
Entwicklung von tumorspezifischen replikations-kompetenten ... · τουτον δη τον...
94
Entwicklung von tumorspezifischen replikations-kompetenten Adenoviren Dissertation zur Erlangung des Grades eines Doktors der Naturwissenschaften an der Fakultät für Chemie der Ruhr-Universität Bochum angefertigt in der Abteilung für Molekulare und Medizinische Virologie, Fakultät für Medizin, Ruhr-Universität Bochum Betreuer: Prof. Dr. med. K. Überla vorgelegt von Diplom-Biochemiker Dennis Hoffmann aus Oberhausen Bochum 2004
Transcript of Entwicklung von tumorspezifischen replikations-kompetenten ... · τουτον δη τον...

Entwicklung von tumorspezifischen
replikations-kompetenten Adenoviren
Dissertation
zur Erlangung des Grades eines Doktors der Naturwissenschaften
an der Fakultät für Chemie
der Ruhr-Universität Bochum
angefertigt in der Abteilung für Molekulare und Medizinische Virologie,
Fakultät für Medizin, Ruhr-Universität Bochum
Betreuer: Prof. Dr. med. K. Überla
vorgelegt von Diplom-Biochemiker
Dennis Hoffmann aus
Oberhausen
Bochum 2004

τουτον δη τον κοινον λογον και θειον και ου κατα µετοην γινοµεθα
λογικοι, κριτηριον αληθειαχς ϕησιν ο. Ηρακλειτος (540 − 480 v. Chr.)

Vielen Dank an...
… PD Dr. Oliver Wildner für die Aufnahme in seine Arbeitsgruppe, die Betrauung mit dem
spannenden Thema und sein Interesse an dem Fortgang der Arbeit.
… Prof. Dr. Klaus Überla für die freundliche Übernahme der Betreuung.
… Prof. Dr. Bernd-Joachim Benecke und Prof. Dr. Sabine Müller für die Übernahme des
Koreferats bzw. die Bereitschaft als dritte Prüferin der Prüfungskommission beizutreten.
… alle Mitarbeiter in meiner Arbeitsgruppe: für unzählige fachliche, politische und private
Diskussionen, Wett-Minipräppen, Biologenwitze und der Suche nach dem idealen
Kaffee. Zu nennen wären Christian Jogler, Wibke Bayer, Anika Ekrut und Sandra
Überberg.
… meine beiden Diplomandinnen Anika und Wibke, die meinen Two-Hybrid-Wahn ausleben
durften (mussten?!) und mit mir unendliche Diskussionen über Vollmilch- oder
Nugatschokolade für die Schublade führten.
… natürlich meine „Lieblings-TA“ Bettina G. Tippler, die schon in der Zellkultur der
„Pharma“ Angst vor meinen Hefen hatte und mir die Geheimnisse der Zell- und
Gewebekultur zeigte.
… die aus der anderen Arbeitsgruppe („die mit den instabilen, langweiligen RNA-Viren“):
für den Spaß, die gemeinsame Kaffeesucht, die Einführung im Labor, die vielen
Schultern, das Erleiden meiner nicht-lentiviralen Papervorschläge. Dazu gehören Dr.
Thomas Grunwald (Erklärbär ;-)), Dr. Alex Stang (rock rules, aber ohne Keyboard!),
Susann Lucke (Autoritäszwerg), Hella Monse (G.O.), Sabine Brandt, Matthias
Tenbusch (nachtragender Axti), Claudius Grossmann, Nicola Ternette, Dr. Seraphin
Kuate, Ph.D. Charles Esimone, Andreia Fernandes, Rosi Bohr, Ulla Vogel, Heike
Seidenstücker, Regina Bütermann, Klaus Sure, Uli Schumacher (der beste
Kaffeekocher!).
… alle die mich ein Stück begleitet haben, egal in welcher Arbeitsgruppe: Vera Siegmund,
Yvonne Klingen, Alexandra Hey, Mieke Sprangers, Daniela Stefanou, Dr. Uli Heindel,
Dr. Godwin Nchinda.
… Bärbel Rölleke für das Entziffern meiner „W“, „A“ und „3.“.
… und natürlich für „critical reviewing“: Vera, Susi, Thomas, Wibke und Mona.
… vielen Dank für die rosa Kaffeetasse!

i
Abkürzungsverzeichnis 5-FU 5-Flurouracil
5-FC 5-Flurocystein
AdV Adenovirus
ATP Adenosintriphosphat
bp Basenpaare
cDNA complementary desoxyribonucleic acid
DNA Desoxyribonukleinsäure
FACS fluorescence activated cell sorting
GCV Ganciclovir
HSV-tk Herpes Simplex Virus-1 Thymidin Kinase
ITR inverted terminal repeat
MCS multiple cloning site
mRNA messenger RNA
orf open reading frame
PCR Polymerase-Kettenreaktion
BRAV bedingt replikations-kompetenter Adenoviraler Vektor
RLU relative light units
RNA Ribonukleinsäure
RT Raumtemperatur

Inhaltsverzeichnis
ii
1. Einleitung ............................................................................................................................... 1 1.1 Tumortherapie mit onkolytischen Viren .......................................................................... 1 1.2 Adenovirale Vektoren als präklinische Modelle zur Tumortherapie ............................... 3 1.3 Adenoviren ....................................................................................................................... 6
1.3.1 Taxonomie, Morphologie und Genom....................................................................... 6 1.3.2 Replikationszyklus ..................................................................................................... 8
2. Zielsetzung ....................................................................................................................... 12 3. Material und Methoden ........................................................................................................ 13
3.1 Material .......................................................................................................................... 13 3.1.1 Chemikalien............................................................................................................. 13 3.1.2 Enzyme .................................................................................................................... 14 3.1.3 Verbrauchsmaterialien............................................................................................ 15 3.1.4 Kits .......................................................................................................................... 15 3.1.5 Plasmide .................................................................................................................. 16 3.1.6 Lösungen, Puffer und Medien ................................................................................. 17
3.1.6.1 Medien für Bakterienkultur .............................................................................. 17 3.1.6.2 Medien und Puffer für die Zellkultur................................................................ 18 3.1.6.3 Puffer und Lösungen für molekularbiologische Arbeiten mit DNA ................. 18
3.1.7 Geräte...................................................................................................................... 19 3.1.8 Bakterien und Zelllinien .......................................................................................... 20
3.1.8.1 Verwendete Bakterienstämme .......................................................................... 20 3.1.8.2 Eukaryotische Zelllinien................................................................................... 21
3.2 Allgemeine Molekularbiologische Methoden................................................................ 22 3.2.1 Gelelektrophorese von DNA.................................................................................... 22 3.2.2 Isolierung von DNA-Fragmenten aus Agarosegelen .............................................. 22 3.2.3 Restriktion von DNA................................................................................................ 22 3.2.4 Auffüllen von 5'-überhängenden DNA-Enden durch Klenow-Behandlung............. 23 3.2.5 Dephosphorylierung von Vektor-DNA mit Calf Intestinal Phosphatase ................ 23 3.2.6 Ligation von DNA-Fragmenten............................................................................... 23 3.2.7 Transformation von Bakterien ................................................................................ 24
3.2.7.1 Herstellung chemisch kompetenter Bakterien.................................................. 24 3.2.7.2 Transformation chemisch kompetenter Bakterien............................................ 24 3.2.7.3 Herstellung elektrokompetenter Bakterien....................................................... 25 3.2.7.4 Transformation elektrokompetenter Bakterien ................................................ 25
3.2.8 DNA-Präparation in kleinem Maßstab (Minipräp) ................................................ 25 3.2.9 DNA-Präparation in großem Maßstab (Maxipräp) ................................................ 26 3.2.10 Fällung .................................................................................................................. 26
3.2.10.1 Fällung von DNA ........................................................................................... 26 3.2.10.2 Fällung von RNA ............................................................................................ 26
3.2.11 Präparation von genomischer DNA aus Zellen .................................................... 27 3.2.12 Polymerase-Kettenreaktion (PCR)........................................................................ 27 3.2.13 TA-Klonierung....................................................................................................... 28
3.2.13.1 Anhängen eines überhängenden Adenosin..................................................... 28 3.2.13.2 TA-Klonierung................................................................................................ 28
3.3 Zytologische Methoden.................................................................................................. 28 3.3.1 Zellpassagen............................................................................................................ 28 3.3.2 Zellen einfrieren und auftauen ................................................................................ 28 3.3.3 DNA-Transfektion von Zellen mit Calcium-Phosphat ............................................ 29 3.3.4 DNA-Transfektion von Zellen mittels Elektroporation ........................................... 29 3.3.5 Quantifizierung GFP-positiver Zellen mittels FACS .............................................. 29 3.3.6 Selektion mit G418 .................................................................................................. 29

Inhaltsverzeichnis
iii
3.3.7 Limiting dilution ...................................................................................................... 30 3.3.8 Luciferase-Assay ..................................................................................................... 30 3.3.9 Virus-Aufreinigung mit Sucrose und CsCl .............................................................. 30 3.3.10 Infektion................................................................................................................. 30
3.3.10.1 Titerbestimmung............................................................................................. 30 3.3.10.2 Infektion.......................................................................................................... 31
4. Ergebnisse ............................................................................................................................ 32 4.1 Konstruktion und Charakterisierung von Kolon- und Pankreas-spezifischen Promotoren ........................................................................................................................... 32 4.2. Abhängigkeit der Promotoren von dem Zellzyklus ...................................................... 34
4.2.1 Zellzyklusanalyse der Zellen ................................................................................... 34 4.2.2 Analyse der Promotorspezifität ............................................................................... 35
4.3 Test von Promotoren auf Transaktivierung durch adenovirale Sequenzen und Genprodukte ......................................................................................................................... 36
4.3.1 Konstruktion der Vektoren zum Testen von Promotoren auf Transaktivierung...... 36 4.3.2 Einfluss des AdV (nt 1-353)-Segmentes auf die Aktivität der getesteten Promotoren; mit und ohne stromaufwärts liegender polyA-Sequenz .............................. 39 4.3.4 Einfluss der frühen adenoviralen Genprodukte auf die Aktivität der getesteten Promotoren....................................................................................................................... 40 4.3.5 Verifizierung im adenoviralen Vektor ..................................................................... 41
4.4 AdV E4 open reading frame Transkomplementierung .................................................. 43 4.4.1 Konstruktion der AdV E4 orfs exprimierenden Plasmide ....................................... 44 4.4.2 Transkomplementierungsbestimmung mit Luciferase-Assay .................................. 44
4.5 Konstruktion des bedingt replikationskompetenten adenoviralen Vektorsystems ........ 47 4.6 Analyse der bedingt replikationskompetenten adenoviralen Vektoren.......................... 49
5. Diskussion ............................................................................................................................ 55 5.1 Promotoranalyse............................................................................................................. 55 5.2 Analyse der bedingt replikationskompetenten adenoviralen Vektoren.......................... 58
6. Ausblick ............................................................................................................................... 61 7. Zusammenfassung................................................................................................................ 62 8. Quellenangaben.................................................................................................................... 64 9. Dissertationsbezogene bibliographische Daten.................................................................... 86 10. Lebenslauf .......................................................................................................................... 87

Abbildungs- und Tabellenverzeichnis
iv
Abbildungsverzeichnis Abb. 1: Schematischer Aufbau des humanen Adenovirus Typ 5 7
Abb. 2: Genomorganisation und Regulation des Adenovirus 8
Abb. 3: Replikationszyklus eines Adenovirus 10
Abb. 4: Plasmidkarten der vorhandenen Plasmide 16
Abb. 5: Promotoranalyse in verschiedenen Zelllinien 34
Abb. 6: Zellzyklusanalyse der genutzten Zelllinien 35
Abb. 7: Analyse der Promotorspezifität 36
Abb. 8: Analysierte Promotoren und AdV (nt 1-353) Segment 37
Abb. 9: Vektor-Konstrukte zur Analyse der Promotor-Transaktivierung 38
Abb. 10: Einfluss des AdV (nt 1-353)-Segments auf die Promotoraktivität, mit
und ohne stromaufwärts liegender polyA-Sequenz 40
Abb. 11: Einfluss von viralen Produkten auf die Promotoraktivität 41
Abb. 12: Verwendete adenovirale Konstrukte 42
Abb. 13: Einfluss auf die Promotoraktivität im adenoviralen Vektor 43
Abb. 14: Ergebnisse des Transkomplementierungstestes 46
Abb. 15: Konstruktion des adenoviralen Vektorsystems 48
Abb. 16: Analyse der Transduktionseffizienz in verschiedenen Zelllinien. 50
Abb. 17: Exemplarische Darstellung von Kristallviolett gefärbten und mit BRAV
infizierten Zellen 51
Abb. 18 A: Replikationsverhalten der 20 getesteten BRAVs 53
Abb. 18 B: Replikationsverhalten der 20 getesteten BRAVs 54
Tabellenverzeichnis Tab 1: Promotorvarianten die im pGL3basic-System charakterisiert wurden 33
Tab. 2: Nummerierungen der orf-Kombinationen 45
Tab. 3: Bedingt replikationskompetente adenovirale Vektoren 49

1. Einleitung
1
1. Einleitung
1.1 Tumortherapie mit onkolytischen Viren
Der erste Bericht über ein replikations-kompetentes Virus, das onkolytische Eigenschaften
besitzt, findet Erwähnung in einer Veröffentlichung von 1912; hier wird über eine
italienische Patientin mit Zervix-Karzinom berichtet 1. Es wurde eine Tumorregression
beobachtet, als der Frau nach einem Hundebiss ein attenuiertes Tollwut-Virus injiziert wurde.
Danach wurden verschiedene Virustypen als antineoplastische Agenzien verwendet, wie zum
Beispiel West Nil-, Ilheus- und Bunyamwera-Virus 2, Egypt 101- 3, Adeno- 4 und Newcastle
Disease-Virus 5-8, Masern- 9, Mumps 10 und bovines Enterovirus 11. Bis 1990 waren alle
Versuche eine replikations-kompetente Virusmutante zur Tumortherapie herzustellen
erfolglos. 1991 stellten Martuza et al. eine Thymidin Kinase (TK)-negative Herpes Simplex-
Virus-1 (HSV-1)-Mutante, genannt dlsptk, her, welche selektiv Tumorzellen lysiert 12. 1996
berichteten Bischoff et al., dass die Adenovirusmutante dl1520 13 in p53-dysfunktionellen
Tumorzellen repliziert und diese selektiv lysiert 14. Zurzeit ist eine schnelle Erweiterung des
Gebietes zur Entwicklung von replikations-kompetenten viralen Agenzien zur Behandlung
von Tumoren zu beobachten.
Die Eigenschaften eines idealen onkolytischen Virus sind: (i) hohe Infektiösität in einem
weiten Feld von Tumoren, (ii) ein kurzer Replikationszyklus in teilenden und post-
mitotischen Zellen, (iii) eine starke Verteilung nach der Lyse, (iv) eine hohe Distribution im
Tumorgewebe und (v) eine hohe physikalische und genetische Stabilität.
Des Weiteren sollte das ideale Agens nicht toxisch in normalen Zellen sein, eine geringe
immunogene Aktivität und eine hohe Tumorspezifität besitzen um eine systemische
Applikation zu ermöglichen. Aus Sicherheitsgründen sollte ein klinisch getestetes Agens
vorhanden sein, damit eine virale Replikation und Ausbreitung ausgeschlossen werden kann.
Verschiedene Mechanismen der viralen Onkolyse sind vorstellbar: Als erstes die direkte Lyse
von Tumorzellen durch Infektion mit lytischen Viren (zum Beispiel HSV-1 und Adenovirus),
welche in Tumorregression resultiert. Als zweiter Mechanismus produzieren verschiedene
onkolytische Viren Proteine während ihres Replikationszyklus, die für die Zellen zytotoxisch
sind. Ein dritter Mechanismus ist die Immunmodulation durch Expression von viralen
Genprodukten, welche die Tumorzellen verändert und für Zytokine zugänglich macht (z.B.
verstärkt die AdV E1A Expression die Sensitivität zum Tumor Nekrose Faktor (TNF)-α 15).
Alternativ führt Xenogenisation von Tumorzellen durch Präsentation von viralen Antigenen

1. Einleitung
2
auf der Zelloberfläche durch MHC-I Proteine zur zytotoxischen T-Lymphozyten (CTL)-
vermittelten Zerstörung von Tumorzellen 16. Ferner wurden Lysate von Virus infizierten
Tumorzellen (Onkolysate) als immuntherapeuisches Agens bei der Behandlung von
Melanomen oder Ovarienkarzinome als Adjuvanz verwendet 17. Als vierter Mechanismus
kann die Expression von viralen Proteinen, die die Sensitivität von Tumorzellen zu
Chemotherapeutika oder Strahlungstherapie erhöhen, in Betracht gezogen werden. Zum
Beispiel sind AdV E1A Genprodukte ein potentieller Chemosensibilisierer, vor allem in p53-
intakten Zellen 18. Verstärkte Chemosensibilität nach viraler Infektion wurde in vivo in einer
klinischen Phase II-Studie mit dem intratumoralen Adenovirus ONYX-015 bei der
Koinjektion mit cis-Platin und 5-Flurouracil (5-FU) bei Patienten mit Krebs im Kopf- und
Nackenbereich beobachtet 19. Ein weiterer Mechanismus, bei dem onkolytische Viren eine
antineoplastische Aktivität induzieren, ist die Expression von therapeutischen Transgenen
(z.B. Suizid-Gene oder Zyktokine cDNAs), welche im viralem Genom inseriert wurden 20-25.
Trotz der viel versprechenden Anfänge zeigten viele klinische Versuche im besten Fall einen
minimalen Gentransfer und eine geringe beobachtbare Veränderung des Tumors 26. Eine
effektive Tumortherapie benötigt eine komplette Auslöschung aller Tumorzellen im Körper.
Ein Hauptproblem der viralen Tumorgentherapie ist das Problem mit den vorhandenen
replikations-defekten Viren mehr als ein paar Schichten eines Tumors in vivo zu infizieren.
Dieses ist dadurch zu erklären, dass im Tumor ein großer hydrostatischer Druck herrscht, der
die Vektorpenetration verringert 27. Ein weiterer Grund ist das Fehlen von Rezeptoren auf den
Tumorzellen, mit denen das Virus zum Eintritt in die Zelle interagiert und Immunantwort
durch den Vektor. Eine Möglichkeit diese Probleme zu lösen ist die Verwendung von
replikations-kompetenten Vektoren, die die Möglichkeit besitzen durch aufeinander folgende
Zyklen der Infektion und Lyse von benachbarten Tumorzellen sich im Tumor zu vermehren
und auszubreiten.
Als Versuch die antineoplasitsche Effizienz der Virustherapie zu erhöhen wurde die virale
Onkolyse kombiniert mit der Prodrug-Suizid-Gen-Therapie. Die Prodrug-Suizid-Gen-
Therapie basiert auf der selektiven Umwandlung eines nicht toxischen Medikaments in eine
zytotoxische Form, z.B. Ganciclovir (GCV) durch HSV-tk zu GCV-Monophosphat und
anschließend durch zelluläre Kinasen in GCV-Diphosphat und abschließend zu GCV-
Triphosphat. Einbau von GCV-Triphosphat in makromolekulare DNA von sich teilenden
Zellen führt zu einer Kettenabbruchreaktion 28, chromosomalen Anomalien, Schwester-
Chromatid-austausch 29,30, und letztendlich zum Zelltod. Ein weiteres weitverbreitetes System
basiert auf der Umwandlung von dem Prodrug 5-Flurocytosin (5-FC) in 5-FU durch ein

1. Einleitung
3
vektorkodiertes Cytosin Deaminase (CD) Gen 31,32. Der virale onkolytische Effekt des E1B-
55K-deletierten Adenovirus konnte signifikant verstärkt werden. Dies wurde in soliden
Xenograft Tumor Modellen, wenn das Prodrug (GCV allein oder in Kombination mit 5-FC)
erst nach maximaler Vektorreplikation und Genexpression gegeben wurde, unter-sucht 33-36.
Jedoch war die Verabreichung von GCV nicht förderlich zur viralen onkolytischen Aktivität
der HSV Vektoren hrR3, G207 sowie des adenoviralen Ad.OW34 (ein AdV, das die
komplette E1-Region und HSV-tk exprimiert, mit einer vielfachen robusteren Replikation als
das E1B-deletierte Ad.TKRC) unter Konditionen, die die Vektorreplikation und –ausbreitung
begünstigen. Dieses resultierte aus der möglichen Verstärkung des HSV-tk/GCV vermittelten
onkolytischen Effekts und des virostatischen Effekt von GCV 37-39. Chase et al. generierten
einen replikations-kompetenten HSV-1-Vektor, welcher das Ratten Cytochrom P450 2B1
(CYP2B1) besitzt, das Cyclophosphamid (CPA) in den aktiven Metaboliten Phosphoamid
konvertiert. Phosphoamid vermittelt intra-Strang Verbindungen, welche dann bei der
Zellteilung für Strangbruch verantwortlich sind. Im Gegensatz zum HSV-tk/GCV und
CD/5-FU Paradigma war die Verwendung des CYP2B1/CPA-Systems nicht für eine
signifikante Inhibition der viralen Replikation verantwortlich, dieses System verstärkte den
onkolytischen Effekt des HSV-1 Vektors 40. Das Fehlen eines antiviralen Agens zur
Behandlung einer Infektion mit in der Tumortherapie eingesetzten Viren macht es
unabdingbar, dass zum klinischen Einsatz von replikations-kompetenten Viren ein
Sicherheitsgen (fail-save gene) zum Abstoppen der viralen Replikation und Ausbreitung
inseriert sein muß.
1.2 Adenovirale Vektoren als präklinische Modelle zur Tumortherapie
Drei verschiedene Ansätze werden zurzeit in der präklinischen Tumortherapie als
adenoviralen replikations-kompetente Vektoren verwendet. Der erste Ansatz verfolgt den
Austausch von viralen Promotoren, die essentiell für die Replikationskontrolle sind, durch
gewebespezifische und/oder zellzyklusabhängige Promotoren. Die E1A-Region kodiert für
zwei wichtige E1A Proteine, die während der viralen Infektion exprimiert werden. Diese
binden an den Tumorsuppressor Retinoblastoma-Protein (pRb). E1A Expression führt zu
einer Freisetzung des Transkriptionsfaktors E2F, welcher die Zellen in die S-Phase bringt um
für die AdV Replikation ein ideales zelluläres Umfeld zu generieren 41. In einem Versuch, die
Virus-Replikation durch das Prostata-Spezifischen Antigen (PSA) zu limitieren, welches im
Prostatakrebs exprimiert wird, platzierten Rodriguez et al. die E1A-Region des CN706

1. Einleitung
4
Vektors unter die Kontrolle des PSA Promotors und Enhancers 42. Um die Selektivität des
Vektors zu erhöhen, wurde ein zweiter PSA Enhancer stromaufwärts der E1B-Region
inseriert 43. Andere Forscher versuchten die E1A-Expression selektiv in anderen Tumoren
unter Verwendung von gewebespezifischen Promotoren zu erhöhen (z.B. α-Fetoprotein und
MUC-1) 44,45. Der Versuch, die virale Replikation durch heterologe Promotorkontrolle der
E1A-Region zu kontrollieren, wird dadurch beeinträchtigt, dass schon eine geringe Menge an
E1A-Genprodukten ausreicht um die adenovirale Replikation zu initiieren 46. Außerdem
beinhaltet die der E1-Region stromaufwärts gelegene Verpackungssequenz zwei Enhancer-
Elemente 47, welche in vielen Geweben aktiv sind und daher zahlreiche Promotoren aktivieren
können 48,49. Diese Elemente können nicht entfernt werden ohne die Verpackungseffizienz des
Virus zu reduzieren. Des Weiteren befindet sich in dem stromaufwärts gelegenen AdV-
Segment eine kryptische Transkriptions-Startstelle 50, welche signifikant mit den heterologen
Promotoren interferiert und somit zur Aufhebung der Spezifität führt.
Da eine Menge an kritischen regulatorischen Proteinen, die während einer adenoviralen
Replikation inaktiviert werden, auch in tumorösem Gewebe inaktiviert sind 51,52, wurde ein
zweiter Ansatz entwickelt, um bedingt replikations-kompetente AdVs herzustellen. Dies
wurde durch die Deletion von den adenoviralen Proteinen erreicht, die mit den zellulären
regulatorischen Proteinen interagieren.
Um virale Replikation zu ermöglichen, bringt das adenovirale Protein E1A die Zellen in die
S-Phase. Dieser abnormale S-Phasen Eintritt aktiviert normalerweise p53, welches die Zellen
in einen Zellzyklus-Arrest oder Apoptose bringt. Die beiden adenoviralen Proteine E1B-55K
und E1B-15K inaktivieren den p53-Signalweg; E1B-55K bindet und inhibiert p53,
wohingegen E1B-15K die p53-abhängige Apoptose blockt 53-55. Daher wählten Barker und
Berk et al. ein E1B-55K-deletiertes Adenovirus, dl1520 56 oder ONYX-015 um Tumore zu
therapieren, die eine p53-Dysfunktion aufweisen. Gesunde Zellen, die keine Veränderung im
p53 Signalweg besitzen, werden nicht beeinflusst 57,58. Jedoch zeigten viele
Veröffentlichungen, dass die Interaktion zwischen E1B-55K und p53 weitaus komplexer ist
als angenommen und die Fähigkeit des dl1520 Vektors zur produktiven Replikation nicht mit
dem p53-Status der Wirtszelle korreliert 59-68. ONYX-015, welches kein therapeutisches oder
fail-save-Gen, wie HSV-tk, besitzt, wurde in klinischen Phasen mit Patienten, die rezidive,
refraktäre Adenokarzinome am Kopf und Nacken hatten, eingesetzt. Wenn es als einziges
Agens verwendet wurde, war nur eine Tumorregression bei 15% der behandelten Patienten zu
beobachten 69. Aber in Kombination mit cis-Platin- und 5-FU-basierender Chemotherapie,
konnte mit dem dl1520 Vektor eine Regression in 25 von 30 untersuchten Patienten

1. Einleitung
5
beobachtet werden. In acht Fällen (27%) wurden eine komplette und in elf Fällen (36%) eine
teilweise Regression erzielt und das ohne nennenswerte Nebeneffekte 70.
Ein weiterer Mutationsansatz basiert auf der Deletion der konservierten-E1A Region 2 (CR2),
welche für die Bindung von pRB verantwortlich ist 71. Das ∆24 E1A-CR2 mutierte Virus ist
fähig, in Zellen effizient zu replizieren, die pRB defizient sind, währenddessen in pRB-
intakten Zellen die Replikation inhibiert ist. Dies konnte in vitro und in vivo gezeigt
werden 72. Eine weitere E1A-CR2 Mutante dl922/947 verstärkt den antitumoralen Effekt
wenn man diesen Vektor mit dem dl1520 Vektor vergleicht. dl922/947 inhibierte den S-
Phasen Eintritt und die virale Replikation in postmitotischen Zellen 73. Um die
Tumorselektivität zu erhöhen setzten Johnson et al. die E4-Region in ihrem Vektor unter die
Kontrolle des E2F-Promotors 74.
Ein dritter Ansatz zur adenoviralen Gentherapie basiert auf der Modifikation des Fiber-
Proteins, welcher kritisch für den ersten Schritt der Virus-Absorption ist, um das Virus von
einem natürlichen Tropismus zu einem Rezeptor, der auf Tumoren überexprimiert ist, zu
lenken 75-78.
Obwohl Viren als onkolytische Agenzien seit ca. 100 Jahren untersucht und verwendet
werden, gibt es nur wenige klinische Versuche und Konzepte zur Tumor-spezifischen
Onkolyse und Replikation. Ergebnisse von klinischen Phase II Studien mit onkolytischen
Adenoviren zeigten ein enttäuschendes Ergebnis, wenn sie als einzelnes Agens zum Einsatz
kamen. Ein viel versprechendes Ergebnis zeigten sie, wenn sie mit Chemotherapien
kombiniert wurden.
Während genetisch veränderte Viren zur Verwendung bei der Tumortherapie entwickelt
wurden, bleiben einige native, unveränderte Viren Kandidaten als onkolytische Agenzien. In
Frage steht weiterhin die Regulation der Replikation. Des Weiteren sind noch viele
Signaltransduktionswege, welche in der Zelle stattfinden und durch virale Proteine verändert
werden, unbekannt. In der weiteren Entwicklung von onkolytischen Vektoren sollten Tumor-
spezifische Infektion und/oder Replikation, die Ausbreitung im Tumor, die Effizienz als
einziges Agens, und Interaktion mit dem Immunsystem berücksichtigt werden. Des Weiteren
muss untersucht werden, in welchen Kombinationen das Virus, Gentherapie, Chirurgie,
Strahlentherapie und/oder Chemotherapie die Überlebensrate und die Lebensqualität bei
verschiedenen malignen Tumorerkrankungen erhöhen.
Obwohl erste Studien vermuten lassen, dass die intratumorale Injektion von replikations-
kompetenten onkolytischen Viren sicher ist, müssen Vektoren entwickelt werden, die die
Effizienz von Tumorlyse, Replikation und Infektion in Tumoren erhöhen. Für den Fall, dass

1. Einleitung
6
eine nicht gewollte lokale oder systemische Infektion sich ausbreitet, sollten onkolytische
Viren die zum klinischen Einsatz gelangen ein fail-save System, wie HSV-tk/GCV besitzen.
1.3 Adenoviren
1.3.1 Taxonomie, Morphologie und Genom
Adenoviren (AdV) gehören zur Familie der Adenoviridae, die in zwei Genera,
Mastadenoviridae (Adenoviren der Säugetiere) und Aviadenoviridae (Adenoviren der Vögel),
unterteilt ist. Die Mastadenoviren beinhalten zahlreiche Subgenera, die neben den humanen
AdV auch Tierpathogene Viren, wie z. B. Rinder-, Hunde- und Vogeladenovirus umfassen.
Die humanen AdV sind auf Grund ihrer Fähigkeit zur Agglutination roter Blutkörperchen in 6
Subgruppen (A bis F) unterteilt, denen 51 Serotypen 41 zugeteilt werden 79-82.
Im Rahmen dieser Arbeit wurde das humane Adenovirus Typ 5 eingesetzt. Dieses ist
endemisch verbreitet und verursacht vor allem Infektionen des Respirationstraktes. Die
Übertragung erfolgt gewöhnlich über Aerosole oder kontaminierte Flüssigkeiten. Etwa 50 %
der Infektionen verlaufen symptomatisch, nach einer Inkubationszeit von sechs Tagen treten
die ersten Anzeichen auf. Die Erkrankung verläuft in der Regel mild, wobei die Pathologie
aus Entzündung und Schädigung epithelialer Zellen besteht.
AdV sind unbehüllte, ikosaedrische Partikel, die einen Durchmesser von 80 – 100 nm
aufweisen 83. Sie beinhalten eine lineare doppelsträngige DNA von ca. 36 kb, die zusammen
mit vier Strukturproteinen (Protein V, VII, µ und TP) das Core bildet 84. Sieben weitere
Strukturproteine bilden das Kapsid, welches aus 252 Untereinheiten zusammengesetzt ist,
nämlich aus 240 Hexonen und 12 Pentonen, welche die Ecken des Ikosaeders bilden. Für die
Fiberproteine, welche antennenartig in die Peripherie reichen, sind die Pentone die
Ankerproteine 85. Die distale C-terminale Domäne des Fiberproteins wird als „Knob“
bezeichnet und interagiert mit dem Coxsacki und Adenovirus-Rezeptor (CAR) der Zielzelle.

