
The role of methionine and α-chymotrypsin-catalysed reactions
11
Biochem. J. (1965) 95, 180 The Role of Methionine in a-Chymotrypsin-Catalysed Reactions By J. R. KNOWLES Dyson Perrins Laboratory, University of Oxford (Received 23 July 1964) 1. The reaction of oc-chymotrypsin with sodium periodate at pH5f0 has been investigated. The enzyme consumes 2 moles of periodate/mole, and there is a concomitant fall in enzymic activity (with respect to L-tyrosine ethyl ester) to 55% of that of the native enzyme. After 3hr. no further change is observed in periodate uptake or in catalytic activity. 2. The oxidized enzyme is a homogeneous preparation of partially active chymotrypsin. 3. In the oxidized enzyme, one of the two methionine residues in the molecule has been converted into its sulphoxide. It is this reaction only that is responsible for the loss of activity. 4. The rate constants for the enzyme-catalysed acylation and deacylation reactions are unaltered by oxidation of the enzyme, both for a non-specific substrate (p-nitro- phenyl acetate), and for three specific substrates: N-acetyl-L-tryptophan ethyl ester, N-acetyl-L-tryptophanamide and N-acetyl-L-valine ethyl ester. 5. The Km values for the aromatic substrates with the oxidized enzyme are twice those with the native enzyme. No change in Michaelis constant is seen for the non-aromatic sub- strate N-acetyl-L-valine ethyl ester. 6. The evidence points to the oxidized methionine residue in the modified enzyme being situated in the locus of the active site at which aromatic (or bulky) side chains of the substrates are bound. Most experiments involving chemical modifica- tion of amino acid residues at active sites of enzymes result in the production of enzymically inactive material. For example, the complete reaction of oc-chymotrypsin with a wide variety of reagents, including phosphoryl halides, sulphonyl halides, carbamoyl halides, halo ketones, acid anhydrides and ultraviolet irradiation, results in the destruction of enzymic activity. The possible deductions from total-inactivation experiments are normally limited to statements that the modified amino acid residue is either directly involved in the catalytic mechan- ism, that it is close enough to the active site sterical- ly to block substrate-binding or reaction, or that the critical tertiary folding around the active site has been modified sufficiently to abolish the activity. If an enzyme can be minimally modified by a chemical reagent so as to decrease the activity (but not destroy it), studies on the residual activity may define the function of the modified residue, as distinct from simply defining its existence at the active site. The usefulness of this approach is nicely exemplified by the work of Vallee and his co-workers on carboxypeptidase A (Vallee, 1964). A report by Jansen, Curl & Balls (1951) on the reaction of oc-chymotrypsin with periodate appeared to satisfy the requirement for a modified but partially active enzyme. The present paper reports a more detailed study of this reaction, and the results of some experiments designed to de- lineate the function of the modified residue in the oxidized enzyme. MATERIALS Three-times-crystallized oc-chymotrypsin (lot no. CDI 6037-38) was purchased from Worthington Biochemical Corp., Freehold, N.J., U.S.A. Assays of purity, moisture content etc. are given below. Sodium metaperiodate was AnalaR grade from British Drug Houses Ltd., Poole, Dorset. L-Tyrosine ethyl ester was purchased from Mann Research Laboratories Inc., New York, N.Y., U.S.A. N-Cinnamoylimidazole was prepared according to the method of Schonbaum, Zerner & Bender (1961), and had m.p. 133-133-5' [Schonbaum et al. (1961) give 133-133.50] (Found: C, 72-6; H, 5-2; N, 14-4. Cale. for C12H,oN20: C, 72-7; H, 5-1; N, 14-2%). N-Acetyl-L-tryptophan was prepared according to the method of du Vigneaud & Sealock (1932), and had m.p. 189-.5190-50, [oc]2+29 0+±005° (c 1 in aq. 40mN-NaOH). du Vigneaud & Sealock (1932) give m.p. 189-190°, [a]3+ 290 (c 1 in aq. NaOH). N-Acetyl-L-tryptophanamide was purchased from Yeda Research and Development Co., Israel (lot no. TR 17), and had m.p. 1970, [a]"'+ 19-50 (c 2 in methanol). Huang & Niemann (1951) give m.p. 192-193°, Dx] + 200 (c 2 in methanol). N-Acetyl-L-tryptophan ethyl ester was prepared by the method of Spies (1948), and had m.p. 109.5_1100, [oc]27+ 7-1 + 0.020 (c 6 in ethanol). Zerner, Bond & Bender (1964) 180
