
4.2. Molecular Properties from VB Theory a) … to 4_3 VB... · 4.2. Molecular Properties from VB...
13
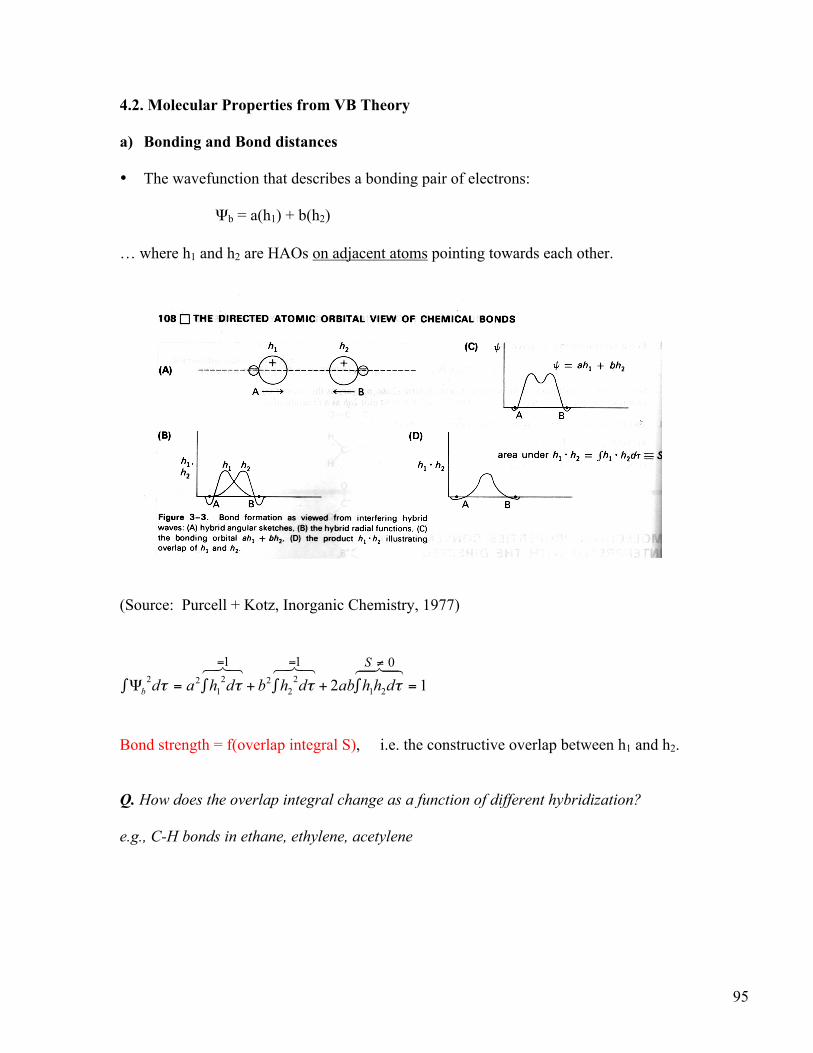
95 4.2. Molecular Properties from VB Theory a) Bonding and Bond distances • The wavefunction that describes a bonding pair of electrons: Ψ b = a(h 1 ) + b(h 2 ) … where h 1 and h 2 are HAOs on adjacent atoms pointing towards each other. (Source: Purcell + Kotz, Inorganic Chemistry, 1977) Bond strength = f(overlap integral S), i.e. the constructive overlap between h 1 and h 2 . Q. How does the overlap integral change as a function of different hybridization? e.g., C-H bonds in ethane, ethylene, acetylene
Transcript of 4.2. Molecular Properties from VB Theory a) … to 4_3 VB... · 4.2. Molecular Properties from VB...
95
4.2. Molecular Properties from VB Theory a) Bonding and Bond distances • The wavefunction that describes a bonding pair of electrons:
Ψb = a(h1) + b(h2) … where h1 and h2 are HAOs on adjacent atoms pointing towards each other.