1. Einleitung
7
Abb. 1: Schematischer Aufbau des humanen Adenovirus Typ 5 Das Viruscapsid besteht aus Hexon- und Pentonbasisproteinen, deren Interaktionen durch die Strukturproteine IIIa, VI, VIII und IX unterstützt werden. Das lineare DNA-Genom ist mit dem terminalen Protein kovalent verbunden und interagiert mit dem Core-Protein VII. Die Interaktion zwischen Genom und Capsid-Proteinen wird durch das Core-Protein V vermittelt. Verändert Nach 86.
Das etwa 36 kb große Genom der AdV enthält überlappende transkriptionale Einheiten auf
beiden DNA-Strängen, die über 50 Polypeptide kodieren 41. Die Gene werden eingeteilt in die
fünf frühen Transkriptionseinheiten (E1A, E1B, E2, E3, E4), die beiden verzögerten
Transkriptionseinheiten (IX, IVa) und in die späte Transkriptionseinheit (L, major late).
Letztere generiert fünf mRNAs und ist verantwortlich für die Synthese der viralen
Strukturproteine (Kapsidbildung) 41. An beiden Enden des viralen Genoms befinden sich
wiederholende Sequenzen, die Inverted Terminal Repeats (ITR) genannt werden und eine
Rolle in der Anfangsphase der Replikation spielen 41. Bei der Transkription werden beide
DNA-Stränge abgelesen, wobei E1A, E1B, IX, E3 und die Major Late Region in sense-, E4,
E2 und IVa dagegen in antisense-Richtung abgelesen werden 87.

1. Einleitung
8
Abb. 2: Genomorganisation und Regulation des Adenovirus Die Genomgröße des humanen Adenovirus Typ 5 beträgt 35.938 bp; an den Enden des Genoms befinden sich invertierte Sequenzwiederholungen (ITR) von 103 bp Länge. Die Gene der E1-Gruppe befinden sich am 5’ Ende des Genoms und umfassen etwa 4.000 bp, die zwei aktiven Transkriptionseinheiten kodieren für die sofort transkribierten E1A- und verzögert transkribierten E1B-, IVa und IX-Proteine. E1A transaktiviert die Expression der anderen frühen Gene. Der für die E2-Proteine kodierende Bereich erstreckt sich über 20.000 bp in der zentralen Region des Genoms und ist in zwei Bereiche, E2A und E2B, unterteilt. E2 ist verantwortlich für die virale Replikation. Die E3-Genregion befindet sich im 3’ Bereich des Genoms, hat eine Größe von etwa 4.000 bp und schützt den Virus vor der Wirtsimmunantwort. Die E4-Region liegt am 3’ Ende des Genoms, umfasst 3.500 bp. Ihre Produkte regulieren die E2-Expression und bilden mit E1B einen regulatorischen Komplex der die L1 – 5 RNA-Translation beschleunigt. Die spät exprimierten Gene liegen in der L-Genregion, diese überspannt etwa 80% des Genoms und ist somit beinahe 30.000 bp lang. Nach 88.
1.3.2 Replikationszyklus
Das Virion wird nach Bindung des C-terminalen Endes des Fiberproteins (Knob) an den
Coxsacki und Adenovirus-Rezeptor (CAR) durch Endozytose in die Zielzelle aufgenommen,
welche durch die Interaktion des Pentonbasisproteins (RGD-Motiv) mit den αvβ5-/αvβ3-
Integrinen vermittelt wird 89. Die Endosomenmembran wird durch eine weitere Interaktion
der αvβ5-Integrine mit anderen Zellfaktoren und dem Pentonbasisprotein aktiviert 90, die
freien Viruspartikel gelangen durch Assoziation mit den Mikrotubuli der Wirtszelle zum
Zellkern 91. Die viruskodierte Protease zerstört während des Transports zu den nukleären
Poren 92,93 das Kapsid durch Proteolyse des Proteins VI. Dieses stellt die Verbindung

1. Einleitung
9
zwischen Kapsid und den Core-Komponenten dar. Die Replikation wird durch die Bindung
des nun freien, an die virale DNA gekoppelten Terminalproteins an die Kernmatrix initiiert 94.
Der Replikationszyklus beginnt mit der sofortigen Transkription des E1A-Gens 41. Die E1A-
Proteine binden an verschiedene zelluläre Transkriptionsfaktoren und Regulatorproteine,
wodurch sie die Transkription der weiteren frühen Gene der viralen DNA induzieren und das
zelluläre Umfeld für die Virusreplikation verbessert 95.
Das E2-Gen kodiert für das prä-terminale Protein und die DNA-Polymerase, die beide unter
Komplexbildung an die ITRs des viralen Chromosoms binden 96, sowie für das Einzelstrang-
DNA-Binde-Protein, welches zusammen mit der Polymerase und dem zellulären Protein NFII
die Elongation des Transkripts ermöglicht. Das prä-terminale Protein dient als Primer für die
DNA-Replikation 97. In der ersten Phase der Replikation dient nur einer der beiden DNA-
Stränge als Template für die Synthese, so dass das Ergebnis der Replikation ein Duplex aus
der originalen Matrix und einem Tochterstrang sowie ein separater Einzelstrang ist. Der
Einzelstrang bildet hierauf eine Kreisform, in der die komplementären ITRs sich einander
annähern („Pfannenstiel“) und somit die gleiche Struktur bilden wie die Termini der viralen
Duplexform. Diese Struktur kann nun wie in der ersten Replikationsphase durch die
Initiationsmaschinerie der DNA-Synthese erkannt werden, und das zweite Duplex, bestehend
aus originaler Matrix und einem Tochterstrang wird generiert 41.
Die E3-Transkriptionseinheit kodiert immunsuppressive Funktionen, die auf zwei
Mechanismen basieren. Das E3-19 kDa-Protein reguliert die Transkription des Major-
Histokompatibilitäts-Komplex Klasse I (MHC-1) herab, verhindert so die Antigenpräsentation
auf der Zelloberfläche und somit die Differenzierung zytotoxischer T-Lymphozyten gegen die
viralen Antigene 98. Das E3-14.7 kDa- und das RIDα/β-Protein unterbinden die Apoptose
infizierter Zellen, was durch Fas/Fas-Liganden und/oder den Tumornekrose-Faktor α (TNFα)
vermittelt wird 99,100. Ein weiteres wesentliches Produkt der E3-Region ist das Adenovirus
Death Protein (ADP; 11.6 kDa-Protein), das die späte Zytolyse der infizierten Zelle fördert
und damit die Effizienz der Freigabe der neu entstandenen Viruspartikel erhöht 101.
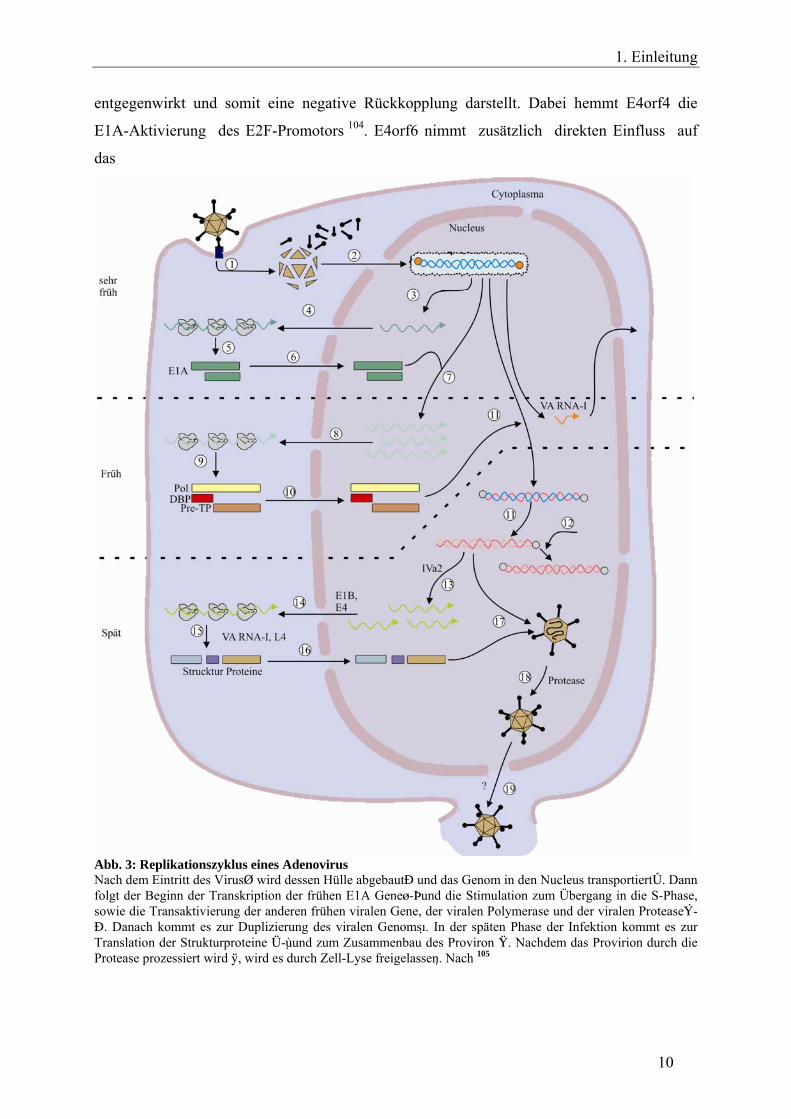
Die E4-Region ist in sechs offene Leserahmen aufgeteilt (open reading frames (orf) 1–4; 6;
6/7) und spielt durch Förderung der selektiven Expression viraler Gene, sowie durch
Herunterregulierung zellulärer Gene eine wichtige Rolle im viralen Zyklus 102. So verhindern
zum Beispiel die Proteine E4orf3 und E4orf6 den Transport der zellulären Transkripte vom
Zellkern ins Zytoplasma und unterstützen den Transport der späten viralen Transkripte, indem
sie die Stelle des zellulären Transportfaktors einnehmen 103. Gleichzeitig aber konnte auch
nachgewiesen werden, dass E4orf4 den E1A-regulierten Mechanismen zum Teil
1. Einleitung
10
entgegenwirkt und somit eine negative Rückkopplung darstellt. Dabei hemmt E4orf4 die
E1A-Aktivierung des E2F-Promotors 104. E4orf6 nimmt zusätzlich direkten Einfluss auf
das
Abb. 3: Replikationszyklus eines Adenovirus Nach dem Eintritt des Virusq wird dessen Hülle abgebautw und das Genom in den Nucleus transportierte. Dann folgt der Beginn der Transkription der frühen E1A Gener-yund die Stimulation zum Übergang in die S-Phase, sowie die Transaktivierung der anderen frühen viralen Gene, der viralen Polymerase und der viralen Proteaseu-o. Danach kommt es zur Duplizierung des viralen Genoms. In der späten Phase der Infektion kommt es zur Translation der Strukturproteine g-jund zum Zusammenbau des Proviron k. Nachdem das Provirion durch die Protease prozessiert wird l, wird es durch Zell-Lyse freigelassen . Nach 105

1. Einleitung
11
zelluläre p53-Protein, indem es dieses mittels einer Zinkfinger-Bindungsdomäne aus dem
Komplex mit E1B-55kDa löst und es nachfolgend destabilisiert 106. E4orf3 relokalisiert das
E1B-55kDa-Protein vom Zytoplasma in den Zellkern und unterbindet somit die Inaktivierung
des p53 durch E1B-55kDa. Hierbei wird in Anwesenheit von E4orf6 die Freisetzung des p53
durch E4orf3 außer Kraft gesetzt 107.
Neuere Untersuchungen haben gezeigt, dass p53 mit dem E1A-regulierten
Transkriptionsfaktor p120-E4F interferiert und dadurch die Aktivität des E4-Promotors
hemmt 108.
Die Expression der späten mRNAs, die aus komplexen Spleißvorgängen resultieren und in
fünf Familien (L1 – L5) eingeteilt werden, wird durch den Major-Late-Promotor (MLP)
kontrolliert. Es entstehen hieraus die viralen Strukturkomponenten, und es folgt die Formation
des zunächst leeren Kapsids 109. Der Eintritt eines viralen DNA-Moleküls wird durch die
Verpackungssequenz, ein AT-reiches DNA-Element, das ca. 260 Basenpaare vom 5’ ITRs
des Genoms entfernt liegt, vermittelt 110. Die reifen Viruspartikel reichern sich 20 bis 24
Stunden nach der Infektion (post infectionem; p.i.) im Zellkern an 41. Zur Aktivierung des
MLP werden mindestens zwei Komponenten benötigt. Nach dem Beginn des
Replikationszyklus kommt es zum einem zur E4orf1 vermittelten Transformation des viralen
Chromatin-Komplexes, welches die Bindung des zellulären Transkriptionsfaktors USF
(MLTF) an den MLP ermöglicht 111. Zum anderen wird die Aktivierung durch die
Transkriptionsfaktoren IVa2 und IX, die durch die verzögerte frühe Transkriptionseinheit
kodiert werden, erreicht 112,113. IV2a bindet hierbei zusätzlich zu USF an alternative
Bindungsdomänen stromabwärts der Major-Late-Startdomäne 114,115, IX steuert zur
Aktivierung des MLP über das TATA-Motiv bei. Diese Vorgänge werden von
Veränderungen der nukleären Infrastruktur und einer Permeabilitätserhöhung der
Kernmembran begleitet 116,117. Der Austritt der Viren in das Zytoplasma wird nach Zerstörung
der Intermediärfiliamente und Disintegration der Zellmembran, gefolgt von der Freilassung
der Viren in den Interzellularraum 2 bis 4 Tage p.i., ermöglicht.

2. Zielsetzung
12
2. Zielsetzung
Das Ziel der vorliegenden Arbeit war es, ein bedingt replikations-kompetentes Adenovirus
zur Behandlung von Kolon- und Pankreaskarzinomen zu konstruieren. Dieses Ziel wurde mit
der transkriptionellen Kontrolle der frühen Gene des Adenovirus verfolgt. Dazu sollten zwei
heterologe Promotoren die Regulation der zur Replikation wichtigen adenoviralen Gene E1
und E4 kontrollieren. Bisher wurde versucht, die transkriptionelle Kontrolle durch einen
heterologen Promotor vor der E1 Region zu erreichen, jedoch war die Replikationsspezifität
des Virus nicht sehr hoch, das heißt, dass das Virus auch in nicht tumorartigem Gewebe
repliziert. Daher sollten zunächst einige Promotoren gefunden werden, die bevorzugt in den
beiden Karzinomen aktiv sind und in gesunden Geweben keine Aktivität aufweisen. Als
nächstes war das Ziel, dass die Promotoren im adenoviralen Kontext nicht transaktiviert
werden. Für den potentiellen E1-Promotor kam noch eine Besonderheit hinzu. Im 5’ITR und
im Verpackungssignal, die sich in unmittelbarer Nähe zum nativen E1-Promotor befinden und
für das Virus essentiell sind, befindet sich eine alternative Transkriptionsstart-Stelle, die den
E1-Promotor oder den heterologen Promotor beeinflusst. Dazu muss ein Lösungsweg
gefunden werden wie diese alternative Start-Stelle von der Promotoraktivität entkoppelt
werden kann.
Ein anderes Problem stellt die Verpackung von übergroßen Genomen im Adenovirus dar. Die
Replikationseffizienz sinkt sobald die Genomlänge über 103% der Wildtyp-Genomlänge
liegt. Liegt die Genomlänge über 106%, wird das Genom nicht verpackt. Um genügend Platz
für die heterologen Promotoren und das therapeutische Gen zu schaffen, sollte mittels
Transkomplementierung untersucht werden, welche open reading frames der E4-Region für
eine effiziente Replikation entbehrlich sind. Des Weiteren wurde die E3-Region, die das
Adenovirus vor der zellulären Immunantwort schützt, deletiert.
Als Grundlage für das onkolytische Adenovirus musste ein Klonierungsvektor konstruiert
werden, der die Möglichkeit bietet, die E4-Region und die E1-Region zu manipulieren. Dazu
sollte zum einem ein adenoviraler backbone, dem pAdEasy1-Plasmid ähnlich, geschaffen
werden, in dem die E4-Region von zwei Homing-Endonukleasen flankiert wird. Die
veränderte E1-Region sollte dann mittels homologer Rekombination in das Gesamtgenom
rekombiniert werden.
Als Abschluss der vorliegenden Arbeit sollte die tumorspezifische virale Replikation in vitro
gezeigt werden. Dazu wurden verschiedene Karzinom-Zelllinien und primäre Zellen mit den
bedingt replikations-kompetenten Vektoren infiziert und die viral-vermittelte Lyse analysiert.

3. Material und Methoden
13
3. Material und Methoden
3.1 Material
3.1.1 Chemikalien
Agar AppliChem
Agarose SeaKem®, Cambrex
Carbencillin AppliChem
Bromphenolblau (0,1%) Fluka
Butanol J. T. Baker
Cäsiumchlorid Fluka, AppliChem
DEPC AppliChem
DMEM Gibco
dNTP-Mix Pharmacia
EDTA Merck
Eisessig J. T. Baker
Ethanol Riedel-de Haёn
Ethidiumbromid-Lösung (1%) AppliChem
FCS Gibco
Formamid, deionisiert Sigma
Formaldehyd J. T. Baker
Geneticindisulfat (G418) AppliChem
Gentamycinsulfat AppliChem
Glycerol J. T. Baker
Glucose J.T Baker
Hefeextrakt AppliChem
HEPES Biomol
Isopropanol Merck
Kaliumacetat J. T. Baker
Kaliumchlorid J. T. Baker
Kaliumdihydrogenphosphat Riedel-de Haën
Kanamycinsulfat AppliChem
Magnesiumchlorid J. T. Baker
Maleinsäure J. T. Baker

3. Material und Methoden
14
MOPS Biomol
NaOH J. T. Baker
Natriumacetat J. T. Baker
Natriumchlorid J. T. Baker
Natriumcitrat J. T. Baker
Natriumdihydrogenphosphat Merck
Natriumdodecylsulfat (SDS) AppliChem
Pepton AppliChem
Phenol AppliChem
Phenol:Chloroform:Isoamylalkohol (25:24:1) AppliChem
RNase A AppliChem
Streptomycinsulfat AppliChem
Tetracyclinhydrochlorid AppliChem
Tris AppliChem
Trypton AppliChem
Tween AppliChem
Wasser BBraun
Alle hier nicht aufgeführten Chemikalien wurden von den Firmen Riedel-de-Haën und Sigma
bezogen.
3.1.2 Enzyme
CIAP MBI Fermentas
Klenow Gibco
T4 DNA Ligase NEB
Taq-Polymerase Amersham Pharmacia Biotech Inc.
Restriktionsendonukleasen NEB, MBI Fermentas

3. Material und Methoden
15
3.1.3 Verbrauchsmaterialien
Centricons Microcon
Cellulose-Säulen Invitrogen
Filter Papier 288 Sartorius
Hybridisierungsröhrchen Schott
Nitrocellulose Schleicher & Schuell
Whatman-Papier Schleicher & Schuell
QIAshredder Qiagen
Zellkulturflaschen (25 cm2, 75 cm2, 175 cm2) Nunc
96-Loch-Platte Nunc
Sterilfilter Roth, Sarstedt
Plastikwaren (Pipettenspitzen, PE-Röhrchen,
Petrischalen, Reaktionsgefäße etc.) Brand, Greiner, Nunc, Sarstedt,
STARLAB
3.1.4 Kits
Jetsorb Gel Extraction Kit Genomed
CombizymeTM (DNA-Polymerase-Mix) Invitek
NucleoSpin Macherey-Nagel
pCR2.1-TA-kloning Kit Invitrogen
pGem-T-Easy Clonig Kit Promega
3. Material und Methoden
16
3.1.5 Plasmide
Abb. 4: Plasmidkarten der vorhandenen Plasmide (A) pCR2.1-TOPO (Invitrogen); (B) pCR3.1 (Invitrogen); (C) pBR322 (Promega); (D) pGL3basic (Promega); (E) pRL-CMV (Promega); (F) pGL3Control (Promega); (G) pGCsamEN (Promega); (H) pShuttle (Qbiogene); (I) pShuttle-CMV (Qbiogene); (J) pAdEasy-1 (Qbiogene); (K) pAdEasy-2 (Qbiogene)

3. Material und Methoden
17
3.1.6 Lösungen, Puffer und Medien
Alle angegebenen Puffer, Lösungen und Medien wurden, soweit nicht anders angegeben, in
Wasser angesetzt.
Zum Ansetzen von Lösungen wurde ausschließlich demineralisiertes Wasser aus der
Reinstwasseranlage Pro 90 CN der Firma Seralpur verwendet. Bei den Medien für die
Zellkultur wurde Reinstwasser von BBraun verwendet.
3.1.6.1 Medien für Bakterienkultur
LB-Medium 1 % (w/v) Trypton
0,5 % (w/v) Hefeextrakt
1 % (w/v) NaCl
in H2O, pH 7,0
LB-Festagar LB-Medium mit 2% (w/v) Agar
TB-Medium 17 mM KH2PO4
72 mM K2HPO4
1.2 % (w/v) Trypton
2.4 % (w/v) Hefeextrakt
0.4 % (w/v) Glycerol
in H2O, pH 7,2
Dem Nährmedium wurde nach dem Autoklavieren ein Antibiotikum zur Selektion zugesetzt
(Endkonzentration des Carbencillins: 100 µg/ml, Endkonzentration des Kanamycins:
50 µg/ml, Endkonzentration des Streptomycins: 30 µg/ml).
SOC-Medium 20 g/l Pepton
5 g/l Hefe-Extrakt
8,6 mM NaCl
2,5 mM KCl
10 mM MgCl2
20 mM Glucose

3. Material und Methoden
18
X-Gal-Lösung 20 % (w/v) 5-Brom-4-chlor-3-indolyl-β-D-
galactopyranosid in N,N-Dimethylformamid
3.1.6.2 Medien und Puffer für die Zellkultur
Kultur-Medium DMEM (Gibco)
10 % FCS (Invitrogen)
0,01 % Gentamycin (AppliChem)
PBS 10x (Gibco) 1,37 M NaCl
0,027 M KCl
0,043 M Na2HPO4 x 7H2O
0,014 M KH2PO4
in H2O, pH 7,4
Trypsin/EDTA 10x (Biochrom KG) 0,05 % (w/v) Trypsin
0,02 % (w/v) EDTA
in PBS
HBS (2x) 1,4 M NaCl
250 mM HEPES
10 mM Na2HPO4
in H2O, pH 7,12; steril filtriert
G418-Lösung 10 mg/ml
in DMEM
3.1.6.3 Puffer und Lösungen für molekularbiologische Arbeiten mit DNA
TE-Puffer 100x (AppliChem) 1 M EDTA x Na2
1 M Tris
pH 8,0

3. Material und Methoden
19
TAE-Puffer 50x (AppliChem) 0,05 M EDTA x Na2 x H2O
1 M Eisessig
2 M Tris
pH 8,5
DNA-Ladepuffer (6x) 0,25 % (w/v) Bromphenolblau
0,25 % (w/v) Xylencyanol FF
30 % (v/v) Glyzerin
autoklavieren
P1 50 mM Tris-Cl, pH 8,0
10 mM EDTA
100 µg/ml RNase A
in bidest. Wasser
P2 200 mM NaOH
1 % (w/v) SDS
in bidest. Wasser
P3 3,0 M Kaliumacetat
in bidest. Wasser
pH 5,5
3.1.7 Geräte
Analysewaage SCALTEC SPB64
Präzisionswaage SCALTEC SBH31
BioPhotometer Eppendorf
Brutschrank HS12 Heraeus Instruments
Dampfsterilisatoren H+P Labortechnik GmbH
Elektrophoresesystem PEQLAB Biotechnologie GmBH
Elektorporator Gene Pulser Excell BioRad
FACS Calibur Becton Dickinson
Geldokumentationskammer Biozym
Labor-pH-Meter Greisinger electronic

3. Material und Methoden
20
Lichtmikroskop CK40 Olympus
Luminometer Hamamatsu Photonics
pH-Meter GHPR 1400A Greisinger electronic
Pipetten Finnpipette
Pipettierhilfe, Repeater Plus Hirschmann Laborgeräte, Eppendorf
Peltier Thermal Cycler PTC 200 MJ Research
Rotoren:
Festwinkelrotor NVT100 Beckman Coulter
SW 28, SW 41 Beckman Coulter
Festwinkelrotor 12,500 Sigma
Rührer IKAMAG RCT
Schüttelapparat Certomat SII B. Braun Biotech International
Schüttelwasserbad 1086 GFL
Thermomixer compact Eppendorf
Tiefkühlschrank -86C ULT Freezer Thermo Forma
Ultraschallbad MERCK eurolab
Vortexer K-550-GE BENDER & HOBEIN AG
Wasseraufbereitungssystem Pro 90 CN Seralpur
Wasserbad E100 LAUDA
Zentrifugen:
Mikrofuge 18 Zentrifuge Beckman Coulter
6-15K Laboratory Zentrifuge Sigma
Optima L-70K Ultrazentrifuge Beckman Coulter
Zentrifuge 5810 Eppendorf
3.1.8 Bakterien und Zelllinien
3.1.8.1 Verwendete Bakterienstämme
DH5α (Invitrogen) F- Φ80lacZ∆M15 ∆( lacZYA-argF)U169 recA1 endA1 hsdR17(rk-,
mk+) phoA supE44 thi-1 gyrA96 relA1 λ-
DH10B (Invitrogen) F- mcrA ∆(mrr-hsdRMS-mcrBC) Φ80dlacZ∆M15 ∆lacX74 endA1
recA1 deoR ∆(ara, leu)7697 araD139 galU galK nupG rpsL

