
α-Synuclein as an intrinsically disordered monomer - fact or...
34
Accepted Article This article has been accepted for publication and undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process, which may lead to differences between this version and the Version of Record. Please cite this article as doi: 10.1111/febs.12471 This article is protected by copyright. All rights reserved. α-Synuclein as an intrinsically disordered monomer: fact or artifact? Eduardo Coelho-Cerqueira 1# , Phelippe do Carmo-Gonçalves 1# , Anderson Sá Pinheiro 2 , Juliana Cortines 3 , Cristian Follmer 1 * 1 Department of Physical Chemistry, Institute of Chemistry, Federal University of Rio de Janeiro, Rio de Janeiro 21941-909, Brazil. 2 Department of Biochemistry, Institute of Chemistry, Federal University of Rio de Janeiro, Rio de Janeiro 21941-909, Brazil. 3 Institute of Microbiology, Federal University of Rio de Janeiro, Rio de Janeiro 21941- 909, Brazil. *Corresponding author: Tel: 55 21 2562 7752. Fax: 55 21 2562 7265. E-mail: [email protected] # E.C.C. and P.C.G. contributed equally to this work. Article type:Original Article Running title: Native oligomeric state of recombinant α-syn
Transcript of α-Synuclein as an intrinsically disordered monomer - fact or...

Acc
epte
d A
rtic
le
This article has been accepted for publication and undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process, which may lead to differences between this version and the Version of Record. Please cite this article as doi: 10.1111/febs.12471 This article is protected by copyright. All rights reserved.
α-Synuclein as an intrinsically disordered monomer: fact or
artifact?
Eduardo Coelho-Cerqueira1#, Phelippe do Carmo-Gonçalves1#, Anderson Sá Pinheiro2,
Juliana Cortines3, Cristian Follmer1*
1Department of Physical Chemistry, Institute of Chemistry, Federal University of Rio de
Janeiro, Rio de Janeiro 21941-909, Brazil. 2Department of Biochemistry, Institute of
Chemistry, Federal University of Rio de Janeiro, Rio de Janeiro 21941-909, Brazil.
3Institute of Microbiology, Federal University of Rio de Janeiro, Rio de Janeiro 21941-
909, Brazil.
*Corresponding author: Tel: 55 21 2562 7752. Fax: 55 21 2562 7265.
E-mail: [email protected]
# E.C.C. and P.C.G. contributed equally to this work.
Article type:Original Article
Running title: Native oligomeric state of recombinant α-syn

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Abbreviation list: BCIP: 5-bromo-4-chloro-3-indolyl-phosphate; BOG: n-octyl-β-
glucopyranoside; BSA: bovine serum albumin; CD: circular dichroism; ESI-MS:
electrospray ionization mass spectrometry; GHD: glutaraldehyde; GST: glutathione S-
transferase; HPLC: high-performance liquid chromatography; HSQC: heteronuclear
single quantum coherence; MALDI-TOF: matrix-assisted laser desorption/ionization –
time of flight; NAC: non-amyloid-β component; NBT: nitro blue tetrazolium; NMR:
nuclear magnetic resonance; SAXS: small-angle X-ray scattering; SDS-PAGE: sodium
dodecyl sulfate polyacrylamide gel electrophoresis; SEC: size exclusion chromatography.
Abstract
Fibrillization of the protein α-synuclein (α-syn) is a hallmark of Parkinson’s disease and
other α-synucleinopathies. The well-established idea that α-syn is a natively disordered
monomer prone to forming fibrils was recently challenged by data showing that the
protein mostly exists in vitro and in vivo as helically folded tetramers that are resistant to
fibrillization. These apparently conflicting findings may be reconciled by the idea that α-
syn exists as a disordered monomer in equilibrium with variable amounts of dynamic
oligomeric species. In this context, varying the approaches used for protein purification
such as the method used to lyse cells or the inclusion of denaturing agents could
dramatically perturb this equilibrium and hence alter the relative abundance of the
disordered monomer. Herein, we investigated how the current methods for α-syn
purification affect the structure and oligomeric state of the protein, discussing the main
pitfalls associated with the production of recombinant α-syn in Escherichia coli. We
demonstrated that α-syn was expressed in E. coli as a disordered monomer independent
of both the cell lysis method and the use of heating/acidification for protein purification.
In addition, we provide convincing evidence that the disordered monomer exists in

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
equilibrium with a dynamic dimer, which is not an artifact of the cross-linking protocol as
previously suggested. Unlike the helically folded tetramer, α-syn dimer is prone to
fibrillate and thus it may be an interesting target for anti-fibrillogenic molecules.
Keywords: α-synuclein; disordered monomer; dimer; purification; Escherichia coli.
Introduction:
The pathological characteristic of Parkinson’s disease (PD) is the degeneration of
dopaminergic neurons in the substantia nigra pars compacta accompanied by the
formation of intracellular fibrillar inclusions, termed Lewy bodies and Lewy neurites,
which are mainly composed of the small protein α-synuclein (α-syn) [1,2]. Besides PD,
α-syn is also involved in the Lewy body variant of Alzheimer’s disease (AD), diffuse
Lewy body disease (LBD) and multiple system atrophy [2-4]. α-Syn is a 14 kDa
presynaptic protein that acts as a non-classical chaperone during the formation of a
soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE)-
complex assembly in presynaptic terminals [5]. The role of α-syn in PD
neuropathogenesis is evidenced by the fact that three missense mutations in this protein,
A53T, A30P and E46K, are linked to rare, early-onset forms of PD [6,7].
α-Syn has long been thought to exist as a natively disordered monomer [8]. For this
reason, it has been postulated that the utilization of denaturing conditions to purify
bacterially expressed α-syn does not affect the native protein structure. However, this
view was challenged by recent data showing that α-syn isolated from neuronal and non-
neuronal cell lines exists predominantly as a putative tetramer that is rich in α-helices and

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
resistant to fibrillization [9]. Corroborating this hypothesis, an N-terminal GST fusion
construct of α-syn expressed in E. coli and purified under non-denaturing conditions
forms a non-fibrillogenic tetramer in addition to the unfolded monomer [10]. Moreover,
α-syn was found in intact cells mostly as tetramers that undergo destabilization and form
monomers after cell lysis [11]. In contrast with these data, Lashuel and colleagues
demonstrated that bacterially expressed recombinant α-syn behaves as a disordered
monomer when purified under denaturing or nondenaturing conditions. α-Syn has also
been found mostly as a disordered monomer in various mammalian cell lines and in
homogenates from rat, mouse and human brains, including human brain homogenates
from AD and LBD patients [12]. Unlike α-syn produced in bacteria, the protein expressed
in mammalian cell lines undergoes N-terminal acetylation, which may affect the
conformation of the protein [13].
In this work, we explored the oligomeric state of recombinant α-syn using different
strategies to lyse cells (osmotic shock versus ultrasound sonication) or to purify the
protein (heating versus acidification). Independent of the method used for protein
purification, α-syn was obtained predominantly as a disordered monomer in equilibrium
with a minor population of fibrillogenic, non-covalent dimers, which display a mixture of
random-coil and β-sheet secondary structures. These data were corroborated by the
analysis of the structure of α-syn using 1H-15N-heteronuclear single quantum coherence
(HSQC) experiments that revealed that neither the method used for cell lysis nor the
exposure to high temperature or low pH during purification changed the conformation of
the protein. Collectively, these data reinforce the idea that α-syn is expressed in E. coli as
an unfolded monomer that likely exists in equilibrium with variable amounts of
oligomeric species, notably a dynamic dimer prone to fibrillization.

