
Synthetic lethality of PARP inhibition in BRCA-network disrupted tumor cells is associated with...
11
ORIGINAL PAPER Synthetic lethality of PARP inhibition in BRCA-network disrupted tumor cells is associated with interferon pathway activation and enhanced by interferon-c Paul Warrener • Sammy Kim • Sybil M. G. Williams • Matthew Biery • Marcia Gordon • Carlo Toniatti • Michele A. Cleary • Peter S. Linsley • Michael Carleton Published online: 6 March 2012 Ó Springer Science+Business Media, LLC 2012 Abstract Tumor suppressor genes BRCA1 and BRCA2 function in a complex gene network that regulates homologous recombination and DNA double-strand break repair. Disruption of the BRCA-network through gene mutation, deletion, or RNAi-mediated silencing can sen- sitize cells to small molecule inhibitors of poly (ADP- ribose) polymerase (PARPi). Here, we demonstrate that BRCA-network disruption in the presence of PARPi leads to the selective induction and enhancement of interferon pathway and apoptotic gene expression in cultured tumor cells. In addition, we report PARPi cytotoxicity in BRCA1- deficient tumor cells is enhanced [ 10-fold when combined with interferon-c. These findings establish a link between synthetic lethality of PARPi in BRCA-network disrupted cells and interferon pathway activation triggered by genetic instability. Keywords PARP inhibition Á BRCA1 Á Synthetic lethality Á Apoptosis Á Interferon response Introduction The search for targeted cancer therapies has benefited from approaches that permit genome-scale assessment of tumor context. A targeted therapy that has generated significant interest is the selective cytotoxicity of poly (ADP ribose) polymerase (PARP) inhibition in tumor cells with a deficit in homologous recombination (HR) resulting from mutation, deletion or RNAi-mediated silencing of BRCA-network genes [1–3]. PARPs consti- tute a multi-gene family of enzymes catalyzing the polymerization and attachment of ADP-ribose units from NAD? onto specific amino acid residues of selected protein substrates [4]. PARP-1 and PARP-2 are DNA repair proteins whose catalytic activity is stimulated by DNA damage and required for repair of DNA single- strand breaks (SSBs) by way of base excision repair [5]. Inhibition of PARP activity causes an accumulation in SSBs resulting in stalled replication forks, and generation of DNA double-strand breaks (DSBs) at the site of the original DNA lesion [2]. Tumor suppressor genes BRCA1 and BRCA2 function within a multi-protein network that, in response to DSBs, plays a role in activating DNA damage responses, cell cycle checkpoints, and DSB repair [6]. SHFM1 (also referred to as DSS1) is a member of the BRCA-network, and its interaction with BRCA2 is required for recruitment of RAD51 to DNA damage foci [7, 8]. Cells harboring a functional BRCA-network can repair DSBs formed in response to PARP inhibition through HR in a RAD51-dependent manner [9]. However, PARP inhibition in the context of HR deficiency caused by BRCA-network disruption is synthetically lethal. This synthetic lethal phenotype results in part from replication fork collapse, accumulation of DSBs, cell cycle arrest [1, 2] and aberrant activation of nonhomologous end joining Electronic supplementary material The online version of this article (doi:10.1007/s10495-012-0707-4) contains supplementary material, which is available to authorized users. P. Warrener Á S. Kim Á M. Biery Á M. Gordon Á P. S. Linsley Á M. Carleton (&) Rosetta Inpharmatics LLC, A Wholly Owned Subsidiary of Merck & Co., Inc, Seattle, WA 98109, USA e-mail: [email protected] S. M. G. Williams Á C. Toniatti Merck Research Laboratories, Boston, MA 02115, USA M. A. Cleary Merck & Co., Inc, West Point, PA 19486, USA 123 Apoptosis (2012) 17:691–701 DOI 10.1007/s10495-012-0707-4
Transcript of Synthetic lethality of PARP inhibition in BRCA-network disrupted tumor cells is associated with...

ORIGINAL PAPER
Synthetic lethality of PARP inhibition in BRCA-networkdisrupted tumor cells is associated with interferon pathwayactivation and enhanced by interferon-c
Paul Warrener • Sammy Kim • Sybil M. G. Williams •
Matthew Biery • Marcia Gordon • Carlo Toniatti •
Michele A. Cleary • Peter S. Linsley • Michael Carleton
Published online: 6 March 2012
� Springer Science+Business Media, LLC 2012
Abstract Tumor suppressor genes BRCA1 and BRCA2
function in a complex gene network that regulates
homologous recombination and DNA double-strand break
repair. Disruption of the BRCA-network through gene
mutation, deletion, or RNAi-mediated silencing can sen-
sitize cells to small molecule inhibitors of poly (ADP-
ribose) polymerase (PARPi). Here, we demonstrate that
BRCA-network disruption in the presence of PARPi leads
to the selective induction and enhancement of interferon
pathway and apoptotic gene expression in cultured tumor
cells. In addition, we report PARPi cytotoxicity in BRCA1-
deficient tumor cells is enhanced[10-fold when combined
with interferon-c. These findings establish a link between
synthetic lethality of PARPi in BRCA-network disrupted
cells and interferon pathway activation triggered by genetic
instability.
Keywords PARP inhibition � BRCA1 � Synthetic
lethality � Apoptosis � Interferon response
Introduction
The search for targeted cancer therapies has benefited
from approaches that permit genome-scale assessment of
tumor context. A targeted therapy that has generated
significant interest is the selective cytotoxicity of poly
(ADP ribose) polymerase (PARP) inhibition in tumor
cells with a deficit in homologous recombination (HR)
resulting from mutation, deletion or RNAi-mediated
silencing of BRCA-network genes [1–3]. PARPs consti-
tute a multi-gene family of enzymes catalyzing the
polymerization and attachment of ADP-ribose units from
NAD? onto specific amino acid residues of selected
protein substrates [4]. PARP-1 and PARP-2 are DNA
repair proteins whose catalytic activity is stimulated by
DNA damage and required for repair of DNA single-
strand breaks (SSBs) by way of base excision repair [5].
Inhibition of PARP activity causes an accumulation in
SSBs resulting in stalled replication forks, and generation
of DNA double-strand breaks (DSBs) at the site of the
original DNA lesion [2]. Tumor suppressor genes BRCA1
and BRCA2 function within a multi-protein network that,
in response to DSBs, plays a role in activating DNA
damage responses, cell cycle checkpoints, and DSB repair
[6]. SHFM1 (also referred to as DSS1) is a member of the
BRCA-network, and its interaction with BRCA2 is
required for recruitment of RAD51 to DNA damage foci
[7, 8]. Cells harboring a functional BRCA-network can
repair DSBs formed in response to PARP inhibition
through HR in a RAD51-dependent manner [9]. However,
PARP inhibition in the context of HR deficiency caused
by BRCA-network disruption is synthetically lethal. This
synthetic lethal phenotype results in part from replication
fork collapse, accumulation of DSBs, cell cycle arrest [1,
2] and aberrant activation of nonhomologous end joining
Electronic supplementary material The online version of thisarticle (doi:10.1007/s10495-012-0707-4) contains supplementarymaterial, which is available to authorized users.
