Genetic Steroid Disorders || Apparent Mineralocorticoid Excess
7
Genetic Steroid Disorders. http://dx.doi.org/10.1016/B978-0-12-416006-4.00017-X Copyright © 2014 Published by Elsevier Inc. 239 BACKGROUND Steroid 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) is one of the key enzymes in maintaining blood pressure homeostasis. It is the metabolic enzyme that confers specificity to the mineralocorticoid receptor. This relationship is evident in the syndrome of apparent mineralocorticoid excess. Steroid 11β-HSD2 is one of two distinct isozymes of 11β-hydroxysteroid dehydrogenases that catalyze the interconversion of hormonally active cortisol to inactive cortisone. Steroid 11β-HSD2 functions unidirectionally and selectively on cortisol. With the use of in situ hybridization and immunohistochemistry, it was found predominantly in mineralocorticoid respon- sive tissues: kidney, colon, and salivary gland. The type 1 enzyme (11β-HSD1) is more widely distributed, with the highest expression in the liver and adipose tissue, and functions to enhance organ-specific effects of cortisol. It functions bidirectionally and has a lower affinity for cortisol [1]. The two isoforms share only 20% sequence homology, suggesting that they belong to different gene families. Apparent mineralocorticoid excess (AME) is the rare inherited form of hypertension caused by 11β-HSD2 deficiency. Clinical manifestations of AME mimic those of excessive mineralocorticoid states, but no elevation of known mineralocorticoids exists in these patients. Although Werder et al. described the clinical features of AME in 1974, the first biochemical and hormonal description of the disorder was made in 1977 by New et al. in a 3-year-old Native American girl with severe hypertension [2]. Endocrine evaluation of the patient did not reveal excessive secretion of known endogenous salt-retaining steroids in mineralocorticoid group. Ini- tially, it was thought that this condition was caused by an unknown mineralocorticoid. By measuring the release of tritiated [11- 3 H] water after tritiated [11- 3 H] cortisol infusion, it was found that the metabolism of cortisol to biologically inactive cortisone was decreased and that the serum cortisol half-life was prolonged in these cases. This finding implicated diminished activity of 11β-HSD2, the enzyme responsible for converting cortisol to cor- tisone. The exploration and elucidation of this disease opened a new area in receptor biology as a result of the demonstration that the specificity of the mineralocorti- coid receptor function depends on a metabolic enzyme (11β-HSD2) rather than the receptor itself [3–7]. The natural ligand of mineralocorticoid receptor, aldosterone, is the primary mineralocorticoid respon- sible for regulating electrolyte excretion and intravascu- lar volume. Aldosterone stimulates increased resorption of sodium from the urine at the mineralocorticoid tar- get tissues, the cortical collecting duct in the kidneys. The mineralocorticoid receptor is non-selective in vitro and binds both aldosterone and cortisol, a glucocorti- coid, with similar affinity [8,9]. The 11β-HSD2 enzyme functions to protect the mineralocorticoid receptor by converting cortisol to its inactive metabolite, cortisone. This inactivation occurs through the conversion of the hydroxyl group at position C 11 to a C 11 oxo group. This is one of the pathways involved in cortisol metabolism. Other enzymatic pathways are shown in Figure 6A.1. Both cortisol and cortisone metabolites are excreted through urine. Since cortisone is unable to bind the mineralocorticoid receptor, this enables aldosterone to selectively interact with the mineralocorticoid receptor in vivo [10,11]. Aldosterone, itself, is not metabolized by 11β-HSD2 because it forms a C 11 –C 18 hemi-ketal group in aqueous solution. Loss of function of the 11β-HSD2 enzyme allows cortisol to bind to the mineralocorticoid receptor and act as a mineralocorticoid. As cortisol is secreted in CHAPTER 6A Apparent Mineralocorticoid Excess Mabel Yau, Saroj Nimkarn New York-Presbyterian Hospital/Weill Cornell Medical College, New York, NY 10065
Transcript of Genetic Steroid Disorders || Apparent Mineralocorticoid Excess

Genetic Steroid Di
C H A P T E R
6AApparent Mineralocorticoid Excess
Mabel Yau, Saroj NimkarnNew York-Presbyterian Hospital/Weill Cornell Medical College, New York, NY 10065
BACKGROUND
Steroid 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) is one of the key enzymes in maintaining blood pressure homeostasis. It is the metabolic enzyme that confers specificity to the mineralocorticoid receptor. This relationship is evident in the syndrome of apparent mineralocorticoid excess. Steroid 11β-HSD2 is one of two distinct isozymes of 11β-hydroxysteroid dehydrogenases that catalyze the interconversion of hormonally active cortisol to inactive cortisone. Steroid 11β-HSD2 functions unidirectionally and selectively on cortisol. With the use of in situ hybridization and immunohistochemistry, it was found predominantly in mineralocorticoid respon-sive tissues: kidney, colon, and salivary gland. The type 1 enzyme (11β-HSD1) is more widely distributed, with the highest expression in the liver and adipose tissue, and functions to enhance organ-specific effects of cortisol. It functions bidirectionally and has a lower affinity for cortisol [1]. The two isoforms share only 20% sequence homology, suggesting that they belong to different gene families.
