
Combined solid-phase microextraction and high-performance liquid chromatography with ultroviolet...
7
Combined solid-phase microextraction and high-performance liquid chromatography with ultroviolet detection for simultaneous analysis of clenbuterol, salbutamol and ractopamine in pig samples Wei Du a , Siruo Zhang b , Qiang Fu a *, Gang Zhao a and Chun Chang a ABSTRACT: A simple and sensitive method based on the combination of solid-phase microextraction (SPME) and high-performance liquid chromatography with ultroviolet detection was developed for the simultaneous determination of clenbuterol, salbutamol and ractopamine in pig samples. Parameters of the SPME procedure affecting extraction efficiency, such as the type of fiber, extraction time, extraction temperature, ion strength, pH of sample and stirring rate, were optimized. The developed method was validated according to the International Conference on Harmonization guidelines. The calibration curves were linear over a range of 0.5–50 μg/L for clenbuterol and ractopamine, and 0.2–20 μg/L for salbutamol. The limits of detection were 0.1 μg/L for clenbuterol, 0.05 μg/L for salbutamol and 0.1μg/L for ractopamine, respectively. The averages of intra- and inter-day accuracy ranged from 79.8 to 92.4%. The intra-day and inter-day precision were below 9.6% for the three analytes. This method exhibited the advantages of simplicity, rapidity and low solvent consumption, and was suitable for the monitoring of β 2 -agonists residue in pig samples. Copyright © 2013 John Wiley & Sons, Ltd. Keywords: solid-phase microextraction; high-performance liquid chromatography–ultraviolet detection; clenbuterol; salbutamol; ractopamine Introduction Veterinary drugs are widely administered to food-producing animals to prevent the outbreak of diseases (Stolker et al., 2007). However, veterinary drug residues in meat products, eggs and milk products can increase the risk of introducing harmful residues into the human food chain. β 2 -Agonists are a class of synthetic phenethanolamine compounds that are designed and used clinically for the treatment of pulmonary disease, especially asthma. They have been found to reduce fat deposition in body and en- hance growth in cattle, sheep and swine if used in high doses (Engeseth et al., 1992; Sawaya et al., 2000; Strydom et al., 2009). A number of β 2 -agonists have been illegally used as the feed additive in animal production to promote animal growth. The prolonged accumulation of β 2 -agonists in human foodstuffs has caused severe threats to human health, leading to muscle tremors, headache and palpitations (Martínea-Navarro, 1990; Brambilla et al., 2000). To protect consumers’ health, β 2 -agonists are banned in the EU (Commission of the European Communities, 1996) and China (The Ministry of Agriculture, 2002; Zhang et al., 2012). Among β 2 -agonists, clenbuterol is the most popular β 2 -agonist for illegal use in animals. Salbutamol and ractopamine are usually illegally used as substitutes of clenbuterol to avoid government examination (Yan et al., 2013). The chemical structures of clenbuterol, salbutamol and ractopamine are showed in Figure 1. Therefore, a rapid, simple and sensitive analytical method is required to simultaneously determine clenbuterol, salbutamol and ractopamine in food samples. Recent studies have shown that many methods can be applied for screening and identification of β 2 -agonists in biological samples, such as high-performance liquid chromatog- raphy (HPLC) with ultraviolet detection (Yan et al., 2012, 2013; Blomgren et al., 2002) and fluorescence detection (Hu et al., 2010), liquid chromatography–tandem mass spectrometry (Shao et al., 2009; Dong et al., 2011), gas chromatography–mass spectrometry (GC-MS; Abukhalaf et al., 2000; He et al., 2007), enzymelinked immunoassay (Johansson and Hellenas, 2004; Posyniak et al., 2003; Lu et al., 2012), capillary electrophoresis with amperometric detection (Chen et al., 2005), and electro- chemical method with differential-pulse voltammetry (Moane et al., 1996). In these articles, the extraction and purification of β 2 -agonists from food samples was mainly performed by solid- * Correspondence to: Q. Fu, School of Pharmaceutical Sciences, Xi'an Jiaotong University, Xi'an, 710061, People's Republic of China. Email: [email protected] a School of Pharmaceutical Sciences, Xi'an Jiaotong University, Xi'an, 710061, People's Republic of China b Department of microbiology, Dalian Medical University, Dalian, 116044, People's Republic of China Abbreviations used: PA, polyacrilate; PDMS/DVB, polydimethylsiloxane- divinylbenzene; SPE, solid-phase extraction; SPME, solid-phase microextraction. Biomed. Chromatogr. 2013; 27: 1775–1781 Copyright © 2013 John Wiley & Sons, Ltd. Research article Received: 9 April 2013, Revised: 22 May 2013, Accepted: 9 June 2013 Published online in Wiley Online Library: 11 July 2013 (wileyonlinelibrary.com) DOI 10.1002/bmc.2993 1775
Transcript of Combined solid-phase microextraction and high-performance liquid chromatography with ultroviolet...

