
TUMOUR MICROENVIRONMENT Laminin 332 in squamous-cell carcinoma · Squamous-cell carcinoma (SCC) is...
11
Squamous-cell carcinoma (SCC) is a highly invasive malignant neoplasm associated with a high risk of metastasis and morbidity. SCCs can arise in a wide range of tissues causing skin, lung, oral, cervical, gastric and colorectal cancers, among others. Given the range of tissues it affects, SCC represents the most common cancer capable of metastatic spread in the USA and worldwide 1,2 . In 1973, approximately 480,000 cases of non-melanoma skin cancer were reported in the USA, whereas in 1994, the number of cases had more than doubled, and was estimated to be between 900,000 and 1,200,000 cases 1 . Cervical SCC also causes significant morbidity and mortality, although it is hoped that the incidence of cervical SCC will be reduced by human papillomavirus (HPV) vac- cines, one of which was recently approved by the US Food and Drug Administration. The risk of SCC is strongly linked with environmental factors, such as sun exposure, viral infections and tobacco use. SCCs are associated with a high risk of recurrence, resulting in significant mortality and often requiring specialized surgical techniques (Mohs’ surgery) for complete exci- sion. SCCs often invade neighbouring tissues, and can also metastasize to the lymph nodes, lung and other distant sites. All of these factors have led to the search for non-surgical means of treating SCCs. Secretion, assembly and remodelling of extracellu- lar matrix proteins into an organized structure termed the basement membrane zone (BMZ) is an essential function of epithelial tissues and cells 3,4 . This process, which also depends on stromal contributions, provides a scaffold on which epithelial cells and tissues can dif- ferentiate, divide, migrate, develop and, in the case of malignant cancers, invade and metastasize 5–8 . In addi- tion to serving as a scaffold, the epithelial BMZ itself can exert a profound influence on cell behaviour. It can dictate whether cells will proliferate, growth arrest, or undergo programmed cell death. The BMZ can influ- ence whether cells will migrate or remain stationary. As will be shown in this Review, the BMZ produced by carcinomas is required for tumour growth and cellular or tissue invasion. In cancer research, BMZs have traditionally been viewed as protective structures to defend against cancer spread, or as obstacles that in situ carcinomas have to overcome to invade and metastasize 9 . The proteolytic cleavage of BMZ molecules by enzymes produced by carcinoma cells was once thought to sim- ply function in the degradation of the host matrix to provide space for tumour extension 10 . Recent studies have shown that the roles of the BMZ and extracellular matrix in cancer invasion and tumour development are more complex. Indeed, we now know that carci- noma cells depend on their own array of BMZ mol- ecules that they preferentially use as a substrate for invasion and proliferation. It is also now appreciated that tumour-associated proteolytic enzymes have a significant effect on modifying tumour-derived BMZ, and that proteolytic modifications of tumour-derived BMZ molecules are a necessary prerequisite to inva- sion and growth. This emerging view recognizes BMZ molecules in carcinomas as potential pro-tumorigenic VA Medical Center, Palo Alto, California, USA, and Program in Epithelial Biology, Stanford University School of Medicine, Stanford, California 94305, USA. e-mail: [email protected] doi:10.1038/nrc2089 Mohs’ surgery A tissue-sparing technique for complete excision of cutaneous SCC tumours, which uses frozen sections of marked excisional tissue boundries to ascertain complete tumour clearance. Laminin 332 in squamous-cell carcinoma M. Peter Marinkovich Abstract | Basement membranes can be a barrier to tumour growth, but basement membrane molecules, including laminins, are also important autocrine factors produced by cancers to promote tumorigenesis. Many studies have shown the importance of laminin 332 (previously known as laminin 5) in this process, especially in squamous cell carcinoma. Through interactions with several cell-surface receptors (including α6β4 and α3β1 integrins, epidermal growth factor receptor and syndecan 1) and other basement membrane components (including type VII collagen), laminin 332 drives tumorigenesis through phosphatidylinositol-3 kinase (PI3K) and RAC1 activation, promoting tumour invasion and cell survival. The extracellular interactions of laminin 332 appear amenable to antibody-mediated therapies. TUMOUR MICROENVIRONMENT REVIEWS 370 | MAY 2007 | VOLUME 7 www.nature.com/reviews/cancer © 2007 Nature Publishing Group
Transcript of TUMOUR MICROENVIRONMENT Laminin 332 in squamous-cell carcinoma · Squamous-cell carcinoma (SCC) is...

Squamous-cell carcinoma (SCC) is a highly invasive malignant neoplasm associated with a high risk of metastasis and morbidity. SCCs can arise in a wide range of tissues causing skin, lung, oral, cervical, gastric and colorectal cancers, among others. Given the range of tissues it affects, SCC represents the most common cancer capable of metastatic spread in the USA and worldwide1,2. In 1973, approximately 480,000 cases of non-melanoma skin cancer were reported in the USA, whereas in 1994, the number of cases had more than doubled, and was estimated to be between 900,000 and 1,200,000 cases1. Cervical SCC also causes significant morbidity and mortality, although it is hoped that the incidence of cervical SCC will be reduced by human papillomavirus (HPV) vac-cines, one of which was recently approved by the US Food and Drug Administration. The risk of SCC is strongly linked with environmental factors, such as sun exposure, viral infections and tobacco use. SCCs are associated with a high risk of recurrence, resulting in significant mortality and often requiring specialized surgical techniques (Mohs’ surgery) for complete exci-sion. SCCs often invade neighbouring tissues, and can also metastasize to the lymph nodes, lung and other distant sites. All of these factors have led to the search for non-surgical means of treating SCCs.
Secretion, assembly and remodelling of extracellu-lar matrix proteins into an organized structure termed the basement membrane zone (BMZ) is an essential function of epithelial tissues and cells3,4. This process, which also depends on stromal contributions, provides
a scaffold on which epithelial cells and tissues can dif-ferentiate, divide, migrate, develop and, in the case of malignant cancers, invade and metastasize5–8. In addi-tion to serving as a scaffold, the epithelial BMZ itself can exert a profound influence on cell behaviour. It can dictate whether cells will proliferate, growth arrest, or undergo programmed cell death. The BMZ can influ-ence whether cells will migrate or remain stationary. As will be shown in this Review, the BMZ produced by carcinomas is required for tumour growth and cellular or tissue invasion.
In cancer research, BMZs have traditionally been viewed as protective structures to defend against cancer spread, or as obstacles that in situ carcinomas have to overcome to invade and metastasize9. The proteolytic cleavage of BMZ molecules by enzymes produced by carcinoma cells was once thought to sim-ply function in the degradation of the host matrix to provide space for tumour extension10. Recent studies have shown that the roles of the BMZ and extracellular matrix in cancer invasion and tumour development are more complex. Indeed, we now know that carci-noma cells depend on their own array of BMZ mol-ecules that they preferentially use as a substrate for invasion and proliferation. It is also now appreciated that tumour-associated proteolytic enzymes have a significant effect on modifying tumour-derived BMZ, and that proteolytic modifications of tumour-derived BMZ molecules are a necessary prerequisite to inva-sion and growth. This emerging view recognizes BMZ molecules in carcinomas as potential pro-tumorigenic
VA Medical Center, Palo Alto, California, USA, and Program in Epithelial Biology, Stanford University School of Medicine, Stanford, California 94305, USA. e-mail: [email protected]:10.1038/nrc2089
Mohs’ surgeryA tissue-sparing technique for complete excision of cutaneous SCC tumours, which uses frozen sections of marked excisional tissue boundries to ascertain complete tumour clearance.
Laminin 332 in squamous-cell carcinomaM. Peter Marinkovich
Abstract | Basement membranes can be a barrier to tumour growth, but basement membrane molecules, including laminins, are also important autocrine factors produced by cancers to promote tumorigenesis. Many studies have shown the importance of laminin 332 (previously known as laminin 5) in this process, especially in squamous cell carcinoma. Through interactions with several cell-surface receptors (including !6"4 and !3"1 integrins, epidermal growth factor receptor and syndecan 1) and other basement membrane components (including type VII collagen), laminin 332 drives tumorigenesis through phosphatidylinositol-3 kinase (PI3K) and RAC1 activation, promoting tumour invasion and cell survival. The extracellular interactions of laminin 332 appear amenable to antibody-mediated therapies.
T U M O U R M I C R O E N V I R O N M E N T
R E V I E W S
370 | MAY 2007 | VOLUME 7 www.nature.com/reviews/cancer
© 2007 Nature Publishing Group
autocrine factors. Although tumour-derived BMZ molecules do not form morphologically recognizable ultrastructural elements equivalent to those found in normal tissues, there are nonetheless some important intramolecular BMZ associations that are crucial in supporting carcinoma development. This will be the focus of this Review.
Laminin 332 structure and functionLaminins are large extracellular glycoproteins that are important components of all BMZs, and are involved in several important biological processes11 including tissue development, wound healing, and, as will be discussed in this Review, tumorigenesis. The main role of laminin 332 (formerly termed laminin 5, kalinin, nicein, ladsin and epiligrin) in normal tissues is in the maintenance of epithelial–mesenchymal cohesion in tissues exposed to external disruptive forces, including the skin, stratified squamous mucosa, the amnion and the cornea12. The crucial role of laminin 332 in epidermal adhesion was highlighted further by the demonstration of its absence in the severe and lethal blistering disease, Herlitz’s junctional epidermolysis bullosa (JEB)13,14, owing to underlying laminin 332 gene mutations15,16.
