Circulating anti-β2-glycoprotein I antibodies of peripheral arterial disease patients trigger a...
10
Circulating anti-b 2 -glycoprotein I antibodies of peripheral arterial disease patients trigger a genomic overexpression of Toll-like receptor 4 in endothelial cells Cesar Varela, MD, a Joaquin de Haro, MD, a Silvia Bleda, MD, a Javier Rodriguez-Padilla, MD, a Antonio Ferruelo, PhD, b and Francisco Acín, MD, PhD, a Madrid, Spain Objective: Circulating anti-b 2 -glycoprotein I (ABGPI) antibodies are associated with peripheral arterial disease (PAD) and induce the expression of leukocyte adhesion molecules and proinflammatory cytokines by endothelial cells. Our aim is to study a transcriptional activation pathway of the innate immune system through the cellular signalling cascade triggered by receptors Toll-like receptor 2 (TLR2) and Toll-like receptor 4 (TLR4) of endothelial cells after the exposure of these cells to seropositive ABGPI human serum obtained from PAD patients. Methods: We obtained serum samples from PAD patients and controls without PAD. ABGPI serum titer was detected using indirect immunofluorescence. Our sample was stratified into three groups: group I (PAD and ABGPI titer $1:100; n [ 15), group II (PAD and ABGPI titer <1:100; n [ 15), and control participants (no PAD; n [ 15). All serum samples were incubated with human aortic endothelial cell (HAEC) culture. Genomic expression of TLR2 and TLR4 receptors and their shared intracellular signalling molecules, myeloid differentiation primary response gene 88 (MyD88), and interleukin (IL)-1 receptor-associated kinase (1IRAK1), were measured after the exposure of HAECs to each serum. Results: HAEC genomic expression of TLR4 was higher after the exposure to group I serum than after the exposure to group II serum (log 10 310-relative quantification [RQ]: 1.80 6 0.42 vs 1.37 6 0.39; P [ .01) or control serum (log 10 310-RQ: 1.80 6 0.42 vs 1.09 6 0.26; P < .01). TLR4 expression was higher in group II than in the control group (log 10 310-RQ: 1.37 6 0.39 vs 1.09 6 0.26; P [ .04). TLR4 expression correlated with MyD88 (r [ 0.54; P < .01) and IRAK1 (r [ 0.55; P < .01) expression. We recorded a positive correlation between MyD88 and IRAK1 genomic expression (r [ 0.58; P < .01). Conclusions: Our results suggest that serum from PAD patients with elevated ABGPI antibodies induces a genomic overexpression of TLR4 and its cellular signalling molecules in endothelial cells. (J Vasc Surg 2014;-:1-9.) Clinical Relevance: Our results suggest that circulating anti-b 2 -glycoprotein I antibodies of peripheral arterial disease patients could trigger an activation of the innate immune system through Toll-like 4 endothelial cell membrane receptors inducing a proatherogenic endothelial environment. The identification of a specific atherosclerotic activation pathway may provide new therapeutic strategies to decrease the progression of the disease or prevent it in its early stages. b 2 -Glycoprotein-I (B2GPI) is a highly glycosylated plasma protein that modulates coagulation and platelet aggregation and has a strong tropism for cell membrane phospholipids, to which it could be attached by the interac- tion between the positive charges of the protein and the anionic cell membrane structures. 1-3 Indeed, B2GPI shifts from a circular plasmatic conformation to an “activated” linear conformation after being bound to a negatively charged surface. This interaction allows the exposure of the cryptic epitope G40-R43 on domain I, to which anti-b 2 - glycoprotein I (ABGPI) antibodies could be directed. 4 Circulating ABGPI activity has also been associated with the B2GPI antigen polymorphism valine/leucine 247 . 5 The oxidative stress associated with the endothelial injury observed in atherosclerosis leads to the exposure of phospholipids to the outer cell surface and could also induce exposition of B2GPI antigen neocryptic epitopes promoting ABGPI formation. 6,7 In this context, several studies of patients with coronary heart disease and periph- eral arterial disease (PAD) have shown elevated ABGPI levels compared with controls. 8-10 B2GPI is overexpressed in the subendothelial space and intima-media interface of arterial samples obtained after a carotid endarterectomy in close relation to the atherosclerotic plaque cluster of dif- ferentiation 4-positive infiltrate. 11 The presence of this circulating autoantibody has been associated with the size and morphology of the atherosclerotic lesions induced in murine models immunized against B2GPI. 12 The endothe- lial dysfunction and inflammatory status of PAD patients From the Department of Angiology and Vascular Surgery a and the Depart- ment of Research, b Hospital Universitario de Getafe. Author conflict of interest: none. The results of this study were presented at the poster session of the Eighty- first European Atherosclerosis Society Congress held in Lyon, France, June 2-5 2013. Additional material for this article may be found online at www.jvascsurg.org. Reprint requests: Cesar Varela, MD, Hospital Universitario de Getafe, Department of Angiology and Vascular Surgery, Carretera de Toledo Km 12.5, Getafe, 28905 Madrid, Spain (e-mail: [email protected]). The editors and reviewers of this article have no relevant financial relationships to disclose per the JVS policy that requires reviewers to decline review of any manuscript for which they may have a conflict of interest. 0741-5214/$36.00 Copyright Ó 2014 by the Society for Vascular Surgery. http://dx.doi.org/10.1016/j.jvs.2013.11.066 1
Transcript of Circulating anti-β2-glycoprotein I antibodies of peripheral arterial disease patients trigger a...

Fromm
AuthThefirJu
AddRepDK
Thetom
0741Cophttp
Circulating anti-b2-glycoprotein I antibodiesof peripheral arterial disease patients trigger agenomic overexpression of Toll-like receptor 4in endothelial cellsCesar Varela, MD,a Joaquin de Haro, MD,a Silvia Bleda, MD,a Javier Rodriguez-Padilla, MD,a
Antonio Ferruelo, PhD,b and Francisco Acín, MD, PhD,a Madrid, Spain
Objective: Circulating anti-b2-glycoprotein I (ABGPI) antibodies are associated with peripheral arterial disease (PAD) andinduce the expression of leukocyte adhesion molecules and proinflammatory cytokines by endothelial cells. Our aim is tostudy a transcriptional activation pathway of the innate immune system through the cellular signalling cascade triggeredby receptors Toll-like receptor 2 (TLR2) and Toll-like receptor 4 (TLR4) of endothelial cells after the exposure of thesecells to seropositive ABGPI human serum obtained from PAD patients.Methods: We obtained serum samples from PAD patients and controls without PAD. ABGPI serum titer was detectedusing indirect immunofluorescence. Our sample was stratified into three groups: group I (PAD and ABGPI titer $1:100;n [ 15), group II (PAD and ABGPI titer <1:100; n [ 15), and control participants (no PAD; n [ 15). All serumsamples were incubated with human aortic endothelial cell (HAEC) culture. Genomic expression of TLR2 and TLR4receptors and their shared intracellular signalling molecules, myeloid differentiation primary response gene 88 (MyD88),and interleukin (IL)-1 receptor-associated kinase (1IRAK1), were measured after the exposure of HAECs to each serum.Results: HAECgenomic expression of TLR4was higher after the exposure to group I serum than after the exposure to groupII serum (log10310-relative quantification [RQ]: 1.806 0.42 vs 1.376 0.39; P[ .01) or control serum (log10310-RQ:1.80 6 0.42 vs 1.09 6 0.26; P < .01). TLR4 expression was higher in group II than in the control group (log10310-RQ:1.376 0.39 vs 1.096 0.26; P[ .04). TLR4 expression correlated withMyD88 (r[ 0.54; P < .01) and IRAK1 (r[ 0.55;P< .01) expression.We recorded a positive correlation betweenMyD88and IRAK1genomic expression (r[ 0.58;P < .01).Conclusions: Our results suggest that serum from PAD patients with elevated ABGPI antibodies induces a genomicoverexpression of TLR4 and its cellular signalling molecules in endothelial cells. (J Vasc Surg 2014;-:1-9.)