Transcript of The role of methionine and α-chymotrypsin-catalysed reactions

Biochem. J. (1965) 95, 180
The Role of Methionine in a-Chymotrypsin-Catalysed Reactions
By J. R. KNOWLESDyson Perrins Laboratory, University of Oxford
(Received 23 July 1964)
1. The reaction of oc-chymotrypsin with sodium periodate at pH5f0 has beeninvestigated. The enzyme consumes 2 moles of periodate/mole, and there is a
concomitant fall in enzymic activity (with respect to L-tyrosine ethyl ester) to55% of that of the native enzyme. After 3hr. no further change is observed inperiodate uptake or in catalytic activity. 2. The oxidized enzyme is a homogeneouspreparation of partially active chymotrypsin. 3. In the oxidized enzyme, one ofthe two methionine residues in the molecule has been converted into its sulphoxide.It is this reaction only that is responsible for the loss of activity. 4. The rateconstants for the enzyme-catalysed acylation and deacylation reactions are
unaltered by oxidation of the enzyme, both for a non-specific substrate (p-nitro-phenyl acetate), and for three specific substrates: N-acetyl-L-tryptophan ethylester, N-acetyl-L-tryptophanamide and N-acetyl-L-valine ethyl ester. 5. The Kmvalues for the aromatic substrates with the oxidized enzyme are twice those with thenative enzyme. No change in Michaelis constant is seen for the non-aromatic sub-strate N-acetyl-L-valine ethyl ester. 6. The evidence points to the oxidizedmethionine residue in the modified enzyme being situated in the locus of the activesite at which aromatic (or bulky) side chains of the substrates are bound.
Most experiments involving chemical modifica-tion ofamino acid residues at active sites ofenzymesresult in the production of enzymically inactivematerial. For example, the complete reaction ofoc-chymotrypsin with a wide variety of reagents,including phosphoryl halides, sulphonyl halides,carbamoyl halides, halo ketones, acid anhydridesand ultraviolet irradiation, results in the destructionof enzymic activity. The possible deductions fromtotal-inactivation experiments are normally limitedto statements that the modified amino acid residueis either directly involved in the catalytic mechan-ism, that it is close enough to the active site sterical-ly to block substrate-binding or reaction, or that thecritical tertiary folding around the active site hasbeen modified sufficiently to abolish the activity.If an enzyme can be minimally modified by achemical reagent so as to decrease the activity(but not destroy it), studies on the residual activitymay define the function of the modified residue, asdistinct from simply defining its existence at theactive site. The usefulness of this approach isnicely exemplified by the work of Vallee and hisco-workers on carboxypeptidase A (Vallee, 1964).A report by Jansen, Curl & Balls (1951) on the
reaction of oc-chymotrypsin with periodate appearedto satisfy the requirement for a modified butpartially active enzyme. The present paperreports a more detailed study of this reaction, and
the results of some experiments designed to de-lineate the function of the modified residue in theoxidized enzyme.
MATERIALS
Three-times-crystallized oc-chymotrypsin (lot no. CDI6037-38) was purchased from Worthington BiochemicalCorp., Freehold, N.J., U.S.A. Assays of purity, moisturecontent etc. are given below.Sodium metaperiodate was AnalaR grade from British
Drug Houses Ltd., Poole, Dorset.L-Tyrosine ethyl ester was purchased fromMann Research
Laboratories Inc., New York, N.Y., U.S.A.N-Cinnamoylimidazole was prepared according to the
method of Schonbaum, Zerner & Bender (1961), and hadm.p. 133-133-5' [Schonbaum et al. (1961) give 133-133.50](Found: C, 72-6; H, 5-2; N, 14-4. Cale. for C12H,oN20:C, 72-7; H, 5-1; N, 14-2%).
N-Acetyl-L-tryptophan was prepared according to themethod of du Vigneaud & Sealock (1932), and had m.p.189-.5190-50, [oc]2+29 0+±005° (c 1 in aq. 40mN-NaOH).du Vigneaud & Sealock (1932) give m.p. 189-190°, [a]3+ 290(c 1 in aq. NaOH).N-Acetyl-L-tryptophanamide was purchased from Yeda
Research and Development Co., Israel (lot no. TR 17), andhad m.p. 1970, [a]"'+ 19-50 (c 2 in methanol). Huang &Niemann (1951) give m.p. 192-193°, Dx] + 200 (c 2 inmethanol).