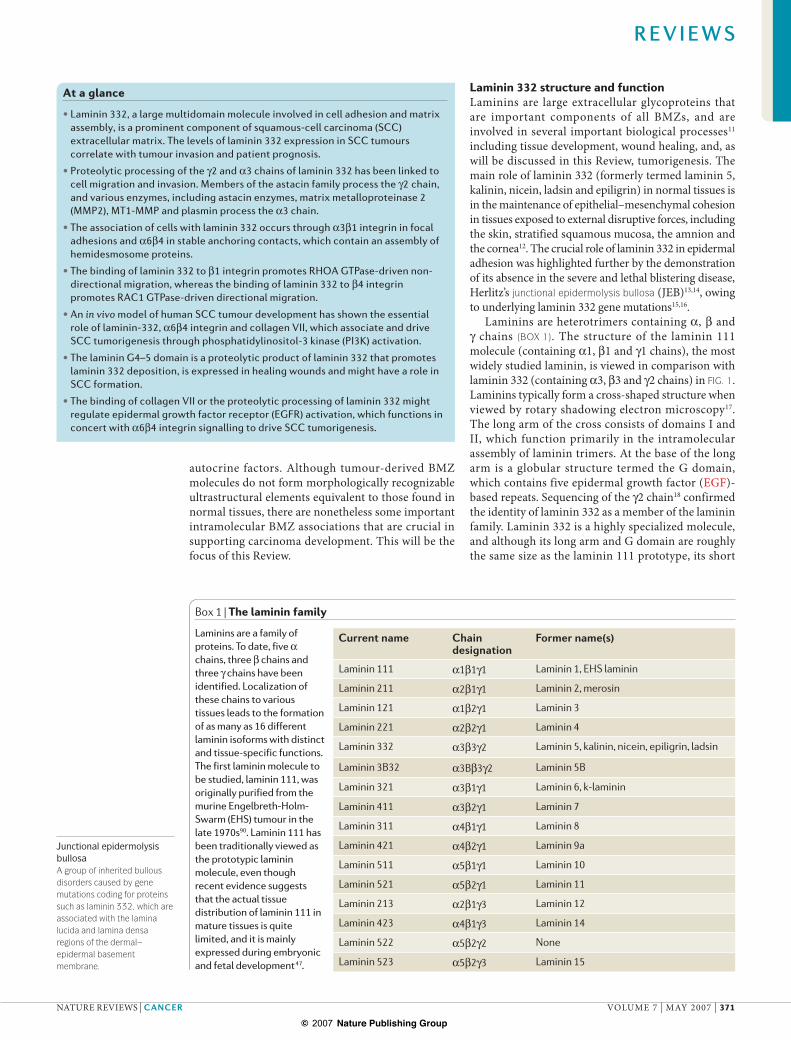
Laminins are heterotrimers containing !, " and # chains (BOX 1). The structure of the laminin 111 molecule (containing !1, "1 and #1 chains), the most widely studied laminin, is viewed in comparison with laminin 332 (containing !3, "3 and #2 chains) in FIG. 1. Laminins typically form a cross-shaped structure when viewed by rotary shadowing electron microscopy17. The long arm of the cross consists of domains I and II, which function primarily in the intramolecular assembly of laminin trimers. At the base of the long arm is a globular structure termed the G domain, which contains five epidermal growth factor (EGF)-based repeats. Sequencing of the #2 chain18 confirmed the identity of laminin 332 as a member of the laminin family. Laminin 332 is a highly specialized molecule, and although its long arm and G domain are roughly the same size as the laminin 111 prototype, its short
At a glance
• Laminin 332, a large multidomain molecule involved in cell adhesion and matrix assembly, is a prominent component of squamous-cell carcinoma (SCC) extracellular matrix. The levels of laminin 332 expression in SCC tumours correlate with tumour invasion and patient prognosis.
• Proteolytic processing of the #2 and !3 chains of laminin 332 has been linked to cell migration and invasion. Members of the astacin family process the #2 chain, and various enzymes, including astacin enzymes, matrix metalloproteinase 2 (MMP2), MT1-MMP and plasmin process the !3 chain.
• The association of cells with laminin 332 occurs through !3"1 integrin in focal adhesions and !6"4 in stable anchoring contacts, which contain an assembly of hemidesmosome proteins.
• The binding of laminin 332 to "1 integrin promotes RHOA GTPase-driven non-directional migration, whereas the binding of laminin 332 to "4 integrin promotes RAC1 GTPase-driven directional migration.
• An in vivo model of human SCC tumour development has shown the essential role of laminin-332, !6"4 integrin and collagen VII, which associate and drive SCC tumorigenesis through phosphatidylinositol-3 kinase (PI3K) activation.
• The laminin G4–5 domain is a proteolytic product of laminin 332 that promotes laminin 332 deposition, is expressed in healing wounds and might have a role in SCC formation.
• The binding of collagen VII or the proteolytic processing of laminin 332 might regulate epidermal growth factor receptor (EGFR) activation, which functions in concert with !6"4 integrin signalling to drive SCC tumorigenesis.
Box 1 | The laminin family
Laminins are a family of proteins. To date, five ! chains, three " chains and three # chains have been identified. Localization of these chains to various tissues leads to the formation of as many as 16 different laminin isoforms with distinct and tissue-specific functions. The first laminin molecule to be studied, laminin 111, was originally purified from the murine Engelbreth-Holm-Swarm (EHS) tumour in the late 1970s90. Laminin 111 has been traditionally viewed as the prototypic laminin molecule, even though recent evidence suggests that the actual tissue distribution of laminin 111 in mature tissues is quite limited, and it is mainly expressed during embryonic and fetal development47.
Current name Chain designation
Former name(s)
Laminin 111 !1"1#1 Laminin 1, EHS laminin
Laminin 211 !2"1#1 Laminin 2, merosin
Laminin 121 !1"2#1 Laminin 3
Laminin 221 !2"2#1 Laminin 4
Laminin 332 !3"3#2 Laminin 5, kalinin, nicein, epiligrin, ladsin
Laminin 3B32 !3B"3#2 Laminin 5B
Laminin 321 !3"1#1 Laminin 6, k-laminin
Laminin 411 !3"2#1 Laminin 7
Laminin 311 !4"1#1 Laminin 8
Laminin 421 !4"2#1 Laminin 9a
Laminin 511 !5"1#1 Laminin 10
Laminin 521 !5"2#1 Laminin 11
Laminin 213 !2"1#3 Laminin 12
Laminin 423 !4"1#3 Laminin 14
Laminin 522 !5"2#2 None
Laminin 523 !5"2#3 Laminin 15
Junctional epidermolysis bullosaA group of inherited bullous disorders caused by gene mutations coding for proteins such as laminin 332, which are associated with the lamina lucida and lamina densa regions of the dermal–epidermal basement membrane.
R E V I E W S
NATURE REVIEWS | CANCER VOLUME 7 | MAY 2007 | 371
© 2007 Nature Publishing Group

1
3
231 1
VI VIVIV VIV IVIII
G domain G domain
III
100 nm
III-V
II
I
II
I
III VIV
IIIa
Laminin 111 Laminin 332
VIVIVbIIIbIVaIIIa
12
345
12
345
IntegrinsIntegrins are transmembrane heterodimeric BMZ receptors consisting of ! and " subunits that comprise a family of 20 currently identified members. Individual members of the integrin family have specific BMZ-recognition profiles and often distinct patterns of signal transduction when activated by ligand binding.
MatrigelA commercially available basement membrane extract derived from the murine Engelbreth-Holm-Swarm (EHS) tumour. Matrigel primarily contains the proteins laminin 111, type IV collagen, nidogen and perlecan.
arms are extremely short11. Despite their relatively small size, certain N-terminal domains of laminin 332 chains are important because of their ability to interact with other BMZ molecules (BOX 2).
Expression of laminin 332 in human tumoursLaminin 332 has been shown to be highly expressed in several types of squamous and other epithelial tumours, including cutaneous, oral, oesophageal, laryngeal, tracheal, cervical and colon carcinomas19–31. In these tumours, laminin 332 often is noted to accumulate at the interface of the tumour with the surrounding stroma. Laminin 332 expression has been shown to correlate well with tumour invasiveness22,25,32,33 and poor patient prognosis25,27,28,34–36 in various SCC sub-types. In general, tumours derived from tissues that normally express laminin 332 show high levels of lam-inin 332 expression, whereas tumours derived from tissues that do not normally express laminin 332, such as sarcomas, do not express laminin 332. However, there are some notable exceptions. For example, there is generally decreased laminin 332 expression in basal cell carcinomas37, as well as in advanced breast38,39
and prostate cancers40, even though the tissues from which these cancers are believed to originate normally do express laminin 332. The expression of laminin receptors, in particular !6"4 integrin, have also been shown to have an important role in SCC progression41. Developmental influences probably also have a role in the regulation of laminin 332 expression, and it is of interest to note that the downregulation of laminin 332 during early hair follicle development42, as well as in basal cell and prostate carcinomas, coincides with activation of the sonic hedgehog signalling pathway. Indeed, forced retroviral overexpression of sonic hedgehog in a human keratinocyte xenograft model of basal cell carcinoma resulted in decreased laminin 332 expression, suggesting that the two processes are linked43.
Laminin 332 in cellular adhesive contactsLaminins act as major BMZ ligands for epithelial and carcinoma cells, supporting spreading, attachment, migration and invasion3. Laminin 111, the laminin contained in matrigel has been well studied44–46, but it is becoming increasingly clear that much of the informa-tion gained with studies of laminin 111 (REF. 47) is not applicable to other laminin isoforms, including laminin 332 (REF. 48).