Clinical Relevance: Our results suggest that circulating anti-b2-glycoprotein I antibodies of peripheral arterial diseasepatients could trigger an activation of the innate immune system through Toll-like 4 endothelial cell membrane receptorsinducing a proatherogenic endothelial environment. The identification of a specific atherosclerotic activation pathwaymay provide new therapeutic strategies to decrease the progression of the disease or prevent it in its early stages.
b2-Glycoprotein-I (B2GPI) is a highly glycosylatedplasma protein that modulates coagulation and plateletaggregation and has a strong tropism for cell membranephospholipids, to which it could be attached by the interac-tion between the positive charges of the protein and theanionic cell membrane structures.1-3 Indeed, B2GPI shiftsfrom a circular plasmatic conformation to an “activated”
the Department of Angiology and Vascular Surgerya and the Depart-ent of Research,b Hospital Universitario de Getafe.or conflict of interest: none.results of this study were presented at the poster session of the Eighty-st European Atherosclerosis Society Congress held in Lyon, France,ne 2-5 2013.itional material for this article may be found online at www.jvascsurg.org.rint requests: Cesar Varela, MD, Hospital Universitario de Getafe,epartment of Angiology and Vascular Surgery, Carretera de Toledom 12.5, Getafe, 28905 Madrid, Spain (e-mail: [email protected]).editors and reviewers of this article have no relevant financial relationshipsdisclose per the JVS policy that requires reviewers to decline review of anyanuscript for which they may have a conflict of interest.-5214/$36.00yright � 2014 by the Society for Vascular Surgery.://dx.doi.org/10.1016/j.jvs.2013.11.066
linear conformation after being bound to a negativelycharged surface. This interaction allows the exposure of thecryptic epitope G40-R43 on domain I, to which anti-b2-glycoprotein I (ABGPI) antibodies could be directed.4
Circulating ABGPI activity has also been associated withthe B2GPI antigen polymorphism valine/leucine247.5
The oxidative stress associated with the endothelialinjury observed in atherosclerosis leads to the exposure ofphospholipids to the outer cell surface and could alsoinduce exposition of B2GPI antigen neocryptic epitopespromoting ABGPI formation.6,7 In this context, severalstudies of patients with coronary heart disease and periph-eral arterial disease (PAD) have shown elevated ABGPIlevels compared with controls.8-10 B2GPI is overexpressedin the subendothelial space and intima-media interface ofarterial samples obtained after a carotid endarterectomyin close relation to the atherosclerotic plaque cluster of dif-ferentiation 4-positive infiltrate.11 The presence of thiscirculating autoantibody has been associated with the sizeand morphology of the atherosclerotic lesions induced inmurine models immunized against B2GPI.12 The endothe-lial dysfunction and inflammatory status of PAD patients
1

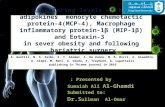
Fig 1. Toll-like receptor (TLR) cellular signaling cascade. ABGPI,Anti-b2-glycoprotein I; B2GPI, b2-glycoprotein I; IL-1,interleukin-1; IL-1R, interleukin-1 receptor; IRAKs, IL-1R-associated kinases; MyD88, myeloid differentiation primaryresponse gene 88; NF-kB, nuclear factor-kB; RQ, relative quanti-fication; TIR domain, Toll/IL-1R homologous domain; TLR2,Toll-like receptor 2; TLR4, Toll-like receptor 4.
JOURNAL OF VASCULAR SURGERY2 Varela et al --- 2014
may be related to high serum antiendothelial cell autoanti-bodies titers that react against B2GPI present in cell mem-branes.10 Moreover, ABGPI antibodies have beenassociated with adverse outcomes of coronary heart diseaseand with PAD progression in patients undergoing lowerextremity revascularization.13,14
The interaction between ABGPI and the endothelialcell membrane induces an overexpression of leukocyteadhesion molecules and increases the release of proinflam-matory cytokines in vitro.15 Nevertheless, the mechanismsby which ABGPI modulates a proinflammatory endothelialphenotype are not fully elucidated.16 In this setting, it hasbeen recently suggested that circulating ABGPI antibodiescould activate the innate immune system through the inter-action with the interleukin-1 receptor (IL-1R)/Toll-likereceptor (TLR) superfamily.17-22 TLRs are transmembranemolecules present in endothelial cells, monocytes, lympho-cytes, and dendritic cells. These proteins are a key compo-nent of the innate immune system, and TLR activation isessential to trigger a rapid immune response against infec-tion in the presence of certain microbial products.23
The members of the TLR family share their cytoplas-matic domain with IL-1R, known as the Toll/IL-1R(TIR) homologous domain. The TIR domain recruits theadaptor protein myeloid differentiation primary responsegene 88 (MyD88), which activates a family of IL-1R-associated kinases (IRAKs). This activation pathway elicitsdownstream signaling via nuclear factor-kB (NF-kB) thattriggers the nuclear transcription of proinflammatorygenes23 (Fig 1). The MyD88-dependent pathway is sharedby all TLRs, with the exception of TLR3. In this context,TLR2 and TLR4 receptor subtypes have been consistentlyrelated to atherosclerosis.24-29 Taking all of these data intoaccount, it is biologically plausible that circulating ABGPI
could modulate the atherogenic process in PAD patientsand activate endothelial cells through the innate immunesystem mediators TLR2 and TLR4.
The aim of this study was to investigate a potentialtranscriptional activation pathway of the innate immunesystem through receptors TLR2 and TLR4 and theirshared intracellular signalling molecules MyD88-IRAK1after the exposure of endothelial cells to seropositiveABGPI human serum from PAD patients to suggest a po-tential atherosclerosis activation pathway.
METHODS
The protocol for this experimental translationalcontrolled study was approved by the Hospital Universi-tario de Getafe Ethics Committee. All participants signedan informed consent form according to the principles ofthe Helsinki Declaration.
