
Autophagy hijacked through viroporin-activated calcium ... · Rotavirus is the causative agent of...
9
Autophagy hijacked through viroporin-activated calcium/calmodulin-dependent kinase kinase-β signaling is required for rotavirus replication Sue E. Crawford, Joseph M. Hyser, Budi Utama, and Mary K. Estes 1 Department of Molecular Virology and Microbiology, Baylor College of Medicine, Houston, TX 77030 Contributed by Mary K. Estes, September 25, 2012 (sent for review May 23, 2012) Autophagy is a cellular degradation process involving an intracel- lular membrane trafficking pathway that recycles cellular compo- nents or eliminates intracellular microbes in lysosomes. Many pathogens subvert autophagy to enhance their replication, but the mechanisms these pathogens use to initiate the autophagy pro- cess have not been elucidated. This study identifies rotavirus as a pathogen that encodes a viroporin, nonstructural protein 4, which releases endoplasmic reticulum calcium into the cytoplasm, thereby activating a calcium/calmodulin-dependent kinase kinase-β and 5′ adenosine monophosphate-activated protein kinase-dependent signaling pathway to initiate autophagy. Rotavirus hijacks this membrane trafficking pathway to transport viral proteins from the endoplasmic reticulum to sites of viral replication to produce infec- tious virus. This process requires PI3K activity and autophagy-initi- ation proteins Atg3 and Atg5, and it is abrogated by chelating cytoplasmic calcium or inhibiting calcium/calmodulin-dependent ki- nase kinase-β. Although the early stages of autophagy are initiated, rotavirus infection also blocks autophagy maturation. These studies identify a unique mechanism of virus-mediated, calcium-activated signaling that initiates autophagy and hijacks this membrane traf- ficking pathway to transport viral proteins to sites of viral assembly. viroplasm | morphogenesis | LC3 | STO-609 V iruses are obligate intracellular parasites that, due to their limited coding capacity, have evolved strategies that usurp cellular processes to facilitate their own propagation. Macro- autophagy (hereafter referred to as autophagy) is a cellular catabolic process used to maintain homeostasis by delivering cytoplasmic material to lysosomes for degradation via an in- tracellular membrane trafficking pathway (1). Autophagy also has intracellular antimicrobial properties and plays a role in the initiation of innate and adaptive immune responses to viral and bacterial infections. Numerous pathogens, including a number of DNA and RNA viruses, have been shown to evade or subvert autophagy (2); however, for most of these viruses, the mecha- nisms used to initiate autophagy and subvert the normal autophagy process have not been elucidated. The formation of autophagy membranes is complex and not completely understood, but the autophagy (Atg) proteins com- prise the core molecular machinery involved in this dynamic membrane rearrangement (3). Autophagy, which is repressed by the mammalian target of rapamycin (mTOR), can be activated by nutrient deprivation; growth factor depletion; or cellular stress, such as hypoxia, energy depletion, endoplasmic reticulum (ER) stress, high temperature, or high cell density conditions (4). Following nutrient deprivation, mTOR is inhibited and a com- plex composed of Atg13/ULK1/FIP200/Atg101 forms to initiate nucleation of an isolation membrane, or phagophore (5). The phagophore elongates and subsequently encloses cytoplasmic components, forming a double-membrane vacuole, the autopha- gosome. The elongation phase requires two ubiquitin-like conju- gation reactions to form the Atg5/Atg12/Atg16 complex and to conjugate phosphatidylethanolamine (PE) onto microtubule- associated protein light chain 3 (LC3). The lipid tail of LC3 is inserted into the forming autophagosome. Finally, autophago- somes are transported in a dynein-dependent manner on micro- tubules to lysosomes, where they fuse to form autolysosomes, and the engulfed material is degraded by lysosomal enzymes. Many important pathogens, including RNA viruses [picorna- viruses (poliovirus, coxsackievirus, rhinovirus, and hepatitis A), coronaviruses (severe acute respiratory syndrome), and flavivi- ruses (hepatitis C virus, yellow fever virus, dengue virus, and West Nile virus)] and some DNA viruses (hepatitis B virus and parvovirus) induce the accumulation of autophagosomes or autolysosomes (6–10). It has been proposed for picornaviruses that these dramatically remodeled autophagic intracellular mem- branes serve as a structural platform for viral replication and as- sembly. However, the mechanism of autophagy induction for most of these viruses is unknown. Rotavirus is the causative agent of severe gastroenteritis and vomiting in young children and animals worldwide (11). We previously reported that the rotavirus nonstructural protein 4 (NSP4), expressed alone or during virus infection, colocalizes with the endogenous autophagy marker protein LC3 in membranes that surround viroplasms, sites of viral replication and particle assembly, but the functional relevance of autophagy in rotavirus infection and the mechanism of autophagy induction remained unknown (12). The current study investigated whether autophagy is required for rotavirus replication and the mechanism used by rotavirus and NSP4 to initiate autophagy. We report an example of a virus-encoded viroporin that mediates the initiation of auto- phagy. We discovered that the rotavirus-encoded viroporin NSP4 releases calcium from the ER into the cytoplasm, activating cal- cium/calmodulin-dependent kinase kinase-β (CaMKK-β) signaling to initiate autophagy. The current study provides insight into a unique mechanism through which rotavirus initiates autophagy Significance This study describes a unique mechanism of virus-initiated autophagy and exploitation of autophagy membranes for virus replication. Autophagy is a highly regulated cellular process in which cells destroy and recycle their own components in lyso- somes. The mechanism most viruses use to induce autophagy is unknown. We show a rotavirus pore-forming protein activates a calcium-dependent signaling pathway to initiate autophagy. Rotavirus hijacks autophagy membranes to transport viral proteins to sites of virus replication for assembly of infectious particles and interferes with autophagy maturation. Inhibition of the signaling pathway blocks virus production, suggesting a therapeutic target to fight infection. Author contributions: S.E.C., J.M.H., B.U., and M.K.E. designed research; S.E.C., J.M.H., and B.U. performed research; S.E.C., J.M.H., B.U., and M.K.E. analyzed data; and S.E.C., J.M.H., and M.K.E. wrote the paper. The authors declare no conflict of interest. 1 To whom correspondence should be addressed. E-mail: [email protected]. This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10. 1073/pnas.1216539109/-/DCSupplemental. www.pnas.org/cgi/doi/10.1073/pnas.1216539109 PNAS | Published online November 26, 2012 | E3405–E3413 MICROBIOLOGY PNAS PLUS
Transcript of Autophagy hijacked through viroporin-activated calcium ... · Rotavirus is the causative agent of...

Autophagy hijacked through viroporin-activatedcalcium/calmodulin-dependent kinase kinase-βsignaling is required for rotavirus replicationSue E. Crawford, Joseph M. Hyser, Budi Utama, and Mary K. Estes1
Department of Molecular Virology and Microbiology, Baylor College of Medicine, Houston, TX 77030
Contributed by Mary K. Estes, September 25, 2012 (sent for review May 23, 2012)
Autophagy is a cellular degradation process involving an intracel-lular membrane trafficking pathway that recycles cellular compo-nents or eliminates intracellular microbes in lysosomes. Manypathogens subvert autophagy to enhance their replication, but themechanisms these pathogens use to initiate the autophagy pro-cess have not been elucidated. This study identifies rotavirus as apathogen that encodes a viroporin, nonstructural protein 4, whichreleases endoplasmic reticulum calcium into the cytoplasm, therebyactivating a calcium/calmodulin-dependent kinase kinase-β and5′ adenosine monophosphate-activated protein kinase-dependentsignaling pathway to initiate autophagy. Rotavirus hijacks thismembrane trafficking pathway to transport viral proteins from theendoplasmic reticulum to sites of viral replication to produce infec-tious virus. This process requires PI3K activity and autophagy-initi-ation proteins Atg3 and Atg5, and it is abrogated by chelatingcytoplasmic calcium or inhibiting calcium/calmodulin-dependent ki-nase kinase-β. Although the early stages of autophagy are initiated,rotavirus infection also blocks autophagy maturation. These studiesidentify a unique mechanism of virus-mediated, calcium-activatedsignaling that initiates autophagy and hijacks this membrane traf-ficking pathway to transport viral proteins to sites of viral assembly.
