
Studies on the role of aminoacyl-tRNA synthesis in the...
230
UNIVERSITY OF PATRAS SCHOOL OF MEDICINE DEPARTMENT OF BIOCHEMISTRY Studies on the role of aminoacyl-tRNA synthesis in the regulation of ribosomal and exo-ribosomal protein synthesis in pathogens DOCTORAL THESIS MARIA APOSTOLIDI BIOCHEMIST & BIOTECHNOLOGIST PATRAS 2015
Transcript of Studies on the role of aminoacyl-tRNA synthesis in the...

UNIVERSITY OF PATRAS
SCHOOL OF MEDICINE
DEPARTMENT OF BIOCHEMISTRY
Studies on the role of aminoacyl-tRNA synthesis in the regulation of
ribosomal and exo-ribosomal protein synthesis in pathogens
DOCTORAL THESIS
MARIA APOSTOLIDI
BIOCHEMIST & BIOTECHNOLOGIST
PATRAS 2015


ΠΑΝΕΠΙΣΗΜΙΟ ΠΑΣΡΩΝ – ΧΟΛΗ ΕΠΙΣΗΜΩΝ ΤΓΕΙΑ
ΣΜΗΜΑ ΙΑΣΡΙΚΗ
ΣΟΜΕΑ ΒΑΙΚΩΝ ΙΑΣΡΙΚΩΝ ΕΠΙΣΗΜΩΝ Ι
ΕΡΓΑΣΗΡΙΟ ΒΙΟΛΟΓΙΚΗ ΧΗΜΕΙΑ
Μελζτεσ επί του ρόλου τθσ ςφνκεςθσ μορίων αμινοάκυλο-tRNA ςτθν
ρφκμιςθ τθσ ριβοςωμικισ και εξω-ριβοςωμικισ πρωτεϊνικισ ςφνκεςθσ
ςε πακογόνα βακτιρια
ΔΙΔΑΚΣΟΡΙΚΗ ΔΙΑΣΡΙΒΗ
ΜΑΡΙΑ ΑΠΟΣΟΛΙΔΗ
ΒΙΟΧΗΜΙΚΟ & ΒΙΟΣΕΧΝΟΛΟΓΟ
ΠΑΣΡΑ 2015




“Δε μπορείσ να ανακαλφψεισ νζουσ ωκεανοφσ αν δεν ζχεισ το κουράγιο να χάςεισ τθν ακτι
από τα μάτια ςου.”
—Πλάτων
“One never notices what has been done; one can only see what remains to be done.”
— Marie Curie Letter to her brother (18 Mar 1894)


ΕΤΧΑΡΚΣΚΕ
Πρϊτον από όλουσ, κα ικελα να ευχαριςτιςω τον Κακθγθτι μου, τον κφριο
Κωνςταντίνο τακόπουλο, για τθν επιςτθμονικι κακοδιγθςι του και τθν άψογθ
ςυνεργαςία. Θταν ο πρϊτοσ κακθγθτισ από τον οποίο διδάχτθκα τθ βιοχθμεία και
τόςο θ διδακτικι του ικανότθτα όςο και θ επιςτθμονικι του ςκζψθ και λόγοσ με
βοικθςαν να επιλζξω το δφςκολο αλλά πάντα ενδιαφζρον κομμάτι τθσ ζρευνασ.
Ευχαριςτϊ τον Κακθγθτι Διονφςιο Δραΐνα για τθν προκυμία του να με
ςυμβουλεφςει κάκε φορά που χρειαηόμουν τθν πλιρωσ καταρτιςμζνθ επιςτθμονικι
του άποψθ όςο και για τθ φιλικι και άψογθ ςυνεργαςία του.
« J’aimerais remercier le Professeur Hubert Becker de l’Université de Strasbourg, pour
sa collaboration et son aide sur la partie de mon travail, concernant la recherche,
ainsi que pour son soutien personnel. En plus je ne dois pas oublier mes sincères
remerciements à tous les collègues du labo de Strasbourg pour leur collaboration
impeccable et intègre. Bruno Senger, Jonathan Huot, Yuhei Araiso, Ludovic Enkler,
Daphné Laporte et Gaetan Bader.»
Ευχαριςτϊ τον Κακθγθτι Γεϊργιο πυροφλια για όλθ του τθ βοικεια και τισ
ςυμβουλζσ του ςε επιςτθμονικό επίπεδο αλλά και για τθ φιλικι του ςυνεργαςία,
όπωσ επίςθσ και για τθν υποςτιριξθ του ςε προςωπικό επίπεδο.
Ια ικελα ακόμα να ευχαριςτιςω ιδιαίτερα όλουσ τουσ κακθγθτζσ του Εργαςτθρίου
Βιολογικισ Χθμείασ, όπωσ επίςθσ και τθν υποψιφια διδάκτορα Κατερίνα
Γραφανάκθ για τθν ςυνεργαςία και τισ φιλικζσ ςυμβουλζσ τουσ όλα αυτά τα χρόνια
που αποτελϊ μζλοσ αυτοφ του εργαςτθρίου.
Ευχαριςτϊ ιδιαίτερα τουσ κακθγθτζσ Γεϊργιο Ντίνο, πφρο Πουρνάρα, Χαράλαμπο
Γϊγο, που δζχτθκαν να είναι μζλθ τθσ επταμελοφσ εξεταςτικισ επιτροπισ.
Επίςθσ κα ικελα ιδιαίτερα να ευχαριςτιςω τον ςυνεργάτθ μου ςτο εργαςτιριο και
ςυμφοιτθτι μου, υποψιφιο διδάκτορα Δθμιτρθ Αναςταςάκθ, όπωσ επίςθσ και τθ
διδάκτορα του τμιματοσ Χρυςαυγι Σουμπζκθ για τισ επιςτθμονικζσ ςυμβουλζσ
τουσ, τθ ςυνεργαςία τουσ και τθν φιλία τουσ. Ια ικελα να ευχαριςτιςω ξεχωριςτά
τισ μεταπτυχιακζσ φοιτιτριεσ Κατερίνα Σςίκα, Δανάθ Γιάνναρθ και Ιζνια
Κωνςταντινίδου για τθ φιλία τουσ και τθν αγάπθ τουσ που κα ικελα να μπορϊ να
τουσ ανταποδίδω πάντα.
Ια ικελα επίςθσ ιδιαίτερα να ευχαριςτιςω τουσ μεταπτυχιακοφσ και
προπτυχιακοφσ φοιτθτζσ του εργαςτθρίου του κυρίου τακόπουλου για τθ
ςυνεργαςία τουσ και το κλίμα φιλίασ και υποςτιριξθσ: τον Ηλία κεπαρνιά, τθν
Δζςποινα Ψυχογιοφ, τθν Κωνςταντίνα Παπακωνςταντίνου, τον Νάςο οκάτ, τον
Κάςων Καρυοφφλλθ και τθν Χριςτιάννα Γενεκλίου.

Δε κα μποροφςα να παραλείψω επίςθσ να ευχαριςτιςω κερμά και ξεχωριςτά όλα
τα μζλθ του εργαςτθρίου του κυρίου Δραΐνα και του κυρίου πυροφλια για τθν
φιλία τουσ και τθν υποςτιριξι τουσ: τθν Μαρία Μπίκου, τθν Χρφςα Κοντοποφλου,
τον Διονφςθ Βοφρτςθ, τον Χριςτο Χαςάπθ και τθν Κατερίνα Αργυρίου.
Ια ικελα να ευχαριςτιςω ιδιαίτερα τουσ διδάκτορεσ Βίκυ ταματοποφλου και
Μάριο Κροκίδθ για τθν ςυνεργαςία τουσ και τθ φιλία τουσ, όπωσ επίςθσ και τουσ
υπόλοιπουσ μεταπτυχιακοφσ και διδακτορικοφσ φοιτθτζσ του εργαςτθρίου
Βιολογικισ Χθμείασ, τθ Ράνια Κωςτοποφλου, τθ Γωγϊ Κουρνοφτου, τον Αντϊνθ
Μπουγά για τθν εξαιρετικι ςυνεργαςία που είχαμε και για το πολφ καλό φιλικό
περιβάλλον.
Ευχαριςτϊ τισ φίλεσ μου τζλλα, Μαριζττα, Μαρία, Νατάςα, Χρφςα, Χρυςαυγι,
Βίκυ, Κατερίνα και Δανάθ, όπωσ και τον φίλο μου Βαςίλθ, γιατί ιταν πάντα κοντά
μου ςε όλεσ τισ ευχάριςτεσ αλλά και δφςκολεσ ςτιγμζσ.
Σζλοσ, δε κα μποροφςα να εκφράςω εφκολα με λόγια τθν ευγνωμοςφνθ που ζχω για
τουσ γονείσ μου που ιταν πάντα ςτο πλευρό μου και με υποςτιριηαν ςε κάκε μου
βιμα. Πιςτεφω ότι τουσ χρωςτάω πολλά, γιατί χωρίσ τθ βοικεια τουσ κανζνα από τα
όνειρα μου δε κα είχε γίνει πραγματικότθτα. Μαηί με αυτοφσ ευχαριςτϊ τον αδερφό
μου, τθ Νονά μου και τον Νονό μου που αποτελοφν ιδιαίτερο κομμάτι τθσ ηωισ μου
και με ςτθρίηουν ςε ότι και να κάνω.

The present thesis was conducted at the department of Biochemistry, School of Medicine, University of Patras,
Greece. Part of the work described in this thesis was conducted at the Department of Génétique Moléculaire,
Génomique, Microbiologie (GMGM), University of Strasbourg, France.
ADVISORY COMMITTEE
1. Professor Constantinos Stathopoulos (Supervisor – School of Medicine, University of Patras)
2. Professor Denis Drainas (Internal Member – School of Medicine, University of Patras)
3. Professor Hubert D. Becker (External Member – GMGM, University of Strasbourg, France)
EXAMINATION COMMITTEE
1. Professor Constantinos Stathopoulos (Supervisor – School of Medicine, University of Patras)
2. Professor Denis Drainas (Internal Member – School of Medicine, University of Patras)
3. Professor Hubert D. Becker (External Member – GMGM, University of Strasbourg, France)
4. Professor Georgios A. Spyroulias (External Member – Department of Pharmacy, University of
Patras)
5. Professor Charalampos Gogos (Internal Member – School of Medicine, University of Patras)
6. Associate Professor George Dinos (Internal Member – School of Medicine, University of Patras)
7. Associate Professor Spyros Pournaras (External Member – School of Medicine, University of
Athens)
The present thesis has been supported by the University of Patras Research Committee “K. Karatheodoris” Grant
(D164) and in part under the "ARISTEIA I" Action of the "OPERATIONAL PROGRAMME EDUCATION AND LIFELONG
LEARNING" which is co-funded by the European Social Fund (ESF) and National Resources (D608). FEBS committee
is also gratefully acknowledged for granting a short term fellowship.


Η παροφςα διδακτορικι διατριβι εκπονικθκε ςτο Εργαςτιριο Βιολογικισ Χθμείασ του Σμιματοσ Ιατρικισ του
Πανεπιςτθμίου Πατρϊν, Ελλάδα. Μζροσ τθσ εργαςίασ εκπονικθκε ςτο Σμιμα Γενετικισ, Γενωμικισ Ανάλυςθσ και
Μικροβιολογίασ του Πανεπιςτθμίου του τραςβοφργου, Γαλλία [Génétique Moléculaire, Génomique,
Microbiologie (GMGM), University of Strasbourg, France].
ΣΡΙΜΕΛΗ ΤΜΒΟΤΛΕΤΣΙΚΗ ΕΠΙΣΡΟΠΗ
1. Κακθγθτισ Κωνςταντίνοσ τακόπουλοσ (Επιβλζπων – Σμιμα Ιατρικισ, Πανεπιςτιμιο Πατρϊν)
2. Κακθγθτισ Διονφςιοσ Δραΐνασ (Σμιμα Ιατρικισ, Πανεπιςτιμιο Πατρϊν)
3. Professor Hubert D. Becker (GMGM, University of Strasbourg, France)
ΕΠΣΑΜΕΛΗ ΕΞΕΣΑΣΙΚΗ ΕΠΙΣΡΟΠΗ
1. Κακθγθτισ Κωνςταντίνοσ τακόπουλοσ (Επιβλζπων – Σμιμα Ιατρικισ, Πανεπιςτιμιο Πατρϊν)
2. Κακθγθτισ Διονφςιοσ Δραΐνασ (Μζλοσ τριμελοφσ – Σμιμα Ιατρικισ, Πανεπιςτιμιο Πατρϊν)
3. Professor Hubert D. Becker (Μζλοσ τριμελοφσ – GMGM, University of Strasbourg, France)
4. Κακθγθτισ Γεϊργιοσ πυροφλιασ (Σμιμα Φαρμακευτικισ, Πανεπιςτιμιο Πατρϊν)
5. Κακθγθτισ Χαράλαμποσ Γϊγοσ (Σμιμα Ιατρικισ, Πανεπιςτιμιο Πατρϊν)
6. Αναπλθρωτισ Κακθγθτισ Γεϊργιοσ Ντίνοσ (Σμιμα Ιατρικισ, Πανεπιςτιμιο Πατρϊν)
7. Αναπλθρωτισ Κακθγθτισ πφροσ Πουρνάρασ (Σμιμα Ιατρικισ, Εκνικό Καποδιςτριακό
Πανεπιςτιμιο Ακθνϊν)
Η παροφςα διδακτορικι διατριβι υποςτθρίχκθκε από τθν Επιτροπι Ερευνϊν του Πανεπιςτιμιου Πατρϊν, *Κ.
Καρακεοδωρισ 2010 (D164)+ και εν μζρει ςτο πλαίςιο του Επιχειρθςιακοφ Προγράμματοσ Εκπαίδευςθσ και Δια
Βίου Μάκθςθσ «Αριςτεία Ι», που ςυγχρθματοδοτείται από το Ευρωπαϊκό Κοινωνικό Σαμείο (ΕΚΣ) και από
Εκνικοφσ Πόρουσ (D608). Επίςθσ θ εργαςία αυτι υποςτθρίχκθκε με τθ χοριγθςθ υποτροφίασ από τθν
Ομοςπονδία Ευρωπαϊκϊν Βιοχθμικϊν Εταιρειϊν (FEBS Short Term Fellowship).
Η ζγκριςθ τθσ διδακτορικισ διατριβισ από το Σμιμα Ιατρικισ δεν υποδθλϊνει αποδοχι των απόψεων του
ςυγγραφζα (Νόμοσ 5343/32, άρκρο 202 §2).




Table of contents
Abstract -Περίληψη 23
Introduction 33
1. The tRNA molecule 35
1.1. The “adaptor hypothesis” and the “second genetic code” 35
1.2. Structure, identity elements and evolution 36
1.3. Biogenesis and processing 39
1.4. tRNA and pathogenesis 43
2. Aminoacyl-tRNA synthetases (aaRSs) 45
2.1. Classes of aaRSs, functional and structural features 45
2.2. tRNA-dependent amidotransferases (AdTs) 49
2.3. Function complexity of eukaryotic aaRSs and their connections to
diseases 52
3. The regulatory role of tRNAs outside translation 55
3.1. Roles of charged tRNAs 56
3.1.1. Cell wall formation and remodeling in pathogens 56
3.1.2. Antibiotic biosynthesis 58
3.1.3. Protein turnover 59
3.1.4. Precursors for tRNA-dependent aa-tRNA formation 60
3.1.5. Tetrapyrrole biosynthesis 60
3.2. Roles of uncharged tRNAs 61
3.2.1. Role of tRNAs in the regulation of gene expression 61
3.2.2. tRNA-derived fragments (tRFs) 63
3.2.3. tRNA-mediated cell death regulation 65
4. Transcriptional attenuation and metabolic regulation in bacteria 67
5. Riboswitches 71
5.1. Origins and mechanism 73
5.2. Riboswitch categories 74

Table of contents
5.3. The T-box regulatory system 78
5.3.1. T-box regulons and mechanism 79
5.3.2. Structural analysis of T-boxes 83
5.3.3. Targeting a T-box riboswitch 85
6. tRNA-dependent exo-ribosomal protein synthesis in pathogens 86
6.1. Interpeptide bridge formation in Staphylococcus aureus cell wall 87
6.1.1. The role of Fem factors 88
6.1.2. Proteinogenic and non-proteinogenic tRNAGly 90
7. Antibiotic resistance regulation in Staplylococcus aureus 92
7.1. RNA-mediated infectivity and resistance modulation 94
7.2. RNAs as Targets for Antimicrobial Drugs 96
Emerging questions and aim of the thesis 97
Materials and Methods 101
1. Materials 103
1.1. Chemicals 103
1.2. Enzymes 104
1.3. Kits 104
1.4. Bacterial strains 105
1.5. Bacterial vectors 105
1.6. Primers 105
2. Methods 108
2.1. Cloning and expression of S. epidermidis GlyRS enzyme 108
2.1.1. Vector cloning and production of bacterial transformants 108
2.1.2. Protein expression and purification 109
2.2. Total tRNA extraction from S. epidermidis 110
2.3. S. aureus glyS T-box and tRNAGly transcripts production and purification 111

Table of contents
2.3.1. Cloning of T-box leader RNA constructs 111
2.3.2. In vitro transcription 112
2.3.3. RNA transcript purification and labeling 114
2.4. In silico analysis 115
2.4.1. Staphylococcal GlyRS protein structural homology modeling 115
2.4.2. S. aureus glyS T-box leader in silico secondary structure prediction 116
2.4.3. glyS T-box:tRNAGly complex Kd determination 117
2.5. In vitro assays for biochemical and structural characterization 117
2.5.1. Aminoacylation assay 117
2.5.2. Electrophoresis mobility shift assay 118
2.5.3. Chemical and enzymatic probing analysis 119
2.5.4. In vitro tRNA directed antitermination assay 122
2.6. In vivo experiments 124
2.6.1. RT- PCR validation of endogenous Sau glyS T-box riboswitch system 124
2.6.2. S. aureus in vivo expression system 126
2.6.3. Construction of T-box:tRNA double expression system in E. coli 127
2.6.4. In vivo anti-termination assay (β-galactosidase activity test) 130
Results 133
1. Cross-species aminoacylation of tRNAGly reveals functional
differences 135
1.1. Cloning and expression of recombinant GlyRS enzyme from
S.epidermidis
135
1.2. In silico structure prediction model of Staphylococcal GlyRS 136
1.3. S. epidermidis GlyRS enzyme charges differentially S. aureus tRNAGly 138
1.4. Proteinogenic and non-proteinogenic tRNAGly isoacceptors in S. aureus 140
1.5. Contribution of NEW-tRNAGly in Staphylococcal cell wall formation 142

Table of contents
2. Identification of the S. aureus glyS mRNA leader sequence 143
2.1. In silico analysis of the glyS 5’UTR sequence 143
2.2. Verification of the predicted 5’UTR glyS regulatory system 146
2.3. Identification of the predicted transcription starting point 147
3. Analysis of the S. aureus glyS T-box structure 148
3.1. In silico analysis of the glyS T-box riboswitch RNA secondary structure 148
3.2. In vitro verification of the predicted structural elements 154
3.2.1. Structural analysis of the stem I conformation 155
3.2.2. Structural analysis of the unusual terminator/antiterminator stem 158
4. Analysis of the regulatory role of S. aureus glyS T-box riboswitch 160
4.1. S. aureus glyS T-box interacts with all tRNAGly isoacceptors 160
4.2. Binding analysis of the glyS T-box:tRNAGly complex formation 162
4.3. Transcription read-through is induced by all tRNAGly isoacceptors 164
4.4. Differential glyS T-box structural changes upon tRNAGly binding 167
5. In vivo function of the S. aureus glyS T-box riboswitch mechanism 171
Discussion 177
1. Glycyl-tRNA synthetase: a structural divergent but functionally
significant enzyme from bacteria to mammals 179
1.1. Eukaryotic GlyRS and its association with disease etiology in humans 181
1.2. Staphylococcal GlyRS involvement in cell wall synthesis 184
2. Staphylococcal glyS gene expression is controlled by a species-specific
T-box regulatory element 185
3. The glyS T-box riboswitch exhibits a divergent structure 188

Table of contents
4. Genetic code-like ambiguity of glycine codon reading by the S. aureus
glyS T-box riboswitch 190
5. A proposed mechanism for synchronization of two essential but
metabolically unrelated pathways in S. aureus 193
6. Conclusions and perspectives 194
Abbreviations 197
Supplementary information 199
References 205
Publications 223




Abstract-Περίλθψθ
25
During the flow of the genetic information, tRNA molecules hold a central position as
adaptors between nucleic acids and proteins. Although until recently it was believed that
tRNAs act only as passive carriers of amino acids, recent discoveries brought them into
spotlight as essential regulators of transcription and translation. It has been recently
established that the functional role of tRNA molecules extends beyond their core cellular
role in translation. This notion triggered a new point of view of tRNAs and established their
involvement in gene expression regulation. The parallel identification of an important
riboswitch class which is highly distributed among bacterial organisms (mostly in pathogens),
termed T-boxes, confirmed the ability of tRNAs to control essential metabolic pathways. T-
box riboswitches are found in the 5’UTR of mRNAs and can utilize either charged or
uncharged tRNA molecules as ligands in order to control the expression of enzymes involved
in amino acid biosynthesis and aminoacyl-tRNA synthesis. It is known that T-boxes can
regulate downstream gene transcription by adopting two alternative conformations termed
"terminator" and "antiterminator”. The uncharged tRNA which is initially recognized by the
specifier loop in stem I region through codon-anticodon complementarity, stabilizes the
antiterminator conformation and as a consequence allows the transcription “read-through”
of the downstream gene or operon. In staphylococci, a T-box riboswitch precedes the glyS
gene encoding glycyl-tRNA synthetase (GlyRS). GlyRS mediates the formation of the Gly-
tRNAGly molecules that serve as substrates for the protein synthesis and for the exo-
ribosomal glycine-mediated stabilization of the bacterial cell wall. Previous work of our
group revealed that in S. aureus there are two encoded proteinogenic tRNAGly isoacceptors
[P1(GCC) and P2(UCC)] and three non-proteinogenic tRNAGly isoacceptors [NP1(UCC),
NP2(UCC) and NEW(UCC)] with extra-translational roles which bind poorly to EF-Tu. In the
present study, we tried to unravel and verify both the glyS T-box structure and the
differential utilization of tRNAGly isoacceptors either in protein synthesis or cell wall
formation. Extended bioinformatic and biochemical analyses revealed the existence of a
functional T-box regulatory element upstream the glyS gene albeit with divergent structural
features in comparison with other known glyQS T-boxes. The most intriguing structural
feature identified is the additional 42 nt long intervening sequence, termed stem Sa, which
is present in both terminator and antiterminator conformations and moreover, seems to be
staphylococci-specific. In vitro binding and transcription readthrough experiments revealed
that this T-box riboswitch can utilize both proteinogenic and non-proteinogenic tRNAGly
isoacceptors through specifier’s codon unconventional reading. Moreover, in vivo
readthrough experiments confirmed this ambiguity and verified the proposed species-

Abstract-Περίλθψθ
26
specific regulatory mechanism. Additional in vivo data suggested that all tRNAGly isoacceptor
presence is essential for growth and viability. In conclusion, specific utilization of different
tRNAGly isoacceptor during pathogen’s life contributes both in regulation and
synchronization of ribosomal and exo-ribosomal peptide synthesis in a species-specific
context. However, the exact regulatory mechanisms that occur during pathogens’s
metabolic adaptation and infection require further experimentation. Finally, this study gives
for the first time evidence to the existence of an elegant mechanism that synchronizes
essential metabolic pathways in pathogens and moreover can be used as an alternative to
the current therapy target for the development of novel antimicrobial drugs. In conclusion,
the present thesis contributes towards the elucidation of the regulatory role of tRNA
molecules, expands our current knowledge on the structure and function of regulatory RNAs
in bacteria and underlines the impressive complexity of networks and components of
translation during regulation of the flow of the genetic information.



Abstract-Περίλθψθ
29
Σα μόρια tRNA αποτελοφν τουσ βαςικοφσ προςαρμογείσ του γενετικοφ κϊδικα ςτθν γλϊςςα
των αμινοξζων. Μζχρι πρόςφατα ο κφριοσ ρόλοσ τουσ φαινόταν να περιορίηεται ςτθν
ςυνειςφορά τουσ ωσ υποςτρϊματα τθσ πρωτεϊνοςυνκετικισ μθχανισ αν και θ ςυμβολι
τουσ ςτθν εξζλιξθ είναι κακοριςτικι. Παρόλα αυτά πρόςφατεσ μελζτεσ ζδειξαν ότι τα μόρια
tRNA εκτόσ τθσ βαςικισ λειτουργίασ που επιτελοφν ςτθν πρωτεϊνικι ςφνκεςθ, δθλαδι
αυτισ τθσ μεταφοράσ αμινοξζων, παρουςιάηουν και επιπρόςκετεσ λειτουργιζσ που
αφοροφν ςτθ ςυμμζτοχθ τουσ ςε άλλεσ ςθμαντικζσ κυτταρικζσ διαδικαςίεσ όπωσ είναι θ
ρφκμιςθ τθσ μεταγραφισ και τθσ μετάφραςθσ. Οι καινοφριεσ αυτζσ λειτουργίεσ των μορίων
tRNA ανζδειξαν μια διαφορετικι προςζγγιςθ του ρόλου τουσ μζςα ςτο κφτταρο,
κακορίηοντάσ τα ωσ ρυκμιςτικοφσ τελεςτζσ τθσ γονιδιακισ ζκφραςθσ. Παράλλθλα θ
ανακάλυψθ μιασ ςθμαντικισ κατθγορίασ ρυκμιςτικϊν ςτοιχείων τθσ γονιδιακισ ζκφραςθσ,
γνωςτά ωσ Σ-box ριβοδιακόπτεσ, επιβεβαίωςε τθν ικανότθτα των μορίων tRNA να ελζγχουν
ςθμαντικζσ μεταβολικζσ οδοφσ. Οι Σ-box ριβοδιακόπτεσ βρίςκονται ςτθν 5' αμετάφραςτθ
περιοχι των μορίων mRNA (5’UTR) και μποροφν να ελζγχουν τθν ζκφραςθ των υπό-
ρφκμιςθ γονιδίων τουσ αλλθλεπιδρϊντασ τόςο με αμινοακυλιωμζνα όςο και με μθ-
αμινοακυλιωμζνα μόρια tRNA. Αυτοφ του τφπου τα ρυκμιςτικά ςτοιχεία βρίςκονται ευρζωσ
διαδεδομζνα ςε προκαρυωτικοφσ οργανιςμοφσ αλλά ειδικότερα ςε πακογόνα βακτιρια.
Επιπρόςκετα τα γονίδια τα οποία υπόκεινται ςε μεταγραφικό ζλεγχο από τουσ Σ-box
ριβοδιακόπτεσ κωδικοποιοφν κυρίωσ ζνηυμα τα οποία εμπλζκονται ςτθ βιοςφνκεςθ
αμινοξζων και ςτθν αμιναακυλίωςθ των μορίων tRNA (αμινοακυλο-tRNA ςυνκετάςεσ). Η
αποκάλυψθ τθσ λειτουργίασ των Σ-box ριβοδιακοπτϊν ανζδειξε τθν φπαρξθ ενόσ
ρυκμιςτικοφ μθχανιςμοφ ςε μεταγραφικό επίπεδο, όπου θ ίδια θ αλλθλουχία του RNA
μπορεί να εναλλάςςεται μεταξφ δφο διαφορετικϊν διαμορφϊςεων χωρίσ τθ ςυμμετοχι
κάποιου πρωτεϊνικοφ παράγοντα και ωσ αποτζλεςμα, να ρυκμίηει τθ γονιδιακι ζκφραςθ.
Αυτζσ οι εναλλακτικζσ διαμορφϊςεισ αποτελοφνται από δφο χαρακτθριςτικζσ ελικοειδείσ
διαμορφϊςεισ οι οποίεσ ονομάηονται βρόχοσ τερματιςμοφ (terminator) και βρόχοσ αντί-
τερματιςμοφ τθσ μεταγραφισ (antiterminator). Ο βρόχοσ τερματιςμοφ αποτελεί τθ
κερμοδυναμικά ευνοοφμενθ διαμόρφωςθ απουςία του tRNA-προςδζτθ ενϊ ο βρόχοσ αντί-
τερματιςμοφ χρειάηεται τθν παρουςία μθ-αμινοακυλιωμζνου tRNA για να ςτακεροποιθκεί.
Αλλθλεπίδραςθ του μορίου tRNA με μια εξειδικευμζνθ δομικι περιοχι του ριβοδιακόπτθ,
που ονομάηεται κθλιά εξειδίκευςθσ (specifier loop), μζςω ςυμπλθρωματικότθτασ τφπου
κωδικωνίου-αντικωδικονίου μπορεί να ςτακεροποιιςει τθ δομι αντί-τερματιςμοφ εφόςον
το μόριο tRNA βρίςκεται ςτθ μθ-αμινοακυλιωμζνθ του μορφι και κατά ςυνζπεια να επάγει
τθ μεταγραφι του υπό-ρφκμιςθ γονιδίου ι οπερονίου. το πακογόνο Staphylococcus, ζνα
τζτοιο ρυκμιςτικό ςτοιχείο Σ-box βρίςκεται ανοδικά του γονιδίου που κωδικοποιεί τθν
αμινοάκυλο-tRNA ςυνκετάςθ τθσ γλυκίνθσ (GlyRS). Η GlyRS μεςολαβεί το ςχθματιςμό
αμινοακυλιωμζνων μορίων Gly-tRNAGly και ακολοφκωσ τα ςυγκεκριμζνα μόρια αποτελοφν

Abstract-Περίλθψθ
30
υποςτρϊματα τόςο τθσ πρωτεϊνικισ ςφνκεςθσ όςο και τθ ςφνκεςθσ πενταπεπτιδίων
γλυκίνθσ θ οποία καταλφεται από τθν οικογζνεια των μθ-ριβοςωμικϊν πεπτιδυλ-
τρανςφεραςϊν (FemXAB). Σα ζνηυμα αυτά ςυμβάλλουν ςτθ ςτακεροποίθςθ τθσ
τριςδιάςτατθσ δομισ του κυτταρικοφ τοιχϊματοσ. Προθγοφμενθ εργαςία τθσ ερευνθτικισ
μασ ομάδασ αποκάλυψε τθν φπαρξθ πζντε διαφορετικϊν ιςοδεκτικϊν μορίων tRNAGly ςτον
S. aureus τα οποία μποροφν να χωριςτοφν ςε δφο κατθγορίεσ. Η πρϊτθ κατθγορία
περιλαμβάνει δφο πρωτεϊνογενετικά ιςοδεκτικά μόρια tRNAGly *Ρ1 (GCC) και P2 (UCC)+, τα
οποία αλλθλεπιδροφν ιςχυρά με τον παράγοντα επιμικυνςθσ EF-Tu και ςυμμετζχουν ςτθν
πρωτεϊνοςφνκεςθ, ενϊ θ δεφτερθ κατθγορία περιλαμβάνει τρία μθ-πρωτεϊνογενετικά
ιςοδεκτικά μόρια *NP1 (UCC), ΝΡ2 (UCC) και NEW (UCC)], τα οποία αλλθλεπιδροφν
αςκενϊσ με τον EF-Tu και ςυμμετζχουν ςτθ ζξω-ριβοςωμικι πρωτεϊνικι ςφνκεςθ που
λαμβάνει χϊρα ςτθ τελικι διαμόρφωςθ του κυτταρικοφ τοιχϊματοσ. Η παροφςα μελζτθ
ζχει ωσ ςκοπό αρχικά τον βιοχθμικό χαρακτθριςμό των υποςτρωμάτων τθσ GlyRS και
ακολοφκωσ τθν διερεφνθςθ και αποςαφινιςθ τθσ δομισ του ρυκμιςτικοφ ςτοιχείου T-box
που βρίςκεται ανοδικά του γονιδίου glyS ςτο πακογόνο S. aureus. Επιπλζον θ μελζτθ
επεκτάκθκε και ςτθ διερεφνθςθ τθσ διαφορικισ χριςθσ τόςο των πρωτεϊνογενετικϊν όςο
και των μθ-πρωτεϊνογενετικϊν ιςοδεκτικϊν μορίων tRNAGly ςτθν πρωτεϊνικι ςφνκεςθ και
ςτο ςχθματιςμό του κυτταρικοφ τοιχϊματοσ. Η ανάλυςθ ζδειξε ότι ανάμεςα ςε ςυγγενι
είδθ ςταφυλόκοκκων και παρά τθν ςθμαντικι ςυντιρθςθ τόςο τθσ δομισ τθσ GlyRS όςο και
των ςτοιχείων ταυτότθτασ των ομόλογων μορίων tRNAGly παρατθρικθκε διαφοροποίθςθ ωσ
προσ τα επίπεδα αμινοακυλίωςθσ. Ακολοφκωσ, λεπτομερισ βιοπλθροφορικι και βιοχθμικι
ανάλυςθ επιβεβαίωςε τθν φπαρξθ ενόσ λειτουργικοφ ρυκμιςτικοφ ςτοιχείου Σ-box ανοδικά
του γονιδίου glyS, αλλά με τθν φπαρξθ επιμζρουσ δομικϊν χαρακτθριςτικϊν που
διαφζρουν ςε ςφγκριςθ με άλλεσ γνωςτζσ δομζσ που ζχουν αποκαλυφκεί για αντίςτοιχα
ρυκμιςτικά ςτοιχεία ςε άλλουσ οργανιςμοφσ. Ο ςυγκεκριμζνοσ ριβοδιακόπτθσ
περιλαμβάνει ζνα χαρακτθριςτικό δομικό ςτοιχείο το οποίο αποτελείται από μια
επιπρόςκετθ αλλθλουχία μικουσ 42 νουκλεοτιδίων, το οποίο και ονομάςτθκε stem Sa
(βρόχοσ Sa, από τα αρχικά Staphylococcus aureus). Σο stem Sa ςυμμετζχει τόςο ςτθ
διαμόρφωςθ του βρόχου τερματιςμοφ όςο και ςτθ διαμόρφωςθ του βρόχου αντί-
τερματιςμοφ τθσ μεταγραφισ. Επιπρόςκετα, το ςυγκεκριμζνο ςτοιχείο αποτελεί μοναδικό
δομικό χαρακτθριςτικό αυτοφ του τφπου ριβοδιακόπτθ (glyS T-box) και εμφανίηεται
ςυντθρθμζνο μόνο ςε ςτελζχθ ςταφυλόκοκκων και ςε κανζνα άλλο βακτιριο. In vitro
μελζτθ τθσ δευτεροταγοφσ διαμόρφωςθσ ςυμπλόκων ριβοδιακόπτθ:tRNA, όςο και
επαγωγισ τθσ μεταγραφισ ζδειξαν ότι ο ςυγκεκριμζνοσ T-box ριβοδιακόπτθσ μπορεί να
χρθςιμοποιεί τόςο τα πρωτεϊνογενετικά όςο και τα μθ-πρωτεϊνογενετικά ιςοδεκτικά μόρια
tRNAGly, μζςω αντιςυμβατικισ ανάγνωςθσ του κωδικονίου τθσ κθλιάσ εξειδίκευςθσ, κάτι το
οποίο αναφζρεται για πρϊτθ φορά ςτθν διεκνι βιβλιογραφία. Παρόμοια αντίςτοιχθ

Abstract-Περίλθψθ
31
αντιςυμβατικι ανάγνωςθ ζχει αναφερκεί και ςτθν αποκωδικοποίθςθ κωδικονίων γλυκίνθσ
κατά τθν ριβοςωμικι πρωτεϊνοςφνκεςθ, κακϊσ τόςο το GCC όςο και το UCC αντικωδικόνιο
μπορεί να αναγνωριςτεί από τθν ίδια κωδικι τριπλζτα (GGC). Πειράματα in vivo επαγωγισ
τθσ μεταγραφισ επαλικευςαν τθν προτεινόμενθ αντιςυμβατικι αναγνϊριςθ τθσ τριπλζτασ
εξειδίκευςθσ και τθ ςυμμετοχι διαφορετικϊν ιςοδεκτικϊν μορίων tRNAGly ςτο
ςυγκεκριμζνο ρυκμιςτικό μθχανιςμό. Πρόςκετεσ in vivo πειραματικζσ διαδικαςίεσ
ανζδειξαν επίςθσ ότι θ παρουςία όλων των ιςοδεκτικϊν μορίων tRNAGly είναι απαραίτθτθ
για τθν ανάπτυξθ και τθ βιωςιμότθτα του πακογόνου. Πιο ςυγκεκριμζνα, επιβεβαιϊκθκε
ότι τα μθ-πρωτεινογενετικά μόρια tRNA όντωσ ςυμμετζχουν ωσ υποςτρϊματα ςτθν
ςφνκεςθ του κυτταρικοφ τοιχϊματοσ και επιπλζον, τόςο τα πρωτεϊνογενετικά όςο και τα
μθ-πρωτεϊνογενετικά μόρια tRNA μποροφν να ελζγχουν τα επίπεδα μεταγραφισ τθσ GlyRS
in vivo ςε διαφορετικά όμωσ επίπεδα. υμπεραςματικά, θ παροφςα διατριβι ςυμβάλει
ςτθν αποςαφινιςθ του ρυκμιςτικοφ ρόλου των μορίων tRNA ςε δφο μεταβολικά αςφνδετεσ
πορείεσ και αποτελεί το πρϊτο παράδειγμα εξειδικευμζνου δομικά ριβοδιακόπτθ ανάμεςα
ςτα είδθ ςταφυλοκόκκων. Η αρχικι υπόκεςθ υποςτθρίχκθκε από δεδομζνα που δείχνουν
διαφορικι εξειδίκευςθ των ιςοδεκτικϊν μορίων tRNAGly ςτθ διάρκεια ηωισ του πακογόνου
θ οποία ςυμβάλλει τόςο ςτθ ρφκμιςθ και όςο και ςτο ςυγχρονιςμό δυο ανεξάρτθτων αλλά
ςθμαντικϊν μεταβολικϊν μονοπατιϊν, τθσ ριβοςωμικισ και τθσ ζξω-ριβοςωμικισ ςφνκεςθσ
και επιπλζον ςε ζνα πλαίςιο εξειδικευμζνο για το ςυγκεκριμζνο είδοσ πακογόνου. Ωςτόςο
οι ακριβείσ ρυκμιςτικοί μθχανιςμοί που λαμβάνουν χϊρα κατά τθ διάρκεια τθσ
προςαρμογισ του πακογόνου ςε μεταβαλλόμενεσ ςυνκικεσ περιβάλλοντοσ όςο και ςε
ςυνκικεσ μόλυνςθσ του ξενιςτι παραμζνουν προσ μελλοντικι διερεφνθςθ και
αποςαφινιςθ. Σζλοσ θ ςυγκεκριμζνθ μελζτθ αναδεικνφει για πρϊτθ φορά τθν φπαρξθ ενόσ
«λεπτοφ» μθχανιςμοφ γονιδιακισ ζκφραςθσ που ςυγχρονίηει απαραίτθτεσ για τθ
βιωςιμότθτα του πακογόνου μεταβολικζσ οδοφσ. Επιπλζον θ ανάδειξθ τθσ λειτουργίασ του
ςυγκεκριμζνου ρυκμιςτικοφ μθχανιςμοφ τον κακιςτά εναλλακτικό ςτόχο για τθν ανάπτυξθ
νζων αντιμικροβιακϊν φαρμάκων που μποροφν να χρθςιμοποιθκοφν ωσ λφςθ ζναντι τθσ
τρζχουςασ κεραπείασ για αυτοφ του είδουσ τα πακογόνα, θ οποία φαίνεται να είναι
υπεφκυνθ για τθν παρουςία ανκεκτικϊν ςτελεχϊν.




Introduction
35
1. The tRNA molecule
All domains of life require intact and functional tRNA molecules as the most
essential components for protein translation. After their aminoacylation with the cognate
amino acid, they are used as substrates for translation and determine the evolutionary
preserved integrity of the genetic code. This ability resides in a precise match of each amino
acid to the corresponding anticodon. The primary reaction of aminoacylation mediated by
the cognate aminoacyl-tRNA synthetase, induces an additional intrinsic proofreading [1].
1.1. The “adaptor hypothesis” and the “second genetic code”
Going back in 1958, Francis Crick in his famous "Adaptor Hypothesis" introduced for
the first time the notion of "adaptor molecules" which must exist to mediate the flow of the
genetic information from DNA to proteins [2]. Each of these molecules could carry a specific
amino acid and should consist of ribonucleotides. Moreover, identification of the codons
could take place through base-pairing. This theory was proved prophetic as well as accurate
in prediction, because later on it was found that the amino acids required for protein
synthesis are transported and esterified at the 3’ end of transport RNA molecules (tRNAs).
Thirty years later, in 1988, Paul Schimmel’s group showed that a simple structural feature, a
single base pair, is the major determinant of the Identity of a tRNA, an observation that
demonstrates the existence of a “second genetic code”, that seems to be older and more
deterministic than the classical genetic code [3, 4+. According to this hypothesis, this “second
code” was represented by a para-codon coding that was used by the oldest ancestors of
tRNA molecules, as a recognizable structural feature for aminoacylation. The anticodons
appeared probably later in evolution, as a new and “younger” structural determinant of
tRNA stereochemistry. These observations also proved that tRNA molecules were the
evolving linkers between the two codes [4].
Finally, an additional proof of the observation that the genetic code evolution was
followed by tRNA adaptation is the fact of genetic code degeneration (20 amino acids are
encoded by 61 triplet codes; Figure 1). In 1966, Francis Crick proposed the “Wobble
Hypothesis”, as tRNAs decoded the genome by recognizing more than one codons [5]. The
discovery of post-transcriptional modifications at tRNA's wobble position 34 lead to “The
Modified Wobble Hypothesis” proposed in 1991 by Paul F. Agris *6]. Decoding involves both
selective hydrogen bonding and stability of the anti-codon stereochemistry, including those
post-transcriptional modifications that are necessary as tRNA’s essential identity elements.

Introduction
36
In conclusion, the involvement of the tRNA anticodon architecture in genetic
decoding seems to enhance the RNA world evolutionary theory and the introduction of new
amino acids into proteins [7]. The latest sustains the basis of the development of synthetic
biology and the recoding of the genetic code to incorporate unusual amino acids in E. coli
[8].
Figure 1. (Left) The standard genetic code and its degeneracy. Grey, green, yellow and blue indicates
two, three, four and six fold degenerate codons respectively. The single codons are in white and stop
codons in red backgrounds. (Right) tRNA anticodon wobble pairing for amino acids with degenerate
codons [7].
1.2. Structure, identity elements and evolution
The first tRNA molecule fully sequenced was yeast tRNAAla by R. Holley (Nobel Prize
Laureate in Physiology or Medicine 1968) and his co-workers [9]. This innovative study
together with the discovery of modern sequencing led in early 2000s to the development of
the tRNA database [10], in which more than 4000 tRNAs from more than 300 organisms
have been included. These numbers progressively increased along with the decoding of
numerous genomes, until nowadays.
Today it is known that tRNA molecules consist of 73 to 93 ribonucleotides, with the
exception of mitochondrial tRNA molecules, which typically have a smaller size. They exhibit
a standard secondary structure (known as “the cloverleaf structure”) that is universal
throughout all three domains of life. This characteristic structure is composed of four double
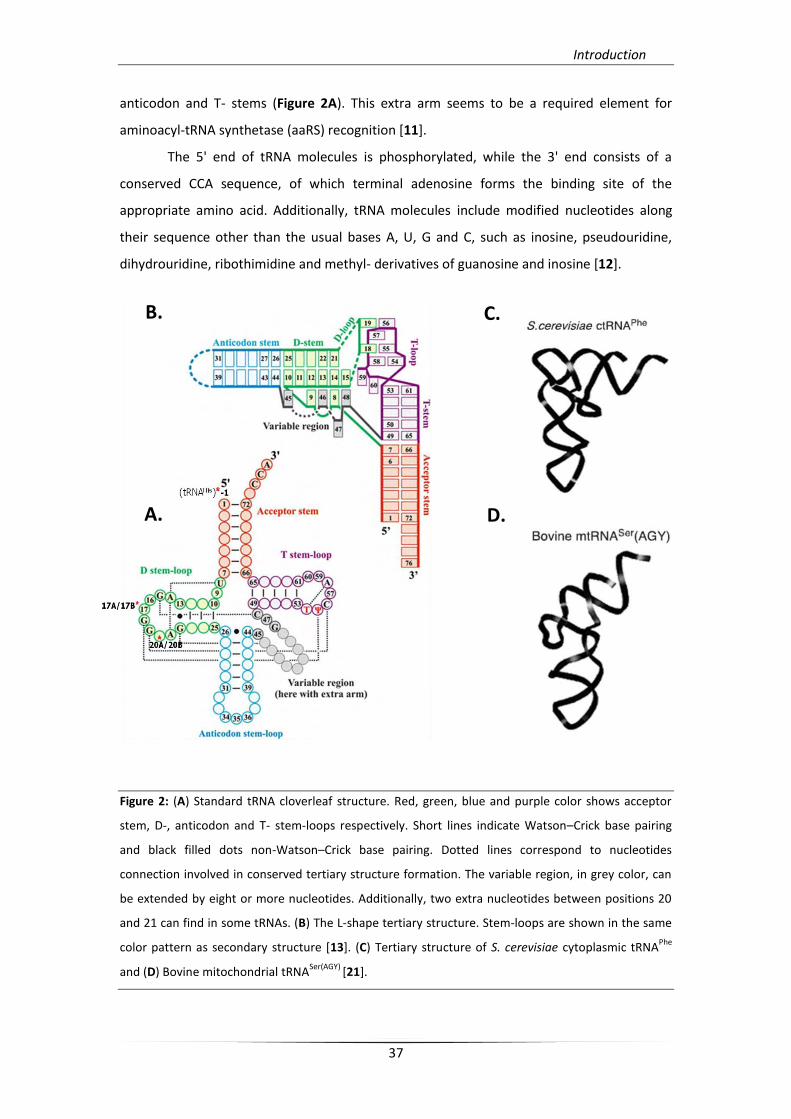
strand regions, the acceptor stem, the D-, anticodon and T- stems-loops (arms). With the
exception of these universal structural elements, some tRNAs (tRNALeu, tRNASer, tRNASec and
tRNATyr) contain an extra stem-loop named variable/extra arm, located between the
Introduction
37
anticodon and T- stems (Figure 2A). This extra arm seems to be a required element for
aminoacyl-tRNA synthetase (aaRS) recognition [11].
The 5' end of tRNA molecules is phosphorylated, while the 3' end consists of a
conserved CCA sequence, of which terminal adenosine forms the binding site of the
appropriate amino acid. Additionally, tRNA molecules include modified nucleotides along
their sequence other than the usual bases A, U, G and C, such as inosine, pseudouridine,
dihydrouridine, ribothimidine and methyl- derivatives of guanosine and inosine [12].
Figure 2: (A) Standard tRNA cloverleaf structure. Red, green, blue and purple color shows acceptor
stem, D-, anticodon and T- stem-loops respectively. Short lines indicate Watson–Crick base pairing
and black filled dots non-Watson–Crick base pairing. Dotted lines correspond to nucleotides
connection involved in conserved tertiary structure formation. The variable region, in grey color, can
be extended by eight or more nucleotides. Additionally, two extra nucleotides between positions 20
and 21 can find in some tRNAs. (B) The L-shape tertiary structure. Stem-loops are shown in the same
color pattern as secondary structure [13]. (C) Tertiary structure of S. cerevisiae cytoplasmic tRNAPhe
and (D) Bovine mitochondrial tRNASer(AGY)
[21].
B.
A.
C.
D.

Introduction
38
In the tertiary structure of the molecule, stems and loops of the secondary structure
are combined to produce an L-shape architecture that is stabilized by various tertiary
interactions. The formation of the two characteristic helical domains of the L-shape
conformation, acceptor/T and D/anticodon, requires the interaction of discrete regions of
secondary structure. These connector regions consist of the acceptor and D- stems
(connector 1) and the anticodon and T- stems (connector 2) (Figure 2B.) [13]. Finally, these
conserved tertiary interactions (with the exception of tRNACys, which is missing the Levitt
triple, 10-25-45) are responsible for exposure over the tRNA structure of the two functional
centers, the anticodon loop (responsible for codon reading) and the acceptor stem
(responsible for amino acid esterification), which also dictate tRNA’s function.
Because tRNA secondary and tertiary structure is crucial for aaRS recognition, its
sequence has to be highly conserved and is subjected to specific rules. Moreover, the bases
that are preserved at the loops and the arms are referred to a specific numbering system
[14] with some exceptions that are observed in D-loop and the variable loop. On the
contrary, some mitochondrial tRNA molecules lack the arm and D-loop and / or the arm and
the T-loop, but nevertheless can form the L-shape (Figure 2D.) [15, 16].
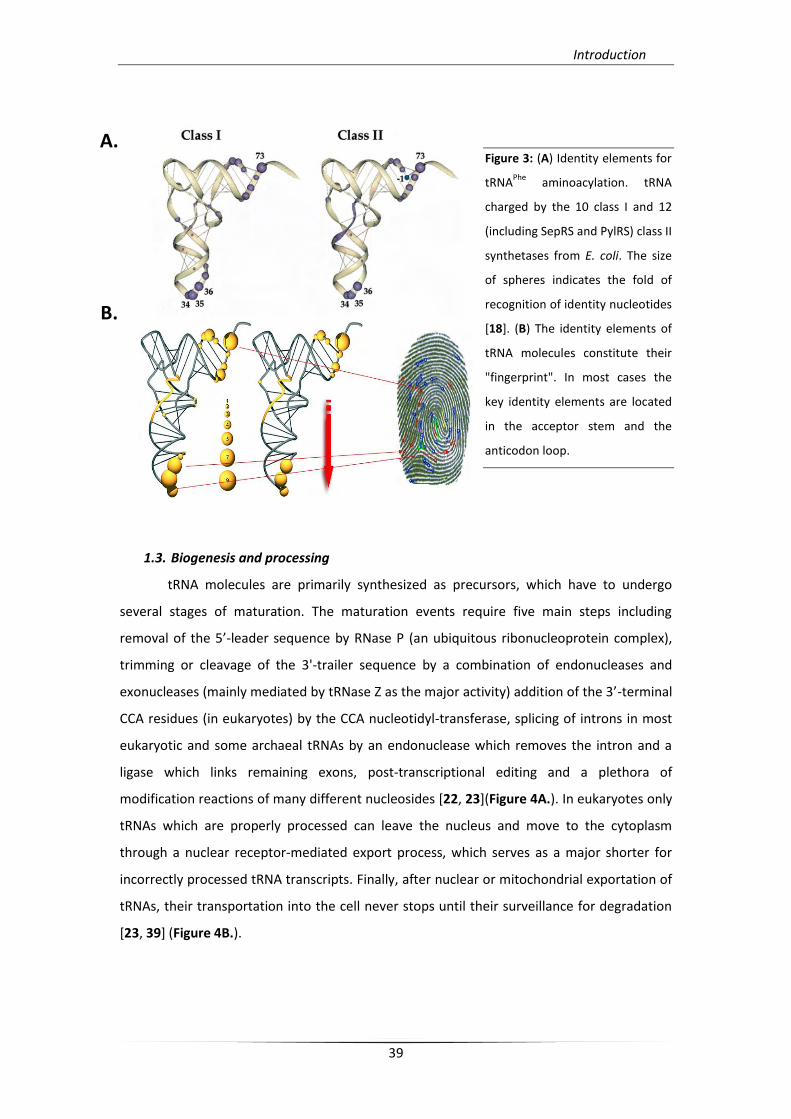
Fidelity of tRNA aminoacylation depends on a specific recognition system that
consists of an operational RNA code that is associated with specific elements (termed
identity elements) derived from co-evolution of catalytic cores of synthetases and RNA
hairpins that served as primordial acceptor stems [17]. From Escherichia coli to higher
eukaryotes including humans, a compilation of tRNA identity elements referring to standard
noucleotite positions have been determined and found in most cases conserved (Figure 3).
Also, modified nucleotides that are exclusively located in the anticodon loop of some tRNAs
are either additional identity elements or negative determinants (anti-determinants), but
their involvement in aminoacylation seems to be scarce [18].
As it is known for most systems, tRNA binding modules of synthetases can recognize
idiosyncratic characteristics that include identity determinants of the tRNA molecule,
especially in the anticodon region. According to the universal nature of the genetic code,
these identity elements are obviously conserved in evolution. Based on these observations,
it has been proposed that origin of tRNA aminoacylation system is tightly connected with the
development of the genetic code [19, 20].
Introduction
39
Figure 3: (A) Identity elements for
tRNAPhe
aminoacylation. tRNA
charged by the 10 class I and 12
(including SepRS and PylRS) class II
synthetases from E. coli. The size
of spheres indicates the fold of
recognition of identity nucleotides
[18]. (B) The identity elements of
tRNA molecules constitute their
"fingerprint". In most cases the
key identity elements are located
in the acceptor stem and the
anticodon loop.
1.3. Biogenesis and processing
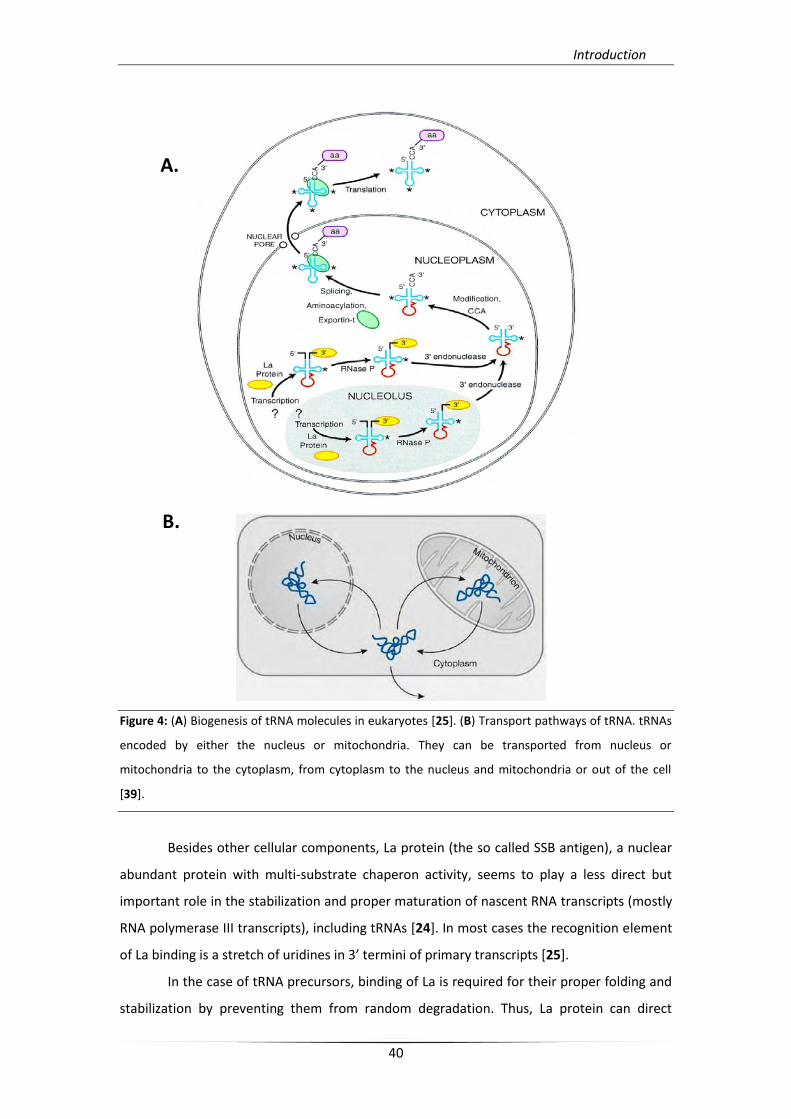
tRNA molecules are primarily synthesized as precursors, which have to undergo
several stages of maturation. The maturation events require five main steps including
removal of the 5’-leader sequence by RNase P (an ubiquitous ribonucleoprotein complex),
trimming or cleavage of the 3'-trailer sequence by a combination of endonucleases and
exonucleases (mainly mediated by tRNase Z as the major activity) addition of the 3’-terminal
CCA residues (in eukaryotes) by the CCA nucleotidyl-transferase, splicing of introns in most
eukaryotic and some archaeal tRNAs by an endonuclease which removes the intron and a
ligase which links remaining exons, post-transcriptional editing and a plethora of
modification reactions of many different nucleosides [22, 23](Figure 4A.). In eukaryotes only
tRNAs which are properly processed can leave the nucleus and move to the cytoplasm
through a nuclear receptor-mediated export process, which serves as a major shorter for
incorrectly processed tRNA transcripts. Finally, after nuclear or mitochondrial exportation of
tRNAs, their transportation into the cell never stops until their surveillance for degradation
[23, 39] (Figure 4B.).
A.
B.
Introduction
40
Figure 4: (A) Biogenesis of tRNA molecules in eukaryotes [25]. (B) Transport pathways of tRNA. tRNAs
encoded by either the nucleus or mitochondria. They can be transported from nucleus or
mitochondria to the cytoplasm, from cytoplasm to the nucleus and mitochondria or out of the cell
[39].
Besides other cellular components, La protein (the so called SSB antigen), a nuclear
abundant protein with multi-substrate chaperon activity, seems to play a less direct but
important role in the stabilization and proper maturation of nascent RNA transcripts (mostly
RNA polymerase III transcripts), including tRNAs [24]. In most cases the recognition element
of La binding is a stretch of uridines in 3’ termini of primary transcripts *25].
In the case of tRNA precursors, binding of La is required for their proper folding and
stabilization by preventing them from random degradation. Thus, La protein can direct
A.
B.
A.
B.
Introduction
41
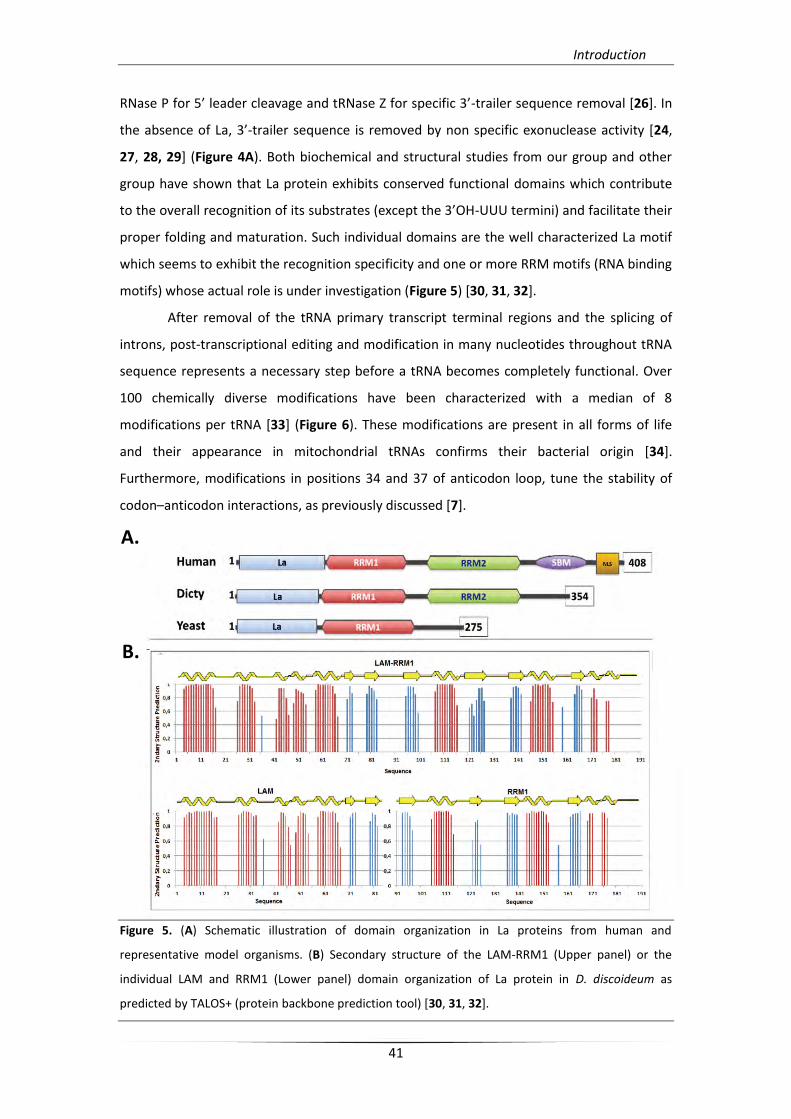
RNase P for 5’ leader cleavage and tRNase Z for specific 3’-trailer sequence removal [26]. In
the absence of La, 3’-trailer sequence is removed by non specific exonuclease activity [24,
27, 28, 29] (Figure 4A). Both biochemical and structural studies from our group and other
group have shown that La protein exhibits conserved functional domains which contribute
to the overall recognition of its substrates (except the 3’OH-UUU termini) and facilitate their
proper folding and maturation. Such individual domains are the well characterized La motif
which seems to exhibit the recognition specificity and one or more RRM motifs (RNA binding
motifs) whose actual role is under investigation (Figure 5) [30, 31, 32].
After removal of the tRNA primary transcript terminal regions and the splicing of
introns, post-transcriptional editing and modification in many nucleotides throughout tRNA
sequence represents a necessary step before a tRNA becomes completely functional. Over
100 chemically diverse modifications have been characterized with a median of 8
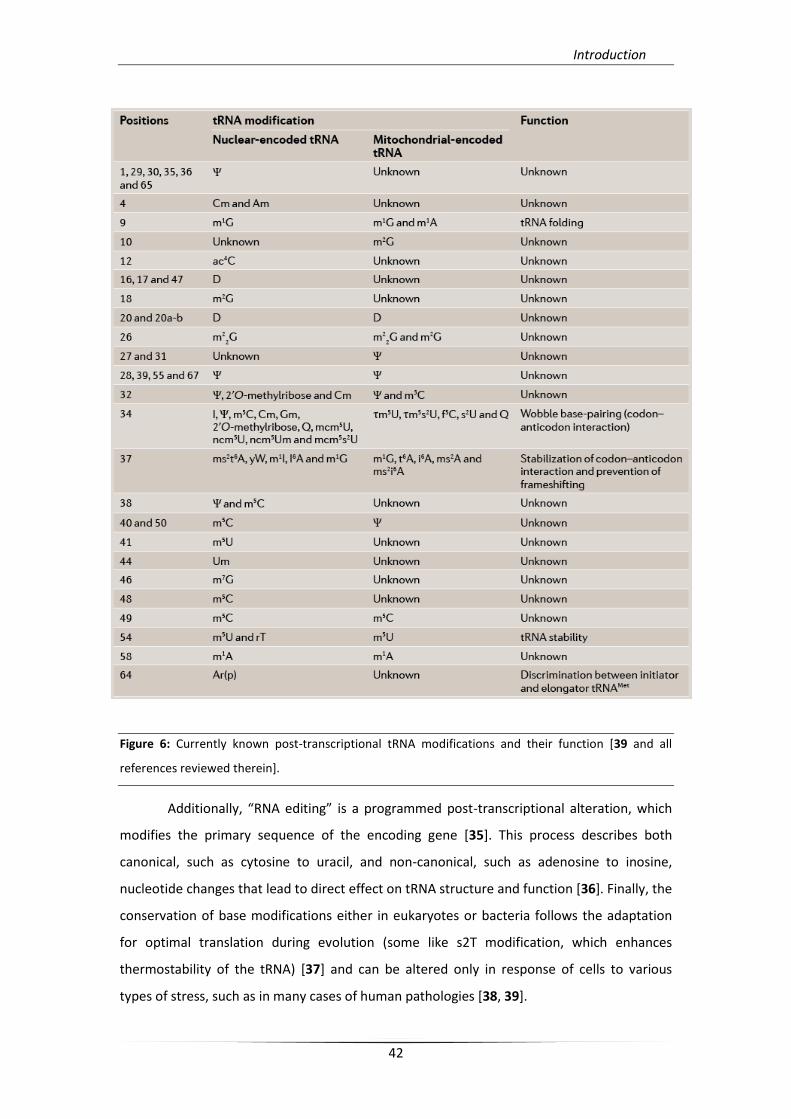
modifications per tRNA [33] (Figure 6). These modifications are present in all forms of life
and their appearance in mitochondrial tRNAs confirms their bacterial origin [34].
Furthermore, modifications in positions 34 and 37 of anticodon loop, tune the stability of
codon–anticodon interactions, as previously discussed [7].
Figure 5. (A) Schematic illustration of domain organization in La proteins from human and
representative model organisms. (B) Secondary structure of the LAM-RRM1 (Upper panel) or the
individual LAM and RRM1 (Lower panel) domain organization of La protein in D. discoideum as
predicted by TALOS+ (protein backbone prediction tool) [30, 31, 32].
A.
B.
Introduction
42
Figure 6: Currently known post-transcriptional tRNA modifications and their function [39 and all
references reviewed therein].
Additionally, “RNA editing” is a programmed post-transcriptional alteration, which
modifies the primary sequence of the encoding gene [35]. This process describes both
canonical, such as cytosine to uracil, and non-canonical, such as adenosine to inosine,
nucleotide changes that lead to direct effect on tRNA structure and function [36]. Finally, the
conservation of base modifications either in eukaryotes or bacteria follows the adaptation
for optimal translation during evolution (some like s2T modification, which enhances
thermostability of the tRNA) [37] and can be altered only in response of cells to various
types of stress, such as in many cases of human pathologies [38, 39].

