AEROSOL FORMATION FROM THE REACTION OF α …carter/epacham/kamens.pdfA GAS PHASE KINETICS-PARTICLE...
10
AEROSOL FORMATION FROM THE REACTION OF α-PINENE AND OZONE USING A GAS PHASE KINETICS-PARTICLE PARTITIONING MODEL R.M. Kamens, M. Jaoui, S. Lee, C.J.Chien Department of Environmental Sciences and Engineering, University of North Carolina, Chapel Hill, NC 27599-7400, USA e-mail: [email protected] fax: 919 966-7911 11-17-99, Bangkok, Thailand ABSTRACT A kinetic mechanism was used to describe the gas-phase reactions of α-pinene with ozone. This reaction scheme produces low vapor pressure reaction products that distribute between gas and particle phases. Partitioning was treated as an equilibrium between the rate of particle uptake and rate of particle loss of semivolatile terpene reaction products. Given estimated liquid vapor pressures and activation energies of desorption, it was possible to calculate gas-particle equilibrium constants and aerosol desorption rate constants at different temperatures. Gas and aerosol phase reactions were linked together in one chemical mechanism and a chemical kinetics solver was used to predict reactant and product concentrations over time. Aerosol formation from the model was then compared with aerosol production observed from outdoor chamber experiments. Approximately 10-40% of the reacted α-pinene carbon appeared in the aerosol phase. Models vs. experimental aerosol yields illustrate that reasonable predictions of secondary aerosol formation are possible from both dark ozone and light/NOx α-pinene systems. INTRODUCTION The atmospheric chemistry of non-methane biogenic hydrocarbons has received much attention because of their significant global emissions, high photochemical reactivity, and their high aerosol forming potential. Although the potential of aerosol formation from terpenes was noted as early as 1960 by Went1, the magnitude of the natural contribution by biogenics to the particulate burden in the atmosphere is still not well characterized. In this paper, we will describe the feasibility of a predictive technique for the formation of secondary aerosols from biogenic hydrocarbons. An advantage of this approach is that it has the ability to embrace the range of different atmospheric chemical and physical conditions that bring about secondary aerosol formation. EXPERIMENTAL SECTION Gas-particle samples for this study were generated in a large outdoor 190 m 3 Teflon film chamber (Kamens et al.,1995, Fan et al., 1996). All experiments were carried out under darkness to exclude photochemical effects. Rural background air was used to charge the chamber without not any additional injections of oxides of nitrogen. Secondary aerosols were created by the
Transcript of AEROSOL FORMATION FROM THE REACTION OF α …carter/epacham/kamens.pdfA GAS PHASE KINETICS-PARTICLE...

AEROSOL FORMATION FROM THE REACTION OF αα-PINENE AND OZONE USINGA GAS PHASE KINETICS-PARTICLE PARTITIONING MODEL
R.M. Kamens, M. Jaoui, S. Lee, C.J.ChienDepartment of Environmental Sciences and Engineering,
University of North Carolina, Chapel Hill, NC 27599-7400, USAe-mail: [email protected]
fax: 919 966-7911
11-17-99, Bangkok, Thailand
ABSTRACT
A kinetic mechanism was used to describe the gas-phase reactions of α-pinene with ozone.This reaction scheme produces low vapor pressure reaction products that distribute between gasand particle phases. Partitioning was treated as an equilibrium between the rate of particle uptakeand rate of particle loss of semivolatile terpene reaction products. Given estimated liquid vaporpressures and activation energies of desorption, it was possible to calculate gas-particleequilibrium constants and aerosol desorption rate constants at different temperatures. Gas andaerosol phase reactions were linked together in one chemical mechanism and a chemical kineticssolver was used to predict reactant and product concentrations over time. Aerosol formation fromthe model was then compared with aerosol production observed from outdoor chamberexperiments. Approximately 10-40% of the reacted α-pinene carbon appeared in the aerosolphase. Models vs. experimental aerosol yields illustrate that reasonable predictions of secondaryaerosol formation are possible from both dark ozone and light/NOx α-pinene systems.