Research article
Received: 9 April 2013, Revised: 22 May 2013, Accepted: 9 June 2013 Published online in Wiley Online Library: 11 July 2013
(wileyonlinelibrary.com) DOI 10.1002/bmc.2993
Combined solid-phase microextraction andhigh-performance liquid chromatography withultroviolet detection for simultaneous analysisof clenbuterol, salbutamol and ractopaminein pig samplesWei Dua, Siruo Zhangb, Qiang Fua*, Gang Zhaoa and Chun Changa
ABSTRACT: A simple and sensitivemethod based on the combination of solid-phasemicroextraction (SPME) and high-performanceliquid chromatography with ultroviolet detection was developed for the simultaneous determination of clenbuterol, salbutamoland ractopamine in pig samples. Parameters of the SPME procedure affecting extraction efficiency, such as the type of fiber,extraction time, extraction temperature, ion strength, pH of sample and stirring rate, were optimized. The developed methodwas validated according to the International Conference on Harmonization guidelines. The calibration curves were linear over arange of 0.5–50 μg/L for clenbuterol and ractopamine, and 0.2–20 μg/L for salbutamol. The limits of detection were 0.1 μg/L forclenbuterol, 0.05 μg/L for salbutamol and 0.1μg/L for ractopamine, respectively. The averages of intra- and inter-day accuracyranged from 79.8 to 92.4%. The intra-day and inter-day precision were below 9.6% for the three analytes. This method exhibitedthe advantages of simplicity, rapidity and low solvent consumption, and was suitable for the monitoring of β2-agonists residue inpig samples. Copyright © 2013 John Wiley & Sons, Ltd.
Keywords: solid-phase microextraction; high-performance liquid chromatography–ultraviolet detection; clenbuterol; salbutamol;ractopamine
* Correspondence to: Q. Fu, School of Pharmaceutical Sciences, Xi'anJiaotong University, Xi'an, 710061, People's Republic of China. Email:[email protected]
a School of Pharmaceutical Sciences, Xi'an Jiaotong University, Xi'an, 710061,People's Republic of China
b Department of microbiology, Dalian Medical University, Dalian, 116044,People's Republic of China
Abbreviations used: PA, polyacrilate; PDMS/DVB, polydimethylsiloxane-divinylbenzene; SPE, solid-phase extraction; SPME, solid-phasemicroextraction.
17
IntroductionVeterinary drugs are widely administered to food-producinganimals to prevent the outbreak of diseases (Stolker et al., 2007).However, veterinary drug residues in meat products, eggs andmilk products can increase the risk of introducing harmful residuesinto the human food chain. β2-Agonists are a class of syntheticphenethanolamine compounds that are designed and usedclinically for the treatment of pulmonary disease, especially asthma.They have been found to reduce fat deposition in body and en-hance growth in cattle, sheep and swine if used in high doses(Engeseth et al., 1992; Sawaya et al., 2000; Strydom et al., 2009). Anumber of β2-agonists have been illegally used as the feed additivein animal production to promote animal growth. The prolongedaccumulation of β2-agonists in human foodstuffs has caused severethreats to human health, leading to muscle tremors, headache andpalpitations (Martínea-Navarro, 1990; Brambilla et al., 2000). Toprotect consumers’ health, β2-agonists are banned in the EU(Commission of the European Communities, 1996) and China (TheMinistry of Agriculture, 2002; Zhang et al., 2012). Among β2-agonists,clenbuterol is the most popular β2-agonist for illegal use in animals.Salbutamol and ractopamine are usually illegally used as substitutesof clenbuterol to avoid government examination (Yan et al., 2013).The chemical structures of clenbuterol, salbutamol and ractopamineare showed in Figure 1. Therefore, a rapid, simple and sensitiveanalytical method is required to simultaneously determineclenbuterol, salbutamol and ractopamine in food samples.
Biomed. Chromatogr. 2013; 27: 1775–1781 Copyright © 2013 John
Recent studies have shown that many methods can beapplied for screening and identification of β2-agonists inbiological samples, such as high-performance liquid chromatog-raphy (HPLC) with ultraviolet detection (Yan et al., 2012, 2013;Blomgren et al., 2002) and fluorescence detection (Hu et al.,2010), liquid chromatography–tandem mass spectrometry (Shaoet al., 2009; Dong et al., 2011), gas chromatography–massspectrometry (GC-MS; Abukhalaf et al., 2000; He et al., 2007),enzymelinked immunoassay (Johansson and Hellenas, 2004;Posyniak et al., 2003; Lu et al., 2012), capillary electrophoresiswith amperometric detection (Chen et al., 2005), and electro-chemical method with differential-pulse voltammetry (Moaneet al., 1996). In these articles, the extraction and purification ofβ2-agonists from food samples was mainly performed by solid-
Wiley & Sons, Ltd.
75

W. Du et al.
1776
phase extraction (SPE; Yan et al., 2012; Shao et al., 2009; Dong et al.,2011). However, these techniques were often time-consuming,laborious and expensive (the use of expensive disposablecartridges); in addition, they could cause environmental pollutionbecause of the associated use and release of abundant organicsolvents. Recently, increasing public concern over environmentalissues has prompted the development of simple and nonpollutingtechniques of sample preparation (Farré et al., 2010).
Solid phase microextraction (SPME) has emerged as an attractivealternative for sample preparation owing to its simplicity, lowsolvent consumption and ease of connection online to either gasor liquid chromatographic systems (Pawliszyn, 1997; Kataoka et al.,2000). SPME incorporates sample extraction, concentration andsample introduction into a single procedure. In SPME, analytes arepartitioned between the sample matrix or sample extract and thephase on the fused silica fiber (Risticevic et al., 2010). In contrast totraditional extraction techniques, SPME is cheap (one fiber can begenerally used for hundreds of extractions) and simple to perform;furthermore, it can reduce the solvent consumption and analyticaltime. SPME is therefore a green technique for sample preparation.Since its inception, SPME has been widely used in different fields,including biology, toxicology, pharmaceuticals and forensics (TorresPadrón et al., 2009; Moller et al., 2010; Bojko et al., 2011; Zhang et al.,2009; Ouyang et al., 2011). Recently, it has emerged as a rapidanalytical tool for the monitoring of contaminants in food samples(Kataoka et al., 2000; Lambropoulou and Albanis, 2003; Zamboninet al., 2004; Ridgway et al., 2007). The first attempt for the analysisof clenbureol in biological samples with SPME was reported byMelwanki et al. (2005), who developed a combined headspaceSPME and GC-MS method for the determination of clenbuterol inhuman urine. However, GC-MS required a time-consumingderivatization steps prior to injection. Aresta et al. (2008) reporteda HPLC-UV method combined with SPME to determine clenbuterolin human urine and serum samples. However, the extraction timewas long (60 min), which was not suitable for routine analysis. Tothe best of our knowledge, the application of SPME forsimultaneous analysis of clenbuterol, salbutamol and ractopaminein pig samples has not been reported.