Introduction
43
1.4. tRNA and pathogenesis
Considering the repertoire of tRNA genes in all known decoded genomes, containing
from 86 in E. coli to 12,794 genes in zebrafish (including genes which encode tRNA
isoacceptors), it is obvious that the differential variability and expression levels are
functionally associated [40]. This variability is even more extensive considering the
observation that the tRNA gene content and expression differs among individuals in humans
[41, 42]. Furthermore, tRNA isodecoders probably have functional roles and their differential
expression depends on cell type and state [43]. It was recently shown that tRNA codon
usage differs between normal and cancer cells [44]. Searching for the molecular basis of
pathologies in humans, it was shown that genome sequencing of patients results in variable
tRNA alteration in many cases. These tRNA-based pathologies can be classified into two
categories, including direct mutation within the tRNAs or genetic disorders that affect
processing and modifying enzymes and have an indirect alteration of tRNAs.
All cases directly related to mutations in tRNAs, include mt‑tRNA alterations. This
observation can be explained by the low content of tRNA genes in the mitochondrial
genome (only a single copy of each 22 mt-tRNAs) and total absence of isodecoder (or
isoacceptor) genes. In addition, it has been suggested that mitochondrial genome undergoes
10- to 17-fold higher rate of mutations than the nuclear genome, probably due to absence of
efficient DNA repair mechanisms [45, 46]. According to MITOMAP database, up to 251
mutations which are located in tRNA genes have been identified within human
mitochondrial genome and are associated to pathogenicity. Clinical phenotypes of such mt-
tRNA alterations can range from lesions of single structures to more severe impairments
such as myopathies, neural disorders, metabolic syndromes or multisystem syndromes
(Figure 7A). Almost 50% of the known pathogenic mutations are identified in tRNALeu(UUR),
tRNALys and tRNAIle genes. The highest pathogenic mutation is observed for tRNALeu(UUR) gene,
while the less polymorphic is the tRNAPro gene. Most of these mutations are base transitions
(between pyrimidines or purines) rather than conversions. Most likely canonical Watson-
Crick base pairs of stem regions can be converted to C·A or G·U mismatches. In addition,
such disease-associated mutations are distributed throughout the tRNA body, but they are
totally absent from the anticodon triplet because these ones would be lethal. Moreover, it
has been proposed that the location of identified mutations is linked to the pathological
significance of a tRNA mutation. However, some pathogenic mutations do affect non-
conserved positions and suggest that the degree of nucleotide conservation cannot be taken
as a threshold for pathology association [47]. Finally, a paradigm of indirect tRNA alteration
Introduction
44
linked to disorders is mutations in CLP1 kinase which decrease pre-tRNA processing in
fibroblasts and neurons [48, 49]. According to the complexity of tRNA-based alterations, we
have to consider both the multi-factor and tissue-specific aspects which may directly affect
the state and development of disease. A representative system of multiple alterations in
composition and concentration of the tRNAome is the tumor cell where decoding of specific
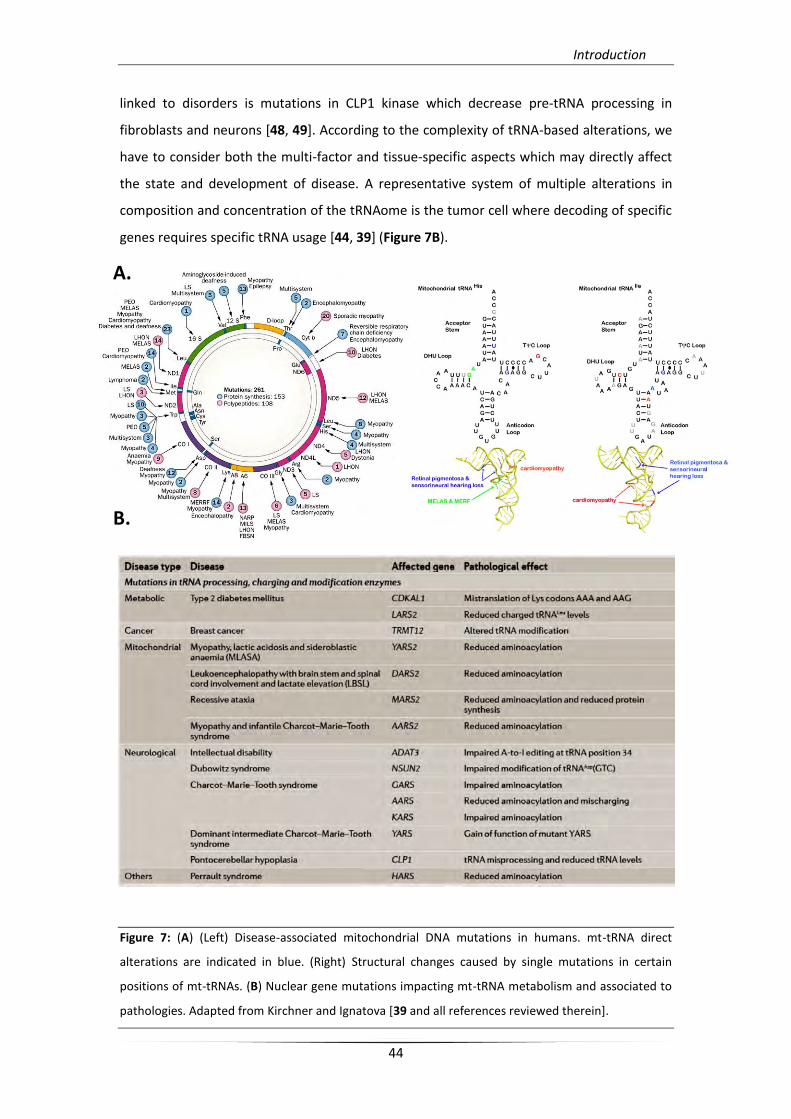
genes requires specific tRNA usage [44, 39] (Figure 7B).
Figure 7: (A) (Left) Disease-associated mitochondrial DNA mutations in humans. mt-tRNA direct
alterations are indicated in blue. (Right) Structural changes caused by single mutations in certain
positions of mt-tRNAs. (B) Nuclear gene mutations impacting mt-tRNA metabolism and associated to
pathologies. Adapted from Kirchner and Ignatova [39 and all references reviewed therein].
A.
B.

Introduction
45
2. Aminoacyl-tRNA synthetases (aaRSs)
In all living cells, accurate transmission of genetic information, resides in correct
synthesis of aminoacyl-tRNAs (aa-tRNAs), mediated by aminoacyl-tRNA synthetases (aaRSs).
Aminoacylation fidelity is based on high substrate selectivity and proofreading mechanisms.
Specific substrate stereochemical recognition by aaRSs, allows the appropriate amino acid
and tRNA coupling. Many aaRSs rely on additional editing proofreading mechanisms that
ensure faithful aa-tRNA release. Furthermore, aa-tRNA synthesis is the first step of the
translation process during which idiosyncratic recognition elements of tRNA architecture are
converted into “protein vocabulary” *1, 50].
2.1. Classes of aaRSs, functional and structural features
Some theories on the evolutionary origin of aaRSs suggest that contemporary aaRSs
replaced RNA-based enzymes (mainly ribozymes), while following the universal nature of the
genetic code which confirms that they have to be of ancient origin [51, 52]. However, recent
data suggest that contemporary aaRSs do not exhibit vertical inheritance from a common
ancestral enzyme. Ancient aaRSs presumably consisted in only the aminoacylation site, and
several appended domains, such as editing domains, were acquired later during evolution as
the amino acid repertoire expanded gradually [53-55].
Figure 8: The two step basic mechanism of tRNA aminoacylation reaction is catalyzed by the cognate
aaRS (with the exception of Glu-, Gln-, Arg- and class I LysRS, which need the cognate tRNA for aa
activation); AS corresponds to aminoacylation active site [50].
Contemporary aaRSs are clustered into two evolutionary conserved and structurally
disparate groups, termed class I and class II, based on their structural, functional, and
evolutionary resemblance [1, 56-58]. Each class in model organisms like E. coli contains 10
enzymes each. However, the last 15 years many functional genomic studies have shown that
this perfect division is rather the exception and not the rule. Therefore nowadays, we
recognize 11 and 13 enzymes respectively. Each aaRS always belongs to a single class
Introduction
46
throughout evolution with the exception of LysRS that can be of class I or class II.
Additionally, each class comprises distinct subclasses based on sequence and structural
similarity. Both classes utilize the same catalytic mechanism, an observation that suggests
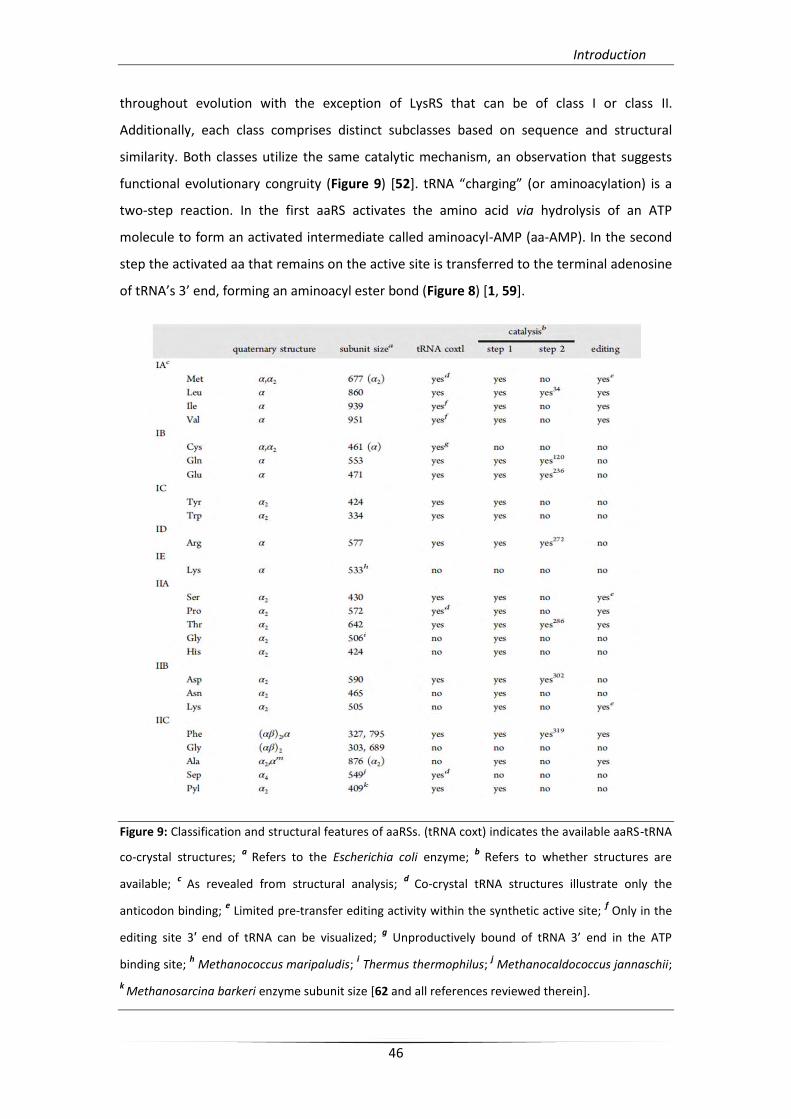
functional evolutionary congruity (Figure 9) [52+. tRNA “charging” (or aminoacylation) is a
two-step reaction. In the first aaRS activates the amino acid via hydrolysis of an ATP
molecule to form an activated intermediate called aminoacyl-AMP (aa-AMP). In the second
step the activated aa that remains on the active site is transferred to the terminal adenosine
of tRNA’s 3’ end, forming an aminoacyl ester bond (Figure 8) [1, 59].
Figure 9: Classification and structural features of aaRSs. (tRNA coxt) indicates the available aaRS-tRNA
co-crystal structures; a Refers to the Escherichia coli enzyme;
b Refers to whether structures are
available; c
As revealed from structural analysis; d
Co-crystal tRNA structures illustrate only the
anticodon binding; e Limited pre-transfer editing activity within the synthetic active site;
f Only in the
editing site 3′ end of tRNA can be visualized; g Unproductively bound of tRNA 3’ end in the ATP
binding site; h Methanococcus maripaludis;
i Thermus thermophilus;
j Methanocaldococcus jannaschii;
k Methanosarcina barkeri enzyme subunit size [62 and all references reviewed therein].

Introduction
47
Regarding the architecture similarity within each class, only the catalytic domains
and some active site motifs are strictly conserved. In class I aaRSs, the aminoacylation
catalytic center has a Rossmann nucleotide-binding fold, including two characteristic motifs,
HIGH and KMSKS [1, 58]. On the contrary, class II aaRSs possess an antiparallel β-sheet
conformation, delineated by I, II, and III representative hydrophobic motifs. Motif I is
involved in dimerization of subunits, while motifs II and III participate to ATP and amino acid
binding, as well as to the editing activity of many aaRSs (Figure 10). Moreover, class I
enzymes bind to the minor groove side of the tRNA acceptor stem, with the exception of
TyrRS [60+, and aminoacylate the 2’-OH of terminal adenosine. Class II enzymes bind to the
major groove side and aminoacylate the 3’-OH, with the exception of PheRS that can charge
either the 2’-OH or the 3'-OH [61]. Finally, class I aaRSs in most cases are found as
monomers (except TyrRS, TrpRS, and MetRS in some species) and, on the other hand, class II
aaRSs are usually dimmers (α2) or tetramers (α2β2).
Figure 10: Characteristic domains of aaRSs primary structures. (a) Class I aaRSs; (orange) Add1 domain
responsible for tRNA D-loop recognition; (green) anticodon-binding domain (ACBD); (yellow) Editing
domains/CP1 post-transfer editing domain; (blue) HIGH and KMSKS motifs. (b) Class IIA aaRSs; (blue
and red) class II representative I, II, and III motifs; (blue) dimer interfaces that include motif I. (c) Class
IIB aaRSs. ACBD domain adopts an N-terminal-beta-barrel domain (OB fold). (d) Class IIC aaRSs; B1
and B5 represent DNA binding-like domains; (αβ)2 tetramers possess two active and two inactive
chains; (pink) C-Ala major tRNA-binding domain serves as a bridge between editing and
aminoacylation functions; PylRS bulge and tRNA binding domains are unique [62, and all references
reviewed therein].

Introduction
48
According to RNA world evolutionary theory, the addition of the tRNA anticodon
module to the primitive acceptor arm allowed the expansion of the repertoire of amino
acids that could be encoded by the genetic code [7]. The presence of phosphoseryl-tRNA
synthetase (SepRS) and pyrrolysyl-tRNA synthetase (PylRS) in some organisms confirms this
adaptation of the genetic code expansion during evolution [63, 64]. Due to their structure
both enzymes are classified to IIC AARS subclass (Figure 10). SepRS possesses subunit
structural organization related to (αβ)2 PheRS of the same class, and is responsible for
phosphoseryl (Sep)-tRNACys formation in Archaeoglobus and in most methanogens [65-67].
By contrast, PylRS harbors unique structural domains that are responsible for tRNA binding
and dimerization. This enzyme charge tRNAPyl suppressor tRNA, which recognize UAG stop
codons with pyrrolysine and is present in Methanosarcina and in some isolated bacterial
groups [68, 69].
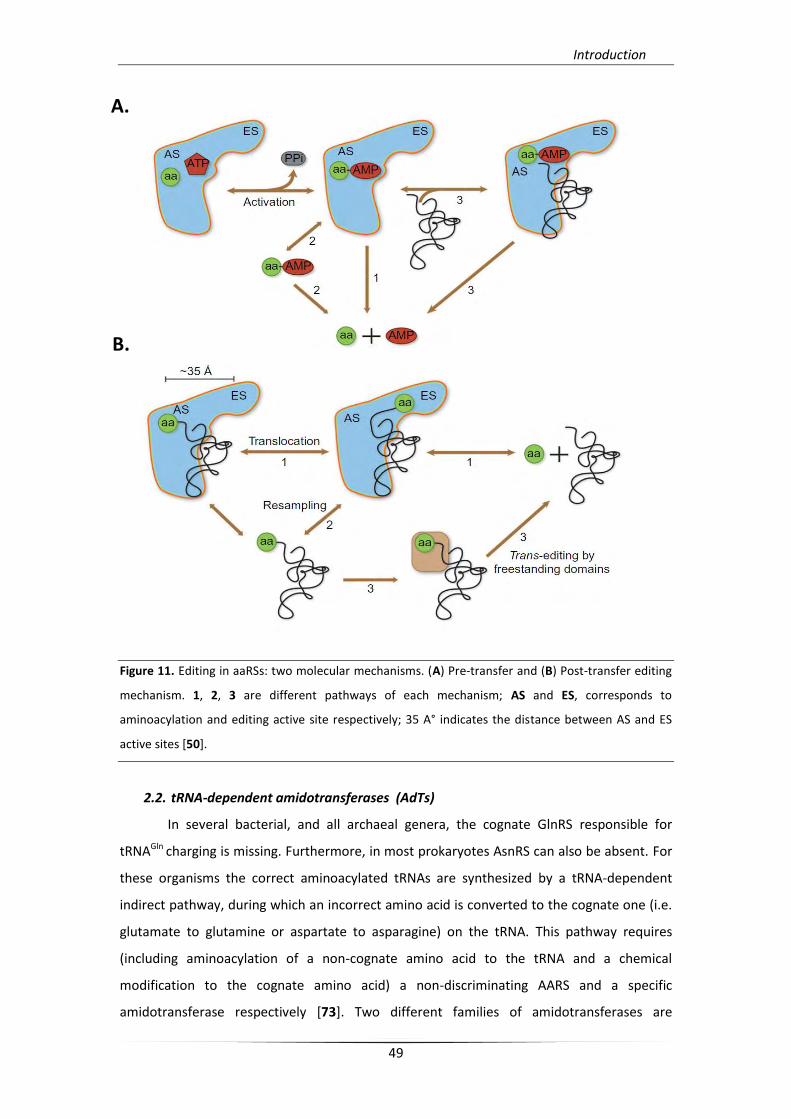
With respect to aaRS of aminoacylation proofreading, two molecular editing
mechanisms have been described [59, 70]. The ‘‘pre-transfer’’ editing, occurs prior to
aminoacyl transfer and the ‘‘post-transfer’’ editing, occurs after transfer reaction. Pre-
transfer editing concerns misactivated aa-AMPs hydrolysis and occurs in three alternative
pathways, two tRNA-independent, by enzyme-catalyzed hydrolysis or spontaneous
hydrolysis after selective release of non-cognate aa-AMP, taking place in the aminoacylation
active site or in solution respectively (Figure 11A; pathways 1, 2), and one tRNA-dependent
hydrolysis of misactivated aa-AMP, taking place in the aminoacylation or editing active site
(Figure 11A; pathway 3). Class II ProRS [71] and SerRS [72] are representative paradigms of
tRNA-independent enzymatic pre-transfer editing. In the case of post-transfer editing, there
is a distinct editing site which has a different topology than that of aminoacylation active
site, and is responsible for non-cognate amino acid deacylation. This kind of editing activities
are present in both classes of aaRSs and can occur in three different pathways, a direct
translocation model (Figure 11B; pathway 1), and/or a dissociation–reassociation model, or
an alternative deacylation reaction involving a supplemental trans-editing factor (Figure
11B; pathways 2, 3). As it is mentioned previously, proofreading properties of aaRSs render
them the first checkpoint for quality control in translation. However, the actual role of
editing in vivo is yet to be resolved [50].
Introduction
49
Figure 11. Editing in aaRSs: two molecular mechanisms. (A) Pre-transfer and (B) Post-transfer editing
mechanism. 1, 2, 3 are different pathways of each mechanism; AS and ES, corresponds to
aminoacylation and editing active site respectively; 35 A° indicates the distance between AS and ES
active sites [50].
2.2. tRNA-dependent amidotransferases (AdTs)
In several bacterial, and all archaeal genera, the cognate GlnRS responsible for
tRNAGln charging is missing. Furthermore, in most prokaryotes AsnRS can also be absent. For
these organisms the correct aminoacylated tRNAs are synthesized by a tRNA-dependent
indirect pathway, during which an incorrect amino acid is converted to the cognate one (i.e.
glutamate to glutamine or aspartate to asparagine) on the tRNA. This pathway requires
(including aminoacylation of a non-cognate amino acid to the tRNA and a chemical
modification to the cognate amino acid) a non-discriminating AARS and a specific
amidotransferase respectively [73]. Two different families of amidotransferases are
A.
B.

Introduction
50
responsible for the amino acid chemical modification of the non-cognate Glu-tRNAGln, and
Asp-tRNAAsn intermediates. In bacteria, the heterotrimeric amidotransferase GatCAB [74] is
able to catalyze both Gln-tRNAGln and/or Asn-tRNAAsn formation in a genome-dependent
context [73]. In Archaea, GatCAB can function exclusively as Asp-AdT, while a distinct
heterotetrameric amidotransferase GatDE is responsible for Gln-tRNAGln formation [75].
Both enzymes share the same catalytic mechanism [73]. Firstly, phosphorylation of the
terminal carboxylate of the mischarged Glu or Asp aminoacyl-tRNA activates the
glutaminase site, which generates ammonia from amide donors such as Gln or Asn and
transfers it to the aminoacyl moiety located in active site. Subsequently, the activated
intermediate is amidated by the amidotransferase active site, using the released ammonia.
GatB or GatE subunit encompasses a tRNA-dependent kinase activity, and GatA or GatD a
glutaminase activity (Figure 12). Crystal structures of GatCAB [76] and GatDE [77] complexes
with their aminoacyl-tRNA substrates suggested the connection of glutaminase and amidase
active sites by a specific formation of an ammonia tunnel connecting the GatA/GatD
amidase site to the GatB/GatE active site. Moreover, the existence of a multicomplex which
consists of the non-discriminating aaRS and the specific AdT that “channels” synthetase and
transamidase activities has been proposed. This notion gives evidence for the existence of a
“transamidosome” apparatus, which protects the non-cognate intermediates from
hydrolysis and most importantly from interaction with EF-Tu, which could have lead to
misincorporation during protein synthesis of Glu or Asp into Gln and Asn codons,
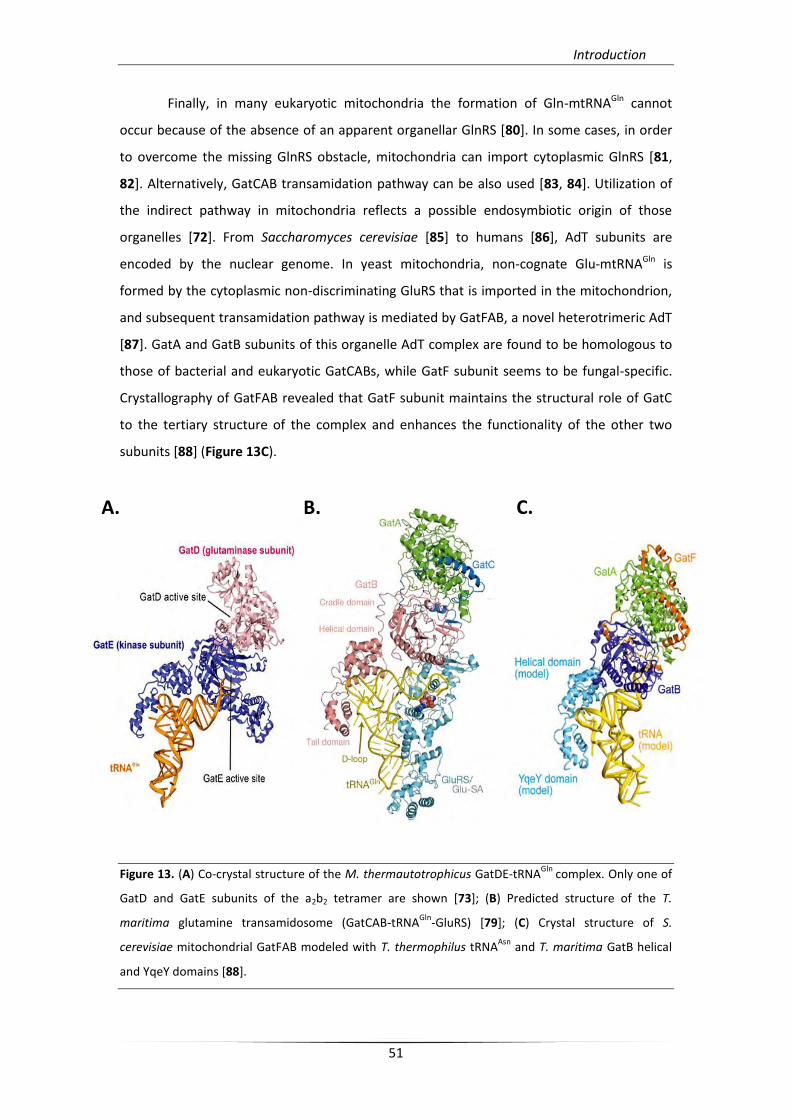
respectively [78, 79] (Figure 13 A, B).
Figure 12. GatCAB (A)/(B), and
GatDE (A) transamidate their
mischarged aa-tRNA substrates
using a three-reaction mechanism;
(i) Phosphorylation of mischarged
aa-tRNA by GatB or GatE kinase
subunit; (ii) Ammonia release by
GatA or GatD glutaminase subunit
using an amide donor (Gln or Asn);
(iii) Amidation of the activated
intermediate by GatB or GatE
subunit using the liberated
ammonia [73].
B.
A.
Introduction
51
Finally, in many eukaryotic mitochondria the formation of Gln-mtRNAGln cannot
occur because of the absence of an apparent organellar GlnRS [80]. In some cases, in order
to overcome the missing GlnRS obstacle, mitochondria can import cytoplasmic GlnRS [81,
82]. Alternatively, GatCAB transamidation pathway can be also used [83, 84]. Utilization of
the indirect pathway in mitochondria reflects a possible endosymbiotic origin of those
organelles [72]. From Saccharomyces cerevisiae [85] to humans [86], AdT subunits are
encoded by the nuclear genome. In yeast mitochondria, non-cognate Glu-mtRNAGln is
formed by the cytoplasmic non-discriminating GluRS that is imported in the mitochondrion,
and subsequent transamidation pathway is mediated by GatFAB, a novel heterotrimeric AdT
[87]. GatA and GatB subunits of this organelle AdT complex are found to be homologous to
those of bacterial and eukaryotic GatCABs, while GatF subunit seems to be fungal-specific.
Crystallography of GatFAB revealed that GatF subunit maintains the structural role of GatC
to the tertiary structure of the complex and enhances the functionality of the other two
subunits [88] (Figure 13C).
Figure 13. (A) Co-crystal structure of the M. thermautotrophicus GatDE-tRNAGln
complex. Only one of
GatD and GatE subunits of the a2b2 tetramer are shown [73]; (B) Predicted structure of the T.
maritima glutamine transamidosome (GatCAB-tRNAGln
-GluRS) [79]; (C) Crystal structure of S.
cerevisiae mitochondrial GatFAB modeled with T. thermophilus tRNAAsn
and T. maritima GatB helical
and YqeY domains [88].
A. B. C.

Introduction
52
2.3. Function complexity of eukaryotic aaRSs and their connections to diseases
In all domains of life, aaRSs retain the minimal architectures, while eukaryotic aaRSs
have often additional domains. Those domains in some cases are required for association
with the large multi-tRNA synthetase complex (MARS) which is formed in higher eukaryotes.
From Drosophila to humans, this complex consists of nine cytoplasmic aaRSs linked to three
non-enzymatic proteins called AIMPs (AIMP 1, 2 and 3). The complex formation plays role in
the protein synthesis efficiency via a specific channeling mechanism [89-92]. Furthermore,
many of these additional domains can also confer novel functions to aaRSs [93, 94], as in the
case of human cytoplasmic GlnRS and LeuRS which function as sensors that trigger apoptosis
or cellular proliferation [95, 96]. They can also impart an altered functionality that involved
in human diseases [97, 98]. However, it is known that these additional domains can also
improve kinetics of aminoacylation [99, 100], whereas canonical domains can participate to
novel functions [73].
The most well-characterized MARS complex is the mammalian, composed of MRS,
DRS, KRS, RRS, LRS, QRS, IRS, EPRS, and three AIMPs; AIMP1/p43, AIMP2/p38, and
AIMP3/p18 [101]. This complex constitutes a platform for releasable aaRSs which can be
subsequently used for regulatory and signaling functions [97, 102]. Besides the cytosolic
anchoring of aaRSs by the MARS complex there are many observations that AARSs can be
released from the complex and relocate to another subcellular compartment or organelle,
such as the nucleus or the mitochondrion [87, 103]. Despite the consideration that these
complexes can function as dynamic platforms for aaRS relocalization, the actual channeling
mechanism and the presence of the whole MARS complex in subcellular compartments
remains to be investigated.
Apart from metazoan species, MARS can be formed also in low-complexity
organisms. Such complexes have been found in prokaryotes, and seem to participate in
more simple functions than their metazoan orthologues, like the enhancement of
aminoacylation efficiency. On the other hand, S. cerevisiae MARS complex is constituted of
two aaRSs, MRS and ERS, and one AIMP (termed Arc1p) [104]. The complex exhibits an
intriguing dual role. In fermenting cells, the cytoplasmic complex formation improves
aminoacylation efficiency of synthetases that take part and when cells need to adapt the
respiration state, the complex mediates ERS release and its relocation in mitochondria.
Subsequently, released MRS relocates in the nucleus and probably modulates gene
expression. Finally, assembly and disassembly of the complex seems to be regulated by a
Snf1/4 depended glucose-sensing pathway, which affects Arc1p expression [105] (Figure 14).

Introduction
53
Figure 14: Schematic model of S. cerevisiae MRS-Arc1p-ERS complex assembly/disassembly upon
Snf1/4 kinase-mediated regulation in respiration state cell adaptation [105].
There are many additional domains and motifs that are encountered in each specific
aaRS during evolution spanning from lower to higher eukaryotes with putative novel
functions (Figure 15). Some of them share structural similarity to those found in other
proteins, such as the WHEP domain which contains a specific helix-turn-helix motif and
function as an anchor for other proteins. Moreover, the EMAPII domain contains an
oligonucleotide binding fold and after cleavage from AARS can function as a cytokine that
binds to a surface receptor [106]. In the case of GluRS–ProRS fusion enzyme (EPRS) a WHEP
domain links the two aaRSs and mediates a specific translation silencing of a subset of genes
that regulate inflammation and iron homeostasis [107]. Furthermore, TyrRS in higher
eukaryotes contains an additional tripeptide ELR motif (Glu-Leu-Arg) and an EMAPII domain,
which in combination participate to angiogenesis [108]. Other additional shared domains are
the Leu-zipper motif and the glutathione S-transferase (GST) domain, which are responsible
for complex formation with other proteins [109] and a specialized N-terminal helix [93].
Finally, there are eight more unique motifs that only one synthetase harbors, and their
addition during evolution are accomplished at distinct points. These motifs reported as UNE
with one additional letter refer to each synthetase e.g. UNe-L corresponds to LeuRS, UNe-S
to SerRS and so on [94].

Introduction
54
Figure 15: Progressive development of aaRSs appeared with novel additional domains from low-
complexity to higher eukaryotes during evolution. As indicated, increasing number of additional
domains in aaRSs has been led to increasing appearance of multi-complexes and novel functions.
UNE-L, UNE-S and so on represent the unique motifs that only one aaRS harbors; GST and LZ
represent glutathione S-transferase domain and Leu-zipper motif respectively [94].
Considering the plethora of novel biochemical pathways where aaRS participate,
present knowledge of aaRS disease association is progressively magnified. This association
can be divided in two categories. In the first, aaRSs are related directly to the disease, such
as in the case of Anti-Synthetase Syndromes like some chronic inflammatory syndromes and
skin/muscle disorders in which patient's serum contains autoantibodies against epitopes of
specific aaRSs [110, 111]. In the second, aaRSs can influence biochemical pathways involved
in the disease, as in many cases of carcinogenesis. There are many observations that link
pathways like aaRSs mediated apoptosis with human cancers. Human TyrRS exhibits a
cytokine-like activity as previously mentioned and seems to have an increased expression in
myeloid leukemic cell lines [112]. Moreover, mitochondrial IleRS is found in high percentage
mutated in colorectal cancers [113]. Finally, it has been proposed that aaRSs or AIMPs that
participate in multi-protein complexes in mammals alone or in combination with other
regulatory factors can control signaling pathways connected to the immune system,
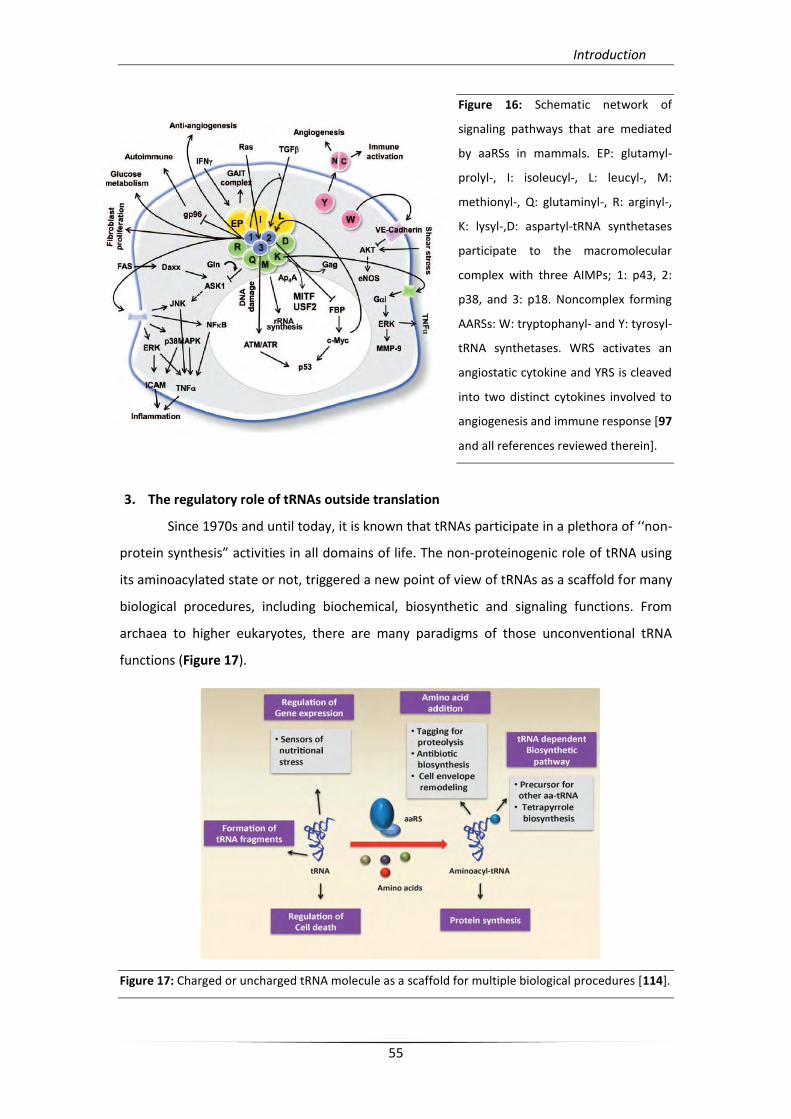
angiogenesis, cell proliferation and metabolism [97] (Figure 16).
Introduction
55
Figure 16: Schematic network of
signaling pathways that are mediated
by aaRSs in mammals. EP: glutamyl-
prolyl-, I: isoleucyl-, L: leucyl-, M:
methionyl-, Q: glutaminyl-, R: arginyl-,
K: lysyl-,D: aspartyl-tRNA synthetases
participate to the macromolecular
complex with three AIMPs; 1: p43, 2:
p38, and 3: p18. Noncomplex forming
AARSs: W: tryptophanyl- and Y: tyrosyl-
tRNA synthetases. WRS activates an
angiostatic cytokine and YRS is cleaved
into two distinct cytokines involved to
angiogenesis and immune response [97
and all references reviewed therein].
3. The regulatory role of tRNAs outside translation
Since 1970s and until today, it is known that tRNAs participate in a plethora of ‘‘non-
protein synthesis” activities in all domains of life. The non-proteinogenic role of tRNA using
its aminoacylated state or not, triggered a new point of view of tRNAs as a scaffold for many
biological procedures, including biochemical, biosynthetic and signaling functions. From
archaea to higher eukaryotes, there are many paradigms of those unconventional tRNA
functions (Figure 17).
Figure 17: Charged or uncharged tRNA molecule as a scaffold for multiple biological procedures [114].

Introduction
56
3.1. Roles of charged tRNAs
In many bacterial and Archaea genera aa-tRNAs serve as precursors in synthesis of
other tRNAs, where some canonical aaRS are missing [65, 73]. Synthesis of bacterial
peptidoglycan and bacterial membrane aminoacylated lipids (aa-PGs) uses aa-tRNAs as
amino acid donors [115, 116]. Furthermore, aa-tRNA serves as an intermediate during
antibiotic synthesis, such as valanimycin synthesis in Streptomyces viridifaciens [117].
Tetrapyrrole biosynthesis, such as heme and chlorophyll, occurs in the presence of a tRNAGlu
intermediate [118]. Selective tRNA-dependent amino acid addition in protein degradation
pathways is catalyzed by the L/F- in prokaryotes and R-transferases in eukaryotes [119] [120]
(Figure 20).
3.1.1. Cell wall formation and remodeling in pathogens
Bacterial cell wall is composed of a multilayer complex structure termed
peptidoglycan (PG) which maintains cell shape and is used as an anchor for attachment of
proteins like the virulence factors [121] (Figure 18). Peptidoglycan polymer is composed of
N-acetyl-glucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) units linked with β (1-4)
glycosidic bonds. Moreover, linkage of PG in the cell wall lipid moiety is carried out by MraY
translocase and MurG transferase, involving a soluble UDP-MurNAc-pentapeptide
intermediate. Every MurNAc residue binds a stem peptide that consists of a general
structure: L-Ala-γ-D-Glu-X-D-Ala-D-Ala. This stem pentapeptide varies among different
bacterial species [122] (Figure 17A).
Figure 18. Bacteria cell wall composition differs between Gram-positive and Gram-negative bacteria.
In Gram-positive bacteria peptidoglycan layer is broader and allows protein attachment via
lipoteichoic acid anchors. Whereas, in Gram-negative bacteria is thicker and is surrounded by an
additional membrane which allows substances to pass through via specific porins.
Introduction
57
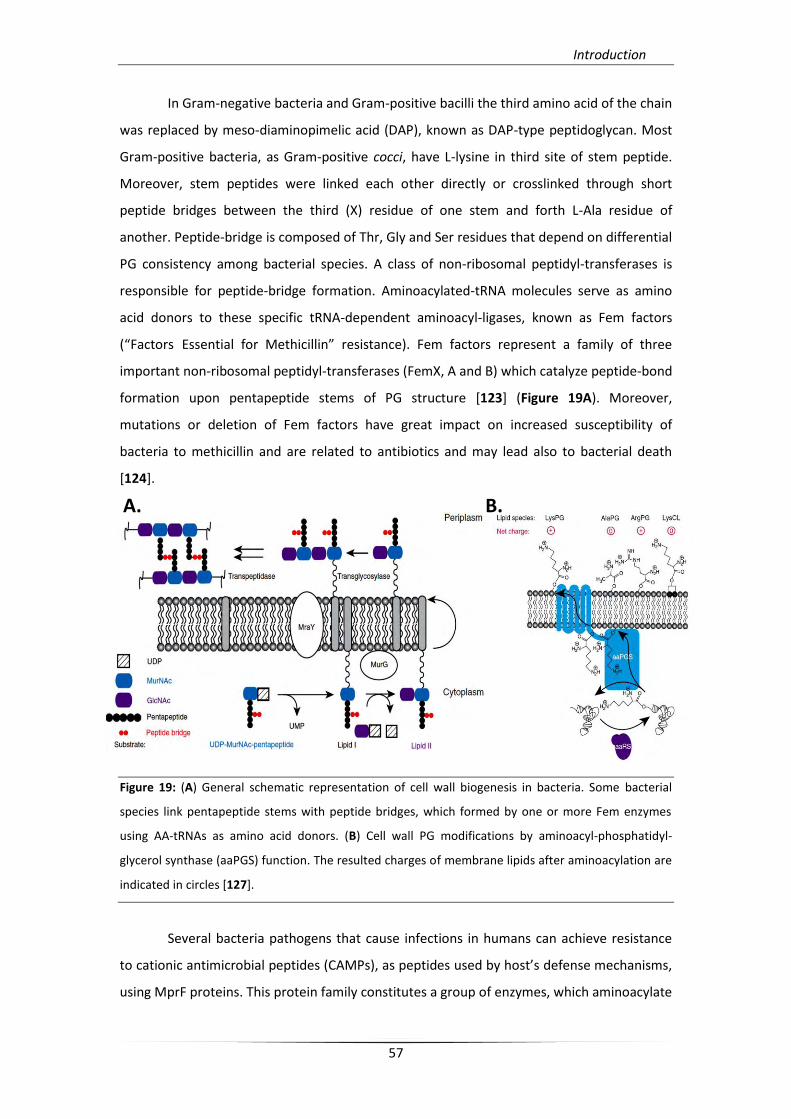
In Gram-negative bacteria and Gram-positive bacilli the third amino acid of the chain
was replaced by meso-diaminopimelic acid (DAP), known as DAP-type peptidoglycan. Most
Gram-positive bacteria, as Gram-positive cocci, have L-lysine in third site of stem peptide.
Moreover, stem peptides were linked each other directly or crosslinked through short
peptide bridges between the third (X) residue of one stem and forth L-Ala residue of
another. Peptide-bridge is composed of Thr, Gly and Ser residues that depend on differential
PG consistency among bacterial species. A class of non-ribosomal peptidyl-transferases is
responsible for peptide-bridge formation. Aminoacylated-tRNA molecules serve as amino
acid donors to these specific tRNA-dependent aminoacyl-ligases, known as Fem factors
(“Factors Essential for Methicillin” resistance). Fem factors represent a family of three
important non-ribosomal peptidyl-transferases (FemX, A and B) which catalyze peptide-bond
formation upon pentapeptide stems of PG structure [123] (Figure 19A). Moreover,
mutations or deletion of Fem factors have great impact on increased susceptibility of
bacteria to methicillin and are related to antibiotics and may lead also to bacterial death
[124].
Figure 19: (A) General schematic representation of cell wall biogenesis in bacteria. Some bacterial
species link pentapeptide stems with peptide bridges, which formed by one or more Fem enzymes
using AA-tRNAs as amino acid donors. (B) Cell wall PG modifications by aminoacyl-phosphatidyl-
glycerol synthase (aaPGS) function. The resulted charges of membrane lipids after aminoacylation are
indicated in circles [127].
Several bacteria pathogens that cause infections in humans can achieve resistance
to cationic antimicrobial peptides (CAMPs), as peptides used by host’s defense mechanisms,
using MprF proteins. This protein family constitutes a group of enzymes, which aminoacylate
A. B.

Introduction
58
negatively charged membrane bacterial lipids, such as phosphatidyl-glycerol (PG) and
cardiolipin (CL), using L-lysine or L-alanine as positive charges to reduce CAMPs’ binding
properties [125]. MprFs (Multiple peptide resistance factors) were first identified in S.
aureus and can use aa-tRNAs as amino acid donors for PG aminoacylation. Moreover, MprF
homologs were identified in most Firmicutes, actinobacteria, and proteobacteria and their
abundance seems to be derived from lateral gene transfer events [126]. Differential
specificity in substrate recognition among MprF homologs resulted in a broader
classification of this enzyme family as aminoacyl-phosphatidyl-glycerol synthases (aaPGS)
[127] (Figure 19B).
3.1.2. Antibiotic biosynthesis
Different aa-tRNAs serve as donors for variable compound transformation yielding
new chemical composition with new functionalities. These aa-tRNA-dependent antibiotic
biosynthetic pathways, including valanimycin, pacidamycin, and cyclodipeptide synthesis
have been identified and characterized in several bacterial species [128]. Valanimycin first
isolated from Streptomyces viridifaciens [117] is an azoxy- compound with potent antitumor
and antibacterial properties. Biosynthesis of this compound involves several steps that
contain multiple enzymatic reactions catalyzed by different enzymes, which are coded by a
gene cluster including 14 genes [129]. Among currently known functions of this biosynthetic
pathway, VlmL, VlmA, VlmJ and VlmK compartments mediate the later steps of valanimycin
formation using an isobutyl-hydroxylamine intermediate. VlmL catalyzes L-seryl-tRNA
formation, while VlmA transfer L-serine to isobutyl-hydroxylamine intermediate in a tRNA-
dependent way for which the actual mechanism of tRNA recognition is still unknown. The
resulted product, O-(L-seryl)-isobutyl-hydroxylamine, is subsequently phosphorylated and
dehydrated by VlmJ and VlmK, respectively, to form valanimycin [130] (Figure 20).
Furthermore, cyclodipeptides (CDP), a secondary metabolite group with a
remarkable clinical use, is an additional example of antibiotics with AA-tRNA-mediated
biosynthesis. Among other CDPs, albonoursin is an antibacterial CDP synthesized by a tRNA-
dependent CDP synthase (AlbC) in Streptomyces noursei [131]. AlbC synthase use two
different aa-tRNAs as donors for cyclo-(L-Phe-L-Leu) formation, an albonoursin precursor
peptide, in a two-step ATP-independent mechanism [132].
Introduction
59
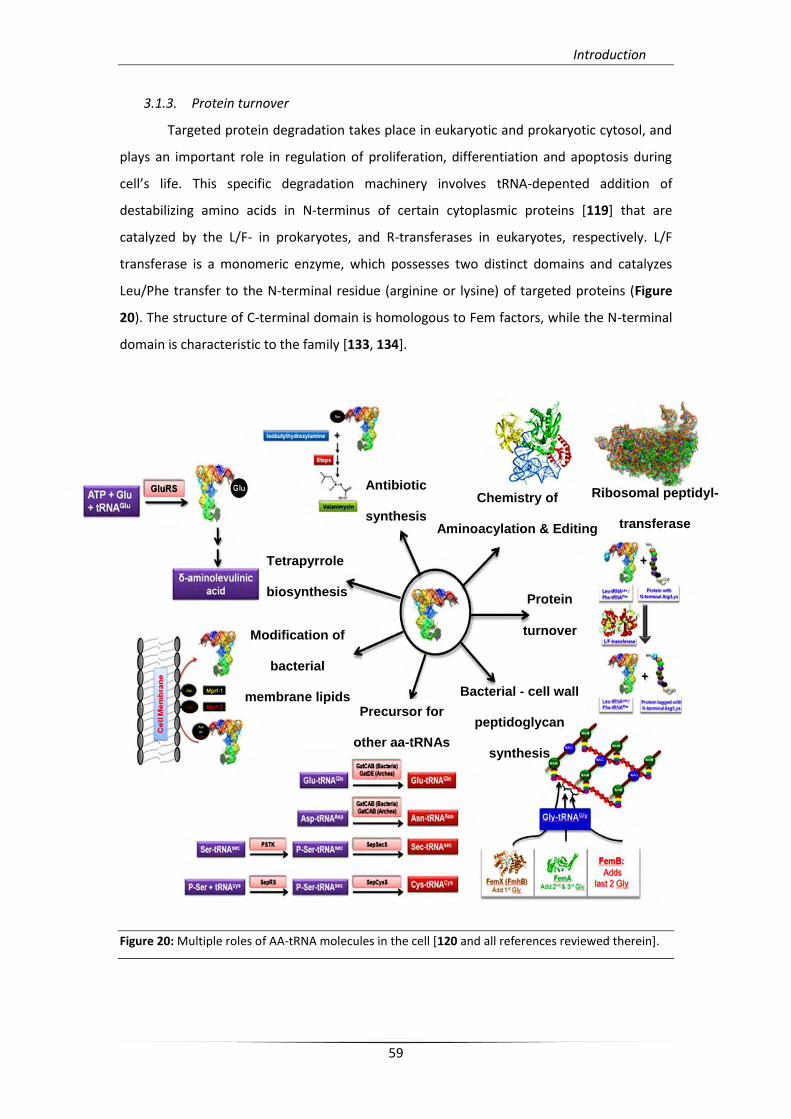
3.1.3. Protein turnover
Targeted protein degradation takes place in eukaryotic and prokaryotic cytosol, and
plays an important role in regulation of proliferation, differentiation and apoptosis during
cell’s life. This specific degradation machinery involves tRNA-depented addition of
destabilizing amino acids in N-terminus of certain cytoplasmic proteins [119] that are
catalyzed by the L/F- in prokaryotes, and R-transferases in eukaryotes, respectively. L/F
transferase is a monomeric enzyme, which possesses two distinct domains and catalyzes
Leu/Phe transfer to the N-terminal residue (arginine or lysine) of targeted proteins (Figure
20). The structure of C-terminal domain is homologous to Fem factors, while the N-terminal
domain is characteristic to the family [133, 134].
Figure 20: Multiple roles of AA-tRNA molecules in the cell [120 and all references reviewed therein].
Antibiotic
synthesis
Chemistry of
Aminoacylation & Editing
Ribosomal peptidyl-
transferase
Tetrapyrrole
biosynthesis
Bacterial - cell wall
peptidoglycan
synthesis
Precursor for
other aa-tRNAs
Modification of
bacterial
membrane lipids
Protein
turnover

Introduction
60
3.1.4. Precursors for tRNA-dependent aa-tRNA formation
As previously mentioned (§2.2.), in the absence of GlnRS or AsnRS in many bacterial
and Archaea genera, an aminoacylated intermediate is converted to the cognate amino acid
by a specific amidotransferase. Similarly, in some cases formation of Cys-tRNACys mediates
an indirect pathway using phosphoseryl-tRNACys as an intermediate, and selenocysteine is
formed only by such an indirect pathway. Indirect pathway of Cys-tRNACys synthesis includes
phosphoseryl-tRNACys formation by SepRS and subsequently intermediate conversion to Cys-
tRNACys by SepCysS [65] (Figure 21B). In bacteria, selenophosphate, which is synthesized by
selenophosphate synthetase (SelD) is used as selenium donor for selenocysteine-tRNASec
formation mediated by Sec synthase (SelA). SerRS, acting as a non-discriminating aaRS in this
case, aminoacylates tRNASec with serine. In eukaryotes and archaea this intermediate is
subsequently converted to Sec-tRNASec by selenocysteine synthase (SepSecS). Ser-tRNASec
intermediate is phosphorylated by the phosphoseryl-tRNA kinase (PSTK), before conversion
to Sec-tRNASec by SepSecS [135] (Figure 21A). Furthermore, selenocysteine incorporation in
protein synthesis mediates a UGA codon specific recognition and a specialized elongation
factor (SelB) involvement.
3.1.5. Tetrapyrrole biosynthesis
In plants, algae, and some bacteria, biosynthesis of hemes, chlorophylls, and bilins
use d-aminolevulinic acid (ALA) as an intermediate. ALA synthesis involves Glu-tRNAGlu
formation and glutamate reduction by glutamyl-tRNA synthetase (GluRS), and Glu-tRNA
reductase (GluTR), respectively. Subsequently, the resulting product, glutamate-
semialdehyde (GSA), is transamidated to ALA by GSA aminomutase [136] (Figure 20). Further
analysis of the aminoacyleted tRNA intermidiate specificity in ALA biosynthesis led to the
notion that separate tRNAGlu species are responsible for translation and heme synthesis. As
an example, Acidithiobacillus ferrooxidans exhibits a dual GluRSs presence. GluRS-1 is able to
charge both tRNAGlu and tRNAGln, and is involved in heme biosynthetic pathway, while GluRS-
2 has non-discriminating function as it preferentially charges tRNAGln. Differential expression
levels of GluRS-1 depend on heme biosynthetic intermediate requirements (Fe2+/ ALA). On
the contrary, GluRS-2 appears to be stable in expression levels as it is essential for protein
synthesis [137].
A.

Introduction
61
Figure 21: Indirect tRNA-
dependent pathways leading
to Sec-tRNASec
and Cys-
tRNACys
formation. (A) The
upper route corresponds to
SelA bacterial pathway, and
the lower to archaeal /
eukaryal PSTK / SepSecS
pathway. (B) Cys-tRNACys
formation is mediated by
SepRS / Sep-tRNA:Cys-tRNA
synthase (SepCysS) pathway
in methanogens [135].
3.2. Roles of uncharged tRNAs
According to the universal conservation of primary sequence and tertiary
architecture, tRNAs appear to be a more solid and stable molecules than mRNA that can be
utilized by cells as potential signaling and regulatory elements for rapid response under
stress conditions. The first considerations for such functions came from the fact that
uncharged tRNAs are able to regulate gene expression in response to nutrition stress during
changes in intracellular amino acid pools. More recent findings revealed a plethora of
regulatory and signaling roles for tRNAs both in eukaryotes and prokaryotes with the
discovery of small RNA molecules derived from tRNA fragmentation (termed tRNA-derived
fragments, tRFs) of, so far, unknown function. Furthermore, it was recently shown that
tRNAs can effectively bind to cytochrome c and as a result they are involved to a signaling
pathway that regulates apoptosis.
3.2.1. Role of tRNAs in the regulation of gene expression
In bacteria, response to environmental stress involves tRNA-dependent pathways
that participate in various adaptation strategies. The most well-known adaptation
mechanism to nutrient deficiency is the one which results in production of 5’-diphosphate
3’-diphosphate guanosine (ppGpp) and 5’-triphosphate 3’-diphosphate guanosine (pppGpp),
molecules known as alarmones. The alarmone synthesis was first described in Escherichia
coli as a response to amino acid starvation [138]. The ppGpp synthesis involves two
pathways that include a ribosome-associated (p)ppGpp synthase (RelA) and a (p)ppGpp
B.
A.

Introduction
62
synthase/hydrolase (SpoT), respectively. In the first pathway, as a result of amino acid
limitation, the presence of uncharged tRNAs to A site of the ribosome activates RelA to
phosphorylate GTP or GDP in order to synthesize pppGpp and ppGpp [139]. Recent findings
revealed the association of ppGpp with transcription regulation since it acts as an allosteric
effector of RNA polymerase. Moreover, this downregulated metabolic pathway seems to
selectively inhibit rRNA and tRNA synthesis [140].
Another well-known mechanism in bacteria that involves uncharged tRNA as the
effector molecule and is related to transcription attenuation is the T-box regulatory system
[141]. In most Gram-positive bacteria, expression of genes encoding enzymes involved in
aminoacyl-tRNA formation and amino acid biosynthesis or uptake, is regulated by such a
tRNA-depended control system [142] (Figure 22). T-boxes are essential riboswitches and as
such are located in the 5’ untranslated region (5’UTR) of mRNAs, upstream the translation
start site of regulated genes. T-boxes can sense amino acid nutrient deficiency by adopting
two alternative conformations with different effect on gene expression, in the absence of
additional protein partners. The first conformation is adopted under high nutrition
conditions, in which charged tRNAs are abundant and includes a stable structural element
that possesses an intrinsic Rho-independent transcriptional terminator (Figure 22). As a
response terminator stem is formed. This conformation blocks RNA polymerase “read-
through” and inhibits transcription of the downstream regulated gene. Under amino acid
starvation conditions, the second conformation is favored by the cognate uncharged tRNA
that binds the 5’-UTR sequence through two distinct interactions; the tRNA anticodon region
binds the complementary codon sequence of the specifier loop (SL) structural element, and
the 3’ acceptor end binds the UGGN complementary sequence found in the T-box region.
This specific interaction stabilizes the antiterminator stem conformation and allows
transcription and expression of the corresponding gene under control [143+. This “on-off”
switch between transcription read-through and transcription stall depends on the
aminocaylation status of the cognate tRNA (charged or uncharged) and is very sensitive to
intracellular amino acid concentrations for the majority of the amino acids. The existence of
T-box riboswitches corresponding to all amino acids indicates the central role of T-box
riboswitches in bacterial metabolism. During T-box:tRNA interaction the role of the SL
structural element seems to be crucial for tRNA recognition and may harbors one or dual
codon specificity as recently proposed for the NT-box riboswitch from Clostridium
acetobutylicum, which regulates the transcription of an operon involved in an essential
tRNA-dependent transamidation pathway [144].

Introduction
63
Finally, in eukaryotes uncharged tRNAs also can be used as effectors of regulatory
mechanisms. Both in yeast and mammalian cells under amino acid starvation conditions,
GCN2 protein kinase was activated after uncharged tRNA binding, in a HisRS like regulatory
domain, and phosphorylates eukaryotic initiation factor 2 (eIF2), which is involved in
translation initiation complex formation [145]. The discrimination mechanism for uncharged
tRNA binding by GCN2 is analogous to RelA protein activation in E. coli, as mentioned above.
GCN2 activation requires uncharged tRNA binding at the A site of the ribosome and also
interaction with a specific GCN1–GCN20 ribosome-associated protein complex, which seems
to increase uncharged tRNA binding at the A site [146].
Figure 22: The T-box regulatory system in bacteria. (Left) When aminacylated tRNA exists in high
levels in the cell, it can bind only the Specifier Loop (SL) region of the 5’UTR riboswitch element and
result in adaptation to the terminator conformation; in contrary (Right) the decreased ratio of aa-
tRNA/ tRNA, enables the uncharged tRNA binding both in SL and T-box (in red) sequences, which
stabilizes the antiterminator conformation; region in blue color indicates the sequester sequence and
the green lines show the specific interaction between tRNA anticodon and acceptor stem regions with
the SL and T-box sequences [142].
3.2.2. tRNA-derived fragments (tRFs)
Recent developments of high-throughput sequencing technologies provided novel
knowledge on the field of small non-coding RNA molecules (sncRNA) besides the well-
characterized miRNAs and siRNAs. As a result, tRNA-derived RNA fragments (tRFs) were
identified as functional regulatory molecules that mediate control of translation and gene
expression under stress conditions [147] (Figure 23). All tRFs derive from functional tRNA
molecules, not as random tRNA degradation products, but rather as products of a short-

Introduction
64
term regulation response mechanism used under stress conditions. The first type of
functional tRFs are the tRNA halves, which are composed of 30 to 35 nucleotides (5’ or 3’
tRNA halves) and are derived from mature full-length tRNA sequence after selective or
random cleavage in the anticodon loop. Generation of tRNA halves in mammalian cells,
occur under stress conditions by angiogenin nuclease activation [148], whereas in yeast
Rny1p nuclease is responsible for the same type of cleavage [149]. These two nucleases can
additionally sense cellular damage and moreover, angiogenin activation can potentially
inhibit tumor formation. Furthermore, tRNA halves were identified also in low complexity
organisms. In bacteria, PrrC, colicin D, and colicin E5 nucleases, appear to be responsible for
tRNA halves production [150].
The second type of tRFs were identified in all domains of life and differ from tRNA
halves in size (13 to 20 nt). They derive from mature and pre-tRNAs which are responsible
for their classification. Four known types have been described so far: 5’ tRFs and 3’ CCA tRFs
derived from mature tRNAs, and 3’ U tRFs or 5’ leader-exon tRFs derived from pre-tRNAs. In
mammalian cells, Dicer mediates 5’ tRFs biogenesis *151]. Among the functions of tRFs as
effectors in various biological processes, is the translation regulation of gene expression
under stress conditions and gene silencing. There are many examples of tRFs function in
such regulating pathways. Under stress conditions angiogenin is activated and can form
tRNA halves, which are able of inhibiting the protein translation initiation machinery [152].
In addition, a 5’ tRF derived from tRNAVal in a stress-dependent manner found in the
archaeon Haloferax volcanii and targets translation by affecting peptide bond formation
[153]. Moreover, tRFs can presumably associate with different Argonaute proteins and
function in the same mode as miRNAs, as in the case of a 3’ tRF derived from tRNAGly in a
Dicer-dependent manner, which after association with Argonaute proteins binds to the 3’
UTR region of an essential gene that involves in DNA repair of mature B cells [154]. However,
the exact type of the Ago proteins that are involved and the exact regulatory mechanism still
remains elusive for the majority of tRFs.
Apart from the already known roles, additional tRF functions are progressively
identified. It has been recently shown that a tRF fragment is probably involved in human T-
cell leukemia virus-type 1 (HTLV-1) reverse transcriptase priming, and enhances viral
infectivity [155]. Furthermore, a possible link has been suggested between tRFs production
and p53-dependent tumor suppressor pathway in response to oxidative stress in CLP1
mutant cells [156]. Nevertheless, the discovery of tRFs was an unexpected finding which
significantly expands our knowledge on gene expression regulation and creates a whole new
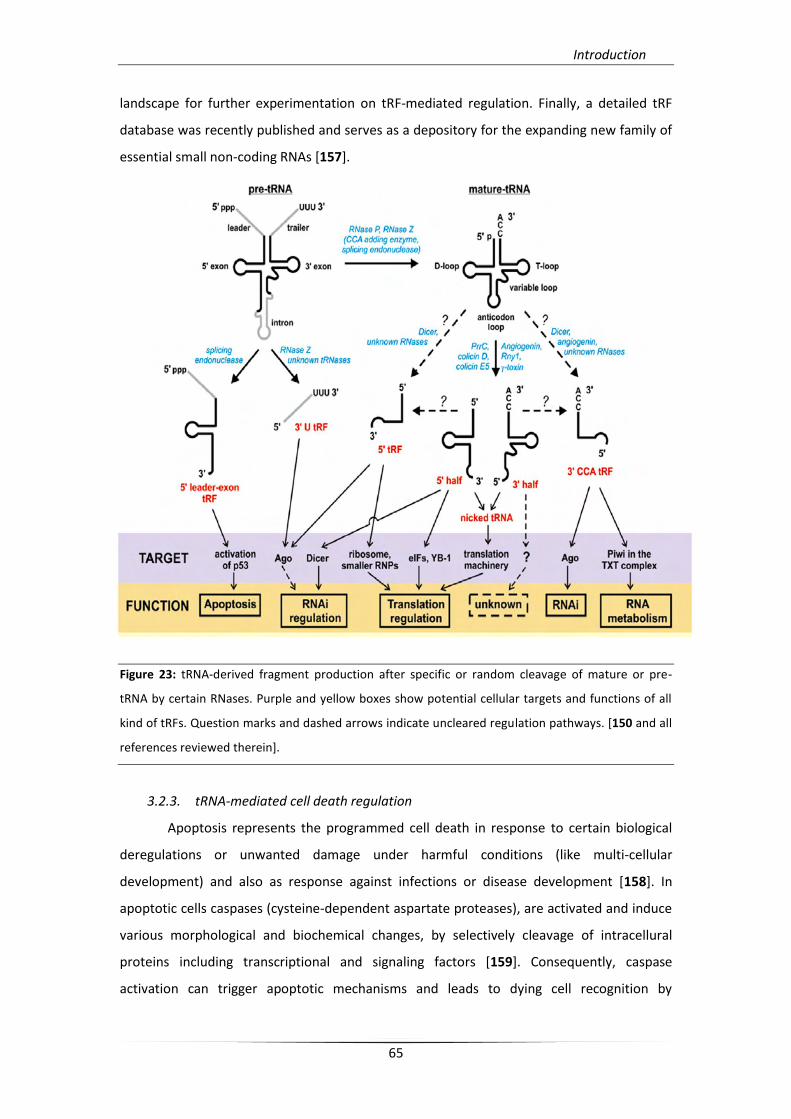
Introduction
65
landscape for further experimentation on tRF-mediated regulation. Finally, a detailed tRF
database was recently published and serves as a depository for the expanding new family of
essential small non-coding RNAs [157].
Figure 23: tRNA-derived fragment production after specific or random cleavage of mature or pre-
tRNA by certain RNases. Purple and yellow boxes show potential cellular targets and functions of all
kind of tRFs. Question marks and dashed arrows indicate uncleared regulation pathways. [150 and all
references reviewed therein].
3.2.3. tRNA-mediated cell death regulation
Apoptosis represents the programmed cell death in response to certain biological
deregulations or unwanted damage under harmful conditions (like multi-cellular
development) and also as response against infections or disease development [158]. In
apoptotic cells caspases (cysteine-dependent aspartate proteases), are activated and induce
various morphological and biochemical changes, by selectively cleavage of intracellural
proteins including transcriptional and signaling factors [159]. Consequently, caspase
activation can trigger apoptotic mechanisms and leads to dying cell recognition by

Introduction
66
phagocytic agents. Among the factors which regulate such apoptotic pathways, the role of
tRNA as a cell death regulator has recently emerged. Recent findings propose that tRNA
inhibits the apoptosome formation, as it is able to interact with the heme moiety of
cytochrome c and reduces its interaction ability with the Apaf-1 complex [160, 161] (Figure
24). Moreover, increased tRNA expression is also found in a variety of tumor cells [162], such
as ovarian, cervical and breast carcinomas. As an example, in breast cancer, overexpression
of tRNAiMet seems to trigger cell proliferation and immortalization [163]. Additionally,
differential expression levels of certain individual tRNAs compared to other tRNAs, showed
that this kind of overexpression cannot be random and considered to be related to
cytochrome c regulation in tumor cells. Finally, it has been suggested that selective tRNA
cleavage by specific RNases can be used as antitumor agents, such as onconase, which
seems to block an apoptosis resistance mechanism that involves cytochrome c release [164].
Figure 24: tRNA-dependent disassembly of the apoptosome in apoptotic pathway is initiated by DNA
damage and p53 activation. Specific uncharged tRNA species binds to the released cytochrome c after
permeabilization of the mitochondrial outer membrane pathway for apoptosis. Blocked cytochrome c
is unable to bind Apaf-1 and form the apoptosome. Subsequently, activation of pro-caspase-9 and
pro-caspase 3 is unable, and apoptotic pathway is prematurely terminated [114].

