
Synthetic, structural, photophysical and computational studies of π-conjugated bis- and...
13
PAPER www.rsc.org/dalton | Dalton Transactions Synthetic, structural, photophysical and computational studies of p-conjugated bis- and tris-1,3,2-benzodiazaboroles and related bis(boryl) dithiophenes† Lothar Weber,* a Vanessa Werner, a Mark A. Fox, b Todd B. Marder,* b Stefanie Schwedler, a Andreas Brockhinke, a Hans-Georg Stammler a and Beate Neumann a Received 11th September 2008, Accepted 5th November 2008 First published as an Advance Article on the web 12th January 2009 DOI: 10.1039/b815931a A series of p-conjugated systems with two and three 1,3-diethyl-1,3,2-benzodiazaborolyl end-groups was synthesised in 58–91% yields using established 1,3,2-diazaborole methodologies. The bis(diazaborolyl) compounds contain thiophene -2,5-C 4 H 2 S- (2a), dithiophene -5,5¢-(2,2¢-C 4 H 2 S) 2 -(2b), phenylene -1,4-C 6 H 4 -(2c), biphenylene -4,4¢-(1,1¢-(C 6 H 4 ) 2 )- (2d) and dioctylfluorene -2,7-(9,9-(C 8 H 7 ) 2 C 11 H 6 )- (2e) bridges. The three-way linkers in the tris(diazaborolyl) assemblies contain a central phenylene unit -1,3,5-C 6 H 3 - linked to the borolyl end groups via thiophene -2,5-C 4 H 2 S- (3a), directly bonded (3b) or via phenylene -1,4-C 6 H 4 -(3c) units. Molecular structures of 2a, 2b, 2c, 3a, 3b and 3c were determined by X-ray crystallographic studies. These borolylated systems show intense blue/violet luminescence with Stokes shifts of 6200–9500 cm -1 and quantum yields of 0.33 to 0.98. The absorption maxima (296–351 nm) of these assemblies are reproduced well by TD-DFT computations (B3LYP/6–31G*), and arise from strong, low energy HOMO–LUMO transitions. From molecular orbital computations on optimised geometries of these diazaborolyl systems, the LUMO is located mainly on the thiophene/benzene bridge (66–92%) while the HOMO is largely benzodiazaborolyl in character (53–83%). The S 1 ←S 0 absorption bands are thus assigned to p(diazaborolyl)–p*(thiophene/ benzene) transitions. Computations on related bis(boryl) dithiophenes [with diarylboryl e.g. Ph 2 B, Mes 2 B, (C 6 F 5 ) 2 B and FMes 2 B (Mes = 2,4,6-Me 3 C 6 H 2 ; FMes = 2,4,6-(CF 3 ) 3 C 6 H 2 ), dioxaborolyl and other diazaborolyl groups] reveal strong, low energy UV-visible absorption bands arising from p(thiophene)–p*(thiophene) transitions, with increasing boron participation in the LUMO of the diarylboryl and especially the highly fluorinated systems. Introduction There is considerable interest in conjugated materials containing three-coordinate boryl substituents as p-acceptors, as the vacant p z -orbital on boron is capable of significant interaction with organic p-systems. 1,2 These boron-containing materials exhibit intriguing fluorescence emission, second and third order nonlinear optical properties, and fluoride ion sensing ability and can serve as emissive and/or electron-transport layers in OLEDs. This decade has seen rapid developments in the chemistry of 1,3,2-diazaboroles. 3,4 Compounds where the 1,3-diethyl-1,3,2- benzodiazaborolyl group, 1,3-Et 2 -1,3,2-N 2 BC 6 H 4 -, (see Chart 1) is attached to p-systems revealed intriguing absorption and emission characteristics. 5–8 We recently reported synthetic and luminescence studies for the 1,3,2-diazaboroles (1a, 1b, 2a and 2d) 6 and here we expand these systems to include six new compounds containing aromatic or heteroaromatic p-systems substituted by two or three diazaborolyl groups, 2b, 2c, 2e, 3a, 3b and 3c. Synthetic, structural, a Fakult¨ atf¨ ur Chemie der Universit¨ at Bielefeld, Universit¨ atsstrasse 25, 33615, Bielefeld, Germany. E-mail: [email protected] b Department of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: [email protected] †CCDC reference numbers 701586–701591. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b815931a Chart 1 UV-visible spectroscopic and computational studies were carried out on the series of compounds in Chart 1 to examine the influence of the 1,3-diethyl-1,3,2-benzodiazaborolyl groups on the (hetero)aromatic p-systems. In addition, DFT calculations were carried out on a series of 5,5¢-(BR 2 ) 2 -(2,2¢-bithiophenes) to explore the influence of boron substituents on the HOMO–LUMO gap and LUMO energy. This journal is © The Royal Society of Chemistry 2009 Dalton Trans., 2009, 1339–1351 | 1339 Published on 12 January 2009. Downloaded by UNIVERSITY OF ALABAMA AT BIRMINGHAM on 22/10/2014 19:56:02. View Article Online / Journal Homepage / Table of Contents for this issue
Transcript of Synthetic, structural, photophysical and computational studies of π-conjugated bis- and...

PAPER www.rsc.org/dalton | Dalton Transactions
Synthetic, structural, photophysical and computational studies ofp-conjugated bis- and tris-1,3,2-benzodiazaboroles and relatedbis(boryl) dithiophenes†
Lothar Weber,*a Vanessa Werner,a Mark A. Fox,b Todd B. Marder,*b Stefanie Schwedler,a
Andreas Brockhinke,a Hans-Georg Stammlera and Beate Neumanna
Received 11th September 2008, Accepted 5th November 2008First published as an Advance Article on the web 12th January 2009DOI: 10.1039/b815931a
A series of p-conjugated systems with two and three 1,3-diethyl-1,3,2-benzodiazaborolyl end-groupswas synthesised in 58–91% yields using established 1,3,2-diazaborole methodologies. Thebis(diazaborolyl) compounds contain thiophene -2,5-C4H2S- (2a), dithiophene -5,5¢-(2,2¢-C4H2S)2- (2b),phenylene -1,4-C6H4- (2c), biphenylene -4,4¢-(1,1¢-(C6H4)2)- (2d) and dioctylfluorene-2,7-(9,9-(C8H7)2C11H6)- (2e) bridges. The three-way linkers in the tris(diazaborolyl) assemblies containa central phenylene unit -1,3,5-C6H3- linked to the borolyl end groups via thiophene -2,5-C4H2S- (3a),directly bonded (3b) or via phenylene -1,4-C6H4- (3c) units. Molecular structures of 2a, 2b, 2c, 3a, 3band 3c were determined by X-ray crystallographic studies. These borolylated systems show intenseblue/violet luminescence with Stokes shifts of 6200–9500 cm-1 and quantum yields of 0.33 to 0.98. Theabsorption maxima (296–351 nm) of these assemblies are reproduced well by TD-DFT computations(B3LYP/6–31G*), and arise from strong, low energy HOMO–LUMO transitions. From molecularorbital computations on optimised geometries of these diazaborolyl systems, the LUMO is locatedmainly on the thiophene/benzene bridge (66–92%) while the HOMO is largely benzodiazaborolyl incharacter (53–83%). The S1←S0 absorption bands are thus assigned to p(diazaborolyl)–p*(thiophene/benzene) transitions. Computations on related bis(boryl) dithiophenes [with diarylboryl e.g. Ph2B,Mes2B, (C6F5)2B and FMes2B (Mes = 2,4,6-Me3C6H2; FMes = 2,4,6-(CF3)3C6H2), dioxaborolyl andother diazaborolyl groups] reveal strong, low energy UV-visible absorption bands arising fromp(thiophene)–p*(thiophene) transitions, with increasing boron participation in the LUMO of thediarylboryl and especially the highly fluorinated systems.
Introduction
There is considerable interest in conjugated materials containingthree-coordinate boryl substituents as p-acceptors, as the vacantpz-orbital on boron is capable of significant interaction withorganic p-systems.1,2 These boron-containing materials exhibitintriguing fluorescence emission, second and third order nonlinearoptical properties, and fluoride ion sensing ability and can serveas emissive and/or electron-transport layers in OLEDs.
This decade has seen rapid developments in the chemistryof 1,3,2-diazaboroles.3,4 Compounds where the 1,3-diethyl-1,3,2-benzodiazaborolyl group, 1,3-Et2-1,3,2-N2BC6H4-, (see Chart 1) isattached to p-systems revealed intriguing absorption and emissioncharacteristics.5–8 We recently reported synthetic and luminescencestudies for the 1,3,2-diazaboroles (1a, 1b, 2a and 2d)6 and here weexpand these systems to include six new compounds containingaromatic or heteroaromatic p-systems substituted by two or threediazaborolyl groups, 2b, 2c, 2e, 3a, 3b and 3c. Synthetic, structural,
aFakultat fur Chemie der Universitat Bielefeld, Universitatsstrasse 25, 33615,Bielefeld, Germany. E-mail: [email protected] of Chemistry, Durham University, South Road, Durham, DH13LE, UK. E-mail: [email protected]† CCDC reference numbers 701586–701591. For crystallographic data inCIF or other electronic format see DOI: 10.1039/b815931a
Chart 1
UV-visible spectroscopic and computational studies were carriedout on the series of compounds in Chart 1 to examine theinfluence of the 1,3-diethyl-1,3,2-benzodiazaborolyl groups on the(hetero)aromatic p-systems. In addition, DFT calculations werecarried out on a series of 5,5¢-(BR2)2-(2,2¢-bithiophenes) to explorethe influence of boron substituents on the HOMO–LUMO gapand LUMO energy.
This journal is © The Royal Society of Chemistry 2009 Dalton Trans., 2009, 1339–1351 | 1339
Publ
ishe
d on
12
Janu
ary
2009
. Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F A
LA
BA
MA
AT
BIR
MIN
GH
AM
on
22/1
0/20
14 1
9:56
:02.
View Article Online / Journal Homepage / Table of Contents for this issue
Results and discussion
Two distinct methods employed in the syntheses of aryldiaz-aboroles are summarised in Scheme 1. Method A, with 1,3-diethyl-1,3,2-benzodiazaborolyl bromide9 (4) as the precursor, was usedin the reported syntheses of 1a and 1b, and herein to give thebis(borolyl) compounds 2b and 2e in high yields (81–91%) andthe tris(borolyl) compounds 3a and 3c in modest yields (58–61%).In method B, diamine N,N¢-diethyl-1,2-diaminobenzene10 (5) wasused as starting material in the published syntheses of 2a and 2d,and herein to prepare 2c and 3b in good yields (71–76%).
