Filip Pawl owski, Simen Reine, Erik Tellgren Stinne Høst...
22
SCF methods for energies and properties of large molecular systems Trygve Helgaker, University of Oslo, Norway Filip Paw lowski, Simen Reine, Erik Tellgren Stinne Høst, Branislav Jans´ ık, Poul Jørgensen, Pekka Manninen, Jeppe Olsen, University of Aarhus, Denmark Sonia Coriani, University of Trieste, Italy Pawe l Sa lek, Royal Institute of Technology, Sweden Electron Correlation for the Whole Periodic Table – a theoretical symposium in honour of Bj¨ orn Roos – International Conference of Computational Methods in Sciences and Engineering (ICCMSE 2006) October 27–November 1, 2006 Hotel Panorama, Chania, Crete, Greece 1
Transcript of Filip Pawl owski, Simen Reine, Erik Tellgren Stinne Høst...

SCF methods for energies and properties of large molecular systems
Trygve Helgaker, University of Oslo, Norway
Filip Paw lowski, Simen Reine, Erik Tellgren
Stinne Høst, Branislav Jansık, Poul Jørgensen, Pekka Manninen, Jeppe Olsen,
University of Aarhus, Denmark
Sonia Coriani, University of Trieste, Italy
Pawe l Sa lek, Royal Institute of Technology, Sweden
Electron Correlation for the Whole Periodic Table
– a theoretical symposium in honour of Bjorn Roos –
International Conference of Computational Methods in Sciences and Engineering
(ICCMSE 2006)
October 27–November 1, 2006
Hotel Panorama, Chania, Crete, Greece
1
Self-consistent field (SCF) theory for large systems
• The development of SCF methods for large systems presents several challenges:
– expensive and difficult Fock/Kohn–Sham evaluation
– expensive diagonalization and difficult SCF convergence
– DFT functionals may perform poorly for large systems
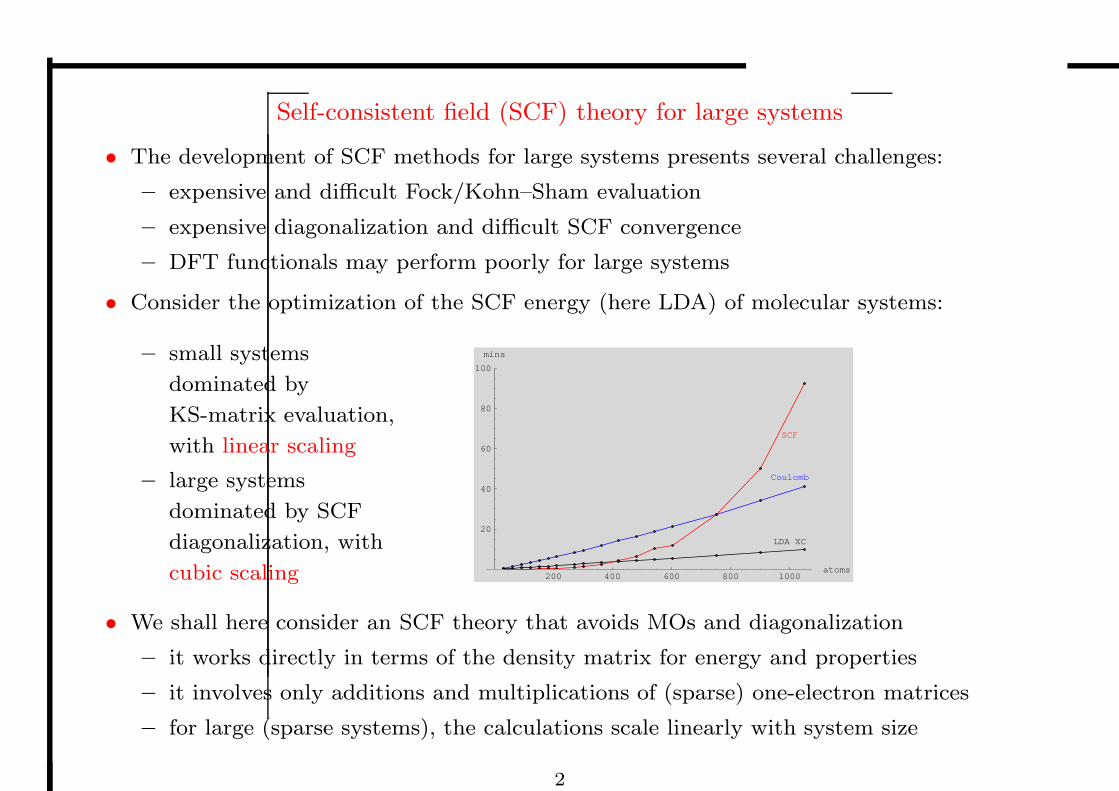
• Consider the optimization of the SCF energy (here LDA) of molecular systems:
– small systems
dominated by
KS-matrix evaluation,
with linear scaling
– large systems
dominated by SCF
diagonalization, with
cubic scaling 200 400 600 800 1000atoms
20
40
60
80
100
mins
SCF
Coulomb
LDA XC
• We shall here consider an SCF theory that avoids MOs and diagonalization
– it works directly in terms of the density matrix for energy and properties
– it involves only additions and multiplications of (sparse) one-electron matrices
– for large (sparse systems), the calculations scale linearly with system size
2

Direct optimization of the density matrix
• Consider the direct optimization of the density matrix:
E(D) = TrDh + 2-el. part
– there are constraints on the density matrix
D = DT| {z }
symmetry
, TrDS = N| {z }trace
, DSD = D| {z }idempotency
• Any other valid density matrix D(X) can then be generated from this matrix:
D(X) = exp(−XS)D exp(SX)| {z }exponential parameterization
, XT = −X| {z }antisymmetric
– Helgaker, Jørgensen and Olsen: Molecular Electronic-Structure Theory (Wiley, 2000)
– Head-Gordon and coworkers, MolPhys 101, 37 (2003), JCP 118, 6144 (2003)
• We can obtain any valid density matrix, in the AO basis, without recourse to MOs!
– in particular, we may optimize the energy by freely varying Xµν with µ > ν:
Emin(X) = minX
[TrD(X)h + 2-el. part]
• Is this use of D(X) a practical proposition?
3

Two questions about D(X) = exp(−XS)D exp(SX)
• Can it be evaluated efficiently?
– we use a generalized Baker–Campbell–Hausdorff (BCH) expansion:
D(X) = D +ˆD,X
˜S
+ 12
ˆˆD,X
˜S,X
˜S
+ · · ·
– we have here introduced the S commutatorˆD,X
˜S
= DSX−XSD
– converges rapidly (purification may be necessary), in about 10 matrix multiplications
• Are redundancies a problem?
– the AO space consists of two parts: the occupied space and the virtual space
P = DS (onto occupied space), Q = I−DS (onto virtual space)
– only rotations between the occupied and virtual spaces are nonredundant:
X = PXPT + QXQT
| {z }redundant
+ PXQT + QXPT
| {z }Xov
– to avoid problems with redundancies, we use the projected parameterization
D(X) = exp(−XovS)D exp(SXov), XT = −X
4

Diagonalization-free Roothaan–Hall SCF optimization
• The SCF (Fock or Kohn–Sham) energy may, in principle, be optimized directly:
Emin = minX
E(X) ⇔ F(D)DS = SDF(D)| {z }stationary condition
– a difficult global minimization problem!
