
Lymphoid Enhancer Factor-1 and Beta-Catenin Inhibit Runx2 ...
41
Lymphoid Enhancer Factor-1 and Beta-Catenin Inhibit Runx2-Dependent Transcriptional Activation of the Osteocalcin Promoter Running Title: LEF1 and β-catenin inhibit Runx2 Rachel A. Kahler and Jennifer J. Westendorf* University of Minnesota Cancer Center, Department of Orthopaedic Surgery, and Graduate Program in Microbiology, Immunology and Cancer Biology, Minneapolis, MN 55455 *Corresponding Author: Jennifer J. Westendorf, PhD University of Minnesota Cancer Center MMC 806, 420 Delaware St. SE Minneapolis, MN 55455 Phone: 612-626-3365, Fax: 612-626-4915 E-mail: [email protected] Copyright 2003 by The American Society for Biochemistry and Molecular Biology, Inc. JBC Papers in Press. Published on January 27, 2003 as Manuscript M211443200 by guest on April 7, 2018 http://www.jbc.org/ Downloaded from
-
Upload
hoangquynh -
Category
Documents
-
view
217 -
download
1
Transcript of Lymphoid Enhancer Factor-1 and Beta-Catenin Inhibit Runx2 ...

Lymphoid Enhancer Factor-1 and Beta-Catenin Inhibit Runx2-Dependent
Transcriptional Activation of the Osteocalcin Promoter
Running Title: LEF1 and β-catenin inhibit Runx2
Rachel A. Kahler and Jennifer J. Westendorf*
University of Minnesota Cancer Center, Department of Orthopaedic Surgery, and Graduate
Program in Microbiology, Immunology and Cancer Biology,
Minneapolis, MN 55455
*Corresponding Author:
Jennifer J. Westendorf, PhD
University of Minnesota Cancer Center
MMC 806, 420 Delaware St. SE
Minneapolis, MN 55455
Phone: 612-626-3365, Fax: 612-626-4915
E-mail: [email protected]
Copyright 2003 by The American Society for Biochemistry and Molecular Biology, Inc.
JBC Papers in Press. Published on January 27, 2003 as Manuscript M211443200 by guest on A
pril 7, 2018http://w
ww
.jbc.org/D
ownloaded from

LEF1 and β-catenin inhibit Runx2
2
Summary
Functional control of the transcription factor, Runx2, is crucial for normal bone
formation. Runx2 is detectable throughout osteoblast development and maturation and
temporally regulates several bone-specific genes. In this study, we identified a novel post-
translational mechanism regulating Runx2-dependent activation of the osteocalcin promoter. A
functional binding site for the high mobility group protein, LEF1, was found adjacent to the
proximal Runx2 binding site in the osteocalcin promoter. In transcription assays, LEF1
repressed Runx2-induced activation of the mouse osteocalcin 2 promoter in several osteoblast-
lineage cell lines. Mutations in the LEF1 binding site increased the basal activity of the
osteocalcin promoter; however, the LEF1 recognition site in the osteocalcin promoter was
surprisingly not required for LEF1 repression. A novel interaction between the DNA binding
domains of Runx2 and LEF1 was identified and found crucial for LEF1-mediated repression of
Runx2. LEF1 is a nuclear effector of the Wnt/LRP5/β-catenin signaling pathway, which is also
essential for osteoblast proliferation and normal skeletal development. A constitutively active β-
catenin enhanced LEF1-dependent repression of Runx2. These data identify a novel mechanism
of regulating Runx2 activity in osteoblasts and link Runx2 transcriptional activity to ß-catenin
signaling.
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LEF1 and β-catenin inhibit Runx2
3
Introduction
Runx2 (Cbfa1, AML-3, PEBP2αA) is one of three mammalian Runt-domain factors and
is essential for bone formation (1). Runx2 deficiency is developmentally lethal because it
inhibits osteoblast development, chondrocyte differentiation in some skeletal elements, and
vascular invasion into cartilage to prevent skeletal ossification (2-4). Moreover, a dominant-
negative Runx2 protein prevents osteoblast differentiation in adult mice without decreasing
osteoblast numbers (5). Runx2-haploinsufficiency hinders intramembranous bone formation and
causes the rare skeletal disorder, cleidocranial dysplasia (6). Interestingly, Runx2 over
expression induces bone fragility by blocking osteoblast differentiation and enhancing bone
resorption (7,8). These genetic studies demonstrate that cellular control of Runx2 expression
levels and function is crucial for skeletal development.
At the molecular level, Runx2 activity is regulated by multiple transcriptional and post-
translational mechanisms (9). Runx proteins activate or repress gene expression by binding the
DNA sequence, TGPuGGTPu (10). Runx binding sites are often necessary but are not sufficient
for transcriptional regulation of tissue-specific genes; thus it has been hypothesized that Runx
proteins are organizers that facilitate the assembly of transcriptional regulatory complexes on
gene regulatory elements (11,12). Runx factors, in fact, interact with numerous transcription
factors (e.g. AP1, C/EBP, Ets1, SMADs) and transcriptional co-factors (e.g. p300, ALY,
mSin3A, TLE, HDAC6) to regulate tissue-specific gene expression (13-16). Runx1-dependent
trans-activation of the T cell receptor enhancer is also increased indirectly by DNA binding
proteins, such as LEF1, which bends DNA to facilitate long-distance cooperative interactions
between Runx1 and Ets1 (17,18).
LEF1 is a high mobility group (HMG) protein and nuclear effector of the canonical Wnt
signaling pathway (19,20). LEF1 binds the consensus DNA sequence C/TCTTTGAA in the
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LEF1 and β-catenin inhibit Runx2
4
minor groove, creates a 130o bend in the double helix and alters of binding of other transcription
factors to neighboring sites (17,21). LEF1 transcriptional activity is regulated by interactions
with transcriptional co-activators and co-repressors. In the absence of Wnt signals, LEF1 binds
to transcriptional co-repressors TLE, CtBP and HDACs to inhibit gene expression (22). Wnts
convert LEF1 into a transcriptional activator by stimulating the disassembly the GSK-3-Axin-
APC multi-protein complex to prevent ubiquitin-mediated degradation of ß-catenin (22,23).
Increased ß-catenin expression facilitates its translocation to the nucleus where it displaces co-
repressors from LEF1, binds LEF1 and recruits co-activators to induce gene transcription
(22,23). LEF1, c-myc and cyclin D1 are among many genes whose promoters are activated by
the Wnt/ß-catenin/LEF1 pathway (24-26). It was recently demonstrated that this canonical
pathway also directly represses gene transcription in a manner dependent upon Wnt
concentrations. In Drosophila, Wnt/wingless (Wg) induces Armadillo (ß-catenin homologue)
and dTCF1/Pangolin (LEF1 homologues) to repress transcription of shavenbaby, stripe,
decapentaplegic and daughterless (27-30). The mechanism of Wnt-induced transcriptional
repression through LEF1 binding sites has not yet been defined but these studies demonstrate
that Wnts stimulate multiple, complex nuclear events.
The canonical Wnt signaling pathway regulates bone density by regulating postnatal
osteoblast proliferation (31). Inactivating mutations in Wnt receptor, LRP5, decrease bone mass
accrual during growth and cause the autosomal-recessive disorder, osteoporosis-pseudoglioma
syndrome (OPPG), in humans and mice by decreasing osteoblast proliferation and without
altering Runx2 mRNA levels (32,33). Conversely, activating mutations in LRP5 are linked to
autosomal-dominant high bone mass traits and increased osteoblast proliferation (34,35). The
downstream effects of Wnt signaling on osteoblast-specific gene transcription have not yet been
described. In this report, we demonstrate that Runx2-dependent activation of the most
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LEF1 and β-catenin inhibit Runx2
5
osteoblast-specific gene, osteocalcin, is repressed by LEF1. A constitutively activated ß-catenin
protein enhances LEF1-mediated repression of Runx2. These data suggest that in addition to
stimulating osteoblast proliferation, the Wnt signaling pathway may slow differentiation by
inhibiting Runx2 activity.
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LEF1 and β-catenin inhibit Runx2
6
Experimental Procedures
Plasmids. Mammalian expression plasmids (pCMV5) for Runx2 (MRIPV and MASNSL
isoforms) were provided by Dr. Scott Heibert and Dr. Gary Stein, respectively. pCMV5-LEF1-
HA was obtained from Dr. Rudolf Grosschedl. LEF1 mutants were generated from pCMV5-
LEF1-HA with gene specific oligonucleotides (primer sequences available upon request) and
PCR. PCR products were cloned into pPCR-Script Amp SK(+) (Stratagene). GAL fusion
proteins were constructed by subcloning LEF1 fragments from pPCR-Script into pENTR-3C
(Invitrogen) with DraI and EcoRI and then recombining them into the pDEST-GAL(M2) vector
with the GATEWAY Clonase Enzyme mix (14). The β-catenin vector Xβ/pcDNA3.1+ was
provided by Dr. Jennifer Hall (36). The osteocalcin promoter reporters mOG2-luc and p6OSE2-
luc were provided by Dr. Gerard Karsenty (37). The GAL-responsive pAH205 was provided by
Dr. Vivian Bardwell. Mutant mOG2-luc reporters were generated using QuikChange
(Stratagene) and gene-specific oligonucleotides containing mutations in the Runt and LEF1
binding sites (mutations are depicted in Figure 1A, primer sequences are available upon request.)