Scheme 1
Compounds with thiophene units at the benzo-1,3,2-diazaborole functions (1a, 1b, 2a, 2b and 3a) give rise to singletsin the range of 26.0 to 26.6 ppm in the 11B{1H} NMR spectra.The 1,3,2-benzodiazaborole derivatives 2c–e and 3b–c with theboron atoms attached to a benzene ring reveal broad singlets inthe narrow range of d = 28.6 to 29.3 ppm. In the 1H-NMR spectra,the peaks at d = 7.02–7.10 and 7.12–7.21 ppm are assigned to H-atoms of the benzodiazaborole unit at positions 5, 8 and 6, 7respectively. The corresponding ring carbon atoms give signalsin the 13C{1H}-NMR spectra at d = 108.8–108.9 and 118.6–118.9 ppm. The peaks for the 13C nuclei at positions 4 and 9were observed at d = 137.1–137.2 ppm.
X-Ray crystallography
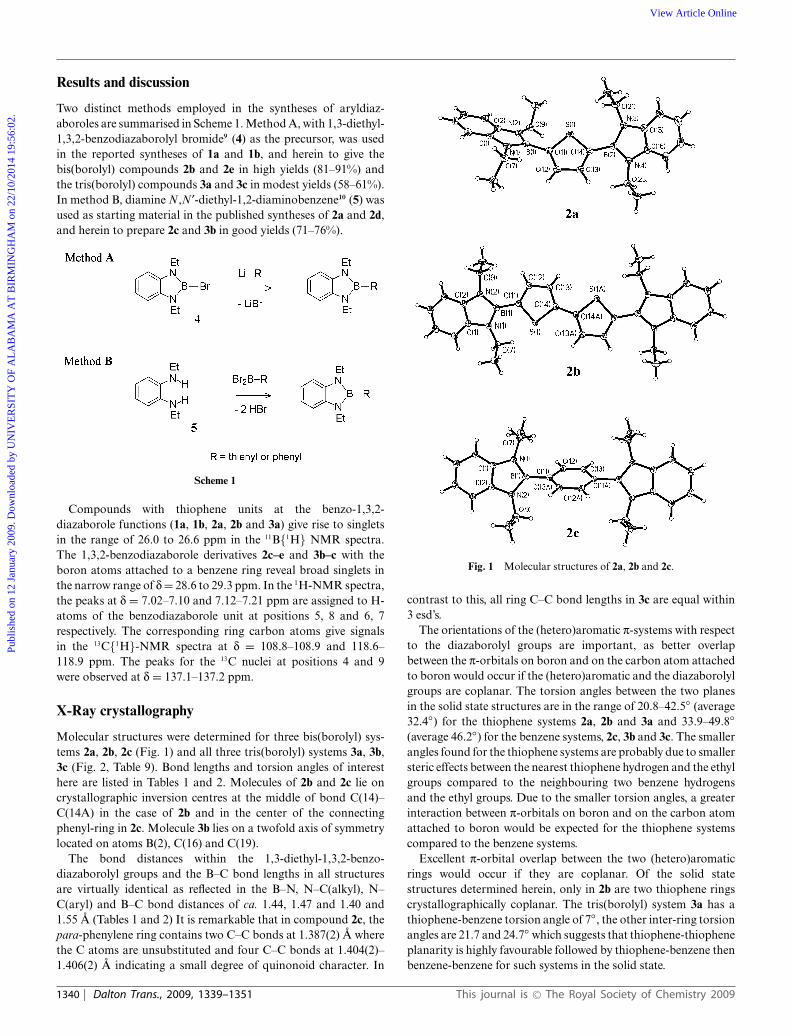
Molecular structures were determined for three bis(borolyl) sys-tems 2a, 2b, 2c (Fig. 1) and all three tris(borolyl) systems 3a, 3b,3c (Fig. 2, Table 9). Bond lengths and torsion angles of interesthere are listed in Tables 1 and 2. Molecules of 2b and 2c lie oncrystallographic inversion centres at the middle of bond C(14)–C(14A) in the case of 2b and in the center of the connectingphenyl-ring in 2c. Molecule 3b lies on a twofold axis of symmetrylocated on atoms B(2), C(16) and C(19).
The bond distances within the 1,3-diethyl-1,3,2-benzo-diazaborolyl groups and the B–C bond lengths in all structuresare virtually identical as reflected in the B–N, N–C(alkyl), N–C(aryl) and B–C bond distances of ca. 1.44, 1.47 and 1.40 and1.55 A (Tables 1 and 2) It is remarkable that in compound 2c, thepara-phenylene ring contains two C–C bonds at 1.387(2) A wherethe C atoms are unsubstituted and four C–C bonds at 1.404(2)–1.406(2) A indicating a small degree of quinonoid character. In
Fig. 1 Molecular structures of 2a, 2b and 2c.
contrast to this, all ring C–C bond lengths in 3c are equal within3 esd’s.
The orientations of the (hetero)aromatic p-systems with respectto the diazaborolyl groups are important, as better overlapbetween the p-orbitals on boron and on the carbon atom attachedto boron would occur if the (hetero)aromatic and the diazaborolylgroups are coplanar. The torsion angles between the two planesin the solid state structures are in the range of 20.8–42.5◦ (average32.4◦) for the thiophene systems 2a, 2b and 3a and 33.9–49.8◦
(average 46.2◦) for the benzene systems, 2c, 3b and 3c. The smallerangles found for the thiophene systems are probably due to smallersteric effects between the nearest thiophene hydrogen and the ethylgroups compared to the neighbouring two benzene hydrogensand the ethyl groups. Due to the smaller torsion angles, a greaterinteraction between p-orbitals on boron and on the carbon atomattached to boron would be expected for the thiophene systemscompared to the benzene systems.
Excellent p-orbital overlap between the two (hetero)aromaticrings would occur if they are coplanar. Of the solid statestructures determined herein, only in 2b are two thiophene ringscrystallographically coplanar. The tris(borolyl) system 3a has athiophene-benzene torsion angle of 7◦, the other inter-ring torsionangles are 21.7 and 24.7◦ which suggests that thiophene-thiopheneplanarity is highly favourable followed by thiophene-benzene thenbenzene-benzene for such systems in the solid state.
1340 | Dalton Trans., 2009, 1339–1351 This journal is © The Royal Society of Chemistry 2009
Publ
ishe
d on
12
Janu
ary
2009
. Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F A
LA
BA
MA
AT
BIR
MIN
GH
AM
on
22/1
0/20
14 1
9:56
:02.
View Article Online

Fig. 2 Molecular structures of 3a, 3b and 3c. In 3b, the methyl groups at C(7) are disordered over two positions [C(8A) and C(8B)] in the crystal.
A structural comparison between the dithiophene 2b and thereported6 molecular structure of the closely related monoborolyldithiophene 1b reveals similar thiophene-thiophene torsion angles(0◦ and 4.6◦, respectively) and bond distances. Comparisonbetween the structures of 3b and a system7 where the threeend groups are 1,3-tert-butyl-1,3,2-diazaborolyl (C2H2(tBuN)2B,Chart 2), reveal the benzene ring in both cases to be similarwith averaged C–C bond distances of 1.399 A. However, thesteric effects of the tert-butyl groups in the reported system resultin larger torsional angles (73.9–87.1◦) between the borolyl andaryl rings. Comparison of compound 2c with the reported crystalstructure of an analogous system7 containing 1,3-tert-butyl-1,3,2-diazaborolidinyl groups (C2H4(tBuN)2B, Chart 2) in place of thebenzoborolyl groups reveals larger ring-ring torsion angles of 86.5and 89.5◦ due to the steric effects of the bulky tert-butyl groups.Compounds 2c and 3b, with smaller borolyl-aryl torsion angles,would be expected to contain greater p-orbital interactions on theboron-carbon links than their tert-butyl analogues.
To our knowledge, this is the largest series of structurallycharacterised bis- and tris-boryl systems containing arenes andthiophenes. Known structural examples, in addition to thosementioned, are a Cl(2,2,6,6-tetramethylpiperidyl)B analogue11 of2a, a (C6F5)2B analogue12 of 2b, a Mes2B analogue of 2e with ethylgroups at the fluorene bridge,13 a C6H4O2B analogue of 2e with
hexyl groups at the fluorene bridge,14 (HO)2B,15 C6H4O2B,16 Br2B,17
tBuB(iPr2P)2B18 and Mes2B19 analogues of 2c, and C2H4O2B,20
Br2B17 and Mes2B21 analogues of 3b (Chart 2 shows the borylgroups). No structural examples containing boryl groups havebeen reported previously for the larger molecules such as 3aand 3c. There is only one other known tris(boryl) exampleof 3a, which contains Mes2B as boryl groups.22 This literaturesearch implies that boryl systems containing the 1,3-diethyl-1,3,2-benzoborolyl group form suitable crystals in many cases for X-raycrystallography studies and, perhaps more importantly, improvethe solubilities of larger molecules such as 3a and 3c. It is notedelsewhere that compounds containing the closely related 1,3-di-iso-propyl-1,3,2-benzoborolyl group also crystallise readily.23
Also of importance for optical device fabrication is the fact thatcompounds such as the Mes2B analogue of 2b (BMB2T) readilyform amorphous films.24
UV-visible and luminescence spectroscopy
Table 3 lists the photophysical data for all compounds shownin Chart 1. Published data6 for 1a, 1b, 2a and 2d are includedin the Table for completeness. The absorption maxima rangefrom 296 to 351 nm, with notably longer wavelengths for theextended thiophene systems 1b, 2b and 3a. These observations
This journal is © The Royal Society of Chemistry 2009 Dalton Trans., 2009, 1339–1351 | 1341
Publ
ishe
d on
12
Janu
ary
2009
. Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F A
LA
BA
MA
AT
BIR
MIN
GH
AM
on
22/1
0/20
14 1
9:56
:02.
