
SPECTRES ELECTRONIQUES I – Spectres...
15
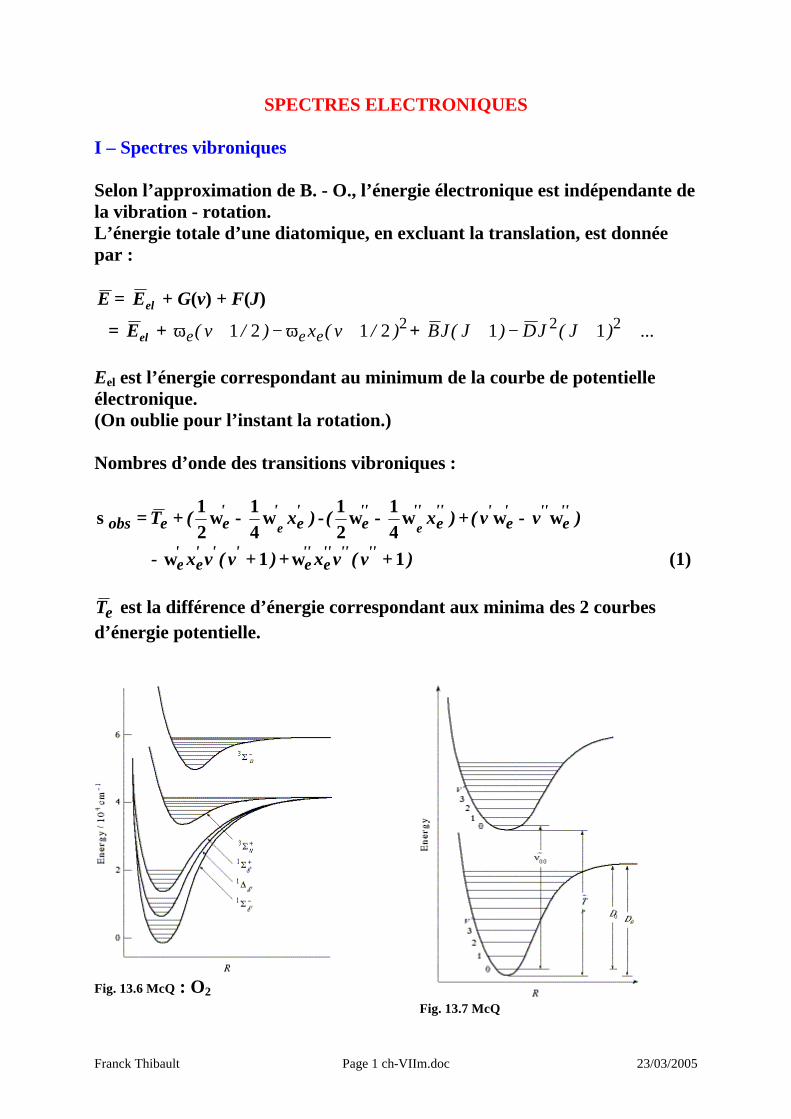
Franck Thibault Page 1 ch-VIIm.doc 23/03/2005 SPECTRES ELECTRONIQUES I – Spectres vibroniques Selon l’approximation de B. - O., l’énergie électronique est indépendante de la vibration - rotation. L’énergie totale d’une diatomique, en excluant la translation, est donnée par : E = el E + G(v) + F(J) = el E + 2 2 1 2 1 ) / v ( x ) / v ( e e e ϖ - ϖ + ... ) J ( J D ) J ( J B - 2 2 1 1 E el est l’énergie correspondant au minimum de la courbe de potentielle électronique. (On oublie pour l’instant la rotation.) Nombres d’onde des transitions vibroniques : ) x ( T ' e ' ' e e obs e w - w + = s 4 1 2 1 - ) x ( ' ' e ' ' ' ' e e w - w 4 1 2 1 + ) v v ( ' ' e ' ' ' e ' w - w - ) v ( v x ' ' ' e ' e 1 + w + ) v ( v x ' ' ' ' ' ' e ' ' e 1 + w (1) e T est la différence d’énergie correspondant aux minima des 2 courbes d’énergie potentielle. Fig. 13.6 McQ : O 2 Fig. 13.7 McQ
Transcript of SPECTRES ELECTRONIQUES I – Spectres...
Franck Thibault Page 1 ch-VIIm.doc 23/03/2005
SPECTRES ELECTRONIQUES I – Spectres vibroniques Selon l’approximation de B. - O., l’énergie électronique est indépendante de la vibration - rotation. L’énergie totale d’une diatomique, en excluant la translation, est donnée par : E = elE + G(v) + F(J)
= elE + 22121 )/v(x)/v( eee +ω−+ω + ...)J(JD)J(JB ++−+ 22 11 Eel est l’énergie correspondant au minimum de la courbe de potentielle électronique. (On oublie pour l’instant la rotation.) Nombres d’onde des transitions vibroniques :
)x(T 'e
''eeobs e
ω−ω+=σ41
21
- )x( ''e
''''e e
ω−ω41
21
+ )vv( ''e
'''e
' ω−ω
- )v(vx '''e
'e 1+ω + )v(vx ''''''
e''e 1+ω (1)
eT est la différence d’énergie correspondant aux minima des 2 courbes
d’énergie potentielle.