Introduction
67
4. Transcriptional attenuation and metabolic regulation in bacteria
Transcriptional attenuation is the result of a group of events which take advantage of
different aspects of RNA polymerase function and modulates numerous of mechanistically
related responses. As transcription proceeds, distinct segments of the newly synthesized
transcript itself enable the formation of different secondary and tertiary local structures,
which can provide signals for transcription pause or termination, in order to modulate
downstream gene expression. Such regulation mechanisms that are used mostly by
prokaryotes, occur after transcription initiation, and influence the RNA polymerase function
alone or in combination to simultaneous response of the translation machinery [165].
Sites responsible for transcription termination activation or inhibition involved in
transcription attenuation mechanisms are located between the promoter and the start
codon of the regulated gene or operon and are related to their function. According to the
currently known genome sequences, 10% of the total bacterial operons appear to be
regulated by transcriptional attenuation which leads to transcription termination. The
bacterial transcription termination mechanisms can be divided into two major categories;
the intrinsic and the factor dependent termination. Intrinsic termination involves the
formation of a stable hairpin followed by a stretch of uridines during the transcription
reaction, which serves as a signal for RNA polymerase release [141]. Furthermore, in some
cases this specific segment of the transcript could give rise to a preceding structure, termed
“the antiterminator” (in comparison to “the terminator” form) *166]. The antiterminator
form is able to induce transcription “read-through”. On the other hand, during factor-
dependent termination, Rho protein can recognize specific sequences of the unstructured
synthesized transcript forming an hexamer architecture and directing the RNA polymerase
release in response to termination signals. Modulation of Rho-dependent termination can
occur by altering Rho protein binding to the transcript, or by controlling the RNA polymerase
response to pause signals [141].
There are several distinguished and well-characterized transcription termination
mechanisms which are used for regulation of gene expression in bacteria. Each paradigm
below involves a unique mechanism that modulates transcription termination or
antitermination, in response to metabolic stimuli [141]. Firstly, transcription antitermination
can respond to tRNA charging levels. This kind of signal can primary monitor the availability
of charged tRNAs, and be translation-mediated, or directly distinguish between charged and
uncharged tRNA, in a tRNA-depended manner [166, 167]. In many bacteria, translation-

Introduction
68
mediated transcription termination, involves a ribosome-mediated mechanism, regulating
amino acid synthesis and utilization, at the transcriptional level. As an example, transcription
regulation of trp operon in E. coli, involves an antiterminator structure formation of the
leader RNA, which is induced by a translating ribosome stalling to a tandem Trp codon in
response to charged tRNATrp deficiency and allows transcription to continue to the
downstream coding regions [168]. On the contrary, tRNA-dependent transcription
antitermination occurs in most Gram-positive bacteria and involves a specific RNA-RNA
interaction between uncharged tRNA and the T-box regulatory element at the leader
sequence, in response to the cognate amino acid limitation [169] (Figure 25).
Figure 25: Translation-mediated and tRNA-dependent transcriptional attenuation. (I) Ribosome-
mediated transcription regulation of trp operon in E. coli; (II) tRNA-dependent transcription regulation
by the T-box control system in most Gram-positive bacteria; B/C and C/D regions correspond to
antiterminator and terminator conformation respectively [170 and all references reviewed therein].
Moreover, there are also some proteins which promote or inhibit transcription
termination by interaction with specific RNA sequences of the leader RNA (Figure 26). Such a
representative mechanism found in B. subtilis, interferes with transcription termination of
the trp operon, in response to tryptophan increased levels. The tryptophan RNA-binding
attenuation protein (TRAP) can be activated by eleven Trp residues bound to eleven
identical subunits. Activation of TRAP protein leads to terminator structure formation after
specific interaction with a stretch of eleven UAG or GAG triplets located in the 5’ leader
sequence of the synthesized transcript [171]. Another paradigm of protein-dependent
transcription termination is the regulation of bgl operon involved in betaglucoside sugars
utilization, by the RNA-binding protein BglG, in E. coli. Under abudance of the sugar
substrate, BglG protein can act as a dimer and bind to the leader RNA sequence in order to
stabilize the antierminator conformation. When the sugar substrate is absent, after
phosphorylation by a phosphoenol-pyruvate sugar phosphotransferase protein (BglF) BglG
protein cannot dimerize to bind the leader RNA, thus inducing stabilization of the terminator
conformation and subsequent transcription termination [172, 173].
I. II.

Introduction
69
Figure 26: Protein-mediated transcriptional attenuation. (I) Transcription regulation of trp operon
mediated by TRAP protein in B. subtilis; (II) Transcription regulation of bgl operon mediated by BglG
protein in E. coli; B/C and C/D regions correspond to antiterminator and terminator conformation
respectively [170 and all references reviewed therein]
Additionally, there are two more representative of the transcription attenuation
mechanisms involving protein factors, which can interact selectively with the transcriptional
unit, and proceed in combination with Rho-dependent transcription termination [174]. The
first paradigm is the N protein-mediated antiterminator formation, found in bacteriophage
λ. According to this mechanism, early in infection synthesized proteins, N proteins (NusA,
NusB, NusG, ribosomal protein S10), can modulate expression of new gene products
required at later stages. Consequently, when N proteins are present, they trigger the
formation of a multi-complex, containing host’s specific proteins, which bind to RNA
polymerase and modifies Rho-dependent terminator formation [175] (Figure 27A).
Furthermore, another paradigm of protein factor-mediated transcriptional attenuation is the
direct interference with Rho-dependent termination. In the case of tryptophanase (tna)
operon regulation in E. coli involved in tryptophan utilization, a peptide signal is coded by a
specific region at leader RNA and can suppress Rho-depended termination by ribosome
stalling. In response to tryptophan increasing levels ribosome binds to the leader RNA and
translates a 24-residues coding region, tnaC, which contains a crucial tryptophan residue.
This Trp residue presence results in TnaC-peptidyl-tRNA stacking and keeps the ribosome
stalled before the stop codon. Subsequently, Rho-dependent transcription termination is
blocked, because Rho protein cannot bind to RNA leader sequence, and downstream coding
regions “read-through” is allowed *176] (Figure 27B).
I. II.

Introduction
70
Figure 27: Protein-mediated regulation of Rho-dependent transcription termination. (A) N protein-
mediated antitermination in bacteriophage λ. (B) TnaC-peptidyl-tRNA stacks into translating ribosome
and promotes transcription antitermination of tna operon in E. coli [141].
The specific molecular mechanisms described above, can be divided (according to
the type of the molecule that is sensed or serve as ligand), as ribosome-, protein-, and tRNA-
mediated transcription attenuation mechanisms. Moreover, the diversity in choice of the
mechanism that would be used in response to the same intracellular signal, is likely to be
species-specific, namely among organisms in which the transcriptional attenuation
mechanism that can sense tryptophan limitation or abundance is different [170]. As
previously described, termination of transcription in response to tryptophan deficiency
mediates a ribosome-dependent mechanism in E. coli and a specific tryptophan-activated
RNA-binding protein function in B. subtilis. Additionally, one more category of transcriptional
attenuation mechanism according to the type of the molecule that can be sensed is
characterized as small-molecule-mediated transcription attenuation. It involves sensing of a
small ligand, and can regulate operons responsible for vitamin, pourine or other small
molecule biosynthesis. These small ligands can bind to specific segments of the leader RNA
and are able to stabilize at different levels exclusive secondary structures that result in
transcription termination or antitermination. For example, in Gram-positive bacteria,
regulation of an operon responsible for vitamin B12 biosynthesis, involves
adenosylcobalamine (Ado-CBL or B12) binding to the leader RNA which in turn mediates
stabilization of a specific aptamer formation and results in transcription termination [177]
(Figure 28).
A.
B.

Introduction
71
Figure 28: Small-molecule-mediated transcription
attenuation. A small ligand, as adenosylcobalamine
(B12), can stabilize an aptamer formation resulting
transcription termination (C/D region indicates the
terminator formation) [170 and all references
reviewed therein].
Finally, transcriptional attenuation is also found in eukaryotes but not as commonly
as in bacteria. The fact that there is only one common molecular mechanism from a variety
of bacterial mechanisms which is also found in eukaryotic cells, led to the suggestion that
eukaryotic cells can tolerate and handle better nutrition deficiency than bacteria. As a result
in higher eukaryotes there are far less genetic systems that sense fundamental metabolites
and more use of second messages and signaling compounds in response to metabolic stress
[178]. Eukaryotic expression control systems, such uORFs (conserved upstream open reading
frames), are able to modulate protein translation and in some cases are involved in human
diseases [179]. Characteristic examples are the mammalian S-adenosyl-decarboxylase uORF
and the human cytomegalovirus gpUL4 uORF2, which use a transcription attenuation control
system to regulate expression of the downstream coding region. Moreover, both eukaryotic
and prokaryotic cells can use a different kind of regulation mechanisms that involve specific
RNA-RNA interactions and interfere with multiple small RNAs, which implicate antisense and
dsRNA regulatory systems [180]. The existence of such mechanism provides further
evidence and bases to the notion that multiple RNA sequences evolved in order to exploit
the regulation of gene expression [141].
5. Riboswitches
Many bacterial, archaeal and eukaryotic organisms use elegant molecular systems
that are able to sense small metabolite concentrations and tune their expression levels in
order to control production and function of essential enzymes involved in deferential
metabolic requirements during cellular life. These small-molecule sensors, termed
“riboswitches”, are located in the 5′ untranslated region (5’UTR) of mRNAs. Their “switching”
ability relies on folding changes formed upon small-molecule binding and their functionality
is protein-independent [181].
There is biochemical evidence supporting “The RNA World” theory. As an example,
the fact that many co-enzymes are of RNA origin or that many contemporary enzymes
harbor RNA subunits crucial for their function, supports the theory of a very ancient RNA
world where no protein or DNA molecule played significant role in the primordial cells [182].

Introduction
72
Similar theories raised the hypothesis that many riboswiches are of a very ancient origin
[183]. Each riboswitch class has evolved in order to sense a specific metabolite and despite
the sequence diversity among species of the same class the core binding pockets of each
riboswitch remain highly conserved (Figure 29). Furthermore, many of the riboswitch classes
are found in bacteria, but there are some classes, such TPP riboswitches, which can sense
thiamin pyrophosphate coenzyme and are found in all domains of life [184]. Finally, there
are some classes of small riboswitches which are rare and of restricted distribution among
species might be of a more recent evolutionary origin.
Figure 29: Schematic models of eight different small-ligand-binding riboswitch classes. Each atomic-
resolution structure model indicates the aptamer formation (ribbon diagrams) after ligand (spheres)
specific binding. The (a) purine [PDB ID: 1Y26, 1Y27, 1U8D], (b) TPP [PDB ID 2GDI, 2HOJ, 2CKY], (c)
SAM-I [PDB ID: 2GIS], (d) SAM-II [PDB ID: 2QWY], (e) SAM-III/SMK [PDB ID: 3E5C, and (f) lysine [PDB
ID: 3D0U, 3DIL] riboswitch aptamers; The (g) GlcN6P-sensing ribozyme [PDB ID: 2HO7, 2N74] and the
(h) ion-sensing (Mg2+
)-box [PDB ID: 2QBZ] [181 and all references reviewed therein].
a. b. c. d.
e. f. g. h.

Introduction
73
5.1. Origins and mechanism
The term “riboswitch” was introduced for the first time in 2002 by Nahvi and
colleagues inspired by the function and name of ribozymes in order to characterize an RNA
segment that shifts its idiosyncratic features like a switch upon small-molecule binding [185].
Following this initial suggestion, the term “riboswitch” was confirmed by many subsequent
studies and is currently used for every RNA motif which exhibits structural “switches” in
response to a physiological signal, such as a small-metabolite and that occurs directly and
without any protein mediation.
Riboswitch regulatory elements are commonly found in bacteria and are almost
exclusively located in 5′ UTRs of the mRNAs in order to control the expression of
downstream coding regions. Riboswitch-mediated modulation of downstream gene
expression involves intrinsic attenuation mechanisms that control transcription termination
(Figure 30A). As transcription proceeds, the newly synthesized mRNA regions which include
the regulatory element segment are able to sense metabolism requirements by structural
diversity and induce or not the production of full-length mRNA.
Figure 30: (A) Riboswich-mediated transcription regulation mechanism. Stabilization of the aptamer
formation upon ligand binding leads to transcription termination as schematically represented
(intrinsic terminator formation followed by a string of U residues). In the absence of ligand, the
altered antiterminator formation allows RNAP to pass and transcribe the downstream region. (B)
Predicted architectures of ligand-mediated gene control systems with simple or mixed specifications.
The expression platforms can also correspond to self-processing ribozymes (riboswitch-ribozyme
cooperative systems) [178].
A. B.

Introduction
74
Apart from riboswitch elements which can bind small metabolites, there are also
some additional categories of such RNA-based regulatory elements. The T-box regulatory
system, which is first described as a transcription attenuation mechanism [169] is able to
bind charged or uncharged tRNAs in response to the cognate amino-acid availability. The
special category of RNA “thermosensors” can adopt alternative secondary structures in
response to selective temperature changes, and functions without binding any small-
molecule or any protein presence [186]. Moreover, there is a variety of riboswitches with
mixed specificities (Figure 30B). An example is the Ado-CBL/SAM mixed-ligand riboswitch, in
which binding of each co-enzyme results in the same modulation of downstream gene
expression [187]. Also, more recent studies revealed the existence of a riboswitch that
functions as an allosteric ribozyme in Clostridium difficile [188]. In this regulatory system, c-
di-GMP metabolite binds to a region upstream the 5′ splice site and stabilizes a specific
aptamer structure, which unmasks the splicing site and enables the ribozyme to proceed.
Consequently, these RNA-based regulators seem to be highly refined sensors, with different
specificities among bacterial species that evolve rapidly and compete with other protein-
based regulatory mechanisms during evolution.
5.2. Riboswitch categories
Riboswitches classification is in many cases difficult because of their broad diversity
and despite the high conservation of their core regions. RNA thermosensors represent the
simplest class of riboswitch RNAs. They are able to sense temperature fluctuations that
result in fine tuning of the secondary structure and release or sequester Shine-Dalgarno (SD)
sequence region [189]. In most cases, under normal temperature growth conditions, mRNA
SD sequence is not accessible for ribosome binding. An increase in temperature can
destabilize the formation of the sequester helix by a melting shift, and set SD sequence into
an accessible mode for the ribosome state. This kind of riboswitch elements often control
expression of genes involved in activation of heat-shock pathways or in induction of
virulence processes after entrance into a mammalian host cells [190, 191] (Figure 31).

Introduction
75
Figure 31: RNA thermosensor regulatory
mechanism. (Left) Sensing of low
temperature results in translation
termination via anti-SD/SD sequence base
pairing; (Right) Increased temperature
disrupts anti-SD/SD helix stabilization and
allows translation initiation [192].
Another category of riboswitch RNAs are the ligand-binding motifs that are found
widespread among species and possess high diversity. In contrast to RNA thermosensors,
they are composed of two distinct structural domains, a binding domain, and a domain
responsible for gene expression modulation. The ligand-binding domain is able to
discriminate between related ligands for the correct one. Interaction with the appropriate
ligand can result in a shifted structure of the binding domain, which promotes a specific
alteration in the gene expression domain. Such structural shifts of gene expression platform
could result in transcription termination or translation sequestration. In most cases, this
gene expression control systems can occur as “on-off switches” for either transcription or
translation processes (Figure 32). Moreover, they are found upstream of operons whose
coding products are involved in biosynthetic pathways of the regulating effector itself, in
order to form a feedback control. A common example is the glycine-binding gcv element, in
which the structural shift upon the effector binding leads to antiterminator helix
stabilization, namely a transcription “switch on”, and a “feed-forward” rearrangement *193].
Figure 32: Metabolite-sensing riboswitch control system. In the presence of the effector molecule (*),
binding domain (L) forms a specific conformation which results in downstream regulatory sequence
rearrangement. (A) Terminator (T) formation leads to transcription pause, and (B) stabilization of the
anti-SD/SD helix leads to ribosome binding site sequestration. Alternatively, when the ligand is
absent, anti-terminator or anti-SD helix formation allows transcription or translation respectively, to
“switch on” *192].
A. B.
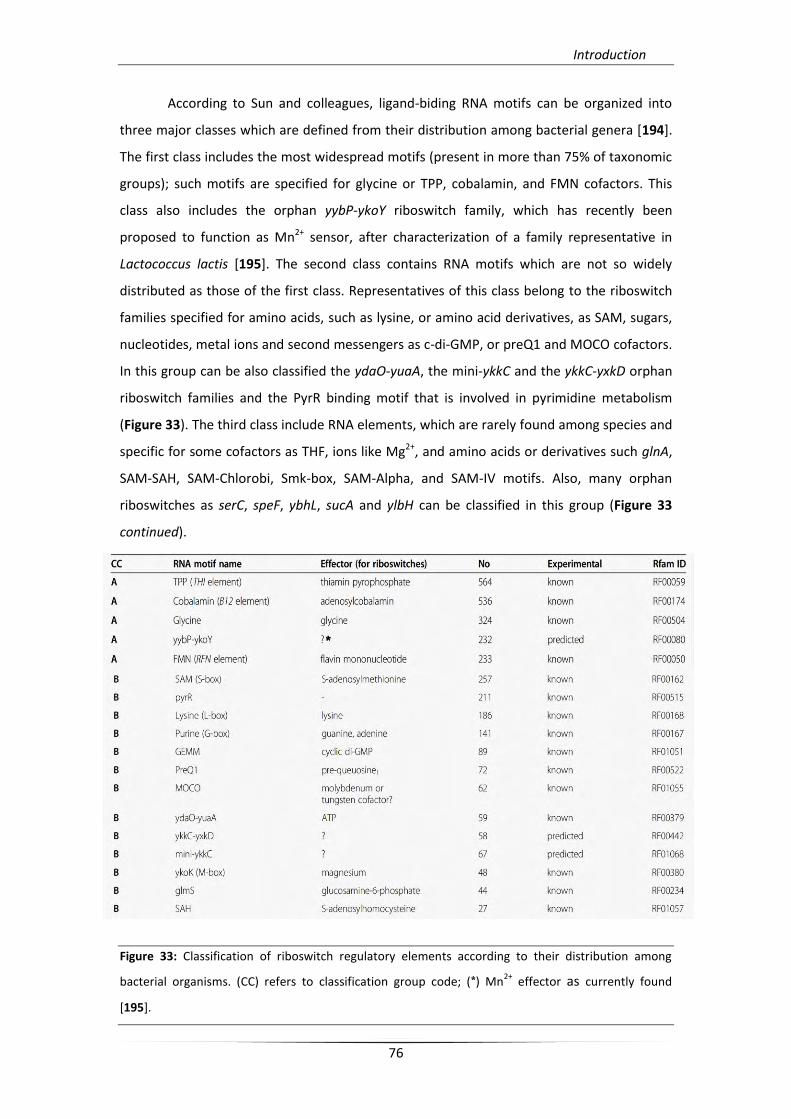
Introduction
76
According to Sun and colleagues, ligand-biding RNA motifs can be organized into
three major classes which are defined from their distribution among bacterial genera [194].
The first class includes the most widespread motifs (present in more than 75% of taxonomic
groups); such motifs are specified for glycine or TPP, cobalamin, and FMN cofactors. This
class also includes the orphan yybP-ykoY riboswitch family, which has recently been
proposed to function as Mn2+ sensor, after characterization of a family representative in
Lactococcus lactis [195]. The second class contains RNA motifs which are not so widely
distributed as those of the first class. Representatives of this class belong to the riboswitch
families specified for amino acids, such as lysine, or amino acid derivatives, as SAM, sugars,
nucleotides, metal ions and second messengers as c-di-GMP, or preQ1 and MOCO cofactors.
In this group can be also classified the ydaO-yuaA, the mini-ykkC and the ykkC-yxkD orphan
riboswitch families and the PyrR binding motif that is involved in pyrimidine metabolism
(Figure 33). The third class include RNA elements, which are rarely found among species and
specific for some cofactors as THF, ions like Mg2+, and amino acids or derivatives such glnA,
SAM-SAH, SAM-Chlorobi, Smk-box, SAM-Alpha, and SAM-IV motifs. Also, many orphan
riboswitches as serC, speF, ybhL, sucA and ylbH can be classified in this group (Figure 33
continued).
Figure 33: Classification of riboswitch regulatory elements according to their distribution among
bacterial organisms. (CC) refers to classification group code; (*) Mn2+
effector as currently found
[195].
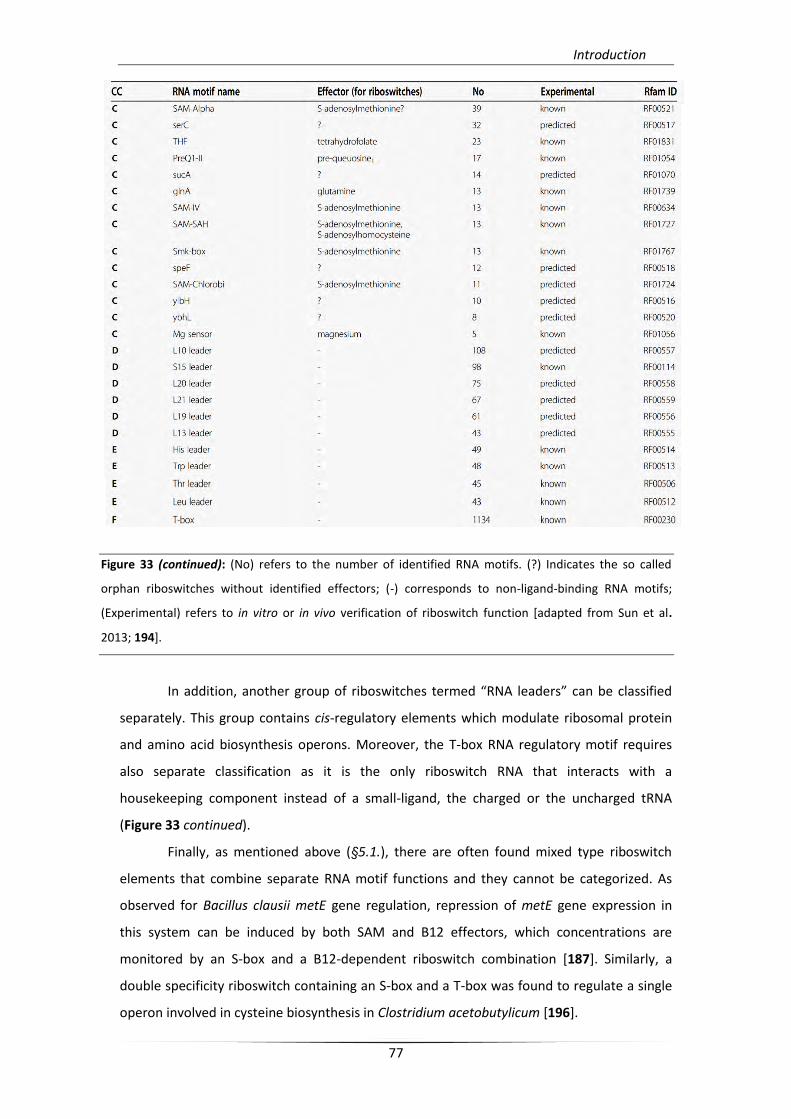
Introduction
77
Figure 33 (continued): (No) refers to the number of identified RNA motifs. (?) Indicates the so called
orphan riboswitches without identified effectors; (-) corresponds to non-ligand-binding RNA motifs;
(Experimental) refers to in vitro or in vivo verification of riboswitch function [adapted from Sun et al.
2013; 194].
In addition, another group of riboswitches termed “RNA leaders” can be classified
separately. This group contains cis-regulatory elements which modulate ribosomal protein
and amino acid biosynthesis operons. Moreover, the T-box RNA regulatory motif requires
also separate classification as it is the only riboswitch RNA that interacts with a
housekeeping component instead of a small-ligand, the charged or the uncharged tRNA
(Figure 33 continued).
Finally, as mentioned above (§5.1.), there are often found mixed type riboswitch
elements that combine separate RNA motif functions and they cannot be categorized. As
observed for Bacillus clausii metE gene regulation, repression of metE gene expression in
this system can be induced by both SAM and B12 effectors, which concentrations are
monitored by an S-box and a B12-dependent riboswitch combination [187]. Similarly, a
double specificity riboswitch containing an S-box and a T-box was found to regulate a single
operon involved in cysteine biosynthesis in Clostridium acetobutylicum [196].

Introduction
78
5.3. The T-box regulatory system
The T-box regulatory element was first identified in Bacillus subtilis as a specific RNA
motif. It was first described to be located in the 5’ UTR region of tyrS mRNA and was
involved in transcription modulation using the transcription attenuation mechanism [197].
Unlike other ligand-binding RNA motifs, T-box RNA can interact with both charged and
uncharged cognate tRNAs thus monitoring the ratio of its substrates and not their absolute
concentrations [198]. Bacterial cells can sense the status of aminoacylated tRNAs in
response to nutrition changes by using this type of regulation systems specific for particular
tRNA species. Most importantly, the T-box system regulates biosynthesis or/and utilization
of the cognate amino acid.
Figure 34: Secondary structure model of the T-box regulatory element. Sequence represents the B.
subtilis tyrS T-box leader RNA. The major structural domains, Stem I, Stem II (plus Stem IIA/Stem IIB
pseudoknot), Stem III, and terminator/antiterminator alternative stems were indicated. Filled colored
circles indicate the conservation among T-box species, including a highly conserved purine (green
open circle) after the specifier sequence (boxed residues) [200].

Introduction
79
Bioinformatics analysis of the T-box riboswitch RNA regions revealed an array of
conserved structural features, besides the highly conserved T-box element [199, 200] (Figure
34). The first region is Stem I, a complex structure which includes distinct functional domains
like the kink-turn (K-turn), the GA motif (in most of the cases), the essential Specifier Loop
(SL) which in some variants is followed by two additional helixes; the Stem II and the Stem
IIA/B pseudoknot elements [201, 202]. Furthermore, following the specified linker region,
two more distinct domains are formed; a third helix (Stem III) and a terminal important helix
which includes the conserved T-box sequence responsible for the formation of the
functional terminator/antiterminator element or a specialized Shine-Dalgarno sequence
sequester/antisequester present in some species like actinobacteria [203]. Conservation of
these structural elements is crucial for T-box regulatory system functionality. Even a simple
single mutation can often lead to insufficient transcription antitermination [204]. Further
biochemical and structural studies of these distinct domains revealed that tRNA binding
specificity depends on the interaction between the tRNA anticodon region and the Specifier
Loop, as well as on the acceptor stem interaction with a conserved UGGNACC sequence at
the terminal stem (N indicates the specific base which interacts with the discriminator base
of the tRNA acceptor stem) that stabilizes the antiterminator formation [198, 205]. In
addition to those crucial interactions there are some observations suggesting that the K-turn
motif and other structural features of the Stem I domain (with the exception of the Specifier
Loop) can be involved in the broader recognition mechanism of cognate tRNAs and possibly
enhance antiterminator formation.
5.3.1. T-box regulons and mechanism
Extensive genomic analyses have identified a variety of T-box regulons among bacterial
genera and revealed that they are widely distributed among the majority of Gram-positive
bacteria, mainly in Firmicutes, but also in Actinobacteria, which include many severe
pathogens. Moreover, T-box regulons can be found in some Gram-negative bacteria (δ-
Proteobacteria) and other groups such as Deinococcales/Thermales, Chloroflexi, and
Dictyoglomi (Figure 35). These regulons can be categorized according to the role and
specificity of the protein product. T-boxes which regulate aminoacyl-tRNA synthetases for
aromatic and branched-chain amino acids, or Asp, Asn, Ser, and Gly are found in most
Firmicutes, but also in actinobacteria, Deinococcales/Thermales, Chloroflexi and
Dictyoglomi. T-boxes which control enzyme operons are involved in amino acid metabolism
and are found mostly in Firmicutes. Such T-boxes are rarely found outside Firmicutes, with
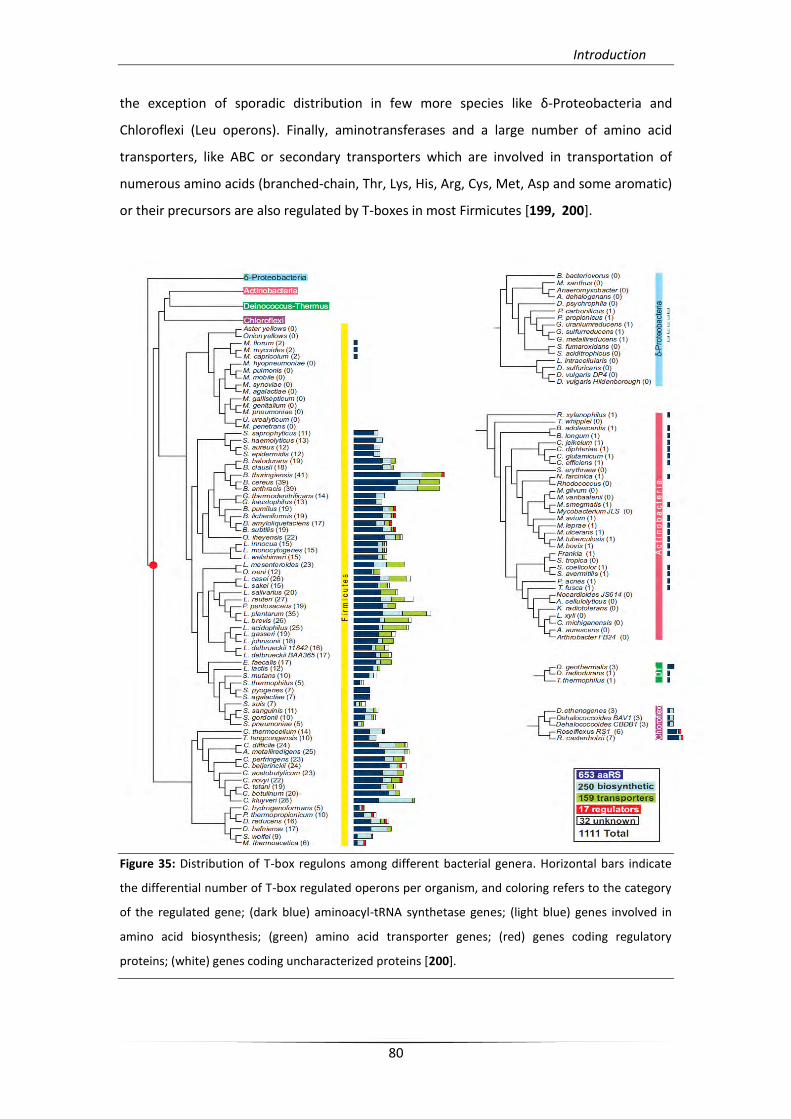
Introduction
80
the exception of sporadic distribution in few more species like δ-Proteobacteria and
Chloroflexi (Leu operons). Finally, aminotransferases and a large number of amino acid
transporters, like ABC or secondary transporters which are involved in transportation of
numerous amino acids (branched-chain, Thr, Lys, His, Arg, Cys, Met, Asp and some aromatic)
or their precursors are also regulated by T-boxes in most Firmicutes [199, 200].
Figure 35: Distribution of T-box regulons among different bacterial genera. Horizontal bars indicate
the differential number of T-box regulated operons per organism, and coloring refers to the category
of the regulated gene; (dark blue) aminoacyl-tRNA synthetase genes; (light blue) genes involved in
amino acid biosynthesis; (green) amino acid transporter genes; (red) genes coding regulatory
proteins; (white) genes coding uncharacterized proteins [200].

Introduction
81
There are also different mechanisms through which T-box RNAs can modulate gene
expression, including mainly transcription antitermination and secondly regulation of
translation initiation. Predominantly, T-box regulation occurs at the transcriptional level as
they can modulate premature transcription termination, by an alternative to terminator
hairpin formation, termed “antiterminator” which is stabilized after specific interaction with
an uncharged cognate tRNA (Figure 36A). This intrinsic mechanism of transcription
termination/antitermination is found in most Firmicutes, but also in δ-Proteobacteria,
Deinococcales/Thermales, Chloroflexi, and Dictyoglomi. Additionally, some T-boxes that
function according to the transcription regulation mechanism seem to possess a different
terminator formation. As an example ileS T-box in S. aureus can form the terminator
structure by base pairing an additional region which extends the antiterminator structure,
without the need of an alternative structure formation [206] (Figure 36B). Such unusual T-
boxes can also be found in some Firmicutes, namely in Streptococcaecae (serS),
Leuconostocaceae (alaS), and Clostridiaceae (glyS).
Figure 36: The T-box transcription regulation mechanism; Canonical (A) and unusual (B)
antiterminator (1/1’-2/2’) or terminator (3-4) helix formation. (P1-P2-P3) corresponds to the T-box
specifier codon and (Y1-Y2-Y3) to the tRNA anticodon nucleotides. (VAR) refers to the variable region
[199].
A.
B.
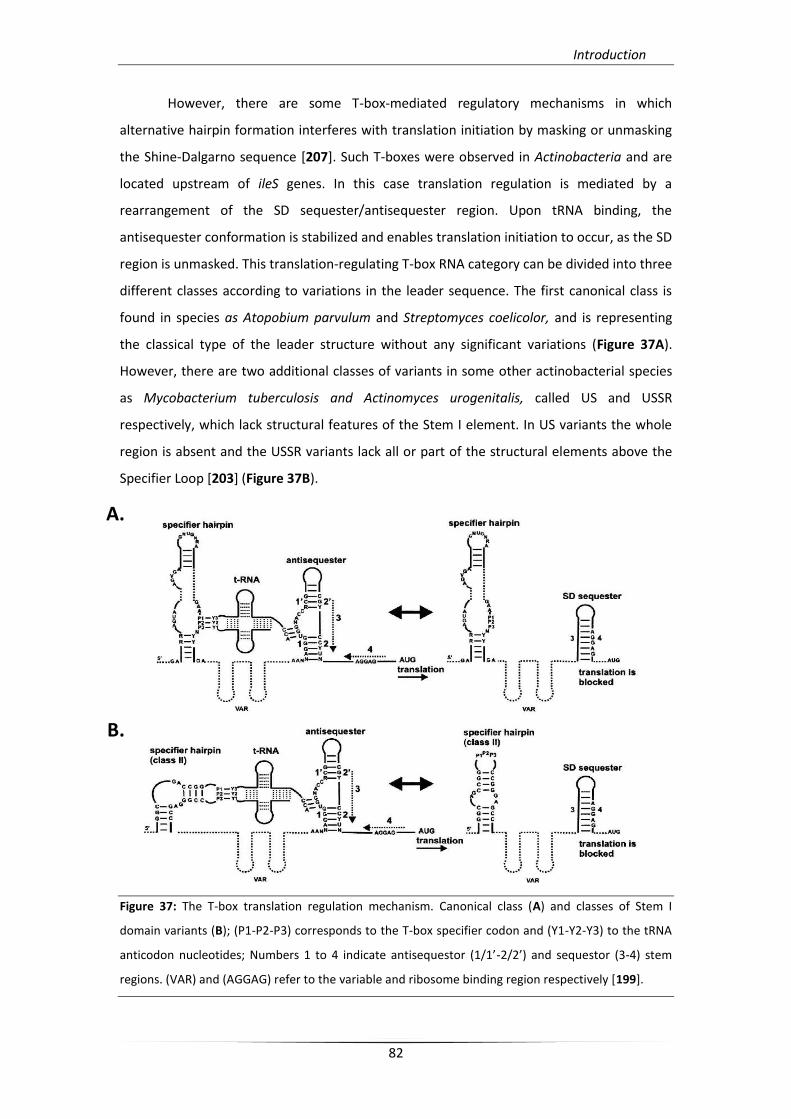
Introduction
82
However, there are some T-box-mediated regulatory mechanisms in which
alternative hairpin formation interferes with translation initiation by masking or unmasking
the Shine-Dalgarno sequence [207]. Such T-boxes were observed in Actinobacteria and are
located upstream of ileS genes. In this case translation regulation is mediated by a
rearrangement of the SD sequester/antisequester region. Upon tRNA binding, the
antisequester conformation is stabilized and enables translation initiation to occur, as the SD
region is unmasked. This translation-regulating T-box RNA category can be divided into three
different classes according to variations in the leader sequence. The first canonical class is
found in species as Atopobium parvulum and Streptomyces coelicolor, and is representing
the classical type of the leader structure without any significant variations (Figure 37A).
However, there are two additional classes of variants in some other actinobacterial species
as Mycobacterium tuberculosis and Actinomyces urogenitalis, called US and USSR
respectively, which lack structural features of the Stem I element. In US variants the whole
region is absent and the USSR variants lack all or part of the structural elements above the
Specifier Loop [203] (Figure 37B).
Figure 37: The T-box translation regulation mechanism. Canonical class (A) and classes of Stem I
domain variants (B); (P1-P2-P3) corresponds to the T-box specifier codon and (Y1-Y2-Y3) to the tRNA
anticodon nucleotides; Numbers 1 to 4 indicate antisequestor (1/1’-2/2’) and sequestor (3-4) stem
regions. (VAR) and (AGGAG) refer to the variable and ribosome binding region respectively [199].
A.
B.

Introduction
83
Moreover, in some cases, tandem copies of the same T-box RNA upstream certain
biosynthetic and transport genes can be found, as it has been described for the thrZ gene in
B. subtilis. In this case, independent binding of uncharged tRNAThr effector molecule at each
riboswitch motif promotes transcription “read-through” of the downstream coding
sequence in response to intracellular decreased concentrations of Thr [208]. This paradigm
is a representative of the first major class of tandem T-boxes, called double T-boxes derived
from complete duplication of the same T-box structure. The second major class is the so-
called partially double T-boxes, namely one specifier hairpin followed by a repeated
terminator/antiterminator structure, which seems to be derived from a duplication of a
complete T-box riboswitch and a subsequent deletion of the second specifier hairpin during
evolution. Despite the fact that the actual role of partially double T-boxes remains unclear,
this element is found highly conserved among some bacterial species as in
Lactobacillaceae/Leuconostocaceae (His ABC transporter genes) [199].
Finally, T-box riboswitch can function in concert with other regulatory systems
upstream a single coding region. This combinatorial system can fine-tune two independent
mechanisms and serve as a dual level of regulation of gene expression by monitoring two
different physiological coditions. As an example the ilv-leu operon containing genes which
are involved in biosynthesis of branched chain amino acid in B. subtilis, can be regulated in
the transcription level by a T-box RNA in response to Leu starvation, and its promoter can be
also repressed by the CodY DNA-binding protein in response to Ile and GTP increased
concentrations [209, 210]. In addition to these observations, it was also found that in B.
subtilis a T-box riboswitch specific for uncharged tRNATyr, can synchronize regulation of both
tyrZ and tyrS genes in transcriptional level, in order to accomplish growth requirements
according to nutrient availability [211].
5.3.2. Structural analysis of T-boxes
The first information on T-box RNA structure came from NMR analysis. A helix–
bulge–helix formation was predicted for the antiterminator stem and in solution interaction
with the tRNA confirmed the affinity for the UGGN sequence of the T-box bulge region (the
region that contains the T-box sequence). The theoretical model of the cognate tRNA
binding was also confirmed by selective mutagenesis and further structural analysis of the
bugle region according to which, the acceptor stem interacts with the unpaired residues of
the bugle and results in stabilization of its overall folding thus allowing the formation of the
antiterminator stem [205]. Moreover, NMR analysis of the tyrS leader RNA in B. subtilis

Introduction
84
revealed that both the predicted K-turn and the Specifier Loop S-turn of Stem I domain can
be formed in solution [212] (Figure 38B). Additionally, similar data for B. subtilis glyQS Stem I
domain has shown that residues of the codon sequence are exposed as Specifier Loop folds,
in order to interact with tRNA, and also proposed that a conserved purine located
downstream the codon sequence interferes with stabilization of the anticodon loop without
base-pairing requirement [213].
The first co-crystal structure recently published shed light on the overall structural
rearrangement of stem I bound to the cognate tRNA. The structure was obtained for Stem I
domain of the glyQS leader RNA of Oceanobacillus iheyensis coupled with the cognate
tRNAGly and showed that conserved residues on different structural domains of Stem I can
interact with the elbow of the tRNA (namely selective nucleotides exposed to tertiary
structure formation of D-loop and T-loop) and as a result they maintain the specificity of
structure recognition of the cognate tRNA [214, 215] (Figure 38A). Interestingly, these
conserved residues which are responsible for the specific recognition can be arranged much
differently in other T-boxes and in some cases, such as ileS T-boxes in Actinobacteria, they
can be totally or partially absent [203]. Structural data for the Stem II, the Stem IIA/B
pseudoknot and the variable Stem III domains are yet elusive.
The most important interaction between Stem I and the cognate tRNA is the base
pairing between the SL codon-like triplet and the anticodon of the tRNA. Structural analysis
revealed that this interaction is secured through an essential local geometry which is favored
by flaking stacked bases (conserved purines located nearby the SL codon or tRNA anticodon
triplet). This local architecture participates also in the recognition that occurs during
decoding of mRNA in ribosome where conserved bases of both mRNA and rRNA molecules
also participate (Figure 38A) [215, 216]. However, the codon reading that occurs in the T-box
SL by the tRNA’s anticodon is EF-Tu independent and no other additional proteins factors
seem to participate. According to the proposed two-checkpoint mechanism tRNA binding
ability is initially checked through the SL:tRNA interaction and subsequently specific identity
elements of tRNA’s overall structure favor essential dynamic associations in order to secure
the correct accommodation of the effector (Figure 36B) [214]. This notion suggests that T-
box regulatory mechanism requires both the correct measurement and recognition of
tRNA’s structure beyond the codon-anticodon complementation that seem to favor a
mutually induced fit [215]. Finally, further investigation of the structural variability of other
T-box leader RNA classes and as well as investigation of tRNA recognition by the full leader
RNA remains to be unraveled.
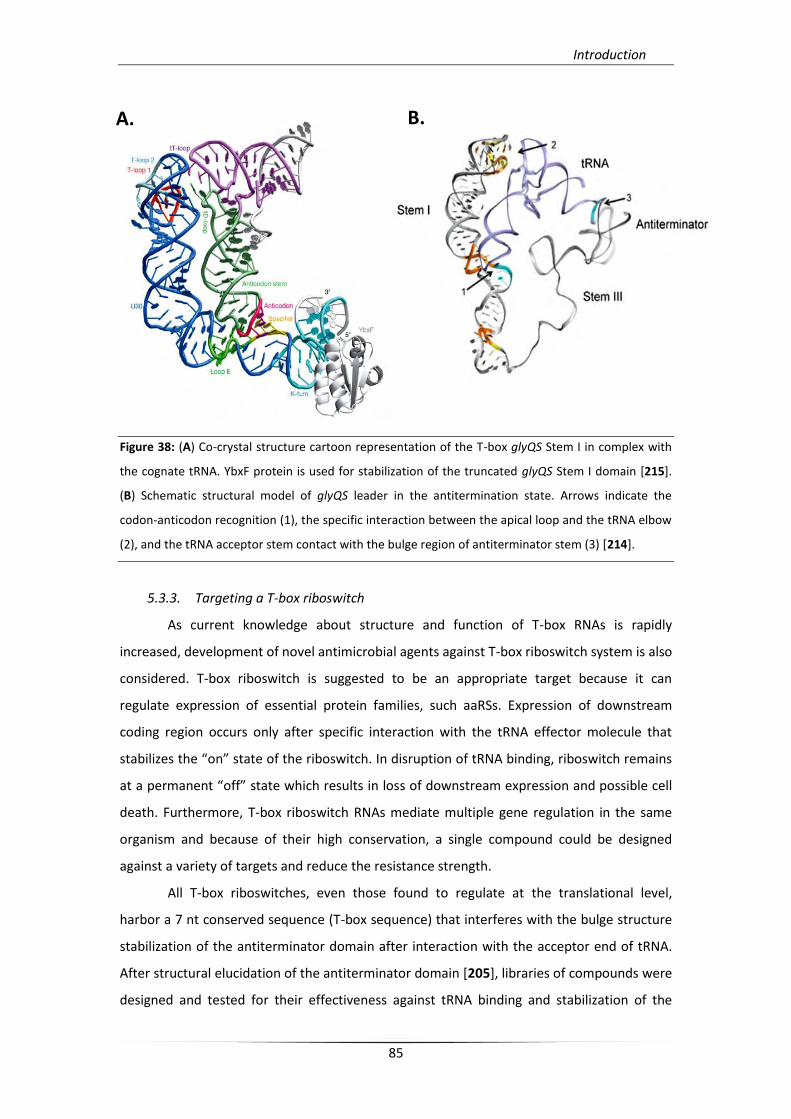
Introduction
85
Figure 38: (A) Co-crystal structure cartoon representation of the T-box glyQS Stem I in complex with
the cognate tRNA. YbxF protein is used for stabilization of the truncated glyQS Stem I domain [215].
(B) Schematic structural model of glyQS leader in the antitermination state. Arrows indicate the
codon-anticodon recognition (1), the specific interaction between the apical loop and the tRNA elbow
(2), and the tRNA acceptor stem contact with the bulge region of antiterminator stem (3) [214].
5.3.3. Targeting a T-box riboswitch
As current knowledge about structure and function of T-box RNAs is rapidly
increased, development of novel antimicrobial agents against T-box riboswitch system is also
considered. T-box riboswitch is suggested to be an appropriate target because it can
regulate expression of essential protein families, such aaRSs. Expression of downstream
coding region occurs only after specific interaction with the tRNA effector molecule that
stabilizes the “on” state of the riboswitch. In disruption of tRNA binding, riboswitch remains
at a permanent “off” state which results in loss of downstream expression and possible cell
death. Furthermore, T-box riboswitch RNAs mediate multiple gene regulation in the same
organism and because of their high conservation, a single compound could be designed
against a variety of targets and reduce the resistance strength.
All T-box riboswitches, even those found to regulate at the translational level,
harbor a 7 nt conserved sequence (T-box sequence) that interferes with the bulge structure
stabilization of the antiterminator domain after interaction with the acceptor end of tRNA.
After structural elucidation of the antiterminator domain [205], libraries of compounds were
designed and tested for their effectiveness against tRNA binding and stabilization of the
A. B.

Introduction
86
antiterminator domain. For example, a series of 4,5-disubstituted oxazolidinones can
modulate T-box transcription antitermination function as they have high affinity for the
antiterminator structure [217]. Furthermore, many aminoglycosides, like neomycin B,
appear to have affinity for residues located in the bulge region of antiterminator via
electrostatic attraction. However, considering that during the formation of the
antiterminator the bulge structure could also act as a metal ion binding-pocket (a possible
explanation for neomycin B affinity) this kind of interaction is not able to force insufficient
tRNA binding. Therefore new ligands, which can target the bulge structure in a non-
electrostatic-dependent manner, could also be designed [218]. Development of such novel
compounds is likely to be useful and promising for improvement of specific agents against
common pathogens such as many Gram-positive bacteria.
6. tRNA-dependent exo-ribosomal protein synthesis in pathogens
As described in previous section (§3.1.1.), the cell wall peptidoglycan layer of Gram-
positive bacteria family (including many severe human pathogens) consists of a chain of
alternating GlcNAc and MurNAc units and a branched pentapeptide stem which is linked
with the MurNAc moiety. The pentapeptide stem includes a partially conserved sequence (L-
Ala-γ-D-Glu-X-D-Ala-D-Ala), where the X residue substitution differs among species, and
plays an important role to antibiotic resistance modulation [219]. Additionally, the
peptidoglycan strand harbors peptide bridges that are formed by Fem non-ribosomal
peptidyl-transferases using AA-tRNAs as amino acid donors. Those peptide bridges are used
by DD-transpeptidases for the final cross-link of peptidoglycan chains. Interpeptide bridge
composition is also species-specific. Furthermore, during cell wall formation, peptidoglycan
chain association with the membrane occurs via specific linkage to lipid I and lipid II
moieties, which are synthesized by MraY and MurG enzymes. MraY translocase transfers the
soluble UDP-MurNAc-pentapeptide precursor to lipid I moiety and subsequently MurG
transferase catalyzes the linkage of an activated UDP-GlcNAc unit, which results in lipid II
formation. Lipid I or lipid II moieties can be used as substrates for peptide bridge synthesis
by Fem factors or other tRNA-dependent ligases (like MurM Ser/Ala adding enzyme in
Streptococcus pneumoniae); the preference for lipid precursor recognition can differ among
species [220].

Introduction
87
Bacterial cell wall integrity and the correct formation of its peptidoglycan layer is
crucial for survival and virulence induction [221]. Many antibiotics have been designed in
order to selectively target cell wall integrants or enzymes involved in peptidoglican
synthesis; as an example, the commonly used β-lactam antibiotics can target DD-
transpeptidases, which are responsible for the final step of peptidoglycan chain cross-
linking, as mentioned above.
Figure 39: S. aureus cell wall biosynthesis. After formation of the lipid II moiety, it can be recognized
specifically by Fem factors in order to synthesize the pentaglycine interpeptide bridge. Subsequently,
each completed peptidoglycan unit is translocated from the cytoplasmic membrane and is
incorporated to the cell wall by a specific DD-transpeptidase [222].
6.1. Interpeptide bridge formation in Staphylococcus aureus cell wall
Staphylococcus aureus cell wall consists of 20 or more glycan chains of GlcNAc-
MurNAc alternating units, where MurNAc is substituted with an L-Ala-D-iGlu-L-Lys-D-Ala-D-
Ala pentapeptide stem. L-Lys residue of one pentapeptide stem linked with D-Ala residue in
position 4 of another neighboring stem via a pentaglycine interpeptide bridge, results in
peptidoglycal three-dimensional cross-linking (Figure 39). This characteristic structure of the
peptidoglycan layer can serve as an anchor for protein attachment, which is important for
pathogen’s infectivity *223]. Furthermore pentaglycine interpeptide formation appears to be
essential for methicillin resistance modulation in MRSA species [224].

Introduction
88
6.1.1. The role of Fem factors
In S. aureus there are three non-ribosomal peptidyl-transferases, namely FemA,
FemB, and FemX, which are responsible for pentaglycine interpeptide bridge formation,
using the lipid II moiety as precursor. The first Gly residue incorporation to the ε-amino
group of L-Lys that is located in the pentapeptide stem of lipid II is catalyzed by FemX
protein [225]. It has been shown that deletion of Fem X leads to lethality of the organism
since it severely affects proper cell wall formation [226]. FemA and FemB proteins are
responsible for incorporation of four additional Gly residues (two Gly residues by each
factor), however deletion or deficiency of either Fem A or Fem B does not seem to be
deleterious for the cell [227]. These factors use Gly-tRNAGly species as substrates for the
amino acid addition reaction in a rate-limiting manner. Each Gly residue incorporation
occurs independently, as recognition of the lipid moiety can be performed by one factor a
time [222] (Figure 40B). From the five tRNAGly isoacceptors encoded in S. aureus only three
of them can be used as substrates for the Fem proteins; these isoacceptors bind poorly to
EF-Tu and therefore they can escape the translational machinery [228]. Moreover, the final
Gly residue of the interpeptide can be linked to the fourth D-Ala residue of the neighboring
pentapeptide stem by DD-transpeptidase, and is independent from FemABX protein
presence.
Recognition of aminoacylated tRNA isoacceptors by Fem proteins differs from that
of AARSs. In S. epidermidis, tRNAGly species, used for peptidoglycan synthesis, feature
differences in sequence that are likely to be essential for their exclusion of translational
machinery. Such differences are found in D-loop, where two G residues in positions 18 and
19 are changed to U or C nucleotides, as well as in anticodon loop, where an additional G-C
base pairing by positions 32 and 38 respectively, exists. Moreover, mutations in Fem factors
in several nosocomial strains of S. epidermidis have shown that Fem factors can affect
susceptibility of S. epidermidis to methicillin and oxacillin [124]. Furthermore, as indentified
in Weissella viridescens two cytosines in positions 71 and 72 of the tRNAAla acceptor stem
seems to be essential for recognition by FemX; in contrary the G30-U70 wobble base pair,
which is important for AlaRS recognition and which does not affect FemX function [229]
(Figure 38A). In addition, specific recognition of the aminoacylated acceptor stem of tRNA by
FemX, which excludes non-canonical amino acid charging, enables a preferential
incorporation of amino acid residues, as the first residue incorporation seems to be crucial
for the interpeptide bridge formation [230].
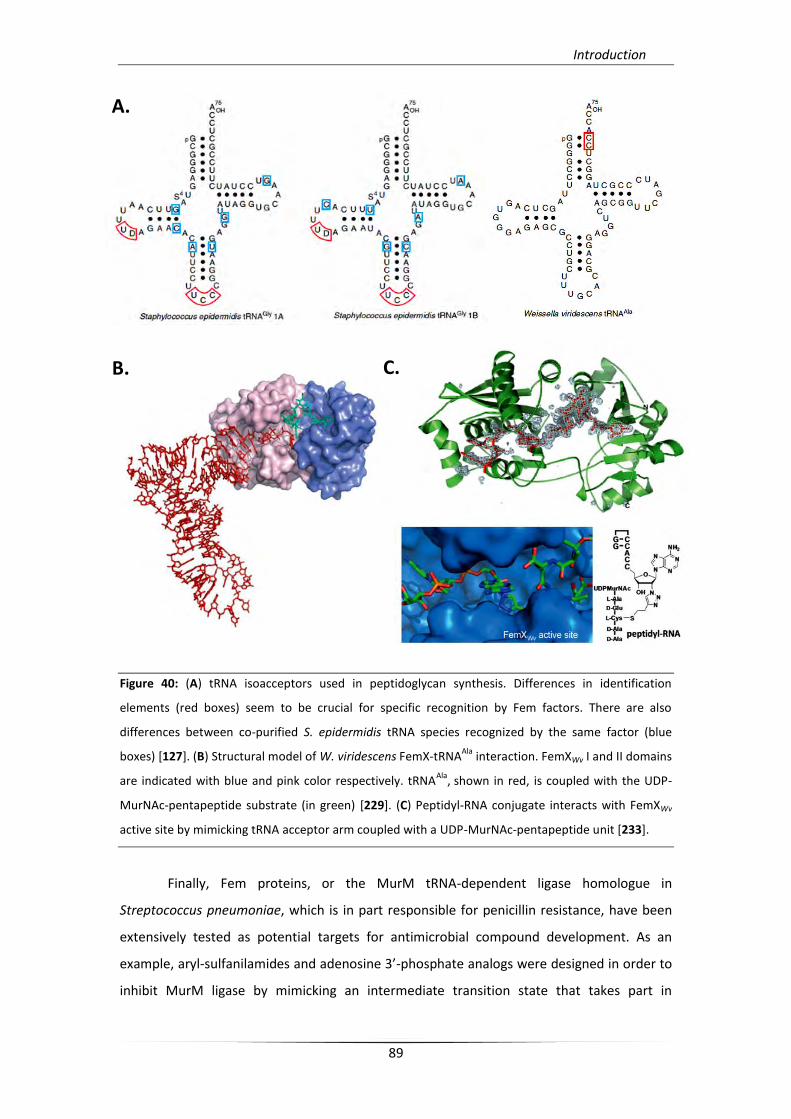
Introduction
89
Figure 40: (A) tRNA isoacceptors used in peptidoglycan synthesis. Differences in identification
elements (red boxes) seem to be crucial for specific recognition by Fem factors. There are also
differences between co-purified S. epidermidis tRNA species recognized by the same factor (blue
boxes) [127]. (B) Structural model of W. viridescens FemX-tRNAAla
interaction. FemXWv I and II domains
are indicated with blue and pink color respectively. tRNAAla
, shown in red, is coupled with the UDP-
MurNAc-pentapeptide substrate (in green) [229]. (C) Peptidyl-RNA conjugate interacts with FemXWv
active site by mimicking tRNA acceptor arm coupled with a UDP-MurNAc-pentapeptide unit [233].
Finally, Fem proteins, or the MurM tRNA-dependent ligase homologue in
Streptococcus pneumoniae, which is in part responsible for penicillin resistance, have been
extensively tested as potential targets for antimicrobial compound development. As an
example, aryl-sulfanilamides and adenosine 3’-phosphate analogs were designed in order to
inhibit MurM ligase by mimicking an intermediate transition state that takes part in
A.
B. C.

Introduction
90
deacylation reaction [231]. Moreover, stable tRNA analogs [232] or specific peptidyl-RNA
conjugates, as currently proposed [233], are found to inhibit FemX protein from Weissella
viridescens, by mimicking the charged acceptor arm of tRNAAla (Figure 40C).
6.1.2. Proteinogenic and non-proteinogenic tRNAGly
As detailed discussed in previous section (§3.1.), tRNA functionality can extend
beyond its role in protein synthesis and this ability relies on the specificity of its structural
element recognition and also on its capacity for amino acid charging and delivery. Among
the abundance in cellular tRNA pool, tRNAGly species are of the most with multiple extra-
translational roles. In many Gram-positive bacteria, charged or uncharged tRNAGly can
regulate its cognate sythetase (GlyRS) expression via specific binding to a T-box leader RNA,
as described [234]. T-box riboswitch discrimination between tRNA substrates depends on
initial recognition of the tRNA anticodon by the Specifier Loop. This specific binding occurs
via codon-anticodon base pairing and an additional interaction between a conserved purine
subsequent to the Specifier codon and the U residue in position 33 of the tRNA [198]. In
most Firmicutes including Bacilli and Staphylococci, the glycyl-T-box leader is specified for
tRNAGly(GCC) isoacceptors as its Specifier region includes a GGC codon. The Specifier GGA
codon is also represented in other Gly-T-box species and predictably would be specified for
tRNAGly(UCC) isoacceptors [199]. However, non-cognate tRNAGlyUCC
isoacceptors cannot be
excluded for GGC codon Specifier recognition.
Moreover, as it was first identified in staphylococci, the so called non-proteinogenic
tRNAGly isoacceptors (NP-tRNAGly) [228, 235] are aminoacylated by the sole cognate GlyRS
which is expressed and preferably participate in peptidoglycan cell wall biosynthesis via
recognition by the FemABX non-ribosomal peptidyl-transferase (Figure 41B). The NP-tRNAGly
isoacceptor sequences include a UUC anticodon and a C residue in position 37, in contrast to
the proteinogenic tRNAGly which bears a purine in the same position. In staphylococcal NP-
tRNAGly species there are also some variations in the T-arm; G49-U65 and G51-C63 base pairs
which are found in proteinogenic tRNAGly, are replaced by A49-U65 and A51-U63 base pairs
respectively. It has been identified that specific G49-U65 and G51-C63 base pairing of T-arm
stabilizes the strong interaction with EF-Tu [236, 237]. As a result this base pair replacement
in NP-tRNAGly sequence appears to affect their participation in ribosomal protein synthesis.
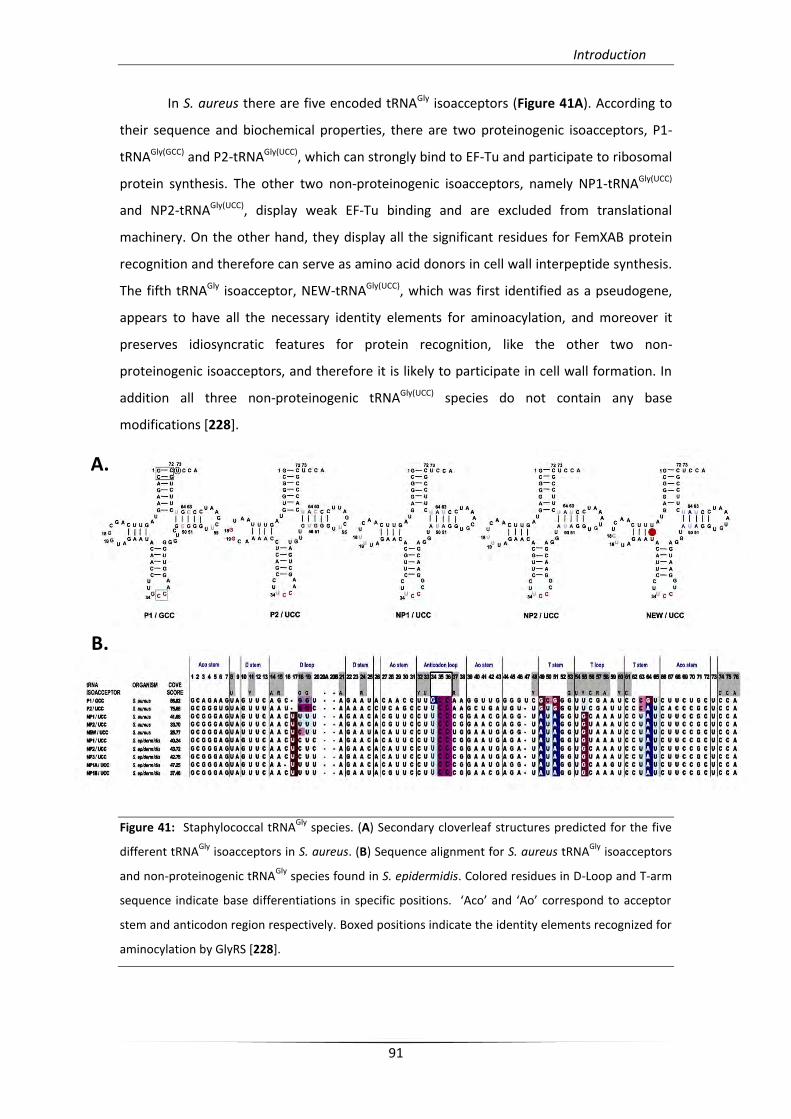
Introduction
91
In S. aureus there are five encoded tRNAGly isoacceptors (Figure 41A). According to
their sequence and biochemical properties, there are two proteinogenic isoacceptors, P1-
tRNAGly(GCC) and P2-tRNAGly(UCC), which can strongly bind to EF-Tu and participate to ribosomal
protein synthesis. The other two non-proteinogenic isoacceptors, namely NP1-tRNAGly(UCC)
and NP2-tRNAGly(UCC), display weak EF-Tu binding and are excluded from translational
machinery. On the other hand, they display all the significant residues for FemXAB protein
recognition and therefore can serve as amino acid donors in cell wall interpeptide synthesis.
The fifth tRNAGly isoacceptor, NEW-tRNAGly(UCC), which was first identified as a pseudogene,
appears to have all the necessary identity elements for aminoacylation, and moreover it
preserves idiosyncratic features for protein recognition, like the other two non-
proteinogenic isoacceptors, and therefore it is likely to participate in cell wall formation. In
addition all three non-proteinogenic tRNAGly(UCC) species do not contain any base
modifications [228].
Figure 41: Staphylococcal tRNAGly
species. (A) Secondary cloverleaf structures predicted for the five
different tRNAGly
isoacceptors in S. aureus. (B) Sequence alignment for S. aureus tRNAGly
isoacceptors
and non-proteinogenic tRNAGly
species found in S. epidermidis. Colored residues in D-Loop and T-arm
sequence indicate base differentiations in specific positions. ‘Aco’ and ‘Ao’ correspond to acceptor
stem and anticodon region respectively. Boxed positions indicate the identity elements recognized for
aminocylation by GlyRS [228].
A.
B.

Introduction
92
Finally, NMR analysis of unmodified tRNAGly anticodon arm revealed that both GCC
and UCC anticodon hairpins cannot form the classical U-turn motif, which is found in many
integrated crystal structures of other tRNA species, but possess differential dynamic
disorders under multivalent ion conditions. These observations proposed a possible
synergistic contribution of idiosyncratic features located at the tRNA overall structure,
except for the anticodon region, in specific protein recognition, namely in GlyRS, EF-Tu and
FemXAB factors, or even in interaction with the T-box leader RNA [238].
7. Antibiotic resistance regulation in Staplylococcus aureus
One of the major threats against antimicrobial treatment in patients is the
uninterrupted spread of antibiotic resistance. Staphylococcal species seem to be responsible
for most of hospital-acquired infections. Many S. aureus strains, predominantly the highly
distributed MRSA strain, are found to be resistant against multiple currently used antibiotic
drugs, which target essential for survival bacterial functions, such as cell wall formation,
protein synthesis and DNA replication.
Figure 42: Currently used antibiotics against cell wall components and enzymes involved in
peptidoglycan synthesis. Scheme illustrates cell wall biosynthesis in S. aureus and blocked arrows
indicate specific inhibition by each compound [248].

Introduction
93
Concerning the antibiotics targeting cell wall formation (Figure 42), S. aureus
resistance to beta-lactams (like methicillin or oxacillin) can occur via two different
mechanisms. The first involves the expression of BlaZ penicillinase that hydrolyzes penicillin
class beta-lactam antibiotics. The second mechanism is broader and mediated by PBP2a
protein (penicillin binding protein 2A), which is encoded by the mecA gene [239]. PBP2a
protein has decreased affinity for beta-lactam binding and reduces their inhibition rate.
Therefore production of this resistant PBP can maintain cell wall formation even in beta-
lactam presence [240]. Moreover, resistant S. aureus isolates in glycopeptides, such as
vancomycin, referred as GISA/VISA isolates, can synthesize a cell wall with different
architecture. This altered formation exhibits an extension of D-ala-D-ala dipeptide targets
located to the external cell wall layer, which prevents glycopeptides molecules from
reaching and acting against their actual targets [241]. Finally, a broader resistance
mechanism triggered by S. aureus exposure to several cell-wall targeting antibiotics,
including beta-lactams and glycopeptides, but also bacitracin, D-cycloserine, mersacidin, and
daptomycin, is the VraS/VraR two-component system (TCS); This is a phosphotransfer-
mediated signaling pathway, which can sense cell wall peptidoglycan damage and enhances
the S. aureus resistance phenotype [242, 243].
Protein synthesis inhibitors, like linezolid which represents the first oxazolidinone
antibiotic drug and interferes with aminoacyl-tRNA binding to the A site of the ribosome, are
also associated to S. aureus and S. epidermidis resistance phenotypes. Such linezolid
resistant isolates are observed with an increasing number of 23S rRNA gene mutations,
which couple resistance accumulation with a parallel loss of fitness [244, 245]. In addition, as
currently identified, an S. epidermidis MLST isolate is found to exhibit a partially linezolid-
dependent growth that seems to be the result of the appearance of a new functional
ribosomal population (Figure 43) [246]. Moreover, a cfr gene, which is first identified in
MRSA isolates and encode a methyl-transferase, also appears to be responsible for
resistance. This element can be transferred among staphylococcal species and can be linked
to other resistance-associated genes, such ermB (erythromycin resistance gene), in order to
confer multiple resistance against antibiotics that have relevant targets [247].