• In MO theory, the Roothaan–Hall SCF scheme works well, especially with DIIS:
F = h + g(D)F⇄
DFC = SCǫ; Dnew = CoccC
Tocc
– each diagonalization is equivalent to minimizing the sum of the (occ.) orbital energies
ε(X) =X
I
ǫI = TrD(X)F
• By analogy with MO theory, we set up the following Roothaan–Hall SCF scheme:
F = h + g(D)F⇄
Dεmin = min
X
TrD(X)F; Dnew = D(X∗)
– at each SCF iteration, we minimize Tr D(X)F with respect to X
– the new density is then obtained by expansion of D(X) with the minimizer X∗
• We thus avoid MOs and diagonalization but retain the SCF iterations
5

Newton minimization of the Roothaan–Hall energy function
• At each SCF iteration, our task is to minimize the Roothaan–Hall energy function
ε(X) = TrD(X)F = TrDF + TrˆD,X
˜SF + 1
2Tr
ˆˆD,X
˜S,X
˜SF + · · ·
• Truncating at second order and setting the gradient to zero, we obtain the Newton step:
H−X+S− + S−X+H− = G− ← the Roothaan–Hall (RH) Newton equation
– where the (negative) gradient and Hessian matrices are given by
G = Fvo − Fov
H = Fvv − Foo F = Foo+Fov+Fvo+Fvv
• Because of their large dimensions, the Newton equations cannot be solved directly
– transformation to orthonormal Lowdin basis: A± = S±1/2AS±1/2
– S−1/2 precalculated: Z0 = I, Zn+1 = 12Zn (3I− λZnSZn) for ‖I− λS‖2 < 1
– solution by the preconditioned conjugate-gradient method (typically 10 iterations)
– elementary (sparse) matrix manipulations (typically less than 100 multiplications)
• A RH diagonalization corresponds to an exact minimization (many Newton steps)
– however, a partial minimization will do (one RH Newton step is sufficient)