Fig. 13.6 McQ : O2
Fig. 13.7 McQ

Franck Thibault Page 2 ch-VIIm.doc 23/03/2005
Tableau 13.5 McQ : Longueurs de liaison à l’équilibre pour divers états de O2. Etat électronique eT / cm-1 Re / pm
−Σg3 0 120.74
g∆1 7918.1 121.55
+Σg1 13195.2 122.67
+Σu3 36096. 142.
−Σu3 49802. 160.
eω et ee xω dépendent de la forme de la courbe. Les 2 premiers termes entre parenthèses sont les énergies au point zéro des états supérieur et inférieur. La grandeur :
)x(T 'e
''ee e
ω−ω+=σ41
21
00 - )x( ''e
''''e e
ω−ω41
21
(2)
correspond donc à la transition vibronique 0 à 0. Et l’Eq. 1 devient :
+σ=σ 00obs )vv( ''e
'''e
' ω−ω - )v(vx '''e
'e 1+ω + )v(vx ''''''
e''e 1+ω (3)
Dans le cas (usuel) où l’état inférieur est v = 0 :
+σ=σ 00obs'e
'v ω - )v(vx '''e
'e 1+ω (4)
Lorsque v’ augmente, l’espacement vibronique devient plus petit jusqu’à l’obtention d’un spectre pratiquement continu.
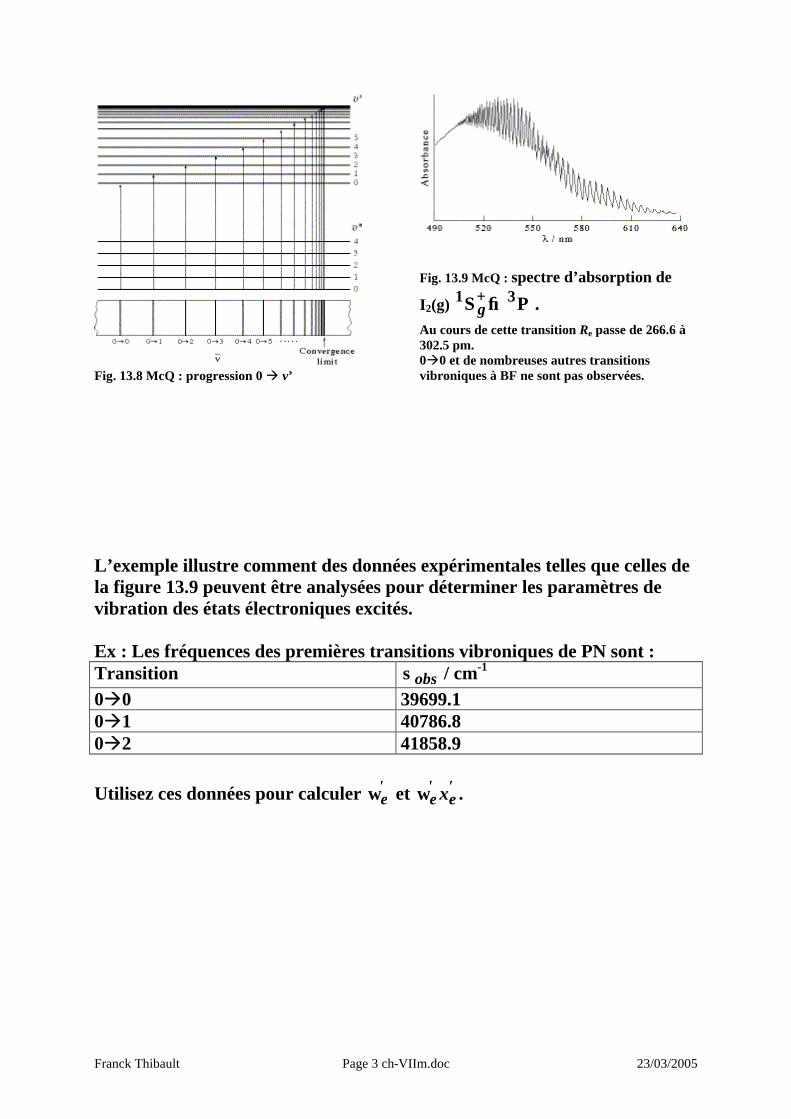
Franck Thibault Page 3 ch-VIIm.doc 23/03/2005
Fig. 13.8 McQ : progression 0 à v’
Fig. 13.9 McQ : spectre d’absorption de
I2(g) Π→Σ+ 31g .
Au cours de cette transition Re passe de 266.6 à 302.5 pm. 0à0 et de nombreuses autres transitions vibroniques à BF ne sont pas observées.
L’exemple illustre comment des données expérimentales telles que celles de la figure 13.9 peuvent être analysées pour déterminer les paramètres de vibration des états électroniques excités. Ex : Les fréquences des premières transitions vibroniques de PN sont : Transition obsσ / cm-1 0à0 39699.1 0à1 40786.8 0à2 41858.9 Utilisez ces données pour calculer '
eω et 'e
'e xω .