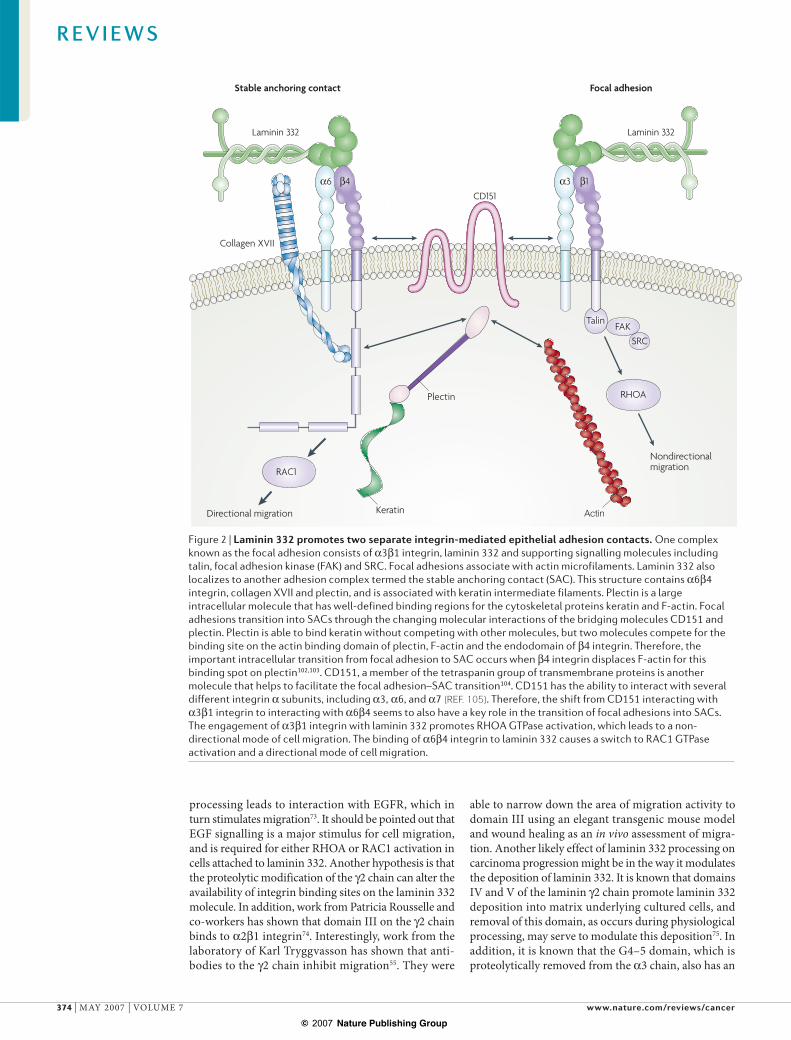
One of the unique functions of laminin 332 in epidermal cells is its ability to interact with two major epithelial integrin receptors, !3"1 and !6"4 (REF. 49), and its ability to promote the formation of two separate types of attachment structures, focal adhesions and stable anchoring contacts (SACs)50. These two structures, and the role that laminin 332 performs in each of them, are illustrated schematically in FIG. 2. Focal adhesions have been well studied in the attachment and migration of normal and malignant cells51, and laminin-332 localizes to these structures through !3"1 integrin49. SACs assem-ble through the interaction of the extracellular domains of !6"4 integrin with laminin 332 and the interaction of the endodomain of "4 integrin with collagen XVII and plectin52,53. In vivo, SACs form hemidesmosomes which, like SACs, link the keratin-containing intermediate fila-ment cytoskeleton to the BMZ. In vitro, SACs provide stable adhesion to epidermal cells. Whereas focal adhe-sions provide short-term adhesion through reversible associations of integrins, SAC-derived stable adhesion in epidermal cells is derived from a more complex assembly of integrins and integrin-associated proteins, and pro-vides an increased resistance to disruptive forces. It is generally believed that focal adhesions change into SACs over time within the same area of deposited laminin 332 (REFS 53,54). Two molecules have been identified that help to facilitate this transition, plectin and CD151.
Laminin 332 in cell migrationThe interaction of laminin 332 with focal adhesions and SACs has distinct roles during the process of cell movement. Laminin 332 is known to stimulate the migration of various cells including carcinoma cells55–57. Before this, Kauro Miyazaki and his group showed that laminin 332 derived from gastric carcinoma cells was
Figure 1 | Comparison between laminin 111 and laminin 332. Laminins form cross-shaped structures as viewed by rotary shadowing electron microscopy and are composed of three chains. The large G domain at the base of the long arm of the cross contains five repeating segments with epidermal growth factor (EGF)-like sequences. The first three of these repeats, termed the G1–3 domains, contain binding sites for cell surface receptors termed integrins98,99. The last two EGF-based repeats on the large laminin globular domain contain heparin binding activity100, and interact with extracellular heparan sulphate proteoglycans, such as !-dystroglycan101. Laminins can also interact with transmembrane proteoglycan receptors, exemplified by the known interaction of the laminin 332 G4 domain with syndecan 1 (REF. 85). Domains I and II in each of the three chains facilitate trimeric assembly. Laminin 332 differs significantly from laminin 111 in that it contains significantly truncated globular domains on the !3, "3 and #2 chains that comprise the short arms of the laminin molecule. These chains differ significantly in their abilities to interact with other extracellular matrix molecules.
R E V I E W S
372 | MAY 2007 | VOLUME 7 www.nature.com/reviews/cancer
© 2007 Nature Publishing Group

Focal adhesionA structure that provides dynamic cell adhesion in vivo, which involves integrin-mediated binding to the extracellular matrix and connections to the actin cytoskeleton.
Stable anchoring contactA structure that provides stable cell adhesion in vivo consisting of an assembly of hemidesmosomal components.
HemidesmosomesSpecialized condensations of basal keratinocyte plasma membrane that contain a number of specialized basement membrane components. These structures provide additional stable adhesion in the face of external disruptive forces.
KeratinocytesSpecialized epithelial cells that make up the stratified squamous epithelium of skin and mucosal tissues.
GalvanotaxisThe responsive movement of cells along the direction of an electrical field.
a scatter factor58. Interestingly, they found that both soluble and deposited laminin 332 could promote migration through a pathway involving both protein kinase C (PKC) and mitogen-activated protein kinase (MAPK) pathway activation59. Although soluble lam-inin can be studied easily during in vitro experiments, it is less clear to what extent laminin 332 exists in a soluble versus insoluble state in vivo during processes such as wound healing and tumour invasion. Work from William Carter and colleagues has shown that during wound healing, keratinocytes migrating along a wound edge in the absence of laminin 332 initially attached to the wound bed through focal adhesions and "1 integrin, which activates the GTPase RHOA60,61. Once laminin 332 started to be produced and deposited by these migrating cells, keratinocytes switched their attachment to this preferred ligand, and the pattern of GTPase activation changed to that of RAC1 activation. This occurred because the deposited laminin 332 was ligated by the !6"4 integrin on the keratinocyte cell surface. Work from Randy Kramer and colleagues has also shown the downregulation of RHOA and its effector molecule Rho-associated coiled-coil contain-ing protein kinase 1 (ROCK1) in cells migrating on laminin 332 (REF. 62). Interestingly, if ligation with !6"4 integrin is prevented, through the site-directed muta-genesis of the ligand binding site (LBS) on the "4 inte-grin exodomain, ligation of !3"1 integrin with laminin 332 promotes migration through RHOA activation63. Therefore, laminin 332 appears to participate in two opposing modes of migration and GTPase activation, depending on whether or not !6"4 integrin engages laminin 332.
The consequences of this switch have profound implications for cell migration. The migration patterns of RHOA- versus RAC1-mediated keratinocyte migra-tion are strikingly different. RHOA-mediated migra-tion appears to be associated with decreased cell–cell contact, whereas keratinocyte migration mediated by RAC1 appears to occur more as multi-cellular epider-mal sheets63. In the presence of !6"4 integrin ligation of laminin 332, RAC1 activation is associated with directional migration as seen in either galvanotaxis64 or time-lapse videomicroscopy assays65. Interestingly, RHOA-mediated migration is non-directional. This is inferred from the observation that the migration of
keratinocytes that lack "4 integrin–laminin 332 inter-action, either through antibody inhibition or through mutation of the "4 integrin LBS, shows a complete lack of directionality64. Interestingly, if constitutively active RAC1 cDNA is overexpressed in LBS mutant cells, direc-tionality of migration is restored64. Therefore, if !6"4 integrin is prevented from binding laminin 332, then RHOA is activated instead of RAC1 and migration is non-directional.
Laminin 332 produced by mammary tumours has been shown to promote cell migration through ligation with !6"4 integrin66. Integrin-driven RAC1 activation requires the Rac-specific guanine nucleotide exchange factor TIAM1 (T-lymphoma invasion and metastasis 1)67. A role for TIAM1 in carcinomas is suggested by the observation that TIAM1-deficient mice are resistant to skin tumours induced by activated HRAS68. It is known that RAC1 and TIAM1 activation significantly affects laminin 332 deposition, and this may occur through cofilin intermediate signalling65,69. Thus, the engage-ment of !6"4 integrin with laminin 332 may control the pattern of subsequent laminin 332 deposition through RAC1 activation.
Proteolytic processing of laminin 332Laminin 332 undergoes extracellular proteolytic process-ing of two of its three chains70. The #2 chain undergoes processing in domain III near the junction with domain IV71. The !3 chain undergoes processing in the G3 domain near the junction with G4. In addition, the !3 chain undergoes another proteolytic cleavage in domain IIIa near the junction with domain II72. In mature, nor-mal tissue, all laminin 332 molecules have a processed #2 chain, and a processed !3 G3 domain; however, only about half contain a processed domain IIIa on the !3 chain70. Therefore, in mature tissues laminin 332 performs its cohesive function in a processed state. By contrast, as mentioned below, unprocessed laminin 332 appears to have an important function in SCC tumorigenesis. Several matrix metalloproteinases (MMPs) have been studied for their effects on laminin 332 cleavage (BOX 3).
One of the effects of processing of laminin 332 is the stimulation of cell migration both in normal and carcinoma cells. It is known that domain III of the #2 chain can directly interact with the EGF receptor (EGFR), and it has been proposed that proteolytic
Box 2 | Interactions of laminins with other molecules
Laminin 332, through interaction of the G1–3 domain of its !3 chain with !6"4 integrin, localizes to hemidesmosomes48, which are condensations at the plasma membrane of basal epithelium72. Hemidesmosomes link with the epithelial cell intermediate filament network. Through these interactions, laminin 332 serves to act as a coupling molecule in the basement membrane zone (BMZ), joining cell surface integrins that bind to its C-terminal G domain, with laminin 311 and collagen VII in the extracellular BMZ, which interact with its N-terminal domains. These extracellular interactions are quite distinct from those of other laminins. For example, although the #1 chain in laminin 111 binds to the basement membrane glycoprotein nidogen with extremely high affinity91, the #2 chain of laminin 332 shows minimal interactions with nidogen. Laminin 111 and most other laminins have the ability to self polymerize, a function that requires a full length ! chain short arm92. The !3 chain is unable to perform this function; however, it contains an unpaired cysteine that has been proposed to act in the disulphide bonding of laminin 332 with laminin 311 (REF. 93). The interaction of the "3 chain with the non-collagenous (NC1) domain of collagen VII is another important BMZ assembly mechanism that is unique from other laminin chains94,95.