P. Warrener � S. Kim � M. Biery � M. Gordon �P. S. Linsley � M. Carleton (&)
Rosetta Inpharmatics LLC, A Wholly Owned Subsidiary
of Merck & Co., Inc, Seattle, WA 98109, USA
e-mail: [email protected]
S. M. G. Williams � C. Toniatti
Merck Research Laboratories, Boston, MA 02115, USA
M. A. Cleary
Merck & Co., Inc, West Point, PA 19486, USA
123
Apoptosis (2012) 17:691–701
DOI 10.1007/s10495-012-0707-4

(NHEJ), an error-prone means of DNA repair that drives
greater genomic instability [10].
A small molecule PARP inhibitor (PARPi) has been
used in the clinic for treatment of BRCA1 and BRCA2
mutant ovarian and breast cancer patients [11]. As BRCA
mutations are rare (*5 to 10% of all breast and ovarian
cancers are hereditary with a majority attributable to germ-
line mutations in either BRCA1 or BRCA2 [12]), there
exists a growing effort to precisely define the molecular
basis of PARPi cytotoxicity in the context of HR defi-
ciency. Notably, the application of the PARPi iniparib to
triple negative breast cancer, hypothesized to contain
tumors exhibiting a ‘‘BRCAness’’ phenotype [13, 14], has
yielded mixed clinical results [15, 16] underscoring ques-
tions around the exact mechanism of action for this com-
pound. Despite the emerging importance of PARPi as
therapeutic agents, the mechanism(s) by which they kill
tumor cells remains poorly understood. Moreover, emerg-
ing PARPi clinical data suggest that expansion of PARPi
responder populations will require a more precise under-
standing of the molecular context that sensitizes HR-defi-
cient tumors to PARPi.
To elucidate PARPi-dependent transcriptional changes
triggered specifically in HR-deficient tumor cells, we
employed lentiviral vector-directed expression of tetracy-
cline-inducible shRNAs to disrupt BRCA-network function
in HT-29 and HeLa cells [17]. We report here that shRNA-
mediated BRCA-network disruption can modify a cells
PARPi phenotype from one of resistance to sensitivity. In
addition, we establish a link between sensitivity to PARPi
and induction of interferon and apoptosis gene expression.
Furthermore, we demonstrate that recombinant IFN-cenhances the cytotoxicity of PARPi in BRCA1-deficient
cells.
Materials and methods
Cell lines and chemicals
Colon carcinoma HT-29 and cervical carcinoma HeLa S3
(HeLa) cells were obtained from the American Type Cul-
ture Collection, Rockville, MD. Cell lines were maintained
in exponential growth in Dulbecco’s modified Eagle’s
medium supplemented with 10% fetal bovine serum
(Invitrogen, Carlsbad, CA). The BRCA1 and SHFM1
inducibly silenced cell lines (HT-29 tts BRCA1 3484,
HT-29 tts BRCA1 1768, HT-29 tts SHFM1 208, HeLa tts
BRCA1 1768) were generated by transduction of cells with
lentiviral vectors expressing one of three shRNA sequen-
ces: BRCA1-3484 (GATCCCCGAAGTAGTTCAGACTG
TTATTCAAGAGATAACAGTCTGAACTACTTCTTTT
TGGAAGCTT), BRCA1-1768 (GATCCCCGGTCAA
GTGATGAATATTATTCAAGAGATAATATTCATCA
CTTGACCTTTTTGGAAGCTT) or SHFM1-208 (GGC
TGGCTTAGATGAAGATTTCAAGAGAATCTTCATCT
AAGCCAGCC) under control of KRAB-tTR [17].
Silencing was induced in these cells by treatment with
500 ng/ml doxycycline (DOX). PARPi compounds were
synthesized according to published reports and are referred
to herein as PARPi-A (#45) [18], PARPi-B (AG14361)
[19] and PARPi-C (KU0058684) [2]. Recombinant inter-
feron-c was purchased from R&D Systems (Cat # 285-IF).
Caspase 3/7 assay
Trypsinized cells were plated ± DOX. 24 h later, cells
were treated with either DMSO or 2uM PARPi-C. 72 h
after addition of PARPi-C, cells were treated with Caspase
Glo 3/7 according to manufacturer’s instructions (Pro-
mega) and read on a luminometer.
Cell proliferation and EC50 determination
Each PARPi was titrated across a nine point, fourfold
dilution series on HT-29 tts BRCA1-3484 cells cultured in
the presence or absence of DOX. Cell growth was deter-
mined by use of Alamar Blue according to manufacturer
specifications (BioSource International, Camarillo, CA.) as
described previously [20], and EC50 values for PARPi
were determined by use of GraphPad Prism software and a
non-linear curve fitting function.
Cell cycle analysis
Cell cycle profiles were generated with a FACS Calibur
flow cytometer (Becton–Dickinson) as described previously
[21].
c-H2AX assays
BRCA1 silencing was induced by DOX treatment of HT-
29 tts BRCA1 3484 cells. After 24 h, DOX (?) and DOX
(-) cells were treated with or without PARPi-C (10 lM)
for 72 h. Cells were then harvested, pelleted, and total cell
lysates extracted. Western immunoblots were performed
with 10 lg protein/lane and probed with anti-c-H2A.X
monoclonal antibody (H2A.X Ser139, Millipore cat#
05-636). Immunoblots were then stripped and re-probed
with polyclonal anti-H2A.X (Millipore cat# 07-627). Pro-
tein levels were normalized to H2A.X signal and quantified
by use of the DC protein assay kit (BioRad, Hercules, CA)
in conjunction with infrared imaging (LI-COR Biosci-
ences, Lincoln, NE).
692 Apoptosis (2012) 17:691–701
123

Gene expression analysis
Cell lysates were homogenized by QIAshredder spin col-
umns and total cellular RNA isolated with the RNAeasy
mini kit (QIAGEN). RNA amplification, labeling and
hybridization to human ink-jet DNA microarrays was
carried out as described previously [22]. Transcript regu-
lations are presented as PARPi (?)/PARPi (-) ratios in
1-dimensional hierarchical clusters in which experimental
samples are ordered and transcripts are clustered. mRNA
levels for BRCA1, SHFM1, MX1, MX2, 2,5 OASL, 2,5
OAS2 and IRF1 were quantified by PCR by use of an ABI
PRISM 7900HT sequence detection system and Assays-on-
Demand gene expression products (Applied Biosystems,
Foster City, CA). mRNA values were normalized relative
to human GUSB (catalogue no. 431088E) mRNA levels.
Drug titration and drug synergy determination
PARPi/IFN-c synergy was assessed according to the
method of Chou-Talalay method [23], and data was ana-
lyzed by CalcuSyn dose effect analyzer software.
Results
Induced repression of BRCA-network genes sensitizes
HT-29 colon adenocarcinoma cells to PARPi
To establish a cell culture model in which the transcrip-
tional basis for PARPi cytotoxicity in BRCA-network
disrupted tumor cells could be investigated, we used len-
tiviral vector-directed expression of tetracycline-inducible
shRNAs to silence BRCA-network transcripts [17].