Franck Thibault Page 4 ch-VIIm.doc 23/03/2005
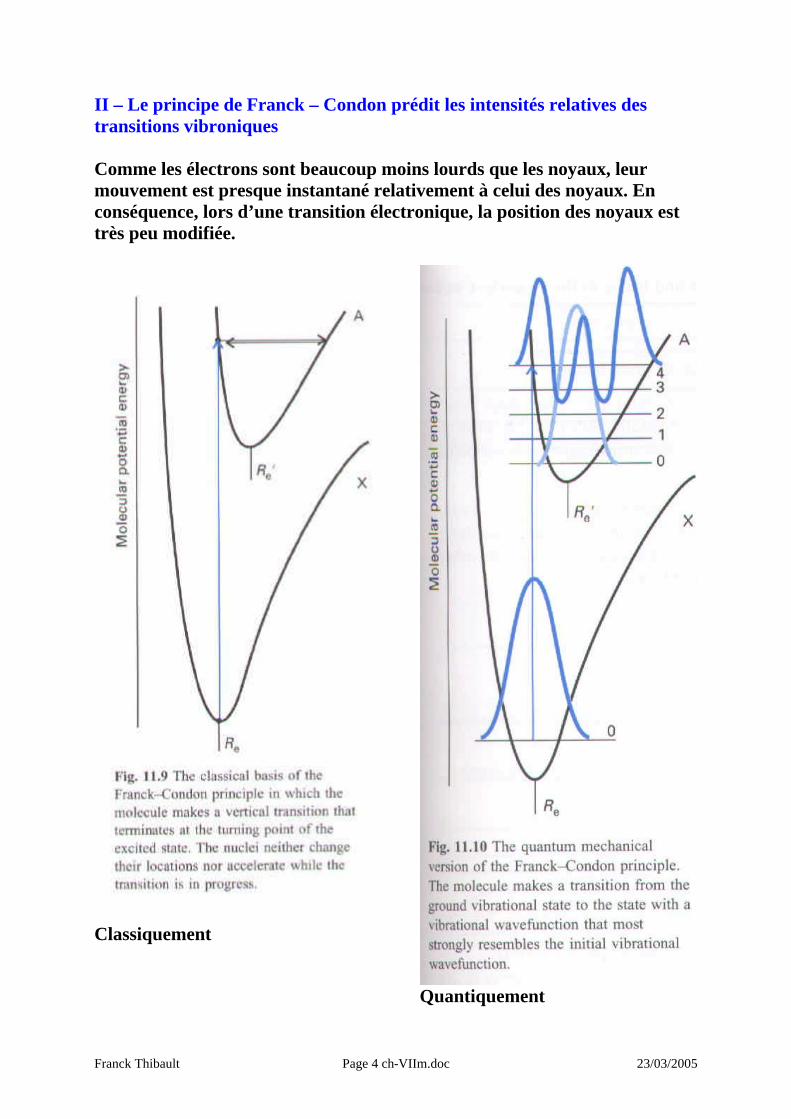
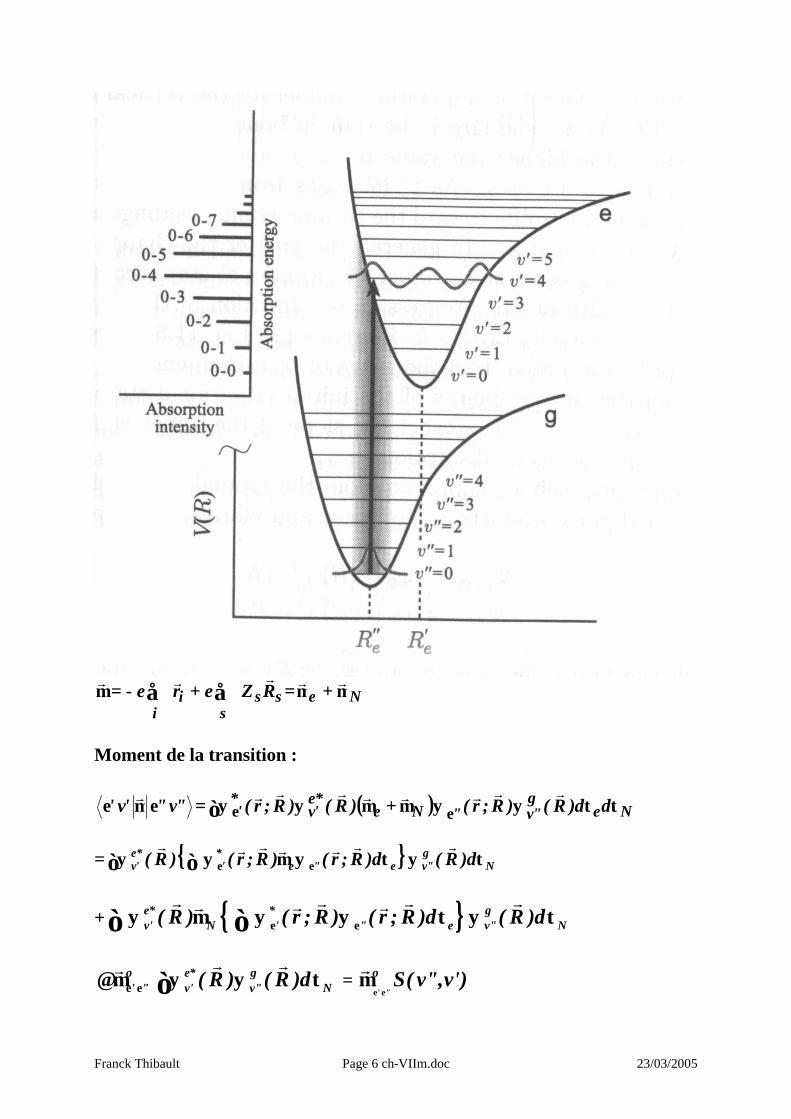
II – Le principe de Franck – Condon prédit les intensités relatives des transitions vibroniques Comme les électrons sont beaucoup moins lourds que les noyaux, leur mouvement est presque instantané relativement à celui des noyaux. En conséquence, lors d’une transition électronique, la position des noyaux est très peu modifiée.
