
Chapter 6. Vibrational Spectroscopy 6.1 Diatomic molecules
21
On the basis of harmonic oscillator approximation, the vibrational energy levels E v are given Eq. (6.1) Eq. (6.2) Eq. (6.3) E v = hν(v + ½) The classical vibrational frequency ν is related to the reduced mass μ [= m 1 m 2 /(m 1 + m 2 )] and the force constant k by 6.1 Diatomic molecules ν = (1/2π)[k/μ] 1/2 Vibrational term values in unit of wavenumber are given where the vibrational quantum number v = 0, 1, 2, … hc Ev = G(v) = ω(v + ½) Chapter 6. Vibrational Spectroscopy where ωis the vibrational wavenumber. wbt 1 Vibraional Spectroscopy
Transcript of Chapter 6. Vibrational Spectroscopy 6.1 Diatomic molecules

On the basis of harmonic oscillator approximation, the vibrational energy levels Ev are given
Eq. (6.1)
Eq. (6.2)
Eq. (6.3)
Ev = hν(v + ½)
The classical vibrational frequency ν is related to the reducedmass μ [= m1m2/(m1 + m2)] and the force constant k by
6.1 Diatomic molecules
ν = (1/2π)[k/μ]1/2
Vibrational term values in unit of wavenumber are given
where the vibrational quantum number v = 0, 1, 2, …
hcEv
= G(v) = ω(v + ½)
Chapter 6. Vibrational Spectroscopy
where ωis the vibrational wavenumber.
wbt 1Vibraional Spectroscopy

6.1.1 Infrared spectraThe transition moment is given
Rv = ∫ dx "*' vv µψψ Eq. (6.4)
where x is (r-re), the displacement of the internuclear distancefrom equilibrium.For homogeneous diatomics, μ = 0, thus, Rv = 0. For hetero-geneous diatomics, μ≠ 0 and μ varies with x as follows:
Eq. (6.5)ee
....dd
!21
dd 2
2
2
+⎟⎟⎠
⎞⎜⎜⎝
⎛+⎟
⎠⎞
⎜⎝⎛+= x
xx
xe
µµµµ
where “e” refers to the equilibrium configuration.
wbt 2

The transition moment becomes
Eq. (6.6)
Eq. (6.7)
Eq. (6.8)
Eq. (6.6) becomes
e
Because the wavefunctions of the lower and upper statesare of the same hamiltonian, they are orthogonal for v’ ≠ v”,namely 0dx "*' =∫ vv ψψ
The first term in Eq. (6.6) is non-zero only ifΔv = ± 1 Eq. (6.9)
which is the vibrational selection rule.
....d"*'ddd"*' +⎟
⎠⎞
⎜⎝⎛+= ∫∫ xx
xx vvvvv e ψψµψψµR
....d"*'dd
+⎟⎠⎞
⎜⎝⎛= ∫ xx
xvvv ψψµR
wbt 3