Quantitative PCR revealed that DOX induction of lentiviral
vector-directed BRCA1 or SHFM1 shRNA in HT-29
repressed targeted transcript levels by C 85% compared to
DOX (-) controls (Supplemental Figure 1). DOX induced
transcript repression combined with PARPi treatment
resulted in a sustained accumulation of cells in G2/M
indicative of DNA damage induced cell cycle arrest
(Fig. 1). In addition, PARPi treatment caused a selective
two to threefold increase in caspase 3/7 activity in shRNA
expressing cells (Fig. 2). Furthermore, using c-H2A.X as a
marker for the accumulation of DSBs and apoptosis [24],
PARPi treatment selectively triggered a threefold increase
in phosphorylation of histone H2A.X (c-H2A.X) in shRNA
expressing cells (Supplemental Figure 2 and data not
shown), and PARPi induced the formation of c-H2A.X foci
in the nucleus of BRCA1 siRNA transfected cells
(unpublished data). Taken together these data demonstrate
that PARPi-mediated cell cycle arrest, DSB accumula-
tion and apoptosis can be induced by repression of
BRCA-network genes repeating, in HT-29, the reported
effect of PARPi in BRCA mutant cells [1, 2].
Isolation of a PARPi induced gene signature
that correlates with increasing PARPi potency
and disruption of BRCA-network function
Distinct changes in gene expression are known to accom-
pany cell cycle disruption and apoptosis. In addition, cell
cycle and apoptotic phenotypes induced by pharmacologi-
cal inhibition can be associated with both mechanism-based
and compound-specific changes in gene expression that
when segregated provide important molecular information
about cellular responses triggered by small molecule-target
engagement. To identify a PARPi mechanism-based gene
signature(s) that exhibits BRCA-network selectivity, we
assembled a panel of three PARPi that differed by potency,
structural class and EC50 in HT-29 tts BRCA1 3484
(Fig. 3) [2, 18, 19]. Each inhibitor was titrated on the HT-29
tts BRCA1 3484 cell line in the presence or absence of
DOX, and full genome transcript profiles were generated
from PARPi treated and untreated cells using Agilent 25K
ink-jet DNA microarrays. All samples were fluor-reversed
and subjected to competitive hybridization against DOX-
matched, PARPi negative controls in order to identify
transcripts whose expression was PARPi-dependent and
DOX-independent. P values were assigned to each gene
represented on the 25K array using a ratio error model for
two-color microarrays [25].
To identify only those genes whose expression was
dependent upon the activity of all three PARPi tested, we
focused our gene expression analysis on transcripts that
displayed each of the following three characteristics: (1)
PARPi dose-dependent expression, (2) altered expression
in response to multiple PARPi, and (3) altered expression
dependent upon BRCA1 transcript repression. Using this
approach we identified an induced set of genes that fulfilled
all three criteria and varied in size depending upon inhib-
itor potency (Fig. 3). Notably, for the least potent inhibitor
tested, PARPi-A, only the strongest signature genes were
induced in BRCA1- cells generating a profile in the 1 lLsample whose magnitude of induction fell between that of
the 10 and 100 nm samples for the 10–60 fold more potent
PARPi-B and C (Fig. 3). Exposure of BRCA1? cells to
1 lM of the more potent inhibitors (PARPi-B, C) triggered
induction of a subset of signature genes observed in the
BRCA1- cells, but the magnitude of induction did not
approach that observed in BRCA1- cells (Fig. 3).
To gain functional insight into PARPi-dependent gene
expression we grouped all signature genes on the basis of
their Gene Ontology (GO) annotation. We defined the
PARPi signature for this analysis as all HT-29 tts BRCA1
3484 transcripts that exhibited PARPi dose dependence
Apoptosis (2012) 17:691–701 693
123

between 1 nM and 1 lM PARPi-C and a C1.5 fold
PARPi-C dependent change in expression (p B 0.01) at the
highest PARPi-C dose (1 lM) in DOX treated cells
(Supplemental Table T1). Using as background all tran-
scripts represented on the microarray, E values were calcu-
lated for GO terms associated with PARPi signature genes.
Fig. 1 Induced silencing of
BRCA1 or SHFM1 triggers cell
cycle arrest in response to
PARPi. HT-29 tts BRCA1 3484
and SHFM1 208 cells were
cultured in the presence
(DOX?, BRCA1-) or absence
(DOX-, BRCA1?) of
500 ng/ml DOX. After 24 h,
both BRCA1? and BRCA1-
cells were treated with 10 lM
of PARPi-C followed by flow
cytometry to generate cell cycle
profiles at the indicated times
post treatment. Data are
presented as the % of total
fluorescence detected with the
mean G2/M population and
standard deviation (SD)
provided for each sample
derived from three separate
experiments
694 Apoptosis (2012) 17:691–701
123
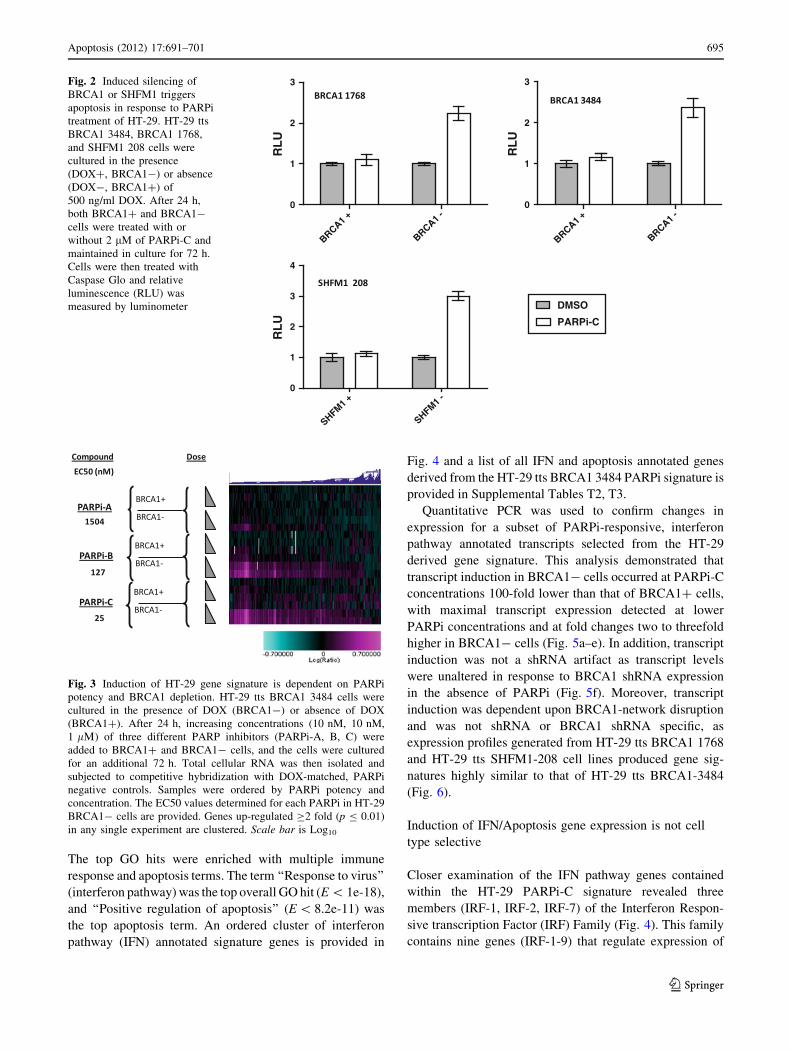
The top GO hits were enriched with multiple immune
response and apoptosis terms. The term ‘‘Response to virus’’
(interferon pathway) was the top overall GO hit (E \ 1e-18),
and ‘‘Positive regulation of apoptosis’’ (E \ 8.2e-11) was
the top apoptosis term. An ordered cluster of interferon
pathway (IFN) annotated signature genes is provided in
Fig. 4 and a list of all IFN and apoptosis annotated genes
derived from the HT-29 tts BRCA1 3484 PARPi signature is
provided in Supplemental Tables T2, T3.