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Results and Discussion
Purification of recombinant α-syn. To verify the effect of the purification protocol on
the conformation of α-syn, we produced recombinant α-syn using different strategies:
osmotic shock versus ultrasound sonication to lyse cells and heating versus acidification
for protein purification. Table 1 compares the α-syn yield obtained using either
ultrasound sonication or osmotic shock to lyse cells. For both methods, the protein
purification was based on two steps: acidification with HCl (pH 3.5) of the cell lysate to
precipitate non-α-syn proteins, followed by precipitation of α-syn with (NH4)2SO4 (50%
saturation). After precipitation of non-α-syn proteins, the supernatant contained 70-74%
α-syn when either ultrasound sonication or osmotic shock was used. Afterwards,
precipitation with 50% saturated (NH4)2SO4 and dialysis resulted in approximately 25-31
mg per L culture of highly pure α-syn, which represented a yield of 58-65% with a purity
of approximately 95-98% as estimated by SDS-PAGE. In this protocol, no
chromatographic steps were used and all purification steps can be easily performed in a
single day. Collectively, our data did not indicate significant differences between osmotic
shock and sonication with respect to the yield or purity of α-syn, though osmotic shock
was previously suggested to be the best choice for release of α-syn-containing periplasm
fraction because the protein is located inside the periplasm instead of the cytoplasm
[14,15]. In addition, the use of MgCl2 in the osmotic shock protocol should be carefully
evaluated because Mg2+ ions were described to interact with α-syn and induce
conformational changes that might inhibit protein aggregation [16].
Most of the current protocols for α-syn purification are based on the boiling of the
cell lysate fraction to remove non-α-syn proteins by precipitation [17,18]. Considering
that α-syn is an unfolded protein, sample heating is not expected to have any effect on the

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
structure of α-syn that remains in solution, whereas natively folded cytoplasmic proteins
thermally denature and precipitate. Thus, heating of the cell lysate has been extensively
used as a simple and high-yield protocol for the purification of α-syn, though the
formation of degradation products after α-syn solution is exposed to 90-100 °C has been
reported [19]. To avoid α-syn degradation by heating, the use of acidification with HCl as
an alternative method to purify α-syn was suggested [20]. Figure 1A shows the SDS-
PAGE analysis of samples from each purification step in which either the acidification or
heating of cell lysate was used to remove the majority of non-α-syn proteins. For both
methods, ultrasound sonication rather than osmotic shock was used. The purified α-syn
[Figure 1A, lanes 4 and 4´] displayed a predominant band with an apparent molecular
mass of ~17 kDa corresponding to α-syn monomers. Although α-syn has a molecular
mass of 14 kDa, it migrates on SDS-PAGE with an apparent mass of ~17 kDa, which is
likely due to the poor binding of the N-terminal acidic tail of α-syn to SDS molecules
[14,19]. Curiously, in our experiments, degradation bands were not observed after heat
treatment.
Table 1 shows that a single step based on acid precipitation resulted in α-syn with
a purity of 70%, whereas heating at 90 °C for 15 min resulted in α-syn with only 39%
purity, as estimated by SDS-PAGE. However, the yield from acid precipitation was
significantly lower than obtained by heating (71% compared with 94% yield,
respectively). Though the protein yield was 20% lower with acid precipitation than with
the heating process, both methods provided highly purified protein samples. After the
ammonium sulfate/dialysis steps, the yields were 58% and 78% for heating and acid
precipitation, respectively. Together, these data indicate that precipitation with acid is

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
more efficient at removing non-α-syn proteins from the cell lysate, although acidification
resulted in a lower yield than the boiling procedure.
Both heat- and acid-purified α-syn displayed a CD spectrum with a negative band
at 200 nm that is typical of disordered structures, although a slight band at approximately
220 nm was verified for the α-syn obtained using the heating protocol (Figure 1B).
Additionally, the α-syn purified by both the acidification and heating methods displayed
similar fibrillogenic properties; typical sigmoidal curves for the kinetics of fibrillization
were observed, with an initial lag phase, associated with the nucleation process, followed
by an exponential growth phase (Figure 1C).
Oligomeric state of α-syn. To determine the oligomeric state of the recombinant α-syn
purified with either the heating or the acidification protocol, the protein was cross-linked
with GHD and then analyzed by SDS-PAGE (Figure 2). After cross-linking, a small
population of α-syn was found as dimers (3-5% of total α-syn) with an apparent
molecular mass of ~34 kDa, as determined using the Totallab software (Figures 2A and
2B). These species were also revealed by western blot using the monoclonal antibody
anti-human α-syn 14H2L1. We also observed a very small population of α-syn running at
~60 kDa. Because α-syn monomers and dimers exhibit respective molecular masses of 18
and 34 kDa according to SDS-PAGE, this 60 kDa protein may be either a tetrameric or
trimeric form of α-syn. Mass spectrometry (MALDI-TOF/TOF) was used to confirm the
oligomeric state of cross-linked α-syn. A molecular mass of 14,458 Da was obtained
from the MALDI-TOF/TOF spectrum of non-cross-linked α-syn purified using the
heating protocol (Figure 2C), and no differences were observed in comparison with α-
syn purified using the acidification protocol (data not shown). After cross-linking, both
heat- and acid-purified α-syn exhibited a minor population of dimers and trimers with the

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
predominant monomer. For the α-syn purified using the heating protocol, a very small
population of tetramers (64,269 Da) was also observed. Together, the analysis of cross-
linked α-syn by both SDS-PAGE and mass spectroscopy indicated that the protein is
mostly monomeric with a small population of oligomers, notably dimers.
Uversky and colleagues demonstrated that a reversible folding of α-syn, with a
gain in β-sheet structure, occurs under high temperature or at low pH, resulting in the
formation of partially folded intermediates with similar conformations [21]. However, the
formation of heat- or acid-induced stable α-syn oligomers has not been reported. Considering that α-syn has a pI=4.7 and. hence, an excess of negative charge at neutral
pH, acidification decreases protein-protein repulsion, thus favoring protein aggregation.
Therefore, one could expect that a small fraction of dimers could be produced by the
acidification of cell lysate during α-syn purification. However, SAXS experiments
indicated that α-syn is monomeric at pH 3.0; hence, the gain in β-sheet structure was not
associated with α-syn oligomerization/fibrillization. More importantly, at pH 3.0,
essentially all of the molecules of the protein rather than just a small fraction behave as
partially folded intermediates. Therefore, the exposure of α-syn to low pH induces a
reversible folding of α-syn mediated by an intramolecular process and not by self-
association/oligomerization of the protein. Collectively, these data indicate that the α-syn
dimers observed here did not result from either HCl or heat treatment. Indeed, dimers
were found at similar concentrations in recombinant α-syn produced by both protocols.
The existence of α-syn dimers in equilibrium with monomers has also been observed
using NMR paramagnetic relaxation enhancement experiments [22] and ESI-MS-ESI
[23,24].