Classiquement
Quantiquement
Franck Thibault Page 5 ch-VIIm.doc 23/03/2005
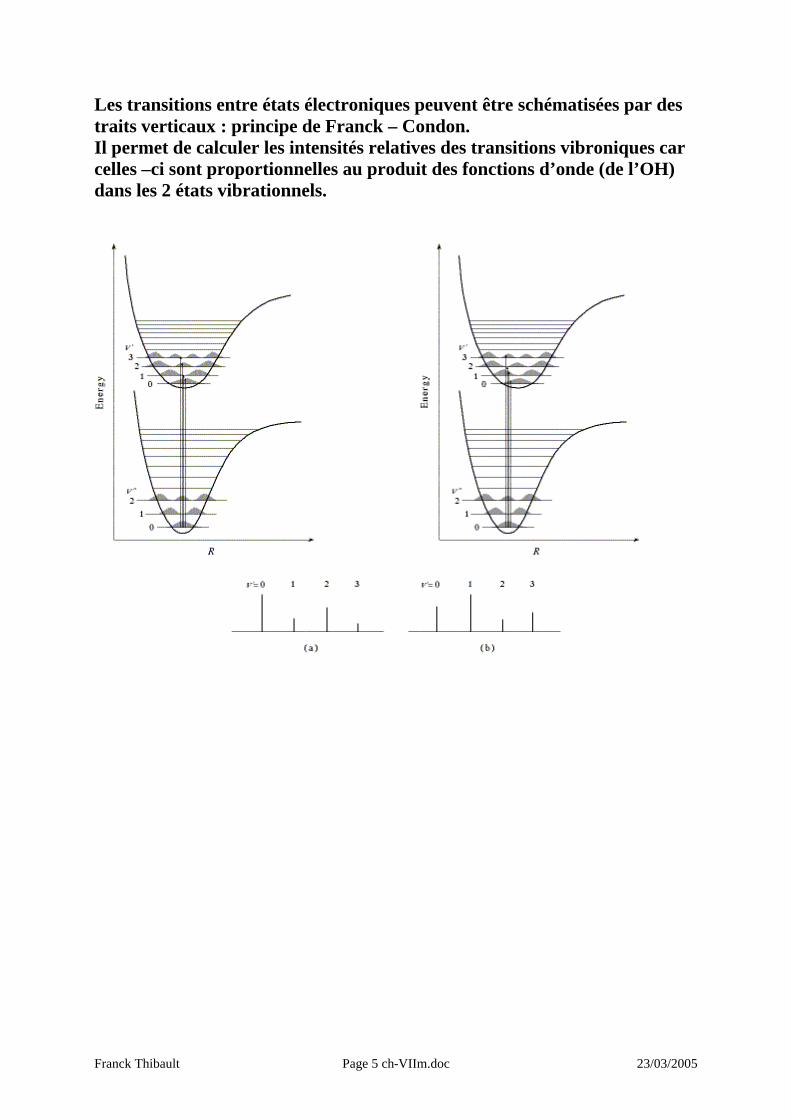
Les transitions entre états électroniques peuvent être schématisées par des traits verticaux : principe de Franck – Condon. Il permet de calculer les intensités relatives des transitions vibroniques car celles –ci sont proportionnelles au produit des fonctions d’onde (de l’OH) dans les 2 états vibrationnels.
Franck Thibault Page 6 ch-VIIm.doc 23/03/2005
ss
si
iRZererrr ∑∑ +−=µ = Ne µ+µ
rr
Moment de la transition :
"v"'v' εµεr
= ( ) Neg"v"Ne
*e'v
*' dd)R()R;r()R()R;r( ττψψµ+µψψ εε∫
rrrrrrrr
= N
g"ve"e
*'
*e'v d)R(d)R;r()R;r()R( τψτψµψψ∫ ∫ εε
rrrrrrr
+ e* * gv' N ' " e v" N( R ) ( r ; R ) ( r ; R )d ( R )dε εψ µ ψ ψ τ ψ τ∫ ∫
r r r rr rr
o e* g' " v' v" N( R ) ( R )dε ε≅ µ ψ ψ τ∫
r rr =
' "
o S( v",v')ε ε
µr

Franck Thibault Page 7 ch-VIIm.doc 23/03/2005
Attention : il n’est pas nécessaire que la molécule possède un moment dipolaire permanent ou que le moment varie au cours de la vibration. Toutes les molécules montrent un spectre électronique.