Subject inclusion criteria. Target patients and controlparticipants were screened at the Angiology and VascularSurgery outpatient clinics. All patients with symptomaticchronic limb ischemia (intermittent claudication or criticallimb ischemia) were eligible for inclusion after hemody-namic confirmation of the disease. Vascular imaging testswere performed before any revascularization attempt.TransAtlantic Inter-Society Consensus clinical criteriawere used for assigning patients as having intermittentclaudication or critical limb ischemia.30
Participants were evaluated for hypertension, dyslipide-mia, diabetesmellitus, chronic renal failure, smoking history,coronary artery disease, and previous stroke or transientischemic attack. Patients with prior lower extremity revascu-larization, infected or extensive ischemic ulcers (Rutherford6 category), chronic renal failure on dialysis, or any active in-fectious or inflammatory processes were excluded. Alsoexcluded were all those individuals who were immunode-pressed, transplant recipients, or with a documented diag-nosis of an autoimmune or rheumatologic disease.
Individuals with normal results on vascular examinationsand no cardiovascular risk factors, and who were notreceiving any ongoing pharmacologic treatment for vasculardisease, were eligible to be included in the control group. Inthis setting, any individual receiving antiplatelet treatment,anticoagulant drugs, lipid-lowering agents, antihypertensivemedications, hemorheologic drugs, or oral hypoglycemicdrugs or insulin therapy, or both, was considered to be phar-macologically treated for vascular disease.
The remaining individuals underwent a serologicscreening for autoimmune, rheumatologic, and neoplasticdisease markers. Antibodies antinuclear antibody, anti-DNA, anti-liver kidney microsomal type 1, anti-mitochondria, and anti-smooth muscle were analyzed bymeans of indirect immunofluorescence and antibodiesanti-Smith, anti-Jo, anti-LA, anti-Scl 70, anti-basementmembrane, anti-RO, and anti-ribonucleoprotein weremeasured using enzyme-linked immunosorbent assay(ELISA). All individuals seropositive to any marker wereexcluded from the analysis.

JOURNAL OF VASCULAR SURGERYVolume -, Number - Varela et al 3
Serum tests. Individuals who met the inclusion criteriawere called to attend our outpatient clinics after fasting for12 hours. We obtained a general sample for basic laboratorydeterminations (glycemia, electrolytes, and renal function),lipid profile, and blood cell count. Using the same puncture,we also obtained an independent blood sample to measuretarget serum variables and to perform the cell culture exper-iment. Samples were stored at�60�C until tests were done.
Assessment of high-sensitivity C-reactive proteinlevels. Levels of high-sensitivity C-reactive protein(hsCRP) were measured using a highly sensitive automatedimmunoassay (Roche Diagnosis, Basel, Switzerland). Thistest provides a lower detection limit of 0.2 mg/L anda variation coefficient of 4.2% in 4 mg/L and 6.3% in1 mg/L. Each test was analyzed in triplicate, and the meanof the three measurements was used for the analysis.
Assessment of ABGPI levels. The titer of circulatingantiendothelial cell antibodies directed against circulatingABGPI31 was detected by indirect immunofluorescenceusing a diagnosis reagent kit from EUROIMMUN(Medizinische Labordiagnostika AG, Luebeck, Germany)with a TITERPLANE technique. Cultivated umbilical veinendothelial cells (UVECs) covered the reaction areas of aBIOCHIP. Slides were incubated with the patient’s dilutedserum samples. Serum dilutions were performed accordingto the manufacturer’s recommendation for immunoglob-ulin G detection. Positive reactions were marked by gran-ular staining in cytoplasm of human UVECs (HUVECs)with fluorescein-labeled antibodies and were visualized byfluorescence microscopy. Titers of <1:10 represent thereference range and the lower detection limit of the test.
Design of study groups. According to previous obser-vations,10 an ABGPI titer of 1:100 was considered abiologically significant cutoff point (10 times the referencerange) and was used to stratify the sample of PAD patientsin two groups: group I (PAD and ABGPI titers $1:100)and group II (PAD and ABGPI<1:100). These two groupswere matched by age (2-year range), sex, and clinical status(intermittent claudication vs critical limb ischemia). Allof the samples obtained from participants without PAD(control group) showed a determination of ABGPI titersunder the lower detection limit of the test (<1:10). Thecontrol group was matched by age (2-year range) and sexwith the cases, resulting in three groups for the cell cultureexperiment: group I, group II, and the control group.
The number of individuals needed to reach an 80%statistical power with an a error of .05 was 45 cases(http://calculator.stat.ucla.edu). Therefore, the numberof cases in each group was 15.
Cell culture experiment. Primary human aortic endo-thelial cells (HAECs; Lonza, Wokingham, UnitedKingdom) were cultured (37�C, 5% CO2) in endothelialcell growth medium-2, consisting of endothelial cell basalmedium supplemented with 2% fetal bovine serum,gentamicin/amphotericin (GA-1000), and growth sup-plements (Lonza). All endothelial cells were studied be-tween passages 2 and 6. HAECs (2 � 104 cells/well) werestimulated during 2 hours using the human serum samples
of the participants included. All serum samples were dilutedwith endothelial cell growth medium-2 (ratio 1:2) beforecell stimulation.32
RNA was isolated using the High Pure RNA IsolationKit (Roche Diagnostics, Indianapolis, Ind) according tothe manufacturer’s protocol. RNA and DNA quantificationwere determined with the NanoDrop ND-1000 Spectro-photometer (NanoDrop Technologies Inc, Wilmington,Del). The optical density 260:280 ratio was used to eval-uate the purity of the nucleic acid samples.
The Transcriptor First Strand cDNA Synthesis Kit(Roche Diagnostics, Indianapolis, Ind) was used followingthe manufacturer’s protocol for reverse transcription with1mg total RNA (Applied Biosystems Inc, Foster City, Calif).The reactions were done in a Verity analyzer (AppliedBiosystems) consisting of incubation at 25�C for 10minutes,followed by 30�C for 55 minutes, 50�C for 60 minutes, and85�C for minutes, and finally chilled to 4�C.
Quantification of the transcripts were performed usingreal-time polymerase chain reaction (PCR) with humantissue-specific TaqMan primer probe mixes purchased asready-to-use assays (TLR4, Id. 90002850; MyD88; Id.90002853; IRAK1, Id. 90002852; and the housekeepinggene RN18S1, Id. 90003161; Roche Diagnostics) andFastStart Universal Probe Master with the 7500 Fast RealTime PCR System (Applied Biosystems Inc). Sampleswere heated to 95�C for 5 minutes for preincubationthen underwent 40 cycles of PCR amplification (denatur-ation at 95�C for 10 seconds and primer annealing at60�C for 60 seconds) in triplicate. Cycle threshold valueswere determined using 7500 Fast System SDS 2.0.1 soft-ware. The relative quantification (RQ) of TLR4, TLR2,MyD88, and IRAK1 gene expression was performed ac-cording to the DDCT comparative method.33 The baselineexpression of each gene in cell culture was determined intriplicate before stimulation with serum samples.