Introduction
94
Figure 43. Characteristic mutations in 23S rRNA which favor the presence of new functional
ribosomal populations in nosocomial strains of Staphylococcus [246]; L3/L4: large subunit ribosomal
proteins associated to the 23S rRNA; LZD: linezolid.
7.1. RNA-mediated infectivity and resistance modulation
In S. aureus, horizontally acquired pathogenicity islands (SaPIs), which are included
in transferable elements as plasmids, phages and transposons, can mediate both resistance
development and adaptation into hosts [249]. Beside genes encoding resistance and
virulence proteins that are included in such SaPIs, these elements are also responsible for
sRNA transportation and expression [250]. sRNAs can be found in multiple copies and
several locations around S. aureus genome, and their presence in SaIPs would suggest their
participation during host infection. For example, the SprD sRNA, which is expressed by the
PIφ island, can interfere with translation initiation of the S. aureus sbi mRNA that encodes
the second immunoglobulin-binding protein Sbi. During infection, Sbi production contributes
to immune evasion and protects the pathogen from defense mechanisms of the host [251].
In addition to expression repression of Sbi factor, SprD sRNA can possibly modulate
expression of many other virulence factors, which implies a pathogen strategy to control its
infection mechanisms (Figure 44A).

Introduction
95
Figure 44: RNA-dependent infectivity and resistance regulation in pathogens (A) S. aureus
SprD sRNA and sbi mRNA ‘loop–single strand’ interaction (in green) prevents translation
initiation via an antisense regulatory mechanism [251]. (B) H2 and H3 hairpins of 5’ RNAIII
structure (in red) can bind to the S. aureus hla mRNA and activate translation initiation using
also an antisense regulatory mechanism [255]. (C) An aminoglycoside-sensing leader RNA
activates translation initiation of resistance gene expression via release of ribosomal binding
site. Nucleotides in blue and in green represent Shine Dalgarno and anti-Shine Dalgarno
sequences respectively. Anti-SD region can swift between SD1 and SD2 pairing [254].
Moreover, multifunctional RNA elements, as the RNAIII effector can control multiple
gene expression. RNAIII element is characterized by its high stability and a complicated
structure of multiple domains, including 14 stem-loops linked with unpaired nucleotides
[252]. Each domain represents a separate regulatory element, which interferes with
expression modulation of multiple mRNA targets, alone or in combination. For instance,
regulatory domains of the RNAIII 5’ end can directly regulate translation of hla that encodes
an alpha hemolysin, by triggering the formation of an intra-molecular SD anti-sequester
[253] (Figure 44B). As a result RNAIII element can function as an sRNA that anneals with
multiple mRNA targets and mediates rapid expression regulation of different virulence
factors in a sequential manner. Interestingly, RNA regulatory elements can also induce
antibiotic resistance in pathogens. As it was recently identified, a riboswitch leader RNA can
bind aminoglycoside compounds. This aminoglycoside-sensing riboswitch is found
widespread among antibiotic-resistant pathogens and located upstream operons encoding
resistance factors, as AAC (aminoglycoside acetyl-transferase) and AAD (aminoglycoside
adenyl-transferase). Such enzymes are involved in specific drug modification [254] (Figure
44C).
A. B. C.

Introduction
96
7.2. RNAs as Targets for Antimicrobial Drugs
The systematic expansion of antibiotic resistance raises the necessity of alternative
antimicrobial strategies. Therefore, the need for novel molecular targets has attracted much
attention at many levels, both in basic and in clinical research. The potential new targets
have to mediate and participate in essential cellular pathways different from what has
previously been targeted, in order to reduce the extensive selective pressure of the
microbiome, which has been the result of the current treatments. So far, new drug
development was focused on virulence mechanisms that cause host cell infection and
appeared efficient [256]. In addition, the increasing knowledge about small regulatory RNAs
and small metabolite-sensing riboswitches point towards alternative targets [257]. In S.
aureus more than 40 genes are under regulation of metabolite sensing riboswitch elements
and are involved in essential metabolic pathways. Such RNA elements can be easily targeted
by specific metabolite analogs. For instance, PC1 (2,5,6-triaminopyrimidin-4-one), a novel
pyrimidine derivative, was designed to target a purine-sensing riboswitch in S. aureus. This
riboswitch controls the expression of GuaA GMP synthase, involved in pathogen’s survival
during infection [258]. Moreover, new strategies could be developed, in a way to interfere
with sRNA mechanism and function. Those RNA-mediated strategies are likely to increase
the specificity of antimicrobial agents.



Emerging questions and aim of the thesis
99
It is obvious from all the accumulated data and current studies that the integrity of
the cell wall plays an important role in infection and pathogenicity of all staphylococci and
especially S. aureus. Because of the increasing rate of β-lactam resistance and the unique
features of the staphylococcal peptidoglycan structure and assembly, Fem transferases,
involved in peptidoglycan pentaglycine side chain synthesis, have been considered among
the most preferred molecular targets for novel antibiotic development. On the other hand, it
has been already demonstrated, that such antimicrobial strategies against components of
the cell wall synthesis machinery can enhance selective pressure of the microbiome, and
result in an integrated anti-antibiotic response. Moreover, strategies against components of
the translational machinery are likely to progressively have the same fate. This notion
supports the needs of alternative strategies which must include pathways that also are
unique in bacterial organisms and crucial for infectivity or survival.
The recent discovery that the functional importance of tRNA molecules extends
beyond their core cellular role in translation came as a surprise. Both in eukaryotes and
prokaryotes (especially in pathogens) tRNA molecules can exhibit unexpected role by
switching from their main role of translation and genetic code adaptation to the role of gene
expression regulation. Based on previous work from our laboratory we focused on the role
of aminoacylated tRNAGly pool in S. aureus and its characteristic clustering into proteinogenic
and non-proteinogenic tRNAGly isoacceptors (NP1, NP2 and NEW) in an effort to extensively
characterize their regulatory role both in ribosomal and exo-ribosomal protein synthesis. As
previously reported, all NP-tRNAGly(UCC) species have reduced affinity to EF-Tu and therefore
they are excluded from translational machinery. Furthermore, they can be recognized by the
sole cognate synthetase for aminoacylation (GlyRS) that exists in S. aureus and in turn they
can participate in cell wall biosynthesis.
Bioinformatics analyses revealed that all staphylococci genomes possess a conserved
T-box leader RNA lying upstream the glycyl-tRNA synthetase gene (glyS), with a predicted
GGC codon specificity. As a result the proteinogenic P1-tRNAGly(GCC) isoacceptor is likely to be
the most preferred effector for promoting this transcription attenuation mechanism. On the
other hand, the utilization of the other four tRNAGly(UCC) (proteinogenic; P2, and non-
proteinogenic; NP1, NP2, and NEW) isoacceptors cannot be excluded from antitermination
control. This observation is based on the fact that all S. aureus tRNAGly species possess all
identity elements necessary for specific recognition from T-box specifier and antiterminator
bulge moieties, except on residue in the wobble position 34, which interferes with the
codon-anticodon pairing.

Emerging questions and aim of the thesis
100
According to these findings the remaining questions are the following: Is S. aureus
glyS regulated by a bona fide T-box, harboring all the necessary structural elements for tRNA
effector recognition? Can all of tRNAGly isoacceptors sufficiently bind to T-box leader and
induce transcription “read-through”? How this glyS T-box RNA can synchronize two distinct
metabolic pathways (translation and cell wall formation) by regulating transcription of
GlyRS?
The present study aims to answer the above questions by setting the following
goals; the major is the clarification of the structural formation and function of the
staphylococcal glyS T-box riboswitch RNA and the identification of its regulatory partners.
Another goal is the confirmation of the different S. aureus tRNAGly isoacceptor
extratranslational roles and their potential synergistic function in modulation of two distinct
pathways, both in vitro and in vivo. Moreover, the completion of this work will give for the
first time evidence for the existence of a regulatory mechanism that is used for differential
proteinogenic and non-proteinogenic functional switching of the tRNAGly pool, according to
nutrient deficiency and growth or infection state adaptation. Finally, the proposed
regulatory mechanism that shifts between two crucial metabolic pathways could be used for
novel antibacterial compound development.


Τλικά & Μζκοδοι

Materials and Methods
103
1. Materials
1.1. Chemicals and reagents
Acetic Acid (MERCK)
Acrylamide 2X (SERVA)
Agar (SERVA)
Agarose (BIORAD)
Amino acids (MERCK)
Ammonium acetate (MERCK)
Ammonium chloride (MERCK)
Ammonium persulfate (SERVA)
Ampicillin (APPLICHEM)
[γ-32P] ΑΣΡ (6000 Ci/mmol) (IZOTOP)
Boric acid (MERCK)
Bis-acrylamide (SERVA)
Bradford reagent
(BIOQUANT®Protein) (MERCK)
Bromophenol blue (SIGMA-ALDRICH)
Calcium chloride (MERCK)
Coomassie R250 (PANREAC)
ddNTPs (JENA BIOSCIENCE)
Dinucleotides (ApU,CpU,UpA) (IBA)
dNTPs (BIOLINE)
DEPC (RESEARCH ORGANICS)
DMS (SIGMA-ALDRICH)
Ethanol Absolute (MERCK)
EDTA (SERVA)
Formamide 99% (MERCK)
Gel Red (BIOTIUM)
D(+)-Glucose (PANREAC)
Glycerol (DUCHEFA)
[14C]-Glycine (2.3 GBq /mmol)
(PERKIN ELMER)
Heparin (SIGMA-ALDRICH)
HEPES (SIGMA-ALDRICH)
Hydrochloric acid (MERCK)
Imidazole (MERCK)
IPTG (DUCHEFA)
Kanamycin (APPLICHEM)
Kethoxal (RESEARCH ORGANICS)
Magnesium acetate (MERCK)
Magnesium chloride (MERCK)
Magnesium sulfate (MERCK)
β-Mercaptoethanol (SERVA)
Methanol (MERCK)
Ni-NTA Agarose (QIAGEN)
ONPG (SIGMA-ALDRICH)
Peptone (SERVA)
Phenol / chloroform / isoamyl-alcohol
(25:24:1) (APPLICHEM)
PMSF (SERVA)
Polyethylene glycol (SERVA)
Potassium acetate (MERCK)
Potassium chloride (MERCK)
Potassium di-hydrogen phosphate
(MERCK)
Ribonucleotides (PROMEGA)
SDS (SERVA)
SMMP Broth (TEKNOVA)
Sodium acetate (MERCK)
Sodium chloride (MERCK)
Sodium di-hydrogen Phosphate di-
hydrate (MERCK)
di-Sodium hydrogen phosphate di-
hydrate (MERCK)
Streptomycin (MERCK)
TCA (PANREAC)

Materials and Methods
104
TEMED (RESEARCH ORGANICS)
Tris (SERVA)
Triton-X-100 (MERCK)
Tryptic Soy broth (BIOLIFE)
Urea (SERVA)
[α-32P] UTP (800 Ci/mmol) (IZOTOP)
Χ-Gal (SERVA)
Xylene cyanol (SERVA)
Yeast extract (SERVA)
1.2. Enzymes
AMV Reverse Transcriptase
(PROMEGA)
Antarctic Phosphatase (NEB)
BamHI (TAKARA)
BstNI (NEB)
DNase I (NEB)
E. coli RNAP, Holoenzyme (NEB)
EcoRI (TAKARA)
2G Fast Taq DNA polymerase (KAPA
BIOSYSTEMS)
HindIII (NEB)
Lysozyme (FLUKA)
NdeI (TAKARA)
PPase (Thermostable Inorganic)
(NEB)
PrimeSTAR® Max DNA Polymerase
(TAKARA)
RNase A (AMBION)
RNase T1 (AMBION)
RNase V1 (AMBION)
RNasin (TAKARA)
SalI (TAKARA)
T4 DNA ligase (NEB)
TNK T4 Nucleotide Kinase (TAKARA)
T7 RNA polymerase (TAKARA)
XhoI (TAKARA)
1.3. Kits
Brilliant III SYBR® Master Mix (AGILENT TECHNOLOGIES)
CloneJETTM PCR Cloning Kit (THERMO SCIENTIFIC)
KAPA HiFi PCR Kit (KAPA BIOSYSTEMS)
Mini Quick Spin RNA Columns (ROCHE)
RNA 6000 Nano Kit (AGILENT TECHNOLOGIES)
MicroPulserTM electroporation system (BIORAD)
NucleoSpin® Gel and PCR Clean-up (MACHEREY-NAGEL)
NucleoSpin® Plasmid DNA Mini Prep or Midi Prep (MACHEREY-NAGEL)
RiboPure™-Bacteria Kit (AMBION)
StrataClone™-PCR Cloning Kit (AGILENT TECHNOLOGIES)
Superdex® 200 10/300 GL chromatography column (GE HEALTHCARE LIFE SCIENCIES)

Materials and Methods
105
1.4. Bacterial strains
DH5α [genotype: E. coli F- φ80lacΖΔΜ15 Δ(lacΖΤ Α-argF) U169 deoR recA1 endA1
hsdR17(rk-, mk
+) phoA supE44 thi-1 gyrA96 rela1 λ-] (INVITROGEN)
BL21 (DE3) Rosetta [genotype: E. coli F- ompT hsdSB (rB-, mB
-) gal dcm pRARE2 (Camr)]
(INVITROGEN)
E. coli M5154 [genotype: F- ΔlacZ39 trpA49(Am) recA11 relA1 rpsL150(Strr) spoT1 λ-]
(kindly provided by Prof. Hubert Becker, Université de Strasbourg, CNRS)
S. aureus WT (kindly provided by Prof. Spyros Pournaras, Medical School, University of
Athens)
S. aureus RN4220 [genotype: Restriction-deficient derivate of 8325-4 (rk-, mk
+)] (a gift
from Dr. Pascale Romby, Université de Strasbourg, CNRS, IBMC)
S. epidermidis WT (kindly provided by Prof. Spyros Pournaras, Medical School,
University of Athens)
SoloPack® Gold Competent Cells [genotype: E. coli F- Tetr ∆(mcrA)183 ∆(mcrCB-
hsdSMR-mrr)173 endA1 supE44 thi-1 recA1 gyrA96 relA1 lac Hte proAB lacIqZ∆M15
Tn10 (Tetr) Amy Camr] (included in StrataCloneTM-PCR Cloning Kit)
1.5. Bacterial vectors
pBAD18-Kan (ATCC® 87397™)
pET-20b(+) (NOVAGEN)
pJET1.2/blunt Cloning Vector (included in CloneJETTM PCR Cloning Kit)
pLacZFT (kindly provided by Prof. Hubert Becker, Université de Strasbourg, CNRS)
pRB382 (ATCC® 77378™)
pSC-A (included in StrataCloneTM-PCR Cloning Kit)
pUC18 (INVITROGEN)
pUC57 (GEN SCRIPT)
1.6. Primers
Primer Name Sequence (5’-3’) Application
Sep_glyS_FW_NdeI GCATATGGTTAAAAATATGGATACAATTG** S. epidermidis
glyS gene cloning in pET-20b Sep_glyS_RV_XhoI CTCGAGAAATTTAACTTTCTCAGCTAA**
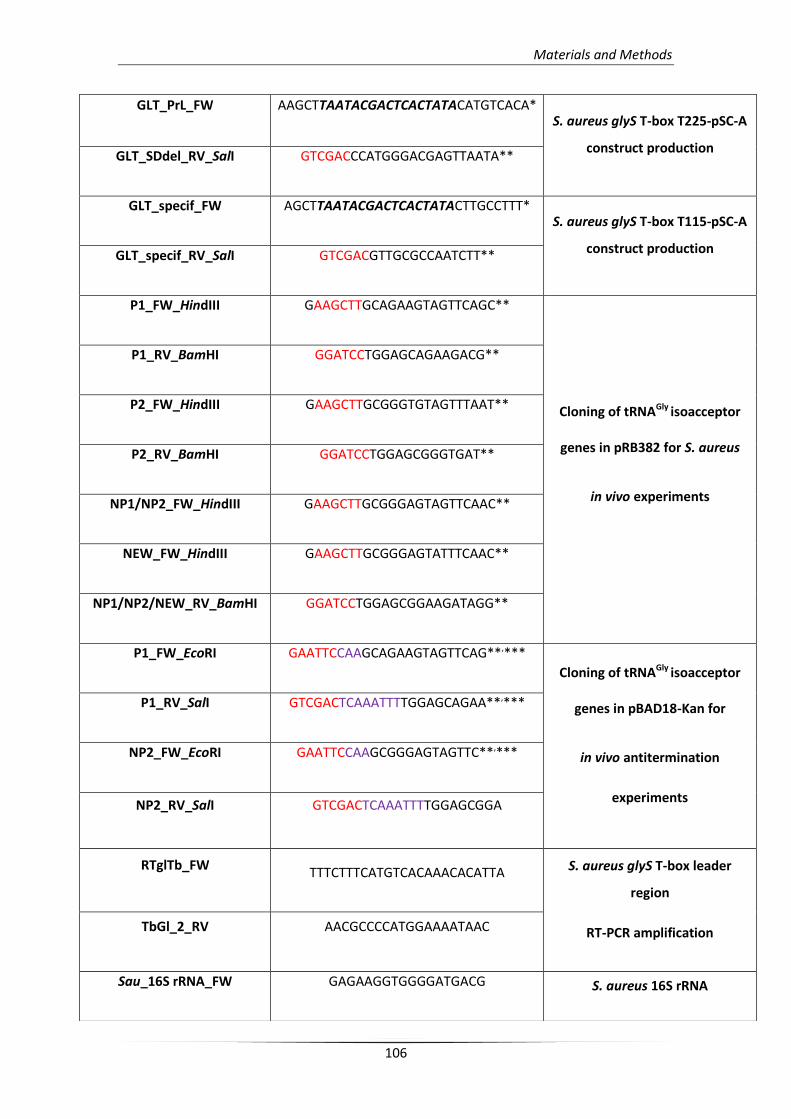
Materials and Methods
106
GLT_PrL_FW AAGCTTAATACGACTCACTATACATGTCACA* S. aureus glyS T-box T225-pSC-A
construct production GLT_SDdel_RV_SalI GTCGACCCATGGGACGAGTTAATA**
GLT_specif_FW AGCTTAATACGACTCACTATACTTGCCTTT* S. aureus glyS T-box T115-pSC-A
construct production GLT_specif_RV_SalI GTCGACGTTGCGCCAATCTT**
P1_FW_HindIII GAAGCTTGCAGAAGTAGTTCAGC**
Cloning of tRNAGly isoacceptor
genes in pRB382 for S. aureus
in vivo experiments
P1_RV_BamHI GGATCCTGGAGCAGAAGACG**
P2_FW_HindIII GAAGCTTGCGGGTGTAGTTTAAT**
P2_RV_BamHI GGATCCTGGAGCGGGTGAT**
NP1/NP2_FW_HindIII GAAGCTTGCGGGAGTAGTTCAAC**
NEW_FW_HindIII GAAGCTTGCGGGAGTATTTCAAC**
NP1/NP2/NEW_RV_BamHI GGATCCTGGAGCGGAAGATAGG**
P1_FW_EcoRI GAATTCCAAGCAGAAGTAGTTCAG**,***
Cloning of tRNAGly isoacceptor
genes in pBAD18-Kan for
in vivo antitermination
experiments
P1_RV_SalI GTCGACTCAAATTTTGGAGCAGAA**,***
NP2_FW_EcoRI GAATTCCAAGCGGGAGTAGTTC**,***
NP2_RV_SalI GTCGACTCAAATTTTGGAGCGGA
RTglTb_FW TTTCTTTCATGTCACAAACACATTA S. aureus glyS T-box leader
region
RT-PCR amplification TbGl_2_RV AACGCCCCATGGAAAATAAC
Sau_16S rRNA_FW GAGAAGGTGGGGATGACG S. aureus 16S rRNA

Materials and Methods
107
Table 1: Primer List; all primers were synthesized by VBC biotech;*Bold and italics fonts
indicate the T7 promoter region; **Sequences in red indicate regions for restriction enzyme
recognition; ***Sequences in purple are designed in order to enhance in vivo
endonucleolytic maturation of the tRNA transcripts. These additional sequence motifs in
tRNA precursors can be recognized by E. coli specific RNases which can cleave the 5’ leader
(RNase P) and the 3’ trailer sequences (several RNases like RNases T, PH, Z and II can cleave
downstream the 3’ CCA sequence).
Sau_16S rRNA_RV ATGGTGTGACGGGCGGTGTG RT-PCR amplification
GLT_EnPr_FW_BamHI GGATCCAGTTCTATAGGTCTTCACCCC** S. aureus glyS T-box
in vitro and in vivo
antitermination RTglTb_RV CCAGGGAACACAAAACCTATG
GLT_2ndSD_RV_SalI GTCGACAATTACCTCTCCTCATACA**
S. aureus glyS T-box
in vivo antitermination
ORF_GLT_2_FW GATATTTCTTTCATGTCACAAACACATT
ORF_GLT_RV GCATAATTACCTCTCCTCATACATGAAA
TbGl_1_(129-148) TGTAGTCACTCGCTTTTATT
Primer extension analysis
TbGl_6_(216-238) TGCTTTACTTCCATGGGACGAGT

Materials and Methods
108
2. Methods
2.1. Cloning and expression of S. epidermidis GlyRS enzyme
2.1.1. Vector cloning and production of bacterial transformants
Specific primers (see Table 1) were designed for S. epidermidis glyS gene (encoding
glycyl-tRNA synthetase; GlyRS) cloning based on the available ORF sequence in KEGG
database (Annotation number: SE1252). S. epidermidis genomic DNA was used for glyS gene
amplification by PCR, with an optimal annealing temperature at 56oC, as follows:
Reagents Final concentration
5x KAPA HiFi Fidelity Buffer 1x
dNTP mix (10 mM each) 0.3 mM
Sep_glyS_FW_NdeI 0.3 μM
Sep_glyS_RV_XhoI 0.3 μM
Sep genomic DNA (template) 100 ng
KAPA HiFi DNA polymerase 0.5 U
PCR grade water up to 25 μL
PCR product was purified by NucleoSpin® PCR Clean-up, and cloned into pSC-A
vector according to StrataClone™-PCR Cloning Kit manufacturer’s protocol. TA cloning Sep
glyS-pSC-A recombinant derivatives used for SoloPack® Gold competent cells transformation
(also according to StrataClone™-PCR Cloning Kit instructions). Positive transformed clones
were selected by blue/white colony screening on LB/Amp agar1 plates, in presence of X-gal
substrate (40 μg/mL final concentration) that was used to obtain β-galactosidase activity.
Moreover, the cloned glyS gene was sequenced by VBC biotech, in order to avoid any
incorporation of random mutations. Plasmid DNA, purified by Sep glyS-pSC-A positive clones
NucleoSpin® Plasmid DNA Mini Prep), were digested with NdeI and XhoI restriction enzymes,
pursuant to standard reaction protocol, and after electrophoresis on 1% agarose gel, the
produced segments, which corresponded to the desired size (1392 bp), were extracted,
purified (NucleoSpin® Gel Clean-up), and inserted into pET20b expression vector, with a T4
DNA ligase reaction as follows. Incubation was performed at 16oC for 14-16 h:

Materials and Methods
109
Reagents Final concentration
10X T4 DNA Ligase Buffer 1x
pET20b linear vector 0.020 pmol
Purified Insert 0.060 pmol
T4 DNA Ligase 400 U
Nuclease-free water up to 20 μL
Subsequently, the resulted Sep glyS-pET20b ligation product was used to transform
E. coli BL21 (DE3) Rosetta electrocompetent cells using MicroPulserTM electroporation
apparatus, by 12.5 kV/cm field strength in 0.2 cm cuvettes. The positive transformed clones
were selected on LB/Amp agar plates, and are likely to produce Sep GlyRS recombinant
protein, which was tagged with six His residues at the C-terminus.
1. LB/Amp agar: 1% Peptone, 1% NaCl, 0.5% yeast extract, agar 12 g/L, ampicillin 100 μg/mL.
2.1.2. Protein expression and purification
E. coli BL21 (DE3) Rosetta cells transformed with Sep glyS-pET20b plasmid construct
were grown in 100 mL LB/Amp medium2 at 37oC under continuous shaking, until they
reached the mid-Log growth phase (A600=0.5 to 0.7). After 10 min incubation on ice they
were divided into four different cultures (25 mL each), without changing the medium, and
tested for protein expression under several induction conditions. Two of them were induced
for expression by the addition of 0.5 and 1 mM IPTG respectively and incubated at 37oC for 5
h. The remaining two were induced also with 0.5 and 1mM IPTG but incubated at 25oC for 16
h. After incubation, cells from all cultures were harvested (4,500xg for 15min at 4oC), and
resuspended in lysis buffer3. Each cell suspension was incubated for 20 min on ice, and
sonicated for 2 min. The cell lysates were centrifuged at 12,000xg for 30 min, and the
resulted supernatants were run on 12% SDS PAGE using as control the lysate of an
uninduced cell sample, and also a resuspension from the corresponding pellet of each
induced sample.
For GlyRS enzyme purification, positive Sep glyS-pET20b transformed clones that are
found to express the His-tagged enzyme in a high yield, under best induction conditions,
were inoculated in 500 mL LB/Amp medium and incubated at 37oC until A600=0.6. After
induction cells were harvested and lysed as described above. The corresponding lysates
were centrifuged successively for 30 min at 12,000xg and for 90 min at 100,000xg. The

Materials and Methods
110
cleared S-100 was incubated with 2 mL Ni-NTA-agarose bits, which have been equilibrated
with the same lysis buffer (without lysozyme), for 1 h at 4oC under rotation. Subsequently,
the S-100/column bits suspension was loaded onto a 5 mL column with adjustable flow, and
the column allowed to drain from the lysate by gravity. After several washing steps with
wash buffer4 containing 10 mM imidazole to avoid insufficient protein binding (at least 10
volumes), His6-tagged recombinant proteins were eluted in the presence of elution buffer
using imidazole concentration gradient (20 to 500 mM; elution buffer 205 and elution buffer
5006, respectively). The purification procedure was performed at 4oC. Samples of fragments
from all purification steps were analyzed by SDS-PAGE electrophoresis and the
corresponding elution fragments that were found with the highest protein purity were
pulled. GlyRS-His6 purified recombinant protein was dialyzed (1:1,000) in the appropriate
GlyRS dialysis buffer7 (approximately, 2 h at 4oC), in order to remove imidazole remains, and
after condensation in PEG-40,000, it was supplemented with 25% glycerol, before it was
stored at -20oC. Protein concentration was measured by Bradford standard method.
2. LB/Amp medium: 1% Peptone, 1% NaCl, 0.5% yeast extract, ampicillin 100 μg/mL.
3. Lysis buffer: 50 mM Tris-HCl, pH 7.5, 0.5 M NaCl, 0.1% Triton-X, 5 mM β-ME, 5 mM MgCl2, 1 mM
PMSF, 10% glycerol, 0.02% w/v lysozyme.
4. Wash buffer: 50 mM Tris-HCl, pH 7.5, 0.5 M NaCl, 10 mM imidazole.
5. Elution buffer 20: 50 mM Tris-HCl, pH 7.5, 0.5 M NaCl, 20 mM imidazole.
6. Elution buffer 500: 50 mM Tris-HCl, pH 7.5, 0.5 M NaCl, 500 mM imidazole.
7. Dialysis buffer: 50 mM Tris-HCl pH 7.5, 25 mM KCl, 15 mM MgCl2, 2 mM β-ME, 10% glycerol.
2.2. Total tRNA extraction from S. epidermidis
S. epidermidis WT strain was inoculated in 1 L LB medium and incubated at 37oC
under shaking until reached the Log growth phase. Cells were harvested (5,000xg for 20
min), resuspended in lysis buffer B8 (5 mL per 1g of cell pellet), and sonicated (up to 5 min)
for complete lysis. An equal volume of phenol, equilibrated with 0.1 M citrate buffer, pH 7,
was added in the produced cell lysate, and the two phases were mixed under vigorous
vortexing for 20 min. After centrifugation at 8,500xg for 20 min, equal volume of chloroform
was added in the upper aqueous phase, and mixed by vortex (5 min). The mix was
centrifuged at 5,000xg in order to be separated into two phases, and the aqueous phase was
precipitated for 30 min at -80oC, by adding 2.5 volumes of ethanol (absolute), and 1/10
volume of 3M sodium acetate, pH 5.2. The produced pellet was received by centrifugation
at 13,000xg, 4oC for 30 min, and supernatant was discarded. Subsequently, the pellet was
washed again with 70% ethanol (13,000xg, 4oC, 15 min). After this step, the resulted pellet

Materials and Methods
111
includes the total RNA extract. For total tRNA separation, the total RNA pellet was
resuspended in 0.2 M Tris-acetate, pH 9.0, and incubated for 30 min at 37oC (tRNA
deacylation). The suspension was precipitated by ethanol and the produced pellet was
resuspended at 4 mL 0.1M NaCl, followed by incubation at 4oC for 16 h, and centrifugation
at 7,000xg, 4oC for 30 min, in order to removed large molecular weight nucleic acids. The
resulted supernatant precipitated by ethanol to produce the final total tRNA pellet, which in
turn was resuspended in 0.1 M Tris-acetate, pH 7.5. Moreover, for quality verification of the
resulted total tRNA product, the final suspension was analyzed on a 10% PAGE/8 M Urea.
8. Lysis buffer B: 20 mM Tris-HCl, pH 7.4, 20 mM Mg(OAc)2.
2.3. S. aureus glyS T-box and tRNAGly transcripts production and purification
2.3.1. Cloning of T-box leader RNA constructs
The T-box riboswitch was predicted to be part of the region starting from the Shine-
Dalgarno sequence and stretching 300 upstream the S. aureus glyS gene nucleotides
according to the available genomic sequence in KEGG database (Accession number: SA1394).
Three T-box leader constructs that differed in length were designed and cloned in order to
verify the functionality and the structural features of the riboswitch. The first construct,
termed as T275, including the whole leader sequence (position +1 to position +275), was
synthesized and cloned into pUC57 vector by GenScript. For the second and third riboswitch
construct, termed as T225 and T115 respectively, specific primers were designed (see Table
1) for PCR amplification using as template the T275-pUC57 plasmid DNA. T225 segment is
including part of the whole T-box sequence from position +10 to position +225, while T115 is
including part of the T-box leader sequence from position +34 to position +115. All
constructs were designed with a T7 promoter leader sequence and a terminal restriction
enzyme recognition site, BstNI for T275, or SalI for T225 and T115. After digestion with the
corresponding restriction enzyme they can be used as template for in vitro transcription.
PCR reactions for T225 and T115 construct preparation were carried out as follows with an
optimal annealing temperature at 62oC:

Materials and Methods
112
Reagents Final concentration
5x KAPA HiFi Fidelity Buffer 1x
dNTP mix (10 mM each) 0.3 mM
GLT_PrL_FW (T225) or GLT_specif_FW (T115) 0.3 μM
GLT_SDdel_RV_SalI (T125) or GLT_specif_RV_SalI (T115) 0.3 μM
T275-pUC57 plasmid DNA (template) 10 ng
KAPA HiFi DNA polymerase 0.5 U
PCR grade water up to 25 μL
PCR products were purified prior to ligation with the pSC-A vector and the resulted
recombinant plasmids were used for E. coli competent cells (SoloPack® Gold)
transformation. The TA cloning procedure was performed according to StrataClone™-PCR
Cloning Kit instruction manual as described in previous section (§2.1.1.). Plasmids from
positive clones were extracted (NucleoSpin® Plasmid DNA Midi Prep) and sequenced (VBC
biotech), in order to verify the accuracy of the constructs. Furthermore, T225-pSCA and
T115-pSCA plasmids were linearized and checked for their ability to produce in vitro
transcripts by T7 RNA polymerase reaction, under several incubation conditions, according
to standard transcription protocol (small scale).
2.3.2. In vitro transcription
Sau tRNAGly genes, which were encoding five different isoacceptors (P1, P2, NP2,
NP1 and NEW), were cloned in pUC18 vector by cassette cloning procedure, as previously
described [228]. Sau glyS T-box T275 segment was cloned in pUC57, while T-box T225, and
T115 segments were cloned in pSC-A vector, as described above (§2.3.1.). All tRNAGly and T-
box plasmid constructs were designed with a T7 promoter leader sequence and a terminal
restriction enzyme recognition site, BstNI for T275-pUC57 and each tRNAGly-pUC18, or SalI
for T225-pSC-A and T115-pSC-A constructs. Digestion reactions were carried out according
to the following general protocol for the production of linear template in high yield:

Materials and Methods
113
Reagents Final concentration
10x Universal digestion buffer9 1x
Plasmid construct 10 μg
BstNI or SalI 50 U
Nuclease-free water up to 50 μL
The reaction mix incubated for 2 h at 37oC. Each produced linear plasmid construct
was extracted by one volume of phenol and one volume of chloroform. Subsequently, the
aqueous phase was precipitated with ethanol, and the received pellet resuspended in
nuclease-free H2O (DEPC treated). Prior to use into in vitro transcription reactions, plasmid
constructs were checked for accurate linearization by 1% agarose gel elactophoresis.
Purified linear plasmid constructs used for in vitro transcript production by T7 RNA
polymerase in a large scale run-off transcription reaction as follows:
Reagents Final concentration
10x Σ7 RNAP buffer10 1x
DTT 5 mM
UTP 2 mM
GTP 2 mM
CTP 2 mM
ATP 2 mM
Linear plasmid 15 μg
RNasin 160 U
T7 RNAP 500 U
PPase 8 U
H2O (DEPC treated) up to 500μL
Transcription reactions for all constructs were carried out at 37oC for 16 h, except T-
box T275, T225, and T115 construct that incubated at 30oC. After addition of 5μl DNase I (2
U/ μL), and 10 μl EDTA (200mM) followed by incubation at 37oC for 30 min, transcripts were
extracted by adding equal volume of phenol/chloroform/IAA (25:24:1) and precipitated with

Materials and Methods
114
ethanol. All transcripts were checked for their functionality according to different
purification methods as described in the following section (§2.3.3.).
9. 10x Universal digestion buffer: 200 mM Tris-acetate, pH 7.9, 500 mM KOAc, 100 mM Mg(OAc)2,
1 mg/mL BSA.
10. 10x Σ7 RNAP buffer: 400 mM Tris-HCl, pH 8.0, 80 mM MgCl2, 20 mM spermidine.
2.3.3. RNA transcript purification and labeling
After several purification conditions tests, the best found were presented wherein.
Both T225 and T115 transcript precipitants were resuspended in i.v.t. loading buffer11 and
purified from nucleotide remains on 8% PAGE 8 M urea. The corresponding transcript bands
were visualized using UV shadowing (PLC filter plate, 254 nm UV lamp), and were eluted by
400 μL i.v.t. elution buffer12 at 4oC for 16 h under rotation. After extraction they were
precipitated again with ethanol and were resuspended in H2O (DEPC treated). T-box T275
and all tRNAGly isoacceptor transcripts were purified from nucleotides and unwanted dimeric
conformations on gel filtration Superdex 200 10/300 GL-ÄKTA FPLC system. T275 and tRNAGly
precipitants were resuspended in 1x RNA FPLC buffer13 and were denaturated at 50oC for 5
min prior loading on gel filtration column. Purification performed at the same 1x RNA FPLC
buffer in a 0.5 ml/min rate flow. After fragmentation procedure, the fragments including
transcripts with the desired molecular weight were pulled, and precipitated with ethanol.
Received pellets were resuspended in H2O (DEPC treated).
For each RNA purified transcript 5’ *γ-32P] labeling was performed according to the
following general protocol. Firstly, 5 to 10 pmol of RNA transcript were dephosphorylated at
5’ end by phosphatase reaction:
Reagents Final concentration
10x Antarctic phosphatase reaction buffer 1x
RNA transcript 5 to 10 pmol
Antarctic phosphatase 7.5 U
H2O (DEPC treated) up to 50 μL
Reaction mix was incubated at 37oC for 30 min. After addition of 0.5 μL EDTA (200
mM) reactions were incubated at 65oC for 5 min to inactivate phosphatase enzyme. RNA
transcripts were extracted by adding equal volume of phenol/chloroform/IAA (25:24:1) and

Materials and Methods
115
precipitated with ethanol. Precipitation derivates were resuspendent in H2O (DEPC treated)
up to 1 pmol/μL. Subsequently, 2.5 to 5 pmol of dephosphorylated transcripts were
phosporylated at the 5’ end with *γ-32P]-ATP by T4 polynucleotide kinase reaction:
Reagents Final concentration
10x T4 PNK reaction buffer 1x
Dephosphorylated RNA transcript 2.5 to 5 pmol
RNasin 10 U
*γ-32P]-ΑΣΡ (6000 Ci/mmol) 15 to 30 pmol
T4 PNK 10 U
H2O (DEPC treated) up to 20 μL
Reaction was carried out at 37oC for 1 h, and stopped by addition of 1 μL EDTA (20
mM). Labeled transcripts were extracted by adding equal volume of phenol / chloroform /
IAA (25:24:1), and purified on size exclusion G25 spin columns (Mini Quick Spin RNA
Columns), for unincorporated [γ-32P]-ATP nucleotide removal, according to manufacturer’s
instruction manual.
11. 2x i.v.t. loading buffer: 90% formamide, 0.025% bromophenol blue, 0.025% xylene cyanol
12. 1x i.v.t. elution buffer: 10 mM Tris-HCl, pH 7.5, 300 mM KCl, 1 mM EDTA.
13. 1x RNA FPLC buffer: 70mM Hepes-KOH, pH 7.8, 10 mM Mg(OAc)2 and 270 mM KOAc, 10%
glycerol.
2.4. In silico analysis
2.4.1. Staphylococcal GlyRS protein structural homology modeling
Information available for the staphylococcal GlyRS protein was found in UniProt
database (http://www.uniprot.org; annotation numbers: Q8CSD5 S. epidermidis ATCC 12228
strain; P67034 S. aureus Mu50 strain). Amino acid sequences of staphylococcal GlyRS
enzymes were aligned in the same server in order to obtain the percentage of similarity
between different strains (S. epidermidis Q8CSD5; S. aureus P67034; S. saprophyticus
Q49Y08; S. carnosus B9DNL1; S. haemolyticus Q4L6R5). Prediction of S. epidermidis GlyRS
tertiary structure was performed in the Expert Protein Analysis System (ExPASy) proteomics
server using the SWISS-MODEL homology modeling tool. Running the prediction tool to the

Materials and Methods
116
automated mode (http://swissmodel.expasy.org/workspace), the protein can be modeled in
3D, according to its amino acid chain organization by similarity matching with another
protein sequence, whose tertiary structure has already been experimentally verified and
annotated. T. thermophilus GlyRS (Uniprot No: P56206; PDB ID: 1ati) [259] represents a Class
II (α2) homolog with characterized structural features and it was chosen for the homology
modeling procedure, possessing 41 % identity in sequence with the S. epidermidis protein.
Results of the structural analysis were downloaded from the SWISS-MODEL workplace as a
PDB file and they were analyzed by the MOLMOL modeling bioinformatic tool
(http://www.mol.biol.ethz.ch/groups/wuthrich_group/softwaremolecular).
2.4.2. S. aureus glyS T-box leader in silico secondary structure prediction
The sequence containing the T-box riboswitch upstream the S. aureus glyS coding
region was identified according to RegPrecise database (http://regprecise.lbl.gov) [260]. The
broader full-length region upstream S. aureus glyS gene (SA1394), which includes the T-box
RNA sequence was further verified according to KEGG database [261] and “The T-box search
pattern” *199]. Moreover, promoter sequence and the transcription start site was predicted
using BPROM online tool [262] in Softberry database. Thermodynamic analysis for the
secondary structure stability of the antiterminator/terminator region was performed using
the Mfold web server [263]. All multiple sequence alignments were created using the T-
Coffee server algorithm [264], based on the full length T-box riboswitch sequences or
separate highly conserved regions of Stem I, Specifier loop, terminator stem and anti-
terminator stem. Furthermore, T-box sequences were used for multiple alignments obtained
in RegPrecise and verified in KEGG database upstream the corresponding glyS or glyQS ORFs.
Annotation numbers: SA1394 S. aureus N315; SE1252 S. epidermidis ATCC 12228;
STACA0001_1907 S. capitis SK14; Sca_1187 S. carnosus TM300; SH1351 S. haemolyticus
JCSC1435; SSP1191 S. saprophyticus ATCC 15305; BSU25270 B. subtilis 168; OB1949 O.
iheyensis HTE831; GK3430 G. kaustophilus HTA426; CAC3195 C. acetobutylicum ATCC 824;
LJ1320 L. johnsonii NCC 533; str0505 S. thermophilus CNRZ1066; Haur_4697 H. aurantiacus
ATCC 23779; Dgeo_1716 D. geothermalis DSM 11300 (see Supplementary data). In addition,
anticodon usage of tRNAGly species in each organism were obtained in Genomic tRNA
database, which contains tRNA gene predictions based on tRNAscan-SE program [265].
Finally, phylogenetic trees of T-boxes were constructed using the ClustalW2 algorithm [266]
and were visualized using Phylogeny.fr online tool [267].

Materials and Methods
117
2.4.3. glyS T-box:tRNAGly complex Kd determination
Binding analysis of glyS T-box:tRNAGly complexes were identified for T-box structural
constructs (T225 or T115) and each tRNAGly isoacceptor interaction after EMSA analysis, as
described in section §2.5.2. After electrophoresis of binding reactions the products (bound
and free radiolabel tRNAGly) were visualized by phosphoimager scanning and band intensity
measurement was performed using AIDA image analyzer software (version 5.0). The results
were exported as the percent integrated raw intensity of the bands corresponding to bound
tRNAGly transcript (the amount of T225 or T115 transcripts that was in complex with each
tRNAGly) as a result of one-site specific binding. GraphPad Prism Software (version 5.00), was
used to plot and analyze binding affinities by non-linear regression (amount of specific
binding vs T225 or T115 increasing concentrations) and to generate the corresponding
Scatchard plots (Bound/free vs Bound). The plots were used to calculate binding constants
(Kd) according to the equation: slope=-1/Kd. All experiments were performed in triplicates
and each point value used in Kd determination was the mean of two discrete measurements.
2.5. In vitro assays for biochemical and structural characterization
2.5.1. Aminoacylation assay
S. epidermidis native total tRNAGly and S. aureus tRNAGly purified transcripts (P1, P2,
NP1, NP2, NEW isoacceptors) were tested for their capacity to charge glycine, using S.
epidermidis GlyRS recombinant enzyme. For tRNAGly aminoacylation reactions 50 pmol of
each S. aureus isoacceptor transcript and 20 μΜ of total S. epidermidis total tRNA were
used. Aminoacylation of all tRNAGly molecules were performed in the following reaction
mixture. Reaction mixtures in the absence of tRNA were used as negative control.
Reagents Final concentration
5x Aminoacylation Reaction Buffer14 1x
[14C]-Gly (0.1 cpm/fmol) 10 μΜ
S. epidermidis GlyRS 0.5 μM
Nuclease-free water up to 20 μL
Reaction mixture was incubated at 37oC. The reaction was stopped on ice at several
time intervals (1, 2, 5, 10, and 20 min) and spotted directly on Whatman 3MM filter pads
(pre-soaked with 10% TCA). After time plot completion filters were washed one time in 10%

Materials and Methods
118
TCA (5 min), two times in 5% TCA (5 min each), and one time in 96% ethanol (2 min), under
gentle shaking at 4oC. Finally, filter pads were dried for 20 to 30 min and radioactivity
corresponding to the labeled glycine which is aminoacylated on tRNA molecules was
measured on a liquid scintillation counter (PHARMACIA-RACKBETA 1209).
14. 5x Aminoacylation Reaction Buffer: 500 mM Hepes-NaOH, pH 7.2, 50 mM MgCl2, 150 mM KCl,
10 mM DTT, 15 mM ATP.
2.5.2. Electrophoresis mobility shift assay
Electrophoretic mobility shift assay was used in order to identify the ability of each
five tRNAGly isoacceptors (P1, P2, NP2, NP1 and NEW) to form complexes with the three glyS
T-box RNA structural constructs (T275, T225, and T115). According to the method’s
principals, the molecules that participate in such complexes can migrate slower than those
possess their free status, when analyzed on a non-denaturing (native) gel matrix, exhibiting
kinetic behavior of high molecular weight complexes.
Each 5’ *γ-32P]-tRNAGly transcript (up to 0.1 μΜ in the presence of unlabeled tRNAGly
transcript) was mixed at the same time with each glyS T-box variant (5 μΜ) in 1x binding
buffer15 and was denatured for 3 min at 65oC. After refolding at RT samples were incubated
at 25oC for 1 to 1.5 h. All reactions were performed in a final volume of 10 μL and were
stopped with glycerol (8% v/v). Moreover, reaction mixtures without glyS T-box were used
as a negative control. Once left on ice for at least 10 min, samples were loaded on 6% non-
denaturing polyacrylamide gel16 containing 0.35x Tris-Borate buffer17. Native PAGE
electrophoresis was carried out at 400 V for 15 min, and at 200 V for 3 h at 4oC using the
same buffer. After electrophoresis, gels were vacuum dried and autoradiographed on a
phosphoimager (Fujifilm FLA 3000 platform).
For kinetic analysis of the complex formation, which was obtained for glyS T-box
structural constructs (T225 or T115) and each tRNAGly isoacceptor interaction additional
EMSA analysis was performed under the same experimental conditions described above. In
this series of reactions, concentration of each tRNAGly transcript was 0.1 μM (labeled and
unlabeled transcript mix), while T225 and T115 transcripts were used in increasing
concentrations of 0.5, 1, 2, 5, and 0.5, 1, 2, 3, 5, 10 μΜ respectively.
15. 1x Binding buffer: 10 mM Tris-HCl, pH 7.1, 100 mM KCl, 10 mM MgCl2.
16. 6% Native PAGE: 0.35x Tris-Borate buffer, 6% v/v acrylamide/bis-acrylamide (29:1), 5 mM
MgCl2.
17. 0.35x Tris-Borate buffer: 31.2 mM Tris base, 31.2 mM boric acid.
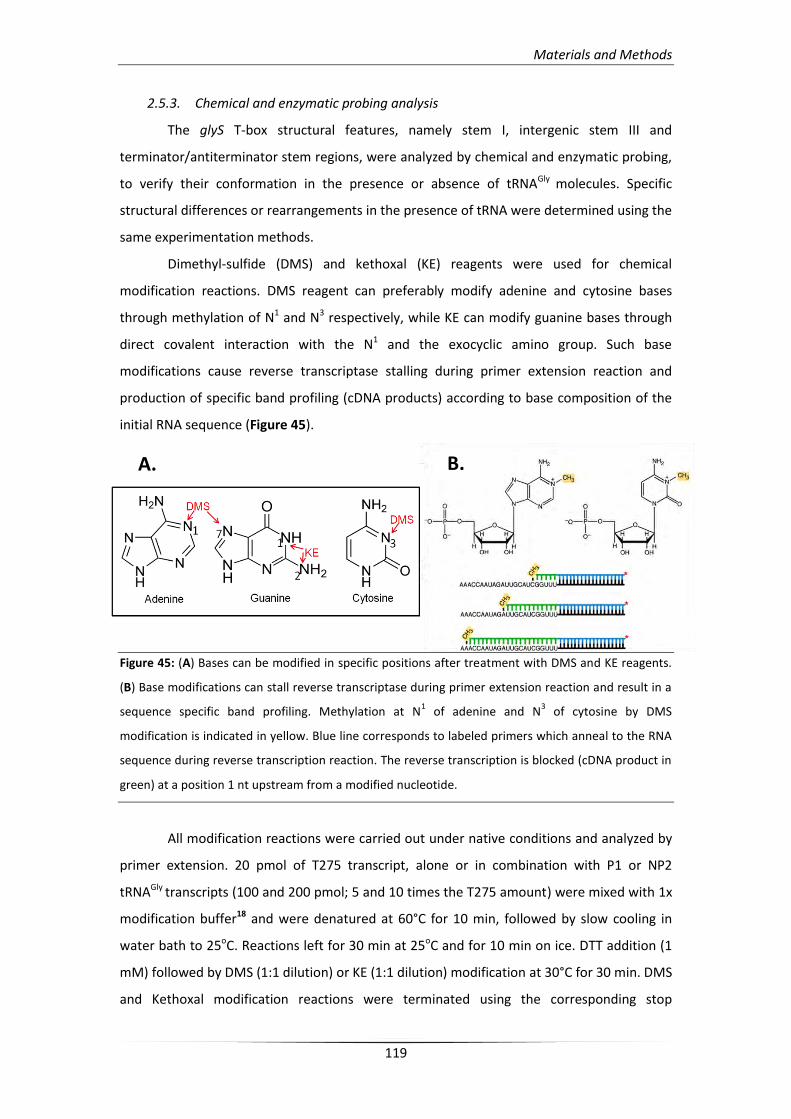
Materials and Methods
119
2.5.3. Chemical and enzymatic probing analysis
The glyS T-box structural features, namely stem I, intergenic stem III and
terminator/antiterminator stem regions, were analyzed by chemical and enzymatic probing,
to verify their conformation in the presence or absence of tRNAGly molecules. Specific
structural differences or rearrangements in the presence of tRNA were determined using the
same experimentation methods.
Dimethyl-sulfide (DMS) and kethoxal (KE) reagents were used for chemical
modification reactions. DMS reagent can preferably modify adenine and cytosine bases
through methylation of N1 and N3 respectively, while KE can modify guanine bases through
direct covalent interaction with the N1 and the exocyclic amino group. Such base
modifications cause reverse transcriptase stalling during primer extension reaction and
production of specific band profiling (cDNA products) according to base composition of the
initial RNA sequence (Figure 45).
Figure 45: (A) Bases can be modified in specific positions after treatment with DMS and KE reagents.
(B) Base modifications can stall reverse transcriptase during primer extension reaction and result in a
sequence specific band profiling. Methylation at N1 of adenine and N
3 of cytosine by DMS
modification is indicated in yellow. Blue line corresponds to labeled primers which anneal to the RNA
sequence during reverse transcription reaction. The reverse transcription is blocked (cDNA product in
green) at a position 1 nt upstream from a modified nucleotide.
All modification reactions were carried out under native conditions and analyzed by
primer extension. 20 pmol of T275 transcript, alone or in combination with P1 or NP2
tRNAGly transcripts (100 and 200 pmol; 5 and 10 times the T275 amount) were mixed with 1x
modification buffer18 and were denatured at 60°C for 10 min, followed by slow cooling in
water bath to 25oC. Reactions left for 30 min at 25oC and for 10 min on ice. DTT addition (1
mM) followed by DMS (1:1 dilution) or KE (1:1 dilution) modification at 30°C for 30 min. DMS
and Kethoxal modification reactions were terminated using the corresponding stop
A. B.

Materials and Methods
120
buffer19,20, followed by phenol/chloroform/IAA (25:24:1) extraction and ethanol
precipitation. Modified transcripts were resuspended in 1x TE buffer21, with the exception of
KE modified transcripts which were resuspended in 1x TE buffer in the presence of 50 mM
potassium borate, pH 7.5.
For primer extension analysis, seven different oligonucleotides were designed which
could hybridize in T275 sequence (every 40 to 50 nucleotides) and they were checked for
their annealing capacity during reverse transcription reactions. Only two of them provided
accurate reverse transcription reactions and were subsequently used for modification
analysis: TbGl_1, which hybridizes at nucleotides 129 to 148; TbGl_6 that hybridizes at
nucleotides 216 to 238 of T275 region (see Table 1) and they can map the Stem I or
terminator / antiterminator stem region of the T-box respectively. Selected probes were
labeled at the 5’ end as described in §2.3.3., without the dephosphorylation step. In primer
extension reactions 2 pmol of each modified transcript were hybridized with 5’ *γ-32P]-
labeled TbGl_1 or TbGl_6 primer in 1x hybridization buffer22 (final volume 10 μL). Reactions
were incubated for 10 min at 80oC, followed by slowly cooling in water bath to 47oC.
Subsequently, extension reactions were induced by addition of 1x primer extension buffer23
(23 μL) in presence of 2U RNasin and 5U AMV reverse transcriptase in a final volume of 42
μL and were incubated at 47oC for 1 h. After addition of 158 μL 1x TE buffer, the prodused
cDNAs were ethanol precipitated, resuspented in 4 μL of 0.1 N NaOH, and 8 μL of f-EDTA
loading buffer24, and were denatured at 80oC for 2 min, prior being analyzed on 6% PAGE/8
M Urea. Finally, four additional primer extension reactions using unmodified T275 template
were performed. The first was used as negative control and the other three were used for
sequencing analysis in presence of 0.17 mM of each ddNTP (ddTTP, ddGTP, ddCTP).
Three different ribonucleases, T1, V1, and A, were used in enzymatic probing
reactions. RNase T1 is an endonuclease that digests RNA molecules after recognition of G
residues, while RNase A digests after recognition of pyrimidines, namely C or U residues, in
single strand regions. RNase V1 digests base-paired nucleotides, without specific residue
recognition. In order to obtain structural conformation of leader Stem I region in presence or
in absence of tRNAGly, digestion reactions of T115 transcript alone were performed, or in
complex with P1 or NP2 tRNAGly as described above. T115 transcript (20 pmol of unlabeled
transcript mixed with 0.2 pmol of *γ-32P]-labeled transcript) mixed with P1 or NP2 (5x and
10x), were denatured at 65oC for 3 min under native conditions (1x Structure buffer25), and
digested with 0.1 U of RNase T1. Moreover, T115 alone was digested by RNase T1 (0.1), V1
(0.01U) and RNase A (0.2 pg) in native conditions or denaturing conditions (1x Sequencing

Materials and Methods
121
buffer26). Digestion reactions in both native and denaturing conditions were incubated at
25oC for 15 min; for denaturing conditions pre-incubation of the transcript was performed at
50oC for 5 min. Two additional reactions were performed. The first was carried out in native
conditions without presence of ribonuclease, and it was used as negative control. The
second was performed under alkaline hydrolysis conditions (1x Alkaline hydrolysis buffer27),
and was used for ladder construction. Alkaline hydrolysis reaction was carried out at 95oC for
10 min. All reactions were stopped on ice; after ethanol precipitation were denaturized at
80oC for 5 min in 10 μL of f-EDTA loading buffer and were analyzed on 10% PAGE/8 M Urea.
The same digestion protocol was used for analysis of NEW tRNAGly isoacceptor in
complex with T225 transcript in order to verify the interaction between anticodon loop and
specifier loop, with minor modifications. NEW tRNAGly transcript (20 pmol of unlabeled
transcript mixed with 0.2 pmol of *γ-32P]-labeled transcript) mixed with T225 transcript (20
and 60 pmol; 1:1 and 1:3 stoichiometry) was denatured at 65oC for 3 min and it was
subsquently incubated for 1 h at 25oC under native conditions. After incubation, reactions
were transferred into cap strips and radiated for 5 min by UV (254 nm) in 8 cm distance.
Once reactions were transferred back into new tubes, they were digested by 0.1 U of RNase
T1 and 0.2 pg of RNase A.
Finally, both primer extension and enzymatic probing analysis were visualized using
phosphoimager. The results that were obtained were used for comparison and verification
of the secondary structure that was proposed based on bioinformatics analysis.
18. 1x Modification buffer: 70mM Hepes-KOH, pH 7.8, 10 mM Mg(OAc)2, 270 mM KOAc.
19. 1x DMS stop buffer: 0.25 M Tris-acetate, pH 7.5, 0.25 M β-ME, 0.3 M sodium acetate, pH 9.2,
0.025 mM EDTA.
20. 1x KE stop buffer: 0.3 M sodium acetate pH 6.0, 25 mM potassium borate pH 6.0.
21. 1x TE buffer: 10 mM Tris-acetate, pH 7.5, 0.1 mM EDTA.
22. 1x hybridization buffer: 2 mM Tris-acetate, pH 7.9, 0.25 M KOAc, 0.2 mM EDTA.
23. 1x primer extension buffer: 20 mM Tris-acetate pH 8.3, 10 mM Mg(OAc)2, 5 mM DTT, 1 mM of
each dNTP.
24. f-EDTA loading buffer: 95% formamide, 18 mM EDTA, 0.025% bromophenol blue, 0.025% xylene
cyanol.
25. 1x Structure buffer: 10 mM Tris-HCl pH 7.1, 100 mM KCl and 10 mM MgCl2.
26. 1x Sequencing buffer: 20 mM sodium citrate pH 5.1 mM EDTA, 7 M urea.
27. 1x Alkaline hydrolysis buffer: 50 mM sodium carbonate pH 9.2, 1 mM EDTA.

Materials and Methods
122
2.5.4. In vitro tRNA directed antitermination assay
In order to confirm the in silico predicted transcription starting point of T-box glyS
leader RNA, dinucleotide-primed initiation of transcription was performed. Transcription
initiation occurs at a unique position (+1) downstream the conserved sequences at positions
-35 and -10 which interact with the E. coli RNA polymerase (RNAP) in vivo. According to
Samuels et al. [268] dinucleotide-prime initiation of transcription can identify this unique
position in vitro. Dinucleotides complementary to sequences which are distributed over the
+1 nucleotide (approximately 16 nucleotides around the transcription start) can adjust the
proper transcription initiation. Transcription start point can be primed by dinucleotides
complementary to positions -4 to +4, although the most efficient initiation can be recorded
by one dinucleotide which anneals at position -1 to +1. For Sau glyS T-box starting point
verification were used three different dinucleotides, ApU, UpA and CpU which can anneal at
positions -1 to +1, +1 to +2 and +6 to +7, according to the in silico predicted initiator
sequence (Figure 46).
Figure 46. Dinucleotide priming of in vitro transcription. The sequence corresponds to the in silico
predicted promoter region (red boxes represent the conserved -35 and -10 sequences) and the
transcription starting point (+1) of Sau glyS leader RNA. Dinucleotide in red (ApU) indicates the most
efficient transcription priming.
The template that was used in in vitro transcription reactions was a 408-bp PCR
fragment, starting 48 nucleotides upstream the transcription start site, and extending to 354
nucleotides downstream, which included the endogenous promoter, the whole T-box leader
region and part of the glyS coding sequence (up to 60 nt). Transcription termination site was
predicted approximately at position +274. For template production specific primers were
designed (see Table 1). Amplification reaction was performed as follows using as template S.
aureus WT genomic DNA, with an optimal annealing temperature at 60oC:

Materials and Methods
123
Reagents Final concentration
5x KAPA HiFi Fidelity Buffer 1x
dNTP mix (10 mM each) 0.3 mM
GLT_EnPr_FW_BamHI 0.3 μM
RTglTb_RV 0.3 μM
Sau genomic DNA (template) 100 ng
KAPA HiFi DNA polymerase 0.5 U
PCR grade water up to 25 μL
The PCR product (template) was purified (NucleoSpin® PCR Clean-up), and
sequenced (VBC biotech), prior to use. Transcription initiation reactions were performed by
omitting the G nucleotide, as follows:
Reagents Final concentration
5x Reaction buffer28 1x
ATP 2.5 μM
CTP 2.5 μM
UTP 0.75 μM
Dinucleotide (ApU or UpA or CpU) 150 μM
[α32P]-UTP (800 Ci/mmol) 0.1 to 0.25 μΜ
GLT PCR product (template) 10 ng
RNasin 1 U
E. coli RNAP 1 U
H2O (DEPC treated) up to 25 μL
Reactions were incubated at 37oC for 15min. The initiation step is paused with
heparin (20μg/ml) and elongation of transcription is allowed by the addition of MgCl2 (28
μΜ), KCl (86 μΜ), and all nucleotides (GUAC mix) in a final concentration of 5 μM each.
Elongation reactions were incubated at 37oC for 15 min in a final volume of 35 μL.
Transcription products were extracted by equal volume of phenol / chloroform / IAA

Materials and Methods
124
(25:24:1). 20 μL of aqueous phase were mixed with 10 μL of 3x stop buffer29, and were
analyzed on 6% PAGE/8 M Urea, after denaturation at 50oC for 5min.
In order to verify whether specific binding of the tRNA ligand in the T-box sequence
can facilitate the RNAP to overrun the terminator conformation, each tRNAGly isoacceptor
was tested for its ability to favor the antiterminator formation and in turn induce
transcription “read-through”. In vitro transcription of the glyS T-box region was performed in
presence of each Sau tRNAGly using the same protocol with minor modifications. Initiation
reaction was carried out as described above in presence of ApU dinucleotide, which was
obtained by transcription initiation priming reactions. Before transcription elongation
induction, tRNAGly transcripts were refolded (5min at 65oC) in the presence of 1 mM MgCl2,
and were added to the reactions in final concentration of 300 nM. Elongation reactions
stopped at various time points (5, 10 and 15 min). An additional elongation reaction was
performed in presence of a eukaryotic precursor tRNAArg(CCU), which was used as a negative
control.
For all in vitro transcription reactions, bands corresponding to termination (T) and
readthrough (RT) transcripts were visualized by scanning with phosphoimager (Fujifilm FLA
3000 platform), and the corresponding “band volumes” were quantified using AIDA image
analyzer software (version 5.0). (%) RT measurement was performed according to the
equation: (%) RT = [(Volume RT - Volume background) / (Volume T + Volume RT - 2*Volume
background)]*100.
28. 5x Reaction buffer: 100 mM Tris-HCl, pH 8.0, 100 mM NaCl, 50 mM MgCl2, 0.5 mM EDTA, 70
mM β-mercaptoethanol (Added immediately before use).
29. 3x Stop buffer: 6 M urea, 80 mM EDTA, 4% glycerol, 0.0125 % bromophenol blue, 0.0125%
xylene cyanol.
2.6. In vivo experiments
2.6.1. RT- PCR validation of endogenous Sau glyS T-box riboswitch system
S. aureus WT strain was used for total RNA generation. 200 μL of overnight cultures
in Tryptic Soy Broth (TSB) were used to inoculate 10 mL of M9 minimal salts medium
cultures in the presence of a full set of amino acids supplement30, 31. Furthermore, glycine
starvation and non starvation conditions were used by addition of 5 μg/mL, and 100 μg/mL
glycine respectively. Cultures were grown at 37°C for 16 h under shaking (250 rpm), and 109
cells were received for lysis and total RNA extraction according to RiboPure™-Bacteria Kit
instruction manual (Protocol for Gram-positive bacteria). All total RNA samples were

Materials and Methods
125
checked for their quality and possible genomic or plasmid DNA contamination (RNA 6000
Nano Kit) and used as templates for the RT-PCR reactions. qRT-PCR experiments were
performed using a one step reaction, for cDNA synthesis and amplification (Brilliant III SYBR®
Master Mix). A specific primer pair was designed for both cDNA synthesis and amplification
reactions (269-bp product, including the region from the transcription start to terminator
region of glyS T-box), as well as an additional primer pair for Sau 16 S rRNA, which was used
as internal control (see Table 1). The reaction mix and the thermal profile were performed
as follows, according to the manufacturer's recommendations:
Reagents Final concentration
2x Brilliant III Master mix 1x
DTT 1 mM
RTglTb_FW 0.2 μΜ
TbGl_2_RV 0.2 μΜ
RNA template 50 ng
RT/RNase block mix 1 μL
Nuclease free water up to 20 μL
qRT-PCR Thermal profile:
Temperature (oC) Time (min) Cycles
50 10:00 1
95 03:00 1
95 00:15 40
60 00:30
Two additional reactions were used as negative control, without template or RT mix
presence. Finally, RT-PCR-amplified fragments were analyzed on 1.5 % agarose gel,
extracted (NucleoSpin® Gel and PCR Clean-up PCR Kit) and sequenced (VBC biotech) for
further validation.