Facteur de Franck – Condon : 2)'v,"v(S)'v,"v(FC = Exos : 1 - Montrer que la somme des facteurs de F – C des transitions à partir de v’’ est égale à 1.
1==∑ "v"v"v'v'v"v'v
2 - Considérez le cas où les deux états électronique ont la même constante de force mais pour lesquels la longueur de la liaison à l’équilibre diffère de ∆R. Trouvez une expression de l’intensité relative de la transition 0 – 0 en fonction de ∆R. On prendra les fonctions d’onde de l’OH :
)/yexp()y(/
2241
0 α−
πα
=ψ avec hµω
=α .
On donne : dye ay2−∞+
∞−∫ =
21 /
a
π
(inutile !)
Dans le cas simple où les courbes de potentiel sont harmoniques et ont la même constante de force, ce résultat peut être généralisé :
)/exp(!v
vFCv
v 22
10 2
220 ∆−
∆== avec )RR( "
e'e −α=∆ .
− ∆2: Il n’est donc pas possible de prédire à partir de la progression FC si la longueur de la liaison est plus grande dans l’état fondamental ou excité. - FC0v = δ0v quand ∆ = 0.

Franck Thibault Page 8 ch-VIIm.doc 23/03/2005
III - Spectre rovibronique Visible, UV Pas de règle de sélection sur v car les fonctions d’onde vibrationnelles appartiennent à des états électroniques différents. Règle de sélection des transitions vibroniques : v∆ entier u ↔ g (homonucléaires) s ↔ s, a ↔ a (homo) + ↔ +, - ↔ - Branches P, Q, R : ∆J = -1, 0, 1 mais pas J = 0 → J = 0 Transitions Σ à Σ (Λ = 0) : ∆J = -1, 1 Transitions où l’un des états électronique au moins a Λ ≠ 0 : (Σ à Π, Π à∆….) : ∆J = -1, 0, 1
10 ±=∆Λ , , (cf plus tard : cas (a) et (b) de Hund) 10 ±=∆Ω , , (cf plus tard : cas (a) de Hund)
∆S = 0, (cas (a) et (b)) : ∆Σ = 0 (s-o faible, cas (a)) ∆J = -1, 0, 1 mais pas J = 0 → J = 0, et pour Ω = 0 → Ω = 0, 0≠∆J . Σ+ à Σ+, Σ- à Σ- Structure fine : (1Σ à 1Σ)
...)''J(''J''B)'J('J'B'v'''v'' ++−++σ=σ ε→ε 11 soit (avec J = J’’, niveau du bas)
)J(J''B)J(J'B'v'''v''P 11 +−−+σ=σ ε→ε
)J(J)''B'B('v'''v''
Q 1+−+σ=σ ε→ε
)J(J''B)J)(J('B'v'''v''R 121 +−+++σ=σ ε→ε
Franck Thibault Page 9 ch-VIIm.doc 23/03/2005
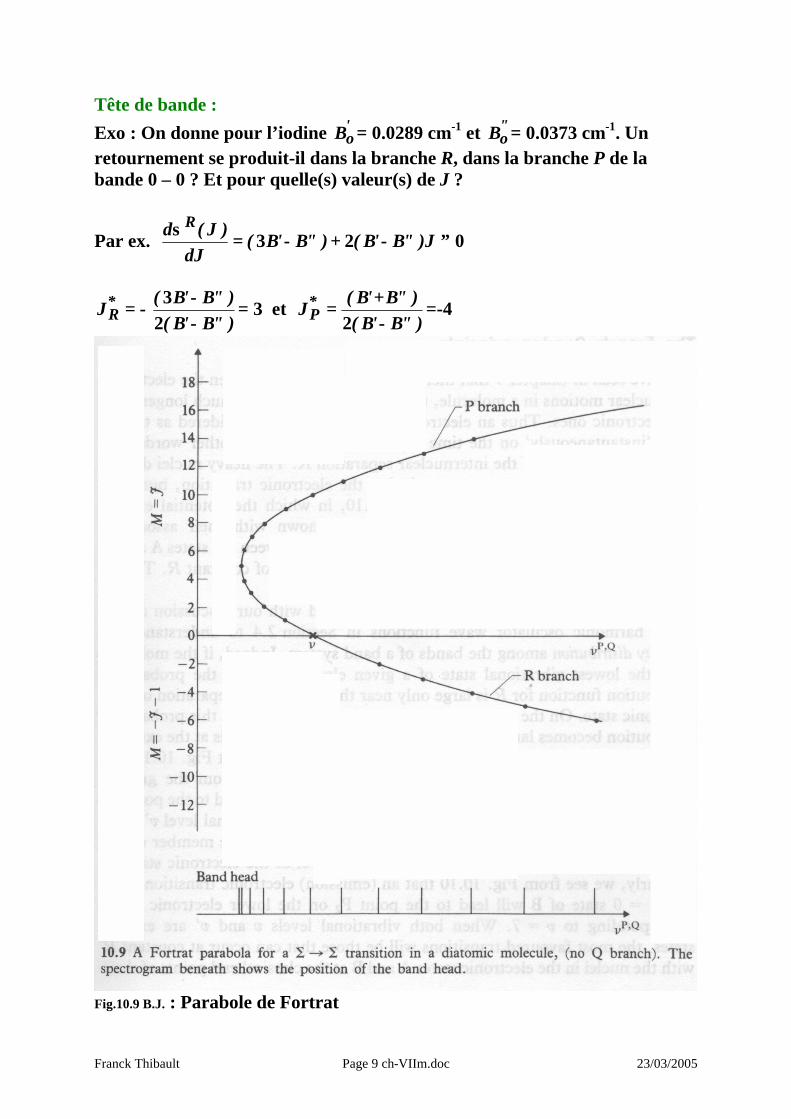
Tête de bande : Exo : On donne pour l’iodine '
oB = 0.0289 cm-1 et "oB = 0.0373 cm-1. Un
retournement se produit-il dans la branche R, dans la branche P de la bande 0 – 0 ? Et pour quelle(s) valeur(s) de J ?
