

Active Site Engineering of ω-Transaminase for Production of ...
44
1 (Enzymology and Protein Engineering) 1 2 Active Site Engineering of ω-Transaminase for Production of Unnatural 3 Amino Acids Carrying a Side Chain Bulkier Than an Ethyl Substituent 4 5 6 Sang-Woo Han, Eul-Soo Park, Joo-Young Dong and Jong-Shik Shin* 7 Department of Biotechnology, Yonsei University, Seoul 120-749, South Korea 8 9 10 *To whom correspondence should be addressed. 11 Engineering Building 2 (Rm 507), Yonsei University, Shinchon-Dong 134, Seodaemun-Gu, 12 Seoul 120-749, South Korea (E-mail: [email protected], Tel: (+82) 2-2123-5884, Fax: 13 (+82) 2-362-7265) 14 15 16 17 Running Title: Engineering substrate specificity of ω-transaminase 18 AEM Accepted Manuscript Posted Online 31 July 2015 Appl. Environ. Microbiol. doi:10.1128/AEM.01533-15 Copyright © 2015, American Society for Microbiology. All Rights Reserved. on February 9, 2018 by guest http://aem.asm.org/ Downloaded from
Transcript of Active Site Engineering of ω-Transaminase for Production of ...

1
(Enzymology and Protein Engineering) 1
2
Active Site Engineering of ω-Transaminase for Production of Unnatural 3
Amino Acids Carrying a Side Chain Bulkier Than an Ethyl Substituent 4
5
6
Sang-Woo Han, Eul-Soo Park, Joo-Young Dong and Jong-Shik Shin* 7
Department of Biotechnology, Yonsei University, Seoul 120-749, South Korea 8
9
10
*To whom correspondence should be addressed. 11
Engineering Building 2 (Rm 507), Yonsei University, Shinchon-Dong 134, Seodaemun-Gu, 12
Seoul 120-749, South Korea (E-mail: [email protected], Tel: (+82) 2-2123-5884, Fax: 13
(+82) 2-362-7265) 14
15
16
17
Running Title: Engineering substrate specificity of ω-transaminase18
AEM Accepted Manuscript Posted Online 31 July 2015Appl. Environ. Microbiol. doi:10.1128/AEM.01533-15Copyright © 2015, American Society for Microbiology. All Rights Reserved.
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from

2
ABSTRACT 19
ω-Transaminase (ω-TA) is a promising enzyme for production of unnatural amino acids from 20
keto acids using cheap amino donors such as isopropylamine. The small substrate binding 21
pocket of most ω-TAs permits entry of substituents no larger than an ethyl group, which 22
presents a significant challenge to the preparation of structurally diverse unnatural amino acids. 23
Here we report engineering of an (S)-selective ω-TA from Ochrobactrum anthropi (OATA) to 24
reduce the steric constraint and thereby allow the small pocket to readily accept bulky 25
substituents. Based on a docking model using L-alanine as a ligand, nine active site residues 26
were selected for alanine scanning mutagenesis. Among the resulting variants, a L57A variant 27
showed dramatic activity improvements for α-keto acids and α-amino acids carrying up to a n-28
butyl substituent (e.g. 48 and 56-fold activity increases for 2-oxopentanoic acid and L-norvaline, 29
respectively). A L57G mutation also relieved the steric constraint but much less than the L57A 30
mutation did. In contrast, a L57V substitution failed to induce the activity improvements for 31
bulky substrates. Molecular modeling suggested that the alanine substitution of L57, located in 32
a large pocket, induces an altered binding orientation of an α-carboxyl group and thereby 33
provides more room to the small pocket. Synthetic utility of the L57A variant was demonstrated 34
by carrying out production of optically pure L and D-norvaline (i.e. ee > 99 %) by asymmetric 35
amination of 2-oxopantanoic acid and kinetic resolution of racemic norvaline, respectively. 36
37
38
39
40
41
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from

3
INTRODUCTION 42
Unnatural amino acids are widely used as essential chiral building blocks for diverse 43
pharmaceutical drugs, agrochemicals and chiral ligands (1-3). In contrast to natural amino acids, 44
a fermentative method is not yet commercially available for production of the unnatural amino 45
acids (4, 5). This has driven development of various biocatalytic approaches to afford scalable 46
processes for preparing enantiopure unnatural amino acids, which includes kinetic resolution of 47
racemic amino acids using acylase (6, 7), amidase (8, 9), hydantoinase (10, 11) and amino acid 48
oxidase (12, 13), and asymmetric reductive amination of keto acids using dehydrogenase (14, 49
15) and transaminase (16, 17). The asymmetric amination is usually favoured over the kinetic 50
resolution because the former affords 100 % yield without racemization of an unwanted 51
enantiomer. 52
In contrast to a mandatory requirement of supply and regeneration of an expensive external 53
cofactor for the dehydrogenase reactions, transaminase catalyzes transfer of an amino group 54
from an amino donor to an acceptor using pyridoxal 5′-phosphate (PLP) as a prosthetic group 55
(18). Nevertheless, industrial implementation of the transaminase-mediated synthesis of 56
unnatural amino acids has lagged behind because of low equilibrium constants, usually close to 57
unity, for the reactions between keto acids and amino acids. However, recent studies have 58
demonstrated that reductive amination of keto acids can be driven to completion without the 59
thermodynamic limitation by employing cheap amines as a cosubstrate when the transfer of an 60
amino group can be mediated by ω-transaminase (ω-TA) that utilizes primary amines as an 61
amino donor (19-21). For example, the equilibrium constant for amination of pyruvic acid by 62
isopropylamine was reported to be 67 (22). Therefore, use of 1.5 molar equivalent of 63
isopropylamine relative to pyruvic acid affords 97 % theoretical conversion of pyruvic acid to L 64
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from

4
or D-alanine, depending on the stereoselectivity of ω-TA. Moreover, acetone (i.e. the 65
deamination product of isopropylamine) is highly volatile and the resulting equilibrium shift by 66
facile evaporation can drive even thermodynamically unfavorable reactions to completion as 67
demonstrated elsewhere with reductive amination of ketones (23-25). Another advantage of 68
using isopropylamine as an amino donor is that most ω-TAs show low activities for acetone 69
(26), which minimizes enzyme inhibition by the ketone product. Taken together, ω-TA-70
catalyzed asymmetric amination of keto acids using isopropylamine is a promising strategy for 71
scalable production of unnatural amino acids. 72
One crucial bottleneck for synthesizing diverse unnatural amino acids using ω-TAs is very 73
narrow substrate specificity of the enzyme toward keto acids. It is generally accepted that both 74
(R) and (S)-selective ω-TAs possess two substrate-binding pockets consisting of a large pocket 75
capable of dual recognition of hydrophobic and carboxyl groups and a small pocket that can 76
accept substituents no larger than an ethyl group (27, 28). The canonical structural features of 77
the active site of ω-TAs render a carboxyl group and a side chain of keto acid substrates bound 78
to the large and small pockets, respectively. This leads all known ω-TAs to accept a limited 79
range of α-keto acids such as glyoxylic acid, pyruvic acid and 2-oxobutyric acid. The only 80
exception to the canonical substrate specificity to date is an (S)-selective ω-TA from 81
Paracoccus denitrificans that can accommodate up to a n-butyl substituent of α-keto acid in the 82
small pocket (29). We demonstrated that a single point mutation in the small pocket (i.e. 83
V153A) endowed the ω-TA with a substantial activity toward even 2-oxooctanoic acid carrying 84
a n-hexyl side chain (29). Despite the non-canonical steric constraint of the ω-TA from P. 85
denitrificans, a low enzyme activity for isopropylamine renders a synthetic potential of the ω-86
TA less optimal for practical amination of the bulky keto acids (21). This led us to set out 87
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from

5
engineering of substrate specificity of the canonical ω-TAs showing a high activity for 88
isopropylamine. To this end, we chose an (S)-selective ω-TA from Ochrobactrum anthropi 89
(OATA) which showed 43 % activity for isopropylamine relative to that for (S)-α-90
methylbenzylamine ((S)-α-MBA) (21). 91
To date, there have been three reports dealing with engineering of the canonical steric 92
constraint in the small pocket of ω-TAs. In the first example, an (R)-selective ω-TA from 93
Arthrobacter sp. was successfully engineered to create a variant that can accept bulky arylalkyl 94
ketones as an amino acceptor (23). In our previous study, substrate specificity of the engineered 95
variant, harboring 27 amino acid substitutions, was examined using structurally diverse α-keto 96
acids (21). In spite of the remarkable activity improvements for bulky arylalkyl ketones, the 97
canonical steric constraint of a parental enzyme toward α-keto acids was not significantly 98
altered in the engineered variant, e.g. only 2 % reactivity of 2-oxopentanoic acid relative to 99
pyruvic acid. This result suggests that engineering of the steric constraint should be guided in 100
the context of a substrate type, especially substituents bound to the large pocket (i.e. 101
hydrophobic vs carboxyl). The second example was carried out with an (S)-selective ω-TA 102
from Vibrio fluvialis to improve an activity toward a β-keto ester (i.e. (R)-ethyl 5-methyl 3-103
oxooctanoate) (30). The resulting variant, carrying eight amino acid substitutions, showed a 60-104
fold activity increase for the target β-keto ester but led to a complete activity loss for keto acids. 105
The third example employed the same ω-TA as the one used in the second example and focused 106
on improving activities for arylalkyl ketones. To the best of our knowledge, active site 107
engineering of a ω-TA to relieve the canonical steric constraint for α-keto acids and α-amino 108
acids has not yet reported. 109
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from

6
In this study, we aimed at creating a variant of OATA capable of accommodating bulky α-110
keto acids. To this end, we performed substrate docking simulations to select key active site 111
residues and carried out alanine scanning mutagenesis to identify a hot spot responsible for the 112
narrow substrate specificity for α-keto acids. The resulting variant carrying only a single point 113
mutation in the large pocket showed desirable relaxation of the canonical steric constraint and 114
afforded efficient stereoselective amination of bulky α-keto acids using isopropylamine as an 115
amino donor. 116
MATERIALS AND METHODS 117
Chemicals 118
Pyruvic acid was obtained from Kanto Chemical Co. (Tokyo, Japan). Isopropylamine was 119
purchased from Junsei Chemical Co. (Tokyo, Japan). L-Alanine was purchased from Acros 120
Organics Co. (Geel, Belgium). All other chemicals were purchased from Sigma Aldrich Co. (St. 121
Louis, MO). Materials used for preparation of culture media including yeast extract, tryptone 122
and agar were purchased from BD Biosciences (Franklin Lakes, NJ). 123
Site-directed mutagenesis of ω-TA 124
Single point mutations of OATA were carried out using a QuikChange Lightning site-directed 125
mutagenesis kit (Agilent Technologies Co., Santa Clara, CA) according to an instruction 126
manual. The template used for the mutagenesis PCR was pET28-OATA which was previously 127
constructed (31). Mutagenesis primers were designed by a primer design program 128
(http://www.agilent.com). The primer sequences are provided in Table 1. Intended mutagenesis 129
was confirmed by DNA sequencing. 130
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from

