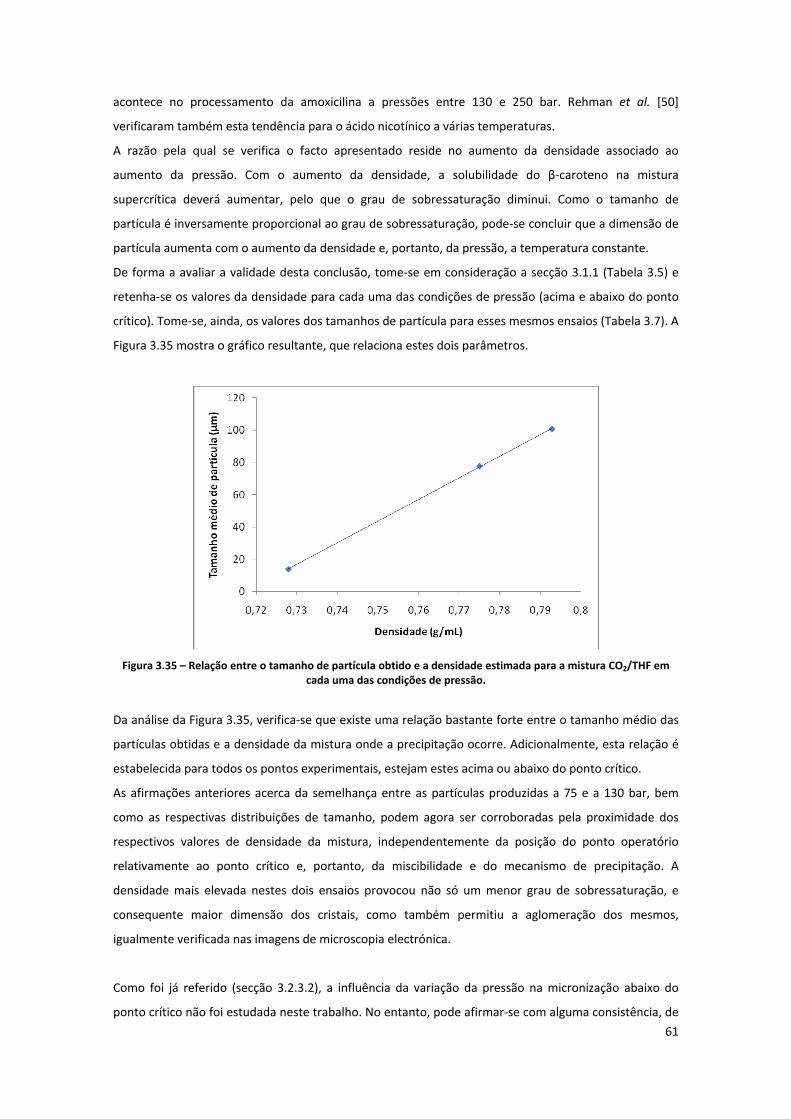
61
acontece no processamento da amoxicilina a pressões entre 130 e 250 bar. Rehman et al. [50]
verificaram também esta tendência para o ácido nicotínico a várias temperaturas.
A razão pela qual se verifica o facto apresentado reside no aumento da densidade associado ao
aumento da pressão. Com o aumento da densidade, a solubilidade do β‐caroteno na mistura
supercrítica deverá aumentar, pelo que o grau de sobressaturação diminui. Como o tamanho de
partícula é inversamente proporcional ao grau de sobressaturação, pode‐se concluir que a dimensão de
partícula aumenta com o aumento da densidade e, portanto, da pressão, a temperatura constante.
De forma a avaliar a validade desta conclusão, tome‐se em consideração a secção 3.1.1 (Tabela 3.5) e
retenha‐se os valores da densidade para cada uma das condições de pressão (acima e abaixo do ponto
crítico). Tome‐se, ainda, os valores dos tamanhos de partícula para esses mesmos ensaios (Tabela 3.7). A
Figura 3.35 mostra o gráfico resultante, que relaciona estes dois parâmetros.
Figura 3.35 – Relação entre o tamanho de partícula obtido e a densidade estimada para a mistura CO2/THF em cada uma das condições de pressão.
Da análise da Figura 3.35, verifica‐se que existe uma relação bastante forte entre o tamanho médio das
partículas obtidas e a densidade da mistura onde a precipitação ocorre. Adicionalmente, esta relação é
estabelecida para todos os pontos experimentais, estejam estes acima ou abaixo do ponto crítico.
As afirmações anteriores acerca da semelhança entre as partículas produzidas a 75 e a 130 bar, bem
como as respectivas distribuições de tamanho, podem agora ser corroboradas pela proximidade dos
respectivos valores de densidade da mistura, independentemente da posição do ponto operatório
relativamente ao ponto crítico e, portanto, da miscibilidade e do mecanismo de precipitação. A
densidade mais elevada nestes dois ensaios provocou não só um menor grau de sobressaturação, e
consequente maior dimensão dos cristais, como também permitiu a aglomeração dos mesmos,
igualmente verificada nas imagens de microscopia electrónica.
Como foi já referido (secção 3.2.3.2), a influência da variação da pressão na micronização abaixo do
ponto crítico não foi estudada neste trabalho. No entanto, pode afirmar‐se com alguma consistência, de

62
acordo com o que foi colocado até aqui, que o aumento da pressão até ao ponto crítico provoca uma
diminuição do tamanho de partícula obtido. Senão, note‐se que, havendo duas fases e ocorrendo a
precipitação na fase líquida (mais densa e rica em solvente orgânico), o aumento da pressão provoca
uma maior expansão volumétrica no solvente e, portanto, a diminuição mais acentuada da sua
densidade. Por sua vez, a solubilidade diminui, o que torna a solução mais sobressaturada, reduzindo
assim o tamanho das partículas obtidas. Trabalhos como o de Reverchon & Della Porta [55], o de
Tenorio et al. [70], ou o de Costa et al. [11], obtiveram este resultado.
3.3. Estudo da precipitação de um extracto natural de β‐caroteno
Com o objectivo de avaliar a micronização com anti‐solvente supercrítico como processo de purificação,
foi realizado um ensaio em que se utilizou como produto de partida um extracto produzido a partir de
microrganismos. Este extracto foi totalmente dissolvido em THF e processado nas condições de pressão
de 130 bar, temperatura de 40°C e caudal de solução de 1 mL/min. O produto final foi analisado por
HPLC e espectrofotometria.
3.3.1. Análise macroscópica
Após processamento, o produto apresenta uma morfologia macroscópica em tudo semelhante à do β‐
caroteno sintético processado nas mesmas condições. Contudo, a coloração dos cristais obtidos é mais
clara, tendo passado do vermelho forte dos cristais sintéticos para o cor‐de‐laranja do produto natural
(Figura 3.36). Este facto demonstra que os cristais contêm, para além do β‐caroteno, outras substâncias
não coradas, tal como seria de prever, visto que o produto de partida não é β‐caroteno puro.
Figura 3.36 – Aspecto do β‐caroteno natural processado.