INTRODUCTION
The atmospheric chemistry of non-methane biogenic hydrocarbons has received muchattention because of their significant global emissions, high photochemical reactivity, and theirhigh aerosol forming potential. Although the potential of aerosol formation from terpenes wasnoted as early as 1960 by Went1, the magnitude of the natural contribution by biogenics to theparticulate burden in the atmosphere is still not well characterized. In this paper, we will describethe feasibility of a predictive technique for the formation of secondary aerosols from biogenichydrocarbons. An advantage of this approach is that it has the ability to embrace the range ofdifferent atmospheric chemical and physical conditions that bring about secondary aerosolformation.
EXPERIMENTAL SECTION
Gas-particle samples for this study were generated in a large outdoor 190 m3 Teflon filmchamber (Kamens et al.,1995, Fan et al., 1996). All experiments were carried out under darknessto exclude photochemical effects. Rural background air was used to charge the chamber withoutnot any additional injections of oxides of nitrogen. Secondary aerosols were created by the

reaction of α-pinene with O3 in the chamber. O3 from an electric discharge ozone generator wasadded to the chamber over the course of an hour with initial concentrations ranging, depending onthe experiment, from 0.25 to 0.65 ppm. It was measured using a Bendex chemilumenesent O3
meter (model 8002, Roncerverte,WV) and calibrated via gas phase titration using a NIST traceableNO tank. O3 addition to the chamber was followed by volatilizing, depending on the experiment,0.4 to 1 mL of liquid α-pinene into the chamber atmosphere. The gas-phase concentration of α-pinene was monitored with an online gas chromatograph (Shimadzu Model 8A, column: 1.5 m, 3.2mm stainless steel packed with Supelco 5% Bentanone 34) using a flame ionization detector, andcalibrated with a known concentration of a mixture of toluene and propylene.
Gas and particle phase α-pinene products were simultaneously collected with a samplingtrain that consisted of an upstream 5-channel annular denuder, followed by a 47mm Teflon glassfiber filter (type T60A20, Pallflex Products Corp., Putnam, CT, USA) and another denuder. In oneof the experiments, a parallel sampling system consisting of a filter, followed bv a denuder wasalso used. The details of the sample workup procedure and quantitative analysis have beenreported in previous manuscripts Kamens et al., 1995 and Fan et al., 1996). Carbonyl products ofα-pinene-O3 aerosols were derivatized by O-(2,3,4,5,6-pentafluorobenzyl)hydroxyl-aminehydrochloride (PFBHA) as described by Yu et al.,1997, and carboxylic acids, usingpentafluorobenzyl bromide (PFBBr) as a derivatizing agent as described by Chien et al., (1998).(±)-α-Pinene, O-(2,3,4,5,6-pentafluorobenzyl)hydroxylamine hydrochloride (PFBHA),pentafluorobenzylbromide (PFBBr), decafluorobibenzyl (internal standard for derivatization), cis-pinonic acid, n-hexanoic acid, n-octanoic acid, hexane-1,6-dionic acid, and heptane-1,7-dionic acidwere all purchased from Aldrich (Milwaukee, WI).
In one of the experiments the particle size distribution and aerosol concentration forparticles ranging from 0.018 to 1.0 µm were monitored by an Electrical Aerosol Analyzer (EAA)(Thermo Systems, Inc., Model 3030, Minneapolis, MN). Total aerosol number concentration wasalso measured by a Condensation Nuclei Counter (CNC, Model Rich 100, Environment OneCorp., Schenectady, NY).
A GAS-PARTICLE MECHANISM
As a result of new aerosol compositional information(Jang and Kamens, 1999, Yu et al., 1998) we havedeveloped an exploratory model for predicting aerosolyields from the reaction of α−pinene with ozone in theatmosphere. Reaction pathways (Scheme1 and 2) wereconstructed from experimentally measured productswhich include: pinonaldehyde, norpinonaldehyde,pinonic acid, norpinonic acid, pinic acid, 2,2-dimethylcyclobutane-1,3-dicarboxylic acid, and hydroxyand aldehyde substituted pionaldehydes and hydroxypinonic acids. To simplify the mechanism in this study,six generalized semivolatile products were defined: 1.“pinald” to represent pinonaldehyde and norpinonaldehyde, 2. “pinacid” to represent pinonic andnorpinonic acids, 3. “diacid” for pinic acid and norpinic acid, (2,2-dimethylcyclobutane1,3-dicarboxylic acid), 4. “oxy-pinald” for hydroxy and aldehyde substituted pinonaldehydes (calledoxo-substituted), 5. “P3” for 2,2-dimethylcyclobutyl-3-acetylcarboxylicacid, (a pinic acidprecursor), and 6. “oxy-pinacid” for hydroxy and aldehyde substituted pinonic acids. A lastgroup, frag, was employed to account for volatile oxygenated products.