Cell lines and transfection. The murine preosteoblast cell line MC3T3 (clone E1,
passage 20 or less), murine mesenchymal precursor cell line C3H10T1/2 and rat osteosarcoma
cell line UMR-106 (clone 106.01) were cultured in minimum essential medium (MEM)
supplemented with 10% FBS, 292 mg/L L-glutamine, 1X non-essential amino acids, and 100 U
penicillin and 100 µg/ml streptomycin. Prior to transfection, MC3T3 and C3H10T1/2 were
seeded in 6-well plates at a density of 1.25 x 105 cells/well. UMR 106 cells were seeded in 12-
well plates at a density of 7 x 104 cells/well. MC3T3, C3H10T1/2, and UMR-106 cells were
transiently transfected with mOG2-luc (300 ng, 6-well; 100 ng, 12-well), pRL-TK (50 ng, 6-
well; 5 ng, 12-well) or pCMV-SEAP (500 ng, 6-well) and expression plasmids pCMV5-LEF1-
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LEF1 and β-catenin inhibit Runx2
7
HA, pCMV5-Runx2 (MRIPV or MASNS isoform) and β-catenin as indicated with either
LipofectAMINE or LipofectAMINE 2000 (Invitrogen) and as directed by the manufacturer.
COS cells were transfected using DEAE-dextran. For differentiation assays, MC3T3 cells were
seeded at a concentration of 2000 cells/ cm2 and cultured in α−MEM supplemented with 10%
FBS, 50 µg/ml ascorbic acid, 10 mM β-glycerol phosphate, 100 U/ml penicillin and 100 µg/ml
streptomycin.
Immunofluorescence. MC3T3 cells grown on coverslips were washed twice with PBS
and fixed at room temperature for 30 minutes in 4% paraformaldehyde. Fixed cells were lysed
for 5 minutes at room temperature in 0.3% Triton X-100 in PBS and blocked for 30 minutes at
room temperature in immunofluorescence (IF) solution (3% BSA, 20mM MgCl2, 0.3% Tween20,
PBS). The cells were then incubated with either anti-LEF-1 (REMB1, Exalpha Biologicals) or
anti-β-catenin (BD Biosciences) diluted to 1 µg/ml in IF solution for one hour at 37°C in a
humidified chamber. Excess primary antibody was removed by washing three times for 5
minutes in PBS + 0.1% TX-100. The cells were incubated for 30 minutes with goat-anti-mouse
IgG conjugated to Alexa 546 (1:50 dilution in IF solution) at 37°C in a humidified chamber.
Excess secondary antibody was removed by washing three times for 5 minutes in PBS + 0.1%
TX-100. The nuclei were stained with 5 µg/ml Hoescht 3258 for 5 minutes. Coverslips were
mounted on glass microscope slides and viewed with fluorescence microscopy.
Transcription assays. Cells were harvested two days post-transfection and luciferase
expression was measured using the Luciferase Assay kit, or Dual Luciferase Assay kit
(Promega). Firefly luciferase activities were normalized to either human placental secreted
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LEF1 and β-catenin inhibit Runx2
8
alkaline phosphatase (SEAP) or renilla luciferase. SEAP values were determined as previously
described (38).
In vitro protein interaction studies. LEF1 proteins were in vitro transcribed and
translated from pPCR-Script-LEF1 in the presence of 35S-labeled amino acids using rabbit
reticulocyte lysates (Promega). GST and GST-Runx fusion proteins were produced in E. coli
and extracted in lysis buffer A (10 mM MES, pH 6.5, 150 mM NaCl, 2 mM MgCl2, 0.5 mM
EDTA, 0.5% Triton X-100, 5 mM DTT, and protease inhibitors (aprotinin, leupeptin, PMSF),
and 50µg/ml ethidium bromide as indicated) with sonication. GST proteins were purified with
glutathione-sepharose beads and incubated with radiolabeled LEF1 proteins in the presence or
absence of 50 µg/ml ethidium bromide. The beads were then washed three times with buffer A,
resuspended in Laemmli buffer, boiled and pelleted by centrifugation. Proteins in the
supernatant were resolved by SDS-8% PAGE. The gel was fixed with 40% methanol and 10%
acetic acid, incubated with Amplify (Amersham Pharmacia Biotech), dried and exposed to film.
Electrophoretic mobility shift assays. Double stranded probes derived from the mOG2
osteocalcin promoter (wild type sequence: 5’-
CAATCACCAACCACAGCATCCTTTGGGTTTGAC-3’) were end-labeled with α32P-dATP
using Klenow DNA polymerase. COS cell lysates (5 µg) expressing Runx2 or LEF1 (HA
tagged) were incubated with an equal volume of 2X probe mix (100 mM HEPES, pH 7.8, 5 mM
MgCl2 0.5 mM EGTA, pH 8.0, 2 mM DTT, 200 mM KCl, 8.25% ficoll, 12.5 µg/ml salmon
sperm DNA, 41.25 pg/µl probe) at room temperature for 30 minutes. For supershift assays, 240
µg/ml of anti-Runx2 (39) or HA antiserum (Sigma) was added to the binding reaction for 30
minutes on ice. Unlabeled consensus binding sites for either Runx (5'-
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LEF1 and β-catenin inhibit Runx2
9
AATTCGAGTATTGTGGTTAATACG-3') or LEF1 (5'-AATTCCGGCCTTTGATCTTTGCTA-
3') were used at a concentration of 100X the probe concentration to compete with binding to the
labeled probe. Protein-DNA complexes were resolved by non-denaturing 4% acrylamide gel
electrophoresis and visualized by autoradiography.
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LEF1 and β-catenin inhibit Runx2
10
Results
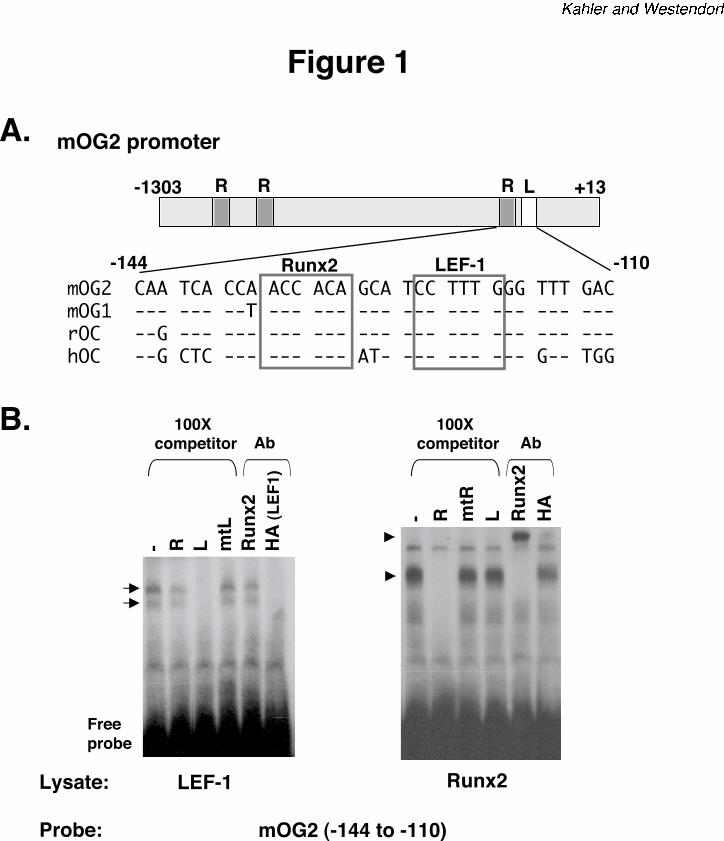
LEF1 binds the osteocalcin promoter. In an effort to discover novel mechanisms that regulate
Runx2-dependent trans-activity, we identified a sequence adjacent to the crucial proximal Runx2
binding site on the murine osteocalcin promoter, mOG2 (37), that resembles the consensus LEF1
recognition site (Figure 1A). The LEF1 recognition sequence was separated from the proximal
Runx binding site by just four base pairs. The LEF1 binding sequence and its relative location to
the Runx site are conserved in mouse, rat and human osteocalcin promoters. To determine if
LEF1 binds the mOG2 promoter, electrophoretic mobility shift assays (EMSAs) were performed
with radiolabeled DNA probes containing mOG2 residues, -144 to -110 (Figure 1A). COS
lysates expressing LEF1-HA produced two complexes that hindered the migration of the probe
(Figure 1B). These bands were specifically competed with either unlabeled double-stranded
oligonucleotides containing a wild type LEF (L) consensus binding site or HA-specific
antibodies. Oligonucleotides containing a mutant LEF binding site (mtL) or a Runx2 antibody
did not alter the LEF complexes (Figure 1B). Runx2 lysates produced a single band when
incubated with the mOG2 probe. This band was specifically competed by the addition of 100-
fold excess unlabeled wild-type Runx (R) consensus binding site and its migration was hindered
by Runx2-specific antisera. The Runx2 complex was not altered by anti-HA antibodies or by
unlabeled oligonucleotides containing a scrambled Runx binding site or the wild-type LEF1
binding site. These data demonstrate that LEF1 specifically interacts with the osteocalcin
promoter.
