
Lecture 7,8 January 24, 2011 CC Bonds -...
148
1 © copyright 2011 William A. Goddard III, all rights reserved Ch120a-Goddard-L07,08 Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy Lecture 7,8 January 24, 2011 CC Bonds William A. Goddard, III, [email protected] 316 Beckman Institute, x3093 Charles and Mary Ferkel Professor of Chemistry, Materials Science, and Applied Physics, California Institute of Technology Teaching Assistants: Wei-Guang Liu <[email protected] > Caitlin Scott <[email protected]>
Transcript of Lecture 7,8 January 24, 2011 CC Bonds -...

1© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Nature of the Chemical Bond with applications to catalysis, materials
science, nanotechnology, surface science, bioinorganic chemistry, and energy
Lecture 7,8 January 24, 2011
CC Bonds
William A. Goddard, III, [email protected] Beckman Institute, x3093
Charles and Mary Ferkel Professor of Chemistry, Materials Science, and Applied Physics,
California Institute of Technology
Teaching Assistants: Wei-Guang Liu <[email protected]>Caitlin Scott <[email protected]>

2© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Course scheduleFriday January 14: L3 and L4Monday January 17: Caltech holiday (MLKing)Wednesday January 19: wag L5 and L6Friday January 21: wag L7 and L8, caught upMonday January 24: wag L7 and L8Wednesday January 26: wag L9 and L10Friday January 28: wag participates in a retreat for our nanotechnology project with UCLAFriday January 28: wag L11 Back on schedule Monday January 31: wag L12
Wag rotator cuff operation

3© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Last time

4© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
The GVB orbitals for the (3s)2 pair of Si atom
Long dashes indicate zero amplitude, solid lines indicate positive amplitude while short dashes indicate negative amplitude. The spacing between contours is 0.05 in atomic units

5© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
analyze the pooched or hybridized orbitals
z z
zx
zx
φ2s + λφpzφ2s - λφpzPooching of the
2s orbitals in opposite
directions leads to a dramatic
increase in the ee distance, reducing eerepulsion.
1-D
2-D
Schematic. The line shows symmetric pairing.Notation: sz and sz bar or ℓ and ℓ bar. Cannot type bars. use zs to show the bar case

6© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Role of pooched or hybridized atomic lobe orbitals in bonding of BeH+
Consider the bonding of H to Be+
The simple VB combination of H1s with the 2s orbital of Be+ leads to a very small overlap and contragradience
In fact optimizing the wavefunction for BeH+ leads to pooching of the 2s toward the H1s with much improved overlap and contragradience.

7© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
The ground state for C atomInstead of the simple (1s)2(2s)2(2p)2 form
Ψyz=A[(sx)(xs)+(xs)(sx)](αβ−βα)(yα)(zα)]
2s pair pooched ±x yz open shell
Ψyz=A[(2sα)(2sβ)(yα)(zα)]
pz
sx
xs
pythe ground state of C is

8© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
The GVB orbitals of Silicon atom
Long dashes indicate zero amplitude, solid lines indicate positive amplitude while short dashes indicate negative amplitude. The spacing between contours is 0.05 in atomic units

9© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
At small R the H can overlap significantly more with sz than with zH, so that we can form a bond pair just like in BeH+. This leads to the wavefunction
Thus the wave function is A{[(sz)(zs)+(zs)(sz)](αβ−βα)(Hα)}
where sz≡(s+λz) and zs ≡(s-λz)
Here the H overlaps slightly more with sz than with zs, but the spin on sz is half the time α
Thus at large R we obtain a slightly repulsive interaction.
Role of pooched or hybridized atomic lobe orbitals in bonding of BeH neutral
At large R the the orbitals of Be are already hybridized
Hszzs
Hszzs
A{[(sz)(H)+(H)(sz)](αβ−βα)(zsα)}In which the zs hybrid must now get orthogonal to the szand H bond pair. This weakens the bond from that of BeH+ by ~ 1 eV

10© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Compare bonding in BeH+ and BeH
BeH+
BeH
3 eV
2 eV
1 eV
Long range Repulsive interaction
with H
Short range Attractive interaction sz with H
1eV Repulsive orthogonalization of
zs with sz H
BeH+ has long range attraction no short range repulsion
TA’s check numbers, all from memory

11© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Now bond 2nd H to BeH
Expect linear bond in H-Be-H and much stronger than the 1st bond
Expect bond energy similar to BeH+, maybe stronger, because the zs orbital is already decoupled from the sz.
1Σ+
Cannot bind 3rd H because no singly occupied orbitals left.
~3.1 eV
12© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
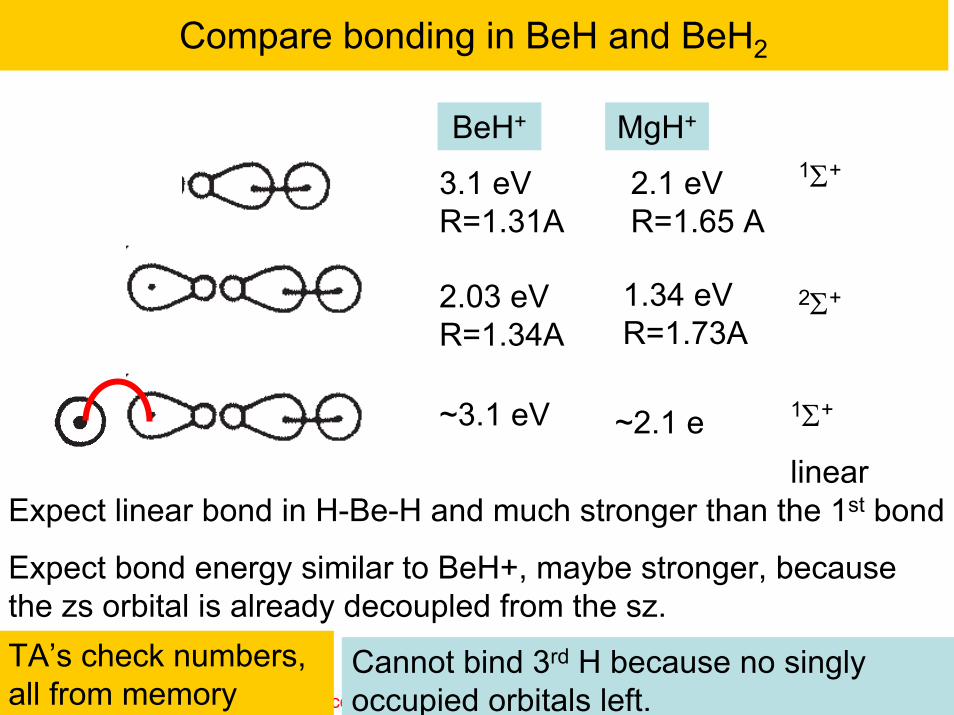
Compare bonding in BeH and BeH2
BeH+
TA’s check numbers, all from memory
MgH+
3.1 eVR=1.31A
1.34 eVR=1.73A
Expect linear bond in H-Be-H and much stronger than the 1st bond
Expect bond energy similar to BeH+, maybe stronger, because the zs orbital is already decoupled from the sz.
1Σ+
2Σ+
1Σ+
linear
Cannot bind 3rd H because no singly occupied orbitals left.
2.1 eVR=1.65 A
2.03 eVR=1.34A
~3.1 eV ~2.1 e

13© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
The GVB orbitals of Silicon atom
Long dashes indicate zero amplitude, solid lines indicate positive amplitude while short dashes indicate negative amplitude. The spacing between contours is 0.05 in atomic units

14© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Bonding H atom to all 3 states of C
Now we can get a bond to the lobe orbital just as for BeH
Bring H1s along z axis to C and consider all 3 spatial states.
(2px)(2pz)
C 2pz singly occupied.
H1s can get bonding
Get S= ½ state,
Two degenerate states, denote as 2Π
(2px)(2py)
(2py)(2pz)

15© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Is the 2Π state actually 2Π?
The presence of the lobe orbitals might seem to complicate the symmetryΨyz=A[(sx)(xs)+(xs)(sx)](αβ−βα)(yα) (zΗ bond)2]
Ψxz=A[(sy)(ys)+(ys)(sy)](αβ−βα)(xα)(zΗ bond)2)]
To see that there is no problem, rewrite in the CI form (and ignore the zH bond)Ψyz=A[(s2 – λ x2)](αβ−βα)(yα)]
Now form a new wavefunction by adding - λ y2 to Ψyz
Φyz ≡ A[s2 – λ x2 – λ y2](αβ−βα)(yα)] But the 3rd term is A[y2](αβ−βα)(yα)]= – λ A[(yα)(yβ)(yα)]=0Thus Φyz = Ψyz and similarly Φxz = A[s2 – λ x2 – λ y2](αβ−βα)(xα)] = Ψxz
Thus the 2s term [s2 – λ x2 – λ y2] is clearly symmetric about the z axis, so that these wavefunctions have 2Π symmetry
skip
16© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
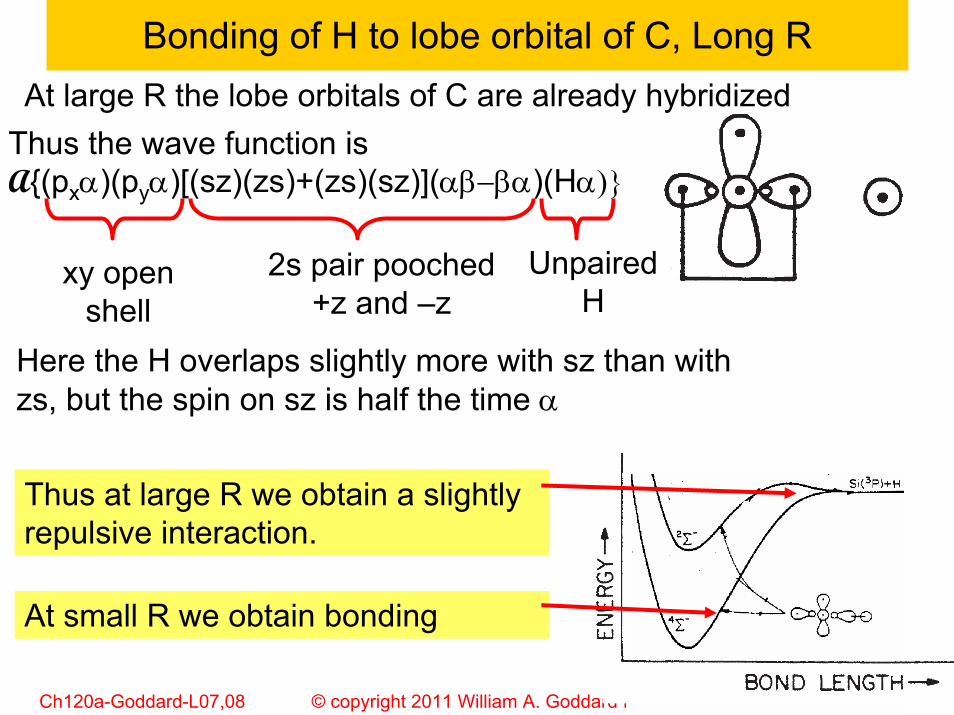
Bonding of H to lobe orbital of C, Long R
Thus the wave function is A{(pxα)(pyα)[(sz)(zs)+(zs)(sz)](αβ−βα)(Hα)}
At large R the lobe orbitals of C are already hybridized
2s pair pooched+z and –z
xy open shell
Unpaired H
Here the H overlaps slightly more with sz than with zs, but the spin on sz is half the time α
Thus at large R we obtain a slightly repulsive interaction.
At small R we obtain bonding

17© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Bonding of H to lobe orbital of C, small R
At small R the H can overlap significantly more with sz than with zH, so that we can form a bond pair just like in BeH+. This leads to the wavefunctionA{[(sz)(H)+(H)(sz)](αβ−βα)(zsα)(pxα)(pyα)}
But now the zs hybrid must now get orthogonal to the sz and H bond pair. This destabilizes the bond by ~ 1 eVThe symmetries of the nonbond orbitals are: zs=σ, px=πx, py=πy
Since the nonbond orbitals, σ, πx, πy are orthogonal to each other the high spin is lowest (S=3/2 or quartet state)We saw for NH that (πxπy –πyπx)(αα) has 3Σ- symmetry. CH has one additional high spin nonbond σ orbital, leading to 4Σ-
Hszzs
pxpy
Sz-H bond pair nonbond orbitals

18© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
The bonding states of CH and SiH
The low-lying state of SiHare shown at the right. Similar results are obtained for CH.
The bond to the p orbital to form the 2Π state is best
CH SiH
De(2Π) 80.0 70.1Kcal/mol
Δ(2Π−4Σ-) De(4Σ-) 62.9 33.9
17.1 36.2
p bond
sz bond
The bond to the lobe orbital is weaker than the p, but it is certainly significant

19© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
GVB orbitals of SiH4Σ- state

20© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
GVB orbitals of SiH 2Πstate
H
sx
xs
pz
py

21© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Analysis of bonding in CH and SiHBond to p orbital is still the best for C and Si but the lobe bond is also quite strong, especially for CHThus hydridization in the atom due to electron correlation leads naturally to the new 4Σ- bonding state.Note that although the (sx)(xs) lobe pair for the atom are at 180º in the atom, they bend to ~104º for CH and SiH
180º
The reason is that as the pH bond is formed, the incoming H orbital overlaps the 2s part of the lobe orbital. To remain orthogonal, the 2s orbital builds in some –z character along with the x character already there. This rotates the lobe orbital away from the incoming H. This destabilizes the lobe pair making it easier for the 2nd H to bond to the lobe pair.
104º

22© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Bonding the 2nd H to CH and SiH
As usual, we start with the ground states of CH or SiH, 2Πxand 2Πy and bond bring an H along some axis, say x.
H
sx
xs
pz
py
2Πy
2Πx

23© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Bonding the 2nd H to CH and SiH
Now we get credible bound states from both components
H
sx
xs
pz
py
2Πy
2Πx
A bond to the sx lobe orbital of CH (2Πy)
A bond to the p orbital of CH (2Πx)
This leads to the 1A1state of CH2 and SiH2that has already been
discussed.
This leads to the 3B1state that is the ground
state of CH2

24© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
The p bond leads to the 1A1 state
GVB orbitals for SiH(1A1)
Ψ=A{[(sy)(ys)+(ys)(sy)(αβ−βα)](SiHLbond)2(SiHRbond)2}The wavefunction is
Applying C2z or σ in the plane interchanges (sy) and (ys) but the (sy)(ys) pair is symmetric under this interchange. Thus the total symmetry is 1A1.

25© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Bonding the 2nd H to the lobe orbital
At large distances the bond to the lobe orbital will be slightly repulsive and at an angle of 128 to the already formed p bond.
However at short distances, we form a strong bond. After forming the bond, each bond pair readjusts to have equivalent character (but an average of lobe bond and p bond).
The wavefunction becomesΨ=A{(SiHLbond)2(SiHRbond)2 [(σℓα)(πxα)]}
σℓπx
Here the two bond pairs and the σℓ orbital have A1 while πx has b1symmetry so that the total spatial symmetry is B1.
This leads to both 3B1 and 1B1 states, but triplet is lower (since σℓand πx are orthogonal).

26© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Analyze Bond angles of CH2 and SiH2θe Re
1A1 state. Optimum bonding, pz orbital points at Hz while
px orbital points at Hx, leading to a bond angle of 90º. We expect that HH repulsion increases this by slightly less thanthe 13.2º for NH2 and 14.5º for OH2 and indeed it increases by 12.4º. But for Si the increase from 90º is only 2º as for P and As.
θe Re3B1 state. Optimum bonding, the two bonds at ~128º.
Here HH orthogonalizationshould increase this a bit but Cmuch less than 12º since H’s are much farther apart. However now the σℓ orbital must get orthogonal to the two bond pairs a bond angle decrease. The lone pair affect dominates for SiH2decreasing the bond angle by 10º to 118º while the HH affect dominates for CH2, increasing the bond angle by 5º to 133º
σℓ

27© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
The GVB orbitals for SiH2 (3B1)

28© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Analysis of bond energies of 1A1 stateConsider the first two p bonds. Ignoring the affect of the bonds on the lobe orbitals, the main difference arises from the exchange terms. For C or Si A[(2s)2(pzα)(pxα)] leads to a term in the energy of the form (Jxz –Kxz) since the x and z orbitals have the same spin. But upon bonding the first H to pz, the wavefunction becomes A{(2s)2[(pzH+Hpx)(αβ−βα)(pxα)}. Now the pz and px orbitals have the same spin only have the time, so that this exchange term is decreased to - ½ Kxz. However in forming the second bond, there is no additional correction.Since Kxz ~ 14 kcal/mol for C and ~9 kcal/mol for Si. This means that the 2nd bond should be stronger than the first by 7 kcal/mol for C and by 4.5 kcal/mol for Si. E(kcal/mol)1st bond 2nd bond
C 80 90Si 70 76.2
This is close to the observed differences.

29© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Analysis of bond energies of CH2 and SiH2 state
2Π
4Σ-62.9ℓ
CH SiH
3P
80.0p
2Π
4Σ-33.9ℓ
70.1p
3B1
1A1 90.0p99.1ℓ
3B11A1
56.9ℓ76.1p
CH2
SiH2
CH Lobe bonds: 63 and 9950% increase
SiH Lobe bonds: 35 and 5750% increase
Assume 50% increase in lobe bond is from the first p bond destabilizing the lone pair

30© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
The Bending potential surface for CH2
3B1
1A1
1B1
3Σg-
1Δg
9.3 kcal/mol
The ground state of CH2 is the 3B1 state not 1A1.

31© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Analysis of bond energies of 3B1 state
For CH the lobe bond is 17 kcal/mol weaker than the p bond while for SiH it is 37 kcal/mol weaker. Forming the lobe bond requires unpairing the lobe pair which is ~1 eV for the C row and ~1.5 eV for the Si row. This accounts for the main differences suggesting that p bonds and lobe bonds are otherwise similar in energy. Forming a lobe bond to CH or SiH should be easier than to C or Si, because the first p bond has already partially destabilized thelobe pair. Since the SiH2(3B1) state is 19 kcal/mol higher than SiH2(1A1) but SiH(4Σ-) is 35 kcal/mol higher than Si(2Π), we conclude that lobe bond has increased in strength by ~16 kcal/mol Indeed for CH the 3B1 state is 9.3 kcal/mol lower than 1A1implying that the lobe bond has increased in strength relative to the p bond by 26 kcal/mol.

32© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
CH2 GVB orbitals
33© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
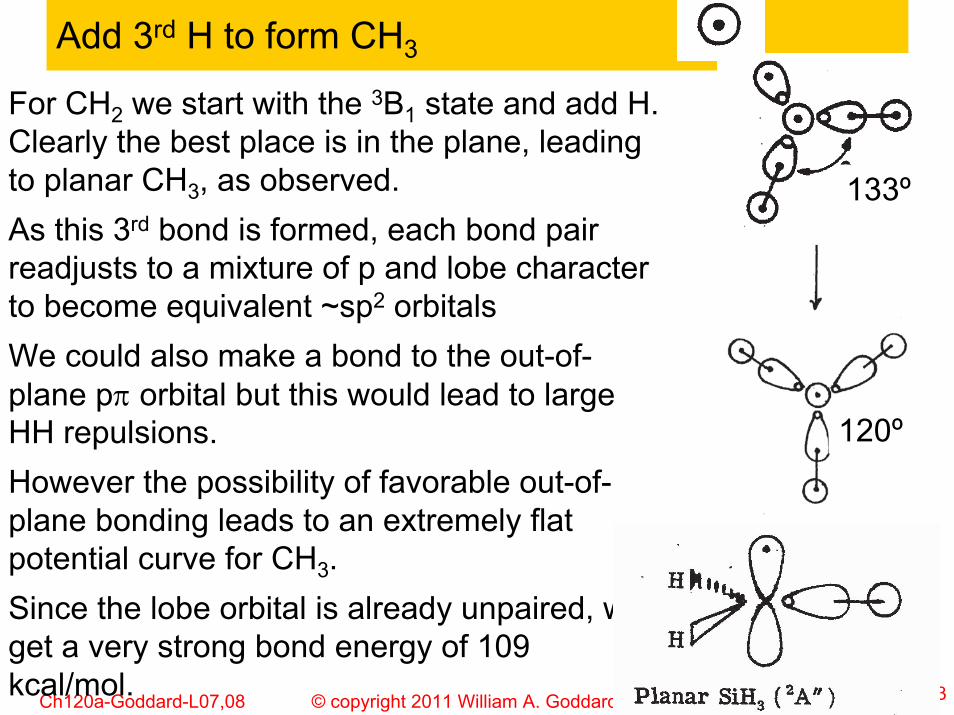
Add 3rd H to form CH3
For CH2 we start with the 3B1 state and add H. Clearly the best place is in the plane, leading to planar CH3, as observed. As this 3rd bond is formed, each bond pair readjusts to a mixture of p and lobe character to become equivalent ~sp2 orbitalsWe could also make a bond to the out-of-plane pπ orbital but this would lead to large HH repulsions. However the possibility of favorable out-of-plane bonding leads to an extremely flat potential curve for CH3. Since the lobe orbital is already unpaired, we get a very strong bond energy of 109 kcal/mol.
120º
133º

34© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Add 3rd H to 1A1 SiH2 to form SiH3.
For SiH2 we start with the 1A1 state and add H to the lobe pair. Clearly this leads to a pyramidal SiH3, with an the optimum bond angle of 111º.Since we must unpair the lobe orbital this 3rd bond is relatively weak, 72 kcal/mol.

35© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Add 4th H to form CH4 and SiH4.For CH3 we start with the planar molecule and bring the H up to the out of plane p orbital. As the new bond forms all four bondsreadjust to become equivalent leading to a tetrahedral CH4molecule. This bond is 105 kcal/mol, slightly weaker than the 3rd
109 kcal/mol) since it is to a p orbital and must interact with the other H’s.For SiH2 we start with the pyramidal geometry (111º bond angle) and add to the remaining lobe orbital. As the new bond forms, all fourbonds readjust to become equivalent, leading to a tetrahedral SiH4 molecule. No unpairing is required a strong bond, 92 kcal/mol (the 3rd bond was 72)

36© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
GVB orbitals of CH3 and CH4

37© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
GVB orbitals of SiH3 SiH bond pair
Dangling bond orbital

38© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
GVB orbitals of SiH4

39© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Hybridization of GVB Orbitals
Orthogonal and point to vertices of a tetrahedron.Rationalizes bonding in CH4.Assumes 75% p character
Idealized case.Tetrahedral: sp3
x Tetr
y
z
o
(s-x+y-z)/2
(s+x+y+z)/2(s-x-y+z)/2
(s+x-y-z)/2
GVB: CH4 is 70% pAtom: lobe is 13%p: total =226/4=57% p

40© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Comparisons of successive bond energies SiHn and CHn
p lobe
p
lobep
p
lobe
lobe

41© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
The ground state for B atom
Ψyz=A[(sx)(xs)+(xs)(sx)](αβ−βα)(zα)] which we visualize as
z
x
Based on our study of C, we expect that the ground state of B is
2s pair pooched ±x(or ±y)
z open shell pz
xs
sx
2s pair pooched ± z x open shell
px
szzs
Ψyx=A[(sz)(zs)+(zs)(sz)](αβ−βα)(xα)] which we visualize as
Ψxz=A[(sz)(zs)+(zs)(sz)](αβ−βα)(yα)] which we visualize as
2s pair pooched ± z y open shell szpy
zs

42© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
form BH and AlH by bonding along the z axis
Ψyz=A[(sx)(xs)pair](zΗ+Ηz)(αβ−βα)]Bonding to the pz state of B we obtain
2s pair pooched ±x(or ±y)
H-z covalent bond
pz
sx
xs
H-sz covalent bond open shell
pxszzs
Ψxz yx=A[(sz)(H)+(H)(sz)](αβ−βα)(yα)(zsα)]
Ψyx=A[(sz)(H)+(H)(sz)](αβ−βα)(xα)(zsα)]
open shell szpy
zs
128º
H
H
HH-sz covalent bond
1Σ+
3Πx
3Πy

43© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
The bonding states of BH and AlH
The ground state of BH and AlHis obtained by bonding to the p orbital (leading to the 1Σ+ state.
However the bond to the lobe orbital (leading to the 3Π state) is also quite strong.
The bond to the lobe orbital is weaker than the p, but it is certainly significant
1Σ+
3Π
1Π
3Σ+
2P+ H

44© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
BH2 and AlH2
128º
Starting from the ground state of BH or AlH, the second bond is to a lobe orbital, to form the 2A1 state.
Just as for the 3B1 state of CH2 and SiH2 the bond for BH2 opens by several degrees to 131º while the bond to the AlH2 closes down by ~9º.
θe Re

45© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
BH3 and AlH3
Bonding the 3rd H to the 2A1 state of BH2, leads to planar BH3 or AlH3.
But there is no 4th bond since there remain no additional unpaired orbitals to bond to.

46© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Re-examine bonding in NH, OH, and FHWhy did we ignore hybridization of the (2s) pair for NH, OH, andFH?
The reason is that the ground state of N atom
A[(2sα)(2sβ)(xα)(yα)(zα)]
Already has occupied px,py,pz orbitals. Thus
Pauli annihilates any hybridization in the 2s orbital.

47© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Re-examine bonding in NH, OH, and FHHowever, the doublet excited state of N can have hybridization, eg
A(2s)2(y)2(zα) A[(sx)(xs)+(xs)(sx)](αβ−βα)(y)2(zα) which leads to the 2A1 excited states of NH2 of the form
θe Re

48© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Bonding to halides (AXn, for X=F,Cl,…
The ground state of F has just one singly-occuped orbital and hence bonding to C is in many ways similar to H, leading to CF, CF2, CF3, and CF4 species. However there are significant differences.
Thus CF leads to two type so bonds, p and lobe just like CH
2Π
4Σ−
p bond
lobe bond
Covalent bond expect (CH)80 kcal/mol
63 kcal/mol
actual bond (CF)
120 kcal/mol
63 kcal/mol

49© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
How can CF lead to such a strong bond, 120 vs 80 kcal/mol?
Consider the possible role of ionic character in the bonding
In the extreme limit
+
C F C+ F-
E (R=∞) = 0 E (R=∞) =IP (C) – EA (F)
=11.3 – 3.4 =7.9 eVIP (C) = 11.3 eV = 260 kcal/mol EA (F) = 3.40 eV = 78.4 kcal/molCan Coulomb attraction make up this difference?

50© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Estimate energy of pure ionic bond for CF
Covalent limit (C + F)
Ionic limit (C+ + F-)7.9 eV14.4/R
Re=1.27A
14.4/1.27 = 11.3 eV
Net bond = 11.3-7.9 = 3.4 eV= 78 kcal/mol
Units for electrostatic interactions E=Q1*Q2/(ε0*R)where ε0 converts units (called permittivity of free space)E(eV) = 14.4 Q1(e units)*Q2(e units)/R(angstrom)E(kcal/mol) = 332.06 Q1(e units)*Q2(e units)/R(angstrom)
Ionic estimate ignores shielding and pauli repulsion for small R. Thus too large
51© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
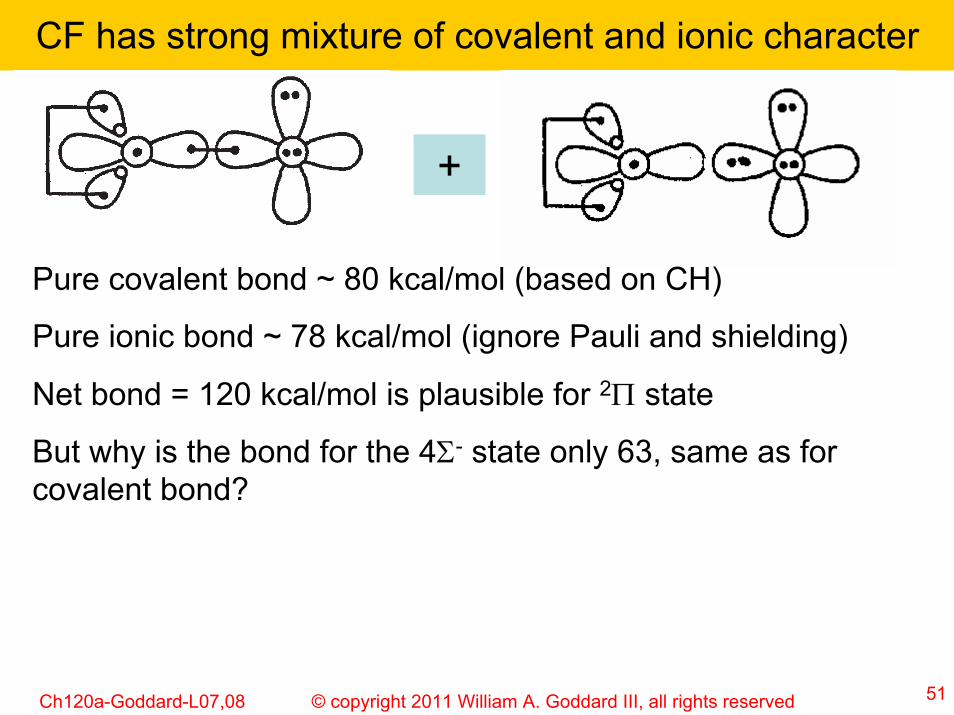
CF has strong mixture of covalent and ionic character
+
Pure covalent bond ~ 80 kcal/mol (based on CH)
Pure ionic bond ~ 78 kcal/mol (ignore Pauli and shielding)
Net bond = 120 kcal/mol is plausible for 2Π state
But why is the bond for the 4Σ- state only 63, same as for covalent bond?

52© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Consider ionic contribution to 4Σ− state
+
C F C+ F-
E (R=∞) = 0 E (R=∞) =IP (C) – EA (F)
=16.6 – 3.4 =13.2 eV
To mix ionic character into the 4Σ− state the electron must be pulled from the sz lobe orbital.
This leads to the (2s)1(2p)2 state of C+ rather than the ground state (2s)2(2p)1 which is 123 kcal/mol = 5.3 eV higher
Thus ionic bond is NEGATIVE (78-123=-45 kcal/mol)

53© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Bonding the 2nd F to CFWith the 4Σ- state at 57 kcal/mol higher than 2Π, we need only consider bonding to 2Π, leading to the 1A1 state.
Bad Pauli repulsion increases FCF angle to 105º
1A1
3B1
57 kcal/mol

54© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Now bond 3rd F to form CF3
Get pyramidal CF3(FCF angle ~ 112º)
In sharp contrast to planar CH3
The 3rd CF bond should be much weaker than 1st two.
This strong preference for CF to use p character makes conjugated flourocarbons much less stabe than corresponding saturated compounds.
Thus C4H6 prefers butadiene but C4F6 prefers cylcobutene
Of course the 4th bond to form CF4 leads to a tetrahedral structure

55© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Summary, bonding to form hydrides
General principle: start with ground state of AHn and add H to form the ground state of AHn+1
Thus use 1A1 AH2 for SiH2 and CF2 get pyramidal AH3
Use 3B1 for CH2 get planar AH3.
For less than half filled p shell, the presence of empty p orbitals allows the atom to reduce electron correlation of the (ns) pair by hybridizing into this empty orbital.
This has remarkable consequences on the states of the Be, B, and C columns.

56© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
New material

57© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Now combine Carbon fragments to form larger molecules (old chapter 7)
Starting with the ground state of CH3 (planar), we bring two together to form ethane, H3C-CH3.
As they come together to bond, the CH bonds bend back from the CC bond to reduce overlap (Pauli repulsion or steric interactions between the CH bonds on opposite C).
At the same time the 2pp radical orbital on each C mixes with 2s character, pooching it toward the corresponding hybrid orbital on the other C
107.7º
111.2º
1.526A
1.095A1.086A120.0º

58© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Bonding (GVB) orbitals of ethane (staggered)
Note nodal planes from
orthogonalizationto CH bonds on
right C

59© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Staggered vs. Eclipsed
There are two extreme cases for the orientation about the CC axis of the two methyl groups
The salient difference between these is the overlap of the CH bonding orbitals on opposite carbons.
To whatever extent they overlap, SCH-CH Pauli requires that they be orthogonalized, which leads to a repulsion that increases exponentially with decreasing distance RCH-CH.
The result is that the staggered conformation is favored over eclipsed by 3.0 kcal/mol

60© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Alternative interpretation
The bonding electrons are distributed over the molecule, but it is useful to decompose the wavefunction to obtain the net charge on each atom.
qH ~ +0.15
qC ~ -0.45
This leads to qH ~ +0.15 and qC ~ -0.45.
These charges do NOT indicate the electrostatic energies within the molecule, but rather the electrostatic energy for interacting with an external field.Even so, one could expect that electrostatics would favor staggered.
The counter example is CH3-C=C-CH3, which has a rotational barrier of 0.03 kcal/mol (favoring eclipsed). Here the CH bonds are ~ 3 times that in CH3-CH3 so that electrostatic effects would decrease by only 1/3. However overlap decreases exponentially.

61© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Propane
Replacing an H of ethane with CH3, leads to propane
Keeping both CH3 groups staggered leads to the unique structure
Details are as shown. Thus the bond angles are
HCH = 108.1 and 107.3 on the CH3
HCH =106.1 on the secondary C
CCH=110.6 and 111.8
CCC=112.4,
Reflecting the steric effects
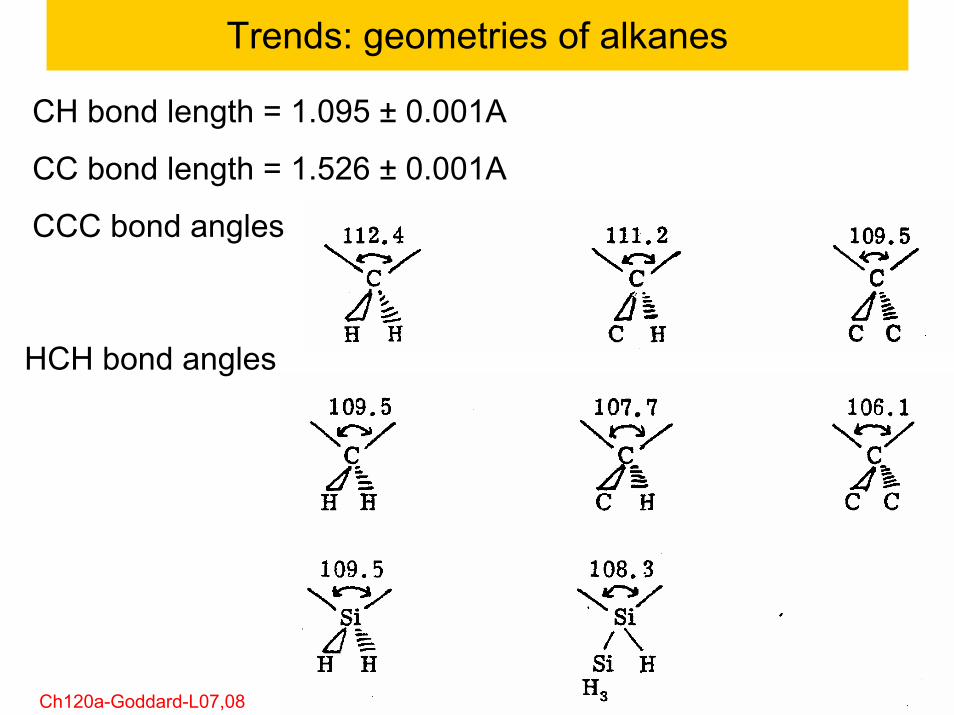
62© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Trends: geometries of alkanes
CH bond length = 1.095 ± 0.001A
CC bond length = 1.526 ± 0.001A
CCC bond angles
HCH bond angles

63© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Bond energiesDe = EAB(R=∞) - EAB(Re)e for equilibrium)Get from QM calculations. Re is distance at minimum energy.

64© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Bond energiesDe = EAB(R=∞) - EAB(Re)Get from QM calculations. Re is distance at minimum energyD0 = H0AB(R=∞) - H0AB(Re)H0=Ee + ZPE is enthalpy at T=0KZPE = Σ(½Ћω) This is spectroscopic bond energy from ground vibrational state (0K)Including ZPE changes bond distance slightly to R0

65© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Bond energiesDe = EAB(R=∞) - EAB(Re)Get from QM calculations. Re is distance at minimum energyD0 = H0AB(R=∞) - H0AB(Re)H0=Ee + ZPE is enthalpy at T=0KZPE = Σ(½Ћω) This is spectroscopic bond energy from ground vibrational state (0K)Including ZPE changes bond distance slightly to R0Experimental bond enthalpies at 298K and atmospheric pressure D298(A-B) = H298(A) – H298(B) – H298(A-B)D298 – D0 = 0∫
298 [Cp(A) +Cp(B) – Cp(A-B)] dT =2.4 kcal/mol if A and B are nonlinear molecules (Cp(A) = 4R). {If A and B are atoms D298 – D0 = 0.9 kcal/mol (Cp(A) = 5R/2)}.(H = E + pV assuming an ideal gas)

66© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Bond energies, temperature corrections
Experimental measurements of bond energies, say at 298K, require an additional correction from QM or from spectroscopy.The experiments measure the energy changes at constant pressure and hence they measure the enthalpy,H = E + pV (assuming an ideal gas)Thus at 298K, the bond energy isD298(A-B) = H298(A) – H298(B) – H298(A-B)D298 – D0 = 0∫
298 [Cp(A) +Cp(B) – Cp(A-B)] dT =2.4 kcal/molif A and B are nonlinear molecules (Cp(A) = 4R). {If A and B are atoms D298 – D0 = 0.9 kcal/mol (Cp(A) = 5R/2)}.

67© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Snap Bond Energy: Break bond without relaxing the fragments
Snap
Adiabatic
ΔErelax = 2*7.3 kcal/mol
DsnapDesnap (109.6 kcal/mol) De (95.0kcal/mol)

68© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Bond energies for ethane
D0 = 87.5 kcal/mol
ZPE (CH3) = 18.2 kcal/mol,
ZPE (C2H6) = 43.9 kcal/mol,
De = D0 + 7.5 = 95.0 kcal/mol (this is calculated from QM)
D298 = 87.5 + 2.4 = 89.9 kcal/mol
This is the quantity we will quote in discussing bond breaking processes

69© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
The snap Bond energy
In breaking the CC bond of ethane the geometry changes from CC=1.526A, HCH=107.7º, CH=1.095A
To CC=∞, HCH=120º, CH=1.079A
Thus the net bond energy involves both breaking the CC bond and relaxing the CH3 fragments.
We find it useful to separate the bond energy into
The snap bond energy (only the CC bond changes, all other bonds and angles of the fragments are kept fixed)
The fragment relaxation energy.
This is useful in considering systems with differing substituents.
For CH3 this relation energy is 7.3 kcal/mol so that
De,snap (CH3-CH3) = 95.0 + 2*7.3 = 109.6 kcal/mol

70© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Substituent effects on Bond energiesThe strength of a CC bond changes from 89.9 to 70 kcal/mol as the various H are replace with methyls.Explanations given include:
•Ligand CC pair-pair repulsions
•Fragment relaxation
•Inductive effects

71© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Ligand CC pair-pair repulsions:
Each H to Me substitution leads to 2 new CH bonds gauche to the original CC bond, which would weaken the CC bond.
Thus C2H6 has 6 CH-CH interactions lost upon breaking the bond,
But breaking a CC bond of propane loses also two addition CC-CH interactions.
This would lead to linear changes in the bond energies in the table, which is approximately true. However it would suggest that the snap bond energies would decrease, which is not correct.

72© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Fragment relaxation
Because of the larger size of Me compared to H, there will be larger ligand-ligand interaction energies and hence a bigger relaxation energy in the fragment upon relaxing form tetrahedral to planar geometries.
In this model the snap bond enegies are all the same.
All the differences lie in the relaxation of the fragments.
This is observed to be approximately correct
Inductive effect
A change in the character of the CC bond orbital due to replacement of an H by the Me.
Goddard believes that fragment relaxation is the correct explanation PUT IN ACTUAL RELAXATION ENERGIES

73© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Bond energies: Compare to CF3-CF3
For CH3-CH3 we found a snap bond energy of
De = 95.0 + 2*7.3 = 109.6 kcal/mol
Because the relaxation of tetrahedral CH3 to planar gains 7.3 kcal/mol
For CF3-CF3, there is no such relaxation since CF3 wants to be pyramidal, FCF~111º
Thus we might estimate that for CF3-CF3 the bond energy would be De = 109.6 kcal/mol, hence D298 ~ 110-5=105
Indeed the experimental value is D298=98.7±2.5 kcal/mol suggesting that the main effect in substituent effects is relaxation (the remaining effects might be induction and steric)

74© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
CH2 +CH2 ethene
Starting with two methylene radicals (CH2) in the ground state (3B1) we can form ethene (H2C=CH2) with both a σ bond and a π bond.
The HCH angle in CH2 was 132.3º, but Pauli Repulsion with the new σ bond, decreases this angle to 117.6º (cf with 120ºfor CH3)

75© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Comparison of The GVB bonding orbitals of ethene
and methylene
76© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
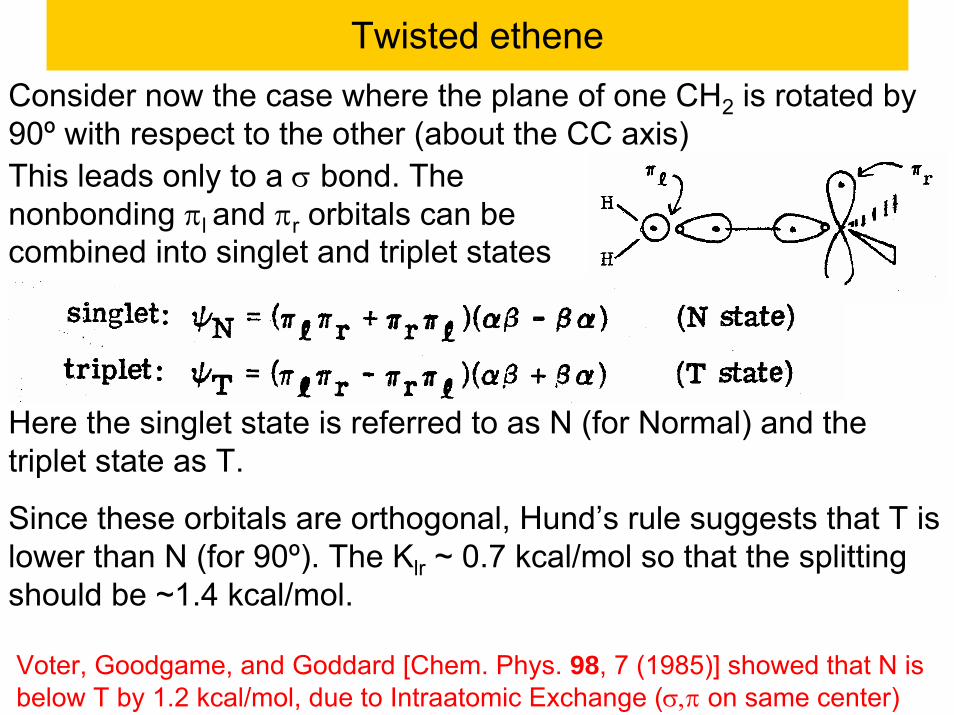
Twisted etheneConsider now the case where the plane of one CH2 is rotated by 90º with respect to the other (about the CC axis)This leads only to a σ bond. The nonbonding πl and πr orbitals can be combined into singlet and triplet states
Here the singlet state is referred to as N (for Normal) and the triplet state as T.
Since these orbitals are orthogonal, Hund’s rule suggests that T is lower than N (for 90º). The Klr ~ 0.7 kcal/mol so that the splitting should be ~1.4 kcal/mol.
Voter, Goodgame, and Goddard [Chem. Phys. 98, 7 (1985)] showed that N is below T by 1.2 kcal/mol, due to Intraatomic Exchange (σ,π on same center)

77© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Twisting potential surface for ethene
The twisting potential surface for ethene is shown below. The N state prefers θ=0º to obtain the highest overlap while the T state prefers θ=90º to obtain the lowest overlap

78© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
geometries
For the N state (planar) the CC bond distance is 1.339A, but this increases to 1.47A for the twisted form with just a single σ bond. This compares with 1.526 for the CC bond of ethane.
Probably the main effect is that twisted ethene has very little CH Pauli Repulsion between CH bonds on opposite C, whereas ethane has substantial interactions.
This suggests that the intrinsic CC single bond may be closer to1.47A
For the T state the CC bond for twisted is also 1.47A, but increases to 1.57 for planar due to Orthogonalization of the triple coupled pπ orbitals.

79© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
CC double bond energies
Breaking the double bond of ethene, the HCH bond angle changes from 117.6º to 132.xº, leading to an increase of 2.35 kcal/mol in the energy of each CH2 so that
Desnap = 180.0 + 4.7 = 184.7 kcal/mol
Since the Desnap = 109.6 kcal/mol, for H3C-CH3,
The π bond adds 75.1 kcal/mol to the bonding.
Indeed this is close to the 65kcal/mol rotational barrier.
For the twisted ethylene, the CC bond is De = 180-65=115 Desnap = 115 + 5 =120. This increase of 10 kcal/mol compared to ethane might indicate the effect of CH repulsions
The bond energies for ethene are
De=180.0, D0 = 169.9, D298K = 172.3 kcal/mol

80© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
bond energy of F2C=CF2
The snap bond energy for the double bond of ethene od
Desnap = 180.0 + 4.7 = 184.7 kcal/mol
As an example of how to use this consider the bond energy of F2C=CF2,
Here the 3B1 state is 57 kcal/higher than 1A1 so that the fragment relaxation is 2*57 = 114 kcal/mol, suggesting that the F2C=CF2 bond energy is Dsnap~184-114 = 70 kcal/mol.
The experimental value is D298 ~ 75 kcal/mol, close to the prediction

81© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Bond energies double bondsAlthough the ground state of CH2 is 3B1 by 9.3 kcal/mol, substitution of one or both H with CH3 leads to singlet ground states. Thus the CC bonds of these systems are weakened because of this promotion energy.

82© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
C=C bond energies

83© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
CC triple bonds
Starting with two CH radicals in the 4Σ- state we can form ethyne (acetylene) with two π bonds and a σ bond.
This leads to a CC bond length of 1.208A compared to 1.339 for ethene and 1.526 for ethane.
The bond energy is
De = 235.7, D0 = 227.7, D298K = 229.8 kcal/mol
Which can be compared to De of 180.0 for H2C=CH2 and 95.0 for H3C-CH3.
84© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
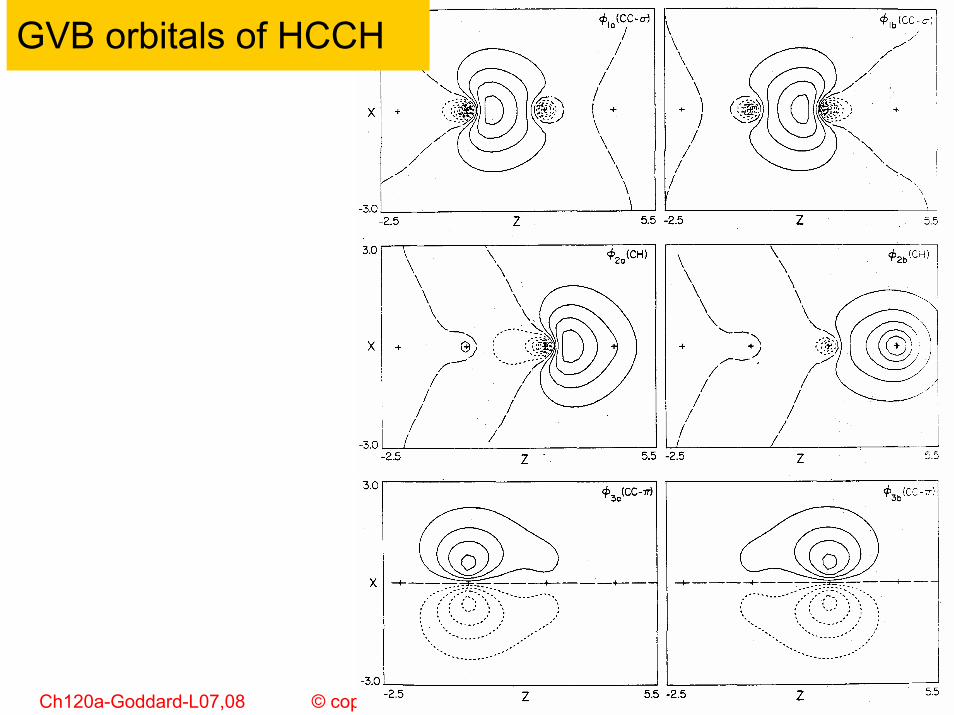
GVB orbitals of HCCH

85© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
GVB orbitals of CH 2Π and 4Σ- state

86© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
CC triple bonds
Since the first CCσ bond is De=95 kcal/mol and the first CCπbond adds 85 to get a total of 180, one might wonder why the CC triple bond is only 236, just 55 stronger.
The reason is that forming the triple bond requires promoting the CH from 2Π to 4Σ-, which costs 17 kcal each, weakening the bond by 34 kcal/mol. Adding this to the 55 would lead to a total 2nd π bond of 89 kcal/mol comparable to the first
2Π
4Σ-

87© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Bond energies

88© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08

89© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
DiamondReplacing all H atoms of ethane and with methyls, leads to with a staggered conformation
Continuing to replace H with methyl groups forever, leads to the diamond crystal structure, where all C are bonded tetrahedrally to four C and all bonds on adjacent C are staggered
A side view is
This leads to the diamond crystal structure. An expanded view is on the next slide

90© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Infinite structure from tetrahedral bonding plus staggered bondson adjacent centers
Chair configuration
of cylco-hexane
Not shown: zero layer just like 2nd layer but above layer 13rd layer just like the 1st layer but below layer 2
2nd layer
1st layer
2nd layer
1st layer
2nd layer
1st layer
1
1
c 1
3 1
02
1
2
1
0
11
20

91© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
The unit cell of diamond crystalAn alternative view of the diamond structure is in terms of cubes of side a, that can be translated in the x, y, and z directions to fill all space.
Note the zig-zag chains c-i-f-i-cand cyclohexane rings (f-i-f)-(i-f-i)
•all 8 corners (but only 1/8 inside the cube): (0,0,0)•all 6 faces (each with ½ in the cube): (a/2,a/2,0), (a/2,0,a/2), (0,a/2,a/2)•plus 4 internal to the cube: (a/4,a/4,a/4), (3a/4,3a/4,a/4), (a/4,3a/4,3a/4), (3a/4,a/4,3a/4), Thus each cube represents 8 atoms.All other atoms of the infinite crystal are obtained by translating this cube by multiples of a in the x,y,z directions
There are atoms at c c
c c
c c
c c
ff
f
f
f
f
ii
ii

92© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Diamond Structure
12
1a
1c1b
32b
2a
Start with C1 and make 4 bonds to form
a tetrahedron.
Now bond one of these atoms, C2, to 3 new C
so that the bond are staggered with respect
to those of C1.
Continue this process.
Get unique structure: diamond
Note: Zig-zag chain
1b-1-2-3-4-5-6
Chair cyclohexane ring: 1-2-3-3b-7-1c
43b
3a
54b
4a
5b
5a
67

93© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Properties of diamond crystals

94© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Properties of group IV molecules (IUPAC group 14)
1.526
There are 4 bonds to each atom, but each bond connects two atoms. Thus to obtain the energy per bond we take the total heat of vaporization and divide by two. Note for Si, that the average bond is much different than for Si2H6

95© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Comparisons of successive bond energies SiHn and CHn
p lobe
p
lobep
p
lobe
lobe

96© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Miller indices A 3D crystal is characterized by a unit cell with axes, a, b, c that can be translated by integer translations along a, b, c to fill all space.
The corresponding points in the translated cells are all equivalent.
Passing a plane through any 3 such equivalent points defines a plane denoted as (h,k,l). An equally space set of parallel to (h,k,l) pass through all equivalent points, which the l,m,ncorrespond to the reciprocal intersections on the unit cell whenone plane passes through the origin. These are called Miller indices
a
b
c
b/k
a/h
c/l

97© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Examples of special planes
a
b
c
b/k
a/h
c/l
To denote all equivalent planes we use {h,k,l} so that
{1,0,0} for cubic includes the 3 cases in the first row)
A number with a bar indicates negative
From Wikipedia

98© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Crystallographic directions
A lattice vector can be written as
Rmnp = m a + n b + p c
where m,n,p are integers. This is denoted as [m,n,p]
The set of equivalent vectors is denoted as <m,n,p>
Examples are shown here.
From Wikipedia
99© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
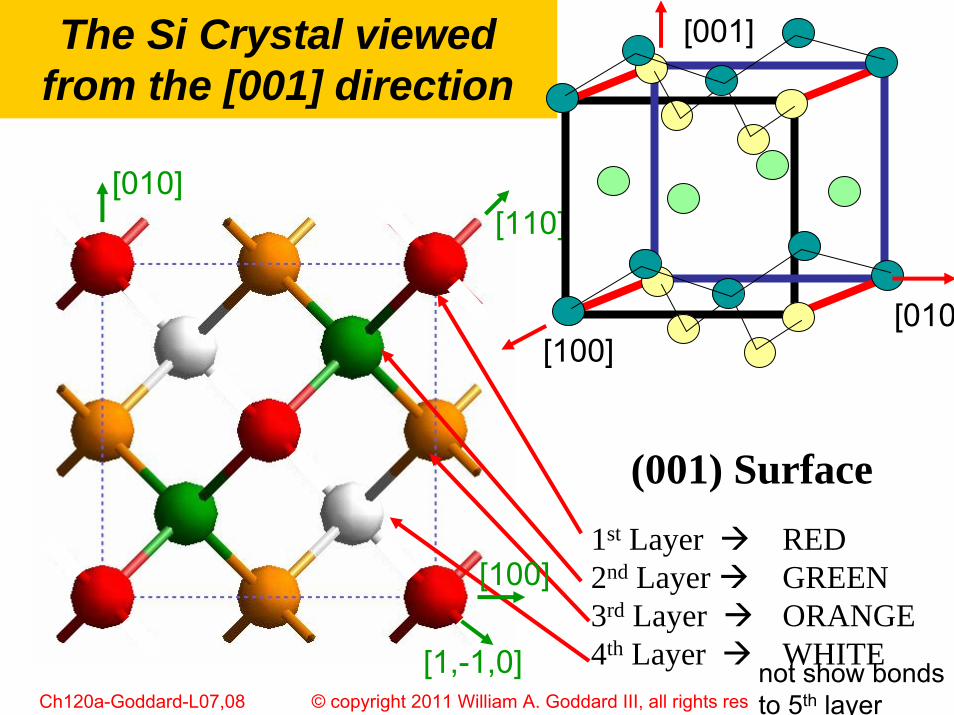
The Si Crystal viewed from the [001] direction
[100]
[010][110]
[1,-1,0] not show bonds to 5th layer
(001) Surface1st Layer RED2nd Layer GREEN3rd Layer ORANGE4th Layer WHITE
[010[100]
[001]

100© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
The Si Crystal (100) surface, unreconstructed
Surface zig-zag row
1st Layer RED2nd Layer GREEN3rd Layer ORANGE4th Layer WHITE
Every red surface atom is bonded to two green 2nd layer atoms, but the other two bonds were to two Si that are now removed. This leaves two non bonding electrons to distribute among the two dangling bond orbitals sticking out of plane (like the 1A1state of SiH2)
Surface unit cell P(1x1)
Projection of bulk cubic cell

101© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Si(100) surface (unreconstructed) viewed (nearly) along the [110] direction
Each surface atom has two dangling bond orbitals pointing to left and right, along [1,-1,0] direction

102© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Get one strong σ bond but leave two dangling bond orbitals on adjacent now bonded atoms (form weak π bond in plane)
The (100) Surface Reconstructionviewed (nearly) along the [110] direction
Spin pair dangling bond orbitals of adjacent atoms in [1,-1,0] direction (originally 2nd near neighbors

103© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Si(100) surface reconstructed (side view)
7.6A 2.4A3.8A 0.7A 0.7A
Lateral displacements
Surface bond
New cell length
orginal cell length
Surface atoms now bond to form dimers (move from 3.8 to 2.4A)Get row of dimes with doubled surface unit cellOne strong σ bond, plus weak π bond in plane

104© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Si(100) surface reconstructed (top view)
New unit cell reconstructed
surface
P(2x1)
original unit cell unreconstructed
surface
P(1x1)
Rows of dimer pairs are parallel
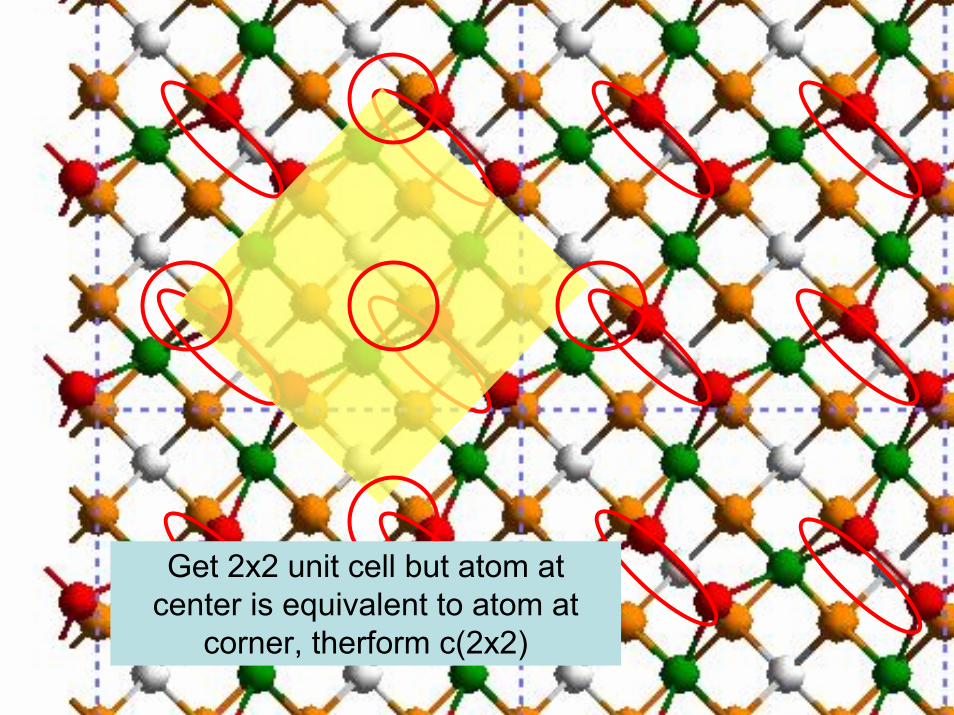
105© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Get 2x2 unit cell but atom at center is equivalent to atom at
corner, therform c(2x2)
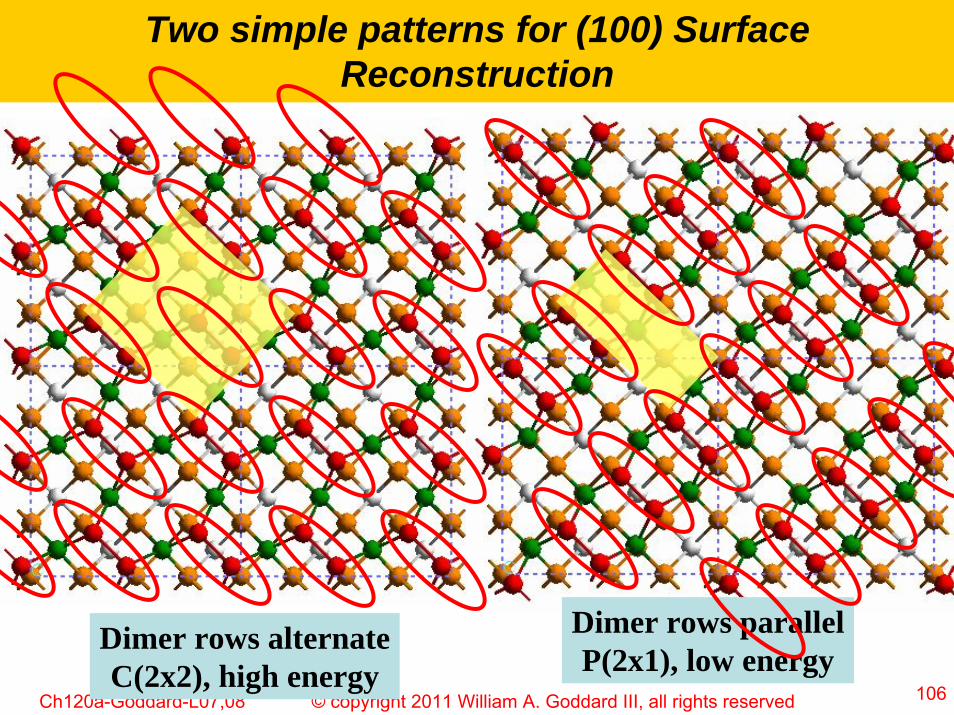
106© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Two simple patterns for (100) Surface Reconstruction
Dimer rows alternateC(2x2), high energy
Dimer rows parallelP(2x1), low energy

107© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
P(2x1) more stable than c(2x2) by ~ 1kcal/mol
110º
110º
120º
120º
The Sisurf-Si2nd-Sisurf bond for c(2x2) opens up to 120ºbecause the Sisurf move opposite directions
For P(2x1) the Sisurf move the same directions and Sisurf-Si2nd-Sisurf bond remains at 110º

108© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
stop

109© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Si(110) surface (top view)
Cut through cubic unit cell
Surface atoms red
surface unit cell P(1x1)

110© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Si(110) surface (side view)Surface
atoms redOne dangling bond
electron per surface atom

111© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
The (111) 1x1 Surface Unit Cell
Unpaired electron
60°
120°

112© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Construct (111) surface using cubic unit cell
4 2
2 0
c 4
4 2
24
2
4
4
2
33
31
Start at diagonal atom #0
Go straight down to atom #1
Atom #1 bonded to 3 atoms #2
Each #2 is bonded straight down to an atom#3
Each atom #3 is bonded to 3 atom#4.
Atoms 2 form a red (111) plane atoms #4 form a green (111) plane

113© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Si(111) surface (alternate construction)Start with red atom on top, bond to 3 green atoms in 2nd layer
Each green atom is bonded to 2 other 1st layer atoms plus a 3rd
atom straight down (not shown)
The 3rd layer atoms bond to 3 4th layer atoms in orange
Surface unit cell P(1x1)

114© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Reconstruction of Si(111) surface
Surface unit cell P(1x1)
Each surface atom has a single dangling bond electron, might guess that there would be some pairing of this with an adjacent atom to form a 2x1 unit cell.
Indeed freshly cleaved Si(111) at low temperature does show 2x1

115© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
LEED experiments (Schlier and Farnsworth, 1959) observed 7th Order Spots 7x7 unit
cellFrom 1959 to 1981 many models proposed to fit various experiments or calculations.
Binnig et al., 1981 did first STM image of Si (7x7) and saw 12 bright spots in 7x7 cell, showed that every previous model was incorrect
Takayanagi et al., 1985, proposed the DAS Model that explained the experiments

116© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
two 7x7 cells
What kind of interactions can go over a 7x7 region, with cell size 26.6 by 26.6 A?

117© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Origin of complex reconstruction of Si(111)
In 49 surface unit cells have 49 dangling bonds. Since cohesive energy of Si crystal is 108 kcal/mol expect average bond energy must be 108/2 = 54 kcal/mol (each atom has 4 bonds, but double count the bonds)
(H3Si-SiH3 bond energy is 74 kcal/mol)
Thus each dangling bond represents ~ 27 kcal/mol of surface energy = 1.1 eV per surface atom
Calculated value = 1.224 eV snap and 1.200 ev relaxed.

118© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
Consider bonding an atom on top of 3 dangling bonds
(a) T4 (b) H3
SIDE TOP SIDE TOP
(a) T4 (b) H3
SIDE TOP SIDE TOP
Two ways to do this. T4 and H3
Bond angle strain (H3)Pauli repulsion (H3)
Bond alignment/linear dependence (T4)*HOMO delocalization (T4)
H3 (not observed)T4 (observed)

119© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
AA
BB
H3 T4
AA
BB
H3 T4
HOMO T4HOMO H3 HOMO T4HOMO H3
T4 versus H3 site bonding to dangling bonds

120© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08

121© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
The (111) 7x7 DAS Surface

122© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
The (111) 7x7 DAS Surface Layers(purple, brown and blue atoms have one dangling bond)
1st 2nd
3rd 4th
First unreconstructed layer

123© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
The (111) 7x7 DAS Surface
12- and 8-membered rings

124© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
The (111) 7x7 DAS Surface
Side view

125© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
The (111) 7x7 DAS Surface Cornerhole

126© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
The (111) 7x7 DAS Layer Positions
1233
4 56
7 7 89
REFREF REF

127© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
The (111) 7x7 DAS Surface Adatoms
(a) T4 (b) H3
SIDE TOP SIDE TOP
(a) T4 (b) H3
SIDE TOP SIDE TOP
Bond angle strain (H3)Pauli repulsion (H3)
Bond alignment/linear dependence (T4)*HOMO delocalization (T4)
H3 (not observed)T4 (observed)

128© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
AA
BB
H3 T4
AA
BB
H3 T4
HOMO T4HOMO H3 HOMO T4HOMO H3
The (111) 7x7 DAS Surface Adatoms

129© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
The (111) 3x3 DAS Surface Unit Cell
Side view Top view12-membered rings

130© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
The (111) 5x5 DAS Surface Unit Cell
Side view

131© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
The (111) 5x5 DAS Surface Unit Cell
Top view12- and 8-membered rings

132© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
The (111) 9x9 DAS Surface Unit Cell
Side view

133© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
The (111) 9x9 DAS Surface Unit Cell
Top view12- and 8-membered rings

134© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
DAS Surface Energies (PBE DFT)
1.04
1.05
1.06
1.07
1.08
1.09
3 5 7 9 11 13DAS Cell Size
Ener
gy, e
V/1x
1 C
ell Regression
Ab Initio
Unreconstructed relaxed surface: 1.200 eV/1x1 cellInfinite DAS model: 1.107 eV/1x1 cell
1.044
1.0551.048
1.0701.068
1.078

135© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
DAS Surface Energies5x5 Surface (9 dangling bonds)
0.00
0.01
0.02
0.03
0.04
0 2 4 6 8 10
Spin Polarization
Ener
gy, e
V/1x
1 C
ell

136© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
DAS Reconstruction Driving Force
• 49 unpaired electrons (1/2 Si-Si bond) per 7x7 cell @ 1.2 eV = 58.8 eV/cell
• DAS 7x7 Surface energy = 51.2 eV/cell (19 unpaired electrons)
• Energy reduction due to reconstruction = 7.6 eV• Difference is due to strain• Bond length range = 2.31 – 2.50 Å (equilibrium 2.35
Å)• Bond angle range = 91 – 117º (Equilibrium 109.4°)
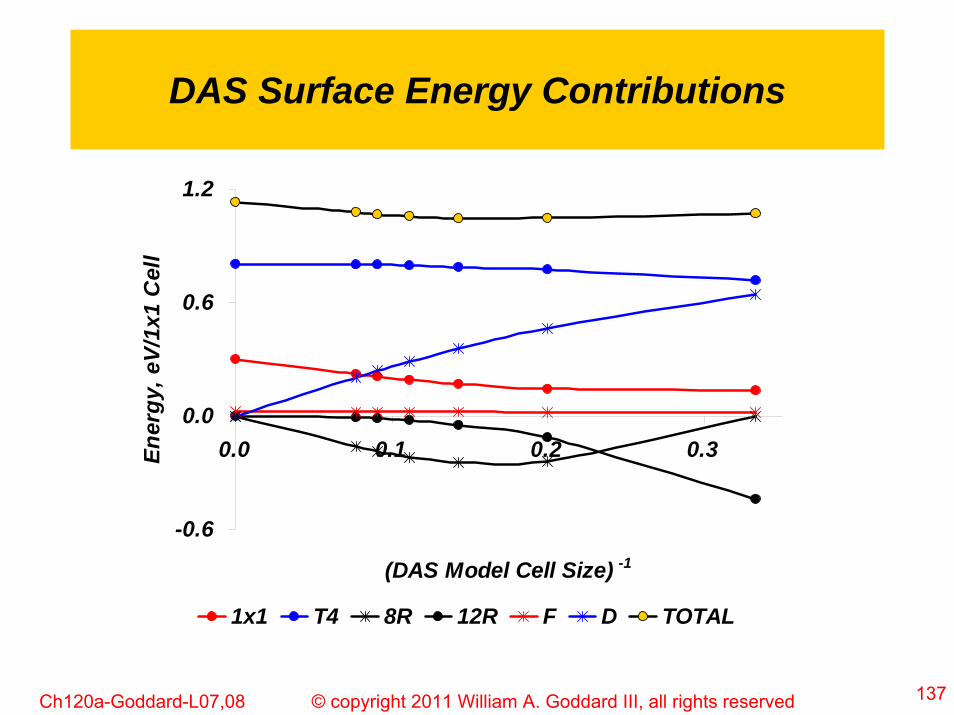
137© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
DAS Surface Energy Contributions
-0.6
0.0
0.6
1.2
0.0 0.1 0.2 0.3
(DAS Model Cell Size) -1
Ener
gy, e
V/1x
1 C
ell
1x1 T4 8R 12R F D TOTAL

138© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
DAS Surface Energies:Sequential Size Change Model
-20
-15
-10
-5
0
5
1 3 5 7 9 11 13
SSC Cell Size
Ener
gy, e
V/16
x16
Cel
l
SSC Irregular-odd and evenSSC regular-odd
Real-time STM by Shimada & Tochihara, 2003

139© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
DAS Surface Energies:Origin of a finite cell size
1.0
1.1
1.2
1.3
1.4
1 3 5 7 9 11 13Cell Size
Ener
gy, e
V/1x
1 C
ellSSC Irregular-odd and evenSSC regular-oddDFT
140© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
DAS 3x3 (side view)

141© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
DAS 3x3 (top view)

142© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
DAS 5x5 (side view)

143© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
DAS 5x5 (top view)

144© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
DAS 7x7 (side view)

145© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
DAS 7x7 (top view)

146© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
DAS 9x9 (side view)

147© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
DAS 9x9 (top view)

148© copyright 2011 William A. Goddard III, all rights reservedCh120a-Goddard-L07,08
stop


















