
Pa I Ca c R a c · Translational Radiobiology Group Robert Hawkins 54 Medical Oncology:Cell Therapy...
92
Paterson Institute for Cancer Research Scientific Report 2009
Transcript of Pa I Ca c R a c · Translational Radiobiology Group Robert Hawkins 54 Medical Oncology:Cell Therapy...

PatersonInstitute for CancerResearchScientific Report 2009

Cover images
Top
Mitotic BPAE cells in anaphase. F-actin is labelled with
Texas Red-x phalloidin. Microtubules, in green, are la-
belled with mouse anti-α-tubulin BODIPY FL goat anti-
mouse IgG. Blue nuclear staining with DAPI. Imaged
on the Spinning Disk Confocal microscope.
Image provided by Achille Dunn, Advanced Imaging Fa-
cility.
Bottom
Immunostaining demonstrating blood vessels
surrounding a tumour. Glut1 immunostaining (red)
specifically labels veinous structures whereas arterial
structures are Glut1 negative. Red blood cells in the
vessels were detected by inherent autofluorescence
(green) and cell nuclei were labelled with DAPI (blue).
Image provided by Darren Roberts, Clinical and Exper-
imental Pharmacology Group.

Paterson Institute for Cancer Research
Scientific Report 2009

Contents
Director’s Introduction 5
Research Highlights 8
Drug Discovery in the Manchester 12
Cancer Research Centre
Research Groups – Paterson Institute
Crispin Miller 16Applied Computational Biology and
Bioinformatics Group
Geoff Margison 18Carcinogenesis Group
Karim Labib 20Cell Cycle Group
Iain Hagan 22Cell Division Group
Nic Jones 24Cell Regulation Group
Angeliki Malliri 26Cell Signalling Group
Caroline Dive and Malcolm Ranson 28Clinical and Experimental Pharmacology Group
Ivan Ahel 30
DNA Damage Response Group
Peter L Stern 32Immunology Group
Nullin Divecha 34Inositide Laboratory
Tim Somervaille 36Leukaemia Biology Group
Georges Lacaud 38
Stem Cell Biology Group
Valerie Kouskoff 40Stem Cell and Haematopoiesis Group
Akira Orimo 42Stromal-Tumour Interaction Group
Research Groups – The University of Manchester
School of Cancer and Enabling Sciences
Robert E Hawkins and Peter L Stern 46Biological, Immune and Gene Therapy Group
Vaskar Saha 48Children’s Cancer Group
Tim Illidge 50
Targeted Therapy Group
Catharine M.L. West 52Translational Radiobiology Group
Robert Hawkins 54Medical Oncology: Cell Therapy Group
Gordon Jayson 56Medical Oncology: Translational
Anti-Angiogenesis Group
Research Services 58
Research Publications 66
Seminar Series 2009 74
Postgraduate Education 76
Contents

Operations 78
Cancer Research UK’s 84
Local Engagement and Development
Acknowledgement for Funding 86
of the Paterson Institute
Career Opportunities at the 87
Paterson Institute
Contact Details 88

4 | Paterson Institute for Cancer Research Scientific Report 2009
Director’s introduction
Welcome to the 2009 Paterson Institute Annual ScientificReport. It has been a particularly important year with thecompletion of the Institute’s Quinquennial Review but alsowith the initiation of the new Drug Discovery Centre.
The Institute is very privileged to receive suchstrong financial core support from CancerResearch UK. In return the Institute has toensure that it is undertaking research of thehighest international quality and that theresearch that we do will impact significantlytowards achievement of CR-UK’s major researchgoals as outlined in their 5-year strategic plan.There are a number of ways of measuring oursuccess which includes periodic review of all theresearch programmes. However, perhaps themost important process is the QuinquennialInstitute Review which is commissioned by CR-UK and involves assembling a team of world-leading scientists experienced in runningresearch organisations or institutes. The reviewteam visits the Institute over a two-day periodand assesses in depth the Institute’s performanceover the last five years and also reviews thestrategic direction and plans for the next five-year period. For CR-UK, such reviews areessential for them to ensure that the ‘return oninvestment’ is meeting or exceeding their
expectations and thereby justifies the long-termcommitment they make to the Institute and tothe Institute Director. For us, it is a strongreminder of the competitive environment wework in and the duty we have to deliver high-quality and relevant research.
The review took place at the end of June with avery successful outcome. The review partypraised the continuing progress we had madeover the last five years and were especiallysupportive of the establishment of theManchester Cancer Research Centre (MCRC),the progress it had made in such a short-periodof time since it was initiated and the significantrole the Institute plays in delivering the MCRC’sgoals and ambitions. I was obviously verypleased that the Institute received such strongsupport and that developments over the last fewyears were positively recognised. However,these reviews are not just about judgement ofpast activities but are also about assessing andadvising on future directions and in this respect
Nic Jones
Director’s Introduction | 5

6 | Paterson Institute for Cancer Research Scientific Report 2009
the review was also supportive and providedvaluable, constructive input into thedevelopment of future programmes which willensure that we can build on the progress wehave made over the next five years. Cancerresearch is at a very exciting stage and driven bynew technologies, new and exciting avenues areopening up especially at the laboratory/clinicalinterface. Given our proximity to The ChristieNHS Foundation Trust and our involvement inthe MCRC, we are well placed to take advantageof these new opportunities.
An exciting development that really began totake off this last year is the Drug DiscoveryCentre, an initiative that is a key component ofCR-UK’s strategic plan to increase its capabilityand activity in the development of smallmolecule drugs. Two new centres are beingdeveloped linked to the Paterson Institute andour sister institute in Glasgow, the BeatsonInstitute. CR-UK is providing significant newfunding in order to develop the centre at thePaterson Institute. Linking the centre to theInstitute has a number of advantages includingthe potential interactions with already existinggroups involved in biomarker research and thetesting of new therapies in early clinical trials, theexploitation of the cancer biology researchongoing with the Institute and the widerinteractions facilitated through the MCRC. Thereare real opportunities for academic drugdiscovery programmes to exploit and add valueto the research that we do and to considerareas of real clinical need that for a variety ofreasons might not be attractive to thepharmaceutical industry. Developing new drugsis very challenging and takes many years but thebenefits to us of having such a Centre within theInstitute will be tangible from the start – it willgreatly enhance the multidisciplinaryenvironment of the Institute, provideleading–edge chemical tools to enhance our
research efforts and instil within the Institute adrug hunting culture. Donald Ogilvie joined usin February 2009 to lead this new development.He previously acquired considerable experiencein leading drug discovery programmes atAstraZeneca having overseen the developmentof at least eight candidates from targetidentification to clinical trials. A chemistry lead(Allan Jordan) has also been recruited as well asa number of chemistry and biology teammembers and refurbishment of laboratoryfacilities suitable for this type of activity has justbeen completed. Thus in 2010 a number ofdiscovery programmes will be initiated and welook forward to experiencing the success of theCentre over the years to come.
Recruitment and retention of internationallycompetitive scientific leaders is essential formaintaining research excellence and buildingareas of research strength. Many of our recruitsare at the Junior Group Leader level and aftersix years they undergo a rigorous evaluation toconsider promotion to Senior Group Leaderlevel and as a consequence long-term andincreased commitment to research support. Thisis a very important quality-control step and theopinions of outside experts in the field arecrucial to the decision that is made. Only thoseleaders who have a demonstrable internationalprofile and have contributed significantly to thefield are expected to be successful. During thelast year, two group leaders were evaluated forsuch promotion – Georges Lacaud and ValerieKouskoff. We are delighted that both weresuccessful and will continue their careers andproductive research programmes in the Institute.They both work on the differentiation ofembryonic stem cells especially down thehaematopoietic lineage. Understanding in detailhow this process is regulated is important tounderstanding a number of haematologicalmalignancies. Another indicator of research

Director’s Introduction | 7
success and reputation is winning individualawards and this year two of our group leadersreceived an award. Iain Hagan was elected as anEMBO member. Membership is a lifelonghonour with new members nominated andelected annually based on proven excellence inresearch. Karim Labib was awarded the HookeMedal by the British Society for Cell Biology. Themedal is awarded to an emerging leader in cellbiology and will be presented to Karim at theannual spring meeting in 2010.
We continue to develop our research services –they are a vital component of the infrastructureof the Institute providing cutting-edge capabilitiesand technologies. The quality of these serviceswas especially praised during the site visit andthere is no doubt that their availabilityprofoundly changes the nature of theexperimental approaches that can be adoptedwithin the various research programmes. Aspart of our continuing investment we establisheda next generation sequencing platform and theadditional computation and analysisinfrastructure that supports this technology. Thistechnology is incredibly powerful and is beingused to address a number of importantbiological questions and will increasingly in thefuture be essential for addressing questions ofhigh clinical importance. This will be an area thatwill require continuous investment over thecoming years as the applications of thetechnology grows and new generation platformsdeveloped.
Inevitably the MCRC was an important themeconsidered by the Institute review party andthere was great enthusiasm for the partnershipand in particular the potential that is has forpromoting translational research and ensuringthat research funding can ultimately benefitcancer patients. Much progress within theMCRC has been made over the last year :investment in breast cancer research continuedwith the appointment to the University MedicalSchool of Professor Jonas Bergh from theKarolinska Institute; further investment was madeto the tumour biobank initiative recognising itsimpressive success; investment in biomarkerresearch by AstraZeneca through the AZ/MCRCalliance was doubled in a new three-yearagreement; development of a strategic plan forinvestment and development of lung cancerresearch. These are just a few examples ofdevelopments in the MCRC. We are building forthe future and 2009 saw great progress in thedevelopment of a new clinical treatment centreby The Christie NHS Foundation Trust. A thirdof this new £35 million facility will be devoted toearly phase clinical trials leading to one of thebiggest dedicated trial centres of its kindworldwide. Completion of this excitingdevelopment is expected in 2010. In addition,work is expected to begin soon on the detailedplanning of a new MCRC research building co-funded by CR-UK and The University ofManchester. This will provide great opportunitiesto increase our overall research efforts in keyareas of cancer research. So there is much tolook forward to. I hope you enjoy reading thisannual report and seeing the advances we aremaking.

In this section we are highlighting some research publicationsfrom 2009 which report significant advances in specific areas.The selected papers demonstrate the breadth and the qualityof the research being undertaken by Cancer Research UK-funded groups in the Paterson Institute.
Woodcock, S.A., Rooney, C., Liontos, M.,
Connolly, Y., Zoumpourlis, V., Whetton, A.D.,
Gorgoulis, V.G. and Malliri, A.
SRC-induced disassembly of adherens junctionsrequires localized phosphorylation anddegradation of the rac activator tiam1. Mol Cell 2009; 33: 639-653.
The Rac activator Tiam1 is required for adherensjunction (AJ) maintenance and its depletionresults in AJ disassembly. Conversely, theoncoprotein Src potently induces AJ disassemblyand epithelial–mesenchymal transition (EMT). Inthis study it was shown that Tiam1 isphosphorylated on Y384 by Src. This occurspredominantly at AJs, is required for Src-inducedAJ disassembly and cell migration, and creates adocking site on Tiam1 for Grb2. It was foundthat Tiam1 is associated with ERK. Followingrecruitment of the Grb2-Sos1 complex, ERKbecomes activated and triggers the localiseddegradation of Tiam1 at AJs, likely involvingcalpain proteases. Furthermore it wasdemonstrated that in human tumours Y384phosphorylation positively correlates with Srcactivity, while total Tiam1 levels are inverselycorrelated. Therefore, these data implicatedTiam1 phosphorylation and consequentdegradation in Src-mediated EMT and resultantcell motility, and established a new paradigm forregulating local concentrations of Rho-GEFs.
Tubbs, J.L., Latypov, V., Kanugula, S., Butt, A.,
Melikishvili, M., Kraehenbuehl, R., Fleck, O.,
Marriott, A., Watson, A.J., Verbeek, B., McGown,
G., Thorncroft, M., Santibanez-Koref, M.F.,
Millington, C., Arvai, A.S., Kroeger, M.D.,
Peterson, L.A., Williams, D.M., Fried, M.G.,
Margison, G.P., Pegg, A.E. and Tainer, J.A.
Flipping of alkylated DNA damage bridges baseand nucleotide excision repair. Nature 2009;
459: 808-813.
A few years ago, the Carcinogenesis Groupdiscovered a new family of proteins thatrecognise certain types of damage in DNAbases. Collaborating with groups in Newcastle,Sheffield, Bangor, Hershey, Minneapolis, Lexingtonand La Jolla, the crystal structure of the proteinfrom Schizosaccharomyces pombe, bound to ashort oligonucleotide containing such damage,has now been published in Nature. The proteinclamps around the damaged base and flips it outof the helix into a binding pocket, generating akink in the DNA. This results in the eliminationof the lesion from DNA, but defining thedetailed molecular mechanism of this process isproving rather a challenge. Nevertheless, if asimilar mechanism occurred in human cells, itcould have important implications not only incancer causation, but also in cancerchemotherapy, where the sensitivity of normalcells to the toxic side effects of treatment, andthe resistance of tumour cells to drugs, arerecurrent problems. The search is on.
Ivanov, A., Beers, S.A., Walshe, C.A.,
Honeychurch, J., Alduaij, W., Cox, K.L., Potter,
K.N., Murray, S., Chan, C.H., Klymenko, T.,
Erenpreisa, J., Glennie, M.J., Illidge, T.M. and
Cragg, M.S.
Monoclonal antibodies directed to CD20 andHLA-DR can elicit homotypic adhesion followedby lysosome-mediated cell death in humanlymphoma and leukemia cells. J Clin Invest 2009; 119: 2143-2159.
After the initial success with Rituximab (anti-CD20) monoclonal antibody (mAb) which hasimproved outcomes for patients with in B cell
Research Highlights
8 | Paterson Institute for Cancer Research Scientific Report 2009

malignancies, mAb are increasingly utilized in thetreatment of many cancers. Although the Fc-FcgR interactions with recruitment of immuneeffector cells such as macrophages and NK cellsare thought to explain much of the therapeuticeffect seen with some mAb like Rituximab, thisdoes not explain why certain mAb specificitiesare more potent than others. An additionaleffector mechanism available to mAb is thedirect induction of cell death. Previously, wedemonstrated that Type II anti-CD20 mAb wereable to evoke a non-apoptotic mode of celldeath that appeared linked with the induction ofhomotypic adhesion and furthermore was ableto overcome resistance to apoptosis in tumourcells. In this publication we reveal that peripheralre-localization of actin is critical for the adhesionand cell death induced by both Type II anti-CD20mAb and HLA DR Class II mAb in bothlymphoma cell lines and primary CLL cells. Themode of cell death engaged is rapid, non-apoptotic, non-autophagic and dependent onboth the integrity of plasma membranecholesterol and activation of the V-type ATPase.This cytoplasmic cell death involves lysosomeswhich swell and then disperse their contents,including cathepsin B, into the cytoplasm andsurrounding environment. The resulting loss ofplasma membrane integrity occurs in theabsence of DNA fragmentation and isindependent of caspase and Bcl-2 control. Theseexperiments provide new insights into how twoclinically relevant mAb elicit cell death and showfor the first time that this occurs through apreviously unrecognized lysosome-dependentpathway.
Somervaille, T.C., Matheny, C.J., Spencer, G.J.,
Iwasaki, M., Rinn, J.L., Witten, D.M., Chang, H.Y.,
Shurtleff, S.A., Downing, J.R. and Cleary, M.L.
Hierarchical maintenance of MLL myeloidleukemia stem cells employs a transcriptionalprogram shared with embryonic rather thanadult stem cells. Cell Stem Cell 2009; 4:
129-140.
Highlighted in:Cell Stem Cell Preview.Cell Stem Cell 2009; 4: 97-98.
An important question in the biology of acutemyeloid leukaemia is whether the leukaemiastem cells (LSCs) that drive expansion of thedisease and which trigger relapse are closer innature to normal haematopoietic stem cells(HSCs) or alternatively more like downstreammyeloid lineage cells that have inappropriatelyacquired an ability to undergo self-renewal. In amouse model of human leukaemia initiated byMLL fusion oncogenes LSCs have biologicalproperties quite distinct from HSCs: they aremetabolically active, proliferating, aberrantly self-renewing, downstream myeloid cells which havea transcriptional programme more akin to thatof embryonic stem cells than adult tissue stemcells. This observation suggests that genes andpathways important in LSCs could be selectivelytargeted by therapies that spare normal HSCs.
Patel, N., Krishnan, S., Offman, M.N., Krol, M.,
Moss, C.X., Leighton, C., van Delft, F.W.,
Holland, M., Liu, J., Alexander, S., Dempsey, C.,
Ariffin, H., Essink, M., Eden, T.O., Watts, C.,
Bates, P.A. and Saha, V.
A dyad of lymphoblastic lysosomal cysteineproteases degrades the antileukemic drug L-asparaginase. J Clin Invest 2009; 119:
1964-1973.
We are now in an unprecedented era where~90% of children with acute lymphoblasticleukaemia can be cured with combinationcytotoxic chemotherapy. The drugs used arenon-specific in action, show a wide interpatientvariability and associated with considerabletoxicity. This makes tailoring therapy difficult. Thispaper shows for the first time how leukaemiccells from some patients produce proteases thatdegrade and inactivate a key antileukaemic drugL-Asparaginase, suggesting that early screeningmay identify patients who do not benefit fromthis drug. By pinpointing and then modifying theexact sites of cleavage, the investigators wereable to produce a protease resistant active L-Asparaginase. In the process, they identified keystructural details that will allow the engineeringof a safer and better drug for all patients. Whilecurrent focus is on identifying smart moleculesfor targeted therapy, this paper shows there isstill life in the old drug yet.
Research Highlights | 9

Morohashi, H., Maculins, T. and Labib, K.
The Amino-Terminal TPR Domain of Dia2Tethers SCF(Dia2) to the Replisome ProgressionComplex. Curr Biol 2009;19: 1943-1949.
E3 ligases for ubiquitin and Sumo play a key role in preserving genome stability duringchromosome replication, by activating orrepressing particular pathways of DNA repair.This study reported that a specific form of theSCF ubiquitin ligase associates with thereplisome in budding yeast. All eukaryotes havemultiple forms of the SCF E3 ligase, distinguishedfrom each other by different ‘F-box’ subunits that target the ligase to specific substrates. Inaddition to the substrate-binding domain, around one third of F-box proteins haveadditional domains at the amino terminus ofunknown function. The association of SCFDia2with the replisome was found to be mediated by a unique TPR domain at the amino terminusof Dia2, which binds two particular componentsof the replisome. The TPR domain of Dia2tethers SCFDia2 to the replisome, probablyincreasing the local concentration of the ligase at forks. This represents a novel form ofregulation of SCF E3 ligases, and becomesimportant when cells accumulate a specific classof stalled fork. It now seems likely that theamino terminal domains of other F-box proteinsmight also control the localisation of theircognate SCF ligases.
Lawrence, C.L., Jones, N. and Wilkinson, C.R.
Stress-Induced Phosphorylation of S. pombe Atf1Abrogates Its Interaction with F Box ProteinFbh1. Curr Biol 2009; 19: 1907-1911.
The Atf1 transcription factor is critical fordirecting stress-induced gene expression infission yeast. Previously we found that uponexposure to stress, Atf1 is hyper-phosphorylatedby the MAP kinase, Sty1, which results in itsstabilization. The resulting increase in Atf1 is vitalfor a robust response to stress. Here, weinvestigated the mechanism by whichphosphorylation stabilizes Atf1 and found thatthis protein is a target for the ubiquitin-proteasome system with its degradationdependent upon an SCF E3 ligase containing theF-box protein Fbh1. F-box proteins usuallytarget phosphorylated substrates forubiquitination. However, stress-inducedphosphorylation serves to inhibit the binding ofAtf1 to Fbh1, thus representing a novel means ofregulating the interaction between an F-boxprotein and its substrate. Atf1 is the firstexample of a substrate for any SCFFbh1complex but it seems likely that Fbh1, incommon with other F-box proteins, will directmultiple targets for ubiquitination via theSCFFbh1. Potential substrates are proteins
involved in the homologous recombinationpathway of DNA repair, as others have shownthat Fbh1 acts downstream of Rad51 in thisprocess. Moreover, the mechanism we havedescribed for regulating Atf1-Fbh1 binding mayapply to other substrates of Fbh1.
Dean, E., Jodrell, D., Connolly, K., Danson, S.,
Jolivet, J., Durkin, J., Morris, S., Jowle, D., Ward,
T., Cummings, J., Dickinson, G., Aarons, L.,
Lacasse, E., Robson, L., Dive, C. and Ranson, M.
Phase I trial of AEG35156 administered as a 7-day and 3-day continuous intravenous infusion inpatients with advanced refractory cancer. J Clin Oncol 2009; 27: 1660-1666.
This paper demonstrates synergistic workingbetween the DCU Early Clinical Trials Unit andthe Clinical and Experimental PharmacologyGroup and reports the ‘first into man’ study ofAEG35156, a second generation antisense to X-linked inhibitor of apoptosis protein (XIAP). Theclinical hypothesis tested was that XIAPinhibition reduces the threshold for apoptosis intumour, exploiting inherent cellular stresses inthe tumour micro-environment. This CR-UKsponsored trial was the first undertakenworldwide for a XIAP targeted drug. Wedetermined the maximum tolerated dose ofAEG35156, and examined a number ofpharmacodynamic circulating and imagingbiomarkers. Knock down of XIAP mRNA wasdemonstrated in PBMCs and drug-inducedchanges in circulating cell death biomarkers wereobserved. The study showed that the drug waswell tolerated with clinical evidence of activity inrefractory lymphoma, melanoma and breastcancer. Dr Dean has since taken up a ClinicalLectureship to continue her research onapoptosis targeted drugs.
Lancrin, C., Sroczynska, P., Stephenson, C., Allen,
T., Kouskoff, V. and Lacaud, G.
The haemangioblast generates haematopoieticcells through a haemogenic endothelium stage. Nature 2009; 457: 892-895.
Highlighted in:Nature News & Views. Nature 2009; 457: 801-803.
Cell Stem Cell Preview. Cell Stem Cell 2009; 4:189-190.
Nature Reports Stem Cells 2009; Mar 12;
doi:10.1038/stemcells.2009.35.
selected by F1000
The cellular origin of blood cells is controversial.One first model proposes that haematopoieticand endothelial cells arise from a commonmesodermal precursor called thehaemangioblast. A conflicting theory insteadassociates the first haematopoietic cells to a
10 | Paterson Institute for Cancer Research Scientific Report 2009

differentiated endothelial cell withhaematopoietic potential, i.e. a haemogenicendothelium. In this paper, we demonstratedthat the emergence of blood cells from thehaemangioblast precursor proceeds through ahaemogenic endothelium intermediate. Theseresults unite the two theories on the origin ofhaematopoietic development into a single lineardevelopmental process. This finding stronglysupports the endothelial origin of some, if not all,haematopoietic cells.
Gandillet, A., Serrano, A.G., Pearson, S., Lie,
A.L.M., Lacaud, G. and Kouskoff, V.
Sox7-sustained expression alters the balancebetween proliferation and differentiation ofhematopoietic progenitors at the onset of bloodspecification. Blood 2009; 114: 4813-4822.
The molecular mechanisms that regulate thebalance between proliferation and differentiationof precursors at the onset of haematopoiesisspecification are poorly understood. We show in
this study that Sox7 is transiently expressed atthe onset of blood specification. While Sox7knockdown decreases the formation ofhaematopoietic progenitors, the enforcedexpression of this transcription factor promotesthe maintenance of multi-potency and self-renewal. Our data demonstrate that thesustained expression of Sox7 is sufficient tocompletely alter the balance betweenproliferation and differentiation ofhaematopoietic precursors. Removal of Sox7-enforced expression fully restores thisequilibrium and leads to the efficientdifferentiation of haematopoietic progenitors.This represent a very attractive characteristic ofSox7 function and might in the future become apowerful molecular tool to allow the expansionof haematopoietic progenitors to be used forpotential cell replacem-ent therapy. From afundamental perspective, it will be veryinteresting to explore the molecular programmethat is either maintained or initiated by Sox7 expression.
Research Highlights | 11

In their recent strategy review, Cancer Research UK decided toincrease significantly their long term investment in smallmolecule drug discovery and to align this additional resourcewith the core-funded cancer research institutes in Glasgow(Beatson) and Manchester (Paterson).
The purpose of co-locating these activities is, ofcourse, to maximise the opportunity fortranslating the ground-breaking basic cancerresearch from these centres of excellence intonovel therapeutic opportunities.
In this article, we will outline our vision for thedrug discovery centre in Manchester and howwe intend to deliver maximum value from thisnew investment.
Our ultimate aim is to identify novel drugtherapies to satisfy the unmet clinical needs ofcancer patients. However, drug discovery andclinical development are long and complexprocesses and we will need to engage with manypartners to achieve this goal.
The first key partner is of course CancerResearch UK who are providing the crucialfunding - £8 million for the first five years. ButCancer Research UK is more than just a sourceof funding for this new venture. As well asindividual programme grants, Cancer ResearchUK already supports major drug discoverycentres in London, Sutton and Newcastleproviding a broad portfolio of projects. The newcentres in Manchester and Glasgow will beseeking to complement one another inextending this portfolio into new areas ofbreaking cancer science and drug discoverytechnology. The leaders of these drug discoverycentres are now meeting regularly to shareexpertise, coordinate their activities and identifyareas of cooperation and collaboration in orderto maximise the effectiveness of CancerResearch UK drug discovery. As one example ofthis cooperation, we will be accessing thecompound collection and screening technologyin the London centre to support our hitidentification projects.
Another important part of the Cancer ResearchUK “family” is Cancer Research Technology(CRT) who provide us with intellectual propertyand business development support. This isparticularly important for the protection of drugdiscovery inventions and, in the longer term, foridentification of partners to take our candidatedrugs into clinical trials.
Our major source of local partnerships is theManchester Cancer Research Centre (MCRC).Within this environment there is a rich pool ofbasic and translational cancer science, cuttingedge technology and clinical expertise.
A key component of the MCRC is the breadthof clinical and drug development expertise atThe Christie Hospital. This provides directinsight into the areas of unmet clinical need andthe hypotheses to address them but also bringsa tangible connection with our ultimatecustomer, the cancer patient. At the other endof the MCRC spectrum are the basic scientists inthe Paterson Institute and more broadly inManchester University who provide insights intothe mechanisms of cancer and how to measurethese in preclinical models. In the middle are thetranslational scientists and clinicians, particularlyin the Clinical and Experimental PharmacologyGroup at the Paterson, who provide theroadmap for initial clinical development,particularly in the validation of novel biomarkers.We are also exploring opportunities to accessother key technologies (e.g. biophysical andcomputational chemistry, biochemistry andprotein structural analysis) through experts inManchester University.
Since drug discovery and development takessuch a long time (10+ years) and many projectsdo not make it to the clinic we need to need tobe able to demonstrate that we are making
Donald Ogilvie &
Allan Jordan
Drug Discovery in the Manchester Cancer Research Centreby Donald Ogilvie and Allan Jordan
12 | Paterson Institute for Cancer Research Scientific Report 2009

progress in the shorter term. In the first fiveyears, this will be primarily through thegeneration of a unique (within Cancer Research UK) portfolio of attractive drugdiscovery projects.
During the last six months we have spent a lotof time developing our target selection strategyinto a “roadshow” that we have been presentingto groups of cancer researchers in the MCRC.These presentations have been followed up withmore detailed target discussions and this hasidentified the highest priority projects that arealready underway (through collaborations).Target review will be an ongoing activity so thatwe can keep abreast of new developments incancer science and fuel the drug discovery“pipeline” with the best opportunities.
Once a target is identified, the aim of the drugdiscovery process is to identify compounds thatmodulate its activity in order to deliver clinicalbenefit. This is an iterative process involving theidentification of initial chemical “hits”, theexploration of their drug potential to create
“leads” and then the optimisation of these leadsto create a clinical candidate for testing in cancer patients.
In parallel with our target selection activities, wehave designed, built and equipped a newlaboratory and have recruited a highly skilledteam of drug discovery biologists and chemists,all of whom have had industrial experience inthe large or smaller (Biotech) pharmaceuticalsectors. This core of expertise will enable us tohit the ground running when the new laboratoryopens in January 2010. An unusual butdeliberate feature of the new facility is the co-location of chemistry and biological scienceactivities in the same laboratory. We believe thatthis will foster closer teamwork between thosewho design and make novel compounds andthose who test their activity.
In 2009 we laid the foundations of this new andexciting venture. By the end of 2010 we willhave a fully functioning team and laboratory andwill have started our first home-grown MCRCdrug discovery projects.
Drug Discovery in the Manchester Cancer Research Centre | 13
Figure 1
The newly completed Drug
Discovery laboratories

14 | Paterson Institute for Cancer Research Scientific Report 2009
Research groupsPaterson Institute for Cancer Research
Research Groups - Paterson Institute for Cancer Research | 15

16 | Paterson Institute for Cancer Research Scientific Report 2009
Applied Computational Biology and Bioinformatics Grouphttp://www.paterson.man.ac.uk/bioinformatics
The Applied Computational Biology and Bioinformatics groupis a computational genomics group focused on developing abetter understanding of the genome and the role it plays incancer. Much of the group’s work is directed at exploring thecomplexities that arise through processes such as alternativesplicing and the expression of non-coding RNAs. We do thisthrough a combination of bench science, computer science,mathematics and statistics, and our work is highly dependenton analysing and integrating the data arising from technologiessuch as next generation sequencing, microarrays andproteomics.
Alternative Splicing
Although generally the most well characterisedparts of the genome, many protein-coding lociare still not fully understood, not least because ofthe additional complexities caused by alternativesplicing. This is the process by which cells canselectively remove different sections of pre-mRNA during RNA processing. It allows theexpression of a set of closely related, butdifferent transcripts from a single locus, isprevalent, and tightly controlled. The majority ofhuman genes are alternatively spliced, increasingthe molecular repertoire of a cell substantially.Given its prevalence, it is not surprising that it isintimately involved in many of the key processesassociated with cancer, including angiogenesis,differentiation and apoptosis, and it has beenshown to be disrupted in many cancers.
Until relatively recently, it has been impossible tostudy alternative splicing in a systematic manner,due to our inability to generate global surveys oftranscription at sufficient levels of detail.However, advances in technology have nowstarted to make this possible. Affymetrix Exon1.0ST arrays aim, for example, to separatelytarget every known and predicted exon in theentire genome by featuring individual probesetsplaced at strategic intervals across each gene. Incollaboration with Professor Adrian Harris inOxford and the Translational RadiobiologyGroup at The University of Manchester, we havebeen using these arrays to consider changes in
splicing as a consequence of tumour hypoxia inHead and Neck Squamous Cell Carcinomas(HNSCCs). To do this, Carla Möller-Levet hasdeveloped novel algorithms for analysing thesignals from each individual exon probesettargeting a given gene in order to identifydifferential splicing events. This work has built onearlier efforts in the group to developannotation databases (http://xmap.picr.man.ac.uk;Yates et al., Nucleic Acids Res 2008; D780) andanalysis software in R/BioConductor (exonmap;Okoniewski et al., Genome Biol 2007; 8: R79).
Through these studies, we were able to identifya set of characteristic splicing events, a subset ofwhich were subsequently validated using realtime PCR (Guy Betts; Translational Radiobiology).This included an isoform of the gene Laminin α3,LAMA3-A, which we were able to show wasprognostic for overall survival, while an alternateisoform of the same gene, LAMA3-B, was not(Möller-Levet et al., 2009).
Massively Parallel Nucleotide Sequencing
(MPNS) and RNA-Seq
A substantial amount of the group’s effort hasbeen directed at handling the billions ofnucleotides generated each week by our ABSOLiD Next Generation Sequencing platform.James Bradford has been exploring how it canbe used to generate global surveys oftranscription through the analysis of total RNAand generating, in collaboration with Yaoyong Li,
Group Leader
Crispin Miller
Postdoctoral Fellows
James BradfordJohn Hall (joint with TranslationalRadiobiology Group)Hui Sun LeongYaoyong LiCarla Möller-Levet (joint withTranslational RadiobiologyGroup; to September 2009)
Scientific Officer
Paul Scutt
Research Applications
Programmers
Tim YatesChris Wirth
Graduate Students
Danny BittonSharmin Naaz (joint with StemCell and Haematopoeisis Group)Andrzej Rutkowski (joint withImmunology Group)
System Administrator
Zhi Cheng Wang (joint with ITdepartment)

Applied Computational Biology and Bioinformatics Group | 17
the analysis techniques and statistical filtersneeded to routinely use the platform for RNA-Seq applications. This has involved extensive useof genome annotation supplied through ourdatabase, X:Map, and BioConductor package,exonmap.
Non coding RNAs
The human genome consists of approximately 3billion nucleotides, of which only about 2%actually code for proteins. This raises afundamental question as to how much of theremaining 98% of the genome is functional, andthe additional roles it might play within a cell.Recent technological developments includingtiling microarrays and next-generationsequencing have led to a substantial increase inour understanding of the non protein-codingcomplement of the human genome, and it isnow known that the majority of it is actuallytranscribed. Many of these loci are now knownto express RNA sequences that are functionalwithin their own right, even though they arenever translated into proteins. A key focus ofthe group is to develop a better understandingof these non-coding RNAs (ncRNAs) and theroles they play in cancer. We are currently usingMPNS technology to generate RNA-Seqdatasets to support these analyses.
Genome annotation
As technology advances further, a commontheme is the ability to generate unbiased surveysof the entire genome at increasingly fine levels ofgranularity. In order to make sense of thesedata, it is necessary to have access to genomeannotation in a form that makes it amenable toalgorithmic analysis and the application of
appropriately robust statistics. We have beendeveloping annotation tools that make thesedata available in the statistical software R, andhave been extending this work to provideintegrated access to DNA, RNA and proteinlevel annotation.
A further challenge with MPNS datasets is theneed to visualise the results of an experiment,which in its raw form consists of millions ofindividual short nucleotide sequences. We aredeveloping extensions to our Google Mapsbased genome browser (Tim Yates, X:Map;http://xmap.picr.man.ac.uk) that allows MPNSdata to be presented alongside genomeannotations (see Figure).
Formalin-Fixed Paraffin-Embedded Tissue
Vast archives of well-annotated clinical materialexist as Formalin-Fixed Paraffin-Embedded(FFPE) tissue. However, this approach to tissuepreservation, which was developed beforetechniques such as microarrays were invented,poses significant challenges if these samples areto be used for expression profiling experiments.In collaboration with Kim Linton (The Universityof Manchester) and Stuart Pepper (MolecularBiology Core Facility) we have been able toshow that the material can be successfullyprofiled on Affymetrix Exon arrays (Linton et al.,2009), and work is also underway incollaboration with the Translational RadiobiologyGroup to apply these arrays to the analysis ofcervix tumours (see also the TranslationalRadiobiology report).
Publications listed on page 66
Figure 1
RNA-Seq data generated from AB
SOLiD and displayed in the X:Map
genome browser. Millions of short
50mer RNA sequences were
generated using the AB SOLiD
sequencer, and aligned to the
genome (green peaks) in order to
provide a very fine-grained measure
of gene expression. These data can
be placed alongside genome
annotation using the X:Map browser.
The blue box in the figure
corresponds to a gene, with individual
exons shown by the smaller red
boxes. The 3’ UTR is shown at the
end of the gene in white, and the
locations of known protein domains
are shown in the orange box below.
Below this, protein domain
annotations are shown.