7
Expression and purification of ω-TAs 131
Overexpression of His6-tagged ω-TAs was carried out as described previously with minor 132
modifications (32). Escherichia coli BL21(DE3) cells carrying the expression vectors (i.e. 133
pET28a(+) harboring the ω-TA gene) were cultivated in LB medium (typically 1 L) containing 134
50 μg/mL kanamycin. Protein expression was induced by IPTG at 0.4 OD600 and the cells were 135
allowed to grow for 10 h. The culture broth was centrifuged and the resulting cell suspension 136
was subjected to ultrasonic disruption. Protein purification was carried out as described 137
previously (32). Protein purity was confirmed by SDS-PAGE (Fig. S1). Molar concentrations 138
of the purified ω-TAs were determined by measuring UV absorbance at 280 nm. 139
Enzyme assay 140
All enzyme assays were carried out at 37 °C and pH 7 (50 mM phosphate buffer). Unless 141
otherwise specified, reaction conditions for the activity assay were 10 mM (S)-α-MBA and 10 142
mM pyruvic acid. Typical reaction volume was 50 μL. The enzyme reaction was stopped after 143
10 min by adding 300 µL acetonitrile. Acetophenone produced from the reactions was analyzed 144
by HPLC. For initial rate measurements, reaction conversions were limited less than 10 %. 145
Substrate specificity 146
To examine substrate specificity of the OATA variants, initial rate measurements (i.e. < 10 % 147
conversion) were independently triplicated. Reaction conditions to examine substrate 148
specificity for amino acceptors were 20 mM α-keto acid and 20 mM (S)-α-MBA in 50 mM 149
phosphate buffer (pH 7). Acetophenone produced was analyzed by HPLC. To measure 150
activities for α-amino acids, 20 mM amino donor and 20 mM propanal were used as substrates 151
and the α-keto acids produced were analyzed by HPLC. To measure amino donor activities for 152
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from

8
isopropylamine, reaction conditions were 20 mM isopropylamine and 20 mM pyruvic acid. L-153
alanine was analyzed by chiral HPLC after derivatization with a Marfey’s reagent (33, 34). 154
Molecular modeling 155
Using four X-ray structures of (S)-selective ω-TAs as templates, a homology model of OATA 156
was constructed by the Modeler module (version 9.8) of the Discovery Studio package (version 157
3.5.0, BIOVIA, San Diego, CA). X-ray structures used as templates were ω-TAs from P. 158
denitrificans (PDB ID: 4GRX) (35), Chromobacterium violaceum (4A6T) (36), Mesorhizobium 159
loti (3GJU) (37) and Rhodobacter sphaeroides (3I5T) (37). The outward-pointing arginine of 160
4GRX was set to be conserved in the homology model, resulting in a dimeric structure of 161
OATA in which each subunit has a different conformation of the active-site arginine. To 162
construct a holoenzyme structure, the PLP moiety was copied from 4A6T. Only 0.4 % non-163
glycine residues were found to lie in the disallowed region by a Ramachandran phi-psi analysis. 164
The active site models of the L57A and L57G variants were built by an amino acid 165
substitution and then energy minimization (2,000 steps; dielectric constant = 4) of the mutation 166
site, the internal aldimine and the neighboring residues within 3 Å from L57 (i.e. S55, G56, 167
L57, W58, S59, F82, H84 and T324) until the RMS gradient reached 0.1 kcal/mol/Å. 168
Docking simulations with L-alanine and L-norleucine as ligands were accomplished using 169
the CDOCKER module under a default setting within the active site defined by the Binding-170
Site module. The most stable docking pose was chosen as a docking model. 171
Kinetic analysis 172
A pseudo-one-substrate kinetic model was used to obtain apparent kinetic parameters for 173
pyruvic acid and 2-oxohexanoic acid as described previously (38). Concentration range of 174
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from

9
pyruvic acid for initial rate measurements was 0.5 - 5 mM. Concentration ranges of 2-175
oxohexanoic acid were 7 - 200 mM and 1 - 30 mM for the wild-type and the L57A variant, 176
respectively. Concentration of the cosubstrate (i.e. (S)-α-MBA) was fixed at 20 mM. 177
Acetophenone produced was analyzed by HPLC to measure the initial rates. The initial rate 178
data were fitted to a Michaelis-Menten equation, and the KM and kcat values were calculated 179
from the slopes and y-intercepts of the double-reciprocal plots. 180
Measurement of enzyme stability 181
To examine the effect of the L57A mutation on enzyme stability, 10 μM enzyme was incubated 182
in 50 mM potassium phosphate buffer (pH 7) at 37 oC. Aliquots of the enzyme solution were 183
sampled at predetermined incubation times and were mixed with reaction buffer containing 10 184
mM pyruvic acid and 10 mM (S)-α-MBA. Initial reaction rates were measured by analyzing 185
acetophenone produced. Inactivation constants were obtained by curve fitting of the residual 186
activity data to a single exponential function. 187
Enzyme reactions to produce unnatural amino acids 188
The reaction volume for the enzyme reactions was 1 mL and the reaction mixture in 50 mM 189
potassium phosphate buffer (pH 7) was incubated at 37 °C. Reaction conditions for asymmetric 190
synthesis of L-norvaline were 50 mM 2-oxopentanoic acid, 100 mM isopropylamine and 40 191
μM ω-TA. Synthesis of L-norleucine was carried out under the reaction conditions of 100 mM 192
2-oxohexanoic acid, 200 mM isopropylamine, 0.1 mM PLP and 200 μM ω-TA. Kinetic 193
resolution of racemic norvaline was performed at 50 mM rac-norvaline, 50 mM glyoxylic acid 194
and 40 μM ω-TA. Aliquots of the reaction mixture (50 μL) were taken at predetermined 195
reaction times and mixed with 10 μL HCl solution (5 N) to stop the reaction. The reaction 196
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from

10
mixtures were subjected to HPLC analysis for measurement of conversion and enantiomeric 197
excess. 198
HPLC analysis 199
All the HPLC analyses were performed on a Waters HPLC system (Milford, MA). Analysis of 200
acetophenone was performed using a Sunfire C18 column (Waters Co.) with isocratic elution of 201
60 % methanol/40 % water/0.1 % trifluoroacetic acid at 1 mL/min. UV detection was done at 202
254 nm. Quantitative chiral analyses of alanine, norvaline and norleucine were carried out using 203
a Crownpak CR(-) column (Daicel Co., Japan) or a Sunfire C18 column after chiral 204
derivatization with a Marfey’s reagent (33, 34). Κeto acids were analyzed using an Aminex 205
HPX-87H column (Bio-Rad, Hercules, CA) with isocratic elution of 5 mM H2SO4 solution at 206
0.5 mL/min. Column oven temperature was set to 40 oC and UV detection was done at 210 nm. 207
RESULTS AND DISCUSSION 208
Substrate docking 209
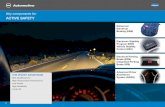
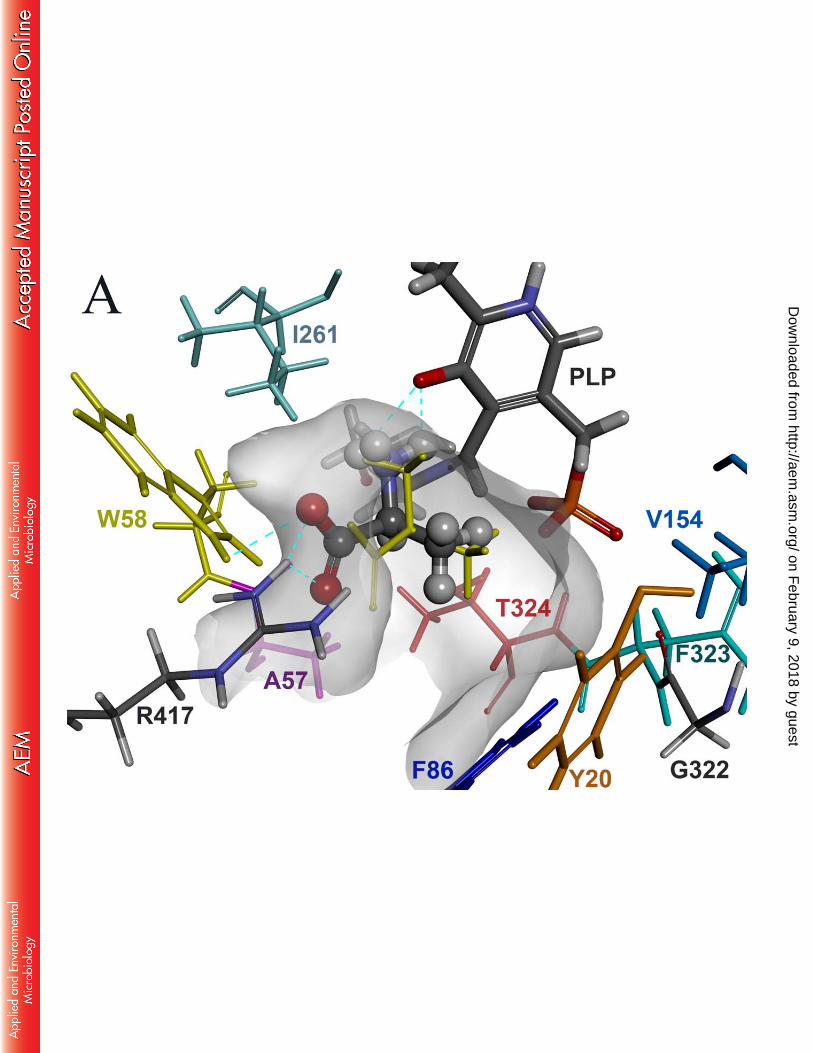
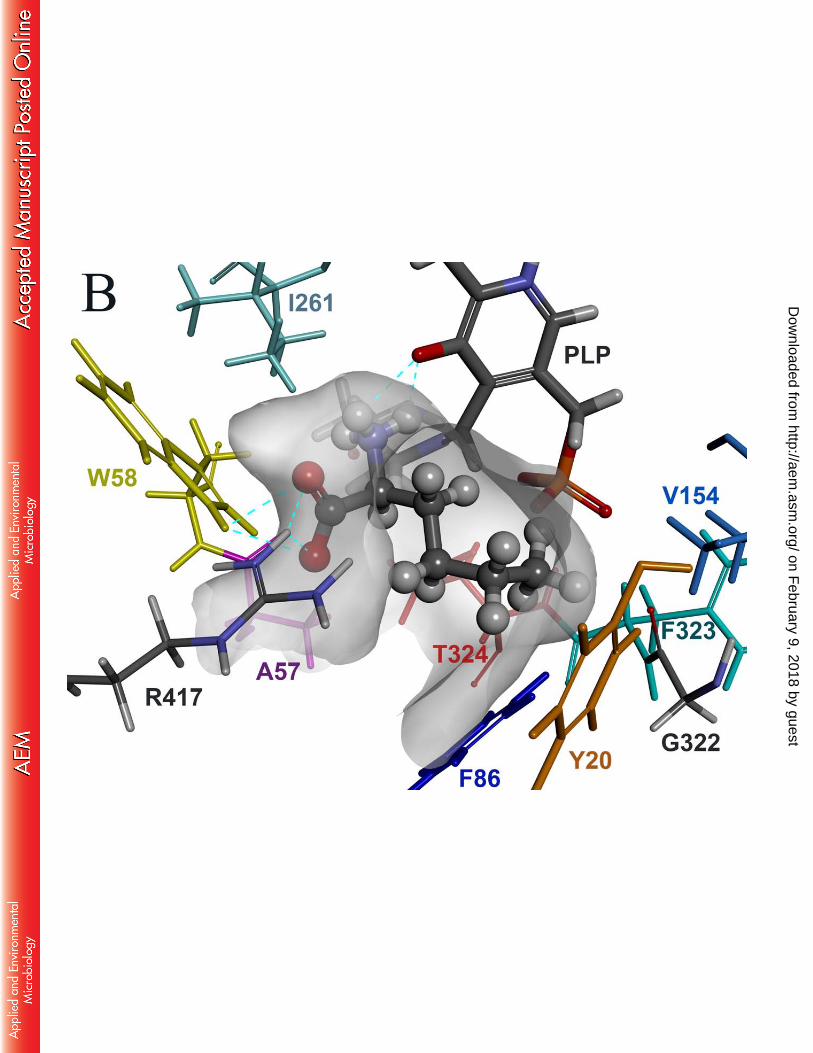
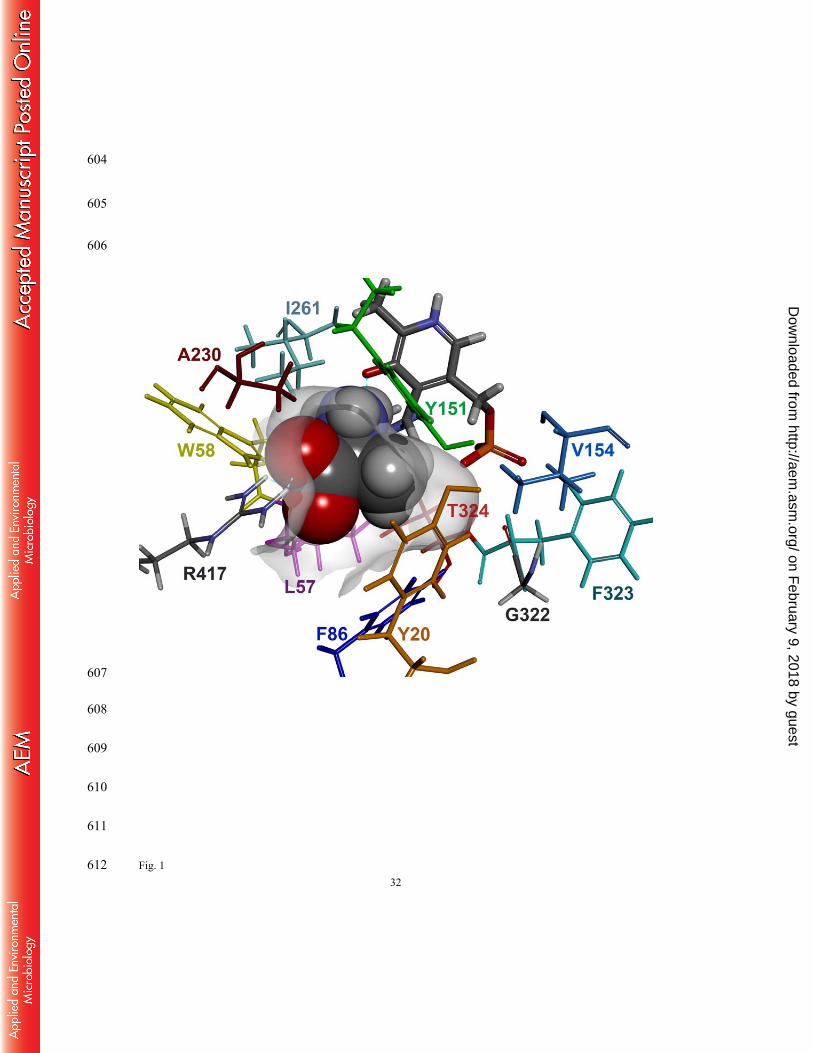
We set out to construct a homology model of OATA to identify key active site residues 210
responsible for rejecting entry of bulky substituents in the small pocket. X-ray structures used 211
as templates for the homology modeling were ω-TAs from P. denitrificans (PDB ID: 4GRX, 212
sequence identity: 41 %) (35), C. violaceum (4A6T, 41 %) (36), M. loti (3GJU, 43 %) (37) and 213
R. sphaeroides (3I5T, 34 %) (37). Sequence alignment of OATA against the template ω-TAs is 214
shown in Fig. S2. The four template ω-TAs adopt a homodimeric structure where both active 215
site arginines assume an inward conformation (i.e. pointing toward the large pocket), except 216
4GRX where one subunit harbors an outward arginine (i.e. pointing toward a solvent side). It is 217
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from