3. Material und Methoden
21
SCS110 (Stratagene) rpsL (StrR) thr leu endA thi-1 lacY galK galT ara tonA tsx dam dcm
supE44 ∆(lac-proAB) [F' tra∆36 proAB lacIqZDM15]
Stabl2 (Gibco BRL) F- mcrA ∆(mcrBC-hsdRMS-mrr) recA1 endA1 lon gyrA96 thi-1 supE44
relA1- λ- (lac-proAB)
BJ5183 (Stratagene) endA1 sbcBC recBC galK met thi-1 bioT hsdR (StrR)
3.1.8.2 Eukaryotische Zelllinien
A375 ATCC# CRL-1619
A549 ATCC# CCL-185
BxPc3 ATCC# CRL-1687
cFPac3 ATCC# CRL-715
DLD-1 ATCC# CCL-221
D54 Dr. Derek Bigner (Duke University, Durham, IL)
HaCaT ATCC# CCL-169
HCT-116 ATCC# CCL-247
HEK-293 Microbix
HeLa ATCC# CCL-2
HT-29 ATCC# HTB-38
JB6 Dr. Kerstin Reimers (MHH, Hannover)
K-1-17 Dr. Kerstin Reimers (MHH, Hannover)
MiaPaca ATCC# CRL-1420
PancTH1 ATCC# CRL-193
Panc1 ATCC# CRL-1469
Sk-Mel2 DSMZ# ACC-303
SNB-19 DSMZ# ACC-325
SW480 ATCC# CCL-228
U373 ATCC# HTB-17
293T ATCC# CRL-11268
888 ATCC# CRL-307
911E4 Crucell Holland B.V.
HEK-293vil∆BH von Bettina Tippler zur Verfügung gestellt

3. Material und Methoden
22
3.2 Allgemeine Molekularbiologische Methoden
Die molekularbiologischen Methoden wurden, soweit nicht anders gekennzeichnet, nach 118
durchgeführt.
3.2.1 Gelelektrophorese von DNA
Die DNA-Fragmente wurden mittels Agarosegelelektrophorese nach ihrer Größe aufgetrennt.
Die verwendeten 1%igen Gele wurden durch Aufkochen der Agarose in TAE-Puffer, Zusatz
von 0,01% Ethidiumbromid-Lösung und Erstarren in der Elektrophoresekammer hergestellt.
Die DNA-Proben wurden mit etwa 1/6 Ladepuffer versetzt und in die Taschen des mit TAE-
Puffer überschichteten Gels geladen.
Als Größenstandard wurden die Marker 1 kb DNA Ladder bzw. 1 kb Plus DNA Ladder von
Invitrogen verwendet, hiervon wurden in kleine 5 µl und in größere Taschen 10 µl
aufgetragen.
3.2.2 Isolierung von DNA-Fragmenten aus Agarosegelen
Für die Isolierung von DNA-Fragmenten (< 8 kb) aus Agarosegelen wurde das NucleoSpin
Kit nach Herstellerangaben verwendet. Bei größeren Fragmenten wurde das Jetsorb Gel
Extraction Kit (Genomed) nach Herstellerangaben verwendet.
3.2.3 Restriktion von DNA
Die verwendeten Typ II-Restriktionsendonukleasen prokaryotischer Herkunft spalten
spezifische DNA-Sequenzen hydrolytisch, wobei entweder glatte (blunt-end) oder
überhängende (sticky-end) Enden entstehen. Je nach Restriktionsenzym werden verschiedene
Pufferbedingungen und optimale Temperaturen (üblicherweise 37 °C) benötigt, die der
Hersteller im Beipackzettel angibt. Folgende Reaktionsansätze wurden standardmäßig
verwendet:
-analytischer Verdau (20 µl): 1-3 µl Plasmid-DNA (0,1-1 µg DNA)
2 µl Reaktionspuffer
1 µl Restriktionsenzym
mit bidest. Wasser auf 20 µl Gesamtvolumen auffüllen

3. Material und Methoden
23
-präparativer Verdau (50 µl): 5-10 µl Plasmid-DNA (1-5 µg DNA)
5 µl Reaktionspuffer
2 µl Restriktionsenzym
mit bidest. Wasser auf 50 µl Gesamtvolumen auffüllen
Der Ansatz wurde für 1 bis 3 h oder auch über Nacht bei 37 °C inkubiert. Anschließend
wurde die Vollständigkeit des Verdaues durch elektrophoretische Auftrennung in einem
Agarosegel kontrolliert.
3.2.4 Auffüllen von 5'-überhängenden DNA-Enden durch Klenow-Behandlung
Für das Auffüllen von 5'-überhängenden DNA-Enden wurde das Enzym DNA-Polymerase I
Large Fragment (Klenow) verwendet. Zum Restriktionsansatz wurde 1 µl einer 10 mM
dNTP-Lösung und 2 µl Klenow gegeben. Dann wurde er für 1½ h bei Raumtemperatur
inkubiert.
3.2.5 Dephosphorylierung von Vektor-DNA mit Calf Intestinal Phosphatase
Die Calf Intestinal Phosphatase (CIAP) katalysiert die Entfernung der 5’-Phosphatgruppe von
DNA, RNA, sowie Ribo- und Desoxyribonucleosidtriphosphaten, wodurch eine Religation
von Vektor-DNA nach erfolgreichem Restriktionsverdau verhindert wird.
Direkt im Anschluß an den Restriktionsverdau wurden 10-20 µl bidest. Wasser, 5 µl CIAP-
Puffer und 1 µl CIAP (10 U) hinzugefügt und für ½ h bei 37°C inkubiert.
Danach wurde die Reaktion durch eine Inkubation für 10 min bei 75 °C gestoppt.
3.2.6 Ligation von DNA-Fragmenten
Die ATP-abhängige T4-DNA-Ligase ist in der Lage, freie 3’-Hydroxylenden mit 5’-
Phosphatenden von doppelsträngiger DNA zu verknüpfen, indem sie die Bildung einer
Phosphodiesterbindung katalysiert. T4-DNA-Ligase kann sowohl kohäsive (sticky-end)
Enden als auch glatte (blunt-end) Enden miteinander ligieren.
Der Klonierungsvektor und das Insert wurden vor der Ligation mit den entsprechenden
Enzymen geschnitten und gereinigt. Der Vektor wurde, wenn nur mit einem
Restriktionsenzym geschnitten wurde, vor der Reinigung dephosphoryliert .

3. Material und Methoden
24
-20 µl Ansatz: 4 µl 5x Ligationspuffer
2 µl T4-Ligase (2 U/µl)
5 µl Insert DNA
2 µl Vektor DNA
7 µl bidest. Wasser
-20 µl Kontrollansatz: 4 µl 5xLigationspuffer
2 µl T4-Ligase (2 U/µl)
2 µl Vektor DNA
12 µl bidest. Wasser
Die Reaktion erfolgte für 1 h bei Raumtemperatur oder über Nacht bei 16°C.
3.2.7 Transformation von Bakterien
3.2.7.1 Herstellung chemisch kompetenter Bakterien
Bakterien einer Übernachtkultur wurden in 2 l SOB verdünnt und bei 30 °C inkubiert bis eine
OD600 von 0,5 +/- 0,2 erreicht war. Die Bakterien wurden 1 h auf Eis geschüttelt und dann bei
3.200 g und 4 °C für 12 min zentrifugiert. Das Zellpellet wurde in 1/3 des ursprünglichen
Kulturvolumens FSB resuspendiert und nach 15 min Inkubation auf Eis erneut bei 3.200 g
und 4 °C für 10 min zentrifugiert. Das Pellet wurde in 1/12,5 des ursprünglichen Volumens
FSB resuspendiert und mit 3,5% des aktuellen Volumens DMSO versetzt. Nach vorsichtigem
Schwenken wurde 5 min auf Eis, nach Zusatz weiterer 3,5% DMSO für weitere 10 min
inkubiert. Nach dieser Inkubationszeit wurden die Bakterien aliquotiert, in flüssigem
Stickstoff eingefroren und bei -80 °C gelagert.
3.2.7.2 Transformation chemisch kompetenter Bakterien
50 µl der Bakteriensuspension wurden in einem 13 ml-Röhrchen auf Eis vorgelegt und mit
der zu transformierenden DNA versetzt. Von den oben angegebenen Ligationsansätzen
wurden hierbei 3 µl verwendet, die DNA-Menge musste unter 1 µg liegen. Nach Inkubation
auf Eis für 30 min erfolgte ein Hitzeschock für 30 sec bei 42,2 °C, es wurden 500 µl SOC
zugegeben, bei 37 °C für 1 h inkubiert und schließlich 250 µl auf Selektionsmedium
ausplattiert.

3. Material und Methoden
25
3.2.7.3 Herstellung elektrokompetenter Bakterien
Eine frische Kolonie oder ein gefrorener Stock der Bakterien wurde über Nacht in einer 3-
5 ml großen Kultur angezogen, diese Übernachtkultur wurde verwendet, um eine 300 ml-
Kultur anzuimpfen. Es wurde bis zu einer OD600 von ~0,8 bei 32 °C oder 37 °C geschüttelt.
Die Zellen wurden in einem Zentrifugationsbecher gesammelt und 10 bis 60 min auf Eis
inkubiert. Die Zellen wurden bei 2.600 g und 4 °C für 10 min pelletiert, dann mit demselben
Volumen eiskaltem Wasser (+ 15% Glycerol) gewaschen und für 30 min pelletiert. Nach
Wiederholen des Waschschrittes wurden die Zellen in 20 ml Restvolumen resuspendiert, in
ein 50 ml-Gefäß überführt und nach erneutem 10-minütigen Pelletieren in 5 ml Restvolumen
resuspendiert. Nach Aliquotieren wurden die Zellen in flüssigem Stickstoff eingefroren und
bei -80 °C gelagert.
3.2.7.4 Transformation elektrokompetenter Bakterien
In einer 2 mm-Küvette wurden 20 µl Bakterien auf Eis vorgelegt und zwischen 0,1 und 1,0 µg
DNA hinzu pipettiert. Die Elektroporation erfolgte bei einer Spannung von 2,5 kV. Nach
Zusatz von 200 µl SOC-Medium wurden die Bakterien auf LB-Platten ausplattiert. Bei einer
Retransformation wurden 50 µl auf einer kleinen Platte (92 mm Durchmesser) ausgestrichen.
Bei einer Transformation vortransformierter Bakterien zur Rekombination der Plasmide
wurde der gesamte Ansatz auf einer großen Platte (150 mm Durchmesser) ausgestrichen.
3.2.8 DNA-Präparation in kleinem Maßstab (Minipräp)
Unter den mit den rekombinanten Plasmiden transformierten Bakterien mussten die Klone
identifiziert werden, die das gewünschte Plasmid korrekt amplifizierten. Hierfür wurden 4 -
48 willkürlich ausgewählte Bakterienkolonien, je nach Transformationseffizienz und dem
Verhältnis der Kolonien auf der Kontroll- und der Ligationsplatte, mit sterilen Pipettenspitzen
gestochen und in etwa 5 ml LB-Medium mit Selektionsantibiotikum in 15 ml verschließbare
15 ml-Plastikreagenzgefäße überführt. Die Minikulturen wurden über Nacht im 37°C-
Schüttler inkubiert.
Je 2 ml der Minikulturen wurden in Eppendorf-Reaktionsgefäßen 1 min lang in einer
Tischzentrifuge bei Höchstgeschwindigkeit zentrifugiert. Das Pellet wurde mit 150 µl kaltem
P1 resuspendiert und mit 150 µl P2 6-mal invertiert. Anschließend wurde 5 min bei
Raumtemperatur inkubiert und nach Zugabe von 300 µl P3 6-mal invertiert und 10 min bei
Höchstgeschwindigkeit zentrifugiert. Der Überstand wurde in ein neues Eppendorf-

3. Material und Methoden
26
Reaktionsgefäß überführt und mit 900 µl kaltem absoluten Ethanol versetzt. Nach 15 min
Zentrifugation bei Höchstgeschwindigkeit wurde der Überstand verworfen und das Pellet
10 min im Vakuum getrocknet. Das getrocknete Pellet wurde in 50 µl bidest. Wasser gelöst.
3.2.9 DNA-Präparation in großem Maßstab (Maxipräp)
300 ml Medium wurden mit einer Kolonie oder 200 µl einer Übernachtkultur angeimpft und
über Nacht bei 37 °C inkubiert. Die Zellen wurden bei 5.000 g und 4 °C für 10 min pelletiert
und anschließend in 10 ml P1 resuspendiert. Nach Zusatz von 10 ml P2 und sechsmaligem
Invertieren wurde für 10 min bei RT inkubiert. Es wurden 10 ml P3 zugesetzt, erneut 6x
invertiert und durch einen Faltenfilter filtriert. Dem Filtrat wurden 0,7 Volumen Isopropanol
zugesetzt und dann für 30 min bei 4 °C bei 12.500 g zentrifugiert. Das erhaltene Pellet wurde
in 4 ml P1 resuspendiert, mit 4,6 g CsCl und 40 µl Ethidiumbromidlösung versetzt und in ein
Ultrazentrifugationsröhrchen überführt. Die Zentrifugation erfolgt bei 20 °C entweder bei
70.000 g für 4,5 h, bei 90.000 g für 2,5 h oder bei 50.000 g über Nacht. Die erhaltenen
Banden wurden abgesaugt, das Volumen auf etwa 4 ml erweitert und mit TE-gesättigtem 2-
Butanol zweimal ausgeschüttelt. Das Volumen wurde anschließend auf 5 ml eingestellt, mit
0,7 Volumen Isopropanol versetzt und bei 20 °C und 12.500 g für 30 min zentrifugiert. Das
erhaltene Pellet wurde mit 70%igem Ethanol gewaschen, im Vakuum getrocknet und in 1x
TE resuspendiert.
3.2.10 Fällung
3.2.10.1 Fällung von DNA
Die DNA-Lösung wurde mit 0,1 Volumen 3 M NaAc, 0,1 Volumen 100 mM EDTA und 3,0-
3,5 Volumen Ethanol versetzt, für 30 min bei -20 °C inkubiert und 30 min lang bei
Höchstgeschwindigkeit zentrifugiert. Das erhaltene Pellet wurde mit 70% Ethanol gewaschen
und nach erneuter Zentrifugation getrocknet und in Wasser resuspendiert.
3.2.10.2 Fällung von RNA
Die RNA-Lösung wurde mit 0,1 Volumen 8 M Lithiumchlorid und 3 Volumen Ethanol
versetzt und 1 h bei -80 °C inkubiert. Es wurde bei 4 °C und 14.500 g für 30 min zentrifugiert

3. Material und Methoden
27
und das erhaltene Pellet mit 75% Ethanol gewaschen. Nach erneuter Zentrifugation wurde das
Pellet getrocknet und schließlich in RNase-freiem Wasser resuspendiert.
3.2.11 Präparation von genomischer DNA aus Zellen
Die Präparation von genomischer DNA wurde mit den Genomed Tissue
3.2.12 Polymerase-Kettenreaktion (PCR)
Zur Amplifikation einzelner DNA-Abschnitte wurde die Polymerase-Ketten-Reaktion
eingesetzt. Ein Standard-PCR-Ansatz wurde wie folgt zusammengesetzt:
10x Puffer 5 µl
2,5 mM dNTPs 4 µl
10 pmol/µl sense-Primer 10 µl
10 pmol/µl antisense-Primer 10 µl
Template 1 µl
2,5 U/µl Taq-Polymerase 0,5 µl
bidest. Wasser 19,5 µl
Die Amplifikation einiger cDNAs erfolgte mit Hilfe einer Proofreading-PCR. Hierzu wurde
das Proofreading-Polymerase-Kit von Combizyme nach Herstellerangaben angewendet.
Für Standard-PCRs wurden folgende Temperatur-Zyklen eingesetzt:
1. 95 °C 4:00 min
2. 95 °C 0:30 min
3. 52 °C 0:30 min
4. 72 °C 1:00 min
30 Wiederholungen Schritt 2.-4.
5. 72 °C 1:00 min
Die Wahl der Annealing-Temperatur wurde an die Schmelztemperatur der verwendeten
Primer angelehnt, die sich nach folgender Formel ermitteln lässt:
Tm = 69,3 °C + 0,41 x (%G+C) °C bei pH 7,0, 165 mM NaCl
Je nach Größe der erwarteten Fragmente wurde die Elongationszeit (72 °C) auf bis zu 5 min
erhöht.

3. Material und Methoden
28
3.2.13 TA-Klonierung
3.2.13.1 Anhängen eines überhängenden Adenosin
Die durch PCR mit einer Proofreading-Polymerase erhaltenen PCR-Produkte wurden vor der
TA-Klonierung mit einem überhängenden Adenosinrest versehen, hierzu wurden die PCR-
Fragmente in Anwesenheit von Taq-Polymerase und 10 mM ATP 15 min bei 72 °C inkubiert.
3.2.13.2 TA-Klonierung
Die TA-Klonierungen wurden mit dem TA-Cloning-Kit von Invitrogen nach
Herstellerangaben durchgeführt.
3.3 Zytologische Methoden
3.3.1 Zellpassagen
Alle verwendeten Zelllinien wurden, je nach Flaschengröße, in 5 ml, 20 ml oder 30 ml des
entsprechenden Mediums bei 37 °C, 5 % CO2 und 90 % Luftfeuchtigkeit kultiviert.
Im Abstand von 2-3 Tagen wurden die Zellen 1:10 passagiert: Zunächst erfolgte ein Waschen
mit 1x PBS, dann ein Abtrypsinieren der Zellen und schließlich das Resuspendieren im
Medium. Danach wurde ein Teil dieser Suspension in eine neue Kulturflasche mit Medium
gegeben.
3.3.2 Zellen einfrieren und auftauen
Um Zellen einzufrieren, wurden diese zunächst abtrypsiniert und sedimentiert (1000 rpm,
10 min, 4 °C). Dann erfolgte eine Aufnahme in DMSO-haltigem Zellkulturmedium (10 %
DMSO). Die Zellen konnten in Aliqots bei -80 °C gelagert werden.
Für weitere Verwendungen wurden die Zellen bei 37 °C aufgetaut und in frischem Medium
aufgenommen. Um das DMSO, welches toxisch für gewisse Zelllinien ist, zu entfernen,
wurden die Zellen zuvor sedimentiert.

3. Material und Methoden
29
3.3.3 DNA-Transfektion von Zellen mit Calcium-Phosphat
Für die Transfektion von Zellen wurden diese subkonfluent in T25-Kulturflaschen ausplattiert.
Ein Mediumwechsel fand noch einmal 4 h vor der Transfektion statt. Die Transfektion wurde
nach der Calcium-Phosphat-Methode durchgeführt: Zuerst wurden 31 µl CaCl2, 5 – 10 µg
DNA und bidest. Wasser vermischt, so dass ein Ansatz mit dem Endvolumen von 250 µl
entstand. Dieser wurde schließlich gemischt und mit 250 µl HBS tröpfchenweise versetzt.
Nach einer 10-minütigen Inkubation bei Raumtemperatur konnte der Transfektionsansatz zu
den ausplattierten 293T-Zellen gegeben werden. Das Medium wurde dann entweder nach 4
oder nach 12 h gewechselt.
3.3.4 DNA-Transfektion von Zellen mittels Elektroporation
Die Transfektion von einigen Zellen ließ sich mittels Elektorporation durchführen. Hierzu
wurden die Zellen einer T75-Kulturflasche gewaschen, abtrypsiniert, pelletiert (1000 rpm, 10
min, 4°C) und in 1 ml PBS aufgenommen. Jeweils 10 µg der zu transfizierenden DNA
wurden mit Zellsuspension (107 Zellen/Puls) vermischt und in eine 1mm-Küvette überführt.
Die Elektroporation erfolgte mit dem BioRad Protokoll, welches nach der jeweiligen Zelllinie
ausgesucht wurde. Anschließend erfolgte jeweils eine Zugabe von 200 µl Kulturmedium. 3 -
5 Elektroporationsansätze wurden dann zusammen in eine neue T75-Kulturflasche mit
Medium gegeben.
3.3.5 Quantifizierung GFP-positiver Zellen mittels FACS
Eine FACS-Analyse transfizierter Zellen wurde ca. 48 h nach der Transfektion durchgeführt.
Nachdem die Zellen mit PBS gewaschen, abtrypsiniert und pelletiert (1000 rpm, 10 min,
4 °C) wurden, sind sie in einem angemessenen Volumen PBS (ca. 1 ml) aufgenommen
worden. Die FACS-Analyse erfolgt nach Angaben des Herstellers mit dem FACS Calibur.
3.3.6 Selektion mit G418
24 h nach einer Transfektion mit einem Plasmid, welches für eine Neomycin-Resistenz
kodiert, wurde dem Zellmedium G418 zugefügt (Endkonzentration: 1000 µg/ml). Die Zellen
wurden je nach Zelllinie 1-4 Wochen unter G418-Selektionsmedium passagiert.

3. Material und Methoden
30
3.3.7 Limiting dilution
Um aus einer Zellsuspension monoklonale Zellkulturen herzustellen, wurde das Verfahren
Limiting dilution angewendet. Hierbei wurden die Zellen so verdünnt, dass sich ca. 4 Zellen
in 1 ml Medium befanden. Diese verdünnte Suspension ließ sich dann in eine 96-Kavitäten-
Kulturplatte verteilen (200 µl/Kavität). Einzelne Klone sind nach einigen Tagen in 24-
Kavitäten-Platten und schließlich in T25-Kulturflaschen ausplattiert worden.
3.3.8 Luciferase-Assay
Das Enzym Luciferase katalysiert die Oxidation von Luciferin unter Abgabe eines Photons.
Der Nachweis der Luciferase-Expression erfolgte mit dem Luciferase Detection Kit
(Promega). Nach Abnehmen des Mediums und Waschen der Zellen mit 1x PBS wurden die
Zellen direkt mit 100 µl Lysispuffer (für eine 96er Kavität) überschichtet. Nach einer
Inkubation von 15 min bei Raumtemperatur und 10 min bei -80 °C wurden die abgelösten
Zellen in ein 1,5 ml Eppendorf-Gefäß überführt und anschließend 15 Sekunden bei 14000 x g
zentrifugiert. 20 µl des Überstandes wurden mit 100 µl Luciferase-Substrat gemischt und die
Luciferase-Aktivität innerhalb von 10 s im Luminometer bestimmt.
3.3.9 Virus-Aufreinigung mit Sucrose und CsCl
Die Virus-Überstände wurden nach dem Auftauen geschüttelt und bei 1000 rpm für 10 min
zentrifugiert. Dann wurden 2 ml CsCl-Lösung (26,5 g CsCl + 43,5 ml 10 mM Tris, pH 7,9)
mit 4 ml Saccharose-Lösung/VTE (30 % oder 50 %) und dem Überstand der Viruspräparation
überschichtet. Es folgte eine Zentrifugation bei 25000 rpm (SW 28-Rotor) und 4°C für 2 h.
Die sedimentierten Viruspartikel wurden geerntet und in PBS-Glycerin aufgenommen.
Anschließend wurde eine Ultrazentrifugation mit CsCl bei 55000 rpm (NVT 100-Rotor) und
4 °C für ca. 16 h durchgeführt.
3.3.10 Infektion
3.3.10.1 Titerbestimmung
Die Titerbestimmung wurde nach der TCID50-Methode durch Infektionen von 911E4-Zellen
in 96-Kavitäten-Platten mit Verdünnungen des Virus durchgeführt. In den Vertiefungen einer
neuen Platte wurden zunächst 120 µl Medium vorgelegt, in die erste Vertiefung jeder Reihe

3. Material und Methoden
31
wurden je 40 µl des Virus-Stocks zugesetzt. 40 µl dieser ersten Verdünnung wurden in die
nächste Vertiefung der Reihe überführt und mit dem vorgelegten Medium vermischt. Durch
sukzessives Überführen der Verdünnungen in die folgenden Vertiefungen erhielt man so eine
Verdünnungsreihe. Das Medium der Zellen wurde entfernt und durch jeweils 100 µl des
virushaltigen Mediums der Verdünnungsreihe ersetzt. Nach 24-36 h wurde der cytopathische
Effekt beurteilt. Es wurde der Quotient der Vertiefungen bestimmt, in denen die Zellen bei
einer bestimmten Verdünnung einen cytopathischen Effekt zeigten, und die TCID50 nach
folgender Formel berechnet:
T = 101+d(S-0,5) mit d = log10 der Verdünnung
S = Summe der Quotienten
Das Ergebnis gibt den Titer des Virus-Stocks in TCID50 für das eingesetzte Volumen an.
3.3.10.2 Infektion
Nach der Titerbestimmug des Virus-Stocks wurden die Zellen infiziert. Zunächst wurde die
Zellzahl bestimmt und dann das Zellmedium mit dem entsprechenden Volumen des Virus-
Stocks versetzt. Der Wechsel des Mediums erfolgte nach 12 h.