N-Acetyl-L-tryptophan ethyl ester was prepared by themethod of Spies (1948), and had m.p. 109.5_1100, [oc]27+ 7-1+ 0.020 (c 6 in ethanol). Zerner, Bond & Bender (1964)
180

ROLE OF METHIONINE IN a-CHYMOTRYPSIN ACTIONgive m.p. 110_111.50, [a]24+7 10 (c 6 in ethanol) (Found:C, 65-2; H, 6-4; N, 10-1. Calc. for C15H18N203: C, 65-7;H, 6-6; N, 10-2%)
N-Acetyl-L-valine ethyl ester was prepared according tothe method of Waite & Niemann (1962), and had m.p.31.5-34-50, []24_-51*7 + 0*150 (c 2 in water). Waite &Niemann (1962) give m.p. 32.5-34-20, [C]25_ 50-30 (c 4-5in water) (Found: C, 57-5; H, 9-0; N, 7-8. Calc. for C9H17N03:C, 57-7; H, 9-0; N, 7.5%).p-Nitrophenyl acetate was given by Mr A. Williams, and
had m.p. 79-5°. Kezdy & Bender (1962) give m.p. 79-5-80.00.
Acetonitrile was purified by the method of Cooper &Waters (1964).
All buffers were made up with AnalaR reagents andfreshly distilled water. pH measurements were made on aRadiometer instrument TTT Ic with scale-expanderPHA630, standardized against British Drug Houses Ltd.standard buffer solutions.
METHODS
Periodate e8timation. Periodate uptake was followed byspectrophotometric estimation of the single equivalent ofI2 liberated on the addition of excess of aq. KI solution tothe periodate solution in borate buffer, pH8-1 (Muller &Friedberger, 1902). Since protein interferes with the assay(by reacting with the 12 formed; see Glazer & Sanger, 1964;Dube, Roholt & Pressman, 1963), it was removed byprecipitation with methanol followed by centrifugation. Atypical uptake curve was obtained as follows. To 0-5 ml. of2mM-enzyme solution in 0-1m-acetate buffer, pH5-0, wasadded at 00, in the dark, 0-5ml. of aq. 10mM-sodium meta-periodate. At suitable intervals, a 50jul. sample waswithdrawn, 0-95ml. of pure methanol added, and thesolution mixed and then centrifuged for 30sec. The clearsupernatant was poured off, and 0-8ml. of it pipetted into astoppered 1cm. optical cell. Then 1-5ml. of 0-1 M-boratebuffer, pH8-1, was added, followed by 0-2ml. of aq. 2%0(w/w) KI solution. The extinction at 352 m,u was measuredon a Unicam SP. 500 spectrophotometer. Extinctionscorresponding to initial periodate concentrations wereobtained by exactly similar assays in the absence of protein.The following checks on the method were made: (i)
excess of iodate does not interfere with the estimation ofperiodate at pH8.1; (ii) neither methanol nor acetate(from the buffer) reacts with periodate at pH5-0; (iii) noperiodate is lost during the precipitation of protein; (iv)neither methanol nor acetate reacts with the 12 produced atpH 8-1 (i.e., the I2 observed after protein removal is equiva-lent to the periodate present in the original sample); (v)protein precipitation by the 20-fold excess of methanol isessentially complete, the observed extinction diminishingonly by approx. 10% over 30min. (the time from initialsampling to extinction reading was commonly less than3min.); (vi) check estimates of periodate plus iodate weremade by adding to 0-8 ml. of the methanol solution, afterremoval of protein, 1-5ml. of 2N-HCI and 0-4ml. ofKI solu-tion. Under these conditions 4moles of 12 are liberated/mole of periodate, and 3moles/mole of iodate. These esti-mates agreed well with the known concentrations of iodateand remaining periodate. Points on the uptake curve (Fig.1) are means from two reactions, duplicate samples beingtaken in each case. Readings from the same run were
within 3%, and those from different runs were within 6%.This is reasonable, since only 0-025-0-015 emole of per-iodate is being assayed.