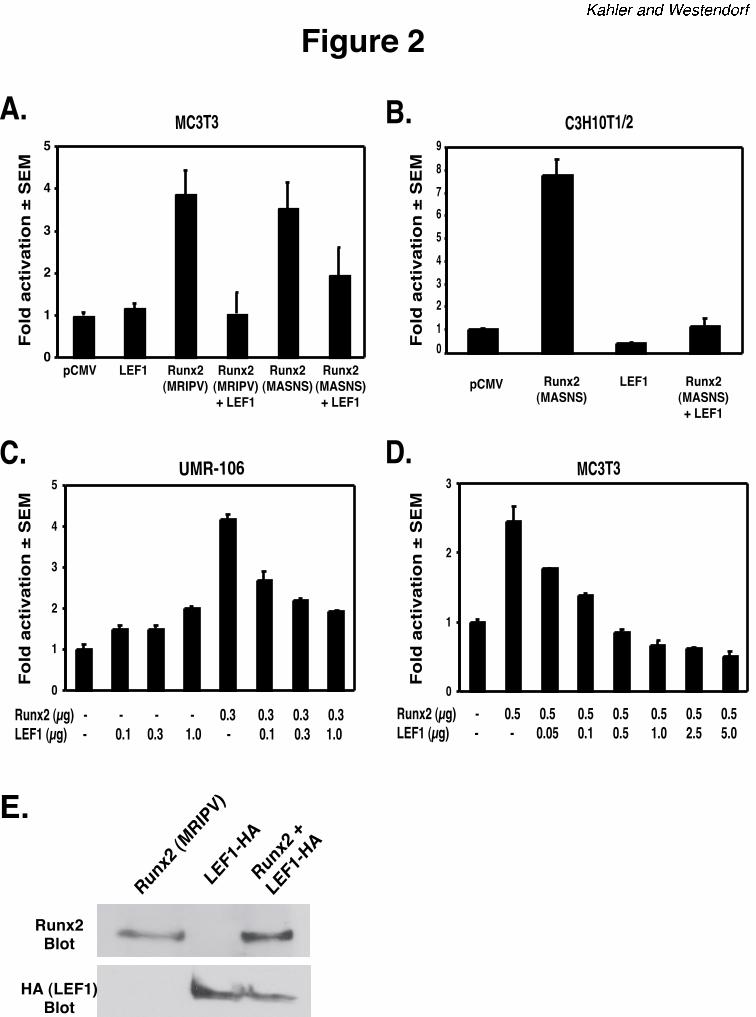
LEF1 inhibits Runx2-dependent activation of the mOG2 promoter. Because the LEF1 and
Runx2 binding sites are separated by only four base pairs, we examined the effects of LEF1 on
the transcriptional activity of Runx2. MC3T3 (murine pre-osteoblasts), C3H10T1/2 (murine
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LEF1 and β-catenin inhibit Runx2
11
mesenchymal precursors), or UMR-106 (rat osteosarcoma) cells were transiently transfected
with the mOG2-luciferase reporter in the presence or absence of Runx2 and/or LEF1 expression
plasmids. Consistent with data previously reported by others (37,40), Runx2 proteins with
amino terminal sequences of either MRIPV or MASNSL activated the mOG2 promoter
approximately four-fold in MC3T3 cells (Figure 2). LEF1 did not significantly alter the basal
activity of the wild-type mOG2 reporter. However, LEF1 blocked the activation induced by
either Runx2 isoform (Figure 2A). LEF1 similarly repressed Runx2-dependent activation of
mOG2 in osteoblast precursor C3H10T1/2 cells (Figure 2B) and in UMR-106 osteosarcoma cells
(Figure 2C). The inhibition was dependent on the amount of LEF1 expression plasmids added to
UMR-106 and MC3T3 cells (Figures 2C and 2D). Complete inhibition was observed when
equal amounts of LEF1 and Runx2 expression plasmids were co-transfected into these cells.
Importantly, co-expression of LEF1 did not suppress Runx2 protein levels (Figure 2E). These
data indicate that LEF1 represses Runx2 activity in osteoblasts, thereby defining a novel
intermolecular mechanism that may regulate Runx2 activity during differentiation and a
functional role for LEF1 in osteoblasts.
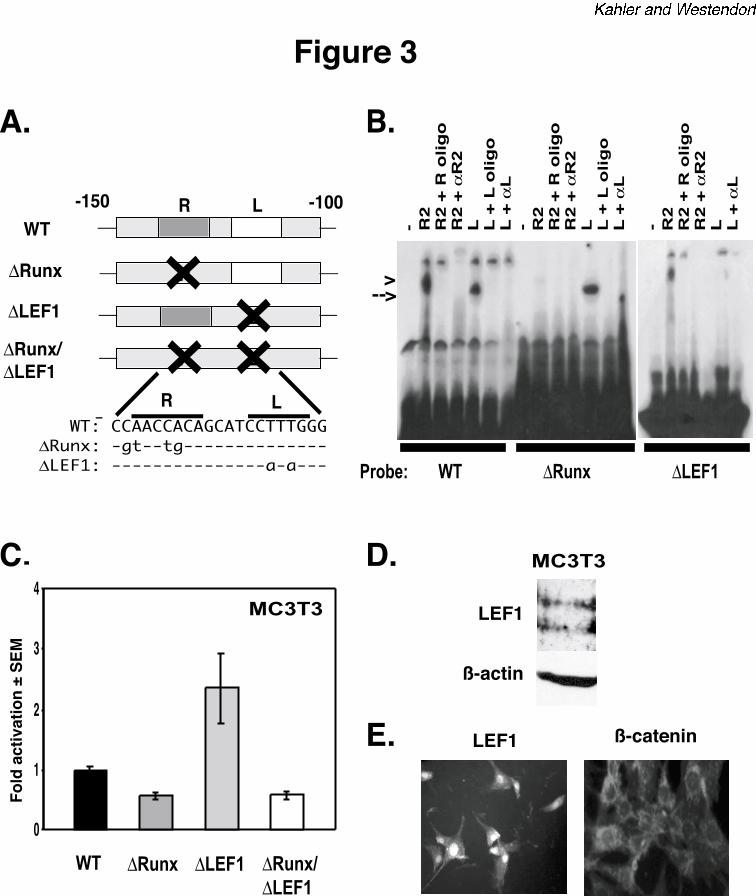
The LEF1 binding site in mOG2 is a repressive element but is not required for repression
of Runx2 activity. To determine the requirement of the LEF1 binding site in osteocalcin
regulation, we made point mutations in the mOG2 EMSA probes and luciferase reporter
constructs that disrupt the binding sites for LEF1, Runx2 or both factors (Figure 3A). The
mutations completely eliminated DNA binding of the respective transcription factor(s) without
affecting the DNA-binding ability of the other (Figure 3B). Mutations in the Runx2 binding site
decreased basal activity of mOG2 (Figure 3C). This result is consistent with reports from other
laboratories (37,40). Interestingly, mutations in the LEF1 binding site increased the basal
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LEF1 and β-catenin inhibit Runx2
12
activity of the mOG2-luciferase reporter in MC3T3 cells by two- to three-fold (Figure 3C).
Immunoblot and in situ immunofluorescence analyses indicated that MC3T3 cells express LEF1
in their nuclei (Figures 3D and 3E). These data indicate endogenous LEF1 suppresses the basal
activity of mOG2 in osteoblasts. This is consistent with LEF1’s role as a transcriptional
repressor in the absence of Wnt signals (22).
We next utilized the mOG2 mutant reporter constructs to begin to determine the
mechanism by which LEF1 represses Runx2. MC3T3 cells were transiently transfected with
wild type or mutant mOG2 reporters in the presence or absence of Runx2 and LEF1 expression
plasmids. Because basal activities of the mutant reporter constructs differed (Figure 3), data
were normalized to the activity of the indicated mOG2 reporter in the absence of Runx2 and
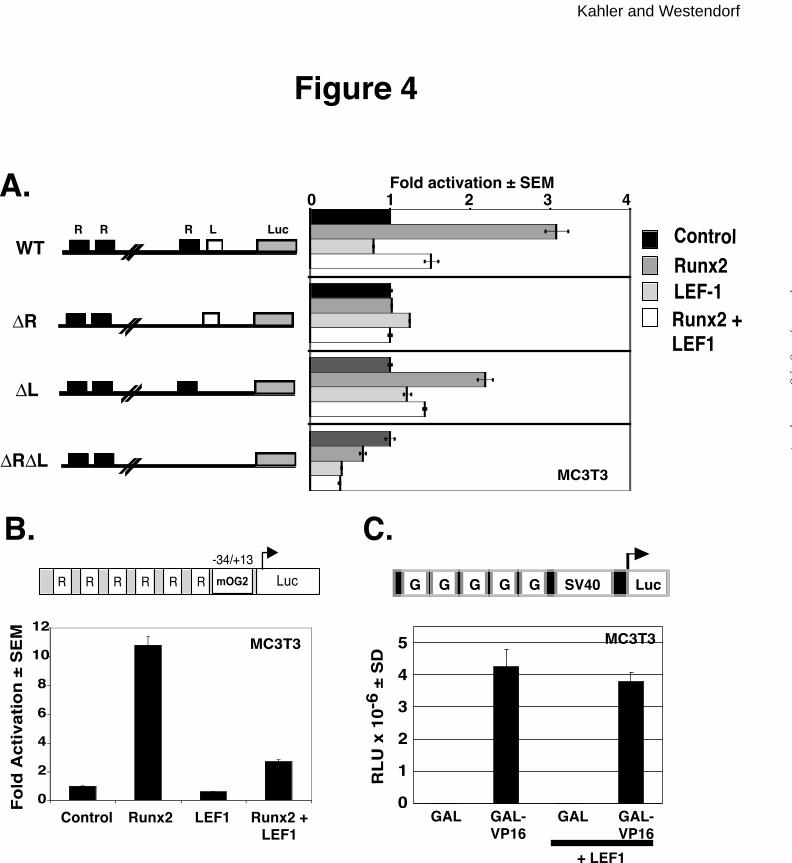
LEF1 expression plasmids (Figure 4). As previously demonstrated, Runx2 activates the wild-
type mOG2 promoter approximately three-fold (Figure 4A, top panel). The Runx2 binding site
is necessary for Runx2 activation (second and fourth panels from top). However, mutations in
the LEF1 binding site did not significantly affect Runx2-dependent activation (third panel from
top). Surprisingly, the LEF1 binding site was also not required for LEF1-mediated repression of
Runx2 (third panel from top). To confirm that LEF1 binding sites are not necessary for
repression, we tested the effects of LEF1 on Runx2-driven expression of an artificial promoter,
p6OSE2 (37), which contains six Runx binding elements upstream of the minimal osteocalcin
promoter (-34/+13) luciferase and no recognizable LEF1 binding sites. Runx2 activated the
p6OSE2 reporter by more than ten-fold (Figure 4B). LEF1 did not affect the basal activity of the
reporter but repressed Runx2-induced activation approximately four-fold. In contrast, LEF1 did
not inhibit GAL or GAL-VP16 activity on a GAL-responsive promoter, pAH205 (Figure 4C).