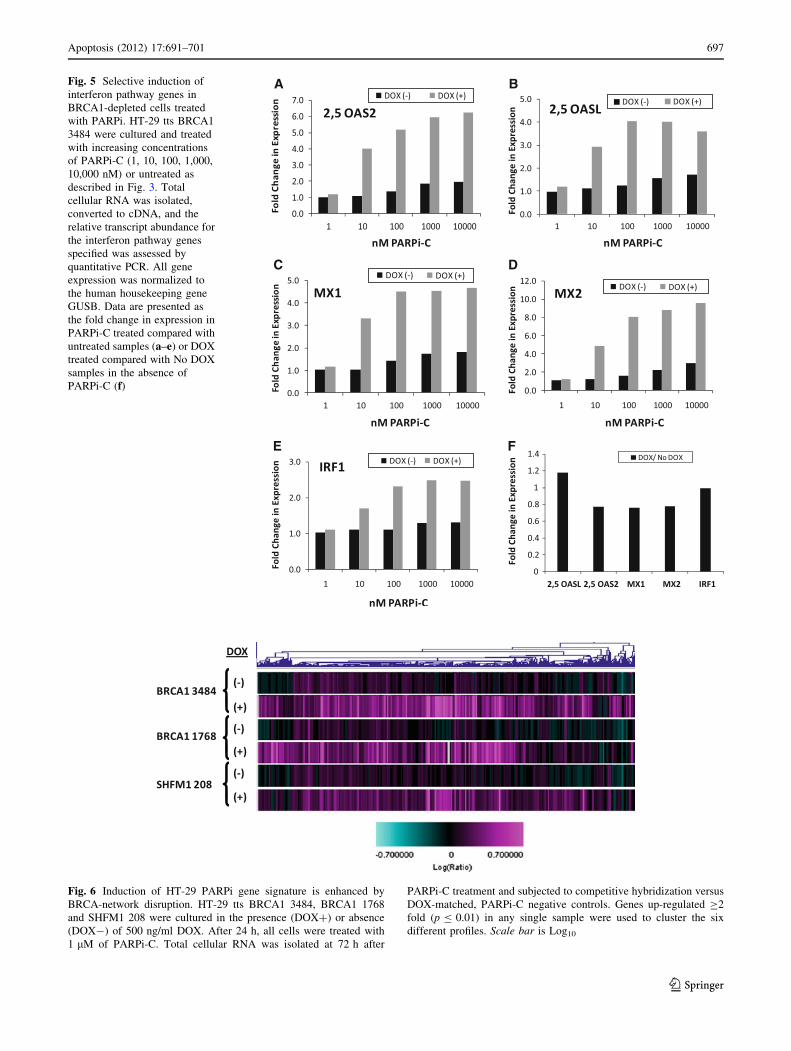
Quantitative PCR was used to confirm changes in
expression for a subset of PARPi-responsive, interferon
pathway annotated transcripts selected from the HT-29
derived gene signature. This analysis demonstrated that
transcript induction in BRCA1- cells occurred at PARPi-C
concentrations 100-fold lower than that of BRCA1? cells,
with maximal transcript expression detected at lower
PARPi concentrations and at fold changes two to threefold
higher in BRCA1- cells (Fig. 5a–e). In addition, transcript
induction was not a shRNA artifact as transcript levels
were unaltered in response to BRCA1 shRNA expression
in the absence of PARPi (Fig. 5f). Moreover, transcript
induction was dependent upon BRCA1-network disruption
and was not shRNA or BRCA1 shRNA specific, as
expression profiles generated from HT-29 tts BRCA1 1768
and HT-29 tts SHFM1-208 cell lines produced gene sig-
natures highly similar to that of HT-29 tts BRCA1-3484
(Fig. 6).
Induction of IFN/Apoptosis gene expression is not cell
type selective
Closer examination of the IFN pathway genes contained
within the HT-29 PARPi-C signature revealed three
members (IRF-1, IRF-2, IRF-7) of the Interferon Respon-
sive transcription Factor (IRF) Family (Fig. 4). This family
contains nine genes (IRF-1-9) that regulate expression of
RL
U
BRCA1 +
BRCA1 -
0
1
2
3
DMSO
PARPi-C
RL
U
BRCA1 +
BRCA1 -
0
1
2
3
RL
U
SHFM1 +
SHFM1 -
0
1
2
3
4
Fig. 2 Induced silencing of
BRCA1 or SHFM1 triggers
apoptosis in response to PARPi
treatment of HT-29. HT-29 tts
BRCA1 3484, BRCA1 1768,
and SHFM1 208 cells were
cultured in the presence
(DOX?, BRCA1-) or absence
(DOX-, BRCA1?) of
500 ng/ml DOX. After 24 h,
both BRCA1? and BRCA1-
cells were treated with or
without 2 lM of PARPi-C and
maintained in culture for 72 h.
Cells were then treated with
Caspase Glo and relative
luminescence (RLU) was
measured by luminometer
Fig. 3 Induction of HT-29 gene signature is dependent on PARPi
potency and BRCA1 depletion. HT-29 tts BRCA1 3484 cells were
cultured in the presence of DOX (BRCA1-) or absence of DOX
(BRCA1?). After 24 h, increasing concentrations (10 nM, 10 nM,
1 lL) of three different PARP inhibitors (PARPi-A, B, C) were
added to BRCA1? and BRCA1- cells, and the cells were cultured
for an additional 72 h. Total cellular RNA was then isolated and
subjected to competitive hybridization with DOX-matched, PARPi
negative controls. Samples were ordered by PARPi potency and
concentration. The EC50 values determined for each PARPi in HT-29
BRCA1- cells are provided. Genes up-regulated C2 fold (p B 0.01)
in any single experiment are clustered. Scale bar is Log10
Apoptosis (2012) 17:691–701 695
123

transcripts involved with anti-viral responses, cell cycle
regulation, and apoptosis [26]. IRF-1 is a critical regulator
of cellular responses induced by IFN-c, and its activation in
response to DNA damage involves an ATM-dependent
signaling pathway [27]. IRF-1 and IRF-5 may function as
tumor suppressor genes [28, 29], and transcription of these
two IRF family members along with IRF-3 and IRF-7 is
stimulated in response to DNA damage [30–33].