R E V I E W S
NATURE REVIEWS | CANCER VOLUME 7 | MAY 2007 | 373
© 2007 Nature Publishing Group
Laminin 332 Laminin 332
Keratin
Nondirectional migration
Focal adhesion
Plectin
CD151
Stable anchoring contact
Collagen XVII
Directional migration
RHOA
RAC1
SRCFAKTalin
Actin
!6 !3"4 "1
processing leads to interaction with EGFR, which in turn stimulates migration73. It should be pointed out that EGF signalling is a major stimulus for cell migration, and is required for either RHOA or RAC1 activation in cells attached to laminin 332. Another hypothesis is that the proteolytic modification of the #2 chain can alter the availability of integrin binding sites on the laminin 332 molecule. In addition, work from Patricia Rousselle and co-workers has shown that domain III on the #2 chain binds to !2"1 integrin74. Interestingly, work from the laboratory of Karl Tryggvasson has shown that anti-bodies to the #2 chain inhibit migration55. They were
able to narrow down the area of migration activity to domain III using an elegant transgenic mouse model and wound healing as an in vivo assessment of migra-tion. Another likely effect of laminin 332 processing on carcinoma progression might be in the way it modulates the deposition of laminin 332. It is known that domains IV and V of the laminin #2 chain promote laminin 332 deposition into matrix underlying cultured cells, and removal of this domain, as occurs during physiological processing, may serve to modulate this deposition75. In addition, it is known that the G4–5 domain, which is proteolytically removed from the !3 chain, also has an
Figure 2 | Laminin 332 promotes two separate integrin-mediated epithelial adhesion contacts. One complex known as the focal adhesion consists of !3"1 integrin, laminin 332 and supporting signalling molecules including talin, focal adhesion kinase (FAK) and SRC. Focal adhesions associate with actin microfilaments. Laminin 332 also localizes to another adhesion complex termed the stable anchoring contact (SAC). This structure contains !6"4 integrin, collagen XVII and plectin, and is associated with keratin intermediate filaments. Plectin is a large intracellular molecule that has well-defined binding regions for the cytoskeletal proteins keratin and F-actin. Focal adhesions transition into SACs through the changing molecular interactions of the bridging molecules CD151 and plectin. Plectin is able to bind keratin without competing with other molecules, but two molecules compete for the binding site on the actin binding domain of plectin, F-actin and the endodomain of "4 integrin. Therefore, the important intracellular transition from focal adhesion to SAC occurs when "4 integrin displaces F-actin for this binding spot on plectin102,103. CD151, a member of the tetraspanin group of transmembrane proteins is another molecule that helps to facilitate the focal adhesion–SAC transition104. CD151 has the ability to interact with several different integrin ! subunits, including !3, !6, and !7 (REF. 105). Therefore, the shift from CD151 interacting with !3"1 integrin to interacting with !6"4 seems to also have a key role in the transition of focal adhesions into SACs. The engagement of !3"1 integrin with laminin 332 promotes RHOA GTPase activation, which leads to a non-directional mode of cell migration. The binding of !6"4 integrin to laminin 332 causes a switch to RAC1 GTPase activation and a directional mode of cell migration.
R E V I E W S
374 | MAY 2007 | VOLUME 7 www.nature.com/reviews/cancer
© 2007 Nature Publishing Group

Recessive dystrophic epidermolysis bullosaAn inherited bullous disorder caused by COL7A1 gene mutations, and a deficiency of collagen VII.
Astacin familyA group of zinc endopeptidases named after the digestive enzyme astacin isolated from the stomach-like cardia of the freshwater crayfish Astacis astacus.
important role in laminin 332 deposition76. It remains to be shown whether the G4–5 domain or domains IV–V of the #2 chain exert their primary effects before or after removal from the rest of the laminin 332 molecule. The level of laminin 332 expression in SCC tumours has been shown to correlate with invasiveness and patient prognosis, and the modulation of laminin 332 deposition through proteolytic processing may be an important factor in this process. Therefore, laminin-332-processing enzymes, as well as laminin 332 itself, represent potential candidates for antibody-mediated cancer therapies.
A model to study laminin 332 in SCC tumoursFurther information on the role of laminin 332 in tumorigenesis has been obtained using an animal model of human SCC in which activated HRAS (Gly12Val mutant, hereafter referred to as HRAS-V12) and I$B! are overexpressed in primary keratinocytes, resulting in transformation, shown schematically in FIG. 3. Abnormal HRAS or KRAS expression has been associated with many carcinomas, including SCC, and in this model, primary human keratinocytes trans-formed through the retrovirally-mediated expression of the cell-cycle-control molecules HRAS-V12 and I$B! gave rise to tumours that were histologically and biochemically indistinguishable from human SCC77. Tumorigenesis in this model was completely inhibited with blocking antibodies to either laminin 332 or "4 integrin77. Similarly, keratinocytes from patients with JEB that did not express laminin 332 or "4 integrin (owing to either LAMB3 or ITGB4 null mutations)
showed a complete lack of tumorigenicity in immu-nodeficient mice after transformation. Interestingly, retrovirally-mediated expression of either laminin "3 or integrin "4 chain expression in these cells restored tumorigenesis completely77. These results confirmed the clinical observations noted above suggesting a correlation between the expression of laminin 332 and "4 integrin, tumour invasion and the prognosis of patients with SCC.
These studies did not address which domains or functions of laminin 332 have a role in SCC tumori-genesis. However, this was recently addressed through a structure–function approach. In one series of experi-ments, laminin "3 mutants that contained deletions of domains VI or III–V were retrovirally expressed in laminin "3-null JEB keratinocytes78. JEB keratinocytes that expressed these deletions showed normal levels of laminin 332 expression, assembly, secretion and depo-sition into extracellular matrix, and showed levels of spreading and migration comparable with wild-type cells. However, keratinocytes that lacked either lam-inin "3 domain VI or domains III–V and VI showed abnormal adhesion, as demonstrated by increased sen-sitivity to trypsin-induced detachment, attributable to the absence of laminin "3 domain VI. In addition, each of these laminin 332 mutants supported !6"4- and !3"1-dependent keratinocyte attachment equivalent to wild-type laminin 332, suggesting that the presence or absence of the "3 chain short arm did not influence integrin binding. Despite the poor stable adhesion, cells that expressed laminin "3 domain VI deletions formed tumours after HRAS-V12 and I$B! transformation78. These studies brought to light the idea that laminin 332 could serve a signalling, rather than a purely adhesive role, in tumorigenesis. Although poorly adherent cells that expressed the laminin "3 mutant lacking domain VI were able to produce tumours, cells that expressed laminin "3 lacking domains III–V and VI had similar adhesion properties but no tumour-forming abilities in the SCC model. The difference was explained by the observation that laminin 332 lacking the "3 domain VI was able to bind collagen VII normally, whereas laminin 332 lacking "3 domain III–V was unable to bind collagen VII, either in solution or in solid phase assays78,79. These studies suggested that the binding of collagen VII to laminin 332 through domain III–V was essential for SCC tumorigenesis.
Collagen VII and laminin 332 in cancerA link between collagen VII and SCC has been appre-ciated for some time. It was originally believed that a complete lack of collagen VII was a predisposing factor towards the development of SCC. This was due to the observation that individuals with the genetic skin condition recessive dystrophic epidermolysis bul-losa (RDEB) suffered from a high frequency of severe invasive SCC, with this cancer actually being the main cause of mortality in these patients after puberty80,81. Although collagen VII expression was thought to be absent in these individuals, Western blot analysis of patient skin cells in culture showed low but detectable
Box 3 | Laminin-332-processing enzymes
Matrix metalloproteinase 2 (MMP2) and MMP14 (also known as membrane metalloproteinase type I) have been shown to cleave the laminin 332 #2 chain at the junction between domain II and domain III56,96. However, these studies were performed with rat laminin 332, and follow-up studies with each of these enzymes on human laminin 332 failed to show cleavage of the #2 chain even at high enzyme:substrate ratios87. This is because there is a complete lack of sequence homology between the rat and human cleavage sites on the laminin #2 chain. Instead, enzymes of the astacin family of MMPs were shown to specifically cleave the human laminin #2 chain near the junction between domains III and IV71,87. One member of this family, bone morphogenic protein 1 (BMP1) is capable of this cleavage event71, but a related enzyme, mammalian tolloid (mTLD), is the predominant astacin expressed in skin and squamous cell carcinoma (SCC) cells87, and thus is the enzyme most probably responsible for cleavage of the laminin #2 chain in normal and malignant human epithelium. Cleavage of the #2 chain can be abrogated by the removal of the astacin consensus cleavage site on the #2 chain or through the use of specific astacin enzyme inhibitors87. A site on the !3 IIIa domain also contains an astacin consensus site and astacin inhibition inhibits cleavage of this domain as well.
Although the #2 chain cleavage specificity appears to be restricted to astacin enzymes, cleavage of the !3 chain G3 domain seems to occur through multiple enzymes. BMP1, mTLD, MMP2, MMP13 and plasmin have all been shown to efficiently cleave the G3 domain87,97. Attempts at abrogating cleavage of the G3 domain by single site-directed mutagenesis of !3 cDNA have been unsuccessful, and this cleavage event has only been disrupted by complete substitution of the entire bridge region between the G3 and G4 domains with the corresponding segment of the laminin !1 chain (which does not undergo proteolysis) 76. Therefore, the cleavage of the laminin !3 G domain appears to proceed through several redundant mechanisms and enzymes.
R E V I E W S
NATURE REVIEWS | CANCER VOLUME 7 | MAY 2007 | 375
© 2007 Nature Publishing Group

Retroviral transfer of HRAS-VI2 and I$B!
Retroviral transfer of engineered laminin 332 or collagen VII cDNA
Cutaneous or subcutaneoustransfer to immunodeficient mice
Laminin 332 or collagen VIIdeficient humanEB keratinocytes
EB keratinocytes expressing engineered laminin 332 or collagen VII
Transformed keratinocytes expressing engineered laminin 332 or collagen VII
Establishment of tumours expressing engineered laminin 332 or collagen VII
Tumour progression and invasion
levels of expression of the collagen VII non-collag-enous (NC1) domain, which appeared to be a stable degradation product of the collagen VII molecule82. Indeed, a very clear correlation was noted between the persistence of collagen VII NC1 expression in a given patient’s skin cells, and the tumorigenic potential after HRAS-V12 transformation. In a small study of 12 patients, 3 did not have detectable NC1 expression, and their keratinocytes were not tumorigenic when transformed by HRAS-V12 and I$B! in the SCC model, but the keratinocytes from the 9 patients with NC1 expression produced tumours in this system82.