View Article Online
Table 1 Selected bond distances and torsion angles for 2a, 2b and 2c
2a 2b 2c
Bond distances (A)B–C B(1)–C(11) 1.552(2), B(2)–C(14) 1.554(2) B(1)–C(11) 1.558(2) B(1)–C(11) 1.563(2)B–N B(1)–N(1) 1.434(2), B(1)–N(2) 1.439(2),
B(2)–N(3) 1.439(2), B(2)–N(4) 1.435(2)B(1)–N(1) 1.443(2), B(1)–N(2) 1.437(2) B(1)–N(1) 1.434(1), B(1)–N(2) 1.437(1)
Aryl linksS–C S(1)–C(11) 1.724(1), S(1)–C(14) 1.727(1) S(1)–C(11) 1.737(1), S(1)–C(14) 1.732(1)C–C C(11)–C(12) 1.381(2), C(12)–C(13)
1.416(2), C(13)–C(14) 1.381(2)C(11)–C(12) 1.384(2), C(12)–C(13)1.416(2), C(13)–C(14) 1.375(2)
C(11)–C(12) 1.404(2), C(12)–C(13)1.387(2), C(11)–C(13A) 1.406(2)
Inter-ring C–C C(14)–C(14A) 1.453(2)Borolyl unitN–C(Et) N(1)–C(7) 1.457(2), N(2)–C(9) 1.462(2),
N(3)–C(21) 1.463(2), N(4)–C(23) 1.459(2)N(1)–C(7) 1.462(2), N(2)–C(9) 1.461(2) N(1)–C(7) 1.457(1), N(2)–C(9) 1.460(1)
N–C(aryl) N(1)–C(1) 1.394(2), N(2)–C(2) 1.393(2),N(3)–C(15) 1.395(1), N(4)–C(16) 1.397(2)
N(1)–C(1) 1.397(1), N(2)–C(2) 1.396(2) N(1)–C(1) 1.394(1), N(2)–C(2) 1.397(1)
C–C C(1)–C(2) 1.413(2), C(15)–C(16) 1.411(2) C(1)–C(2) 1.408(2) C(1)–C(2) 1.411(2)Torsion Angles (◦)N–B–C–C N(1)–B(1)–C(11)–C(12) 42.5,
N(4)–B(2)–C(14)–C(13) 33.3N(2)–B(1)–C(11)–C(12)–20.8 N(1)–B(1)–C(11)–C(12) 49.7
C–C–C–C C(13)–C(14)–C(14A)–C(13A) 180.0
Chart 2
suggest extended p-conjugation interactions occur between thetwo thiophene rings for 1b and 2b and between the benzene ringand the three thiophene rings for 3a.
Fig. 3 Bis(borolyl) derivatives 2a–e (ca. 0.01M in THF) under UV-radi-ation (355 nm).
Table 3 Absorption and emission data
lmax (abs, nm) e (mol-1cm-1dm3) lmax (em, nm)Stokesshift (cm-1) U
1aa 296 — 382 7600 —1ba 326 — 433 7600 0.292aa 316 — 404 6900 0.592bb 351 33000 448 6200 0.522cb 300 20200 385 7400 0.982da 304 — 427 9500 0.522eb 318 32300 399 6400 0.873ab 330 52900 446 7900 0.333bb 296 27700 364 6300 0.453cb 300 54600 417 9400 0.71
a Reference 6, solvent THF, against standard coumarin 120 (U = 0.50).b This work, solvent THF, against standard POPOP (U = 0.93).
All bis- and tris(borolyl) molecules listed in Table 3 emitblue or violet luminescence under ultraviolet irradiation inTHF solutions (Fig. 3 and 4). The emission maxima for these
1342 | Dalton Trans., 2009, 1339–1351 This journal is © The Royal Society of Chemistry 2009
Publ
ishe
d on
12
Janu
ary
2009
. Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F A
LA
BA
MA
AT
BIR
MIN
GH
AM
on
22/1
0/20
14 1
9:56
:02.
View Article Online

Table 2 Selected bond distances and torsion angles for 3a, 3b and 3c
3a 3b 3c
Bond distances (A)B–C B(1)–C(11) 1.555(4), B(2)–C(25)
1.556(3), B(3)–C(39) 1.544(4)B(1)–C(17) 1.561(3), B(2)–C(19) 1.565(5) B(1)–C(11) 1.562(4), B(2)–C(27)
1.565(4), B(3)–C(43) 1.564(4)B–N B(1)–N(1) 1.429(3), B(1)–N(2)
1.440(3), B(2)–N(3) 1.427(3),B(2)–N(4) 1.435(3), B(3)–N(5)1.434(4), B(3)–N(6) 1.433(3)
B(1)–N(1) 1.424(4), B(1)–N(2) 1.435(3),B(2)–N(3) 1.443(3)
B(1)–N(1) 1.433(4), B(1)–N(2)1.434(4), B(2)–N(3) 1.436(4),B(2)–N(4) 1.436(4), B(3)–N(5)1.435(4), B(3)–N(6) 1.435(4)
Aryl linksS–C S(1)–C(11) 1.732(2), S(1)–C(14)
1.735(2), S(2)–C(25) 1.728(2),S(2)–C(28) 1.728(2), S(3)–C(39)1.731(2), S(3)–C(42) 1.736(2)
C–C C(11)–C(12) 1.371(3), C(12)–C(13)1.410(3), C(13)–C(14) 1.364(3),C(25)–C(26) 1.374(3), C(26)–C(27)1.407(3), C(27)–C(28) 1.367(3),C(39)–C(40) 1.374(3), C(40)–C(41)1.410(3), C(41)–C(42) 1.369(3)
C(16)–C(17) 1.395(3), C(17)–C(18)1.398(3), C(18)–C(19) 1.402(3)
C(11)–C(12) 1.403(4), C(12)–C(13)1.389(4), C(13)–C(14) 1.403(4),C(14)–C(15) 1.397(4), C(15)–C(16)1.389(4), C(11)–C(16) 1.408(4),C(27)–C(28) 1.405(4), C(28)–C(29)1.391(4), C(29)–C(30) 1.399(4),C(30)–C(31) 1.400(4), C(31)–C(32)1.389(4), C(27)–C(32) 1.400(4),C(43)–C(44) 1.407(4), C(44)–C(45)1.393(4), C(45)–C(46) 1.400(4),C(46)–C(47) 1.401(4), C(47)–C(48)1.389(4), C(43)–C(48) 1.402(4)
Inter-ring C–C C(14)–C(43) 1.472(3), C(28)–C(45)1.468(3), C(42)–C(47) 1.463(3)
C(14)–C(49) 1.492(3), C(30)–C(51)1.489(3), C(46)–C(53) 1.496(3)
Central ring C(43)–C(48) 1.395(3), C(43)–C(44)1.398(3), C(44)–C(45) 1.396(3),C(45)–C(46) 1.395(3), C(46)–C(47)1.395(3), C(47)–C(48) 1.396(3)
C(49)–C(50) 1.397(4), C(50)–C(51)1.397(3), C(51)–C(52) 1.396(4),C(52)–C(53) 1.397(3), C(53)–C(54)1.395(3), C(49)–C(54) 1.394(4)
Borolyl unitN–C(Et) N(1)–C(7) 1.461(3), N(2)–C(9)
1.462(3), N(3)–C(21) 1.462(3),N(4)–C(23) 1.465(3), N(5)–C(35)1.462(3), N(6)–C(37) 1.462(3)
N(1)–C(9) 1.469(3), N(2)–C(7) 1.454(3),N(3)–C(14) 1.454(3)
N(1)–C(7) 1.469(3), N(2)–C(9)1.465(3), N(3)–C(23) 1.462(4),N(4)–C(25) 1.462(3), N(5)–C(41)1.460(3), N(6)–C(39) 1.458(3)
N–C(aryl) N(1)–C(1) 1.394(3), N(2)–C(6)1.405(3), N(3)–C(15) 1.395(3),N(4)–C(20) 1.396(3), N(5)–C(29)1.401(3), N(6)–C(34) 1.403(3)
N(1)–C(1) 1.397(3), N(2)–C(2) 1.394(3),N(3)–C(11) 1.393(3)
N(1)–C(1) 1.396(3), N(2)–C(6)1.393(3), N(3)–C(17) 1.395(3),N(4)–C(22) 1.392(3), N(5)–C(33)1.393(3), N(6)–C(38) 1.397(3)
C–C C(1)–C(6) 1.405(3), C(15)–C(20)1.409(3), C(29)–C(34) 1.402(3)
C(1)–C(2) 1.398(4), C(11)–C(11A) 1.406(4) C(1)–C(6) 1.403(4), C(17)–C(22)1.411(4)
Torsion Angles (◦)N–B–C–C N(1)–B(1)–C(11)–C(12) 38.2,
N(3)–B(2)–C(25)–C(26) 35.5,N(6)–B(3)–C(39)–C(40) 35.9
N(1)–B(1)–C(17)–C(16) 49.9,N(3)–B(2)–C(19)–C(18)–33.9
N(1)–B(1)–C(11)–C(16) 49.8,N(3)–B(2)–C(27)–C(32) 42.5,N(5)–B(3)–C(43)–C(48) 44.2
C–C–C–C C(13)–C(14)–C(43)–C(48) 21.7,C(27)–C(28)–C(45)–C(44) 7.6,C(41)–C(42)–C(47)–C(48)–24.7
C(13)–C(14)–C(49)–C(50)–31.7,C(29)–C(30)–C(51)–C(52)–33.2,C(45)–C(46)–C(53)–C(54)–28.1
molecules generally follow the trends in the shifts found for theabsorption maxima, with the thiophene systems containing thelongest wavelengths. The normalised emission spectra for thenew compounds are shown in Fig. 5. The Stokes shifts for thesemolecules range between 6200 and 9500 cm-1 for all systems.The largest values observed for 2d and 3c, may result fromplanarisation of the biphenyl cores in the excited state, whereasthe fluorenyl compound 2e, in which the biphenyl core is alreadyheld planar in the ground state, has one of the smallest Stokesshifts.
The phenylene bridged compound 2c has an exceptionally highquantum yield (U) of 0.98, whereas the fluorene derivative 2e hasthe next highest quantum yield of 0.87. The thiophene systemsgenerally have lower quantum yields than the phenylene systems.
This is presumably due to the excited singlet state, S1, for thethiophenes undergoing more rapid intersystem crossing (ISC)to the triplet state, T1, than the benzenes. ISC is faster withintroduction of sulfur which is relatively heavy thus enhancingspin–orbit coupling in the molecule.25 The medium to highquantum yields and the strong blue or violet luminescence forthese borolyl compounds suggest that these systems may havepractical use as precursors for organic devices e.g. organic lightemitting diodes (OLEDs).