Apparent mineralocorticoid excess (AME) is the rare inherited form of hypertension caused by 11β-HSD2 deficiency. Clinical manifestations of AME mimic those of excessive mineralocorticoid states, but no elevation of known mineralocorticoids exists in these patients. Although Werder et al. described the clinical features of AME in 1974, the first biochemical and hormonal description of the disorder was made in 1977 by New et al. in a 3-year-old Native American girl with severe hypertension [2]. Endocrine evaluation of the patient did not reveal excessive secretion of known endogenous salt-retaining steroids in mineralocorticoid group. Ini-tially, it was thought that this condition was caused by an unknown mineralocorticoid. By measuring the release
sorders. http://dx.doi.org/10.1016/B978-0-12-416006-4.00017-X 23
of tritiated [11-3H] water after tritiated [11-3H] cortisol infusion, it was found that the metabolism of cortisol to biologically inactive cortisone was decreased and that the serum cortisol half-life was prolonged in these cases. This finding implicated diminished activity of 11β-HSD2, the enzyme responsible for converting cortisol to cor-tisone. The exploration and elucidation of this disease opened a new area in receptor biology as a result of the demonstration that the specificity of the mineralocorti-coid receptor function depends on a metabolic enzyme (11β-HSD2) rather than the receptor itself [3–7].
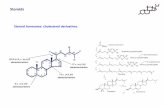
The natural ligand of mineralocorticoid receptor, aldosterone, is the primary mineralocorticoid respon-sible for regulating electrolyte excretion and intravascu-lar volume. Aldosterone stimulates increased resorption of sodium from the urine at the mineralocorticoid tar-get tissues, the cortical collecting duct in the kidneys. The mineralocorticoid receptor is non-selective in vitro and binds both aldosterone and cortisol, a glucocorti-coid, with similar affinity [8,9]. The 11β-HSD2 enzyme functions to protect the mineralocorticoid receptor by converting cortisol to its inactive metabolite, cortisone. This inactivation occurs through the conversion of the hydroxyl group at position C11 to a C11 oxo group. This is one of the pathways involved in cortisol metabolism. Other enzymatic pathways are shown in Figure 6A.1.
Both cortisol and cortisone metabolites are excreted through urine. Since cortisone is unable to bind the mineralocorticoid receptor, this enables aldosterone to selectively interact with the mineralocorticoid receptor in vivo [10,11]. Aldosterone, itself, is not metabolized by 11β-HSD2 because it forms a C11–C18 hemi-ketal group in aqueous solution.
Loss of function of the 11β-HSD2 enzyme allows cortisol to bind to the mineralocorticoid receptor and act as a mineralocorticoid. As cortisol is secreted in
Copyright © 2014 Published by Elsevier Inc.9

6A. APPARENT MINERALOCORTICOID EXCESS240
5α-dihydrocortisol
5β-dihydrocortisone
6β-hydroxylase5β-reductase
5β-reductase
20β-oxoreductase
Cortisone
Cortisol
6β-hydroxycortisone
6β-hydroxycortisol6β-hydroxylase/CYP3A
20β-dihydrocortisone
5β-dihydrocortisol
Tetrahydrocortisone
5β-tetrahydrocortisol
20β-dihydrocortisol20β-oxoreductase
5α-reductase
3α-hydroxysteroiddehydrogenase
3α-hydroxysteroiddehydrogenase
3α-hydroxysteroiddehydrogenase
11β-hydroxysteroiddehydrogenase
5α-tetrahydrocortisol
FIGURE 6A.1 Metabolization of cortisol. Cortisol is predominantly metabolized in the liver. The main pathway of cortisol inactivation oc-curs through the conversion of the hydroxyl group at position C11 to a C11 oxo group by the 11β-hydroxysteroid dehydrogenase type 2 enzyme. This functions to protect the mineralocorticoid receptor by converting cortisol to its inactive metabolite, cortisone. Other pathways of cortisol metabolism are shown. The majority of excreted cortisol appears in the urine as 5α-tetrahydrocortisol, 5β-tetrahydrocortisol, and tetrahydrocor-tisone. Adapted from Williams [14].