Preparation of oxidized oc-chymotrypsin. Oxidizedoc-chymotrypsin was prepared as follows. A 1g. sample ofcz-chymotrypsin was dissolved in 20ml. of 0-1M-acetatebuffer, pH5-0, at 00, and added to 20ml. ofaq. 10mM-sodiummetaperiodate solution. The mixture was stirred slowly for3hr. in the dark at 00, and solid (NH4)2SO4 was then addedto give 75% saturation during 1 hr. at 00. The protein wasspun down and dissolved in the minimum of 20mN-H2SO4,and solid (NH4)2SO4 added to induce crystallization. After2 days at 40 the material had crystallized, the diamond-shaped crystals being identical in form with those obtainedfrom az-chymotrypsin itself under these conditions. Thecrystalline oxidized a-chymotrypsin was spun down,dissolved in 20mmN-H2SO4 and dialysed exhaustivelyagainst distilled water for 3 days at 4°. After centrifugationto remove a very slight cloudiness, the material was freeze-dried.Samples required for assay of enzymic activity during the
course of the oxidation were treated as described above,though the crystallization step was omitted in these casesand the (NH4)2S04 was added over 5min.
Activity assays. The 'all-or-none' assay employed bySchonbaum et al. (1961) with N-cinnamoylimidazole wasused to obtain operational normalities of enzyme solutions,and as a check that no total inactivation was occurringduring the oxidation. In general the commercial enzymecontains about 86% (on a weight basis, assuming amolecular weight of 25000) of 'titratable' active sites bythis all-or-none assay, a moisture content of 6-9% and anash weight corresponding to 0-4%. The 'rate' assayemployed by Schwert & Takenaka (1955) with L-tyrosineethyl ester was used as a measure of enzymic activity. Forboth these methods a Unicam SP. 500 spectrophotometer[with a temperature-controlled (Adkins thermostat) cellcompartment, fitted with a photomultiplier and coupled toa pen recorder] was used.Amino acid analyses. Acid-catalysed hydrolysates were
obtained by heating 1 ml. of constant-boiling HCI (twiceredistilled)/mg. of protein in a sealed tube, 02-free, at 1100for 17 hr. Insignificant amounts of cysteic acid and chloro-tyrosines were observed. Base-catalysed hydrolysates wereobtained by the procedure of Ray & Koshland (1962).The hydrolysates were analysed with a two-column aminoacid analyser (Evans Electroselenium Ltd., Halstead,Essex).
Sedimentation. Sedimentation experiments were per-formed in a Spinco model E analytical ultracentrifuge at59 780rev./min. at 200. The proteins were in concentrationsof approx. 8mg./ml. in 0-1 m-acetate buffer, pH4-85.
Kinetic determinations. The rates of reaction of z-chymo-trypsin and oxidized a-chymotrypsin with the followingsubstrates were determined according to the methodsdescribed.
(a) p-Nitrophenyl acetate. The rate of the enzyme-catalysed hydrolysis of this substrate under conditions ofenzyme saturation was followed spectrophotometricallyby the method of Kezdy & Bender (1962). A typical run isdescribed. To 3-Oml. of 0-1 M-phosphate buffer in alem. quartz cell was added 50l. of substrate solutionin acetonitrile. The change in extinction at 400m,u dueto spontaneous hydrolysis of the substrate was followed for
Vol. 95 181

J. R. KNOWLES4min., and 50,ul. of aqueous enzyme solution was thenadded. The change in extinction was followed for a further6min. Both parts of the extinction-time plot were linearover the range studied. Enzyme concentrations werecalculated from the immediate increase in extinction on theaddition ofenzyme, assuming 6400 to be 18350 and pK to be7.04 for p-nitrophenol (Kezdy & Bender, 1962). Theseconcentrations agreed very closely with those obtained bythe titration procedure of Schonbaum et al. (1961) withN-cinnamoylimidazole, and with the concentrationscalculated on a weight basis, assuming that 86% of thepreparation as weighed out is active. Extinction-timecurves were obtained in the Unicam SP. 500 spectrophoto.meter.
(b) N-Cinnamoylimidazole. The acylation ofa-chymo-trypsin and of oxidized enzyme by this reagent was followedspectrophotometrically at 355m, according to the methodof Schonbaum et al. (1961). Low concentrations of reagent(approx. 10.tM) and high instrument sensitivities wereemployed to detect the difference in acylation rates.