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Using an in vitro cross-linking protocol, α-syn dimers were detected in HEK293T
cells expressing high levels of the protein [12]. However, α-syn from diverse cell lines
migrates as a single band in native electrophoresis gels. In this context, Lashuel and
colleagues suggested that the α-syn dimers detected in intact cells using a cross-linking
protocol may be the result of a diffusion-controlled process rather than actual oligomers
[12]. Although a random association among α-syn monomers that specifically produces
dimers but not other oligomeric forms appears improbable, a series of experiments were
performed to rule out the diffusion-controlled hypothesis. Although only the α-syn
purified by the acidification protocol was used for these experiments, the presence of
dimers was also found in the protein purified using the heating protocol. First, we
evaluated the effect of the concentration of GHD on the pattern of α-syn oligomers.
Notably, the formation of non-specific oligomers induced by cross-linking was expected
to be highly dependent on the concentration of cross-linker. Figure 3A shows an increase
in the band running at the expected molecular weight of dimers when the GHD
concentration was increased from 0.1 to 5 mM (i.e., a protein:GHD ratio of 1:2 to 1:100).
Tetramers were visualized only at high GHD concentrations (25 mM). By fixing the
GHD concentration (5 mM) and varying the protein concentration from 10-100 µM, α-
syn dimers appeared with an intensity proportional to the protein concentration.
Importantly, a ladder-like pattern of α-syn oligomers such as what would be produced by
non-specific cross-linking were not observed at high concentrations of either GHD or α-
syn. A ladder-like pattern of α-syn oligomers, including dimers, was reported for
recombinant α-syn subjected to in vitro cross-linking with discuccinimidyl glutarate
(DSG) [11]. In this case, the cross-linking of α-syn in the presence of lysozyme resulted
in the formation of hetero-oligomers, suggesting that the dimers were products of
diffusion-controlled cross-linking. However, a direct interaction between lysozyme and

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
α-syn that may perturb the equilibrium among the diverse oligomeric states of α-syn in
solution cannot be discounted. It should be kept in mind that lysozyme is capable of
forming fibrils in solution similarly to α-syn [25]. In our experiments, we cross-linked α-
syn with GHD in the presence of bovine serum albumin (BSA). Figure 3C shows that
BSA did not interfere with the oligomeric pattern of α-syn. Cross-linking the protein in
the presence of BSA trapped monomers and dimers similarly to α-syn cross-linked alone.
The additional bands correspond to BSA (66 kDa) and its higher-order oligomers.
Therefore, α-syn dimers trapped by GHD are likely actual oligomers rather than an
artifact associated with diffusion-controlled cross-linking.
To verify whether the cross-linking of α-syn produced higher-order molecular
species not detected by SDS-PAGE, the protein was cross-linked with different
concentrations of GHD and then analyzed by SEC in a Superdex 200 column. Due to its
disordered structure, the α-syn monomer has a larger Stokes radius than a folded globular
protein with the same molecular weight, and it thus elutes in SEC as a 60-70 kDa protein
[26]. SEC analysis of non-crosslinked α-syn revealed two distinct populations: a major
population corresponding to the unfolded monomer (elution volume 14.3 mL) along with
the presence of a small population (12.3 mL) that eluted at close proximity to the
unfolded monomer (Figure 3D). The A140C α-syn dimeric mutant exhibits two distinct
peaks in SEC that correspond to the monomeric and the dimeric forms with elution
volumes nearly identical to those observed here [12]. Higher-order oligomers (elution
volume ~7.5 mL) are eluted in or just after the void volume, and they were only
visualized at high GHD concentrations. The low proportion of dimers in the non-cross-
linked sample may be attributed in part to the dilution-induced dissociation of the dimers.
Thus, an increase in GHD concentration resulted in an increase in peak area

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
corresponding to the dimer (Figure 3D, inset). Interestingly, cross-linking increased the
elution volume of both the monomeric and the dimeric forms (Figure 3E), which might
be associated with a compaction of the structure of these conformers. It has been
described that monomers and dimers exhibit dynamic conformations and that they may
exist in both extended and compact conformations [24]. For the monomer, a slight
compaction occurs due the contact between the C-terminal region and both the NAC and
N-terminal portions of the protein [27]. In this context, cross-linking with GHD appeared
either to trap compact forms of the monomer and dimer or, more likely, to compact their
structures by linkage-induced approximating amine groups that are distant of each other.
To verify whether α-syn dimers exist in equilibrium with monomers, we isolated
the putative dimer by SEC (elution at 12.3 mL) and then cross-linked with GHD (Figure
3F). Whereas the sample immediately cross-linked appeared as a single ~34 kDa species
by western blot using the monoclonal antibody anti human-α-syn (14H2L1), the sample
that was not cross-linked exhibited a molecular mass corresponding to α-syn monomers
(~17 kDa). Importantly, the presence of the monomer (elution volume of 14.3 mL) with
the dimer was verified when the fraction collected at 12.3 mL was re-chromatographed in
SEC, indicating that the dimers readily dissociated to monomers upon dilution.
Collectively, these data provide convincing evidence that α-syn dimers are highly
dynamic and exist in equilibrium with the monomeric species.
To determine whether α-syn dimers are irreversibly dissociated by heating as
found for the helicoidal tetramer [9], α-syn samples were boiled for 5 min, cooled in an
ice bath and then cross-linked immediately (labeled as 1 min) or 10 or 30 min after the
boiling procedure (Figure 3G). The cross-linking was performed at 25 °C. Whereas α-

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
syn boiled but not cross-linked appeared as a single band corresponding to the monomer,
α-syn dimers were visualized in all cross-linked samples, and their intensity was
enhanced with increased recovery time. These data support the idea that the dimers are
not products of non-specific cross-linking and, more importantly, that they are reversibly
dissociated by heat treatment. This effect contrasts with that of the α-syn helicoidal
tetramer, which irreversibly dissociates into unfolded monomers after heating [9].
Structural and fibrillogenic properties of α-syn dimers. To verify whether α-syn
dimers exist on a pathway to fibril formation, aliquots were withdrawn at specific times
from an α-syn solution under aggregation conditions, cross-linked with GHD and then
analyzed by SDS-PAGE (Figure 4A). Only the α-syn purified using the acidification
protocol was used for these experiments. We verified that α-syn dimers were readily
consumed during fibrillization. In fact, the initial population of dimers completely
disappeared after 3 days of aggregation, suggesting that they are an intermediate of the
fibrillization pathway. Unlike the putative helicoidal tetramer that resists fibril formation,
the population of tetramers/trimers found at the initial time appeared to be substrates for
fibrillization because they were consumed during aggregation. In addition, these data
suggest that these species were not produced by diffusion-controlled cross-linking. It is
worth mentioning that a GST-tagged construct of α-syn expressed in E. coli under non-
denaturing conditions fails to form fibrils [10]. However, this construct retained an extra
10 residues at the N-terminus after the removal of the GST tag, and the impact of this
additional sequence on fibrillization behavior was not determined. Only the similarity
between the 1H-15N-HSQC fingerprint of this construct and in vivo α-syn [28] was used to
confirm that this extra sequence did not influence the structural properties of α-syn.
Although our findings agree, in part, with previous works that indicated that existence of
an in vitro and in vivo equilibrium between the disordered monomer and different

