
THESE DE DOCTORAT Présentée par Céline GALANT NOUVEAUX ...doxa.u-pec.fr/theses/th0211123.pdf ·...
313
UNIVERSITE PARIS XII – VAL DE MARNE THESE DE DOCTORAT Pour obtenir le grade de DOCTEUR DE L’UNIVERSITE PARIS XII Spécialité : Physico-Chimie des Polymères Présentée par Céline GALANT NOUVEAUX COMPLEXES POLYELECTROLYTES IMPLIQUANT UN POLYMERE DE β-CYCLODEXTRINE, UN TENSIOACTIF CATIONIQUE ET UN POLYANION Soutenue le 19 Décembre 2003 devant le jury composé de : Mme Monique AXELOS (rapporteur) M. Christophe TRIBET (rapporteur) M. Jean-François JOANNY M. Karsten HAUPT M. Loïc AUVRAY Mme Catherine AMIEL (directeur de thèse)
Transcript of THESE DE DOCTORAT Présentée par Céline GALANT NOUVEAUX ...doxa.u-pec.fr/theses/th0211123.pdf ·...

UNIVERSITE PARIS XII – VAL DE MARNE
THESE DE DOCTORAT
Pour obtenir le grade de
DOCTEUR DE L’UNIVERSITE PARIS XII
Spécialité : Physico-Chimie des Polymères
Présentée par Céline GALANT
NOUVEAUX COMPLEXES POLYELECTROLYTES
IMPLIQUANT UN POLYMERE DE ββββ-CYCLODEXTRINE,
UN TENSIOACTIF CATIONIQUE
ET UN POLYANION
Soutenue le 19 Décembre 2003 devant le jury composé de :
Mme Monique AXELOS (rapporteur)
M. Christophe TRIBET (rapporteur)
M. Jean-François JOANNY
M. Karsten HAUPT
M. Loïc AUVRAY
Mme Catherine AMIEL (directeur de thèse)

- 2 -
TABLE DES MATIERES
INTRODUCTION GÉNÉRALE .....................................................................................................................- 6 -
PARTIE I. ETUDE BIBLIOGRAPHIQUE..................................................................................................- 10 -
CHAPITRE I. LES COMPLEXES D’INCLUSION CYCLODEXTRINE/TENSIOACTIF (CD/TA) EN SOLUTION AQUEUSE .................................................................................................................................- 10 - I- GÉNÉRALITÉS SUR LES CYCLODEXTRINES (CDS) ......................................................................- 10 -
1- PRÉSENTATION DES CDS........................................................................................................................... - 10 - 2- MÉCANISME ET PROPRIÉTÉS DU COMPLEXE D’INCLUSION ......................................................................... - 12 - 3- APPLICATIONS .......................................................................................................................................... - 19 -
II- ETUDE DES COMPLEXES CD/TA EN SOLUTION AQUEUSE.......................................................- 25 - 1- PROPRIÉTÉS DES TAS EN SOLUTION AQUEUSE .......................................................................................... - 25 - 2- MISE EN ÉVIDENCE DE LA FORMATION DES COMPLEXES CD/TA............................................................... - 28 - 3- STOECHIOMÉTRIES ET CONSTANTES D’ASSOCIATION DES COMPLEXES CD/TA ......................................... - 33 -
a) Origines de la dispersion des constantes d’association.................................. Erreur ! Signet non défini. b) Influence de la nature des composants................................................................................................- 40 - c) Influence de la température, de la force ionique et du pH ..................................................................- 42 -
III- CONCLUSION.........................................................................................................................................- 45 -
CHAPITRE II. LES COMPLEXES POLYÉLECTROLYTES HYDROSOLUBLES.............................- 47 - I- GÉNÉRALITÉS SUR LES POLYÉLECTROLYTES (PELS) ..............................................................- 47 -
1- DÉFINITION ET CLASSIFICATION................................................................................................................ - 47 - 2- PROPRIÉTÉS DES PELS EN SOLUTION AQUEUSE......................................................................................... - 50 - 3- APPLICATIONS .......................................................................................................................................... - 52 -
II- ETUDE DES COMPLEXES POLYÉLECTROLYTES (PECS) SOLUBLES DANS L’EAU ...........- 54 - 1- FORMATION ET STRUCTURE DES PECS SOLUBLES ..................................................................................... - 55 - 2- INFLUENCE DE DIVERS PARAMÈTRES SUR LA STRUCTURE DES PECS SOLUBLES ........................................ - 60 -
a) Influence de la concentration ..............................................................................................................- 60 - b) Influence du degré de conversion........................................................................................................- 64 - c) Influence du sel....................................................................................................................................- 73 - d) Influence de la densité de charges et du pH........................................................................................- 86 - e) Influence de la température .................................................................................................................- 95 - f) Influence de la nature des composants PELs .......................................................................................- 98 -
3- BILAN ..................................................................................................................................................... - 101 - III- CAS PARTICULIER: LES COMPLEXES PEL/TA DE CHARGES OPPOSÉES.........................- 102 -
1- FORMATION ET STRUCTURE DES COMPLEXES .......................................................................................... - 103 - 2- INFLUENCE DE DIVERS PARAMÈTRES....................................................................................................... - 106 -
IV- APPLICATIONS POSSIBLES DES PECS SOLUBLES ...................................................................- 118 - V- CONCLUSION ........................................................................................................................................- 122 -

- 3 -
PARTIE II. ETUDE EXPÉRIMENTALE..................................................................................................- 124 -
CHAPITRE III. ETUDE DU COMPLEXE BINAIRE POLY(CD)/TA IONIQUE EN SOLUTION DILUÉE .........................................................................................................................................................- 124 -
I- CONDITIONS EXPÉRIMENTALES.....................................................................................................- 124 - 1- LES COMPOSANTS UTILISÉS..................................................................................................................... - 124 -
a) Le polymère de CD............................................................................................................................- 124 - b) Le TA ionique ....................................................................................................................................- 127 -
2- LES TECHNIQUES EMPLOYÉES ................................................................................................................. - 128 - a) Mesures de tension de surface...........................................................................................................- 128 -
i) Principe .........................................................................................................................................- 128 - ii) Appareillage .................................................................................................................................- 129 -
b) Conductimétrie ..................................................................................................................................- 129 - i) Principe .........................................................................................................................................- 129 - ii) Appareillage .................................................................................................................................- 130 -
c) Viscosimétrie capillaire.....................................................................................................................- 130 - i) Principe .........................................................................................................................................- 130 - ii) Appareillage .................................................................................................................................- 131 -
d) Diffusion de neutrons aux petits angles (Small Angle Neutron Scattering, SANS) ...........................- 132 - i) Principe .........................................................................................................................................- 132 - ii) Appareillage .................................................................................................................................- 137 - iii) Choix des configurations d’étude ................................................................................................- 138 - iv) Traitement des données................................................................................................................- 139 -
e) Fluorimétrie.......................................................................................................................................- 139 - i) Principe .........................................................................................................................................- 139 - ii) Appareillage .................................................................................................................................- 141 -
II- MISE EN ÉVIDENCE DE LA COMPLEXATION BINAIRE POLY(CD)/TA IONIQUE ET DÉTERMINATION DE LA CONSTANTE D’ASSOCIATION..............................................................- 141 -
1- MISE EN ÉVIDENCE DE LA COMPLEXATION BINAIRE ................................................................................ - 141 - a) Par mesures de tension de surface ....................................................................................................- 141 - b) Par conductimétrie............................................................................................................................- 144 - c) Par viscosimétrie ...............................................................................................................................- 150 - d) Par SANS...........................................................................................................................................- 154 - e) Bilan ..................................................................................................................................................- 159 -
2- DÉTERMINATION DE LA CONSTANTE D’ASSOCIATION DU COMPLEXE BINAIRE ........................................ - 160 - a) Par fluorimétrie.................................................................................................................................- 160 -
i) Principe .........................................................................................................................................- 160 - ii) Résultats .......................................................................................................................................- 163 -
b) Par mesures de tension de surface ....................................................................................................- 167 - i) Principe .........................................................................................................................................- 167 - ii) Résultats .......................................................................................................................................- 169 -
c) Par conductimétrie ............................................................................................................................- 170 - i) Principe .........................................................................................................................................- 170 - ii) Résultats .......................................................................................................................................- 172 -
d) Bilan ..................................................................................................................................................- 173 - III- ETUDE DES PROPRIÉTÉS STRUCTURALES DU COMPLEXE BINAIRE POLY(CD)/TA IONIQUE.......................................................................................................................................................- 174 -
1- INFLUENCE DE LA FORCE IONIQUE........................................................................................................... - 174 - 2- INFLUENCE DE LA CONCENTRATION EN TA............................................................................................. - 180 -
IV- CONCLUSION.......................................................................................................................................- 188 -

- 4 -
CHAPITRE IV. ETUDE DU COMPLEXE TERNAIRE POLY(CD)/TA CATIONIQUE/POLYANION EN SOLUTION DILUÉE ..................................................................- 190 -
I- CONDITIONS EXPÉRIMENTALES DE L’ÉTUDE EN SOLUTION ...............................................- 190 - 1- LES NOUVEAUX COMPOSANTS UTILISÉS .................................................................................................. - 190 -
a) Les différents polyanions...................................................................................................................- 190 - i) Le polystyrène sulfonate (NaPSS) .................................................................................................- 191 - ii) Le dextrane sulfate (NaDxS).........................................................................................................- 191 - iii) L’ADN .........................................................................................................................................- 192 -
b) Les différents compétiteurs................................................................................................................- 192 - i) L’hydroxypropyl(β-cyclodextrine) (HP(β-CD)) ............................................................................- 192 - ii) Le dodécyl-β-D-maltoside (DM) ..................................................................................................- 193 -
2- UNE TECHNIQUE DE CARACTÉRISATION SUPPLÉMENTAIRE: LA DIFFUSION DYNAMIQUE DE LA LUMIÈRE (DYNAMIC LIGHT SCATTERING, DLS) ........................................................................................................ - 193 -
a) Principe .............................................................................................................................................- 193 - b) Appareillage ......................................................................................................................................- 197 -
II- MISE EN ÉVIDENCE DE LA FORMATION DE COMPLEXES TERNAIRES POLY(CD)/TA CATIONIQUE/POLYANION SOLUBLES DANS L’EAU......................................................................- 198 -
1- DÉTERMINATION DES DOMAINES DE SOLUBILITÉ .................................................................................... - 198 - 2- MISE EN ÉVIDENCE DE L’ASSOCIATION TERNAIRE PAR VISCOSIMÉTRIE ................................................... - 205 - 3- MISE EN ÉVIDENCE DE L’ASSOCIATION TERNAIRE PAR SANS................................................................. - 213 -
a) Complexes à base de NaDxS 1 ..........................................................................................................- 213 - b) Complexes à base d’ADN..................................................................................................................- 216 - c) Complexes à base de NaPSS .............................................................................................................- 218 -
4- BILAN ..................................................................................................................................................... - 221 - III- ETUDE DES PROPRIÉTÉS STRUCTURALES DES COMPLEXES TERNAIRES EN SOLUTION . - 222 -
1- INFLUENCE DE LA CONCENTRATION EN TA............................................................................................. - 222 - a) Etude par SANS.................................................................................................................................- 222 -
i) Complexes à base de NaDxS 1 ......................................................................................................- 222 - ii) Complexes à base de NaDxS 2 et d’ADN .....................................................................................- 226 - ii) Complexes à base de NaPSS ........................................................................................................- 232 -
b) Etude par DLS et par viscosimétrie...................................................................................................- 238 - i) Par DLS .........................................................................................................................................- 238 - ii) Par viscosimétrie ..........................................................................................................................- 243 -
c) Bilan ..................................................................................................................................................- 245 - 2- INFLUENCE DE LA FORCE IONIQUE........................................................................................................... - 246 -
a) Influence de la dilution......................................................................................................................- 246 - b) Influence de la concentration en sel ..................................................................................................- 247 -
3- INFLUENCE DE LA CONCENTRATION EN COMPÉTITEURS .......................................................................... - 252 - a) Influence de l’HP(β-CD)...................................................................................................................- 252 - b) Influence du docécylmaltoside (DM).................................................................................................- 255 -
IV- CONCLUSION.......................................................................................................................................- 257 -
CONCLUSION GÉNÉRALE ......................................................................................................................- 259 -
RÉFÉRENCES BIBLIOGRAPHIQUES....................................................................................................- 262 -
ANNEXE 1.....................................................................................................................................................- 280 - DÉTERMINATION PAR LA MÉTHODE FLUORIMÉTRIQUE COMPÉTITIVE DES CONSTANTES D’ASSOCIATION DE DIFFÉRENTS COMPLEXES D’INCLUSION ββββ-CD/DTAC ...........................- 280 -

- 5 -
I- DANS L’EAU PURE.................................................................................................................................- 280 - 1- CAS DE LA β-CD NATIVE ........................................................................................................................ - 280 - 2- CAS DE L’HP(β-CD) ............................................................................................................................... - 282 -
II- EN PRÉSENCE DE 0,5 M DE NACL ...................................................................................................- 284 - 1- CAS DE LA β-CD NATIVE ........................................................................................................................ - 284 - 2- CAS DE L’HP(β-CD) ............................................................................................................................... - 285 - 3- CAS DU POLY(β-CD)............................................................................................................................... - 287 -
ANNEXE 2.....................................................................................................................................................- 290 -
ETUDE PAR MESURES DE TENSION DE SURFACE DU COMPLEXE BINAIRE POLY(ββββ-CD)/DTAC EN PRÉSENCE DE SEL ....................- 290 -
ANNEXE 3.....................................................................................................................................................- 294 -
CALCUL DES DENSITÉS DE LONGUEUR DE DIFFUSION ρρρρ...........................................................- 294 -
ANNEXE 4.....................................................................................................................................................- 296 -
ETUDE PAR MESURES DE TENSION DE SURFACE DU COMPLEXE BINAIRE POLY(ββββ-CD)/DM DANS L’EAU PURE ....................- 296 -
ANNEXE 5.....................................................................................................................................................- 299 -
ETUDE PAR QCM DES INTERACTIONS POLY(ββββ-CD)/POLYANION .............................................- 299 - I- CONDITIONS EXPÉRIMENTALES.....................................................................................................- 299 -
1- LE POLYMÈRE DE CD UTILISÉ ................................................................................................................. - 299 - 2- LA QCM (QUARTZ CRYSTAL MICROBALANCE) ..................................................................................... - 300 -
a) Principe .............................................................................................................................................- 300 - b) Appareillage ......................................................................................................................................- 302 -
II- RÉSULTATS ...........................................................................................................................................- 304 - III- CONCLUSION.......................................................................................................................................- 308 -
ANNEXE 6.....................................................................................................................................................- 309 -
ETUDE VISCOSIMÉTRIQUE DES SYSTÈMES POLY(ββββ-CD)/NADXS 2 ET POLY(ββββ-CD)/ADN EN SOLUTION DANS L’EAU .....................................- 309 -
ANNEXE 7.....................................................................................................................................................- 311 -
ETUDE PAR SANS DE MÉLANGES POLY(ββββ-CD)/NADXS 1 ET POLY(ββββ-CD)/ADN EN PRÉSENCE DE SEL ............................................................- 311 -

- 6 -
Introduction générale
Les complexes polyélectrolytes (PECs) représentent une catégorie d’assemblages
macromoléculaires qui, depuis près de cinquante ans, suscitent un intérêt permanent et
croissant, tant d’un point de vue théorique que pratique. Ils sont le résultat d’une agrégation
spontanée induite par la mise en présence en solution aqueuse de deux polyélectrolytes
(PELs) de charges opposées. Cette agrégation est contrôlée principalement par de fortes
interactions coulombiennes qui peuvent conduire à la formation de complexes colloïdaux, à la
formation de coacervats ou bien à la formation de précipités ou de gels selon les conditions de
réaction et la nature des composants utilisés. Les propriétés de gélification des PECs sont par
exemple souvent exploitées dans l’industrie alimentaire. Leur tendance à précipiter est quant à
elle à la base des processus de séparation et de purification couramment employés dans le
domaine biologique (purification des protéines, par exemple). Les PECs colloïdaux, pour leur
part, servent de modèles pour mieux comprendre les phénomènes d’association entre acides
nucléiques et protéines survenant au sein des cellules. Sur le plan pratique, ces systèmes
solubles sont très souvent utilisés pour leur aptitude à faire floculer des suspensions de
particules solides, très appréciée en particulier dans le domaine du traitement des eaux
usagées. Ils montrent également un fort potentiel à la microencapsulation de molécules
insolubles dans l’eau comme certains pigments (stabilisation des peintures) ou certains
médicaments. Ils entrent alors dans la composition de nombreuses formulations
pharmaceutiques. Notons enfin que depuis quelques années, les PECs colloïdaux font l’objet
d’un vif engouement dans le domaine de la thérapie génique. De nombreux efforts sont en
effet déployés pour développer des systèmes non viraux capables d’acheminer de l’ADN
(polyanionique) au sein des cellules défaillantes.
Un autre type d’assemblages macromoléculaires a récemment fait son apparition, basé
sur le mélange en solution aqueuse d’un polymère de cyclodextrine (CD) hydrosoluble et d’un
polymère amphiphile. L’association des deux polymères est gouvernée, dans ce cas, par la
formation de complexes d’inclusion réversibles entre les cavités de CD peu polaires de l’un et
les groupements hydrophobes de l’autre. Suivant les conditions de réaction fixées, ces
interactions d’inclusion peuvent engendrer une variété de structures supramoléculaires, dont
des agrégats colloïdaux ou des gels transitoires. Jusqu’à présent, ces systèmes associatifs à
base de polymères de CD sont voués principalement à la microencapsulation de médicaments.

- 7 -
Dans ce travail de thèse, nous nous proposons d’élaborer de nouveaux assemblages
macromoléculaires combinant les deux processus d’association décrits précédemment, à
savoir par interactions coulombiennes entre charges opposées et par interactions d’inclusion.
Pour cela, nous comptons mélanger en solution aqueuse un polymère neutre de CD, un
polymère anionique et des petites molécules complexantes servant de connecteurs entre les
deux polymères. Pour jouer ce rôle de connecteur, nous avons choisi un tensioactif (TA)
cationique possédant un chaînon alkyle susceptible de s’inclure dans les cavités de CD et un
groupement chargé positivement capable d’interagir avec les charges négatives du polyanion.
Figure 1. Principe de l’association ternaire basée, d’une part, sur la formation de complexes
d’inclusion entre un polymère de CD (à gauche) et un TA cationique (au centre) et, d’autre
part, sur la formation de complexes par charges opposées entre ce TA et un polyanion (à
droite).
Appliqués à l’ADN (avec l’ADN comme polyanion), ces nouveaux assemblages
constituent des vecteurs potentiels pour la thérapie génique. Comparés aux systèmes
polymères déjà existants (PECs) dont le mode d’association repose essentiellement sur des
interactions électrostatiques entre deux PELs de charges opposées, ces assemblages à base de
polymères de CD nous semblent présenter différents avantages. Ils affichent tout d’abord une
plus grande versatilité puisque le contrôle des interactions entre les deux polymères peut
s’effectuer simplement en modulant la concentration en TA incorporée dans le mélange, sans
faire intervenir de nouvelle synthèse de polymères. Dans le cas de l’ADN, la compaction
induite par la formation des complexes (condition nécessaire pour le passage de l’ADN au
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕

- 8 -
travers des membranes cellulaires) pourra donc apparemment être contrôlée avec une plus
grande dextérité. Ensuite, ces nouveaux systèmes présentent une réversibilité accrue: ils
peuvent être dissociés ou bien, de façon classique, par addition de sel (écrantage des
interactions électrostatiques), ou bien par addition d’un compétiteur capable de dissocier les
complexes d’inclusion CD/TA. Le relargage de l’ADN dans la cellule en sera donc, à priori,
facilité.
Ce manuscrit est divisé en deux parties.
La première partie, basée sur une étude bibliographique, expose certains éléments
théoriques relatifs aux deux processus d’association mis en jeu et considérés séparément:
l’association par interactions d’inclusion impliquant les CDs, d’une part, et l’association par
interactions électrostatiques impliquant les PELs, d’autre part. Nous avons voulu donner un
aperçu des nombreux travaux consacrés aux assemblages résultants de ces associations et à
leurs applications potentielles.
Le premier chapitre a pour objet de présenter les CDs et le processus de
complexation par inclusion. Ce chapitre est consacré, en particulier, à l’étude aussi bien
qualitative que quantitative des complexes d’inclusion formés en solution aqueuse entre les
CDs et les TAs. Nous décrirons notamment comment les propriétés physico-chimiques des
solutions de TAs sont modifiées par la formation de complexes avec les CDs ou, en d’autres
termes, comment celle-ci peut-elle être détectée en solution (au moyen de quelles techniques).
Les principaux facteurs affectant le mode d’association et la constante d’équilibre des
complexes CDs/TAs seront également répertoriés.
Le deuxième chapitre rappelle quelques notions fondamentales sur les PELs et leurs
solutions. Il présente les mécanismes d’association par charges opposées, premièrement, entre
deux PELs antagonistes, deuxièmement, entre un PEL et un TA ionique. Les paramètres
déterminant la solubilité des complexes formés seront particulièrement mis en avant. Nous
attacherons surtout de l’importance à décrire les propriétés structurales des agrégats
hydrosolubles, en fonction de nombreux facteurs tels que la force ionique du milieu ou la
nature des composants. La grande complexité des systèmes et la difficulté de prévoir leur
comportement en solution seront alors mises en évidence.
La deuxième partie regroupe les résultats expérimentaux obtenus, d’une part, pour les
complexes binaires réversibles constitués d’un polymère de CD et d’un TA cationique et,
d’autre part, pour les complexes ternaires formés en rajoutant un polyanion aux systèmes
précédents, solubles dans l’eau.

- 9 -
Le troisième chapitre vise à caractériser les agrégats formés dans l’eau par
interactions d’inclusion entre un polymère de CD et un TA cationique. Nous commencerons
par une présentation des composants utilisés et des techniques expérimentales choisies pour
mener à bien cette étude. Les techniques de viscosimétrie capillaire et de diffusion de
neutrons aux petits angles (SANS) seront, entre autres, décrites. Des preuves de l’association
poly(CD)/TA seront apportées avant d’estimer quantitativement l’affinité des composants l’un
envers l’autre. Les propriétés structurales des agrégats seront ensuite examinées et discutées
en fonction de la force ionique et de la stoechiométrie des mélanges.
La mise en évidence de la formation de complexes hydrosolubles par association
ternaire poly(CD)/TA cationique/polyanion fera l’objet du quatrième et dernier chapitre.
Les différents polyanions destinés à cette étude seront présentés, ainsi qu’une technique de
caractérisation supplémentaire: la diffusion dynamique de la lumière (DLS). Un des objectifs
de ce chapitre sera de déterminer si la formation de complexes ternaires dépend uniquement
des interactions d’inclusion et électrostatiques combinées, indépendamment de la nature et de
la structure des polymères impliqués. L’influence de ces deux paramètres sur les propriétés
structurales des assemblages formés sera évaluée en fonction de la concentration en TA
incorporée dans les mélanges. La stabilité et la réversibilité des agrégats ternaires seront
également étudiées en faisant varier la force ionique du milieu et la concentration en
compétiteurs.
Nous terminerons sur un résumé des principaux résultats obtenus et sur les problèmes
restant à traiter. Les perspectives dans l’étude de ces nouveaux systèmes seront alors
exposées.

- 10 -
PARTIE I. ETUDE BIBLIOGRAPHIQUE
Chapitre I. Les complexes d’inclusion
cyclodextrine/tensioactif (CD/TA) en solution aqueuse
I- Généralités sur les cyclodextrines (CDs)
1- Présentation des CDs
Les cyclodextrines (CDs) sont le produit de la dégradation de l’amidon par une
enzyme, la CGTase (« Cyclodextrin Glycosyl Transferase »), présente dans certains micro-
organismes1. L’amidon, extrait essentiellement des céréales (blé, maïs) et de la pomme de
terre, est un polysaccharide linéaire constitué d’unités glucose associées par des liaisons α(1-
4). (Dans la conformation α, l’oxygène de la liaison α(1-x) entre les carbones C(1) et C(x) de
deux cycles glucose est placé dans des positions axiale et équatoriale respectivement par
rapport à chacun des cycles). L’enzyme découpe l’amidon en plusieurs fragments de
longueurs inégales et joint leurs deux extrémités pour former des molécules cycliques. Les
CDs résultantes comportent principalement 6, 7 ou 8 unités glucose et sont appelées,
respectivement, α-, β- et γ-CD (figure I-1), la β-CD étant le résidu formé en majorité (à 80 %
environ).
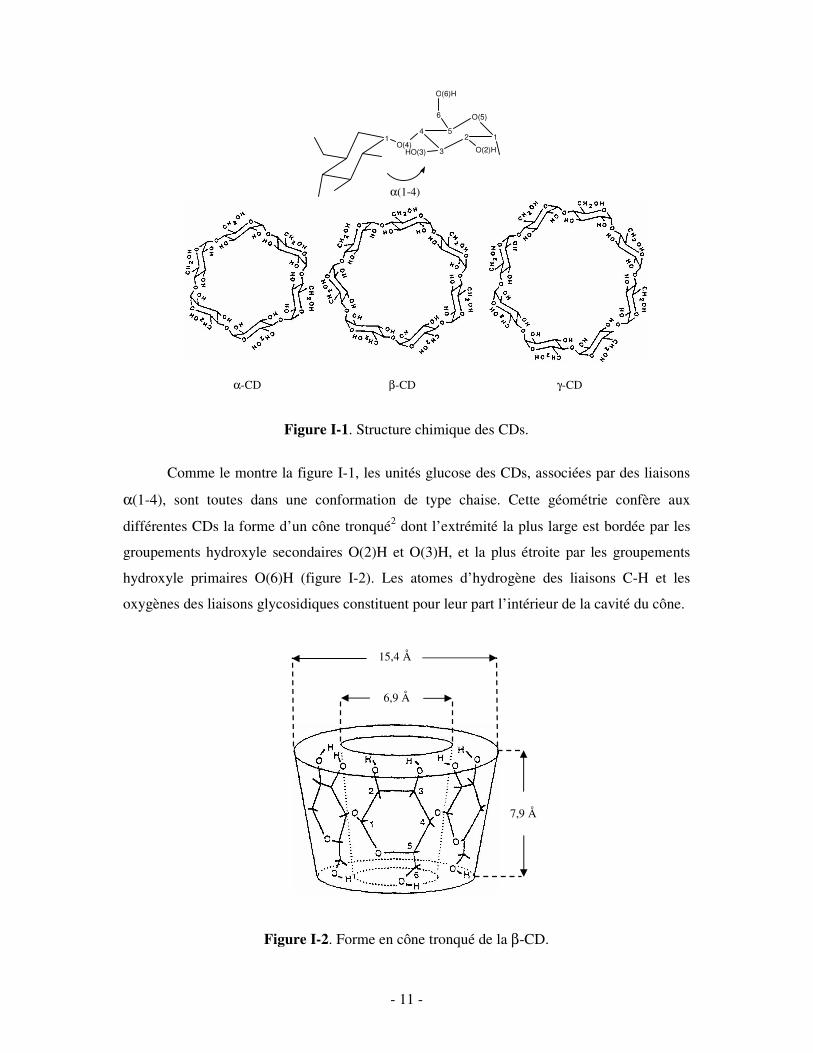
- 11 -
Figure I-1. Structure chimique des CDs.
Comme le montre la figure I-1, les unités glucose des CDs, associées par des liaisons
α(1-4), sont toutes dans une conformation de type chaise. Cette géométrie confère aux
différentes CDs la forme d’un cône tronqué2 dont l’extrémité la plus large est bordée par les
groupements hydroxyle secondaires O(2)H et O(3)H, et la plus étroite par les groupements
hydroxyle primaires O(6)H (figure I-2). Les atomes d’hydrogène des liaisons C-H et les
oxygènes des liaisons glycosidiques constituent pour leur part l’intérieur de la cavité du cône.
Figure I-2. Forme en cône tronqué de la β-CD.
4
3
2 1
O(5)
5
O(4)1
6
O(6)H
O(2)HHO(3)
α(1-4)
α-CD β-CD γ-CD
15,4 Å
6,9 Å
7,9 Å
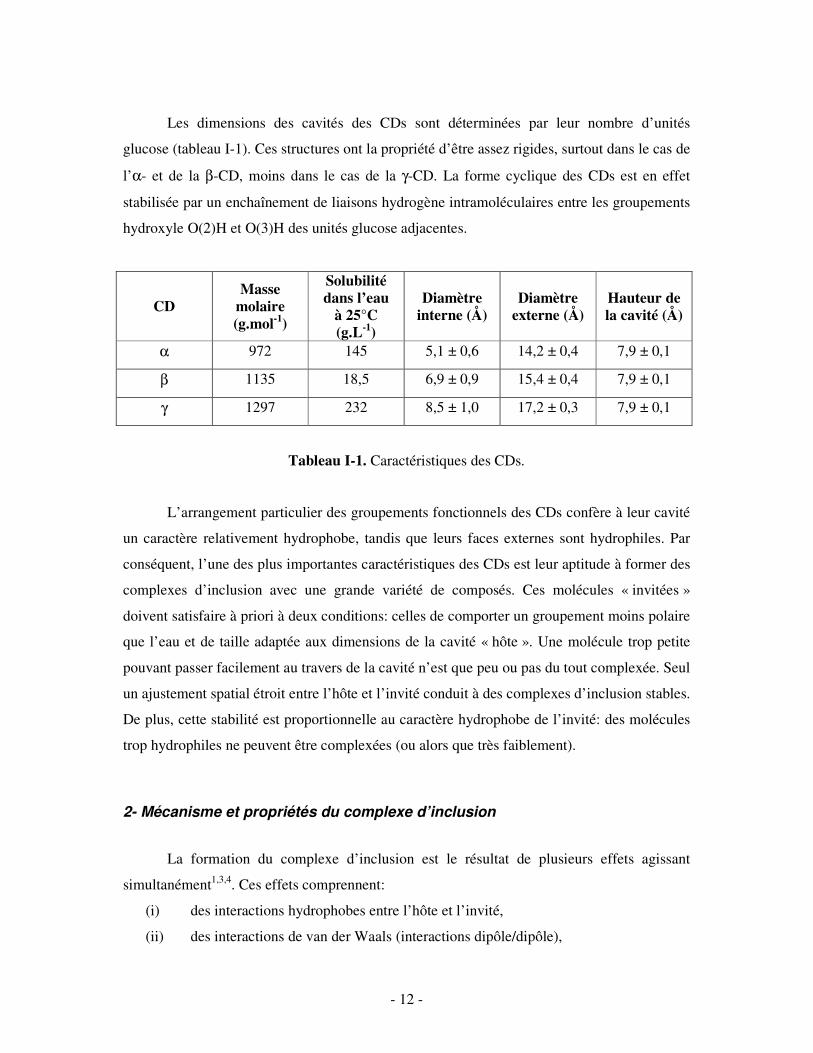
- 12 -
Les dimensions des cavités des CDs sont déterminées par leur nombre d’unités
glucose (tableau I-1). Ces structures ont la propriété d’être assez rigides, surtout dans le cas de
l’α- et de la β-CD, moins dans le cas de la γ-CD. La forme cyclique des CDs est en effet
stabilisée par un enchaînement de liaisons hydrogène intramoléculaires entre les groupements
hydroxyle O(2)H et O(3)H des unités glucose adjacentes.
CD Masse
molaire (g.mol-1)
Solubilité dans l’eau
à 25°C (g.L-1)
Diamètre interne (Å)
Diamètre externe (Å)
Hauteur de la cavité (Å)
α 972 145 5,1 ± 0,6 14,2 ± 0,4 7,9 ± 0,1
β 1135 18,5 6,9 ± 0,9 15,4 ± 0,4 7,9 ± 0,1
γ 1297 232 8,5 ± 1,0 17,2 ± 0,3 7,9 ± 0,1
Tableau I-1. Caractéristiques des CDs.
L’arrangement particulier des groupements fonctionnels des CDs confère à leur cavité
un caractère relativement hydrophobe, tandis que leurs faces externes sont hydrophiles. Par
conséquent, l’une des plus importantes caractéristiques des CDs est leur aptitude à former des
complexes d’inclusion avec une grande variété de composés. Ces molécules « invitées »
doivent satisfaire à priori à deux conditions: celles de comporter un groupement moins polaire
que l’eau et de taille adaptée aux dimensions de la cavité « hôte ». Une molécule trop petite
pouvant passer facilement au travers de la cavité n’est que peu ou pas du tout complexée. Seul
un ajustement spatial étroit entre l’hôte et l’invité conduit à des complexes d’inclusion stables.
De plus, cette stabilité est proportionnelle au caractère hydrophobe de l’invité: des molécules
trop hydrophiles ne peuvent être complexées (ou alors que très faiblement).
2- Mécanisme et propriétés du complexe d’inclusion
La formation du complexe d’inclusion est le résultat de plusieurs effets agissant
simultanément1,3,4. Ces effets comprennent:
(i) des interactions hydrophobes entre l’hôte et l’invité,
(ii) des interactions de van der Waals (interactions dipôle/dipôle),

- 13 -
(iii) des liaisons hydrogène entre les groupements hydroxyle des CDs et certaines
molécules invitées.
L’importance de la contribution de chacune de ces forces dépend de la nature de l’invité. Le
rôle primordial tenu par l’eau dans le processus d’association peut toutefois être souligné.
En solution aqueuse, les CDs renferment plusieurs molécules d’eau qui
s’accommodent tant bien que mal du caractère peu polaire et de la taille limitée de leur
environnement: leur capacité à former des liaisons hydrogène tétraédrales n’est pas satisfaite.
Les molécules d’eau incluses dans les cavités des CDs n’ont donc pas la cohésion présentée
par le reste de la solution. Elles acquièrent, de ce fait, une énergie enthalpique plus
importante. Par conséquent, l’inclusion d’un composé hydrophobe, impliquant l’expulsion de
ces molécules d’eau, est énergétiquement favorable (∆H < 0). Le système perd de l’énergie en
raison de l’augmentation des interactions solvant/solvant: le contact entre le solvant et la
cavité des CDs, d’une part, et le contact entre le solvant et la molécule invitée hydrophobe,
d’autre part, sont réduits. Un gain entropique (∆S > 0) peut, de surcroît, contribuer à la
stabilité du complexe d’inclusion. En effet, les molécules d’eau expulsées acquièrent plus de
degrés de liberté au sein de la solution que dans l’espace restreint de la cavité. Il a cependant
été démontré que la formation du complexe d’inclusion est le plus souvent gouvernée par une
variation d’entropie négative (ou légèrement positive)3. La contribution des interactions de
van der Waals, par exemple, accompagnée d’une variation négative de l’enthalpie et de
l’entropie, fait qu’il ne s’agit pas d’un processus hydrophobe classique.

- 14 -
Figure I-3. Représentation schématique de la formation d’un complexe d’inclusion.
Compte tenu du rôle essentiel joué par l’eau, le mécanisme d’inclusion peut être divisé
en plusieurs étapes5:
(i) rapprochement entre l’hôte et l’invité,
(ii) déstructuration du solvant au sein de la cavité impliquant l’expulsion de plusieurs
molécules d’eau,
(iii) déstructuration de l’eau autour de l’invité et transfert de plusieurs molécules d’eau
vers le reste de la solution,
(iv) interaction entre les substituants de l’invité et les groupements fonctionnels de la CD
en bordure ou à l’intérieur de la cavité (interactions de van der Waals),
(v) formation possible de liaisons hydrogène entre l’invité et la CD,
(vi) restructuration de l’eau autour des parties de l’invité exposées au solvant après son
inclusion.
Ce processus est très rapide4, mais la vitesse de formation du complexe (de même que
sa stabilité) dépend néanmoins de la géométrie des molécules impliquées5. Les étapes (i), (iv)
et (v) peuvent en effet être retardées par des gênes stériques. Cependant, il est important de
préciser qu’aucune liaison covalente n’est établie entre l’hôte et l’invité. La formation du
complexe d’inclusion est un processus d’équilibre dynamique qui peut être illustré par
l’équation (1), où G représente la molécule invitée et CD-G, le complexe d’inclusion:
CD
molécule d’eau
partie polaire
partie apolaire

- 15 -
CD + G CD-G (1) La stabilité du complexe est alors décrite quantitativement par la constante d’association:
GCD
GCDass NN
NK
⋅= − (M-1) (2)
où Ni désigne la concentration molaire du composé i.
Remarque: les équations (1) et (2) sont valables généralement en solution diluée, lorsque les
complexes hôte:invité sont formés en majorité avec une stoechiométrie 1:1. Des
stoechiométries 1:2, 2:1 ou 2:2 peuvent toutefois être observées à concentrations plus élevées,
ou bien avec des invités spécifiques.
Plusieurs facteurs peuvent influencer la valeur de Kass. La géométrie et la polarité de la
molécule invitée en font partie. Lorsque celle-ci est ionisable, la constante d’association peut
également dépendre du pH. Le tableau I-2 présente par exemple l’influence du pH sur la
constante d’équilibre du complexe formé entre la β-CD et le 2,6-anilinonaphtalène sulfonate
(2,6-ANS). A mesure que le pH augmente, la fonction amine du 2,6-ANS est de plus en plus
ionisée, ce qui réduit la tendance du groupement naphtalène à vouloir résider dans la cavité de
β-CD, d’où la diminution de la valeur de Kass observée.
pH 2,5 4,0 6,0 9,5 11,0
Kass (M-1) 2650 2560 2500 2270 1860
Tableau I-2. Influence du pH sur la constante d’association du système β-CD/2,6-ANS
mesurée à 25°C par fluorimétrie6.
De même, il a été montré que la constante d’association des complexes formés entre la β-CD
et l’acide benzoïque7 est plus élevée lorsque celui-ci est sous forme protonée (Kass = 1380 M-1
-O3S
N
H
2,6-ANS + β-CD

- 16 -
à pH = 10,2) que sous forme ionisée (Kass = 174 M-1 à pH = 2,2). D’une façon générale, la
plus forte hydratation des espèces chargées est défavorable à leur complexation.
La température est également déterminante pour la stabilité du complexe. En général,
Kass diminue à température plus élevée (voir le tableau I-3 pour l’exemple de la β-CD et du
2,6-ANS). En effet, comme déjà mentionné, la formation du complexe d’inclusion est bien
souvent caractérisée par une variation d’enthalpie négative. Or, l’énergie libre de la réaction
s’écrit:
∆G° = -RT ln K = ∆H° - T ∆S°,
soit ln K =
°∆−°∆TH
SR1
.
Donc si ∆H° < 0, Kass diminue lorsque T augmente.
T (°C) 5 15 25 35 45
Kass (M-1) 3010 2570 2080 1610 1260
Tableau I-3. Influence de la température sur la constante d’association du système β-CD/2,6-
ANS (pH = 7,0)6.
Il est aussi possible d’altérer les propriétés complexantes des CDs en les modifiant
chimiquement. Les atomes d’hydrogène des groupements –OH, ou bien les groupements –OH
eux-mêmes, peuvent être substitués par exemple par d’autres groupements spécifiques.
Plusieurs livres et revues sont ainsi dédiés à la synthèse et aux applications des dérivés des
CDs1,3,8-10.
Parmi ces dérivés figurent les polymères de CD qui, selon les voies de synthèse
utilisées, peuvent présenter deux types de structure différents: des structures linéaires où les
unités de CD sont localisées dans des groupements pendants par rapport à la chaîne
principale, et des structures généralement branchées dans lesquelles les CDs font partie
intégrante du squelette. Des polymères appartenant à la première catégorie ont été préparés,
par exemple, par polymérisation radicalaire de monomères monofonctionnels de CDs
acryloyle11, ou par modification chimique de polymères préexistants comme la
poly(vinylamine)12 (greffée dans ce cas par des dérivés de β-CD monotosyle) ou le

- 17 -
poly[(anhydride maléique)-alt-(isobutène)]13. Des polymères du second type ont par contre été
obtenus par polycondensation de monomères de β-CD et d’épichlorhydrine14. Comparés aux
CDs naturelles, ces polymères de CD solubles dans l’eau peuvent engendrer des complexes
d’inclusion différents en termes de solubilité et de stabilité15. D’une façon générale, les CDs
polymérisées perdent leur capacité à former des liaisons hydrogène entre elles (liaisons à
l’origine de la faible solubilité de la β-CD dans l’eau, cf. tableau I-1). Les complexes
d’inclusion générés sont par conséquent protégés d’une éventuelle cristallisation. D’un autre
côté, les substituants des CDs polymérisées peuvent induire des interférences stériques,
gênant l’inclusion des molécules invitées dans les cavités hôtes, de la même façon que pour
un monomère dérivé de CD. La stabilité des complexes résultants peut alors devenir plus
faible que dans le cas des CDs naturelles, en particulier pour des molécules invitées
comportant un unique groupement hydrophobe. Pour des invités bifonctionnels comme le 2-p-
toluidinylnaphtalène-6-sulfonate de potassium (TNS), Harada et coll.16 ont montré qu’un
polymère de CD de type poly(acryloyl-β-cyclodextrine) (poly(β-CD-A)) pouvait se montrer
par contre plus efficace que la β-CD native dans le processus de complexation. Ils ont attribué
cet effet favorable du polymère à une coopération des unités de CD le long de la chaîne après
avoir déterminé par fluorimétrie une stoechiométrie des complexes CD:TNS exclusivement
de type 2:1 (alors que les complexes impliquant de la β-CD native présentent une
stoechiométrie 1:1 à faibles concentrations en CDs, et une stoechiométrie 2:1 à plus fortes
concentrations).

- 18 -
Figure I-4. Modes d’association des complexes β-CD/TNS (a) et poly(β-CD-A)/TNS (b),
d’après Harada16.
Les auteurs ont de plus mesuré les constantes d’association des systèmes. Comme
l’indique le tableau I-4, les complexes 2:1 obtenus dans le cas du poly(β-CD-A) sont
largement plus stables que les complexes 2:1 formés avec la β-CD native. Il s’avère donc que
la dissociation des complexes 2:1 est rendue plus difficile si les deux unités de CDs
coopérantes sont reliées par une même chaîne.
CD:TNS Kass (M-1)
1:1 4000 ββββ-CD
2:1 20
poly(ββββ-CD-A) 2:1 10000
Tableau I-4. Constantes d’association des complexes β-CD/TNS et poly(β-CD-A)/TNS,
déterminées par fluorimétrie16.
H3C N
H
SO3-
CH2 CH CH CH2
CC OO
OO
H3C N
H
SO3-
H3C N
H
SO3-
1:1 à faible concentration
2:1 à forte concentration
2:1
a)
b)

- 19 -
3- Applications
Les CDs et leurs complexes d’inclusion trouvent de nombreuses applications dans les
domaines de l’industrie chimique, pharmaceutique, cosmétique et alimentaire. Ces
applications tirent avantage des diverses conséquences possibles du processus
d’ « encapsulation » de la molécule invitée au sein de la CD1:
(1) modification de l’activité chimique de la molécule invitée
(i) certaines substances réactives sont protégées par l’inclusion et peuvent ainsi
être mélangées à d’autres substances à moindre risque,
(ii) des réactions chimiques peuvent être réalisées sélectivement par inclusion
de certains groupements fonctionnels (les CDs jouent alors le rôle de
catalyseurs),
(iii) certaines réactions peuvent être favorisées ou, à l’opposé, supprimées.
(2) modification des propriétés physico-chimiques de la molécule invitée
(i) des substances peu solubles dans l’eau voient leur solubilité s’accroître par
addition de CDs,
(ii) la couleur de certaines substances peut être altérée puisque, généralement,
l’inclusion induit un changement des propriétés spectrales de l’invité,
(iii) certains goûts déplaisants peuvent être éliminés.
(3) stabilisation de substances sensibles à la lumière ou à l’oxygène
(4) fixation de substances très volatiles
(i) le stockage et le maniement de certaines substances toxiques peuvent être
améliorés,
(ii) la quantité de substance volatile peut être économisée puisque l’évaporation
est réduite,
(iii) les quantités de substances aromatiques et actives physiologiquement
peuvent être facilement quantifiées.
Dans le domaine pharmaceutique, en particulier, l’aptitude des CDs naturelles et de
leurs dérivés synthétiques à améliorer la solubilité, la stabilité et la biocompatibilité de
certains médicaments a été considérablement étudiée17-19. Une amélioration de l’activité
thérapeutique, un ciblage plus sélectif et une réduction des effets secondaires ont pu être
observés dans certains cas17.

- 20 -
De récentes études, répertoriées par Bibby et coll.20, ont montré que la présence de
CDs permet de contrôler la libération de principes actifs par des matrices polymères
(l’utilisation de polymères sous forme de gels, microsphères ou nanosphères est souvent
nécessaire pour franchir certaines barrières physiologiques ou pour réduire certaines
propriétés indésirables des médicaments employés). La formation de complexes d’inclusion
avec les CDs permet par exemple d’accroître le poids moléculaire des molécules invitées et,
par conséquent, de réduire leur diffusion au sein de la matrice.
Il existe plusieurs façons d’incorporer les CDs au sein des systèmes polymères: par
simple mélange, par interactions électrostatiques (entre un polycation et une CD anionique,
par exemple21) ou par formation de liaisons covalentes. Comme vu précédemment, les CDs
peuvent être liées chimiquement aux polymères préexistants ou bien être polymérisées entre
elles22. Des copolymères de CDs linéaires et cationiques ont ainsi été synthétisés par
Gonzalez et coll.23, dans le but de délivrer in vivo des macromolécules thérapeutiques comme
l’ADN ou certaines protéines (l’association procède alors par interactions hydrophobes et par
interactions électrostatiques). Avec l’émergence de la thérapie génique depuis quelques
années (cf. chapitre II, paragraphe IV), un vif intérêt est en effet porté aux propriétés
particulières des CDs et de leurs dérivés, en vertu de leur capacité à interagir avec les
membranes de certaines cellules et, de ce fait, à favoriser la pénétration d’oligonucléotides au
sein des cellules24. L’utilisation des CDs n’est donc pas exclusivement réservée à la
complexation de molécules de petite taille. Comme déjà évoqué, la formation de complexes
d’inclusion avec des molécules beaucoup plus larges que la cavité des CDs est également
possible. Dans ce cas, seuls certains groupes ou morceaux de chaîne de la molécule invitée
pénètrent dans la cavité.
L’association spécifique entre des CDs hôtes et des groupements invités liés de façon
covalente à des chaînes de polymères permet de créer différents types de structures
supramoléculaires. Lorsque les CDs interagissent directement avec le squelette du polymère
invité, des structures en collier de perles appelées polyrotaxanes25 (ou pseudo polyrotaxanes)
peuvent être observées. Dans cette configuration, plusieurs unités de CDs sont enfilées le long
de la chaîne invitée (cf. figures I-5 et I-6). Des complexes cristallins ont ainsi été obtenus par
Harada et coll. avec une variété de polymères neutres tels que le poly(éthylène glycol)
(PEG)26,27, le poly(propylène glycol) (PPG)28, le poly(méthyl vinyl éther) (PMVE)29 ou
certains polyesters aliphatiques30 –[O(CH2)xOCO(CH2)4CO]n- (avec x = 2, 3 ou 4) comme le
poly(éthylène adipate) (PEA, x = 2).
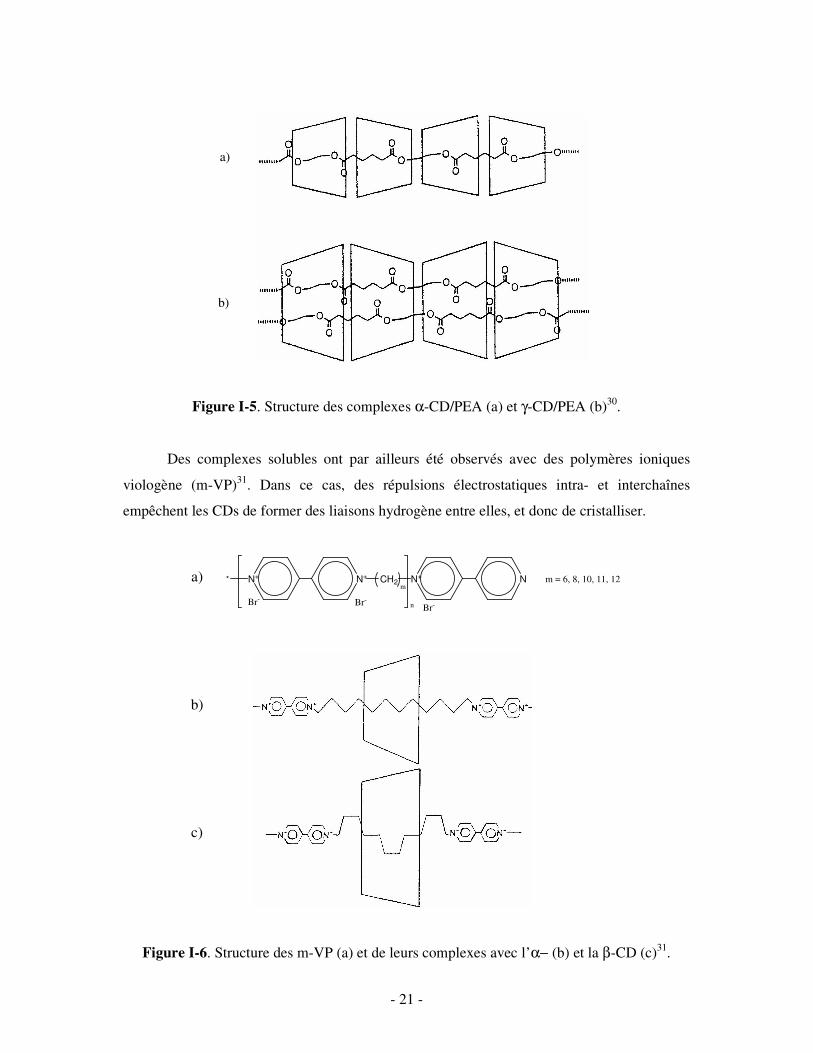
- 21 -
Figure I-5. Structure des complexes α-CD/PEA (a) et γ-CD/PEA (b)30.
Des complexes solubles ont par ailleurs été observés avec des polymères ioniques
viologène (m-VP)31. Dans ce cas, des répulsions électrostatiques intra- et interchaînes
empêchent les CDs de former des liaisons hydrogène entre elles, et donc de cristalliser.
Figure I-6. Structure des m-VP (a) et de leurs complexes avec l’α− (b) et la β-CD (c)31.
a)
b)
* N+ N+ CH2
Br-
N+ N
Br-Br-
m
n
m = 6, 8, 10, 11, 12a)
b)
c)

- 22 -
Les CDs peuvent être utilisées comme agents déstructurants lorsqu’elles interagissent,
cette fois-ci, avec les groupements secondaires du polymère invité. Dans le cas de polymères
amphiphiles (c’est-à-dire de polymères hydrosolubles comportant quelques groupements
hydrophobes capables de s’autoassocier), Zhang et coll.32 ont montré que l’ajout de CDs
permet de dissocier complètement les agrégats hydrophobes issus d’associations
intermoléculaires. La formation de complexes d’inclusion est alors accompagnée par une forte
réduction de la viscosité des systèmes.
Figure I-7. Représentation schématique des interactions entre une β-CD et un PEG modifié
par un groupement terminal fluorocarboné C6F13, d’après Zhang32.
Dans le cas particulier d’un polyélectrolyte associé à un tensioactif de charge opposée
(cf. chapitre II, paragraphe III), Tsianou et Alexandridis33 ont pour leur part démontré
l’aptitude des CDs à contrôler les propriétés rhéologiques du système. L’inclusion des
groupements hydrophobes des molécules de tensioactif dans les cavités de CDs permet, d’une
part, d’inverser l’augmentation de viscosité induite par l’association
polyélectrolyte/tensioactif et, d’autre part, de détruire le gel éventuellement formé. Les CDs
offrent donc la possibilité de dissocier des gels physiques à volonté.
+ +
PEG
C6F13

- 23 -
Figure I-8. Représentation schématique des interactions entre un polymère cationique, un
tensioactif anionique et des molécules de CD: (a) association entre le polymère et des micelles
de tensioactif donnant lieu à un enchevêtrement physique des chaînes et à un accroissement
de viscosité; (b) formation de complexes d’inclusion entre les molécules de tensioactif et les
CDs induisant une dissociation des micelles et une réduction de la viscosité.
D’autres structures supramoléculaires peuvent être obtenues en faisant interagir en
milieu aqueux un polymère de CD et un polymère amphiphile. Weickenmeier et coll.13 ont
par exemple rapporté une forte augmentation de viscosité accompagnant l’association entre
deux polymères linéaires dérivés de l’acide poly(maléique), l’un comportant des unités de β-
CD pendantes, l’autre des groupements butylaniline hydrophobes, également de type pendant.
Amiel et coll.34-39 se sont quant à eux intéressés aux interactions entre un polymère de β-CD
branché et une variété de poly(amphiphiles) neutres d’architectures différentes: des
poly(oxyde d’éthylène)s (PEO) linéaires ou de forme étoilée, modifiés par des groupements
naphtalène35 ou adamantane36 terminaux, et des polymères en peigne dérivés du
poly(diméthylacrylamide)37 ou du dextrane38. Les auteurs ont montré que la force des
interactions entre le polymère hôte et le polymère invité est principalement déterminée par le
nombre de groupements hydrophobes portés par le chaîne amphiphile (nH). Selon la force des
⊕ ⊕
⊕
a)
⊕
b)
⊕
+
⊕
⊕
⊕
+
+
enchevêtrement physique

- 24 -
interactions, deux phénomènes peuvent être observés: une agrégation en solution pouvant
donner lieu à la formation d’un réseau transitoire et une séparation de phases associative (pour
nH 3). D’après les auteurs, ce dernier phénomène peut être évité en remplaçant le
poly(amphiphile) neutre par un autre de type polyélectrolyte40. Les propriétés d’association
entre polymères hôte et invité deviennent dans ce cas dépendantes du pH (selon la nature des
groupements chargés) et de la force ionique du milieu. La charge globale de l’invité, dont
dépend la conformation des chaînes, est en effet déterminée par ces deux paramètres. Or c’est
suivant la conformation des chaînes (compacte ou étirée) que sont rendus plus ou moins
accessibles les groupements hydrophobes susceptibles de s’inclure dans les cavités de CDs.
Ces systèmes basés sur la formation de complexes d’inclusion entre un polymère de
CD et un poly(amphiphile) (neutre ou ionique) présentent donc deux sortes de propriétés:
(i) des propriétés épaississantes relatives au phénomène d’agrégation entre les deux
polymères,
(ii) des propriétés d’inclusion spécifique relatives à la présence d’unités de CDs. De
nombreuses CDs restent en effet disponibles pour complexer des molécules
invitées supplémentaires, sans que les agrégats ne soient pour autant dissociés.
La combinaison de ces propriétés fait que ces systèmes sont de bons candidats pour la
délivrance de médicaments39.
Figure I-9. Représentation schématique des associations entre les polymères de CD et les
polymères amphiphiles39 (les boules noires greffées le long des chaînes représentent les
groupements hydrophobes).

- 25 -
L’utilisation d’agents tensioactifs est une méthode alternative pour solubiliser les
composés hydrophobes, fréquemment rencontrée dans les systèmes vecteurs de
médicaments41. Cependant, les formulations pharmaceutiques comportent de plus en plus
souvent des combinaisons de CDs et de tensioactifs non ioniques. Le mélange de ces deux
types d’agents solubilisants permet en effet de réduire la concentration en chacun des
composants, et donc de rester en dessous des seuils de toxicité.
Dans ces mélanges, le cœur des micelles de tensioactif et la cavité des CDs offrent des
sites de solubilisation pour les composés hydrophobes. La capacité solubilisante de ces
mélanges peut toutefois diminuer si les tensioactifs eux-mêmes forment des complexes
d’inclusion avec les CDs, entrant dans ce cas en compétition avec les molécules invitées
hydrophobes.
Comme déjà mentionné, le rôle des molécules invitées peut être endossé par des
médicaments. Beaucoup d’entre eux sont en effet tensioactifs et, par conséquent, souvent
toxiques lorsqu’ils interagissent avec les membranes cellulaires. L’ajout de CDs dans la
formulation de tels médicaments permet de pallier au problème.
Donc, aussi bien d’un point de vue pratique que fondamental, l’étude des interactions
entre les CDs et les tensioactifs s’avère être d’un grand intérêt.
II- Etude des complexes CD/TA en solution aqueuse
1- Propriétés des TAs en solution aqueuse
Une molécule de TA est caractérisée par un groupement hydrophile qui la solubilise et
un groupement hydrophobe (le plus souvent une chaîne hydrocarbonée) qui a tendance à
fortement minimiser le contact avec les molécules d’eau42. Pour éviter ce contact avec l’eau,
le groupement hydrophobe va s’adsorber à la surface de la solution et/ou s’associer avec les
autres entités hydrophobes éventuellement présentes en solution. Lorsque tous les sites
d’adsorption sont occupés, les groupements hydrophobes des TAs en excès s’autoassocient
pour éviter le contact avec le solvant. La concentration en TA à laquelle survient cette
formation d’agrégats hydrophobes (micelles) est appelée concentration micellaire critique
(cmc).

- 26 -
En s’adsorbant à la surface de la solution, les groupements hydrophobes des TAs
prennent la place des molécules d’eau initialement présentes à l’interface solution/air. Puisque
les interactions entre les molécules d’eau et les groupements hydrophobes sont moins fortes
que les interactions entre les molécules du solvant elles-mêmes (forte énergie cohésive due
aux liaisons hydrogène), l’adsorption des TAs à l’interface s’accompagne d’une réduction
marquée de la tension de surface, γ, de la solution.
En solution diluée, les variations de γ s’expriment en fonction de la concentration en
soluté selon l’équation de Gibbs:
NdRTd lnΓ−=γ (1)
où Γ, appelé excès de surface, est le nombre de moles de soluté adsorbé par unité de surface à
l’interface solution/air et N, la concentration molaire en soluté.
Remarque: l’équation (1) est valable dans le cas des TAs neutres et pour des TAs ioniques en
présence d’un excès de sel.
Typiquement, en traçant la tension de surface d’une solution de TA en fonction du
logarithme de la concentration, une décroissance significative des valeurs est observée, suivie
d’une brisure de pente au-delà de laquelle γ reste essentiellement constante (figure I-10). Cette
brisure de pente correspond à la cmc du système: au-delà, les molécules de TA rajoutées sont
consommées pour former des micelles et la concentration en TA libre n’est que très peu
modifiée (les variations de γ sont attribuées aux variations de la concentration en TA libre).
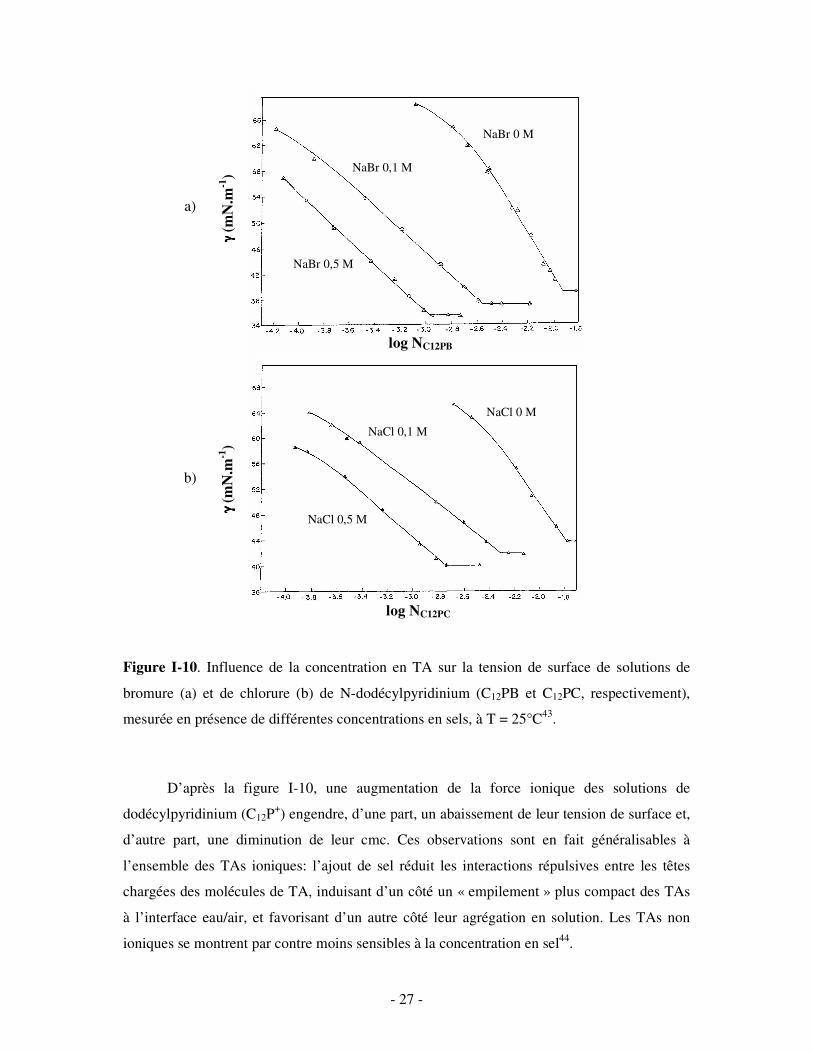
- 27 -
Figure I-10. Influence de la concentration en TA sur la tension de surface de solutions de
bromure (a) et de chlorure (b) de N-dodécylpyridinium (C12PB et C12PC, respectivement),
mesurée en présence de différentes concentrations en sels, à T = 25°C43.
D’après la figure I-10, une augmentation de la force ionique des solutions de
dodécylpyridinium (C12P+) engendre, d’une part, un abaissement de leur tension de surface et,
d’autre part, une diminution de leur cmc. Ces observations sont en fait généralisables à
l’ensemble des TAs ioniques: l’ajout de sel réduit les interactions répulsives entre les têtes
chargées des molécules de TA, induisant d’un côté un « empilement » plus compact des TAs
à l’interface eau/air, et favorisant d’un autre côté leur agrégation en solution. Les TAs non
ioniques se montrent par contre moins sensibles à la concentration en sel44.
NaBr 0 M
NaBr 0,1 M
NaBr 0,5 M
NaCl 0 M
NaCl 0,1 M
NaCl 0,5 M
γγ γγ (m
N.m
-1)
γγ γγ (m
N.m
-1)
log NC12PB
log NC12PC
a)
b)

- 28 -
En comparant les figures I-10 a) et I-10 b), il peut aussi être constaté que le C12PB est
plus tensioactif que le C12PC. Dans les mêmes conditions de force ionique, le C12PB se
montre plus efficace pour réduire la tension de surface du solvant et, de surcroît, il présente
des valeurs de cmc plus faibles. Le C12PB est donc plus enclin à former des micelles. Ceci est
cohérent avec la tendance générale selon laquelle les TAs cationiques comportant un ion
bromure comme contre-ion sont plus tensioactifs en solution aqueuse que les composés
correspondants dotés d’un ion chlorure. Le fait que les processus d’adsorption et de
micellisation (tous deux gouvernés principalement par des contributions entropiques)
impliquent une déshydratation du contre-ion est à l’origine de cette différence. Il s’avère en
effet que l’ion chlorure possède un degré d’hydratation supérieur à celui du bromure.
2- Mise en évidence de la formation des complexes CD/TA
L’incorporation d’une molécule de TA au sein d’une cavité de CD peut être
directement mise en évidence en comparant les spectres RMN 1H de la CD, en absence et en
présence du TA45: les résonances des protons H(3) et H(5) situés à l’intérieur de la cavité sont
déplacées à champ plus élevé en présence du TA, tandis que les signaux des autres protons de
la CD ne subissent aucune variation. De plus, la structure des complexes d’inclusion formés
peut être déterminée en comparant, cette fois-ci, les signaux des différents protons du TA en
absence et en présence de la CD46. Dans ce cas, il est généralement démontré que seule la
partie hydrophobe du TA est insérée à l’intérieur de la cavité.
Du fait que son groupement hydrophobe soit protégé par la CD contre tout contact
avec le milieu aqueux environnant, la molécule de TA complexée voit ses activités
thermodynamiques considérablement réduites. Son activité de surface, en particulier, est
largement affectée par la complexation.
Une remontée de la tension de surface d’une solution de TA en présence de CDs
(reflet d’une réduction de l’activité de surface du TA) est une preuve indirecte de la formation
de complexes d’inclusion46-50. La figure I-11 met ainsi en évidence la formation de complexes
d’inclusion entre la β-CD et le dodécylsulfate de sodium C12H25OSO3Na (SDS): la tension de
surface de la solution de SDS augmente avec la concentration en β-CD jusqu’à atteindre une
valeur maximale proche de la tension de surface de l’eau pure (γ = 72 mN.m-1 à 25°C) (ceci
démontre que ni la β-CD, ni les complexes d’inclusion ne présentent d’activité de surface).
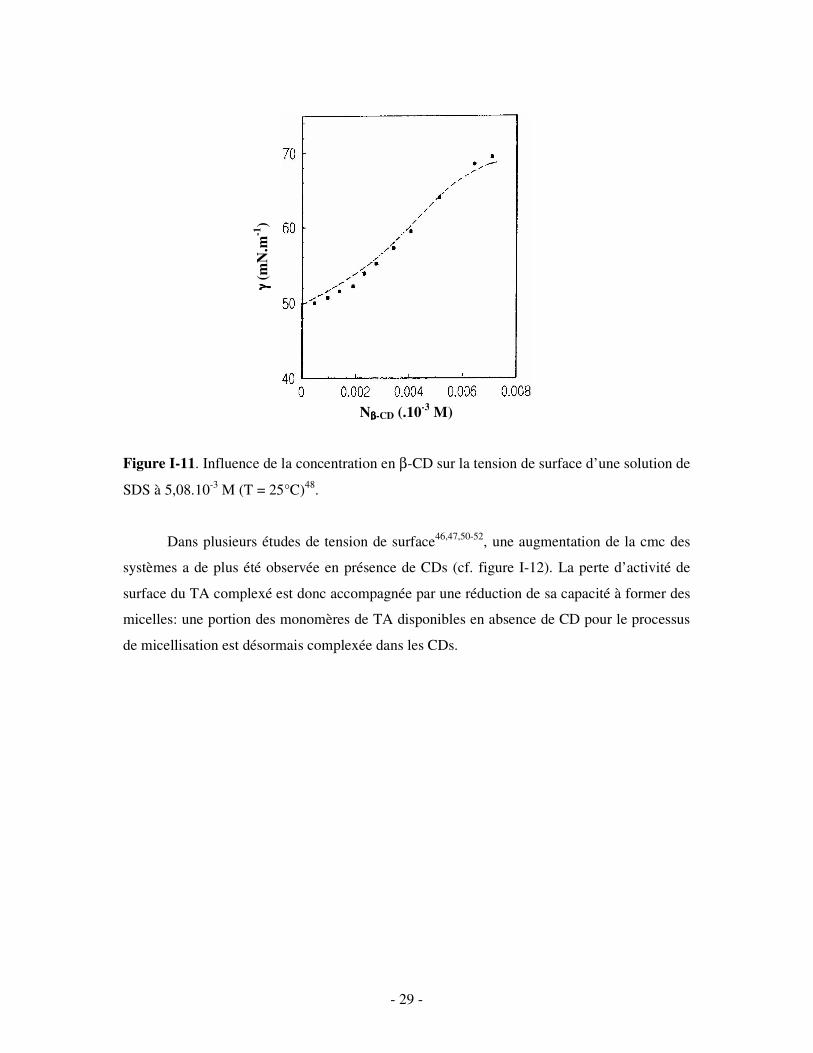
- 29 -
Figure I-11. Influence de la concentration en β-CD sur la tension de surface d’une solution de
SDS à 5,08.10-3 M (T = 25°C)48.
Dans plusieurs études de tension de surface46,47,50-52, une augmentation de la cmc des
systèmes a de plus été observée en présence de CDs (cf. figure I-12). La perte d’activité de
surface du TA complexé est donc accompagnée par une réduction de sa capacité à former des
micelles: une portion des monomères de TA disponibles en absence de CD pour le processus
de micellisation est désormais complexée dans les CDs.
Nββββ-CD (.10-3 M)
γγ γγ (m
N.m
-1)
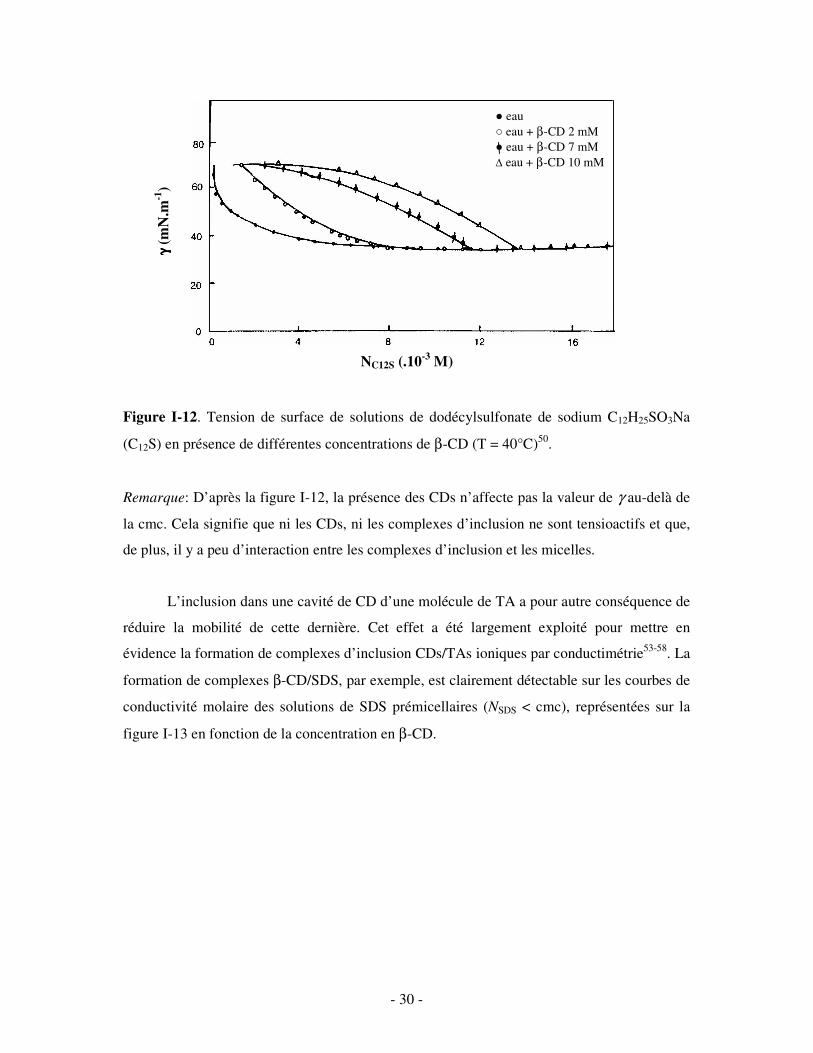
- 30 -
Figure I-12. Tension de surface de solutions de dodécylsulfonate de sodium C12H25SO3Na
(C12S) en présence de différentes concentrations de β-CD (T = 40°C)50.
Remarque: D’après la figure I-12, la présence des CDs n’affecte pas la valeur de γ au-delà de
la cmc. Cela signifie que ni les CDs, ni les complexes d’inclusion ne sont tensioactifs et que,
de plus, il y a peu d’interaction entre les complexes d’inclusion et les micelles.
L’inclusion dans une cavité de CD d’une molécule de TA a pour autre conséquence de
réduire la mobilité de cette dernière. Cet effet a été largement exploité pour mettre en
évidence la formation de complexes d’inclusion CDs/TAs ioniques par conductimétrie53-58. La
formation de complexes β-CD/SDS, par exemple, est clairement détectable sur les courbes de
conductivité molaire des solutions de SDS prémicellaires (NSDS < cmc), représentées sur la
figure I-13 en fonction de la concentration en β-CD.
NC12S (.10-3 M)
γγ γγ (m
N.m
-1)
eau eau + β-CD 2 mM eau + β-CD 7 mM eau + β-CD 10 mM
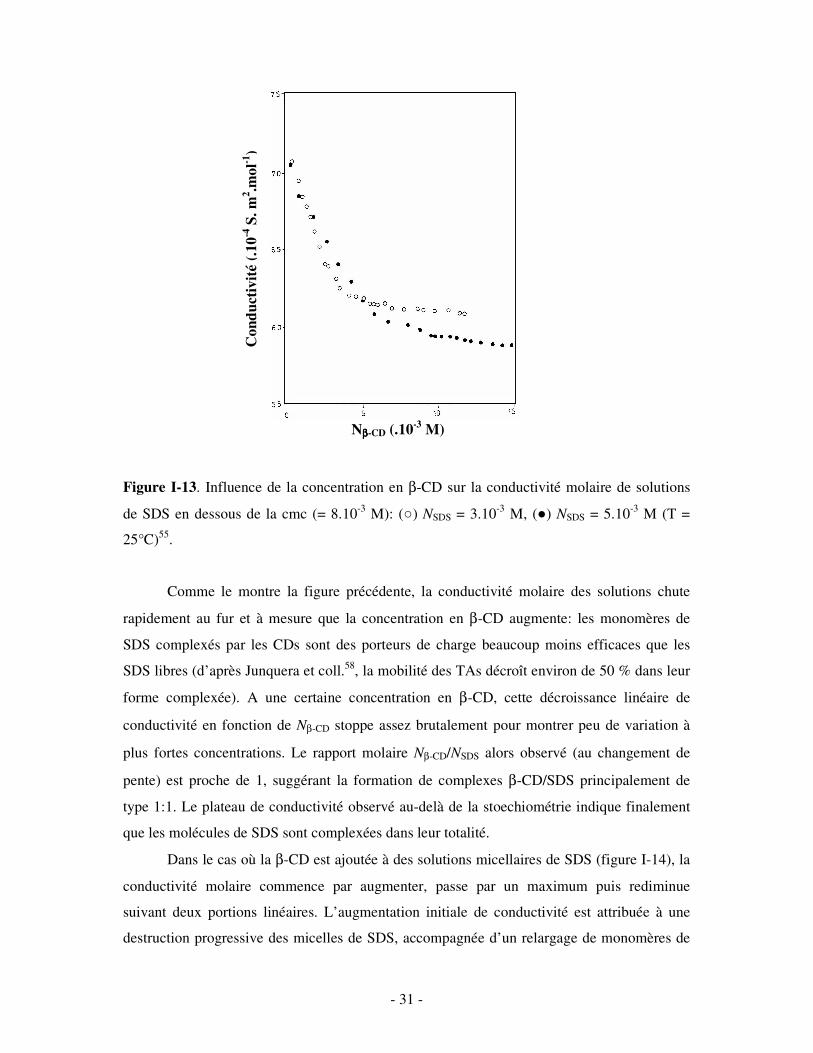
- 31 -
Figure I-13. Influence de la concentration en β-CD sur la conductivité molaire de solutions
de SDS en dessous de la cmc (= 8.10-3 M): () NSDS = 3.10-3 M, () NSDS = 5.10-3 M (T =
25°C)55.
Comme le montre la figure précédente, la conductivité molaire des solutions chute
rapidement au fur et à mesure que la concentration en β-CD augmente: les monomères de
SDS complexés par les CDs sont des porteurs de charge beaucoup moins efficaces que les
SDS libres (d’après Junquera et coll.58, la mobilité des TAs décroît environ de 50 % dans leur
forme complexée). A une certaine concentration en β-CD, cette décroissance linéaire de
conductivité en fonction de Nβ-CD stoppe assez brutalement pour montrer peu de variation à
plus fortes concentrations. Le rapport molaire Nβ-CD/NSDS alors observé (au changement de
pente) est proche de 1, suggérant la formation de complexes β-CD/SDS principalement de
type 1:1. Le plateau de conductivité observé au-delà de la stoechiométrie indique finalement
que les molécules de SDS sont complexées dans leur totalité.
Dans le cas où la β-CD est ajoutée à des solutions micellaires de SDS (figure I-14), la
conductivité molaire commence par augmenter, passe par un maximum puis rediminue
suivant deux portions linéaires. L’augmentation initiale de conductivité est attribuée à une
destruction progressive des micelles de SDS, accompagnée d’un relargage de monomères de
Con
duct
ivité
(.10
-4 S
. m2 .m
ol-1
)
Nββββ-CD (.10-3 M)
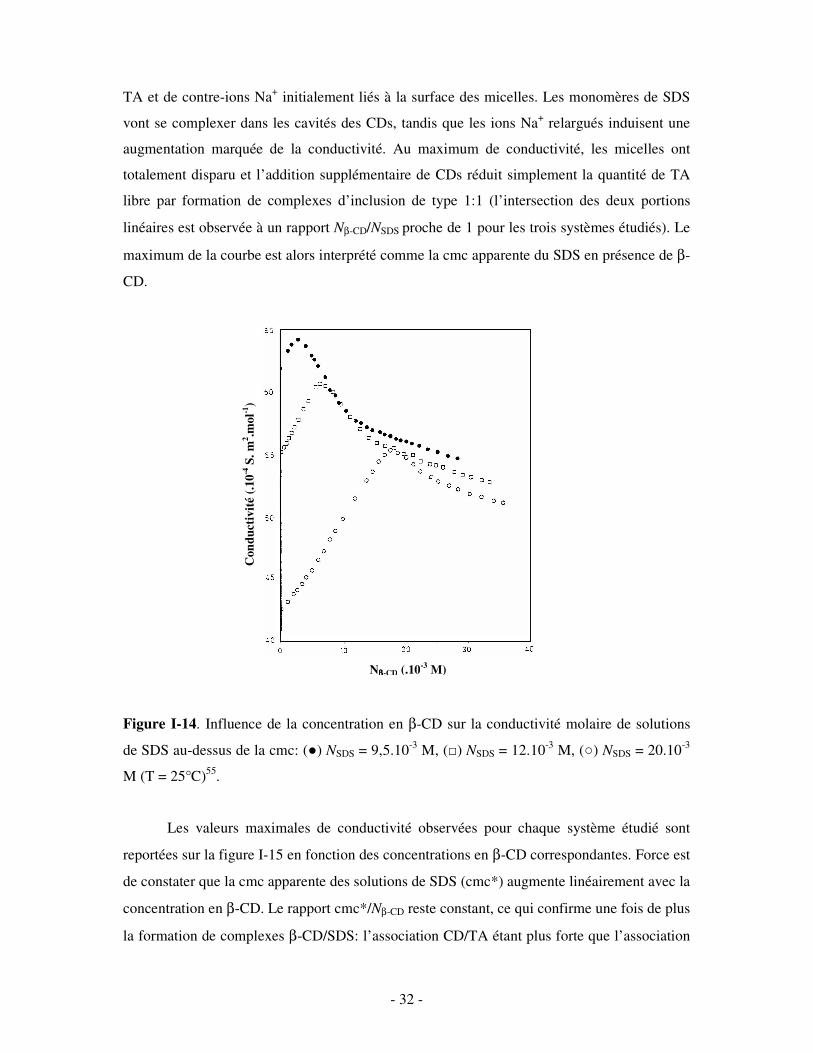
- 32 -
TA et de contre-ions Na+ initialement liés à la surface des micelles. Les monomères de SDS
vont se complexer dans les cavités des CDs, tandis que les ions Na+ relargués induisent une
augmentation marquée de la conductivité. Au maximum de conductivité, les micelles ont
totalement disparu et l’addition supplémentaire de CDs réduit simplement la quantité de TA
libre par formation de complexes d’inclusion de type 1:1 (l’intersection des deux portions
linéaires est observée à un rapport Nβ-CD/NSDS proche de 1 pour les trois systèmes étudiés). Le
maximum de la courbe est alors interprété comme la cmc apparente du SDS en présence de β-
CD.
Figure I-14. Influence de la concentration en β-CD sur la conductivité molaire de solutions
de SDS au-dessus de la cmc: () NSDS = 9,5.10-3 M, () NSDS = 12.10-3 M, () NSDS = 20.10-3
M (T = 25°C)55.
Les valeurs maximales de conductivité observées pour chaque système étudié sont
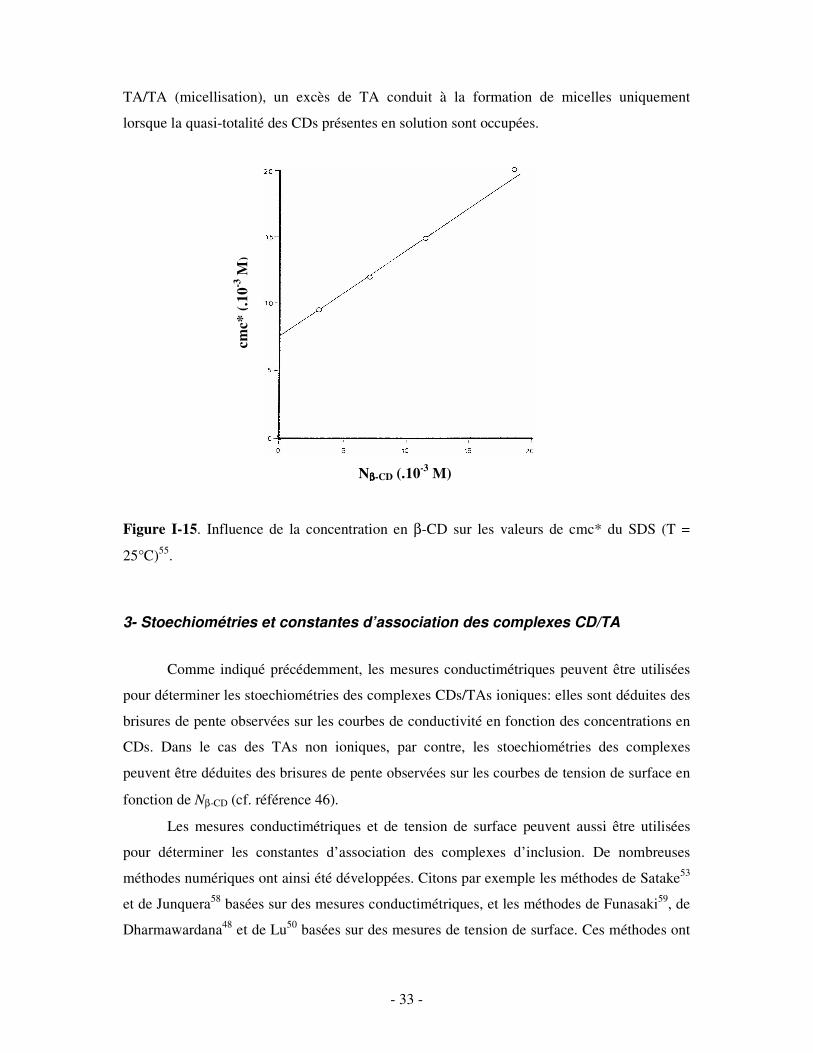
reportées sur la figure I-15 en fonction des concentrations en β-CD correspondantes. Force est
de constater que la cmc apparente des solutions de SDS (cmc*) augmente linéairement avec la
concentration en β-CD. Le rapport cmc*/Nβ-CD reste constant, ce qui confirme une fois de plus
la formation de complexes β-CD/SDS: l’association CD/TA étant plus forte que l’association
Nββββ-CD (.10-3 M)
Con
duct
ivité
(.10
-4 S
. m2 .m
ol-1
)
- 33 -
TA/TA (micellisation), un excès de TA conduit à la formation de micelles uniquement
lorsque la quasi-totalité des CDs présentes en solution sont occupées.
Figure I-15. Influence de la concentration en β-CD sur les valeurs de cmc* du SDS (T =
25°C)55.
3- Stoechiométries et constantes d’association des complexes CD/TA
Comme indiqué précédemment, les mesures conductimétriques peuvent être utilisées
pour déterminer les stoechiométries des complexes CDs/TAs ioniques: elles sont déduites des
brisures de pente observées sur les courbes de conductivité en fonction des concentrations en
CDs. Dans le cas des TAs non ioniques, par contre, les stoechiométries des complexes
peuvent être déduites des brisures de pente observées sur les courbes de tension de surface en
fonction de Nβ-CD (cf. référence 46).
Les mesures conductimétriques et de tension de surface peuvent aussi être utilisées
pour déterminer les constantes d’association des complexes d’inclusion. De nombreuses
méthodes numériques ont ainsi été développées. Citons par exemple les méthodes de Satake53
et de Junquera58 basées sur des mesures conductimétriques, et les méthodes de Funasaki59, de
Dharmawardana48 et de Lu50 basées sur des mesures de tension de surface. Ces méthodes ont
Nββββ-CD (.10-3 M)
cmc*
(.10
-3 M
)
- 34 -
principalement été appliquées à l’étude des complexes d’inclusion formés entre les CDs
naturelles (α-, β- et γ-) et les TAs ioniques de type alcanesulfonate50,53,54,57 (CnH2n+1SO3-),
alkylsulfate48,53-55,57 (CnH2n+1OSO3-), alkyltriméthylammonium48,53-56,58,60 (CnH2n+1N(CH3)3
+,
noté CnTA+) et alkylpyridinium48,53,54,61 (CnP+), ainsi que les TAs non ioniques de type
dodécylmaltoside59. Les résultats de ces études (stoechiométries et constantes d’association)
sont reportés dans le tableau I-5. Dans ce tableau figurent également des résultats obtenus au
moyen d’autres techniques: des méthodes fluorimétriques utilisant des compétiteurs
fluorescents62,63 (car ni les CDs, ni les TAs ne comportent de chromophore), des titrages
potentiométriques à l’aide d’électrodes sélectives aux TAs64-68 et des titrages calorimétriques
isothermiques67.
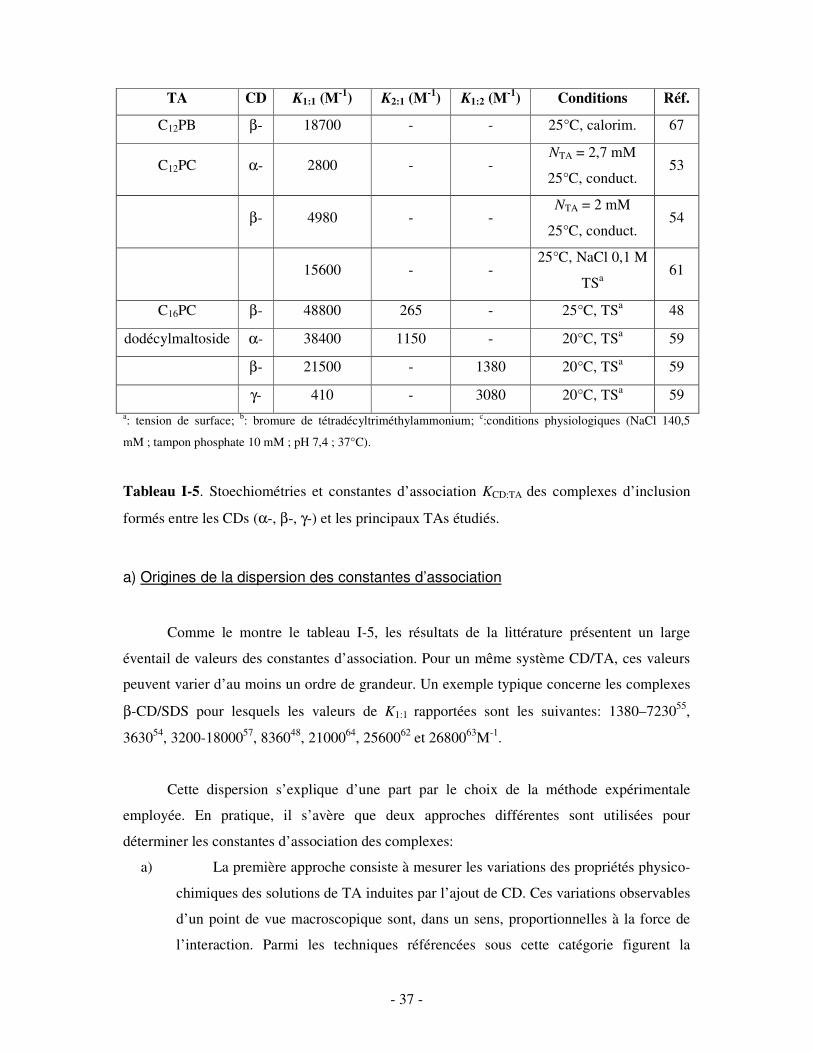
TA CD K1:1 (M-1) K2:1 (M-1) K1:2 (M-1) Conditions Réf.
CnH2n+1SO3Na
n = 6 α- 450 - - 25°C, conduct. 57
538 - - 25°C, conduct. 53
β- 140 - - 25°C, conduct. 57
163 - - 25°C, fluo. 62
177 - - 25°C, conduct. 54
n = 8 α- 1080 - - 25°C, conduct. 53
β- 1030 - - 25°C, conduct. 54
1180 - - 25°C, fluo. 62
n = 10 α- 2020 - - 25°C, conduct. 53
β- 4090 - - 25°C, conduct. 54
5360 16 - 25°C, fluo. 62
n = 12 α- 2140 - - 25°C, conduct. 53
β- 4340 - - 25°C, conduct. 54
16100 40 - 25°C, fluo. 62
15100 - - 40°C, TSa 50
14700 - - 45°C, TSa 50
11000 - - 50°C, TSa 50
n = 16 β- 44900 - - 50°C, TSa 50
27900 - - 55°C, TSa 50
- 35 -
TA CD K1:1 (M-1) K2:1 (M-1) K1:2 (M-1) Conditions Réf.
CnH2n+1OSO3Na
n = 8 α- 1400 - - 25°C, conduct. 57
β- 1610-565 - - NTA = 2,5-10 mM
25°C, conduct. 57
2560 13 - 25°C, fluo. 62
n = 10 α- 1890 - - 25°C, conduct. 57
β- 5400-2310 - - NTA = 2,5-5 mM
25°C, conduct. 57
8750 58 - 25°C, fluo. 62
n = 12 (SDS) α- 754 - - NTA = 2 mM
25°C, conduct. 55
1120 - - NTA = 2,7 mM
25°C, conduct. 53
2480 NTA = 2,5 mM
25°C, conduct. 57
β- 1380-7230 - - NTA = 5-1 mM
25°C, conduct. 55
3630 - - NTA = 2,6 mM
25°C, conduct. 54
3200-18000 - - NTA = 5-1 mM
25°C, conduct. 57
8360 - - 25°C, TSa 48
21000 210 - 25°C, potentio. 64
25600 200 - 25°C, fluo. 62
26800 440 - 25°C, fluo. 63
γ- 830 - - NTA = 2 mM
25°C, conduct. 55
TTABb α- 4500 - - NTA = 0,8 mM
25°C, conduct. 55
45000-42100 4340-2390 - NCD = 1-12 mM
25°C, potentio. 66
61000 7000 - 25°C, potentio. 67
- 36 -
TA CD K1:1 (M-1) K2:1 (M-1) K1:2 (M-1) Conditions Réf.
β- 11700-9610 - - NTA = 0,8-5 mM
25°C, conduct. 55
39750 3060 - 25°C, potentio. 67
39810 56 - 25°C, potentio. 64
49100-51300 - - NCD = 1-12 mM
25°C, potentio. 66
γ- 500-570 - 4450-6290 NCD = 3-12 mM
25°C, potentio. 66
2300 - - NTA = 0,8 mM
25°C, conduct. 55
C12TAB α- 17000 1000 - 25°C, potentio. 65
17000 - - cond. physio.c,
TSa 60
β- 4880 - - NTA = 2 mM
25°C, conduct. 54
17000 - - cond. physio.c,
TSa 60
18100 - - 25°C, potentio. 65
23700 - - 25°C, calorim. 67
γ- 110 - 1820 cond. physio.c,
TSa 60
C12TAC α- 2480 - - 25°C, conduct. 53
C16TAB α- 46510-57230 2140-2530 - NCD = 1-10 mM
NaBr 1 mM 30°C, potentio.
68
99200 20400 - 25°C, potentio. 67
β- 20000 - - NTA = 5 mM
25°C, conduct. 56
65500 398 - 25°C, TSa 48
67700 9600 - 25°C, potentio. 67
70790 126 - 25°C, potentio. 64
46350-74930 7010-5490 - NCD = 1-10 mM
NaBr 1 mM 30°C, potentio.
68
- 37 -
TA CD K1:1 (M-1) K2:1 (M-1) K1:2 (M-1) Conditions Réf.
C12PB β- 18700 - - 25°C, calorim. 67
C12PC α- 2800 - - NTA = 2,7 mM
25°C, conduct. 53
β- 4980 - - NTA = 2 mM
25°C, conduct. 54
15600 - - 25°C, NaCl 0,1 M
TSa 61
C16PC β- 48800 265 - 25°C, TSa 48
dodécylmaltoside α- 38400 1150 - 20°C, TSa 59
β- 21500 - 1380 20°C, TSa 59
γ- 410 - 3080 20°C, TSa 59 a: tension de surface; b: bromure de tétradécyltriméthylammonium; c:conditions physiologiques (NaCl 140,5
mM ; tampon phosphate 10 mM ; pH 7,4 ; 37°C).
Tableau I-5. Stoechiométries et constantes d’association KCD:TA des complexes d’inclusion
formés entre les CDs (α-, β-, γ-) et les principaux TAs étudiés.
a) Origines de la dispersion des constantes d’association
Comme le montre le tableau I-5, les résultats de la littérature présentent un large
éventail de valeurs des constantes d’association. Pour un même système CD/TA, ces valeurs
peuvent varier d’au moins un ordre de grandeur. Un exemple typique concerne les complexes
β-CD/SDS pour lesquels les valeurs de K1:1 rapportées sont les suivantes: 1380–723055,
363054, 3200-1800057, 836048, 2100064, 2560062 et 2680063M-1.
Cette dispersion s’explique d’une part par le choix de la méthode expérimentale
employée. En pratique, il s’avère que deux approches différentes sont utilisées pour
déterminer les constantes d’association des complexes:
a) La première approche consiste à mesurer les variations des propriétés physico-
chimiques des solutions de TA induites par l’ajout de CD. Ces variations observables
d’un point de vue macroscopique sont, dans un sens, proportionnelles à la force de
l’interaction. Parmi les techniques référencées sous cette catégorie figurent la

- 38 -
conductimétrie et les mesures de tension de surface. La première conduit
généralement à de plus faibles valeurs de constantes car les calculs sont fondés sur la
présomption d’une association exclusivement de type 1:1. Les valeurs de K1:1
rapportées au moyen de ces techniques pour le système β-CD/SDS varient alors entre
1380 et 8360 M-1.
b) La seconde approche est basée sur des mesures directes des concentrations en
TA libre et complexé dans une solution contenant une quantité connue de CD. Les
techniques employées appartenant à cette catégorie comprennent les mesures
potentiométriques à l’aide d’une électrode sélective au TA et les études d’association
compétitive utilisant une sonde fluorescente. Les valeurs de K1:1 obtenues par ces
deux méthodes pour les complexes β-CD/SDS varient dans ce cas entre 21000 et
26800 M-1.
D’autre part, une forte dépendance des constantes d’association vis-à-vis des
concentrations en CD62,66,67 et en TA55,57 a été rapportée, surtout pour les TAs dotés d’une
chaîne hydrocarbonée trop longue pour pouvoir être insérée entièrement dans la cavité de CD
considérée. Par exemple, pour les TAs de type alkylsulfate de sodium (CnH2n+1OSO3Na)
possédant un nombre d’atomes de carbone n supérieur ou égal à 8, Saint Aman et Serve57 ont
montré par conductimétrie que la constante d’association K1:1 des complexes formés avec la
β-CD (celle-ci pouvant accommoder au maximum 8 atomes de carbone d’une chaîne
aliphatique62) augmente plus ou moins rapidement selon le composant lorsque la
concentration totale en TA diminue (figure I-16). Pour le SDS (n = 12), en particulier, les
auteurs ont montré que K1:1 passe de 3200 à 18000 M-1 lorsque la concentration totale en SDS
est réduite de 5 à 1 mM.
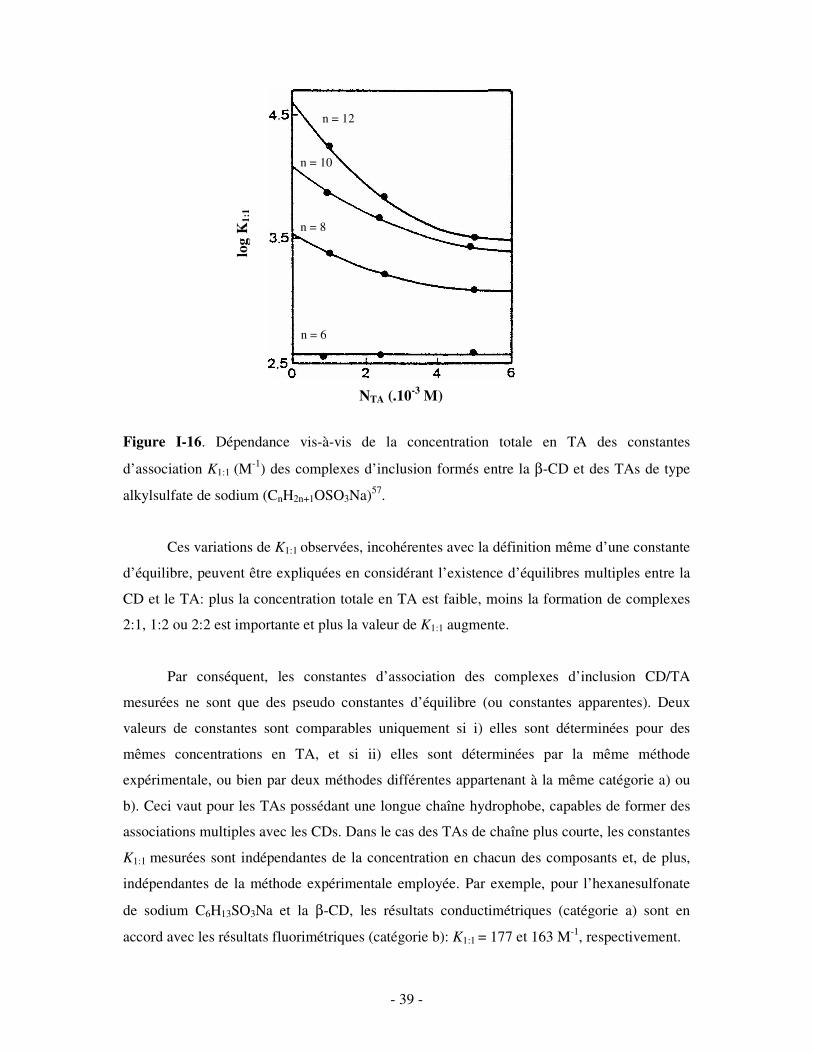
- 39 -
Figure I-16. Dépendance vis-à-vis de la concentration totale en TA des constantes
d’association K1:1 (M-1) des complexes d’inclusion formés entre la β-CD et des TAs de type
alkylsulfate de sodium (CnH2n+1OSO3Na)57.
Ces variations de K1:1 observées, incohérentes avec la définition même d’une constante
d’équilibre, peuvent être expliquées en considérant l’existence d’équilibres multiples entre la
CD et le TA: plus la concentration totale en TA est faible, moins la formation de complexes
2:1, 1:2 ou 2:2 est importante et plus la valeur de K1:1 augmente.
Par conséquent, les constantes d’association des complexes d’inclusion CD/TA
mesurées ne sont que des pseudo constantes d’équilibre (ou constantes apparentes). Deux
valeurs de constantes sont comparables uniquement si i) elles sont déterminées pour des
mêmes concentrations en TA, et si ii) elles sont déterminées par la même méthode
expérimentale, ou bien par deux méthodes différentes appartenant à la même catégorie a) ou
b). Ceci vaut pour les TAs possédant une longue chaîne hydrophobe, capables de former des
associations multiples avec les CDs. Dans le cas des TAs de chaîne plus courte, les constantes
K1:1 mesurées sont indépendantes de la concentration en chacun des composants et, de plus,
indépendantes de la méthode expérimentale employée. Par exemple, pour l’hexanesulfonate
de sodium C6H13SO3Na et la β-CD, les résultats conductimétriques (catégorie a) sont en
accord avec les résultats fluorimétriques (catégorie b): K1:1 = 177 et 163 M-1, respectivement.
n = 12
n = 10
n = 8
n = 6
log
K1:
1
NTA (.10-3 M)
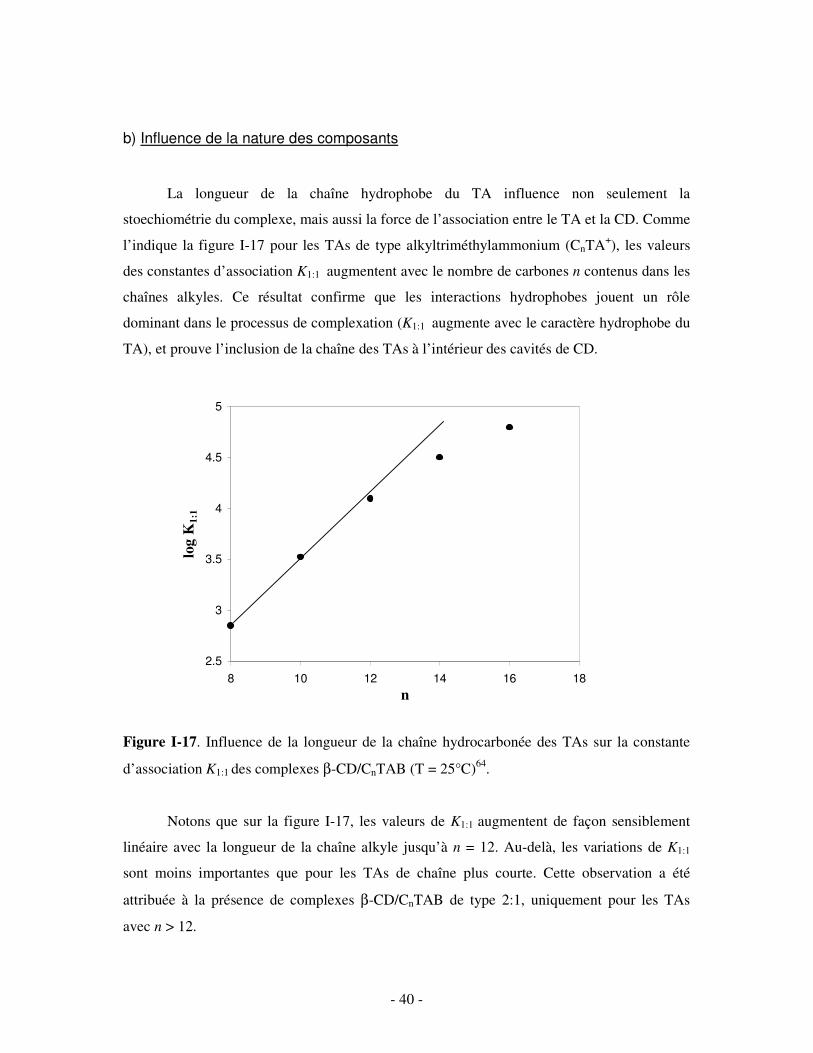
- 40 -
b) Influence de la nature des composants
La longueur de la chaîne hydrophobe du TA influence non seulement la
stoechiométrie du complexe, mais aussi la force de l’association entre le TA et la CD. Comme
l’indique la figure I-17 pour les TAs de type alkyltriméthylammonium (CnTA+), les valeurs
des constantes d’association K1:1 augmentent avec le nombre de carbones n contenus dans les
chaînes alkyles. Ce résultat confirme que les interactions hydrophobes jouent un rôle
dominant dans le processus de complexation (K1:1 augmente avec le caractère hydrophobe du
TA), et prouve l’inclusion de la chaîne des TAs à l’intérieur des cavités de CD.
2.5
3
3.5
4
4.5
5
8 10 12 14 16 18n
log
K1:
1
Figure I-17. Influence de la longueur de la chaîne hydrocarbonée des TAs sur la constante
d’association K1:1 des complexes β-CD/CnTAB (T = 25°C)64.
Notons que sur la figure I-17, les valeurs de K1:1 augmentent de façon sensiblement
linéaire avec la longueur de la chaîne alkyle jusqu’à n = 12. Au-delà, les variations de K1:1
sont moins importantes que pour les TAs de chaîne plus courte. Cette observation a été
attribuée à la présence de complexes β-CD/CnTAB de type 2:1, uniquement pour les TAs
avec n > 12.

- 41 -
Comme déjà mentionné, la β-CD peut accommoder jusqu’à 8 atomes de carbone d’une
chaîne hydrocarbonée aliphatique62. Donc, sur de simples considérations géométriques, il n’y
a aucune raison pour que le C12TAB ne forme pas de complexes 1:1 et 2:1 avec elle (le SDS
qui comporte lui aussi 12 atomes de carbone forme bien des complexes 1:1 et 2:1 avec la β-
CD62-64). En fait, il s’avère que selon la tête polaire du TA, des répulsions stériques et
dipolaires peuvent entrer en jeu, défavorisant la formation de complexes 2:1 même si la
chaîne alkyle est assez longue. Dans la configuration 1:1, la molécule de CD est positionnée
sur la chaîne hydrocarbonée du TA de façon à éviter un maximum d’interactions
défavorables. La formation du complexe 2:1 implique que la seconde molécule de CD soit
ajoutée bout à bout à la première sur la chaîne alkyle. Si les CDs ont suffisamment de liberté
de mouvement (ajustement pas trop étroit entre les CDs et la chaîne du TA), la première
molécule de CD peut alors être poussée vers la tête hydrophile chargée du TA. Si celle-ci
présente un encombrement comme c’est le cas pour le C12TAB, des répulsions dipolaires sont
créées, non seulement entre l’extrémité de la première CD et les groupements massifs de la
tête du TA, mais aussi entre les deux molécules de CDs enfilées sur la chaîne alkyle. C’est
pourquoi le C12TAB forme uniquement des complexes d’inclusion 1:1 avec la β-CD. Par
contre, il peut former des complexes 2:1 avec l’α-CD65. Le diamètre interne de la cavité de
cette dernière étant du même ordre que celui de la coupe transversale de la chaîne aliphatique
(soit de 5 Å environ, contre 6-7 Å pour la cavité de β-CD), l’ajustement produit entre l’α-CD
et le TA est suffisamment étroit pour que les forces répulsives soient réduites. Néanmoins,
bien que la formation de complexes 2:1 soit plus favorable avec l’ α-CD, la complexation 1:1
est toujours préférée. En effet, d’après le tableau I-5, la constante d’association K1:1 du
système α-CD/C12TAB reste supérieure à K2:1 d’au moins un ordre de grandeur (K1:1 = 17000
M-1 contre K2:1 = 1000 M-1, d’après une étude potentiométrique65). Par ailleurs, il est à noter,
toujours d’après le tableau I-5, que les constantes K1:1 mesurées par conductimétrie pour l’α-
CD et différents TAs possédant la même chaîne dodécyle (soit le C12S, le SDS, le C12TAC et
le C12PC) présentent des valeurs équivalentes (soit 2140, 2480, 2480 et 2800 M-1,
respectivement). Ceci montre que l’interaction entre la tête polaire du TA et la molécule d’α-
CD joue un rôle mineur dans le processus d’association, renforçant l’argument selon lequel
seule la chaîne hydrophobe des TAs est complexée au sein des cavités de CD.
Ainsi donc, la différence en diamètre interne des cavités des CDs est la principale
cause des différents modes d’association observés. Avec la plupart des TAs possédant une

- 42 -
longue chaîne alkyle, l’α-CD forme des complexes 1:1 et 2:1 (α-CD:TA), la β-CD forme
principalement des complexes 1:1 (K2:1 observée dans certains cas est plus faible que K1:1
d’au moins deux ordres de grandeur) et la γ-CD forme des complexes 1:1 et 1:2 (γ-CD:TA).
L’explication repose sur l’ajustement adéquat de la chaîne alkyle des TAs à l’intérieur des
cavités des CDs. La formation de complexes α-CD/TA de type 2:1 est favorisée parce que le
diamètre interne de la cavité d’α-CD (5 Å) et le diamètre en coupe transversale de la chaîne
alkyle (4,5 Å) sont très proches. Dans le cas de la β-CD, le diamètre interne est tel (6-7 Å)
que l’ajustement du TA au sein de sa cavité est trop lâche pour former des complexes 2:1
stables. Enfin, le diamètre interne de la γ-CD (8,5 Å) est environ deux fois plus grand que la
section de la chaîne alkyle, rendant les complexes 1:1 très instables. Par contre, la cavité de γ-
CD est capable d’inclure convenablement deux molécules de TA. Les valeurs des constantes
K2:1 sont donc croissantes dans l’ordre β-CD < α-CD mais restent néanmoins plus faibles que
K1:1 d’au moins un ordre de grandeur (voir l’exemple du TTAB dans le tableau I-5). Celles
des constantes K1:2 sont dans l’ordre β-CD < γ-CD et sont beaucoup plus élevées que K1:1 (voir
l’exemple du dodécylmaltoside). Par conséquent, pour la plupart des TAs possédant une
longue chaîne alkyle, les valeurs des constantes d’association des complexes d’inclusion 1:1
sont, pour les CDs, dans l’ordre γ-CD << α-CD β-CD.
Enfin, notons qu’une modification chimique des CDs peut influencer leur aptitude à
complexer les TAs. Par exemple, il a été démontré que l’hydroxypropylation de la β-CD
renforce son association avec le SDS69: K1:1 = 1380 M-1 avec la β-CD, contre K1:1 = 7940 M-1
avec l’ hydroxypropyl(β-CD) (HP(β-CD)), d’après des mesures conductimétriques réalisées à
25°C avec NSDS = 5 mM. Cet effet a été attribué à une forte interaction entre le groupement
ionique du TA et les groupements en bordure de la cavité d’HP(β-CD), ceci via des liaisons
hydrogène.
c) Influence de la température, de la force ionique et du pH
La plupart des constantes d’association présentées dans le tableau I-5 ont été
déterminées à 25°C. Pour quelques TAs, toutefois, les constantes d’équilibre ont été évaluées
en fonction de la température. Pour le dodécanesulfonate de sodium C12H25SO3Na, par
exemple, il peut être constaté que les valeurs de K1:1 des complexes formés avec la β-CD

- 43 -
diminuent lorsque la température augmente. L’interaction entre la β-CD et le TA est donc
affaiblie à température plus élevée. Cette observation est en fait une tendance générale qui
montre que le processus d’inclusion est une réaction exogène: l’enthalpie de complexation
∆H° est toujours négative, et ce quelque soit la nature de la CD68,69 ou celle du TA70.
La force de l’interaction CD/TA peut aussi être influencée par le pH et la force ionique
du milieu. C’est par exemple ce qu’ont pu constater Tunçay et Christian52 en étudiant par une
méthode de tension de surface la formation de complexes d’inclusion entre la β-CD et l’oxyde
de diméthyldodécylamine (DDAO), à différents pH, en absence et en présence de 0,1 M de
NaCl. Il faut préciser qu’à pH 8, les molécules de DDAO sont pour la plupart totalement
sous la forme neutre N-O, tandis qu’à pH faible (~ 2), l’atome d’oxygène est quasiment 100
% protoné. A pH intermédiaire, les formes N-O et N-OH+ sont toutes deux présentes à
l’équilibre.
Les auteurs ont tout d’abord observé que l’ajout de sel réduit de façon significative
non seulement la cmc du TA pur, mais aussi celle des systèmes mixtes β-CD/DDAO, en
particulier à pH 2 (figure I-18). A pH 8, pour lequel les molécules de DDAO sont pour la
plupart complètement neutres, l’ajout de sel a par contre peu ou pas d’influence sur les
valeurs de cmc. Par ailleurs, quelque soit le système (en absence ou en présence de β-CD), les
valeurs de cmc diminuent à mesure que le pH augmente. Ces observations ont été attribuées à
une réduction des répulsions électrostatiques par augmentation de la force ionique et/ou par
augmentation du pH, ayant pour effet de favoriser l’agrégation des molécules de TA en
solution.
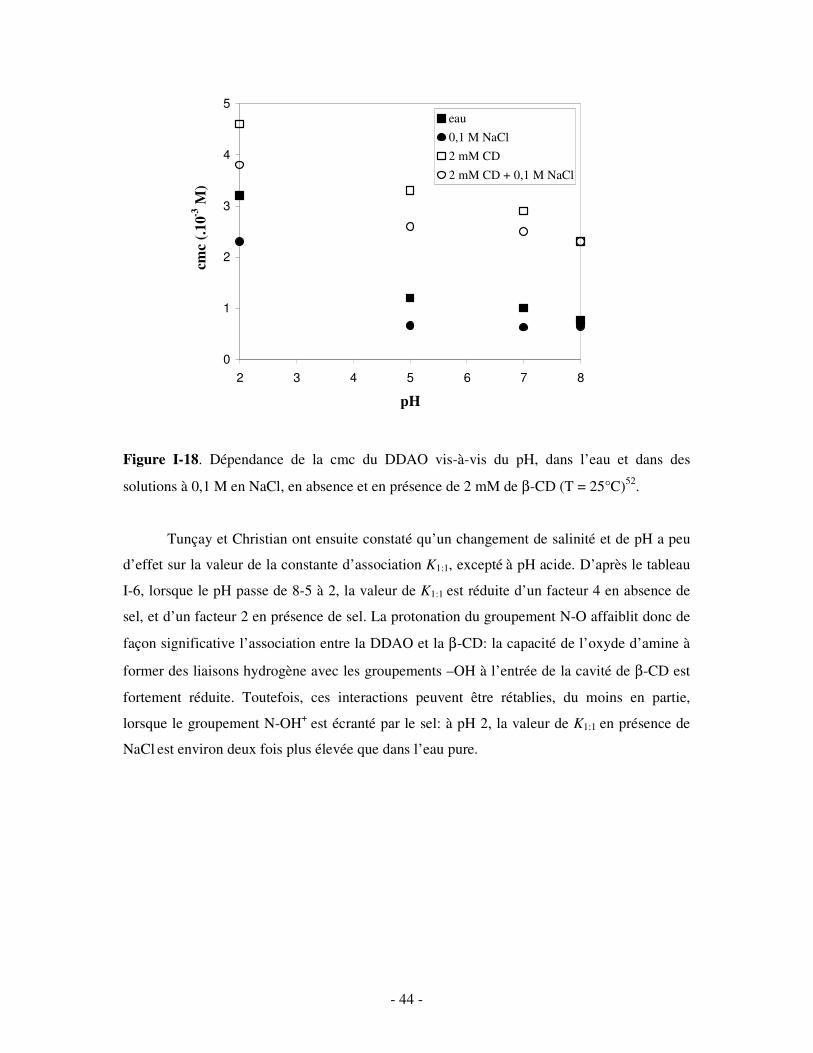
- 44 -
0
1
2
3
4
5
2 3 4 5 6 7 8
pH
cmc
(.10-3
M)
eau0,1 M NaCl2 mM CD
2 mM CD + 0,1 M NaCl
Figure I-18. Dépendance de la cmc du DDAO vis-à-vis du pH, dans l’eau et dans des
solutions à 0,1 M en NaCl, en absence et en présence de 2 mM de β-CD (T = 25°C)52.
Tunçay et Christian ont ensuite constaté qu’un changement de salinité et de pH a peu
d’effet sur la valeur de la constante d’association K1:1, excepté à pH acide. D’après le tableau
I-6, lorsque le pH passe de 8-5 à 2, la valeur de K1:1 est réduite d’un facteur 4 en absence de
sel, et d’un facteur 2 en présence de sel. La protonation du groupement N-O affaiblit donc de
façon significative l’association entre la DDAO et la β-CD: la capacité de l’oxyde d’amine à
former des liaisons hydrogène avec les groupements –OH à l’entrée de la cavité de β-CD est
fortement réduite. Toutefois, ces interactions peuvent être rétablies, du moins en partie,
lorsque le groupement N-OH+ est écranté par le sel: à pH 2, la valeur de K1:1 en présence de
NaCl est environ deux fois plus élevée que dans l’eau pure.
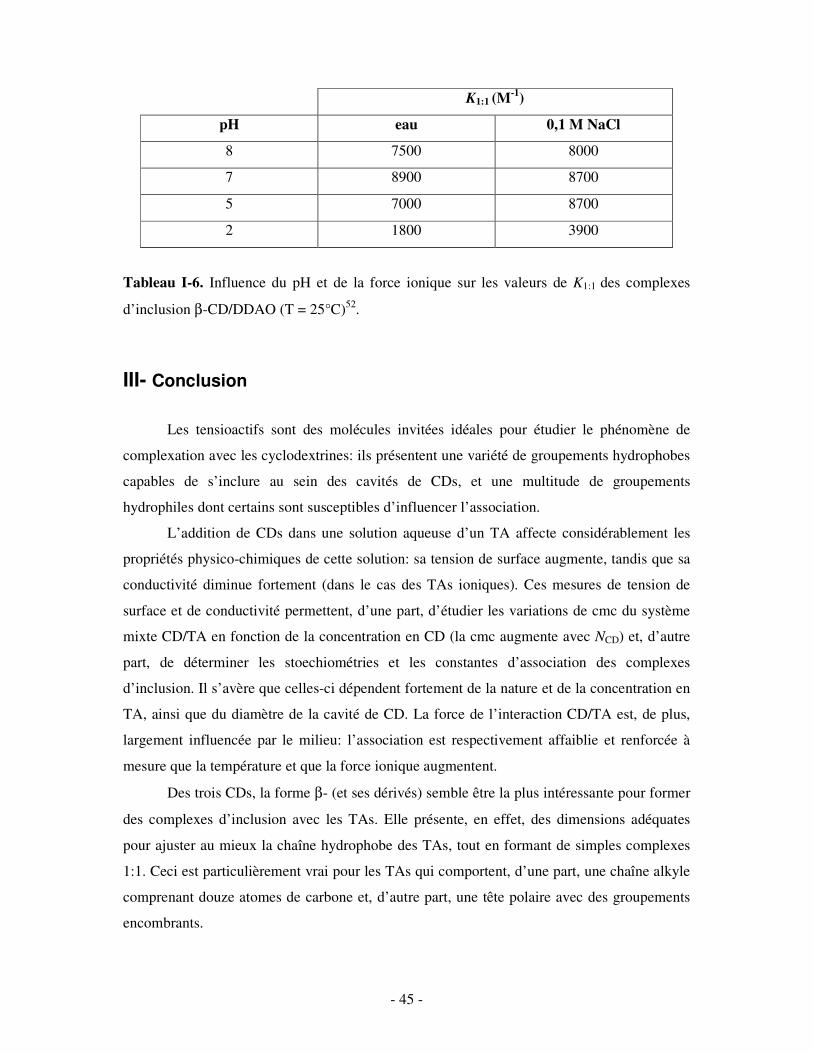
- 45 -
K1:1 (M-1)
pH eau 0,1 M NaCl
8 7500 8000
7 8900 8700
5 7000 8700
2 1800 3900
Tableau I-6. Influence du pH et de la force ionique sur les valeurs de K1:1 des complexes
d’inclusion β-CD/DDAO (T = 25°C)52.
III- Conclusion
Les tensioactifs sont des molécules invitées idéales pour étudier le phénomène de
complexation avec les cyclodextrines: ils présentent une variété de groupements hydrophobes
capables de s’inclure au sein des cavités de CDs, et une multitude de groupements
hydrophiles dont certains sont susceptibles d’influencer l’association.
L’addition de CDs dans une solution aqueuse d’un TA affecte considérablement les
propriétés physico-chimiques de cette solution: sa tension de surface augmente, tandis que sa
conductivité diminue fortement (dans le cas des TAs ioniques). Ces mesures de tension de
surface et de conductivité permettent, d’une part, d’étudier les variations de cmc du système
mixte CD/TA en fonction de la concentration en CD (la cmc augmente avec NCD) et, d’autre
part, de déterminer les stoechiométries et les constantes d’association des complexes
d’inclusion. Il s’avère que celles-ci dépendent fortement de la nature et de la concentration en
TA, ainsi que du diamètre de la cavité de CD. La force de l’interaction CD/TA est, de plus,
largement influencée par le milieu: l’association est respectivement affaiblie et renforcée à
mesure que la température et que la force ionique augmentent.
Des trois CDs, la forme β- (et ses dérivés) semble être la plus intéressante pour former
des complexes d’inclusion avec les TAs. Elle présente, en effet, des dimensions adéquates
pour ajuster au mieux la chaîne hydrophobe des TAs, tout en formant de simples complexes
1:1. Ceci est particulièrement vrai pour les TAs qui comportent, d’une part, une chaîne alkyle
comprenant douze atomes de carbone et, d’autre part, une tête polaire avec des groupements
encombrants.