6
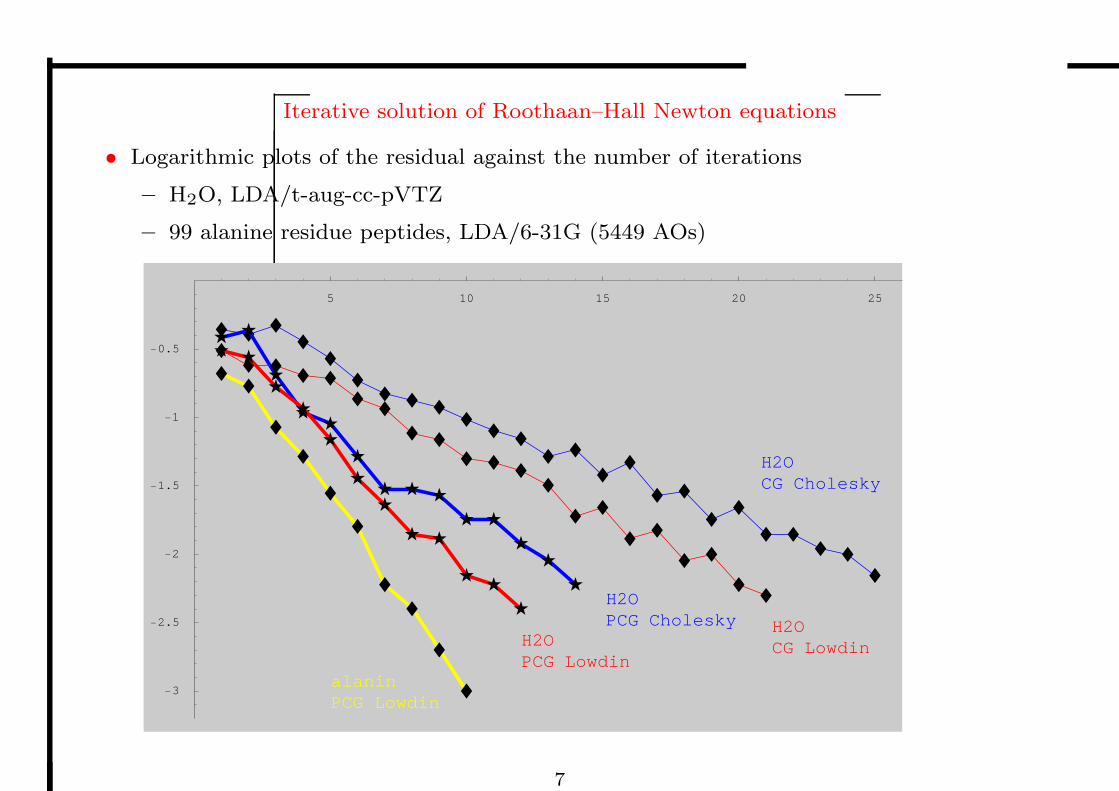
Iterative solution of Roothaan–Hall Newton equations
• Logarithmic plots of the residual against the number of iterations
– H2O, LDA/t-aug-cc-pVTZ
– 99 alanine residue peptides, LDA/6-31G (5449 AOs)
5 10 15 20 25
-3
-2.5
-2
-1.5
-1
-0.5
H2OPCG Lowdin
H2OPCG Cholesky H2O
CG Lowdin
H2OCG Cholesky
alaninPCG Lowdin
7
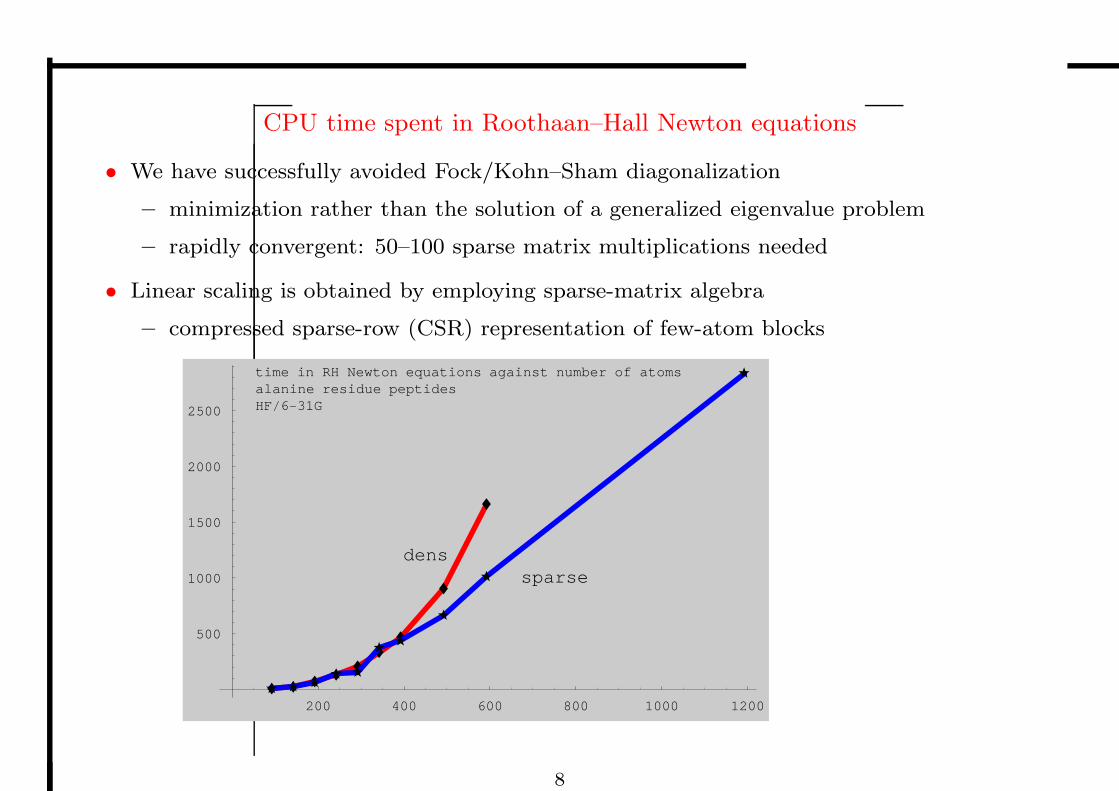
CPU time spent in Roothaan–Hall Newton equations
• We have successfully avoided Fock/Kohn–Sham diagonalization
– minimization rather than the solution of a generalized eigenvalue problem
– rapidly convergent: 50–100 sparse matrix multiplications needed
• Linear scaling is obtained by employing sparse-matrix algebra
– compressed sparse-row (CSR) representation of few-atom blocks
200 400 600 800 1000 1200
500
1000
1500
2000
2500
time in RH Newton equations against number of atomsalanine residue peptidesHF�6-31G
sparsedens
8
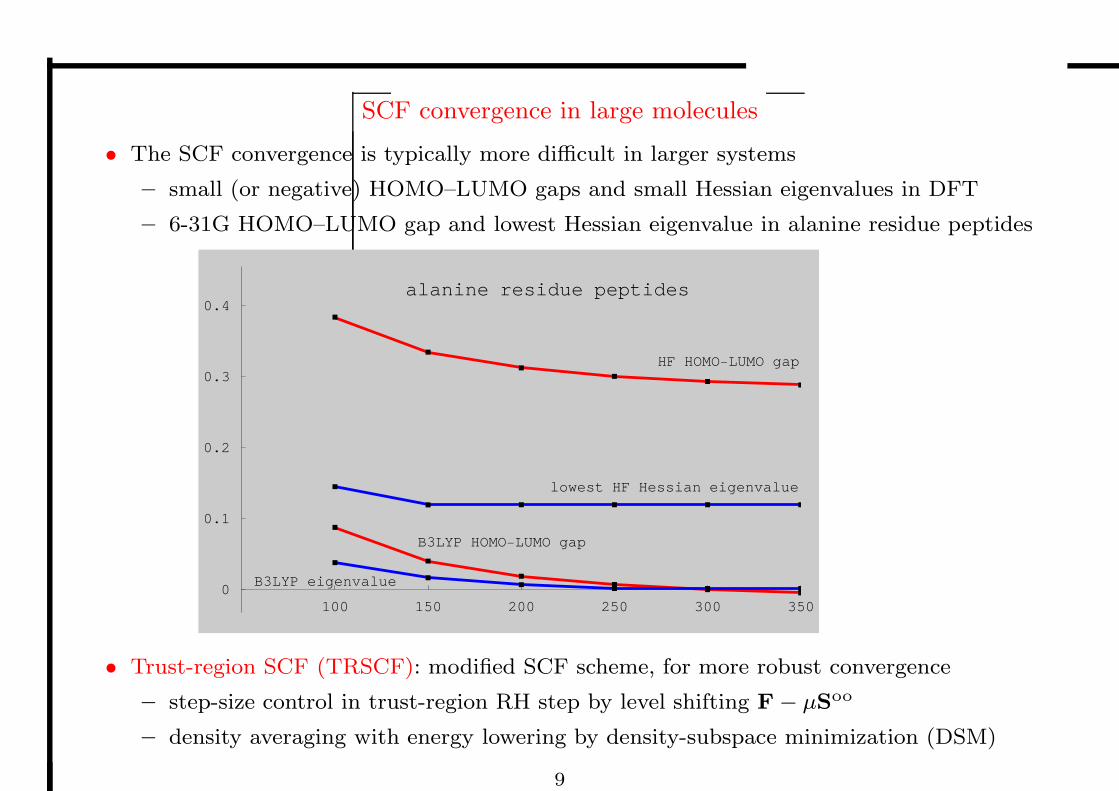
SCF convergence in large molecules
• The SCF convergence is typically more difficult in larger systems
– small (or negative) HOMO–LUMO gaps and small Hessian eigenvalues in DFT
– 6-31G HOMO–LUMO gap and lowest Hessian eigenvalue in alanine residue peptides
100 150 200 250 300 3500
0.1
0.2
0.3
0.4alanine residue peptides
HF HOMO-LUMO gap
B3LYP HOMO-LUMO gap
lowest HF Hessian eigenvalue
B3LYP eigenvalue
• Trust-region SCF (TRSCF): modified SCF scheme, for more robust convergence
– step-size control in trust-region RH step by level shifting F− µSoo
– density averaging with energy lowering by density-subspace minimization (DSM)