Thus LEF1 specifically inhibits Runx2 and the LEF1 DNA binding is not required for repression
of Runx2-dependent activation of the mOG2 promoter.
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LEF1 and β-catenin inhibit Runx2
13
LEF1 interacts with the RHD. To determine how LEF1 is suppressing Runx2, we considered
five potential mechanisms of transcriptional repression: (1) competition for DNA binding sites,
(2) squelching of the basal transcriptional machinery, (3) quenching of the activation properties
of DNA-bound Runx2, (4) indirect transcriptional repression by complexing with Runx2 and
preventing DNA binding and (5) co-repressor recruitment (41-43). Competition is unlikely
because a DNA probe containing just the Runx binding element does not compete with a mOG2
probe for LEF1 (Figure 1B) and LEF1 does not bind to a Runx2-specific probe (data not shown).
Squelching also does not appear to be the mechanism because ectopic LEF1 did not repress the
basal activity of the wild-type mOG2 promoter (Figure 2). To further explore the possibility that
LEF1 quenches or indirectly represses Runx2, we asked if LEF1 physically interacts with
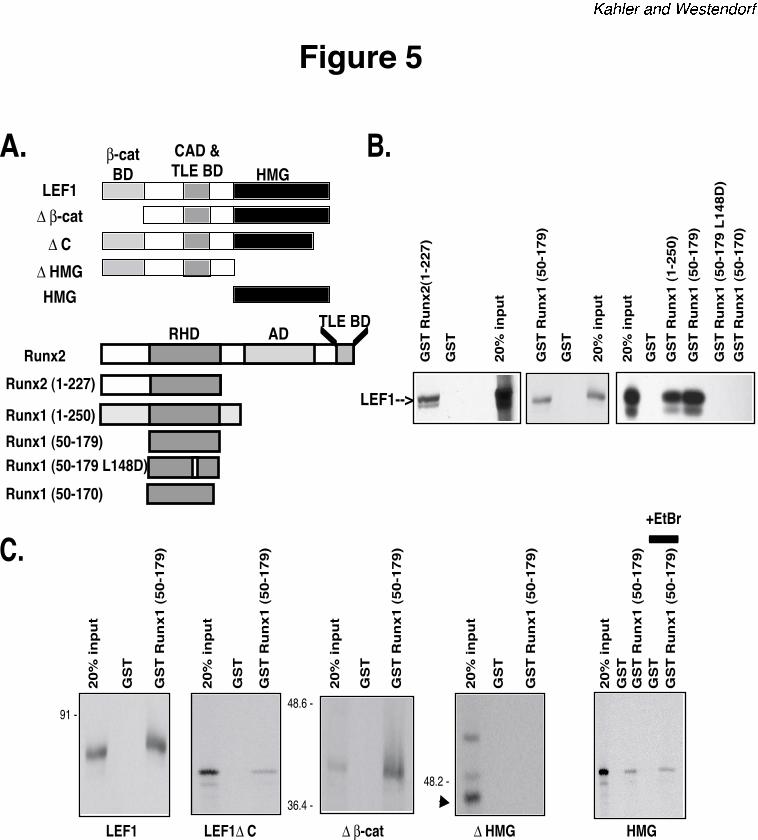
Runx2. Using a panel of Runx2 and Runx1 GST-fusion proteins (Figure 5A), we found that
LEF1 interacts with the amino-termini of both Runt-domain proteins. The Runt homology
domain (RHD; Runx1 residues 50 to 179), which mediates DNA binding, is necessary and
sufficient to interact with full-length LEF1 in vitro (Figure 5B). A single point mutation, L148D,
in the Runx1 RHD that eliminates DNA binding (44) also prevented LEF1 interactions. We then
mapped the Runx binding site in LEF1 with a panel of LEF1 truncation mutants (Figure 5A).
Neither the ß-catenin-binding domain nor the extreme carboxy-terminus was necessary for LEF1
to interact with the RHD (residues 50-179) (Figure 5C). The HMG domain however was
required and sufficient to interact with the RHD. This interaction persisted in the presence of
ethidium bromide, indicating that intermolecular association between respective DNA binding
domains of LEF1 and Runx2 is not mediated by DNA present in the reactions (Figure 5C, right
panel). These data demonstrate a novel interaction between the nuclear LEF1 and Runx2 and
suggest that LEF1 indirectly represses Runx2 activity by binding its DNA binding domain.
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LEF1 and β-catenin inhibit Runx2
14
The HMG and ß-catenin-interacting domains of LEF1 contribute to repression of Runx2.
To determine the regions of LEF1 required for repressing Runx2 activity and to test the
fifth potential mechanism of repression (i.e. co-repressor recruitment), we assayed the LEF1 and
Runx2 truncation mutants in the mOG2 transcription assay. The LEF1 and Runx2 proteins used
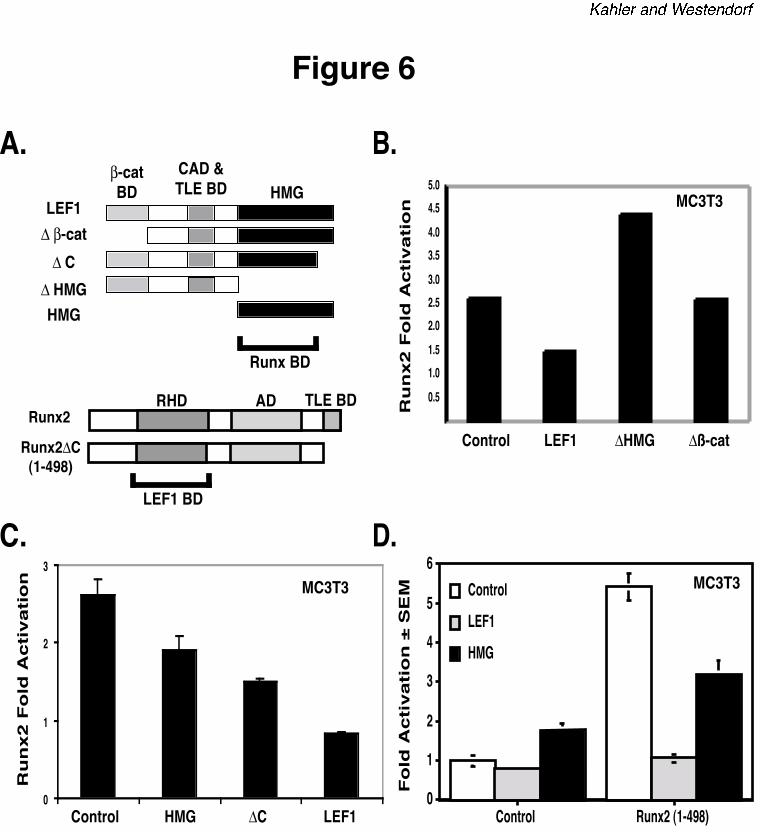
in these experiments are diagrammed in Figure 6A. All LEF1 and Runx2 proteins localized to
cell nuclei (data not shown). The LEF1-HMG domain, which interacts with Runx2, was
necessary for repression of Runx2 transcriptional activity (Figure 6B). The HMG and a LEF1
protein lacking the extreme carboxy-terminus (∆C) also partially blocked Runx2-dependent
activation, but was not as effective as full length LEF1 (Figure 6C). These results suggest that
additional regions of LEF1 are required for maximal repression. This hypothesis was supported
by experiments with a LEF1 protein lacking the ß-catenin binding domain (∆ß-cat), which did
not repress Runx2-dependent transcription (Figure 6B). Thus, the HMG is required for LEF1 to
repress transcription, but the ß-catenin binding domain also contributes to repression.