- 46 -
A notre connaissance, aucune publication ne porte, à ce jour, sur l’association pouvant
exister entre un TA ionique et un polymère de CD. C’est à l’étude d’un tel système que sera
dédié le chapitre III de ce mémoire. Dans ce chapitre, il sera question de mettre en évidence la
formation de complexes d’inclusion entre un polymère de β-CD et un TA cationique de type
dodécyltriméthylammonium, ces composants ayant été choisis par souci de simplification:
seule la formation de complexes 1:1 est attendue dans ce cas. Une étude de l’influence de la
force ionique et de la concentration en TA sera également envisagée. Dans un dernier
chapitre, enfin, nous estimerons l’aptitude d’un tel système à former des structures
supramoléculaires, en mélangeant en solution aqueuse le polymère de β-CD, le C12TA+ et
différents polyanions.

- 47 -
Chapitre II. Les complexes polyélectrolytes hydrosolubles
I- Généralités sur les polyélectrolytes (PELs)
1- Définition et classification
Le terme « polyélectrolyte » est employé pour tout système polymère comportant des
groupements ionisables71. En solution aqueuse, un PEL se dissocie d’une part en
« macroion », c’est-à-dire en une macromolécule constituée d’un enchaînement de
groupements ioniques liés de façon covalente, et d’autre part, en contre-ions assurant
l’électroneutralité de l’ensemble. Du fait de leur nature électrostatique, les PELs font preuve
d’une grande solubilité dans l’eau et peuvent être classés en « polycation », « polyanion » ou
« polyampholyte » selon que les groupements du macroion sont respectivement tous chargés
positivement, tous chargés négativement ou bien les deux. De plus, une distinction est faite
entre PELS « faibles » et PELS « forts » : les PELS dits « faibles » forment un ensemble
macroion/contre-ions uniquement dans un domaine limité de pH tandis que la dissociation des
PELS dits « forts » est indépendante du pH de la solution. Par exemple, le poly(styrène)
sulfonate de sodium (polyanion fort) et le chlorure de poly(diallyldiméthylammonium)
(polycation fort) dont les formules chimiques sont représentées dans le tableau II-1, sont
dissociés en macroions et contre-ions en solution aqueuse quelque soit le pH compris entre 0
et 14. Par contre, l’acide poly(acrylique) (polyacide faible) et la poly(éthylènimine) (polybase
faible) restent non dissociés respectivement dans les domaines acide et alcalin72. La
dissociation de ces PELS faibles est présentée en figure II-1.
* CH2 CH *
COOH
* CH2 CH *
COO-
+ H+a)pH > 7
* CH2 CH2 NH * CH2 CH2b)pH < 7
+ H2O NH2+ + OH-
Figure II-1. Dissociation de l’acide poly(acrylique) (a) et de la poly(éthylènimine) (b).
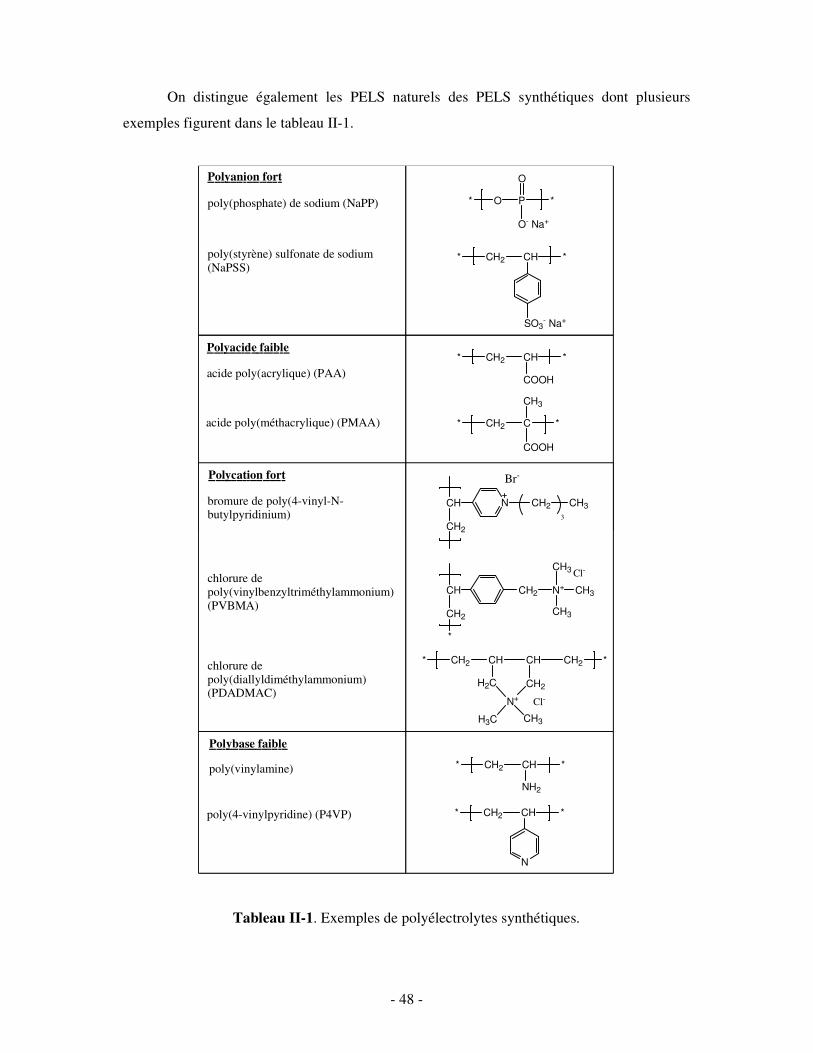
- 48 -
On distingue également les PELS naturels des PELS synthétiques dont plusieurs
exemples figurent dans le tableau II-1.
Polyanion fort
poly(styrène) sulfonate de sodium (NaPSS)
poly(phosphate) de sodium (NaPP)
* CH2 CH *
SO3- Na+
* O P
O
O- Na+
*
Polyacide faible
acide poly(acrylique) (PAA)
acide poly(méthacrylique) (PMAA)
* CH2 CH *
COOH
* CH2 C *
CH3
COOH
Polycation fort
bromure de poly(4-vinyl-N-butylpyridinium)
chlorure de poly(vinylbenzyltriméthylammonium) (PVBMA)
chlorure de poly(diallyldiméthylammonium) (PDADMAC)
CH
CH2
N CH2 CH33
Br-
CH
CH2
*
CH2 N+
CH3
CH3
CH3
* CH2 CH CH CH2 *
H2C CH2
N+
CH3H3C
Cl-
Cl-
Polybase faible
poly(vinylamine)
poly(4-vinylpyridine) (P4VP)
* CH2 CH *
NH2
* CH2 CH *
N
Tableau II-1. Exemples de polyélectrolytes synthétiques.

- 49 -
Les synthèses de certains de ces polymères tels que le chlorure de
poly(diallyldiméthylammonium) (PDADMAC) ou la poly(éthylènimine) (PEI) sont
présentées dans la revue de Mortimer73 consacrée aux PELs synthétiques et à leurs
applications industrielles. Les PELs naturels sont quant à eux essentiellement représentés par
les protéines et les acides nucléiques qui, d’une façon générale, sont polyampholytes: la
charge globale portée par le macroion dépend du pH de la solution. La gélatine74, par
exemple, qui est globalement neutre à pH 4,9 (pH isoélectrique), est respectivement positive
ou négative à pH inférieur ou supérieur à cette valeur. La catégorie des PELs naturels
regroupe également de nombreux PELs synthétisés à partir de biopolymères neutres.
Dautzenberg72 consacre ainsi tout un chapitre à la synthèse des PELs dits « naturels », dont
beaucoup dérivent de polysaccharides tels la cellulose, l’amidon ou le dextrane. Comme
l’illustre la figure II-2, ces polysaccharides sont constitués d’unités hexose ou pentose (5 ou 6
atomes de carbone) enchaînées par des liaisons glucosidiques entre l’atome C1 d’un
monomère et l’atome C2, 3, 4 ou 6 du monomère voisin. Dans l’exemple de la cellulose
donné en figure II-2, il s’agit de liaisons de type β-(1-4). Dans cette conformation β,
l’oxygène de la liaison entre les carbones C1 et C4 de deux cycles glucose est placé en
position équatoriale par rapport aux deux cycles, ce qui confère une grande rigidité à la
cellulose et à ses dérivés.
OHOH2C
O*
HOOH
OO
HOOH
CH2OH
*
n
4
1 4
1
β−(1−4)
Figure II-2. Structure chimique de la cellulose.
Enfin, on distingue les PELS linéaires des PELS de structure branchée, ainsi que les
PELs de type « intégral » (c’est-à-dire dont les charges sont intégrées au squelette) des PELs
de type « pendant » (dont les charges sont portées par des groupements ou des chaînons
pendants). La figure II-3 présente par exemple la structure branchée de la PEI qui appartient à
la catégorie des PELs de type « intégral ». Comme exemples de PELs de type « pendant », on
peut entre autres citer le NaPSS et la poly(vinylamine) qui figurent dans le tableau II-1.
- 50 -
*NH2
+NH+
+H3N
NH2+
+HNNH2
+*
NH2+
+H3N
8 Cl-
Figure II-3. Structure branchée de l’hydrochlorure de poly(éthylènimine).
2- Propriétés des PELs en solution aqueuse
La présence de groupements chargés confère aux chaînes PELs des propriétés uniques
en solution. Alors qu’en milieu dilué, les polymères neutres adoptent une conformation en
« pelote statistique » (figure II-4 a), les PELs sont quant à eux soumis à des répulsions intra-
(et aussi inter-) moléculaires qui résultent en des conformations étirées (figure II-4 b).
Figure II-4. Conformation en pelote statistique (a), conformation étirée d’un PEL (b),
influence du sel (c).
Br
⊕ ⊕
⊕ ⊕ ⊕
⊕
⊕ ⊕
⊕
⊕
⊕
⊕
⊕
Br
ajout de sel K⊕ Br
(a) (b)
Br
Br
K⊕
K⊕
K⊕
K⊕
(c)
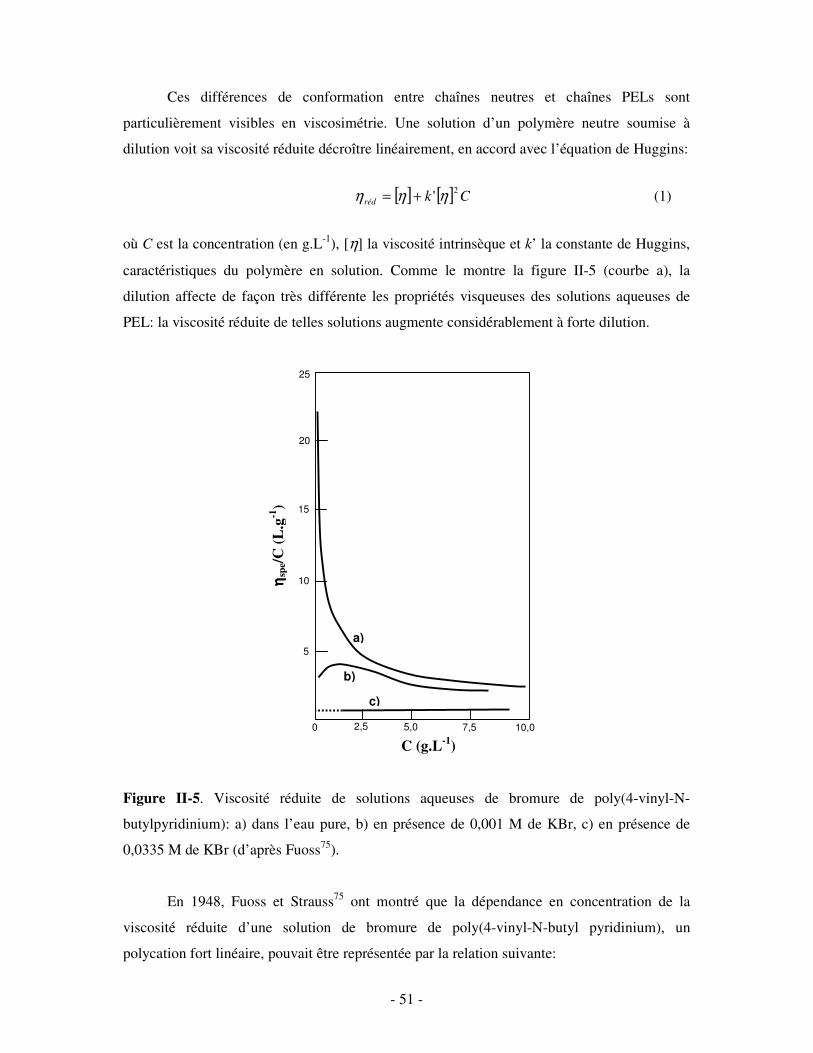
- 51 -
Ces différences de conformation entre chaînes neutres et chaînes PELs sont
particulièrement visibles en viscosimétrie. Une solution d’un polymère neutre soumise à
dilution voit sa viscosité réduite décroître linéairement, en accord avec l’équation de Huggins:
[ ] [ ] Ckréd
2'ηηη += (1)
où C est la concentration (en g.L-1), [η] la viscosité intrinsèque et k’ la constante de Huggins,
caractéristiques du polymère en solution. Comme le montre la figure II-5 (courbe a), la
dilution affecte de façon très différente les propriétés visqueuses des solutions aqueuses de
PEL: la viscosité réduite de telles solutions augmente considérablement à forte dilution.
Figure II-5. Viscosité réduite de solutions aqueuses de bromure de poly(4-vinyl-N-
butylpyridinium): a) dans l’eau pure, b) en présence de 0,001 M de KBr, c) en présence de
0,0335 M de KBr (d’après Fuoss75).
En 1948, Fuoss et Strauss75 ont montré que la dépendance en concentration de la
viscosité réduite d’une solution de bromure de poly(4-vinyl-N-butyl pyridinium), un
polycation fort linéaire, pouvait être représentée par la relation suivante:
0 2,5 5,0 7,5 10,0
a)
b)
c)
5
10
15
20
25
ηη ηη spe
/C (L
.g-1
)
C (g.L-1)

- 52 -
2/11 BCA
red +=η (2)
où A, la valeur limite de ηred à dilution infinie, est identifiée comme la viscosité intrinsèque du
PEL dans sa conformation la plus étirée, c’est-à-dire sous forme d’un bâtonnet rigide. La forte
augmentation de ηred observée par dilution a été attribuée à une expansion des chaînes de
polymère. A forte dilution (c’est-à-dire à faible concentration), les contre-ions forment un
nuage très diffus autour de la macromolécule, ce qui a pour effet de renforcer les forces
coulombiennes entre les charges portées par la chaîne et, lorsque celles-ci sont toutes de
même signe, il s’ensuit une dilatation de la chaîne (interactions répulsives intrachaînes). A
plus forte concentration, par contre, le nuage de contre-ions est plus dense et les interactions
électrostatiques entre les charges portées par la chaîne sont écrantées sur des distances plus
courtes. L’intensité des répulsions et, par conséquent, les dimensions de la macromolécule
sont donc réduites à faible dilution.
Les propriétés des PELs en solution dépendent fortement de la présence ou non de sel:
l’ajout d’un électrolyte fort tel que le bromure de potassium induit une contraction
considérable de la conformation des chaînes (figure II-4 c). Preuve en est la chute de viscosité
observée sur la figure II-5 à mesure que la concentration en KBr augmente. Les ions ajoutés
créent un effet d’écran entre les groupes chargés de la macromolécule, ce qui réduit les
répulsions électrostatiques et donc le phénomène d’allongement de la chaîne. Pour 0,0335 M
de KBr ajouté, le PEL adopte une conformation telle que la viscosité observée devient
analogue à celle d’un polymère neutre: ηred diminue au fur et à mesure de la dilution.
3- Applications
De par leur aptitude à complexer des macromolécules, des tensioactifs ou bien des
particules colloïdales de charges opposées (par interactions coulombiennes principalement),
les PELs offrent un large champ d’applications possibles, et ceci dans de nombreux domaines
(industries, médecine…). Ici, nous ne nous intéresserons qu’au cas particulier des complexes
PEL/colloïde, les complexes PEL/PEL et PEL/tensioactif de charges opposées faisant l’objet
des paragraphes suivants. Nous précisons que, parmi les nombreuses applications possibles

- 53 -
des PELs, seules les principales sont ici brièvement présentées. Pour plus de détails, nous
renvoyons le lecteur aux œuvres de Dautzenberg72 et Mortimer73.
La plupart des particules en suspension dans l’eau portent des charges négatives en
surface, ce qui induit des répulsions à faible distance entre particules et assure ainsi une
certaine stabilité de la suspension (les particules ne s’agrègent pas). Or, d’importants
problèmes liés à l’environnement tels que la production d’eau potable et le traitement des
eaux usagées nécessitent de faire sédimenter ces particules, ceci afin d’obtenir une eau
limpide. Les PELs cationiques peuvent alors être utilisés comme agents de floculation. Leur
fonction dans le processus de séparation de phases solide-liquide consiste à vaincre les forces
répulsives existant entre les particules colloïdales, soit en neutralisant les charges de ces
dernières, soit en s’adsorbant à leur surface et en les rassemblant sous forme d’agrégats. Le
PDADMAC, par exemple, s’avère être un agent floculant très efficace, couramment utilisé
pour résoudre ces problèmes de traitement des eaux.
L’industrie du papier fait également appel aux propriétés floculantes des PELs. Par un
processus d’adsorption-floculation, ces macromolécules contribuent en effet à une meilleure
rétention des petites fibres, charges et colorants qui interviennent dans la fabrication du
papier. La PEI, notamment, tient une place importante comme agent de rétention.
Contrairement aux PELs cationiques généralement employés comme agents
floculants, les polyanions sont quant à eux couramment utilisés pour leurs propriétés
dispersantes. Les polyanions d’origine naturelle, entre autres, fréquemment employés comme
agents épaississants, permettent également de stabiliser les suspensions colloïdales, aussi bien
en contribuant à une augmentation de la viscosité des solutions qu’en interagissant avec les
particules en suspension. Citons notamment le rôle important tenu par la gélatine dans le
processus de stabilisation de solutions aqueuses à usage alimentaire.
Enfin, les PELs sont aussi largement utilisés pour leur aptitude à modifier les
propriétés rhéologiques de systèmes aqueux. Ils sont ainsi couramment employés comme
agents épaississants ou gélifiants dans de nombreux domaines tels l’industrie alimentaire, les
industries pharmaceutique et cosmétique, l’industrie du pétrole et celle de la peinture. Les
agents épaississants, généralement d’origine naturelle, appartiennent à la famille des
polysaccharides (exception faite de la gélatine). La gomme arabique, par exemple, issue d’une
variété d’acacia, améliore la tenue des encres et de la peinture à l’eau, aussi bien par ses
propriétés dispersantes que par ses propriétés épaississantes.

- 54 -
II- Etude des complexes polyélectrolytes (PECs) solubles dans l’eau
La formation de PECs est le résultat quasi instantané de la mise en présence en
solution de deux PELs de charges opposées, comme illustré en figure II-6.
Figure II-6. Formation de PECs: a) entre PELs forts, b) entre polybase et polyacide faibles.
Ce processus d’association, principalement gouverné par de fortes attractions
électrostatiques entre les deux antagonistes, s’explique également d’un point de vue
entropique76. Dans une solution ne comportant qu’un seul PEL, l’existence d’interactions
attractives coulombiennes induit une concentration en contre-ions plus élevée au voisinage du
polyion. Cette distribution non uniforme des contre-ions représente une perte en entropie par
rapport au cas où ils seraient répartis de façon homogène dans le milieu. Si dans cette même
solution est introduit un polyion de charges opposées (accompagné de ses n contre-ions), une
distribution uniforme des 2n contre-ions peut-être obtenue par association des deux polyions.
Ce « relargage » des contre-ions dans le milieu représente un gain en entropie et fait que la
formation des PECs est favorable. Ceci explique aussi pourquoi, comme nous le verrons par
la suite, l’association a tendance à disparaître en présence de sel: la distribution des contre-
ions devient plus uniforme même sans association des polyions.
C+ C+ C+
A- A- A-
HB+ HB+ HB+
A- A- A-
C+ C+ C+
Cl- Cl- Cl-
A- A- A- Na+ Na+ Na+
HB+ HB+ HB+
A- A- A- H+ H+ H+
OH- OH- OH-
+ n NaCl
+ n H2O
a)
b)

- 55 -
D’un point de vue macroscopique, les interactions polyanion/polycation peuvent
engendrer trois types de systèmes différents77:
(i) des systèmes limpides dans lesquels des agrégats de petite taille sont répartis de
façon homogène,
(ii) des systèmes troubles où les agrégats en suspension sont à la limite de la
séparation de phases,
(iii) des systèmes bi-phasiques comportant un surnageant et un précipité qui, après
rinçage et séchage, permet d’obtenir des complexes à l’état solide.
C’est aux systèmes appartenant à la catégorie i), à savoir les PECs solubles dans l’eau,
qu’est dédié principalement ce chapitre. Dans un premier temps, nous nous intéresserons aux
différentes façons de préparer ces systèmes ainsi qu’à la structure des agrégats formés. Puis
nous étudierons l’influence de divers paramètres sur ces structures: l’influence des conditions
de réaction telles que la concentration, la stoechiométrie du mélange (degré de conversion), la
force ionique, le pH et la température du milieu ainsi que les caractéristiques des composants
PELs (type et position des groupements ioniques, densité de charges, rigidité de la
structure…).
1- Formation et structure des PECs solubles
En 1972, Tsuchida et coll.78 se sont intéressés aux interactions pouvant exister entre un
poly(styrène) sulfonate (NaPSS) et un ionène aromatique dénommé 3X (figure II-7).
* N+
CH3
CH3
CH2 N+
CH3
CH3
CH2 CH2 *3Cl- Cl-
n
Figure II-7. Structure chimique du ionène aromatique 3X.
Les auteurs ont montré que des systèmes macroscopiquement homogènes pouvaient
être obtenus si l’un des PELs était mis en présence de son antagoniste en plus faible quantité,
un mélange de stoechiométrie 1:1 ( 1 mole de NaPSS pour 1 mole de 3X) donnant lieu à un
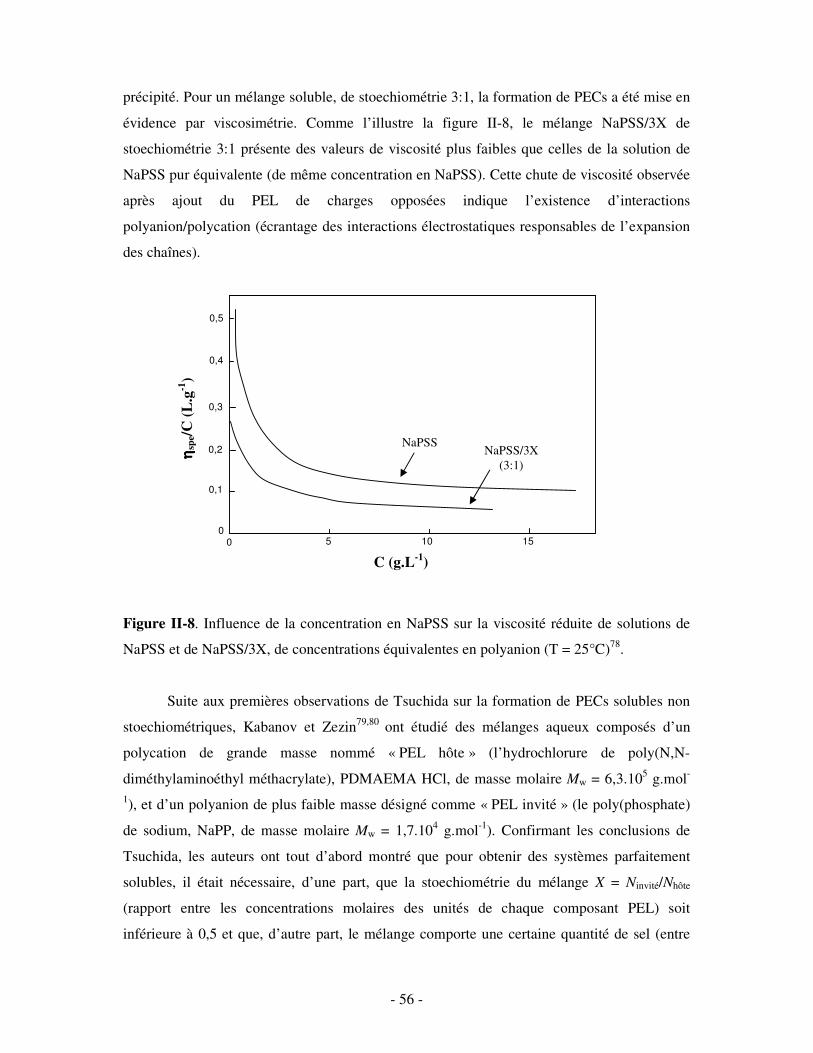
- 56 -
précipité. Pour un mélange soluble, de stoechiométrie 3:1, la formation de PECs a été mise en
évidence par viscosimétrie. Comme l’illustre la figure II-8, le mélange NaPSS/3X de
stoechiométrie 3:1 présente des valeurs de viscosité plus faibles que celles de la solution de
NaPSS pur équivalente (de même concentration en NaPSS). Cette chute de viscosité observée
après ajout du PEL de charges opposées indique l’existence d’interactions
polyanion/polycation (écrantage des interactions électrostatiques responsables de l’expansion
des chaînes).
Figure II-8. Influence de la concentration en NaPSS sur la viscosité réduite de solutions de
NaPSS et de NaPSS/3X, de concentrations équivalentes en polyanion (T = 25°C)78.
Suite aux premières observations de Tsuchida sur la formation de PECs solubles non
stoechiométriques, Kabanov et Zezin79,80 ont étudié des mélanges aqueux composés d’un
polycation de grande masse nommé « PEL hôte » (l’hydrochlorure de poly(N,N-
diméthylaminoéthyl méthacrylate), PDMAEMA HCl, de masse molaire Mw = 6,3.105 g.mol-
1), et d’un polyanion de plus faible masse désigné comme « PEL invité » (le poly(phosphate)
de sodium, NaPP, de masse molaire Mw = 1,7.104 g.mol-1). Confirmant les conclusions de
Tsuchida, les auteurs ont tout d’abord montré que pour obtenir des systèmes parfaitement
solubles, il était nécessaire, d’une part, que la stoechiométrie du mélange X = Ninvité/Nhôte
(rapport entre les concentrations molaires des unités de chaque composant PEL) soit
inférieure à 0,5 et que, d’autre part, le mélange comporte une certaine quantité de sel (entre
0 0
5 10 15
0,1
0,2
0,3
0,4
0,5
NaPSS NaPSS/3X
(3:1)
C (g.L-1)
ηη ηη spe
/C (L
.g-1
)
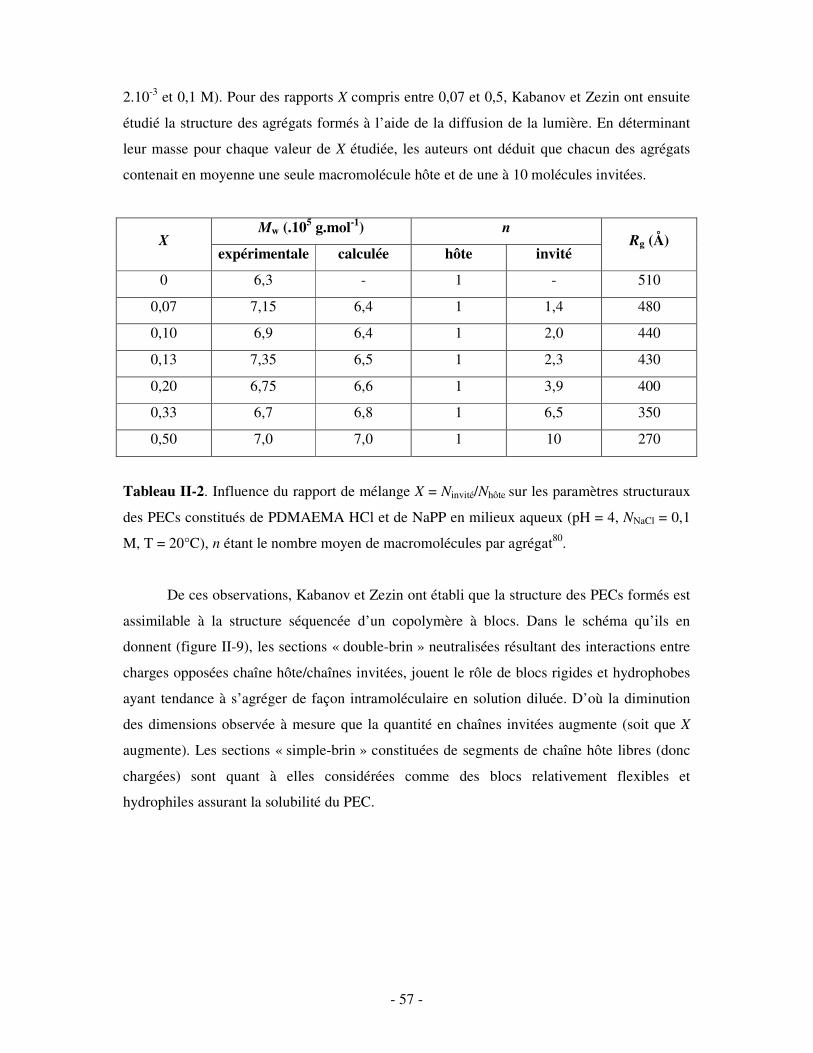
- 57 -
2.10-3 et 0,1 M). Pour des rapports X compris entre 0,07 et 0,5, Kabanov et Zezin ont ensuite
étudié la structure des agrégats formés à l’aide de la diffusion de la lumière. En déterminant
leur masse pour chaque valeur de X étudiée, les auteurs ont déduit que chacun des agrégats
contenait en moyenne une seule macromolécule hôte et de une à 10 molécules invitées.
Mw (.105 g.mol-1) n X
expérimentale calculée hôte invité Rg (Å)
0 6,3 - 1 - 510
0,07 7,15 6,4 1 1,4 480
0,10 6,9 6,4 1 2,0 440
0,13 7,35 6,5 1 2,3 430
0,20 6,75 6,6 1 3,9 400
0,33 6,7 6,8 1 6,5 350
0,50 7,0 7,0 1 10 270
Tableau II-2. Influence du rapport de mélange X = Ninvité/Nhôte sur les paramètres structuraux
des PECs constitués de PDMAEMA HCl et de NaPP en milieux aqueux (pH = 4, NNaCl = 0,1
M, T = 20°C), n étant le nombre moyen de macromolécules par agrégat80.
De ces observations, Kabanov et Zezin ont établi que la structure des PECs formés est
assimilable à la structure séquencée d’un copolymère à blocs. Dans le schéma qu’ils en
donnent (figure II-9), les sections « double-brin » neutralisées résultant des interactions entre
charges opposées chaîne hôte/chaînes invitées, jouent le rôle de blocs rigides et hydrophobes
ayant tendance à s’agréger de façon intramoléculaire en solution diluée. D’où la diminution
des dimensions observée à mesure que la quantité en chaînes invitées augmente (soit que X
augmente). Les sections « simple-brin » constituées de segments de chaîne hôte libres (donc
chargées) sont quant à elles considérées comme des blocs relativement flexibles et
hydrophiles assurant la solubilité du PEC.

- 58 -
Figure II-9. Structure schématique des PECs solubles séquencés, d’après Kabanov79,80.
Notons que pour un rapport X supérieur à 0,5, le nombre de blocs hydrophobes devient
suffisamment élevé pour pouvoir provoquer une ségrégation intermoléculaire des sections
« double-brin », et donc une agglomération des particules individuelles de PECs jusqu’à
l’obtention d’une séparation de phases. Les auteurs ont montré qu’une dissociation des
agglomérats pouvait ensuite être retrouvée par simple addition de molécules hôtes, cette
dissociation étant accompagnée d’une redistribution des chaînes invitées parmi les chaînes
hôtes. En jouant sur la balance hydrophile/hydrophobe des agrégats, Kabanov et Zezin ont
ainsi mis en évidence le caractère réversible de la transformation complexes
solubles/complexes insolubles et vice-versa.
Une manière différente d’obtenir des PECs solubles a été décrite par Dautzenberg et
coll.72, en faisant interagir cette fois des PELs antagonistes de masses molaires (soit de
longueurs de chaîne) comparables, mais ce à très faible concentration (entre 10-2 et 1 g.L-1). Il
a en effet été démontré que le degré d’agrégation des particules individuelles de PECs est
principalement déterminé par la concentration des polymères. En mélangeant progressivement
deux solutions de PELs de charges opposées, quatre étapes différentes peuvent être
successivement rencontrées à mesure que la concentration en polymères augmente:
(i) la persistance d’un système homogène à une seule phase après mélange des deux
solutions,
hôte
invité
bloc hydrophobe
bloc hydrophile

- 59 -
(ii) la formation d’une dispersion trouble constituée de particules colloïdales de grande
taille pour des concentrations comprises entre 0,1 et 1% en poids,
(iii) une rapide séparation de phases avec l’obtention d’un précipité et d’une phase
surnageante limpide pour des concentrations de l’ordre de 1%,
(iv) la formation d’un système à une seule phase à concentrations plus élevées ( 5%
en poids).
Ces observations ont conduit Kötz81 à schématiser la formation des PECs selon le
modèle à deux étapes présenté en figure II-10. La première étape, observable à faible
concentration, consiste en la formation d’« agrégats primaires » relativement petits et
stabilisés par des charges en surface, pouvant contenir entre 10 et 1000 chaînes de polymère.
La seconde étape, à concentration plus élevée, correspond à la formation d’agrégats de plus
grande taille, composés de nombreux agrégats primaires. Dans la plupart des cas, cette
agrégation secondaire induit une séparation de phases plus ou moins rapide (floculation), mais
l’obtention d’une suspension stable est possible si la masse molaire des composés est plutôt
faible et/ou si les agrégats primaires possèdent une structure diffuse défavorable à une
séparation de phases. On note qu’une dissociation des agrégats primaires est observable par
augmentation de la concentration d’un seul composant au-delà d’une certaine valeur critique.

- 60 -
Figure II-10. Modèle à deux étapes de la formation des PECs, d’après Kötz81.
2- Influence de divers paramètres sur la structure des PECs solubles
a) Influence de la concentration
Comme mentionné dans le paragraphe précédent, la formation des PECs peut être
stabilisée à un niveau colloïdal si le milieu est suffisamment dilué, la floculation du système
ne survenant que pour un certain degré de conversion X (rapport molaire entre charges
⊕
⊕
⊕
⊕
⊕ ⊕
polyanion + polycation
⊕
⊕
⊕
étape 1 agrégats primaires
(stabilisés par des charges en surface)
étape 2 agrégats secondaires

- 61 -
positives et négatives dans le milieu). Toutefois, la structure des particules en solution diluée
peut également dépendre de la concentration des polymères.
Philipp et coll.77 ont par exemple déterminé par diffusion de lumière la masse et la
taille des particules formées entre un copolymère de N-méthyl-N,N-diéthyl-
ammonioéthylacrylate/acrylamide (polycation) et un copolymère d’acrylate de
sodium/acrylamide (polyanion), à différentes concentrations mais à degré de conversion X
constant. Le tableau II-3 présente les résultats obtenus: les paramètres structuraux des
agrégats formés augmentent avec la concentration.
Cpolycation (g.L-1) Mw (g.mol-1) Rg (nm)
1 1,2.109 188
0,5 1,5.109 195
0,25 9,8.108 143
0,1 8,9.107 63
0,05 6,6.107 63
Tableau II-3. Influence de la concentration en polymère sur les paramètres structuraux des
particules de PECs formées pour un degré de conversion X = N-/N+ fixé à 0,4.
Les auteurs en ont alors déduit l’existence d’une compétition entre deux processus différents
de neutralisation des charges: par changements conformationnels des chaînes et par
incorporation de nouvelles macromolécules au sein des complexes, le second processus étant
favorisé à concentrations plus élevées.
La conformation des chaînes elle-même dépend de la concentration, ce qui peut aussi
se répercuter sur la structure des PECs. Il est à noter que le processus de complexation est un
mécanisme à deux étapes impliquant, premièrement, un rapprochement des deux macroions
de charges opposées, suivi d’une neutralisation plus ou moins rapide des charges. La vitesse
de cette seconde étape dépend de la structure des molécules, de leur densité de charges et de
la mobilité des contre-ions lors de leur relargage pendant la complexation. Il arrive qu’au
cours d’une neutralisation rapide, certaines charges restent « piégées » sur les macroions et ne
soient compensées au sein du complexe que par les contre-ions. La composition du complexe

- 62 -
dévie alors de la stoechiométrie 1:1, ce qui peut être observé pour des chaînes dans des
conformations peu étirées.
Vishalakshi et coll.82 ont étudié par viscosimétrie et par spectroscopie d’absorption
UV des mélanges constitués de carboxyméthylcellulose (CMC) et de iodure de poly(vinyl-N-
méthylpyridinium) (PVMP), de masses molaires et de degrés de substitution équivalents, à
différentes concentrations. Dans cette étude, les auteurs se sont particulièrement intéressés au
degré de conversion critique qui se traduit par la floculation du système, et qui donc
correspond à des minima de viscosité (mesurée sur phase limpide) et d’absorbance des
mélanges. En augmentant la concentration des polymères, ils ont observé une déviation du
degré de conversion critique, de la stoechiométrie 1:1 à la valeur X = 0,67. Ce résultat a été
attribué à des changements de configuration différents pour les deux polyions suivant leur
concentration. Les deux PELs étudiés présentent des flexibilités différentes: le PVMP est un
polymère relativement flexible tandis que la CMC est un polysaccharide rigide. Par
conséquent, les dimensions du PVMP sont beaucoup plus sensibles à la force ionique du
milieu, et donc à la dilution, que celles de la CMC. A concentration plus élevée, les chaînes de
PVMP sont moins étendues que celles de la CMC et le processus de complexation conduit au
« piégeage » de certaines des charges positives du PVMP (figure II-11 a), ce qui se traduit par
une composition N-/N+ = 0,67 pour les complexes. A faible concentration, les deux polyions
sont par contre supposés adopter des conformations plus rigides du fait d’une augmentation
des répulsions électrostatiques. Dans ces conditions, le processus de complexation entre les
chaînes de conformation étendue résulte en une composition proche de la stoechiométrie 1:1
(figure II-11 b).

- 63 -
Figure II-11. Représentation schématique de l’influence de la concentration sur l’expansion
des chaînes de CMC (polyanion) et de PVMP (polycation), et sur la formation des PECs82.
Notons enfin que l’influence de la concentration sur la composition des PECs est
d’autant plus importante si l’un des composés est un PEL faible. Le degré de neutralisation α’
de ce dernier (ou bien son degré de dissociation α), dont dépend la composition du PEC, est
en effet fonction de la concentration.
Abe et coll.83 ont étudié l’influence de la concentration sur la composition des PECs
formés entre un acide poly(méthacrylique) (PMAA) et un ionène aromatique nommé 2X, dont
la formule chimique s’apparente à celle présentée en figure II-7 (le squelette comporte dans ce
cas un enchaînement de deux groupements –CH2 et non trois). La figure II-12 montre l’effet
de la concentration sur la composition des PECs formés, pour α’ = 1 (100 % des groupements
carboxyle du PMAA dissociés) et pour α’ = 0 (le degré de dissociation α du PMAA augmente
de quelques % en présence du 2X), les compositions présentées ayant été déterminées par
viscosimétrie à faibles concentrations (PECs solubles pour N < 10-3 M), et par analyse
élémentaire à plus fortes concentrations.
⊕
⊕ ⊕
⊕
⊕ ⊕
⊕
⊕
⊕
⊕
⊕
⊕
⊕
⊕
a) b)
à concentration relativement élevée à faible concentration
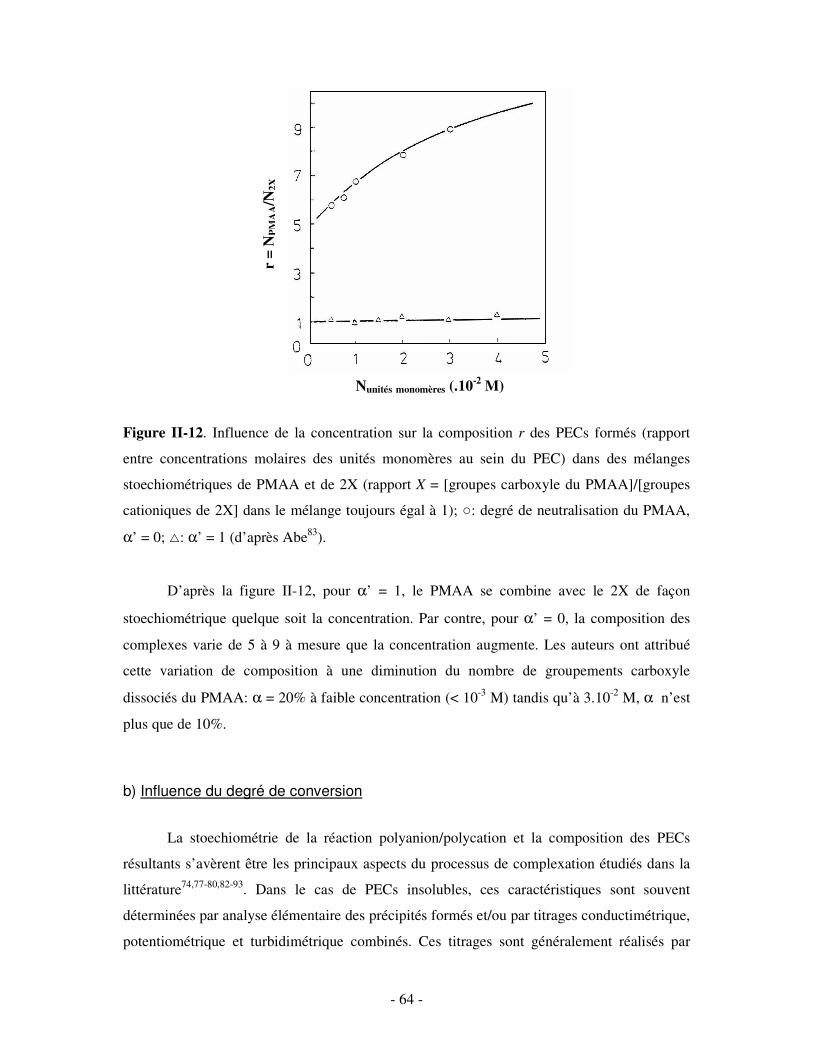
- 64 -
Figure II-12. Influence de la concentration sur la composition r des PECs formés (rapport
entre concentrations molaires des unités monomères au sein du PEC) dans des mélanges
stoechiométriques de PMAA et de 2X (rapport X = [groupes carboxyle du PMAA]/[groupes
cationiques de 2X] dans le mélange toujours égal à 1); : degré de neutralisation du PMAA,
α’ = 0; : α’ = 1 (d’après Abe83).
D’après la figure II-12, pour α’ = 1, le PMAA se combine avec le 2X de façon
stoechiométrique quelque soit la concentration. Par contre, pour α’ = 0, la composition des
complexes varie de 5 à 9 à mesure que la concentration augmente. Les auteurs ont attribué
cette variation de composition à une diminution du nombre de groupements carboxyle
dissociés du PMAA: α = 20% à faible concentration (< 10-3 M) tandis qu’à 3.10-2 M, α n’est
plus que de 10%.
b) Influence du degré de conversion
La stoechiométrie de la réaction polyanion/polycation et la composition des PECs
résultants s’avèrent être les principaux aspects du processus de complexation étudiés dans la
littérature74,77-80,82-93. Dans le cas de PECs insolubles, ces caractéristiques sont souvent
déterminées par analyse élémentaire des précipités formés et/ou par titrages conductimétrique,
potentiométrique et turbidimétrique combinés. Ces titrages sont généralement réalisés par
r =
NPM
AA/N
2X
Nunités monomères (.10-2 M)
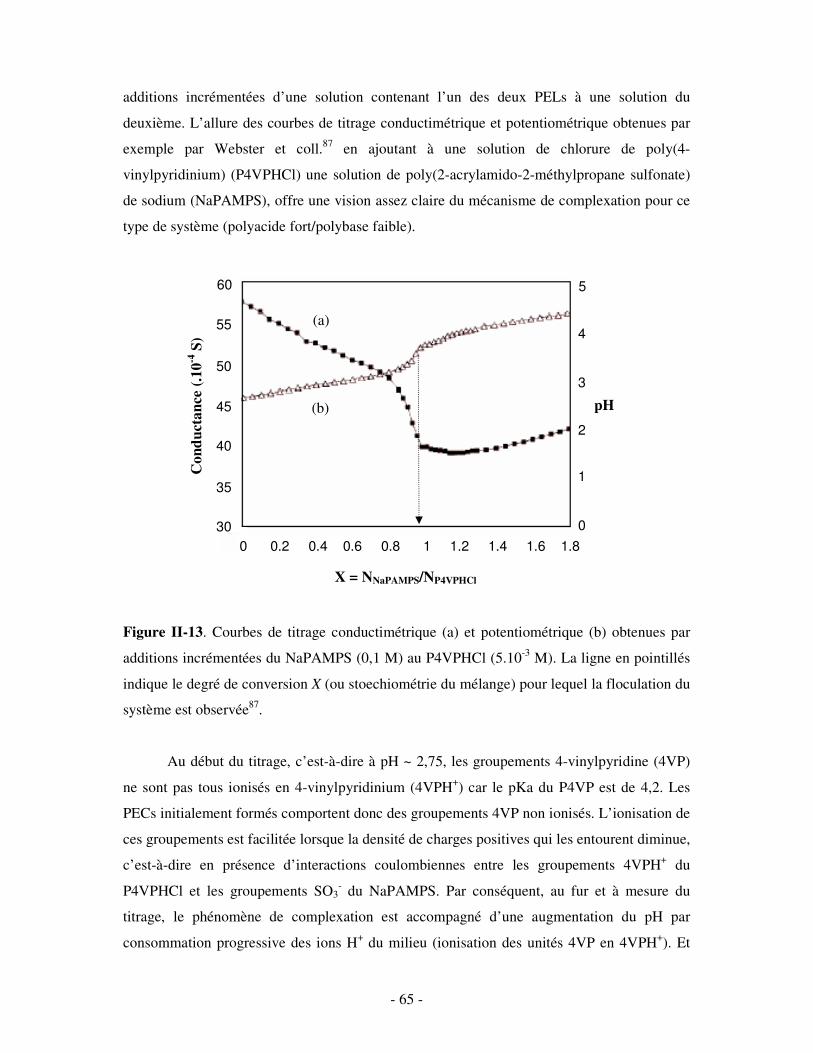
- 65 -
additions incrémentées d’une solution contenant l’un des deux PELs à une solution du
deuxième. L’allure des courbes de titrage conductimétrique et potentiométrique obtenues par
exemple par Webster et coll.87 en ajoutant à une solution de chlorure de poly(4-
vinylpyridinium) (P4VPHCl) une solution de poly(2-acrylamido-2-méthylpropane sulfonate)
de sodium (NaPAMPS), offre une vision assez claire du mécanisme de complexation pour ce
type de système (polyacide fort/polybase faible).
Figure II-13. Courbes de titrage conductimétrique (a) et potentiométrique (b) obtenues par
additions incrémentées du NaPAMPS (0,1 M) au P4VPHCl (5.10-3 M). La ligne en pointillés
indique le degré de conversion X (ou stoechiométrie du mélange) pour lequel la floculation du
système est observée87.
Au début du titrage, c’est-à-dire à pH ~ 2,75, les groupements 4-vinylpyridine (4VP)
ne sont pas tous ionisés en 4-vinylpyridinium (4VPH+) car le pKa du P4VP est de 4,2. Les
PECs initialement formés comportent donc des groupements 4VP non ionisés. L’ionisation de
ces groupements est facilitée lorsque la densité de charges positives qui les entourent diminue,
c’est-à-dire en présence d’interactions coulombiennes entre les groupements 4VPH+ du
P4VPHCl et les groupements SO3- du NaPAMPS. Par conséquent, au fur et à mesure du
titrage, le phénomène de complexation est accompagné d’une augmentation du pH par
consommation progressive des ions H+ du milieu (ionisation des unités 4VP en 4VPH+). Et
pH
X = NNaPAMPS/NP4VPHCl
(a)
(b)
0 0.2 0.4 0.6 0.8 1 1.2 1.4 1.6 1.8
5
4
3
2
1
0
60
55
50
45
40
35
30
Con
duct
ance
(.10
-4 S
)

- 66 -
puisque la conductivité ionique des H+ est au moins trois fois plus élevée que celle des contre-
ions relargués au cours de la complexation, la conductance du mélange diminue
simultanément. A faible degré de conversion (X < 1), les particules de PEC comportent des
charges positives (P4VPHCl) en excès et sont donc réparties de façon homogène dans le
milieu (suspension stabilisée par des répulsions électrostatiques). La floculation des PECs ne
survient qu’une fois la neutralisation atteinte. D’après la figure II-13, la floculation est
observée pour un degré de conversion X égal à 0,95, ce qui indique une réaction quasi
stoechiométrique. Une analyse élémentaire effectuée sur les précipités formés à différents
degrés de conversion (jusqu’à X = 1,5) a cependant montré que la composition des PECs
dévie de la stoechiométrie 1:1. Cette déviation a été attribuée à un excès de groupements 4VP
non ionisés au sein des PECs. Notons que des mesures turbidimétriques (mesures de densité
optique) ont montré que la taille des particules formées pour X compris entre 0,3 et 0,6 reste
quasi constante: les particules croissent en nombre plutôt qu’en taille au fur et à mesure de la
complexation.
Dans le cas de PECs solubles, la composition des complexes en fonction du degré de
conversion peut être estimée par viscosimétrie. Brand et Dautzenberg88 ont ainsi évalué la
stoechiométrie et la densité des agrégats formés dans des solutions très diluées (10-2-1 g.L-1)
de poly(styrène sulfonate) de sodium (NaPSS) et de copolymères de chlorure de
diallyldiméthylammonium/acrylamide (DADMAC/AAM).
*
N+
*
OH2N
Cl-
DADMAC AAM
Figure II-14. Structure chimique des copolymères DADMAC/AAM.
La figure II-15 présente l’influence du degré de conversion sur la viscosité spécifique
des mélanges obtenus en ajoutant des solutions de copolymères DADMAC/AAM (de
concentration initiale NP+0 et de composition molaire en DADMAC comprise entre 35 et 100
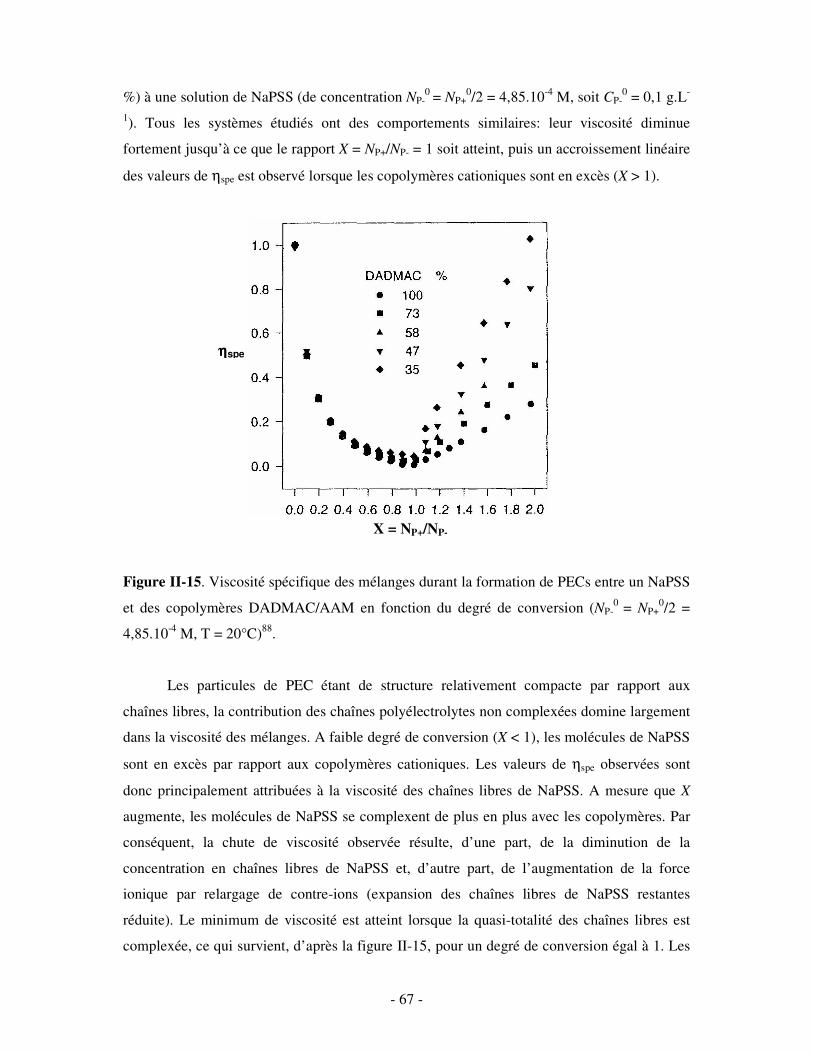
- 67 -
%) à une solution de NaPSS (de concentration NP-0
= NP+0/2 = 4,85.10-4 M, soit CP-
0 = 0,1 g.L-
1). Tous les systèmes étudiés ont des comportements similaires: leur viscosité diminue
fortement jusqu’à ce que le rapport X = NP+/NP- = 1 soit atteint, puis un accroissement linéaire
des valeurs de ηspe est observé lorsque les copolymères cationiques sont en excès (X > 1).
Figure II-15. Viscosité spécifique des mélanges durant la formation de PECs entre un NaPSS
et des copolymères DADMAC/AAM en fonction du degré de conversion (NP-0
= NP+0/2 =
4,85.10-4 M, T = 20°C)88.
Les particules de PEC étant de structure relativement compacte par rapport aux
chaînes libres, la contribution des chaînes polyélectrolytes non complexées domine largement
dans la viscosité des mélanges. A faible degré de conversion (X < 1), les molécules de NaPSS
sont en excès par rapport aux copolymères cationiques. Les valeurs de ηspe observées sont
donc principalement attribuées à la viscosité des chaînes libres de NaPSS. A mesure que X
augmente, les molécules de NaPSS se complexent de plus en plus avec les copolymères. Par
conséquent, la chute de viscosité observée résulte, d’une part, de la diminution de la
concentration en chaînes libres de NaPSS et, d’autre part, de l’augmentation de la force
ionique par relargage de contre-ions (expansion des chaînes libres de NaPSS restantes
réduite). Le minimum de viscosité est atteint lorsque la quasi-totalité des chaînes libres est
complexée, ce qui survient, d’après la figure II-15, pour un degré de conversion égal à 1. Les
ηηηηspe
X = NP+/NP-
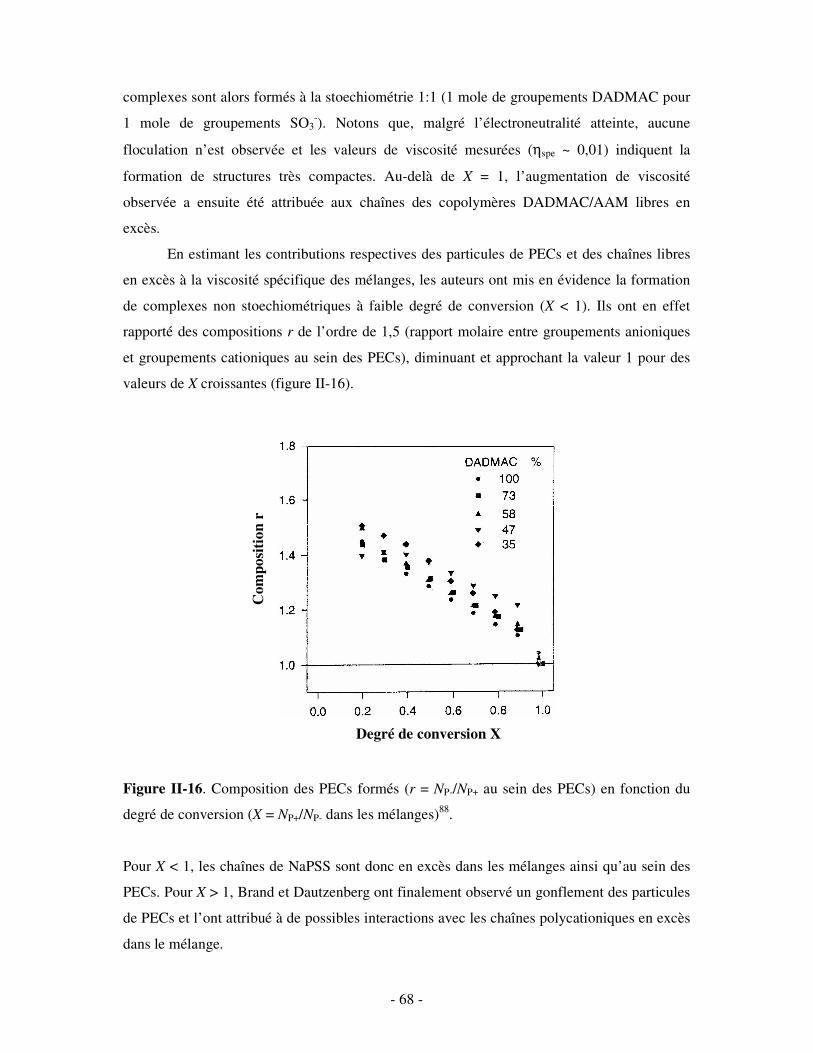
- 68 -
complexes sont alors formés à la stoechiométrie 1:1 (1 mole de groupements DADMAC pour
1 mole de groupements SO3-). Notons que, malgré l’électroneutralité atteinte, aucune
floculation n’est observée et les valeurs de viscosité mesurées (ηspe ~ 0,01) indiquent la
formation de structures très compactes. Au-delà de X = 1, l’augmentation de viscosité
observée a ensuite été attribuée aux chaînes des copolymères DADMAC/AAM libres en
excès.
En estimant les contributions respectives des particules de PECs et des chaînes libres
en excès à la viscosité spécifique des mélanges, les auteurs ont mis en évidence la formation
de complexes non stoechiométriques à faible degré de conversion (X < 1). Ils ont en effet
rapporté des compositions r de l’ordre de 1,5 (rapport molaire entre groupements anioniques
et groupements cationiques au sein des PECs), diminuant et approchant la valeur 1 pour des
valeurs de X croissantes (figure II-16).
Figure II-16. Composition des PECs formés (r = NP-/NP+ au sein des PECs) en fonction du
degré de conversion (X = NP+/NP- dans les mélanges)88.
Pour X < 1, les chaînes de NaPSS sont donc en excès dans les mélanges ainsi qu’au sein des
PECs. Pour X > 1, Brand et Dautzenberg ont finalement observé un gonflement des particules
de PECs et l’ont attribué à de possibles interactions avec les chaînes polycationiques en excès
dans le mélange.
Degré de conversion X
Com
posi
tion
r
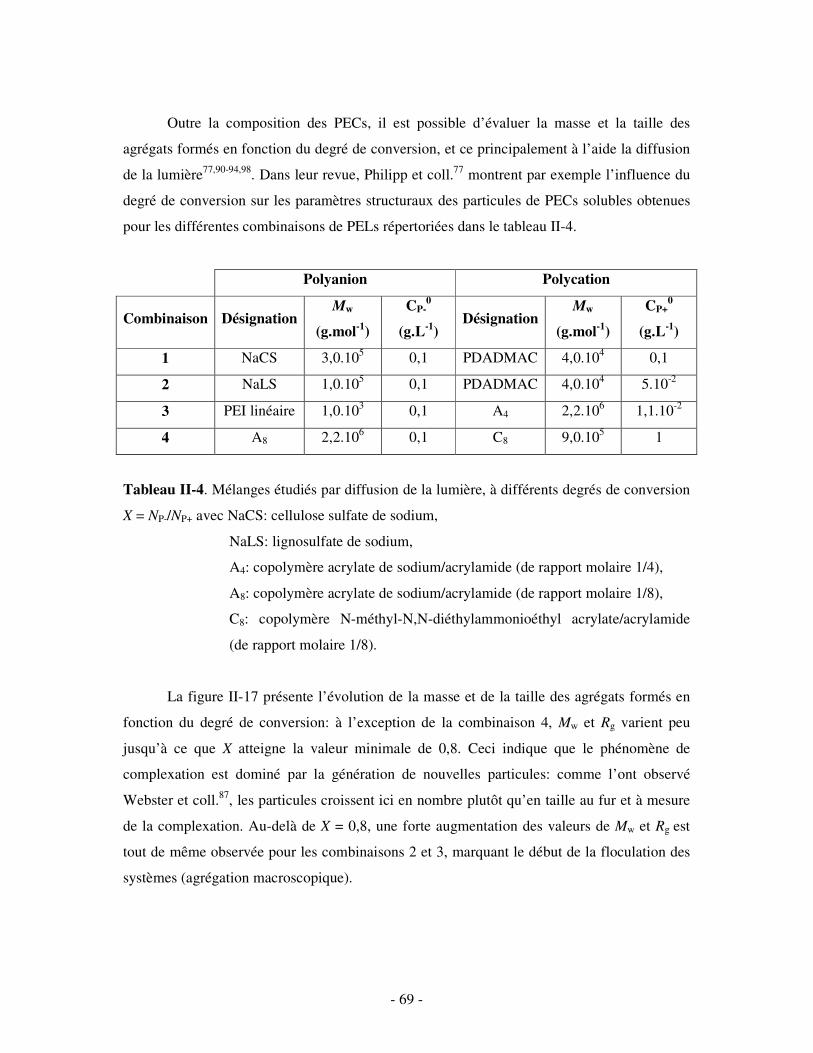
- 69 -
Outre la composition des PECs, il est possible d’évaluer la masse et la taille des
agrégats formés en fonction du degré de conversion, et ce principalement à l’aide la diffusion
de la lumière77,90-94,98. Dans leur revue, Philipp et coll.77 montrent par exemple l’influence du
degré de conversion sur les paramètres structuraux des particules de PECs solubles obtenues
pour les différentes combinaisons de PELs répertoriées dans le tableau II-4.
Polyanion Polycation
Combinaison Désignation Mw
(g.mol-1)
CP-0
(g.L-1) Désignation
Mw
(g.mol-1)
CP+0
(g.L-1)
1 NaCS 3,0.105 0,1 PDADMAC 4,0.104 0,1
2 NaLS 1,0.105 0,1 PDADMAC 4,0.104 5.10-2
3 PEI linéaire 1,0.103 0,1 A4 2,2.106 1,1.10-2
4 A8 2,2.106 0,1 C8 9,0.105 1
Tableau II-4. Mélanges étudiés par diffusion de la lumière, à différents degrés de conversion
X = NP-/NP+ avec NaCS: cellulose sulfate de sodium,
NaLS: lignosulfate de sodium,
A4: copolymère acrylate de sodium/acrylamide (de rapport molaire 1/4),
A8: copolymère acrylate de sodium/acrylamide (de rapport molaire 1/8),
C8: copolymère N-méthyl-N,N-diéthylammonioéthyl acrylate/acrylamide
(de rapport molaire 1/8).
La figure II-17 présente l’évolution de la masse et de la taille des agrégats formés en
fonction du degré de conversion: à l’exception de la combinaison 4, Mw et Rg varient peu
jusqu’à ce que X atteigne la valeur minimale de 0,8. Ceci indique que le phénomène de
complexation est dominé par la génération de nouvelles particules: comme l’ont observé
Webster et coll.87, les particules croissent ici en nombre plutôt qu’en taille au fur et à mesure
de la complexation. Au-delà de X = 0,8, une forte augmentation des valeurs de Mw et Rg est
tout de même observée pour les combinaisons 2 et 3, marquant le début de la floculation des
systèmes (agrégation macroscopique).
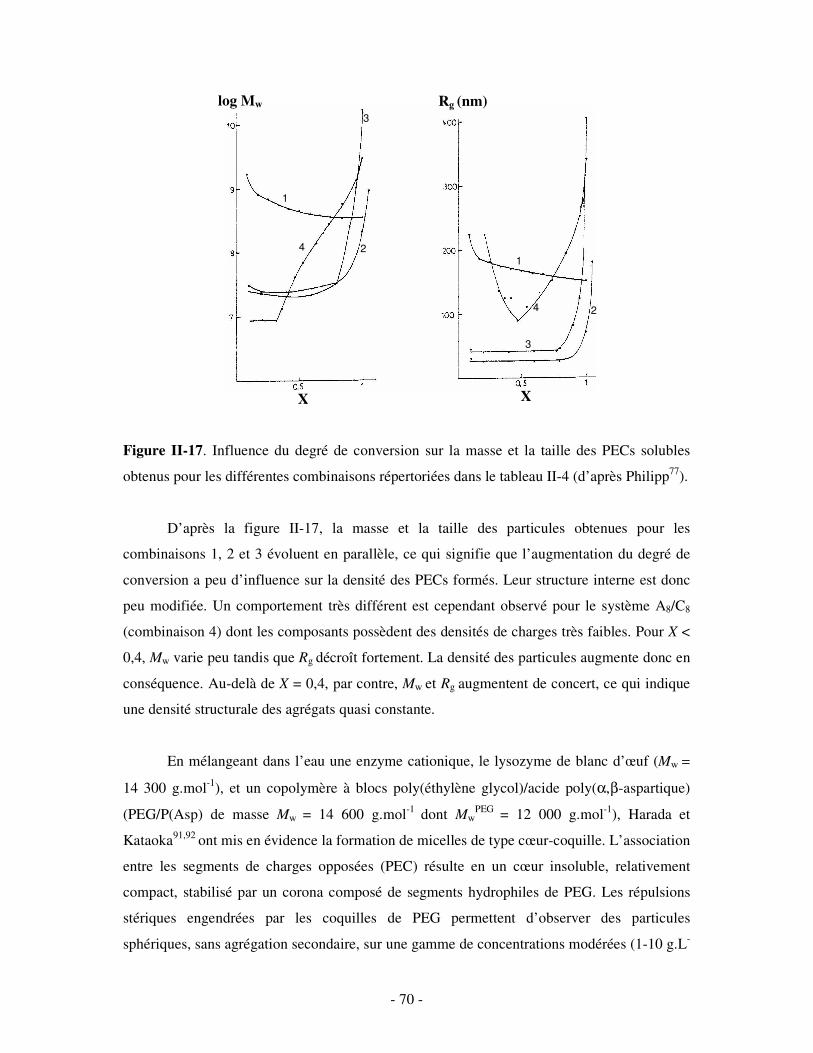
- 70 -
Figure II-17. Influence du degré de conversion sur la masse et la taille des PECs solubles
obtenus pour les différentes combinaisons répertoriées dans le tableau II-4 (d’après Philipp77).
D’après la figure II-17, la masse et la taille des particules obtenues pour les
combinaisons 1, 2 et 3 évoluent en parallèle, ce qui signifie que l’augmentation du degré de
conversion a peu d’influence sur la densité des PECs formés. Leur structure interne est donc
peu modifiée. Un comportement très différent est cependant observé pour le système A8/C8
(combinaison 4) dont les composants possèdent des densités de charges très faibles. Pour X <
0,4, Mw varie peu tandis que Rg décroît fortement. La densité des particules augmente donc en
conséquence. Au-delà de X = 0,4, par contre, Mw et Rg augmentent de concert, ce qui indique
une densité structurale des agrégats quasi constante.
En mélangeant dans l’eau une enzyme cationique, le lysozyme de blanc d’œuf (Mw =
14 300 g.mol-1), et un copolymère à blocs poly(éthylène glycol)/acide poly(α,β-aspartique)
(PEG/P(Asp) de masse Mw = 14 600 g.mol-1 dont MwPEG
= 12 000 g.mol-1), Harada et
Kataoka91,92 ont mis en évidence la formation de micelles de type cœur-coquille. L’association
entre les segments de charges opposées (PEC) résulte en un cœur insoluble, relativement
compact, stabilisé par un corona composé de segments hydrophiles de PEG. Les répulsions
stériques engendrées par les coquilles de PEG permettent d’observer des particules
sphériques, sans agrégation secondaire, sur une gamme de concentrations modérées (1-10 g.L-
X
log Mw Rg (nm)
1
1 4
4 2
2
3
3
X

- 71 -
1), et ceci même sous condition d’électroneutralité (1 mole de groupements aspartiques pour 1
mole de lysine et d’arginine). A l’aide de la diffusion de la lumière, les auteurs ont relié le
degré de conversion (X = [Asp du PEG/P(Asp)]/[Lys et Arg du lysozyme]) aux propriétés
physicochimiques des micelles telles que le nombre d’association, la taille du cœur et
l’épaisseur de la coquille. Pour X 1, le processus de formation des micelles s’avère être
coopératif: toutes les chaînes de PEG/P(Asp) présentes en solution s’associent au lysozyme
pour former des micelles stoechiométriques (de composition 1:1), de diamètre quasi constant
(~ 50 nm), les chaînes de lysozyme en excès restant sous forme non complexée. Par contre,
pour X > 1, la micellisation procède de façon non coopérative: les micelles comportent un
excès de PEG/P(Asp). Dans cette région (X > 1), une augmentation linéaire du diamètre des
micelles (entre 50 et 80 nm) a été observée pour des valeurs de X croissantes. Les auteurs ont
par ailleurs estimé le nombre d’association de chacun des polymères. Pour X > 1, le nombre
d’association des molécules de PEG/P(Asp) passe de 60 à 120 pour X compris entre 1 et 2,7.
Le nombre d’association des molécules de lysozyme contenues dans le cœur reste par contre
plus ou moins égal à 50. De plus, Harada et Kataoka ont montré que dans cette région le
rayon du cœur est indépendant de X. D’après la figure II-18, Rcoeur conserve en effet une
valeur proche de 7 nm. Seule l’épaisseur de la coquille, Rcoquille, augmente à mesure que X
s’approche de la valeur 2,7. Cette variation de l’épaisseur du corona avec X a été attribuée à
l’augmentation de densité observée (augmentation du nombre d’association des chaînes de
PEG/P(Asp)), poussant les segments de PEG à adopter des conformations plus étendues.
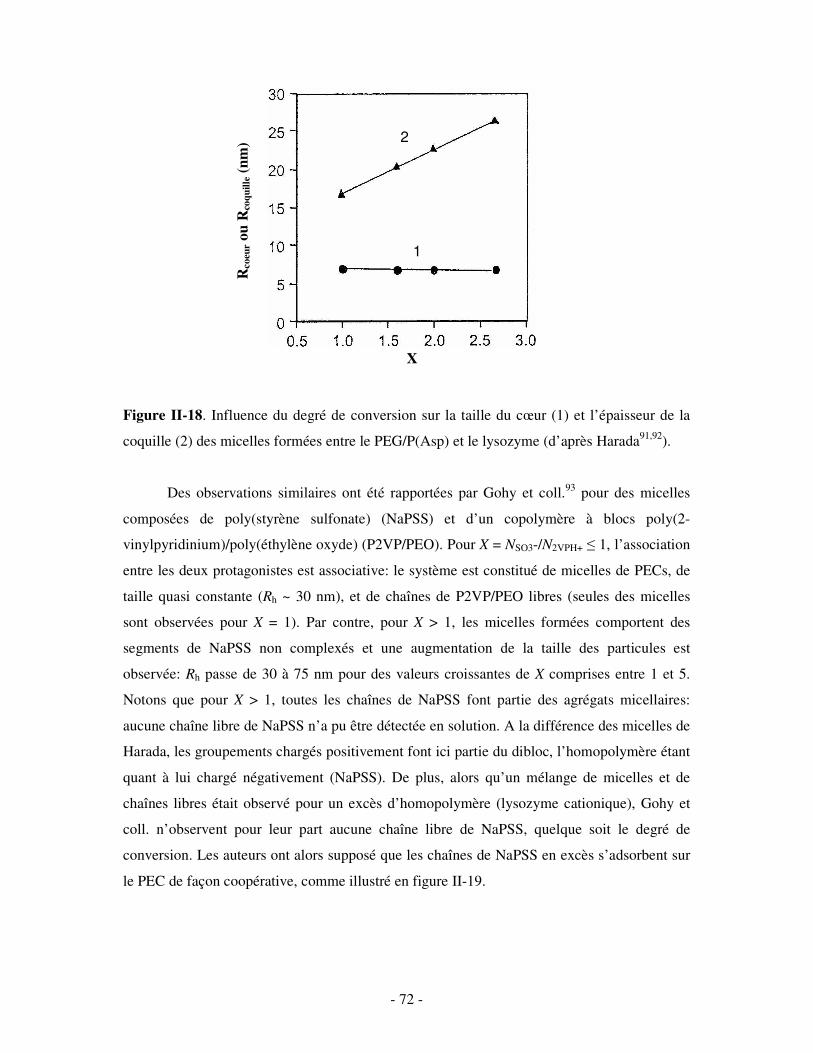
- 72 -
Figure II-18. Influence du degré de conversion sur la taille du cœur (1) et l’épaisseur de la
coquille (2) des micelles formées entre le PEG/P(Asp) et le lysozyme (d’après Harada91,92).
Des observations similaires ont été rapportées par Gohy et coll.93 pour des micelles
composées de poly(styrène sulfonate) (NaPSS) et d’un copolymère à blocs poly(2-
vinylpyridinium)/poly(éthylène oxyde) (P2VP/PEO). Pour X = NSO3-/N2VPH+ 1, l’association
entre les deux protagonistes est associative: le système est constitué de micelles de PECs, de
taille quasi constante (Rh ~ 30 nm), et de chaînes de P2VP/PEO libres (seules des micelles
sont observées pour X = 1). Par contre, pour X > 1, les micelles formées comportent des
segments de NaPSS non complexés et une augmentation de la taille des particules est
observée: Rh passe de 30 à 75 nm pour des valeurs croissantes de X comprises entre 1 et 5.
Notons que pour X > 1, toutes les chaînes de NaPSS font partie des agrégats micellaires:
aucune chaîne libre de NaPSS n’a pu être détectée en solution. A la différence des micelles de
Harada, les groupements chargés positivement font ici partie du dibloc, l’homopolymère étant
quant à lui chargé négativement (NaPSS). De plus, alors qu’un mélange de micelles et de
chaînes libres était observé pour un excès d’homopolymère (lysozyme cationique), Gohy et
coll. n’observent pour leur part aucune chaîne libre de NaPSS, quelque soit le degré de
conversion. Les auteurs ont alors supposé que les chaînes de NaPSS en excès s’adsorbent sur
le PEC de façon coopérative, comme illustré en figure II-19.
Rco
eur o
u R
coqu
ille (
nm)
X
1
2

- 73 -
Figure II-19. Schématisation des structures micellaires en fonction du degré de conversion93.
c) Influence du sel
Comme déjà vu au paragraphe II-1, le mélange non stoechiométrique de PELs de
charges opposées, portant des groupements ioniques faibles et de masses molaires très
différentes, résulte en la formation de PECs solubles79,80,94, le PEL de grande masse étant
l’« hôte », celui de plus petite masse l’« invité ». La structure des PECs formés est
schématisée en figure II-9: les blocs hydrophobes résultant de l’association entre segments de
chaînes hôtes/invités sont rassemblés dans des domaines compacts, stabilisés par les segments
hôtes non complexés et hydrophiles (chargés). Kabanov et Zezin79,80 ont évalué l’influence du
sel sur la structure de différents systèmes formés de la sorte. Les PECs constitués d’acide
poly(acrylique) (NaPA, Mw = 3.105 g.mol-1) et de PEI linéaire (Mw = 1,6.104 g.mol-1), deux
PELs linéaires flexibles, en sont un exemple. La figure II-20 présente la courbe de titrage
turbidimétrique d’une solution aqueuse de ces complexes par une solution de NaCl.
- - - -
- -
+ +
+ +
+ + +
+ +
+ + +
- -
- -
- -
- -
- -
- - --
+ +
- - - -
- - - -
- - - -
- -
- - -
- - -
- - - -
- - - -
- -
: bloc PEG : bloc P2VP : NaPSS
X < 1 X > 1
+ +
+ +
+ + +
+ +
+
+ +
- -
- -
- -
- -
- -
- - - -
+ +

- 74 -
Figure II-20. Courbe de titrage turbidimétrique d’une solution de PECs NaPA/PEI, de
composition X = Ninvité/Nhôte = 0,5, par une solution de NaCl (NPEC = 2.10-3 M, pH = 7, T =
20°C)79.
Sur cette figure, trois régions correspondant à des états de phase différents du système
sont distinguables. Dans la région I, le système est totalement homogène. Lorsque la force
ionique atteint la valeur critique I1, le système devient turbide. La turbidité augmente avec la
concentration en sel et le système reste hétérogène dans toute la région II. Puis, pour une
certaine force ionique I2, le système redevient homogène et le reste dans toute la région III. A
l’aide de la diffusion de la lumière, Kabanov et Zezin ont montré que dans la région I le
complexe subit d’importants changements conformationnels: pour des valeurs croissantes de
NNaCl comprises entre 0,05 et 0,2 M, le rayon de giration des particules chute de 700 à 270 Å,
alors que leur masse reste pratiquement constante (Mw ~ 3.105 g.mol-1, soit environ 1 chaîne
hôte par agrégat). Cette augmentation de compacité a été attribuée à une redistribution des
liaisons électrostatiques réversibles hôte/invité au sein des particules de PECs. En provoquant
la dissociation d’une certaine fraction des liaisons interchaînes, l’ajout de sel favorise la
réalisation de structures contenant de nombreuses boucles, comme celles schématisées en
figure II-21b. Ces boucles sont le résultat d’une compaction de grands segments de la chaîne
hôte le long des chaînes invitées plus courtes et, par conséquent, contribuent largement à la
chute des dimensions du PEC observée.
Tur
bidi
té
NNaCl (M)
I II III

- 75 -
Figure II-21. Influence du sel sur la structure des PECs séquencés, selon Kabanov79.
Dans la région II (figure II-20), une séparation de phases est observée: un précipité
constitué de PECs stoechiométriques coexiste avec une solution de PECs non
stoechiométriques. Le passage de la région I à la région II est donc marqué par une
redistribution des chaînes invitées entre les particules de PECs. Ceci met en évidence le
caractère réversible des interactions à l’origine de la formation des PECs. En présence de sel,
les chaînes invitées peuvent migrer le long des chaînes hôtes et se déplacer d’une chaîne à
l’autre.
Enfin, dans la région III, le système redevient homogène suite à un écrantage complet
de l’ensemble des liaisons électrostatiques intermoléculaires. A concentration en sel
suffisamment élevée (I > I2), les PECs sont ainsi totalement dissociés en leurs composants
individuels.
Les auteurs ont observé un comportement similaire pour des PECs non
stoechiométriques composés de poly(méthacrylate) de sodium (NaPMA) et de bromure de
poly(N-éthyl-4-vinylpyridinium) (PVP)80: le système passe successivement par les régions I,
II et III à mesure que la concentration en NaBr augmente. Il est à noter, cependant, que la
position des frontières entre les trois régions (c’est-à-dire les valeurs I1 et I2) dépend fortement
de la nature des composants PELs et de celle des contre-ions ajoutés95. Dans le cas du système
NaPA/PEI, I2 correspond à NNaCl = 1,1 M contre NNaBr = 0,45 M pour le système
NaPMA/PVP. En mélangeant dans l’eau un alcool poly(vinylique) partiellement
hôte
invité
hôte
invité
COO- +NH2
NaCl
a) b)
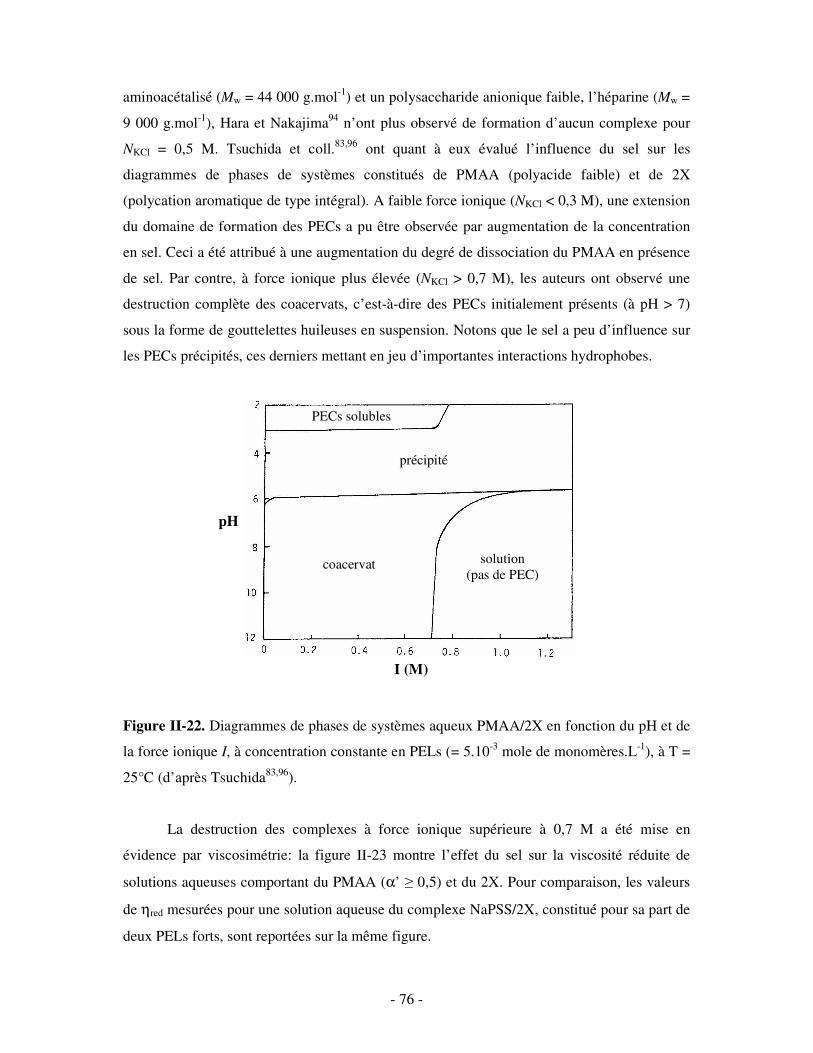
- 76 -
aminoacétalisé (Mw = 44 000 g.mol-1) et un polysaccharide anionique faible, l’héparine (Mw =
9 000 g.mol-1), Hara et Nakajima94 n’ont plus observé de formation d’aucun complexe pour
NKCl = 0,5 M. Tsuchida et coll.83,96 ont quant à eux évalué l’influence du sel sur les
diagrammes de phases de systèmes constitués de PMAA (polyacide faible) et de 2X
(polycation aromatique de type intégral). A faible force ionique (NKCl < 0,3 M), une extension
du domaine de formation des PECs a pu être observée par augmentation de la concentration
en sel. Ceci a été attribué à une augmentation du degré de dissociation du PMAA en présence
de sel. Par contre, à force ionique plus élevée (NKCl > 0,7 M), les auteurs ont observé une
destruction complète des coacervats, c’est-à-dire des PECs initialement présents (à pH > 7)
sous la forme de gouttelettes huileuses en suspension. Notons que le sel a peu d’influence sur
les PECs précipités, ces derniers mettant en jeu d’importantes interactions hydrophobes.
Figure II-22. Diagrammes de phases de systèmes aqueux PMAA/2X en fonction du pH et de
la force ionique I, à concentration constante en PELs (= 5.10-3 mole de monomères.L-1), à T =
25°C (d’après Tsuchida83,96).
La destruction des complexes à force ionique supérieure à 0,7 M a été mise en
évidence par viscosimétrie: la figure II-23 montre l’effet du sel sur la viscosité réduite de
solutions aqueuses comportant du PMAA (α’ 0,5) et du 2X. Pour comparaison, les valeurs
de ηred mesurées pour une solution aqueuse du complexe NaPSS/2X, constitué pour sa part de
deux PELs forts, sont reportées sur la même figure.
pH
I (M)
PECs solubles
précipité
coacervat solution (pas de PEC)
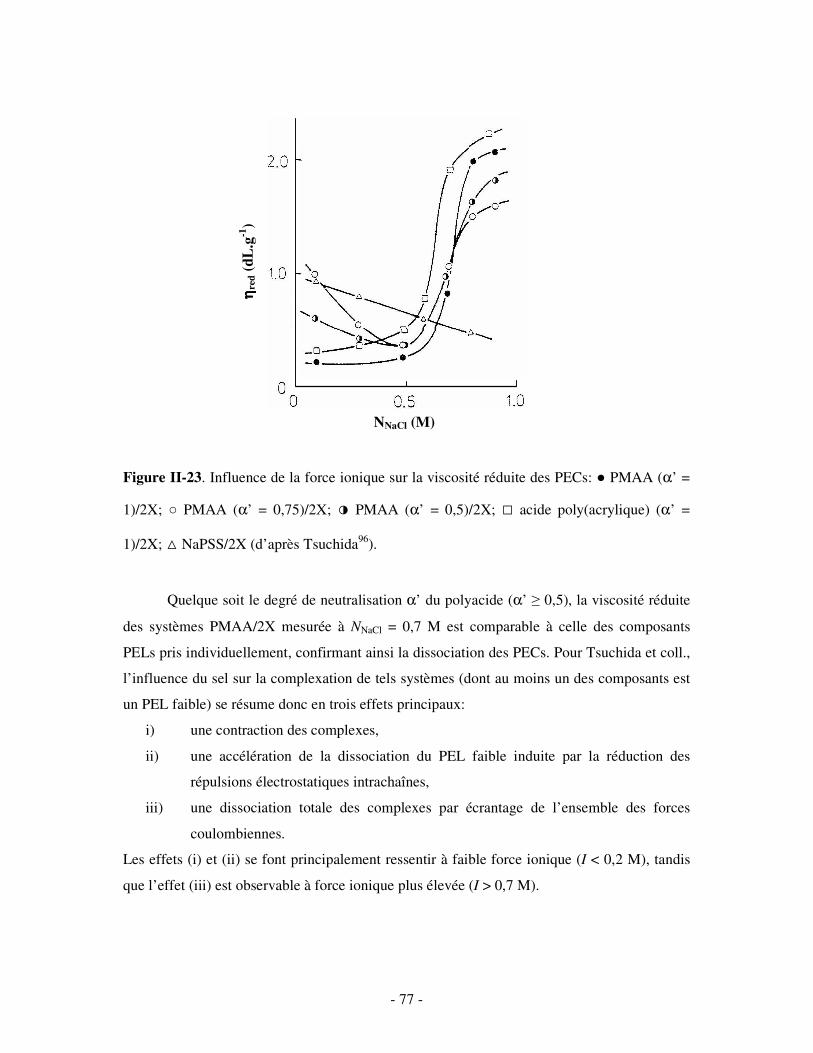
- 77 -
Figure II-23. Influence de la force ionique sur la viscosité réduite des PECs: PMAA (α’ =
1)/2X; PMAA (α’ = 0,75)/2X; PMAA (α’ = 0,5)/2X; acide poly(acrylique) (α’ =
1)/2X; NaPSS/2X (d’après Tsuchida96).
Quelque soit le degré de neutralisation α’ du polyacide (α’ 0,5), la viscosité réduite
des systèmes PMAA/2X mesurée à NNaCl = 0,7 M est comparable à celle des composants
PELs pris individuellement, confirmant ainsi la dissociation des PECs. Pour Tsuchida et coll.,
l’influence du sel sur la complexation de tels systèmes (dont au moins un des composants est
un PEL faible) se résume donc en trois effets principaux:
i) une contraction des complexes,
ii) une accélération de la dissociation du PEL faible induite par la réduction des
répulsions électrostatiques intrachaînes,
iii) une dissociation totale des complexes par écrantage de l’ensemble des forces
coulombiennes.
Les effets (i) et (ii) se font principalement ressentir à faible force ionique (I < 0,2 M), tandis
que l’effet (iii) est observable à force ionique plus élevée (I > 0,7 M).
ηη ηη red
(dL
.g-1
)
NNaCl (M)

- 78 -
De même que la viscosimétrie, la fluorimétrie est une technique efficace pour mettre
en évidence l’effet (iii). Dans cette optique, Itoh et coll.97 ont attaché de façon covalente des
groupements fluorescents (anthryle) sur trois polyanions de type pendant: le PMAA
(polyacide faible), le NaPAMPS et le NaPSS (polyacides forts).
* CH2 CH CH2 C *
CH3
C O
NH
C CH3H3C
CH2
SO3- Na+
C O
O
CH2
99,8 0,2
Figure II-24. Structure chimique du NaPAMPS modifié à 0,2 % en moles par des
groupements anthrylméthyl méthacrylate (AMMA).
Les auteurs ont pu suivre la formation de PECs entre ces polyanions modifiés et une
série de polycations dont le 3X, de type intégral, et le PVBMA, de type pendant (cf. tableau
II-1). En favorisant la formation de microdomaines hydrophobes et en réduisant les
mouvements moléculaires de la sonde, le phénomène de complexation se traduit en effet par
une augmentation de l’intensité de fluorescence, If, émise par cette dernière.
Pour les systèmes PMAA/3X et PMAA/PVBMA, un retour aux valeurs initiales de If
(celles correspondantes aux polyanions seuls en solution) a pu être observé pour NNaCl 0,7
M, indiquant une dissociation totale des PECs en leurs composants individuels. Par contre,
une augmentation de la force ionique a peu d’effet sur les systèmes comportant le NaPAMPS
et le NaPSS: If reste quasiment constante même pour NNaCl = 0,9 M. Ces résultats sont donc
cohérents avec ceux obtenus par Tsuchida et Abe96 par viscosimétrie: les PECs constitués de
poly(carboxylates) (polyacides faibles) et des polycations cités précédemment sont
dissociables pour des forces ioniques supérieures à 0,7 M, tandis que les PECs constitués de
polyacide et polybase forts sont stables à force ionique relativement élevée (cf. figure II-23).
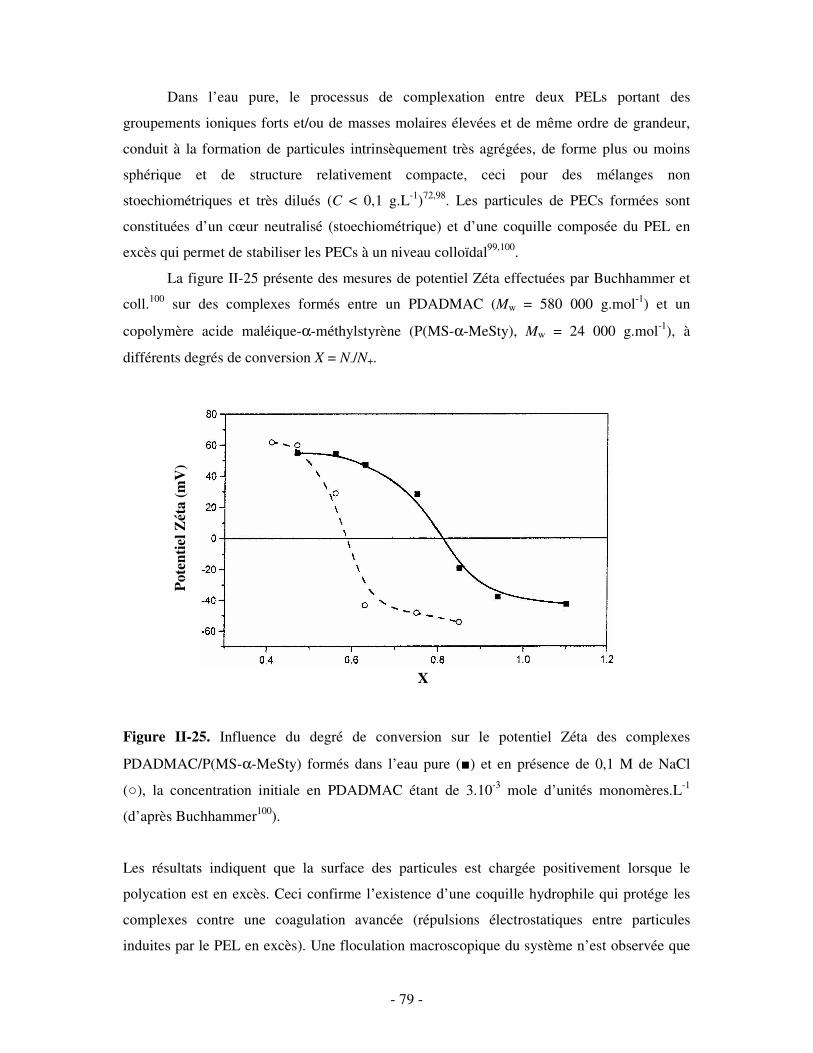
- 79 -
Dans l’eau pure, le processus de complexation entre deux PELs portant des
groupements ioniques forts et/ou de masses molaires élevées et de même ordre de grandeur,
conduit à la formation de particules intrinsèquement très agrégées, de forme plus ou moins
sphérique et de structure relativement compacte, ceci pour des mélanges non
stoechiométriques et très dilués (C < 0,1 g.L-1)72,98. Les particules de PECs formées sont
constituées d’un cœur neutralisé (stoechiométrique) et d’une coquille composée du PEL en
excès qui permet de stabiliser les PECs à un niveau colloïdal99,100.
La figure II-25 présente des mesures de potentiel Zéta effectuées par Buchhammer et
coll.100 sur des complexes formés entre un PDADMAC (Mw = 580 000 g.mol-1) et un
copolymère acide maléique-α-méthylstyrène (P(MS-α-MeSty), Mw = 24 000 g.mol-1), à
différents degrés de conversion X = N-/N+.
Figure II-25. Influence du degré de conversion sur le potentiel Zéta des complexes
PDADMAC/P(MS-α-MeSty) formés dans l’eau pure () et en présence de 0,1 M de NaCl
(), la concentration initiale en PDADMAC étant de 3.10-3 mole d’unités monomères.L-1
(d’après Buchhammer100).
Les résultats indiquent que la surface des particules est chargée positivement lorsque le
polycation est en excès. Ceci confirme l’existence d’une coquille hydrophile qui protége les
complexes contre une coagulation avancée (répulsions électrostatiques entre particules
induites par le PEL en excès). Une floculation macroscopique du système n’est observée que
X
Pote
ntie
l Zét
a (m
V)

- 80 -
pour un degré de conversion proche de 1 (X = 1,1 dans l’eau pure), soit lorsque les particules
de PEC sont quasiment neutralisées.
Selon Dautzenberg, le niveau d’agrégation des PECs peut être contrôlé par la
concentration des composants PELs72, mais également par la force ionique du milieu98,99. La
formation de PECs solubles entre un NaPSS (Mw = 66 000 g.mol-1) et un PDADMAC (Mw =
250 000 g.mol-1) en milieu très dilué (CNaPSS < 5.10-2 g.L-1), a été suivie par l’auteur en
fonction du degré de conversion et à différentes forces ioniques, au moyen de la diffusion de
la lumière. Les résultats de cette étude peuvent être résumés comme suit. Dans l’eau pure, un
niveau d’agrégation très élevé, avec plus de 1000 chaînes par complexe, a pu être constaté, la
floculation du système survenant pour X = 1. En présence d’une faible quantité de sel (I 10-
2 M), le niveau d’agrégation des complexes chute d’un facteur 30. Puis, pour NNaCl = 0,1 M, le
niveau d’agrégation augmente à nouveau et augmente avec X jusqu’à ce que la floculation du
système soit observée pour X = 0,8. Notons que, dans ce cas, la formation des complexes
dépend peu de l’ordre du mélange (PDADMAC/NaPSS ou NaPSS/PDADMAC).
L’influence du sel sur les paramètres structuraux du système PDADMAC/NaPSS a été
étudiée plus en détails pour un degré de conversion fixé à 0,5. La figure II-26 présente ainsi
l’évolution de la masse et de la taille des particules de PECs formées en fonction de la force
ionique.
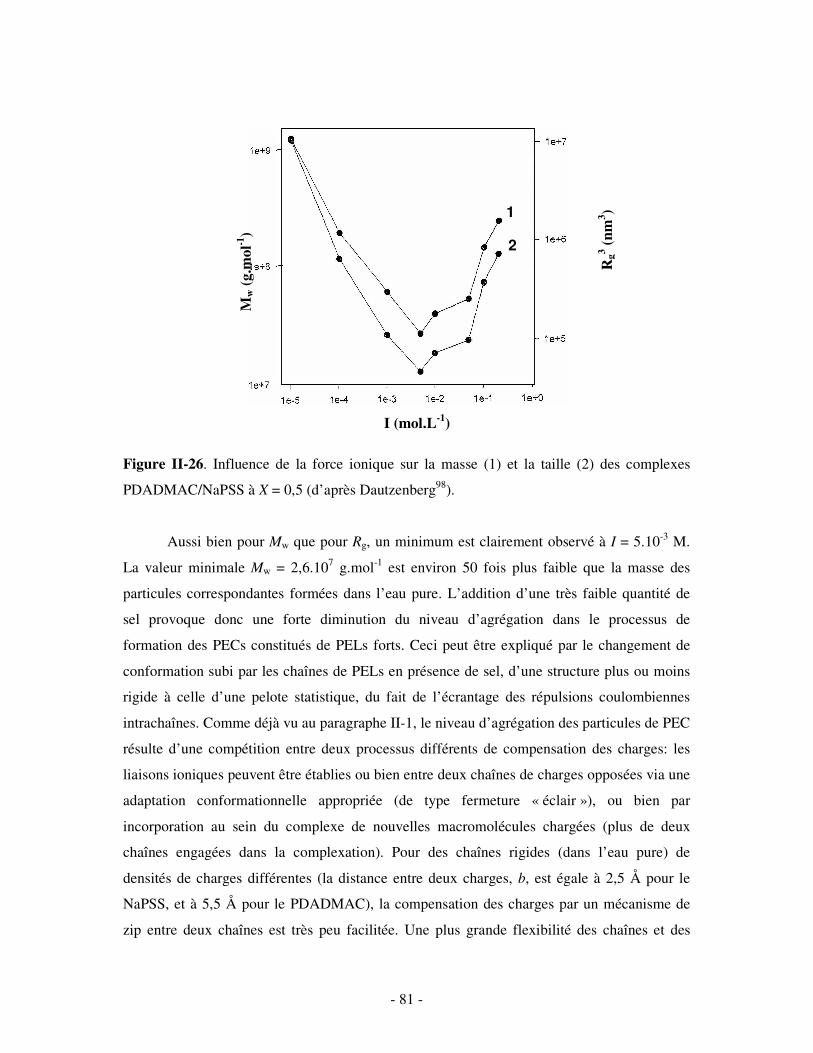
- 81 -
Figure II-26. Influence de la force ionique sur la masse (1) et la taille (2) des complexes
PDADMAC/NaPSS à X = 0,5 (d’après Dautzenberg98).
Aussi bien pour Mw que pour Rg, un minimum est clairement observé à I = 5.10-3 M.
La valeur minimale Mw = 2,6.107 g.mol-1 est environ 50 fois plus faible que la masse des
particules correspondantes formées dans l’eau pure. L’addition d’une très faible quantité de
sel provoque donc une forte diminution du niveau d’agrégation dans le processus de
formation des PECs constitués de PELs forts. Ceci peut être expliqué par le changement de
conformation subi par les chaînes de PELs en présence de sel, d’une structure plus ou moins
rigide à celle d’une pelote statistique, du fait de l’écrantage des répulsions coulombiennes
intrachaînes. Comme déjà vu au paragraphe II-1, le niveau d’agrégation des particules de PEC
résulte d’une compétition entre deux processus différents de compensation des charges: les
liaisons ioniques peuvent être établies ou bien entre deux chaînes de charges opposées via une
adaptation conformationnelle appropriée (de type fermeture « éclair »), ou bien par
incorporation au sein du complexe de nouvelles macromolécules chargées (plus de deux
chaînes engagées dans la complexation). Pour des chaînes rigides (dans l’eau pure) de
densités de charges différentes (la distance entre deux charges, b, est égale à 2,5 Å pour le
NaPSS, et à 5,5 Å pour le PDADMAC), la compensation des charges par un mécanisme de
zip entre deux chaînes est très peu facilitée. Une plus grande flexibilité des chaînes et des
Rg3 (n
m3 )
Mw (g
.mol
-1)
I (mol.L-1)
1
2

- 82 -
structures en pelote statistique (conformation adoptée en présence de sel) favorisent
l’adaptation entre deux chaînes et, par conséquent, conduisent à des niveaux d’agrégation plus
faibles.
A mesure que la force ionique augmente, les répulsions entre les coquilles constituées
du PEL en excès autour des particules de PEC sont écrantées, ce qui favorise une agrégation
secondaire pour X < 1. Sur la figure II-26, cette coagulation se traduit par une forte
augmentation de la masse et de la taille des agrégats au-delà de I = 5.10-3 M. A concentration
élevée en sel, la floculation du système peut donc être observée pour des degrés de conversion
inférieurs à 1. D’après la figure II-25, les complexes PDADMAC/P(MS-α-MeSty) floculent
par exemple pour X 0,8 en présence de 0,1 M de NaCl. Pour des systèmes constitués d’un
polycation de type intégral comportant des unités de chlorure de N,N-diméthyl-2-
hydroxypropylène ammonium (le PCA5 présenté en figure II-27), et d’un poly(acrylate) de
sodium (NaPA), Dragan et Cristea95 ont également observé une séparation de phases du
système à des degrés de conversion X = N-/N+ de plus en plus faibles pour des concentrations
en sel de plus en plus élevées.
* N+
CH3
CH3
CH2Cl-
CH
OH
CH2 N CH2
CH2
N+
3
H3C CH3
CH2 CH CH2
OH
Cl-
CH CH2
OH
*
0,95 0,05
n
Figure II-27. Structure chimique du PCA5.
Selon les auteurs, l’effet d’écran des contre-ions en excès induit une réduction des
sites ioniques disponibles pour former des liaisons électrostatiques entre les chaînes de
polymères. Par conséquent, plus la concentration en sel est élevée, plus la quantité de NaPA
inclus dans le PEC diminue et plus la neutralisation est observée à Xsép faible (Xsép est le degré
de conversion correspondant à la séparation de phases). La décroissance de Xsép avec
l’augmentation de la force ionique est observée tant que NNaCl reste inférieure à 2 M. Au-delà,
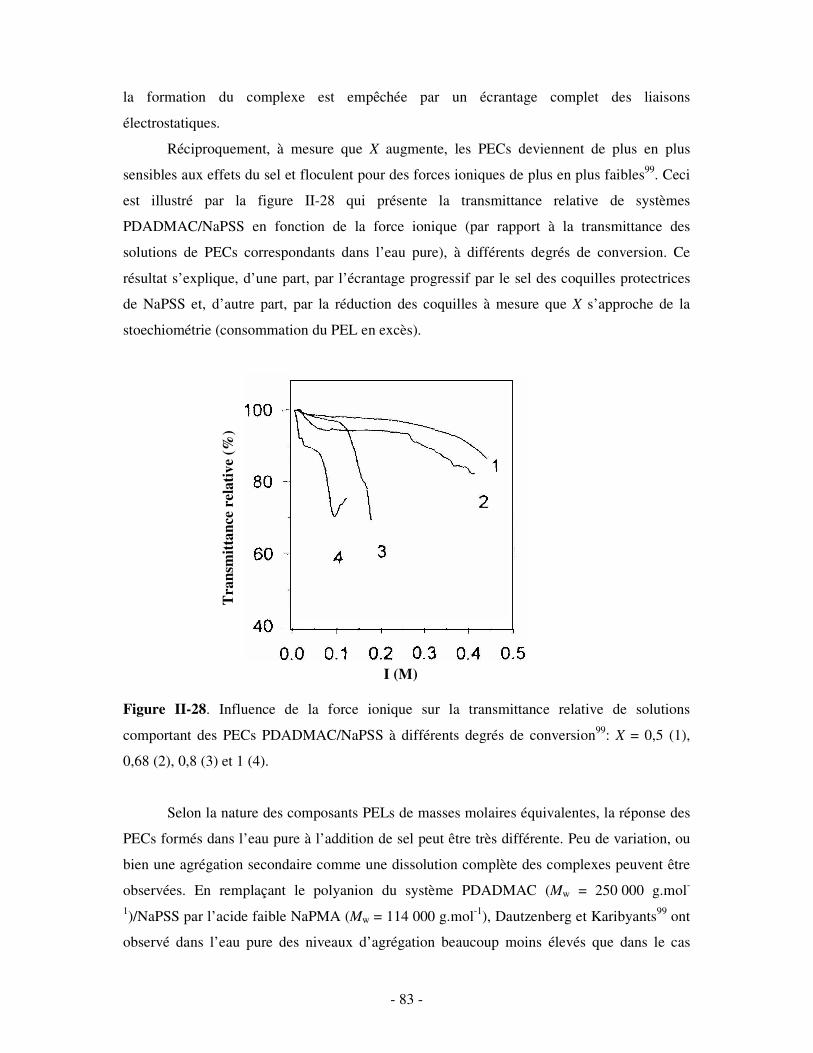
- 83 -
la formation du complexe est empêchée par un écrantage complet des liaisons
électrostatiques.
Réciproquement, à mesure que X augmente, les PECs deviennent de plus en plus
sensibles aux effets du sel et floculent pour des forces ioniques de plus en plus faibles99. Ceci
est illustré par la figure II-28 qui présente la transmittance relative de systèmes
PDADMAC/NaPSS en fonction de la force ionique (par rapport à la transmittance des
solutions de PECs correspondants dans l’eau pure), à différents degrés de conversion. Ce
résultat s’explique, d’une part, par l’écrantage progressif par le sel des coquilles protectrices
de NaPSS et, d’autre part, par la réduction des coquilles à mesure que X s’approche de la
stoechiométrie (consommation du PEL en excès).
Figure II-28. Influence de la force ionique sur la transmittance relative de solutions
comportant des PECs PDADMAC/NaPSS à différents degrés de conversion99: X = 0,5 (1),
0,68 (2), 0,8 (3) et 1 (4).
Selon la nature des composants PELs de masses molaires équivalentes, la réponse des
PECs formés dans l’eau pure à l’addition de sel peut être très différente. Peu de variation, ou
bien une agrégation secondaire comme une dissolution complète des complexes peuvent être
observées. En remplaçant le polyanion du système PDADMAC (Mw = 250 000 g.mol-
1)/NaPSS par l’acide faible NaPMA (Mw = 114 000 g.mol-1), Dautzenberg et Karibyants99 ont
observé dans l’eau pure des niveaux d’agrégation beaucoup moins élevés que dans le cas
I (M)
Tra
nsm
ittan
ce r
elat
ive
(%)
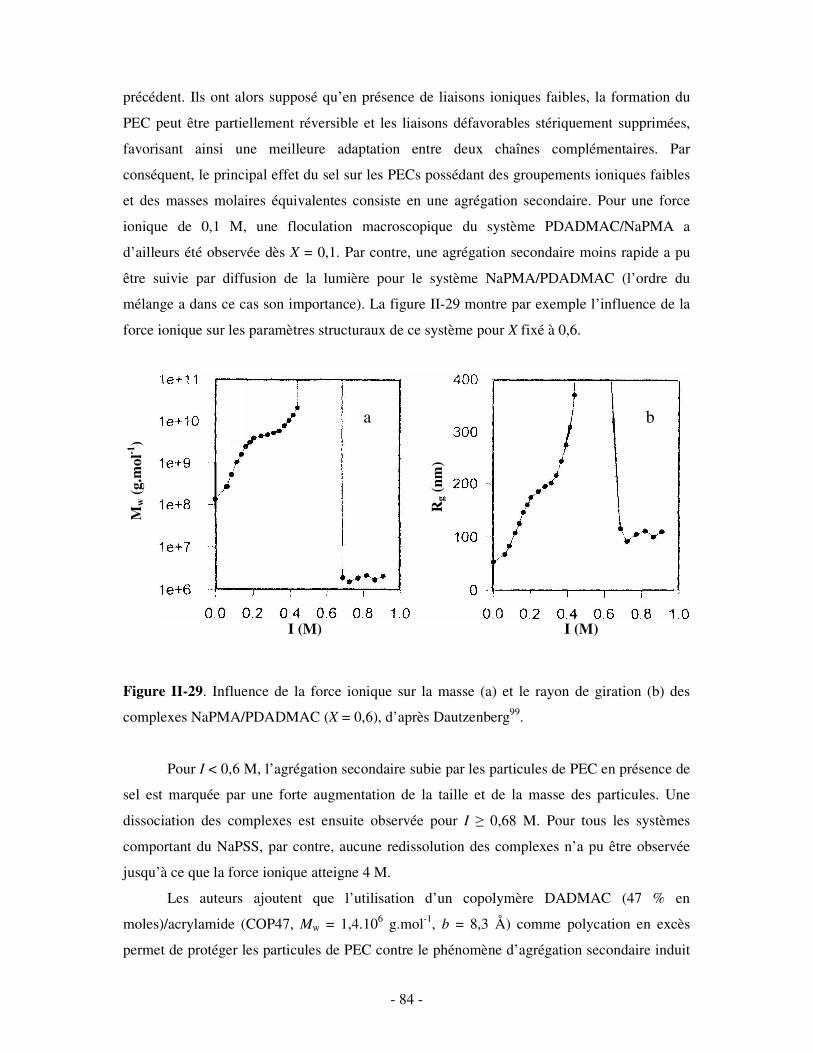
- 84 -
précédent. Ils ont alors supposé qu’en présence de liaisons ioniques faibles, la formation du
PEC peut être partiellement réversible et les liaisons défavorables stériquement supprimées,
favorisant ainsi une meilleure adaptation entre deux chaînes complémentaires. Par
conséquent, le principal effet du sel sur les PECs possédant des groupements ioniques faibles
et des masses molaires équivalentes consiste en une agrégation secondaire. Pour une force
ionique de 0,1 M, une floculation macroscopique du système PDADMAC/NaPMA a
d’ailleurs été observée dès X = 0,1. Par contre, une agrégation secondaire moins rapide a pu
être suivie par diffusion de la lumière pour le système NaPMA/PDADMAC (l’ordre du
mélange a dans ce cas son importance). La figure II-29 montre par exemple l’influence de la
force ionique sur les paramètres structuraux de ce système pour X fixé à 0,6.
Figure II-29. Influence de la force ionique sur la masse (a) et le rayon de giration (b) des
complexes NaPMA/PDADMAC (X = 0,6), d’après Dautzenberg99.
Pour I < 0,6 M, l’agrégation secondaire subie par les particules de PEC en présence de
sel est marquée par une forte augmentation de la taille et de la masse des particules. Une
dissociation des complexes est ensuite observée pour I 0,68 M. Pour tous les systèmes
comportant du NaPSS, par contre, aucune redissolution des complexes n’a pu être observée
jusqu’à ce que la force ionique atteigne 4 M.
Les auteurs ajoutent que l’utilisation d’un copolymère DADMAC (47 % en
moles)/acrylamide (COP47, Mw = 1,4.106 g.mol-1, b = 8,3 Å) comme polycation en excès
permet de protéger les particules de PEC contre le phénomène d’agrégation secondaire induit
Mw (g
.mol
-1)
Rg (
nm)
I (M) I (M)
a b
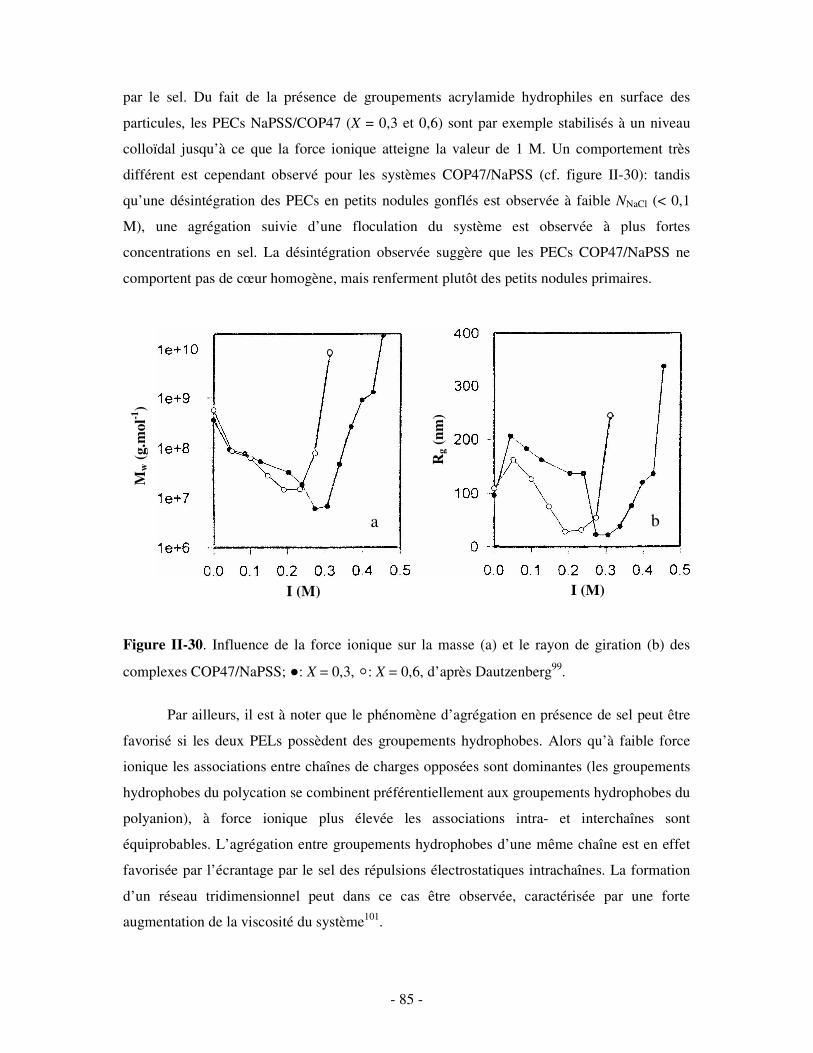
- 85 -
par le sel. Du fait de la présence de groupements acrylamide hydrophiles en surface des
particules, les PECs NaPSS/COP47 (X = 0,3 et 0,6) sont par exemple stabilisés à un niveau
colloïdal jusqu’à ce que la force ionique atteigne la valeur de 1 M. Un comportement très
différent est cependant observé pour les systèmes COP47/NaPSS (cf. figure II-30): tandis
qu’une désintégration des PECs en petits nodules gonflés est observée à faible NNaCl (< 0,1
M), une agrégation suivie d’une floculation du système est observée à plus fortes
concentrations en sel. La désintégration observée suggère que les PECs COP47/NaPSS ne
comportent pas de cœur homogène, mais renferment plutôt des petits nodules primaires.
Figure II-30. Influence de la force ionique sur la masse (a) et le rayon de giration (b) des
complexes COP47/NaPSS; : X = 0,3, : X = 0,6, d’après Dautzenberg99.
Par ailleurs, il est à noter que le phénomène d’agrégation en présence de sel peut être
favorisé si les deux PELs possèdent des groupements hydrophobes. Alors qu’à faible force
ionique les associations entre chaînes de charges opposées sont dominantes (les groupements
hydrophobes du polycation se combinent préférentiellement aux groupements hydrophobes du
polyanion), à force ionique plus élevée les associations intra- et interchaînes sont
équiprobables. L’agrégation entre groupements hydrophobes d’une même chaîne est en effet
favorisée par l’écrantage par le sel des répulsions électrostatiques intrachaînes. La formation
d’un réseau tridimensionnel peut dans ce cas être observée, caractérisée par une forte
augmentation de la viscosité du système101.
I (M) I (M)
Mw (g
.mol
-1)
Rg (
nm)
a b
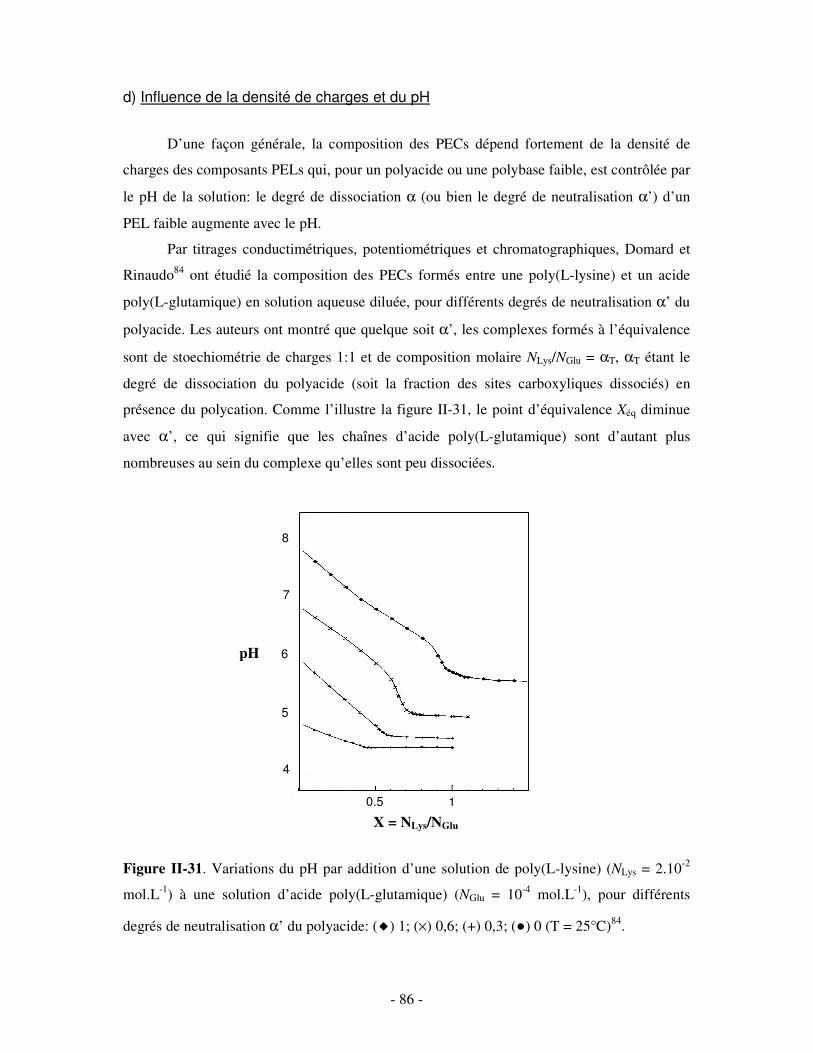
- 86 -
d) Influence de la densité de charges et du pH
D’une façon générale, la composition des PECs dépend fortement de la densité de
charges des composants PELs qui, pour un polyacide ou une polybase faible, est contrôlée par
le pH de la solution: le degré de dissociation α (ou bien le degré de neutralisation α’) d’un
PEL faible augmente avec le pH.
Par titrages conductimétriques, potentiométriques et chromatographiques, Domard et
Rinaudo84 ont étudié la composition des PECs formés entre une poly(L-lysine) et un acide
poly(L-glutamique) en solution aqueuse diluée, pour différents degrés de neutralisation α’ du
polyacide. Les auteurs ont montré que quelque soit α’, les complexes formés à l’équivalence
sont de stoechiométrie de charges 1:1 et de composition molaire NLys/NGlu = αT, αT étant le
degré de dissociation du polyacide (soit la fraction des sites carboxyliques dissociés) en
présence du polycation. Comme l’illustre la figure II-31, le point d’équivalence Xéq diminue
avec α’, ce qui signifie que les chaînes d’acide poly(L-glutamique) sont d’autant plus
nombreuses au sein du complexe qu’elles sont peu dissociées.
Figure II-31. Variations du pH par addition d’une solution de poly(L-lysine) (NLys = 2.10-2
mol.L-1) à une solution d’acide poly(L-glutamique) (NGlu = 10-4 mol.L-1), pour différents
degrés de neutralisation α’ du polyacide: () 1; (×) 0,6; (+) 0,3; () 0 (T = 25°C)84.
pH
4
5
6
7
8
0.5 1
X = NLys/NGlu
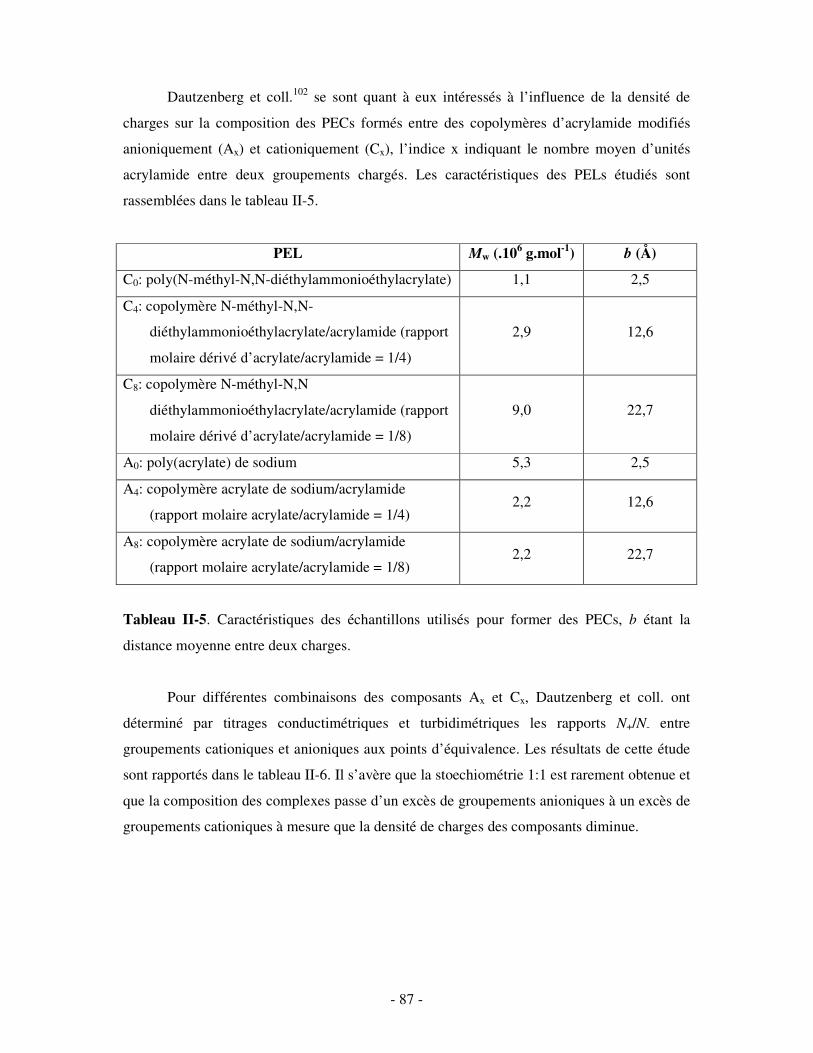
- 87 -
Dautzenberg et coll.102 se sont quant à eux intéressés à l’influence de la densité de
charges sur la composition des PECs formés entre des copolymères d’acrylamide modifiés
anioniquement (Ax) et cationiquement (Cx), l’indice x indiquant le nombre moyen d’unités
acrylamide entre deux groupements chargés. Les caractéristiques des PELs étudiés sont
rassemblées dans le tableau II-5.
PEL Mw (.106 g.mol-1) b (Å)
C0: poly(N-méthyl-N,N-diéthylammonioéthylacrylate) 1,1 2,5
C4: copolymère N-méthyl-N,N-
diéthylammonioéthylacrylate/acrylamide (rapport
molaire dérivé d’acrylate/acrylamide = 1/4)
2,9 12,6
C8: copolymère N-méthyl-N,N
diéthylammonioéthylacrylate/acrylamide (rapport
molaire dérivé d’acrylate/acrylamide = 1/8)
9,0 22,7
A0: poly(acrylate) de sodium 5,3 2,5
A4: copolymère acrylate de sodium/acrylamide
(rapport molaire acrylate/acrylamide = 1/4) 2,2 12,6
A8: copolymère acrylate de sodium/acrylamide
(rapport molaire acrylate/acrylamide = 1/8) 2,2 22,7
Tableau II-5. Caractéristiques des échantillons utilisés pour former des PECs, b étant la
distance moyenne entre deux charges.
Pour différentes combinaisons des composants Ax et Cx, Dautzenberg et coll. ont
déterminé par titrages conductimétriques et turbidimétriques les rapports N+/N- entre
groupements cationiques et anioniques aux points d’équivalence. Les résultats de cette étude
sont rapportés dans le tableau II-6. Il s’avère que la stoechiométrie 1:1 est rarement obtenue et
que la composition des complexes passe d’un excès de groupements anioniques à un excès de
groupements cationiques à mesure que la densité de charges des composants diminue.

