
A Novel Combination Immunotherapy for Cancer by IL-13Ra2
13
of January 5, 2019. This information is current as Models Vaccine and Immunotoxin in Murine Tumor Targeted DNA - 2 α Cancer by IL-13R A Novel Combination Immunotherapy for Syed R. Husain and Raj K. Puri Hideyuki Nakashima, Masaki Terabe, Jay A. Berzofsky, http://www.jimmunol.org/content/187/10/4935 doi: 10.4049/jimmunol.1102095 October 2011; 2011; 187:4935-4946; Prepublished online 17 J Immunol References http://www.jimmunol.org/content/187/10/4935.full#ref-list-1 , 23 of which you can access for free at: cites 45 articles This article average * 4 weeks from acceptance to publication Fast Publication! • Every submission reviewed by practicing scientists No Triage! • from submission to initial decision Rapid Reviews! 30 days* • Submit online. ? The JI Why Subscription http://jimmunol.org/subscription is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/About/Publications/JI/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/alerts Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. All rights reserved. 1451 Rockville Pike, Suite 650, Rockville, MD 20852 The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on January 5, 2019 http://www.jimmunol.org/ Downloaded from by guest on January 5, 2019 http://www.jimmunol.org/ Downloaded from
Transcript of A Novel Combination Immunotherapy for Cancer by IL-13Ra2

of January 5, 2019.This information is current as
ModelsVaccine and Immunotoxin in Murine Tumor
Targeted DNA−2αCancer by IL-13RA Novel Combination Immunotherapy for
Syed R. Husain and Raj K. PuriHideyuki Nakashima, Masaki Terabe, Jay A. Berzofsky,
http://www.jimmunol.org/content/187/10/4935doi: 10.4049/jimmunol.1102095October 2011;
2011; 187:4935-4946; Prepublished online 17J Immunol
Referenceshttp://www.jimmunol.org/content/187/10/4935.full#ref-list-1
, 23 of which you can access for free at: cites 45 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. All rights reserved.1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on January 5, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on January 5, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
A Novel Combination Immunotherapy for Cancer byIL-13Ra2–Targeted DNA Vaccine and Immunotoxin inMurine Tumor Models
Hideyuki Nakashima,* Masaki Terabe,† Jay A. Berzofsky,† Syed R. Husain,* and
Raj K. Puri*
Optimum efficacy of therapeutic cancer vaccines may require combinations that generate effective antitumor immune responses,
as well as overcome immune evasion and tolerance mechanisms mediated by progressing tumor. Previous studies showed that
IL-13Ra2, a unique tumor-associated Ag, is a promising target for cancer immunotherapy. A targeted cytotoxin composed of
IL-13 and mutated Pseudomonas exotoxin induced specific killing of IL-13Ra2+ tumor cells. When combined with IL-13Ra2 DNA
cancer vaccine, surprisingly, it mediated synergistic antitumor effects on tumor growth and metastasis in established murine
breast carcinoma and sarcoma tumor models. The mechanism of synergistic activity involved direct killing of tumor cells and cell-
mediated immune responses, as well as elimination of myeloid-derived suppressor cells and, consequently, regulatory T cells.
These novel results provide a strong rationale for combining immunotoxins with cancer vaccines for the treatment of patients with
advanced cancer. The Journal of Immunology, 2011, 187: 4935–4946.
Therapeutic cancer vaccines and adoptive T cell immuno-therapy have been considered very attractive therapeuticapproaches for the treatment of human cancer. However,
despite the identification of a number of tumor-associated Ags thatare recognized by the immune system, clinical trials of differentcancer vaccines performed in recent years demonstrated subop-timal clinical efficacy (1, 2). Recently, an APC-based therapeuticcancer vaccine has been licensed for patients with minimallysymptomatic androgen-resistant prostate cancer (3). In general,cancer vaccines face a number of scientific challenges. They in-clude the ability of cancer vaccines to generate potent immuneresponses given the presence of numerous cancer-induced immu-nosuppressive factors, the correct choice and characterization ofAg for immunization, dose of Ag, and frequency of vaccination. Inaddition, it has become apparent that therapeutic cancer vaccinesgiven as a single agent may not produce substantial clinical ben-efits, and combination with conventional and novel methods oftreatment will be necessary. Several strategies are being tested toenhance immune response in patients. For example, cancer vac-cines are being combined with other immunotherapy, with stan-dard cancer drugs, targeted small-molecule drugs, and local andsystemic radiation of tumors, or laser therapy (4, 5). These recent
approaches show that combination therapy may be more effectivethan either modality alone in animal models of cancer, as well asin the clinical trials (5). However, cancer vaccines have never beencombined with immunotoxins/cytotoxins, which are highly spe-cific and very potent molecules that kill cancer cells directly atvery low concentrations (6, 7). Various immunotoxins/cytotoxinsare being tested in the clinic for cancer therapy and have showntherapeutic anticancer effects in various human cancers (8, 9).For a successful cancer vaccine, identification of a specific
tumor-associated Ag is critical. A variety of cancer-associatedtumor cell surface-associated Ags have been identified (10). Wediscovered that the IL-13R a2 chain, which is overexpressedon a variety of human cancers, including glioblastoma, head andneck, kidney, ovarian, breast, pancreatic, and Kaposi’s sarcoma, isa potent tumor Ag (7, 11–15). IL-13Ra2 is one of the two subunitsof the receptor for IL-13, a Th2 cell-derived pleiotropic immuneregulatory cytokine (16). We recently demonstrated that IL-13Ra2is directly involved in cancer invasion and metastasis in humanpancreatic cancer models (17). These findings suggested that IL-13Ra2 can modulate certain antitumor immune responses. Indeed,we showed that immunization with a DNA vaccine encoding IL-13Ra2 chain inhibited IL-13Ra2–expressing D5 murine mela-noma tumor growth in both prophylactic and therapeutic models(18). Furthermore, we demonstrated that IL-13Ra2 DNA vaccine,followed by extracellular domain of IL-13Ra2 protein boost,worked effectively and induced stronger antitumor activity inmurine tumor models (19). Because IL-13R–directed immuno-toxin IL-13-PE (composed of IL-13 and Pseudomonas exotoxin)has been shown to kill cancer cells directly (7), we hypothesizedthat IL-13Ra2 DNA vaccination, when combined with IL13-PEtreatment, may induce an enhanced antitumor response to tumorsexpressing IL-13Ra2.In the cancer vaccine field, myeloid-derived suppressor cells
(MDSCs) and regulatory T cells (Tregs) have been shown to playa major role in modulating the therapeutic effect (20). MDSCs area heterogeneous population of undifferentiated cells that expressmarkers of monocytes (CD11b) and neutrophils (Gr-1) and cause
*Tumor Vaccines and Biotechnology Branch, Division of Cellular and Gene Thera-pies, Center for Biologics Evaluation and Research, U.S. Food and Drug Adminis-tration, Bethesda, MD 20892; and †Vaccine Branch, Center for Cancer Research,National Cancer Institute, National Institutes of Health, Bethesda, MD 20892
Received for publication July 19, 2011. Accepted for publication August 25, 2011.
Address correspondence and reprint requests to Dr. Raj K. Puri, Tumor Vaccines andBiotechnology Branch, Division of Cellular and Gene Therapies, Center for Bio-logics Evaluation and Research, U.S. Food and Drug Administration, National In-stitutes of Health, Building 29B, Room 2NN20, 29 Lincoln Drive, Bethesda, MD20892. E-mail address: [email protected]
Abbreviations used in this article: IL13-PE, IL-13 and mutated Pseudomonas exo-toxin; iNOS, inducible NO synthase; IP-10, IFN-g–inducible protein-10; MDSC,myeloid-derived suppressor cell; MIG, monocyte induced by IFN-g; NIH, NationalInstitutes of Health; OST, overall sacrifice time; REML, restricted maximum likeli-hood; Treg, regulatory T cell.
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1102095
by guest on January 5, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

T cell dysfunction in tumor-bearing mice and humans. They re-duce Ag-specific CD8+ T cell proliferation, increase T cell deathby apoptosis, foster T cell tolerance, and change the profile ofcytokines secreted by activated T lymphocytes (21–24). MDSCsadditionally inhibit T cell function by inducing the developmentof Tregs in vitro and in vivo (25). Importantly, increased levels ofcirculating MDSCs were recently correlated with disease stageand extensive metastatic tumor burden in patients with breastcancer (26). Because MDSCs are one of the main immunosup-pressive factors in cancer and other pathological conditions, sev-eral therapeutic strategies that target these cells are being explored(27).In this study, we sought to determine the effect of IL13-PE
combined with IL-13Ra2 DNA vaccine in murine 4T1 breastcarcinoma and MCA304 sarcoma tumors, which naturally over-express IL-13Ra2 Ag. Our results indicated that the combinationtherapy with IL13-PE and IL-13Ra2 DNA vaccine (VRa2) me-diated a synergistic effect in primary and metastatic lesions. Inaddition, the combination therapy prolonged the survival of ani-mals with advanced disease. This combination therapy generatedantitumor CD4+ and CD8+ T cell immune responses and elimi-nated MDSCs and Tregs, further enhancing the antitumor immu-nity.
Materials and MethodsCell lines, DNA vaccine, and reagents
MCA304 murine sarcoma and D5 melanoma cell lines were kind giftsfrom Dr. Bernard A. Fox (Earle A. Chiles Research Institute, Portland,OR), and the 4T1 breast carcinoma cell line (28) was purchased from theAmerican Type Culture Collection. The CT-26 colon cancer cell line wasgrown in our laboratory from a stock originally provided by Dr. NicholasRestifo (National Cancer Institute, National Institutes of Health [NIH]).Both MCA304 and 4T1 tumors naturally express IL-13Ra2, as determinedby RT-PCR (19). Both D5 and CT-26 tumors, which express low or un-detectable levels of IL-13Ra2 mRNA, served as negative controls. PM-RCC, a renal cell carcinoma cell line, served as a positive control. cDNAvaccine encoding the murine IL-13Ra2 was cloned into the VR1012mammalian expression vector (a kind gift from Vical, San Diego, CA),according to the procedure described in an earlier study (19). We usedVR1012 vector (VR mock) with no insert gene as a negative control. Wealso constructed the irrelevant cDNA plasmid vector encoding human IL-2Rg–chain. The resulting constructs were expanded in Escherichia coliand purified using an endotoxin-free EndoFree Giga kit (Qiagen, Valencia,CA). Anti-CD4 (GK1.5) and anti-CD8 (2.43) Abs were grown as ascites,purified on protein G columns, and titered for in vivo efficacy. Rabbit anti-asialo GM1 Ab was purchased from WAKO (Osaka, Japan). RecombinantIL13-PE38 was generated following the procedure previously described(29) and diluted in PBS containing 0.2% human serum albumin.