Materials and Methods
126
30. 5x M9 salts: 197 mM Na2HPO4, 110 mM KH2PO4, 43 mM NaCl, 93 mM NH4Cl (adjust to pH 7.4).
31. M9 broth plus amino acids: 1x M9 salts, 2 mM MgSO4, 0.1 mM CaCl2, 0.2 % glucose, 0.2 mg/mL
L-proline; L-phenylalanine; L-tryptophan; L-histidine; L-tyrosine, 0.1 mg/mL L-glutamine; L-
glutamic acid; L-cysteine; L-arginine; L-methionine; L-threonine; L-leucine.
2.6.2. S. aureus in vivo expression system
S. aureus RN4220 strain was used for construction of a homologous in vivo
expression system, to investigate the potential growth-depended regulation in production
and utilization of different tRNAGly isoacceptors. Specific primers were designed for cloning
of each S. aureus tRNAGly in pRB382 expression vector cloning (see Table 1). tRNAGly-pUC18
plasmid constructs (derived from cassette cloning; §2.3.2.) were used as template for tRNA
genes PCR amplification according to the following general reaction protocol with optimal
annealing temperature at 56oC.
Reagents Final concentration
2X PrimeSTAR Max Premix 1x
Primer FW 0.3 μM
Primer RV 0.3 μM
tRNAGly-pUC18 plasmid DNA 10 ng
PCR grade water up to 50 μL
The corresponding primer pair was used for each tRNA gene: P1 (P1_FW_HindIII/
P1_RV_BamHI), P2 (P2_FW_HindIII/P2_RV_BamHI), NP1 or NP2 (NP1/NP2_FW_HindIII/
NP1/NP2/NEW_RV_BamHI), NEW (NEW_FW_HindIII/NP1/NP2/NEW_RV_BamHI).
PCR product was gel extracted (NucleoSpin® Gel Clean-up), and cloned into pJET1.2
/blunt vector according to CloneJETTM PCR Cloning Kit instruction manual. Ligated tRNAGly-
pJET1.2 plasmid derivates were used for E. coli DH5α chemical competent cells
transformation. 5 μL of ligation reaction were mixed with 50 μL of cells and incubated for 20
min on ice. Cells were heat shocked at 42oC for 1 min and left on ice for 2 min prior to being
inoculated to 1 mL LB broth. After 1 h incubation at 37oC, cells were plated on LB/Amp agar
for positive tranformants selection. tRNAGly-pJET1.2 plasmids were extracted from positive
cell colonies (NucleoSpin® Plasmid DNA Mini Prep) and sequenced by VBC biotech, to avoid
incorporation of any random mutations. Subsequently, plasmid constructs were digested
with HindIII and BamHI restriction enzymes, and derived tRNAGly gene fragments were

Materials and Methods
127
ligated into pRB382 expression vector by T4 DNA ligase reaction, prior to E. coli DH5α
transformation, as described in §2.1.1. Positive clones were selected on LB/Amp agar plates.
The obtained constructs were able to initiate tRNAGly isoacceptors in vivo transcription both
in E. coli and S. aureus strains, as they were designed to be under control of the vegII shuttle
promoter.
S. aureus RN4220 electrocompetent cells were prepared as described before by
Kraemer et al. [269] 2 mL of overnight cultured cells were used to inoculate 100 mL TSB
medium and incubated at 37oC under shaking (250 rpm) on 1 L flask until they reached an
OD600 0.5 to 0.7. Cell growth was paused for 10 min on ice and culture was centrifuged at
8,000xg (10 min). Cell pellet was resuspended in 100 mL 500 mM ice cold sucrose water
solution (filter sterilized) and was repeatedly centrifuged (8,000xg for 10 min). Subsequently,
cells were washed with 50 mL of the same sucrose solution and were placed on ice for 15
min prior to being collected by centrifugation, and they were resuspended in 10 mL 500 mM
sucrose. Resulted suspension was aliquoted into 1.5 mL tubes (200 μL each), which were
immediately frozen at -80oC.
For S. aureus RN4220 transformation, electrocompetent cells were left to thaw at RT
and were divided into aliquots of 40 μL on ice. 0.5 μg of each tRNAGly-pRB382 plasmid
construct mixed with S. aureus cells (40 μL) and placed on 0.2 cm cuvettes. After 1 min
incubation on ice the mix were pulsed once in MicroPulserTM electroporation apparatus (1.8
kV; 2.5 msec). Two additional transformation reactions, one with empty pRB382 plasmid and
one without plasmid presence, were used as positive and negative control respectively.
Transformed cells were inoculated immediately at 1 mL SMMP broth32 plus 50 μg/mL
kanamycin, and were incubated for 45 min at 37oC. After harvesting (11,000xg, 1 min) they
resuspended in 5 mL TSB/ kanamycin (100 μg/mL), and they were incubated at 37oC for 72 h
under shaking (250 rpm). Culture samples were collected at several time points, namely, 0,
16, 20, 24, 40, 44, 48, 64, 72 h, and cell growth measured at A600.
32. SMMP broth: 28g/L Antibiotic medium #3, 20 mM maleic acid, 20 mM MgCl2, 187.9 g/L sucrose,
0.5% BSA.
2.6.3. Construction of T-box:tRNA double expression system in E. coli
E. coli M5154 strains transformed with vectors carrying the T-box and each tRNA
were produced in order to investigate in vivo the S. aureus glyS T-box dependent
transcription regulation capacity in the presence of tRNA, in a heterologous enviroment.
Therefore, an expression system which could host the expression of two vectors (with the T-
box and each tRNA, respectively) was constructed to synchronize tRNAGly isoacceptor in vivo

Materials and Methods
128
expression in concert with the reporter gene (β-galactosidase) to be fused and under the
control of the T-box. The whole region of Sau glyS T-box riboswitch (FL, Full-Length), from
position -48 (including the endogenous promoter) to position +294, was cloned into pLacZFT
expression vector (FLSD_1). In addition the same T-box region starting from position -2 was
cloned into pRB382 expression vector under control of vegII promoter instead the
endogenous promoter region (FLSD_2). P1 and NP2 tRNAGly genes were cloned into pBAD18-
Kan with additional 5’ leader and 3’ trailer sequences to boost in vivo endonucleolytic
maturation of the tRNA transcripts in E. coli. Specific primers were designed for each T-box
region vector cloning upstream the ORF sequence of β-galactosidase gene, while for tRNAGly
cloning specific oligonucleotides were synthesized including the additional 5’ and 3’ regions
(see Table 1). In order to increase the specificity of the PCR reaction, T-box and tRNAGly PCR
products from amplification reactions described in sections §2.5.4. and §2.6.2., were used as
template for second amplification. Nested PCR reactions for glyS T-box regions amplification
(FLSD_1 and FLSD_2) were carried out as follows with an optimal annealing temperature at
60oC and 63oC for FLSD_1 and FLSD_2 cloning, using the corresponding primer pair:
GLT_EnPr_FW_BamHI/ GLT_2ndSD_RV_SalI or ORF_GLT_2_FW / ORF_GLT_RV.
Reagents Final concentration
5x KAPA HiFi Fidelity Buffer 1x
dNTP mix (10 mM each) 0.3 mM
FW primer 0.3 μM
RV primer 0.3 μM
1st PCR product 1 ng
KAPA HiFi DNA polymerase 0.5 U
PCR grade water up to 25 μL
For the second amplification of the tRNAGly gene sequences the same general
reaction was used with optimal annealing temperature at 62oC using the corresponding
primer pairs: P1 (P1_FW_EcoRI / P1_RV_SalI; NP2 (NP2_FW_EcoRI / NP2_RV_SalI). FLSD_1,
P1, and NP2 tRNAGly PCR products were gel extracted (NucleoSpin® Gel Clean-up), cloned
into pJET1.2 /blunt vector, and used for E. coli DH5α chemical competent cells
transformation according to the CloneJETTM PCR Cloning Kit instruction manual (§2.6.2.).
After selection of positive clones on LB/Amp agar plates the recombinant plasmids were

Materials and Methods
129
extracted (NucleoSpin® Plasmid DNA Mini Prep) and sequenced (VBC biotech). FLSD_1-
pJET1.2 plasmid construct was digested with BamHI and SalI, while tRNAGly-pJET1.2
constructs were digested with EcoRI and SalI restriction enzymes. Derived FLSD_1 segment
was cloned into pLacZFT expression vector, while tRNAGly gene segments were ligated into
pBAD18-Kan expression vector by T4 DNA ligase reaction, prior to transformation in E. coli
DH5α competent cells. Positive clones were selected against the appropriate antibiotics
(Ampicillin or Kanamycin), on LB/Amp for FLSD_1-pLacZFT construct and on LB/Kan (50
μg/mL) agar plates for tRNAGly-pBAD18-Kan constructs. FLSD_2 PCR product was directly
ligated with pRB382 linear vector (digested with HindIII and BamHI restriction enzymes,
pursuant to standard digestion protocol) according blunt end ligation protocol, using
CloneJETTM PCR Cloning Kit components, as follows:
1st Step:
Reagents Final concentration/amount
2X Reaction Buffer 1x
Linear pRB382 0.05 pmol
DNA Blunting Enzyme 1 μL
Incubation for 5 min at 70oC and place on ice
2nd Step:
Reagents Final concentration/amount
FLSD_2 PCR product 0.15 pmol
T4 DNA Ligase 5 U
Nuclease free water up to 20 μL
Ligation reactions were incubated at 25oC for 15 min and the resulted FLSD_2-
pRB382 plasmid constructs were used for E. coli DH5α chemical competent cells
transformation. Positive E. coli clones were selected based on blue/white colony screening,
on LB/Amp agar plus 40 μg/mL X-gal and 0.2 mM IPTG. Moreover, FLSD_1-pLacZFT and
FLSD_2-pRB382 resulted constructs were able to initiate β-galactosidase reporter gene in
vivo expression by transcription antitermination modulation upon tRNAGly isoacceptor
binding. tRNAGly-pBAD18-Kan constructs are able to initiate P1 or NP2 tRNAGly isoacceptor in
vivo transcription after induction with L-arabinose.

Materials and Methods
130
Finally 0.5 μg of each FLSD_1-pLacZFT and FLSD_2-pRB382 constructs were mixed
with 0.5 μg of each tRNAGly-pBAD18-Kan constructs (P1 or NP2), and after being transferred
in 50 μL of E. coli M5154 chemical competent cells, they were incubated for 30 min on ice.
Cells were heat shocked at 42oC for 1 min and placed on ice for 2 min prior to inoculation of
0.5 mL LB broth. After 1 h incubation at 37oC, half amount of cells was selected on LB/Amp
(100 μg/mL)-Kan (50 μg/mL) agar. Positive clones bearing both plasmids were confirmed by
colony PCR for each construct [glyS T-box (FLSD_1 or FLSD_2), and tRNAGly (P1 or NP2)] by
using 0.5 U 2G Fast Taq DNA polymerase.
2.6.4. In vivo anti-termination assay (β-galactosidase activity test)
E. coli M5154 clones bearing both S. aureus glyS T-box-pLacZFT or pRB382
constructs and P1 or NP2 tRNAGly-pBAD18-Kan constructs were used for in vivo
antitermination assays: FLSD_1/P1 and FLSD_1/NP2 or FLSD_2/P1 and FLSD_2/NP2, using as
control a simple transformed strain bearing only the T-box construct (FLSD_1 or FLSD_2) but
not the tRNA plasmids. All strains were grown under amino acid limitation conditions. 2 mL
of overnight cultures in LB medium were used to inoculate minimal medium (M9 broth plus
25 μg/mL L-tryptophan), and they were incubated at 37oC until the early Log growth phase
(OD600= 0.4) was reached. Cells were collected by centrifugation at 6,000xg for 10 min, and
resuspended in 60 mL of minimal medium. Cultures were divided into two different batches
(30 mL each). 50μg/mL of glycine was added to the first one for starvation conditions, and
5μg/mL to the second for non-starvation conditions. Both cultures under starvation and
non-starvation condition were induced for tRNAGly expression by adding 0.1 % w/v L-
arabinose. Cultures were incubated at 37oC for 16 h and samples were collected for cell
growth and β-galactosidase measurement at several time points (0, 1, 2, 3, 4, 5, and 16 h).
Culture samples were measured at A595 and pelleted by centrifugation at 14,000xg for 5 min
prior storage at -80oC.
Measurement of β-galactosidase reporter gene expression in E. coli M5154 double
vector transformed strains was performed as described before by Henkin T. M. [270]. Cell
pellets from each time point were suspended in 1 mL Z buffer33 and lysed by addition of 10
μL Toluene, under vigorous shaking in vortex apparatus (up to 1 min). Samples were
incubated at fume hood with open lids for 10 min to allow toluene evaporation.
Subsequently, 0.4 mL of the lysate were transferred into new tubes (2 mL), and mixed with
0.6 mL Z buffer. β-galactosidase reaction was induced by addition of 0.2 mL ONPG
substrate34, and mixtures were incubated at RT until they reached a light yellow color where

Materials and Methods
131
they were stopped by addition of 0.5 mL Na2CO3 (1 M). Cell debris were pelleted by
centrifugation at 14,000xg for 5 min and supernatants were measured (A420) using as blank a
lysed cell sample without addition of ONPG substrate (negative control). β-galactosidase
activity was calculated in Miller units [271] according to the following equation: (A420*1,000)
/ (A595*0.4 mL*min), where A595 is indicating the cell growth of each time point and min is
representing the recorded time of β-galactosidase reaction after substrate addition.
33. Z buffer: 40 mM NaH2PO4, 60 mM Na2HPO4, 10 mM KCl, 1 mM MgSO4 (adjust to pH 7.0), 38 mM
β-mercaptoethanol (Added immediately before use).
34. ONPG substrate: 4 mg/mL ONPG in Z buffer without β-ME.


Αποτελζςματα


Results
135
1. Cross-species aminoacylation of tRNAGly reveals functional differences
1.1. Cloning and expression of recombinant GlyRS enzyme from S. epidermidis
The glycyl-tRNA synthetase (GlyRS) from S. epidermidis was cloned into pET20b
vector for expression in high yield (Methods §2.1.1.). According to the available glyS ORF in
KEGG genomic database, the corresponding nucleotide sequence (1392 bp) was amplified
using genomic DNA from S. epidermidis WT strain and was initially cloned in pSC-A vector via
TA-cloning for plasmid replication in E. coli. After sequencing verification of the cloned
region the glyS gene was cloned into pET20b expression vector downstream a T7 promoter
using specific restriction enzyme cleavage (NdeI/XhoI: 5’-3’ orientation) (Figure 47). The E.
coli expression system that was used [BL21 (DE3) Rosetta strain / pET20b expression vector]
can produce adequate amount of recombinant proteins, which are tagged with six histidine
residues (His6) in the C-terminal after induction of gene expression by adding the
appropriate concentration of IPTG in the culture medium.
Figure 47. Cloning of S. epidermidis
glyS gene in pET-20b expression
vector. Lane 1 indicates the
amplified glyS gene sequence. L
corresponds to molecular weight
DNA ladder.
S. epidermidis GlyRS recombinant enzyme was assessed for induction of protein
expression under various growth conditions and various IPTG induction concentrations
(Methods §2.1.2.). Conditions that allowed significant protein production were observed in
the presence of 0.5 mM IPTG after overnight growth at 25oC (Figure 48A). Purification of the
recombinant proteins was performed onto a Ni-NTA affinity chromatography column for
selective isolation of His6-tagged S. epidermidis GlyRS enzyme from the E. coli protein pool.
As illustrated in Figure 48B column derived fractions were analyzed for purity evaluation and
protein concentration of each fraction. Fractions E 3, 4, 5 which were found with the best
purity and yield of recombinant proteins were pulled together and were dialyzed in the
appropriate storage buffer. Each purification procedure resulted in efficient yield of
recombinant proteins (90 μM, as measured with Bradford standard method) which were
also of high purity (95%). The purified S. epidermidis GlyRS enzyme was examined for its
aminoacylation ability in presence of native total tRNA from S. epidermidis and subsequently
pET-20b
Sep glyS
NdeI XhoI
L 1
1500 bp
1000 bp
800 bp
1392 bp
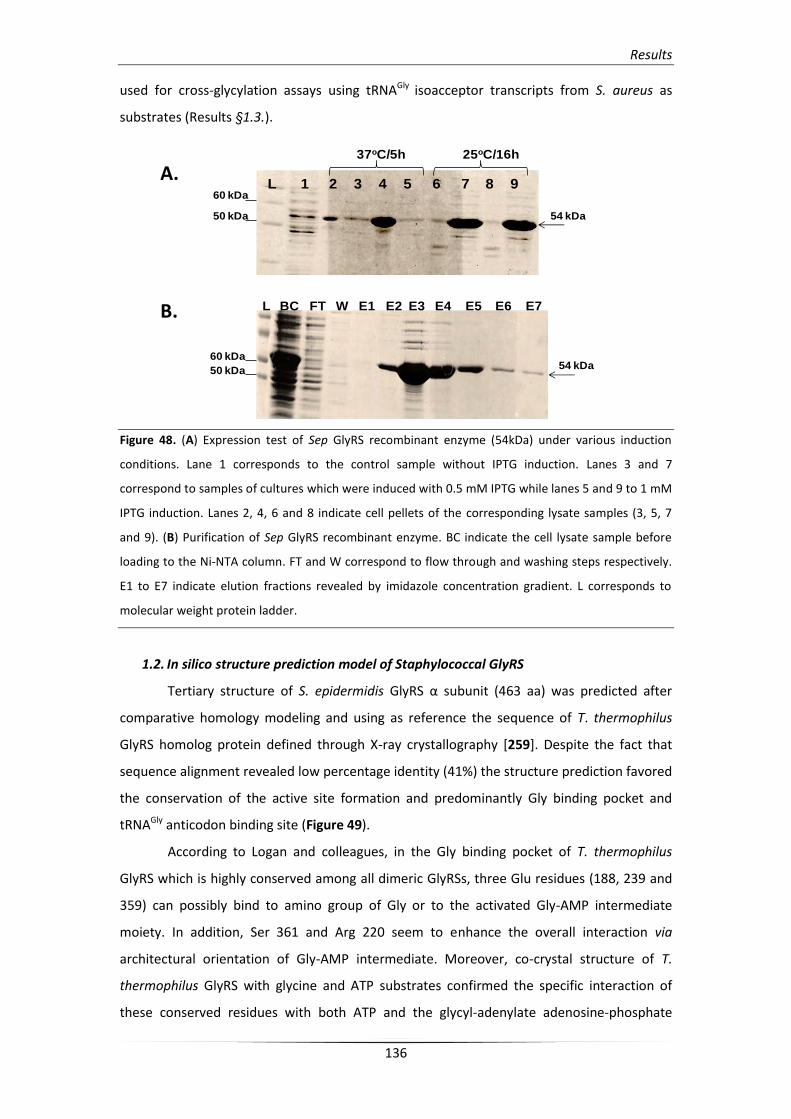
Results
136
used for cross-glycylation assays using tRNAGly isoacceptor transcripts from S. aureus as
substrates (Results §1.3.).
Figure 48. (A) Expression test of Sep GlyRS recombinant enzyme (54kDa) under various induction
conditions. Lane 1 corresponds to the control sample without IPTG induction. Lanes 3 and 7
correspond to samples of cultures which were induced with 0.5 mM IPTG while lanes 5 and 9 to 1 mM
IPTG induction. Lanes 2, 4, 6 and 8 indicate cell pellets of the corresponding lysate samples (3, 5, 7
and 9). (B) Purification of Sep GlyRS recombinant enzyme. BC indicate the cell lysate sample before
loading to the Ni-NTA column. FT and W correspond to flow through and washing steps respectively.
E1 to E7 indicate elution fractions revealed by imidazole concentration gradient. L corresponds to
molecular weight protein ladder.
1.2. In silico structure prediction model of Staphylococcal GlyRS
Tertiary structure of S. epidermidis GlyRS α subunit (463 aa) was predicted after
comparative homology modeling and using as reference the sequence of T. thermophilus
GlyRS homolog protein defined through X-ray crystallography [259]. Despite the fact that
sequence alignment revealed low percentage identity (41%) the structure prediction favored
the conservation of the active site formation and predominantly Gly binding pocket and
tRNAGly anticodon binding site (Figure 49).
According to Logan and colleagues, in the Gly binding pocket of T. thermophilus
GlyRS which is highly conserved among all dimeric GlyRSs, three Glu residues (188, 239 and
359) can possibly bind to amino group of Gly or to the activated Gly-AMP intermediate
moiety. In addition, Ser 361 and Arg 220 seem to enhance the overall interaction via
architectural orientation of Gly-AMP intermediate. Moreover, co-crystal structure of T.
thermophilus GlyRS with glycine and ATP substrates confirmed the specific interaction of
these conserved residues with both ATP and the glycyl-adenylate adenosine-phosphate
60 kDa
50 kDa 54 kDa
L 1 2 3 4 5 6 7 8 9
37oC/5h 25oC/16h
50 kDa
60 kDa54 kDa
L BC FT W E1 E2 E3 E4 E5 E6 E7
A.
B.
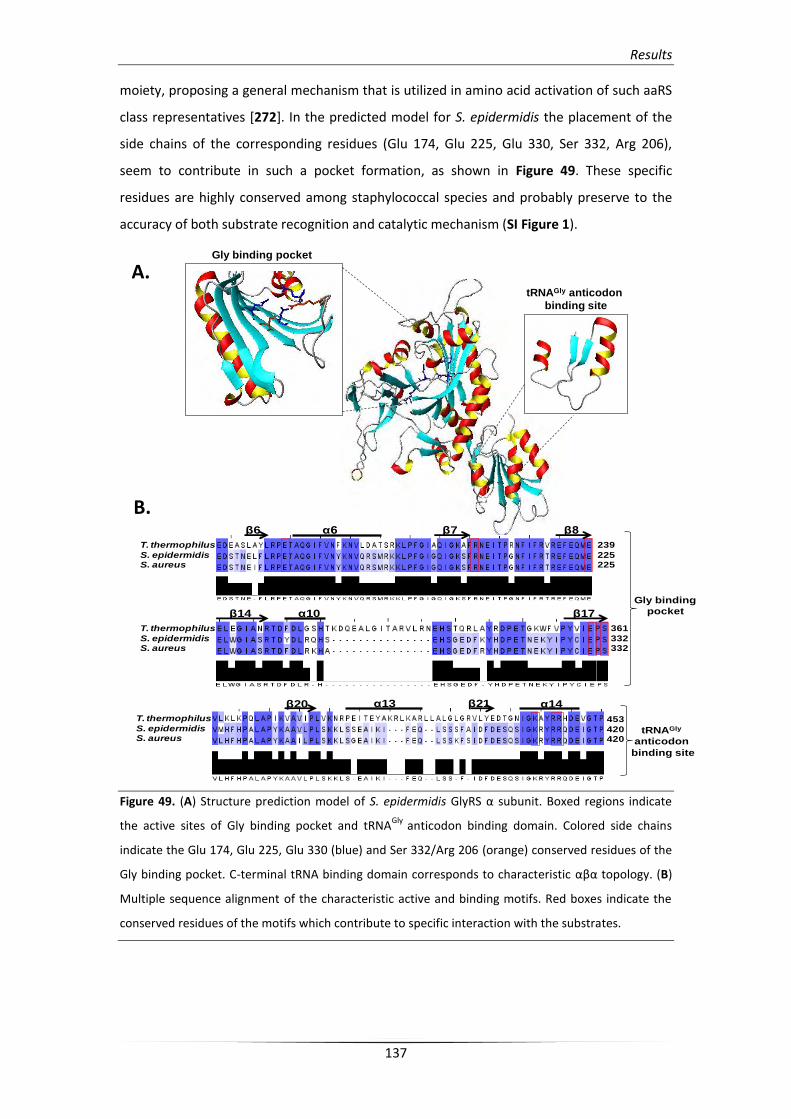
Results
137
moiety, proposing a general mechanism that is utilized in amino acid activation of such aaRS
class representatives [272]. In the predicted model for S. epidermidis the placement of the
side chains of the corresponding residues (Glu 174, Glu 225, Glu 330, Ser 332, Arg 206),
seem to contribute in such a pocket formation, as shown in Figure 49. These specific
residues are highly conserved among staphylococcal species and probably preserve to the
accuracy of both substrate recognition and catalytic mechanism (SI Figure 1).
Figure 49. (A) Structure prediction model of S. epidermidis GlyRS α subunit. Boxed regions indicate
the active sites of Gly binding pocket and tRNAGly
anticodon binding domain. Colored side chains
indicate the Glu 174, Glu 225, Glu 330 (blue) and Ser 332/Arg 206 (orange) conserved residues of the
Gly binding pocket. C-terminal tRNA binding domain corresponds to characteristic αβα topology. (B)
Multiple sequence alignment of the characteristic active and binding motifs. Red boxes indicate the
conserved residues of the motifs which contribute to specific interaction with the substrates.
Gly binding pocket
tRNAGly anticodon
binding site
α6 β7 β8
239
225225
T. thermophilus
S. epidermidisS. aureus
β6
β14 α10 β17
α13 α14β21β20
T. thermophilus
S. epidermidisS. aureus
361
332332
453
420420
T. thermophilus
S. epidermidisS. aureus
Gly binding
tRNAGly
anticodon
binding site
A.
B.

Results
138
On the other hand, little information about the tRNAGly overall structure recognition
has been defined by the crystal structure of T. thermophilus. Therefore, only minor
observations about the characteristic anticodon binding domain of the C-terminal part were
proposed, including a characteristic topology (αβα; Η13/B20/H14) that confers specific
orientation of the anticodon phosphate backbone, which seem to contribute in further
anticodon base recognition. Similarly, in our S. epidermidis prediction model, the tRNAGly
anticodon region can be recognized by the C-terminal domain base, as residues 386-416
have been predicted to form the characteristic αβα topology (Figure 49). In addition, the
second helix of this motif is found to bear positively charged arginine residues which can
potentially interact with the negatively charged phosphate backbone. Moreover, conserved
positively charged and aromatic residues of the C-terminal base among GlyRSs from
different staplylococcal species (SI Figure 1) presume in general tRNAGly substrate
recognition due to architectural favored specific orientation.
These observations could support the proposed cross-charging ability of GlyRS
homologs from different or relative species by specific recognition of idiosyncratic elements
of tRNAGly isoacceptors. Despite the high conservation among staplyloccocal GlyRSs it is
possible that minor divergences in sequence presumably contribute to insufficient cross-
glycylation of tRNAGly substrates from different species. This notion can be also supported by
the fact that the function of glycylation systems differs in substrate recognition even in
relative species despite the conservation of tRNA’s identity elements and is probably
attributed to structural and evolutionary complexity [273].
1.3. S. epidermidis GlyRS enzyme charges differentially S. aureus tRNAGly
In order to verify the potential ability of S. epidermidis GlyRS to cross-charge tRNAGly
isoacceptors that are encoded by S. aureus genome, proteinogenic and non-proteinogenic
tRNAGly transcripts were aminoacylated in vitro in the presence of recombinant GlyRS
enzyme (Methods §2.5.1.). Endogenous total tRNA from S. epidermidis was used as a
positive control for aminoacylation reaction (Methods §2.2.) and S. aureus tRNAGly
isoacceptors were in vitro transcribed without any post-transcriptional base modification
(Methods §2.3.2). Both S. aureus proteinogenic (P1) and non-proteinogenic (NP2 and NEW)
tRNAGly transcripts can be aminoacylated by S. epidemidis GlyRS with differences in the level
of charging capacity (Figure 50B). P1 proteinogenic isoacceptor presents the lowest charging
ability, which is likely due to post-transcriptional modifications absence (like the wide
distributed modified N6-dimethylallyl-adenosine in position 37 of anticodon loop), as it is
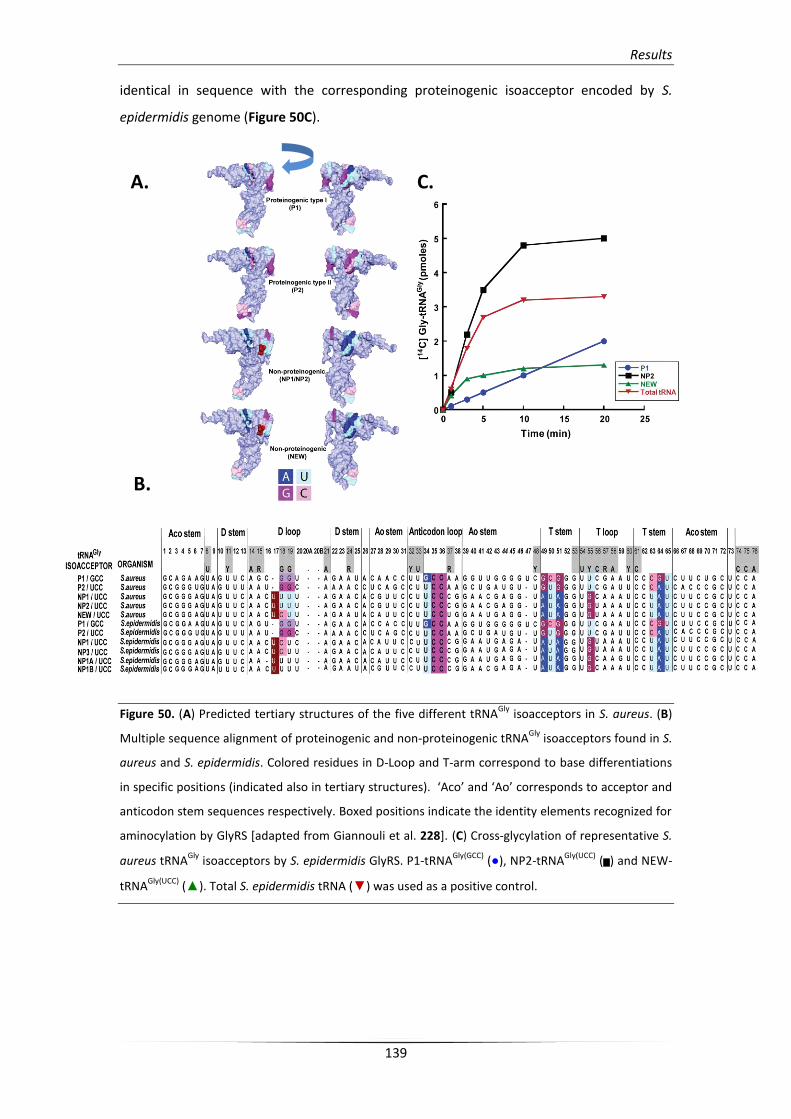
Results
139
identical in sequence with the corresponding proteinogenic isoacceptor encoded by S.
epidermidis genome (Figure 50C).
Figure 50. (A) Predicted tertiary structures of the five different tRNAGly
isoacceptors in S. aureus. (B)
Multiple sequence alignment of proteinogenic and non-proteinogenic tRNAGly
isoacceptors found in S.
aureus and S. epidermidis. Colored residues in D-Loop and T-arm correspond to base differentiations
in specific positions (indicated also in tertiary structures). ‘Aco’ and ‘Ao’ corresponds to acceptor and
anticodon stem sequences respectively. Boxed positions indicate the identity elements recognized for
aminocylation by GlyRS [adapted from Giannouli et al. 228]. (C) Cross-glycylation of representative S.
aureus tRNAGly
isoacceptors by S. epidermidis GlyRS. P1-tRNAGly(GCC)
(●), NP2-tRNAGly(UCC)
(▅) and NEW-
tRNAGly(UCC)
(▲). Total S. epidermidis tRNA (▼) was used as a positive control.
A.
B.
C.

Results
140
Interestingly, both NP2 and NEW non-proteinogenic isoacceptors preserve their
charging capacity without any base modification presence, where NP2 isoacceptor
conferring the most favorable aminoacylation ability. Non-proteinogenic tRNAGly anticodon
loop were found totally unmodified in S. epidermidis according to MODOMICS server
(http://modomics.genesilico.pl) albeit U34 of the anticodon triplet is mostly modified in 5-
methoxycarbonyl-methyl-2-thiouridine. This observation can support the high efficient
discrimination and cross-glycylation of S. aureus non-proteinogenic isoacceptors. Moreover,
both S. aureus and proteinogenic and non-proteinogenic tRNAGly isoacceptors, which were
used in cross-glycylation assays, retain all the important identity recognition elements for
GlyRS discrimination, namely G1-C72 base pair, G10 (except NEW tRNA which bears a U
residue in the same position), C35, and C36 (Figure 50A and 50B). These residues constitute
highly conserved identity elements among different glycylation systems during evolution
and seem to be responsible for such cross charging inter-relations [273].
On the other hand, despite the fact that identity elements of tRNAGly isoacceptors
from both species (S. aureus and S. epidermidis) are identical, minor structural divergences
in GlyRS enzymes can probably contribute in differential cross-charging of each isoacceptor.
This observation can be the reason why the Sau P1 tRNA cannot be efficiently aminoacylated
by Sep GlyRS and can be also supported by the fact that tRNAGly isoacceptors participate in
cell wall synthesis which differs among staphylococcal species (glycine peptide in S. aureus
and glycine/ L-alanine or glycine/L-serine peptide in S. epidermidis; [127]).
1.4. Proteinogenic and non-proteinogenic tRNAGly isoacceptors in S. aureus
All publicly available S. aureus genomes include five tRNAGly genes which encode for
five different isoacceptors, termed P1 (P for Proteinogenic), P2, NP1 (NP for Non-
Proteinogenic), NP2, and NEW. P1 and P2 (termed as proteinogenic) bear a GCC and a UCC
anticodon respectively and participate in ribosomal protein synthesis. NP1, NP2, and NEW,
(termed as non-proteinogenic) bear a UCC anticodon and are used by FemXAB factors as
glycine donors in cell wall pentaglycine interpeptide synthesis.
In order to investigate the differential utilization of each tRNAGly isoacceptor in a
growth phase-depended manner an endogenous expression system were constructed in S.
aureus RN4220 strain (Methods §2.6.2.). All tRNAGly genes were cloned into pRB382
expression vector under the control of a vegII promoter, which is active in S. aureus, using
specific restriction enzyme sites (HindIII/BamHI: 5’-3’ orientation; Figure 51). Each S. aureus
RN4220 strain clone which was transformed with the corresponding tRNAGly-pRB382 plasmid

Results
141
could enhance the endogenous expression of each one of the five isoacceptors and as a
result, could increase the level of its aminoacylation status.
Figure 51. Cloning of S. aureus tRNAGly
genes in pRB382 expression vector. Lanes 1 to 5 indicate the
amplified gene sequences of each isoacceptor (P1, P2, NP1, NP2 and NEW). L corresponds to
molecular weight DNA ladder.
All clones that transcribe each isoacceptor exhibited reduced growth ability. Early
growth phase was extended approximately to 48 hours in contrast to the control S. aureus
strain which reached the Log growth phase after 24 hours. Most transformed strain cultures
could rapidly transited to mid-Log phase and subsequently could reach the stationary phase
after 60 hours, except NP2-pRB382 transformed strain that failed to reach the mid-Log
phase even after 72 hours. Moreover, the NEW-pRB382 transformed strain exhibited an
obvious reduced growth in comparison with P1-, P2-, and NP1-pRB382 transformed strains.
In addition, the control strain which did not exhibit resistance in kanamycin exhibited the
same reduced growth pattern comparable with the clones that are expressing each
isoacceptor (Figure 52).
These findings suggest the existence of a potential growth phase-dependent
mechanism which synchronizes the utilization of each aminoacylated tRNAGly isoacceptor. All
isoacceptors can and must be present in different growth phases to be used for both protein
and cell wall synthesis. Absence or over-production of each isoacceptor, in either charged or
uncharged status, can lead to divergent growth profiles or significant loss of fitness.
pRB382
tRNAGly
HindIII BamHI
L 1 2 3 4 5
150 bp
100 bp
50 bp76 bp
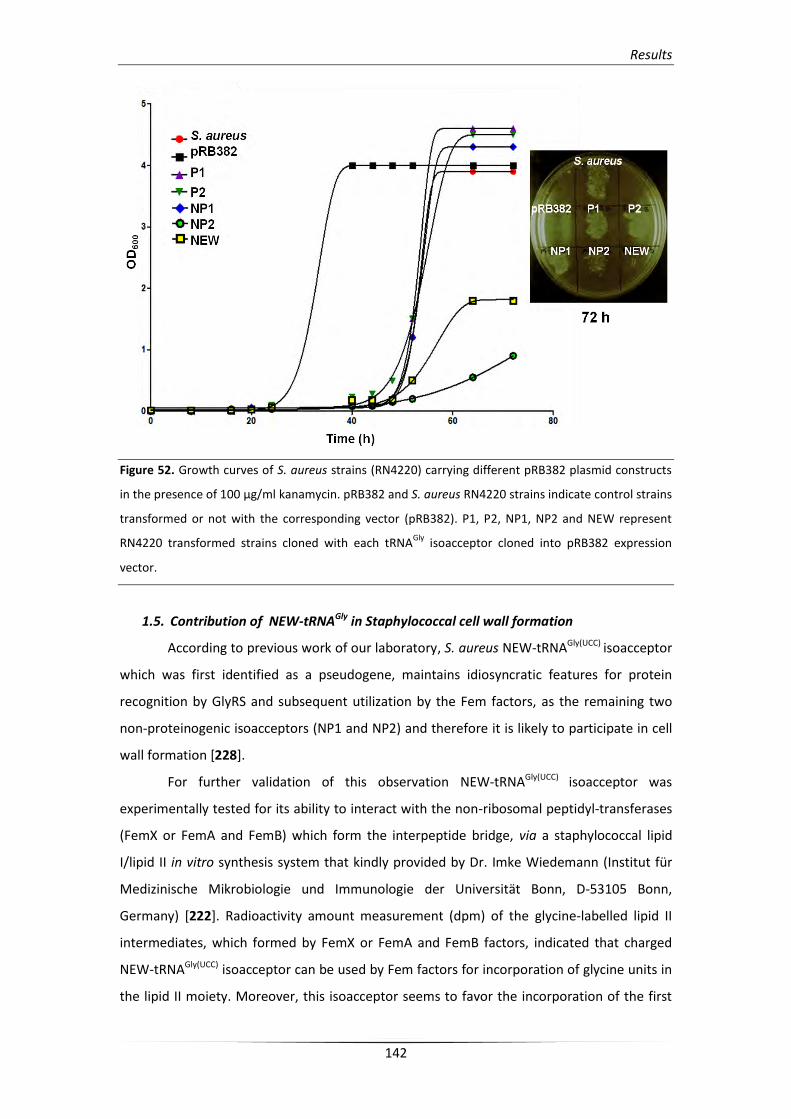
Results
142
Figure 52. Growth curves of S. aureus strains (RN4220) carrying different pRB382 plasmid constructs
in the presence of 100 μg/ml kanamycin. pRB382 and S. aureus RN4220 strains indicate control strains
transformed or not with the corresponding vector (pRB382). P1, P2, NP1, NP2 and NEW represent
RN4220 transformed strains cloned with each tRNAGly
isoacceptor cloned into pRB382 expression
vector.
1.5. Contribution of NEW-tRNAGly in Staphylococcal cell wall formation
According to previous work of our laboratory, S. aureus NEW-tRNAGly(UCC) isoacceptor
which was first identified as a pseudogene, maintains idiosyncratic features for protein
recognition by GlyRS and subsequent utilization by the Fem factors, as the remaining two
non-proteinogenic isoacceptors (NP1 and NP2) and therefore it is likely to participate in cell
wall formation [228].
For further validation of this observation NEW-tRNAGly(UCC) isoacceptor was
experimentally tested for its ability to interact with the non-ribosomal peptidyl-transferases
(FemX or FemA and FemB) which form the interpeptide bridge, via a staphylococcal lipid
I/lipid II in vitro synthesis system that kindly provided by Dr. Imke Wiedemann (Institut für
Medizinische Mikrobiologie und Immunologie der Universität Bonn, D-53105 Bonn,
Germany) [222]. Radioactivity amount measurement (dpm) of the glycine-labelled lipid II
intermediates, which formed by FemX or FemA and FemB factors, indicated that charged
NEW-tRNAGly(UCC) isoacceptor can be used by Fem factors for incorporation of glycine units in
the lipid II moiety. Moreover, this isoacceptor seems to favor the incorporation of the first
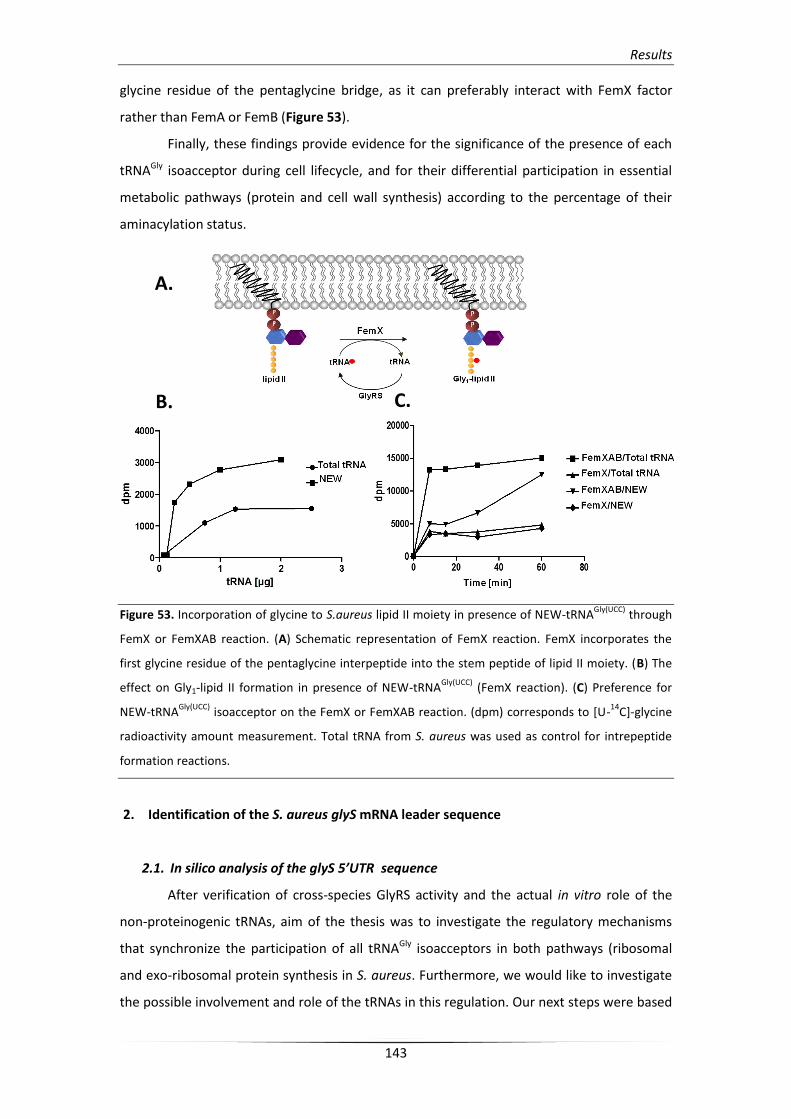
Results
143
glycine residue of the pentaglycine bridge, as it can preferably interact with FemX factor
rather than FemA or FemB (Figure 53).
Finally, these findings provide evidence for the significance of the presence of each
tRNAGly isoacceptor during cell lifecycle, and for their differential participation in essential
metabolic pathways (protein and cell wall synthesis) according to the percentage of their
aminacylation status.
Figure 53. Incorporation of glycine to S.aureus lipid II moiety in presence of NEW-tRNAGly(UCC)
through
FemX or FemXAB reaction. (A) Schematic representation of FemX reaction. FemX incorporates the
first glycine residue of the pentaglycine interpeptide into the stem peptide of lipid II moiety. (B) The
effect on Gly1-lipid II formation in presence of NEW-tRNAGly(UCC)
(FemX reaction). (C) Preference for
NEW-tRNAGly(UCC)
isoacceptor on the FemX or FemXAB reaction. (dpm) corresponds to [U-14
C]-glycine
radioactivity amount measurement. Total tRNA from S. aureus was used as control for intrepeptide
formation reactions.
2. Identification of the S. aureus glyS mRNA leader sequence
2.1. In silico analysis of the glyS 5’UTR sequence
After verification of cross-species GlyRS activity and the actual in vitro role of the
non-proteinogenic tRNAs, aim of the thesis was to investigate the regulatory mechanisms
that synchronize the participation of all tRNAGly isoacceptors in both pathways (ribosomal
and exo-ribosomal protein synthesis in S. aureus. Furthermore, we would like to investigate
the possible involvement and role of the tRNAs in this regulation. Our next steps were based
A.
B. C.
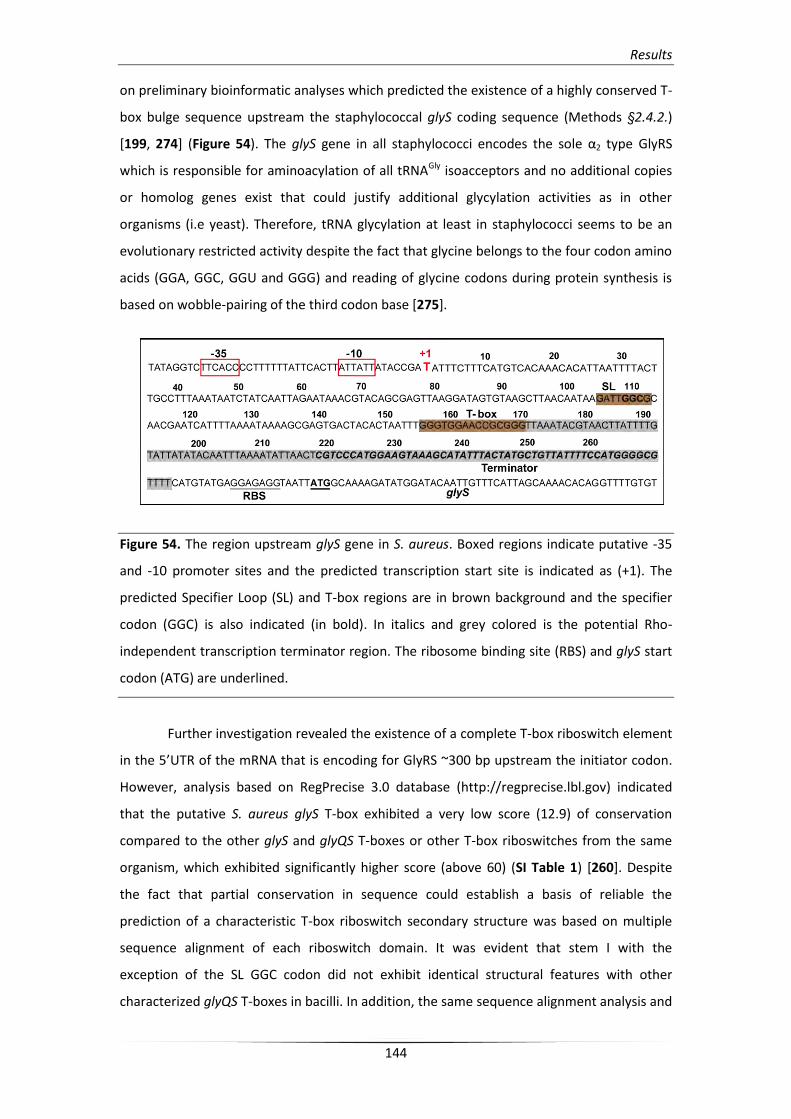
Results
144
on preliminary bioinformatic analyses which predicted the existence of a highly conserved T-
box bulge sequence upstream the staphylococcal glyS coding sequence (Methods §2.4.2.)
[199, 274] (Figure 54). The glyS gene in all staphylococci encodes the sole α2 type GlyRS
which is responsible for aminoacylation of all tRNAGly isoacceptors and no additional copies
or homolog genes exist that could justify additional glycylation activities as in other
organisms (i.e yeast). Therefore, tRNA glycylation at least in staphylococci seems to be an
evolutionary restricted activity despite the fact that glycine belongs to the four codon amino
acids (GGA, GGC, GGU and GGG) and reading of glycine codons during protein synthesis is
based on wobble-pairing of the third codon base [275].
Figure 54. The region upstream glyS gene in S. aureus. Boxed regions indicate putative -35
and -10 promoter sites and the predicted transcription start site is indicated as (+1). The
predicted Specifier Loop (SL) and T-box regions are in brown background and the specifier
codon (GGC) is also indicated (in bold). In italics and grey colored is the potential Rho-
independent transcription terminator region. The ribosome binding site (RBS) and glyS start
codon (ATG) are underlined.
Further investigation revealed the existence of a complete T-box riboswitch element
in the 5’UTR of the mRNA that is encoding for GlyRS ~300 bp upstream the initiator codon.
However, analysis based on RegPrecise 3.0 database (http://regprecise.lbl.gov) indicated
that the putative S. aureus glyS T-box exhibited a very low score (12.9) of conservation
compared to the other glyS and glyQS T-boxes or other T-box riboswitches from the same
organism, which exhibited significantly higher score (above 60) (SI Table 1) [260]. Despite
the fact that partial conservation in sequence could establish a basis of reliable the
prediction of a characteristic T-box riboswitch secondary structure was based on multiple
sequence alignment of each riboswitch domain. It was evident that stem I with the
exception of the SL GGC codon did not exhibit identical structural features with other
characterized glyQS T-boxes in bacilli. In addition, the same sequence alignment analysis and
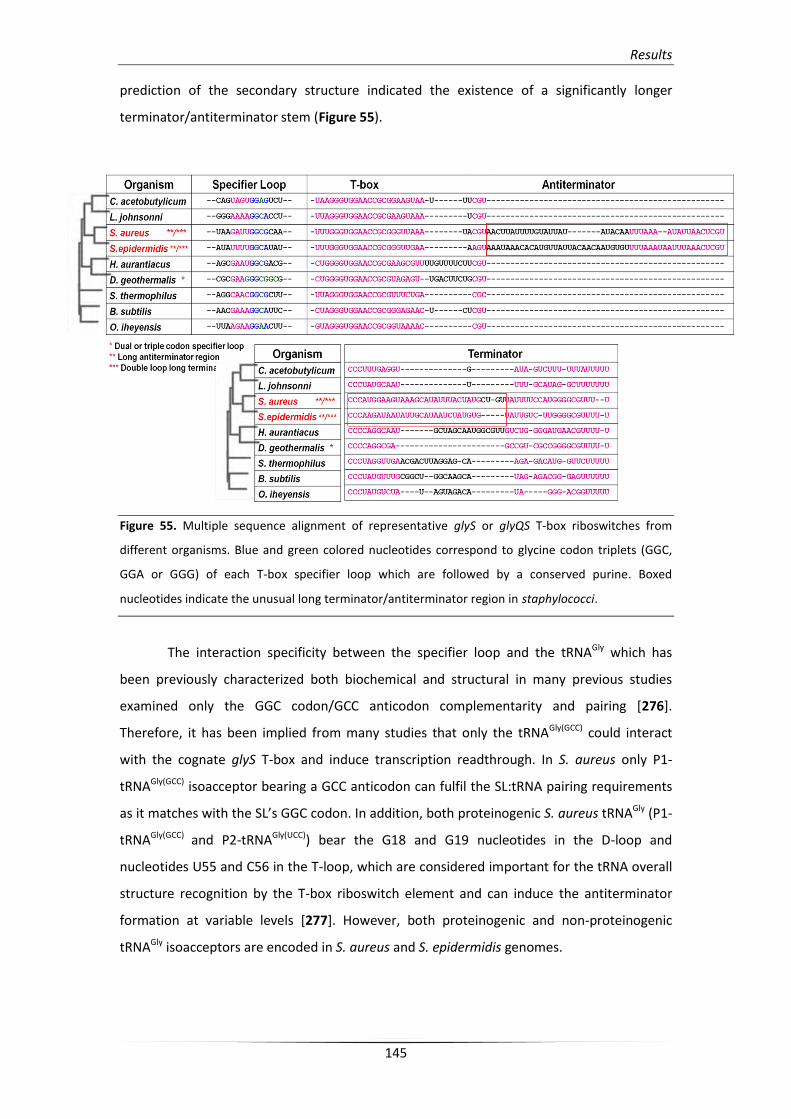
Results
145
prediction of the secondary structure indicated the existence of a significantly longer
terminator/antiterminator stem (Figure 55).
Figure 55. Multiple sequence alignment of representative glyS or glyQS T-box riboswitches from
different organisms. Blue and green colored nucleotides correspond to glycine codon triplets (GGC,
GGA or GGG) of each T-box specifier loop which are followed by a conserved purine. Boxed
nucleotides indicate the unusual long terminator/antiterminator region in staphylococci.
The interaction specificity between the specifier loop and the tRNAGly which has
been previously characterized both biochemical and structural in many previous studies
examined only the GGC codon/GCC anticodon complementarity and pairing [276].
Therefore, it has been implied from many studies that only the tRNAGly(GCC) could interact
with the cognate glyS T-box and induce transcription readthrough. In S. aureus only P1-
tRNAGly(GCC) isoacceptor bearing a GCC anticodon can fulfil the SL:tRNA pairing requirements
as it matches with the SL’s GGC codon. In addition, both proteinogenic S. aureus tRNAGly (P1-
tRNAGly(GCC) and P2-tRNAGly(UCC)) bear the G18 and G19 nucleotides in the D-loop and
nucleotides U55 and C56 in the T-loop, which are considered important for the tRNA overall
structure recognition by the T-box riboswitch element and can induce the antiterminator
formation at variable levels [277]. However, both proteinogenic and non-proteinogenic
tRNAGly isoacceptors are encoded in S. aureus and S. epidermidis genomes.

Results
146
Multiple sequence alignment of the staphylococcal tRNAGly isoacceptors with tRNAGly
genes encoded by bacilli genomes (B. subtilis, O. iheyensis, and G. kaustophilus) which have
been previously used in many biochemical and structural studies to elucidate and establish
the regulatory role of glyS T-boxes, revealed that all bacilli tRNAGly gene sequences were
identical exhibiting minor variations with the staphylococcal proteinogenic isoacceptors
(Figure 56). As a result, proteinogenic tRNAGly(UCC) isoacceptors, which differ only in wobble
position 34 cannot be excluded for GGC codon SL recognition. Moreover, the remaining non-
proteinogenic tRNAs can be used as controls, since their sequence varies in the anticodon
loop (position 37 in addition to wobble position 34), as well as in the D-arm/–loop and the
Tarm/-loop (Figure 56).
Figure 56. Multiple sequence alignment of both proteinogenic or non-proteinogenic tRNAGly
isoacceptors bearing GCC or UCC anticodons found in staphylococcal and bacilli species.
Colored panels indicate D-loop and T-stem/loop sequence differentiations in specific
positions. Boxed and in grey background positions refer to identity elements recognized for
aminocylation by GlyRS. ‘Aco’ and ‘Ao’ correspond to acceptor and anticodon stems
respectively.
2.2. Verification of the predicted 5’UTR glyS regulatory system
To clarify in vivo the specific recognition properties and the regulatory role of 5’UTR
glyS leader region its expression was tested under minimal growth conditions in presence or
absence of glycine. Total RNA extracts from S. aureus N315 strain overnight cultures in the
corresponding culture medium were evaluated for their quality (Figure 57A) and analyzed
via qRT-PCR (Methods §2.6.1.). Primers that were used (Table 1) were designed to anneal in
sequence region of the in silico predicted T-box riboswitch region upstream the glyS gene
initiator codon (269-bp; Figure 57B).
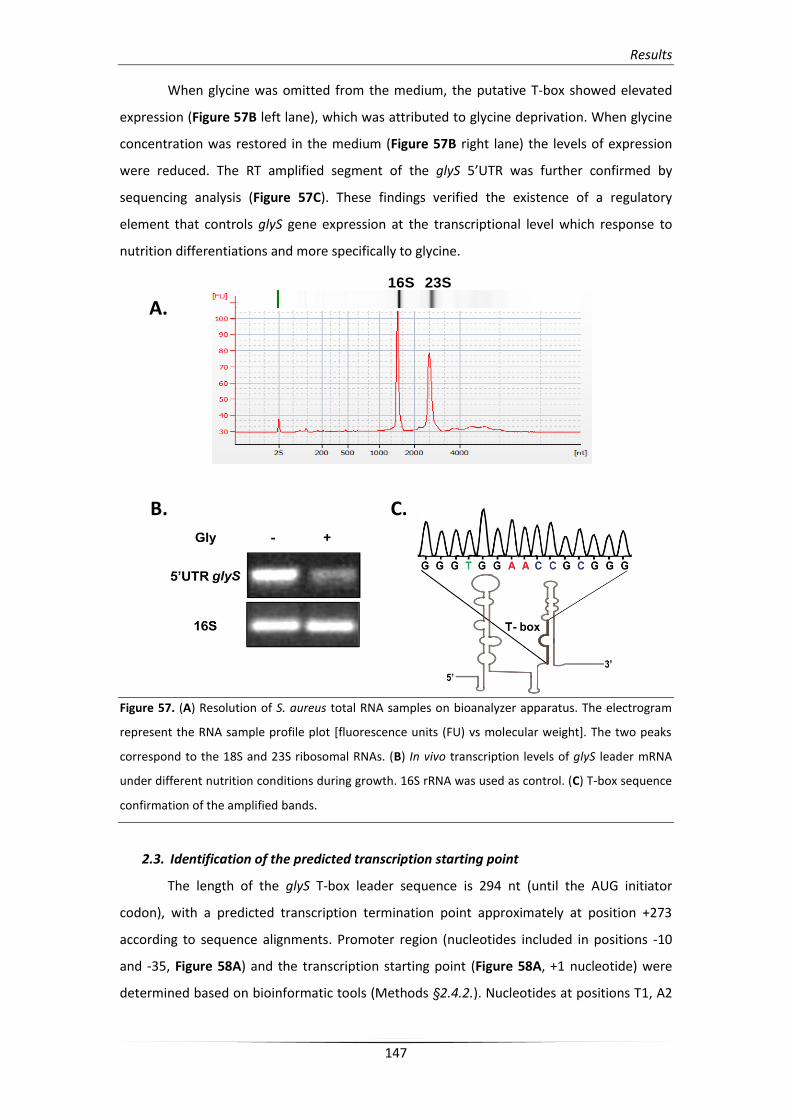
Results
147
When glycine was omitted from the medium, the putative T-box showed elevated
expression (Figure 57B left lane), which was attributed to glycine deprivation. When glycine
concentration was restored in the medium (Figure 57B right lane) the levels of expression
were reduced. The RT amplified segment of the glyS 5’UTR was further confirmed by
sequencing analysis (Figure 57C). These findings verified the existence of a regulatory
element that controls glyS gene expression at the transcriptional level which response to
nutrition differentiations and more specifically to glycine.
Figure 57. (A) Resolution of S. aureus total RNA samples on bioanalyzer apparatus. The electrogram
represent the RNA sample profile plot [fluorescence units (FU) vs molecular weight]. The two peaks
correspond to the 18S and 23S ribosomal RNAs. (B) In vivo transcription levels of glyS leader mRNA
under different nutrition conditions during growth. 16S rRNA was used as control. (C) T-box sequence
confirmation of the amplified bands.
2.3. Identification of the predicted transcription starting point
The length of the glyS T-box leader sequence is 294 nt (until the AUG initiator
codon), with a predicted transcription termination point approximately at position +273
according to sequence alignments. Promoter region (nucleotides included in positions -10
and -35, Figure 58A) and the transcription starting point (Figure 58A, +1 nucleotide) were
determined based on bioinformatic tools (Methods §2.4.2.). Nucleotides at positions T1, A2
23S16S
A.
B. C.

Results
148
and T7 were predicted as possible starting points [261]. Those putative starting points were
tested using the dinucleotide-primed transcription initiation reaction and three different
dinucleotides, ApU, UpA, and CpU to elongate transcription (Methods §2.5.4.; [268]).
Initiation reactions were performed by omitting the G nucleotide which was used
subsequently for elongation reaction induction. As revealed, transcription initiation could be
induced only in the presence of ApU (Figure 58B), while all three dinucleotides could be
used as substrates for the elongation reaction. These results showed that only position T1
could fulfill this observation and it was considered further as the transcription starting point
of the T-box riboswitch.
Figure 58. Dinucleotide priming of Sau glyS leader RNA in vitro transcription. (A) Sequence in red
boxes represent the conserved -35 and -10 regions of the in silico predicted promoter. The predicted
transcription starting point (+1) is also indicated. (B)The upper panel indicates the initiation reaction
and the lower panel the elongation reaction. ApU, UpA, and CpU corresponds to dinucleotides used
for each reaction.
3. Analysis of the S. aureus glyS T-box structure
3.1. In silico analysis of the glyS T-box riboswitch RNA secondary structure
Prediction of the putative secondary structure of the S. aureus glyS T-box was
initially performed by using bioinformatics analysis (Methods §2.4.2.). Multiple sequence
alignment of stem I, terminator/antiterminator stem, and intervening regions, in
combination with thermodynamic analysis of the secondary structure stability (Mfold web
server) were considered for the proposed secondary structure model as illustrated in Figure
A.
B.