- 88 -
C0 C4 C8
A0 0,58 0,72 1,11
A4 1,29 1,19 2,14
A8 0,95 1,60 2,11
Tableau II-6. Rapports stoechiométriques N+/N- aux points d’équivalence des courbes de
titrages conductimétriques et turbidimétriques (les polyanions sont les composants en excès
au sein des mélanges).
Afin d’obtenir des informations quantitatives sur la structure des particules de PECs
formées, des mesures de diffusion de lumière ont été effectuées en milieu très dilué ( 0,1
g.L-1) pour plusieurs combinaisons Cx/Ax. Pour les systèmes C0/A0 et C4/A4, par exemple, peu
de variations des paramètres structuraux (Mw et Rg) ont été observées77 jusqu’à ce que le degré
de conversion atteigne la valeur X = 0,8. Par contre, ceux du système C8/A8 qui implique les
densités de charges les plus faibles, s’avèrent être fortement dépendants du degré de
conversion, comme l’indique la figure II-17. Les trois systèmes C0/A0, C4/A4 et C8/A8 ont été
comparés dans des conditions identiques de concentration et de conversion: le tableau II-7
indique une réduction de la densité structurale des particules, soit une augmentation du degré
de gonflement, à mesure que la densité de charges des composants diminue.
Combinaison ρρρρ (.103 g.L-1) NNaClcrit(mol.L-1)
C0/A0 0,13-0,17 0,45
C4/A4 0,07-0,13 0,065-0,09
C8/A8 0,02-0,08 0-0,02
Tableau II-7. Densité structurale des agrégats (ρ) et concentration critique en sel
correspondant à la dissociation totale des PECs.
Il est à noter que sous l’influence du sel, ces trois systèmes se comportent de façon
identique. Comme le montre la figure II-32 pour le système C4/A4, une agrégation
accompagnée d’un gonflement des particules de PECs est tout d’abord observée à mesure que
la force ionique augmente. Puis, pour une certaine concentration critique en sel, les complexes
sont totalement dissociés. D’après le tableau II-7, cette concentration critique dépend
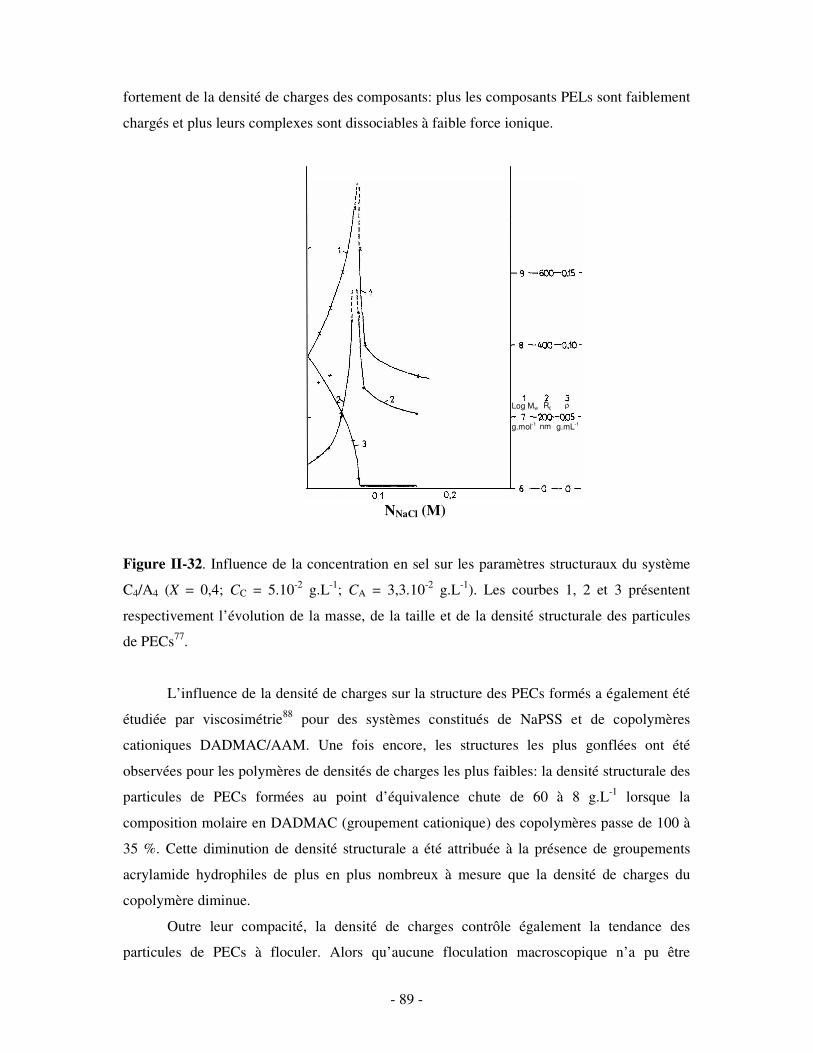
- 89 -
fortement de la densité de charges des composants: plus les composants PELs sont faiblement
chargés et plus leurs complexes sont dissociables à faible force ionique.
Figure II-32. Influence de la concentration en sel sur les paramètres structuraux du système
C4/A4 (X = 0,4; CC = 5.10-2 g.L-1; CA = 3,3.10-2 g.L-1). Les courbes 1, 2 et 3 présentent
respectivement l’évolution de la masse, de la taille et de la densité structurale des particules
de PECs77.
L’influence de la densité de charges sur la structure des PECs formés a également été
étudiée par viscosimétrie88 pour des systèmes constitués de NaPSS et de copolymères
cationiques DADMAC/AAM. Une fois encore, les structures les plus gonflées ont été
observées pour les polymères de densités de charges les plus faibles: la densité structurale des
particules de PECs formées au point d’équivalence chute de 60 à 8 g.L-1 lorsque la
composition molaire en DADMAC (groupement cationique) des copolymères passe de 100 à
35 %. Cette diminution de densité structurale a été attribuée à la présence de groupements
acrylamide hydrophiles de plus en plus nombreux à mesure que la densité de charges du
copolymère diminue.
Outre leur compacité, la densité de charges contrôle également la tendance des
particules de PECs à floculer. Alors qu’aucune floculation macroscopique n’a pu être
Log Mw Rg ρ
g.mol-1 nm g.mL-1
NNaCl (M)
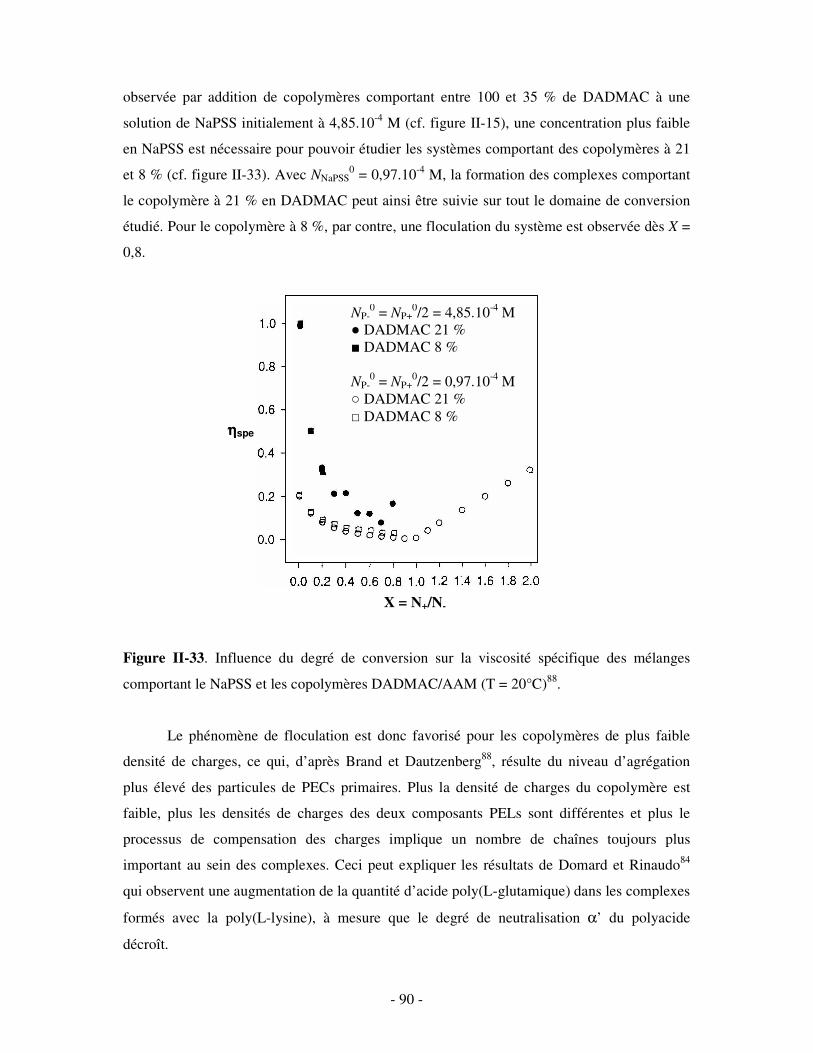
- 90 -
observée par addition de copolymères comportant entre 100 et 35 % de DADMAC à une
solution de NaPSS initialement à 4,85.10-4 M (cf. figure II-15), une concentration plus faible
en NaPSS est nécessaire pour pouvoir étudier les systèmes comportant des copolymères à 21
et 8 % (cf. figure II-33). Avec NNaPSS0 = 0,97.10-4 M, la formation des complexes comportant
le copolymère à 21 % en DADMAC peut ainsi être suivie sur tout le domaine de conversion
étudié. Pour le copolymère à 8 %, par contre, une floculation du système est observée dès X =
0,8.
Figure II-33. Influence du degré de conversion sur la viscosité spécifique des mélanges
comportant le NaPSS et les copolymères DADMAC/AAM (T = 20°C)88.
Le phénomène de floculation est donc favorisé pour les copolymères de plus faible
densité de charges, ce qui, d’après Brand et Dautzenberg88, résulte du niveau d’agrégation
plus élevé des particules de PECs primaires. Plus la densité de charges du copolymère est
faible, plus les densités de charges des deux composants PELs sont différentes et plus le
processus de compensation des charges implique un nombre de chaînes toujours plus
important au sein des complexes. Ceci peut expliquer les résultats de Domard et Rinaudo84
qui observent une augmentation de la quantité d’acide poly(L-glutamique) dans les complexes
formés avec la poly(L-lysine), à mesure que le degré de neutralisation α’ du polyacide
décroît.
NP-0 = NP+
0/2 = 4,85.10-4 M DADMAC 21 % DADMAC 8 % NP-
0 = NP+0/2 = 0,97.10-4 M
DADMAC 21 % DADMAC 8 %
X = N+/N-
ηηηηspe
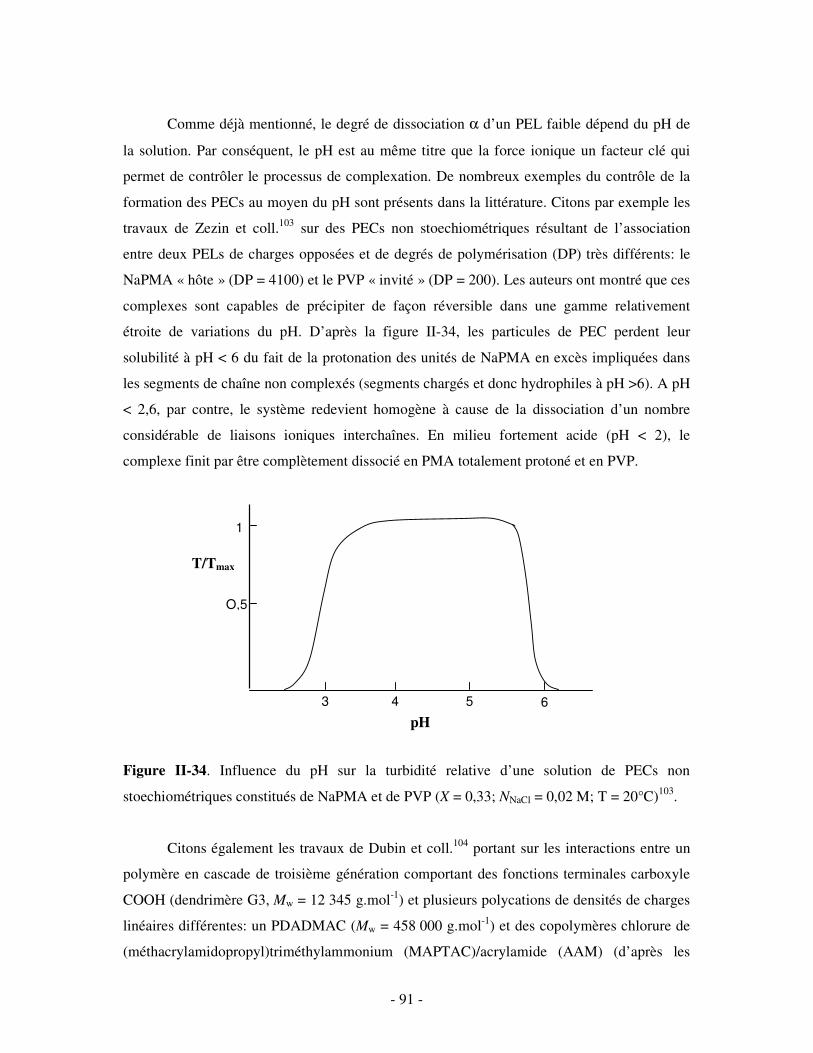
- 91 -
Comme déjà mentionné, le degré de dissociation α d’un PEL faible dépend du pH de
la solution. Par conséquent, le pH est au même titre que la force ionique un facteur clé qui
permet de contrôler le processus de complexation. De nombreux exemples du contrôle de la
formation des PECs au moyen du pH sont présents dans la littérature. Citons par exemple les
travaux de Zezin et coll.103 sur des PECs non stoechiométriques résultant de l’association
entre deux PELs de charges opposées et de degrés de polymérisation (DP) très différents: le
NaPMA « hôte » (DP = 4100) et le PVP « invité » (DP = 200). Les auteurs ont montré que ces
complexes sont capables de précipiter de façon réversible dans une gamme relativement
étroite de variations du pH. D’après la figure II-34, les particules de PEC perdent leur
solubilité à pH < 6 du fait de la protonation des unités de NaPMA en excès impliquées dans
les segments de chaîne non complexés (segments chargés et donc hydrophiles à pH >6). A pH
< 2,6, par contre, le système redevient homogène à cause de la dissociation d’un nombre
considérable de liaisons ioniques interchaînes. En milieu fortement acide (pH < 2), le
complexe finit par être complètement dissocié en PMA totalement protoné et en PVP.
Figure II-34. Influence du pH sur la turbidité relative d’une solution de PECs non
stoechiométriques constitués de NaPMA et de PVP (X = 0,33; NNaCl = 0,02 M; T = 20°C)103.
Citons également les travaux de Dubin et coll.104 portant sur les interactions entre un
polymère en cascade de troisième génération comportant des fonctions terminales carboxyle
COOH (dendrimère G3, Mw = 12 345 g.mol-1) et plusieurs polycations de densités de charges
linéaires différentes: un PDADMAC (Mw = 458 000 g.mol-1) et des copolymères chlorure de
(méthacrylamidopropyl)triméthylammonium (MAPTAC)/acrylamide (AAM) (d’après les
T/Tmax
pH
1
O,5
3 4 5 6
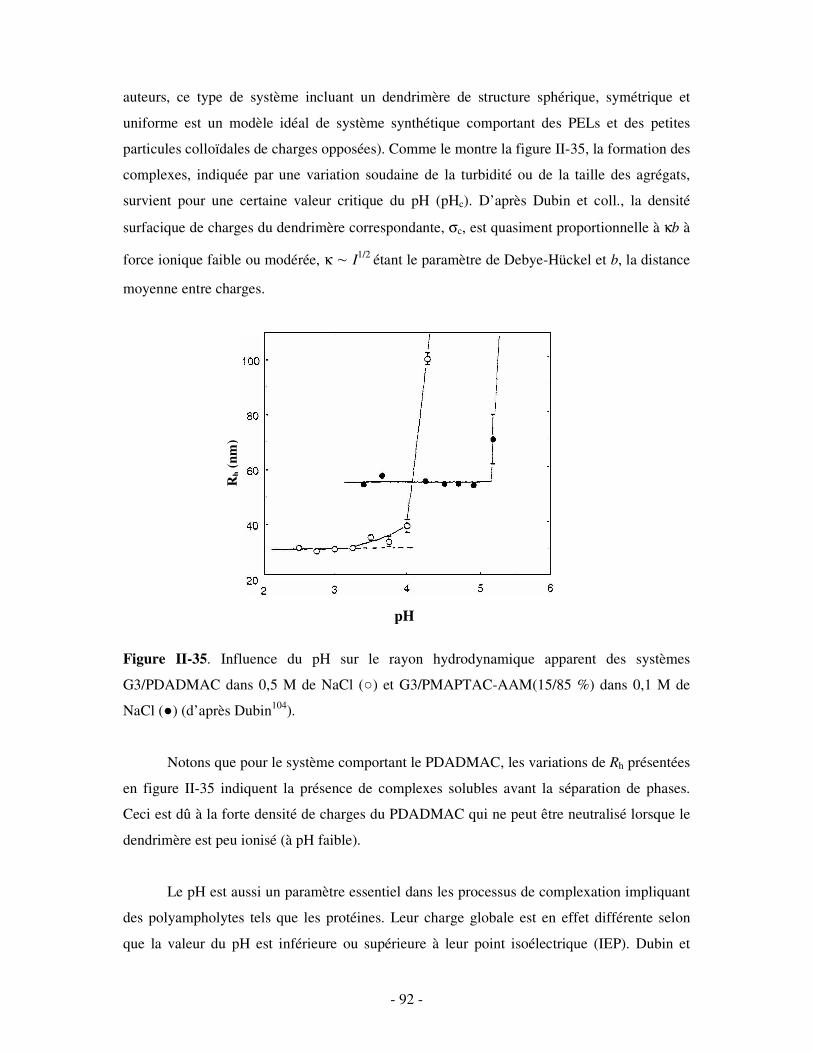
- 92 -
auteurs, ce type de système incluant un dendrimère de structure sphérique, symétrique et
uniforme est un modèle idéal de système synthétique comportant des PELs et des petites
particules colloïdales de charges opposées). Comme le montre la figure II-35, la formation des
complexes, indiquée par une variation soudaine de la turbidité ou de la taille des agrégats,
survient pour une certaine valeur critique du pH (pHc). D’après Dubin et coll., la densité
surfacique de charges du dendrimère correspondante, σc, est quasiment proportionnelle à κb à
force ionique faible ou modérée, κ I1/2 étant le paramètre de Debye-Hückel et b, la distance
moyenne entre charges.
Figure II-35. Influence du pH sur le rayon hydrodynamique apparent des systèmes
G3/PDADMAC dans 0,5 M de NaCl () et G3/PMAPTAC-AAM(15/85 %) dans 0,1 M de
NaCl () (d’après Dubin104).
Notons que pour le système comportant le PDADMAC, les variations de Rh présentées
en figure II-35 indiquent la présence de complexes solubles avant la séparation de phases.
Ceci est dû à la forte densité de charges du PDADMAC qui ne peut être neutralisé lorsque le
dendrimère est peu ionisé (à pH faible).
Le pH est aussi un paramètre essentiel dans les processus de complexation impliquant
des polyampholytes tels que les protéines. Leur charge globale est en effet différente selon
que la valeur du pH est inférieure ou supérieure à leur point isoélectrique (IEP). Dubin et
Rh (
nm)
pH

- 93 -
coll.105,106 ont par exemple étudié les interactions entre des PELs synthétiques tels que le
PDADMAC ou le NaPSS, et trois protéines de structure globulaire: l’albumine de sérum
bovin (BSA, IEP = 4,9), le lysozyme d’œuf de poule (IEP = 11,0) et la ribonucléase de
pancréas bovin (RNAse, IEP = 9,0). Pour chaque paire polymère/protéine, les auteurs ont tout
d’abord observé par turbidimétrie et diffusion de la lumière la formation de PECs solubles
pour une valeur de pH bien définie (pHc), les complexes étant alors stabilisés contre une
agrégation plus avancée de par leur charge globale non nulle. Dans le cas particulier de la
BSA et de polyanions synthétiques, Dubin et coll. ont observé la formation de complexes
même pour une charge globale de la protéine négative (pH > IEP). Les auteurs ont alors
supposé l’existence de régions riches en groupements basiques, interagissant fortement avec
le polyanion malgré une charge globale négative (distribution inhomogène des charges
positives au sein de la protéine). En faisant varier le pH (augmentation du pH dans le cas des
polycations, diminution dans le cas des polyanions), Dubin et coll. ont ensuite observé une
agrégation secondaire, induisant une séparation de phases pour une deuxième valeur de pH
bien définie (pHφ). A mesure que le pH varie, les protéines acquièrent en effet une charge
globale opposée à celle des polyions, la charge du complexe approchant alors
l’électroneutralité.
A l’aide de la diffusion de neutrons aux petits angles, Hone et coll.107 se sont
particulièrement intéressés aux interactions pouvant exister entre deux polyanions, le
NaPAMPS et le NaPSS, et un polyampholyte de conformation de type pelote statistique, la
gélatine. Alors que la diffusion de la lumière renseigne sur la taille globale des agrégats, la
diffusion de neutrons aux petits angles permet quant à elle d’accéder à leur structure interne.
Cette technique est en effet sensible à des structures de taille comparable à 1/q (soit à des
tailles comprises entre 10 et 100 Å dans cette étude), le vecteur de diffusion q étant égal à
2sin
4 θλπ
, où λ est la longueur d’onde des neutrons et θ, l’angle de diffusion. La formation
dans l’eau pure de complexes polyanion/gélatine se traduit par un accroissement considérable
de l’intensité diffusée (comparée à celle des composants pris séparément) et par l’apparition
d’un pic. L’augmentation de l’intensité, attribuée à une forte augmentation de la « densité »
des corps diffusants, résulte d’une interaction entre les deux espèces présentes en solution.
L’observation d’un pic, caractéristique en général d’un polyélectrolyte seul en solution,
indique ici qu’une structure assez ordonnée résulte des interactions entre la gélatine et les
polyanions.
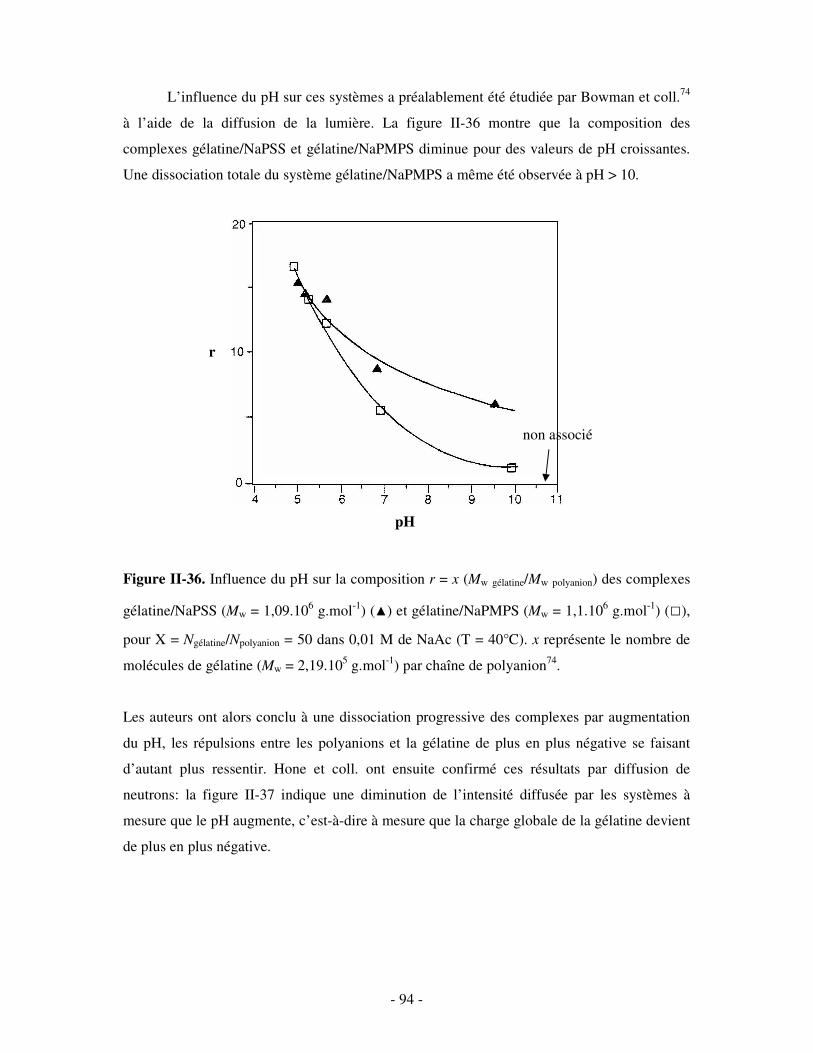
- 94 -
L’influence du pH sur ces systèmes a préalablement été étudiée par Bowman et coll.74
à l’aide de la diffusion de la lumière. La figure II-36 montre que la composition des
complexes gélatine/NaPSS et gélatine/NaPMPS diminue pour des valeurs de pH croissantes.
Une dissociation totale du système gélatine/NaPMPS a même été observée à pH > 10.
Figure II-36. Influence du pH sur la composition r = x (Mw gélatine/Mw polyanion) des complexes
gélatine/NaPSS (Mw = 1,09.106 g.mol-1) () et gélatine/NaPMPS (Mw = 1,1.106 g.mol-1) (),
pour X = Ngélatine/Npolyanion = 50 dans 0,01 M de NaAc (T = 40°C). x représente le nombre de
molécules de gélatine (Mw = 2,19.105 g.mol-1) par chaîne de polyanion74.
Les auteurs ont alors conclu à une dissociation progressive des complexes par augmentation
du pH, les répulsions entre les polyanions et la gélatine de plus en plus négative se faisant
d’autant plus ressentir. Hone et coll. ont ensuite confirmé ces résultats par diffusion de
neutrons: la figure II-37 indique une diminution de l’intensité diffusée par les systèmes à
mesure que le pH augmente, c’est-à-dire à mesure que la charge globale de la gélatine devient
de plus en plus négative.
pH
non associé
r
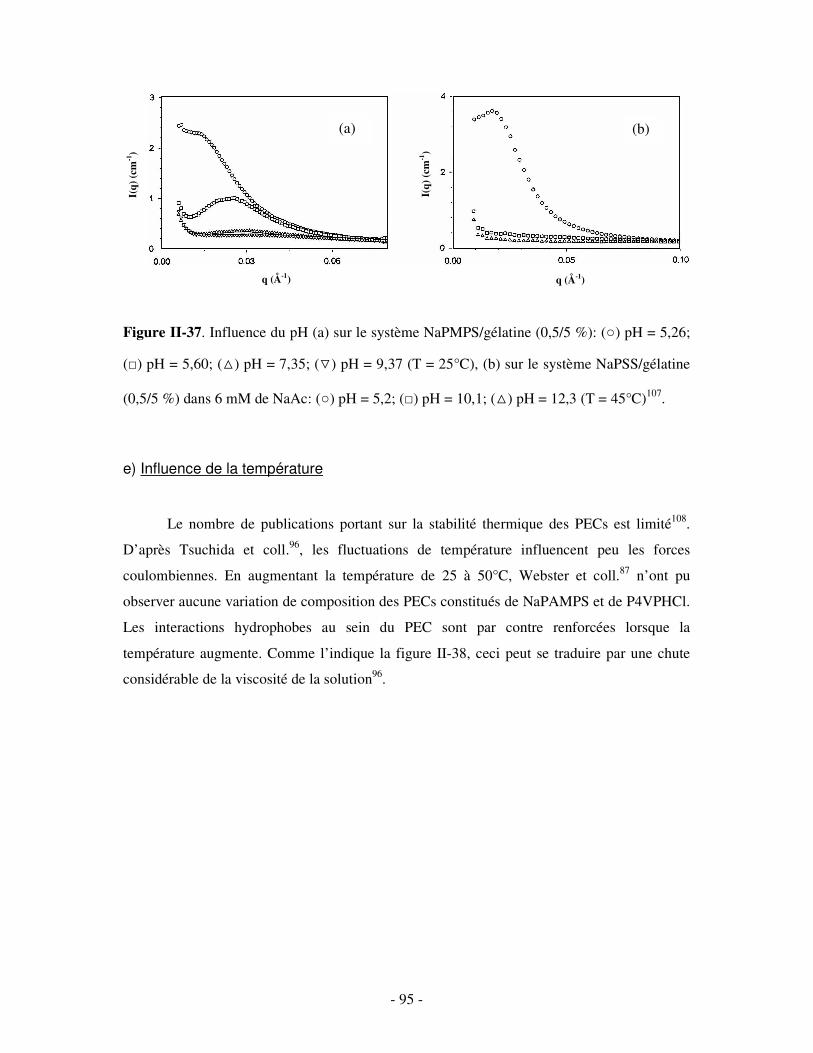
- 95 -
Figure II-37. Influence du pH (a) sur le système NaPMPS/gélatine (0,5/5 %): () pH = 5,26;
() pH = 5,60; () pH = 7,35; () pH = 9,37 (T = 25°C), (b) sur le système NaPSS/gélatine
(0,5/5 %) dans 6 mM de NaAc: () pH = 5,2; () pH = 10,1; () pH = 12,3 (T = 45°C)107.
e) Influence de la température
Le nombre de publications portant sur la stabilité thermique des PECs est limité108.
D’après Tsuchida et coll.96, les fluctuations de température influencent peu les forces
coulombiennes. En augmentant la température de 25 à 50°C, Webster et coll.87 n’ont pu
observer aucune variation de composition des PECs constitués de NaPAMPS et de P4VPHCl.
Les interactions hydrophobes au sein du PEC sont par contre renforcées lorsque la
température augmente. Comme l’indique la figure II-38, ceci peut se traduire par une chute
considérable de la viscosité de la solution96.
I(q)
(cm
-1)
I(q)
(cm
-1)
q (Å-1) q (Å-1)
(a) (b)
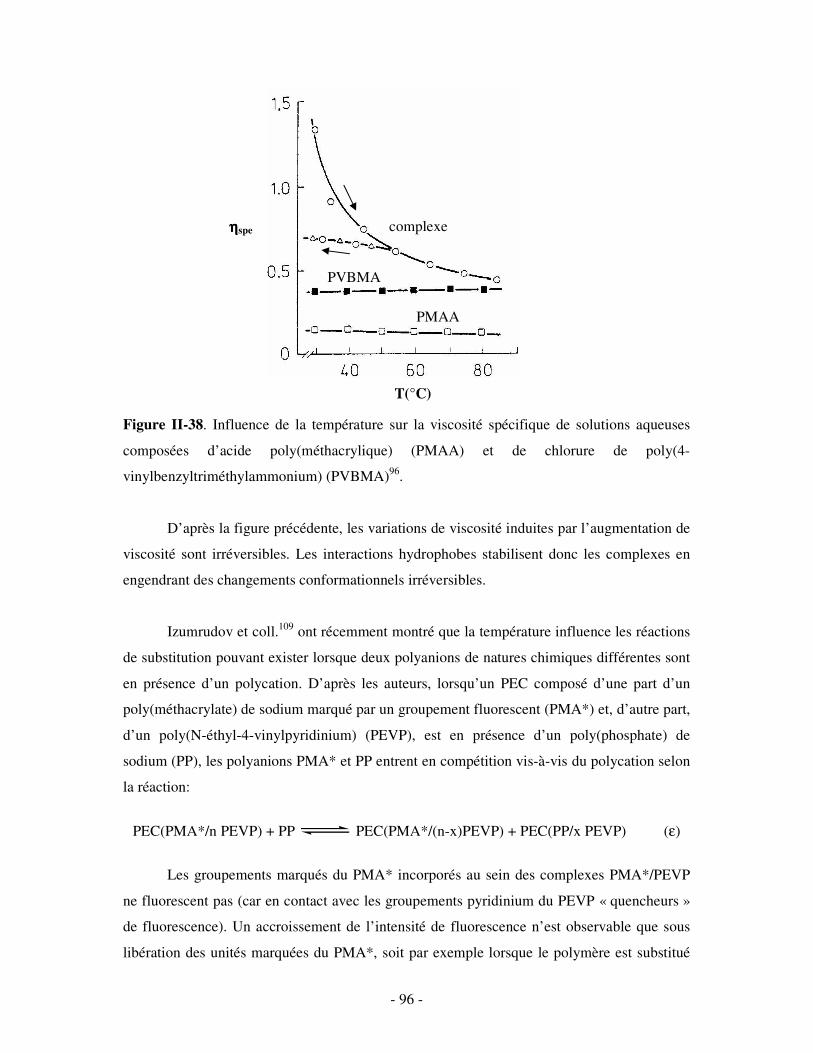
- 96 -
Figure II-38. Influence de la température sur la viscosité spécifique de solutions aqueuses
composées d’acide poly(méthacrylique) (PMAA) et de chlorure de poly(4-
vinylbenzyltriméthylammonium) (PVBMA)96.
D’après la figure précédente, les variations de viscosité induites par l’augmentation de
viscosité sont irréversibles. Les interactions hydrophobes stabilisent donc les complexes en
engendrant des changements conformationnels irréversibles.
Izumrudov et coll.109 ont récemment montré que la température influence les réactions
de substitution pouvant exister lorsque deux polyanions de natures chimiques différentes sont
en présence d’un polycation. D’après les auteurs, lorsqu’un PEC composé d’une part d’un
poly(méthacrylate) de sodium marqué par un groupement fluorescent (PMA*) et, d’autre part,
d’un poly(N-éthyl-4-vinylpyridinium) (PEVP), est en présence d’un poly(phosphate) de
sodium (PP), les polyanions PMA* et PP entrent en compétition vis-à-vis du polycation selon
la réaction:
PEC(PMA*/n PEVP) + PP PEC(PMA*/(n-x)PEVP) + PEC(PP/x PEVP) (ε)
Les groupements marqués du PMA* incorporés au sein des complexes PMA*/PEVP
ne fluorescent pas (car en contact avec les groupements pyridinium du PEVP « quencheurs »
de fluorescence). Un accroissement de l’intensité de fluorescence n’est observable que sous
libération des unités marquées du PMA*, soit par exemple lorsque le polymère est substitué
T(°C)
ηηηηspe complexe
PVBMA
PMAA
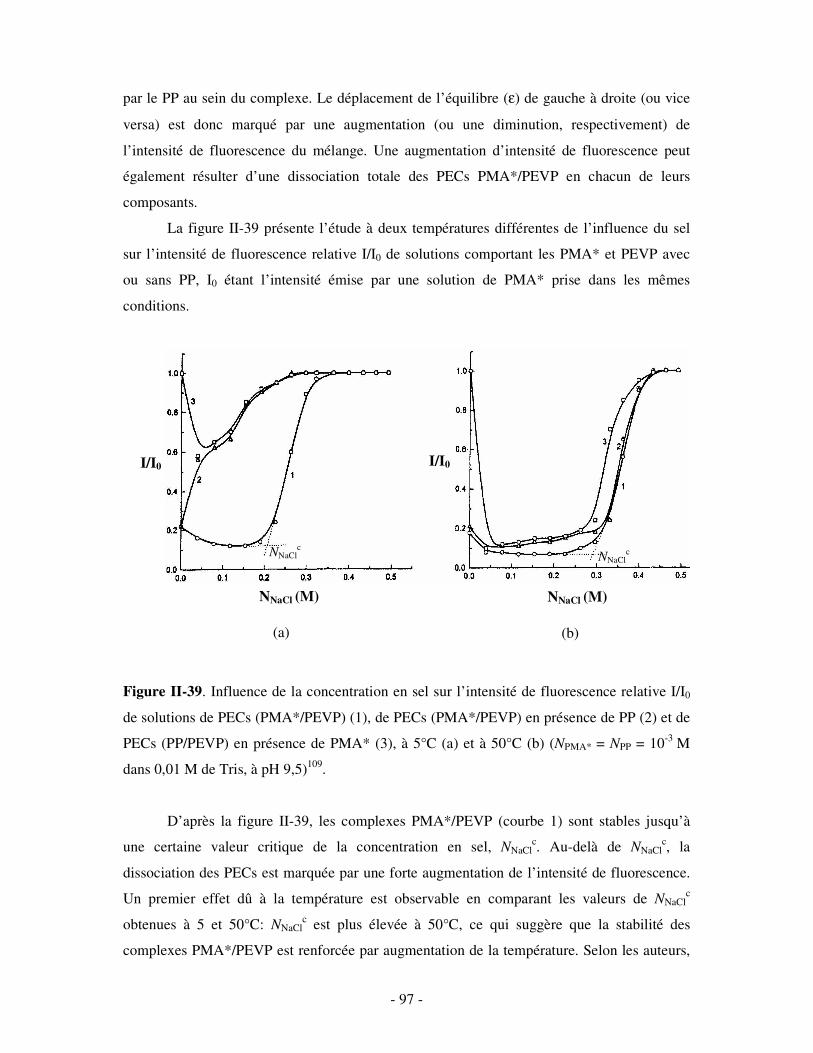
- 97 -
par le PP au sein du complexe. Le déplacement de l’équilibre (ε) de gauche à droite (ou vice
versa) est donc marqué par une augmentation (ou une diminution, respectivement) de
l’intensité de fluorescence du mélange. Une augmentation d’intensité de fluorescence peut
également résulter d’une dissociation totale des PECs PMA*/PEVP en chacun de leurs
composants.
La figure II-39 présente l’étude à deux températures différentes de l’influence du sel
sur l’intensité de fluorescence relative I/I0 de solutions comportant les PMA* et PEVP avec
ou sans PP, I0 étant l’intensité émise par une solution de PMA* prise dans les mêmes
conditions.
Figure II-39. Influence de la concentration en sel sur l’intensité de fluorescence relative I/I0
de solutions de PECs (PMA*/PEVP) (1), de PECs (PMA*/PEVP) en présence de PP (2) et de
PECs (PP/PEVP) en présence de PMA* (3), à 5°C (a) et à 50°C (b) (NPMA* = NPP = 10-3 M
dans 0,01 M de Tris, à pH 9,5)109.
D’après la figure II-39, les complexes PMA*/PEVP (courbe 1) sont stables jusqu’à
une certaine valeur critique de la concentration en sel, NNaClc. Au-delà de NNaCl
c, la
dissociation des PECs est marquée par une forte augmentation de l’intensité de fluorescence.
Un premier effet dû à la température est observable en comparant les valeurs de NNaClc
obtenues à 5 et 50°C: NNaClc est plus élevée à 50°C, ce qui suggère que la stabilité des
complexes PMA*/PEVP est renforcée par augmentation de la température. Selon les auteurs,
NNaClc
NNaClc
I/I0
NNaCl (M) NNaCl (M)
(a)
I/I0
(b)

- 98 -
la stabilisation observée par chauffage des particules de PEC est due en grande partie à une
réduction de la fraction des contre-ions liés fortement au polyanion (Na+ dans ce cas) et, dans
une moindre mesure, à un accroissement des interactions hydrophobes (stabilisation
également observée dans le cas des PECs comportant le PP inorganique). Un second effet dû
à la température est observable en considérant les valeurs d’intensité obtenues pour les
systèmes à trois composants (courbes 2 et 3 dans les intervalles 0,05 M < NNaCl < 0,2 M (a) et
0,05 M < NNaCl < 0,35 M (b)): les valeurs d’intensité sont plus faibles à température élevée, ce
qui signifie que l’équilibre (ε) a été déplacé vers la gauche par chauffage. Izumrudov et coll.
ont finalement conclu au caractère réversible des réactions de substitution: les groupements
du PEVP sont de préférence en contact avec ceux du PP lorsque la température diminue ou
avec ceux du PMA* lorsque la température augmente.
f) Influence de la nature des composants PELs
Les complexes composés de PELs forts et flexibles réagissent généralement de façon
stoechiométrique110,111 (composition équimolaire en chacun des groupements ionisés). Un
comportement différent peut être observé lorsque les polymères présentent des conformations
compliquées tels que certains polysaccharides ou polypeptides (chaînes rigides). Vishalakshi
et coll.82 ont par exemple observé des compositions déviant de la stoechiométrie 1:1 dans le
cas de PECs constitués de PVMP flexible et de CMC rigide. Selon les auteurs, les
compositions non stoechiométriques observées sont attribuables aux différences d’extension
des deux types de chaînes (vis-à-vis de la concentration et de la force ionique du milieu). Il
faut souligner que dans ce cas, mais aussi d’une façon générale, la composition des PECs est
indépendante de la masse molaire des composants. Par exemple, pour des PECs constitués de
PDADMAC linéaire et de dextrane sulfate branché (NaDxS, figure II-40), Möller et coll.89
ont toujours observé une stoechiométrie 2:1, quelque soit la masse molaire du PDADMAC
(240 000 ou 500 000 g.mol-1) ou celle du NaDxS (5000 ou 40 000 g.mol-1).

- 99 -
O
CH2
HH
NaO3SOOSO3Na
H
H
OSO3Na
H
OO
CH2
HH
NaO3SOOSO3Na
H
H
OH
H
O
*
O
CH2
HH
OHOSO3Na
H
H
OH
H
OO
CH2
HH
NaO3SO
H
H
OSO3Na
H
O
*
O
CH2
HH
NaO3SOOSO3Na
H
H
OSO3Na
H
OO
CH2
HH
OHOSO3Na
H
H
OSO3Na
H
O*
n
Figure II-40. Formule chimique du NaDxS, de structure branchée.
La formation de PECs non stoechiométriques peut également résulter de l’association
entre un PEL de type pendant et un PEL de type intégral97,112 (deux PELs de type pendant,
tels que le PVBMA et le NaPSS, ont tendance à former des PECs stoechiométriques
insolubles96). En mesurant le potentiel Zéta de systèmes composés d’une part d’un
copolymère acide maléique/styrène (MS-St, figure II-41) et, d’autre part, d’un PDADMAC ou
d’une PEI, Kramer et coll.113,114 ont observé un déplacement du point isoélectrique (charge
globale nulle) de N-/N+ = 0,85 pour le système MS-St/PDADMAC à N-/N+ = 0,6 dans le cas
MS-St/PEI. Les auteurs ont alors supposé que la flexibilité du PDADMAC linéaire ainsi que
le caractère « pendant » de ses groupements ioniques favorisent un arrangement adéquat entre
les sites de charges opposées, d’où une composition proche de la stoechiométrie. Par contre,
la PEI très branchée (assimilable à une particule sphérique rigide) et de type intégral offre peu
de réarrangement possible durant le processus de compensation des charges. Le polyanion se
lie préférentiellement en surface des complexes MS-St/PEI et, de ce fait, les particules
correspondantes non chargées (potentiel Zéta nul) comportent toujours des charges positives
en leur sein, ce qui explique la déviation de stoechiométrie observée.

- 100 -
* CH CH CH CH2 *
CO
OH
C O
OH
Figure II-41. Formule chimique du copolymère acide maléique/styrène (MS-St).
Comme déjà vu précédemment (section II-2-d), la stoechiométrie 1:1 n’est pas
toujours maintenue dans le cas de PECs constitués d’un polyacide ou d’une polybase
faible96,110, ou plus généralement, lorsque les deux composants PELs (faibles ou forts) sont de
densité de charges différentes77. L’influence de la densité de charges sur la déviation de la
stoechiométrie est corrélée à une augmentation des gênes stériques (dans le processus de
neutralisation entre chaînes de charges opposées) avec la longueur moyenne des portions de
chaînes non chargées. Plus la densité de charges d’un composant diminue, c’est-à-dire plus la
longueur de ses séquences non chargées augmente, plus la composition des complexes
s’éloigne de la stoechiométrie 1:1. La compacité des objets formés est aussi fortement
affectée par les variations de densité de charges. Alors que des agrégats très compacts sont
obtenus dans le cas de PELs de densités de charges comparables, des structures plus gonflées
sont observables avec des composants de densités de charges très différentes.
Enfin, la composition des PECs dépend également de la nature hydrophile des
polymères. Bowman et coll.74 ont par exemple observé des degrés de complexation de la
gélatine plus importants avec un NaPSS (comportant des groupements styrène hydrophobes)
plutôt qu’avec un NaPAMPS (comportant des groupements acrylamide hydrophiles). Pour un
degré de conversion Ngélatine/Npolyanion = 200, la composition en gélatine des complexes
gélatine/NaPSS est en effet plus élevée que celle des complexes gélatine/NaPAMPS: r = 18,5
soit 1 molécule de gélatine (Mw = 2,19.105 g.mol-1) pour 57 monomères de NaPSS (Mw =
1,09.106 g.mol-1), contre r = 15,0 soit 1 molécule de gélatine pour 64 monomères de
NaPAMPS (Mw = 1,10.106 g.mol-1). D’après les auteurs, les différences de conformation entre
les deux polyanions ont un impact direct sur leurs interactions avec la gélatine. En
déterminant la viscosité intrinsèque de chacun des polymères (dans 0,01 M de NaAC, à
25°C), Bowman et coll. ont montré que les chaînes de NaPSS sont moins étendues que celles
du NaPAMPS (volume hydrodynamique plus faible): [η] = 0,65 L.g-1 pour le NaPSS (Mw =
1,09.106 g.mol-1, soit DP = 5400) contre [η] = 0,71 L.g-1 pour le NaPAMPS (Mw = 1,10.106

- 101 -
g.mol-1, soit DP = 4800). C’est la nature hydrophobe des groupements styrène qui confère aux
chaînes de NaPSS une conformation plus compacte, et donc un nombre de charges par unité
de longueur plus élevé. Par conséquent, le nombre de molécules de gélatine liées à une chaîne
de polymère donnée est plus élevé pour le NaPSS que pour le NaPAMPS. Notons qu’une fois
de plus la stoechiométrie des complexes (gélatine/NaPSS dans ce cas) est indépendante de la
longueur de chaîne des composants (Mw = 1,09.106 g.mol-1 ou 5,05.105 g.mol-1 pour le
NaPSS).
3- Bilan
L’agrégation spontanée induite par les fortes interactions électrostatiques existant entre
deux PELs de charges opposées donne lieu à la formation de PECs solubles dans l’eau:
i) pour des mélanges non stoechiométriques de PELs faibles (ou du moins l’un des
deux composants est faible) et de masses molaires très différentes,
ii) pour des mélanges non stoechiométriques de PELs forts et/ou de masses molaires
équivalentes, et ce dans des solutions très diluées.
La structure et la stabilité des particules colloïdales formées dépendent principalement
des caractéristiques des composants PELs (nature des groupements ioniques, densité de
charges…) et de leur concentration, du rapport molaire entre groupements anioniques et
cationiques (degré de conversion) et de la force ionique du milieu. Par exemple, une
augmentation de la concentration engendre la formation de complexes très agrégés. Un niveau
d’agrégation élevé est également obtenu lorsque les deux composants PELs possèdent des
densités de charges différentes. La densité de charges des PELs contrôle aussi la compacité
des particules: plus les composants sont de densité de charges faible, plus les particules de
PECs sont gonflées. Les agrégats alors formés présentent une structure fortement dépendante
du degré de conversion et sont dissociables à faible force ionique.
En général, l’effet du sel sur la stabilité et les propriétés structurales des PECs est
difficile à prévoir, étant donnés les différents processus pouvant interagir selon la nature des
composants PELs impliqués. Pour des mélanges non stoechiométriques de PELs faibles (ou
pour des mélanges PEL faible/PEL fort) de masses molaires très différentes, l’ajout d’une
faible quantité de sel peut engendrer une augmentation de compacité des particules de PECs.
Une séparation de phases, suivie d’une dissociation totale des complexes, est ensuite
généralement observée à force ionique plus élevée. Dans le cas de mélanges non

- 102 -
stoechiométriques et très dilués de PELs forts et/ou de masses molaires élevées et de même
ordre de grandeur, le principal effet observé à faible force ionique consiste en une réduction
considérable du niveau d’agrégation des complexes. Ceci offre la possibilité supplémentaire
(en plus des variations de concentrations en PELs) de contrôler la masse et la taille des
particules de PECs. Des forces ioniques plus élevées induisent une agrégation secondaire et
une floculation macroscopique pour des degrés de conversion inférieurs à 1. Notons que dans
l’eau pure, les niveaux d’agrégation des PECs constitués d’au moins un PEL faible sont
beaucoup moins élevés. Pour ces systèmes, le principal effet du sel consiste donc en une
agrégation secondaire uniquement. Dans tous les cas, l’agrégation induite par le sel est
favorisée si les PELs possèdent des groupements hydrophobes et, au contraire, défavorisée si
l’un des composants possède des groupements hydrophiles. La force ionique critique
correspondant par la suite à la dissociation totale des PECs dépend de la nature chimique des
PELs et s’avère être spécifique à chaque paire polyanion/polycation (en général, les PECs
constitués de deux PELs forts ne sont dissociables qu’à forces ioniques très élevées). Une
augmentation de la température peut toutefois renforcer la stabilité des PECs contre une
dissociation totale induite par le sel.
III- Cas particulier: les complexes PEL/TA de charges opposées
Lorsque le tensioactif (TA) est sous forme de micelles, les interactions PEL/TA de
charges opposées peuvent être considérées comme un cas particulier de formation de
PECs72,77. De par leur taille et leur nombre de sites ioniques, les micelles présentent en effet
des similitudes avec les polymères chargés. Tout comme les complexes polyanion/polycation,
les complexes PEL/TA peuvent être de composition stoechiométrique ou non, et peuvent être
solubles dans l’eau ou former un précipité. Plusieurs études, notamment celles de Thalberg et
coll.115-117 et Hansson et Almgren118, ont consisté à établir des diagrammes de phases de
systèmes aqueux comportant différents PELs et tensioactifs de charges opposées. Pour un
grand nombre de ces systèmes, les précipités obtenus peuvent être redissous en ajoutant du
TA en excès, ce qui n’est pas toujours le cas lorsque, par exemple, la densité de charges du
polymère est trop élevée. Il s’avère que les systèmes PEL/TA ont un comportement en
solution assez compliqué qui dépend fortement du polymère utilisé.
Dans cette partie, nous nous intéresserons principalement à la nature de l’association
PEL/TA de charges opposées, ainsi qu’à la structure des agrégats formés. Nous verrons que

- 103 -
dans ce cas, les interactions hydrophobes tiennent un rôle beaucoup plus important que pour
la plupart des complexes polyanion/polycation. Nous verrons également quelle peut être
l’influence des conditions de réaction (force ionique, pH, concentration…) et celle de la
nature du PEL employé (structure, densité de charges, masse molaire…) sur le processus
d’association.
1- Formation et structure des complexes
De nombreux travaux ont montré que la formation des complexes PEL/TA de charges
opposées est accompagnée de changements conformationnels des chaînes de polymère.
Plusieurs techniques expérimentales ont été utilisées pour observer ces changements
structuraux, dont la fluorimétrie, la diffusion de la lumière et la viscosimétrie. Mel’nikov et
coll.119, de même que Mel’nikova et Lindman120, ont par exemple utilisé la microscopie de
fluorescence pour mettre en évidence la transition structurale subie par les molécules d’ADN
en présence de bromure de cétyltriméthylammonium ou d’oxyde de dodécyldiméthylamine
protonée: les chaînes d’ADN passent de l’état de pelote (conformation plutôt étendue) à celui
de globule (particule compacte) lorsqu’elles sont associées aux tensioactifs cationiques. Abuin
et Scaiano121, ainsi que Thalberg et coll.116, ont pour leur part effectué des mesures
viscosimétriques pour rendre compte de la compaction subie par des PELs synthétiques
(NaPSS et NaPA) en présence de bromure de dodécyltriméthylammonium (C12TAB). En
milieu dilué, la viscosité reflète en effet les dimensions de la chaîne de polymère, une
contraction de cette dernière se manifestant par une forte chute de la viscosité (cf. figure II-43,
par exemple). La diffusion de neutrons aux petits angles122,123, ainsi que les techniques
fluorimétriques répertoriées par Françoise M. Winnik dans les références 124 et 125, ont aussi
largement contribué à la description de la structure des complexes PEL/TA. L’image du
« collier de perles » où des agrégats de type micelles sont adsorbés sur la chaîne de polymère
est généralement employée pour décrire la structure de tels systèmes en milieu dilué. Dans
quelques publications, des structures différentes ont cependant été proposées. Pour un
polymère plutôt rigide dérivé de la cellulose, Leung et coll.126 ont par exemple suggéré la
formation de structures constituées de chaînes enchevêtrées au niveau des molécules de TA
liées. De telles structures interchaînes impliquant de la PEI très branchée et du SDS ont été
mises en évidence par Mitchell A. Winnik et coll.127 à l’aide de la diffusion de la lumière. En
faisant interagir de l’amidon cationique (de forme hélicoïdale) avec différents TAs de charges

- 104 -
opposées, Merta et coll.128 ont quant à eux rapporté la formation d’agrégats cylindriques de
type cœur-coquille (avec un cœur formé par les chaînes de TA et une coquille constituée de
l’amidon).
La structure en « collier de perles » fréquemment rencontrée dans la littérature et
représentée en figure II-42 est le résultat d’un processus très coopératif, impliquant des forces
coulombiennes et des interactions hydrophobes. En réduisant efficacement les répulsions
entre les molécules de TA (répulsions entre les têtes chargées), les interactions
électrostatiques entre les groupements ioniques du PEL et le TA de charges opposées
favorisent les interactions hydrophobes entre les chaînons de TAs liés au polymère.
Figure II-42. Formation coopérative d’agrégats de tensioactif sur la chaîne de polyélectrolyte.
Ce processus coopératif prend effet dès que la concentration en TA excède une
certaine valeur critique, la cac (concentration critique d’agrégation) qui, typiquement, est de
un à trois ordres de grandeur plus faible que la cmc des micelles libres correspondantes. Via
ce processus d’agrégation, les molécules de TA génèrent une contraction des chaînes de
polymère. La figure II-43 présente par exemple l’influence de la concentration en C12TAB sur
⊕
⊕ ⊕
⊕ ⊕ ⊕ ⊕
⊕ ⊕
⊕ +
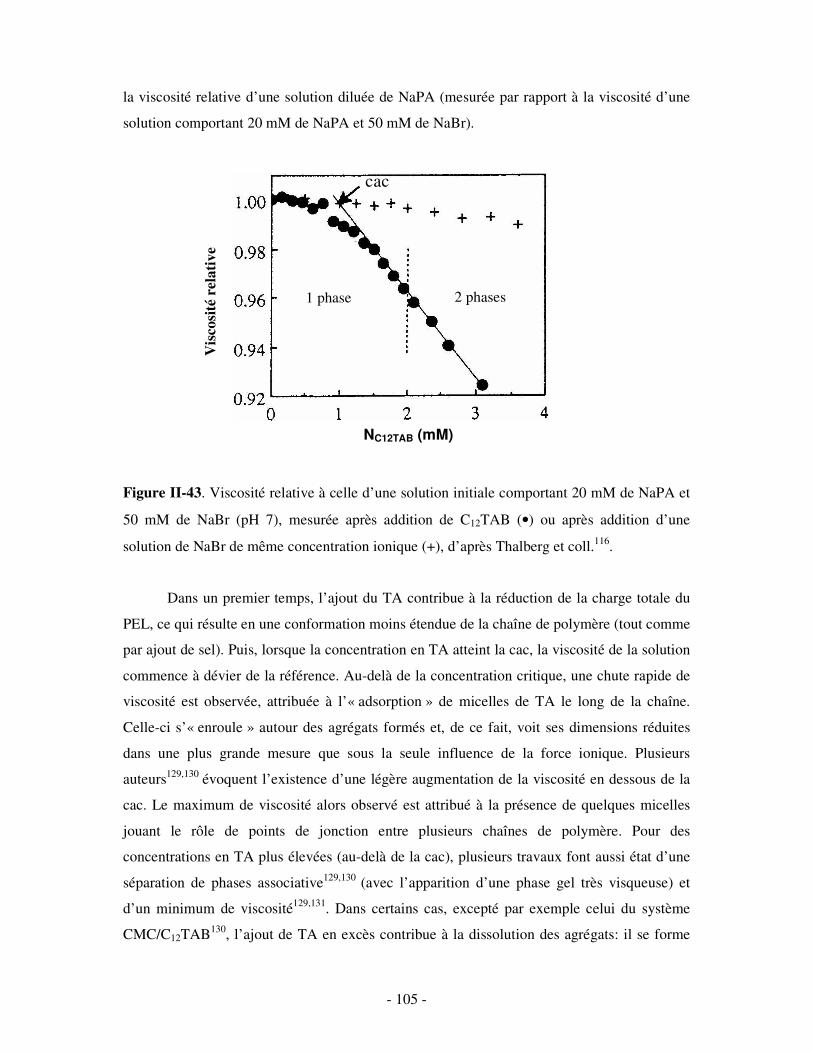
- 105 -
la viscosité relative d’une solution diluée de NaPA (mesurée par rapport à la viscosité d’une
solution comportant 20 mM de NaPA et 50 mM de NaBr).
Figure II-43. Viscosité relative à celle d’une solution initiale comportant 20 mM de NaPA et
50 mM de NaBr (pH 7), mesurée après addition de C12TAB (•) ou après addition d’une
solution de NaBr de même concentration ionique (+), d’après Thalberg et coll.116.
Dans un premier temps, l’ajout du TA contribue à la réduction de la charge totale du
PEL, ce qui résulte en une conformation moins étendue de la chaîne de polymère (tout comme
par ajout de sel). Puis, lorsque la concentration en TA atteint la cac, la viscosité de la solution
commence à dévier de la référence. Au-delà de la concentration critique, une chute rapide de
viscosité est observée, attribuée à l’« adsorption » de micelles de TA le long de la chaîne.
Celle-ci s’« enroule » autour des agrégats formés et, de ce fait, voit ses dimensions réduites
dans une plus grande mesure que sous la seule influence de la force ionique. Plusieurs
auteurs129,130 évoquent l’existence d’une légère augmentation de la viscosité en dessous de la
cac. Le maximum de viscosité alors observé est attribué à la présence de quelques micelles
jouant le rôle de points de jonction entre plusieurs chaînes de polymère. Pour des
concentrations en TA plus élevées (au-delà de la cac), plusieurs travaux font aussi état d’une
séparation de phases associative129,130 (avec l’apparition d’une phase gel très visqueuse) et
d’un minimum de viscosité129,131. Dans certains cas, excepté par exemple celui du système
CMC/C12TAB130, l’ajout de TA en excès contribue à la dissolution des agrégats: il se forme
1 phase 2 phases
cac
NC12TAB (mM)
Vis
cosi
té r
elat
ive

- 106 -
assez de micelles pour que les chaînes de polymère n’aient plus besoin de les partager. De
plus, l’addition excessive de TA (au-delà de l’équimolarité) contribue à la formation de
micelles libres, et donc à une augmentation du nombre de colloïdes dans le système. D’où
l’accroissement de viscosité observé dans certains cas pour des concentrations en TA élevées.
2- Influence de divers paramètres
Alors que la température a peu d’effet sur l’association PEL/TA de charges
opposées132,133, la balance entre forces coulombiennes et interactions hydrophobes peut être
largement influencée par le pH et la force ionique du milieu. De même que pour les
complexes polyanion/polycation, l’ajout de sel affaiblit l’association en écrantant les
interactions électrostatiques. Ceci a pour conséquence d’augmenter la valeur de la cac134,135.
En effet, les liaisons électrostatiques entre le PEL et les molécules de TA étant réduites, la
concentration locale en TA nécessaire à son autoagrégation sur la chaîne de polymère est
atteinte pour une concentration totale en TA plus élevée, d’où une plus grande cac (la
coopérativité persiste même si les interactions coulombiennes sont réduites par le sel).
Plusieurs auteurs115,116,118,131 mentionnent également une réduction du domaine de séparation
de phases et une redissolution des complexes par ajout de sel. Ceci confirme que l’association
PEL/TA de charges opposées est d’abord de nature électrostatique. Une forte influence du pH
a été observée par Chandar et coll.134 pour des systèmes composés d’acide poly(acrylique) et
de C12TAB. La figure II-44 présente les valeurs de cac mesurées pour ces systèmes en
fonction du pH, en présence ou non de sel.
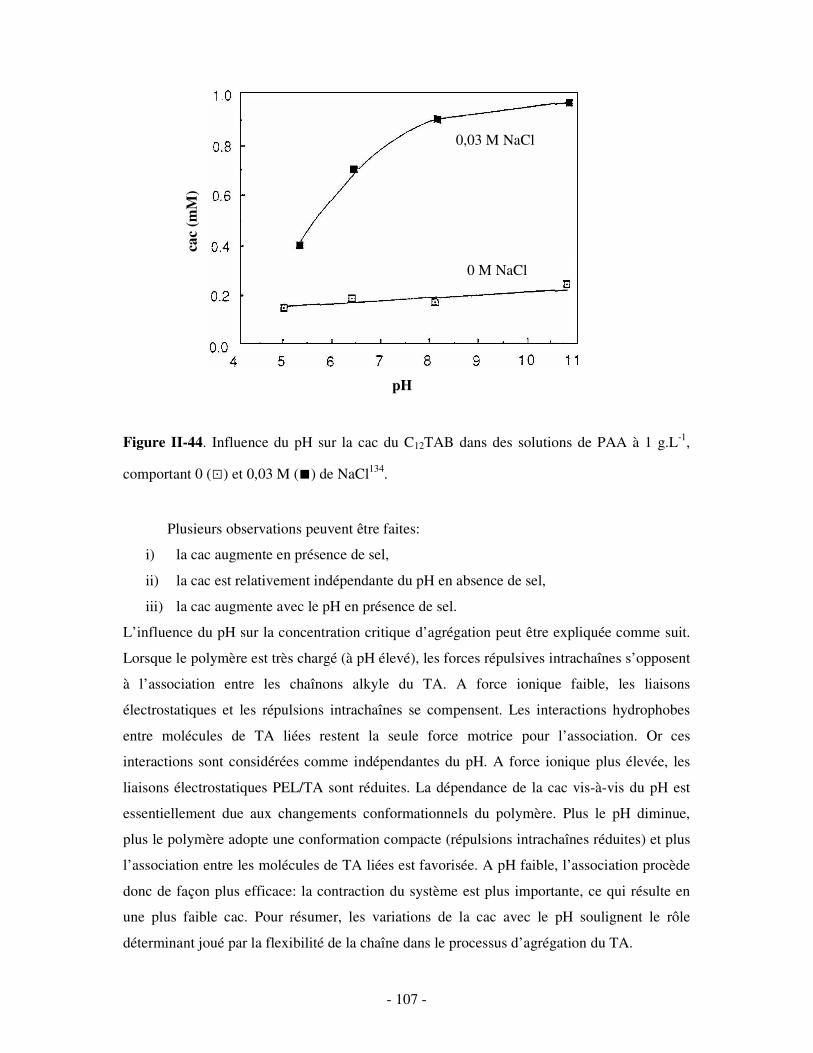
- 107 -
Figure II-44. Influence du pH sur la cac du C12TAB dans des solutions de PAA à 1 g.L-1,
comportant 0 () et 0,03 M () de NaCl134.
Plusieurs observations peuvent être faites:
i) la cac augmente en présence de sel,
ii) la cac est relativement indépendante du pH en absence de sel,
iii) la cac augmente avec le pH en présence de sel.
L’influence du pH sur la concentration critique d’agrégation peut être expliquée comme suit.
Lorsque le polymère est très chargé (à pH élevé), les forces répulsives intrachaînes s’opposent
à l’association entre les chaînons alkyle du TA. A force ionique faible, les liaisons
électrostatiques et les répulsions intrachaînes se compensent. Les interactions hydrophobes
entre molécules de TA liées restent la seule force motrice pour l’association. Or ces
interactions sont considérées comme indépendantes du pH. A force ionique plus élevée, les
liaisons électrostatiques PEL/TA sont réduites. La dépendance de la cac vis-à-vis du pH est
essentiellement due aux changements conformationnels du polymère. Plus le pH diminue,
plus le polymère adopte une conformation compacte (répulsions intrachaînes réduites) et plus
l’association entre les molécules de TA liées est favorisée. A pH faible, l’association procède
donc de façon plus efficace: la contraction du système est plus importante, ce qui résulte en
une plus faible cac. Pour résumer, les variations de la cac avec le pH soulignent le rôle
déterminant joué par la flexibilité de la chaîne dans le processus d’agrégation du TA.
0,03 M NaCl
0 M NaCl
cac
(mM
)
pH

- 108 -
D’une façon générale, la cac dépend également de la concentration en
polymère116,118,136,137 (le cas du C12TAB ou de la dodécylamine en présence d’ADN est
l’exception à la règle138). Plus la concentration en polymère est élevée, plus les molécules de
TA sont distribuées parmi un nombre de chaînes plus important. La concentration locale en
TA proche d’une chaîne de polyion est donc d’autant plus faible. Par conséquent, le degré
d’association critique pour lequel les agrégats micellaires commencent à se former correspond
à une plus grande cac. La concentration en polymère influence aussi largement le
comportement des complexes en solution. Pour le système NaPA/C12TAB116 par exemple, les
chaînes de polymère en très faible concentration ( 1 mM) peuvent s’associer à une quantité
considérable de TA tout en restant en solution (avec plus de 60 % de molécules de C12TAB
liées). Un précipité n’est observé que pour des concentrations en TA proches de la
neutralisation des charges. Pour des concentrations en PEL plus élevées (> 2 mM dans ce
cas), une séparation de phases commence à apparaître pour des concentrations en TA pouvant
être beaucoup plus faibles que celles correspondant à l’équimolarité (avec seulement moins de
5 % de TA lié). Selon Dubin et coll.139-141, c’est une transition entre complexes intra- et
complexes interchaînes qui est à l’origine de la séparation de phases associative observée.
La cac dépend enfin de la longueur du chaînon alkyle du TA. Plus le chaînon est long,
c’est-à-dire plus le nombre n de groupements –CH2 est grand, plus les interactions entre le
PEL et le TA sont fortes. Ceci induit une cac plus faible129,136,137,142 et, d’après Thalberg et
coll.117, une tendance à la séparation de phases plus importante. Pour exemple, l’influence de
la longueur du chaînon alkyle sur la cac de tensioactifs de type bromure
d’alkyltriméthylammonium (CnTAB) en présence de PMAA136 est représentée en figure II-45.
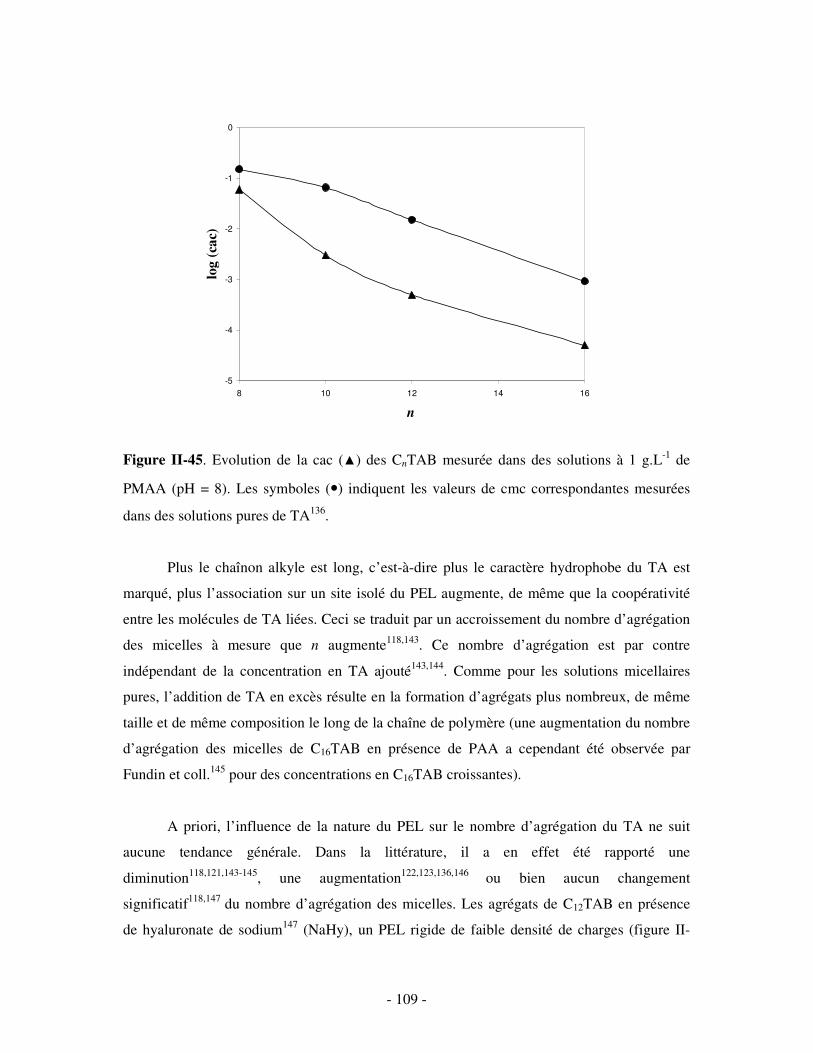
- 109 -
-5
-4
-3
-2
-1
0
8 10 12 14 16
n
log
(cac
)
Figure II-45. Evolution de la cac () des CnTAB mesurée dans des solutions à 1 g.L-1 de
PMAA (pH = 8). Les symboles (•) indiquent les valeurs de cmc correspondantes mesurées
dans des solutions pures de TA136.
Plus le chaînon alkyle est long, c’est-à-dire plus le caractère hydrophobe du TA est
marqué, plus l’association sur un site isolé du PEL augmente, de même que la coopérativité
entre les molécules de TA liées. Ceci se traduit par un accroissement du nombre d’agrégation
des micelles à mesure que n augmente118,143. Ce nombre d’agrégation est par contre
indépendant de la concentration en TA ajouté143,144. Comme pour les solutions micellaires
pures, l’addition de TA en excès résulte en la formation d’agrégats plus nombreux, de même
taille et de même composition le long de la chaîne de polymère (une augmentation du nombre
d’agrégation des micelles de C16TAB en présence de PAA a cependant été observée par
Fundin et coll.145 pour des concentrations en C16TAB croissantes).
A priori, l’influence de la nature du PEL sur le nombre d’agrégation du TA ne suit
aucune tendance générale. Dans la littérature, il a en effet été rapporté une
diminution118,121,143-145, une augmentation122,123,136,146 ou bien aucun changement
significatif118,147 du nombre d’agrégation des micelles. Les agrégats de C12TAB en présence
de hyaluronate de sodium147 (NaHy), un PEL rigide de faible densité de charges (figure II-

- 110 -
46), possèdent par exemple le même nombre d’agrégation que les micelles de C12TAB libres
formées dans les mêmes condition de force ionique (soit <n>C12TAB pur ~ 65 d’après la
référence 118).
OHOH2C
HO*
ONHCOCH3
OO
HOOH
COO- Na+
*
n Figure II-46. Formule chimique du NaHy.
Il est à noter que l’association entre le C12TAB et le NaHy est assez faible, marquée
par une valeur de cac relativement élevée: en présence de NaHy, la cac du C12TAB équivaut à
la moitié de la cmc du TA pur. Il s’avère, en fait, que la constante d’association et la
coopérativité sont généralement corrélées à la densité de charges du PEL. Plus la densité de
charges linéaire est élevée (c’est-à-dire plus la distance moyenne b entre deux charges est
faible), plus les interactions PEL/TA sont fortes116,144 et plus la coopérativité est importante148:
la cac diminue et le nombre d’agrégation des micelles liées augmente avec 1/b144. Les cas du
NaDxS et de l’ADN, de formules chimiques respectives représentées en figures II-40 et II-47,
échappent toutefois à cette règle. La coopérativité de l’association ADN/C12TAB est en effet
plus faible que pour le système NaPA/C12TAB, bien que la densité de charges de l’ADN
(bADN = 1,7 Å) soit supérieure à celle du NaPA (bNaPA = 2,5 Å). Dans le cas du NaDxS, par
contre, la coopérativité est plus élevée qu’avec le NaPA, et ce malgré des densités de charges
équivalentes (bNaDxS = 2,55 Å). L’association entre le C12TAB et le NaDxS est alors
caractérisée par une plus faible cac (~ un ordre de grandeur plus faible qu’avec le NaPA) et un
nombre d’agrégation plus élevé (supérieur à celui obtenu avec le NaPA, et même supérieur à
celui des solutions pures de C12TAB)146. En fait, comme déjà mentionné, la flexibilité et la
structure du polymère s’avèrent être elles aussi des facteurs déterminants dans le processus
d’association PEL/TA de charges opposées. Par exemple, la structure branchée du NaDxS
facilite ses interactions avec les micelles, tandis que la grande rigidité de l’ADN l’empêche de
s’enrouler facilement autour d’elles149. Dans des solutions de faible force ionique, l’ADN
adopte en effet une conformation de type bâtonnet rigide, avec une longueur de persistance de
plus de 50 nm150. Néanmoins, en solution diluée, les molécules de TA s’associent de façon
- 111 -
coopérative avec l’ADN: des agrégats de type micelles viennent s’adsorber à la surface de la
macromolécule.
N
O
O
H3C
NO
HO
HH
HH
P
O
O
N
N
O
NO
HO
HH
H
H2C
H
P-O
O
O
O
P-O
O
O
H3C
-O
N
N
NO
N
N
O
H
HH
H
H2C
HO
P-O
O
O
N
N
N
N
N
O
HO
HH
H
H2C
H
P-O
O
O
O
H O
H H
H
CH2
H
P O-
O
ON
N
N
NN
H
H
H
O
H
H H
H
CH2
HO
P O-
O
ON
N
NO
N
NH
H
H
H
H
O
H O
H H
H
CH2
H
P O-
O
ON
N
O
N
H
H
H
H
H
O
H O
H H
H
CH2
H
P O-
O
ON
O
O
CH3
N
H
H
H
Thymine
Cytosine
Guanine
Adénine
Adénine
Guanine
Cytosine
Thymine
Figure II-47. Structure chimique de l’ADN (« double-brin »).
D’une façon générale, à faible concentration en TA, l’utilisation de PELs rigides tels
que la plupart des dérivés de la cellulose privilégie les associations intermoléculaires126 (via
des interactions entre des molécules de TA liées à différentes chaînes de polymère). Par
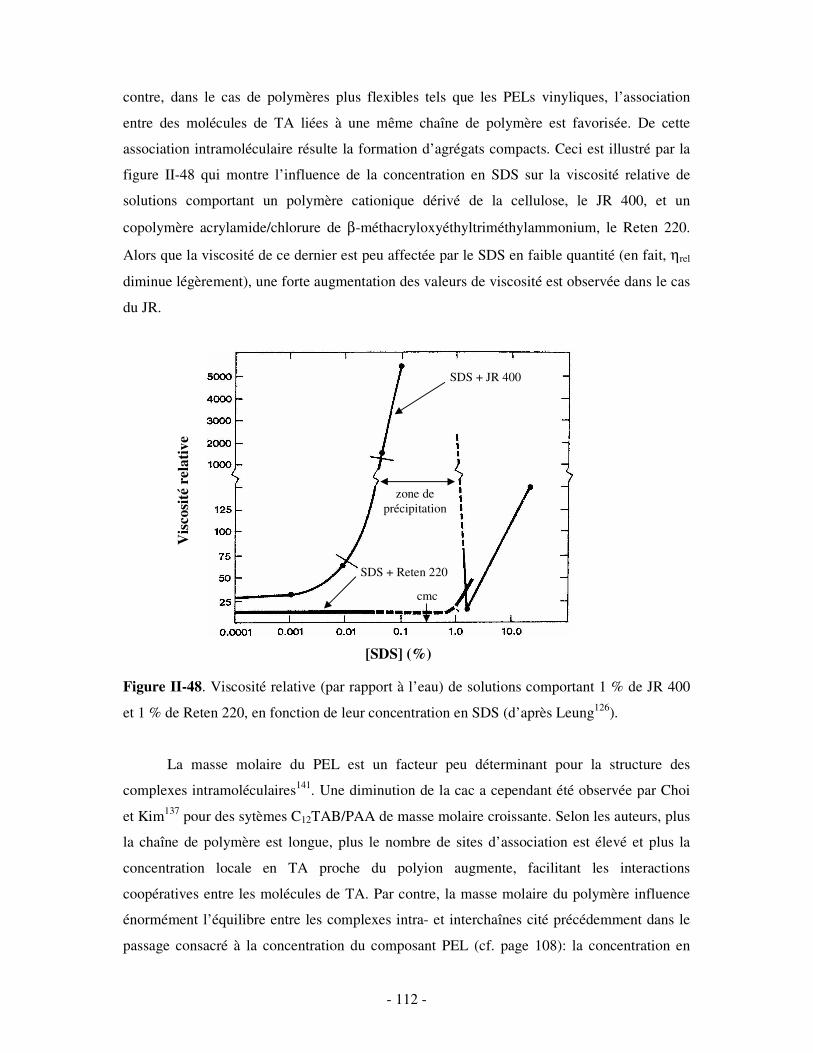
- 112 -
contre, dans le cas de polymères plus flexibles tels que les PELs vinyliques, l’association
entre des molécules de TA liées à une même chaîne de polymère est favorisée. De cette
association intramoléculaire résulte la formation d’agrégats compacts. Ceci est illustré par la
figure II-48 qui montre l’influence de la concentration en SDS sur la viscosité relative de
solutions comportant un polymère cationique dérivé de la cellulose, le JR 400, et un
copolymère acrylamide/chlorure de β-méthacryloxyéthyltriméthylammonium, le Reten 220.
Alors que la viscosité de ce dernier est peu affectée par le SDS en faible quantité (en fait, ηrel
diminue légèrement), une forte augmentation des valeurs de viscosité est observée dans le cas
du JR.
Figure II-48. Viscosité relative (par rapport à l’eau) de solutions comportant 1 % de JR 400
et 1 % de Reten 220, en fonction de leur concentration en SDS (d’après Leung126).
La masse molaire du PEL est un facteur peu déterminant pour la structure des
complexes intramoléculaires141. Une diminution de la cac a cependant été observée par Choi
et Kim137 pour des sytèmes C12TAB/PAA de masse molaire croissante. Selon les auteurs, plus
la chaîne de polymère est longue, plus le nombre de sites d’association est élevé et plus la
concentration locale en TA proche du polyion augmente, facilitant les interactions
coopératives entre les molécules de TA. Par contre, la masse molaire du polymère influence
énormément l’équilibre entre les complexes intra- et interchaînes cité précédemment dans le
passage consacré à la concentration du composant PEL (cf. page 108): la concentration en
cmc
SDS + JR 400
SDS + Reten 220
zone de précipitation
[SDS] (%)
Vis
cosi
té r
elat
ive
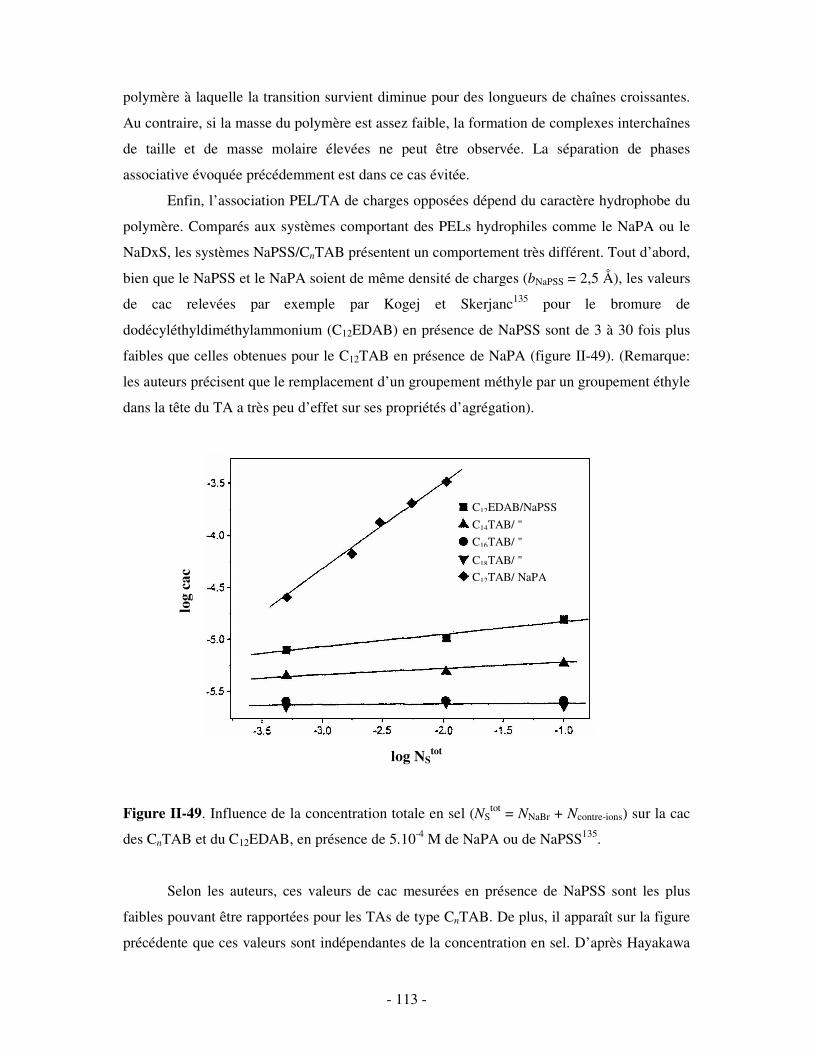
- 113 -
polymère à laquelle la transition survient diminue pour des longueurs de chaînes croissantes.
Au contraire, si la masse du polymère est assez faible, la formation de complexes interchaînes
de taille et de masse molaire élevées ne peut être observée. La séparation de phases
associative évoquée précédemment est dans ce cas évitée.
Enfin, l’association PEL/TA de charges opposées dépend du caractère hydrophobe du
polymère. Comparés aux systèmes comportant des PELs hydrophiles comme le NaPA ou le
NaDxS, les systèmes NaPSS/CnTAB présentent un comportement très différent. Tout d’abord,
bien que le NaPSS et le NaPA soient de même densité de charges (bNaPSS = 2,5 Å), les valeurs
de cac relevées par exemple par Kogej et Skerjanc135 pour le bromure de
dodécyléthyldiméthylammonium (C12EDAB) en présence de NaPSS sont de 3 à 30 fois plus
faibles que celles obtenues pour le C12TAB en présence de NaPA (figure II-49). (Remarque:
les auteurs précisent que le remplacement d’un groupement méthyle par un groupement éthyle
dans la tête du TA a très peu d’effet sur ses propriétés d’agrégation).
Figure II-49. Influence de la concentration totale en sel (NStot = NNaBr + Ncontre-ions) sur la cac
des CnTAB et du C12EDAB, en présence de 5.10-4 M de NaPA ou de NaPSS135.
Selon les auteurs, ces valeurs de cac mesurées en présence de NaPSS sont les plus
faibles pouvant être rapportées pour les TAs de type CnTAB. De plus, il apparaît sur la figure
précédente que ces valeurs sont indépendantes de la concentration en sel. D’après Hayakawa
log
cac
C12EDAB/NaPSS C14TAB/ " C16TAB/ "
C18TAB/ " C12TAB/ NaPA
log NStot

- 114 -
et Kwak151, l’effet coopératif de l’association NaPSS/C12TAB est par ailleurs moins prononcé
que pour le système NaDxS/C12TAB, et ce malgré des densités de charges une fois de plus
équivalentes (bNaDxS = 2,55 Å). Les auteurs indiquent néanmoins une constante d’association
K du C12TAB avec un site isolé du polyanion beaucoup plus forte dans le cas du NaPSS: la
valeur de K estimée pour le système NaPSS/C12TAB est environ 3,5RT fois plus élevée que
celle du système NaDxS/C12TAB (pour des concentrations en NaCl comprises entre 0,02 et
0,1 M). Hansson et Almgren118 ajoutent que les complexes NaPSS/C12TAB sont stables même
après addition de grandes quantités de sel. Thalberg et coll.116 concluent quant à eux à une
stabilisation des complexes contre une séparation de phases associative tant que le NaPSS est
en excès. Tous ces résultats ont été attribués aux fortes interactions hydrophobes existant
entre le NaPSS et les micelles: les groupements phényle du polyanion sont incorporés à la
surface des micelles, ce qui induit un contact plus rapproché entre les charges du PEL et celles
des molécules de TA. Il en résulte la formation d’agrégats très stables et compacts (de faible
énergie électrostatique interne), peu sensibles à la force ionique du milieu: la hausse des
valeurs de cac observée à mesure que la force ionique augmente n’est pas très prononcée
(figure II-49). Notons que ces interactions hydrophobes entre le TA lié et le squelette du
NaPSS ne contribuent pas à l’effet coopératif global et, par conséquent, sont à l’origine de la
réduction de coopérativité observée. Ceci se traduit par des nombres d’agrégation assez
faibles. Hansson et Almgren118 annoncent par exemple un nombre d’agrégation du C12TAB
réduit environ d’un facteur 2 par rapport aux micelles libres (soit <n> = 38 pour NC12TAB
comprise entre 1,3 et 3,6 mM en présence de 5 mM de NaPSS), tandis que Abuin et
Scaiano121 décrivent des « minimicelles » comportant entre 7 et 10 molécules de C12TAB
(pour 14 mM de C12TAB et 28 mM de NaPSS).
En résumé, pour les systèmes PEL hydrophile/TA, l’association est gouvernée (i) par
des attractions électrostatiques entre le polyion et les groupements chargés du TA et (ii) par
des attractions hydrophobes entre les chaînons alkyle des molécules de TA. Cette association
de nature coopérative est caractérisée par l’existence d’une cac et par un nombre d’agrégation
indépendant de la concentration en TA. Pour des PELs à caractère hydrophobe, les
interactions PEL/TA sont renforcées mais, d’un autre côté, la coopérativité du processus
d’association est restreinte. Ceci est clairement observable pour des systèmes constitués de
polymères associatifs hydrosolubles (ou polysavons) contenant une petite fraction d’unités
hydrophobes (typiquement < à 5 % en moles) capables de former des microdomaines
intramoléculaires en absence de TA. Par exemple, pour des systèmes composés de C12TAC et
de copolymères acide maléique/alkylvinyléther désignés par PSX (où X 6 est le nombre de

- 115 -
carbone par chaîne alkyle), Anthony et Zana152 évoquent une disparition de la cac et de la
coopérativité. Dans ce cas, les interactions (i) et (ii) citées précédemment sont largement
dominées par des attractions hydrophobes entre le TA et les chaînes alkyles du PSX associées
en microdomaines. D’après les auteurs, aucun nouvel agrégat ou microdomaine n’est formé
lorsque le C12TAC est ajouté aux PSX: les molécules de TA viennent simplement gonfler les
microdomaines préexistants (figure II-50). Le nombre d’agrégation du TA fixé augmente
alors linéairement avec sa concentration.
Figure II-50. Représentation schématique de l’association polysavon/TA: les groupements
ioniques des molécules de TA sont proches des charges du polymère (ces dernières
remplaçant les contre-ions du TA à la surface des agrégats), tandis que leurs chaînons alkyles
gonflent les microdomaines préexistants152.
En général, les différences de comportement entre les systèmes comportant des PELs
hydrophiles ou des PELs à caractère hydrophobe se manifestent au travers de leurs propriétés
viscosimétriques. A concentration en polymère constante, la viscosité des mélanges PEL
hydrophile/TA a tendance à diminuer pour des concentrations en TA croissantes (ce qui a été
attribué à un enroulement du polymère autour des agrégats micellaires), tandis que les
systèmes PEL associatif/TA présentent un maximum de viscosité152-154. La figure II-51
+ ⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕ ⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕ ⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕ ⊕⊕⊕⊕
⊕⊕⊕⊕
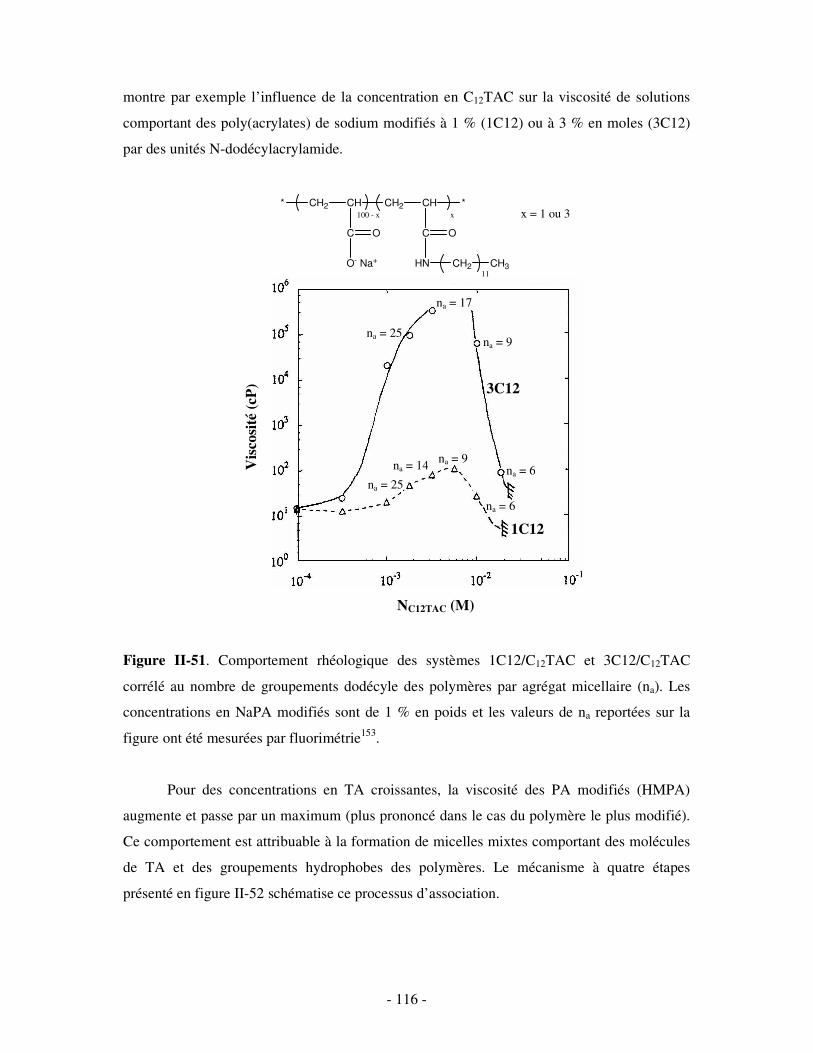
- 116 -
montre par exemple l’influence de la concentration en C12TAC sur la viscosité de solutions
comportant des poly(acrylates) de sodium modifiés à 1 % (1C12) ou à 3 % en moles (3C12)
par des unités N-dodécylacrylamide.
Figure II-51. Comportement rhéologique des systèmes 1C12/C12TAC et 3C12/C12TAC
corrélé au nombre de groupements dodécyle des polymères par agrégat micellaire (na). Les
concentrations en NaPA modifiés sont de 1 % en poids et les valeurs de na reportées sur la
figure ont été mesurées par fluorimétrie153.
Pour des concentrations en TA croissantes, la viscosité des PA modifiés (HMPA)
augmente et passe par un maximum (plus prononcé dans le cas du polymère le plus modifié).
Ce comportement est attribuable à la formation de micelles mixtes comportant des molécules
de TA et des groupements hydrophobes des polymères. Le mécanisme à quatre étapes
présenté en figure II-52 schématise ce processus d’association.
NC12TAC (M)
Vis
cosi
té (c
P)
na = 25
na = 14 na = 9
na = 6
na = 6
na = 9
na = 17
na = 25
3C12
1C12
* CH2 CH CH2
C O
O- Na+
CH *
C
HN
O
CH2 CH3
100 - x x
11
x = 1 ou 3

- 117 -
Figure II-52. Représentation schématique de l’association HMPA/C12TAC, d’après
Magny153.
En absence de C12TAC et à concentration modérée (figure a), pas ou peu
d’autoassociation des groupements dodécyle des HMPA n’est observée. Lorsque la
concentration en TA s’approche de la cac du système (figure b), les molécules de TA
commencent à s’associer avec les chaînes de polymère, de préférence au voisinage de leurs
groupements dodécyle. Quelques agrégats mixtes sont formés et jouent le rôle de points de
jonction entre plusieurs chaînes de polymère. Au-delà de la cac (figure c), le nombre de
micelles mixtes augmente avec la concentration en TA. Le nombre de groupements dodécyle
appartenant au HMPA diminue au sein de chaque micelle mais, petit à petit, la totalité des ces
groupements est inclue dans des agrégats. Un réseau tridimensionnel est progressivement
formé, marqué par une forte augmentation de la viscosité du mélange. Un excès en TA a deux
conséquences (figure d). Les molécules de C12TAC continuent d’être impliquées dans la
formation des micelles mixtes, mais le rapport [groupements dodécyle]/[TA] de ces dernières
diminue. Les micelles finissent par ne plus contenir que quelques groupements dodécyle
tensioactif
tensioactif
tensioactif
groupement hydrophobe
micelle mixte
micelle liée
a b
c d

- 118 -
appartenant à la même chaîne de polymère. Par conséquent, le réseau tridimensionnel est
détruit et la viscosité du mélange diminue à nouveau. Des micelles pures de C12TAC
commencent à apparaître et viennent s’« adsorber » par interactions électrostatiques sur les
chaînes de polymère. S’ensuit une séparation de phases qui procède de façon similaire au
système PA non modifié/TA. Magny et coll.153 ajoutent que la cac du système diminue
lorsque le degré de modification ou la longueur des chaînes alkyles du HMPA augmente. De
plus, des quantités croissantes de TA sont nécessaires pour produire une séparation de phases.
L’affinité du TA pour le PEL de charges opposées augmente donc considérablement avec le
caractère hydrophobe du polymère.
IV- Applications possibles des PECs solubles
L’aptitude des PECs solubles à modifier la surface de substrats inorganiques a fait
l’objet de nombreuses études. Kramer et coll.113,114 ont par exemple montré que l’adsorption
de PECs non stoechiométriques comportant un excès de charges cationiques permet
d’inverser la charge en surface des poudres de silice (de charge initiale négative). Ce
processus d’inversion de charge du substrat est à la base de diverses applications possibles.
Décher et son équipe155 ont pu ainsi développer une technique permettant de construire des
films multicomposites (ou multicouches) par adsorption consécutive de polyanions et de
polycations sur une surface solide chargée. Petzold et coll.100,156,157 ont quant à eux démontré
l’efficacité des PECs non stoechiométriques à faire floculer des suspensions de silice, de
cellulose ou d’argile selon le mécanisme (b) présenté en figure II-53.
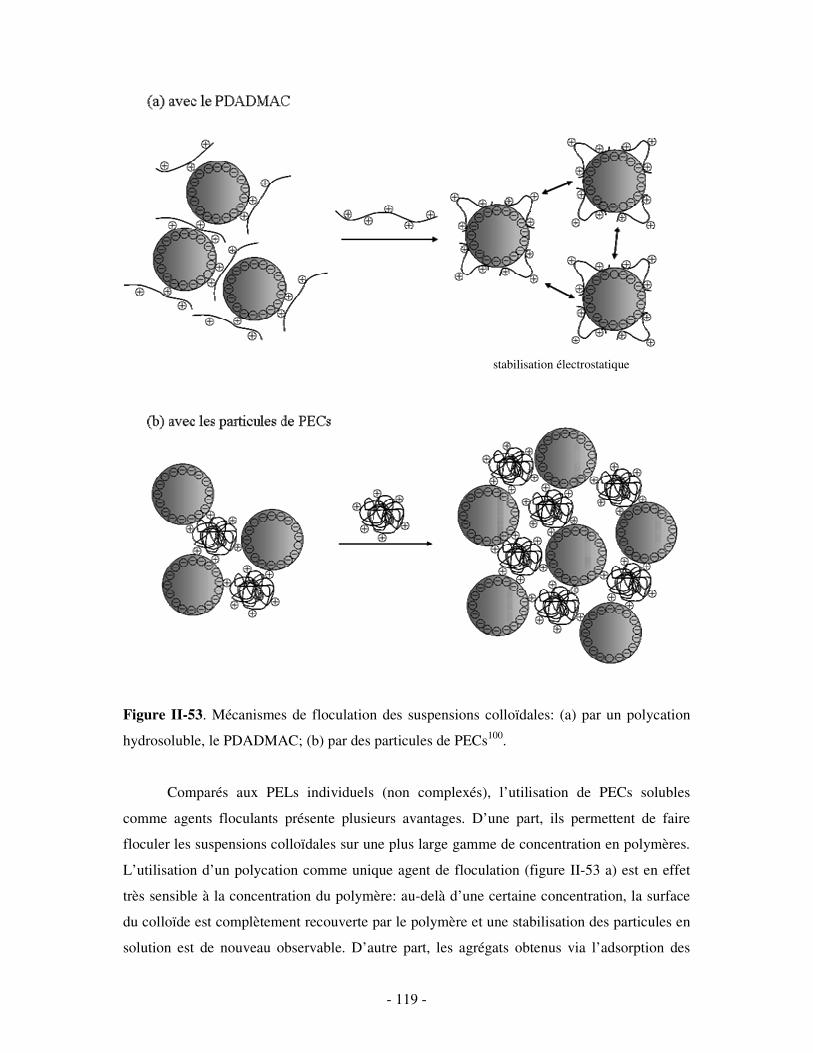
- 119 -
Figure II-53. Mécanismes de floculation des suspensions colloïdales: (a) par un polycation
hydrosoluble, le PDADMAC; (b) par des particules de PECs100.
Comparés aux PELs individuels (non complexés), l’utilisation de PECs solubles
comme agents floculants présente plusieurs avantages. D’une part, ils permettent de faire
floculer les suspensions colloïdales sur une plus large gamme de concentration en polymères.
L’utilisation d’un polycation comme unique agent de floculation (figure II-53 a) est en effet
très sensible à la concentration du polymère: au-delà d’une certaine concentration, la surface
du colloïde est complètement recouverte par le polymère et une stabilisation des particules en
solution est de nouveau observable. D’autre part, les agrégats obtenus via l’adsorption des
stabilisation électrostatique

- 120 -
PECs sont de plus grande taille et, par conséquent, présentent une vitesse de sédimentation
plus rapide, ce qui peut être appréciable dans les domaines de la chimie du papier et du
traitement des eaux usées.
Les PECs solubles trouvent aussi de nombreuses applications dans le domaine
médical. En combinant un polymère anionique et une protéine thérapeutique cationique, ils
peuvent par exemple participer à la préparation de vaccins oraux158. Ceux comportant de
l’ADN sont pour leur part de plus en plus sollicités pour les progrès de la thérapie génique. Ce
type de traitement qui fait l’objet de recherches considérables depuis plus de vingt ans,
consiste à administrer des gènes aux cellules du patient afin de stopper la production de
protéines défaillantes, ou bien de produire de nouvelles protéines. Les gènes en question sont
contenus dans les molécules d’ADN qui doivent être condensées et vectorisées pour pouvoir
traverser la membrane des cellules cibles. En effet, l’ADN acquiert en solution aqueuse une
charge globale négative (par ionisation des groupements phosphate), de même que les
phospholipides qui constituent les membranes cellulaires. Par conséquent, l’introduction de
l’ADN dans les cellules est défavorisée aussi bien par sa conformation très étendue que par
les répulsions électrostatiques existant avec les membranes.
Jusqu’à présent, l’emploi de virus « désactivés » est le moyen le plus fréquemment
utilisé pour vectoriser des gènes au sein des cellules. Ils se montrent en effet très performants
pour franchir les barrières que constituent les membranes des cellules. Leur utilisation
présente cependant quelques limitations liées à leur toxicité, à leur capacité à ne transporter
qu’une petite quantité de matériel génétique, ainsi qu’à leur coût élevé159.
Pour pallier à ces inconvénients, des efforts considérables ont été déployés pour
concevoir des vecteurs synthétiques capables de remplir les fonctions biologiques requises, à
savoir moduler la taille et la charge électrique de l’ADN pour faciliter son transport vers les
cellules cibles, tout en le protégeant de la dégradation par les globules blancs du patient. Du
fait de la grande coopérativité observée lors de l’association entre polyélectrolytes et
tensioactifs de charges opposées, l’utilisation de TAs cationiques comme « empaquetage » de
l’ADN a parfois été suggérée160. Seuls les TAs cationiques sont en effet capables de
complexer l’ADN en particules de petite taille, condition nécessaire pour pouvoir le transférer
efficacement au travers des pores membranaires (les pores nucléaires font 10 nm de diamètre
environ159). D’après Clamme et coll.161, le transfert de gènes au moyen de TAs cationiques est
toutefois limité par le « piégeage » des complexes ADN/TA sur la membrane cellulaire: les
complexes restent adsorbés à la surface de la cellule via des interactions hydrophobes

- 121 -
impliquant les micelles de TA. Un transfert efficace de l’ADN a néanmoins été observé par
Pinnaduwage et coll.162 avec des TAs de type CnTAB associés en liposomes cationiques,
c’est-à-dire en vésicules microscopiques de diamètre compris entre 20 nm et 4µm, constitués
d’une ou plusieurs couche(s) bilipidique(s). Selon Safinya et coll.163-165, ce type d’association
ADN/liposomes cationiques résulte en la formation d’assemblages multicouches comportant
des molécules d’ADN prises en sandwich entre deux membranes bilipidiques. La taille des
structures résultantes peut parfois devenir conséquente mais, d’une façon générale, la taille
des liposomes elle-même limite leur application en tant que vecteurs de gènes in vivo166. Leur
densité de charges trop élevée peut aussi devenir un facteur limitant lorsque l’excès de
charges positives induit de fortes interactions avec l’environnement biologique167.
Au vu de ces difficultés, les PECs constitués d’ADN et de polymères cationiques
synthétiques apparaissent de plus en plus attractifs en terme de souplesse d’utilisation. De
nombreux polymères semblent en effet capables d’optimiser la charge électrique des vecteurs
et d’adapter la taille de ces derniers en fonction de la cellule cible. Parmi ceux susceptibles de
vectoriser l’ADN de façon efficace figurent la poly(L-ornithine)168-170, les poly(4-
vinylpyridines) quaternisées171,172, la poly(éthylènimine) (linéaire ou branchée)173-176 et les
dendrimères de type poly(amidoamine) (PAMAM)177-179. Il est cependant reconnu que
l’utilisation de polycations linéaires comme la poly(L-lysine) et la poly(L-arginine) peut
induire une forte contraction des molécules d’ADN et poser des problèmes de floculation et
de précipitation des systèmes174,180. Un moyen d’y remédier consiste alors à introduire des
groupements hydrophiles le long de la chaîne polycationique (formation d’un corona
protecteur autour des complexes). En synthétisant des copolymères de poly(L-lysine)
comportant des greffons de dextrane, Maruyama et coll.181,182 ont ainsi pu observer une
amélioration de la solubilité des complexes ADN/polycation. Un effet similaire a pu être
constaté en utilisant des copolymères à blocs poly(éthylène glycol)/poly(L-lysine) (PEG-b-
PLL)180,183-185 ou bien des copolymères en peigne composés de PLL et de PEG greffé (PEG-g-
PLL)186,187. La présence des segments hydrophiles a de plus l’avantage d’écranter
partiellement les charges en surface des complexes qui, de ce fait, voient leur biocompatibilité
et donc leur efficacité de transfection s’améliorer.
Malgré ces résultats prometteurs, d’énormes progrès restent encore à accomplir. Il ne
faut en effet pas moins de 100 000 molécules d'ADN par cellule cible pour qu'une seule
séquence parvienne à pénétrer dans le noyau. Or, à cette concentration, commencent à se
poser des problèmes de toxicité du matériau polymère. De plus, il faut que l'ADN soit libéré
de son vecteur, ce qui n’est pas toujours aisé étant donné le caractère partiellement réversible
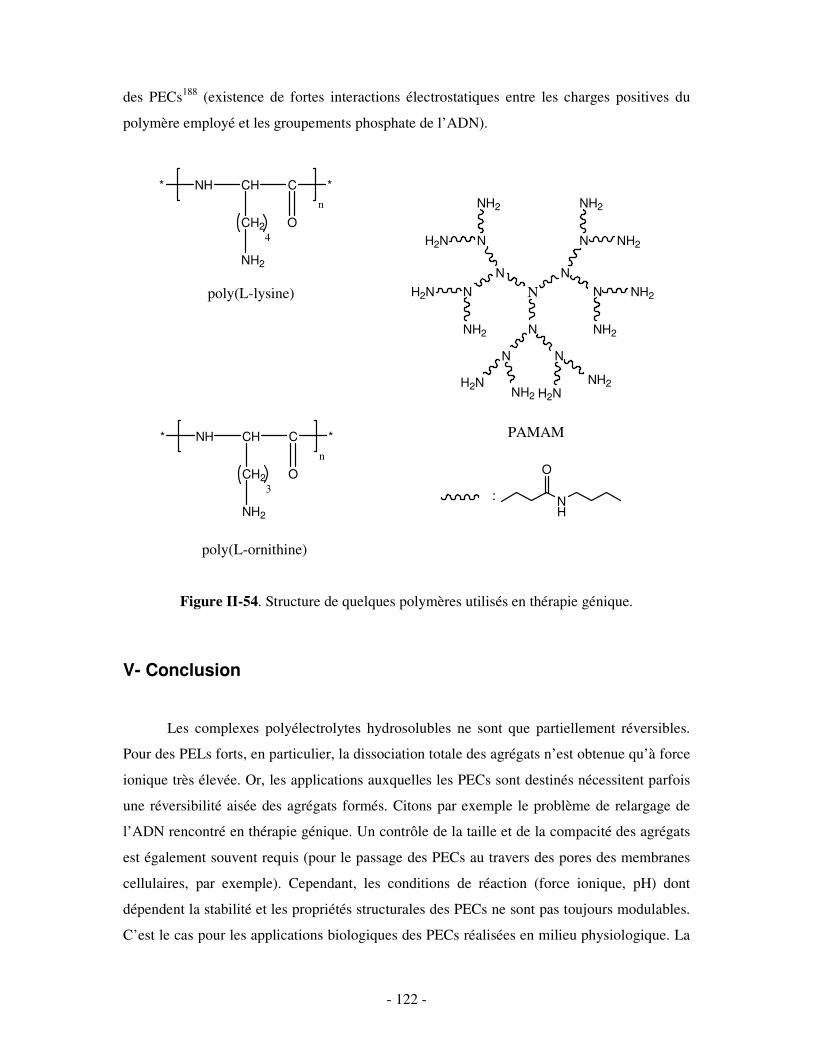
- 122 -
des PECs188 (existence de fortes interactions électrostatiques entre les charges positives du
polymère employé et les groupements phosphate de l’ADN).
* NH CH C *
OCH2
NH2
4
n
* NH CH C *
OCH2
NH2
3
n
poly(L-lysine)
poly(L-ornithine)
NNN
N
N
N
NN
N
N
NH2
NH2
NH2
NH2
NH2H2NNH2
H2N
NH2
H2N
H2N
NH2
PAMAM
: NH
O
Figure II-54. Structure de quelques polymères utilisés en thérapie génique.
V- Conclusion
Les complexes polyélectrolytes hydrosolubles ne sont que partiellement réversibles.
Pour des PELs forts, en particulier, la dissociation totale des agrégats n’est obtenue qu’à force
ionique très élevée. Or, les applications auxquelles les PECs sont destinés nécessitent parfois
une réversibilité aisée des agrégats formés. Citons par exemple le problème de relargage de
l’ADN rencontré en thérapie génique. Un contrôle de la taille et de la compacité des agrégats
est également souvent requis (pour le passage des PECs au travers des pores des membranes
cellulaires, par exemple). Cependant, les conditions de réaction (force ionique, pH) dont
dépendent la stabilité et les propriétés structurales des PECs ne sont pas toujours modulables.
C’est le cas pour les applications biologiques des PECs réalisées en milieu physiologique. La

- 123 -
compacité des agrégats peut alors être contrôlée uniquement en faisant varier la densité de
charges des PELs (par modification chimique, par exemple).
Pour tenter d’apporter des solutions à ces problèmes, nous nous proposons d’élaborer
un nouveau type de PEC basé sur l’association entre un polymère neutre de cyclodextrine, un
tensioactif ionique et un PEL de charge opposée. Cette association ternaire implique, d’une
part, des complexes d’inclusion entre les cavités de CD et les molécules de TA et, d’autre
part, des interactions électrostatiques entre les charges de ces dernières et celles de signe
opposé du polyion. Dans cette configuration, le TA joue le rôle de connecteur entre les deux
polymères.
Comparé aux PECs classiques basés principalement sur des interactions
électrostatiques, ce nouveau modèle de complexe présente à priori une réversibilité accrue: la
dissociation du système ternaire peut être obtenue par ajout de sel (écrantage classique des
interactions électrostatiques) et/ou par ajout d’un compétiteur capable de dissocier les
complexes d’inclusion CD/TA. Le système proposé offre également une plus grande
versatilité: la densité de charges du PEL réversible induit par l’association poly(CD)/TA
ionique est aisément contrôlable en ajustant la concentration en TA dans le mélange, sans
qu’aucune nouvelle synthèse de polymère soit nécessaire. Un contrôle plus efficace de la
taille et de la compacité des agrégats formés est alors attendu.
Un des objectifs de cette thèse est la mise en évidence de la formation d’un tel
complexe ternaire en solution aqueuse. Des études de viscosimétrie et de diffusion de
neutrons dédiées à ce sujet seront rapportées dans le chapitre IV de ce manuscrit. Mesurer la
viscosité d’une solution est en effet une méthode simple pour observer l’apparition d’un
phénomène macroscopique comme la formation d’agrégats. En milieu dilué, les mesures
viscosimétriques sont sensibles à la compacité de ces derniers. La diffusion de neutrons, quant
à elle, est une technique qui permet en plus d’étudier la structure interne des agrégats de façon
directe.
Le chapitre IV visera aussi à discuter la dépendance de la stabilité et des propriétés
structurales des complexes éventuellement formés vis-à-vis de plusieurs facteurs: du rapport
molaire entre charges opposées du TA et du polyanion (à concentrations en cavités de CD et
en groupements chargés du polyion fixées), des caractéristiques du polyion (nature chimique,
caractère hydrophile/hydrophobe, masse molaire et flexibilité), de la force ionique du milieu
et de la concentration en compétiteurs.

