
Total-molecular-weight-dependent Rouse dynamic of ultra ...hosting03.snu.ac.kr/~eco/file/115.pdf ·...
8
Total-molecular-weight-dependent Rouse dynamic of ultra-small branched star poly(ε-caprolactone)s as a single coarse-grain unit Woohyuk Choi a , Jae Woo Chung b, ** , Seung-Yeop Kwak a, * a Department of Materials Science and Engineering, Seoul National University, 599 Gwanak-ro, Gwanak-gu, Seoul,151-744, Republic of Korea b Department of Organic Materials and Fiber Engineering, Soongsil University, 369 Sangdo-ro, Dongjak-gu, Seoul, 156-743, Republic of Korea article info Article history: Received 7 April 2015 Received in revised form 1 September 2015 Accepted 2 September 2015 Available online 9 September 2015 Keywords: Molecular dynamics Star poly(ε-caprolactone) Ultra-small branches abstract The extremely small branched effects on molecular dynamics are investigated using well-defined star poly(ε-caprolactone)s containing ultra-small branches (USB-SPCLs) as a model system. USB-SPCLs interestingly show total-molecular-weight-dependent glass transitions regardless of the molecular ar- chitecture parameters, such as the number and length of branches, whereas typical star polymers with polymeric large branches show the end-group-concentration-dependent glass transitions. The visco- elasticity of USB-SPCLs does not depend exponentially on the individual branched molecular weight, as observed in typical star polymers, and instead follows the modified MarkeHouwink power law and the Bueche-modified Rouse model for unentangled linear polymers. The flow activation energy and the longest Rouse relaxation time of USB-SPCLs show that the individual branches of USB-SPCL are dynamically equivalent and that a whole USB-SPCL molecule moves with a simple uni-motion. These results suggest that a whole USB-SPCL molecule presumably acts as a dynamically-equivalent single coarse-grain unit because of the extremely small branches on the scale of 20e40 atoms. © 2015 Elsevier Ltd. All rights reserved. 1. Introduction Over past decades, branched polymers (i.e., polymers with more than two chain ends per molecule) have attracted significant attention in varied scientific and industrial fields due to their distinctive properties, including low melting point, low melt vis- cosity, and rapid molecular motion, that differ from those of linear polymers [1e5]. Among various branched polymers, star-shaped polymers (also known as ‘star polymers’) are the simplest branched polymers having only one central tie point, and the un- derstanding of their dynamic properties can provide fundamental insight into the development and characterization of more complex branched polymers, such as hyperbranched, dendritic, H-shaped, comb-shaped, and pomepom polymers [4e7]. Thus, these struc- tureeproperty relationships have exhaustively been investigated, especially in comparison with linear polymers. Kraus et al. first re- ported that the increase in the viscosity of star polymers with mo- lecular weight did not occur according to a power law, as observed in linear polymers, but instead occurred exponentially [8]. Quack et al. showed that the viscosity of star polymers did not depend on the total molecular weight but only on the molecular weights of individual branched segments [9]. Similar findings were made by Mykhaylyk et al., who observed that the zero-shear-rate viscosity of hydrogenated star polybutadienes was exponentially dependent on the molecular weights of the arms rather than on the total molec- ular weights of the stars [10]. Acebo et al. also reported that the viscosity of multiarm star block copolymers (hyperbranched poly(- ethyleneimine)-b-poly(ε-caprolactone)) increased exponentially as the arm length (or arm molecular weight) of the star block copol- ymer increased [11]. These phenomena were attributed to the fact that star polymers are not able to reptate as linear polymers because of their central tie point that suppresses the translational motion of the branched segments [12]. Instead, star polymers can relax by contour-length fluctuations of branched segments. This contour- length fluctuation allows the branched segments to retract some distance down the confining tube toward the central tie point and then explore a new path by extending back in another direction [12,13]. Since deep retractions of branched segments in star poly- mers are not favored entropically and are exponentially unlikely, thus the viscoelastic properties of star polymers depend exponen- tially on the molecular weights of individual branching segments [12,13]. These studies clearly showed that the dynamics of star * Corresponding author. ** Corresponding author. E-mail addresses: [email protected] (J.W. Chung), [email protected] (S.-Y. Kwak). Contents lists available at ScienceDirect Polymer journal homepage: www.elsevier.com/locate/polymer http://dx.doi.org/10.1016/j.polymer.2015.09.009 0032-3861/© 2015 Elsevier Ltd. All rights reserved. Polymer 79 (2015) 91e98
Transcript of Total-molecular-weight-dependent Rouse dynamic of ultra ...hosting03.snu.ac.kr/~eco/file/115.pdf ·...

lable at ScienceDirect
Polymer 79 (2015) 91e98
Contents lists avai
Polymer
journal homepage: www.elsevier .com/locate/polymer
Total-molecular-weight-dependent Rouse dynamic of ultra-smallbranched star poly(ε-caprolactone)s as a single coarse-grain unit
Woohyuk Choi a, Jae Woo Chung b, **, Seung-Yeop Kwak a, *
a Department of Materials Science and Engineering, Seoul National University, 599 Gwanak-ro, Gwanak-gu, Seoul, 151-744, Republic of Koreab Department of Organic Materials and Fiber Engineering, Soongsil University, 369 Sangdo-ro, Dongjak-gu, Seoul, 156-743, Republic of Korea
a r t i c l e i n f o
Article history:Received 7 April 2015Received in revised form1 September 2015Accepted 2 September 2015Available online 9 September 2015
Keywords:Molecular dynamicsStar poly(ε-caprolactone)Ultra-small branches
* Corresponding author.** Corresponding author.
E-mail addresses: [email protected] (J.W.(S.-Y. Kwak).
http://dx.doi.org/10.1016/j.polymer.2015.09.0090032-3861/© 2015 Elsevier Ltd. All rights reserved.
a b s t r a c t
The extremely small branched effects on molecular dynamics are investigated using well-defined starpoly(ε-caprolactone)s containing ultra-small branches (USB-SPCLs) as a model system. USB-SPCLsinterestingly show total-molecular-weight-dependent glass transitions regardless of the molecular ar-chitecture parameters, such as the number and length of branches, whereas typical star polymers withpolymeric large branches show the end-group-concentration-dependent glass transitions. The visco-elasticity of USB-SPCLs does not depend exponentially on the individual branched molecular weight, asobserved in typical star polymers, and instead follows the modified MarkeHouwink power law and theBueche-modified Rouse model for unentangled linear polymers. The flow activation energy and thelongest Rouse relaxation time of USB-SPCLs show that the individual branches of USB-SPCL aredynamically equivalent and that a whole USB-SPCL molecule moves with a simple uni-motion. Theseresults suggest that a whole USB-SPCL molecule presumably acts as a dynamically-equivalent singlecoarse-grain unit because of the extremely small branches on the scale of 20e40 atoms.