OO O
CHOOO
CH3
OO
O
Criegee2
αα-pinene
COOHCOOH
pinonic acid
CHOO
Criegee1COOHO
O3
pinic acid
+ otherproducts
+ CO, HO2, OH,H2
COOHO
norpinonaldehyde
norpinonic acid
Scheme 1

A kinetic mechanism was used todescribe the gas-phase reactions of α-pinenewith ozone. One of the products, pinic acid,a dicarboxylic acid, has a subcooled liquidvapor pressure of ~10-6 mmHg at 295K,which may be low enough to initiate selfnucleation. More volatile products such aspinonic acid and pinonaldehyde, will notself-nucleate, but will partition onto existingparticles. Gas and aerosol phase reactions arelinked together in one chemical mechanismand a chemical kinetics solver (Jeffries,1991) is used to predict reactant and productconcentrations over time. Gas phase rateconstants are available for a number of these reactions (Atkinson, 1997). These reactions were thenadded to the inorganic chemistry reactions of the Carbon Bond 4 photochemical mechanism [Geryet al., 1989). Reactions of Criegee 1 and 2 biradicals in Scheme 1 & 2, and OH attack on α-pinene are partially described below as:
Criegee1 à 0.3 pinonic aicd+ 0.3 pinald + 0.25 oxy-pinaldehyde + 0.15 stablecrieg1 + 0.8OH + 0.5HO2 (1)
stablecrieg1 + H2O à pinonic acid (2)
pinald + OH {+O2} à pinald-oo (3)
pinald-oo + HO2 à pinonic acid + { O3} (4)
α-pinene + OH à ap-oo (5a)
ap-oo + ap-oo à 0.6*pinald + HCHO + 0.2*oxypinald (5b)
Criegee2 à 0.15 stabcrieg2 +0.3 crgprod2 + 0.3 HCHO (6)+ 0.55 oxy-pinald + 0.8 OH + 0.5 HO2
crgprod2 + HO2 à diacid + HCHO (7)
stabcrieg2 + H2O à P3 + CH3OH (8)
oxy-pinald + OHà oxy-prepinacid (9)
oxy-prepinacid + HO2 à oxy-pinonic acid + { O3} (10)
Partitioning is treated as an equilibrium between the rate of particle up-take and the rate ofparticle loss of individual semivolatile α-pinene reaction products. The ratio of the rate constantsfor the forward and backward reactions ikon/
ikoff is equal to the equilibrium constant, iKp, for the
CHOOO
CH3
CHO
COOH CHO
COOHCOOH
CHOCOCH 3
O
COOHO
hydroxypinonaldehyde
O
-O3
A
C
+H2O
Pinic acid
O
CHO
OH
*
COOH
OH
P3
D
+ HO2
COOH
Criegee2
O
COOH
+OH,O 2,HO2
-O3
-CH 3OH
C8 diacid*
Scheme 2
CHO
OH
*
O
hydroxypinonic acid
O2
OOH
CHO
O
oxo-pinonaldehyde
-OH-OH
-HCHO

gas-particle equilibrium of a given partitioning compound i.
iKp = ikon/ikoff (11)
It is assumed that secondary aerosols are liquid-like, and iKp can be calculated from gas-liquid(absorptive) partitioning theory, as described by Pankow (1994).
log iKp = - log iPoL- log iγom +constant (12)
From estimated vapor pressures, iPoL (Kamens et al., 1999) and calculated activity
coefficients, iγom, which are close to one in these aerosols (Jang et al., 1997) iKp can be calculatedfor the reaction products. The rate constant, koff, for loss of a compound from the liquid particlephase to the gas phase has the form koff = β e-Ea/R. β can be evaluated and the activation energy,Ea estimated (Kamens et al., 1999). This then permits an estimate of the rate of absorption ikon
from the gas phase.