Animals and antitumor studies
All animal experiments were carried out in accordance with the NIHGuidelines for the Care and Use of Laboratory Animals. Four-week-old(∼20 g body weight) female C57BL/6 and BALB/c mice were obtainedfrom the Frederick Cancer Center Animal Facilities (National CancerInstitute-Frederick, Frederick, MD). Female B6.129S6-Rag2 knockout(C57BL/6 background) mice were obtained from Taconic Farms (Ger-mantown, NY). MCA304 tumor models were established in C57BL/6 miceby s.c. injection of 0.5 3 106 cells in 150 ml PBS into the dorsal flank.Orthotopic 4T1 tumor models were established in BALB/c mice by in-jection of 0.5 3 106 cells in 150 ml PBS into the right side of a mammaryfat pad. Palpable tumors developed within 5 d. Tumors were measured, asdescribed earlier (19). Five to six mice were used for each group. Animalswere injected with excipient (0.2% human serum albumin in PBS) orIL13-PE (50 or 250 mg/ kg) once a day on every third day intratumorally(30 ml using a Hamilton microinjection syringe). Animals were immu-nized i.m. in right (50 mg) and left (50 mg) thighs with IL-13Ra2 DNAvaccine (VRa2) or control plasmid vector (VR mock) on the indicateddays using a 50-ml Hamilton syringe (total 100 mg/vaccination). In somecases, IL-2Rg–chain cDNA plasmid was used as an irrelevant negativecontrol.
Protein synthesis-inhibition assay
In vitro cytotoxic activity of IL13-PE was measured by the inhibition ofprotein synthesis, as described (7).
India ink assay
Pulmonary metastases were enumerated by intratracheal injection of indiaink (15% india Ink, 85% water, three drops NH4OH/100 ml). India ink-injected lungs were washed in Feket’s solution (300 ml 70% EtOH, 30 ml37% formaldehyde, 5 ml glacial acetic acid) and then placed in freshFeket’s solution overnight. White tumor nodules against a blue lung back-ground were counted.
Depletion experiments
Mice were injected i.p. with 0.25 mg anti-CD4 Ab (GK1.5) and/or anti-CD8Ab (2.43) on days 22, 21, +5, +11, +17, and +23 relative to the tumorimplantation. The control mice were injected i.p. with rat IgG (SigmaAldrich, St. Louis, MO). Depletion of CD4+ and/or CD8+ T cells (.95%)was confirmed by FACS analysis using blood samples from the treatedmice. In the same experiments, NK cells were also depleted with rabbitanti–asialo-GM1 Ab (50 mg/ injection).
IFN-g assay by ELISA
For IFN-g release, splenocytes harvested from each group of mice wererestimulated with mitomycin C-treated MCA304 or 4T1 tumor cells for48 h and then the culture supernatant was collected and assayed by ELISAkit (e-Bioscience, San Diego, CA), according to the procedure described inan earlier study (18).
CTL assay
Splenocytes from the immunized mice (4 3 106 per well) were restimu-lated with 23 105 mitomycin C-treated MCA304 or 4T1 tumor cells in thepresence of IL-2 (20 IU/ml) for 1 wk in 24-well plates and then used aseffector cells for [51Cr]-release assay, according to the procedure described(18)
Immunohistochemistry and immunofluorescence assay
Tumor samples were harvested and fixed with 10% formalin. Paraffin-embedded sections were deparaffinized by xylene treatment and washedsuccessively with alcohol (100–50%) and PBS. Slides were incubated withrat anti-mouse CD4 Ab (1 mg/ml; MCA1767; Serotec, Oxford, U.K.); ratanti-mouse CD8 Ab (1 mg/ml; MCA1108G; Serotec); goat anti-mousemonokine induced by IFN-g (MIG/CXCL9) (1 mg/ml; R&D Systems)Ab; rabbit anti-mouse IFN-g–inducible protein-10 (IP-10/CXCL10) (1 mg/ml; PeproTech, Rocky Hill, NJ) Ab; rat anti-CXCR3, CD11b, and Gr-1 Ab(5 mg/ml; eBioscience); mouse anti–arginase-1 Ab (5 mg/ml; BD Bio-science); rabbit anti–NO synthase-2 Ab (5 mg/ml; Santa Cruz Bio-technology); or isotype control for 18 h at 4˚C. The slides were analyzedwith laser scanning by fluorescence microscopy (Cascade II 1024, Pho-tometrics). Automated image processing and analysis software (Meta-morph; Universal Imaging, Downingtown, PA) was used to identify andquantitate the number of respective positive cells.
Quantification of autoantibody against IL-13Ra2 by ELISAassay
Ninety-six-well plates were coated with a recombinant mouse IL-13Ra2–Fc (human IgG1) chimera protein (10 mg/ml; R&D Systems) overnight at4˚C. Serum samples (100 ml/well) diluted 1:1000 in blocking solutionwere assayed in duplicate and incubated at 4˚C for 12 h. Anti mouse IL-13Ra2 Ab (R&D Systems) was used as the standard curve for determiningAb titers in the mouse serum. Wells were washed and then incubated withbiotinylated anti-mouse IgG Ab (0.1 mg/ml; vector) for an additional hour.This was followed by incubation with streptavidin-HRP conjugated andsubstrate solution (e-Bioscience) at room temperature for 20 min each.Absorbance was read at 450 nm.
Cell-proliferation assay
4T1 or MCA304 cells (5 3 103 per well) were cultured for 48 h with orwithout various concentrations of serum collected from each group ofmice before cell counter kit-8 solution (Dojindo) was added to eachwell. Cells were cultured for an additional hour. Absorbance was read at450 nm.
4936 COMBINATION OF IL-13Ra2 VACCINE AND IL13-PE REGRESS TUMORS
by guest on January 5, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
Flow cytometric analysis
To evaluate CD4+CD25+Foxp3+ Tregs in splenocytes, cells (1 3 106) werefirst stained with allophycocyanin-conjugated anti-CD3, FITC-conjugatedanti-CD4, and PE-conjugated anti-CD25 Abs (e-Bioscience). Cells werethen permeabilized and stained using PE-Cy5–conjugated anti-Foxp3 Ab,according to the manufacturer’s instructions (e-Bioscience). A rat IgG2aPE-Cy5 Ab was used as an isotype control. To evaluate MDSCs (CD11b+
GR-1+) in splenocytes, cells (1 3 106) were stained with FITC-conjugatedanti-CD11b and PerCP-Cy5.5–conjugated anti–Gr-1 Abs (e-Bioscience).All flow cytometric analyses were performed on a FACSCanto II (Becton
Dickinson, San Jose, CA) flow cytometer. FACSDiva software was usedfor acquisition. Cytometry Setup and Tracking beads kit (BD Biosciences)was used to initialize PMT setting. Individual compensation controls wereprepared for each reagent. FlowJo software version 8.8.6 (Tree Star,Ashland, OR) was used for data analysis and display.
Statistical analysis
Statistical analyses for tumor volume, lung nodules, and other assays wereanalyzed by one-way ANOVA, and synergism was analyzed by the least-
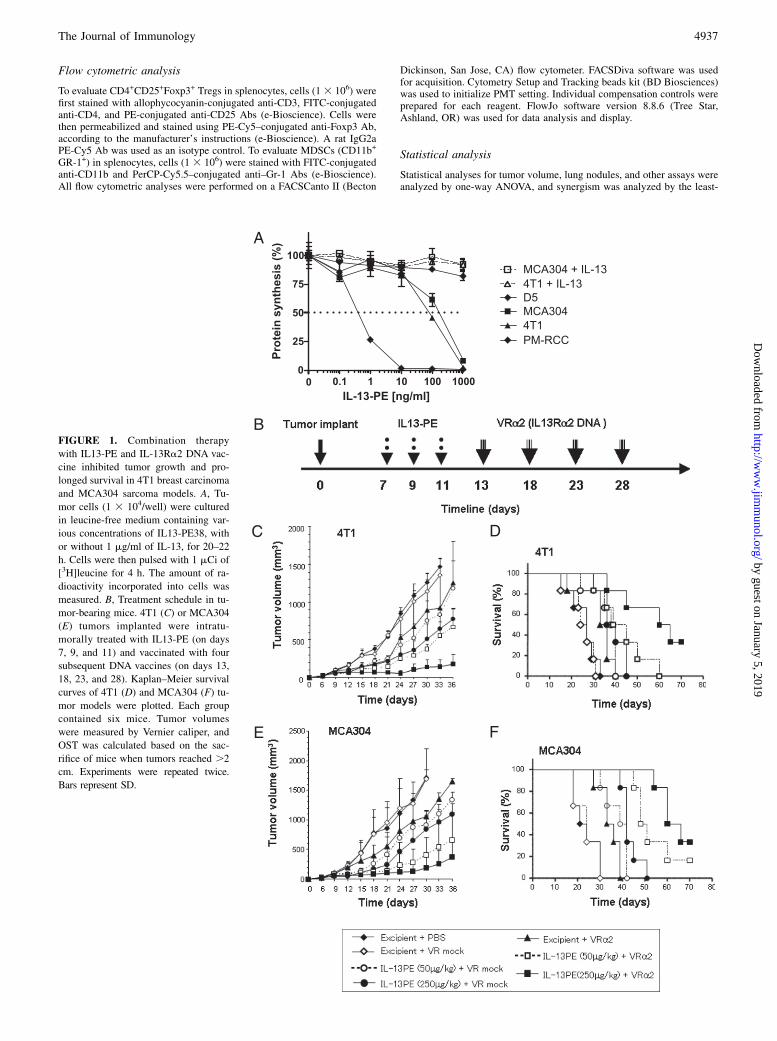
FIGURE 1. Combination therapy
with IL13-PE and IL-13Ra2 DNA vac-
cine inhibited tumor growth and pro-
longed survival in 4T1 breast carcinoma
and MCA304 sarcoma models. A, Tu-
mor cells (1 3 104/well) were cultured
in leucine-free medium containing var-
ious concentrations of IL13-PE38, with
or without 1 mg/ml of IL-13, for 20–22
h. Cells were then pulsed with 1 mCi of
[3H]leucine for 4 h. The amount of ra-
dioactivity incorporated into cells was
measured. B, Treatment schedule in tu-
mor-bearing mice. 4T1 (C) or MCA304
(E) tumors implanted were intratu-
morally treated with IL13-PE (on days
7, 9, and 11) and vaccinated with four
subsequent DNA vaccines (on days 13,
18, 23, and 28). Kaplan–Meier survival
curves of 4T1 (D) and MCA304 (F) tu-
mor models were plotted. Each group
contained six mice. Tumor volumes
were measured by Vernier caliper, and
OST was calculated based on the sac-
rifice of mice when tumors reached .2
cm. Experiments were repeated twice.
