
Wissenschaftliche Ausrichtung der Arbeitsgruppe von Prof. … · 2011-01-12 · Methylpyruvat zu...
32
1 Wissenschaftliche Ausrichtung der Arbeitsgruppe von Prof. Dr. Carsten Bolm Institut für Organische Chemie der RWTH Aachen University Landoltweg 1 52056 Aachen Deutschland Tel.: (int) +49 241 809 4675 FAX.: (int) +49 241 809 2391 E-Mail: [email protected] - Dezember 2008 – zusammengestellt von Dr. I. Schiffers und Prof. Dr. C. Bolm Inhalt Seite - Darstellung der Arbeitsrichtung der Gruppe sowie bisheriger Ergebnisse 2 - Zusammenstellung der Veröffentlichungen der Arbeitsgruppe Bolm ab 2004 5 a) Originalpublikationen (chronologische Nummerierung ab 1986) 5 b) Übersichten (chronologische Nummerierung ab 1986) 12 c) Bücher und Zeitschrifteneditorials (chronologische Nummerierung ab 1986) 14 - Auswahl aktueller, laufender Forschungsprojekte 15 - Wissenschaftlicher Werdegang von Prof. Dr. Carsten Bolm 30
Transcript of Wissenschaftliche Ausrichtung der Arbeitsgruppe von Prof. … · 2011-01-12 · Methylpyruvat zu...

1
Wissenschaftliche Ausrichtung der Arbeitsgruppe von
Prof. Dr. Carsten Bolm
Institut für Organische Chemie der RWTH Aachen University Landoltweg 1 52056 Aachen Deutschland Tel.: (int) +49 241 809 4675 FAX.: (int) +49 241 809 2391 E-Mail: [email protected]
- Dezember 2008 – zusammengestellt von Dr. I. Schiffers und Prof. Dr. C. Bolm
Inhalt Seite - Darstellung der Arbeitsrichtung der Gruppe sowie bisheriger Ergebnisse 2
- Zusammenstellung der Veröffentlichungen der Arbeitsgruppe Bolm ab 2004 5
a) Originalpublikationen (chronologische Nummerierung ab 1986) 5
b) Übersichten (chronologische Nummerierung ab 1986) 12
c) Bücher und Zeitschrifteneditorials (chronologische Nummerierung ab 1986) 14
- Auswahl aktueller, laufender Forschungsprojekte 15
- Wissenschaftlicher Werdegang von Prof. Dr. Carsten Bolm 30

2
Darstellung der Arbeitsrichtung der Gruppe sowie bisheriger Ergebnisse Die Arbeitsgruppe von Prof. Dr. Carsten Bolm befaßt sich überwiegend mit dem Design, der
Entwicklung, der Synthese und dem Einsatz neuer Katalysatoren für selektive Umsetzungen in
organischen Reaktionen. In den letzten Jahren lagen dabei im Bereich stereoselektiver
Synthesen Schwerpunkte einerseits auf der Optimierung asymmetrischer Aryltransferreaktionen
auf aromatische Aldehyde, wobei die Ausarbeitung eines preiswerten, leicht durchführbaren,
hochenantioselektiven Verfahrens unter Verwendung von Boronsäuren als Arylquelle gelang,
andererseits auf der Untersuchung von Sulfoximinen als chirale Liganden in enantioselektiven
Katalysen, wobei die Entwicklung hocheffizienter Systeme zur Etablierung dieser neuen
Ligandenklasse in Diels-Alder-, Hetero Diels-Alder-, Carbonyl-En-Reaktionen sowie in enantio-
selektiven Hydrierungen führte. Zudem wurde ein einfacher Zugang zu β-Hydroxysulfoximin-
derivaten gefunden, bei deren Einsatz als Liganden in diphenylzinkvermittelten Arylierungen
verschieden substituierter Benzaldehyde die entsprechenden Diarylmethanole mit bis zu 93%
Enantiomerenüberschuss (ee) isoliert werden konnten, ein Resultat, das auf einer
Zusammenführung der auf beiden Forschungsgebieten gesammelten Erfahrungen beruht.
Weiterhin wurde in jüngster Zeit eine äußerst variable Methode zum Aufbau von C1-
symmetrischen Oxazolinylsulfoximinen basierend auf kommerziell erhältlichen Startmaterialien
ausgearbeitet, so daß eine ganze Reihe verschiedener Derivate schnell synthetisiert werden
konnte, deren Screening als Liganden in der kupferkatalysierten Mukaiyama Aldolreaktion mit
Methylpyruvat zu α-Hydroxyestern mit bis zu 94% ee führte. Zudem wurde der Einsatz von
neuartigen, sulfoximinbasierten P,N-Liganden in asymmetrischen Hydrierungsreaktionen
eingehend untersucht und dabei kationische Iridiumkomplexe hergestellt, die sich als
hochenantio- und chemoselektive Katalysatoren bei der Hydrierung von linearen α,β-
ungesättigten Ketonen erwiesen. Bei 2-Methylchinolinderivaten als Substrate konnten mit dem
Katalysatorsystem Enantiomerenüberschüsse von bis zu 92% erreicht werden.
Die Herstellung der Sulfoximinliganden war zudem von der Entwicklung neuer palladium-,
kupfer- und eisenkatalysierter N-Arylierungsmethoden begleitet. Letzteres System hat ein
besonderes Interesse erfahren, da Eisen als Katalysatorvorläufer sehr preiswert,
umweltfreundlich und im Vergleich zu vielen anderen Metallen nicht toxisch ist. Daher wurde in
den letzten zwei Jahren verstärkt das Potential von Eisensalzen als Katalysatoren in
Kreuzkupplungen untersucht und dabei hocheffiziente Methoden zur N-, O- und S-Arylierung
von Pyrazol-, Acetanilid-, Phenol- und Thiophenolderivaten unter Verwendung von 10 Mol-%
Eisen(III)chlorid erarbeitet. Eine intramolekulare Version der O-Arylierung wird derzeit zur
Synthese von Benzoxazolen genutzt, ein Protokoll, das aufgrund der geringen Kosten des leicht

3
handhabbaren FeCl3-Katalysators auch für eine industrielle Applikation geeignet wäre. Kürzlich
wurde zudem das in den vorherigen Studien optimierte Katalysatorsystem in der Arbeitsgruppe
erfolgreich auf Sonogashira Reaktionen übertragen. Bei Verwendung von 2-Iodphenol und
Phenylacetylen resultierte dabei in moderater Ausbeute 2-Phenylbenzofuran, was offensichtlich
durch Domino-Sonogashirakupplung gefolgt von einer intramolekularen Hydroalkoxylierung
entsteht und nach bisherigem Wissensstand das erste Beispiel einer exklusiv eisenkatalysierten
Hydroalkoxylierung eines Alkens darstellt.
Auch beim metallkatalysierten Nitrentransfer zur Iminierung von Sulfoxiden oder Sulfiden, dem
Schlüsselschritt zur Herstellung von Sulfoximinbausteinen, gelang die Optimierung des in der
Arbeitsgruppe entwickelten Rhodiumkatalysatorsystems zunächst zu einer kostengünstigeren
Methode, die auf Silbernitrat in Kombination mit einem Terpyridinliganden basierte, und
schließlich zu einem Verfahren, in dem Eisen(III)acetylacetonat die Iminierung unter milden
Reaktionsbedingungen in Gegenwart einer in situ generierten Nitrenquelle katalysiert.
Hervorzuheben ist dabei, daß die Iminierung von Sulfoxiden stereospezifisch unter Retention
der Konfiguration am Schwefel erfolgt und die N-Nosylprodukte unter Standardbedingungen
epimerisierungsfrei entschützt werden können, so daß unter Einsatz eines preiswerten
Eisenkatalysators hochenantiomerenangereicherte, vielseitig modifizierbare, freie NH-
Sulfoximinbausteine leicht herstellbar sind, die früher nur durch Einsatz toxischer und explosiver
Iminierungsreagenzien wie Stickstoffwasserstoffsäure oder O-Mesitylensulfonylhydroxylamin
zugänglich waren. Solch ein eisenkatalysierter Nitrentransfer konnte anschließend auch
erfolgreich auf Aziridinierungsreaktionen von Enolsilylethern und Styrolderivaten übertragen
werden. Hierbei ist zu bemerken, daß kürzlich der zunächst für akzeptable Ausbeuten
notwendige, hohe Styrolüberschuß durch Zusatz katalytischer Mengen einer ionischen
Flüssigkeit und 2-Chinolincarbonsäure auf ein Substrat zu Iminoiodinan Verhältnis von 1:1 bei
Ausbeuten von bis zu 95% reduziert werden konnte.
Weiterhin wurden in der Arbeitsgruppe metallkatalysierte Oxidationsreaktion, wie z.B. Baeyer-
Villiger Reaktionen, Kharasch-Sosnovski-Reaktionen, Anelli-Oxidationen und Sulfidoxidationen
bearbeitet. Hier konnte u.a. gezeigt werden, daß die Kombination einer leicht zugänglichen
Schiff'schen Base mit einem Eisensalz zu einem Katalysator führt, der in Gegenwart von
Wasserstoffperoxid Sulfide hochenantioselektiv zu Sulfoxiden oxidiert. Den präparativen Nutzen
dieses neuen Protokolls demonstrierte die Arbeitsgruppe bei der asymmetrischen Synthese der
beiden Vorläufer zu den jeweiligen Enantiomeren von Sulindac, einem Antirheumatikum. Ein
weiterer Schwerpunkt wird auf die metallkatalysierte Funktionalisierung reaktionsträger
Kohlenwasserstoffe gelegt. Hier wurde gezeigt, daß das System Eisen/Wasserstoffperoxid auch

4
unter sehr milden Reaktionsbedingungen Cyclohexan zu Cyclohexanol und Cyclohexanon
oxidiert.
Ein weiteres Teilgebiet der Katalyse, auf dem in den letzten vier Jahren in der Arbeitsgruppe
intensiver als zuvor gearbeitet wurde, untersucht das Potential rein organischer Moleküle
Reaktionen zu beschleunigen. Da ein Metall entweder im Katalysator nicht vorhanden ist oder
zumindest nicht aktiv am Katalysecyclus teilnimmt, hat sich die Bezeichnung Organokatalyse für
solche Reaktionen etabliert. Bereits 1998 wurde im Arbeitskreis ein äußerst effizientes Protokoll
zur alkaloidvermittelten, hochselektiven Desymmetrisierung von meso-Anhydriden erarbeitet,
das anschließend weiter optimiert und von einigen anderen Arbeitsgruppen als Schlüsselschritt
in asymmetrischen Synthesen eingesetzt wurde. Kürzlich konnte in einem völlig neuartigen
Ansatz zur Reaktionsführung organokatalytischer Umsetzungen gezeigt werden, daß die
stereoselektive Desymmetrisierung auch lösungsmittelfrei mittels Mahlens der Substratfeststoffe
in einer Kugelmühle möglich ist. Diese Reaktionsführung konnte auch äußerst erfolgreich auf
prolinkatalysierte Mannich-Reaktionen übertragen werden. Daneben ermöglichte der Einsatz der
Mikrowellensynthese bei letzterer Reaktion die Reduktion der Prolinmenge auf 0.5 Mol-% ohne
Einfluss auf die Stereoselektivität, eine Größenordnung der Katalysatorbeladung, die sonst bei
Organokatalysen nur selten erreicht wird. In einem weiteren, im Ansatz völlig neuartigen
Konzept der Aktivierung eines Elektrophils gegenüber einem nukleophilen Angriff mittels einer
nicht-kovalenten Wechselwirkung wurde untersucht, ob Halogenbrückenbindungsdonoren als
Organokatalysatoren fungieren können. Hier konnte gezeigt werden, daß die Reduktion von 2-
Methylchinolinderivaten zu den entsprechenden Tetrahydrochinolinen in Gegenwart von
Hantzsch-Ester durch perfluorierte Halogenalkane katalysiert wird.