In the model seed nuclei provide initial liquid volumes for gas-liquid partitioning. Theproduct compounds that actually participate in the self nucleation process are as yet unknown.One possibility is that the stabilized Criegee radical (stabcrieg1 and stabcrieg2) reacts with thecarbonyl portion of product compounds to produce an extremely low pressure secondary ozonideproduct. These types of products have been observed from an O3- propylene system by Niki andco-workers (1997) and more recently by Neeb et al., 1995) for larger molecules]. It is alsopossible to form an anhydride from the reaction of the Criegee with a dicarboxylic acid (Neeb etal., 1996). In the α−pinene case, these products would have molecular weights of ~350, and be ofextremely low volatility. By techniques described previously (Kamens et al., 1999) vaporpressures lower than 10-15 torr are estimated, and ~one second after the start of the experiments inthis study, would exceed supersaturation by ~1000 fold. This suggests these compounds wouldmost certainly self nucleate. In our mechanism these are called “seed1” particles. We assumedthat stabilized Criegees would react with all carbonyl products. For example:
stabcrieg1 + pinald à seed1 (13)
This process was not very important for the experiments in this study, given thebackground nucleation levels that resulted during the injection of α-pinene into the chamber.Predictions, using the model developed here, however, suggest a strong influence of these seeds onparticle formation when α-pinene has concentrations of ~5 ppb.Gas phase products migrate to theparticle phase and then create more mass for additional partitioning.
Once seed surfaces are available, the migration of gas phase products such as pinonic acid(pinacidgas) on to seed1 particles to give pinacid particle phase (pinacidpart) and diacid gas producton to the newly formed pinacidpart to form pinic diacid particle phase (diacidpart) can thus berepresented as:
pinacidgas+ seed1 à pinacidpart+ seed1 koff = 19.4 min-1 (14)pinacidpart+ diacidgas à pinacidpart+ diacidpart koff = 68 min-1 (15)
To conserve mass, the amount of diacidpart and pinacidpart mass appears on both sides of theequation. To maintain equilibrium, both diacidpart and pinacidpart back reacts or “off-gases” fromthe particle to give back gas phase pinonic acid. A similar set of reactions can be written for all of

the partitioning products.
pinacidpart → pinacidgas koff = 3.73 × 1014 exp (-9525/T) min-1 (16)
diacidpart → diacidgas koff = 3.73 × 1014 exp (-10285/T) min-1 (17)
MODEL AND CHAMBER DATA COMPARISONS
Aerosol formation from the model is compared with aerosol production determined from190 m3 dark outdoor chamber experiments. The actual rate constants and mechanism used aresuimmarized in Table 1. The main updates since our first publication of the mechanism (Kamenset al, 1999) are:
1. An increase in the rate constant for the OH attack on pinonaldehyde from 2.4x10-12 cm3
molecule-1 sec-1 (as per to Kwok and Atkinson, 1995) to their more recent value of ~5x10-
12 cm3 molecule-1 sec-1.
2. We replaced the reaction
α-pinene + OH à 0.6 pinonald + 0.1 oxy-pinald + 0.3 frag
with
α-pinene + OH à ap-ooap-oo + ap-oo à 0.6*pinald + HCHO + 0.2*oxypinald
This was done to more easily add NO oxidation reactions by the hydroxy-peroxy-radical,ap-oo. In addition, the previously used lumped reaction may over estimate thepinonaldehyde yield from OH attack on αα-pinene in the absence of NOx.
3. We lowered the kon values for all absorbing species because it is more appropriated touse the the average molecular weight of the aerosol in converting from Kp units of
Given the offsetting nature of the above changes, model simulations vs. experimental datafor α-pinene, O3 and aerosol yields for a warm (295K) and a cool outdoor experiment (285K)showed little difference (Figure 1) between our originally published simulations and the updatedmechanism (Kamens et al, 1999.) Approximately 20-40% of the reacted α-pinene carbon appearsin the aerosol phase. Under cool conditions (285K), both measurements and model predictionssuggest that pinonaldehyde is the major gas phase product, while pinic acid, pinonic acid, hydroxy-pinonaldehydes, and hydroxy-pinonic acids are the major aerosol phase products. Under warmconditions, the predicted major gas phase product are pinonaldehyde, hydroxy-pinonaldehydes,some pinonic acid, and a trace of dicarboxylic acid. The particle phase was dominated by pinicacid followed by pinonic acids, hydroxy-pinonaldehydes, and hydroxypinonic acids. Theimportant observation is that the aerosol composition seems to differ radically between warm andcool experiments and this is consistent with the observations of Jang and Kamens (1999).