concentrations approximately 1000-fold higher than aldosterone, cortisol saturates the mineralocorticoid receptor in patients with 11β-HSD2 deficiency. This results in the clinical phenotype of excess mineralo-corticoid activity. Thus, AME illustrates an important “pre-receptor” pathway in the analysis of corticosteroid hormone action [12].
The role that 11β-HSD2 plays in mineralocorti-coid receptor specificity was also noted in the study of the effects of the licorice plant, Glycyrrhiza glabra, extracts, and its derived drug, carbenoxolone. Licorice and carbenoxolone are used in the treatment of gas-tric ulcers. Licorice ingestion in large amounts (greater than 50 g/day) can cause blood pressure elevation, sodium retention, and potassium wasting, similar to symptoms seen in AME. These symptoms tend to be milder than those of AME. Endocrine evaluation of patients with licorice ingestion also shows low plasma renin activity and low aldosterone concentrations. This effect was first documented in the 1940s [1]. The active components in licorice, glycyrrhizic acid and its hydrolytic product glycyrrhetinic acid, have a low affinity for the mineralocorticoid receptor. However, they act as potent competitive inhibitors of 11β-HSD2. This condition is reversible with discontinuation of licorice ingestion. In addition to licorice, glycyrrhizic acid can also be found in chewing gums and tobacco as a sweetening agent.
AME was thought to exist only in a severe form until 1998 when an unusual patient with a mild form of AME was reported by Wilson et al. [13]. The patient was a 12-year-old American girl with asymptomatic, mild, low
renin hypertension diagnosed during a sport physical. A moderately abnormal ratio of cortisol-to-cortisone metabolites and a mildly increased half-life of cortisol by measurement of tritiated water release after tritiated cor-tisol infusion led to the diagnosis. The findings of milder phenotype patients with partial functional 11β-HSD2 suggest the existence of a continuum between a caricatu-ral form of AME due to mutations drastically affecting the activity of the enzyme and more subtle genetic alter-ations resulting in milder phenotypes, which are labeled “essential hypertension”.
It has been speculated that a mild form of 11β-HSD2 defect may exist in a subgroup of individuals with essen-tial hypertension since a substantial percentage demon-strate low renin activity and/or are salt-sensitive. Indeed, several studies showed that reduced 11β-HSD2 activity might be a factor in a subset of patients diagnosed with essential hypertension.
MOLECULAR GENETICS
AME is an autosomal recessive monogenic disorder. The syndrome is caused by non-functional mutations in the HSD11B2 gene. The gene is located on the long arm of chromosome 16 (16q22) and is approximately 6 kb in length containing five exons. In 1995, the first muta-tion in the HSD11B2 gene was discovered in a consan-guineous Iranian family with three siblings suffering from AME [15]. Subsequently, more than 40 causative mutations in the HSD11B2 gene have been identified in patients affected with AME [16]. As seen in Figure 6A.2,

CliniCAl PREsEnTATion And diAgnosis 241
R74GP75,∆ 1nt
C-T, nt14A-G, nt1
771C>GL114,∆6nt
C1393T
1 2 3 4 5
A221VL170RS180FR186CR208C
R208H;R337H,∆3ntR213C
D223NY226NP227LY232C
Y232L,∆9ntA237VD244N
F246,+1ntL250RL250PL251SC771G
R279C∆299
N286∆1ntG305∆11nt
A328VV332,ins9nt
R337CR337H, ∆Y338
E356,∆1ntR359WR374XL376P
FIGURE 6A.2 Steroid 11β-HSD2 gene and its mutations. Mutations leading to AME in the HSD11B2 gene on the long arm of chromosome 16 are distributed in all five exons and also in the intron regions of the gene. Most mutations are located in exons 3,4, and 5. Modified from ref. [17].
the mutations are distributed in all five exons and also in the intron regions of the gene.