3.3.2. Análise da pureza do produto
O extracto de β‐caroteno micronizado obtido foi analisado por espectrofotometria e por HPLC e
comparado com o β‐caroteno obtido no efluente líquido, de forma a concluir acerca da purificação
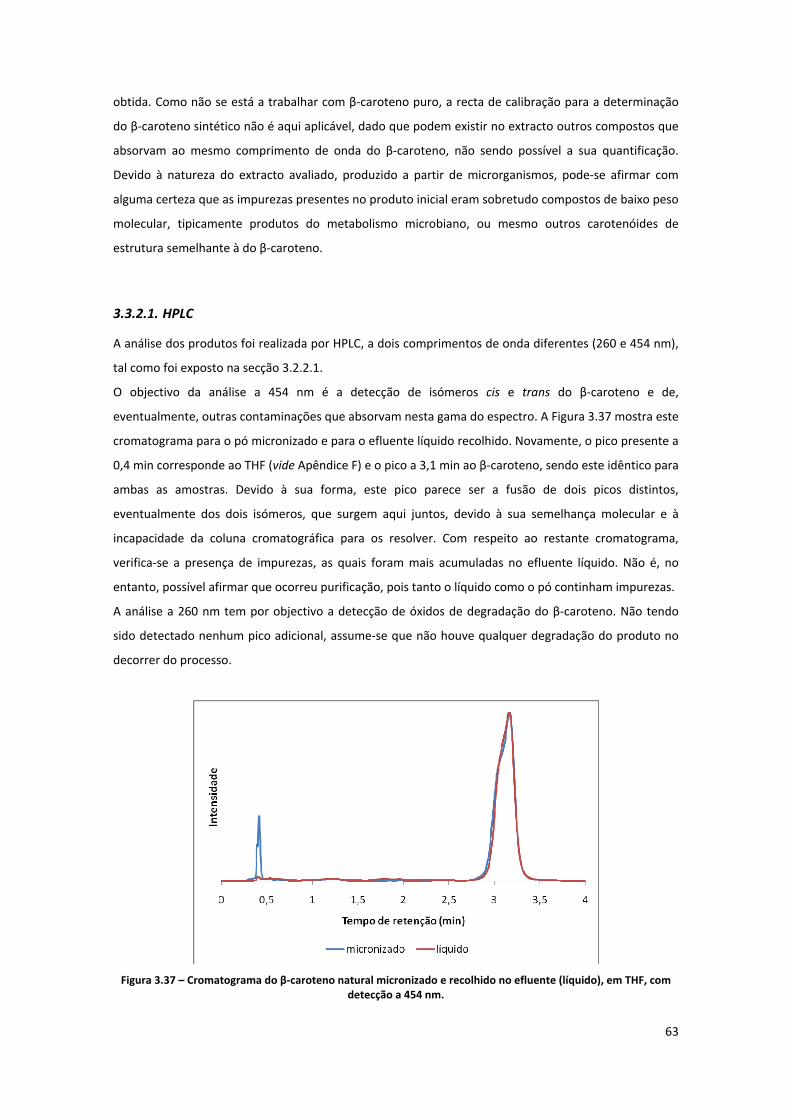
63
obtida. Como não se está a trabalhar com β‐caroteno puro, a recta de calibração para a determinação
do β‐caroteno sintético não é aqui aplicável, dado que podem existir no extracto outros compostos que
absorvam ao mesmo comprimento de onda do β‐caroteno, não sendo possível a sua quantificação.
Devido à natureza do extracto avaliado, produzido a partir de microrganismos, pode‐se afirmar com
alguma certeza que as impurezas presentes no produto inicial eram sobretudo compostos de baixo peso
molecular, tipicamente produtos do metabolismo microbiano, ou mesmo outros carotenóides de
estrutura semelhante à do β‐caroteno.
3.3.2.1. HPLC
A análise dos produtos foi realizada por HPLC, a dois comprimentos de onda diferentes (260 e 454 nm),
tal como foi exposto na secção 3.2.2.1.
O objectivo da análise a 454 nm é a detecção de isómeros cis e trans do β‐caroteno e de,
eventualmente, outras contaminações que absorvam nesta gama do espectro. A Figura 3.37 mostra este
cromatograma para o pó micronizado e para o efluente líquido recolhido. Novamente, o pico presente a
0,4 min corresponde ao THF (vide Apêndice F) e o pico a 3,1 min ao β‐caroteno, sendo este idêntico para
ambas as amostras. Devido à sua forma, este pico parece ser a fusão de dois picos distintos,
eventualmente dos dois isómeros, que surgem aqui juntos, devido à sua semelhança molecular e à
incapacidade da coluna cromatográfica para os resolver. Com respeito ao restante cromatograma,
verifica‐se a presença de impurezas, as quais foram mais acumuladas no efluente líquido. Não é, no
entanto, possível afirmar que ocorreu purificação, pois tanto o líquido como o pó continham impurezas.
A análise a 260 nm tem por objectivo a detecção de óxidos de degradação do β‐caroteno. Não tendo
sido detectado nenhum pico adicional, assume‐se que não houve qualquer degradação do produto no
decorrer do processo.
Figura 3.37 – Cromatograma do β‐caroteno natural micronizado e recolhido no efluente (líquido), em THF, com detecção a 454 nm.
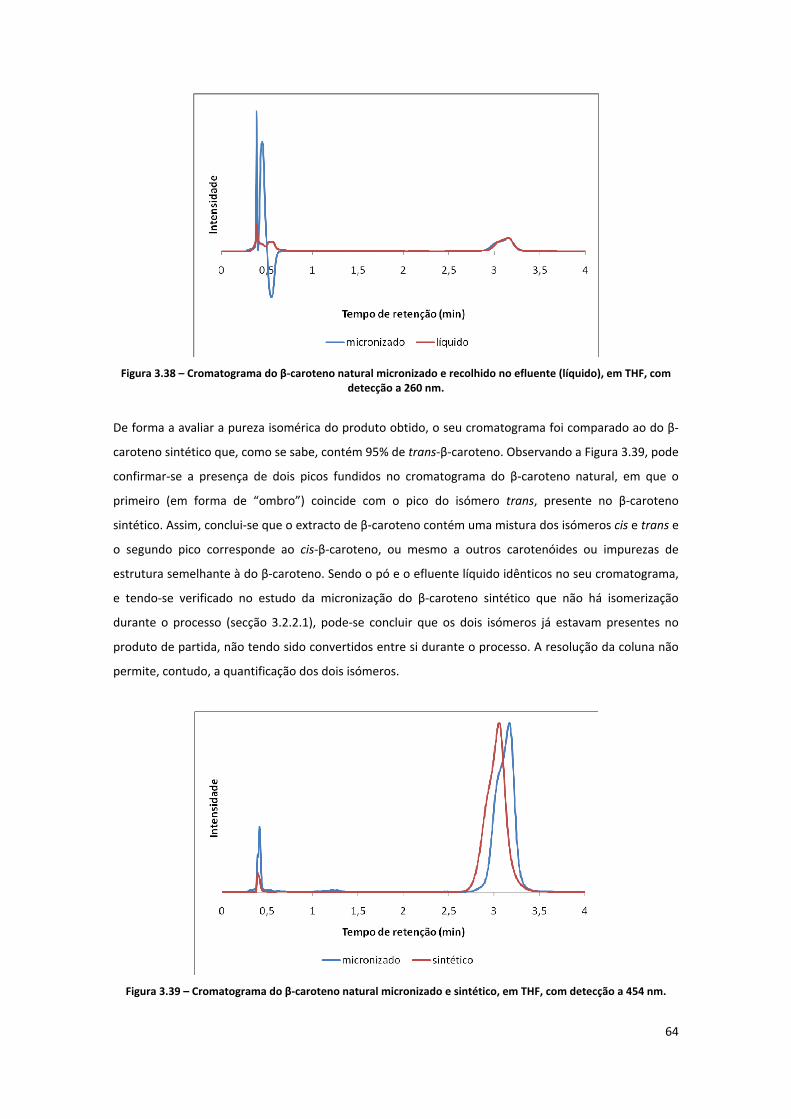
64
Figura 3.38 – Cromatograma do β‐caroteno natural micronizado e recolhido no efluente (líquido), em THF, com detecção a 260 nm.