4. Ergebnisse
32
4. Ergebnisse
4.1 Konstruktion und Charakterisierung von Kolon- und Pankreas-
spezifischen Promotoren
Zur Auswahl eines Kolon- bzw. Pankreas-spezifischen Promotors zur Kontrolle der E1- und
E4-Region des AdV wurden 17 verschiedene Promotoren in insgesamt 23 verschiedenen
Varianten getestet. Dazu wurden alle Promotorvarianten mittels nested PCR aus genomischer
DNA mit einer Pwo-Polymerase amplifiziert und zur weiteren Bearbeitung in pCR2.1 TA
kloniert. Mittels Overlap-Extension-PCR wurden Modifikationen an den einzelnen
Promotoren zur Verkleinerung der Gesamtlänge vorgenommen. Außerdem wurden SacI und
XhoI Restriktionsschnittstellen 5’ und 3’ der Promotorsequenzen durch die Wahl der Primer
bei der Overlap-Extension-PCR angehängt Anschließend wurden alle erhaltenen PCR-
Produkte mittels SacI und XhoI in das Luciferase-Expressions-Plasmid pGL3basic
subkloniert. Die beiden Promotorvarianten von COX-2 wurden von Dr. Inoue und Dr. Tanabe
(National Cardiovascular Center Research Institute, Osaka, Japan) zur Verfügung gestellt und
ohne weitere Modifikationen auch mittels SacI und XhoI in pGL3basic subkloniert. Dabei
entstanden die in Tabelle 1 aufgeführten Promotorkonstrukte.
Die Integrität der Konstrukte wurde durch Sequenzierungen überprüft.
Anschließend wurden die Konstrukte mit pRL-CMV als interne Kontrolle der
Transfektionseffizienz in verschiedenen Zelllinien mittels Luciferase-Assay analysiert. Je
10µg der Konstrukte und ein Zehntel der äquimolaren Menge von pRL-CMV wurden mit der
Calciumchlorid Transfektionsmethode oder mit dem kationischen Lipid DOTAP in 2 x 105
Zellen pro Kavität einer 24-Kavitäten Platte transfiziert. Der Versuch wurde in zwei Ansätze
pro Konstrukt und Transfektionsmethode durchgeführt. Als humane Zelllinien wurden dabei
verschiedene Kolonkarzinome (DLD-1, HCT116, HT-29 und SW480), Glioblastome (D54,
SNB19 und U373), Pankreaskarzinome (BxPc3, cFPac3, MiaPaca, Panc1 und PancTH1),
Melanome (888, A375 undSk-Mel2) sowie embryonale Nierenzellen (HEK-293),
Lungenepithelzellen (A549), Cervixkarzinomzellen (HeLa) und eine Keratinozytenzelllinie
(HaCaT) verwendet. 48 h nach der Transfektion wurden die Zellen lysiert und je ein Fünftel
des Lysates wurde verwendet, um die Luciferase-Aktivität zu bestimmen. Die
Promotoraktivität wurde über die Quantenausbeute von Firefly-Luciferin bestimmt. Bei der
Messung der internen Transfektionskontrolle wurde Renilla reniformis Luciferin verwendet.
4. Ergebnisse
33
Gemessen wurde die Quantenausbeute in RLU/s. Die Promotoraktivität wurde in
Relation zu der zehnfachen
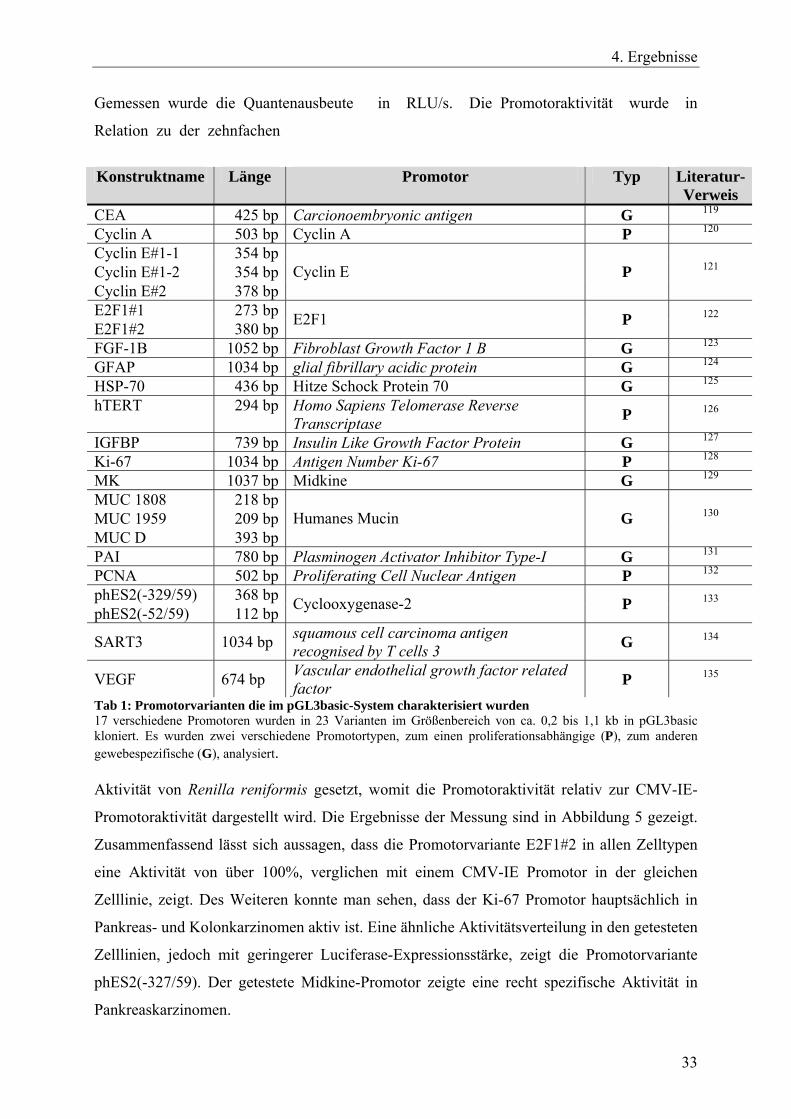
Konstruktname Länge Promotor Typ Literatur-
Verweis CEA 425 bp Carcionoembryonic antigen G 119 Cyclin A 503 bp Cyclin A P 120 Cyclin E#1-1 354 bp Cyclin E#1-2 354 bp Cyclin E#2 378 bp
Cyclin E P 121
E2F1#1 273 bp E2F1#2 380 bp E2F1 P 122
FGF-1B 1052 bp Fibroblast Growth Factor 1 B G 123 GFAP 1034 bp glial fibrillary acidic protein G 124 HSP-70 436 bp Hitze Schock Protein 70 G 125 hTERT 294 bp Homo Sapiens Telomerase Reverse
Transcriptase P 126
IGFBP 739 bp Insulin Like Growth Factor Protein G 127 Ki-67 1034 bp Antigen Number Ki-67 P 128 MK 1037 bp Midkine G 129 MUC 1808 218 bp MUC 1959 209 bp MUC D 393 bp
Humanes Mucin G 130
PAI 780 bp Plasminogen Activator Inhibitor Type-I G 131 PCNA 502 bp Proliferating Cell Nuclear Antigen P 132 phES2(-329/59) 368 bp phES2(-52/59) 112 bp Cyclooxygenase-2 P 133
SART3 1034 bp squamous cell carcinoma antigen recognised by T cells 3 G 134
VEGF 674 bp Vascular endothelial growth factor related factor P 135
Tab 1: Promotorvarianten die im pGL3basic-System charakterisiert wurden 17 verschiedene Promotoren wurden in 23 Varianten im Größenbereich von ca. 0,2 bis 1,1 kb in pGL3basic kloniert. Es wurden zwei verschiedene Promotortypen, zum einen proliferationsabhängige (P), zum anderen gewebespezifische (G), analysiert. Aktivität von Renilla reniformis gesetzt, womit die Promotoraktivität relativ zur CMV-IE-
Promotoraktivität dargestellt wird. Die Ergebnisse der Messung sind in Abbildung 5 gezeigt.
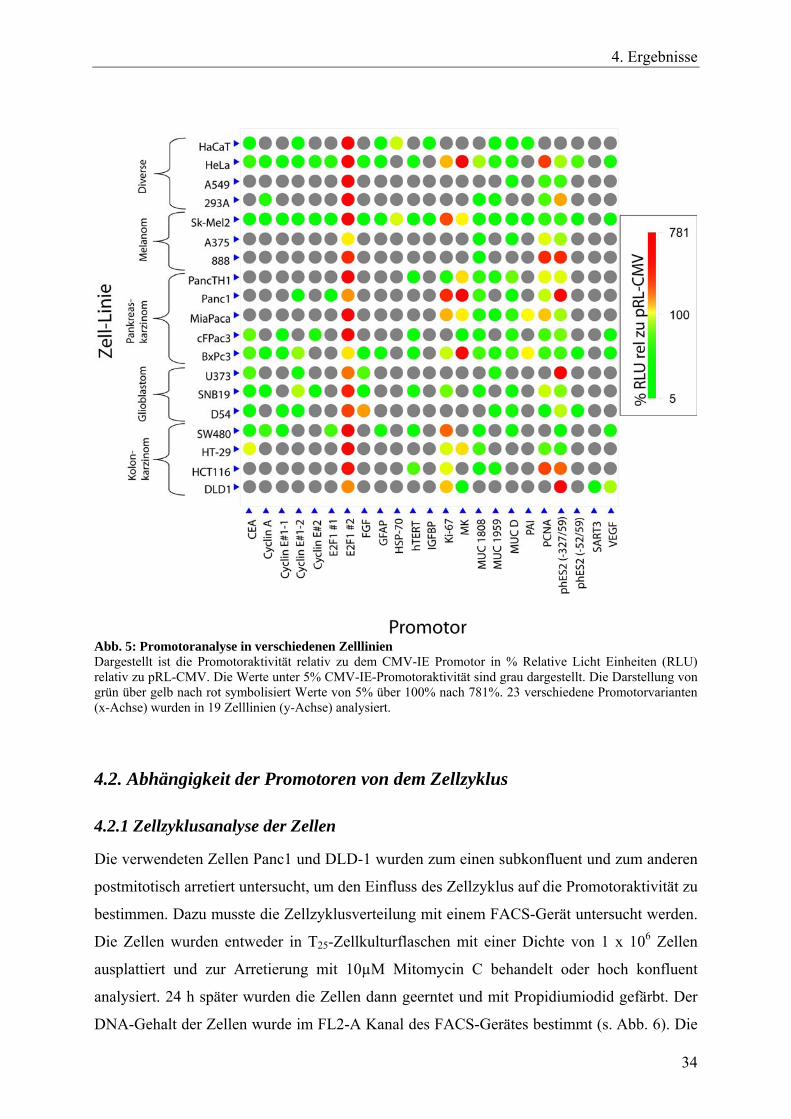
Zusammenfassend lässt sich aussagen, dass die Promotorvariante E2F1#2 in allen Zelltypen
eine Aktivität von über 100%, verglichen mit einem CMV-IE Promotor in der gleichen
Zelllinie, zeigt. Des Weiteren konnte man sehen, dass der Ki-67 Promotor hauptsächlich in
Pankreas- und Kolonkarzinomen aktiv ist. Eine ähnliche Aktivitätsverteilung in den getesteten
Zelllinien, jedoch mit geringerer Luciferase-Expressionsstärke, zeigt die Promotorvariante
phES2(-327/59). Der getestete Midkine-Promotor zeigte eine recht spezifische Aktivität in
Pankreaskarzinomen.
4. Ergebnisse
34
Abb. 5: Promotoranalyse in verschiedenen Zelllinien Dargestellt ist die Promotoraktivität relativ zu dem CMV-IE Promotor in % Relative Licht Einheiten (RLU) relativ zu pRL-CMV. Die Werte unter 5% CMV-IE-Promotoraktivität sind grau dargestellt. Die Darstellung von grün über gelb nach rot symbolisiert Werte von 5% über 100% nach 781%. 23 verschiedene Promotorvarianten (x-Achse) wurden in 19 Zelllinien (y-Achse) analysiert.
4.2. Abhängigkeit der Promotoren von dem Zellzyklus
4.2.1 Zellzyklusanalyse der Zellen
Die verwendeten Zellen Panc1 und DLD-1 wurden zum einen subkonfluent und zum anderen
postmitotisch arretiert untersucht, um den Einfluss des Zellzyklus auf die Promotoraktivität zu
bestimmen. Dazu musste die Zellzyklusverteilung mit einem FACS-Gerät untersucht werden.
Die Zellen wurden entweder in T25-Zellkulturflaschen mit einer Dichte von 1 x 106 Zellen
ausplattiert und zur Arretierung mit 10µM Mitomycin C behandelt oder hoch konfluent
analysiert. 24 h später wurden die Zellen dann geerntet und mit Propidiumiodid gefärbt. Der
DNA-Gehalt der Zellen wurde im FL2-A Kanal des FACS-Gerätes bestimmt (s. Abb. 6). Die
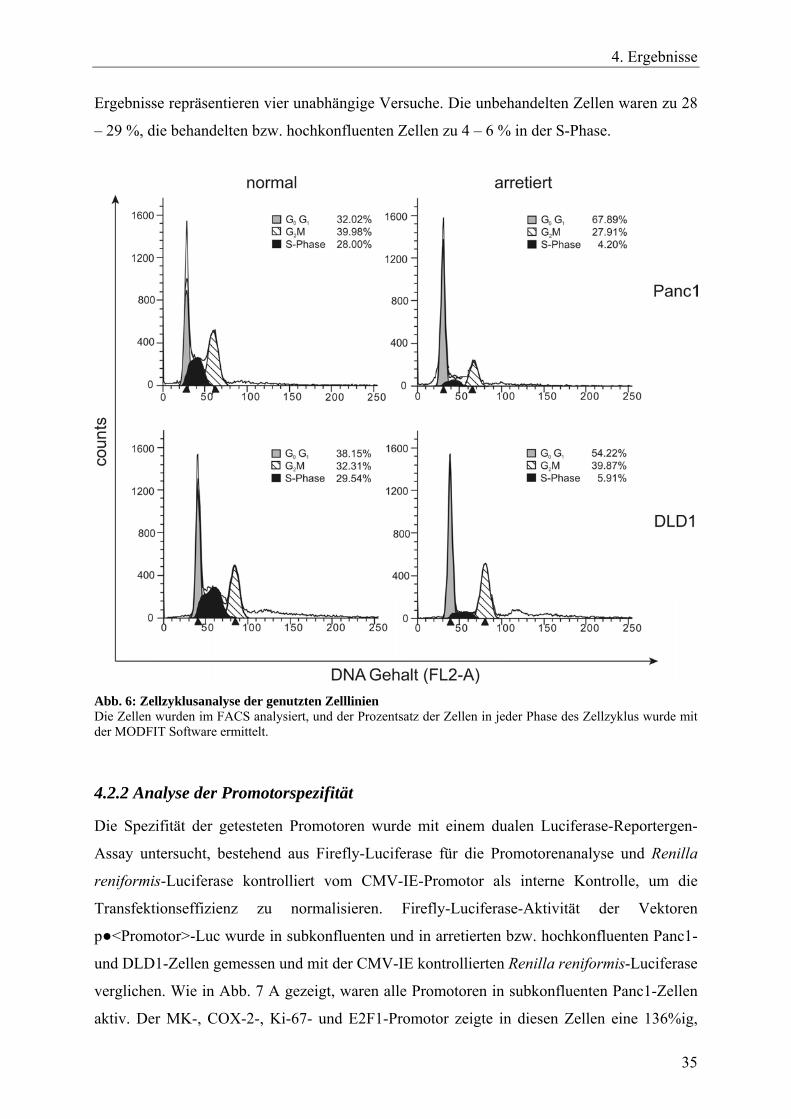
4. Ergebnisse
35
Ergebnisse repräsentieren vier unabhängige Versuche. Die unbehandelten Zellen waren zu 28
– 29 %, die behandelten bzw. hochkonfluenten Zellen zu 4 – 6 % in der S-Phase.
Abb. 6: Zellzyklusanalyse der genutzten Zelllinien Die Zellen wurden im FACS analysiert, und der Prozentsatz der Zellen in jeder Phase des Zellzyklus wurde mit der MODFIT Software ermittelt.
4.2.2 Analyse der Promotorspezifität
Die Spezifität der getesteten Promotoren wurde mit einem dualen Luciferase-Reportergen-
Assay untersucht, bestehend aus Firefly-Luciferase für die Promotorenanalyse und Renilla
reniformis-Luciferase kontrolliert vom CMV-IE-Promotor als interne Kontrolle, um die
Transfektionseffizienz zu normalisieren. Firefly-Luciferase-Aktivität der Vektoren
p●<Promotor>-Luc wurde in subkonfluenten und in arretierten bzw. hochkonfluenten Panc1-
und DLD1-Zellen gemessen und mit der CMV-IE kontrollierten Renilla reniformis-Luciferase
verglichen. Wie in Abb. 7 A gezeigt, waren alle Promotoren in subkonfluenten Panc1-Zellen
aktiv. Der MK-, COX-2-, Ki-67- und E2F1-Promotor zeigte in diesen Zellen eine 136%ig,
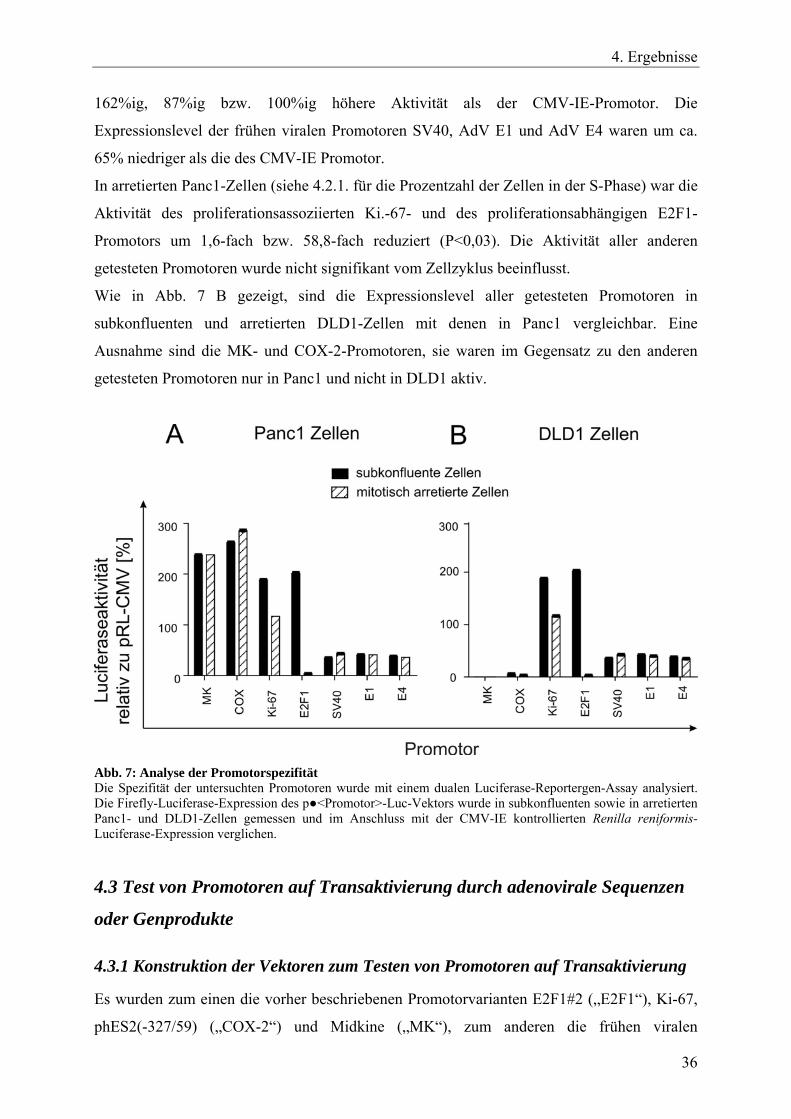
4. Ergebnisse
36
162%ig, 87%ig bzw. 100%ig höhere Aktivität als der CMV-IE-Promotor. Die
Expressionslevel der frühen viralen Promotoren SV40, AdV E1 und AdV E4 waren um ca.
65% niedriger als die des CMV-IE Promotor.
In arretierten Panc1-Zellen (siehe 4.2.1. für die Prozentzahl der Zellen in der S-Phase) war die
Aktivität des proliferationsassoziierten Ki.-67- und des proliferationsabhängigen E2F1-
Promotors um 1,6-fach bzw. 58,8-fach reduziert (P<0,03). Die Aktivität aller anderen
getesteten Promotoren wurde nicht signifikant vom Zellzyklus beeinflusst.
Wie in Abb. 7 B gezeigt, sind die Expressionslevel aller getesteten Promotoren in
subkonfluenten und arretierten DLD1-Zellen mit denen in Panc1 vergleichbar. Eine
Ausnahme sind die MK- und COX-2-Promotoren, sie waren im Gegensatz zu den anderen
getesteten Promotoren nur in Panc1 und nicht in DLD1 aktiv.
Abb. 7: Analyse der Promotorspezifität Die Spezifität der untersuchten Promotoren wurde mit einem dualen Luciferase-Reportergen-Assay analysiert. Die Firefly-Luciferase-Expression des p●<Promotor>-Luc-Vektors wurde in subkonfluenten sowie in arretierten Panc1- und DLD1-Zellen gemessen und im Anschluss mit der CMV-IE kontrollierten Renilla reniformis-Luciferase-Expression verglichen.
4.3 Test von Promotoren auf Transaktivierung durch adenovirale Sequenzen
oder Genprodukte
4.3.1 Konstruktion der Vektoren zum Testen von Promotoren auf Transaktivierung
Es wurden zum einen die vorher beschriebenen Promotorvarianten E2F1#2 („E2F1“), Ki-67,
phES2(-327/59) („COX-2“) und Midkine („MK“), zum anderen die frühen viralen

4. Ergebnisse
37
Promotoren Simian Virus 40 minimal-Promotor („SV40“) und die frühen adenoviralen
Promotoren E1 und E4 verwendet. Für den Simian Virus 40 (SV40) frühen Promotor ohne
Enhancer diente das XhoI – HindIII Fragment aus dem pGL3-Control-Vector. Zur Kontrolle
wurden die getesteten Promotoren in die Multiple-Klonierungsstelle (MCS) stromabwärts
einer synthetischen Polyadenylierungssequenz (polyA), bezeichnet mit „●“, und
stromaufwärts der Firefly Luciferase-cDNA des pGL3basic-Vektors mit XhoI und SacI
kloniert. Die resultierenden Vektoren wurden p●MK-Luc, p●COX2-Luc, p●Ki67-Luc,
p●E2F1-Luc, p●SV40-Luc, p●E1-Luc, and p●E4-Luc (Abb. 7 A) genannt. Das polyA diente
zur Untersuchung, ob es sich bei den beobachteten Ergebnissen um die Auswirkung eines
Enhancers oder einer alternativen Startstelle handelt, da das polyA keinen Einfluss auf die
Aktivitätsänderung eines Enhancers hat, jedoch auf die einer alternativen Startstelle.
Abb. 8: Analysierte Promotoren und AdV (nt 1-353) Segment Der Cyclooxygenase-2 (cox2)-Promotor wurde von -327 bis +59 inklusive zweier NF-κB Bindestellen kloniert. Der E2F1-Promotor beinhaltet die Basenpaare von -229 bis +66 mit drei Sp-1 Erkennungstellen, zwei E2F-Bindestellen und einer GGGCGG-Box in der translatierten Region. Der Midkine Promotor wurde mit vier GC-Boxen von -1009 bis +25 und der Ki-67-Promotor von -1121 bis +7 mit den zwei MRE- Erkennungstellen und einer E2A-Bindungstelle zur Analyse kloniert. Der adenovirale E1-Promotor beinhaltet die Basenpaare -99/+2 und der E4-Promotor die Basenpaaren -328/+4 und enthält eine E4F1-Bindestelle. Als Enhancer Element wurden die ersten 322 Basenpaare der wtAdV genomischen DNA verwendet, die auch den 5’ inverted terminal repeat und alle Verpackungssignale (PI und PII) beinhaltet und die mit den Enhancer-Elementen EEI und EEII überlappen.

4. Ergebnisse
38
Das mittels PCR amplifizierte AdV Segment (nt 1-353) wurde mit AflII und SpeI
stromaufwärts des synthetischen polyA und des Promotors eingesetzt, die resultierenden
Vektoren wurden pAd353●<Promotor>-Luc genannt (Abb. 7 B); <Promotor> wird als
Platzhalter für den verwendeten Promotor verwendet. Um den positionellen Effekt des AdV
(nt 1-353) Segmentes zu untersuchen, wurde dieses in die BamHI-SalI-Stelle des
p●<Promotor>-Luc kloniert und p●<Promotor>-Luc-Ad353 genannt (Abb. 7 C). Des Weiteren
wurde das synthetische polyA in die Vektoren pAd353●<Promotor> und p●<Promotor>-Luc-
Ad353 mit SacI/AflII-Restriktion, Nukleaseverdau und abschließender Religation entfernt,
wodurch die Vektoren pAd353<Promotor> und p<Promotor>-Luc-Ad353 entstanden (Abb. 7
D & E).
Abb. 9: Vektor-Konstrukte zur Analyse der Promotor-Transaktivierung Die folgenden Vektoren wurden konstruiert: Zur Kontrolle wurde der zu testende Promotor in die MCS des pGL3basic-Vektors 5’ der Luciferase-cDNA kloniert (A). Insertion des AdV (nt 1-353)-Segments stromaufwärts des Promotors und der polyA-Sequenz (B) und stromabwärts der Luciferase Expressionskassette (C). Die Vektoren D und E wurden durch die Deletion der polyA-Sequenz 5’ der Promotoren der Konstrukte B und C erhalten.
Je 10 µg der Vektoren und ein Zehntel der äquimolaren Menge von pRL-CMV wurden mit
der Calciumchlorid-Transfektionsmethode in 1 x 104 Zellen pro Kavität einer 24-Kavitäten

4. Ergebnisse
39
Platte transfiziert. Der Versuch wurde in zwei unabhängige Ansätze pro Konstrukt und
Transfektionsmethode durchgeführt. 48 h nach der Transfektion wurde das Zelllysat
analysiert. Alle Versuche wurden in vier unabhängigen Experimenten durchgeführt.
4.3.2 Einfluss des AdV (nt 1-353)-Segmentes auf die Aktivität der getesteten
Promotoren; mit und ohne stromaufwärts liegender polyA-Sequenz
Im nächsten Schritt wurde der Effekt des AdV (nt 1-353)-Segmentes analysiert. Um
festzustellen, ob der Einfluss des AdV (nt 1-353)-Segmentes positionsabhängig ist, wurde das
Fragment stromaufwärts und stromabwärts der Luciferase-Expressionskassette in den
Reportergen-Vektor gesetzt. Des Weiteren wurde eine synthetische polyA-Sequenz
stromaufwärts des Promotors eingesetzt um die Aktivität der kryptischen Initiations-
Startstelle zu blockieren, die im AdV (nt 1-353)-Segment lokalisiert ist. Die Firefly-
Luciferase-Aktivität der Vektoren, die das AdV (nt 1-353)-Segment enthalten, wurde mit der
Expression der korrespondierenden Vektoren p●<Promotor>-Luc ohne AdV (nt 1-353)-
Segment verglichen. Wie in Abb. 10 A gezeigt, war die Aktivität der COX-2-, E2F1-, SV40-,
AdV E1- und AdV E4-Promotoren um 146%, 149%, 99%, 116% bzw. 96% erhöht, wenn das
AdV (nt 1-353)-Segment vor der Luciferase-Expressionskassette eingesetzt wurde und keine
polyA-Sequenz vorhanden war. Wenn das AdV-Segment stromabwärts der Luciferase-
Expressionskassette lokalisiert war, wurde die Aktivität der getesteten Promotoren, mit
Ausnahme von COX-2, nicht beeinflusst. Die Aktivität des MK- und Ki-67-Promotors wurde
durch das AdV-Segment, unabhängig von der Position, nicht signifikant beeinflusst, wenn
man es mit den korrespondierenden Kontroll-Vektoren vergleicht.
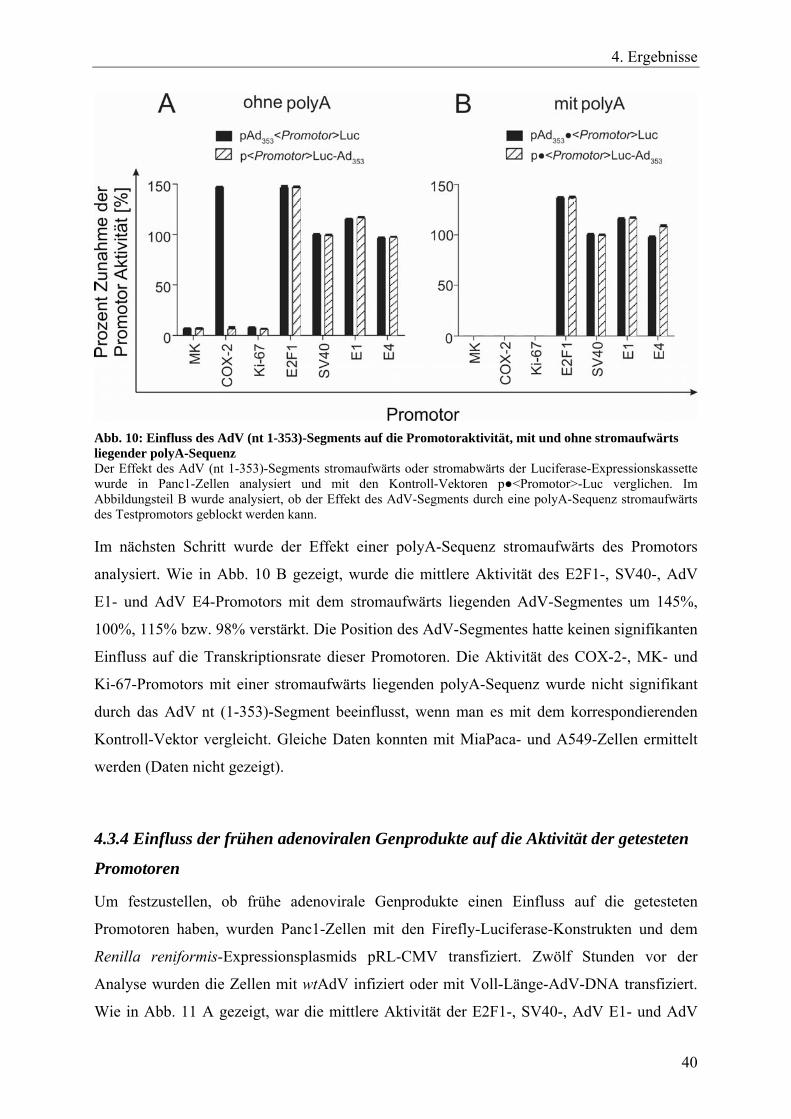
4. Ergebnisse
40
Abb. 10: Einfluss des AdV (nt 1-353)-Segments auf die Promotoraktivität, mit und ohne stromaufwärts liegender polyA-Sequenz Der Effekt des AdV (nt 1-353)-Segments stromaufwärts oder stromabwärts der Luciferase-Expressionskassette wurde in Panc1-Zellen analysiert und mit den Kontroll-Vektoren p●<Promotor>-Luc verglichen. Im Abbildungsteil B wurde analysiert, ob der Effekt des AdV-Segments durch eine polyA-Sequenz stromaufwärts des Testpromotors geblockt werden kann. Im nächsten Schritt wurde der Effekt einer polyA-Sequenz stromaufwärts des Promotors
analysiert. Wie in Abb. 10 B gezeigt, wurde die mittlere Aktivität des E2F1-, SV40-, AdV
E1- und AdV E4-Promotors mit dem stromaufwärts liegenden AdV-Segmentes um 145%,
100%, 115% bzw. 98% verstärkt. Die Position des AdV-Segmentes hatte keinen signifikanten
Einfluss auf die Transkriptionsrate dieser Promotoren. Die Aktivität des COX-2-, MK- und
Ki-67-Promotors mit einer stromaufwärts liegenden polyA-Sequenz wurde nicht signifikant
durch das AdV nt (1-353)-Segment beeinflusst, wenn man es mit dem korrespondierenden
Kontroll-Vektor vergleicht. Gleiche Daten konnten mit MiaPaca- und A549-Zellen ermittelt
werden (Daten nicht gezeigt).
4.3.4 Einfluss der frühen adenoviralen Genprodukte auf die Aktivität der getesteten
Promotoren
Um festzustellen, ob frühe adenovirale Genprodukte einen Einfluss auf die getesteten
Promotoren haben, wurden Panc1-Zellen mit den Firefly-Luciferase-Konstrukten und dem
Renilla reniformis-Expressionsplasmids pRL-CMV transfiziert. Zwölf Stunden vor der
Analyse wurden die Zellen mit wtAdV infiziert oder mit Voll-Länge-AdV-DNA transfiziert.
Wie in Abb. 11 A gezeigt, war die mittlere Aktivität der E2F1-, SV40-, AdV E1- und AdV
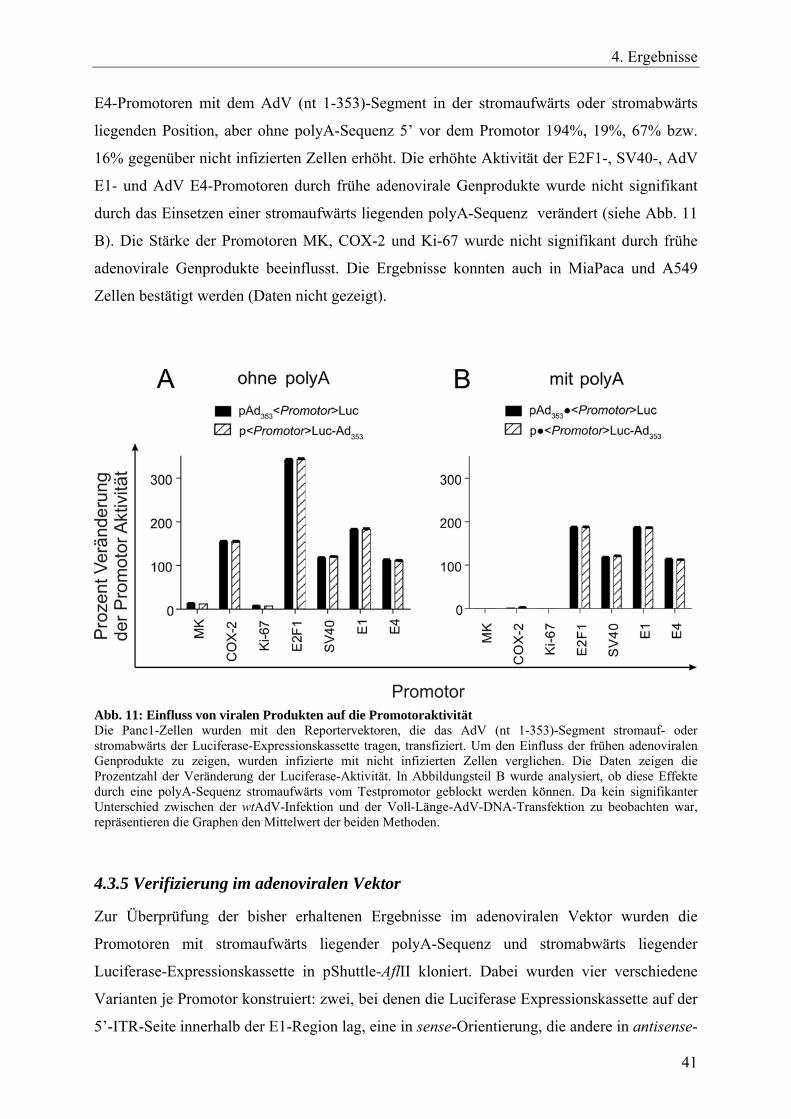
4. Ergebnisse
41
E4-Promotoren mit dem AdV (nt 1-353)-Segment in der stromaufwärts oder stromabwärts
liegenden Position, aber ohne polyA-Sequenz 5’ vor dem Promotor 194%, 19%, 67% bzw.
16% gegenüber nicht infizierten Zellen erhöht. Die erhöhte Aktivität der E2F1-, SV40-, AdV
E1- und AdV E4-Promotoren durch frühe adenovirale Genprodukte wurde nicht signifikant
durch das Einsetzen einer stromaufwärts liegenden polyA-Sequenz verändert (siehe Abb. 11
B). Die Stärke der Promotoren MK, COX-2 und Ki-67 wurde nicht signifikant durch frühe
adenovirale Genprodukte beeinflusst. Die Ergebnisse konnten auch in MiaPaca und A549
Zellen bestätigt werden (Daten nicht gezeigt).
Abb. 11: Einfluss von viralen Produkten auf die Promotoraktivität Die Panc1-Zellen wurden mit den Reportervektoren, die das AdV (nt 1-353)-Segment stromauf- oder stromabwärts der Luciferase-Expressionskassette tragen, transfiziert. Um den Einfluss der frühen adenoviralen Genprodukte zu zeigen, wurden infizierte mit nicht infizierten Zellen verglichen. Die Daten zeigen die Prozentzahl der Veränderung der Luciferase-Aktivität. In Abbildungsteil B wurde analysiert, ob diese Effekte durch eine polyA-Sequenz stromaufwärts vom Testpromotor geblockt werden können. Da kein signifikanter Unterschied zwischen der wtAdV-Infektion und der Voll-Länge-AdV-DNA-Transfektion zu beobachten war, repräsentieren die Graphen den Mittelwert der beiden Methoden.
4.3.5 Verifizierung im adenoviralen Vektor
Zur Überprüfung der bisher erhaltenen Ergebnisse im adenoviralen Vektor wurden die
Promotoren mit stromaufwärts liegender polyA-Sequenz und stromabwärts liegender
Luciferase-Expressionskassette in pShuttle-AflII kloniert. Dabei wurden vier verschiedene
Varianten je Promotor konstruiert: zwei, bei denen die Luciferase Expressionskassette auf der
5’-ITR-Seite innerhalb der E1-Region lag, eine in sense-Orientierung, die andere in antisense-