Correlations between the severity of the enzymatic defects and clinical presentation have been found [18,19]. Most of these mutations have been characterized by in vitro expression studies and have been shown to have varying degrees of enzyme activity. The mutations can be classified as “severe” or “mild,” as shown in Table 6A.1. Severe mutations have a very low enzyme activity, of broadly <10%. Two mild mutations were identified in patients with the clinical markers of AME (low aldoste-rone, hypertension, abnormal ratio of urinary free corti-sol [UFF]/urinary free cortisone [UFE]); in vitro studies demonstrated a very mild alteration in enzyme activity [4,20].
CLINICAL PRESENTATION AND DIAGNOSIS
AME typically presents early in life. In addition to hypertension, clinical features may include failure to thrive, poor growth, polydipsia, polyuria, and muscle weakness. Also, birthweights are significantly lower than those of their unaffected siblings [31]. Biochemical pro-files demonstrate metabolic alkalosis and severe hypo-kalemia. Insidious end-organ damage may arise at an early age because of chronic elevation of blood pressure and metabolic derangements, particularly hypokalemia. Renal, neurological, cardiovascular, and ocular systems
are particularly sensitive to damage. Left-ventricular hypertrophy is the most common cardiac abnormality seen. Renal manifestations include nephrocalcinosis, hypercalciuria, and renal insufficiency. Severe hypoka-lemia can lead to nephrogenic diabetes insipidus mani-festing as polydipsia and polyuria. Paralysis, respiratory failure, muscle weakness, and cardiac arrhythmias can also result from hypokalemia.
Pubertal delay is observed in all severe AME patients where data is available. Puberty is delayed secondary to poor weight gain, growth failure, and delayed bone age. The delayed puberty observed in patients with AME is unrelated to the genetic disorder, but is attributable to failure to thrive that is the result of the genetic disorder.
In evaluating a hypertensive child, it is important for clinicians to utilize proper tools to measure and inter-pret the blood pressure (BP) readings. The preferred method of BP measurement is auscultation using a mer-cury sphygmomanometer connected to the appropriate size cuff. Systolic blood pressure (SBP) is determined by the onset of the “tapping” Korotkoff sounds (K1) while diastolic blood pressure (DBP) is defined as the fifth Korotkoff sound (K5), or the disappearance of Korotkoff sounds. Use of automated devices can be used for BP measurement in newborns and young infants, in whom auscultation is difficult. An elevated BP reading obtained with an oscillometric device should be repeated with aus-cultation. To determine percentile of BP, values are com-pared to normal BP in children and adults adjusted for age, sex, and height. Hypertension is defined as average

6A. APPARENT MINERALOCORTICOID EXCESS242
TABLE 6A.1 Mutations in 11β-Hsd2 gene Classified as severe or Mild
Mutation
In vitro percentage activity conversion of cortisol to cortisone
Expression system Ref
SEVERE MUTATIONS AS DEFINED BY EXPRESSION STUDIES
E356-1 frameshift
0 CHO cell [21]
R337H, ΔY338 0 CHO cell [22]
R186C 0 CHO cell [21]
L250P, L251S
0 CHO cell [21,22]
R208C 7 CHO cell [22]
R337C 0 CHO cell [22,23]
Δ299 0 CHOP cell [24]
Y226N 2 HEK-293 cell [25]
L114, Δ6nt Near 0 HEK-293 cell lysate
[26]
L179R 0 CHO whole cell [19]
R208H 0 CHO whole cell [19]
S180F 1.8 CHO cell lysate [19]
A237V 3.1 CHO cell lysate [19]
A328V 0.6 CHO cell lysate [19]
R208H 0 HeLa cells and COS-1 cells
[27]
R213C <5 CHO cell [28]
A328V <5 CHO cell [28]
R208C 4 CHO cell [22]
R213C 7 CHO cell [22]
Y232C 1.5 HEK-293 [29]
R359W 5 HEK-293 [29]
L376P 9 HEK-293 [29]
SEVERE MUTATIONS AS BASED ON PREDICTION MODELS
R74G Predicted to generate stop codon leading to a truncated protein
ND [12]
R75Δ1nt Predicted to generate stop codon leading to a truncated protein
ND [12]
A221V Prediction of pre-mRNA splicing site interference
ND [12]
Ivs3+1G>A Destroy donor splice site sequence
ND [29]
Y232Δ9nt Completely inactive due to abolish catalytic site
ND [22]
SBP and/or DBP that is ≥95th percentile for gender, age, and height on three or more occasions. Elevated BP must be confirmed on repeated visits before characterizing a child as having hypertension.