The intensities of transition bands in a vibrational spectrumusually decrease rapidly as v” increases because thepopulation Nv of the vth vibrational level is related to N0 byBoltzmann factor
)exp(0 kT
ENN vv −= Eq. (6.10)
All bands with v” ≠ 0 are called “hot bands” whosepopulation Nv increases with temperature.
Note: 1. transition intensity ∝ |Rv|2, i.e. ∝ (dμ/dx)e
2
2. μ varies with internuclear distance re in diatomics.
wbt 4
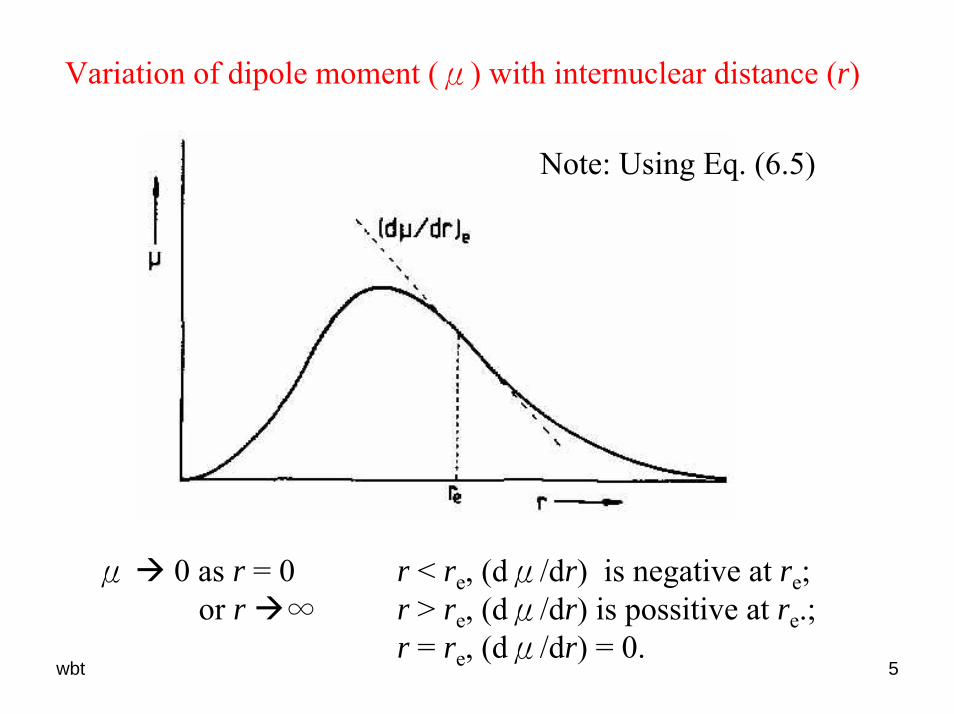
Variation of dipole moment (μ) with internuclear distance (r)
Note: Using Eq. (6.5)
μ 0 as r = 0 or r ∞
wbt 5
r < re, (dμ/dr) is negative at re;r > re, (dμ/dr) is possitive at re.;r = re, (dμ/dr) = 0.

6.1.2 Raman spectra
The vibrational Raman effect is similar to the rotational Ramaneffect. With Eq.(5.46), the dipole moment μ of a moleculeinduced by intense monochromatic radiation is given by
μ = α0,vA sin 2πc t - ½α1,v Acos 2πc ( + ω)t+ ½α1,vA cos 2πc( - ω)t
wbt 6
ν~ ν~ν~
Eq. (6.11)
where α0,v is the average polarizability during vibration,α1,v is the amplitude of the change of polarization due tovibration, A is the amplitude of the oscillating electric fieldof the incident radiation, and ω is the vibrational wavenumber.All three terms in Eq. (6.11) represent scattering of the radiation.The first term corresponds to Rayleigh scattering, where thewavenumber remains unchanged. The second and third correspond to the anti-Stokes and Stokes Raman scattering withwavenumbers of ( + ω) and ( - ω), respectively.
ν~
ν~ ν~