4. Ergebnisse
42
Orientierung. Zum anderen wurden zwei Vektoren konstruiert, bei denen die
Expressionskassette nahe des 3’-ITRs innerhalb der E4-Region in sense und antisense-
Orientierung eingesetzt wurde. Als Kontrolle wurde ein pShuttle-Plasmid konstruiert, welches
eine CMV-IE-Promotor-regulierte Luciferase-cDNA in der E1-Region in sense-Orientierung
enthielt. Anschließend wurden alle Konstrukte mit pAdEasy1 rekombiniert, so dass volllange
adenovirale DNA entstand. Mit dieser DNA wurden dann replikationsdefekte Adenoviren in
Zellkultur hergestellt.
Abb. 12: Verwendete adenovirale Konstrukte Die folgenden Vektoren wurden konstruiert: Zur Kontrolle wurde in die E1A-Region des AdV eine mit CMV-IE Promotor kontrollierte Luciferase-cDNA kloniert (A). Insertion des Promotors mit der Luciferase cDNA in die E1A-Region des AdV zum einen in sense-Orientierung (B) und in antisense-Orientierung (C). Insertion der Expressionskassette zwischen der E4-Region und den 3’ITR ergab in sense-Orientierung den Vektor 3’ <Promotor S>-Luc (D) und in antisense-Orientierung den Vektor 3’ <Promotor AS>-Luc (E).
Zur Analyse der Promotoraktivität und -spezifität wurden zuerst die pShuttle-Konstrukte im
dualen Luciferase-Assay untersucht. Dazu wurden, wie vorher schon beschrieben, 10µg je
pShuttle-Konstrukt mit einem Zehntel der äquimolaren Menge an pRL-CMV kotransfiziert.
Zum einen wurde ein Test mit einer Zelllinie, in der der Promotor aktiv ist, und zum anderen
mit einer Zelllinie, in der er inaktiv ist, durchgeführt. 48h nach der Transfektion wurde der
duale Luciferase-Assay durchgeführt und die Werte relativ zur CMV-IE-Aktivität gesetzt (s.
Abb. 13 A). Im zweiten Schritt wurden dieselben Zelllinien mit den replikationsdefekten
Adenoviren mit einer MOI von 5 infiziert und nach 24 h wurde die Promotoraktivität wieder
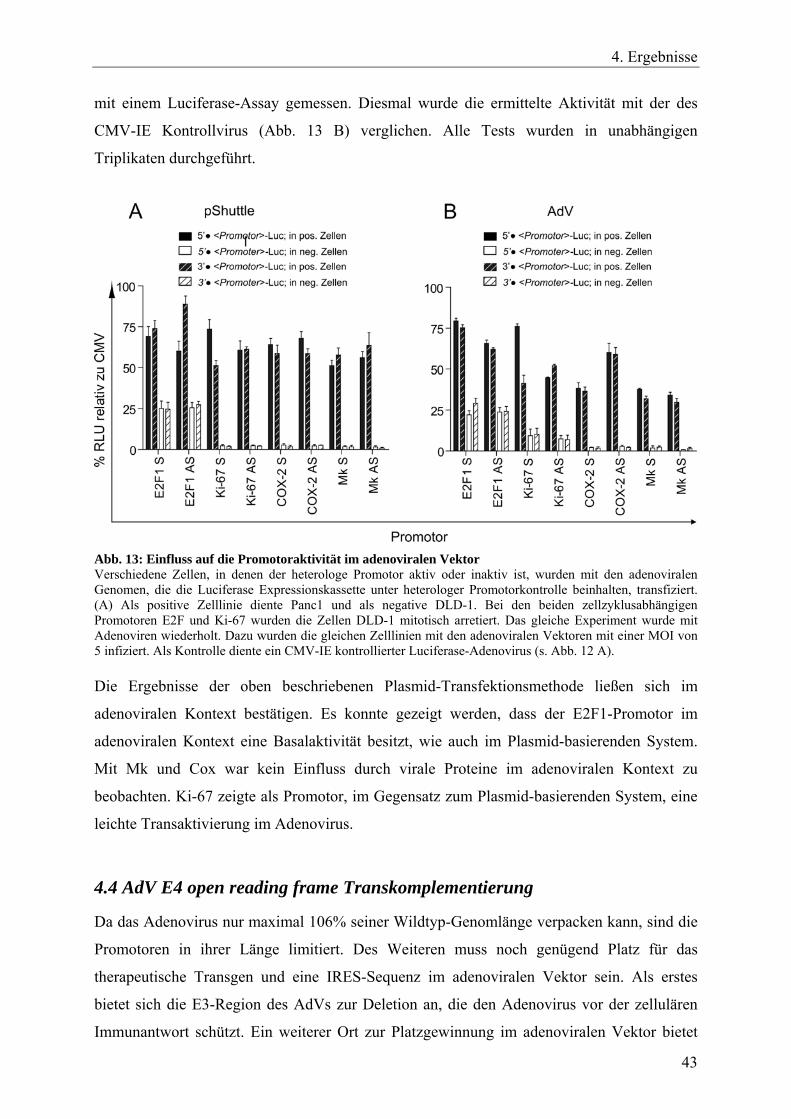
4. Ergebnisse
43
mit einem Luciferase-Assay gemessen. Diesmal wurde die ermittelte Aktivität mit der des
CMV-IE Kontrollvirus (Abb. 13 B) verglichen. Alle Tests wurden in unabhängigen
Triplikaten durchgeführt.
Abb. 13: Einfluss auf die Promotoraktivität im adenoviralen Vektor Verschiedene Zellen, in denen der heterologe Promotor aktiv oder inaktiv ist, wurden mit den adenoviralen Genomen, die die Luciferase Expressionskassette unter heterologer Promotorkontrolle beinhalten, transfiziert. (A) Als positive Zelllinie diente Panc1 und als negative DLD-1. Bei den beiden zellzyklusabhängigen Promotoren E2F und Ki-67 wurden die Zellen DLD-1 mitotisch arretiert. Das gleiche Experiment wurde mit Adenoviren wiederholt. Dazu wurden die gleichen Zelllinien mit den adenoviralen Vektoren mit einer MOI von 5 infiziert. Als Kontrolle diente ein CMV-IE kontrollierter Luciferase-Adenovirus (s. Abb. 12 A). Die Ergebnisse der oben beschriebenen Plasmid-Transfektionsmethode ließen sich im
adenoviralen Kontext bestätigen. Es konnte gezeigt werden, dass der E2F1-Promotor im
adenoviralen Kontext eine Basalaktivität besitzt, wie auch im Plasmid-basierenden System.
Mit Mk und Cox war kein Einfluss durch virale Proteine im adenoviralen Kontext zu
beobachten. Ki-67 zeigte als Promotor, im Gegensatz zum Plasmid-basierenden System, eine
leichte Transaktivierung im Adenovirus.
4.4 AdV E4 open reading frame Transkomplementierung
Da das Adenovirus nur maximal 106% seiner Wildtyp-Genomlänge verpacken kann, sind die
Promotoren in ihrer Länge limitiert. Des Weiteren muss noch genügend Platz für das
therapeutische Transgen und eine IRES-Sequenz im adenoviralen Vektor sein. Als erstes
bietet sich die E3-Region des AdVs zur Deletion an, die den Adenovirus vor der zellulären
Immunantwort schützt. Ein weiterer Ort zur Platzgewinnung im adenoviralen Vektor bietet

4. Ergebnisse
44
die E4-Region mit den verschiedenen offenen Leserahmen (orf). Diese orfs besitzen in der
frühen und späten Phase des viralen Infektionszyklus verschiedene Aufgaben. Daher soll
untersucht werden, wie sich die Deletion verschiedener E4 orfs auf die virale Replikation
auswirkt.
4.4.1 Konstruktion der AdV E4 orfs exprimierenden Plasmide
Um zu untersuchen, welche E4 orfs transkomplementierend wirken, wurden alle sechs
verschiedenen orfs der E4 Region in eukaryotische Expressionsplasmide kloniert. Dazu
wurden die orfs jeweils mittels Proofreading-Polymerase amplifiziert und durch die Primer
am 5’- und am 3’-Ende mit den Restriktionsenzym-Erkennungssequenzen von NotI und XhoI
erweitert. Abschließend wurden diese Produkte mittels NotI und XhoI in pCR3.1 subkloniert.
Die Integrität der Konstrukte wurde durch Sequenzierungen überprüft.
4.4.2 Transkomplementierungsbestimmung mit Luciferase-Assay
Es wurde mit Hilfe von Transfektionen und nachfolgenden Infektionen untersucht, welche E4
orfs transkomplementierend wirken; wodurch gezeigt werden kann, welche E4 orfs die
Replikation eines E4-deletierten replikationsdefekten Adenovirus unterstützen. Hierzu
wurden HEK-293-MLV-Luc3-Zellen zunächst mit einem oder mehreren orfs transfiziert.
Insgesamt gibt es 63 Möglichkeiten, die sechs orfs zu kombinieren, dies zeigt sich anhand der
Formel:
⎟⎟⎠
⎞⎜⎜⎝
⎛=
kn
Ckn
Die Kombinationen wurden nummeriert (Tab. 2).
Als Transkomplementierungskontrolle wurden Zellen mit einem Voll-Länge-Konstrukt
transfiziert, das alle adenoviralen Genregionen umfasst.
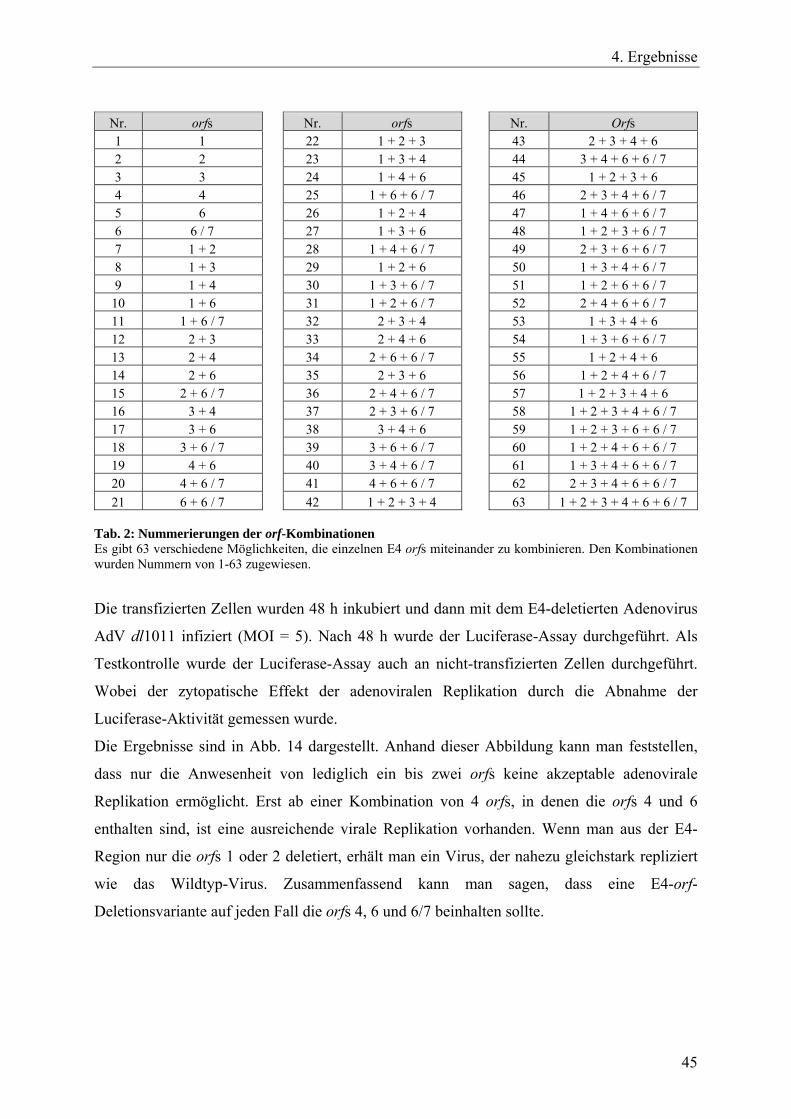
4. Ergebnisse
45
Nr. orfs Nr. orfs Nr. Orfs 1 1 22 1 + 2 + 3 43 2 + 3 + 4 + 6 2 2 23 1 + 3 + 4 44 3 + 4 + 6 + 6 / 7 3 3 24 1 + 4 + 6 45 1 + 2 + 3 + 6 4 4 25 1 + 6 + 6 / 7 46 2 + 3 + 4 + 6 / 7 5 6 26 1 + 2 + 4 47 1 + 4 + 6 + 6 / 7 6 6 / 7 27 1 + 3 + 6 48 1 + 2 + 3 + 6 / 7 7 1 + 2 28 1 + 4 + 6 / 7 49 2 + 3 + 6 + 6 / 7 8 1 + 3 29 1 + 2 + 6 50 1 + 3 + 4 + 6 / 7 9 1 + 4 30 1 + 3 + 6 / 7 51 1 + 2 + 6 + 6 / 7
10 1 + 6 31 1 + 2 + 6 / 7 52 2 + 4 + 6 + 6 / 7 11 1 + 6 / 7 32 2 + 3 + 4 53 1 + 3 + 4 + 6 12 2 + 3 33 2 + 4 + 6 54 1 + 3 + 6 + 6 / 7 13 2 + 4 34 2 + 6 + 6 / 7 55 1 + 2 + 4 + 6 14 2 + 6 35 2 + 3 + 6 56 1 + 2 + 4 + 6 / 7 15 2 + 6 / 7 36 2 + 4 + 6 / 7 57 1 + 2 + 3 + 4 + 6 16 3 + 4 37 2 + 3 + 6 / 7 58 1 + 2 + 3 + 4 + 6 / 7 17 3 + 6 38 3 + 4 + 6 59 1 + 2 + 3 + 6 + 6 / 7 18 3 + 6 / 7 39 3 + 6 + 6 / 7 60 1 + 2 + 4 + 6 + 6 / 7 19 4 + 6 40 3 + 4 + 6 / 7 61 1 + 3 + 4 + 6 + 6 / 7 20 4 + 6 / 7 41 4 + 6 + 6 / 7 62 2 + 3 + 4 + 6 + 6 / 7 21 6 + 6 / 7 42 1 + 2 + 3 + 4 63 1 + 2 + 3 + 4 + 6 + 6 / 7
Tab. 2: Nummerierungen der orf-Kombinationen Es gibt 63 verschiedene Möglichkeiten, die einzelnen E4 orfs miteinander zu kombinieren. Den Kombinationen wurden Nummern von 1-63 zugewiesen.
Die transfizierten Zellen wurden 48 h inkubiert und dann mit dem E4-deletierten Adenovirus
AdV dl1011 infiziert (MOI = 5). Nach 48 h wurde der Luciferase-Assay durchgeführt. Als
Testkontrolle wurde der Luciferase-Assay auch an nicht-transfizierten Zellen durchgeführt.
Wobei der zytopatische Effekt der adenoviralen Replikation durch die Abnahme der
Luciferase-Aktivität gemessen wurde.
Die Ergebnisse sind in Abb. 14 dargestellt. Anhand dieser Abbildung kann man feststellen,
dass nur die Anwesenheit von lediglich ein bis zwei orfs keine akzeptable adenovirale
Replikation ermöglicht. Erst ab einer Kombination von 4 orfs, in denen die orfs 4 und 6
enthalten sind, ist eine ausreichende virale Replikation vorhanden. Wenn man aus der E4-
Region nur die orfs 1 oder 2 deletiert, erhält man ein Virus, der nahezu gleichstark repliziert
wie das Wildtyp-Virus. Zusammenfassend kann man sagen, dass eine E4-orf-
Deletionsvariante auf jeden Fall die orfs 4, 6 und 6/7 beinhalten sollte.
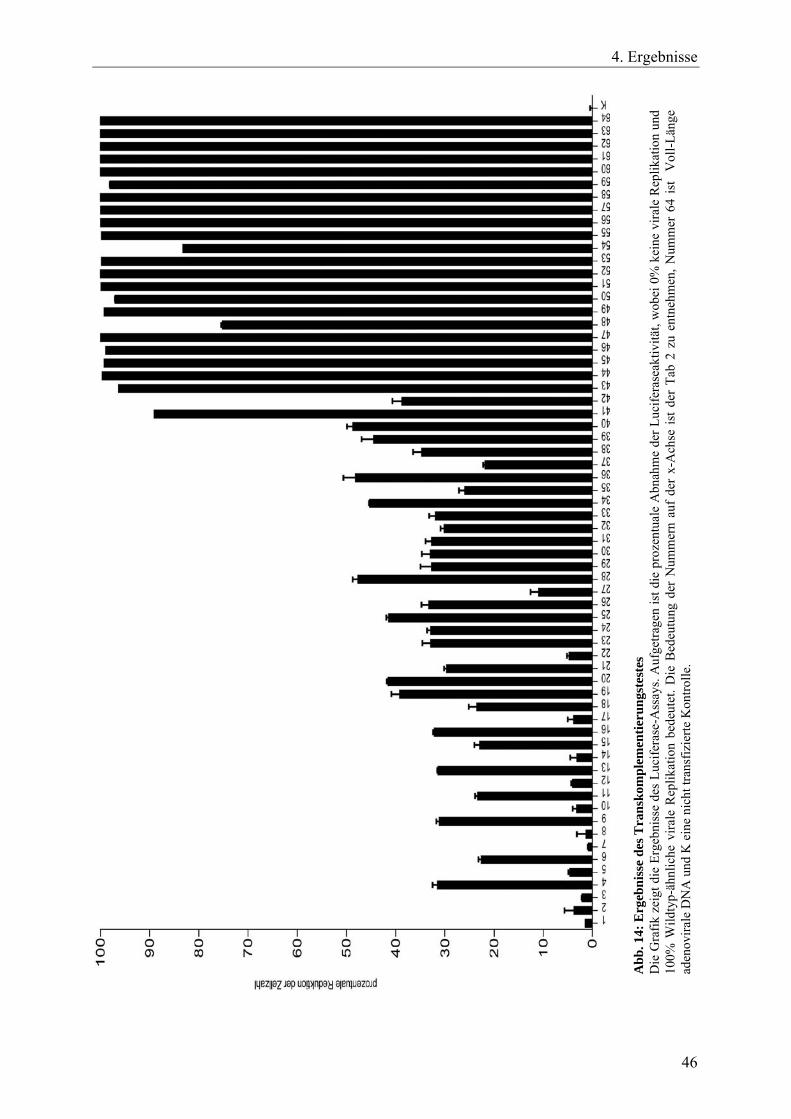
4. Ergebnisse
46
Abb
. 14:
Erg
ebni
sse
des T
rans
kom
plem
entie
rung
stes
tes
Die
Gra
fik z
eigt
die
Erg
ebni
sse
des
Luci
fera
se-A
ssay
s. A
ufge
trage
n is
t die
pro
zent
uale
Abn
ahm
e de
r Luc
ifera
seak
tivitä
t, w
obei
0%
kei
ne v
irale
Rep
likat
ion
und
100%
Wild
typ-
ähnl
iche
vira
le R
eplik
atio
n be
deut
et. D
ie B
edeu
tung
der
Num
mer
n au
f de
r x-
Ach
se is
t der
Tab
2 z
u en
tneh
men
, Num
mer
64
ist
Vol
l-Län
ge
aden
ovira
le D
NA
und
K e
ine
nich
t tra
nsfiz
ierte
Kon
trolle
.