In this study, the simultaneous determination of clenbuterol,salbutamol and ractopamine residues in pig samples, usingSPME combined with HPLC-UV (SPME-HPLC-UV) was presentedfor the first time. The SPME parameters affecting the extractionefficiency, such as type of fiber, extraction time, extractiontemperature, ion strength, stirring rate, pH and desorption time,were optimized. The proposed method was validated over awide range of concentrations for the three analytes. Itsapplication to real samples demonstrated that this method issimple, rapid, organic solvent-free, and suitable for themonitoring of β2-agonists residue in pig samples.
Experimental
Chemicals and reagents
Clenbuterol was obtained from Jinhe Pharmaceutical Co. (Wuhan, China).Ractopamine was purchased from Sigma-Aldrich (NJ, USA). Salbutamolwas purchased from Cunyi Chemical Co. (Jiangsu, China). HPLC-gradeacetonitrile and methanol were obtained from Kermel Co. (Tianjin,China). Analytical-grade sodium chloride, sodium dihydrogen phos-phate, disodium hydrogen phosphate and phosphoric acid were pur-chased from Yaohua Chemical Reagent Co. (Tianjin, China). Ultrapurewater was prepared using a Molelement 1805b unit (Shanghai, China).
Copyright © 2013 John Wiwileyonlinelibrary.com/journal/bmc
Apparatus
HPLC analysis was performed using a Shimadzu HPLC system equippedwith an LC-10ATVP pump, a SPD-10AVP UV detector, a CTO-20A columnoven and an LC Solution workstation software (Shimadzu, Kyoto, Japan).The separation was performed on a Shimadzu VP-ODS column (4.6 × 250mm, i.d., 5.0 μm particle size; Dalian, China) at 25°C. The mobile phasewas methanol–0.1% aqueous formic acid (40:60, v/v) at a flow rate of1.0 mL/min. The injection volume was 20 μL and the detectionwavelength of the detector was set at 225 nm.
The SPME–HPLC interface consisted of a six-port Rheodyneinjection valve, equipped with a desorption chamber (total volume60 μL; Supelo, Bellenfonte, PA, USA). The SPME vials (4.0 mL), afiber holder for manual sampling and three types of SPME fibers(65 μm polydimethylsiloxane-divinylbenzene (PDMS/DVB), 85 μmpolyacrilate (PA) and 100 μm PDMS) were supplied by Supelo(Bellenfonte, PA, USA). Agitation was performed by magnetic stirbars and a magnetic stirring apparatus (Henan, China). The pH ofthe solution was tested using a Mettler-Toledo FE 20 pH meter(Shanghai, China). The High Speed Refrigerated Centrifuge Genius16K-R was supplied by Xinao Instrument Co. (Changsha, China).
Sample pretreatment
Porcine muscle and liver were chosen as the biological matrix toevaluate the usefulness of the developed method. The pretreatmentof porcine muscle and liver was performed according to the methodpublished by Hu et al. (2011). Briefly, 2.0 g of tested samples werecrushed up and mixed with 1 mL of ultrapure water. After homoge-nization, the samples were extracted with 2 mL acetonitrile and thenultrasonicated for 5 min. After centrifuging at 8000 rpm for 5 min,the supernatant was collected and dried under nitrogen stream.Finally, the residual was dissolved in 4.0 mL of 0.1 mol/L phosphatebuffer (pH 10) for further SPME processing.
Standard and sample solution preparation
The stock solutions of clenbuterol, salbutamol and ractopamine wereseparately prepared by dissolving each compound in ultrapure waterto give a final concentration 10 mg/mL. Mixed standard solutions ofthe analytes (20–2000 μg/mL for clenbuterol and ractopamine, and8–800 μg/L for salbutamol) were prepared daily by diluting the stocksolutions with the mobile phase,
For the spiking analysis, 2.0 g of porcine muscle and liver samples,determined to be free of the analyte, were mixed with 100 μL of mixedstandard solutions, prior to the sample pretreatment. The spikedcalibration concentration of calibrates was 0.5–50 μg/L for clenbuteroland ractopamine, and 0.2–20 μg/L for salbutamol. Quality controlsamples (0.5, 5 and 20 μg/L) were also prepared in the same way asthe spiked calibration and stored at 4°C until analysis.
SPME procedure
For SPME procedure, 4.0 mL of pre-treated sample was put into a 4.0 mLSPME vial followed by adding to 1.0 g sodium chloride. After addition ofa stirring bar, the vial was closed and placed into a thermostatted bathadjusted at 30°C. Under the magnetic agitation (800 rpm), the SPME fiberwas direct-immersed into the sample solution for 20 min. After eachextraction, the fiber was removed from the vial and desorbed for 3 minin the desorption chamber with the six-port injection valve in the ‘load’position. The chamber was previously filled with methanol–0.1%aqueous formic acid (40:60, v/v). Following the static desorption, theinjection valve was switched from the ‘load’ to the ‘injection’ position,allowing the mobile phase to pass through the chamber for 1 min. Inorder to avoid carryover, the fiber was removed and soaked in 4 mL ofmethanol–water (80:20, v/v) for 2 min after each analysis.
Biomed. Chromatogr. 2013; 27: 1775–1781ley & Sons, Ltd.