5
Zusammenstellung der Veröffentlichungen der Arbeitsgruppe Bolm ab 2004 a) Originalpublikationen (chronologische Nummerierung ab 1986): 119. Boronic acids as aryl sources for the catalyzed enantioselective aryl transfer to aldehydes J. Rudolph, C. Bolm in Catalysts for Fine Chemical Synthesis, Vol. 3 (Eds.: S. M. Roberts,
J. Xiao, J. Whittall, T. Pickett), Wiley/VCH, 2004, 161 – 164. 120. General Synthesis of Unsymmetrical Norbornane Scaffolds as Inducers for Hydrogen Bond
Interactions in Peptides C. P. R. Hackenberger, I. Schiffers, J. Runsinck, C. Bolm, J. Org. Chem. 2004, 69, 739 –
743. 121. The MPEG effect: Improving asymmetric catalyses by simple additives J. Rudolph, N. Hermanns, C. Bolm, J. Org. Chem. 2004, 69, 3997 – 4000. 122. Synthetic and Spectroscopic Investigations of N-Acylated Sulfoximines C. P. R. Hackenberger, G. Raabe, C. Bolm, Chem. Eur. J. 2004 10, 2942 – 2952. 123. Vaulted Biaryls: Efficient Ligands for the Aluminum-Catalyzed Asymmetric Baeyer-Villiger
Reaction C. Bolm, J.-C. Frison, Y. Zhang, W. D. Wulff, Synlett 2004, 1619 – 1621. 124. Synthesis of Chiral Cyrhetrenes and their Application in Asymmetric Catalysis C. Bolm, L. Xiao, L. Hintermann, T. Focken, G. Raabe Organometallics 2004, 23, 2362 –
2369. 125. Synthesis of Chiral 2,2’-Dipyridylamines and their Use in the Copper-Catalyzed
Asymmetric Allylic Oxidation of Cyclohexene C. Bolm, J.-C. Frison, J. Le Paih, C. Moessner, Tetrahedron Lett. 2004, 45, 5019 – 5021. 126. Rhodium-catalyzed imination of sulfoxides and sulfides: Efficient preparation of N-
unsubstituted sulfoximines and sulfilimines H. Okamura, C. Bolm, Org. Lett. 2004, 6, 1305 – 1308. 127. Synthesis of Iridium Complexes with new Planar Chiral Chelating Phosphinyl-
Imidazolylidene Ligands and their Application in Asymmetric Hydrogenation T. Focken, G. Raabe, C. Bolm, Tetrahedron: Asymmetry 2004, 15, 1693 – 1706. 128. Highly enantioselective synthesis of secondary alcohols using triphenyl borane J. Rudolph, F. Schmidt, C. Bolm, Adv. Synth. Catal. 2004, 346, 867 – 872. 129. A Highly Enantioselective Iron-Catalyzed Sulfide Oxidation with Aqueous Hydrogen
Peroxide under Simple Reaction Conditions J. Legros, C. Bolm, Angew. Chem. 2004, 116, 4321 – 4324; Angew. Chem. Int. Ed. 2004,
43, 4225 – 4228. 130. Synthesis of C2-Symmetric and Unsymmetrically Substituted 2,2’-Dipyridylamines and
Crystal Structure of a Chiral 2,2’-Dipyridylamine Copper(II) Complex C. Bolm, J.-C. Frison, J. Le Paih, C. Moessner, G. Raabe, J. Organomet. Chem. 2004,
689, 3767 – 3777.

6
131. New Chiral Ligands from Mandelic Acid: Synthesis and Application in the Asymmetric
Phenyl Transfer Reaction to an Aromatic Aldehyde C. Bolm, L. Zani, J. Rudolph, I. Schiffers, Synthesis 2004, 2173 – 2180. 132. C1-symmetric sulfoximines as ligands in copper-catalyzed asymmetric Mukaiyama-type
aldol reactions M. Langner, C. Bolm, Angew. Chem. 2004, 116, 6110 – 6113; Angew. Chem. Int. Ed.
2004, 43, 5984 – 5987. (Highlighted by Dr. M. Zanda in Lett. Org. Chem. 2005, 2, 392.) 133. Copper-mediated cross coupling reactions of N-unsubstituted sulfoximines and aryl halides G. Y. Cho, P. Rémy, J. Jansson, C. Moessner, C. Bolm, Org. Lett. 2004, 6, 3293 – 3296. 134. Asymmetric Synthesis of Sulindac by Iron-Catalzed Sulfoxidation A. Korte, J. Legros, C. Bolm, Synlett 2004, 2397 – 2399. 135. Palladium-catalyzed N-vinylation of sulfoximines J. R. Dehli, C. Bolm, J. Org. Chem. 2004, 69, 8518 – 8520. 136. Planar-Chiral Imidazolium Salts in the Asymmetric Rhodium-catalyzed 1,2-Addition of Aryl
Boronic Acids to Aldehydes T. Focken, J. Rudolph, C. Bolm, Synthesis 2005 429 – 436. 137. ZnMe2 mediated addition of phenylacetylene to aldehydes and ketones P. G. Cozzi, J. Rudolph, C. Bolm, P.-O. Norrby, C. Tomasini, J. Org. Chem. 2005, 70, 5733
– 5736. 138. Boronic acids as versatile aryl sources for the highly enantioselective synthesis of chiral
diarylmethanols on multi-gram scale (PSP) J. Rudolph, F. Schmidt, C. Bolm, Synthesis 2005, 840 – 842. 139. Asymmetric Diethyl- and Diphenylzinc Additions to Aldehydes Using a Fluorine Containing
Chiral Amino Alcohol: A Striking Temperature Effect on the Enantioselectivity, a Minimal Amino Alcohol Loading, and an Efficient Recycling of the Amino Alcohol
J. K. Park, H. G. Lee, C. Bolm, B. M. Kim, Chem. Eur. J. 2005, 11, 945 – 950. 140. New insights into the stereoselectivity of the aryl zinc addition to aldehydes J. Rudolph, C. Bolm, P.-O. Norrby, J. Am. Chem. Soc. 2005, 117, 1548 – 1552. 141. Iron-catalysed C-H oxidation of hydrocarbons with hydrogen peroxide under mild
conditions C. Pavan, J. Legros, C. Bolm, Adv. Synth. Catal. 2005, 347, 703-705. 142. Investigations on the Iron-Catalyzed Asymmetric Sulfide Oxidation J. Legros, C. Bolm, Chem. Eur. J. 2005, 11, 1086 – 1092. 143. A General Copper-Promoted Coupling of Sulfoximines with Vinyl Bromides J. R. Dehli, C. Bolm, Adv. Synth. Catal. 2005, 347, 239 – 242. 144. Enantiopure α-Silyl-Substituted α-Hydroxyacetic Acids using O-H Insertion Methodology
and Boron-Based Asymmetric Reductions C. Bolm, S. Saladin, A. Claßen, A. Kasyan, E. Veri, G. Raabe, Synlett 2005, 461 – 464.

7
145. Synthesis and Palladium Catalyzed Coupling Reactions of Enantiopure p-Bromo-
phenylmethyl Sulfoximine G. Y. Cho, H. Okamura, C. Bolm, J. Org. Chem. 2005, 70, 2346 – 2349. 146. Highly symmetrical amino acid-derived N,N'-diacylated sulfodiimines J. R. Dehli, C. Bolm, Synthesis 2005, 1058 – 1060. 147. Synthesis of New Chiral Hydroxy Oxazolines and their Use in the Catalyzed Asymmetric
Phenyl Transfer to Aldehydes C. Bolm, F. Schmidt, L. Zani, Tetrahedron: Asymmetry 2005, 16, 1367 – 1376. 148. C1-Symmetric Aminosulfoximines as Ligands in Copper-Catalyzed Carbonyl-Ene
Reactions Langner, P. Rémy, C. Bolm, Synlett 2005, 781 – 784. 149. Palladium-catalyzed α-arylation of sulfoximines G. Y. Cho, C. Bolm, Org. Lett. 2005, 70, 1351 – 1354. 150. Organosilanols as Catalysts in Asymmetric Aryl Transfer Reactions S. Özçubukçu, F. Schmidt, C. Bolm, Org. Lett. 2005, 7, 1407 – 1410. 151. Ring Closing Metathesis (RCM) for the Synthesis of Cyclic Sulfoximines C. Bolm, H. Villar, Synthesis 2005, 1421 – 1424. 152. A high-throughput screening approach for the determination of additive effects in
organozinc addition reactions to aldehydes J. Rudolph, M. Lormann, C. Bolm, S. Dahmen, Adv. Synth. Catal. 2005, 347, 1361 – 1368. 153. Synthesis of novel silylated 1,2,4-triazin-5-ones C. Bolm, A. Kasyan, S. Saladin, Tetrahedron Lett. 2005, 46, 4049 – 4051. 154. Ethylene-Bridged Bissulfoximines in Copper-Catalyzed Enantioselective Hetero-Diels-
Alder Reactions C. Bolm, M. Verrucci, O. Simic, C. P. R. Hackenberger, Adv. Synth. Catal. 2005, 347, 1696
– 1700. 155. Cu(OAc)2-catalyzed N-Arylations of Sulfoximines with Aryl Boronic Acids C. Moessner, C. Bolm, Org. Lett. 2005, 7, 2667 – 2669. 156. Novel Rhodium Complexes with Ferrocene-Based N-Heterocylic Carbenes: Synthesis,
Structure and Catalysis Y. Yuan, G. Raabe, C. Bolm, J. Orgamomet. Chem. 2005, 690, 5747 – 5752. 157. Iron(III)-Catalyzed Tandem Sequential Methanol Oxidation/Aldol Coupling V. Lecomte, C. Bolm, Adv. Synth. Catal. 2005, 347, 1666 – 1672. 158. Ionic liquids as additives in the pig liver esterase (PLE) catalysed synthesis of chiral
disubstituted malonates S. Wallert, K. Drauz, I. Grayson, H. Gröger, P. Dominguez de Maria, C. Bolm, Green
Chem. 2005, 7, 602 – 605.

8
159. Highly Modular Synthesis of C1-Symmetric Aminosulfoximines and their Use as Ligands in Copper-Catalyzed Asymmetric Mukaiyama-Aldol Reactions
M. Langner, P. Rémy, C. Bolm, Chem. Eur. J. 2005, 11, 6254 – 6265. 160. BINOL-derived N-phosphino sulfoximines as ligands for asymmetric catalysis M. T. Reetz, O. G. Bondarev, H.-J. Gais, C. Bolm, Tetrahedron Lett. 2005, 46, 5643 –
5646. 161. Efficient Copper-Catalyzed N-Arylation of Sulfoximines with Aryl Iodides and Aryl Bromides J. Sedelmeier, C. Bolm, J. Org. Chem. 2005, 70, 6904 – 6906. 162. Diphenylphosphinosulfoximines as Ligands in Iridium-catalyzed Asymmetric Imine
Hydrogenations C. Moessner, C. Bolm, Angew. Chem. 2005, 117, 7736 – 7739; Angew. Chem. Int. Ed.
2005, 44, 7564 – 7567. 163. Planar-Chiral Cyrhetrenes for the Rhodium-Catalyzed Asymmetric 1,4-Addition and the
Hydrogenation of Enamides R. T. Stemmler, C. Bolm, J. Org. Chem. 2005, 70, 9925 – 9931. 164. Silver-catalyzed imination of sulfoxides and sulfides G. Y. Cho, C. Bolm, Org. Lett. 2005, 7, 4983 – 4985. 165. Metal-free imination of sulfoxides and sulfides G. Y. Cho, C. Bolm, Tetrahedron Lett. 2005, 46, 8007 – 8008. 166. Asymmetric alcoholysis of meso-anhydrides mediated by alkaloids C. Bolm, I. Atodiresei, I. Schiffers, Org. Synth. 2005, 82, 120 – 125. 167. Proton affinities and relative basicities of two 1,4,7-triazacyclononanes, Me3TACN and TP-
TACN. Quantum-chemical ab initio calculations, solution measurements, and the sructure of [TP-TACN•2H]2+ in the solid state
N. C. Meyer, C. Bolm, G. Raabe, U. Kölle, Tetrahedron 2005, 61, 12371 – 12376. 168. Dimethylzinc-mediated alkynylation of imines L. Zani, S. Alesi, P. G. Cozzi, C. Bolm, J. Org. Chem. 2006, 71, 1558 – 1562. 169. Asymmetric synthesis of chiral bisoxazolines and their use as ligands in metal catalysis I. Atodiresei, I. Schiffers, C. Bolm, Tetrahedron: Asymmetry 2006, 17, 620 – 633. 170. Resolution of Racemic 2-Aminocyclohexanol Derivatives and Their Application as Ligands
in Asymmetric Catalysis I. Schiffers, T. Rantanen, F. Schmidt, W. Bergmans, L. Zani, C. Bolm, J. Org. Chem. 2006,
71, 2320 – 2331. 171. Ligand effects in aluminium-catalyzed asymmetric Baeyer-Villiger reactions J.-C. Frison, C. Palazzi, C. Bolm, Tetrahedron 2006, 62, 6700 – 6706. 172. C1-Symmetric Sulfoximines as Ligands in Copper-Catalyzed Asymmetric Vinylogous
Mukaiyama-Type Aldol Reactions of Silyl Vinyl Keteneketals and Pyruvate Esters P. Rémy, M. Langner, C. Bolm, Org. Lett. 2006, 8, 1209 – 1211. 173. Thermal effects in Organocatalytic Asymmetric Mannich Reactions B. Rodríguez, C. Bolm, J. Org. Chem 2006, 71, 2888 – 2891.