The established link between IRF expression and DNA
damage induced apoptosis led us to speculate that inter-
feron pathway gene expression could extend to PARPi
treatment of additional tumor cell types modified to repress
BRCA1. To test this hypothesis, we generated the tetra-
cycline-inducible BRCA1 shRNA cell line, HeLa tts
BRCA1 1768. Treatment of this cell line to DOX caused a
75% reduction in BRCA1 mRNA as determined by quan-
titative PCR (Supplemental Figure 1), and BRCA1
silencing increased HeLa sensitivity to PARPi-C (Supple-
mental Figure 3). In addition, PARPi-C treatment selec-
tively triggered G2/M cell cycle arrest in BRCA1-depleted
HeLa (Supplemental Figure 4) and caused the formation of
gamma H2AX nuclear foci (unpublished data). Expression
profiles of PARPi treated HeLa tts BRCA1 1768 yielded a
DOX-dependent PARPi gene signature that, like HT-29,
was enriched with transcripts having IFN pathway or
apoptosis GO annotation (Fig. 7, Supplemental Tables
T4-7). In addition, 88% of the IFN/apoptosis annotated
transcripts induced by PARPi in BRCA1-deficient HeLa
were contained in the HT-29 signature indicating that
PARPi-dependent IFN/apoptosis gene expression in the
context of BRCA-network disruption is not cell type
specific (Fig. 7, Supplemental Table T7). Furthermore,
40% of the HeLa PARPi-C signature was contained within
the larger HT-29 signature with 17% of both signatures
associated with IFN pathway or apoptosis annotation
(Fig. 7, Supplemental Table T7). The PARPi-dependent
signature in BRCA1-depleted HeLa is one-fourth the size of
the HT-29 PARPi signature generated with the same con-
centration of inhibitor (PARPi-C). This is likely a function
of gene dosage as silencing of BRCA1 mRNA is less in the
HeLa line (75%) compared to that of both HT-29 BRCA1
shRNA lines ([85%). This is consistent with a [50-fold
reduction of PARPi-C potency in HeLa tts BRCA1 1768
compared to HT-29 tts BRCA1 lines and HeLa cells in
which BRCA1 mRNA is suppressed in excess of 85% by
siRNA transfection (unpublished data), and the reduced
potency of PARPi-C in all shRNA expressing cell lines
when compared to PARPi-C potency in BRCA1 mutant
cells (Fig. 3, Supplemental Figure 3) [2].
IFN-c enhances the cytoxicity of PARPi in BRCA1-
depleted cells
Because IRF family members regulate growth inhibitory
and apoptotic responses triggered by DNA damage, we
hypothesized that recombinant IFNs might enhance the
cytotoxicity of PARPi. In preliminary HT-29 experiments,
we found that recombinant IFN-c enhanced PARPi cyto-
toxicity in BRCA1-depleted cells. Recombinant IFN-b had
a smaller effect and recombinant IFN-a had no effect
(unpublished data). Therefore, IFN-c was chosen for a
detailed assessment of Type II IFN pathway activation on
Fig. 4 PARPi triggers
enhanced IFN pathway gene
induction in BRCA1-depleted
cells. HT-29 tts BRCA1 3484
were cultured and treated with
increasing concentrations of
PARPi-C (1, 10, 100,
1,000 nM) as described in
Fig. 3. Total cellular RNA was
isolated and subjected to
competitive hybridization
versus DOX-matched, PARPi
negative controls All transcripts
associated with GO Biological
Process term ‘‘Response to
virus’’ (interferon pathway) that
were up-regulated C1.5 fold
(p B 0.01) in any single
experiment were used to cluster
samples ordered by BRCA1
status. Scale bar is Log10
696 Apoptosis (2012) 17:691–701
123
A B
C D
E F
Fig. 5 Selective induction of
interferon pathway genes in
BRCA1-depleted cells treated
with PARPi. HT-29 tts BRCA1
3484 were cultured and treated
with increasing concentrations
of PARPi-C (1, 10, 100, 1,000,
10,000 nM) or untreated as
described in Fig. 3. Total
cellular RNA was isolated,
converted to cDNA, and the
relative transcript abundance for
the interferon pathway genes
specified was assessed by
quantitative PCR. All gene
expression was normalized to
the human housekeeping gene
GUSB. Data are presented as
the fold change in expression in
PARPi-C treated compared with
untreated samples (a–e) or DOX
treated compared with No DOX
samples in the absence of
PARPi-C (f)
BRCA1 3484
BRCA1 1768
SHFM1 208
DOX
(-)
(-)
(-)
(+)
(+)
(+)
Fig. 6 Induction of HT-29 PARPi gene signature is enhanced by
BRCA-network disruption. HT-29 tts BRCA1 3484, BRCA1 1768
and SHFM1 208 were cultured in the presence (DOX?) or absence
(DOX-) of 500 ng/ml DOX. After 24 h, all cells were treated with
1 lM of PARPi-C. Total cellular RNA was isolated at 72 h after
PARPi-C treatment and subjected to competitive hybridization versus
DOX-matched, PARPi-C negative controls. Genes up-regulated C2
fold (p B 0.01) in any single sample were used to cluster the six
different profiles. Scale bar is Log10
Apoptosis (2012) 17:691–701 697
123
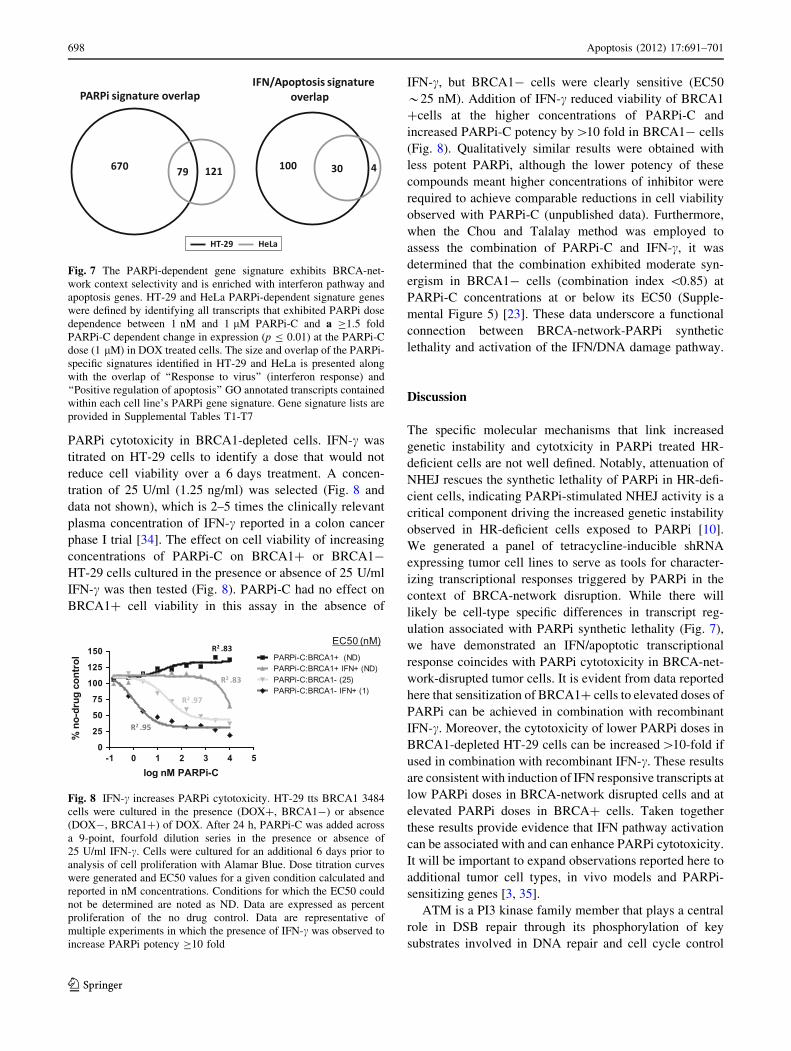
PARPi cytotoxicity in BRCA1-depleted cells. IFN-c was
titrated on HT-29 cells to identify a dose that would not
reduce cell viability over a 6 days treatment. A concen-
tration of 25 U/ml (1.25 ng/ml) was selected (Fig. 8 and
data not shown), which is 2–5 times the clinically relevant
plasma concentration of IFN-c reported in a colon cancer
phase I trial [34]. The effect on cell viability of increasing
concentrations of PARPi-C on BRCA1? or BRCA1-
HT-29 cells cultured in the presence or absence of 25 U/ml
IFN-c was then tested (Fig. 8). PARPi-C had no effect on
BRCA1? cell viability in this assay in the absence of
IFN-c, but BRCA1- cells were clearly sensitive (EC50
*25 nM). Addition of IFN-c reduced viability of BRCA1
?cells at the higher concentrations of PARPi-C and
increased PARPi-C potency by[10 fold in BRCA1- cells
(Fig. 8). Qualitatively similar results were obtained with
less potent PARPi, although the lower potency of these
compounds meant higher concentrations of inhibitor were
required to achieve comparable reductions in cell viability
observed with PARPi-C (unpublished data). Furthermore,
when the Chou and Talalay method was employed to
assess the combination of PARPi-C and IFN-c, it was
determined that the combination exhibited moderate syn-
ergism in BRCA1- cells (combination index \0.85) at
PARPi-C concentrations at or below its EC50 (Supple-
mental Figure 5) [23]. These data underscore a functional
connection between BRCA-network-PARPi synthetic
lethality and activation of the IFN/DNA damage pathway.