18 | Paterson Institute for Cancer Research Scientific Report 2009
Carcinogenesis Grouphttp://www.paterson.man.ac.uk/carcinogenesis
We have been involved in investigating the mechanism of thebiological effects of a class of chemical agents called thealkylating agents. Our interest is based on the observationsthat agents of this type are mutagenic, and are probably humancarcinogens, and on the fact that they are toxic, a characteristicthat is exploited in their use in the treatment of certain typesof cancer. Both mutation and toxicity can be explained by thereaction of these agents with the purine and pyrimidine basesin DNA. Although there are more than a dozen known typesof DNA damage that can be generated, one of these, O6-alkylguanine, which often constitutes only about 6% of the totaldamage, seems to be the most important. Our current focusis on how this damage is processed and the impact that thishas on the biological effects of these agents.
recognised by the post replication mismatchrepair system, which results in a series of eventsthat can culminate in cell death or DNArecombination. MGMT can therefore protectcells against both the mutagenic (both pointmutations and recombinations) and toxic effectsof alkylating agents.
In relation to cancer chemotherapy, that sometumours do not respond to dacarbazine orTemozolomide treatment has been attributed tothe protective effect of MGMT. In our attemptsto circumvent this, we have previously describedthe drug Lomeguatrib (LM), originally known asPaTrin-2 and one of the products of a veryfruitful collaboration with Prof Brian McMurryand the late Dr Stanley McElhinney (and theirgroup at the Chemistry Department, TrinityCollege, Dublin). LM is a very potent inactivatorof MGMT and in a range of preclinical studies iteffectively sensitised human cells and humantumour xenografts to the killing effect ofTemozolomide and other agents of that type.Clinical trials of LM in combination withTemozolomide have been completed and thedose required to inactivate MGMT in severaltumour types has been established.
Group Leader
Geoff Margison
Postdoctoral Fellow
Vitaly Latypov
Scientific Officers
Gail McGown Mary Thorncroft Mandy Watson
Graduate Student
Andrew Marriott
Undergraduate Students
Alison Bennett (From Sept)James Ding (June-Aug)Michael Morten (July-Sept)Sonia McNichol (From Aug)
Volunteer worker
Jonathan Doyle (to Feb)
Background
The simplest representatives of the alkylatingagents are the methylating agents. These includepotent toxins and mutagens such as N-methyl-N-nitrosoguanidine (MNNG) andchemotherapeutic agents such as dacarbazine,which is used in the treatment of malignantmelanoma and the Cancer Research UK drugTemozolomide, which is used in the treatment ofmelanoma and glioma. All of these agentsgenerate O6-methylguanine in DNA and thisappears to be responsible for their biologicaleffects. Our current perception of themechanisms of these effects is summarised inFigure 1. The most critical factor in whether ornot these effects are manifested is probably thedamage reversal protein O6-methylguanine-DNAmethyltransferase (MGMT), which can simplyremove the methyl group and restore the DNAto its predamaged state (Figure 1) in a reactionthat also results in the inactivation of the protein.If this does not happen, the DNA can bereplicated and a mispair, either O6meG:T orO6meG:C, can be generated. If the formerundergoes further replication, a G:C to A:Ttransition mutation is generated, and this is themost characteristic mutational hallmark of theseagents. However, both mismatches can be

Carcinogenesis Group | 19
Some organisms do not have an MGMT gene,and based on our original amino acid sequencehomology searches we have established thatthese possess a different mechanism for dealingwith O6-alkylguanine damage in their DNA.Efforts directed towards the characterisation ofthis novel repair pathway are ongoing.
Clinical trials of Lomeguatrib
Lomeguatrib, has now completed phase II clinicaltrials in combination with Temozolomide inmalignant melanoma. Unfortunately, LM did notimprove the response of melanoma toTemozolomide, but actually this is not unusual,melanoma being one of the most difficult andchemotherapy-resistant tumours to treatsuccessfully. Further work in this area hasinvolved defining the dose of LM that isnecessary to inactivate MGMT in other humantumour types including glioma, prostate andcolorectal tumours. This study involvedadministration of LM to patients who wereabout to undergo surgical removal of theirprimary tumour. Resected tumours wereanalysed for active and inactivated MGMT andthis allowed calculation of the amount of MGMTthat was inactivated. In subsequent patients,doses were escalated until complete inactivationwas obtained. In principle this knowledge cannow be exploited to design phase II trials inthese diseases.
In addition, we have published an assessment ofthe levels of MGMT in peripheral blood cells inrelation to the toxic effects of Temozolomide inthe bone marrow. This has highlighted thepossibility that blood samples might be used toindicate which patients may be more affected bythe myelosuppressive effects of Temozolomide,and hence be more closely monitored, but alsowhich patients should be more resistant, andhence might tolerate higher doses of the drug.
These possibilities have yet to be put intopractice clinically.
Alkyltransferase-like proteins
MGMT genes are present in prokaryotes, archeaand eukaryotes and are characterised by thepresence in the active site domain of a cysteineresidue that accepts the alkyl group from the O6-position of guanine. A few years ago wereported the presence of what we calledalkyltransferase-like (ATL) proteins inprokaryotes and some simple eukaryotes. Thekey difference is that ATL proteins do not havecysteine, but usually tryptophan in this position.The evolution of these proteins is itself intriguing,inasmuch as E. coli expresses in fact two MGMT-like and one ATL protein, that we have namedeATL, whereas Saccharomyces cerevisiae
expresses only an MGMT protein andSchizosaccharomyces pombe expresses only anATL protein, that we have named Atl1. ATLproteins are much smaller than MGMT proteinsand the tryptophan residue is not the onlyreason for their inability to transfer alkyl groups:in our initial studies, we showed that mutation oftryptophan to cysteine in the Atl1 protein didnot confer MGMT activity.
Work in this area continues and in onecollaboration with David Williams (University ofSheffield) and John Tainer (Scripp’s ResearchInstitute, La Jolla), crystal structures of Atl1bound to short duplex oligonucleotidescontaining O6-methylguanine or O6-pyridyloxobutylguanine have been obtained.From these, the similarity in the ways that bothAtl1 and MGMT bind to DNA, flip out the O6-alkylguanine from the base stack using anarginine “finger” and accommodate the base in abinding pocket are very clear. We have alsoprovided further evidence, in the form ofadditional epistasis analysis, to support ourprevious suggestion that processing of thedamage proceeds following the binding of Atl1,via the nucleotide excision repair pathway, andnot the base excision repair or double strandbreak repair pathways. Thus there is increasingevidence that Atl1 is a damage sensing proteinthat signals to downstream factors. We arecurrently exploring if this is the global genomeor transcription-coupled branch of thismechanism and, using various methodologies,what proteins might be involved in theseinteractions.
Publications listed on page 66
Figure 1
Possible fates of O6-methylguanine in
DNA. SCE, sister chromatid
exchange; TDG, thymine-DNA
glycosylase; BER, base excision repair ;
MGMT, O6-methylguanine-DNA
methyltransferase; me, methyl group;
Top1, indicates binding of
topoisomerase 1 to O6-
methylguanine present in Top1
cleavage sites; HR, homologous
recombination; NHEJ, non-
homologous end joining; Y-family,
translesion DNA polymerases; N,
Cytosine or Thymine; ,
phosphorylation; S1, S2, first and
second S-phase following exposure to
methylating agents and formation of
DNA damage; Exo1, Exonuclease 1.
Black and red horizontal dashed
lines generally indicate parent and
template DNA strands. Question
marks indicate a degree of
uncertainty.
p

20 | Paterson Institute for Cancer Research Scientific Report 2009
Cell Cycle Grouphttp://www.paterson.man.ac.uk/cellcycle
Our group studies the mechanisms that drive the eukaryoticcell cycle. During 2009 we described novel aspects of thestructure and function of the eukaryotic replisome, the multi-protein machine that mediates chromosome replication atDNA replication forks. Co-ordination between the replicativehelicase that unwinds the parental duplex, and the DNApolymerases that act on the leading and lagging strands, isimportant to minimise the exposure of single-strand DNA andthus preserve genome integrity. We found that the Ctf4protein and the GINS complex play a key role in coupling thehelicase complex to DNA polymerase alpha that acts on thelagging strand. In addition, we found that two proteinsassociated with the helicase at forks serve to recruit aparticular E3 ubiquitin ligase, which acts to preserve genomeintegrity during chromosome replication.
breakage of chromosomes during replication.The eukaryotic replisome is poorlycharacterised, and is likely to be significantlymore complex than its prokaryotic counterparts.It seems likely that the formation of thereplisome will involve factors that physically linkthe MCM2-7 helicase to the three replicativepolymerases that mediate DNA synthesis on theleading and lagging strands.
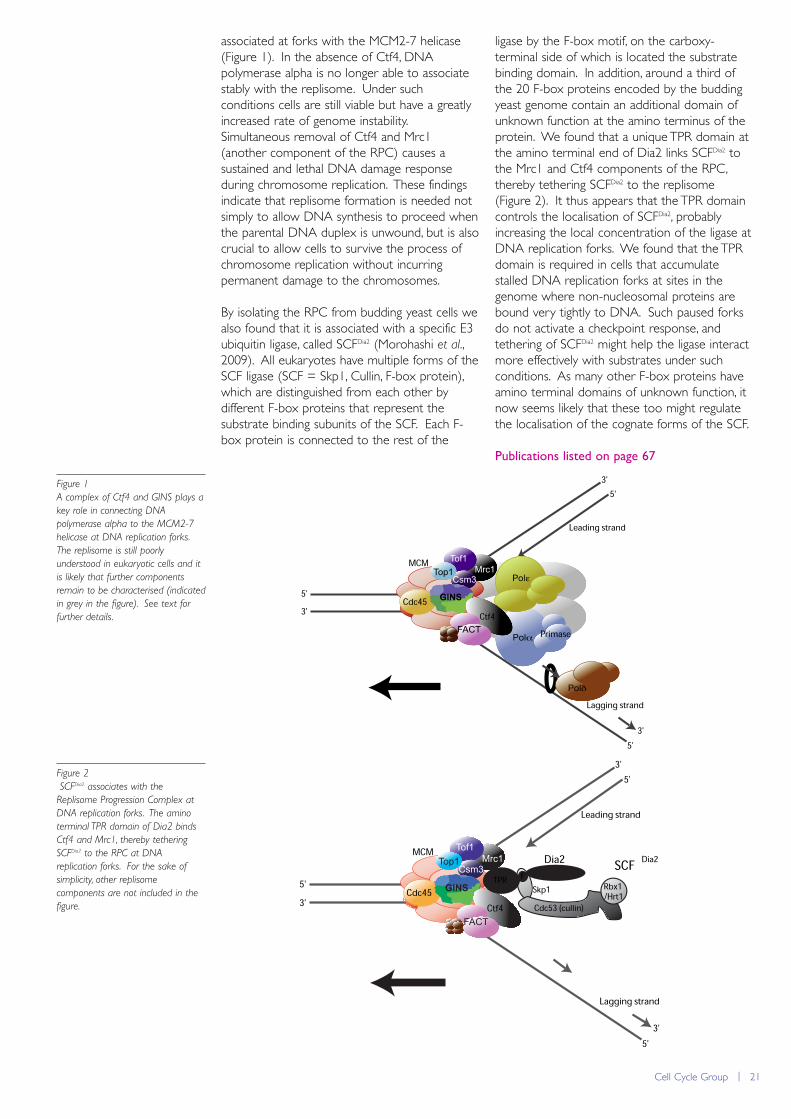
Previously we found that a set of regulatoryfactors assemble around the MCM2-7 helicase atreplication forks to form what we called theReplisome Progression Complex or RPC(Gambus et al., Nat Cell Biol 2006; 8: 358).Formation and maintenance of the RPC requiresthe GINS complex, which together with Cdc45is likely to be an essential component of theactive MCM2-7 helicase (Cdc45-MCM-GINStogether form the CMG complex). This year wereported that the Ctf4 protein plays a key rolein connecting MCM2-7 helicase at forks to DNApolymerase alpha that acts on the lagging strand(Gambus et al., 2009). We found that Ctf4 bindsdirectly to the amino terminus of the catalyticsubunit of DNA polymerase alpha, as well asbinding directly to the GINS complex that is
Group Leader
Karim Labib
Postdoctoral Fellows
Giacomo de PiccoliLuis Garcia-RodriguezAlberto Sanchez-DiazSugopa Sengupta
Scientific Officers
Frederick van DeursenPedro Junior Nkosi
Graduate Students
Asli DevrekanliMagdalena FoltmanTim Maculins
Chromosome replication is a highly complexprocess in eukaryotic cells, about which muchremains to be discovered. Part of thecomplexity comes from the fact that replicationis regulated very carefully to try to ensure that asingle perfect copy of the genome is made ineach round of the cell cycle. Additionalcomplexity comes from the fact that DNAsynthesis at replication forks is coupled to otherinteresting processes such as the reproduction ofepigenetic chromatin marks throughout thegenome, the establishment of cohesion betweenthe nascent sister chromatids, and the activationof checkpoint signaling pathways in response toproblems at forks (e.g. caused by DNA damage).
By analogy with prokaryotes it seems very likelythat a subset of replication factors acting atDNA replication forks will interact to form alarge multi-protein machine called the replisome.The formation of the replisome ensures thatunwinding of the DNA duplex by the replicativehelicase is co-ordinated with synthesis of theleading and lagging strands. This co-ordination isvery important as it serves to reduce theexposure of single-strand DNA that mightotherwise be attacked by nucleases and lead to
Cell Cycle Group | 21
associated at forks with the MCM2-7 helicase(Figure 1). In the absence of Ctf4, DNApolymerase alpha is no longer able to associatestably with the replisome. Under suchconditions cells are still viable but have a greatlyincreased rate of genome instability.Simultaneous removal of Ctf4 and Mrc1(another component of the RPC) causes asustained and lethal DNA damage responseduring chromosome replication. These findingsindicate that replisome formation is needed notsimply to allow DNA synthesis to proceed whenthe parental DNA duplex is unwound, but is alsocrucial to allow cells to survive the process ofchromosome replication without incurringpermanent damage to the chromosomes.
By isolating the RPC from budding yeast cells wealso found that it is associated with a specific E3ubiquitin ligase, called SCFDia2 (Morohashi et al.,2009). All eukaryotes have multiple forms of theSCF ligase (SCF = Skp1, Cullin, F-box protein),which are distinguished from each other bydifferent F-box proteins that represent thesubstrate binding subunits of the SCF. Each F-box protein is connected to the rest of the
ligase by the F-box motif, on the carboxy-terminal side of which is located the substratebinding domain. In addition, around a third ofthe 20 F-box proteins encoded by the buddingyeast genome contain an additional domain ofunknown function at the amino terminus of theprotein. We found that a unique TPR domain atthe amino terminal end of Dia2 links SCFDia2 tothe Mrc1 and Ctf4 components of the RPC,thereby tethering SCFDia2 to the replisome(Figure 2). It thus appears that the TPR domaincontrols the localisation of SCFDia2, probablyincreasing the local concentration of the ligase atDNA replication forks. We found that the TPRdomain is required in cells that accumulatestalled DNA replication forks at sites in thegenome where non-nucleosomal proteins arebound very tightly to DNA. Such paused forksdo not activate a checkpoint response, andtethering of SCFDia2 might help the ligase interactmore effectively with substrates under suchconditions. As many other F-box proteins haveamino terminal domains of unknown function, itnow seems likely that these too might regulatethe localisation of the cognate forms of the SCF.
Publications listed on page 67
Figure 1
A complex of Ctf4 and GINS plays a
key role in connecting DNA
polymerase alpha to the MCM2-7
helicase at DNA replication forks.
The replisome is still poorly
understood in eukaryotic cells and it
is likely that further components
remain to be characterised (indicated
in grey in the figure). See text for
further details.
5'3'
5'3'
Leading strand
PriPrimase
5'
Lagging strand
3'
Cdc45
MCMMrc1
Tof1
Csm3Top1
GINS
Ctf4FACT
5'3'
5'3'
Leading strand
5'
Lagging strand
3'
Cdc45
MCMMrc1
Tof1
Csm3Top1
GINS
Ctf4FACT
Dia2
Skp1Cdc53 (cullin)
Rbx1/Hrt1
FTPRSCF Dia2
Figure 2
SCFDia2 associates with the
Replisome Progression Complex at
DNA replication forks. The amino
terminal TPR domain of Dia2 binds
Ctf4 and Mrc1, thereby tethering
SCFDia2 to the RPC at DNA
replication forks. For the sake of
simplicity, other replisome
components are not included in the
figure.

22 | Paterson Institute for Cancer Research Scientific Report 2009
Cell Division Grouphttp://www.paterson.man.ac.uk/celldivision
Development and health rely upon the controlled balance ofcell growth and division. The size of each organ at each stageof our lives is a product of the number of cells in that tissueand the size of each cell. Cancer arises from an imbalance ofcell proliferation. It is becoming increasingly apparent thaterrors in the ability to integrate growth control and celldivision can lead to this imbalance. Thus, understanding the co-ordination between growth and division lies at the heart ofunderstanding the basis of many cancers. Because theregulatory networks that control the timing of cell division andchromosome segregation are highly conserved, studying thecomplexities of cell division in the relatively simple unicellularyeasts greatly accelerates the analysis of the more complexissue of the control of cell division in man.
positive feedback loop to promote the furtheractivation of Cdc25 and inhibition of Wee1 todrive complete commitment to mitosis.
Chromosome segregation by the mitotic
spindle
In addition to their role in the feedbackactivation of MPF, the mitotic kinases promotethe assembly of the bipolar mitotic spindle thatphysically segregates the duplicated genomesinto each daughter cell. The principalcomponents of the mitotic spindle are the twosets of microtubules that extend from the twospindle poles. After attachment to spindlemicrotubules, the chromosomes align midwaybetween the two poles (figure 2) before eachchromosome splits in two and each half movesto either pole. Subsequent ingression of theplasma membrane between these segregatedgenomes (figure 3) completes cell division aseach daughter cell inherits one genome and onespindle pole.
Group Leader
Iain Hagan
Postdoctoral Fellows
Marisa Alonso-Nuñez Marisa Madrid
Scientific Officer
Torsten Geerlings
Graduate Students
Elvan BokeDorota FeretAvinash PatelYisu Wang
Activation of Mitosis Promoting Factor (MPF)
regulates the timing of cell division
We study cell division in the fission yeastSchizosaccharomyces pombe because it is asimple, unicellular organism with excellentgenetics that is cheap to grow and dividesrapidly. Commitment to mitosis in S. pombe, asin all eukaryotes, is regulated by the activity of aprotein kinase complex called MPF. MPF iscomposed of a catalytic sub-unit encoded by thecdc2+ gene and a regulatory sub-unit calledcyclin B. Prior to mitosis MPF is inhibited viaphosphorylation by the protein kinase Wee1 ona residue (tyrosine 15) that lies in the ATPbinding pocket of Cdc2. This phosphate can beremoved by a protein phosphatase encoded bythe cdc25+ gene. The balance of activitybetween Cdc25 and Wee1 is the critical factor indetermining when MPF will be activated to drivemitotic commitment. Once a critical thresholdlevel of MPF is activated a positive feedback loopis promoted to boost Cdc25 activity andsuppress Wee1 activity, thereby driving full-scalecommitment to mitosis (figure 1). Fully activatedMPF then activates a number of highlyconserved kinases that are named after thefounder members of each group Polo, auroraand NIMA. These kinases participate in the

Cell Division Group | 23
Characterisation of Cut12 suggests that events
on the spindle pole regulate MPF activation
during mitotic commitment
The characterisation of the cut12.1 mutation inour laboratory a number of years ago uncoveredan unanticipated link between the spindle poleand MPF activation. We found that this loss offunction mutation in the spindle pole componentCut12 blocked spindle formation as cells formedmonopolar, rather than bipolar spindles.Surprisingly, we also found that the cut12+ genewas identical to the stf1+ gene. The gain-of-function stf1 mutations had been identifiedbecause they enabled cells to live without themitotic promoter Cdc25 (suppressor of twentyfive). Subsequent work in our laboratory hadestablished that this influence of Cut12 uponmitotic control involved modulation of theactivity of the feedback loop kinase polo at thespindle pole, however, the basis for the cut12.1
monopolar spindle phenotype has remainedobscure. By exploiting the light- and electron-microscopy core facilities within the PatersonInstitute we have established that cut12.1 cellsare unable to form a bipolar spindle becausethey fail to activate the newer of the two mitoticspindle poles. Thus, the cut12.1 loss-of-functionphenotype arises from a local deficiency in MPFactivation while the gain-of-function stf1mutations lead to inappropriate global activationof MPF throughout the entire cell. Theseobservations put the Cut12/Stf1 molecule andthe spindle pole at the heart of the controls thatdetermine the timing of cell division.
From yeast to man
Our findings in fission yeast suggest that thefeedback loop that provides the priming impetusto promote global MPF activation emanatesfrom the spindle pole. The demonstration thatactive MPF first appears on human spindle poles
in human cells by Jon Pines’ group (CR-UKfunded, Gurdon Institute Cambridge) supportsthe view that the networks we are studying inyeast are representative of those operating inman. Thus, continued exploitation of themalleability of fission yeast to interrogate Cut12function will shed light on conservedmechanisms operating in the more intractablehuman cells.
Lessons from yeast
The ability to manipulate genes at will in a simpleorganism whose primary purpose is simply togrow and divide is enabling us to explore thefiner points of the pathways that co-ordinategrowth with spatial and environmental cues. Thisinformation informs studies in higher systemsthat, in turn, raise models that can be mostreadily tested in yeast. This re-iterative cycle ofcomparative studies ensures that great stridesare being made in understanding the molecularbasis of cell division and growth.
Publications listed on page 67
Cdc25
Wee1cyclinB
Cdc2
cyclinB
Cdc2
T14 Y15
P P
Positive feedback loop
Positive feedback loop
Figure 1
Figure 2
Microtubules extend from the two
spindle poles (purple spheres) to
either interdigitate with microtubules
extending from the opposite poles
(black lines) or bind to specialised
regions on the chromosomes (brown
bars) called kinetochores (green
circles).
Figure 3
After the chromosomes have moved
towards the poles along kinetochore
associated microtubules the plasma
membrane (thick blue line) is pulled
in by the cytokinetic, actomyosin ring
(green hoops) to initiate the
separation of the cell into two
daughters.
Figure 1 Figure 2

24 | Paterson Institute for Cancer Research Scientific Report 2009
Cell Regulation Grouphttp://www.paterson.man.ac.uk/groups/cellreg.jsp
The AP-1 transcription factor is activated in response to manyextracellular signals including growth factors, cytokines andvarious stress conditions. As a result it is essential for a widevariety of biological activities which in mammalian cells rangefrom cell proliferation and differentiation to regulation ofapoptosis. Deregulation of AP-1 activity has been associatedwith numerous disease conditions such as inflammation andcancer.
Group Leader
Nic Jones
Associate Scientists
Wolfgang BreitwieserCaroline Wilkinson
Postdoctoral Fellows
Yujun DiClare Lawrence (until Sept)
Scientific Officers
Keren DawsonSteve Lyons
Graduate Students
Orestis Mavroudis-Chocholis(until Dec)Malgorzata GozdeckaEmily Holmes (from Oct)Jacek WalczynskiLu Zhang
We are characterising the potential role of ATF2in B-cell lymphomas since a number of reportshave shown that JNK is highly active in culturedB lymphoma cell lines, and that JNK is critical fortumour cell growth and survival. Interestingly wefound that the levels of active ATF2 is alsoelevated in B lymphoma lines (e.g. Burkitt’slymphoma, Follicular lymphoma) and to addressthe functional significance of this increasedactivity we currently analyse a set of tumour celllines in which ATF2 has been targeted by RNAknockdown. In a complementary approach wehave generated a B-cell specific ATF2 knockoutmouse and crossed them to transgenic mice thatexpress the B-cell tumour inducing Eμ-Myctransgene. Differences in the number of Mycinduced lymphomas or the timing of lymphomaonset will establish the importance of ATF2 inthis tumour type.
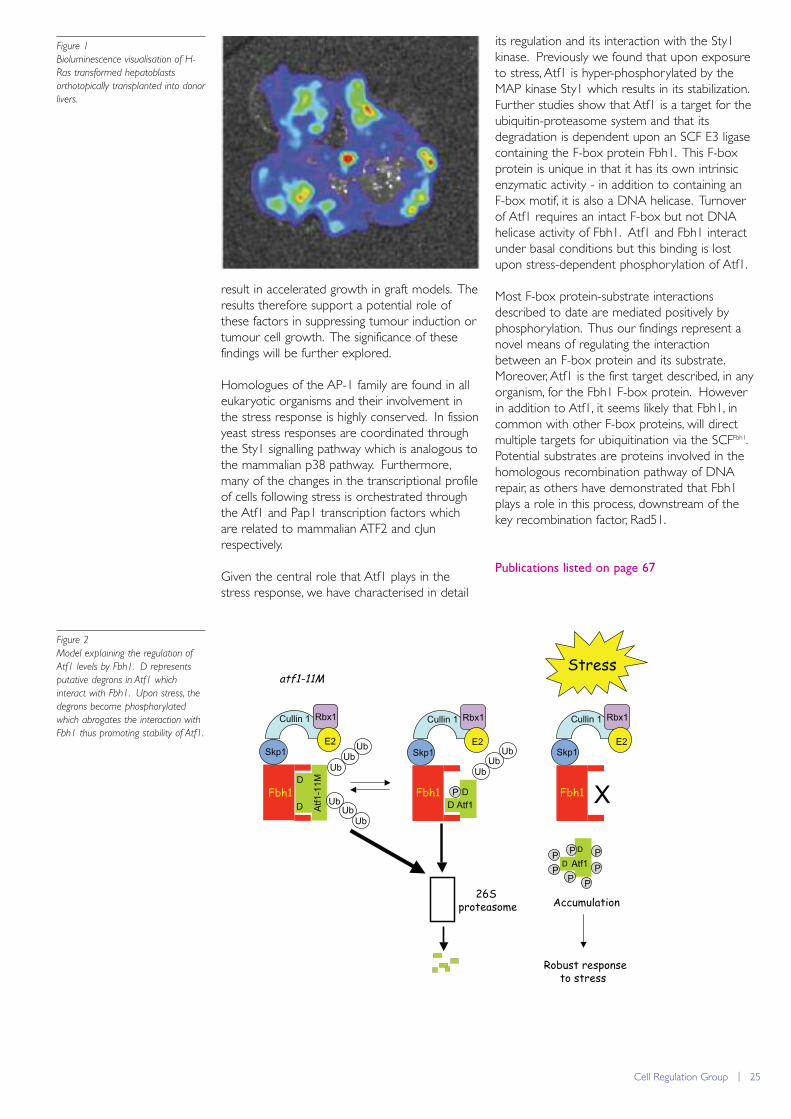
We had previously established that ATF2 andATF7 were essential for the survival ofhepatoblasts in the developing embryo throughcoordinating negative regulating feedbackmechanisms that restrict the activity of the stressactivated kinase p38. Cultured hepatoblasts canbe used to study the onset of hepatocellularcarcinoma (HCC) through transformation withoncogenes and reintroduction into recipientlivers via orthotopic transplantation. We utilisedthis technique to address a possible role forATF2 and ATF7 in HCC. We found that doubleknockout hepatoblasts transformed with theHRas oncogene (HRasG12D) produced moreand significantly larger tumours in recipient liverscompared to hepatoblasts that were normal forATF2. In addition, deletion of ATF2 and ATF7 incells isolated from established liver tumours
The AP-1 factor comprises a diverse array ofhomo- and heterodimeric complex combinationsinvolving proteins from the Jun, Fos, ATF and Maftranscription factor families. These combinationsvary from one cell type to another and differentcombinations recognise distinct DNA elementsand are differentially regulated. Much of thework in our laboratory has focused on two ofthese AP-1 proteins, the transcription factorsATF2 and ATF7, which are both activated byphosphorylation mediated by the stress activatedMAP (mitogen activated protein) Kinases p38and JNK. ATF2 and ATF7 have been specificallyimplicated in stress responses, cell cycleprogression, apoptosis and DNA damageresponse. Germ line mutation of Atf2 leads topost-natal lethality and simultaneous deletion ofboth Atf2 and Atf7 leads to embryonic lethalityas a result of massive apoptosis in the embryonicliver involving both developing hepatocytes andhaematopoietic cells.
Evidence is accumulating that ATF2 can promoteoncogene activation or tumour suppressionactivity depending on the tissue context. We areusing different mouse models to investigate thepotential role of ATF2 and ATF7 intumourigenesis. A tumour suppressor role hasbeen revealed in a skin tumourigenesis modelusing a mutant mouse where ATF2 is specificallydeleted in keratinocytes (in collaboration with ZRonai, Burnham Institute for Medical Research).Upon tumour initiation and promotion, themutant animals demonstrate a significantly earlieronset of papillomas as well as greater numbers.Likewise we have shown that irradiation of micewith ATF2 specifically deleted in T-cells results inearlier onset of T-cell lymphomas.
Cell Regulation Group | 25
Figure 1
Bioluminescence visualisation of H-
Ras transformed hepatoblasts
orthotopically transplanted into donor
livers.
result in accelerated growth in graft models. Theresults therefore support a potential role ofthese factors in suppressing tumour induction ortumour cell growth. The significance of thesefindings will be further explored.
Homologues of the AP-1 family are found in alleukaryotic organisms and their involvement inthe stress response is highly conserved. In fissionyeast stress responses are coordinated throughthe Sty1 signalling pathway which is analogous tothe mammalian p38 pathway. Furthermore,many of the changes in the transcriptional profileof cells following stress is orchestrated throughthe Atf1 and Pap1 transcription factors whichare related to mammalian ATF2 and cJunrespectively.
Given the central role that Atf1 plays in thestress response, we have characterised in detail
its regulation and its interaction with the Sty1kinase. Previously we found that upon exposureto stress, Atf1 is hyper-phosphorylated by theMAP kinase Sty1 which results in its stabilization.Further studies show that Atf1 is a target for theubiquitin-proteasome system and that itsdegradation is dependent upon an SCF E3 ligasecontaining the F-box protein Fbh1. This F-boxprotein is unique in that it has its own intrinsicenzymatic activity - in addition to containing anF-box motif, it is also a DNA helicase. Turnoverof Atf1 requires an intact F-box but not DNAhelicase activity of Fbh1. Atf1 and Fbh1 interactunder basal conditions but this binding is lostupon stress-dependent phosphorylation of Atf1.
Most F-box protein-substrate interactionsdescribed to date are mediated positively byphosphorylation. Thus our findings represent anovel means of regulating the interactionbetween an F-box protein and its substrate.Moreover, Atf1 is the first target described, in anyorganism, for the Fbh1 F-box protein. Howeverin addition to Atf1, it seems likely that Fbh1, incommon with other F-box proteins, will directmultiple targets for ubiquitination via the SCFFbh1.Potential substrates are proteins involved in thehomologous recombination pathway of DNArepair, as others have demonstrated that Fbh1plays a role in this process, downstream of thekey recombination factor, Rad51.
Publications listed on page 67
Atf1
26S proteasome
atf1-11M
Skp1
Rbx1Cullin 1
E2
Fbh1
Atf1
-11M
Ub
Ub
UbSkp1
Rbx1Cullin 1
E2
P
P
Skp1
Rbx1Cullin 1
E2
PPPP P
XAtf1PFbh1 Fbh1
Accumulation
UbUb
Ub
Ub
UbUb
D
DD
D
Stress
Robust response to stress
D
D
Figure 2
Model explaining the regulation of
Atf1 levels by Fbh1. D represents
putative degrons in Atf1 which
interact with Fbh1. Upon stress, the
degrons become phosphorylated
which abrogates the interaction with
Fbh1 thus promoting stability of Atf1.

26 | Paterson Institute for Cancer Research Scientific Report 2009
Cell Signalling Grouphttp://www.paterson.man.ac.uk/cellsignalling
Tumour initiation and progression result from inappropriateactivation of intracellular signalling cascades. Rho-like GTPasesare molecular switches in signalling pathways that regulatecytoskeletal and junctional organisation, as well as genetranscription. In this way, Rho proteins influence cellmorphology, adhesion, motility, as well as cell cycle progressionand cell survival. Rho proteins are transforming in vitro and areessential for Ras-mediated in vitro transformation. Moreover,data have emerged to directly implicate Rho proteins intumour initiation and progression in vivo. Our groupinvestigates how the activities of certain regulators of the Rhoprotein Rac are controlled. We are also identifying signallingevents and cellular processes downstream of Rac thatmodulate tumour susceptibility and disease progression.
number of mesenchymal cell lines (Malliri &Collard, Curr Opin Cell Biol 2003; 15: 583).Moreover, using both RNA interference and cellsderived from Tiam1-deficient mice, it has beenshown that endogenous Tiam1 is required forboth the formation as well as the maintenanceof cadherin-based adhesions (Malliri et al., J BiolChem 2004; 279: 30092). The oncoprotein Src,a non-receptor tyrosine kinase implicated inmalignant progression, potently inducesepithelial–mesenchymal transition (EMT) bytargeting AJs for dissassembly. We recentlyshowed that direct phosphorylation of Tiam1 bySrc is required for the initial stages of Src-induced EMT. Moreover, we identified a novelpost-translational mechanism of regulating Tiam1levels. We showed that Src phosphorylatesTiam1 on tyrosine 384 (Y384). This occurspredominantly at AJs during the initial stages ofSrc-induced EMT and creates a docking site onTiam1 for Grb2. We found that Tiam1 isconstitutively associated with extracellular signal-regulated kinase (ERK). Following recruitment ofthe Grb2-Sos1 complex, ERK becomes activatedand triggers the localised degradation of Tiam1at AJs through activating calpain proteases.Significantly, we demonstrated that in humanlung, colon, and head and neck cancers
Group Leader
Angeliki Malliri
Postdoctoral Fellows
Sonia Castillo-LluvaClaire Rooney (until March2009)Helen Rushton Simon Woodcock
Scientific Officer
Gavin White
Graduate Students
Lucy Dalton (until October2009)Natalie ReevesChong Tan
Tiam1 (for T-lymphoma invasion and metastasisprotein) is a guanine nucleotide exchange factor(GEF) that selectively activates Rac. Tiam1-deficient cells are resistant to Ras-inducedcellular transformation (Malliri et al., Nature2002; 417: 867). Mice deficient for Tiam1 areresistant to the formation of skin tumoursinduced by chemical carcinogens and consequentoncogenic activation of the c-Ha-Ras gene(Malliri et al., Nature 2002; 417: 867).Nonetheless, the few skin tumours arising inTiam1-deficient mice progressed morefrequently to malignancy than those in wild-typemice, suggesting that Tiam1 deficiency promotesmalignant conversion (Malliri et al., Nature 2002;417: 867). Thus, while Tiam1/Rac co-operatewith Ras in establishing tumours, they antagonizeRas during tumour invasion.
Tiam1/Rac signalling is targeted by Src during
the epithelial–mesenchymal transition.
One mechanism by which Tiam1 and Racsuppress malignant progression is throughpromoting cell–cell adhesion. In vitro studieshave shown that over-expression of activatedRac or Tiam1 promotes the formation ofadherens junctions (AJs) and the accompanyinginduction of an epithelial-like phenotype in a

Cell Signalling Group | 27
phosphorylation of Y384 of Tiam1 positivelycorrelated with Src activity, while total levels ofTiam1 were inversely correlated with Src activity,consistent with the above-mentioned post-translational regulatory mechanism operating inmalignancies. Abrogating Tiam1 phosphorylationand degradation suppressesed Src-induced AJdisassembly. As a consequence, cells expressinga non-phosphorylatable Tiam1 showed a markeddecrease in wound closure in response to Src(Woodcock et al., 2009b).
A distinct role for the homologue of Tiam1,
STEF, in regulating focal adhesions.
The mechanisms underlying focal adhesiondisassembly, required for optimal cell migration,are poorly understood. Microtubules are criticalmediators of this process; direct targeting offocal adhesions by microtubules coincides withtheir disassembly. Re-growth of microtubules,induced by removal of the microtubuledestabiliser nocodazole, activates the Rho-likeGTPase Rac, concomitant with focal adhesiondisassembly. Recently we have shown that theRac guanine nucleotide exchange factor (GEF)STEF (for Sif and Tiam1-like exchange factor) isresponsible for activation of Rac duringmicrotubule re-growth. Importantly we alsoshowed that STEF is required for multipletargeting of focal adhesions by microtubules. Asa result, focal adhesions in STEF knock-downcells have a reduced rate of disassembly and areconsequently enlarged. This leads to a reducedspeed of migration in these cells. Taken together,these findings reveal a novel role for the Rac-
GEF STEF in focal adhesion disassembly and cellmigration via microtubule-mediated mechanisms.
Tiam1 interacting proteins.
It is increasingly apparent that Rho GEFs domore than simply activate Rho molecules; severalstudies now point to their role in influencing thechoice of biological response elicited by a givenRho protein. GEFs have been shown to bind toeffectors directly or to scaffold proteins thatcomplex with components of effector pathways.Thus Tiam1 interacts with IB2/JIP2, a scaffold thatpromotes Rac activation of p38 kinase cascadeover JNK MAP kinase cascade (Buchsbaum et al.,Mol Cell Biol 2002; 22: 4073), and also withspinophilin, a scaffold that promotes Racactivation of p70 S6K over Pak1, a different Raceffector (Buchsbaum et al., J Biol Chem 2003;278: 18833). In our lab, we are usingbiochemical approaches to identify Rac and RacGEF interacting proteins involved in differentaspects of transformation including malignantprogression (acquisition of invasiveness). Towardthis end, we recently reported a modifiedtandem affinity purification method that enrichesfor transient protein interactions. Using thistechnique, we identified 14-3-3 proteins as Tiam1binding partners. The interaction of Tiam1 with14-3-3 proteins was largely dependent on theN-terminal region of Tiam1; within this region,there are four putative phospho-serine-containing 14-3-3 binding motifs, and weconfirmed that two of them (Ser172 andSer231) are phosphorylated in cells using massspectrometry. Moreover, we showed thatphosphorylation at three of these motifs(containing Ser60, Ser172 and Ser231) isrequired for the binding of 14-3-3 proteins tothis region of Tiam1. We showed also thatphosphorylation of these sites does not affectTiam1 activity; significantly however, wedemonstrated that phosphorylation of theSer60-containing motif is required for thedegradation of Tiam1 (Woodcock et al., 2009a).
Publications listed on page 67
Figure 1
MDCK cells treated with sodium
pervanadate (PV), an irreversible
protein-tyrosine phosphatase
inhibitor, display endogenous Tiam1
phosphorylated at Tyrosine 384
specifically at cell-cell adhesions.
Figure 2
Model: Src phosphorylates Tiam1 at
sites of cell-cell adhesions.
Phosphorylated Tiam1 recruits the
Grb2-Sos complex and, via MEK,
increases activation of the ERK
associated with Tiam1, and hence
the local activation of calpain
proteases at cell–cell adhesions.
Calpain mediated proteolysis of
Tiam1 results in its inactivation,
reducing the activity of Rac that is
necessary to maintain cadherin
adhesions.