De forma a avaliar a pureza isomérica do produto obtido, o seu cromatograma foi comparado ao do β‐
caroteno sintético que, como se sabe, contém 95% de trans‐β‐caroteno. Observando a Figura 3.39, pode
confirmar‐se a presença de dois picos fundidos no cromatograma do β‐caroteno natural, em que o
primeiro (em forma de “ombro”) coincide com o pico do isómero trans, presente no β‐caroteno
sintético. Assim, conclui‐se que o extracto de β‐caroteno contém uma mistura dos isómeros cis e trans e
o segundo pico corresponde ao cis‐β‐caroteno, ou mesmo a outros carotenóides ou impurezas de
estrutura semelhante à do β‐caroteno. Sendo o pó e o efluente líquido idênticos no seu cromatograma,
e tendo‐se verificado no estudo da micronização do β‐caroteno sintético que não há isomerização
durante o processo (secção 3.2.2.1), pode‐se concluir que os dois isómeros já estavam presentes no
produto de partida, não tendo sido convertidos entre si durante o processo. A resolução da coluna não
permite, contudo, a quantificação dos dois isómeros.
Figura 3.39 – Cromatograma do β‐caroteno natural micronizado e sintético, em THF, com detecção a 454 nm.
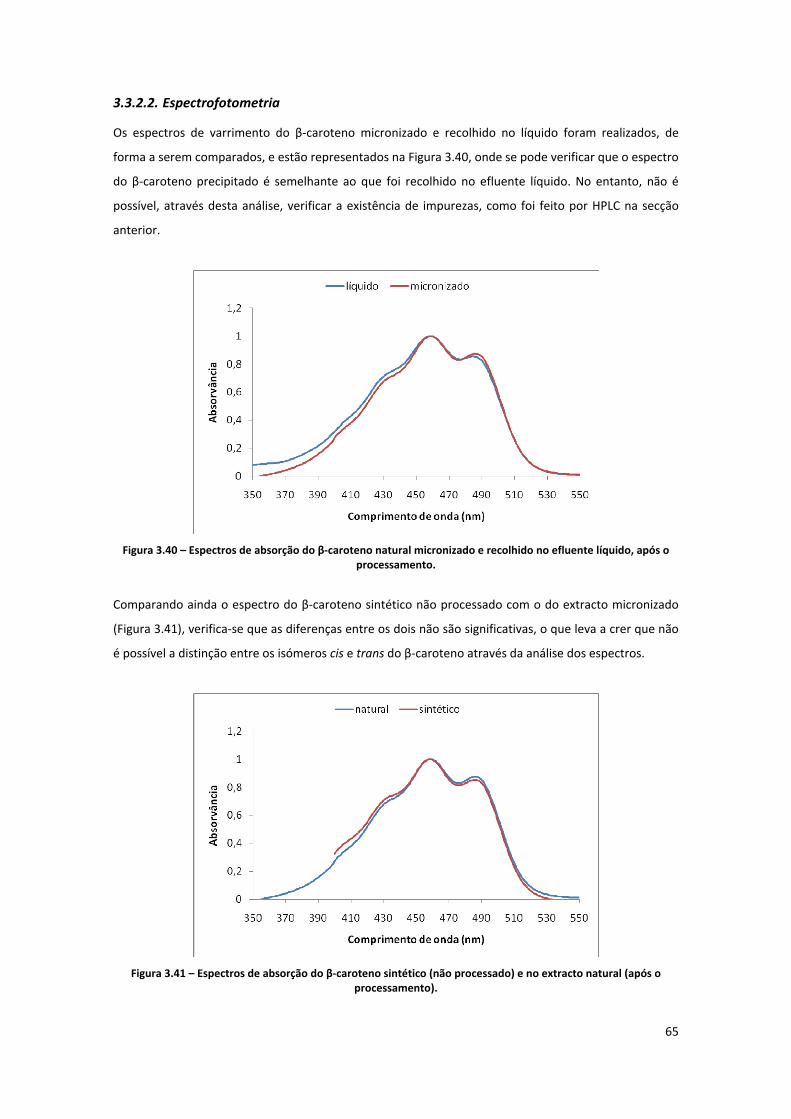
65
3.3.2.2. Espectrofotometria
Os espectros de varrimento do β‐caroteno micronizado e recolhido no líquido foram realizados, de
forma a serem comparados, e estão representados na Figura 3.40, onde se pode verificar que o espectro
do β‐caroteno precipitado é semelhante ao que foi recolhido no efluente líquido. No entanto, não é
possível, através desta análise, verificar a existência de impurezas, como foi feito por HPLC na secção
anterior.
Figura 3.40 – Espectros de absorção do β‐caroteno natural micronizado e recolhido no efluente líquido, após o processamento.
Comparando ainda o espectro do β‐caroteno sintético não processado com o do extracto micronizado
(Figura 3.41), verifica‐se que as diferenças entre os dois não são significativas, o que leva a crer que não
é possível a distinção entre os isómeros cis e trans do β‐caroteno através da análise dos espectros.
Figura 3.41 – Espectros de absorção do β‐caroteno sintético (não processado) e no extracto natural (após o processamento).

66
3.3.3. Conclusão acerca da precipitação do β‐caroteno natural
Este estudo permitiu verificar que é possível a micronização de um extracto de β‐caroteno produzido
biologicamente e contendo impurezas. O aspecto macroscópico destes cristais é em tudo semelhante ao
obtido com o β‐caroteno puro, à excepção da cor que, devido à presença de outros compostos que não
absorvem na mesma gama do espectro, se apresenta mais clara.
A avaliação do grau de pureza do produto de partida e do produto final não foi possível, devido à falta
de informação acerca da verdadeira composição deste extracto. A recta de calibração determinada para
o β‐caroteno puro não pode ser aqui aplicada, dado que não se pode quantificar as substâncias que
absorvem na mesma gama do espectro que o β‐caroteno. A análise dos cromatogramas permite
verificar que existem de facto substâncias no extracto que absorvem a cerca de 450 nm. Algumas destas
surgem acumuladas no líquido recolhido, embora este facto não seja suficientemente evidente para se
poder concluir que existiu purificação.
3.4. Conclusões e perspectivas futuras
Após a realização deste trabalho, é possível tecer algumas conclusões acerca do processo de
micronização utilizado e da sua aplicação ao composto estudado.
A micronização com anti‐solvente supercrítico foi aplicada com sucesso na formação de micropartículas
de β‐caroteno, utilizando como solvente o tetrahidrofurano e como anti‐solvente o dióxido de carbono
supercrítico. Mostrou‐se que é possível micronizar este carotenóide em condições experimentais abaixo
ou acima do ponto crítico, sendo a pressão mínima para que a precipitação ocorra, a 40°C, igual a 75
bar.
Dependendo da pressão imposta no sistema, foram conseguidas micropartículas com dimensões médias
entre 14 e 100 μm. A morfologia dos cristais obtidos e a sua agregação é também dependente da
pressão de trabalho. Estabeleceu‐se, ainda, uma relação linear entre a densidade da mistura onde
ocorre a precipitação e o tamanho médio das partículas obtidas. O controlo do tamanho das partículas
é, portanto, possível, através da manipulação das condições experimentais.