(Source: Purcell + Kotz, Inorganic Chemistry, 1977)
Bond strength = f(overlap integral S), i.e. the constructive overlap between h1 and h2. Q. How does the overlap integral change as a function of different hybridization? e.g., C-H bonds in ethane, ethylene, acetylene
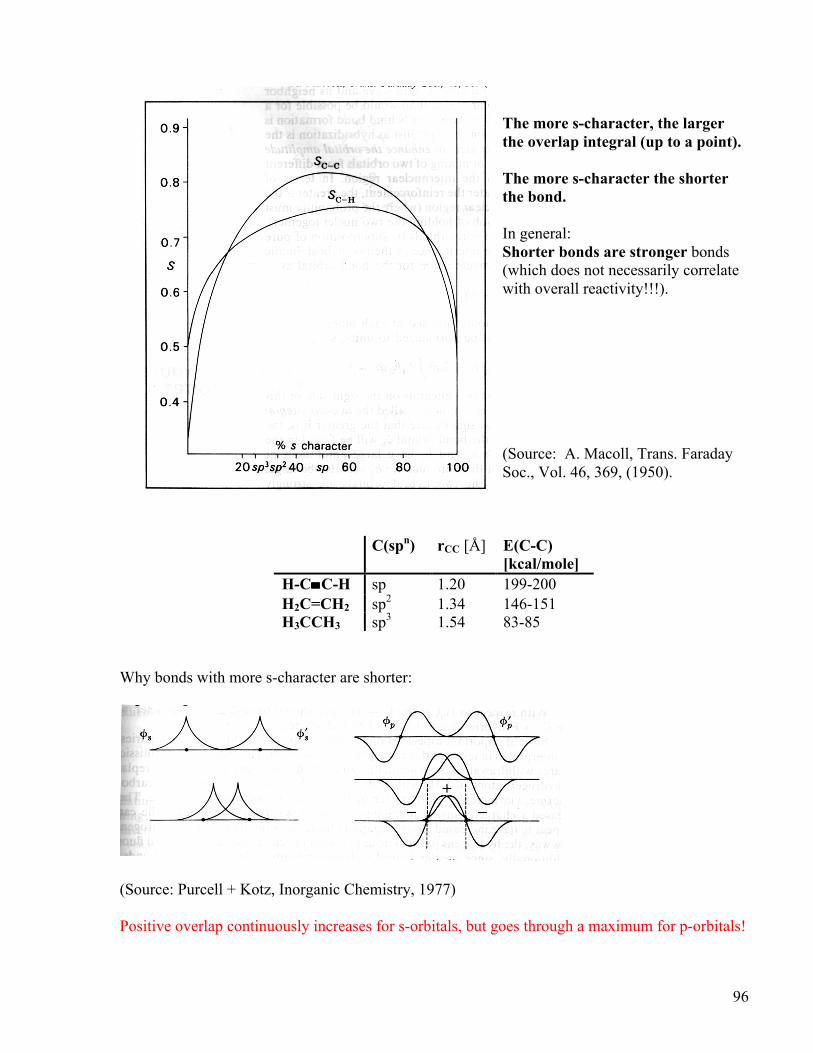
96
The more s-character, the larger the overlap integral (up to a point). The more s-character the shorter the bond. In general: Shorter bonds are stronger bonds (which does not necessarily correlate with overall reactivity!!!). (Source: A. Macoll, Trans. Faraday Soc., Vol. 46, 369, (1950).
C(spn) rCC [Å] E(C-C)
[kcal/mole] H-C≡C-H sp 1.20 199-200 H2C=CH2 sp2 1.34 146-151 H3CCH3 sp3 1.54 83-85
Why bonds with more s-character are shorter:
(Source: Purcell + Kotz, Inorganic Chemistry, 1977) Positive overlap continuously increases for s-orbitals, but goes through a maximum for p-orbitals!