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
oligomeric forms of α-syn, the inability of these oligomers to evolve to fibrils defended
by some studies remains obscure.
A conversion of α-syn monomer from random-coil to β-rich conformation is a
critical step during protein fibrillization. To verify the secondary structure of α-syn
dimers, dimers were isolated by SEC and analyzed using CD spectroscopy. Figure
4B shows that α-syn dimers displayed a mix of random-coil and β-sheet structure.
The negative band at 200 nm associated with disordered structure was reduced for
the dimer compared with the monomer and accompanied by a rising signal at 218
nm, which corresponds to β-sheet conformation. Based on the CD data, the
hypothesis that α-syn dimers are intermediates in the formation of α-helix-rich
tetramers appears to be improbable, even though our data also suggested the presence
of a minor population of tetramers/trimers in equilibrium with the unfolded
monomers and dimers. Unfortunately, we were not able to isolate and characterize
the secondary structure and fibrillogenic properties of these higher-order oligomers
because of their very low abundance in our experiments.
An important rate-limiting step in fibrillization is the formation of stable covalent
dimers of α-syn, which is favored by oxidative modifications of α-syn such as tyrosine
nitration and the formation of di-tyrosines [29]. In our studies, we observed the presence
of a population of non-covalent dimers of α-syn that was consumed prior to the monomer
during the formation of fibrils. Regarding these data, we speculate that the reversible
dimerization of α-syn may represent an important step in the formation of fibrils, similar
to that reported for the formation of stable covalent dimers under oxidative conditions.

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Conformation of α-syn expressed into E. coli. To determine whether different protocols
for α-syn purification produced conformational changes in the protein, we compared the
1H-15N-HSQC spectra of the protein under different conditions. A similar strategy was
used by Lashuel and colleagues to confirm that α-syn purified using either non-
denaturing or denaturing conditions present nearly identical conformations [12].
However, for these studies, 15N-α-syn (denaturing protocol) was prepared using reverse-
phase HPLC but not the boiling protocol. In our work, cell lysate obtained from either
sonication or osmotic shock was subjected to two sequential precipitations with
ammonium sulfate, at 30% and 50% saturation, to provide an α-syn-rich fraction in
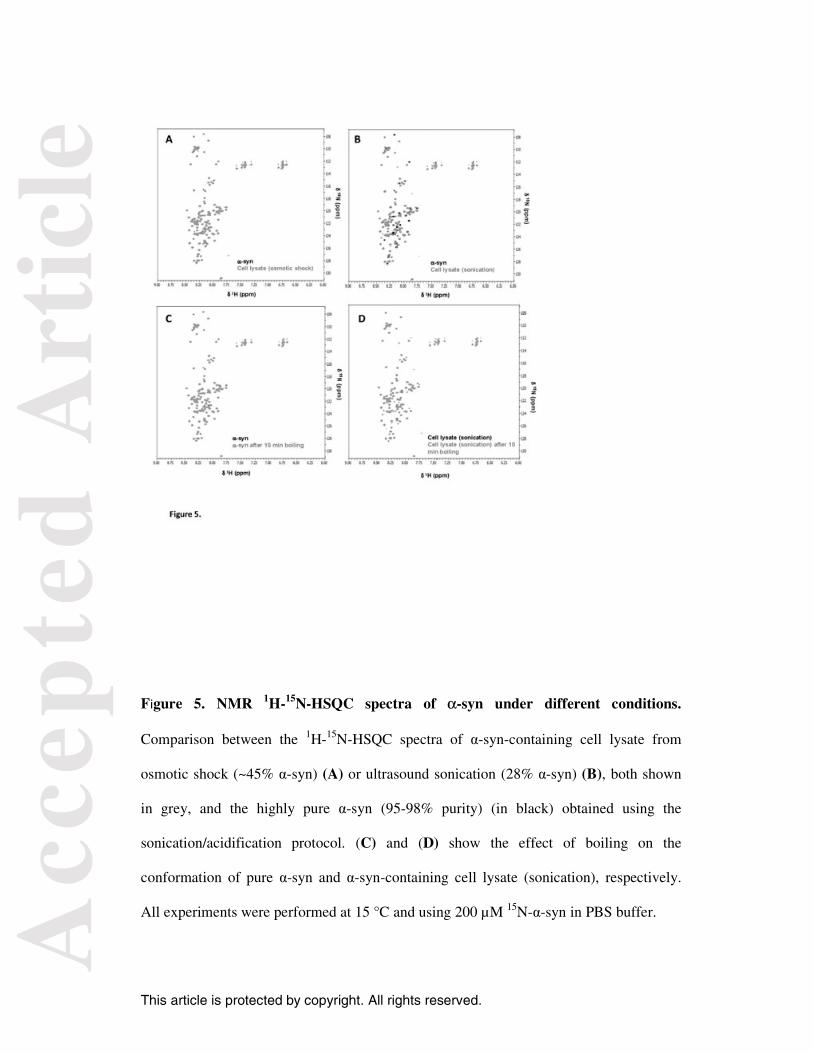
crowded conditions. Figure 5A shows that the 1H-15N-HSQC spectrum of α-syn-
containing cell lysate from osmotic shock (in grey) superimposed completely with the
spectrum of highly pure α-syn (95-98% purity) (in black), which was obtained using the
sonication/acidification protocol. Although α-syn in crowded conditions has 45-50%
purity, the contaminants did not appear to perturb the conformation of the protein. For the
cell lysate obtained by ultrasound sonication, a number of deviations were observed when
its spectrum was compared with that of the pure protein (Figure 5B). These deviations
may be due to either the lower proportion of α-syn in the cell lysate from sonication
compared with osmotic shock (28% vs. 45%) or minor conformational changes of the
protein in the crowded conditions. Nevertheless, we observed that the NMR spectra of α-
syn from the cell lysates and pure protein are similar, and both exhibit a dense cluster of
cross-peaks over a narrow range that indicates a highly disordered conformation. The
Selkoe group demonstrated that the α-syn folded tetramer may undergo destabilization
and the formation of unfolded monomers after cell lysis, which was attributed to the heat
and/or shearing induced by sonication [11]. However, no significant differences in α-syn
structure were observed when the α-syn-containing cell lysates obtained by osmotic