O estudo de solubilidades realizado neste trabalho demonstrou igualmente alguns pontos importantes
na termodinâmica do sistema estudado, com interesse em aplicações futuras deste sistema na
micronização ou noutros processos envolvendo fluidos supercríticos. Os dois resultados de maior
relevância mostram a diminuição exponencial da solubilidade do β‐caroteno na mistura supercrítica
CO2/THF com o aumento da temperatura, como consequência da diminuição da densidade do sistema, e
o aumento exponencial da solubilidade do mesmo composto com a fracção de THF na mistura, devido à
afinidade do soluto para este solvente orgânico. Provou‐se, igualmente, que a presença do co‐solvente
numa composição de aproximadamente 5% aumenta a solubilidade do β‐caroteno em cerca de duas
ordens de grandeza, relativamente ao CO2 puro em condições semelhantes de pressão e temperatura.
Verificou‐se, ainda, que o efeito da fracção molar de THF se sobrepõe ao efeito da pressão: ao

67
representar os resultados obtidos a 100 e a 130 bar, observou‐se que a solubilidade dependia apenas da
composição da mistura.
Um extracto de β‐caroteno produzido biologicamente foi também micronizado com sucesso. Este facto
demonstra que o processo pode ser aplicado na precipitação de produtos biológicos com aplicações nas
indústrias alimentar e farmacêutica, dado que o processamento é realizado com um gás inerte, não
tóxico e de baixo custo, e o produto final está isento de solventes orgânicos. No entanto, não se
constatou que houvesse purificação, dado que os cromatogramas do produto precipitado e recolhido no
líquido eram praticamente idênticos. Por outro lado, não existe forma de determinar a concentração
dos produtos inicial e final, de forma a compará‐los.
As perspectivas futuras da micronização com anti‐solvente supercrítico são várias, embora seja
necessário conhecer melhor o processo. No processamento do β‐caroteno, impõe‐se um estudo mais
abrangente da influência das condições experimentais sobre a dimensão das partículas obtidas. A
temperatura, a concentração e o caudal de solução e de CO2 são variáveis que, noutros estudos, já
provaram poder influenciar fortemente a forma e a dimensão das partículas resultantes. É
particularmente interessante que encontrem as condições óptimas de processamento para obtenção de
partículas ainda mais pequenas. Deve‐se também avaliar o desempenho de outros solventes menos
tóxicos e voláteis do que o tetrahidrofurano, dado que a sua utilização a nível industrial deverá estar
comprometida devido às desvantagens apontadas, que limitam o seu manuseamento.
Em termos de purificação de produtos biológicos, as condições de trabalho deverão igualmente ser alvo
de estudo, para que seja optimizado também o grau de pureza obtido. Dependendo das aplicações do
produto, pode‐se ainda ponderar as condições de processamento segundo os dois objectivos que o
processo pode ter: a purificação e a formação de micropartículas.
Este trabalho foi realizado num aparelho de micronização à escala piloto. Estudos sobre a
hidrodinâmica, os fenómenos de transferência de calor e massa e a termodinâmica do sistema em causa
deverão ser efectuados, para que a modelação deste tipo de sistema seja realizada, só assim sendo
possível um aumento de escala eficaz. Ao nível industrial, seria indispensável a recirculação do dióxido
de carbono, de forma a aumentar o rendimento, rentabilizar economicamente o processo e reduzir a
quantidade de efluente gasoso a tratar, e ainda a automatização dos sistemas de recolha do produto
precipitado e do solvente.

68
REFERÊNCIAS BIBLIOGRÁFICAS
[1] Alessi, P., Álvarez, L., Lafuente, B., Kikic, I., Vecchione, F.. Drug delivery systems with SC process:
precipitation of β‐carotene by SAS process and supercritical carbon dioxide impregnation. 5th
International Symposium on High Pressure Process Technology and Chemical Engineering. June
24‐27, 2007; Sergovia (Spain).
[2] Bakhbakhi, Y., Charpentier, P. A., Rohami, S., 2006. Experimental study of the GAS process for
producing microparticles of beclomethasone‐17,21‐dipropionate suitable for pulmonary delivery.
International Journal of Pharmaceutics, 309, 71‐80.
[3] Besenfelder, U., Solti, L., Seregi, J., Müller, M., Brem, G., 1996. Different roles for β‐carotene and
vitamin A in the reproduction of rabbits. Theriogeneology, 45, 1583‐1591.
[4] Bristow, S., Shekunov, T., Shekunov, B. Y., York, P., 2001. Analysis of the supersaturation and
precipitation process with supercritical CO2. Journal of Supercritical Fluids, 21, 257‐271.
[5] Bruno, T. J., Nieto de Castro, C. A., Hamel, J‐F. P., Palavra, A. M. F., 1993. Supercritical fluid
extraction of biological products. In Recovery processes for biological materials, John Wiley &
Sons Ltd.
[6] Carpenter, K. L. H., 2006. β‐Carotene: A coloruful killer of cancer cells? A commentary on “ROS‐
triggered caspase 2 activation and feedback amplification loop in β‐carotene‐induced apoptosis”.
Free Radical Biology & Medicine, 41, 418‐421.
[7] Cháfer, A., Fornari, T., Berna, A., Ibañez, E., Reglero, G., 2005. Solubility of solid carnosic acid in
supercritical CO2 with ethanol as co‐solvent. Journal of Supercritical Fluids, 34, 323‐329.
[8] Chang, C. J., Randolph, A. D., 1991. Separation of β‐carotene mixtures precipitated from liquid
solvents with high pressure CO2. Biotechnol. Prog., 7, 275‐278.
[9] Chow, A. H. L., Tong, H. H. Y., Chattopadhyay, P., Shekunov, B. Y., 2007. Particle engineering for
pulmonary drug delivery. Pharmaceutical Research, Vol. 24, No. 3, 411‐437.
[10] Cocero, M. J., Ferrero, S., 2002. Crystallization of β‐carotene by a GAS process in batch; effect of
operating conditions. Journal of Supercritical Fluids, 22, 237‐245.
[11] Costa, M. S., Duarte, A. R. C., Cardoso, M. M., Duarte, C. M. M., 2007. Supercritical antisolvent
precipitation of PHBV microparticles. International Journal of Pharmaceutics, 328, 72‐77.
[12] Cross, A. M., Gunter, M. J., Wood, R. J., Pietinen, P., Taylor, P. R., Virtamo, J., Albanes, D., Sinha,
R., 2006. Iron and colorectal cancer risk in the α‐tocopherol, β‐carotene cancer prevention study.
Int. J. Cancer, 118, 3147‐3152.
[13] De Marco, I., Reverchon, E., 2006. Supercritical antisolvent micronization of α‐ciclodextrin. Eighth
Conference on Supercritical Fluids and Their Applications.
[14] Delgado‐Vargas, F., Jiménez, A. R., Paredes‐López, O., 2000. Natural pigments: Carotenoids,
Anthocianins, and Betalains – Characteristics, biosynthesis, processing, and stability. Critical
Reviews in Food Science and Nutrition, 40 (3), 173‐289.

69
[15] Dukhin, S. S., Shen, Y., Dave, R., Pfeffer, R., 2005. Droplet mass transfer, intradroplet nucleation
and submicron particle production in two‐phase flow of solvent‐supercritical antisolvent
emusion. Colloids and Surfaces A: Physicochem. Eng. Aspects, 261, 163‐176.
[16] Edge, R., McGarvey, D. J., Truscott, T. G., 1997. The carotenoids as anti‐oxidants – a review.
Journal of Photochemistry and Photobiology B: Biology, 41, 189‐200.
[17] Faber, M., van Jaarsveld, P. J., 2007. The production of provitamin A‐rich vegetables in home‐
gardens as a means of addressing vitamin A deficiency in rural African communities. Journal of
the Science of Food and Agriculture, 87, 366‐377.