97
Concept of isovalent hybdridization: BENT’S RULES (cf. H.A. Bent, Chem. Rev., 61, 275, (1961)) • So far we’ve only considered high symmetry systems EXn or XnE-EXn.
What happens in EXmYn? • The more electronegative an atom X or Y bound to a central atom E, the greater will be its
demand for electron density from the central atom. • From the concept of orbital electronegativity: EN(s) > EN(p) > EN(d)
…the more the s-character in a hybrid orbital, the greater its electronegativity. Recall: Mulliken electronegativities of carbon: (sp)2π2 10.4 eV 5.7 eV (sp2)3π1 8.8 eV 5.6 eV (sp3)4 8.0 eV
• Central atom E will direct more p (and less s) character towards the more electronegative atom
(and vice versa) • Lone pairs will be in orbitals with more s-character.
(Think of a lone pair as a “bonding pair to a very electro-positive atom”). → fine tuning of VSEPR structures… e.g. PCl5:
Note that phosphorus pentachloride exists as a neutral PCl5 molecule in the gas phase and as ionic [PCl4
+][PCl6-] in the solid state.
Q. Can you predict the structure of SbF2Br3 based on isovalent hybridization? Q. Can you predict the structure of SF4?

98
b) Molecular Dipole Moments Rationalized by VB Theory • Need to find the centers of electronic (-ve) and nuclear (+ve) charge in the molecule as
determined by the molecular structure and e--distribution. (… hmm, wasn’t there another concept that we used for this as well?)
• Consider each bond and lone-pair separately (→ essence of VB theory).
Def. of dipole: µ = n × r where n = number of charges r= distance between charges
Lone pair dipoles:
µ lp = 2e<r>
<r> = average value of r
Lone pair dipoles are fairly large. Absolute size varies with hybrid character:
sp > sp2 > sp3 4.4 → 3.7 Debye
Note: Dipole moments are expressed in Debye units. 1 Debye = 10-8pm·esu Example: Two charges equal in magnitude to the charge of an electron (4.8 x 10-10 electrostatic units esu) and separated by a distance of 91.7 pm (interatomic distance of HF molecule) give a dipole of 4.4 Debye. The value 4.4 Debye represents the expected dipole moment for 100% ionic H+F-. The observed dipole moment for HF is 1.98 Debye. Therefore, the H-F bond has 1.98/4.4 = 45% ionic character. • Okay, now let’s look at how HAO’s are useful for explaining lone pair dipole moments …

99
One of the quantum mechanical postulates is that the average or expectation value of any observable property O is given by:
where is the operator that corresponds to the property O (e.g. the Hamiltonian for the energy of the system, or the distance between charge centroids) … …and ψ is the state function (i.e. wavefunction) of the system. Consider an sp hybrid orbital: Total orbital: φn = csφs + cpφp Average (= quantum mechanical expectation) value of r for an sp hybrid orbital:
The last equality makes the approximation: <r>p ≈ <r>s Furthermore because of normalization: cs
2 + cp2 = 1
Thus:
Because the s-orbital is centrosymmetric (i.e., <rs> = 0), the only dipole contribution comes from the hybrid orbital and the dipole is simply …
µ = 2 × e × <r>h = 4 × e × cscp × <r>sp
i.e., only the interference integral that describes the hybrid contributes to the dipole. POINT: s and p orbitals are centrosymmetric. HAO’s are not and therefore can be invoked in the explanation of dipoles.

100
Bond pair dipoles: … more difficult to estimate • Need to identify the charge centers along a bond: +ve: In 1st approximation simply the halfway point of the bond -ve: Need to know the electron density distribution along the bond as f(r) Consider bond H-E, where E = any element except hydrogen:
; φEh = sp-hybrid orbital on E
Expectation value of r (as before):
Physical meaning of all the terms: a2<r>H electron density contribution of the hydrogen 1s AO to center of –ve charge b2<r>E electron density contribution of the E sp-hybrid orbital to center of –ve charge 2ab<r>HE electron density contribution of the overlap density to center of –ve charge
“2+” = positive charge midpoint (because we are dealing with electron pairs, we must count up the positive charge in pairs as well).
R = distance between nuclei

101
From figure: - The electron density center for hydrogen is at <r>H = -(R/2) - The electron density center for the hybrid orbital is at (using the result for rsp from the derivatization for the lone pair):
<r>E = <r>s + 2cscp<r>sp = R/2 + 2 cscp<r>sp For the complete electron pair:
where the substitution i = b2 - a2 is a measure of the ionic character of the bond: a2 = b2 → i = 0 purely covalent bond (also: a2 + b2 = 1) a2 or b2 = 1 i = 1 purely ionic bond In summary: i(R/2) orientation of the bond-dipole to the more electronegative atom 2ab<r>HE contribution from the overlap density of both atoms in the bond to the center of
negative charge along the bond 2b2cscp<r>sp contribution from the hybrid orbital itself on E to the charge center; orientation
depends on the radial distribution of electron density in φE. (non-existent for φ1s because of radial symmetry) • The orientation and size of a bond-dipole moment cannot solely be predicted on the basis
of the difference in EN values of the two atoms involved !!!
• Have to consider the charge distribution contributions from the orbitals involved, which can lead to a dipole that is reversed from the one expected based on ΔEN!!!