SPME-HPLC for analysis of clenbuterol, salbutamol and ractopamine
Method validation
The method validation was performed in terms of specificity, linearity,accuracy, precision, limit of detection (LOD) and limit of quantitation(LOQ), following the recommendations of the International Conferenceon Harmonization (ICH, 2005). Specificity was assessed by the absence ofinterferences at the retention time of analytes. The linearity of the methodwas tested by preparing the spiked calibration curve for each analyte. Thetested concentration ranges were 0.5–50 μg/L for clenbuterol andractopamine, and 0.2–20 μg/L for salbutamol. The LOD and LOQ werecalculated from the injection of the spiked sample that provided asignal-to-noise ratio of 3 and 10, respectively.
Intra-day precision (repeatability) and accuracy of the assay wereevaluated by determining the QC samples in five replicates during a singleday. The inter-day (intermediate) precision and accuracy were calculated bydetermining QC samples in five replicates on five consecutive days. Theprecision was expressed as percentage relative standard deviation (RSD) ofthe replicate measurements, with acceptable values for the RSD being<15%. Accuracy was expressed as a percentage of the recovery.
Results and discussion
Optimization of chromatographic conditions
In this study, the mobile phase composition, wavelength fordetection, type of column and flow rate were selected to obtainthe best chromatographic conditions. The initial mobile phase wasmethanol–water (40:60, v/v). A buffer solution was added to themobile phase to reduce the ionization and polarity of somecompounds. Three concentrations (0.01, 0.1 and 0.5%) of formic acidsolution with methanol were tested. A mixture of methanol–0.1%aqueous formic acid (40:60, v/v) at a flow rate of 1.0 mL/min wasobserved to provide acceptable resolution and symmetric peakshapes for three analytes. Therefore it was selected as the mobilephase. The wavelength selected for detection was 225 nm, becauseof the maximum absorption of three analytes.
To achieve baseline separation and good resolution, C18columns from various manufacturers were evaluated, includingAgela Technologies Promosil C18 (150 × 4.6 mm i.d, 5 μm),Shimadzu VP-ODS (150 × 4.6 mm i.d., 5 μm), Tianhe Kromasil(150 × 4.6 mm i.d., 5 μm), Shimadzu VP-ODS (250 mm × 4.6mm i.d., 5 μm) and Tianhe Kromasil (250 × 4.6 mm i.d., 5 μm).The Shimadzu VP-ODS (250 mm × 4.6 mm i.d., 5 μm) was foundto provide satisfactory peak symmetries, resolutions and runtimes for three analytes.
Optimization of SPME conditions
In the SPME procedure, various parameters affecting theextraction efficiency were optimized to obtain high extractionefficiency. These parameters include the type of fiber, ionstrength, extraction temperature, extraction time, pH of sample,stirring rate and desorption conditions. The mixed standardssolution (200 μg/mL for clenbuterol and ractopamine, and80 μg/L for salbutamol) was extracted with the SPME fiber. Theperformance for each parameter was analyzed in triplicate.
Figure 1. Chemical structures: (a) clenbuterol; (b) salbutamol; and(c) ractopamine.
177
Type of fiber
Selection of the fiber is the first step in SPME methoddevelopment. The type of fiber is an important parameteraffecting the extraction efficiency. Therefore, three differentfibers (85 μm PA, 100 μm PDMS and 65 μm PDMS/DVB) weretested during the optimization. Among the tested fibers,
Biomed. Chromatogr. 2013; 27: 1775–1781 Copyright © 2013 John
65 μm PDMS/DVB exhibited the greatest extraction efficiencyfor all analytes. This could be explained by the fact that 65 μmPDMS/DVB is a mixed-phase fiber coating, which provides ahigher distribution constant than PDMS and PA, since theadsorption process on porous particles is suitable for more polarcompounds (Risticevic et al., 2010). Thus, 65 μm PDMS/DVB wasselected as the SPME fiber. This choice is consistent with theobservation of other workers for clenbuterol analysis (Arestaet al., 2008).
Effect of ionic strength
The addition of salt increases the ionic strength of the samplesolution, and then enhances the extraction efficiency (Risticevicet al., 2010). In this study, the effect of ionic strength on extractionefficiency was investigated by adding NaCl to sample solution atconcentrations from 5 to 35% (w/v). As shown in Fig. 2(a), theextraction efficiencies for three analytes increased with the NaClconcentration from 5 to 25%, and then decreased with the NaClconcentration from 25 to 35%. This result could be explained bythe high sodium chloride concentration leading to the ionizationof analyte and causing the analyte molecules to pass more readilyfrom the sample onto the fiber coating (Risticevic et al., 2010).However, at the saturation state (35%), the analyte moleculesmight participate in electrostatic interactions with the salt ions,reducing their ability to move onto the fiber coatings (Quintoet al., 2009; Perestrelo et al., 2011). Thus, 25% of sodium chlorideconcentration was chosen as the optimal level of ion strength.
Effect of extraction temperature
Extraction temperature affects the mass transfer rate and theanalyte partition coefficient. Increasing extraction temperaturecan accelerate the mass transfer and raise the extractionefficiency (Risticevic et al., 2010). In our study, the extractiontemperature was optimized in the range from 20 to 60°C. Asseen in Fig. 2(b), the extraction efficiencies for three analytesincreased with the temperature from 20 to 30°C, and thendecreased with the temperature from 30 to 60°C. This resultindicated that the adsorption on the fiber was an exothermicprocess, and the amounts of the three analytes adsorbeddecreased with the increasing temperature (Risticevic et al.,2010). Therefore, the extraction was carried out at 30°C.