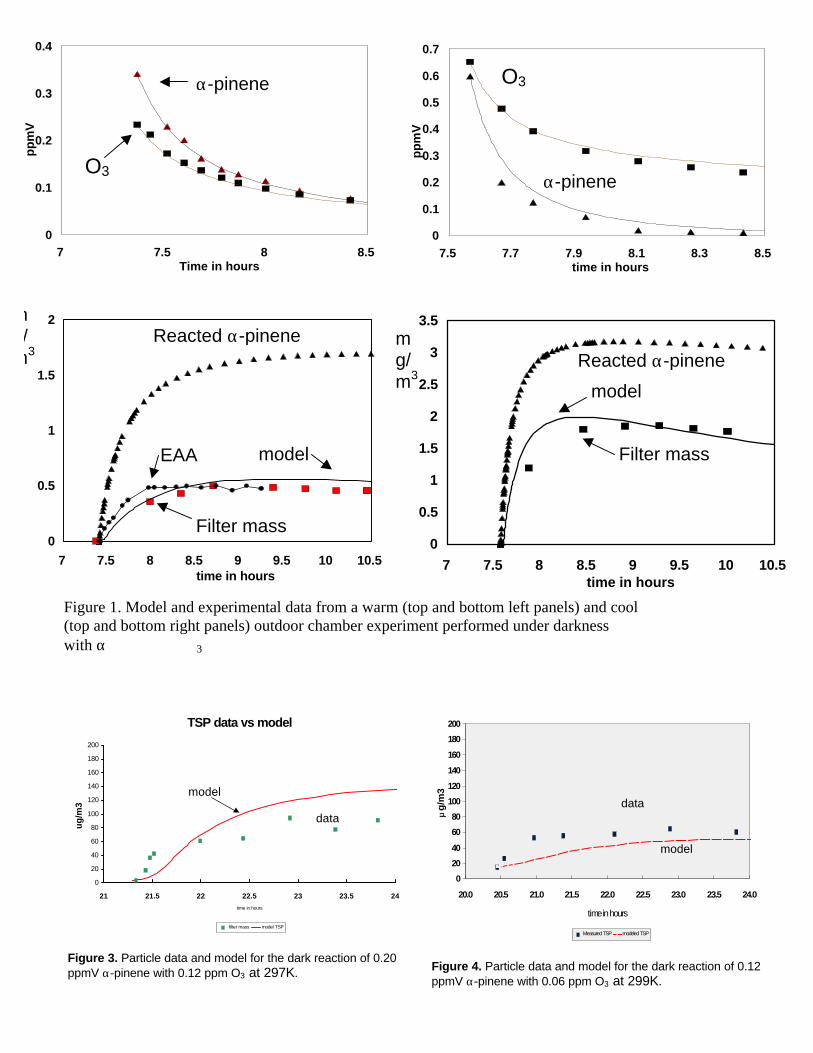
TSP data vs model
0
20
40
60
80
100
120
140
160
180
200
21 21.5 22 22.5 23 23.5 24
time in hours
ug
/m3
filter mass model TSP
model
data
Figure 3. Particle data and model for the dark reaction of 0.20ppmV α-pinene with 0.12 ppm O3 at 297K.
0
0.5
1
1.5
2
2.5
3
3.5
7 7.5 8 8.5 9 9.5 10 10.5 time in hours
0
0.5
1
1.5
2
7 7.5 8 8.5 9 9.5 10 10.5 time in hours
0
0.1
0.2
0.3
0.4 p
pm
V
7 7.5 8 8.5 Time in hours
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
pp
mV
7.5 7.7 7.9 8.1 8.3 8.5 time in hours
α-pinene O3
α-pineneO3
Reacted α-pineneReacted α-pinene
EAA model
model
Filter mass
Filter mass
mg/m3
m/
m3
Figure 1. Model and experimental data from a warm (top and bottom left panels) and cool(top and bottom right panels) outdoor chamber experiment performed under darknesswith α 3
0
20
40
60
80
100
120
140
160
180
200
20.0 20.5 21.0 21.5 22.0 22.5 23.0 23.5 24.0
time in hours
µµg
/m3
Measured TSP modeled TSP
model
data
Figure 4. Particle data and model for the dark reaction of 0.12ppmV α-pinene with 0.06 ppm O3 at 299K.