viroplasm | morphogenesis | LC3 | STO-609
Viruses are obligate intracellular parasites that, due to theirlimited coding capacity, have evolved strategies that usurp
cellular processes to facilitate their own propagation. Macro-autophagy (hereafter referred to as autophagy) is a cellularcatabolic process used to maintain homeostasis by deliveringcytoplasmic material to lysosomes for degradation via an in-tracellular membrane trafficking pathway (1). Autophagy alsohas intracellular antimicrobial properties and plays a role in theinitiation of innate and adaptive immune responses to viral andbacterial infections. Numerous pathogens, including a number ofDNA and RNA viruses, have been shown to evade or subvertautophagy (2); however, for most of these viruses, the mecha-nisms used to initiate autophagy and subvert the normalautophagy process have not been elucidated.The formation of autophagy membranes is complex and not
completely understood, but the autophagy (Atg) proteins com-prise the core molecular machinery involved in this dynamicmembrane rearrangement (3). Autophagy, which is repressed bythe mammalian target of rapamycin (mTOR), can be activated bynutrient deprivation; growth factor depletion; or cellular stress,such as hypoxia, energy depletion, endoplasmic reticulum (ER)stress, high temperature, or high cell density conditions (4).Following nutrient deprivation, mTOR is inhibited and a com-plex composed of Atg13/ULK1/FIP200/Atg101 forms to initiatenucleation of an isolation membrane, or phagophore (5). Thephagophore elongates and subsequently encloses cytoplasmiccomponents, forming a double-membrane vacuole, the autopha-gosome. The elongation phase requires two ubiquitin-like conju-gation reactions to form the Atg5/Atg12/Atg16 complex and toconjugate phosphatidylethanolamine (PE) onto microtubule-associated protein light chain 3 (LC3). The lipid tail of LC3 is
inserted into the forming autophagosome. Finally, autophago-somes are transported in a dynein-dependent manner on micro-tubules to lysosomes, where they fuse to form autolysosomes, andthe engulfed material is degraded by lysosomal enzymes.Many important pathogens, including RNA viruses [picorna-
viruses (poliovirus, coxsackievirus, rhinovirus, and hepatitis A),coronaviruses (severe acute respiratory syndrome), and flavivi-ruses (hepatitis C virus, yellow fever virus, dengue virus, andWest Nile virus)] and some DNA viruses (hepatitis B virus andparvovirus) induce the accumulation of autophagosomes orautolysosomes (6–10). It has been proposed for picornavirusesthat these dramatically remodeled autophagic intracellular mem-branes serve as a structural platform for viral replication and as-sembly. However, the mechanism of autophagy induction for mostof these viruses is unknown.Rotavirus is the causative agent of severe gastroenteritis and
vomiting in young children and animals worldwide (11). Wepreviously reported that the rotavirus nonstructural protein 4(NSP4), expressed alone or during virus infection, colocalizes withthe endogenous autophagy marker protein LC3 in membranesthat surround viroplasms, sites of viral replication and particleassembly, but the functional relevance of autophagy in rotavirusinfection and the mechanism of autophagy induction remainedunknown (12). The current study investigated whether autophagyis required for rotavirus replication and the mechanism used byrotavirus and NSP4 to initiate autophagy. We report an exampleof a virus-encoded viroporin that mediates the initiation of auto-phagy. We discovered that the rotavirus-encoded viroporin NSP4releases calcium from the ER into the cytoplasm, activating cal-cium/calmodulin-dependent kinase kinase-β (CaMKK-β) signalingto initiate autophagy. The current study provides insight intoa unique mechanism through which rotavirus initiates autophagy
Significance
This study describes a unique mechanism of virus-initiatedautophagy and exploitation of autophagy membranes for virusreplication. Autophagy is a highly regulated cellular process inwhich cells destroy and recycle their own components in lyso-somes. The mechanism most viruses use to induce autophagy isunknown. We show a rotavirus pore-forming protein activatesa calcium-dependent signaling pathway to initiate autophagy.Rotavirus hijacks autophagy membranes to transport viralproteins to sites of virus replication for assembly of infectiousparticles and interferes with autophagy maturation. Inhibitionof the signaling pathway blocks virus production, suggestinga therapeutic target to fight infection.
Author contributions: S.E.C., J.M.H., B.U., and M.K.E. designed research; S.E.C., J.M.H., andB.U. performed research; S.E.C., J.M.H., B.U., and M.K.E. analyzed data; and S.E.C., J.M.H.,and M.K.E. wrote the paper.
The authors declare no conflict of interest.1To whom correspondence should be addressed. E-mail: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1216539109/-/DCSupplemental.
www.pnas.org/cgi/doi/10.1073/pnas.1216539109 PNAS | Published online November 26, 2012 | E3405–E3413
MICRO
BIOLO
GY
PNASPL
US
and hijacks this membrane trafficking pathway to transport viralproteins from the ER to sites of virus replication for assembly ofinfectious virus.
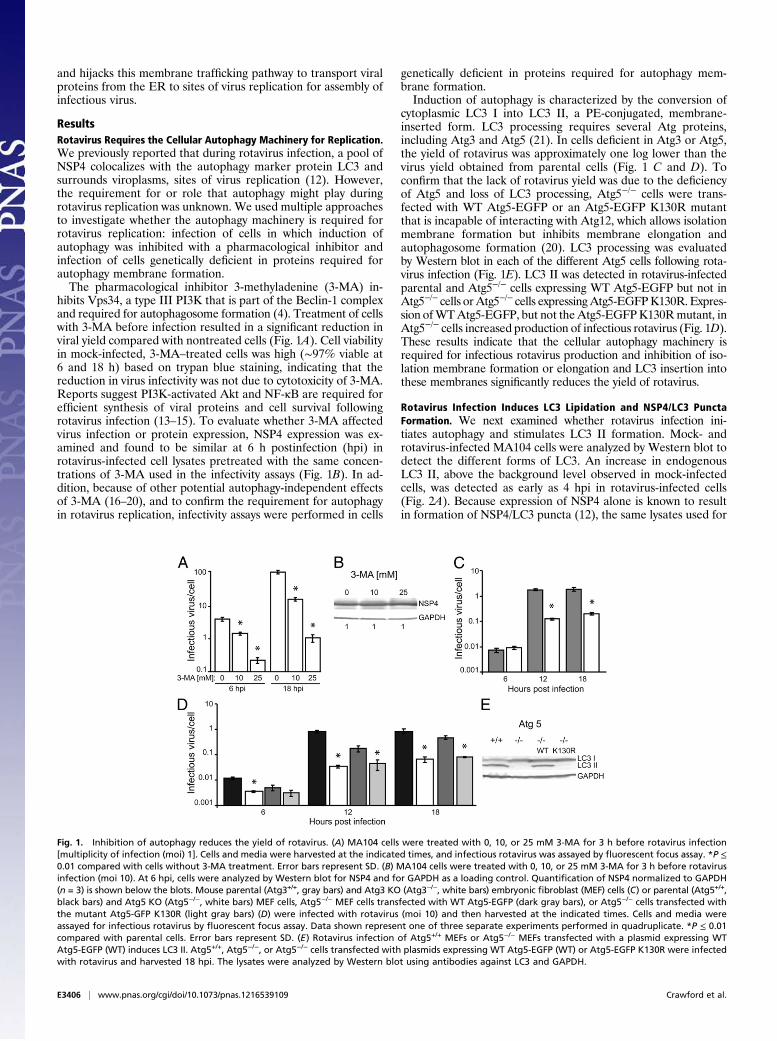
ResultsRotavirus Requires the Cellular Autophagy Machinery for Replication.We previously reported that during rotavirus infection, a pool ofNSP4 colocalizes with the autophagy marker protein LC3 andsurrounds viroplasms, sites of virus replication (12). However,the requirement for or role that autophagy might play duringrotavirus replication was unknown. We used multiple approachesto investigate whether the autophagy machinery is required forrotavirus replication: infection of cells in which induction ofautophagy was inhibited with a pharmacological inhibitor andinfection of cells genetically deficient in proteins required forautophagy membrane formation.The pharmacological inhibitor 3-methyladenine (3-MA) in-
hibits Vps34, a type III PI3K that is part of the Beclin-1 complexand required for autophagosome formation (4). Treatment of cellswith 3-MA before infection resulted in a significant reduction inviral yield compared with nontreated cells (Fig. 1A). Cell viabilityin mock-infected, 3-MA–treated cells was high (∼97% viable at6 and 18 h) based on trypan blue staining, indicating that thereduction in virus infectivity was not due to cytotoxicity of 3-MA.Reports suggest PI3K-activated Akt and NF-κB are required forefficient synthesis of viral proteins and cell survival followingrotavirus infection (13–15). To evaluate whether 3-MA affectedvirus infection or protein expression, NSP4 expression was ex-amined and found to be similar at 6 h postinfection (hpi) inrotavirus-infected cell lysates pretreated with the same concen-trations of 3-MA used in the infectivity assays (Fig. 1B). In ad-dition, because of other potential autophagy-independent effectsof 3-MA (16–20), and to confirm the requirement for autophagyin rotavirus replication, infectivity assays were performed in cells
genetically deficient in proteins required for autophagy mem-brane formation.Induction of autophagy is characterized by the conversion of
cytoplasmic LC3 I into LC3 II, a PE-conjugated, membrane-inserted form. LC3 processing requires several Atg proteins,including Atg3 and Atg5 (21). In cells deficient in Atg3 or Atg5,the yield of rotavirus was approximately one log lower than thevirus yield obtained from parental cells (Fig. 1 C and D). Toconfirm that the lack of rotavirus yield was due to the deficiencyof Atg5 and loss of LC3 processing, Atg5−/− cells were trans-fected with WT Atg5-EGFP or an Atg5-EGFP K130R mutantthat is incapable of interacting with Atg12, which allows isolationmembrane formation but inhibits membrane elongation andautophagosome formation (20). LC3 processing was evaluatedby Western blot in each of the different Atg5 cells following rota-virus infection (Fig. 1E). LC3 II was detected in rotavirus-infectedparental and Atg5−/− cells expressing WT Atg5-EGFP but not inAtg5−/− cells orAtg5−/− cells expressingAtg5-EGFPK130R. Expres-sion ofWTAtg5-EGFP, but not theAtg5-EGFPK130Rmutant, inAtg5−/− cells increased production of infectious rotavirus (Fig. 1D).These results indicate that the cellular autophagy machinery isrequired for infectious rotavirus production and inhibition of iso-lation membrane formation or elongation and LC3 insertion intothese membranes significantly reduces the yield of rotavirus.