Effect of extraction time
The extraction time is the time-limiting step of the SPMEprocedure. SPME is an equilibrium process, in which analyte isdistributed between the fiber and sample matrix (Risticevic
Wiley & Sons, Ltd. wileyonlinelibrary.com/journal/bmc
7
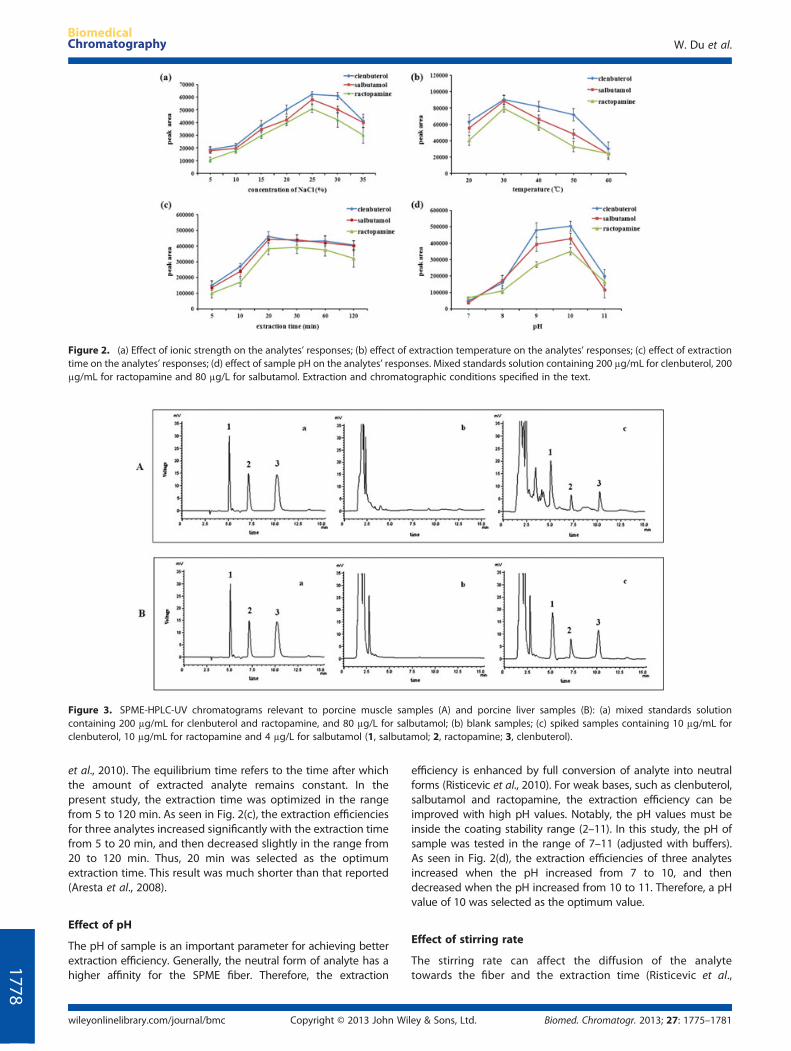
Figure 2. (a) Effect of ionic strength on the analytes’ responses; (b) effect of extraction temperature on the analytes’ responses; (c) effect of extractiontime on the analytes’ responses; (d) effect of sample pH on the analytes’ responses. Mixed standards solution containing 200 μg/mL for clenbuterol, 200μg/mL for ractopamine and 80 μg/L for salbutamol. Extraction and chromatographic conditions specified in the text.
Figure 3. SPME-HPLC-UV chromatograms relevant to porcine muscle samples (A) and porcine liver samples (B): (a) mixed standards solutioncontaining 200 μg/mL for clenbuterol and ractopamine, and 80 μg/L for salbutamol; (b) blank samples; (c) spiked samples containing 10 μg/mL forclenbuterol, 10 μg/mL for ractopamine and 4 μg/L for salbutamol (1, salbutamol; 2, ractopamine; 3, clenbuterol).
W. Du et al.
1778
et al., 2010). The equilibrium time refers to the time after whichthe amount of extracted analyte remains constant. In thepresent study, the extraction time was optimized in the rangefrom 5 to 120 min. As seen in Fig. 2(c), the extraction efficienciesfor three analytes increased significantly with the extraction timefrom 5 to 20 min, and then decreased slightly in the range from20 to 120 min. Thus, 20 min was selected as the optimumextraction time. This result was much shorter than that reported(Aresta et al., 2008).Effect of pH
The pH of sample is an important parameter for achieving betterextraction efficiency. Generally, the neutral form of analyte has ahigher affinity for the SPME fiber. Therefore, the extraction
Copyright © 2013 John Wiwileyonlinelibrary.com/journal/bmc
efficiency is enhanced by full conversion of analyte into neutralforms (Risticevic et al., 2010). For weak bases, such as clenbuterol,salbutamol and ractopamine, the extraction efficiency can beimproved with high pH values. Notably, the pH values must beinside the coating stability range (2–11). In this study, the pH ofsample was tested in the range of 7–11 (adjusted with buffers).As seen in Fig. 2(d), the extraction efficiencies of three analytesincreased when the pH increased from 7 to 10, and thendecreased when the pH increased from 10 to 11. Therefore, a pHvalue of 10 was selected as the optimum value.
Effect of stirring rate
The stirring rate can affect the diffusion of the analytetowards the fiber and the extraction time (Risticevic et al.,
Biomed. Chromatogr. 2013; 27: 1775–1781ley & Sons, Ltd.

SPME-HPLC for analysis of clenbuterol, salbutamol and ractopamine
2010). The optimization of stirring rate was carried out at200, 400, 600, 800 and 1000 rpm. The results showed thatthe extraction efficiencies for three analytes increased withthe stirring rate from 200 to 800 rpm, and then kept constantwith the stirring rate from 800 to 1000 rpm. Therefore, 800rpm was the optimal stirring rate.