Statistical analysis. Data were processed using SPSS15.0 software (SPSS Inc, Chicago, Ill). Differences be-tween groups were considered statistically significant for avalue of P < .05 in two-tailed tests. Log transformation(log10) of all quantitative variables was determined to adjustthem to a normal distribution. The log transformation of thegenomic expression data and ankle-brachial index (ABI)measurements showed a great number of negative values.For a better interpretation of the results, these variables weremultiplied by 10 before their transformation (�10-log10).The association between continuous variables was analyzedusing the Student t-test for independent samples and theircorrelation by the Pearson correlation coefficient. The as-sociation between categoric variables was studied using thec2 test and the Fisher exact test when required. Continuousvariables are expressed as the mean 6 standard deviation.Categoric data are expressed as percentages.
RESULTS
During 8 months, 495 patients were screened at ouroutpatient clinic, but only 30 PAD patients and 15 individ-uals without PADwere recruited. Twelve control individuals

Table. Baseline characteristics of participants enrolled by study group
Variablea
Study group
Group I (n ¼ 15) Group II (n ¼ 15) P
PAD and ABGPIantibodies titer $1:100
PAD and ABGPIantibodies ABGPI titer <1:100 Group I vs group II
Control(n ¼ 15)
Age, years 67 6 6.40 65 6 7.22 .35 66 6 7.62Male sex 13 (87) 13 (87) .50 13 (87)Diabetes mellitus 4 (27) 3 (20) .50 NASmoking history 6 (40) 7 (47) .71 NACAD 3 (20) 5 (33) .68 NAHypertension 9 (60) 10 (67) .70 NADyslipidemia 9 (60) 8 (53) .71 NAAcute stroke 1 (7) 2 (13) .50 NACRF 0 (0) 1 (7) .50 NAClaudication 10 (67) 10 (67) .65 NACLI 5 (33) 5 (33) .65 NAABI, �10-log10 0.78 6 0.09 0.79 6 0.11 .82 0.98 6 0.01
ABGPI, anti-b2-glycoprotein I; ABI, ankle-brachial index; CAD, coronary artery disease; CLI, critical limb ischemia; CRF, chronic renal failure; NA, notapplicable; PAD, peripheral arterial disease.aContinuous data are shown as mean 6 standard deviation and categoric data as number (%).
JOURNAL OF VASCULAR SURGERY4 Varela et al --- 2014
were referred because of walking limb pain but were diag-nosed of lumbar and lower limb osteoarthrosis. Two individ-uals complained of a pulsatile abdominal mass with normalresults on abdominal echography assessment. One controlindividual attended our outpatient clinic because of vertigo,but no pathologic findings were observed in a cranialcomputed tomography scan or in duplex scanning of thesupra-aortic vessels (Table).
Relation of hsCRP levels and ABI values to PADand circulating ABGPI titers. Serum hsCRP levels werehigher in PAD patients than in subjects without PAD(0.92 6 0.37 vs 0.46 6 0.04 log10-mg/dL; P < .01) and inpatients with critical limb ischemia compared with claudi-cant patients (1.27 6 0.36 vs 0.74 6 0.23 log10-mg/dL;P ¼ .01). ABI values were lower in PAD patients than inthose without PAD (0.78 6 0.10 vs 0.98 6 0.01 �10-log10; P < .01) and in patients with critical limb ischemiathan in claudicant patients (0.65 6 0.04 vs 0.83 60.07 �10-log10; P < .01).
Group I patients showed higher hsCRP levels thangroup II patients (1.06 6 0.43 vs 0.77 6 0.25 log10-mg/dL; P ¼ .03) and controls (1.06 6 0.43 vs 0.47 6 0.00log10-mg/dL; P < .01). Serum hsCRP levels were higherin group II than in the control group (0.77 6 0.25 vs0.47 6 0.00 log10-mg/dL; P < .01). Group I and groupII patients presented similar ABI values (0.78 6 0.09 vs0.79 6 0.11 �10-log10; P ¼ .82). We observed lower ABIvalues in group I (0.78 6 0.09 vs 0.98 6 0.01 �10-log10;P < .01) and in group II (0.79 6 0.11 vs 0.98 60.01 �10-log10; P < .01) than in the control group.
A negative correlation between serum hsCRP levelsand ABI values was found (r ¼ �0.61; P < .01).
Genomic expression of TLR4 by HAECs afterserum exposure. TLR4 genomic expression values ob-tained after HAEC stimulation with PAD patients’ serum
(�10-log10-RQ: 1.59 6 0.45 vs 1.11 6 0.35; P < .01),group I serum (�10-log10-RQ: 1.806 0.42 vs 1.116 0.35;P< .01), or group II serum (�10-log10-RQ: 1.376 0.39 vs1.11 6 0.35; P ¼ .04) were higher compared with thebaseline cell culture expression of TLR4. Control seruminduced similar TLR4 expression values as found in baselinelevels (�10-log10-RQ:1.0960.26 vs 1.1160.35;P¼ .75).
TLR4 expression was higher after HAEC stimulationwith PAD patients’ serum than after exposure to serumof individuals without PAD (log10�10-RQ: 1.59 6 0.45vs 1.09 6 0.26; P < .01). No statistical differences wereobserved according to the clinical status (log10�10-RQ:1.81 6 0.47 [critical limb ischemia] vs 1.50 6 0.42 [inter-mittent claudication]; P ¼ .16). However, we found atrend toward a weak negative correlation between ABIvalues and TLR4 expression (r ¼ e0.31; P ¼ .09). SerumhsCRP levels did not correlate with the expression TLR4by HAECs (r ¼ 0.26; P ¼ .16).
The expression of TLR4 was higher in group I than ingroup II (log10�10-RQ: 1.80 6 0.42 vs 1.37 6 0.39; P ¼.01) or in the control group (log10�10-RQ: 1.80 6 0.42vs 1.09 6 0.26; P < .01). Group II serum modulated ahigher gene expression than the control serum (log10�10-RQ: 1.376 0.39 vs 1.096 0.26; P ¼ .04; Fig 2).
Genomic expression of TLR2 by HAECs afterserum exposure. TLR2 genomic expression values ob-tained after HAEC stimulation with PAD patients’ serum(�10-log10-RQ: 0.67 6 0.58 vs 0.50 6 0.20; P ¼ .33),group I serum (�10-log10-RQ: 0.85 6 0.57 vs 0.50 60.20; P ¼ .10), group II serum (�10-log10-RQ: 0.45 60.53 vs 0.50 6 0.20; P ¼ .79), or control serum (�10-log10-RQ: 0.45 6 0.37 vs 0.50 6 0.20; P ¼ .78) weresimilar to the baseline cell culture expression of TLR2.