Discussion
The specific molecular mechanisms that link increased
genetic instability and cytotxicity in PARPi treated HR-
deficient cells are not well defined. Notably, attenuation of
NHEJ rescues the synthetic lethality of PARPi in HR-defi-
cient cells, indicating PARPi-stimulated NHEJ activity is a
critical component driving the increased genetic instability
observed in HR-deficient cells exposed to PARPi [10].
We generated a panel of tetracycline-inducible shRNA
expressing tumor cell lines to serve as tools for character-
izing transcriptional responses triggered by PARPi in the
context of BRCA-network disruption. While there will
likely be cell-type specific differences in transcript reg-
ulation associated with PARPi synthetic lethality (Fig. 7),
we have demonstrated an IFN/apoptotic transcriptional
response coincides with PARPi cytotoxicity in BRCA-net-
work-disrupted tumor cells. It is evident from data reported
here that sensitization of BRCA1? cells to elevated doses of
PARPi can be achieved in combination with recombinant
IFN-c. Moreover, the cytotoxicity of lower PARPi doses in
BRCA1-depleted HT-29 cells can be increased[10-fold if
used in combination with recombinant IFN-c. These results
are consistent with induction of IFN responsive transcripts at
low PARPi doses in BRCA-network disrupted cells and at
elevated PARPi doses in BRCA? cells. Taken together
these results provide evidence that IFN pathway activation
can be associated with and can enhance PARPi cytotoxicity.
It will be important to expand observations reported here to
additional tumor cell types, in vivo models and PARPi-
sensitizing genes [3, 35].
ATM is a PI3 kinase family member that plays a central
role in DSB repair through its phosphorylation of key
substrates involved in DNA repair and cell cycle control
Fig. 7 The PARPi-dependent gene signature exhibits BRCA-net-
work context selectivity and is enriched with interferon pathway and
apoptosis genes. HT-29 and HeLa PARPi-dependent signature genes
were defined by identifying all transcripts that exhibited PARPi dose
dependence between 1 nM and 1 lM PARPi-C and a C1.5 fold
PARPi-C dependent change in expression (p B 0.01) at the PARPi-C
dose (1 lM) in DOX treated cells. The size and overlap of the PARPi-
specific signatures identified in HT-29 and HeLa is presented along
with the overlap of ‘‘Response to virus’’ (interferon response) and
‘‘Positive regulation of apoptosis’’ GO annotated transcripts contained
within each cell line’s PARPi gene signature. Gene signature lists are
provided in Supplemental Tables T1-T7
Fig. 8 IFN-c increases PARPi cytotoxicity. HT-29 tts BRCA1 3484
cells were cultured in the presence (DOX?, BRCA1-) or absence
(DOX-, BRCA1?) of DOX. After 24 h, PARPi-C was added across
a 9-point, fourfold dilution series in the presence or absence of
25 U/ml IFN-c. Cells were cultured for an additional 6 days prior to
analysis of cell proliferation with Alamar Blue. Dose titration curves
were generated and EC50 values for a given condition calculated and
reported in nM concentrations. Conditions for which the EC50 could
not be determined are noted as ND. Data are expressed as percent
proliferation of the no drug control. Data are representative of
multiple experiments in which the presence of IFN-c was observed to
increase PARPi potency C10 fold
698 Apoptosis (2012) 17:691–701
123

[36, 37]. ATM is required to prevent DSB accumulation
during DNA replication as it functions in concert with ATR to
promote repair and restart of collapsed replication forks [38].
Stable siRNA-mediated silencing of ATM in HeLa cells
induced the expression of a thirty-five gene signature con-
taining IFN response genes IRF-7, OASL, OAS1, IFITM1
and STAT1 [39]. These transcripts were also upregulated in
ATM mutant cells derived from ataxia telangiectasia patients
[39]. There is overlap between the HeLaATM601 gene sig-
nature and the PARPi-dependent signature detected in HeLa
and HT-29 (Supplemental Table T8). This overlap in gene
expression is interesting when considered in the context of
recent reports demonstrating ATM deficiency sensitizing
tumor cells to PARPi [40, 41]. Taken together, these data
support the conclusion that PARPi-induced activation of the
interferon pathway is not a cell-type specific effect, but is a
general response to increased genetic instability resulting
from the accumulation of DSBs.
Our observation that PARPi cytotoxicity in BRCA-net-
work disrupted cells corresponds with induction of IRF
family member transcripts suggests that these transcription
factors may be driving apoptosis in our culture models. IRF
family members were originally identified as regulators of
IFN pathway gene expression. IRFs 1, 3, 5 and 7 are
activated in response to DNA damage [30–33], and IRF-1
has been shown to regulate TP53-independent DNA dam-
age-induced apoptosis [31]. In addition, BRCA1 can indi-
rectly increase IRF-7 expression by way of its regulation of
STAT1 transcriptional activity and the expression of type I
interferons [42]. However, the increased expression of
IRF-7 in BRCA1-depleted HT-29 and HeLa cells in response
to PARPi indicates an alternative mechanism is responsible
for elevated IRF-7 expression reported here. It should be
possible to evaluate the role of IRF family members and
other IFN-responsive genes in regulating PARPi cytotoxic-
ity by use of targeted siRNA libraries to screen for PARPi
suppressor genes whose depletion reduces or eliminates
PARPi cytotoxicity. In addition, HR-deficient cells in which
the IFN response has been attenuated either through RNAi-
mediated silencing or pharmacological inhibition could be
used to further define a requirement for IFN pathway acti-
vation in triggering PARPi cytotoxicity.