Desorption conditions
There are two different modes of desorption in the SPMEprocedure: dynamic desorption and static desorption. In thedynamic desorption, the analytes are desorbed in a movingstream of mobile phase. In the static mode, the fiber wasdesorbed in the desorption chamber with the six-port injectionvalve in the ‘load’ position. After desorption for some time, thevalve of SPME assembly was switched into the ‘inject’ position,allowing the mobile phase to pass through the chamber.(Risticevic et al., 2010). Since the dynamic desorption couldproduce a broad chromatographic peak, the static desorptionwas adopted in this study.
Different desorption time (from 0.5 to 10 min) wasstudied to optimize the desorption condition. The resultsshowed that the peak areas of three analytes increased withthe desorption time from 0.5 to 3 min, and then remainedalmost constant in the range from 3 to 10 min. Thus, inorder to implement a more expeditious method, 3 min ofdesorption time was selected.
Carry-over
In contrast to SPE procedure, which use disposable cartridge,SPME requires the evaluation of potential carry-over, as itinvolves the reuse of the fiber. Carry-over was tested by
Table 1. The linear ranges, correlation coefficients and limits of d
Analyte Linear range (μg/L) Correla
Muscle samplesClenbuterol 0.5–50Salbutamol 0.2–20Ractopamine 0.5–50Liver samplesClenbuterol 0.5–50Salbutamol 0.2–20Ractopamine 0.5–50
Table 2. Accuracy and precision (n = 5)
Muscle samples
Intra-day Inter-day
Accuracy(%)
Precision(RSD%)
Accuracy(%)
Prec(RSD
Clenbuterol 80.6 7.9 79.8 8Sabutamol 80.0 5.8 82.3 5Ractopamine 87.5 8.1 86.4 9
Biomed. Chromatogr. 2013; 27: 1775–1781 Copyright © 2013 John
analyzing the fiber blank directly after the initial injection andexpressed as the percentage of the initial peak area (Changet al., 2003). Therefore, the SPME fiber was was removed andsoaked in 4 mL of methanol-water (80:20, v/v) for 2 min betweentwo extractions. The carry-over values of clenbuterol, salbutamoland ractopamine were 0.9, 1.2 and 0.6%.
Method validation
Matrix effects are the major problems in immersion SPMEprocedure. In the present study, a blank of real samples wasrun to verify the absence of peaks at the retention time of thecompounds under the optimized SPME-HPLC-UV conditions.The representative chromatograms of a mixed standardsolution, blank and spiked muscle and liver samples are com-pared in Fig. 3. As can be observed from the chromatograms,the target analytes were well resolved from matrix components.Moreover, to observe the feasibility of SPME-HPLC-UV method
in real muscle and liver samples, calibration was carried out usingspiked samples. Under the optimum conditions, a good linearrelationship between the corresponding peak areas and the con-centrations was obtained for three analytes, with the correlationcoefficients (r2) being>0.9821. The LODs of the proposed methodranged from 0.05 to 0.1 μg/L, whereas the LOQs ranged from 0.2to 0.5 μg/L (Table 1). These results were lower than those achievedwith the conventional preparation techniques, such as SPE (Huet al., 2010).Method accuracy and precision were assessed by performing
replicate analyses of QC samples on the same day and threeconsecutive days. The averages of intra- and inter-day accuracyranged from 79.8 to 92.4%. The intra-day precision expressedas the RSD ranged from 5.0 to 8.1%, and the inter-day precisionranged from 5.7 to 9.6% (Table 2). The results indicated that the
etection (LOD) and quanitation (LOQ)
tion coefficient LOD (μg/L) LOQ (μg/L)
0.9979 0.1 0.50.9968 0.05 0.20.9883 0.1 0.5
0.9910 0.1 0.50.9842 0.05 0.20.9821 0.1 0.5
Liver samples
Intra-day Inter-day
ision%)
Accuracy(%)
Precision(RSD%)
Accuracy(%)
Precision(RSD%)
.5 89.3 6.6 81.4 8.2
.7 92.4 5.0 90.7 7.3
.2 79.6 7.5 89.9 9.6
Wiley & Sons, Ltd. wileyonlinelibrary.com/journal/bmc
1779

W. Du et al.
1780
proposed method could be applied for the determination ofβ2-agonists in pig samples with satisfactory accuracy and preci-sion. Notably, a significant increase in terms of sensitivity couldbe easily achieved with this method, replacing the UV detectorwith MS or MS/MS (Aresta et al., 2010).
Real sample analysis
Real pig samples (muscle and liver) were analyzed by the proposedSPME-HPLC-UVmethod to test the effectiveness of themethod. Tenporcine muscle and liver samples were purchased from localmarkets and were analyzed by SPME-HPLC-UVmethod. Clenbuterolwas detected in two liver samples with the concentrations of 0.62and 0. 81 μg/kg. Salbutamol and ractopamine were not detectedin all samples. These results suggested that clenbuterol was stillbeing illegally used in local food-producing animals.
ConclusionsIn the present study, a combined SPME and HPLC-UV method wasdeveloped and validated for the simultaneous determination ofclenbuterol, salbutamol and ractopamine in porcine muscle andliver samples. Under the optimized conditions, the precision,accuracy and recovery of the proposed method were good andconvenient for the routine analysis. Moreover, the proposedmethod is simple to perform, and requires less consumption oforganic solvents. The SPME fiber can be used several times, avoidingdisposal of the expensive SPE cartridges. Currently, work is inprogress to extend the application of the proposedmethod to otherreal matrices, and to perform in vivo SPME sampling in tissues.
AcknowledgmentThis work was financially supported by National Natural ScienceFoundations of China (nos 30873193, 812278063 and 81173024).