The TLR2 expression observed after HAEC stimula-tion with PAD patients’ serum was similar to the expression

Fig 2. A, Toll-like receptor 4 (TLR4) expression by human aortic endothelial cell (HAEC) culture after stimulationwith serum from peripheral arterial disease (PAD) patients and healthy controls without PAD. B, TLR4 expression byHAECs according to anti-b2-glycoprotein I (ABGPI) antibody titers. Group I, PAD and ABGPI titer $1:100;group II, PAD and ABGPI titer <1:100; RQ, relative quantification. Variables are expressed as mean 6 standarddeviation (error bars). *Indicates statistically different bars and statistical trends.
Fig 3. A, Toll-like receptor 2 (TLR2) expression by human aortic endothelial cell (HAEC) culture after stimulationwith serum from peripheral arterial disease (PAD) patients and healthy controls without PAD. B, TLR2 expression byHAEC culture according to anti-b2-glycoprotein I (ABGPI) antibody titers. Group I, PAD and ABGPI titer $1:100;group II, PAD and ABGPI titer <1:100; RQ, relative quantification. Variables are expressed as mean 6 standarddeviation (error bars). *Indicates statistically different bars and statistical trends.
JOURNAL OF VASCULAR SURGERYVolume -, Number - Varela et al 5
obtained after the exposure to serum of individuals withoutPAD (log10�10-RQ: 0.67 6 0.58 vs 0.45 6 0.37; P ¼.22), and no statistical differences were seen according tothe clinical status (log10�10-RQ: 0.84 6 0.69 [criticallimb ischemia] vs 0.59 6 0.52 [intermittent claudication];P ¼ .38). ABI values (r ¼ �0.10; P ¼ .60) and serumhsCRP levels (r ¼ 0.23; P ¼ .21) did not correlate withthe expression of TLR2.
We recorded a trend toward a higher genomic expres-sion of TLR2 in group I compared with group II(log10�10-RQ: 0.85 6 0.57 vs 0.45 6 0.53; P ¼ .08) orwith the control group (log10�10-RQ: 0.85 6 0.57 vs0.45 6 0.37; P ¼ .06). No differences were observed inTLR2 expression between group II and the control group(log10�10-RQ: 0.456 0.53 vs 0.456 0.37;P¼ .99; Fig 3).
Genomic expression of MyD88 by HAECs afterserum exposure. MyD88 genomic expression values ob-tained after HAEC stimulation with PAD patients’ serum(�10-log10-RQ: 0.68 6 0.45 vs 0.17 6 0.15; P < .01),group I serum (�10-log10-RQ: 0.886 0.39 vs 0.176 0.15;P< .01), or group II serum (�10-log10-RQ: 0.486 0.43 vs0.17 6 0.15; P ¼ .04) were higher compared with thebaseline cell culture expression of MyD88. Control seruminduced similar MyD88 expression values as baseline levels(�10-log10-RQ: 0.186 0.29 vs 0.176 0.15; P ¼ .92).
MyD88 expression was higher after HAEC stimulationwith PAD patients’ serum than after exposure to serumfrom individuals without PAD (log10�10-RQ: 0.68 60.45 vs 0.18 6 0.29; P < .01), and no differences wereobserved according to the clinical status (log10�10-RQ:

Fig 4. A, Myeloid differentiation primary response gene 88 (MyD88) expression by human aortic endothelial cell(HAEC) culture is shown after stimulationwith serum fromperipheral arterial disease (PAD) patients andhealthy controlswithout PAD. B, MyD88 expression by HAEC culture according to anti-b2-glycoprotein I (ABGPI) antibody titers.Group I, PADandABGPI titer$1:100; group II,PADandABGPI titer<1:100;RQ, relative quantification. Variables areexpressed as mean6 standard deviation (error bars). *Indicates statistically different bars and statistical trends.
JOURNAL OF VASCULAR SURGERY6 Varela et al --- 2014
0.82 6 0.34 [critical limb ischemia] vs 0.61 6 0.49 [inter-mittent claudication]; P ¼ .19). ABI values (r ¼ �0.22;P ¼ .17) and serum hsCRP levels (r ¼ 0.27; P ¼ .10)did not correlate with the expression of MyD88.
The expression of MyD88 was higher in group I thanin group II (log10�10-RQ: 0.88 6 0.39 vs 0.48 6 0.43;P ¼ .01) or in the control group (log10�10-RQ: 0.88 60.39 vs 0.18 6 0.29; P < .01). Group II modulateda higher gene expression than the control serum(log10�10-RQ: 0.486 0.43 vs 0.186 0.29;P¼ .03; Fig 4).
Genomic expression of IRAK1 by HAECs afterserum exposure. IRAK1 genomic expression values ob-tained after HAEC stimulation with PAD patients’ serum(�10-log10-RQ: 0.59 6 0.35 vs 0.33 6 0.05; P < .01),group I serum (�10-log10-RQ: 0.65 6 0.37 vs 0.33 60.05; P < .01), or group II serum (�10-log10-RQ: 0.52 60.33 vs 0.33 6 0.05; P ¼ .05) were higher than thebaseline cell culture expression of IRAK1. Serum fromcontrol individuals induced similar IRAK1 expressionvalues as baseline levels (�10-log10-RQ: 0.31 6 0.29 vs0.33 6 0.05; P ¼ .85).
IRAK1 expression was higher after HAEC stimulationwith PAD patients’ serum than after exposure to serumfrom participants without PAD (log10�10-RQ: 0.59 60.35 vs 0.316 0.29; P < .01), and no statistical differenceswere observed according to the clinical status (log10�10-RQ: 0.65 6 0.39 [critical limb ischemia] vs 0.56 6 0.34[intermittent claudication]; P ¼ .54). ABI values(r ¼ �0.22; P ¼ .18) and serum hsCRP levels (r ¼ 0.13;P ¼ .41) did not correlate with the expression of IRAK1.
No statistical differences in IRAK1 gene expressionwere recorded between group I and group II (log10�10-RQ: 0.65 6 0.37 vs 0.52 6 0.33; P ¼ .30). However,we observed a higher IRAK1 expression after HAEC stimu-lation with group I serum than after the exposure to controlgroup serum (log10�10-RQ: 0.65 6 0.37 vs 0.31 6 0.29;
P¼ .01). There was a trend toward a higher IRAK1 expres-sion in group II than in the control group (log10�10-RQ:0.526 0.33 vs 0.31 6 0.29; P ¼ .08; Fig 5).
Correlation between the different studied genesexpressed by HAECs. We observed a positive correlationbetween TLR4 and TLR2 genomic expression by HAECs(r ¼ 0.49; P ¼ .01). TLR4 expression correlated withMyD88 (r ¼ 0.54; P < .01) and IRAK1 (r ¼ 0.55; P <.01) expression. TLR2 expression correlated with MyD88(r ¼ 0.69; P < .01) expression. There was a trend towardthe correlation between TLR2 and IRAK1 expression (r ¼0.31; P ¼ .07). We recorded a positive correlation betweenthe genomic expression of MyD88 and IRAK1 (r ¼ 0.58;P < .01; Supplementary Fig, online only).