© 2015 Elsevier Ltd. All rights reserved.
1. Introduction
Over past decades, branched polymers (i.e., polymers with morethan two chain ends per molecule) have attracted significantattention in varied scientific and industrial fields due to theirdistinctive properties, including low melting point, low melt vis-cosity, and rapid molecular motion, that differ from those of linearpolymers [1e5]. Among various branched polymers, star-shapedpolymers (also known as ‘star polymers’) are the simplestbranched polymers having only one central tie point, and the un-derstanding of their dynamic properties can provide fundamentalinsight into the development and characterization of more complexbranched polymers, such as hyperbranched, dendritic, H-shaped,comb-shaped, and pomepom polymers [4e7]. Thus, these struc-tureeproperty relationships have exhaustively been investigated,especially in comparison with linear polymers. Kraus et al. first re-ported that the increase in the viscosity of star polymers with mo-lecular weight did not occur according to a power law, as observed
Chung), [email protected]
in linear polymers, but instead occurred exponentially [8]. Quacket al. showed that the viscosity of star polymers did not depend onthe total molecular weight but only on the molecular weights ofindividual branched segments [9]. Similar findings were made byMykhaylyk et al., who observed that the zero-shear-rate viscosity ofhydrogenated star polybutadienes was exponentially dependent onthe molecular weights of the arms rather than on the total molec-ular weights of the stars [10]. Acebo et al. also reported that theviscosity of multiarm star block copolymers (hyperbranched poly(-ethyleneimine)-b-poly(ε-caprolactone)) increased exponentially asthe arm length (or arm molecular weight) of the star block copol-ymer increased [11]. These phenomena were attributed to the factthat star polymers are not able to reptate as linear polymers becauseof their central tie point that suppresses the translational motion ofthe branched segments [12]. Instead, star polymers can relax bycontour-length fluctuations of branched segments. This contour-length fluctuation allows the branched segments to retract somedistance down the confining tube toward the central tie point andthen explore a new path by extending back in another direction[12,13]. Since deep retractions of branched segments in star poly-mers are not favored entropically and are exponentially unlikely,thus the viscoelastic properties of star polymers depend exponen-tially on the molecular weights of individual branching segments[12,13]. These studies clearly showed that the dynamics of star

W. Choi et al. / Polymer 79 (2015) 91e9892
polymers were determined by the individual branched length andthe central tie point. However, most of these previous studiesaddressed the dynamic behaviors of star polymers with polymericlarge branches. Star polymers with extremely small branches andtheir dynamic behaviors have not been studied sufficiently althoughstar polymers with extremely small branches are highly attractivedue to the ease of control over degradation, low softening point, andgood miscibility with other materials, compared with typical starpolymers [14e17]. Thus, from both the scientific and industrial pointof view, the various investigations into dynamic phenomena in starpolymers with extremely small branches are positively necessary touse these highly attractive advantages in an extremely smallbranched system.
Star poly(ε-caprolactone)s (SPCLs) can be synthesized with anarrow molecular weight distribution (MWD) and the target mo-lecular architecture (e.g., degree of branching and branchinglength) by a simple ring-opening polymerization [18,19]. Moreover,SPCLs are highly biocompatible and biodegradable polyesters, andhave thus been widely investigated for pharmacological, biomed-ical, agricultural, and environmental purposes [16,19e22]. Despitesuch advantageous properties of SPCLs, SPCLs with extremely smallbranches have hardly been studied owing to the failure to obtainwell-controlled SPCLs. The chronic backbiting problem occurswhen attempting to synthesize and purify SPCLs, which results inthe collapse of the PCL structure and the formation of undesirablecyclic PCLs, and eventually makes impossible the synthesis andisolation of the well-controlled SPCLs. We have already overcomethese synthetic problems via the manipulating monomer-to-coreratio, adjusting the monomer-to-polymer conversion, end-capping the terminal hydroxyl groups, and vacuum purificationusing a facile pseudo-one-pot process on a pilot scale, resulting inprecisely controlled SPCLs with extremely small branches (degreeof polymerization (DP) in an extremely small branch �5) [14,15].Furthermore, since the terminal hydroxyl groups of our SPCLs arecompletely capped with small acetate groups, it is anticipated thatthe dynamic behaviors of extremely small branches in SPCLs can beinvestigated without hydrogen-bonding effects.
In this study, we investigated the effects of extremely smallbranches on the dynamic behavior of a star polymer using well-defined ultra-small-branched star poly(ε-caprolactone)s (USB-SPCLs) as a model system. The glass transition, viscoelasticity, flowactivation energy, and longest Rouse relaxation time of USB-SPCLsinterestingly depended on total molecular weight regardless ofmolecular architecture parameters, such as the number and lengthof branched segments. Moreover, these dynamic phenomena ofUSB-SPCLs followed the modified MarkeHouwink power law andthe Bueche-modified Rouse model for unentangled linear polymersrather than traditional star polymer models that depend expo-nentially on the individual branched molecular weight. This sug-gests that a whole USB-SPCL molecule acts as a dynamically-equivalent single coarse-grain unit with one Rouse-segmentalmotion. These results broaden and deepen the understanding ofthe molecular dynamic behaviors of star polymers with extremelysmall branches, and further this enables new applications for starpolymers as well as more complex branched polymers with uniquedynamic properties.
2. Experimental
2.1. Materials
Ultra-small-branched star poly(ε-caprolactone)s (USB-SPCLs)were synthesized by ring-opening polymerization (ROP) of ε-cap-rolactone (CL) monomers via manipulating the monomer-to-coreratio, adjusting the monomer-to-polymer conversion, end-
capping the terminal hydroxyl groups, and vacuum purification ina facile pseudo-one-pot bulk process without organic solvents[14,15]. Eight different USB-SPCLs were synthesized in which thenumber of branches, Nnumber, and the average lengths of the indi-vidual branches (the average DP per branch), Nlength, were varied.These polymers are denoted as USB-SPCL Nnumber � Nlength, and thespecific polymers treated here are named USB-SPCL3-2 (whereNnumber ¼ 3 and Nlength ¼ 2), USB-SPCL3-3 (where Nnumber ¼ 3 andNlength ¼ 3), USB-SPCL3-4 (where Nnumber ¼ 3 and Nlength ¼ 4), USB-SPCL3-5 (Nnumber ¼ 3 and Nlength ¼ 5), USB-SPCL6-2 (Nnumber ¼ 6and Nlength ¼ 2), USB-SPCL6-3 (Nnumber ¼ 6 and Nlength ¼ 3), USB-SPCL6-4 (Nnumber ¼ 6 and Nlength ¼ 4), and USB-SPCL6-5(Nnumber ¼ 6 and Nlength ¼ 5). The chemical structures of the syn-thesized USB-SPCLs, including Nnumber and Nlength, were analyzedusing 1H nuclear magnetic resonance (NMR) spectroscopy. Thenumber-average molecular weights, Mn, the weight-average mo-lecular weights, Mw, and the molecular weight distributions(MWDs) were measured using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS).More detailed information about the syntheses and general char-acterizations of USB-SPCLs are provided in our previous publica-tions [14,15].