Reported photophysical data5 show violet-blue luminescence inDMF for the analogues of 2c shown in Fig. 6. Direct comparisonwith our data here may not be appropriate as different solventswere used. Nevertheless, the reported systems I-IV have quantumyields (U) of 0.66–0.99 comparable to the values shown in Table 3.
This journal is © The Royal Society of Chemistry 2009 Dalton Trans., 2009, 1339–1351 | 1343
Publ
ishe
d on
12
Janu
ary
2009
. Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F A
LA
BA
MA
AT
BIR
MIN
GH
AM
on
22/1
0/20
14 1
9:56
:02.
View Article Online
Fig. 4 Tris(borolyl) derivatives 3a–c (ca. 0.01M in THF) under UV-radi-ation (355 nm).
The extinction coefficients (e) are in the region of 45000 in thesesystems, which are below the values corresponding to our largersystems 3a and 3c.
Blue emissive materials containing boryl groups other than the1,3,2-diazaborolyl groups are known.7,12,13,14,22,24,26 Closely relatedto our systems are the Mes2B,24 (C6F5)2B12 and (p-tBuC6H4)2B12
analogues of 2b, a Br2B analogue7 of 2d, a Mes2B analogue13
of 2e with ethyl groups at the fluorene bridge and a Mes2Banalogue22 of 3a (see Chart 2 for boryl groups discussed here).Only detailed photophysical data are quoted12 for the dithiopheneswith (C6F5)2B and (p-tBuC6H4)2B groups. Values of U = 0.03 ande = 31400 were determined for the (C6F5)2B compound and U =0.79 and e = 48300 for the (p-tBuC6H4)2B analogue in DCM. Thirdorder NLO properties of the Mes2B analogues of 2c and 2d havealso been reported.19
Fig. 6 Reported analogues of 2c with violet-blue luminescence properties.
DFT calculations
On the basis of the experimental UV-visible and diffraction datahere for all borolyl molecules, dithiophene 2b contains the mosteffective extended p-conjugation within the molecule. Thus initialgeometry optimisations were carried out on the dithiophene 2band a model compound 2b¢ with methyl groups in place of ethylgroups at the B3LYP/6–31G* level of theory. Very good agreementbetween geometrical parameters for the X-ray determined geome-try of 2b and both optimised geometries were found (Table 4). TD-DFT computations carried out on the optimised geometries for 2band 2b¢ gave identical lowest energies of 390 nm for allowed S1 ← S0
electronic absorptions with comparable oscillator strengths (f =0.944 and 0.980 for 2b and 2b¢, respectively) corresponding to theHOMO–LUMO transitions. These computed values differ fromthe observed absorption maxima by 39 nm. This is typical ofcomputed static ‘gas-phase’ geometries for which p-conjugationis favoured in planar forms such as in 2b and 2b¢, whereas in theexperimental solution state there is only a small rotational barrier.The systems exist as an ensemble of rotational isomers and thusa blue shift and a broadening is expected in the experimentalsolution phase spectrum. By constraining the torsion angle of thethiophene-thiophene link to 90◦ in the optimised geometry of 2b¢,a computed value of 315 nm was found.
It is clear here from the DFT and TD-DFT data for 2b thatoptimised geometries of our systems depicted in Chart 1 can be
Fig. 5 Emission spectra of the quadrupolar molecules 2b (green), 2c (red) and 2e (blue) on left graph and octupolar (trigonal) molecules 3a (green), 3b(blue) and 3c (red) on right.
1344 | Dalton Trans., 2009, 1339–1351 This journal is © The Royal Society of Chemistry 2009
Publ
ishe
d on
12
Janu
ary
2009
. Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F A
LA
BA
MA
AT
BIR
MIN
GH
AM
on
22/1
0/20
14 1
9:56
:02.
View Article Online
Table 4 Comparison of experimental and computed geometrical param-eters for the bis(benzodiazaborolyl) dithiophenes
2b expt 2b calc 2b¢ calc
Bond distances (A)B–C 1.558 1.555 1.555B–N 1.440 1.443 1.442S–C 1.737 1.755 1.754
1.732 1.752 1.754C–C(thiophene) 1.384 1.382 1.381
1.416 1.418 1.4181.375 1.380 1.380
Inter-ring C–C 1.453 1.448 1.448N–C(Et/Me) 1.462 1.458 1.451N–C(aryl) 1.397 1.397 1.396C–C(aryl) 1.408 1.416 1.415
1.390 1.394 1.3931.392 1.399 1.4001.396 1.399 1.399
Torsion angles (◦)N–B–C–C -20.8 -37.7 -44.5C–C–C–C 180.0 173.4 179.9
successfully modelled using 1,3-dimethyl-1,3,2-benzodiazaborolylgroups. As for 2b and 2b¢, very good agreement in the geometricalparameters between experimental data for 1b, 2a, 2c, 3a, 3b and3c and optimised geometries for 1b¢, 2a¢, 2c¢, 3a¢, 3b¢ and 3c¢ werefound, as shown in Table 5 for B–C bond distances and torsionangles between rings.
The calculated absorption maxima from TD-DFT computa-tions for the model geometries are listed in Table 6. Systems whereno (hetero)aromatic ring links exist show calculated absorptionmaxima values in very good agreement with observed data. Asexpected, those with (hetero)aromatic ring links give differencesof ca. 36 nm for thiophene-thiophene, 31 nm for thiophene-benzene and 27 nm for benzene-benzene links between observedand computed absorption maxima values.
Selected data from molecular orbital computations on themodel geometries are summarised in Table 7. The HOMO–LUMOgaps (HLGs) are in line with observed low-energy absorptiondata, as would be expected. The energies for the HOMOs areconsistent within the -5.11 to -5.33 eV region, whereas theenergies for the LUMOs vary between -0.63 and -1.54 eV. TheLUMOs are mainly thienyl/aryl in character (92% to 66%).The LUMO generally increases in thienyl/aryl character at theexpense of the borolyl group character as its orbital energy islowered. The HOMOs, on the other hand, are mainly borolyl
Table 6 Observed and computed (TD-DFT) UV absorption data
lmax (calc)(abs, nm)
lmax (expt)a
(abs, nm)lmax(calc) -lmax(expt) (nm) f (calc)
1a¢ 298 296b 2 0.281b¢ 360 326b 34 0.522a¢ 327 316b 11 0.622b¢ 390 351 39 0.982c¢ 307 300 7 0.582d¢ 332 304b 28 0.722e¢ 341c 318 23 0.783a¢ 361 330 31 0.703b¢ 296 296 0 0.403c¢ 327 300 27 0.51
a From compounds containing ethyl groups instead of methyl groups.b Reference 6. c Methyl groups used instead of octyl groups at the fluorenefragment in the computed geometry.
Table 7 Orbital energies and compositions for model geometries
HLG(eV) Orbital E (eV)
C6H4(NMe)2
(%) B (%) (hetero)aryl (%)
1a¢ 4.66 LUMO -0.63 14 11 75HOMO -5.29 79 9 12
1b¢ 3.82 LUMO -1.38 5 4 91HOMO -5.20 55 5 40
2a¢ 4.31 LUMO -0.91 17 15 68HOMO -5.22 77 7 16
2b¢ 3.57 LUMO -1.54 8 6 87HOMO -5.11 53 4 44
2c¢ 4.56 LUMO -0.71 18 16 66HOMO -5.27 83 9 8
2d¢ 4.18 LUMO -1.08 8 8 84HOMO -5.26 78 9 13
2e¢ 4.07 LUMO -1.13 7 7 85HOMO -5.20 72 8 20
3a¢ 3.84 LUMOa -1.44 5 4 92HOMOb -5.28 66 7 27
3b¢ 4.72 LUMOa -0.61 17 13 70HOMOb -5.33 83 10 7
3c¢ 4.21 LUMOa -1.11 4 4 91HOMOb -5.32 80 10 10
a Degenerate with LUMO + 1. b Degenerate with HOMO-1 and HOMO-2.
group in character (83% to 53%). Fig. 7 shows the frontier orbitalsfor 2b¢ and 2c¢. They contain the highest and lowest borolylgroup contribution to the HOMOs in the series. The nature ofthe HOMO–LUMO transitions in all compounds is essentially
Table 5 Comparison of parameters relevant to p-conjugation in experimental and optimised geometries of benzodiazaboroles. Values from optimisedgeometries are in parentheses
1b 2a 2b 2c 3a 3b 3c
(1b¢) (2a¢) (2b¢) (2c¢) (3a¢) (3b¢) (3c¢)
Bond distance (A)B–C 1.550a 1.553 1.558 1.563 1.552 1.563 1.564
(1.555) (1.556) (1.555) (1.565) (1.555) (1.565) (1.564)Torsion Angles (◦)N–B–C–C 38.7a 37.9 20.8 49.7 36.5 41.9 45.5
(43.0) (43.5) (44.5) (50.4) (43.9) (51.1) (50.8)C–C–C–C 175.4a — 180.0 — 18.0 — 31.0
(162.4) — (179.9) — (27.0) — (38.0)
a Reference.
This journal is © The Royal Society of Chemistry 2009 Dalton Trans., 2009, 1339–1351 | 1345
Publ
ishe
d on
12
Janu
ary
2009
. Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F A
LA
BA
MA
AT
BIR
MIN
GH
AM
on
22/1
0/20
14 1
9:56
:02.