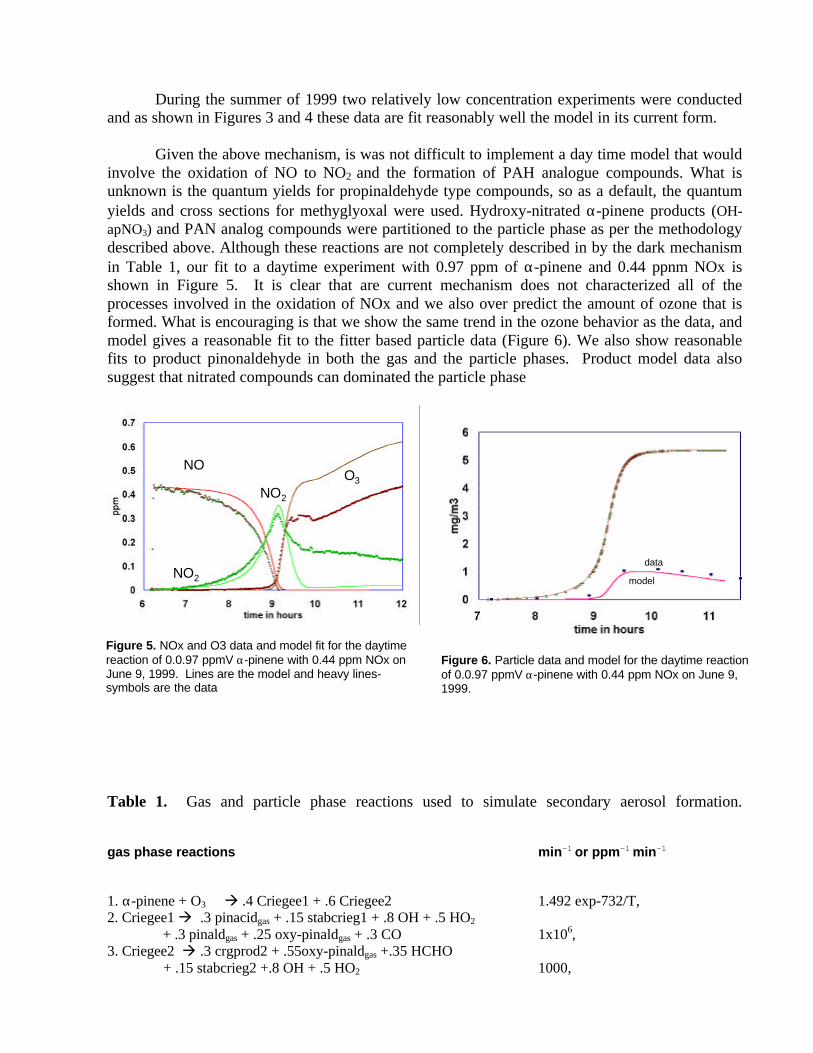
During the summer of 1999 two relatively low concentration experiments were conductedand as shown in Figures 3 and 4 these data are fit reasonably well the model in its current form.
Given the above mechanism, is was not difficult to implement a day time model that wouldinvolve the oxidation of NO to NO2 and the formation of PAH analogue compounds. What isunknown is the quantum yields for propinaldehyde type compounds, so as a default, the quantumyields and cross sections for methyglyoxal were used. Hydroxy-nitrated α-pinene products (OH-apNO3) and PAN analog compounds were partitioned to the particle phase as per the methodologydescribed above. Although these reactions are not completely described in by the dark mechanismin Table 1, our fit to a daytime experiment with 0.97 ppm of α-pinene and 0.44 ppnm NOx isshown in Figure 5. It is clear that are current mechanism does not characterized all of theprocesses involved in the oxidation of NOx and we also over predict the amount of ozone that isformed. What is encouraging is that we show the same trend in the ozone behavior as the data, andmodel gives a reasonable fit to the fitter based particle data (Figure 6). We also show reasonablefits to product pinonaldehyde in both the gas and the particle phases. Product model data alsosuggest that nitrated compounds can dominated the particle phase
Table 1. Gas and particle phase reactions used to simulate secondary aerosol formation.