2.2. Characterization
The glass transitions of USB-SPCLs were measured by differen-tial scanning calorimetry (DSC) using a Netzsch DSC 200 F3 with aheating rate of 20 �C min�1 over the temperature range of �110 to140 �C under a nitrogen atmosphere. The glass transition temper-ature, Tg, was determined in each case according to the inflectionpoint in the second DSC curve. The viscoelastic behaviors of USB-SPCLs were observed by dynamic mechanical spectrometry usinga stress-controlled rheometer (TA Instruments Inc. AR2000). TheAR2000was operated in a cone-and-plate geometrywith a 1� angleand a 60-mm-diameter cone. The gap between the cone and theplate was 80 mm in all measurements. Dynamic frequency sweepswere performed over an angular frequency range of0.5e100 rad s�1 and the temperature range 30e90 �C, measured in10 �C intervals. The strain values were determined using a dynamicstrain sweep to lie within the linear viscoelastic region.
3. Results and discussion
3.1. Glass transition behaviors of USB-SPCLs
Three- and six-branched USB-SPCLs were prepared with variousnumber of CL repeating units (DP ¼ 2, 3, 4, or 5) in each individualbranch initiated from trimethylolpropane (TMP) and dipentaery-thritol (DPTOL) cores (Fig. 1). From 1H NMR and MALDI-TOF-MSmeasurements (Table 1), it was confirmed that the USB-SPCLswere well synthesized with the desired Nnumber, Nlength, and nar-row MWDs (Mw/Mn �1.2). Then, the glass transitions of theresulting USB-SPCLs were observed through DSC. As shown inFig. 2(a), the Tg values for TMP-cored USB-SPCLs decreased withdecreasing Nlength, i.e., USB-SPCL3-5 (�64.4 �C) > USB-SPCL3-4(�64.9 �C) > USB-SPCL3-3 (�65.4 �C) > USB-SPCL3-2 (�65.8 �C),and those for DPTOL-cored USB-SPCLs also had the same tendency(USB-SPCL6-5 (�62.7 �C) > USB-SPCL6-4 (�63.2 �C) > USB-SPCL6-3(�64.0 �C) > USB-SPCL6-2 (�64.6 �C)). It is generally accepted thata star polymer with a greater number of branches is known to yielda smaller pervaded volume (V z R3) and faster molecular mobilityat a given total molecular weight [23,24]. In addition, Roovers et al.reported that the Tg value of a star polymer decreased as the end-group concentration increased due to an increase in the free vol-ume [25,26]. Thus, the end-group concentrations of USB-SPCLs
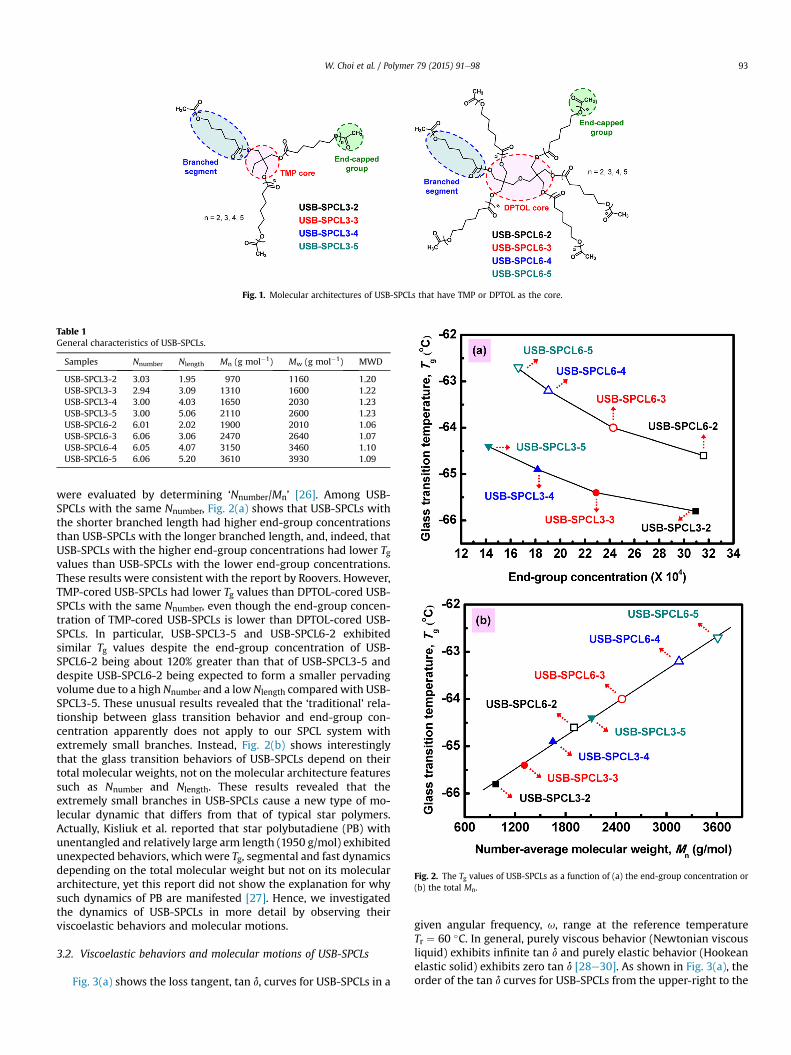
Fig. 1. Molecular architectures of USB-SPCLs that have TMP or DPTOL as the core.
Table 1General characteristics of USB-SPCLs.
Samples Nnumber Nlength Mn (g mol�1) Mw (g mol�1) MWD
USB-SPCL3-2 3.03 1.95 970 1160 1.20USB-SPCL3-3 2.94 3.09 1310 1600 1.22USB-SPCL3-4 3.00 4.03 1650 2030 1.23USB-SPCL3-5 3.00 5.06 2110 2600 1.23USB-SPCL6-2 6.01 2.02 1900 2010 1.06USB-SPCL6-3 6.06 3.06 2470 2640 1.07USB-SPCL6-4 6.05 4.07 3150 3460 1.10USB-SPCL6-5 6.06 5.20 3610 3930 1.09
Fig. 2. The Tg values of USB-SPCLs as a function of (a) the end-group concentration or(b) the total Mn.