28 | Paterson Institute for Cancer Research Scientific Report 2009
Clinical and Experimental Pharmacology Grouphttp://www.paterson.man.ac.uk/cep
Development of molecularly targeted anticancer drugsmandates parallel development of biomarkers to achieve theright drug at the right dose for the right patient. CEP researchis predicated on novel agents entering clinical trial within theDerek Crowther Early Clinical Trials Unit (DCU) at TheChristie Hospital. DCU typically supports c100-120 trials withc6400 patient visits p.a. A new £35M Cancer TreatmentCentre will open in 2010 to provide comprehensive clinicaltrials facilities, one of the largest early clinical trials centresworldwide, and the incorporated and enhanced biomarkerlaboratory facilities will strengthen further the CEP-DCU axis.In 2009, biomarker research highlights included investigations ofthe collective migration of circulating tumour cells, the utility ofcirculating cell death biomarkers in B lymphoma, drugcombination studies for BH-3 mimetic drug that targetapoptosis control, and developments in clinical proteomics,tissue biomarkers and biostatistics approaches related tobiomarker driven clinical trials.
Radford and Dr Kim Linton, circulatingbiomarkers of cell death (nucleosomal DNA(nDNA) and cytokeratin 18 (CK18)), togetherwith circulating FLT3 ligand, a potentialmyelosuppression biomarker, were assessed atbaseline and serially after standardchemotherapy. CK18 is not expressed inlymphoma cells and thus reports drug-inducedepithelial toxicity. Baseline nDNA level washigher in all lymphoma subtypes compared tohealthy controls and was prognostic for overallsurvival in Diffuse Large B Cell Lymphoma.Baseline nDNA also predicted extent of tumourshrinkage post chemotherapy. Circulating CK18increased within 48h of chemotherapy and wassignificantly higher in patients experiencingepithelial toxicity graded >3 by CTCAE scoringcriteria. FLT3 ligand was elevated within 3-8days of chemotherapy and predicted subsequentdevelopment of neutropenic sepsis. These earlypromising data suggest that inexpensivecirculating biomarkers could contribute usefulinformation regarding tumour response and
Circulating Tumour Cells (CTCs).
Within the Lung Cancer Disease Focus Group(Lead, F Blackhall), a filter-based approach wastested to isolate CTCs. CTCs were foundcirculating both as tumour microemboli (clustersof cells, Figure 1) and as single cells thatexpressed the lung diagnostic biomarker TTF1.Considerable cellular heterogeneity wasapparent regarding epithelial (cytokeratins) andmesenchymal (vimentin, N-cadherin) markers inboth single CTCs and microemboli suggestive ofpartial Epithelial Mesenchymal Transition (EMT).Initial analysis suggests that CTC survival isenhanced during collective migration (asmicroemboli) and the relevance of this topatient’s response to therapy and survival isbeing sought.
CK18 based circulating biomarkers of cell
death in lymphoma
Despite recent improvement in lymphomatreatment, chemoresistance and toxicity risk arehard to predict. In collaboration with Prof John
Group Leader
Caroline Dive andMalcolm Ranson
Disease Focus Team LeadersFiona BlackhallGuy MakinAndrew Renehan
Staff ScientistsJeff CummingsTim Ward
Associate ScientistKathryn Simpson
Postdoctoral Fellows Ivona Baricevic-JonesLuke HarrisonSarah Holt (AZ secondment)Jian Mei HouTetanya KlymenkoFlavia Moreira LeiteLee LancashireDavid MooreChristopher MorrowDarren Roberts
Clinical Research FellowsKyaw AungRuth BoardEmma DeanAlastair GreystokeSarah HughesMatthew KrebsLeila KhojaGireesh KumaranKalena Marti Marti

Clinical and Experimental Pharmacology Group | 29
toxicity for lymphoma patients on standardchemotherapy and have potential utility indevelopment of mechanism-based therapeutics.These biomarkers are being incorporated withinmulticentre phase III trials in order to progress tobiomarker qualification.
The Colorectal (CRC) Disease Focus Group
Our focus on CRC (lead: Dr Andrew Renehan)has developed in 2009 with respect tobiomarker-driven trials (and their statisticalconsiderations) and the influence of obesity onanti-cancer therapy response. Key landmarkswere commencement of the biomarker-linked E-SCOUT trial of cetuximab combination therapy(encompassing CTCs, detection of KRAS
mutations in circulating free DNA and circulatingbiomarkers of cell death); expansion of ourblood and tissue collection portfolio (to includeStepping Hill Hospital); appointment of a CR-UKAZ clinical fellow with specific interest in anti-angiogenic therapies and biomarkers in CRC;and establishment of the Manchester-BirminghamBiostatistical Biomarkers Collaboration (MBBBC)with Professors Cindy Billingham and PhilipJohnson. With the appointment of a furtherpostdoctoral scientist, studies commenced usingan in vitro model of K-Ras dependentchemoresistance to chronic insulin exposure andof CRC progression in obese mice.
Clinical proteomics
In collaboration with Prof Tony Whetton, theclinical proteomics team substantially improvedits capability to detect and relatively quantify, lowabundance proteins in human plasma followingupgrade of our MALDI-ToFToF massspectrometer. The new ABI 5800 instrumentenables 10x faster data acquisition withimproved resolution and sensitivity. Variousoperating methods involved in ion selection anddata processing were optimised for maximalprotein identification with minimal false discoveryrates. The throughput of MALDI plate spottingwas increased and processing of 8-channeliTRAQ samples of human plasma is now twiceas fast with 2-3 times the number of proteinidentifications. Performing at internationallycompetitive levels to detect and quantify ~400proteins in human plasma, we now detect
proteins in the low abundance range, a probablesource of tissue leakage molecules that includesdisease-specific biomarkers. Our first clinicalstudy assessing the intra-and inter-personreproducibility and experimental variation ofsamples from the PACER-TRANS clinical trial inpancreatic cancer is underway in collaborationwith Prof. Catherine West.
BH-3 Mimetics, Hypoxia and Drug
Combinations
The preclinical pharmacology team examineseffects of tumour micro-environment on drugsensitivity and evaluates drug combinations, witha particular emphasis on apoptosis-targeteddrugs. Hypoxia occurs in regions of solidtumours distant from blood supply where cellsexhibit reduced sensitivity to standardchemotherapy and radiotherapy. However,hypoxia sensitised CRC and SCLC cells toapoptosis induced by the Bcl-2 family targetedBH-3 mimetic ABT-737 via reduced expressionof Mcl-1, an established ABT-737 resistancefactor. Increased ABT-737-induced apoptosisoccurred in hypoxic regions of SCLC tumourxenografts. This suggests that, if deliveredsufficiently, ABT-737 could target hypoxic tumourcells that are chemo-refractive that in patientscause tumour repopulation during relapse. InCRC cancer cells, ABT-737 induced cell deathwas also enhanced when combined withinhibitors of PI3-kinase signalling. This was notrecapitulated by inhibition of PKB or mTOR thatact downstream of PI3-kinase. PI3-kinaseinhibition was associated with increased bindingof the pro-apoptotic BH-3 only protein BIM tothe ABT-737 target Bcl-xL, suggesting a possiblemechanism for this drug interaction.
Tissue Biomarkers
Our validated tissue biomarker assay portfolioincreased in 2009 to include Ca IX, GLUT-1 1and pimonidazole to report hypoxia. With tissuebiomarker studies increasing, automatedhistology has been incorporated into the GCLPlaboratory base. With ECMC funding, an Ariolimage analysis platform (Applied ImagingGenetix) has been installed and is beingcustomised for CEP GCLP activity allowing high-throughput and automated image analysis forbiomarker quantification. This system supports‘slide-link’ a software package linking serial tissuesections. CEP has scanned and analysed >800slides from a number of preclinical in vivo studiesThis approach allows analysis of co-localisedmultiple markers without the need for complexmultiple staining techniques and was used to thisapproach to quantify cell death biomarkers inboth normoxic and hypoxic regions of tumourxenografts treated with ABT-737.
Publications listed on page 67
Figure 1
The collective migration of SCLC
circulating tumour microemboli
(CTM) identified using ISET
technology. Dark circles are pores in
the filter, brown staining for CD45
identifies contaminant white blood
cells (WBC).
Scientific OfficersKaren BrookesDamien BrownFouziah ButtMartin DawsonOlive DennenyMaciej DolniakMartin GreavesGrace HampsonCassandra HodgkinsonMatthew LancashireDaniel MorrisKaren MorrisLyndsey PriestRobert SloaneNigel SmithHannah TurpinZaira Yunis
Graduate StudentsMartin BrandenburgCristina Martin-FernandezDimitra MichaElizabeth HitchmanShaun Villa
Scientific AdministratorAileen Jardine

30 | Paterson Institute for Cancer Research Scientific Report 2009
DNA Damage Response Grouphttp://www.paterson.man.ac.uk/dnadamage
The genetic material (DNA) is constantly exposed to damage,both from endogenous and exogenous sources, and the livingorganisms had to evolve a variety of DNA repair mechanismsto maintain the genome stability. Inadequate or abnormalDNA repair can cause diseases that are in man associated withcancer, neurodegeneration, immunodeficiency ordevelopmental abnormalities. Our laboratory is studyingmolecular pathways employed in mammalian DNA damageresponse in order to gain a better understanding of themechanisms underlying human disease, which could potentiallyprovide a basis for the development of new therapies.
DNA serves as a platform for specificrecruitment and scaffolding of DNA repaircomplexes. In addition, the damage-inducedpoly(ADP-ribosyl)ation has been known to havea role in relaxation of the chromatin structure, aswell as in apoptotic signalling. The recentdevelopment of potent PARP1/2 inhibitorsprovided powerful tools to study pathwaysregulated by poly(ADP-ribose), as well asproviding one of the very promising class ofdrugs for cancer treatment. Specifically, selectiveinhibition of the single-strand break repairpathway using permeable PARP inhibitors hasbeen proven highly effective against breast andovarian cancers (Bryant et al., Nature 2005; 434:913). Thus, understanding the molecular basis ofPAR-dependent DNA repair processes is likelyof vital importance for selecting and developingefficient therapies.
Identification and characterization of novel
poly(ADP-ribose)-regulated factors
Our laboratory is particularly interested inidentification of new pathways and proteinfunctions regulated by poly(ADP-ribosyl)ation.Recently, in screening for proteins with the abilityto bind poly(ADP-ribose), we discovered apoly(ADP-ribose)-binding zinc finger motif(PBZ). PBZ is a structurally distinctive, atypicaltype of zinc finger that is associated with severalproteins involved in response to DNA damage(Ahel et al., Nature, 2008; 451: 81) (Figure 1).One of the human proteins containing a PBZ
Group Leader
Ivan Ahel
Postdoctoral Fellows
Dragana AhelRolf KraehenbuehlMarianna Rossi
Scientific Officer
Ria Weston
Graduate Student
Pawan Mehrotra
Genomic instability is the driving force of cancerdevelopment, which requires multiple DNAmutations resulting in loss of cellular growthcontrol. To accelerate the accumulation ofgenetic changes, cancers often sacrifice specificDNA repair pathways, which in turn can beexploited as an Achilles heel of cancer. Thismeans that the genetic damage that builds up incancer cells makes them susceptible totreatment with certain DNA-damaging agents, orto inhibitors that block alternative DNA repairpathways, while normal cells with a full repairrepertoire are readily much less sensitive. Onespecific class of anticancer drugs that work in thisway are inhibitors of family of enzymes calledpoly(ADP-ribose) polymerases (PARPs). PARPsuse cellular NAD as a substrate to synthesize apeculiar type of posttranslational proteinmodification consisting of long chains ofrepeating ADP-ribose units (poly(ADP-ribose) orPAR) and regulate a variety of importantprocesses including DNA repair, chromatinstructure, mitosis, transcription, checkpoint andapoptosis (Hakme et al., EMBO rep 2009; 9:1094).
Poly(ADP-ribosyl)ation in regulation of
DNA repair
The role of poly(ADP-ribosyl)ation is bestunderstood in the regulation of DNA repair,which is controlled by the two PARPs responsiveto DNA strand breaks (PARP1 and PARP2).Poly(ADP-ribose) arising at the sites of damaged
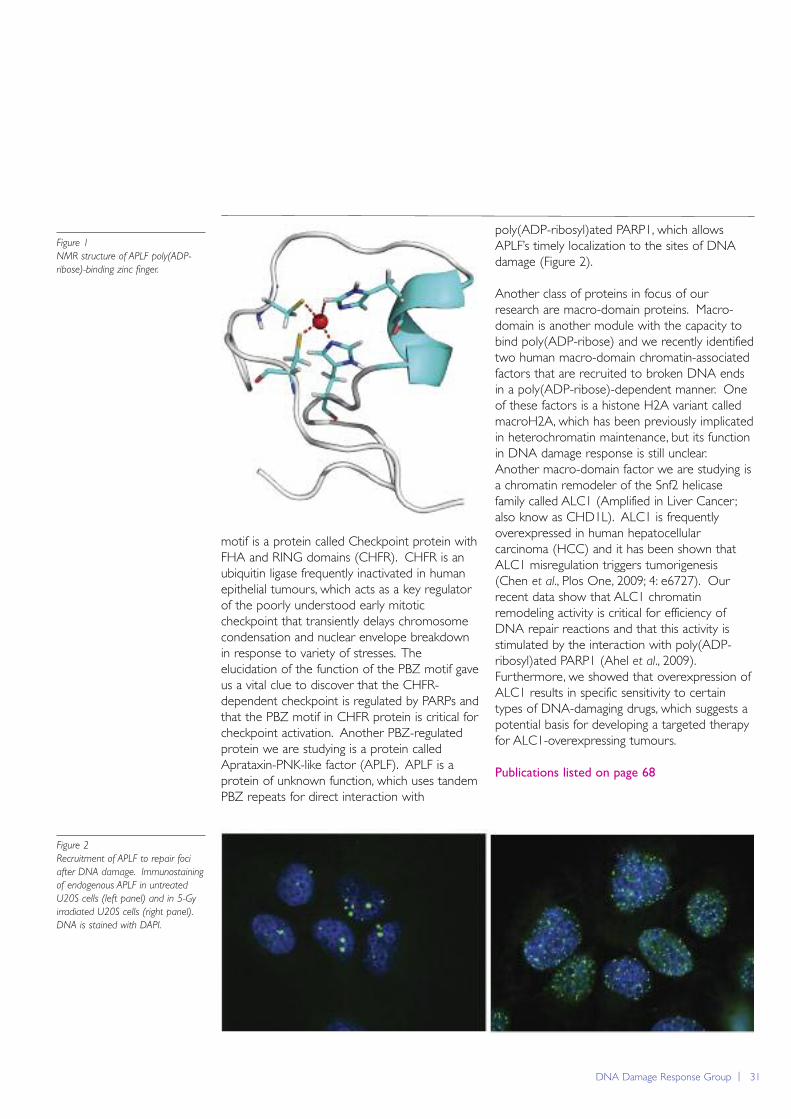
DNA Damage Response Group | 31
motif is a protein called Checkpoint protein withFHA and RING domains (CHFR). CHFR is anubiquitin ligase frequently inactivated in humanepithelial tumours, which acts as a key regulatorof the poorly understood early mitoticcheckpoint that transiently delays chromosomecondensation and nuclear envelope breakdownin response to variety of stresses. Theelucidation of the function of the PBZ motif gaveus a vital clue to discover that the CHFR-dependent checkpoint is regulated by PARPs andthat the PBZ motif in CHFR protein is critical forcheckpoint activation. Another PBZ-regulatedprotein we are studying is a protein calledAprataxin-PNK-like factor (APLF). APLF is aprotein of unknown function, which uses tandemPBZ repeats for direct interaction with
poly(ADP-ribosyl)ated PARP1, which allowsAPLF’s timely localization to the sites of DNAdamage (Figure 2).
Another class of proteins in focus of ourresearch are macro-domain proteins. Macro-domain is another module with the capacity tobind poly(ADP-ribose) and we recently identifiedtwo human macro-domain chromatin-associatedfactors that are recruited to broken DNA endsin a poly(ADP-ribose)-dependent manner. Oneof these factors is a histone H2A variant calledmacroH2A, which has been previously implicatedin heterochromatin maintenance, but its functionin DNA damage response is still unclear.Another macro-domain factor we are studying isa chromatin remodeler of the Snf2 helicasefamily called ALC1 (Amplified in Liver Cancer;also know as CHD1L). ALC1 is frequentlyoverexpressed in human hepatocellularcarcinoma (HCC) and it has been shown thatALC1 misregulation triggers tumorigenesis(Chen et al., Plos One, 2009; 4: e6727). Ourrecent data show that ALC1 chromatinremodeling activity is critical for efficiency ofDNA repair reactions and that this activity isstimulated by the interaction with poly(ADP-ribosyl)ated PARP1 (Ahel et al., 2009).Furthermore, we showed that overexpression ofALC1 results in specific sensitivity to certaintypes of DNA-damaging drugs, which suggests apotential basis for developing a targeted therapyfor ALC1-overexpressing tumours.
Publications listed on page 68
Figure 2
Recruitment of APLF to repair foci
after DNA damage. Immunostaining
of endogenous APLF in untreated
U20S cells (left panel) and in 5-Gy
irradiated U20S cells (right panel).
DNA is stained with DAPI.
Figure 1
NMR structure of APLF poly(ADP-
ribose)-binding zinc finger.

32 | Paterson Institute for Cancer Research Scientific Report 2009
Immunology Grouphttp://www.paterson.man.ac.uk/immunology
The basic research of the Immunology Group is directed atunderstanding and exploiting the function and/or expression of5T4 oncofoetal molecules in the context of cancer associatedchanges in motility and adhesion contributing to metastasis.We also have significant collaborations within the MCRCfocused on more translational objectives for 5T4-directedimmunotherapies with the Cellular Therapy Group (REHawkins), Gynaecological Oncology (HC Kitchener, St Mary’sHospital) and the Children’s Cancer Group (V Saha). Some ofour studies relating to successful translation of 5T4 directedimmunotherapies are reported elsewhere (BIGT). This reportwill focus on our work utilizing our 5T4 KO mouse and theinvestigation of modulation of immune regulatory processes in5T4 tumour immunity.
influence on motility in embryonic and cancer cells.
5T4 immunotherapy
We have utilized the 5T4 KO mice to generateseveral monoclonal antibodies specific for m5T4(B3F1 (IgG2a); B5C9 (IgG1); B1C3 (IgG2a); P1C9(IgG2b) and P1H10 (IgG2b), mapped the locationof the different epitopes recognized within them5T4 glycoprotein and measured their affinities.One aim is to assess the functional and possiblytherapeutic impact of treatment with theseantibodies in different autologous tumourtherapy models (e.g. m5T4B16, CT26). Thefocus on antibodies has been prompted byresults from several clinical studies of the 5T4-MVA vaccine (TroVax) which have associatedprolonged survival with an antibody response to5T4, and an immunotherapy based on targetingof a modified bacterial superantigen withantibodies against 5T4 with efficacy in renalcarcinoma patients (Elkord et al., 2009b). Modelsystems have also been established investigatingchimaeric immune receptor (CIR) T-cells directedto human 5T4 in combination with vaccination(Jiang et al., J Immunol 2006; 177: 4288). We arenow exploring aspects of T-cell and antibodytherapy in our C57BL/6 m5T4 tumour modelsystem utilizing the 5T4 null mouse to generate
Group Leader
Peter L. Stern
Postdoctoral Fellows
Fernanda Castro Owen McGinn Tom SouthgateAurelia Gallego (July-September)
Clinical Research Fellow
Christy Ralph (with RobertHawkins)
Scientific Officers
Kate MulryanDebbie Burt (seconded toCIMML)
Graduate Students
Mariam Al-Muftah (with DavidGilham)Andrzej Rutkowski (with CrispinMiller)Georgi Marinov
Undergraduate student
Stefanie Frick (October toDecember)
5T4 knock out (KO) mouse
We have constructed a 5T4 KO mouse byreplacing the second exon of 5T4, whichencodes the entire protein, with an IRES-LacZneo reporter gene in ES cells. These cellswere used to produce chimaeric mice andgermline progeny; 5T4 KO heterozygote micewere backcrossed to the C57BL/6 background(> 10 generations). KO 5T4 C57BL/6 animalsare viable but adult animals show somestructural disorganization within the brain andexhibit a high frequency of hydrocephalus (13%,at median age 49 days). Preliminary studiessuggest that lack of 5T4 expression in vivo caninfluence the rate of wound healing and thatpolyp formation by null 5T4-MIN mice issignificantly reduced. Previous studies haveshown a role for 5T4 influencing theorganization of the actin cytoskeleton relating toaltered motility relevant in development andcancer. We have now used primary KO 5T4mouse embryonic fibroblasts (MEF) from day 13embryos as a tool for further investigation of5T4 function. For example, Figure 1 shows thatin 5T4KO MEFs, the organization of the actincytoskeleton is disrupted compared to the WT-MEF. Current studies are focused on themolecular mechanisms underpinning 5T4
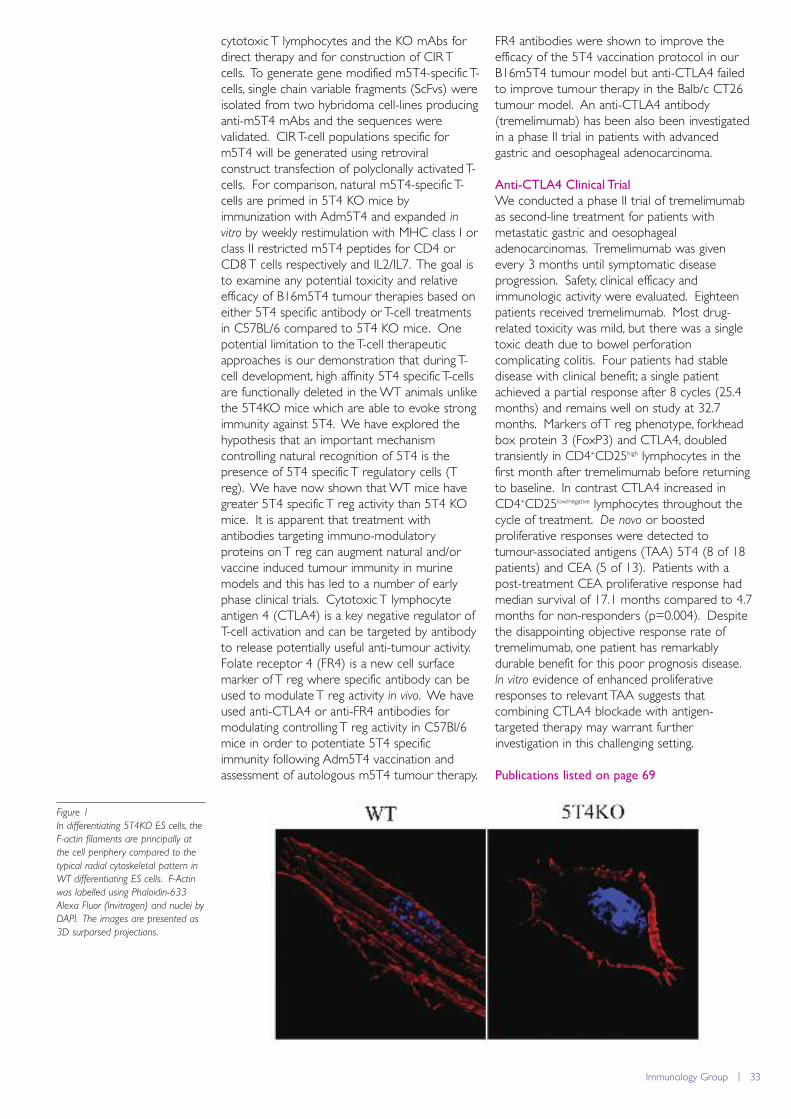
Immunology Group | 33
cytotoxic T lymphocytes and the KO mAbs fordirect therapy and for construction of CIR Tcells. To generate gene modified m5T4-specific T-cells, single chain variable fragments (ScFvs) wereisolated from two hybridoma cell-lines producinganti-m5T4 mAbs and the sequences werevalidated. CIR T-cell populations specific form5T4 will be generated using retroviralconstruct transfection of polyclonally activated T-cells. For comparison, natural m5T4-specific T-cells are primed in 5T4 KO mice byimmunization with Adm5T4 and expanded invitro by weekly restimulation with MHC class I orclass II restricted m5T4 peptides for CD4 orCD8 T cells respectively and IL2/IL7. The goal isto examine any potential toxicity and relativeefficacy of B16m5T4 tumour therapies based oneither 5T4 specific antibody or T-cell treatmentsin C57BL/6 compared to 5T4 KO mice. Onepotential limitation to the T-cell therapeuticapproaches is our demonstration that during T-cell development, high affinity 5T4 specific T-cellsare functionally deleted in the WT animals unlikethe 5T4KO mice which are able to evoke strongimmunity against 5T4. We have explored thehypothesis that an important mechanismcontrolling natural recognition of 5T4 is thepresence of 5T4 specific T regulatory cells (Treg). We have now shown that WT mice havegreater 5T4 specific T reg activity than 5T4 KOmice. It is apparent that treatment withantibodies targeting immuno-modulatoryproteins on T reg can augment natural and/orvaccine induced tumour immunity in murinemodels and this has led to a number of earlyphase clinical trials. Cytotoxic T lymphocyteantigen 4 (CTLA4) is a key negative regulator ofT-cell activation and can be targeted by antibodyto release potentially useful anti-tumour activity.Folate receptor 4 (FR4) is a new cell surfacemarker of T reg where specific antibody can beused to modulate T reg activity in vivo. We haveused anti-CTLA4 or anti-FR4 antibodies formodulating controlling T reg activity in C57Bl/6mice in order to potentiate 5T4 specificimmunity following Adm5T4 vaccination andassessment of autologous m5T4 tumour therapy.
FR4 antibodies were shown to improve theefficacy of the 5T4 vaccination protocol in ourB16m5T4 tumour model but anti-CTLA4 failedto improve tumour therapy in the Balb/c CT26tumour model. An anti-CTLA4 antibody(tremelimumab) has been also been investigatedin a phase II trial in patients with advancedgastric and oesophageal adenocarcinoma.
Anti-CTLA4 Clinical Trial
We conducted a phase II trial of tremelimumabas second-line treatment for patients withmetastatic gastric and oesophagealadenocarcinomas. Tremelimumab was givenevery 3 months until symptomatic diseaseprogression. Safety, clinical efficacy andimmunologic activity were evaluated. Eighteenpatients received tremelimumab. Most drug-related toxicity was mild, but there was a singletoxic death due to bowel perforationcomplicating colitis. Four patients had stabledisease with clinical benefit; a single patientachieved a partial response after 8 cycles (25.4months) and remains well on study at 32.7months. Markers of T reg phenotype, forkheadbox protein 3 (FoxP3) and CTLA4, doubledtransiently in CD4+CD25high lymphocytes in thefirst month after tremelimumab before returningto baseline. In contrast CTLA4 increased inCD4+CD25low/negative lymphocytes throughout thecycle of treatment. De novo or boostedproliferative responses were detected totumour-associated antigens (TAA) 5T4 (8 of 18patients) and CEA (5 of 13). Patients with apost-treatment CEA proliferative response hadmedian survival of 17.1 months compared to 4.7months for non-responders (p=0.004). Despitethe disappointing objective response rate oftremelimumab, one patient has remarkablydurable benefit for this poor prognosis disease.In vitro evidence of enhanced proliferativeresponses to relevant TAA suggests thatcombining CTLA4 blockade with antigen-targeted therapy may warrant furtherinvestigation in this challenging setting.
Publications listed on page 69
Figure 1
In differentiating 5T4KO ES cells, the
F-actin filaments are principally at
the cell periphery compared to the
typical radial cytoskeletal pattern in
WT differentiating ES cells. F-Actin
was labelled using Phaloidin-633
Alexa Fluor (Invitrogen) and nuclei by
DAPI. The images are presented as
3D surparsed projections.

34 | Paterson Institute for Cancer Research Scientific Report 2009
Inositide Laboratoryhttp://www.paterson.man.ac.uk/inositide
Phosphoinositides are a family of lipid second messengers thatare regulated by a network of kinases and phosphatases inresponse to environmental changes. Alterations inphosphoinositide levels can regulate many different cancer-relevant pathways including survival, proliferation, migration, cellsubstratum interactions and transcription. PtdIns(4,5)P2 is atthe heart of phosphoinositide signalling in cancer as it is thesubstrate for phosphatidylinositol-3-kinase (PI3K) andphospholipase C (PIC) both of which are deregulated inhuman tumours. Furthermore PtdIns(4,5)P2 is itself aregulator of cytoskeletal dynamics, cell survival and cell polarity.
cellular stressors. PIP5Kβ is modified byphosphorylation at multiple sites and one ofthese sites is phosphorylated in response to PKCactivation, although it is not clear if this is direct.PKC is stimulated in response to PLC signalling,suggesting that phosphorylation of PIP5K mayregulate the its activity or localisationdownstream of phospholipase signalling. Tounderstand how PIP5K isoforms regulate specificsignalling pathways we are identifying proteinsthat interact with them using affinity purificationand mass spectrometry and yeast two hybridanalysis. To define how PtdIns(4,5)P2 regulatescellular function we have identified nuclear andcytosolic proteins, that interact specifically withthis lipid (collaboration with Dr. C. D’Santos CRICambridge) and will study how their interactionwith PtdIns(4,5)P2 is able to modulate theirfunction in vivo.
PIP4K and PtdIns5P
There are three isoforms of PIP4Ks (α, β and γ)of which α is cytosolic, β is cytosolic and nuclearand γ localises to internal membranecompartments. PIP4Ks can regulate the cellularlevels of PtdIns5P however in vitro studiesmeasuring their activity showed thatα>>>β>>>>γ. Analysis of the molecular sizeof PIP4Kβ showed that it exists as a dimer.PIP4Kβ forms homodimers and heterodimerswith the α isoform and heterodimerisation cantarget the α isoform to the nucleus.Furthermore PIP4Kα is required for functions
Group Leader
Nullin Divecha
Associate Scientist
David Jones
Postdoctoral Fellows
Daniel Fitzgerald (fromNovember)Maria Carla MottaIman van den Bout
Scientific Officer
Yvette Bultsma
Graduate Students
Julian Blaser (Joint with TimSomervaille)Xiaowen Hu Willem-Jan KeuneLilly Sommer
PIP5Ks and PtdIns(4,5)P2PtdIns(4,5)P2 is present in the plasma membraneand in the nucleus where each pool can beregulated separately. PtdIns(4,5)P2 can besynthesised by two different families of kinasesusing two different substrates (see figure 1). It islikely that PIP5Ks are the major regulators ofPtdIns(4,5)P2 synthesis while the PIP4Ks regulatePtdIns5P levels and perhaps a minor pool ofPtdIns(4,5)P2.
There are four genes coding for PIP5K (α, β andγ and L) of which α, β and γ are active while L isinactive but can interact with and may regulatethe localisation and activity of other PIP5Kisoforms. We are using RNAi mediatedsuppression of PIP5Ks to define isoform specificregulation of PtdIns(4,5)P2 pools and whetherPIP5K isoforms are linked to specific downstreampathways. For example suppression of α or βexpression in N1E-115 neuroblastoma cells leadsto their differentiation and a cessation inproliferation, while the suppression of γ doesnot. Furthermore suppression of α reducedPtdIns(4,5)P2 levels by approximately 50% whilesuppression of β and γ had little effect on cellularPtdIns(4,5)P2 levels. How a cell maintainsdifferent pools of PtdIns(4,5)P2 and how thesepools signal differentially is not clear. PIP5Kβ isupregulated in a variety of cancer cell lines andoverexpression of PIP5Kβ can induce dramaticchanges in cell morphology, increase migratorycapacity and attenuate apoptosis in response to
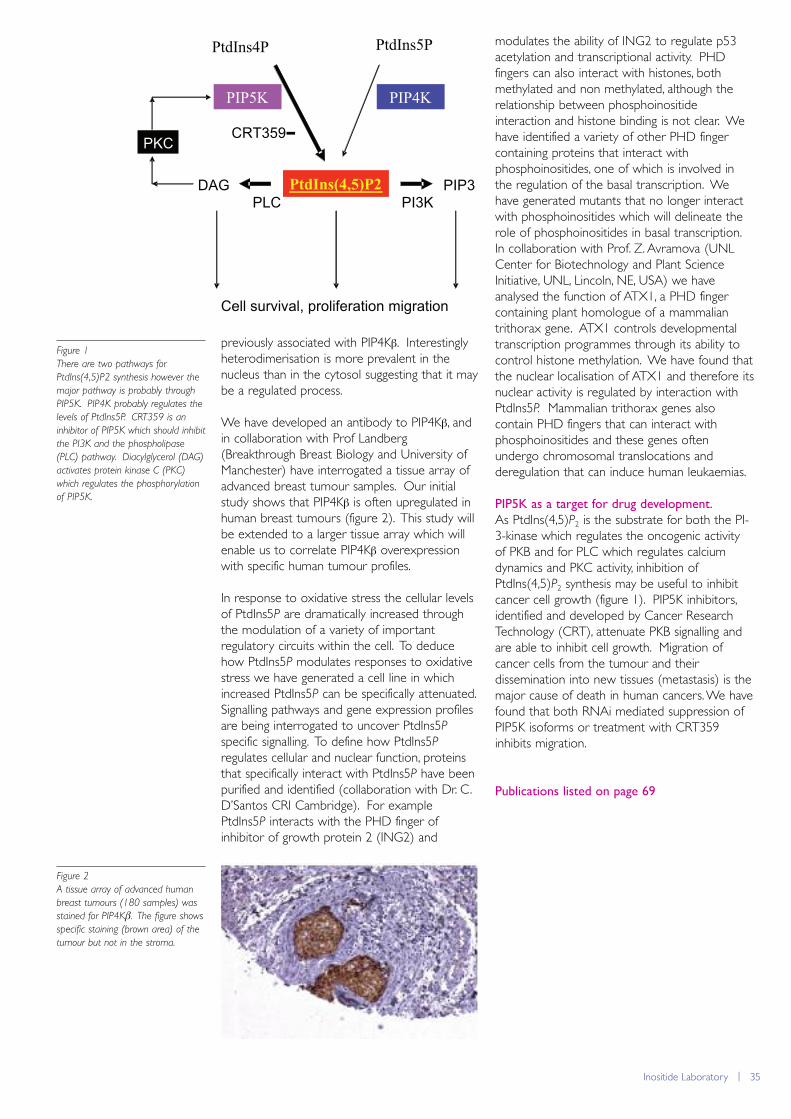
previously associated with PIP4Kβ. Interestinglyheterodimerisation is more prevalent in thenucleus than in the cytosol suggesting that it maybe a regulated process.
We have developed an antibody to PIP4Kβ, andin collaboration with Prof Landberg(Breakthrough Breast Biology and University ofManchester) have interrogated a tissue array ofadvanced breast tumour samples. Our initialstudy shows that PIP4Kβ is often upregulated inhuman breast tumours (figure 2). This study willbe extended to a larger tissue array which willenable us to correlate PIP4Kβ overexpressionwith specific human tumour profiles.
In response to oxidative stress the cellular levelsof PtdIns5P are dramatically increased throughthe modulation of a variety of importantregulatory circuits within the cell. To deducehow PtdIns5P modulates responses to oxidativestress we have generated a cell line in whichincreased PtdIns5P can be specifically attenuated.Signalling pathways and gene expression profilesare being interrogated to uncover PtdIns5P
specific signalling. To define how PtdIns5P
regulates cellular and nuclear function, proteinsthat specifically interact with PtdIns5P have beenpurified and identified (collaboration with Dr. C.D’Santos CRI Cambridge). For examplePtdIns5P interacts with the PHD finger ofinhibitor of growth protein 2 (ING2) and
modulates the ability of ING2 to regulate p53acetylation and transcriptional activity. PHDfingers can also interact with histones, bothmethylated and non methylated, although therelationship between phosphoinositideinteraction and histone binding is not clear. Wehave identified a variety of other PHD fingercontaining proteins that interact withphosphoinositides, one of which is involved inthe regulation of the basal transcription. Wehave generated mutants that no longer interactwith phosphoinositides which will delineate therole of phosphoinositides in basal transcription.In collaboration with Prof. Z. Avramova (UNLCenter for Biotechnology and Plant ScienceInitiative, UNL, Lincoln, NE, USA) we haveanalysed the function of ATX1, a PHD fingercontaining plant homologue of a mammaliantrithorax gene. ATX1 controls developmentaltranscription programmes through its ability tocontrol histone methylation. We have found thatthe nuclear localisation of ATX1 and therefore itsnuclear activity is regulated by interaction withPtdIns5P. Mammalian trithorax genes alsocontain PHD fingers that can interact withphosphoinositides and these genes oftenundergo chromosomal translocations andderegulation that can induce human leukaemias.
PIP5K as a target for drug development.
As PtdIns(4,5)P2 is the substrate for both the PI-3-kinase which regulates the oncogenic activityof PKB and for PLC which regulates calciumdynamics and PKC activity, inhibition ofPtdIns(4,5)P2 synthesis may be useful to inhibitcancer cell growth (figure 1). PIP5K inhibitors,identified and developed by Cancer ResearchTechnology (CRT), attenuate PKB signalling andare able to inhibit cell growth. Migration ofcancer cells from the tumour and theirdissemination into new tissues (metastasis) is themajor cause of death in human cancers. We havefound that both RNAi mediated suppression ofPIP5K isoforms or treatment with CRT359inhibits migration.
Publications listed on page 69
PtdIns(4,5)P2
PtdIns4P
PIP5K
PtdIns5P
PIP4K
DAGPLC PI3K
PIP3
PKCCRT359-
Cell survival, proliferation migration
Figure 1
There are two pathways for
PtdIns(4,5)P2 synthesis however the
major pathway is probably through
PIP5K. PIP4K probably regulates the
levels of PtdIns5P. CRT359 is an
inhibitor of PIP5K which should inhibit
the PI3K and the phospholipase
(PLC) pathway. Diacylglycerol (DAG)
activates protein kinase C (PKC)
which regulates the phosphorylation
of PIP5K.
Inositide Laboratory | 35
Figure 2
A tissue array of advanced human
breast tumours (180 samples) was
stained for PIP4Kβ. The figure shows
specific staining (brown area) of the
tumour but not in the stroma.