The NC1 domain itself was shown to be required for SCC tumour formation, as expression of this protein fragment in NC1-null RDEB cells restored tumour-genic potential. Even in trans, when the NC1 domain was expressed in fibroblasts that were co-injected into immunodeficient mice with collagen VII null, HRAS-V12-transformed keratinocytes, the NC1 domain was still able to rescue tumorigenesis82.
Therefore the collagen VII NC1 domain and the col-lagen VII NC1 domain binding site on the laminin 332 molecule both appear to be required for SCC tumori-genesis. Thus, a laminin 332 molecule that supports col-lagen VII binding, rather than one that supports stable adhesion, seems to be required for HRAS-V12-driven SCC tumorigenesis. This notion that the function of laminin 332 in SCC tumorigenesis is not directly linked to its well known function in supporting adhesion is surprising, but is consistent with other observations that do not support a clear association between matrix adhesion and tumorigenicity. For example, collagen VII null cells show normal stable adhesion in vitro, but show a complete lack of tumorigenicity in vivo82. Although expression of the collagen VII NC1 domain is sufficient to induce tumorigenesis, it is clear that this molecular fragment does not increase epithelial adhesion. Either in the case of the laminin "3 domain VI mutation shown above, or in the case of RDEB-associated SCC in collagen VII NC1-expressing patients, there is a persistence of tumorigenesis in the face of defective adhesive function of the laminin 332–collagen VII complex.
Laminin 332 associated signalling pathwaysThe evidence to date suggests that laminin-332-derived signalling is an important component of SCC tumori-genesis. After Ras transformation, cells that lacked col-lagen VII–laminin 332 interaction, either through the deletion of collagen VII or the collagen VII binding site on laminin 332 ("3 domain III–V), also lacked activa-tion of the phosphatidylinositol-3 kinase (PI3K) path-way78. The constitutive activation of the PI3K pathway in these cells, using an activated PI3K p110 subunit, restored tumorigenesis in the absence of collagen VII–laminin 332 binding78. These rescue studies suggested that HRAS-V12 transformation and downregulation of the nuclear factor $B (NF$B) pathway by I$B! is not by itself sufficient to promote tumorigenesis in the SCC model, but that additional input from the BMZ is essential. Furthermore, this contribution seems to arise specifically from laminin 332 and its association with collagen VII, and results in the activation of the PI3K signalling pathway. Interestingly, as mentioned before, the presence or absence of the collagen VII NC1 bind-ing site on laminin 332 did not affect the availability of !6"4 or !3"1 integrin binding sites on laminin 332. These observations suggest that laminin 332–collagen VII interaction may instead affect integrin-associated receptors that complement "4 integrin signalling. Associated cell surface receptors that are known to synergize or associate with "4 integrin include EGFR, the EGFR family member ERBB2 (REF. 83) or MET (the receptor for hepatocyte growth factor (HGF))84.
Figure 3 | A model of human squamous cell carcinoma tumorigenesis. In this model, primary human keratinocytes are isolated from skin biopsies of patients with epidermolysis bullosa (EB) who have genetic null mutations of genes coding for one of the laminin 332 chains, or the collagen VII !1 chain. Primary EB keratinocytes are then infected with retroviruses carrying either wild-type or mutant laminin 332 or collagen VII cDNA, resulting in cells expressing wild-type or recombinantly engineered laminin 332 or collagen VII molecules. Next, the cells are transformed with HRAS-V12 and I$B! and transferred, either through skin grafting or subcutaneous injection, to immunodeficient mice. Subsequent tumour growth and invasion can be analysed.
R E V I E W S
376 | MAY 2007 | VOLUME 7 www.nature.com/reviews/cancer
© 2007 Nature Publishing Group
Laminin 332 ligation
Laminin 332 deposition
Survival Migration and invasionPI3K RAC1
EGFR
FYNFYNP
T T T
FA to SACtransition
Collagen VII
Laminin 332NC1
1 2 3 4 5
Syndecan 1
!6 "4
3
3
V
VVI
III IVIII
IIIa
2
Another non-integrin cell surface receptor that has been shown to interact with laminin 332 is syndecan 1, a heparan sulphate proteoglycan85. The !3 chain of laminin 332 contains heparin-binding residues on its G4 domain that mediate this interaction. This domain and the G5 domain are proteolytically removed from the laminin 332 molecule by extracellular processing as described above86. In vitro studies have shown that at least one function of the laminin !3 G4–5 domain is to promote the deposition of laminin 332 into the extracellular matrix76. It is tempting to speculate that syndecan 1 might have a role in this process, as it is known to specficially bind to the laminin 332 !3 G4–5 domain74.
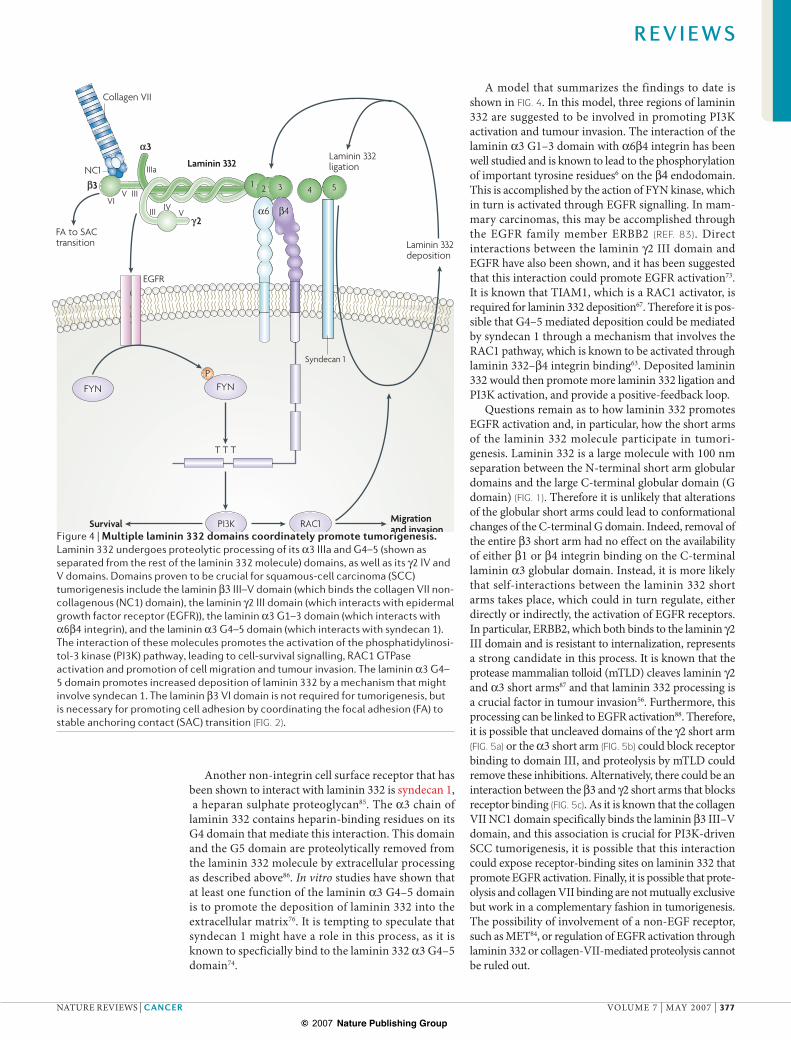
A model that summarizes the findings to date is shown in FIG. 4. In this model, three regions of laminin 332 are suggested to be involved in promoting PI3K activation and tumour invasion. The interaction of the laminin !3 G1–3 domain with !6"4 integrin has been well studied and is known to lead to the phosphorylation of important tyrosine residues6 on the "4 endodomain. This is accomplished by the action of FYN kinase, which in turn is activated through EGFR signalling. In mam-mary carcinomas, this may be accomplished through the EGFR family member ERBB2 (REF. 83). Direct interactions between the laminin #2 III domain and EGFR have also been shown, and it has been suggested that this interaction could promote EGFR activation73. It is known that TIAM1, which is a RAC1 activator, is required for laminin 332 deposition67. Therefore it is pos-sible that G4–5 mediated deposition could be mediated by syndecan 1 through a mechanism that involves the RAC1 pathway, which is known to be activated through laminin 332–"4 integrin binding63. Deposited laminin 332 would then promote more laminin 332 ligation and PI3K activation, and provide a positive-feedback loop.