Interferons are important modulators of innate and
adaptive immune responses as they possess antiviral and
anti-proliferative activity in addition to being able to trigger
apoptosis [43]. Type I (IFN-a/b), type II (IFN-c) and type III
(IL-28A, IL-28B, and IL-29) interferons are grouped in part
on the basis of their distinct receptor complexes [44, 45]. The
utility of interferons as therapeutics for a whole host of
diseases, ranging from autoimmune disorders, viral infection
and cancer has and continues to be explored. Notably, IFN-chas been used clinically in combination with certain che-
motherapeutic regimes [34, 46], and it was recently shown
that IFN-c but not IFN-a/b could restore the responsiveness
of an anti-estrogen-resistant ER-positive breast cancer cell
line to fulvestrant through the induction of IRF-1 [47]. There
is also increasing interest in the therapeutic potential of type
III interferons, because their receptor expression is restricted
primarily to cells of epithelial origin, suggesting clinical use
of type III interferons, unlike IFN-a, may not be plagued by
systemic toxicity [45]. Signaling through type III receptors
was reported to produce more robust apoptosis in HT-29
cells than signaling through either type I or type II receptors,
and IFN-c treatment further sensitized HT-29 cells to type III
mediated apoptosis [48]. PARPi treatment of BRCA-net-
work disrupted HT-29 caused elevation in transcript levels
for all type III interferons, and this may in part explain the
IFN-c sensitization of HT-29 to PARPi. Furthermore, we
found that type I and type II interferons exhibited differences
in their ability to enhance PARPi cytotoxicity in BRCA-
network disrupted HT-29. This could be a cell type specific
effect or a more general phenomenon resulting from dif-
ferences in how type I and type II mediated responses
interact with PARPi-dependent IFN pathway activation. It
will be important to extend the observations reported here to
combinations of PARPi and type III interferons in addition to
defining molecular differences between various PARPi/IFN
combinations.
Our findings suggest several avenues for continued
investigation in pre-clinical animal models with the potential
to impact clinical application of PARPi. Detection of IFN
pathway activation may be a useful biomarker for assessing
in vivo responses to PARPi. Several cytokine transcripts are
contained within the PARPi signature (Fig. 5, Supplemental
Table T1, T4) suggesting PARPi-dependent elevation of
these cytokines in plasma might correlate with PARPi
response. Alternatively, interferon gene expression analysis
of circulating tumor cells or pre- and post dose tumor
biopsies, when retrievable, could be utilized to assess PARPi
response. Our observation that IFN-c can enhance PARPi
toxicity in BRCA1-deficient cells, while not predictive of
clinical benefit, does identify a possible therapeutic method
for increasing PARPi efficacy. Realization of any clinical
benefit from the combination of selected interferons with
PARPi will likely depend upon tumor type, genetic context,
and limiting interferon associated toxicity. In this context,
maturation of methods being developed to target cytokine
secreting dendritic cells directly to solid tumors could pro-
vide a more precise, less toxic method for enhancing PARPi
efficacy [49, 50].
Acknowledgments We wish to thank our colleagues at Rosetta and
Merck for support and stimulating discussions. We thank Kenzie
MacIsaac for help with GO annotation of PARPi signature gene lists.
We thank Jim Roberts, David Wiest, and Brett Kaiser for critical
reading of this manuscript. The authors state that there exists no
conflict of interest with the publication of this manuscript.
Apoptosis (2012) 17:691–701 699
123

References
1. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez
E et al (2005) Specific killing of BRCA2-deficient tumours with
inhibitors of poly(ADP-ribose) polymerase. Nature 434(7035):
913–917
2. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson
TB et al (2005) Targeting the DNA repair defect in BRCA mutant
cells as a therapeutic strategy. Nature 434(7035):917–921
3. McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift
S et al (2006) Deficiency in the repair of DNA damage by
homologous recombination and sensitivity to poly(ADP-ribose)
polymerase inhibition. Cancer Res 66(16):8109–8115
4. Kim MY, Zhang T, Kraus WL (2005) Poly(ADP-ribosyl)ation by
PARP-1: ‘PAR-laying’ NAD? into a nuclear signal. Genes Dev
19(17):1951–1967
5. Schreiber V, Dantzer F, Ame JC, de Murcia G (2006) Poly(ADP-
ribose): novel functions for an old molecule. Nat Rev Mol Cell
Biol 7(7):517–528
6. O’Donovan PJ, Livingston DM (2010) BRCA1 and BRCA2:
breast/ovarian cancer susceptibility gene products and partici-
pants in DNA double-strand break repair. Carcinogenesis 31(6):
961–967
7. Marston NJ, Richards WJ, Hughes D, Bertwistle D, Marshall CJ,
Ashworth A (1999) Interaction between the product of the breast
cancer susceptibility gene BRCA2 and DSS1, a protein func-
tionally conserved from yeast to mammals. Mol Cell Biol 19(7):
4633–4642
8. Gudmundsdottir K, Lord CJ, Witt E, Tutt AN, Ashworth A
(2004) DSS1 is required for RAD51 focus formation and geno-
mic stability in mammalian cells. EMBO Rep 5(10):989–993
9. Arnaudeau C, Lundin C, Helleday T (2001) DNA double-strand
breaks associated with replication forks are predominantly
repaired by homologous recombination involving an exchange
mechanism in mammalian cells. J Mol Biol 307(5):1235–
1245
10. Patel AG, Sarkaria JN, Kaufmann SH (2011) Nonhomologous
end joining drives poly(ADP-ribose) polymerase (PARP) inhib-
itor lethality in homologous recombination-deficient cells. Proc
Natl Acad Sci USA 108(8):3406–3411
11. Yap TA, Sandhu SK, Carden CP, de Bono JS (2011) Poly(ADP-
ribose) polymerase (PARP) inhibitors: exploiting a synthetic
lethal strategy in the clinic. CA Cancer J Clin 61(1):31–49
12. Zhang J, Powell SN (2005) The role of the BRCA1 tumor sup-
pressor in DNA double-strand break repair. Mol Cancer Res
3(10):531–539
13. Turner N, Tutt A, Ashworth A (2004) Hallmarks of ‘BRCAness’
in sporadic cancers. Nat Rev Cancer 4(10):814–819
14. Rastelli F, Biancanelli S, Falzetta A, Martignetti A, Casi C,
Bascioni R et al (2010) Triple-negative breast cancer: current
state of the art. Tumori 96(6):875–888
15. O’Shaughnessy J, Osborne C, Pippen JE, Yoffe M, Patt D, Rocha
C et al (2011) Iniparib plus chemotherapy in metastatic triple-
negative breast cancer. N Engl J Med 364(3):205–214
16. Guha M (2011) Parp inhibitors stumble in breast cancer. Nat
Biotechnol 29(5):373–374
17. Wiznerowicz M, Trono D (2003) Conditional suppression of
cellular genes: lentivirus vector-mediated drug-inducible RNA
interference. J Virol 77(16):8957–8961
18. Pescatore G, Branca D, Fiore F, Kinzel O, Bufi LL, Muraglia E et al
(2010) Identification and SAR of novel pyrrolo[1,2-a]pyrazin-
1(2H)-one derivatives as inhibitors of poly(ADP-ribose) poly-
merase-1 (PARP-1). Bioorg Med Chem Lett 20(3):1094–1099
19. Smith LM, Willmore E, Austin CA, Curtin NJ (2005) The novel
poly(ADP-Ribose) polymerase inhibitor, AG14361, sensitizes
cells to topoisomerase I poisons by increasing the persistence of
DNA strand breaks. Clin Cancer Res 11(23):8449–8457
20. Bartz SR, Zhang Z, Burchard J, Imakura M, Martin M, Palmieri
A et al (2006) Small interfering RNA screens reveal enhanced
cisplatin cytotoxicity in tumor cells having both BRCA network
and TP53 disruptions. Mol Cell Biol 26(24):9377–9386
21. Carleton M, Mao M, Biery M, Warrener P, Kim S, Buser C et al
(2006) RNA interference-mediated silencing of mitotic kinesin
KIF14 disrupts cell cycle progression and induces cytokinesis
failure. Mol Cell Biol 26(10):3853–3863
22. Hughes TR, Mao M, Jones AR, Burchard J, Marton MJ, Shannon
KW et al (2001) Expression profiling using microarrays fabri-
cated by an ink-jet oligonucleotide synthesizer. Nat Biotechnol
19(4):342–347
23. Chou TC, Talalay P (1984) Quantitative analysis of dose-effect
relationships: the combined effects of multiple drugs or enzyme
inhibitors. Adv Enzyme Regul 22:27–55
24. Kinner A, Wu W, Staudt C, Iliakis G (2008) Gamma-H2AX in
recognition and signaling of DNA double-strand breaks in the
context of chromatin. Nucleic Acids Res 36(17):5678–5694
25. Weng L, Dai H, Zhan Y, He Y, Stepaniants SB, Bassett DE
(2006) Rosetta error model for gene expression analysis. Bioin-
formatics 22(9):1111–1121
26. Tanaka N, Taniguchi T (2000) The interferon regulatory factors
and oncogenesis. Semin Cancer Biol 10(2):73–81
27. Pamment J, Ramsay E, Kelleher M, Dornan D, Ball KL (2002)
Regulation of the IRF-1 tumour modifier during the response to
genotoxic stress involves an ATM-dependent signalling pathway.