102
• In general: LP dipole moments >> BP dipole moments
• In bonds of the type E1-E2 (E1 ≠ E2) with hybrid orbitals from both atoms forming the bond,
the hybrid contributions to the dipole moment tend to cancel. Examples: NH3 vs. NF3
µ = 1.5 D µ = 0.2 D ΧN > ΧH ΧN < ΧF no hybrid dipole moment on H overall weak BP dipole moment dominated by LP dipole moment
Bent’s rule requires more s-character on NLP F atoms have LP aligned with bond overall weak NLP dipole moment dominated by BPNF and LPF overall dipole moment orientation ??
Carbon monoxide: measured dipole moment µ = 0.1 D, i.e. very small Lone pairs Bond pairs
From Bent’s rule: both sp lone pairs have more than 50 % s-character with carbon more so than oxygen → dipole towards oxygen but: C(sp) more diffuse (i.e. spreads further out) → dipole towards carbon net effect µ ≈ 0
Dominated by ΔEN: More compact oxygen orbitals → dipole towards oxygen but: +ve charge center closer to the oxygen (higher nuclear charge) net effect µ ≈ 0
… and now you understand why CO is a gas !!!

103
c) Bond Energies and VB theory
VB Theory Thermodynamics
microscopic picture/molecular scale macroscopic picture/bulk scale isolated, localized electron pairs form bonds bond energies within a molecule are additive
Diatomic molecules: Experimentally, bond energy (D) is simply the internal energy change required to separate the gas phase molecule into its two constituent atoms (also in the gas phase.) A-B(g) → A(g) + B(g) DAB = ΔE (Note: D is discrete bond energy, E is average) Polyatomic molecules: 1) Removal of one terminal atom to leave a fragment ABn-1 alters the bonding between A and the remaining B atoms (changes hybridization / multiple bonding). 2) Ground state electron configuration of the dissociated B atom may be quite different from its valence state when bound to A. The valence state promotion energy for B may make a significant contribution to the bond energy D. Consider a simple binary compound ABn. ABn(g) → ABn-1(g) + B(g) DAB = ?? DAB is a function of: • Hybridization state of ABn and ABn-1
• Changes in multiple bonding between AB caused by removal of one B
• Electrostatic configuration of B “alone” vs. B in ABn (in particular of the promotion energy
Bgroundstate → Bhybrid). Example: Stepwise dissociation of CO2 (gasphase) Step 1: O=C=O → C≡O + O DCO = 127 kcal/mol Step 2: C≡O → C + O DCO = 256 kcal/mol In step 1: Breaking of one bond followed by configuration relaxation: O: (sp2)1(sp2)2(sp2)2π1 (= s5/3p13/3)→ s2p4 (3P) from hybrid to ground state atomic C: (sp)1(sp)1(π)1(π)1 (= s1p3) → (sp)1(sp)2π1 (= s1.5p2.5) from double to triple bonding In step 2: Breaking of second bond and relaxation of both atoms into g. s. atomic configuration DO THIS FOR YOURSELF AT HOME