DISCUSSION
In the present research, we have observed that serumfrom PAD patients induced transcriptional activation ofthe innate immune system through the overexpression ofTLR4 receptors and its cellular signaling molecule MyD88in HAECs. This genomic overexpression was associatedwith high ABGPI titers in the serum of PAD patients.
TLR2 and TLR4 transmembrane receptors have beenconsistently related to atherosclerosis. During atheroscle-rotic lesion development, TLR2 and TLR4 expression in-creases in atherosclerotic plaques.24 Genetic deficiency ofMyD88, TLR2, and TLR4 in apolipoprotein E�/� miceresults in a 55% to 75% reduction of atherosclerotic lesiondevelopment and macrophage infiltration.25,26 Further-more, inactivation by mutation of the cytoplasmic portionof TLR4 in mice confers resistance to diet-induced athero-sclerosis. However, bone marrow transplantation fromthese mice to other apolipoprotein E�/� nonmutation car-riers does not alter atherosclerosis progression.27 In addi-tion, TLR stimulation through several microbial productsmay lead to atherosclerosis development.28,29 These data

Fig 5. A, Interleukin-1 receptor-associated kinase 1 (IRAK1) expression by human aortic endothelial cell (HAEC)culture after stimulation with serum from peripheral arterial disease (PAD) patients and healthy controls without PAD.B, IRAK1 expression by HAEC culture according to anti-b2-glycoprotein I (ABGPI) antibody titers. Group I, PADand ABGPI titer $1:100; group II, PAD and ABGPI titer <1:100; RQ, relative quantification. Variables are expressedas mean 6 standard deviation (error bars). *Indicates statistically different bars and statistical trends.
JOURNAL OF VASCULAR SURGERYVolume -, Number - Varela et al 7
suggest that endothelial TLR expression could play a keyrole in the atherogenic process.
Our study has documented that serum from PAD pa-tients induces a genomic overexpression of TLR4 receptor,MyD88 adaptor protein and one of its associated kinases(IRAK1), in human endothelial cells. There was a tendencytoward a weak correlation between TLR4 genomic expres-sion and ABI values. These results are consistent with pre-viously published data.
However, the main finding of our research was an asso-ciation between the expression of TLR receptors and circu-lating ABGPI titers. These autoantibodies are elevated inpatients with atherosclerosis and have been related to dis-ease progression.29,30 The interaction between ABGPIand the endothelial cell membrane induces an overexpres-sion of leukocyte adhesion molecules and increases therelease of proinflammatory cytokines through an NF-kB-dependent pathway.8-12 The circulating ABGPI antibodiesendothelial activation pattern is similar to that obtained af-ter lipopolysaccharides stimulation (main exogenous ligandof TLR4). In fact, autogenous vein bypasses performed inmouse models exposed to lipopolysaccharide could upre-gulate adipose TLR4, leading to an early inflammatoryresponse of the vein graft wall.28 In this context, ABGPIantibodies show certain molecular mimicry with variousmicrobial products that are able to trigger TLR receptoractivation.17,34,35 Furthermore, several studies have shownthat ABGPI antibodies may act as possible endogenous li-gands for TLR2-TLR4 receptors.17-22 Taking all these datainto account, our results are consistent with the hypothesisthat circulating ABGPI antibodies present in PAD patients’serum could activate endothelial cells through TLR4 recep-tors. According to this hypothesis, the association observedin this study and others between TLR4 and atherosclerosismay be partly explained by the presence of circulatingABGPI antibodies in the patients’ blood.
Endothelial TLR4 genomic overexpression was associ-ated with serum ABGPI titers and with PAD. However,the TLR2 receptor was overexpressed only after the expo-sure of endothelial cells to the serum of patients with highABGPI titers but was not associated with PAD. In thiscontext, it has been documented that lipopolysaccharidesstimulate cells via TLR4 and interact with interferon-g toupregulate TLR2 expression.20 Therefore, it is possiblethat an increased TLR4 expression in our cell cultureexperiment stimulated TLR2 upregulation. The findingof a correlation between TLR2 and TLR4 genomic expres-sion supports this argument.
The genomic expression of TLR2 and TLR4 showed apositive correlation with MyD88 and IRAK1 levels, sug-gesting that the upregulation of TLR membrane receptorstriggered the overexpression of its intracytoplasmaticpathway effectors. It is noteworthy that no associationwas seen between IRAK1 gene expression and ABGPI ti-ters. IRAK1 levels were only related to PAD. These resultscould be explained by other extracellular or intracellularbiologic mechanisms associated with atherosclerosis or tothe TLR activation pathway that could have collaboratedin the modulation of endothelial IRAK1 genomicexpression.
This hypothesis is feasible taking into account thatour HAEC culture was stimulated using unfractionatedhuman serum samples. In this setting, it is possible thatIRAK1 autophosphorylation, which occurs 45 minutesafter the stimulation with ABGPI or lipopolysaccharides,initiates early as a result of the presence of high IL-1 levelsin some serum samples. IL-1 is able to trigger IRAKphosphorylation within 10 minutes.36 After a sustainedstimulus, IRAK1 could be degraded through an IRAK4-dependent negative feedback mechanism.37 This negativefeedback may partly explain why IRAK1 expression levelswere not associated with circulating ABGPI titers.

JOURNAL OF VASCULAR SURGERY8 Varela et al --- 2014
Nowadays, it is commonly accepted that atherosclerosisis a chronic inflammatory vascular disease. Plasma levels ofCRP have been related with the clinical severity of PAD.38
In this context, ABGPI antibodies could be also associatedwith an increase in CRP levels in patients with atheroscle-rosis.10 In the present research, we observed higher CRPlevels in patients with advanced-stage PAD (critical limbischemia) and in those with high circulating ABGPI titers.Meanwhile, serum hsCRP levels were correlated with ABIvalues. These findings are consistent with previously pub-lished data and suggest that an ABGPI-dependent autoim-mune mechanism could cooperate with the inflammatoryprocess that surrounds the disease. However, ABI valueswere not associated with the ABGPI antibodies titer, aswould be expected. Unfractionated human serum couldcontain a wide variety of exogenous and endogenousproinflammatory vectors able to trigger the TIR homolo-gous domain. These vectors could also serve as ABGPI in-termediaries, affecting TLR activation. This fact mayexplain why ABGPI titers were not significantly associatedwith the degree of PAD.
In the cell culture set, we found that ABGPI titerswere associated with a genomic overexpression of TLR4and MyD88 and observed a trend toward a weak negativecorrelation between ABI values and TLR4. ABI values didnot correlate with other transcriptional variables. In thesame manner, hsCRP levels were not correlated with theexpression of the studied genes. Nevertheless, weobserved higher nonsignificant genomic overexpressionof TLR4, TLR2, MyD88, and IRAK1 after the exposureof HAECs to serum from critical limb ischemia patients.These results are consistent with the hypothesis that thetranscriptional activation of the TLR endothelial-signalling cascade associated with ABGPI titer occursindependently of the degree of PAD. However, TLR acti-vation could be also associated with PAD severity throughother serum factors related to the intensity of the proin-flammatory status. In fact, IL-1 is able to activate theTIR homologous domain leading to a NF-kB nucleartranslocation.