ReferencesAbukhalaf IK, Deutsch DA, Parks BA, Wineski L, Paulsen D, Aboul-Enein HY
and Potter DE. Comparative analytical quantitation of clenbuterol inbiological matrices using GC-MS and EIA. Biomedical Chromatography2000; 14: 99–105.
Aresta A, Calvano CD, Palmisano F and Zambonin CG, Determination ofclenbuterol in human urine and serum by solid-phase microextractioncoupled to liquid chromatography. Journal of Pharmaceutical andBiomedical Analysis 2008; 47: 641–645.
Aresta A, Bianchi D, Calvano CD and Zambonin CG. Solid phasemicroextraction-Liquid chromatography (SPME-LC) determination ofchloramphenicol in urine and environmental water samples. Journalof Pharmaceutical and Biomedical Analysis 2010; 53:440–444.
Blomgren A, Berggren C, Holmberg A, Larsson F, Sellergren B and EnsingK. Extraction of clenbuterol from calf urine using a molecularlyimprinted polymer followed by high-peformance liquid chromatog-raphy with UV detection. Journal of Chromatography. A 2002;975:157–164.
Bojko B, Vuckovic D, Cudjoe E, HoqueME, Mirnaghi F, Wasowicz M, Jerath Aand Pawliszyn J. Determiantion of traneamic acid concentration bysolid phase microextraction and liquid chromatography-tandem massspectrometry: first step to in vivo analysis. Journal of ChromatographyB 2011; 879: 3871–3787.
Brambilla G, Cenci T, Franconi F, Galarini R, Macri A, Rondoni F, Strozzie Mand Loizzo A. Clinical and pharmacological profile in a clenbuterolepidemic poisoning of contaminated beef meat in Italy. ToxicologyLetters 2000, 114, 47–53.
Chang WY, Sun YH and Duan SD. Analysis of carcinogenic aromaticamines in water samples by solid-phase microextraction coupledwith high-performance liquid chromatography. Analytica ChimicaActa 2003; 495: 109–122.
Copyright © 2013 John Wiwileyonlinelibrary.com/journal/bmc
Chen YC, Wang W, Duan J, Chen H and Chen G. Separation anddetermination of clenbuterol, cimaterol and salbutamol by capillaryelectrophoresis with amperometric detection. Electroanalysis 2005;17: 706–712.
Commission of the European Communities. Council Directive 96/22/EC onthe prohibition of the use of certain substances having a hormonal andthyreostatic action and β2-agonists in animal husbandry. OfficialJournal of the European Communities 1996; L 125: 3–9.
Dong YC, Xia X, Wang X, Ding SY, Li XW, Zhang SX, JiangHY, Liu JF, Li JC, FengZZ, Ye N, ZhouMX and Shen JZ. Validation of an ultra-performance liquidchromatography–tandem mass spectrometry method for determinationof ractopamine: application to residue depletion study in swine. FoodChemistry 2011; 127: 327–332.
Engeseth NJ, Lee KO, Bergen WG, Helferich WH, Knudson BK and MerkelRA. Fatty acid profiles of lipid depots and cholesterol concentration inmuscle tissue of finishing pig fed ractopamine. Journal of FoodScience, 1992; 57: 1060–1062.
Farré M, Pérez S, Gonçalves C, Alpendurada MF and Barceló D. Greenanalytical chemistry in the determination of organic pollutants inthe aquatic environment. Trends in Analytical Chemistry 2010; 29:1347–1362.
He LM, Su YJ, Zeng ZL, Liu YH and Huang XH. Determination ofractopamine and clenbuterol in feeds by gas chromatography–mass spectrometry. Animal Feed Science and Technology 2007;132: 316–323.
Hu YL, Liu RJ, Li YW and Li GK. Investigation of ractopamine-imprintedpolymer for dispersive solid-phase extraction of trace β-agonists inpig tissues. Journal of Separation Science 2010; 33:2017–2025.
Hu YL, Li YW, Liu RJ, Tan W and Li GK. Magnetic molecularly imprintedpolymer beads prepared by microwave heating for selectiveenrichment of beta-agonists in pork and pig liver samples. Talanta2011; 84:462–470.
ICH. International Conference on Harmonization of TechnicalRequirements for Registration of Pharmaceuticals for Human Use.Validation of Analytical Procedures: Text and Methodology Q2 (R1).ICH Expert Working Group, ICH Harmonised Tripartite Guideline:Geneva, 2005; 1–13.
Johansson MA and Hellenas KE. Immunobiosensor determination ofbeta-agonists in urine using integrated immunofiltration clean-up.International Journal of Food Science and Technology (2004); 39:891–898.
Kataoka H, Lord HL and Pawliszyn J. Applications of solid-phasemicroextraction in food analysis. Journal of Chromatography. A2000; 880: 35–62.
Lambropoulou DA and Albanis TA. Headspace solid-phasemicroextraction incombination with gas chromatography–mass spectrometry for the rapidscreening of organophosphorus insecticide residues in strawberries andcherries. Journal of Chromatography. A 2003; 993: 197–203.
Lu X, Zheng H, Li XQ, Yuan XX, Li H, Deng LG, Zhang H, Wang WZ, YangGS, Meng M, Xi RM and Aboul-Enein HY. Detection of ractopamineresidues in pork by surface plasmon resonance-based biosensorinhibition immunoassay. Food Chemistry 2012; 130:1061–1065.
Martínea-Navarro JF. Food poisoning related to consumption of illicitβ-agonists in liver. Lancet 1990; 336:1311.
Melwanki MB, Hsu WH and Huang SD. Determination of clenbuterol in urineusing headspace solid phase microextraction or liquid–liquid–liquidmicroextraction. Analytica Chimica Acta 2005; 552: 67–75.