Rotavirus Infection Induces LC3 Lipidation and NSP4/LC3 PunctaFormation. We next examined whether rotavirus infection ini-tiates autophagy and stimulates LC3 II formation. Mock- androtavirus-infected MA104 cells were analyzed by Western blot todetect the different forms of LC3. An increase in endogenousLC3 II, above the background level observed in mock-infectedcells, was detected as early as 4 hpi in rotavirus-infected cells(Fig. 2A). Because expression of NSP4 alone is known to resultin formation of NSP4/LC3 puncta (12), the same lysates used for
Fig. 1. Inhibition of autophagy reduces the yield of rotavirus. (A) MA104 cells were treated with 0, 10, or 25 mM 3-MA for 3 h before rotavirus infection[multiplicity of infection (moi) 1]. Cells and media were harvested at the indicated times, and infectious rotavirus was assayed by fluorescent focus assay. *P ≤0.01 compared with cells without 3-MA treatment. Error bars represent SD. (B) MA104 cells were treated with 0, 10, or 25 mM 3-MA for 3 h before rotavirusinfection (moi 10). At 6 hpi, cells were analyzed by Western blot for NSP4 and for GAPDH as a loading control. Quantification of NSP4 normalized to GAPDH(n = 3) is shown below the blots. Mouse parental (Atg3+/+, gray bars) and Atg3 KO (Atg3−/−, white bars) embryonic fibroblast (MEF) cells (C) or parental (Atg5+/+,black bars) and Atg5 KO (Atg5−/−, white bars) MEF cells, Atg5−/− MEF cells transfected with WT Atg5-EGFP (dark gray bars), or Atg5−/− cells transfected withthe mutant Atg5-GFP K130R (light gray bars) (D) were infected with rotavirus (moi 10) and then harvested at the indicated times. Cells and media wereassayed for infectious rotavirus by fluorescent focus assay. Data shown represent one of three separate experiments performed in quadruplicate. *P ≤ 0.01compared with parental cells. Error bars represent SD. (E) Rotavirus infection of Atg5+/+ MEFs or Atg5−/− MEFs transfected with a plasmid expressing WTAtg5-EGFP (WT) induces LC3 II. Atg5+/+, Atg5−/−, or Atg5−/− cells transfected with plasmids expressing WT Atg5-EGFP (WT) or Atg5-EGFP K130R were infectedwith rotavirus and harvested 18 hpi. The lysates were analyzed by Western blot using antibodies against LC3 and GAPDH.
E3406 | www.pnas.org/cgi/doi/10.1073/pnas.1216539109 Crawford et al.
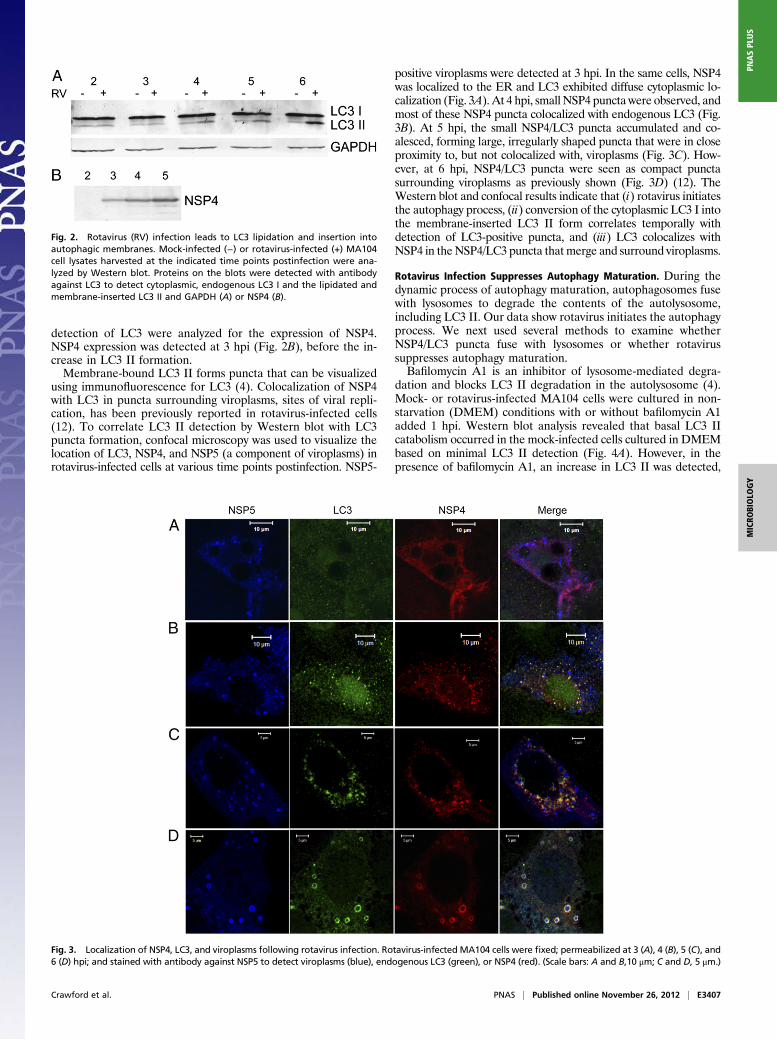
detection of LC3 were analyzed for the expression of NSP4.NSP4 expression was detected at 3 hpi (Fig. 2B), before the in-crease in LC3 II formation.Membrane-bound LC3 II forms puncta that can be visualized
using immunofluorescence for LC3 (4). Colocalization of NSP4with LC3 in puncta surrounding viroplasms, sites of viral repli-cation, has been previously reported in rotavirus-infected cells(12). To correlate LC3 II detection by Western blot with LC3puncta formation, confocal microscopy was used to visualize thelocation of LC3, NSP4, and NSP5 (a component of viroplasms) inrotavirus-infected cells at various time points postinfection. NSP5-
positive viroplasms were detected at 3 hpi. In the same cells, NSP4was localized to the ER and LC3 exhibited diffuse cytoplasmic lo-calization (Fig. 3A). At 4 hpi, small NSP4 puncta were observed, andmost of these NSP4 puncta colocalized with endogenous LC3 (Fig.3B). At 5 hpi, the small NSP4/LC3 puncta accumulated and co-alesced, forming large, irregularly shaped puncta that were in closeproximity to, but not colocalized with, viroplasms (Fig. 3C). How-ever, at 6 hpi, NSP4/LC3 puncta were seen as compact punctasurrounding viroplasms as previously shown (Fig. 3D) (12). TheWestern blot and confocal results indicate that (i) rotavirus initiatesthe autophagy process, (ii) conversion of the cytoplasmic LC3 I intothe membrane-inserted LC3 II form correlates temporally withdetection of LC3-positive puncta, and (iii) LC3 colocalizes withNSP4 in the NSP4/LC3 puncta thatmerge and surround viroplasms.
Rotavirus Infection Suppresses Autophagy Maturation. During thedynamic process of autophagy maturation, autophagosomes fusewith lysosomes to degrade the contents of the autolysosome,including LC3 II. Our data show rotavirus initiates the autophagyprocess. We next used several methods to examine whetherNSP4/LC3 puncta fuse with lysosomes or whether rotavirussuppresses autophagy maturation.Bafilomycin A1 is an inhibitor of lysosome-mediated degra-
dation and blocks LC3 II degradation in the autolysosome (4).Mock- or rotavirus-infected MA104 cells were cultured in non-starvation (DMEM) conditions with or without bafilomycin A1added 1 hpi. Western blot analysis revealed that basal LC3 IIcatabolism occurred in the mock-infected cells cultured in DMEMbased on minimal LC3 II detection (Fig. 4A). However, in thepresence of bafilomycin A1, an increase in LC3 II was detected,
Fig. 2. Rotavirus (RV) infection leads to LC3 lipidation and insertion intoautophagic membranes. Mock-infected (−) or rotavirus-infected (+) MA104cell lysates harvested at the indicated time points postinfection were ana-lyzed by Western blot. Proteins on the blots were detected with antibodyagainst LC3 to detect cytoplasmic, endogenous LC3 I and the lipidated andmembrane-inserted LC3 II and GAPDH (A) or NSP4 (B).
Fig. 3. Localization of NSP4, LC3, and viroplasms following rotavirus infection. Rotavirus-infected MA104 cells were fixed; permeabilized at 3 (A), 4 (B), 5 (C), and6 (D) hpi; and stained with antibody against NSP5 to detect viroplasms (blue), endogenous LC3 (green), or NSP4 (red). (Scale bars: A and B,10 μm; C and D, 5 μm.)