9
Featured on the ACS Publications website http://pubs.acs.org/journals/joceah/promo/most/most_cited/2006.html
as a 2006 Most-Cited Article based on citation data obtained from Thomson ISI 174. Iron-catalyzed imination of sulfoxides and sulfides O. García Mancheño, C. Bolm, Org. Lett. 2006, 8, 2349 – 2352. 175. The Synthesis of Pseudo-Geminal, Pseudo-Ortho and Ortho Phosphinyl-Oxazolinyl-
[2.2]Paracyclophanes for Use as Ligands in Asymmetric Catalysis D. K. Whelligan, C. Bolm, J. Org. Chem. 2006, 71, 4609 – 4618. 176. Palladium-Catalyzed N-Arylations of 1,4,7-Triazacyclononanes
M. Nakanishi, C. Bolm, Adv. Synth. Catal. 2006, 348, 1823 – 1825. 177. The Synthesis of Pseudo-Geminal, Pseudo-Ortho and Ortho Hydroxy-Oxazolinyl-
[2.2]Paracyclophanes for Use as Ligands in Asymmetric Catalysis D. K. Whelligan, C. Bolm, Adv. Synth. Catal. 2006, 348, 2093 – 2100. 178. Computational Study of the Electronic Properties of the SN Bond in Sulfoximines E. Voloshina, I. Atodiresei, C. Bolm, J. Fleischhauer, G. Raabe, submitted for publication. 179. Solvent-free asymmetric organocatalysis in a ball mill B. Rodriguez, T. Rantanen, C. Bolm, Angew. Chem. 2006, 118, 7078 – 7080; Angew.
Chem. Int. Ed. 2006, 45, 6924 – 6926. 180. Catalyzed Enantioselective Synthesis of Allyl Alcohols from Aldehydes and Alkenyl Boronic
Acids F. Schmidt, J. Rudolph, C. Bolm, Synthesis 2006, 3625-3630. 181. A General and Efficient Synthesis of Nitrogen-Substituted Ferrocenes S. Özçubukçu, E. Schmitt, A. Leifert, C. Bolm, Synthesis 2006, 389 – 392. 182. Diaryl methanols by Catalyzed Asymmetric Aryl Transfer Reactions onto Aldehydes Using
Boronic Acids as Aryl Source F. Schmidt, J. Rudolph, C. Bolm, Adv. Synth. Catal. 2007, 349, 703 – 708. 183. Dimethylzinc-Mediated, Enantioselective Synthesis of Propargylic Amines L. Zani, T. Eichhorn, C. Bolm, Chem. Eur. J. 2007, 13, 2587 – 2600. 184. Mechanistic Insights into Stereoselective Catalysis – The Effects of Counterions in a
Cu(II)Bissulfoximine Catalyzed Diels-Alder Reaction C. Bolm, M. Martin, G. Gescheidt, C. Palivan, T. Stanoeva, H. Bertagnolli, M. Feth, A.
Schweiger, G. Mitrikas, J. Harmer, Chem. Eur. J. 2007, 13, 1842 – 1850. 185. Regio- and Stereoselective Copper-Catalyzed Carbozincation Reactions of Alkynyl
Sulfoximines and Sulfones G. Sklute, C. Bolm, I. Marek, Org. Lett. 2007, 9, 1259 – 1261.
186. Efficient Synthesis of Sulfonimidoylguanidines by Coupling of Sulfonimidamides with
Uronium Reagents C. Worch, C. Bolm, Synthesis 2007, 1355 – 1358.
187. Solvent-free asymmetric anhydride opening in a ball mill
T. Rantanen, I. Schiffers, C. Bolm, Org. Proc. Res. Develop. 2007, 11, 592 – 597.

10
188. Iron-catalyzed benzylic oxidation with aqueous tert-butyl hydroperoxide M. Nakanishi, C. Bolm, Adv. Synth. Catal. 2007, 349, 861 – 864. 189. Unprecedented Rhodium-Catalyzed Asymmetric Intermolecular Hydroacylation Reaction
with Salicylaldehydes R. T. Stemmler, C. Bolm, Adv. Synth. Catal. 2007, 349, 1185 – 1198 190. A Highly Efficient Asymmetric Organocatalytic Aldol Reaction in a Ball Mill
B Rodríguez, A. Bruckmann, C. Bolm, Chem. Eur. J. 2007, 13, 4710 – 4722. 191. Synthesis of Novel Chiral Phosphine-Olefin Complexes and their Evaluation as Ligands in
the Rhodium-Catalyzed Asymmetric 1,4-Addition R. Stemmler, C. Bolm, Synlett 2007, 1365 – 1370. 192. Anaerobic oxidation of propane and butane by novel marine sulphete-reducing bacteria
O. Kniemeyer, F. Musat, S. M. Sievert, K. Knittel, H. Wilkes, M. Blumenberg, W. Michaelis, A. Classen, C. Bolm, S. B. Joye, F. Widdel, Nature 2007, 449, 898 – 901.
194. Comparative Study of Metal–Catalyzed Iminations of Sulfoxides and Sulfides O. García Mancheño, C. Bolm, Chem. Eur. J. 2007, 13, 6674 – 6681. 195. Redox-neutral synthesis of β-amino aldehydes from imines by an alkynylation/hydration
sequence A. Labonne, L. Zani, L. Hintermann, C. Bolm, J. Org. Chem. 2007, 72, 5704 – 5708. 196. NR Transfer Reactivity of Azo-Compound I of P450 – How Does the Nitrogen Substituent
Tune the Reactivity of the Species Towards C-H and C=C Activation? Y. Moreau, H. Chen, E. Derat, H. Hirao, C. Bolm, S. Shaik, J. Phys. Chem. B 2007,111, 10288 – 10299.
197. Synthesis of N-(1H)-Tetrazole Sulfoximines
O. García Mancheño, C. Bolm, Org. Lett. 2007, 9, 2951 – 2954. 198. Ligand-Free Copper-Catalyzed N-Arylations of Nitrogen Nucleophiles
A. Correa, C. Bolm, Adv. Synth. Catal. 2007, 349, 2673 – 2676. 199. Asymmetric Enamide Hydrogenation Using Planar Chiral Cyrhetrenes
R. Stemmler, C. Bolm, Tetrahedron Lett. 2007, 48, 6189 – 6191. 200. Iodinane- and Metal-free Synthesis of N-Cyano Sulfilimines: Novel and Easy Access to
NH-Sulfoximines O. García Mancheño, O. Bistri, C. Bolm, Org. Lett. 2007, 9, 3809 – 3811.
201. Iron-Catalyzed N-Arylations of Nitrogen Nucleophiles
A. Correa, C. Bolm, Angew. Chem. 2007, 119, 9018 – 9021; Angew. Chem. Ind. Ed. 2007, 46, 8862 – 8865. Being highlighted by C&EN, December 3, 2007 (C&EN 2007, 85, 52).
202. Application of β-Hydroxysulfoximines in Catalytic Asymmetric Phenyl Transfer Reaction for
the Synthesis of Diarylmethanols J. Sedelmeier, C. Bolm, J. Org. Chem. 2007, 72, 8859 – 8862.

11
203. The use of chiral lithium amides in the desymmetrisation of N-trialkylsilyl dimethyl sulfoximines M. J. McGrath, C. Bolm, Beilstein J. Org. Chem. 2007, 3, 33.
204. Synthesis of Sulfonimidamides from Sulfinamides by Oxidation with N-Chlorosuccinimide
García Mancheño, C. Bolm, Beilstein J. Org. Chem. 2007, 3, 25. 205. Synthesis and resolution of racemic trans-2-(N-benzyl)amino-1-cyclohexanol: Enantiomer
separation by sequential use of (R)- and (S)-mandelic acid I. Schiffers, C. Bolm, Org. Synth. 2008, 85, 106 – 107.
206. Iron-Catalyzed C-O Cross-Coupling of Phenols with Aryl Iodides
O. Bistri, A. Correa, C. Bolm, Angew. Chem. 2008, 120, 596 – 598; Angew. Chem. Int. Ed. 2008, 47, 586 – 588.
207. Synthesis of Sulfoximidoyl-Substituted Triazoles by Huisgen 1,3-Dipolar Cycloaddition
B. Füger, G. Sklute, I. Marek, G. Y. Bolm, C. Bolm, Synlett 2008, 116 – 118. 208. Copper-mediated Cross-Coupling Reactions of N-Protected Sulfonimidamides and Aryl
Halides C. Worch, C. Bolm, Synthesis 2008, 739 – 742.
209. Iron-Catalyzed C-N Cross-Coupling of Sulfoximines with Aryl Iodides
A. Correa, C. Bolm, Adv. Synth. Catal. 2008, 350, 391 – 394. 210. Iron-Catalyzed S-Arylation of Thiols with Aryl Iodides
A. Correa, M. Carril, C. Bolm, Angew. Chem. 2008, 120, 2922 – 2925; Angew. Chem. Int. Ed. 2008, 47, 2880 – 2883.
211. Synthesis of Sulfoximine-Derived P,N-Ligands and Their Applications in Asymmetric
Hydrogenation Reactions S.-M. Lu, C. Bolm, Adv. Synth. Catal. 2008, 350, 1101 – 1105.
212. Iron-Catalyzed aziridination reactions
M. Nakanishi, A.-F. Salit, C. Bolm, Adv. Synth. Catal. 2008, 350, 1835 – 1840. 213. C1-Symmetric Oxazolinyl-Sulfoximines as Ligands in Copper-Catalyzed Asymmetric
Mukaiyama Aldol Reactions J. Sedelmeier, J. Hammerer, C. Bolm, Org. Lett. 2008, 10, 917 – 920.
214. Iron-Catalyzed N-arylations of amides
A. Correa, S. Elmore, C. Bolm, Chem. Eur. J. 2008, 14, 3527 – 3529. 215. Organocatalysis Through Halogen Bond Activation
A. Bruckmann, M. A. Pena, C. Bolm, Synlett 2008, 900 – 902. 216. Iron-Catalyzed Sonogashira Reactions
M. Carril, A. Correa, C. Bolm, Angew. Chem. 2008, 120, 4940 – 4943; Angew. Chem. Int. Ed. 2008, 47, 4862 – 4865.
217. Iron-Catalyzed Intramolecular O-Arylations: Synthesis of 2-Aryl Benzoxazoles
J. Bonnamour, C. Bolm, Org. Lett. 2008, 10, 2665 – 2667.

12
218. Highly Chemo- and Enantioselective Hydrogenation of Linear α,β-Unsaturated Ketones S.-M. Lu, C. Bolm, Chem. Eur. J. 2008, 14, 7513 – 7516.
219. Highly Enantioselective Synthesis of Optically Active Ketones by Iridium-Catalyzed
Asymmetric Hydrogenation S.-M. Lu, C. Bolm, Angew. Chem. 2008, 120, 9052-9055; Angew. Chem. Int. Ed. 2008, 47, 8920-8923.
220. Iron-catalysed aziridination reactions promoted by an ionic liquid
A. C. Mayer, A.-F. Salit, C. Bolm, Chem. Commun. 2008, 5975 – 5977. 221. Synthesis of diarylamines catalyzed by iron salts
A. Correa, M. Carril, C. Bolm, Chem. Eur. J., early view, DOI: 10.1002/chem.200802018. 222. Light-Induced Synthesis of π-Extended Tetrathiafulvalenes Incorporating Ferrocenes. An
Efficient Route for the Synthesis of Electron Donor Materials A. A. O. Sarhan, C. Bolm, Synthesis, accepted for publication.
b) Übersichten (chronologische Nummerierung ab 1986): 34. Oxidations of Carbonyl Compounds (Review)
Le Paih, J.-C. Frison, C. Bolm, in Oxidation Chemisty, (Ed.: J.-E. Bäckvall), Wiley/VCH, Weinheim, 2004, 253 - 294.
35. Metal-Catalyzed Baeyer-Villiger Reactions (Review) C. Bolm, C. Palazzi, O. Beckmann, in Transition Metals For Organic Chemistry: Building Blocks and Fine Chemicals, Vol. 2 (Eds.: M. Beller, C. Bolm), Wiley/VCH, Weinheim, 2. ed, 2004, 267 - 274.
36. Kharasch-Sosnovsky Type Allylic Oxidations (Review)
J. Le Paih, G. Schlingloff, C. Bolm in Transition Metals For Organic Chemistry: Building Blocks and Fine Chemicals, Vol. 2 (Eds.: M. Beller, C. Bolm), Wiley/VCH, Weinheim, 2. ed, 2004, 256 - 266.
37. Manganese-catalyzed Epoxidations (Review)
K. Muñiz, C. Bolm in Transition Metals For Organic Chemistry: Building Blocks and Fine Chemicals, Vol. 2 (Eds.: M. Beller, C. Bolm), Wiley/VCH, Weinheim, 2. ed, 2004, 344 - 356.
38. Catalyzed Asymmetric Aziridinations (Review)
C. Mößner, C. Bolm in Transition Metals For Organic Chemistry: Building Blocks and Fine Chemicals, Vol. 2 (Eds.: M. Beller, C. Bolm), Wiley/VCH, Weinheim, 2. ed, 2004, 389 - 402.
39. Sulfoximines: Synthesis and Catalytic Application (Highlight Review) H. Okamura, C. Bolm, Chem. Lett. 2004, 33, 482 - 487. 40. Iron-catalyzed reactions in organic synthesis (Review) C. Bolm, J. Legros, J. Le Paih, L. Zani, Chem. Rev. 2004, 104, 6217 - 6254. 41. Applications of Catalytic Asymmetric Sulfide Oxidations to the Syntheses of Biologically
Active Sulfoxides (Review) J. Legros, J. R. Dehli, C. Bolm, Adv. Synth. Catal. 2005, 347, 19 - 31.