Bars represent SD.
The Journal of Immunology 4937
by guest on January 5, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
squares regression test (GraphPad Prism software version 5.02) and re-stricted maximum likelihood (REML) approach. Survival curves weregenerated by the Kaplan–Meier method and compared using the log-ranktest.
ResultsCytotoxicity of IL13-PE in murine tumor cell lines
Sensitivity of murine tumor cell lines to IL13-PE was tested bymeasuring protein synthesis inhibition, which was shown to bedirectly proportional to cell death (29). As shown in Fig. 1A, IL-13Ra2+ MCA304 and 4T1 cell lines were moderately sensitive toIL13-PE in a concentration-dependent manner (IC50 was 80 and110 ng/ml in 4T1 and MCA304 cell lines, respectively). An excessof IL-13 abrogated the cytotoxic effect of IL13-PE in both celllines, indicating receptor specificity. Consistent with the low orundetectable expression levels of IL-13Ra2 RNA in D5 mela-noma cells, we did not find significant cytotoxicity in this cell line.PM-RCC, a renal cell carcinoma cell line served as a positivecontrol (7).
Combination therapy with IL13-PE and IL-13Ra2 DNAvaccination inhibited tumor growth and prolonged survival ofanimals in established 4T1 breast carcinoma and MCA304sarcoma tumor models
To evaluate the efficacy of IL13-PE combined with IL-13Ra2 DNAvaccination, we tested two models of established murine tumors:4T1 breast carcinoma and MCA304 sarcoma. First, we evaluatedthe 4T1 tumor model, in which mice with established tumors (50–70 mm3) received intratumoral injection of IL13-PE (100 or 250mg/kg) or excipient on days 7, 9, and 11 and then were vaccinatedwith IL-13Ra2 DNA by i.m. injection (100 mg/injection) on days13, 18, 23, and 28 posttumor implantation (Fig. 1B). As shown inFig. 1C, treatment of mice with IL13-PE combined with VR mockvaccine delayed 4T1 tumor growth following tumor implantationcompared with control. However, the tumor in these groups ofmice started to grow again after day 21. In contrast, the tumorgrowth in mice treated with IL13-PE and IL-13Ra2 DNA vaccinecontinued to be reduced during the treatment. On day 30, thetumor volumes of 4T1 tumors in mice receiving IL13-PE (250mg/kg) combined with the IL-13Ra2 DNA vaccine (142 6 93mm3) were significantly smaller than those of mice receiving theIL-13Ra2 DNA vaccine alone (898 6 481 mm3; p , 0.01) orIL13-PE alone (514 6 197 mm3; p , 0.05) (Fig. 1C). IrrelevantcDNA plasmid vector encoding human IL-2Rgc did not inhibittumor growth, which was the same as for the VR mock-vaccinatedgroup (data not shown). As shown in Fig. 1D, overall sacrificetime (OST) of animals (tumor-bearing mice were sacrificed whentumor volume reached 2 cm in diameter, according to NIH animalguidelines) was 31 and 30 d in PBS- and VR mock-vaccinatedcontrol groups, whereas it was increased to 40 and 45 d in the IL-13Ra2 DNA vaccine-alone group and IL13-PE–alone group, re-spectively. A significantly prolonged OST was observed in thecombination-therapy group (65 d), which was .2-fold that ofthe control group. Similar results were observed in the MCA304sarcoma model (Fig. 1E, 1F). Furthermore, complete responses(tumor-free survival) were observed in two of six mice in both theMCA304 and 4T1 models. These results indicated that combina-tion therapy with IL13-PE and IL-13Ra2 DNA vaccination couldbe effective in significantly reducing tumor burden and prolongingsurvival in 4T1 and MCA304 tumor-bearing mice. Our calculationby least-squares regression test and REML approach indicatedthat the combination therapy synergistically enhanced the effectof antitumor activity in the 4T1 tumor model (p , 0.001).
Combination therapy inhibited lung and lymph node metastasisin orthotopic 4T1 breast tumor model
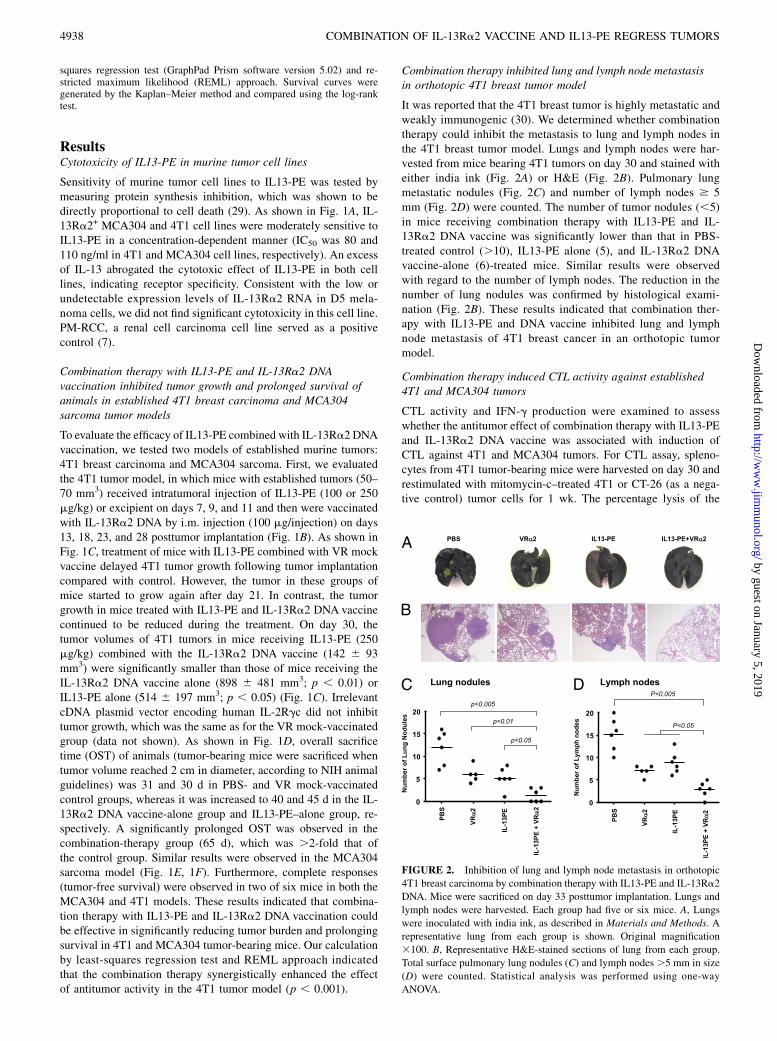
It was reported that the 4T1 breast tumor is highly metastatic andweakly immunogenic (30). We determined whether combinationtherapy could inhibit the metastasis to lung and lymph nodes inthe 4T1 breast tumor model. Lungs and lymph nodes were har-vested from mice bearing 4T1 tumors on day 30 and stained witheither india ink (Fig. 2A) or H&E (Fig. 2B). Pulmonary lungmetastatic nodules (Fig. 2C) and number of lymph nodes $ 5mm (Fig. 2D) were counted. The number of tumor nodules (,5)in mice receiving combination therapy with IL13-PE and IL-13Ra2 DNA vaccine was significantly lower than that in PBS-treated control (.10), IL13-PE alone (5), and IL-13Ra2 DNAvaccine-alone (6)-treated mice. Similar results were observedwith regard to the number of lymph nodes. The reduction in thenumber of lung nodules was confirmed by histological exami-nation (Fig. 2B). These results indicated that combination ther-apy with IL13-PE and DNA vaccine inhibited lung and lymphnode metastasis of 4T1 breast cancer in an orthotopic tumormodel.
Combination therapy induced CTL activity against established4T1 and MCA304 tumors
CTL activity and IFN-g production were examined to assesswhether the antitumor effect of combination therapy with IL13-PEand IL-13Ra2 DNA vaccine was associated with induction ofCTL against 4T1 and MCA304 tumors. For CTL assay, spleno-cytes from 4T1 tumor-bearing mice were harvested on day 30 andrestimulated with mitomycin-c–treated 4T1 or CT-26 (as a nega-tive control) tumor cells for 1 wk. The percentage lysis of the
FIGURE 2. Inhibition of lung and lymph node metastasis in orthotopic
4T1 breast carcinoma by combination therapy with IL13-PE and IL-13Ra2
DNA. Mice were sacrificed on day 33 posttumor implantation. Lungs and
lymph nodes were harvested. Each group had five or six mice. A, Lungs
were inoculated with india ink, as described in Materials and Methods. A
representative lung from each group is shown. Original magnification
3100. B, Representative H&E-stained sections of lung from each group.
Total surface pulmonary lung nodules (C) and lymph nodes.5 mm in size
(D) were counted. Statistical analysis was performed using one-way
ANOVA.