[18] Foster, N., Mammucari, R., Dehghani, F., Barrett, A., Bezanehtak, K., Coen, E., Combes, G., Meure,
L., Ng, A., Regtop, H. L., Tandya, A., 2003. Processing pharmaceutical compounds using dense gas
technology. Ind. Eng. Chem. Res., 42, 6476‐6493.
[19] Fraser, P. D., Bramley, P. M., 2004. The biosynthesis and nutritional uses of carotenoids. Progress
in Lipid Research, 43, 228‐265.
[20] Giry, K., Péan, J. M., Giraud, L., Marsas, S., Rolland, H., Wüthrich, P., 2006. Drug/lactose co‐
micronization by jet milling to improve aerosolization properties of a powder for inhalation.
International Journal of Pharmaceuticals, 321, 162‐166.
[21] Goralczyk, R., Wertz, K., Lenz, B., Riss, G., Hunziker, P. B., Geatrix, B., Aebischer, C‐P., Bachmann,
H., 2005. β‐Carotene interaction with NNK in the AJ‐mouse model: Effects on the cell
proliferation, tumor formation and retinoic acid responsive genes. Biochimica et Biophysics Acta,
1740, 179‐188.
[22] Herceg, Z., Lelas, V., Brnčič, M., Tripalo, B., Ježek, D., 2004. Fine milling and micronization of
organic and inorganic materials under dynamic conditions. Powder Technology, 139, 111‐117.
[23] Hu, J., Johnston, K. P., Williams III, R. O., 2003. Spray freezing into liquid (SFL) particle engineering
technology to enhance dissolution of poorly water soluble drugs: organic solvent versus
organic/aqueous co‐solvent systems. European Journal of Pharmaceutical Sciences, 20, 295‐303.
[24] Hülsmann, S., Backensfeld, T., Keitel, S., Bodmeier, R., 2000. European Journal of Pharmaceutics
and Biopharmaceutics, 49, 237‐242.
[25] Jung, J., Perrut, M., 2001. Particle design using supercritical fluids: Literature and patent survey.
Journal of Supercritical Fluids, 20, 179‐219.
[26] Kalogiannis, C., Pavlidou, E., Panayiotou, G., 2005. Production of amoxicillin microparticles by
supercritical antisolvent precipitation. Ind. Eng. Chem. Res., 44, 9339‐9346.
[27] Kopcak, U., Mohamed, R. S., 2005. Caffeine solubility in supercritical carbon dioxide/co‐solvent
mixtures. Journal of Supercritical Fluids, 34, 209‐214.
[28] Kordikowski, A., Schenk, A. P., Van Nielsen, R. M., Peters, C. J., 1995. Volume expansions and
vapor‐liquid equilibria of binary mixtures of a variety of polar solvents and certain near‐critical
solvents. Journal of Supercritical Fluids, 8, 205‐216.
[29] Krukonis, V. J., 1986. Supercritical fluid nucleation of difficult to comminute solids. AIChE Fall
Meeting, San Francisco. CA paper 1408.

70
[30] Latscha, T., 1990. Carotenoids – Their nature and significance in animal feeds. F. Hoffmann‐La
Roche Ltd.
[31] Leach, G., Oliveira, G., Morais, R., 1998. Production of a carotenoid‐rich product by alginate
entrapment and fluid‐bed drying of Dunaliella salina. J. Sci. Food. Agric., 76, 298‐302.
[32] Leuenberger, H., 2002. Spray freeze‐drying – the process of choice for low water soluble drugs?.
Journal of Nanoparticle Research, 4, 111‐119.
[33] Li, J., Rodrigues, M., Paiva, A., Matos, H. A., Azevedo, E. G., 2007. Vapor‐liquid equilibria and
volume expansion of the tetrahydrofuran/CO2 system: application to a SAS‐atomization process.
Journal of Supercritical Fluids, 41 (3), 343‐351.
[34] Majerik, V., Charbit, G., Badens, E., Horváth, G., Szokonya, L., Bosc, N., Teillaud, E., 2007.
Bioavaibility enhancement of an active substance by supercritical antisolvent precipitation.
Journal of Supercritical Fluids, 40, 101‐110.
[35] Mammucari, R, Dehghabu, F., Foster, N. R., 2006. Dense gas processing of micron‐sized drug
formulations incorporating hydroxypropylated and methylated beta‐cyclodextrin. Pharmaceutical
Research, Volume 23, No. 2, 429‐437.
[36] Martín, A., Cocero, M. J., 2004. Numerical modeling of jet hydrodynamics, mass transfer, and
crystalization kinetics in the supercritical antisolvent (SAS) process. Journal of Supercritical Fluids,
32, 203‐219.
[37] Martín, A., Gutiérrez, L., Mattea, F., Cocero, M. J., 2007. Precipitation of mandelic acid with a
supercritical antisolvent process: experimental and theoretical analysis, optimization and
scaleup. Ind. Eng. Chem. Res., 46, 1552‐1562.
[38] Martín, A., Mattea, F., Gutiérrez, L., Miguel, F., Cocero, M. J., 2007. Co‐precipitation of
carotenoids and bio‐polymers with the supercritical anti‐solvent process. Journal of Supercritical
Fluids, 41, 138‐147.
[39] Maschke, A., Calí, N., Appel, B., Kiermaier, J., Blunk, T., Göpferich, A., 2006. Micronization of
insulin by high pressure homogeneization. Pharmaceutical Research, Volume 23, No. 9, 2220‐
2229.
[40] Mendes, R. L., Nobre, B. P., Cardoso, M. T., Pereira, A. P., Palavra, A. F., 2003. Supercritical carbon
dioxide extraction of compounds with pharmaceutical importance from microalgae. Inorganica
Chimica Acta, 356, 328‐334.
[41] Mendes, R. L., Nobre, B. P., Coelho, J. P., Palavra, A. F., 1999. Solubility of β‐carotene in
supercritical carbon dioxide and ethane. Journal of Supercritical Fluids, 16, 99‐106.
[42] Miguel, F., Martín, A., Gamse, T., Cocero, 2006. Supercritical anti solvent precipitation of
lycopene; effect of operating parameters. Journal of Supercritical Fluids, 36, 225‐235.
[43] Murthy, K. N. C., Vanitha, A., Rajesha, J., Swamy, M. M., Sowmya, P. R., Ravishankar, A., 2005. In
vivo antioxidant activity of carotenoids from Dunaliella salina – a green microalga. Life Sciences,
76, 1381‐1390.

71
[44] Muzandu, K., Ishizuka, M., Sakamoto, K. Q., Shaban, Z., El Bohi, K., Kazusaka, A., Fujita, S., 2006.
Effect of lycopene and β‐carotene on peroxynitrite‐mediated cellular modifications. Toxicology
and Applied Pharmacology, 215, 330‐340.
[45] Omenn, G., 1998. Chemoprevention of lung cancer: The rise and demise of beta‐carotene. Annu.
Rev. Public Health, 19, 73‐99.
[46] Palozza, P., Serini, S., Torsello, A., Di Nicuolo, F., Piccioni, E., Ubaldi, V., Pioli, C., Wolf, F. I.,
Calviello, G., 2003. β‐Carotene regulates NF‐κB DNA‐binding activity by a redox mechanism on
human leukaemia and colon adenocarcinoma cells. Journal of Nutrition, 133, 381‐388.
[47] Park, H. J., Kim, M‐S., Lee, S., Kim, J‐S., Woo, J‐S., Park, J‐S‐, Hwang, S‐J., 2007. Recrystallization of
fluconazole using the supercritical antisolvent (SAS) process. International Journal of
Pharmaceutics, 328, 152‐160.