Crawford et al. PNAS | Published online November 26, 2012 | E3407
MICRO
BIOLO
GY
PNASPL
US
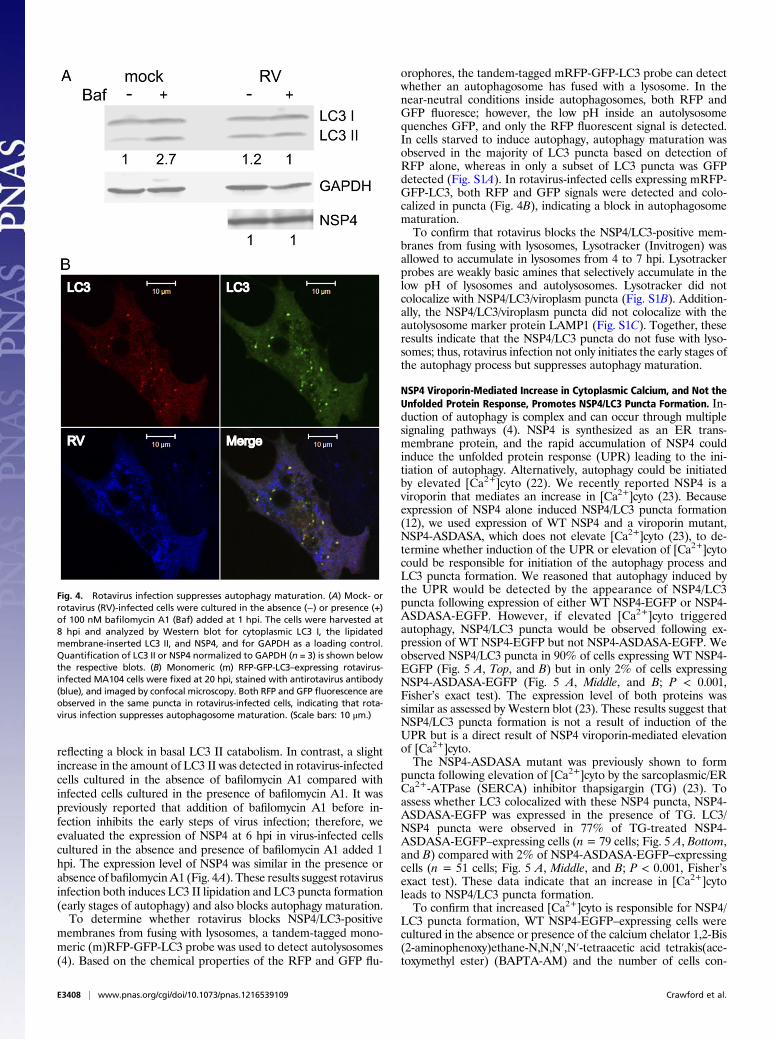
reflecting a block in basal LC3 II catabolism. In contrast, a slightincrease in the amount of LC3 II was detected in rotavirus-infectedcells cultured in the absence of bafilomycin A1 compared withinfected cells cultured in the presence of bafilomycin A1. It waspreviously reported that addition of bafilomycin A1 before in-fection inhibits the early steps of virus infection; therefore, weevaluated the expression of NSP4 at 6 hpi in virus-infected cellscultured in the absence and presence of bafilomycin A1 added 1hpi. The expression level of NSP4 was similar in the presence orabsence of bafilomycinA1 (Fig. 4A). These results suggest rotavirusinfection both induces LC3 II lipidation and LC3 puncta formation(early stages of autophagy) and also blocks autophagy maturation.To determine whether rotavirus blocks NSP4/LC3-positive
membranes from fusing with lysosomes, a tandem-tagged mono-meric (m)RFP-GFP-LC3 probe was used to detect autolysosomes(4). Based on the chemical properties of the RFP and GFP flu-
orophores, the tandem-tagged mRFP-GFP-LC3 probe can detectwhether an autophagosome has fused with a lysosome. In thenear-neutral conditions inside autophagosomes, both RFP andGFP fluoresce; however, the low pH inside an autolysosomequenches GFP, and only the RFP fluorescent signal is detected.In cells starved to induce autophagy, autophagy maturation wasobserved in the majority of LC3 puncta based on detection ofRFP alone, whereas in only a subset of LC3 puncta was GFPdetected (Fig. S1A). In rotavirus-infected cells expressing mRFP-GFP-LC3, both RFP and GFP signals were detected and colo-calized in puncta (Fig. 4B), indicating a block in autophagosomematuration.To confirm that rotavirus blocks the NSP4/LC3-positive mem-
branes from fusing with lysosomes, Lysotracker (Invitrogen) wasallowed to accumulate in lysosomes from 4 to 7 hpi. Lysotrackerprobes are weakly basic amines that selectively accumulate in thelow pH of lysosomes and autolysosomes. Lysotracker did notcolocalize with NSP4/LC3/viroplasm puncta (Fig. S1B). Addition-ally, the NSP4/LC3/viroplasm puncta did not colocalize with theautolysosome marker protein LAMP1 (Fig. S1C). Together, theseresults indicate that the NSP4/LC3 puncta do not fuse with lyso-somes; thus, rotavirus infection not only initiates the early stages ofthe autophagy process but suppresses autophagy maturation.
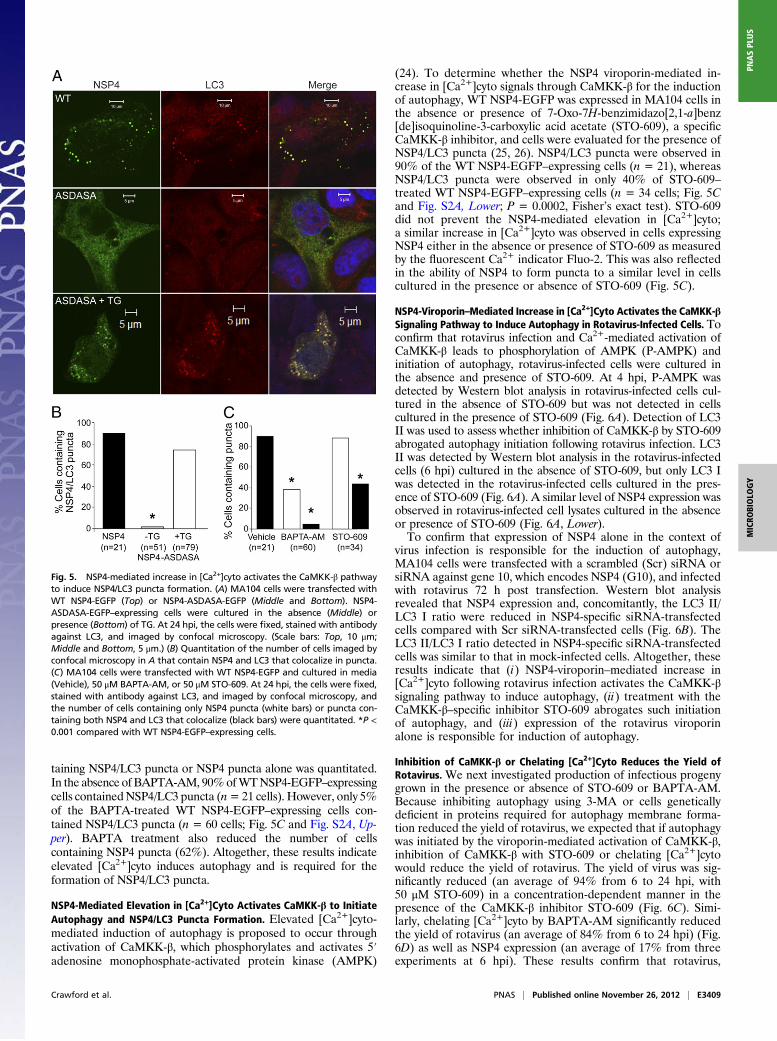
NSP4 Viroporin-Mediated Increase in Cytoplasmic Calcium, and Not theUnfolded Protein Response, Promotes NSP4/LC3 Puncta Formation. In-duction of autophagy is complex and can occur through multiplesignaling pathways (4). NSP4 is synthesized as an ER trans-membrane protein, and the rapid accumulation of NSP4 couldinduce the unfolded protein response (UPR) leading to the ini-tiation of autophagy. Alternatively, autophagy could be initiatedby elevated [Ca2+]cyto (22). We recently reported NSP4 is aviroporin that mediates an increase in [Ca2+]cyto (23). Becauseexpression of NSP4 alone induced NSP4/LC3 puncta formation(12), we used expression of WT NSP4 and a viroporin mutant,NSP4-ASDASA, which does not elevate [Ca2+]cyto (23), to de-termine whether induction of the UPR or elevation of [Ca2+]cytocould be responsible for initiation of the autophagy process andLC3 puncta formation. We reasoned that autophagy induced bythe UPR would be detected by the appearance of NSP4/LC3puncta following expression of either WT NSP4-EGFP or NSP4-ASDASA-EGFP. However, if elevated [Ca2+]cyto triggeredautophagy, NSP4/LC3 puncta would be observed following ex-pression of WT NSP4-EGFP but not NSP4-ASDASA-EGFP. Weobserved NSP4/LC3 puncta in 90% of cells expressing WT NSP4-EGFP (Fig. 5 A, Top, and B) but in only 2% of cells expressingNSP4-ASDASA-EGFP (Fig. 5 A, Middle, and B; P < 0.001,Fisher’s exact test). The expression level of both proteins wassimilar as assessed by Western blot (23). These results suggest thatNSP4/LC3 puncta formation is not a result of induction of theUPR but is a direct result of NSP4 viroporin-mediated elevationof [Ca2+]cyto.The NSP4-ASDASA mutant was previously shown to form
puncta following elevation of [Ca2+]cyto by the sarcoplasmic/ERCa2+-ATPase (SERCA) inhibitor thapsigargin (TG) (23). Toassess whether LC3 colocalized with these NSP4 puncta, NSP4-ASDASA-EGFP was expressed in the presence of TG. LC3/NSP4 puncta were observed in 77% of TG-treated NSP4-ASDASA-EGFP–expressing cells (n= 79 cells; Fig. 5 A, Bottom,and B) compared with 2% of NSP4-ASDASA-EGFP–expressingcells (n = 51 cells; Fig. 5 A, Middle, and B; P < 0.001, Fisher’sexact test). These data indicate that an increase in [Ca2+]cytoleads to NSP4/LC3 puncta formation.To confirm that increased [Ca2+]cyto is responsible for NSP4/
LC3 puncta formation, WT NSP4-EGFP–expressing cells werecultured in the absence or presence of the calcium chelator 1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(ace-toxymethyl ester) (BAPTA-AM) and the number of cells con-
Fig. 4. Rotavirus infection suppresses autophagy maturation. (A) Mock- orrotavirus (RV)-infected cells were cultured in the absence (−) or presence (+)of 100 nM bafilomycin A1 (Baf) added at 1 hpi. The cells were harvested at8 hpi and analyzed by Western blot for cytoplasmic LC3 I, the lipidatedmembrane-inserted LC3 II, and NSP4, and for GAPDH as a loading control.Quantification of LC3 II or NSP4 normalized to GAPDH (n = 3) is shown belowthe respective blots. (B) Monomeric (m) RFP-GFP-LC3–expressing rotavirus-infected MA104 cells were fixed at 20 hpi, stained with antirotavirus antibody(blue), and imaged by confocal microscopy. Both RFP and GFP fluorescence areobserved in the same puncta in rotavirus-infected cells, indicating that rota-virus infection suppresses autophagosome maturation. (Scale bars: 10 μm.)