13
42. Copper- and Palladium-Catalyzed Allylic Acyloxydations (Review)
J.-C. Frison, J. Legros, C. Bolm in Handbook of CH Transformations (Ed.: G. Dyker), Wiley/VCH, Weinheim, 2005, 445 - 454.
43. Synthesis of enamines, enol ethers and related compounds by cross-coupling reactions
(Review) J. R. Dehli, J. Legros, C. Bolm, Chem. Commun. 2005, 973 - 986. 44. Protonated Chrial Catalysts: New and Versatile Tools for Asymmetric Synthesis (Highlight)
C. Bolm, I. Schiffers, L. Zani, T. Rantanen, Angew. Chem. 2005, 117, 1788 – 1793; Angew. Chem. Int. Ed. 2005, 44, 1758 - 1763.
45. Catalytic Asymmetric Approaches towards Enantiomerically Enriched Diarylmethanols and Diarylmethylamines
F. Schmidt, R. T. Stemmler, J. Rudolph, C. Bolm, Chem. Soc. Rev. 2006, 35, 454 – 470. (one of the top 10 2006 most cited articles based on citation data obtained from Thomson ISI)
46. Asymmetric Baeyer-Villiger Reactions (Review) C. Bolm in Asymmetric Synthesis - The Essentials (Ed.: M. Christmann, S. Bräse), Wiley/VCH, Weinheim, 2006, 57 - 61.
47. Direct Addition of Alkynes to Imines and Related C=N Electrophiles: a Convenient Access to
Propargylamines L. Zani, C. Bolm, Chem. Commun. 2006, 4263 - 4275. 48. Sulfoximines as ligands in asymmetric metal catalysis (Personal Account)
C. Bolm, in Asymmetric Synthesis with Chemical and Biological Methods (Eds.: D. Enders, K.-E. Jaeger), Wiley/VCH, Weinheim, 2007, 149 - 176.
49. Catalyzed asymmetric aryl transfer reactions (Personal Account)
C. Bolm, in Asymmetric Synthesis with Chemical and Biological Methods (Eds.: D. Enders, K.-E. Jaeger), Wiley/VCH, Weinheim, 2007, 176 - 196.
50. Ring closure enyne metathesis: A powerful tool for the synthesis of heterocycles (Review) H. Villar, M. Frings, C. Bolm, Chem. Soc. Rev. 2007, 36, 55 - 66. 51. Solvent-Free Carbon-Carbon-Bond Formations in Ball Mills
B. Rodriguez, T. Rantanen, A. Bruckmann, C. Bolm, Adv. Synth. Catal. 2007, 349, 2213 - 2233.
52. Asymmetric Anhydride Openings I. Atodiresei, I. Schiffers, C. Bolm, Chem. Rev. 2007, 107, 5683 - 5712. 53. New chiral catalysts for C-C-bond formations
C. Bolm, in New Methodologies and Techniques for a Sustainable Chemistry (Eds.: A. Mordini, F. Faigl), Springer, 2008, 85 - 98.
54. Oxidation of Heteroatoms (N and S)
O. García Mancheño, C. Bolm, in Iron Catalysis in Organic Chemistry (Ed.: B. Plietker), Wiley-VCH, 2008, 109 - 123.
55. Oxidations of C,H- and C=C-bonds

14
A. Mayer, C. Bolm, in Iron Catalysis in Organic Chemistry (Ed.: B. Plietker), Wiley-VCH, 2008, 73 - 92.
56. Synthesis and use of chiral sulfoximines
C. Worch, A. C. Mayer, C. Bolm, in Organosulfur Chemistry in Asymmetric Synthesis (Eds.: T. Toru, C. Bolm), Wiley/VCH, Weinheim, 2008, 209- 229.
57. Iron-Catalysed Carbon-Heteroatom and Heteroatom-Heteroatom Bond Forming Processes A. Correa, O. García Mancheño, C. Bolm, Chem. Soc. Rev. 2008, 37, 1108 - 1117. 58. Directed C-H functionalizations
C. Bolm, in Activating Unreactive Substrates - The Role of Secondary Interactions (Eds.: C. Bolm, E. Hahn), Wiley-VCH, in press.
59. Organocatalytic Reactions: Effects of Ball Milling, Microwave and Ultrasound Irradiation A. Bruckmann, A. Krebs, C. Bolm, Green Chem. 2008, 10, 1131 - 1141. 60. Synthesis and Applications of Silicon-Containing α-Amino Acids M. Mortensen, R. Husmann, E. Veri, C. Bolm, Chem. Soc. Rev. accepted for publication. c) Bücher und Zeitschrifteneditorials (chronologische Nummerierung ab 1986): 3. Editorship of the 2-volume book Transition Metals For Organic Chemistry: Building Blocks
and Fine Chemicals, 2nd edition (co-editorship: M. Beller), Wiley/VCH, Weinheim, 2004 (>1000 pages).
4. Commentary in Organic Molecules: a Success Story in Modern Catalytic Chemistry
Editorship) C. Bolm, Adv. Synth. Catal. 2004, 346, 1022.
5. Organometallics in heterocyclic chemistry (Editorship) H. Davies, C. Bolm (Guest Editors), Chem. Soc. Rev. 2007, 36, 1035. 6. Editorship of Organosulfur Chemistry in Asymmetric Synthesis (Eds.: T. Toru, C. Bolm),
Wiley-VCH, Weinheim, Germany 2008. (432 pages) 7. Editorship of Activating Unreactive Substrates - The Role of Secondary Interactions (Eds.:
C. Bolm, E. Hahn), Wiley-VCH, Weinheim, Germany, in press.

15
Auswahl aktueller, laufender Forschungsprojekte
Von den zur Zeit in der Forschungsgruppe von Prof. Dr. Carsten Bolm untersuchten Projekten
wird hier nur eine Auswahl von Promotionsthemen vorgestellt, wobei die folgenden Schwer-
punkte eingehender beschrieben werden: Herstellung und Untersuchung neuartiger Sulfoximin-
und Sulfonimidamidverbindungen, Kreuzkupplungsreaktionen und Organokatalyse. (Unmittel-
bare Ansprechpartner, die sich im Rahmen ihrer Doktorarbeiten mit diesen Themen befassen,
sind im Text rot markiert.)
Bei den Studien zum Einsatz von Sulfoximinen als Liganden in asymmetrischen Katalysen sind
von der Arbeitsgruppe bereits hocheffiziente Systeme entwickelt worden,1 so daß basierend auf
diesen Ergebnissen zukünftig weiter optimierte Katalysatoren synthetisiert und eine deutliche
Erweiterung des Applikationsspektrums erarbeitet werden soll. Beispielsweise ist in der
Arbeitsgruppe in den vergangenen Jahren eine modulare Synthese C1-symmetrischer
Aminosulfoximine des Typs 4 ausgearbeitet worden, die einen einfachen Zugang zu einer
Vielzahl arylverbrückter Derivate mit äußerst variablem Substitutionsmuster gestattet. Diese
Verbindungen erwiesen sich als effiziente Liganden in kupferkatalysierten Mukaiyama-
Aldolreaktionen,2 so daß beim Screening einer kleinen Ligandenbibliothek ein System gefunden
wurde, das bei Verwendung von Tetrahydrofuran als Lösungsmittel, 2,2,2-Trifluorethanol als
Additiv bei Tieftemperaturreaktionen und Kupfer(II)triflat als Metallquelle Silylenolether 1 mit
Pyruvatestern 2 zu den entsprechenden Aldolprodukten in sehr guten Ausbeuten mit
ausgezeichneter Enantioselektivität von bis zu 99% ee umsetzt.3
R
O
OR''
O
HO R'
R'OR''
O
O
OSiMe3
R
HNNS
O
R1
R2
Ar
R3 R4
R5
4
1 2 3
Cu(OTf)2 (10 Mol-%)Sulfoximin 4 (10 Mol-%)
THF, CF3CH2OH+
71-90%, 89-99% ee
In folgenden Studien konnte gezeigt werden, daß dieses System auch hocheffizient vinyloge
asymmetrische Mukaiyama-Aldolreaktionen katalysiert, wobei erstmals der Einsatz von
Pyruvatestern in Kombination mit Silylvinylketenacetalen beschrieben wurde. In einem Schritt werden so γ,δ-ungesättigte α-Hydroxydiester mit quarternärem Stereozentrum mit bis zu 99% ee
generiert, die als wichtige Bausteine für Naturstoffsynthesen fungieren können.4
Derzeit wird in der Arbeitsgruppe das Substratspektrum eingehend untersucht und erste
Ergebnisse belegen, daß auch cyclische Dienolsilane vom Katalysatorsystem hochselektiv mit

16
Pyruvatestern umgesetzt werden. (Ansprechpartner: Marcus Frings und Anke Krebs) Diese
Substratkombination ist in der Literatur bislang nur selten beschrieben worden,5 da die simultane
Generierung zweier Stereozentren im Produkt neben hoher Regio- und Enantioselektivität als
zusätzliche Herausforderung ein hohes Maß an Diastereoselektivität für einen Einsatz des
Protokolls in einer Synthese bedingt. Bei einem Einsatz von 10 Mol-% des in den Primärstudien entwickelten Katalysatorsystems konnten hier γ-Butenolide des Typs 7 hochenantio- und
diastereoselektiv in sehr guten Ausbeuten hergestellt werden.6
EtO
O O
O
OH
O OSiMe3
EtO
O
O
HNNS
O
Me
Ph
8
5 6 7
Cu(OTf)2 (10 Mol-%)
Sulfoximin 8 (10 Mol-%)
Et2O, CF3CH2OH+
99%, 99% de, 99% ee
Zwar war durch die Arbeiten von Evans auf diesem Gebiet bekannt, daß bei Einsatz von 2-
Trimethylsilyloxyfuran in Mukaiyama-Aldolreaktionen das anti-Diastereomer meist bevorzugt
gebildet wird,4a doch gestatteten die publizierten analytischen Daten keine eindeutige Zuordnung
der Absolutkonfiguration der hier synthetisierten Aldolprodukte. Letztere gelang am Beispiel der
Verbindung 7 durch NMR- und CD-spektroskopische Untersuchungen in Kombination mit
quantenchemischen Berechnungen. Dazu wurde 7 ohne chirale Induktion synthetisiert, beide
Diastereomere säulenchromatographisch getrennt und zunächst in Entkopplungs-experimenten
im 1H-NMR Spektrum die Intensitätsänderungen ihrer Signale gemessen. Bei beiden Diastereomeren wurden NOE-Effekte zwischen dem zur Carbonylgruppe β-ständigigen
olefinischen CH-Proton und dem CH-Proton der Isopropylgruppe beobachtet. Um diese in
Relation zueinander setzen zu können, wurde für jedes Diastereomer die Intensität des
beschriebenen NOE-Effekts durch die des zwischen den beiden benachbarten olefinischen
Protonen auftretenden NOE-Effekts dividiert und dabei für ein Diastereomer das Verhältnis 0.8
erhalten, was dementsprechend für eine starke Wechselwerkung zwischen Isopropylproton zum
olefinischen Butenolidproton steht, für das andere Diastereomer wurde 0.4 berechnet,
gleichbedeutend mit einer schwächeren Interaktion, da der NOE-Effekt zwischen den beiden
Olefinprotonen aufgrund des gleichen Abstands in beiden Verbindungen konstant bleibt. Dann
wurden die energetisch günstigsten Konformationen für beide Diastereomere in Lösung