Similar to the case for dipole moment, the change ofpolarizability with vibrational displacement x can be expressed as a Taylor series:
....dd
!21
dd 2
2
2
+⎟⎟⎠
⎞⎜⎜⎝
⎛+⎟
⎠⎞
⎜⎝⎛+= x
xx
xe
αααα Eq. (6.12)ee e
Then, the vibrational Raman transition moment Rvis given by
....d"*'dd
+⎟⎠⎞
⎜⎝⎛= ∫ xxA
xvvv ψψαR
eEq. (6.13)
The first term in Eq. (6.13) is non-zero only if
Eq. (6.14)Δv = ± 1
Which constitutes the vibrational Raman selection rule.wbt 7
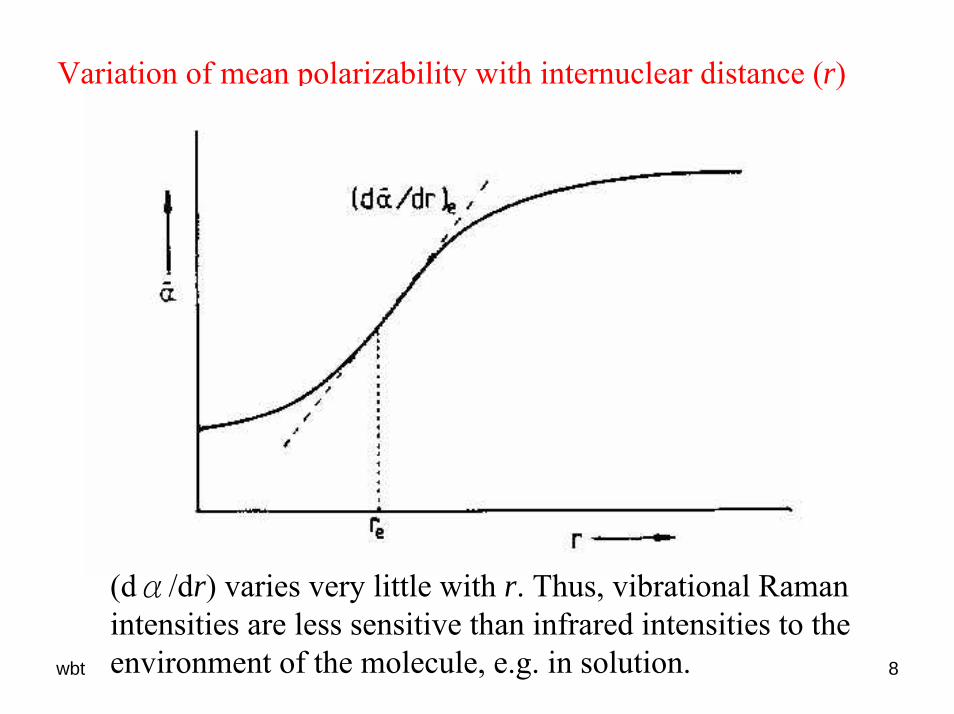
wbt 8
Variation of mean polarizability with internuclear distance (r)
(dα/dr) varies very little with r. Thus, vibrational Ramanintensities are less sensitive than infrared intensities to the environment of the molecule, e.g. in solution.
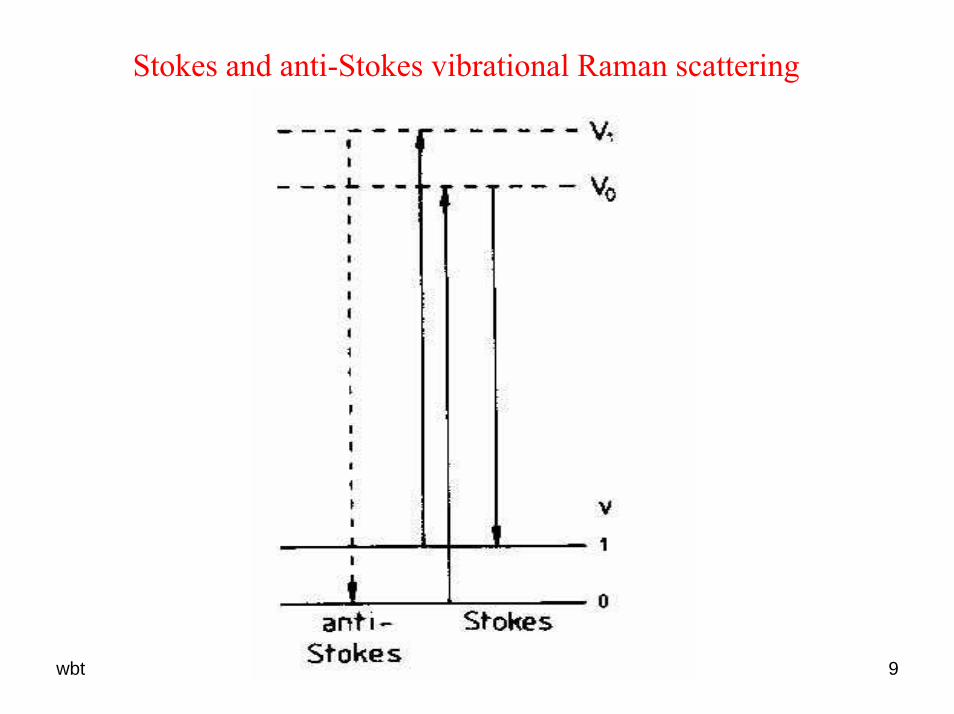
wbt 9
Stokes and anti-Stokes vibrational Raman scattering