E3408 | www.pnas.org/cgi/doi/10.1073/pnas.1216539109 Crawford et al.
taining NSP4/LC3 puncta or NSP4 puncta alone was quantitated.In the absence of BAPTA-AM, 90%ofWTNSP4-EGFP–expressingcells containedNSP4/LC3 puncta (n=21 cells). However, only 5%of the BAPTA-treated WT NSP4-EGFP–expressing cells con-tained NSP4/LC3 puncta (n = 60 cells; Fig. 5C and Fig. S2A, Up-per). BAPTA treatment also reduced the number of cellscontaining NSP4 puncta (62%). Altogether, these results indicateelevated [Ca2+]cyto induces autophagy and is required for theformation of NSP4/LC3 puncta.
NSP4-Mediated Elevation in [Ca2+]Cyto Activates CaMKK-β to InitiateAutophagy and NSP4/LC3 Puncta Formation. Elevated [Ca2+]cyto-mediated induction of autophagy is proposed to occur throughactivation of CaMKK-β, which phosphorylates and activates 5′adenosine monophosphate-activated protein kinase (AMPK)
(24). To determine whether the NSP4 viroporin-mediated in-crease in [Ca2+]cyto signals through CaMKK-β for the inductionof autophagy, WT NSP4-EGFP was expressed in MA104 cells inthe absence or presence of 7-Oxo-7H-benzimidazo[2,1-a]benz[de]isoquinoline-3-carboxylic acid acetate (STO-609), a specificCaMKK-β inhibitor, and cells were evaluated for the presence ofNSP4/LC3 puncta (25, 26). NSP4/LC3 puncta were observed in90% of the WT NSP4-EGFP–expressing cells (n = 21), whereasNSP4/LC3 puncta were observed in only 40% of STO-609–treated WT NSP4-EGFP–expressing cells (n = 34 cells; Fig. 5Cand Fig. S2A, Lower; P = 0.0002, Fisher’s exact test). STO-609did not prevent the NSP4-mediated elevation in [Ca2+]cyto;a similar increase in [Ca2+]cyto was observed in cells expressingNSP4 either in the absence or presence of STO-609 as measuredby the fluorescent Ca2+ indicator Fluo-2. This was also reflectedin the ability of NSP4 to form puncta to a similar level in cellscultured in the presence or absence of STO-609 (Fig. 5C).
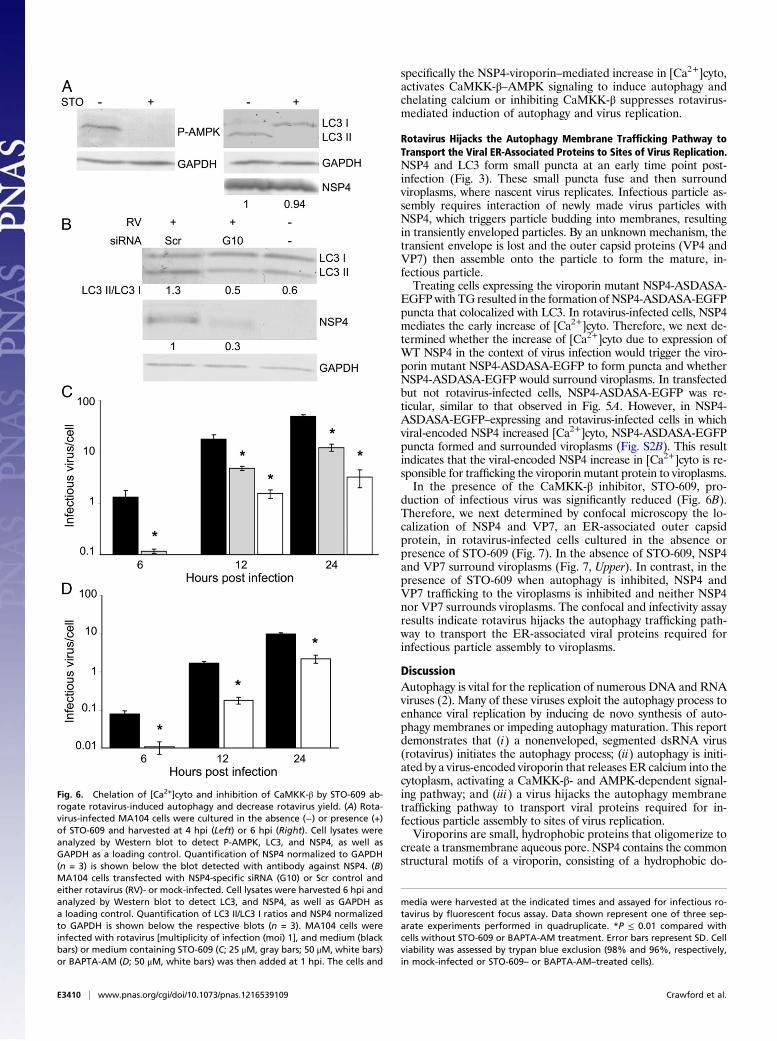
NSP4-Viroporin–Mediated Increase in [Ca2+]Cyto Activates the CaMKK-βSignaling Pathway to Induce Autophagy in Rotavirus-Infected Cells. Toconfirm that rotavirus infection and Ca2+-mediated activation ofCaMKK-β leads to phosphorylation of AMPK (P-AMPK) andinitiation of autophagy, rotavirus-infected cells were cultured inthe absence and presence of STO-609. At 4 hpi, P-AMPK wasdetected by Western blot analysis in rotavirus-infected cells cul-tured in the absence of STO-609 but was not detected in cellscultured in the presence of STO-609 (Fig. 6A). Detection of LC3II was used to assess whether inhibition of CaMKK-β by STO-609abrogated autophagy initiation following rotavirus infection. LC3II was detected by Western blot analysis in the rotavirus-infectedcells (6 hpi) cultured in the absence of STO-609, but only LC3 Iwas detected in the rotavirus-infected cells cultured in the pres-ence of STO-609 (Fig. 6A). A similar level of NSP4 expression wasobserved in rotavirus-infected cell lysates cultured in the absenceor presence of STO-609 (Fig. 6A, Lower).To confirm that expression of NSP4 alone in the context of
virus infection is responsible for the induction of autophagy,MA104 cells were transfected with a scrambled (Scr) siRNA orsiRNA against gene 10, which encodes NSP4 (G10), and infectedwith rotavirus 72 h post transfection. Western blot analysisrevealed that NSP4 expression and, concomitantly, the LC3 II/LC3 I ratio were reduced in NSP4-specific siRNA-transfectedcells compared with Scr siRNA-transfected cells (Fig. 6B). TheLC3 II/LC3 I ratio detected in NSP4-specific siRNA-transfectedcells was similar to that in mock-infected cells. Altogether, theseresults indicate that (i) NSP4-viroporin–mediated increase in[Ca2+]cyto following rotavirus infection activates the CaMKK-βsignaling pathway to induce autophagy, (ii) treatment with theCaMKK-β–specific inhibitor STO-609 abrogates such initiationof autophagy, and (iii) expression of the rotavirus viroporinalone is responsible for induction of autophagy.