11
known that the active site arginine, responsible for recognition of a carboxylate of an incoming 218
substrate, undergoes a gross movement and assumes the outward conformation when the large 219
pocket is taken up by a hydrophobic substituent (39). 220
We performed substrate docking simulations using the subunit structure where the active 221
site arginine (i.e. R417) adopts the inward conformation (Fig. 1). For the docking simulation, 222
we used L-alanine, instead of pyruvic acid, as a ligand because all the active site lysines in the 223
template ω-TAs form a Schiff base with PLP (i.e. an internal aldimine capable of deamination 224
of an amino donor) and thereby the resulting homology model adopts the internal aldimine 225
structure. It is notable that binding of L-alanine is coordinated by a hydrogen bond between the 226
amino group of the substrate and the phenolic oxygen of the PLP moiety as well as multiple 227
hydrogen bonds between the α-carboxylate and the active site arginine. Compared to the active 228
site structure where R417 adopts an outward conformation, the large pocket becomes 229
contracted owing to the inward conformation of R417 to recognize the carboxyl group. The 230
small pocket accommodating the methyl group of L-alanine is wrapped by side chains of the 231
five active site residues (i.e. Y20, F86, Y151, V154 and T324), a phosphate group of the PLP 232
and a backbone chain ranging from G322 to T324. The Connolly surface of the active site 233
clearly explains why the small pocket can accept only up to an ethyl substituent. 234
235
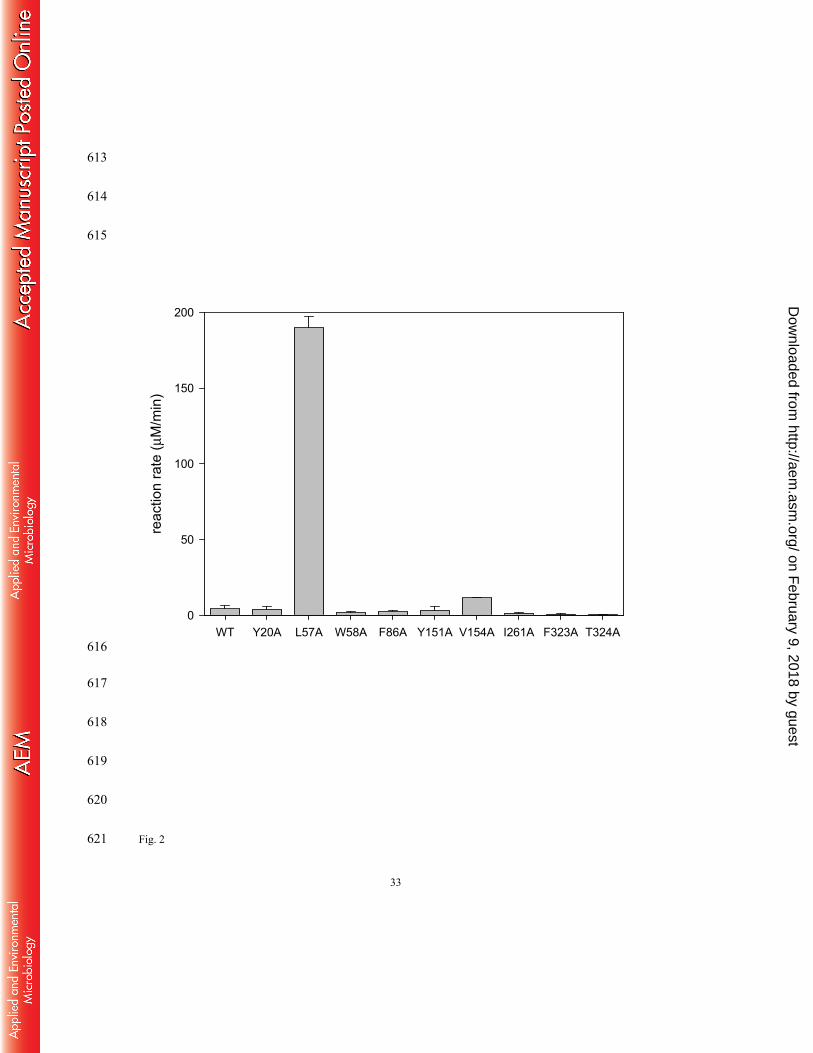
Alanine scanning mutagenesis of the active site residues 236
The docking model indentified thirteen active site residues that participate in the formation of 237
the active site surface and lie within a 7 Å distance from the bound L-alanine (i.e. Y20, L57, 238
W58, F86, Y151, V154, A230, I261, K287, G322, F323, T324 and R417). We carried out 239
alanine substitution of the active site residues one by one to examine size reduction of which 240
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from