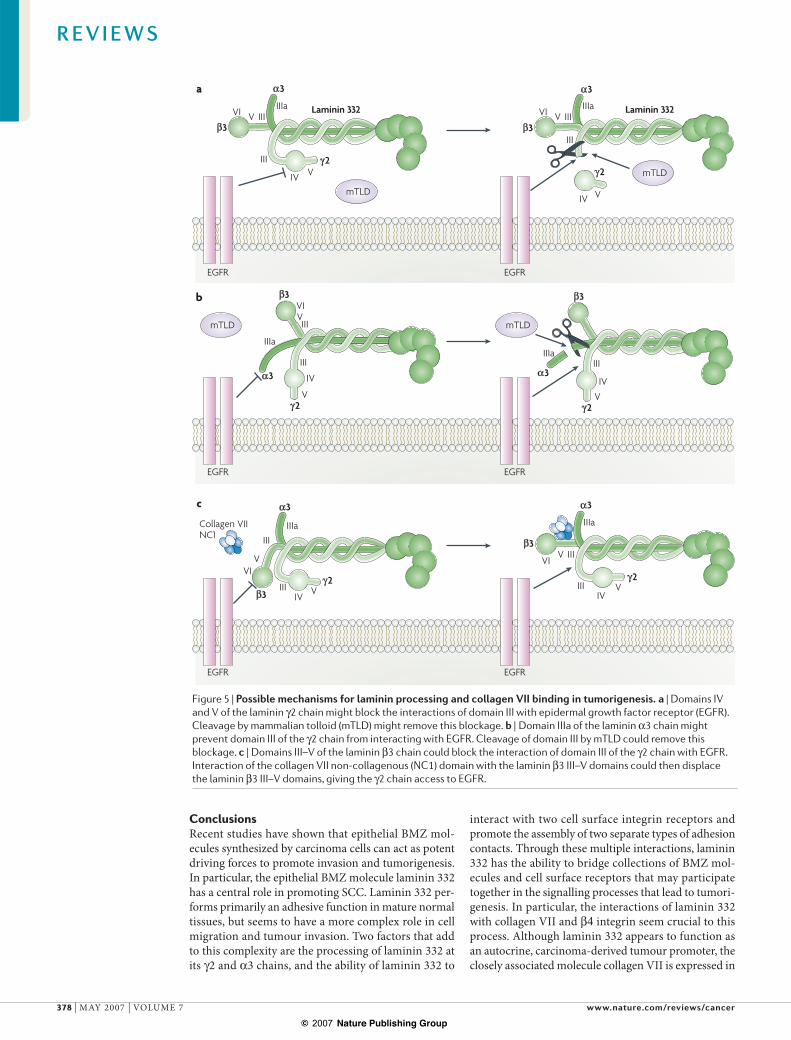
Questions remain as to how laminin 332 promotes EGFR activation and, in particular, how the short arms of the laminin 332 molecule participate in tumori-genesis. Laminin 332 is a large molecule with 100 nm separation between the N-terminal short arm globular domains and the large C-terminal globular domain (G domain) (FIG. 1). Therefore it is unlikely that alterations of the globular short arms could lead to conformational changes of the C-terminal G domain. Indeed, removal of the entire "3 short arm had no effect on the availability of either "1 or "4 integrin binding on the C-terminal laminin !3 globular domain. Instead, it is more likely that self-interactions between the laminin 332 short arms takes place, which could in turn regulate, either directly or indirectly, the activation of EGFR receptors. In particular, ERBB2, which both binds to the laminin #2 III domain and is resistant to internalization, represents a strong candidate in this process. It is known that the protease mammalian tolloid (mTLD) cleaves laminin #2 and !3 short arms87 and that laminin 332 processing is a crucial factor in tumour invasion56. Furthermore, this processing can be linked to EGFR activation88. Therefore, it is possible that uncleaved domains of the #2 short arm (FIG. 5a) or the !3 short arm (FIG. 5b) could block receptor binding to domain III, and proteolysis by mTLD could remove these inhibitions. Alternatively, there could be an interaction between the "3 and #2 short arms that blocks receptor binding (FIG. 5c). As it is known that the collagen VII NC1 domain specifically binds the laminin "3 III–V domain, and this association is crucial for PI3K-driven SCC tumorigenesis, it is possible that this interaction could expose receptor-binding sites on laminin 332 that promote EGFR activation. Finally, it is possible that prote-olysis and collagen VII binding are not mutually exclusive but work in a complementary fashion in tumorigenesis. The possibility of involvement of a non-EGF receptor, such as MET84, or regulation of EGFR activation through laminin 332 or collagen-VII-mediated proteolysis cannot be ruled out.
Figure 4 | Multiple laminin 332 domains coordinately promote tumorigenesis. Laminin 332 undergoes proteolytic processing of its !3 IIIa and G4–5 (shown as separated from the rest of the laminin 332 molecule) domains, as well as its #2 IV and V domains. Domains proven to be crucial for squamous-cell carcinoma (SCC) tumorigenesis include the laminin "3 III–V domain (which binds the collagen VII non-collagenous (NC1) domain), the laminin #2 III domain (which interacts with epidermal growth factor receptor (EGFR)), the laminin !3 G1–3 domain (which interacts with !6"4 integrin), and the laminin !3 G4–5 domain (which interacts with syndecan 1). The interaction of these molecules promotes the activation of the phosphatidylinosi-tol-3 kinase (PI3K) pathway, leading to cell-survival signalling, RAC1 GTPase activation and promotion of cell migration and tumour invasion. The laminin !3 G4–5 domain promotes increased deposition of laminin 332 by a mechanism that might involve syndecan 1. The laminin "3 VI domain is not required for tumorigenesis, but is necessary for promoting cell adhesion by coordinating the focal adhesion (FA) to stable anchoring contact (SAC) transition (FIG. 2).
R E V I E W S
NATURE REVIEWS | CANCER VOLUME 7 | MAY 2007 | 377
© 2007 Nature Publishing Group
mTLD
EGFR
VI VIIIa
IIIa
IIIa
III
III
III
III
III
III
IV
IV
IV
V
V
V
VI
VI
V
V
IV
IV
IV
V
V
V
mTLD
mTLDmTLD
VI VIIIa
IIIa
III
III
EGFR EGFR
EGFR
EGFR
IIILaminin 332 Laminin 332
3
3
3
3
2
2
2
3
3
3
3
VIV
IIIa
III
III
33
22
2
3
3
a
b
c
EGFR
Collagen VII NC1
ConclusionsRecent studies have shown that epithelial BMZ mol-ecules synthesized by carcinoma cells can act as potent driving forces to promote invasion and tumorigenesis. In particular, the epithelial BMZ molecule laminin 332 has a central role in promoting SCC. Laminin 332 per-forms primarily an adhesive function in mature normal tissues, but seems to have a more complex role in cell migration and tumour invasion. Two factors that add to this complexity are the processing of laminin 332 at its #2 and !3 chains, and the ability of laminin 332 to
interact with two cell surface integrin receptors and promote the assembly of two separate types of adhesion contacts. Through these multiple interactions, laminin 332 has the ability to bridge collections of BMZ mol-ecules and cell surface receptors that may participate together in the signalling processes that lead to tumori-genesis. In particular, the interactions of laminin 332 with collagen VII and "4 integrin seem crucial to this process. Although laminin 332 appears to function as an autocrine, carcinoma-derived tumour promoter, the closely associated molecule collagen VII is expressed in
Figure 5 | Possible mechanisms for laminin processing and collagen VII binding in tumorigenesis. a | Domains IV and V of the laminin #2 chain might block the interactions of domain III with epidermal growth factor receptor (EGFR). Cleavage by mammalian tolloid (mTLD) might remove this blockage. b | Domain IIIa of the laminin !3 chain might prevent domain III of the #2 chain from interacting with EGFR. Cleavage of domain III by mTLD could remove this blockage. c | Domains III–V of the laminin "3 chain could block the interaction of domain III of the #2 chain with EGFR. Interaction of the collagen VII non-collagenous (NC1) domain with the laminin "3 III–V domains could then displace the laminin "3 III–V domains, giving the #2 chain access to EGFR.
R E V I E W S
378 | MAY 2007 | VOLUME 7 www.nature.com/reviews/cancer
© 2007 Nature Publishing Group

both epithelial and mesenchymal cells89. In a similar manner, the processing enzyme for laminin 332, mTLD, is also derived both from epithelial and mes-enchymal sources87. These observations suggest that the surrounding host mesenchyme may also have a key role in cooperating with tumour cells in promot-ing their tumorigenesis, through the production of these laminin-332-associated proteins.
As these laminin-associated interactions take place extracellularly, they may be particularly amenable to antibody intervention as a mode of cancer therapy, with the potential for synergy with currently available therapies such as EGFR antibody treatment. Inhibitory therapy for the laminin !3 G4–5 domain could offer theoretical advantages in that this domain, although not required for maintenance of epithelial–mesenchymal cohesion and not present in normal tissues, nonetheless has the ability to control laminin 332 deposition.
Therefore, antibodies to this domain would not affect processed laminin 332 in normal mature tissues, but rather would target newly synthesized laminin 332, and it is known that ongoing laminin 332 expression occurs in SCC tumours. Therefore, the potential exists to exploit this observation in the production of an anti-tumour therapy that does not cause the side effect of blistering in normal tissues. Identification of other domains in laminin 332 or collagen VII, which selectively affect SCC progres-sion and not normal tissue adhesion, would be an important goal in designing future cancer therapies. In addition, as processing of the laminin #2 chain appears to be a crucial factor in carcinoma progres-sion, the targeting of astacin-like laminin-processing enzymes or of domains IV–V of the #2 chain, which are removed by processing, may also be a promis-ing avenue of research in the development of future anticancer therapies.
1. Miller, D. L. & Weinstock, M. A. Nonmelanoma skin cancer in the United States: incidence. J. Am. Acad. Dermatol. 30, 774–778 (1994).
2. Czarnecki, D., Staples, M., Mar, A., Giles, G. & Meehan, C. Metastases from squamous cell carcinoma of the skin in southern Australia. Dermatology 189, 52–54 (1994).
3. McGowan, K. A. & Marinkovich, M. P. Laminins and human disease. Microsc. Res. Tech. 51, 262–279 (2000).
4. Keene, D. R., Marinkovich, M. P. & Sakai, L. Y. Immunodissection of the connective tissue matrix in human skin. Microsc. Res. Tech. 38, 394–406 (1997).
5. Beauvais, D. M. & Rapraeger, A. C. Syndecans in tumor cell adhesion and signaling. Reprod. Biol. Endocrinol. 2, 3 (2004).
6. Guo, W. & Giancotti, F. G. Integrin signalling during tumour progression. Nature Rev. Mol. Cell Biol. 5, 816–826 (2004).
7. Hornebeck, W. & Maquart, F. X. Proteolyzed matrix as a template for the regulation of tumor progression. Biomed. Pharmacother. 57, 223–230 (2003).
8. Mareel, M. & Leroy, A. Clinical, cellular, and molecular aspects of cancer invasion. Physiol. Rev. 83, 337–376 (2003).
9. Liotta, L. A., Rao, C. N. & Wewer, U. M. Biochemical interactions of tumor cells with basement membrane. Ann. Rev. Biochem. 55, 1037–1057 (1986).
10. Liotta, L. A. et al. Metastatic potential correlates with enzymatic degradation of basement membrane collagen. Nature 284, 67–68 (1980).
11. Sasaki, T., Fassler, R. & Hohenester, E. Laminin: the crux of basement membrane assembly. J. Cell Biol. 164, 959–963 (2004).
12. Ryan, M. C. et al. The functions of laminins: lessons from in vivo studies. Matrix Biol. 15, 369–381 (1996).
13. Marinkovich, M. P. et al. The basement membrane proteins kalinin and nicein are structurally and immunologically identical. Lab. Invest. 69, 295–299 (1993).