4. Ergebnisse
47
4.5 Konstruktion des bedingt replikationskompetenten adenoviralen
Vektorsystems
Um ein Vektorsystem zu erhalten, in dem man zum einem die E1A-Region, zum anderen die
E4-Region unter heterologer Promotorkontrolle stellen kann, musste ein neuer adenoviraler
backbone-Vektor konstruiert werden, in dem es möglich ist, die E4-Region zu manipulieren.
Daher wurde der pAdEasy-Vektor als Grundlage verwendet, damit die manipulierte E1
Region mittels pShuttle hineinrekombiniert werden kann. Durch die homologe
Rekombination entsteht eine zirkuläre rekombinante Adenovirus-DNA, die durch PacI-
Restriktion linearisiert und dann in HEK-293- oder 911E4-Zellen zur Vektorproduktion
transfiziert werden kann (s. Abb. 15).
Zuerst wurde in die EcoRI- und NdeI-Restriktionsschnittstelle des pBR322-Vektors ein
synthetisches Oligonukleotid eingesetzt. Dieses Oligonukleotid war so konstruiert, dass es
verschiedene Restriktionsschnittstellen zur weiteren Klonierung beinhaltet (s. Abb 15).
Anschließend wurde mit einer Proofreading-Polymerase ein linker homologer Arm (LHA)
mit eine Länge von 3052 bp 5’ der E4-Region bis zur SpeI-Seite aus dem Adenovirus
amplifiziert. Durch die Primer wurden die Restriktionsschnittstellen NheI und KpnI eingefügt,
und mit diesen wurde das LHA-Fragment dann in den modifizierten pBR322-Vektor
eingesetzt. Der nächste Schritt war die Klonierung des rechten homologen Arms (RHA) in
das Plasmid. Dazu wurde wiederum mit einer Proofreading-Polymerase ein Fragment aus
dem pAdEasy-1-Plasmid amplifiziert. Dieses besitzt eine Länge von 1179 bp und liegt
zwischen dem 3’ Ende des E4-Promotors und dem Vektor-backbone von pAdEasy-1. Durch
die Primer wurden die Restriktionsschnittstellen XbaI und MluI eingefügt. Zum Abschluss der
Grundkonstruktion wurde das Kanamycin-Resistenzgen aus dem pCR3.1-Plasmid mit NotI /
SacII in das Grundkonstrukt subkloniert.
Jetzt können alle Promotoren, die die E4-Region kontrollieren sollen, mit den
Restriktionsenzymen XhoI und SacI in das Vektorsystem kloniert werden. Die E4-Region
oder andere (therapeutische) Gene können über ClaI und SalI in dieses Vektorsystem kloniert
werden. Anschließend kann diese modifizierte E4-Region mit SpeI und PacI oder durch
homologe Rekombination in pAdEasy-1 kloniert oder rekombinert werden. Als
Selektionsmarker dient Kanamycin. Nach erfolgreicher Klonierung kann das Kanamycin-Gen
mit PI-PspI oder SwaI deletiert werden.
Der Promotor zur Kontrolle der E1-Region wurde mit MluI und XhoI in pShuttle-AflII-HSV-
tk-IRES subkloniert. Die E1-Region aus dem Plasmid pOW127 wurde in das pShuttle-
Plasmid mit AflII und NotI kloniert.
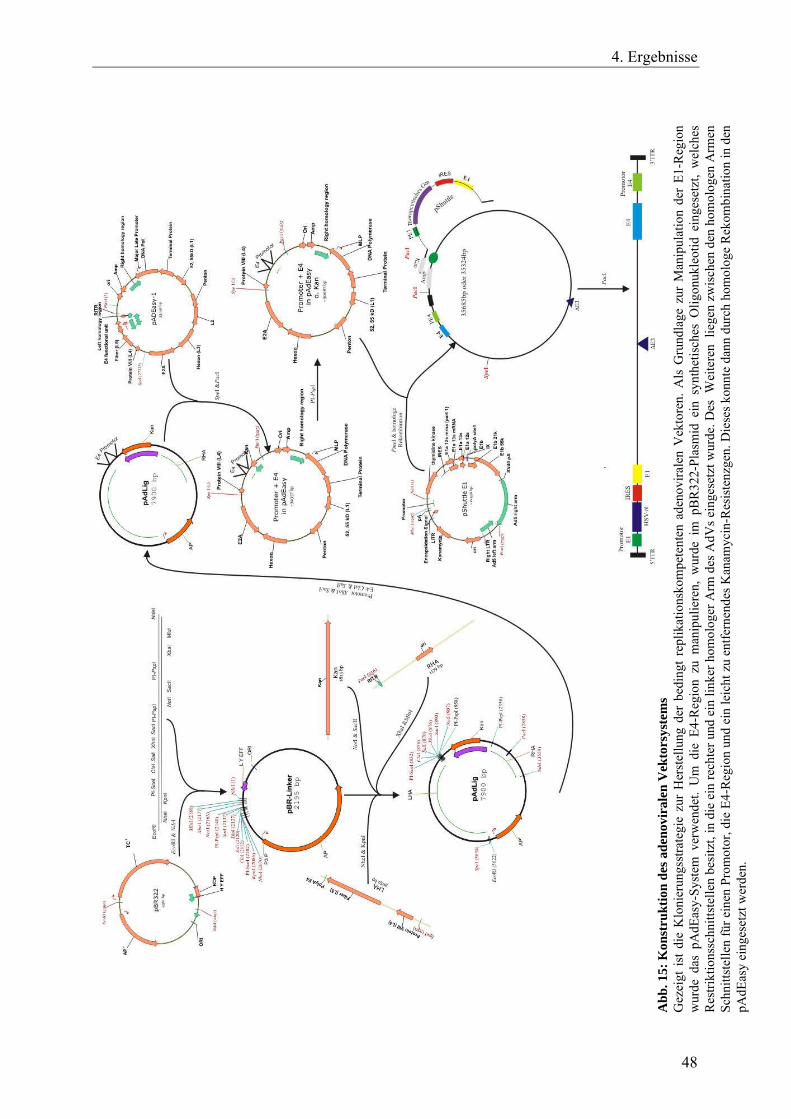
4. Ergebnisse
48
Abb
. 15:
Kon
stru
ktio
n de
s ade
novi
rale
n V
ekto
rsys
tem
s G
ezei
gt is
t die
Klo
nier
ungs
stra
tegi
e zu
r H
erst
ellu
ng d
er b
edin
gt r
eplik
atio
nsko
mpe
tent
en a
deno
vira
len
Vek
tore
n. A
ls G
rund
lage
zur
Man
ipul
atio
n de
r E1
-Reg
ion
wur
de d
as p
AdE
asy-
Syst
em v
erw
ende
t. U
m d
ie E
4-R
egio
n zu
man
ipul
iere
n, w
urde
im
pB
R32
2-Pl
asm
id e
in s
ynth
etis
ches
Olig
onuk
leot
id e
inge
setz
t, w
elch
es
Res
trikt
ions
schn
ittst
elle
n be
sitz
t, in
die
ein
rech
ter u
nd e
in li
nker
hom
olog
er A
rm d
es A
dVs e
inge
setz
t wur
de. D
es W
eite
ren
lieg
en z
wis
chen
den
hom
olog
en A
rmen
Sc
hnitt
stel
len
für e
inen
Pro
mot
or, d
ie E
4-R
egio
n un
d ei
n le
icht
zu
entfe
rnen
des K
anam
ycin
-Res
iste
nzge
n. D
iese
s kon
nte
dann
dur
ch h
omol
oge
Rek
ombi
natio
n in
den
pA
dEas
y ei
nges
etzt
wer
den.
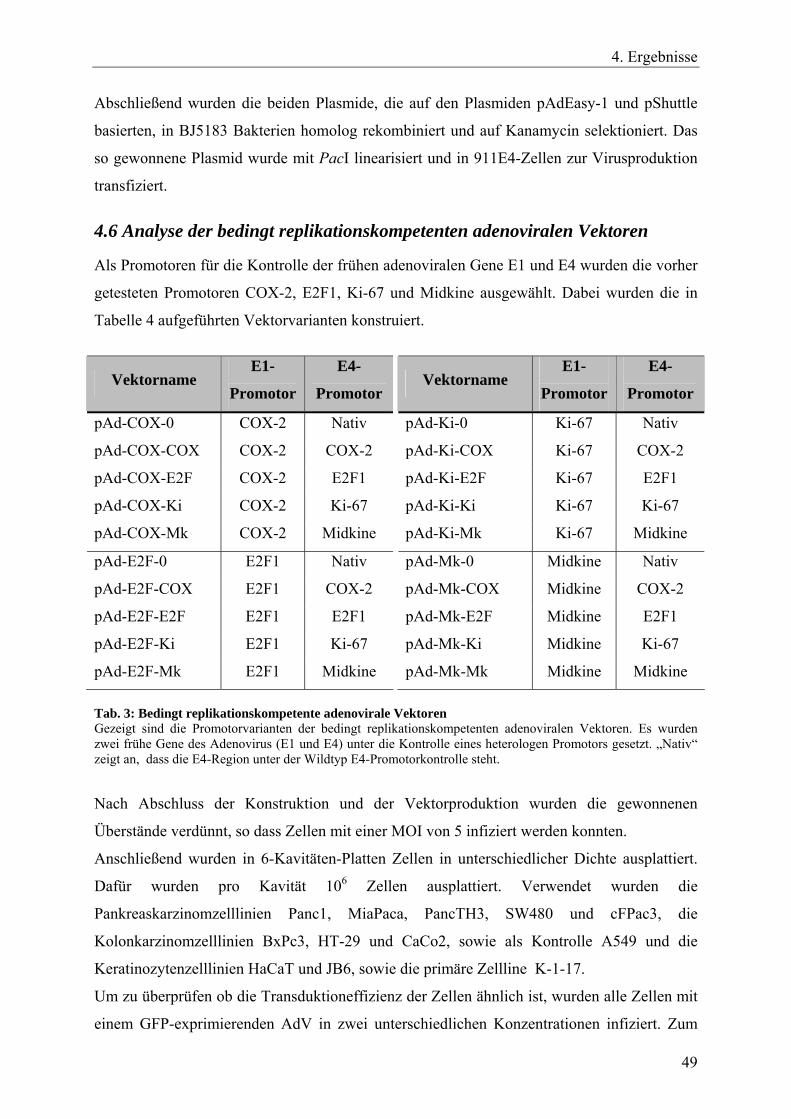
4. Ergebnisse
49
Abschließend wurden die beiden Plasmide, die auf den Plasmiden pAdEasy-1 und pShuttle
basierten, in BJ5183 Bakterien homolog rekombiniert und auf Kanamycin selektioniert. Das
so gewonnene Plasmid wurde mit PacI linearisiert und in 911E4-Zellen zur Virusproduktion
transfiziert.
4.6 Analyse der bedingt replikationskompetenten adenoviralen Vektoren
Als Promotoren für die Kontrolle der frühen adenoviralen Gene E1 und E4 wurden die vorher
getesteten Promotoren COX-2, E2F1, Ki-67 und Midkine ausgewählt. Dabei wurden die in
Tabelle 4 aufgeführten Vektorvarianten konstruiert.
Tab. 3: Bedingt replikationskompetente adenovirale Vektoren Gezeigt sind die Promotorvarianten der bedingt replikationskompetenten adenoviralen Vektoren. Es wurden zwei frühe Gene des Adenovirus (E1 und E4) unter die Kontrolle eines heterologen Promotors gesetzt. „Nativ“ zeigt an, dass die E4-Region unter der Wildtyp E4-Promotorkontrolle steht.
Nach Abschluss der Konstruktion und der Vektorproduktion wurden die gewonnenen
Überstände verdünnt, so dass Zellen mit einer MOI von 5 infiziert werden konnten.
Anschließend wurden in 6-Kavitäten-Platten Zellen in unterschiedlicher Dichte ausplattiert.
Dafür wurden pro Kavität 106 Zellen ausplattiert. Verwendet wurden die
Pankreaskarzinomzelllinien Panc1, MiaPaca, PancTH3, SW480 und cFPac3, die
Kolonkarzinomzelllinien BxPc3, HT-29 und CaCo2, sowie als Kontrolle A549 und die
Keratinozytenzelllinien HaCaT und JB6, sowie die primäre Zellline K-1-17.
Um zu überprüfen ob die Transduktioneffizienz der Zellen ähnlich ist, wurden alle Zellen mit
einem GFP-exprimierenden AdV in zwei unterschiedlichen Konzentrationen infiziert. Zum
Vektorname E1-
Promotor
E4-
Promotor
pAd-COX-0 COX-2 Nativ
pAd-COX-COX COX-2 COX-2
pAd-COX-E2F COX-2 E2F1
pAd-COX-Ki COX-2 Ki-67
pAd-COX-Mk COX-2 Midkine
pAd-E2F-0 E2F1 Nativ
pAd-E2F-COX E2F1 COX-2
pAd-E2F-E2F E2F1 E2F1
pAd-E2F-Ki E2F1 Ki-67
pAd-E2F-Mk E2F1 Midkine
Vektorname E1-
Promotor
E4-
Promotor
pAd-Ki-0 Ki-67 Nativ
pAd-Ki-COX Ki-67 COX-2
pAd-Ki-E2F Ki-67 E2F1
pAd-Ki-Ki Ki-67 Ki-67
pAd-Ki-Mk Ki-67 Midkine
pAd-Mk-0 Midkine Nativ
pAd-Mk-COX Midkine COX-2
pAd-Mk-E2F Midkine E2F1
pAd-Mk-Ki Midkine Ki-67
pAd-Mk-Mk Midkine Midkine
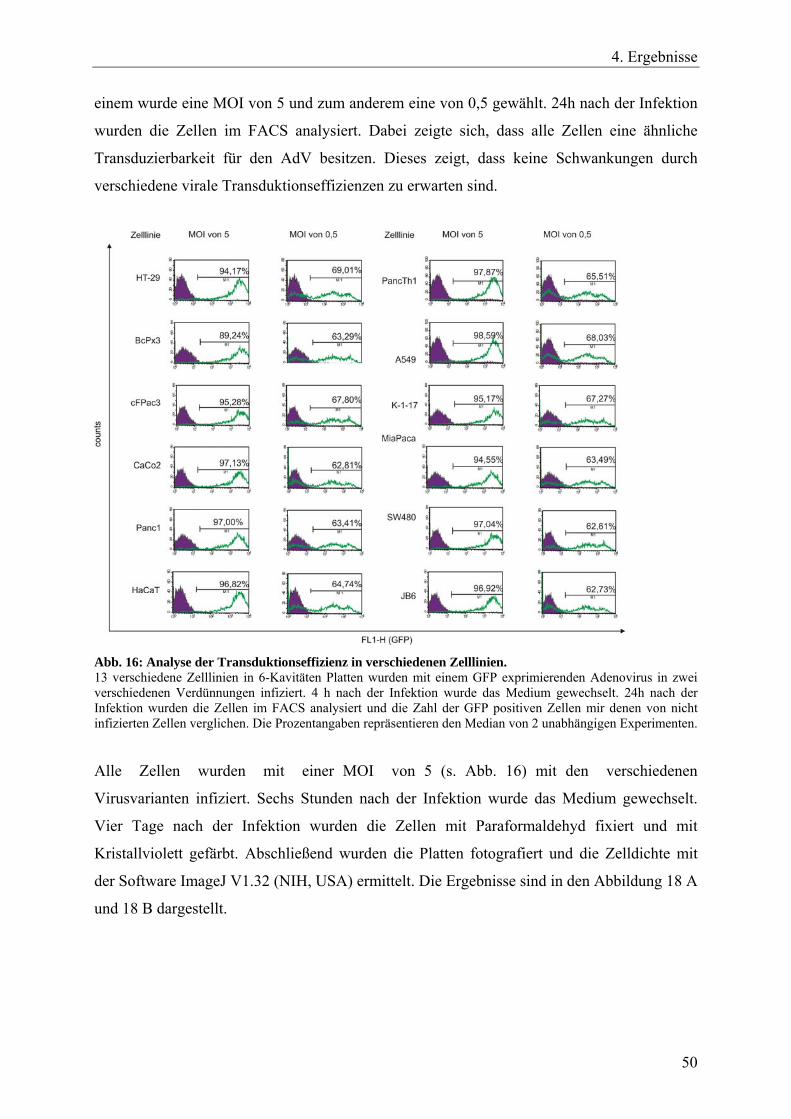
4. Ergebnisse
50
einem wurde eine MOI von 5 und zum anderem eine von 0,5 gewählt. 24h nach der Infektion
wurden die Zellen im FACS analysiert. Dabei zeigte sich, dass alle Zellen eine ähnliche
Transduzierbarkeit für den AdV besitzen. Dieses zeigt, dass keine Schwankungen durch
verschiedene virale Transduktionseffizienzen zu erwarten sind.
Abb. 16: Analyse der Transduktionseffizienz in verschiedenen Zelllinien. 13 verschiedene Zelllinien in 6-Kavitäten Platten wurden mit einem GFP exprimierenden Adenovirus in zwei verschiedenen Verdünnungen infiziert. 4 h nach der Infektion wurde das Medium gewechselt. 24h nach der Infektion wurden die Zellen im FACS analysiert und die Zahl der GFP positiven Zellen mir denen von nicht infizierten Zellen verglichen. Die Prozentangaben repräsentieren den Median von 2 unabhängigen Experimenten. Alle Zellen wurden mit einer MOI von 5 (s. Abb. 16) mit den verschiedenen
Virusvarianten infiziert. Sechs Stunden nach der Infektion wurde das Medium gewechselt.
Vier Tage nach der Infektion wurden die Zellen mit Paraformaldehyd fixiert und mit
Kristallviolett gefärbt. Abschließend wurden die Platten fotografiert und die Zelldichte mit
der Software ImageJ V1.32 (NIH, USA) ermittelt. Die Ergebnisse sind in den Abbildung 18 A
und 18 B dargestellt.
4. Ergebnisse
51
Vektor / Platte A B
Kontrolle
wtAdV
pAd-COX
pAd-COX-E2F
pAd-COX-COX
pAd-COX-Ki
pAd-COX-Mk
Legende
Abb. 17: Exemplarische Darstellung von Kristallviolett gefärbten und mit BRAV infizierten Zellen Gezeigt sind zwei 6-Kavitäten Platten in denen in jeder Kavität eine andere Zelllinie ausplattiert wurde. Diese Zellen wurden mit pAd-COX-E2F mit einer MOI von 5 infiziert. Nach 4 Tagen wurden die Zellen fixiert und gefärbt. Auf Grund der Menge wurde darauf verzichtet alle Konstrukte als Lichtbild zu zeigen. Im unteren Teil des Bildes wurde in den skizzierten 6-Kavitäten Platten die Anordnung der Zelllinien dargestellt.

4. Ergebnisse
52
Dabei konnte gezeigt werden, dass das AdV, das E2F1 als Promotor in der E1-Region enthält,
in allen Tumoren repliziert, jedoch nicht in primären Zellen und Keratinozyten. Dies gilt aber
nur für die Konstrukte in denen der E4-Promotor durch COX-2, Ki-67 oder Midkine ersetzt
wurde.
Bei den Konstrukten mit Mk und Ki-67 wurde eine höhere Spezifität des Virus erreicht, wenn
auch die E4-Region unter heterologe Promotorkontrolle gestellt wurde. Jedoch brachte die
Kombination des gleichen heterologen Promotors zur Kontrolle der E1- und der E4-Region
nur eine leichte Veränderung in der Spezifität des Virus. In ein paar Fällen replizierte das
Virus stärker als in einer Kombination mit zwei verschiedenen heterologen Promotoren. Alle
Viren, deren E1-Region durch COX-2-, Ki-67- oder Midkine-Promotoren kontrolliert wurden,
zeigten eine Spezifität für Pankreas- und Kolonkarzinome, jedoch replizierten sie auch in den
Lungenepithelzellen A549, jedoch nicht in den Keratinozyten, die als Kontrolle dienten. Die
Kombination von COX-2 zur Regulation der E1-Region und Ki-67 zur Regulation der E4-
Region zeigt eine erhöhte Spezifität zu Kolon- statt zu Pankreaskarzinomen.
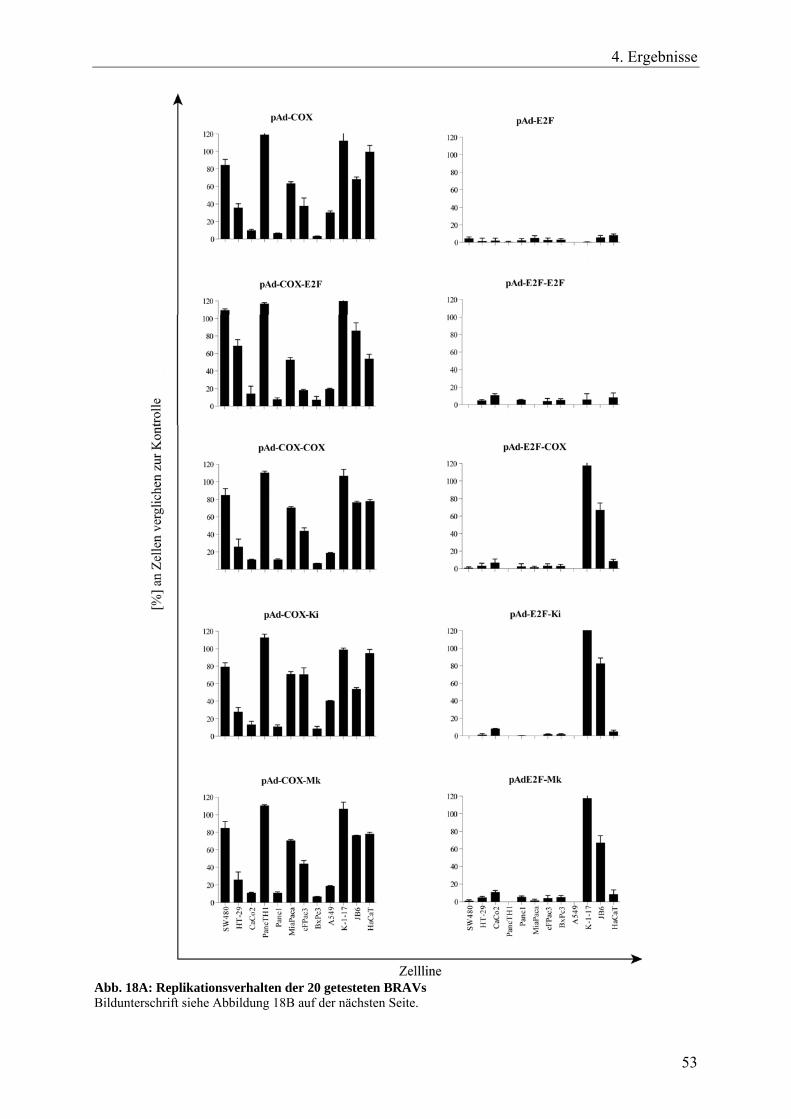
4. Ergebnisse
53
Abb. 18A: Replikationsverhalten der 20 getesteten BRAVs Bildunterschrift siehe Abbildung 18B auf der nächsten Seite.
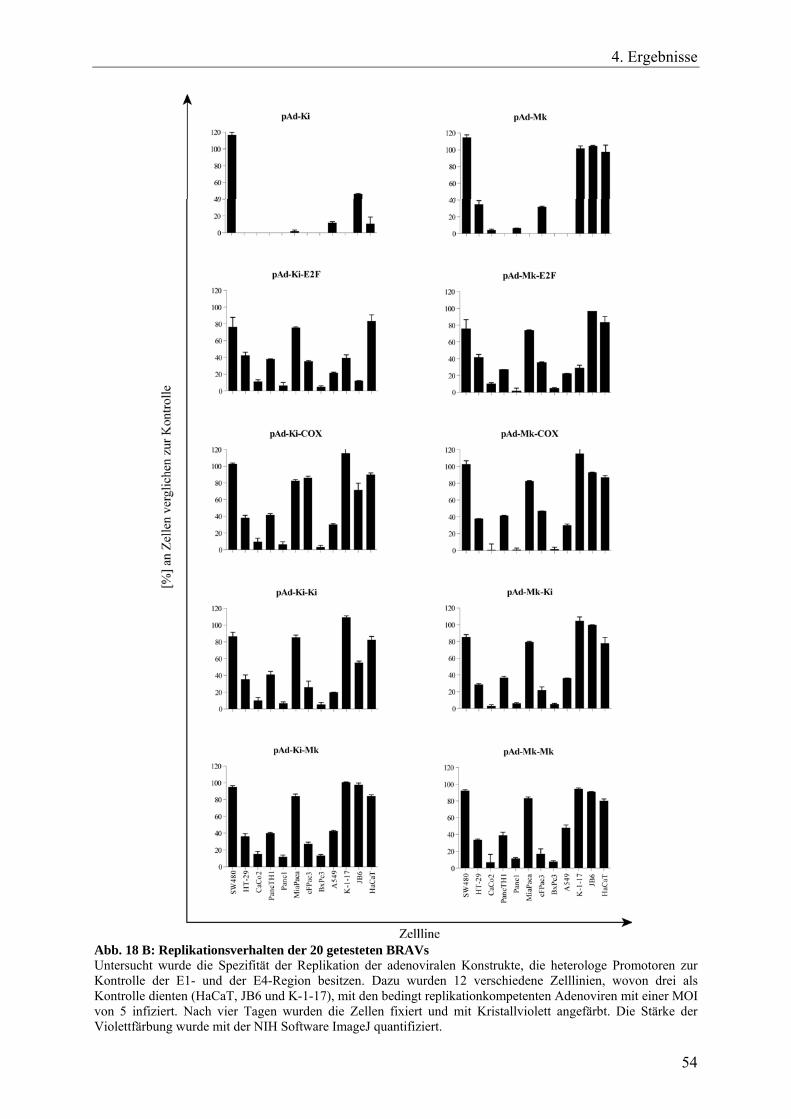
4. Ergebnisse
54
Abb. 18 B: Replikationsverhalten der 20 getesteten BRAVs Untersucht wurde die Spezifität der Replikation der adenoviralen Konstrukte, die heterologe Promotoren zur Kontrolle der E1- und der E4-Region besitzen. Dazu wurden 12 verschiedene Zelllinien, wovon drei als Kontrolle dienten (HaCaT, JB6 und K-1-17), mit den bedingt replikationkompetenten Adenoviren mit einer MOI von 5 infiziert. Nach vier Tagen wurden die Zellen fixiert und mit Kristallviolett angefärbt. Die Stärke der Violettfärbung wurde mit der NIH Software ImageJ quantifiziert.

5. Diskussion
55
5. Diskussion
5.1 Promotoranalyse
Um bedingt replikationskompetente adenovirale Vektoren (BRAV) zur Onkolyse
herzustellen, die auf der transkriptionellen Ebene kontrolliert werden, ist es unverzichtbar
Promotoren zu finden, die in dem Zielgewebe, in diesem Fall in Pankreas- und
Kolonkarzinomen, aktiv sind und in anderem, gesunden Gewebe nicht. Dazu wurden
verschiedene gewebespezifische und proliferationsabhängige Promotorfragmente getestet.
Auf Grund der limitierten Verpackungskapazität eines Adenovirus musste die Wahl der
Fragmente ein Kompromiss aus Erhalt der Spezifität des Promotors und Fragmentlänge sein.
Das Ziel war es, Promotoren mit einer maximalen Größe von 1 kb einzusetzen. Da die
Promotoren im Genom häufig eine Länge von mehreren kb und nicht selten eine Länge von
10 kb besitzen ist von vorneherein nicht die beschriebene Promotorspezifität zu erreichen. Für
Pankreas- und Kolonkarzinome konnten drei Promotorfragmente gefunden werden, die in
ihrer Schnittmenge hauptsächlich in den beiden Gewebetypen aktiv sind und in anderen keine
Aktivität aufweisen. Zu diesen Promotoren gehörten der COX-2-, Midkine- und Ki-67-
Promotor. Zu weiteren Untersuchungszwecken wurde auch noch der E2F1-Promotor
analysiert, denn die Tumore befinden sich meist, im Gegensatz zum umliegenden gesunden
Gewebe, in S-Phase, in welcher dieser Promotor aktiv wäre.
In einer größeren Zahl von Veröffentlichungen wurde festgestellt , dass gewebe- oder
tumorspezifische Promotoren in ihrer Stärke oder Spezifität beeinflusst werden, wenn sie in
einen adenoviralen Vektor gesetzt werden 136-139, währenddessen andere diesen Effekt nicht
beobachteten 140-146. In den meisten dieser Veröffentlichungen wurden verschiedene
Reportergen-Assays verwendet. Die Wahl des Reportergen-Assays kann aber die Ergebnisse
beeinflussen. Zwei hauptsächlich verwendete Assayformen in Genexpressionsstudien sind
CAT und Luciferase. CAT besitzt einen engen dynamischen Messbereich und eine
Halbwertszeit von 50 h in Säugetierzellen, was nützlich zur Messung von kumulativen
Änderungen in der Genexpression ist 147,148. Im Gegensatz dazu hat Luciferase eine
Halbwertszeit von nur 3 h, welches eine Messung einer Promotoraktivität an einem
diskreteren Zeitpunkt ermöglicht 149. Die CAT-Assay-Prozedur produziert im Allgemeinen
eine größere Differenz in der Promotoraktivität als der Luciferase-Assay. In der vorliegenden
Arbeit wurde ein transienter Luciferase-Reporter-Assay verwendet, wobei ein Plasmid,
welches Elemente zur Analyse von möglichen regulatorischen Sequenzen enthält, eingesetzt
wurde. Dieses kann zur Analyse des Einflusses von adenoviralen Sequenzen und

5. Diskussion
56
Genprodukten auf heterologe Promotoren mit wtAdV überinfiziert werden. Da die
Herstellung von adenoviralen Vektoren, die einen zu testenden heterologen Promotor tragen,
ein sehr zeitaufwändiger Prozess und die exakte Bestimmung von infektiösem adenoviralen
Titer problematisch ist, wurde das vorliegende Testverfahren ausgewählt, da dieses die
Probleme umgeht 150.
Plasmid-basierende Transfektions-Assays für Promotorstudien, welche einen internen
Standard nutzen, sind schnell und hoch reproduzierbar. Durch Kalziumphosphat-basierende
Transfektions-methoden gelangen ca. 50 Kopien der DNA in eine Zelle 151, währenddessen
durch adenovirale Infektionen ca. 104 Kopien DNA in eine Zelle gelangen 41. Der Effekt des
AdV (nt 1-353)-Segmentes und/oder der frühen adenoviralen Genprodukte, welche in dieser
Arbeit untersucht worden sind, könnten im adenoviralen System stärker ausgeprägt sein.
Im Einklang mit früheren Veröffentlichungen 152 wurde die Anwesenheit von cis-
aktivierenden Elementen in dem AdV (nt1- 353)-Segment, welche die Transkription von allen
getesteten viralen Promotoren und die des zellzyklusabhängigen E2F1-Promotors
beeinflussten, gezeigt. Dieser Effekt wurde von der Position des AdV-Segmentes zur RNA-
Startseite oder durch die Anwesenheit einer Polyadenylisierungs-Sequenz stromaufwärts vom
Promotor nicht beeinflusst. Dies deutet darauf hin, dass ein Enhancer im diesem AdV-
Segment vorhanden ist.
Des Weiteren ist die Aktivität der COX-2-Promotoren erhöht, wenn das AdV (nt 1-353)-
Segment stromaufwärts des Promotors lokalisiert ist. Der Effekt verschwindet, wenn dieses
Fragment stromabwärts vom Promotor oder eine polyA-Sequenz vor dem Promotor eingesetzt
ist. Dieses deutet auf eine kryptische Transkriptionsstartseite in dem AdV-Segment hin. Die
Aktivitäten des MK- und des Ki-67-Promotors wurden nicht durch das AdV (nt 1-353)-
Segment verändert. Diese Daten deuten darauf hin, dass die adenovirale E1A stromaufwärts
liegende regulatorische Region (URR) bevorzugt virale und zellzyklusabhängige Promotoren
transaktiviert. Schon in vorangegangenen Veröffentlichungen wurde gezeigt, dass die E1A
URR auf einige, aber nicht auf alle, Promotoren einen Einfluss hat. Die Beobachtung ist im
Einklang mit dem aktuellen Verständnis von Enhancern. Enhancer können über eine große
Distanz wirken und interagieren mit nahezu jedem Typ von Promotor, es ist aber wichtig für
die Genregulation, dass ein Enhancer nur die ihm zugehörigen Promotoren beeinflusst, nicht
jedoch andere Promotoren. Ein Modell, genannt „enhancer Promotor-selectivity“, sagt aus,
dass inhärente Eigenschaften vom Promotor und Enhancer eine Interaktion ermöglichen,
während andere keinen Einfluss haben. Diese Promotor-Enhancer-Interaktionen werden durch
Proteine, die an der DNA an Enhancer und core-Promotor binden, gesteuert 153,154. Um diesen

5. Diskussion
57
Effekt zu überwinden, wurden Insulatoren im adenoviralen Kontext analysiert, jedoch mit
wechselhaftem Erfolg 155,156.
Die Aktivität des E2F1 und der viralen Promotoren wurde auch durch die frühen adenoviralen
Genprodukte, wie schon vorher beschrieben, beeinflusst 157. Verantwortlich für diesen Effekt
ist die frühe Expression der adenoviralen Proteine der E1A-Region, welche die Zellen durch
Bindung von pRB und andauernde Freisetzung von E2F-Transkriptionsfaktoren in die S-
Phase bringt. Dies hat eine Zellprogression und die verstärkte Aktivität von Promotoren, die
E2F-Bindungsmotive enthalten, zur Folge 158,159. Des Weiteren aktiviert E1A durch direkte
Bindung an verschiedene zelluläre Faktoren, z.B. an Proteine der ATF-Familie 160 und an
YY1 161, die Transkription. Innerhalb der adenoviralen E1A URR wurden verschiedene
Bindungsstellen für Transkriptionsfaktoren, die im Zellzyklus involviert sind, wie z.B. zwei
E2F-Bindestellen, identifiziert 162,163. Die E1A URR ist verantwortlich für die Autoregulation
der E1A-Gene auf der Ebene der Transkription 164.
Diese Daten sind nicht nur relevant für das transkriptionelle Targeting von
replikationsdefekten adenoviralen Vektoren, sondern auch für replikationskompetente
adenovirale Vektoren in der Tumor-Gentherapie. Das transkriptionelle Targeting von bedingt
replikationkompetenten adenoviralen Vektoren scheint nur bedingt eine gute Wahl zu sein,
denn drei Gründe sprechen gegen diese Vektorvariante: Als erstes können die cis-agierenden
adenoviralen regulatorischen Elemente, die weiter oben beschrieben wurden, die Spezifität
der heterologen Promotoren beeinflussen. Als zweites kann genannt werden, dass schon
geringe Mengen an E1A-Genprodukten ausreichend sind um die adenovirale Replikation zu
initiieren 165. Als dritter Grund sollte auch die positive Rückkopplungsschleife („feedback
loop“) genannt werden, denn bei dieser wird der heterologe Promotor, der die E1A-Region
kontrolliert, durch die Genprodukte der E1A-Region transaktiviert, was dann zu einem
Verlust an Spezifität führt 166-169.
Die wichtigste Rolle im BRAV kommt dem heterologen E1A-Promotor zu, dieser Promotor
muss auch im adenoviralen Umfeld noch immer seine Spezifität bewahren und nicht durch
adenovirale Produkte transaktiviert werden. Dieser heterologe Promotor darf auch keine
Basalaktivität besitzen, da, wie schon oben erwähnt, eine geringe Menge an E1A-
Genprodukten zur viralen Replikation ausreicht.
In einem Versuch, Bindungsstellen in den Promotorsequenzen zu finden, welches
Vorhersagen auf Elemente, welche die Aktivität des heterologen Promotors verändern,
zulässt, wurden diese mit der „Transkriptionsfaktor-Bindungsstellen“-Suchmaschine
„TFSEARCH“ Version 1.3 (URL: http://cbrc.jp/research/db /TFSEARCH.html) analysiert 170.