- 124 -
PARTIE II. ETUDE EXPERIMENTALE
Chapitre III. Etude du complexe binaire poly(CD)/TA ionique
en solution diluée
Comme il l’a été mentionné auparavant, ce travail porte sur l’élaboration et l’étude
d’un nouveau modèle de PEC basé sur l’association ternaire entre un polymère de CD, un TA
ionique et un PEL de charges opposées en solution aqueuse. Pour qu’un tel système soit
effectivement formé, il est nécessaire qu’au sein du mélange le TA ionique et le PEL de
charges opposées s’associent par interactions électrostatiques, d’une part, et que le TA et le
poly(CD) forment des complexes d’inclusion, d’autre part.
Ce chapitre vise à s’assurer tout d’abord que la mise en présence dans l’eau du
poly(CD) et du TA ionique seuls conduit bien à la formation de complexes d’inclusion. Dans
une première partie, nous commencerons par présenter les composants et les différentes
techniques de caractérisation utilisés. Puis, nous tenterons d’apporter plusieurs preuves de la
complexation binaire avant de caractériser les agrégats formés. Enfin, nous nous intéresserons
aux propriétés structurales de ces derniers en fonction de deux paramètres: la force ionique du
milieu et la concentration en TA.
I- Conditions expérimentales
1- Les composants utilisés
a) Le polymère de CD
Le polymère de CD employé dans cette étude a été synthétisé par E. Renard (LRP),
par polycondensation de la β-cyclodextrine (β-CD) et de l’épichlorhydrine (EP) en milieu
fortement alcalin14. C’est un copolymère de structure branchée189, constitué de molécules de
β-CD pouvant être modifiées par des séquences plus ou moins longues de poly(2-
hydroxypropyl)éther libres à une extrémité, ou bien jouant le rôle de pontages entre plusieurs
CDs.

- 125 -
O
O*
RO(3) O(2)RO
*
O(6)R
7
* CH2 CH CH2O H
OHm
* CH2 CH CH2O glucose
OHn
H
R =
Figure III-1. Structure schématique des unités de β-CD dans le copolymère β-CD/EP.
La structure exacte du copolymère est difficile à déterminer étant donnés la
polyfonctionnalité des unités de CD (avec trois positions possibles O(2), O(3) et O(6) pour les
substitutions, soit 21 au total pour une molécule de β-CD) et le fait que l’EP peut aussi bien
réagir avec les groupements hydroxyle d’une CD qu’avec ceux des substituants. Dans ce
dernier cas, la polymérisation peut donner lieu à des longueurs de chaîne plus longues ou bien
à la formation de ponts. La structure du copolymère peut devenir très compacte et peut être
schématisée comme sur la figure III-2.
Figure III-2. Structure schématique du copolymère β-CD/EP.
L’échantillon utilisé tout au long de ce travail est soluble dans l’eau. Sa masse molaire
moyenne déterminée par chromatographie d’exclusion stérique couplée à un détecteur de
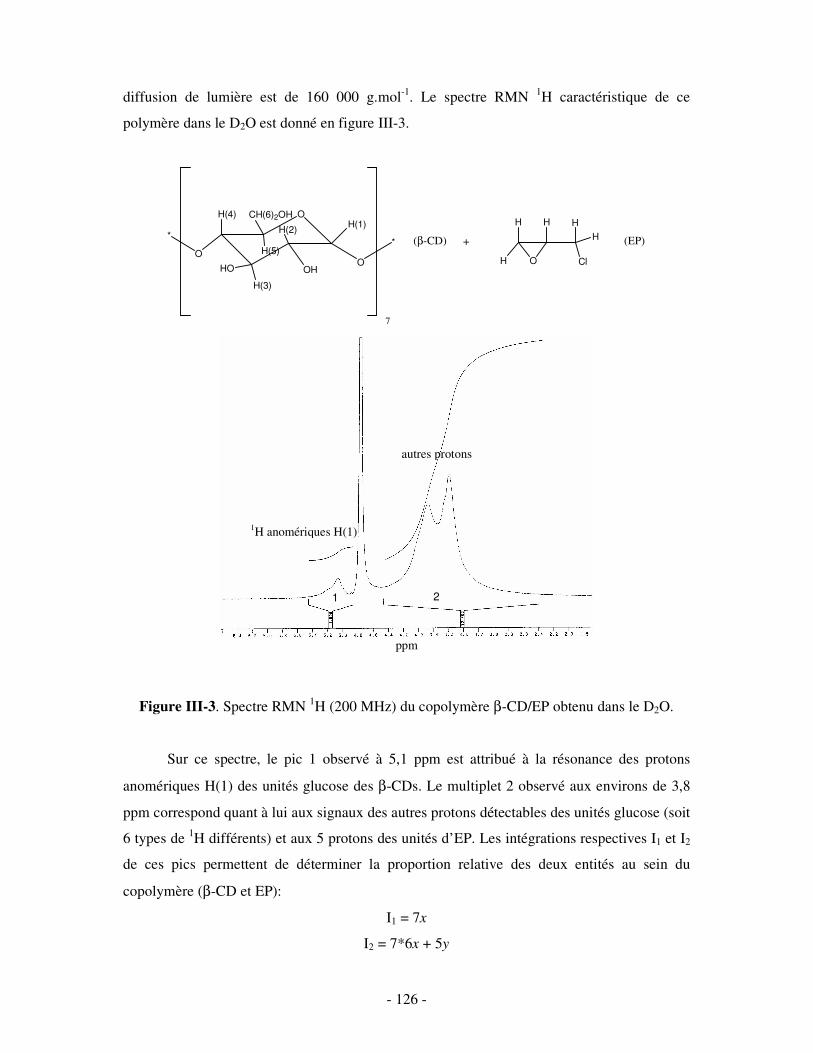
- 126 -
diffusion de lumière est de 160 000 g.mol-1. Le spectre RMN 1H caractéristique de ce
polymère dans le D2O est donné en figure III-3.
Figure III-3. Spectre RMN 1H (200 MHz) du copolymère β-CD/EP obtenu dans le D2O.
Sur ce spectre, le pic 1 observé à 5,1 ppm est attribué à la résonance des protons
anomériques H(1) des unités glucose des β-CDs. Le multiplet 2 observé aux environs de 3,8
ppm correspond quant à lui aux signaux des autres protons détectables des unités glucose (soit
6 types de 1H différents) et aux 5 protons des unités d’EP. Les intégrations respectives I1 et I2
de ces pics permettent de déterminer la proportion relative des deux entités au sein du
copolymère (β-CD et EP):
I1 = 7x
I2 = 7*6x + 5y
1 2
1H anomériques H(1)
autres protons
O
O
*
HO OHO
*
CH(6)2OH
7
H(1)H(2)
H(5)
H(3)
H(4)
(β-CD) +
H H
H O
HH
Cl
(EP)
ppm

- 127 -
où x représente le nombre de moles de β-CD et y, le nombre de moles d’EP (sans les atomes
de chlore) dans l’échantillon. Le pourcentage massique de β-CD dans le copolymère est alors
donné par:
EPCD
CD
yMxM
xMCDmassique
+=−
−
−
β
ββ% ,
avec Mβ-CD = 1135 g.mol-1 et MEP = 57 g.mol-1 (sans Cl). Un taux de 59 % en poids de β-CD
est obtenu, ce qui représente environ 83 cavités de β-CD par chaîne de polymère
(160000*0,59/1135). Le nombre moyen de greffons d’épichlorhydrine par cavité de β-CD est
alors estimé à 14 (= (0,41/57)/(0,59/1135)).
b) Le TA ionique
Le TA choisi est le chlorure de dodécyltriméthylammonium (DTAC) dont la formule
chimique est représentée sur la figure suivante:
CH3 CH2 N+
CH3
CH3
CH3
11Cl-
Figure III-4. Formule chimique du DTAC.
Sa masse molaire est égale à 263,9 g.mol-1 et sa concentration micellaire critique dans
l’eau pure est de 2.10-2 M à 25°C44.
L’échantillon de DTAC utilisé dans cette étude a été fourni par Acros Organics
(France). Il n’a subi aucune repurification de notre part et a donc été utilisé tel quel, c’est-à-
dire pur à 98 %. Etant donné le caractère hygroscopique du produit, le lot de DTAC a
toutefois été conservé dans un dessicateur sous vide.
Un bromure de dodécyltriméthylammonium deutéré (dDTAB) a aussi été employé
pour certaines expériences de diffusion de neutrons. Sa synthèse, réalisée par V. Wintgens
(LRP), est décrite dans la référence 190.

- 128 -
Toutes les solutions ont été préparées avec de l’eau ultra-pure fournie par un système
de purification et de déionisation Millipore (exceptés les échantillons de SANS préparés dans
du D2O). Elles ont été préparées par pesée du solvant et des solutés. Leurs concentrations sont
donc exprimées en g.g-1 d’eau (exceptées celles des échantillons de SANS, exprimées en g.L-1
de D2O).
Les échantillons ont été préparés au moins 12 heures avant chaque mesure.
2- Les techniques employées
a) Mesures de tension de surface
i) Principe
Cette technique repose sur le fait que l’adsorption de molécules tensioactives à la
surface d’une solution s’accompagne d’une réduction marquée de la tension de surface γ de
cette dernière (cf. chapitre I, paragraphe II-1). Pour des TAs ioniques A+B- se dissociant
complètement en ions positifs et négatifs en solution, les variations de γ peuvent être reliées
aux variations des concentrations en solutés selon l’équation de Gibbs42:
( )−−++ Γ+Γ−=
BBAANdNdRTd lnlnγ , (1)
où Γi est le nombre de moles de soluté i adsorbé par unité de surface à l’interface solution/air
et Ni, la concentration molaire en soluté i. Pour maintenir l’électroneutralité à la surface de la
solution, il est admis que ΓA+ = ΓB- = Γ. Par ailleurs, on a NA+ = NB- = N. Par conséquent,
l’équation (1) peut être réécrite sous la forme:
NdRTd ln2Γ−=γ . (2)
Il est à noter qu’en présence d’un excès constant de sel possédant un ion commun avec
le TA (NaCl, par exemple, pour le DTAC), la variation de la concentration en contre-ions
dans l’équation (2) devient négligeable. L’équation (2) s’écrit alors:
NdRTd lnΓ−=γ . (3)

- 129 -
La forme de l’équation de Gibbs pour des TAs ioniques en présence d’un excès de sel est
donc identique à celle définie pour des TAs neutres (cf. p.26).
Il est important de préciser que les variations de γ observées sont attribuables aux
variations de la concentration en TA libre uniquement (l’équation de Gibbs n’est valable
qu’en absence de micelles). Dès l’apparition de micelles dans le milieu, les valeurs de γ
deviennent généralement constantes. L’observation d’un plateau permet donc de déduire la
valeur de la cmc du TA en solution.
ii) Appareillage
Les mesures de tension de surface ont été réalisées au laboratoire MPI (Université
d’Evry), à l’aide d’un tensiomètre NIMA ST 9000. Des feuilles de papier filtre de largeur
10,25 mm et d’épaisseur 0,25 mm ont été utilisées pour effectuer les mesures. Ces dernières
ont été réalisées à température ambiante, après calibration du tensiomètre: pour une masse de
100 mg (déposée sur une petite nacelle de 21 mm de diamètre suspendue au tensiomètre), une
valeur équivalente de tension de surface de 46,7 mN.m-1 est attendue.
b) Conductimétrie
i) Principe
La conductivité d’une solution est reliée linéairement aux concentrations des espèces
ioniques présentes. Soit un ion i, de charge zi et de concentration molaire Ni. Cet ion participe
à la conductivité totale de la solution en quantité λiziNi où λi, appelée conductivité équivalente
de l’ion i, est une caractéristique de l’ion considéré, proportionnelle à sa mobilité (λCl- = 76,3
S.cm2.mol-1 et λNa+ = 50,1 S.cm2.mol-1, par exemple). La conductivité totale de la solution est
alors donnée par191:
iii
i Nz= λσ ,
exprimée en -1.cm-1 ou en S.cm-1 (1 S = 1 -1).

- 130 -
Dans la pratique, il est mesuré généralement non pas la conductivité σ de la solution,
mais la conductance κ d’une colonne d’électrolyte dont la section et la longueur sont
caractéristiques de la cellule de mesure employée. La conductance κ d’une solution est dans
ce cas reliée à sa conductivité σ selon:
κ iii
i Nzkk == λσ 1
,
où k est une constante caractéristique de la cellule (en cm-1). κ est alors exprimée en -1 ou en
S.
ii) Appareillage
Les mesures conductimétriques ont été effectuées à l’aide d’un conductimètre de type
CD 6N fourni par TACUSSEL. La cellule de mesure de cet appareil comporte deux électrodes
de platine, de surface égale à 0,25 cm2, espacées de 0,5 cm. Sa constante caractéristique k est
égale à 2 cm-1 (= 0,5/0,25).
Tous les échantillons ont été placés dans un bain thermostaté à 25°C avant chaque
mesure.
c) Viscosimétrie capillaire
i) Principe
La viscosité spécifique d’une solution suffisamment diluée est exprimée par la relation
suivante, déduite de la loi de Poiseuille:
0
0
ttt
spe
−=η ,
où t0 est le temps d’écoulement du solvant et t, le temps d’écoulement de la solution.
Pour une solution simple (ne comportant qu’un soluté), la viscosité réduite est définie
par:

- 131 -
Cspe
red
ηη = ,
où C est la concentration en g.L-1 du soluté. Par contre, pour une solution comportant
plusieurs composants, la viscosité réduite du mélange est définie par:
tot
spered C
ηη = ,
où Ctot est la concentration totale en g.L-1.
Par ailleurs, la viscosité réduite d’une solution d’un polymère neutre est exprimée par
l’équation de Huggins:
[ ] [ ] Λ++= CAred
2ηηη ,
et celle d’un polymère ionique par l’équation de Fuoss:
[ ] [ ] Λ++= 2111
CB
red ηηη .
Dans ces deux équations, A et B sont des constantes et [η], désignée comme la viscosité
intrinsèque du polymère en solution, caractérise la compacité de ce dernier. En effet, [η] est
proportionnelle au rapport volume/masse du polymère selon:
[ ] fM
V ah ℵ⋅=
25η ,
où Vh est le volume hydrodynamique d’une chaîne du polymère, M, sa masse molaire, aℵ , le
nombre d’Avogadro et f, une fonction décrivant la perméabilité de la chaîne (f → 1 lorsque la
chaîne est imperméable, c’est-à-dire lorsque les molécules de solvant au sein de la pelote se
déplacent avec elle).
ii) Appareillage
La viscosité des solutions a été mesurée à l’aide d’un viscosimètre à capillaire de type
Ubbelohde à niveau suspendu, placé dans un bain thermostaté à 25°C.

- 132 -
Les échantillons ont été préalablement filtrés à l’aide de verres frittés de porosité 2
(diamètre des pores compris entre 40 et 100 µm). Toutes les dilutions nécessaires ont été
effectuées en ajoutant de l’eau ultra-pure directement dans le viscosimètre.
d) Diffusion de neutrons aux petits angles (Small Angle Neutron Scattering, SANS)
i) Principe
Une expérience de diffusion de neutrons consiste à envoyer sur un échantillon un
faisceau de neutrons de longueur d’onde λ, et à mesurer les variations de l’intensité diffusée
I(q) en fonction de la norme du vecteur de diffusion q . Celui-ci est défini comme la
différence vectorielle 0kk − (figure III-5) entre le vecteur d’onde diffusé k et le vecteur
d’onde incident 0k . Sa norme est donnée par la relation:
=2
sin4 θλπ
q , (1)
où θ est l’angle de diffusion défini par les vecteurs 0k et k .
Figure III-5. Diffusion d’une onde plane.
O
M
θ
détecteur
r k
0k
q

- 133 -
Sur la figure III-5, une onde plane vient frapper un noyau de l’échantillon au point O,
défini comme l’origine des coordonnées et de la phase. Elle est ensuite diffusée dans toutes
les directions de l’espace. L’onde plane incidente frappe ainsi de nombreux noyaux au sein de
l’échantillon, chacun d’entre eux induisant une nouvelle onde sphérique. Considérons l’une de
ces ondes générée au point M, dont la position par rapport à O est repérée par le vecteur r .
Au niveau du détecteur, la différence de phase enregistrée entre l’onde issue du point O et
celle issue du point M est égale à rq ⋅ (d’après la référence 192). L’amplitude complexe de
l’onde diffusée au point M dans une direction x peut être exprimée par rapport à l’onde
diffusée en O de la façon suivante:
( ) ( )( )rqtiatxA ⋅−= ωexp,
( ) ( )rqitia ⋅−= expexp ω , (2)
où a, nommée longueur de diffusion, est l’amplitude de l’onde diffusée relative à celle de
l’onde incidente, est la fréquence angulaire et t, le temps. Lorsque l’interaction du
rayonnement avec la matière est de nature élastique, c’est-à-dire sans transfert d’énergie (soit
0kk = ), le terme de fréquence t peut être omis dans l’équation (2) (ce terme n’est variable
que si les neutrons subissent des fluctuations d’énergie). L’amplitude relative de l’onde
diffusée par le noyau au point M dépend alors uniquement du terme ( )rqia ⋅−exp .
Pour un système composé de N noyaux identiques (de même longueur de diffusion a),
l’amplitude totale diffusée s’écrit:
( ) ( )=
⋅−=N
iirqiaqA
1
exp (3)
et l’intensité détectée est donnée par:
( ) ( ) ( ) ( ) ( )==
⋅⋅−==N
jj
N
ii rqirqiaqAqAqI
11
2* expexp
( )( )= =
−⋅−=N
i
N
jji rrqia
1 1
2 exp . (4)

- 134 -
L’intensité diffusée par l’ensemble des noyaux dépend ainsi de leurs positions relatives au
sein du système. Les propriétés de I(q) sont donc interprétables en termes d’arrangement
structural de l’échantillon.
Il est important de souligner que la valeur de a dépend de la nature du noyau et varie
de façon importante lorsque l’atome considéré est substitué par l’un de ses isotopes193. Par
exemple, la longueur de diffusion de l’hydrogène (aH = -0,374.10-12 cm) est très différente de
celle du deutérium (aD = +0,667.10-12 cm). Le remplacement d’un proton par un deutérium se
traduit donc par un changement conséquent de l’intensité diffusée. Notons que la substitution
isotopique engendre cependant peu de modifications des propriétés physico-chimiques du
système. Les cortèges électroniques des atomes sont en effet conservés.
Considérons maintenant une section σ de l’échantillon telle que tout rayonnement
passant par cette section soit diffusé. Le nombre de neutrons n(θ) (en s-1) diffusé par
l’échantillon dans un angle solide ∆, à un certain angle de diffusion θ, est donné par:
( ) ( ) ( ) ∆ΩΩ
Θ=d
dn
λθσλθ , , (5)
où Θ(λ) est le flux de neutrons incident (en cm-2.s-1) et Ωd
dσ, la section efficace différentielle
de diffusion (en cm-2) qui est une représentation quantitative de l’interaction
rayonnement/matière.
Figure III-6. Principe d’une expérience de diffusion de neutrons.
n(θ)
θ
∆
Θ(λ)

- 135 -
L’intensité diffusée mesurée par le détecteur est alors reliée à la section efficace différentielle
de diffusion par unité de volume de l’échantillon, ( ) ( )Ω
=Σd
qdV
qσ1
(en cm-1), selon:
( ) ( )qdTAqI ΣΩΘ= ε0 , (6)
où 0 est l’angle solide défini par le détecteur, ε, l’efficacité du détecteur, A, la section du
faisceau incident, T, la transmission de l’échantillon et d, l’épaisseur de l’échantillon.
La spécificité de la SANS (θ < 10°, 3 < λ < 20 Å) provient de l’échelle de distances
(entre 2 et 500 Å) que cette technique permet d’explorer. A cette échelle, il est pratique de
considérer comme diffuseurs non plus les noyaux des atomes, mais des objets de plus grande
taille appelés diffuseurs élémentaires, telles que les molécules de solvant ou les unités de
répétition d’un polymère. La longueur de diffusion d’un diffuseur élémentaire, a, est dans ce
cas égale à la somme des longueurs de diffusion des atomes qui le composent.
Pour un système comportant n particules homogènes, constituées chacune de N
diffuseurs élémentaires d’une même espèce chimique, et dispersées dans un solvant de
volume V (solution d’un homopolymère, par exemple), les relations fondamentales de la
diffusion aux petits angles sont les suivantes194:
( )
−⋅=Σ
nn NN
jiji rrqib
Vq
,
,
,
,
2 exp1
βα
βα
( )qSb 2= , (7)
00 v
vaab −=
( )0ρρ −ℵ
=a
v , (8)
avec ava ℵ=ρ et av
aℵ=
0
00ρ ,
où b (en cm) est la longueur de contraste du diffuseur élémentaire (monomère), reliée aux
longueurs de diffusion a ou aux densités de longueurs de diffusion des diffuseurs
élémentaires de volumes molaires v (le solvant est repéré par l’indice 0), aℵ étant le nombre
d’Avogadro. S(q) (en cm-3) est la fonction de structure des particules homogènes

- 136 -
(homopolymère). C’est une combinaison linéaire d’une fonction de corrélation
intramoléculaire, P(q), appelée facteur de forme, caractéristique de la taille et de la forme des
particules, et d’une fonction de corrélation intermoléculaire, Q(q):
( ) ( ) ( )[ ]qQNnqPnNV
qS 2221 += . (9)
Aux grandes valeurs de q (caractéristiques de petites distances dans l’espace réel), le terme
P(q) domine tandis qu’à plus faibles valeurs de q (distances plus grandes), c’est le terme
interactionnel Q(q) qui est prépondérant, sauf généralement en milieu très dilué pour les
polymères neutres. Lorsque les particules n’interagissent pas entre elles (Q(q) négligeable),
leur taille moyenne, Rg, peut être déduite de l’approximation de Guinier, valable à faibles
valeurs de q uniquement (qRg 1):
( ) ( )qPnNV
qS 21=
−=
3exp
122
2 gRqnN
V . (10)
A plus grandes valeurs de q, par contre, S(q) varie en q-α où l’exposant α permet d’identifier
la forme des particules et, en quelque sorte, de renseigner sur leur compacité:
α = 1 pour des bâtonnets rigides,
α = 2 pour des pelotes gaussiennes (chaînes idéales),
α = 4 pour des sphères.
Pour n particules hétérogènes composées chacune de Ni diffuseurs élémentaires
d’espèces chimiques i différentes, la relation de base (7) se généralise sous la forme:
( ) ( )qSbbq ijj
jii=Σ
,
(11)
avec ( )0ρρ −ℵ
= ia
ii
vb (12)
où bi et bj sont les longueurs de contraste des diffuseurs élémentaires i et j, et Sij(q), des
facteurs de structure partiels qui peuvent aussi être séparés en fonctions de diffusion intra- et

- 137 -
interparticules. Par exemple, pour des particules hétérogènes ne comportant que deux espèces
différentes de diffuseurs élémentaires, l’équation (11) s’écrit:
( ) ( ) ( ) ( )qSbqSbbqSbq 22
22122111
21 2 ++=Σ (13)
où S11 correspond à l’intensité que diffuseraient les espèces 1 en absence des espèces 2 (et
inversement pour S22),
S12 représente le terme de diffusion lié aux interactions croisées entre les diffuseurs 1 et 2.
L’interprétation des spectres de diffusion pose alors de sérieuses difficultés qui peuvent
cependant être contournées en utilisant la substitution isotopique ou bien la méthode de
variation de contraste. Celle-ci consiste à utiliser comme solvant un mélange H2O/D2O
caractérisé par un pourcentage volumique xD2O en D2O. La densité de longueur de diffusion
du solvant s’écrit alors195:
( ) OHODODOD xx 22220 1 ρρρ −+= . (14)
En faisant varier la composition xD2O du solvant, il est ainsi possible de modifier, voire même
d’annuler la contribution d’un diffuseur élémentaire à la diffusion du rayonnement
neutronique (cf. équation 12). En ce qui concerne les mélanges de poly(β-CD) et de TA
cationique dans le D2O pur, l’utilisation de TA deutéré ( = 6,95.1010 cm-2), de densité de
longueur de diffusion proche de celle du solvant (D2O = 6,41.1010 cm-2), a permis d’observer
au sein des mélanges uniquement la contribution à la diffusion du polymère (poly(β-CD) =
2,29.1010 cm-2).
ii) Appareillage
Les mesures de SANS ont été réalisées au laboratoire Léon Brillouin (CEA-CNRS)
sur le spectromètre PACE, en collaboration avec Loïc Auvray. Comme le montre la figure III-
7, le faisceau de neutrons fourni par le réacteur Orphée (par fission d’atomes d’uranium 235),
est collimaté avant de rencontrer l’échantillon. La collimation est définie par des diaphragmes
Φ1 et Φ2 de 0,7 et 1 cm de diamètre, respectivement.

- 138 -
Figure III-7. Principe de montage du spectromètre PACE.
La détection des neutrons diffusés s’effectue via absorption par du gaz BF3. Les
électrons générés par ionisation de ce dernier sont comptabilisés par un multidétecteur
constitué de 30 anneaux concentriques, de 1 cm de largeur chacun.
Les échantillons ont été préparés dans des cellules en quartz de type Hellma
caractérisées par une épaisseur de 2 mm. La température du porte-échantillons a été
maintenue à 25°C à l’aide d’une circulation d’eau et d’un bain thermostaté.
iii) Choix des configurations d’étude
Le choix des configurations d’étude s’appuie sur le type d’informations désirées à
propos de l’échantillon. Au cours de ce travail, deux configurations présentées dans le tableau
III-1 ont été utilisées. Chacune de ces configurations est caractérisée par une longueur d’onde
λ et une distance échantillon-détecteur D correspondant à une gamme de valeurs de q bien
définie.
Configuration λλλλ (Å) D (m) qmin (Å-1) qmax (Å-1)
Petits q 13 4,6 3,2.10-3 3,4.10-2
Grands q 6 3 1,1.10-2 1,2.10-1
Tableau III-1. Configurations d’étude des expériences de SANS.
La gamme des petits q permet d’explorer des distances comprises entre 30 et 310 Å,
tandis que celle des grands q vise une échelle de distances comprises entre 8 et 90 Å. Ainsi,
monochromateur
guide du faisceau
collimation échantillon
θ
multidétecteur
D

- 139 -
des informations sur la taille et sur la structure interne des particules présentes en solution
sont attendues en utilisant, respectivement, la gamme des petits q et celle des grands q.
iv) Traitement des données
Pour pouvoir raccorder les spectres de diffusion sur les deux gammes de q considérées
et, de même, pour pouvoir comparer différents spectres entre eux, les intensités détectées ont
été normalisées à l’échelle absolue (en cm-1) à l’aide du logiciel Pasidur développé par le
LLB. Dans ce procédé, les spectres sont divisés par leur transmission correspondante (rapport
de l’intensité transmise par l’échantillon à l’intensité incidente). La diffusion de la cellule de
mesure (cellule vide) et celle de l’eau légère (H2O) sont soustraites de celle de l’ensemble
pour s’affranchir de contributions parasites. De plus, les spectres sont corrigés de la diffusion
du solvant (D2O) afin de ne considérer que la diffusion du soluté. Enfin, les données sont
corrigées d’un facteur de normalisation finale:
−=
CVOH TTdF
114
1
2π
où d est l’épaisseur de l’échantillon, TH2O et TCV les transmissions de l’eau et de la cellule
vide, respectivement.
e) Fluorimétrie
i) Principe
La fluorimétrie a été essentiellement utilisée pour déterminer la constante
d’association K des complexes poly(β-CD)/DTAC (le principe de détermination de K sera
exposé au paragraphe II-2-a). Etant donné que ni le poly(β-CD) ni le DTAC ne comportent de
chromophore, l’utilisation d’une sonde fluorescente capable de former des complexes
d’inclusion avec les cavités de CD s’est avérée nécessaire. La sonde 4-amino-N-
tertiobutylphtalimide (synthétisée par V. Wintgens (LRP)) s’est ainsi vue attribuer le rôle de
compétiteur vis-à-vis de l’inclusion du DTAC dans les cavités de CD.

- 140 -
N
H2N
C
CH3
CH3
CH3
O
O
Figure III-8. Formule chimique de la sonde 4-amino-N-tertiobutylphtalimide.
La complexation par les cavités de CD ayant tendance à bloquer les phénomènes non
radiatifs dissipateurs d’énergie, l’intensité de fluorescence émise par la sonde augmente en
présence de CD. Les mesures fluorimétriques ont alors consisté à suivre l’évolution de
l’intensité émise par la sonde en présence d’une quantité connue de poly(β-CD), en fonction
de la concentration en DTAC.
Avant chaque série de mesures, nous avons procédé à la recherche d’un point
isobestique sur les spectres d’absorption UV de la sonde en absence et en présence du poly(β-
CD), c’est-à-dire à la recherche d’une longueur d’onde d’excitation pour laquelle la densité
optique des solutions (proportionnelle au nombre de photons absorbés) reste constante
quelque soit la concentration en cavités de β-CD. C’est cette longueur d’onde qui, par la suite,
a été utilisée comme longueur d’onde d’excitation pour les expériences de fluorimétrie. Le
nombre de photons absorbés par les solutions étant supposé rester constant pour cette
longueur d’onde, il n’est pas nécessaire d’effectuer de corrections correspondantes sur les
intensités de fluorescence émises (nombre de photons émis).
Nous avons également procédé à une comparaison des spectres d’émission de la sonde
en absence et en présence du poly(β-CD), afin de déterminer la longueur d’onde d’émission
pour laquelle la différence d’intensité entre les deux spectres (∆I) est maximale. C’est à cette
longueur d’onde que les intensités de fluorescence ont toutes par la suite été mesurées, de
façon à obtenir des valeurs de ∆I les plus précises possible (ces valeurs interviennent dans le
calcul de K).

- 141 -
ii) Appareillage
La recherche d’un point isobestique a été effectuée à l’aide d’un spectromètre
VARIAN CARY 50. Les mesures fluorimétriques ont quant à elles été réalisées à l’aide d’un
spectrofluorimètre SLM AMINCO 8100 équipé d’une lampe à xénon et d’un
monochromateur.
Les échantillons ont été préparés dans des cuves en quartz de 1 cm de côtés. Ils ont été
maintenus à 25°C grâce à un circuit d’eau alimenté par un bain thermostaté.
II- Mise en évidence de la complexation binaire poly(CD)/TA ionique
et détermination de la constante d’association
1- Mise en évidence de la complexation binaire
a) Par mesures de tension de surface
La figure III-9 présente les variations de tension de surface d’une solution de DTAC
seul, ainsi que celles d’un mélange aqueux de poly(β-CD) et de DTAC de concentration en
cavités de β-CD fixée à 14.10-3 M. Ces résultats ont été obtenus en ajoutant respectivement
des volumes croissants d’une solution mère de DTAC (NDTACi = 40.10-3 M) dans de l’eau
ultra-pure (Veaui = 10 mL), et des volumes croissants d’un mélange poly(β-CD)/DTAC (Nβ-CD
= 14.10-3 M, NDTACi = 80.10-3 M) dans une solution de poly(β-CD) (Nβ-CD = 14.10-3 M, Vi = 10
mL).
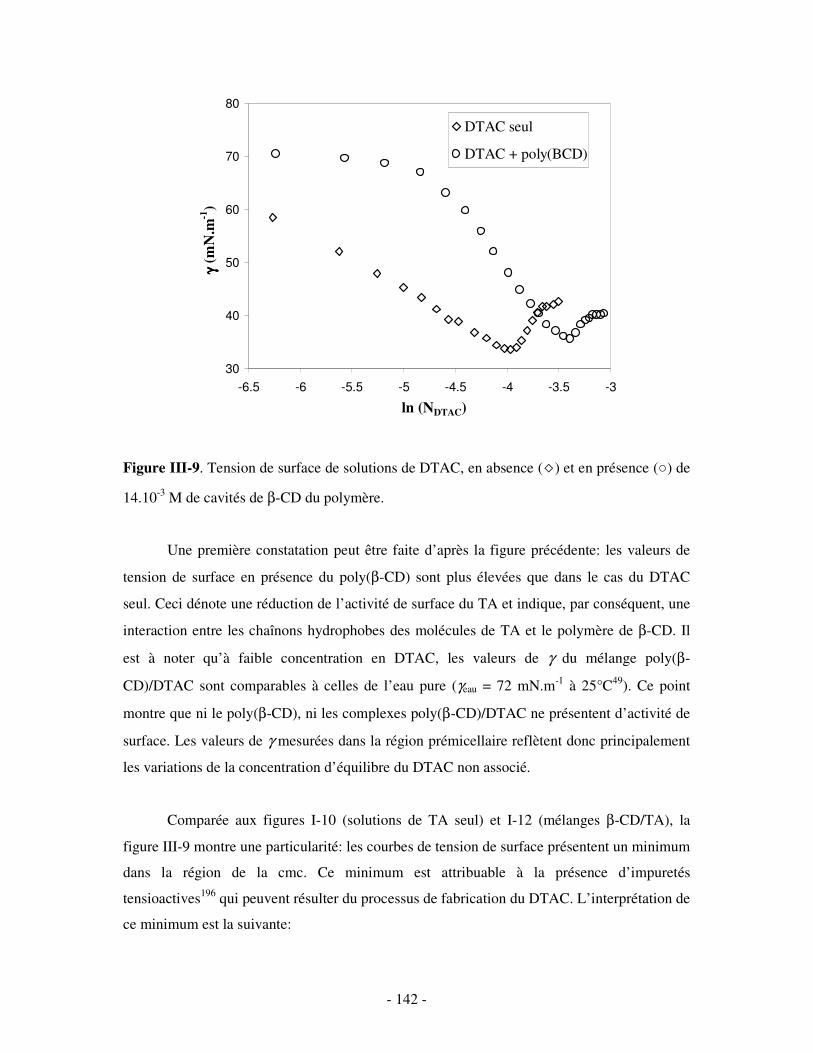
- 142 -
30
40
50
60
70
80
-6.5 -6 -5.5 -5 -4.5 -4 -3.5 -3
ln (NDTAC)
γ γ γ γ (m
N.m
-1)
DTAC seul
DTAC + poly(BCD)
Figure III-9. Tension de surface de solutions de DTAC, en absence ( ) et en présence () de
14.10-3 M de cavités de β-CD du polymère.
Une première constatation peut être faite d’après la figure précédente: les valeurs de
tension de surface en présence du poly(β-CD) sont plus élevées que dans le cas du DTAC
seul. Ceci dénote une réduction de l’activité de surface du TA et indique, par conséquent, une
interaction entre les chaînons hydrophobes des molécules de TA et le polymère de β-CD. Il
est à noter qu’à faible concentration en DTAC, les valeurs de γ du mélange poly(β-
CD)/DTAC sont comparables à celles de l’eau pure (γeau = 72 mN.m-1 à 25°C49). Ce point
montre que ni le poly(β-CD), ni les complexes poly(β-CD)/DTAC ne présentent d’activité de
surface. Les valeurs de γ mesurées dans la région prémicellaire reflètent donc principalement
les variations de la concentration d’équilibre du DTAC non associé.
Comparée aux figures I-10 (solutions de TA seul) et I-12 (mélanges β-CD/TA), la
figure III-9 montre une particularité: les courbes de tension de surface présentent un minimum
dans la région de la cmc. Ce minimum est attribuable à la présence d’impuretés
tensioactives196 qui peuvent résulter du processus de fabrication du DTAC. L’interprétation de
ce minimum est la suivante:

- 143 -
- pour des concentrations en DTAC inférieures à la cmc, les impuretés s’adsorbent à la
surface des solutions parmi les molécules de DTAC. Cela a pour conséquence de
réduire la tension de surface à des valeurs plus faibles que ce qu’elles seraient si le
DTAC était pur.
- pour des concentrations supérieures à la cmc, les impuretés sont progressivement
solubilisées dans les micelles de DTAC, provoquant une remontée de la tension de
surface vers la valeur de γ qui serait observée dans le cas d’un DTAC pur.
Quoiqu’il en soit, la présence de 2 % au maximum d’impuretés (Acros Organics indique un
DTAC pur à 98 %) a peu d’influence sur la valeur de la cmc du TA. En effet, les mesures de
tension de surface effectuées sur la solution de DTAC seul (sans poly(β-CD)) conduisent à
une valeur de cmc de 19.10-3 M, en accord avec les données de la littérature (cmcDTAC pur =
20.10-3 M dans l’eau, à 25°C197).
La figure III-9 montre également une nette augmentation de la cmc du système en
présence du poly(β-CD) (Nβ-CD = 14.10-3 M): la cmc apparente, cmc*, atteint dans ce cas la
valeur de 34.10-3 M. Comme l’ont affirmé de nombreux auteurs46,47,50-52, cette observation est
une preuve de la formation de complexes d’inclusion entre les molécules de TA et les cavités
de CD présentes en solution. Les monomères de TA complexés n’étant plus disponibles pour
former des micelles, celles-ci n’apparaissent qu’à plus forte concentration en TA, une fois
l’interface saturée et toutes les cages de CD occupées.
Selon Junquera et coll.198, l’augmentation de la cmc d’un TA induite par la présence
de CD s’exprime sous la forme:
cmc* = cmc + R
NCD ,
où R est le rapport stoechiométrique NCD/NTA dans lequel les complexes d’inclusion sont
formés. Etant donné qu’un TA de type dodécyltriméthylammonium forme uniquement des
complexes d’inclusion 1:1 avec la β-CD native (cf. chapitre I, paragraphe II-3-b), une valeur
de R peu différente de 1 peut être attendue dans le cas du poly(β-CD). Un rapport R égal à
0,93 est obtenu en présence des 14.10-3 M de cavités de β-CD, ce qui tend à prouver que ni la
polymérisation de ces dernières, ni la présence des chaînons hydroxypropyléther (second
composant du poly(β-CD)) n’affectent le processus d’interaction. Dans la région
prémicellaire, chaque molécule de DTAC vient s’inclure de façon spécifique dans une cavité

- 144 -
de β-CD inoccupée. (Si les molécules de TA s’adsorbaient en plus sur les chaînes
hydroxypropyléther, la cmc* serait observée à une concentration en DTAC encore plus élevée
que 34.10-3 M Nβ-CD + cmc).
b) Par conductimétrie
La figure III-10 présente les résultats d’une étude préalable réalisée en mélangeant le
DTAC avec des monomères d’hydroxypropyl(β-cyclodextrine) (HP(β-CD)) dans l’eau (voir
les caractéristiques de l’ HP(β-CD) p. 192). Les trois courbes présentées sur cette figure ont
été obtenues:
() en titrant un mélange aqueux HP(β-CD)/DTAC (Nβ-CDi = 65.10-3 M, NDTAC = 30.10-3 M)
par une solution de DTAC à 30.10-3M,
() en titrant un mélange aqueux HP(β-CD)/DTAC (Nβ-CDi = 70.10-3 M, NDTAC = 40.10-3 M)
par une solution de DTAC à 40.10-3M,
() en titrant un mélange aqueux HP(β-CD)/DTAC (Nβ-CDi = 80.10-3 M, NDTAC = 50.10-3 M)
par une solution de DTAC à 50.10-3M.
Elles montrent les variations de conductance de mélanges comportant du DTAC en quantités
supérieures à sa cmc, en fonction de la teneur en HP(β-CD).
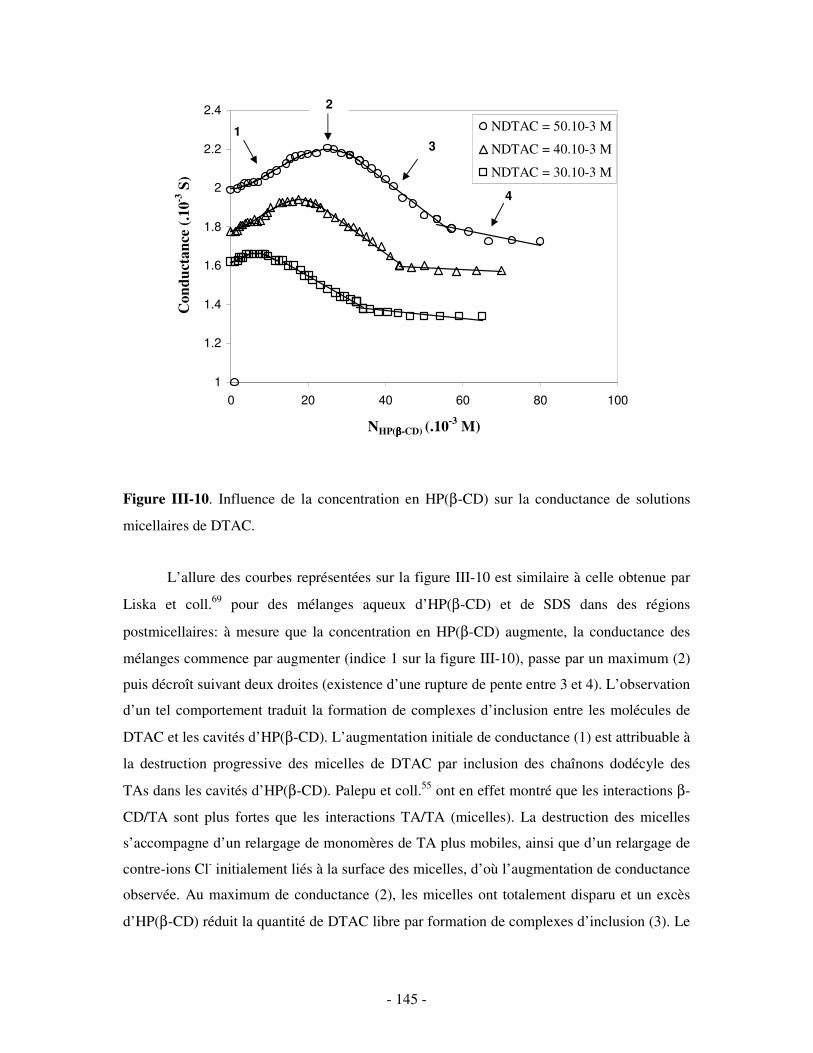
- 145 -
1
1.2
1.4
1.6
1.8
2
2.2
2.4
0 20 40 60 80 100
NHP(ββββ-CD) (.10-3 M)
Con
duct
ance
(.10
-3 S
)
NDTAC = 50.10-3 M
NDTAC = 40.10-3 M
NDTAC = 30.10-3 M
1
2
3
4
Figure III-10. Influence de la concentration en HP(β-CD) sur la conductance de solutions
micellaires de DTAC.
L’allure des courbes représentées sur la figure III-10 est similaire à celle obtenue par
Liska et coll.69 pour des mélanges aqueux d’HP(β-CD) et de SDS dans des régions
postmicellaires: à mesure que la concentration en HP(β-CD) augmente, la conductance des
mélanges commence par augmenter (indice 1 sur la figure III-10), passe par un maximum (2)
puis décroît suivant deux droites (existence d’une rupture de pente entre 3 et 4). L’observation
d’un tel comportement traduit la formation de complexes d’inclusion entre les molécules de
DTAC et les cavités d’HP(β-CD). L’augmentation initiale de conductance (1) est attribuable à
la destruction progressive des micelles de DTAC par inclusion des chaînons dodécyle des
TAs dans les cavités d’HP(β-CD). Palepu et coll.55 ont en effet montré que les interactions β-
CD/TA sont plus fortes que les interactions TA/TA (micelles). La destruction des micelles
s’accompagne d’un relargage de monomères de TA plus mobiles, ainsi que d’un relargage de
contre-ions Cl- initialement liés à la surface des micelles, d’où l’augmentation de conductance
observée. Au maximum de conductance (2), les micelles ont totalement disparu et un excès
d’HP(β-CD) réduit la quantité de DTAC libre par formation de complexes d’inclusion (3). Le

- 146 -
maximum de conductance observé est alors interprétable en termes de cmc apparente (cmc*)
du DTAC en présence d’HP(β-CD)55,69.
Sur la figure III-10, la rupture de pente (observée entre les domaines linéaires 3 et 4)
survient pour un rapport R = NHP(β-CD)/NDTAC égal à 1,1 pour les trois systèmes étudiés,
indiquant la formation de complexes d’inclusion de type 1:1. Au-delà de la stoechiométrie, le
plateau de conductance observé est attribuable uniquement aux complexes d’inclusion
chargés et aux contre-ions Cl-.
Pour chaque système étudié, la valeur de cmc* lue est reportée sur la figure III-11 en
fonction de la concentration en HP(β-CD) correspondante. Comme cela a déjà été montré
dans la littérature46,47,50-53,55,57,59,69, l’addition de CDs induit une augmentation de la cmc du
TA en solution, ce qui constitue une preuve supplémentaire de la formation de complexes
d’inclusion: un excès de DTAC conduit à la formation de micelles uniquement lorsque la
totalité des cavités d’HP(β-CD) sont occupées. Preuve en est la valeur de la cmc* lue sur la
figure III-11 pour une concentration en cavités d’HP(β-CD) égale, par exemple, à 14.10-3 M:
cmc* = 38.10-3 M, ce qui correspond quasiment à la concentration en HP(β-CD) ajoutée à la
cmc du DTAC seul. Notons que cette dernière peut être obtenue en extrapolant les résultats de
la figure III-11 à concentration en HP(β-CD) nulle. Une valeur en accord avec les données de
la littérature est alors obtenue: cmcDTAC = 22.10-3 M.
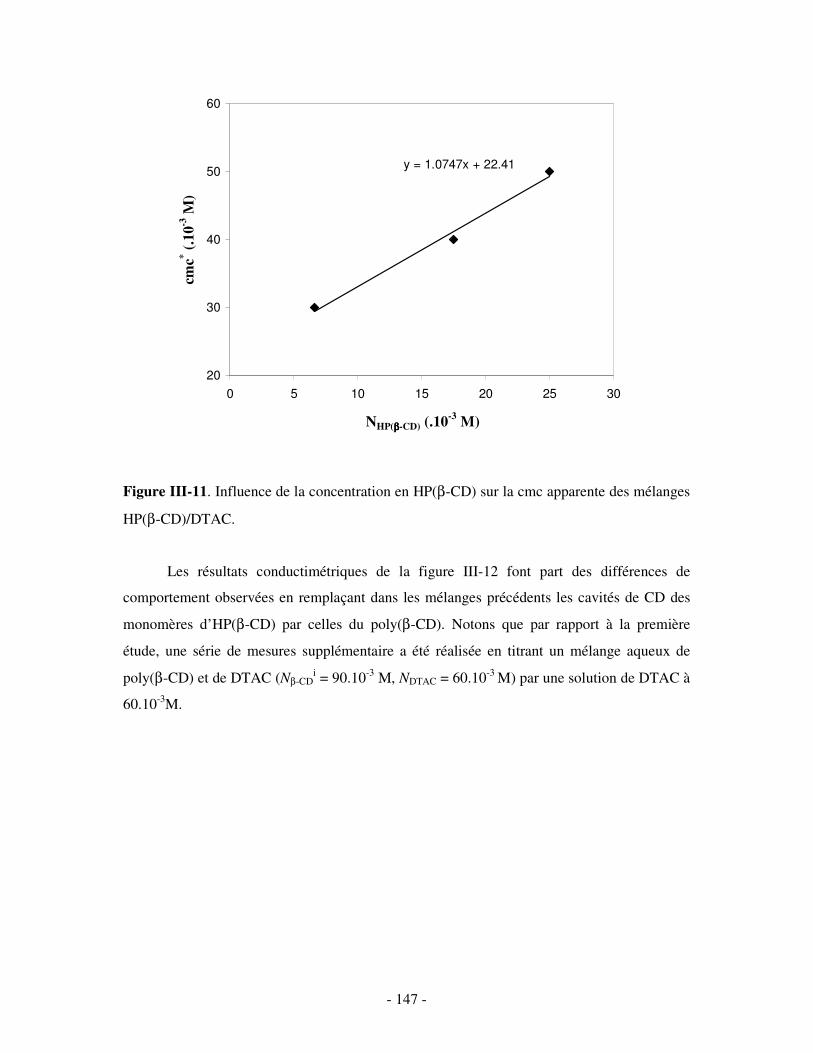
- 147 -
y = 1.0747x + 22.41
20
30
40
50
60
0 5 10 15 20 25 30
NHP(ββββ-CD) (.10-3 M)
cmc* (.
10-3
M)
Figure III-11. Influence de la concentration en HP(β-CD) sur la cmc apparente des mélanges
HP(β-CD)/DTAC.
Les résultats conductimétriques de la figure III-12 font part des différences de
comportement observées en remplaçant dans les mélanges précédents les cavités de CD des
monomères d’HP(β-CD) par celles du poly(β-CD). Notons que par rapport à la première
étude, une série de mesures supplémentaire a été réalisée en titrant un mélange aqueux de
poly(β-CD) et de DTAC (Nβ-CDi = 90.10-3 M, NDTAC = 60.10-3 M) par une solution de DTAC à
60.10-3M.
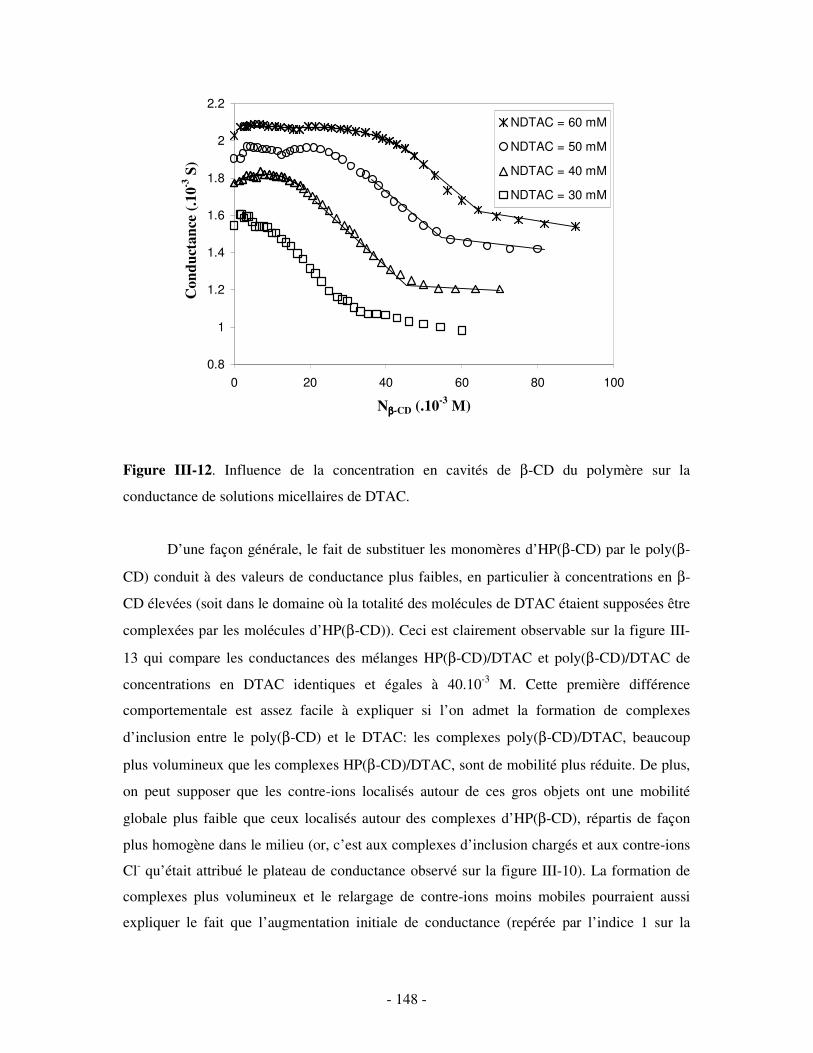
- 148 -
0.8
1
1.2
1.4
1.6
1.8
2
2.2
0 20 40 60 80 100
Nββββ-CD (.10-3 M)
Con
duct
ance
(.10
-3 S
)
NDTAC = 60 mM
NDTAC = 50 mM
NDTAC = 40 mM
NDTAC = 30 mM
Figure III-12. Influence de la concentration en cavités de β-CD du polymère sur la
conductance de solutions micellaires de DTAC.
D’une façon générale, le fait de substituer les monomères d’HP(β-CD) par le poly(β-
CD) conduit à des valeurs de conductance plus faibles, en particulier à concentrations en β-
CD élevées (soit dans le domaine où la totalité des molécules de DTAC étaient supposées être
complexées par les molécules d’HP(β-CD)). Ceci est clairement observable sur la figure III-
13 qui compare les conductances des mélanges HP(β-CD)/DTAC et poly(β-CD)/DTAC de
concentrations en DTAC identiques et égales à 40.10-3 M. Cette première différence
comportementale est assez facile à expliquer si l’on admet la formation de complexes
d’inclusion entre le poly(β-CD) et le DTAC: les complexes poly(β-CD)/DTAC, beaucoup
plus volumineux que les complexes HP(β-CD)/DTAC, sont de mobilité plus réduite. De plus,
on peut supposer que les contre-ions localisés autour de ces gros objets ont une mobilité
globale plus faible que ceux localisés autour des complexes d’HP(β-CD), répartis de façon
plus homogène dans le milieu (or, c’est aux complexes d’inclusion chargés et aux contre-ions
Cl- qu’était attribué le plateau de conductance observé sur la figure III-10). La formation de
complexes plus volumineux et le relargage de contre-ions moins mobiles pourraient aussi
expliquer le fait que l’augmentation initiale de conductance (repérée par l’indice 1 sur la
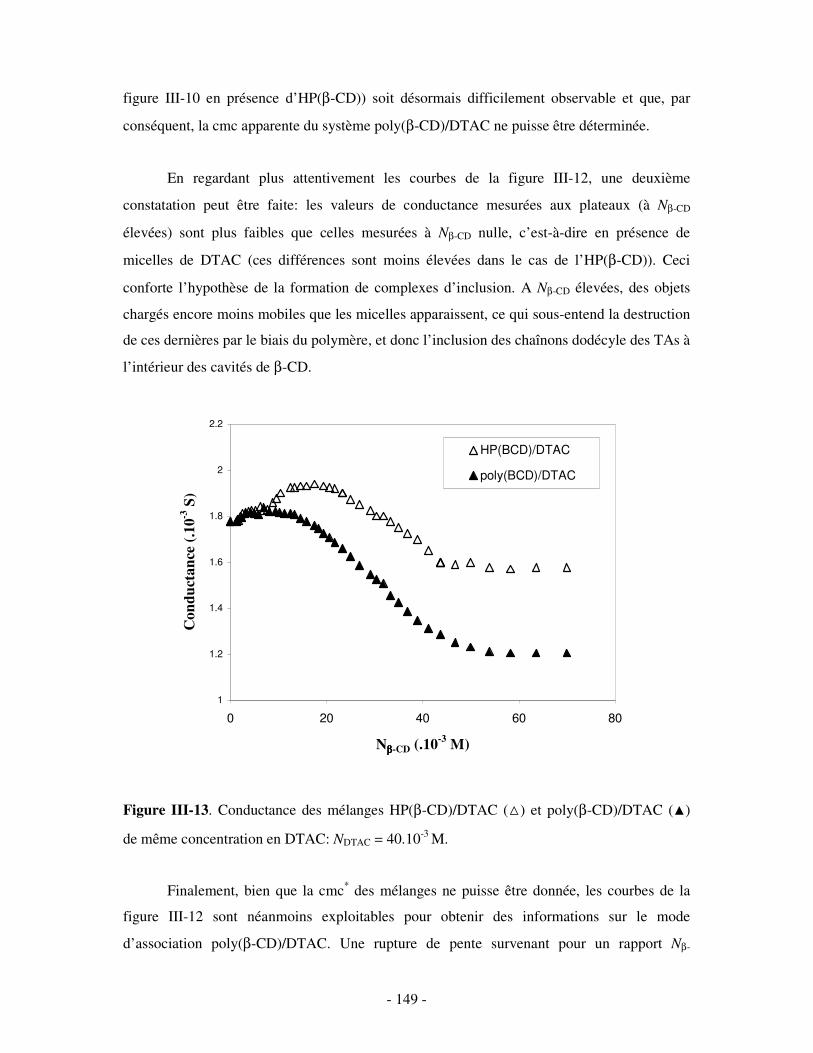
- 149 -
figure III-10 en présence d’HP(β-CD)) soit désormais difficilement observable et que, par
conséquent, la cmc apparente du système poly(β-CD)/DTAC ne puisse être déterminée.
En regardant plus attentivement les courbes de la figure III-12, une deuxième
constatation peut être faite: les valeurs de conductance mesurées aux plateaux (à Nβ-CD
élevées) sont plus faibles que celles mesurées à Nβ-CD nulle, c’est-à-dire en présence de
micelles de DTAC (ces différences sont moins élevées dans le cas de l’HP(β-CD)). Ceci
conforte l’hypothèse de la formation de complexes d’inclusion. A Nβ-CD élevées, des objets
chargés encore moins mobiles que les micelles apparaissent, ce qui sous-entend la destruction
de ces dernières par le biais du polymère, et donc l’inclusion des chaînons dodécyle des TAs à
l’intérieur des cavités de β-CD.
1
1.2
1.4
1.6
1.8
2
2.2
0 20 40 60 80
Nββββ-CD (.10-3 M)
Con
duct
ance
(.10
-3 S
)
HP(BCD)/DTAC
poly(BCD)/DTAC
Figure III-13. Conductance des mélanges HP(β-CD)/DTAC () et poly(β-CD)/DTAC ()
de même concentration en DTAC: NDTAC = 40.10-3 M.
Finalement, bien que la cmc* des mélanges ne puisse être donnée, les courbes de la
figure III-12 sont néanmoins exploitables pour obtenir des informations sur le mode
d’association poly(β-CD)/DTAC. Une rupture de pente survenant pour un rapport Nβ-

- 150 -
CD/NDTAC égal à 1,1 est observable pour chacun des mélanges étudiés. Il s’en déduit que les
complexes poly(β-CD)/DTAC sont majoritairement formés avec une stoechiométrie 1:1,
c’est-à-dire avec une molécule de DTAC par cavité de β-CD.
Remarque: Les molécules d’HP(β-CD) ont l’avantage d’être environ 20 fois plus solubles que
les molécules de β-CD native (ces dernières sont insolubles dans l’eau dès 16.10-3 M, soit
dans la majeure partie des domaines de concentration étudiés en conductimétrie). Par contre,
elles présentent une activité de surface199, rendant les complexes d’inclusion HP(β-
CD)/DTAC eux aussi tensioactifs. C’est pourquoi l’étude comparative entre HP(β-CD) et
poly(β-CD) n’a pu être réalisée par mesures de tension de surface.
c) Par viscosimétrie
La viscosité réduite d’une solution du poly(β-CD) est représentée sur la figure III-14
(courbe a) en fonction de sa concentration. En extrapolant à concentration nulle les valeurs
observées (cf. équation de Huggins), la viscosité intrinsèque du polymère branché est
obtenue:
[η]poly(β-CD) = (6,3 ± 0,5).10-3 L.g-1 .
Comparée à la viscosité intrinsèque d’un poly(oxyde d’éthylène) linéaire (POE), par exemple,
déterminée dans les mêmes conditions (même solvant, même température et même masse
molaire), cette valeur obtenue pour le poly(β-CD) est environ 10 fois plus faible ([η]POE =
62,4.10-3 L.g-1), indiquant une structure très compacte pour ce dernier.
Sur la même figure sont reportées les valeurs de viscosité réduite d’une solution
comportant initialement (avant dilution) 14.10-3 M de cavités de β-CD et 6.10-3 M de
molécules de DTAC (courbe b).
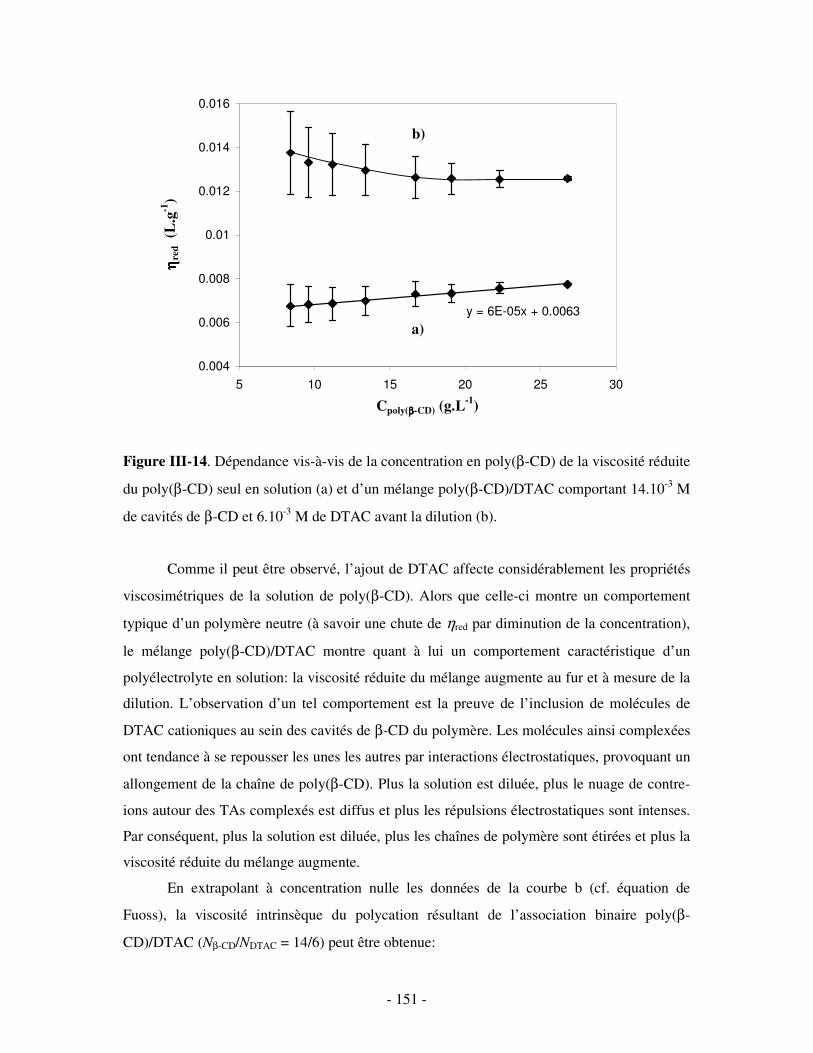
- 151 -
y = 6E-05x + 0.0063
0.004
0.006
0.008
0.01
0.012
0.014
0.016
5 10 15 20 25 30
Cpoly(ββββ-CD) (g.L-1)
ηη ηη red
(L
.g-1
)
a)
b)
Figure III-14. Dépendance vis-à-vis de la concentration en poly(β-CD) de la viscosité réduite
du poly(β-CD) seul en solution (a) et d’un mélange poly(β-CD)/DTAC comportant 14.10-3 M
de cavités de β-CD et 6.10-3 M de DTAC avant la dilution (b).
Comme il peut être observé, l’ajout de DTAC affecte considérablement les propriétés
viscosimétriques de la solution de poly(β-CD). Alors que celle-ci montre un comportement
typique d’un polymère neutre (à savoir une chute de ηred par diminution de la concentration),
le mélange poly(β-CD)/DTAC montre quant à lui un comportement caractéristique d’un
polyélectrolyte en solution: la viscosité réduite du mélange augmente au fur et à mesure de la
dilution. L’observation d’un tel comportement est la preuve de l’inclusion de molécules de
DTAC cationiques au sein des cavités de β-CD du polymère. Les molécules ainsi complexées
ont tendance à se repousser les unes les autres par interactions électrostatiques, provoquant un
allongement de la chaîne de poly(β-CD). Plus la solution est diluée, plus le nuage de contre-
ions autour des TAs complexés est diffus et plus les répulsions électrostatiques sont intenses.
Par conséquent, plus la solution est diluée, plus les chaînes de polymère sont étirées et plus la
viscosité réduite du mélange augmente.
En extrapolant à concentration nulle les données de la courbe b (cf. équation de
Fuoss), la viscosité intrinsèque du polycation résultant de l’association binaire poly(β-
CD)/DTAC (Nβ-CD/NDTAC = 14/6) peut être obtenue:

- 152 -
[η]poly(β-CD)/DTAC = (15,3 ± 0,5).10-3 L.g-1 .
Cette valeur permet de rendre compte du gonflement subi par la chaîne de poly(β-CD)
décorée de molécules de DTAC complexées. En effet, on a:
[ ]
[ ] 4,2)(
)(
/)(
/)(
)(
/)( =⋅≈−
−
−
−
−
−
CDpolyh
CDpoly
DTACCDpoly
DTACCDpolyh
CDpoly
DTACCDpoly
V
M
M
V
β
β
β
β
β
β
ηη
(1)
soit )(
/)(
)(
/)( 4,2CDpoly
DTACCDpoly
CDpolyh
DTACCDpolyh
M
M
V
V
−
−
−
− ⋅≈β
β
β
β . (2)
En considérant qu’une chaîne de poly(β-CD) contient 83 cavités de β-CD en moyenne, et en
supposant que toutes les molécules de DTAC présentes en solution sont complexées, Mpoly(β-
CD)/DTAC = Mpoly(β-CD) + 83*6/14 MDTAC, avec Mpoly(β-CD) = 160 000 g.mol-1 et MDTAC =263,9
g.mol-1. Il s’en déduit que Mpoly(β-CD)/DTAC = 169 390 g.mol-1 et que, d’après l’équation (2), le
volume hydrodynamique d’une chaîne du polycation est 2,5 fois plus élevé que celui d’une
chaîne de poly(β-CD) libre correspondante.
Notons que les résultats précédents ont été obtenus pour un rapport Nβ-CD/NDTAC fixé à
14/6. La figure III-15 présente cette fois-ci les valeurs de viscosité réduite du système binaire
déterminées en faisant varier la stoechiométrie du mélange, soit en diluant une solution de
poly(β-CD) (Nβ-CDi = 10-2 M) par une solution de DTAC (NDTAC
i = 10-2 M).
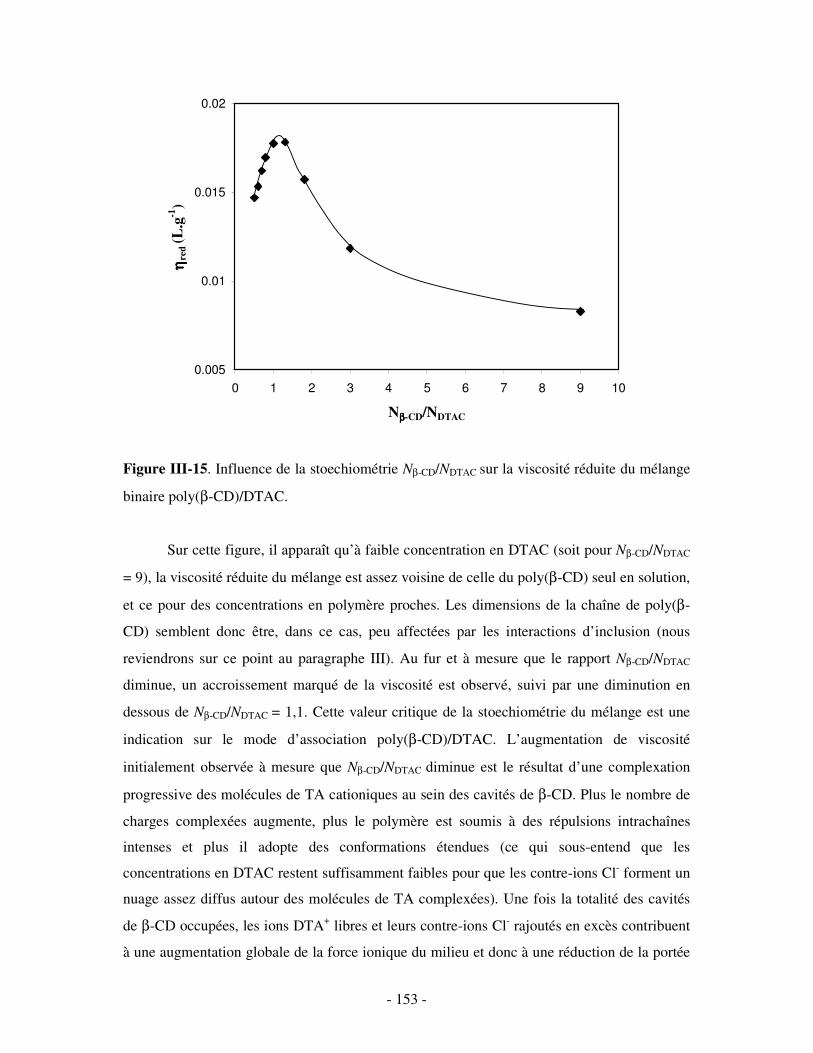
- 153 -
0.005
0.01
0.015
0.02
0 1 2 3 4 5 6 7 8 9 10
Nββββ-CD/NDTAC
ηη ηη red
(L.g
-1)
Figure III-15. Influence de la stoechiométrie Nβ-CD/NDTAC sur la viscosité réduite du mélange
binaire poly(β-CD)/DTAC.
Sur cette figure, il apparaît qu’à faible concentration en DTAC (soit pour Nβ-CD/NDTAC
= 9), la viscosité réduite du mélange est assez voisine de celle du poly(β-CD) seul en solution,
et ce pour des concentrations en polymère proches. Les dimensions de la chaîne de poly(β-
CD) semblent donc être, dans ce cas, peu affectées par les interactions d’inclusion (nous
reviendrons sur ce point au paragraphe III). Au fur et à mesure que le rapport Nβ-CD/NDTAC
diminue, un accroissement marqué de la viscosité est observé, suivi par une diminution en
dessous de Nβ-CD/NDTAC = 1,1. Cette valeur critique de la stoechiométrie du mélange est une
indication sur le mode d’association poly(β-CD)/DTAC. L’augmentation de viscosité
initialement observée à mesure que Nβ-CD/NDTAC diminue est le résultat d’une complexation
progressive des molécules de TA cationiques au sein des cavités de β-CD. Plus le nombre de
charges complexées augmente, plus le polymère est soumis à des répulsions intrachaînes
intenses et plus il adopte des conformations étendues (ce qui sous-entend que les
concentrations en DTAC restent suffisamment faibles pour que les contre-ions Cl- forment un
nuage assez diffus autour des molécules de TA complexées). Une fois la totalité des cavités
de β-CD occupées, les ions DTA+ libres et leurs contre-ions Cl- rajoutés en excès contribuent
à une augmentation globale de la force ionique du milieu et donc à une réduction de la portée

- 154 -
des répulsions électrostatiques intrachaînes. En effet, la distance κ -1 au-delà de laquelle l’effet
des charges complexées est totalement écranté (soit la distance de Debye) est reliée à la force
ionique I selon:
( ) 21
1
8
1
Il aB ℵ=−
πκ ,
où lB est la longueur de Bjerrum (en dessous de laquelle un phénomène de condensation des
contre-ions peut être observé) et aℵ , le nombre d’Avogadro. L’écrantage des répulsions
coulombiennes induit alors une réduction des dimensions de la chaîne de polymère, ce qui se
traduit par une chute de la viscosité. D’après la figure III-15, la contribution des molécules de
DTAC à l’effet d’écran survient pour un rapport Nβ-CD/NDTAC égal à 1,1. Ceci suggère que les
cavités du poly(β-CD) et le DTAC ont tendance à former des complexes d’inclusion
essentiellement de type 1:1.
d) Par SANS
Les premières expériences de SANS ont porté sur la diffusion du poly(β-CD) seul en
solution (Nβ-CD = 14.10-3 M dans le D2O). Comme le montre la figure III-16, l’intensité
diffusée par celui-ci décroît de façon monotone pour des valeurs de q croissantes, ce qui est
caractéristique d’un polymère neutre en solution200,201.
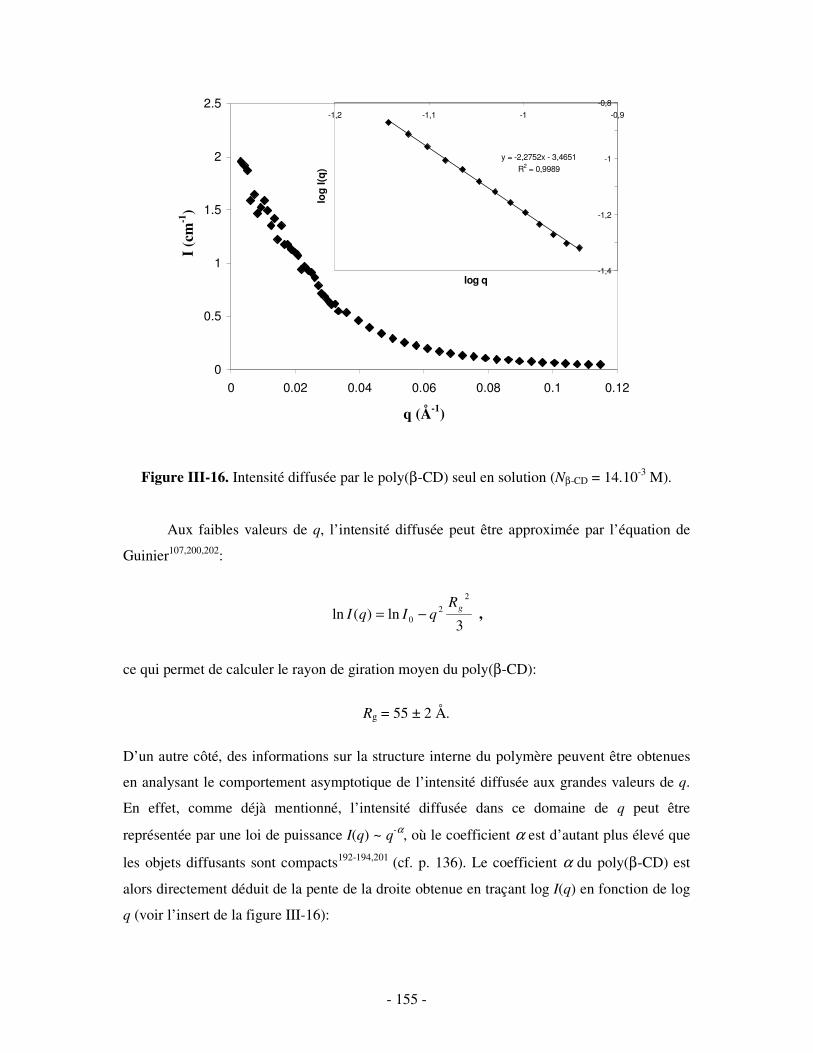
- 155 -
0
0.5
1
1.5
2
2.5
0 0.02 0.04 0.06 0.08 0.1 0.12
q (Å-1)
I (cm
-1)
Figure III-16. Intensité diffusée par le poly(β-CD) seul en solution (Nβ-CD = 14.10-3 M).
Aux faibles valeurs de q, l’intensité diffusée peut être approximée par l’équation de
Guinier107,200,202:
3ln)(ln
22
0gR
qIqI −= ,
ce qui permet de calculer le rayon de giration moyen du poly(β-CD):
Rg = 55 ± 2 Å.
D’un autre côté, des informations sur la structure interne du polymère peuvent être obtenues
en analysant le comportement asymptotique de l’intensité diffusée aux grandes valeurs de q.
En effet, comme déjà mentionné, l’intensité diffusée dans ce domaine de q peut être
représentée par une loi de puissance I(q) ~ q-α, où le coefficient α est d’autant plus élevé que
les objets diffusants sont compacts192-194,201 (cf. p. 136). Le coefficient α du poly(β-CD) est
alors directement déduit de la pente de la droite obtenue en traçant log I(q) en fonction de log
q (voir l’insert de la figure III-16):
y = -2,2752x - 3,4651R2 = 0,9989
-1,4
-1,3
-1,2
-1,1
-1
-0,9
-0,8-1,2 -1,1 -1 -0,9
log q
log
I(q)

- 156 -
αpoly(β-CD) = 2,25 ± 0,05.
Le fait que cette valeur de α soit supérieure à celle attendue pour une chaîne idéale (soit α =
2) indique une certaine compacité de la structure interne du poly(β-CD). Cette compacité est
attribuable au caractère branché du polymère, ce qui est confirmé par l’accord quasi parfait
obtenu entre αpoly(β-CD) et la valeur prédite par le modèle de Daoud et Joanny203 pour les
polymères de structure branchée. En effet, si pour un polymère branché en bon solvant ce
modèle prévoit une dimension fractale égale à 2, une valeur de 2,28 est attendue pour un
polymère branché en solvant θ. Or, des mesures de diffusion de lumière40 réalisées sur un
échantillon de poly(β-CD) (de masse molaire plus élevée) ont montré que ce polymère est
proche des conditions θ en solution aqueuse (second coefficient du viriel très faible).
Ainsi donc, les résultats de diffusion de neutrons et ceux obtenus par viscosimétrie
mettent respectivement en évidence une compacité locale et macroscopique du poly(β-CD)
étudié, en corrélation avec la nature branchée de son architecture (une structure plus diffuse
constituée de plusieurs nodules très compacts est observable pour des échantillons de masse
molaire beaucoup plus élevée40). Ces résultats sont rassemblés dans le tableau III-2 qui
présente l’ensemble des caractéristiques du poly(β-CD) considéré tout au long de cette étude.
% CD (g.g-1)a Mw (g.mol-1)b [ηηηη] (L.g-1) Rg (Å)c ααααc
59 160 000 0,0063 55 2,25 a : déterminé par RMN; b : déterminée par SEC couplée à de la diffusion de la lumière; c : déterminé par SANS.
Tableau III-2. Caractéristiques du poly(β-CD).
La figure III-17 montre l’effet produit en ajoutant 6.10-3 M de DTAC à la solution de
poly(β-CD) précédente (Nβ-CD = 14.10-3 M): un pic de diffusion caractéristique d’un
comportement polyélectrolyte200-202,204-206 apparaît à q* = 2,5.10-2 Å-1.
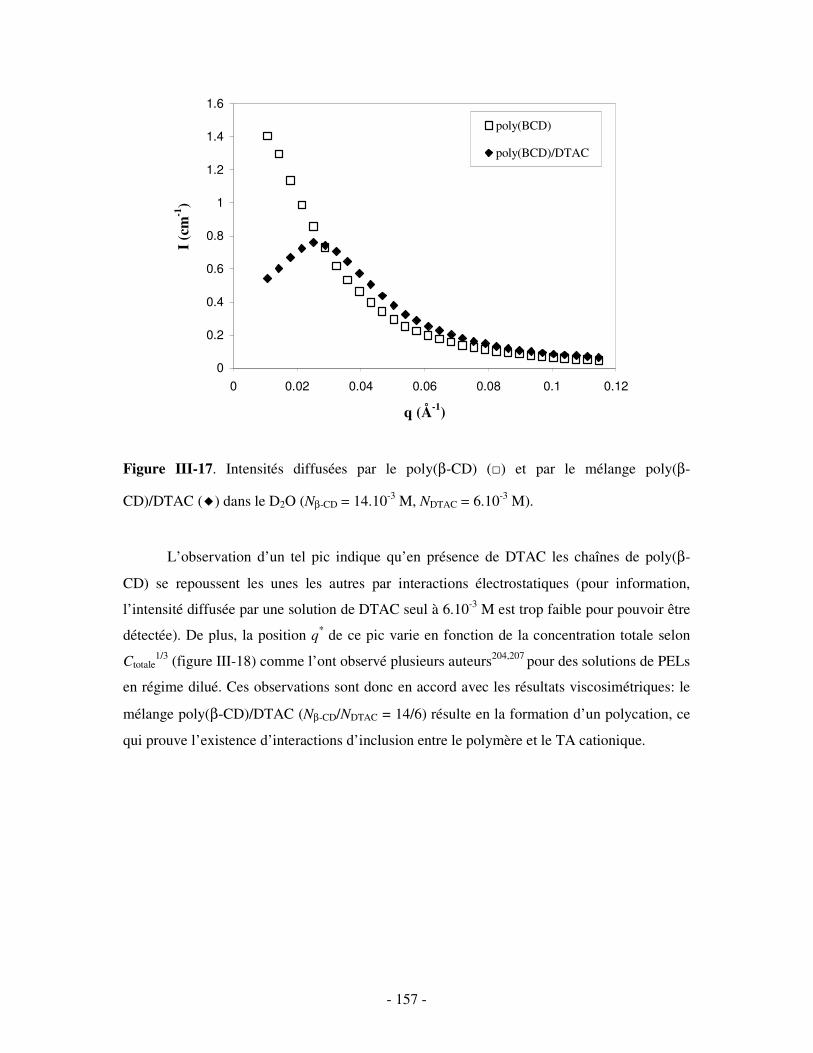
- 157 -
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
0 0.02 0.04 0.06 0.08 0.1 0.12
q (Å-1)
I (cm
-1)
poly(BCD)
poly(BCD)/DTAC
Figure III-17. Intensités diffusées par le poly(β-CD) () et par le mélange poly(β-
CD)/DTAC () dans le D2O (Nβ-CD = 14.10-3 M, NDTAC = 6.10-3 M).
L’observation d’un tel pic indique qu’en présence de DTAC les chaînes de poly(β-
CD) se repoussent les unes les autres par interactions électrostatiques (pour information,
l’intensité diffusée par une solution de DTAC seul à 6.10-3 M est trop faible pour pouvoir être
détectée). De plus, la position q* de ce pic varie en fonction de la concentration totale selon
Ctotale1/3 (figure III-18) comme l’ont observé plusieurs auteurs204,207 pour des solutions de PELs
en régime dilué. Ces observations sont donc en accord avec les résultats viscosimétriques: le
mélange poly(β-CD)/DTAC (Nβ-CD/NDTAC = 14/6) résulte en la formation d’un polycation, ce
qui prouve l’existence d’interactions d’inclusion entre le polymère et le TA cationique.
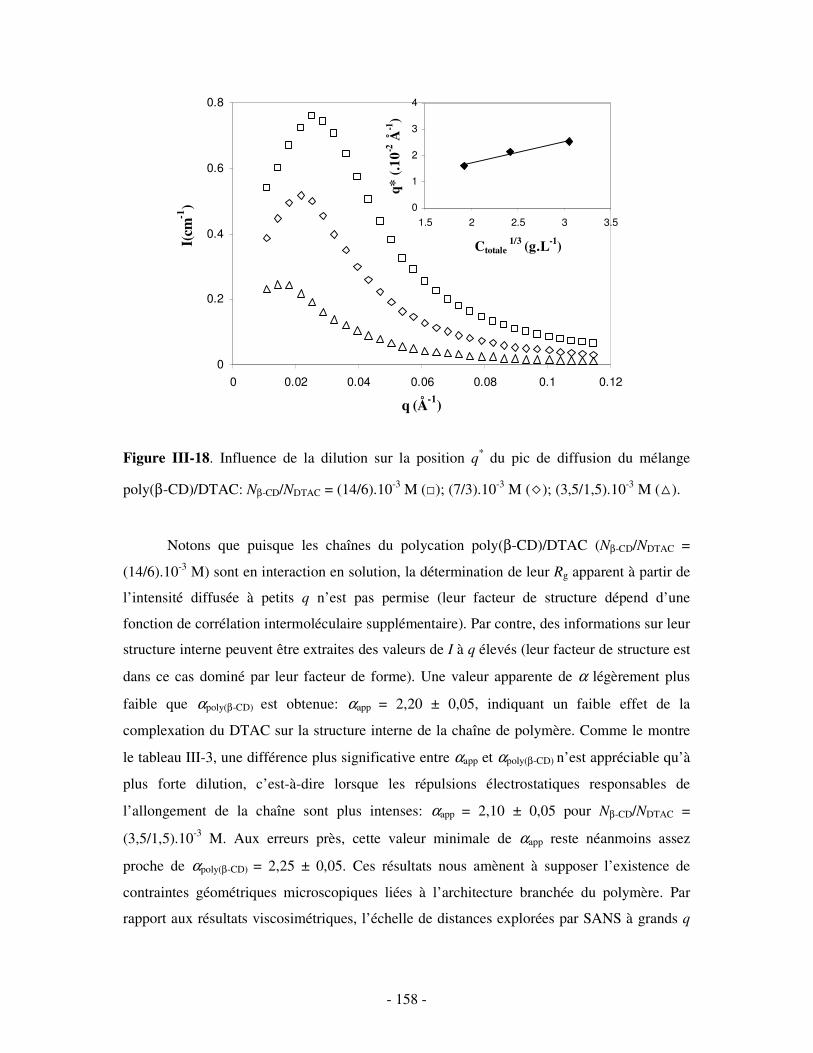
- 158 -
Figure III-18. Influence de la dilution sur la position q* du pic de diffusion du mélange
poly(β-CD)/DTAC: Nβ-CD/NDTAC = (14/6).10-3 M (); (7/3).10-3 M ( ); (3,5/1,5).10-3 M ().
Notons que puisque les chaînes du polycation poly(β-CD)/DTAC (Nβ-CD/NDTAC =
(14/6).10-3 M) sont en interaction en solution, la détermination de leur Rg apparent à partir de
l’intensité diffusée à petits q n’est pas permise (leur facteur de structure dépend d’une
fonction de corrélation intermoléculaire supplémentaire). Par contre, des informations sur leur
structure interne peuvent être extraites des valeurs de I à q élevés (leur facteur de structure est
dans ce cas dominé par leur facteur de forme). Une valeur apparente de α légèrement plus
faible que αpoly(β-CD) est obtenue: αapp = 2,20 ± 0,05, indiquant un faible effet de la
complexation du DTAC sur la structure interne de la chaîne de polymère. Comme le montre
le tableau III-3, une différence plus significative entre αapp et αpoly(β-CD) n’est appréciable qu’à
plus forte dilution, c’est-à-dire lorsque les répulsions électrostatiques responsables de
l’allongement de la chaîne sont plus intenses: αapp = 2,10 ± 0,05 pour Nβ-CD/NDTAC =
(3,5/1,5).10-3 M. Aux erreurs près, cette valeur minimale de αapp reste néanmoins assez
proche de αpoly(β-CD) = 2,25 ± 0,05. Ces résultats nous amènent à supposer l’existence de
contraintes géométriques microscopiques liées à l’architecture branchée du polymère. Par
rapport aux résultats viscosimétriques, l’échelle de distances explorées par SANS à grands q
0
0.2
0.4
0.6
0.8
0 0.02 0.04 0.06 0.08 0.1 0.12
q (Å-1)
I(cm
-1) 0
1
2
3
4
1.5 2 2.5 3 3.5
Ctotale 1/3 (g.L-1)
q* (.
10-2
Å-1
)
- 159 -
semble donc trop petite pour permettre d’apprécier pleinement les conséquences des
répulsions électrostatiques intrachaînes (étirement de la chaîne).
Nββββ-CD/NDTAC (.10-3 M) 14/6 7/3 3,5/1,5
ααααapp (± 0,05) 2,20 2,15 2,10
Tableau III-3. Influence de la dilution sur le coefficient apparent αapp mesuré à grands q pour
le système poly(β-CD)/DTAC.
e) Bilan
Au cours de ces différentes études, plusieurs preuves de la formation de complexes
d’inclusion entre le polymère de β-CD et le DTAC ont été apportées. En présence du poly(β-
CD), une réduction de l’activité de surface du tensioactif, de même qu’une réduction de ses
propriétés de micellisation, ont d’une part été constatées. Une diminution de la conductance
du DTAC (aussi bien sous forme de monomère que sous forme de micelles) a d’autre part été
rapportée. De plus, des mesures viscosimétriques et de diffusion de neutrons ont clairement
mis en évidence la formation d’un polycation en mélangeant le poly(β-CD) et le DTAC dans
un rapport Nβ-CD/NDTAC = 14/6: un accroissement de viscosité réduite sous l’effet de la
dilution, ainsi que l’apparition d’un pic de diffusion ont été observées.
Des informations sur la structure des complexes binaires ont également été fournies.
Les résultats de tension de surface, de conductimétrie et de viscosimétrie laissent supposer
que le DTAC et les cavités de β-CD du polymère forment principalement des complexes
d’inclusion de type 1:1 (une molécule de DTAC par cavité de β-CD). D’après l’étude
viscosimétrique, ces interactions d’inclusion confèrent aux agrégats formés une structure
globalement gonflée pour un rapport Nβ-CD/NDTAC = 14/6 (via des répulsions électrostatiques
intrachaînes). Néanmoins, comme l’indiquent les résultats de diffusion de neutrons, leur
structure locale apparaît peu différente de celle du poly(β-CD) neutre, ce qui a été attribué à la
nature branchée de ce dernier.
La suite de cette partie II consiste à caractériser de façon quantitative l’association
binaire poly(β-CD)/DTAC, c’est-à-dire à déterminer la constante d’association K1:1. Comme
il l’a été montré au paragraphe II-3-a du chapitre I, K1:1 peut dépendre fortement des

- 160 -
concentrations en cavités de CD et en TA, mais aussi de la méthode expérimentale employée,
surtout si le TA considéré est capable de former des associations multiples avec les CDs (1:1,
2:1 et 1:2, par exemple). Aussi nous a-t-il semblé intéressant de comparer les valeurs de K1:1
obtenues au moyen des diverses techniques à notre disposition. Parmi ces techniques figure la
fluorimétrie qui permet de mesurer directement les concentrations en DTAC libre et complexé
dans des solutions contenant des quantités connues de poly(β-CD). Figurent également les
techniques conductimétrique et de tension de surface qui, pour leur part, permettent de suivre
les variations des propriétés physico-chimiques des solutions de DTAC induites par l’ajout du
poly(β-CD).
2- Détermination de la constante d’association du complexe binaire
a) Par fluorimétrie
i) Principe
La détermination de Kpoly(β-CD)/DTAC au moyen de la technique fluorimétrique nécessite
l’utilisation d’une sonde fluorescente dont la constante d’affinité pour les cavités de β-CD est
notée Ksonde. En supposant qu’en absence de DTAC cette sonde forme des complexes
d’inclusion 1:1 avec les β-CDs selon:
sonde + β-CD β-CD/sonde , (1)
Ksonde est définie par:
CDsonde
complsonde NN
NK
−⋅=
β
1 , (2)
où Ncompl1, Nsonde et Nβ-CD sont les concentrations molaires d’équilibre en complexes β-
CD/sonde, en sonde et en cavités de β-CD, respectivement.
D’après la loi de conservation de la matière, les concentrations totales en sonde et en
cavités de β-CD en absence de DTAC s’écrivent:
Nsonde
0 = Nsonde + Ncompl1 , (3)

- 161 -
Nβ-CD 0 = Nβ-CD + Ncompl1 . (4)
L’équation (2) peut donc être transcrite sous la forme:
))(( 10
10
1
complCDcomplsonde
complsonde
NNNN
NK
−−=
−β
. (5)
Puisque les cavités de β-CD ne sont pas fluorescentes, l’intensité de fluorescence If émise par
un mélange β-CD/sonde est la somme des contributions de la sonde libre et complexée, ce qui
peut se traduire par l’expression suivante:
If = Φsonde Nsonde + Φcompl1 Ncompl1 ,
soit encore If = Φsonde (Nsonde0 - Ncompl1) + Φcompl1 Ncompl1
= Φsonde Nsonde0 + (Φcompl1 - Φsonde) Ncompl1. (6)
où Φsonde et Φcompl1 sont les rendements quantiques de fluorescence respectifs de la sonde et
des complexes β-CD/sonde (constantes caractéristiques des entités).
Soit I0 l’intensité de fluorescence émise par la sonde libre en absence de β-CD et I,
l’intensité de fluorescence émise lorsque toutes les molécules sondes sont complexées (Nsonde
= 0 dans ce cas):
I0 = Φsonde Nsonde
0 , (7)
I = Φcompl1 Ncompl1
= Φcompl1 Nsonde0 . (8)
L’expression (6) devient:
If = I0 + 00
sondeN
II −∞ Ncompl1 ,
soit Ncompl1 = 0
0
II
II f
−−
∞
Nsonde0 . (9)
En combinant les équations (5) et (9), une équation du second degré est obtenue:
( )( ) 0
11)( 0
000
0
202
0
0
=+−
++
−−−
− −−∞∞
CDfsonde
CDsondefsonde NII
KNN
IIII
II
Nββ . (10)

- 162 -
La résolution de cette équation conduit à l’expression finale:
−
++−++
−+= −−−
∞2
1
002
00000
00 4
112
CDsondesonde
CDsondesonde
CDsonde
sonde
f NNK
NNK
NNN
IIII βββ
Les paramètres Ksonde et I sont dès lors accessibles en traçant l’intensité de fluorescence
mesurée, If, en fonction de Nβ-CD 0.
En présence de DTAC, un second équilibre vient s’établir en plus de l’équilibre (1):
DTAC + β-CD β-CD/DTAC , (12)
CDDTAC
complDTACCD NN
NK
−− ⋅
=β
β2
/ , (13)
où Ncompl2 est la concentration molaire en complexes β-CD/DTAC à l’équilibre.
Dans ce cas, la loi de conservation de la matière donne:
NDTAC
0 = NDTAC + Ncompl2 , (14)
et Nβ-CD0 = Nβ-CD + Ncompl1 + Ncompl2
ou bien Ncompl2 = Nβ-CD0 - Nβ-CD - Ncompl1 . (15)
Dans cette dernière équation, Nβ-CD représente la concentration d’équilibre en cavités de β-CD
libres qui, d’après (2), s’écrit:
sonde
compl
sonde
libreCDCD N
N
KNN 11 ⋅== −− ββ ,
avec Nsonde = Nsonde
0 – Ncompl1
et Ncompl1 = ⋅−−
∞ 0
0
II
II f Nsonde0 ,
soit ⋅=−sonde
libreCD K
N1
βf
f
II
II
−−
∞
0 . (16)
(11)

- 163 -
Par contre, Ncompl2 représente la concentration d’équilibre en cavités de β-CD liées au DTAC
(Ncompl2 = Nβ-CDliée). L’équation (15) peut donc être réécrite de la façon suivante:
Nβ-CD
liée = Nβ-CD0 - Nβ-CD
libre - Ncompl1 , (17)
tandis que l’équation (13) devient:
DTACDTACCDlibreCD
liéeCD NK
N
N⋅= −
−
−:β
β
β . (18)
La combinaison des équations (14) et (18) donne finalement:
( )liéeCDDTACDTACCDlibre
CD
liéeCD NNK
N
N−−
−
− −⋅= βββ
β 0: . (19)
En traçant libre
CD
liéeCD
N
N
−
−
β
β en fonction de ( )liéeCDDTAC NN −− β
0 , on s’attend donc à obtenir une
droite de pente égale à Kβ-CD/DTAC.
ii) Résultats
La figure III-19 présente les valeurs d’intensité de fluorescence mesurées en ajoutant
des volumes croissants d’un mélange poly(β-CD)/sonde (Nβ-CD = 10-2 M, Nsonde = 6,5.10-5 M)
dans une solution de la sonde à 6,5.10-5 M (Vsondei = 2 mL). Ces valeurs de If ont été
déterminées à une longueur d’onde d’émission de 500 nm, la longueur d’onde d’excitation
ayant été fixée à 412 nm.
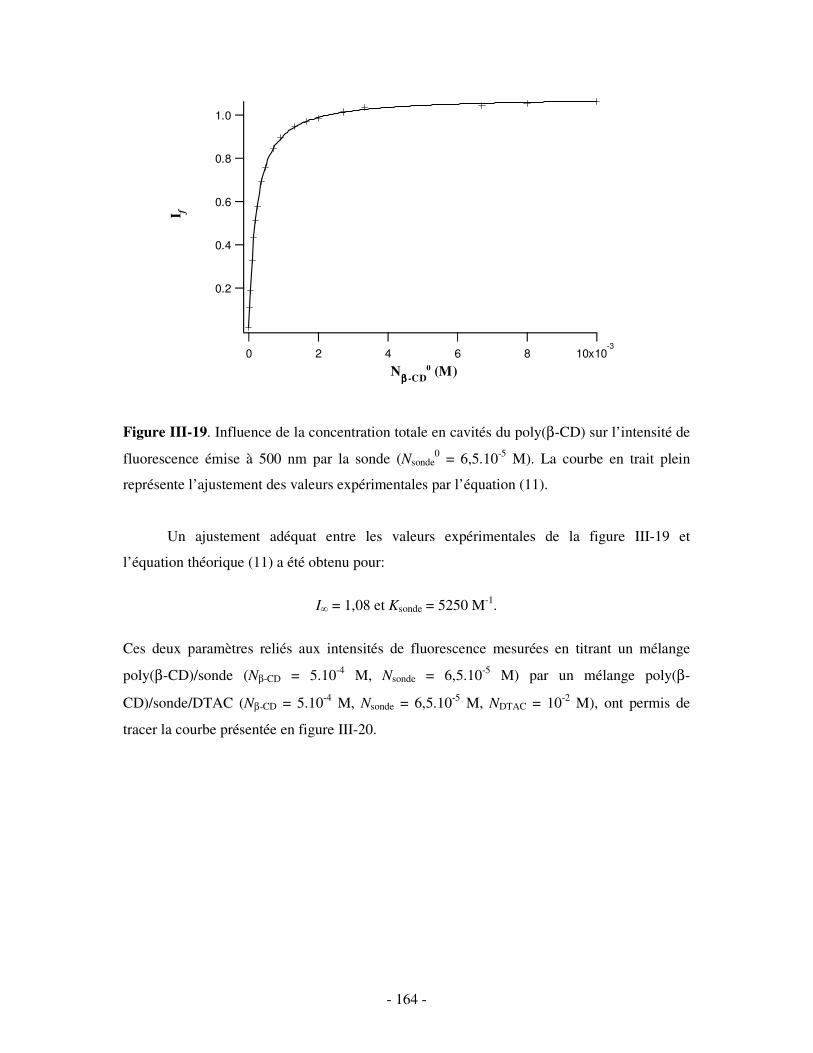
- 164 -
1.0
0.8
0.6
0.4
0.2
I f
10x10-3
86420
Nββββ -CD0 (M)
Figure III-19. Influence de la concentration totale en cavités du poly(β-CD) sur l’intensité de
fluorescence émise à 500 nm par la sonde (Nsonde0 = 6,5.10-5 M). La courbe en trait plein
représente l’ajustement des valeurs expérimentales par l’équation (11).
Un ajustement adéquat entre les valeurs expérimentales de la figure III-19 et
l’équation théorique (11) a été obtenu pour:
I = 1,08 et Ksonde = 5250 M-1.
Ces deux paramètres reliés aux intensités de fluorescence mesurées en titrant un mélange
poly(β-CD)/sonde (Nβ-CD = 5.10-4 M, Nsonde = 6,5.10-5 M) par un mélange poly(β-
CD)/sonde/DTAC (Nβ-CD = 5.10-4 M, Nsonde = 6,5.10-5 M, NDTAC = 10-2 M), ont permis de
tracer la courbe présentée en figure III-20.
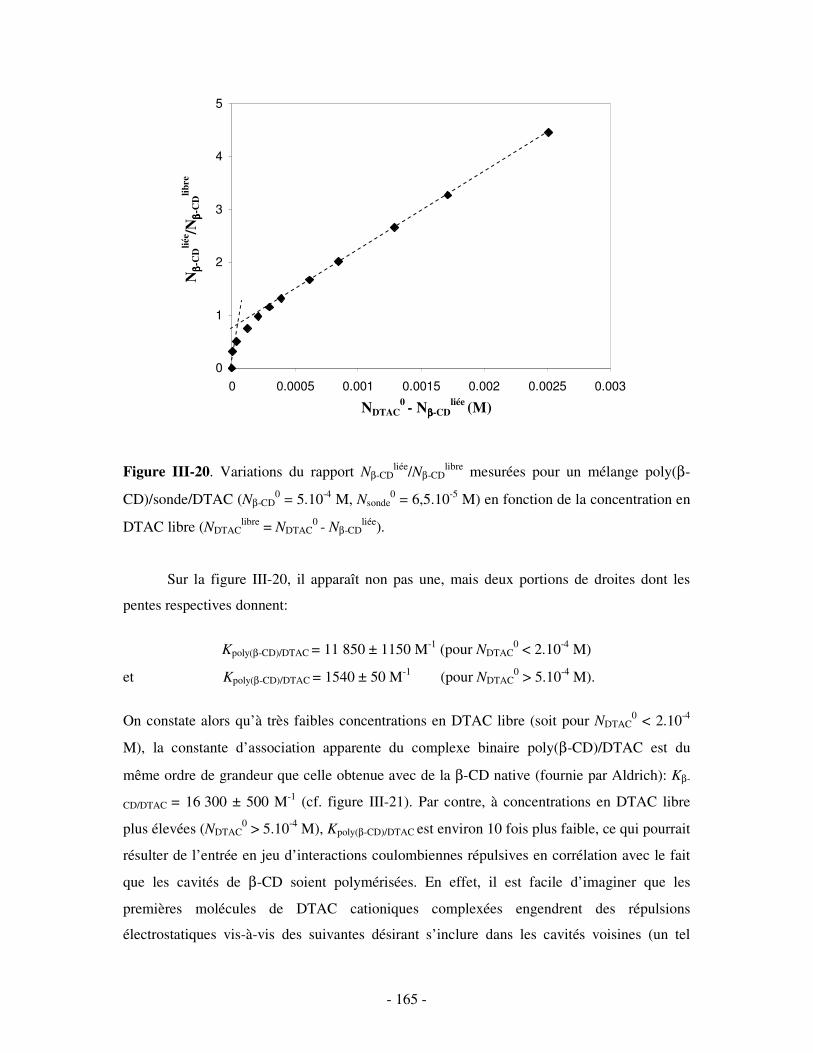
- 165 -
0
1
2
3
4
5
0 0.0005 0.001 0.0015 0.002 0.0025 0.003
NDTAC0
- Nββββ-CDliée
(M)
Nββ ββ-
CD
liée /N
ββ ββ-C
Dlib
re
Figure III-20. Variations du rapport Nβ-CDliée/Nβ-CD
libre mesurées pour un mélange poly(β-
CD)/sonde/DTAC (Nβ-CD0 = 5.10-4 M, Nsonde
0 = 6,5.10-5 M) en fonction de la concentration en
DTAC libre (NDTAClibre = NDTAC
0 - Nβ-CDliée).
Sur la figure III-20, il apparaît non pas une, mais deux portions de droites dont les
pentes respectives donnent:
Kpoly(β-CD)/DTAC = 11 850 ± 1150 M-1 (pour NDTAC
0 < 2.10-4 M)
et Kpoly(β-CD)/DTAC = 1540 ± 50 M-1 (pour NDTAC0 > 5.10-4 M).
On constate alors qu’à très faibles concentrations en DTAC libre (soit pour NDTAC
0 < 2.10-4
M), la constante d’association apparente du complexe binaire poly(β-CD)/DTAC est du
même ordre de grandeur que celle obtenue avec de la β-CD native (fournie par Aldrich): Kβ-
CD/DTAC = 16 300 ± 500 M-1 (cf. figure III-21). Par contre, à concentrations en DTAC libre
plus élevées (NDTAC0 > 5.10-4 M), Kpoly(β-CD)/DTAC est environ 10 fois plus faible, ce qui pourrait
résulter de l’entrée en jeu d’interactions coulombiennes répulsives en corrélation avec le fait
que les cavités de β-CD soient polymérisées. En effet, il est facile d’imaginer que les
premières molécules de DTAC cationiques complexées engendrent des répulsions
électrostatiques vis-à-vis des suivantes désirant s’inclure dans les cavités voisines (un tel
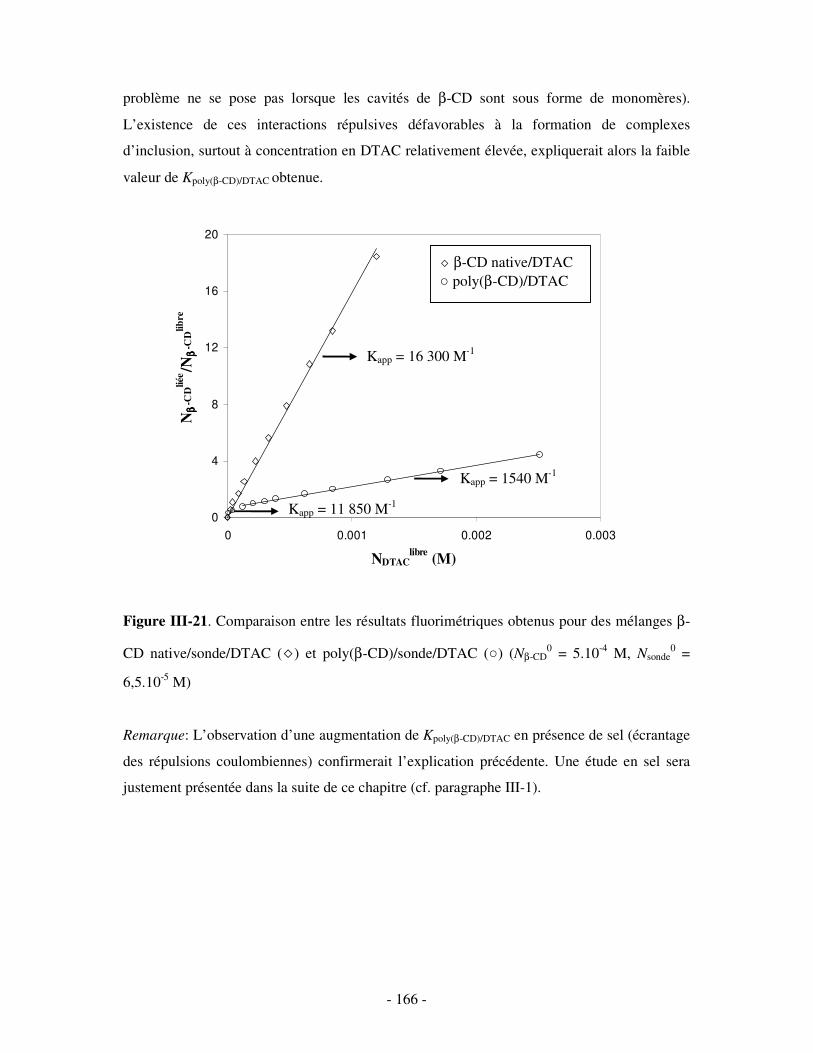
- 166 -
problème ne se pose pas lorsque les cavités de β-CD sont sous forme de monomères).
L’existence de ces interactions répulsives défavorables à la formation de complexes
d’inclusion, surtout à concentration en DTAC relativement élevée, expliquerait alors la faible
valeur de Kpoly(β-CD)/DTAC obtenue.
Figure III-21. Comparaison entre les résultats fluorimétriques obtenus pour des mélanges β-
CD native/sonde/DTAC ( ) et poly(β-CD)/sonde/DTAC () (Nβ-CD0 = 5.10-4 M, Nsonde
0 =
6,5.10-5 M)
Remarque: L’observation d’une augmentation de Kpoly(β-CD)/DTAC en présence de sel (écrantage
des répulsions coulombiennes) confirmerait l’explication précédente. Une étude en sel sera
justement présentée dans la suite de ce chapitre (cf. paragraphe III-1).
0
4
8
12
16
20
0 0.001 0.002 0.003
NDTAClibre (M)
Nββ ββ
-CD
liée /N
ββ ββ-C
Dlib
re
Kapp = 16 300 M-1
Kapp = 1540 M-1
Kapp = 11 850 M-1
β-CD native/DTAC poly(β-CD)/DTAC