View Article Online
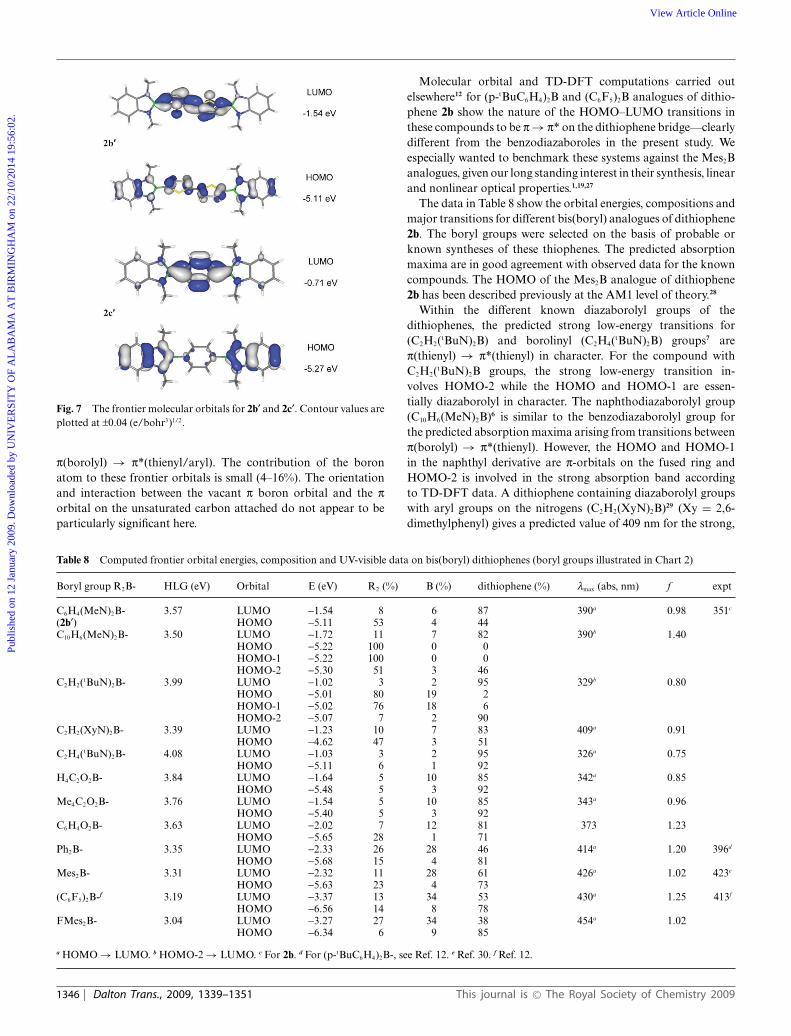
Fig. 7 The frontier molecular orbitals for 2b¢ and 2c¢. Contour values areplotted at ±0.04 (e/bohr3)1/2.
p(borolyl) → p*(thienyl/aryl). The contribution of the boronatom to these frontier orbitals is small (4–16%). The orientationand interaction between the vacant p boron orbital and the porbital on the unsaturated carbon attached do not appear to beparticularly significant here.
Molecular orbital and TD-DFT computations carried outelsewhere12 for (p-tBuC6H4)2B and (C6F5)2B analogues of dithio-phene 2b show the nature of the HOMO–LUMO transitions inthese compounds to be p → p* on the dithiophene bridge—clearlydifferent from the benzodiazaboroles in the present study. Weespecially wanted to benchmark these systems against the Mes2Banalogues, given our long standing interest in their synthesis, linearand nonlinear optical properties.1,19,27
The data in Table 8 show the orbital energies, compositions andmajor transitions for different bis(boryl) analogues of dithiophene2b. The boryl groups were selected on the basis of probable orknown syntheses of these thiophenes. The predicted absorptionmaxima are in good agreement with observed data for the knowncompounds. The HOMO of the Mes2B analogue of dithiophene2b has been described previously at the AM1 level of theory.28
Within the different known diazaborolyl groups of thedithiophenes, the predicted strong low-energy transitions for(C2H2(tBuN)2B) and borolinyl (C2H4(tBuN)2B) groups7 arep(thienyl) → p*(thienyl) in character. For the compound withC2H2(tBuN)2B groups, the strong low-energy transition in-volves HOMO-2 while the HOMO and HOMO-1 are essen-tially diazaborolyl in character. The naphthodiazaborolyl group(C10H6(MeN)2B)6 is similar to the benzodiazaborolyl group forthe predicted absorption maxima arising from transitions betweenp(borolyl) → p*(thienyl). However, the HOMO and HOMO-1in the naphthyl derivative are p-orbitals on the fused ring andHOMO-2 is involved in the strong absorption band accordingto TD-DFT data. A dithiophene containing diazaborolyl groupswith aryl groups on the nitrogens (C2H2(XyN)2B)29 (Xy = 2,6-dimethylphenyl) gives a predicted value of 409 nm for the strong,
Table 8 Computed frontier orbital energies, composition and UV-visible data on bis(boryl) dithiophenes (boryl groups illustrated in Chart 2)
Boryl group R2B- HLG (eV) Orbital E (eV) R2 (%) B (%) dithiophene (%) lmax (abs, nm) f expt
C6H4(MeN)2B- 3.57 LUMO -1.54 8 6 87 390a 0.98 351c
(2b¢) HOMO -5.11 53 4 44C10H6(MeN)2B- 3.50 LUMO -1.72 11 7 82 390b 1.40
HOMO -5.22 100 0 0HOMO-1 -5.22 100 0 0HOMO-2 -5.30 51 3 46
C2H2(tBuN)2B- 3.99 LUMO -1.02 3 2 95 329b 0.80HOMO -5.01 80 19 2HOMO-1 -5.02 76 18 6HOMO-2 -5.07 7 2 90
C2H2(XyN)2B- 3.39 LUMO -1.23 10 7 83 409a 0.91HOMO -4.62 47 3 51
C2H4(tBuN)2B- 4.08 LUMO -1.03 3 2 95 326a 0.75HOMO -5.11 6 1 92
H4C2O2B- 3.84 LUMO -1.64 5 10 85 342a 0.85HOMO -5.48 5 3 92
Me4C2O2B- 3.76 LUMO -1.54 5 10 85 343a 0.96HOMO -5.40 5 3 92
C6H4O2B- 3.63 LUMO -2.02 7 12 81 373 1.23HOMO -5.65 28 1 71
Ph2B- 3.35 LUMO -2.33 26 28 46 414a 1.20 396d
HOMO -5.68 15 4 81Mes2B- 3.31 LUMO -2.32 11 28 61 426a 1.02 423e
HOMO -5.63 23 4 73(C6F5)2B-f 3.19 LUMO -3.37 13 34 53 430a 1.25 413f
HOMO -6.56 14 8 78FMes2B- 3.04 LUMO -3.27 27 34 38 454a 1.02
HOMO -6.34 6 9 85
a HOMO → LUMO. b HOMO-2 → LUMO. c For 2b. d For (p-tBuC6H4)2B-, see Ref. 12. e Ref. 30. f Ref. 12.
1346 | Dalton Trans., 2009, 1339–1351 This journal is © The Royal Society of Chemistry 2009
Publ
ishe
d on
12
Janu
ary
2009
. Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F A
LA
BA
MA
AT
BIR
MIN
GH
AM
on
22/1
0/20
14 1
9:56
:02.
View Article Online

low-energy HOMO → LUMO transition. This wavelength is19 nm longer than for the benzodiazaborolyl groups. The arylgroups on the nitrogens of the borolyl groups raise the HOMOenergy compared to other boryl groups. Examination of theoptimised structure reveals an intriguing near-planar geometrywith torsion angles of 8.2◦ and 170.7◦ for N–B–C–C and C–C–C–C respectively and short B–C bond distances of 1.548 A whichmay be attributed to steric interactions of the methyl groupsand/or the favourable S/C–H ◊ ◊ ◊ p(aryl) interactions. It remainsto be seen whether p-systems with N-aryl diazaborolyl groups canbe synthesised from N-aryl diazaboroles.
Dioxaborolyl groups are shown from Table 8 to contribute littleto the frontier orbitals of thiophenes and the predicted HOMO–LUMO low energy transitions are (thienyl)p → (thienyl)p* incharacter. This is in spite of obvious planarity and short C–B bond distances of 1.529 A in the optimised geometries.The benzodioxaborolyl group, C6H4O2B, influences the HOMOcharacter (28%) and lowers the LUMO energy compared to non-aromatic dioxaborolyl groups (5%).
The thiophenes with Ar2B groups have the lowest LUMOenergies and smallest HOMO–LUMO gaps in Table 8. Strongabsorption maxima at around 425 nm arising from HOMO–LUMO transitions are computed for these systems and thesetransitions are (thienyl)p → (thienyl)p* in character with theLUMOs containing substantial contributions from the boronatoms (28–34%). The effect of the fluorines on the methyl groupsof the dimesitylboryl group (FMes2B)31 results in the smallestHOMO–LUMO gap and the longest predicted wavelength of454 nm for the strong, low-energy HOMO–LUMO transition inthe series listed in Table 8. Attempts to synthesise p-systems withFMes2B groups from fluorinated dimesitylboranes are underway.Such compounds are expected to be efficient luminophores andelectron transporters, as well as being very stable, glass-formingsystems.
While the absorption maxima may be predicted with someconfidence for p-systems containing boryl groups by DFT compu-tations, the emission maxima and quantum yields cannot reliablybe determined as yet from computations. The nature of an S0
← S1 transition requires an accurate geometry for S1 whichis notoriously difficult to obtain computationally. However, thepredicted absorption maxima from these dithiophenes combinedwith observed Stokes shifts of ca. 7500 cm-1 indicate that strongblue/violet-green luminescence are expected (and observed insome cases)12,24 from these dithiophenes.
Conclusions
The new bis- and tris(diazaborolyl) assemblies 2b, 2c, 2e,3a, 3b and 3c were prepared from 2-bromo-1,3-diethyl-1,3,2-benzodiazaborole or N,N¢-diethyl-1,2-diaminobenzene in modestto very good yields. Molecular structures of 2a, 2b, 2c, 3a,3b and 3c determined by X-ray crystallographic studies showvery good agreement with optimised geometries, with thiophene-containing systems being more planar and containing shorterB–C bonds thus increasing p conjugation between the borolyland thiophene rings. These borolylated systems show intenseblue/violet luminescence, with the thiophenes containing longerwavelength absorption and emission maxima than the ben-zenes. The benzene systems, however, have higher fluorescence
quantum yields than the thiophenes. TD-DFT computationsindicate that the absorption maxima of these assemblies arisefrom strong, low energy HOMO–LUMO transitions which areassigned to p(diazaborolyl)–p*(thiophene/benzene) transitions.Computations on related bis(boryl) dithiophenes with diarylboryl,dioxaborolyl and other diazaborolyl groups, reveal that strong, lowenergy UV-visible absorption bands are expected and assignedas arising from p(thiophene)–p*(thiophene) transitions. The pre-dicted longest absorption wavelength from a diazaborolyl group isfor the 1,3-diaryl-1,3,2-diazaborolyl group and the longest overallis from a fluorinated dimesitylboryl group, [1,3,5-(CF3)3C6H2]2B.