Previous studies have pointed out that oxidized low-density lipoprotein (ox-LDL) could act by itself as anendogenous ligand for TLR4.39 However, ox-LDL isalso able to modulate the oxidation of B2GPI favoringthe exposition of neocryptic epitopes of the protein that in-duces ABGPI formation.6,7 Furthermore, a recent studysuggested that ABGPI and ox-LDL (anti-oxLDL-B2GPI)could form immunocomplexes that promote the intracel-lular accumulation of ox-LDL in macrophages, enhancingfoam cell formation.13 The relation between atherosclerosisand innate immunity is highly complex. In this sense, thepresent research was not designed to explore the associa-tion between PAD severity and TLR transcriptional activa-tion. Therefore, it is not possible to clearly concluderegarding a possible relationship between ABGPI titersand degree of PAD (ABI), degree of PAD (ABI), andTLR genomic expression and between hsCRP levels andTLR genomic expression due to an eventual type II error.
This research has several limitations. The main limita-tion is that TLR receptor protein expression could not beassessed. Second, proinflammatory cytokine release byHAECs after serum stimulation was also not explored.Third, there was a preponderance of men in our sample;therefore, our results should be interpreted with cautionon women. Last, the use of unfractionated human serumdoes not allow us to establish a direct causal relationshipbetween circulating ABGPI antibodies and the overex-pressed genes. Nevertheless, we argue that the unfractio-nated serum used for this study and obtained during ourdaily clinical practice improves the representativeness ofthe results. Despite the multiple stimulation factors con-tained in the studied serum samples to which HAECsmay have been exposed, we found that the exposure ofHAECs to serum with high ABGPI titers was associatedwith a genomic overexpression of TLR4 receptors and itsintracellular adaptor protein MyD88.
CONCLUSIONS
Our results are consistent with the hypothesis that thecirculating ABGPI antibodies of PAD patients couldtrigger a transcriptional activation of the innate immunesystem through TLR4 endothelial cell membrane receptorsand the downstream signaling molecule MyD88. Howev-er, the pathophysiologic relation between these moleculesshould be confirmed in future research.
AUTHOR CONTRIBUTIONS
Conception and design: CV, JH, FAAnalysis and interpretation: CV, JH, SB, JR, AFData collection: CV, SB, JR, AFWriting the article: CVCritical revision of the article: JH, SB, FAFinal approval of the article: CV, JH, FAStatistical analysis: CV, JHObtained funding: Not applicableOverall responsibility: CV
REFERENCES
1. Schousboe I. Beta2-glycoprotein: a plasma inhibitor of the contactactivation of the intrinsic blood coagulation pathway. Blood 1985;66:1086-91.
2. Nimpf J, Wurm H, Kostner GM. Beta2-glycoprotein I (apo H) inhibitsthe release reaction of human platelets during ADP-induced aggrega-tion. Atherosclerosis 1987;63:109-14.
3. Tincani A, Spatola L, Prati E, Allegri F, Ferremi P, Cattaneo R, et al.The anti-beta2-glicoprotein I activity in human anti-phospholipidsyndrome sera is due to monoreactive low-affinity autoantibodiesdirected to epitopes located on native beta2-glycoprotein I and pre-served during species evolution. J Immunol 1996;157:5732-8.
4. Agar C, van Os GM, Mörgelin M, Sprenger RR, Marquat JA,Urbanus RT, et al. Beta2-glycoprotein I can exist in 2 conformations:implications for our understanding of the antiphospholipid syndrome.Blood 2010;116:1336-43.
5. Lee YH, Choi SJ, Ji JD, Song GG. Association between the valine/leucine247 polymorphism of b2-glycoprotein I and susceptibility toanti-phospholipid syndrome: a meta-analysis. Lupus 2012;21:865-71.
6. Broder A, Chan JJ, Putterman C. Dendritic cells: an important linkbetween antiphospholipid antibodies, endothelial dysfunction, and

JOURNAL OF VASCULAR SURGERYVolume -, Number - Varela et al 9
atherosclerosis in autoimmune and non-autoimmune diseases. ClinImmunol 2013;146:197-206.
7. Buttari B, Profumo E, Capozzi A, Sorice M, Rigano R. Oxidized hu-man beta2-glycoprotein I: its impact on innate immune cells. Curr MolMed 2011;11:719-25.
8. Aslim E, Hakki Akay T, Bastürk B, Ozkan S, Gültekin B,Ozcobanoglu S, et al. The role of antiendothelial cell antibodies in thedevelopment and follow-up of coronary and peripheral arterial diseases.Angiology 2008;59:209-13.
9. Franck M, Staub HL, Petracco JB, Norman GL, Lassen AJ, Schiavo N,et al. Autoantibodies to the atheroma component beta2-glycoprotein Iand risk of symptomatic peripheral artery disease. Angiology 2007;58:295-302.
10. Varela C, De Haro J, Bleda S, Esparza L, López de Maturana I, Acín F.Anti-endothelial cell antibodies are associated with peripheral arterialdisease and markers of endothelial dysfunction and inflammation.Interact Cardiovasc Thorac Surg 2011;13:463-567.
11. George J, Harats D, Gilburd B, Afek A, Levy Y, Schneiderman J, et al.Immunolocalization of beta2-glycoprotein I (apolipoprotein H) tohuman atherosclerotic plaques: potential implications for lesion pro-gression. Circulation 1999;99:2227-30.
12. George J, Afek A, Gilburd B, Blank M, Levy Y, Aron-Maor A, et al.Induction of early atherosclerosis in LDL-receptor deficient miceimmunized with beta2-glycoprotein I. Circulation 1998;98:1108-15.
13. Greco TP, Conti-Kelly AM, Greco T Jr, Doyle R, Matsuura E,Anthony JR, et al. Newer antiphospholipid antibodies predict adverseoutcomes in patients with acute coronary syndrome. Am J Clin Pathol2009;132:613-20.
14. Lam EY, Taylor LM Jr, Landry GJ, Porter JM, Moneta GL. Rela-tionship between anti-phospholipid antibodies and progression of thelower extremity arterial occlusive disease after lower extremity bypassoperations. J Vasc Surg 2001;33:976-82.
15. Meroni PL, Raschi E, Testoni C, Borgui MO. Endothelial cell activa-tion by antiphospholipid antibodies. Clin Immunol 2004;112:169-74.
16. Meroni PL, Tincani A, Sepp N, Raschi E, Testoni C, Corsini E, et al.Endothelium and the brain in CNS lupus. Lupus 2012;21:919-28.