4938 COMBINATION OF IL-13Ra2 VACCINE AND IL13-PE REGRESS TUMORS
by guest on January 5, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
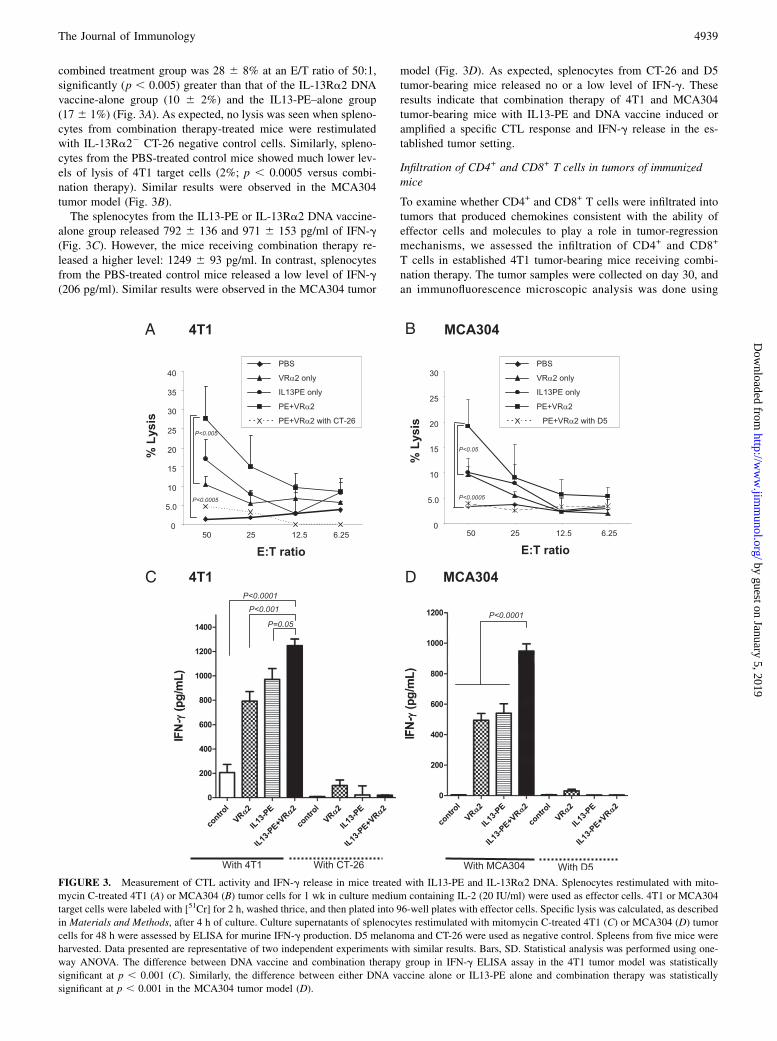
combined treatment group was 28 6 8% at an E/T ratio of 50:1,significantly (p , 0.005) greater than that of the IL-13Ra2 DNAvaccine-alone group (10 6 2%) and the IL13-PE–alone group(17 6 1%) (Fig. 3A). As expected, no lysis was seen when spleno-cytes from combination therapy-treated mice were restimulatedwith IL-13Ra22 CT-26 negative control cells. Similarly, spleno-cytes from the PBS-treated control mice showed much lower lev-els of lysis of 4T1 target cells (2%; p , 0.0005 versus combi-nation therapy). Similar results were observed in the MCA304tumor model (Fig. 3B).The splenocytes from the IL13-PE or IL-13Ra2 DNA vaccine-
alone group released 792 6 136 and 971 6 153 pg/ml of IFN-g(Fig. 3C). However, the mice receiving combination therapy re-leased a higher level: 1249 6 93 pg/ml. In contrast, splenocytesfrom the PBS-treated control mice released a low level of IFN-g(206 pg/ml). Similar results were observed in the MCA304 tumor
model (Fig. 3D). As expected, splenocytes from CT-26 and D5tumor-bearing mice released no or a low level of IFN-g. Theseresults indicate that combination therapy of 4T1 and MCA304tumor-bearing mice with IL13-PE and DNA vaccine induced oramplified a specific CTL response and IFN-g release in the es-tablished tumor setting.
Infiltration of CD4+ and CD8+ T cells in tumors of immunizedmice
To examine whether CD4+ and CD8+ T cells were infiltrated intotumors that produced chemokines consistent with the ability ofeffector cells and molecules to play a role in tumor-regressionmechanisms, we assessed the infiltration of CD4+ and CD8+
T cells in established 4T1 tumor-bearing mice receiving combi-nation therapy. The tumor samples were collected on day 30, andan immunofluorescence microscopic analysis was done using
FIGURE 3. Measurement of CTL activity and IFN-g release in mice treated with IL13-PE and IL-13Ra2 DNA. Splenocytes restimulated with mito-
mycin C-treated 4T1 (A) or MCA304 (B) tumor cells for 1 wk in culture medium containing IL-2 (20 IU/ml) were used as effector cells. 4T1 or MCA304
target cells were labeled with [51Cr] for 2 h, washed thrice, and then plated into 96-well plates with effector cells. Specific lysis was calculated, as described
in Materials and Methods, after 4 h of culture. Culture supernatants of splenocytes restimulated with mitomycin C-treated 4T1 (C) or MCA304 (D) tumor
cells for 48 h were assessed by ELISA for murine IFN-g production. D5 melanoma and CT-26 were used as negative control. Spleens from five mice were
harvested. Data presented are representative of two independent experiments with similar results. Bars, SD. Statistical analysis was performed using one-
way ANOVA. The difference between DNA vaccine and combination therapy group in IFN-g ELISA assay in the 4T1 tumor model was statistically
significant at p , 0.001 (C). Similarly, the difference between either DNA vaccine alone or IL13-PE alone and combination therapy was statistically
significant at p , 0.001 in the MCA304 tumor model (D).
The Journal of Immunology 4939
by guest on January 5, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
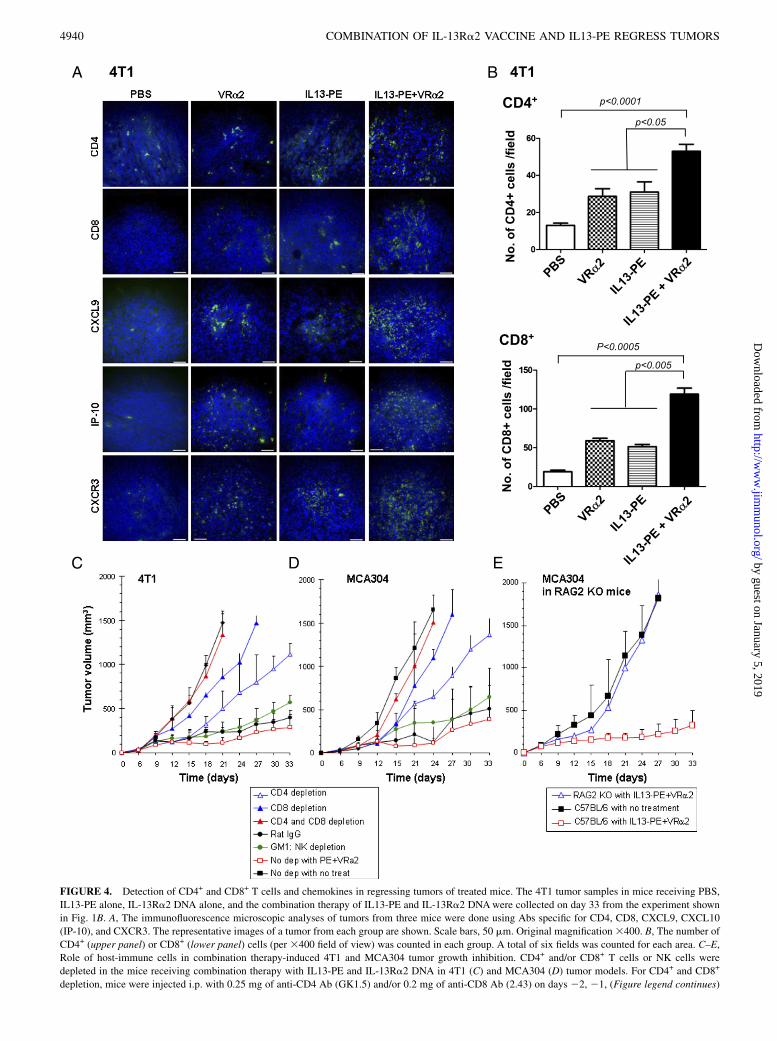
FIGURE 4. Detection of CD4+ and CD8+ T cells and chemokines in regressing tumors of treated mice. The 4T1 tumor samples in mice receiving PBS,
IL13-PE alone, IL-13Ra2 DNA alone, and the combination therapy of IL13-PE and IL-13Ra2 DNAwere collected on day 33 from the experiment shown
in Fig. 1B. A, The immunofluorescence microscopic analyses of tumors from three mice were done using Abs specific for CD4, CD8, CXCL9, CXCL10
(IP-10), and CXCR3. The representative images of a tumor from each group are shown. Scale bars, 50 mm. Original magnification3400. B, The number of
CD4+ (upper panel) or CD8+ (lower panel) cells (per 3400 field of view) was counted in each group. A total of six fields was counted for each area. C–E,
Role of host-immune cells in combination therapy-induced 4T1 and MCA304 tumor growth inhibition. CD4+ and/or CD8+ T cells or NK cells were
depleted in the mice receiving combination therapy with IL13-PE and IL-13Ra2 DNA in 4T1 (C) and MCA304 (D) tumor models. For CD4+ and CD8+
depletion, mice were injected i.p. with 0.25 mg of anti-CD4 Ab (GK1.5) and/or 0.2 mg of anti-CD8 Ab (2.43) on days 22, 21, (Figure legend continues)
4940 COMBINATION OF IL-13Ra2 VACCINE AND IL13-PE REGRESS TUMORS
by guest on January 5, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

specific Abs. As shown in Fig. 4A, a greater density of CD4 andCD8+ cells was identified in tumor samples from the mice re-
ceiving combination therapy compared with control tumors. The
number of CD4 cells in the combination-therapy group (53 6 4)
was higher than in the control group (13 6 2; p , 0.0001), the
DNA vaccine-alone group (29 6 4; p , 0.05), and the IL13-PE–
alone group (31 6 6; p , 0.05) (Fig. 4B). The number of CD8+
cells in the combination-therapy group (1196 8) was significantly
higher than in the control group (19 6 2; p = 0.0002), the DNA
vaccine-alone group (59 6 3; p , 0.005), and the IL13-PE–alonegroup (51 6 3; p , 0.005) (Fig. 4B).Tumor samples were also stained with anti-MIG/CXCL9, anti–
IP-10/CXCL10, and CXCR3 Abs (Fig. 4A). CXCL9 and CXCL10chemokines were selected because they were shown to be in-volved in CTL-induced tumor regression (31–33). In addition, themajority of activated T lymphocytes express CXCR3 (34), whichbelongs to the CXCR subfamily and has three endogenousligands: CXCL9 (MIG), CXCL10 (IP-10), and CXCL11 (IFN-inducible T cell a chemoattractant). Tumor samples collectedfrom the mice receiving combination therapy were positive forCXCL9 and CXCL10, whereas control tumor samples were neg-ative for these chemokines. CXCR3, CXCL9, and CXCL10 werehighly expressed in the tumors of mice that received combinationtherapy compared with the other groups. Similar results wereobserved in the MCA304 tumor model (data not shown). Theseresults suggested that combination therapy with IL13-PE and IL-13Ra2 DNA vaccine induced regression of 4T1 and MCA304tumors and involved infiltration of CD4+ and CD8+ T cells andproduction of proinflammatory chemokines in tumors.