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
shock and sonication were compared. Osmotic shock does not produce heat and is hence
a less destabilizing method to for cell lysis than sonication.
Figures 5C and 5D show the effect of boiling on the conformation of highly pure
α-syn and α-syn-containing cell lysate, respectively. Therefore, unlike the previous
experiments described by Lashuel and colleagues [12], the effect of heating on protein
conformation was evaluated on both α-syn alone or in crowded conditions. We observed
that boiling the sample did not produce any effect on the structure of either α-syn sample
as verified by the superposition of the 1H-15N-HSQC spectra before (black) and after
(grey) the treatment. Studies on the effect of macromolecular crowding on α-syn structure
by NMR experiments in vitro and in living E. coli revealed that the crowded environment
in the bacterial periplasm stabilizes the disordered monomer, preventing structural
changes of the protein induced by temperature [28]. Because conformational differences
were not observed between α-syn-containing cell lysate and purified α-syn after heating
to 100 °C and then cooling, we suggest that the conformational changes produced by
heating are most likely completely reversible when the protein returns to 15 °C.
Supporters of the idea that α-syn exists as a helically folded tetramer under native
conditions have noted that the unfolded monomer may be a product of either problems in
generating a correctly folded protein in E. coli or the use of denaturing conditions such as
heating or other chemical denaturants in the purification protocol. However, an
alternative non-denaturing protocol for the production of α-syn generated the helically
folded α-syn tetramer in E. coli [10], suggesting that the reasons for the structural
differences were associated with the protocol used for protein purification rather than its
expression in bacteria. We have shown that the purified α-syn obtained using either

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
heating or acidic conditions has essentially the same conformation as the α-syn found in
cell lysate, independent of the use of ultrasound sonication or osmotic shock to lyse
bacteria.
Divergences in the α-syn native structure. Since the helical tetramer was first proposed
to be the predominant physiological species of α-syn in neuronal and non-neuronal cells
[9], a number of studies corroborating this idea have been published in parallel with other
studies contradicting it [10-13]. Curiously, all of these studies are based on well-
established protocols and present compelling evidence to sustain their conclusions. Table
2 summarizes some of the data about the oligomeric state of native α-syn. In light of
these findings, we attempted to examine the source of these contradictions. Upon
purification under non-denaturing conditions and using an in vitro cross-linking protocol,
native α-syn was found to be predominantly a helically folded tetramer in neuronal
(M17D) and non-neuronal cells (HEK293, HeLa, COS-7), frontal cortex mouse tissue
and human red blood cells [9]. An in vivo cross-linking protocol was used to demonstrate
that endogenous α-syn forms an ~60 kDa species in intact neuronal and non-neuronal
cells (HEL cells, M17D, rat primary neurons) [11]. In this same study, it was suggested
that cell lysis (ultrasound sonication) results in a destabilization of the putative 60 kDa
tetramer to form monomers. Therefore, according to this hypothesis, the production of
unfolded monomers may be associated with a lysis sensitivity of α-syn folded tetramers.
Conversely, several lines of evidence support the idea of α-syn as a disordered
monomer. First, the native state of α-syn, alone or tagged with GST, in E. coli was found
to be a disordered monomer even with the use of non-denaturing conditions to purify the
protein. The same results were obtained for α-syn from diverse mammalian cell lines and

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
homogenates of mouse, rat and human brains [12]. Second, in-cell NMR studies indicated
that α-syn behaves as a monomer in intact E. coli cells [30], reinforcing the idea that the
disordered monomer is not an artifact associated with either the use of denaturing
methods or cell lysis. It is worth noting that Selkoe and colleagues also identified varying
amounts of monomers, dimers and trimers in certain types of cells [9]. These apparent
conflicting findings may be reconciled by the idea of α-syn monomers existing in
equilibrium with variable amounts of oligomeric species, including the helical tetramer.
In this context, the differences observed in the native structure of α-syn in different cell
lines might arise from the relative abundance of the helical fraction at the time of analysis
as well as the influence of the purification protocols on this subtle equilibrium (see reply
of Bartels and Selkoe in [31]).
Another aspect that may be relevant for understanding the divergence of findings
about the native structure of α-syn is the use of bacterially expressed α-syn instead of α-
syn isolated from higher organisms. Post-translational modifications of α-syn may occur
in mammalian cells that are not present when the protein is bacterially expressed that may
interfere with the native structure of the protein [32]. For example, α-syn purified from
human erythrocytes is N-α-acetylated, a modification commonly found on human
proteins. The acetylation increases the hydrophobicity of the N-terminal portion of the
protein, facilitating protein-protein interactions, and it is also considered a stabilizing
factor for the α-helical structure of proteins [33,34]. However, the impact of N-
acetylation of α-syn (NAc-α-syn) on its physical and functional properties, including
fibrillization kinetics, interaction with lipid membranes and secondary structure, remains
unclear.

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
It was reported that the binding properties of α-syn to small unilamellar vesicles are
strongly impacted by its acetylation; NAc-α-syn has an approximately 2-fold higher
affinity for the negatively charged lipid vesicles than the non-acetylated protein does
[35]. However, the N-terminal acetylation appears to not have any effect on the ability of
α-syn to bind to synaptosomal membranes in vitro and in HeLa cells [36]. Differences
were also observed for the role of N-acetylation on the kinetics of fibrillization; some
studies have indicated no effect [35,36], whereas others pointed to a slower growth rate
for the formation of fibrils with NAc-α-syn [37]. Additionally, some studies suggest that
NAc-α-syn exhibits an increased transient helical propensity in comparison with the
intrinsically disordered monomer [35, 37], whereas other studies indicate that N-terminal
acetylation does not significantly affect the protein structure in vivo and in intact cells
[36]. Moreover, the influence of the current protocols for α-syn purification on the
properties of NAc-α-syn is still under investigation. In this context, NAc-α-syn, but not
unmodified α-syn, purified under mild conditions in the presence of the non-ionic
detergent n-octyl-β-glucopyranoside (BOG), forms α-helical tetramers along with
monomers [13]. However, NAc-α-syn behaves as an unfolded monomer when it is
purified in the absence of BOG, even if the detergent is subsequently added to pure α-
syn. These data suggest that the N-acetylation of α-syn alone is not sufficient to promote
the stabilization of helically folded oligomers.
In conclusion, our findings strongly indicate that the disordered monomer is the
major α-syn form expressed in E. coli, although the existence of less-stable oligomeric
forms cannot be ruled out. Importantly, the α-syn disordered monomer is not an artifact
of the production of the protein in E. coli at least in relation to either the method of lysis
or the use of heating/acidification to purify the protein. Additionally, we demonstrated