Oncogene 21(51):7776–7785
28. Bouker KB, Skaar TC, Riggins RB, Harburger DS, Fernandez
DR, Zwart A et al (2005) Interferon regulatory factor-1 (IRF-1)
exhibits tumor suppressor activities in breast cancer associated
with caspase activation and induction of apoptosis. Carcinogen-
esis 26(9):1527–1535
29. Hu G, Barnes BJ (2006) Interferon regulatory factor-5-regulated
pathways as a target for colorectal cancer therapeutics. Expert
Rev Anticancer Ther 6(5):775–784
30. Hu G, Mancl ME, Barnes BJ (2005) Signaling through IFN regu-
latory factor-5 sensitizes p53-deficient tumors to DNA damage-
induced apoptosis and cell death. Cancer Res 65(16):7403–7412
31. Tamura T, Ishihara M, Lamphier MS, Tanaka N, Oishi I, Aizawa
S et al (1995) An IRF-1-dependent pathway of DNA damage-
induced apoptosis in mitogen-activated T lymphocytes. Nature
376(6541):596–599
32. Kim T, Kim TY, Song YH, Min IM, Yim J, Kim TK (1999)
Activation of interferon regulatory factor 3 in response to DNA-
damaging agents. J Biol Chem 274(43):30686–30689
33. Kim TK, Kim T, Kim TY, Lee WG, Yim J (2000) Chemother-
apeutic DNA-damaging drugs activate interferon regulatory
factor-7 by the mitogen-activated protein kinase kinase-4-cJun
NH2-terminal kinase pathway. Cancer Res 60(5):1153–1156
34. Turner PK, Houghton JA, Petak I, Tillman DM, Douglas L,
Schwartzberg L et al (2004) Interferon-gamma pharmacokinetics
and pharmacodynamics in patients with colorectal cancer. Cancer
Chemother Pharmacol 53(3):253–260
35. Turner NC, Lord CJ, Iorns E, Brough R, Swift S, Elliott R et al
(2008) A synthetic lethal siRNA screen identifying genes medi-
ating sensitivity to a PARP inhibitor. EMBO J 27(9):1368–1377
36. Elson A, Wang Y, Daugherty CJ, Morton CC, Zhou F, Campos-
Torres J et al (1996) Pleiotropic defects in ataxia-telangiectasia
protein-deficient mice. Proc Natl Acad Sci USA 93(23):13084–
13089
37. Smith J, Tho LM, Xu N, Gillespie DA (2010) The ATM-Chk2
and ATR-Chk1 pathways in DNA damage signaling and cancer.
Adv Cancer Res 108:73–112
700 Apoptosis (2012) 17:691–701
123

38. Trenz K, Smith E, Smith S, Costanzo V (2006) ATM and ATR
promote Mre11 dependent restart of collapsed replication forks
and prevent accumulation of DNA breaks. EMBO J 25(8):
1764–1774
39. Chen S, Wang G, Makrigiorgos GM, Price BD (2004) Stable
siRNA-mediated silencing of ATM alters the transcriptional
profile of HeLa cells. Biochem Biophys Res Commun 317(4):
1037–1044
40. Weston VJ, Oldreive CE, Skowronska A, Oscier DG, Pratt G,
Dyer MJ et al (2010) The PARP inhibitor olaparib induces sig-
nificant killing of ATM-deficient lymphoid tumor cells in vitro
and in vivo. Blood 116(22):4578–4587
41. Williamson CT, Muzik H, Turhan AG, Zamo A, O’Connor MJ,
Bebb DG et al (2010) ATM deficiency sensitizes mantle cell
lymphoma cells to poly(ADP-ribose) polymerase-1 inhibitors.
Mol Cancer Ther 9(2):347–357
42. Buckley NE, Hosey AM, Gorski JJ, Purcell JW, Mulligan JM,
Harkin DP et al (2007) BRCA1 regulates IFN-gamma signaling
through a mechanism involving the type I IFNs. Mol Cancer Res
5(3):261–270
43. Parmar S, Platanias LC (2003) Interferons: mechanisms of action
and clinical applications. Curr Opin Oncol 15(6):431–439
44. Samuel CE (2001) Antiviral actions of interferons. Clin Micro-
biol Rev 14(4):778–809 (table of contents)
45. Donnelly RP, Kotenko SV (2010) Interferon-lambda: a new
addition to an old family. J Interferon Cytokine Res 30(8):
555–564
46. Schwartzberg LS, Petak I, Stewart C, Turner PK, Ashley J,
Tillman DM et al (2002) Modulation of the Fas signaling path-
way by IFN-gamma in therapy of colon cancer: phase I trial and
correlative studies of IFN-gamma, 5-fluorouracil, and leucovorin.
Clin Cancer Res 8(8):2488–2498
47. Ning Y, Riggins RB, Mulla JE, Chung H, Zwart A, Clarke R
(2010) IFNgamma restores breast cancer sensitivity to fulvestrant
by regulating STAT1, IFN regulatory factor 1, NF-kappaB, BCL2
family members, and signaling to caspase-dependent apoptosis.
Mol Cancer Ther 9(5):1274–1285
48. Li W, Lewis-Antes A, Huang J, Balan M, Kotenko SV (2008)
Regulation of apoptosis by type III interferons. Cell Prolif
41(6):960–979
49. Komita H, Zhao X, Katakam AK, Kumar P, Kawabe M, Okada H
et al (2009) Conditional interleukin-12 gene therapy promotes
safe and effective antitumor immunity. Cancer Gene Ther
16(12):883–891
50. Huang FP, Chen YX, To CK (2011) Guiding the ‘‘misguided’’—
functional conditioning of dendritic cells for the DC-based
immunotherapy against tumours. Eur J Immunol 41(1):18–25
Apoptosis (2012) 17:691–701 701
123


