104
A reverse example: Cl-HgII-Cl → HgI-Cl + Cl → Hg0 + 2 Cl DHgCl = 81 kcal/mol DHgCl = 25 kcal/mol - Can you write down the electron configurations of this example? - Why is the second step energetically easier? Important conclusion: Average bond energies and individual bond energies in ABn systems are not the same!!! Average bond energies (thermodynamics): ABn(g) → A(g) + n B(g) EA = 1/n ΔE Expect major consequences on stability/reactivity of molecules in general!!! Consider the reaction PCl5 → PCl4 + Cl • What are the structures of PCl5 and PCl4?
• Based on VB are all the P-Cl bonds equally strong?
• Thought experiment: No matter which P-Cl is broken ΔE is always the same. This is a
requirement of the macroscopically observed real experimental value – but how can that be?? In summary: • Experimental bond energies are composed of the actual bond dissociation energy and the
energy terms that account for changes in the electronic configurations of the participating atoms and/or molecular fragments.
(… to be really correct would also have to consider vibrational and rotational states …)
105
More examples: • Series of EHn molecules with n = 2, 3, or 4 (NOTE: These are called hydrides.)
Average <EEH> [kcal/mole]
Promotion energy [kcal/mole] s2pn (n = 2,3,4) → 4 × sp3
CH4 99 150 SiH4 76 115 NH3 93 228 PH3 78 133 OH2 111 194 SH2 88 109
Compare CH4 vs. SiH4: 4 <ECH> = 4 × 99 = 4 × E*
CH – PC 396 = 4 × E*
CH – 150 E*CH = 137 kcal/mole
4 <ESiH> = 4 × 76 = 4 × E*
SiH – PSi 304 = 4 × E*
Si – 115 E*Si = 105 kcal/mole
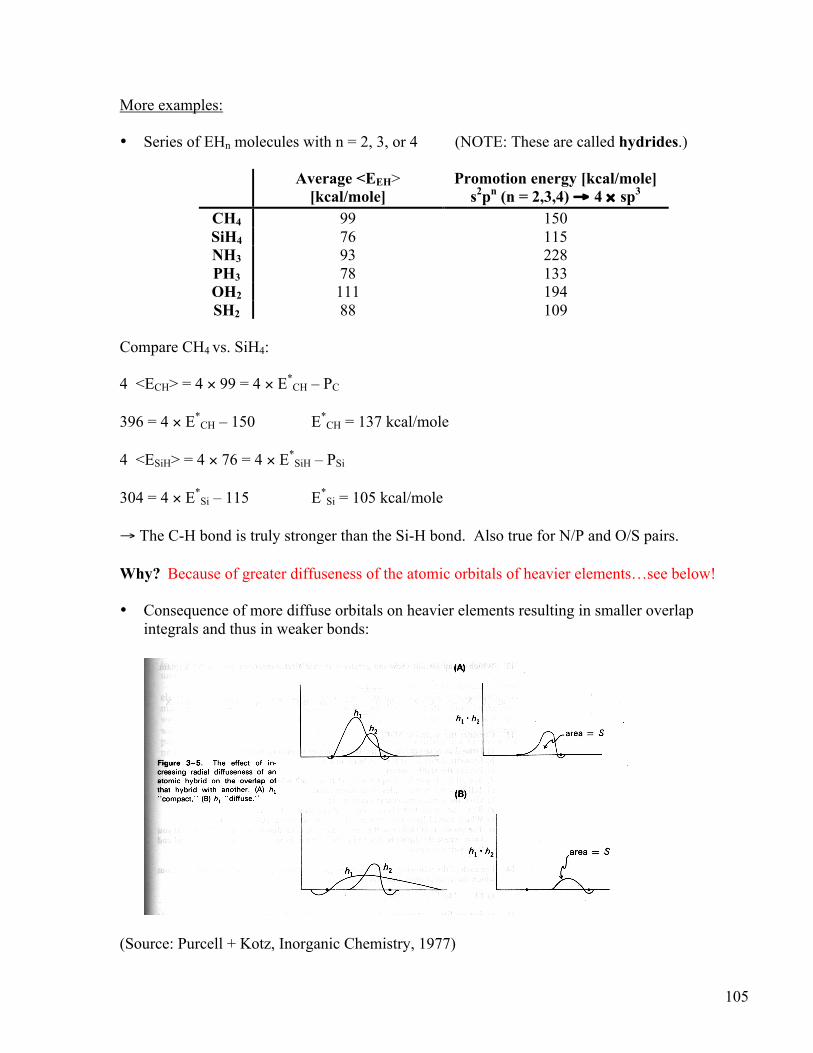
→ The C-H bond is truly stronger than the Si-H bond. Also true for N/P and O/S pairs. Why? Because of greater diffuseness of the atomic orbitals of heavier elements…see below! • Consequence of more diffuse orbitals on heavier elements resulting in smaller overlap
integrals and thus in weaker bonds:
(Source: Purcell + Kotz, Inorganic Chemistry, 1977)

106
4.3 Concept of Isolobality (Nobel laureate Roald Hoffman) See: Angew. Chem. Int. Ed. 1982, 21, 711. • Contributes to the understanding of parallels between organic and inorganic chemistry. • Structurally analogous fragments of molecules are described as isolobal. • Studying isolobal molecular fragments can help suggest patterns of bonding / reactivity. • Concept of hybridization can be used to identify isolobality. Def. Two molecular fragments are said to “isolobal” if the number, symmetry properties, approximated energy and shape of the frontier orbitals and the number of electrons in them are similar (NOT identical, but similar). Example: The following orbitals have σ-type symmetry, are similar in energy, and are occupied by one electron…therefore we can expect analogous bonding properties.
We can also make the isolobal analogy with the following two and three orbitals examples:
NOTE: Although the isolobal analogy can give clues about what kind of compounds might be expected to exist, it is only a useful tool and not a hard and fast rule. HOMEWORK: Exercise 2.25

107
4.4. Limitations of VB Theory • Valence shell expansion is often required to draw satisfactory Lewis diagrams, e.g. for PCl5
and SF6, even though the incorporation of d-orbitals does energetically not make sense the process: s2pnd0 → s2pn-mdm Costs too much hybridization energy. So how can it be that e.g. sulfur can bind six fluorine atoms???
• Why is O2(g) paramagnetic (i.e., has unpaired electrons)?
• How can we describe the bonding in B2H6 using only H1s orbitals?
• How can we explain these bond angles through hybridization?
No hybridization can explain bond angles < 90°!!
→ We need a new theory !


