(c) N-Acetyl-L-valine ethyl ester. The kinetics of hydro-lysis of this ester catalysed byc-chymotrypsin and byoxidized enzyme were examined with a RadiometerTitrator (TTTlc) coupled to a Titrigraph (SBR2c). Thetitration cells were lOml.-capacity scintillation bottles,which fittedsnugly into a thermostatically controlled jacket.Afresh bottle was used for each run. The cellwas covered bya polythene cap fitted with the two miniature electrodes(Radiometer G222B and K4312), the titration-base feedcapillary, the nitrogen inlet and a 1mm. hole for the strong-base feed capillary. The electrode assembly could beraised from and lowered into the titration cell rapidly on aratcheted rack and pinion. Stirring was achieved magnetic-ally. Nitrogen was blown over the surface of the reactionsolution, having been bubbled through distilled water in athermostatically controlled container. A typical run wasperformed as follows. The titration cell (commonly contain-ing 3-Oml. of substrate solution and 0'5ml. of 09m-NaClsolution), which had been thermostatically controlled at thetemperature of the run for at least lOmin., was transferredto the jacket, and 1 ml. of enzyme solution, also thermo-statically controlled at the temperature of the run, wasadded. The final volume in the cell was thus 4-5ml. Theelectrode assembly was lowered, stirring started, and strongbase (1.Ox- or 01IN-NaOH) was added through the strong-base feed capillary with the Titrator coupled to a magneticvalve (Radiometer MNVlc) which controlledthe addition ofstrong base. In this way the pH of the solution was broughtup to the pHrequiredforthe titration quickly and accurately(the pH of aqueous solutions of a-chymotrypsin is between3 and 5). This method is to be preferred to (a) storing theenzyme at the pH of the run, since a-chymotrypsin isdenatured irreversibly fairly rapidly at alkaline pH values(Bender, Clement, Kezdy & Heck, 1964a) and later runswill be in error if denaturation has been significant, or (b)raising the pH of the enzyme solution before addition byusing a different meter. The method described eliminatesinconsistencies between meters, and avoids the wastage ofenzyme consequent on raising the pH of enzyme solutionsbatchwise. When the pH of the solution was that required,the Titrator was coupled to a 0O5ml. syringe burette (Radio-meter SBUla with B101/0.5) (containing OOlw-NaOH,standardized against potassium hydrogen phthalate) andthe Titrigrph, for pH-$tsat oporation. The time taken from
initial enzyme addition to the start of the titration properwas commonly less than 30sec. In every experiment thereaction time was at least 8 min.
Since the Titrigraph traces are slightly curved, initialvelocities are difficult to estimate visually with muchprecision. A computer programme has been written (inAutocode, for a Ferranti Mercury digital computer) alongthe general lines of those of Abrash, Kurtz & Niemann(1960) and of Elmore, Kingston & Shields (1963). From theexperimental results (the co-ordinates of at least ninepoints on a titration curve) the coefficients of the poly-nomial best fitting these results are computed. The value ofthe gradient at zero time is then readily calculated. Unlikethe authors mentioned above, I do not use Student'st test to decide on the significance of the coefficient for thenth-order term, but limit the polynomial to one of ordertwo. By so doing the restriction is placed on the equationthat it will only 'fit' non-re-entrant curves. The plot iscurved for a variety of reasons (e.g., product inhibition,autolysis of the enzyme, change of volume, change of ionicstrength etc.), but I believe that the arbitrary restriction ofthe best-fit polynomial to a second-order equation elimin-ates possibly serious errors inthe calculated initialvelocities,arising from random fluctuations in the titration curves.With as few as nine points (this is also about the numbertreated by the two groups cited above) this limitationbecomes even more necessary. Ideally, perhaps, one shouldhave an nth-order polynomial derived from a large (say,more than 30) number of points for each run, but this, forthe 14 or so runs necessary for one Lineweaver-Burk plot,is unduly laborious. The computer programme, havingobtained all the initial velocities, now calculates the kineticparameters of the Michaelis-Menten equation:
vo = ko[Eo][So]/(Km+ [So])from a least-squares treatment of a Lineweaver-Burk plot.All values of l/vo and 1/[So] are printed out, with values ofko and Ki, and the probable errors in these two parameters(Margenau & Murphy, 1956). As a check against trulyerrant points in the Lineweaver-Burk plot (e.g. teleprintererrors, pipetting errors etc.), the programme then calculatesthe most deviant point, discards it, and recalculates koand Km and their errors. This process is repeated until thedeviation of each point (in 1/vo) is less than 5% of the valueof l/vo. In this way any really deviant point becomesimmediately apparent. Normally, however, the values ofko and K. calculated from all points are accepted (seerelevant Lineweaver-Burk plots).The errors quoted in values of ko and Km are probable
errors from the Lineweaver-Burk plot. They are not (andare not meant to be) measures of accuracy, only of theprecision of the experimentation.