12
residue led to generation of more room required for accepting bulky substituents in the small 241
pocket. A230 and G322 were excluded from the mutagenesis because alanine substitution of the 242
two residues could not generate more room in the active site. K287 and R417 were also 243
excluded because these are essential residues responsible for catalytic turnover and recognition 244
of a carboxyl group, respectively. Therefore, nine active site residues were subjected to the 245
alanine scanning mutagenesis and the enzyme activities of the resulting variants toward 2-246
oxopentanoic acid were measured (Fig. 2). Among the nine variants, a L57A variant showed an 247
exceptionally improved activity for 2-oxopentanoic acid (i.e. a 48-fold activity increase relative 248
to the wild-type enzyme). This was an unexpected result because the L57 residue participates in 249
the large pocket. Besides L57A, V154A was the only alanine substitution that allowed activity 250
improvement for 2-oxopentanoic acid (i.e. a 3-fold increase). This result is in line with our 251
previous observation where alanine substitution at the same position of the ω-TA from P. 252
denitrificans achieved excavation of the small pocket (29). 253
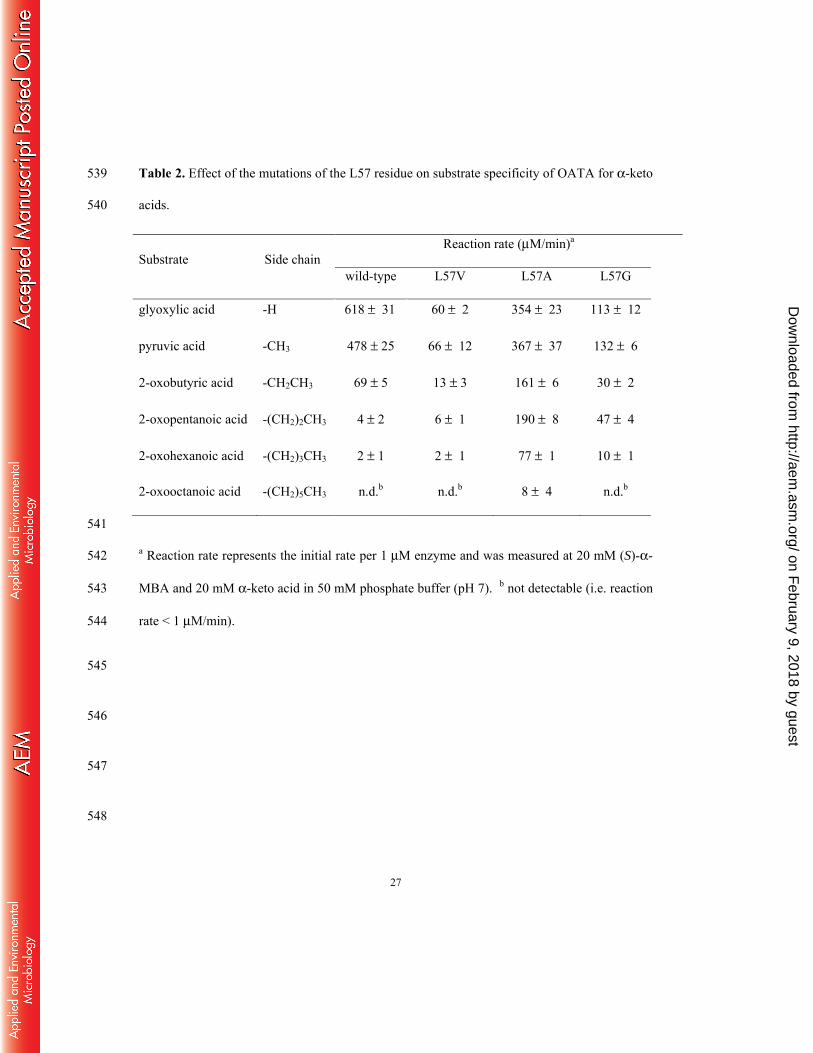
Site-directed mutagenesis of L57 254
We presumed that the remarkable activity improvement for 2-oxopentanoic acid by the L57A 255
mutation would result from altered binding of a carboxyl group owing to generation of more 256
room in the large pocket by the size reduction of L57. To examine whether a higher activity 257
improvement could be induced by substitution of L57 with small amino acids other than alanine, 258
we prepared L57V and L57G variants by site-directed mutagenesis. Enzyme activities for 259
various α-keto acids were measured in comparison with the wild-type enzyme and the L57A 260
variant (Table 2). The wild-type OATA showed negligible activities (i.e. < 1 % relative to 261
pyruvic acid) for bulky α-keto acids carrying a side chain larger than an ethyl group. The L57V 262
mutation did not induce any activity improvement for the bulky α-keto acids but caused drastic 263
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from

13
activity losses toward native substrates (i.e. from glyoxylic acid to 2-oxobutanoic acid). In 264
contrast, the L57A variant showed activity improvements for α-keto acids carrying side chains 265
larger than a methyl group (i.e. 2, 48 and 39-fold activity increases for 2-oxobutyric acid, 2-266
oxopentanoic acid and 2-oxohexanoic acid, respectively). The activity improvements for bulky 267
α-keto acids were so remarkable that the L57A variant exhibited a substantial activity even for 268
2-oxooctanoic acid (i.e. 2 % relative to pyruvic acid). The L57G substitution also induced the 269
activity improvements for bulky α-keto acids but to a much lesser degree than those observed 270
with the L57A substitution. Despite the activity improvements toward α-keto acids carrying 271
bulky linear alkyl groups, neither L57A nor L57G variant displayed substantial activities for 272
branched-chain α-keto acids, including 3-methyl-2-oxobutyric acid, 3-methyl-2-oxopentanoic 273
acid, 4-methyl-2-oxopentanoic acid and trimethylpyruvic acid (data not shown). 274
As shown in Table 3, the relaxed steric constraint for α-keto acids was also observed for 275
α-amino acids (i.e. 56 and 5-fold activity improvements of the L57A and L57G variants, 276
respectively, for L-norvaline). Despite the nondetectable activity of the wild-type OATA for L-277
norleucine, the relaxed steric constraint allowed the L57A and L57G variants to show 278
substantial activities for L-norleucine. The L57V mutation led to drastic activity decreases for 279
α-amino acids as also observed with α-keto acids. Interestingly, all the OATA variants in 280
addition to the wild-type showed very low activities for glycine despite the high activities for 281
the cognate keto acid. These extremely opposite activities for glycine and glyoxylic acid seem 282
to be ascribable to the thermodynamically unfavorable conversion of glycine to its keto acid 283
(40). As observed with branched-chain α-keto acids, both L57A and L57G variants showed 284
nondetectable activities for α-amino acids carrying branched-chain substituents, including L-285
valine, L-leucine, L-isoleucine and L-tert-leucine (data not shown). 286
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from

14
The synthetic utility of the L57A variant for asymmetric amination of bulky keto acids is 287
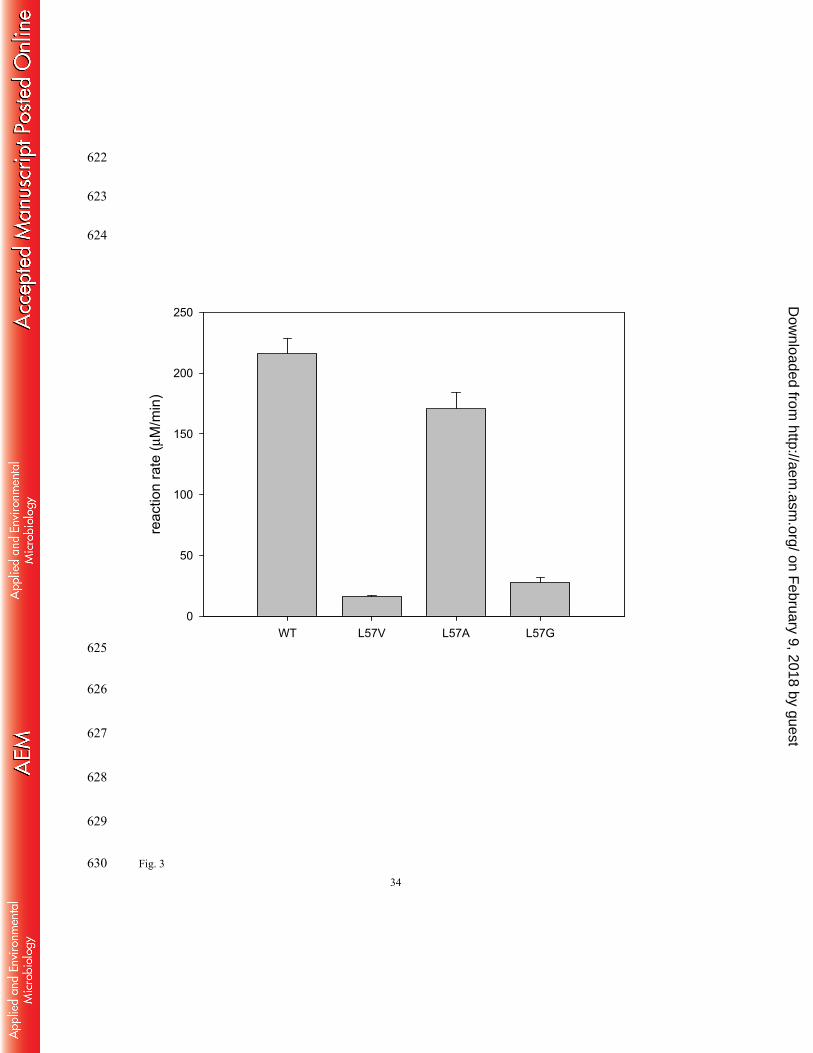
contingent upon high activity for isopropylamine as well as no loss in the parental 288
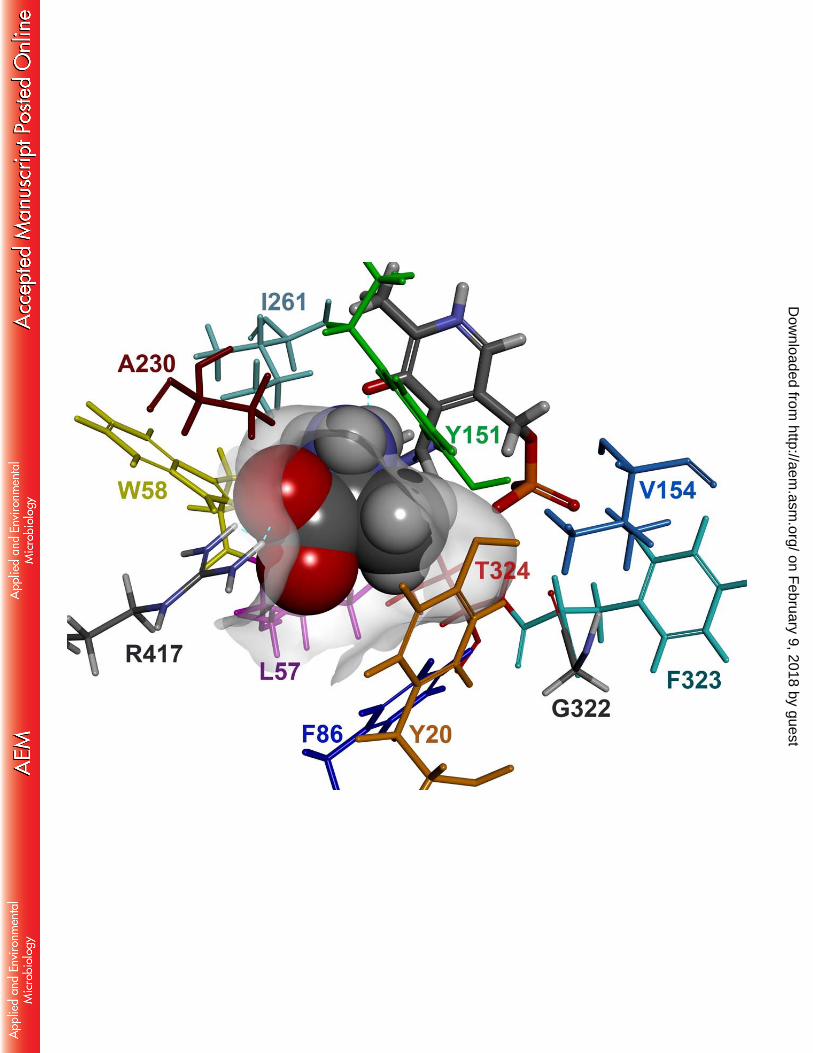
stereoselectivity. We measured amino donor reactivity of isopropylamine with the L57 variants 289
(Fig. 3). In contrast to L57V and L57G variants, the L57A variant conserved 80 % of a parental 290
activity for isopropylamine. In addition to the wild-type OATA, all the L57 variants showed 291
nondetectable formation of D-alanine during the transamination between isopropylamine and 292
pyruvic acid (i.e. ee of L-alanine produced > 99.9 %). This result indicates that the three amino 293
acid substitutions of L57 do not affect the stringent stereoselectivity of the parental enzyme. 294
295
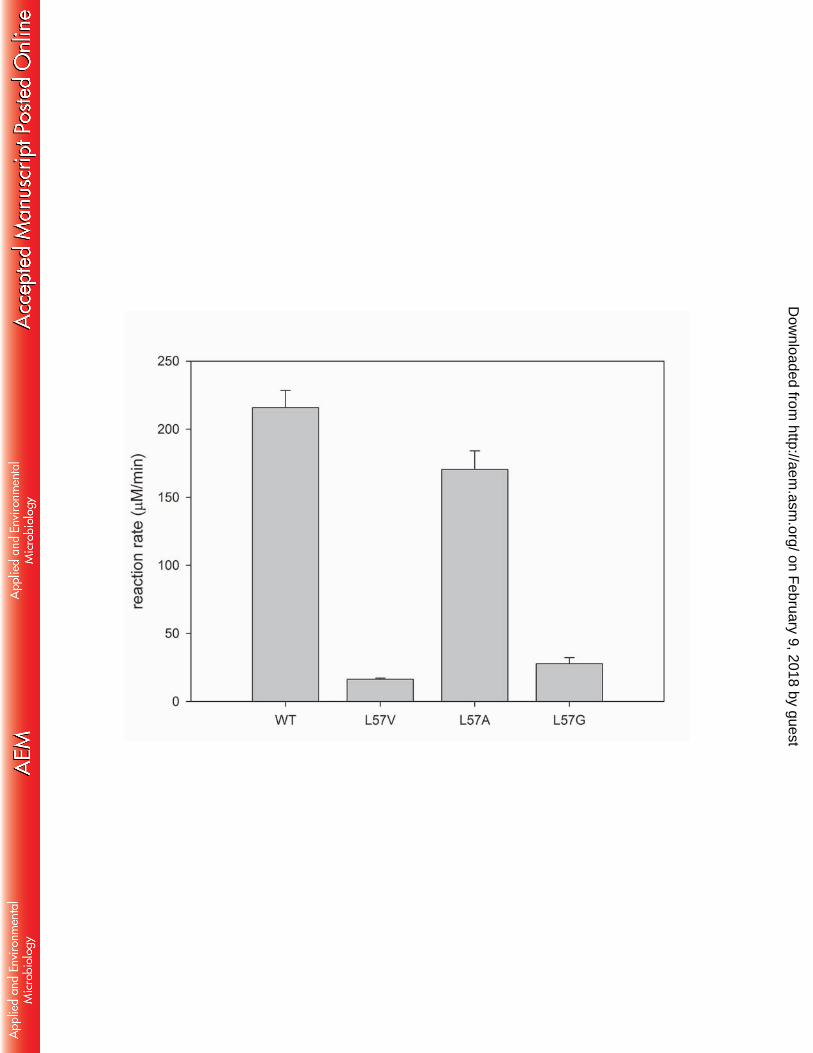
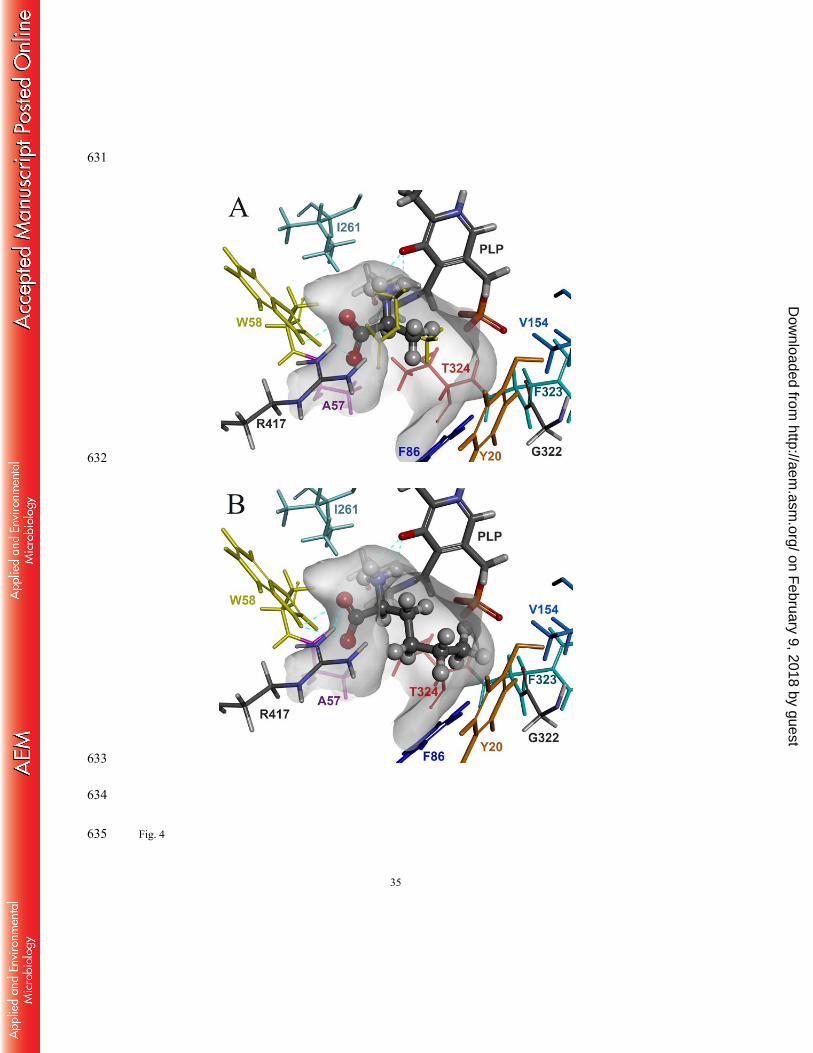
Structural modeling of the L57A variant 296
To provide a structural insight into how the L57A mutation relieved the steric constraint in the 297
small pocket, we performed docking simulation of the L57A variant using L-alanine as a ligand 298
(Fig. 4A). Compared to the docking pose of L-alanine in the wild-type active site, the L57A 299
mutation allowed docking of L-alanine slightly away from the small pocket with 0.71 and 0.73 300
Å translocations of the C-α and C-β of the substrate, respectively, to the large pocket. These 301
translocations are attributed to a 1.35 Å movement of the α-carboxyl carbon to the W58 residue 302
owing to the room created by the L57A substitution. The altered binding orientation of the α-303
carboxylate in the large pocket permits an additional hydrogen bond formation with the indole 304
group of W58, which led to a concurrent loss in one of the pre-existing hydrogen bonds with 305
R417. 306
The altered binding of the α-carboxylate in the active site of the L57A variant led to the 307
substrate docking more close to the large pocket, which provides more effective room to the 308
small pocket even without any structural change in the small pocket. In the substrate specificity 309
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from