W. Choi et al. / Polymer 79 (2015) 91e98 93
were evaluated by determining ‘Nnumber/Mn’ [26]. Among USB-SPCLs with the same Nnumber, Fig. 2(a) shows that USB-SPCLs withthe shorter branched length had higher end-group concentrationsthan USB-SPCLs with the longer branched length, and, indeed, thatUSB-SPCLs with the higher end-group concentrations had lower Tgvalues than USB-SPCLs with the lower end-group concentrations.These results were consistent with the report by Roovers. However,TMP-cored USB-SPCLs had lower Tg values than DPTOL-cored USB-SPCLs with the same Nnumber, even though the end-group concen-tration of TMP-cored USB-SPCLs is lower than DPTOL-cored USB-SPCLs. In particular, USB-SPCL3-5 and USB-SPCL6-2 exhibitedsimilar Tg values despite the end-group concentration of USB-SPCL6-2 being about 120% greater than that of USB-SPCL3-5 anddespite USB-SPCL6-2 being expected to form a smaller pervadingvolume due to a high Nnumber and a lowNlength comparedwith USB-SPCL3-5. These unusual results revealed that the ‘traditional’ rela-tionship between glass transition behavior and end-group con-centration apparently does not apply to our SPCL system withextremely small branches. Instead, Fig. 2(b) shows interestinglythat the glass transition behaviors of USB-SPCLs depend on theirtotal molecular weights, not on the molecular architecture featuressuch as Nnumber and Nlength. These results revealed that theextremely small branches in USB-SPCLs cause a new type of mo-lecular dynamic that differs from that of typical star polymers.Actually, Kisliuk et al. reported that star polybutadiene (PB) withunentangled and relatively large arm length (1950 g/mol) exhibitedunexpected behaviors, which were Tg, segmental and fast dynamicsdepending on the total molecular weight but not on its moleculararchitecture, yet this report did not show the explanation for whysuch dynamics of PB are manifested [27]. Hence, we investigatedthe dynamics of USB-SPCLs in more detail by observing theirviscoelastic behaviors and molecular motions.
3.2. Viscoelastic behaviors and molecular motions of USB-SPCLs
Fig. 3(a) shows the loss tangent, tan d, curves for USB-SPCLs in a
given angular frequency, u, range at the reference temperatureTr ¼ 60 �C. In general, purely viscous behavior (Newtonian viscousliquid) exhibits infinite tan d and purely elastic behavior (Hookeanelastic solid) exhibits zero tan d [28e30]. As shown in Fig. 3(a), theorder of the tan d curves for USB-SPCLs from the upper-right to the
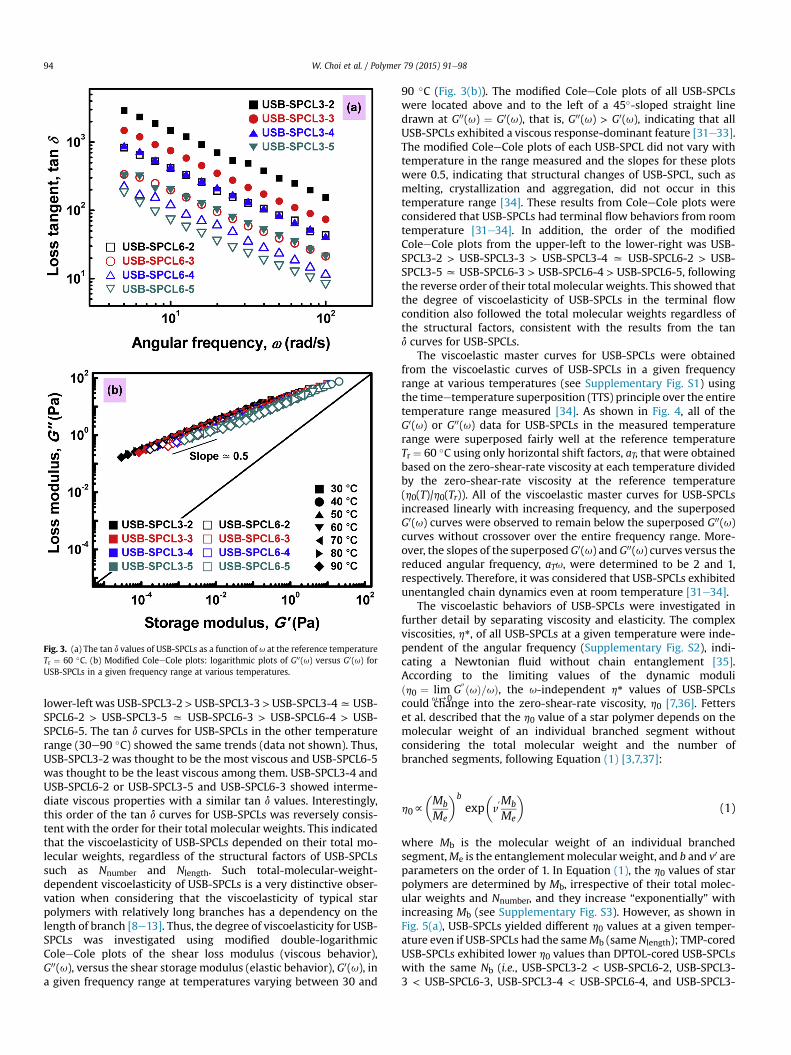
Fig. 3. (a) The tan d values of USB-SPCLs as a function of u at the reference temperatureTr ¼ 60 �C. (b) Modified ColeeCole plots: logarithmic plots of G00(u) versus G0(u) forUSB-SPCLs in a given frequency range at various temperatures.
W. Choi et al. / Polymer 79 (2015) 91e9894
lower-left was USB-SPCL3-2>USB-SPCL3-3 >USB-SPCL3-4xUSB-SPCL6-2 > USB-SPCL3-5 x USB-SPCL6-3 > USB-SPCL6-4 > USB-SPCL6-5. The tan d curves for USB-SPCLs in the other temperaturerange (30e90 �C) showed the same trends (data not shown). Thus,USB-SPCL3-2 was thought to be the most viscous and USB-SPCL6-5was thought to be the least viscous among them. USB-SPCL3-4 andUSB-SPCL6-2 or USB-SPCL3-5 and USB-SPCL6-3 showed interme-diate viscous properties with a similar tan d values. Interestingly,this order of the tan d curves for USB-SPCLs was reversely consis-tent with the order for their total molecular weights. This indicatedthat the viscoelasticity of USB-SPCLs depended on their total mo-lecular weights, regardless of the structural factors of USB-SPCLssuch as Nnumber and Nlength. Such total-molecular-weight-dependent viscoelasticity of USB-SPCLs is a very distinctive obser-vation when considering that the viscoelasticity of typical starpolymers with relatively long branches has a dependency on thelength of branch [8e13]. Thus, the degree of viscoelasticity for USB-SPCLs was investigated using modified double-logarithmicColeeCole plots of the shear loss modulus (viscous behavior),G00(u), versus the shear storage modulus (elastic behavior), G0(u), ina given frequency range at temperatures varying between 30 and
90 �C (Fig. 3(b)). The modified ColeeCole plots of all USB-SPCLswere located above and to the left of a 45�-sloped straight linedrawn at G00(u) ¼ G0(u), that is, G00(u) > G0(u), indicating that allUSB-SPCLs exhibited a viscous response-dominant feature [31e33].The modified ColeeCole plots of each USB-SPCL did not vary withtemperature in the range measured and the slopes for these plotswere 0.5, indicating that structural changes of USB-SPCL, such asmelting, crystallization and aggregation, did not occur in thistemperature range [34]. These results from ColeeCole plots wereconsidered that USB-SPCLs had terminal flow behaviors from roomtemperature [31e34]. In addition, the order of the modifiedColeeCole plots from the upper-left to the lower-right was USB-SPCL3-2 > USB-SPCL3-3 > USB-SPCL3-4 x USB-SPCL6-2 > USB-SPCL3-5 x USB-SPCL6-3 > USB-SPCL6-4 > USB-SPCL6-5, followingthe reverse order of their total molecular weights. This showed thatthe degree of viscoelasticity of USB-SPCLs in the terminal flowcondition also followed the total molecular weights regardless ofthe structural factors, consistent with the results from the tand curves for USB-SPCLs.