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
that the disordered monomer exists in equilibrium with a dynamic dimer, which is not a
product of diffusion-controlled cross-linking as previously suggested. Moreover, the α-
syn dimer is prone to fibrillate, and thus it might be considered an interesting target to
anti-fibrillogenic molecules.
Materials and Methods
Expression and purification of α-syn. The pT7-7-wt plasmid containing wildtype
human α-syn (kindly provided by D. Foguel) was transformed into BL21(DE3)pLysS E.
coli, and when the absorbance at 600 nm reached 0.5, its expression was induced with the
addition of 0.5 mM isopropyl β-D-thiogalactopyranoside (IPTG) for 15-18 h at 20 °C.
Cells were harvested by centrifugation (8,000 g) for 30 min, resuspended in 20 mM Tris-
HCl, 5 mM EDTA and 1 mM PMSF pH 8.0 and then lysed by ultrasonication for 1 min
intervals from 1 min to 30 min at 300 Watts. Alternatively, α-syn from the periplasm was
released by osmotic shock as previously described [14]. The non-α-syn proteins in the
crude extract were removed using two well-established protocols: acidic or heat
treatment. In the former, the pH of the crude extract was adjusted to 3.5 with 9% HCl and
then centrifuged to 16,000 g for 20 min. The pH of the supernatant was then raised to 7.5.
In the heating protocol, crude extract was heated at 90 °C for 15 min and then centrifuged
at 16,000 g for 20 min. Then, α-syn was precipitated by the addition of ammonium
sulfate to 50% saturation, followed by centrifugation (16,000 g, 20 min). The pellet was
resuspended in 20 mM Tris buffer pH 8.0 with 1 mM EDTA and dialyzed overnight
against 2 L of the same buffer. After a dialysis against MilliQ water (2 L, 2 times), α-syn
was lyophilized and characterized by circular dichroism, SDS-PAGE and size-exclusion
chromatography (SEC) in a Superdex 200 10/300 GL column (GE Healthcare, Uppsala,

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Sweden). The percentage of α-syn in each fraction of the purification was estimated by
the relative intensity of its band after SDS-PAGE using the Totallab program.
Cross-linking. To determine the α-syn oligomeric state, 50 µM of protein in 20 mM
HEPES pH 7.0 was cross-linked by incubation with varying concentrations of
glutaraldehyde (GHD) for 10 min (25 °C), and the reaction was stopped by the addition
of 1 M Tris buffer.
Immunoblotting. A total of 5-10 μg per lane of protein, cross-linked or untreated, was
loaded onto a 12% SDS-PAGE gel, blotted onto a nitrocellulose membrane and incubated
with rabbit monoclonal anti-human α-syn (14H2L1) (1:1,000) (Life Technologies, USA)
followed by incubation with the secondary antibody anti-rabbit IgG tagged with alkaline
phosphatase; the bands were visualized using Sigma-Fast™ BCIP/NBT (Sigma-Aldrich,
USA).
α-Syn fibrillization. α-Syn fibrils were formed by the incubation of 50 μM α-syn in 10
mM sodium phosphate buffer (NaPB) with 100 mM NaCl pH 7.5 in a 96-well plate
(Corning NBS 96-well white plate) at 37 °C under stirring (350 rpm) in a Thermomixer
Comfort apparatus (Eppendorf, Germany) in the presence of one 3-mm glass bead per
well. The fibrillization was monitored in situ by performing the assay in the presence of 5
µM Thioflavin-T (Thio-T). Thio-T binding was evaluated by fluorescence in a Cary
Eclypse Fluorimeter (Varian Inc.) by exciting at 446 nm and collecting the emission at
485 nm (slits: 5 nm).

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Circular dichroism (CD) spectroscopy. CD spectra were recorded in a Jasco-715
spectropolarimeter (Jasco, Tokyo). A buffer containing 10 mM NaPB and 100 mM NaCl
pH 7.5 was used as baseline and subtracted from the protein spectra. The spectra were
recorded from 195 to 260 nm.
Mass spectrometry analysis of GHD-cross-linked α-syn. Mass spectra were obtained
using a Bruker Daltonics Ultraflex Speed MALDI-TOF/TOF. Samples were diluted to
approximately 10 mg/mL in 0.1% TFA and mixed 1:1 with sinapinic acid at 10 mg/mL.
Samples from cross-linked α-syn (from the heating or the acidification protocol) were
centrifuged at 13,400 rpm for 5 min to pellet insoluble material. Non-cross-linked α-syn
was diluted with 3 M guanidine hydrochloride with 0.1% TFA and cleaned using a
ZipTip (Millipore, USA) prior to mixing with sinapinic acid. Appropriate calibrants
were measured every four unknown samples.
Two-dimensional NMR. NMR measurements were performed at 15 °C on a Bruker
Avance III (Bruker Biospin GmbH, Reinstetten, Germany) operating at a 1H frequency of
800 MHz. The sample contained 200 µM of purified 15N-α-syn in 10 mM NaPB pH 7.5
and 100 mM NaCl [10% D2O (v/v)]. For the analysis of cell lysate containing 15N-α-syn,
the fraction from the sonication step was subjected to two sequential precipitations with
ammonium sulfate: the first at 30% saturation to precipitate non-α-syn proteins, followed
by a second precipitation at 50% saturation in which α-syn was recovered in the pellet.
This procedure provided α-syn with approximately 60-70% purity. The 1H-15N-HSQC
experiments were acquired with echo-antiecho using 1,024 points in the direct dimension

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
and 320 points in the indirect dimension. Data were processed with TOPSPIN 3.1
(Bruker Biospin Corporation, USA) and analyzed using the CARA software package
(http://www.nmr.ch). The 1H-15N-HSQC spectra of α-syn were assigned according to Rao
et al. [38].
Acknowledgements:
This work was supported by Fundação de Amparo à Pesquisa do Estado do Rio de
Janeiro (FAPERJ) and the International Foundation for Science (IFS).
References
1. Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM, Trojanowsky JQ &
Iwatsubo T (1998) Aggregation of alpha-synuclein in Lewy bodies of sporadic
Parkinson's disease and dementia with Lewy bodies. Am J Pathol 152, 879-884.
2. Goedert M (2001) Alpha-synuclein and neurodegenerative diseases. Nat Rev
Neurosci 2, 492 -501.
3. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R & Goedert M (1997)
Alpha-synuclein in Lewy bodies. Nature 388, 839-840.
4. Wakabayashi K, Yoshimoto M, Tsuji S & Takahashi H (1998) Alpha-synuclein
immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy.
Neuroscience 249, 180-182.