6.1.3 AnharmonicityElectrical anharmonicityAs seen in Eqs. (6.5) and (6.12), μ and α can be expressed in terms of Taylor series. If μ and α vary linearly with x, it is said to be harmonic and the selection rule is Δv = ± 1 as applied in infrared and Raman spectroscopy. If the higher terms are concerned, it is referred to as anharmonicity. When electrical properties of a molecule is concerned, it is called electrical anharmonicity, which has little effect on the spectral band intensity resulting from Δv = ± 2, ± 3, …transitions, which are known as vibrational overtones.Mechanical anharmonicity
wbt 10
The vibrational motion of a diatomic molecule can be treated by a mechanical ball-and-spring model. Hooke’s law [restoring force = -dV(x)/dx = -kx] holds only when x is small (r ≈ re).
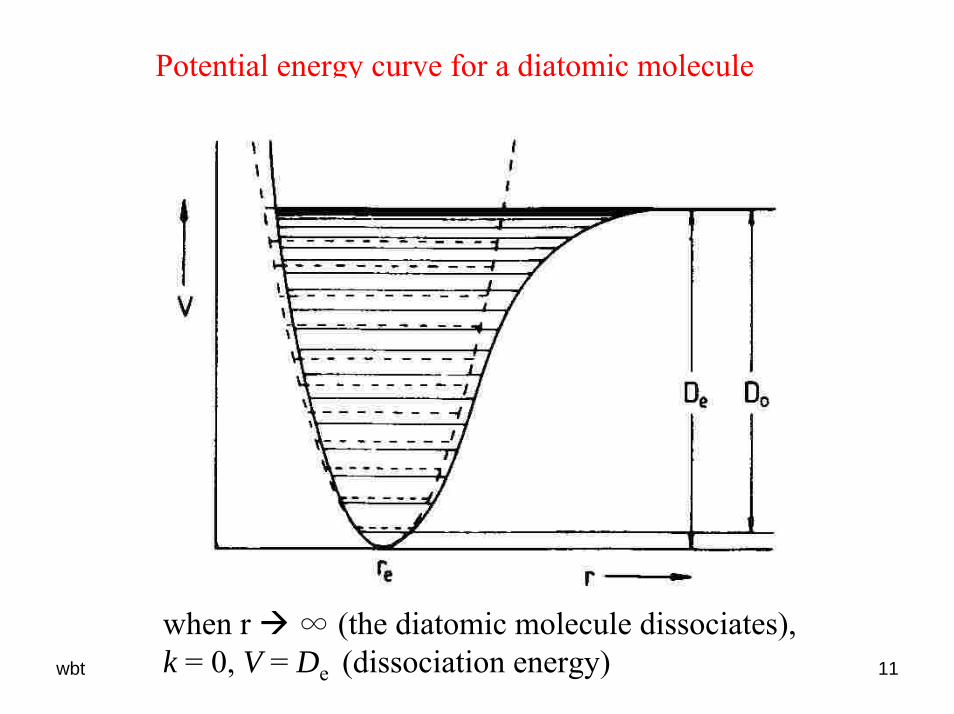
wbt 11
Potential energy curve for a diatomic molecule
when r ∞ (the diatomic molecule dissociates), k = 0, V = De (dissociation energy)

The vibrational motion of a diatomic molecule can be treated by a mechanical ball-and-spring model. Hooke’s law [restoring force = -dV(x)/dx = -kx] holds only when x is small (r ≈ re).The mechanical anharmonicity modifies the Δv = ± 1 IR and Raman selection rule to Δv = ± 1, ±2, ±3,.. but the overtone transitions with Δv = ±2, ±3,.. .The harmonic oscillator term values of Eq. (6.3) can be modified to G(v) = ωe(v + ½) – ωexe(v + ½)2 + ωeye(v + ½)3 + … Eq. (6.16)
where ωe is the vibration wavenumber which a classical oscillator with x = ∞ . ωexe, ωeye, ..are anharmonicconstants and are always positive for diatomics.
wbt 12
E.g. for 1H35Cl, ωe = 2990.946 cm-1, ωexe = 52.8186 cm-1, ωeye = 0.2244 cm-1, ωeze = -0.0122 cm-1.