All of the AME patients reported until 1998 had the characteristic signs of severe AME. In 1998 the first patient with a mild form of AME [32] associated with mutations that resulted in an 11β-HSD2 protein with attenuated activity was reported. Subsequently, addi-tional patients have been reported [29]. Table 6A.2 indi-cates the difference in phenotype in the mild and severe forms of AME. Only three clinical features of mild AME are the same as in severe AME: low renin activity, low aldosterone concentrations, and elevated ratio of tetra-hydrocortisol (THF) + allo-THF/tetrahydrocortisone (THE).
In AME, low plasma renin activity (PRA) suggests a volume-expansion hypertension, which responds to dietary sodium restriction. All steroid concentrations, including aldosterone, are very low. The biochemical diagnosis of AME is made by indirectly measuring the ratio of cortisol to cortisone through the ratios of their uri-nary metabolites. As shown in Figure 6A.1, cortisol can be metabolized to tetrahydrocortisol and allotetrahydro-cortisol independent of 11β-HSD2 activity and excreted in urine. Cortisone is further metabolized to tetrahydro-cortisone by 5β-reductase and 3α-hydroxysteroid dehy-drogenase. The metabolite ratios are calculated, each reflecting a different aspect of enzyme function. The ratio of tetrahydrocortisol (THF) + allo-THF/tetrahy-drocortisone (THE) represents global function of HSD [33]. The ratio of urinary free cortisol (UFF)/urinary free cortisone (UFE) represents kidney HSD function [34].
Mutation
In vitro percentage activity conversion of cortisol to cortisone
Expression system Ref
771 C>G Aberrant splicing ND [29]
R374X Premature stop codon ND [30]
N286-1 frameshift
Extrapolated from E356-1 frameshift
ND [31]
D224N Algorithm developed by Chou and Fasman prediction
ND [31]
MILD MUTATIONS AS BASED ON EXPRESSION STUDIES
P227L Altered enzyme kinetics (Km increased by 5)
CHOP cell and homogenate
[15]
R279C Altered enzyme kinetics in homogenate (Vmax reduced by 33%)
CHOP homogenate
[20]
TABLE 6A.1 Mutations in 11β-Hsd2 gene Classified as severe or Mild — (cont’d)

ARy 243
suMMElevated ratios are consistent with 11β-HSD2 enzyme dysfunction or deficiency.
Originally, AME was described through the plasma half-life of tritiated [11-3H] cortisol which, when metabo-lized by 11β-HSD2, yields tritiated water and cortisone. The plasma cortisol half-life is prolonged at 120–190 min-utes compared to a half-life of 70–90 minutes in controls. This may have more accurately reflected renal 11β-HSD2 activity [35]. However, this diagnostic method, which required administration of radioisotopes, has been dis-continued in clinical practice.
TREATMENT
The treatment of AME is focussed on the correction of hypokalemia and hypertension. Spironolactone, a min-eralocorticoid receptor antagonist, is the medication of choice. Pharmacologically, it binds competitively and protects the receptors against any excess mineralocorti-coid activity. This competitive inhibition can be overcome by increased cortisol concentrations secreted in response to stress. Follow-up studies of AME patients treated with
TABLE 6A.2 Clinical Parameters Classifying severe Versus Mild Forms of AME
Clinical parameter Severe AME Mild AME
Birth weight (kg) Low (1.8–2.6) Normal (2.7–3.6)
Age at diagnosis (years)
1–14 12.6–36
Height and weight Less than the 3rd percentile for age
Normal growth parameters
Blood pressure (mmHg)
Very elevated (systolic155–250/ diastolic 90–180)
Slightly elevated (systolic 148–155/diastolic 80–92)
Kidney function Abnormal Normal
Puberty Delayed Normal
BIOCHEMICAL FEATURES
Serum potassium (mmol/L)
1.7–3.5 3.1–5.0
Urinary cortisol/cortisone metabolites [THF+5αTHF/THE]
Markedly elevated (6.7–33.0)
Moderately elevated (3.0–5.6)
Percentage conversion of F to E*
0–6.0 31–58.4
Renin Low renin (<0.12–7.9 ng/mL/h)
Low renin (0.14–0.7 ng/mL/h)
Aldosterone Low to absent aldosterone (undetectable –6 ng/dL)
Low to absent aldosterone (undetectable)
* Test no longer done.