36 | Paterson Institute for Cancer Research Scientific Report 2009
Leukaemia Biology Grouphttp://www.paterson.man.ac.uk/leukaemia
The development of malignant disease is thought to requirethe accumulation of multiple collaborating genetic mutations ina long lived cell. In leukaemogenesis, one hypothesis is thatmutations accumulate over time in the self-renewinghaematopoietic stem cell (HSC) compartment, perhapsconferring a selective proliferative or survival advantage in themutated HSCs by comparison with unmutated ones. Thiseventually results in full-fledged leukaemia stem cells (LSCs)that share many of the properties of their normal HSCcounterparts. A complementary hypothesis is thatleukaemogenic mutations confer the capacity to undergo self-renewal on progenitor cells downstream of the HSC, whichunder normal circumstances are irreversibly committed toterminal differentiation. Aberrantly self-renewing downstreamprogenitor cells, which may proliferate more frequently thatthe relatively quiescent HSC pool, represent an importantpotential reservoir for the acquisition and fixation of furthercooperating mutations required for development of fullpotency LSCs.
block is incomplete. Interestingly terminaldifferentiation of MLL transformed cells takesplace in the presence of continued high levelexpression of Hoxa/Meis1 suggesting that, whileinappropriate expression of these genes may besufficient to establish the leukaemia cell hierarchy,alternative mechanisms may regulate exit ofLSCs from the self-renewing compartment. It isinteresting to speculate that the cellular effect ofHoxa/Meis1 expression in MLL leukaemias maybe to introduce at the level of the GMP ormyeloblast a finite probability of a self-renewaldivision where none previously existed, althoughthe molecular circuitry of this phenomenonremains unclear. The accumulation of aninappropriately self-renewing, proliferatingpopulation of pre-LSC downstream of the HSCwould provide a long-lived target population ofcells receptive to secondary mutations in, forexample, Ras or Flt3 which are often mutated inhuman MLL leukaemias, or for accretion of
Group Leader
Tim Somervaille
Postdoctoral Fellows
Xu Huang James Lynch
Scientific Officer
Gary Spencer
Clinical Research Fellow
Brigit Greystoke
Graduate Students
William Harris Filippo Ciceri Julian Blaser (co-supervised withNullin Divecha)
In work published in Cell Stem Cell earlier thisyear, members of the Leukaemia Biology Group(Tim Somervaille and Gary Spencer) reportedfindings pertinent to this question. A retroviraltransduction and transplantation model thatfaithfully recapitulates many of the pathologicalfeatures of human acute myeloid leukaemias(AMLs) associated with chromosomaltranslocations of the MLL gene was used in thestudy. The MLL gene is mutated bychromosomal translocation in approximately 5-10% of human acute leukaemias. The study leadto the conclusion that MLL fusion oncogenesconvert a normal haematopoietic hierarchy intoa leukaemia cell hierarchy, sustained at its apexby a population of LSCs most similar in natureto inappropriately self-renewing downstreammyeloid cells (Figure 1). Critically, MLL LSCsretain the capacity to undergo spontaneousdifferentiation in vivo to terminally differentiatedneutrophils, emphasising that the differentiation
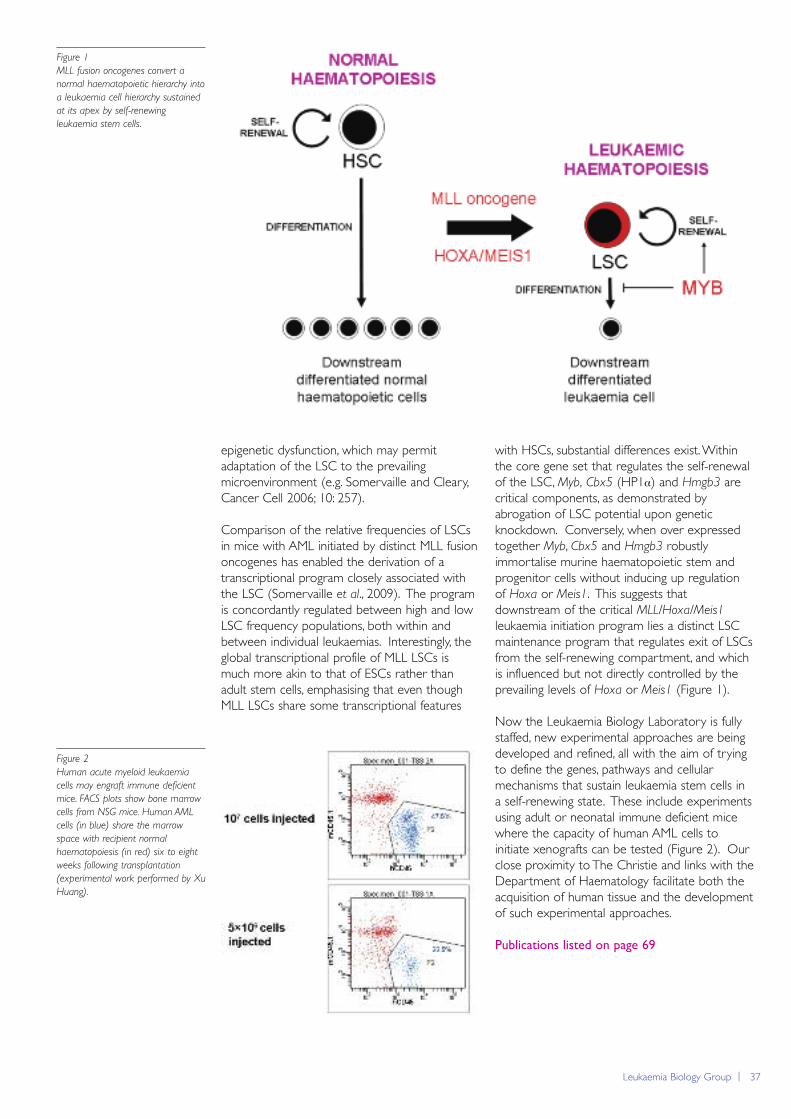
Leukaemia Biology Group | 37
epigenetic dysfunction, which may permitadaptation of the LSC to the prevailingmicroenvironment (e.g. Somervaille and Cleary,Cancer Cell 2006; 10: 257).
Comparison of the relative frequencies of LSCsin mice with AML initiated by distinct MLL fusiononcogenes has enabled the derivation of atranscriptional program closely associated withthe LSC (Somervaille et al., 2009). The programis concordantly regulated between high and lowLSC frequency populations, both within andbetween individual leukaemias. Interestingly, theglobal transcriptional profile of MLL LSCs ismuch more akin to that of ESCs rather thanadult stem cells, emphasising that even thoughMLL LSCs share some transcriptional features
with HSCs, substantial differences exist. Withinthe core gene set that regulates the self-renewalof the LSC, Myb, Cbx5 (HP1α) and Hmgb3 arecritical components, as demonstrated byabrogation of LSC potential upon geneticknockdown. Conversely, when over expressedtogether Myb, Cbx5 and Hmgb3 robustlyimmortalise murine haematopoietic stem andprogenitor cells without inducing up regulationof Hoxa or Meis1. This suggests thatdownstream of the critical MLL/Hoxa/Meis1
leukaemia initiation program lies a distinct LSCmaintenance program that regulates exit of LSCsfrom the self-renewing compartment, and whichis influenced but not directly controlled by theprevailing levels of Hoxa or Meis1 (Figure 1).
Now the Leukaemia Biology Laboratory is fullystaffed, new experimental approaches are beingdeveloped and refined, all with the aim of tryingto define the genes, pathways and cellularmechanisms that sustain leukaemia stem cells ina self-renewing state. These include experimentsusing adult or neonatal immune deficient micewhere the capacity of human AML cells toinitiate xenografts can be tested (Figure 2). Ourclose proximity to The Christie and links with theDepartment of Haematology facilitate both theacquisition of human tissue and the developmentof such experimental approaches.
Publications listed on page 69
Figure 1
MLL fusion oncogenes convert a
normal haematopoietic hierarchy into
a leukaemia cell hierarchy sustained
at its apex by self-renewing
leukaemia stem cells.
Figure 2
Human acute myeloid leukaemia
cells may engraft immune deficient
mice. FACS plots show bone marrow
cells from NSG mice. Human AML
cells (in blue) share the marrow
space with recipient normal
haematopoiesis (in red) six to eight
weeks following transplantation
(experimental work performed by Xu
Huang).

38 | Paterson Institute for Cancer Research Scientific Report 2009
Stem Cell Biology Grouphttp://www.paterson.man.ac.uk/stemcellbiology
The major interest of our lab is to decipher the cellular andmolecular mechanisms controlling the development andmaintenance of the haematopoietic system. In this context, westudy the functions of the transcription factor AML1/RUNX1and the transcriptional co-activator MOZ. AML1/RUNX1 isone of the most frequent targets of gene rearrangements andmutations in acute leukaemia. Similarly the gene MOZ isinvolved in myeloid chromosomal translocations.Understanding the function of these transcription regulatorsduring normal haematopoiesis should result in a bettercomprehension of how perturbations of their functions leadthe development of leukaemia.
generates haematopoietic cells through theformation of a haemogenic endotheliumintermediate, providing the first direct linkbetween these two precursor populations. Thishaemogenic endothelial cell population istransiently generated during blast developmentand is also detected in gastrulating embryos. Atthe molecular level, we have demonstrated thatthe transcription factor SCL/TAL1 isindispensable for the establishment of thishaemogenic endothelium cell population fromthe haemangioblast whereas RUNX1/AML1 iscritical for the generation of haematopoietic cellsfrom this haemogenic endothelium. Theseresults indicate that the two a priori conflictingtheories on the origin of haematopoieticdevelopment, haemangioblast and haemogenicendothelium, can be merged into a single linear developmental process leading to the formation of the first committed haematopoietic precursors.
Transcriptional targets of
Runx1/AML1:AI467606
Our initial studies revealed a profound defect inthe potential of the Runx1-/- ES cells to generateblast colonies and therefore RUNX1 is likely toregulate the expression of an important set ofgenes at this stage of development. To identifythese genes, we compared gene expression inhaemangioblast-derived cell populations
Group Leader
Georges Lacaud
Postdoctoral Fellows
Cristina FerrerasChristophe LancrinMichael Lie-A-LingFlor Perez-Campo
Scientific Officer
Ting Zheng
Graduate Students
Patrycja SroczynskaOlga TsoulakiMonika AntkiewiczMilena Mazan
Generation of blood cells
The earliest site of blood cell development inthe mouse embryo is the yolk sac where bloodislands, consisting of haematopoietic cellssurrounded by a layer of angioblasts, develop atapproximately day 7.5 of gestation. The paralleldevelopment of these two lineages in closeassociation provided the basis for the hypothesisthat they arise from a common precursor, a cellcalled the haemangioblast. A conflicting theoryhowever associates the first haematopoietic cellsto a phenotypically differentiated endothelial cellwith haematopoietic potential, i.e. a haemogenicendothelium. Support for the haemangioblastconcept was initially provided by theidentification during embryonic stem (ES) cellsdifferentiation of a clonal precursor, the blastcolony-forming cell (BL-CFC), which gives riseafter 4 days to blast colonies with bothendothelial and haematopoietic potential.Although recent studies have now providedevidence for the presence of this bipotentialprecursor in vivo, the precise mechanism ofgeneration of haematopoietic cells from the haemangioblast still remains completely unknown.
We performed a series of studies to determinethe cellular and molecular events leading to thegeneration of blast colony from BL-CFC. Ourdata demonstrate that the haemangioblast

Stem Cell Biology Group | 39
generated from either Runx1 deficient or Runx1
competent ES cells. Such comparisonshighlighted a candidate, AI467606 (AI), apreviously uncharacterized gene, for furtherstudies. This novel gene encodes a protein of225 amino acids with a putative trans-membranedomain. We first demonstrated by chromatinimmuno-precipitation the binding of Runx1 tothe promoter of AI during blast colonydevelopment and by luciferase reporter assay,the regulation of AI transcription by Runx1.Furthermore we observed that re-expression ofRunx1 in Runx1-/- cells led to a rapid upregulationof AI expression. Altogether these resultssuggest that AI is a direct transcriptional target ofRunx1. To track the cells expressing AI, wegenerated a transgenic ES cell line with amodified bacterial artificial chromosome (BAC)containing a GFP reporter gene knocked-in theAI locus. We demonstrated in vivo and in vitro
the emergence of AI expressing cells from theCD41+ cell population. In foetal and adult mice,AI was specifically and highly expressed inhaematopoietic organs as indicated by immuno-histochemistry and flow cytometry analysis.Furthermore, the AI+ cell population containedall haematopoietic potential. Altogether theseresults suggest that AI expression markshaematopoietic cells throughout development,from their emergence in the yolk sac, throughfoetal liver and finally to adult haematopoieticorgans. We are currently establishing knock-outES cells and conditional knock-out mouse line toevaluate the potential function of this geneduring haematopoiesis.
Transcriptional activities of Runx1 promoters
In vertebrates, the transcription of the Runx1
gene is under the control of two alternativepromoters, distal P1 and proximal P2, which
generate specific Runx1 transcripts. Weinvestigated the activities of distal and proximalRunx1 promoters at the single cell level andtracked the cell populations expressing the tworespective isoforms. We demonstrated that atthe onset of haematopoiesis both in vitro and invivo the activity of the proximal promoter marksa haemogenic endothelium cell population,whereas the subsequent activation of the distalRunx1 promoter defines fully committeddefinitive haematopoietic progenitors.Interestingly, haematopoietic commitment indistal Runx1 knock-out embryos appears normal,suggesting that the proximal isoform plays acritical role in the generation of haematopoieticcells from haemogenic endothelium. Altogether,our data demonstrate that the differentialactivities of the two Runx1 promoters definemilestones of haematopoietic development.Identification and access to the discrete stages of haematopoietic development defined by theactivities of the Runx1 promoters will providethe opportunity to further explore the cellular and molecular mechanisms ofblood development.
Chromatin remodelling and Runx1 at the onset
of haematopoietic development
We have examined in collaboration with thegroup of Prof. Bonifer (Institute for MolecularMedicine, Leeds University) the molecular eventsmediated by Runx1 leading to the transcriptionalactivation of two master myeloid genes Pu.1 andCsf1r. We demonstrated that although Runx1
binds to both loci, it is sufficient to initiatechromatin remodelling events at the Pu.1 locusbut not at the Csf1r locus. We also found thatRunx1 directly interacts with its target sequencesin haemangioblasts, but that this interaction isinitially transient (“hit and run” fashion).Accordingly, a single pulse of Runx1 expression indeveloping Runx1-/- blast colony is sufficient torescue the expression of these genes and thedevelopment of definitive haematopoieticprecursors. These results suggest that Runx1 iscrucial for the initial chromatin remodelingevents at loci encoding transcription factors thatare regulators of specific blood cell lineages.Our data suggest that once these transcriptionfactors are expressed, stable transcription factorcomplexes are formed on these genes, activechromatin is maintained and an epigeneticmemory for active gene expression isestablished. Once this has occurred, Runx1
becomes less important and is used only incertain genomic contexts as one of many other transcription factors. This could explainwhy Runx1 is not generally essential for adult haematopoiesis.
Publications listed on page 69
Figure 1
Model of sequential activation of
Runx1 promoters.
Runx1 independent
Haemangioblast HaemogenicEndothelium Ⅰ
Flk1⁺ Proximal⁺Tie2⁺cKit⁺CD41⁻Distal⁻
Runx1 dependent
HaemogenicEndothelium Ⅱ
DefinitiveHaematopoietic
Progenitors
Proximal⁺Tie2⁺cKit⁺
CD41⁺Distal⁻
Proximal⁺Tie2low
cKit⁺CD41⁺Distal⁺
Figure 2
Binding of Runx1 to the promoter of
AI467606 detected by ChIP-seq.

40 | Paterson Institute for Cancer Research Scientific Report 2009
Stem Cell and Haematopoiesis Grouphttp://www.paterson.man.ac.uk/sch
Understanding the molecular mechanisms that control theformation of blood precursors from the mesodermal germlayer is the major focus of our laboratory. Several lines ofevidence suggest that leukemogenesis may result from the re-initiation of an embryonic programme during adult life orfrom the inappropriate expression of genes controlling criticalsteps of the haematopoietic embryonic programme. A clearunderstanding of the molecular mechanisms orchestrating theonset of haematopoietic specification should help us to betterdefine the basis of de-regulated proliferation and differentiationobserved in haematological malignancies.
narrow window of development betweenhaemangioblast specification and haematopoieticcommitment.
We also undertook classical loss- and gain-of-function approaches to define a possible role forSox7 in haematopoiesis. As a mean to achieveloss of function, we made use of the shRNAtechnology to knockdown Sox7 expression invitro. Knocking down Sox7 expression during thespecification of mesodermal precursors tohaematopoiesis significantly decreased theformation of haematopoietic progenitors as wellas endothelial progenitors. Sox7 knockdown ledto a substantial increase in precursor cell death,pointing toward a possible role for Sox7 in cellsurvival during haematopoiesis specification.Sox7 expression is naturally down regulated ashaematopoietic differentiation progresses to theformation mature blood lineages. It seemedtherefore of interest to address the biologicalrelevance of this down-regulation forhaematopoietic maturation. Gain-of-functionstudies were performed using an induciblesystem to generate ES cell lines in which theexpression of Sox7 was inducible upondoxycycline addition. Surprisingly, Sox7-sustainedexpression in the earliest committedhaematopoietic precursors resulted in themaintenance of their multi-potent and self-renewing status. Sox7-expressing cells rapidlyaccumulated in the culture, showing extensiveproliferative potential but no sign of maturation.
Group Leader
Valerie Kouskoff
Postdoctoral Fellows
Arnaud GandilletAlicia Gonzalez-Serrano
Scientific Officer
Stella Pearson
Graduate Students
Katalin BorosGuilherme CostaSarah LewisAndrzej Mazan
Sox7-sustained expression alters the balance
between proliferation and differentiation at the
onset of blood development
Based on its differential expression profilebetween mesoderm and haemangioblast, weselected Sox7 as a potential candidate geneinvolved in the regulation of blood lineageformation. A thorough analysis of Sox7
expression at the onset of blood specificationrevealed that this transcription factor wastransiently expressed in Flk1+ precursors as theyunderwent specification to the haematopoieticprogramme. To identify the cells that transientlyexpress Sox7, we established an ES cell linecarrying a bacterial artificial chromosome (BAC)transgene that contains the complete Sox7 locuswith the first exon of Sox7 replaced by a GFP-reporter cassette. Upon the in vitro
differentiation of these ES cells, we were able totrack and analyse Sox7-expressing cells. Resultsobtained using this ES cell line revealed that Sox7
was transiently expressed in a small subset ofFlk1+ cells that subsequently became committedto CD41+ haematopoietic precursors of bothprimitive and definitive lineages. The expressionof CD41 and Sox7 was inversely correlated asSox7 expression was down regulated as soon asthe cells acquired CD41 expression. Using amouse transgenic line derived from these Sox7-GFP BAC ES cells, we also showed that a smallsubpopulation of Flk1+ cells co-expressed Sox7
transiently in gastrulating embryos. Altogether,these data suggest that Sox7 expression defines a

Stem Cell and Haematopoiesis Group | 41
Removal of this differentiation block driven bySox7-enforced expression led to the efficientdifferentiation of haematopoietic progenitors toall erythroid and myeloid lineages.
Altogether, our study identify Sox7 as a novel andimportant player in the molecular regulation ofthe first committed blood precursors. Our datademonstrate that decreased Sox7 expressionaffects the formation of blood precursors whileits sustained expression is sufficient to alterdrastically the balance between proliferation anddifferentiation.
Contrasting effects of Sox17 and Sox18-
sustained expression at the onset of blood
specification
Given the known redundant roles of Sox7, Sox17
and Sox18 in cardiovascular development andthe importance of Sox7 at the onset ofhaematopoiesis, we decided to explore howSox17 and Sox18 enforced expression mayinfluence blood specification. To determine theimpact of Sox17 and Sox18 ectopic expressionon blood precursor development, we establishedES clones carrying either Sox17 or Sox18
inducible constructs using the doxycyclineinducible system described for Sox7. Withoutdoxycycline, cells derived from day 5 EB formedprimitive and definitive haematopoietic colonies,whereas with doxycycline few primitive ordefinitive colonies could be detected. UponSox18-enforced expression, a large number ofcolonies, blastic in appearance, developedwhereas very few blastic colonies wereobserved upon Sox17-enforced expression.Little evidence of maturation was detected inSox18-expressing blast colonies, with most cellsretaining an immature immuno-phenotype andmorphology. Upon removal of doxycycline anddown-regulation of Sox18 expression, individualSox18+ colonies gave rise to all erythroid andmyeloid lineages. Overall, the outcome ofSox18-enforced expression was reminiscent ofthe phenotype observed upon Sox7-enforcedexpression: in both cases, we observed a block inblood lineage maturation and the accumulationof blast-like colonies containing immature multi-potent blood progenitors.
Considering the high degree of homology amongthe three Sox F genes and their knownfunctional redundancy, it was surprising thatSox17-enforced expression led to an almostcomplete absence of colonies, either mature orimmature. Intrigued by these results, we set outto define the basis for such a difference. Wepreviously showed that Sox7-enforcedexpression led to the enhanced proliferation ofblood precursors. Similarly, Sox18 induced themassive proliferation of CD41+ bloodprecursors. In contrast, Sox17 did not confer anyproliferative advantage. Indeed, cell deathanalysis revealed that Sox17-enforced expressioncaused an increase in apoptosis. However, thisapoptosis was cell-context dependent andspecific to early haematopoietic precursors, asSox17 induction in undifferentiated ES cells didnot impair proliferation or increased apoptosis.To compare and contrast enforced-expressionphenotype to expression pattern, we analysedendogenous expression of Sox7, Sox17 andSox18 at the onset of haematopoieticspecification. Absolute quantification of the threetranscripts during haemangioblast differentiationrevealed that Sox7 was the most abundanttranscript in Flk1+ cells and at day 1 of blastdevelopment. Sox18 expression waspredominant at day 2 and 3 while Sox17
expression was only marginally present at alltime points analyzed.
Overall, our data suggest that despite theredundant roles generally observed for Sox Ffactors in several aspects of cardiovasculardevelopment, their expression patterns andfunction upon enforced expression at the onsetof blood development are rather different. Sox7
and Sox18 have partially overlapping expressionpatterns and their sustained expressionpromotes the proliferation of early bloodprecursors while blocking further differentiation.In contrast, Sox17 expression is marginal at theonset of blood development and its ectopicexpression appears detrimental to the survival ofthese early precursors.
Publications listed on page 70
Figure 1
Haemangioblast
EndotheliumPrimitiveErythrocytes
Macrophages Mast
Definitive Precursors
Mesoderm
DefinitiveErythrocytes
Smooth muscle
Haemogenic Endothelium
Sox7
Sox18

42 | Paterson Institute for Cancer Research Scientific Report 2009
Stromal-Tumour Interaction Grouphttp://www.paterson.man.ac.uk/stromal/
Human tumours are highly complex tissues and the non-neoplastic cell compartment of tumours, which is often termedthe “stroma”, is itself quite complex histologically. Carcinomacells initially recruit and/or activate these various stromal non-neoplastic cells, including fibroblasts, myofibroblasts, immunecells, endothelial cells and bone marrow-derived cells. Theresulting stromal cells reciprocate by fostering carcinoma cellgrowth and survival during the course of tumour progression.Studying the heterotypic interactions between the neoplasticcells and the supporting stroma is believed to be essential forunderstanding nature of a bulk of carcinoma mass. We focuson studying 1) how tumour-associated stromal fibroblastsbecome altered and co-evolve with tumour cells during thecourse of tumour progression, 2) how the stromal fibroblastsfacilitate tumour progression, and 3) what specific stroma-derived signal is crucial in promoting tumour invasion and metastasis.
between tumour stroma and the stroma presentin sites of wound healing, both of which containconsiderable numbers of myofibroblasts.
Stromal fibroblasts and myofibroblasts,collectively termed carcinoma-associatedfibroblasts (CAFs), were extracted from varioushuman carcinomas. CAFs, in comparison withtheir control fibroblasts, when coinjected withcarcinoma cells into immunodeficient mice, areknown to substantially promote carcinomagrowth and neoangiogenesis.
Evolution of tumour stromal myofibroblasts in
tumour
CAFs retain their myofibroblastic properties andtumour-promoting phenotypes, after they havebeen passaged for ten population doublings(PDs) in vitro in the absence of ongoing contactwith carcinoma cells. Accordingly, even thoughthe CAFs appear to have initially acquired theirunique phenotypes under the influence ofcarcinoma cells, once it is acquired, they might
Group Leader
Akira Orimo
Postdoctoral Fellows
Yasushi KojimaUrszula Polanska
Scientific Officer
Kieran Mellody
Graduate Student
Ahmet Acar
Tumour-promoting roles of carcinoma-
associated fibroblasts (CAFs)
Neoplastic epithelial cells coexist in carcinomaswith a stroma composed of various types ofmesenchymal cells as well as extracellular matrix(ECM), both of which create the complexity ofthe tumour microenvironment. Noticeablenumbers of myofibroblasts, which arecharacterized by their production of α-smoothmuscle actin (α-SMA), have been observedrepeatedly in the stroma of the majority ofinvasive human breast cancers (Figure 1).However, the specific contributions of these cellsto tumour progression are poorly defined.Myofibroblasts also exist in areas of woundhealing and chronic inflammation, and are oftenportrayed as “activated fibroblasts” that playcrucial roles in wound repair ; myofibroblastspossess greatly increased contractile ability,promote angiogenesis, and stimulate epithelialcell growth through the production of ECM andthe secretion of growth factor and cytokines.The striking histological resemblance is observed

Figure 1.
Tumour-associated stroma includes
large numbers of α-smooth muscle
actin (α-SMA)-positive myofibroblasts
in human breast cancers.
Immunostaining of human breast
tissues by an anti-α-SMA antibody
(a, c) and also staining with H&E (b,
d). The tumour region (c, d) and
non-tumour region (a, b) dissected
from the breast tissue of the human
breast cancer patient are shown.
Myofibroblasts (indicated by an
arrow) in the tumour region (c) and
myoepithelial cells in non-tumour
breast region (a) are strongly positive
for α-SMA. Scale bar, 75 μm.
Stromal-Tumour Interaction Group | 43
display this trait independent of further signallingfrom the carcinoma cells. Unanswered by theseobservations are (i) how do CAFs acquire andmaintain their activated, tumour-enhancingphenotypes? (ii) might CAFs harbour geneticand/or epigenetic alterations that act to conferthe unique phenotypes?
Some reports indicate that stromal regionsmicrodissected from human breast cancersexhibit a high frequency of genetic alterations,such as chromosomal regions of loss ofheterozygosity (LOH) and somatic mutations. A recent report also suggests that stromalfibroblasts that have undergone p53 loss areclonally selected during tumour progression,yielding a highly proliferative stroma. However,others indicate that tumour-associated stromaand CAFs exhibit no detectable geneticalterations, as gauged by array CGH and SNParray analyses; this suggests that any stablymaintained phenotype may depend onepigenetic modifications of the genome, such asDNA methylation. Alternatively, the stabilizationof their phenotype may depend on some typeof positive-feedback signalling of the sort createdby autocrine signalling loops. We note that ourCAFs show no detectable aneuploidy asdetermined by karyotype analysis, no anchorage-independent growth in culture, and notumourigenicity in vivo. Moreover, some of theCAFs begin to senesce after 15 population
doublings (PDs) in culture, similar to thebehaviour of normal human stromal fibroblasts.
Various cell types are thought to act as a sourcefor the emergence of tumour-promoting CAFmyofibroblasts (Figure 2). Resident fibroblastsare likely a major source of myofibroblasts, whilstother mesenchymal cell types includingendothelial cells, pericytes, smooth muscle cellsand preadipocytes are also capable of convertinginto myofibroblasts. Bone marrow-derivedprogenitors, such as fibrocytes and mesenchymalstem cells (MSCs), are also reported todifferentiate into myofibroblasts within thetumour. It is also possible that a small number ofmyofibroblasts present within the normalfibroblast populations are clonally expanded inresponse to selective pressure imposed by thetumour microenvironment. In addition, thestromal cells that have acquired epigenetic orgenetic alterations may differentiate intomyofibroblasts and these cells are then also likelyto undergo clonal expansion.
The tumour stroma continues to remodel itselfduring tumour progression and actively recruitsvarious cell types into the tumour mass wherethey act as different sources for myofibroblasts.It is not known, however, if different cells oforigins in the myofibroblast populations in thestroma of tumour exhibit different tumour-promoting properties. Once generated,myofibroblasts could maintain their ability tosubstantially promote tumour growth andprogression in many aspects via their interactionswith carcinoma cells and other host stromal cells.
Studying cross-talk between tumour cells andmesenchymal cells during tumour progressioncould help understand nature of biology ofhuman carcinomas and facilitate to developnovel stroma-targeted therapeutic approaches.
Publications listed on page 70
Figure 2.
Schematic representation of cellular
origins of CAF myofibroblasts during
tumour progression; 1) myofibroblasts
are proposed to be derived from
various mesenchymal cell types, such
as pre-existing fibroblasts, pericytes
and/or endothelial cells through
trans-differentiation. 2) Bone
marrow-derived progenitor cells
(BMDCs) including mesenchymal
stem cells (MSCs) and fibrocytes also
differentiate into myofibroblasts. 3
and 4) A small population of residual
myofibroblasts or mesenchymal cells
that carry genetic alterations may be
clonally selected during tumour
progression. The genetically altered
mesenchymal cells may or may not
differentiate into myofibroblasts.
44 | Paterson Institute for Cancer Research Scientific Report 2009
Research Groups - The University of Manchester School of Cancer and Enabling Sciences | 45
Research groupsThe University of Manchester School of Cancerand Enabling Sciences

46 | Paterson Institute for Cancer Research Scientific Report 2009
Biological, Immune and Gene Therapy Group
The Biological Immune and Gene Therapy (BIGT) groupfocuses on translational and clinical research in this field. TheClinical Immune and Molecular Monitoring Laboratory(CIMML) in TRF2 was established at the end of 2008 and isnow functional in respect to GCLP evaluation. A range ofvalidated immune and molecular based assays for monitoringclinical trials have been established including primary, secondaryand research related assays in several different studies. There isa separate but linked development of a GMP cell processingfacility which will open in March 2010 at the Incubator Building.
• TroVax® is a recombinant viral vaccine basedon MVA delivering 5T4
• Anyara® is an antibody targeted super-antigenwhich acts to target T cells to tumours
Both are progressing through Phase II to Phase IIIstudies and the BIGT group continues to play amajor role in their evaluation. Both studies wereadopted by the UK NCRN.
Vaccination of metastatic renal cancer patients
with TroVax given alongside interferon-alpha
We conducted an open-label phase I/II trial inwhich TroVax was administered alongsideinterferon-alpha (IFNα) to 11 patients withmetastatic renal cell carcinoma. Antigen-specificcellular and humoral responses were monitoredthroughout the study, and clinical responses wereassessed by measuring the changes in tumorburden. Treatment with TroVax plus IFNα waswell tolerated with no serious adverse eventsattributed to TroVax. All 11 patients mounted5T4-specific antibody responses and 5 (45%)mounted cellular responses. No objectivetumour responses were seen, but the overallmedian time to progression of 9 months waslonger than expected for IFNα alone and themedian overall survival was >26 months which isalso much longer than expected. The highfrequency of 5T4-specific immune responses andapparent long term benefit provided part of thejustification for a large Phase III trial (TRIST).
Group Leader
Robert E Hawkinsand Peter L Stern
Senior Clinical Research Fellow
Fiona Thistlethwaite
Senior Research Fellow
David Gilham
Postdoctoral Fellows
Eyad Elkord (CellularImmunology)Dominic Rothwell (MolecularMonitoring)Ryan Guest (at GMP CellularTherapy Unit)
Clinical Research Fellows
Christy Ralph (to Dec 2009)Alaaeldin Shablak Saladin Sawan (with CatherineHolland)Smita Sharma (from Oct 2009)
Research Student
Sameena Khan
GCLP validation
The CIMML was built through Christie GeneTherapy appeal funding and the staffing issupported by CR-UK following our 2005 sitevisit. The GCLP compliance of CIMML wasaudited in November 2009 by CR-UK with anextremely positive outcome. The GCLP primaryand secondary assays of the ongoing genemodified T-cell clinical trials have now beentransferred from the National Blood Service.Molecular assays developed include DNA/RNAisolation from clinical samples, T-cellspectratyping, real-time PCR, LAM-PCR, SNPanalysis, mutational analysis and gene expressionstudies. Immunological assays developed includePBMC isolation, multicolour flow cytometricassays for detection of surface and intracellularmarkers, ELISPOT, cellular proliferation, cellularcytotoxicity, ELISA and cytokine release assays.These are broadly applicable to a range of trialsand are critical for engineered T-cell therapy trials(see below).
Trials targeting 5T4
5T4 is a trophoblast antigen found to beselectively expressed on many common cancers.It was discovered by Professor Stern and he andthe BIGT group have been involved at all stagesof pre-clinical and clinical development. Thiswork links with companies who have licensedthe antigen from CR-UK. There are two currentapproaches to targeting 5T4:

Biological, Immune and Gene Therapy Group | 47
A randomised, double-blind, placebo-controlled
phase III study of MVA-5T4 (TroVax) in
metastatic renal cancer patients
The TRIST trial (TroVax Renal ImmunotherapySurvival Trial; 2006-001246-13), whichinvestigated whether TroVax added to first-linestandard of care, prolonged the survival ofpatients with metastatic clear cell renal cancer,has completed recruitment. 700 patients wererandomised 1:1 to receive up to 13immunisations of MVA-5T4 or placebo incombination with either Sunitinib, low-dose IL-2or IFN-α. Only preliminary results are availableat present but Professor Hawkins presentedthese for the first time at ECCO 2009 in Berlin.The treatment was generally well tolerated withno significant toxicity related to the vaccine.Early survival analysis demonstrated no significantimprovement in overall survival (20.1 vs 19.2months; HR: 1.07; 95% CI: 0.86, 1.33; p = 0.55).However, a prospectively planned analysisdemonstrated a significant survival advantage ingood prognosis patients treated with IL-2 +Trovax (not reached vs 19.5 months; HR: 0.54;95% CI: 0.30, 0.98; p = 0.04). Antibodyresponses against 5T4 were induced in mostMVA-5T4-treated patients and were associatedwith enhanced survival. The group of goodprognosis patients comprises those who benefitmost from immunotherapy and consideration isbeing given how to take this positive findingforward in the context of changing standardtreatments for renal cancer.
5T4 antibody targeted superantigen therapy
A Phase II/III study of ABR-217620 (ANYARA)combined with IFN-α compared to IFN-α alonein patients with advanced renal cell cancer hasbeen undertaken. The interim analysis satisfiedpre-defined criteria and the study was expandedfrom a Phase II to a Phase III study. Recruitmentis now completed and CIMML has completedthe validation and undertaken an extendedimmune analysis of a subset the patients treatedwith ANYARA and IFNα. The analysis includesflow cytometric assays for the expansion ofspecific VβTCR cells driven by the ANYARA, andalso the evaluation of different lymphocytesubpopulations.
Cellular Therapy
Adoptive cellular therapy for cancer is a verypromising and potentially general approach tothe treatment of cancer. The Manchester Groupis the leading European Centre and co-ordinatestwo large European programmes in this area andis the only centre with two engineered celltherapy trials on-going. The ATTACK consortium(www.ATTACK-cancer.org; Coordinator: RobertHawkins) is a €12M FP6 preclinical project whichhas had a major impact in technologydevelopment in this field. An FP7 training
network (ATTRACT) is commencing (end 2009)with a more clinical/translational focus. Theoverall aim is to efficiently evaluate this promisingapproach to cancer treatment.
Engineered T-cell Trials
Two products developed by the Cellular TherapyGroup are in CR-UK-NAC and Kay Kendallprogramme funded Phase I/II trials targeting CEAin gastrointestinal and CD19 in B-cellmalignancies. They both combinepreconditioning chemotherapy and IL-2 inaddition to cell therapy as pre-clinical modelsand clinical trials at the NCI in melanomapatients suggest this is advantageous. The trialtargeting CD19 is at early stage but alreadyencouraging clinical effects have been seen andtreatment is well tolerated. The trial targetingCEA is nearing the maximum doses and wehope to expand these cohorts to look forefficacy and to optimize the cellular productionmethods. These trials are clinically complex andNHS R&D funding supports ward facilities wherethese types of specialist treatment can beundertaken. The clinical team led by ProfessorHawkins and Dr Fiona Thistlethwaite issupported by experienced research nurse/trialscoordinators to ensure effective management ofthese complex patients. Recruitment to thesetrials will continue next year.
GMP Cell Therapy Unit
To further develop this area we are developing anew GMP Cell Therapy Unit. This is being fittedand equipped with funding (£700,000) from TheChristie Charity and has University and EUsupport via the ATTRACT training network aswell as specific clinical trials grants. The unit isanticipated to produce its first GMP product forthe current CR-UK trial in March 2010.
Based on the strategy defined at our ATTACKorganized international workshop we are alsodeveloping clinical trials in melanoma linking withthe NCI. After initial national feasibility studiesmulti-centre European studies are planned. Initialmelanoma studies should commence mid 2010and already key preliminary studies have beencompleted with the melanoma group.
Publications listed on page 70
Figure 1
Specifically designed isolator being
installed in the Cellular Therapy Unit
at The University of Manchester
Incubator Building. The innovative
design of the unit, devised with
MHRA and CR-UK involvement, will
facilitate the ambitious plans to
develop a core multi-user facility
which can deliver multiple cellular
therapy products in an efficient
manner.
Figure 1
Scientific Officers
Debbie Burt (seconded fromImmunology) Hayley BathaNatalia Krillova (at GMP CellularTherapy Unit)Sam Mowbray (at GMP CellularTherapy Unit)Lidan Chrisitie (at GMP CellularTherapy Unit)
Research Nurse
Andrea Byatt
Administration
Anvi Wadke – Clinical TrialsAdministrationNicola Hudson –ATTACK/ATTRACT projectmanager

48 | Paterson Institute for Cancer Research Scientific Report 2009
Children’s Cancer Group
Our research focuses on understanding the biological basis forthe differences in the therapeutic response in children withacute lymphoblastic leukaemia (ALL). Our clinical data isobtained from a number of international clinical trials which wecoordinate and we obtain clinical material nationwide fromchildren with ALL undergoing therapy on national protocols.The material linked with clinical data, provides the basis for ourlaboratory research. This has been a busy year for us as thegroup is finally at full strength. We moved into new premiseslast summer and the laboratory is now fully kitted andexpanding.
Laboratory
We reported last year lymphoblasts frompatients with high risk ALL express the lysosomalcysteine proteases Cathepsin B (CTSB) andasparaginyl endopeptidase (AEP) and degradethe drug asparaginase (2). Ashish Masurekar isinvestigating this in context of a prospectivenational biomarker trial in patients undergoingtherapy. AEP has been reported to beoverexpressed at the leading edge of epithelialcancers of poor prognosis with increasedinvasive and metastatic potential. In ourobservations active AEP was localised to adiscrete endocytic compartment contiguous withthe plasma membrane (Patel et al., 2009). Wespeculated that lymphoblast adhesion andinvasion into the CNS could be facilitated byAEP. In our initial experiments we found in vitro
evidence of increased invasive potential in AEPproducing leukaemic cell lines. However, suchinvasion was only modestly inhibited by an AEPinhibitor or by knocking down AEP. To furtherinvestigate the novel changes in the plasmamembrane proteome of invasive cells, MarkHolland used a comparative semiquantitativemass spectrometry (MS) approach. Almost halfthe proteins on the plasma membrane of theinvasive cell type were associated with adhesion,invasion and cytoskeleton re-organisation.Pathway analyses suggested a pivotal role forRAC. Seema Alexander showed that specificinhibition of RAC completely abrogated invasion
Group Leader
Vaskar Saha
Postdoctoral Fellows
John BridgemanClare DempseyMark Holland Jizhong Liu
Clinical Research Fellows
Shekhar KrishnanAshish Masurekar
Scientific Officers
Seema AlexanderNaina Patel
Graduate Student
Eva Diffner
Clinical Trials Manager
Catriona Parker
Administrator
Charlotte O’Horo
Clinical Trials
We reported on the results of the clinical trials,managed by Catriona Parker, last year. Whiletherapy is highly successful in childhood ALL,relapses within the central nervous system(CNS) are a poorly understood peculiarity ofthe disease. Opinion is divided as to themechanisms of these relapses. As this feature istherapy related, Shekhar Krishnan investigatedthe temporal changes and incidence in thepattern of CNS relapses in children with ALLtreated on four consecutive national trials from1985 – 2001. During this period we abandonedcranial irradiation and progressively intensifiedsystemic therapy. Shekhar’s critical analysisshowed that not only have overall relapseshalved but both isolated and combined (withbone marrow) CNS relapses have dramaticallydecreased since 1985 (Krishnan et al., Leukaemiain press). This suggests that systemic therapyrather than directed CNS therapy has been thekey to the decline in all types of relapses.However, over time the proportionate numberof CNS relapses has increased and bonemarrow relapses have declined. This supportsthe view that cells which have an increasedability to migrate into extramedullary sites mayhave a survival advantage giving rise to relapseslater on. These findings have influenced thedesign of the forthcoming frontline trial,ALL2010.
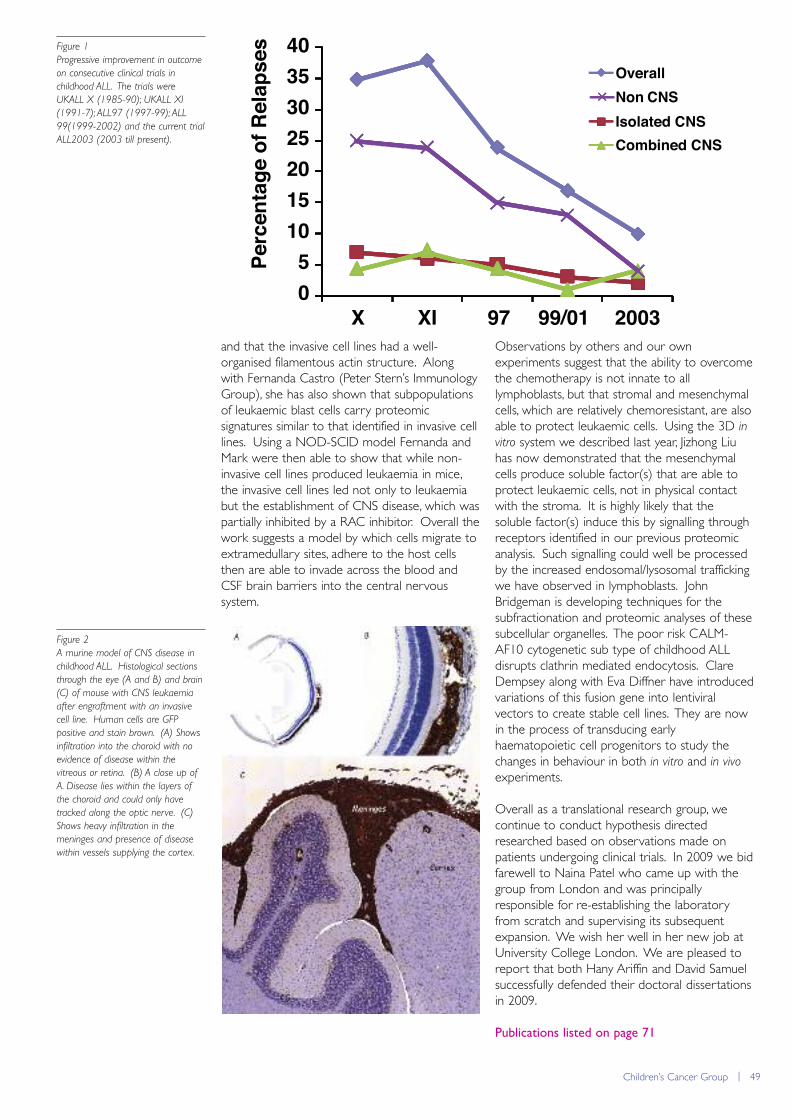
Children’s Cancer Group | 49
and that the invasive cell lines had a well-organised filamentous actin structure. Alongwith Fernanda Castro (Peter Stern’s ImmunologyGroup), she has also shown that subpopulationsof leukaemic blast cells carry proteomicsignatures similar to that identified in invasive celllines. Using a NOD-SCID model Fernanda andMark were then able to show that while non-invasive cell lines produced leukaemia in mice,the invasive cell lines led not only to leukaemiabut the establishment of CNS disease, which waspartially inhibited by a RAC inhibitor. Overall thework suggests a model by which cells migrate toextramedullary sites, adhere to the host cellsthen are able to invade across the blood andCSF brain barriers into the central nervoussystem.
Observations by others and our ownexperiments suggest that the ability to overcomethe chemotherapy is not innate to alllymphoblasts, but that stromal and mesenchymalcells, which are relatively chemoresistant, are alsoable to protect leukaemic cells. Using the 3D invitro system we described last year, Jizhong Liuhas now demonstrated that the mesenchymalcells produce soluble factor(s) that are able toprotect leukaemic cells, not in physical contactwith the stroma. It is highly likely that thesoluble factor(s) induce this by signalling throughreceptors identified in our previous proteomicanalysis. Such signalling could well be processedby the increased endosomal/lysosomal traffickingwe have observed in lymphoblasts. JohnBridgeman is developing techniques for thesubfractionation and proteomic analyses of thesesubcellular organelles. The poor risk CALM-AF10 cytogenetic sub type of childhood ALLdisrupts clathrin mediated endocytosis. ClareDempsey along with Eva Diffner have introducedvariations of this fusion gene into lentiviralvectors to create stable cell lines. They are nowin the process of transducing earlyhaematopoietic cell progenitors to study thechanges in behaviour in both in vitro and in vivo
experiments.
Overall as a translational research group, wecontinue to conduct hypothesis directedresearched based on observations made onpatients undergoing clinical trials. In 2009 we bidfarewell to Naina Patel who came up with thegroup from London and was principallyresponsible for re-establishing the laboratoryfrom scratch and supervising its subsequentexpansion. We wish her well in her new job atUniversity College London. We are pleased toreport that both Hany Ariffin and David Samuelsuccessfully defended their doctoral dissertationsin 2009.
Publications listed on page 71
Figure 1
Progressive improvement in outcome
on consecutive clinical trials in
childhood ALL. The trials were
UKALL X (1985-90); UKALL XI
(1991-7); ALL97 (1997-99); ALL
99(1999-2002) and the current trial
ALL2003 (2003 till present).
05
10152025303540
X XI 97 99/01 2003
Perc
enta
ge o
f Rel
apse
s
Overall
Isolated CNS Combined CNS
Non CNS
Figure 1
Figure 2
A murine model of CNS disease in
childhood ALL. Histological sections
through the eye (A and B) and brain
(C) of mouse with CNS leukaemia
after engraftment with an invasive
cell line. Human cells are GFP
positive and stain brown. (A) Shows
infiltration into the choroid with no
evidence of disease within the
vitreous or retina. (B) A close up of
A. Disease lies within the layers of
the choroid and could only have
tracked along the optic nerve. (C)
Shows heavy infiltration in the
meninges and presence of disease
within vessels supplying the cortex.

50 | Paterson Institute for Cancer Research Scientific Report 2009
Targeted Therapy Group
The past year has been a highly successful one for the TargetedTherapy Group. The highlights have included a successfulquinquennial review of the CR-UK programme grant, theaward of a Leukaemia Research Fund project grant, highimpact factor publications, the appointment of two new post-doctoral research fellows namely Drs Ellie Cheadle and SimonDovedi (arriving in January 2010) and the development of CR-UK/AZ clinical training fellowships in conjunction withProfessor Stratford (School of Pharmacy).
The overarching goal of the Targeted TherapyGroup is to define the optimal way to combineradiotherapy (RT) with immunotherapy in thetreatment of cancer by enhancing ourunderstanding of the underlying mechanisms ofaction. The specific objectives of the group are i)to investigate the mechanisms of action ofradioimmunotherapy ii) to investigate how therecognition of RT-induced tumour cell death bydifferent antigen presenting cells in the tumourmicroenvironment can impact on the ensuingimmune response; iii) to investigate the role ofmyeloid derived suppressor cells (MDSC) intumour regrowth after RT and to developstrategies to enhance RT tumour control bymodifying host immune response after RT andiii) to translate our experimental researchfindings into developing early phase clinical trials.Aspects of progress made with these researchprojects are outlined below.
Mechanisms of action of radioimmunotherapy
Our recent work, in collaboration with Dr MarkCragg’s group in Southampton, has investigated anew form of mAb induced cell death in B-cellleukaemias and lymphomas. Using bothlymphoma cell lines and primary chroniclymphocytic leukaemia (CLL) cells, we havedemonstrated for the first time the importanceof lysosome-mediated cell death for antibodytherapy elicited using clinically relevant mAbdirected against two different target antigensnamely CD20 and HLA DR. We havedetermined that death is preceded by
homotypic adhesion with both adhesion anddeath being dependent upon actin redistribution (Figure 1).
The ability of functionally different mAb targetedto different antigens namely, CD20 and HLA DRantigens suggests that the phenomena observedmay be a more general mechanism of lymphomacell killing by activating antibodies. The latter ispotentially of great interest as it provides apotential means to bypass the often dysregulatedapoptotic death pathways of tumour cellsallowing for effective tumour cell killing in thepresence of apoptotic inhibition. Interestinglyboth of the mAbs studied evoke lysosomal non-apoptotic cell death pathway and this is likely togo some way to explain their efficacy in vivo. Thiswork was recently published in the Journal ofClinical Investigation. (Ivanov et al., 2009). Wehave built on this success recently and WaleedAlduaij in collaboration with Andrei Ivanov andMark Cragg’s group has focused on the new-generation humanised anti-CD20 MonoclonalAntibody (GA101). GA101 has been shown toinitiate large amounts of this non-apoptoticmode of cell death in B-lymphoma cell lines incontrast to rituximab. Inhibitors of actinpolymerization (latrunculin B and cytochalasin D)inhibited cell death elicited by GA101. The roleof lysosomal activity in GA101-inducedprogrammed cell death (PCD) was assessedusing the inhibitor of the lysosomal protease,cathepsin B, which significantly inhibited celldeath induced by GA101 (Figure 2). To confirmthat this mode of death is non-apoptotic, we
Group Leader
Tim Illidge
Preclinical Group
Postdoctoral Fellows
Jamie HoneychurchEllie Cheadle
Academic Clinical Fellow
Nick Brown
Graduate Students
Waleed AlduaijMonique Melis
Senior Lecturer
Yong Du
Clinical Radioimmunotherapy
Group
Senior Clinical Scientist
Maureen Zivanovic
Clinical Scientist (ECMC
funded)
Jill Tipping
Research Nurses
Susan NeesonCaroline Hamer

Targeted Therapy Group | 51
demonstrated that GA101-induced PCD wasindependent of the antiapoptotic BCL-2oncoprotein and caspases. Taken together, thesefindings demonstrate that GA101 is the firsthumanized anti-CD20 mAb with Type IIproperties, potently eliciting a novel mode of celldeath in B-cell malignancies, which potentially canlead to improved B-cell depletion over rituximab.We are currently investigating this in vivo usinghuman CD20 transgenic mice and this work willbe presented as an oral presentation at theAmerican Society of Hematologists 2009.
Immune response to RT-induced dying
tumour cells
Our recent work in this area has focused onunderstanding the nature of the host immuneresponse to RT-induced tumour cell death. Wehave focused our attentions on two types ofAntigen Presenting Cells (APC), namelymacrophages (MΦ) and Dendritic cells (DC).This work, carried out by Jamie Honeychurch,has demonstrated that by manipulating MΦwithin the tumour microenvironment protectiveanti-tumour CD8 T-cell responses with anti-CD40 against irradiated lymphoma cells can beinduced. In these studies the potentialimportance of MΦ in cellular vaccination hasbeen demonstrated. Depletion of MΦ usingclodronate-encapsulated liposomes has beenshown to considerably enhance primaryvaccination efficacy in the presence of adjuvantanti-CD40 mAb. Our results demonstrate thatin order to induce a protective immuneresponse, additional host immune stimulation is required and that depletion of MΦpopulations can improve tumour cellularvaccination strategies.
Monique Melis in collaboration with KathrynSimpson (Clinical Experimental Pharmacology)has developed a Doxycycline-regulated Caspase-3 death switch in a number of tumour models.These models will enable us to titrate theproportion of ‘death switch’ cells and define thelevels of apoptotic cell death required in tumourto provoke immune responses as well asassessing the T-cell responses to apoptotic celldeath and how this changes with depletion ofselective APC.
Clinical translational applications of the
laboratory research programme
There have been a number of major successes intranslational research that have resulted directlyfrom, or are closely related to this CR-UKlaboratory programme of work.
A serum rituximab ELISA assay has now beenvalidated to GCLP in collaboration with theClinical and Experimental Pharmacology Group(CEP), through a CR-UK TRICC grant (incollaboration with Professor Dive) to fund Grace
Hampson. A robust, reliable and reproducibleELISA which can accurately establish serumrituximab concentrations accurately has beenestablished as a national reference laboratoryresource (Hampson et al., – submitted). Work isongoing analyzing serum rituximab levels fromsamples collected as part of the NCRI Phase IISCHRIFT study.
Early phase clinical trials of
radioimmunotherapy (RIT)
The clinical RIT group has made considerableprogress over the last few years in leading earlyphase clinical trial design both nationally andinternationally, and has a substantial portfolio ofearly phase clinical trials. A highlight of the yearwas the publication of the Phase I/II doseescalation RIT study using 131-rituximab. Thisstudy was the first to investigate the effect thatinduction therapy (4 weekly infusions of 375mg/m2 rituximab) and the subsequent efficacyand toxicity of anti-CD20 RIT in relapsedindolent B cell Lymphoma (Illidge et al., 2009).Induction therapy with rituximab was found tosignificantly increase the effective half-life of 131I-rituximab. An important observation from thiswork was that induction therapy with multipledoses of rituximab did not compromise theclinical efficacy or increase toxicity of subsequent131I-rituximab RIT. The overall response rate(ORR) was 94%, with a complete response (CR)rate 50%. The median time to progression was20 months, significantly longer than for the lastqualifying chemotherapy with ongoing durableremission of more than 60 months.
The phase II trial of Fractionated Zevalin (FIZZstudy) is the first study to be performed usingtwo fractions of RIT in previously untreatedfollicular lymphoma. It is a multicentreinvestigator-led study using fractionated RIT andis progressing well and 54 of the 70 patientsrequired have been recruited. A further novelinvestigator-led trial uses abbreviatedchemotherapy followed by RIT in relapsedfollicular lymphoma. The SCHRIFT study (ShortCHemotherapy Radioimmunotherapy InFollicular lymphoma Trial) is a NCRI lymphomagroup study and has recruited 32 of the 60required patients with widespread interest acrossthe UK. The SCHRIFT study is the first RIT studyever to be conducted across the UK making RITwidely available to more patients than has everbeen previously possible within the UK.Translational science associated with all of thesetrials include the measurement of rituximabpharmacokinetics, Human Anti-Mouse Antibody(HAMA) responses and circulating biomarkers ofcell death nucleosomal DNA (nDNA) incollaboration with Professor Dive’s CEP group.
Publications listed on page 71
Figure 1
Actin relocalization and cell death
following treatment with GA101
Figure 2
GA101 induced-PCD is dependent
on lysosomes

52 | Paterson Institute for Cancer Research Scientific Report 2009
Translational Radiobiology Group
The expansion of knowledge through sequencing the humangenome has been accompanied by the growth of highthroughput technologies for genome-wide analyses. Thesedevelopments increase the possibility of future personalisedtherapy based on molecular profiling. The TranslationalRadiobiology Group aims to exploit high throughputtechnologies to develop molecular profiles that predict theresponse of cancer patients to radiation therapy.
expression of the gene was associated with lowSF2 (p=0.0038) and a good outcome followingradiotherapy (p=0.024). This workdemonstrates that RNA extracted from archivalFFPE samples (up to 23 years old) can be usedto accurately identify genes which aredifferentially expressed at the protein level.These initial findings endorse the methods usedand allay fears concerning sample degradation.Continuing work is focussing on validating thegene signature and investigating the rolecandidate genes play in determining the intrinsicradiosensitivity of cervix tumours.
Tumour hypoxia
In collaboration with Prof Adrian Harris and DrFrancesca Buffa in Oxford and the AppliedComputational Biology and Bioinformatics(ACBB) Group, a tumour hypoxia-associatedgene signature was derived. Last year thesignature was streamlined and reduced from 99to 26 genes, and an MRC grant obtained tovalidate and qualify the multiplex 26-genehypoxia biomarker. Guy Betts liaised withApplied Biosystems to produce customised 384-well TaqMan Low Density Array (TLDA) cardsfor a quantitative real-time PCR (qRT-PCR)application. An endogenous control (18Sribosomal RNA) and five reference genes areincluded with the signature so that four samplescan be run in triplicate per array. One of thefirst experiments carried out by Guy Betts andFabian Zanella showed an excellent correlationbetween TLDA and Affymetrix HGU133 Plus 2array data (r=0.91; figure B) in a series of humanhead and neck squamous cell carcinomas(HNSCCs). An achievement for 2009 was
Group Leader
Catharine M.L.West
Postdoctoral Fellows
John Hall (joint withApplied Computational Biology& Bioinformatics)Carla Möller Levet (joint withApplied Computational Biology& Bioinformatics; to September2009)Fabian Zanella (from August2009)
Clinical Research Fellows
Guy BettsAhmad Mirza (from February2009)
Scientific Officers
Joely Irlam-JonesHelen Valentine
Graduate Student
Stephanie Donaldson (joint withImaging Science)
Scientific Administrator
Rebecca Elliott
Tumour radiosensitivity
Some tumours respond well to radiotherapy,whereas others do not. The underlying biologythat account for differences in response toradiotherapy is poorly understood. There isevidence that intrinsic sensitivity to radiation,hypoxia and proliferation are important. Workcarried out by the group several years agoshowed measurements of tumour radiosensitivitydetermined as the surviving fraction of cells after2 Gy of irradiation (SF2) in an in vitro clonogenicassay was an independent prognostic factor forthe outcome of radiotherapy. However, theclonogenic assay took four weeks to performand was technically demanding, precluding its useas a clinical test. John Hall is carrying out aproject exploring the potential of usingAffymetrix Human Exon 1.0 arrays inconjunction with archival specimens (aged 15-23years) to derive a molecular profile associatedwith tumour radiosensitivity (SF2). Incollaboration with Carla Möller-Levet, a tumourradiosensitivity associated gene expressionsignature was generated using 49 formalin-fixed,paraffin-embedded (FFPE) cervix tumours forwhich SF2 data were available. Differentialexpression analysis compared 11 samples withhigh and 11 with low SF2 values. A signaturewas derived comprising 1073 probesets targeting 1025 genes (false discovery rate, FDR0.1; figure A).
One of the genes predicted to be linked withradiosensitive tumours from the array analysis(p=0.001, FDR-adj.) was verified at the proteinlevel by immunohistochemistry in 74 cervixtumours (Helen Valentine). High protein
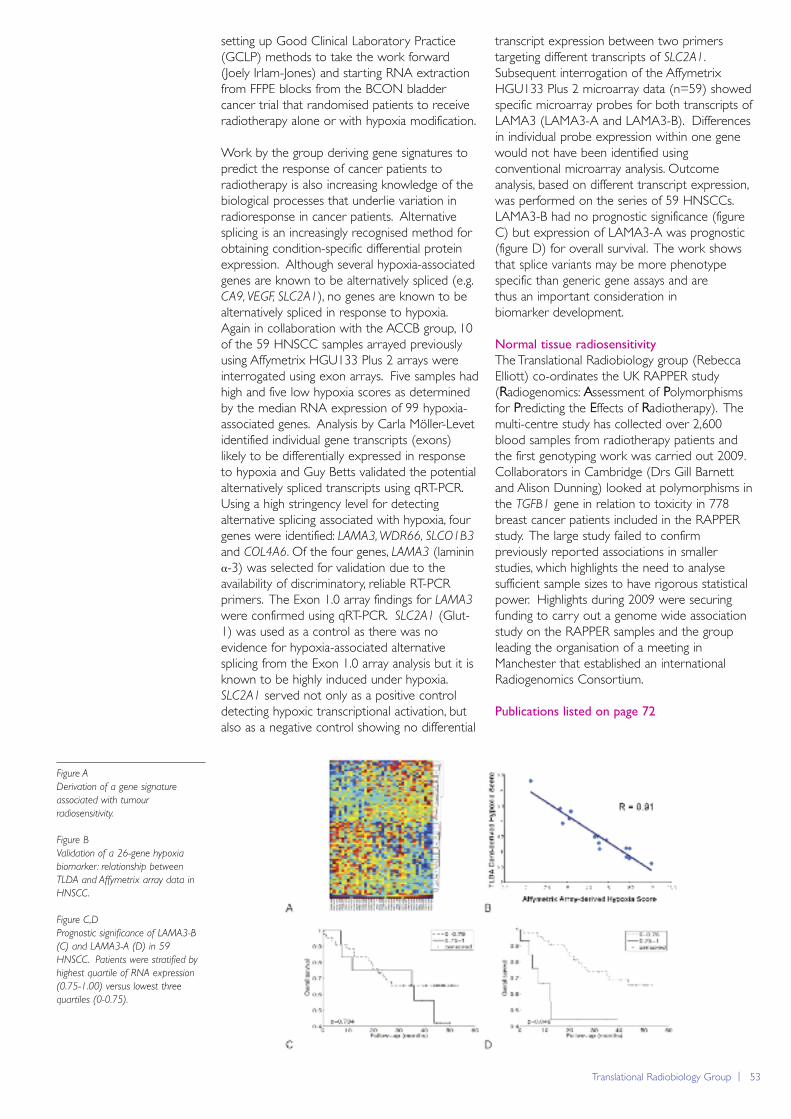
Translational Radiobiology Group | 53
setting up Good Clinical Laboratory Practice(GCLP) methods to take the work forward(Joely Irlam-Jones) and starting RNA extractionfrom FFPE blocks from the BCON bladdercancer trial that randomised patients to receiveradiotherapy alone or with hypoxia modification.
Work by the group deriving gene signatures topredict the response of cancer patients toradiotherapy is also increasing knowledge of thebiological processes that underlie variation inradioresponse in cancer patients. Alternativesplicing is an increasingly recognised method forobtaining condition-specific differential proteinexpression. Although several hypoxia-associatedgenes are known to be alternatively spliced (e.g.CA9, VEGF, SLC2A1), no genes are known to bealternatively spliced in response to hypoxia.Again in collaboration with the ACCB group, 10of the 59 HNSCC samples arrayed previouslyusing Affymetrix HGU133 Plus 2 arrays wereinterrogated using exon arrays. Five samples hadhigh and five low hypoxia scores as determinedby the median RNA expression of 99 hypoxia-associated genes. Analysis by Carla Möller-Levetidentified individual gene transcripts (exons)likely to be differentially expressed in responseto hypoxia and Guy Betts validated the potentialalternatively spliced transcripts using qRT-PCR.Using a high stringency level for detectingalternative splicing associated with hypoxia, fourgenes were identified: LAMA3, WDR66, SLCO1B3
and COL4A6. Of the four genes, LAMA3 (lamininα-3) was selected for validation due to theavailability of discriminatory, reliable RT-PCRprimers. The Exon 1.0 array findings for LAMA3
were confirmed using qRT-PCR. SLC2A1 (Glut-1) was used as a control as there was noevidence for hypoxia-associated alternativesplicing from the Exon 1.0 array analysis but it isknown to be highly induced under hypoxia.SLC2A1 served not only as a positive controldetecting hypoxic transcriptional activation, butalso as a negative control showing no differential
transcript expression between two primerstargeting different transcripts of SLC2A1.Subsequent interrogation of the AffymetrixHGU133 Plus 2 microarray data (n=59) showedspecific microarray probes for both transcripts ofLAMA3 (LAMA3-A and LAMA3-B). Differencesin individual probe expression within one genewould not have been identified usingconventional microarray analysis. Outcomeanalysis, based on different transcript expression,was performed on the series of 59 HNSCCs.LAMA3-B had no prognostic significance (figureC) but expression of LAMA3-A was prognostic(figure D) for overall survival. The work showsthat splice variants may be more phenotypespecific than generic gene assays and are thus an important consideration in biomarker development.
Normal tissue radiosensitivity
The Translational Radiobiology group (RebeccaElliott) co-ordinates the UK RAPPER study(Radiogenomics: Assessment of Polymorphismsfor Predicting the Effects of Radiotherapy). Themulti-centre study has collected over 2,600blood samples from radiotherapy patients andthe first genotyping work was carried out 2009.Collaborators in Cambridge (Drs Gill Barnettand Alison Dunning) looked at polymorphisms inthe TGFB1 gene in relation to toxicity in 778breast cancer patients included in the RAPPERstudy. The large study failed to confirmpreviously reported associations in smallerstudies, which highlights the need to analysesufficient sample sizes to have rigorous statisticalpower. Highlights during 2009 were securingfunding to carry out a genome wide associationstudy on the RAPPER samples and the groupleading the organisation of a meeting inManchester that established an internationalRadiogenomics Consortium.
Publications listed on page 72
Figure A
Derivation of a gene signature
associated with tumour
radiosensitivity.
Figure B
Validation of a 26-gene hypoxia
biomarker: relationship between
TLDA and Affymetrix array data in
HNSCC.
Figure C,D
Prognostic significance of LAMA3-B
(C) and LAMA3-A (D) in 59
HNSCC. Patients were stratified by
highest quartile of RNA expression
(0.75-1.00) versus lowest three
quartiles (0-0.75).

54 | Paterson Institute for Cancer Research Scientific Report 2009
Medical Oncology: Cell Therapy Group
Tumours actively avoid recognition by the immune system.This avoidance takes many forms but includes the loss of keyimmune cell recognition proteins on the tumour cell surfaceeffectively making the cancer invisible to T-cells, a critical part ofthe immune system. We have been working on methods tomodify T-cells so that they can recognise and then specificallytarget tumours.
ability to modify primary T-cells. Our preliminarystudies indicate that the approach is feasible andT-cells expressing CAR’s can be generated usingthis approach.
In order to select the gene-modified T-cells, wehave employed multi-gene expression systems.To this end, a selection marker (e.g. thetruncated CD34 protein) and the CAR are co-expressed from a single vector by means of bi-cistronic expression motifs. We have tested theeffect that one such motif (the Foot and MouthDisease virus 2A proteocleavage sequence) andidentified that the orientation of the transgenesrelative to the 2A sequence may be importantand especially so with respect to secretedproteins (Rothwell et al., submitted to HumGene Ther). Further optimisation of the 2A andCAR sequence has now generated vectors thatdrive higher levels of protein expression andthese are now being tested in primary T-cells todetermine whether this enhanced expressioncorrelates with improved tumour killing.
Identification of CAR – host protein
interactions on the T-cell surface: implications
for optimal CAR function.
In last years report, we documented ourpreliminary observations showing that the CARassociates with the host T-cell receptor and thatthis interaction was important for the optimalfunction for CAR’s bearing a CD3ζ signallingdomain. These observations have beenstrengthened during this past year (Bridgeman et
al., J Immunol, under revision) with the technicaldevelopment of a novel bead-based flowcytometry immuno-precipitation method(Bridgeman, Blaylock et al., Cytometry part A, inpress; the result of a collaborative effort with
Group Leader
Robert Hawkins
Senior Research Fellow
David Gilham
Postdoctoral Fellows
Vivien HansonJohn Bridgeman (until October)Eleanor Cheadle (until October)Jennifer Loconto (until October)
Scientific Officers
Vicky SheardMarzieh Kamjoo (fromNovember)Allison Robinson (until October)
Graduate Students
Grazyna Lipowska-BhallaErik Alcantar OrozcoMariam Al-Muftah (withImmunology)Anissa Cucchi (visiting student)
ATTACK Project Officer
Nikki Hudson
Chimeric Antigen Receptors
Our central approach has been the expressionof a chimeric antigen receptor (CAR) on T-cellswhich uses antibody-based technology to targettumour cells directly thereby circumventing someof the immune avoidance mechanisms employedby the tumour. The CAR also consists of a T-cellsignalling domain which, as a result of binding ofthe antibody to tumour, results in activation ofthe T-cell and subsequent immune mediateddestruction of the target tumour cell. Whilst theapproach is feasible and now being tested inclinical trials in Manchester (see BIGT report),there are significant improvements that arerequired in order to increase the therapeuticpotential of this approach. Delivering theseimprovements is the focus of the researchactivity of the Cell Therapy group.
Improving the expression of CAR’s in T-cells:
Gene-transfer and vector engineering.
For the CAR T-cell approach to succeed, thegenetic material encoding the receptor needs tobe efficiently introduced and strongly expressedin primary T-cells. The most commonly usedapproach involves retroviral gene transfer.However, to achieve high level geneticmodification, T-cells need to be activated – aprocess that may reduce the ability of the T-cellto optimally function within the patient.Lentiviral vectors can transduce non-dividingcells; however, these vectors are less efficient in T-cells. During the past year, we have optimisedmethods that permit lentiviral vectors totransduce primary mouse T-cells (Gilham et al., JGene Med, in press) thereby allowing us to fullyexplore the benefits of this vector in modelsystems. As an alternative approach, we are alsotesting non-viral nucleofection systems for their

Medical Oncology: Cell Therapy Group | 55
Morgan Blaylock) being critical to proving thebiochemical interaction of CAR and T-cellreceptor.
Mouse models of CAR T-cell function: the role
of the autologous antigen.
In order to understand how CAR T-cells functionand to prove that the modifications weintroduce result in an increased anti-tumourpotency, mouse models are critical. We havepreviously shown that human T-cells canchallenge the growth of B-cell lymphoma inimmuno-compromised models when combinedwith chemotherapy. Furthermore, mouse T-cellsbearing a CD19 specific CAR can eradicate a 13day established mouse B-cell lymphomaexpressing human CD19 (Cheadle et al., 2009).
However, these systems do not accurately modelthe natural situation where the tumour antigenmay be expressed on normal healthy tissues.We have generated a mouse CD19 specificCAR and used this to target mouse celllymphoma in the immuno-compromised mouse.In this system, the tumour antigen is naturallyexpressed at high level on host B-cells. As such,this model questions whether the CAR T-cellscan target tumour cells while examining anypotential side-effects on normal cells. Similar toour previous studies, these T-cells can efficientlyreject long-term (13 day) established syngeneiclymphoma; however, the CAR T-cells persistwithin the animal’s peripheral circulation for aperiod of 40 – 50 days after which theydisappear. During this time, the number ofcirculating B-cells remains depressed but returnsto normal levels shortly after the CAR T-cellsdisappear (Cheadle et al., J Immunol, in press,Figure 1). Taken together, these observationssuggest that the CAR T-cells are eliminating hostB-cells and finally become exhausted in the faceof the continuous re-population of B-cells by thehost and are themselves eliminated. In thismodel, there is no apparent toxicity associatedwith this transient absence of B-cells. Questionsremain concerning the potential impact of CART-cells that possess more powerful signallingdomains and whether targeting of tumour
antigens that are expressed on ‘more sensitive’organs by CAR T-cells results in more severetoxicities remains an important area toinvestigate. As such, the development of theseautologous model systems is important and amajor focus of the group’s work.
Malignant Melanoma: pre-clinical development
of cell therapy.
Following the lead of workers in the USA (Dr SRosenberg), we are looking to develop celltherapy protocols to treat malignant melanomain Manchester. Our initial work focused uponisolating tumour infiltrating lymphocytes (TIL) foruse in adoptive cell therapy and this work is nowmoving on to clinical development withproposals to test the approach both at a smallscale local level and also to test TIL therapy inmulti-centre EU phase III trials as the majorimpetus of a major EU grant initiative lead byour group. We are also working on a genetherapy approach using a T-cell receptorprovided by Dr Rosenberg to re-direct T-cells tomelanoma tumours.
The ‘ATTACK’ consortium
The ATTACK consortium is an EU FP6 fundedintegrated project (lead by Professor Hawkins)involving 16 laboratories focusing upon the pre-clinical optimisation of gene-modified T-celltherapy. The second ATTACK-organised ‘CellularTherapy of Cancer’ symposium was held inMilan, Italy this year and attended by over 150delegates from the EU, USA and Australia. Areport summarised the meeting has beenpublished (Bridgeman et al., 2009); however, acritical output of the meeting was a clinicallyfocused discussion meeting where it was agreedthat multi-centre clinical trials of cell therapywere clearly needed in order to test the abilityto deliver and the potency of cell therapies inthe wider context than that currently achievedwithin small-scale phase I clinical trials. Allparticipants agreed that this was an importantfocus for the community to work towardsdelivering in the near future.
Publications listed on page 72
Figure 1
In vivo imaging of mice bearing
systemic A20 B-cell lymphoma
labelled with a luciferase marker
gene (day 0) were treated with a
single of CD19z specific T-cells. Six
days later, imaging confirmed that
the B-cell lymphoma had been
eradicated due to targeted T-cell
activity.