Results
149
59. The putative structure forms an extended T-box riboswitch RNA element with unusual
structural features.
In its initial part, Sau glyS T-box consists of a conventional stem I however exhibits
variations in its characteristic individual structural elements. In the base of stem I
conformation, a K-turn motif (also known as GA motif) which exhibits very low conservation
can be identified. K-turn motifs are characteristic RNA structural domains found also in other
riboswitch leaders (S-boxes) as well as in eukaryal and archaeal RNAs like several
ribonucleoprotein units (U1, U2, U5, and U4/U6 snRNPs) and snoRNAs which play essential
role in rRNA modification and processing. Moreover, equal important K-turn-like motifs have
been identified in archaeal 16S and 23S rRNAs however their actual function remains elusive
[201].
The K-turn is followed by the specifier loop (SL) which includes a GGC codon triplet
that is presumably recognized by the GCC anticodon of the P1-tRNAGly(GCC) ligand. Moreover,
Sau glyS T-box SL element seems to belong to the 8 nt class. This type of T-box SL elements
has been described to exhibit dual-specificity depending on their downstream regulated
genes (as in the case of NT-box from C. acetobutylicum [144]). Despite the fact that in the
case of Sau glyS T-box the downstream sequence encodes only a single gene and not an
operon, which excludes the possibility of dual-specificity, a second codon sequence can be
identified for tRNATrp(CCA) (U108, G109, G110). In addition, SL contains the conserved
nucleotides G105 and A106 according to the previously characterized pattern [276] and the
SL codon is followed by a highly conserved purine, which is guanosine instead of adenosine
in B. subtilis and G. kaustophilus.
In the upper helix region loop E can be found. Loop E although quite conserved it
appears displaced (not opposite to the SL loop as in the case of B. subtilis) and is followed by
a potential bulge region and a partially conserved AG bulge. The apical (or distal) loop on the
top of the helix contains 6 additional nucleotides and is adequately conserved. Most
interestingly, the intervening region extending to terminator/antiterminator stem includes a
very short variable linker and a quite conserved stem III (shorter compared to that from B.
subtilis; 4bp instead of 7), but the characteristic stem II and pseudoknot stem IIA/B are
absent as has been also described for the B. subtilis glyQS T-box [278].
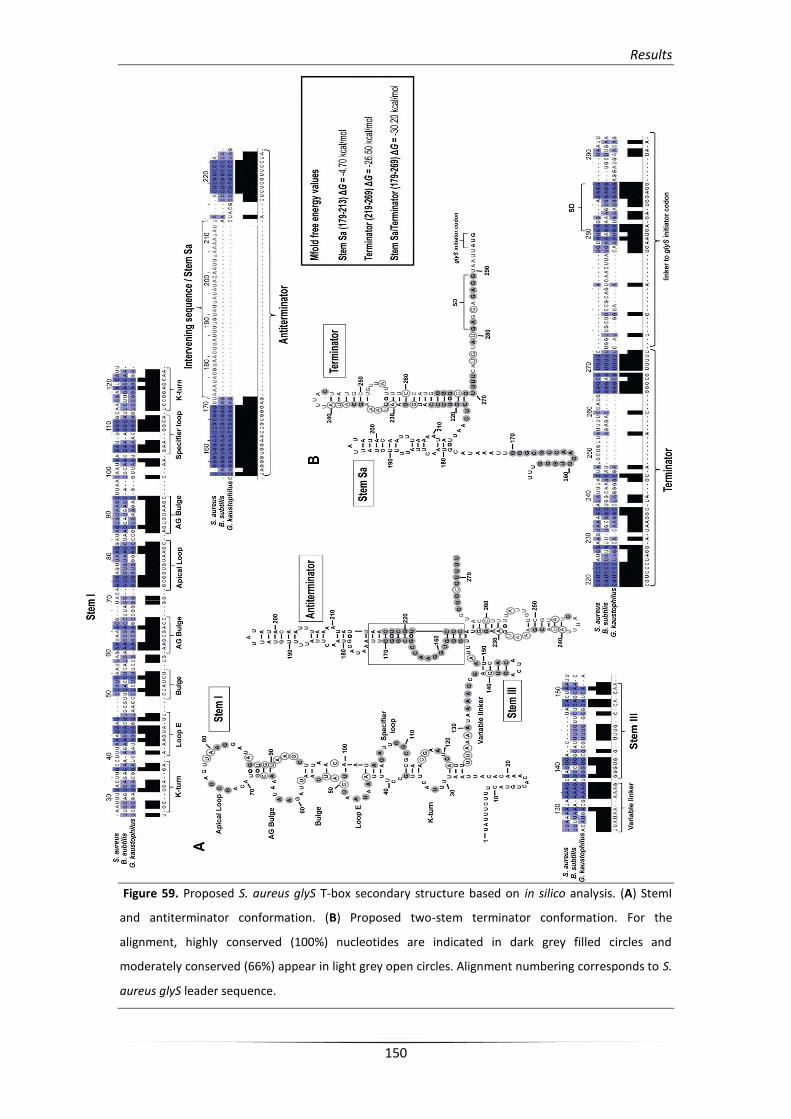
Results
150
Figure 59. Proposed S. aureus glyS T-box secondary structure based on in silico analysis. (A) StemI
and antiterminator conformation. (B) Proposed two-stem terminator conformation. For the
alignment, highly conserved (100%) nucleotides are indicated in dark grey filled circles and
moderately conserved (66%) appear in light grey open circles. Alignment numbering corresponds to S.
aureus glyS leader sequence.
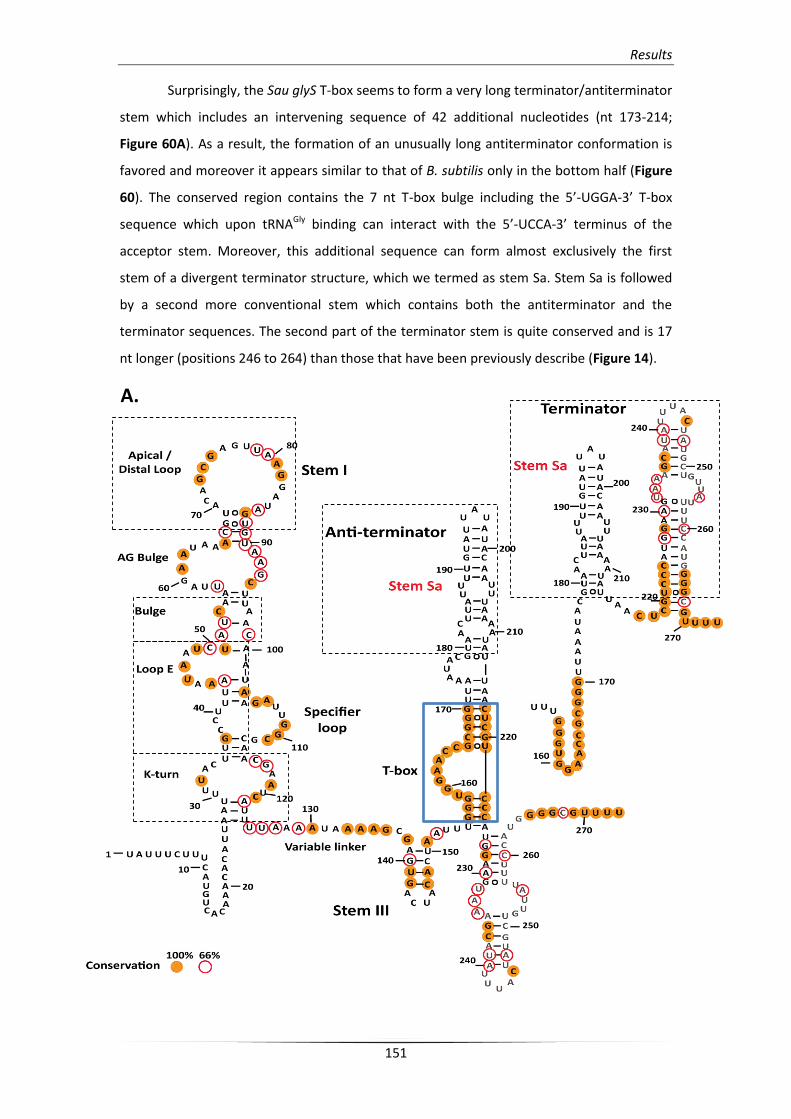
Results
151
Surprisingly, the Sau glyS T-box seems to form a very long terminator/antiterminator
stem which includes an intervening sequence of 42 additional nucleotides (nt 173-214;
Figure 60A). As a result, the formation of an unusually long antiterminator conformation is
favored and moreover it appears similar to that of B. subtilis only in the bottom half (Figure
60). The conserved region contains the 7 nt T-box bulge including the 5’-UGGA-3’ T-box
sequence which upon tRNAGly binding can interact with the 5’-UCCA-3’ terminus of the
acceptor stem. Moreover, this additional sequence can form almost exclusively the first
stem of a divergent terminator structure, which we termed as stem Sa. Stem Sa is followed
by a second more conventional stem which contains both the antiterminator and the
terminator sequences. The second part of the terminator stem is quite conserved and is 17
nt longer (positions 246 to 264) than those that have been previously describe (Figure 14).
A.
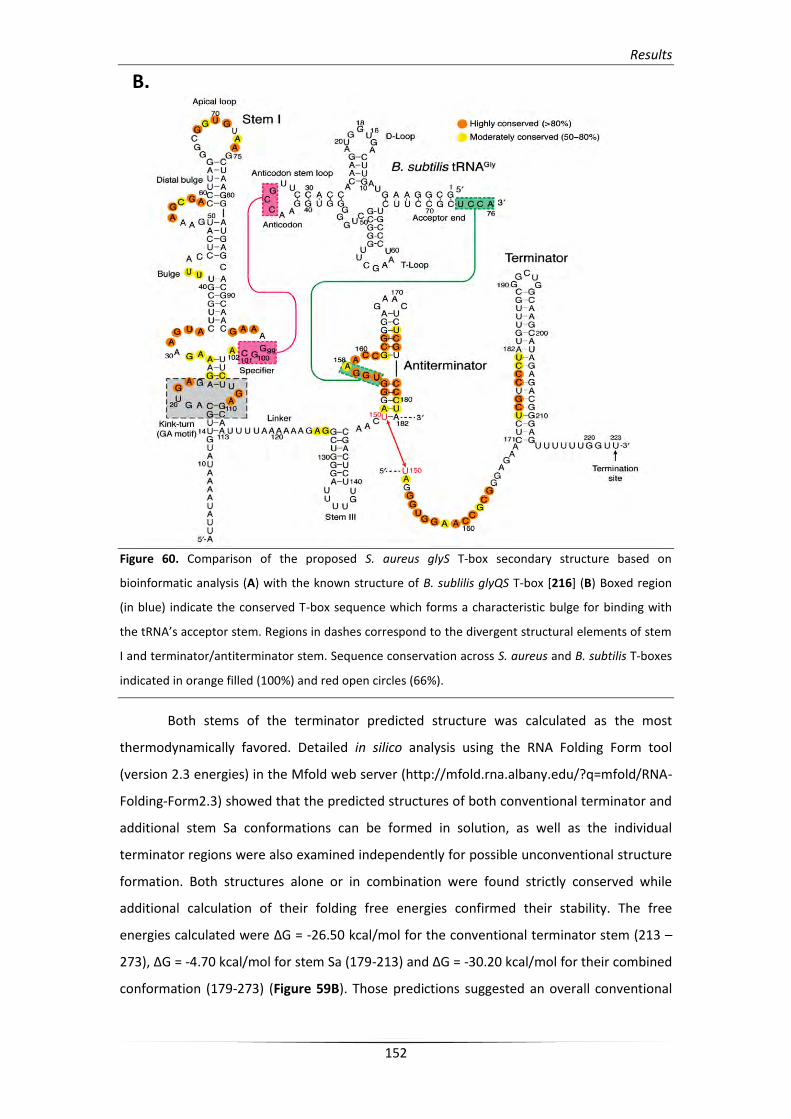
Results
152
Figure 60. Comparison of the proposed S. aureus glyS T-box secondary structure based on
bioinformatic analysis (A) with the known structure of B. sublilis glyQS T-box [216] (B) Boxed region
(in blue) indicate the conserved T-box sequence which forms a characteristic bulge for binding with
the tRNA’s acceptor stem. Regions in dashes correspond to the divergent structural elements of stem
I and terminator/antiterminator stem. Sequence conservation across S. aureus and B. subtilis T-boxes
indicated in orange filled (100%) and red open circles (66%).
Both stems of the terminator predicted structure was calculated as the most
thermodynamically favored. Detailed in silico analysis using the RNA Folding Form tool
(version 2.3 energies) in the Mfold web server (http://mfold.rna.albany.edu/?q=mfold/RNA-
Folding-Form2.3) showed that the predicted structures of both conventional terminator and
additional stem Sa conformations can be formed in solution, as well as the individual
terminator regions were also examined independently for possible unconventional structure
formation. Both structures alone or in combination were found strictly conserved while
additional calculation of their folding free energies confirmed their stability. The free
energies calculated were ΔG = -26.50 kcal/mol for the conventional terminator stem (213 –
273), ΔG = -4.70 kcal/mol for stem Sa (179-213) and ΔG = -30.20 kcal/mol for their combined
conformation (179-273) (Figure 59B). Those predictions suggested an overall conventional
B.
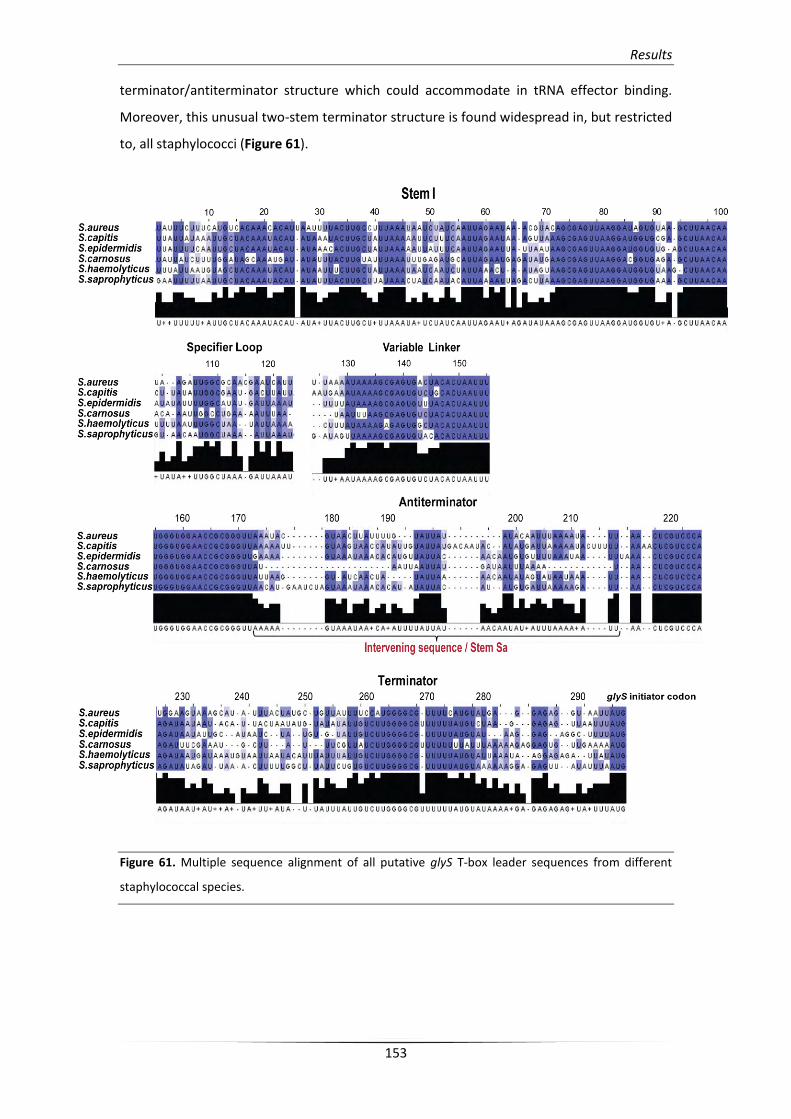
Results
153
terminator/antiterminator structure which could accommodate in tRNA effector binding.
Moreover, this unusual two-stem terminator structure is found widespread in, but restricted
to, all staphylococci (Figure 61).
Figure 61. Multiple sequence alignment of all putative glyS T-box leader sequences from different
staphylococcal species.

Results
154
3.2. In vitro verification of the predicted structural elements
For further verification of the in silico predicted Sau glyS T-box secondary structure
three different structural variants were constructed (Methods §2.3.1.). The first construct
(termed T275) included the whole sequence upstream the glyS gene (position +1 to position
+275). The second variant (termed T225) included part of the full length T-box which is
predicted for antiterminator conformation (position +10 to position +225). The third
construct was a truncated version of stem I without the K-turn motif (position +34 to
position +115) and was termed T115. T275 variant was cloned in pUC57 while T225 and
T115 were cloned in pSC-A vector under the control of a T7 promoter (Figure 62). An
additional terminal restriction enzyme recognition site (BstNI for T275 construct and SalI for
T225 and T115 constructs) was designed for in vitro transcription template preparation
(Methods §2.3.2.).
Figure 62. Cloning of S. aureus glyS T-box structural variants. T275 construct were cloned into pUC57
vector while T225 and T115 into pSC-A vector. In each construct a T7 promoter and a restriction
enzyme recognition site were inserted at the 5’ and 3’ end respectively. Lanes 1, 2 and 3 indicate
the amplified T275, T225 and T115 sequences. L corresponds to molecular weight DNA ladder.
Both T225 and T115 transcripts were purified on PAGE, after in vitro transcription
under optimized conditions. T275 construct was purified using a gel filtration
chromatography column (Methods §2.3.3.). All constructs had very good yield during
preparation (40 to 60 μM; Figure 63) and appeared intact and without any degradation
traces by random ribonuclease activity. All the constructs were used for subsequent
experimentation.
pSC-A
T7pr-T225/T115
SalI
L 1 2 3
300 bp
200 bp
100 bp T115
T225
T275
pUC57
T7pr-T275
BstNI
T275 T225 T115
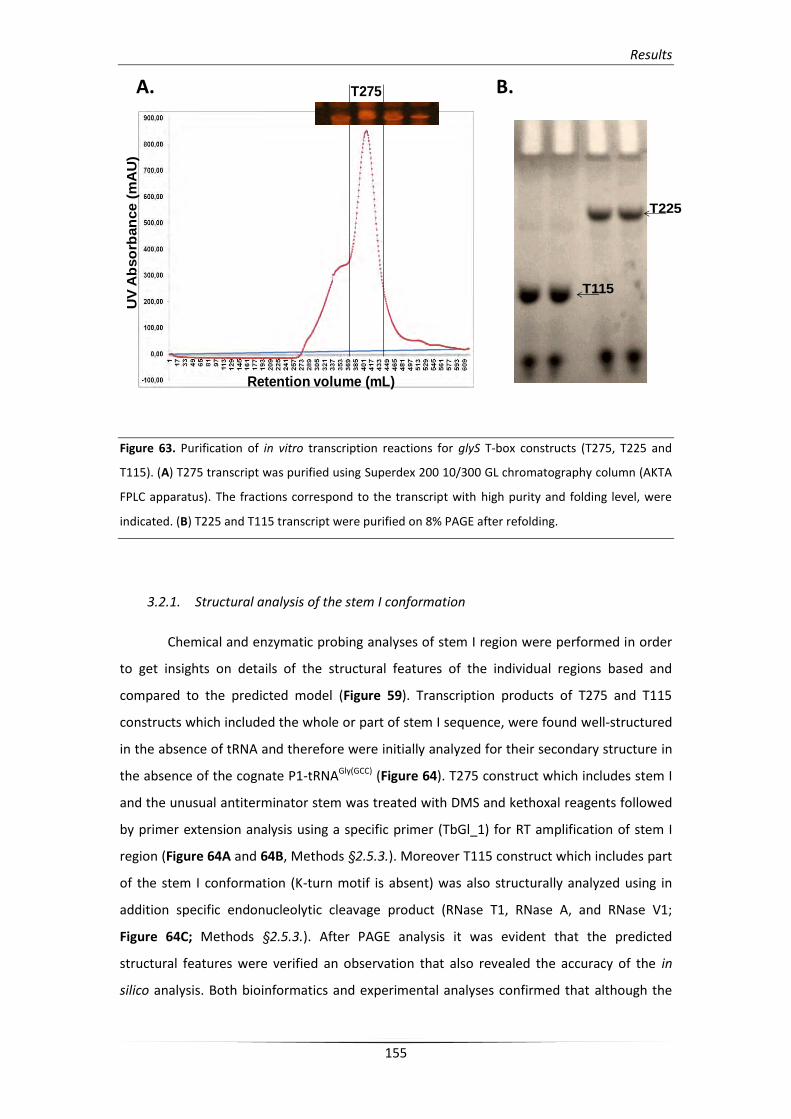
Results
155
Figure 63. Purification of in vitro transcription reactions for glyS T-box constructs (T275, T225 and
T115). (A) T275 transcript was purified using Superdex 200 10/300 GL chromatography column (AKTA
FPLC apparatus). The fractions correspond to the transcript with high purity and folding level, were
indicated. (B) T225 and T115 transcript were purified on 8% PAGE after refolding.
3.2.1. Structural analysis of the stem I conformation
Chemical and enzymatic probing analyses of stem I region were performed in order
to get insights on details of the structural features of the individual regions based and
compared to the predicted model (Figure 59). Transcription products of T275 and T115
constructs which included the whole or part of stem I sequence, were found well-structured
in the absence of tRNA and therefore were initially analyzed for their secondary structure in
the absence of the cognate P1-tRNAGly(GCC) (Figure 64). T275 construct which includes stem I
and the unusual antiterminator stem was treated with DMS and kethoxal reagents followed
by primer extension analysis using a specific primer (TbGl_1) for RT amplification of stem I
region (Figure 64A and 64B, Methods §2.5.3.). Moreover T115 construct which includes part
of the stem I conformation (K-turn motif is absent) was also structurally analyzed using in
addition specific endonucleolytic cleavage product (RNase T1, RNase A, and RNase V1;
Figure 64C; Methods §2.5.3.). After PAGE analysis it was evident that the predicted
structural features were verified an observation that also revealed the accuracy of the in
silico analysis. Both bioinformatics and experimental analyses confirmed that although the
UV
Ab
so
rban
ce (
mA
U)
Retention volume (mL)
T275
T225
T115
A. B.
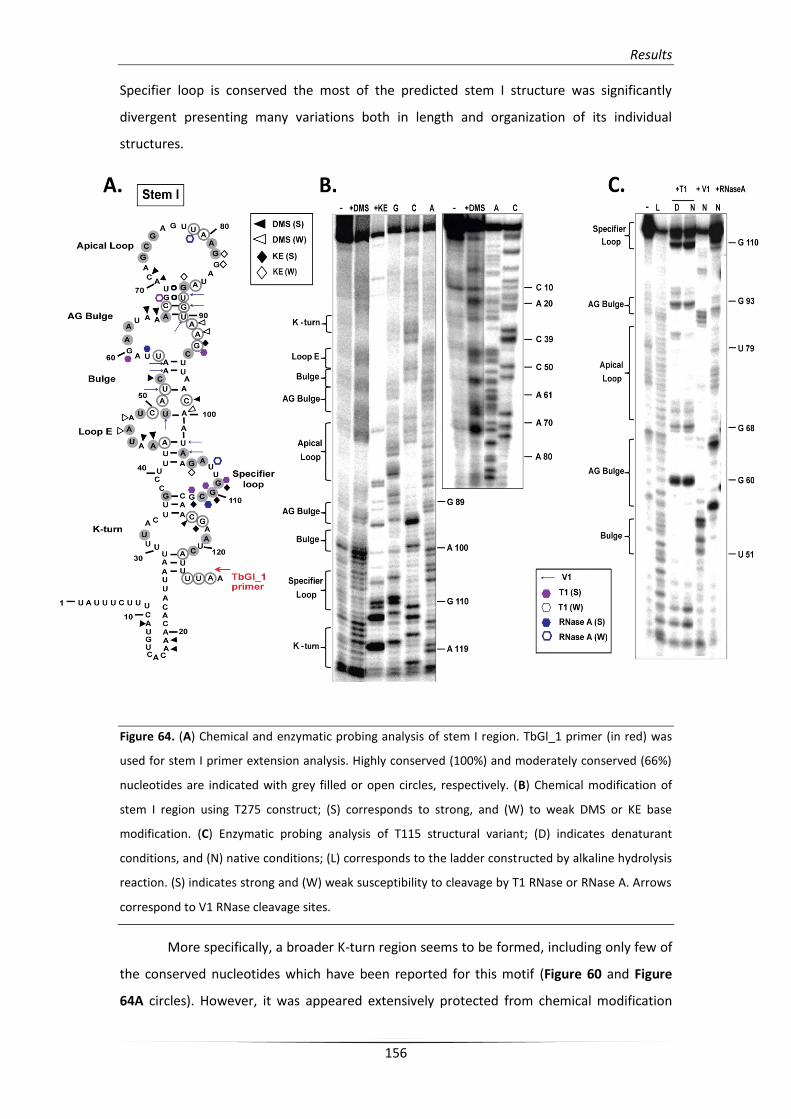
Results
156
Specifier loop is conserved the most of the predicted stem I structure was significantly
divergent presenting many variations both in length and organization of its individual
structures.
Figure 64. (A) Chemical and enzymatic probing analysis of stem I region. TbGl_1 primer (in red) was
used for stem I primer extension analysis. Highly conserved (100%) and moderately conserved (66%)
nucleotides are indicated with grey filled or open circles, respectively. (B) Chemical modification of
stem I region using T275 construct; (S) corresponds to strong, and (W) to weak DMS or KE base
modification. (C) Enzymatic probing analysis of T115 structural variant; (D) indicates denaturant
conditions, and (N) native conditions; (L) corresponds to the ladder constructed by alkaline hydrolysis
reaction. (S) indicates strong and (W) weak susceptibility to cleavage by T1 RNase or RNase A. Arrows
correspond to V1 RNase cleavage sites.
More specifically, a broader K-turn region seems to be formed, including only few of
the conserved nucleotides which have been reported for this motif (Figure 60 and Figure
64A circles). However, it was appeared extensively protected from chemical modification

Results
157
with an exception in C116 and G117 positions even if does not include the typical
arrangement that has been previously described [213, 215]. Therefore, partly conserved
nucleotides that constitute the region (i.e. U32/A33 and A119/U120 cross interaction) can
be favor the formation of the necessary K-turn bent into the helical path. In addition, the K-
turn motif that was detected does not contain a typical GA motif as for the majority of the
organisms in Firmicutes phylum (widespread in Bacillus and Clostridium T-box leaders;
[201]). Although K-turn is presumably required for in vivo antitermination its distortion does
not essentially affect the tRNA binding ability of stem I in vitro, as it is also observed via
EMSA analysis (Results §4.1.).
Another striking difference is the displaced loop E. Positions A44 and A45 (at its
initial part) appear exposed and more modification-prone than positions A47 and A48. Loop
E is responsible for an S-turn which contributes to helical formation of stem I and as a result
can expose the SL element for interaction with the tRNA’s anticodon. In S. aureus glyS stem
I, the S-turn can be formed via interactions with the A101 which was found protected by
DMS modification. Most of the E-loop region includes conserved nucleotides (A44, U46, A47
and U49) followed by a short bulge which contains the conserved C54. Stem I structure
continues with a wider AG bulge which contains a 9 nt loop instead of 8 nt in Bacilli.
Moreover, the AG bulge contains the conserved A61, A62, and A66 nucleotides and was
observed resistant to endonucleolytic cleavage. The A62 appears protected from either
chemical modification or enzymatic digestion and can potentially interact with A64 or A65,
since the conserved A66 was found paired with U90. This interaction is essential, as has been
previously observed and can bring the AG bulge in proximity to the apical loop (A72-A76) in
the absence of tRNA [214, 215].
The distal loop is also broader compared to the 11 nt apical loop in bacilli, spanning
from A70 to A86 (17 nt), and exhibits different arrangement in terms of sequence. On the
other hand, it includes conserved nucleotides in positions G73, C74, G75, A81, and G82. The
latest is the only nucleotide that is found susceptible to modification. This observation
suggests that the apical loop can probably form long-range interactions with the AG bulge
and eventually shape the necessary conformation to interact with tRNA’s elbow (D-loop and
T-loop). This specific interaction is used as an additional checkpoint which maintains the
specificity of structure recognition of the tRNA ligand and moreover seems to accommodate
the subsequent antiterminator conformation fold [214, 215]. Moreover, both chemical
modification and enzymatic probing analyses using the T115 variant indicated that a possible
interaction can be formed between G77 and A81 nucleotides and that the region A72-A81 is
inaccessible to modifying agents and enzymatic cleavage (Figure 64C). As a result, this region

Results
158
appears extensively protected more likely because of a possible interaction with the AG
bulge. These findings reconcile the available structural data and in addition, both the
observed differences in the AG bulge and the distal loop support the previously described
ambiguity over the exact interactions that take place between the upper part of stem I and
the tRNA ligand [215, 279]. Most of the SL region appears exposed containing the G109,
G110 and C111 codon sequence which is able to interact with the corresponding anticodon
sequence of the P1-tRNAGly(GCC) isoacceptor. The codon sequence is followed by the
conserved purine (G112) which is characteristic of all T-boxes, and is also crucial for the
interaction with the anticodon loop of the tRNA.
3.2.2. Structural analysis of the unusual terminator/antiterminator stem
The predicted Sau glyS T-box terminator/antiterminator region was further
experimentally verified taking into account the unusual 42-nt intervening sequence (nt 173-
214) that exists between the conserved T-box sequence (nt 156-170) and the well conserved
terminator sequence (nt 219-269) (Figure 60). This intervening sequence that is termed as
stem Sa forms an additional to the terminator stem conformation (Figure 65A). Attempts to
successfully map both stems using a specific primer which is designed to anneal at the 3’ end
of the terminator stem (nt 252-269) failed possibly due to the complexity of the region.
The secondary structure of the terminator stem was initially obtained using the
Mfold software, which predicts the most thermodynamic favorable conformation via
calculation of the free energy value based on the conservation of the region (Figure 59;
Methods §2.4.2.). After modification of the T275 variant with DMS and kethoxal, followed by
primer extension using a specific primer (TbGl_6), designed against the short intergenic
region between stem Sa and the terminator stem, the predicted model was also verified
(Methods §2.5.3.). In the absence of the tRNA ligand, the intervening sequence forms a very
rigid stem which exhibits limited accessibility to modification. In the same line of
experiments the secondary structure of part of the intergenic sequence between the stem I
and the terminator/antiterminator region were also confirmed. This region includes the
stem III structural element which appears almost inaccessible to modifying agents (Figure
65A).
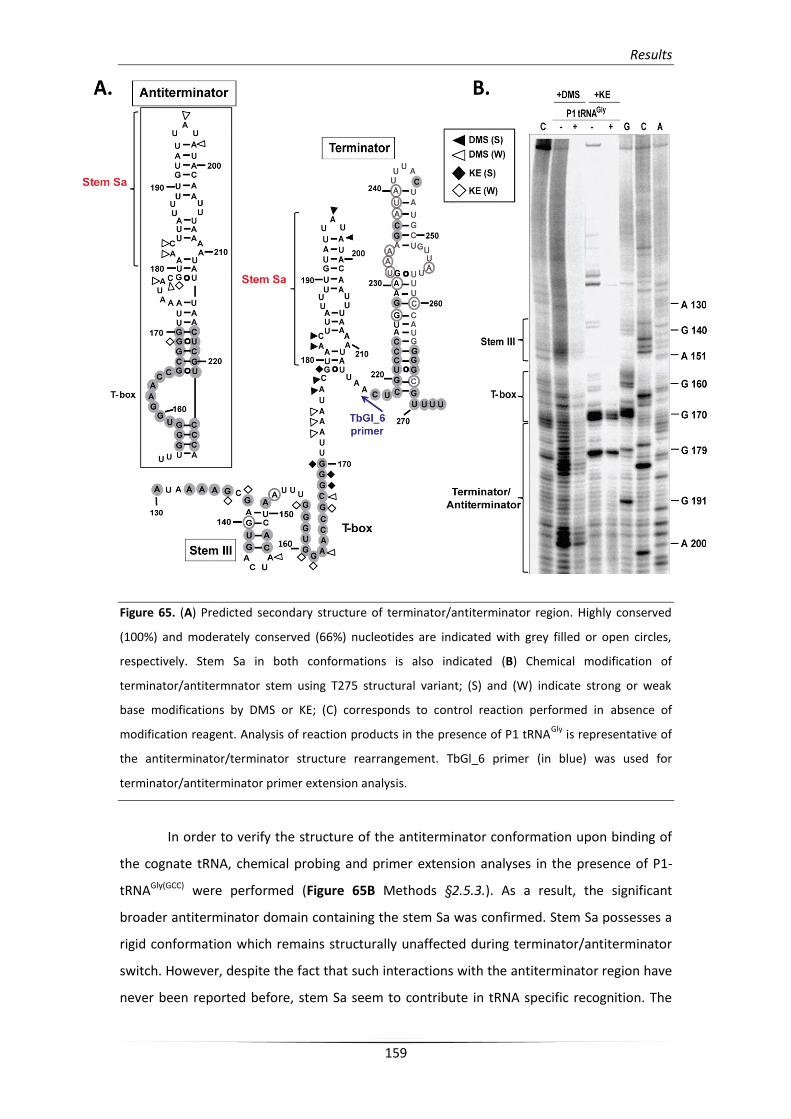
Results
159
Figure 65. (A) Predicted secondary structure of terminator/antiterminator region. Highly conserved
(100%) and moderately conserved (66%) nucleotides are indicated with grey filled or open circles,
respectively. Stem Sa in both conformations is also indicated (B) Chemical modification of
terminator/antitermnator stem using T275 structural variant; (S) and (W) indicate strong or weak
base modifications by DMS or KE; (C) corresponds to control reaction performed in absence of
modification reagent. Analysis of reaction products in the presence of P1 tRNAGly
is representative of
the antiterminator/terminator structure rearrangement. TbGl_6 primer (in blue) was used for
terminator/antiterminator primer extension analysis.
In order to verify the structure of the antiterminator conformation upon binding of
the cognate tRNA, chemical probing and primer extension analyses in the presence of P1-
tRNAGly(GCC) were performed (Figure 65B Methods §2.5.3.). As a result, the significant
broader antiterminator domain containing the stem Sa was confirmed. Stem Sa possesses a
rigid conformation which remains structurally unaffected during terminator/antiterminator
switch. However, despite the fact that such interactions with the antiterminator region have
never been reported before, stem Sa seem to contribute in tRNA specific recognition. The

Results
160
predicted model suggested that the antiterminator region can presumably shape a helix turn
in its upper part which accommodates this specific interaction and it will be discussed below
(Results§ 4.4). Moreover, the nucleotides between positions A173 and C183 clearly undergo
conformational changes which stabilize the structure of the T-box upon tRNA effector
binding (Figure 65A). Finally, these observations suggest a potential involvement of the
intervening sequence in additional tRNA:antiterminator interactions. However, such
interactions were difficult to detect because of the increased flexibility that is favored by the
unusual length of the terminator/antiterminator region.
4. Analysis of the regulatory role of S. aureus glyS T-box riboswitch
4.1. S. aureus glyS T-box interacts with all tRNAGly isoacceptors
As it has been previously described, all the available S. aureus genomes encode
genes for five different tRNAGly isoacceptors, two proteinogenic (termed as P1 and P2) and
three non-proteinogenic (termed as NP1, NP2, and NEW) (Figure 50A and 50B). Based on
their affinity for binding to the homologous EF-Tu, each tRNAGly isoacceptor can potentially
serve different roles during the S. aureus lifecycle. P1 and P2 proteinogenic isoacceptors,
bearing a GCC and a UCC anticodon respectively, can participate in ribosomal protein
synthesis. However, all three non-proteinogenic isoacceptors, bearing UCC anticodons, after
their aminoacylation can potentially escape protein synthesis due to their weak binding to
EF-Tu, and can serve as substrates for the pentaglycine interpeptide formation that stabilizes
the 3D cell wall structure [228].
As discussed in previous section (Results §2.1.) P1-tRNAGly(GCC) is the only tRNAGly that
can fulfill the T-box SL codon:tRNA anticodon pairing. The P2-tRNAGly(UCC) isoacceptor, which
bear all the conserved nucleotides for D-loop and T-loop recognition and differ only in
wobble position 34, cannot be excluded for interaction with the T-box region. The remaining
three non-proteinogenic isoacceptors appear many variations in the anticodon loop, the D-
arm/–loop and the T-arm/-loop sequence and can be used as controls. Therefore, all five
tRNAGly species were tested for binding to each of glyS T-box structural constructs via EMSA
analysis (Methods §2.5.2.). Prior of being used in gel shift assays tRNAGly isoacceptors were
in vitro transcribed and were purified under optimized conditions via gel filtration
chromatography in order to avoid unfavorable conformations (like potential bulky dimmer
conformations) that could reduce the level of the appropriate folding (Methods §2.3.3.)
(Figure 66).
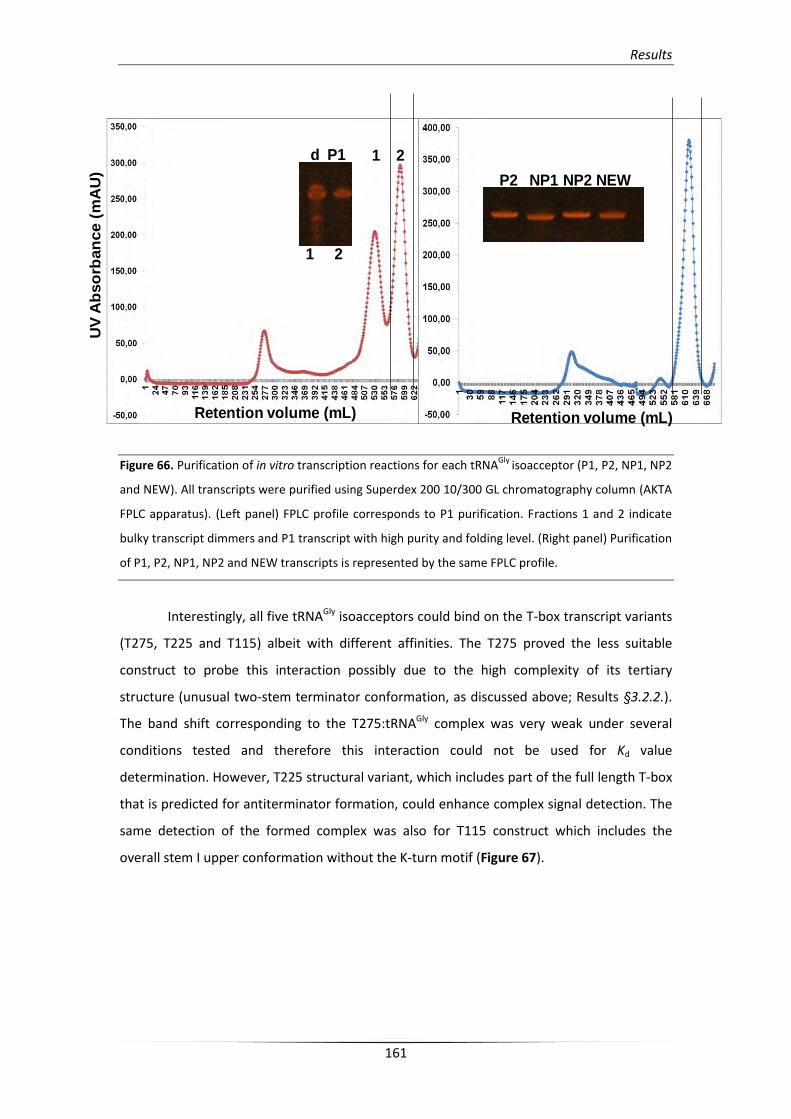
Results
161
Figure 66. Purification of in vitro transcription reactions for each tRNAGly
isoacceptor (P1, P2, NP1, NP2
and NEW). All transcripts were purified using Superdex 200 10/300 GL chromatography column (AKTA
FPLC apparatus). (Left panel) FPLC profile corresponds to P1 purification. Fractions 1 and 2 indicate
bulky transcript dimmers and P1 transcript with high purity and folding level. (Right panel) Purification
of P1, P2, NP1, NP2 and NEW transcripts is represented by the same FPLC profile.
Interestingly, all five tRNAGly isoacceptors could bind on the T-box transcript variants
(T275, T225 and T115) albeit with different affinities. The T275 proved the less suitable
construct to probe this interaction possibly due to the high complexity of its tertiary
structure (unusual two-stem terminator conformation, as discussed above; Results §3.2.2.).
The band shift corresponding to the T275:tRNAGly complex was very weak under several
conditions tested and therefore this interaction could not be used for Kd value
determination. However, T225 structural variant, which includes part of the full length T-box
that is predicted for antiterminator formation, could enhance complex signal detection. The
same detection of the formed complex was also for T115 construct which includes the
overall stem I upper conformation without the K-turn motif (Figure 67).
UV
Ab
so
rban
ce (
mA
U)
Retention volume (mL) Retention volume (mL)
1 2d P1
1 2
P2 NP1 NP2 NEW
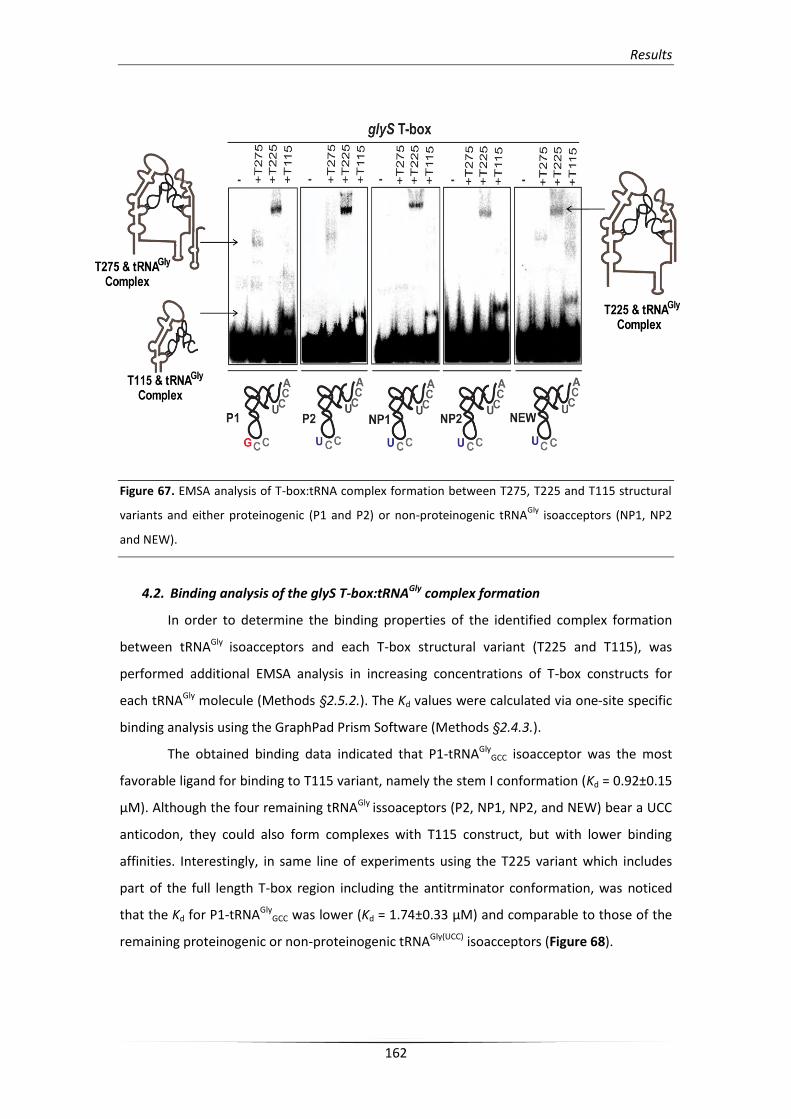
Results
162
Figure 67. EMSA analysis of T-box:tRNA complex formation between T275, T225 and T115 structural
variants and either proteinogenic (P1 and P2) or non-proteinogenic tRNAGly
isoacceptors (NP1, NP2
and NEW).
4.2. Binding analysis of the glyS T-box:tRNAGly complex formation
In order to determine the binding properties of the identified complex formation
between tRNAGly isoacceptors and each T-box structural variant (T225 and T115), was
performed additional EMSA analysis in increasing concentrations of T-box constructs for
each tRNAGly molecule (Methods §2.5.2.). The Kd values were calculated via one-site specific
binding analysis using the GraphPad Prism Software (Methods §2.4.3.).
The obtained binding data indicated that P1-tRNAGlyGCC isoacceptor was the most
favorable ligand for binding to T115 variant, namely the stem I conformation (Kd = 0.92±0.15
μΜ). Although the four remaining tRNAGly issoaceptors (P2, NP1, NP2, and NEW) bear a UCC
anticodon, they could also form complexes with T115 construct, but with lower binding
affinities. Interestingly, in same line of experiments using the T225 variant which includes
part of the full length T-box region including the antitrminator conformation, was noticed
that the Kd for P1-tRNAGlyGCC was lower (Kd = 1.74±0.33 μΜ) and comparable to those of the
remaining proteinogenic or non-proteinogenic tRNAGly(UCC) isoacceptors (Figure 68).
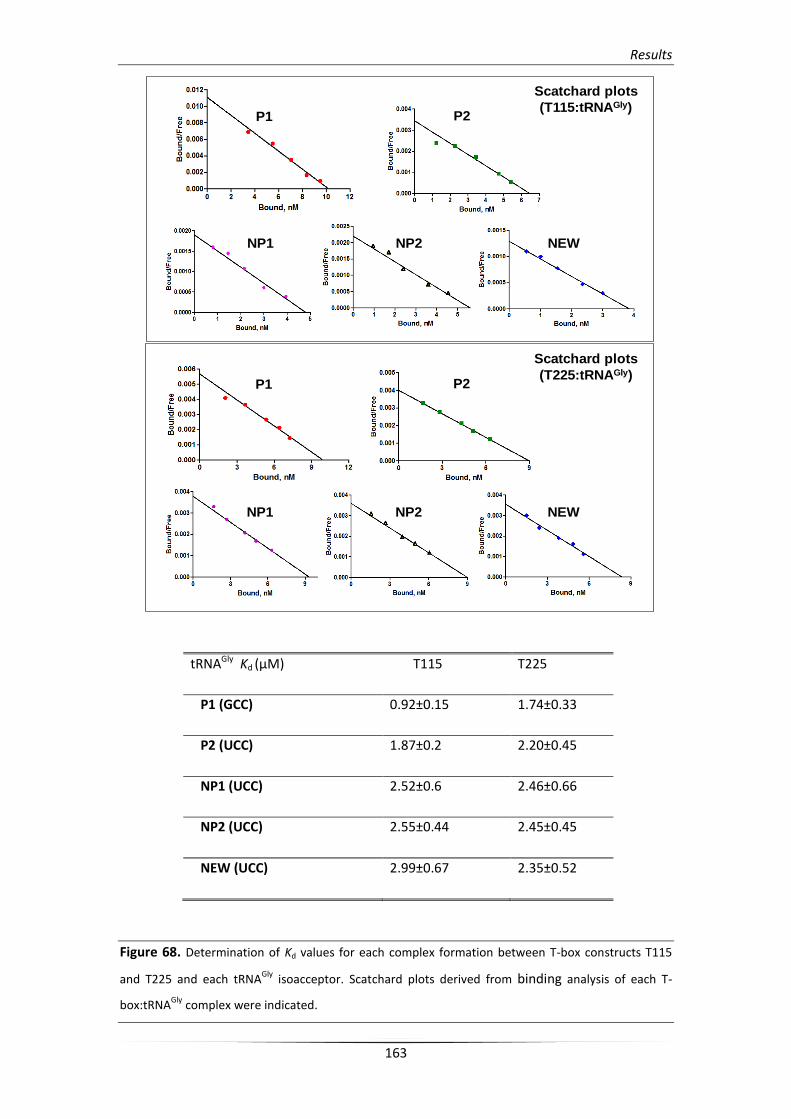
Results
163
tRNAGly Kd (μM) T115 T225
P1 (GCC) 0.92±0.15 1.74±0.33
P2 (UCC) 1.87±0.2 2.20±0.45
NP1 (UCC) 2.52±0.6 2.46±0.66
NP2 (UCC) 2.55±0.44 2.45±0.45
NEW (UCC) 2.99±0.67 2.35±0.52
Figure 68. Determination of Kd values for each complex formation between T-box constructs T115
and T225 and each tRNAGly
isoacceptor. Scatchard plots derived from binding analysis of each T-
box:tRNAGly
complex were indicated.
P1 P2
NP1 NP2 NEW
Scatchard plots
(T225:tRNAGly)
P1 P2
NP1 NP2 NEW
Scatchard plots
(T115:tRNAGly)
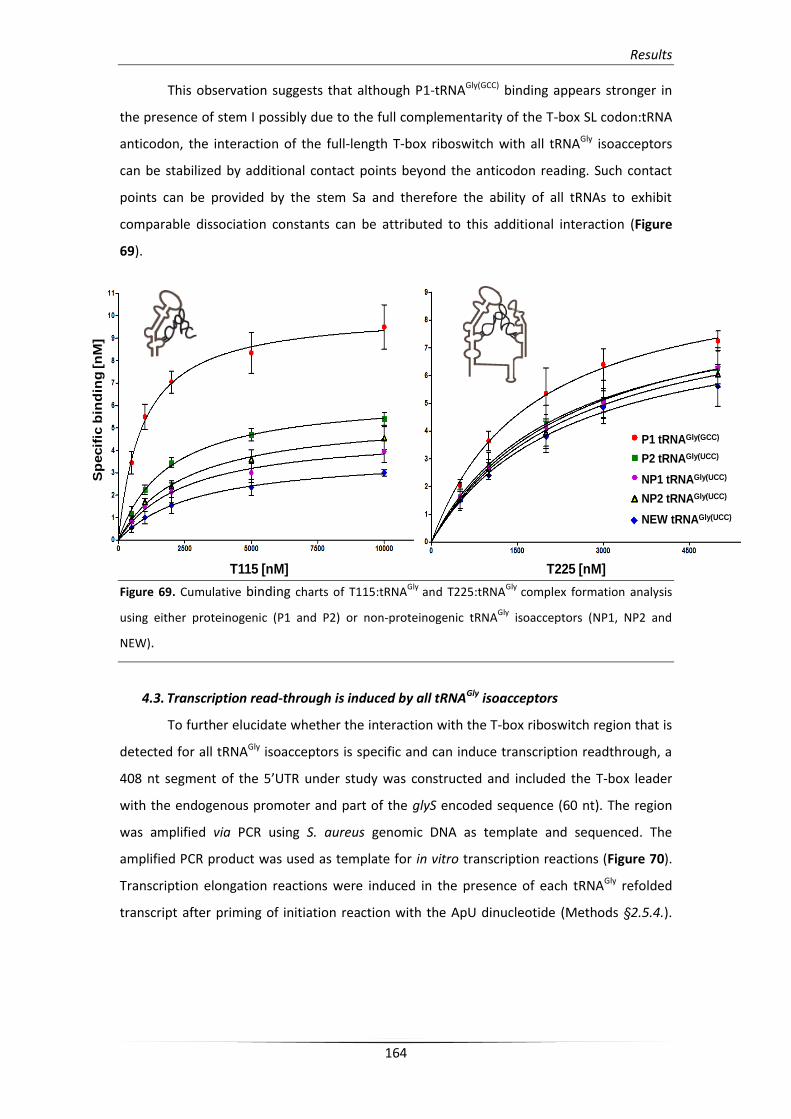
Results
164
This observation suggests that although P1-tRNAGly(GCC) binding appears stronger in
the presence of stem I possibly due to the full complementarity of the T-box SL codon:tRNA
anticodon, the interaction of the full-length T-box riboswitch with all tRNAGly isoacceptors
can be stabilized by additional contact points beyond the anticodon reading. Such contact
points can be provided by the stem Sa and therefore the ability of all tRNAs to exhibit
comparable dissociation constants can be attributed to this additional interaction (Figure
69).
Figure 69. Cumulative binding charts of T115:tRNAGly
and T225:tRNAGly
complex formation analysis
using either proteinogenic (P1 and P2) or non-proteinogenic tRNAGly
isoacceptors (NP1, NP2 and
NEW).
4.3. Transcription read-through is induced by all tRNAGly isoacceptors
To further elucidate whether the interaction with the T-box riboswitch region that is
detected for all tRNAGly isoacceptors is specific and can induce transcription readthrough, a
408 nt segment of the 5’UTR under study was constructed and included the T-box leader
with the endogenous promoter and part of the glyS encoded sequence (60 nt). The region
was amplified via PCR using S. aureus genomic DNA as template and sequenced. The
amplified PCR product was used as template for in vitro transcription reactions (Figure 70).
Transcription elongation reactions were induced in the presence of each tRNAGly refolded
transcript after priming of initiation reaction with the ApU dinucleotide (Methods §2.5.4.).
T115 [nM] T225 [nM]
P1 tRNAGly(GCC)
P2 tRNAGly(UCC)
NP1 tRNAGly(UCC)
NP2 tRNAGly(UCC)
NEW tRNAGly(UCC)
Sp
ecif
ic b
ind
ing
[n
M]

Results
165
Figure 70. 5’UTR T-box leader sequence including the endogenous promoter and part of the glyS
encoded sequence (boxed regions). The amplified region was used as template for in vitro
transcription reactions. Arrows indicate the predicted transcription termination (+273) and
readthrough (+354) sites.
Interestingly, all tRNAGly isoacceptors could adequately induce RNA polymerase
transcription readthrough (RT; 354 nt) overcoming the terminator conformation (T; 273 nt).
Although the percentage of the transcription readthrough (%RT) differed between
elongation reactions of each tRNAGly isoacceptor it was time-dependent and the effect was
independent of the anticodon sequence that each tRNA had. The maximum (%RT) was
recorded for the P1-tRNAGly(GCC) (19%) which was significant lower than those that obtained
for in vitro readthrough reactions from other glyQS T-boxes. The tRNAGly(UCC) isoacceptors
revealed lower (%RT) than P1 tRNA but also notable. P2 and NP1 exhibited the highest
recorded value (15-16%) while NP2 and NEW exhibited lower (12% and 9% respectively).
However, all tRNAGly isoacceptors can equal efficiently bind to the whole T-box region as
revealed from the kinetic analyses of the formed complex between T225 and each
isoacceptor (Results §4.2.) (Figure 71).
In addition, the very strong termination conformation signal (T) that was observed
suggests that the Sau glyS T-box can behave in a different manner compared to its
previously characterized counterpart from B. subtilis, possibly due to the very rigid two-stem
termination conformation. Furthermore, this unusual conformation could also explain the
lower readthrough band volume (RT) or the additional intermediate bands that was
observed and possibly suggest the existence of differential conformations which are favored
upon tRNA binding.
L 1
400 bp
300 bp
200 bp
408bp
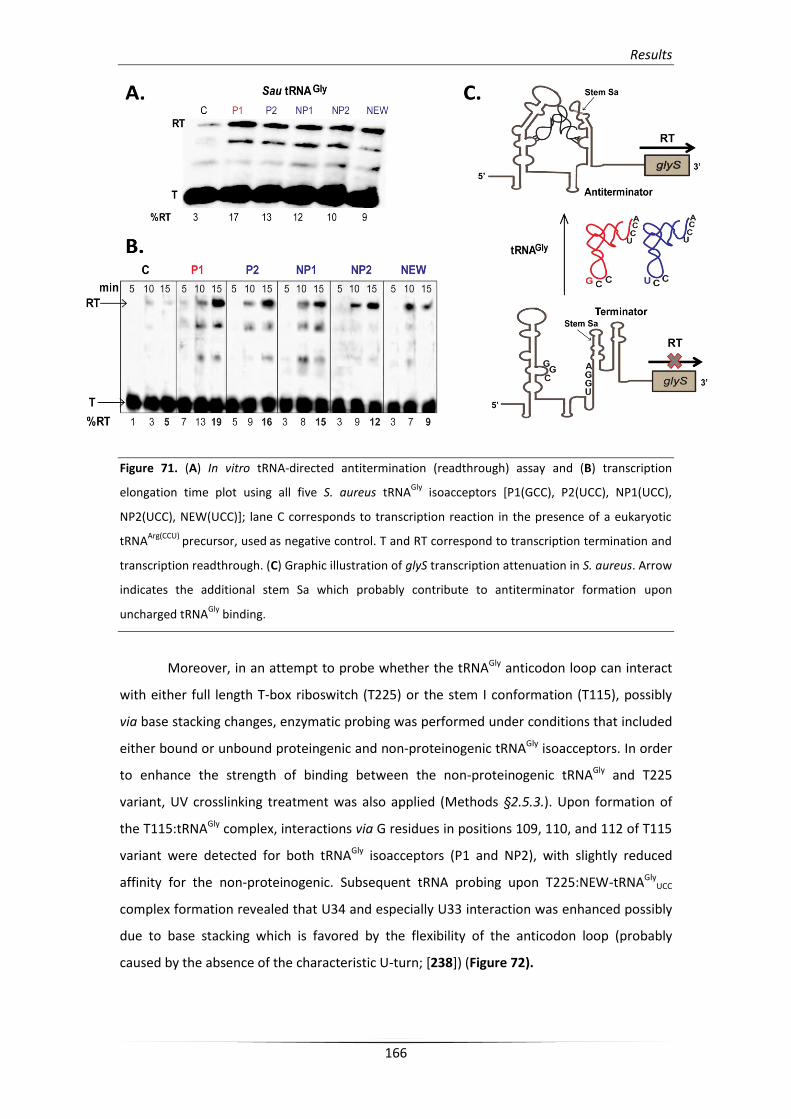
Results
166
Figure 71. (A) In vitro tRNA-directed antitermination (readthrough) assay and (B) transcription
elongation time plot using all five S. aureus tRNAGly
isoacceptors [P1(GCC), P2(UCC), NP1(UCC),
NP2(UCC), NEW(UCC)]; lane C corresponds to transcription reaction in the presence of a eukaryotic
tRNAArg(CCU)
precursor, used as negative control. T and RT correspond to transcription termination and
transcription readthrough. (C) Graphic illustration of glyS transcription attenuation in S. aureus. Arrow
indicates the additional stem Sa which probably contribute to antiterminator formation upon
uncharged tRNAGly
binding.
Moreover, in an attempt to probe whether the tRNAGly anticodon loop can interact
with either full length T-box riboswitch (T225) or the stem I conformation (T115), possibly
via base stacking changes, enzymatic probing was performed under conditions that included
either bound or unbound proteingenic and non-proteinogenic tRNAGly isoacceptors. In order
to enhance the strength of binding between the non-proteinogenic tRNAGly and T225
variant, UV crosslinking treatment was also applied (Methods §2.5.3.). Upon formation of
the T115:tRNAGly complex, interactions via G residues in positions 109, 110, and 112 of T115
variant were detected for both tRNAGly isoacceptors (P1 and NP2), with slightly reduced
affinity for the non-proteinogenic. Subsequent tRNA probing upon T225:NEW-tRNAGlyUCC
complex formation revealed that U34 and especially U33 interaction was enhanced possibly
due to base stacking which is favored by the flexibility of the anticodon loop (probably
caused by the absence of the characteristic U-turn; [238]) (Figure 72).

Results
167
Figure 72. (A) Enzymatic probing of the NEW tRNAGly
:T225 complex. The region illustrated in
corresponds to anticodon loop sequence and RNase A cleavage was performed either under
denaturing (lane D) or native (lane N) conditions, with or without UV crosslink treatment. Lane L
indicates alkaline hydrolysis products (ladder). (B) Enzymatic probing of P1 tRNAGly
:T115 or NP2
tRNAGly
:T115 complexes. The region illustrated in corresponds to SL triplet followed by the conserved
purine (G) of the T115 variant. Lanes (RNA) and (T1 seq) indicate the negative control without T1
RNase cleavage and the T1 RNase sequencing under denaturing conditions used for ladder
construction. T1 RNase probing reactions were performed under native conditions.
Despite the fact that a higher specificity is achieved through the SL:anticodon
complementarity, additional base interactions that were detected for the formed T-
box:tRNAGly complex, in combination with the lower affinity of P1 in the presence of the
T225 variant (Results §4.2.) implies that in the presence of the antiterminator conformation
a more extensive rearrangement which favors anticodon base stacking must exists. Such an
induced fit can accommodate in specificity of tRNA structural recognition and can be
achieved possibly through additional “sealing” by stem Sa conformation. Based on the above
observations a model of the putative mechanism can be proposed and includes additional
interactions of tRNA upper part with stem Sa (Figure 71C).
4.4. Differential glyS T-box structural changes upon tRNAGly binding
The observation that all tRNAGly isoacceptors can bind to the T-box riboswitch region
and in turn can efficiently induce transcription readthrough was further examined for
possible additional interaction specificities in the structural level. For this reason changes in
the pattern of the protected bases in overall T-box structure upon binding of either P1-
tRNAGly(GCC) (exhibiting the highest binding affinity) or NP2-tRNAGly(UCC) which was used as a
representative of non-proteinogenic isoacceptors, were examined after chemical
modification followed by primer extension analysis (Methods §2.4.3.). The detailed analysis
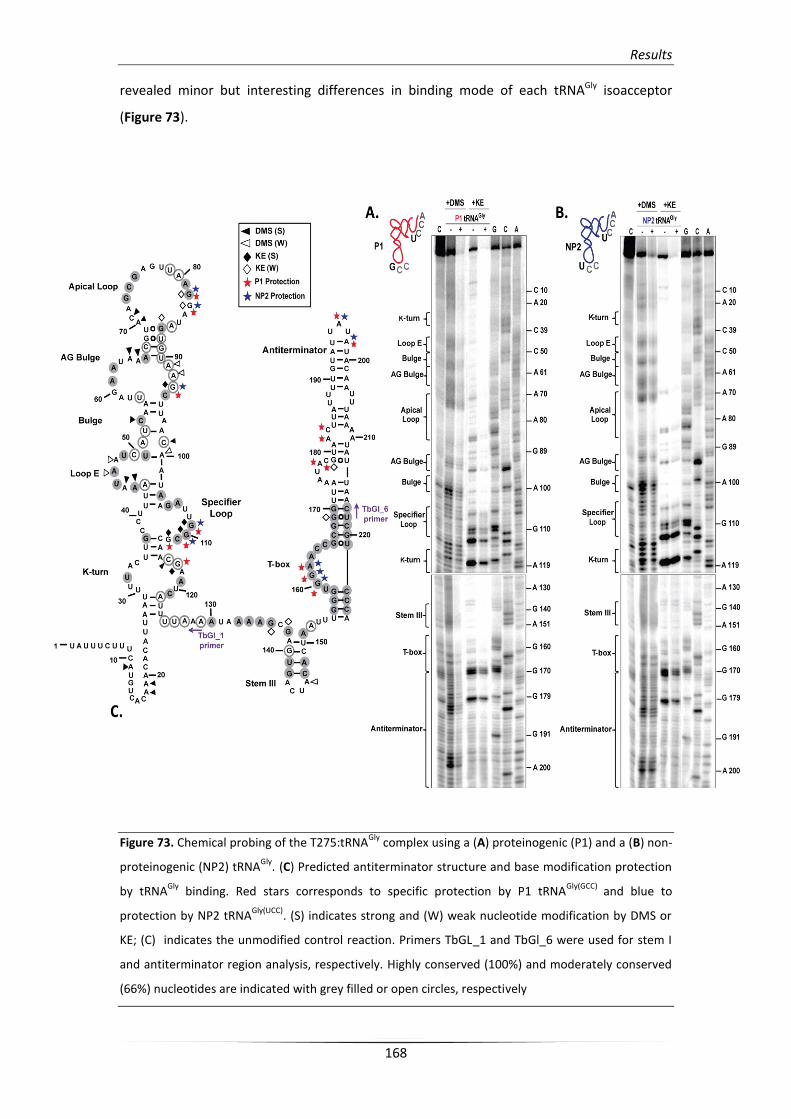
Results
168
revealed minor but interesting differences in binding mode of each tRNAGly isoacceptor
(Figure 73).
Figure 73. Chemical probing of the T275:tRNAGly
complex using a (A) proteinogenic (P1) and a (B) non-
proteinogenic (NP2) tRNAGly
. (C) Predicted antiterminator structure and base modification protection
by tRNAGly
binding. Red stars corresponds to specific protection by P1 tRNAGly(GCC)
and blue to
protection by NP2 tRNAGly(UCC)
. (S) indicates strong and (W) weak nucleotide modification by DMS or
KE; (C) indicates the unmodified control reaction. Primers TbGL_1 and TbGl_6 were used for stem I
and antiterminator region analysis, respectively. Highly conserved (100%) and moderately conserved
(66%) nucleotides are indicated with grey filled or open circles, respectively
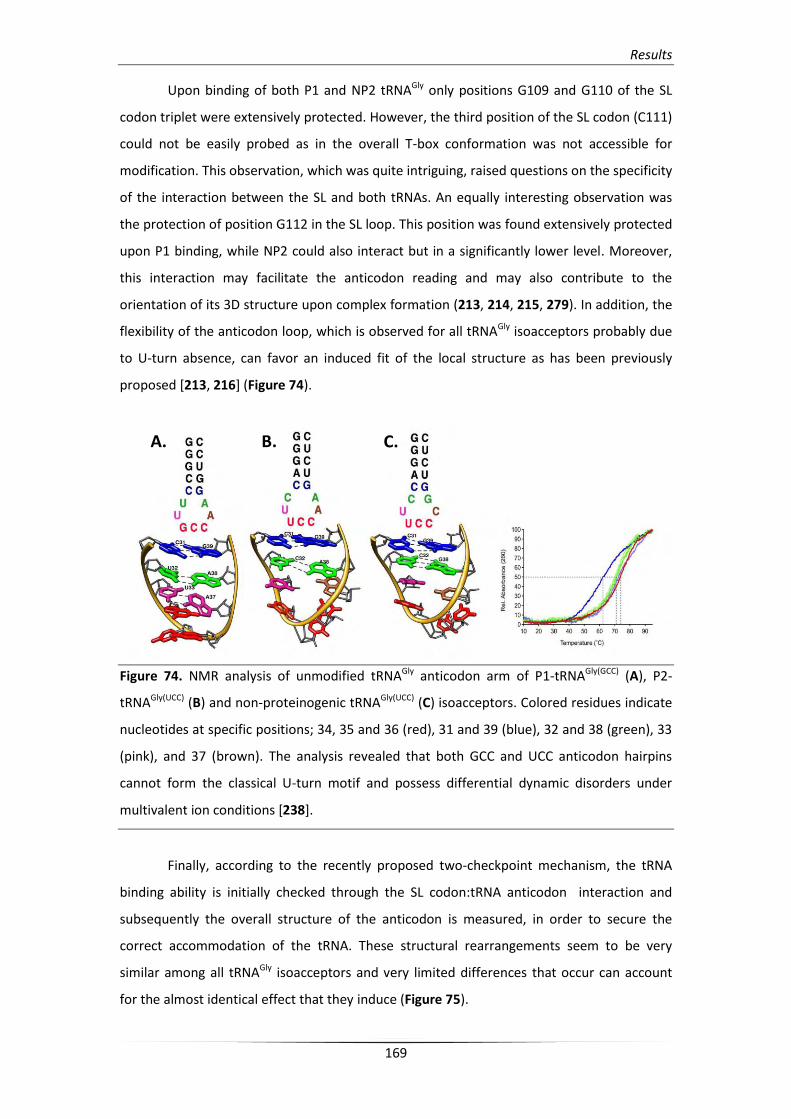
Results
169
Upon binding of both P1 and NP2 tRNAGly only positions G109 and G110 of the SL
codon triplet were extensively protected. However, the third position of the SL codon (C111)
could not be easily probed as in the overall T-box conformation was not accessible for
modification. This observation, which was quite intriguing, raised questions on the specificity
of the interaction between the SL and both tRNAs. An equally interesting observation was
the protection of position G112 in the SL loop. This position was found extensively protected
upon P1 binding, while NP2 could also interact but in a significantly lower level. Moreover,
this interaction may facilitate the anticodon reading and may also contribute to the
orientation of its 3D structure upon complex formation (213, 214, 215, 279). In addition, the
flexibility of the anticodon loop, which is observed for all tRNAGly isoacceptors probably due
to U-turn absence, can favor an induced fit of the local structure as has been previously
proposed [213, 216] (Figure 74).
Figure 74. NMR analysis of unmodified tRNAGly anticodon arm of P1-tRNAGly(GCC) (A), P2-
tRNAGly(UCC) (B) and non-proteinogenic tRNAGly(UCC) (C) isoacceptors. Colored residues indicate
nucleotides at specific positions; 34, 35 and 36 (red), 31 and 39 (blue), 32 and 38 (green), 33
(pink), and 37 (brown). The analysis revealed that both GCC and UCC anticodon hairpins
cannot form the classical U-turn motif and possess differential dynamic disorders under
multivalent ion conditions [238].
Finally, according to the recently proposed two-checkpoint mechanism, the tRNA
binding ability is initially checked through the SL codon:tRNA anticodon interaction and
subsequently the overall structure of the anticodon is measured, in order to secure the
correct accommodation of the tRNA. These structural rearrangements seem to be very
similar among all tRNAGly isoacceptors and very limited differences that occur can account
for the almost identical effect that they induce (Figure 75).
A. B. C.