Experimental
All manipulations were performed under an atmosphere ofdry, oxygen-free argon using standard Schlenk techniques. Allsolvents were dried by standard methods and freshly dis-tilled prior to use. The compounds 2-bromo-1,3-diethyl-1,3,2-benzodiazaborole 4,9 N,N¢-diethyl-1,2-diaminobenzene 5,10 1,4-bis(dibromoboryl)benzene,17 1,3,5-tris(dibromoboryl)benzene,17
1,3,5-tris(4¢-bromophenyl)benzene32 and 1,3,5-tris(2¢-thienyl)-benzene33 were prepared according to literature methods. 9,9¢-Dioctyl-2,7-dibromo-fluorene and 5,5¢-dibromodithiophene werepurchased from commercial sources. All experiments were con-ducted in glass vessels which were flame-dried prior to use andfilled with argon. NMR spectra were recorded from solutionsat room temperature in CDCl3 (unless otherwise stated) on aBruker AM Avance DRX500 spectrometer (1H, 11B, 13C) withSiMe4 (1H, 13C) and BF3·OEt2 (11B) as external standards. Theexpected broad 13C peaks corresponding to the carbon attachedto boron were not detected above the noise levels. Mass spectrawere obtained with a VG Autospec sector field mass spectrometer(Micromass). Absorption is measured with a UV/VIS double-beam spectrometer (Shimadzu UV-2550), using the solvent as areference. The setup used to acquire excitation-emission spectra(EES) was similar to that employed in commercial static fluo-rimeters: the output of a continuous Xe-lamp (75 W, LOT Oriel)was wavelength-separated by a first monochromator (Spectra ProARC-175, 1800 l/mm grating, Blaze 250 nm) and then usedto irradiate a sample. The fluorescence was collected by mirroroptics at right angles and imaged on the entrance slit of asecond spectrometer while compensating astigmatism at the sametime. The signal was detected by a back-thinned CCD camera(RoperScientific, 1024 ¥ 256 pixels) in the exit plane of thespectrometer. The resulting images were spatially and spectrallyresolved. As the next step, one averaged fluorescence spectrumwas calculated from the raw images and stored in the computer.This process was repeated for different excitation wavelengths.The result is a two-dimensional fluorescence pattern with they-axis corresponding to the excitation, and the x-axis to theemission wavelength. Fig. 8 shows sample spectra obtained withthis technique. Here, the wavelength range is lex = 230–450 nm(in 1 nm increments) for the UV light and lem = 100–700 nm forthe detector. The time to acquire a complete EES is typically lessthan 15 min. Post-processing of the EES includes subtraction ofthe dark current background, conversion of pixel to wavelengthscales, and multiplication with a reference file to take the varyinglamp intensity as well as grating and detection efficiency intoaccount. For all measurements, samples were contained in quartz
This journal is © The Royal Society of Chemistry 2009 Dalton Trans., 2009, 1339–1351 | 1347
Publ
ishe
d on
12
Janu
ary
2009
. Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F A
LA
BA
MA
AT
BIR
MIN
GH
AM
on
22/1
0/20
14 1
9:56
:02.
View Article Online

Fig. 8 Example of an EES spectrum.
cuvettes of 10 ¥ 10 mm2 (Hellma type 111-QS, suprasil, opticalprecision). They were prepared with distilled and dried THF, withconcentrations varying from 1 to 8 mM according to their opticaldensity. The quantum yields were determined against POPOP (p-bis-5-phenyl-oxazolyl(2)-benzene) (U = 0.93) as the standard.
The poor values of C and N in elemental analysis may be a resultof incomplete combustion due to formation of ceramic boroncarbides or nitrides. In keeping with this, varying amounts of solidresidues were found after combustion. No significant impuritiescould be detected in the NMR/MS and EAS-spectra, which canbe seen as a typical fingerprint of the molecules and would showany other fluorescent species (for example molecules with only twoborolyl functions in the case of 3a–c).
5,5¢-Bis(1¢¢,3¢¢-diethyl-1¢¢,3¢¢,2¢¢-benzodiazaborol-2¢¢-yl)-2,2¢-dithiophene 2b
A chilled solution (-78 ◦C) of 5,5¢-dibromo-2,2¢-dithiophene(1.0 g, 3.1 mmol) in THF (50 ml) was treated with 4.3 ml of a1.6 M n-hexane solution of n-butyllithium (6.5 mmol). Stirring wascontinued at -78 ◦C for 1 h and then at 0 ◦C for another h. Afterre-cooling the mixture to -78 ◦C, a neat sample of 2-bromo-1,3-diethyl-1,3,2-benzodiazaborole 4 (1.56 g, 6.2 mmol) was added viasyringe. After 1 h at -78 ◦C, stirring of the mixture was continuedovernight at ambient temperature. Solvents were then removedin vacuo. The dry residue was dissolved in dichloromethane andfiltered. The solvent was evaporated from the filtrate in vacuo andthe residue was distilled at 10-3 bar at 300 ◦C. Product 2b (1.44 g,91%) was collected as a yellow solid.
Found C, 60.45; H, 6.26; N, 9.75%; C28H32N4B2S2 requires C,65.90; H, 6.32; N, 10.98%; 1H-NMR: d 1.4 (t, 12H, 3J = 6.9 Hz,CH3), 3.96 (q, 8H, 3J = 6.9 Hz, CH2), 7.05 (m, 4H, C5,8H), 7.12(m, 4H, C6,7H), 7.36 (d, 2H, 3J = 3.4 Hz, thienyl-H), 7.43 (d, 2H,3J = 3.4 Hz, thienyl-H); 13C{1H}-NMR: d 16.3 (s, CH3), 37.9 (s,CH2), 108.8 (s, C5,8), 118.9 (s, C6,7), 125.4 (s, thienyl C3), 134.7 (s,thienyl C4), 137.1 (s, C4,9), 141.6 (s, thienyl C2); 11B{1H}-NMR:d 26.0 (s); MS/EI: m/z = 510 [M+].
1,4-Bis(1¢,3¢-diethyl-1¢,3¢,2¢-benzodiazaborol-2¢-yl)benzene 2c
To a cold slurry (0 ◦C) of calcium hydride (0.9 g, 21.4 mmol)in toluene (50 ml) were added, dropwise and simultaneously,solutions of 1,4-bis(dibromoboryl)benzene (2.00 g, 4.8 mmol) andN,N¢-diethyl-1,2-diaminobenzene 5 (1.59 g, 9.7 mmol) in 20 mlof toluene each. Stirring was continued for 1 h at 0 ◦C andthen overnight at room temperature. After filtration, the volatilecomponents were removed in vacuo. The solid residue was distilledat 10-3 bar. The distillate, collected at ca. 280 ◦C, was recrystallised
from THF to afford 1.43 g (71% yield) of product 2c as colourlesscrystals.
Found C, 72.67; H, 7.63; N, 12.89%; C26H32B2N4 requires C,73.89; H, 7.64; N, 13.26%; 1H-NMR: d 1.38 (t, 12H, 3J = 6.9 Hz,CH3), 3.85 (q, 8H, 3J = 6.9 Hz, CH2), 7.06 (m, 4H, C5,8H), 7.13 (m,4H, C6,7H), 7.65 (s, 4H, C6H4); 13C{1H}-NMR: d 16.3 (s, CH3),37.7 (s, CH2), 108.9 (s, C5,8), 118.6 (s, C6,7), 133.0 (s, C6H4), 137.2(s, C4,9); 11B{1H}-NMR: d 28.7 (s); MS/EI: m/z = 422 [M+].
9,9-Di(n-octyl)-2,7-bis(1¢,3¢-diethyl-1¢,3¢,2¢-benzodiazaborol-2¢-yl)fluorene 2e
A volume of 2.50 mL of a 1.6 M n-butyllithium solution inn-hexane (4.0 mmol) was added dropwise to a chilled solution(-78 ◦C) of 9,9-di-n-octyl-2,7-dibromofluorene (1.0 g, 1.80 mmol)in 50 ml of THF and the reaction mixture was stirred for90 min. Then a sample of 0.9 g (3.6 mmol) of 2-bromo-1,3,2,-benzodiazaborole 4 was slowly added. Stirring at -78 ◦C wascontinued for 1 h before the reaction mixture was allowed toreach room temperature over 12 h. The solvent was evaporatedin vacuo and the residue was dissolved in dichloromethane (30 ml).Filtration was followed by the complete removal of volatiles andrecrystallisation of the solid residue from n-pentane to give product2e as thin colourless needles (yield: 1.07 g, 1.45 mmol, 81%).
Found C, 73.84; H, 9.41; N, 6.34%; C49H68B2N4 requires C,80.10; H, 9.33; N, 7.63%; 1H-NMR: d 0.83–1.24 (m, 30 H, octyl-H), 1.40 (t, 12 H, 3J = 6.9 Hz, NCH2CH3), 2.05 (m, 4H, octyl-H)3.88 (q, 8H, 3J = 6.9 Hz, NCH2), 7.10 (m, 4H, C5,8H), 7.18 (m,4H, C6,7H), 7.60 (d, 2H, 3J = 7.2 Hz, aryl-H), 7.62 (s, 2H, aryl-H),7.88 (d, 2H, 3J = 7.2 Hz, aryl-H); 13C{1H}-NMR (C6D6): d 14.1 (s,octyl-CH3), 16.3 (s, NCH2CH3), 22.6, 24.1, 29.2, 29.3, 30.1, 31.8(s, octyl-CH2), 37.7 (s, NCH2), 40.4 (s, C-CH2C7H15), 55.0 (C9-fluorene), 108.8 (s, C5,8), 118.6 (s, C6,7), 119.5 (s), 128.1 (s), 131.9(s), 137.2 (s, C4,9), 141.5 (s), 150.2 (s); 11B{1H}-NMR: d 29.3 (s);MS/EI: m/z = 734 [M+].
1,3,5-Tris{5¢(1¢¢,3¢¢-diethyl-1¢¢,3¢¢,2¢¢-benzodiazaborol-2¢¢-yl)-2¢-thienyl}benzene 3a
To a chilled solution (-78 ◦C) of 1,3,5-tris(2-thienyl)benzene (0.5 g,1.54 mmol) in 50 ml of THF was added dropwise a 1.5 M hexanesolution of n-butyllithium (3.1 ml, 4.65 mmol) and the mixturewas stirred for 1.5 h. After stirring for 0.5 h at 0 ◦C, the solutionwas cooled to -78 ◦C again and three equiv. of 2-bromo-1,3,2-diazaborole 4 (1.17 g, 4.63 mmol) were added before stirringwas continued for another 2 h. After stirring overnight at roomtemperature, the mixture was filtered and the volatile componentswere removed in vacuo. The solid residue was extracted withdichloromethane and then the filtered extract was evaporated todryness. Purification of the solid residue was effected by columnchromatography (silica 60, Merck; hexane/CH2Cl2 1:1) to afford0.69 g (53%) of 3a as a colourless solid.