56 | Paterson Institute for Cancer Research Scientific Report 2009
Medical Oncology: Translational Anti-Angiogenesis Group
Heparan sulfate (HS) is essential for the biological activity ofthe majority of angiogenic cytokines. Using novel syntheticchemistry we have generated a range of oligosaccharides thathave allowed us to determine the structures that inhibit theactivity of several angiogenic growth factors. One issue that iscritical to the development of these molecules is theelucidation of proof of mechanism biomarkers and to that endwe are developing novel assays of HS and HS proteoglycans,which will complement our existing portfolio of serological andimaging biomarkers that have been implemented in the clinic.
enhanced the sensitivity of the xenografts toplatinum chemotherapy through enhancedapoptosis and reductions in the number of cellsentering the cell cycle. These data identifyFGFR2IIIb and its ligands as new targets for thetreatment of EOC. However, in preliminarystudies we have demonstrated that inhibition ofother FGF receptors can have the converseeffect implying that caution should be exhibitedwhen introducing broad spectrum FGF receptorinhibitors into the clinic.
Imaging biomarkers
As the above programme moves towards theclinic it is critical that we develop biomarkersthat can detect proof of mechanism andprinciple in phase I evaluation. In a longstandingprogramme that aims to identify imagingbiomarkers that could be used to evaluate anti-angiogenic agents we have demonstrated inseveral different clinical trials and clinical settingsthat the vascular enhancing fraction is ofprognostic significance. We therefore sought todetermine whether anti-angiogenic agents impacton enhancing fraction and have demonstratedthat bevacizumab does reduce enhancingfraction and subsequently that cediranibmaintains a reduction in enhancing fraction inpatients who continue to benefit from VEGFinhibitors.
We have reported the potential of oxygen-enhanced imaging as a novel biomarker for thefunction of the vasculature in tumours allowing
Group Leader
Gordon Jayson
Senior Fellow
Egle Avizienyte
Postdoctoral fellows
Alison BackenClaire ColeSteen HansenGavin Miller
Clinical Research Fellows
Ying Kiat Zee Gireesh KumaranClaire MitchellNish Murukesh
Scientific Officers
Graham RushtonKaren Brookes
Heparan sulfate and angiogenesis
Heparan sulfate (HS) is a linearglycosaminoglycan that is essential for thebiological activity of most angiogenic cytokines.Previously we demonstrated that heparinoligosaccharides inhibited angiogenesis in vivo andtherefore undertook an organic chemistryprogramme to synthesise HS oligosaccharides todetermine structure-activity relationships. Wehave generated a range of HS oligosaccharidescontaining up to 12 saccharide residues and havedemonstrated that specific sulfation patterns areassociated with a broader range of activityagainst several angiogenic cytokines that havebeen implicated in ovarian cancer (see figure).These molecules demonstrate the capacity toinhibit cytokine-induced endothelial migration,proliferation and tube formation and further invivo evaluation is planned.
FGF and ovarian cancer
We previously demonstrated that humanepithelial ovarian cancer (EOC) endotheliumexpresses heparan sulfate that can activate FGF2.We therefore undertook a comprehensiveevaluation of FGFs in EOC and have identified areceptor isotype switch where transformation isassociated with the expression of FGFR2 IIIb; anisoform that confers on EOC cells the ability torespond to FGFs 3, 7 and 10. Further in vitro
and subsequently in vivo evaluationdemonstrated that RNAi-mediated knock downof FGFR2 IIIb in human EOC xenograftsimpaired tumour growth and significantly
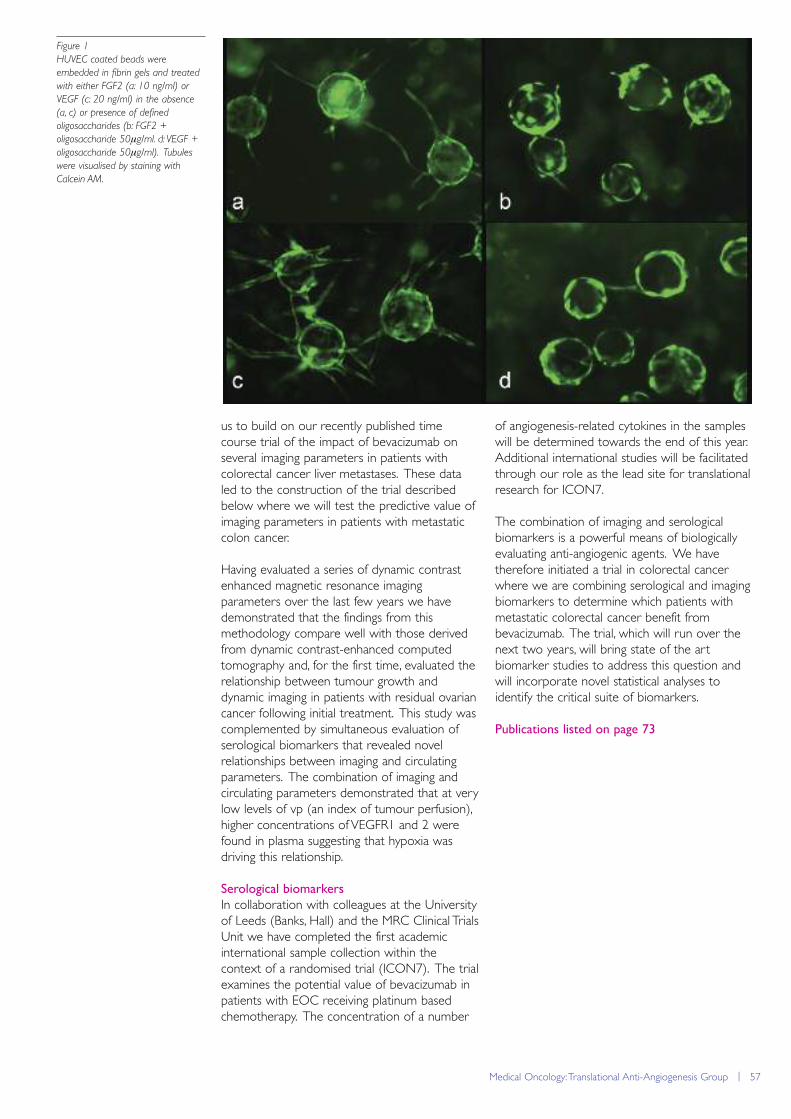
Medical Oncology: Translational Anti-Angiogenesis Group | 57
us to build on our recently published timecourse trial of the impact of bevacizumab onseveral imaging parameters in patients withcolorectal cancer liver metastases. These dataled to the construction of the trial describedbelow where we will test the predictive value ofimaging parameters in patients with metastaticcolon cancer.
Having evaluated a series of dynamic contrastenhanced magnetic resonance imagingparameters over the last few years we havedemonstrated that the findings from thismethodology compare well with those derivedfrom dynamic contrast-enhanced computedtomography and, for the first time, evaluated therelationship between tumour growth anddynamic imaging in patients with residual ovariancancer following initial treatment. This study wascomplemented by simultaneous evaluation ofserological biomarkers that revealed novelrelationships between imaging and circulatingparameters. The combination of imaging andcirculating parameters demonstrated that at verylow levels of vp (an index of tumour perfusion),higher concentrations of VEGFR1 and 2 werefound in plasma suggesting that hypoxia wasdriving this relationship.
Serological biomarkers
In collaboration with colleagues at the Universityof Leeds (Banks, Hall) and the MRC Clinical TrialsUnit we have completed the first academicinternational sample collection within thecontext of a randomised trial (ICON7). The trialexamines the potential value of bevacizumab inpatients with EOC receiving platinum basedchemotherapy. The concentration of a number
of angiogenesis-related cytokines in the sampleswill be determined towards the end of this year.Additional international studies will be facilitatedthrough our role as the lead site for translationalresearch for ICON7.
The combination of imaging and serologicalbiomarkers is a powerful means of biologicallyevaluating anti-angiogenic agents. We havetherefore initiated a trial in colorectal cancerwhere we are combining serological and imagingbiomarkers to determine which patients withmetastatic colorectal cancer benefit frombevacizumab. The trial, which will run over thenext two years, will bring state of the artbiomarker studies to address this question andwill incorporate novel statistical analyses toidentify the critical suite of biomarkers.
Publications listed on page 73
Figure 1
HUVEC coated beads were
embedded in fibrin gels and treated
with either FGF2 (a: 10 ng/ml) or
VEGF (c: 20 ng/ml) in the absence
(a, c) or presence of defined
oligosaccharides (b: FGF2 +
oligosaccharide 50μg/ml. d: VEGF +
oligosaccharide 50μg/ml). Tubules
were visualised by staining with
Calcein AM.

58 | Paterson Institute for Cancer Research Scientific Report 2009
Research Serviceshttp://www.paterson.man.ac.uk/research/
The quality of our Research Services was rightly highlighted forpraise during the Institute Site Visit in 2009 and the enthusiasmand knowledge of all the service unit heads was acknowledged.The Research Services underpin the investigations of all groupsin the Paterson, and we continue to invest in both the staff andequipment to ensure the services are properly resourced.
chamber systems are not ideal as they are notamenable to allowing the researcher to alter theenvironment around the cell whilst maintainingconstant focus of the protein or cell structureunder study. Over this year, micro-fluidic systemshave been examined as a viable technique forthe researcher to change the liquid phasearound cells for tasks such as cell maintenanceand drug screening. Both the cell media andtreatment can be altered around the cell in acontrolled manner whilst maintaining a fixedpoint of focus. These techniques are in theprocess of being introduced into the laboratoryfor both short and long term time lapse and aresuitable for quantifying changes in cell contentand response in both mammalian cell cultureand model cell systems.
The Förster Resonance Energy Transfer (FRET)technique allows the researcher to investigatemolecular interactions at the level ofprotein/protein interaction (4-6nm resolution).
Head of Research Services
Jenny VarleyAdvanced Imaging Facility
Head: Steve Bagley
The Advanced Imaging Facility provides access tostate-of-the-art imaging tools, analysis of images,training researchers and consultancy inexperimental design, all of which is undertakenwith an array of microscopes and computationaltools for both imaging live cellular processes andtissues. To achieve this staff within the facilitydevelop and integrate a range of techniques fordetecting cellular and molecular interactions, toimprove the spatial and temporal resolution ofthe microscope and to progress environmentalcontrol so that cells remain in a physiologicallyviable state whilst imaging.
One of the major advances this year has been toinvestigate and refine how the cells under studyare presented to the microscope. There is arequirement for changing the aqueousenvironment around the cells which is essentialwhen setting up drug screening investigations orwhen considering how the cells respond tochanges in the surrounding environment, forexample when they undergo a stress response.Conventionally, to image biological activity overtime, cells in media are imaged between twoglass coverslips, on a multi-well plate, Petri dishor culture flask. The coverslip/cell chambermethod, which is by far the most accuratemethod of both imaging and environmentalcontrol, has a major flaw in that if a liquid such ascell media is introduced over time, a smallchange in pressure occurs which leads to one ofthe coverslips acting as a diaphragm. This axialmovement of the coverslip leads to the objectunder study going in and out of focus which inturn requires the biological sample to besampled more frequently. As biology occurs infour dimensions, in 3D space over time, cell
Figure 1
Mitotic BPAE cells in anaphase.
F-actin is labelled with Texas
Red-x phalloidin. Microtubules,
in green, are labelled with
mouse anti-α-tubulin BODIPY
FL goat anti-mouse IgG. Blue
nuclear staining with DAPI.
Imaged on the Spinning Disk
Confocal microscope.

Research Services | 59
Currently in the laboratory, filters are utilised torecord three waveforms and then a calculation ismade of the level of interaction between twoproteins. An examination of spectral unmixingtechniques for the analysis of FRET has beenundertaken; a spectrophotometer attached to amicroscope records across a field of view, notjust for intensity as with camera based systems,but also for wavelength (from 500-1000nm), andthen by gating the resultant wavelengths a levelof the amount of interaction between twoproteins can be assessed. Over the coming year this will be developed further to allowstandardisation of the technique anddevelopment of computation analysis. Thespectrophotometer is also be utilised forquantum dot multiplexing and for spectrallyunmixing fluorescent proteins which will allow for the visualisation of up to five molecular interactions.
Over the last year several approaches have beenmade to improve both the image quality and thestandardisation of the equipment. With theappointment of a new member of staff it is nowpossible for all of the equipment to bemonitored on a weekly basis for defects in theway the equipment is set up that would lead tochanges in photo-sensitivity, faithfulness of theimaging method and highlights any problems thatcould arise in the future. Additional checks arealso made to the quality of data being producedby histological imaging. As many of thetechniques utilised as standard are becomingmore complex and multifaceted, support andadvice at the microscope is now easier to obtain.
The facility is working closer than ever with theIT department, consequently storage and archiveare now maintained and managed in a moresuitable fashion. In 2009 15-17 Terabytes of rawdata were generated. Analysis of images(volumes of data over time) is becoming moreprevalent and at the end of the year a separatecomputer room has been provided with 64-bitworkstations running a variety of software toolsto achieve visualisation and numerical analysis.Due to the amount of data being generatedthere are problems that ensue when trying tolocate data and to marry both the image withthe information about how the cells weretreated in the laboratory, consequently imagedatabase techniques have been examined as atechnique for the archival of raw data andlaboratory method. Over the coming there will be a trial of the software in the laboratory environment.
The advances in techniques, equipment andtraining have allowed more scientists to use themicroscopes as a tool in their research. Overthe last year forty five scientists have beentrained to use the microscopes and to analyse
their results in an appropriate manner. Tenresearch papers have been published bymembers of the Institute where equipment fromthe facility has been integral part of the studywhich cover diverse subjects such as defects incell division, proliferation, differentiation,haematopoiesis, hypoxia, drug resistance, celladhesion and cell migration.
During 2009, 150 members of the public beenshown the imaging techniques used in thelaboratory. Over two days, school leavers andsixth form students have used the microscopesto investigate cell division and had hands onexperience in the analysis of histological images.
Over the coming year priority will be given torefinement of the FRET technique, introductionof micro-fluidics as a routine practice, controllinglasers for inducing DNA damage in live cells tostudy the DNA repair and stress responses, anddeveloping analysis software.
Biological Resources Unit
The Paterson animal facility is now running at fullcapacity with all 3000-plus cages in use, and wehave seen a steady increase in both thetransgenic and experimental areas whichsupports 13 project licence holders and 65personal licence holders.
Transgenic production remains at the forefrontwith approximate 140 lines in active breedingprogrammes. To ensure that stocks are availablefor experimental use, transgenic mice areproduced under a centralised service licencewhich provides strict control with respect tominimum wastage, batch production and optimalanimal husbandry.
The facility is regulated by the Home Office instrict accordance with the Animal ScientificProcedures Act 1986 and locally the EthicalReview Process is managed through acommittee which reviews new applications,amendments and grant applications.
Transgenic Services
During 2009 we have been developingtechniques for in vitro fertilisation (IVF) usingfresh sperm. We are achieving a 50-60% successrate which is comparable with the JacksonLaboratories. We have also continued tooptimise the IVF technique using cryopreservedsperm – a technique which can be problematic.When using cryopreserved sperm the thaw rateof the straws is critical as is the use of theadditive methyl-β-cyclodextrin (MBCD) in thehuman tubule fluid (HTF) media. MBCD is usedto sequester cholesterol from the sperm

60 | Paterson Institute for Cancer Research Scientific Report 2009 Research Services | 60
membrane which aids capacitation before theaddition of the oocytes to the IVF culture dish.Chemical thinning of the oocyte zona pellucidaand selection of motile sperm is also employedto increase fertilisation rates. Throughrefinement of these techniques ourcryopreserved sperm fertilisation rates haveachieved rates as high as 40% which iscomparable to other transgenic facilities withinthe UK.
The microinjection service has carried out over25 Embryonic Stem cell injections and associatedtechniques such as critically timed superovulationregimes in female mice, and the production ofsterile male mice. To date eleven geneticallyaltered mouse strains have been produced, withsome others still in the pipeline.
Resource sharing of genetically altered animalshas resulted in a number of lines beingtransferred and received to facilitate the researchboth within the UK and worldwide. Sharingstrains in this way ensures the application of the3R’s and as such the IVF technique will lead to amajor refinement, reduction and replacement forarchiving of strains.
The rederivation process of new strains at thePaterson has included the following
• MC4R and tubby strains. These are both well characterised models of obesity and will be studied to better understand the biological mechanisms underpinning the clinical observation that obesity is associated with increased cancer risk and adverse treatment outcome
• MLL conditional mice. This strain will be used to ascertain whether MLL is required for leukemogenesis by MLL fusion oncogenes or other leukaemia-initiating oncogenes
• B6 FVB Tg (ITGAM) mice. These provide a model system for the conditional ablation of macrophages and will be used to understand how macrophages influence the immune response to radiation-induced tumour cell death and contribute to the activity of anti-CD20 monoclonal antibody therapy
• Rt TA2S-M2 mice. This line ubiquitously expresses rtTA which is controlled by an 8kb genomic fragment from the methylation-free CpG island of the human hnRNPA2B1-CBX3 housekeeping gene locus. This strain will be crossed with a number of established lines thatcarry the tetracycline reverse transcriptional activator rtTA
A dedicated Quarantine area using flexible filmisolators running at negative pressureaccommodates ‘live mice’ with unknown healthstatus to ensure the specific pathogen free (SPF)status of the facility is maintained.
Experimental Services
In vivo technical support has been providedoffering a range of surgical and non surgicalprocedures. Many of these procedures are wellestablished, and range from routine oral dosing,subcutaneous cell implantation, venepuncture viathe lateral tail vein, intra-tumoural injections,radiolabelled antibody delivery, intramuscularinjections and X-ray irradiations. Health andwelfare for animals under procedure remainsparamount and extensive monitoring has beenperformed including calliper measurements forsubcutaneous tumours, body weights, abdominalpalpations and routine tissue sampling post
mortem at the completion of studies.
New in vivo techniques developed have included,
• Intra-facial vein injection of mouse neonatesfollowing irradiation
• Bone marrow cells transplanted to formheterotopic ossicles
• Subcutaneous wound healing
Equipment Purchase
• Two replacement cage changing stationsfor carrying out routine husbandry tasks and animal manipulation
Cancer Research UK GeneChip Microarray
Service
Head: Stuart Pepper
The Molecular Biology Core Facility at thePaterson hosts a microarray service which isavailable not just to staff on site but to allCancer Research UK funded groups. The facilityhas two full Affymetrix systems which allow usto handle a high throughput of samples. Despitethe appearance of competing technologies,demand for expression profiling has remainedhigh and this year we have processed severalhundred samples.
The main difference we have seen over the lastyear is that more samples require specialhandling: small samples resulting from FACSprotocols have become more common as havearchival samples extracted from formalin-fixedparaffin-embedded material.
Last year we reported a collaboration with DrKim Linton (Christie Hospital) determining geneexpression profiles in archival sarcoma samples.This year we have published a follow-up papershowing that by using exon arrays and newlyreleased reagents, it is now possible to obtainbetter results than previous methods allowed(Linton et al., Biotechniques 2009; 47: 587).

Research Services | 61
Since this work was completed we have hadseveral large projects exploiting this applicationbrought to the facility.
Samples derived from FACS sorting are oftenlimited in the number of cells available. Usingthe latest amplification protocols we have beenable to return good quality data to users wherepopulations have been limited to a few hundredcells rather than the more usual several millionused as a start point. Ideally we would want tooffer single-cell profiling, but whilst this is possiblethere is no currently available protocol thatwould allow it to be offered as a routine service.In the meantime we will continue to evaluateany approaches to single-cell profiling thatbecome available.
Flow Cytometry Facility
Head: Morgan Blaylock
The Flow Cytometry Facility at the PatersonInstitute provides state-of-the-artinstrumentation, education and expert technicalassistance to investigators for the successfulperformance of flow cytometry-based studies.The goal of the facility is both to supportcurrent research applications and to continuouslyextend the repertoire of flow cytometricmethods available to users to facilitateresearchers in finding answers for the treatment,prevention and understanding of cancer.
Flow cytometry can be viewed as a specialisedform of fluorescence microscopy and is a meansof measuring the physical and chemicalcharacteristics of cells or particles. Any aspect ofa cell which can be labelled or detected with afluorescent marker can be identified andquantified by flow cytometry. We can assess: cellphenotype by looking for expression of cellsurface, cytoplasmic or nuclear antigens, cellularDNA or RNA content, fluorescent proteinexpression, functional aspects of the cell such asenzyme activity, apoptotic status, ion flux or pH.In addition, any population identified on ananalytical flow cytometer can be retrieved byusing a cell sorter which has the ability tophysically separate cells of interest from a mixedpopulation.
Analytical Cytometry
Early this year we relocated analytical cytometryto our new dedicated lab which offers a verystable optimal environment for the systems. Thefacility currently has four bench top cytometersincluding one plate-based bead reader. Theseare all user-operated systems which we offerbasic training in a group setting which issupplemented with one to one training forspecific applications.
BD FACScan - 3 colour single laser (blue)BD FACSCalibur - 4 colours dual laser (blue and red)BD FACSArray - 4 colours, dual laser (green and red)BD LSRII - 17 colours, quadruple laser (UV,violet, blue and red)
Cell sorters
With the removal of the analytical cytometersfrom the main cytometry lab we haveestablished a sorting suite which has minimisedthe traffic through the room providing a muchmore stable environment for the sorters. Thesorting suite currently houses three cell sorterswhich are able to retrieve up to four specificallydefined populations so that cells may berecovered for further study including re-culture,RNA or DNA extraction or use in functional cellassays. The cell sorters are operated solely bythe Flow Cytometry team on a daily basis
BD FACSVantage SE - 2 way sorting, 5 colours,dual laser (blue and red)BD FACSAria - 4 way sorting 12 colours, triplelaser (violet, blue and red)BD InFlux - 4 way sorting, 14 colours, quadruplelaser (UV, violet, blue red or orange)
Other services
Our lab offers a full range of educational andcytometric services. We are able to advise on awide variety of cytometry related subjectsincluding experimental design, selection ofreagents, data analysis and interpretation, we canact as a beta test site for novel cytometryapplications and we also advise on data
Figure 2
A snapshot of sorting in action -
each of the droplets contains a single
cell, the sorter decides on their fate.

62 | Paterson Institute for Cancer Research Scientific Report 2009
presentation. The latter is becoming more andmore important as journals require cytometricdata to be more transparent.
We have been involved in the continuation anddevelopment of a number of projects this year.We have continued our collaboration with theLeukaemia Biology Group, who are studying thedevelopmental control of human haematopoieticstem cells and their progenitors. They are usinghuman bone marrow to isolate differentpopulations of human haematopoietic cells. They have been using flow cytometry toimmunophenotype and sort cells, producingpurified populations highly enriched forpreviously defined cellular potentials. By applyingmicroarray and proteomic analysis to these cellsthey are expecting to generate a detailed geneand protein expression map in humanhaematopoiesis. In addition we have beeninvolved in studies which have yielded novelapplications of flow cytometry such as workconducted with John Bridgeman and DavidGilham. Together we have developed a flowcytometric immunoprecipitation method capableof investigating protein-protein interactionsspecifically the analysis of the T-cell receptor.
Histology
Head: Garry Ashton
Workloads have continued to increase, but therecruitment and training of a new scientificofficer has given the unit the capacity tocontinue to offer a comprehensive and flexibleservice in all of our heavily used key services.We have also been able to continue to focus onthe unit’s development over the last twelvemonths.
In early 2009 the existing Arcturus PixCell II lasercapture microdissection (LCM) system wasreplaced with the Leica LMD6000 system. Thesystem relies on gravity for sample collection anda UV optics driven laser. Evaluation of varioussample preparation techniques has continued.At present the Clinical and ExperimentalPharmacology Group are looking at the feasibilityof utilising LCM for the purification of circulatingtumour cells (CTCs) enriched by either theCellSearch or ISET technologies, and thesubsequent RNA evaluation for gene expressionanalysis. Proof of principle spiking experimentshave shown promising results. Several othergroups are using the system including the BreastBiology Group who are looking for differences ingene expression within breast tissue frompatients before and after an intermittent diet. Inparticular, gene expression differences betweenmicrodissected stromal and epithelial cells arebeing investigated.
Arrival of the automated tissue microarrayplatform (Beecher ATA27) is imminent. Speedof construction will be increased, whilst mappingof an H&E to the donor block will allow moreaccurate core acquisition. Chemically-inducedmouse liver tumours, head/neck, gastric, lung andbreast tumour TMA’s have been constructedmanually over the last 12 months. NumerousTMA’s have also been constructed for antibodyvalidation studies and the evaluation of imageanalysis systems.
The Manchester Cancer Research CentreBiobank has continued to expand, collectingmatched blood, urine and tissue samples fromfive collaborating Trusts across Manchester. Allthe samples are centrally processed within thelab. To date, samples from almost 800 patientshave been collected with over 40% containingboth fixed and frozen tumour and matchednormal pairs.
Last year the Biobank was expanded to includethe collection of blood and bone marrow fromhaematological malignancies including acutemyeloid leukaemia (AML), acute lymphoblasticleukaemia (ALL), chronic myeloid leukaemia(CML), myelodysplasia (MDS) and chroniclymphocytic leukaemia (CLL). This methodologyhas now been fully optimised and samples from52 patients (with consent from a further 50),representing 100 bone marrow and peripheralblood samples have been banked and availablefor use by researchers. To date, 10 applicationshave been made to the Biobank for samples.
Figure 3
The photomicrographs show
normal human breast tissue
before (upper panel) and after
(lower panel) laser capture
microdissection of epithelial
cells. The frozen section has
been stained with Gills
haematoxylin which highlights
the terminal ductal lobular unit
(centre of view) from which
epithelial cells are captured for
RNA isolation in order to study
gene expression.

Research Services | 63
Development of the immunohistochemistry(IHC) service has continued. The i6000automated platform and the antigen retrievalstations allow for high throughput andstandardisation. These are multi-user pieces ofequipment currently running at full capacity.Optimisation and multiple labelling studiestogether with alternative fixation anddecalcification regimes have continued to beexplored.
In collaboration with the Stromal TumourInteraction Group, IHC has been used in orderto understand the paracrine and/or autocrinemechanisms responsible for trans-differentiationof normal fibroblasts into tumour-promotingstably maintained carcinoma-associatedfibroblasts (CAFs). The characterisation of α-SMA-positive myofibroblasts and the validatedmyofibroblast-associated markers from in vitro
findings in human ductal invasive breastcarcinoma samples have been performed.
IHC has also been used in the development amouse model for acute lymphoblastic leukaemia(ALL). To achieve this GFP/luciferase-positivehuman leukaemic cell lines were injected intoimmunodeficient mice and once metastasisedtissue samples were isolated. A rapiddecalcification and extended retrieval methodwas developed to allow visualisation in wholehead samples without destroying themorphology of fragile structures.The Translational Radiobiology Group is usingIHC to investigate the expression of genes whichare implicated in radiosensitivity. This is a keyavenue of exploration as validation at theprotein level has substantiated Affymetrix datagenerated from RNA extracted from 15-25 yearold FFPE samples. The protein expression of anumber of different markers which canpotentially be used to define histologicalsubgroups such as squamous carcinoma andadenocarcinoma of the cervix is also beingstudied.
Kostoris Library
Head: Steve Glover
Working in partnership with The University ofManchester, the Kostoris Library provides on siteand online access to resources for all Patersonstaff and students. In 2009, the library launcheda DSpace Institutional Repository capturing theresearch output of The Christie and PatersonInstitute for Cancer Research. The repositorypresents the information within an organisationalcontext allowing papers to be mapped togroups and departments. The database is fullysearchable by both keyword and free textsearching and also has a comprehensive authorindex which allows the work of the individual
scientist to be captured. The repository holdsrecords from 2009 dating back to 2001 and it isplanned to add historical records dating back asfar as 1950.
The library offers a comprehensive portfolio toInstitute members including access to onlinejournals and databases provided jointly by theUniversity and The Christie. Databases includePubMed, EMBASE, BIOSIS, Scopus, ScienceCitation Index, and Journal Citation Reportsfrom Thomson ISI’s Web of Knowledge.
The library also offers a number of servicesaround literature searching, training and alertingservices. Literature search requests will aim tobe turned around within two working days andresults can be delivered in a variety of formats.In addition to ad hoc over the counter searchrequests the librarians will also set-up a numberof automated systematic searches of thedatabases on a monthly or bi-weekly schedule.These will help users to keep on top of thelatest published research on a particular topic,gene, protein or molecule. Training is alsoavailable and the library staff can deliver trainingin small groups, one-to-one, or even outreachsessions to suit the clients’ needs.
Where papers, textbooks or documents cannotbe readily accessed online or via the library afast and efficient inter-library lending anddocument supply service is available. Urgentpapers can usually be sourced same-day if therequest is received before 3pm and the libraryhas a 98% success rate on sourcing papers in thepublic domain.
In addition to access to resources the library canprovide a place to study and has recentlyembarked on an upgrade to the studyenvironment which will include a new readingarea, classroom, IT training room, and computerdrop-in suite. This work should be taking placein early 2010 and is expected to be completedby April.
Laboratory Services
Head: Mark Craven
The Laboratory Services department has threemain roles within the Institute. Firstly weprovide the Institute with a bulk liquid media anda separate Agar plate pouring service. We canproduce over 1000 litres of sterile liquid media amonth and we work with the laboratory groupsand can produce new types of media as andwhen required. We aim to deliver the mediawithin 3 days of the initial request

64 | Paterson Institute for Cancer Research Scientific Report 2009
Secondly, we supply sterile glassware and plasticsto the laboratories each day and arrange for thereturn of dirty glassware back to LaboratoryServices for washing and autoclaving. We alsosupply sterile water and PBS.
Finally, Laboratory Services provide thelaboratories with a Laboratory Aide to performa range of housekeeping duties requested byeach laboratory at fixed time points each week.
During 2009 the Institute has expanded to takeon new groups. This has increased our workloadand we have increased our staff numbers toenable us to meet this higher demand whilstmaintaining the flexibility to provide all of theabove requirements.
Logistics
Head: Maurice Cowell
A modern and efficient Logistics facility providesa comprehensive and vital role in supporting theresearch carried out at the institute. This groupundertakes a wide range of duties including theaccurate and efficient receipt, checking, bookingin and distribution of goods ordered bypersonnel in the Institute. The logistics team isalso responsible for the collection and removalof waste, be it general rubbish, yellow bags orGM waste, and for the collection of liquidnitrogen containers from laboratories andtransportation to the loading bay for refilling andreturning.
Ordering and distribution of the Central Storesstock via the intranet email has been updated tobecome more user-friendly and it is our duty toensure adequate stock levels are maintained atall times. This also includes maintaining andmonitoring the media and enzymes stored in theInstitute freezers (Sigma, Invitrogen, Roche,Promega and Qiagen), and recently a NewEngland Biolab freezer has been introduced tosave on delivery charges and number ofdeliveries, again the Logistics department isresponsible for the ordering, distribution andstock levels of these items. Currently we arealso looking at the gas cylinder usage and areawaiting estimates for the installation of tanks (tosave money and avoid handling cylinders). Thedepartment works closely with all groups andhelps out where necessary, be it tracing andconfirming delivery of goods with suppliers, anddealing with missing, damaged or wrong items.We also assist or manage the moving of heavyequipment or furniture, and setting up variousmeeting rooms for numerous events.
Molecular Biology Core Facility
Head: Stuart Pepper
The Molecular Biology Core Facility covers awide range of technologies including two selfcontained service areas - advanced massspectrometric analysis of proteins and microarraybased expression profiling - which are detailedseparately in this section.
There are three core services that have acontinuous level of demand; these are plasmidDNA preparation, DNA sequencing and PCR-based genotyping. These services have allcontinued this year with similar or slightlyincreased throughput. Typically we run around2000 PCR reactions, 600-800 DNA extractionsand anywhere from 400 to 800 sequences perweek.
The core facility also provides support forexpression profiling by quantitative PCR. AnABI7900 in conjunction with an EppendorfepMotion allows qPCR to be carried out in 384-well formats, allowing large projects to beprocessed efficiently. Demand for this servicehas been particularly high over the last fewmonths and as the year draws to a close we arelooking into adding more equipment to supportthis application.
The most exciting development in the corefacility this year has been the arrival of a highthroughput clonal sequencer. These platformswere initially designed to facilitate whole genomesequencing projects and have the capacity togenerate a staggering amount of data; we aregenerating around 300 million sequences perrun. Although these systems were designed forDNA sequencing they are now proving to beflexible tools for many other types of analysis.Our main interest is in evaluating the use ofsequence data sets for high resolutionexpression analysis as this may have someadvantages over traditional microarray profiling.These systems are highly complex, presentingchallenges both for the laboratory work anddata analysis. MBCF is working very closely withthe Crispin Miller’s Applied ComputationalBiology and Bioinformatics Group to ensure thatresearch groups within the Paterson will be ableto access this cutting edge technology.
Biological Mass Spectrometry Facility
Head: Duncan Smith
The Biological Mass Spectrometry Facility at thePaterson Institute enables the use of cutting-edge LCMS technology to groups for a multitudeof protein characterisation needs. Ourapplications encompass both proteinidentification and post-translational modification

Research Services | 65
analysis in both qualitative and quantitativeenvironments. Our remit involves both routineservice provision and novel applicationdevelopment in conjunction with researchgroups in the Institute. These applicationdevelopments are key contributors to themaintenance of the Institute’s presence at theresearch cutting-edge. Our routine serviceportfolio has benefitted from the addition offour new applications (delivered from ourdevelopment pipeline in 2009) and these arediscussed below.
In the area of post-translational modificationanalysis, we have developed and implemented alinear ion trap based approach to allowresearchers to reliably and confidently map sitesof ubiquitination. This approach has involvedsignificant tuning of both the mass spectrometricand database searching protocols to facilitate thesuccessful analysis of isopeptides which carry theanalytically useful information about the site ofUbl modification. This work was carried out incollaboration with the Cell Signalling Group whohave now successfully mapped sites of Ublmodifications on Tiam1 utilising our newapproach. With respect to mapping sites ofphosphorylation, we have developed a set ofprotocols which has massively improved typicalsequence coverage of protein molecules key incomprehensive PTM analysis. This has beenachieved by utilising ‘low specificity’ proteaseswith the intention of generating overlappingpeptide fragments not usually associated with‘high specificity’ digest agents. The work hasfocussed on both the generation of these ‘lowspecificity’ fragments and the most appropriateinformatics route to ensure efficient database
searching parameters are able to map sites ofphosphorylation. This development work hasbeen a collaborative effort with the Cell DivisionGroup who have recently been able to doublethe number of phosphorylation sites mapped ona target as a consequence of the utility of thisapproach.
In the area of protein quantitation, we havedeveloped a ‘label-free- Fourier Transform MS’based approach to facilitate the relativequantitation of specific phosphopeptides definedin previous qualitative mapping experiments.This work has been performed in collaborationwith the Cell Division Group and has facilitatedthe quantitative comparison of multiple sites ofphosphorylation on Cut12 between differentgenetic background strains, helping to definewhich sites of phosphorylation are likelyimplicated in key biological processes. A similar‘label-free- Fourier Transform MS’ protocol hasalso been applied to the quantitative study ofcomplex proteomes (whole human cell extracts)in collaboration with the Molecular Pathologyand Genito-Urinary Groups. This has demandedbespoke developments of (1) ultra highperformance nano-LC (ultra high resolutionseparations), (2) gas phase fractionation(essentially approximating to 2 dimensionalperformance from a 1 dimensional LCMS run)and (3) advanced informatics tools for dataanalysis (purchase of LC-MS data package).
All the new LCMS applications (Ubl mapping,phospho-mapping with overlapping fragments,label-free quantitation of phosphopeptides andcomplex proteome label-free quantitation) arenow available to all groups.