- 167 -
b) Par mesures de tension de surface
i) Principe
La détermination de Kpoly(β-CD)/DTAC à partir de mesures de tension de surface fait ici
appel à la méthode numérique développée par Lu et coll.50. L’utilisation de cette méthode
nécessite que ni le poly(β-CD), ni les complexes poly(β-CD)/DTAC ne présentent d’activité
de surface, ce qui a été démontré au paragraphe II-1-a.
D’après Lu et coll., lorsque la tension de surface γ d’un mélange CD/TA est
attribuable au TA libre uniquement, la concentration en TA libre au sein du mélange (NTAlibre)
peut être déduite de la relation existant entre γ et la concentration en TA en absence de CD
(NTA). Comme l’indique la figure III-22, à chaque concentration totale en TA en présence de
CD (NTA0) correspond une valeur de γ qui elle-même correspond à une valeur de NTA
(concentration en TA en absence de CD). NTA représente alors la concentration en TA libre
dans le mélange CD/TA, de concentration totale en TA égale à NTA0 (NTA = NTA
libre en
présence de CD).
Remarque: Nous avons vu auparavant (cf. p. 142) que la valeur de γ mesurée pour le DTAC
« seul » en solution tient compte de la présence d’impuretés à l’interface solution/air. Pour
que les variations de γ du mélange poly(β-CD)/DTAC ne soient attribuables qu’aux variations
de la concentration en DTAC libre (condition de base pour pouvoir déterminer Kpoly(β-CD)/DTAC
par la méthode de Lu), il est nécessaire que l’activité de surface de l’impureté ne soit pas
modifiée en présence du polymère (sa contribution à la valeur de γ mesurée en absence et en
présence du poly(β-CD) doit être identique). Par conséquent, nous émettons ici l’hypothèse
que l’impureté n’est pas solubilisée dans les cavités de β-CD du polymère.
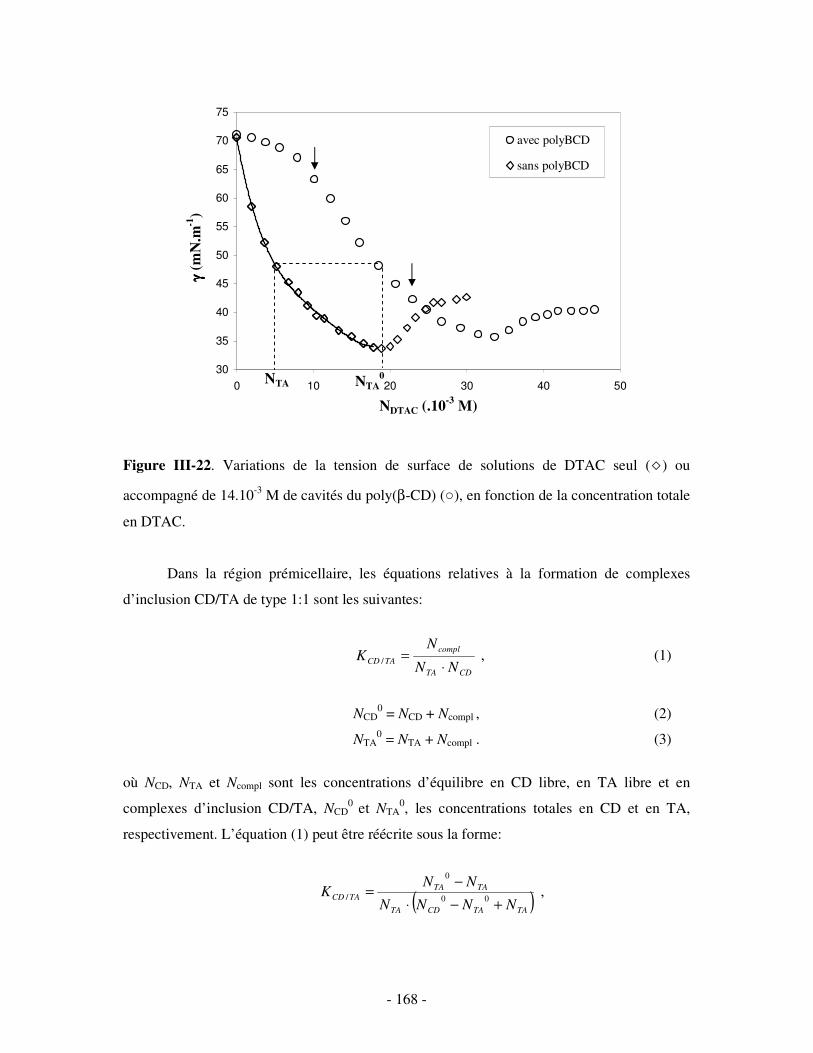
- 168 -
30
35
40
45
50
55
60
65
70
75
0 10 20 30 40 50
NDTAC (.10-3 M)
γγ γγ (m
N.m
-1)
avec polyBCD
sans polyBCD
NTA NTA0
Figure III-22. Variations de la tension de surface de solutions de DTAC seul ( ) ou
accompagné de 14.10-3 M de cavités du poly(β-CD) (), en fonction de la concentration totale
en DTAC.
Dans la région prémicellaire, les équations relatives à la formation de complexes
d’inclusion CD/TA de type 1:1 sont les suivantes:
CDTA
complTACD NN
NK
⋅=/ , (1)
NCD
0 = NCD + Ncompl , (2)
NTA0 = NTA + Ncompl . (3)
où NCD, NTA et Ncompl sont les concentrations d’équilibre en CD libre, en TA libre et en
complexes d’inclusion CD/TA, NCD0 et NTA
0, les concentrations totales en CD et en TA,
respectivement. L’équation (1) peut être réécrite sous la forme:
( )TATACDTA
TATATACD
NNNN
NNK
+−⋅−
= 00
0
/ ,

- 169 -
soit TATA
TATACDTA
TACD NN
NNNN
K −+−
⋅= 0
00
:
1
10
00
−
+−=
TA
TA
TATACD
NN
NNN . (4)
D’où l’expression finale:
00
/
0 11
CDTA
TA
TACDTATA N
NN
KNN +
−−=− . (5)
En traçant ( TATA NN −0 ) en fonction de
−1
0
TA
TA
NN
(valeurs déduites des mesures de tension
de surface), il est donc possible de déterminer la constante d’association KCD/TA des complexes
d’inclusion (inverse de la pente).
ii) Résultats
La figure III-23 présente les résultats obtenus en appliquant la méthode de Lu aux
valeurs expérimentales de la figure III-22 délimitées par les flèches. C’est en effet dans ce
domaine de concentrations que les variations de γ induites par les variations de la
concentration en DTAC libre sont les plus significatives. (Pour comparaison avec les mesures
fluorimétriques, une faible quantité de DTAC suffisait à déplacer la sonde fluorescente des
cavités de β-CD. Les variations de If étaient donc significatives même à faible NDTAC0).
La constante d’association apparente des complexes poly(β-CD)/DTAC est finalement
calculée par régression linéaire des données de la figure III-23. De la pente de la droite (cf.
équation 5) est extraite:
Kpoly(β-CD)/DTAC = 1820 ± 310 M-1 .
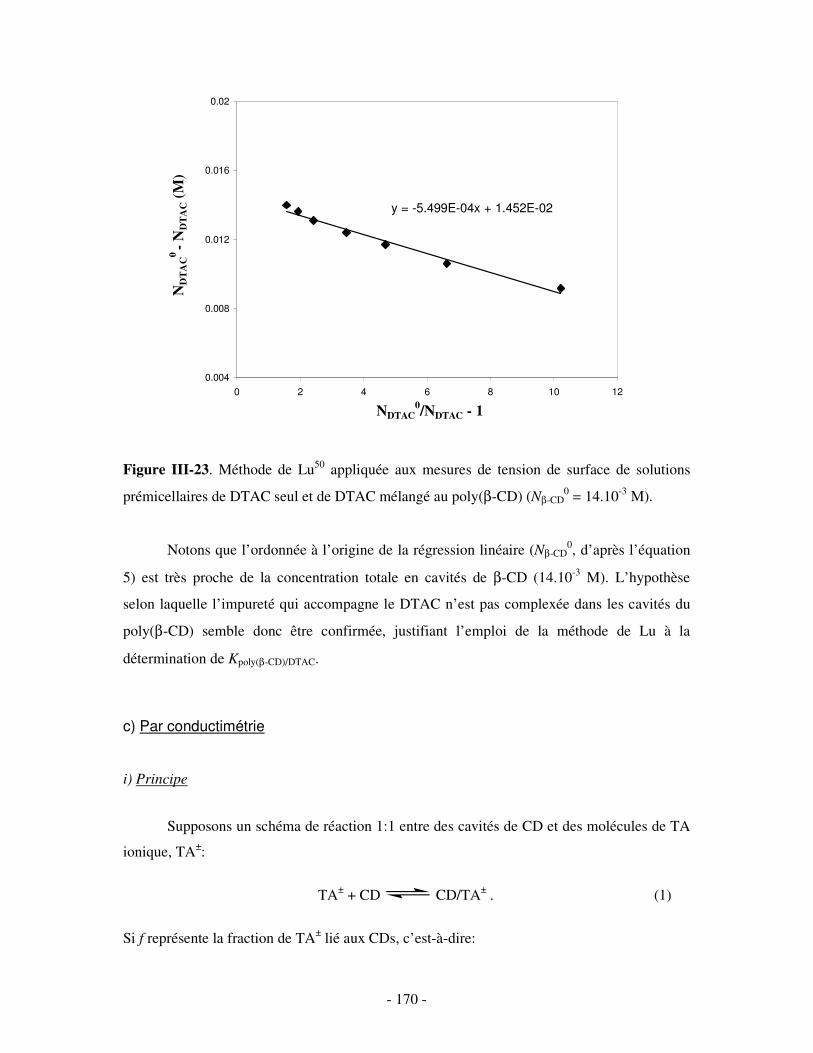
- 170 -
y = -5.499E-04x + 1.452E-02
0.004
0.008
0.012
0.016
0.02
0 2 4 6 8 10 12
NDTAC0/NDTAC - 1
ND
TA
C0 - N
DT
AC (M
)
Figure III-23. Méthode de Lu50 appliquée aux mesures de tension de surface de solutions
prémicellaires de DTAC seul et de DTAC mélangé au poly(β-CD) (Nβ-CD0 = 14.10-3 M).
Notons que l’ordonnée à l’origine de la régression linéaire (Nβ-CD0, d’après l’équation
5) est très proche de la concentration totale en cavités de β-CD (14.10-3 M). L’hypothèse
selon laquelle l’impureté qui accompagne le DTAC n’est pas complexée dans les cavités du
poly(β-CD) semble donc être confirmée, justifiant l’emploi de la méthode de Lu à la
détermination de Kpoly(β-CD)/DTAC.
c) Par conductimétrie
i) Principe
Supposons un schéma de réaction 1:1 entre des cavités de CD et des molécules de TA
ionique, TA±:
TA± + CD CD/TA± . (1)
Si f représente la fraction de TA± lié aux CDs, c’est-à-dire:

- 171 -
0TA
compl
N
Nf = , (2)
avec Ncompl et NTA
0 la concentration d’équilibre en complexes et la concentration totale en TA±
respectivement, la loi de conservation donne:
NTA0 = NTA + fNTA
0 (3)
et NCD0 = NCD + fNTA
0 . (4)
La constante d’association K des complexes d’inclusion CD/TA± s’écrit alors:
( ) ( )001 TACD fNNf
fK
−⋅−= , (5)
où NCD
0 est la concentration totale en cavités de CD.
Soient Λ0 et Λ les conductivités équivalentes (en S.cm2.mol-1) d’une solution diluée du
TA± en absence et en présence de CD respectivement. D’après Satake et coll.53,
( )λλ −=Λ−Λ 00 f , (6)
où λ0 et λ sont les conductivités ioniques équivalentes respectives du TA± et du complexe
CD/TA±. Une combinaison des équations (5) et (6) conduit à l’expression finale:
( ) ( )
−++−++−
=Λ−Λ 002200000
00 411
2CDTACDTACDTA
TA
NNKNNKNNKKN
λλ , (7)
ou bien
κ0 – κ ( ) ( )
−++−++−
= 002200000 4112 CDTACDTACDTA NNKNNKNNK
kKλλ
, (8)
avec κ0 et κ, les conductances (en S) de la solution de TA± en absence et en présence de CD,
et k, la constante caractéristique de la cellule d’analyse. Les valeurs de K et de λ0 - λ peuvent
donc être évaluées à partir des courbes de κ0 - κ vs. NCD0 et de l’équation (8).
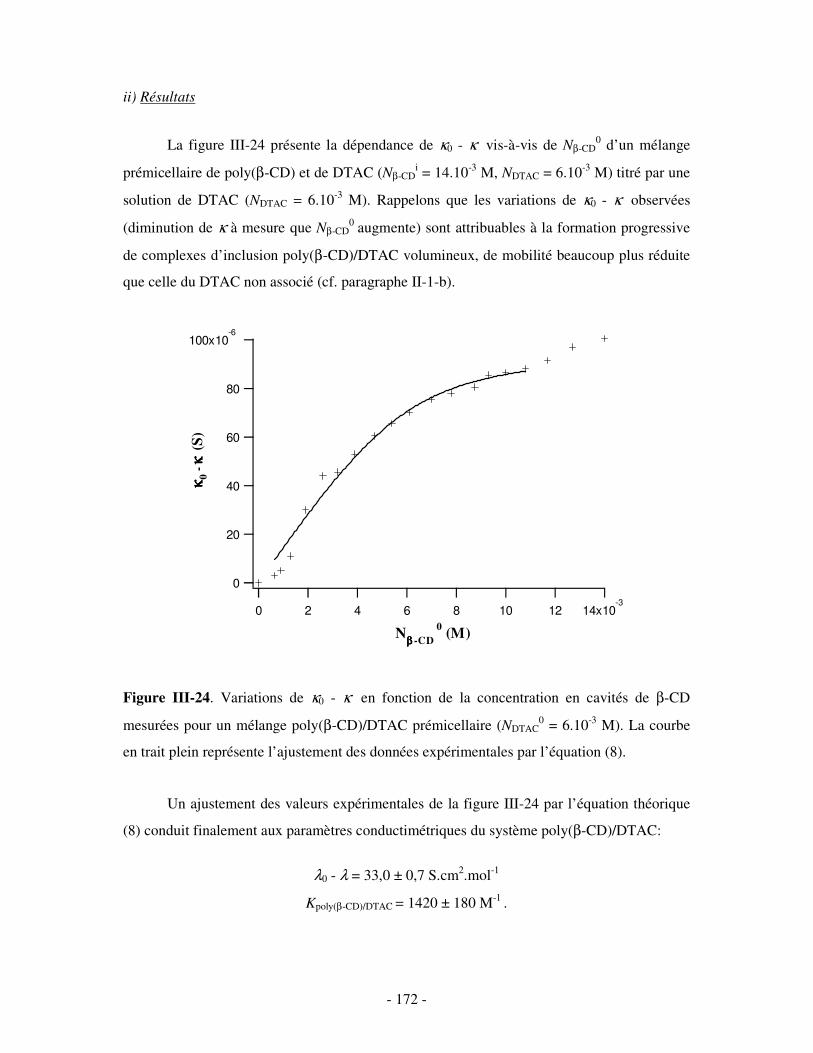
- 172 -
ii) Résultats
La figure III-24 présente la dépendance de κ0 - κ vis-à-vis de Nβ-CD0 d’un mélange
prémicellaire de poly(β-CD) et de DTAC (Nβ-CDi = 14.10-3 M, NDTAC = 6.10-3 M) titré par une
solution de DTAC (NDTAC = 6.10-3 M). Rappelons que les variations de κ0 - κ observées
(diminution de κ à mesure que Nβ-CD0 augmente) sont attribuables à la formation progressive
de complexes d’inclusion poly(β-CD)/DTAC volumineux, de mobilité beaucoup plus réduite
que celle du DTAC non associé (cf. paragraphe II-1-b).
100x10-6
80
60
40
20
0
κκ κκ 0 - κ κ κ κ
(S)
14x10-3
121086420
Nββββ -CD 0 (M)
Figure III-24. Variations de κ0 - κ en fonction de la concentration en cavités de β-CD
mesurées pour un mélange poly(β-CD)/DTAC prémicellaire (NDTAC0 = 6.10-3 M). La courbe
en trait plein représente l’ajustement des données expérimentales par l’équation (8).
Un ajustement des valeurs expérimentales de la figure III-24 par l’équation théorique
(8) conduit finalement aux paramètres conductimétriques du système poly(β-CD)/DTAC:
λ0 - λ = 33,0 ± 0,7 S.cm2.mol-1
Kpoly(β-CD)/DTAC = 1420 ± 180 M-1 .

- 173 -
Il est à noter que la différence obtenue entre les conductivités ioniques équivalentes du DTAC
et du complexe poly(β-CD)/DTAC est plus importante que celle déterminée par Satake et
coll.54 pour le système β-CD/DTAB: λ0 - λ = 12,5 S.cm2.mol-1 (NDTAB0 = 2.10-3 M). Ceci
conforte l’hypothèse de la formation de complexes d’inclusion moins mobiles avec le
polymère plutôt qu’avec les monomères de β-CD. D’autre part, l’association poly(β-
CD)/DTAC apparaît moins forte (Kβ-CD/DTAB = 4880 M-1), ce qui appuie l’argument selon
lequel l’architecture polymérisée des β-CDs est défavorable à l’association.
Remarque: L’influence de la nature du contre-ion (Cl- ou Br-) sur l’association CD/TA a très
peu été explicitée dans la littérature. On peut toutefois supposer que l’influence du contre-ion
est mineure étant donné que seule la chaîne hydrophobe du TA pénètre au sein des cavités des
CDs.
d) Bilan
Les résultats des études quantitatives de l’association binaire poly(β-CD)/DTAC sont
résumés dans le tableau III-4.
Fluorimétrie
Tension de
surface Conductimétrie
Concentrations NDTAC
0 < 0,2 mM
Nβ-CD0 = 0,5 mM
NDTAC0 > 0,5 mM
Nβ-CD0 = 0,5 mM
10 < NDTAC0 (mM) < 23
Nβ-CD0 = 14 mM
NDTAC0 = 6 mM
2 < Nβ-CD0 (mM) < 11
K1:1 (M-1) 11 850
(± 1150)
1540
(± 50)
1820
(± 310)
1420
(± 180)
Tableau III-4. Valeurs de K1:1 du système poly(β-CD)/DTAC en fonction de la méthode
expérimentale employée et des domaines de concentrations en β-CD et en DTAC explorés.
Excepté pour des concentrations en DTAC très faibles et inférieures à Nβ-CD, les
valeurs de Kpoly(β-CD)/DTAC obtenues sont équivalentes (K ~ 1600 M-1), et ce malgré les
différences concernant, d’une part, les approches selon lesquelles K est déterminée (en suivant
les variations des propriétés physico-chimiques des solutions, ou bien en mesurant
directement les concentrations d’équilibre en DTAC libre et complexé) et concernant, d’autre

- 174 -
part, les domaines de concentrations explorés. Cette première constatation tend à confirmer
que les cavités du poly(β-CD) et les molécules de DTAC forment exclusivement des
complexes d’inclusion de type 1:1, l’existence d’équilibres multiples conduisant généralement
à une dispersion des valeurs de K1:1.
Le fait que Kpoly(β-CD)/DTAC soit plus faible d’environ un ordre de grandeur que la
constante d’équilibre des complexes β-CD native/DTAC (K ~ 16 000 M-1) montre ensuite que
l’appartenance des cavités de β-CD à une chaîne de polymère est relativement défavorable à
l’association binaire. L’existence de contributions électrostatiques répulsives, en corrélation
avec cette architecture polymère, a alors été supposée: Kpoly(β-CD)/DTAC ne s’approche de 16 000
M-1 qu’en présence d’une très faible quantité de DTAC, soit lorsque les répulsions
électrostatiques sont dans l’ensemble minimisées.
La concentration en DTAC a donc une influence relative sur la force de l’association
poly(β-CD)/DTAC. Etant donnée la nature ionique du tensioactif, NDTAC doit surtout
constituer un paramètre clé pour les propriétés structurales des agrégats, au même titre que la
force ionique du milieu (répulsions coulombiennes intrachaînes plus ou moins intenses). C’est
ce que nous allons étudier dans la dernière partie de ce chapitre.
III- Etude des propriétés structurales du complexe binaire
poly(CD)/TA ionique
1- Influence de la force ionique
Les figures III-25 et III-26 présentent respectivement l’influence de la force ionique
sur la viscosité réduite et le spectre de diffusion neutronique d’un mélange comportant le
poly(β-CD) et le DTAC dans un rapport stoechiométrique Nβ-CD/NDTAC = 14/6. Comme il peut
être observé, l’effet du sel se manifeste par une chute marquée des valeurs de viscosité et par
une perte du comportement PEL: la viscosité du mélange diminue par dilution en présence de
sel (figure III-25), tandis que le pic de diffusion disparaît à mesure que la force ionique
augmente (figure III-26).
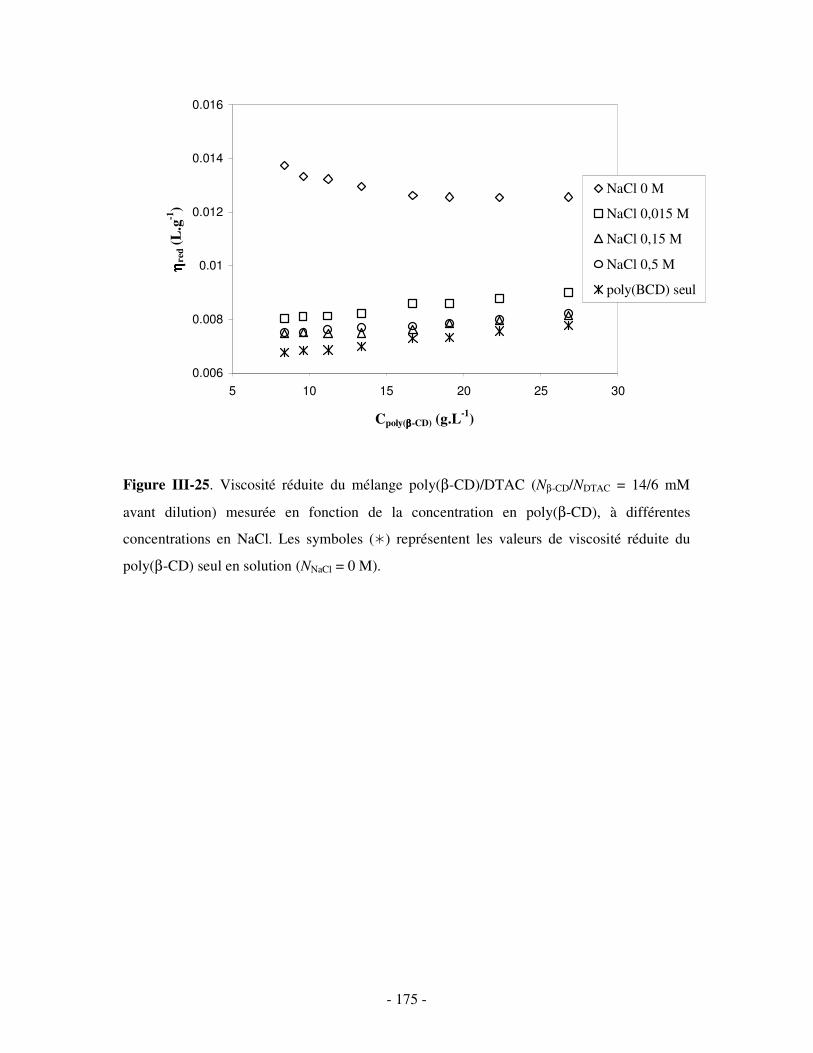
- 175 -
0.006
0.008
0.01
0.012
0.014
0.016
5 10 15 20 25 30
Cpoly(ββββ-CD) (g.L-1)
ηη ηη red
(L.g
-1)
NaCl 0 M
NaCl 0,015 M
NaCl 0,15 M
NaCl 0,5 M
poly(BCD) seul
Figure III-25. Viscosité réduite du mélange poly(β-CD)/DTAC (Nβ-CD/NDTAC = 14/6 mM
avant dilution) mesurée en fonction de la concentration en poly(β-CD), à différentes
concentrations en NaCl. Les symboles () représentent les valeurs de viscosité réduite du
poly(β-CD) seul en solution (NNaCl = 0 M).
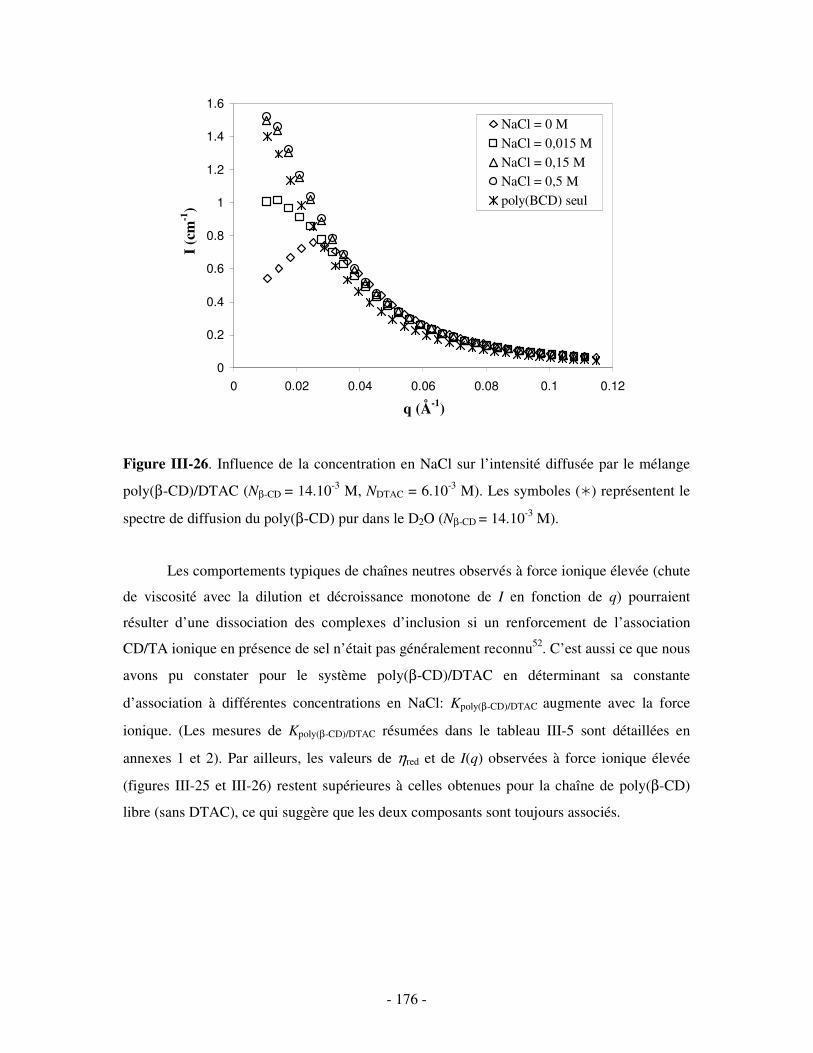
- 176 -
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
0 0.02 0.04 0.06 0.08 0.1 0.12
q (Å-1)
I (cm
-1)
NaCl = 0 MNaCl = 0,015 MNaCl = 0,15 MNaCl = 0,5 Mpoly(BCD) seul
Figure III-26. Influence de la concentration en NaCl sur l’intensité diffusée par le mélange
poly(β-CD)/DTAC (Nβ-CD = 14.10-3 M, NDTAC = 6.10-3 M). Les symboles () représentent le
spectre de diffusion du poly(β-CD) pur dans le D2O (Nβ-CD = 14.10-3 M).
Les comportements typiques de chaînes neutres observés à force ionique élevée (chute
de viscosité avec la dilution et décroissance monotone de I en fonction de q) pourraient
résulter d’une dissociation des complexes d’inclusion si un renforcement de l’association
CD/TA ionique en présence de sel n’était pas généralement reconnu52. C’est aussi ce que nous
avons pu constater pour le système poly(β-CD)/DTAC en déterminant sa constante
d’association à différentes concentrations en NaCl: Kpoly(β-CD)/DTAC augmente avec la force
ionique. (Les mesures de Kpoly(β-CD)/DTAC résumées dans le tableau III-5 sont détaillées en
annexes 1 et 2). Par ailleurs, les valeurs de ηred et de I(q) observées à force ionique élevée
(figures III-25 et III-26) restent supérieures à celles obtenues pour la chaîne de poly(β-CD)
libre (sans DTAC), ce qui suggère que les deux composants sont toujours associés.

- 177 -
K1:1 (M-1)
NNaCl (M) Fluorimétrie
(NDTAC0 > 0,5 mM, Nβ-CD
0 = 0,5 mM)
Tension de surface (9 < NDTAC
0 (mM) < 23, Nβ-CD0 = 14 mM)
0 1540 ± 50 1820 ± 310
0,015 - 2480 ± 440
0,15 - 5350 ± 410
0,5 9050 ± 640 7680 ± 160
Tableau III-5. Influence du sel sur les valeurs apparentes de Kpoly(β-CD)/DTAC déterminées par
fluorimétrie (cf. annexe 1) et par mesures de tension de surface (cf. annexe 2).
Par conséquent, la seule explication possible aux chutes de viscosité et disparition du
comportement PEL observées est un écrantage par le sel des répulsions électrostatiques intra-
et interchaînes. En d’autres termes, l’influence du sel se traduit par une réduction de la
longueur de Debye κ -1, celle-ci correspondant à l’épaisseur du nuage électronique autour
d’une charge complexée, soit à la distance au-delà de laquelle l’effet de cette charge est
complètement écranté.
Par définition, κ -1 est reliée à la force ionique I du milieu selon:
( ) 21
1
8
1
Il aB ℵ=−
πκ , (1)
où lB = 7,135 Å est la longueur de Bjerrum dans l’eau à 25°C, aℵ = 6,023.1023 mol-1 est le
nombre d’Avogadro et I, la concentration molaire en espèces ioniques mobiles. Il est
important de préciser qu’en présence de 14.10-3 M de cavités de β-CD et 6.10-3 M de DTAC,
les molécules de TA peuvent toutes être considérées comme inclues au sein des cavités du
polymère, et ce quelque soit la salinité. En effet, comme l’indique le tableau III-6, la cmc
apparente du système mixte (cmc*) est loin d’être atteinte aux concentrations en β-CD, DTAC
et NaCl choisies.
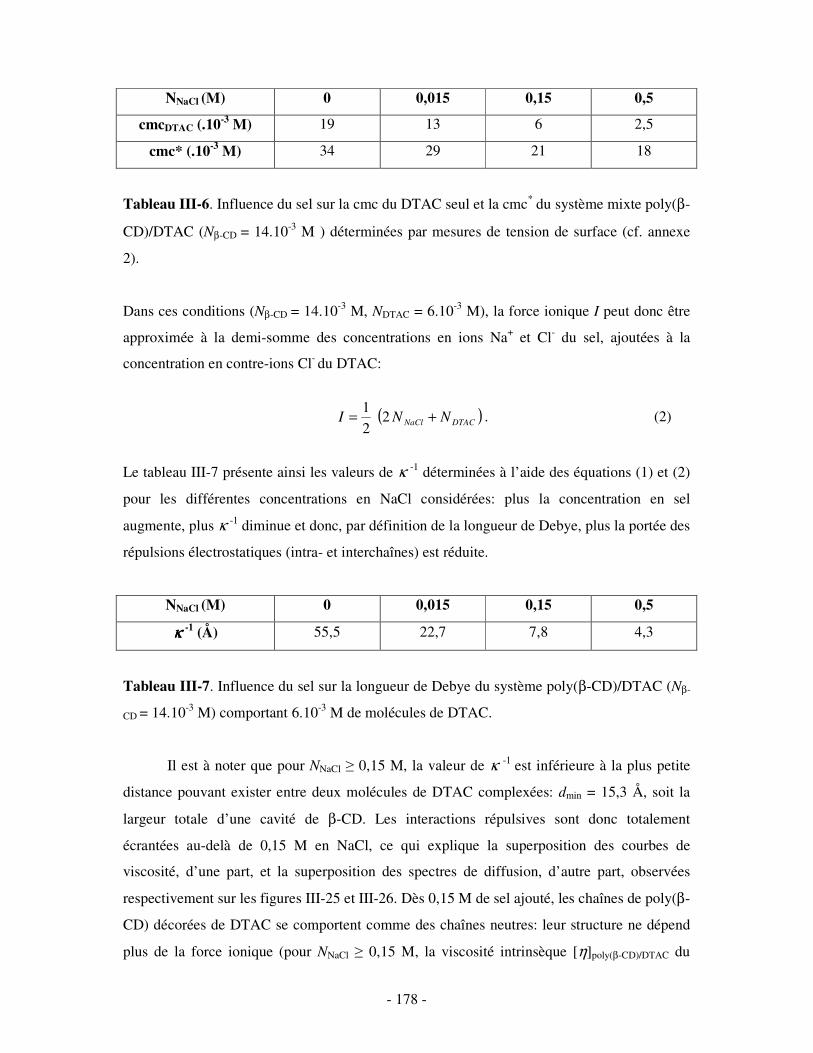
- 178 -
NNaCl (M) 0 0,015 0,15 0,5
cmcDTAC (.10-3 M) 19 13 6 2,5
cmc* (.10-3 M) 34 29 21 18
Tableau III-6. Influence du sel sur la cmc du DTAC seul et la cmc* du système mixte poly(β-
CD)/DTAC (Nβ-CD = 14.10-3 M ) déterminées par mesures de tension de surface (cf. annexe
2).
Dans ces conditions (Nβ-CD = 14.10-3 M, NDTAC = 6.10-3 M), la force ionique I peut donc être
approximée à la demi-somme des concentrations en ions Na+ et Cl- du sel, ajoutées à la
concentration en contre-ions Cl- du DTAC:
( )DTACNaCl NNI += 221
. (2)
Le tableau III-7 présente ainsi les valeurs de κ -1 déterminées à l’aide des équations (1) et (2)
pour les différentes concentrations en NaCl considérées: plus la concentration en sel
augmente, plus κ -1 diminue et donc, par définition de la longueur de Debye, plus la portée des
répulsions électrostatiques (intra- et interchaînes) est réduite.
NNaCl (M) 0 0,015 0,15 0,5
κκκκ -1 (Å) 55,5 22,7 7,8 4,3
Tableau III-7. Influence du sel sur la longueur de Debye du système poly(β-CD)/DTAC (Nβ-
CD = 14.10-3 M) comportant 6.10-3 M de molécules de DTAC.
Il est à noter que pour NNaCl 0,15 M, la valeur de κ -1 est inférieure à la plus petite
distance pouvant exister entre deux molécules de DTAC complexées: dmin = 15,3 Å, soit la
largeur totale d’une cavité de β-CD. Les interactions répulsives sont donc totalement
écrantées au-delà de 0,15 M en NaCl, ce qui explique la superposition des courbes de
viscosité, d’une part, et la superposition des spectres de diffusion, d’autre part, observées
respectivement sur les figures III-25 et III-26. Dès 0,15 M de sel ajouté, les chaînes de poly(β-
CD) décorées de DTAC se comportent comme des chaînes neutres: leur structure ne dépend
plus de la force ionique (pour NNaCl 0,15 M, la viscosité intrinsèque [η]poly(β-CD)/DTAC du
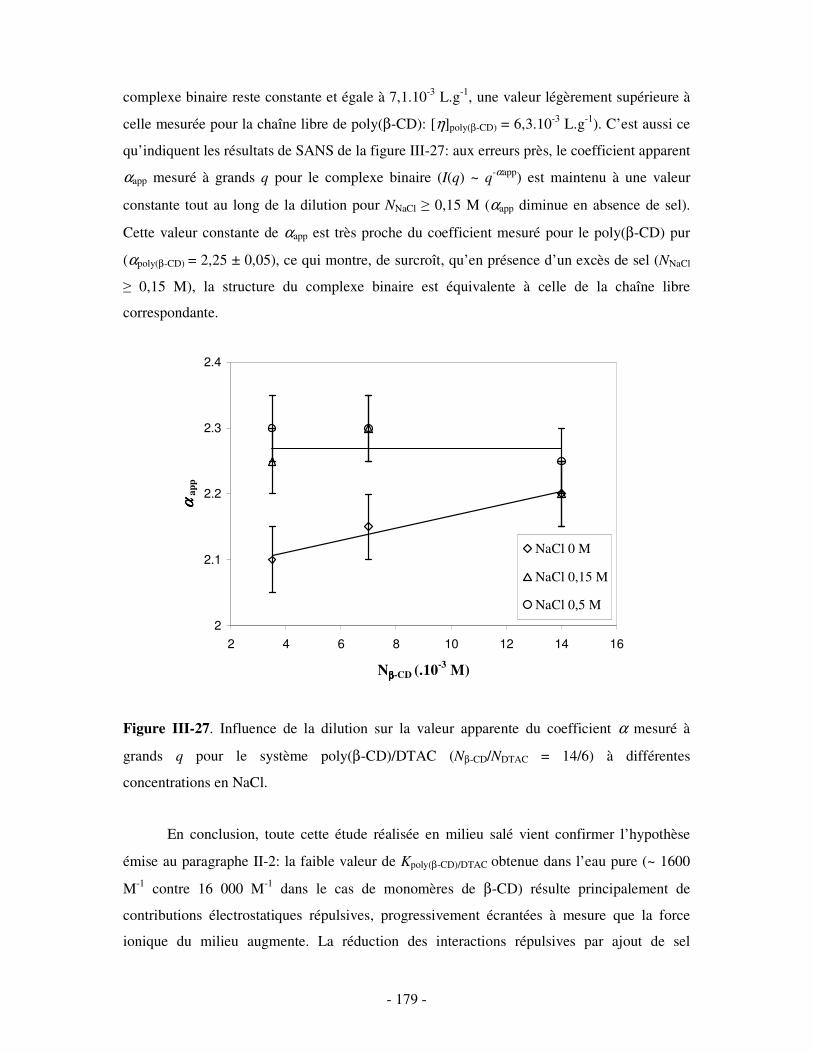
- 179 -
complexe binaire reste constante et égale à 7,1.10-3 L.g-1, une valeur légèrement supérieure à
celle mesurée pour la chaîne libre de poly(β-CD): [η]poly(β-CD) = 6,3.10-3 L.g-1). C’est aussi ce
qu’indiquent les résultats de SANS de la figure III-27: aux erreurs près, le coefficient apparent
αapp mesuré à grands q pour le complexe binaire (I(q) ~ q-αapp) est maintenu à une valeur
constante tout au long de la dilution pour NNaCl 0,15 M (αapp diminue en absence de sel).
Cette valeur constante de αapp est très proche du coefficient mesuré pour le poly(β-CD) pur
(αpoly(β-CD) = 2,25 ± 0,05), ce qui montre, de surcroît, qu’en présence d’un excès de sel (NNaCl
0,15 M), la structure du complexe binaire est équivalente à celle de la chaîne libre
correspondante.
2
2.1
2.2
2.3
2.4
2 4 6 8 10 12 14 16
Nββββ-CD (.10-3 M)
αα ααap
p
NaCl 0 M
NaCl 0,15 M
NaCl 0,5 M
Figure III-27. Influence de la dilution sur la valeur apparente du coefficient α mesuré à
grands q pour le système poly(β-CD)/DTAC (Nβ-CD/NDTAC = 14/6) à différentes
concentrations en NaCl.
En conclusion, toute cette étude réalisée en milieu salé vient confirmer l’hypothèse
émise au paragraphe II-2: la faible valeur de Kpoly(β-CD)/DTAC obtenue dans l’eau pure (~ 1600
M-1 contre 16 000 M-1 dans le cas de monomères de β-CD) résulte principalement de
contributions électrostatiques répulsives, progressivement écrantées à mesure que la force
ionique du milieu augmente. La réduction des interactions répulsives par ajout de sel

- 180 -
s’accompagne d’une compaction globale du complexe poly(β-CD)/DTAC (marquée par une
chute de viscosité), et d’une faible compaction de sa structure interne (en rapport avec
l’architecture branchée du polymère).
Cependant, bien qu’un écrantage complet des répulsions coulombiennes ait été
démontré pour NNaCl 0,15 M (κ -1 < dmin), la valeur de Kpoly(β-CD)/DTAC déterminée pour NNaCl
= 0,5 M (cf. tableau III-5) reste inférieure à celle déterminée dans les mêmes conditions pour
les complexes impliquant de la β-CD native (Kβ-CD/DTAC0,5 M = 17 085 ± 680 M-1, cf. annexe 1-
II-1). Kpoly(β-CD)/DTAC apparaît plutôt du même ordre de grandeur que la constante d’association
des complexes composés d’HP(β-CD), un monomère de β-CD modifié par 5-6 chaînons
hydroxypropyle en moyenne (KHP(β-CD)/DTAC0,5 M = 9645 ± 215 M-1, d’après l’annexe 1-II-2).
Ceci nous amène à supposer l’existence supplémentaire de gênes stériques induites par la
présence des chaînes hydroxypropyléther en bordure des cavités du poly(β-CD). (On
considère que la structure des β-CDs au sein du polymère est bien modélisée par les
monomères d’HP(β-CD)).
2- Influence de la concentration en TA
La figure III-28 montre l’influence de la concentration molaire en TA sur la viscosité
réduite de mélanges aqueux poly(β-CD)/DTAC comportant 14.10-3 M de cavités de β-CD.
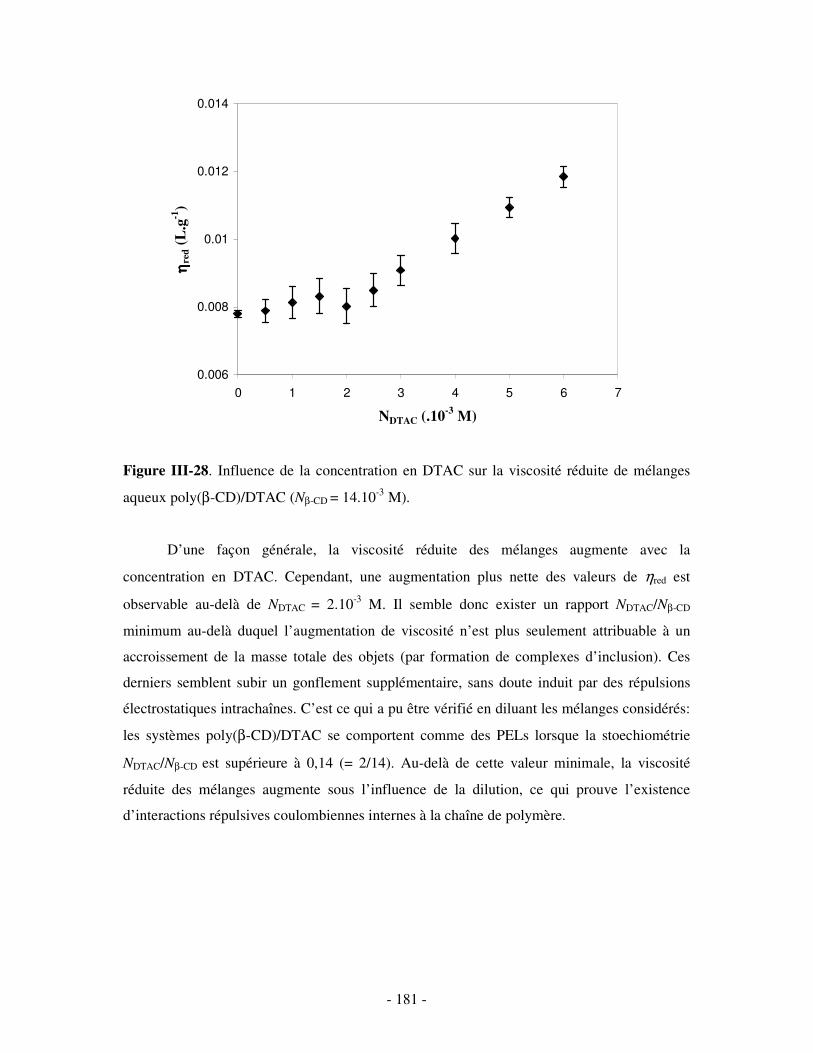
- 181 -
0.006
0.008
0.01
0.012
0.014
0 1 2 3 4 5 6 7
NDTAC (.10-3 M)
ηη ηη red
(L.g
-1)
Figure III-28. Influence de la concentration en DTAC sur la viscosité réduite de mélanges
aqueux poly(β-CD)/DTAC (Nβ-CD = 14.10-3 M).
D’une façon générale, la viscosité réduite des mélanges augmente avec la
concentration en DTAC. Cependant, une augmentation plus nette des valeurs de ηred est
observable au-delà de NDTAC = 2.10-3 M. Il semble donc exister un rapport NDTAC/Nβ-CD
minimum au-delà duquel l’augmentation de viscosité n’est plus seulement attribuable à un
accroissement de la masse totale des objets (par formation de complexes d’inclusion). Ces
derniers semblent subir un gonflement supplémentaire, sans doute induit par des répulsions
électrostatiques intrachaînes. C’est ce qui a pu être vérifié en diluant les mélanges considérés:
les systèmes poly(β-CD)/DTAC se comportent comme des PELs lorsque la stoechiométrie
NDTAC/Nβ-CD est supérieure à 0,14 (= 2/14). Au-delà de cette valeur minimale, la viscosité
réduite des mélanges augmente sous l’influence de la dilution, ce qui prouve l’existence
d’interactions répulsives coulombiennes internes à la chaîne de polymère.
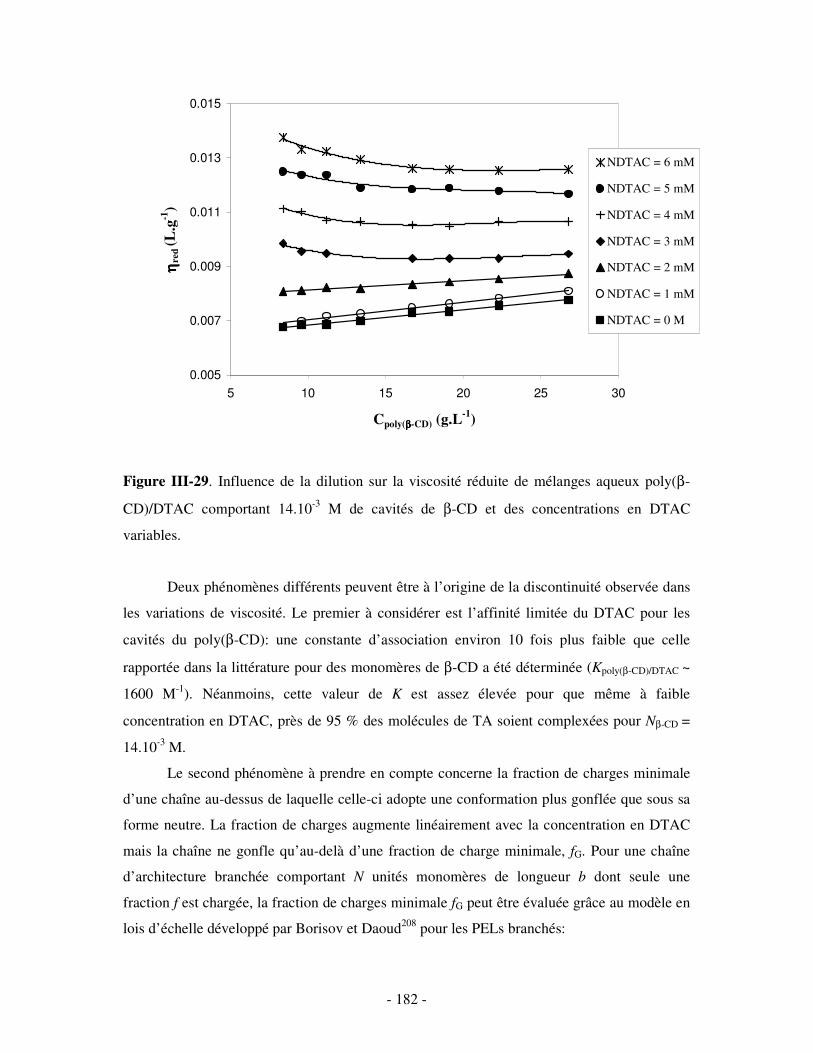
- 182 -
0.005
0.007
0.009
0.011
0.013
0.015
5 10 15 20 25 30
Cpoly(ββββ-CD) (g.L-1)
ηη ηη red
(L.g
-1)
NDTAC = 6 mM
NDTAC = 5 mM
NDTAC = 4 mM
NDTAC = 3 mM
NDTAC = 2 mM
NDTAC = 1 mM
NDTAC = 0 M
Figure III-29. Influence de la dilution sur la viscosité réduite de mélanges aqueux poly(β-
CD)/DTAC comportant 14.10-3 M de cavités de β-CD et des concentrations en DTAC
variables.
Deux phénomènes différents peuvent être à l’origine de la discontinuité observée dans
les variations de viscosité. Le premier à considérer est l’affinité limitée du DTAC pour les
cavités du poly(β-CD): une constante d’association environ 10 fois plus faible que celle
rapportée dans la littérature pour des monomères de β-CD a été déterminée (Kpoly(β-CD)/DTAC ~
1600 M-1). Néanmoins, cette valeur de K est assez élevée pour que même à faible
concentration en DTAC, près de 95 % des molécules de TA soient complexées pour Nβ-CD =
14.10-3 M.
Le second phénomène à prendre en compte concerne la fraction de charges minimale
d’une chaîne au-dessus de laquelle celle-ci adopte une conformation plus gonflée que sous sa
forme neutre. La fraction de charges augmente linéairement avec la concentration en DTAC
mais la chaîne ne gonfle qu’au-delà d’une fraction de charge minimale, fG. Pour une chaîne
d’architecture branchée comportant N unités monomères de longueur b dont seule une
fraction f est chargée, la fraction de charges minimale fG peut être évaluée grâce au modèle en
lois d’échelle développé par Borisov et Daoud208 pour les PELs branchés:
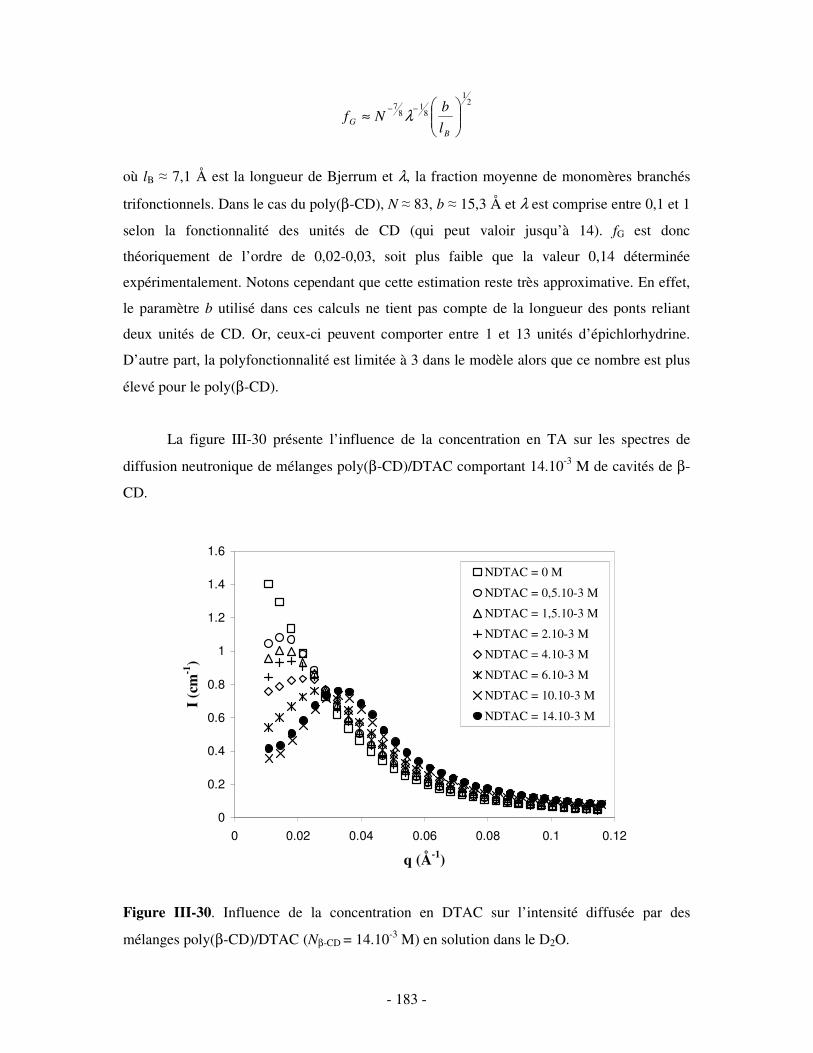
- 183 -
21
81
87
≈ −−
BG l
bNf λ
où lB 7,1 Å est la longueur de Bjerrum et λ, la fraction moyenne de monomères branchés
trifonctionnels. Dans le cas du poly(β-CD), N 83, b 15,3 Å et λ est comprise entre 0,1 et 1
selon la fonctionnalité des unités de CD (qui peut valoir jusqu’à 14). fG est donc
théoriquement de l’ordre de 0,02-0,03, soit plus faible que la valeur 0,14 déterminée
expérimentalement. Notons cependant que cette estimation reste très approximative. En effet,
le paramètre b utilisé dans ces calculs ne tient pas compte de la longueur des ponts reliant
deux unités de CD. Or, ceux-ci peuvent comporter entre 1 et 13 unités d’épichlorhydrine.
D’autre part, la polyfonctionnalité est limitée à 3 dans le modèle alors que ce nombre est plus
élevé pour le poly(β-CD).
La figure III-30 présente l’influence de la concentration en TA sur les spectres de
diffusion neutronique de mélanges poly(β-CD)/DTAC comportant 14.10-3 M de cavités de β-
CD.
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
0 0.02 0.04 0.06 0.08 0.1 0.12
q (Å-1)
I (cm
-1)
NDTAC = 0 M
NDTAC = 0,5.10-3 M
NDTAC = 1,5.10-3 M
NDTAC = 2.10-3 M
NDTAC = 4.10-3 M
NDTAC = 6.10-3 M
NDTAC = 10.10-3 M
NDTAC = 14.10-3 M
Figure III-30. Influence de la concentration en DTAC sur l’intensité diffusée par des
mélanges poly(β-CD)/DTAC (Nβ-CD = 14.10-3 M) en solution dans le D2O.
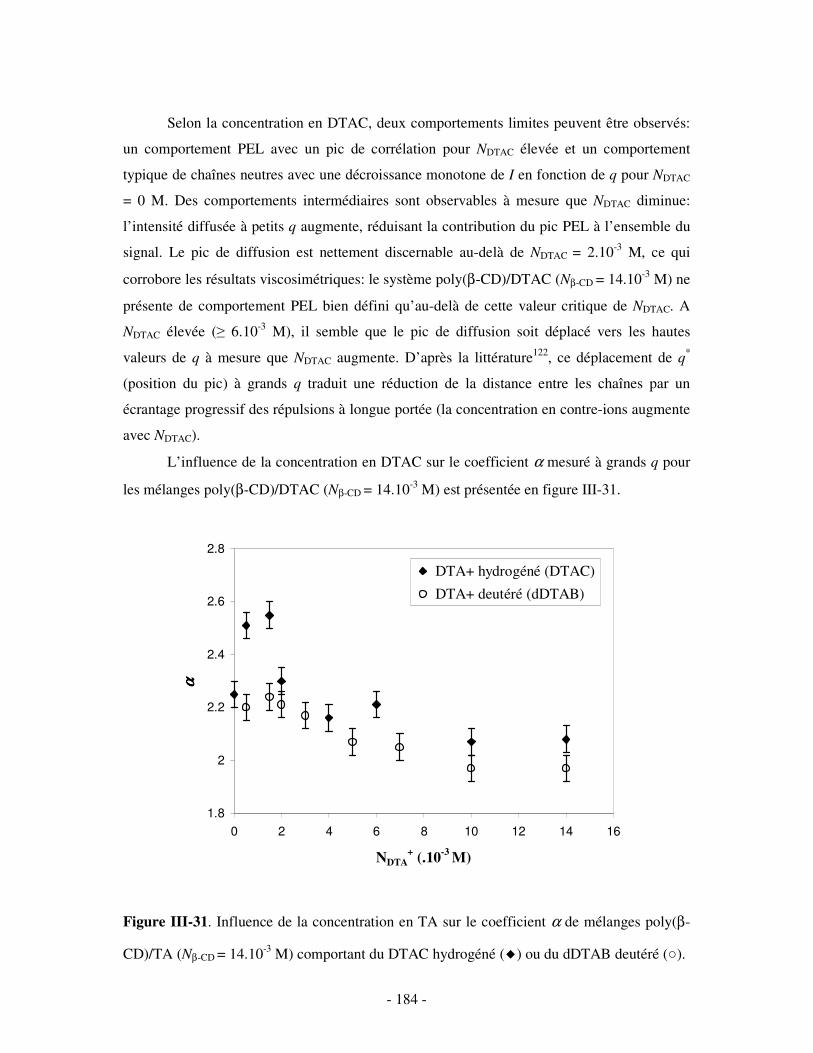
- 184 -
Selon la concentration en DTAC, deux comportements limites peuvent être observés:
un comportement PEL avec un pic de corrélation pour NDTAC élevée et un comportement
typique de chaînes neutres avec une décroissance monotone de I en fonction de q pour NDTAC
= 0 M. Des comportements intermédiaires sont observables à mesure que NDTAC diminue:
l’intensité diffusée à petits q augmente, réduisant la contribution du pic PEL à l’ensemble du
signal. Le pic de diffusion est nettement discernable au-delà de NDTAC = 2.10-3 M, ce qui
corrobore les résultats viscosimétriques: le système poly(β-CD)/DTAC (Nβ-CD = 14.10-3 M) ne
présente de comportement PEL bien défini qu’au-delà de cette valeur critique de NDTAC. A
NDTAC élevée ( 6.10-3 M), il semble que le pic de diffusion soit déplacé vers les hautes
valeurs de q à mesure que NDTAC augmente. D’après la littérature122, ce déplacement de q*
(position du pic) à grands q traduit une réduction de la distance entre les chaînes par un
écrantage progressif des répulsions à longue portée (la concentration en contre-ions augmente
avec NDTAC).
L’influence de la concentration en DTAC sur le coefficient α mesuré à grands q pour
les mélanges poly(β-CD)/DTAC (Nβ-CD = 14.10-3 M) est présentée en figure III-31.
1.8
2
2.2
2.4
2.6
2.8
0 2 4 6 8 10 12 14 16
NDTA+ (.10-3 M)
αα αα
DTA+ hydrogéné (DTAC)
DTA+ deutéré (dDTAB)
Figure III-31. Influence de la concentration en TA sur le coefficient α de mélanges poly(β-
CD)/TA (Nβ-CD = 14.10-3 M) comportant du DTAC hydrogéné () ou du dDTAB deutéré ().

- 185 -
Par comparaison avec les résultats viscosimétriques, il apparaît clairement que NDTAC =
2.10-3 M est aussi une concentration critique pour laquelle les agrégats formés subissent une
transition structurale microscopique. Pour NDTAC < 2.10-3 M, les valeurs de α augmentent avec
la concentration en TA ce qui, théoriquement, est synonyme d’une compaction des objets
diffusants. Par contre, α diminue pour NDTAC 2.10-3 M, ce qui suggère que les chaînes sont
de moins en moins compactes à mesure que NDTAC augmente.
Dans la partie précédente, le comportement viscosimétrique critique observé pour
NDTAC = 2.10-3 M a été attribué à la fraction de charges minimale fG du complexe, au-delà de
laquelle la chaîne de polymère adopte une conformation gonflée. Donc, à priori, même si les
résultats de SANS obtenus à NDTAC élevée semblent corroborer cette interprétation
(gonflement de la chaîne), ceux obtenus à faible NDTAC apparaissent contradictoires:
l’augmentation de α observée (bien au-dessus des barres d’erreur) suggère une compacité
grandissante du complexe dans ce domaine de concentrations. En fait, les résultats
viscosimétriques et de diffusion de neutrons ne sont pas en contradiction. Au contraire, ils
offrent un aperçu de la distribution de charges au sein du complexe binaire selon la valeur de
NDTAC.
Si les charges (molécules de DTAC cationiques) étaient réparties de façon homogène
le long de la chaîne de polymère, le facteur de structure global du complexe serait dominé par
la diffusion du poly(β-CD). La concentration en DTAC (1,6 g.L-1 lorsque NDTAC = 6.10-3 M)
est en effet trop faible comparée à celle du polymère (26,8 g.L-1 lorsque Nβ-CD = 14.10-3 M).
Au vu des résultats viscosimétriques, le coefficient α mesuré pour le système binaire serait
donc maintenu à une valeur proche de 2,25 pour NDTAC < 2.10-3 M (soit à la valeur de α du
poly(β-CD) neutre, non gonflé), puis diminuerait pour NDTAC > 2.10-3 M. Le fait que α
augmente avec NDTAC à faibles concentrations vient contredire, par conséquent, l’hypothèse
d’une distribution de charges homogène le long de la chaîne de poly(β-CD). Etant données les
répulsions électrostatiques induites entre les molécules de TA complexées, une distribution de
charges « sphérique » est donc envisageable pour NDTAC inférieure à la valeur critique. Il est
en effet très probable qu’en faible quantité, le DTAC vienne se complexer en périphérie de
l’architecture branchée du poly(β-CD), définissant un corona extérieur comme illustré en
figure III-32-a). Dans cette configuration, le facteur de structure global impliquerait celui du
polymère variant en q-2,25 à grands q, celui du corona sphérique variant en q-4, plus un terme
de diffusion lié aux interactions croisées entre le corona de DTAC et le polymère. D’où les

- 186 -
valeurs intermédiaires de α (comprises entre 2,25 et 4) observées pour NDTAC < 2.10-3 M
(figure III-31).
a) b)
Figure III-32. Structure schématique du complexe binaire poly(β-CD)/DTAC suivant la
concentration en TA: (a) NDTAC < 2.10-3 M, les charges sont réparties selon un corona
« sphérique »; (b) NDTAC 2.10-3 M, les charges sont distribuées de façon homogène le long
de la chaîne de polymère.
Dans le but de confirmer notre interprétation concernant une distribution de charges
inhomogène à faible NDTAC, les facteurs de structure du poly(β-CD), d’une part, et celui du
TA, d’autre part, ont été mesurés au sein du complexe binaire. Pour cela, une méthode de
marquage et de variation de contraste a été appliquée en utilisant un TA deutéré, le dDTAB.
Les coefficients α mesurés pour des mélanges poly(β-CD)/dDTAB dans du D2O pur
(diffusion du TA « matchée ») sont reportés sur la figure III-31. Aux erreurs expérimentales
près, le coefficient α du poly(β-CD) au sein du complexe binaire montre une valeur constante
et égale à 2,25 tant que NdDTAB reste inférieure à 2.10-3 M. Aucune compaction ni aucun
gonflement de la chaîne n’est donc observable dans ce domaine de concentration. Ce résultat
est la preuve indirecte d’une distribution inhomogène des molécules de TA le long de la
chaîne de polymère: l’augmentation des valeurs de α observée en présence de TA hydrogéné
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
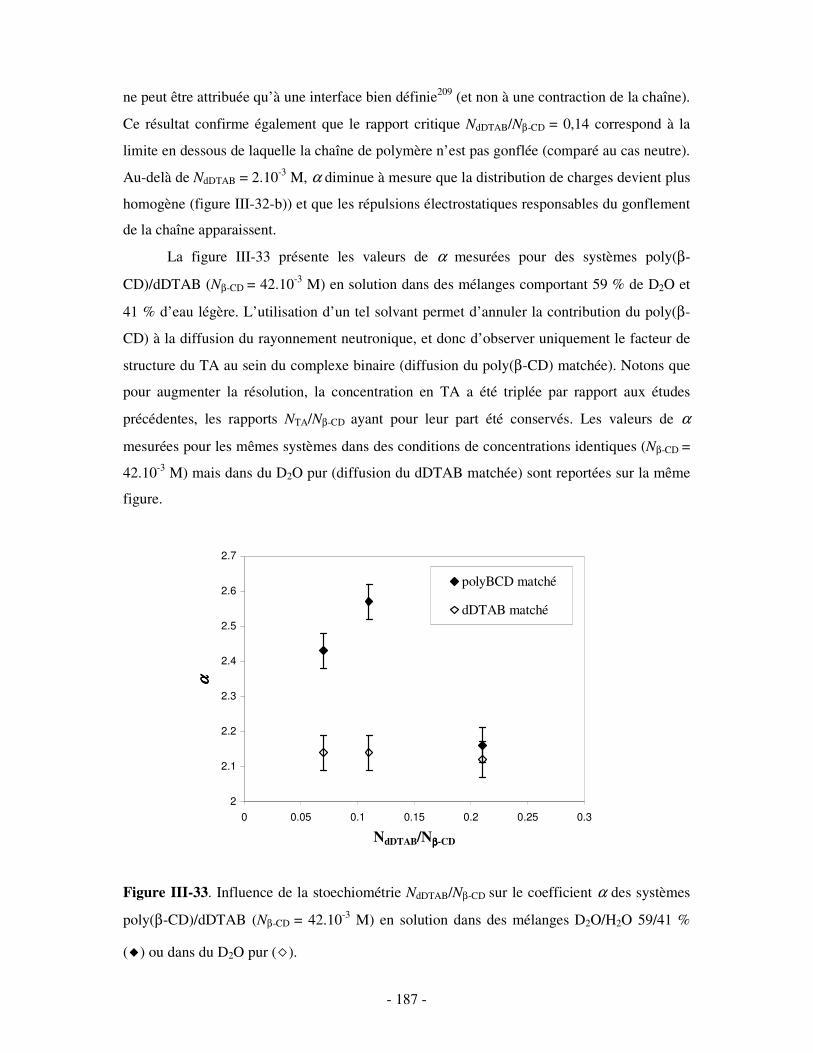
- 187 -
ne peut être attribuée qu’à une interface bien définie209 (et non à une contraction de la chaîne).
Ce résultat confirme également que le rapport critique NdDTAB/Nβ-CD = 0,14 correspond à la
limite en dessous de laquelle la chaîne de polymère n’est pas gonflée (comparé au cas neutre).
Au-delà de NdDTAB = 2.10-3 M, α diminue à mesure que la distribution de charges devient plus
homogène (figure III-32-b)) et que les répulsions électrostatiques responsables du gonflement
de la chaîne apparaissent.
La figure III-33 présente les valeurs de α mesurées pour des systèmes poly(β-
CD)/dDTAB (Nβ-CD = 42.10-3 M) en solution dans des mélanges comportant 59 % de D2O et
41 % d’eau légère. L’utilisation d’un tel solvant permet d’annuler la contribution du poly(β-
CD) à la diffusion du rayonnement neutronique, et donc d’observer uniquement le facteur de
structure du TA au sein du complexe binaire (diffusion du poly(β-CD) matchée). Notons que
pour augmenter la résolution, la concentration en TA a été triplée par rapport aux études
précédentes, les rapports NTA/Nβ-CD ayant pour leur part été conservés. Les valeurs de α
mesurées pour les mêmes systèmes dans des conditions de concentrations identiques (Nβ-CD =
42.10-3 M) mais dans du D2O pur (diffusion du dDTAB matchée) sont reportées sur la même
figure.
2
2.1
2.2
2.3
2.4
2.5
2.6
2.7
0 0.05 0.1 0.15 0.2 0.25 0.3
NdDTAB/Nββββ-CD
αα αα
polyBCD matché
dDTAB matché
Figure III-33. Influence de la stoechiométrie NdDTAB/Nβ-CD sur le coefficient α des systèmes
poly(β-CD)/dDTAB (Nβ-CD = 42.10-3 M) en solution dans des mélanges D2O/H2O 59/41 %
() ou dans du D2O pur ( ).

- 188 -
Pour des rapports NdDTAB/Nβ-CD inférieurs à 0,14, le coefficient α mesuré pour le TA au
sein du système binaire (poly(β-CD) matché) présente des valeurs beaucoup plus élevées que
pour NdDTAB/Nβ-CD > 0,14. Ce résultat est bien la preuve directe qu’à faible concentration en
TA, les charges sont distribuées de façon beaucoup moins homogène le long de la chaîne de
poly(β-CD). Cependant, les valeurs de α obtenues suggèrent une répartition des charges
nettement moins régulière qu’une distribution sphérique: α n’atteint qu’une valeur maximale
de 2,6 alors que pour une sphère parfaite, une valeur de l’ordre de 4 est attendue. Deux
explications sont possibles. La première concerne les faibles valeurs du rapport
stoechiométrique NdDTAB/Nβ-CD auxquelles une distribution de charges inhomogène est
observée. Il est en effet peu concevable qu’avec une si petite quantité de TA disponible (1
mole de TA pour 14 moles de β-CDs, par exemple), des coronas parfaitement sphériques
puissent être formés. La deuxième explication repose sur le fait que l’architecture branchée du
poly(β-CD) ne puisse être vraiment assimilée à une boule comme illustrée en figure III-32. En
effet, la structure globale de ce dernier n’est pas définissable de façon précise189.
IV- Conclusion
Le mélange du poly(β-CD) et du DTAC en solution aqueuse résulte en la formation de
complexes solubles, impliquant des interactions d’inclusion entre les cavités de β-CD et les
chaînons alkyle des molécules de TA. Comme dans le cas des monomères de β-CD54,65, les
complexes d’inclusion poly(β-CD)/DTAC sont formés exclusivement avec une
stoechiométrie 1:1 (une molécule de DTAC par cavité de β-CD). Le fait que les cavités de β-
CD soient interconnectées via des chaînes hydroxypropyléther n’a donc aucune conséquence
sur leur mode d’association avec le TA cationique. Par contre, une affinité plus faible a pu
être observée: Kpoly(β-CD)/DTAC ~ 1600 M-1 contre 16 000 M-1 pour la β-CD native dans l’eau
pure. Ceci a été attribué à l’existence de contraintes stériques et, surtout, à l’existence de
répulsions électrostatiques induites le long de la chaîne de polymère. Des mesures de
fluorescence et de tension de surface ont en effet montré une association binaire plus
favorable en présence de sel (écrantage des répulsions coulombiennes).
Selon la concentration en DTAC, ces complexes binaires présentent des propriétés
structurales différentes. Les résultats viscosimétriques et de diffusion de neutrons (SANS)

- 189 -
mettent en évidence l’existence d’un rapport critique NDTAC/Nβ-CD égal à 0,14 en dessous
duquel la chaîne de poly(β-CD) décorée de DTAC est dans la même conformation que sous sa
forme libre (c’est-à-dire neutre). Par contre, au-delà de ce rapport critique, la chaîne de
polymère adopte une conformation plus gonflée et se comporte comme un PEL. Des mesures
de SANS impliquant des variations de contraste ont permis d’attribuer ces comportements
différents à un changement dans la répartition des charges le long de la chaîne. Lorsque
NDTAC/Nβ-CD est inférieur à la valeur critique de 0,14, les molécules de DTAC sont distribuées
en périphérie de l’architecture branchée du poly(β-CD), formant un corona pseudo-sphérique
et laissant la structure du polymère inchangée. Par contre, au-delà de NDTAC/Nβ-CD = 0,14, la
distribution des charges devient plus homogène au sein de la structure branchée du poly(β-
CD), provoquant un gonflement de la chaîne (par interactions coulombiennes répulsives).

- 190 -
Chapitre IV. Etude du complexe ternaire
poly(CD)/TA cationique/polyanion en solution diluée
Dans le chapitre précédent, la formation de complexes d’inclusion hydrosolubles entre
un polymère de β-CD et un TA cationique de type dodécyltriméthylammonium a été mise en
évidence et étudiée en fonction de divers paramètres (force ionique et concentration en TA).
Désormais, il s’agit d’évaluer l’aptitude du système poly(CD)/TA cationique à former des
assemblages supramoléculaires de type PECs, solubles dans l’eau, lorsque mélangé à un
polyanion. Les complexes d’inclusion poly(CD)/TA cationique sont-ils dissociés au profit de
la formation de complexes binaires TA/polyanion (de charges opposées), ou bien subsistent-
ils, le TA jouant alors le rôle de connecteur entre les deux polymères? C’est à cette question
que nous tenterons de répondre dans un premier temps. Puis, dans l’hypothèse d’une réelle
association ternaire poly(CD)/TA cationique/polyanion, nous verrons quelle peut être
l’importance de la nature du polyanion impliqué et comment peuvent se comporter les
structures formées en solution selon la concentration en TA, la force ionique du milieu et la
concentration en compétiteurs vis-à-vis des interactions d’inclusion CD/TA. Nous vérifierons
que notre modèle de PEC à trois composants montre bien les avantages escomptés par rapport
aux systèmes classiques polycation/polyanion, à savoir une taille et une compacité des
agrégats mieux contrôlables et une réversibilité accrue (en général, les PECs classiques
constitués de PELs forts ne sont dissociables qu’à force ionique très élevée).
I- Conditions expérimentales de l’étude en solution
1- Les nouveaux composants utilisés
a) Les différents polyanions
Dans le but de discuter l’influence de la nature du polyanion sur la formation et les
propriétés structurales du complexe ternaire, plusieurs polyanions de natures chimiques,
masses molaires et rigidités différentes ont été utilisés: un polystyrène sulfonate (NaPSS),
linéaire et très flexible, des échantillons de dextrane sulfate (NaDxS), de structure branchée et

- 191 -
plus rigide que celle du NaPSS mais dont le squelette est plus hydrophile, et des fragments
d’ADN double brin très rigides.
i) Le polystyrène sulfonate (NaPSS)
Le polystyrène sulfonate de sodium utilisé nous a été fourni par Alfa Aesar (Karlsruhe,
Allemagne). Sa composition en soufre (déterminée par analyse élémentaire) est d’environ 10
%, ce qui correspond à un taux moyen de sulfonation de 80 %. Sa masse molaire moyenne est
de 70 000 g.mol-1.
Un échantillon de NaPSS deutéré (dNaPSS), de masse molaire moyenne égale à
37 000 g.mol-1, a aussi été employé pour certaines expériences de SANS. Il nous a
aimablement été offert par F. Cousin (LLB, Saclay). Sa densité de longueur de diffusion, ρ,
est de 6,48.1010 cm-2 sans les contre-ions Na+ (cf. annexe 3), soit proche de ρD2O = 6,41.1010
cm-2, tandis que celle du PSS hydrogéné est égale à 2,49.1010 cm-2.
ii) Le dextrane sulfate (NaDxS)
Deux échantillons de dextrane sulfate de sodium ont été utilisés dans cette étude. Le
premier, NaDxS 1, nous a été fourni par Sigma (France). Sa masse molaire moyenne est de
10 000 g.mol-1. Le second, NaDxS 2, de masse molaire plus élevée, nous a été fourni par ICN
Biomedicals (Aurora, Ohio) avec une masse molaire moyenne de 40 000 g.mol-1. Tous deux
présentent une teneur en soufre d’environ 17 %, ce qui correspond à une moyenne de deux
groupements sulfate par unité glucose. Ils sont constitués approximativement de 5 % d’unités
glucose branchées, avec des branchements comportant en moyenne 3 unités glucose. Pour le
NaDxS 1 comportant 27 motifs, cela correspond à un seul branchement. La structure du
polymère peut donc être considérée comme quasi linéaire. Pour le NaDxS 2 comportant 110
motifs, cela correspond à 6 branchements en moyenne. Mais selon les fournisseurs, les
échantillons de NaDxS de plus grande masse molaire présentent un taux de motifs branchés
légèrement plus élevé que 5 %. Le nombre de branchements moyen du NaDxS 2 est donc
certainement plus élevé que 6.
La densité de longueur de diffusion de NaDxS 1 et NaDxS 2 (sans les contre-ions) est
égale à 1,75.1010 cm-2.

- 192 -
iii) L’ADN
Des fragments d’ADN extraits de sperme de hareng, fournis par Sigma (France), ont
été utilisés avec des masses molaires moyennes de l’ordre de 10 000 - 30 000 g.mol-1. La
composition des brins nous étant inconnue (composition relative en adénine, guanine,
cytosine et thymine non précisée), la densité de longueur de diffusion, ρADN, n’a pu être
calculée. Pour information, une valeur théorique de 4,11.1010 cm-2 est rapportée dans la
littérature pour de l’ADN d’érythrocyte de poulet210.
b) Les différents compétiteurs
Deux types différents de compétiteurs ont été utilisés: un type capable de complexer le
TA cationique (monomère de β-CD), un autre capable de s’inclure dans les cavités de β-CD
du polymère (TA neutre).
i) L’hydroxypropyl(β-cyclodextrine) (HP(β-CD))
Les monomères d’HP(β-CD) nous ont été fournis par Aldrich (France) avec un degré
de substitution égal à 0,8 (soit 0,8 groupement hydroxypropyle par unité glucose), ce qui
correspond à une masse molaire moyenne de 1460 g.mol-1.
Leur constante d’association avec les molécules de DTAC dans l’eau pure a été
déterminée par fluorimétrie (cf. annexe 1-I-2): KHP(β-CD)/DTAC = 6470 ± 55 M-1 (contre 1540 ±
50 M-1 dans le cas du poly(β-CD)).
O
O*
RO ORO
*
OR
7
R = * CH2 CH OH
CH3
H
Figure IV-1. Formule chimique de l’HP(β-CD).

- 193 -
ii) Le dodécyl-β-D-maltoside (DM)
Fourni par Aldrich (France), le DM est un TA neutre dont la masse molaire est égale à
510,6 g.mol-1.
O O
O OCH2(CH2)10CH3
OHHO
HOH2CHOH2C
OHHO
HO
Figure IV-2. Formule chimique du DM.
D’après les mesures de tension de surface rapportées en annexe 4, la cmc du DM dans
l’eau pure est de l’ordre de 1,5.10-4 M, en accord avec les données de la littérature47. Sa
constante d’affinité pour les cavités du poly(β-CD), Kpoly(β-CD)/DM, est égale à 35 370 ± 3360
M-1, soit environ 20 fois plus élevée que Kpoly(β-CD)/DTAC déterminée dans les mêmes
conditions.
2- Une technique de caractérisation supplémentaire: la diffusion dynamique de
la lumière (Dynamic Light Scattering, DLS)
a) Principe
Une expérience de DLS consiste à envoyer un faisceau de lumière monochromatique
sur un échantillon et à enregistrer les fluctuations temporelles de l’intensité diffusée par ce
dernier à un angle θ donné. Ceci permet de corréler l’intensité I(t) diffusée à un temps t
arbitraire à l’intensité I(t + τ) diffusée au temps t + τ ou, en d’autres termes, de construire la
fonction d’autocorrélation de l’intensité diffusée )()( τ+tItI pour des valeurs discrètes de
τ, où désigne la moyenne au cours du temps. Cette opération est rendue possible d’un

- 194 -
point de vue expérimental grâce à la présence de 500 canaux de mesure indépendants sur le
détecteur.
Soit g(τ) la fonction d’autocorrélation normalisée du champ électrique diffusé, définie
par:
)()(
)()()(
*
*
tEtE
tEtEg
ττ
+= , (1)
où E(t) et E(t + τ) sont les champs électriques diffusés aux temps t et t + τ , respectivement, et * indique leurs complexes conjugués. La grandeur mesurée expérimentalement est la fonction
d’autocorrélation de l’intensité diffusée )()0()( ττ IIC = qui peut être reliée à g(τ) selon:
( )2)(1)( ττ gBAC += , (2)
où A est une amplitude dépendant de la norme du vecteur d’onde de diffusion
( )λ
θπ 2/sin4 0nq = (n0 étant l’indice de réfraction du milieu) et B, un facteur expérimental
compris entre 0 et 1.
Pour des solutions diluées et monodisperses de particules n’interagissant pas entre
elles, g(τ) peut se mettre sous la forme:
( )ττ ⋅Γ−= exp)(g . (3)
La vitesse de relaxation Γ qui intervient dans cette relation dépend du coefficient de diffusion
D des particules selon:
2qD ⋅=Γ , (4)
D étant lui-même relié à la taille des particules par la loi de Stokes-Einstein:
h
B
RTk
Dηπ6
= , (5)

- 195 -
avec kB, la constante de Boltzmann, T, la température, η, la viscosité du milieu et Rh, le rayon
hydrodynamique des particules (Rh est le rayon d’une sphère hypothétique soumise à la même
résistance hydrodynamique que la particule).
En combinant les équations (2), (3), (4) et (5), on obtient:
−+=
2
2
6exp1)( τ
ηπτ q
RTk
BACh
B . (6)
Par conséquent, un ajustement des valeurs expérimentales de C(τ) par l’équation théorique (6)
permet de déterminer la taille des objets diffusants. En pratique, celle-ci doit être comprise
entre 10 et 1000 Å environ pour pouvoir être mesurée par DLS.
Lorsque des agrégats de tailles différentes coexistent en solution, g(τ) n’est plus une
simple exponentielle. Elle dépend de tous les modes de relaxation individuels et peut être
exprimée sous la forme:
( ) Γ⋅Γ−Γ= ∞
dGg0
exp)()( ττ , (7)
où G(Γ), la distribution supposée continue des vitesses de relaxation, est définie par:
1)(0
=ΓΓ∞
dG . (8)
Dans ce cas, il est usuel de traiter les données par la méthode des cumulants211, qui consiste à
développer g(τ) en termes de moments de la distribution des tailles. La base de ce
développement repose sur l’expression:
( ) ( ) [ ]τττ )(expexpexp Γ−Γ−⋅Γ−=⋅Γ− , (9)
où Γ est la valeur moyenne de Γ, définie par:
ΓΓΓ=Γ ∞
dG0
)( . (10)
En développant la seconde exponentielle à droite de l’égalité dans l’équation (9), on obtient:

- 196 -
( ) ( ) ( ) ( ) ( )
+Γ−Γ−Γ−Γ+Γ−Γ−⋅Γ−=⋅Γ− Λ3
3
2
2
!3!21expexp τττττ (11)
puis, d’après l’équation (7),
+−+−⋅Γ−= Λ3322
1 !3!21)(exp)( τµτµτµττg (12)
où ΓΓΓ−Γ= ∞
dGnn
0
)()(µ (13)
sont les moments de la distribution. Etant donnée la définition de Γ (cf. équation 10), µ1 = 0
et l’équation (12) peut être réécrite sous la forme:
+−+⋅Γ−= Λ3322
!3!21)(exp)( τµτµττg (14)
En prenant le logarithme népérien de l’équation (14) et en utilisant le développement
( ) xx ≈+1ln valable à x faible (c’est-à-dire lorsque τ tend vers 0), on obtient finalement:
Λ+−+Γ−= 3322
!3!2)(ln τµτµττg . (15)
L’ajustement des données de DLS par cette dernière équation permet de déterminer Γ qui,
via la relation de Stokes-Einstein, conduit à une valeur moyenne du rayon hydrodynamique
Rh.
La distribution des vitesses de relaxation G(Γ) est calculée à partir de la transformée
de Laplace inverse (ILT) de la fonction d’autocorrélation et d’un algorithme de calcul (NNLS,
Non Negatively Constrained Least-Squares) qui minimise la somme des différences au carré
entre les fonctions C(τ) calculées et expérimentales.

- 197 -
b) Appareillage
Les mesures de DLS ont été réalisées au laboratoire MPI (Université d’Evry), à l’aide
d’un appareillage fourni par Brookhaven Instruments Corp. comprenant un corrélateur BI-
9000 AT, un goniomètre BI-200 SM et un laser produisant un faisceau de lumière
monochromatique de longueur d’onde λ = 514,5 nm. Le dispositif expérimental est
schématisé en figure IV-3.
Figure IV-3. Schéma du montage des expériences de DLS.
Dans ce montage, l’échantillon à analyser (V ~ 2 mL) est contenu dans un tube
cylindrique en verre immergé dans une cuve thermostatée à 25°C, placée sur l’axe du
goniomètre. La cuve est remplie de décaline, un liquide qui permet de minimiser l’écart
d’indice entre la cellule de mesure (verre) et le milieu environnant, et donc de minimiser les
réflexions parasites du faisceau incident (réflexions importantes dans le cas d’une interface
air/verre). L’intensité diffusée est mesurée à l’angle θ = 90°, pour des temps τ compris entre
100 ns et 10 ms.
Les échantillons ont été introduits dans les cellules de mesure à l’aide de seringues
munies de filtres comportant des pores de 0,45 µm de diamètre.
détecteur
cuve
θ = 90°
laser

- 198 -
II- Mise en évidence de la formation de complexes ternaires
poly(CD)/TA cationique/polyanion solubles dans l’eau
1- Détermination des domaines de solubilité
De nombreux échantillons aqueux comportant entre 1 et 18.10-3 M de cavités du
poly(β-CD), entre 2 et 12.10-3 M de DTAC et entre 1 et 45.10-3 M de groupements chargés
négativement d’un polyanion ont été soigneusement mélangés et mis à équilibrer durant 48
heures dans un bain thermostaté à 25 ± 0,5°C.
La figure IV-4 rend compte des observations effectuées à l’œil nu au bout des 48
heures pour les mélanges comportant le NaDxS 1 comme polyanion. Notons qu’aucune
évolution des échantillons n’a pu être constatée durant les 7 jours qui ont suivi ces
observations.
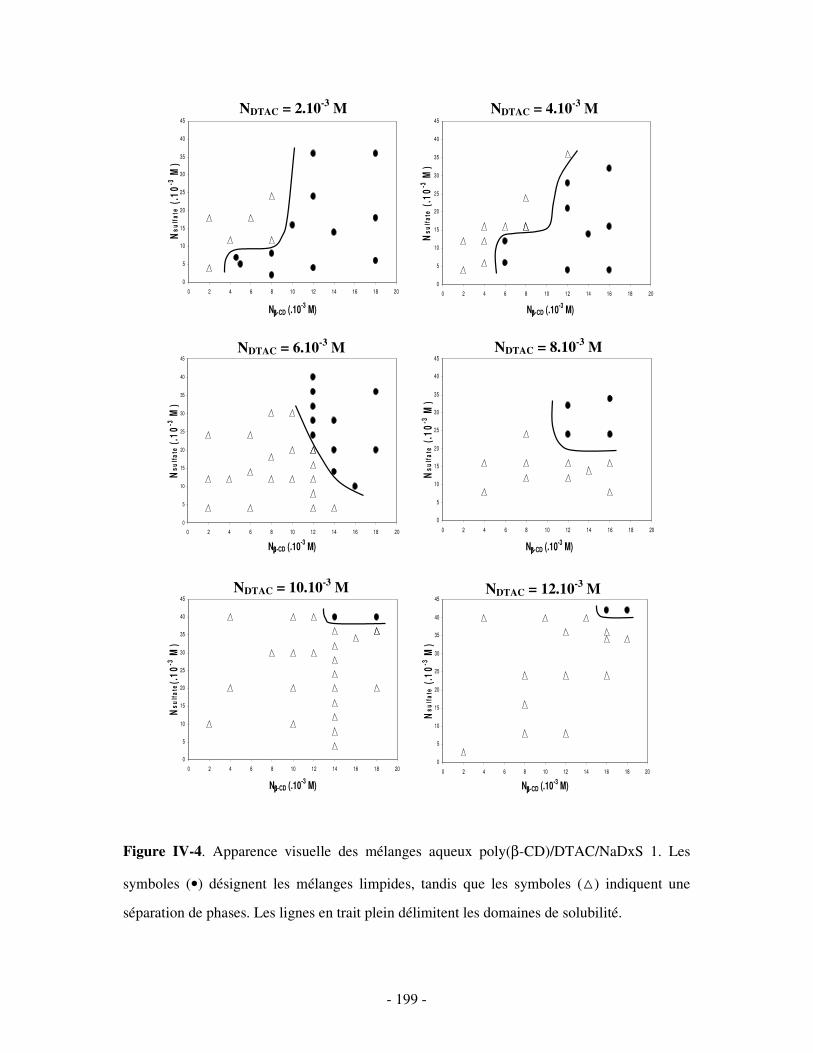
- 199 -
Figure IV-4. Apparence visuelle des mélanges aqueux poly(β-CD)/DTAC/NaDxS 1. Les
symboles (•) désignent les mélanges limpides, tandis que les symboles () indiquent une
séparation de phases. Les lignes en trait plein délimitent les domaines de solubilité.
0
5
10
15
20
25
30
35
40
45
0 2 4 6 8 10 12 14 16 18 20
Nββββ-CD (.10-3 M)
Nsu
lfat
e(.1
0-3 M
)
0
5
10
15
20
25
30
35
40
45
0 2 4 6 8 10 12 14 16 18 20
Nββββ -CD (.10-3 M)
Nsu
lfat
e (.
10-3
M)
0
5
10
15
20
25
30
35
40
45
0 2 4 6 8 10 12 14 16 18 20
Nββββ-CD (.10-3 M)
Nsu
lfat
e (.1
0-3 M
)
0
5
10
15
20
25
30
35
40
45
0 2 4 6 8 10 12 14 16 18 20
Nββββ-CD (.10-3 M)
Nsu
lfat
e (.
10-3
M)
0
5
10
15
20
25
30
35
40
45
0 2 4 6 8 10 12 14 16 18 20
Nββββ-CD (.10-3 M)
Nsu
lfat
e (.
10-3
M)
0
5
10
15
20
25
30
35
40
45
0 2 4 6 8 10 12 14 16 18 20
Nββββ-CD (.10-3 M)
Nsu
lfat
e (
.10-3
M)
NDTAC = 2.10-3 M NDTAC = 4.10-3 M
NDTAC = 6.10-3 M
NDTAC = 8.10-3 M
NDTAC = 10.10-3 M
NDTAC = 12.10-3 M
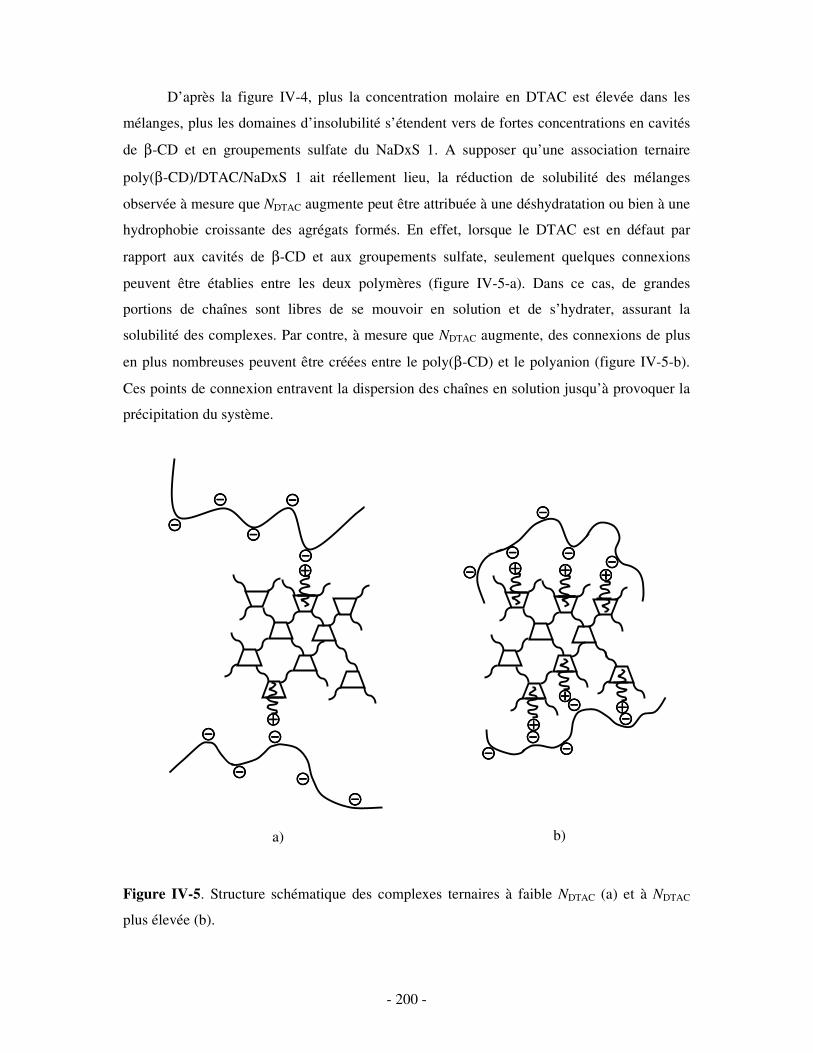
- 200 -
D’après la figure IV-4, plus la concentration molaire en DTAC est élevée dans les
mélanges, plus les domaines d’insolubilité s’étendent vers de fortes concentrations en cavités
de β-CD et en groupements sulfate du NaDxS 1. A supposer qu’une association ternaire
poly(β-CD)/DTAC/NaDxS 1 ait réellement lieu, la réduction de solubilité des mélanges
observée à mesure que NDTAC augmente peut être attribuée à une déshydratation ou bien à une
hydrophobie croissante des agrégats formés. En effet, lorsque le DTAC est en défaut par
rapport aux cavités de β-CD et aux groupements sulfate, seulement quelques connexions
peuvent être établies entre les deux polymères (figure IV-5-a). Dans ce cas, de grandes
portions de chaînes sont libres de se mouvoir en solution et de s’hydrater, assurant la
solubilité des complexes. Par contre, à mesure que NDTAC augmente, des connexions de plus
en plus nombreuses peuvent être créées entre le poly(β-CD) et le polyanion (figure IV-5-b).
Ces points de connexion entravent la dispersion des chaînes en solution jusqu’à provoquer la
précipitation du système.
Figure IV-5. Structure schématique des complexes ternaires à faible NDTAC (a) et à NDTAC
plus élevée (b).
⊕⊕⊕⊕
⊕⊕⊕⊕
b)
a)
⊕⊕⊕⊕ ⊕⊕⊕⊕
⊕⊕⊕⊕ ⊕⊕⊕⊕ ⊕⊕⊕⊕
⊕⊕⊕⊕

- 201 -
Un comportement similaire est observable pour les mélanges ternaires comportant le
NaPSS (cf. figure IV-6): plus la concentration en DTAC est élevée, plus les domaines de
solubilité sont réduits. Ces derniers apparaissent cependant beaucoup plus vastes que dans le
cas du NaDxS 1. Pour une même concentration en DTAC, le domaine de solubilité des
systèmes constitués de NaPSS s’étend vers des concentrations en β-CD et en groupements
anioniques plus faibles que dans le cas précédent. Il est constaté, de plus, que les mélanges
ternaires comportant le NaPSS sont solubles, d’une part, tant que les groupements sulfonate
sont en excès comparés au DTAC, quelque soit la concentration en β-CD (une observation
similaire a été rapportée dans la littérature pour des mélanges binaires composés de NaPSS et
de DTAB116,212), et d’autre part, tant que les cavités de β-CD sont en excès comparées au
DTAC, quelque soit la concentration en groupements sulfonate. Ceci n’est pas toujours vrai
dans le cas du NaDxS 1, bien que celui-ci possède un squelette plus hydrophile que celui du
NaPSS. Par conséquent, la nature hydrophile/hydrophobe du polyanion ne semble pas jouer
un rôle déterminant dans la solubilité des complexes ternaires.
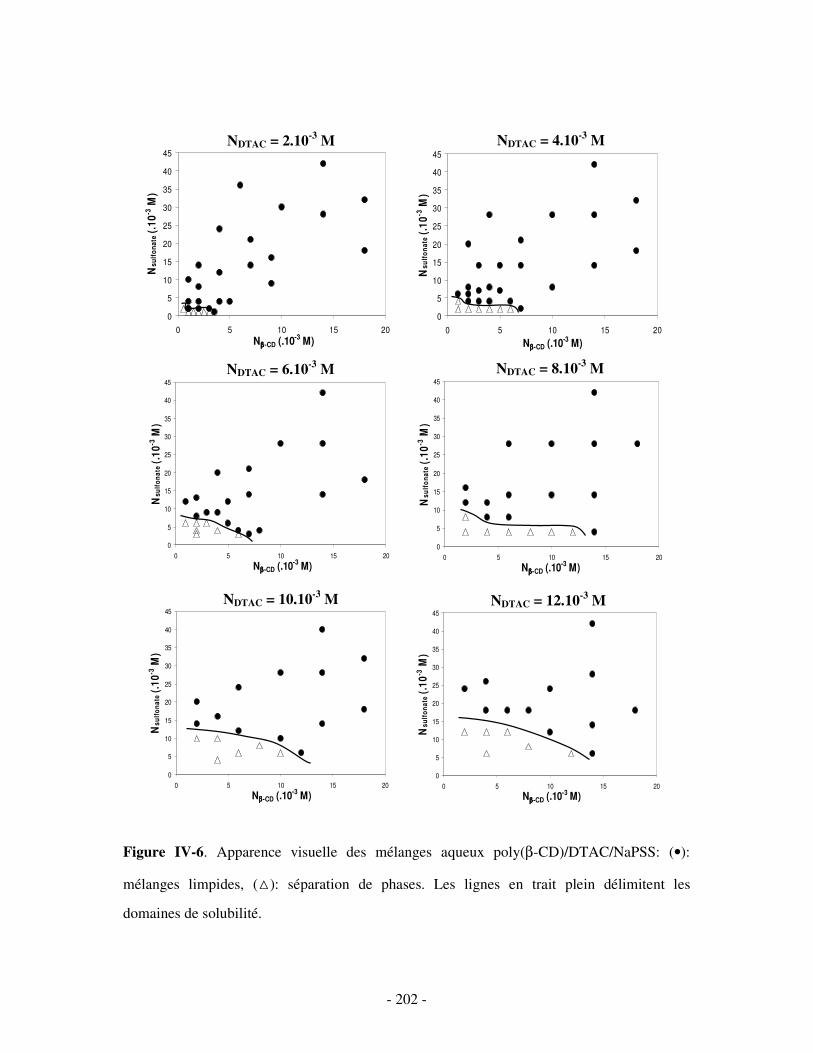
- 202 -
Figure IV-6. Apparence visuelle des mélanges aqueux poly(β-CD)/DTAC/NaPSS: (•):
mélanges limpides, (): séparation de phases. Les lignes en trait plein délimitent les
domaines de solubilité.
0
5
10
15
20
25
30
35
40
45
0 5 10 15 20
Nββββ -CD (.10-3 M)
Nsu
lfon
ate (
.10-3
M)
0
5
10
15
20
25
30
35
40
45
0 5 10 15 20
Nββββ -CD (.10-3 M)
Nsu
lfon
ate (
.10-3
M)
0
5
10
15
20
25
30
35
40
45
0 5 10 15 20Nββββ -CD (.10-3 M)
Nsu
lfon
ate (
.10-3
M)
0
5
10
15
20
25
30
35
40
45
0 5 10 15 20Nββββ-CD (.10-3 M)
Nsu
lfon
ate (
.10-3
M)
0
5
10
15
20
25
30
35
40
45
0 5 10 15 20
Nββββ -CD (.10-3 M)
Nsu
lfon
ate (
.10-3
M)
0
5
10
15
20
25
30
35
40
45
0 5 10 15 20
Nββββ-CD (.10-3 M)
Nsu
lfon
ate (
.10-3
M)
NDTAC = 2.10-3 M NDTAC = 4.10-3 M
NDTAC = 6.10-3 M NDTAC = 8.10-3 M
NDTAC = 10.10-3 M NDTAC = 12.10-3 M

- 203 -
Notons que le NaPSS et le NaDxS 1 présentent des longueurs de chaîne très
différentes: le degré de polymérisation, DP, du NaPSS (Mw = 70 000 g.mol-1) est de 340,
tandis que celui du NaDxS 1 (Mw = 10 000 g.mol-1) est de 27 seulement. Un effet de la masse
molaire du composant polyanionique sur la solubilité des complexes ternaires peut donc être
envisagé. Cependant, dans le cas du NaDxS, l’augmentation de la masse molaire du polyanion
se traduit par une diminution de la solubilité des complexes: les mélanges ternaires constitués
du NaDxS 2 (Mw = 40 000 g.mol-1, DP = 110) montrent des domaines de solubilité plus
réduits que pour le NaDxS 1 (cf. figure IV-7). Il a été mentionné auparavant que le degré de
branchement du NaDxS augmente avec sa masse molaire. La rigidité de la chaîne de NaDxS
est donc d’autant plus importante que sa masse molaire est élevée. Par conséquent, il peut être
supposé que la solubilité des complexes ternaires de NaDxS est contrôlée en partie par la
rigidité du polyanion. Pour séparer les contributions éventuelles de la masse molaire et de la
rigidité de la chaîne polyanionique, il faudrait étudier l’influence de la masse molaire du
NaPSS sur les diagrammes de solubilité des complexes ternaires correspondants. Cependant,
les résultats obtenus avec les différents polyanions montrent une interdépendance entre la
flexibilité des polyanions et la solubilité des complexes: plus le polyanion est rigide (le
NaPSS est plus flexible que le NaDxS 1, qui lui présente une structure moins branchée que le
NaDxS 2), moins les systèmes ternaires sont solubles. Ces différences de solubilité sont
illustrées par le tableau IV-1 qui présente les plus faibles valeurs de NDTAC pour lesquelles une
séparation de phases est observée dans les mélanges, à concentrations en poly(β-CD) et
polyanion fixées.
polyanion NaPSS NaDxS 1 NaDxS 2 ADN
NDTACsép (.10-3 M) 14 7 6 5
Tableau IV-1. Concentration en DTAC minimale correspondant à une séparation de phases
(NDTACsép) dans les mélanges aqueux poly(β-CD)/DTAC/polyanion (Nβ-CD = Nanion = 14.10-3
M).
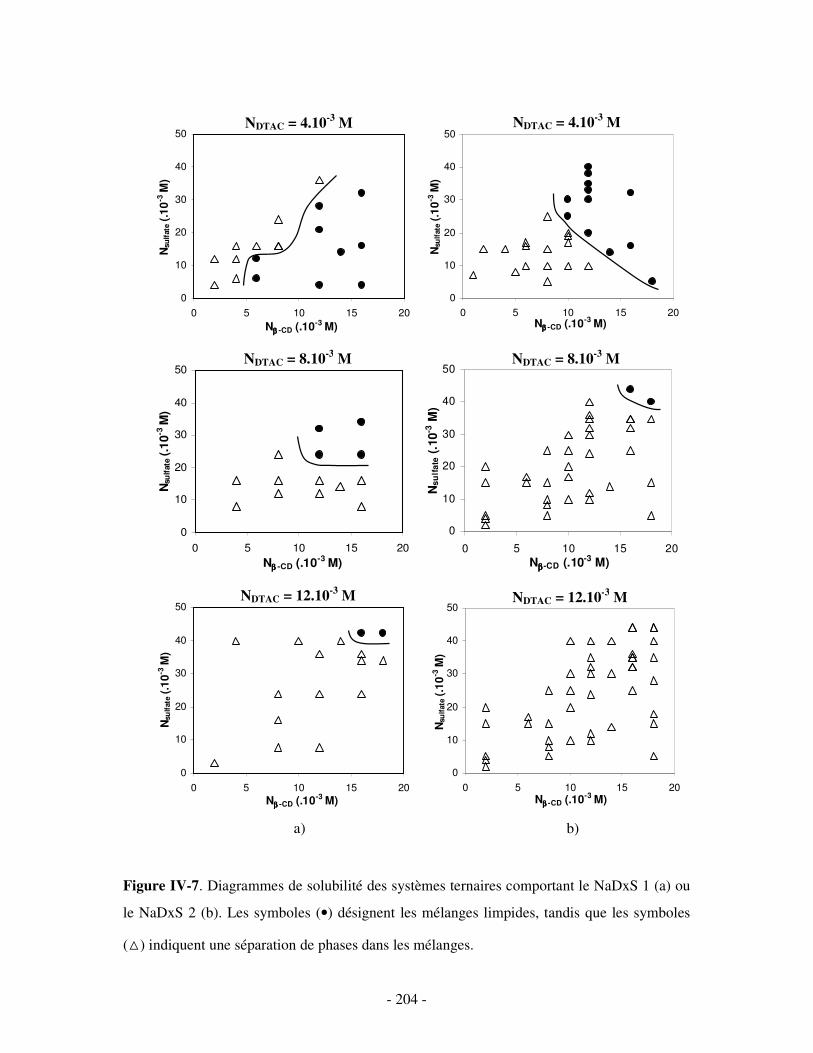
- 204 -
0
10
20
30
40
50
0 5 10 15 20Nββββ -CD (.10-3 M)
Nsu
lfat
e (.1
0-3 M
)
0
10
20
30
40
50
0 5 10 15 20Nββββ -CD (.10-3 M)
Nsu
lfat
e (.1
0-3 M
)
0
10
20
30
40
50
0 5 10 15 20Nββββ -CD (.10-3 M)
Nsu
lfat
e (.1
0-3 M
)
0
10
20
30
40
50
0 5 10 15 20Nββββ -CD (.10-3 M)
Nsu
lfate
(.10
-3 M
)
0
10
20
30
40
50
0 5 10 15 20Nββββ -CD (.10-3 M)
Nsu
lfate
(.10
-3 M
)
0
10
20
30
40
50
0 5 10 15 20Nββββ -CD (.10-3 M)
Nsu
lfate
(.10
-3 M
)
Figure IV-7. Diagrammes de solubilité des systèmes ternaires comportant le NaDxS 1 (a) ou
le NaDxS 2 (b). Les symboles (•) désignent les mélanges limpides, tandis que les symboles
() indiquent une séparation de phases dans les mélanges.
NDTAC = 4.10-3 M NDTAC = 4.10-3 M
NDTAC = 8.10-3 M NDTAC = 8.10-3 M
NDTAC = 12.10-3 M NDTAC = 12.10-3 M
a) b)
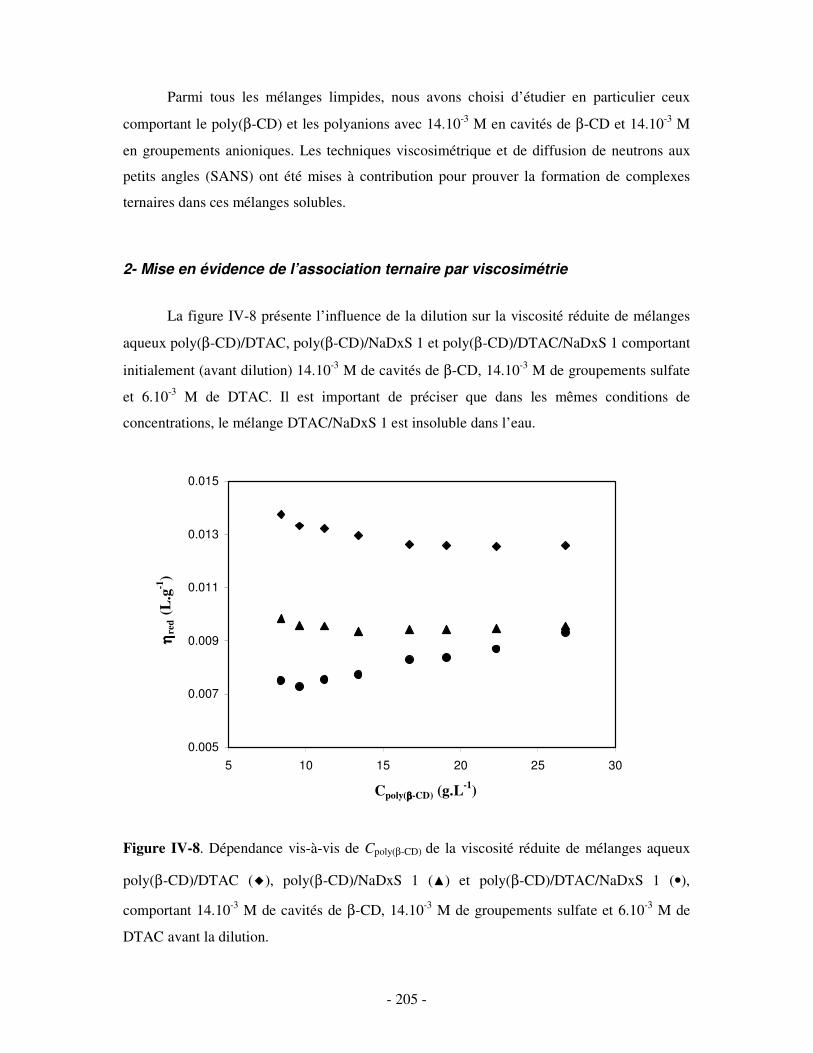
- 205 -
Parmi tous les mélanges limpides, nous avons choisi d’étudier en particulier ceux
comportant le poly(β-CD) et les polyanions avec 14.10-3 M en cavités de β-CD et 14.10-3 M
en groupements anioniques. Les techniques viscosimétrique et de diffusion de neutrons aux
petits angles (SANS) ont été mises à contribution pour prouver la formation de complexes
ternaires dans ces mélanges solubles.
2- Mise en évidence de l’association ternaire par viscosimétrie
La figure IV-8 présente l’influence de la dilution sur la viscosité réduite de mélanges
aqueux poly(β-CD)/DTAC, poly(β-CD)/NaDxS 1 et poly(β-CD)/DTAC/NaDxS 1 comportant
initialement (avant dilution) 14.10-3 M de cavités de β-CD, 14.10-3 M de groupements sulfate
et 6.10-3 M de DTAC. Il est important de préciser que dans les mêmes conditions de
concentrations, le mélange DTAC/NaDxS 1 est insoluble dans l’eau.
0.005
0.007
0.009
0.011
0.013
0.015
5 10 15 20 25 30
Cpoly(ββββ-CD) (g.L-1)
ηη ηη red
(L
.g-1
)
Figure IV-8. Dépendance vis-à-vis de Cpoly(β-CD) de la viscosité réduite de mélanges aqueux
poly(β-CD)/DTAC (), poly(β-CD)/NaDxS 1 () et poly(β-CD)/DTAC/NaDxS 1 (•),
comportant 14.10-3 M de cavités de β-CD, 14.10-3 M de groupements sulfate et 6.10-3 M de
DTAC avant la dilution.