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
5. Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR & Südhof TC (2010)
Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science
329, 1663-1667.
6. Polymeropoulos MH, Lavedan CLeroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root
H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A,
Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI
& Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in
Parkinson's disease. Science 276, 2045–2047.
7. Fink AL (2006) The aggregation and fibrillation of alpha-synuclein, Acc Chem Res
39, 628-634.
8. Weinreb PH, Zhen Z, Poon AW, Conway KA & Lansbury PT Jr (1996) NACP, a
protein implicated in Alzheimer's disease and learning, is natively unfolded.
Biochemistry 35, 13709-13715.
9. Bartels T, Choi JG & Selkoe DJ (2011) α-Synuclein occurs physiologically as a
helically folded tetramer that resists aggregation. Nature 477, 107-110.
10. Wang W, Perovic I, Chittuluru J, Kaganovich A, Nguyen LT, Liao J, Auclair JR,
Johnson D, Landeru A, Simorellis AK, Ju S, Cookson MR, Asturias FJ, Agar JN,
Webb BN, Kang C, Ringe D, Petsko GA, Pochapsky TC & Hoang QQ (2011) A
soluble α-synuclein construct forms a dynamic tetramer. Proc Natl Acad Sci USA
108, 17797-17802.
11. Dettmer U, Newman AJ, Luth ES, Bartels T & Selkoe D (2013) In vivo cross-linking
reveals principally oligomeric forms of α-synuclein and β-synuclein in neurons and
non-neural cells. J Biol Chem 288, 6371-6385.
12. Fauvet B, Mbefo MK, Fares MB, Desobry C, Michael S, Ardah MT, Tsika E, Coune
P, Prudent M, Lion N, Eliezer D, Moore DJ, Schneider B, Aebischer P, El-Agnaf

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
OM, Masliah E & Lashuel HA (2012) Alpha-synuclein in the central nervous system
and from erythrocytes, mammalian cells and Escherichia coli exists predominantly as
a disordered monomer. J Biol Chem 287, 15345-15364.
13. Trexler AJ & Rhoades E (2012) N-Terminal acetylation is critical for forming α-
helical oligomer of α-synuclein. Protein Sci 21, 601-605.
14. Huang C, Ren G, Zhou H & Wang CC (2005) A new method for purification of
recombinant human alpha-synuclein in Escherichia coli. Protein Expr Purif 42,
173-177.
15. Ren G, Wang X, Hao S, Hu H & Wang CC (2007). Translocation of alpha-
synuclein expressed in Escherichia coli. J Bacteriol 189, 2777-2786.
16. Golts N, Snyder H, Frasier M, Theisler C, Choi P &Wolozin B (2002)
Magnesium inhibits spontaneous and iron-induced aggregation of alpha-
synuclein. J Biol Chem 277, 16116-16123.
17. Giasson BI, Uryu K, Trojanowski JQ & Lee VM (1999) Mutant and wild type
human alpha-synucleins assemble into elongated filaments with distinct
morphologies in vitro. J Biol Chem 274, 7619-7622.
18. Hoyer W, Antony T, Cherny D, Heim G, Jovin TM & Subramaniam V (2002)
Dependence of alpha-synuclein aggregate morphology on solution conditions. J
Mol Biol 322, 383-393.
19. Giehm L, Lorenzen N & Otzen DE (2011) Assays for α-synuclein aggregation.
Methods 53, 295-305.
20. Narhi L, Wood SJ, Steavenson S, Jiang Y, Wu GM, Anafi D, Kaufman SA,
Martin F, Sitney K, Denis P, Louis JC, Wypych J, Biere AL & Citron M (1999)

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Both familial Parkinson's disease mutations accelerate alpha-synuclein
aggregation. J Biol Chem 274, 9843-9846.
21. Uversky NV, Li J & Fink AL (2001) Evidence for a partially folded intermediate
in alpha-synuclein fibril formation. J Biol Chem 276, 10737-10744.
22. Wu KP & Baum J (2010) Detection of transient interchain interactions in the
intrinsically disordered protein alpha-synuclein by NMR paramagnetic relaxation
enhancement. J Am Chem Soc 132, 5546-5547.
23. Frimpong AK, Abzalimov RR, Uversky VN & Kaltashov IA (2010)
Characterization of intrinsically disordered proteins with electrospray ionization
mass spectrometry: conformational heterogeneity of alpha-synuclein. Proteins
Struct Funct Bioinform 78, 714–722.
24. Bernstein SL, Liu DF, Wyttenbach T, Bowers MT, Lee JC, Gray HB & Winkler JR
(2004) Alpha-synuclein: stable compact and extended monomeric structures and pH
dependence of dimer formation. J Am Soc Mass Spectrom 15, 1435–1443.
25. Buell AK, Dhulesia A, Mossuto MF, Cremades N, Kumita JR, Dumoulin M,
Welland ME, Knowles TP, Salvatella X & Dobson CM (2011) Population of
nonnative states of lysozyme variants drives amyloid fibril formation. J Am Chem
Soc 133, 7737-7743.
26. Uversky VN (1993) Use of fast protein size-exclusion liquid chromatography to
study the unfolding of proteins which denature through the molten globule.
Biochemistry 32, 13288-13298.
27. Wu KP, Weinstock DS, Narayanan C, Levy RM & Baum J (2009) Structural
reorganization of alpha-synuclein at low pH observed by NMR and REMD
simulations. J Mol Biol 391, 784–796.

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
28. McNulty BC, Young GB & Pielak GJ (2006) Macromolecular crowding in the
Escherichia coli periplasm maintains alpha-synuclein disorder. J Mol Biol 355, 893-
897.
29. Krishnan S, Chi EY, Wood SJ, Kendrick BS, Li C, Garzon-Rodriguez W, Wypych J,
Randolph TW, Narhi LO, Biere AL, Citron M & Carpenter JF (2003) Oxidative
dimer formation is the critical rate-limiting step for Parkinson's disease alpha-
synuclein fibrillogenesis. Biochemistry 42, 829-837.
30. Binolfi A, Theillet FX & Selenko P (2012) Bacterial in-cell NMR of human alpha-
synuclein: a disordered monomer by nature? Biochem Soc Trans 40, 950-954.
31. Burré J, Vivona S, Diao J, Sharma M, Brunger AT & Südhof TC (2013. Properties of
native brain α-synuclein. Nature 498, E4-6; discussion E6-7.
32. Arnesen T, Van Damme P, Polevoda B, Helsens K, Evjenth R, Colaert N, Varhaug
JE, Vandekerckhove J, Lillehaug JR, Sherman F & Gevaert K (2009) Proteomics
analyses reveal the evolutionary conservation and divergence of N-terminal
acetyltransferases from yeast and humans. Proc Natl Acad Sci USA 106, 8157–8162.
33. Chakrabartty A, Doig AJ & Baldwin RL (1993) Helix capping propensities in
peptides parallel those in proteins. Proc Natl Acad Sci USA 90, 11332–11336.
34. Scott DC, Monda JK, Bennett EJ, Harper JW & Schulman BA (2011) N-Terminal
acetylation acts as an avidity enhancer within an interconnected multiprotein
complex. Science 334, 674–678.
35. Maltsev AS, Ying J & Bax A (2012) Impact of N-terminal acetylation of α-synuclein
on its random coil and lipid binding properties. Biochemistry 51, 5004-5013.
36. Fauvet B, Fares MB, Samuel F, Dikiy I, Tandon A, Eliezer D & Lashuel HA (2012)
Characterization of semisynthetic and naturally N-α-acetylated α-synuclein in vitro