14. Meneguzzi, G. et al. Kalinin is abnormally expressed in epithelial basement membranes of Herlitz’s junctional epidermolysis bullosa patients. Exp. Dermatol. 1, 221–229 (1992).
15. Pulkkinen, L. et al. A homozygous nonsense mutation in the beta 3 chain gene of laminin 5 (LAMB3) in Herlitz junctional epidermolysis bullosa. Genomics 24, 357–360 (1994).
16. Aberdam, D. et al. Herlitz’s junctional epidermolysis bullosa is linked to mutations in the gene (LAMC2) for the gamma 2 subunit of nicein/kalinin (LAMININ-5). Nature Genet. 6, 299–304 (1994).
17. Martin, G. R. & Timpl, R. Laminin and other basement membrane components. Annu. Rev. Cell Biol. 3, 57–85 (1987).
18. Kallunki, P. et al. A truncated laminin chain homologous to the B2 chain: structure, spacial
expression, and chromosomal assignment. J. Cell Biol. 119, 679–694 (1992).Identification of laminin 332 as a member of the laminin family.
19. Mizushima, H. et al. Differential expression of laminin-5/ladsin subunits in human tissues and cancer cell lines and their induction by tumor promoter and growth factors. J. Biochem. (Tokyo) 120, 1196–1202 (1996).
20. Pyke, C. et al. Laminin-5 is a marker of invading cancer cells in some human carcinomas and is coexpressed with the receptor for urokinase plasminogen activator in budding cancer cells in colon adenocarcinomas. Cancer Res. 55, 4132–4139 (1995).Early extensive survey of laminin 332 in human carcinomas.
21. Tani, T. et al. Pancreatic carcinomas deposit laminin-5, preferably adhere to laminin-5, and migrate on the newly deposited basement membrane. Am. J. Pathol. 151, 1289–1302 (1997).
22. Berndt, A., Hyckel, P., Könneker, A., Katenkamp, D. & Kosmehl, H. Oral squamous cell carcinoma invasion is associated with a laminin-5 matrix re-organization but independent of basement membrane and hemidesmosome formation. Clues from an in vitro invasion model. Invasion Metastasis 17, 251–258 (1997).
23. Orian-Rousseau, V. et al. Human colonic cancer cells synthesize and adhere to laminin-5. Their adhesion to laminin-5 involves multiple receptors among which is integrin alpha2beta1. J. Cell Sci. 111, 1993–2004 (1998).
24. Skyldberg, B. et al. Laminin-5 as a marker of invasiveness in cervical lesions. J. Natl Cancer Inst. 91, 1882–1887 (1999).
25. Ono, Y. et al. Clinocopathologic significance of laminin-5 #2 chain expression in squamous cell carcinoma of the tongue: immunohistochemical analysis of 67 lesions. Cancer 85, 2315–2321 (1999).
26. Lohi, J. et al. Basement membrane laminin-5 is deposited in colorectal adenomas and carcinomas and serves as a ligand for !3"1 integrin. APMIS 108, 161–172 (2000).
27. Nordemar, S. et al. Laminin-5 as a predictor of invasiveness in cancer in situ lesions of the larynx. Anticancer Res. 21, 509–512 (2001).
28. Lenander, C. et al. Laminin-5 gamma 2 chain expression correlates with unfavorable prognosis in colon carcinomas. Anal. Cell. Pathol. 22, 201–209 (2001).
29. Calaluce, R. et al. Laminin-5-mediated gene expression in human prostate carcinoma cells. Mol. Carcinog. 30, 119–129 (2001).
30. Nordemar, S., Hogmo, A., Lindholm, J., Auer, G. & Munck-Wikland, E. Laminin-5 gamma 2: a marker to identify oral mucosal lesions at risk for tumor development? Anticancer Res. 23, 4985–4989 (2003).
31. Giannelli, G., Fransvea, E., Bergamini, C., Marinosci, F. & Antonaci, S. Laminin-5 chains are expressed differentially in metastatic and nonmetastatic hepatocellular carcinoma. Clin. Cancer Res. 9, 3684–3691 (2003).
32. Yamamoto, H., Itoh, F., Iku, S., Hosokawa, M. & Imai, K. Expression of the # (2) chain of laminin-5 at the invasive front is associated with recurrence and poor prognosis in human esophageal squamous cell carcinoma. Clin. Cancer Res. 7, 896–900 (2001).
33. Nordstrom, B. et al. Laminin-5 gamma 2 chain as an invasivity marker for uni- and multifocal lesions in the lower anogenital tract. Int. J. Gynecol. Cancer 12, 105–109 (2002).
34. Giannelli, G. & Antonaci, S. Biological and clinical relevance of Laminin-5 in cancer. Clin. Exp. Metastasis 18, 439–443 (2000).
35. Katayama, M., Sanzen, N., Funakoshi, A. & Sekiguchi, K. Laminin #2-chain fragment in the circulation: a prognostic indicator of epithelial tumor invasion. Cancer Res. 63, 222–229 (2003).
36. Shinto, E. et al. Prognostic implication of laminin-5 gamma 2 chain expression in the invasive front of colorectal cancers, disclosed by area-specific four-point tissue microarrays. Lab. Invest. 85, 257–266 (2005).
37. Savoia, P., Trusolino, L., Pepino, E. & Marchisio, P. C. Expression and topography of integrins and basement membrane proteins in epidermal carcinomas: basal but not squamous cell carcinomas display loss of alpha 6 beta 4 and BM-600/nicein. J. Invest. Dermatol. 101, 352–358 (1993).
38. Martin, K. J. et al. Down-regulation of laminin-5 in breast carcinoma cells. Mol. Med. 4, 602–613 (1998).
39. Holler, E. Laminin isoform expression in breast tumors. Breast Cancer Res. 7, 166–167 (2005).
40. Hao, J. et al. Investigation into the mechanism of the loss of laminin 5 (!3"3#2) expression in prostate cancer. Am. J. Pathol. 158, 1129–135. (2001).
41. Janes, S. M. & Watt, F. M. New roles for integrins in squamous-cell carcinoma. Nature Rev. Cancer 3, 175–183 (2006).
42. Li, J. et al. Laminin-10 is crucial for hair morphogenesis. EMBO J. 22, 2400–2410 (2003).
43. Fan, H. & Khavari, P. A. Sonic hedgehog opposes epithelial cell cycle arrest. J. Cell Biol. 147, 71–76 (1999).
44. Shaw, L. M. Tumor cell invasion assays. Methods Mol. Biol. 294, 97–105 (2005).
45. Malinda, K. M. In vivo matrigel migration and angiogenesis assays. Methods Mol. Med. 78, 329–335 (2003).
46. Mullen, P. The use of Matrigel to facilitate the establishment of human cancer cell lines as xenografts. Methods Mol. Med. 88, 287–292 (2004).
47. Ekblom, P., Lonai, P. & Talts, J. F. Expression and biological role of laminin-1. Matrix Biol. 22, 35–47 (2003).
R E V I E W S
NATURE REVIEWS | CANCER VOLUME 7 | MAY 2007 | 379
© 2007 Nature Publishing Group

48. Rousselle, P., Lunstrum, G. P., Keene, D. R. & Burgeson, R. E. Kalinin: an epithelium-specific basement membrane adhesion molecule that is a component of anchoring filaments. J. Cell Biol. 114, 567–576 (1991).Characterization of the role of laminin 332 in epidermal adhesion.
49. Carter, W. G., Ryan, M. C. & Gahr, P. J. Epiligrin, a new cell adhesion ligand for integrin-3–1 in epithelial basement membranes. Cell 65, 559–610 (1991).Characterization of laminin 332 integrin specificity.
50. Carter, W. G., Kaur, P., Gil, S. G., Gahr, P. J. & Wayner, E. A. Distinct functions for integrins alpha 3 beta 1 in focal adhesions and alpha 6 beta 4/bullous pemphigoid antigen in a new stable anchoring contact (SAC) of keratinocytes: relation to hemidesmosomes. J. Cell Biol. 111, 3141–3154 (1990).
51. Hood, J. D. & Cheresh, D. A. Role of integrins in cell invasion and migration. Nature Rev. Cancer 2, 91–100 (2002).
52. Nievers, M. G., Schaapveld, R. Q. & Sonnenberg, A. Biology and function of hemidesmosomes. Matrix Biol. 18, 5–17 (1999).
53. Litjens, S. H., de Pereda, J. M. & Sonnenberg, A. Current insights into the formation and breakdown of hemidesmosomes. Trends Cell Biol. 16, 376–383 (2006).
54. Geuijen, C. A. & Sonnenberg, A. Dynamics of the !6"4 Integrin in keratinocytes. Mol. Biol. Cell 13, 3845–3858 (2002).
55. Salo, S. et al. Laminin-5 promotes adhesion and migration of epithelial cells: identification of a migration-related element in the #2 chain gene (LAMC2) with activity in transgenic mice. Matrix Biol. 18, 197–210 (1999).
56. Giannelli, G., Falk-Marzillier, J., Schiraldi, O., Stetler-Stevenson, W. G. & Quaranta, V. Induction of cell migration by matrix metalloprotease-2 cleavage of laminin-5. Science 277, 225–228 (1997).Shows the association between laminin 332 processing and carcinoma cell migration.
57. Zhang, K. & Kramer, R. H. Laminin 5 deposition promotes keratinocyte motility. Exp. Cell Res. 227, 309–322 (1996).
58. Miyazaki, K., Kikkawa, Y., Nakamura, A., Yasumitsu, H. & Umeda, M. A large cell-adhesive scatter factor secreted by human gastric carcinoma cells. Proc. Natl Acad. Sci. USA 90, 11767–1171 (1993).Demonstration of the role of laminin 332 in tumour-cell migration.
59. Kariya, Y. & Miyazaki, K. The basement membrane protein laminin-5 acts as a soluble cell motility factor. Exp. Cell Res. 297, 508–520 (2004).
60. Nguyen, B. P., Gil, S. G. & Carter, W. G. Deposition of laminin 5 by keratinocytes regulates integrin adhesion and signaling. J. Biol. Chem. 275, 31896–31907 (2000).
61. Nguyen, B. P., Ren, X. D., Schwartz, M. A. & Carter, W. G. Ligation of integrin !3"1 by laminin 5 at the wound edge activates Rho-dependent adhesion of leading keratinocytes on collagen. J. Biol. Chem. 276, 43860–43870 (2001).