Par ex. 023 ≡−+−=σ
J)"B'B()"B'B(dJ
)J(d R
)"B'B()"B'B(
J*R −
−−=
23
= 3 et )"B'B()"B'B(
J*P −
+=
2=-4
Fig.10.9 B.J. : Parabole de Fortrat
Franck Thibault Page 10 ch-VIIm.doc 23/03/2005
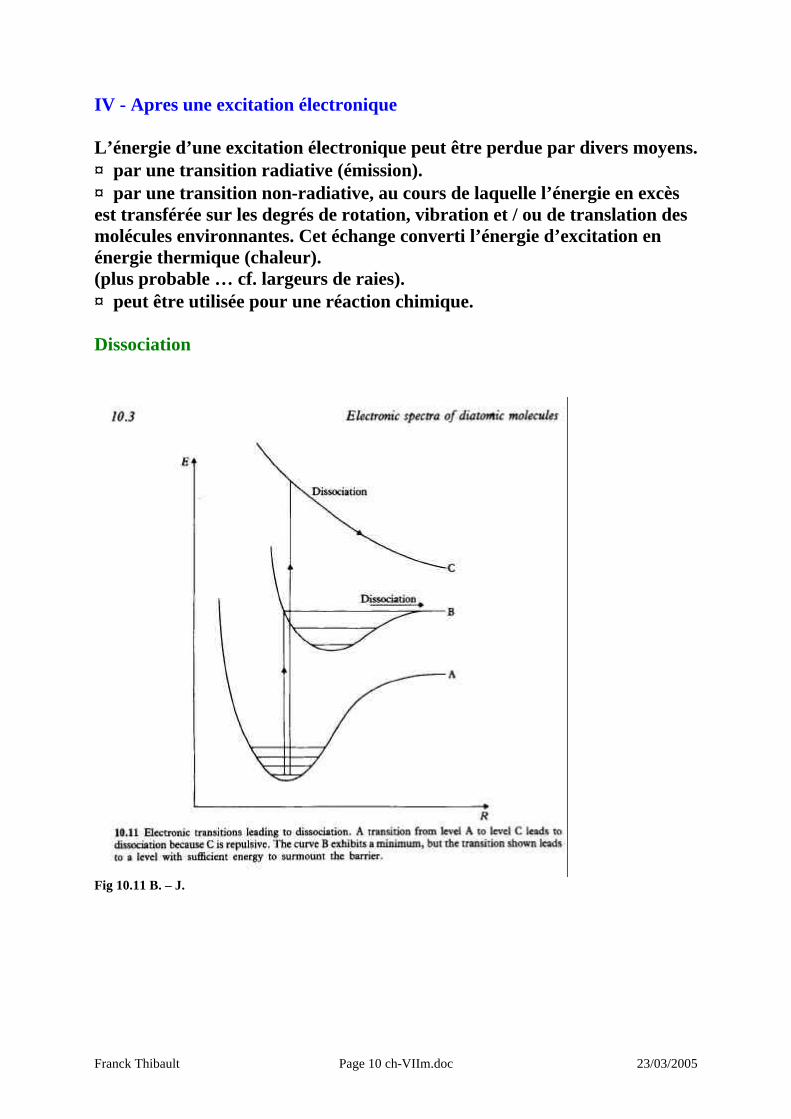
IV - Apres une excitation électronique L’énergie d’une excitation électronique peut être perdue par divers moyens. ♦ par une transition radiative (émission). ♦ par une transition non-radiative, au cours de laquelle l’énergie en excès est transférée sur les degrés de rotation, vibration et / ou de translation des molécules environnantes. Cet échange converti l’énergie d’excitation en énergie thermique (chaleur). (plus probable … cf. largeurs de raies). ♦ peut être utilisée pour une réaction chimique. Dissociation
Fig 10.11 B. – J.