5. Diskussion
58
Der „Threshold score“ wurde bei der Analyse auf 90% gesetzt. Es konnten aber keine
Übereinstimmungen von vorhergesagten Transkriptionsfaktor-Bindungsstellen in den
heterologen Promotoren und dem cis- oder trans-aktivierendem AdV-Element gefunden
werden.
Das System aus transienten Plasmid-basierenden Luciferase-Assays und adenoviralen
Überinfektionen, ist ein Werkzeug um eine schnelle Identifikation von Promotoren, die ihre
Spezifität im adenoviralen Umfeld behalten, zu erreichen.
Diese Beobachtungen, die im Plasmid-basierenden Luciferase-Assay gemacht wurden,
konnten auch im adenoviralen Umfeld bestätigt werden. Dazu wurden die Promotoren mit
polyA und Luciferase-Expresionskassette in den pShuttle-Vektor kloniert und dann mittels
homologer Rekombination mit dem pAdEasy1-Vektor zu Volllänge adenoviralen Genomen
rekombiniert. Mit den davon hergestellten AdVs konnte ein Luciferase-Assay durchgeführt
werden, der dem oben beschriebenen sehr ähnlich ist. Dieser Assay zeigte die
Vergleichbarkeit der beiden Systeme, da die Ergebnisse qualitativ miteinander vergleichbar
sind. Jedoch ist das AdV-basierende System zeitlich um ein vielfaches langsamer als das
Plasmid-basierende System.
5.2 Analyse der bedingt replikationskompetenten adenoviralen Vektoren
Um einen bedingt replikationskompetenten adenoviralen Vektor für die Onkolyse von
Pankreas- oder Kolonkarzinomen zu erhalten, wurde die transkriptionelle Kontrolle gewählt.
Da in mehreren Veröffentlichungen gezeigt wurde, dass die heterologe Promotorspezifität im
adenoviralen System beeinflusst wird 138,171-173, musste eine weitere Möglichkeit zur
Genregulation und zum Erhalt der heterologen Promotorspezifität im adenoviralen Kontext
gezeigt werden. Als erstes wurde die Wirkung des Enhancereffektes und der alternativen
Startstelle im AdV auf die heterologen Promotoren untersucht. Dabei wurde festgestellt, dass
die Wahl des heterologen Promotors, der die E1-Region des AdVs kontrollieren soll, ein
wichtiger Schritt ist. Als erstes darf dieser Promotor durch den adenoviralen Enhancer nicht
induzierbar sein. Als grober Richtwert für die Wahl des heterologen Promotor gilt: Es sollte
kein Promotor sein, der durch S-Phasen-Eintritt aktiviert wird, da das Virus selbst die Zelle in
die S-Phase bringt und es sollten im Promotor keine DNA-Bindungstellen für regulatorische
Proteine vorhanden sein, die im Zellzyklus involviert sind. Als nächstes konnte festgestellt
werden, dass die Basalaktivität des heterologen Promotors im adenoviralen Umfeld durch ein,
dem Promotor vorangestelltes, synthetisches Polyadenylierungs-Signal stark reduziert werden

5. Diskussion
59
kann. Bisherige Ansätze verfolgten die transkriptionelle Kontrolle der adenoviralen
Replikation nur durch einen heterologen Promotor zur Regulation der E1A-Region. Häufig
wurde die E1A-Region mittels einer IRES-Sequenz mit einem therapeutischen Gen gekoppelt,
so dass es bei einer Transkription von E1A direkt zur Translation des therapeutischen Gens
kommt. Jedoch besitzen viele dieser Vektoren den Nachteil, dass die Gewebespezifität nicht
gegeben ist, da die Promotoren eine geringe Basalaktivität besitzen oder durch virale
Sequenzen oder Genprodukte transaktiviert werden. Daher kam es zum Aufschaukeln der
E1A-Genprodukte, welche durch die positive Rückkopplung entstand 174. Ein in letzter Zeit
verfolgter Ansatz war die heterologe Regulation eines zweiten frühen Gens, der E4-Region.
Dabei wurden, in zwei verschiedenen Ansätzen, zwei Promotoren in das adenovirale Genom
zur Regulation integriert 175,176. Beide Ansätze verfolgten aber nur die Möglichkeit jeweils
den gleichen heterologen Promotor zur Kontrolle der E1A- und der E4-Region zu verwenden.
Wenn aber der Promotor der E1A-Region durch die adenoviralen Genprodukte oder durch
den Enhancereffekt des AdV (nt 1-353)-Segmentes beeinflusst wird, so wird auch derselbe
Effekt für den gleichen heterologen Promotor der E4-Region zu beobachten sein. Daher wird
nur eine Spezifitätserhöhung des BRAV wegen der fehlenden Beeinflussung des heterologen
E4-Promotors durch die alternative Startseite, die nur im 5’ITR vorhanden ist und nicht im
3’ITR, erwartet.
In der vorliegenden Arbeit wurde daher der Ansatz der doppelten heterologen
Promotorkontrolle der E1A- und E4-Region gewählt. Vor beide Promotoren wurde jeweils
ein synthetisches polyA-Signal gesetzt um die Basalaktivität der heterologen Promotoren zu
minimieren. Es wurden zwei unterschiedliche Promotoren zur Kontrolle der E1A- und der E4-
Region verwendet. Als therapeutisches Gen wurde das in unserer Arbeitgruppe analysierte
Gen HSV-tk verwendet. Aus den vorangegangenen Experimenten wurden die Promotoren
COX-2, Midkine, Ki-67 und E2F1 verwendet. Dadurch entstanden vier Vektoren mit nur
einem heterologen Promotor zur Regulation der E1A-Region und 16 Vektoren, in denen
jeweils zwei heterologe Promotoren zur Kontrolle der E1A- und E4-Region verwendet
wurden.
Als erstes sollen nur die BRAVs mit einem E2F1-Promotor zur Kontrolle der E1A-Region
betrachtet werden. Deregulation des Retinoblastoma-Proteins (pRB) und des E2F-
regulatorischen Weges sind eine Grundeigenschaft von humanen Tumoren. Daher wurde
unter anderem der E2F1-Promotor, der die frühen adenoviralen Gene kontrolliert, zur
Replikationskontrolle ausgesucht. Wie schon vorher erwähnt, konnte E2F1-Promotoraktivität
in der G0-Phase oder in mitotisch arretierten Zellen nachgewiesen werden. Des Weiteren

5. Diskussion
60
konnte gezeigt werden, dass adenovirale Sequenzen und frühe adenovirale Genprodukte die
Aktivität des E2F1-Promotors erhöhen. Die Beobachtungen mit Vektoren, in denen die E1A-
Region mit E2F1 kontrolliert wurde, stimmen mit diesen Befunden überein. Die Vektoren
zeigten keine Gewebespezifität, sondern replizierten in nahezu jedem Gewebe, unabhängig
davon, ob es sich um Keratinozyten oder um tumoröses Gewebe handelte. Resultierend kann
zu den E2F1-Varianten gesagt werden, dass diese Vektoren zur spezifischen Onkolyse nicht
geeignet sind, sondern nur unspezifisch in Tumore replizieren. Dieselben Beobachtungen
machten auch die Arbeitsgruppe um Johnson et al. an Ihren Vektor in dem die E1A- und E4
Region durch identische E2F1-Promotoren kontrolliert werden 177.
Bei den anderen Vektoren, bei denen E1A unter der Kontrolle von COX-2, Midkine und Ki-
67 steht, konnte eine Tumorspezifität gezeigt werden. Dabei stellte sich heraus, das die
Funktion des heterologen E1A-Promotors eine größere Rolle spielt als die des heterologen
E4-Promotors. Dieses korrespondiert mit den vorangegangenen Veröffentlichungen und
Beobachtungen, denn die Rolle des E4-Promotors ist nur eine untergeordnete. Wenn der E1A-
Promotor eine Basalaktivität besitzt, kommt es zum Aufschaukeln von frühen viralen
Genprodukten, vor allem E1A, und somit zur viralen Replikation. Der heterologe Promotor
der E4-Region kann nur die Spezifität des Vektors effektiv erhöhen, wenn der E1A-Promotor
nicht von adenoviralen Sequenzen oder frühen Genprodukten beeinflusst wird. Bei dem
Einsatz von gleichen heterologen Promotoren zur Regulation der E1- und E4-Regionen
kommt es zu einem ähnlichen Replikationsmuster in den verschiedenen Geweben wie mit nur
einem heterologen Promotor vor der E1A-Region. Daher führt der Ansatz mit zwei
verschiedenen heterologen Promotoren zur Kontrolle der E1- und E4-Region zu einer
Verbesserung der Tumorspezifität von BRAV. Ähnliche folgerungen konnten Hernandez-
Alcoceba et al. in ihrem adenoviralem Vektor mit dualem Promotor zeigen, dieser Vektor
besitzt ein PSA spezifisches Promotor-System 178.
Aus den in vitro-Experimenten kann gefolgert werden, dass Viren, deren heterologe
Promotoren aus COX-2, Midkine oder Ki-67 bestehen, präferentiell in Pankreas- und
Kolonkarzinome replizieren und das BRAV pAd-COX-Ki Pankreaskarzinom- spezifisch ist.
Des Weiteren ist das Adenovirus mit dem E1 heterologen Promotor E2F1 und Cox-2, Ki-67
oder Midkine als E4 heterologen Promotor, als nicht tumorspezifischer onkolytischer Vektor
geeignet. Weitere Aussagen lassen sich erst durch ein in vivo-Experiment im permissiven
Tiermodell treffen.

6. Ausblick
61
6. Ausblick
Die Ergebnisse, die im in vitro-Experiment gezeigt werden konnten, müssen noch im
Xenograft-Tiermodell bestätigt werden. Dazu müssen die bedingt replikationkompetenten
adenoviralen Vektoren (BRAV) amplifiziert und gereinigt werden. Anschließend können
diese im Tiermodell durch subkutane, intraperitoneale oder in den Pankreas gesetzte Tumore
getestet werden. Dazu wurden verschiedene Tumorzelllinien, die stabil Luciferase
exprimieren, entwickelt. Diese kann man dann mittels einer hochsensitiven Kamera im Tier
direkt durch i.p. Gabe von D-Luciferin quantitativ erfassen, wodurch eine nicht invasive
Beobachtung der Tumorregression möglich ist. Des Weiteren soll versucht werden, die
Tumorspezifität durch einen dritten heterologen Promotor weiter zu erhöhen. Gleichzeitig soll
die transkripionelle Kontrolle mit der transduktionellen Kontrolle kombiniert werden. Mit
dieser BRAV-Variante wird eine noch höhere Tumorspezifität erwartet.
Im Anschluss soll versucht werden, die in vivo-Experimente mit primären humanen Zellen,
sowohl Tumoren, als auch gesundem Gewebe, zu wiederholen. Dazu müssen aber geeignete
und charakterisierte Gewebeproben vorliegen.
In unserer Arbeitsgruppe wird zurzeit ein permissives Tiermodel für das Adenovirus
untersucht. Sollte ein geeignetes gefunden werden, dann sollten die BRAV auch in diesem
eingesetzt und eine Biodistributionsstudie durchgeführt werden.

7. Zusammenfassung
62
7. Zusammenfassung
In der vorliegenden Arbeit sollten bedingt replikationskompetente adenovirale Vektoren
(BRAV) zur Onkolyse von Pankreas- und Kolonkarzinomen entwickelt werden. Der Ansatz
dazu war, die adenovirale E1- und die E4-Region unter heterologe Promotorkontrolle zu
setzen. Diese Promotoren sollen das Adenovirus nur in Pankreas- und in Kolonkarzinomen
replizieren lassen.
Als erstes wurden dazu verschiedene Promotoren ausgesucht und auf deren Gewebespezifität
mit einem transienten Luciferase-Assay untersucht. Anschließend wurde ein System
entwickelt, in dem diese Promotoren leicht und schnell auf ihre Transaktivierbarkeit im
adenoviralen Umfeld und der Einfluss des AdV (nt 1-353)-Segmentes, welches eine
alternative Transkriptionsstartstelle und nicht-spezifische Enhancer-Aktivität besitzt, getestet
werden können. Dieses System zeigte auf, dass die Basalaktivität eines heterologen
Promotors, der den adenoviralen E1-Promotor ersetzen soll, stark reduziert werden muss.
Ansonsten käme es durch die transaktivierende Eigenschaft von E1A-Genprodukten zu einem
Aufschaukeln der Expression und damit zur Replikation des Virus. Als Promotoren kamen
drei Promotoren für einen BRAV infrage, COX-2, Ki-67 und Midkine. Des Weiteren wurde
der unspezifische, aber proliferationsabhängige Promotor E2F1 für weitere Untersuchungen
in Betracht gezogen, da sich Tumore in ihrer Proliferation von dem gesunden, benachbarten
Gewebe unterscheiden.
Als nächster Schritt musste ein Vektorsystem entwickelt werden, welches die Manipulation
der E1- und der E4-Region erlaubt. Dazu wurde ein auf dem AdEasy-System basierendes
Plasmidsystem kloniert. Da zum Erhalt der Spezifität der heterologen Promotoren die
Fragmentlänge größer ist, als die der viralen Promotoren und im BRAV ein therapeutisches
Gen mit IRES-Sequenz untergebracht werden soll, musste versucht werden durch weitere
Deletionen, außer in der E3-Region, mehr Platz im viralen Genom zu erhalten. Dazu wurde
ein Transkomplementierungstest mit einem E4-deletierten Adenovirus (dl1011) und
transfizierten E4-orf-Expressionsplasmiden in Luciferase-exprimierenden HEK-293-Zellen
etabliert. Dabei wurde festgestellt, dass eine E4-Region, die die orfs 3, 4, 6 und 6/7 enthält,
eine ausreichende Replikation ermöglicht. Da aber die untersuchten und weiter verwendeten
heterologen Promotoren relativ klein waren, war es nicht nötig die E4-Region zu verkleinern.
Im abschließenden Experiment wurden die BRAVs in vitro getestet und die Replikation
mittels Kristallviolett-Färbung analysiert. Dabei wurde festgestellt, dass Viren, deren
heterologe Promotoren aus COX-2, Midkine oder Ki-67 bestehen, Pankreas- und

7. Zusammenfassung
63
Kolonkarzinomspezifisch replizieren und das BRAV pAd-COX-Ki Pankreaskarzinom-
spezifisch ist. Des Weiteren konnte ein Tumortyp unspezifischer BRAV mit dem E1A
heterologen Promotor E2F1 entwickelt werden, der im in vitro-Experiment viel versprechende
Ergebnisse lieferte.

8. Quellenangaben
64
8. Quellenangaben
1. DePage NG. Sulla scomparsa di un enorme canero vegetante del callo dell'utero senza
cura chirurgica. Ginecologia 1912; 9: 82-88.
2. SOUTHAM CM, MOORE AE. West Nile, Ilheus, and Bunyamwera virus infections
in man. Am.J.Trop.Med.Hyg. 1951; 31: 724-741.
3. SOUTHAM CM, MOORE AE. Clinical studies of viruses as antineoplastic agents
with particular reference to Egypt 101 virus. Cancer 1952; 5: 1025-1034.
4. HUEBNER RJ, ROWE WP, SCHATTEN WE, SMITH RR, THOMAS LB. Studies
on the use of viruses in the treatment of carcinoma of the cervix. Cancer 1956; 9:
1211-1218.
5. Reichard KW, Lorence RM, Cascino CJ et al. Newcastle disease virus selectively kills
human tumor cells. J.Surg.Res. 1992; 52: 448-453.
6. Lorence RM, Rood PA, Kelley KW. Newcastle disease virus as an antineoplastic
agent: induction of tumor necrosis factor-alpha and augmentation of its cytotoxicity.
J.Natl.Cancer Inst. 1988; 80: 1305-1312.
7. Cassel WA, Murray DR, Phillips HS. A phase II study on the postsurgical
management of Stage II malignant melanoma with a Newcastle disease virus
oncolysate. Cancer 1983; 52: 856-860.
8. Cassel WA, Garrett RE. NEWCASTLE DISEASE VIRUS AS AN
ANTINEOPLASTIC AGENT. Cancer 1965; 18: 863-868.
9. Gross S. Measles and leukaemia. Lancet 1971; 1: 397-398.

8. Quellenangaben
65
10. Asada T. Treatment of human cancer with mumps virus. Cancer 1974; 34: 1907-1928.
11. Taylor MW, Cordell B, Souhrada M, Prather S. Viruses as an aid to cancer therapy:
regression of solid and ascites tumors in rodents after treatment with bovine
enterovirus. Proc.Natl.Acad.Sci.U.S.A 1971; 68: 836-840.
12. Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Experimental therapy of
human glioma by means of a genetically engineered virus mutant. Science 1991; 252:
854-856.
13. Bischoff JR, Kirn DH, Williams A et al. An adenovirus mutant that replicates
selectively in p53-deficient human tumor cells. Science 1996; 274: 373-376.
14. Yew PR, Berk AJ. Inhibition of p53 transactivation required for transformation by
adenovirus early 1B protein. Nature 1992; 357: 82-85.
15. Gooding LR. Regulation of TNF-mediated cell death and inflammation by human
adenoviruses. Infect.Agents Dis. 1994; 3: 106-115.
16. Toda M, Rabkin SD, Kojima H, Martuza RL. Herpes simplex virus as an in situ cancer
vaccine for the induction of specific anti-tumor immunity. Hum.Gene Ther. 1999; 10:
385-393.
17. Savage HE, Rossen RD, Hersh EM, Freedman RS, Bowen JM, Plager C. Antibody
development to viral and allogeneic tumor cell-associated antigens in patients with
malignant melanoma and ovarian carcinoma treated with lysates of virus-infected
tumor cells. Cancer Res. 1986; 46: 2127-2133.
18. Lowe SW, Bodis S, McClatchey A et al. p53 status and the efficacy of cancer therapy
in vivo. Science 1994; 266: 807-810.

8. Quellenangaben
66
19. Khuri FR, Nemunaitis J, Ganly I et al. a controlled trial of intratumoral ONYX-015, a
selectively-replicating adenovirus, in combination with cisplatin and 5-fluorouracil in
patients with recurrent head and neck cancer. Nat.Med. 2000; 6: 879-885.
20. Freytag SO, Rogulski KR, Paielli DL, Gilbert JD, Kim JH. A novel three-pronged
approach to kill cancer cells selectively: concomitant viral, double suicide gene, and
radiotherapy. Hum.Gene Ther. 1998; 9: 1323-1333.
21. Wildner O, Morris JC, Vahanian NN, Ford H, Jr., Ramsey WJ, Blaese RM.
Adenoviral vectors capable of replication improve the efficacy of HSVtk/GCV suicide
gene therapy of cancer. Gene Ther. 1999; 6: 57-62.
22. Kruyt FA, Curiel DT. Toward a new generation of conditionally replicating
adenoviruses: pairing tumor selectivity with maximal oncolysis. Hum.Gene Ther.
2002; 13: 485-495.
23. Andreansky S, He B, van CJ et al. Treatment of intracranial gliomas in
immunocompetent mice using herpes simplex viruses that express murine interleukins.
Gene Ther. 1998; 5: 121-130.
24. Parker JN, Gillespie GY, Love CE, Randall S, Whitley RJ, Markert JM. Engineered
herpes simplex virus expressing IL-12 in the treatment of experimental murine brain
tumors. Proc.Natl.Acad.Sci.U.S.A 2000; 97: 2208-2213.
25. Hermiston TW, Kuhn I. Armed therapeutic viruses: strategies and challenges to
arming oncolytic viruses with therapeutic genes. Cancer Gene Ther. 2002; 9: 1022-
1035.
26. Roth JA, Cristiano RJ. Gene therapy for cancer: what have we done and where are we
going? J.Natl.Cancer Inst. 1997; 89: 21-39.

8. Quellenangaben
67
27. Jain RK. Physiological barriers to delivery of monoclonal antibodies and other
macromolecules in tumors. Cancer Res. 1990; 50: 814s-819s.
28. Ilsley DD, Lee SH, Miller WH, Kuchta RD. Acyclic guanosine analogs inhibit DNA
polymerases alpha, delta, and epsilon with very different potencies and have unique
mechanisms of action. Biochemistry 1995; 34: 2504-2510.
29. Thust R, Schacke M, Wutzler P. Cytogenetic genotoxicity of antiherpes virostatics in
Chinese hamster V79-E cells. I. Purine nucleoside analogues. Antiviral Res. 1996; 31:
105-113.
30. Haynes P, Lambert TR, Mitchell ID. Comparative in-vivo genotoxicity of antiviral
nucleoside analogues; penciclovir, acyclovir, ganciclovir and the xanthine analogue,
caffeine, in the mouse bone marrow micronucleus assay. Mutat.Res. 1996; 369: 65-74.
31. Huber BE, Austin EA, Good SS, Knick VC, Tibbels S, Richards CA. In vivo
antitumor activity of 5-fluorocytosine on human colorectal carcinoma cells genetically
modified to express cytosine deaminase. Cancer Res. 1993; 53: 4619-4626.
32. Mullen CA, Coale MM, Lowe R, Blaese RM. Tumors expressing the cytosine
deaminase suicide gene can be eliminated in vivo with 5-fluorocytosine and induce
protective immunity to wild type tumor. Cancer Res. 1994; 54: 1503-1506.
33. Freytag SO, Rogulski KR, Paielli DL, Gilbert JD, Kim JH. A novel three-pronged
approach to kill cancer cells selectively: concomitant viral, double suicide gene, and
radiotherapy. Hum.Gene Ther. 1998; 9: 1323-1333.
34. Freytag SO, Khil M, Stricker H et al. Phase I study of replication-competent
adenovirus-mediated double suicide gene therapy for the treatment of locally recurrent
prostate cancer. Cancer Res. 2002; 62: 4968-4976.

8. Quellenangaben
68
35. Wildner O, Blaese RM, Morris JC. Therapy of colon cancer with oncolytic adenovirus
is enhanced by the addition of herpes simplex virus-thymidine kinase. Cancer Res.
1999; 59: 410-413.
36. Wildner O, Morris JC, Vahanian NN, Ford H, Jr., Ramsey WJ, Blaese RM.
Adenoviral vectors capable of replication improve the efficacy of HSVtk/GCV suicide
gene therapy of cancer. Gene Ther. 1999; 6: 57-62.
37. Carroll NM, Chase M, Chiocca EA, Tanabe KK. The effect of ganciclovir on herpes
simplex virus-mediated oncolysis. J.Surg.Res. 1997; 69: 413-417.
38. Todo T, Rabkin SD, Martuza RL. Evaluation of ganciclovir-mediated enhancement of
the antitumoral effect in oncolytic, multimutated herpes simplex virus type 1 (G207)
therapy of brain tumors. Cancer Gene Ther. 2000; 7: 939-946.
39. Wildner O, Morris JC. The role of the E1B 55 kDa gene product in oncolytic
adenoviral vectors expressing herpes simplex virus-tk: assessment of antitumor
efficacy and toxicity. Cancer Res. 2000; 60: 4167-4174.
40. Chase M, Chung RY, Chiocca EA. An oncolytic viral mutant that delivers the
CYP2B1 transgene and augments cyclophosphamide chemotherapy. Nat.Biotechnol.
1998; 16: 444-448.
41. Horwitz M. Fields Virology. Fields, BN. 2. 1996. Philadelphia, New York,
Lippincott-Raven.
Ref Type: Generic
42. Rodriguez R, Schuur ER, Lim HY, Henderson GA, Simons JW, Henderson DR.
Prostate attenuated replication competent adenovirus (ARCA) CN706: a selective

8. Quellenangaben
69
cytotoxic for prostate-specific antigen-positive prostate cancer cells. Cancer Res.
1997; 57: 2559-2563.
43. Yu DC, Sakamoto GT, Henderson DR. Identification of the transcriptional regulatory
sequences of human kallikrein 2 and their use in the construction of calydon virus 764,
an attenuated replication competent adenovirus for prostate cancer therapy. Cancer
Res. 1999; 59: 1498-1504.
44. Hallenbeck PL, Chang YN, Hay C et al. A novel tumor-specific replication-restricted
adenoviral vector for gene therapy of hepatocellular carcinoma. Hum.Gene Ther.
1999; 10: 1721-1733.
45. Kurihara T, Brough DE, Kovesdi I, Kufe DW. Selectivity of a replication-competent
adenovirus for human breast carcinoma cells expressing the MUC1 antigen.
J.Clin.Invest 2000; 106: 763-771.
46. Hitt MM, Graham FL. Adenovirus E1A under the control of heterologous promoters:
wide variation in E1A expression levels has little effect on virus replication. Virology
1990; 179: 667-678.
47. Grable M, Hearing P. cis and trans requirements for the selective packaging of
adenovirus type 5 DNA. J.Virol. 1992; 66: 723-731.
48. Hearing P, Shenk T. The adenovirus type 5 E1A enhancer contains two functionally
distinct domains: one is specific for E1A and the other modulates all early units in cis.
Cell 1986; 45: 229-236.
49. Vassaux G, Hurst HC, Lemoine NR. Insulation of a conditionally expressed transgene
in an adenoviral vector. Gene Ther. 1999; 6: 1192-1197.