(d) N-Acetyl-L-tryptophanamide. The enzyme-catalysedhydrolysis of this substrate was followed spectrophoto-metrically at 306mu according to the method of Benderet al. (1964a). A Beckman DB recording spectrophoto-meter (with a thermostatically controlled cellcompartment)was used. In the 'reference' beam of the instrument weretwo 1cm. cells in tandem, containing respectively 2-8ml. of0OIM-tris-HCI buffer, pH7.87, with 0-2ml. of aqueousenzyme solution, and 2-8ml. of substrate solution in bufferwith 0-2 ml. of water. In the 'solution' beam were also twocells in tandem, containing respectively 2-8ml. of substratesolution in buffer with 0-2ml. of enzyme solution, and
182 1965
ROLE OF METHIONINE INa-CHYMOTRYPSIN ACTION2-8ml. of buffer solution with 0-2ml. of water. The spectro-photometer was run on the 80-100% transmission range.The value of Ae for the conversion of N-acetyl-L-trypto-phanamide into N-acetyl-L-tryptophan (73-8) was takenfrom Bender et at. (1964a). Transmission percentageswere converted into extinctions, and plotted out to obtaininitial velocities. These values, with the correspondingsubstrate concentrations, were fed into a computer pro-gramme that just performs the least-squares treatment onthe Lineweaver-Burk plot (i.e., the latter half of theprogramme described above) to yield values of ko and Km.
(e) N-Acetyl-L-tryptophan ethyl ester. To obtain resultsunder as similar conditions as possible, the method ofZerner et al. (1964) for measuring the kinetics of hydro-lysis of this substrate was modified to eliminate acetonitrilefrom the reaction solution. N-Acetyl-L-tryptophan ethylester (approx. 1 mm) can be dissolved in water or buffersolution by heating at 800 in a water bath for 2min. Thefollowing checks were made to ensure that none of the esteris hydrolysed during the dissolution. A solution of the esterin water (prepared as described above) was brought topH8-0 in the pH-stat, and then placed in a boiling-waterbath for10min. The solution was cooled and then returnedto the pH-stat titration cell. Under these conditions lessthan 2% of the ester was hydrolysed. This was furtherchecked by the addition of 10,1d. ofl1uM-a-chymotrypsinsolution and measuring the total uptake of base. The baserequired for complete hydrolysis was within 2% of thatcalculated on the basis of the known weight of N-acetyl-L-tryptophan ethyl ester in solution. Extinction coefficientsof N-acetyl-L-tryptophan ethyl ester and N-acetyl-L-tryp-tophan at 300m,u under the conditions used (in 0-1M-tris-HCI buffer, pH7-90) were determined in duplicate. Thesewere: N-acetyl-L-tryptophan ethyl ester: 567-7, 566-9; N-acetyl-L-tryptophan: 779-2, 782-3. The solutions of the ethylester were hydrolysed completely to N-acetyl-L-tryptophan(IO,l. of 1LM-ce-chymotrypsin was added to 10ml. samplesof the ethyl ester and left overnight) to check with thevalues obtained from the authentic samples of N-acetyl-L-tryptophan. This method gave, for N-acetyl-L-tryptophan:782-5, 787-8. The values used were: N-acetyl-L-tryptophanethyl ester: 567; N-acetyl-L-tryptophan: 781; i.e., Ac300:214. The kinetic runs were performed on the UnicamSP. 500 spectrophotometer as described above. The75-100% transmission range was used. Instrument driftduring the time of a single run was negligible. To 3-Oml.of substrate solution maintained at a thermostaticallycontrolled temperature in a lem. optical cell was added10l,1. of enzyme solution (approx. 10pM) in water. Record-ing was started less than 10sec. after mixing. Serialdilutions of the enzyme solution were avoided (Jones &Niemann, 1963) and check experiments were carried out toensure that the [Eo] (determined from vo) was indeedproportional (at these high dilutions) to the volume of stocksolution taken. The extinction-time plots were curved.This is not unexpected since the reaction is so fast, and theester soluble with such difficulty, that it is impossible tostudy only the first few percent of the reaction. For themost dilute substrate solutions hydrolysis was essentiallycomplete within 3min., and even at the highest substrateconcentrations about a quarter of the substrate had beenused up after this time. The experimental results weretherefore fed directly into a computer programme (adaptedto accept 75-100%-transmission readings) similar to that
used for the runs with N-acetyl-L-valine ethyl ester. Theinitial velocities were thus computed from the 'best-fit'binomial expressing the experimental curve, and theseresults subjected to a Lineweaver-Burk treatment toobtain ko and Km. Because of the rapidity of the reaction,these results are not as accurate as those obtained for theother substrates.