15
experiments, we could test neither α-keto acid nor α-amino acid substrates carrying a n-pentyl 310
side chain because these substrates were not commercially available. To examine how large 311
substituent the extended small pocket can accommodate, we carried out docking simulation 312
using L-norleucine as a ligand (Fig. 4B). The docking model is consistent with the significant 313
enzyme activity for L-norleucine and predicts that the small pocket cannot accept a n-pentyl 314
group. The n-butyl group of the bound L-norleucine does not assume a staggered conformation 315
due to a steric interference with the small pocket residues, resulting in an intramolecular steric 316
strain of the bound substrate. This could explain the 87 % activity loss for L-norleucine 317
compared to L-alanine. 318
Contrary to the expectation, the L57G variant did not show higher activities toward 319
bulky substrates than the L57A variant did. To examine how much the L57G mutation allowed 320
the altered binding of the α-carboxyl group to the large pocket, we performed docking 321
simulation of L-norleucine in the active site of the L57G variant (Fig. S3). The docking pose of 322
L-norleucine in the L57G variant turned out very close to that generated with the L57A variant, 323
indicating that creation of a bigger room in the large pocket by the L57G mutation did not elicit 324
larger translocation of the α-carboxyl group toward the large pocket. This is in agreement with 325
the docking model of L-norleucine in the L57A variant where the α-carboxyl group is already 326
in a close contact with W58 and thereby further translocation of the α-carboxyl group is 327
prohibited because of a steric clash against W58 (Fig. 4B). It is presumable that the lower 328
activities of the L57G variant for bulky substrates, compared to the L57A variant, result from 329
increased conformation flexibility of the backbone chain which might be deleterious to a 330
catalytic step after formation of a Michaelis complex. This is supported by the drastic activity 331
losses of the L57G variant even for the native substrates such as glyoxylic acid, pyruvic acid 332
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from

16
and L-alanine. Note that two consecutive glycine residues happen to be created immediately 333
before W58 in the L57G variant because the 56th residue is glycine (Fig. S2). 334
Kinetic analysis of the L57A variant 335
To provide mechanistic understanding of how the L57A mutation improves activities for bulky 336
substrates, we compared kinetic parameters of the wild-type and the L57A variant for pyruvic 337
acid and 2-oxohexanoic acid (Table 4). Compared with the wild-type enzyme, the L57A variant 338
showed a 50 % reduction in the specificity constant (i.e. kcat/KM) for pyruvic acid. This activity 339
loss for the native substrate is caused by a 2.8-fold decrease in the binding affinity although the 340
L57A mutation induced a 1.4-fold increase in the turnover number. The weakened binding to 341
pyruvic acid by the L57A mutation seems to result from the loss of a native hydrogen bond 342
between R417 and the α-carboxylate of pyruvic acid (Fig. 1 and Fig. 4B). 343
As the side chain of the α-keto acid substrate extends from a methyl to a n-butyl group 344
(i.e. from pyruvic acid to 2-oxohexanoic acid), the wild-type enzyme showed a 290- and 110-345
fold decreases in the binding affinity and the turnover number, respectively. The deterioration 346
in both binding and catalytic steps renders the wild-type enzyme near inactive toward 2-347
oxohexanoic acid. The L57A mutation turned out to promote both steps (i.e. 6- and 48-fold 348
increases in the binding affinity and the catalytic turnover, respectively), leading to a 290-fold 349
increase in the specificity constant for 2-oxohexanoic acid. It is notable that the fold-change in 350
kcat is even larger than that in KM, indicating that the L57A-induced alleviation of the steric 351
interference in the small pocket benefits the catalytic step much more than the binding step. 352
This result is in accordance with the structural model of a quinonoid intermediate (i.e. the most 353
unstable reaction intermediate) where formation of a covalent linkage between substrate and 354
PLP rearranges the substrate moiety close to the PLP side and thereby results in stronger steric 355
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from