gas phase reactions min-1 or ppm-1 min-1
1. α-pinene + O3 à .4 Criegee1 + .6 Criegee2 1.492 exp-732/T,2. Criegee1 à .3 pinacidgas + .15 stabcrieg1 + .8 OH + .5 HO2
+ .3 pinaldgas + .25 oxy-pinaldgas + .3 CO 1x106,3. Criegee2 à .3 crgprod2 + .55oxy-pinaldgas +.35 HCHO + .15 stabcrieg2 +.8 OH + .5 HO2 1000,
O3
NO
NO2
NO2
Figure 5. NOx and O3 data and model fit for the daytimereaction of 0.0.97 ppmV α-pinene with 0.44 ppm NOx onJune 9, 1999. Lines are the model and heavy lines-symbols are the data
data
model
Figure 6. Particle data and model for the daytime reactionof 0.0.97 ppmV α-pinene with 0.44 ppm NOx on June 9,1999.

4. stabcrieg1 + H2O à pinacidgas 1x106,5. stabcrieg2 + H2O àP3gas (pinalic acid) + CH3OH 6x10-3,6. P3gas + OH à predi-oo 72380,7. oxy-pinald + OH à oxy-prepinacid 72380,8. predi-oo + HO2 à diacidgas 677 exp1040/T,9. crgprod2 + HO2 à diacidgas 677 exp1040/T,10.oxy-prepinacid +HO2 à oxy-pinacid 677 exp1040/T11. OH + apine à ap-oo 17873 @444,12. ap-oo + NO à 0.85*NO2 + 0.67*pinald+ 0.13*oxypinald + 0.65*HO2 + 0.17*HCHO + 0.15*OH-apNO3+0.07*acetone 5482.1 @ 242,13. ap-oo + ap-oo à 0.6*pinald + HCHO + 0.2*oxypinald 4000,14. α−pinene + NO3 à 0.7*pinald + 0.2*OH-apNO3 {+ 0.05*oxydiNO3} 544 @818,15. pinald + OH à pinald-oo 72380,15. pinald-oo + HO2 à pinacidgas 677 exp1040/T,17. diacidgas {walls} à 6x10-7 exp2445/T,18. oxy-pinacidgas {walls} à 6x10-7 exp2445/T,19. pinacidgas {walls} à 4x10-7 exp2445/T,20. oxy-pinaldgas {walls} à 4x10-7 exp2445/T,21. pinaldgas {walls} à 2.5x10-7 exp2445/T,
partitioning reactions
22. stabcrieg1 + pinaldgas à seed1 29.5,23. stabcrieg2 + oxy-pinaldgas à seed1 29.524. stabcrieg2 + HCHO à oxy-pinald 29.5,25. pinacidgas + seed1 à seed1 + pinacidpart 19.4,26. pinacidgas + diacidpart à diacidpart + pinacidpart 19.4,27. pinacidgas + seed à seed + pinacidpart 19.4,28. pinacidgas + pinacidpart à pinacidpart + pinacidpart 19.4,29. pinacidgas + pinacidpart à pinacidpart + pinacidpart 19.4,30. pinacidgas + oxy-pinaldpart à oxy-pinaldpart + pinacidpart 19.4,31. pinacidgas + P3part à P3part + pinacidpart 19.4,32. pinacidgas + oxy-pinacidpart à oxy-pinacidpart + pinacidpart 19.4,33. pinacidpart à pinacidgas 3.73x1014 exp-9525/T34. diacidgas + pinacidpart --> pinacidpart + diacidpart 6835. diacidpart à diacidgas 3.73x1014 exp-10285/T,36. pinaldgas + oxy-pinacidpart à oxy-pinacidpart + pinaldpart 4.2,37. pinaldpartàpinaldgas 3.73x1014 exp-8598/T,38. oxy-pinaldgas + P3part à P3part + oxy-pinaldpart 14.1,39. oxy-pinaldpart à oxy-pinaldgas 3.73x1014 exp-9341/T,40. p3gas + oxy-pinacidpart à oxy-pinacidpart + p3part 13,41. p3part à p3gas 3.73x1014 exp-9282/T,42. oxy-pinacidgas + seed1 à seed1 + oxy-pinacidpart 74,43 oxy-pinaldpart à oxy-pinacidgas 3.73x1014 exp-10353,/T44. diacidpart {walls} à 0.0008,45 O3 {walls)} à 0.0005,