9
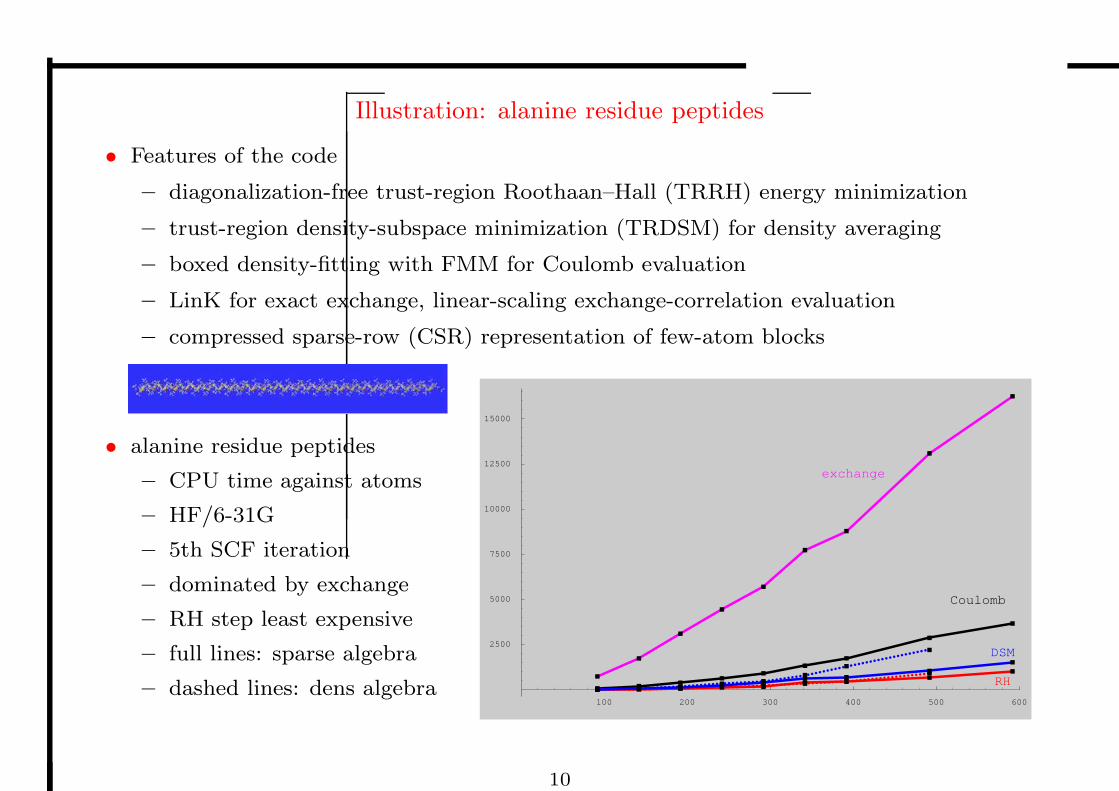
Illustration: alanine residue peptides
• Features of the code
– diagonalization-free trust-region Roothaan–Hall (TRRH) energy minimization
– trust-region density-subspace minimization (TRDSM) for density averaging
– boxed density-fitting with FMM for Coulomb evaluation
– LinK for exact exchange, linear-scaling exchange-correlation evaluation
– compressed sparse-row (CSR) representation of few-atom blocks
• alanine residue peptides
– CPU time against atoms
– HF/6-31G
– 5th SCF iteration
– dominated by exchange
– RH step least expensive
– full lines: sparse algebra
– dashed lines: dens algebra100 200 300 400 500 600
2500
5000
7500
10000
12500
15000
exchange
Coulomb
DSM
RH
10
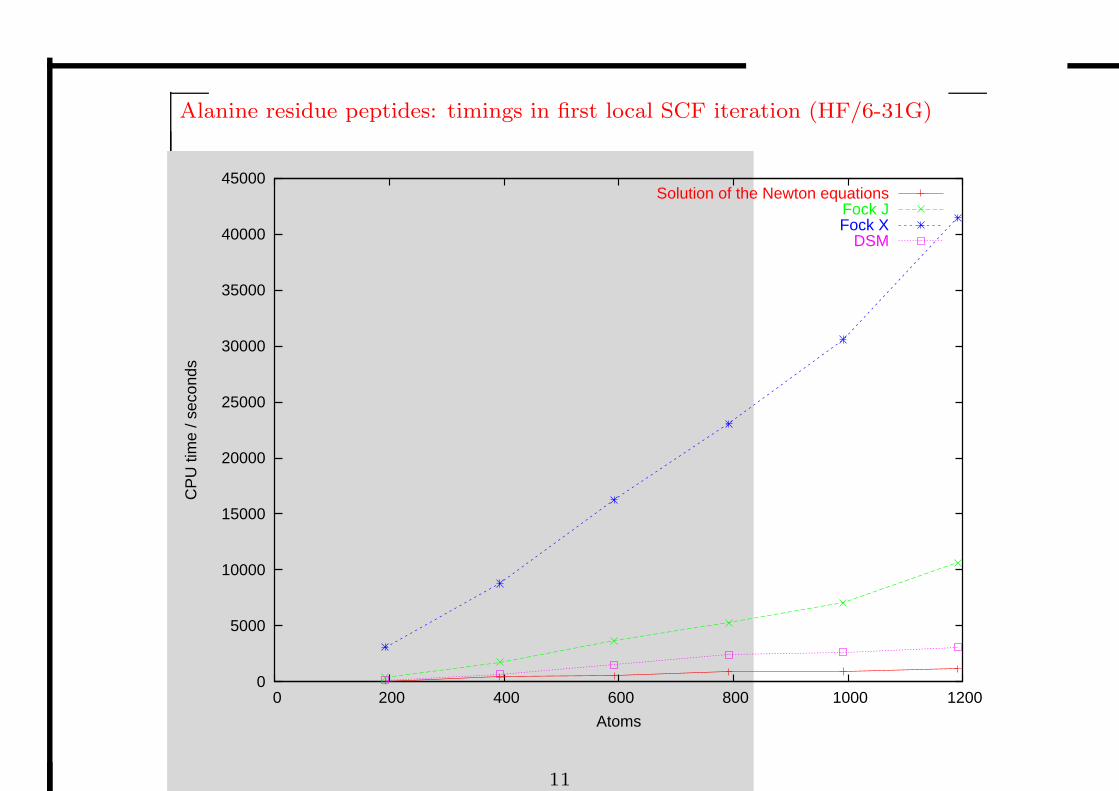
Alanine residue peptides: timings in first local SCF iteration (HF/6-31G)
0
5000
10000
15000
20000
25000
30000
35000
40000
45000
0 200 400 600 800 1000 1200
CP
U ti
me
/ sec
onds
Atoms
Solution of the Newton equationsFock JFock X
DSM
11
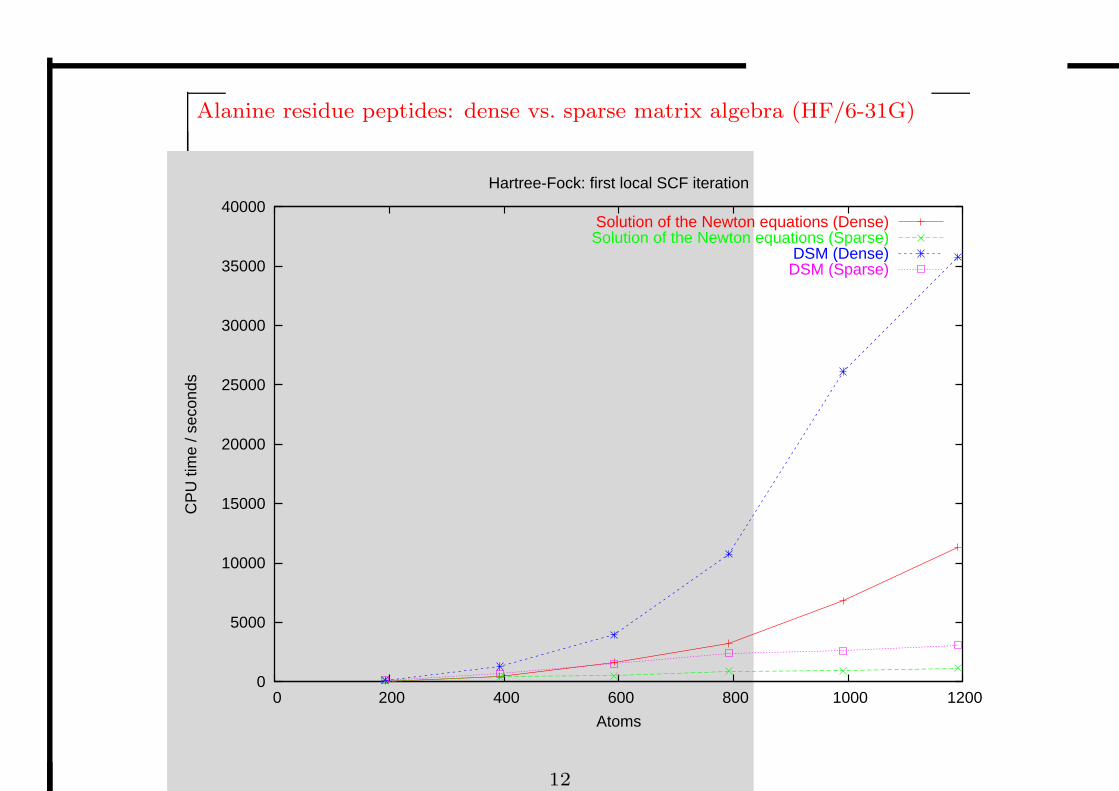
Alanine residue peptides: dense vs. sparse matrix algebra (HF/6-31G)
0
5000
10000
15000
20000
25000
30000
35000
40000
0 200 400 600 800 1000 1200
CP
U ti
me
/ sec
onds
Atoms
Hartree-Fock: first local SCF iteration
Solution of the Newton equations (Dense)Solution of the Newton equations (Sparse)
DSM (Dense)DSM (Sparse)
12

Response theory
• The expectation value of A in the presence of a perturbation Vω of frequency ω:
˙t˛A
˛t¸
=˙0
˛A
˛0
¸+
Z〈〈A; Vω〉〉ω exp (−iωt) dω + · · ·
– the linear-response function 〈〈A; Vω〉〉ω carries information about the first-order