- 206 -
Comme il peut être constaté, la viscosité réduite des mélanges binaires poly(β-
CD)/DTAC et poly(β-CD)/NaDxS 1 augmente avec la dilution, ce qui est typique de
comportements PELs: plus les solutions sont diluées, plus les répulsions coulombiennes entre
les charges portées par les chaînes polyanioniques (NaDxS 1) ou polycationiques (poly(β-
CD)/DTAC) sont intenses, et plus celles-ci adoptent des conformations étirées. Il a en effet
été démontré (cf. chapitre III) que l’ajout de DTAC au poly(β-CD) neutre dans une
stoechiométrie 6/14 résulte en la formation d’un polycation, comme une conséquence des
interactions d’inclusion entre les chaînes dodécyle des TAs et les cavités de β-CD du
polymère. Notons que dans le cas du mélange poly(β-CD)/NaDxS 1, les valeurs de viscosité
réduite n’augmentent que faiblement avec la dilution, à cause de la très faible quantité de
polyanion présente en solution: la concentration initiale de 14.10-3 M en groupements sulfate
correspond à 2,6 g.L-1 de NaDxS 1 seulement.
Dans le cas du mélange ternaire poly(β-CD)/DTAC/NaDxS 1, par contre, les valeurs
de viscosité observées diminuent avec la dilution. Ceci indique un écrantage des répulsions
électrostatiques intrachaînes à la fois pour le polycation poly(β-CD)/DTAC et pour le
polyanion NaDxS 1. Ce résultat suggère, par conséquent, la formation de complexes par
interactions entre charges opposées entre le DTAC et le NaDxS 1. Puisqu’en absence du
troisième composant (poly(β-CD)), les complexes DTAC/NaDxS 1 se montrent insolubles
dans l’eau et que, dans le cas du mélange ternaire, la solution est très limpide, cela signifie
que les molécules de DTAC sont en plus associées au poly(β-CD): les complexes
DTAC/NaDxS 1 sont solubilisés par inclusion des chaînes dodécyle des TAs dans les cavités
de β-CD.
Ainsi donc, l’étude viscosimétrique des systèmes composés de poly(β-CD), de DTAC
et de NaDxS 1 laisse supposer la formation de complexes ternaires solubles dans l’eau, d’une
part, par interactions électrostatiques entre le DTAC et le polyanion et, d’autre part, par
interactions d’inclusion entre le DTAC et le poly(β-CD). Les études viscosimétriques de
mélanges comportant du NaDxS 2 (figure IV-9) ou de l’ADN (figure IV-10) aboutissent aux
mêmes conclusions: en présence des trois composants (poly(β-CD), DTAC et polyanion), les
comportements PELs disparaissent (par interactions DTAC/polyanion de charges opposées),
et les complexes DTAC/NaDxS 2 ou DTAC/ADN donnant lieu à un précipité en absence du
poly(β-CD) sont solubilisés (par interactions d’inclusion DTAC/poly(β-CD)).
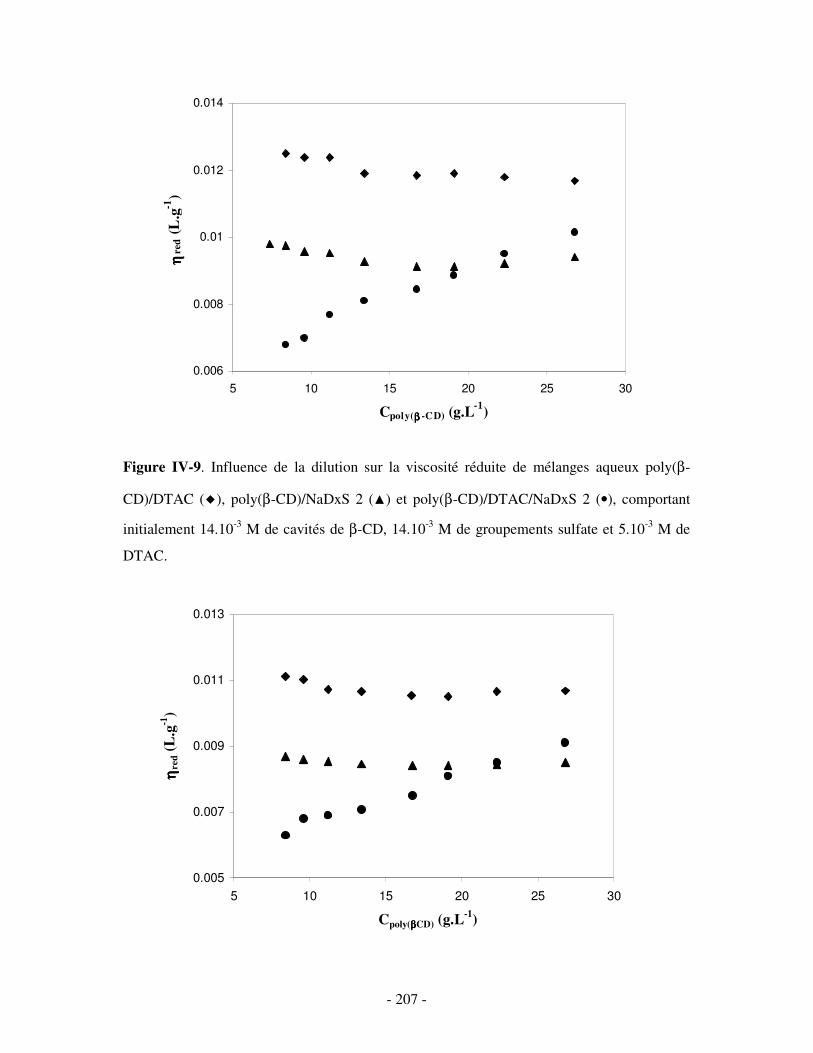
- 207 -
0.006
0.008
0.01
0.012
0.014
5 10 15 20 25 30
Cpoly(ββββ -CD) (g.L-1)
ηη ηηre
d (L
.g-1
)
Figure IV-9. Influence de la dilution sur la viscosité réduite de mélanges aqueux poly(β-
CD)/DTAC (), poly(β-CD)/NaDxS 2 () et poly(β-CD)/DTAC/NaDxS 2 (•), comportant
initialement 14.10-3 M de cavités de β-CD, 14.10-3 M de groupements sulfate et 5.10-3 M de
DTAC.
0.005
0.007
0.009
0.011
0.013
5 10 15 20 25 30
Cpoly(ββββCD) (g.L-1)
ηη ηη red
(L.g
-1)

- 208 -
Figure IV-10. Influence de la dilution sur la viscosité réduite de mélanges aqueux poly(β-
CD)/DTAC (), poly(β-CD)/ADN () et poly(β-CD)/DTAC/ADN (•), comportant
initialement 14.10-3 M de cavités de β-CD, 14.10-3 M de groupements phosphate (CADN = 6,1
g.L-1) et 4.10-3 M de DTAC.
Les comportements viscosimétriques de systèmes comportant du NaPSS ont
également été étudiés: la figure IV-11 présente les valeurs de ηred mesurées pour des mélanges
aqueux à 2 ou 3 composants comprenant le poly(β-CD), le DTAC et le NaPSS, avec Nβ-CDi =
Nsulfonatei = 14.10-3 M et NDTAC
i = 6.10-3 M avant la dilution. Il est à noter que, contrairement
aux cas précédents, le mélange DTAC/polyanion est parfaitement soluble, et ce malgré la
nature hydrophobe du squelette du NaPSS. Ce comportement différent peut être attribué au
manque de coopérativité qui caractérise l’association TA cationique/NaPSS151. Il a été vu au
paragraphe III-1 du chapitre II que dans le cas général des systèmes TA ionique/PEL de
charges opposées, un effet coopératif résulte d’une action mutuelle des attractions
électrostatiques TA ionique/PEL, et des attractions hydrophobes entre les chaînes alkyles des
TAs liés au polymère. Pour le système DTAB/NaDxS, cet effet coopératif est très prononcé146
et se traduit par la formation de grandes micelles de DTAB le long de la chaîne de NaDxS,
avec un nombre d’agrégation, <n>, de l’ordre de 107 (les nombres d’agrégation semblent être
peu dépendants de la nature du contre-ion: <n> ~ 65 pour le DTAB pur118 et <n> ~ 55 pour le
DTAC pur144). Par contre, dans le cas du système DTAB/NaPSS, l’existence d’interactions
hydrophobes entre le DTAB lié et le squelette du NaPSS conduit à des nombres d’agrégation
plus faibles, compris entre 7 et 38 d’après la littérature118,121. C’est donc la formation
d’agrégats hydrophobes plus petits qui pourrait expliquer la meilleure solubilité dans l’eau du
complexe DTAC/NaPSS (comparé aux complexes DTAC/NaDxS).
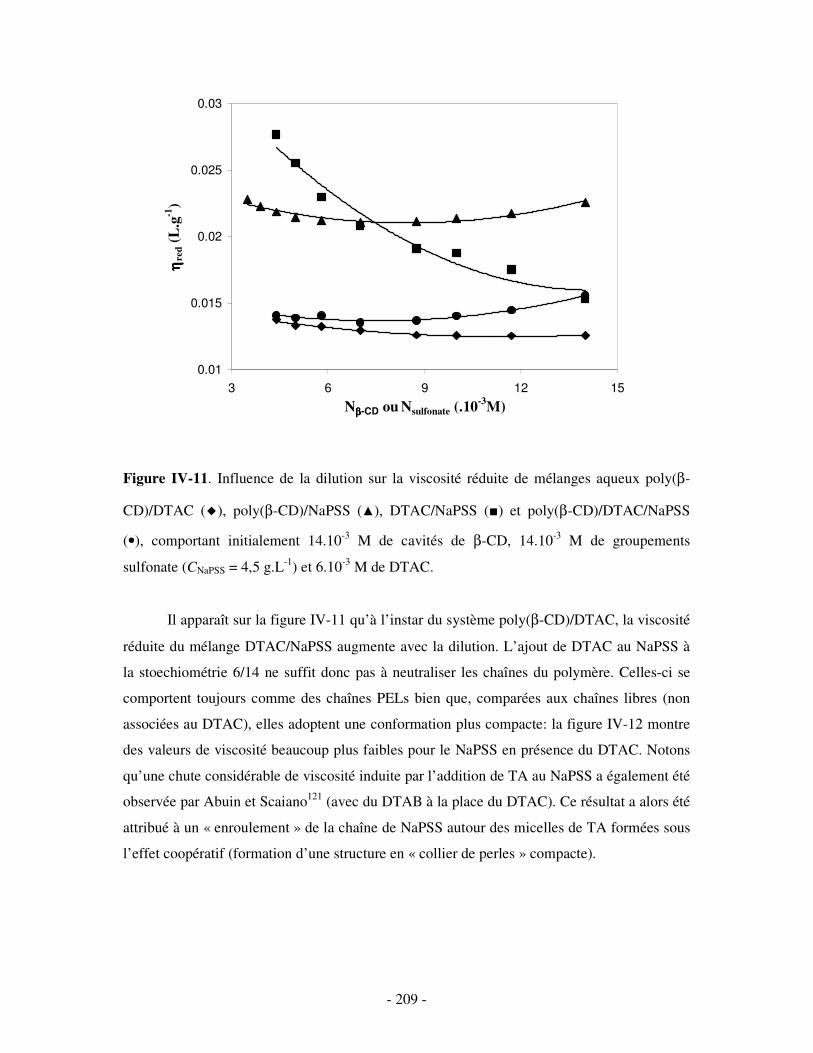
- 209 -
0.01
0.015
0.02
0.025
0.03
3 6 9 12 15Nββββ-CD ou Nsulfonate (.10-3M)
ηη ηη red
(L.g
-1)
Figure IV-11. Influence de la dilution sur la viscosité réduite de mélanges aqueux poly(β-
CD)/DTAC (), poly(β-CD)/NaPSS (), DTAC/NaPSS () et poly(β-CD)/DTAC/NaPSS
(•), comportant initialement 14.10-3 M de cavités de β-CD, 14.10-3 M de groupements
sulfonate (CNaPSS = 4,5 g.L-1) et 6.10-3 M de DTAC.
Il apparaît sur la figure IV-11 qu’à l’instar du système poly(β-CD)/DTAC, la viscosité
réduite du mélange DTAC/NaPSS augmente avec la dilution. L’ajout de DTAC au NaPSS à
la stoechiométrie 6/14 ne suffit donc pas à neutraliser les chaînes du polymère. Celles-ci se
comportent toujours comme des chaînes PELs bien que, comparées aux chaînes libres (non
associées au DTAC), elles adoptent une conformation plus compacte: la figure IV-12 montre
des valeurs de viscosité beaucoup plus faibles pour le NaPSS en présence du DTAC. Notons
qu’une chute considérable de viscosité induite par l’addition de TA au NaPSS a également été
observée par Abuin et Scaiano121 (avec du DTAB à la place du DTAC). Ce résultat a alors été
attribué à un « enroulement » de la chaîne de NaPSS autour des micelles de TA formées sous
l’effet coopératif (formation d’une structure en « collier de perles » compacte).
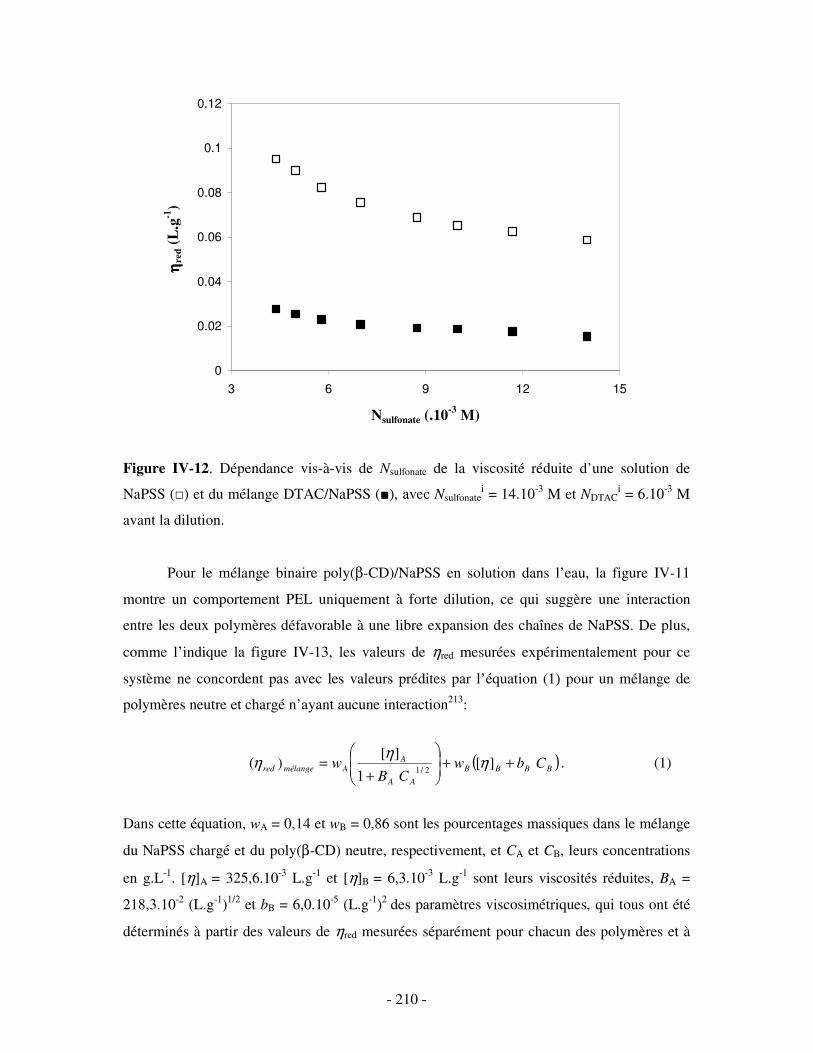
- 210 -
0
0.02
0.04
0.06
0.08
0.1
0.12
3 6 9 12 15
Nsulfonate (.10-3 M)
ηη ηη red
(L.g
-1)
Figure IV-12. Dépendance vis-à-vis de Nsulfonate de la viscosité réduite d’une solution de
NaPSS () et du mélange DTAC/NaPSS (), avec Nsulfonatei = 14.10-3 M et NDTAC
i = 6.10-3 M
avant la dilution.
Pour le mélange binaire poly(β-CD)/NaPSS en solution dans l’eau, la figure IV-11
montre un comportement PEL uniquement à forte dilution, ce qui suggère une interaction
entre les deux polymères défavorable à une libre expansion des chaînes de NaPSS. De plus,
comme l’indique la figure IV-13, les valeurs de ηred mesurées expérimentalement pour ce
système ne concordent pas avec les valeurs prédites par l’équation (1) pour un mélange de
polymères neutre et chargé n’ayant aucune interaction213:
( )BBBB
AA
AAmélangered Cbw
CBw ++
+= ][
1
][)( 2/1 ηηη . (1)
Dans cette équation, wA = 0,14 et wB = 0,86 sont les pourcentages massiques dans le mélange
du NaPSS chargé et du poly(β-CD) neutre, respectivement, et CA et CB, leurs concentrations
en g.L-1. [η]A = 325,6.10-3 L.g-1 et [η]B = 6,3.10-3 L.g-1 sont leurs viscosités réduites, BA =
218,3.10-2 (L.g-1)1/2 et bB = 6,0.10-5 (L.g-1)2 des paramètres viscosimétriques, qui tous ont été
déterminés à partir des valeurs de ηred mesurées séparément pour chacun des polymères et à
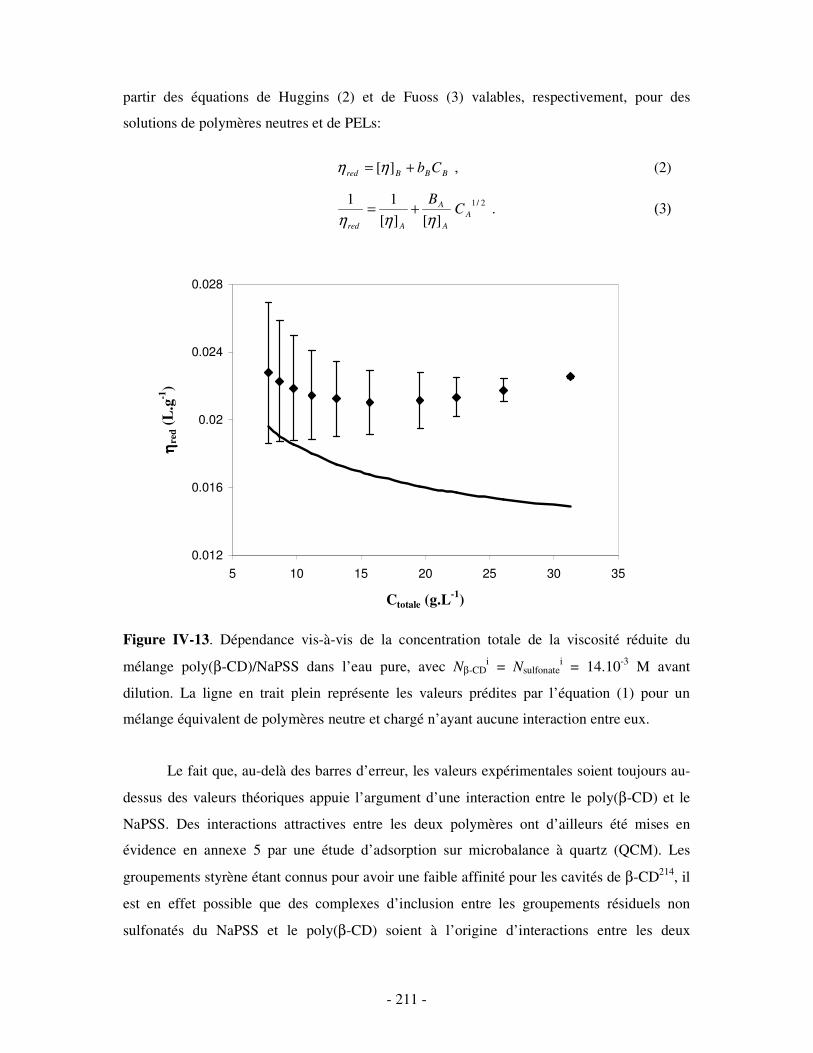
- 211 -
partir des équations de Huggins (2) et de Fuoss (3) valables, respectivement, pour des
solutions de polymères neutres et de PELs:
BBBred Cb+= ][ηη , (2)
2/1
][][11
AA
A
Ared
CBηηη
+= . (3)
0.012
0.016
0.02
0.024
0.028
5 10 15 20 25 30 35
Ctotale (g.L-1)
ηη ηη red
(L.g
-1)
Figure IV-13. Dépendance vis-à-vis de la concentration totale de la viscosité réduite du
mélange poly(β-CD)/NaPSS dans l’eau pure, avec Nβ-CDi = Nsulfonate
i = 14.10-3 M avant
dilution. La ligne en trait plein représente les valeurs prédites par l’équation (1) pour un
mélange équivalent de polymères neutre et chargé n’ayant aucune interaction entre eux.
Le fait que, au-delà des barres d’erreur, les valeurs expérimentales soient toujours au-
dessus des valeurs théoriques appuie l’argument d’une interaction entre le poly(β-CD) et le
NaPSS. Des interactions attractives entre les deux polymères ont d’ailleurs été mises en
évidence en annexe 5 par une étude d’adsorption sur microbalance à quartz (QCM). Les
groupements styrène étant connus pour avoir une faible affinité pour les cavités de β-CD214, il
est en effet possible que des complexes d’inclusion entre les groupements résiduels non
sulfonatés du NaPSS et le poly(β-CD) soient à l’origine d’interactions entre les deux

- 212 -
polymères. Pour information, un bon accord entre valeurs de viscosité expérimentales et
valeurs théoriques a été obtenu en annexe 6 pour les systèmes poly(β-CD)/NaDxS 2 et
poly(β-CD)/ADN. Donc, à priori, ni le NaDxS, ni l’ADN n’interagissent avec le poly(β-CD),
ce qui a également été vérifié par QCM (annexe 5).
Lorsque le DTAC est ajouté au mélange aqueux poly(β-CD)/NaPSS, une chute
significative des valeurs de viscosité est observée sur la figure IV-11, indiquant une
compaction des chaînes (par écrantage des répulsions électrostatiques intrachaînes). Ceci
suggère une association ternaire en solution, tout d’abord par interactions entre charges
opposées DTAC/NaPSS qui induisent une compaction des chaînes de NaPSS, ensuite par
interactions entre le poly(β-CD) et le complexe DTAC/NaPSS qui réduisent considérablement
le comportement de ce dernier: l’ajout de poly(β-CD) au mélange DTAC/NaPSS induit une
réduction marquée de la viscosité, surtout à forte dilution. Notons que le comportement PEL
du système ne semble pas pour autant être totalement éliminé. En effet, une faible
augmentation de la viscosité du mélange à 3 composants est observée à forte dilution,
indiquant un effet de charge. Le mélange poly(β-CD)/DTAC/NaPSS à la stoechiométrie
14/6/14 (Nβ-CD/NDTAC/Nsulfonate) semble donc résulter en la formation d’agrégats portant une
charge globale non nulle (en supposant que l’augmentation de viscosité observée ne soit pas
simplement due à la présence de chaînes de NaPSS libres dans le mélange). Notons, de plus,
que le phénomène d’association ternaire doit être accompagné d’une disparition plus ou
moins totale de la structure en collier de perles des complexes DTAC/NaPSS. La formation de
complexes d’inclusion entre des cavités du poly(β-CD) et des molécules de DTAC liées au
NaPSS implique, en effet, qu’une partie (sinon la totalité) des agrégats micellaires présents le
long de la chaîne polyanionique soient dissociés (micelles de DTAC engendrées par l’effet
coopératif de l’association DTAC/NaPSS).
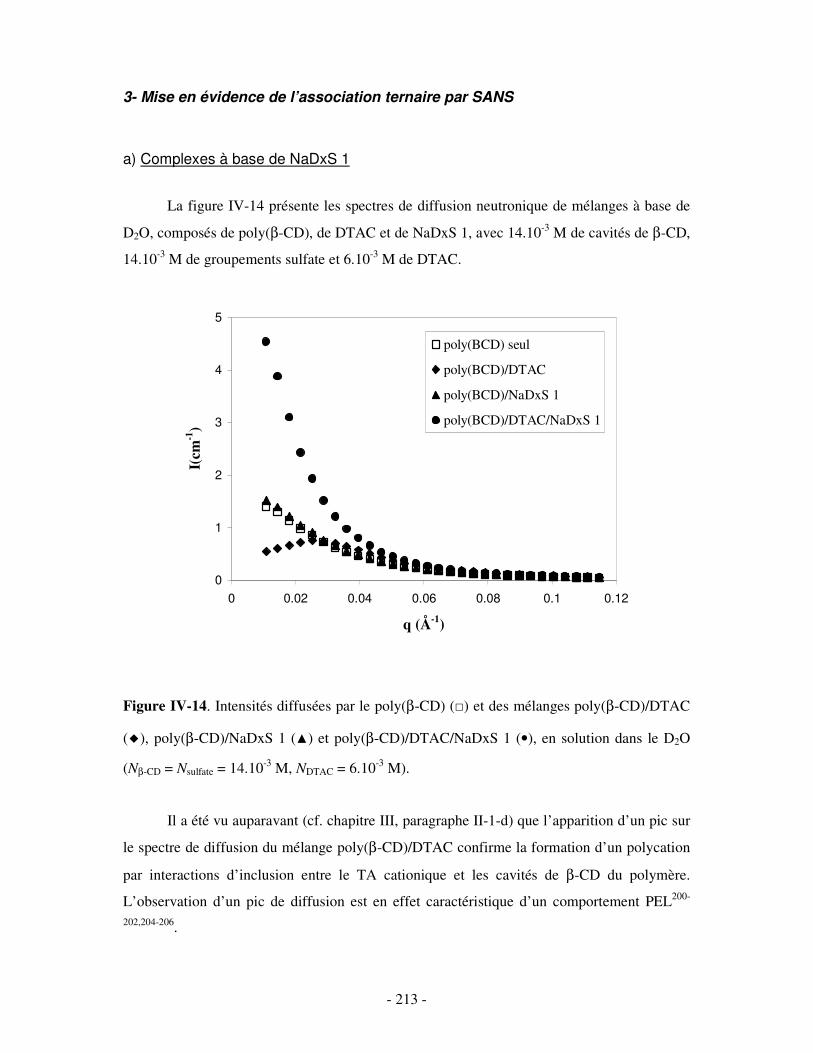
- 213 -
3- Mise en évidence de l’association ternaire par SANS
a) Complexes à base de NaDxS 1
La figure IV-14 présente les spectres de diffusion neutronique de mélanges à base de
D2O, composés de poly(β-CD), de DTAC et de NaDxS 1, avec 14.10-3 M de cavités de β-CD,
14.10-3 M de groupements sulfate et 6.10-3 M de DTAC.
0
1
2
3
4
5
0 0.02 0.04 0.06 0.08 0.1 0.12
q (Å-1)
I(cm
-1)
poly(BCD) seul
poly(BCD)/DTAC
poly(BCD)/NaDxS 1
poly(BCD)/DTAC/NaDxS 1
Figure IV-14. Intensités diffusées par le poly(β-CD) () et des mélanges poly(β-CD)/DTAC
(), poly(β-CD)/NaDxS 1 () et poly(β-CD)/DTAC/NaDxS 1 (•), en solution dans le D2O
(Nβ-CD = Nsulfate = 14.10-3 M, NDTAC = 6.10-3 M).
Il a été vu auparavant (cf. chapitre III, paragraphe II-1-d) que l’apparition d’un pic sur
le spectre de diffusion du mélange poly(β-CD)/DTAC confirme la formation d’un polycation
par interactions d’inclusion entre le TA cationique et les cavités de β-CD du polymère.
L’observation d’un pic de diffusion est en effet caractéristique d’un comportement PEL200-
202,204-206.

- 214 -
Bien que le NaDxS 1 soit un PEL, aucun pic n’est observé sur la figure IV-14 pour le
mélange poly(β-CD)/NaDxS 1. La diffusion de ce dernier est d’ailleurs quasiment superposée
à celle du poly(β-CD) seul en solution. Il s’avère que le NaDxS 1 n’est pas suffisamment
concentré pour que sa diffusion puisse être détectée: Nsulfate = 14.10-3 M correspond à CNaDxS 1
= 2,6 g.L-1, alors que Nβ-CD = 14.10-3 M correspond à 26,8 g.L-1 de poly(β-CD). Les résultats
de SANS collectés pour les systèmes comportant le poly(β-CD) et le NaDxS 1 (Nβ-CD = Nsulfate
= 14.10-3 M) peuvent donc être interprétés en termes de diffusion du poly(β-CD) uniquement
(même en présence de 6.10-3 M de DTAC car, dans ce cas, la diffusion par le DTAC est
également négligeable). Il peut être considéré, par exemple, que la valeur du coefficient α des
comportements asymptotiques I(q) ~ q-α observés à grands q caractérise la seule compacité
des chaînes de poly(β-CD) au sein des systèmes. Dans le cas du mélange binaire poly(β-
CD)/NaDxS 1, la valeur du coefficient α apparent mesurée est identique à celle du poly(β-
CD) seul en solution: αapp = 2,25 ± 0,05, ce qui indique que la structure du poly(β-CD) n’est
pas affectée par la présence du polyanion.
D’après la figure IV-14, l’ajout du NaDxS 1 au système poly(β-CD)/DTAC a deux
conséquences importantes. La première est une disparition du pic PEL, indiquant un écrantage
des répulsions coulombiennes entre les chaînes polycationiques poly(β-CD)/DTAC. La
deuxième est une forte augmentation de l’intensité diffusée, à des valeurs plus élevées que
celles obtenues par ajout de sel au système poly(β-CD)/DTAC (cf. figure III-26, p. 176) (les
14.10-3 M de groupements sulfate introduits sont accompagnés par 14.10-3 M de contre-ions
Na+), mais aussi à des valeurs plus élevées que celles observées pour le mélange poly(β-
CD)/NaDxS 1. Ceci confirme la formation de complexes ternaires suggérée par les résultats
viscosimétriques: la disparition du pic PEL résulte d’interactions entre charges opposées entre
les chaînes du complexe poly(β-CD)/DTAC et celles du NaDxS 1. Ces interactions induisent
une agrégation multichaîne en solution, comme l’indique la forte augmentation d’intensité
observée (en général, plus les objets diffusants sont agrégés, c’est-à-dire de masse de plus en
plus importante, et plus les valeurs d’intensité diffusée sont élevées200,201). Il s’en déduit que
les agrégats formés par complexation ternaire poly(β-CD)/DTAC/NaDxS 1 comportent plus
d’une chaîne de poly(β-CD), étant donné que les chaînes de NaDxS 1 diffusent peu (tout du
moins à CNaDxS 1 considérée). Mais nous reviendrons plus en détails sur la structure des
complexes ternaires dans la suite de ce chapitre (cf. paragraphe III-1).

- 215 -
La valeur de α mesurée à grands q pour le mélange ternaire poly(β-
CD)/DTAC/NaDxS 1, αapp = 2,39 ± 0,05, indique également une interaction
macromoléculaire. Le fait qu’elle soit supérieure à celle mesurée pour le poly(β-CD) pur
(αpoly(β-CD) = 2,25 ± 0,05) et celles mesurées pour les autres mélanges (αapp = 2,20 pour le
mélange poly(β-CD)/DTAC et 2,25 pour le mélange poly(β-CD)/NaDxS 1), suggère que les
chaînes de poly(β-CD) subissent une compaction de leur structure interne uniquement
lorsqu’elles sont mélangées à la fois au DTAC et au NaDxS 1 (plus les objets diffusants sont
compacts, plus α est élevé192-194,201).
Il est important de souligner que toutes les observations concernant les systèmes
constitués de 14.10-3 M de cavités du poly(β-CD), 14.10-3 M de sulfate du NaDxS 1 et 6.10-3
M de DTAC sont transposables aux mêmes systèmes dilués d’un facteur 4 (figure IV-15).
Même à faibles concentrations (Nβ-CD = Nsulfate = 3,5.10-3 M et NDTAC = 1,5.10-3 M), le pic PEL
du mélange poly(β-CD)/DTAC disparaît par addition du NaDxS 1 et l’intensité diffusée par le
système ternaire est près de deux fois supérieure à celle diffusée par le mélange poly(β-
CD)/NaDxS 1. D’autre part, une valeur de αapp égale à 2,43 ± 0,05 est mesurée en présence
des trois composants (contre 2,10 pour le mélange poly(β-CD)/DTAC et 2,25 pour le mélange
poly(β-CD)/NaDxS 1). Ceci indique que la complexation ternaire subsiste sur tout le domaine
de concentrations exploré par dilution en viscosimétrie (cf. figure IV-8).
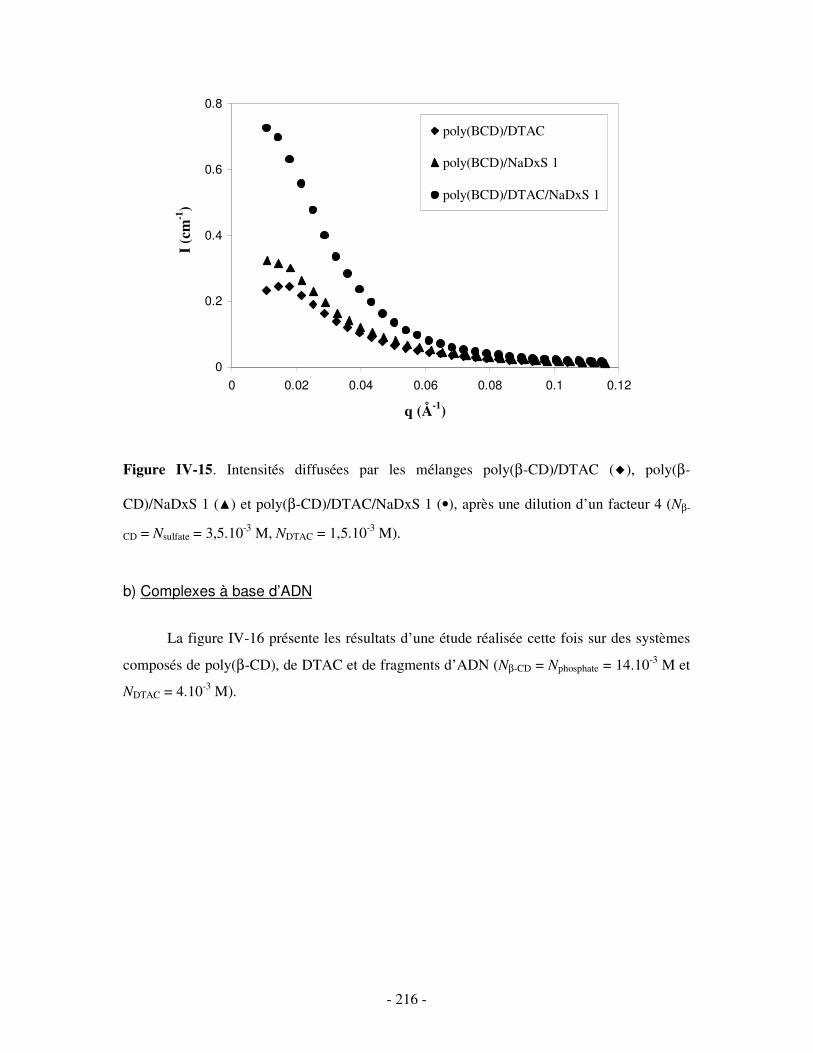
- 216 -
0
0.2
0.4
0.6
0.8
0 0.02 0.04 0.06 0.08 0.1 0.12
q (Å-1)
I (cm
-1)
poly(BCD)/DTAC
poly(BCD)/NaDxS 1
poly(BCD)/DTAC/NaDxS 1
Figure IV-15. Intensités diffusées par les mélanges poly(β-CD)/DTAC (), poly(β-
CD)/NaDxS 1 () et poly(β-CD)/DTAC/NaDxS 1 (•), après une dilution d’un facteur 4 (Nβ-
CD = Nsulfate = 3,5.10-3 M, NDTAC = 1,5.10-3 M).
b) Complexes à base d’ADN
La figure IV-16 présente les résultats d’une étude réalisée cette fois sur des systèmes
composés de poly(β-CD), de DTAC et de fragments d’ADN (Nβ-CD = Nphosphate = 14.10-3 M et
NDTAC = 4.10-3 M).
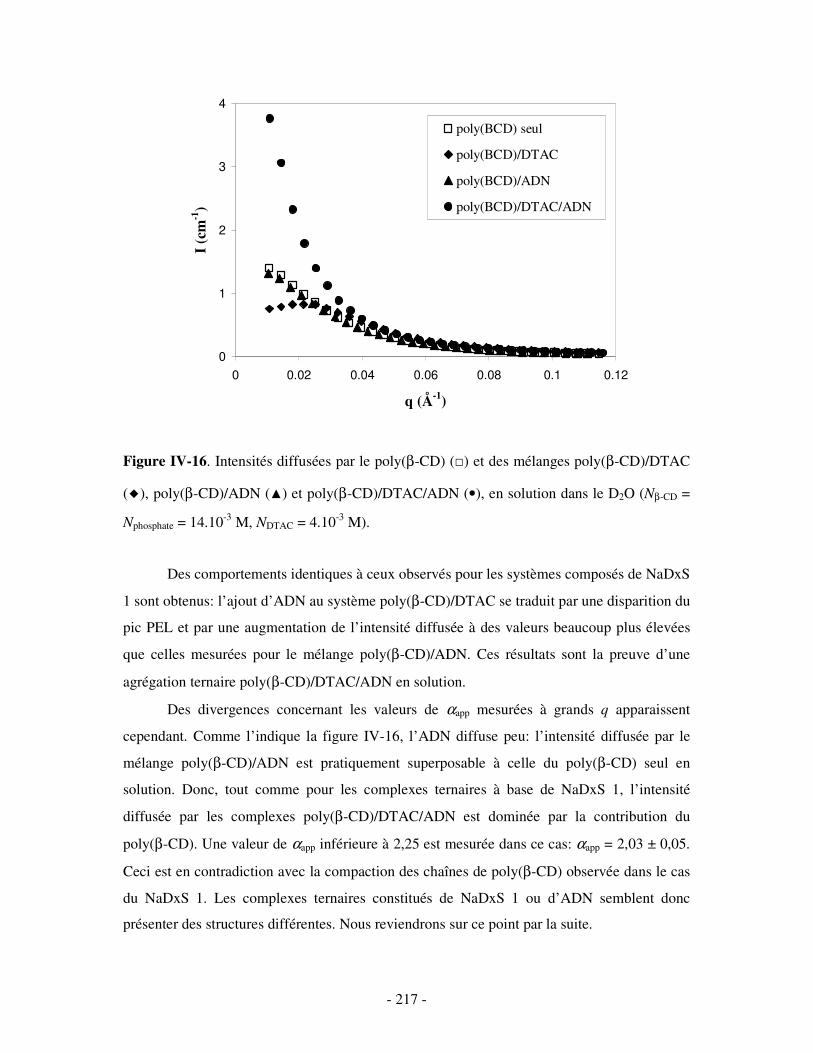
- 217 -
0
1
2
3
4
0 0.02 0.04 0.06 0.08 0.1 0.12
q (Å-1)
I (cm
-1)
poly(BCD) seul
poly(BCD)/DTAC
poly(BCD)/ADN
poly(BCD)/DTAC/ADN
Figure IV-16. Intensités diffusées par le poly(β-CD) () et des mélanges poly(β-CD)/DTAC
(), poly(β-CD)/ADN () et poly(β-CD)/DTAC/ADN (•), en solution dans le D2O (Nβ-CD =
Nphosphate = 14.10-3 M, NDTAC = 4.10-3 M).
Des comportements identiques à ceux observés pour les systèmes composés de NaDxS
1 sont obtenus: l’ajout d’ADN au système poly(β-CD)/DTAC se traduit par une disparition du
pic PEL et par une augmentation de l’intensité diffusée à des valeurs beaucoup plus élevées
que celles mesurées pour le mélange poly(β-CD)/ADN. Ces résultats sont la preuve d’une
agrégation ternaire poly(β-CD)/DTAC/ADN en solution.
Des divergences concernant les valeurs de αapp mesurées à grands q apparaissent
cependant. Comme l’indique la figure IV-16, l’ADN diffuse peu: l’intensité diffusée par le
mélange poly(β-CD)/ADN est pratiquement superposable à celle du poly(β-CD) seul en
solution. Donc, tout comme pour les complexes ternaires à base de NaDxS 1, l’intensité
diffusée par les complexes poly(β-CD)/DTAC/ADN est dominée par la contribution du
poly(β-CD). Une valeur de αapp inférieure à 2,25 est mesurée dans ce cas: αapp = 2,03 ± 0,05.
Ceci est en contradiction avec la compaction des chaînes de poly(β-CD) observée dans le cas
du NaDxS 1. Les complexes ternaires constitués de NaDxS 1 ou d’ADN semblent donc
présenter des structures différentes. Nous reviendrons sur ce point par la suite.
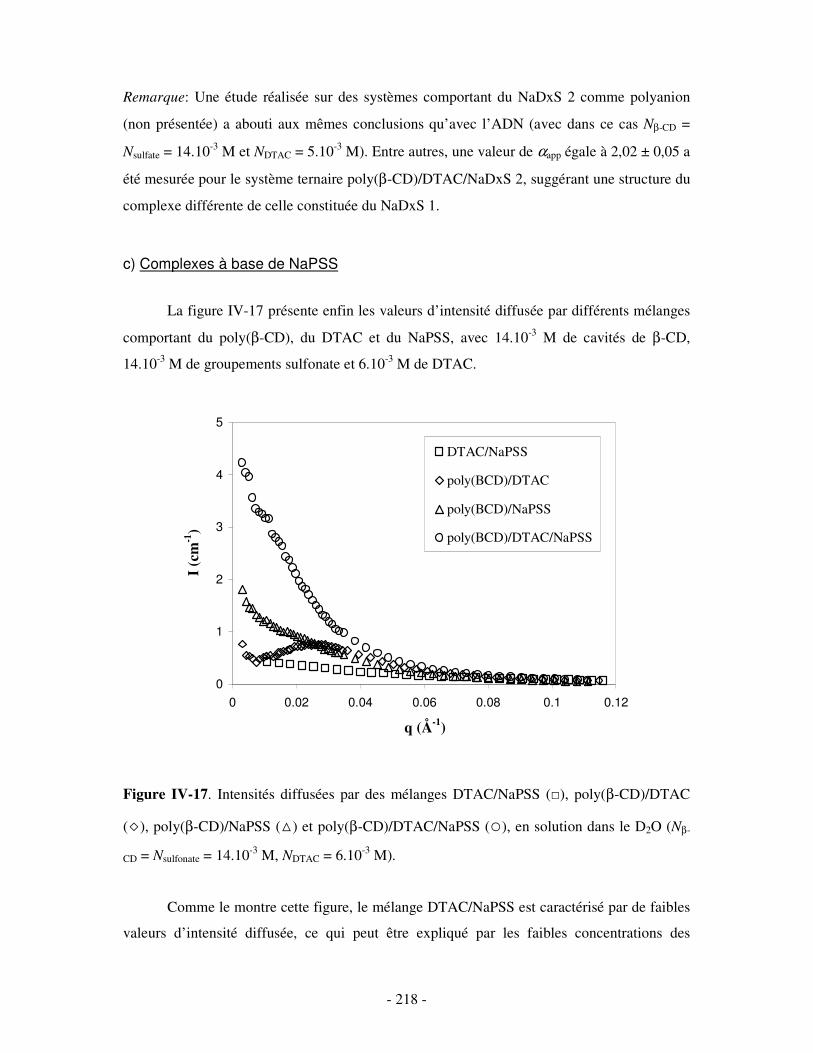
- 218 -
Remarque: Une étude réalisée sur des systèmes comportant du NaDxS 2 comme polyanion
(non présentée) a abouti aux mêmes conclusions qu’avec l’ADN (avec dans ce cas Nβ-CD =
Nsulfate = 14.10-3 M et NDTAC = 5.10-3 M). Entre autres, une valeur de αapp égale à 2,02 ± 0,05 a
été mesurée pour le système ternaire poly(β-CD)/DTAC/NaDxS 2, suggérant une structure du
complexe différente de celle constituée du NaDxS 1.
c) Complexes à base de NaPSS
La figure IV-17 présente enfin les valeurs d’intensité diffusée par différents mélanges
comportant du poly(β-CD), du DTAC et du NaPSS, avec 14.10-3 M de cavités de β-CD,
14.10-3 M de groupements sulfonate et 6.10-3 M de DTAC.
0
1
2
3
4
5
0 0.02 0.04 0.06 0.08 0.1 0.12
q (Å-1)
I (cm
-1)
DTAC/NaPSS
poly(BCD)/DTAC
poly(BCD)/NaPSS
poly(BCD)/DTAC/NaPSS
Figure IV-17. Intensités diffusées par des mélanges DTAC/NaPSS (), poly(β-CD)/DTAC
( ), poly(β-CD)/NaPSS () et poly(β-CD)/DTAC/NaPSS (), en solution dans le D2O (Nβ-
CD = Nsulfonate = 14.10-3 M, NDTAC = 6.10-3 M).
Comme le montre cette figure, le mélange DTAC/NaPSS est caractérisé par de faibles
valeurs d’intensité diffusée, ce qui peut être expliqué par les faibles concentrations des
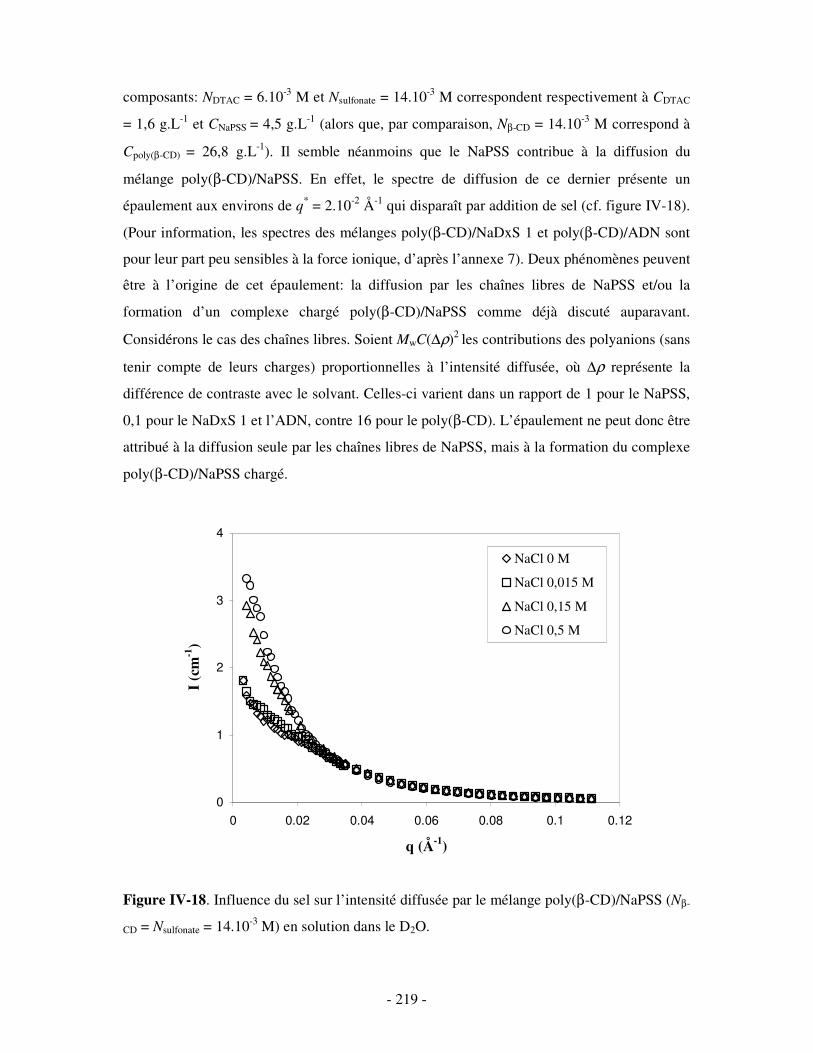
- 219 -
composants: NDTAC = 6.10-3 M et Nsulfonate = 14.10-3 M correspondent respectivement à CDTAC
= 1,6 g.L-1 et CNaPSS = 4,5 g.L-1 (alors que, par comparaison, Nβ-CD = 14.10-3 M correspond à
Cpoly(β-CD) = 26,8 g.L-1). Il semble néanmoins que le NaPSS contribue à la diffusion du
mélange poly(β-CD)/NaPSS. En effet, le spectre de diffusion de ce dernier présente un
épaulement aux environs de q* = 2.10-2 Å-1 qui disparaît par addition de sel (cf. figure IV-18).
(Pour information, les spectres des mélanges poly(β-CD)/NaDxS 1 et poly(β-CD)/ADN sont
pour leur part peu sensibles à la force ionique, d’après l’annexe 7). Deux phénomènes peuvent
être à l’origine de cet épaulement: la diffusion par les chaînes libres de NaPSS et/ou la
formation d’un complexe chargé poly(β-CD)/NaPSS comme déjà discuté auparavant.
Considérons le cas des chaînes libres. Soient MwC(∆ρ)2 les contributions des polyanions (sans
tenir compte de leurs charges) proportionnelles à l’intensité diffusée, où ∆ρ représente la
différence de contraste avec le solvant. Celles-ci varient dans un rapport de 1 pour le NaPSS,
0,1 pour le NaDxS 1 et l’ADN, contre 16 pour le poly(β-CD). L’épaulement ne peut donc être
attribué à la diffusion seule par les chaînes libres de NaPSS, mais à la formation du complexe
poly(β-CD)/NaPSS chargé.
0
1
2
3
4
0 0.02 0.04 0.06 0.08 0.1 0.12
q (Å-1)
I (cm
-1)
NaCl 0 M
NaCl 0,015 M
NaCl 0,15 M
NaCl 0,5 M
Figure IV-18. Influence du sel sur l’intensité diffusée par le mélange poly(β-CD)/NaPSS (Nβ-
CD = Nsulfonate = 14.10-3 M) en solution dans le D2O.
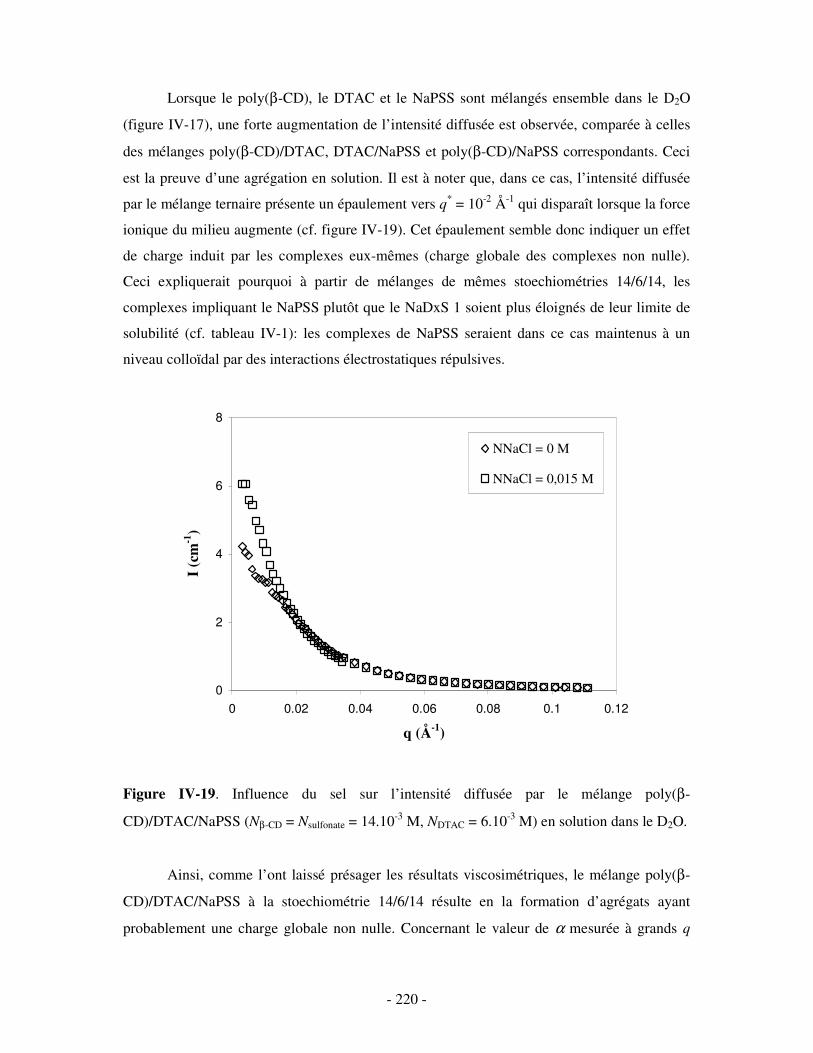
- 220 -
Lorsque le poly(β-CD), le DTAC et le NaPSS sont mélangés ensemble dans le D2O
(figure IV-17), une forte augmentation de l’intensité diffusée est observée, comparée à celles
des mélanges poly(β-CD)/DTAC, DTAC/NaPSS et poly(β-CD)/NaPSS correspondants. Ceci
est la preuve d’une agrégation en solution. Il est à noter que, dans ce cas, l’intensité diffusée
par le mélange ternaire présente un épaulement vers q* = 10-2 Å-1 qui disparaît lorsque la force
ionique du milieu augmente (cf. figure IV-19). Cet épaulement semble donc indiquer un effet
de charge induit par les complexes eux-mêmes (charge globale des complexes non nulle).
Ceci expliquerait pourquoi à partir de mélanges de mêmes stoechiométries 14/6/14, les
complexes impliquant le NaPSS plutôt que le NaDxS 1 soient plus éloignés de leur limite de
solubilité (cf. tableau IV-1): les complexes de NaPSS seraient dans ce cas maintenus à un
niveau colloïdal par des interactions électrostatiques répulsives.
0
2
4
6
8
0 0.02 0.04 0.06 0.08 0.1 0.12
q (Å-1)
I (cm
-1)
NNaCl = 0 M
NNaCl = 0,015 M
Figure IV-19. Influence du sel sur l’intensité diffusée par le mélange poly(β-
CD)/DTAC/NaPSS (Nβ-CD = Nsulfonate = 14.10-3 M, NDTAC = 6.10-3 M) en solution dans le D2O.
Ainsi, comme l’ont laissé présager les résultats viscosimétriques, le mélange poly(β-
CD)/DTAC/NaPSS à la stoechiométrie 14/6/14 résulte en la formation d’agrégats ayant
probablement une charge globale non nulle. Concernant le valeur de α mesurée à grands q

- 221 -
pour ce mélange, une valeur proche de celle du poly(β-CD) pur est obtenue: αapp = 2,23 ±
0,05. Néanmoins, il est difficile d’en tirer des conclusions étant donné qu’en plus du poly(β-
CD), le NaPSS contribue à la diffusion des neutrons par le mélange.
4- Bilan
Des diagrammes de solubilité ont été établis en mélangeant dans l’eau le poly(β-CD),
le DTAC et plusieurs polyanions de natures et de rigidités différentes. Il s’avère que tous les
systèmes montrent une réduction de leurs domaines de solubilité à concentrations en DTAC
élevées. D’un autre côté, la solubilité des mélanges dépend à la fois de la nature chimique du
polyanion impliqué et, dans le cas du NaDxS, de la longueur de la chaîne. Ces différentes
sensibilités semblent être corrélées à un paramètre caractéristique des polyanions: la rigidité
de leur chaîne. En effet, plus les chaînes polyanioniques sont rigides (dans l’ordre NaPSS <
NaDxS 1 < NaDxS 2 < ADN), moins les mélanges ternaires sont solubles.
Pour des mélanges de stoechiométrie Nβ-CD/NDTAC/Nanion égale à 1/0,3-0,4/1 (Nβ-CD et
Nanion étant respectivement les concentrations molaires en cavités du poly(β-CD) et en
groupements chargés négativement du polyanion), des études viscosimétriques couplées à des
études de diffusion de neutrons ont prouvé la formation de complexes ternaires solubles dans
l’eau, quelque soient la nature et la rigidité du polyanion utilisé (NaDxS 1 ou 2, NaPSS ou
ADN). Dans le cas des NaDxS et de l’ADN, l’association ternaire se manifeste, d’une part,
par la solubilisation des complexes binaires DTAC/polyanion (précipités en absence du
poly(β-CD)) et, d’autre part, par la perte du comportement PEL du complexe poly(β-
CD)/DTAC (disparition du pic de diffusion et chute de la viscosité par dilution en présence
des trois composants). L’agrégation en solution se manifeste également par une forte
augmentation des valeurs d’intensité diffusée. Dans le cas du NaPSS, si un fort accroissement
de l’intensité diffusée a pu aussi été observé, des différences de comportement ont cependant
être relevées. Tout d’abord, le mélange DTAC/NaPSS s’avère être parfaitement soluble dans
l’eau (à la stoechiométrie NDTAC/Nsulfonate = 0,4). Ensuite, les comportements PELs observés
pour les mélanges binaires poly(β-CD)/DTAC et poly(β-CD)/NaPSS ne sont pas totalement
écrantés en présence des trois composants: le spectre de diffusion du mélange poly(β-
CD)/DTAC/NaPSS n’est pas une fonction monotone de q (existence d’un épaulement) et sa
viscosité augmente légèrement à forte dilution. Cet effet de charge manifesté par le mélange

- 222 -
ternaire à la stoechiométrie 1/0,4/1 laisse entendre que les agrégats formés dans ce cas
(impliquant le NaPSS) portent une charge globale non nulle.
L’un des objectifs de la partie III sera d’analyser la structure des complexes ternaires.
Dans le cas particulier du NaPSS, un échantillon deutéré du polyanion sera mélangé au
poly(β-CD) et au tensioactif dans le D2O, de façon à annuler sa contribution à la diffusion.
Dans cette partie, il sera aussi question d’évaluer l’influence de la stoechiométrie NDTAC/Nanion
sur la structure des complexes, en faisant varier la concentration en DTAC dans des mélanges
comportant des quantités fixes de poly(β-CD) et de polyanion. Enfin, nous étudierons la
stabilité et la réversibilité des complexes en faisant varier la force ionique du milieu et la
concentration en compétiteurs.
III- Etude des propriétés structurales des complexes ternaires en
solution
1- Influence de la concentration en TA
a) Etude par SANS
i) Complexes à base de NaDxS 1
L’influence de la concentration en DTAC sur l’intensité diffusée par des systèmes
poly(β-CD)/DTAC/NaDxS 1 (Nβ-CD = Nsulfate = 14.10-3 M) en solution dans le D2O est
illustrée en figure IV-20.
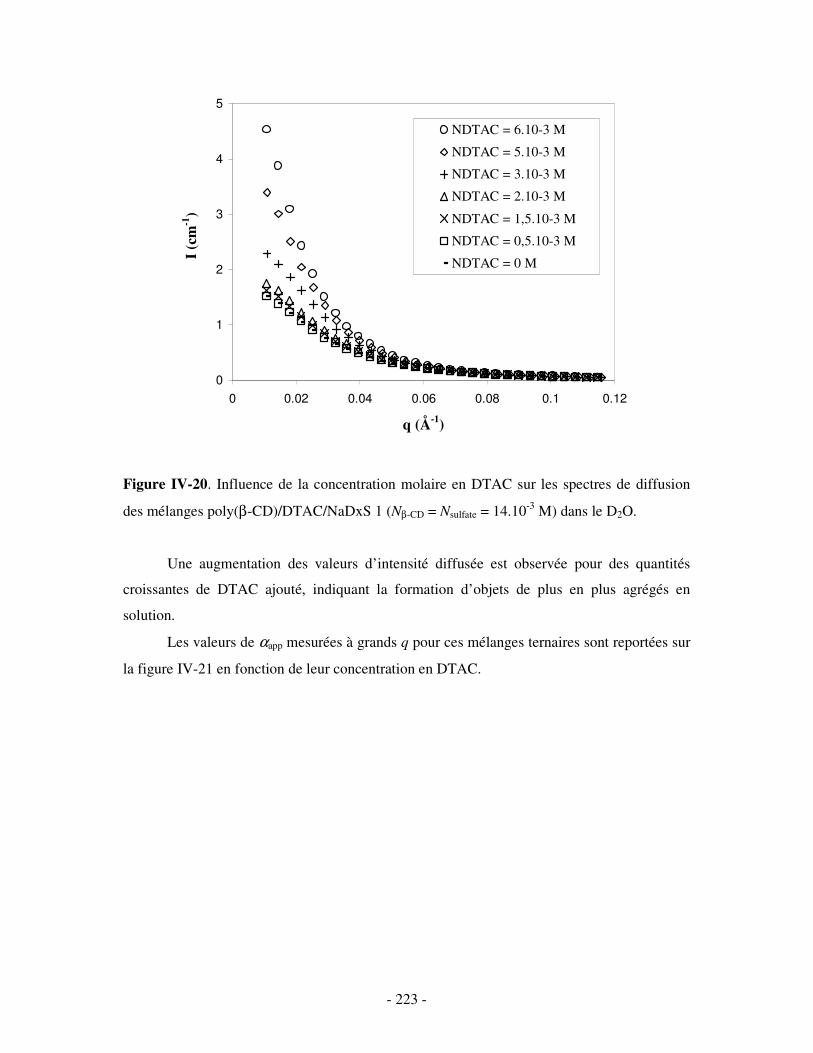
- 223 -
0
1
2
3
4
5
0 0.02 0.04 0.06 0.08 0.1 0.12
q (Å-1)
I (cm
-1)
NDTAC = 6.10-3 M
NDTAC = 5.10-3 M
NDTAC = 3.10-3 M
NDTAC = 2.10-3 M
NDTAC = 1,5.10-3 M
NDTAC = 0,5.10-3 M
NDTAC = 0 M
Figure IV-20. Influence de la concentration molaire en DTAC sur les spectres de diffusion
des mélanges poly(β-CD)/DTAC/NaDxS 1 (Nβ-CD = Nsulfate = 14.10-3 M) dans le D2O.
Une augmentation des valeurs d’intensité diffusée est observée pour des quantités
croissantes de DTAC ajouté, indiquant la formation d’objets de plus en plus agrégés en
solution.
Les valeurs de αapp mesurées à grands q pour ces mélanges ternaires sont reportées sur
la figure IV-21 en fonction de leur concentration en DTAC.
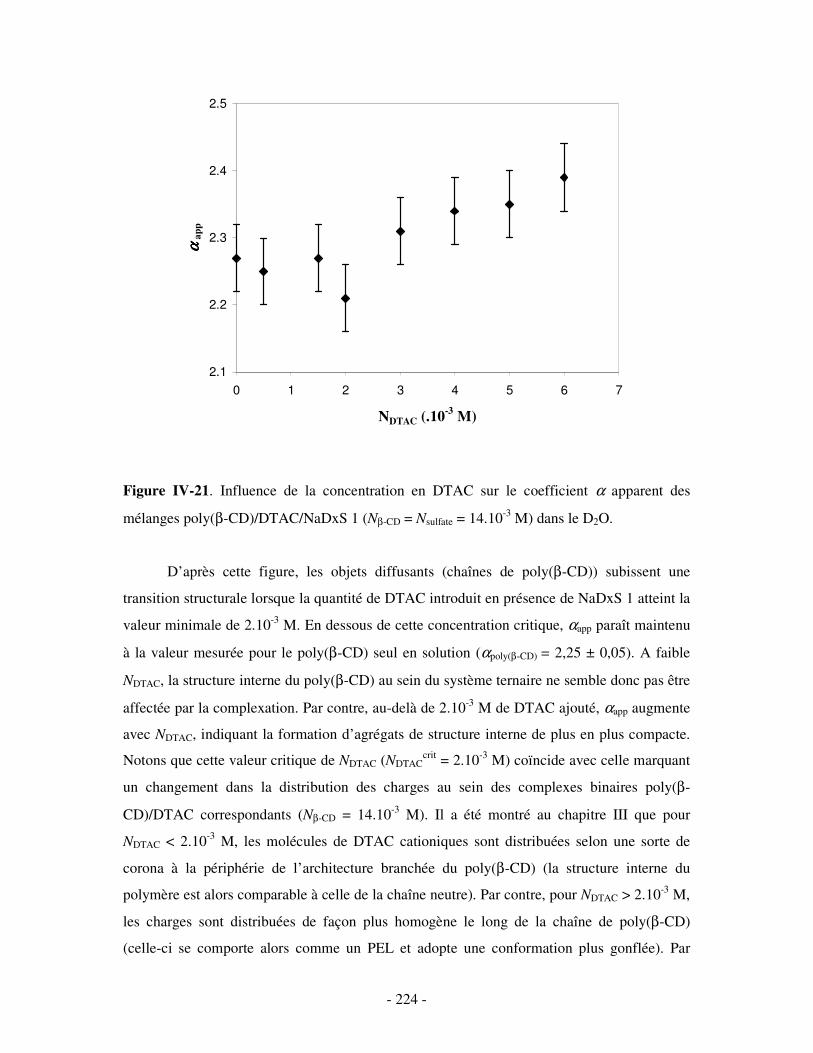
- 224 -
2.1
2.2
2.3
2.4
2.5
0 1 2 3 4 5 6 7
NDTAC (.10-3 M)
αα ααap
p
Figure IV-21. Influence de la concentration en DTAC sur le coefficient α apparent des
mélanges poly(β-CD)/DTAC/NaDxS 1 (Nβ-CD = Nsulfate = 14.10-3 M) dans le D2O.
D’après cette figure, les objets diffusants (chaînes de poly(β-CD)) subissent une
transition structurale lorsque la quantité de DTAC introduit en présence de NaDxS 1 atteint la
valeur minimale de 2.10-3 M. En dessous de cette concentration critique, αapp paraît maintenu
à la valeur mesurée pour le poly(β-CD) seul en solution (αpoly(β-CD) = 2,25 ± 0,05). A faible
NDTAC, la structure interne du poly(β-CD) au sein du système ternaire ne semble donc pas être
affectée par la complexation. Par contre, au-delà de 2.10-3 M de DTAC ajouté, αapp augmente
avec NDTAC, indiquant la formation d’agrégats de structure interne de plus en plus compacte.
Notons que cette valeur critique de NDTAC (NDTACcrit = 2.10-3 M) coïncide avec celle marquant
un changement dans la distribution des charges au sein des complexes binaires poly(β-
CD)/DTAC correspondants (Nβ-CD = 14.10-3 M). Il a été montré au chapitre III que pour
NDTAC < 2.10-3 M, les molécules de DTAC cationiques sont distribuées selon une sorte de
corona à la périphérie de l’architecture branchée du poly(β-CD) (la structure interne du
polymère est alors comparable à celle de la chaîne neutre). Par contre, pour NDTAC > 2.10-3 M,
les charges sont distribuées de façon plus homogène le long de la chaîne de poly(β-CD)
(celle-ci se comporte alors comme un PEL et adopte une conformation plus gonflée). Par

- 225 -
conséquent, en corrélant les résultats de la figure IV-21 à ceux du chapitre III, deux schémas
de la structure interne des complexes poly(β-CD)/DTAC/NaDxS 1 peuvent être proposés
suivant la valeur de la concentration en DTAC ajouté. Pour NDTAC < 2.10-3 M (figure IV-22-
a), l’addition de DTAC peut résulter en la formation de structures de type cœur-coquille, avec
un cœur constitué de poly(β-CD), un corona intermédiaire de DTAC et une coquille
extérieure de NaDxS 1. Dans cette configuration, la structure interne du poly(β-CD) semble
pouvoir rester intacte, ce qui expliquerait les valeurs de αapp observées: ~ 2,25. Pour NDTAC >
2.10-3 M (figure IV-22-b), le fait que αapp soit supérieur à 2,25 suggère, par contre, que les
interactions électrostatiques entre le NaDxS 1 et les molécules de DTAC réparties
régulièrement le long de la chaîne de poly(β-CD) conduisent à des structures plus compactes,
où les deux polymères sont intimement mêlés. Ceci implique que le NaDxS 1, de rayon de
giration de l’ordre de 20 Å en solvant θ (à force ionique élevée), est capable de pénétrer au
sein de la structure branchée du poly(β-CD), de rayon de giration égal à 55 Å.
Figure IV-22. Schéma de la structure interne des complexes poly(β-CD)/DTAC/NaDxS 1
(Nβ-CD = Nsulfate = 14.10-3 M), selon la concentration en DTAC ajouté dans les mélanges.
NDTAC < 2.10-3 M
⊕⊕⊕⊕
⊕⊕⊕⊕ ⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
NDTAC > 2.10-3 M
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
a) b)
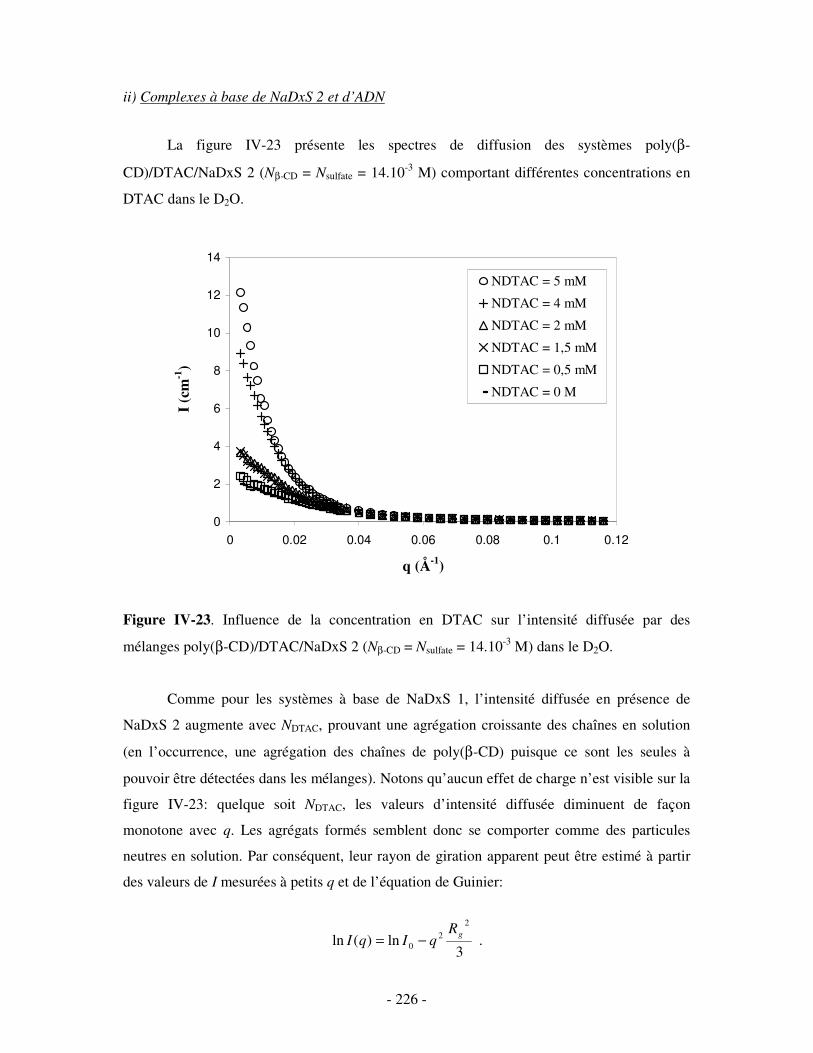
- 226 -
ii) Complexes à base de NaDxS 2 et d’ADN
La figure IV-23 présente les spectres de diffusion des systèmes poly(β-
CD)/DTAC/NaDxS 2 (Nβ-CD = Nsulfate = 14.10-3 M) comportant différentes concentrations en
DTAC dans le D2O.
0
2
4
6
8
10
12
14
0 0.02 0.04 0.06 0.08 0.1 0.12
q (Å-1)
I (cm
-1)
NDTAC = 5 mM
NDTAC = 4 mM
NDTAC = 2 mM
NDTAC = 1,5 mM
NDTAC = 0,5 mM
NDTAC = 0 M
Figure IV-23. Influence de la concentration en DTAC sur l’intensité diffusée par des
mélanges poly(β-CD)/DTAC/NaDxS 2 (Nβ-CD = Nsulfate = 14.10-3 M) dans le D2O.
Comme pour les systèmes à base de NaDxS 1, l’intensité diffusée en présence de
NaDxS 2 augmente avec NDTAC, prouvant une agrégation croissante des chaînes en solution
(en l’occurrence, une agrégation des chaînes de poly(β-CD) puisque ce sont les seules à
pouvoir être détectées dans les mélanges). Notons qu’aucun effet de charge n’est visible sur la
figure IV-23: quelque soit NDTAC, les valeurs d’intensité diffusée diminuent de façon
monotone avec q. Les agrégats formés semblent donc se comporter comme des particules
neutres en solution. Par conséquent, leur rayon de giration apparent peut être estimé à partir
des valeurs de I mesurées à petits q et de l’équation de Guinier:
3ln)(ln
22
0gR
qIqI −= .
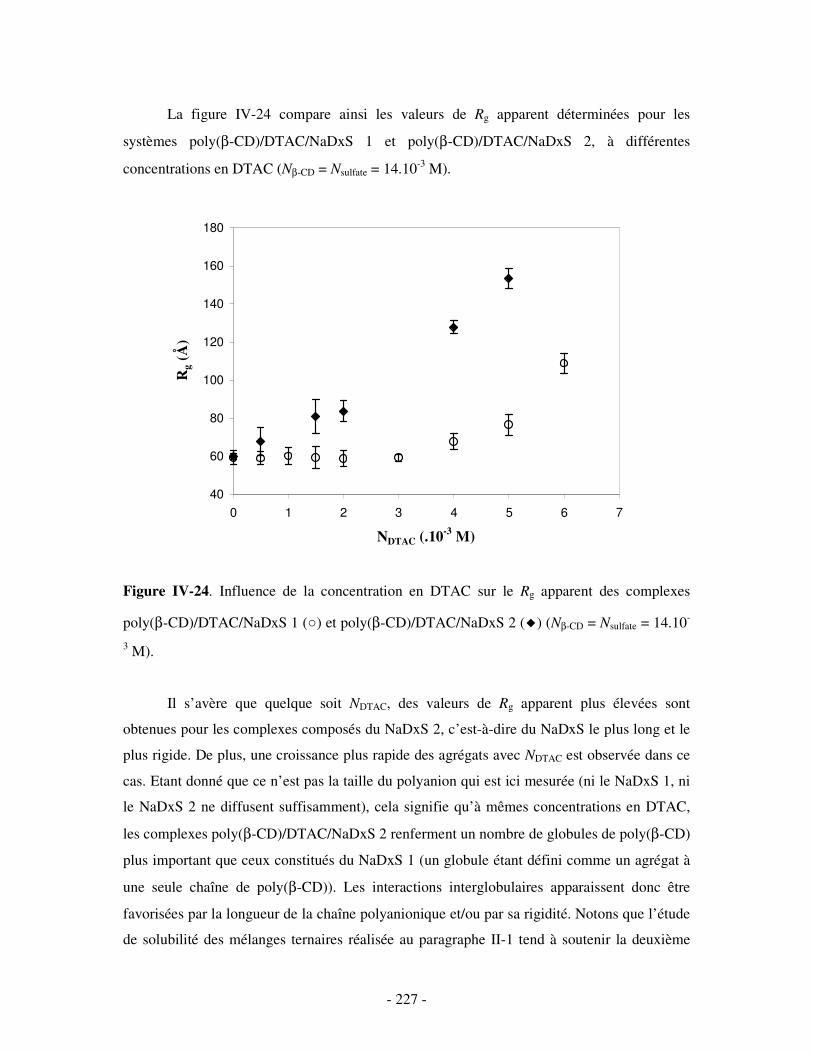
- 227 -
La figure IV-24 compare ainsi les valeurs de Rg apparent déterminées pour les
systèmes poly(β-CD)/DTAC/NaDxS 1 et poly(β-CD)/DTAC/NaDxS 2, à différentes
concentrations en DTAC (Nβ-CD = Nsulfate = 14.10-3 M).
40
60
80
100
120
140
160
180
0 1 2 3 4 5 6 7
NDTAC (.10-3 M)
Rg (
Å)
Figure IV-24. Influence de la concentration en DTAC sur le Rg apparent des complexes
poly(β-CD)/DTAC/NaDxS 1 () et poly(β-CD)/DTAC/NaDxS 2 () (Nβ-CD = Nsulfate = 14.10-
3 M).
Il s’avère que quelque soit NDTAC, des valeurs de Rg apparent plus élevées sont
obtenues pour les complexes composés du NaDxS 2, c’est-à-dire du NaDxS le plus long et le
plus rigide. De plus, une croissance plus rapide des agrégats avec NDTAC est observée dans ce
cas. Etant donné que ce n’est pas la taille du polyanion qui est ici mesurée (ni le NaDxS 1, ni
le NaDxS 2 ne diffusent suffisamment), cela signifie qu’à mêmes concentrations en DTAC,
les complexes poly(β-CD)/DTAC/NaDxS 2 renferment un nombre de globules de poly(β-CD)
plus important que ceux constitués du NaDxS 1 (un globule étant défini comme un agrégat à
une seule chaîne de poly(β-CD)). Les interactions interglobulaires apparaissent donc être
favorisées par la longueur de la chaîne polyanionique et/ou par sa rigidité. Notons que l’étude
de solubilité des mélanges ternaires réalisée au paragraphe II-1 tend à soutenir la deuxième
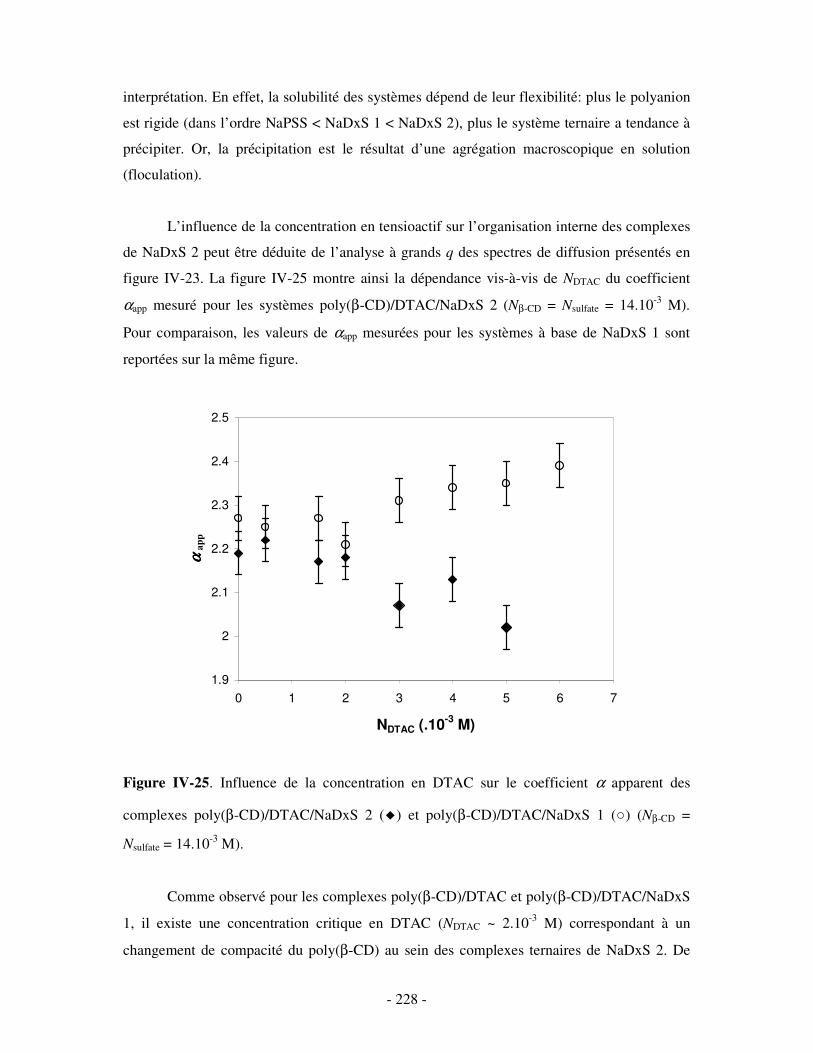
- 228 -
interprétation. En effet, la solubilité des systèmes dépend de leur flexibilité: plus le polyanion
est rigide (dans l’ordre NaPSS < NaDxS 1 < NaDxS 2), plus le système ternaire a tendance à
précipiter. Or, la précipitation est le résultat d’une agrégation macroscopique en solution
(floculation).
L’influence de la concentration en tensioactif sur l’organisation interne des complexes
de NaDxS 2 peut être déduite de l’analyse à grands q des spectres de diffusion présentés en
figure IV-23. La figure IV-25 montre ainsi la dépendance vis-à-vis de NDTAC du coefficient
αapp mesuré pour les systèmes poly(β-CD)/DTAC/NaDxS 2 (Nβ-CD = Nsulfate = 14.10-3 M).
Pour comparaison, les valeurs de αapp mesurées pour les systèmes à base de NaDxS 1 sont
reportées sur la même figure.
1.9
2
2.1
2.2
2.3
2.4
2.5
0 1 2 3 4 5 6 7
NDTAC (.10-3 M)
αα ααap
p
Figure IV-25. Influence de la concentration en DTAC sur le coefficient α apparent des
complexes poly(β-CD)/DTAC/NaDxS 2 () et poly(β-CD)/DTAC/NaDxS 1 () (Nβ-CD =
Nsulfate = 14.10-3 M).
Comme observé pour les complexes poly(β-CD)/DTAC et poly(β-CD)/DTAC/NaDxS
1, il existe une concentration critique en DTAC (NDTAC ~ 2.10-3 M) correspondant à un
changement de compacité du poly(β-CD) au sein des complexes ternaires de NaDxS 2. De

- 229 -
même que pour les complexes de NaDxS 1, αapp reste quasi constant et proche de 2,25 à faible
NDTAC (NDTAC < 2.10-3 M). Par contre, à NDTAC plus élevées, αapp diminue suggérant que les
chaînes de poly(β-CD) sont de moins en moins compactes à mesure que NDTAC augmente. Ce
comportement est difficile à comprendre d’un point de vue où le nombre de connexions entre
le poly(β-CD) et le NaDxS 2 est supposé augmenter avec NDTAC, induisant un enchevêtrement
entre les deux polymères et une compaction de leur structure. La seule façon d’expliquer le
gonflement du poly(β-CD) au sein du système ternaire est donc de supposer la présence de
DTAC non neutralisé le long de sa chaîne (existence de répulsions coulombiennes au sein de
la structure branchée du polymère). Ceci conduit à une nouvelle schématisation de la structure
interne du complexe ternaire pour NDTAC > 2.10-3 M (figure IV-26-b): une structure de type
cœur-coquille, avec un cœur constitué de poly(β-CD) et de DTAC, et une coquille de NaDxS
2 interagissant avec la surface du cœur par association ternaire groupement sulfate/TA
cationique/cavité de β-CD. En effet, il paraît raisonnable que les chaînes de NaDxS 2 soient
trop encombrantes pour pouvoir pénétrer dans le cœur de poly(β-CD): leur rayon de giration
(estimé en supposant une chaîne linéaire) est de l’ordre de 40 Å en solvant θ, ce qui
représente près des ¾ de la taille du poly(β-CD) (Rg = 55 Å).
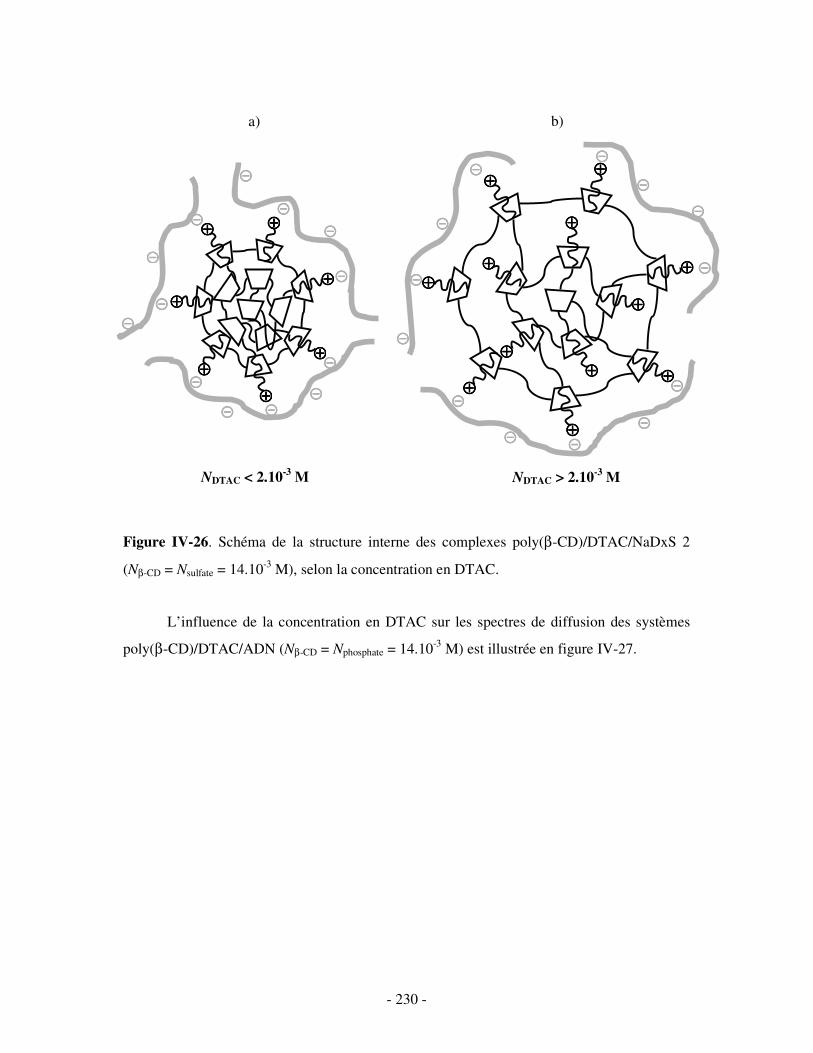
- 230 -
Figure IV-26. Schéma de la structure interne des complexes poly(β-CD)/DTAC/NaDxS 2
(Nβ-CD = Nsulfate = 14.10-3 M), selon la concentration en DTAC.
L’influence de la concentration en DTAC sur les spectres de diffusion des systèmes
poly(β-CD)/DTAC/ADN (Nβ-CD = Nphosphate = 14.10-3 M) est illustrée en figure IV-27.
NDTAC < 2.10-3 M
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
NDTAC > 2.10-3 M
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
⊕⊕⊕⊕
b) a)
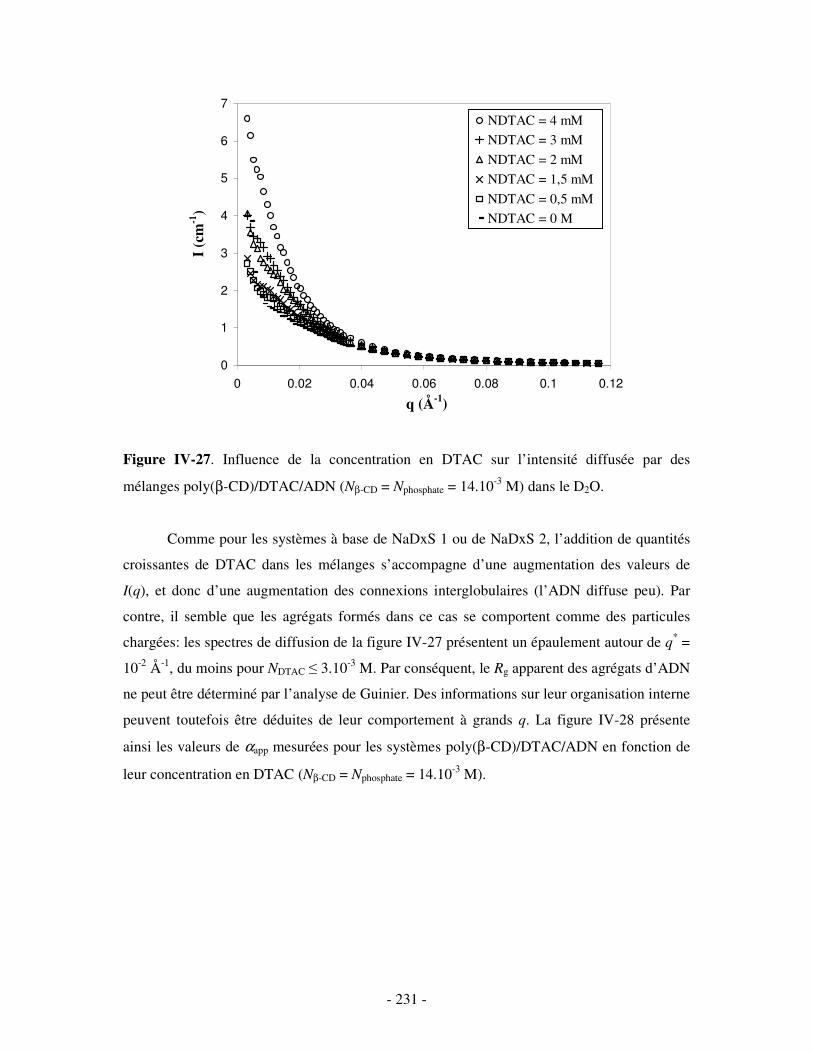
- 231 -
0
1
2
3
4
5
6
7
0 0.02 0.04 0.06 0.08 0.1 0.12
q (Å-1)
I (cm
-1)
NDTAC = 4 mMNDTAC = 3 mMNDTAC = 2 mMNDTAC = 1,5 mMNDTAC = 0,5 mMNDTAC = 0 M
Figure IV-27. Influence de la concentration en DTAC sur l’intensité diffusée par des
mélanges poly(β-CD)/DTAC/ADN (Nβ-CD = Nphosphate = 14.10-3 M) dans le D2O.
Comme pour les systèmes à base de NaDxS 1 ou de NaDxS 2, l’addition de quantités
croissantes de DTAC dans les mélanges s’accompagne d’une augmentation des valeurs de
I(q), et donc d’une augmentation des connexions interglobulaires (l’ADN diffuse peu). Par
contre, il semble que les agrégats formés dans ce cas se comportent comme des particules
chargées: les spectres de diffusion de la figure IV-27 présentent un épaulement autour de q* =
10-2 Å-1, du moins pour NDTAC 3.10-3 M. Par conséquent, le Rg apparent des agrégats d’ADN
ne peut être déterminé par l’analyse de Guinier. Des informations sur leur organisation interne
peuvent toutefois être déduites de leur comportement à grands q. La figure IV-28 présente
ainsi les valeurs de αapp mesurées pour les systèmes poly(β-CD)/DTAC/ADN en fonction de
leur concentration en DTAC (Nβ-CD = Nphosphate = 14.10-3 M).
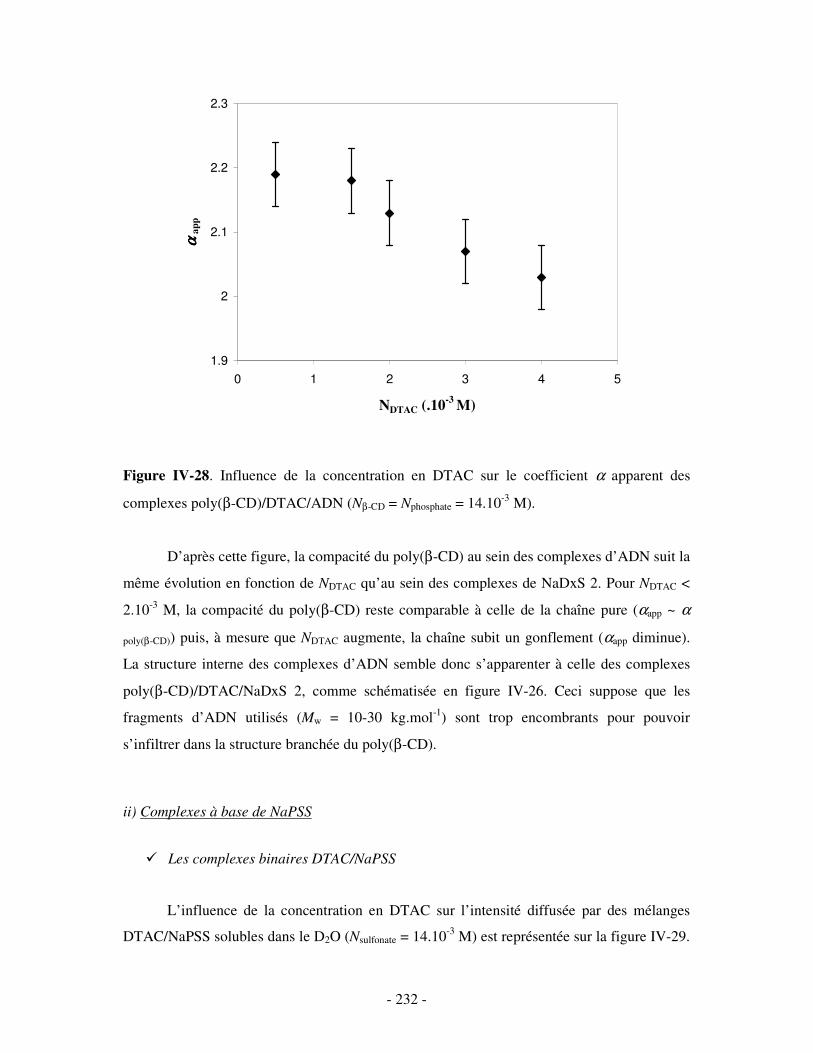
- 232 -
1.9
2
2.1
2.2
2.3
0 1 2 3 4 5
NDTAC (.10-3 M)
αα ααap
p
Figure IV-28. Influence de la concentration en DTAC sur le coefficient α apparent des
complexes poly(β-CD)/DTAC/ADN (Nβ-CD = Nphosphate = 14.10-3 M).
D’après cette figure, la compacité du poly(β-CD) au sein des complexes d’ADN suit la
même évolution en fonction de NDTAC qu’au sein des complexes de NaDxS 2. Pour NDTAC <
2.10-3 M, la compacité du poly(β-CD) reste comparable à celle de la chaîne pure (αapp ~ α
poly(β-CD)) puis, à mesure que NDTAC augmente, la chaîne subit un gonflement (αapp diminue).
La structure interne des complexes d’ADN semble donc s’apparenter à celle des complexes
poly(β-CD)/DTAC/NaDxS 2, comme schématisée en figure IV-26. Ceci suppose que les
fragments d’ADN utilisés (Mw = 10-30 kg.mol-1) sont trop encombrants pour pouvoir
s’infiltrer dans la structure branchée du poly(β-CD).
ii) Complexes à base de NaPSS
Les complexes binaires DTAC/NaPSS
L’influence de la concentration en DTAC sur l’intensité diffusée par des mélanges
DTAC/NaPSS solubles dans le D2O (Nsulfonate = 14.10-3 M) est représentée sur la figure IV-29.
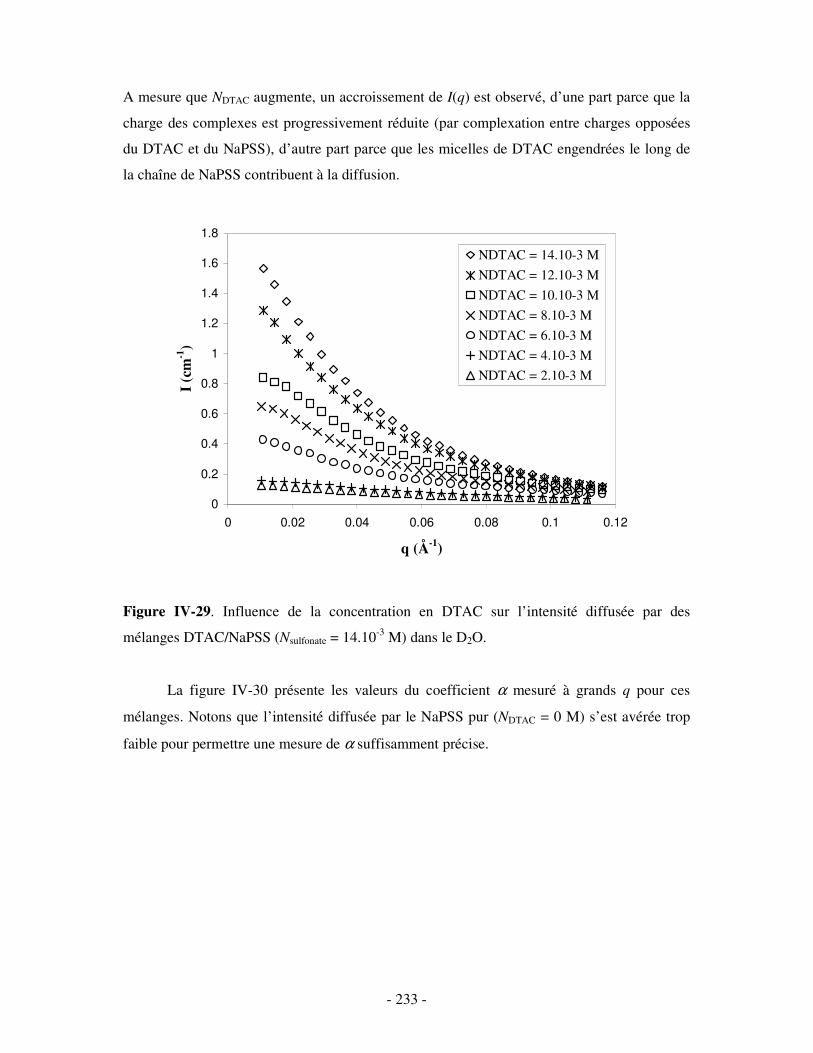
- 233 -
A mesure que NDTAC augmente, un accroissement de I(q) est observé, d’une part parce que la
charge des complexes est progressivement réduite (par complexation entre charges opposées
du DTAC et du NaPSS), d’autre part parce que les micelles de DTAC engendrées le long de
la chaîne de NaPSS contribuent à la diffusion.
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
0 0.02 0.04 0.06 0.08 0.1 0.12
q (Å-1)
I (cm
-1)
NDTAC = 14.10-3 MNDTAC = 12.10-3 MNDTAC = 10.10-3 MNDTAC = 8.10-3 MNDTAC = 6.10-3 MNDTAC = 4.10-3 MNDTAC = 2.10-3 M
Figure IV-29. Influence de la concentration en DTAC sur l’intensité diffusée par des
mélanges DTAC/NaPSS (Nsulfonate = 14.10-3 M) dans le D2O.
La figure IV-30 présente les valeurs du coefficient α mesuré à grands q pour ces
mélanges. Notons que l’intensité diffusée par le NaPSS pur (NDTAC = 0 M) s’est avérée trop
faible pour permettre une mesure de α suffisamment précise.
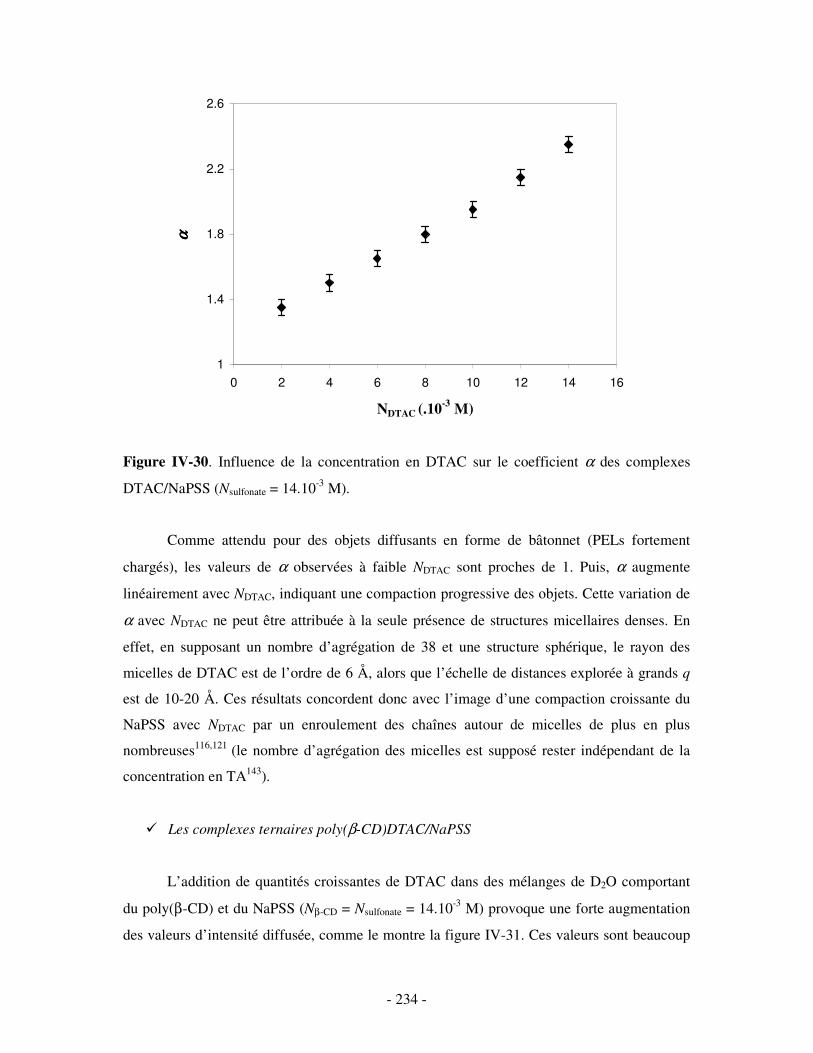
- 234 -
1
1.4
1.8
2.2
2.6
0 2 4 6 8 10 12 14 16
NDTAC (.10-3 M)
αα αα
Figure IV-30. Influence de la concentration en DTAC sur le coefficient α des complexes
DTAC/NaPSS (Nsulfonate = 14.10-3 M).
Comme attendu pour des objets diffusants en forme de bâtonnet (PELs fortement
chargés), les valeurs de α observées à faible NDTAC sont proches de 1. Puis, α augmente
linéairement avec NDTAC, indiquant une compaction progressive des objets. Cette variation de
α avec NDTAC ne peut être attribuée à la seule présence de structures micellaires denses. En
effet, en supposant un nombre d’agrégation de 38 et une structure sphérique, le rayon des
micelles de DTAC est de l’ordre de 6 Å, alors que l’échelle de distances explorée à grands q
est de 10-20 Å. Ces résultats concordent donc avec l’image d’une compaction croissante du
NaPSS avec NDTAC par un enroulement des chaînes autour de micelles de plus en plus
nombreuses116,121 (le nombre d’agrégation des micelles est supposé rester indépendant de la
concentration en TA143).
Les complexes ternaires poly(β-CD)DTAC/NaPSS
L’addition de quantités croissantes de DTAC dans des mélanges de D2O comportant
du poly(β-CD) et du NaPSS (Nβ-CD = Nsulfonate = 14.10-3 M) provoque une forte augmentation
des valeurs d’intensité diffusée, comme le montre la figure IV-31. Ces valeurs sont beaucoup
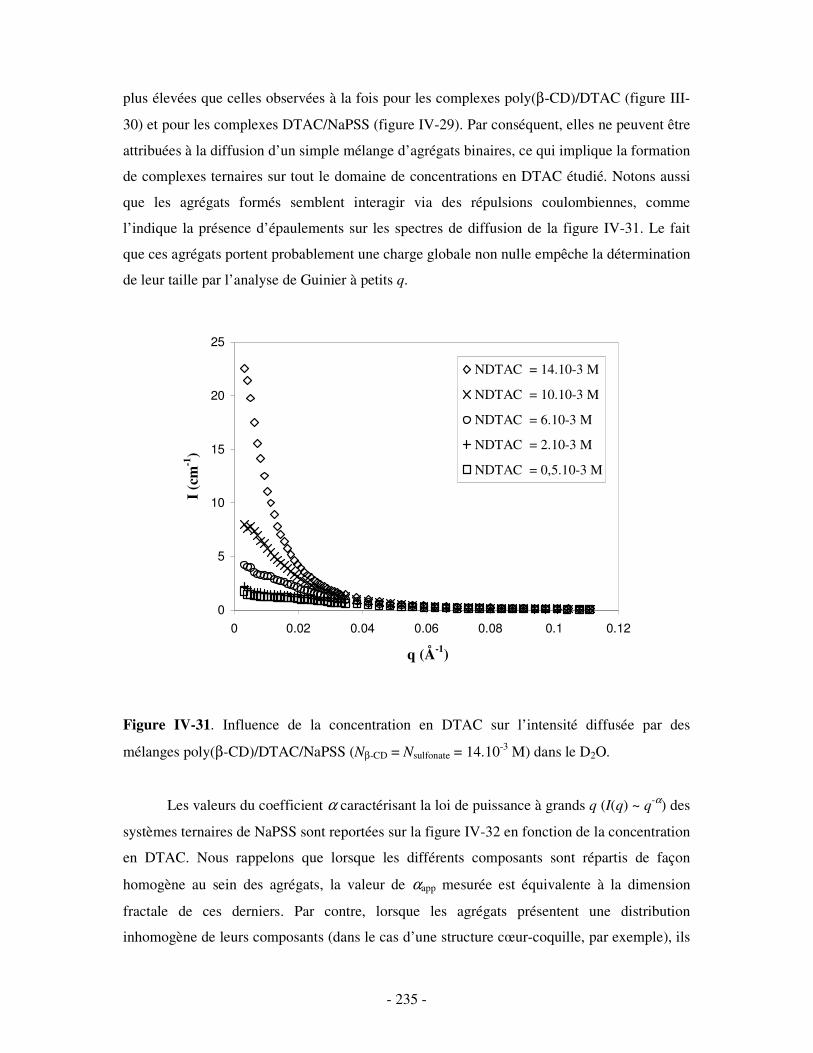
- 235 -
plus élevées que celles observées à la fois pour les complexes poly(β-CD)/DTAC (figure III-
30) et pour les complexes DTAC/NaPSS (figure IV-29). Par conséquent, elles ne peuvent être
attribuées à la diffusion d’un simple mélange d’agrégats binaires, ce qui implique la formation
de complexes ternaires sur tout le domaine de concentrations en DTAC étudié. Notons aussi
que les agrégats formés semblent interagir via des répulsions coulombiennes, comme
l’indique la présence d’épaulements sur les spectres de diffusion de la figure IV-31. Le fait
que ces agrégats portent probablement une charge globale non nulle empêche la détermination
de leur taille par l’analyse de Guinier à petits q.
0
5
10
15
20
25
0 0.02 0.04 0.06 0.08 0.1 0.12
q (Å-1)
I (cm
-1)
NDTAC = 14.10-3 M
NDTAC = 10.10-3 M
NDTAC = 6.10-3 M
NDTAC = 2.10-3 M
NDTAC = 0,5.10-3 M
Figure IV-31. Influence de la concentration en DTAC sur l’intensité diffusée par des
mélanges poly(β-CD)/DTAC/NaPSS (Nβ-CD = Nsulfonate = 14.10-3 M) dans le D2O.
Les valeurs du coefficient α caractérisant la loi de puissance à grands q (I(q) ~ q-α) des
systèmes ternaires de NaPSS sont reportées sur la figure IV-32 en fonction de la concentration
en DTAC. Nous rappelons que lorsque les différents composants sont répartis de façon
homogène au sein des agrégats, la valeur de αapp mesurée est équivalente à la dimension
fractale de ces derniers. Par contre, lorsque les agrégats présentent une distribution
inhomogène de leurs composants (dans le cas d’une structure cœur-coquille, par exemple), ils
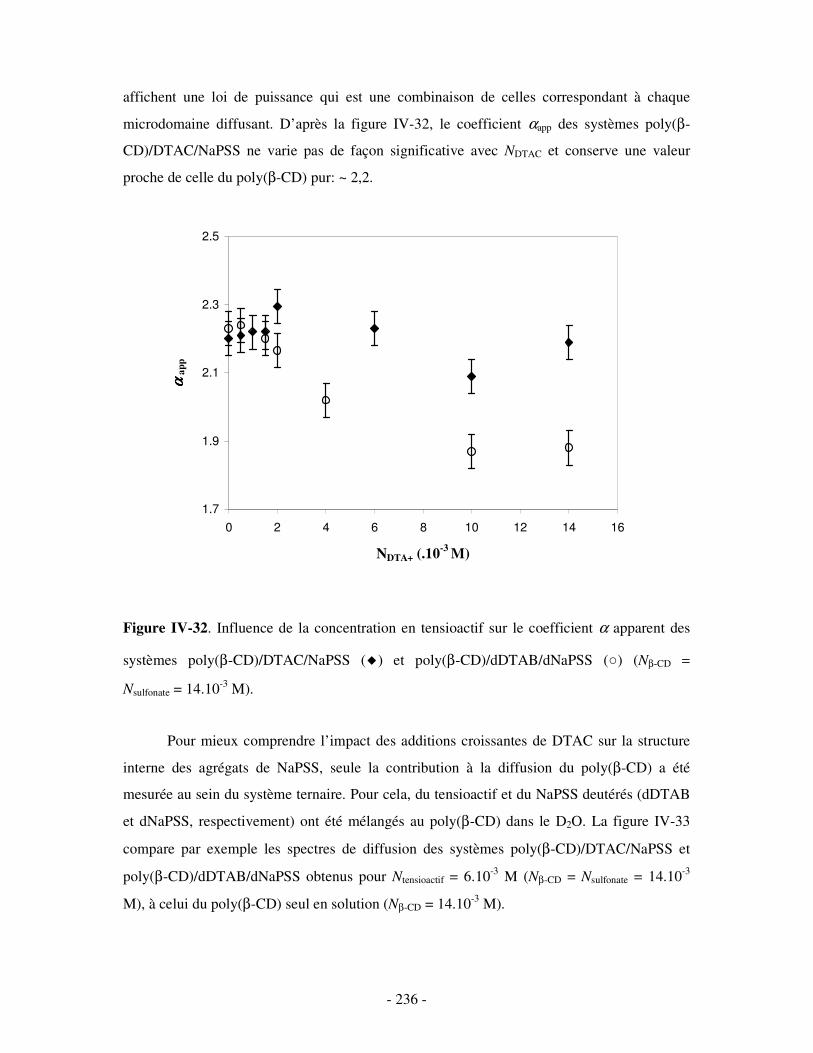
- 236 -
affichent une loi de puissance qui est une combinaison de celles correspondant à chaque
microdomaine diffusant. D’après la figure IV-32, le coefficient αapp des systèmes poly(β-
CD)/DTAC/NaPSS ne varie pas de façon significative avec NDTAC et conserve une valeur
proche de celle du poly(β-CD) pur: ~ 2,2.
1.7
1.9
2.1
2.3
2.5
0 2 4 6 8 10 12 14 16
NDTA+ (.10-3 M)
αα ααap
p
Figure IV-32. Influence de la concentration en tensioactif sur le coefficient α apparent des
systèmes poly(β-CD)/DTAC/NaPSS () et poly(β-CD)/dDTAB/dNaPSS () (Nβ-CD =
Nsulfonate = 14.10-3 M).
Pour mieux comprendre l’impact des additions croissantes de DTAC sur la structure
interne des agrégats de NaPSS, seule la contribution à la diffusion du poly(β-CD) a été
mesurée au sein du système ternaire. Pour cela, du tensioactif et du NaPSS deutérés (dDTAB
et dNaPSS, respectivement) ont été mélangés au poly(β-CD) dans le D2O. La figure IV-33
compare par exemple les spectres de diffusion des systèmes poly(β-CD)/DTAC/NaPSS et
poly(β-CD)/dDTAB/dNaPSS obtenus pour Ntensioactif = 6.10-3 M (Nβ-CD = Nsulfonate = 14.10-3
M), à celui du poly(β-CD) seul en solution (Nβ-CD = 14.10-3 M).
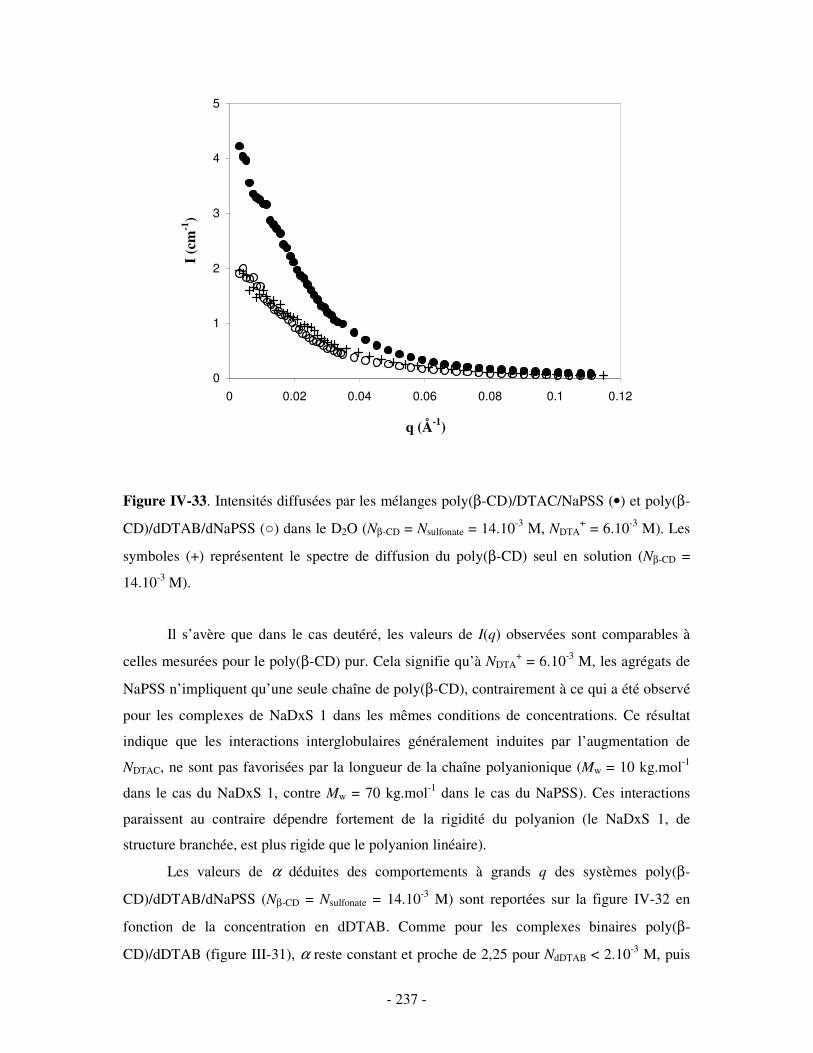
- 237 -
0
1
2
3
4
5
0 0.02 0.04 0.06 0.08 0.1 0.12
q (Å-1)
I (cm
-1)
Figure IV-33. Intensités diffusées par les mélanges poly(β-CD)/DTAC/NaPSS (•) et poly(β-
CD)/dDTAB/dNaPSS () dans le D2O (Nβ-CD = Nsulfonate = 14.10-3 M, NDTA+ = 6.10-3 M). Les
symboles (+) représentent le spectre de diffusion du poly(β-CD) seul en solution (Nβ-CD =
14.10-3 M).
Il s’avère que dans le cas deutéré, les valeurs de I(q) observées sont comparables à
celles mesurées pour le poly(β-CD) pur. Cela signifie qu’à NDTA+ = 6.10-3 M, les agrégats de
NaPSS n’impliquent qu’une seule chaîne de poly(β-CD), contrairement à ce qui a été observé
pour les complexes de NaDxS 1 dans les mêmes conditions de concentrations. Ce résultat
indique que les interactions interglobulaires généralement induites par l’augmentation de
NDTAC, ne sont pas favorisées par la longueur de la chaîne polyanionique (Mw = 10 kg.mol-1
dans le cas du NaDxS 1, contre Mw = 70 kg.mol-1 dans le cas du NaPSS). Ces interactions
paraissent au contraire dépendre fortement de la rigidité du polyanion (le NaDxS 1, de
structure branchée, est plus rigide que le polyanion linéaire).
Les valeurs de α déduites des comportements à grands q des systèmes poly(β-
CD)/dDTAB/dNaPSS (Nβ-CD = Nsulfonate = 14.10-3 M) sont reportées sur la figure IV-32 en
fonction de la concentration en dDTAB. Comme pour les complexes binaires poly(β-
CD)/dDTAB (figure III-31), α reste constant et proche de 2,25 pour NdDTAB < 2.10-3 M, puis