Since ω cannot be directly determined, one defines ΔGv+½for (v+1) – (v) transitions as follows,ΔGv+½ = G(v+1) –G(v)
= ωe – ωexe(2v + 2) + ωeye(3v2 + 6v + 13/4) + …Eq. (6.18)
In order to determine ωe and ωexe, at least two transition wavenumbers, e.g. G(1) – G(0) = ω0 and G(2) – G(1) = ω1must be obtained. Then, the dissociation energy can be approximated to be
Eq. (6.19)De ≈ (ωe)2/[4ωexe]
Experimentalists can only determine the dissociation energy relative to the zero-point level, i.e. D0
Note that De is isotope-independent whereas D0 isotope-dependent.
wbt 13
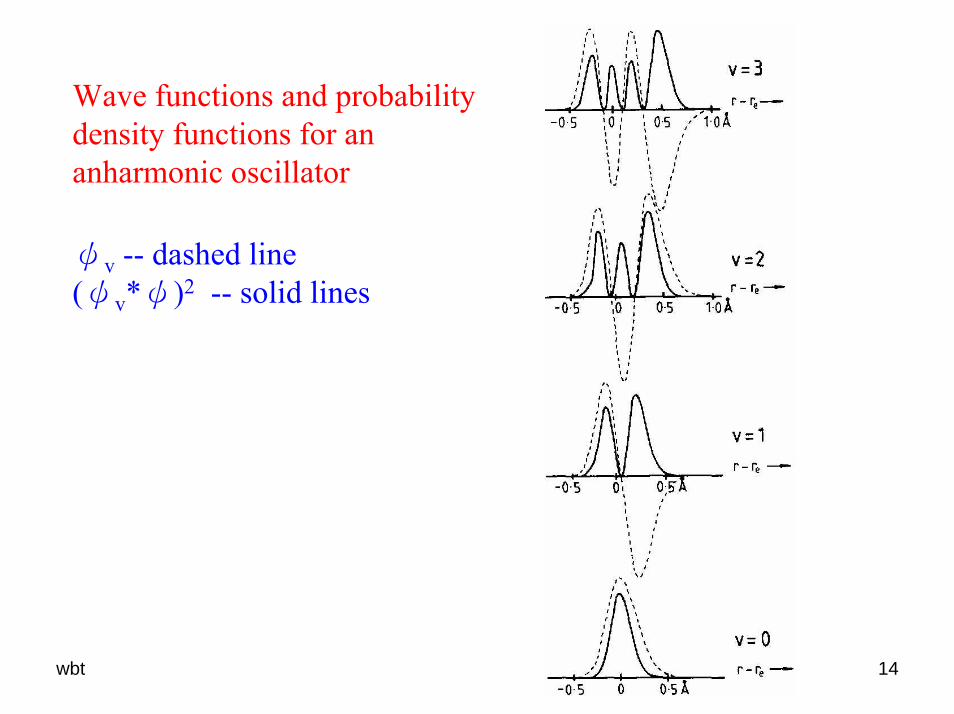
wbt 14
Wave functions and probability density functions for an anharmonic oscillator
ψv -- dashed line (ψv*ψ)2 -- solid lines

The zero-point energy corresponds to the term value G(0)G(0) = ½ωe – ¼ωexe + (1/8)ωeye + … Eq. (6.21)
D0(2H2) > D0(1H2) Eq. (6.22)
In 1929, Morse suggestedV(x) = De[1 – exp(-ax)]2 Eq. (6.23)
as a potential function relating to the behavior of an anharmonic oscillator.In the Morse function, x = r - re,
V(x) De as x ∞
Since ωe for 2H2 is less than for 1H2, so that
wbt 15