17
quantenmechanisch berechnet und das höhere NOE-Verhältnis dem anti-Diastereomer
aufgrund der darin größeren räumlichen Nähe der analysierten Protonen zugeordnet. Eine
Röntgenkristallstrukturanalyse verifizierte zudem kürzlich die postulierte Zuordnung.
Dementsprechend wird in der Katalyse vorzugsweise das enantiomerenangereicherte anti-
Produkt 7 gebildet. Dessen absolute Konfiguration wurde durch Vergleich eines kalkulierten CD-
Spektrums mit dem vom isolierten Produkt experimentell aufgenommenen bestimmt, wobei sich
zeigte, daß der Sulfoximinligand mit (S)-Konfiguration zum (R,R)-Butenolid führt. Derzeit wird
das Substratspektrum um andere (hetero)cyclische Dienolderivate erweitert, wobei die
resultierenden Produkte in vielen Fällen erstmals enantioselektiv hergestellt werden. Da nicht
auszuschließen ist, daß im Substrat enthaltene Heteroatome wie Schwefel und Stickstoff bzw.
deren Substitutenten wie beispielsweise eine N-Boc-Schutzgruppe den Übergangszustand
maßgeblich beeinflussen, sollen auch hier die absoluten Konfigurationen nach den
beschriebenen Methoden ermittelt werden, auch um aussagekräftige Rückschlüsse zum
Mechanismus ziehen zu können.
In einem weiteren Projekt werden C1-symmetrische Diarylphosphinylsulfoximine als Liganden in
enantioselektiven Hydrierungsreaktionen getestet. Nachdem sich benzolverbrückte Sulfoximin-
liganden in iridiumkatalysierten Hydrierungen von Alkyl-Arly-Iminen bewährt hatten und die
entsprechenden Amine mit bis zu 98% ee erhalten wurden,7 konnte kürzlich eine optimierte
Syntheseroute zu den analogen P,N-Liganden mit Naphthalinrückgrat erarbeitet werden.8 Dabei
wird 1,8-Diiodonahpthalin mit n-Butyllithium lithiiert, mittels Diarylchlorphosphin mono-
phosphoryliert und zum Iodphosphinoylnaphthalin 10 oxidiert. Letzteres wird kupferkatalysiert
mit dem Sulfoximin gekuppelt, das Phosphinoxid reduziert und das resultierende
Phosphanylsulfoximin 13 mit einem Iridiumkatalysatorvorläufer und einem Tetraarylborat zum
entsprechenden Iridiumkomplex umgesetzt.

18
BArF
(COD)
NSO
PhR
PHAr2
Ir
NSO
PhR
PAr2O=PAr2NSO
PhR
OS
NH
Ph RO=PAr2II I
9 10
12
11
13
14
1) n-BuLi, THF, –78 °C
2) Ar2PCl, –78 °C3) H2O2, Aceton
+CuI, Cs2CO3, DMEDA
Toluol, 110 °C
HSiCl3, Et3N
Toluol, 105 °C
[Ir(COD)Cl]2,
NaBArF
CH2Cl2, RT
Der modulare Aufbau der Liganden durch unabhängige Einführung der Diarylphosphin- und der
Sulfoximineinheit gestattet ein schnelles Screening unterschiedlich substituierter Analoga, was
die Optimierung des Katalysatorsystems auf das jeweilige Substrat vereinfacht. Beispielsweise
konnten in einer ersten Studie bei der Hydrierung von substituierten Chinolinen die
resultierenden chiralen Piperidinderivate mit bis zu 92% ee erhalten werden, wobei letztere
Produkte von großem Interesse für die asymmetrische Synthese von Alkaloiden sind.9 Bei der enantioselektiven Hydrierung acyclischer α,β-ungesätigter Ketone wurden kürzlich mit dem
entwickelten System bei β,β-disubstituierten Enonen 15 Enantioselektivitäten von bis zu 97% ee
erzielt, wohingegen bei α,β-substituierten Substraten des Typs 17 nur moderate Enantiomeren-
überschüsse beobachtet wurden.10 Dabei ist anzumerken, daß mit analogen Phosphinooxazolin-
Iridiumkomplexen des Typs 19 ein dem entgegengesetzter Trend zu beobachten war, so daß
letztlich nun für beide Arten von Substraten Katalysatorsysteme mit hoher Chemo- und
Enantioselektivität für die asymmetrische Synthese der entsprechenden gesättigten Ketone zur
Verfügung stehen.10b
H2R R2
O
R2
R R2
O
R1
H2
R1
R R2
OR1
R R2
O
BArF
(COD)
Ph2P N
O
Ir
19
*(60 bar)
Ir-Komplex 14
(1 Mol-%)
Toluol, RT+
70-94%, 75-97% ee
*(2 bar)
Ir-Komplex 19
(1 Mol-%)
Toluol, RT+
84-96%, 86-99% ee
15 16
17 18

19
Schon vor einigen Jahren wurde in der Arbeitsgruppe im Rahmen der Untersuchung zum
Einfluß des Arylrückgrates in Bissulfoximinliganden eine Synthese des Naphthalinderivates mit
zwei chiralen Methylphenylsulfoximineinheiten ausgehend von 1,8-Dibromonapthalin angestrebt,
die jedoch auch bei einem Überschuß der Sulfoximinkomponente mittels paladiumkatalysierter
Kupplung unter Standardbedingungen nicht gelang. Zwar erfolgte die erste N-Arylierung
offensichtlich glatt, doch war dann die intramolekulare Kupplung der Sulfoximinmethylgruppe
unter Bildung des heterocyclischen Derivats gegenüber einer zweiten N-Arylierung deutlich
bevorzugt.11 Erst mit den in der Arbeitsgruppe entwickelten Protokollen zur kupferkatalysierten
Sulfoximinarylierungen konnte eine Synthese der Verbindungen des Typs 20 ausgearbeitet
werden.8b,12 (Ansprechpartnerin: Marianne Steurer) Aufgrund der strukturellen Ähnlichkeit zu
aromatischen Diaminen, die aufgrund der räumlichen Wechselwirkung der eng benachbarten
basischen Zentren ungewöhnlich hohe Basizitätskonstanten aufweisen und daher als
Protonenschwämme bezeichnet werden,13 wird derzeit untersucht, ob das vorliegende
Bissulfoximin 20 vergleichbare Eigenschaften aufweist.
NS
O
PhMe
N SO
PhMe
OS
NH
Ph MeI I
9 2011
+CuI, Cs2CO3
DMSO, 90 °C
90%
Wichtige Strukturmerkmale sind bei Protonenschwämmen meist einerseits eine starke sterische
Spannung und die destabilisierend wirkende Überlappung der Stickstoffelektronenpaare der
neutralen Diamine und andererseits die starke N-H-N-Wasserstoffbrücke, die bei
Monoprotonierung gebildet wird und zu einer deutlichen Verringerung der sterischen Spannung
führt. Neben der Strukturaufklärung geben die thermodynamische Basizität des unprotonierten
und die kinetische Aktivität des protonierten Diamins Aufschluss. Daher wurde in der laufenden
Untersuchung zunächst die Protonenaffinität des Bissulfoximins in der Gasphase und in
Acetonitril quantenmechanisch berechnet. (Ansprechpartnerin: Steffanie Mersmann) Erste
NMR-spektroskopische Experimente zum Protonenaustausch und der Bestimmung der Basizität
wurden in Kooperation mit der Arbeitsgruppe von Herrn Prof. J. Sundermeyer (Philipps-
Universität Marburg) durchgeführt, die über die notwendige Expertise hinsichtlich der
Untersuchung von Protonenschwämmen an Hochfeldspektrometern verfügen.
Moleküle mit Sulfoximineinheiten erfahren aber in jüngster Zeit auch stark zunehmendes
Interesse von Seiten der Medizinalchemie und des Pflanzenschutzes, da eine Reihe von
Derivaten mit bemerkenswerten biologischen Aktivitäten identifiziert wurden.14,15 In diesem
Zusammenhang untersucht die Arbeitsgruppe von Prof. Dr. Carsten Bolm Verfahren zur
Synthese neuartiger Sulfoximinheterozyklen16 (Ansprechpartner: Bernhard Füger und Ankur

20
Pandey) wie z.B. 21-23 sowie die Inkorporation von Sulfxoximinen in kurze Peptidketten.17 In
Kooperation mit Prof. Dr. Elmar Weinhold wurde ein ungewöhnlicher enzymatischer Abbau des
Pseudotripeptids 24 in Gegenwart von Peptidase A gefunden, ein Effekt, der unmittelbar von der
zentralen Sulfoximineinheit ausgehen muß.18
H2NN
S
Ph
O O Ph
HN
O
OH
O
Ph
N
S PhOR
S
NX
OAr
N
SO
PhX = CH2
X = C(O)
21 22 23 24
Der wachsende Bedarf an enantiomerenangereicherten Sulfoximinbausteinen innerhalb der
Gruppe für die Entwicklung neuer Wirkstoffe und Liganden gab zudem Anstoß, verbesserte
Syntheseverfahren zu entwickeln. Dabei kann die Herstellung prinzipiell über zwei Wege
erfolgen: zum einen lassen sich Sulfide mit verschiedenen Methoden durch Nitren-Transfer zu
den entsprechenden Sulfiminen umsetzen, die anschließend zu Sulfoximinen oxidiert werden.1
Diese Vorgehensweise birgt jedoch den entscheidenden Nachteil, daß keine
hochenantioselektiven Iminierungsverfahren existieren, während chirale Sulfoxide nach
etablierten Verfahren leicht zugänglich sind und stereospezifisch iminiert werden können.1 In der
Arbeitsgruppe wurde ein effizientes System zur asymmetrischen Oxidation von Sulfiden
entwickelt,19 so daß daran anschließend die Untersuchungen auf den Iminierungsschritt
fokussiert wurden. Da für letztere Umsetzung in der Regel toxische und explosive Reagentien
wie Stickstoffwasserstoffsäure (in situ aus Schwefelsäure und Natriumazid generiert) oder O-
Mesitylensulfonylhydroxylamin (MSH) verwendet werden,20 die für Reaktionsansätze in großem
Maßstab wenig geeignet erscheinen, sind metallkatalysierte Nitrentransferreaktionen in jüngster
Vergangenheit intensiv untersucht worden und neue Methoden unter Einsatz von Kupfersalzen
oder Magnesium- wie auch Rutheniumkatalysatoren wurden vorgestellt.21 Jedoch führen diese
Iminierungsprotokolle überwiegend zu N-tosylgeschützten Produkten, die sich nur schwer zum
benötigten NH-Sulfoximin entschützen lassen. Hier gelang aber der Arbeitsgruppe die
Ausarbeitung eines sehr milden, katalytischen Verfahrens, bei dem ein Sulfonamid in
Kombination mit Iodosylbenzol nicht nur eine unbedenkliche Stickstoffquelle (ein in situ
generiertes Iminoiodinan) bildet, sondern auch bei Verwendung von p-Nitrobenzolsulfonamid
(NsNH2) die Einführung einer leicht abspaltbaren Schutzgruppe ermöglicht.22

21
S
O
Me
NHS
O
Me
N-NsS
O
Me
(S)-25
Fe(acac)3 (5 Mol-%),NsNH2, PhI=O
CH3CN, RT88-96%
(S)-26
PhSH, Cs2CO3
CH3CN, RT76-83%
(S)-11
Eisenkomplexe sind aufgrund ihrer sehr geringen Toxizität, ihrer Umweltverträglichkeit und des
geringen Preises als Katalysatorvorläufer stets von besonderem Interesse, so daß die hohe
Aktivität des Eisenacetylacetonats hier einen weiteren entscheidenden Vorteil bietet. Zwar
wurde für den Katalysezyklus ein Arbeitsmodell postuliert, das die Bildung eines intermediären
Eisen-Nitren-Komplexes beinhaltet, doch fehlen bislang experimentelle Daten, mit denen sich
ein konkreter Mechanismusvorschlag ausarbeiten läßt. Da zudem in diesem Prozeß Sulfide
schneller als Sulfoxide iminiert werden,22b könnte die Kenntnis des Mechanismus Hinweise für
die Entwicklung einer enantioselektiven Sulfiminsynthese liefern.
Aus atomökonomischer Sicht stellt aber auch dieses ausgereifte Verfahren keine befriedigende
Lösung dar, da ja für den Iminierungsschritt hypervalente Iodinane und Schutzgruppen im
Nitrenvorläufer mit recht hohem Molekulargewicht notwendig sind. Doch gelang der
Arbeitsgruppe kürzlich eine metallfreie Synthese von N-Cyanosulfiminen 28 durch Iminierung
von Sulfiden mittels Cyanamid in Gegenwart einer Base und NBS bzw. Iod als Halogenierungs-
reagenz.23 Diese Verbindungen können unter basischen Bedingungen leicht zu den
entsprechenden N-Cyanosulfoxminen oxidiert werden, die sich wiederum durch Behandlung mit
Trifluoressigsäureanhydrid und anschließender Methanolyse in die synthehisch wertvollen,
freien NH-Sulfoximine überführen lassen. Die N-Cyanosulfoxmine können aber auch direkt als
Substrate für die Synthese N-heterocyclischer Sulfoximine eingesetzt werden, so lassen sie sich
beispielsweise mit Natriumazid in Gegenwart von Zinkbromid direkt zu den entsprechenden N-
(1H)-Tetrazolderivaten 30 umsetzen.23b
ZnBr2
NaN3
RS
R'
O N
NN
NH
N
RS
R'
O NH
RS
R'
O N CN
RS
R'
NCN
TFAA
RS
R'
NBS oder I2
H2NCN, t-BuOM
M = Na, K
m-CPBA
27 28 29
29
11
29
30