- 238 -
diminue jusqu’à des valeurs voisines de 1,9 à NdDTAB plus élevées. En d’autres termes, les
chaînes de poly(β-CD) impliquées dans le complexe ternaire de dNaPSS et dans le complexe
binaire poly(β-CD)/dDTAB sont sujettes au même gonflement à NdDTAB élevées. Il s’en
déduit que pour NdDTAB < 2.10-3 M, la structure du complexe ternaire de dNaPSS s’apparente
à la structure cœur-coquille schématisée en figure IV-26-a qui n’implique aucune compaction
ni aucun gonflement du poly(β-CD): les molécules de dDTAB forment un corona autour du
cœur de poly(β-CD) et les chaînes de dNaPSS interagissent avec le cœur uniquement via ce
corona cationique. Pour NdDTAB > 2.10-3 M, le complexe ternaire semble par contre adopter
une structure cœur-coquille similaire à celle de la figure IV-26-b, où le cœur de poly(β-CD)
est gonflé par les molécules de dDTAB non neutralisées. Ceci suppose, dans ce cas, que la
chaîne de dNaPSS (Rg ~ 35 Å en solvant θ) est trop grande pour pouvoir pénétrer dans la
structure branchée du cœur de poly(β-CD) (Rg ~ 55 Å).
b) Etude par DLS et par viscosimétrie
i) Par DLS
La figure IV-34 présente par exemple la fonction d’autocorrélation mesurée pour le
mélange aqueux poly(β-CD)/DTAC/NaDxS 1 (Nβ-CD = Nsulfate = 14.10-3 M, NDTAC = 6.10-3
M). Comme il peut être constaté, un ajustement adéquat est obtenu entre les données
expérimentales et les données calculées par la méthode des cumulants avec une relaxation
monoexponentielle caractérisée par un temps de relaxation moyen Γ . Notons qu’un
comportement comparable a été observé pour tous les mélanges poly(β-CD)/DTAC/NaDxS 1
et poly(β-CD)/DTAC/NaDxS 2 étudiés (avec NDTAC variables). Le fait d’obtenir un bon
ajustement des valeurs expérimentales avec une relaxation monoexponentielle montre que la
diffusion est essentiellement due à une seule espèce présente en solution: les complexes
ternaires. Leurs rayons hydrodynamiques moyens ont donc été calculés à partir des valeurs de
Γ et de la relation de Stokes-Einstein (équation 5).
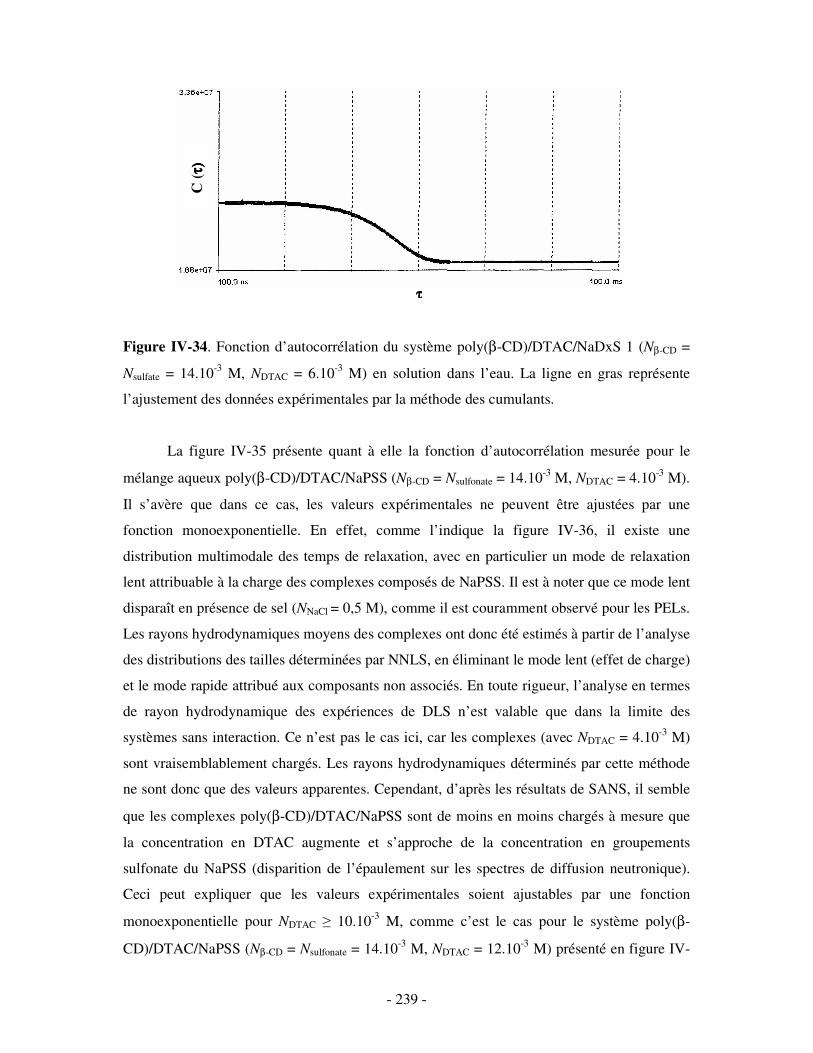
- 239 -
Figure IV-34. Fonction d’autocorrélation du système poly(β-CD)/DTAC/NaDxS 1 (Nβ-CD =
Nsulfate = 14.10-3 M, NDTAC = 6.10-3 M) en solution dans l’eau. La ligne en gras représente
l’ajustement des données expérimentales par la méthode des cumulants.
La figure IV-35 présente quant à elle la fonction d’autocorrélation mesurée pour le
mélange aqueux poly(β-CD)/DTAC/NaPSS (Nβ-CD = Nsulfonate = 14.10-3 M, NDTAC = 4.10-3 M).
Il s’avère que dans ce cas, les valeurs expérimentales ne peuvent être ajustées par une
fonction monoexponentielle. En effet, comme l’indique la figure IV-36, il existe une
distribution multimodale des temps de relaxation, avec en particulier un mode de relaxation
lent attribuable à la charge des complexes composés de NaPSS. Il est à noter que ce mode lent
disparaît en présence de sel (NNaCl = 0,5 M), comme il est couramment observé pour les PELs.
Les rayons hydrodynamiques moyens des complexes ont donc été estimés à partir de l’analyse
des distributions des tailles déterminées par NNLS, en éliminant le mode lent (effet de charge)
et le mode rapide attribué aux composants non associés. En toute rigueur, l’analyse en termes
de rayon hydrodynamique des expériences de DLS n’est valable que dans la limite des
systèmes sans interaction. Ce n’est pas le cas ici, car les complexes (avec NDTAC = 4.10-3 M)
sont vraisemblablement chargés. Les rayons hydrodynamiques déterminés par cette méthode
ne sont donc que des valeurs apparentes. Cependant, d’après les résultats de SANS, il semble
que les complexes poly(β-CD)/DTAC/NaPSS sont de moins en moins chargés à mesure que
la concentration en DTAC augmente et s’approche de la concentration en groupements
sulfonate du NaPSS (disparition de l’épaulement sur les spectres de diffusion neutronique).
Ceci peut expliquer que les valeurs expérimentales soient ajustables par une fonction
monoexponentielle pour NDTAC 10.10-3 M, comme c’est le cas pour le système poly(β-
CD)/DTAC/NaPSS (Nβ-CD = Nsulfonate = 14.10-3 M, NDTAC = 12.10-3 M) présenté en figure IV-
ττττ
C ( τ
)τ) τ)τ)
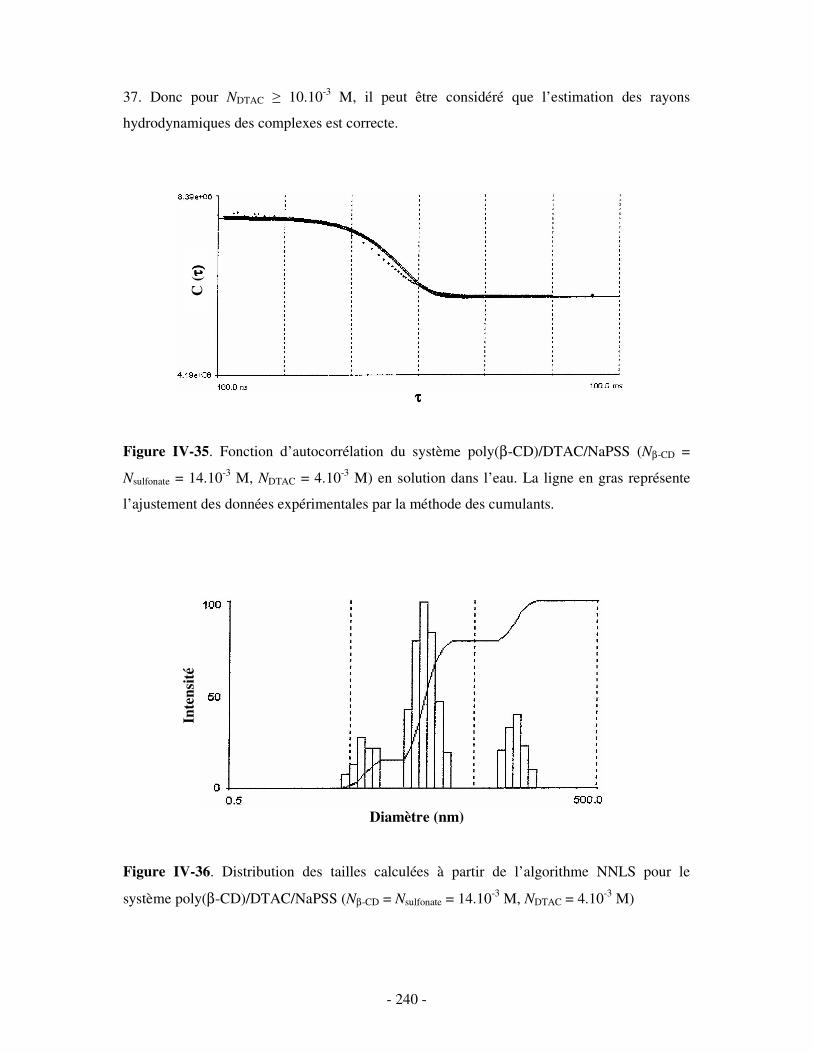
- 240 -
37. Donc pour NDTAC 10.10-3 M, il peut être considéré que l’estimation des rayons
hydrodynamiques des complexes est correcte.
Figure IV-35. Fonction d’autocorrélation du système poly(β-CD)/DTAC/NaPSS (Nβ-CD =
Nsulfonate = 14.10-3 M, NDTAC = 4.10-3 M) en solution dans l’eau. La ligne en gras représente
l’ajustement des données expérimentales par la méthode des cumulants.
Figure IV-36. Distribution des tailles calculées à partir de l’algorithme NNLS pour le
système poly(β-CD)/DTAC/NaPSS (Nβ-CD = Nsulfonate = 14.10-3 M, NDTAC = 4.10-3 M)
ττττ
C ( τ
)τ) τ)τ)
Diamètre (nm)
Inte
nsité
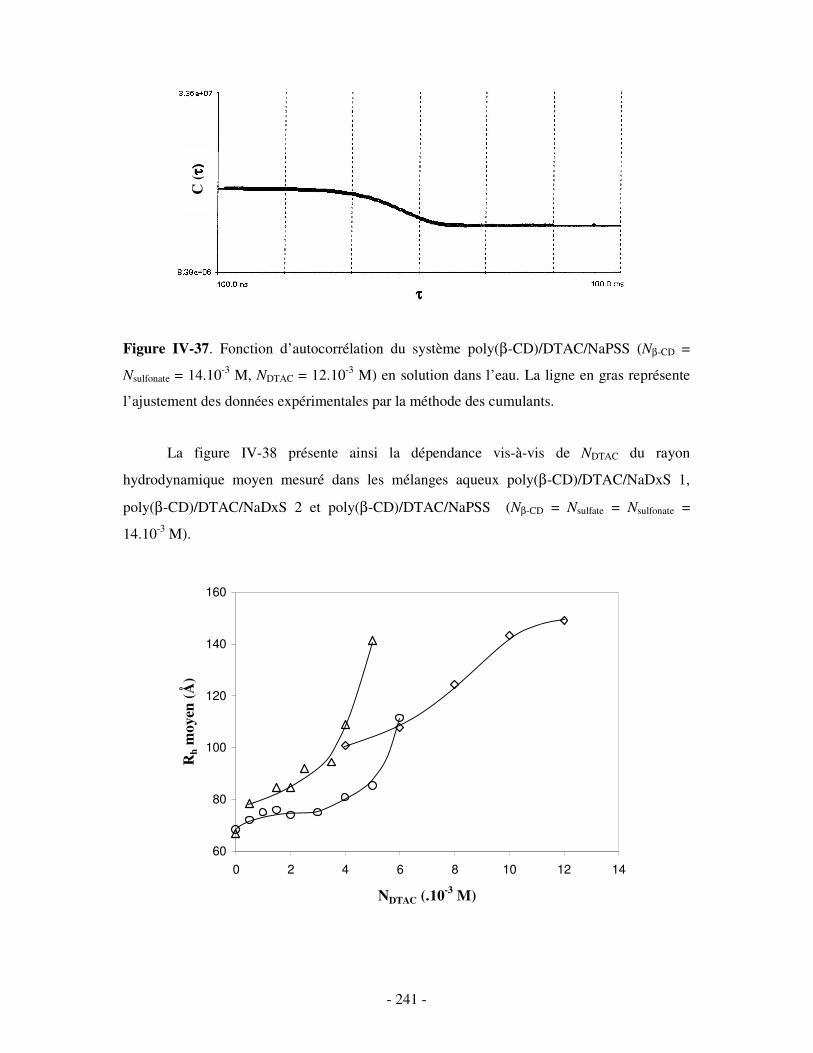
- 241 -
Figure IV-37. Fonction d’autocorrélation du système poly(β-CD)/DTAC/NaPSS (Nβ-CD =
Nsulfonate = 14.10-3 M, NDTAC = 12.10-3 M) en solution dans l’eau. La ligne en gras représente
l’ajustement des données expérimentales par la méthode des cumulants.
La figure IV-38 présente ainsi la dépendance vis-à-vis de NDTAC du rayon
hydrodynamique moyen mesuré dans les mélanges aqueux poly(β-CD)/DTAC/NaDxS 1,
poly(β-CD)/DTAC/NaDxS 2 et poly(β-CD)/DTAC/NaPSS (Nβ-CD = Nsulfate = Nsulfonate =
14.10-3 M).
60
80
100
120
140
160
0 2 4 6 8 10 12 14
NDTAC (.10-3 M)
Rh m
oyen
(Å)
C ( τ
)τ) τ)τ)
ττττ

- 242 -
Figure IV-38. Influence de NDTAC sur le Rh moyen des systèmes poly(β-CD)/DTAC/NaDxS 1
(), poly(β-CD)/DTAC/NaDxS 2 () et poly(β-CD)/DTAC/NaPSS ( ) (Nβ-CD = Nsulfate =
Nsulfonate = 14.10-3 M). Les lignes en trait plein sont un guide pour l’œil.
Comme il l’a été évoqué auparavant, une agrégation en solution est observée à mesure
que la concentration en DTAC connecteur augmente. Cette agrégation paraît plus importante
et plus rapide dans le cas du NaDxS 2 qui, parmi les trois polyanions utilisés, s’avère être le
plus rigide (le cas de l’ADN n’a pas été traité). D’après Dautzenberg et coll.77,98, le niveau
d’agrégation des particules de PECs classiques (constitués d’un polyanion et d’un polycation)
résulte d’une compétition entre deux processus différents de neutralisation des charges: par
une adaptation conformationnelle appropriée entre deux chaînes de charges opposées, ou par
incorporation au sein du complexe de plusieurs macromolécules chargées. Il apparaît, d’après
la figure IV-38, que la compensation des charges par des changements conformationnels soit
très peu facilitée dans le cas du NaDxS 2 rigide (niveaux d’agrégation élevés). Au contraire,
ce mécanisme semble être favorisé dans le cas du NaDxS 1 moins branché (niveaux
d’agrégation plus faibles) et, surtout, dans le cas du NaPSS flexible: à mêmes concentrations
en DTAC (NDTAC = 6.10-3 M), les agrégats de NaDxS 1 comportent plusieurs chaînes de
poly(β-CD) (d’après les résultats de SANS), alors que ceux composés de NaPSS n’en
comportent qu’une seule. Il est à noter que dans ce cas (NDTAC = 6.10-3 M), le Rh moyen
mesuré pour le système poly(β-CD)/DTAC/NaPSS (à une seule chaîne de poly(β-CD)) est
peu éloigné de celui du système poly(β-CD)/DTAC/NaDxS 1 correspondant (à plusieurs
chaînes de poly(β-CD)). Dans la structure cœur-coquille du complexe ternaire de NaPSS
(figure IV-26-b), le cœur de poly(β-CD) paraît donc entouré d’une coquille de NaPSS très
gonflée. Ceci rend plus compréhensible le fait que, comparés aux complexes de NaDxS 1 ou
de NaDxS 2, les agrégats impliquant le NaPSS (dont le squelette est de nature hydrophobe)
soient solubles sur une plus large gamme de valeurs de NDTAC: les répulsions électrostatiques
engendrées par les coquilles gonflées de NaPSS les aident à se maintenir à l’état colloïdal
jusqu’à ce que l’électroneutralité soit atteinte (NDTAC = Nsulfonate = 14.10-3 M).
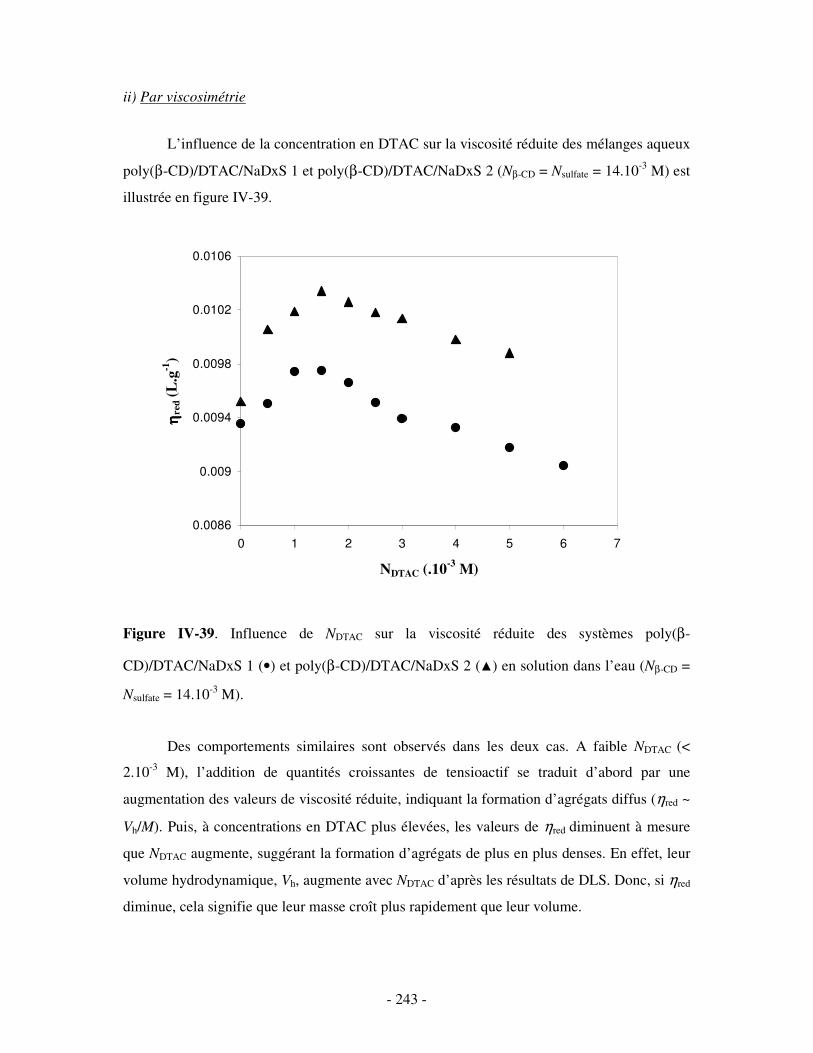
- 243 -
ii) Par viscosimétrie
L’influence de la concentration en DTAC sur la viscosité réduite des mélanges aqueux
poly(β-CD)/DTAC/NaDxS 1 et poly(β-CD)/DTAC/NaDxS 2 (Nβ-CD = Nsulfate = 14.10-3 M) est
illustrée en figure IV-39.
0.0086
0.009
0.0094
0.0098
0.0102
0.0106
0 1 2 3 4 5 6 7
NDTAC (.10-3 M)
ηη ηη red
(L.g
-1)
Figure IV-39. Influence de NDTAC sur la viscosité réduite des systèmes poly(β-
CD)/DTAC/NaDxS 1 (•) et poly(β-CD)/DTAC/NaDxS 2 () en solution dans l’eau (Nβ-CD =
Nsulfate = 14.10-3 M).
Des comportements similaires sont observés dans les deux cas. A faible NDTAC (<
2.10-3 M), l’addition de quantités croissantes de tensioactif se traduit d’abord par une
augmentation des valeurs de viscosité réduite, indiquant la formation d’agrégats diffus (ηred ~
Vh/M). Puis, à concentrations en DTAC plus élevées, les valeurs de ηred diminuent à mesure
que NDTAC augmente, suggérant la formation d’agrégats de plus en plus denses. En effet, leur
volume hydrodynamique, Vh, augmente avec NDTAC d’après les résultats de DLS. Donc, si ηred
diminue, cela signifie que leur masse croît plus rapidement que leur volume.
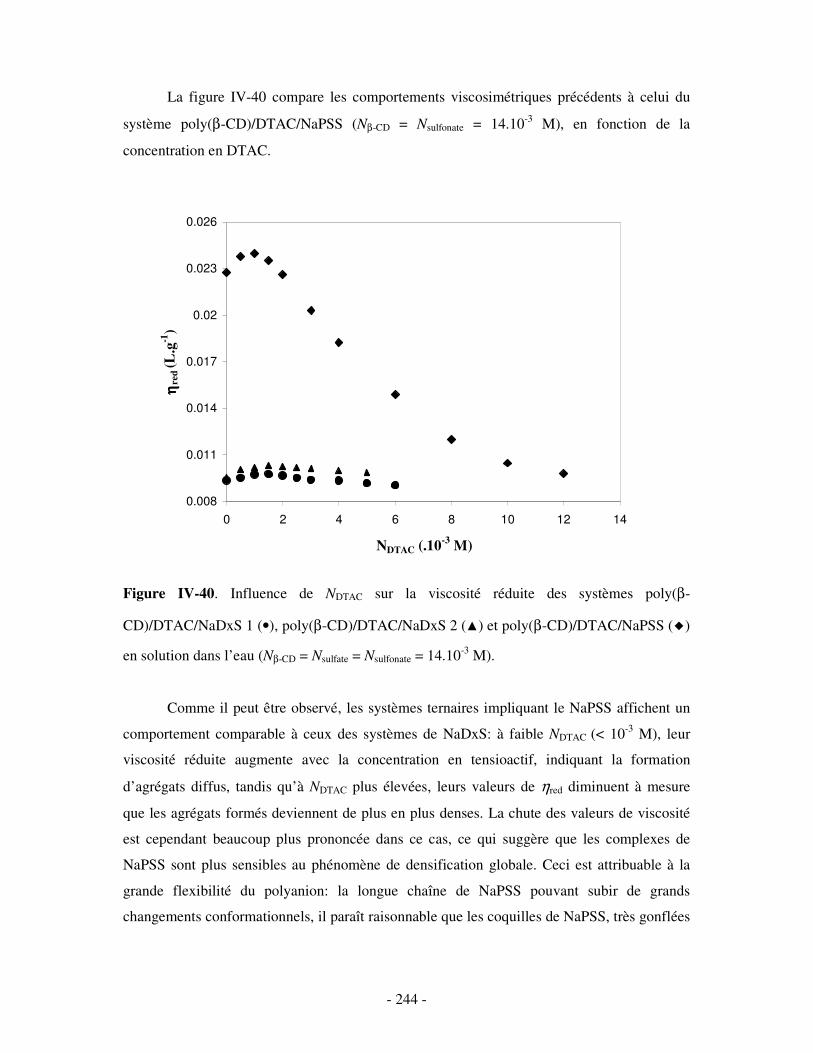
- 244 -
La figure IV-40 compare les comportements viscosimétriques précédents à celui du
système poly(β-CD)/DTAC/NaPSS (Nβ-CD = Nsulfonate = 14.10-3 M), en fonction de la
concentration en DTAC.
0.008
0.011
0.014
0.017
0.02
0.023
0.026
0 2 4 6 8 10 12 14
NDTAC (.10-3 M)
ηη ηηre
d (L
.g-1
)
Figure IV-40. Influence de NDTAC sur la viscosité réduite des systèmes poly(β-
CD)/DTAC/NaDxS 1 (•), poly(β-CD)/DTAC/NaDxS 2 () et poly(β-CD)/DTAC/NaPSS ()
en solution dans l’eau (Nβ-CD = Nsulfate = Nsulfonate = 14.10-3 M).
Comme il peut être observé, les systèmes ternaires impliquant le NaPSS affichent un
comportement comparable à ceux des systèmes de NaDxS: à faible NDTAC (< 10-3 M), leur
viscosité réduite augmente avec la concentration en tensioactif, indiquant la formation
d’agrégats diffus, tandis qu’à NDTAC plus élevées, leurs valeurs de ηred diminuent à mesure
que les agrégats formés deviennent de plus en plus denses. La chute des valeurs de viscosité
est cependant beaucoup plus prononcée dans ce cas, ce qui suggère que les complexes de
NaPSS sont plus sensibles au phénomène de densification globale. Ceci est attribuable à la
grande flexibilité du polyanion: la longue chaîne de NaPSS pouvant subir de grands
changements conformationnels, il paraît raisonnable que les coquilles de NaPSS, très gonflées

- 245 -
à faibles NDTAC, subissent une forte compaction à mesure que la concentration en DTAC
connecteur augmente (d’autant plus que la force ionique augmente avec NDTAC).
c) Bilan
Les résultats de SANS à petits q, de DLS et de viscosimétrie mettent en évidence des
comportements macroscopiques comparables pour les différents complexes poly(β-
CD)/DTAC/polyanion étudiés. Quelque soit le polyanion impliqué, une augmentation de la
concentration en DTAC dans les systèmes ternaires se manifeste, d’une part, par une
agrégation en solution (marquée par une augmentation des valeurs d’intensité diffusée et des
valeurs de Rg et Rh) et, d’autre part, par une transition structurale entre des agrégats diffus et
des agrégats de plus en plus denses (transition marquée par une chute de viscosité pour NDTAC
> 2.10-3 M).
Des différences comportementales apparaissent cependant selon la rigidité du
polyanion utilisé. Plus la chaîne polyanionique est rigide (dans l’ordre NaPSS < NaDxS 1 <
NaDxS 2), plus l’agrégation en solution est favorisée, conduisant à une précipitation plus
rapide des systèmes: les complexes de NaDxS 2, NaDxS 1 et NaPSS précipitent
respectivement pour NDTAC égale à 6, 7 et 14.10-3 M (Nβ-CD = Nsulfate = Nsulfonate = 14.10-3 M).
Des niveaux d’agrégation très différents ont ainsi été observés à NDTAC = 6.10-3 M pour les
complexes ternaires impliquant le NaDxS 1 et le NaPSS: les premiers renferment plusieurs
chaînes de poly(β-CD), tandis que les seconds n’en contiennent qu’une seule. Au contraire,
plus le polyanion est flexible, plus les agrégats subissent une densification de leur structure
globale à mesure que NDTAC augmente. Les complexes composés de NaPSS montrent ainsi
une chute relative de viscosité plus conséquente à NDTAC élevées.
L’influence de la concentration en DTAC sur la structure interne des complexes
ternaires a pu aussi être étudiée par SANS, à l’aide de l’analyse à grands q et, le cas échéant, à
l’aide d’une méthode de variation de contraste (en utilisant du DTAB et du NaPSS deutérés).
Il s’avère que, quelque soit le polyanion employé (NaDxS 1 ou 2, ADN, NaPSS), les agrégats
formés par association ternaire à NDTAC < 2.10-3 M présentent une structure interne de type
cœur-coquille, avec un cœur de poly(β-CD) et une coquille polyanionique. A NDTAC plus
élevées, par contre, la structure interne des agrégats est fortement influencée par la longueur
et l’architecture du polyanion, selon sa capacité à pénétrer ou non le cœur branché de poly(β-
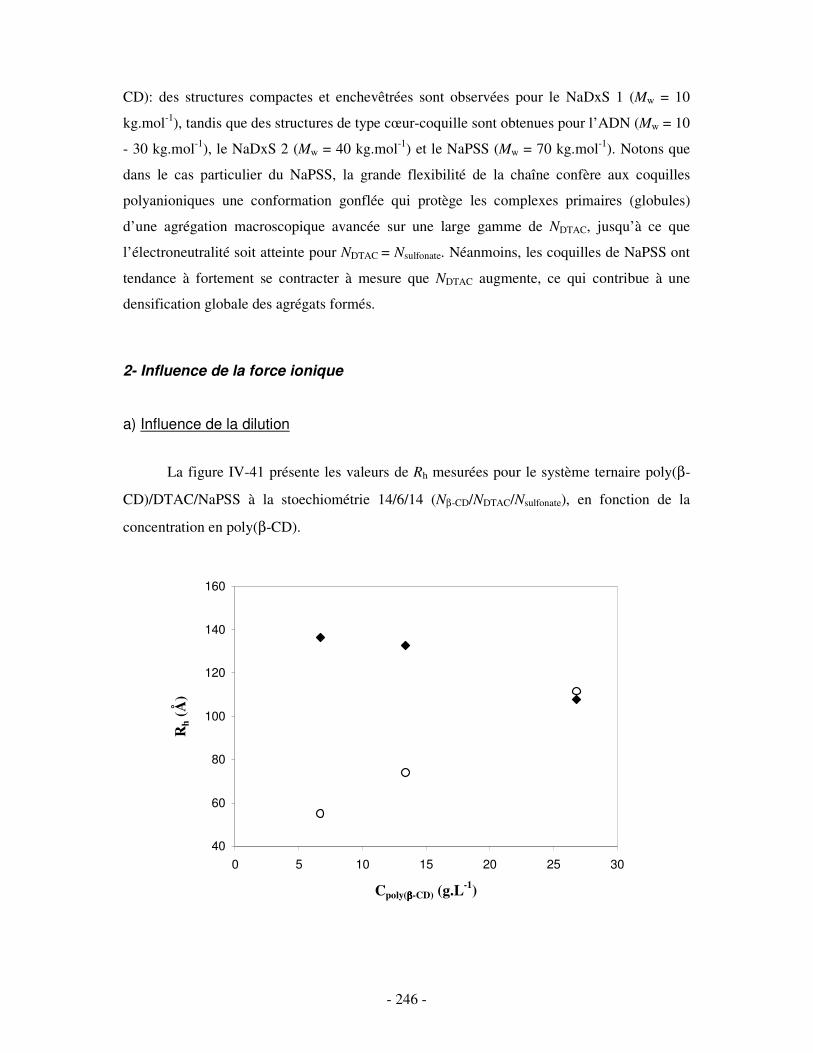
- 246 -
CD): des structures compactes et enchevêtrées sont observées pour le NaDxS 1 (Mw = 10
kg.mol-1), tandis que des structures de type cœur-coquille sont obtenues pour l’ADN (Mw = 10
- 30 kg.mol-1), le NaDxS 2 (Mw = 40 kg.mol-1) et le NaPSS (Mw = 70 kg.mol-1). Notons que
dans le cas particulier du NaPSS, la grande flexibilité de la chaîne confère aux coquilles
polyanioniques une conformation gonflée qui protège les complexes primaires (globules)
d’une agrégation macroscopique avancée sur une large gamme de NDTAC, jusqu’à ce que
l’électroneutralité soit atteinte pour NDTAC = Nsulfonate. Néanmoins, les coquilles de NaPSS ont
tendance à fortement se contracter à mesure que NDTAC augmente, ce qui contribue à une
densification globale des agrégats formés.
2- Influence de la force ionique
a) Influence de la dilution
La figure IV-41 présente les valeurs de Rh mesurées pour le système ternaire poly(β-
CD)/DTAC/NaPSS à la stoechiométrie 14/6/14 (Nβ-CD/NDTAC/Nsulfonate), en fonction de la
concentration en poly(β-CD).
40
60
80
100
120
140
160
0 5 10 15 20 25 30
Cpoly(ββββ-CD) (g.L-1)
Rh (
Å)

- 247 -
Figure IV-41. Influence de la dilution sur les valeurs de Rh mesurées pour les systèmes
poly(β-CD)/DTAC/NaPSS () et poly(β-CD)/DTAC/NaDxS 1 () (Nβ-CD/NDTAC/Nanion =
14/6/14) en solution dans l’eau.
Comme il peut être constaté, la taille des agrégats de NaPSS augmente par dilution,
indiquant un effet de charge à mesure que la force ionique du milieu diminue. Ce résultat
concorde bien avec la description du complexe ternaire de NaPSS donnée précédemment: le
gonflement observé peut être attribué à la coquille de NaPSS qui est supposée entourer le
cœur de poly(β-CD) et protéger le globule résultant contre une agrégation macroscopique (il a
été démontré qu’à la stoechiométrie 14/6/14, le complexe de NaPSS ne comporte qu’une
seule chaîne de poly(β-CD)).
Pour comparaison, les valeurs de Rh mesurées pour le complexe poly(β-
CD)/DTAC/NaDxS 1 à la même stoechiométrie 14/6/14 sont reportées sur la figure IV-41. Un
comportement totalement différent est observé dans ce cas: la taille des agrégats de NaDxS 1
décroît par dilution, indiquant une diminution du nombre de composants au sein des agrégats.
Ce résultat est aussi en accord avec l’image décrite pour la structure globale du complexe de
NaDxS 1. En effet, la chute des valeurs de Rh observée par dilution apparaît comme la
conséquence de la réduction du nombre de chaînes de poly(β-CD) dans le système. Ceci
suggère qu’à Cpoly(β-CD) = 26,8 g.L-1(avant la dilution), le complexe de NaDxS 1 implique
plusieurs chaînes de poly(β-CD), comme il l’a été supposé. Notons qu’à forte dilution (Cpoly(β-
CD) = 6,7 g.L-1), le complexe ternaire reste tout de même associé, comme indiqué par les
résultats de SANS présentés en figure IV-15.
b) Influence de la concentration en sel
Complexes à base de NaDxS et d’ADN
Les figures IV-42, IV-43 et IV-44 montrent respectivement l’influence du sel sur les
spectres de diffusion des systèmes ternaires composés de NaDxS 1 (avec NDTAC = 6.10-3 M),
de NaDxS 2 (avec NDTAC = 5.10-3 M) et d’ADN (avec NDTAC = 4.10-3 M), en solution dans le
D2O (Nβ-CD = Nanion = 14.10-3 M).
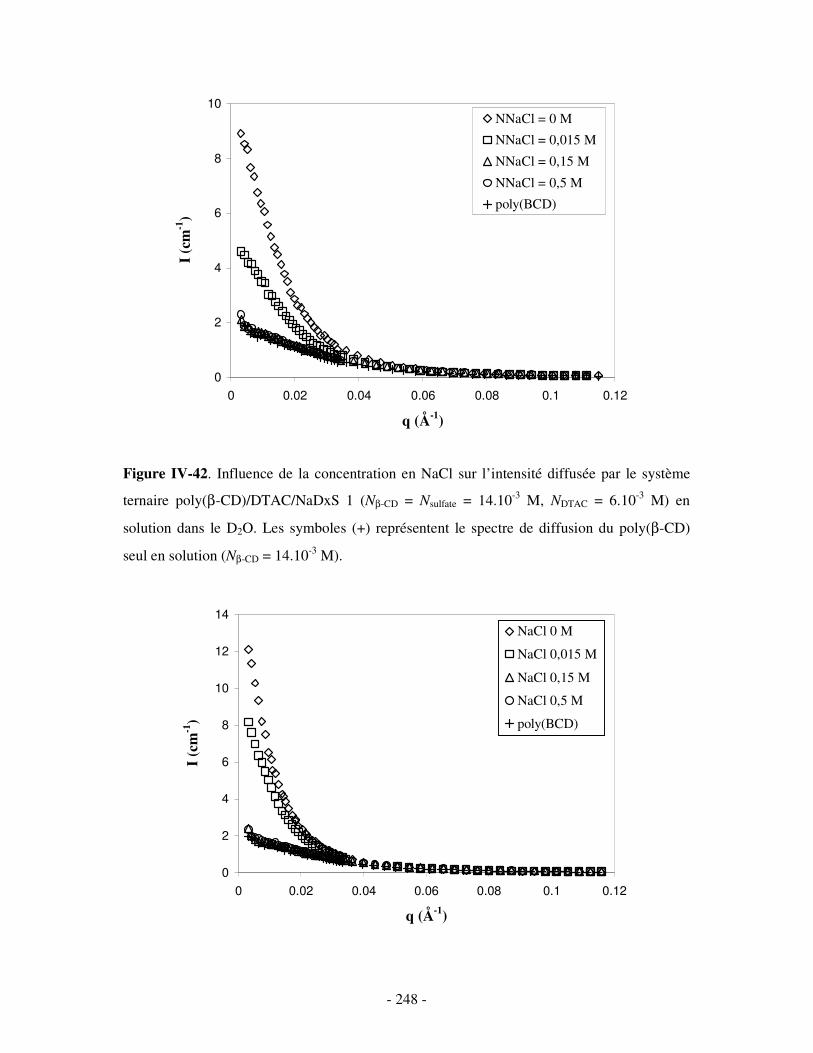
- 248 -
0
2
4
6
8
10
0 0.02 0.04 0.06 0.08 0.1 0.12
q (Å-1)
I (cm
-1)
NNaCl = 0 M
NNaCl = 0,015 M
NNaCl = 0,15 M
NNaCl = 0,5 M
poly(BCD)
Figure IV-42. Influence de la concentration en NaCl sur l’intensité diffusée par le système
ternaire poly(β-CD)/DTAC/NaDxS 1 (Nβ-CD = Nsulfate = 14.10-3 M, NDTAC = 6.10-3 M) en
solution dans le D2O. Les symboles (+) représentent le spectre de diffusion du poly(β-CD)
seul en solution (Nβ-CD = 14.10-3 M).
0
2
4
6
8
10
12
14
0 0.02 0.04 0.06 0.08 0.1 0.12
q (Å-1)
I (cm
-1)
NaCl 0 M
NaCl 0,015 M
NaCl 0,15 M
NaCl 0,5 M
poly(BCD)
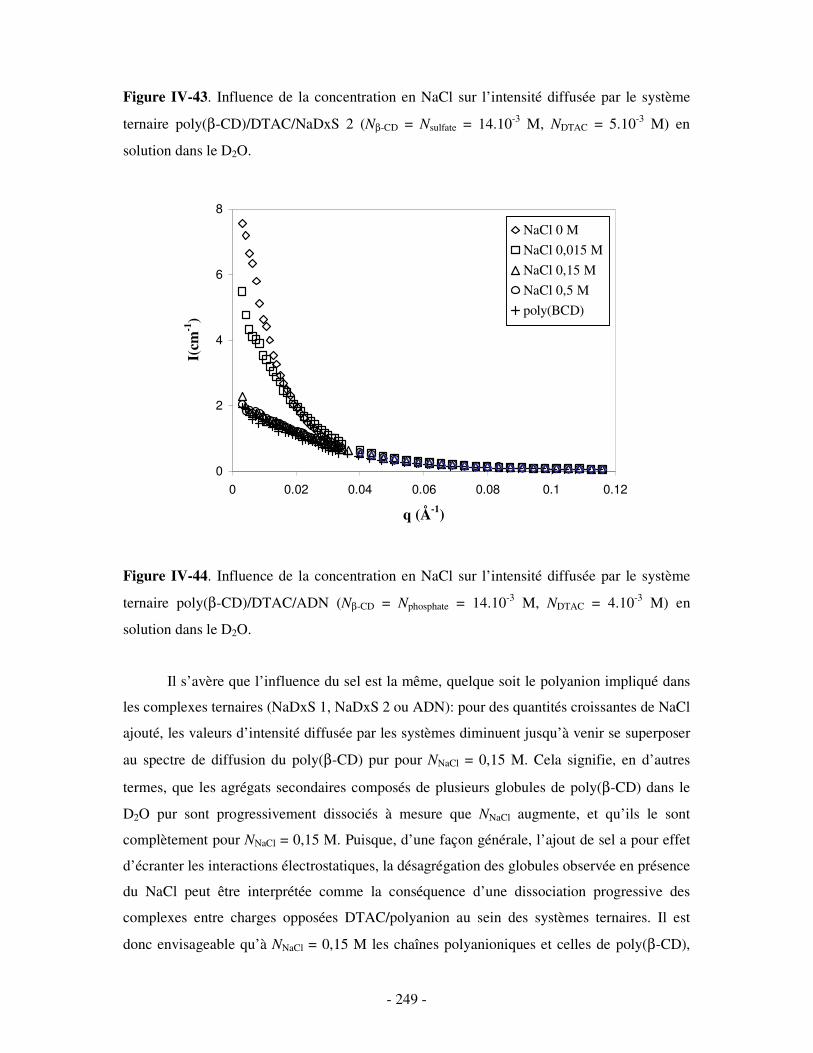
- 249 -
Figure IV-43. Influence de la concentration en NaCl sur l’intensité diffusée par le système
ternaire poly(β-CD)/DTAC/NaDxS 2 (Nβ-CD = Nsulfate = 14.10-3 M, NDTAC = 5.10-3 M) en
solution dans le D2O.
0
2
4
6
8
0 0.02 0.04 0.06 0.08 0.1 0.12
q (Å-1)
I(cm
-1)
NaCl 0 MNaCl 0,015 MNaCl 0,15 MNaCl 0,5 Mpoly(BCD)
7.83E-01
0.67586
Figure IV-44. Influence de la concentration en NaCl sur l’intensité diffusée par le système
ternaire poly(β-CD)/DTAC/ADN (Nβ-CD = Nphosphate = 14.10-3 M, NDTAC = 4.10-3 M) en
solution dans le D2O.
Il s’avère que l’influence du sel est la même, quelque soit le polyanion impliqué dans
les complexes ternaires (NaDxS 1, NaDxS 2 ou ADN): pour des quantités croissantes de NaCl
ajouté, les valeurs d’intensité diffusée par les systèmes diminuent jusqu’à venir se superposer
au spectre de diffusion du poly(β-CD) pur pour NNaCl = 0,15 M. Cela signifie, en d’autres
termes, que les agrégats secondaires composés de plusieurs globules de poly(β-CD) dans le
D2O pur sont progressivement dissociés à mesure que NNaCl augmente, et qu’ils le sont
complètement pour NNaCl = 0,15 M. Puisque, d’une façon générale, l’ajout de sel a pour effet
d’écranter les interactions électrostatiques, la désagrégation des globules observée en présence
du NaCl peut être interprétée comme la conséquence d’une dissociation progressive des
complexes entre charges opposées DTAC/polyanion au sein des systèmes ternaires. Il est
donc envisageable qu’à NNaCl = 0,15 M les chaînes polyanioniques et celles de poly(β-CD),
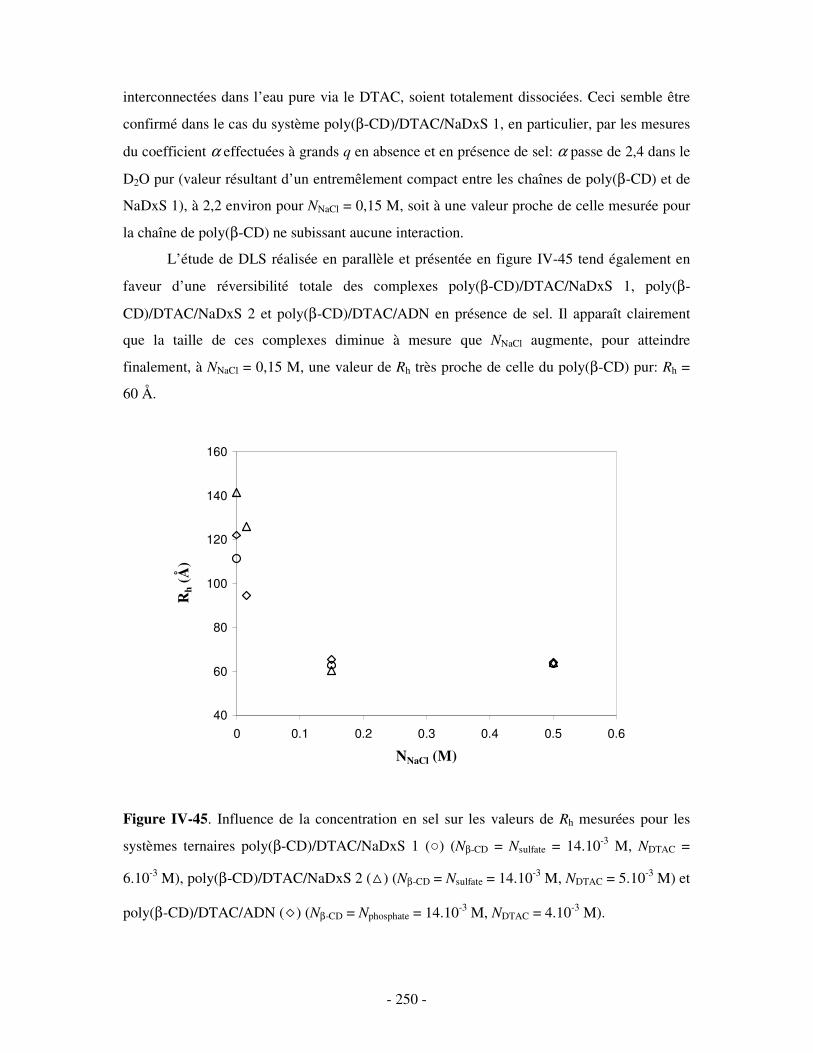
- 250 -
interconnectées dans l’eau pure via le DTAC, soient totalement dissociées. Ceci semble être
confirmé dans le cas du système poly(β-CD)/DTAC/NaDxS 1, en particulier, par les mesures
du coefficient α effectuées à grands q en absence et en présence de sel: α passe de 2,4 dans le
D2O pur (valeur résultant d’un entremêlement compact entre les chaînes de poly(β-CD) et de
NaDxS 1), à 2,2 environ pour NNaCl = 0,15 M, soit à une valeur proche de celle mesurée pour
la chaîne de poly(β-CD) ne subissant aucune interaction.
L’étude de DLS réalisée en parallèle et présentée en figure IV-45 tend également en
faveur d’une réversibilité totale des complexes poly(β-CD)/DTAC/NaDxS 1, poly(β-
CD)/DTAC/NaDxS 2 et poly(β-CD)/DTAC/ADN en présence de sel. Il apparaît clairement
que la taille de ces complexes diminue à mesure que NNaCl augmente, pour atteindre
finalement, à NNaCl = 0,15 M, une valeur de Rh très proche de celle du poly(β-CD) pur: Rh =
60 Å.
40
60
80
100
120
140
160
0 0.1 0.2 0.3 0.4 0.5 0.6
NNaCl (M)
Rh (
Å)
Figure IV-45. Influence de la concentration en sel sur les valeurs de Rh mesurées pour les
systèmes ternaires poly(β-CD)/DTAC/NaDxS 1 () (Nβ-CD = Nsulfate = 14.10-3 M, NDTAC =
6.10-3 M), poly(β-CD)/DTAC/NaDxS 2 () (Nβ-CD = Nsulfate = 14.10-3 M, NDTAC = 5.10-3 M) et
poly(β-CD)/DTAC/ADN ( ) (Nβ-CD = Nphosphate = 14.10-3 M, NDTAC = 4.10-3 M).
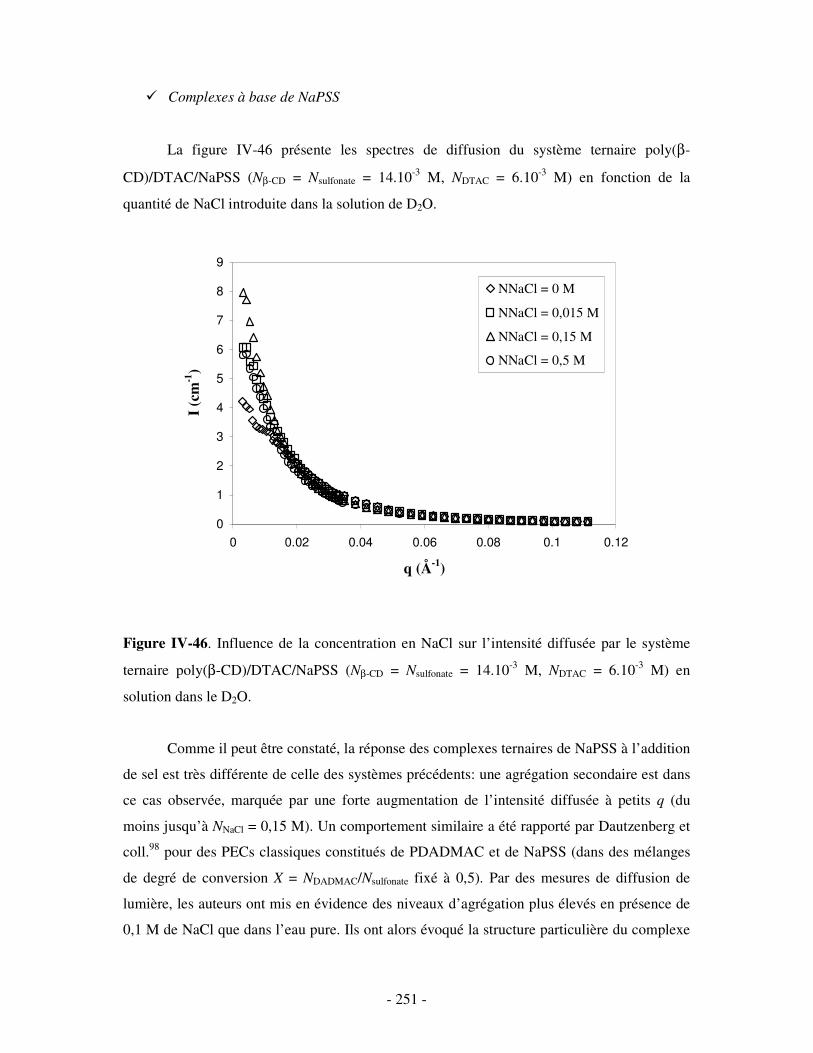
- 251 -
Complexes à base de NaPSS
La figure IV-46 présente les spectres de diffusion du système ternaire poly(β-
CD)/DTAC/NaPSS (Nβ-CD = Nsulfonate = 14.10-3 M, NDTAC = 6.10-3 M) en fonction de la
quantité de NaCl introduite dans la solution de D2O.
0
1
2
3
4
5
6
7
8
9
0 0.02 0.04 0.06 0.08 0.1 0.12
q (Å-1)
I (cm
-1)
NNaCl = 0 M
NNaCl = 0,015 M
NNaCl = 0,15 M
NNaCl = 0,5 M
Figure IV-46. Influence de la concentration en NaCl sur l’intensité diffusée par le système
ternaire poly(β-CD)/DTAC/NaPSS (Nβ-CD = Nsulfonate = 14.10-3 M, NDTAC = 6.10-3 M) en
solution dans le D2O.
Comme il peut être constaté, la réponse des complexes ternaires de NaPSS à l’addition
de sel est très différente de celle des systèmes précédents: une agrégation secondaire est dans
ce cas observée, marquée par une forte augmentation de l’intensité diffusée à petits q (du
moins jusqu’à NNaCl = 0,15 M). Un comportement similaire a été rapporté par Dautzenberg et
coll.98 pour des PECs classiques constitués de PDADMAC et de NaPSS (dans des mélanges
de degré de conversion X = NDADMAC/Nsulfonate fixé à 0,5). Par des mesures de diffusion de
lumière, les auteurs ont mis en évidence des niveaux d’agrégation plus élevés en présence de
0,1 M de NaCl que dans l’eau pure. Ils ont alors évoqué la structure particulière du complexe

- 252 -
PDADMAC/NaPSS: une structure cœur-coquille, avec un cœur formé par le PEC et une
coquille constituée du NaPSS en excès qui, dans l’eau pure, induit des répulsions
électrostatiques vis-à-vis des autres particules de PEC. C’est un écrantage de ces répulsions
qui, selon les auteurs, est à l’origine de l’agrégation observée en présence de sel. Puisque la
structure du complexe ternaire poly(β-CD)/DTAC/NaPSS est également de type cœur-
coquille, avec un cœur de poly(β-CD) et une coquille gonflée de NaPSS non neutralisé
(NDTAC/Nsulfonate = 6/14), une interprétation similaire à celle de Dautzenberg peut être donnée
aux résultats de la figure IV-46: à mesure que la force ionique augmente, les répulsions entre
les coquilles de NaPSS sont écrantées, ce qui favorise l’agrégation des globules (chacun
constitué d’une seule chaîne de poly(β-CD)). Notons que l’interaction poly(β-CD)/NaPSS
mise en évidence par viscosimétrie (cf. figure IV-13) pourrait elle aussi contribuer au
phénomène d’agrégation observé. En effet, d’après les expériences de QCM décrites en
annexe 5, cette interaction est favorisée en présence de sel. Néanmoins, comme l’indique la
figure IV-46, les agrégats ternaires semblent pouvoir être redissous au moins partiellement
pour NNaCl = 0,5 M. Une rediminution des valeurs de I(q) est en effet observée à cette
concentration en sel.
3- Influence de la concentration en compétiteurs
a) Influence de l’HP(β-CD)
Les figures IV-47 et IV-48 présentent les résultats d’une étude préliminaire consacrée
au comportement du système ternaire poly(β-CD)/DTAC/NaDxS 1 (Nβ-CD = Nsulfate = 14.10-3
M, NDTAC = 5.10-3 M) sous l’influence d’additions croissantes d’HP(β-CD), une molécule
susceptible de complexer le DTAC. Dans un cas extrême, un mélange de chaînes libres de
poly(β-CD) et de complexes HP(β-CD)/DTAC/NaDxS 1 est attendu. (Cette étude n’a pu
malheureusement être menée à terme, par manque de temps). Les spectres de diffusion
neutronique du système et les valeurs de Rg apparent correspondantes (déterminées à petits q)
sont respectivement présentés sur les figures IV-47 et IV-48 en fonction de la concentration
en HP(β-CD) ajoutée. Nous précisons que dans la gamme de NHP(β-CD) étudiée, la contribution
des monomères d’HP(β-CD) à la diffusion par les mélanges s’avère être très peu significative.
En effet, les valeurs d’intensité diffusée, par exemple, par un mélange NaDxS 1/HP(β-CD)
comportant 14.10-3 M de groupements sulfate et 14.10-3 M de monomères d’HP(β-CD) sont
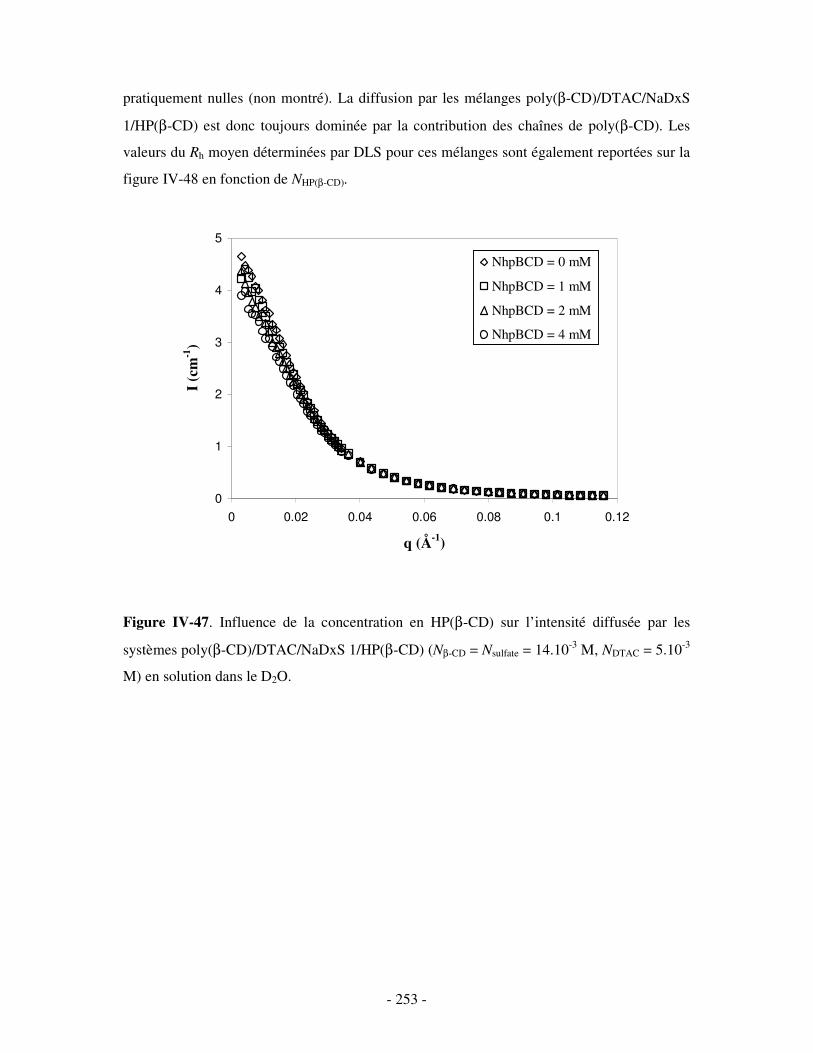
- 253 -
pratiquement nulles (non montré). La diffusion par les mélanges poly(β-CD)/DTAC/NaDxS
1/HP(β-CD) est donc toujours dominée par la contribution des chaînes de poly(β-CD). Les
valeurs du Rh moyen déterminées par DLS pour ces mélanges sont également reportées sur la
figure IV-48 en fonction de NHP(β-CD).
0
1
2
3
4
5
0 0.02 0.04 0.06 0.08 0.1 0.12
q (Å-1)
I (cm
-1)
NhpBCD = 0 mM
NhpBCD = 1 mM
NhpBCD = 2 mM
NhpBCD = 4 mM
Série3
Série7
Série1
Figure IV-47. Influence de la concentration en HP(β-CD) sur l’intensité diffusée par les
systèmes poly(β-CD)/DTAC/NaDxS 1/HP(β-CD) (Nβ-CD = Nsulfate = 14.10-3 M, NDTAC = 5.10-3
M) en solution dans le D2O.
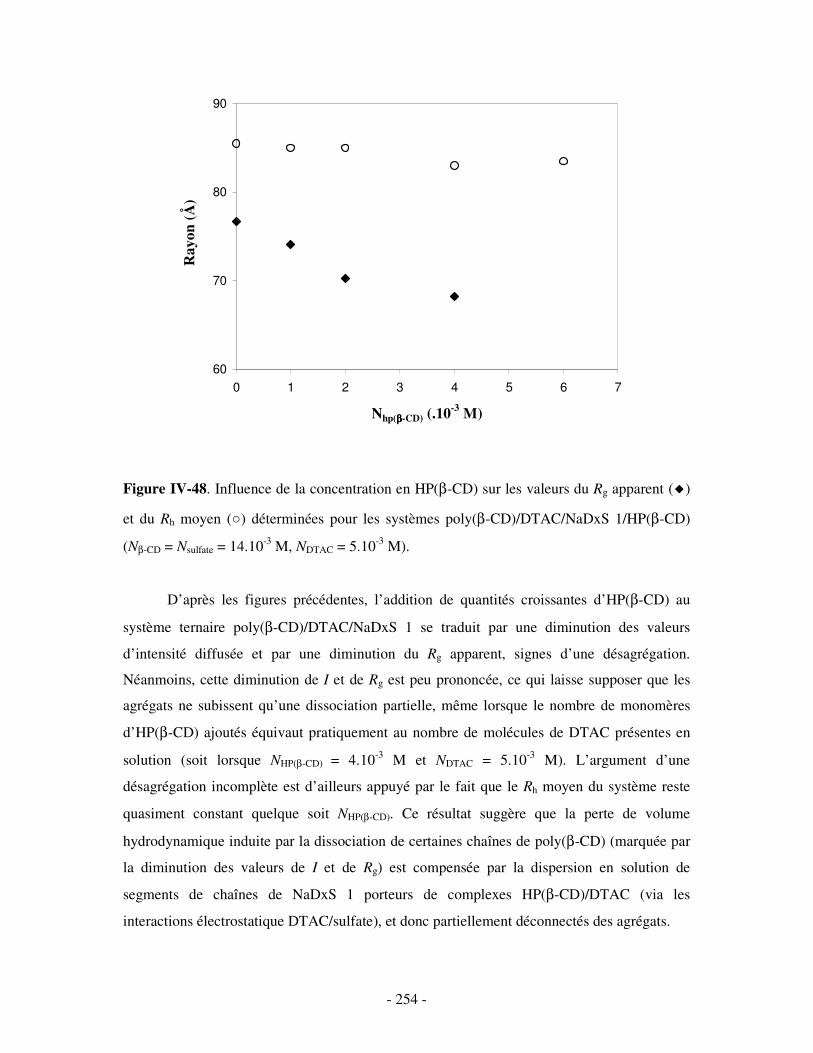
- 254 -
60
70
80
90
0 1 2 3 4 5 6 7
Nhp(ββββ-CD) (.10-3 M)
Ray
on (Å
)
Figure IV-48. Influence de la concentration en HP(β-CD) sur les valeurs du Rg apparent ()
et du Rh moyen () déterminées pour les systèmes poly(β-CD)/DTAC/NaDxS 1/HP(β-CD)
(Nβ-CD = Nsulfate = 14.10-3 M, NDTAC = 5.10-3 M).
D’après les figures précédentes, l’addition de quantités croissantes d’HP(β-CD) au
système ternaire poly(β-CD)/DTAC/NaDxS 1 se traduit par une diminution des valeurs
d’intensité diffusée et par une diminution du Rg apparent, signes d’une désagrégation.
Néanmoins, cette diminution de I et de Rg est peu prononcée, ce qui laisse supposer que les
agrégats ne subissent qu’une dissociation partielle, même lorsque le nombre de monomères
d’HP(β-CD) ajoutés équivaut pratiquement au nombre de molécules de DTAC présentes en
solution (soit lorsque NHP(β-CD) = 4.10-3 M et NDTAC = 5.10-3 M). L’argument d’une
désagrégation incomplète est d’ailleurs appuyé par le fait que le Rh moyen du système reste
quasiment constant quelque soit NHP(β-CD). Ce résultat suggère que la perte de volume
hydrodynamique induite par la dissociation de certaines chaînes de poly(β-CD) (marquée par
la diminution des valeurs de I et de Rg) est compensée par la dispersion en solution de
segments de chaînes de NaDxS 1 porteurs de complexes HP(β-CD)/DTAC (via les
interactions électrostatique DTAC/sulfate), et donc partiellement déconnectés des agrégats.
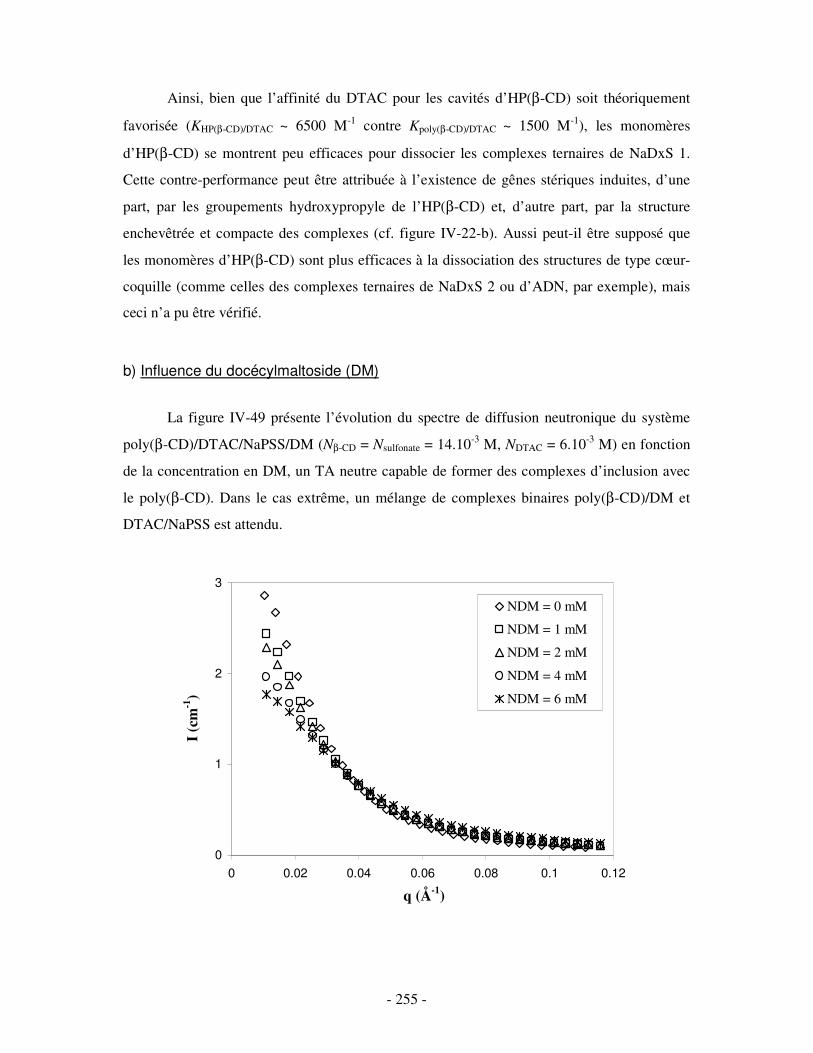
- 255 -
Ainsi, bien que l’affinité du DTAC pour les cavités d’HP(β-CD) soit théoriquement
favorisée (KHP(β-CD)/DTAC ~ 6500 M-1 contre Kpoly(β-CD)/DTAC ~ 1500 M-1), les monomères
d’HP(β-CD) se montrent peu efficaces pour dissocier les complexes ternaires de NaDxS 1.
Cette contre-performance peut être attribuée à l’existence de gênes stériques induites, d’une
part, par les groupements hydroxypropyle de l’HP(β-CD) et, d’autre part, par la structure
enchevêtrée et compacte des complexes (cf. figure IV-22-b). Aussi peut-il être supposé que
les monomères d’HP(β-CD) sont plus efficaces à la dissociation des structures de type cœur-
coquille (comme celles des complexes ternaires de NaDxS 2 ou d’ADN, par exemple), mais
ceci n’a pu être vérifié.
b) Influence du docécylmaltoside (DM)
La figure IV-49 présente l’évolution du spectre de diffusion neutronique du système
poly(β-CD)/DTAC/NaPSS/DM (Nβ-CD = Nsulfonate = 14.10-3 M, NDTAC = 6.10-3 M) en fonction
de la concentration en DM, un TA neutre capable de former des complexes d’inclusion avec
le poly(β-CD). Dans le cas extrême, un mélange de complexes binaires poly(β-CD)/DM et
DTAC/NaPSS est attendu.
0
1
2
3
0 0.02 0.04 0.06 0.08 0.1 0.12
q (Å-1)
I (cm
-1)
NDM = 0 mM
NDM = 1 mM
NDM = 2 mM
NDM = 4 mM
NDM = 6 mM
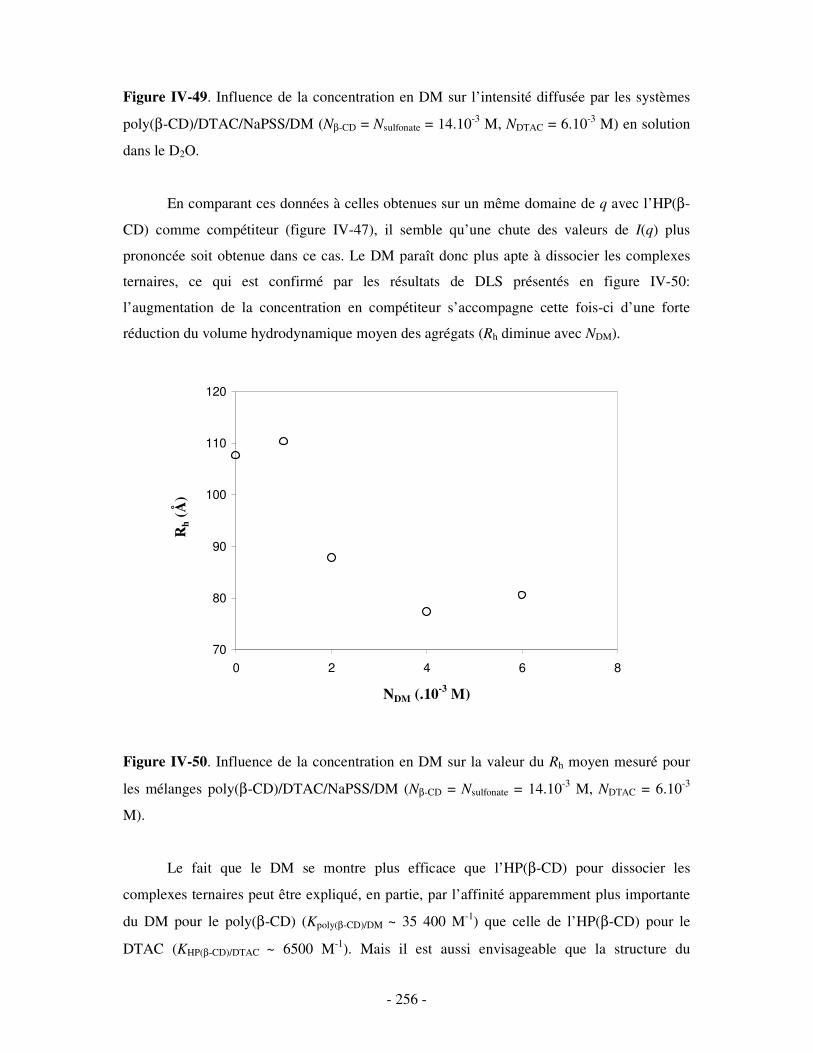
- 256 -
Figure IV-49. Influence de la concentration en DM sur l’intensité diffusée par les systèmes
poly(β-CD)/DTAC/NaPSS/DM (Nβ-CD = Nsulfonate = 14.10-3 M, NDTAC = 6.10-3 M) en solution
dans le D2O.
En comparant ces données à celles obtenues sur un même domaine de q avec l’HP(β-
CD) comme compétiteur (figure IV-47), il semble qu’une chute des valeurs de I(q) plus
prononcée soit obtenue dans ce cas. Le DM paraît donc plus apte à dissocier les complexes
ternaires, ce qui est confirmé par les résultats de DLS présentés en figure IV-50:
l’augmentation de la concentration en compétiteur s’accompagne cette fois-ci d’une forte
réduction du volume hydrodynamique moyen des agrégats (Rh diminue avec NDM).
70
80
90
100
110
120
0 2 4 6 8
NDM (.10-3 M)
Rh (
Å)
Figure IV-50. Influence de la concentration en DM sur la valeur du Rh moyen mesuré pour
les mélanges poly(β-CD)/DTAC/NaPSS/DM (Nβ-CD = Nsulfonate = 14.10-3 M, NDTAC = 6.10-3
M).
Le fait que le DM se montre plus efficace que l’HP(β-CD) pour dissocier les
complexes ternaires peut être expliqué, en partie, par l’affinité apparemment plus importante
du DM pour le poly(β-CD) (Kpoly(β-CD)/DM ~ 35 400 M-1) que celle de l’HP(β-CD) pour le
DTAC (KHP(β-CD)/DTAC ~ 6500 M-1). Mais il est aussi envisageable que la structure du

- 257 -
complexe ternaire considéré influe sur les propriétés « décomplexantes » du compétiteur. En
effet, il paraît raisonnable que les points de connexion poly(β-CD)/DTAC/polyanion soient
plus faciles d’accès pour le compétiteur en présence d’une structure cœur-coquille (comme
c’est le cas ici pour le complexe de NaPSS) qu’en présence d’une structure enchevêtrée
(comme celle du complexe de NaDxS 1). Le compétiteur n’a alors nul besoin de pénétrer au
sein de l’architecture branchée du poly(β-CD) pour jouer son rôle. Enfin, il peut être
considéré que dans le cas du DM, l’inclusion du compétiteur dans les cavités du poly(β-CD)
soit favorisée par la tendance du DTAC à s’autoassocier avec le NaPSS. Hayakawa et
Kwak151 ont par exemple mis en évidence une très forte affinité entre le DTAB et le NaPSS,
plus forte que celle du DTAB pour le NaDxS.
IV- Conclusion
La formation de complexes ternaires hydrosolubles entre un polymère neutre de β-CD,
un TA cationique (le DTAC) et un polyanion a été prouvée pour un polystyrène sulfonate
(NaPSS), pour deux échantillons de dextrane sulfate (NaDxS 1 et NaDxS 2, de masses
molaires et de rigidités croissantes) et pour des fragments d’ADN. Le processus de
complexation ternaire ne dépend donc pas de la nature chimique du polyanion utilisé. De plus,
quelque soit la nature de celui-ci, des comportements macroscopiques similaires ont été
observés pour les complexes ternaires en faisant varier la concentration en DTAC dans le
milieu. D’une façon générale, l’augmentation de NDTAC se traduit par une agrégation en
solution et par une densification des structures formées.
L’utilisation de polyanions différents a toutefois permis de mettre en évidence des
propriétés structurales des complexes spécifiques à la rigidité et à la longueur de chaîne de
leur composant polyanionique. Par exemple, plus le polyanion employé est rigide, plus les
niveaux d’agrégation atteints en augmentant NDTAC sont élevés, favorisant la précipitation du
système: les complexes ternaires de NaPSS, de NaDxS 1, de NaDxS 2 et d’ADN sont, dans
cet ordre, de solubilité décroissante dans l’eau (cela signifie qu’à mêmes concentrations en
poly(β-CD) et en polyanion, ces systèmes précipitent pour des valeurs de NDTAC de moins en
moins élevées). Au contraire, plus le polyanion est flexible, plus la densification structurale
induite par l’augmentation de NDTAC est prononcée: les complexes ternaires de NaPSS se
montrent les plus sensibles à ce phénomène.

- 258 -
D’un point de vue nanoscopique, les variations de NDTAC conduisent à des structures
internes des agrégats qui peuvent s’avérer très différentes selon la rigidité et la taille des
chaînes polyanioniques impliquées. Si pour NDTAC < 2.10-3 M, des structures de type cœur de
poly(β-CD)-coquille polyanionique ont été mises en évidence quelque soit le polyanion
employé (avec Nanion = Nβ-CD = 14.10-3 M), pour NDTAC > 2.10-3 M, des structures compactes
et enchevêtrées ont été observées pour le NaDxS 1 (de masse molaire Mw = 10 kg.mol-1),
contre des structures de type cœur-coquille pour le NaDxS 2 (Mw = 40 kg.mol-1), l’ADN (Mw
= 10-30 kg.mol-1) et le NaPSS (Mw = 70 kg.mol-1). Ces derniers ont été supposés trop
encombrants pour pouvoir pénétrer au sein de l’architecture branchée des cœurs de poly(β-
CD).
Les structures cœur-coquille observées montrent elles aussi des propriétés différentes
selon le polyanion considéré. Dans le complexe ternaire de NaPSS, par exemple, la coquille
constituée du polyanion le plus long et le plus flexible parmi les quatre étudiés, adopte une
conformation très gonflée dans l’eau pure et à NDTAC modérée. Tant que l’électroneutralité
n’est pas atteinte (soit pour NDTAC < Nsulfonate), cette coquille protectrice aide à maintenir les
complexes à l’état colloïdal mais subit tout de même une forte compaction à mesure que la
concentration en DTAC connecteur augmente (d’où la forte densification structurale observée
dans ce cas). Cette propriété de gonflement des coquilles de NaPSS est également fortement
réduite en présence de sel, favorisant une agrégation secondaire des complexes ternaires. Ceci
explique en grande partie pourquoi les agrégats de NaPSS n’apparaissent que partiellement
dissociés pour NNaCl 0,5 M, alors que ceux constitués de NaDxS 1 ou 2 ou d’ADN sont
totalement réversibles dès 0,15 M de NaCl ajouté.
Notons, enfin, que des niveaux d’agrégation plus faibles ont été observés dans l’eau
pure en additionnant aux systèmes ternaires poly(β-CD)/DTAC/polyanion un compétiteur
capable de dissocier les complexes d’inclusion poly(β-CD)/DTAC. Une désagrégation quasi-
totale de la structure cœur-coquille des complexes de NaPSS a par exemple été obtenue en
ajoutant presque autant de TA neutre (NDM = 4.10-3 M) que de DTAC présent dans le milieu
(NDTAC = 6.10-3 M).

- 259 -
Conclusion générale
Ce travail a permis d’élaborer un nouveau modèle de complexe polyélectrolyte basé,
d’une part, sur la formation de complexes d’inclusion entre un polymère neutre de
cyclodextrine et des molécules de tensioactif cationique et, d’autre part, sur la formation de
complexes par charges opposées entre ces molécules de tensioactif et un polyanion.
Expérimentalement, ce modèle à trois composants (complexe ternaire) a été étudié avec un
polymère de β-cyclodextrine de structure branchée (poly(β-CD)), un tensioactif de type
dodécyltriméthylammonium (DTAC) et divers polyanions de natures et de rigidités
différentes.
Des études viscosimétriques couplées à des études de diffusion de neutrons aux petits
angles (SANS) ont tout d’abord permis de mettre en évidence pour les complexes binaires
poly(β-CD)/DTAC des propriétés structurales particulières, relatives à l’architecture branchée
du polymère et à la stoechiométrie NDTAC/Nβ-CD (NDTAC et Nβ-CD étant respectivement les
concentrations molaires en DTAC et en cavités de β-CD du polymère dans le milieu). Pour un
rapport NDTAC/Nβ-CD inférieur à 2/14, les molécules de DTAC cationiques ne sont complexées
qu’en périphérie de la structure branchée du poly(β-CD) qui, doté de ce corona, conserve la
même conformation que sous sa forme libre (sans DTAC). Par contre, pour NDTAC/Nβ-CD >
2/14, les charges sont réparties de façon plus homogène le long de la chaîne de polymère,
induisant un gonflement de cette dernière (via des répulsions coulombiennes). Le poly(β-CD)
se comporte alors comme un polyélectrolyte dont la densité de charges et le gonflement sont
contrôlés par les variations de NDTAC.
Il a ensuite été démontré que le mélange de ces complexes poly(β-CD)/DTAC à du
polystyrène sulfonate (NaPSS), à du dextrane sulfate (NaDxS) ou à des fragments d’ADN
résulte en des assemblages macromoléculaires formés par interactions électrostatiques entre
les molécules de DTAC complexées dans les cavités du poly(β-CD) et le polyanion
additionné. Le mécanisme de complexation ternaire en lui-même n’est donc pas influencé par
la nature chimique du composant polyanionique. Par contre, les propriétés de solubilité des
assemblages résultants sont fortement dépendantes de la rigidité du polyanion: les agrégats
impliquant le NaPSS linéaire et flexible sont plus solubles dans l’eau que ceux composés de

- 260 -
NaDxS de structure branchée, eux-mêmes plus solubles que les agrégats composés d’ADN
double brin très rigide.
La structure des assemblages formés a été analysée par SANS en fonction de la
quantité de DTAC incorporée dans les mélanges (avec Nβ-CD et Nanion fixées à 14.10-3 M). Pour
NDTAC < 2.10-3 M, des structures de type cœur-coquille ont été mises en évidence quelque soit
le polyanion impliqué, en corrélation avec la structure des complexes binaires poly(β-
CD)/DTAC correspondants: les chaînes polyanioniques sont connectées au cœur de poly(β-
CD) uniquement via le corona de DTAC. Pour NDTAC > 2.10-3 M, des structures différentes
ont par contre été observées selon la longueur et la flexibilité du polyanion utilisé. Pour un
échantillon de NaDxS de faible masse (Mw = 10 kg.mol-1), capable d’interagir avec les
molécules de DTAC complexées à l’intérieur du cœur de poly(β-CD), des structures
compactes et enchevêtrées ont été observées. L’utilisation d’un NaDxS de masse plus élevée
(Mw = 40 kg.mol-1) ou de fragments d’ADN (Mw = 10-30 kg.mol-1) trop encombrants pour
pénétrer l’architecture branchée du poly(β-CD) résulte, par contre, en des structures cœurs-
coquilles. Enfin, pour le NaPSS flexible (Mw = 70 kg.mol-1), une structure cœur-coquille a
aussi été mise en évidence avec, dans ce cas, des propriétés de gonflement de la coquille
modulées par la quantité de DTAC ajouté (et par la force ionique du milieu).
Les propriétés structurales des complexes en fonction de NDTAC ont également été
étudiées d’un point de vue macroscopique. Des mesures de diffusion dynamique de la lumière
(DLS) corrélées à des mesures viscosimétriques ont révélé une taille et une compacité des
agrégats contrôlables par un ajustement de NDTAC: plus NDTAC est élevée, plus les complexes
ternaires sont agrégés et plus les structures résultantes sont denses. Ce sont les complexes
composés du NaDxS le plus rigide (de plus grande masse molaire) qui se sont avérés les plus
sensibles au phénomène d’agrégation (la comparaison avec l’ADN n’a pas été établie). Les
agrégats composés du NaPSS flexible, quant à eux, ont montré les plus grandes propriétés de
densification structurale.
Enfin, la stabilité et la réversibilité des complexes ternaires ont été étudiées en faisant
varier la force ionique du milieu et la concentration en compétiteurs tels que le
dodécylmaltoside (DM), un tensioactif neutre. Pour les complexes composés de NaDxS ou
d’ADN, l’ajout de sel en faible quantité (NNaCl = 0,015 M) conduit à un abaissement des
niveaux d’agrégation et à une désagrégation totale à force ionique plus élevée (NNaCl 0,15
M). Par contre, pour les complexes impliquant le NaPSS, l’ajout de sel en quantité inférieure
à 0,5 M se traduit par une agrégation secondaire des complexes. Pour NNaCl = 0,5 M, les

- 261 -
complexes de NaPSS n’ont alors montré qu’une dissociation partielle. La désagrégation des
systèmes poly(β-CD)/DTAC/NaPSS a pu toutefois être obtenue dans l’eau pure, par addition
de 4.10-3 M de DM dans le mélange (avec Nβ-CD = Nsulfonate = 14.10-3 M et NDTAC = 6.10-3 M).
D’une façon générale, il a été montré que l’ajout d’un compétiteur en faible quantité (<
NDTAC) induit des niveaux d’agrégation moindres dans les systèmes ternaires.
En conclusion, la compréhension de ces nouveaux complexes polyélectrolytes a
beaucoup progressé au cours de ce travail de thèse. Cependant, de nombreuses questions sont
encore sans réponse, notamment celle concernant la charge globale portée par les complexes
en fonction de la quantité de DTAC et de la nature des composants polyanioniques. Une
caractérisation des complexes à l’aide d’un zétamètre est donc à envisager. D’autre part, une
forte dépendance de la structure des complexes poly(β-CD)/DTAC et poly(β-
CD)/DTAC/polyanion vis-à-vis de l’architecture branchée du polymère de CD a été
démontré. Quelles seront les structures formées avec un polymère de CD linéaire? Le choix
du DTAC comme connecteur entre le poly(β-CD) et le polyanion est également
problématique. Il a en effet été montré que, de par sa nature tensioactive, le DTAC peut
former des agrégats micellaires le long de la chaîne polyanionique, conduisant à une grande
complexité des systèmes ternaires. Comment se comporteront ces systèmes en présence d’une
molécule connectrice non tensioactive (comportant, par exemple, un groupement cationique et
un groupement adamantane)? De la même façon, comment seront-ils modifiés par l’utilisation
d’un connecteur moins toxique, de nature lipidique par exemple? La réponse à cette question
paraît essentielle pour ce qui est des applications possibles de ces nouveaux systèmes dans le
domaine médical. L’une de ces applications concerne la thérapie génique. En effet, même
s’ils ont été étudiés de façon moins extensive, des complexes ternaires solubles dans l’eau et
impliquant des fragments d’ADN ont été mis en évidence. En extrapolant les résultats obtenus
avec les autres polyanions de nature synthétique (à savoir une taille, une compacité et une
réversibilité des agrégats modulables avec une relative facilité), les complexes ternaires
d’ADN se présentent effectivement comme de bons candidats à la vectorisation de gènes
thérapeutiques. Reste à évaluer leur comportement in vivo.

- 262 -
Références bibliographiques
(1) Saenger W. Cyclodextrin inclusion compounds in research and industry, Angew.
Chem. Int. Ed. Engl. 19, 344 (1980).
(2) Li S., Purdy W. C. Cyclodextrins and their applications in analytical chemistry,
Chem. Rev. 92, 1457 (1992).
(3) Bender M. L., Komiyama M. Cyclodextrin chemistry, Springer-Verlag: Berlin
Heidelberg (1978).
(4) Szejtli J. Cyclodextrin technology, Editor: Davies J. E. D., Kluwer academic:
Dordrecht (1988).
(5) Cramer F., Saenger W., Spatz H.-C. Inclusion compounds. XIX. The formation of
inclusion compounds of α-cyclodextrin in aqueous solutions. Thermodynamics
and kinetics, J. Am. Chem. Soc. 89, 14 (1967).
(6) Catena G. C., Bright F. V. Thermodynamic study on the effects of β-cyclodextrin
inclusion with anilinonaphtalenesulfonates, Anal. Chem. 61, 905 (1989).
(7) Pitchumani K., Vellayappan M. Complex formation of nitrobenzoic acids and
some naphthalene derivatives with β-cyclodextrin, J. Incl. Phenom. Molecular
Recogn. Chem. 14, 157 (1992).
(8) Cyclodextrins and their industrial uses, Editor: Duchêne D., Editions de santé:
Paris (1987).
(9) New trends in cyclodextrins and derivatives, Editor: Duchêne D., Editions de
santé: Paris (1991).
(10) Irie T., Uekama K. Pharmaceutical applications of cyclodextrins. III.
Toxicological issues and safety evaluation, J. Pharm. Sci. 86, 147 (1997).
(11) Harada A., Furue M., Nozakura S. Cyclodextrin-containing polymers. 1.
Preparation of polymers, Macromolecules 9, 701 (1976).
(12) Crini G., Torri G., Guerrini M., Martel B., Lekchiri Y., Morcellet M. Linear
cyclodextrin-poly(vinylamine): Synthesis and NMR characterization, Eur. Polym.
J. 33, 1143 (1997).
(13) Weickenmeier M., Wenz G., Huff J. Association thickener by host guest
interaction of a β-cyclodextrin polymer and a polymer with hydrophobic side-
groups, Macromol. Rapid. Commun. 18, 1117 (1997).

- 263 -
(14) Renard E., Deratani A., Volet G., Sébille B. Preparation and characterization of
water soluble high molecular weight β-cyclodextrin-epichlorohydrin polymers,
Eur. Polym. J. 33, 49 (1997).
(15) Harada A., Furue M., Nozakura S. Inclusion of aromatic compounds by a β-
cyclodextrin-epichlorohydrin polymer, Polymer J. 13, 777 (1981).
(16) Harada A., Furue M., Nozakura S. Interaction of cyclodextrin-containing
polymers with fluorescent compounds, Macromolecules 10, 676 (1977).
(17) Frömming K.-H., Szejtli J. Cyclodextrins in pharmacy, Kluwer academic:
Dordrecht (1994).
(18) Ammar H. O., Ghorab M., El-Nahhas S. A., Omar S. M., Ghorab M. M.
Improvement of some pharmaceutical properties of drugs by cyclodextrin
complexation, Pharmazie 51, 42 (1996).
(19) Loftsson T., Brewster M. E. Pharmaceutical applications of cyclodextrins. 1. Drug
solubilization and stabilization, J. Pharm. Sci. 85, 1017 (1996).
(20) Bibby D. C., Davies N. M., Tucker I. G. Mechanisms by which cyclodextrins
modify drug release from polymeric drug delivery systems, Int. J. Pharm. 197, 1
(2000).
(21) Loftsson T., Leeves N., Sigurjonsdottir J. F., Sigurdsson H. H., Masson M.
Sustained drug delivery system based on a cationic polymer and an anionic
drug/cyclodextrin complex, Pharmazie 56, 746 (2001).
(22) Friedman R. B. Cyclodextrin-containing polymers, in New trends in cyclodextrins
and derivatives, Editor: Duchêne D., Editions de santé: Paris, p. 159 (1991).
(23) Gonzalez H., Hwang S. J., Davis M. E. New class of polymers for the delivery of
macromolecular therapeutics, Bioconjugate Chem. 10, 1068 (1999).
(24) Redenti E., Pietra C., Gerloczy A., Szente L. Cyclodextrins in nucleotide delivery,
Adv. Drug Deliv. Rev. 53, 235 (2001).
(25) Harada A., Li J., Nakamitsu T., Kamachi M. Preparation and characterization of
polyrotaxanes containing many threaded α-cyclodextrins, J. Org. Chem. 58, 7524
(1993).
(26) Harada A., Kamachi M. Complex formation between poly(ethylene glycol) and α-
cyclodextrin, Macromolecules 23, 2821 (1990).
(27) Harada A., Li J., Kamachi M. Preparation and properties of inclusion complexes
of poly(ethylene glycol) with α-cyclodextrin, Macromolecules 26, 5698 (1993).

- 264 -
(28) Harada A., Okada M., Li J., Kamachi M. Preparation and characterization of
inclusion complexes of poly(propylene glycol) with cyclodextrins,
Macromolecules 28, 8406 (1995).
(29) Harada A., Li J., Kamachi M. Complex formation between poly(methyl vinyl
ether) and γ-cyclodextrin, Chem. Lett. 237 (1993).
(30) Harada A., Nishiyama T., Kawaguchi Y., Okada M., Kamachi M. Preparation and
characterization of inclusion complexes of aliphatic polyesters with cyclodextrins,
Macromolecules 30, 7115 (1997).
(31) Harada A., Adachi H., Kawaguchi Y., Okada M., Kamachi M. Complex
formation of cyclodextrins with cationic polymers, Polymer J. 28, 159 (1996).
(32) Zhang H., Hogen-Esch T. E., Boschet F., Margaillan A. Complex formation of β-
cyclodextrin- and perfluorocarbon-modified water-soluble polymers, Langmuir
14, 4972 (1998).
(33) Tsianou M., Alexandridis P. Control of the rheological properties in solutions of a
polyelectrolyte and an oppositely charged surfactant by the addition of
cyclodextrins, Langmuir 15, 8105 (1999).
(34) Amiel C., Sébille B., Association between amphiphilic poly(ethylene oxide) and
β-cyclodextrin polymers: aggregation and phase separation, Adv. Coll. Interface
Sci. 79, 105 (1999).
(35) Amiel C., Sandier A., Sébille B., Valat P., Wintgens V. Associations between
hydrophobically end-capped polyethylene oxide and water soluble β-cyclodextrin
polymers, Int. J. Polymer Anal. Characterization 1, 289 (1995).
(36) Sandier A., Brown W., Mays H., Amiel C. Interaction between an adamantane
end-capped poly(ethylene oxide) and a β-cyclodextrin polymer, Langmuir 16,
1634 (2000).
(37) Gosselet N. M., Beucler F., Renard E., Amiel C., Sébille B. Association of
hydrophobically modified poly(N,N-dimethylacrylamide
hydroxyethylmethacrylate) with water soluble β-cyclodextrin polymers, Colloids
Surf. A 155, 177 (1999).
(38) Amiel C., David C., Renard E., Sébille B. Macromolecular assemblies generated
by inclusion complexes between amphipatic polymers and β-cyclodextrin
polymers in aqueous media, Polym. Prep. 40, 207 (1999).

- 265 -
(39) Amiel C., Moine L., Sandier A., Brown W., David C., Hauss F., Renard E.,
Gosselet M., Sébille B. Macromolecular assemblies generated by inclusion
complexes between amphipatic polymers and β-cyclodextrin polymers in aqueous
media, in Stimuli-responsive water-soluble and amphipatic polymers, Editor:
McCormick, ACS: Symposium series 780, chapter 4, p. 58 (2000).
(40) Moine L., Amiel C., Brown W., Guérin P. Associations between a
hydrophobically modified, degradable, poly(malic acid) and a β-cyclodextrin
polymer in solution, Polym. Int. 50, 663 (2001).
(41) Florence A. T., Attwood D. Pharmaceutical physical chemistry, The Macmillan:
Hong Kong (1994).
(42) Tsuji K. Surface activity: principles, phenomena, and applications, Academic
press: San Diego (1998).
(43) Rosen M. J., Dahanayake M., Cohen A. W. Relationship of structure to properties
in surfactants. 11. Surface and thermodynamic properties of n-dodecylpyridinium
bromide and chloride, Colloids Surf. 5, 159 (1982).
(44) Ananthapadmanabhan K. P. Surfactant solutions: adsorption and aggregation
properties, in Interactions of surfactants with polymers and proteins, Editors:
Goddard E. D., Ananthapadmanabhan K. P., CRC Press: Boca Raton, chapter 2
(1993).
(45) Sunamoto J., Okamoto H., Taira K., Murakami Y. NMR study on the competitive
inclusion of substrate and detergent into cycloheptamose cavity, Chem. Lett. 371
(1975).
(46) Saito Y., Ueda H., Abe M., Sato T., Christian S. D. Inclusion complexation of
Triton X-100 with α-, β- and γ−cyclodextrins, Colloids Surf. A 135, 103 (1998).
(47) Saenger W., Müller-Fahrnow A. Cyclodextrins increase surface tension and
critical micelle concentrations of detergent solutions, Angew. Chem. Int. Ed. Engl.
27, 393 (1988).
(48) Dharmawardana U. R., Christian S. D., Tucker E. E., Taylor R. W., Scamehorn J.
F. A surface tension method for determining binding constants for cyclodextrin
inclusion complexes of ionic surfactants, Langmuir 9, 2258 (1993).
(49) Saito Y., Abe M., Sato T., Scamehorn J. F., Christian S. D. Surface-tension
studies of inclusion complexes of hexanediols with cyclodextrins in aqueous
solutions, Colloids Surf. A 119, 149 (1996).

- 266 -
(50) Lu R., Hao J., Wang H., Tong L. Determination of association constants for
cyclodextrin-surfactant inclusion complexes: a numerical method based on
surface tension measurements, J. Colloid. Interface Sci. 192, 37 (1997).
(51) Okubo T., Kitano H., Ise N. Conductometric studies on association of
cyclodextrin with colloidal electrolytes, J. Phys. Chem. 80, 2661 (1976).
(52) Tunçay M., Christian S. D. A study of the binding of dimethyldodecylamine oxide
by β-cyclodextrin using surface tension measurements, J. Colloid. Interface Sci.
167, 181 (1994).
(53) Satake I., Ikenoue T., Takeshita T., Hayakawa K., Maeda T. Conductometric and
potentiometric studies of the association of α-cyclodextrin with ionic surfactants
and their homologs, Bull. Chem. Soc. Jpn. 58, 2746 (1985).
(54) Satake I., Yoshida S., Hayakawa K., Maeda T., Kusumoto Y. Conductometric
determination of the association constants of β-cyclodextrin with amphiphilic
ions, Bull. Chem. Soc. Jpn. 59, 3991 (1986).
(55) Palepu R., Reinsborough V. C. Surfactant-cyclodextrin interactions by
conductance measurements, Can. J. Chem. 66, 325 (1988).
(56) Palepu R., Richardson J. E., Reinsborough V. C. Binding constants of β-
cyclodextrin/surfactant inclusion by conductivity measurements, Langmuir 5, 218
(1989).
(57) Saint Aman E., Serve D. A conductimetric study of the association between
cyclodextrins and surfactants – Application to the electrochemical study of a
mixed aqueous system: substrate, cyclodextrin, surfactant, J. Colloid Interface
Sci. 138, 365 (1990).
(58) Junquera E., Peña L., Aicart E. A conductimetric study of the interaction of β-
cyclodextrin or hydroxypropyl-β-cyclodextrin with dodecyltrimethylammonium
bromide in water solution, Langmuir 11, 4685 (1995).
(59) Funasaki N., Yodo H., Hada S., Neya S. Stoichiometries and equilibrium
constants of cyclodextrin-surfactant complexations, Bull. Chem. Soc. Jpn. 65,
1323 (1992).
(60) Funasaki N., Ohigashi M., Hada S., Neya S. Surface tensiometric study of
multiple complexation and hemolysis by mixed surfactants and cyclodextrins,
Langmuir 16, 383 (2000).

- 267 -
(61) Czapkiewicz J., Tutaj B. Surface tension studies on the complexation of
dodecylpyridinium chloride by β-cyclodextrin in aqueous electrolyte solutions, J.
Incl. Phenom. Molecular Recogn. Chem. 16, 377 (1993).
(62) Park J. W., Song H. J. Association of anionic surfactants with β-cyclodextrin.
Fluorescence-probed studies on the 1:1 and 1:2 complexation, J. Phys. Chem. 93,
6454 (1989).
(63) Shen X., Belletête M., Durocher G. Studies of the inclusion complexation
between a 3H-indole and β-cyclodextrin in the presence of urea, sodium dodecyl
sulfate, and 1-propanol, Langmuir 13, 5830 (1997).
(64) Jezequel D., Mayaffre A., Letellier P. Etude potentiométrique de la stabilité de
complexes tensioactif-β-cyclodextrine, Can. J. Chem. 69, 1865 (1991).
(65) Wan Yunus W. M. Z., Taylor J., Bloor D. M., Hall D. G., Wyn-Jones E.
Electrochemical measurements on the binding of sodium dodecyl sulfate and
dodecyltrimethylammonium bromide with α- and β- cyclodextrins, J. Phys.
Chem. 96, 8979 (1992).
(66) Tominaga T., Hachisu D., Kamado M. Interactions between the
tetradecyltrimethylammonium ion and α-, β-, and γ-cyclodextrin in water as
studied by a surfactant-selective electrode, Langmuir 10, 4676 (1994).
(67) Mwakibete H., Cristantino R., Bloor D. M., Wyn-Jones E., Holzwarth J. F.
Reliability of the experimental methods to determine equilibrium constants for
surfactant/cyclodextrin inclusion complexes, Langmuir 11, 57 (1995).
(68) Gharibi H., Jalili S., Rajabi T. Electrochemical studies of interaction between
cetyltrimethylammonium bromide and α-, β-cyclodextrins at various temperature,
Colloids Surf. A 175, 361 (2000).
(69) Liska T., Richardson J., Palepu R. Inclusion complexes of anionic surfactants with
hydroxypropyl β-cyclodextrins, in Minutes of the sixth international symposium
on cyclodextrins. Chicago, 21 to 24 April 1992, Editor: Hedges A. R., Editions de
santé: Paris (1992), p. 253.
(70) Ortona O., Paduano L., Costantino L., Vitagliano V. Physico-chemical
characterization of some surfactants in aqueous solution and their interaction with
α-cyclodextrin, J. Molecular Liq. 63, 291 (1995).
(71) Encyclopedia of polymer science and technology, Interscience Publishers, 10, 781
(1969).

- 268 -
(72) Dautzenberg H., Jaeger W., Kötz J., Philipp B., Seidel Ch., Stscherbina D.
Polyelectrolytes: formation, characterization and application, Hanser Publishers:
Cincinnati (1994).
(73) Mortimer D. A. Synthetic polyelectrolytes- A review, Polym. Int. 25, 29 (1991).
(74) Bowman W. A., Rubinstein M., Tan J. S. Polyelectrolyte-gelatin complexation:
light-scattering study, Macromolecules 30, 3262 (1997).
(75) Fuoss R. M., Strauss U. P. Electrostatic interaction of polyelectrolytes and simple
electrolytes, J. Polym. Sci. 3, 602 (1948).
(76) Piculell L., Lindman B. Association and segregation in aqueous polymer/polymer,
polymer/surfactant, and surfactant/surfactant mixtures: similarities and
differences, Adv. Colloid Interface Sci. 41, 149 (1992).
(77) Philipp B., Dautzenberg H., Linow K.-J., Kötz J., Dawydoff W. Polyelectrolyte
complexes - Recent developments and open problems, Prog. Polym. Sci. 14, 91
(1989).
(78) Tsuchida E., Osada Y., Sanada K. Interaction of poly(styrene sulfonate) with
polycations carrying charges in the chain backbone, J. Polym. Sci. 10, 3397
(1972).
(79) Kabanov V. A., Zezin A. B. Soluble interpolymeric complexes as a new class of
synthetic polyelectrolytes, Pure Appl. Chem. 56, 343 (1984).
(80) Kabanov V. A., Zezin A. B. A new class of complex water-soluble
polyelectrolytes, Makromol. Chem. 6, 259 (1984).
(81) Kötz J. Phase behavior of polyanion-polycation aggregates and possibilities of
utilization, Nord. Pulp Paper Res. J. 1, 11 (1993).
(82) Vishalakshi B., Ghosh S., Kalpagam V. The effects of charge density and
concentration on the composition of polyelectrolyte complexes, Polymer 34, 3270
(1993).
(83) Abe K., Ohno H., Tsuchida E. Phase changes of polyion complex between
poly(methacrylic acid) and a polycation carrying charges in the chain backbone,
Makromol. Chem. 178, 2285 (1977).
(84) Domard A., Rinaudo M. Polyelectrolyte complexes. Interaction of poly(L-lysine)-
poly(L-glutamic acid) in dilute aqueous solution, Macromolecules 13, 898 (1980).
(85) Huglin M. B., Webster L., Robb I. D. Complex formation between poly(4-
vinylpyridinium chloride) and poly[sodium (2-acrylamido-2-methyl propane
sulfonate)] in dilute aqueous solution, Polymer 37, 1211 (1996).

- 269 -
(86) Webster L., Huglin M. B., Robb I. D. Observations on complex formation
between polyelectrolytes in dilute aqueous solution, Eur. Polym. J. 33, 1173
(1997).
(87) Webster L., Huglin M. B., Robb I. D. Complex formation between
polyelectrolytes in dilute aqueous solution, Polymer 38, 1373 (1997).
(88) Brand F., Dautzenberg H. Structural analysis in interpolyelectrolyte complex
formation of sodium poly(styrenesulfonate) and diallyldimethylammonium
chloride-acrylamide copolymers by viscometry, Langmuir 13, 2905 (1997).
(89) Möller M., Nordmeier E. Polyelectrolyte complexes formed by poly(diallyl-
N,Ndimethylammoniumchloride) and oligo(dextansulphate), Eur. Polym. J. 38,
445 (2002).
(90) Harada A., Kataoka K. Formation of polyion complex micelles in an aqueous
milieu from a pair of oppositely-charged block copolymers with poly(ethylene
glycol) segments, Macromolecules 28, 5294 (1995).
(91) Harada A., Kataoka K. Novel polyion complex micelles entrapping enzyme
molecules in the core. 2. Characterization of the micelles prepared at
nonstoechiometric mixing ratios, Langmuir 15, 4208 (1999).
(92) Harada A., Kataoka K. Polyion complex micelles with core-shell structure: their
physicochemical properties and utilities as functional materials, Macromol. Symp.
172, 1 (2001).
(93) Gohy J.-F., Varshney S. K., Antoun S., Jérôme R. Water-soluble complexes
formed by sodium poly(4-styrenesulfonate) and a poly(2-vinylpyridinium)-block-
poly(ethyleneoxide) copolymer, Macromolecules 33, 9298 (2000).
(94) Hara M., Nakajima A. Light-scattering study of polyelectrolyte complexes:
heparin with partially aminoacetalized poly(vinyl alcohol) in aqueous solution, J.
Polym. Sci: Part. B 27, 1043 (1989).
(95) Dragan S., Cristea M. Influence of low-molecular-weight salts on the formation of
polyelectrolyte complexes based on polycations with quaternary ammonium salt
groups in the main chain and poly(sodium acrylate), Eur. Polym. J. 37, 1571
(2001).
(96) Tsuchida E., Abe K. Interactions between macromolecules in solution and
intermacromolecular complexes, Adv. Polym. Sci. 45, 1 (1982).

- 270 -
(97) Itoh Y., Negishi K., Iizuka E., Abe K., Kaneko M. Fluorescence study of
polyelectrolyte complex formation: 2. Effect of acidity of polyanions on
complexation, Polymer 33, 3016 (1992).
(98) Dautzenberg H. Polyelectrolyte complex formation in highly aggregating systems.
1. Effect of salt: polyelectrolyte complex formation in the presence of NaCl,
Macromolecules 30, 7810 (1997).
(99) Dautzenberg H., Karibyants N. Polyelectrolyte complex formation in highly
aggregating systems. Effect of salt: response to subsequent addition of NaCl,
Macromol. Chem. Phys. 200, 118 (1999).
(100) Buchhammer H.-M., Petzold G., Lunkwitz K. Salt effect on formation and
properties of interpolyelectrolyte complexes and their interactions with silica
particles, Langmuir 15, 4306 (1999).
(101) Thuresson K., Nilsson S., Lindman B. Effect of hydrophobic modification on
phase behavior and rheology in mixtures of oppositely charged polyelectrolytes,
Langmuir 12, 530 (1996).
(102) Dautzenberg H., Koetz J., Linow K.-J., Philipp B., Rother G. Formation and
structure of polyelectrolyte complexes between polyanions and polycations of
high molecular weight, Polymer Prep. 32, 594 (1991).
(103) Zezin A. B., Izumrudov V. A., Kabanov V. A. Interpolyelectrolyte complexes as a
new family of enzyme carriers, Makromol. Chem., Macromol. Symp. 26, 249
(1989).
(104) Miura N., Dubin P. L., Moorefield C. N, Newkome G. R. Complex formation by
electrostatic interaction between carboxyl-terminated dendrimers and oppositely
charged polyelectrolytes, Langmuir 15, 4245 (1999).
(105) Park J. M., Muhoberac B. B., Dubin P. L., Xia J. Effects of protein charge
heterogeneity in protein-polyelectrolyte complexation, Macromolecules 25, 290
(1992).
(106) Xia J., Dubin P. L., Kim Y., Muhoberac B. B., Klimkowski V. J. Electrophoretic
and quasi-elastic light scattering of soluble protein-polyelectrolyte complexes, J.
Phys. Chem. 97, 4528 (1993).
(107) Hone J. H. E., Howe A. M., Cosgrove T. A small-angle neutron scattering study
of the structure of gelatine/polyelectrolyte complexes, Macromolecules 33, 1206
(2000).

- 271 -
(108) Dragan S., Dranca I., Ghimici L., Cristea M., Funduianu G., Lupascu T. Thermal
behaviour of some cationic polyelectrolytes and polyelectrolyte complexes, Eur.
Polym. J. 34, 733 (1998).
(109) Izumrudov V. A., Ortiz H. O., Zezin A. B., Kabanov V. A. Temperature
controllable interpolyelectrolyte substitutions reactions, Macromol. Chem. Phys.
199, 1057 (1998).
(110) Tsuchida E., Osada Y., Ohno H. Formation of interpolymer complexes, J.
Macromol. Sci.-Phys. B 17, 683 (1980).
(111) Nakajima A. Formation of polyelectrolyte complexes, J. Macromol. Sci.-Phys. B
17, 715 (1980).
(112) Itoh Y., Negishi K., Iizuka E., Abe K. Fluorescence study of polyelectrolyte
complex formation I: complexation of anthracene-loaded poly(methacrylic acid)
with polycations, Polym. Adv. Tech. 1, 225 (1990).
(113) Kramer G., Buchhammer H.-M., Lunkwitz K. Surface modification by
polyelectrolyte complexes: influence of different polyelectrolyte components and
substrates, Colloids Surf. A 122, 1 (1997).
(114) Kramer G., Buchhammer H.-M., Lunkwitz K. Surface modification by
polyelectrolyte complexes: influence of modification procedure, polyelectrolyte
components, and substrates, J. Appl. Polym. Sci. 65, 41 (1997).
(115) Thalberg K., Lindman B., Karlström G. Phase diagram of a system of cationic
surfactant and anionic polyelectrolyte: tetradecyltrimethylammonium bromide-
hyaluronan-water, J. Phys. Chem. 94, 4289 (1990).
(116) Thalberg K., Lindman B., Bergfeldt K. Phase behavior of a systems of
polyacrylate and cationic surfactants, Langmuir 7, 2893 (1991).
(117) Thalberg K., Lindman B., Karlström G. Phase behavior of systems of cationic
surfactant and anionic polyelectrolyte: influence of surfactant chain length and
polyelectrolyte molecular weight, J. Phys. Chem. 95, 3370 (1991).
(118) Hansson P., Almgren M. Interaction of alkyltrimethylammonium surfactants with
polyacrylate and poly(styrenesulfonate) in aqueous solution: phase behavior and
surfactant aggregation numbers, Langmuir 10, 2115 (1994).
(119) Mel’nikov S. M., Sergeyev V. G., Yoshikawa K. Discrete coil-globule transition
of large DNA induced by cationic surfactant, J. Am. Chem. Soc. 117, 2401 (1995).
(120) Mel’nikova Y. S., Lindman B. PH-controlled DNA condensation in the presence
of dodecyldimethylamine oxide, Langmuir 16, 5871 (2000).

- 272 -
(121) Abuin E. B., Scaiano J. C. Exploratory study of the effect of polyelectrolyte-
surfactant aggregates on photochemical behavior, J. Am. Chem. Soc. 106, 6274
(1984).
(122) Cosgrove T., White S. J., Zarbakhsh A., Heenan R. K., Howe A. M. Small-angle
scattering studies of sodium dodecyl sulfate interactions with gelatin. 1, Langmuir
11, 744 (1995).
(123) Watanabe Y., Otomo T., Shimizu S., Adachi T., Sano Y., Furusaka M.
Characterization of a surfactant-protein complex by small-angle neutron
scattering, J. Phys. Chem. Solids 60, 1383 (1999).
(124) Winnik F. M. Applications of fluorescence spectroscopy to the study of polymer-
surfactant interactions, in Interactions of surfactants with polymers and proteins,
Editors: Goddard E. D., Ananthapadmanabhan K. P., CRC Press: Boca Raton,
chapter 9, p. 367 (1993).
(125) Winnik F. M., Regismond S. T. A. Fluorescence methods in the study of the
interactions of surfactants with polymers, Colloids Surf. A 118, 1 (1996).
(126) Leung P. S., Goddard E. D. A study of polycation-anionic-surfactant systems,
Colloids Surf. 13, 47 (1985).
(127) Winnik M. A., Bystryak S. M., Chassenieux C. Study of interaction of
poly(ethylene imine) with sodium dodecyl sulfate in aqueous solution by light
scattering, conductometry, NMR, and microcalorimetry, Langmuir 16, 4495
(2000).
(128) Merta J., Garamus V. M., Kuklin A. I., Willumeit R., Stenius P. Determination of
the structure of complexes formed by a cationic polymer and mixed anionic
surfactants by small-angle neutron scattering, Langmuir 16, 10061 (2000).
(129) Merta J., Stenius P. Interactions between cationic starch and anionic surfactants 2.
Viscosity and aggregate size in dilute solutions, Colloids Surf. A 122, 243 (1997).
(130) Guillot S., Delsanti M., Désert S., Langevin D. Surfactant-induced collapse of
polymer chains and monodisperse growth of aggregates near the precipitation
boundary in carboxymethylcellulose-DTAB aqueous solutions, Langmuir 19, 230
(2003).
(131) Herslöf Å, Sundelöf L.-O., Edsman K. Interaction between polyelectrolyte and
surfactant of opposite charge. Hydrodynamic effects in the sodium
hyaluronate/tetradecyltrimethylammonium bromide/sodium chloride/water
system, J. Phys. Chem. 96, 2345 (1992).

- 273 -
(132) Santerre J.-P., Hayakawa K., Kwak J. C. T. A study of the temperature
dependence of the binding of a cationic surfactant to an anionic polyelectrolyte,
Colloids Surf. 13, 35 (1985).
(133) Cosgrove T., White S. J., Zarbakhsh A., Heenan R. K., Howe A. M. Small-angle
scattering studies of sodium dodecyl sulfate interactions with gelatin. Part 2.
Effect of temperature and pH, J. Chem. Soc., Faraday Trans. 92, 595 (1996).
(134) Chandar P., Somasundaran P., Turro N. J. Fluorescence probe investigation of
anionic polymer-cationic surfactant interactions, Macromolecules 21, 950 (1988).
(135) Kogej K., Skerjanc J. Fluorescence and conductivity studies of polyelectrolyte-
induced aggregation of alkyltrimethylammonium bromides, Langmuir 15, 4251
(1999).
(136) Chu D.-Y., Thomas J. K. Effect of cationic surfactants on the conformational
transition of poly(methacrylic acid), J. Am. Chem. Soc. 108, 6270 (1986).
(137) Choi L.-S., Kim O.-K. Fluorescence probe studies of poly(acrylic acid)-cationic
surfactant interactions following high shear, Langmuir 10, 57 (1994).
(138) Petrov A. I., Khalil D. N., Kazaryan R. L., Savintsev I. V., Sukhorukov B. I.
Structural and thermodynamic features of complexes formed by DNA and
synthetic polynucleotides with dodecylamine and dodecyltrimethylammonium
bromide, Bioelectrochem. 58, 75 (2002).
(139) Dubin P. L., Thé S. S., Gan L. M., Chew C. H. Static light scattering of
polyelectrolyte-micelle complexes, Macromolecules 23, 2500 (1990).
(140) Xia J., Zhang H., Rigsbee D. R., Dubin P. L., Shaikh T. Structural elucidation of
soluble polyelectrolyte-micelle complexes: intra- vs interpolymer association,
Macromolecules 26, 2759 (1993).
(141) Li Y., Xia J., Dubin P. L. Complex formation between polyelectrolyte and
oppositely charged mixed micelles: static and dynamic light scattering study of
the effect of polyelectrolyte molecular weight and concentration, Macromolecules
27, 7049 (1994).
(142) Moriyama Y., Takeda K., Murakami K. Electrophoretic behavior of complexes
between sodium dextran sulfate and cationic surfactants, Langmuir 16, 7629
(2000).
(143) Almgren M., Hansson P., Mukhtar E., Van Stam J. Aggregation of
alkyltrimethylammonium surfactants in aqueous poly(styrenesulfonate) solutions,
Langmuir 8, 2405 (1992).

- 274 -
(144) Anthony O., Zana R. Interactions between water-soluble polymers and
surfactants: effect of the polymer hydrophobicity. 1. Hydrophilic polyelectrolytes,
Langmuir 12, 1967 (1996).
(145) Fundin J., Hansson P., Brown W., Lidegran I. Poly(acrylic acid)-
cetyltrimethylammonium bromide interactions studied using dynamic and static
light scattering and time-resolved fluorescence quenching, Macromolecules 30,
1118 (1997).
(146) Hansson P., Almgren M. Large C12TAB micelles formed in complexes with
polyvinylsulfate and dextran sulfate, J. Phys. Chem. 99, 16694 (1995).
(147) Thalberg K., Van Stam J., Lindblad C., Almgren M., Lindman B. Time-resolved
fluorescence and self-diffusion studies in systems of a cationic surfactant and an
anionic polyelectrolyte, J. Phys. Chem. 95, 8975 (1991).
(148) Hayakawa K., Santerre J.-P., Kwak J. C. T. Study of surfactant-polyelectrolyte
interactions. Binding of dodecyl- and tetradecyltrimethylammonium bromide by
some carboxylic polyelectrolytes, Macromolecules 16, 1642 (1983).
(149) Buckin V., Kudryashow E., Morrissey S., Kapustina T., Dawson K. Do
surfactants form micelles on the surface of DNA?, Progr. Colloid. Polym. Sci.
110, 214 (1998).
(150) Barrat J.-L., Joanny J.-F. Theory of polyelectrolyte solutions, Adv. Chem. Phys.
94, 1 (1996).
(151) Hayakawa K., Kwak J. C. T. Surfactant-polyelectrolyte interactions. 1. Binding of
dodecyltrimethylammonium ions by sodium dextran sulfate and sodium
poly(styrenesulfonate) in aqueous solution in the presence of sodium chloride, J.
Phys. Chem. 86, 3866 (1982).
(152) Anthony O., Zana R. Interactions between water-soluble polymers and
surfactants: effect of the polymer hydrophobicity. 2. Amphiphilic polyelectrolytes
(polysoaps), Langmuir 12, 3590 (1996).
(153) Magny B., Iliopoulos I., Zana R., Audebert R. Mixed micelles formed by cationic
surfactants and anionic hydrophobically modified polyelectrolytes, Langmuir 10,
3180 (1994).
(154) Guillemet F., Piculell L., Nilsson S., Djabourov M., Lindman B. Interactions
between hydrophobically modified polyelectrolyte and oppositely charged
surfactant. Mixed micelle formation, Prog. Colloid. Polym. Sci. 98, 47 (1995).

- 275 -
(155) Decher G. Fuzzy nanoassemblies: toward layered polymeric multicomposites,
Science 277, 1232 (1997).
(156) Petzold G., Lunkwitz K. The interaction between polyelectrolyte complexes made
from poly(dimethyldiallylammonium chloride) (PDMDAAC) and poly(maleic
acid-co-α-methylstyrene) (P(MS-α-MeSty)) and cellulosic materials, Colloids
Surf A 98, 225 (1995).
(157) Petzold G., Nebel A., Buchhammer H.-M., Lunkwitz K. Preparation and
characterization of different polyelectrolyte complexes and their application as
flocculants, Colloid. Polym. Sci. 276, 125 (1998).
(158) Kissel T., Jung T., Kamm W., Breitenbach A. Biodegradable graft polyesters
based on copolymer of lactic acid and glycolic acids grafted onto poly(vinyl
alcohol) for oral vaccine delivery, Macromol. Symp. 172, 113 (2001).
(159) Luo D., Saltzman W. M. Synthetic DNA delivery systems, Nature biotech. 18, 33
(2000).
(160) Kuhn P. S., Levin Y., Barbosa M. C. Complex formation between polyelectrolytes
and ionic surfactants, Chem. Phys. Lett. 298, 51 (1998).
(161) Clamme J. P., Bernacchi S., Vuilleumier C., Duportail G., Mély Y. Gene transfer
by cationic surfactants is essentially limited by the trapping of the surfactant/DNA
complexes onto the cell membrane: a fluorescence investigation, Biochem.
Biophys. Acta 1467, 347 (2000).
(162) Pinnaduwage P., Schmitt L., Huang L. Use of a quaternary ammonium detergent
in liposome mediated DNA transfection of mouse L-cells, Biochem. Biophys. Acta
985, 33 (1989).
(163) Rädler J. O., Koltover I., Salditt T., Safinya C. R. Structure of DNA-cationic
liposome complexes: DNA intercalation in multilamellar membranes in distinct
interhelical packing regimes, Science 275, 810 (1997).
(164) Koltover I., Salditt T., Rädler J. O., Safinya C. R. An inverted hexagonal phase of
cationic liposome-DNA complexes related to DNA release and delivery, Science
281, 78 (1998).
(165) Safinya C. R. Structures of lipid-DNA complexes: supramolecular assembly and
gene delivery, Curr. Opin. Struct. Biol. 11, 440 (2001).
(166) Chirila T. V., Rakoczy P. E., Garrett K. L., Lou X., Constable I. J. The use of
synthetic polymers for delivery of therapeutic antisense oligodeoxynucleotides,
Biomaterials 23, 321 (2002).