17
interference of a bulky substituent in the reaction intermediate than that in the Michaelis 356
complex (38). 357
Synthetic utility of the L57A variant 358
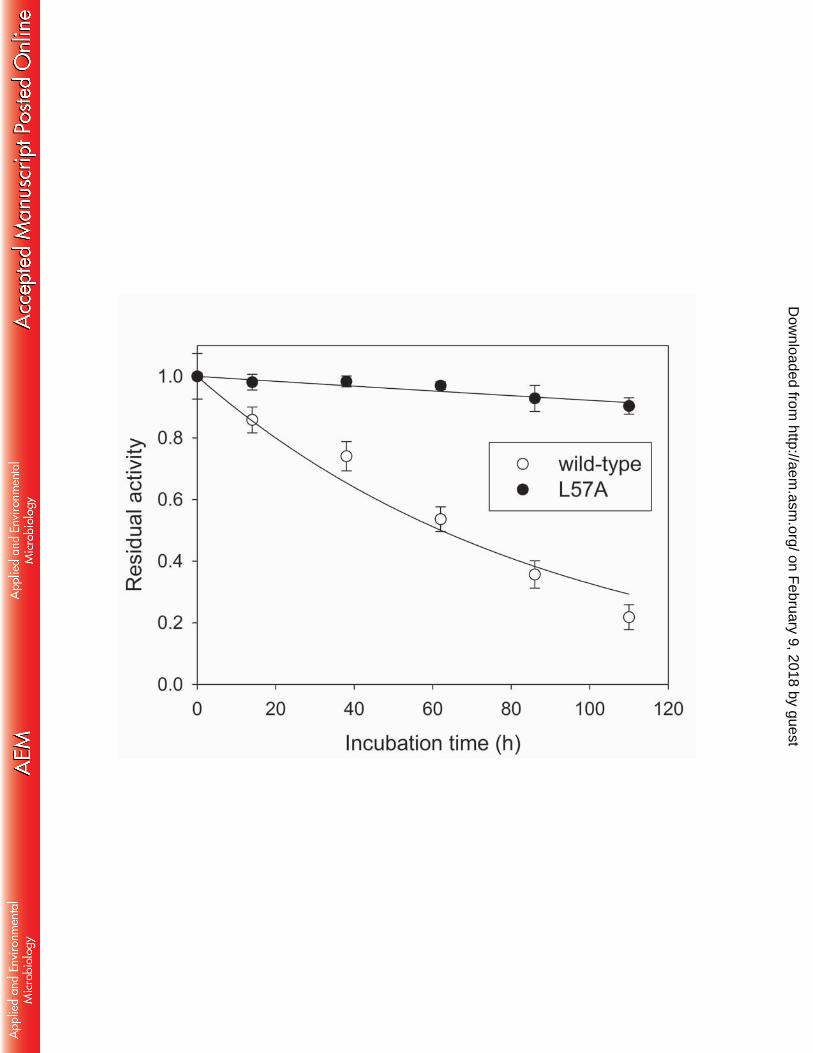
For practical applicability of enzyme variants engineered to attain a desirable functionality, it is 359
essential for the amino acid substitution to disturb the protein stability in a minimal way. We 360
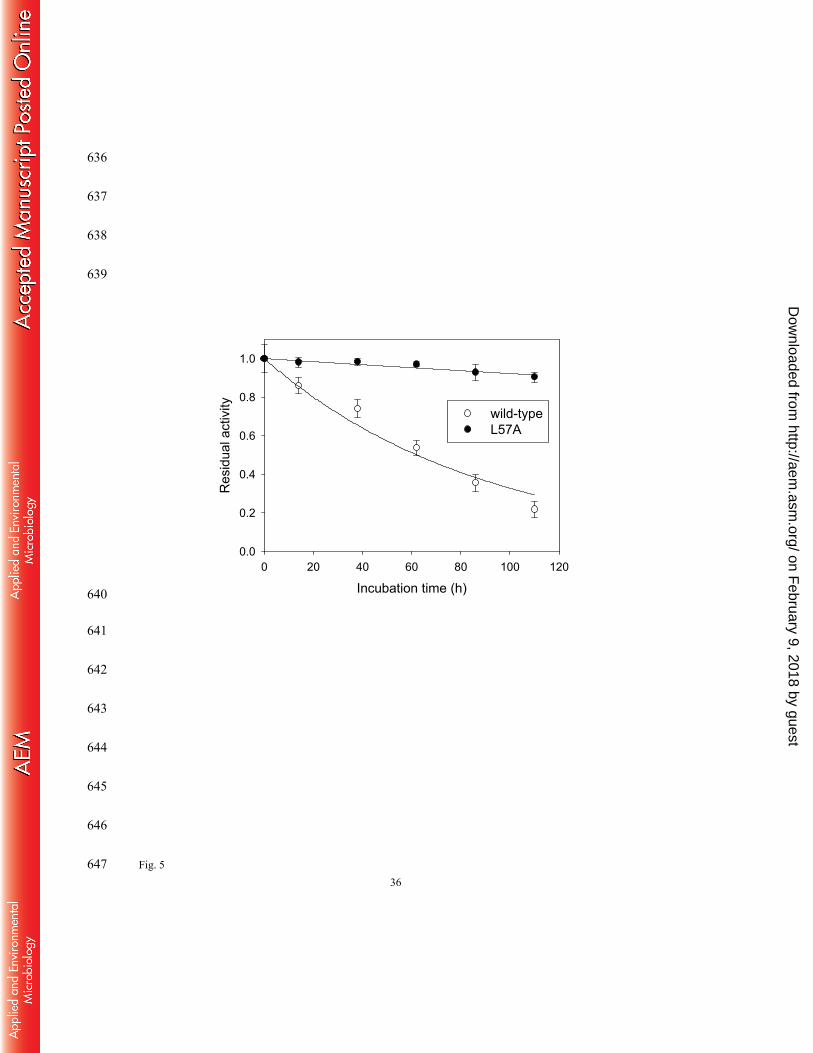
performed time-course monitoring of the enzyme activities under the incubation conditions of 361
50 mM phosphate buffer (pH 7.0) and 37 °C which were the same as those used in the reaction 362
mixture for a preparative purpose (Fig. 5). Intriguingly, we found that the L57A mutation 363
remarkably increased enzyme stability. Inactivation constants obtained from curve fitting of the 364
residual activity data to a single exponential function were 1.1×10-2 h-1 (r2 = 0.97, half life = 62 365
h) and 8.1×10-4 h-1 (r2 = 0.89, 860 h) for the wild-type and the L57A variant, respectively. The 366
striking stability improvement renders the L57A variant highly promising for industrial 367
applications. 368
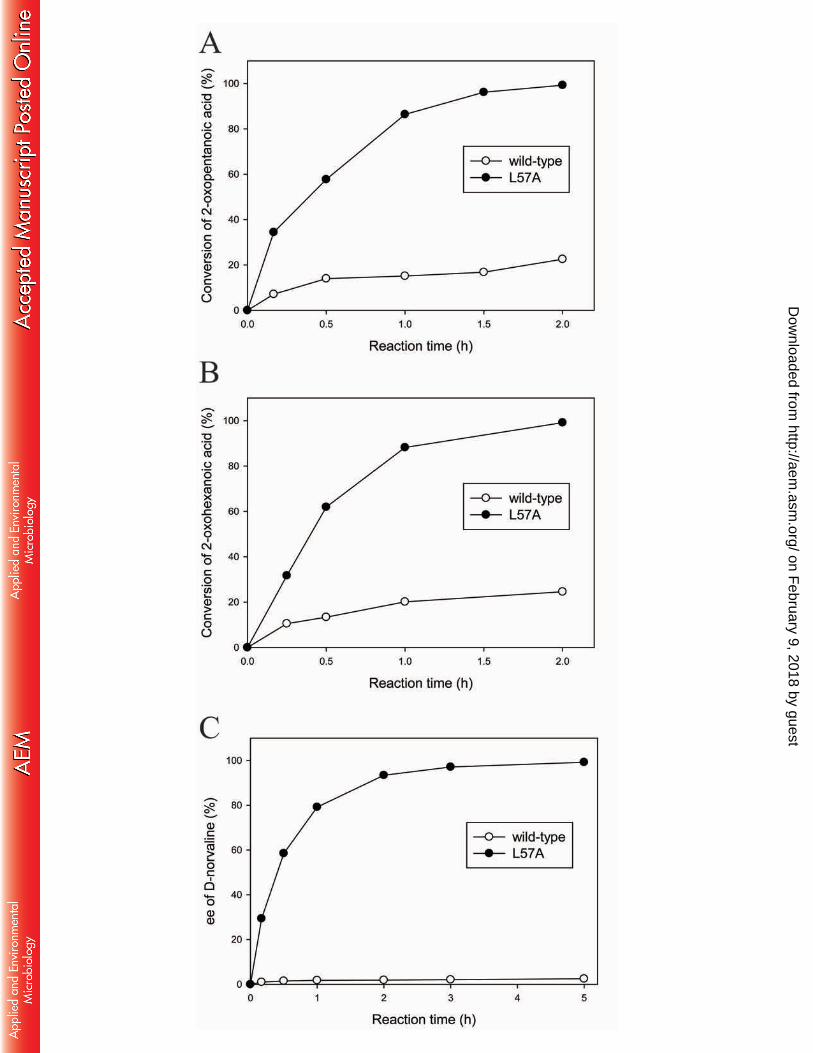
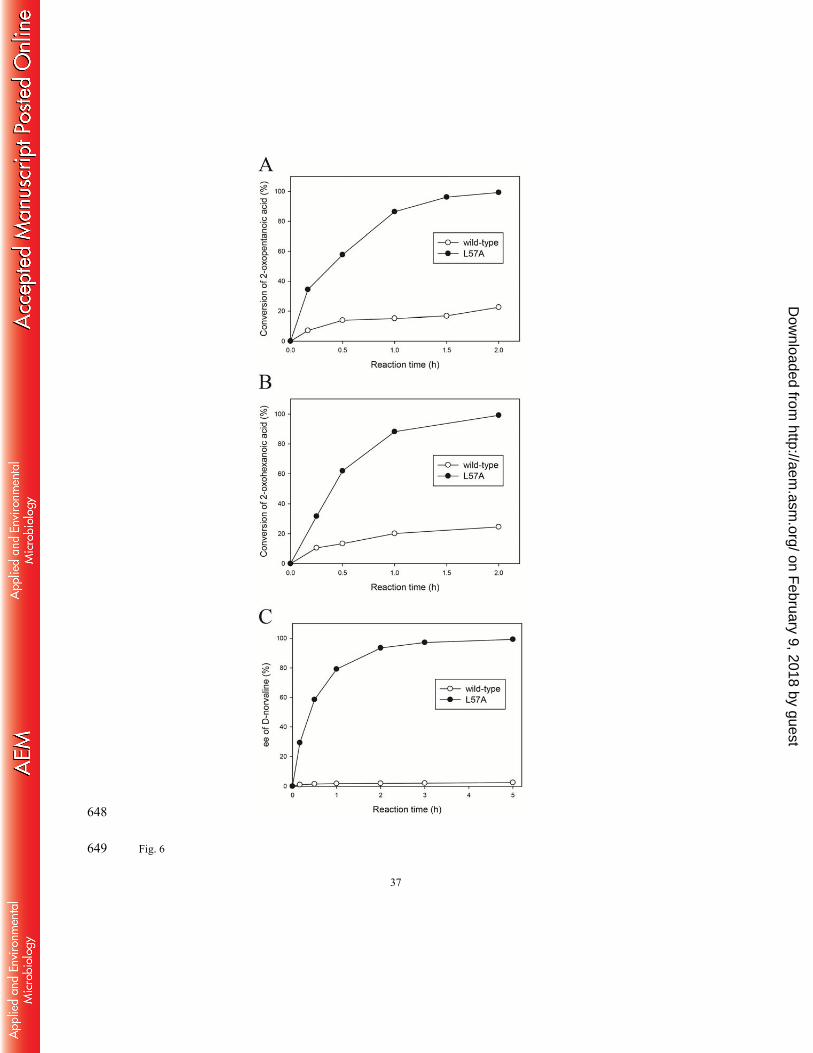
To examine how the L57A mutation improves the catalytic potential of OATA for 369
production of bulky unnatural amino acids, we carried out asymmetric synthesis of L-norvaline 370
which is a key intermediate of Perindopril (i.e. an ACE inhibitor) (41) and a potential inhibitor 371
of arginase (42). As expected, the L57A variant afforded much faster synthesis of L-norvaline 372
from 50 mM 2-oxopentanoic acid and 100 mM isopropylamine than its parental enzyme did at 373
the same reaction conditions (Fig. 6A). Conversion reached 99.3 % at 2 h with > 99.9 % ee of 374
the resulting L-norvaline using the L57A variant, whereas the wild-type OATA permitted only 375
22.6 % conversion at 2 h. As shown in Fig. 6B, we also performed asymmetric synthesis of L-376
norleucine from 100 mM 2-oxohexanoic acid and 200 mM isopropylamine using a 5-fold 377
higher enzyme concentration than that used in Fig. 6A. The L57A variant completed the 378
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from