62. Zhou, H. & Kramer, R. H. Integrin engagement differentially modulates epithelial cell motility by RhoA/ROCK and PAK1. J. Biol. Chem. 280, 10624–10635 (2005).
63. Russell, A. J. et al. Alpha 6 beta 4 integrin regulates keratinocyte chemotaxis through differential GTPase activation and antagonism of alpha 3 beta 1 integrin. J. Cell Sci. 116, 3543–3556 (2003).
64. Pullar, C. E. et al. "4 integrin and epidermal growth factor coordinately regulate electric field-mediated directional migration via Rac1. Mol. Biol. Cell 17, 4925–4935 (2006).
65. Sehgal, B. U. et al. Integrin "4 regulates migratory behavior of keratinocytes by determining laminin-332 organization. J. Biol. Chem. 46, 35487–35498 (2006).
66. Zahir, N. et al. Autocrine laminin-5 ligates alpha6beta4 integrin and activates RAC and NF$B to mediate anchorage-independent survival of mammary tumors. J. Cell Biol. 163, 1397–1407 (2003).
67. Hamelers, I. H. et al. The Rac activator Tiam1 is required for !3"1-mediated laminin-5 deposition, cell spreading, and cell migration. J. Cell Biol. 171, 871–881 (2005).
68. Malliri, A. et al. Mice deficient in the Rac activator Tiam1 are resistant to Ras-induced skin tumours. Nature 417, 867–871 (2002).
69. Danen, E. H. et al. Integrins control motile strategy through a Rho-cofilin pathway. J. Cell Biol. 169, 515–526 (2005).
70. Marinkovich, M. P., Lunstrum, G. P. & Burgeson, R. E. The anchoring filament protein kalinin is synthesized and secreted as a high molecular weight precursor. J. Biol. Chem. 267, 17900–17906 (1992).Characterization of the proteolytic processing of laminin 332.
71. Amano, S. et al. Bone morphogenetic protein 1 is an extracellular processing enzyme of the laminin 5 # 2 chain. J. Biol. Chem. 275, 22728–22735 (2000).
72. Gerecke, D. R., Gordon, M. K., Wagman, W. W., Champliaud, M. F. & Burgeson, R. E. in Extracellular matrix assembly and structure (eds Mecham, R. P., Birk, D. E. & Yurchenko, P. D.) 417–439 (Academic Press, San Diego, USA, 1994).
73. Schenk, S. et al. Binding to EGF receptor of a laminin-5 EGF-like fragment liberated during MMP-dependent mammary gland involution. J. Cell Biol. 161, 197–209 (2003).
74. Decline, F. & Rousselle, P. Keratinocyte migration requires alpha2beta1 integrin-mediated interaction with the laminin 5 #2 chain. J. Cell Sci. 114, 811–823 (2001).
75. Gagnoux-Palacios, L. et al. The short arm of the laminin #2 chain plays a pivotal role in the incorporation of laminin 5 into the extracellular matrix and in cell adhesion. J. Cell Biol. 153, 835–850. (2001).
76. Sigle, R. O. et al. Globular domains 4/5 of the laminin !3 chain mediate deposition of precursor laminin 5. J. Cell Sci. 117, 4481–4494 (2004).
77. Dajee, M. et al. NF$B blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature 421, 639–643 (2003).Demonstration of the requirement for laminin 332 and !6"4 integrin in human SCC tumours.
78. Waterman, E. A. et al. A laminin-collagen complex drives human epidermal carcinogenesis through phosphoinositol-3 kinase activation. Cancer Res. (in the press 2007).
79. Nakashima, Y., Kariya, Y., Yasuda, C. & Miyazaki, K. Regulation of cell adhesion and type VII collagen binding by the "3 chain short arm of laminin-5: effect of its proteolytic cleavage. J. Biochem. (Tokyo) 138, 539–552 (2005).
80. McGrath, J. A., Schofield, O. M., Mayou, B. J., McKee, P. H. & Eady, R. A. Epidermolysis bullosa complicated by squamous cell carcinoma: report of 10 cases. J. Cut. Pathol. 19, 116–123 (1992).
81. Newman, C., Wagner, R. F., Jr., Tyring, S. K. & Spigel, G. T. Squamous cell carcinoma secondary to recessive dystrophic epidermolysis bullosa. A report of 4 patients with 17 primary cutaneous alignancies. J. Dermatol. Surg. Oncol. 18, 301–305 (1992).
82. Ortiz-Urda, S. et al. Type VII collagen is required for Ras-driven human epidermal tumorigenesis. Science 307, 1773–1776 (2005).Demonstration of collagen VII requirement in human SCC tumours.
83. Guo, W. et al. "4 integrin amplifies ErbB2 signaling to promote mammary tumorigenesis. Cell 126, 489–502 (2006).
84. Trusolino, L., Bertotti, A. & Comoglio, P. M. A signaling adapter function for !6"4 integrin in the control of HGF-dependent invasive growth. Cell 5, 643–654 (2001).
85. Okamoto, O. et al. Normal human keratinocytes bind to the !3LG4/5 domain of unprocessed laminin-5 through the receptor syndecan-1. J. Biol. Chem. 278, 44168–44177 (2003).
86. Tsubota, Y. et al. Isolation and activity of proteolytic fragment of laminin-5 !3 chain. Biochem. Biophys. Res. Commun. 278, 614–620 (2000).
87. Veitch, D. P. et al. Mammalian tolloid metalloproteinase, and not matrix metalloprotease 2 or membrane type 1 metalloprotease, processes laminin-5 in keratinocytes and skin. J. Biol. Chem. 278, 15661–15668 (2003).
88. Hintermann, E. & Quaranta, V. Epithelial cell motility on laminin-5: regulation by matrix assembly, proteolysis, integrins and erbB receptors. Matrix Biol. 23, 75–85 (2004).
89. Marinkovich, M. P., Keene, D. R., Rimberg, C. S. & Burgeson, R. E. Cellular origin of the dermal-epidermal basement membrane. Dev. Dyn. 197, 255–267 (1993).
90. Timpl, R. et al. Laminin-a glycoprotein from basement membranes. J. Biol. Chem. 254, 9933–9937 (1979).
91. Takagi, J., Yang, Y., Liu, J. H., Wang, J. H. & Springer, T. A. Complex between nidogen and laminin fragments reveals a paradigmatic "-propeller interface. Nature 424, 969–974 (2003).
92. Cheng, Y. S., Champliaud, M. F., Burgeson, R. E., Marinkovich, M. P. & Yurchenco, P. D. Self-assembly of laminin isoforms. J. Biol. Chem. 272, 31525–31532 (1997).
93. Champliaud, M. F. et al. Human amnion contains a novel laminin variant, laminin 7, which like laminin 6, covalently associates with laminin 5 to promote stable epithelial-stromal attachment. J. Cell Biol. 132, 1189–1198 (1996).
94. Rousselle, P. et al. Laminin 5 binds the NC-1 domain of type VII collagen. J. Cell Biol. 138, 719–728 (1997).
95. Chen, M. et al. Interactions of the amino-terminal noncollagenous (NC1) domain of type VII collagen with extracellular matrix components. J. Biol. Chem. 272, 14516–14522 (1997).
96. Koshikawa, N., Giannelli, G., Cirulli, V., Miyazaki, K. & Quaranta, V. Role of cell surface metalloprotease MT1-MMP in epithelial cell migration over laminin-5. J. Cell Biol. 148, 615–624 (2000).
97. Goldfinger, L. E., Stack, M. S. & Jones, J. C. Processing of laminin-5 and its functional consequences: role of plasmin and tissue-type plasminogen activator. J. Cell Biol. 141, 255–265 (1998).
98. Timpl, R. et al. Structure and function of laminin LG modules. Matrix Biol. 19, 309–317. (2000).
99. Hynes, R. O. Integrins: bidirectional, allosteric signaling machines. Cell 110, 673–687 (2002).
100. Sung, U., O’Rear, J. J. & Yurchenco, P. D. Localization of heparin binding activity in recombinant laminin G domain. Eur. J. Biochem. 250, 138–143 (1997).
101. Henry, M. D. & Campbell, K. P. A role for dystroglycan in basement membrane assembly. Cell 95, 859–870 (1998).
102. Geerts, D. et al. Binding of integrin !6"4 to plectin prevents plectin association with F-actin but does not interfere with intermediate filament binding. J. Cell Biol. 147, 417–434 (1999).
103. Nievers, M. G., Kuikman, I., Geerts, D., Leigh, I. M. & Sonnenberg, A. Formation of hemidesmosome-like structures in the absence of ligand binding by the !6"4 integrin requires binding of HD1/plectin to the cytoplasmic domain of the "4 integrin subunit. J. Cell Sci. 113, 963–973 (2000).
104. Sterk, L. M. et al. The tetraspan molecule CD151, a novel constituent of hemidesmosomes, associates with the integrin !6"4 and may regulate the spatial organization of hemidesmosomes. J. Cell Biol. 149, 969–982 (2000).
105. Sterk, L. M. et al. Association of the tetraspanin CD151 with the laminin-binding integrins !3"1, !6"1, !6"4 and !7"1 in cells in culture and in vivo. J. Cell Sci. 115, 1161–1173 (2002).
AcknowledgementsThis work was supported by the Veterans Affairs Office of Research and Development, and by the US National Institutes of Health and National Institute of Arthritis and Musculoskeletal and Skin Diseases.
Competing interests statementThe author declares no competing financial interests.
DATABASESThe following terms in this article are linked online to:Entrez Gene: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=geneCD151 | collagen XVII | EGF | EGFR | ERBB2 | HGF | HRAS | "1 integrin | "4 integrin | KRAS | LAMB3 | MET | NF$B | PI3K | plectin | RAC1 | RHOA | ROCK | syndecan 1 | TIAM1 | TLL1National Cancer Institute: http://www.cancer.govSCC
FURTHER INFORMATIONPeter Marinkovich’s laboratory homepage: http://bmz.stanford.eduAccess to this links box is available online.
R E V I E W S
380 | MAY 2007 | VOLUME 7 www.nature.com/reviews/cancer
© 2007 Nature Publishing Group


