*Rate constants are at 298K. To convert the 2nd order rate constant in cm3 molecule-1 sec-1 in thetext to ppm-1 min-1, divide the rate constant in cm3 molecule-1 sec-1 by 6.77x10-16 . **exp in thetemperature dependent reactions is the natural base e, in the rate equation, k = B e -A/T. The gasconstant R is imbedded in A so that A = Ea/R. ***The partitioning reactions for pinald, oxy-pinald,diacid, and oxy-pinacid, are the same as for the pinacid, but to save space, are only given for koff
and one particle species. stabcrieg1 and stabcrieg2 reactions are illustrated for pinacidgas, and oxy-

pinacidgas and HCHO, but are the same for the other carbonyls. Loss of product gases to thechamber walls were estimated from observed pyrene loss at 297 and 271K. References forreactions are give in Kamens et al., (1999) were determined in this study. All reactants werediluted from the chamber at a rate determined with an SF6 tracer. A typical loss was 0.0005 min-1,Other specific measured or estimated losses like O3 , particles and α-pinene product to the walls,were adjusted for the SF6 loss rate.

REFERENCES
Atkinson, R. J. Phys. Chem. Ref. Data, 1997 , 26, 215 -290
Kwok, E.; Atkinson, R. Atmos. Environ, 1995, 29, 1685-1695.
Chien, C. J.; Charles, M. J.; Sexton, K. G.; Jeffries, H. E. Environ. Sci. Technol. 1998, 32, 299-309.
Fan, Z.; Kamens, R. M.; Hu, J.; Zhang, J. Environ. Sci. Technol. 1996, 30, 1359-1364.
Grosjean, D.; Willams II, E. L.; Grosjean, E. Environ. Sci. Technol. 1993, 27, 830-840.
Jang, M.; Kamens R. M. Atmos. Environ. 1999 , 33, 459-474
Jeffries, H. E. “PC-Photochemical Kinetics Simulation System (PC-PKSS), Software Version 3.0”,1991, Department of Environmental Sciences and Engineering, School of Public Health,University of North Carolina, Chapel Hill, NC 27599.
Gery, M. W.; Whitten, G. Z.; Killus, J. P.; Dodge, M. C. J. Geophys. Res. 1989, 94, 12925-12956.
Jang, M.; Kamens, R. M.; Leach, K.; Strommen, M. R. Environ. Sci. Technol. 1997, 31, 2805-2811.
Kamens, R. M.; Odum, J.; Fan, Z. Environ. Sci. Technol. 1995, 29, 43-50.
Kamens, R. M.; Jang, M.; Leach, B. K.;Chien,C. “An Exploratory Kinetics and Gas-ParticlePartitioning Model for Aerosol Formation From α-pinene-O3 systems”, Environ Sci andTechnol., in press May, 1999.
Lyman, W. J.; Reehl, W. F.; Rosenblatt, D.H; Handbook of Chemical Property EstimationMethods, Environmental Behavior of Organic Compounds, American Chemical Society,Wash. D.C., 1990
Moortgat,G.K.; Veyret, B.; Lesclaux,R. Chem. Phys. Let., 1989, 160,443-447.
Niki, H.; Maker, P. D.; Savage, C. M., Breitenbach, L. P. Chem. Phys. Let, 1977, 46, 327-330.
Neeb, P.; Horie,O.; Moorgot, G. K. Tetrahedron Let., 1996, 37, 9297-9300.
Neeb.P.; Horie,O.; Moortgat. G.K. Chem. Phys Lett., 1995,150-156
Pankow, J. F. Atmos. Environ. 1994, 28, 185-188.
Yu, J.; Jeffries, H. E.; Le Lacheur, R. M. Environ. Sci. Technol. 1995, 100, 1923-1932.
Went, F. W., Nature 1960, 187, 641 -643.
Yu, J.; Flagan, R.C.; Seinfeld J.H. Envrion. Sci. Technol. 1998, 32, 2357-2370.