22
Weiterhin sind in jüngster Zeit die den Sulfoximinen verwandten Sulfonimidamide (die Aza-
Analoga der Sulfonamide) zunehmend in die laufenden Projekte einbezogen worden und erste
Studien zur Kupplung mit Uronium-Reagentien zu Sulfonimidoylguanidinen oder kupfer-
vermittelte Kreuzkupplungen mit Aryliodiden deuten bereits auf vielseitige Einsatzmöglichkeiten
hin.24 (Ansprechpartnerin: Christin Worch) In einem kürzlich begonnenen Projekt soll
untersucht werden, ob die Kombination aus Oxazolinfunktion und Sulfonimidamideinheit zu einer
neuen Klasse von Verbindungen führt, die als Liganden in asymmetrischen Katalysen fungieren
können. Dazu wurde ein enantiomerenreines, N-geschütztes Sulfonimidoylchlorid 31 mit einem
optisch aktiven Aminooxazolin 32 umgesetzt,25,26 wobei ein Hauptprodukt aus der resultierenden
Reaktionsmischung in 75% Ausbeute sauber isoliert werden konnte. (Ansprechpartnerin: Marianne Steurer) Dessen Struktur wird derzeit intensiv untersucht (im Feststoff mittels
Röntgenkristallstrukturanalyse, in Lösung über eine NMR-Techniken Kombination von 1H-, 13C-,
COSY-, HMBC-, HMQC-, TOCSY- und NOESY-Spektren), da verschiedene Tautomere (33-36)
entstehen können.
SNBzO
p-Tol N
OHN
t-BuSNHBzO
p-Tol
NO
N
t-Bu
SNBzO
p-Tol
NHO
N
t-Bu
Cs2CO3
SNBzO
p-Tol
NO
NH
t-BuNO
NH2
t-Bu
S NBz
O
p-TolCl
31
32
33 34 35 36
Beim eisenkatalysierten Iminierungsverfahren wurde ein Eisen-Nitren-Komplex als
stickstoffübertragendes Intermediat postuliert, daher lag es nahe, das System auf weitere
Transferreaktionen zu untersuchen und in Primärstudien zeigte sich, daß mit nur geringen
Änderungen der Versuchsparameter Aziridinierung von Enolsilanen und Olefinen in guten
Ausbeuten durchgeführt werden können.27 Die Aziridinierungsreaktion gehört zu den
bedeutendsten synthetischen Methoden der organischen Chemie,28 da die Produkte als
wertvolle Bausteine für die Herstellung einer beeindruckenden Vielzahl an Verbindungen mit
einer Stickstoff-Funktionalität genutzt werden.29 Wie bei den meisten derzeit bekannten
katalytischen Verfahren war jedoch auch hier ein hoher Olefinüberschuß notwendig, um
akzeptable Aziridinausbeuten zu erreichen, was den Protokollen eine breite Anwendung
verwehrt. Durch systematische Untersuchung von Additiveffekten und Variation des
Iminoiodinans konnte eine Methode entwickelt werden, bei der Styrolderivate eisenkatalysiert in
bis zu 95%iger Ausbeute äquimolar aziridiniert werden können. Dabei werden 2-Chinolin-
carbonsäure (40) sowie Ethylmethylimidazolium-bis[(trifluormethyl)sulfonyl]-amid (41) in

23
katalytischen Mengen zugesetzt, wobei derzeit noch völlig unklar ist, in welcher Weise die
Additive am Reaktionsmechanismus beteiligt sind. (Ansprechpartnerin: Agathe Mayer)
N(SO2CF3)2
N N
NHOOC
NSNPhI
O O
R
NS
O ON
R
Fe(OTf)2 (5 Mol-%)
40 (15 Mol-%)
41 (8 Mol-%)
37 38
39
4041
CH3CN, MS 4A+
55-95%
Allgemein kann man in Bezug auf die Eisenkatalysen feststellen, dass sie in der Arbeitsgruppe
von Professor Bolm in den letzten Jahren sehr intensiv untersucht worden sind und die rasche
Entwicklung einer ganzen Reihe von effizienten Protokollen auf ein enormes Potential in einer
Vielzahl von Anwendungen deutet, die im Zentrum weiter Untersuchungen stehen werden.
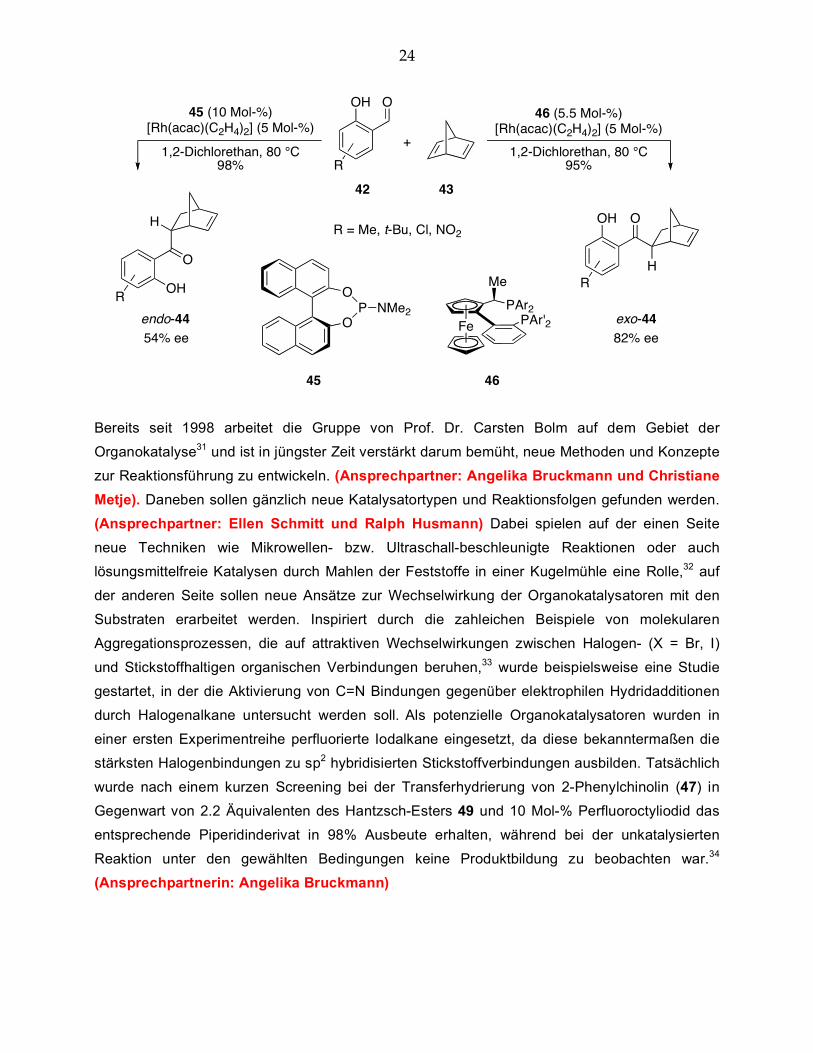
(Ansprechpartner: Isabelle Thomé und Julien Bonnamour) Eine weitere Transferreaktion, die in der Arbeitsgruppe von Prof. Dr. Carsten Bolm untersucht
wurde und für die bislang keine Intermediate zur Verifizierung des postulierten Modells detektiert
werden konnten, ist die rhodiumkatalysierte Hydroacylierung. Hier zeigte sich, daß in der
Reaktion von Salicylaldehyden 42 mit Norbornadien (43) je nach eingesetztem Liganden selektiv
das entsprechende endo- oder das exo-Produkt in hohen Ausbeuten hergestellt werden kann.
Der Einsatz zweizähniger Liganden, wie z.B. vom Walphos-Typ 46, führt vorzugsweise zum exo-
Isomer, wohingegen mit einzähnigen Phosphoramidit-Liganden, wie z.B. 45, hauptsächlich das
endo-Keton 44 gebildet wird.30 Während bereits hohe Diastereoselektivitäten von bis zu 99:1
erreicht werden können, sollen weitere Untersuchungen eine Optimierung der bislang nur
moderaten Enantiomerenüberschüsse erzielen.
Basierend auf den langjährigen Erfahrungen auf dem Gebiet zinkvermittelter Reaktionen wird
zudem derzeit eine enantioselektive Variante der Alkinylierung von Iminen entwickelt
(Ansprechpartnerin: Anne Nijs), und auch die Synthese neuartiger Phosphanliganden für
asymmetrische Hydrierungen bildet einen Schwerpunkt (Ansprechpartner: Andreas Hergesell), der intensiv bearbeitet wird.
24
4645
Me
PAr2PAr'2
O
OP NMe2
exo-44endo-44
4342
RR
R
OH O
Fe
H
OOHH
O
OH
R = Me, t-Bu, Cl, NO2
+
46 (5.5 Mol-%)[Rh(acac)(C2H4)2] (5 Mol-%)
1,2-Dichlorethan, 80 °C95%
45 (10 Mol-%)
[Rh(acac)(C2H4)2] (5 Mol-%)
1,2-Dichlorethan, 80 °C98%
54% ee 82% ee
Bereits seit 1998 arbeitet die Gruppe von Prof. Dr. Carsten Bolm auf dem Gebiet der
Organokatalyse31 und ist in jüngster Zeit verstärkt darum bemüht, neue Methoden und Konzepte
zur Reaktionsführung zu entwickeln. (Ansprechpartner: Angelika Bruckmann und Christiane Metje). Daneben sollen gänzlich neue Katalysatortypen und Reaktionsfolgen gefunden werden. (Ansprechpartner: Ellen Schmitt und Ralph Husmann) Dabei spielen auf der einen Seite
neue Techniken wie Mikrowellen- bzw. Ultraschall-beschleunigte Reaktionen oder auch
lösungsmittelfreie Katalysen durch Mahlen der Feststoffe in einer Kugelmühle eine Rolle,32 auf
der anderen Seite sollen neue Ansätze zur Wechselwirkung der Organokatalysatoren mit den
Substraten erarbeitet werden. Inspiriert durch die zahleichen Beispiele von molekularen
Aggregationsprozessen, die auf attraktiven Wechselwirkungen zwischen Halogen- (X = Br, I)
und Stickstoffhaltigen organischen Verbindungen beruhen,33 wurde beispielsweise eine Studie
gestartet, in der die Aktivierung von C=N Bindungen gegenüber elektrophilen Hydridadditionen
durch Halogenalkane untersucht werden soll. Als potenzielle Organokatalysatoren wurden in
einer ersten Experimentreihe perfluorierte Iodalkane eingesetzt, da diese bekanntermaßen die
stärksten Halogenbindungen zu sp2 hybridisierten Stickstoffverbindungen ausbilden. Tatsächlich
wurde nach einem kurzen Screening bei der Transferhydrierung von 2-Phenylchinolin (47) in
Gegenwart von 2.2 Äquivalenten des Hantzsch-Esters 49 und 10 Mol-% Perfluoroctyliodid das
entsprechende Piperidinderivat in 98% Ausbeute erhalten, während bei der unkatalysierten
Reaktion unter den gewählten Bedingungen keine Produktbildung zu beobachten war.34
(Ansprechpartnerin: Angelika Bruckmann)