Results
170
Interaction with both tRNAs could also protect positions G82, G83 and G92 of the
apical loop:AG bulge region (Figure 73). The observed protections indicate possible
interaction with the elbow, namely T- and D-loop, of the tRNA [214, 215]. However, it must
be noted that in contrast with the P1 for NP2 tRNA the G residues in positions 18 and 19 are
replaced by U residues as discussed above (Results §2.2.) (Figure 75). Therefore, protection
by both tRNAs is supportive of a non-specific interaction due to tRNA’s induced fit and is also
in line with the recent crystallographic analyses which show extensive distortion of tRNA
upon association with stem I conformation [215].
Figure 75. Schematic representation of the proposed S. aureus glyS T-box:tRNAGly
recognition model.
(Left panel) S. aureus glyS T-box structural rearrangement upon P1 tRNAGly(GCC)
isoacceptor binding.
Nucleotides in red that are presented on T-box structure indicate additional interactions with P1
isoacceptor, according to the data presented above (see Figure 73). (Right panel) The corresponding
T-box structural rearrangement upon binding of the remaining tRNAGly
UCC isoacceptors. Nucleotides in
grey represent common interaction with either GCC or UCC bearing tRNAs. Colored residues on tRNAs
correspond to major sequence differences among isoacceptors in positions 18, 19, 33-36, 55, 56, 73-
76 (see Figure 56).
Additional probing analysis of the terminator/antiterminator conformation upon
effector binding (P1 or NP2), showed that both tRNAs can stabilize the antiterminator
structural element. The conserved G160, G161 and A162 residues of the T-box bulge region
were extensively protected in both cases as previously reported. Moreover, an interesting

Results
171
observation was the protection of two A residues in positions 196 and 198 of the upper loop
which is formed by the additional stem Sa during antitermination (Figure 73, Figure 75). It
has to be noted that this residues of the stem Sa sequence are highly conserved among all
staphylococcal species (Figure 61).
However, the interaction between the antiterminator stem and the P1 isoacceptor
revealed additional positions that seem to be protected (A177 and C178 as well as A182 and
C183). This observation implies that the higher affinity of P1 tRNA compared to the other
tRNAs can be attributed in part to those additional contacts that can stabilize the tRNA
binding upon the induced fit that takes place (considering also the full recognition of the
anticodon region). Moreover, the strength of the binding can possibly be enhanced for both
isoacceptors by an additional bend and turn of the antiterminator stem because of the
existence of the extended stem Sa (positions A177-U213; Figure 75). Such a conformational
change in the overall antiterminator structure it would be likely to facilitate a more stable fit
which overcomes the steric hindrance that prevents transcription readthrough by RNA
polymerase. However, the exact role of the antiterminator conformation and its role on such
an induced fit model require further experimentation.
5. In vivo function of the S. aureus glyS T-box riboswitch mechanism
The intriguing observation that both proteinogenic and non-proteinogenic tRNAGly
isoacceptors can induce transcription readthrough via interaction with the glyS T-box
regulatory element was extensively investigated by detailed in vitro analysis and showed a
functional and structural association as it has been initially predicted. The regulatory
interaction of the glyS T-box riboswitch with the tRNA ligands were further examined using
an in vivo tRNA-dependent antitermination system that was applied in E. coli and has been
used in previous similar studies described by Saad et al. [280]. Therefore, we produced an E.
coli strain which could carry two vectors under differential antibiotic selection for expression
and interaction of their corresponding gene products. The strain that was produced
synchronizes the T-box-depended β-galactosidase expression and production with the
inducible expression of either proteinogenic (P1) or non-proteinogenic (NP2) tRNAGly
isoacceptor.
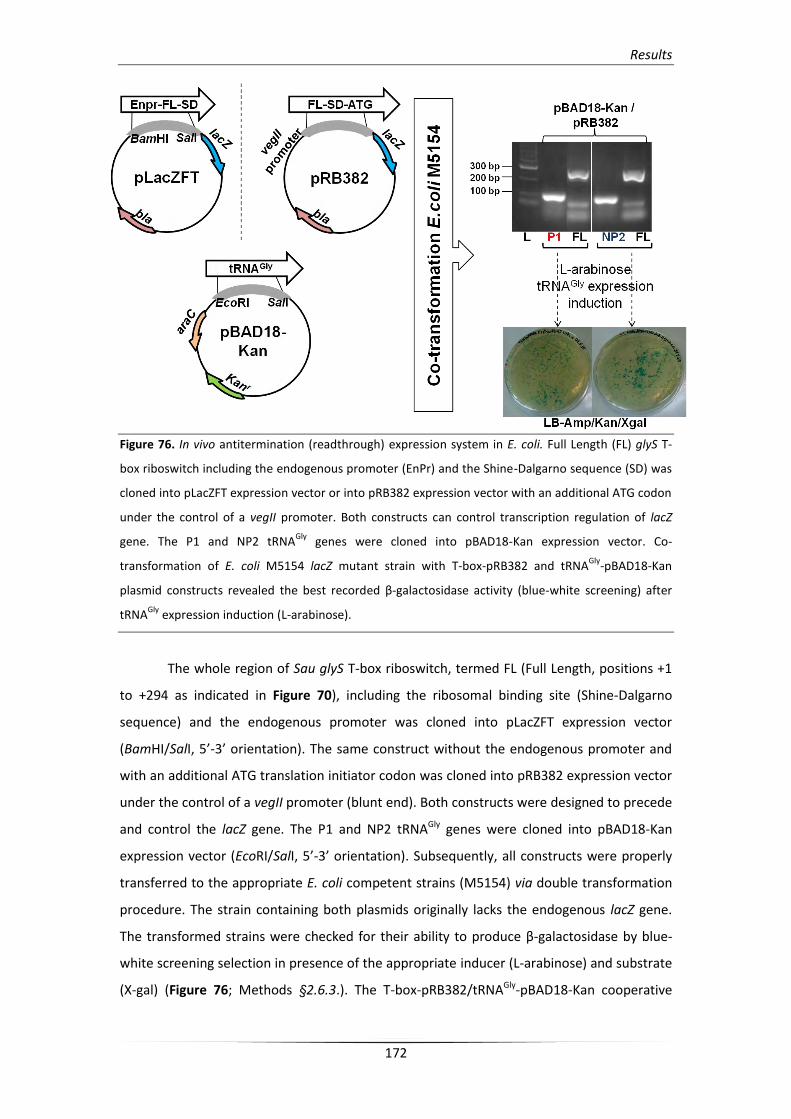
Results
172
Figure 76. In vivo antitermination (readthrough) expression system in E. coli. Full Length (FL) glyS T-
box riboswitch including the endogenous promoter (EnPr) and the Shine-Dalgarno sequence (SD) was
cloned into pLacZFT expression vector or into pRB382 expression vector with an additional ATG codon
under the control of a vegII promoter. Both constructs can control transcription regulation of lacZ
gene. The P1 and NP2 tRNAGly
genes were cloned into pBAD18-Kan expression vector. Co-
transformation of E. coli M5154 lacZ mutant strain with T-box-pRB382 and tRNAGly
-pBAD18-Kan
plasmid constructs revealed the best recorded β-galactosidase activity (blue-white screening) after
tRNAGly
expression induction (L-arabinose).
The whole region of Sau glyS T-box riboswitch, termed FL (Full Length, positions +1
to +294 as indicated in Figure 70), including the ribosomal binding site (Shine-Dalgarno
sequence) and the endogenous promoter was cloned into pLacZFT expression vector
(BamHI/SalI, 5’-3’ orientation). The same construct without the endogenous promoter and
with an additional ATG translation initiator codon was cloned into pRB382 expression vector
under the control of a vegII promoter (blunt end). Both constructs were designed to precede
and control the lacZ gene. The P1 and NP2 tRNAGly genes were cloned into pBAD18-Kan
expression vector (EcoRI/SalI, 5’-3’ orientation). Subsequently, all constructs were properly
transferred to the appropriate E. coli competent strains (M5154) via double transformation
procedure. The strain containing both plasmids originally lacks the endogenous lacZ gene.
The transformed strains were checked for their ability to produce β-galactosidase by blue-
white screening selection in presence of the appropriate inducer (L-arabinose) and substrate
(X-gal) (Figure 76; Methods §2.6.3.). The T-box-pRB382/tRNAGly-pBAD18-Kan cooperative

Results
173
system was found as the more dynamic and therefore was used in all subsequent in vivo
antitermination experiments.
All currently described in vivo antitermination experiments were performed using
the endogenous Gram-positive environment (B. subtilis strains bearing mutations to the
endogenous lacZ gene; [270]). The “transplantation” of this T-box dependent transcription
regulatory system in Gram-negative environment was first reported for the C.
acetobutylicum NT-box [280]. Based on previous studies we expected that the
corresponding in vivo system used for observing T-box:tRNA interaction could very likely be
effective. Our prediction was based on the facts that in the case of Sau glyS T-box the E. coli
RNAP can transcribe T-box riboswitch even by using the S. aureus endogenous promoter
(Results §4.3.) and moreover can overrun the terminator rigid conformation after co-
operative induction of the antiterminator conformation by tRNAGly effector.
The glyS T-box can in principle trigger transcription of downstream gene in response
to glycine deprivation upon uncharged tRNAGly binding. Therefore, in vivo antitermination
assays (β-galactosidase activity measurement assays; Methods §2.6.4.) were performed
under glycine starvation conditions. The reporter gene expression system (β-galactosidase)
was initially normalized by a control transformed strain bearing a construct which included
only the T-box region (T-box-pRB382 construct) in order to avoid measurement of non-
specific transcription induction possibly due to minor interactions with the endogenous
tRNAGly isoacceptors. Interestingly, the β-galactosidase activity for the control strain was
lower but significantly equal to that of the strain which was induced for P1-tRNAGly(GCC)
expression. However, it was higher than the activity that recorded for the strain which
expressed the NP2-tRNAGly(UCC) isoacceptor (Figure 77B). These observations indicate that the
tRNAGly(GCC) isoacceptor from E. coli can efficiently interact with the glyS T-box region and
can compete with the expressed S. aureus tRNAGly for binding. This suggestion can also be
supported by the fact that E. coli tRNAGly(GCC) isoacceptor is almost identical in sequence with
the P1 tRNA (Figure 77C). Furthermore, the reduced β-galactosidase activity can be
explained by the fact that the non-proteinogenic isoacceptor (NP2) is an unusual tRNA for
the E. coli environment and as a result the percentage of the functional well-folded in vivo
transcript was probably reduced.
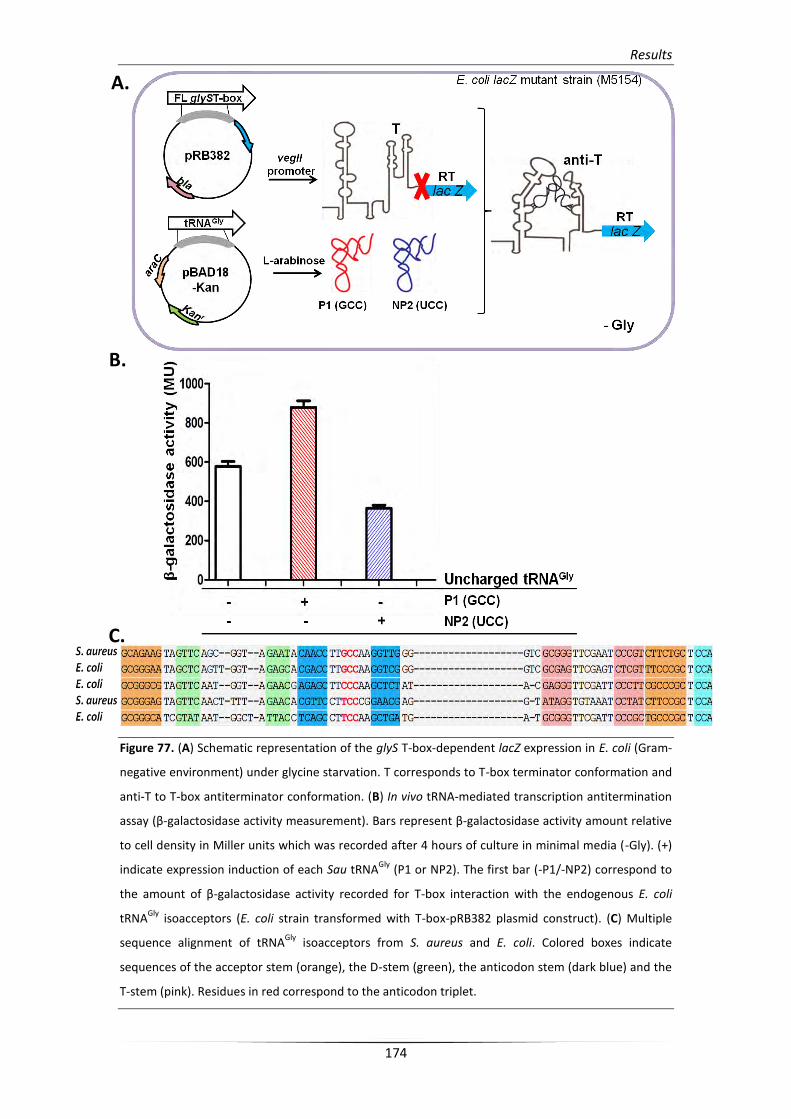
Results
174
Figure 77. (A) Schematic representation of the glyS T-box-dependent lacZ expression in E. coli (Gram-
negative environment) under glycine starvation. T corresponds to T-box terminator conformation and
anti-T to T-box antiterminator conformation. (B) In vivo tRNA-mediated transcription antitermination
assay (β-galactosidase activity measurement). Bars represent β-galactosidase activity amount relative
to cell density in Miller units which was recorded after 4 hours of culture in minimal media (-Gly). (+)
indicate expression induction of each Sau tRNAGly
(P1 or NP2). The first bar (-P1/-NP2) correspond to
the amount of β-galactosidase activity recorded for T-box interaction with the endogenous E. coli
tRNAGly
isoacceptors (E. coli strain transformed with T-box-pRB382 plasmid construct). (C) Multiple
sequence alignment of tRNAGly
isoacceptors from S. aureus and E. coli. Colored boxes indicate
sequences of the acceptor stem (orange), the D-stem (green), the anticodon stem (dark blue) and the
T-stem (pink). Residues in red correspond to the anticodon triplet.
A.
B.
C.

Results
175
On the other hand, as experimentally verified (Results §1.4.) over-production of NP2
isoacceptor in S. aureus can lead to significant loss of fitness of the transformed stains. This
observation indicates the presence of a possible mechanism which synchronizes the
availability of substrates for both protein synthesis and cell wall formation according to the
growth state. This mechanism can presumably include the interaction with the T-box leader
but its exact functional role needs further experimentation.
Moreover, in the absence of glycine and in the presence of the inducer for P1-
tRNAGly(GCC) expression (-Gly, +L-arabinose) the measured β-galactosidase activity was
increased. This result is supportive of the fact that glyS T-box requires uncharged tRNAGly to
induce transcription readthrough. However the fact that both E. coli and S. aureus tRNAGly
isoacceptors can efficiently interact with glyS T-box riboswitch indicates a more relaxed
preference among isoacceptors of the tRNAGly pool. When glycine was restored in the
medium (+Gly, - L-arabinose), no significant change in β- galactosidase activity could be
measured. This result implies that in the particular E. coli environment probably needs
excess concentrations of glycine (Figure 77).
Finally, these observations can verify in part the expected regulatory T-box
machinery as the maximum transcription readthrough was detected for the strain which
overproduce the P1-tRNAGly(GCC) isoacceptor and moreover suggest the usage of all available
effectors, namely every uncharged tRNAGly isoacceptor.
A.
B.
C.


υηιτθςθ
A.
B. C.


Discussion
179
1. Glycyl-tRNA synthetase: a structural divergent but functionally significant enzyme
from bacteria to mammals
Glycyl-tRNA synthetase (GlyRS) represents and evolutionary old amino acyl-tRNA
synthetase and is responsible for incorporation into proteins of the simplest amino acid.
Structural and phylogenetic analyses of GlyRS from different organisms of all kingdoms of
life revealed that it possesses the most intriguing divergences among class II aaRSs. In
eukaryotes, archaea and bacteria GlyRS presents an oligomeric structure (α2 dimeric
organization) exhibiting very low conservation in sequence among species, while in few
bacterial organisms (like E. coli) exhibits a tertameric α2β2 organization. Moreover, in some
organisms the characteristic signature motifs of class II synthetases (motif I, II, and III) are
degenerated or absent. T. thermophilus GlyRS structural determination indicated an
intriguing substitution in position 66 of motif I, which participate in subunit communication,
bearing a Ser residue instead of the conserved Pro residue, which is evident for an atypical
motif formation [259]. In addition, in tetrameric GlyRSs, motifs I and II cannot be detected
[273].
This structural diversity of GlyRSs probably presumes in species-specific
functionality, as prokaryotic and eukaryotic enzymes have differential substrate preference
according to their discrimination for tRNAGly identity elements (U discriminator base in
prokaryotes and A in archaea and eukaryotes, with an exception for plant tRNAGly which
bears a C base [281]). However, in some cases (T. thermophilus and S. cerevisiae) species
specificity is deprived. T. thermophilus GlyRS can acylate tRNAGly species with either A or U
discriminator base, while yeast GlyRS can acylate various tRNAGly species, namely
prokaryotic, eukaryotic, and archaebacterial, with differential specificities (Figure 78). These
functional divergences suggesting that the conservation in discriminating elements is absent
from most glycylation systems and are presumably attributed to evolutionary complexity
[273].
The in silico analysis of staphylococcal GlyRS presented herein (Results §1.2.) showed a well-
organized active site in both individual structures of the Gly binding pocket and the tRNAGly
anticodon binding site and the conservancy of specific residues which contribute to the
catalytic mechanism was also indicated. However, although the overall structural
organization (α2 dimeric) remains accurate in comparison with the T. thermophilus GlyRS
crystal structure, multiple sequence alignment revealed a very low percentage identity
(41%). Moreover, despite the fact that GlyRSs from different staphylococcal species exhibit
high identity in sequence (82%, SI Figure 1) the cross-glycylation efficiency that was tested
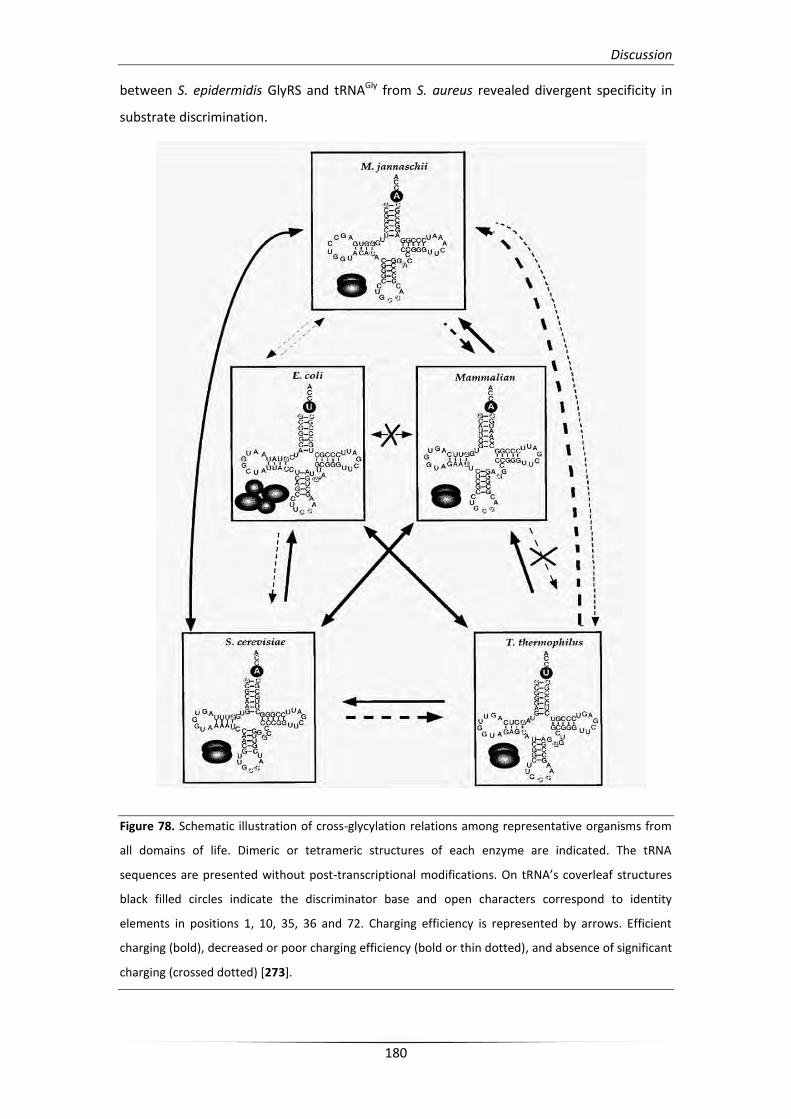
Discussion
180
between S. epidermidis GlyRS and tRNAGly from S. aureus revealed divergent specificity in
substrate discrimination.
Figure 78. Schematic illustration of cross-glycylation relations among representative organisms from
all domains of life. Dimeric or tetrameric structures of each enzyme are indicated. The tRNA
sequences are presented without post-transcriptional modifications. On tRNA’s coverleaf structures
black filled circles indicate the discriminator base and open characters correspond to identity
elements in positions 1, 10, 35, 36 and 72. Charging efficiency is represented by arrows. Efficient
charging (bold), decreased or poor charging efficiency (bold or thin dotted), and absence of significant
charging (crossed dotted) [273].

Discussion
181
Identity elements responsible for efficient cross-glycylation in bacterial systems
(except the discriminator base which is a U base as mentioned above) are the G1-C72 base
pair of the acceptor stem, as well as positions G10, C35, and C36 in the D-stem and the
anticodon loop which constitute the highly conserved identity set during evolution. In
addition the conserved positions G10, C(U)25 and G45 involved in proper tRNA folding. In S.
aureus and S. epidermidis both proteinogenic and non-proteinogenic tRNAGly isoacceptors
retain all the important identity recognition elements for GlyRS discrimination with an
exception for S. aureus NEW and S. epidermidis NP1B tRNAs which bear a U residue in
position 10 (Figure 50B). However, the deprived specificity obtained for S. aureus-
S.epidermidis cross-charging system suggests the existence of a different mechanism
involving interaction with broader substrate recognition elements with a basis in
discrimination properties, but also an involvement of overall structure recognition.
Moreover, this notion can be supportive for the recognition and cross-aminoacylation by
heterologous systems such as glycylation of eukaryotic substrates by prokaryotic GlyRSs and
the opposite (Figure 78).
Finally, despite the fact that P1-tRNAGly(GCC) is identical in sequence with the relative
isoacceptor of S. epidermidis, it cannot be efficiently charged by the cognate S. epidermidis
GlyRS. This observation is presumably attributed to the absence of specific modifications. On
the other hand, unmodified P1-tRNAGly(GCC) transcript is efficiently recognized by the glyS T-
box structure and moreover, can induce transcription readthough. These findings are also
supportive for the existence of an alternative discrimination mechanism with very ancient
origin where protein function does not occur. During evolution such mechanisms were
evolved separately or in combination with relative protein mechanisms and in some cases
like the aminoacylation systems, this co-evolution probably contribute to a more complex
recognition and function.
1.1. Eukaryotic GlyRS and its association with disease etiology in humans
Eukaryotic GlyRS generally possesses a homodimeric structure (α2) while α2β2
heterotetramer is identified only in chloroplasts. In S. cerevisiae can be found two distinct
GlyRS genes, namely GRS1 and GRS2. Both cytoplasmic and mitochondrial enzymes are
encoded by GRS1 via alteration in initiation of translation, while GRS2 is a stress-inducible
gene and its expression is suppressed under normal conditions [282, 283]. Human GlyRS (a2
type) exhibits three additional insertion domains in the N-terminal part which are interposed
the characheristic motifs I, II, and III, and contribute in more complicated three-dimensional
structure and dimeric organization [284] (Figure 79A).
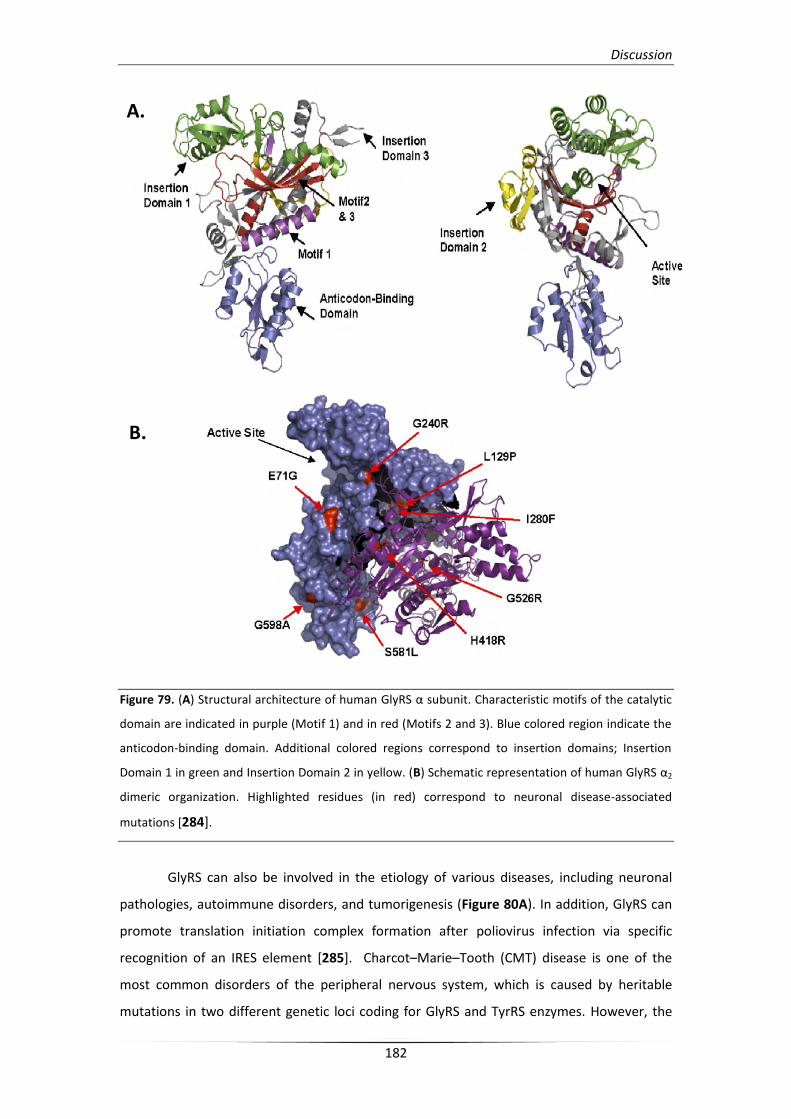
Discussion
182
Figure 79. (A) Structural architecture of human GlyRS α subunit. Characteristic motifs of the catalytic
domain are indicated in purple (Motif 1) and in red (Motifs 2 and 3). Blue colored region indicate the
anticodon-binding domain. Additional colored regions correspond to insertion domains; Insertion
Domain 1 in green and Insertion Domain 2 in yellow. (B) Schematic representation of human GlyRS α2
dimeric organization. Highlighted residues (in red) correspond to neuronal disease-associated
mutations [284].
GlyRS can also be involved in the etiology of various diseases, including neuronal
pathologies, autoimmune disorders, and tumorigenesis (Figure 80A). In addition, GlyRS can
promote translation initiation complex formation after poliovirus infection via specific
recognition of an IRES element [285]. Charcot–Marie–Tooth (CMT) disease is one of the
most common disorders of the peripheral nervous system, which is caused by heritable
mutations in two different genetic loci coding for GlyRS and TyrRS enzymes. However, the
B.
A.

Discussion
183
mutant proteins are fully functional for aminoacylation [286]. Currently, 13 distinct
mutations of GlyRS in patients have been reported [287] that cause CMT2D subtype of the
disease also known as GlyRS-associated axonal neuropathy (Figure 79B). Moreover, the
additional domain that is indentified for the N-terminal part of human GlyRS is presented as
an antigenic epitope in autoimmune disorders known as ‘‘antisynthetase syndrome’’, which
also has been reported for multiple synthetase epitopes for the 30% of the autoimmune
patients. In addition, there has been identified potential involvement of GlyRS in the etiology
of carcinogenesis. For instance GlyRS was found up-regulated in papillary thyroid carcinoma
(PTC) [97]. However, GlyRS can also induce apoptosis as its soluble status in serum, which is
secreted from macrophages in response to Fas ligand signaling pathway, can mediate
suppression of ERK signaling upon tumor formation [288] (Figure 80B).
Figure 80. (A) Schematic representation of aaRSs’s linkage with various human diseases. GRS is
implicated in neuron and autoimmune diseases and is found overexpressed in human cancers [97].
(B) Implication of GRS in anti-tumor activity in the tumor-macrophage microenvironment. Secreted
GRS can bind to a K-cadherin molecule (CDH6) and triggers the release of PP2A which in turn results in
tumor-cell death [288].
Finally, both catalytic mechanism and structural organization seems to be crucial for
GlyRS involvement in certain pathologies in humans. However, despite the fact that crystal
structure of human GlyRS is known, the exact contribution of each identified mutation in its
tertiary organization is not yet verified due to the high structural complexity. On the other
hand, GlyRSs from lower organisms which possess less complex structural organization but
retain the highly conserved active sites (like the staphylococcal α2 GlyRS) can be used for
such studies and moreover for development of novel pharmaceutical agents against GlyRS-
associated disorders.
A. B.

Discussion
184
1.2. Staphylococcal GlyRS involvement in cell wall synthesis
In Staphylococcus spp., GlyRS enzyme, which possesses an α2 type homodimeric
tertiary structure, is involved in synthesis of the cell wall, since it responsible for the
production of substrates for the formation of interpeptide bridges (glycine pentapeptide in
S. aureus and glycine/ L-alanine or glycine/L-serine peptide in S. epidermidis; [127]), which
results in peptidoglycal three-dimensional cross-linking. The first Gly residue incorporation in
lipid II moiety is catalyzed by FemX protein while FemA and FemB proteins are responsible
for incorporation of four additional Gly residues (two Gly residues by each factor) [225, 227].
It has been shown that deletion of Fem X leads to lethality of the organism since it severely
affects proper cell wall formation [226]. However, deletion or deficiencies of either Fem A or
Fem B do not seem to be deleterious for the cell [227]. Moreover, this characteristic cell wall
peptidoglycan layer is important for pathogen’s infectivity, since mutations in Fem factors in
several nosocomial strains of S. epidermidis have shown that Fem factors can affect
susceptibility of S. epidermidis to methicillin and oxacillin [124] (Figure 81).
Figure 81. Peptidoglycan synthesis in S. aureus and targets of cell wall-active antibiotics (in red).
Differential inhibition mechanisms are indicted by arrows (pentaglycine bridge cleavage), blocked
arrows (enzymatic reactions) and half-moon symbols (antibiotic binding) [289].
Both GlyRS enzymes from S. aureus and S. epidermidis are responsible for the
aminoacylation of tRNAGly molecules (non-proteinogenic isoacceptors) (Figure 50B) which
participate mainly in cell wall synthesis. As it has been shown previously, in S. aureus
aminoacylated NP1, NP2, and NEW tRNAGly(UCC) isoacceptors serve as glycine donors for

Discussion
185
FemXAB non-ribosomal peptidyl-transferases which are responsible for pentaglycine bridge
formation [228] (Figure 81). In vitro Fem-mediated lipid II formation revealed that NEW
tRNAGly is a preferable substrate for Fem X, which incorporates the first glycine residue of
the interpeptide bridge (Results §1.5.). This result verifies the essential role of the non-
proteinogenic tRNAs in cell wall formation. Moreover, it provides a new basis for
inactivation of GlyRS as target for inhibition of cell wall formation by novel antibacterial
agents.
In vitro aminoacylation results showed that either proteinogenic or non-
proteinogenic tRNAGly gene transcripts can be charged by both S. aureus and S. epidermidis
GlyRS enzymes with differences in their accepting capacity. Although all tRNAGly can be
aminoacylated by S. aureus enzyme with minor differences in accepting capacity, cross-
glycylation by S. epidermidis enzyme revealed high efficient discrimination for non-
proteinogenic isoacceptors compared with the proteinogenic (Results §1.3.; [228]). Further
in silico analysis based on the available T. thermophilus GlyRS crystal structure is shown high
homology among the enzymes from all staphylococcal species (Results §1.2.), supporting the
proposed cross-charging ability of GlyRS homologues from relative species to discriminate
for highly conserved identity elements of tRNAGly (namely the discriminator base, which is
the same for both S. aureus and S. epidermidis tRNAGly species; Figure 58). However, the two
enzymes exhibit different charging preferences which are presumably depend on the origin
and identity of the tRNAGly substrates including also specific base modifications. Moreover,
investigation of the utilization of each tRNAGly isoacceptor during cell lifecycle using a
recombinant in vivo expression system in S. aureus revealed that all isoacceptors have to be
present in different growth phases in either charged or uncharged status. Alterations in
availability of each tRNAGly substrate for protein synthesis and /or cell wall formation
contribute to divergent growth profiles and a significant loss of fitness (Results §1.4.).
2. Staphylococcal glyS gene expression is controlled by a species-specific T-box
regulatory element
T-box riboswitches represent a highly distributed class of RNA regulatory elements
among bacterial species (up to 1134 T-box regulons have been detected; [194]). T-boxes can
sense amino acid deprivation and modulate gene expression via tRNA-depended
transcriptional or translational attenuation mechanisms. The T-box domain presents a
variability of conserved features which reassure proper interaction with the tRNA ligand. An
appropriate conformational switch upon effector binding, which favors antiterminator or SD
antisequester structure formation, can induce gene expression at the transcriptional or the

Discussion
186
translational level respectively [199, 203]. The recognition of tRNA relies exclusively on
specific structural features of this regulatory element without participation of any protein
partner [290]. It has been proposed that identity elements of tRNA’s overall structure favor
essential dynamic associations by which the T-box can sense either the charged or
uncharged status of the tRNA [216]. This notion suggests that T-box regulatory mechanism
requires both the correct measurement and recognition of tRNA’s structure beyond the SL
codon: tRNA anticodon complementation, that seem to favor a mutually induced fit [216,
278, 291]. The discovery of a T-box regulon in Clostridium acetobutylicum which provide
dual-effector specificity suggests that T-boxes can possibly regulate and synchronize
different metabolic pathways. The NT-box regulatory element is found upstream an operon
which encodes all the necessary enzymes for the transamidation pathway (the GatCAB
amidotransferase and the non-discriminating AspRS). In this specific case, the same
enzymes, besides protein synthesis can be also used for the synthesis of asparagine during
adaptation to metabolic changes [144, 280]. In addition, all the recent bioinformatic,
biochemical, and structural data suggest that species-specific T-box regulon particularities
must exist due to specific metabolic demands or evolutionary complexities (such as regulon
duplications combined with vertical inheritance). For instance, it was currently reported that
in B. subtilis two different T-box regulons, which are specified for uncharged tRNATyr, can
synchronize regulation of both tyrZ (coding a second TyrRS, which is highly selective for L-
Tyr) and tyrS genes, which are found in different genetic loci, in transcriptional level to
accomplish requirements of bacterial growth in response to nutrients availability [211].
Moreover, was recently shown that T-boxes in Actinobacteria phylum, which are located
upstream ileS genes, can specifically provide gene regulation at the translational level via
rearrangement of the SD sequester / antisequester region, and lack structural features of
the highly conserved Stem I element [203].
Bioinformatics analyses, which have been used in order to determine the
phylogenetic distribution or the structure and functionality of the predicted T-box regulons,
is mainly based on the presence of few conserved patterns in regulon sequence like the T-
box region [199, 200, 274, 292]. Therefore, the more riboswitches are characterized the
clearer is the picture of their diversity among different organisms which is probably linked to
special metabolic and/or phylogenetic origin. The glyQS T-box riboswitches that have been
studied so far exhibit structural variations compared to other T-boxes in the same or
different organisms. Furthermore, glyQS T-box regulons from Bacilli were extensively
characterized (both in structural and biochemical level), but such information for the same
type of riboswitches that are identified in other classes or phylum are yet to be elucidated.

Discussion
187
Additional phylogenetic analysis of glyS or glyQS T-box riboswitch based on the
whole mRNA leader region and not restricted to individual conserved sequences like the T-
box sequence, revealed clade diversity among species from the same phylum (Figure 82). As
a result relative species from Firmicutes phylum (Gram-positive bacteria) can be divided into
three separated subgroups, the bacilli-streptococci group which is clustered with
Deinococcales-Thermales and Chloroflexi phylum (including Gram-negative species),
Clostridium-Lactobacilli and staphylococci groups. The last two subgroups exhibit
divergences in sequence where the most are indicated for the group of staphylococci. This
observation suggests a species-specific structural organization of the same T-box regulatory
element which is presumably attributed to metabolic particularities. Moreover, in both S.
aureus and S. epidermidis species GlyRS involved in two metabolically unrelated but
essential for cell viability pathways, protein synthesis and cell wall formation. Those
pathways occur mostly in different growth states during cell lifecycle and can use either
proteinogenic or non-proteinogenic isoacceptors. This notion is supportive for
staphylococcal glyS T-box riboswitch separated evolution since the non-proteinogenic
tRNAGly usage occurs only in such species. In addition T-box specifier loop codon usage or
GlyRS structural organization (dimeric or tetramaric) cannot be related with clade diversity.
In the present study, such a T-box regulatory element in S. aureus was identified
upstream the coding region of glyS gene (Results §2.1.). Initial bioinformatics analysis
showed that it deviates from the usual structural pattern that has been described for other
glyQS T-boxes (Results §3.1.). Moreover, this glyS T-box seem to preserve structural
particularities, which show remarkable conservation within the staphylococcal species, such
as the additional intervening sequence of the terminator/antiterminator stem (also termed
as stem Sa; Figure 60).

Discussion
188
Figure 82. Phylogeny of the predicted glyS or glyQS T-boxes based on the whole mRNA
leader region in different organisms. Online tools: RegPrecise (http://regprecise.lbl.gov),
Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo).
3. The glyS T-box riboswitch exhibits a divergent structure
Both bioinformatics and experimental analyses of the S. aureus glyS T-box mRNA
riboswitch revealed that although the specifier loop/codon region and T-box sequence are
conserved, most of the predicted riboswitch structure was significantly divergent (Results
§3; Figure 60). The most striking differences are located in stem I region, which lacks
conservation in sequence, that forming the characteristic K-turn motif, and possesses a
displaced E-loop and a wider apical loop. According to the suggestion that K-turn motif can
presumably combine specifier loop recognition and antiterminator formation resulting in
higher T box–tRNA affinity, transcription read-through seems to depend on its conservation
[212]. In S. aureus glyS T-box K-turn motif two crucial G-A pairs that contribute in its tertiary
structure formation are absent [201]. This notion can probably explain along to the very high
thermodynamic stability of the terminator conformation, the lower ability of antiterminator
formation that is indicated in in vitro readthrough assays (Results §4.3.). However, all tRNAGly
can induce transcription via antiterminator formation even in presence of the divergent K-
turn motif. Moreover, the observation that such a conserved region, also known as GA
motif, cannot be identified in staplylococcal T-box leaders [201] suggests that in this case
antiterminator formation can probably occur in a species-specific context. In addition,
bioinformatics analysis showed that the SL belongs to the 8nt class. This type of T-boxes

Discussion
189
exhibits a dual-specificity that depend on downstream coding regions, via recognition of
overlapped codon sequences, as in the case of NT-box from C. acetobutylicum [144].
Although in the case of S. aureus glyS T-box the downstream sequence encodes only a single
gene, an additional codon sequence (U108, G109, G110) can be identified for tRNATrp(CCA)
anticodon recognition. This observation is also intriguing and confirms the characteristic
deviation of stem I region. However, it’s possible contribution in specificity needs further
experimentation. On the other hand, the Specifier loop contains the conserved nucleotides
G105 and A106 and the conserved purine (G) in position 112 which follow the pattern of
previously characterized T-boxes [200, 276]. Further extensive mutagenic analysis will
illuminate the details of the role of each T-box structural domain.
Most interestingly, the S. aureus glyS T-box domain, which switches between the
terminator and the antiterminator conformation upon binding of uncharged tRNA, contains
a 42 nt long intervening sequence that is termed stem Sa. This unusual feature, which is
reported for the first time, seems to be glycine-specific and it is also restricted only in
staphylococci (Figure 55 and Figure 61). Extensive chemical and enzymatic probing analyses
(Results §3.2.2.) revealed that stem Sa seem to contribute in the formation of the identified
two-stem rigid terminator conformation. Moreover, it can possibly favor a local
rearrangement through a helix turn, which can lead to interaction with the tRNA effector
during antitermination, via interaction with specific nucleotides that are exposed (Results
§4.4.). This interaction possesses an additional check point for either proteinogenic or non-
proteinogenic tRNAGly isoacceptors recognition (of either T-arm or acceptor stem) and
possibly confers an enhanced structural specificity (Figure 75). Finally, as it has been also
described for other glyQS T-boxes, stem II and pseudoknot stem IIA/B conformations are
absent [278]. The absence of this structural elements, which is characteristic of other T-box
leaders (as tyrS T-box leader in B. subtilis; [197]), indicates that no additional trans-acting
factors mediate the T-box:tRNAGly interaction [293].
In conclusion, the divergent structural and species-specific features of the S. aureus
glyS T-box can be the reason why the available bioinformatic tools failed to provide a high
score for the putative T-box regulon (RegPrecise Score:12.9%) although its presence has
been previously predicted [199, 200]. Moreover, this T-box leader possibly adopts different
conformations in solution that account for the differential binding affinity of each structural
construct, namely T275, T225, and T115, which are used in the presence study (Results
§4.1.).

Discussion
190
4. Genetic code-like ambiguity of glycine codon reading by the S. aureus glyS T-box
riboswitch
The fact that both proteinogenic and non-proteinogenic tRNAGly isoacceptors can
interact with T-box leader region and moreover can induce transcription readthrough it was
the most intriguing result of the present study. Detailed chemical and enzymatic probing
analysis reveled that the interaction between the SL and the anticodon is stronger with the
two of the three bases of the SL anticodon (G109, G110) for both P1-tRNAGlyGCC and NP2-
tRNAGlyUCC as the third position of the SL codon (C111) cannot be easily probed (Results
§4.4.). A similar observation has been recently reported in the case of B. cereus lysK
(encoding a Class I LysRS) T-box, which can responds to both tRNALys and tRNAAsn molecules
bearing GUU and UUU anticodons via interaction with the same specifier codon triplet (AAA)
[194]. However, it should be noted that all the tRNAGly isoacceptors that have been tested so
far in previous studies, bearing a GCC anticodon, and can form full complementarity with the
SL triplet. In addition, a previous study investigating the interaction of a tyrS mRNA leader, in
which the UAC specifier triplet was replaced by a GGC glycyl-codon, failed to detect binding
when tRNAGly(UCC) was used [213]. This observation can be attributed to the nature of the
truncated T-box sequence which is possibly inducing different conformational changes.
Moreover, the existence of GGA SL triplets in some glyQS T-boxes suggest that in other
organisms binding of tRNAGly(UCC) can possibly contribute to the regulation of glyQS but also
recognition of the GGC triplet cannot be excluded (Figure 55; [238]).
The unconventional reading of the GGC triplet of the specifier loop by different
tRNAGly isoacceptors also occur during protein synthesis. According to the genetic code
degeneracy, glycine belongs to a four-fold degenerated codon family (GGU, GGC, GGA,
GGG), where the first two nucleotides can match the C35 and C36 of the tRNAGly anticodon.
In bacteria, three different tRNAGly isoacceptors, bearing UCC, GCC, and CCC anticodon
triplets can read all four codons, while in eukaryotes can be used all four cytoplasmic
isoacceptors. Moreover, in Mycoplasma mycoides, as well as in mitochondria and
chloroplasts, the tRNAGly(UCC) recognizes all four codons with equal efficiency. In Bacillus and
Staphylococcus species two tRNAGly isoacceptors can be used, bearing UCC and GCC
anticodons, where U is a modified. Modifications of U in position 34 of the anticodon triplet
can enhance the ability of wobbling discrimination. According to MODOMICS server
(http://modomics.genesilico.pl), in B. subtilis tRNAGly(UCC) species U34 is mostly modified in 5-
methoxycarbonyl-methyl-2-thiouridine and as a result codon discrimination is increased.
However, this post-transcriptional modification is absent in S. epidermidis tRNAGlyUCC
isoacceptors, which encodes non-proteinogenic tRNAGly molecules, and they are also

Discussion
191
identical to those of S. aureus. It has to be noted that in the present study unmodified
tRNAGly(UCC) transcripts were used, and are evident for unconventional Specifier GGC codon
reading under in vitro experimental conditions. Moreover, it has been suggested that the
ability of the tRNAs to discriminate the third codon position also depends in a certain extend
on the identity of positions 32 and 38, and the composition of positions 35 and 36.
Therefore, the ambiguity that is observed in the S. aureus glyS T-box Specifier codon triplet
can be also attributed to this intrinsic idiosyncrasy of the tRNAGly isoacceptors [238, 275]. On
the other hand, it is still unknown whether this ambiguity plays significant role in the cellular
environment during bacterial growth.
An additional interesting interaction which was identified upon T-box:tRNA complex
formation involves the conserved purine (G) in position 112. Previous structural probing
analyses proposed that this conserved purine can possible interact with the conserved U33
of tRNA anticodon via base pairing [198]. However, subsequent extensive structural analyses
of the T-box stem I:tRNAGly complex establish that the proposed fourth base pair cannot
exist. This conserved purine seems to stack below the codon-anticodon base pairs providing
further stabilization of the specific interaction as it is known that purines can generally
contribute to such base-stacking interactions. Moreover, conserved tRNA purine in position
37 stacks also on the top of codon:anticodon moiety and facilitates the completion of the
local geometry. This specific geometry was also observed for the ribosomal P site, where the
C1400 residue of the ribosomal RNA stacks underneath the mRNA codon-tRNA anticodon
paired triplet [295]. This flanking stacked base geometry seems to be necessary for specific
discrimination among different substrates (Figure 83) [216].

Discussion
192
Figure 83. Conserved base
geometry of the codon-
anticodon reading. (A)
Structural arrangement of
the T-box specifier codon
region during anticodon
reading. (B) Local
structural arrangements
of the ribosomal P-site
that decode tRNA’s
anticodon. (C) Schematic
illustration of the specific
geometry responsible for
tRNA anticodon decoding
[216].
In the case of the S. aureus T-box, binding of P1 isoacceptor to the full-length
riboswitch region seems to affect G112 stacking, while upon NP2 binding this interaction can
be identified in a significant lower level. On the other hand, the same interaction for both P1
and NP2 seems to be enhanced in presence of the stem I structural variant T115, albeit in a
lower level for NP2 (Results §4.1.). As a result specific base stacking upon full-length T-
box:tRNA complex formation can shift towards additional local structural rearrangements
that are favored by the flexibility of the anticodon loop (which is characteristic for all tRNAGly
isoacceptors; [238]). These conformational changes are possibly enhanced by additional
contacts provided most likely by stem Sa (Figure 75).
In conclusion, transcription regulation by S. aureus glyS T-box possibly occurs not
only in the context of the GGC SL triplet recognition but also in an overall synchronization of
additional interactions, which are induced by structural features either in T-box
conformation or tRNA effector structure.
A. B.
C.

Discussion
193
5. A proposed mechanism for synchronization of two essential but metabolically
unrelated pathways in S. aureus
It has been previously proposed that in S. aureus the five encoded tRNAGly molecules
can potentially serve different roles during pathogen’s lifecycle, which are based on their
binding affinity to the homologous EF-Tu. Two of them, P1 and P2 bearing GCC and UCC
anticodons respectively, can serve as glycine carriers for incorporation into nascent proteins
during ribosomal protein synthesis due to their strong binding to EF-Tu. The remaining three
molecules, NP1, NP2, and NEW, bearing UCC anticodons, can potentially escape protein
synthesis due to their weak binding to EF-Tu, and serve as substrates for the formation of
characteristic pentaglycine peptides by Fem non-ribosomal peptidyl-transferases (Figure 84).
This pentaglycine bridges can stabilize three-dimensional structure of the staphylococcal cell
wall by connecting stem peptides of lipid II moiety, a process which is essential for cell
viability [124, 226].
Both in vitro and in vivo tRNA-mediated antitermination assays revealed that either
proteinogenic or non-proteinogenic tRNAGly species can induce transcription read-through
via conventional or unconventional reading of the Specifier GGC codon triplet (Results §4.
and §5.). In such context all tRNAGly isoacceptors can potentially participate in transcriptional
regulation of GlyRS according to the availability of their charged or uncharged status. Most
likely stabilization of the interaction between each isoacceptor and the T-box riboswitch
region can be enhanced through an induced fit. Such structural specificity can presumably
be provided through additional contacts with the stem Sa. This unusual structural feature is
glycine-specific and strictly conserved only among staphylococcal species. These
observations revealed that T-box regulons can also adopt species-specific patterns which
facilitate the overall metabolic strategy of each organism.
According to the regulatory mechanism, which is proposed in the present study, S.
aureus glyS T-box can regulate two essential but metabolically unrelated pathways. The first
is the incorporation of glycine during ribosomal protein synthesis and the second is the exo-
ribosomal pentaglycine peptide bridge synthesis that contributes to cell wall formation. Both
pathways can utilize all available isoacceptors of the aminoacylated tRNAGly pool. Moreover,
all tRNAGly isoacceptors, either in their charged or uncharged status, seem to contribute in
synchronization of each pathway regulation according to nutrient deficiency and growth or
infection state adaptation (Figure 84).
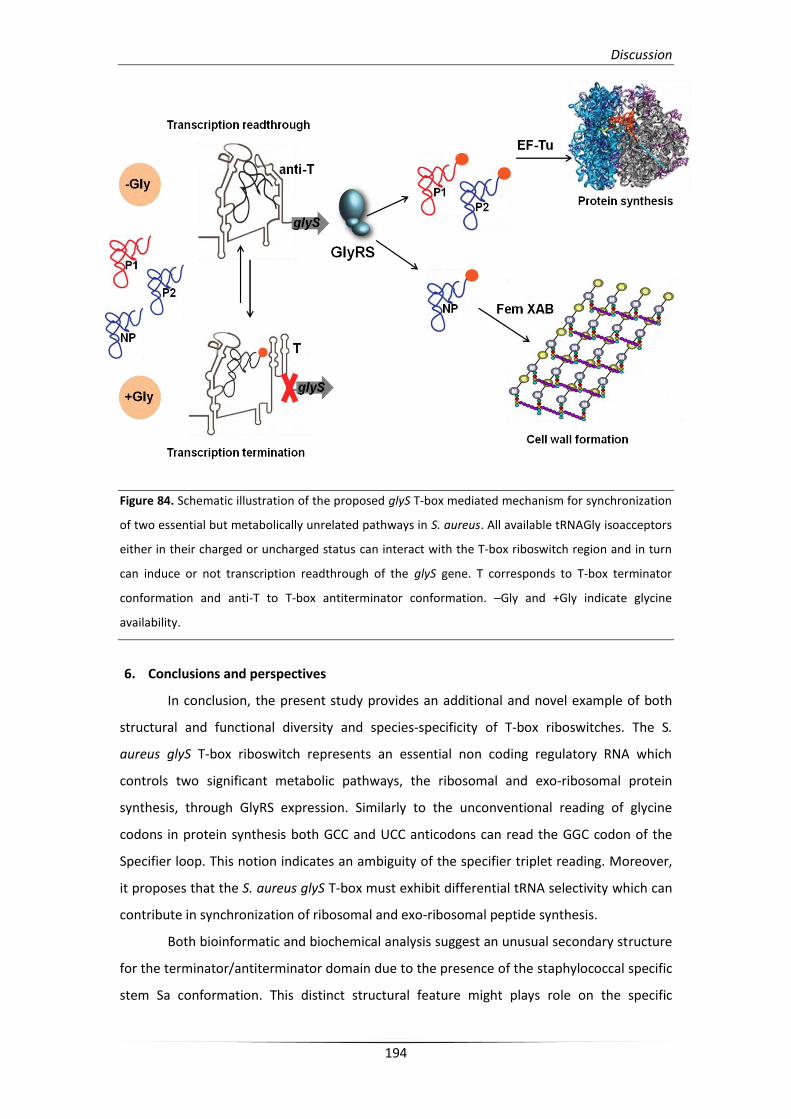
Discussion
194
Figure 84. Schematic illustration of the proposed glyS T-box mediated mechanism for synchronization
of two essential but metabolically unrelated pathways in S. aureus. All available tRNAGly isoacceptors
either in their charged or uncharged status can interact with the T-box riboswitch region and in turn
can induce or not transcription readthrough of the glyS gene. T corresponds to T-box terminator
conformation and anti-T to T-box antiterminator conformation. –Gly and +Gly indicate glycine
availability.
6. Conclusions and perspectives
In conclusion, the present study provides an additional and novel example of both
structural and functional diversity and species-specificity of T-box riboswitches. The S.
aureus glyS T-box riboswitch represents an essential non coding regulatory RNA which
controls two significant metabolic pathways, the ribosomal and exo-ribosomal protein
synthesis, through GlyRS expression. Similarly to the unconventional reading of glycine
codons in protein synthesis both GCC and UCC anticodons can read the GGC codon of the
Specifier loop. This notion indicates an ambiguity of the specifier triplet reading. Moreover,
it proposes that the S. aureus glyS T-box must exhibit differential tRNA selectivity which can
contribute in synchronization of ribosomal and exo-ribosomal peptide synthesis.
Both bioinformatic and biochemical analysis suggest an unusual secondary structure
for the terminator/antiterminator domain due to the presence of the staphylococcal specific
stem Sa conformation. This distinct structural feature might plays role on the specific

Discussion
195
interaction with the T-arm or the acceptor stem of uncharged tRNAs and favors an induced
fit due to the plasticity of the local geometry. As a result, binding of each isoacceptor
triggers tRNA-dependent antitermination which appears to be species-specific. Moreover,
according to the proposed regulatory model interaction between T-box riboswitch and each
tRNAGly isoacceptors is under kinetic control according to an additional check-point
mechanism which is probably attributed to the existence of stem Sa (Figure 75). Finally, both
proteinogenic and non-proteinogenic isoacceptors can induce transcription read-through in
vivo. Although this observation can in part confirm the proposed model, the exact
mechanism that occur in order to synchronize two metabolically unrelated pathways, needs
further experimentation and is currently under investigation.
Staphylococcal species seem to be responsible for most of the hospital-acquired
infections. The major threat against such bacterial infection treatment is the uninterrupted
spread of antibiotic resistance. The highly distributed MRSA strain is found to be resistant
against multiple currently used antibiotic drugs such as beta-lactam antibiotics (methicillin,
flucloxacillin and carbapenems). In addition, alternative to beta-lactam antibiotic drugs such
as inhibitors of protein synthesis are associated also with staplylococcal resistance
phenotypes. Linezolid (the first oxazolidinone antibiotic, also known as Zyvoxid) is
responsible for the existence of additional resistant strains such as MLST nosocomial
isolates. Those particular S. epidermidis isolates are observed with an increasing number of
23S rRNA gene mutations and as currently found, they exhibit linezolid-dependent growth
that is probably linked with the appearance of a new functional ribosomal population [246].
Furthermore, it is evident that such staphylococcal resistant strains can cause severe
infections and their expansion emerge the development of alternative treatments.
The potential new targets have to mediate and participate in essential cellular
pathways different to what has previously targeted. A paradigm of such regulatory
mechanism which control essential metabolic pathways is presented herein. Staphylococcal
glyS T-box riboswitch represents a potential novel target for development of antimicrobial
agents against Gram-positive pathogens since it possesses a highly specialized strategy
which is alternative to currently used antibiotics.

Abbreviations
197
Abbreviations
Amp Ampicillin
DEPC Diethylpyrocarbonate
DMS Dimethyl sulfate
EF-Tu Elongation factor Tu
Gly Glycine
GRS Glycyl-trna synthetase (GlyRS)
IAA Isoamyl alcohol
IPTG Isopropyl β-D-1-thiogalactopyranoside
Kan Kanamycin
KE Kethoxal
NP Non-Proteinogenic
ONPG Ortho-Nitrophenyl-β-galactoside
P Proteinogenic
RNAP RNA polymerase
RT Transcription Read-through
Sau Staphylococcus aureus
Sep Staphylococcus epidermidis
SD Shine-Dalgarno sequence
T Transcription Termination
Xgal 5-bromo-4-chloro-3-indolyl-beta-D-galacto-pyranoside



Supplementary Information
201
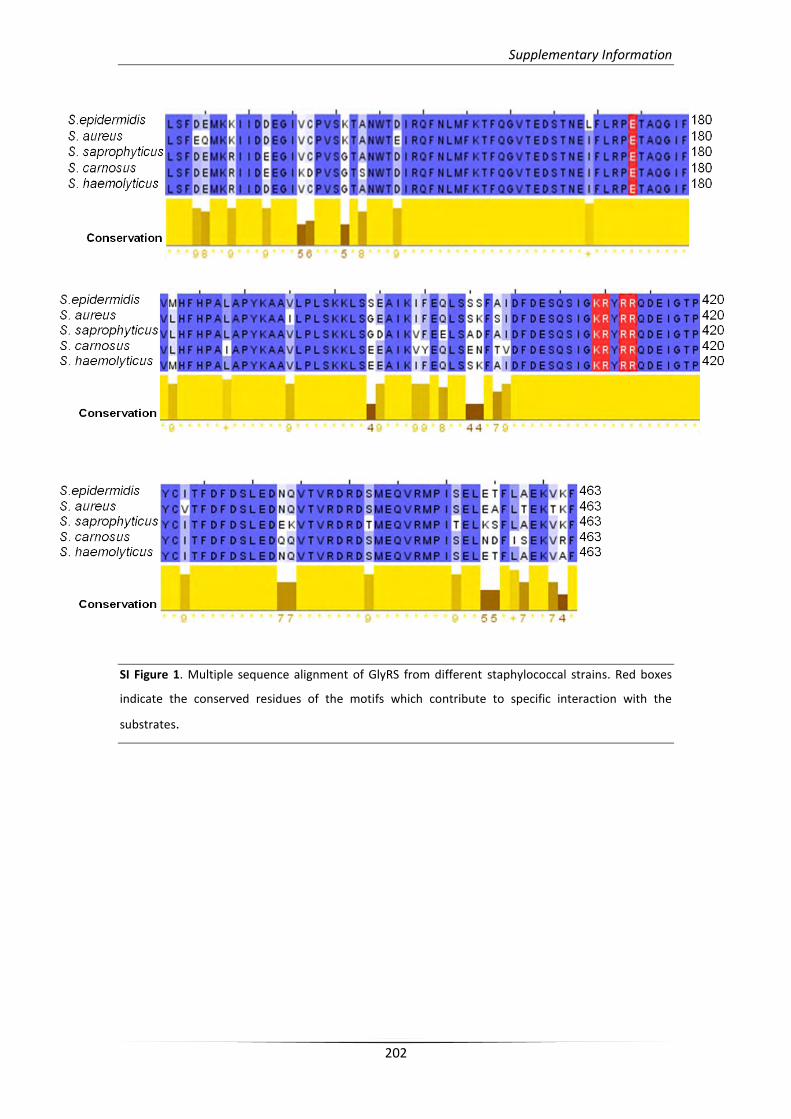
Supplementary Information
202
SI Figure 1. Multiple sequence alignment of GlyRS from different staphylococcal strains. Red boxes
indicate the conserved residues of the motifs which contribute to specific interaction with the
substrates.
Supplementary Information
203
Regulated gene RegPrecise Score
alaS
90.6
hisS-aspS 49.8
cysE-cysS 68.3
glyS 12.9
IleS 80.9
leuS 63.2
metI-metC-metF-metE-mdh (1) 74.8
metI-metC-metF-metE-mdh (2) 35.7
pheS-pheT
75.8
serS 97.2
thrS 92.1
trpE-trpG-trpD-trpC-trpF-trpB-trpA (1) 70.6
trpE-trpG-trpD-trpC-trpF-trpB-trpA (2) 66.6
tyrS
80.5
valS
70.4
SI Table 1. Identification of T-box riboswitches based on T-box sequence pattern according
to RegPrecise database (http://regprecise.lbl.gov) in Staphylococcus aureus N315

F


Βιβλιογραφία

References
207
1. Ibba M. & Söll D. (2000) Aminoacyl-tRNA Synthesis. Annu. Rev. Biochem. 69: 617-650.
2. Crick F. H. C. (1958) On Protein Synthesis. Symp. Soc. Exp. Biol. 12:139-163.
3. Hou Y.-M. & Schimmel P. (1988) A Simple Structural Feature is a Major Determinant of the Identity of a
Transfer RNA. Nature 333:140-145.
4. de Duve C. (1988) Transfer RNAs : The Second Genetic Code. Nature 333:117-118
5. Crick F.H.C. (1966) Codon – anticodon pairing: the wobble hypothesis. J. Mol. Biol. 19:548–555.
6. Agris P. F. (1991) Wobble position modified nucleosidesevolved to select transfer RNA codon
recognition:a modified-wobble hypothesis. Biochimie 73: 1345–1349.
7. Agris P. F., Vendeix F. A. P., Graham W. D. (2007) tRNA’s Wobble Decoding of the Genome: 40 Years of
Modification. J. Mol. Biol. 366: 1–13
8. Rovner A. J., Haimovich A. D., Katz S. R., Li Z., Grome M. W., Gassaway B. M., Amiram M., Patel J. R.,
Gallagher R. R., Rinehart J., Isaacs F. J. (2015) Recoded organisms engineered to depend on synthetic
amino acids. Nature 518: 89-93
9. Holley R. W., Everett G. A., Madison J. T., Zamir A. (1965) Nucleotide sequences in the yeast alanine
transfer ribonucleic acid. J. Biol. Chem. 240: 2122-2128
10. Sprinzl M., Horn C., Brown M., Ioudovitch A., Steinberg S. (1998) Compilation of tRNA sequences and
sequences of tRNA genes. Nucleic Acids Res. 26:148-153.
11. Korencic D., Polycarpo C., Weygand-Durasevic I., Söll D. (2004) Differential modes of transfer RNASer
recognition in Methanosarcina barkeri. J. Biol. Chem. 279: 48780-48786.
12. Bjork G. R., Ericson J. U., Gustafsson C. E. D., Hagervall T. G., Jonsson H., Wikstrom, P. M. (1987) Transfer
RNA modification. Annu. Rev. Biochem. 56: 263-287.
13. Lang B. F., Lavrov D., Beck N., Steinberg S. V. (2012) Mitochondrial tRNA Structure, Identity, and
Evolution of the Genetic Code. Organelle Genetics pp. 431-474.
14. Sprinzl M. & Vassilenko K. S. (2005) Compilation of tRNA sequences and sequences of tRNA genes.
Nucleic Acids Res. 33: 139-140.
15. De Bruijn M. H. & Klug A. (1983) A model for the tertiary structure of mammalian mitochondrial transfer
RNAs lacking the entire 'dihydrouridine' loop and stem. EMBO J. 2: 1309–1321.
16. Helm M., Brule H., Friede D., Giege R., Putz D., Florentz C. (2000) Search for characteristic structural
features of mammalian mitochondrial tRNAs. RNA 6: 1356-1379.
17. Schimmel P., Giegé R., Moras D., Yokoyama S. (1993) Proc. Natl. Acad. Sci. 90: 8763–8768.
18. Giegé R., Sissler M., Florentz C. (1998) Universal rules and idiosyncratic features in tRNA identity. Nucleic
Acids Res. 26: 5017-5035.
19. Ribas de Pouplana L. & Schimmel P. (2001) Aminoacyl-tRNA synthetases: potential markers of genetic
code development. Trends Biochem Sci. 26:591–596.
20. Giegé R. & Frugier M. (2000) Transfer RNA Structure and Identity; Madame Curie Bioscience Database
[Internet]. Austin (TX): Landes Bioscience
21. Frazer-Abel A. A. & Hagerman P. J. (2008) Core flexibility of a truncated metazoan mitochondrial tRNA.
Nucleic Acids Res. 36:5472-5481
22. Hopper A. K. and Phizicky E. M. (2003) tRNA transfers to the limelight. Genes Dev. 17: 162-180.
23. Hopper A. K. and Phizicky E. M. (2010) tRNA biology charges to the front. Genes Dev. 24: 1832-1860
24. Yoo C. J., Wolin S. L. (1997) The yeast La protein is required for the 3’ endonucleolytic cleavage that
matures tRNA precursors. Cell 89: 393–402.
25. Wolin S. L. & Matera A. G. (1999) The trials and travels of tRNA. Genes Dev. 13: 1-10.
26. Bayfield M. A., Maraia R. J. (2009) Precursor-product discrimination by La protein during tRNA
metabolism. Nat. Struct. Mol. Biol. 16: 430-437.

References
208
27. Wolin S. L., Cedervall T. (2002) The La protein. Annu. Rev. Biochem. 71: 375–403.
28. Van Horn D. J., Yoo C. J., Xue D., Shi H., Wolin S. L. (1997) The La protein in Schizosaccharomyces pombe:
a conserved yet dispensable phosphoprotein that functions in tRNA maturation. RNA 3: 1434–1443.
29. Fan H., Goodier J. L., Chamberlain J. R., Engelke D. R., Maraia R. J. (1998) 5' processing of tRNA
precursors can be modulated by the human La antigen phosphoprotein. Mol. Cell. Biol. 18: 3201–3211.
30. Apostolidi M., Vourtsis D. J., Chasapis C. T., Stathopoulos C., Bentrop D., Spyroulias G. A. (2014) ¹H, ¹⁵N,
¹³C assignment and secondary structure determination of two domains of La protein from D. discoideum.
Biomol. NMR Assign. 8: 47-51.
31. Chasapis C. T., Argyriou A. I., Apostolidi M., Konstantinidou P., Bentrop D., Stathopoulos C., Spyroulias G.
A. (2015) 1H, 13C and 15N backbone and side-chain resonance assignment of the LAM-RRM1 N-terminal
module of La protein from Dictyostelium discoideum. Biomol. NMR Assign. DOI: 10.1007/s12104-015-
9597-z.
32. Argyriou A. I., Chasapis C. T., Apostolidi M., Konstantinidou P., Stathopoulos C., Bentrop D., Spyroulias G.
A. (2015) Backbone and side chain NMR assignment, along with the secondary structure prediction of
RRM2 domain of La protein from a lower eukaryote exhibiting identical structural organization with its
human homolog. Biomol. NMR Assign. 9: 219-222.
33. Phizicky E. M., Alfonzo J. D. (2009) Do all modifications benefit all tRNAs? FEBS Lett. 584: 265–271.
34. Suzuki T. (2014) A complete landscape of post-transcriptional modifications in mammalian
mitochondrial tRNAs. Nucleic Acids Res. 42: 7346–7357.
35. Benne R., Van den Burg J., Brakenhoff J. P., Sloof P., Van Boom J. H., Tromp M. C. (1986) Major transcript
of the frameshifted coxII gene from trypanosome mitochondria contains four nucleotides that are not
encoded in the DNA. Cell 46: 819–826.
36. Spears J. L., Rubio M. A., Sample P. J., Alfonzo J. D. (2012) tRNA Biogenesis and Processing. RNA
Metabolism in Trypanosomes pp. 99-121.
37. Novoa E. M., Pavon-Eternod M., Pan T. Ribas de Pouplana L. (2012) A role for tRNA modifications in
genome structure and codon usage. Cell 149: 202–213.
38. Torres A. G., Batlle E., Ribas de Pouplana L. (2014) Role of tRNA modifications in human diseases. Trends
Mol. Med. 20: 306-314.
39. Kirchner S. & Ignatova Z. (2015) Emerging roles of tRNA in adaptive translation, signalling dynamics and
disease. Nat. Rev. Genet. 16: 98-112.
40. Chan P. P. & Lowe T. M. (2009) GtRNAdb: a database of transfer RNA genes detected in genomic
sequence. Nucleic Acids Res. 37: D93–D97.
41. Iben J. R. & Maraia R. J. (2014) tRNA gene copy number variation in humans. Gene 536: 376–384.
42. Parisien, M., Wang, X. & Pan, T. (2013) Diversity of human tRNA genes from the 1000‑genomes project.
RNA Biol. 10: 1853–1867.
43. Kutter C. et al. (2011) Pol III binding in six mammals shows conservation among amino acid isotypes
despite divergence among tRNA genes. Nature Genet. 43: 948–955.
44. Gingold H., Tehler D., Christoffersen N. R., Nielsen M. M., Asmar F., Kooistra S. M., Christophersen N. S.,
Christensen L. L., Borre M., Sørensen K. D., Andersen L. D., Andersen C. L., Hulleman E., Wurdinger T.,
Ralfkiær E., Helin K., Grønbæk K., Orntoft T., Waszak S. M., Dahan O., Pedersen J. S., Lund A. H., Pilpel Y.
(2014) A dual program for translation regulation in cellular proliferation and differentiation. Cell 158:
1281-1292.
45. Pesole G., Gissi C., De Chirico A., Saccone C. (1999) Nucleotide substitution rate of mammalian
mitochondrial genomes. J. Mol. Evol. 48: 427-34.