change in the expectation value
• The linear-response function may be represented compactly as:
〈〈A; V ω〉〉ω = −A[1]T`E[2] − ωS[2]
´−1V
[1]ω| {z }
linear equations
←
8><>:
E[2] electronic Hessian
S[2] metric matrix
A[1] = vec
`
ADS − SDA´
• In practice, the response functions are evaluated by solving a set of linear equations`E[2] − ωS[2]
´N[1] = −V
[1]ω
〈〈A; V ω〉〉ω = A[1]TN[1]
• Excitation energies as poles of linear response function (RPA):`E[2] − ωS[2]
´X = 0
• Can these tasks be accomplished efficiently in the AO basis?
13

Solution of the response equations
• The response equations are solved in the same manner as the RH Newton equations:`E[2] − ωS[2]
´x = V[1]
– generation of an iterative subspace until the residual is sufficiently small
R =`E[2] − ωS[2]
´x−V[1]
• Key step: multiplication of Hessian and metric matrices with trial vectors (matrices)
E[2](X) = (Fvv − Foo)XS + SX(Fvv − Foo) + gvo([D,X]S)− gov([D,X]S)
S[2](X) = SooXSvv − SvvXSoo
– requires recalculation of Fock/Kohn–Sham matrix with modified AO density matrix
– trial matrices always added in pairs X and XT
• For rapid convergence, the residual vector is preconditioned
eR = M−1R, M = E[2] − ωS[2] (but without red part!)
– transformation to orthogonal basis (Cholesky or Lowdin)
– nondiagonal preconditioning requires typically 7 conjugate-gradient steps
• With this preconditioner, the response equations converge in 5–10 iterations
– indeed, this is the same convergence as in the canonical MO basis
14
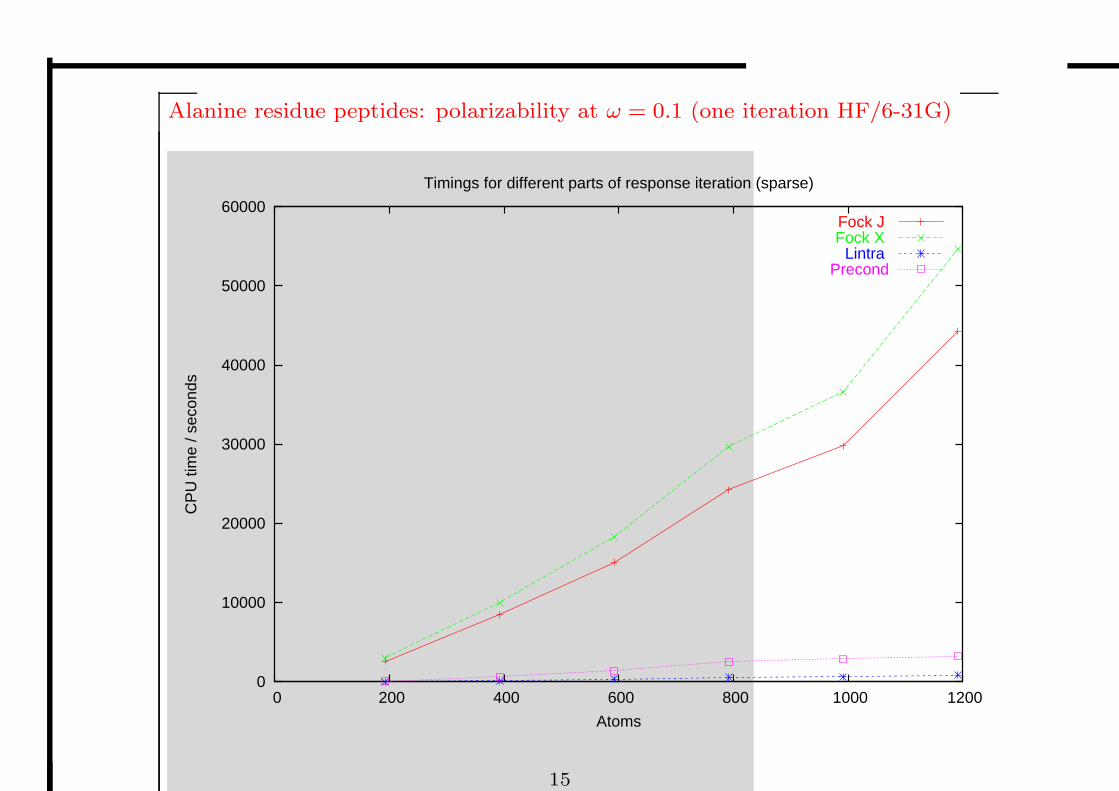