18
reaction within 2 h (i.e. 99.1 % conversion and > 99.9 % ee of L-norleucine), while use of the 379
wild-type OATA led to only 24.6 % conversion at 2 h. 380
To demonstrate the catalytic utility of the L57A variant to access production of D-amino 381
acids, we performed kinetic resolution of racemic norvaline to prepare D-norvaline that can be 382
used as a chiral building block of macrolide drugs such as Pamamycin-607 (43) and 383
Epilachnene (44) (Fig. 6C). Starting with 50 mM rac-norvaline and 50 mM glyoxylic acid 384
using the same enzyme concentration as the one in Fig. 6A, the kinetic resolution was 385
completed by the L57A variant within 5 h (i.e. 51.6 % conversion and 99.2 % ee of D-386
norvaline). In contrast, the same reaction catalyzed by the wild-type enzyme led to only 0.8 % 387
conversion and 2.5 % ee at 5 h. 388
CONCLUSIONS 389
To expand the synthetic utility of ω-TAs for production of diverse unnatural amino acids via 390
either asymmetric synthesis or kinetic resolution, it is indispensable to overcome the canonical 391
steric constraint in the small pocket. To the best of our knowledge, this study is the first 392
example to engineer the canonical substrate specificity of a ω-TA for α-keto acids and α-amino 393
acids. Our results indicate that a single point mutation introduced in the large pocket rather than 394
the small pocket could be more effective in relieving the steric constraint. However, a future 395
direction to further engineer the substrate specificity toward more structurally demanding α-396
keto acids should be accompanied by additional mutations in the small pocket of the L57A 397
variant, e.g. the V154A substitution identified beneficial for relieving the steric constraint. 398
399
400
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from

19
Supplemental Materials 401
Supplemental Figures S1 - S3. 402
403
Acknowledgements 404
This work was supported by the Advanced Biomass R&D Center (ABC-2011-0031358) 405
through the National Research Foundation of Korea and the R&D grant (S2173394) funded by 406
the Small and Medium Business Administration of Korea. 407
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from

20
References 408
1. Patel R. 2013. Biocatalytic synthesis of chiral alcohols and amino acids for 409
development of pharmaceuticals. Biomolecules 3:741-777. 410
2. Gotor-Fernández V, Gotor V. 2009. Biocatalytic routes to chiral amines and amino 411
acids. Curr Opin Drug Discovery Dev 12:784-797. 412
3. Breuer M, Ditrich K, Habicher T, Hauer B, Kesseler M, Sturmer R, Zelinski T. 413
2004. Industrial methods for the production of optically active intermediates. Angew 414
Chem Int Ed 43:788-824. 415
4. Leuchtenberger W, Huthmacher K, Drauz K. 2005. Biotechnological production of 416
amino acids and derivatives: Current status and prospects. Appl Microbiol Biotechnol 417
69:1-8. 418
5. Maier THP. 2003. Semisynthetic production of unnatural L-α-amino acids by 419
metabolic engineering of the cysteine-biosynthetic pathway. Nat Biotechnol 21:422-427. 420
6. Bommarius AS, Schwarm M, Drauz K. 2001. Comparison of different 421
chemoenzymatic process routes to enantiomerically pure amino acids. Chimia 55:50-59. 422
7. Chenault HK, Dahmer J, Whitesides GM. 1989. Kinetic resolution of unnatural and 423
rarely occurring amino acids: Enantioselective hydrolysis of N-acyl amino acids 424
catalyzed by acylase I. J Am Chem Soc 111:6354-6364. 425
8. Krieg L, Ansorge-Schumacher MB, Kula MR. 2002. Screening for amidases: 426
Isolation and characterization of a novel D-amidase from Variovorax paradoxus. Adv 427
Synth Catal 344:965-973. 428
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from

21
9. Komeda H, Ishikawa N, Asano Y. 2003. Enhancement of the thermostability and 429
catalytic activity of D-stereospecific amino-acid amidase from Ochrobactrum anthropi 430
SV3 by directed evolution. J Mol Catal B: Enzym 21:283-290. 431
10. Clemente-Jiménez JM, Martínez-Rodríguez S, Rodríguez-Vico F, Heras-Vázquez 432
FJL. 2008. Optically pure α-amino acids production by the "Hydantoinase Process". 433
Recent Pat Biotechnol 2:35-46. 434
11. Altenbuchner J, Siemann-Herzberg M, Syldatk C. 2001. Hydantoinases and related 435
enzymes as biocatalysts for the synthesis of unnatural chiral amino acids. Curr Opin 436
Biotechnol 12:559-563. 437
12. Caligiuri A, D'Arrigo P, Rosini E, Tessaro D, Molla G, Servi S, Pollegioni L. 2006. 438
Enzymatic conversion of unnatural amino acids by yeast D-amino acid oxidase. Adv 439
Synth Catal 348:2183-2190. 440
13. Singh S, Gogoi BK, Bezbaruah RL. 2011. Racemic resolution of some DL-amino 441
acids using Aspergillus fumigatus L-amino acid oxidase. Curr Microbiol 63:94-99. 442
14. Gonçalves LPB, Antunes OAC, Oestreicher EG. 2006. Thermodynamics and kinetic 443
aspects involved in the enzymatic resolution of (R,S)-3-fluoroalanine in a coupled 444
system of redox reactions catalyzed by dehydrogenases. Org Process Res Dev 10:673-445
677. 446
15. Hummel W, Kuzu M, Geueke B. 2003. An efficient and selective enzymatic oxidation 447
system for the synthesis of enantiomerically pure D-tert-Leucine. Org Lett 5:3649-3650. 448
16. Li T, Kootstra AB, Fotheringham IG. 2002. Nonproteinogenic α-amino acid 449
preparation using equilibrium shifted transamination. Org Process Res Dev 6:533-538. 450
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from

22
17. Taylor PP, Pantaleone DP, Senkpeil RF, Fotheringham IG. 1998. Novel 451
biosynthetic approaches to the production of unnatural amino acids using transaminases. 452
Trends Biotechnol 16:412-418. 453
18. Stewart JD. 2001. Dehydrogenases and transaminases in asymmetric synthesis. Curr 454
Opin Chem Biol 5:120-129. 455
19. Park ES, Dong JY, Shin JS. 2014. Active site model of (R)-selective ω-transaminase 456
and its application to the production of D-amino acids. Appl Microbiol Biotechnol 457
98:651-660. 458
20. Park ES, Dong JY, Shin JS. 2013. Biocatalytic asymmetric synthesis of unnatural 459
amino acids through the cascade transfer of amino groups from primary amines onto 460
keto acids. ChemCatChem 5:3538-3542. 461
21. Park ES, Dong JY, Shin JS. 2013. ω-Transaminase-catalyzed asymmetric synthesis of 462
unnatural amino acids using isopropylamine as an amino donor. Org Biomol Chem 463
11:6929-6933. 464
22. Park E-S, Malik MS, Dong J-Y, Shin J-S. 2013. One-pot production of enantiopure 465
alkylamines and arylalkylamines of opposite chirality catalyzed by ω-transaminase. 466
ChemCatChem 5:1734-1738. 467
23. Savile CK, Janey JM, Mundorff EC, Moore JC, Tam S, Jarvis WR, Colbeck JC, 468
Krebber A, Fleitz FJ, Brands J, Devine PN, Huisman GW, Hughes GJ. 2010. 469
Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin 470
manufacture. Science 329:305-309. 471
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from

23
24. Mutti FG, Fuchs CS, Pressnitz D, Sattler JH, Kroutil W. 2011. Stereoselectivity of 472
four (R)-selective transaminases for the asymmetric amination of ketones. Adv Synth 473
Catal 353:3227-3233. 474
25. Cassimjee KE, Branneby C, Abedi V, Wells A, Berglund P. 2010. Transaminations 475
with isopropylamine: Equilibrium displacement with yeast alcohol dehydrogenase 476
coupled to in situ cofactor regeneration. Chem Comm 46:5569-5571. 477
26. Iwasaki A, Matsumoto K, Hasegawa J, Yasohara Y. 2012. A novel transaminase, 478
(R)-amine:pyruvate aminotransferase, from Arthrobacter sp. KNK168 (FERM BP-479
5228): Purification, characterization, and gene cloning. Appl Microbiol Biotechnol 480
93:1563-1573. 481
27. Shin JS, Kim BG. 2002. Exploring the active site of amine:pyruvate aminotransferase 482
on the basis of the substrate structure-reactivity relationship: How the enzyme controls 483
substrate specificity and stereoselectivity. J Org Chem 67:2848-2853. 484
28. Malik MS, Park ES, Shin JS. 2012. Features and technical applications of ω-485
transaminases. Appl Microbiol Biotechnol 94:1163-1171. 486
29. Park ES, Park SR, Han SW, Dong JY, Shin JS. 2014. Structural determinants for the 487
non-canonical substrate specificity of the ω-transaminase from Paracoccus denitrificans. 488
Adv Synth Catal 356:212-220. 489
30. Midelfort KS, Kumar R, Han S, Karmilowicz MJ, McConnell K, Gehlhaar DK, 490
Mistry A, Chang JS, Anderson M, Villalobos A, Minshull J, Govindarajan S, 491
Wong JW. 2013. Redesigning and characterizing the substrate specificity and activity 492
of Vibrio fluvialis aminotransferase for the synthesis of imagabalin. Protein Eng Des Sel 493
26:25-33. 494
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from