spironolactone revealed significant improvement in clinical symptoms over a 13-year period. This suggests that early diagnosis and adequate treatment are impor-tant [4,31]. A reduction in dietary sodium and potassium supplementation has also been shown to be beneficial. Although other antihypertensive medications can be used, spironolactone and potassium supplement remain the main treatments for AME. In patients with hypercal-ciuria and/or nephrocalcinosis, a thiazide diuretic may be added. Loop diuretics should be used cautiously as they can aggravate hypokalemia and alkalosis.
Additional treatment may be required to address hypertensive end-organ damage. However, despite treatment that lowers blood pressure, some patients suf-fer severe consequences of AME, within years of diag-nosis. The mortality is more than 10%, due to stroke, cerebral hemorrhage, and infarction [33].
Dexamethasone has been used, but is not effective in all. Dexamethasone is a synthetic glucocorticoid similar to cortisol and therefore suppresses endogenous cortisol production by the adrenal glands. It lowers blood pres-sure to normal in 60% of patients. However, dexametha-sone has been shown to aggravate AME in some patients by worsening hypertension and potassium wasting.
Renal transplantation successfully reversed the clini-cal symptoms of AME and normalized cortisol metab-olism in two AME patient [36,37]. In 2013, a patient followed from birth through childhood with AME received a renal transplant from his wife at age 33. The patient was cured of AME with reduction in blood pres-sure, remission of hypokalemic alkalosis, and reversal of the ratio of cortisol/cortisone metabolites [37]. Although not considered standard of care, renal transplantation in early adolescence should be considered as a therapeutic strategy to prevent terminal renal failure.
SUMMARY
Steroid 11β-HSD2 is an important enzyme in blood pressure homeostasis. It is encoded by the HSD11B2 gene, located on chromosome 16 (16q22) and consisted of five exons spanning approximately 6.2 kb. This enzyme, which converts cortisol (F) to inactive cortisone (E), is typically expressed in tissues where the mineralo-corticoid receptor is present. Impairment in 11β-HSD2 enzymatic activity results in an inefficient conversion of F to E, triggering hypertension through cortisol activa-tion of mineralocorticoid receptor. Severe AME is a well described but rare syndrome. The hallmark of AME is a state of mineralocorticoid excess, in the absence of aldo-sterone and the presence of an abnormal urinary steroid profile with an increase in the F:E ratio.
Hypertension is a very common disease worldwide and is an important public health challenge. One of

Proc Natl Acad Sci,. USA 1998;95:10200–5.
6A. APPARENT MINERALO244
the main factors contributing to its development is the amount of salt intake. Nevertheless, not all individuals respond to a salt load in the same way; some develop hypertension while others do not, suggestive of genetic variation in salt sensitivity. AME is a form of salt- sensitive monogenic hypertension and a mild form has been described. Further, in view of the critical role of 11β-HSD2 in ensuring selectivity of the mineralocorticoid receptor in the distal nephron, it has been suggested that minor abnormalities of this enzyme may explain salt sensitivity and pathogenesis in essential hypertension.
References [1] Quinkler M, Stewart PM. Hypertension and the cortisol–cortisone
shuttle. J Clin Endocrinol Metab 2003 Jun;88(6):2384–92. [2] New MI, Levine LS, Biglieri EG, Pareira J, Ulick S. Evidence for
an unidentified steroid in a child with apparent mineralocorticoid hypertension. J Clin Endocrinol Metab 1977;44(5):924–33.
[3] New MI. The prismatic case of apparent mineralocorticoid excess. J Clin Endocrinol Metab 1994;79(1):1–3.
[4] Wilson R, Nimkarn S, New M. Apparent mineralocorticoid excess. Trends Endocrinol Metab 2001;12(3):104–11.