In vivo depletion of CD4+ and CD8+ T cells significantlyimpaired the antitumor effects of combination therapy
We examined the involvement of CD4+ T cells, CD8+ T cells, orNK cells in antitumor effects associated with combination therapy.Each cell type was depleted in vivo using specific Abs. The flowcytometric analysis confirmed depletion of the appropriate cellpopulation by $95%. BALB/c mice with 4T1 tumors and C57BL/6 mice with MCA304 tumors receiving combination therapy weredepleted of NK cells, CD4+ T cells, CD8+ T cells, or CD4+ andCD8+ T cells. In the 4T1 model, the depletion of CD4+ or CD8+
T cells partially reversed the protective effect of the combinationtherapy (CD8 more so than CD4) (Fig. 4C). Depletion of bothCD4+ and CD8+ T cells completely abrogated protection, becausethe tumor size did not decrease (1335 6 272 mm3), and itremained similar in size to the untreated tumors (14746 104 mm3)on day 21. NK cell depletion showed no effect on the tumorgrowth. Similar results were observed in the MCA304 tumormodel (Fig. 4D). Taken together, these results suggested thatCD8+ T cells, with help from CD4+ T cells, but not NK cells, arecritical for the reduction in 4T1 and MCA304 tumor growth seenwith combination therapy.To further confirm the role of adaptive immunity, RAG-2
knockout mice with MCA304 tumors were treated with combi-nation therapy. It was shown that RAG-2 knockout mice are de-ficient in both T and B cells (35). As shown in Fig. 4E, thecombination therapy failed to inhibit the growth of MCA304tumors in RAG-2 knockout mice. MCA304 tumor volume inRAG-2 knockout mice (1861 6 197 mm3) was similar to that in
C57BL/6 mice without combination therapy (1820 6 287 mm3) atday 27. These results suggested that T cells and possibly B cellsare required to achieve inhibition of 4T1 and MCA304 tumordevelopment.
Ab generated by combination therapy modestly inhibitedproliferation of tumor cells
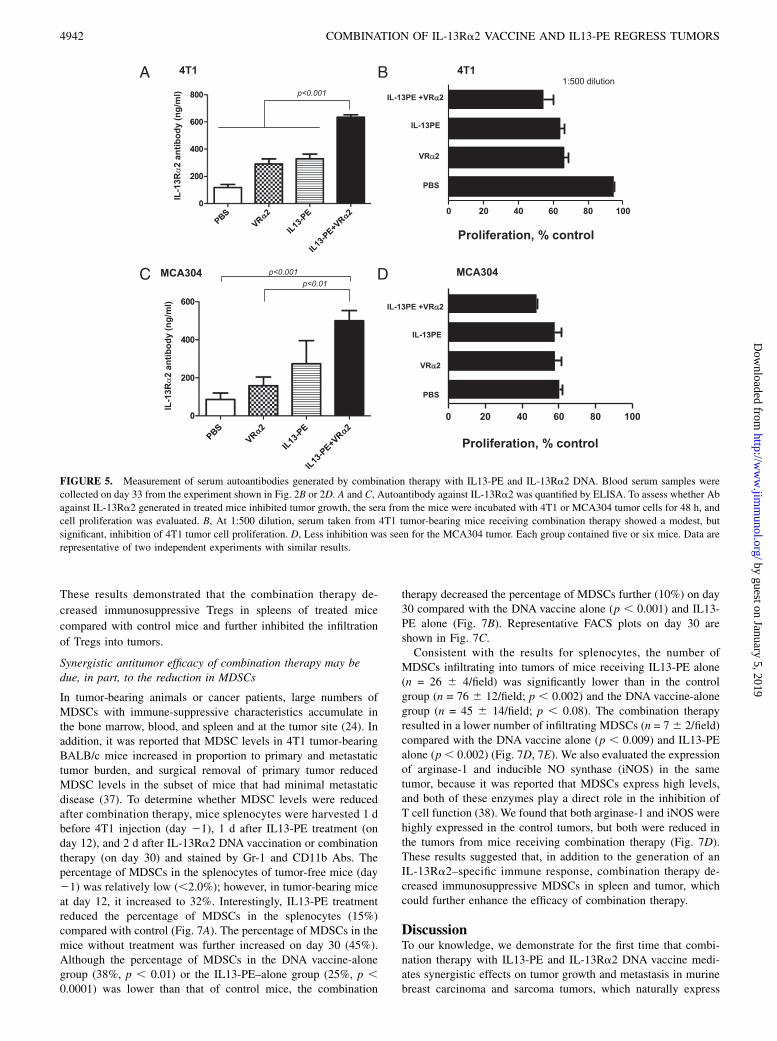
We also determined whether Abs against IL-13Ra2 are generatedin animals receiving combination therapy. Blood serum sampleswere collected on day 33 from 4T1 and MCA304 tumor-bearingmice. An ELISA assay was performed to measure the Ab levelsagainst IL-13Ra2. As shown in Fig. 5A, mice vaccinated with IL-13Ra2 cDNA generated Ab to IL-13Ra2. The level of Ab wassignificantly increased in mice receiving combination therapy(635 6 40 ng/ml) compared with IL13-PE alone (328 6 78 ng/ml), DNA vaccine alone (290 6 82 ng/ml), or control (118 6 51ng/ml) in the 4T1 tumor model. Similar results were obtained inthe MCA304 tumor model (Fig. 5C). To assess the effect of Abon tumor growth, the serum from vaccinated mice was incubatedwith 4T1 or MCA304 tumor cells for 48 h, and cell proliferationwas evaluated. Three different dilutions of serum were tested. At1:500 dilution, no nonspecific cytotoxicity was observed. At thisdilution, serum from 4T1 tumor-bearing mice receiving combi-nation therapy showed a modest, but significant, inhibition of4T1 tumor cell proliferation (57% compared with PBS control[95%]) (Fig. 5B). In the MCA304 tumor model, the inhibition oftumor cell proliferation was modest compared with PBS control(Fig. 5D). These results suggested that Ab against IL-13Ra2 hasa modest growth-inhibitory effect on IL-13Ra2–expressing tumorcells.
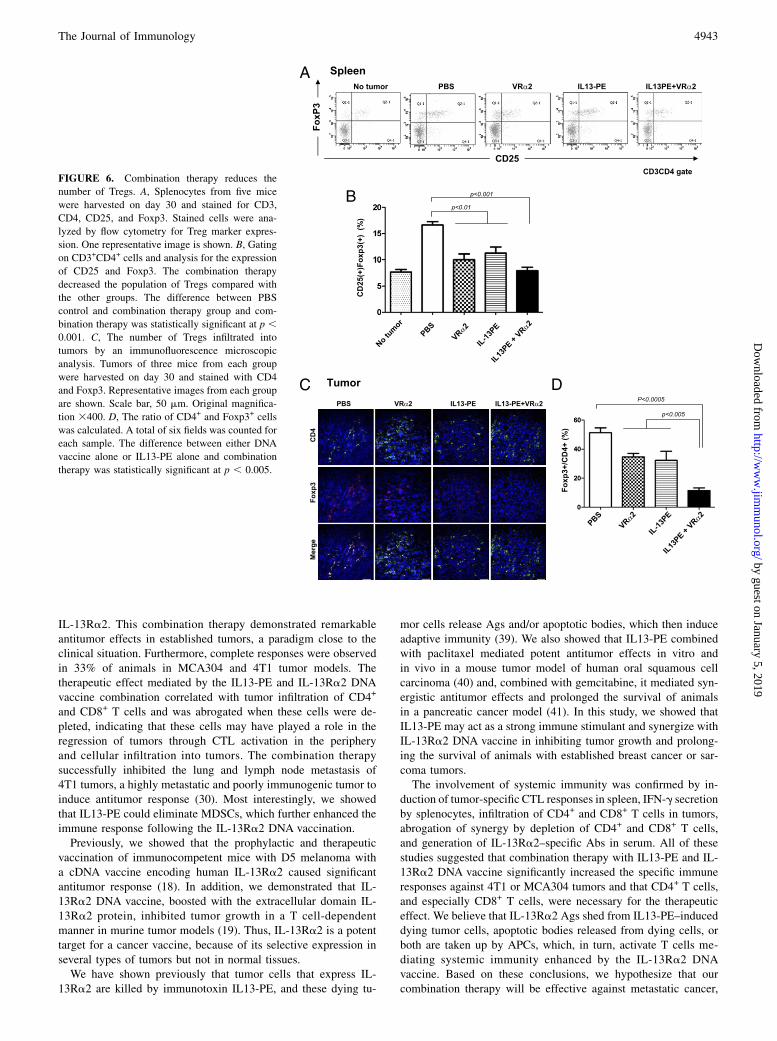
Enhancement of antitumor efficacy by combination therapymay be due, in part, to the reduction in Tregs
It was reported that CD4+CD25+Foxp3+ Tregs contribute to theimmune suppression that occurs in cancer patients (36). We hy-pothesized that the enhancement of the efficacy by combinationtherapy may be due to the reduction in the number of Tregs intumor-bearing mice. To test the hypothesis, mice splenocytes wereharvested on day 30 and stained for CD3, CD4, CD25, and Foxp3(five mice in each group, representative Fig. 6A). Gating on CD3+
CD4+ cells and analyzing for the expression of CD25 and Foxp3,we found that the percentage of CD4+CD25+Foxp3+ cells in thesplenocytes of tumor-free mice was relatively low (7.5%). Therewas a significant increase in the Treg population in tumor-bearingmice without treatment (17%) on day 30. Although the proportionof Tregs in vaccine-alone (10%) or IL13-PE–alone (12%) micewas lower than that of control mice (p , 0.01), the combinationtherapy further decreased the Treg (8%) population (p , 0.001)
(Fig. 6B). We also examined the number of Tregs infiltrating into
tumors by an immunofluorescence microscopic analysis. Tumors
were harvested from each group of mice on day 30 and stained for
CD4 and Foxp3 (Fig. 6C). Consistent with the results of spleno-
cytes, the ratio of Foxp3+/CD4+ cells in tumors of mice receiving
IL13-PE alone (32%) or DNA vaccine alone (33%) was signifi-
cantly lower than in the control group (51%) (p , 0.05). In-
terestingly, Tregs in mice receiving combination therapy were
significantly lower (11.5%) compared with the IL13-PE–alone
group or the DNA vaccine-alone group (p , 0.005) (Fig. 6D).