The viscoelastic master curves for USB-SPCLs were obtainedfrom the viscoelastic curves of USB-SPCLs in a given frequencyrange at various temperatures (see Supplementary Fig. S1) usingthe timeetemperature superposition (TTS) principle over the entiretemperature range measured [34]. As shown in Fig. 4, all of theG0(u) or G00(u) data for USB-SPCLs in the measured temperaturerange were superposed fairly well at the reference temperatureTr ¼ 60 �C using only horizontal shift factors, aT, that were obtainedbased on the zero-shear-rate viscosity at each temperature dividedby the zero-shear-rate viscosity at the reference temperature(h0(T)/h0(Tr)). All of the viscoelastic master curves for USB-SPCLsincreased linearly with increasing frequency, and the superposedG0(u) curves were observed to remain below the superposed G00(u)curves without crossover over the entire frequency range. More-over, the slopes of the superposed G0(u) and G00(u) curves versus thereduced angular frequency, aTu, were determined to be 2 and 1,respectively. Therefore, it was considered that USB-SPCLs exhibitedunentangled chain dynamics even at room temperature [31e34].
The viscoelastic behaviors of USB-SPCLs were investigated infurther detail by separating viscosity and elasticity. The complexviscosities, h*, of all USB-SPCLs at a given temperature were inde-pendent of the angular frequency (Supplementary Fig. S2), indi-cating a Newtonian fluid without chain entanglement [35].According to the limiting values of the dynamic moduliðh0 ¼ lim
u/0G
00 ðuÞ=uÞ, the u-independent h* values of USB-SPCLscould change into the zero-shear-rate viscosity, h0 [7,36]. Fetterset al. described that the h0 value of a star polymer depends on themolecular weight of an individual branched segment withoutconsidering the total molecular weight and the number ofbranched segments, following Equation (1) [3,7,37]:
h0f
�MbMe
�b
exp�v0MbMe
�(1)
where Mb is the molecular weight of an individual branchedsegment,Me is the entanglementmolecular weight, and b and v0 areparameters on the order of 1. In Equation (1), the h0 values of starpolymers are determined by Mb, irrespective of their total molec-ular weights and Nnumber, and they increase “exponentially” withincreasing Mb (see Supplementary Fig. S3). However, as shown inFig. 5(a), USB-SPCLs yielded different h0 values at a given temper-ature even if USB-SPCLs had the sameMb (sameNlength); TMP-coredUSB-SPCLs exhibited lower h0 values than DPTOL-cored USB-SPCLswith the same Nb (i.e., USB-SPCL3-2 < USB-SPCL6-2, USB-SPCL3-3 < USB-SPCL6-3, USB-SPCL3-4 < USB-SPCL6-4, and USB-SPCL3-
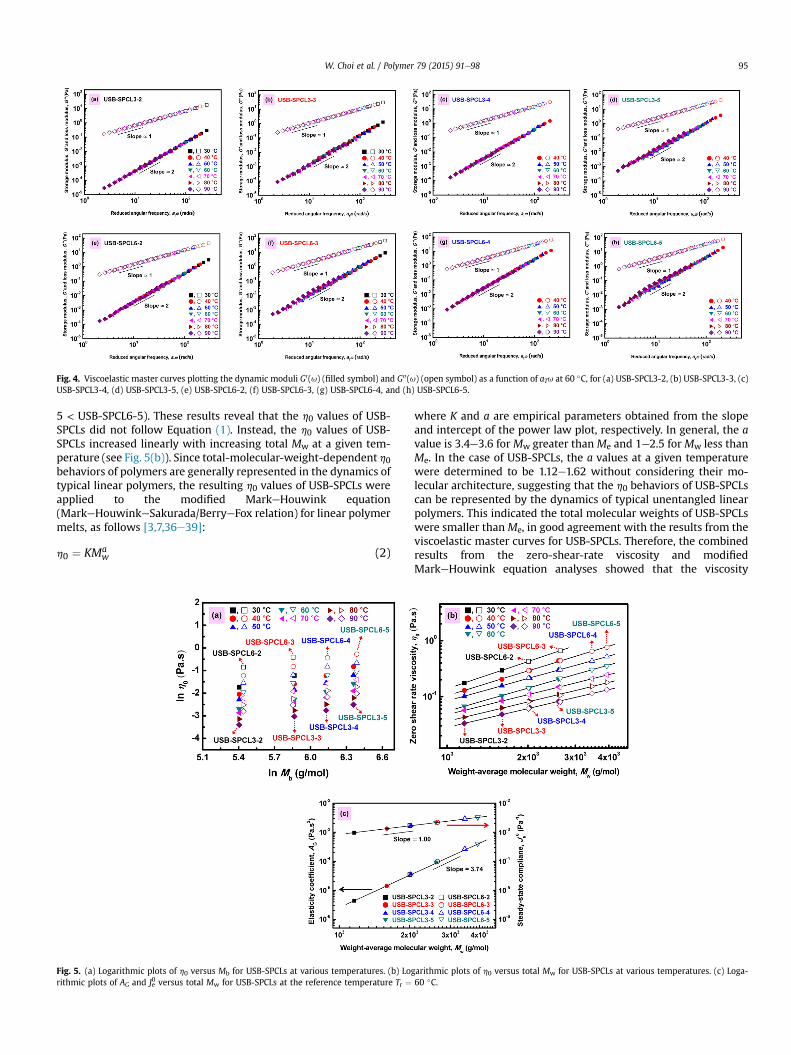
Fig. 4. Viscoelastic master curves plotting the dynamic moduli G0(u) (filled symbol) and G00(u) (open symbol) as a function of aTu at 60 �C, for (a) USB-SPCL3-2, (b) USB-SPCL3-3, (c)USB-SPCL3-4, (d) USB-SPCL3-5, (e) USB-SPCL6-2, (f) USB-SPCL6-3, (g) USB-SPCL6-4, and (h) USB-SPCL6-5.