[5] New MI. Apparent mineralocorticoid excess: a personal history. Steroids 1994;59(2):66–8.
[6] New MI, Stoner E, Di Martino-Nardi J. Apparent mineralocorti-coid excess causing hypertension and hypokalemia in children. Clin Exp Hypertens [A] 1986;8(4-5):751–72.
[7] New MI, Oberfield SE, Carey RM, Greig F, Ulick S, Levine LS. A genetic defect in cortisol metabolism as the basis for the syn-drome of apparent mineralocorticoid excess. In: Mantero F, Bigl-ieri EG, Edwards ERW, editors. Endocrinology of Hypertension, Serono Symposia No 50. New York: Academic Press; 1982. p. 85–101.
[8] Krozowski ZS, Funder JW. Renal mineralocorticoid recep-tors and hippocampal corticosterone-binding species have identical intrinsic steroid specificity. Proc Natl Acad Sci (USA) 1983;80(19):6056–60.
[9] Arriza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, Housman DE, et al. Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science 1987;237(4812):268–75.
[10] Edwards C, Stewart P, Burt D, Brett L, McIntyre M, Sutanto W, et al. Localisation of 11 beta-hydroxysteroid dehydrogenase – tis-sue specific protector of the mineralocorticoid receptor. Lancet 1988;2(8618):p986–9.
[11] Funder J, Pearce P, Smith R, Smith A. Mineralocorticoid action: target tissue specificity is enzyme, not receptor, mediated. Science 1988;242(4878):583–5.
[12] Quinkler M, Bappal B, Draper N, Atterbury AJ, Lavery GG, Walker EA, et al. Molecular basis for the apparent mineralocorti-coid excess syndrome in the Oman population. Mol Cell Endocri-nol 2004 Mar 31;217(1-2):143–9.
[13] Wilson RC, Dave-Sharma S, Wei JQ, Obeyesekere VR, Li K, Ferrari P, et al. A genetic defect resulting in mild low-renin hypertension. Proc Natl Acad Sci USA 1998 Aug 18;95(17):10200–5.
[14] Stewart P. The adrenal cortex. In: Larsen P, Kronenberg H, Melmed S, Polonsky K, editors. Williams Textbook of Endocrinol-ogy. 10th ed. Philadelphia: Saunders; 2002. p. 491–551.
[15] Wilson RC, Krozowski ZS, Li K, Obeyesekere VR, Razzaghy-Azar M, Harbison MD, et al. A mutation in the HSD11B2 gene in a family with apparent mineralocorticoid excess. J Clin Endocrinol Metab 1995 Jul;80(7):2263–6.
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
CORTICOID EXCESS
16] Stenson PD, Mort M, Ball EV, Howells K, Phillips AD, Thomas NS, et al. The Human Gene Mutation Database: 2008 update. Genome Med 2009;1(1):13.
17] New MI, Geller DS, Fallo F, Wilson RC. Monogenic low renin hypertension. Trends Endocrinol Metab 2005 Apr;16(3):92–7.
18] Mune T, White PC. Apparent mineralocorticoid excess: genotype is correlated with biochemical phenotype. Hypertension 1996 Jun;27(6):1193–9.
19] Nunez BS, Rogerson FM, Mune T, Igarashi Y, Nakagawa Y, Phil-lipov G, et al. Mutants of 11beta-hydroxysteroid dehydrogenase (11-HSD2) with partial activity: improved correlations between genotype and biochemical phenotype in apparent mineralocorti-coid excess. Hypertension 1999 Oct;34(4 Pt 1):638–42.
20] Li A, Tedde R, Krozowski ZS, Pala A, Li KX, Shackleton CH, et al. Molecular basis for hypertension in the "type II variant" of appar-ent mineralocorticoid excess. Am J Hum Genet 1998;63(2):370–9.
21] Ferrari P, Obeyesekere VR, Li K, Wilson RC, New MI, Funder JW, et al. Point mutations abolish 11 beta-hydroxysteroid dehydro-genase type II activity in three families with the congenital syn-drome of apparent mineralocorticoid excess. Mol Cell Endocrinol 1996 May 17;119(1):21–4.
22] Mune T, Rogerson FM, Nikkila H, Agarwal AK, White PC. Human hypertension caused by mutations in the kidney iso-zyme of 11β-hydroxysteroid dehydrogenase. Nat Genet 1995;10: 394–9.