5, 11, and 17 relative to the tumor implantation. NK cells were depleted with rabbit anti–asialo-GM1 Ab (50 mg/injection) on the same days as CD4+, CD8+
T cell depletion (E). RAG-2 knockout or wild-type C57BL/6 mice bearing MCA304 tumor received combination therapy. Each group contained five or
six mice.
The Journal of Immunology 4941
by guest on January 5, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
These results demonstrated that the combination therapy de-
creased immunosuppressive Tregs in spleens of treated mice
compared with control mice and further inhibited the infiltration
of Tregs into tumors.
Synergistic antitumor efficacy of combination therapy may bedue, in part, to the reduction in MDSCs
In tumor-bearing animals or cancer patients, large numbers ofMDSCs with immune-suppressive characteristics accumulate inthe bone marrow, blood, and spleen and at the tumor site (24). Inaddition, it was reported that MDSC levels in 4T1 tumor-bearingBALB/c mice increased in proportion to primary and metastatictumor burden, and surgical removal of primary tumor reducedMDSC levels in the subset of mice that had minimal metastaticdisease (37). To determine whether MDSC levels were reducedafter combination therapy, mice splenocytes were harvested 1 dbefore 4T1 injection (day 21), 1 d after IL13-PE treatment (onday 12), and 2 d after IL-13Ra2 DNA vaccination or combinationtherapy (on day 30) and stained by Gr-1 and CD11b Abs. Thepercentage of MDSCs in the splenocytes of tumor-free mice (day21) was relatively low (,2.0%); however, in tumor-bearing miceat day 12, it increased to 32%. Interestingly, IL13-PE treatmentreduced the percentage of MDSCs in the splenocytes (15%)compared with control (Fig. 7A). The percentage of MDSCs in themice without treatment was further increased on day 30 (45%).Although the percentage of MDSCs in the DNA vaccine-alonegroup (38%, p , 0.01) or the IL13-PE–alone group (25%, p ,0.0001) was lower than that of control mice, the combination
therapy decreased the percentage of MDSCs further (10%) on day30 compared with the DNA vaccine alone (p , 0.001) and IL13-PE alone (Fig. 7B). Representative FACS plots on day 30 areshown in Fig. 7C.Consistent with the results for splenocytes, the number of
MDSCs infiltrating into tumors of mice receiving IL13-PE alone(n = 26 6 4/field) was significantly lower than in the controlgroup (n = 76 6 12/field; p , 0.002) and the DNA vaccine-alonegroup (n = 45 6 14/field; p , 0.08). The combination therapyresulted in a lower number of infiltrating MDSCs (n = 76 2/field)compared with the DNA vaccine alone (p , 0.009) and IL13-PEalone (p, 0.002) (Fig. 7D, 7E). We also evaluated the expressionof arginase-1 and inducible NO synthase (iNOS) in the sametumor, because it was reported that MDSCs express high levels,and both of these enzymes play a direct role in the inhibition ofT cell function (38). We found that both arginase-1 and iNOS werehighly expressed in the control tumors, but both were reduced inthe tumors from mice receiving combination therapy (Fig. 7D).These results suggested that, in addition to the generation of anIL-13Ra2–specific immune response, combination therapy de-creased immunosuppressive MDSCs in spleen and tumor, whichcould further enhance the efficacy of combination therapy.
DiscussionTo our knowledge, we demonstrate for the first time that combi-nation therapy with IL13-PE and IL-13Ra2 DNA vaccine medi-ates synergistic effects on tumor growth and metastasis in murinebreast carcinoma and sarcoma tumors, which naturally express
FIGURE 5. Measurement of serum autoantibodies generated by combination therapy with IL13-PE and IL-13Ra2 DNA. Blood serum samples were
collected on day 33 from the experiment shown in Fig. 2B or 2D. A and C, Autoantibody against IL-13Ra2 was quantified by ELISA. To assess whether Ab
against IL-13Ra2 generated in treated mice inhibited tumor growth, the sera from the mice were incubated with 4T1 or MCA304 tumor cells for 48 h, and
cell proliferation was evaluated. B, At 1:500 dilution, serum taken from 4T1 tumor-bearing mice receiving combination therapy showed a modest, but
significant, inhibition of 4T1 tumor cell proliferation. D, Less inhibition was seen for the MCA304 tumor. Each group contained five or six mice. Data are
representative of two independent experiments with similar results.
4942 COMBINATION OF IL-13Ra2 VACCINE AND IL13-PE REGRESS TUMORS
by guest on January 5, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
IL-13Ra2. This combination therapy demonstrated remarkableantitumor effects in established tumors, a paradigm close to theclinical situation. Furthermore, complete responses were observedin 33% of animals in MCA304 and 4T1 tumor models. Thetherapeutic effect mediated by the IL13-PE and IL-13Ra2 DNAvaccine combination correlated with tumor infiltration of CD4+
and CD8+ T cells and was abrogated when these cells were de-pleted, indicating that these cells may have played a role in theregression of tumors through CTL activation in the peripheryand cellular infiltration into tumors. The combination therapysuccessfully inhibited the lung and lymph node metastasis of4T1 tumors, a highly metastatic and poorly immunogenic tumor toinduce antitumor response (30). Most interestingly, we showedthat IL13-PE could eliminate MDSCs, which further enhanced theimmune response following the IL-13Ra2 DNA vaccination.Previously, we showed that the prophylactic and therapeutic
vaccination of immunocompetent mice with D5 melanoma witha cDNA vaccine encoding human IL-13Ra2 caused significantantitumor response (18). In addition, we demonstrated that IL-13Ra2 DNA vaccine, boosted with the extracellular domain IL-13Ra2 protein, inhibited tumor growth in a T cell-dependentmanner in murine tumor models (19). Thus, IL-13Ra2 is a potenttarget for a cancer vaccine, because of its selective expression inseveral types of tumors but not in normal tissues.We have shown previously that tumor cells that express IL-
13Ra2 are killed by immunotoxin IL13-PE, and these dying tu-
mor cells release Ags and/or apoptotic bodies, which then induceadaptive immunity (39). We also showed that IL13-PE combinedwith paclitaxel mediated potent antitumor effects in vitro andin vivo in a mouse tumor model of human oral squamous cellcarcinoma (40) and, combined with gemcitabine, it mediated syn-ergistic antitumor effects and prolonged the survival of animalsin a pancreatic cancer model (41). In this study, we showed thatIL13-PE may act as a strong immune stimulant and synergize withIL-13Ra2 DNA vaccine in inhibiting tumor growth and prolong-ing the survival of animals with established breast cancer or sar-coma tumors.The involvement of systemic immunity was confirmed by in-
duction of tumor-specific CTL responses in spleen, IFN-g secretionby splenocytes, infiltration of CD4+ and CD8+ T cells in tumors,abrogation of synergy by depletion of CD4+ and CD8+ T cells,and generation of IL-13Ra2–specific Abs in serum. All of thesestudies suggested that combination therapy with IL13-PE and IL-13Ra2 DNA vaccine significantly increased the specific immuneresponses against 4T1 or MCA304 tumors and that CD4+ T cells,and especially CD8+ T cells, were necessary for the therapeuticeffect. We believe that IL-13Ra2 Ags shed from IL13-PE–induceddying tumor cells, apoptotic bodies released from dying cells, orboth are taken up by APCs, which, in turn, activate T cells me-diating systemic immunity enhanced by the IL-13Ra2 DNAvaccine. Based on these conclusions, we hypothesize that ourcombination therapy will be effective against metastatic cancer,
FIGURE 6. Combination therapy reduces the
number of Tregs. A, Splenocytes from five mice
were harvested on day 30 and stained for CD3,
CD4, CD25, and Foxp3. Stained cells were ana-
lyzed by flow cytometry for Treg marker expres-
sion. One representative image is shown. B, Gating
on CD3+CD4+ cells and analysis for the expression
of CD25 and Foxp3. The combination therapy
decreased the population of Tregs compared with
the other groups. The difference between PBS
control and combination therapy group and com-
bination therapy was statistically significant at p ,0.001. C, The number of Tregs infiltrated into
tumors by an immunofluorescence microscopic
analysis. Tumors of three mice from each group
were harvested on day 30 and stained with CD4
and Foxp3. Representative images from each group
are shown. Scale bar, 50 mm. Original magnifica-
tion 3400. D, The ratio of CD4+ and Foxp3+ cells
was calculated. A total of six fields was counted for
each sample. The difference between either DNA
vaccine alone or IL13-PE alone and combination
therapy was statistically significant at p , 0.005.