[48] Parvin, S. G., Bhaskaram, P., Sivakumar, B., 2000. A novel pharmacokinetic approach to
determine the relative efficiency of conversion of β‐carotene to vitamin A in rats and children.
Nutrition Research, Vol. 20, No. 5, 633‐645.
[49] Rasenack, N., Müller, B. W., 2004. Micron‐size drug particles: Common and novel techniques.
Pharmaceutical Development and Technology, Vol. 9, No. 1, 1‐13.
[50] Rehman, M., Shekunov, B. Y., York, P., Colthorpe, P., 2001. Solubility and precipitation of nicotinic
acid in supercritical carbon dioxide. Journal of Pharmaceutical Sciences, Vol. 90, 1570‐1582.
[51] Reverchon, E., 1999. Supercritical antisolvent precipitation of micro‐ and nano‐particles. Journal
of Supercritical Fluids, 15, 1‐21.
[52] Reverchon, E., Caputo, G., De Marco, I., 2003. Role of phase behavior and atomization in
supercritical antisolvent precipitation. Ind. Eng. Chem. Res., 42, 6406‐6414.
[53] Reverchon, E., De Marco, I., 2006. Supercritical antisolvent precipitation of Cephalosporins.
Powder Technology, 164, 139‐146.
[54] Reverchon, E., De Marco, I., Caputo, G., Della Porta, G., 2003. Pilot scale micronization of
amoxillin by supercritical antisolvent precipitation. Journal of Supercritical Fluids, 26, 1‐7.
[55] Reverchon, E., Della Porta, G., 1999. Production of antibiotic micro‐ and nano‐particles by
supercritical antisolvent precipitation. Powder Technology, 106, 23‐29.
[56] Reverchon, E., Della Porta, G., 2003. Micronization of antibiotics by supercritical assisted
atomization. Journal of Supercritical Fluids, 26, 243‐252.
[57] Reverchon, E., Della Porta, G., De Rosa, I., Subra, P., Letourneur, D., 2000. Supercritical
antisolvent micronization of some biopolymers. Journal of Supercritical Fluids 18, 239‐245.
[58] Reverchon, E., Della Porta, G., Di Trolio, A., Pace, S., 1998. Supercritical antisolvent precipitation
of nanoparticles of superconductor precursors. Ind. Eng. Chem. Res., 37, 952‐958.
[59] Roelands, C., Horst, J., Kramer, H., Jansens, P., 2006. Analysis of nucleation rate measurements in
precipitation processes. Crystal Growth & Design, Vol.6, No.6, 1380‐1392.

72
[60] Rogers, T. L., Johnston, K. P., Williams, R. O., 2001. Solution‐based particle formation of
pharmaceutical powders by supercritical or compressed fluid CO2 and cryogenic spray‐freezing
technologies. Drug Development and Industrial Pharmacy, 27 (10), 1003‐1015.
[61] Romanchik, J. E., Harrison, E. H., Morel, D. W., 1997. Addition of lutein, lycopene, or b‐carotene
to LDL or serum in vitro: Effects on carotenoid distribution, LDL composition, and LDL oxidation.
Nutritional Biochemistry, 8, 681‐688.
[62] Ruckenstein, E., Djikaev, Y. S., 2005. Recent developments in the kinetic theory of nucleation.
Advances in Colloid and Interface Science, 118, 51‐72.
[63] Schierle, J., Schellenberger, T., Fizet, C., Betz, R., 2002. A simple spectrophotometric
determination of total β‐carotene in food additives with varying E/Z‐isomer ratios using an
isobestic wavelength. Eur. Food. Res. Technol., 215, 268‐274.
[64] Shekunov, B. Y., Chattopadhyay, P., Tong, H. H. Y., Chow, A. H. L., 2007. Particle size analysis in
pharmaceutics: Principles, methods and applications. Pharmaceutical Research, Vol. 24, No. 2,
203‐227.
[65] Shekunov, B. Y., York, P., 2000. Crystallization processes in pharmaceutical technology and drug
design. Journal of Crystal Growth, 211, 122‐136.
[66] Steckel, H., Brandes, H. G., 2004. A novel spray‐drying technique to produce low density particles
for pulmonary delivery. International Journal of Pharmaceutics, 278, 187‐195.
[67] Steckel, H., Markefka, P., teWierik, H., Kammelar, R., 2006. Effect of milling and sieving on
functionality of dry powder inhalation products. International Journal of Pharmaceutical, 309, 51‐
59.
[68] Subra, P., Laudani, C‐G., Vega‐González, A., Reverchon, E., 2005. Precipitation and phase behavior
of theophylline in solvent‐supercritical CO2 mixtures. Journal of Supercritical Fluids, 35, 95‐105.
[69] Tavares Cardoso, M. A., Monteiro, G. A., Cardoso, J. P., Prazeres, T. J. V., Figueiredo, J. M.F.,
Martinho, J. M. G., Palavra, A. M. F. Supercritical antisolvent micronization of minocycline
hydrochloride. Aceite pela revista Journal of Supercritical Fluids.
[70] Tenorio, A., Gordillo, M. D., Pereyra, C. M., Martínez de la Ossa, E. J., 2007. Relative importance
of the operating conditions involved in the formation of nanoparticles of ampicillin by
supercritical antisolvent precipitation. Ind. Eng. Chem. Res., 46, 114‐123.
[71] Tenorio, A., Gordillo, M. D., Pereyra, C., Martínez de la Ossa, E. J., 2007. Controlled submicro
particle formation of ampicillin by supercritical antisolvent precipitation. Journal of Supercritical
Fluids, 40, 308‐316.
[72] Törnwall, M. E., Virtamo, J., Korhonen, P. A., Virtanen, M. J., Albanes, D., Huttunen, J. K., 2004.
Postintervention effect of alpha tocopherol and beta carotene on different strokes. Stroke, 35,
1908‐1913.
[73] Vági, E., Simándi, B., Vásárhelyiné, K. P., Daood, H., Kéry, Á., Doleschall, F., Nagy, B., 2007.
Supercritical carbon dioxide extraction of carotenoids, tocopherols and sitosterols from industrial
tomato by‐products. Journal of Supercritical Fluids, 40, 218‐226.

I
APÊNDICE A
Cálculo da quantidade de solvente a injectar para atingir o estado
estacionário
Exemplo de cálculo para o ensaio nas seguintes condições: P = 130 bar
T = 40°C
QL = 1 mL/min
[β‐caroteno/THF] = 6 mg/mL
Caudal de saída de CO2 = 6,56 L/min (PTN)
Dados:
ρCO2 (130 bar, 40°C) = 0,74304 g/mL
ρCO2 (1 bar, 25°C) = 0,0017842 g/mL
ρTHF = 0,88 g/mL
Vcélula = 300 mL
Base de cálculo = 1 min
Balanço mássico ao CO2:
(CO2)entra = (CO2)sai = 6,56 L/min × 1,7842 g/L = 11,704 g/min
Cálculo da massa de CO2 na célula:
(CO2)célula = 0,74304 g/mL × 300 mL = 222,912 g
Balanço mássico ao THF:
(THF)entra = (THF)sai = 1 mL/min × 0,88 g/mL = 0,88 g/min
Cálculo da composição dentro da célula:
[THF/CO2] = 0,88 g THF / 11,704 g CO2 = 0,075188 g THF/g CO2
Cálculo da massa e do volume de THF na célula:
(THF)célula = 0,75188 g THF/g CO2 × 222,912 g CO2 = 16,76 g THF
VTHF = 16,76 g / 0,88 g/mL = 19 mL

II
APÊNDICE B
Cálculo do tempo de secagem
O tempo de lavagem do produto para remoção do solvente da célula é calculado segundo o método
proposto por Reverchon [58].