Alanine residue peptides: polarizability at ω = 0.1 (one iteration HF/6-31G)
0
10000
20000
30000
40000
50000
60000
0 200 400 600 800 1000 1200
CP
U ti
me
/ sec
onds
Atoms
Timings for different parts of response iteration (sparse)
Fock J Fock X
Lintra Precond
15
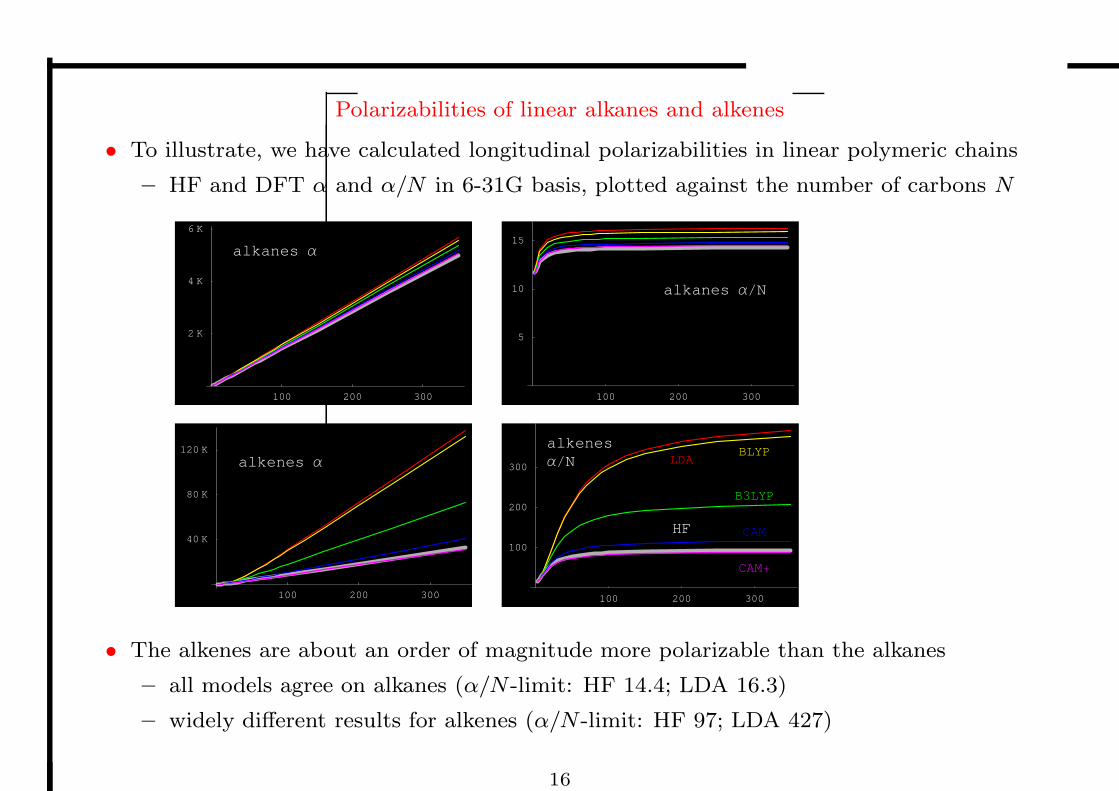
Polarizabilities of linear alkanes and alkenes
• To illustrate, we have calculated longitudinal polarizabilities in linear polymeric chains
– HF and DFT α and α/N in 6-31G basis, plotted against the number of carbons N
100 200 300
40 K
80 K
120 Kalkenes Α
100 200 300
100
200
300LDA
BLYP
B3LYP
CAMHF
CAM+
alkenesΑ�N
100 200 300
2 K
4 K
6 K
alkanes Α
100 200 300
5
10
15
alkanes Α�N
• The alkenes are about an order of magnitude more polarizable than the alkanes
– all models agree on alkanes (α/N -limit: HF 14.4; LDA 16.3)
– widely different results for alkenes (α/N -limit: HF 97; LDA 427)
16
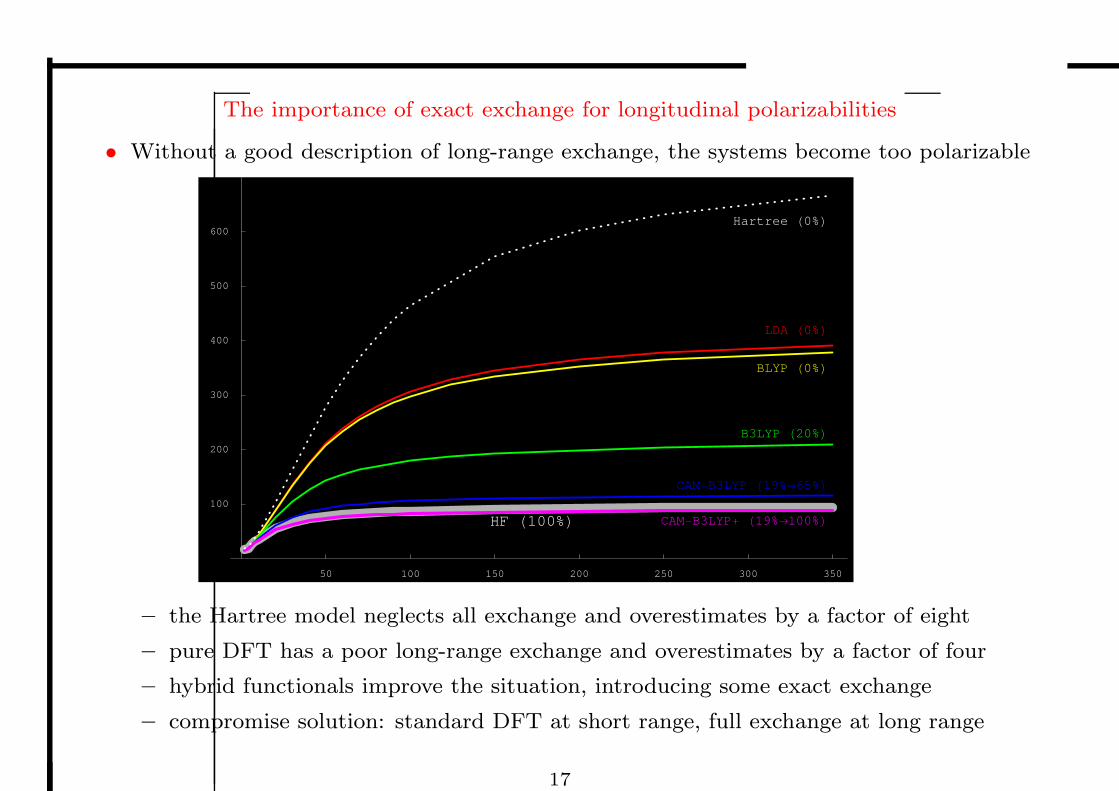
The importance of exact exchange for longitudinal polarizabilities
• Without a good description of long-range exchange, the systems become too polarizable
50 100 150 200 250 300 350
100
200
300
400
500
600
LDA H0%L
BLYP H0%L
B3LYP H20%L
CAM-B3LYP H19%®65%L
HF H100%L CAM-B3LYP+ H19%®100%L
Hartree H0%L
– the Hartree model neglects all exchange and overestimates by a factor of eight
– pure DFT has a poor long-range exchange and overestimates by a factor of four
– hybrid functionals improve the situation, introducing some exact exchange
– compromise solution: standard DFT at short range, full exchange at long range
17
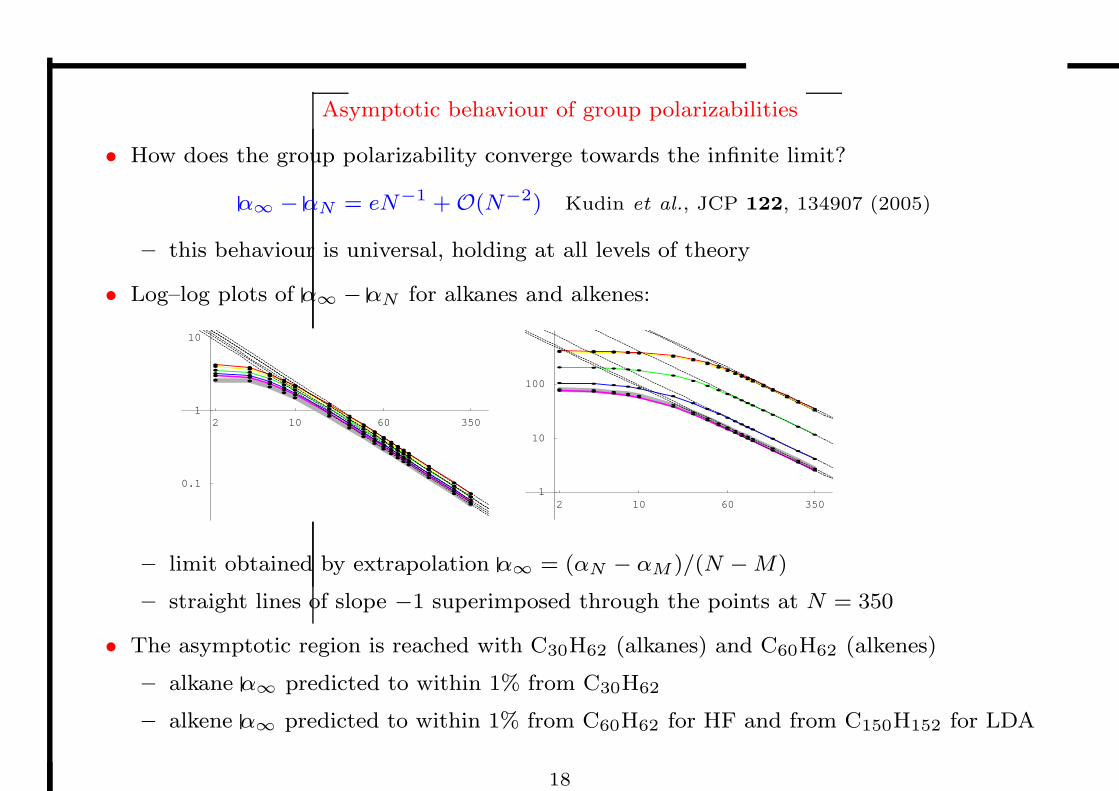