The Journal of Immunology 4943
by guest on January 5, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
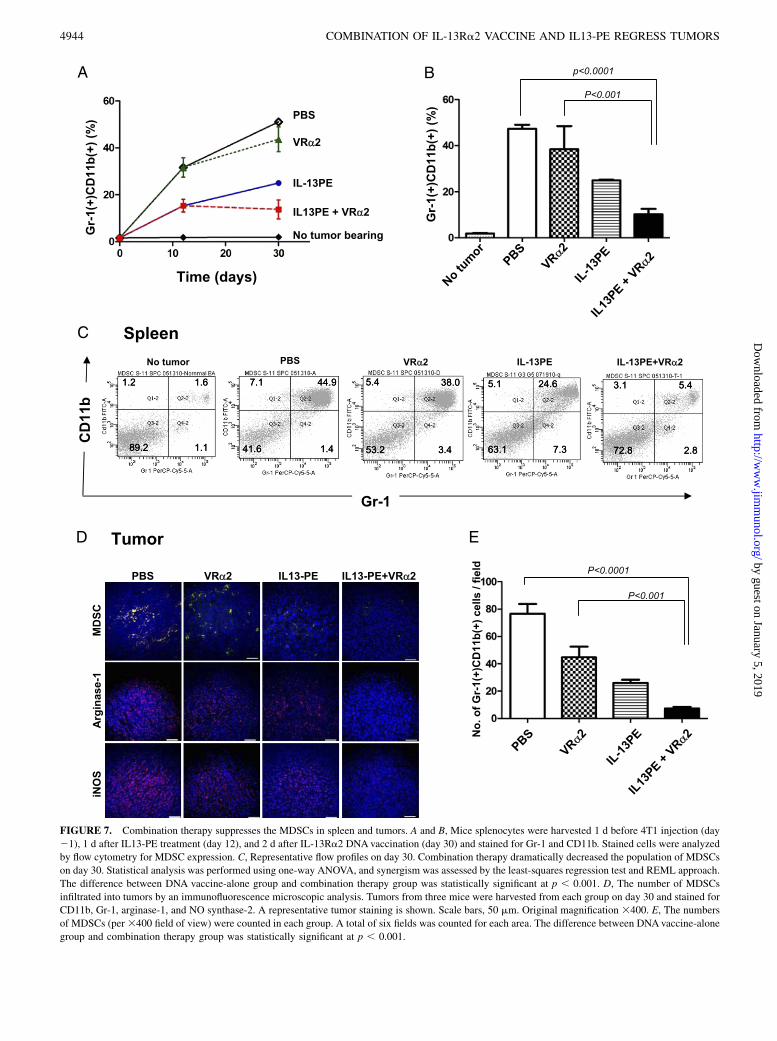
FIGURE 7. Combination therapy suppresses the MDSCs in spleen and tumors. A and B, Mice splenocytes were harvested 1 d before 4T1 injection (day
21), 1 d after IL13-PE treatment (day 12), and 2 d after IL-13Ra2 DNAvaccination (day 30) and stained for Gr-1 and CD11b. Stained cells were analyzed
by flow cytometry for MDSC expression. C, Representative flow profiles on day 30. Combination therapy dramatically decreased the population of MDSCs
on day 30. Statistical analysis was performed using one-way ANOVA, and synergism was assessed by the least-squares regression test and REML approach.
The difference between DNA vaccine-alone group and combination therapy group was statistically significant at p , 0.001. D, The number of MDSCs
infiltrated into tumors by an immunofluorescence microscopic analysis. Tumors from three mice were harvested from each group on day 30 and stained for
CD11b, Gr-1, arginase-1, and NO synthase-2. A representative tumor staining is shown. Scale bars, 50 mm. Original magnification 3400. E, The numbers
of MDSCs (per3400 field of view) were counted in each group. A total of six fields was counted for each area. The difference between DNAvaccine-alone
group and combination therapy group was statistically significant at p , 0.001.
4944 COMBINATION OF IL-13Ra2 VACCINE AND IL13-PE REGRESS TUMORS
by guest on January 5, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

even when tumors are treated with IL13-PE locally and vaccine isgiven systemically.Recent studies showed that some chemokines mediate T lym-
phocyte recruitment in a selective fashion. Experimental modelsdemonstrated that CXCL9 (MIG) and CXCL10 (IP-10) haveantitumorigenic activities through CD4+ and CD8+ T lymphocyterecruitment (42) and are involved in CTL-induced tumor regres-sion in mice. In our study, CXCR3, CXCL9, and CXCL10 wereexpressed at higher levels in tumors derived from mice receivingIL13-PE combined with IL-13Ra2 DNA vaccination comparedwith either control or single-therapy mice. These results suggestedthat CXC chemokine receptors and their ligands are most likelyproduced by infiltrating immune cells causing antitumor effect,because these chemokines act as potent T cell chemoattractantsand angiogenesis inhibitors through their interaction with CXCR3(31). Taken together, our results indicated that these chemokinesplay an important role in recruiting effector T lymphocytes intothe tumor to polarize and amplify Th-1–mediated antitumor cel-lular immunity in murine tumor models.Although the role of immunosurveillance to inhibit tumor pro-
gression is well documented, other obstacles prevent the elimi-nation of advanced tumors by immune cells, one of which is MDSCaccumulation. MDSCs in mice are heterogeneous myeloid cellsprimarily composed of CD11b+/Gr-1+ cells (43). Tumor-mediatedgeneration of MDSCs and their suppressive role against tumor-specific T cells have been well described in many tumor models(44). Recently, drugs, including vitamin D3 (45), sildenafil (46),and gemcitabine (27), have been used to ameliorate the MDSC-mediated suppressive environment. We hypothesized that becauseMDSCs express an IL-13R, they may be killed by IL13-PE. In-deed, in this study we found that combination therapy with IL13-PE and IL-13Ra2 DNA vaccine dramatically decreased MDSCsin both splenocytes and tumor compared with the control group.It was reported that increased levels of circulating MDSCs cor-related with disease stage and extent of metastatic tumor burden(26). Indeed, in our study, the number of MDSCs in both spleno-cytes and tumor correlated with the size of tumor burdens.Moreover, because MDSCs can induce Tregs, it is possible thatthe reduction in Tregs that we observed following combinationtherapy can be explained by the effect on MDSCs. Both effectsmay contribute to the increase in tumor immune response. Addi-tional studies are ongoing to further explore the mechanism bywhich IL13-PE and the IL-13Ra2 DNA vaccine synergize to re-duce these immunosuppressive elements.Cancer is a complex disease, and successful vaccine therapy
will require a multimodal or combinatorial treatment approach. Inthis article, we described the antitumor efficacy of combinationtherapy with IL13-PE and IL-13Ra2 DNA vaccine via directkilling of tumor cells by IL13-PE and the induction of a specificT cell-immune response against tumors, as well as likely throughthe inhibition of immunosuppressive elements, such as Tregs andMDSCs. Thus, our strategy identified a novel mechanism bywhich IL-13Ra2–expressing MDSCs can be eliminated inpathological settings using a combination therapy with IL13-PE.The results provide a strong rationale for combining these mo-dalities targeting IL-13Ra2 Ag on both tumors and MDSCs forthe treatment of patients with advanced cancers that express thisreceptor.
AcknowledgmentsWe thank Drs. Suzanne Epstein and Cheng-Hong Wei of the Center for
Biologics Evaluation and Research, U.S. Food and Drug Administration
for critical reading of the manuscript. We are grateful to Dr. Bharat
Joshi for IL13-PE, Dr. Heba Degheidy for FACS analysis, Dr. Samir Lab-
abidi for statistical analysis, and Pamela Dover for general support and
procuring reagents.
DisclosuresThe authors have no financial conflicts of interest.
References1. Rosenberg, S. A., J. C. Yang, and N. P. Restifo. 2004. Cancer immunotherapy:
moving beyond current vaccines. Nat. Med. 10: 909–915.2. Rice, J., C. H. Ottensmeier, and F. K. Stevenson. 2008. DNA vaccines: precision
tools for activating effective immunity against cancer. Nat. Rev. Cancer 8: 108–120.
3. Cheever, M. A., and C. S. Higano. 2011. PROVENGE (Sipuleucel-T) in prostatecancer: the first FDA-approved therapeutic cancer vaccine. Clin. Cancer Res. 17:3520–3526.
4. Emens, L. A., and E. M. Jaffee. 2005. Leveraging the activity of tumor vaccineswith cytotoxic chemotherapy. Cancer Res. 65: 8059–8064.
5. Gulley, J. L., R. A. Madan, and P. M. Arlen. 2007. Enhancing efficacy of ther-apeutic vaccinations by combination with other modalities. Vaccine 25 (Suppl.2): B89–B96.
6. Pastan, I., R. Hassan, D. J. FitzGerald, and R. J. Kreitman. 2007. Immunotoxintreatment of cancer. Annu. Rev. Med. 58: 221–237.
7. Puri, R. K., P. Leland, N. I. Obiri, S. R. Husain, R. J. Kreitman, G. P. Haas,I. Pastan, and W. Debinski. 1996. Targeting of interleukin-13 receptor on humanrenal cell carcinoma cells by a recombinant chimeric protein composed ofinterleukin-13 and a truncated form of Pseudomonas exotoxin A (PE38QQR).Blood 87: 4333–4339.
8. Pastan, I., R. Hassan, D. J. Fitzgerald, and R. J. Kreitman. 2006. Immunotoxintherapy of cancer. Nat. Rev. Cancer 6: 559–565.
9. Kunwar, S., M. D. Prados, S. M. Chang, M. S. Berger, F. F. Lang,J. M. Piepmeier, J. H. Sampson, Z. Ram, P. H. Gutin, R. D. Gibbons, et al;Cintredekin Besudotox Intraparenchymal Study Group. 2007. Direct in-tracerebral delivery of cintredekin besudotox (IL13-PE38QQR) in recurrentmalignant glioma: a report by the Cintredekin Besudotox IntraparenchymalStudy Group. J. Clin. Oncol. 25: 837–844.
10. Romero, P., M. Pittet, V. Dutoit, A. Zippelius, D. Lienard, F. Lejeune,P. Guillaume, D. Rimoldi, D. Valmori, D. E. Speiser, and J.-C. Cerottini. 2002.Therapeutic cancer vaccines based on molecularly defined human tumor anti-gens. Vaccine 20 (Suppl. 4): A2–A7.