W. Choi et al. / Polymer 79 (2015) 91e98 95
5 < USB-SPCL6-5). These results reveal that the h0 values of USB-SPCLs did not follow Equation (1). Instead, the h0 values of USB-SPCLs increased linearly with increasing total Mw at a given tem-perature (see Fig. 5(b)). Since total-molecular-weight-dependent h0behaviors of polymers are generally represented in the dynamics oftypical linear polymers, the resulting h0 values of USB-SPCLs wereapplied to the modified MarkeHouwink equation(MarkeHouwinkeSakurada/BerryeFox relation) for linear polymermelts, as follows [3,7,36e39]:
h0 ¼ KMaw (2)
Fig. 5. (a) Logarithmic plots of h0 versus Mb for USB-SPCLs at various temperatures. (b) Logrithmic plots of AG and Je
0 versus total Mw for USB-SPCLs at the reference temperature Tr ¼
where K and a are empirical parameters obtained from the slopeand intercept of the power law plot, respectively. In general, the avalue is 3.4e3.6 for Mw greater than Me and 1e2.5 for Mw less thanMe. In the case of USB-SPCLs, the a values at a given temperaturewere determined to be 1.12e1.62 without considering their mo-lecular architecture, suggesting that the h0 behaviors of USB-SPCLscan be represented by the dynamics of typical unentangled linearpolymers. This indicated the total molecular weights of USB-SPCLswere smaller than Me, in good agreement with the results from theviscoelastic master curves for USB-SPCLs. Therefore, the combinedresults from the zero-shear-rate viscosity and modifiedMarkeHouwink equation analyses showed that the viscosity
arithmic plots of h0 versus total Mw for USB-SPCLs at various temperatures. (c) Loga-60 �C.
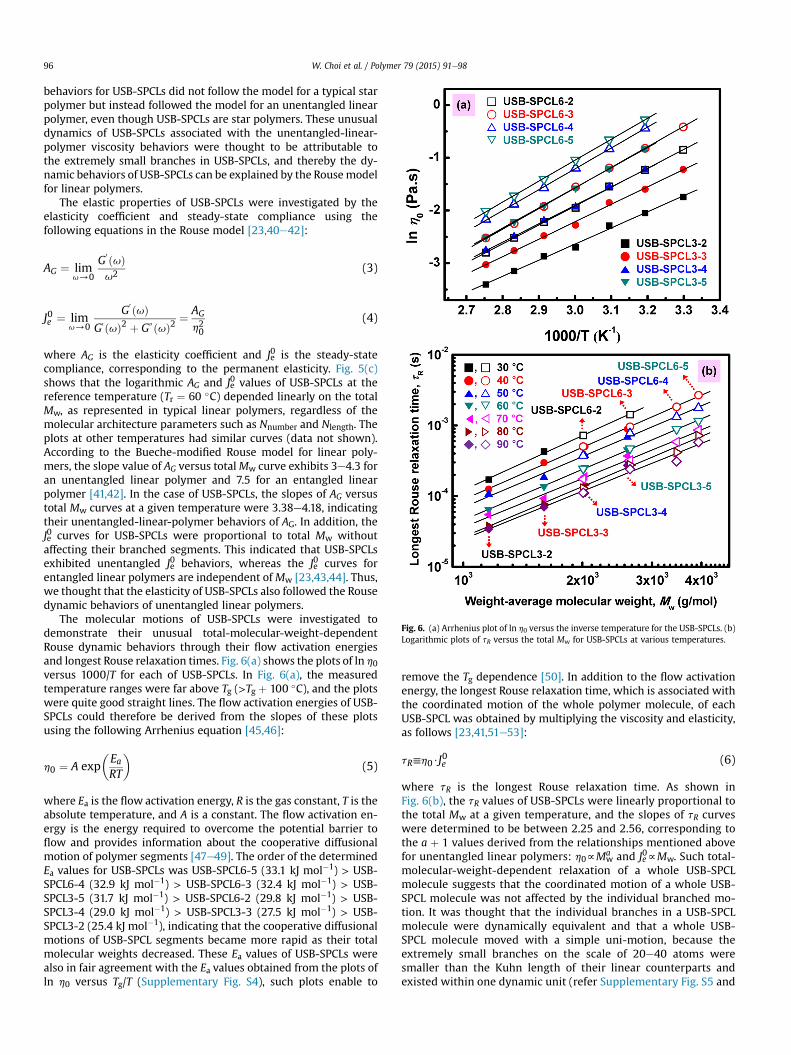
Fig. 6. (a) Arrhenius plot of ln h0 versus the inverse temperature for the USB-SPCLs. (b)Logarithmic plots of tR versus the total Mw for USB-SPCLs at various temperatures.
W. Choi et al. / Polymer 79 (2015) 91e9896
behaviors for USB-SPCLs did not follow the model for a typical starpolymer but instead followed the model for an unentangled linearpolymer, even though USB-SPCLs are star polymers. These unusualdynamics of USB-SPCLs associated with the unentangled-linear-polymer viscosity behaviors were thought to be attributable tothe extremely small branches in USB-SPCLs, and thereby the dy-namic behaviors of USB-SPCLs can be explained by the Rousemodelfor linear polymers.
The elastic properties of USB-SPCLs were investigated by theelasticity coefficient and steady-state compliance using thefollowing equations in the Rouse model [23,40e42]:
AG ¼ limu/0
G0 ðuÞu2 (3)
J0e ¼ limu/0
G0 ðuÞ
G0 ðuÞ2 þ G00 ðuÞ2¼ AG
h20
(4)
where AG is the elasticity coefficient and Je0 is the steady-state
compliance, corresponding to the permanent elasticity. Fig. 5(c)shows that the logarithmic AG and Je
0 values of USB-SPCLs at thereference temperature (Tr ¼ 60 �C) depended linearly on the totalMw, as represented in typical linear polymers, regardless of themolecular architecture parameters such as Nnumber and Nlength. Theplots at other temperatures had similar curves (data not shown).According to the Bueche-modified Rouse model for linear poly-mers, the slope value of AG versus totalMw curve exhibits 3e4.3 foran unentangled linear polymer and 7.5 for an entangled linearpolymer [41,42]. In the case of USB-SPCLs, the slopes of AG versustotal Mw curves at a given temperature were 3.38e4.18, indicatingtheir unentangled-linear-polymer behaviors of AG. In addition, theJe0 curves for USB-SPCLs were proportional to total Mw withoutaffecting their branched segments. This indicated that USB-SPCLsexhibited unentangled Je
0 behaviors, whereas the Je0 curves for
entangled linear polymers are independent ofMw [23,43,44]. Thus,we thought that the elasticity of USB-SPCLs also followed the Rousedynamic behaviors of unentangled linear polymers.