23] Obeyesekere VR, Ferrari P, Andrews RK, Wilson RC, New MI, Funder JW, et al. The R337C mutation generates a high Km 11 beta-hydroxysteroid dehydrogenase type II enzyme in a family with apparent mineralocorticoid excess. J Clin Endocrinol Metab 1995;80(11):3381–3.
24] Lin-Su K, Zhou P, Arora N, Betensky BP, New MI, Wilson RC. In vitro expression studies of a novel mutation delta299 in a patient affected with apparent mineralocorticoid excess. J Clin Endocrinol Metab 2004 May;89(5):2024–7.
25] Lavery GG, Ronconi V, Draper N, Rabbitt EH, Lyons V, Chapman KE, et al. Late-onset apparent mineralocorticoid excess caused by novel compound heterozygous mutations in the HSD11B2 gene. Hypertension 2003 Aug;42(2):123–9.
26] Odermatt A, Dick B, Arnold P, Zaehner T, Plueschke V, Deregi-bus MN, et al. A mutation in the cofactor-binding domain of 11beta-hydroxysteroid dehydrogenase type 2 associated with mineralocorticoid hypertension. J Clin Endocrinol Metab 2001 Mar;86(3):1247–52.
27] Kitanaka S, Katsumata N, Tanae A, Hibi I, Takeyama K, Fuse H, et al. A new compound heterozygous mutation in the 11β-hydroxysteroid dehydrogenase type 2 gene in a case of appar-ent mineralocorticoid excess. J Clin Endo Metab 1997;82:4054–8.
28] Morineau G, Marc JM, Boudi A, Galons H, Gourmelen M, Corvol P, et al. Genetic, biochemical, and clinical studies of patients with A328V or R213C mutations in 11betaHSD2 causing apparent min-eralocorticoid excess. Hypertension 1999;34(3):435–41.
29] Lavery G, Ronconi V, Draper N, Rabbitt E, Lyons V, Chapman K, et al. Late-onset apparent mineralocorticoid excess caused by novel compound heterozygous mutations in the HSD11B2 gene. Hypertension 2003;42(2):123–9.
30] Stewart PM, Krozowski ZS, Gupta A, Milford DV, Howie JA, Sheppard MC. Hypertension in the syndrome of apparent min-eralocorticoid excess due to mutation of the 11β-hydroxysteroid dehydrogenase type 2 gene. Lancet 1996;347:88–91.
31] Dave-Sharma S, Wilson RC, Harbison MD, Newfield R, Azar MR, Krozowski ZS, et al. Examination of genotype and pheno-type relationships in 14 patients with apparent mineralocorticoid excess. J Clin Endocrinol Metab 1998 Jul;83(7):2244–54.
32] Wilson RC, Dave-Sharma S, Wei J, Obeyesekere VR, Li K, Ferrari P, et al. A genetic defect resulting in mild low-renin hypertension.

REFERE
[33] Palermo M, Quinkler M, Stewart PM. Apparent mineralocorticoid excess syndrome: an overview. Arq Bras Endocrinol Metabol 2004 Oct;48(5):687–96.
[34] Palermo M, Shackleton CH, Mantero F, Stewart PM. Urinary free cortisone and the assessment of 11 beta-hydroxysteroid dehydroge-nase activity in man. Clin Endocrinol (Oxf) 1996 Nov;45(5):605–11.
[35] Ulick S, Levine LS, Gunczler P, Zanconato G, Ramirez LC, Rauh W, et al. A syndrome of apparent mineralocorticoid excess associ-ated with defects in the peripheral metabolism of cortisol. J Clin Endocrinol Metab 1979;49(5):757–64.
nCEs 245
[36] Palermo M, Delitala G, Sorba G, Cossu M, Satta R, Tedde R, et al. Does kidney transplantation normalise cortisol metabolism in apparent mineralocorticoid excess syndrome? J Endocrinol Invest 2000 Jul-Aug;23(7):457–62.
[37] Khattab AM, Shackleton CHL, Hughes BA, Bodalia JB, New MI. Remission of hypertension and electrolyte abnormalities fol-lowing renal transplantation in a patient with apparent mine-ralocorticoid excess well documented throughout childhood. J Pediatr Endocr Met 2013.