References
209
46. Allen J. F., Raven J. A. (1996) Free-radical-induced mutation vs redox regulation: Costs and benefits of
genes in organelles. J. Mol. Evol. 42: 482-92.
47. Zifa E., Giannouli S., Theotokis P., Stamatis C., Mamuris Z., Stathopoulos C. (2007) Mitochondrial tRNA
mutations: clinical and functional perturbations. RNA Biol. 4: 38-66.
48. Karaca E. et al. (2014) Human CLP1 mutations alter tRNA biogenesis, affecting both peripheral and
central nervous system function. Cell 157: 636–650.
49. Schaffer, A. E. et al. (2014) CLP1 founder mutation links tRNA splicing and maturation to cerebellar
development and neurodegeneration. Cell 157: 651–663.
50. Yadavalli S. S. & Ibba M. (2012) Quality control in aminoacyl-tRNA synthesis: its role in translational
fidelity. Adv. Protein Chem. Struct. Biol. 86: 1−43.
51. Woese C. R., Olsen G. J., Ibba M., Söll D. (2000) Aminoacyl-tRNA synthetases, the genetic code, and the
evolutionary process. Microbiol. Mol. Biol. Rev. 64: 202−236.
52. O’Donoghue P. & Luthey-Schulten Z. (2003) On the evolution of structure in aminoacyl-tRNA
synthetases. Microbiol. Mol. Biol. Rev. 67: 550−573.
53. Schimmel P., Ribas De Pouplana L. (1995) Transfer RNA: from minihelix to genetic code. Cell 81: 983–
986.
54. Schimmel P., Giegé R., Moras D., Yokoyama S. (1993) An operational RNA code for amino acids and
possible relationship to genetic code. Proc. Natl. Acad. Sci. USA 90: 8763–8768.
55. Ahel I., Korencic D., Ibba M., Söll D. (2003) Trans-editing of mischarged tRNAs. Proc. Natl. Acad. Sci. USA
100: 15422–15427.
56. Cusack S. (1993) Sequence, structure and evolutionary relationships between class 2 aminoacyl-tRNA
synthetases: an update. Biochimie 75: 1077–1081.
57. Burbaum J. J. & Schimmel, P. (1991) Structural relationships and the classification of aminoacyl-tRNA
synthetases. J. Biol. Chem. 266: 16965–16968.
58. Eriani G., Delarue M., Poch O., Gangloff J., Moras D. (1990) Partition of tRNA synthetases into two
classes based on mutually exclusive sets of sequence motifs. Nature 347: 203–206.
59. Mascarenhas A. P., An S., Rosen A. E., Martinis S. A., Musier-Forsyth K. (2009) Fidelity mechanisms of the
aminoacyl-tRNA synthetases. Protein Engineering pp. 155–203. Springer-Verlag, Berlin, Heidelberg.
60. Yaremchuk A., Kriklivyi I., Tukalo M., Cusack S. (2002). Class I tyrosyl-tRNA synthetase has a class II mode
of cognate tRNA recognition. EMBO J. 21: 3829–3840.
61. Carter C. W. (1993). Cognition, mechanism, and evolutionary relationships in aminoacyl-tRNA
synthetases. Annu. Rev. Biochem. 62: 715–748.
62. Perona J. J. & Hadd A. (2012) Structural diversity and protein engineering of the aminoacyl-tRNA
synthetases. Biochemistry 51: 8705-8729.
63. Blight S. K., Larue R. C., Mahapatra A., Longstaff D. G., Chang E., Zhao G., Kang P. T., Green-Church K. B.,
Chan M. K., Krzycki, J. A. (2004) Direct charging of tRNA (CUA) with pyrrolysine in vitro and in vivo.
Nature 431: 333−335.
64. Park H. S., Hohn M. J., Umehara T., Guo L. T., Osborne E. M., Benner J., Noren C. J., Rinehart J., Söll D.
(2011) Expanding the genetic code of Escherichia coli with phosphoserine. Science 333: 1151−1154.
65. Sauerwald A., Zhu W., Major T. A., Roy H., Palioura S., Jahn, D., Whitman W. B., Yates J. R. III, Ibba M.,
Söll D. (2005) RNA-dependent cysteine biosynthesis in archaea. Science 307: 1969−1972.
66. Fukunaga R., & Yokoyama S. (2007) Structural insights into the first step of RNA-dependent cysteine
biosynthesis in archaea. Nat. Struct. Mol. Biol. 14: 272−279.
67. Mosyak L., Reshetnikova L., Goldgur Y., Delarue M., Safro, M. G. (1995) Structure of phenylalanyl-tRNA
synthetase from Thermus thermophilus. Nat. Struct. Biol. 2: 537−547.

References
210
68. Srinivasan G., James C. M., Krzycki J. A. (2002) Pyrrolysine encoded by UAG in Archaea: Charging of a
UAG-decoding specialized tRNA. Science 296: 1459−1462.
69. Polycarpo C., Ambrogelly A., Berube A., Winbush S. M., McCloskey J. A., Crain P. F., Wood J. L., Söll D.
(2004) An aminoacyl-tRNA synthetase that specifically activates pyrrolysine. Proc. Natl. Acad. Sci. U.S.A.
101: 12450−12454.
70. Ling J., Reynolds N., Ibba M. (2009) Aminoacyl-tRNA synthesis and translational quality control. Annu.
Rev. Microbiol. 63: 61–78.
71. Splan K. E., Ignatov M. E., Musier-Forsyth K. (2008) Transfer RNA modulates the editing mechanism used
by class II prolyl-tRNA synthetase. J. Biol. Chem. 283: 7128–7134.
72. Gruic-Sovulj I., Rokov-Plavec J., Weygand-Durasevic I. (2007) Hydrolysis of non-cognate aminoacyl-
adenylates by a class II aminoacyl-tRNA synthetase lacking an editing domain. FEBS Lett. 581: 5110–
5114.
73. Sheppard K., Yuan J., Hohn M. J., Jester B., Devine K. M., Söll D. (2008). From one amino acid to another:
tRNA-dependent amino acid biosynthesis. Nucleic Acids Res. 36: 1813–1825.
74. Curnow A. W., Hong K., Yuan R., Kim S., Martins O., Winkler W., Henkin T. M., Söll D. (1997) Glu-tRNAGln
amidotransferase: a novel heterotrimeric enzyme required for correct decoding of glutamine codons
during translation. Proc. Natl. Acad. Sci. USA 94: 11819–11826.
75. Tumbula D. L., Becker H. D., Chang W. Z., Söll D. (2000) Domain-specific recruitment of amide amino
acids for protein synthesis. Nature 407: 106–110.
76. Nakamura A., Yao M., Chimnaronk S., Sakai N., Tanaka I. (2006) Ammonia channel couples glutaminase
with transamidase reactions in GatCAB. Science 312: 1954–1958.
77. Oshikane H., Sheppard K., Fukai S., Nakamura Y., Ishitani R., Numata T., Sherrer R. L., Feng L., Schmitt E.,
Panvert M., Blanquet S., Mechulam Y., Söll D., Nureki O. (2006) Structural basis of RNA-dependent
recruitment of glutamine to the genetic code. Science 312: 1950–1954.
78. Bailly M., Blaise M., Lorber B., Becker H. D., Kern D. (2007) The transamidosome: a dynamic
ribonucleoprotein particle dedicated to prokaryotic tRNA-dependent asparagine biosynthesis. Mol. Cell
28: 228–239.
79. Ito T., Yokoyama S. (2010) Two enzymes bound to one transfer RNA assume alternative conformations
for consecutive reactions. Nature. 467: 612-616.
80. Frechin M., Duchêne A. M., Becker H. D. (2009) Translating organellar glutamine codons: a case by case
scenario? RNA Biol. 6: 31–34.
81. Rinehart J., Krett B., Rubio M. A., Alfonzo J. D., Söll D. (2005) Saccharomyces cerevisiae imports the
cytosolic pathway for Gln-tRNA synthesis into the mitochondrion. Genes Dev. 19: 583–592.
82. Rinehart J., Horn E. K., Wei D., Söll D., Schneider A. (2004) Non-canonical eukaryotic glutaminyl- and
glutamyl-tRNA synthetases form mitochondrial aminoacyl-tRNA in Trypanosoma brucei. J. Biol. Chem.,
279: 1161–1166.
83. Nagao A., Suzuki T., Katoh T., Sakaguchi Y., Suzuki,T. (2009) Biogenesis of glutaminyl-mt tRNAGln in
human mitochondria. Proc. Natl. Acad. Sci. U.S.A. 106: 16209–16214.
84. Pujol C., Bailly M., Kern D., Maréchal-Drouard L., Becker H., Duchêne A. M. (2008) Dual-targeted tRNA-
dependent amidotransferase ensures both mitochondrial and chloroplastic Gln-tRNAGln synthesis in
plants. Proc. Natl. Acad. Sci. U.S.A. 105: 6481–6485.
85. Kim S. I., Stange-Thomann N., Martins O., Hong K. W., Söll D., Fox T. D. (1997) A nuclear genetic lesion
affecting Saccharomyces cerevisiae mitochondrial translation is complemented by a homologous Bacillus
gene. J. Bact. 179: 5625–5627.

References
211
86. Sheppard K. & Söll D. (2008) On the evolution of the tRNA-dependent amidotransferases, GatCAB and
GatDE. J. Mol. Biol. 377: 831-44
87. Frechin M., Senger B., Brayé M., Kern D., Martin R. P., Becker H. D. (2009) Yeast mitochondrial Gln-
tRNA(Gln) is generated by a GatFAB-mediated transamidation pathway involving Arc1p-controlled
subcellular sorting of cytosolic GluRS. Genes Dev. 23: 1119–1130.
88. Araiso Y., Huot J. L., Sekiguchi T., Frechin M., Fischer F., Enkler L., Senger B., Ishitani R., Becker H. D.,
Nureki O. (2014) Crystal structure of Saccharomyces cerevisiae mitochondrial GatFAB reveals a novel
subunit assembly in tRNA-dependent amidotransferases. Nucleic Acids Res. 42: 6052-6063.
89. Negrutskii, B. S. & Deutscher M. P. (1991) Channeling of aminoacyl-tRNA for protein synthesis in vivo.
Proc. Natl. Acad. Sci. U.S.A. 88: 4991−4995.
90. Kerjan P., Cerini C., Semeriva M., Mirande M. (1994) The multienzyme complex containing nine
aminoacyl-tRNA synthetases is ubiquitous from Drosophila to mammals. Biochim. Biophys. Acta 1199:
293−297.
91. Kyriacou S. V. & Deutscher M. P. (2008) An important role for the multienzyme aminoacyl-tRNA
synthetase complex in mammalian translation and cell growth. Mol. Cell 29: 419−427.
92. Mirande M. (2010) Processivity of translation in the eukaryote cell: Role of aminoacyl-tRNA synthetases.
FEBS Lett. 584: 443−447.
93. Guo M., Schimmel P., Yang X. L. (2010) Functional expansion of human tRNA synthetases achieved by
structural inventions. FEBS Lett. 584: 434−442.
94. Guo M., Yang X. L., Schimmel P. (2010) New functions of aminoacyl-tRNA synthetases beyond
translation. Nat. Rev. Mol. Cell Biol. 11: 668−674.
95. Ko Y. G., Kim E. Y., Kim T., Park H., Park H. S., Choi E. J., Kim S. (2001) Glutamine-dependent
antiapoptotic interaction of human glutaminyl-tRNA synthetase with apoptosis signal-regulating kinase
1. J. Biol. Chem. 276: 6030−6036.
96. Han J. M., Jeong S. J., Park M. C., Kim G., Kwon N. H., Kim H. K., Ha S. H., Ryu S. H., Kim S. (2012) Leucyl-
tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell 149: 410−424.
97. Park S. G., Schimmel P., Kim S. (2008) Aminoacyl tRNA synthetases and their connections to disease.
Proc. Natl. Acad. Sci. U.S.A. 105: 11043−11049.
98. Kim S., You S., Hwang D. (2011) Aminoacyl-tRNA synthetases and tumorigenesis: More than
housekeeping. Nat. Rev. Cancer 11: 708−718.
99. Kaminska M., Shalak V., Mirande M. (2001) The appended C-domain of human methionyl-tRNA
synthetase has a tRNA-sequestering function. Biochemistry 40: 14309−14316.
100. Liu C., Gamper H., Shtivelband S., Hauenstein S., Perona J. J., Hou Y. M. (2007) Kinetic quality control of
anticodon recognition by a eukaryotic aminoacyl-tRNA synthetase. J. Mol. Biol. 367: 1063−1078.
101. Park S. G., Ewalt K. L., Kim S. (2005) Functional expansion of aminoacyl tRNA-synthetases and their
interacting factors: new perspectives on housekeepers. Trends Biochem. Sci. 30: 569–574.
102. Ray P. S., Arif A., Fox P. L. (2007) Macromolecular complexes as depots for releasable regulatory
proteins. Trends Biochem. Sci. 32: 158–164.
103. Ko Y. G., Kang Y. S., Kim E. K., Park S. G., Kim S. (2000) Nucleolar localization of human methionyl-tRNA
synthetase and its role in ribosomal RNA synthesis. J. Cell Biol. 149: 567–574.
104. Simos G., Segref A., Fasiolo F., Hellmuth K., Shevchenko A., Mann M., Hurt E. C. (1996) The yeast protein
Arc1p binds to tRNA and functions as a cofactor for the methionyl- and glutamyl-tRNA synthetases.
EMBO J. 15: 5437–5448.
105. Frechin M., Enkler L., Tetaud E., Laporte D., Senger B., Blancard C., Hammann P., Bader G., Clauder-
Münster S., Steinmetz L. M., Martin R. P., di Rago J. P., Becker H. D. (2014) Expression of nuclear and

References
212
mitochondrial genes encoding ATP synthase is synchronized by disassembly of a multisynthetase
complex. Mol Cell. 56: 763-776.
106. Kao J., Houck K., Fan Y., Haehnel I., Libutti S. K., Kayton M. L., Grikscheit T., Chabot J., Nowygrod R.,
Greenberg S., et al. (1994) Characterization of a novel tumor-derived cytokine. Endothelial-monocyte
activating polypeptide II. J. Biol. Chem. 269: 25106–25119.
107. Mukhopadhyay R., Jia J., Arif A., Ray P. S., Fox P. L. (2009) The GAIT system: a gatekeeper of
inflammatory gene expression. Trends. Biochem. Sci. 34: 324–331.
108. Wakasugi K., Slike B. M., Hood J., Ewalt K. L., Cheresh D. A., Schimmel P. (2002) Induction of
angiogenesis by a fragment of human tyrosyl-tRNA synthetase. J. Biol. Chem. 277: 20124–20126.
109. Rho S. B., Kim M. J., Lee J. S., Seol W., Motegi H., Kim S., Shiba K. (1999) Genetic dissection of protein–
protein interactions in multi-tRNA synthetase complex. Proc. Natl Acad. Sci. USA 96: 4488–4493.
110. Targoff I. N., Trieu E. P., Miller F. W. (1993) Reaction of anti-OJ autoantibodies with component of the
multienzyme complex of aminoacyl-tRNA synthetases in addition to isoleucyl-tRNA synthetase. J. Clin.
Invest. 91: 25556–25564.
111. Hirakata M., Suwa A., Takeda Y., Matsuoka Y., Irimajiri S., Targoff I. N., Hardin J. A., Craft J. (1996)
Autoantibodies to glycl-transfer RNA synthetase in myositis. Association with dermatomyositis and
immunologic heterogeneity. Arthritis Rheum. 39: 146–151
112. Kleeman T. A., Wei D., Simpson K. L., First E. A. (1997) Human tyrosyl-tRNA synthetase shares amino acid
sequence homology with a putative cytokine. J. Biol. Chem. 272: 14420–14425.
113. Miyaki M., Iijima T., Shiba K., Aki T., Kita Y., Yasuno M., Mori T., Kuroki T., Iwama T. (2001) Alternations in
repeated sequences in 5' upstream and coding regions in colorectal tumors from patients with
hereditary nonpolyposis colorectal cancer and Turcot syndrome. Oncogene 20: 5215–5218.
114. Raina M. & Ibba M. (2014) tRNAs as regulators of biological processes. Front Genet. 5: 171.
115. Navarre W. W. & Schneewind O. (1999) Surface proteins of gram-positive bacteria and mechanisms of
their targeting to the cell wall envelope. Microbiol. Mol. Biol. Rev. 63: 174–229.
116. Roy H. & Ibba M. (2008) RNA-dependent lipid remodeling by bacterial multiple peptide resistance
factors. Proc. Natl Acad. Sci. USA 105: 4667–4672.
117. Yamato M., Iinuma H., Naganawa H., Yamagishi Y., Hamada M., Masuda T., Umezawa H., Abe V., Hori M.
(1986) Isolation and properties of valanimycin, a new azoxy antibiotic. J. Antibiot. 39: 184–191.
118. Jahn D., Verkamp E. & Soll D. (1992) Glutamyl-transfer RNA: a precursor of heme and chlorophyll
biosynthesis. Trends Biochem. Sci. 17: 215–218.
119. Mogk A., Schmidt R., Bukau B. (2007) The N-end rule pathway for regulated proteolysis: prokaryotic and
eukaryotic strategies. Trends Cell Biol. 17: 165–172.
120. Francklyn C. S. & Minajigi A. (2010) tRNA as an active chemical scaffold for diverse chemical
transformations FEBS Lett. 584: 366–375.
121. Royet J. & Dziarski R. (2007) Peptidoglycan recognition proteins: pleiotropic sensors and effectors of
antimicrobial defences. Nat. Rev. Microbiol. 5: 264–277.
122. Vollmer W., Blanot D., de Pedro M. A. (2008) Peptidoglycan structure and architecture. FEMS Microbiol.
Rev. 32: 149–167.
123. Vetting M. W., S de Carvalho L. P., Yu M., Hegde S. S., Magnet S., Roderick S. L., Blanchard J. S. (2005)
Structure and functions of the GNAT superfamily of acetyltransferases. Arch. Biochem. Biophys. 433:
212–226.
124. Giannouli S., Labrou M., Kyritsis A., Ikonomidis A., Pournaras S., Stathopoulos C., Tsakris A. (2010)
Detection of mutations in the FemXAB protein family in oxacillin-susceptible mecA-positive
Staphylococcus aureus clinical isolates. J. Antimicrob. Chemother. 65: 626-33.

References
213
125. Ernst C. M. & Peschel A. (2011) Broad-spectrum antimicrobial peptide resistance by MprF-mediated
aminoacylation and flipping of phospholipids. Mol. Microbiol. 80: 290–299.
126. Roy H. & Ibba M. (2009) Broad range amino acid specificity of RNA-dependent lipid remodeling by
multiple peptide resistance factors. J. Biol. Chem. 284: 29677–29683.
127. Dare K. & Ibba M. (2012) Roles of tRNA in cell wall biosynthesis. Wiley Interdiscip. Rev. RNA 3: 247–264.
128. Shepherd J. & Ibba M. (2013) Direction of aminoacylated transfer RNAs into antibiotic synthesis and
peptidoglycan-mediated antibiotic resistance. FEBS Lett. 587: 2895–2904.
129. Garg R. P., Ma Y., Hoyt J. C., Parry R. J. (2002) Molecular characterization and analysis of the biosynthetic
gene cluster for the azoxy antibiotic valanimycin. Mol. Microbiol. 46: 505–517.
130. Garg R. P. & Parry R. J. (2010) Regulation of valanimycin biosynthesis in Streptomyces viridifaciens:
characterization of VlmI as a Streptomyces antibiotic regulatory protein (SARP). Microbiology 156: 472–
483.
131. Lautru S., Gondry M., Genet R., Pernodet J. L.(2002).The albonoursin gene cluster of S. noursei
biosynthesis of diketopiperazine metabolites independent of nonribosomal peptide synthetases. Chem.
Biol. 9: 1355–1364.
132. Sauguet L., Moutiez M., Li Y., Belin P., Seguin J., Le Du M. H., Thai R., Masson C., Fonvielle M., Pernodet J.
L., Charbonnier J. B., Gondry M. (2011). Cyclodipeptide synthases,a family of class-Iaminoacyl-tRNA
synthetase-like enzymes involved in non-ribosomal peptide synthesis. Nucleic Acids Res. 39: 4475–4489.
133. Tobias J. W., Shrader T. E., Rocap G., Varshavsky A. (1991) The N-end rule in bacteria. Science 254: 1374–
1377.
134. Suto K., Shimizu Y., Watanabe K., Ueda T., Fukai S., Nureki O., Tomita K. (2006) Crystal structures of
leucyl/phenylalanyl-tRNA-protein transferase and its complex with an aminoacyl-tRNA analog. EMBO J.
25: 5942–5950.
135. Yuan J., Palioura S., Salazar J. C., Su D., O'Donoghue P., Hohn M. J., Cardoso A. M., Whitman W. B., Söll D.
(2006) RNA-dependent conversion of phosphoserine forms selenocysteine in eukaryotes and archaea.
Proc. Natl. Acad. Sci. USA 103: 18923–18927.
136. Huang D. D., Wang W. Y., Gough S. P., Kannangara C. G. (1984) delta-Aminolevulinic acid-synthesizing
enzymes need an RNA moiety for activity. Science 225: 1482–1484.
137. Levican G., Katz A., de Armas M., Nunez H., Orellana O. (2007) Regulation of a glutamyl-tRNA synthetase
by the heme status. Proc. Natl. Acad. Sci. USA 104: 3135–3140.
138. Cashel M. & Gallant J. (1969) Two compounds implicated in the function of the RC gene of Escherichia
coli. Nature 221: 838–841.
139. Haseltine W. A. & Block R. (1973). Synthesis of guanosine tetra- and pentaphosphate requires the
presence of a codon-specific, uncharged transfer ribonucleic acid in the acceptor site of ribosomes.
Proc.Natl.Acad.Sci.U.S.A. 70: 1564–1568.
140. Ross W., Vrentas C. E., Sanchez-Vazquez P., Gaal T., Gourse, R. L. (2013). The magic spot: a ppGpp
binding site on E. coli RNA polymerase responsible for regulation of transcription initiation. Mol.Cell 50:
420–429.
141. Henkin T. M. & Yanofsky C. (2002). Regulation by transcription attenuation in bacteria: how RNA
provides instructions for transcription termination/antitermination decisions. Bioessays 24: 700–707.
142. Green N. J., Grundy F. J., Henkin T. M. (2010) The T box mechanism: tRNA as a regulatory molecule. FEBS
Lett. 584: 318–324.
143. Grundy F. J., Yousef M. R., Henkin T. M. (2005) Monitoring uncharged tRNA during transcription of the
Bacillus subtilis glyQS gene. J. Mol. Biol. 346: 73–81.

References
214
144. Saad N. Y., Stamatopoulou V., Brayé M., Drainas D., Stathopoulos C., Becker, H. D. (2013) Two-codon T-
box riboswitch binding two tRNAs. Proc. Natl. Acad. Sci. U.S.A. 110: 12756–12761.
145. Wek S. A., Zhu S., Wek R. C. (1995) The histidyl-tRNA synthetase-related sequence in the eIF2 alpha
protein kinase GCN2 interacts with tRNA and is required for activation in response to starvation for
different amino acids. Mol.Cell Biol. 15: 4497–4506.
146. Marton M. J., Vazquez de Aldana C. R., Qiu H., Chakraburtty K., Hinnebusch A. G. (1997) Evidence that
GCN1 and GCN20, translational regulators of GCN4, function on elongating ribosomes in activation of
eIF2 alpha kinase GCN2. Mol. Cell. Biol. 17: 4474–4489.
147. Lee Y. S., Shibata Y., Malhotra A., Dutta A. (2009) A novel class of small RNAs: tRNA-derived RNA
fragments (tRFs). Genes Dev. 23: 2639–2649.
148. Fu H., Feng J., Liu Q., Sun F., Tie Y., Zhu J., Xing R., Sun Z., Zheng X. (2009) Stress induces tRNA cleavage
by angiogenin in mammalian cells. FEBS Lett. 583: 437–442.
149. Thompson D. M. & Parker R. (2009) The RNase Rny1p cleaves tRNAs and promotes cell death during
oxidative stress in Saccharomyces cerevisiae. J. Cell Biol. 185: 43–50.
150. Gebetsberger J. & Polacek N. (2013) Slicing tRNAs to boost functional ncRNA diversity. RNA Biol. 10:
1798–1806.
151. Cole C., Sobala A., Lu C., Thatcher S. R., Bowman A., Brown J. W., Green P. J., Barton G. J., Hutvagner G.
(2009) Filtering of deep sequencing data reveals the existence of abundant Dicer-dependent small RNAs
derived from tRNAs. RNA 15: 2147–2160.
152. Ivanov P., Emara M. M., Villen J., Gygi S. P., Anderson P. (2011) Angiogenin-induced tRNA fragments
inhibit translation initiation. Mol. Cell 43: 613–623.
153. Gebetsberger J., Zywicki M., Künzi A., Polacek N. (2012). tRNA-derived fragments target the ribosome
and function as regulatory non-coding RNA in Haloferax volcanii. Archaea 2012: 260909.
154. Maute R. L., Schneider C., Sumazin P., Holmes A., Califano A., Basso K., Dalla-Favera R. (2013). tRNA-
derived microRNA modulates proliferation and the DNA damage response and is down-regulated in
Bcell lymphoma. Proc. Natl. Acad. Sci. U.S.A. 110: 1404–1409.
155. Ruggero K., Guffanti A., Corradin A., Sharma V. K., De Bellis G., Corti G., Grassi A., Zanovello P., Bronte V.,
Ciminale V., D'Agostino D. M. (2014) Small non coding RNAs in cells transformed by human T-cell
leukemia virus type 1: a role for a tRNA fragment as a primer for reverse transcriptase. J. Virol. 88: 3612–
3622.
156. Hanada T., Weitzer S., Mair B., Bernreuther C., Wainger B. J., Ichida J., Hanada R., Orthofer M., Cronin S.
J., Komnenovic V., Minis A., Sato F., Mimata H., Yoshimura A., Tamir I., Rainer J., Kofler R., Yaron A.,
Eggan K. C., Woolf C. J., Glatzel M., Herbst R., Martinez J., Penninger J. M. (2013) CLP1 links tRNA
metabolism to progressive motor-neuron loss. Nature 495: 474–480.
157. Kumar P., Mudunuri S. B., Anaya J., Dutta A. (2015) tRFdb: a database for transfer RNA fragments.
Nucleic Acids Res. DOI: 10.1093/nar/gku1138.
158. Thompson C. B. (1995) Apoptosis in the pathogenesis and treatment of disease. Science 267: 1456–
1462.
159. Li J. & Yuan J. (2008) Caspases in apoptosis and beyond. Oncogene 27: 6194–6206.
160. Mei Y., Yong J., Liu H., Shi Y., Meinkoth J., Dreyfuss G., Yang X. (2010). tRNA binds to cytochrome c and
inhibits caspase activation. Mol.Cell 37: 668–678.
161. Gorla M. & Sepuri N. B. (2014) Perturbation of apoptosis upon binding of tRNA to the heme domain of
cytochrome c. Apoptosis 19: 259–268.
162. Marshall L. & White R. J. (2008) Non-coding RNA production by RNA polymerase III is implicated in
cancer. Nat. Rev. Cancer 8: 911–914.

References
215
163. Pavon-Eternod M., Gomes S., Geslain R., Dai Q., Rosner M. R., Pan T. (2009) tRNA over-expression in
breast cancer and functional consequences. Nucleic Acids Res. 37: 7268–7280.
164. Hou Y. M. & Yang X. (2013). Regulation of cell death by transfer RNA. Antioxid. Redox. Signal. 19: 583–
594.
165. Yanofsky C. (1988) Transcription Attenuation. J. Biol. Chem. 263: 609-612.
166. Henkin T. M. (1996) Control of transcription termination in prokaryotes. Annu. Rev. Genet. 30: 35–57.
167. Henkin T. M. (2000) Transcription termination control in bacteria. Curr. Opin. Microbiol. 3: 149–153.
168. Yanofsky C. (1981) Attenuation in the control of expression of bacterial operons. Nature 289: 751–758.
169. Grundy F. J. & Henkin T. M. (1993) tRNA as a positive regulator of transcription antitermination in
Bacillus subtilis. Cell 74: 475–482.
170. Merino E. & Yanofsky C. (2005) Transcription attenuation: a highly conserved regulatory strategy used
by bacteria. Trends Genet. 21: 260-264.
171. Antson A. A., Otridge J., Brzozowski A. M., Dodson E. J., Dodson G. G., Wilson K. S., Smith T. M., Yang M.,
Kurecki T., Gollnick P. (1995) The structure of the trp RNA attenuation protein. Nature 374: 693–700.
172. Amster-Choder O. & Wright A. (1993) Transcriptional regulation of the bgl operon of Escherichia coli
involves phosphotransferase system-mediated phosphorylation of a transcriptional antiterminator. J.
Cell Biochem. 51: 83–90.
173. Chen Q., Postma P. W., Amster-Choder O. (2000) Dephosphorylation of the Escherichia coli
transcriptional antiterminator BglG by the sugar sensor BglF is the reversal of its phosphorylation. J.
Bacteriol 182: 2033–2036.
174. Weisberg R. A. & Gottesman M. E. (1999) Processive antitermination. J. Bacteriol 181: 359–367.
175. DeVito J. & Das A. (1994) Control of transcription proccessivity in phage lambda: Nus factors strengthen
the termination-resistant state of RNA polymerase induced by N antiterminator. Proc Natl Acad Sci USA
91: 8660–8664.
176. Gong F., Ito K., Nakamura Y., Yanofsky C. (2001) The mechanism of tryptophan induction of
tryptophanase operon expression: tryptophan inhibits release factor-mediated cleavage of TnaC-
peptidyl-tRNAPro. Proc Natl Acad Sci USA 98: 8997–9001.
177. Vitreschak A. G., Rodionov D. A., Mironov A. A., Gelfand M. S. (2003) Regulation of the vitamin B12
metabolism and transport in bacteria by a conserved RNA structural element. RNA 9: 1084–1097.
178. Breaker R. R. (2011) Prospects for riboswitch discovery and analysis. Mol Cell. 43: 867-879.
179. Wethmar K., Smink J. J., Leutz A. (2010) Upstream open reading frames: molecular switches in
(patho)physiology. Bioessays 32: 885–893.
180. Eddy S. R. (2001) Non-coding RNA genes and the modern RNA world. Nat Rev Genet 2: 919–929.
181. Roth A. & Breaker R. R. (2009) The structural and functional diversity of metabolite-binding
riboswitches. Annu Rev Biochem. 78: 305-334.
182. Gilbert W. (1986) Origin of life: The RNA world. Nature 319: 618.
183. Breaker R. R. (2006) Riboswitches and the RNA world. The RNA World (3rd Edition). Gesteland R.F., Cech
T.R., Atkins J. F. (Eds). Cold Spring Harbor Laboratory Press, NY, USA, pp. 89-107.
184. Sudarsan N., Barrick J. E., Breaker R. R. (2003) Metabolite-binding RNA domains are present in the genes
of eukaryotes. RNA 9: 644-647.
185. Nahvi A., Sudarsan N., Ebert M. S., Zou X., Brown K. L., Breaker R. R. (2002) Genetic control by a
metabolite binding mRNA. Chem. Biol. 9: 1043–1049.
186. Klinkert B. & Narberhaus F. (2009) Microbial thermosensors. Cell. Mol. Life Sci. 66: 2661–2676.
187. Sudarsan N., Hammond M. C., Block K. F., Welz R., Barrick J. E., Roth A., Breaker R. R. (2006). Tandem
riboswitch architectures exhibit complex gene control functions. Science 314: 300–304.

References
216
188. Lee E. R., Baker J. L., Weinberg Z., Sudarsan N., Breaker R. R. (2010) An allosteric self-splicing ribozyme
triggered by a bacterial second messenger. Science 329: 845–848.
189. Narberhaus F., Waldminghaus T., Chowdbury S. (2006) RNA thermometers. FEMS Microbiol. Rev. 30: 3–
16.
190. Morita M. T., Tanaka Y., Kodama T. T., Kyogoku Y., Yanagi H., Yura T. (1999) Translational induction of
heat shock transcription factor ς32: Evidence for a built-in RNA thermosensor. Genes Dev. 13: 655–665.
191. Johansson J., Mandin P., Renzoni A., Chiaruttini C., Springer M., Cossart P. (2002) An RNA thermosensor
controls expression of virulence genes in Listeria monocytogenes. Cell 110: 551–561.
192. Henkin T. M. (2008) Riboswitch RNAs: using RNA to sense cellular metabolism. Genes Dev. 22:3383-
3390.
193. Mandal M., Lee M., Barrick J. E., Weinberg Z., Emilsson G. M., Ruzzo W. L., Breaker R. R. (2004) A glycine-
dependent riboswtch that uses cooperative binding to control gene expression. Science 306: 275–279.
194. Sun E. I., Leyn S. A., Kazanov M. D., Saier M. H. Jr., Novichkov P. S., Rodionov D. A. (2013) Comparative
genomics of metabolic capacities of regulons controlled by cis-regulatory RNA motifs in bacteria. BMC
Genomics 14: 597.
195. Price I. R., Gaballa A., Ding F., Helmann J. D., Ke A. (2015) Mn(2+)-Sensing Mechanisms of yybP-ykoY
Orphan Riboswitches. Mol Cell. 57: 1110-1123.
196. Andre G., Even S., Putzer H., Burquiere P., Croux C., Danchin A., Martin-Verstraete I., Soutourina O.
(2008) S-box and T-box riboswitches and antisense RNA control a sulfur metabolic operon of Clostridium
acetobutylicum. Nucleic Acids Res. 36: 5955–5969.
197. Henkin T. M., Glass B. L., Grundy F. J. (1992) Analysis of the Bacillus subtilis tyrS gene: conservation of a
regulatory sequence in multiple tRNA synthease genes. J. Bacteriol. 174: 1299–1306.
198. Yousef M. R., Grundy F. J., Henkin T.M. (2005) Structural transitions induced by the interaction between
tRNAGly and the Bacillus subtilis glyQS T box leader RNA. J. Mol. Biol. 349: 273–287.
199. Vitreschak A. G., Mironov A. M., Lyubetsky V. A., Gelfand M. S. (2008) Comparative genomic analysis of
T-box regulatory systems in bacteria. RNA 14: 717–735.
200. Gutierrez-Preciado A., Henkin T. M., Yanofsky C., Merino E. (2009) RNA-based T box regulation: new
insights revealed by comparative genomics. Microbiol. Mol. Biol. Rev. 73: 36–61.
201. Winkler W. C., Grundy F. J., Murphy B. A., Henkin T. M. (2001) The GA motif: an RNA element common
to bacterial antitermination systems, rRNA, and eukaryotic RNAs. RNA 7: 1165–1172.
202. Rollins S. M., Grundy F. J., Henkin T. M. (1997) Analysis of cis-acting sequence and structural elements
required for antitermination of the Bacillus subtilis tyrS gene. Mol. Microbiol. 25: 411–421.
203. Sherwood A. V., Grundy F. J., Henkin T. M. (2015) T box riboswitches in Actinobacteria: translational
regulation via novel tRNA interactions. Proc. Natl. Acad. Sci. U S A 112: 1113-1118.
204. Grundy F. J., Moir T. R., Haldeman M. T., Henkin T. M. (2002) Sequence requirements for terminators
and antiterminators in the T-box transcription antitermination system: disparity between conservation
and functional requirements. Nucleic Acids Res. 30: 1646–1655.
205. Gerdeman M. S., Henkin T. M., Hines J. V. (2003) Solution structure of the B. subtilis T box
antiterminator RNA: seven-nucleotide bulge characterized by stacking and flexibility. J. Mol. Biol. 326:
189–201.
206. Grundy F. J., Haldeman M. T., Hornblow G. M., Ward J. M., Chalker A. F., Henkin T. M. (1997) The
Staphylococcus aureus ileS gene, encoding isoleucyl-tRNA synthetase, is a member of the T-box family. J.
Bacteriol. 179: 3767–3772.
207. Seliverstov A. V., Putzer H., Gelfand M. S., Lyubetsky V. A. (2005) Comparative analysis of RNA regulatory
elements of amino acid metabolism genes in Actinobacteria. BMC Microbiol. 5: 54.

References
217
208. Gendron N., Putzer H., Grunberg-Manago M. (1994) Expression of both Bacillus subtilis threonyl-tRNA
synthetase genes is autogenously regulated. J. Bacteriol. 176: 486–494.
209. Garrity D. B. & Zahler S. A. (1994) Mutations in the gene for a tRNA that functions as a regulator of a
transcriptional attenuator in Bacillus subtilis. Genetics 137: 627–636.
210. Shivers R. P. & Sonenshein A. L. (2005) Bacillus subtilis ilvB operon: An intersection of global regulons.
Mol. Microbiol. 56: 1549–1559.
211. Williams-Wagner R. N., Grundy F. J., Raina M., Ibba M., Henkin T. M. (2015) The Bacillus subtilis tyrZ
gene encodes a highly selective tyrosyl-tRNA synthetase and is regulated by a MarR regulator and T Box
riboswitch. J Bacteriol. 197: 1624-1631.
212. Wang J. & Nikonowicz E. P. (2011) Solution structure of the K-turn and Specifier Loop domains from the
Bacillus subtilis tyrS T box leader RNA. J. Mol. Biol. 408: 99–117.
213. Chang A. T. & Nikonowicz E. P. (2013) Solution NMR determination of hydrogen bonding and base
pairing between the glyQS T box riboswitch Specifier domain and the anticodon loop of tRNAGly. FEBS
Lett. 587: 3495–3499.
214. Grigg J. C., Chen Y., Grundy F. J., Henkin T. M., Pollack L., Ke A. (2013) T box RNA decodes both the
information content and geometry of tRNA to affect gene expression. Proc. Natl. Acad. Sci. U. S. A. 110:
7240–7245.
215. Zhang J. & Ferre-D'Amaré A. R. (2013) Co-crystal structure of a T-box riboswitch stem I domain in
complex with its cognate tRNA. Nature 500: 363–366.
216. Zhang J. & Ferré-D'Amaré A. R. (2015) Structure and mechanism of the T-box riboswitches. Wiley
Interdiscip. Rev. RNA DOI: 10.1002/wrna.1285.
217. Anupam R., Nayek A., Green N. J., Grundy F. J., Henkin T. M., Means J. A., Bergmeier S. C., Hines J. V.
(2008) 4,5-Disubstituted oxazolidinones: High affinity molecular effectors of RNA function. Bioorg. Med.
Chem. Lett. 18: 3541-3544.
218. Anupam R., Denapoli L., Muchenditsi A., Hines J. V. (2008) Identification of neomycin B-binding site in T
box antiterminator model RNA. Bioorg. Med. Chem. 16: 4466-4470.
219. Mahapatra S., Crick D. C., McNeil M. R., Brennan P. J. (2008) Unique structural features of the
peptidoglycan of Mycobacterium leprae. J. Bacteriol. 190: 655–661.
220. Lloyd A. J., Gilbey A. M., Blewett A. M., De Pascale G., El Zoeiby A., Levesque R. C., Catherwood A. C.,
Tomasz A., Bugg T. D. H., Roper D. I., Dowson C. G. (2008) Characterization of tRNA-dependent peptide
bond formation by MurM in the synthesis of Streptococcus pneumoniae peptidoglycan. J. Biol. Chem.
283: 6402–6417.
221. Dramsi S., Magnet S., Davison S., Arthur M. (2008) Covalent attachment of proteins to peptidoglycan.
FEMS Microbiol. Rev. 32: 307–320.
222. Schneider T., Senn M. M., Berger-Bächi B., Tossi A., Sahl H. G., Wiedemann I. (2004) In vitro assembly of
a complete, pentaglycine interpeptide bridge containing cell wall precursor (lipid II-Gly5) of
Staphylococcus aureus. Mol. Microbiol. 53: 675-685.
223. Perry A. M., Ton-That H., Mazmanian S. K., Schneewind O. (2002) Anchoring of surface proteins to the
cell wall of Staphylococcus aureus. III. Lipid II is an in vivo peptidoglycan substrate for sortase-catalyzed
surface protein anchoring. J. Biol. Chem. 277: 16241–16248.
224. Maidhof H., Reinicke B., Blumel P., Berger-Bächi B., Labischinski H. (1991) femA, which encodes a factor
essential for expression of methicillin resistance, affects glycine content of peptidoglycan in methicillin-
resistant and methicillin-susceptible Staphylococcus aureus strains. J. Bacteriol. 173: 3507–3513.

References
218
225. Rohrer S., Ehlert K., Tschierske M., Labischinski H., Berger-Bachi B. (1999) The essential Staphylococcus
aureus gene fmhB is involved in the first step of peptidoglycan pentaglycine interpeptide formation.
Proc. Natl. Acad. Sci. U S A 96: 9351–9356.
226. Senn M. M., Bischoff M., von Eiff C., Berger-Bächi B. (2005) sigmaB activity in a Staphylococcus aureus
hemB mutant. J. Bacteriol. 187: 7397-406.
227. Ehlert K, Schroder W, Labischinski H. (1997) Specificities of FemA and FemB for different glycine
residues: FemB cannot substitute for FemA in staphylococcal peptidoglycan pentaglycine side chain
formation. J. Bacteriol. 179: 7573–7576.
228. Giannouli S., Kyritsis A., Malissovas N., Becker H. D., Stathopoulos C. (2009) On the role of an unusual
tRNAGly isoacceptor in Staphylococcus aureus. Biochimie 91: 344–351.
229. Villet R., Fonvielle M., Busca P., Chemama M., Maillard A. P., Hugonnet J. E., Dubost L., Marie A.,
Josseaume N., Mesnage S., Mayer C., Valéry J. M., Ethève-Quelquejeu M., Arthur M. (2007) Idiosyncratic
features in tRNAs participating in bacterial cell wall synthesis. Nucleic Acids Res. 35: 6870–6883.
230. Fonvielle M., Chemama M., Villet R., Lecerf M., Bouhss A., Valery J. M., Etheve-Quelquejeu M., Arthur M.
(2009) Aminoacyl-tRNA recognition by the FemXWv transferase for bacterial cell wall synthesis. Nucleic
Acids Res. 37: 1589–1601.
231. Cressina E., Lloyd A. J., De Pascale G., James Mok B., Caddick S., Roper D. I., Dowson C. G., Bugg T. D.
(2009) Inhibition of tRNA-dependent ligase MurM from Streptococcus pneumoniae by phosphonate and
sulfonamide inhibitors. Bioorg. Med. Chem. 17: 3443–3455.
232. Chemama M., Fonvielle M., Arthur M., Valery J. M., Etheve-Quelquejeu M. (2009) Synthesis of stable
aminoacyl-tRNA analogues containing triazole as a bioisoster of esters. Chemistry 15: 1929–1938.
233. Fonvielle M., Li de La Sierra-Gallay I., El-Sagheer A. H., Lecerf M., Patin D., Mellal D., Mayer C., Blanot D.,
Gale N., Brown T., van Tilbeurgh H., Ethève-Quelquejeu M., Arthur M. (2013) The structure of FemX(Wv)
in complex with a peptidyl-RNA conjugate: mechanism of aminoacyl transfer from Ala-tRNA(Ala) to
peptidoglycan precursors. Angew. Chem. Int. Ed. Engl. 52: 7278-7281.
234. Grundy F. J., Winkler W. C., Henkin T. M. (2002) tRNA-mediated transcription antitermination in vitro:
codon-anticodon pairing independent of the ribosome. Proc. Natl. Acad. Sci. U S A 99: 11121-11126.
235. Roberts R. J. (1974) Staphylococcal transfer ribonucleic acids. II. Sequence analysis of isoaccepting
glycine transfer ribonucleic acids IA and IB from Staphylococcus epidermidis. J. Biol. Chem. 249: 4787-
4796.
236. Asahara H. & Uhlenbeck O. C. (2002) The tRNA specificity of Thermus thermophilus EF-Tu. Proc. Natl.
Acad. Sci. USA 99: 3499-3504.
237. Sanderson L. E. & Uhlenbeck O. C. (2007) The 51-63 base pair of tRNA confers specificity for binding by
EF-Tu. RNA 13: 835-840.
238. Chang A. T. & Nikonowicz E. P. (2012) Solution nuclear magnetic resonance analyses of the anticodon
arms of proteinogenic and nonproteinogenic tRNA(Gly). Biochemistry 51: 3662-3674.
239. Hartman B. J. & Tomasz A. (1984) Low-affinity penicillin-binding protein associated with beta-lactam
resistance in Staphylococcus aureus. J. Bacteriol. 158: 513–516.
240. Lim D. & Strynadka N. C. (2002) Structural basis for the beta-lactam resistance of PBP2a from methicillin-
resistant Staphylococcus aureus. Nat. Struct. Biol. 9: 870–876.
241. Pereira P. M., Filipe S. R., Tomasz A., Pinho M. G. (2007) Fluorescence ratio imaging microscopy shows
decreased access of vancomycin to cell wall synthetic sites in vancomycin-resistant Staphylococcus
aureus. Antimicrob. Agents Chemother. 51: 3627–3633.

References
219
242. Utaida S., Dunman P. M., Macapagal D., Murphy E., Projan S. J., Singh V. K., Jayaswal R. K., Wilkinson B. J.
(2003) Genome-wide transcriptional profiling of the response of Staphylococcus aureus to cell-wall-
active antibiotics reveals a cell-wall-stress stimulon. Microbiology 149: 2719–2732.
243. Belcheva A. & Golemi-Kotra D. (2008) A close-up view of the VraSR two-component system. A mediator
of Staphylococcus aureus response to cell wall damage. J Biol Chem. 283: 12354-12364.
244. Pournaras S., Ntokou E., Zarkotou O., Ranellou K., Themeli-Digalaki K., Stathopoulos C., Tsakris A. (2013)
Linezolid dependence in Staphylococcus epidermidis bloodstream isolates. Emerg. Infect. Dis. 19: 129-
32.
245. Tsakris A., Pillai S. K., Gold H. S., Thauvin-Eliopoulos C., Venkataraman L., Wennersten C., Moellering Jr.
R. C., Eliopoulos G. M. (2007) Persistence of rRNA operon mutated copies and rapid re-emergence of
linezolid resistance in Staphylococcus aureus. J. Antimicrob. Chemother. 60: 649–651.
246. Kokkori S., Apostolidi M., Tsakris A., Pournaras S., Stathopoulos C., Dinos G. (2014) Linezolid-dependent
function and structure adaptation of ribosomes in a Staphylococcus epidermidis strain exhibiting
linezolid dependence. Antimicrob. Agents Chemother. 58: 4651-4656.
247. Toh S. M., Xiong L., Arias C. A., Villegas M. V., Lolans K., Quinn J., Mankin A. S. (2007) Acquisition of a
natural resistance gene renders a clinical strain of methicillin-resistant Staphylococcus aureus resistant
to the synthetic antibiotic linezolid. Mol. Microbiol. 64: 1506–1514.
248. McCallum N., Berger-Bächi B., Senn M. M. (2010) Regulation of antibiotic resistance in Staphylococcus
aureus. Int. J. Med. Microbiol. 300: 118-129.
249. Novick R. P. & Subedi A. (2007) The SaPIs: mobile pathogenicity islands of Staphylococcus. Chem.
Immunol. Allergy 93: 42–57.
250. Pichon C. & Felden B. (2005) Small RNA genes expressed from Staphylococcus aureus genomic and
pathogenicity islands with specific expression among pathogenic strains. Proc. Natl. Acad. Sci. U S A 102:
14249–14254.
251. Chabelskaya S., Gaillot O., Felden B. (2010) A Staphylococcus aureus small RNA is required for bacterial
virulence and regulates the expression of an immune evasion molecule. PLoS Pathog. 6: e1000927.
252. Benito Y., Kolb F. A., Romby P., Lina G., Etienne J., Vandenesch F. (2000) Probing the structure of RNAIII,
the Staphylococcus aureus agr regulatory RNA, and identification of the RNA domain involved in
repression of protein A expression. RNA 6: 668–679.
253. Morfeldt E., Taylor D., von Gabain A., Arvidson S. (1995) Activation of alpha-toxin translation in
Staphylococcus aureus by the trans-encoded antisense RNA, RNAIII. EMBO J. 14: 4569–4577.
254. Jia X., Zhang J., Sun W., He W., Jiang H., Chen D., Murchie A. I. (2013) Riboswitch control of
aminoglycoside antibiotic resistance. Cell 152: 68-81.
255. Felden B., Vandenesch F., Bouloc P., Romby P. (2011) The Staphylococcus aureus RNome and its
commitment to virulence. PLoS Pathog. 7: e1002006.
256. Lynch S. V. & Wiener-Kronish J. P. (2008) Novel strategies to combat bacterial virulence. Curr. Opin. Crit.
Care 14: 593–599.
257. Blount K. F. & Breaker R. R. (2006) Riboswitches as antibacterial drug targets. Nat. Biotechnol. 24: 1558–
1564.
258. Mulhbacher J., Brouillette E., Allard M., Fortier L. C., Malouin F, Lafontaine D. A. (2010) Novel riboswitch
ligand analogs as selective inhibitors of guanine-related metabolic pathways. PLoS Pathog. 6: e1000865.
259. Logan D. T., Mazauric M. H., Kern D., Moras D. (1995) Crystal structure of glycyl-tRNA synthetase from
Thermus thermophilus. EMBO J. 14: 4156-4167.

References
220
260. Novichkov P. S., Kazakov A. E., Ravcheev D. A., Leyn S. A., Kovaleva G. Y., Sutormin R. A., Kazanov M. D.,
Riehl W., Arkin A. P., Dubchak I., Rodionov D. A. (2013) RegPrecise 3.0--a resource for genome-scale
exploration of transcriptional regulation in bacteria. BMC Genomics 14: 745.
261. Aoki K. F. & Kanehisa M. (2005) Using the KEGG database resource. Curr. Protoc. Bioinformatics. Chapter
1: Unit 1.12.
262. Solovyev V. & Salamov A. (2011) Automatic Annotation of Microbial Genomes and Metagenomic
Sequences. In Metagenomics and its Applications in Agriculture, Biomedicine and Environmental Studies
(Ed. RW Li), pp. 61-78. Nova Science Publishers.
263. Zuker M. (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids
Res. 31: 3406-3415.
264. Notredame C., Higgins D. G., Heringa J. (2000) T-Coffee: A novel method for fast and accurate multiple
sequence alignment. J. Mol. Biol. 302: 205-217.
265. Lowe T. M. & Eddy S. R. (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in
genomic sequence. Nucleic Acids Res. 25: 955-964.
266. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F.,
Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Clustal W and Clustal
X version 2.0. Bioinformatics 23: 2947-2948.
267. Dereeper A., Guignon V., Blanc G., Audic S., Buffet S., Chevenet F., Dufayard J. F., Guindon S., Lefort V.,
Lescot M. Claverie J. M., Gascuel O. (2008) Phylogeny.fr: robust phylogenetic analysis for the non-
specialist. Nucleic Acids Res. 36: W465-469.
268. Samuels M., Fire A., Sharp P. A. (1984) Dinucleotide priming of transcription mediated by RNA
polymerase II. J. Biol. Chem. 259: 2517-2525.
269. Kraemer G. R. & Iandolo J. J. (1990) High-frequency transformation of Staphylococcus aureus by
electroporation. Current Microbiology Volume 21, Issue 6, pp. 373-376.
270. Henkin T. M. (2009) Analysis of tRNA-directed transcription antitermination in the T box system in vivo.
Methods Mol. Biol. 540: 281-290.
271. Miller J. H. (1972) Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring
Harbor, NY.
272. Arnez J. G., Dock-Bregeon A. C., Moras D. (1999) Glycyl-tRNA synthetase uses a negatively charged pit
for specific recognition and activation of glycine. J. Mol. Biol. 286: 1449-1459.
273. Mazauric M. H., Roy H., Kern D. (1999) tRNA glycylation system from Thermus thermophilus. tRNAGly
identity and functional interrelation with the glycylation systems from other phylae. Biochemistry. 38:
13094-13105.
274. Wels M., Groot Kormelink T., Kleerebezem M., Siezen R. J., Francke C. (2008) An in silico analysis of T-
box regulated genes and T-box evolution in prokaryotes, with emphasis on prediction of substrate
specificity of transporters. BMC Genomics. 14: 330.
275. Samuelsson T., Axberg T., Borén T., Lagerkvist U. (1983) Unconventional reading of the glycine codons. J.
Biol. Chem. 258: 13178-13184.
276. Yousef M. R., Grundy F. J., Henkin T. M. (2003) tRNA requirements for glyQS antitermination: a new
twist on tRNA. RNA. 9:1148-1156.
277. Wang M. R., Grundy F. J., Henkin T. M. (2005) Structural transitions induced by the interaction between
tRNA(Gly) and the Bacillus subtilis glyQS T box leader RNA. J. Mol. Biol. 349: 273-287.
278. Henkin T. M. (2014) The T box riboswitch: A novel regulatory RNA that utilizes tRNA as its ligand.
Biochim. Biophys. Acta. 1839: 959-963.

References
221
279. Grigg J. C. & Ke A. (2013) Structural determinants for geometry and information decoding of tRNA by T
box leader RNA. Structure 21: 2025-2032.
280. Saad N. Y., Schiel B., Brayé M., Heap J. T., Minton N. P., Dürre P., Becker H. D. (2012) Riboswitch (T-box)-
mediated control of tRNA-dependent amidation in Clostridium acetobutylicum rationalizes gene and
pathway redundancy for asparagine and asparaginyl-tRNA(Asn) synthesis. J. Biol. Chem. 287: 20382-
20394.
281. Sprinzl M., Steegborn C., Hübel F., Steinberg S. (1996) Compilation of tRNA sequences and sequences of
tRNA genes. Nucleic Acids Res. 24:68-72.
282. Chen S. J., Lee C. Y., Lin S. T., Wang C. C. (2011) Rescuing a dysfunctional homologue of a yeast glycyl-
tRNA synthetase gene. ACS Chem. Biol. 6: 1182–1187.
283. Turner R. J., Lovato M., Schimmel P. (2000) One of two genes encoding glycyl-tRNA synthetase in
Saccharomyces cerevisiae provides mitochondrial and cytoplasmic functions. J. Biol. Chem. 275: 27681–
27688.
284. Cader M. Z., Ren J., James P. A., Bird L. E., Talbot K., Stammers D. K. (2007) Crystal structure of human
wildtype and S581L-mutant glycyl-tRNA synthetase, an enzyme underlying distal spinal muscular
atrophy. FEBS Lett. 581: 2959-2964.
285. Andreev D. E., Hirnet J., Terenin I. M., Dmitriev S. E., Niepmann M., Shatsky I. N. (2012) Glycyl-tRNA
synthetase specifically binds to the poliovirus IRES to activate translation initiation. Nucleic Acids Res.
40: 5602-5614.
286. Nangle L. A., Zhang W., Xie W., Yang X. L., Schimmel P. (2007) Charcot-Marie-Tooth disease-associated
mutant tRNA synthetases linked to altered dimer interface and neurite distribution defect. Proc. Natl.
Acad. Sci. USA.104: 11239–11244.
287. Griffin L. B., Sakaguchi R., McGuigan D., Gonzalez M. A., Searby C., Züchner S., Hou Y. M., Antonellis A.
(2014) Impaired function is a common feature of neuropathy-associated glycyl-tRNA synthetase
mutations. Hum. Mutat. 35: 1363-1371
288. Park M. C., Kang T., Jin D., Han J. M., Kim S. B., Park Y. J., Cho K., Park Y. W., Guo M., He W., Yang X. L.,
Schimmel P., Kim S. (2012) Secreted human glycyl-tRNA synthetase implicated in defense against ERK-
activated tumorigenesis. Proc. Natl. Acad. Sci. U S A. 109: E640-647.
289. McCallum N., Meier P. S., Heusser R., Berger-Bächi B. (2011)Mutational analyses of open reading frames
within the vraSR operon and their roles in the cell wall stress response of Staphylococcus aureus.
Antimicrob. Agents Chemother. 55: 1391-1402.
290. Zhang J. & Ferré-D'Amaré A. R. (2014) Direct evaluation of tRNA aminoacylation status by the T-box
riboswitch using tRNA-mRNA stacking and steric readout. Mol Cell. 55:148-155.
291. Grigg J. C., Ke A. (2013) Sequence, structure, and stacking: specifics of tRNA anchoring to the T box
riboswitch. RNA Biol. 10:1761-1764.
292. Sun E. I., Rodionov D. A. (2014) Computational analysis of riboswitch-based regulation. Biochim. Biophys.
Acta. 1839: 900-907.
293. Grundy F. J. & Henkin T. M. (2004) Kinetic analysis of tRNA-directed transcription antitermination of the
Bacillus subtilis glyQS gene in vitro. J. Bacteriol. 186: 5392-5399.
294. Foy N., Jester B., Conant G. C., Devine K. M. (2010) The T box regulatory element controlling expression
of the class I lysyl-tRNA synthetase of Bacillus cereus strain 14579 is functional and can be partially
induced by reduced charging of asparaginyl-tRNAAsn. BMC Microbiol. 22: 196.
295. Korostelev A., Trakhanov S., Laurberg M., Noller H. (2006) Crystal structure of a 70S ribosome-tRNA
complex reveals functional interactions and rearrangements. Cell 126: 1065–10




Publications
225
RNA 2015 Oct;21(10):1790-806. doi: 10.1261/rna.052712.115. Epub 2015 Aug 14.
A glyS T-box riboswitch with species-specific structural features responding to both
proteinogenic and non-proteinogenic tRNAGly isoacceptors.
Apostolidi M., Saad N. Y., Drainas D., Pournaras S., Becker H. D., Stathopoulos C.
Abstract
In Staphylococcus aureus, a T-box riboswitch exists upstream of the glyS gene to regulate
transcription of the sole glycyl-tRNA synthetase, which aminoacylates five tRNAGly
isoacceptors bearing GCC or UCC anticodons. Subsequently, the glycylated tRNAs serve as
substrates for decoding glycine codons during translation, and also as glycine donors for
exoribosomal synthesis of pentaglycine peptides during cell wall formation. Probing of the
predicted T-box structure revealed a long stem I, lacking features previously described for
similar T-boxes. Moreover, the antiterminator stem includes a 42-nt long intervening
sequence, which is staphylococci-specific. Finally, the terminator conformation adopts a
rigid two-stem structure, where the intervening sequence forms the first stem followed by
the second stem, which includes the more conserved residues. Interestingly, all five tRNAGly
isoacceptors interact with S. aureus glyS T-box with different binding affinities and they all
induce transcription readthrough at different levels. The ability of both GCC and UCC
anticodons to interact with the specifier loop indicates ambiguity during the specifier triplet
reading, similar to the unconventional reading of glycine codons during protein synthesis.
The S. aureus glyS T-box structure is consistent with the recent crystallographic and NMR
studies, despite apparent differences, and highlights the phylogenetic variability of T-boxes
when studied in a genome-dependent context. Our data suggest that the S. aureus glyS T-
box exhibits differential tRNA selectivity, which possibly contributes toward the regulation
and synchronization of ribosomal and exoribosomal peptide synthesis, two essential but
metabolically unrelated pathways.
KEYWORDS: Staphylococcus aureus; T-box riboswitch; glycyl-tRNA synthetase; tRNA;
transcription regulation
© 2015 Apostolidi et al.; Published by Cold Spring Harbor Laboratory Press for the RNA
Society.

Publications
226
Biomol NMR Assign 2015 Oct;9(2):303-7. doi: 10.1007/s12104-015-9597-z. Epub 2015 Feb 18.
1H, 13C and 15N backbone and side-chain resonance assignment of the LAM-RRM1 N-terminal
module of La protein from Dictyostelium discoideum.
Chasapis C.T., Argyriou A. I., Apostolidi M., Konstantinidou P., Bentrop D., Stathopoulos C.,
Spyroulias G. A.
Abstract
The N-terminal half of La protein consists of two concatenated motifs: La motif (LAM) and the
N-terminal RNA recognition motif (RRM1) both of which are responsible for poly(U) RNA
binding. Here, we present the backbone and side-chain assignments of the (1)H, (13)C and
(15)N resonances of the 191-residue LAM-RRM1 region of the La protein from the lower
eukaryote Dictyostelium discoideum and its secondary structure prediction.
KEYWORDS: La protein; NMR spectroscopy; RNA binding; RNA polymerase III; Recombinant
protein expression
© Springer International Publishing AG

Publications
227
Biomol NMR Assign 2015 Apr;9(1):219-22. doi: 10.1007/s12104-014-9578-7. Epub 2014 Oct 4.
Backbone and side chain NMR assignment, along with the secondary structure prediction of
RRM2 domain of La protein from a lower eukaryote exhibiting identical structural
organization with its human homolog.
Argyriou A. I., Chasapis C. T., Apostolidi M., Konstantinidou P., Stathopoulos C., Bentrop D.,
Spyroulias G. A.
Abstract
The La protein (Lupus antigen), a key mediator during biogenesis of RNA polymerase III
transcripts, contains a characteristic La motif and one or two RNA recognition motif (RRM)
domains, depending on the organism of origin. The RRM1 domain is conserved in higher
eukaryotes and located in the N-terminal region, whereas the C-terminal RRM2 domain is
absent in most lower eukaryotes and its specific role remains, so far, uncharacterized. Here,
we present the backbone and side-chain assignment of the 1H, 13C and 15N resonances of
RRM2 of La protein from Dictyostelium discoideum. Interestingly, the La protein in this lower
eukaryote, exhibits high homology to its human counterpart. Moreover, it contains two RRM
domains, instead of one, raising questions on its evolutionary origin and the putative role of
RRM2 in vivo. We also provide its secondary structure as predicted by the TALOS+ online tool.
KEYWORDS: La protein ; RNA polymerase III; RNA binding; Recombinant protein expression;
NMR spectroscopy
© Springer International Publishing AG

Publications
228
Antimicrob Agents Chemother 2014 Aug;58(8):4651-6. doi: 10.1128/AAC.02835-14.
Epub 2014 Jun 2.
Linezolid-dependent function and structure adaptation of ribosomes in a Staphylococcus
epidermidis strain exhibiting linezolid dependence.
Kokkori S., Apostolidi M., Tsakris A., Pournaras S., Stathopoulos C., Dinos G.
Abstract
Linezolid-dependent growth was recently reported in Staphylococcus epidermidis clinical
strains carrying mutations associated with linezolid resistance. To investigate this
unexpected behavior at the molecular level, we isolated active ribosomes from one of the
linezolid-dependent strains and we compared them with ribosomes isolated from a wild-
type strain. Both strains were grown in the absence and presence of linezolid. Detailed
biochemical and structural analyses revealed essential differences in the function and
structure of isolated ribosomes which were assembled in the presence of linezolid. The
catalytic activity of peptidyltransferase was found to be significantly higher in the ribosomes
derived from the linezolid-dependent strain. Interestingly, the same ribosomes exhibited an
abnormal ribosomal subunit dissociation profile on a sucrose gradient in the absence of
linezolid, but the profile was restored after treatment of the ribosomes with an excess of the
antibiotic. Our study suggests that linezolid most likely modified the ribosomal assembly
procedure, leading to a new functional ribosomal population active only in the presence of
linezolid. Therefore, the higher growth rate of the partially linezolid-dependent strains could
be attributed to the functional and structural adaptations of ribosomes to linezolid.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.

Publications
229
Biomol NMR Assign 2014 Apr;8(1):47-51. doi: 10.1007/s12104-012-9450-6. Epub 2012 Dec 14.
¹H, ¹⁵N, ¹³C assignment and secondary structure determination of two domains of La protein
from D. discoideum.
Apostolidi M., Vourtsis D. J., Chasapis C. T., Stathopoulos C., Bentrop D., Spyroulias G. A.
Abstract
Biosynthesis of RNA polymerase III transcripts requires binding of the La protein at their 3' end.
La is an abundant nuclear RNA-binding protein which protects the nascent transcripts from 3'
exonuclease degradation. Here, we report the high yield expression and preliminary structural
analysis through NMR spectroscopy of two recombinant RNA binding domains (La motif and
NRRM) from the La protein of Dictyostelium discoideum. Both recombinant protein constructs
were well-folded and allowed for an almost complete sequence-specific assignment of the 15N
and 13C labeled domains and their secondary structure prediction using PECAN online tool.
KEYWORDS: La protein RNA; binding protein; Rheumatic diseases; Recombinant protein
expression; NMR spectroscopy
© Springer International Publishing AG