Inhibition of CaMKK-β or Chelating [Ca2+]Cyto Reduces the Yield ofRotavirus. We next investigated production of infectious progenygrown in the presence or absence of STO-609 or BAPTA-AM.Because inhibiting autophagy using 3-MA or cells geneticallydeficient in proteins required for autophagy membrane forma-tion reduced the yield of rotavirus, we expected that if autophagywas initiated by the viroporin-mediated activation of CaMKK-β,inhibition of CaMKK-β with STO-609 or chelating [Ca2+]cytowould reduce the yield of rotavirus. The yield of virus was sig-nificantly reduced (an average of 94% from 6 to 24 hpi, with50 μM STO-609) in a concentration-dependent manner in thepresence of the CaMKK-β inhibitor STO-609 (Fig. 6C). Simi-larly, chelating [Ca2+]cyto by BAPTA-AM significantly reducedthe yield of rotavirus (an average of 84% from 6 to 24 hpi) (Fig.6D) as well as NSP4 expression (an average of 17% from threeexperiments at 6 hpi). These results confirm that rotavirus,
Fig. 5. NSP4-mediated increase in [Ca2+]cyto activates the CaMKK-β pathwayto induce NSP4/LC3 puncta formation. (A) MA104 cells were transfected withWT NSP4-EGFP (Top) or NSP4-ASDASA-EGFP (Middle and Bottom). NSP4-ASDASA-EGFP–expressing cells were cultured in the absence (Middle) orpresence (Bottom) of TG. At 24 hpi, the cells were fixed, stained with antibodyagainst LC3, and imaged by confocal microscopy. (Scale bars: Top, 10 μm;Middle and Bottom, 5 μm.) (B) Quantitation of the number of cells imaged byconfocal microscopy in A that contain NSP4 and LC3 that colocalize in puncta.(C) MA104 cells were transfected with WT NSP4-EGFP and cultured in media(Vehicle), 50 μM BAPTA-AM, or 50 μM STO-609. At 24 hpi, the cells were fixed,stained with antibody against LC3, and imaged by confocal microscopy, andthe number of cells containing only NSP4 puncta (white bars) or puncta con-taining both NSP4 and LC3 that colocalize (black bars) were quantitated. *P <0.001 compared with WT NSP4-EGFP–expressing cells.
Crawford et al. PNAS | Published online November 26, 2012 | E3409
MICRO
BIOLO
GY
PNASPL
US
specifically the NSP4-viroporin–mediated increase in [Ca2+]cyto,activates CaMKK-β–AMPK signaling to induce autophagy andchelating calcium or inhibiting CaMKK-β suppresses rotavirus-mediated induction of autophagy and virus replication.
Rotavirus Hijacks the Autophagy Membrane Trafficking Pathway toTransport the Viral ER-Associated Proteins to Sites of Virus Replication.NSP4 and LC3 form small puncta at an early time point post-infection (Fig. 3). These small puncta fuse and then surroundviroplasms, where nascent virus replicates. Infectious particle as-sembly requires interaction of newly made virus particles withNSP4, which triggers particle budding into membranes, resultingin transiently enveloped particles. By an unknown mechanism, thetransient envelope is lost and the outer capsid proteins (VP4 andVP7) then assemble onto the particle to form the mature, in-fectious particle.Treating cells expressing the viroporin mutant NSP4-ASDASA-
EGFPwith TG resulted in the formation of NSP4-ASDASA-EGFPpuncta that colocalized with LC3. In rotavirus-infected cells, NSP4mediates the early increase of [Ca2+]cyto. Therefore, we next de-termined whether the increase of [Ca2+]cyto due to expression ofWT NSP4 in the context of virus infection would trigger the viro-porin mutant NSP4-ASDASA-EGFP to form puncta and whetherNSP4-ASDASA-EGFP would surround viroplasms. In transfectedbut not rotavirus-infected cells, NSP4-ASDASA-EGFP was re-ticular, similar to that observed in Fig. 5A. However, in NSP4-ASDASA-EGFP–expressing and rotavirus-infected cells in whichviral-encoded NSP4 increased [Ca2+]cyto, NSP4-ASDASA-EGFPpuncta formed and surrounded viroplasms (Fig. S2B). This resultindicates that the viral-encoded NSP4 increase in [Ca2+]cyto is re-sponsible for trafficking the viroporin mutant protein to viroplasms.In the presence of the CaMKK-β inhibitor, STO-609, pro-
duction of infectious virus was significantly reduced (Fig. 6B).Therefore, we next determined by confocal microscopy the lo-calization of NSP4 and VP7, an ER-associated outer capsidprotein, in rotavirus-infected cells cultured in the absence orpresence of STO-609 (Fig. 7). In the absence of STO-609, NSP4and VP7 surround viroplasms (Fig. 7, Upper). In contrast, in thepresence of STO-609 when autophagy is inhibited, NSP4 andVP7 trafficking to the viroplasms is inhibited and neither NSP4nor VP7 surrounds viroplasms. The confocal and infectivity assayresults indicate rotavirus hijacks the autophagy trafficking path-way to transport the ER-associated viral proteins required forinfectious particle assembly to viroplasms.
DiscussionAutophagy is vital for the replication of numerous DNA and RNAviruses (2). Many of these viruses exploit the autophagy process toenhance viral replication by inducing de novo synthesis of auto-phagy membranes or impeding autophagy maturation. This reportdemonstrates that (i) a nonenveloped, segmented dsRNA virus(rotavirus) initiates the autophagy process; (ii) autophagy is initi-ated by a virus-encoded viroporin that releases ER calcium into thecytoplasm, activating a CaMKK-β- and AMPK-dependent signal-ing pathway; and (iii) a virus hijacks the autophagy membranetrafficking pathway to transport viral proteins required for in-fectious particle assembly to sites of virus replication.Viroporins are small, hydrophobic proteins that oligomerize to
create a transmembrane aqueous pore. NSP4 contains the commonstructural motifs of a viroporin, consisting of a hydrophobic do-
Fig. 6. Chelation of [Ca2+]cyto and inhibition of CaMKK-β by STO-609 ab-rogate rotavirus-induced autophagy and decrease rotavirus yield. (A) Rota-virus-infected MA104 cells were cultured in the absence (−) or presence (+)of STO-609 and harvested at 4 hpi (Left) or 6 hpi (Right). Cell lysates wereanalyzed by Western blot to detect P-AMPK, LC3, and NSP4, as well asGAPDH as a loading control. Quantification of NSP4 normalized to GAPDH(n = 3) is shown below the blot detected with antibody against NSP4. (B)MA104 cells transfected with NSP4-specific siRNA (G10) or Scr control andeither rotavirus (RV)- or mock-infected. Cell lysates were harvested 6 hpi andanalyzed by Western blot to detect LC3, and NSP4, as well as GAPDH asa loading control. Quantification of LC3 II/LC3 I ratios and NSP4 normalizedto GAPDH is shown below the respective blots (n = 3). MA104 cells wereinfected with rotavirus [multiplicity of infection (moi) 1], and medium (blackbars) or medium containing STO-609 (C; 25 μM, gray bars; 50 μM, white bars)or BAPTA-AM (D; 50 μM, white bars) was then added at 1 hpi. The cells and
media were harvested at the indicated times and assayed for infectious ro-tavirus by fluorescent focus assay. Data shown represent one of three sep-arate experiments performed in quadruplicate. *P ≤ 0.01 compared withcells without STO-609 or BAPTA-AM treatment. Error bars represent SD. Cellviability was assessed by trypan blue exclusion (98% and 96%, respectively,in mock-infected or STO-609– or BAPTA-AM–treated cells).
E3410 | www.pnas.org/cgi/doi/10.1073/pnas.1216539109 Crawford et al.

main that forms an amphipathic α-helix and a cluster of basic(positively charged) residues that electrostatically interact withnegatively charged phospholipids to aid in membrane insertion.The NSP4 viroporin domain is responsible for the release of ERcalcium into the cytoplasm, and NSP4 expression alone is suffi-cient to initiate NSP4/LC3 puncta formation (12). Mutation of theamphipathic α-helix of NSP4 that forms the pore lumen on olig-omerization abrogates both ER calcium release and the formationof NSP4/LC3 puncta; however, treatment of viroporin mutant-expressing cells with TG triggered puncta formation. Althoughthe NSP4 viroporin and TG both elevate cytosolic calcium, themechanisms by which this occurs are very different. NSP4 increasesER membrane permeability to calcium without blocking SERCA-mediated calcium uptake (27, 28). In contrast, Michelangeli et al.(29) reported that TG treatment reduces virus yield due to im-proper VP7 assembly onto particles, which was attributed to TGblocking calcium uptake rather than the indirect elevation in cy-tosolic calcium. Our data show that NSP4 viroporin-mediatedincreases in cytoplasmic calcium activate a specific calcium-inducedsignaling pathway to initiate autophagy, which enables traffickingof NSP4 and VP7 to viroplasms for subsequent virus assembly.Rotavirus-initiated autophagy involves the release of ER cal-
cium by ER-localized NSP4 to elevate [Ca2+]cyto, thereby acti-vating CaMKK-β that phosphorylates AMPK to initiate autophagy.Phosphorylated AMPK can either negatively regulate the mTORcomplex (mTORC1) or directly phosphorylate ULK1, both ofwhich can initiate the autophagy process (30, 31). Formation ofendogenous LC3-containing autophagy-like membranes requiresPI3K activity, as well as the autophagy-initiation proteins Atg3 andAtg5, indicating that these membranes are autophagy membranes.By confocal microscopy, NSP4 and LC3 form small and sub-sequently larger NSP4/LC3 puncta that eventually surround viro-plasms. Our results are consistent with reports that the ER isa source of autophagy membranes (32) because NSP4 is initiallysynthesized as an ER transmembrane glycoprotein.Rotavirus-induced autophagosome-like vesicles fail to fuse
with lysosomes in rotavirus-infected cells, indicating that rotavi-rus infection not only initiates the autophagy process but sup-presses autophagy maturation. Inhibiting autophagy maturationmay help rotavirus evade the antiviral function of autophagy.Suppression of autophagy vesicle maturation has been reportedfor influenza A virus and HIV (33, 34). Influenza A M2 and HIVNef viral proteins interact with beclin to suppress the fusion ofautophagosomes and lysosomes. Although it is unknown whetherrotavirus proteins interact with autophagy proteins to suppressautophagy maturation, rotavirus encodes two proteins, NSP4 andNSP2, that depolymerize the microtubule network (35, 36).