6.1.4 Vibration-rotation spectroscopy6.1.4.1 Infrared spectra
In vibration-rotation spectroscopy one observes transitions between stacks of rotational levels associated with two different vibrational states. The vibration-rotation total term values S are the sum of the rotational term value (Eq. 5.23) and the vibrational term value (Eq. 6.16)
S = G(v) + Fv(J)= ωe(v + ½) – ωexe(v + ½)2 + ••• + BvJ(J + 1) – DvJ2(J + 1)2
Eq. (6.24)
Recall: allowed vibrational transition: Δv = ± 1allowed rotational transition: ΔJ = ± 1
The location corresponding to ΔJ is called the band center.
wbt 16
Note: NO is exceptional. The rotational selection rule for NO is ΔJ = 0, ±1. [why?]
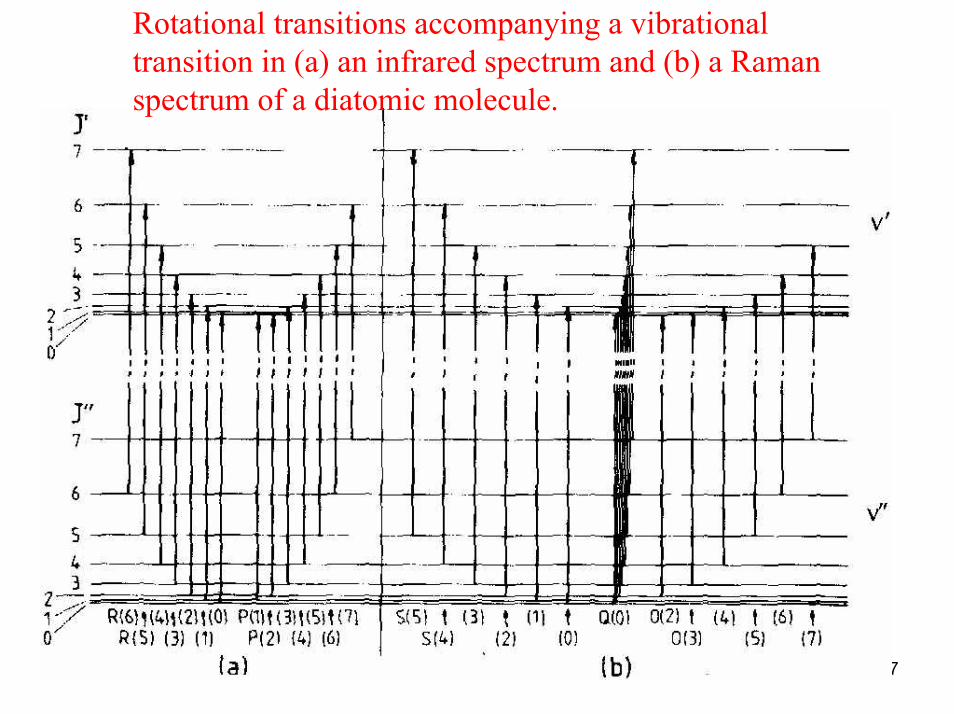
wbt 17
Rotational transitions accompanying a vibrationaltransition in (a) an infrared spectrum and (b) a Raman spectrum of a diatomic molecule.
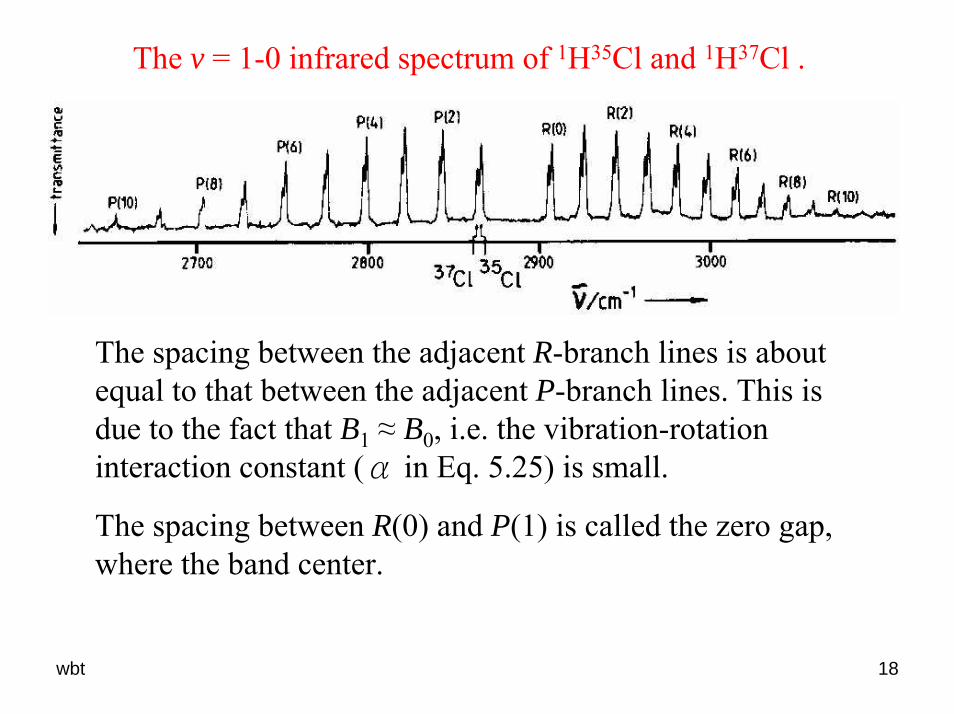
The v = 1-0 infrared spectrum of 1H35Cl and 1H37Cl .
The spacing between the adjacent R-branch lines is about equal to that between the adjacent P-branch lines. This is due to the fact that B1 ≈ B0, i.e. the vibration-rotation interaction constant (α in Eq. 5.25) is small.
The spacing between R(0) and P(1) is called the zero gap, where the band center.
wbt 18