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
and in intact cells: implications for aggregation and cellular properties of α-synuclein.
J. Biol. Chem. 287, 28243-28262.
37. Kang L, Moriarty GM, Woods LA, Ashcroft AE, Radford SE & Baum J (2012) N-
terminal acetylation of α-synuclein induces increased transient helical propensity and
decreased aggregation rates in the intrinsically disordered monomer. Protein Sci 21,
911-917.
38. Rao JN, Kim YE, Park LS & Ulmer TS (2009) Effect of Pseudorepeat
Rearrangement on alpha-Synuclein Misfolding, Vesicle Binding, and Micelle
Binding. J Mol Biol 390, 516-529.
Table 1. Purification of α-syn from E. coli.
Osmotic shock/acidification Sonication/ acidification Sonication/BoilingPurification steps
Total protein, mg
α-syn purity, %
α-syn, mg
Yield, %
Total protein
α-syn purity, %
α-syn, mg
Yield, %
Total protein
α-syn purity, %
α-syn, mg
Yield, %
Crude extract 85 45 38 100 191 28 53 100 195 26 51 100
Acid/ boiling 45 74 33 86 55 70 38 71 123 39 48 94
(NH4)2SO4 31 90 28 74 38 91 35 66 56 80 45 88
Dialysis 27 95 25 65 32 98 31 58 42 95 40 78

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Table 2. Summary of some of the main findings about the native oligomeric state of α-syn.
Source Experimental approach α-Syn oligomeric state Reference
Neuronal (M17D) and non-neuronal cells (HEK293, HeLa, COS-7); frontal cortex mouse tissue; human red blood cells
Native gel electrophoresis; analytical ultracentrifugation; in vitro cell cross-linking
Mostly helical folded tetramer 50-60 kDa; variable amounts of monomer
[9]
Bacterially expressed recombinant α-syn (E. coli; GST-tag)
Mild-purification conditions (use of BOG); NMR; in vitro cross-linking
Stable helical tetramers [10]
Intact neuronal and non-neuronal cells (HEL cells, M17D, rat primary neurons)
In vivo cross-linking Mostly oligomers of ~ 60 kDa; free monomers produced by cell lysis
[11]
Bacterially expressed recombinant α-syn (E. coli; with or without GST-tag); mammalian cell lines; homogenates of mouse, rat and human brains
Native and denaturing electrophoresis; oligomer-specific ELISA
Mostly disordered monomers [12]
Bacterially expressed recombinant α-syn (intact E. coli cells)
In-cell NMR Mostly disordered monomers [30]
Bacterially expressed recombinant NAc-α-syn Mild-purification conditions (use of BOG); analytical ultracentrifugation
α-helical tetramers and monomers [13]
Semisynthetic and bacterially expressed recombinant NAc-α-syn
Native and denaturing electrophoresis; in-cell NMR
Disordered monomers [36]

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Figure 1. Recombinant α-syn produced using either a heating or acidification
protocol. (A) The fractions from each step of purification of α-syn were analyzed by
denaturing electrophoresis (12% SDS-PAGE): 1: crude cell extract; 2 and 2´:
supernatants after boiling (10 min) or acidification (pH 3.5), respectively; 3 and 3’:
pellets from 50% (NH4)2SO4 saturation (precipitation of α-syn) of fractions from boiling

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
and acidification procedures, respectively; 4 and 4´ correspond to the fractions 3 and 3´,
respectively, after the dialysis step. (B) and (C) display the CD spectra and the kinetics of
fibrillization, respectively, of α-syn purified using the heating or acidification protocol.
The kinetic assays were performed in sextuplicates and the results are represented as
mean ± standard deviations.

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Figure 2. Oligomeric states of α-syn purified using either heating (A) or acidification
(B). α-Syn at a concentration of 50 µM, boiled for 5 min or untreated with heat, was in
vitro cross-linked with 5 mM GHD, run on a 12% SDS-PAGE gel and stained by silver
staining (on the left). The corresponding immunoblots (on the right) were performed
using a monoclonal human α-syn antibody (14H2L1) (Life Technologies, USA). The
MALDI-TOF/TOF analysis of α-syn before and after cross-linking with GHD is shown in
(C). Observed molecular weights are depicted above their respective peaks.

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Figure 3. The α-syn dimer is not produced by diffusion-controlled cross-linking. (A)
Pure recombinant α-syn (50 µM) was cross-linked with GHD concentrations of 0, 0.1,
0.5, 1 and 5 mM and run on a 12% SDS-PAGE gel (revealed by silver staining). (B) In
vitro cross-linking was performed by fixing the GHD concentration at 5 mM and using α-
syn concentrations of 10, 20, 50, 70 and 100 µM. (C) α-Syn at a concentration of 50 µM
was cross-linked with 5 mM GHD in the absence or presence of an equimolar
concentration of BSA; on the right, the cross-linking of 50 µM α-syn was performed in
the presence of varying concentrations of BSA (25, 50 and 100 µM). (D) 50 µM α-syn
was in vitro cross-linked with 0, 1, 5 or 25 mM GHD and then run in a Superdex 200
10/300 GL column. The elution volumes of the unfolded monomer and dimer, cross-
linked or left untreated, were plotted in (E); a standard curve was generated using the
proteins lysozyme (14.4 kDa), carbonic anhydrase (30 kDa), ovalbumin (45 kDa), BSA
(66 kDa) and phosphorylase B (97 kDa). (F) The fraction corresponding to the α-syn
dimer (elution at 12.3 mL) was separated from the monomer (elution at 14.3 mL) using
SEC (Superdex 200 10/300 GL column), cross-linked with 5 mM GDH or left untreated
and then analyzed by western blotting using monoclonal human α-syn (14H2L1). In
addition, the non-cross-linked dimer fraction was re-chromatographed in SEC (showed in
red, on the right). (G) To verify whether α-syn dimers were irreversibly dissociated by
heating, 50 µM of protein was boiled for 5 min, cooled in an ice bath and then cross-
linked immediately (labeled as 1 min), 10 or 30 min after the boiling procedure. The non-
cross-linked protein is shown on the left.

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Figure 4. The α-syn dimer displays a mix of random coil and β-sheet secondary
structure and is a precursor of fibril formation. (A) To verify whether α-syn dimers
exist on the fibril formation pathway, aliquots were withdrawn at 0, 1 and 3 days from 50
µM α-syn solution under aggregation conditions (37 °C, 350 rpm), cross-linked with
GHD and then analyzed on a 12% SDS-PAGE gel. (B) α-Syn dimers were isolated by
SEC and analyzed by CD spectroscopy.
Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Figure 5. NMR 1H-15N-HSQC spectra of α-syn under different conditions.
Comparison between the 1H-15N-HSQC spectra of α-syn-containing cell lysate from
osmotic shock (~45% α-syn) (A) or ultrasound sonication (28% α-syn) (B), both shown
in grey, and the highly pure α-syn (95-98% purity) (in black) obtained using the
sonication/acidification protocol. (C) and (D) show the effect of boiling on the
conformation of pure α-syn and α-syn-containing cell lysate (sonication), respectively.
All experiments were performed at 15 °C and using 200 µM 15N-α-syn in PBS buffer.


