Este cálculo considera a célula como um tanque perfeitamente agitado (CSTR) com 300 mL de volume e
caudal de CO2 de 6,56 L/min PTN (estabelecido no decorrer do ensaio) e cujo balanço mássico se
representa na equação (A.1).
dtdC
VCQCQ sssee += (A.1)
Depois de parada a adição de solvente, a concentração de entrada, Ce, é nula. Resolvendo a equação
(A.1), obtém‐se a equação (A.2).
⎟⎟⎠
⎞⎜⎜⎝
⎛=
f
i
s CC
QV
t ln (A.2)
Para as condições de 100 bar e 40°C (ρCO2 = 0,62861 g/mL), Qs = 18,62 mL/min.
O tempo necessário para que a concentração final de THF na célula passe para 1% do seu valor inicial é,
então, cerca de 75 minutos.
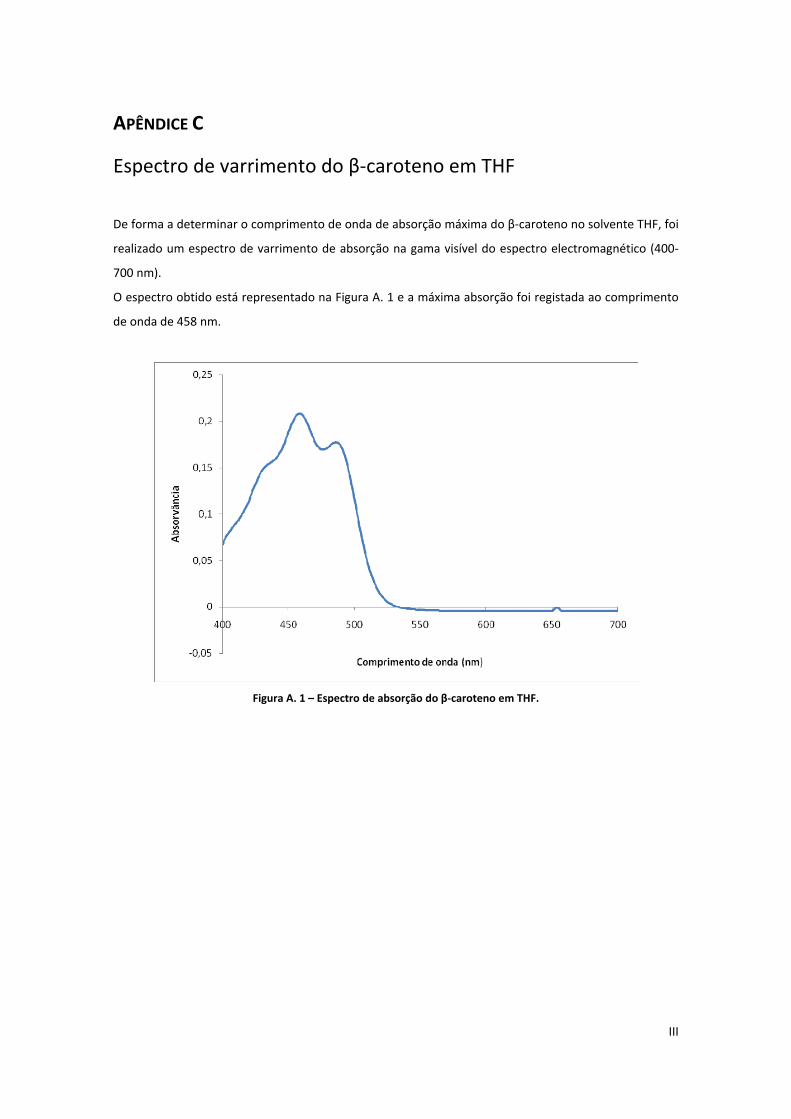
III
APÊNDICE C
Espectro de varrimento do β‐caroteno em THF
De forma a determinar o comprimento de onda de absorção máxima do β‐caroteno no solvente THF, foi
realizado um espectro de varrimento de absorção na gama visível do espectro electromagnético (400‐
700 nm).
O espectro obtido está representado na Figura A. 1 e a máxima absorção foi registada ao comprimento
de onda de 458 nm.
Figura A. 1 – Espectro de absorção do β‐caroteno em THF.
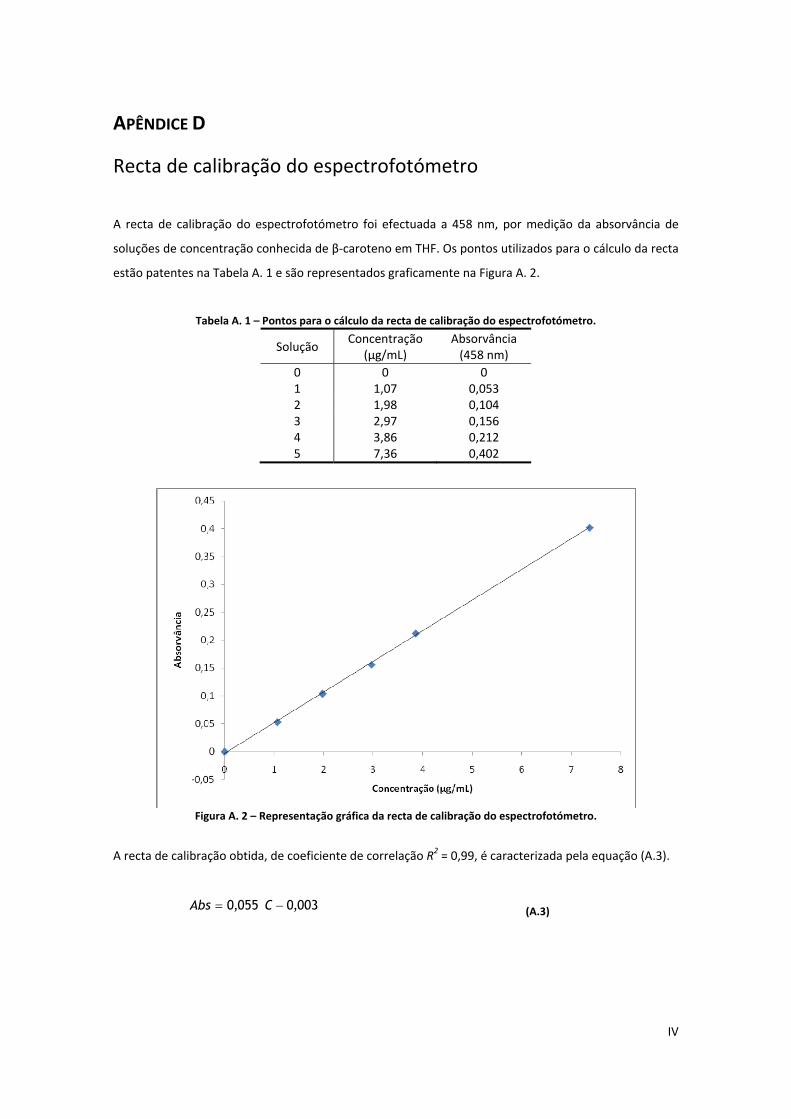
IV
APÊNDICE D
Recta de calibração do espectrofotómetro
A recta de calibração do espectrofotómetro foi efectuada a 458 nm, por medição da absorvância de
soluções de concentração conhecida de β‐caroteno em THF. Os pontos utilizados para o cálculo da recta
estão patentes na Tabela A. 1 e são representados graficamente na Figura A. 2.