Both Runx2 and LEF1 bind the TLE-co-repressors (45). To determine if LEF1 interacts
with TLE proteins that are recruited to the promoter by the Runx2 carboxy-terminus to repress
transcription, we determined the effects of LEF1 proteins on a truncated Runx2 protein (1-498)
that lacks the TLE binding domain. Runx2 (1-498) retains the DNA binding and activation
domains and thus activated mOG2 by five-fold (Figure 6D). LEF1 and LEF1-HMG had similar
effects on Runx2 (1-498) as they did on wild-type Runx2. Thus LEF1 completely blocked
Runx2 (1-498) activity and the LEF1-HMG, which lacks the TLE binding domain, partially
repressed it (Figure 6D). These data indicate that TLE-recruitment is not required for LEF1-
mediated repression of Runx2; however, the possibility that TLE may contribute to LEF1-
mediated repression of Runx2 cannot be excluded.
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LEF1 and β-catenin inhibit Runx2
15
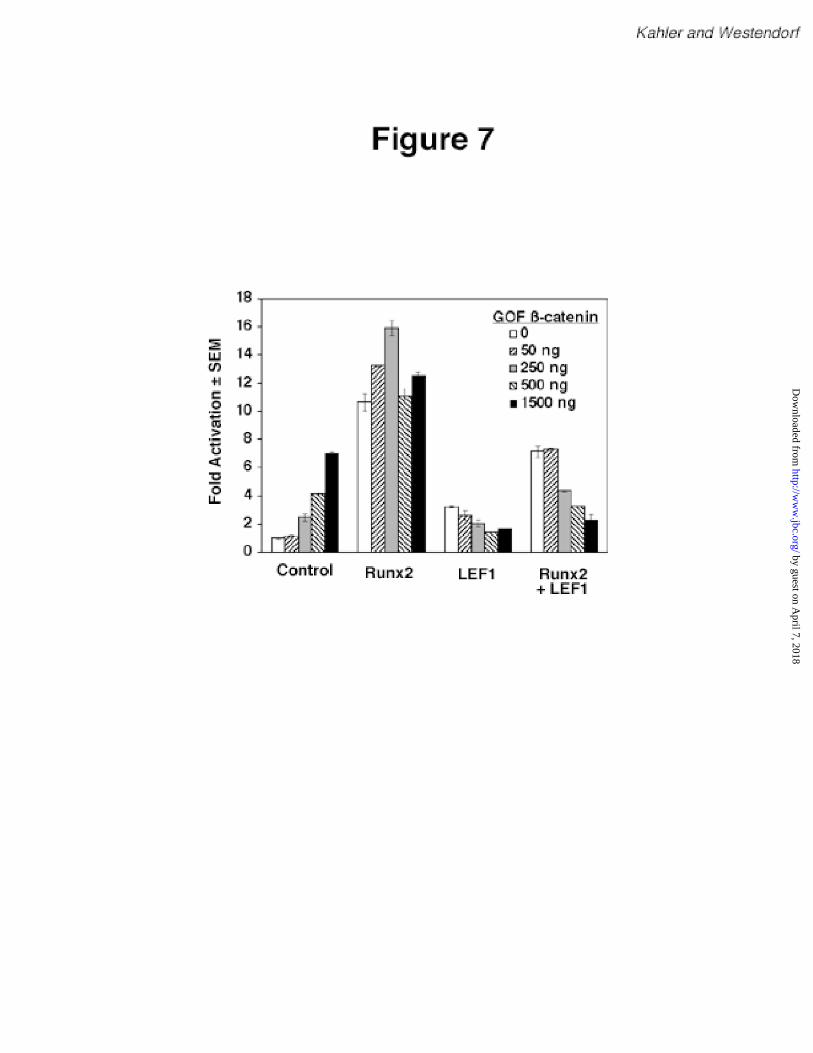
Gain-of-function ß-catenin enhances LEF1-mediated repression of Runx2. Because LEF1
mutants lacking the ß-catenin-binding domain were less effective at inhibiting Runx2 than wild-
type LEF1, we hypothesized that ß-catenin may participate in LEF1-mediated repression of
Runx2. To test this hypothesis, we co-transfected an expression plasmid for a gain-of-function
(GOF) ß-catenin. This cDNA contains point mutations that are found in cancer cells and prevent
ß-catenin from being degraded, thereby increasing its levels and facilitating its transport into the
nucleus (46,47). Interestingly, GOF ß-catenin augmented the basal activity of the mOG2
promoter in a concentration-dependent manner (Figure 7). In contrast, GOF ß-catenin did not
affect Runx2-dependent activation of the mOG2 promoter, but it increased LEF1-mediated
repression of Runx2 in a concentration-dependent manner. These data suggest that LEF1 and ß-
catenin cooperatively repress Runx2-dependent activation. In addition, the observation that GOF
ß-catenin activates mOG2 suggests that ß-catenin activation may stimulate this promoter via
multiple mechanisms.
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LEF1 and β-catenin inhibit Runx2
16
Discussion
Runx2 is a crucial transcription factor for osteoblast development. Runx2 expression and
activity are controlled by transcriptional, translational and post-translational mechanisms (9).
Runx2 activity is further modulated by interactions with other proteins, including coactivators,
corepressors and other transcription factors (13-16). Genetic alterations that decrease Runx2
expression or cause structural changes demonstrate that Runx2 is required for osteoblast
development from mesenchymal stem cells, the terminal differentiation of committed osteoblasts
to osteocytes, and the expression of late-osteoblast genes like osteocalcin (2,3,6,48,49). Runx2
is expressed in proliferating osteoblasts; however, it does not appear to play a crucial role in
regulating osteoblast number (2,3,5). This suggests that Runx2 activity may be suppressed or
controlled in proliferating osteoblasts. Transgenic models support this hypothesis as increased
expression of Runx2 in proliferative osteoblasts causes osteopenia and bone fractures (7,8). The
mechanisms regulating Runx2 activity during osteoblast growth phases are not understood.
In this report we describe novel functional and physical interactions between Runx2 and
LEF1 that may contribute to the regulation of Runx2 activity in osteoblasts. LEF1 is a nuclear
effector of the canonical Wnt signaling pathway, which is commonly thought to convert LEF1 to
an activator by interactions with ß-catenin (22,23). We identified a functional LEF1 recognition
sequence in close proximity to the crucial Runx binding site in the osteocalcin promoter. The
LEF1 site is a repressive element and ectopic expression of LEF1 proteins inhibited Runx2-
dependent activation of the osteocalcin promoter in osteoblast lineage cells. This result is
consistent with LEF1 being a repressor of transcription. We initially hypothesized that activation
of the canonical Wnt signaling pathway would reverse LEF1-mediated repression. Surprisingly,
a constitutively-active ß-catenin mutant that mimics Wnt activation did not prevent LEF1-
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LEF1 and β-catenin inhibit Runx2
17
mediated repression and in fact enhanced suppression. These data suggest that activation of
LEF1 by Wnt signaling represses Runx2-dependent activation of the osteocalcin promoter.
Recent data from developmental models indicate that the Wnt signaling pathway indeed directly
represses transcription of some genes (27-29,50-52). Interestingly, LEF1-mediated repression of
Runx2 on the osteocalcin promoter is remarkably similar to Wg (Wnt homologue) and dTCF
(LEF1)-mediated repression of Drosophila stripe. Thus, Wg and dTCF repress Cubitus
interruptus (Ci)-mediated activation of the stripe promoter where the dTCF binding site is only
four base pairs from a crucial Ci recognition sequence (29). These data indicate that repression
mechanisms used by LEF1/TCF factors may be conserved.
The mechanisms by which Wnt signaling pathways repress transcription are not as
clearly defined. Our data suggest that LEF1 indirectly represses transcription by masking the
DNA binding domain of Runx2. We found that LEF1 interacts with Runt-domain proteins and
that LEF1 binding sites in DNA are not necessary for repression. LEF1 did not repress GAL-
VP16-dependent activation of the GAL-responsive promoter. Thus, LEF1 specifically inhibits
the action of Runx2 (indirect repression), but does not repress transcription by interacting with
the basal transcriptional complex (direct repression). This is a potentially powerful means of
repression because it predicts that a LEF1 binding site is not necessary to suppress Runx2 trans-
activity. Alterations in LEF1 expression levels could therefore regulate Runx2 activity in
osteoblasts. Interestingly, LEF1 mRNA levels are decreased in osteoblasts lacking the Wnt
receptor LRP5 (33). These data suggest that an auto-feedback loop may regulate LEF1
expression in osteoblasts. Additional data in support of this autofeedback mechanism are that
the canonical Wnt signaling pathway stimulates LEF1 expression (53). High levels of the Wg
(Wnt) receptor, Fzd2, stabilize Wg in Drosophila but in turn decrease the Fzd2 expression
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LEF1 and β-catenin inhibit Runx2
18
(54,55). Thus local concentrations of Wnts and/or soluble Wnt antagonists may autoregulate
intracellular signals and gene transcription in vivo.
The physical and functional interactions between LEF1 and Runx2 may link two
pathways that are crucial for osteoblast maturation. As described above, Runx2 is required for
terminal osteoblast differentiation. The role of LEF1 in osteoblasts has not yet been directly
studied; however, LEF1 is expressed in these cells and is thus potential nuclear effector of the
Wnt signaling pathway in osteoblasts. Mutations in the Wnt receptor LRP5 have significant
effects on bone density. Autosomal recessive and inactivating mutations in LRP5 decrease bone
density in mice and humans (32,33,56). In contrast, activating mutations in LRP5 cause
autosomal dominant high bone mass traits (57-59). The effects of these mutations on Runx2
activity have not yet been determined. Because LRP5 activates the canonical Wnt pathway and
stabilizes ß-catenin (60), which enhances LEF1-mediated repression of Runx2, it is possible that
Wnt signals transmitted through LRP5 may regulate Runx2 activity.