25
NH
COOEtEtOOC
N Ph NH
Ph
(2.2 � q.)
CF3(CF2)7I (10 Mol-%)
CH2Cl2, RT
98%47 48
49
Um die postulierten N-Halogen-Wechselwirkungen nachzuweisen, wurden NMR-Experimente
durchgeführt (mit einer CD2Cl2 Lösung des Substrates 47, Hantzsch-Ester 49 und
Perfluorooktyliodid als Katalysator). Nach 3 Stunden wurde das Produkt 48 in der
Reaktionsmischung identifiziert und kompletter Reaktionsumsatz nach 24 Stunden festgestellt.
Im 1H-NMR-Spektrum konnte keine Signalverschiebunge beobachtet werden, doch wiesen die
Chinolinsignale unter Reaktionsbedingungen im Vergleich zu denen der reinen Verbindung im 13C-NMR-Spektrum einen Tieffeldshift auf (um 0.01-0.06 ppm). Diese Veränderung der
chemischen Verschiebung wird als Anzeichen für eine schwache Wechselwirkung zwischen
dem Chinolin-Stickstoff und dem Iodatom der perfluorierten Verbindung interpretiert. Diese
Annahme wurde durch die Aufnahme eines 19F-NMR Spektrums bestärkt, in dem das Signal der CF2I-Gruppe Hochfeld-verschoben war (Δδ = 0.1 ppm). Für die CF2 und CF3 Signale wurde ein
Hochfeldshift von ca. 0.06 ppm beobachtet. Über ähnliche Veränderungen der chemischen
Verschiebungen wurde bereits im Zusammenhang mit anderen Wechselwirkungen zwischen
perfluorierten Halogenalkanen und stickstoffhaltigen Kohlenwasserstoffen berichtet.35 In
Übereinstimmung mit dieser Literatur waren alle chemischen Verschiebungen zudem stark
konzentrationsabhängig. 13C und 19F NMR Studien, die in Abwesenheit des Reduktanden 49 mit
einer 1:1 Mischung von 1-Iodoperfluorooctan und Chinolin 47 in CD2Cl2 (~ 0.07 M) durchgeführt
wurden, zeigten ein ähnliches Verhalten der chemischen Verschiebungen. Dies weist wiederum
auf die postulierten N....I Wechselwirkungen hin. Höher konzentrierte Lösungen (z. B. 1 M)
wiesen eine größere Veränderung der chemischen Verschiebungen auf (1.1 ppm für CF2I und
0.15-0.44 ppm für CF3 und CF2). Jedoch müssen letztere Daten kritisch betrachtet werden, da
unter diesen Bedingungen das perfluorierte Halogenalkan nicht vollständig löslich war.
Erstaunlicherweise führten Versuche, NMR Experimente mit Mischungen des Hantzsch-Esters
und 1-Iodoperfluorooctans in Abwesenheit des Substrates durchzuführen, zur Zersetzung des
Haloperfluoroalkanes innerhalb weniger Stunden. Demnach scheint die Gegenwart des
Chinolins das Perfluorohalogenalkan vor einem Angriff durch den Reduktanden zu schützen.

26
Referenzen: 1. a) C. Bolm, in Asymmetric Synthesis with Chemical and Biological Methods (Eds.: D.
Enders, K.-E. Jaeger), Wiley-VCH, Weinheim, 2007, 149 – 176; b) C. Bolm, in Asymmetric
Synthesis with Chemical and Biological Methods (Eds.: D. Enders, K.-E. Jaeger), Wiley-
VCH, Weinheim 2007, 149 – 176; c) C. Worch, A. C. Mayer, C. Bolm, in Organosulfur
Chemistry in Asymmetric Synthesis (Eds.: T. Toru, C. Bolm), Wiley-VCH, Weinheim, 2008,
209 – 229.
2. Für Übersichten zum Thema enantioselektive Mukaiyama Aldolreaktionen siehe: a) H.
Gröger, E. M. Vogel, M. Shibasaki, Chem. Eur. J. 1998, 4, 1137 – 1141; b) S. G. Nelson,
Tetrahedron: Asymmetry 1998, 9, 357 – 389; c) C. Palomo, M. Oiarbide, J. M. Garía, Chem.
Eur. J. 2002, 8, 36 – 44; d) E. M. Carreira, in Comprehensive Asymmetric Catalysis (Eds.:
E. N. Jacobsen, A. Pfaltz, H. Yamamoto), Springer-Verlag, Berlin, 1999, Vol. 1, 997 – 1065.
3. a) M. Langner, C. Bolm, Angew. Chem. 2004, 116, 6110 – 6113; Angew. Chem. Int. Ed.
2004, 43, 5984 – 5987; b) M. Langner, P. Rémy, C. Bolm, Chem. Eur. J. 2005, 11, 6254 –
6265; c) für den Einsatz dieser Liganden in Kupfer(II)-katalysierten Carbonyl-En-Reaktionen
siehe auch: M. Langner, P. Rémy, C. Bolm, Synlett 2005, 781 – 784; d) für den Einsatz von
Oxazolinsulfoximinliganden in der asymmetrischen Mukaiyama Aldolreaktion siehe: J.
Sedelmeier, T. Hammerer, C. Bolm, Org. Lett. 2008, 10, 917 – 920.
4. P. Rémy, M. Langner, C. Bolm, Org. Lett. 2006, 8, 1209 – 1211.
5. a) D. A. Evans, M. C. Kozlowski, J. A. Murry, C. S. Burgey, K. R. Campos, B. T. Connell,
Richard J. Staples, J. Am. Chem. Soc. 1999, 121, 669 – 685; b) D. A. Evans, C. S. Burgey,
M. C. Kozlowski, S. W. Tregay, J. Am. Chem. Soc. 1999, 121, 686 – 699; c) S. Naito, M.
Escobar, P. R. Kym, S. Liras, S. F. Martin, J. Org. Chem. 2002, 67, 4200 – 4208; d) M. De
Rosa, L. Citro, A. Soriente, Tetrahedron Lett. 2006, 47, 8507 – 8510; e) K. Kong, D. Romo,
Org. Lett. 2006, 8, 2909 – 2912; f) F. Zanardi, C. Curti, A. Sartori, G. Rassu, A. Roggio, L.
Battistini, P. Burreddu, L. Pinna, G. Pelosi, G. Casiraghi, Eur. J. Org. Chem. 2008, 2273 –
2287;
6. a) M. Frings, I. Atodiresei, J. Runsink, G. Raabe, C. Bolm, Chem. Eur. J., zur Publikation
akzeptiert; b) Details und Referenzen zu den quantenmechanischen und den CD-
spektroskopischen Kalkulationen können den Supporting Information zu Ref. 6a
entnommen werden.
7. C. Moessner, C. Bolm, Angew. Chem. 2005, 117, 7736 – 7739; Angew. Chem. Int. Ed.
2005, 44, 7564 – 7567.
8. a) S.-M. Lu, C. Bolm, Adv. Synth. Catal. 2008, 350, 1101 – 1105; b) für die erste Synthese
eines naphthalenverbrückten P,N-Sulfoximinliganden siehe: P. Rémy, Dissertation, RWTH-
Aachen University, 2006.

27
9. M. Rueping, A. P. Antonchick, T. Theissmann, Angew. Chem. 2006, 118, 3765 – 3768;
Angew. Chem. Int. Ed. 2006, 45, 3683 – 3686.
10. a) S.-M. Lu, C. Bolm, Chem. Eur. J. 2008, 14, 7512 – 7516; b) S.-M. Lu, C. Bolm, Angew.
Chem. 2008, 120, 9052 – 9055; ; Angew. Chem. Int. Ed. 2008, 47, 8920 – 8923.
11. C. Bolm, M. Martin, L. Gibson, Synlett 2002, 832 – 834.
12. a) G. Y. Cho, P. Rémy, J. Jansson, C. Moessner, C. Bolm, Org. Lett. 2004, 6, 3293 – 3296;
b) C. Moessner, C. Bolm, Org. Lett. 2005, 7, 2667 – 2669; c) J. Sedelmeier, C. Bolm, J.
Org. Chem. 2005, 70, 6904 – 6906
13. Für Übersichten siehe: a) A. F. Pozharskii, V. A. Ozeryanskii, in Chemistry of Anilines (Ed.:
Z. Rappoport), J. Wiley & Sons: Chichester 2007, 2, 931-1139; b) A. L. Llamas-Saiz, C.
Foces-Foces, J. Elguero, J. Mol. Struct. 1994, 328, 297-323; c) R. W. Alder, Chem. Rev.
1989, 89, 1215-1223; d) H. A. Staab, T. Saupe, Angew. Chem. 1988, 100, 895-909; Angew.
Chem. Int. Ed. Engl. 1988, 27, 865-879.
14. a) A. Meister, Biochem. Biophys. Acta 1995, 35; b) H. H. Bailey, Chem.-Biol. Interact. 1998,
111-112, 239; c) E. Obrador, J. Navarro, J. Mompo, M. Asensi, J. A. Pellicier, J. M. Estrela,
BioFactors 1998, 8, 23; d) M. E. Anderson, Chem.-Biol. Interact. 1998, 111, 1; e) L. L.
Muldoon, L. S. L. Walker-Rosenfeld, C. Hale, S. E. Purcell, L. C. Bennett, E. A. Neuwelt J.
Pharmacol. Exp. Ther. 2001, 296, 797; f) M. Kahraman, S. Sinishtaj, P. M. Dolan, T. W.
Kensler, S. Peleg, U. Saha, S. S. Chuang, G. Bernstein, B. Korczak, G. H. Posner, J. Med.
Chem. 2004, 47, 6854; g) W. L. Mock, J.-T. Tsay, J. Am. Chem. Soc. 1989, 111, 4467; h)
W. L. Mock, J. Z. Zhang, J. Biol. Chem. 1991, 266, 6393; i) H. Kawanishi, H. Morimoto, T.
Nakano, T. Watanabe, K. Oda, K. Tsujihara, Heterocycles 1998, 49, 181.
15. Patente: a) Y. Zhu, R. B. Rogers, J. X. Huang, US 2005/228027 A1 (Dow Agroscience); b)
A. Jeanguenat, A. C. O’Sullivan, WO 2006/032462 A1 (Syngenta); c) A. Jeanguenat, A. C.
O’Sullivan, WO 2006/061200 A1 (Syngenta); d) A. Plant, J. E. Boehmer, A. L. Peace, WO
2006/037945 A1 (Syngenta); e) for another sulfoximine-based herbicide, see: S. Kajita, H.
Ohmura, M. Akashi, S. Kojima, A. Satoh, K. Tomida, WO 2004/052849 (Nippon Soda Co.,
Ltd).
16. a) C. Bolm, M. Martin, L. Gibson, Synlett 2002, 832; b) C. Bolm, H. Okamura, M. Verrucci, J.
Organometal. Chem. 2003, 687, 444; c) C. Bolm, H. Villar, Synthesis 2005, 1421.
17. a) C. Bolm, J. D. Kahmann, G. Moll, Tetrahedron Lett. 1997, 38, 1169 – 1172; b) C. Bolm,
G. Moll, J. D. Kahmann, Chem. Eur. J. 2001, 7, 1118 – 1128; c) C. Bolm, D. Müller, C. P. R.
Hackenberger, Org. Lett. 2002, 4, 893 – 896; d) C. P. R. Hackenberger, G. Raabe, C. Bolm,
Chem. Eur. J. 2004 10, 2942 – 2952.
18. C. Bolm, D. Müller, C. Dalhoff, C. P. R. Hackenberger, E. Weinhold, Bioorg. Med. Chem.
Lett. 2003, 13, 3207 – 3211.