Microtubules are required for autophagosome trafficking (37).The poliovirus 3A protein disrupts microtubules, which inhibitsautophagosome-like vesicle trafficking (7). However, poliovirusinduces autophagosome-like vesicles that are decorated by lipi-dated LC3 and the lysosomal marker protein LAMP1, indicatingthat these vesicles have fused with lysosomes (6). Future studieswill determine whether disruption of microtubules or anothermechanism is responsible for the lack of fusion between theautophagosome-like puncta surrounding viroplasms and lyso-somes in rotavirus-infected cells.RNA viruses, such as picornaviruses (poliovirus, coxsackievirus,
and rhinovirus) and flaviviruses (hepatitis C virus and dengue vi-rus), induce autophagy to rearrange membranes as a scaffold forgenome replication (8, 38, 39). Induction of autophagy by rapa-mycin, the pharmacological inhibitor of mTOR, favors replicationof these viruses (6, 40, 41). In contrast, rotavirus RNA replication,which occurs in viroplasms, does not use autophagy membranes asa surface for genome replication, and treatment of cells withrapamycin does not affect virus replication. Instead, rotavirus ac-tively induces autophagy membrane formation and hijacks themembrane trafficking pathway of autophagy to transport the ER-associated proteins NSP4 and VP7 to viroplasms, the site ofgenome replication and immature particle formation. The in-teraction of NSP4 with immature particles at the interface be-tween viroplasms and NSP4/LC3-containing membranes triggersparticle budding through these membranes that facilitates the as-sembly of outer capsid proteins to form the mature infectiousparticles (11). Thus, rotavirus manipulates the autophagy mem-brane trafficking process to acquire membranes required for in-fectious particle assembly, yet the mechanisms and factors thatfacilitate NSP4/LC3 trafficking remain unknown. This is a uniqueexample of a virus hijacking autophagy membranes for capsidprotein trafficking to the site of virus assembly.A number of viruses (picornaviruses: poliovirus, rhinovirus,
coxsackievirus, and hepatitis B virus) induce autophagy as well asincrease [Ca2+]cyto, leading to the question: Is virus-mediatedelevation in [Ca2+]cyto a common mechanism for the inductionof autophagy? The mechanism(s) used by these viruses to induceautophagy is largely unknown; however, all encode one or moreproteins that increase [Ca2+]cyto from ER or Golgi [Ca2+]stores, or increase storage-operated calcium entry (42, 43). Thus,CaMKK-β activation through elevated [Ca2+]cyto may be acommon mechanism for these viruses to induce autophagy. Fu-ture studies will determine if these pathogens induce autophagyby activating CaMKK-β and the potential for STO-609 to bea broadly acting antiviral drug.
Materials and MethodsCells and Virus. Rotavirus SA114F (G3P6[1]) was cultivated in fetal rhesusmonkey kidney (MA104) cells in the presence of trypsin as previously de-scribed (44). Immortalized Atg5−/− and Atg5+/+ (45) or Atg3−/− and Atg3+/+
mouse embryonic fibroblasts (46) were cultivated in DMEM containing 10%FBS, as well as 1× essential and nonessential amino acids (Invitrogen). Virusin cells and media was titered by fluorescent focus assay as previously de-scribed (47).
Antibodies and Chemicals. The antibodies against NSP4 used in this study weremouse monoclonal antibody B4-2/55/17(1)/13 and rabbit peptide-specificantibody αNSP4114–135 (48). A guinea pig hyperimmune serum preparedagainst NSP5 purified from Escherichia coli as previously described (49) wasgenerated by Cocalico Biologicals, Inc. Monoclonal antibody to VP7 (mAb 60)was kindly provided by H. B. Greenberg (Stanford University School ofMedicine, Palo Alto, CA). LC3 antibody was obtained from Novus, andGAPDH antibody was obtained from Chemicon (mouse mAb 374). SecondaryAlexa Fluor 488-, 568-, and 633-conjugated antibodies were obtained fromInvitrogen. Lysotracker, BAPTA-AM, and TG were obtained from Invitrogen;bafilomycin A was obtained from Santa Cruz Biotechnology; 3-MA wasobtained from Sigma; and STO-609 was obtained from Tocris.
Fig. 7. Inhibition of autophagy by STO-609 hinders NSP4 and VP7 traf-ficking to viroplasms. Rotavirus-infected MA104 cells were cultured in theabsence [(−)STO-609, Upper] or presence [(+)STO-609, Lower] of 50 μM STO-609. At 7 hpi, cells were fixed, permeabilized, and stained with antibodyagainst NSP5 to detect viroplasms (teal), VP7 (green), or NSP4 (red). Thenuclei were detected by DAPI. (Scale bar: 5 μm.)
Crawford et al. PNAS | Published online November 26, 2012 | E3411
MICRO
BIOLO
GY
PNASPL
US

Transient Transfection. MA104 cells were transfected with the mRFP-GFP-LC3plasmid (Addgene), or Atg5−/− cells were transfected with the plasmids (p)pEGFP-C1-mApg5 (WT Atg5-EGFP) expressing mouse WT Atg5 or withpEGFP-C1-mApg5(K130R) (Atg5-EGFP K130R) expressing a mutant Atg5(Addgene) (20) as previously described (12). The transfection efficiencieswere between 50% and 60% as assessed by EGFP expression before in-fection. Cells were infected with SA114F rotavirus 24 h posttransfection.
For siRNA experiments, 75 pmol of annealed duplex siRNA (DharmaconResearch) for the SA11 clone 3 gene 10 siRNA sequence, AAGCCACAGU-CAGCCAUAUCG, or for Scr control, AAGCGGCCCUCCAAAGCCAAA, wastransfected intoMA104 cells using Lipofectamine 2000 (Invitrogen) (50). Cellswere infected 72 h after transfection with SA11 clone 3 rotavirus orwere mock-infected.
Western Blot Analysis. Samples were analyzed by Western blot as previouslydescribed (47). Quantification of bands on Western blots was performedusing ImageJ (National Institutes of Health).
Confocal Microscopy. Confocal microscopy was performed as previously de-scribed (51).
Puncta Formation Assay. MA104 cells on coverslips were transfected withplasmids expressing WT NSP4-EGFP or NSP4-ASDASA-EGFP (23) as previouslydescribed (12). MA104 cells were either loaded with 50 μM BAPTA-AM at 4 hposttransfection or maintained in normal medium. At ∼20 h posttransfection,
a subset of the transfected cells was treated with 1 μM TG for 3 h, and all thecells were then fixed, permeabilized, and stained as described above. Toquantitate the number of cells containing diffuse or punctate NSP4-EGFP orNSP4-ASDASA-EGFP and colocalization with LC3, random fields from twoexperiments were imaged by confocal microscopy and the images werecollected. Cells with a reticular NSP4-EGFP distribution were scored as havingno puncta. The majority of cells with either NSP4 alone or NSP4/LC3 punctacontained between 7 and 35 puncta; however, NSP4-ASDASA-EGFP–expressingcells with the presence of even a single punctate structure were scored ashaving puncta.
Trypan Blue Staining. To check cell viability, cells were stained with 0.4%trypan blue at room temperature for 5 min.
Statistical Analysis. Statistical differences between groups were determinedusing a two-tailed Student t test or Fisher’s exact test. P values of <0.05 wereconsidered significant.
ACKNOWLEDGMENTS. This study was supported by National Institutes ofHealth Public Health Service Award R01 AI080656. Funding for imaging wasprovided by Specialized Cooperative Centers Program in Reproduction(SCCPR) Grant U54 HD-007495 (to B. W. O’Malley); Grant P30 DK-56338,which funds the Texas Medical Center Digestive Diseases Center (to M.K.E.);and Grant P30 CA-125123 (to C. K. Osborne). Funding was also provided bythe Dan L. Duncan Cancer Center of Baylor College of Medicine.
1. Levine B, Mizushima N, Virgin HW (2011) Autophagy in immunity and inflammation.Nature 469(7330):323–335.
2. Dreux M, Chisari FV (2010) Viruses and the autophagy machinery. Cell Cycle 9(7):1295–1307.
3. Levine B, Kroemer G (2008) Autophagy in the pathogenesis of disease. Cell 132(1):27–42.
4. Mizushima N, Yoshimori T, Levine B (2010) Methods in mammalian autophagyresearch. Cell 140(3):313–326.
5. Itakura E, Mizushima N (2010) Characterization of autophagosome formation site bya hierarchical analysis of mammalian Atg proteins. Autophagy 6(6):764–776.