Asymptotic behaviour of group polarizabilities
• How does the group polarizability converge towards the infinite limit?
α∞ − αN = eN−1 +O(N−2) Kudin et al., JCP 122, 134907 (2005)
– this behaviour is universal, holding at all levels of theory
• Log–log plots of α∞ − αN for alkanes and alkenes:
2 10 60 350
0.1
1
10
2 10 60 3501
10
100
– limit obtained by extrapolation α∞ = (αN − αM )/(N −M)
– straight lines of slope −1 superimposed through the points at N = 350
• The asymptotic region is reached with C30H62 (alkanes) and C60H62 (alkenes)
– alkane α∞ predicted to within 1% from C30H62
– alkene α∞ predicted to within 1% from C60H62 for HF and from C150H152 for LDA
18
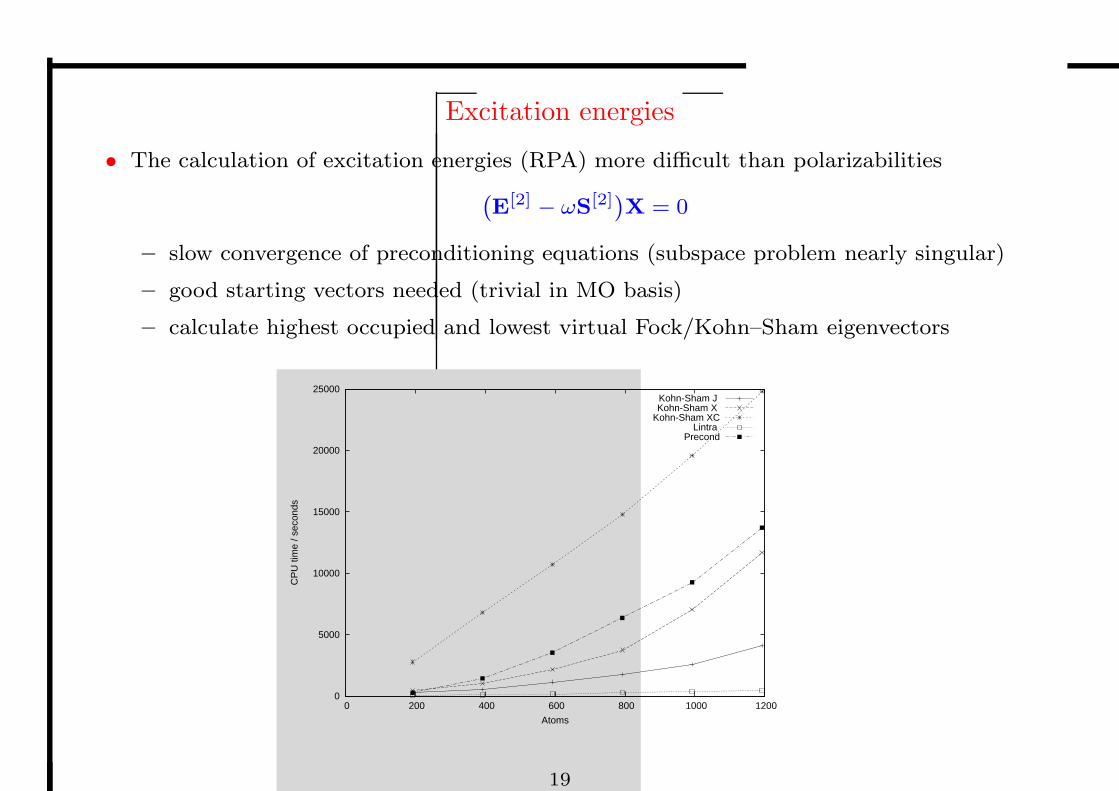
Excitation energies
• The calculation of excitation energies (RPA) more difficult than polarizabilities
`E[2] − ωS[2]
´X = 0
– slow convergence of preconditioning equations (subspace problem nearly singular)
– good starting vectors needed (trivial in MO basis)
– calculate highest occupied and lowest virtual Fock/Kohn–Sham eigenvectors
0
5000
10000
15000
20000
25000
0 200 400 600 800 1000 1200
CP
U ti
me
/ sec
onds
Atoms
Kohn-Sham J Kohn-Sham X
Kohn-Sham XCLintra
Precond
19
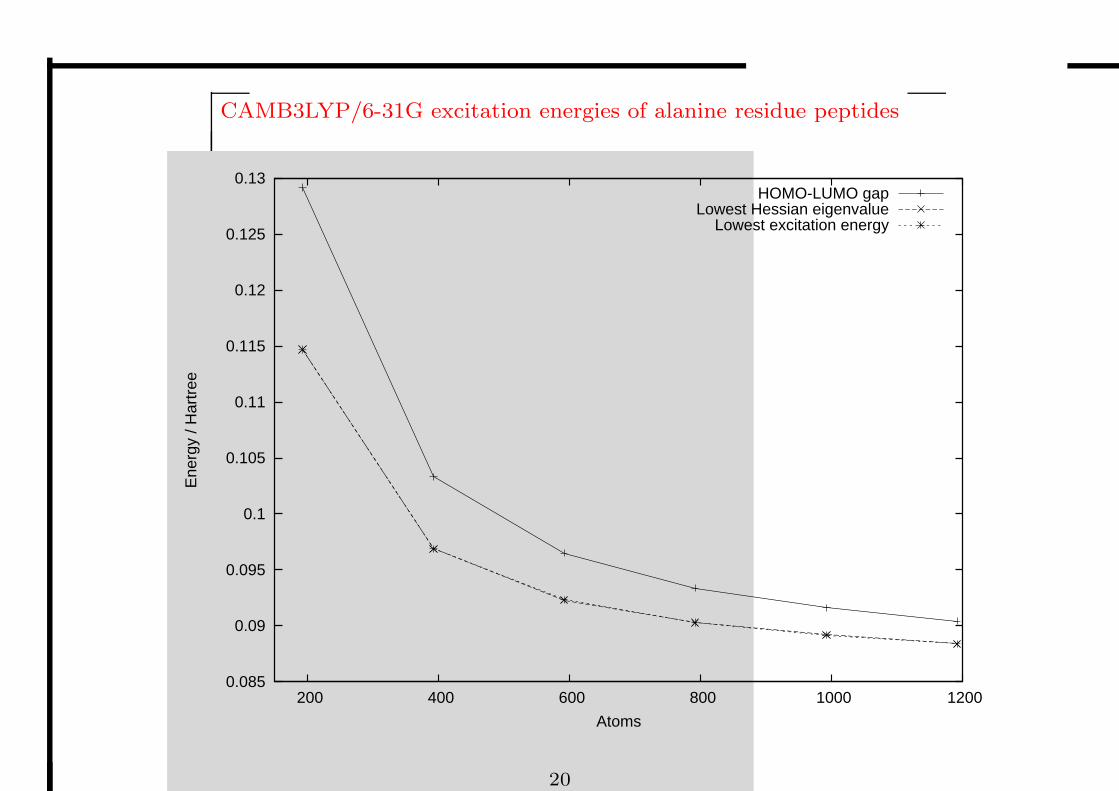
CAMB3LYP/6-31G excitation energies of alanine residue peptides
0.085
0.09
0.095
0.1
0.105
0.11
0.115
0.12
0.125
0.13
200 400 600 800 1000 1200
Ene
rgy
/ Har
tree
Atoms
HOMO-LUMO gapLowest Hessian eigenvalue
Lowest excitation energy
20
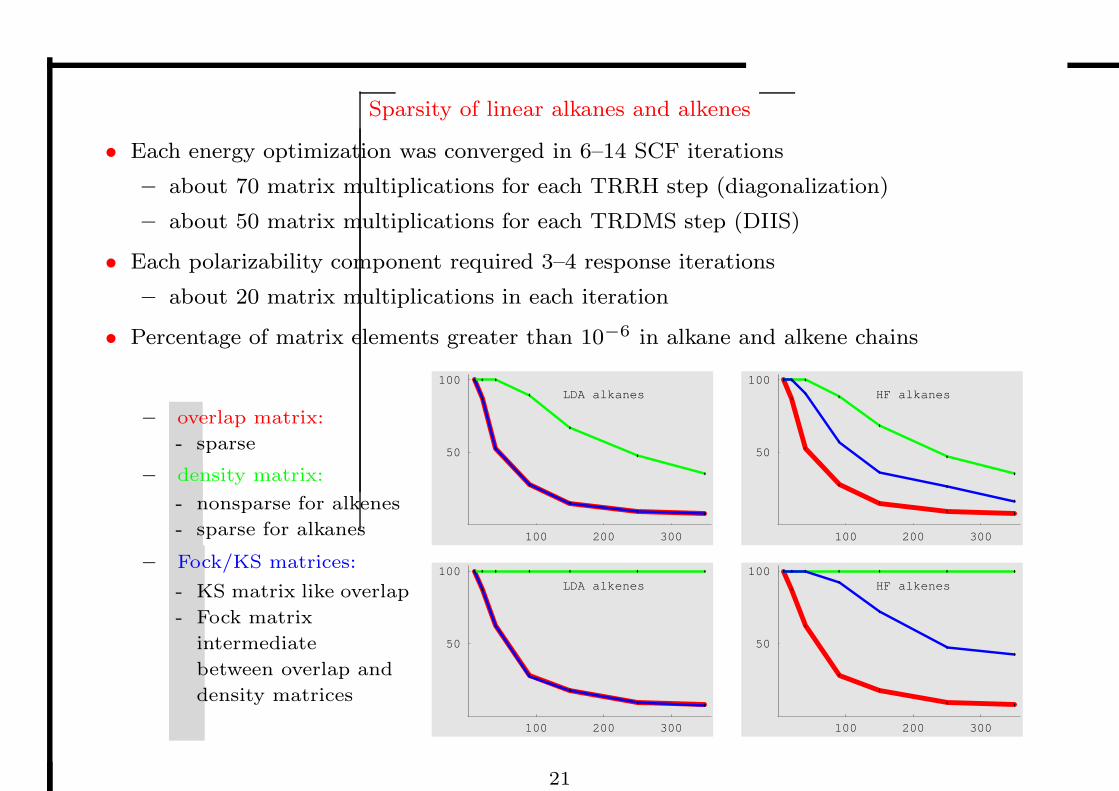
Sparsity of linear alkanes and alkenes
• Each energy optimization was converged in 6–14 SCF iterations