Found C, 67.76; H, 4.86; N, 6.12%; C48H51N6B3S3 requires C,68.59; H, 6.12; N, 10.00%; 1H-NMR: d 1.43 (t, 18H, 3J = 6.9 Hz,CH3), 4.0 (q, 12 H, 3J = 6.9 Hz, CH2), 7.08 (m, 6H, C5,8H), 7.20(m, 6H, C6,7H), 7.48 (d, 3H, 3J = 3.4 Hz, thienyl-H), 7.63 (d, 3H,3J = 3.4 Hz, thienyl-H), 7.93 (s, 3H, C6H3); 13C{1H}-NMR: d 16.4(s, CH3), 38.0 (s, CH2), 108.9 (s, C5,8), 111.4 (s), 118.9 (s, C6,7),
1348 | Dalton Trans., 2009, 1339–1351 This journal is © The Royal Society of Chemistry 2009
Publ
ishe
d on
12
Janu
ary
2009
. Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F A
LA
BA
MA
AT
BIR
MIN
GH
AM
on
22/1
0/20
14 1
9:56
:02.
View Article Online

123.3 (s,), 125.4 (s), 134.9 (s), 135.7 (s), 137.2 (s, C4,9), 148.0 (s);11B{1H}-NMR: d 26.1 (s); MS/EI: m/z = 840 [M+].
1,3,5-Tris(1¢,3¢-diethyl-1¢,3¢,2¢-benzodiazaborol-2¢-yl)benzene 3b
Solutions of 1,3,5-tris(dibromoboryl)benzene (2.0 g, 3.4 mmol)in toluene (50 ml) and of N,N¢-diethyl-1,2-diaminobenzene 5(1.7 g, 10.4 mmol) in toluene (50 ml) were added separately toa well stirred, chilled slurry (0 ◦C) of calcium hydride (3.0 g,71.5 mmol) in toluene (100 ml). Stirring at 0 ◦C was continuedfor 1 h and then overnight at room temperature. The mixture wasfiltered and the solvent was evaporated from the filtrate in vacuo.Recrystallisation of the residue from methylcyclohexane afforded1.52 g (76% yield) of colourless, crystalline 3b. Found C, 64.23;H, 7.62; N, 10.48%; C36H45N6B3 requires C, 72.77; H, 7.63; N,14.14%; 1H-NMR (CD2Cl2): d 1.36 (t, 18H, 3J = 6.9 Hz, CH3),3.90 (q, 12H, 3J = 6.9 Hz, CH2), 7.02 (m, 6H, C5,8H), 7.13 (m,6H, C6,7H), 7.91 (s, 3H, C6H3); 13C{1H}-NMR: d 16.4 (s, CH3),37.7 (s, CH2), 108.9 (s, C5,8), 118.7 (s, C6,7), 133.7 (s, C6H3), 137.2(s, C4,9); 11B{1H}-NMR: d 28.9 (s); MS/EI: m/z = 594 [M+].
1,3,5-Tris{4¢-(1¢¢,3¢¢-diethyl-1¢¢,3¢¢,2¢¢-benzodiazaborol-2¢¢-yl)phenyl}benzene 3c
A chilled solution (-78 ◦C) of 1,3,5-tris(4¢-bromophenyl)benzene(2.00 g, 3.7 mmol) in diethylether (50 ml) was treated dropwisewith 7.47 ml of a 1.6 M solution of n-butyllithium in hexane.Stirring was continued for 90 min at -78 ◦C and then for 30 minat room temperature. The reaction was re-cooled to -78 ◦C before2.82 g (11.2 mmol) of 2-bromo-1,3,2-benzodiazaborole 4 wasadded via a syringe. The obtained slurry was stirred for 2 h at-78 ◦C and then overnight at room temperature. It was filtered
and the filter-cake was extracted with hot dichloromethane. Thecombined filtrates were evaporated to dryness in vacuo to yield1.74 g (58%) of colourless solid 3c. Found C, 68.23; H, 6.96; N,8.63%; C54H57B3N6 requires C, 78.85; H, 6.99; N, 10.22; 1H-NMR:d 1.45 (t, 18H, 3J = 6.9 Hz, CH3), 3.94 (q, 12 H, 3J = 6.9 Hz,CH2), 7.10 (m, 6H, C5,8H), 7.21 (m, 6H, C6,7H), 7.75 (d, 6H,3J = 8.2 Hz, C6H4), 7.88 (d, 6H, 3J = 8.2 Hz, C6H4), 8.00 (s, 3H,C6H3); 13C{1H}-NMR: d 16.3 (s, CH3), 37.7 (s, CH2), 108.9, (s,C5,8), 118.7 (s, C6,7), 125.2 (s), 126.9 (s), 134.1 (s), 137.1 (s, C4,9),141.2 (s), 142.3 (s); 11B{1H}-NMR: d 28.6 (s); MS/EI: m/z =822 [M+].
X-Ray crystallography
Single crystals of 2a, 2b, 2c, 3a, 3b and 3c were coated with alayer of hydrocarbon oil, attached to a glass fiber and cooled to100 K for data collection. Crystallographic data were collectedwith a Nonius KappaCCD diffractometer with Mo-Ka radiation(graphite monochromator, l = 0.71073 A). Crystallographicprogrammes used for structure solution and refinement werefrom SHELXS-97 and SHELXL-97.34 The structures were solvedby direct methods and were refined by using full-matrix leastsquares on F2 of all unique reflections with anisotropic thermalparameters for all non-hydrogen atoms. Hydrogen atoms wereincluded at calculated positions with U(H) = 1.2 Ueq for CH2
groups and U(H) = 1.5 Ueq for CH3 groups. Crystal data of thecompounds are listed in Table 1. CCDC 701586 (2a), CCDC701587 (2b), CCDC 701588 (2c), CCDC 701589 (3a), CCDC701590 (3b) and CCDC 701591 (3c) contain the supplementarycrystallographic data for this paper. These data can be obtainedfree of charge from The Cambridge Crystallographic Data Centrevia www.ccdc.cam.ac.uk/data_request/cif or as ESI.†
Table 9 Crystallographic data for 2a–2c and 3a–3c
Compound 2a 2b 2c 3a 3b 3c
Empirical formula C24H30B2N4S C28H32B2N4S2 C26H32B2N4 C48H51B3N6S3 C36H45B3N6.C7H14 C54H57B3N6.4CH2Cl2
Mr [g mol-1] 428.20 510.32 422.18 840.56 692.39 1162.19Crystal dimensions [mm] 0.30 ¥ 0.27 ¥ 0.20 0.30 ¥ 0.26 ¥ 0.24 0.30 ¥ 0.28 ¥ 0.14 0.30 ¥ 0.30 ¥ 0.02 0.25 ¥ 0.24 ¥ 0.22 0.27 ¥ 0.17 ¥ 0.10Crystal system triclinic monoclinic monoclinic triclinic monoclinic triclinicSpace group P-1 P21/c P 21/n P-1 C 2/c P-1a [A] 10.0540(1) 8.9220(2) 9.8944(2) 9.5930(4) 27.1960(11) 12.3528(12)b [A] 11.4630(2) 8.2980(1) 7.2114(2) 15.6949(6) 16.9590(8) 14.9195(14)c [A] 11.9850(2) 18.0240(4) 16.7304(3) 16.9783(8) 8.7780(4) 18.265(2)a [◦] 73.5560(7) 117.317(2) 114.039(7)b [◦] 68.3920(7) 103.1130(11) 105.6255(13) 96.944(3) 92.737(3) 96.110(9)g [◦] 66.5310(7) 91.638(3) 101.902(9)V [A3] 1162.79(3) 1299.61(4) 1149.64(4) 2244.27(17) 4043.9(3) 2939.1(5)Z 2 2 2 2 4 2rcalc [g cm-3] 1.223 1.304 1.220 1.244 1.137 1.313m [mm-1] 0.158 0.230 0.071 0.207 0.066 0.427F (000) 456 540 452 888 1496 1212H [◦] 3–30 3-27.5 3–30 3–25 3–30 3–27.5No. refl. collected 54448 18514 26453 30208 60259 60087No. refl. unique 6766 2966 3345 7847 5877 13396R (int) 0.039 0.030 0.041 0.0423 0.081 0.0495No. Refl [I >2s(I)] 5874 2695 2731 5814 2851 9667Refined parameters 284 165 147 547 247 699GOF 1.035 1.036 1.038 1.019 1.025 1.036Rf [I > 2s(I)] 0.0355 0.0319 0.0423 0.0428 0.0806 0.0624wRF2(all data) 0.0948 0.0818 0.1138 0.1057 0.2532 0.1706Drmax/min [e A-3]CCDC-number
0.347/-0.263 0.300/-0.233 0.335/-0.208 0.487/-0.302 0.628/-0.332 0.893/-1.030
This journal is © The Royal Society of Chemistry 2009 Dalton Trans., 2009, 1339–1351 | 1349
Publ
ishe
d on
12
Janu
ary
2009
. Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F A
LA
BA
MA
AT
BIR
MIN
GH
AM
on
22/1
0/20
14 1
9:56
:02.
View Article Online

Single crystals suitable for X-ray crystallography were obtainedfrom THF for 2a, from methylcyclohexane for 3b and fromsolvent mixtures for 2b (THF/dichloromethane at -20 ◦C), 2c(n-pentane/dichloromethane), 3a (THF/dichloromethane) and3c (methylcyclohexane/dichloromethane at -4 ◦C). A methyl-cyclohexane molecule was present per molecule of 3b and fourdichloromethane molecules were present for each molecule of 3c.Crystallographic data for 2a–2c and 3a–3c are listed in Table 9. For3b, there is disorder of C(8) on 2 positions (64:36) and disorder ofC(20) to C(26) on an inversion centre. These bonds were restrainedto become equal. For 3c, there is disorder of two dichloromethanemolecules, C(15), C(16) and C(57) on three positions (34:29:37),disorder of C(18) on two positions (71:29). Atoms C(15A), C(57C)and Cl(18) were refined isotropically.
Computational Section
All ab initio computations were carried out with the Gaussian03 package.35 The model and full geometries discussed here wereoptimised using the B3LYP/6–31G* level of theory36,37 with nosymmetry constraints. Frequency calculations carried out onthese optimised geometries showed no imaginary frequencies.The electronic structure and TD-DFT computations were alsocarried out at the same level of theory. The MO diagrams andorbital contributions were generated with the aid of Gabedit38 andGaussSum39 packages respectively.