Franck Thibault Page 11 ch-VIIm.doc 23/03/2005
Prédissociation
Fig 10.12 B. - J
Franck Thibault Page 12 ch-VIIm.doc 23/03/2005
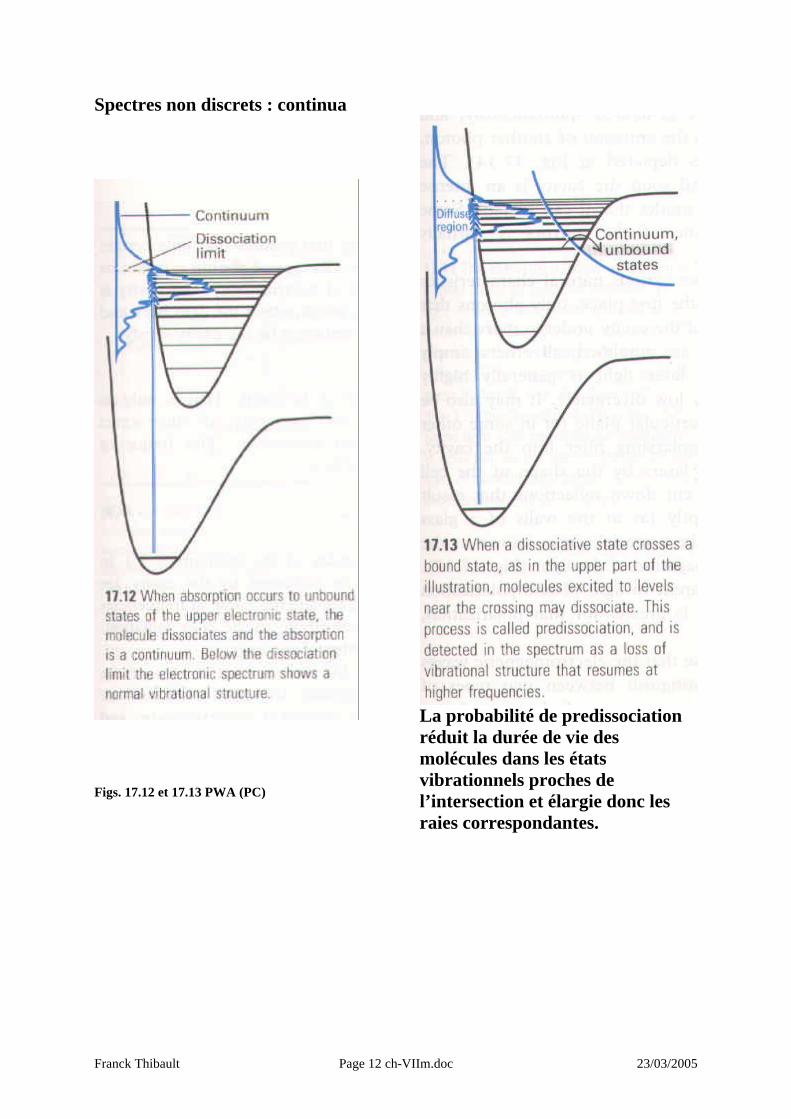
Spectres non discrets : continua
Figs. 17.12 et 17.13 PWA (PC)
La probabilité de predissociation réduit la durée de vie des molécules dans les états vibrationnels proches de l’intersection et élargie donc les raies correspondantes.
Franck Thibault Page 13 ch-VIIm.doc 23/03/2005
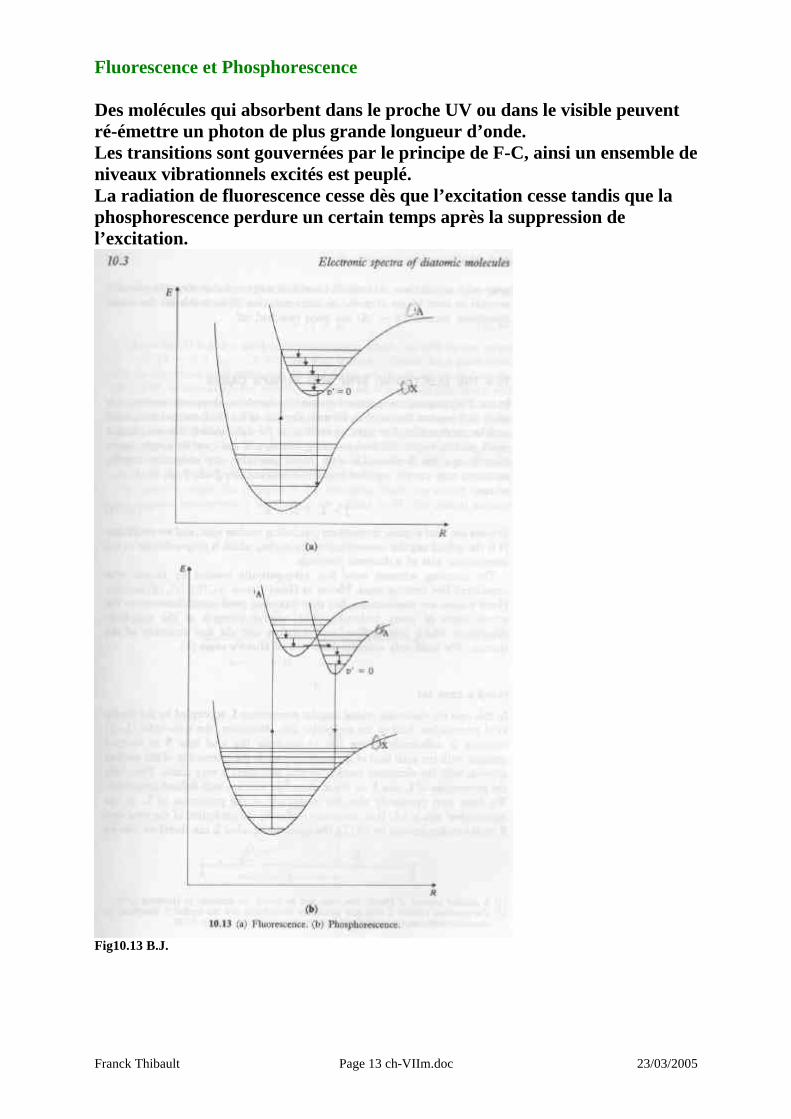
Fluorescence et Phosphorescence Des molécules qui absorbent dans le proche UV ou dans le visible peuvent ré-émettre un photon de plus grande longueur