6. Jackson WT, et al. (2005) Subversion of cellular autophagosomal machinery by RNAviruses. PLoS Biol 3(5):e156.
7. Taylor MP, Burgon TB, Kirkegaard K, Jackson WT (2009) Role of microtubules inextracellular release of poliovirus. J Virol 83(13):6599–6609.
8. Wong J, et al. (2008) Autophagosome supports coxsackievirus B3 replication in hostcells. J Virol 82(18):9143–9153.
9. DreuxM,Gastaminza P,Wieland SF, Chisari FV (2009) The autophagymachinery is requiredto initiate hepatitis C virus replication. Proc Natl Acad Sci USA 106(33):14046–14051.
10. Sir D, et al. (2008) Induction of incomplete autophagic response by hepatitis C virusvia the unfolded protein response. Hepatology 48(4):1054–1061.
11. Estes MK, Kapikian AZ (2007) Fields Virology, eds Knipe DM, Howley PM (LippincottWilliam & Wilkins, Philadelphia), pp 1917–1974.
12. Berkova Z, et al. (2006) Rotavirus NSP4 induces a novel vesicular compartmentregulated by calcium and associated with viroplasms. J Virol 80(12):6061–6071.
13. Bagchi P, et al. (2010) Rotavirus nonstructural protein 1 suppresses virus-inducedcellular apoptosis to facilitate viral growth by activating the cell survival pathwaysduring early stages of infection. J Virol 84(13):6834–6845.
14. Dutta D, et al. (2009) The molecular chaperone heat shock protein-90 positivelyregulates rotavirus infection. Virology 391(2):325–333.
15. Halasz P, Holloway G, Coulson BS (2010) Death mechanisms in epithelial cellsfollowing rotavirus infection, exposure to inactivated rotavirus or genometransfection. J Gen Virol 91(Pt 8):2007–2018.
16. Caro LH, Plomp PJ, Wolvetang EJ, Kerkhof C, Meijer AJ (1988) 3-Methyladenine, aninhibitor of autophagy, has multiple effects on metabolism. Eur J Biochem 175(2):325–329.
17. Punnonen EL, Marjomäki VS, Reunanen H (1994) 3-Methyladenine inhibits transportfrom late endosomes to lysosomes in cultured rat and mouse fibroblasts. Eur J CellBiol 65(1):14–25.
18. Herbst S, Schaible UE, Schneider BE (2011) Interferon gamma activated macrophageskill mycobacteria by nitric oxide induced apoptosis. PLoS ONE 6(5):e19105.
19. Xue L, Borutaite V, Tolkovsky AM (2002) Inhibition of mitochondrial permeabilitytransition and release of cytochrome c by anti-apoptotic nucleoside analogues.Biochem Pharmacol 64(3):441–449.
20. Mizushima N, et al. (2001) Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol 152(4):657–668.
21. Geng J, Klionsky DJ (2008) The Atg8 and Atg12 ubiquitin-like conjugation systems inmacroautophagy. ‘Protein modifications: Beyond the usual suspects’ review series.EMBO Rep 9(9):859–864.
22. Høyer-Hansen M, Jäättelä M (2007) Connecting endoplasmic reticulum stress to auto-phagy by unfolded protein response and calcium. Cell Death Differ 14(9):1576–1582.
23. Hyser JM, Collinson-Pautz MR, Utama B, Estes MK (2010) Rotavirus disrupts calciumhomeostasis by NSP4 viroporin activity. MBio 1(5):pii: e00265-10.
24. Høyer-Hansen M, et al. (2007) Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell 25(2):193–205.
25. Tokumitsu H, et al. (2002) STO-609, a specific inhibitor of the Ca(2+)/calmodulin-dependent protein kinase kinase. J Biol Chem 277(18):15813–15818.
26. Tokumitsu H, Inuzuka H, Ishikawa Y, Kobayashi R (2003) A single amino acid differencebetween alpha and beta Ca2+/calmodulin-dependent protein kinase kinase dictatessensitivity to the specific inhibitor, STO-609. J Biol Chem 278(13):10908–10913.
27. Tian P, et al. (1994) The nonstructural glycoprotein of rotavirus affects intracellularcalcium levels. J Virol 68(1):251–257.
28. Tian P, et al. (1995) The rotavirus nonstructural glycoprotein NSP4 mobilizes Ca2+from the endoplasmic reticulum. J Virol 69(9):5763–5772.
29. Michelangeli F, Liprandi F, Chemello ME, Ciarlet M, Ruiz MC (1995) Selectivedepletion of stored calcium by thapsigargin blocks rotavirus maturation but not thecytopathic effect. J Virol 69(6):3838–3847.
30. Mizushima N (2010) The role of the Atg1/ULK1 complex in autophagy regulation. CurrOpin Cell Biol 22(2):132–139.
31. Kim J, Kundu M, Viollet B, Guan KL (2011) AMPK and mTOR regulate autophagythrough direct phosphorylation of Ulk1. Nat Cell Biol 13(2):132–141.
32. Axe EL, et al. (2008) Autophagosome formation from membrane compartmentsenriched in phosphatidylinositol 3-phosphate and dynamically connected to theendoplasmic reticulum. J Cell Biol 182(4):685–701.
33. Kyei GB, et al. (2009) Autophagy pathway intersects with HIV-1 biosynthesis andregulates viral yields in macrophages. J Cell Biol 186(2):255–268.
34. Gannagé M, et al. (2009) Matrix protein 2 of influenza A virus blocks autophagosomefusion with lysosomes. Cell Host Microbe 6(4):367–380.
35. Yang W, McCrae MA (2012) The rotavirus enterotoxin (NSP4) promotes re-modelingof the intracellular microtubule network. Virus Res 163(1):269–274.
36. Martin D, Duarte M, Lepault J, Poncet D (2010) Sequestration of free tubulinmolecules by the viral protein NSP2 induces microtubule depolymerization duringrotavirus infection. J Virol 84(5):2522–2532.
37. Jahreiss L, Menzies FM, Rubinsztein DC (2008) The itinerary of autophagosomes: Fromperipheral formation to kiss-and-run fusion with lysosomes. Traffic 9(4):574–587.
38. Kirkegaard K (2009) Subversion of the cellular autophagy pathway by viruses. CurrTop Microbiol Immunol 335:323–333.
39. Prentice E, Jerome WG, Yoshimori T, Mizushima N, Denison MR (2004) Coronavirusreplication complex formation utilizes components of cellular autophagy. J Biol Chem279(11):10136–10141.
40. McLean JE, Wudzinska A, Datan E, Quaglino D, Zakeri Z (2011) Flavivirus NS4A-induced autophagy protects cells against death and enhances virus replication. J BiolChem 286(25):22147–22159.
41. Huang SC, Chang CL, Wang PS, Tsai Y, Liu HS (2009) Enterovirus 71-induced autophagydetected in vitro and in vivo promotes viral replication. J Med Virol 81(7):1241–1252.
42. de Jong AS, et al. (2008) Functional analysis of picornavirus 2B proteins: Effectson calcium homeostasis and intracellular protein trafficking. J Virol 82(7):3782–3790.
43. Yang B, Bouchard MJ (2012) The hepatitis B virus X protein elevates cytosolic calciumsignals by modulating mitochondrial calcium uptake. J Virol 86(1):313–327.
44. Estes MK, Graham DY, Gerba CP, Smith EM (1979) Simian rotavirus SA11 replication incell cultures. J Virol 31(3):810–815.
45. Alexander DE, Ward SL, Mizushima N, Levine B, Leib DA (2007) Analysis of the role ofautophagy in replication of herpes simplex virus in cell culture. J Virol 81(22):12128–12134.
46. Sou YS, et al. (2008) The Atg8 conjugation system is indispensable for proper develop-ment of autophagic isolation membranes in mice. Mol Biol Cell 19(11):4762–4775.
47. Crawford SE, et al. (2001) Trypsin cleavage stabilizes the rotavirus VP4 spike. J Virol75(13):6052–6061.
E3412 | www.pnas.org/cgi/doi/10.1073/pnas.1216539109 Crawford et al.

48. Hyser JM, Zeng CQ, Beharry Z, Palzkill T, Estes MK (2008) Epitope mapping and use ofepitope-specific antisera to characterize the VP5* binding site in rotavirus SA11 NSP4.Virology 373(1):211–228.
49. Jiang X, et al. (2006) Cryoelectron microscopy structures of rotavirus NSP2-NSP5and NSP2-RNA complexes: Implications for genome replication. J Virol 80(21):10829–10835.
50. Berkova Z, Crawford SE, Blutt SE, Morris AP, Estes MK (2007) Expression of rotavirusNSP4 alters the actin network organization through the actin remodeling proteincofilin. J Virol 81(7):3545–3553.
51. Hyser JM, Utama B, Crawford SE, Estes MK (2012) Genetic divergence of rotavirusnonstructural protein 4 results in distinct serogroup-specific viroporin activity andintracellular punctate structure morphologies. J Virol 86(9):4921–4934.
Crawford et al. PNAS | Published online November 26, 2012 | E3413
MICRO
BIOLO
GY
PNASPL
US


