24
31. Park ES, Kim M, Shin JS. 2012. Molecular determinants for substrate selectivity of ω-495
transaminases. Appl Microbiol Biotechnol 93:2425-2435. 496
32. Park ES, Shin JS. 2014. Deracemization of amino acids by coupling transaminases of 497
opposite stereoselectivity. Adv Synth Catal 356:3505-3509. 498
33. B'Hymer C, Montes-Bayon M, Caruso JA. 2003. Marfey's reagent: Past, present, and 499
future uses of 1-fluoro-2,4-dinitrophenyl-5-L-alanine amide. J Sep Sci 26:7-19. 500
34. Bhushan R, Brückner H. 2004. Marfey’s reagent for chiral amino acid analysis: A 501
review. Amino Acids 27:231-247. 502
35. Rausch C, Lerchner A, Schiefner A, Skerra A. 2013. Crystal structure of the ω-503
aminotransferase from Paracoccus denitrificans and its phylogenetic relationship with 504
other class III aminotransferases that have biotechnological potential. Proteins 81:774-505
787. 506
36. Humble MS, Cassimjee KE, Hãkansson M, Kimbung YR, Walse B, Abedi V, 507
Federsel HJ, Berglund P, Logan DT. 2012. Crystal structures of the 508
Chromobacterium violaceum ω-transaminase reveal major structural rearrangements 509
upon binding of coenzyme PLP. FEBS J 279:779-792. 510
37. Fabian Steffen-Munsberg CV, Ahmad Thontowi, Sebastian Sch tzle,, Tony 511
Tumlirsch MSH, Henrik Land, Per Berglund,, Uwe T. Bornscheuer aMH. 2013. 512
Connecting unexplored protein crystal structures to enzymatic function. ChemCatChem 513
5:150-153. 514
38. Han SW, Park ES, Dong JY, Shin JS. 2015. Mechanism-guided engineering of ω-515
transaminase to accelerate reductive amination of ketones. Adv Synth Catal 357:1732-516
1740. 517
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from

25
39. Malashkevich VN, Onuffer JJ, Kirsch JF, Jansonius JN. 1995. Alternating arginine-518
modulated substrate specificity in an engineered tyrosine aminotransferase. Nat Struct 519
Mol Biol 2:548-553. 520
40. Cellini B, Bertoldi M, Montioli R, Paiardini A, Borri Voltattorni C. 2007. Human 521
wild-type alanine:glyoxylate aminotransferase and its naturally occurring G82E variant: 522
Functional properties and physiological implications. Biochem J 408:39-50. 523
41. Michel V, Jean B, Bernard M, Georges R. 1990. US Patent No. 4,902,817 524
42. Ming XF, Rajapakse AG, Carvas JM, Ruffieux J, Yang Z. 2009. Inhibition of S6K1 525
accounts partially for the anti-inflammatory effects of the arginase inhibitor L-norvaline. 526
BMC Cardiovasc Disord 9: 12. 527
43. Fraser BH, Mulder RJ, Perlmutter P. 2006. The total synthesis of pamamycin-607. 528
Part 2: Synthesis of the C6-C18 domain. Tetrahedron 62:2857-2867. 529
44. Farmer JJ, Attygalle AB, Smedley SR, Eisner T, Meinwald J. 1997. Absolute 530
configuration of insect-produced epilachnene. Tetrahedron Lett 38:2787-2790. 531
532
533
534
535
536
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from

26
Table 1. PCR primers used for preparing variants of OATA. 537
Mutation Mutagenesis primersa
Y20A Forward: 5'-CGCGATATCCGTTATCATCTCCATTCTGCGACCGATGCTGTCCG-3' Reverse: 5'-CGGACAGCATCGGTCGCAGAATGGAGATGATAACGGATATCGCG-3'
L57A Forward: 5'-CGAAGCGATGTCAGGAGCGTGGAGTGTTGGCGTG-3' Reverse: 5'-CACGCCAACACTCCACGCTCCTGACATCGCTTCG-3'
L57V Forward: 5'-CGAAGCGATGTCAGGAGTGTGGAGTGTTGGC-3' Reverse: 5'-GCCAACACTCCACACTCCTGACATCGCTTCG-3'
L57G Forward: 5'-TATCGAAGCGATGTCAGGAGGCTGGAGTGTTGGCGTGG-3' Reverse: 5'-CCACGCCAA CACTCCAGCCTCCTGACATCGCTTCGATA-3'
W58A Forward: 5'-GCGATGTCAGGACTGGCGAGTGTTGGCGTGGG-3' Reverse: 5'-CCCACGCCAACACTCGCCAGTCCTGACATCGC-3'
F86A Forward: 5'-AGATGAAGAAGCTGCCTTTCTACCATACAGCGTCCTACCGTTCGCAT-3' Reverse: 5'-ATGCGAACGGTAGGACGCTGTATGGTAGAAAGGCAGCTTCTTCATCT-3'
Y151A Forward: 5'-CTCACGCAAGCGCGGCGCGCACGGTGTGACGATTG-3' Reverse: 5'-CAATCGTCACACCGTGCGCGCCGCGCTTGCGTGAG-3'
V154A Forward: 5'-CGGCTATCACGGTGCGACGATTGCCTCTG-3' Reverse: 5'-CAGAGGCAATCGTCGCACCGTGATAGCCG-3'
I261A Forward: 5'-TCTGCTGATCGCCGACGAGGTTGCGTGCGGCTTCGGA-3' Reverse: 5'-TCCGAAGCCGCACGCAACCTCGTCGGCGATCAGCAGA-3'
F323A Forward: 5'-ACGCTTGGCACGGGCGCGACGGCATCTGGCCAT-3' Reverse: 5'-ATGGCCAGATGCCGTCGCGCCCGTGCCAAGCGT-3'
T324A Forward: 5'-GGCACGGGCTTCGCGGCATCTGGCC-3' Reverse: 5'-GGCCAGATGCCGCGAAGCCCGTGCC-3'
a The mutation sites are underlined.538
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from
27
Table 2. Effect of the mutations of the L57 residue on substrate specificity of OATA for α-keto 539
acids. 540
Substrate Side chain Reaction rate (μM/min)a
wild-type L57V L57A L57G
glyoxylic acid -H 618 ± 31 60 ± 2 354 ± 23 113 ± 12
pyruvic acid -CH3 478 ± 25 66 ± 12 367 ± 37 132 ± 6
2-oxobutyric acid -CH2CH3 69 ± 5 13 ± 3 161 ± 6 30 ± 2
2-oxopentanoic acid -(CH2)2CH3 4 ± 2 6 ± 1 190 ± 8 47 ± 4
2-oxohexanoic acid -(CH2)3CH3 2 ± 1 2 ± 1 77 ± 1 10 ± 1
2-oxooctanoic acid -(CH2)5CH3 n.d.b n.d.b 8 ± 4 n.d.b
541
a Reaction rate represents the initial rate per 1 μM enzyme and was measured at 20 mM (S)-α-542
MBA and 20 mM α-keto acid in 50 mM phosphate buffer (pH 7). b not detectable (i.e. reaction 543
rate < 1 μM/min). 544
545
546
547
548
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from

28
Table 3. Effect of the mutations of the L57 residue on substrate specificity of OATA for α-549
amino acids. 550
Substrate Side chain Reaction rate (μM/min)a
wild-type L57V L57A L57G
Glycine -H 18 ± 1 3 ± 2 9 ± 4 3 ± 2
L-alanine -CH3 559 ± 4 75 ± 5 203 ± 26 39 ± 5
L-homoalanine -CH2CH3 42 ± 4 2 ± 1 52 ± 1 9 ± 1
L-norvaline -(CH2)2CH3 2 ± 1 n.d.b 111 ± 7 14 ± 1
L-norleucine -(CH2)3CH3 n.d.b n.d.b 26 ± 2 3 ± 1
551
a Reaction rate represents the initial rate per 1 μM enzyme and was measured at 20 mM amino 552
acid and 20 mM propanal in 50 mM phosphate buffer (pH 7). b not detectable (i.e. reaction rate 553
< 1 μM/min). 554
555
556
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from

29
Table 4. Kinetic parameters of the wild-type and the L57A OATA for α-keto acids.a 557
α-keto acid wild-type
L57A
KM
(mM)
kcat
(s-1) kcat/KM
(M-1 s-1)
KM
(mM) kcat
(s-1) kcat/KM
(M-1 s-1)
pyruvic acid 0.12 ± 0.01b 2.6 ± 0.1b 22000 ± 3000b
0.34 ± 0.05 3.7 ± 0.2 11000 ± 2000
2-oxohexanoic acid 35 ± 1 0.023 ± 0.001 0.66 ± 0.05 5.7 ± 0.3 1.1 ± 0.1 190 ± 20
a Kinetic parameters represent the apparent rate constants determined at a fixed concentration of (S)-α-MBA. b These kinetic parameters 558
were taken from a previous study (38). 559
560
561
562
563
564
565
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from

30
Figure legends. 566
Fig.1 Docking model of L-alanine in the active site of OATA. L-alanine is shown in CPK 567
representation. The internal aldimine, formed between PLP and K287, is shown as 568
thick sticks. K287 is positioned right behind the bound substrate. The cyan dotted lines 569
represent hydrogen bonds. Color use of the active site residues is consistent with that 570
of labels. The active site is visualized by a Connolly surface. 571
Fig.2 Enzyme activities of the alanine scanning mutants for 2-oxopentanoic acid. Reaction 572
rate represents the initial rate per 1 μM enzyme and was measured at 20 mM (S)-α-573
MBA and 20 mM 2-oxopentanoic acid. WT represents the wild-type OATA. 574
Fig. 3 Effect of the mutations of the L57 residue on amino donor activity for isopropylamine. 575
Reaction rate represents the initial rate per 1 μM enzyme and was measured at 20 mM 576
isopropylamine and 20 mM pyruvic acid. 577
Fig. 4 Docking models of the L57A variant using (A) L-alanine and (B) L-norleucine as 578
ligands. The ligands docked in the active site, visualized by a Connolly surface, are 579
represented by ball-and-stick models. The purple sticks and the cyan dotted lines 580
represent the mutation site and the hydrogen bonds, respectively. The yellow sticks in 581
the binding pocket of the upper figure represent the docking pose of L-alanine in the 582
wild-type OATA as shown in Fig. 1. 583
Fig. 5 Effect of the L57A mutation on the enzyme stability. Purified enzymes were incubated 584
in 50 mM potassium phosphate buffer (pH 7.0) at 37 oC. 585
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from

31
Fig. 6 Enzymatic reactions to produce unnatural amino acids using the L57A variant in 586
comparison with the parental ω-TA. (A) Asymmetric synthesis of L-norvaline. 587
Reaction conditions were 50 mM 2-oxopentanoic acid, 100 mM isopropylamine and 588
40 μM ω-TA. (B) Asymmetric synthesis of L-norleucine. Reaction conditions were 589
100 mM 2-oxohexanoic acid, 200 mM isopropylamine and 200 μM ω-TA. (C) Kinetic 590
resolution of rac-norvaline. Reaction conditions were 50 mM rac-norvaline, 50 mM 591
glyoxylic acid and 40 μM ω-TA. 592
593
594
595
596
597
598
599
600
601
602
603
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from
32
604
605
606
607
608
609
610
611
Fig. 1 612
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from
33
613
614
615
WT Y20A L57A W58A F86A Y151A V154A I261A F323A T324A
reac
tion
rate
(μM
/min
)
0
50
100
150
200
616
617
618
619
620
Fig. 2 621
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from
34
622
623
624
WT L57V L57A L57G
reac
tion
rate
(μM
/min
)
0
50
100
150
200
250
625
626
627
628
629
Fig. 3 630
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from
35
631
632
633
634
Fig. 4 635
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from
36
636
637
638
639
Incubation time (h)0 20 40 60 80 100 120
Res
idua
l act
ivity
0.0
0.2
0.4
0.6
0.8
1.0
wild-typeL57A
640
641
642
643
644
645
646
Fig. 5 647
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from
37
648
Fig. 6 649
on February 9, 2018 by guest
http://aem.asm
.org/D
ownloaded from