Our data identify LEF1 as a posttranscriptional regulator of Runx2. This is an
underexplored mechanism of controlling the activity of a transcription factor crucial for bone
development. Previous studies have identified numerous proteins that either enhance Runx2
function at the posttranscriptional level (13-16) or modulate Runx2 activity by regulating the
expression/transcription of the Runx2 promoter (61-65). Our data indicate that additional
mechanisms that negatively regulate Runx2 at the posttranscriptional level also exist.
Repression of Runx2-dependent trans-activation of the osteocalcin is likely only one
consequence of LEF1 expression and/or ß-catenin activation in osteoblasts. In the absence of
ectopic LEF1 or Runx2, GOF ß-catenin activated the osteocalcin promoter. These data indicate
that ß-catenin may regulate osteocalcin expression via multiple mechanisms. It remains to be
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LEF1 and β-catenin inhibit Runx2
19
determined if the LEF1-independent ß-catenin activation mimics LRP5 activation or if other
stimuli regulate ß-catenin function. In addition to the osteocalcin promoter, we have identified
potential LEF1 recognition sites in other Runx2-regulated osteoblast promoters (e.g. bone
sialoprotein and collagenase 3 (data not shown)). Studies that focus on how Wnt, LRP5, ß-
catenin and LEF1 regulate Runx2-dependent and -independent transcription of these genes are
necessary to gain a full understanding of the effects of these proteins on gene expression and the
maturation of osteoblasts. It is also important to consider the possibility that LEF1 may respond
to Wnt-independent stimuli (e.g. Notch (66)) in osteoblasts. LEF1 was previously shown to
facilitate Runx1-dependent activation of the T cell receptor enhancer in a ß-catenin-independent
manner (67,68). Finally, the activities of Runx factors and LEF1 are regulated by interactions
with the nuclear matrix (69,70). Thus in addition to extracellular stimuli, gene structure and
tissue-specific factors are also likely to influence how and when LEF1 and Runx factors interact
in vivo. by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LEF1 and β-catenin inhibit Runx2
20
Acknowledgments
We thank Drs. Scott Hiebert, Gary Stein, Rudolf Grosschedl, Randall Moon, Jeffrey Miller,
Jennifer Hall, Vivian Bardwell, and Gerard Karsenty for kindly providing expression plasmids.
We thank Xiaodong Li for technical assistance. This work was supported by the American
Cancer Society (IRG -58-001-43-17), the University of Minnesota Cancer Center, and the Office
of the Vice President for Research and Dean of the Graduate School at the University of
Minnesota.
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LEF1 and β-catenin inhibit Runx2
21
References
1. Ducy, P. (2000) Dev Dyn 219, 461-471.
2. Komori, T., Yagi, H., Nomura, S., Yamaguchi, A., Sasaki, K., Deguchi, K., Shimizu, Y.,
Bronson, R. T., Gao, Y. H., Inada, M., Sato, M., Okamoto, R., Kitamura, Y., Yoshiki, S.,
and Kishimoto, T. (1997) Cell 89, 755-764
3. Otto, F., Thornell, A. P., Crompton, T., Denzel, A., Gilmour, K. C., Rosewell, I. R.,
Stamp, G. W., Beddington, R. S., Mundlos, S., Olsen, B. R., Selby, P. B., and Owen, M.
J. (1997) Cell 89, 765-771
4. Takeda, S., Bonnamy, J. P., Owen, M. J., Ducy, P., and Karsenty, G. (2001) Genes Dev
15, 467-481.
5. Ducy, P., Starbuck, M., Priemel, M., Shen, J., Pinero, G., Geoffroy, V., Amling, M., and
Karsenty, G. (1999) Genes Dev 13, 1025-1036
6. Mundlos, S., Otto, F., Mundlos, C., Mulliken, J. B., Aylsworth, A. S., Albright, S.,
Lindhout, D., Cole, W. G., Henn, W., Knoll, J. H., Owen, M. J., Mertelsmann, R., Zabel,
B. U., and Olsen, B. R. (1997) Cell 89, 773-779
7. Liu, W., Toyosawa, S., Furuichi, T., Kanatani, N., Yoshida, C., Liu, Y., Himeno, M.,
Narai, S., Yamaguchi, A., and Komori, T. (2001) J Cell Biol 155, 157-166
8. Geoffroy, V., Kneissel, M., Fournier, B., Boyde, A., and Matthias, P. (2002) Mol Cell
Biol 22, 6222-6233
9. Komori, T. (2002) J Cell Biochem 87, 1-8
10. Meyers, S., Downing, J. R., and Hiebert, S. W. (1993) Mol Cell Biol 13, 6336-6345
11. Redondo, J. M., Pfohl, J. L., Hernandez-Munain, C., Wang, S., Speck, N. A., and
Krangel, M. S. (1992) Mol Cell Biol 12, 4817-4823
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LEF1 and β-catenin inhibit Runx2
22
12. Wang, S. W., and Speck, N. A. (1992) Mol. Cell. Biol. 12, 89-102
13. Hess, J., Porte, D., Munz, C., and Angel, P. (2001) J Biol Chem 276, 20029-20038
14. Westendorf, J. J., Zaidi, S. K., Cascino, J. E., Kahler, R., Van Wijnen, A. J., Lian, J. B.,
Yoshida, M., Stein, G. S., and Li, X. (2002) Mol Cell Biol 22, 7982-7992.
15. Zhang, Y. W., Yasui, N., Ito, K., Huang, G., Fujii, M., Hanai, J., Nogami, H., Ochi, T.,
Miyazono, K., and Ito, Y. (2000) Proc Natl Acad Sci U S A 97, 10549-10554
16. Westendorf, J. J., and Hiebert, S. W. (1999) J Cell Biochem Suppl, 51-58
17. Giese, K., Kingsley, C., Kirshner, J. R., and Grosschedl, R. (1995) Genes Dev 9, 995-
1008
18. Mayall, T. P., Sheridan, P. L., Montminy, M. R., and Jones, K. A. (1997) Genes Dev 11,
887-899
19. Hsu, S. C., Galceran, J., and Grosschedl, R. (1998) Mol Cell Biol 18, 4807-4818
20. Porfiri, E., Rubinfeld, B., Albert, I., Hovanes, K., Waterman, M., and Polakis, P. (1997)
Oncogene 15, 2833-2839
21. Giese, K., Amsterdam, A., and Grosschedl, R. (1991) Genes Dev 5, 2567-2578.
22. van Noort, M., and Clevers, H. (2002) Dev Biol 244, 1-8
23. Brantjes, H., Barker, N., van, E. J., and Clevers, H. (2002) Biol Chem 383, 255-261
24. He, T. C., Sparks, A. B., Rago, C., Hermeking, H., Zawel, L., da Costa, L. T., Morin, P.
J., Vogelstein, B., and Kinzler, K. W. (1998) Science 281, 1509-1512
25. Hovanes, K., Li, T. W. H., Munguia, J. E., Truong, T., Milovanovic, T., Marsh, J. L.,
Holcombe, R. F., and Waterman, M. (2001) Nat Genet 28, 53-57
26. Shtutman, M., Zhurinsky, J., Simcha, I., Albanese, C., D'Amico, M., Pestell, R., and Ben-
Ze'ev, A. (1999) Proc Natl Acad Sci U S A 96, 5522-5527
27. Cadigan, K. M., Jou, A. D., and Nusse, R. (2002) Development 129, 3393-3402.
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LEF1 and β-catenin inhibit Runx2
23
28. Payre, F., Vincent, A., and Carreno, S. (1999) Nature 400, 271-275.
29. Piepenburg, O., Vorbruggen, G., and Jackle, H. (2000) Mol Cell 6, 203-209.
30. Yang, X., van Beest, M., Clevers, H., Jones, T., Hursh, D. A., and Mortin, M. A. (2000)
Development 127, 3695-3702.
31. Moon, R. T., Bowerman, B., Boutros, M., and Perrimon, N. (2002) Science 296, 1644-
1646.