17. Raschi E, Testoni C, Bosisio D, Borghi MO, Koike T, Mantovani A,et al. Role of the MyD88 transduction signalling pathway in endothelialactivation by antiphospholipid antibodies. Blood 2003;101:3495-500.
18. Sorice M, Longo A, Capozzi A, Garofalo T, Misasi R, Alessandri C,et al. Anti-beta2-glycoprotein I antibodies induce monocyte release oftumor necrosis factor alpha and tissue factor by signal transductionpathways involving lipid rafts. Arthritis Rheum 2007;56:2687-97.
19. Satta N, Dunoyer-Geindre S, Reber G, Fish RJ, Boehlen F,Kruithof EK, et al. The role of TLR2 in the inflammatory activation ofmouse fibroblasts by human antiphospholipid antibodies. Blood2007;109:1507-14.
20. Alard JE, Gaillard F, Daridon C, Shoenfeld Y, Jamin C, Youinou P.TLR2 is one of the endothelial receptors for beta 2-glycoprotein I.J Immunol 2010;185:1550-7.
21. Zhou H, Yan Y, Xu G, Zhou B, Wen H, Guo D, et al. Toll-like re-ceptor (TLR)-4 mediates anti-B2GPI/B2GPI-induced tissue factorexpression in THP-1 cells. Clin Exp Immunol 2011;163:189-98.
22. Satta N, Kruithof EK, Fickentscher C, Dunoyer-Geindre S, Boehlen F,Reber G, et al. Toll-like receptor 2 mediates the activation of humanmonocytes and endothelial cells by antiphospholipid antibodies. Blood2011;117:5523-31.
23. Akira S, Takeda K, Kaisho T. Toll-Like receptors: critical proteinslinking innate and acquired immunity. Nat Immunol 2001;2:675-80.
24. Schoneveld AH, Hoefer I, Sluijter JP, Laman JD, de Kleijn DP,Pasterkamp G. Atherosclerosis lesion development and Toll likereceptor 2 and 4 responsiveness. Atherosclerosis 2008;197:95-104.
25. MichelsenKS,WongMH, ShahPK,ZhangW,Yano J,DohertyTM, et al.Lack of Toll-like receptor 4 or myeloid differentiation factor88 reduces atherosclerosis and alters plaque phenotype inmice deficientin apolipoprotein E. Proc Natl Acad Sci U S A 2004;101:10679-84.
26. Mullik AE, Tobias PS, Curtiss LK. Modulation of atherosclerosis inmice by toll-like receptor 2. J Clin Invest 2005;115:3149-56.
27. Shi W, Wang NJ, Shih DM, Sun VZ, Wang X, Lusis AJ. Determinantsof atherosclerosis susceptibility in the C3H and C57BL/6 mousemodel: evidence for involvement of endothelial cells but not blood cellsor cholesterol metabolism. Circ Res 2000;86:1078-84.
28. Nguyen BT, Yu P, Tao M, Hao S, Jiang T, Ozaki CK. Perivascularinnate immune events modulate early murine vein graft adaptations.J Vasc Surg 2013;53:486-92.
29. Hayashi C, Gudino CV, Gibson FC 3rd, Genco CA. Review:pathogen-induced inflammation at sites distant from oral infection:bacterial persistence and induction of cell-specific innate immune in-flammatory pathways. Mol Oral Microbiol 2010;25:305-16.
30. Norgren L, Hiatt WR, Dormandy JA, Nehler MR, Harris KA,Fowkes FG, et al. Inter-Society Consensus for the Management ofPeripheral Arterial Disease (TASC II). Eur J Vasc Endovasc Surg2007;33(Suppl 1):S1-75.
31. Del Papa N, Guidali L, Spatola L, Bonara P, Borghi MO, Tincani A,et al. Relationship between anti-phospholipid and anti-endothelial cellantibodies III: beta 2 glycoprotein I mediates the antibody binding toendothelial membranes and induces the expression of adhesion mole-cules. Clin Exp Rheumatol 1995;13:179-85.
32. Oude Nijhuis CS, Vellenga E, Daenen SM, Kamps WA, De Bont ES.Endothelial cells are main producers of interleukin 8 through Toll-likereceptor 2 and 4 signaling during bacterial infection in leukopeniccancer patients. Clin Diagn Lab Immunol 2003;10:558-63.
33. Livak KJ, Schmittgen TD. Analysis of relative gene expression datausing real-time quantitative PCR and the 2(-Delta Delta C(T))Method. Methods 2001;25:402-8.
34. Gharavi AE, Pierangeli SS, Colden-Stanfield M, Liu XW, Espinola RG,Harris EN. GDKV-induced antiphospholipid antibodies enhancethrombosis and activate endothelial cells in vivo and in vitro.J Immunol 1999;163:2922-7.
35. Gharavi AE, Pierangeli SS, Espinola RG, Liu X, Colden-Stanfield M,Harris EN. Antiphospholipid antibodies induced in mice by immuni-zation with a cytomegalovirus-derived peptide cause thrombosis andactivation of endothelial cells in vivo. Arthritis Rheum 2002;46:545-52.
36. Bosisio D, Polentarutti N, Sironi M, Bernasconi S, Miyake K,Webb GR, et al. Stimulation of Toll-like receptor 4 expression in hu-man mononuclear phagocytes by interferon-gamma: a molecular basisfor priming and synergism with bacterial lipopolysaccharide. Blood2002;99:3427-31.
37. Kubo-Murai M, Hazeki K, Nigorikawa K, Omoto T, Inoue N,Hazeki O. IRAK-4-dependent degradation of IRAK-1 is a negativefeedback signal for TLR-mediated NF-kappaB activation. J Biochem2008;143:295-302.
38. De Haro J, Acin F, Medina FJ, Lopez-Quintana A, March JR. Rela-tionship between the plasma concentration of C-reactive protein andthe severity of peripheral arterial disease. Clin Med Cardiol 2008;3:1-7.
39. Falck-Hansen M, Kassiteridi C, Monaco C. Toll-like receptors inatherosclerosis. Int J Mol Sci 2013;14:14008-23.
Submitted Aug 21, 2013; accepted Nov 19, 2013.
Additional material for this article may be found onlineat www.jvascsurg.org.

Supplementary Fig (online only). A, Correlation between Toll-like receptor (TLR4) and Toll-like receptor 2(TLR2) genomic expression. B, Correlation between TLR4 and myeloid differentiation primary response gene 88(MyD88) genomic expression. C, Correlation between MyD88 and IRAK1 genomic expression. RQ, Relativequantification.
JOURNAL OF VASCULAR SURGERY9.e1 Varela et al --- 2014






![Soluble αKlotho downregulates Orai1-mediated store ......Klotho is an aging-suppressor gene that encodes type 1 transmembrane glycoprotein called αKlotho [22, 23]. Klotho-deficient](https://static.fdocument.org/doc/165x107/613b4592f8f21c0c8268e811/soluble-klotho-downregulates-orai1-mediated-store-klotho-is-an-aging-suppressor.jpg)