28
19. a) J. Legros, C. Bolm, Angew. Chem. 2003, 115, 5675 – 5647; Angew. Chem. Int. Ed. 2003,
42, 5487 – 5489; b) J. Legros, C. Bolm, Angew. Chem. 2004, 116, 4321 – 4324; Angew.
Chem. Int. Ed. 2004, 43, 4225 – 4228; c) J. Legros, C. Bolm, Chem. Eur. J. 2005, 11, 1086
– 1092.
20. a) R. Fusco, F. Tenconi, Chim. Ind. (Milan) 1965, 47, 61– 62; b) C. R. Johnson, C. W.
Schroeck, J. Am. Chem. Soc. 1973, 95, 7418 – 7423; c) Y. Tamura, J. Minamikawa, K.
Sumoto, S. Fujii, M. Ikeda, J. Org. Chem. 1973, 38, 1239 – 1241; d) C. R. Johnson, R. A.
Kirchhoff, H. G. Corkins, J. Org. Chem. 1974, 39, 2458 – 2459; e) Y. Tamura, H.
Matushima, J. Minamikawa, M. Ikeda, K. Sumoto, Tetrahedron 1975, 31, 3035 – 3040; f) M.
Fieser, L. F. Fieser, Reagents for Organic Synthesis, Vol. 5, Wiley, New York, 1975, p. 430.
21. a) J. F. K. Müller, P. Vogt, Tetrahedron Lett. 1998, 39, 4805; b) E. Lacôte, M. Amatore, L.
Fensterbank, M. Malacria Synlett 2002, 116; c) S. Cren, T. C. Kinahan, C. L. Skinner, H.
Tye, Tetrahedron Lett. 2002, 43, 2749; d) C. S. Tomooka, E. M. Carreira, Helv. Chim. Acta
2003, 85, 3773; e) H. Takada, K. Ohe, S. Uemura, Angew. Chem. 1999, 111, 1367; Angew.
Chem., Int. Ed. 1999, 38, 1288; f) H. Nishikori, C. Ohta, E. Oberlin, R. Irie, T. Katsuki,
Tetrahedron 1999, 55, 13937; g) C. Ohta, T. Katsuki, Tetrahedron Lett. 2001, 42, 3885; M.
Murakami, T. Uchida, T. Katsuki, Tetrahedron Lett. 2001, 42, 7071. h) Y. Tamura, T.
Uchida, T. Katsuki, Tetrahedron Lett. 2003, 44, 3301; i) M. Murakami,T. Uchida, B. Saito, T.
Katsuki, Chirality 2003, 15, 116. j) T. Uchida, Y. Tamura, M. Ohba, T. Katsuki, Tetrahedron
Lett. 2003, 44, 7965.
22. a) O. García Mancheño, C. Bolm, Org. Lett. 2006, 8, 2349 – 2352; b) O. García Mancheño,
C. Bolm, Chem. Eur. J. 2007, 13, 6674 – 6681.
23. a) O. García Mancheño, O. Bistri, C. Bolm, Org. Lett. 2007, 9, 3809 – 3811; b) O. García
Mancheño, C. Bolm, Org. Lett. 2007, 9, 2951 – 2954.
24. a) C. Worch, C. Bolm, Synthesis 2007, 1355 – 1358; b) C. Worch, C. Bolm, Synthesis 2008,
739 – 742.
25. Zur Synthese des Sulfonimidoylchlorids siehe: a) B. J. Backes, D. R. Dragoli, J. A. Ellman,
J. Org. Chem. 1999, 64, 5472 – 5478; b) C. K. Savile, V. P. Magloire, R. J. Kazlauskas, J.
Am. Chem. Soc. 2005, 127, 2104 – 2113. Zur Synthese des Aminooxazolins siehe: c) G.
Poos, J. Carson, J. Rosenau, A. Roszkowski, N. Kelley, J. McGowin, J. Med. Chem. 1963,
6, 266 – 272; d) M. Glos, O. Reiser, Org. Lett. 2000, 2, 2045 – 2048.
26. Zur Verwendung von Sulfonimidamiden in der Metallkatalyse als Stickstoffquellen: a) D.
Leca, A. Toussaint, C. Mareau, L. Fensterbank, E. Lacôte, M. Malacria, Org. Lett. 2004, 6,
3573 – 3575 und dort zitierte Literatur; b) C. R. Johnson, E. U. Jonsson, C. C. Bacon, J.
Org. Chem. 1979, 44, 2055 – 2061.
27. M. Nakanishi, A.-F. Salit, C. Bolm, Adv. Synth. Catal. 2008, 350, 1835 – 1840.

29
28. Für Übersichten siehe: a) Aziridines and Epoxides in Organic Synthesis (Ed.: A. K. Yudin),
Wiley-VCH, Weinheim, 2006, 1 – 180; b) Catalyzed Asymmetric Aziridinations, C. Mößner,
C. Bolm in Transition Metals For Organic Chemistry: Building Blocks and Fine Chemicals,
Vol. 2 (Eds.: M. Beller, C. Bolm), Wiley/VCH, Weinheim, 2. ed, 2004, 389 – 402; c) P.
Duban, H. Dodd, Synlett 2003, 1571 – 1586.
29. I. D. G. Watson, L. Yu, A. K. Yudin, Acc. Chem. Res. 2006, 39, 194 – 206.
30. R. T. Stemmler, C. Bolm, Adv. Synth. Catal. 2007, 349, 1185 – 1198.
31. Für Übersichten zur Organokatalyse siehe: a) Enantioselective Organocatalysis (Ed.: P. I.
Dalko), Wiley, Weinheim, 2007; b) Asymmetric Organocatalysis (Eds.: A. Berkessel, H.
Gröger) Wiley, Weinheim, 2005.
32. a) B. Rodríguez, C. Bolm, J. Org. Chem 2006, 71, 2888 – 2891; b) B. Rodriguez, T.
Rantanen, C. Bolm, Angew. Chem., 2006, 118, 7078 – 7080; Angew. Chem. Int. Ed., 2006,
45, 6924 – 6926; c) T. Rantanen, I. Schiffers, C. Bolm, Org. Proc. Res. Develop., 2007, 11,
592 – 597; d) B. Rodríguez, T. Rantanen, A. Bruckmann, C. Bolm, Adv. Synth. Catal., 2007,
349, 2213 – 2233; e) B. Rodríguez, A. Bruckmann, C. Bolm, Chem. Eur. J., 2007, 13, 4710
– 4722; f) A. Bruckmann, A. Krebs, C. Bolm, Green Chem. 2008, 10, 1131 - 1141.
33. Ausgewählte Beispiele: a) V. Amico, S. V. Meille, E. Corradi, M. T. Messina, G. Resnati, J.
Am. Chem. Soc. 1998, 120, 8261 – 8262; b) E. Corradi, S. V. Meille, M. T. Messina, P.
Metrangolo, G. Resnati, Angew. Chem. 2000, 112, 1852 – 1856; Angew. Chem. Int. Ed.,
2000, 39, 1782 – 1786; c) D. D. Burton, F. Fontana, P. Metrangolo, T. Pilati, G. Resnati,
Tetrahedron Lett. 2003, 44, 645 – 648.
34. A. Bruckmann, M. A. Pena, C. Bolm, Synlett 2008, 900 – 902.
35. a) M. T. Messina, P. Metrangolo, W. Panzeri, E. Ragg, G. Resnati, Tetrahedron Lett. 1998,
39, 9069 – 9072; b) G. V. D. Tiers, J. Fluorine Chem. 2000, 102, 175 – 184.

30
Wissenschaftlicher Werdegang von Prof. Dr. Carsten Bolm 03/1960 geboren in Braunschweig, Deutschland
1978 - 1984 TH Braunschweig (Studium; Chemie Diplom)
1983 – 1984 University of Wisconsin, Madison, USA
09/1984 Abschluß als: 'Master of Science, Chemistry' (University of Wisconsin, Madison,
USA; Prof. Dr. H.-J. Reich)
12/1984 Abschluß der 'Diplom-Chemiker-Hauptprüfung' (TH Braunschweig, Deutschland;
Prof. Dr. H. Hopf)
1985 – 1987 Doktorarbeit an der Philipps-Universität Marburg, Deutschland
07/1987 Abschluß als 'Dr. rer. nat.' (Philipps-Universität Marburg; Prof. Dr. M. T. Reetz)
1987-1988 Postdoktorat (M.I.T., Cambridge, USA; Prof. Dr. K. B. Sharpless)
1988 – 1993 Habilitation (TU Darmstadt, Deutschland, und Universität Basel, Schweiz; bei
Prof. Dr. B. Giese)
1993 Angebote auf C3-Professuren an den Universitäten von Würzburg (abgeleht),
Hannover (abgeleht) und Marburg
10/1993 C3-Professor für Organische Chemie an der Philipps-Universität Marburg,
Deutschland
03/1996 Lehrstuhl und C4-Professur für Organische Chemie an der RWTH Aachen,
Deutschland
02/2000 Angebot einer C4-Professur für Organische Chemie an der Universität in
Freiburg, Deutschland (abgelehnt)
08/2005 Angebot einer W3-Professur für Organische Chemie an der Ruprecht-Karls
University in Heidelberg, Deutschland (abgelehnt)
04/2006 W3 Professor an der RWTH Aachen, Deutschland
Stipendien und Auszeichnungen 1983-1984 Fulbright-Stipendium
1985-1987 Kekulé-Stipendium
1987-1988 Nato-Stipendium des DAAD
1988-1990 Liebig-Stipendium
1990-1992 Treubel-Fonds Stipendium
06/1991 Heinz-Meier-Leibnitz-Preis
03/1992 ADUC-Jahrespreis 1992 für Habilitanden
02/1993 Dozenten-Stipendium

31
11/1993 Jahrespreis für Chemie der "Akademie der Wissenschaften zu Göttingen“
12/1996 Otto-Klung-Preis 1996
12/1998 Otto-Bayer-Preis
10/2002 AstraZeneca European Lecturer
09/2003 Stipendium der Japan Society for the Promotion of Science (JSPS)
07/2006 Prix Franco-Allemand derSociété Francaise de Chimie
11/2007 CaRLa Fellow (an der Universität Heidelberg in Zusammenarbeit mit BASF)
Gastprofessuren 09-12/1992 Visiting Professor an der University of Wisconsin, Madison, USA
03/1998 Invited Visiting Professorship an der Université Pierre et Marie Curie’ in Paris,
Frankreich
02/2000 Invited Visiting Professorship an der Università di Firenze’ in Florence, Italien
02/2002 Invited Visiting Professorship an der‘Università degli studi di Milano’ in Milan,
Italien
2002 – 2003 Professeur Visiteur am Département de Chimie der Universität ‘Notre-Dame De
La Paix‘ in Namur, Belgien
10/2006 Invited Visiting Professorship an der Tokyo University of Science, Japan
Forschungsinteressen Enantioselektive Katalyse
Asymmetrische Katalyse mit metallorganischen Reagenzien
Pseudopeptide
Zeitschriften 1998 – 2002 Mitglied im International Advisory Editorial Board vom New Journal of Chemistry
1999 - Mitglied im Editorial Advisory Board von Synthesis
2000 - Mitglied im Honorary Advisory Board von Synlett
2001 - Mitglied im Academic Advisory Board von Advanced Synthesis and Catalysis
2003 - Mitglied im Board of Consulting Editors von Tetrahedron Letters
2003 - Mitglied im Board of Consulting Editors von Tetrahedron
2003 - Mitglied im Editorial Board von Letters in Organic Chemistry

32
2003 - Commissioning Editor von Chemical Society Reviews
2005 - Mitglied im Editorial Board von Chemical Reviews
2005 - 2007 Mitglied im Editorial Advisory Board vom Journal of Organic Chemistry
2005 - Mitglied im Editorial Advisory Board vom Beilstein Journal of Organic Chemistry
2007 - Mitglied im Editorial Board von Current Chemical Biology
2008 - Mitglied im Editorial Advisory Board von Organic Letters
2008 - Mitglied im Editorial Advisory Board von Catalysis Communications
2008 - Associate Editor für Journal of Organic Chemistry
Andere Aktivitäten 1998 – 2001 Mitglied im DAAD Auswahlausschuß „Doktorandenstipendien“
2002 - Mitglied im DAAD Auswahlausschuß „PostdocProgramm“
2002 - 2007 Mitglied im „Senats- and Bewilligungsausschuss für die Graduiertenkollegs“ der
DFG
1998 - Mitglied im „Beirat Technologietransfer und Arbeitskreis Technologietransfer
RWTH-IHK“
2000 - Mitglied im Management Commitee der COST-Action D24 „Sustainable chemical
processes: Stereoselective transition-metal catalyzed reactions“
2001- Mitglied im International Scientific Committee von ESOC (European Symposium
on Organic Chemistry)
2001 - 2005 Mitglied im „Wissenschaftlichen Beirat des Leibniz-Instituts für Organische
Katalyse an der Universität Rostock e.V.“
2003- Mitglied im International Advisory Committee von ICHAC (International
Conferrence on Heteroatom Chemistry)
2006 - Vorsitz des „Wissenschaftlichen Beirats des Leibniz-Instituts für Katalyse an der
Universität Rostock e.V.“
2002 - 2006 Mitglied im Scientific Advisory Board von DEGUSSA Fine Chemicals
2002 -2004 Wahlsenator an der RWTH Aachen
2003 - 2007 Federführung des „DFG Schwerpunktprogramms 1118"
2006 - 2008 Mitglied des GDCh-Auswahlgremiums für die "August-Wilhelm-von-Hofmann-
Denkmünze"
2006 - 2008 Sprecher der Fachgruppe Chemie an der RWTH Aachen
2007 - Mitglied im VCI Fachausschuss Forschungs- und Bildungspolitik
2008 - Rektoratsbeauftragter für Korea
![[ger] Eisen und Stahl : 2 1991 Monatlich [eng] Iron and ...aei.pitt.edu/72441/1/1991.2.pdf · En janvier 1991, la production communautaire d'acier brut a enregistré, avec 11,3 mio.t,](https://static.fdocument.org/doc/165x107/5e6c265a64902218da4d23f6/ger-eisen-und-stahl-2-1991-monatlich-eng-iron-and-aeipittedu72441119912pdf.jpg)


![[spa] Siderurgia : Anuario estadístico 1988 [dan] Jern …aei.pitt.edu/70105/1/1988.pdf · SIDERURGIA Anuario estadístico JERN OG STÅL Statistisk årbog EISEN UND STAHL Statistisches](https://static.fdocument.org/doc/165x107/5ba3b74809d3f2c0278c00fb/spa-siderurgia-anuario-estadistico-1988-dan-jern-aeipittedu7010511988pdf.jpg)