Moane S, Smyth MR and O'Keeffe M. Differential-pulse voltammetricdetermination of clenbuterol in bovine urine using a nafion-modifiedcarbon paste electrode. Analyst 1996; 121: 779–784.
Moller M, Aleksa K, Walasek P, Karaskov T and Koren G. Solid-phasemicroextraction for the determination of codeine, morphine and6-monoacetylmorphine in human hair by gas chromatography-massspectrometry. Forensic Science International 2010; 196: 64–69.
Ouyang GF, Oakes KD, Bragg L, Wang S, Liu H, Cui SF, Servos MR, GeorgeDixon D and Pawliszyn J. Sampling-rate calibration for rapid andnonlethal monitoring of organic contaminants in fish muscle bysolid-phase microextraction. Environmental Science and Technology2011; 45: 7792–7798.
Pawliszyn J. Solid Phase Microextraction. Theory and Practice. Wiley-WCH:New York, 1997; 133.
Perestrelo R, Barros AS, Rocha SM and Câmara JS. Optimisation of solid-phase microextraction combined with gas chromatography–massspectrometry based methodology to establish the global volatile
Biomed. Chromatogr. 2013; 27: 1775–1781ley & Sons, Ltd.

SPME-HPLC for analysis of clenbuterol, salbutamol and ractopamine
signature in pulp and skin of Vitis vinifera L. grape varieties. Talanta2011; 85: 1483–1493.
Posyniak A, Zmudzki J and Niedzielska J. Screening procedures forclenbuterol residue determination in bovine urine and liver matricesusing enzyme-linked immunosorbent assay and liquid chromatography.Analytical Chimica Acta 2003; 483: 61–67.
Quinto M, Spadaccino G, Palermo C and Centonze D. Determination ofaflatoxins in cereal flours by solid-phase microextraction coupledwith liquid chromatography and post-column photochemicalderivatization-fluorescence detection. Journal of Chromatography. A2009; 1216: 8636–8641.
Ridgway K, Lalljie SPD and Smith RM. Sample preparation techniques forthe determination of trace residues and contaminants in foods.Journal of Chromatography. A 2007; 1153: 36–53.
Risticevic S, Lord HL, Gόrecki T, Arthur CL and Pawliszyn J. Protocol forsolid-phase microextraction method development. Nature Protocols2010; 5:122–139.
SawayaWN, Lone K, Husain A, Dashti B and Saeed T. Screening for β-agonistsin sheep urine and eyes by an enzyme-linked immunosorbent assay inthe state of Kuwait. Food Control 2000, 11: 1–5.
Shao B, Jia XF, Zhang J, Meng J, Wu YN, Duan HJ and Tu XM. Multi-residualanalysis of 16 b-agonists in pig liver, kidney and muscle by ultraperformance liquid chromatography tandem mass spectrometry. FoodChemistry 2009; 114:1115–1121.
Stolker AAM, Zuidema T and Nielen MWF. Residue analysis of veterinarydrugs and growth-promoting agents. Trends in Analytical Chemistry2007; 26: 967–979.
Strydom PE, Frylinck L, Montgomery JL and Smith MF. The comparison ofthree β-agonists for growth performance, carcass characteristics andmeat quality of feedlot cattle. Meat Science 2009, 81: 557–564.
Biomed. Chromatogr. 2013; 27: 1775–1781 Copyright © 2013 John
The Ministry of Agriculture. Regulation No. 193. People's Republic ofChina, 2002.
Torres Padrón ME, Sosa Ferrera Z and Santana Rodríguez JJ. Coupling ofsolid-phase microextraction with micellar desorption and highperformance liquid chromatography for the determination ofpharmaceutical residues in environmental liquid samples. BiomedicalChromatography 2009; 23: 1175–1185.
Yan HY, Wang RL, Han YH and Liu ST. Screening, recognition andquantitation of salbutamol residues in ham sausages by molecularlyimprinted solid phase extraction coupled with high-performanceliquid chromatography-ultraviolet detection. Journal of Chromatogra-phy B 2012; 900:18–23.
Yan HY, Liu ST, Gao MM and Sun N. Ionic liquids modified dummymolecularly imprinted microspheres as solid phase extractionmaterials for the determination of clenbuterol and clorprenalinein urine. Journal of Chromatography. A 2013; 1294: 10–16.
Zambonin CG, Quinto M, Vietro ND and Palmisano F. Solid-phasemicroextraction-gas chromatography mass spectrometry: a fast andsimple screening method for the assessment of organophosphoruspesticides residues in wine and fruit juices. Food Chemistry 2004;86: 269–274.
Zhang HC, Liu GY and Chai CY. A novel amperometric sensor based onscreen-printed electrode modified with multi-walled carbonnanotubes and molecularly imprinted membrane for rapiddetermination of ractopamine in pig urine. Sensors and Actuators B2012, 168:103–110.
Zhang ZM, Duan HB, Zhang L, Chen X, Liu W and Chen GN. Direct deter-mination of anabolic in pig urine by a new SPME-GC-MS method.Talanta 2009; 78:1083–1089.
Wiley & Sons, Ltd. wileyonlinelibrary.com/journal/bmc
1781








![FARM 2219 : Pharmacologie spéciale · Metabolisme hépatique du paracétamol et toxicité ... HN COCH3 OH HN COCH3 OH SG N COCH3 O O O ... (salbutamol = exception [voir dia suivante])](https://static.fdocument.org/doc/165x107/5c81865109d3f263728c62b5/farm-2219-pharmacologie-spe-metabolisme-hepatique-du-paracetamol-et-toxicite.jpg)