The molecular motions of USB-SPCLs were investigated todemonstrate their unusual total-molecular-weight-dependentRouse dynamic behaviors through their flow activation energiesand longest Rouse relaxation times. Fig. 6(a) shows the plots of ln h0versus 1000/T for each of USB-SPCLs. In Fig. 6(a), the measuredtemperature ranges were far above Tg (>Tg þ 100 �C), and the plotswere quite good straight lines. The flow activation energies of USB-SPCLs could therefore be derived from the slopes of these plotsusing the following Arrhenius equation [45,46]:
h0 ¼ A exp�EaRT
�(5)
where Ea is the flow activation energy, R is the gas constant, T is theabsolute temperature, and A is a constant. The flow activation en-ergy is the energy required to overcome the potential barrier toflow and provides information about the cooperative diffusionalmotion of polymer segments [47e49]. The order of the determinedEa values for USB-SPCLs was USB-SPCL6-5 (33.1 kJ mol�1) > USB-SPCL6-4 (32.9 kJ mol�1) > USB-SPCL6-3 (32.4 kJ mol�1) > USB-SPCL3-5 (31.7 kJ mol�1) > USB-SPCL6-2 (29.8 kJ mol�1) > USB-SPCL3-4 (29.0 kJ mol�1) > USB-SPCL3-3 (27.5 kJ mol�1) > USB-SPCL3-2 (25.4 kJ mol�1), indicating that the cooperative diffusionalmotions of USB-SPCL segments became more rapid as their totalmolecular weights decreased. These Ea values of USB-SPCLs werealso in fair agreement with the Ea values obtained from the plots ofln h0 versus Tg/T (Supplementary Fig. S4), such plots enable to
remove the Tg dependence [50]. In addition to the flow activationenergy, the longest Rouse relaxation time, which is associated withthe coordinated motion of the whole polymer molecule, of eachUSB-SPCL was obtained by multiplying the viscosity and elasticity,as follows [23,41,51e53]:
tR≡h0$J0e (6)
where tR is the longest Rouse relaxation time. As shown inFig. 6(b), the tR values of USB-SPCLs were linearly proportional tothe total Mw at a given temperature, and the slopes of tR curveswere determined to be between 2.25 and 2.56, corresponding tothe a þ 1 values derived from the relationships mentioned abovefor unentangled linear polymers: h0fMw
a and Je0fMw. Such total-
molecular-weight-dependent relaxation of a whole USB-SPCLmolecule suggests that the coordinated motion of a whole USB-SPCL molecule was not affected by the individual branched mo-tion. It was thought that the individual branches in a USB-SPCLmolecule were dynamically equivalent and that a whole USB-SPCL molecule moved with a simple uni-motion, because theextremely small branches on the scale of 20e40 atoms weresmaller than the Kuhn length of their linear counterparts andexisted within one dynamic unit (refer Supplementary Fig. S5 and

Fig. 7. Schematic illustration of the proposed molecular behavior for a star polymer with extremely small branches.
W. Choi et al. / Polymer 79 (2015) 91e98 97
descriptions). If several dynamic boundaries were involved in theindividual branch and the branches in a USB-SPCL molecule werenot dynamically equivalent, USB-SPCL might show the molecularmotion depending on the branching length not the total molec-ular weight, as is represented in typical star polymers [54]. Thus,we presumed that a whole USB-SPCL molecule acts as adynamically-equivalent unit, i.e., a single coarse-grain unit. In ourpresumption, the interesting dynamic phenomena of USB-SPCLcan be demonstrated by comparison with a typical star polymerwith polymeric large branched segments. In Fig. 7, the typical starpolymer has several branches, conceptually divided into multiplecoarse-grain units, each of which is composed of several mono-meric segments. The monomeric segments in a coarse-grain unitare dynamically equivalent and do not represent a valid dynamicmotion, and the coarse-grain unit displays an individual Rouse-segmental motion. Thus, the individual branched motion inthe typical star polymer is determined by the assembly of indi-vidual Rouse-segmental motions, and hence the whole molecularmotion of the typical star polymer depends on the severalbranches moving individually of each other with one central tiepoint. On the other hand, an individual branch of USB-SPCL isextremely small with only 20e40 atoms and is composed ofseveral monomeric segments without a valid dynamic unit.Instead, a whole USB-SPCL molecule is regarded as a singlecoarse-grain unit with dynamically-equivalent branches. There-fore, the whole molecular motion of USB-SPCL represents oneRouse-segmental motion of a single coarse-grain unit, resultingin the total-molecular-weight-dependent Rouse dynamics ofUSB-SPCL.
4. Conclusions
This study demonstrated the unique molecular dynamic be-haviors of USB-SPCLs used as model star polymers with extremelysmall branches. The glass transition, viscoelasticity, viscosity, elas-ticity, flow activation energy, and longest Rouse relaxation time ofUSB-SPCLs exhibited the total-molecular-weight-dependent Rousedynamic behaviors that followed unentangled linear polymermodels, instead of typical star polymer behaviors being affected bythe number and length of branched segments. This suggests that anextremely small branch in USB-SPCLs is composed of severalmonomeric segments without a valid dynamic unit and that awhole USB-SPCL molecule represents one Rouse-segmental motionof a single coarse-grain unit with dynamically-equivalent branches.We believe that the unusual results from this study provide new
insights into the molecular dynamic behaviors in an extremelysmall branched system, and this may pave the way for designingnew types of branched polymers with unique dynamic behaviors aswell as with attractive advantages of high end-group concentration,such as controlling polymer degradation, lowering softening point,and providing miscibility.
Acknowledgment
This work was supported by Research Institute of AdvancedMaterials, Department of Materials Science and Engineering inSeoul National University.
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.polymer.2015.09.009.