8. Quellenangaben
70
50. Kirn D. Replication-selective oncolytic adenoviruses: virotherapy aimed at genetic
targets in cancer. Oncogene 2000; 19: 6660-6669.
51. Wildner O, Morris JC, Vahanian NN, Ford H, Jr., Ramsey WJ, Blaese RM.
Adenoviral vectors capable of replication improve the efficacy of HSVtk/GCV suicide
gene therapy of cancer. Gene Ther. 1999; 6: 57-62.
52. Yew PR, Kao CC, Berk AJ. Dissection of functional domains in the adenovirus 2 early
1B 55K polypeptide by suppressor-linker insertional mutagenesis. Virology 1990;
179: 795-805.
53. Yew PR, Berk AJ. Inhibition of p53 transactivation required for transformation by
adenovirus early 1B protein. Nature 1992; 357: 82-85.
54. Yew PR, Kao CC, Berk AJ. Dissection of functional domains in the adenovirus 2 early
1B 55K polypeptide by suppressor-linker insertional mutagenesis. Virology 1990;
179: 795-805.
55. Roulston A, Marcellus RC, Branton PE. Viruses and apoptosis. Annu.Rev.Microbiol.
1999; 53: 577-628.
56. Barker DD, Berk AJ. Adenovirus proteins from both E1B reading frames are required
for transformation of rodent cells by viral infection and DNA transfection. Virology
1987; 156: 107-121.
57. Bischoff JR, Kirn DH, Williams A et al. An adenovirus mutant that replicates
selectively in p53-deficient human tumor cells. Science 1996; 274: 373-376.
58. Heise C, Sampson-Johannes A, Williams A, McCormick F, Von Hoff DD, Kirn DH.
ONYX-015, an E1B gene-attenuated adenovirus, causes tumor-specific cytolysis and

8. Quellenangaben
71
antitumoral efficacy that can be augmented by standard chemotherapeutic agents.
Nat.Med. 1997; 3: 639-645.
59. Goldsmith KT, Dion LD, Curiel DT, Garver RI, Jr. trans E1 component requirements
for maximal replication of E1-defective recombinant adenovirus. Virology 1998; 248:
406-419.
60. Goodrum FD, Ornelles DA. Roles for the E4 orf6, orf3, and E1B 55-kilodalton
proteins in cell cycle-independent adenovirus replication. J.Virol. 1999; 73: 7474-
7488.
61. Harada JN, Berk AJ. p53-Independent and -dependent requirements for E1B-55K in
adenovirus type 5 replication. J.Virol. 1999; 73: 5333-5344.
62. Hay JG, McElvaney NG, Herena J, Crystal RG. Modification of nasal epithelial
potential differences of individuals with cystic fibrosis consequent to local
administration of a normal CFTR cDNA adenovirus gene transfer vector. Hum.Gene
Ther. 1995; 6: 1487-1496.
63. Morris JC, Wildner O. Therapy of head and neck squamous cell carcinoma with an
oncolytic adenovirus expressing HSV-tk. Mol.Ther. 2000; 1: 56-62.
64. Rothmann T, Hengstermann A, Whitaker NJ, Scheffner M, zur HH. Replication of
ONYX-015, a potential anticancer adenovirus, is independent of p53 status in tumor
cells. J.Virol. 1998; 72: 9470-9478.
65. Wildner O, Morris JC. The role of the E1B 55 kDa gene product in oncolytic
adenoviral vectors expressing herpes simplex virus-tk: assessment of antitumor
efficacy and toxicity. Cancer Res. 2000; 60: 4167-4174.

8. Quellenangaben
72
66. Turnell AS, Grand RJ, Gallimore PH. The replicative capacities of large E1B-null
group A and group C adenoviruses are independent of host cell p53 status. J.Virol.
1999; 73: 2074-2083.
67. Vollmer CM, Ribas A, Butterfield LH et al. p53 selective and nonselective replication
of an E1B-deleted adenovirus in hepatocellular carcinoma. Cancer Res. 1999; 59:
4369-4374.
68. Wildner O, Morris JC. Therapy of peritoneal carcinomatosis from colon cancer with
oncolytic adenoviruses. J.Gene Med. 2000; 2: 353-360.
69. Khuri FR, Nemunaitis J, Ganly I et al. a controlled trial of intratumoral ONYX-015, a
selectively-replicating adenovirus, in combination with cisplatin and 5-fluorouracil in
patients with recurrent head and neck cancer. Nat.Med. 2000; 6: 879-885.
70. Khuri FR, Nemunaitis J, Ganly I et al. a controlled trial of intratumoral ONYX-015, a
selectively-replicating adenovirus, in combination with cisplatin and 5-fluorouracil in
patients with recurrent head and neck cancer. Nat.Med. 2000; 6: 879-885.
71. Whyte P, Williamson NM, Harlow E. Cellular targets for transformation by the
adenovirus E1A proteins. Cell 1989; 56: 67-75.
72. Fueyo J, Gomez-Manzano C, Alemany R et al. A mutant oncolytic adenovirus
targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene 2000; 19: 2-
12.
73. Heise C, Hermiston T, Johnson L et al. An adenovirus E1A mutant that demonstrates
potent and selective systemic anti-tumoral efficacy. Nat.Med. 2000; 6: 1134-1139.

8. Quellenangaben
73
74. Johnson L, Shen A, Boyle L et al. Selectively replicating adenoviruses targeting
deregulated E2F activity are potent, systemic antitumor agents. Cancer Cell 2002; 1:
325-337.
75. Kruyt FA, Curiel DT. Toward a new generation of conditionally replicating
adenoviruses: pairing tumor selectivity with maximal oncolysis. Hum.Gene Ther.
2002; 13: 485-495.
76. Wickham TJ. Targeting adenovirus. Gene Ther. 2000; 7: 110-114.
77. Gu DL, Gonzalez AM, Printz MA et al. Fibroblast growth factor 2 retargeted
adenovirus has redirected cellular tropism: evidence for reduced toxicity and enhanced
antitumor activity in mice. Cancer Res. 1999; 59: 2608-2614.
78. Doukas J, Hoganson DK, Ong M et al. Retargeted delivery of adenoviral vectors
through fibroblast growth factor receptors involves unique cellular pathways. FASEB
J. 1999; 13: 1459-1466.
79. Mei YF, Wadell G. Epitopes and hemagglutination binding domain on subgenus B:2
adenovirus fibers. J.Virol. 1996; 70: 3688-3697.
80. Wadell G, Allard A, Johansson M, Svensson L, Uhnoo I. Enteric adenoviruses. Ciba
Found.Symp. 1987; 128: 63-91.
81. Eiz B, Pring-Akerblom P. Molecular characterization of the type-specific gamma-
determinant located on the adenovirus fiber. J.Virol. 1997; 71: 6576-6581.
82. Hierholzer JC. Further subgrouping of the human adenoviruses by differential
hemagglutination. J.Infect.Dis. 1973; 128: 541-550.

8. Quellenangaben
74
83. Nemerow GR, Stewart PL. Role of alpha(v) integrins in adenovirus cell entry and
gene delivery. Microbiol.Mol.Biol.Rev. 1999; 63: 725-734.
84. Stewart PL, Burnett RM, Cyrklaff M, Fuller SD. Image reconstruction reveals the
complex molecular organization of adenovirus. Cell 1991; 67: 145-154.
85. Norrby E. The structural and functional diversity of Adenovirus capsid components.
J.Gen.Virol. 1969; 5: 221-236.
86. Modrow S FDTU. Molekulare Virology. Spektrum 2003.
87. Russell WC. Update on adenovirus and its vectors. J.Gen.Virol. 2000; 81: 2573-2604.
88. Tauber B DT. Molecular regulation and biological function of adenovirus early genes:
the E4 ORFs. Gene 278, 1-23. 2001.
Ref Type: Journal (Full)
89. Mathias P, Wickham T, Moore M, Nemerow G. Multiple adenovirus serotypes use
alpha v integrins for infection. J.Virol. 1994; 68: 6811-6814.
90. Cotten M, Weber JM. The adenovirus protease is required for virus entry into host
cells. Virology 1995; 213: 494-502.
91. Saphire AC, Guan T, Schirmer EC, Nemerow GR, Gerace L. Nuclear import of
adenovirus DNA in vitro involves the nuclear protein import pathway and hsc70.
J.Biol.Chem. 2000; 275: 4298-4304.
92. Hay JG, McElvaney NG, Herena J, Crystal RG. Modification of nasal epithelial
potential differences of individuals with cystic fibrosis consequent to local
administration of a normal CFTR cDNA adenovirus gene transfer vector. Hum.Gene
Ther. 1995; 6: 1487-1496.

8. Quellenangaben
75
93. Heinemeyer T, Wingender E, Reuter I et al. Databases on transcriptional regulation:
TRANSFAC, TRRD and COMPEL. Nucleic Acids Res. 1998; 26: 362-367.
94. Angeletti PC, Engler JA. Adenovirus preterminal protein binds to the CAD enzyme at
active sites of viral DNA replication on the nuclear matrix. J.Virol. 1998; 72: 2896-
2904.
95. Russell WC. Update on adenovirus and its vectors. J.Gen.Virol. 2000; 81: 2573-2604.
96. Hay JG, McElvaney NG, Herena J, Crystal RG. Modification of nasal epithelial
potential differences of individuals with cystic fibrosis consequent to local
administration of a normal CFTR cDNA adenovirus gene transfer vector. Hum.Gene
Ther. 1995; 6: 1487-1496.
97. Rekosh DM, Russell WC, Bellet AJ, Robinson AJ. Identification of a protein linked to
the ends of adenovirus DNA. Cell 1977; 11: 283-295.
98. Wold WS, Tollefson AE, Hermiston TW. E3 transcription unit of adenovirus.
Curr.Top.Microbiol.Immunol. 1995; 199 ( Pt 1): 237-274.
99. Lukashok SA, Tarassishin L, Li Y, Horwitz MS. An adenovirus inhibitor of tumor
necrosis factor alpha-induced apoptosis complexes with dynein and a small GTPase.
J.Virol. 2000; 74: 4705-4709.
100. Tollefson AE, Hermiston TW, Lichtenstein DL et al. Forced degradation of Fas
inhibits apoptosis in adenovirus-infected cells. Nature 1998; 392: 726-730.
101. Tollefson AE, Ryerse JS, Scaria A, Hermiston TW, Wold WS. The E3-11.6-kDa
adenovirus death protein (ADP) is required for efficient cell death: characterization of
cells infected with adp mutants. Virology 1996; 220: 152-162.

8. Quellenangaben
76
102. Halbert DN, Cutt JR, Shenk T. Adenovirus early region 4 encodes functions required
for efficient DNA replication, late gene expression, and host cell shutoff. J.Virol.
1985; 56: 250-257.
103. Ornelles DA, Shenk T. Localization of the adenovirus early region 1B 55-kilodalton
protein during lytic infection: association with nuclear viral inclusions requires the
early region 4 34-kilodalton protein. J.Virol. 1991; 65: 424-429.
104. Mannervik M, Fan S, Strom AC, Helin K, Akusjarvi G. Adenovirus E4 open reading
frame 4-induced dephosphorylation inhibits E1A activation of the E2 promoter and
E2F-1-mediated transactivation independently of the retinoblastoma tumor suppressor
protein. Virology 1999; 256: 313-321.
105. Flint. Principles of Virology. ASM Press 2000.
106. Boyer JL, Ketner G. Genetic analysis of a potential zinc-binding domain of the
adenovirus E4 34k protein. J.Biol.Chem. 2000; 275: 14969-14978.
107. Konig C, Roth J, Dobbelstein M. Adenovirus type 5 E4orf3 protein relieves p53
inhibition by E1B-55-kilodalton protein. J.Virol. 1999; 73: 2253-2262.
108. Sandy P, Gostissa M, Fogal V et al. p53 is involved in the p120E4F-mediated growth
arrest. Oncogene 2000; 19: 188-199.
109. Philipson L. Structure and assembly of adenoviruses. Curr.Top.Microbiol.Immunol.
1984; 109: 1-52.
110. Hearing P, Samulski RJ, Wishart WL, Shenk T. Identification of a repeated sequence
element required for efficient encapsidation of the adenovirus type 5 chromosome.
J.Virol. 1987; 61: 2555-2558.

8. Quellenangaben
77
111. Toth M, Doerfler W, Shenk T. Adenovirus DNA replication facilitates binding of the
MLTF/USF transcription factor to the viral major late promoter within infected cells.
Nucleic Acids Res. 1992; 20: 5143-5148.
112. Lutz P, Rosa-Calatrava M, Kedinger C. The product of the adenovirus intermediate
gene IX is a transcriptional activator. J.Virol. 1997; 71: 5102-5109.
113. Lutz P, Kedinger C. Properties of the adenovirus IVa2 gene product, an effector of
late-phase-dependent activation of the major late promoter. J.Virol. 1996; 70: 1396-
1405.
114. Leong K, Lee W, Berk AJ. High-level transcription from the adenovirus major late
promoter requires downstream binding sites for late-phase-specific factors. J.Virol.
1990; 64: 51-60.
115. Mansour SL, Grodzicker T, Tjian R. Downstream sequences affect transcription
initiation from the adenovirus major late promoter. Mol.Cell Biol. 1986; 6: 2684-
2694.
116. Rao L, Perez D, White E. Lamin proteolysis facilitates nuclear events during
apoptosis. J.Cell Biol. 1996; 135: 1441-1455.
117. Tollefson AE, Ryerse JS, Scaria A, Hermiston TW, Wold WS. The E3-11.6-kDa
adenovirus death protein (ADP) is required for efficient cell death: characterization of
cells infected with adp mutants. Virology 1996; 220: 152-162.
118. Sambrook J, Russel WD. Molecular Cloning. CSHL Press 2001.

8. Quellenangaben
78
119. Cao G, Kuriyama S, Gao J et al. Comparison of carcinoembryonic antigen promoter
regions isolated from human colorectal carcinoma and normal adjacent mucosa to
induce strong tumor-selective gene expression. Int.J.Cancer 1998; 78: 242-247.
120. Henglein B, Chenivesse X, Wang J, Eick D, Brechot C. Structure and cell cycle-
regulated transcription of the human cyclin A gene. Proc.Natl.Acad.Sci.U.S.A 1994;
91: 5490-5494.
121. Parr MJ, Manome Y, Tanaka T et al. Tumor-selective transgene expression in vivo
mediated by an E2F-responsive adenoviral vector. Nat.Med. 1997; 3: 1145-1149.
122. Neuman E, Flemington EK, Sellers WR, Kaelin WG, Jr. Transcription of the E2F-1
gene is rendered cell cycle dependent by E2F DNA-binding sites within its promoter.
Mol.Cell Biol. 1994; 14: 6607-6615.
123. Kaname T, Kadomatsu K, Aridome K et al. The expression of truncated MK in human
tumors. Biochem.Biophys.Res.Commun. 1996; 219: 256-260.
124. Lothian C, Lendahl U. An evolutionarily conserved region in the second intron of the
human nestin gene directs gene expression to CNS progenitor cells and to early neural
crest cells. Eur.J.Neurosci. 1997; 9: 452-462.
125. Nylandsted J, Rohde M, Brand K, Bastholm L, Elling F, Jaattela M. Selective
depletion of heat shock protein 70 (Hsp70) activates a tumor-specific death program
that is independent of caspases and bypasses Bcl-2. Proc.Natl.Acad.Sci.U.S.A 2000;
97: 7871-7876.
126. Takakura M, Kyo S, Kanaya T et al. Cloning of human telomerase catalytic subunit
(hTERT) gene promoter and identification of proximal core promoter sequences

8. Quellenangaben
79
essential for transcriptional activation in immortalized and cancer cells. Cancer Res.
1999; 59: 551-557.
127. O'brien RM, Streeper RS, Ayala JE, Stadelmaier BT, Hornbuckle LA. Insulin-
regulated gene expression. Biochem.Soc.Trans. 2001; 29: 552-558.
128. Duchrow M, Schluter C, Wohlenberg C, Flad HD, Gerdes J. Molecular
characterization of the gene locus of the human cell proliferation-associated nuclear
protein defined by monoclonal antibody Ki-67. Cell Prolif. 1996; 29: 1-12.
129. Uehara K, Matsubara S, Kadomatsu K, Tsutsui J, Muramatsu T. Genomic structure of
human midkine (MK), a retinoic acid-responsive growth/differentiation factor.
J.Biochem.(Tokyo) 1992; 111: 563-567.
130. Perrais M, Pigny P, Ducourouble MP et al. Characterization of human mucin gene
MUC4 promoter: importance of growth factors and proinflammatory cytokines for its
regulation in pancreatic cancer cells. J.Biol.Chem. 2001; 276: 30923-30933.
131. Westerhausen DR, Jr., Hopkins WE, Billadello JJ. Multiple transforming growth
factor-beta-inducible elements regulate expression of the plasminogen activator
inhibitor type-1 gene in Hep G2 cells. J.Biol.Chem. 1991; 266: 1092-1100.
132. Pietrzkowski Z, Alder H, Chang CD, Ku DH, Baserga R. Characterization of an
enhancer-like structure in the promoter region of the proliferating cell nuclear antigen
(PCNA) gene. Exp.Cell Res. 1991; 193: 283-290.
133. Inoue H, Tanabe T, Umesono K. Feedback control of cyclooxygenase-2 expression
through PPARgamma. J.Biol.Chem. 2000; 275: 28028-28032.

8. Quellenangaben
80
134. Stanek D, Rader SD, Klingauf M, Neugebauer KM. Targeting of U4/U6 small nuclear
RNP assembly factor SART3/p110 to Cajal bodies. J.Cell Biol. 2003; 160: 505-516.
135. Silins G, Grimmond S, Egerton M, Hayward N. Analysis of the promoter region of the
human VEGF-related factor gene. Biochem.Biophys.Res.Commun. 1997; 230: 413-
418.
136. Babiss LE, Friedman JM, Darnell JE, Jr. Cellular promoters incorporated into the
adenovirus genome: effects of viral regulatory elements on transcription rates and cell
specificity of albumin and beta-globin promoters. Mol.Cell Biol. 1986; 6: 3798-3806.
137. Imler JL, Dupuit F, Chartier C et al. Targeting cell-specific gene expression with an
adenovirus vector containing the lacZ gene under the control of the CFTR promoter.
Gene Ther. 1996; 3: 49-58.
138. Ring CJ, Harris JD, Hurst HC, Lemoine NR. Suicide gene expression induced in
tumour cells transduced with recombinant adenoviral, retroviral and plasmid vectors
containing the ERBB2 promoter. Gene Ther. 1996; 3: 1094-1103.
139. Shi Q, Wang Y, Worton R. Modulation of the specificity and activity of a cellular
promoter in an adenoviral vector. Hum.Gene Ther. 1997; 8: 403-410.
140. Babiss LE, Friedman JM, Darnell JE, Jr. Cellular promoters incorporated into the
adenovirus genome: effects of viral regulatory elements on transcription rates and cell
specificity of albumin and beta-globin promoters. Mol.Cell Biol. 1986; 6: 3798-3806.
141. Chen Y, DeWeese T, Dilley J et al. CV706, a prostate cancer-specific adenovirus
variant, in combination with radiotherapy produces synergistic antitumor efficacy
without increasing toxicity. Cancer Res. 2001; 61: 5453-5460.

8. Quellenangaben
81
142. Harris JD, Gutierrez AA, Hurst HC, Sikora K, Lemoine NR. Gene therapy for cancer
using tumour-specific prodrug activation. Gene Ther. 1994; 1: 170-175.
143. Kaneko S, Hallenbeck P, Kotani T et al. Adenovirus-mediated gene therapy of
hepatocellular carcinoma using cancer-specific gene expression. Cancer Res. 1995;
55: 5283-5287.
144. Kim S, Lin H, Barr E, Chu L, Leiden JM, Parmacek MS. Transcriptional targeting of
replication-defective adenovirus transgene expression to smooth muscle cells in vivo.
J.Clin.Invest 1997; 100: 1006-1014.
145. Rothmann T, Hengstermann A, Whitaker NJ, Scheffner M, zur HH. Replication of
ONYX-015, a potential anticancer adenovirus, is independent of p53 status in tumor
cells. J.Virol. 1998; 72: 9470-9478.
146. Siders WM, Halloran PJ, Fenton RG. Transcriptional targeting of recombinant
adenoviruses to human and murine melanoma cells. Cancer Res. 1996; 56: 5638-5646.
147. de W, Jr., Wood KV, DeLuca M, Helinski DR, Subramani S. Firefly luciferase gene:
structure and expression in mammalian cells. Mol.Cell Biol. 1987; 7: 725-737.
148. Thompson JF, Hayes LS, Lloyd DB. Modulation of firefly luciferase stability and
impact on studies of gene regulation. Gene 1991; 103: 171-177.
149. de W, Jr., Wood KV, DeLuca M, Helinski DR, Subramani S. Firefly luciferase gene:
structure and expression in mammalian cells. Mol.Cell Biol. 1987; 7: 725-737.
150. Nyberg-Hoffman C, Shabram P, Li W, Giroux D, guilar-Cordova E. Sensitivity and
reproducibility in adenoviral infectious titer determination. Nat.Med. 1997; 3: 808-
811.

8. Quellenangaben
82
151. Swenarchuk L, Harakidas P, Cochrane A. Regulated expression of HIV-1 Rev
function in mammalian cell lines. Can.J.Microbiol. 1999; 45: 480-490.
152. Hearing P, Shenk T. The adenovirus type 5 E1A enhancer contains two functionally
distinct domains: one is specific for E1A and the other modulates all early units in cis.
Cell 1986; 45: 229-236.
153. Blackwood EM, Kadonaga JT. Going the distance: a current view of enhancer action.
Science 1998; 281: 61-63.
154. Merli C, Bergstrom DE, Cygan JA, Blackman RK. Promoter specificity mediates the
independent regulation of neighboring genes. Genes Dev. 1996; 10: 1260-1270.
155. Buvoli M, Langer SJ, Bialik S, Leinwand LA. Potential limitations of transcription
terminators used as transgene insulators in adenoviral vectors. Gene Ther. 2002; 9:
227-231.
156. Steinwaerder DS, Lieber A. Insulation from viral transcriptional regulatory elements
improves inducible transgene expression from adenovirus vectors in vitro and in vivo.
Gene Ther. 2000; 7: 556-567.
157. Pajalunga D, Tognozzi D, Tiainen M et al. E2F activates late-G1 events but cannot
replace E1A in inducing S phase in terminally differentiated skeletal muscle cells.
Oncogene 1999; 18: 5054-5062.
158. Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998; 12: 2245-
2262.

8. Quellenangaben
83
159. Howe JA, Mymryk JS, Egan C, Branton PE, Bayley ST. Retinoblastoma growth
suppressor and a 300-kDa protein appear to regulate cellular DNA synthesis.
Proc.Natl.Acad.Sci.U.S.A 1990; 87: 5883-5887.
160. Liu F, Green MR. A specific member of the ATF transcription factor family can
mediate transcription activation by the adenovirus E1a protein. Cell 1990; 61: 1217-
1224.
161. Shi Y, Seto E, Chang LS, Shenk T. Transcriptional repression by YY1, a human GLI-
Kruppel-related protein, and relief of repression by adenovirus E1A protein. Cell
1991; 67: 377-388.
162. Yoshida K, Narita M, Fujinaga K. Binding sites of HeLa cell nuclear proteins on the
upstream region of adenovirus type 5 E1A gene. Nucleic Acids Res. 1989; 17: 10015-
10034.
163. Bruder JT, Hearing P. Nuclear factor EF-1A binds to the adenovirus E1A core
enhancer element and to other transcriptional control regions. Mol.Cell Biol. 1989; 9:
5143-5153.
164. Hearing P, Samulski RJ, Wishart WL, Shenk T. Identification of a repeated sequence
element required for efficient encapsidation of the adenovirus type 5 chromosome.
J.Virol. 1987; 61: 2555-2558.
165. Hitt MM, Graham FL. Adenovirus E1A under the control of heterologous promoters:
wide variation in E1A expression levels has little effect on virus replication. Virology
1990; 179: 667-678.
166. Alemany R, Balague C, Curiel DT. Replicative adenoviruses for cancer therapy.
Nat.Biotechnol. 2000; 18: 723-727.

8. Quellenangaben
84
167. Hallenbeck PL, Chang YN, Hay C et al. A novel tumor-specific replication-restricted
adenoviral vector for gene therapy of hepatocellular carcinoma. Hum.Gene Ther.
1999; 10: 1721-1733.
168. Parr MJ, Manome Y, Tanaka T et al. Tumor-selective transgene expression in vivo
mediated by an E2F-responsive adenoviral vector. Nat.Med. 1997; 3: 1145-1149.
169. Rodriguez R, Schuur ER, Lim HY, Henderson GA, Simons JW, Henderson DR.
Prostate attenuated replication competent adenovirus (ARCA) CN706: a selective
cytotoxic for prostate-specific antigen-positive prostate cancer cells. Cancer Res.
1997; 57: 2559-2563.
170. Heinemeyer T, Wingender E, Reuter I et al. Databases on transcriptional regulation:
TRANSFAC, TRRD and COMPEL. Nucleic Acids Res. 1998; 26: 362-367.
171. Babiss LE, Friedman JM, Darnell JE, Jr. Cellular promoters incorporated into the
adenovirus genome: effects of viral regulatory elements on transcription rates and cell
specificity of albumin and beta-globin promoters. Mol.Cell Biol. 1986; 6: 3798-3806.
172. Imler JL, Dupuit F, Chartier C et al. Targeting cell-specific gene expression with an
adenovirus vector containing the lacZ gene under the control of the CFTR promoter.
Gene Ther. 1996; 3: 49-58.
173. Shi Q, Wang Y, Worton R. Modulation of the specificity and activity of a cellular
promoter in an adenoviral vector. Hum.Gene Ther. 1997; 8: 403-410.
174. Shi Q, Wang Y, Worton R. Modulation of the specificity and activity of a cellular
promoter in an adenoviral vector. Hum.Gene Ther. 1997; 8: 403-410.

8. Quellenangaben
85
175. Hernandez-Alcoceba R, Pihalja M, Nunez G, Clarke MF. Evaluation of a new dual-
specificity promoter for selective induction of apoptosis in breast cancer cells. Cancer
Gene Ther. 2001; 8: 298-307.
176. Banerjee NS, Rivera AA, Wang M et al. Analyses of melanoma-targeted oncolytic
adenoviruses with tyrosinase enhancer/promoter-driven E1A, E4, or both in
submerged cells and organotypic cultures. Mol.Cancer Ther. 2004; 3: 437-449.
177. Johnson L, Shen A, Boyle L et al. Selectively replicating adenoviruses targeting
deregulated E2F activity are potent, systemic antitumor agents. Cancer Cell 2002; 1:
325-337.
178. Hernandez-Alcoceba R, Pihalja M, Nunez G, Clarke MF. Evaluation of a new dual-
specificity promoter for selective induction of apoptosis in breast cancer cells. Cancer
Gene Ther. 2001; 8: 298-307.

9. Dissertationsbezogene bibliographische Daten
86
9. Dissertationsbezogene bibliographische Daten
Publikationen:
Hoffmann D, Jogler C, Wildner O (2004). Effects of the Ad5 upstream E1 region and Early
Gene Products on Heterologous Promoters. J. Gene Med. Im Review.
Jogler C, Hoffmann D, Uberla K, Wildner O (2004). Evaluation of Swine as an Animal
Model for Replication-Competent Human Adenoviral Vectors. Hum. Gene Ther. Im Review.
Kuate S, Stefanou D, Hoffmann D, Wildner O, Uberla K (2004). Production of lentiviral
vectors by transient expression of minimal packaging genes from recombinant adenoviruses. J
Gene Med. 6, 1197-1205.
Wildner O, Hoffmann D, Jogler C, Uberla K (2003). Comparison of HSV-1 thymidine kinase-
dependent and -independent inhibition of replication-competent adenoviral vectors by a panel
of drugs. Cancer Gene Ther. 10, 791-802.
Präsentationen:
Development of tumor-specific, oncolytic adenoviral vectors for treatment of pancreatic
carcinoma.
Jahrestagung 2004 der Gesellschaft für Virologie in Tübingen, 17.-20. März 2003
Comparison of HSV-1 thymidine kinase dependent and independent inhibition of replication-
competent adenoviral vectors by a panel of drugs.
Jahrestagung 2004 der Gesellschaft für Virologie in Tübingen, 17.-20. März 2003
Comperative in vitro and in vivo antiviral activity of nucleoside analogs against replication-
competent adenovirus vector expressiong HSV-tk.
Jahrestagung 2003 der Gesellschaft für Virologie in Berlin, 26.-29. März 2003

10. Lebenslauf
87
10. Lebenslauf
Persönliche Daten:
Name: Dennis Hoffmann
Geburtsdatum: 24. März 1973
Geburtsort: Oberhausen
Schulausbildung:
1980 – 1984 Hegelschule in Oberhausen
1984 – 1994 Abtei-Gymnasium in Duisburg-Hamborn
Wehrdienst
1994 – 1995 1. RakArtBat 150 in Wesel
Studium:
WS 1995/96 - SS 2001 Studium der Biochemie an der Ruhr-Universität Bochum
7. Januar - Diplomarbeit in der Pharmakologie und Toxikologie der Ruhr-
7. September 2001 Universität Bochum über „Identifizierung von mit der lös-
lichen Guanylyl-Cyclase interagierenden Proteine“
Promotion:
seit 1. Oktober 2001 Wissenschaftlicher Mitarbeiter der Abteilung für Molekulare
und Medizinische Virologie an der Ruhr-Universität Bochum
18. Juni 2002 Sophia & Fritz Heinemann Preis zur Förderung der
Entwicklung von Gentherapien mittels Adenoviren gegen
Glioblastome


