32. Gong, Y., Slee, R. B., Fukai, N., Rawadi, G., Roman-Roman, S., Reginato, A. M., Wang,
H., Cundy, T., Glorieux, F. H., Lev, D., Zacharin, M., Oexle, K., Marcelino, J., Suwairi,
W., Heeger, S., Sabatakos, G., Apte, S., Adkins, W. N., Allgrove, J., Arslan-Kirchner,
M., Batch, J. A., Beighton, P., Black, G. C., Boles, R. G., Boon, L. M., Borrone, C.,
Brunner, H. G., Carle, G. F., Dallapiccola, B., De Paepe, A., Floege, B., Halfhide, M. L.,
Hall, B., Hennekam, R. C., Hirose, T., Jans, A., Juppner, H., Kim, C. A., Keppler-
Noreuil, K., Kohlschuetter, A., LaCombe, D., Lambert, M., Lemyre, E., Letteboer, T.,
Peltonen, L., Ramesar, R. S., Romanengo, M., Somer, H., Steichen-Gersdorf, E.,
Steinmann, B., Sullivan, B., Superti-Furga, A., Swoboda, W., van den Boogaard, M. J.,
Van Hul, W., Vikkula, M., Votruba, M., Zabel, B., Garcia, T., Baron, R., Olsen, B. R.,
and Warman, M. L. (2001) Cell 107, 513-523
33. Kato, M., Patel, M. S., Leasseuer, R., Lobov, I., Chang, B. H.-J., Glass, D. A. I.,
Hartmann, C., Li, L., Hwang, T.-H., Brayton, C. F., Lang, R. A., Karsenty, G., and Chan,
L. (2002) J Cell Biol 157, 303-314
34. Boyden, L. M., Mao, J., Belsky, J., Mitzer, L., Farhi, A., Mitnick, M. A., Wu, D.,
Insogna, K., and Lifton, R. P. (2002) N Engl J Med 346, 1513-1521
35. Little, R. D., Caruli, J. P., Del Mastro, R. G., Dupuis, J., Osborne, M. A., Folz, C.,
Manning, S. P., Swain, P. M., Zhao, S., Eustace, B., Lappe, M. M., Spitzer, L., Zweier,
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LEF1 and β-catenin inhibit Runx2
24
S., Braunschweiger, K., Benchekroun, Y., Hu, X., Adair, R., Chee, L., FitzGerald, M. G.,
Tulig, C., Caruso, A. M., Tzellas, N., Bawa, A., Franklin, B., McGuire, S., Nogues, X.,
Gong, G., Allen, K. M., Anisowicz, A., Morales, A. J., Lomedico, P. R., Recker, S. M.,
Van Eerdewegh, P., Recker, R. R., and Johnson, M. L. (2002) Am J Hum Genet 70, 11-19
36. Hetman, M., Cavanaugh, J. E., Kimelman, D., and Xia, Z. (2000) J Neurosci 20, 2567-
2574
37. Ducy, P., and Karsenty, G. (1995) Mol Cell Biol 15, 1858-1869
38. Westendorf, J. J., Yamamoto, C. M., Lenny, N., Downing, J. R., Selsted, M. E., and
Hiebert, S. W. (1998) Mol Cell Biol 18, 322-333
39. Meyers, S., Lenny, N., Sun, W., and Hiebert, S. W. (1996) Oncogene 13, 303-312
40. Banerjee, C., Hiebert, S. W., Stein, J. L., Lian, J. B., and Stein, G. S. (1996) Proc Natl
Acad Sci USA 93, 4968-4973
41. Latchman, D. S. (1996) Int J Biochem Cell Biol 28, 965-974.
42. Mannervik, M., Nibu, Y., Zhang, H., and Levine, M. (1999) Science 284, 606-609.
43. Ezashi, T., Ghosh, D., and Roberts, R. M. (2001) Mol Cell Biol 21, 7883-7891.
44. Lenny, N., Meyers, S., and Hiebert, S. W. (1995) Oncogene 11, 1761-1769
45. Levanon, D., Goldstein, R. E., Bernstein, Y., Tang, H., Goldenberg, D., Stifani, S.,
Paroush, Z., and Groner, Y. (1998) Proc Natl Acad Sci U S A 95, 11590-11595
46. Morin, P. J., Sparks, A. B., Korinek, V., Barker, N., Clevers, H., Vogelstein, B., and
Kinzler, K. W. (1997) Science 275, 1787-1790
47. Rubinfeld, B., Robbins, P., El-Gamil, M., Albert, I., Porfiri, E., and Polakis, P. (1997)
Science 275, 1790-1792
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LEF1 and β-catenin inhibit Runx2
25
48. Choi, J. Y., Pratap, J., Javed, A., Zaidi, S. K., Xing, L., Balint, E., Dalamangas, S.,
Boyce, B., van Wijnen, A. J., Lian, J. B., Stein, J. L., Jones, S. N., and Stein, G. S. (2001)
Proc Natl Acad Sci U S A 98, 8650-8655.
49. Lee, B., Thirunavukkarasu, K., Zhou, L., Pastore, L., Baldini, A., Hecht, J., Geoffroy, V.,
Ducy, P., and Karsenty, G. (1997) Nat Genet 16, 307-310
50. Bejsovec, A. (1999) Curr Biol 9, R684-687.
51. Peifer, M. (1999) Nature 400, 213-215.
52. Willert, J., Epping, M., Pollack, J. R., Brown, P. O., and Nusse, R. (2002) BMC Dev Biol
2, 8.
53. Filali, M., Cheng, N., Abbott, D., Leontiev, V., and Engelhardt, J. F. (2002) J Biol Chem
277, 33398-33410.
54. Zhang, J., and Carthew, R. W. (1998) Development 125, 3075-3085.
55. Cadigan, K. M., Fish, M. P., Rulifson, E. J., and Nusse, R. (1998) Cell 93, 767-777.
56. Patel, M. S., and Karsenty, G. (2002) N Engl J Med 346, 1572-1574
57. Boyden, L. M., Mao, J., Belsky, J., Mitzner, L., Farhi, A., Mitnick, M. A., Wu, D.,
Insogna, K., and Lifton, R. P. (2002) N Engl J Med 346, 1513-1521.
58. Little, R. D., Carulli, J. P., Del Mastro, R. G., Dupuis, J., Osborne, M., Folz, C.,
Manning, S. P., Swain, P. M., Zhao, S. C., Eustace, B., Lappe, M. M., Spitzer, L.,
Zweier, S., Braunschweiger, K., Benchekroun, Y., Hu, X., Adair, R., Chee, L.,
FitzGerald, M. G., Tulig, C., Caruso, A., Tzellas, N., Bawa, A., Franklin, B., McGuire,
S., Nogues, X., Gong, G., Allen, K. M., Anisowicz, A., Morales, A. J., Lomedico, P. T.,
Recker, S. M., Van Eerdewegh, P., Recker, R. R., and Johnson, M. L. (2002) Am J Hum
Genet 70, 11-19.
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LEF1 and β-catenin inhibit Runx2
26
59. Van Hul, E., Gram, J., Bollerslev, J., Van Wesenbeeck, L., Mathysen, D., Andersen, P.
E., Vanhoenacker, F., and Van Hul, W. (2002) J Bone Miner Res 17, 1111-1117.
60. Tamai, K., Semenov, M., Kato, Y., Spokony, R., Liu, C., Katsuyama, Y., Hess, F., Saint-
Jeannet, J. P., and He, X. (2000) Nature 407, 530-535.
61. Drissi, H., Luc, Q., Shakoori, R., Chuva De Sousa Lopes, S., Choi, J. Y., Terry, A., Hu,
M., Jones, S., Neil, J. C., Lian, J. B., Stein, J. L., Van Wijnen, A. J., and Stein, G. S.
(2000) J Cell Physiol 184, 341-350
62. Xiao, Z. S., Liu, S. G., Hinson, T. K., and Quarles, L. D. (2001) J Cell Biochem 82, 647-
659
63. Gilbert, L., He, X., Farmer, P., Rubin, J., Drissi, H., van Wijnen, A. J., Lian, J. B., Stein,
G. S., and Nanes, M. S. (2002) J Biol Chem 277, 2695-2701.
64. Drissi, H., Pouliot, A., Koolloos, C., Stein, J. L., Lian, J. B., Stein, G. S., and van Wijnen,
A. J. (2002) Exp Cell Res 274, 323-333
65. Drissi, H., Pouliot, A., Stein, J. L., van Wijnen, A. J., Stein, G. S., and Lian, J. B. (2002)
J Cell Biochem 86, 403-412
66. Ross, D. A., and Kadesch, T. (2001) Mol Cell Biol 21, 7537-7544
67. Bruhn, L., Munnerlyn, A., and Grosschedl, R. (1997) Genes Dev 11, 640-653
68. Giese, K., Pagel, J., and Grosschedl, R. (1997) Proc Natl Acad Sci U S A 94, 12845-
12850.
69. Sachdev, S., Bruhn, L., Sieber, H., Pichler, A., Melchior, F., and Grosschedl, R. (2001)
Genes & Dev 15, 3088-3103
70. Zeng, C., van Wijnen, A. J., Stein, J. L., Meyers, S., Sun, W., Shopland, L., Lawrence, J.
B., Penman, S., Lian, J. B., Stein, G. S., and Hiebert, S. W. (1997) Proc. Natl. Acad. Sci.
U.S.A. 94, 6746-6751
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LEF1 and β-catenin inhibit Runx2
27
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LEF1 and β-catenin inhibit Runx2
28
Abbreviations
LEF1, lymphoid enhancer-binding factor 1; HMG, high mobility group; GST, glutathione S-
transferase; HA, hemagglutinin; GOF, gain-of-function; EMSA, electrophoretic mobility shift
assay; RHD, runt homology domain; LRP5, low density lipoprotein receptor-related protein 5;
OPPG, osteoporosis-pseudoglioma syndrome; MEM, minimum essential medium; mOG2,
mouse osteocalcin 2
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LEF1 and β-catenin inhibit Runx2
29
Figure Legends
Figure 1. LEF1 binds the mOG2 promoter. A. Multiple sequence alignment indicates that the
sequence and positioning of Runx and LEF1 binding sites are conserved in the osteocalcin
promoters of mouse, rat and human (mOG1, NCB accession #4204979; mOG2, NCB accession
#4204980; rat, NCB accession #205865; human, NCB accession #22947853.) The Runx and
LEF1 recognition sites are boxed. Dashes represent conserved nucleotides. The sequence of the
mOG2 EMSA oligonucleotide probe (-144 to –110) is depicted in the multiple sequence
alignment. B. LEF1 interacts with the mOG2 promoter. Electrophoretic mobility shift assay
using the wild-type EMSA oligonucleotide and 5 µg protein from COS cell lysates expressing
either LEF1-HA (left panel) or Runx2 (right panel). LEF1 complexes are indicated with arrows.