References
[1] G. Odian, Principles of Polymerization, fourth ed., Wiley, Hoboken, NJ, 2004.[2] M. Gaborieau, P. Castignolles, Anal. Bioanal. Chem. 399 (2011) 1413e1423.[3] H. Claesson, E. Malmstr€om, M. Johansson, A. Hult, Polymer 43 (2002)
3511e3518.[4] F. Sanda, H. Sanada, Y. Shibasaki, T. Endo, Macromolecules 35 (2002) 680e683.[5] J. Choi, I.-K. Kim, S.-Y. Kwak, Polymer 46 (2005) 9725e9735.[6] E. van Ruymbeke, E.B. Muliawan, D. Vlassopoulos, H. Gao, K. Matyjaszewski,
Eur. Polym. J. 47 (2011) 746e751.[7] L.J. Fetters, A.D. Kiss, D.S. Pearson, G.F. Quack, F.J. Vitus, Macromolecules 26
(1993) 647e654.[8] G. Kraus, J.T.J. Gruver, Polym. Sci. Part A Gen. Pap. 3 (1965) 105e122.[9] G. Quack, L.J. Fetters, Polym. Prepr. (Am. Chem. Soc. Div. Polym. Chem.) 18
(1977) 558e565.[10] O.O. Mykhaylyk, C.M. Fernyhough, M. Okura, J.P.A. Fairclough, A.J. Ryan,
R. Graham, Eur. Polym. J. 47 (2011) 447e464.[11] C. Acebo, X. Fern�andez-Francos, F. Ferrando, �A. Serra, J.M. Salla, X. Ramis,
React. Funct. Polym. 73 (2013) 431e441.[12] A.L. Frischknecht, S.T. Milner, A. Pryke, R.N. Young, R. Hawkins, T.C.B. McLeish,
Macromolecules 35 (2002) 4801e4820.[13] S.T. Milner, T.C.B. McLeish, Macromolecules 31 (1998) 7479e7482.[14] W. Choi, J.W. Chung, S.-Y. Kwak, ACS Appl. Mater. Interfaces 6 (2014)
11118e11128.[15] W. Choi, J.W. Chung, S.-Y. Kwak, J. Polym. Sci. Part A Polym. Chem. 53 (2015)
1134e1142.[16] H. Fukuzaki, M. Yoshida, M. Asano, M. Kumakura, T. Mashimo, H. Yuasa,
K. Imai, H. Yamanaka, Polymer 31 (1990) 2006e2014.[17] S.-Y. Lin, K.-S. Chen, H.-H. Teng, M.-J. Li, J. Microencapsul. 17 (2000) 577e586.[18] E. Martin, P. Dubois, R. J�erome, Macromolecules 33 (2000) 1530e1535.[19] N. Noroozi, J.A. Thomson, N. Noroozi, L.L. Schafer, S.G. Hatzikiriakos, Rheol.
Acta 51 (2012) 179e192.[20] J. Xu, W. Shi, Polymer 47 (2006) 5161e5173.[21] G. Shi, D.G. Cooper, M. Maric, Polym. Degrad. Stabil. 96 (2011) 1639e1647.[22] W. Xie, Z. Gan, Polym. Degrad. Stabil. 94 (2009) 1040e1046.

W. Choi et al. / Polymer 79 (2015) 91e9898
[23] J.D. Ferry, Viscoelastic Properties of Polymers, third ed., Wiley, New York,1980.
[24] C. Sun, J.E. Ritchie, J. Phys. Chem. 115 (2011) 8381e8389.[25] T.G. Fox, P.J. Flory, J. Appl. Phys. 21 (1950) 581e591.[26] J.E.L. Roovers, P.M. Toporowski, J. Appl. Polym. Sci. 18 (1974) 1685e1691.[27] A. Kisliuk, Y. Ding, J. Hwang, J.S. Lee, B.K. Annis, M.D. Foster, A.P. Sokolov,
J. Polym. Sci. Part B Polym. Phys. 40 (2002) 2431e2439.[28] J.A. Walberer, A.J. McHugh, J. Rheol. 45 (2001) 187e201.[29] C.M. Macosko, Rheology: Principles, Measurements and Applications, Wiley-
VCH Publishers, New York, 1994.[30] H.A. Barnes, J.F. Hutton, K. Walters, An Introduction to Rheology, Elsevier,
Amsterdam, 1989.[31] E.R. Harrell, B.F. Nakajima, J. Appl. Polym. Sci. 29 (1984) 995e1010.[32] S.-Y. Kwak, J. Choi, H.J. Song, Chem. Mater. 17 (2005) 1148e1156.[33] J.W. Chung, K.S. Oh, S.-Y. Kwak, Macromol. Mater. Eng. 292 (2007) 627e633.[34] Z. Yang, C.D. Han, Macromolecules 41 (2008) 2104e2118.[35] S. Uppuluri, S.E. Keinath, D.A. Tomalia, P.R. Dvornic, Macromolecules 31
(1998) 4498e4510.[36] R.K. Gupta, Polymer and Composite Rheology, Revised and Expanded ed.,
second ed., Marcel Dekker, Inc., New York, 2000.[37] D. Auhl, F.J. Stadler, H. Münstedt, Macromolecules 45 (2012) 2057e2065.[38] R. Koopmans, J. den Doelder, J. Molenaar, Polymer Melt Fracture, CRC Press,
Taylor & Francis Group, Boca Raton, FL, 2011.
[39] G.C. Berry, T.G. Fox, Adv. Polym. Sci. 5 (1968) 261e357.[40] W.W. Graessley, J. Roovers, Macromolecules 12 (1979) 959e965.[41] S. Onogi, T. Masuda, K. Kitagawa, Macromolecules 3 (1970) 109e116.[42] T. Masuda, K. Kitagawa, T. Inoue, S. Onogi, Macromolecules 3 (1970) 116e125.[43] P.E. Rouse, J. Chem. Phys. 21 (1953) 1272e1280.[44] F. Bueche, J. Chem. Phys. 20 (1952) 1959e1964.[45] P. Lomellini, Polymer 33 (1992) 4983e4989.[46] C.-Y. Liu, J. He, R. Keunings, C. Bailly, Macromolecules 39 (2006) 8867e8869.[47] M. Siline, A.I. Leonov, Polymer 43 (2002) 5521e5525.[48] D. Ratna, S. Divekar, A.B. Samui, B.C. Chakraborty, A.K. Banthia, Polymer 47
(2006) 4068e4074.[49] H. Shinzawa, M. Nishida, W. Kanematsu, T. Tanaka, K. Suzuki, I. Noda, Analyst
137 (2012) 1913e1921.[50] D.W. van Krevelen, P.J. Hogtyzer, Angew. Makromol. Chem. 52 (1976)
101e109.[51] M. Rubinstein, R.H. Colby, Polymer Physics, Oxford Univ. Press, New York,
2003.[52] J. Roovers, Macromolecules 24 (1991) 5895e5896.[53] J.M. Dealy, R.G. Larson, Structure and Rheology of Molten Polymers, Hanser,
Munich, 2006.[54] Liu Li, Multiscale Simulations of Star Polymer Melts (Ph.D. thesis), University
of Twente, The Netherlands, 2014.

![JEREMY ROUSE arXiv:1105.4824v1 [math.NT] 24 May 2011](https://static.fdocument.org/doc/165x107/625b5e537ffc5834cc0abdc4/jeremy-rouse-arxiv11054824v1-mathnt-24-may-2011.jpg)







![1,2 3 Vaclav Vetvicka 4,* and Vincent Ferrières 1,2, · frequency of side branches [17]. Removing those re sidues causes the polysaccharide to precipitate [18]. Finally, high molecular](https://static.fdocument.org/doc/165x107/5fc821e89fa30043ac1bf1de/12-3-vaclav-vetvicka-4-and-vincent-ferrires-12-frequency-of-side-branches.jpg)