d’onde. Les transitions sont gouvernées par le principe de F-C, ainsi un ensemble de niveaux vibrationnels excités est peuplé. La radiation de fluorescence cesse dès que l’excitation cesse tandis que la phosphorescence perdure un certain temps après la suppression de l’excitation.
Fig10.13 B.J.
Franck Thibault Page 14 ch-VIIm.doc 23/03/2005
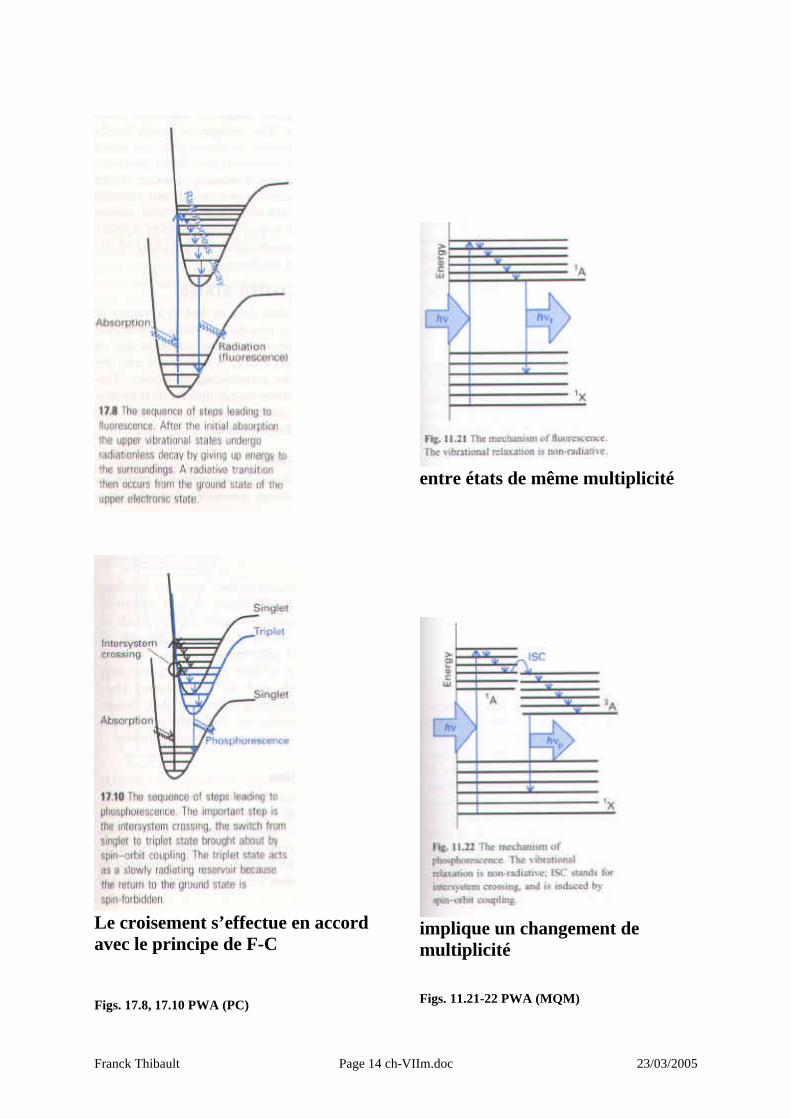
Le croisement s’effectue en accord avec le principe de F-C Figs. 17.8, 17.10 PWA (PC)
entre états de même multiplicité
implique un changement de multiplicité Figs. 11.21-22 PWA (MQM)

Franck Thibault Page 15 ch-VIIm.doc 23/03/2005


