11. Husain, S. R., N. I. Obiri, P. Gill, T. Zheng, I. Pastan, W. Debinski, andR. K. Puri. 1997. Receptor for interleukin 13 on AIDS-associated Kaposi’ssarcoma cells serves as a new target for a potent Pseudomonas exotoxin-basedchimeric toxin protein. Clin. Cancer Res. 3: 151–156.
12. Husain, S. R., B. H. Joshi, and R. K. Puri. 2001. Interleukin-13 receptor asa unique target for anti-glioblastoma therapy. Int. J. Cancer 92: 168–175.
13. Joshi, B. H., G. E. Plautz, and R. K. Puri. 2000. Interleukin-13 receptor alphachain: a novel tumor-associated transmembrane protein in primary explants ofhuman malignant gliomas. Cancer Res. 60: 1168–1172.
14. Caput, D., P. Laurent, M. Kaghad, J. M. Lelias, S. Lefort, N. Vita, and P. Ferrara.1996. Cloning and characterization of a specific interleukin (IL)-13 bindingprotein structurally related to the IL-5 receptor alpha chain. J. Biol. Chem. 271:16921–16926.
15. Kawakami, M., K. Kawakami, S. Takahashi, M. Abe, and R. K. Puri. 2004.Analysis of interleukin-13 receptor a2 expression in human pediatric braintumors. Cancer 101: 1036–1042.
16. Kawakami, K., and R. K. Puri. 2003. Interleukin-13 and cancer. In Interleukin-13. F. Brombacher, ed. Landes Bioscience, Austin, TX, p. 65–78.
17. Fujisawa, T., B. Joshi, A. Nakajima, and R. K. Puri. 2009. A novel role ofinterleukin-13 receptor alpha2 in pancreatic cancer invasion and metastasis.Cancer Res. 69: 8678–8685.
18. Kawakami, K., M. Terabe, M. Kawakami, J. A. Berzofsky, and R. K. Puri. 2006.Characterization of a novel human tumor antigen interleukin-13 receptor alpha2chain. Cancer Res. 66: 4434–4442.
19. Nakashima, H., T. Fujisawa, S. R. Husain, and R. K. Puri. 2010. Interleukin-13receptor a2 DNA prime boost vaccine induces tumor immunity in murine tumormodels. J. Transl. Med. 8: 116.
20. Herber, D. L., S. Nagaraj, J. Y. Djeu, and D. I. Gabrilovich. 2007. Mechanismand therapeutic reversal of immune suppression in cancer. Cancer Res. 67:5067–5069.
21. Frey, A. B. 2006. Myeloid suppressor cells regulate the adaptive immune re-sponse to cancer. J. Clin. Invest. 116: 2587–2590.
22. Zea, A. H., P. C. Rodriguez, M. B. Atkins, C. Hernandez, S. Signoretti,J. Zabaleta, D. McDermott, D. Quiceno, A. Youmans, A. O’Neill, et al. 2005.Arginase-producing myeloid suppressor cells in renal cell carcinoma patients:a mechanism of tumor evasion. Cancer Res. 65: 3044–3048.
23. Gallina, G., L. Dolcetti, P. Serafini, C. De Santo, I. Marigo, M. P. Colombo,G. Basso, F. Brombacher, I. Borrello, P. Zanovello, et al. 2006. Tumors inducea subset of inflammatory monocytes with immunosuppressive activity on CD8+T cells. J. Clin. Invest. 116: 2777–2790.
24. Gabrilovich, D. I., and S. Nagaraj. 2009. Myeloid-derived suppressor cells asregulators of the immune system. Nat. Rev. Immunol. 9: 162–174.
25. Huang, B., P. Y. Pan, Q. Li, A. I. Sato, D. E. Levy, J. Bromberg, C. M. Divino,and S. H. Chen. 2006. Gr-1+CD115+ immature myeloid suppressor cells me-diate the development of tumor-induced T regulatory cells and T-cell anergy intumor-bearing host. Cancer Res. 66: 1123–1131.
The Journal of Immunology 4945
by guest on January 5, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

26. Diaz-Montero, C. M., M. L. Salem, M. I. Nishimura, E. Garrett-Mayer,D. J. Cole, and A. J. Montero. 2009. Increased circulating myeloid-derivedsuppressor cells correlate with clinical cancer stage, metastatic tumor burden,and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol. Immun-other. 58: 49–59.
27. Suzuki, E., V. Kapoor, A. S. Jassar, L. R. Kaiser, and S. M. Albelda. 2005.Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressorcells in tumor-bearing animals and enhances antitumor immune activity. Clin.Cancer Res. 11: 6713–6721.
28. Aslakson, C. J., and F. R. Miller. 1992. Selective events in the metastatic processdefined by analysis of the sequential dissemination of subpopulations of a mousemammary tumor. Cancer Res. 52: 1399–1405.
29. Joshi, B. H., and R. K. Puri. 2005. Optimization of expression and purificationof two biologically active chimeric fusion proteins that consist of humaninterleukin-13 and Pseudomonas exotoxin in Escherichia coli. Protein Expr.Purif. 39: 189–198.
30. Ostrand-Rosenberg, S., S. Baskar, N. Patterson, and V. K. Clements. 1996.Expression of MHC Class II and B7-1 and B7-2 costimulatory moleculesaccompanies tumor rejection and reduces the metastatic potential of tumor cells.Tissue Antigens 47: 414–421.
31. Liao, F., R. L. Rabin, J. R. Yannelli, L. G. Koniaris, P. Vanguri, and J. M. Farber.1995. Human Mig chemokine: biochemical and functional characterization. J.Exp. Med. 182: 1301–1314.
32. Di Carlo, E., A. Comes, A. M. Orengo, O. Rosso, R. Meazza, P. Musiani,M. P. Colombo, and S. Ferrini. 2004. IL-21 induces tumor rejection by specificCTL and IFN-gamma-dependent CXC chemokines in syngeneic mice. J.Immunol. 172: 1540–1547.
33. Inngjerdingen, M., B. Rolstad, and J. C. Ryan. 2003. Activating and inhibitoryLy49 receptors modulate NK cell chemotaxis to CXC chemokine ligand (CXCL)10 and CXCL12. J. Immunol. 171: 2889–2895.
34. Qin, S., J. B. Rottman, P. Myers, N. Kassam, M. Weinblatt, M. Loetscher,A. E. Koch, B. Moser, and C. R. Mackay. 1998. The chemokine receptorsCXCR3 and CCR5 mark subsets of T cells associated with certain inflammatoryreactions. J. Clin. Invest. 101: 746–754.
35. Shinkai, Y., G. Rathbun, K. P. Lam, E. M. Oltz, V. Stewart, M. Mendelsohn,J. Charron, M. Datta, F. Young, A. M. Stall, et al. 1992. RAG-2-deficient micelack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell68: 855–867.
36. Nomura, T., and S. Sakaguchi. 2005. Naturally arising CD25+CD4+ regulatoryT cells in tumor immunity. Curr. Top. Microbiol. Immunol. 293: 287–302.
37. Sinha, P., V. K. Clements, and S. Ostrand-Rosenberg. 2005. Reduction ofmyeloid-derived suppressor cells and induction of M1 macrophages facilitate therejection of established metastatic disease. J. Immunol. 174: 636–645.
38. Bronte, V., and P. Zanovello. 2005. Regulation of immune responses by L-arginine metabolism. Nat. Rev. Immunol. 5: 641–654.
39. Kawakami, K., M. Terabe, M. Kioi, J. A. Berzofsky, and R. K. Puri. 2006.Intratumoral therapy with IL13-PE38 results in effective CTL-mediated sup-pression of IL-13Ralpha2-expressing contralateral tumors. Clin. Cancer Res. 12:4678–4686.
40. Kioi, M., T. Shimamura, H. Nakashima, M. Hirota, I. Tohnai, S. R. Husain, andR. K. Puri. 2009. IL-13 cytotoxin has potent antitumor activity and synergizeswith paclitaxel in a mouse model of oral squamous cell carcinoma. Int. J. Cancer124: 1440–1448.
41. Fujisawa, T., H. Nakashima, A. Nakajima, B. H. Joshi, and R. K. Puri. 2011.Targeting IL-13Ra2 in human pancreatic ductal adenocarcinoma with com-bination therapy of IL-13-PE and gemcitabine. Int. J. Cancer 128: 1221–1231.
42. Narvaiza, I., G. Mazzolini, M. Barajas, M. Duarte, M. Zaratiegui, C. Qian,I. Melero, and J. Prieto. 2000. Intratumoral coinjection of two adenoviruses,one encoding the chemokine IFN-gamma-inducible protein-10 and anotherencoding IL-12, results in marked antitumoral synergy. J. Immunol. 164:3112–3122.
43. Rabinovich, G. A., D. Gabrilovich, and E. M. Sotomayor. 2007. Immunosup-pressive strategies that are mediated by tumor cells. Annu. Rev. Immunol. 25:267–296.
44. Gabrilovich, D. I., M. P. Velders, E. M. Sotomayor, and W. M. Kast. 2001.Mechanism of immune dysfunction in cancer mediated by immature Gr-1+myeloid cells. J. Immunol. 166: 5398–5406.
45. Kusmartsev, S., F. Cheng, B. Yu, Y. Nefedova, E. Sotomayor, R. Lush, andD. Gabrilovich. 2003. All-trans-retinoic acid eliminates immature myeloid cellsfrom tumor-bearing mice and improves the effect of vaccination. Cancer Res.63: 4441–4449.
46. Serafini, P., K. Meckel, M. Kelso, K. Noonan, J. Califano, W. Koch, L. Dolcetti,V. Bronte, and I. Borrello. 2006. Phosphodiesterase-5 inhibition augments en-dogenous antitumor immunity by reducing myeloid-derived suppressor cellfunction. J. Exp. Med. 203: 2691–2702.
4946 COMBINATION OF IL-13Ra2 VACCINE AND IL13-PE REGRESS TUMORS
by guest on January 5, 2019http://w
ww
.jimm
unol.org/D
ownloaded from


