66 | Paterson Institute for Cancer Research Scientific Report 2009
Research publications
Crispin Miller (page 16)
Applied Computational Biology and Bioinformatics Group
Refereed Research Papers
Barenco, M., Papouli, E., Shah, S., Brewer, D., Miller,
C.J. and Hubank, M. (2009)
rHVDM: an R package to predict the activity andtargets of a transcription factor. Bioinformatics, 25,419-420.
Chan, C.W., Wong, N.A., Liu, Y., Bicknell, D., Turley,
H., Hollins, L., Miller, C.J., Wilding, J.L. and Bodmer,
W.F. (2009)
Gastrointestinal differentiation marker Cytokeratin20 is regulated by homeobox gene CDX1. Proc
Natl Acad Sci U S A, 106, 1936-1941.
Linton, K., Hey, Y., Dibben, S., Miller, C., Freemont,
A., Radford, J. and Pepper, S. (2009)
Methods comparison for high-resolutiontranscriptional analysis of archival material onAffymetrix Plus 2.0 and Exon 1.0 microarrays.Biotechniques, 47, 587-596.
Moller-Levet, C.S., Betts, G.N., Harris, A.L., Homer,
J.J., West, C.M. and Miller, C.J. (2009)
Exon Array Analysis of Head and Neck CancersIdentifies a Hypoxia Related Splice Variant ofLAMA3 Associated with a Poor Prognosis. PLoS
Comput Biol, 5, e1000571.
Geoff Margison (page 18)
Carcinogenesis Group
Refereed Research Papers
Billson, H., Harrison, K., Lees, N., Hall, C., Margison,
G. and Povey, A. (2009)
Dietary variables associated with DNA-N7methylguanine levels and O6-alkylguanine DNA-alkyltransferase activity in human colorectal mucosa.Carcinogenesis, 30, 615-620.
Crosbie, P.A., Barber, P.V., Harrison, K.L., Gibbs,
A.R., Agius, R.M., Margison, G.P. and Povey, A.C.
(2009)
GSTM1 copy number and lung cancer risk. Mutat
Res, 664, 1-5.
Kefford, R.F., Thomas, N.P., Corrie, P.G., Palmer, C.,
Abdi, E., Kotasek, D., Beith, J., Ranson, M., Mortimer,
P., Watson, A.J., Margison, G.P. and Middleton, M.R.
(2009)
A phase I study of extended dosing withlomeguatrib with temozolomide in patients withadvanced melanoma. Br J Cancer, 100, 1245-1249.
Sabharwal, A., Waters, R., Danson, S., Clamp, A.,
Lorigan, P., Thatcher, N., Margison, G.P. and
Middleton, M.R. (2009)
Predicting the myelotoxicity of chemotherapy: theuse of pretreatment O6-methylguanine-DNAmethyltransferase determination in peripheral bloodmononuclear cells. Melanoma Res, epub Jun 25.
Tubbs, J.L., Latypov, V., Kanugula, S., Butt, A.,
Melikishvili, M., Kraehenbuehl, R., Fleck, O.,
Marriott, A., Watson, A.J., Verbeek, B., McGown, G.,
Thorncroft, M., Santibanez-Koref, M.F., Millington,
C., Arvai, A.S., Kroeger, M.D., Peterson, L.A.,
Williams, D.M., Fried, M.G., Margison, G.P., Pegg,
A.E. and Tainer, J.A. (2009)
Flipping of alkylated DNA damage bridges base andnucleotide excision repair. Nature, 459, 808-813.
Watson, A.J., Middleton, M.R., McGown, G.,
Thorncroft, M., Ranson, M., Hersey, P., McArthur,
G., Davis, I.D., Thomson, D., Beith, J., Haydon, A.,
Kefford, R., Lorigan, P., Mortimer, P., Sabharwal, A.,
Hayward, O. and Margison, G.P. (2009)
O(6)-methylguanine-DNA methyltransferasedepletion and DNA damage in patients withmelanoma treated with temozolomide alone or withlomeguatrib. Br J Cancer, 100, 1250-1256.
Active Patents
O6-Substituted guanine derivatives, a process for
their preparation and their use in treating tumour
cells. McMurry, T.B.H., McElhinney, R.S., McCormick,
J.E., Elder, R.H., Kelly, J., Margison, G.P., Rafferty, J.A.,
Watson, A.J. and Willington, M.A.
International Patent Application PCT/IE94/0031,published (WO 94/29312, 70 pages) through CRTechnology Ltd. (Chem. Abs. 1995, 122, 239458e).
Pyrimidine derivatives and guanine derivatives, and
their use in treating tumour cells. McMurry, T.B.H.,
McElhinney, R.S., McCormick, J.E., Donnelly, D.J.,
Murray, P., Carola, C., Elder, R.H., Kelly, J., Margison,
G.P., Watson, A.J., Rafferty, J.A., Willington, M.A. and
Willington, M.R.

Research Publications | 67
International Patent Application PCT/IE96/00084,published (WO97/20843, 129 pages) through CRTechnology Ltd. (Chem. Abs., 1997, 127, 108802t).
Potentiation of temozolomide in human tumourcells. Baer, J., Freeman, A.A., Newlands, E.S., Watson, A.J.,
Rafferty, J.A., and Margison, G.P. (USP: 5731304)through CR Technology Ltd.
Karim Labib (page 20)
Cell Cycle Group
Refereed Research Papers
Gambus, A., van Deursen, F., Polychronopoulos, D.,
Foltman, M., Jones, R.C., Edmondson, R.D., Calzada,
A. and Labib, K. (2009)
A key role for Ctf4 in coupling the MCM2-7helicase to DNA polymerase alpha within theeukaryotic replisome. Embo J, 28, 2992-3004.
Morohashi, H., Maculins, T. and Labib, K. (2009)
The Amino-Terminal TPR Domain of Dia2 TethersSCF(Dia2) to the Replisome Progression Complex.Curr Biol, 19, 1943-1949.
Iain Hagan (page 22)
Cell Division group
Refereed Research Paper
Tallada, V.A., Tanaka, K., Yanagida, M. and Hagan, I.M.
(2009)
The S. pombe mitotic regulator Cut12 promotesspindle pole body activation and integration into thenuclear envelope. J Cell Biol, 185, 875-888.
Nic Jones (page 24)
Cell Regulation Group
Refereed Research Paper
Lawrence, C.L., Jones, N. and Wilkinson, C.R.
(2009)
Stress-Induced Phosphorylation of S. pombe Atf1Abrogates Its Interaction with F Box Protein Fbh1.Curr Biol, 19, 1907-1911.
Angeliki Malliri (page 26)
Cell Signalling Group
Refereed Research Papers
Woodcock, S.A., Jones, R.C., Edmondson, R.D. and
Malliri, A. (2009a)
A Modified Tandem Affinity Purification TechniqueIdentifies That 14-3-3 Proteins Interact with Tiam1,an Interaction Which Controls Tiam1 Stability. JProteome Res, 8, 5629-5641.
Woodcock, S.A., Rooney, C., Liontos, M., Connolly,
Y., Zoumpourlis, V., Whetton, A.D., Gorgoulis, V.G.
and Malliri, A. (2009b)
SRC-induced disassembly of adherens junctionsrequires localized phosphorylation and degradationof the rac activator tiam1. Mol Cell, 33, 639-653.
Caroline Dive and Malcolm Ranson (page 28)
Clinical and Experimental Pharmacology
Refereed Research Papers
Backen, A.C., Cummings, J., Mitchell, C., Jayson, G.,
Ward, T.H. and Dive, C. (2009)

68 | Paterson Institute for Cancer Research Scientific Report 2009
'Fit-for-purpose' validation of SearchLight multiplexELISAs of angiogenesis for clinical trial use. JImmunol Methods, 342, 106-114.
Blankley, R.T., Gaskell, S.J., Whetton, A.D., Dive, C.,
Baker, P.N. and Myers, J.E. (2009)
A proof-of-principle gel-free proteomics strategy forthe identification of predictive biomarkers for theonset of pre-eclampsia. Bjog, 116, 1473-1480.
Board, R.E., Ellison, G., Orr, M.C., Kemsley, K.R.,
McWalter, G., Blockley, L.Y., Dearden, S.P., Morris,
C., Ranson, M., Cantarini, M.V., Dive, C. and
Hughes, A. (2009)
Detection of BRAF mutations in the tumour andserum of patients enrolled in the AZD6244 (ARRY-142886) advanced melanoma phase II study. Br J
Cancer, 101, 1724-1730.
Dean, E., Jodrell, D., Connolly, K., Danson, S., Jolivet,
J., Durkin, J., Morris, S., Jowle, D., Ward, T.,
Cummings, J., Dickinson, G., Aarons, L., Lacasse, E.,
Robson, L., Dive, C. and Ranson, M. (2009)
Phase I trial of AEG35156 administered as a 7-dayand 3-day continuous intravenous infusion inpatients with advanced refractory cancer. J Clin
Oncol, 27, 1660-1666.
Dean, E.J., Ward, T., Pinilla, C., Houghten, R., Welsh,
K., Makin, G., Ranson, M. and Dive, C. (2009)
A small molecule inhibitor of XIAP inducesapoptosis and synergises with vinorelbine andcisplatin in NSCLC. Br J Cancer, epub Nov 10.
Hou, J.M., Greystoke, A., Lancashire, L., Cummings,
J., Ward, T., Williamson, A., Board, R., Amir, E.,
Hughes, S., Krebs, M., Hughes, A., Ranson, M.,
Lorigan, P., Dive, C. and Blackhall, F. (2009)
Evaluation of circulating tumor cells and serologicalcell death markers in small cell lung cancer patientsundergoing chemotherapy. Am J Pathol, 175, 808-816.
Hussein, D., Holt, S.V., Brookes, K.E., Klymenko, T.,
Adamski, J.K., Hogg, A., Estlin, E.J., Ward, T., Dive, C.
and Makin, G.W. (2009)
Preclinical efficacy of the bioreductive alkylatingagent RH1 against paediatric tumours. Br J Cancer,101, 55-63.
Martin-Fernandez, C., Bales, J., Hodgkinson, C.,
Welman, A., Welham, M.J., Dive, C. and Morrow,
C.J. (2009)
Blocking PI3-kinase activity in colorectal cancer cellsreduces proliferation but does not increaseapoptosis alone or in combination with cytotoxicdrugs. Mol Cancer Res, 7, 955-965.
Olofsson, M.H., Cummings, J., Fayad, W., Brnjic, S.,
Herrmann, R., Berndtsson, M., Hodgkinson, C.,
Dean, E., Odedra, R., Wilkinson, R.W., Mundt, K.E.,
Busk, M., Dive, C. and Linder, S. (2009)
Specific demonstration of drug-induced tumour cell
apoptosis in human xenografts models using aplasma biomarker. Cancer Biomark, 5, 117-125.
Roberts, D.L., Williams, K.J., Cowen, R.L., Barathova,M., Eustace, A.J., Brittain-Dissont, S., Tilby, M.J.,Pearson, D.G., Ottley, C.J., Stratford, I.J. and Dive, C.(2009)Contribution of HIF-1 and drug penetrance tooxaliplatin resistance in hypoxic colorectal cancercells. Br J Cancer, 101, 1290-1297.
Simpson, K.L., Whetton, A.D. and Dive, C. (2009)
Quantitative mass spectrometry-based techniquesfor clinical use: Biomarker identification andquantification. J Chromatogr B Analyt Technol Biomed
Life Sci, 877, 1240-1249.
Tennant, D.A., Frezza, C., MacKenzie, E.D., Nguyen,
Q.D., Zheng, L., Selak, M.A., Roberts, D.L., Dive, C.,
Watson, D.G., Aboagye, E.O. and Gottlieb, E.
(2009)
Reactivating HIF prolyl hydroxylases under hypoxiaresults in metabolic catastrophe and cell death.Oncogene, 28, 4009-4021.
Other Publications
Roberts, D.L., Dive, C. and Renehan, A.G. (2009)
Biological Mechanisms Linking Obesity and CancerRisk: New Perspectives. Annu Rev Med, 61, 301-316.
Ivan Ahel (page 30)
DNA Damage Response Group
Refereed Research Paper
Ahel, D., Horejsi, Z., Wiechens, N., Polo, S.E.,
Garcia-Wilson, E., Ahel, I., Flynn, H., Skehel, M.,
West, S.C., Jackson, S.P., Owen-Hughes, T. and
Boulton, S.J. (2009)
Poly(ADP-ribose)-dependent regulation of DNArepair by the chromatin remodeling enzyme ALC1.Science, 325, 1240-1243.

Biomolecular modelling | 69Research publications | 69
Peter Stern (page 32)
Immunology Group
Refereed Research Papers
Elkord, E., Dangoor, A., Burt, D.J., Southgate, T.D.,
Daayana, S., Harrop, R., Drijfhout, J.W., Sherlock, D.,
Hawkins, R.E. and Stern, P.L. (2009a)
Immune evasion mechanisms in colorectal cancerliver metastasis patients vaccinated with TroVax(MVA-5T4). Cancer Immunol Immunother, 58,1657-1667.
Karanam, B., Gambhira, R., Peng, S., Jagu, S., Kim,
D.J., Ketner, G.W., Stern, P.L., Adams, R.J. and
Roden, R.B. (2009)
Vaccination with HPV16 L2E6E7 fusion protein inGPI-0100 adjuvant elicits protective humoral andcell-mediated immunity. Vaccine, 27, 1040-1049.
Other Publications
Elkord, E., Shablak, A., Stern, P.L. and Hawkins, R.E.
(2009b)
5T4 as a target for immunotherapy in renal cellcarcinoma. Expert Rev Anticancer Ther, 9, 1705-1709.
Marignol, L., Foley, R., Southgate, T.D., Coffey, M.,
Hollywood, D. and Lawler, M. (2009)
Hypoxia response element-driven cytosinedeaminase/5-fluorocytosine gene therapy system: ahighly effective approach to overcome the dynamicsof tumour hypoxia and enhance the radiosensitivityof prostate cancer cells in vitro. J Gene Med, 11,169-179.
Stern, P.L. (2009)
CervarixTM helps protect women against cervicalcancer. Pulse, March 2009.
Active Patents
Stern, P.L. and Hole, N. (1989).
Improvement relating to antigens. Publicationinformation EP0336562.
Stern PL and Hole N (1999).
5T4 antigen from human trophoblasts Publicationinformation US5869053.
Ward C.M and Stern (2005)
5T4 antigen expression as an indicator of stem celldifferentiation. Publication informationUS2005260591.
Nullin Divecha (page 34)
Inositide Laboratory
Refereed Research Paper
van den Bout, I. and Divecha, N. (2009)
PIP5K-driven PtdIns(4,5)P2 synthesis: regulation andcellular functions. J Cell Sci, 122, 3837-3850.
Other Publications
Halstead, J.R., Snel, M.H., Meeuws, S., Jones, D.R.
and Divecha, N. (2009)
Assaying endogenous phosphatidylinositol-4-phosphate 5-kinase (PIP5K) activities. Methods Mol
Biol, 462, 391-402.
Jones, D.R., Bultsma, Y., Keune, W.J. and Divecha, N.
(2009)
Methods for the determination of the mass ofnuclear PtdIns4P, PtdIns5P, and PtdIns(4,5)P2.Methods Mol Biol, 462, 75-88.
Tim Somervaille (page 36)
Leukaemia Biology Group
Refereed Research Paper
Somervaille, T.C., Matheny, C.J., Spencer, G.J.,
Iwasaki, M., Rinn, J.L., Witten, D.M., Chang, H.Y.,
Shurtleff, S.A., Downing, J.R. and Cleary, M.L.
(2009)
Hierarchical maintenance of MLL myeloid leukemiastem cells employs a transcriptional program sharedwith embryonic rather than adult stem cells. Cell
Stem Cell, 4, 129-140.
Other Publication
Somervaille, T.C. and Cleary, M.L. (2009)
Mutant CEBPA: priming stem cells for myeloidleukemogenesis. Cell Stem Cell, 5, 453-454.
Georges Lacaud (page 38)
Stem Cell Biology Group
Refereed Research Papers
Gandillet, A., Serrano, A.G., Pearson, S., Lie, A.L.M.,
Lacaud, G. and Kouskoff, V. (2009)
Sox7-sustained expression alters the balancebetween proliferation and differentiation ofhematopoietic progenitors at the onset of bloodspecification. Blood, 114, 4813-4822.
Hoogenkamp, M., Lichtinger, M., Krysinska, H.,
Lancrin, C., Clarke, D., Williamson, A., Mazzarella,
L., Ingram, R., Jorgensen, H., Fisher, A., Tenen, D.G.,
Kouskoff, V., Lacaud, G. and Bonifer, C. (2009)
Early chromatin unfolding by RUNX1 - a molecularexplanation for differential requirements duringspecification versus maintenance of thehematopoietic gene expression program. Blood,114, 299-309.
Lancrin, C., Sroczynska, P., Stephenson, C., Allen, T.,
Kouskoff, V. and Lacaud, G. (2009)
The haemangioblast generates haematopoietic cellsthrough a haemogenic endothelium stage. Nature,457, 892-895.
Landry, J.R., Bonadies, N., Kinston, S., Knezevic, K.,
Wilson, N.K., Oram, S.H., Janes, M., Piltz, S.,

70 | Paterson Institute for Cancer Research Scientific Report 2009
Hammett, M., Carter, J., Hamilton, T., Donaldson,
I.J., Lacaud, G., Frampton, J., Follows, G., Kouskoff, V.
and Gottgens, B. (2009)
Expression of the leukaemia oncogene Lmo2 iscontrolled by an array of tissue specific elementsdispersed over 100kb and bound by Tal1/Lmo2, Etsand Gata factors. Blood, 113, 5783-5792.
Perez-Campo, F.M., Borrow, J., Kouskoff, V. and
Lacaud, G. (2009)
The histone acetyl transferase activity of monocyticleukemia zinc finger is critical for the proliferation ofhematopoietic precursors. Blood, 113, 4866-4874.
Sroczynska, P., Lancrin, C., Kouskoff, V. and Lacaud,
G. (2009)
The differential activities of Runx1 promoters definemilestones during embryonic hematopoiesis. Blood,114, 5279-5289.
Other Publications
Lancrin, C., Sroczynska, P., Serrano, A.G., Gandillet,
A., Ferreras, C., Kouskoff, V. and Lacaud, G. (2009)
Blood cell generation from the hemangioblast. J Mol
Med, epub Oct 25.
Sroczynska, P., Lancrin, C., Pearson, S., Kouskoff, V.
and Lacaud, G. (2009)
In vitro differentiation of mouse embryonic stemcells as a model of early hematopoieticdevelopment. Methods Mol Biol, 538, 317-334.
Valerie Kouskoff (page 40)
Stem Cell and Haematopoiesis Group
Refereed Research Papers
Gandillet, A., Serrano, A.G., Pearson, S., Lie, A.L.M.,
Lacaud, G. and Kouskoff, V. (2009)
Sox7-sustained expression alters the balancebetween proliferation and differentiation ofhematopoietic progenitors at the onset of bloodspecification. Blood, 114, 4813-4822.
Hoogenkamp, M., Lichtinger, M., Krysinska, H.,
Lancrin, C., Clarke, D., Williamson, A., Mazzarella,
L., Ingram, R., Jorgensen, H., Fisher, A., Tenen, D.G.,
Kouskoff, V., Lacaud, G. and Bonifer, C. (2009)
Early chromatin unfolding by RUNX1 - a molecularexplanation for differential requirements duringspecification versus maintenance of thehematopoietic gene expression program. Blood,114, 299-309.
Lancrin, C., Sroczynska, P., Stephenson, C., Allen, T.,
Kouskoff, V. and Lacaud, G. (2009)
The haemangioblast generates haematopoietic cellsthrough a haemogenic endothelium stage. Nature,457, 892-895.
Landry, J.R., Bonadies, N., Kinston, S., Knezevic, K.,
Wilson, N.K., Oram, S.H., Janes, M., Piltz, S.,
Hammett, M., Carter, J., Hamilton, T., Donaldson,
I.J., Lacaud, G., Frampton, J., Follows, G., Kouskoff, V.
and Gottgens, B. (2009)
Expression of the leukaemia oncogene Lmo2 iscontrolled by an array of tissue specific elementsdispersed over 100kb and bound by Tal1/Lmo2, Etsand Gata factors. Blood, 113, 5783-5792.
Perez-Campo, F.M., Borrow, J., Kouskoff, V. and
Lacaud, G. (2009)
The histone acetyl transferase activity of monocyticleukemia zinc finger is critical for the proliferation ofhematopoietic precursors. Blood, 113, 4866-4874.
Sroczynska, P., Lancrin, C., Kouskoff, V. and Lacaud,
G. (2009)
The differential activities of Runx1 promoters definemilestones during embryonic hematopoiesis. Blood,114, 5279-5289.
Other Publications
Lancrin, C., Sroczynska, P., Serrano, A.G., Gandillet,
A., Ferreras, C., Kouskoff, V. and Lacaud, G. (2009)
Blood cell generation from the hemangioblast. J Mol
Med, epub Oct 25.
Sroczynska, P., Lancrin, C., Pearson, S., Kouskoff, V.
and Lacaud, G. (2009)
In vitro differentiation of mouse embryonic stemcells as a model of early hematopoieticdevelopment. Methods Mol Biol, 538, 317-334.
Akira Orimo (page 42)
Stromal-Tumour Interaction Group
Other Publication
Shimoda, M., Mellody, K.T. and Orimo, A. (2009)
Carcinoma-associated fibroblasts are a rate-limitingdeterminant for tumour progression. Semin Cell Dev
Biol, epub Oct 24.
Robert Hawkins and Peter Stern (page 46)
Biological, Immune and Gene Therapy
Refereed Research Papers
Borghaei, H., Alpaugh, K., Hedlund, G., Forsberg, G.,
Langer, C., Rogatko, A., Hawkins, R., Dueland, S.,
Lassen, U. and Cohen, R.B. (2009)
Phase I dose escalation, pharmacokinetic andpharmacodynamic study of naptumomabestafenatox alone in patients with advanced cancerand with docetaxel in patients with advanced non-small-cell lung cancer. J Clin Oncol, 27, 4116-4123.
Chau, I., Norman, A.R., Cunningham, D., Oates, J.,
Hawkins, R., Iveson, T., Nicolson, M., Harper, P.,
Seymour, M. and Hickish, T. (2009)
The impact of primary tumour origins in patientswith advanced oesophageal, oesophago-gastricjunction and gastric adenocarcinoma--individual

Biomolecular modelling | 71Research publications | 71
patient data from 1775 patients in four randomisedcontrolled trials. Ann Oncol, 20, 885-891.
Cheadle, E.J., Hawkins, R.E., Batha, H., Rothwell,
D.G., Ashton, G. and Gilham, D.E. (2009)
Eradication of established B-cell lymphoma byCD19-specific murine T cells is dependent on hostlymphopenic environment and can be mediated byCD4+ and CD8+ T cells. J Immunother, 32, 207-218.
Elkord, E. (2009)
Frequency of human T regulatory cells in peripheralblood is significantly reduced by cryopreservation. JImmunol Methods, 347, 87-90.
Elkord, E., Dangoor, A., Burt, D.J., Southgate, T.D.,
Daayana, S., Harrop, R., Drijfhout, J.W., Sherlock, D.,
Hawkins, R.E. and Stern, P.L. (2009)
Immune evasion mechanisms in colorectal cancerliver metastasis patients vaccinated with TroVax(MVA-5T4). Cancer Immunol Immunother, 58,1657-1667.
Hawkins, R.E., Macdermott, C., Shablak, A., Hamer,
C., Thistlethwaite, F., Drury, N.L., Chikoti, P.,
Shingler, W., Naylor, S. and Harrop, R. (2009)
Vaccination of patients with metastatic renal cancerwith modified vaccinia Ankara encoding the tumorantigen 5T4 (TroVax) given alongside interferon-alpha. J Immunother, 32, 424-429.
Karanam, B., Gambhira, R., Peng, S., Jagu, S., Kim,
D.J., Ketner, G.W., Stern, P.L., Adams, R.J. and
Roden, R.B. (2009)
Vaccination with HPV16 L2E6E7 fusion protein inGPI-0100 adjuvant elicits protective humoral andcell-mediated immunity. Vaccine, 27, 1040-1049.
Shablak, A., Hawkins, R.E., Rothwell, D.G. and
Elkord, E. (2009)
T cell-based immunotherapy of metastatic renal cellcarcinoma: modest success and future perspective.Clin Cancer Res, 15, 6503-6510.
Other Publications
Bridgeman, J.S., Gilham, D.E., Hawkins, R.E. and
Cheadle, E.J. (2009)
The second cellular therapy of cancer symposium,27-29 March 2009, Milan, Italy. Cancer Immunol
Immunother, epub Aug 8.
Elkord, E., Shablak, A., Stern, P.L. and Hawkins, R.E.
(2009)
5T4 as a target for immunotherapy in renal cellcarcinoma. Expert Rev Anticancer Ther, 9, 1705-1709.
Marignol, L., Foley, R., Southgate, T.D., Coffey, M.,
Hollywood, D. and Lawler, M. (2009)
Hypoxia response element-driven cytosinedeaminase/5-fluorocytosine gene therapy system: ahighly effective approach to overcome the dynamics
of tumour hypoxia and enhance the radiosensitivityof prostate cancer cells in vitro. J Gene Med, 11,169-179.
Stern, P.L. (2009)
CervarixTM helps protect women against cervicalcancer. Pulse, March 2009.
Vaskar Saha (page 48)
Children’s Cancer Group
Refereed Research Papers
Krishnan, S., Wade, R., Moorman, A., Kinsey, S.,
Eden, T.O.B., Parker, C., Mitchell, C., Vora, A.,
Richards, S. and Saha, V. (2009)
Temporal changes in the incidence and pattern ofcentral nervous system relapses in children withacute lymphoblastic leukaemia treated on fourconsecutive Medical Research Council trials, 1985-2001. Leukemia, epub Dec 17.
Patel, N., Krishnan, S., Offman, M.N., Krol, M., Moss,
C.X., Leighton, C., van Delft, F.W., Holland, M., Liu,
J., Alexander, S., Dempsey, C., Ariffin, H., Essink, M.,
Eden, T.O., Watts, C., Bates, P.A. and Saha, V. (2009)
A dyad of lymphoblastic lysosomal cysteineproteases degrades the antileukemic drug L-asparaginase. J Clin Invest, 119, 1964-1973.
Tim Illidge (page 50)
Targeted Therapy Group
Refereed Research Papers
Illidge, T. and Du, Y. (2009)
When is a predose a dose too much? Blood, 113,6034-6035.
Illidge, T.M., Bayne, M., Brown, N.S., Chilton, S.,
Cragg, M.S., Glennie, M.J., Du, Y., Lewington, V.,
Smart, J., Thom, J., Zivanovic, M. and Johnson, P.W.
(2009)
Phase 1/2 study of fractionated (131)I-rituximab inlow-grade B-cell lymphoma: the effect of priorrituximab dosing and tumor burden on subsequentradioimmunotherapy. Blood, 113, 1412-1421.
Ivanov, A., Beers, S.A., Walshe, C.A., Honeychurch,
J., Alduaij, W., Cox, K.L., Potter, K.N., Murray, S.,
Chan, C.H., Klymenko, T., Erenpreisa, J., Glennie,
M.J., Illidge, T.M. and Cragg, M.S. (2009)
Monoclonal antibodies directed to CD20 and HLA-DR can elicit homotypic adhesion followed bylysosome-mediated cell death in human lymphomaand leukemia cells. J Clin Invest, 119, 2143-2159.
Other Publications
Alduaij, W. and Illidge, T.M. (2009)
Radioimmunotherapy: strategies for the future inindolent and aggressive lymphoma. Curr Oncol Rep,11, 363-370.

72 | Paterson Institute for Cancer Research Scientific Report 2009
Illidge, T., Ivanov, A. and Du, Y. (2009)
Safety and efficacy of (131) Tositumomab in thetreatment of non-Hodgkin’s lymphoma. Clinical
Medicine: Therapeutics, 1, 621-631.
Illidge, T., Cowan, R. and Parry, E. (2009)
Management of Cutaneous T-Cell Lymphoma. BMJonline.
Catharine West (page 52)
Translational Radiobiology Group
Refereed Research Papers
Donaldson, S.B., Buckley, D.L., O'Connor, J.P.,
Davidson, S.E., Carrington, B.M., Jones, A.P. and
West, C.M. (2009)
Enhancing fraction measured using dynamiccontrast-enhanced MRI predicts disease-free survivalin patients with carcinoma of the cervix. Br J Cancer,epub Nov 17.
Hedman, M., Bjork-Eriksson, T., Mercke, C., West,
C., Hesselius, P. and Brodin, O. (2009)
Comparison of predicted and clinical response toradiotherapy: a radiobiology modelling study. Acta
Oncol, 48, 584-590.
Kotz, B., West, C., Saleem, A., Jones, T. and Price, P.
(2009)
Blood flow and Vd (water): both biomarkersrequired for interpreting the effects of vasculartargeting agents on tumor and normal tissue. Mol
Cancer Ther, 8, 303-309.
Moller-Levet, C.S., Betts, G.N., Harris, A.L., Homer,
J.J., West, C.M. and Miller, C.J. (2009)
Exon Array Analysis of Head and Neck CancersIdentifies a Hypoxia Related Splice Variant ofLAMA3 Associated with a Poor Prognosis. PLoS
Comput Biol, 5, e1000571.
O'Connor, J.P., Naish, J.H., Parker, G.J., Waterton,
J.C., Watson, Y., Jayson, G.C., Buonaccorsi, G.A.,
Cheung, S., Buckley, D.L., McGrath, D.M., West,
C.M., Davidson, S.E., Roberts, C., Mills, S.J., Mitchell,
C.L., Hope, L., Ton, N.C. and Jackson, A. (2009)
Preliminary study of oxygen-enhanced longitudinalrelaxation in MRI: a potential novel biomarker ofoxygenation changes in solid tumors. Int J Radiat
Oncol Biol Phys, 75, 1209-1215.
Sillah, K., Griffiths, E.A., Pritchard, S.A., Swindell, R.,
West, C.M., Page, R. and Welch, I.M. (2009)
Clinical impact of tumour involvement of theanastomotic doughnut in oesophagogastric cancersurgery. Ann R Coll Surg Engl, 91, 195-200.
Sillah, K., Pritchard, S.A., Watkins, G.R., McShane, J.,
West, C.M., Page, R. and Welch, I.M. (2009)
The degree of circumferential tumour involvementas a prognostic factor in oesophageal cancer. Eur J
Cardiothorac Surg, 36, 368-373.
Other Publications
Barnett, G.C., West, C.M., Dunning, A.M., Elliott,
R.M., Coles, C.E., Pharoah, P.D. and Burnet, N.G.
(2009)
Normal tissue reactions to radiotherapy: towardstailoring treatment dose by genotype. Nat Rev
Cancer, 9, 134-142.
Davidson, S.E., Hendry, J.H. and West, C.M. (2009)
Point: why choose pulsed-dose-rate brachytherapyfor treating gynecologic cancers? Brachytherapy, 8,269-272.
Martin, C.J., Sutton, D.G., West, C.M. and Wright,
E.G. (2009)
The radiobiology/radiation protection interface inhealthcare. J Radiol Prot, 29, A1-A20.
West, C.M. and Davidson, S.E. (2009)
Measurement tools for gastrointestinal symptoms inradiation oncology. Curr Opin Support Palliat Care, 3,36-40.
West, C.M., Martin, C.J., Sutton, D.G. and Wright,
E.G. (2009)
21st L H Gray Conference: theradiobiology/radiation protection interface. Br J
Radiol, 82, 353-362.
Robert Hawkins (page 54)
Medical Oncology: Cell Therapy Group
Refereed Research Papers
Cheadle, E.J., Hawkins, R.E., Batha, H., Rothwell,
D.G., Ashton, G. and Gilham, D.E. (2009)
Eradication of established B-cell lymphoma byCD19-specific murine T cells is dependent on hostlymphopenic environment and can be mediated byCD4+ and CD8+ T cells. J Immunother, 32, 207-218.
Elkord, E., Dangoor, A., Burt, D.J., Southgate, T.D.,
Daayana, S., Harrop, R., Drijfhout, J.W., Sherlock, D.,
Hawkins, R.E. and Stern, P.L. (2009)
Immune evasion mechanisms in colorectal cancerliver metastasis patients vaccinated with TroVax(MVA-5T4). Cancer Immunol Immunother, 58,1657-1667.
Hawkins, R.E., Macdermott, C., Shablak, A., Hamer,
C., Thistlethwaite, F., Drury, N.L., Chikoti, P.,
Shingler, W., Naylor, S. and Harrop, R. (2009)
Vaccination of patients with metastatic renal cancerwith modified vaccinia Ankara encoding the tumorantigen 5T4 (TroVax) given alongside interferon-alpha. J Immunother, 32, 424-429.
Shablak, A., Hawkins, R.E., Rothwell, D.G. and
Elkord, E. (2009)
T cell-based immunotherapy of metastatic renal cellcarcinoma: modest success and future perspective.Clin Cancer Res, 15, 6503-6510.