Acknowledgements
We thank Durham University for access to its High PerformanceComputing facility and the Deutsche Forschungsgemeinschaft,Bonn, Germany for financial support.
References
1 C. D. Entwistle and T. B. Marder, Angew. Chem., Int. Ed., 2002, 41,2927; C. D. Entwistle and T. B. Marder, Chem. Mater., 2004, 16, 4574.
2 M. Elbing and G. C. Bazan, Angew. Chem., Int. Ed., 2008, 47, 834.3 L. Weber, Coord. Chem. Rev., 2008, 252, 1.4 For recent chemistry of 1,3,2-diazaboroles see: L. Weber, I. Domke, J.
Kahlert and H.-G. Stammler, Eur. J. Inorg. Chem., 2006, 3419; L. Weber,M. Schnieder, T. C. Maciel, H. B. Wartig, M. Schimmel, R. Boese andD. Blaser, Organometallics, 2000, 19, 5791; T. B. Marder, Science, 2006,314, 69; Y. Segawa, M. Yamashita and K. Nozaki, Science, 2006, 314,113; J. M. Murphy, J. D. Lawrence, K. Kawamura, C. Incarvito andJ. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 13684; L. Weber, H. B.Wartig, H.-G. Stammler and B. Neumann, Organometallics, 2001, 20,5248; M. Suginome, A. Yamamoto and M. Murakami, Angew. Chem.,Int. Ed., 2005, 44, 2380; L. Weber, I. Domke, W. Greschner, K. Miqueu,A. Chrostowska and P. Baylere, Organometallics, 2005, 24, 5455.
5 S. Maruyama and Y. Kawanishi, J. Mater. Chem., 2002, 12, 2245.6 L. Weber, V. Werner, I. Domke, H.-G. Stammler and B. Neumann,
Dalton Trans., 2006, 3777.7 L. Weber, I. Domke, C. Schmidt, T. Braun, H.-G. Stammler and B.
Neumann, Dalton Trans., 2006, 2127.8 L. Weber, A. Penner, I. Domke, H.-G. Stammler and B. Neumann,
Z. Anorg. Allg. Chem., 2007, 633, 563.9 L. Weber, H. B. Wartig, H.-G. Stammler and B. Neumann, Z. Anorg.
Allg. Chem., 2001, 627, 2663.10 U. T. Mueller-Westerhoff, B. Vance and D. I. Yoon, Tetrahedron, 1991,
47, 909; C. H. Raeder and A. R. Day, J. Org. Chem., 1941, 6, 25.11 T. Kohler, H. Pritzkow and W. Siebert, Z. Naturforsch. B: Chem. Sci.,
2002, 57, 1101.12 A. Sunderaraman, K. Venkatasubbaiah, M. Victor, L. N. Zakharov,
A. L. Rheingold and F. Jakle, J. Am. Chem. Soc., 2006, 128, 16554.
13 D. X. Cao, Z. Q. Liu, G. B. Xu, G. Xue, G. Q. Liu and W. T. Yu,J. Organomet. Chem., 2004, 689, 2201.
14 W. Niu, M. D. Smith and J. J. Lavigne, J. Am. Chem. Soc., 2006, 128,16466.
15 P. Rodrıguez-Cuamatzi, G. Vargas-Dıaz, T. Maris, J. D. Wuest and H.Hopfl, Acta Crystallogr., 2004, E60, o1316; P. Rodrıguez-Cuamatzi, G.Vargas-Dıaz and H. Hopfl, Angew. Chem., Int. Ed., 2004, 43, 3041.
16 W. Niu, B. Rambo, M. D. Smith and J. J. Lavigne, Chem. Commun.,2005, 5166.
17 M. C. Haberecht, J. B. Heilmann, A. Haghiri, M. Bolte, J. W. Bats, H.-W. Lerner, M. C. Holthausen and M. Wagner, Z. Anorg. Allg. Chem.,2004, 630, 904.
18 A. Rodriguez, F. S. Tham, W. W. Schoeller and G. Bertrand, Angew.Chem., Int. Ed., 2004, 43, 4876.
19 Z. Yuan, N. J. Taylor, R. Ramachandran and T. B. Marder, Appl.Organomet. Chem., 1996, 10, 305.
20 L. H. Tong, S. I. Pascu, T. Jarrosson and J. K. M. Sanders, Chem.Commun., 2006, 1085.
21 K. Okada, T. Sugawa and M. Oda, J. Chem. Soc., Chem. Commun.,1992, 74.
22 M. Kinoshita and Y. Shirota, Chem. Lett., 2001, 614.23 T. Habereder and H. Noth, Appl. Organomet. Chem., 2003, 17, 525.24 T. Noda, H. Ogawa and Y. Shirota, Adv. Mater., 1999, 11, 283.25 J. S. Siddle, R. M. Ward, J. C. Collings, S. R. Rutter, L. Porres, L.
Applegarth, A. Beeby, A. S. Batsanov, A. L. Thompson, J. A. K.Howard, A. Boucekkine, K. Costuas, J. F. Halet and T. B. Marder,New J. Chem., 2007, 31, 841.
26 For examples seeM. S. Yuan, Q. Fang, Z. Q. Liu, J. P. Guo, H. Y. Chen,W. T. Yu, G. Xue and D. S. Liu, J. Org. Chem., 2006, 71, 7858; D. Cao,Z. Liu, G. Li, G. Liu and G. Zhang, J. Mol. Struct., 2008, 874, 46.
27 G. Zhou, C.-L. Ho, W.-Y. Wong, Q. Wang, D. Ma, L. Wang, Z. Lin,T. B. Marder and A. Beeby, Adv. Funct. Mater., 2008, 18, 499; Z.Yuan, C. D. Entwistle, J. C. Collings, D. Albesa-Jove, A. S. Batsanov,J. A. K. Howard, H. M. Kaiser, D. E. Kaufmann, S.-Y. Poon, W.-Y.Wong, C. Jardin, S. Fathallah, A. Boucekkine, J.-F. Halet, N. J. Taylorand T. B. Marder, Chem.–Eur. J., 2006, 12, 2758; L. Porres, M. Charlot,C. D. Entwistle, A. Beeby, T. B. Marder and M. Blanchard-Desce, Proc.SPIE, 2005, 5934, 92; M. Charlot, L. Porres, C. D. Entwistle, A. Beeby,T. B. Marder and M. Blanchard-Desce, Phys. Chem. Chem. Phys., 2005,7, 600; C. D. Entwistle, A. S. Batsanov, J. A. K. Howard, M. A. Foxand T. B. Marder, Chem. Commun., 2004, 702; C. D. Entwistle, T. B.Marder, P. S. Smith, J. A. K. Howard, M. A. Fox and S. A. Mason,J. Organomet. Chem., 2003, 680, 165; Z. Yuan, J. C. Collings, N. J.Taylor, T. B. Marder, C. Jardin and J.-F. Halet, J. Solid State Chem.,2000, 154, 5; Z. Yuan, N. J. Taylor, Y. Sun, T. B. Marder, I. D. Williamsand L.-T. Cheng, J. Organomet. Chem., 1993, 449, 27; Z. Yuan, N. J.Taylor, T. B. Marder, I. D. Williams, S. K. Kurtz and L.-T. Cheng,J. Chem. Soc., Chem. Commun., 1990, 1489.
28 G. P. Kushto, N. J. Watkins, A. J. Makinen and Z. H. Kafafi, J. Phys.Chem. B, 2007, 111, 5794.
29 L. Weber, J. Forster, H.-G. Stammler and B. Neumann, Eur. J. Inorg.Chem., 2006, 5048.
30 Y. Shirota, J. Mater. Chem., 2000, 10, 1.31 S. M. Cornet, K. B. Dillon, C. D. Entwistle, M. A. Fox, A. E. Goeta,
H. P. Goodwin, T. B. Marder and A. L. Thompson, Dalton Trans.,2003, 4395.
32 M. J. Plater, M. McKay and T. Jackson, J. Chem. Soc., Perkin Trans. 1,2000, 2695.
33 C. H. Zhu, J. Q. Pu, J. Y. Huang and J. Y. Wu, Acta Crystallogr., 2006,E62, o4312.
34 G. M. Sheldrick, SHELX-97, program for crystal structure refinement,University of Gottingen, 1997; G. M. Sheldrick, Acta Crystallogr., Sect.A, 2008, 64, 112.
35 M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb,J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C.Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci,M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M.Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T.Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E.Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo,R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi,C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P.Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D.Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K.Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S.
1350 | Dalton Trans., 2009, 1339–1351 This journal is © The Royal Society of Chemistry 2009
Publ
ishe
d on
12
Janu
ary
2009
. Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F A
LA
BA
MA
AT
BIR
MIN
GH
AM
on
22/1
0/20
14 1
9:56
:02.
View Article Online

Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz,I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y.Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson,W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03(Revision C.02), Gaussian, Inc., Wallingford, CT, 2004.
36 A. D. Becke, J. Chem. Phys., 1993, 98, 5648; C. Lee, W. Yang and R. G.Parr, Phys. Rev. B, 1988, 37, 785.
37 G. A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081;G. A. Petersson, A. Bennett, T. G. Tensfeldt, M. A. Al-Laham, W. A.Shirley and J. Mantzaris, J. Chem. Phys., 1988, 89, 2193.
38 A. R. Allouche, Gabedit 2.1.0, CNRS et Universite Claude BernardLyon1, 2007. Available at http://gabedit.sourceforge.net.
39 N. M. O’Boyle and J. G. Vos, GaussSum 1.0, Dublin City University,2005. Available at http://gausssum.sourceforge.net.
This journal is © The Royal Society of Chemistry 2009 Dalton Trans., 2009, 1339–1351 | 1351
Publ
ishe
d on
12
Janu
ary
2009
. Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F A
LA
BA
MA
AT
BIR
MIN
GH
AM
on
22/1
0/20
14 1
9:56
:02.
View Article Online
















![s3-eu-west-1.amazonaws.com › itempdf...doi.org/10.26434/chemrxiv.12931703.v1 π-Extended Helical Nanographenes: Synthesis and Photophysical Properties of Naphtho[1,2-a]pyrenes Paban](https://static.fdocument.org/doc/165x107/60c3c00561a0c4660a64dd7f/s3-eu-west-1-a-itempdf-doiorg1026434chemrxiv12931703v1-extended-helical.jpg)