Assume that B1 = B0 = B and neglect centrifugal distortion, the wavenumbers of the R-branch transitions are given by
[R(J)] = 0 + B(J + 1)(J + 2) – BJ(J + 1)= 0 + 2BJ + 2B
ν~ ν~ν~
Eq. (6.27)
where 0 is the wavenumbers of the pure vibrationaltransition.
ν~
Similarly, the wavenumbers of the P-branch transitions are given by
[P(J)] = 0 + B(J -1)J – BJ(J + 1)= 0 - 2BJν~ν~
Eq. (6.28)
Eqs. (6.27) and (6.28) give the zero gap value of 4B and the spacing between the R-branch and P-branch adjacent lines of 2B.
ν~
wbt 19

6.1.4.2 Raman spectra
The rotational selection rule for vibration-rotation Ramantransitions in diatomic molecules is ΔJ = 0, ±2, giving a Q(ΔJ = 0), an S(ΔJ = +2), and an O(ΔJ = -2) branch. .Assume that B1 = B0 = B and neglect centrifugal distortion, the wavenumbers of the S-, O-, and Q-branch transitions are given by
[S(J)] = 0 + B(J + 2)(J + 3) – BJ(J + 1)= 0 + 4BJ + 6Bν~ν~
ν~ Eq. (6.36)
[O(J)] = 0 + B(J - 2)(J - 1) – BJ(J + 1)= 0 - 4BJ + 2Bν~ν~
ν~ Eq. (6.37)
[Q(J)] = 0ν~ν~ Eq. (6.38)
wbt 20
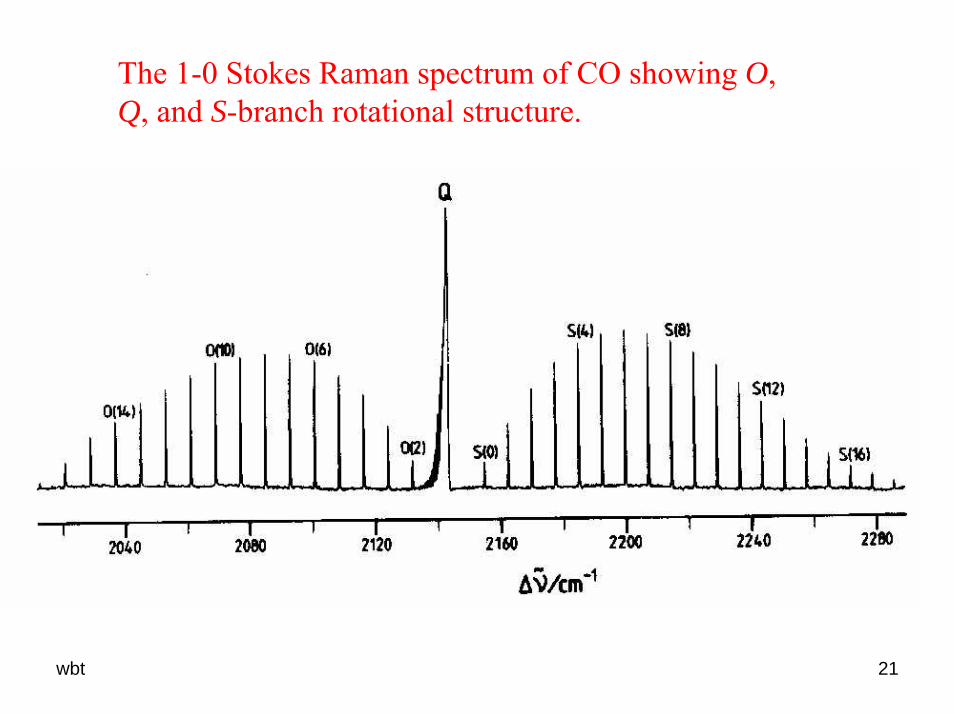
The 1-0 Stokes Raman spectrum of CO showing O, Q, and S-branch rotational structure.
wbt 21


