– about 70 matrix multiplications for each TRRH step (diagonalization)
– about 50 matrix multiplications for each TRDMS step (DIIS)
• Each polarizability component required 3–4 response iterations
– about 20 matrix multiplications in each iteration
• Percentage of matrix elements greater than 10−6 in alkane and alkene chains
– overlap matrix:
- sparse
– density matrix:
- nonsparse for alkenes
- sparse for alkanes
– Fock/KS matrices:
- KS matrix like overlap
- Fock matrix
intermediate
between overlap and
density matrices
100 200 300
50
100LDA alkenes
100 200 300
50
100HF alkenes
100 200 300
50
100LDA alkanes
100 200 300
50
100HF alkanes
21

Conclusions
• We have discussed the optimization of SCF energies without orbital generation
– direct optimization of density matrix in the AO basis
– in each SCF iteration, we replace diagonalization by minimization
– one Newton step is usually enough
– 50–100 matrix multiplications typically required
– linear scaling achieved with sparse matrix algebra
• Large molecules represent a more difficult minimization problem
– small Hessian eigenvalues for pure DFT
– trust-region SCF: careful step-size control
– revert to second-order if necessary
• Linear-response calculations are straightforward in the AO basis
– one Fock/Kohn–Sham matrix build and 20 matrix multiplications pr. iteration
– stable convergence in 5–10 iterations
– preconditioning requires typically 7 iterations
• The calculation of excitation energies more difficult
– near singular subspace problem for preconditioning
– good starting vectors needed
22