- 276 -
(167) Couvreur P., Malvy C., Dubernet C., Fattal E. Liposomes for the delivery of
oligonucleotides, in Pharmaceutical aspects of oligonucleotides, Editors:
Couvreur P., Malvy C., Taylor and Francis: Philadelphia, chapter 7, p. 146 (2000).
(168) Farber F. E., Melnick J. L., Butel J. S. Optimal conditions for uptake of
exogenous DNA by chinese hamster lung cells deficient in hypoxanthine-guanine
phosphoribosyltransferase, Biochem. Biophys. Acta 390, 298 (1975).
(169) Pouton C. W., Lucas P., Thomas B. J., Uduehi A. N., Milroy D. A., Moss S. H.
Polycation-DNA complexes for gene delivery: a comparison of the
biopharmaceutical properties of cationic polypeptides and cationic lipids, J.
Controlled Release 53, 289 (1998).
(170) Ramsay E., Hadgraft J., Birchall J., Gumbleton M. Examination of the biophysical
interaction between plasmid DNA and the polycations, polylysine and
polyornithine, as a basis for their differential gene transfection in-vitro, Int. J.
Pharm. 210, 97 (2000).
(171) Kabanov V. A., Kabanov A. V., Astafieva I. N. Complexes of DNA with
synthetic polycations for cell transformation, Polymer Prep. 32, 592 (1991).
(172) Kabanov A. V., Astafieva I. V., Maksimova I. V., Lukanidin E. M., Georgiev G.
P., Kabanov V. A. Efficient transformation of mammalian cells using DNA
interpolyelectrolyte complexes with carbon chain polycations, Bioconjugate
Chem. 4, 448 (1993).
(173) Boussif O., Lezoualc’h F., Zanta M. A., Mergny M. D., Scherman D., Demeneix
B., Behr J.-P. A versatile vector for gene and oligonucleotide transfer into cells in
culture and in vivo: polyethylenimine, Proc. Natl. Acad. Sci. USA 92, 7297
(1995).
(174) Tang M. X., Szoka F. C. The influence of polymer structure on the interactions of
cationic polymers with DNA and morphology of the resulting complexes, Gene
Ther. 4, 823 (1997).
(175) Pollard H., Remy J.-S., Loussouarn G., Demolombe S., Behr J.-P., Escande D.
Polyethyleneimine but not cationic lipids promotes transgene delivery to the
nucleus in mammalian cells, J. Biol. Chem. 273, 7507 (1998).
(176) Wightman L., Kircheis R., Rössler V., Carotta S., Ruzicka R., Kursa M., Wagner
E. Different behavior of branched and linear polyethylenimine for gene delivery
in vitro and in vivo, J. Gene Med. 3, 362 (2001).

- 277 -
(177) Haensler J., Szoka F. C. Jr. Polyamidoamine cascade polymers mediate efficient
transfection of cells in culture, Bioconjugate Chem. 4, 372 (1993).
(178) Bielinska A., Kukowska-Latallo J. F., Johnson J., Tomalia D. A., Baker J. R. Jr.
Regulation of in vitro gene expression using antisense oligonucleotides or
antisense expression plasmids transfected using starburst PAMAM dendrimers,
Nucleic Acids Res. 24, 2176 (1996).
(179) Tang M. X., Redemann C. T., Szoka F. C. Jr. In vitro gene delivery by degraded
polyamidoamine dendrimers, Bioconjugate Chem. 7, 703 (1996).
(180) Dash P. R., Toncheva V., Schacht E., Seymour L. W. Synthetic polymers for
vectorial delivery of DNA: characterization of polymer-DNA complexes by
photon correlation spectroscopy and stability to nuclease degradation and
disruption by polyanions in vitro, J. Controlled. Release 48, 269 (1997).
(181) Maruyama A., Ishihara T., Kim J., Kim S. W., Akaike T. Nanoparticle DNA
carrier with poly(L-lysine) grafted polysaccharide copolymer and poly(D, L-lactic
acid), Bioconjugate Chem. 8, 735 (1997).
(182) Maruyama A., Watanabe H., Ferdous A., Katoh M., Ishihara T., Akaike T.
Characterization of interpolyelectrolyte complexes between double-stranded DNA
and polylysine comb-type copolymers having hydrophilic side chains,
Bioconjugate Chem. 9, 292 (1998).
(183) Wolfert M. A., Schacht E. H., Toncheva V., Ulbrich K., Nazarova O., Seymour L.
W. Characterization of vectors for gene therapy formed by self-assembly of DNA
with synthetic block co-polymers, Hum. Gene Ther. 7, 2123 (1996).
(184) Katayose S., Kataoka K. Water-soluble polyion complex associates of DNA and
poly(ethylene glycol)-poly(L-lysine) block copolymer, Bioconjugate Chem. 8,
702 (1997).
(185) Choi J. S, Lee E. J., Choi Y. H., Jeong Y. J., Park J. S. Poly(ethylene glycol)-
block-poly(L-lysine) dendrimer: novel linear polymer/dendrimer block copolymer
forming a spherical water-soluble polyionic complex with DNA, Bioconjugate
Chem. 10, 62 (1999).
(186) Choi Y. H., Liu F., Kim J.-S, Choi Y. K., Park J. S., Kim S. W. Polyethylene
glycol-grafted poly-L-lysine as polymeric gene carrier, J. Controlled Release 54,
39 (1998).
(187) Toncheva V., Wolfert M. A., Dash P. R., Oupicky D., Ulbrich K., Seymour L. W.,
Schacht E. H. Novel vectors for gene delivery formed by self-assembly of DNA

- 278 -
with poly(L-lysine) grafted with hydrophilic polymers, Biochem. Biophys. Acta
1380, 354 (1998).
(188) Kurisawa M., Yokoyama M., Okano T. Transfection efficiency increases by
incorporating hydrophobic monomer units into polymeric gene carriers, J.
Controlled Release 68, 1 (2000).
(189) Renard E., Barnathan G., Deratani A., Sébille B. Polycondensation of
cyclodextrins with epichlorohydrin. Influence of reaction conditions on the
polymer structure, Macromol. Symp. 122, 229 (1997).
(190) Voeks J. F., Tartar H. V. The electrical conductance of aqueous solutions of
dodecyltrimethylammonium sulfate at 25°, J. Phys. Chem. 59, 1190 (1955).
(191) Charlot G., Les méthodes de la chimie analytique, Masson: Paris (1961).
(192) Higgins J. S., Benoît H. C. Polymers and neutron scattering, Clarendon Press:
Oxford (1994).
(193) Cotton J.-P. Introduction to scattering experiments, in Neutron, X-ray and light
scattering, Editors: Lindner P. and Zemb T., North Holland Delta series:
Amsterdam, p. 3 (1991).
(194) Cotton J.-P. DNPA: introduction et variations sur le contraste, J. Phys. IV France
9, 21 (1999).
(195) Elaïssari A., Chevalier Y., Ganachaud F., Delair T., Pichot C. Structure of
adsorbed and grafted single-stranded DNA fragments on aminated latex particles:
a small-angle neutron scattering study, Langmuir 16, 1261 (2000).
(196) Miles G. D., Shedlovsky L. Minima in surface tension-concentration curves of
solutions of sodium alcohol sulfates, J. Phys. Chem. 48, 57 (1944).
(197) Tanford C. The hydrophobic effect: formation of micelles and biological
membranes, 2nd Ed., John Wiley and Sons: New York (1980).
(198) Junquera E., Tardajos G., Aicart E. Effect of the presence of β-cyclodextrin on the
micellization process of sodium dodecyl sulfate or sodium perfluorooctanoate in
water, Langmuir 9, 1213 (1993).
(199) Müller B. W., Brauns U. Hydroxypropyl-β-cyclodextrin derivatives: Influence of
average degree of substitution on complexing ability and surface activity, J.
Pharm. Sci. 75, 571 (1986).
(200) Borsali R., Nguyen H., Pecora R. Small-angle neutron scattering and dynamic
light scattering from a polyelectrolyte solution: DNA, Macromolecules 31, 1548
(1998).

- 279 -
(201) Zeghal M., Auvray L. Structure of polymer complexes in water, Europhys. Lett.
45, 482 (1999).
(202) Matsuoka H., Schwahn D., Ise N. Observation of cluster formation in
polyelectrolyte solutions by small-angle neutron scattering. 1. A steep upturn of
the scattering curves from solutions of sodium poly(styrenesulfonate) at scattering
vectors below 0,01 Å-1, Macromolecules 24, 4227 (1991).
(203) Daoud M., Joanny J.-F. Conformation of branched polymers, J. Physique 42,
1359 (1981).
(204) Maier E. E., Krause R., Deggelmann M., Hagenbüchle M., Weber R. Liquidlike
order of charged rodlike particule solutions, Macromolecules 25, 1125 (1992).
(205) Milas M., Rinaudo M., Duplessix R., Borsali R., Lindner P. Small angle neutron
scattering from polyelectrolyte solutions: From disordered to ordered xanthan
chain conformation, Macromolecules 28, 3119 (1995).
(206) Ermi B. D., Amis E. J. Influence of backbone solvation on small angle neutron
scattering from polyelectrolyte solutions, Macromolecules 30, 6937 (1997).
(207) Wang L., Bloomfield V. A. Small-angle X-ray scattering of semidilute rodlike
DNA solutions: Polyelectrolyte behavior, Macromolecules 24, 5791 (1991).
(208) Borisov O. V., Daoud M., Scaling theory of branched polyelectrolytes,
Macromolecules 34, 8286 (2001).
(209) Auvray L., Auroy P. in Neutron, X-ray and light scattering, Editors: Lindner P.
and Zemb T., North Holland Delta series: Amsterdam, p. 199 (1991).
(210) Kassapidou K., Jesse W., Kuil M. E., Lapp A., Egelhaaf S., van der Maarel J. R.
C. Structure and charge distribution in DNA and poly(styrenesulfonate) aqueous
solutions, Macromolecules 30, 2671 (1997).
(211) Koppel D. E. Analysis of macromolecular polydispersity in intensity correlation
spectroscopy: The method of cumulants, J. Chem. Phys. 57, 4814 (1972).
(212) Lindman B., Thalberg K. Polymer-surfactant interactions-Recent developments,
in Interactions of surfactants with polymers and proteins, Editors: Goddard E. D.,
Ananthapadmanabhan K. P., CRC Press: Boca Raton, chapter 5 (1993).
(213) Garcia R., Gomez C. M., Porcar I., Figueruelo J. E., Campos A. Viscometric
study of mixtures of neutral and charged polymers in aqueous solution, Eur.
Polym. J. 33, 1723 (1997).
(214) Iijima T., Uemura T., Tsuzuku S., Komiyama J. Diffusion of cyclodextrins in
aqueous polymer solutions, J. Polym. Sci. 16, 793 (1978).
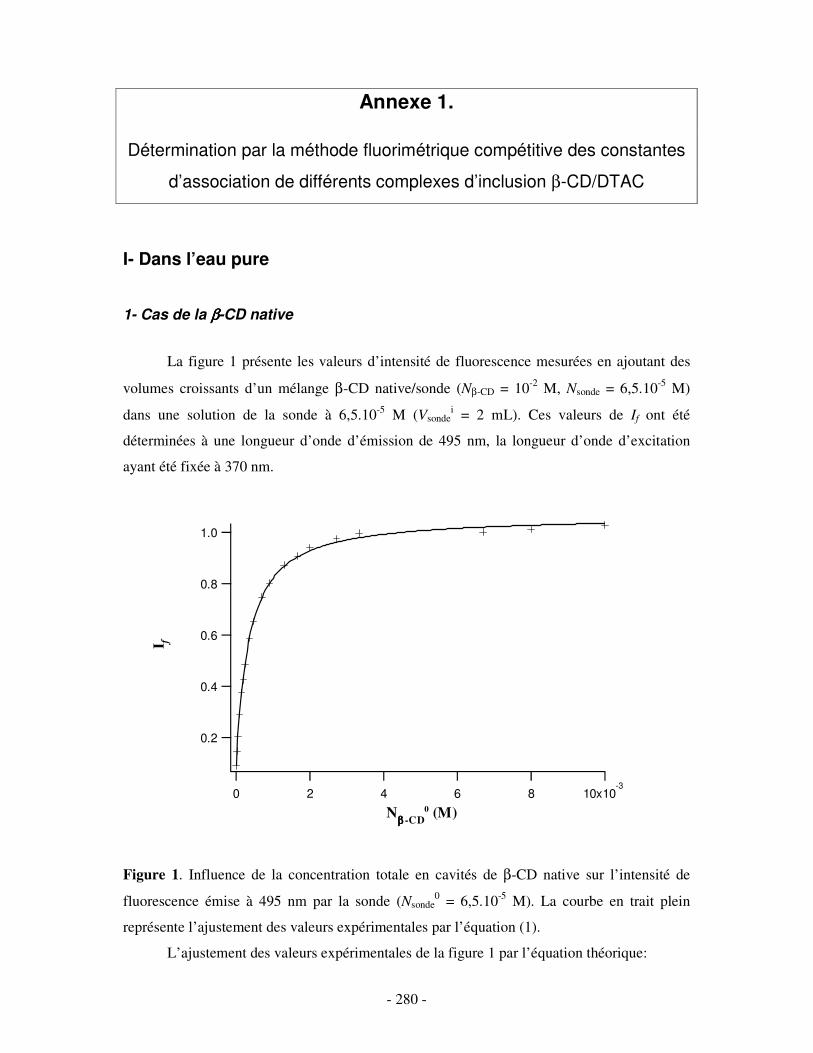
- 280 -
Annexe 1.
Détermination par la méthode fluorimétrique compétitive des constantes
d’association de différents complexes d’inclusion β-CD/DTAC
I- Dans l’eau pure
1- Cas de la ββββ-CD native
La figure 1 présente les valeurs d’intensité de fluorescence mesurées en ajoutant des
volumes croissants d’un mélange β-CD native/sonde (Nβ-CD = 10-2 M, Nsonde = 6,5.10-5 M)
dans une solution de la sonde à 6,5.10-5 M (Vsondei = 2 mL). Ces valeurs de If ont été
déterminées à une longueur d’onde d’émission de 495 nm, la longueur d’onde d’excitation
ayant été fixée à 370 nm.
1.0
0.8
0.6
0.4
0.2
I f
10x10-3
86420
Nββββ -CD0 (M)
Figure 1. Influence de la concentration totale en cavités de β-CD native sur l’intensité de
fluorescence émise à 495 nm par la sonde (Nsonde0 = 6,5.10-5 M). La courbe en trait plein
représente l’ajustement des valeurs expérimentales par l’équation (1).
L’ajustement des valeurs expérimentales de la figure 1 par l’équation théorique:
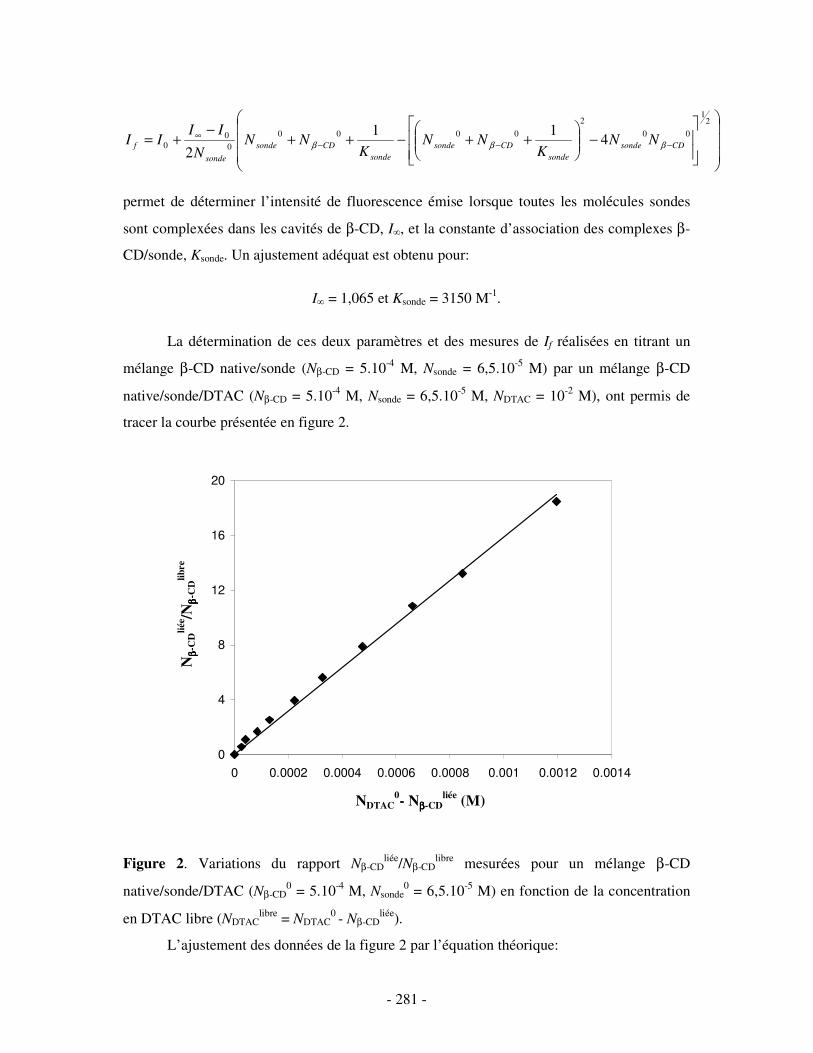
- 281 -
−
++−++
−+= −−−
∞2
1
002
00000
00 4
112
CDsondesonde
CDsondesonde
CDsonde
sonde
f NNK
NNK
NNN
IIII βββ
permet de déterminer l’intensité de fluorescence émise lorsque toutes les molécules sondes
sont complexées dans les cavités de β-CD, I, et la constante d’association des complexes β-
CD/sonde, Ksonde. Un ajustement adéquat est obtenu pour:
I = 1,065 et Ksonde = 3150 M-1.
La détermination de ces deux paramètres et des mesures de If réalisées en titrant un
mélange β-CD native/sonde (Nβ-CD = 5.10-4 M, Nsonde = 6,5.10-5 M) par un mélange β-CD
native/sonde/DTAC (Nβ-CD = 5.10-4 M, Nsonde = 6,5.10-5 M, NDTAC = 10-2 M), ont permis de
tracer la courbe présentée en figure 2.
0
4
8
12
16
20
0 0.0002 0.0004 0.0006 0.0008 0.001 0.0012 0.0014
NDTAC0- Nββββ-CD
liée (M)
Nββ ββ-
CD
liée /N
ββ ββ-C
Dlib
re
Figure 2. Variations du rapport Nβ-CDliée/Nβ-CD
libre mesurées pour un mélange β-CD
native/sonde/DTAC (Nβ-CD0 = 5.10-4 M, Nsonde
0 = 6,5.10-5 M) en fonction de la concentration
en DTAC libre (NDTAClibre = NDTAC
0 - Nβ-CDliée).
L’ajustement des données de la figure 2 par l’équation théorique:
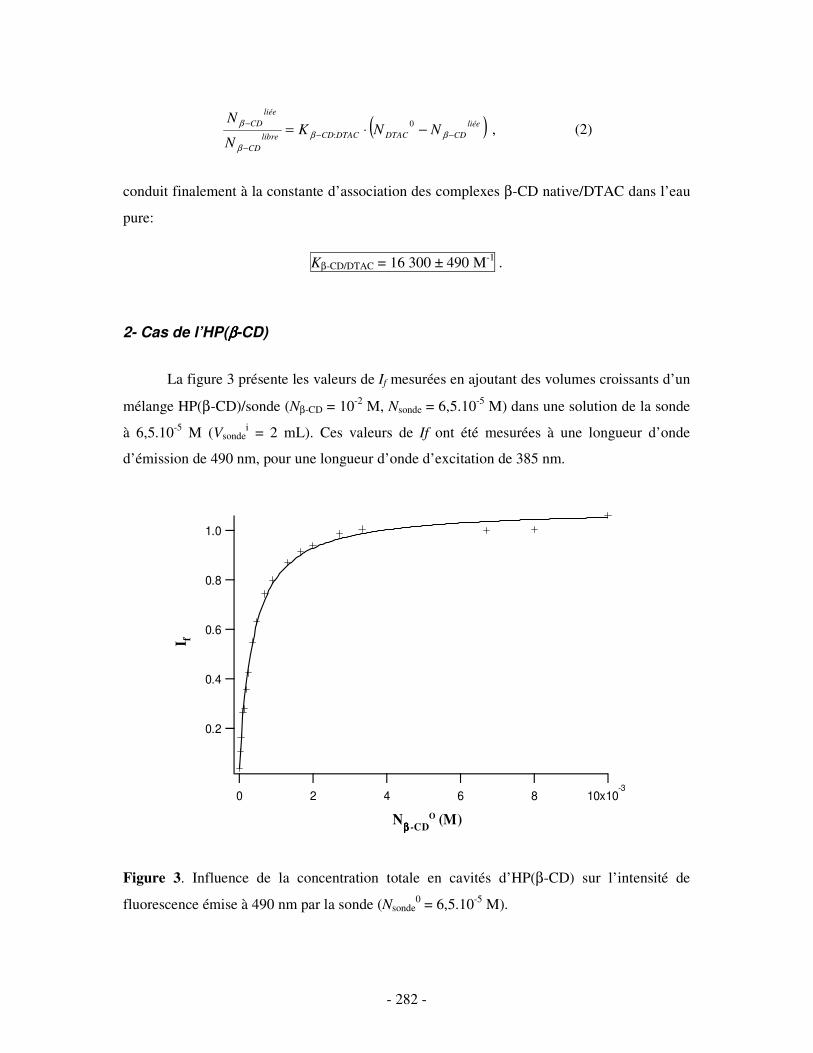
- 282 -
( )liéeCDDTACDTACCDlibre
CD
liéeCD NNK
N
N−−
−
− −⋅= βββ
β 0: , (2)
conduit finalement à la constante d’association des complexes β-CD native/DTAC dans l’eau
pure:
Kβ-CD/DTAC = 16 300 ± 490 M-1 .
2- Cas de l’HP(ββββ-CD)
La figure 3 présente les valeurs de If mesurées en ajoutant des volumes croissants d’un
mélange HP(β-CD)/sonde (Nβ-CD = 10-2 M, Nsonde = 6,5.10-5 M) dans une solution de la sonde
à 6,5.10-5 M (Vsondei = 2 mL). Ces valeurs de If ont été mesurées à une longueur d’onde
d’émission de 490 nm, pour une longueur d’onde d’excitation de 385 nm.
1.0
0.8
0.6
0.4
0.2
I f
10x10-3
86420
Nββββ -CDO (M)
Figure 3. Influence de la concentration totale en cavités d’HP(β-CD) sur l’intensité de
fluorescence émise à 490 nm par la sonde (Nsonde0 = 6,5.10-5 M).
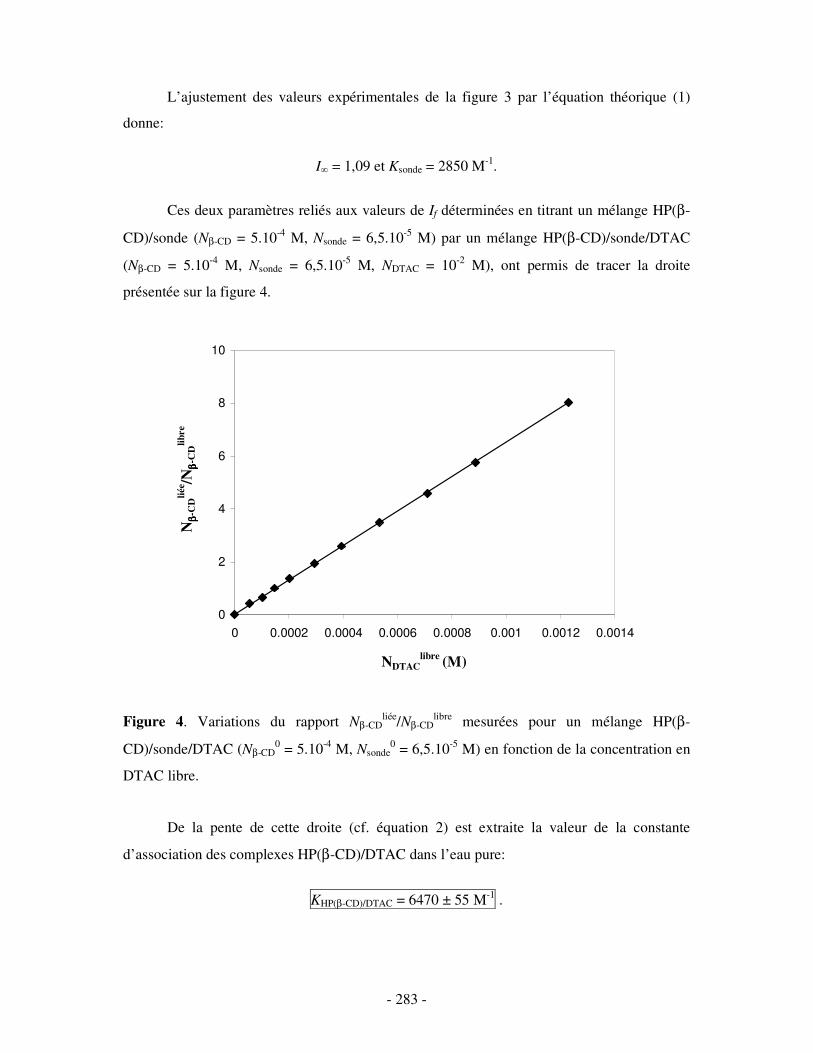
- 283 -
L’ajustement des valeurs expérimentales de la figure 3 par l’équation théorique (1)
donne:
I = 1,09 et Ksonde = 2850 M-1.
Ces deux paramètres reliés aux valeurs de If déterminées en titrant un mélange HP(β-
CD)/sonde (Nβ-CD = 5.10-4 M, Nsonde = 6,5.10-5 M) par un mélange HP(β-CD)/sonde/DTAC
(Nβ-CD = 5.10-4 M, Nsonde = 6,5.10-5 M, NDTAC = 10-2 M), ont permis de tracer la droite
présentée sur la figure 4.
0
2
4
6
8
10
0 0.0002 0.0004 0.0006 0.0008 0.001 0.0012 0.0014
NDTAClibre
(M)
Nββ ββ-
CD
liée /N
ββ ββ-C
Dlib
re
Figure 4. Variations du rapport Nβ-CDliée/Nβ-CD
libre mesurées pour un mélange HP(β-
CD)/sonde/DTAC (Nβ-CD0 = 5.10-4 M, Nsonde
0 = 6,5.10-5 M) en fonction de la concentration en
DTAC libre.
De la pente de cette droite (cf. équation 2) est extraite la valeur de la constante
d’association des complexes HP(β-CD)/DTAC dans l’eau pure:
KHP(β-CD)/DTAC = 6470 ± 55 M-1 .
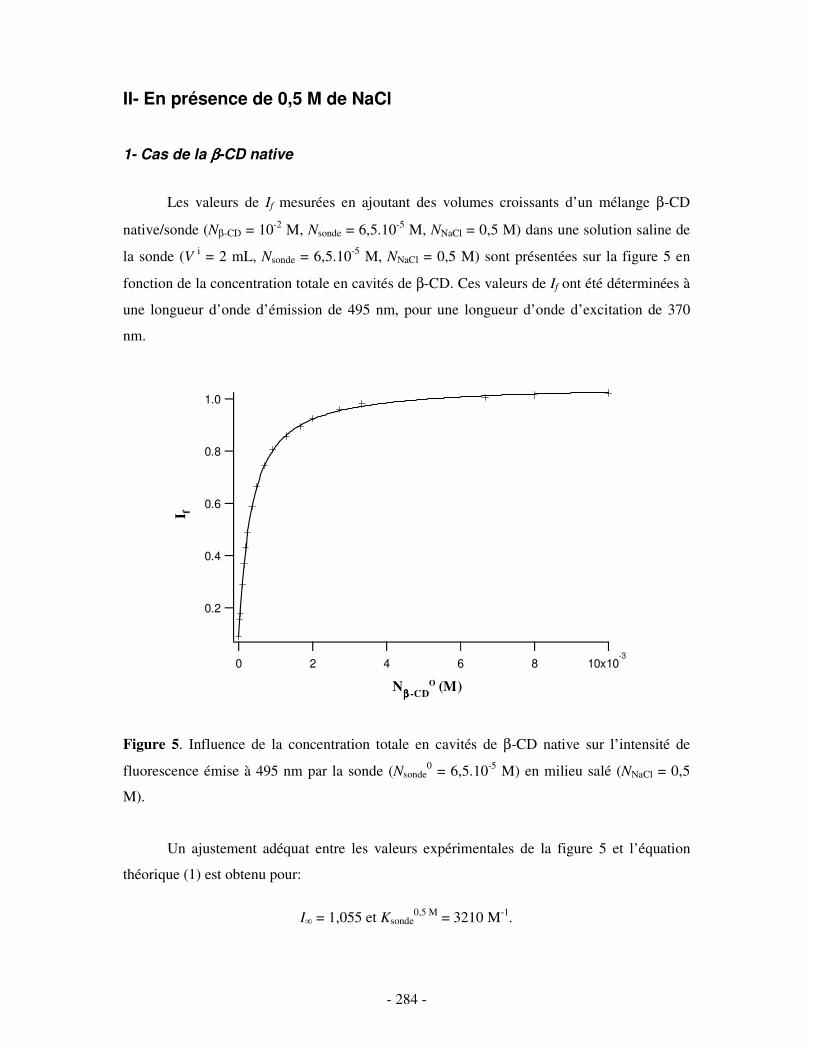
- 284 -
II- En présence de 0,5 M de NaCl
1- Cas de la ββββ-CD native
Les valeurs de If mesurées en ajoutant des volumes croissants d’un mélange β-CD
native/sonde (Nβ-CD = 10-2 M, Nsonde = 6,5.10-5 M, NNaCl = 0,5 M) dans une solution saline de
la sonde (V i = 2 mL, Nsonde = 6,5.10-5 M, NNaCl = 0,5 M) sont présentées sur la figure 5 en
fonction de la concentration totale en cavités de β-CD. Ces valeurs de If ont été déterminées à
une longueur d’onde d’émission de 495 nm, pour une longueur d’onde d’excitation de 370
nm.
1.0
0.8
0.6
0.4
0.2
I f
10x10-3
86420
Nββββ -CDO (M)
Figure 5. Influence de la concentration totale en cavités de β-CD native sur l’intensité de
fluorescence émise à 495 nm par la sonde (Nsonde0 = 6,5.10-5 M) en milieu salé (NNaCl = 0,5
M).
Un ajustement adéquat entre les valeurs expérimentales de la figure 5 et l’équation
théorique (1) est obtenu pour:
I = 1,055 et Ksonde
0,5 M = 3210 M-1.
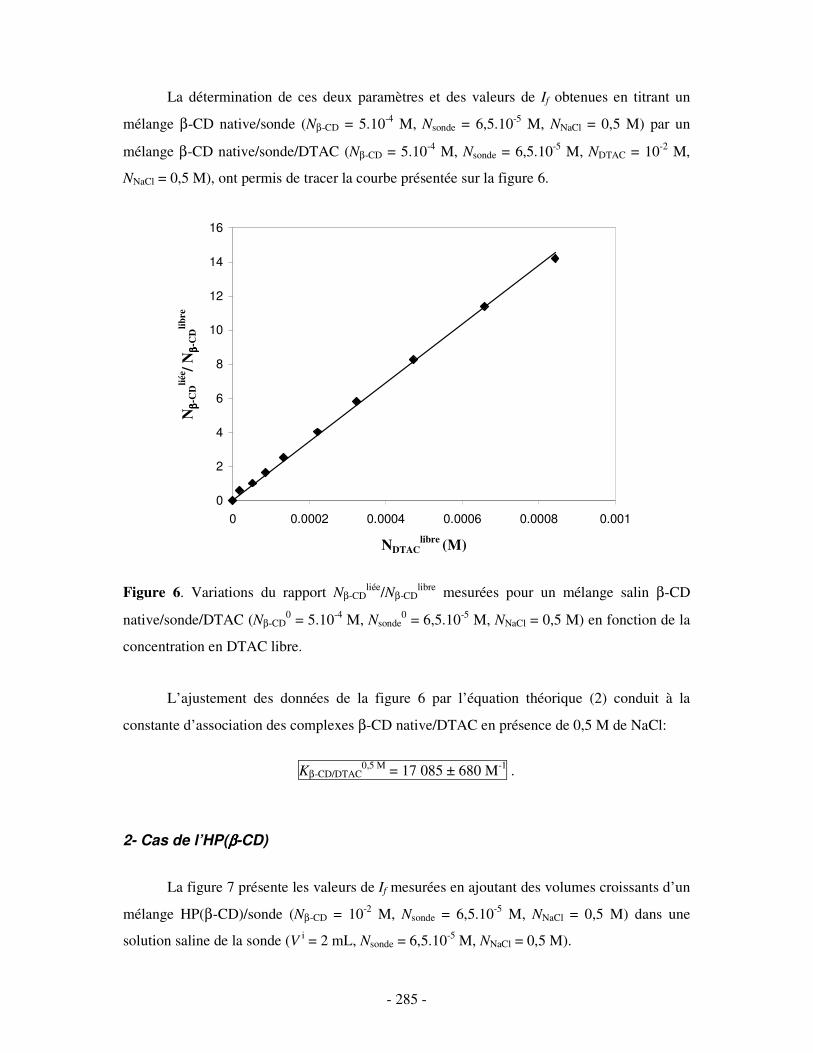
- 285 -
La détermination de ces deux paramètres et des valeurs de If obtenues en titrant un
mélange β-CD native/sonde (Nβ-CD = 5.10-4 M, Nsonde = 6,5.10-5 M, NNaCl = 0,5 M) par un
mélange β-CD native/sonde/DTAC (Nβ-CD = 5.10-4 M, Nsonde = 6,5.10-5 M, NDTAC = 10-2 M,
NNaCl = 0,5 M), ont permis de tracer la courbe présentée sur la figure 6.
0
2
4
6
8
10
12
14
16
0 0.0002 0.0004 0.0006 0.0008 0.001
NDTAClibre
(M)
Nββ ββ-
CD
liée / N
ββ ββ-C
Dlib
re
Figure 6. Variations du rapport Nβ-CDliée/Nβ-CD
libre mesurées pour un mélange salin β-CD
native/sonde/DTAC (Nβ-CD0 = 5.10-4 M, Nsonde
0 = 6,5.10-5 M, NNaCl = 0,5 M) en fonction de la
concentration en DTAC libre.
L’ajustement des données de la figure 6 par l’équation théorique (2) conduit à la
constante d’association des complexes β-CD native/DTAC en présence de 0,5 M de NaCl:
Kβ-CD/DTAC
0,5 M = 17 085 ± 680 M-1 .
2- Cas de l’HP(ββββ-CD)
La figure 7 présente les valeurs de If mesurées en ajoutant des volumes croissants d’un
mélange HP(β-CD)/sonde (Nβ-CD = 10-2 M, Nsonde = 6,5.10-5 M, NNaCl = 0,5 M) dans une
solution saline de la sonde (V i = 2 mL, Nsonde = 6,5.10-5 M, NNaCl = 0,5 M).
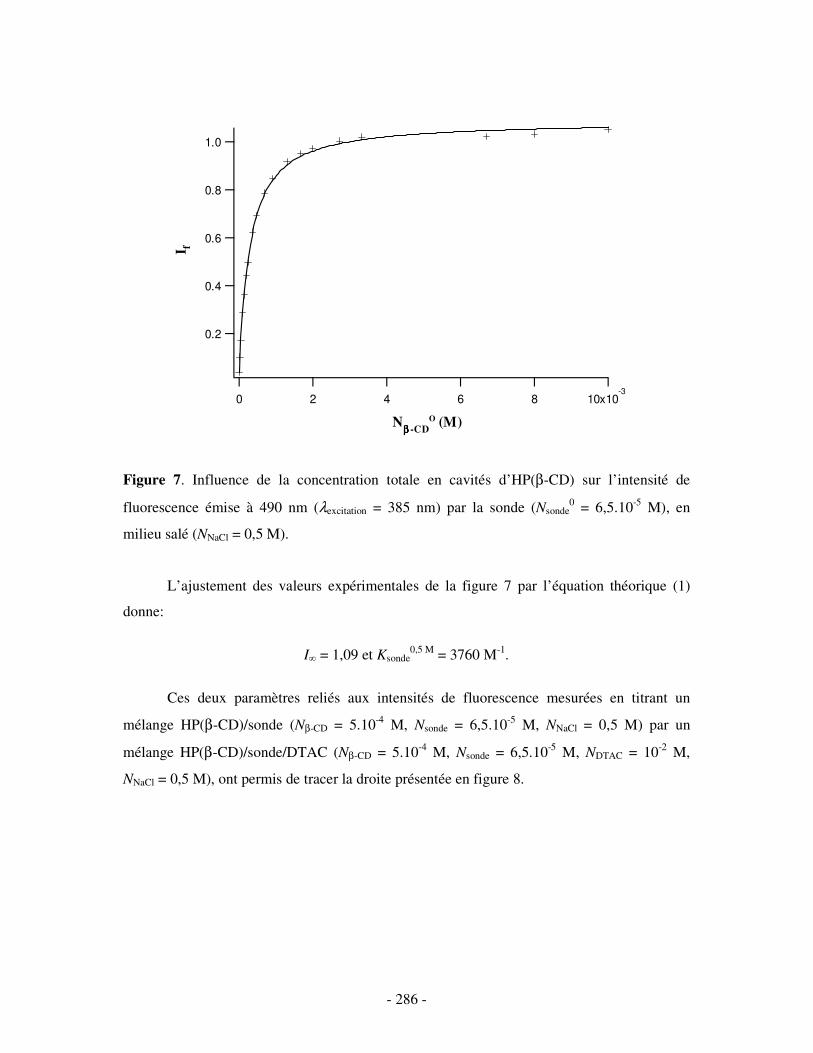
- 286 -
1.0
0.8
0.6
0.4
0.2
I f
10x10-3
86420
Nββββ -CDO (M)
Figure 7. Influence de la concentration totale en cavités d’HP(β-CD) sur l’intensité de
fluorescence émise à 490 nm (λexcitation = 385 nm) par la sonde (Nsonde0 = 6,5.10-5 M), en
milieu salé (NNaCl = 0,5 M).
L’ajustement des valeurs expérimentales de la figure 7 par l’équation théorique (1)
donne:
I = 1,09 et Ksonde
0,5 M = 3760 M-1.
Ces deux paramètres reliés aux intensités de fluorescence mesurées en titrant un
mélange HP(β-CD)/sonde (Nβ-CD = 5.10-4 M, Nsonde = 6,5.10-5 M, NNaCl = 0,5 M) par un
mélange HP(β-CD)/sonde/DTAC (Nβ-CD = 5.10-4 M, Nsonde = 6,5.10-5 M, NDTAC = 10-2 M,
NNaCl = 0,5 M), ont permis de tracer la droite présentée en figure 8.
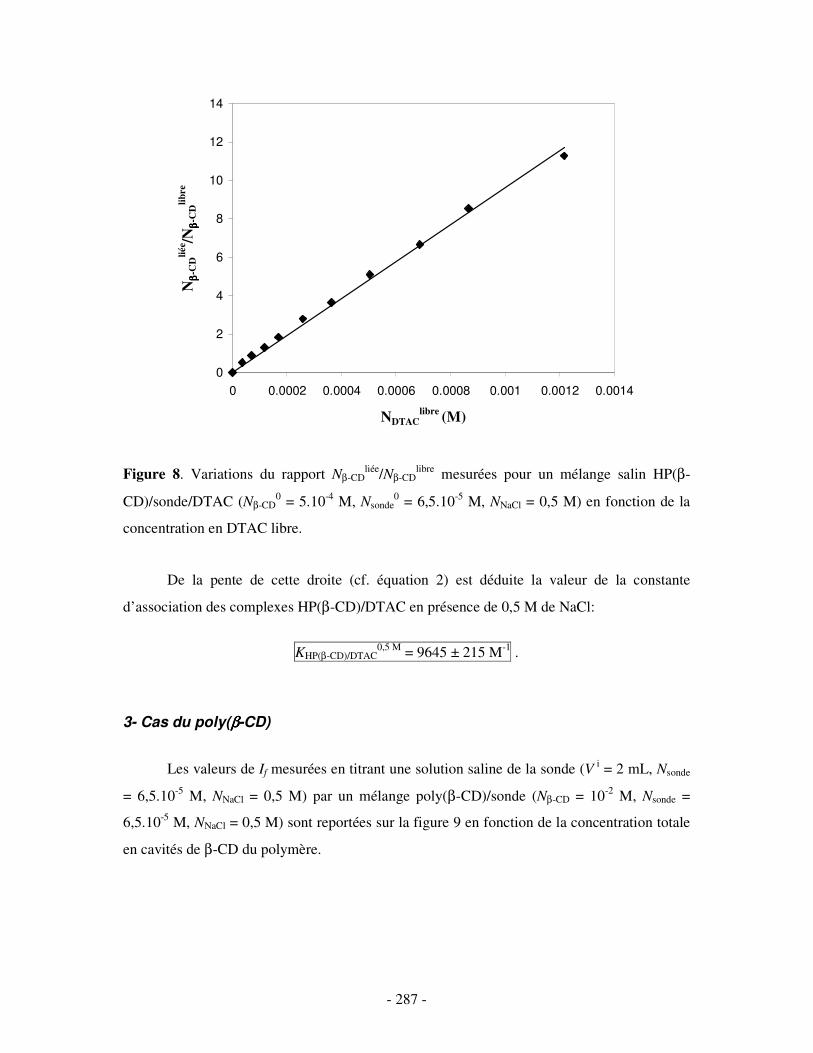
- 287 -
0
2
4
6
8
10
12
14
0 0.0002 0.0004 0.0006 0.0008 0.001 0.0012 0.0014
NDTAClibre (M)
Nββ ββ-
CD
liée /N
ββ ββ-C
Dlib
re
Figure 8. Variations du rapport Nβ-CDliée/Nβ-CD
libre mesurées pour un mélange salin HP(β-
CD)/sonde/DTAC (Nβ-CD0 = 5.10-4 M, Nsonde
0 = 6,5.10-5 M, NNaCl = 0,5 M) en fonction de la
concentration en DTAC libre.
De la pente de cette droite (cf. équation 2) est déduite la valeur de la constante
d’association des complexes HP(β-CD)/DTAC en présence de 0,5 M de NaCl:
KHP(β-CD)/DTAC
0,5 M = 9645 ± 215 M-1 .
3- Cas du poly(ββββ-CD)
Les valeurs de If mesurées en titrant une solution saline de la sonde (V i = 2 mL, Nsonde
= 6,5.10-5 M, NNaCl = 0,5 M) par un mélange poly(β-CD)/sonde (Nβ-CD = 10-2 M, Nsonde =
6,5.10-5 M, NNaCl = 0,5 M) sont reportées sur la figure 9 en fonction de la concentration totale
en cavités de β-CD du polymère.
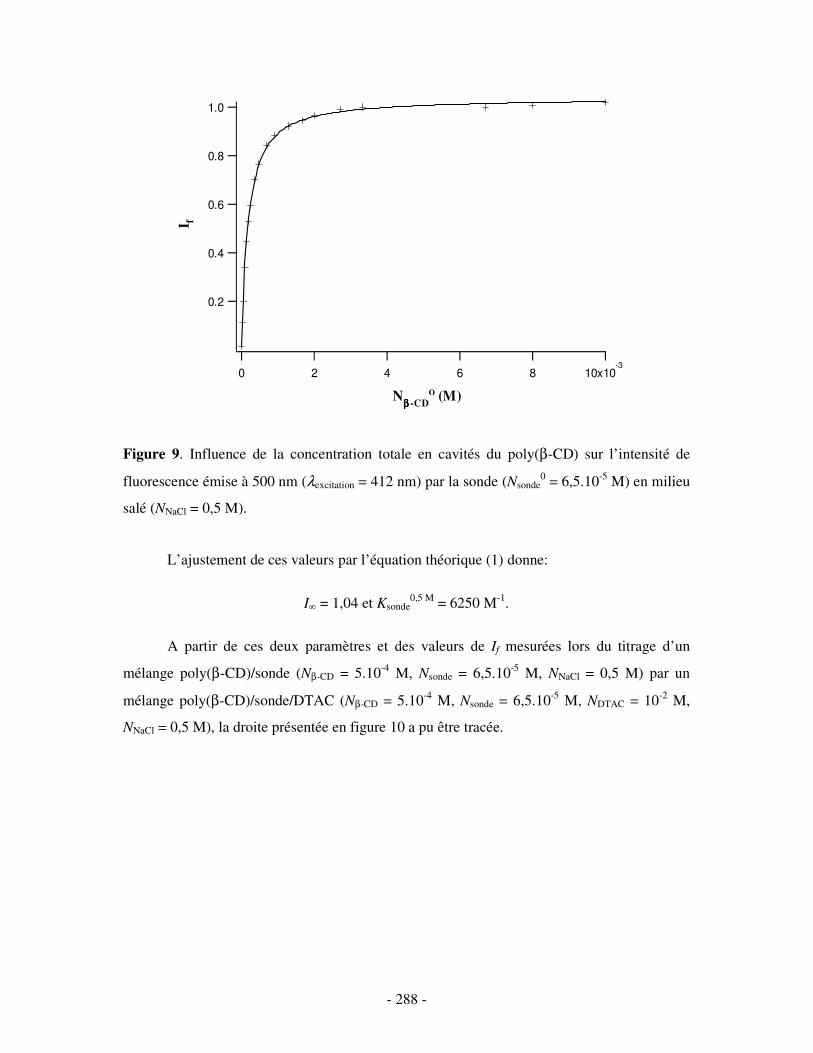
- 288 -
1.0
0.8
0.6
0.4
0.2
I f
10x10-3
86420
Nββββ -CDO (M)
Figure 9. Influence de la concentration totale en cavités du poly(β-CD) sur l’intensité de
fluorescence émise à 500 nm (λexcitation = 412 nm) par la sonde (Nsonde0 = 6,5.10-5 M) en milieu
salé (NNaCl = 0,5 M).
L’ajustement de ces valeurs par l’équation théorique (1) donne:
I = 1,04 et Ksonde
0,5 M = 6250 M-1.
A partir de ces deux paramètres et des valeurs de If mesurées lors du titrage d’un
mélange poly(β-CD)/sonde (Nβ-CD = 5.10-4 M, Nsonde = 6,5.10-5 M, NNaCl = 0,5 M) par un
mélange poly(β-CD)/sonde/DTAC (Nβ-CD = 5.10-4 M, Nsonde = 6,5.10-5 M, NDTAC = 10-2 M,
NNaCl = 0,5 M), la droite présentée en figure 10 a pu être tracée.
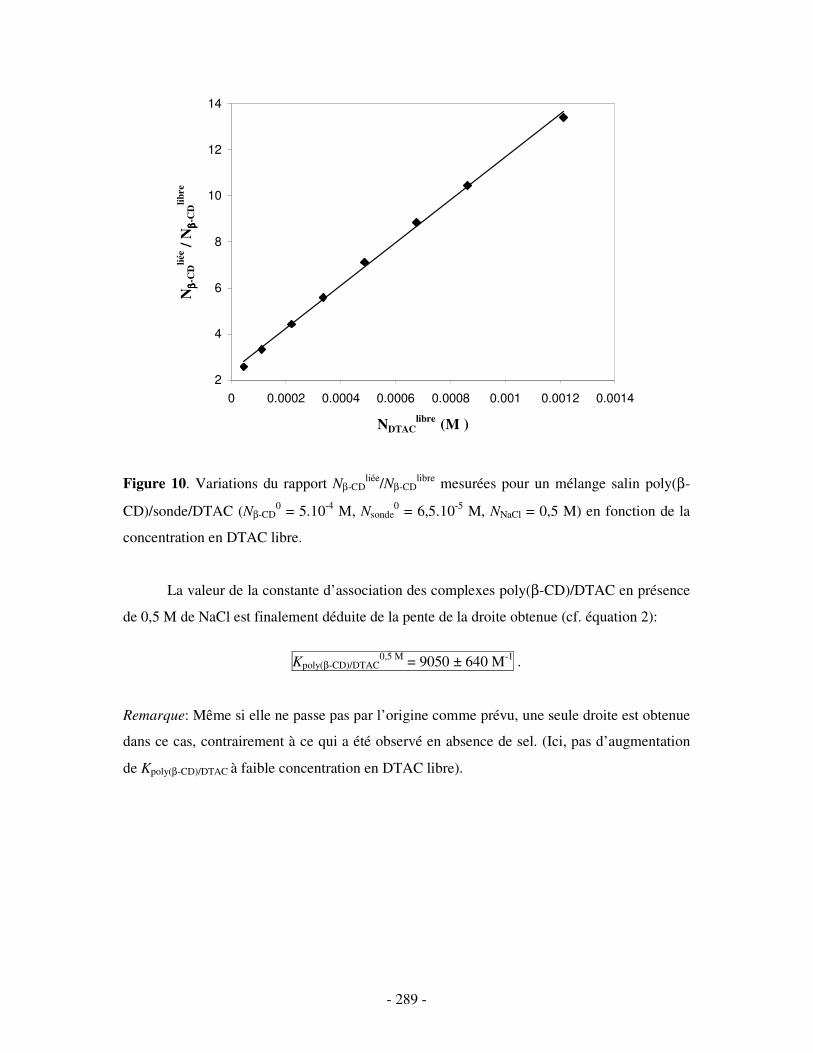
- 289 -
2
4
6
8
10
12
14
0 0.0002 0.0004 0.0006 0.0008 0.001 0.0012 0.0014
NDTAClibre
(M )
Nββ ββ-
CD
liée /
Nββ ββ-
CD
libre
Figure 10. Variations du rapport Nβ-CDliée/Nβ-CD
libre mesurées pour un mélange salin poly(β-
CD)/sonde/DTAC (Nβ-CD0 = 5.10-4 M, Nsonde
0 = 6,5.10-5 M, NNaCl = 0,5 M) en fonction de la
concentration en DTAC libre.
La valeur de la constante d’association des complexes poly(β-CD)/DTAC en présence
de 0,5 M de NaCl est finalement déduite de la pente de la droite obtenue (cf. équation 2):
Kpoly(β-CD)/DTAC
0,5 M = 9050 ± 640 M-1 .
Remarque: Même si elle ne passe pas par l’origine comme prévu, une seule droite est obtenue
dans ce cas, contrairement à ce qui a été observé en absence de sel. (Ici, pas d’augmentation
de Kpoly(β-CD)/DTAC à faible concentration en DTAC libre).

- 290 -
Annexe 2.
Etude par mesures de tension de surface du complexe binaire
poly(β-CD)/DTAC en présence de sel
Les figures 1 et 2 présentent respectivement l’influence de la force ionique sur les
variations de tension de surface de solutions de DTAC seul et de mélanges poly(β-
CD)/DTAC (Nβ-CD = 14.10-3 M).
Les courbes de la figure 1 ont été obtenues en ajoutant des volumes croissants:
( ) d’une solution mère de DTAC (NDTACi = 40.10-3 M) dans de l’eau pure (Veau
i = 10 mL),
() d’une solution saline de DTAC (NDTACi = 30.10-3 M, NNaCl = 0,015 M) dans une solution
de NaCl (Vi = 10 mL, NNaCl = 0,015 M),
() d’une solution saline de DTAC (NDTACi = 20.10-3 M, NNaCl = 0,15 M) dans une solution de
NaCl (Vi = 10 mL, NNaCl = 0,15 M),
() d’une solution saline de DTAC (NDTACi = 15.10-3 M, NNaCl = 0,5 M) dans une solution de
NaCl (Vi = 10 mL, NNaCl = 0,5 M).
Les courbes de la figure 2, quant à elles, ont été tracées après avoir ajouté des volumes
croissants:
( ) d’un mélange poly(β-CD)/DTAC (Nβ-CD = 14.10-3 M, NDTACi = 80.10-3 M) dans une
solution de poly(β-CD) (V i = 10 mL, Nβ-CD = 14.10-3 M),
() d’un mélange salin de poly(β-CD) et de DTAC (Nβ-CD = 14.10-3 M, NDTACi = 80.10-3 M,
NNaCl = 0,015 M) dans une solution saline de poly(β-CD) (V i = 10 mL, Nβ-CD = 14.10-3 M,
NNaCl = 0,015 M),
() d’un mélange salin de poly(β-CD) et de DTAC (Nβ-CD = 14.10-3 M, NDTACi = 60.10-3 M,
NNaCl = 0,15 M) dans une solution saline de poly(β-CD) (V i = 10 mL, Nβ-CD = 14.10-3 M, NNaCl
= 0,15 M),
() d’un mélange salin de poly(β-CD) et de DTAC (Nβ-CD = 14.10-3 M, NDTACi = 60.10-3 M,
NNaCl = 0,5 M) dans une solution saline de poly(β-CD) (V i = 10 mL, Nβ-CD = 14.10-3 M, NNaCl
= 0,5 M).
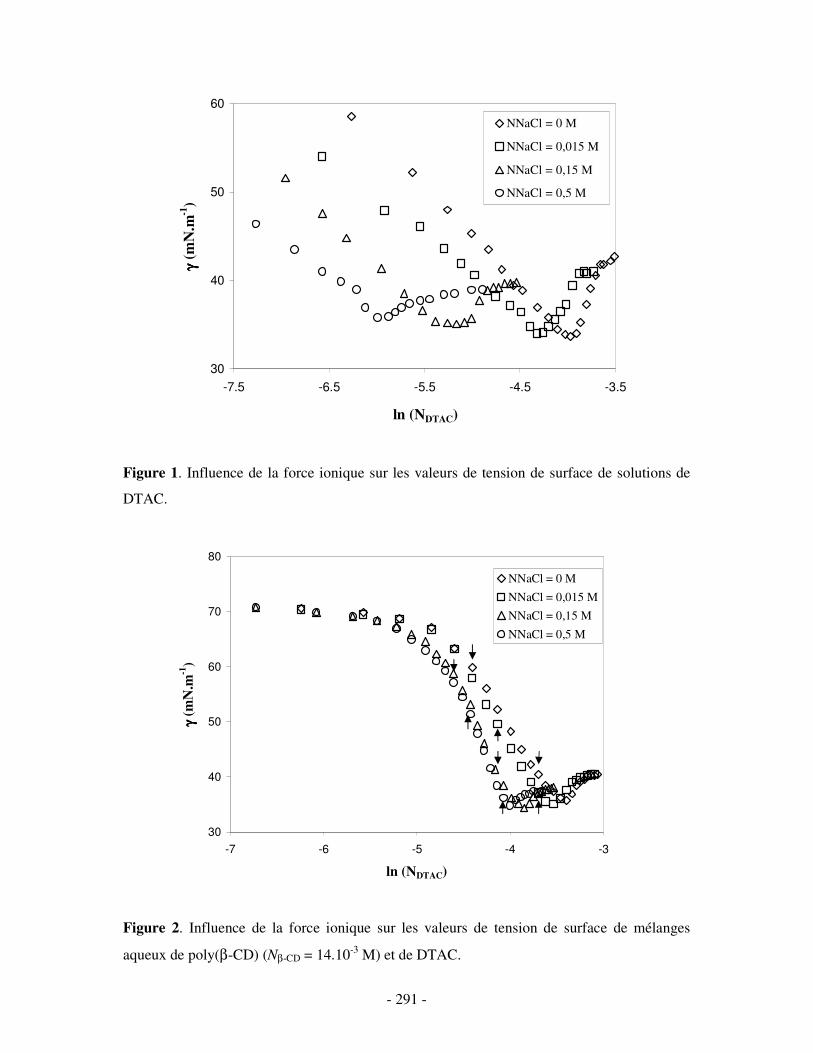
- 291 -
30
40
50
60
-7.5 -6.5 -5.5 -4.5 -3.5
ln (NDTAC)
γ γ γ γ (m
N.m
-1)
NNaCl = 0 M
NNaCl = 0,015 M
NNaCl = 0,15 M
NNaCl = 0,5 M
Figure 1. Influence de la force ionique sur les valeurs de tension de surface de solutions de
DTAC.
30
40
50
60
70
80
-7 -6 -5 -4 -3
ln (NDTAC)
γ γ γ γ (m
N.m
-1)
NNaCl = 0 M
NNaCl = 0,015 MNNaCl = 0,15 M
NNaCl = 0,5 M
Figure 2. Influence de la force ionique sur les valeurs de tension de surface de mélanges
aqueux de poly(β-CD) (Nβ-CD = 14.10-3 M) et de DTAC.

- 292 -
Les valeurs de cmc relevées sur les figures 1 et 2 en fonction des différentes
concentrations en NaCl sont reportées dans le tableau 1. Comme il l’a déjà été constaté dans
la littérature52, l’ajout de sel réduit non seulement la cmc du TA pur, mais aussi celle du
système mixte poly(β-CD)/DTAC. En écrantant les répulsions électrostatiques entre les
groupements ioniques des molécules de TA, le sel favorise l’agrégation de ces dernières en
solution. Par conséquent, la formation de micelles en présence de sel ne nécessite pas une
concentration en monomères de TA aussi élevée que dans l’eau pure.
NNaCl (M) cmcDTAC cmc* cmc*- cmc R
0 19 34 15 0,93
0,015 13 29 16 0,875
0,15 6 21 15 0,93
0,5 2,5 18 15,5 0,90
Tableau 1. Influence du sel sur la cmc du DTAC seul et celle du système mixte poly(β-
CD)/DTAC (Nβ-CD = 14.10-3 M). R désigne la stoechiométrie du complexe poly(β-CD)/DTAC
déterminée à partir de la relation de Junquera198.
Le tableau 1 indique également que l’augmentation de cmc due à la présence des
cavités de β-CD (cmc*- cmc) reste quasiment constante quelque soit la concentration en NaCl.
Le sel n’a donc aucune influence sur le mode d’association poly(β-CD)/DTAC. Les cavités de
β-CD du polymère forment toujours des complexes d’inclusion de type 1:1 avec les molécules
de DTAC, comme en témoignent les valeurs de la stoechiométrie R présentées dans le tableau
1, calculées à partir de la relation de Junquera198:
cmc* = cmc + R
N CD−β .
L’application de la méthode de Lu50 aux données expérimentales des figures 1 et 2
dans les domaines prémicellaires (délimités par les flèches) permet de tracer les segments de
droite présentés sur la figure suivante:
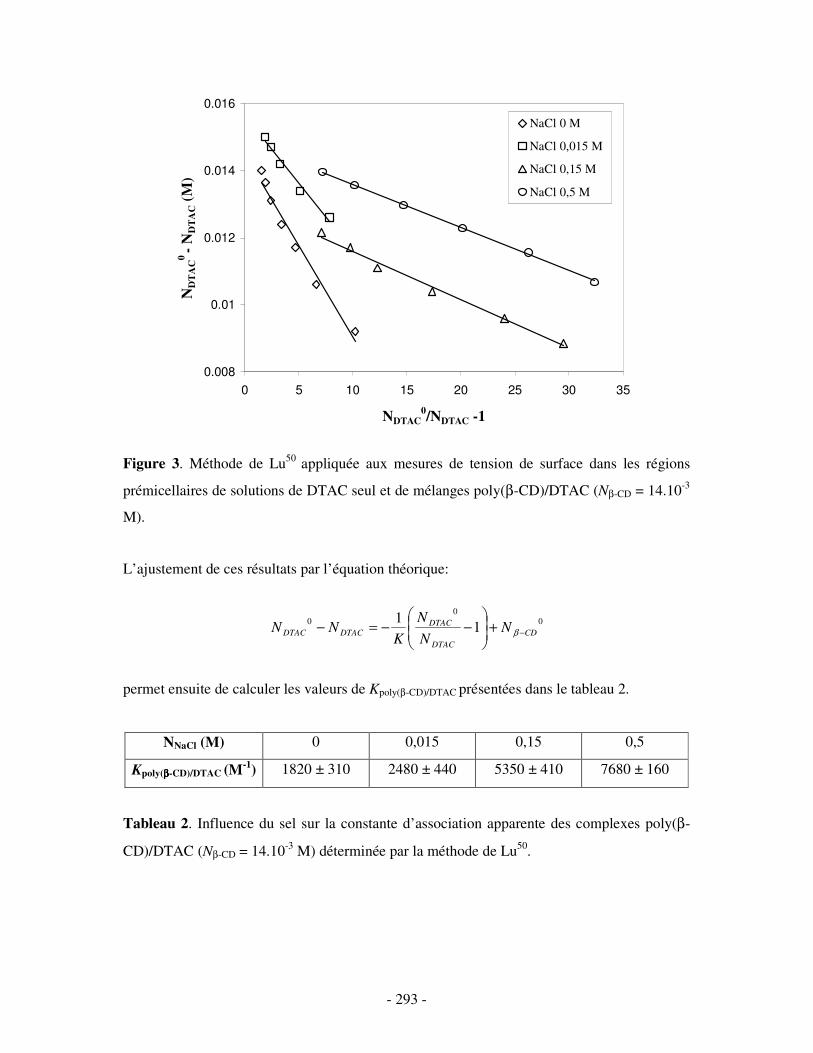
- 293 -
0.008
0.01
0.012
0.014
0.016
0 5 10 15 20 25 30 35
NDTAC0/NDTAC -1
ND
TA
C0 - N
DT
AC (M
)
NaCl 0 M
NaCl 0,015 M
NaCl 0,15 M
NaCl 0,5 M
Figure 3. Méthode de Lu50 appliquée aux mesures de tension de surface dans les régions
prémicellaires de solutions de DTAC seul et de mélanges poly(β-CD)/DTAC (Nβ-CD = 14.10-3
M).
L’ajustement de ces résultats par l’équation théorique:
00
0 11
CDDTAC
DTACDTACDTAC N
NN
KNN −+
−−=− β
permet ensuite de calculer les valeurs de Kpoly(β-CD)/DTAC présentées dans le tableau 2.
NNaCl (M) 0 0,015 0,15 0,5
Kpoly(ββββ-CD)/DTAC (M-1) 1820 ± 310 2480 ± 440 5350 ± 410 7680 ± 160
Tableau 2. Influence du sel sur la constante d’association apparente des complexes poly(β-
CD)/DTAC (Nβ-CD = 14.10-3 M) déterminée par la méthode de Lu50.

- 294 -
Annexe 3.
Calcul des densités de longueur de diffusion ρ
La densité de longueur de diffusion ρ des composants (exceptée celle du poly(β-CD)
déterminée expérimentalement par la méthode de variation de contraste) a été calculée à l’aide
de la relation:
monomère
a
Mda ℵ
=ρ
où aℵ = 6,023.1023 mol-1 est le nombre d’Avogadro, d, la densité du composant dans l’eau et
ii
i ana = , sa longueur de diffusion (ou celle de l’unité de répétition dans le cas d’un
polymère), soit la somme des longueurs de diffusion des N atomes qui le composent
( =i
inN , ni étant le nombre d’atomes de l’espèce i). Les valeurs ai de différents atomes
sont présentées dans le tableau 1.
i ai (.10-12 cm)
H -0,374
D 0,667
C 0,665
O 0,5805
N 0,930
Cl 1,160
Na 0,362
S 0,288
Tableau 1. Longueur de diffusion de différents atomes.
A partir des données du tableau 1, les valeurs de a de différents composants utilisés
dans ce travail de thèse ont pu être déterminées (tableau 2).
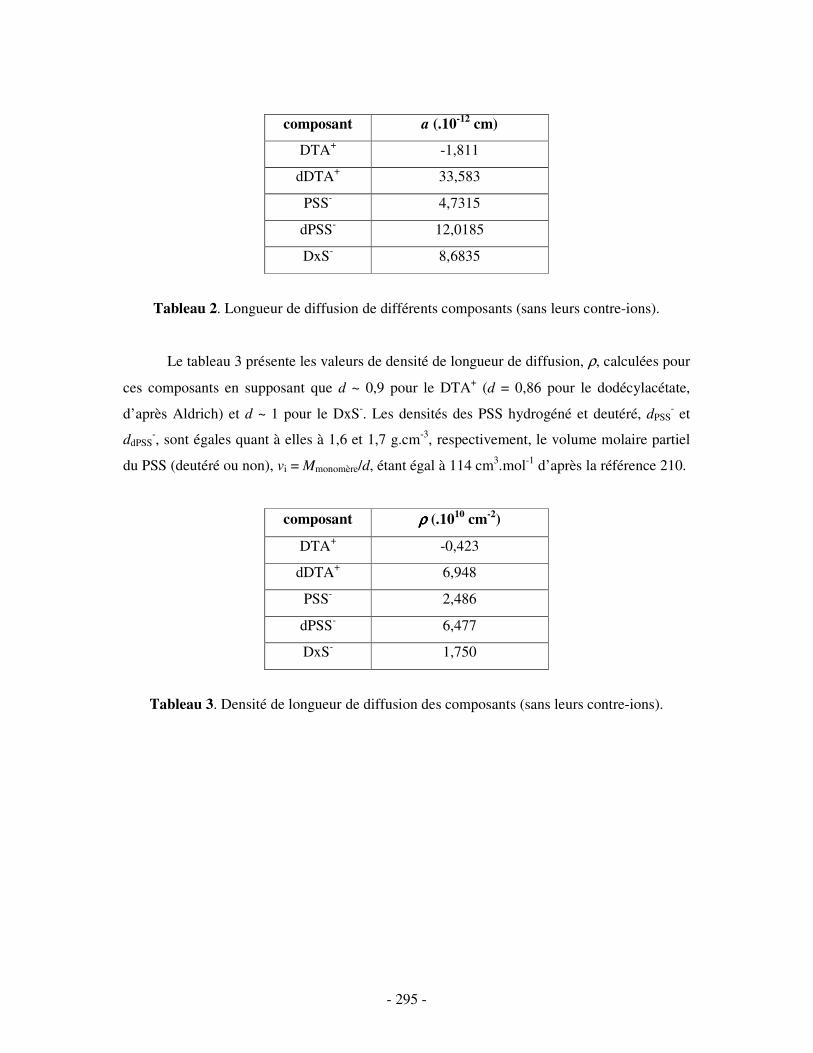
- 295 -
composant a (.10-12 cm)
DTA+ -1,811
dDTA+ 33,583
PSS- 4,7315
dPSS- 12,0185
DxS- 8,6835
Tableau 2. Longueur de diffusion de différents composants (sans leurs contre-ions).
Le tableau 3 présente les valeurs de densité de longueur de diffusion, ρ, calculées pour
ces composants en supposant que d ~ 0,9 pour le DTA+ (d = 0,86 pour le dodécylacétate,
d’après Aldrich) et d ~ 1 pour le DxS-. Les densités des PSS hydrogéné et deutéré, dPSS- et
ddPSS-, sont égales quant à elles à 1,6 et 1,7 g.cm-3, respectivement, le volume molaire partiel
du PSS (deutéré ou non), vi = Mmonomère/d, étant égal à 114 cm3.mol-1 d’après la référence 210.
composant ρρρρ (.1010 cm-2)
DTA+ -0,423
dDTA+ 6,948
PSS- 2,486
dPSS- 6,477
DxS- 1,750
Tableau 3. Densité de longueur de diffusion des composants (sans leurs contre-ions).
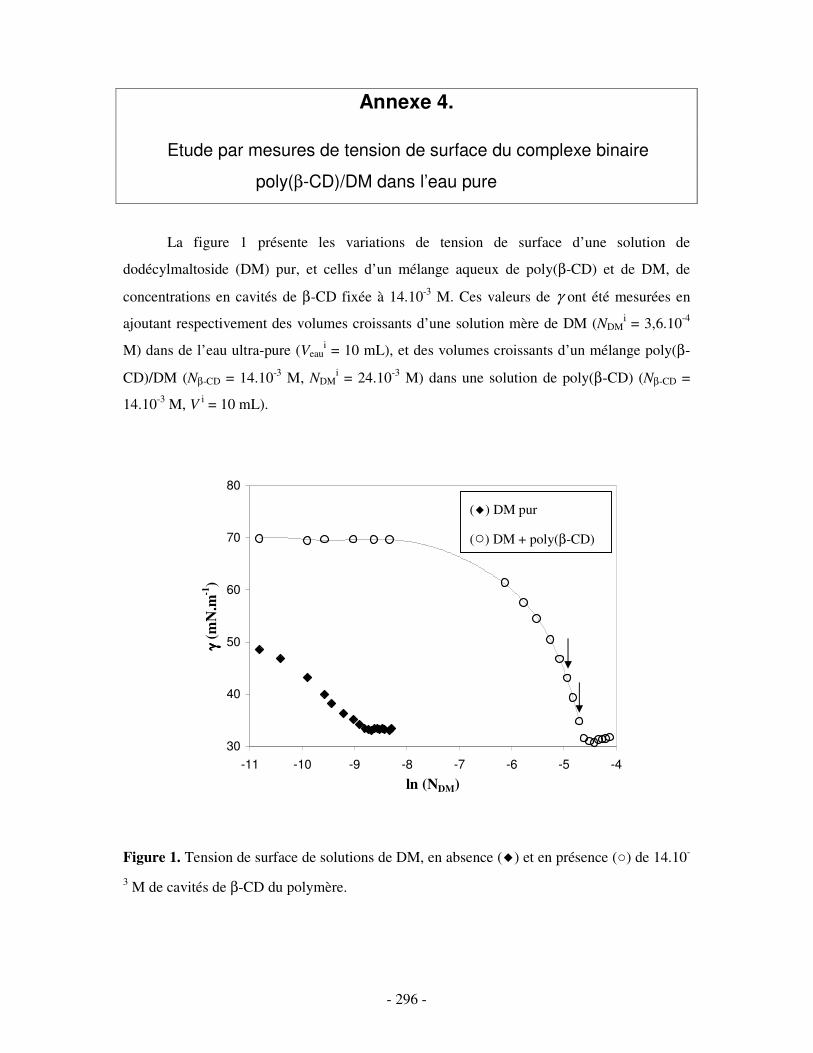
- 296 -
30
40
50
60
70
80
-11 -10 -9 -8 -7 -6 -5 -4
ln (NDM)
γγ γγ (m
N.m
-1)
DM pur
DM +poly(BCD)
Annexe 4.
Etude par mesures de tension de surface du complexe binaire
poly(β-CD)/DM dans l’eau pure
La figure 1 présente les variations de tension de surface d’une solution de
dodécylmaltoside (DM) pur, et celles d’un mélange aqueux de poly(β-CD) et de DM, de
concentrations en cavités de β-CD fixée à 14.10-3 M. Ces valeurs de γ ont été mesurées en
ajoutant respectivement des volumes croissants d’une solution mère de DM (NDMi = 3,6.10-4
M) dans de l’eau ultra-pure (Veaui = 10 mL), et des volumes croissants d’un mélange poly(β-
CD)/DM (Nβ-CD = 14.10-3 M, NDMi = 24.10-3 M) dans une solution de poly(β-CD) (Nβ-CD =
14.10-3 M, V i = 10 mL).
Figure 1. Tension de surface de solutions de DM, en absence () et en présence () de 14.10-
3 M de cavités de β-CD du polymère.
() DM pur
() DM + poly(β-CD)
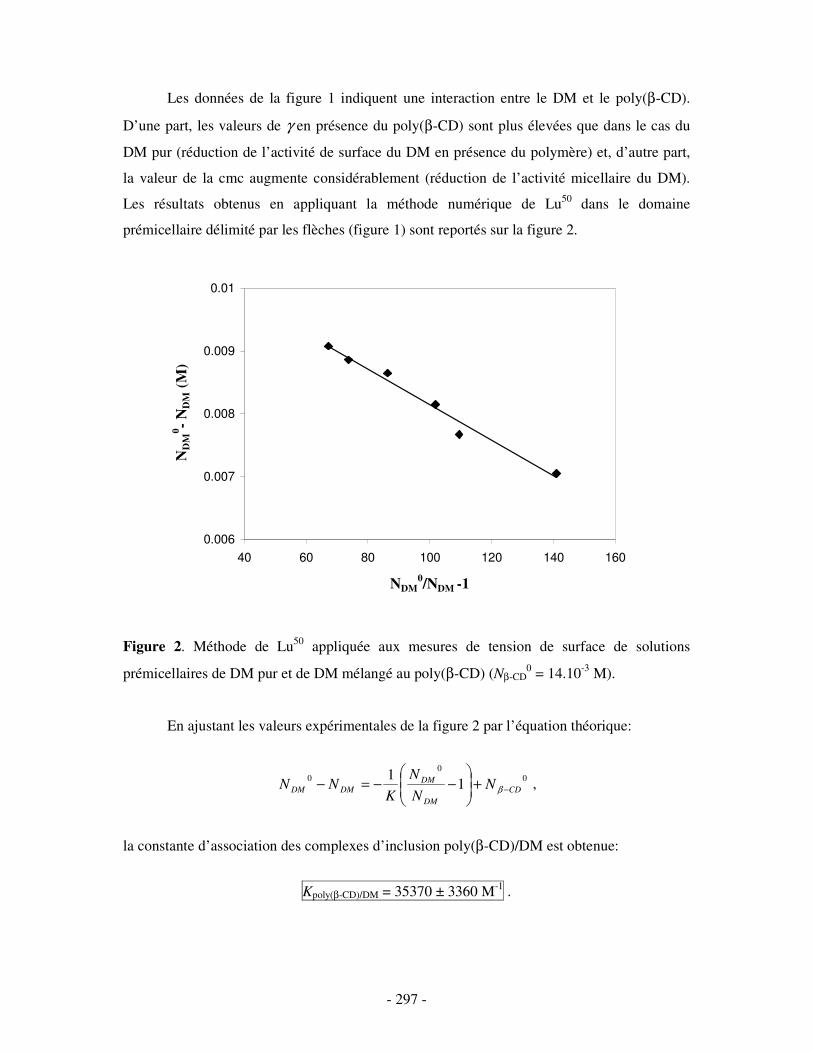
- 297 -
Les données de la figure 1 indiquent une interaction entre le DM et le poly(β-CD).
D’une part, les valeurs de γ en présence du poly(β-CD) sont plus élevées que dans le cas du
DM pur (réduction de l’activité de surface du DM en présence du polymère) et, d’autre part,
la valeur de la cmc augmente considérablement (réduction de l’activité micellaire du DM).
Les résultats obtenus en appliquant la méthode numérique de Lu50 dans le domaine
prémicellaire délimité par les flèches (figure 1) sont reportés sur la figure 2.
0.006
0.007
0.008
0.009
0.01
40 60 80 100 120 140 160
NDM0/NDM -1
ND
M0 - N
DM
(M)
Figure 2. Méthode de Lu50 appliquée aux mesures de tension de surface de solutions
prémicellaires de DM pur et de DM mélangé au poly(β-CD) (Nβ-CD0 = 14.10-3 M).
En ajustant les valeurs expérimentales de la figure 2 par l’équation théorique:
00
0 11
CDDM
DMDMDM N
NN
KNN −+
−−=− β ,
la constante d’association des complexes d’inclusion poly(β-CD)/DM est obtenue:
Kpoly(β-CD)/DM = 35370 ± 3360 M-1 .

- 298 -
Cette valeur est comparable à celle déterminée par Funasaki et coll.59 pour les complexes β-
CD/DM: Kβ-CD/DM = 26200 ± 1900 M-1 (à 20°C, pour Nβ-CD comprise entre 0,1 et 1,7.10-3 M).

- 299 -
Annexe 5.
Etude par QCM des interactions poly(β-CD)/polyanion
I- Conditions expérimentales
1- Le polymère de CD utilisé
Le poly(β-CD) utilisé dans cette étude est un copolymère cationique (poly(β-CD)⊕)
obtenu par réaction entre du 2,3-époxypropyltriméthylammonium (Fluka, France) et un
polymère neutre de β-CD. Ce dernier a été obtenu par polycondensation entre des monomères
de β-CD et d’épichlorhydrine. Sa masse molaire moyenne, déterminée par chromatographie
d’exclusion par la taille, est de 33 kg.mol-1 en équivalent pullulane et sa composition en β-
cyclodextrine est de 65% en masse.
O
O
O
O
OO
O
O
O
O
OO
O
O
HO
HO
OH
OHOH
OH
HO
OH
OH
HO
HO
OH
OH
OH H2C
CH2OH
CH2CH2OH
HOH2C
HOH2C
O CH2 CH
OH
CH2 OH2C
O
O O
HO
OH
H2CHC
OH
H2C
O
O
O
OH
HO
O CH2
O (CH2)3 N(CH3)3
Cl
Figure 1. Formule chimique du poly(β-CD)⊕.

- 300 -
La composition en groupements ammonium du poly(β-CD)⊕ a été déterminée par titrage
potentiométrique des contre-ions Cl- et correspond en moyenne à 0,6 charge positive par
monomère de β-CD.
2- La QCM (Quartz Crystal Microbalance)
a) Principe
La QCM est une technique de pesée ultra-sensible basée sur le principe de l’oscillateur
électromécanique piézoélectrique.
Un matériau piézoélectrique a la propriété de se polariser lorsqu’il subit une contrainte
mécanique. A l’inverse, ce même matériau subit une déformation lorsqu’il est soumis à un
champ électrique et cette déformation a la particularité d’être réversible.
Une QCM est constituée d’un fin disque de quartz - le quartz est la forme cristalline
piézoélectrique du dioxyde de silicium (SiO2) - , recouvert d’une électrode métallique (en or
généralement) sur chacune de ses faces. Lorsque ses électrodes sont soumises à un champ
électrique alternatif, le cristal peut être amené à vibrer de manière stable à sa fréquence de
résonance, f. La condition de résonance du cristal est la suivante:
qtnv
f2
= , (1)
où v est la vitesse des ondes longitudinales engendrées par les vibrations du quartz, tq,
l’épaisseur du cristal de quartz et n, un nombre entier impair (1,3,5…).
L’adsorption d’une masse sur l’une des électrodes du cristal (ou sur les deux
électrodes) induit une variation de la fréquence de résonance du quartz, ∆f. Si la masse
adsorbée, ∆m, est faible comparée à celle du cristal, si elle est répartie de façon uniforme sur
la (ou les) électrode(s) et qu’elle y est rigidement attachée (pas de glissement, ni de
déformation dus au mouvement oscillatoire), alors elle est proportionnelle à la variation de
fréquence de résonance, ∆f, selon la relation de Sauerbrey:
nfC
m∆−=∆ , (2)

- 301 -
avec 0f
tC qqρ
= (3)
où ρq est la densité spécifique du quartz, tq, l’épaisseur du cristal et f0, la fréquence de
résonance fondamentale. Avec ρq = 2648 kg.m-3, tq = 0,33 mm et f0 = 5 MHz, la constante
caractéristique du cristal est de l’ordre de 17,7 ng.cm-2.Hz-1. Il est possible de mesurer de très
faibles variations de fréquence de résonance (de l’ordre de 1 Hz dans l’eau à 25°C), et donc
de déterminer de très faibles masses (∼ 5 ng.cm-2) puisque le cristal oscille généralement de
façon très stable.
Outre la masse adsorbée, la QCM permet de mesurer simultanément les variations du
facteur de dissipation, D, proportionnel à l’énergie dissipée dans le système:
éeemmaga
dissipée
E
ED
sin2π= , (4)
où Edissipée est l’énergie dissipée durant une période de l’oscillation du cristal et Eemmagasinée,
l’énergie emmagasinée dans le système oscillant. D est déterminé à partir du temps
caractéristique de la décroissance du signal, τ, mesuré lorsque le champ électrique n’est plus
appliqué. Une fois le champ électrique éteint, l’amplitude du signal varie effectivement selon
)2sin( ϕπτ +−
ftet
, τ étant fonction de la fréquence f. Ainsi, pour la fréquence fondamentale f0,
le facteur de dissipation mesuré dépend de τ selon la relation:
τπ 0
1f
D = . (5)
Pour un film adsorbé en couche mince, l’énergie est principalement dissipée au travers
du couplage avec le solvant, ce qui correspond à un faible taux de dissipation. Par contre, pour
des structures plus épaisses et visqueuses, l’énergie est dissipée au travers des mouvements
oscillatoires induits dans le film par les vibrations du cristal, ces mouvements pouvant induire
une forte dissipation de l’énergie. En mesurant les variations du facteur de dissipation, ∆D, il
est donc possible d’obtenir des informations supplémentaires sur les propriétés
viscoélastiques de la couche adsorbée. Notons que dans les conditions de solutions diluées, les
effets dus aux changements de viscosité et de densité au sein de la solution sont négligeables.
Seules les variations de dissipation proches de la surface sont prises en compte. Les valeurs de

- 302 -
∆D mesurées comprennent d’une part la dissipation d’énergie au sein de la couche adsorbée
et, d’autre part, les différences de couplage entre solvant/surface nue et solvant/surface
recouverte de la couche adsorbée.
Remarque: La relation de Sauerbrey (équation 2) reliant la masse adsorbée au changement de
fréquence de résonance, n’est valide que pour des variations de dissipation nulles (de l’ordre
de 10-6 unités).
b) Appareillage
Ces études de surface ont été réalisées lors d’un stage d’un mois effectué au
laboratoire du Surface Force Group KTH/YKI dirigé par le Prof. Claesson, au Royal Institute
of Technology de Stockholm (Suède). Les mesures ont été effectuées à l’aide d’un instrument
QCM-DTM (Quartz Crystal Microbalance - Dissipation) fourni par Q-Sense (Gothenburg,
Suède). Dans l’appareillage utilisé, les électrodes du cristal sont connectées à un boîtier
électronique qui excite d’abord le quartz à sa fréquence de résonance, puis mesure la
fréquence et la vitesse de décroissance du signal une fois l’application du champ électrique
interrompue. Le boîtier électronique est contrôlé par un ordinateur qui enregistre les mesures
et permet de suivre quantitativement les phénomènes interfaciaux en temps réel.

- 303 -
Figure 2. Schéma simplifié de la QCM-DTM.
Comme l’indique l’équation (1), le cristal résonne lorsqu’il est excité à des multiples
impairs de la fréquence de résonance f0 = 5 MHz. L’instrument QCM-DTM fonctionne avec le
fondamental (n = 1, f = 5 MHz) et le troisième harmonique (n = 3, f = 15 MHz) mais les
mesures réalisées avec ce dernier s’avèrent être plus reproductibles et plus sensibles à la
région en surface du cristal. C’est pourquoi toutes les expériences de cette étude ont été
réalisées en ne considérant que le troisième harmonique.
Seule l’une des deux faces du cristal a été utilisée comme « surface active », l’autre
surface ayant été utilisée en tant qu’électrode uniquement. Après avoir subi une modification
préalable par une fine couche de silice (SiO2), la surface active comporte des groupements
siloxane (Si2O) relativement hydrophobes et des groupements silanol (SiOH) plus
hydrophiles. Dans l’eau ultra-pure (pH ∼ 5,6), la surface active présente ainsi un excès de
charges négatives (groupes SiO-).
chambre de mesure
cristal de quartz
boîtier électronique
entrée sortie

- 304 -
Figure 3. Schéma du cristal de quartz. Les parties grisées représentent les électrodes en or. La
patte de connexion au bas de la face inférieure du cristal est celle de la face « active ».
Toutes les expériences ont été réalisées à température ambiante. Les surfaces de silice
utilisées ont été préalablement traitées à l’acide bichromosulfurique (dépôt d’une goutte
d’acide sur la face active durant quelques minutes), puis rincées à l’eau ultra-pure et à
l’éthanol. Une surface est parfaitement propre, c’est-à-dire parfaitement hydrophile,
lorsqu’une goutte d’eau déposée s’y étale complètement (formation d’un film d’eau à la
surface du cristal). Le matériel utilisé pour la préparation des solutions a quant à lui été
préalablement nettoyé avec une solution de Deconex (tensioactif), puis rincé à l’eau ultra-
pure.
II- Résultats
La première étape a consisté en l’adsorption du polymère de β-CD (chargé
positivement) sur la surface de silice (chargée négativement). Après avoir obtenu une ligne de
base stable avec de l’eau ultra-pure, une solution de poly(β-CD)⊕ à 2 ppm a donc été injectée
dans la chambre de mesure (injection 1, figure 4). Il est à noter que toute nouvelle injection
dans la chambre de mesure induit instantanément une chute de fréquence du fait du
changement brusque de température et surtout de pression.
Face inférieure
pattes de connexion à
l’électronique
Face supérieure « active »
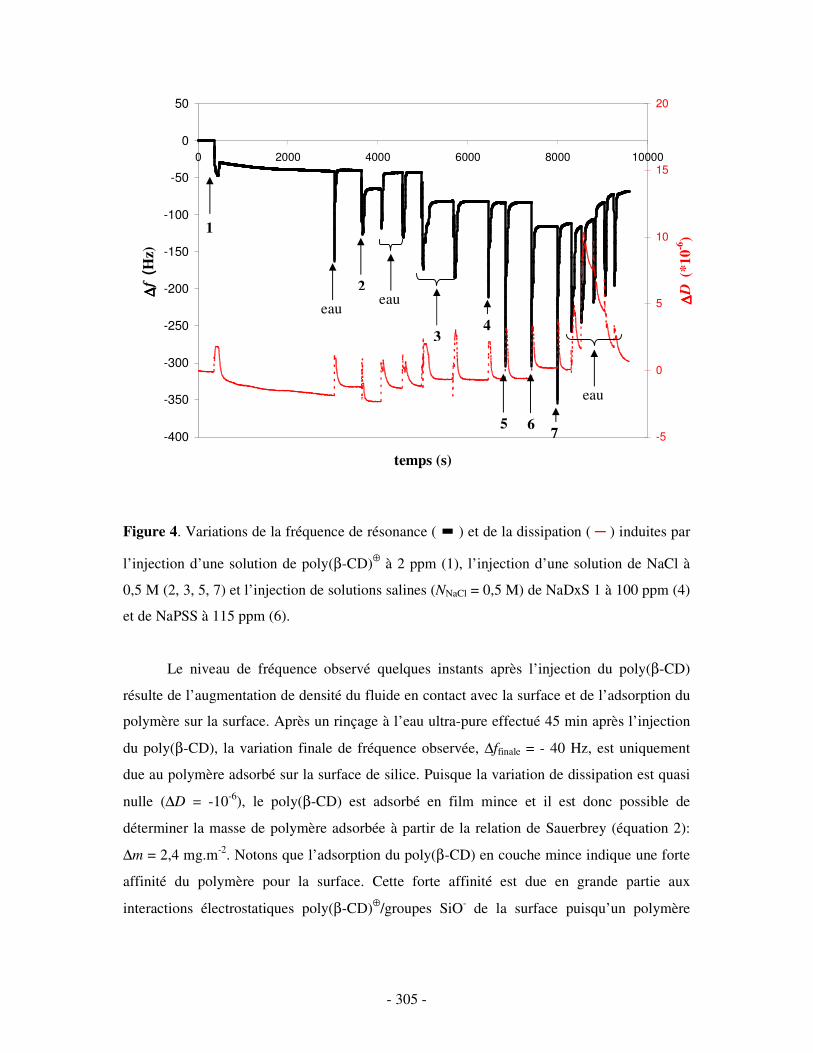
- 305 -
-400
-350
-300
-250
-200
-150
-100
-50
0
50
0 2000 4000 6000 8000 10000
temps (s)
∆∆ ∆∆f
( Hz)
-5
0
5
10
15
20
∆∆ ∆∆D
(*10
-6)
Figure 4. Variations de la fréquence de résonance ( ) et de la dissipation ( ) induites par
l’injection d’une solution de poly(β-CD)⊕ à 2 ppm (1), l’injection d’une solution de NaCl à
0,5 M (2, 3, 5, 7) et l’injection de solutions salines (NNaCl = 0,5 M) de NaDxS 1 à 100 ppm (4)
et de NaPSS à 115 ppm (6).
Le niveau de fréquence observé quelques instants après l’injection du poly(β-CD)
résulte de l’augmentation de densité du fluide en contact avec la surface et de l’adsorption du
polymère sur la surface. Après un rinçage à l’eau ultra-pure effectué 45 min après l’injection
du poly(β-CD), la variation finale de fréquence observée, ∆ffinale = - 40 Hz, est uniquement
due au polymère adsorbé sur la surface de silice. Puisque la variation de dissipation est quasi
nulle (∆D = -10-6), le poly(β-CD) est adsorbé en film mince et il est donc possible de
déterminer la masse de polymère adsorbée à partir de la relation de Sauerbrey (équation 2):
∆m = 2,4 mg.m-2. Notons que l’adsorption du poly(β-CD) en couche mince indique une forte
affinité du polymère pour la surface. Cette forte affinité est due en grande partie aux
interactions électrostatiques poly(β-CD)⊕/groupes SiO- de la surface puisqu’un polymère
1
eau 2
eau
3 4
5 6 7
eau

- 306 -
neutre de β-CD (Mw = 40 kg.mol-1 en équivalent pullulane, soit Mw = 160 kg.mol-1) s’adsorbe
en plus faible quantité: ∆m = 0,6 mg.m-2, ce qui correspond à une ∆ffinale de -10 Hz.
Afin de mettre en évidence d’éventuelles interactions non coulombiennes entre le
polymère de β-CD et les différents polyanions utilisés, ces derniers ont été injectés en
présence de sel (NNaCl = 0,5 M). L’injection d’une simple solution de NaCl à 0,5 M (injection
2, figure 4), suivie par deux rinçages à l’eau ultra-pure, a tout d’abord permis de montrer que
le poly(β-CD) n’est pas désorbable par ajout de sel ni par dilution. La variation finale de
fréquence obtenue (∆ffinale = - 43 Hz) est en effet quasiment identique à celle observée avant
l’injection de NaCl, ce qui prouve la forte affinité du polymère pour la surface.
Afin d’établir une référence en sel pour les injections des polyanions en solutions
salines (NNaCl = 0,5 M), deux nouvelles injections de la solution de NaCl à 0,5 M ont été
effectuées (injections 3, figure 4): l’augmentation de densité due à la simple présence du sel
induit une chute de fréquence de - 42 Hz. Une solution saline de NaDxS 1 à 100 ppm (NNaCl =
0,5 M) a ensuite été introduite (injection 4) induisant de très faibles variations de fréquence et
de dissipation. Après un rinçage avec la solution de NaCl à 0,5 M (injection 5), la variation
finale de fréquence observée est de - 2 Hz seulement par rapport à la référence en sel, ce qui
suggère que le dextrane sulfate ne s’est pas adsorbé (ou alors en très faible quantité) sur la
couche de poly(β-CD). Par QCM, aucune interaction poly(β-CD)/NaDxS 1 n’est donc été
mise en évidence de façon probante.
Une solution saline de NaPSS à 115 ppm (NNaCl = 0,5 M) a ensuite été introduite
(injection 6), suivie par un rinçage avec la solution de NaCl à 0,5 M (injection 7). La chute de
fréquence observée par rapport à la ligne de base en sel, ∆ffinale = - 28 Hz, est cette fois
significative d’une adsorption non négligeable du polymère sur la couche de poly(β-CD). La
variation de dissipation étant très faible, il est possible, d’une part, de calculer l’excès de
masse correspondant à l’adsorption du PSS: ∆m = 1,7 mg.m-2 et, d’autre part, de donner une
première indication sur la force de l’interaction entre les deux polymères. Le fait que le PSS
s’adsorbe en couche mince sur le poly(β-CD) indique en effet une forte affinité entre les deux
polymères: de nombreuses connexions sont établies entre eux de sorte qu’aucune portion de
chaîne du PSS ne puisse s’écarter de la surface et s’étendre vers le reste de la solution (pas de
gonflement des chaînes de PSS). Ensuite, le fait que cinq rinçages successifs à l’eau pure ne
suffisent à désorber le PSS (niveau de fréquence observé pour le poly(β-CD) seul non
retrouvé), est une seconde indication de la forte interaction existant entre les deux polymères.
Notons qu’une augmentation de ∆D est observée au cours des premiers rinçages à l’eau pure,
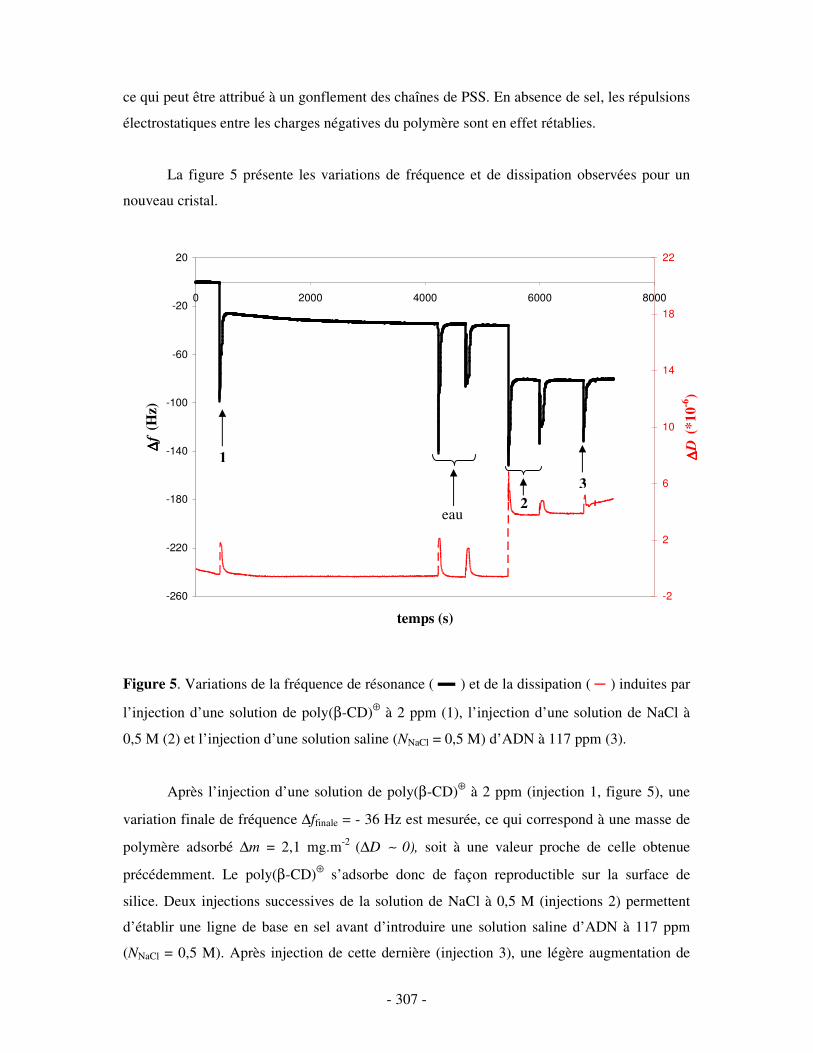
- 307 -
ce qui peut être attribué à un gonflement des chaînes de PSS. En absence de sel, les répulsions
électrostatiques entre les charges négatives du polymère sont en effet rétablies.
La figure 5 présente les variations de fréquence et de dissipation observées pour un
nouveau cristal.
-260
-220
-180
-140
-100
-60
-20
20
0 2000 4000 6000 8000
temps (s)
∆∆ ∆∆f
(Hz)
-2
2
6
10
14
18
22
∆∆ ∆∆D
(*10
-6)
Figure 5. Variations de la fréquence de résonance ( ) et de la dissipation ( ) induites par
l’injection d’une solution de poly(β-CD)⊕ à 2 ppm (1), l’injection d’une solution de NaCl à
0,5 M (2) et l’injection d’une solution saline (NNaCl = 0,5 M) d’ADN à 117 ppm (3).
Après l’injection d’une solution de poly(β-CD)⊕ à 2 ppm (injection 1, figure 5), une
variation finale de fréquence ∆ffinale = - 36 Hz est mesurée, ce qui correspond à une masse de
polymère adsorbé ∆m = 2,1 mg.m-2 (∆D ∼ 0), soit à une valeur proche de celle obtenue
précédemment. Le poly(β-CD)⊕ s’adsorbe donc de façon reproductible sur la surface de
silice. Deux injections successives de la solution de NaCl à 0,5 M (injections 2) permettent
d’établir une ligne de base en sel avant d’introduire une solution saline d’ADN à 117 ppm
(NNaCl = 0,5 M). Après injection de cette dernière (injection 3), une légère augmentation de
1
2 3
eau

- 308 -
dissipation est observée mais aucune variation de fréquence n’est détectée. L’ADN ne
s’adsorbe donc pas (ou très peu) sur la couche de poly(β-CD), ce qui signifie qu’en présence
de sel, il n’existe pas d’interaction majeure entre ces deux polymères.
III- Conclusion
Au cours de cette étude, nous avons prouvé qu’en présence de sel le NaPSS interagit
fortement avec le polymère de β-CD, et ceci sans intervention du DTAC connecteur. Il
semble donc que sous l’effet du sel (écrantage des interactions électrostatiques), l’inclusion du
groupement styrène dans les cages de β-CD soit favorisée. Par contre, aucune interaction
entre le poly(β-CD) et le NaDxS 1 d’une part, et entre le poly(β-CD) et l’ADN d’autre part,
n’a pu vraiment être détectée par QCM.
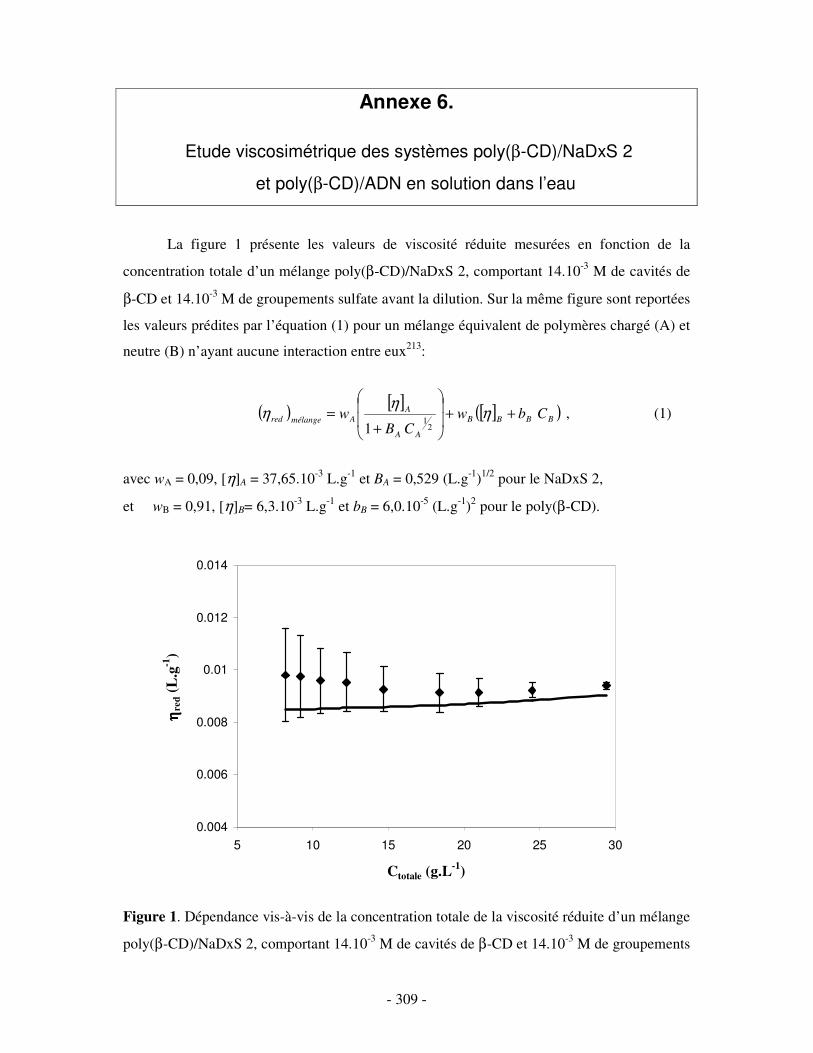
- 309 -
Annexe 6.
Etude viscosimétrique des systèmes poly(β-CD)/NaDxS 2
et poly(β-CD)/ADN en solution dans l’eau
La figure 1 présente les valeurs de viscosité réduite mesurées en fonction de la
concentration totale d’un mélange poly(β-CD)/NaDxS 2, comportant 14.10-3 M de cavités de
β-CD et 14.10-3 M de groupements sulfate avant la dilution. Sur la même figure sont reportées
les valeurs prédites par l’équation (1) pour un mélange équivalent de polymères chargé (A) et
neutre (B) n’ayant aucune interaction entre eux213:
( ) [ ] [ ]( )BBBB
AA
AAmélangered Cbw
CBw ++
+= ηηη
21
1 , (1)
avec wA = 0,09, [η]A = 37,65.10-3 L.g-1 et BA = 0,529 (L.g-1)1/2 pour le NaDxS 2,
et wB = 0,91, [η]B= 6,3.10-3 L.g-1 et bB = 6,0.10-5 (L.g-1)2 pour le poly(β-CD).
0.004
0.006
0.008
0.01
0.012
0.014
5 10 15 20 25 30
Ctotale (g.L-1)
ηη ηη red
(L.g
-1)
Figure 1. Dépendance vis-à-vis de la concentration totale de la viscosité réduite d’un mélange
poly(β-CD)/NaDxS 2, comportant 14.10-3 M de cavités de β-CD et 14.10-3 M de groupements
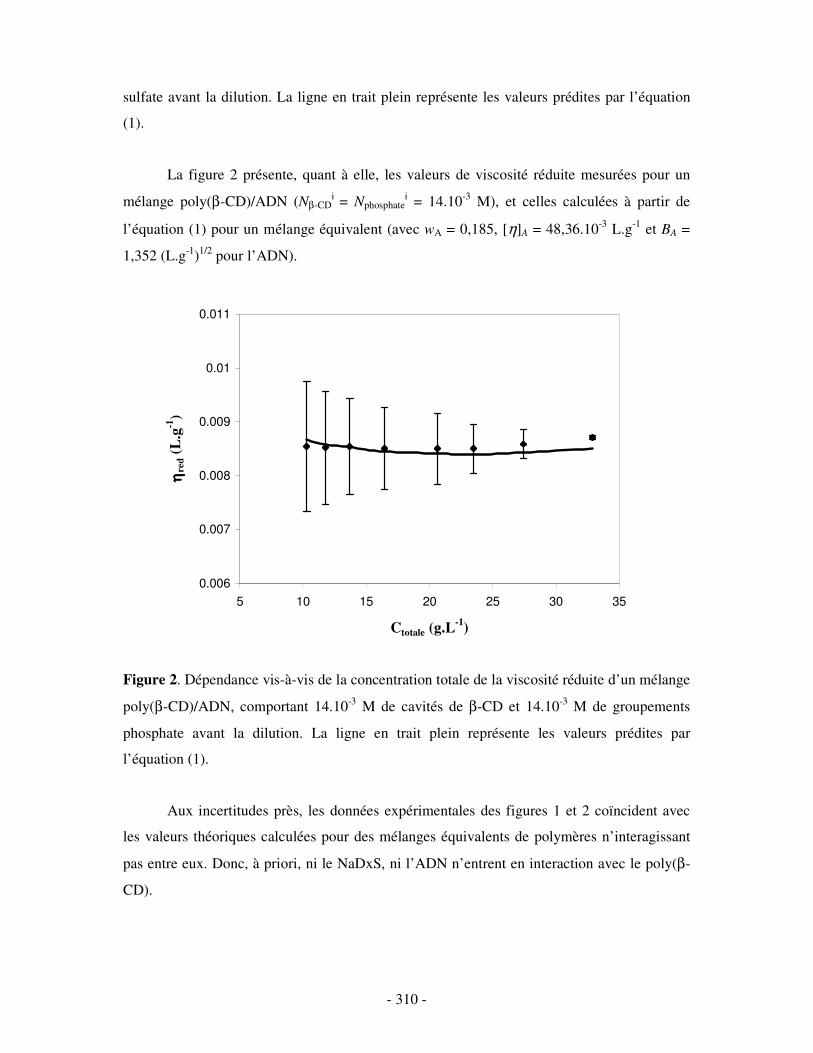
- 310 -
sulfate avant la dilution. La ligne en trait plein représente les valeurs prédites par l’équation
(1).
La figure 2 présente, quant à elle, les valeurs de viscosité réduite mesurées pour un
mélange poly(β-CD)/ADN (Nβ-CDi = Nphosphate
i = 14.10-3 M), et celles calculées à partir de
l’équation (1) pour un mélange équivalent (avec wA = 0,185, [η]A = 48,36.10-3 L.g-1 et BA =
1,352 (L.g-1)1/2 pour l’ADN).
0.006
0.007
0.008
0.009
0.01
0.011
5 10 15 20 25 30 35
Ctotale (g.L-1)
ηη ηη red
(L.g
-1)
Figure 2. Dépendance vis-à-vis de la concentration totale de la viscosité réduite d’un mélange
poly(β-CD)/ADN, comportant 14.10-3 M de cavités de β-CD et 14.10-3 M de groupements
phosphate avant la dilution. La ligne en trait plein représente les valeurs prédites par
l’équation (1).
Aux incertitudes près, les données expérimentales des figures 1 et 2 coïncident avec
les valeurs théoriques calculées pour des mélanges équivalents de polymères n’interagissant
pas entre eux. Donc, à priori, ni le NaDxS, ni l’ADN n’entrent en interaction avec le poly(β-
CD).
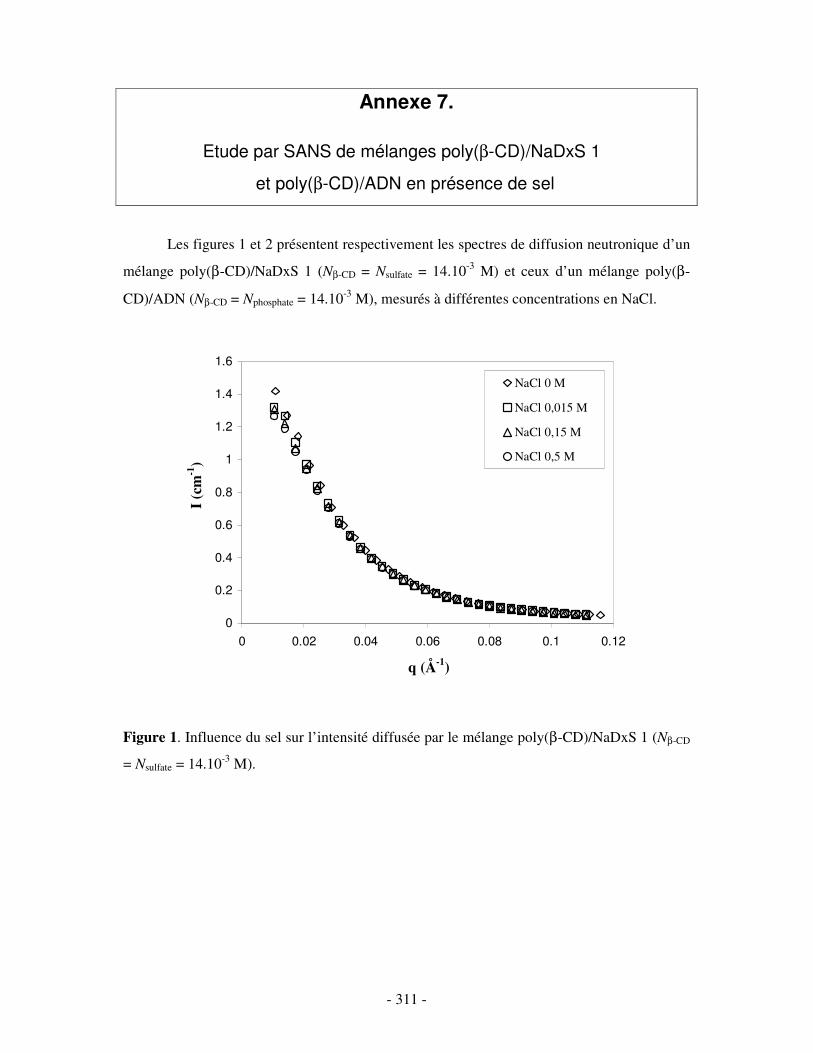
- 311 -
Annexe 7.
Etude par SANS de mélanges poly(β-CD)/NaDxS 1
et poly(β-CD)/ADN en présence de sel
Les figures 1 et 2 présentent respectivement les spectres de diffusion neutronique d’un
mélange poly(β-CD)/NaDxS 1 (Nβ-CD = Nsulfate = 14.10-3 M) et ceux d’un mélange poly(β-
CD)/ADN (Nβ-CD = Nphosphate = 14.10-3 M), mesurés à différentes concentrations en NaCl.
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
0 0.02 0.04 0.06 0.08 0.1 0.12
q (Å-1)
I (cm
-1)
NaCl 0 M
NaCl 0,015 M
NaCl 0,15 M
NaCl 0,5 M
Figure 1. Influence du sel sur l’intensité diffusée par le mélange poly(β-CD)/NaDxS 1 (Nβ-CD
= Nsulfate = 14.10-3 M).
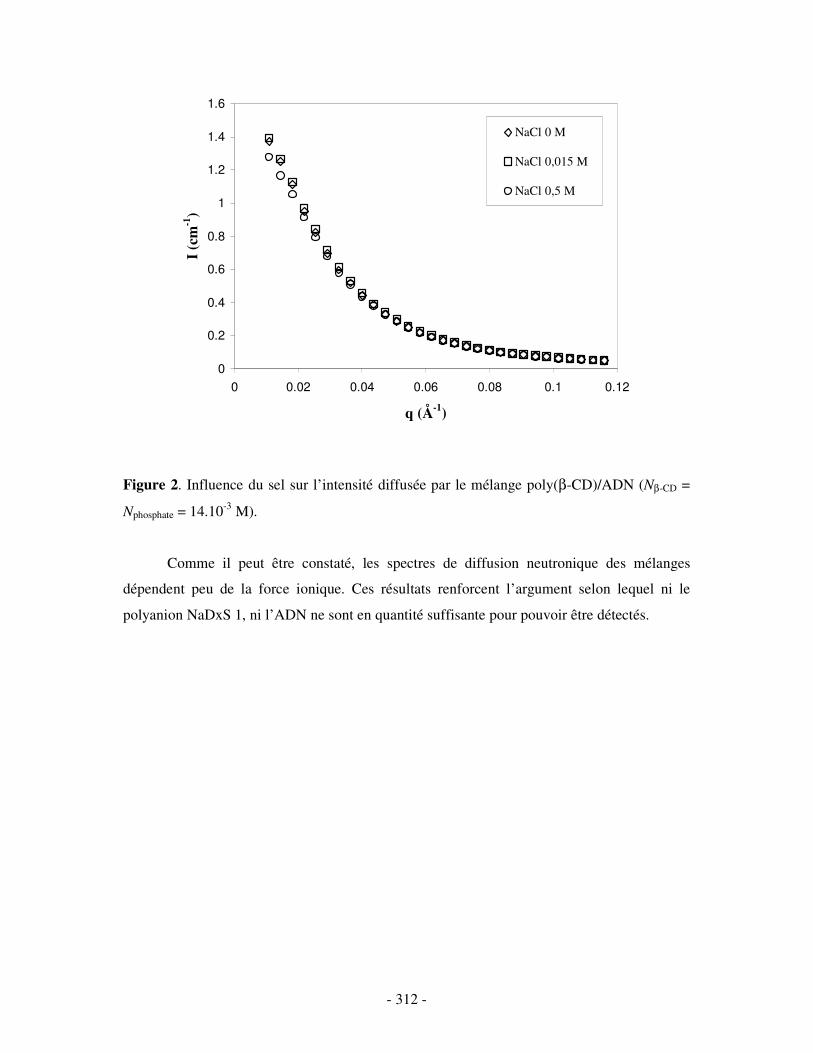
- 312 -
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
0 0.02 0.04 0.06 0.08 0.1 0.12
q (Å-1)
I (cm
-1)
NaCl 0 M
NaCl 0,015 M
NaCl 0,5 M
Figure 2. Influence du sel sur l’intensité diffusée par le mélange poly(β-CD)/ADN (Nβ-CD =
Nphosphate = 14.10-3 M).
Comme il peut être constaté, les spectres de diffusion neutronique des mélanges
dépendent peu de la force ionique. Ces résultats renforcent l’argument selon lequel ni le
polyanion NaDxS 1, ni l’ADN ne sont en quantité suffisante pour pouvoir être détectés.

Résumé: Un nouveau modèle de complexe polyélectrolyte a été élaboré, basé d’une part sur des complexes d’inclusion entre un polymère neutre de β-CD et un tensioactif cationique (DTAC) et, d’autre part, sur des complexes par charges opposées entre le DTAC et un polyanion.
Des interactions d’inclusion entre le poly(β-CD) et le DTAC seuls en solution ont tout d’abord été mises en évidence et évaluées au moyen de plusieurs techniques incluant des mesures conductimétriques et fluorimétriques et des mesures de tension de surface. La structure des agrégats résultants a été étudiée par viscosimétrie et par SANS en fonction de la force ionique et de la stoechiométrie du mélange. La formation de complexes ternaires solubles dans l’eau a ensuite été prouvée lors de l’addition d’un polyanion au mélange poly(β-CD)/DTAC. Pour trois polyanions de natures et d’architectures différentes (NaPSS, NaDxS et ADN), les propriétés structurales des complexes ont été analysées par SANS, par viscosimétrie et par DLS en fonction de la concentration en DTAC. La stabilité et la réversibilité des complexes ont également été étudiées en faisant varier la force ionique du milieu et la concentration en compétiteurs.
Mots clés: complexes polyélectrolytes, polymère de β-cyclodextrine, tensioactif cationique, viscosimétrie, diffusion de neutrons aux petits angles (SANS), diffusion dynamique de la lumière (DLS). Title: New polyelectrolyte complexes involving a β-cyclodextrin polymer, a cationic surfactant and a polyanion Abstract: A new model of polyelectrolyte complex has been elaborated, based on inclusion complexes between a neutral polymer of β-CD and a cationic surfactant (DTAC), and complexes of opposite charges between DTAC and a polyanion.
Inclusion interactions between poly(β-CD) and DTAC alone in solution have first been characterized with several techniques including conductimetric and fluorimetric measurements and surface tension measurements. Structure of the resulting aggregates has been studied by viscosimetry and SANS as a function of the ionic strength and stoechiometry of the mixture. Then, the addition of a polyanion to the poly(β-CD)/DTAC mixture has been shown to form water soluble ternary complexes. For three polyanions of different natures and architectures (NaPSS, NaDxS and DNA), the structural properties of the complexes have been analyzed by SANS, viscosimetry and DLS as a function of the DTAC concentration. Stability and reversibility of the complexes have also been studied by varying the ionic strength of the medium and the concentration in competitors. Key words: polyelectrolyte complexes, polymer of β-cyclodextrin, cationic surfactant, viscosimetry, Small Angle Neutron Scattering (SANS), Dynamic Light Scattering (DLS). Adresse: Laboratoire de Recherche sur les Polymères (LRP), CNRS UMR 7581
2 à 8 rue Henri Dunant 94320 Thiais (France)