Tabela A. 1 – Pontos para o cálculo da recta de calibração do espectrofotómetro.
Solução Concentração
(µg/mL) Absorvância (458 nm)
0 0 01 1,07 0,0532 1,98 0,1043 2,97 0,1564 3,86 0,2125 7,36 0,402
Figura A. 2 – Representação gráfica da recta de calibração do espectrofotómetro.
A recta de calibração obtida, de coeficiente de correlação R2 = 0,99, é caracterizada pela equação (A.3).
003,0055,0 −= CAbs (A.3)
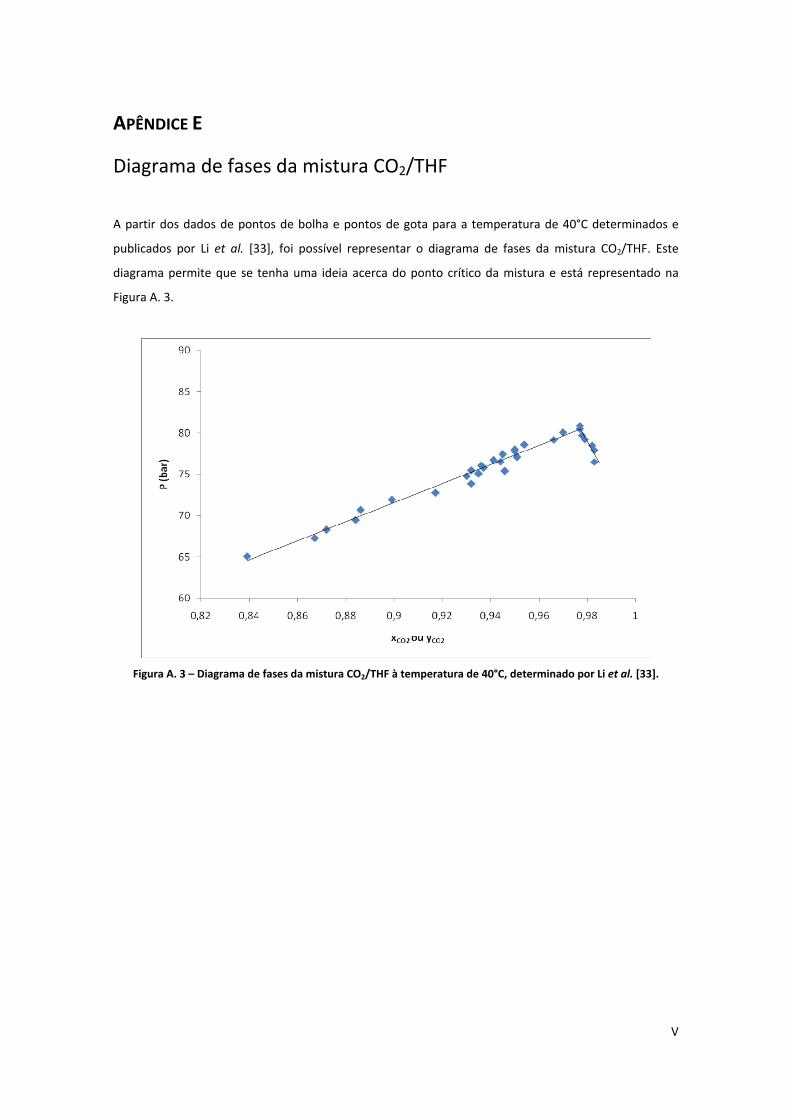
V
APÊNDICE E
Diagrama de fases da mistura CO2/THF
A partir dos dados de pontos de bolha e pontos de gota para a temperatura de 40°C determinados e
publicados por Li et al. [33], foi possível representar o diagrama de fases da mistura CO2/THF. Este
diagrama permite que se tenha uma ideia acerca do ponto crítico da mistura e está representado na
Figura A. 3.
Figura A. 3 – Diagrama de fases da mistura CO2/THF à temperatura de 40°C, determinado por Li et al. [33].
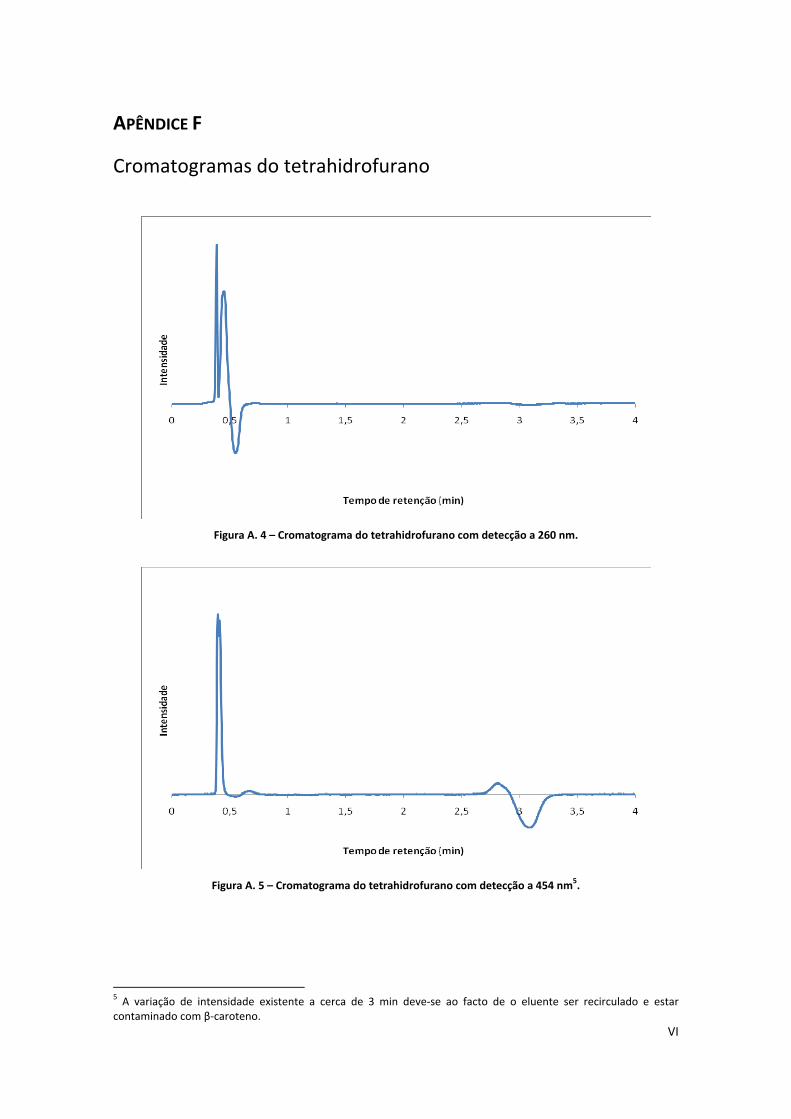
VI
APÊNDICE F
Cromatogramas do tetrahidrofurano
Figura A. 4 – Cromatograma do tetrahidrofurano com detecção a 260 nm.
Figura A. 5 – Cromatograma do tetrahidrofurano com detecção a 454 nm5.
5 A variação de intensidade existente a cerca de 3 min deve‐se ao facto de o eluente ser recirculado e estar contaminado com β‐caroteno.

![WeightedHurwitznumbers andhypergeometric -functions ... · Certain of these may also be shown to satisfy differential constraints, the so-called Vira-soro constraints [33,37,52],](https://static.fdocument.org/doc/165x107/5f07152a7e708231d41b372e/weightedhurwitznumbers-andhypergeometric-functions-certain-of-these-may-also.jpg)
