Runx2 complexes are marked with arrowheads. Cold competitors containing wild-type Runx
consensus site (R), wild type LEF1 consensus site (L), mutant Runx binding site (mtR), and
mutant LEF1 binding site (mtL) were added in 100-fold excess. Anti-Runx2 or anti-HA (LEF1)
antibodies were added at 240 µg/ml. Free probe is visible at the bottom of each panel.
Figure 2. LEF1 represses Runx2-mediated mOG2-luc transcription. A. LEF1 represses
activation of the osteocalcin promoter by type I (MASNS) and type II (MRIPV) Runx2 isoforms
in preosteoblasts. MC3T3 cells were transfected with mOG2-luc (300 ng), pCMV-SEAP (500
ng) and pCMV5-Runx2 (MRIPV) (500 ng) and/or pCMV5-LEF1-HA (500 ng) as indicated.
Normalized luciferase is represented as fold activation over control. Luciferase values represent
the mean of triplicate samples ± standard error of the mean (SEM). B. LEF1 represses Runx2-
mediated activation in mesenchymal precursors. C3H10T1/2 cells were transfected with mOG2-
luc (300 ng), pRL-TK (50 ng) and pCMV5-Runx2 (MASNS) (500 ng) and/or pCMV5-LEF1-HA
(500 ng) as indicated. C. LEF1-mediated repression of Runx2 is concentration dependent.
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LEF1 and β-catenin inhibit Runx2
30
MC3T3 cells were transfected as in A, but with increasing concentrations of pCMV5-LEF1-HA
as indicated. D. UMR-106 cells were transfected with mOG2-luc reporter plasmid (100 ng),
pRL-TK (5 ng), pCMV5-Runx2 (MRIPV) (100 ng) and increasing concentrations of pCMV5-
LEF1-HA as indicated. E. LEF1 does not decrease Runx2 expression. COS cells were
transiently transfected with pCMV5-LEF1-HA and/or pCMV5-Runx2 (MRIPV). Whole cell
extracts were resolved by SDS-PAGE. Runx2 and LEF1 were detected by immunoblotting with
Runx2 and HA antisera, respectively.
Figure 3. The LEF1 site is not required for LEF1-mediated transcriptional repression. A.
Schematic of mutations introduced into the mOG2 reporter plasmids and EMSA probes by site-
directed mutagenesis. The indicated changes were made in either the Runx recognition sequence
or LEF1 recognition sequence by site-directed mutagenesis. Dashes represent non-mutated
nucleotides. Electrophoretic mobility shift assay probes were synthesized to correspond to the
mutations made in the reporter plasmids. B. Mutations in the Runx2 and LEF1 binding sites
specifically eliminate binding of the respective transcription factor. The indicated wild type or
mutant radiolabeled probes were incubated with COS lysates expressing Runx2 or LEF1-HA.
Runx2 complexes are marked with an arrowhead; LEF1-HA complexes are marked with an
arrow. C. Deletion of the LEF1 binding site increases the basal activity of the mOG2 promoter.
MC3T3 cells were transfected as described in Figure 2A using the indicated mOG2-luc mutants.
Normalized luciferase values are represented as fold activity over wild type mOG2-luc. D. LEF1
is detectable as a doublet in MC3T3 cells. MC3T3 cell lysates were resolved by SDS-PAGE and
immunoblotted with anti-LEF1 or anti-β-catenin antibodies. E. LEF1 resides in the nucleus of
MC3T3 cells. MC3T3 were grown on coverslips and fixed in paraformaldehyde. The cells were
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LEF1 and β-catenin inhibit Runx2
31
lysed with 0.3% Triton X-100 in PBS. LEF1 and β-catenin were detected with their respective
monoclonal primary antibody followed by a goat anti-mouse secondary antibody conjugated to
Alexa-546. The proteins were visualized with fluorescence microscopy.
Figure 4. LEF1 binding site is not required for LEF1-mediated repression of Runx2. A.
LEF1 represses Runx2-mediated activation of a mOG2-luc reporter lacking the LEF1 DNA
binding site. MC3T3 cells were transfected with 300 ng each of the indicated mOG2-luc
reporter, pCMV-SEAP (500 ng), and pCMV5-Runx2 (MRIPV) (500 ng) and/or pCMV5-LEF1-
HA (500 ng) as indicated. Specific mutations are shown in Figure 3A. In the schematic on the
left, black boxes represent Runx2 (R) binding sites and open boxes represent LEF1 (L) binding
sites. B. LEF1 represses Runx2-mediated activation of a Runx-responsive synthetic reporter.
MC3T3 cells were transfected in 6-well plates with 300 ng p6OSE2-luc, 500 ng pCMV-SEAP
and 500 ng pCMV5-Runx2 and/or pCMV5-LEF1-HA. Luciferase values were normalized to
SEAP activity and represented as fold activation over p6OSE2-luc alone. A schematic of the
p6OSE-luc reporter is represented above. Runx binding sites (R) are indicated with white boxes.
C. LEF1 repression is specific to Runx2-mediated activation. MC3T3 cells were transfected
with pAH205-luc (300 ng), Gal-VP16 (300 ng) and/or pCMV5-LEF1-HA (500 ng). Gal DNA
binding sites (G) are represented as white boxes.
Figure 5. LEF1 binds the RHD of Runx2 and Runx1. A. Schematic representation of the
LEF1, GST-Runx2 and GST-Runx2 proteins used in protein interaction assays. β-cat BD: β-
catenin binding domain; CAD: transactivation domain; TLE BD: transducin-like enhancer of
split binding domain; HMG: high mobility group domain; NLS: nuclear localization signal;
RHD: runt homology domain; AD: activation domain. B. LEF1 specifically binds the RHD of
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LEF1 and β-catenin inhibit Runx2
32
Runx2 and Runx1. In vitro transcribed and translated, 35S-radiolabeled full-length LEF1 was
incubated with the indicated GST-Runx fusion proteins. Proteins interacting with GST or GST-
Runx2, or 20% of the 35S-LEF1 input were resolved by SDS-PAGE and detected by
autoradiography. C. The HMG domain is necessary and sufficient to interact with the RHD.
GST pulldowns mapping the Runx-interaction site on LEF1. Ethidium bromide (EtBr, 50 µg/ml)
was added to some reactions to disrupt DNA-mediated binding. The arrowhead indicates the
appropriately sized band representing LEF1∆HMG.
Figure 6. LEF1 HMG domain and β-catenin binding domain are required for full
repression. A. Schematic of the LEF1 and Runx2 proteins used for transcriptional assays. B.
The LEF1-HMG and β-catenin binding domains are necessary for complete repression of Runx2.
MC3T3 cells were transfected with mOG2-luc (300 ng), pCMV-SEAP (500 ng), pCMV5-Runx2
(MASNS) (500 ng) and/or 500 ng of the indicated pCMV-Gal-LEF1 protein. Luciferase values
were normalized to SEAP activity. The effects of the indicated LEF1 protein on Runx2 activity
are plotted. C. The HMG domain is not sufficient for full repression by LEF1. MC3T3 cells
were transfected as in (B) with the indicated LEF1 plasmids. Luciferase values were normalized
to SEAP activity and expressed as fold activation of mOG2-luc alone. D. The TLE binding
domains of Runx2 and LEF1 are not required for LEF1-mediated repression of the osteocalcin
promoter. MC3T3 cells were transfected as in (B). Luciferase values were normalized to SEAP
activity and are depicted as fold activation over mOG2-luc.
Figure 7. Constitutively activated β-catenin increases LEF1-mediated repression of Runx2.
Constitutively activated β-catenin enhances LEF1-mediated repression in a concentration-
dependent manner. MC3T3 cells were transfected with mOG2-luc (300 ng), pCMV-SEAP (500
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LEF1 and β-catenin inhibit Runx2
33
ng) and pCMV5-Runx2 (MPIRV) (500 ng) and/or the indicated amounts of constitutively
activated β-catenin plasmid (GOF β-catenin).
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from
Kahler and Westendorf
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from
Kahler and Westendorf by guest on A
pril 7, 2018http://w
ww
.jbc.org/D
ownloaded from
Kahler and Westendorf by guest on A
pril 7, 2018http://w
ww
.jbc.org/D
ownloaded from
Kahler and Westendorf by guest on A
pril 7, 2018http://w
ww
.jbc.org/D
ownloaded from
Kahler and Westendorf by guest on A
pril 7, 2018http://w
ww
.jbc.org/D
ownloaded from
Kahler and Westendorf by guest on A
pril 7, 2018http://w
ww
.jbc.org/D
ownloaded from

Rachel A. Kahler and Jennifer J. Westendorftranscriptional activation of the osteocalcin promoter
Lymphoid enhancer factor-1 and beta-catenin inhibit Runx2-dependent
published online January 27, 2003J. Biol. Chem.
10.1074/jbc.M211443200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on April 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from















![a of nt β catenin signaling pathway in hepatocellulaR ......Nkd, Idax, Frodo, Dapper, GBP/Frat, Stbm, Daam1 and Pricle proteins, which may activate or inhibit Wnt signaling [25].](https://static.fdocument.org/doc/165x107/608470f08c2b48044335f18a/a-of-nt-catenin-signaling-pathway-in-hepatocellular-nkd-idax-frodo.jpg)