Biomolecular modelling | 73Research publications | 73
Other Publications
Bridgeman, J.S., Gilham, D.E., Hawkins, R.E. and
Cheadle, E.J. (2009)
The second cellular therapy of cancer symposium,27-29 March 2009, Milan, Italy. Cancer Immunol
Immunother, epub Aug 8.
Elkord, E., Shablak, A., Stern, P.L. and Hawkins, R.E.
(2009)
5T4 as a target for immunotherapy in renal cellcarcinoma. Expert Rev Anticancer Ther, 9, 1705-1709.
Gordon Jayson (page 56)
Medical Oncology: Translational Anti-Angiogenesis Group
Refereed Research Papers
Backen, A.C., Cummings, J., Mitchell, C., Jayson, G.,
Ward, T.H. and Dive, C. (2009)
'Fit-for-purpose' validation of SearchLight multiplexELISAs of angiogenesis for clinical trial use. JImmunol Methods, 342, 106-114.
Hansen, S.U., Barath, M., Salameh, B.A., Pritchard,
R.G., Stimpson, W.T., Gardiner, J.M. and Jayson, G.C.
(2009)
Scalable synthesis of L-iduronic acid derivatives viastereocontrolled cyanohydrin reaction for synthesisof heparin-related disaccharides. Org Lett, 11,4528-4531.
O'Connor, J.P., Carano, R.A., Clamp, A.R., Ross, J.,
Ho, C.C., Jackson, A., Parker, G.J., Rose, C.J., Peale,
F.V., Friesenhahn, M., Mitchell, C.L., Watson, Y.,
Roberts, C., Hope, L., Cheung, S., Reslan, H.B., Go,
M.A., Pacheco, G.J., Wu, X., Cao, T.C., Ross, S.,
Buonaccorsi, G.A., Davies, K., Hasan, J., Thornton,
P., del Puerto, O., Ferrara, N., van Bruggen, N. and
Jayson, G.C. (2009)
Quantifying antivascular effects of monoclonalantibodies to vascular endothelial growth factor :insights from imaging. Clin Cancer Res, 15, 6674-6682.
O'Connor, J.P., Naish, J.H., Jackson, A., Waterton,
J.C., Watson, Y., Cheung, S., Buckley, D.L., McGrath,
D.M., Buonaccorsi, G.A., Mills, S.J., Roberts, C.,
Jayson, G.C. and Parker, G.J. (2009)
Comparison of normal tissue R1 and R*2modulation by oxygen and carbogen. Magn Reson
Med, 61, 75-83.
O'Connor, J.P., Naish, J.H., Parker, G.J., Waterton,
J.C., Watson, Y., Jayson, G.C., Buonaccorsi, G.A.,
Cheung, S., Buckley, D.L., McGrath, D.M., West,
C.M., Davidson, S.E., Roberts, C., Mills, S.J., Mitchell,
C.L., Hope, L., Ton, N.C. and Jackson, A. (2009)
Preliminary study of oxygen-enhanced longitudinalrelaxation in MRI: a potential novel biomarker ofoxygenation changes in solid tumors. Int J Radiat
Oncol Biol Phys, 75, 1209-1215.
Other Publications
Collinson, F. and Jayson, G. (2009)
New therapeutic agents in ovarian cancer. Curr Opin
Obstet Gynecol, 21, 44-53.
Kumaran, G.C., Jayson, G.C. and Clamp, A.R.
(2009)
Antiangiogenic drugs in ovarian cancer. Br J Cancer,100, 1-7.
Active Patent
Jayson G, Gardiner J, Hansen S.Synthesis route for multioligomeric heparan sulfatemolecules, PCT Application: PCT/ GB2009/ 000300,2009-02-04
Additional Publications
Doyle, A., Martin-Garcia, R., Coulton, A.T., Bagley, S.
and Mulvihill, D.P. (2009)
Fission yeast Myo51 is a meiotic spindle pole bodycomponent with discrete roles during cell fusion andspore formation. J Cell Sci, 122, 4330-4340.
Leverentz, M.K., Campbell, R.N., Connolly, Y.,
Whetton, A.D. and Reece, R.J. (2009)
Mutation of a phosphorylatable residue in Put3paffects the magnitude of rapamycin-induced PUT1activation in a Gat1p-dependent manner. J Biol
Chem, 284, 24115-24122.
Hey, Y. and Pepper, S.D. (2009)
Interesting times for microarray expression profiling.Brief Funct Genomic Proteomic, 8, 170-173.

74 | Paterson Institute for Cancer Research Scientific Report 2009
Seminar Series 2009
Kari Alitalo
University of Helsinki, Finland
Facundo Batista
CR-UK London Research Institute, London
Spencer Collis
University of Sheffield Medical School
Janine Erler
The Institute of Cancer Research, London
Martin Glennie
University of Southampton
Frank Grosveld
University Medical Centre Rotterdam, Netherlands
Thanos Halazonetis
University of Geneva, Switzerland
Martin Humphries
University of Manchester
Masato Kanemaki
Osaka University, Japan
Nicholas Ktistakis
Babraham Institute, Cambridge
Rolf Marschalek
Goethe University, Frankfurt, Germany
Fernando Martin-Belmonte
Centro de Biologia Molecular Severo Ochoa, Madrid
Angel Nebreda
Spanish National Cancer Research Centre (CNIO),Madrid
Chris Proud
University of Southampton
Tim Schroeder
German Research Centre for Environmental Health,Munich
Len Stephens
Babraham Institute, Cambridge
Charles Swanton
CR-UK London Research Institute (LRI), London
Simon Tavare
University of Cambridge
Marc Timmers
University Medical Centre, Utrecht, Netherlands
Marcos Vidal
CR-UK Beatson Institute, Glasgow, Scotland
Paresh Vyas
University of Oxford
A variety of national and international speakers visited the Instituteover the year, to make the seminar series the most varied it has been.The series is complimented by seminars held within The Christie andat The University of Manchester. For the first time the series was alsomerged with speakers arranged through the Breakthrough BreastCancer Unit. The postdoctoral weekly seminar series also proved tobe popular.

Biomolecular modelling | 75Seminar Series 2009 | 75
Breakthrough Breast Cancer Research Unit
Seminar Series 2009
John Bartlett
Edinburgh Cancer Research Centre
Gabriela Dontu
Department of Academic Oncology, Guy’s Hospital
Doug Easton
University of Cambridge
Nadia Harbeck
Technical University of Munich, Germany
Rita Falcioni
Regina Elena Cancer Institute, Rome
Jorge Reis-Filho
Breakthrough Breast Cancer, London
Margaret C Frame
Institute of Genetics & Molecular Medicine,University of Edinburgh
Andy Gescher
Department of Cancer Studies, University ofLeicester
Rudolf Kaaks
Division of Cancer Epidemiology, German CancerResearch Centre
Charlotte Kuperwasser
Tufts University School of Medicine, Boston
Rob Michalides
Professor of Cell Biology, The Netherlands CancerInstitute
Bill Muller
McGill University, Canada
Salvatore Pece
University of Milan
Derek Radisky
Mayo Clinic Cancer Center, USA

76 | Paterson Institute for Cancer Research Scientific Report 2009
Postgraduate Educationhttp://www.paterson.man.ac.uk/education
The Paterson Graduate Programme
The goal of the graduate programme is foreach student to receive training in scientificresearch, through a project that is bothachievable and intellectually demanding. Eachproject is peer-reviewed in advance, andmonitored throughout the PhD through amixture of talks, written reports and progressand planning meetings. These are designed notonly to provide formal points at which progress(of both the student and the project) can beassessed and goals discussed, but also to helpdevelop the presentation skills that are sofundamental to the majority of careers inscience. Graduate training is monitored by anEducation Committee, which features GroupLeaders, senior clinicians and scientists, andstudent representatives. Each student is alsoassigned an advisor (similar to a personal tutoron an undergraduate programme) whose roleis to provide impartial support and advice,while further support is also available from thePostgraduate Tutor and a Student WelfareGroup.
The Paterson runs an external seminar seriesfeaturing talks from many of the key players incancer research, and students are alsoexpected to attend postdoctoral researchseminars and to present their own work in labmeetings within the institute.
PhD Studentships
All our CR-UK funded studentships are fouryears long, and consist of an extended research
project in one or more of our research groups.We also offer rotation projects in which thefirst year consists of three shorter projects,each in a different lab, before focusing on amore extended project for the final three yearsof the PhD. Recruitment is highly competitiveand the majority of interviews are typicallyconducted over a 2-day period in lateNovember or early December.
Students benefit from access to state-of-the-art-facilities including Advanced Imaging, MassSpectrometry, Microarrays, Flow Cytometry,Histology and Next Generation Sequencing(see section on Research Services). All ourresearch groups offer PhD studentships, andprojects cover the entire breadth of researchwithin the institute.
Fellowships in Clinical Pharmacology Research
In order to help train the next generation ofclinical pharmacologists with expertise inoncology, in 2007 the Paterson Institute, incollaboration with the MCRC and AstraZeneca,established a fellowship scheme in ClinicalPharmacology Research. The fellowships areopen to applicants who have obtained, or areclose to obtaining, their Completed Certificateof Specialist Training (CCST) in MedicalOncology.
Each Clinical Pharmacology Research Fellowundertakes a three-year PhD project, whichprovides training in biomarker discovery,method development/validation, and in clinical
A thriving Graduate Programme is a fundamental aspect of aresearch Institute such as the Paterson, not simply to train theresearchers of tomorrow, but also for the valuable contributionmade by our students to the labs they are working in. In 2009,we welcomed another ten graduate students from around theworld to join our four-year PhD programme, working in fieldsas diverse as yeast genetics, stem cells DNA repair,computational genomics and clinical research.
Postgraduate Education
Manager
Julie Edwards
Postgraduate Tutor
Crispin Miller

Biomolecular modelling | 77Postgraduate Education | 77
trial methodology. During tenure at TheChristie/Paterson the post holders receive clinicalsupervision from Malcolm Ranson, and laboratory-based training from Caroline Dive in CEP (incollaboration with MCRC colleagues); atAstraZeneca they receive training in clinical trialsmanagement, regulatory interaction, translationalresearch through project management andattendance at investigator meetings, congresses andmanagement meetings. Clinical training includes oneresearch clinic per week, training in clinical trialdesign and methodology, ICH-GCP, EU Directivesand research governance. Biomarker methoddevelopment and application take place on bothsites in all projects with mutual benefit as eachFellow brings newly acquired knowledge to eachsite. Regular meetings take place between theFellows, their supervisors, as well as other staffmembers involved in the project, ensuring truecollaboration and a ‘joined up’ approach.
Education Committee 2009
Iain Hagan – ChairFiona Blackhall Richard Cowan Caroline Dive Julie Edwards David GilhamIan Hampson Tim Illidge Gordon Jayson Valerie Kouskoff Karim Labib Crispin Miller Vaskar SahaTim Somervaille Jenny Varley Catharine WestCaroline Wilkinson
Student Representatives
Monique MelisAndrzej Rutkowski

78 | Paterson Institute for Cancer Research Scientific Report 2009
Operations
The aim of the work was to brighten up theInstitute and give staff a sense of pride in theirworkplace, whilst at the same time ensuring agreater degree of sustainability. The corridorswere given new daylight lighting and a freshcoat of paint and artistic scientific images wereblown up and created on acrylic and thenaffixed to the stairwells. An artwork project –‘the Art meets Science Challenge’ was createdin conjunction with Sale Grammar Schoolwhich culminated in a prize-giving ceremony forthe winning pupils attended by the local press.The School was so pleased with the projectthat they have suggested refreshing the art intwo years time.
The Site Visit itself lasted over 2 days and wasgruelling for all concerned but the behind-the-scenes organisation was second to none andthis was due to the smooth organisational skillsof Amy Weatheritt and Esther Walker. TheReview party commended the Paterson for itsorganisation of the visit.
The Annual Staff Meeting was particularly wellattended this year as staff came to listen to theDirector and Director of Operations talk aboutthe Institute’s financial situation. The budgetreceived from CR-UK had been cut by 5%,which did not take into account the annual payaward and incremental rises, resulting in realterms of a cut of 8%. Various cost savings sub-committees were formed andrecommendations were actioned to ensure thatsavings were made. The Scientific OfficersWorking Group (SOWG) proved to beinvaluable, sitting on some of the cost-savingsub-committees and working closely with theProcurement department to negotiate the bestdeals for the Institute. It is gratifying to see thatprudent management of the budgets has
resulted in a slight surplus being projected forthe year end. This money is going to be usedto improve the ventilation facilities in two of thelaboratories that are located in the oldest partof the building. The real test of good budgetmanagement will be in 2010/2011 when thePaterson will be faced with more real termcuts.
The Operations team was joined by some newstaff during the year – Julie Jarratt, a part-timerecruitment administrator who is based withinthe HR department; Martyn Bottomley, aBusiness Manager employed by CancerResearch Technology (CRT), CR-UK’s oncologyfocused development and commercialisationcompany and Julia Wright, CR-UK’s new HighValue Donor Manager. Julia’s post is a new oneand it is the first time that CR-UK hasemployed a fundraiser to be directly basedwithin one of its Institutes. Amy Weatheritt,who had previously been employed as anadministration assistant was very deservedlypromoted into the post of PA to the Directorand Director of Operations. TheReception/security function of the Paterson hadbeen undertaken by a national company but itwas decided to bring the service back in-houseand so negotiations are currently underway toensure that the new service becomesoperational at the beginning of January 2010.
The HR department reorganised the staffinduction programme, resulting in a morestreamlined process and are going to beworking on introducing a mentoring scheme fornew starters during 2010.
The Paterson’s website was redesigned duringthe year, providing a more up-to-date websitewhich is easily accessible. The IT team have also
The first half of 2009 was focused upon the Paterson’sQuinquennial Review (Site Visit) which took place at the end ofJune. Discussions for the review started eighteen monthsearlier with the planning of the refurbishment of the Paterson’scorridors, stairways and facilities.
Director of Operations
Pippa McNichol

Biomolecular modelling | 79Operations | 79
been working with an external supplier to provide anew intranet (it has been named PICRaboo after acompetition was held amongst the staff andstudents to name it). It is anticipated that theintranet will launch in the New Year and will providemuch functionality for staff to manage their ownleave, look up polices and procedures and eacharea/interest group will have its own section; forexample there will be sections for the students, theunion, the Scientific Officers Working Group, each ofthe Service Units and Operations departments.
CR-UK introduced a new pay and grading schemefor non-scientific staff towards the end of 2009 andso the Paterson has started the formal consultationprocess with the unions. It is anticipated that thescheme will be implemented in the New Year.
It has been a really productive year and I would liketo extend my thanks to every member of theOperations team for their hard work anddedication. Particular thanks must go to MargaretLowe, my deputy, who has combined managing avery busy department, with implementing theUniversity’s new Procurement to Pay scheme, whilstably assisting me in the running of the operationsdepartment.
Admin and Reception Services
Manager: Amy Weatheritt
Over the year the department’s focus has shiftedfrom providing a catering and document productionfacility to spending more time and energies on eventsupport and organisation. The administration teamplayed a key role in the organisation of the EMBOcourse which was held in Manchester throughoutJune and also the annual Paterson Colloquium inSeptember.
The Site Visit has been the main focus of thedepartment, working alongside the rest of theoperations team to ensure that all ‘behind thescenes’ details ran smoothly. The departmentcontinues to evolve and improve, providing the bestservice possible to the Institute. Over the comingyear we hope to maintain the standards andincrease confidence in the stability of thedepartment.
The Director’s Office
PA to the Director and Director of Operations:
Amy Weatheritt
Throughout 2009, the Director’s office hascontinued to organise the Paterson Seminar Seriesto provide a varied programme of national andinternational speakers, serving to fostercollaboration and encourage positive interactionwithin the wider scientific community. The 2010series has now been confirmed and will againprovide great opportunities for the staff within theInstitute. A list of speakers for 2010 can be found atwww.paterson.man.ac.uk/seminars
The department, like the rest of the Institute hasbeen occupied with the Site Visit which was a greatsuccess. It involved a lot of hard work over the fewdays the review party were at the Institute and alsoin the run up to the event. After the Site Visit theoffice was finally able to move into the new office inTRF2, this has been a positive change and enabled afresh new start for the office coinciding with thenew term.
The office is currently working on recruitment andthe forthcoming tenure reviews due to be held atthe end of December. The Director’s office will

80 | Paterson Institute for Cancer Research Scientific Report 2009
carry on providing a supportive role to the Instituteand its Group Leaders as well as aiding the MCRCwith various meeting arrangements and hopes tocontinue building on relationships throughout theInstitute and manage the busy workload.
Estates
Manager: Steve Alcock
June 2009 saw the successful completion of therefurbishment of the Institute’s thoroughfares andmain stairwells (including the toilet facilities) in timefor the Institute Site Visit. The dated facilities havebeen given a professional look and designed to bemore sustainable with presence detectors installedto lower the lighting levels when the area has notbeen occupied for a while.
Since the review, two capital projects areprogressing and are due to be handed over to theInstitute in December 2009. These are the newDrug Discovery laboratory, which is located on theground floor opposite the administration offices, andthe replacement generator, (a 1.5MVA unit), whichwill eventually be able to supply the whole of theInstitute in the event of mains failure.
A number of minor projects have also beencompleted during 2009 which have improved theenvironment and the services for the BiologicalResearch Unit, the DNA Damage Response Group,Breast Breakthrough, the Clinical and Experimental
Pharmacology Group, the Logistics Department,Medical Oncology and the Director’saccommodation.
The Estates team has been pro-active in 2009,keeping the backlog maintenance in the Institute toa manageable level. The team has also identifiedareas around the Institute that will require somerefurbishment to be carried out in the near future.
Steve Alcock has collaborated with RichardSandland from the Estates team in the Faculty ofMedical and Human Sciences to write a report onthe activities and procedures that the Paterson’sEstates team undertake. This is to ensure that thePaterson’s Estates department is adhering to TheUniversity of Manchester’s rules and regulations. Aline manager from the University’s Estatesdepartment will be providing professional guidanceto the Paterson’s Estates’ department. This will alsosatisfy the University’s governance requirements.
The team as always endeavour to provide a qualityservice that is value for money within budgetconstraints in these challenging times of recession.
Finance and Purchasing
Manager: Margaret Lowe
Last year the University, in partnership with DeloitteConsulting, undertook a review of its procurementand payment processes. This review made a rangeof recommendations, with each one aimed atimproving the purchasing experience both for

Biomolecular modelling | 81Operations | 81
requestors of goods and services and for theUniversity’s suppliers. The Procurement to Payment(P2P) Project has been implemented throughout theUniversity over the second half of this year.Procurement has made a number of changes to theway that goods and services are bought andAccounts Payable have moved to a single sharedfunction.
As the Institute was already using internetprocurement the end users would have noticed littleor no difference and additional staff have receivedtraining as required. However, as with all newsystems this has brought its own problems and thestaff are working hard to resolve the queries thatare coming through from the University to ensuresuppliers are paid promptly.
In light of the current economic climate andreduced funding the Procurement department andthe Scientific Officer Working Group (SOWG) havebeen working together to identify savings inconsumable spending. It is envisaged that thesemeetings will continue over the coming year.
Apart from the everyday purchasing and financeprocedures the Finance department has continuedto support the research groups by providingeffective and efficient professional advice whencosting new research proposals and administeringexisting grants. As the funding to the Institute hasbeen reduced we have strived to ensure thataccurate management information is provided on atimely basis to assist group leaders with budgetcontrol.
Health and Safety
Manager: Colin Gleeson
Safeguarding the health and safety of the Paterson’sstaff is a continuing priority. Initiatives have includedthe improvement of the building access controlsystem with the introduction of proximity cardreaders and restriction of access to some areas ofthe Institute. Also the provision of OccupationalHealth was transferred to The University ofManchester, identifying employees and students andthe health surveillance programmes in which theyare enrolled.
Health and safety training has been provided in anumber of key areas including induction, riskassessment, work with biological agents andgenetically modified (GM) organisms, and COSHH.These are presented quarterly and are wellattended. Local laboratory induction training wasreviewed and developed with employeerepresentatives in the form of the Scientific OfficersWorking Group (SOWG), to standardise laboratoryinduction across the Institute. Other work with thesame group has lead to the development of animproved user friendly risk assessment form. Thiswill be rolled out in early 2010. It is hoped that
employee participation in health and safety matterswill develop further, encouraging participation andownership of health and safety issues and improvethe Institute’s health and safety culture.
Informal and formal safety inspections have beenundertaken in a number of areas, in a rollingprogramme covering the Institute. There has alsobeen a review of the Institute’s transgenic work.Both projects have revealed relatively minor issuesof non-compliance which have been or will beaddressed. The Environment Agency inspected theInstitute’s arrangements for accumulation anddisposal of radioactive substances. The Institutedemonstrated compliance with the legislativerequirements and there were no recommendationsfor any remedial action.
Some early work has been carried out in order toprepare the Institute for the introduction of theSingle Regulatory Framework for work withbiological agents. No doubt this will continue atsome pace in 2010.
HR
Manager: Rachel Powell
Over the past year the HR Department hascontinued to provide a professional proactive HRservice which has been delivered through the use ofeffective systems and processes.
The main focus for this year has been onstreamlining the recruitment and selection process.The department has expanded with the recruitmentof an HR Administrator which has enabled thedepartment to continue to meet its objectives interms of providing a high quality, effectiverecruitment service to the Institute. This hasresulted in the successful recruitment of 32 highlyskilled individuals throughout the year. Thedepartment has also had to become familiar withthe new points-based system for immigration inorder to advise and provide guidance to managersand employees on the new legislation.
Joint partnership working with the unions hascontinued throughout the year which has resulted inthe agreement of several revised policies includingPay Protection Policy and Probation Policy.
The HR department has recently been involvedwith the successful transfer of the Paterson’sOccupational Health services to The University ofManchester. This involved consulting with theOccupational Health providers and the staff toensure a smooth transition without any interruptionto the service.
Moving forward, the department is due to launch anew HR section on the Institute’s Intranet which willenhance and complement the high class service thatthe HR department currently provides.

82 | Paterson Institute for Cancer Research Scientific Report 2009
IT
Manager: Malik Pervez
Information technology, electronic communicationand network connectivity have become crucial tothe success of the Paterson Institute. Over theyears a substantial investment in IT has been madeto provide a world class IT infrastructure. TheInstitute has an enviable platform and range oftechnologies.
The IT team are highly trained to support allapproved systems and provide excellent support tothe researchers within the organisation. Thesystems are protected 24/7 by robust technologiesensuring that data is not compromised and adisaster recovery solution ensures that in the eventof a catastrophic event systems can be reinstatedrapidly.
Continuing to build upon this success year on yearensures that the Institute stays ahead of the game interms of the technology required to support theresearch and development and 2009 has seen theInstitute add to the level of technology via a numberof investments and developments.
A wireless network has been implemented acrossthe organisation to access and provide a muchmore flexible working environment. Furthermore,additional storage and archiving facilities have beenprocured and deployed to meet the growth in datarequirement. The Institute’s website has recentlyundergone a major facelift. The Institute also has
plans to develop a new Intranet for staff to improveinternal communication and access to shareddocuments which will be developed during 2010.
A further development planned for 2010 will be toimplement video conferencing facilities to improveexternal communication with other scientificorganisations. This feature will also create savings asit will reduce the need for staff to travel to somemeetings as these can be undertaken from theInstitute.
Cancer Research Technology (CRT)
Manager: Martyn Bottomley
Cancer Research Technology is a specialist oncology-focused development and commercialisationcompany. CRT aims to maximise patient benefitfrom publicly funded research worldwide byadvancing research discoveries into developmentwith pharmaceutical and biotechnology parties.CRT is wholly owned by Cancer Research UK.
CRT bridges the fundamental gap between cuttingedge academic research and industrial developmentof cancer therapeutics and diagnostics. This isachieved by working closely with prestigious,international research institutes, such as thePaterson, and funding bodies to develop, protectand commercialise oncology related discoveries.Core activities of business development and drugdiscovery are supported by specialists, integrated inthe business with expertise in patents, legal, financeand marketing. CRT’s exclusive focus in oncology

Biomolecular modelling | 83Operations | 83
provides an unrivalled depth of knowledge andexperience in cancer-specific translationaldevelopment and commercialisation. Byarrangement with The University of Manchester,CRT owns and is responsible for the developmentand commercialisation of intellectual property arisingfrom Cancer Research UK funded research at TheUniversity of Manchester (including the Paterson).Proceeds are shared with inventors and theUniversity, whilst surplus CRT revenues are returnedto Cancer Research UK to support further cancerresearch. The company is well resourced to achieveits mission, with headquarters and established drugdiscovery labs in London and Cambridge (UK), a USsubsidiary in Boston and a collaborative drugdiscovery vehicle in Australia.
CRT has an enviable reputation for the productivenature of its academic and commercial partnerships.CRT partnered therapeutics are currently marketedby AstraZeneca (Tomudex), Schering-Plough(Temozolomide) and a third product wasprogressed to market by Pfizer (Zinecard). Morethan 20 partnered therapeutics are currently inclinical development, with 5 in Phase III clinical trialsincluding ANYARA, being developed by ActiveBiotech (Phase III) and stemming from CancerResearch UK funded work at the Paterson(Professor Peter Stern).
CRT’s relationship with the Paterson reflects thespecific requirements of the scientist, the Paterson,Cancer Research UK and the individual project. Toeffectively facilitate these requirements andinteractions CRT has a Business Manager (MartynBottomley) based at the Paterson dedicated toworking closely with the staff there. CRT has alarge and broad portfolio of developmentprogrammes and robust licensing opportunities(including some originating from the Paterson) thatcontinue to attract commercial partners who excelin their field.
CR-UK Fundraising
Manager: Julia Wright
This year Cancer Research UK expanded itsfundraising capacity in the North West byintroducing a new fundraising role, based at thePaterson, to generate large private donations forcancer research projects in the North West. Thisnew role will focus on raising money from localpeople and charitable trusts as part of CR-UK'sinitiative to establish 20 'Centres of Excellence'across the UK. The Paterson has been fortunate toreceive a number of high-value donations fromindividuals and organisations in 2009 and this newpost will enable CR-UK to secure more support ofthis kind.

84 | Paterson Institute for Cancer Research Scientific Report 2009
Cancer Research UK’s Local Engagement andDevelopment
In Manchester the Local Engagement andDevelopment (LEAD) department has workedhard to support our fundraising teams byfacilitating interactions between the Instituteand Cancer Research UK’s supporters. LEADplays an important role in helping to retain andbuild continuing loyalty with these supportersby showing them tangible examples of howtheir money is being spent and the impact thatit is having.
Over the last 12 months there has beenaround fifty engagement activities supportedby researchers from the Institute. Theseinclude attendance at a wide variety ofexternal fundraising events (such as Race forLife, Relay for Life and fundraising committeeAGMs) as well as hosting events within theInstitute. Following one of our monthly labtours Roslyn Burgess (a Cancer Research UKshop manager) said:
“It has made me more determined to beat ourshop target each week in order to be able tofund such brilliant work”.
The Institute also hosts an annual schools dayand the aim of this is to engage with localschools and colleges, giving students a practicalinsight into a potential career in cancerresearch. This is the fifth year the event hastaken place with eight local schools bringingfifty students to the Institute. Nick Snowden,biology head at Manchester College Schoolsaid: "I think it’s fantastic for the students tohave the opportunity to visit such a researchfacility and it provided excellent support forvarious parts of the A-Level syllabus. A mostuseful day."
Involvement in Cancer Research UK activitiesalso extended to hands-on fundraising with theInstitute raising £4,000 through theirparticipation in Stockport Relay for Life and
Researchers at the Paterson Institute have once again excelledin engaging the public with science. Through a series of labtours, supporter open days and other activities in support ofCancer Research UK’s fundraising events, researchers talked toand inspired thousands of people about their work and how itimpacts on cancer.
LEAD Manager
James Dunphy
Figure 1
Cancer Research UK Chief
Executive Harpal Kumar with
Andrew Lansley MP, Shadow
Secretary of State for Health,
and Mark Simmonds MP,
Shadow Minister of State for
Health at the Paterson Institute

the arduous 40 mile Keswick to Barrow walk.The Relay team donned lab coats for the full 24hours of the event and thoroughly enjoyed theirexperience. During the afternoon they tooktime to conduct strawberry DNA extractionexperiments for the other participants. This is aninnovative way of engaging a wide audience inscience and provides an opportunity for peopleto hear more about the work at the Institute.This successful engagement activity wasreplicated at Manchester Race for Life in HeatonPark. Here a team of postdoctoral fellows andgraduate students delivered an impressive 110demonstrations to around 450 supporters -including a four year old and the Lord Mayor ofManchester.
This year the Institute also helped CancerResearch UK’s Policy and Public Affairs teamorganise a tour for two senior Conservative MPs(Andrew Lansley MP, Shadow Secretary of Statefor Health, and Mark Simmonds MP, ShadowMinister of State for Health). They visited duringthe Conservative Party Conference inManchester and met with Harpal Kumar, ChiefExecutive of Cancer Research UK, and ProfessorNic Jones. Harpal Kumar said:
“It is crucial that politicians and decision makersare aware of Britain’s place at the forefront of
medical research worldwide and that they helpus maintain our research excellence. I am verypleased therefore that Andrew Lansley and MarkSimmonds have been able to visit our Instituteto find out not just about the work weundertake but also about how we’re planning forthe future.”
The success of local engagement activity inManchester has resulted in the city beingselected to host an exciting new fundraisinginitiative. In 2010, Manchester will be hostingCancer Research UK’s first ever night-timewalking marathon called ‘Shine’. It is expected5000 men and women will take part to raiseover £1 million for the Charity. The route willtake participants past the Institute and plans arein place to ensure that all participants are giventhe opportunity to engage with their localresearchers and learn more about the researchundertaken in Manchester.
LEAD is a key strand in Cancer Research UK'sCentres Initiative, which is an important strategiccomponent of the Charity’s drive to achieve its2020 Goals. The excellent activity undertaken inManchester is now being replicated across theUK, with LEAD Managers now in Glasgow,Belfast, Cardiff, Birmingham, Southampton andNewcastle.
Cancer Research UK’s Local Engagement and Development | 85
Figure 2
A team of Paterson researchers
helping out at Manchester’s
Race for Life
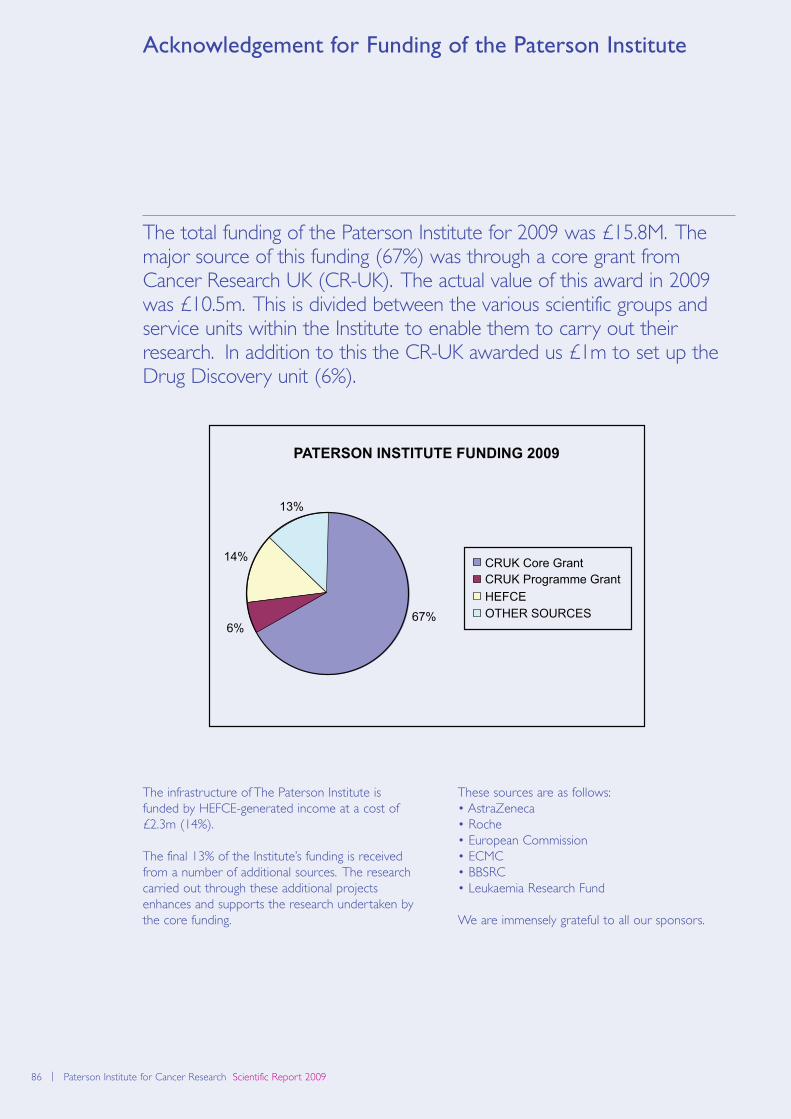
86 | Paterson Institute for Cancer Research Scientific Report 2009
Acknowledgement for Funding of the Paterson Institute
The total funding of the Paterson Institute for 2009 was £15.8M. Themajor source of this funding (67%) was through a core grant fromCancer Research UK (CR-UK). The actual value of this award in 2009was £10.5m. This is divided between the various scientific groups andservice units within the Institute to enable them to carry out theirresearch. In addition to this the CR-UK awarded us £1m to set up theDrug Discovery unit (6%).
The infrastructure of The Paterson Institute isfunded by HEFCE-generated income at a cost of£2.3m (14%).
The final 13% of the Institute’s funding is receivedfrom a number of additional sources. The researchcarried out through these additional projectsenhances and supports the research undertaken bythe core funding.
These sources are as follows:• AstraZeneca• Roche• European Commission• ECMC• BBSRC• Leukaemia Research Fund
We are immensely grateful to all our sponsors.
13%
14%
6%67%
PATERSON INSTITUTE FUNDING 2009
CRUK Core GrantCRUK Programme GrantHEFCEOTHER SOURCES

Biomolecular modelling | 87Career Opportunities at the Paterson Institute | 87
The Manchester Cancer Research Centre (MCRC)was created nearly four years ago with partnersincluding the Paterson Institute, The ChristieHospital NHS Foundation Trust, The University ofManchester and Cancer Research UK. This is anextremely exciting development which is enhancingall aspects of cancer research, education andtreatment. The Institute offers excellent laboratoryfacilities and outstanding core facilities, includingmolecular services, a microarray platform,proteomics, flow cytometry, histology, the productionof knock-in/knock-out animal models, real-time PCRand advanced imaging. Details of all groups andfacilities are given throughout this report, and canguide interested parties to the appropriate contacts.Opportunities exist at a number of levels in theInstitute. We have a well-established programme ofdegrees by research which is described in thesection on Postgraduate Education. We encourageapplications from suitable qualified graduates toapply to join either the PhD or MD programmes.Graduates with a first or 2.1 honours degree in abiological science can apply each year to train for afour-year PhD in one of our research laboratories.First year students will complement their laboratoryskills by attending a small number of specialisedpostgraduate taught and training courses allowingthem to gain a sound knowledge base of the latestdevelopments in cancer treatment and research.The Institute also has a well-developed process forensuring suitable pastoral care and mentoring for allstudents.
Postdoctoral applicants of high calibre are regularlysought. Although post docs will be encouraged toapply for their own fellowships, funded positions areavailable for outstanding candidates. Interestedapplicants should contact the Group Leadersdirectly, with details of their area of interest andrecent experience.
In addition to postgraduate and postdoctoralopportunities, the Institute is still seeking to recruitoutstanding candidates to the positions of Junior andSenior Group Leaders. The packages provided areextremely attractive and commensurate with theexperience of the applicant, with significant fundingfor personnel, recurrent expenditure andequipment. Junior Group Leaders are appointed foran initial six-year period with a review at five yearsfor consideration for promotion to Senior GroupLeader, with Senior Group Leaders appointed tonon-time limited positions.
Specific vacancies can be found on our web pages(http://www.paterson.man.ac.uk/jobs/index.asp), butsuitably qualified and enthusiastic individuals shouldcontact the Institute at any time to enquire aboutcareer possibilities.
The Paterson Institute is located alongside The Christie NHS FoundationTrust, and has a strong programme of basic and translational research.There are very close links with clinical and translational research groupsthroughout The Christie Hospital site.
Career Opportunities at the Paterson Institute
88 | Paterson Institute for Cancer Research Scientific Report 2009
Contact details
Paterson Institute for Cancer Research
Wilmslow RoadManchester M20 4BXUnited Kingdom
Tel +44(0) 161 446 3156Fax +44(0) 161 446 3109
www.paterson.man.ac.uk
Electronic version of this report can befound at: www.paterson.man.ac.uk

ISSN 1479-0378
Copyright © 2009 Cancer Research UK
Printed by Paramount Print

Paterson Institute for Cancer Research
Wilmslow Road
Manchester
M20 4BX
Tel +44(0) 161 446 3156
www.paterson.man.ac.uk


