RESULTS
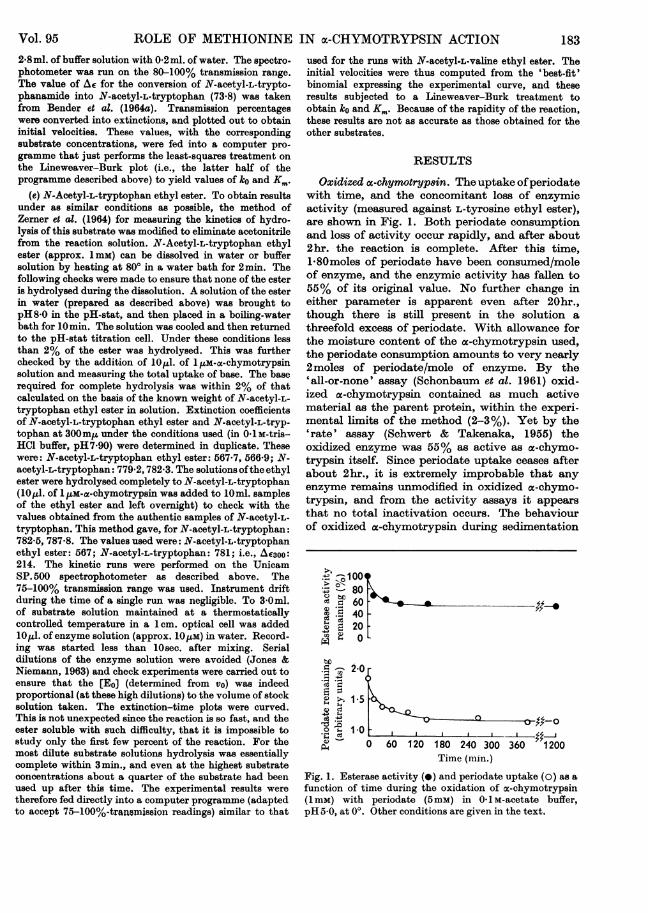
Oxidizedac-chymotryp8in. The uptake ofperiodatewith time, and the concomitant loss of enzymicactivity (measured against L-tyrosine ethyl ester),are shown in Fig. 1. Both periodate consumptionand loss of activity occur rapidly, and after about2hr. the reaction is complete. After this time,1-80moles of periodate have been consumed/moleof enzyme, and the enzymic activity has fallen to55% of its original value. No further change ineither parameter is apparent even after 20hr.,though there is still present in the solution athreefold excess of periodate. With allowance forthe moisture content of the a-chymotrypsin used,the periodate consumption amounts to very nearly2moles of periodate/mole of enzyme. By the'all-or-none' assay (Schonbaum et al. 1961) oxid-izedoc-chymotrypsin contained as much activematerial as the parent protein, within the experi-mental limits of the method (2-3%). Yet by the'rate' assay (Schwert & Takenaka, 1955) theoxidized enzyme was 55% as active as a-chymo-trypsin itself. Since periodate uptake ceases afterabout 2hr., it is extremely improbable that anyenzyme remains unmodified in oxidized a-chymo-trypsin, and from the activity assays it appearsthat no total inactivation occurs. The behaviourof oxidized a-chymotrypsin during sedimentation
.>- 10100
80
eZ 60
T 40
-) E 20
-- 2-0Sn
;:^1-5
SCa-+.
S- c34)
1-0 I I , ,0 60 120 180 240 300 360 1200
Time (min.)Fig. 1. Esterase activity (-) and periodate uptake (o) as afunction of time during the oxidation of oc-chymotrypsin(1 mM) with periodate (5mM) in 0-1 M-acetate buffer,pH 5-0, at 00. Other conditions are given in the text.
Vol. 95 183
0-#-0
J. R. KNOWLES
in the ultracentrifuge is identical with that of theparent enzyme.On analysis of the acid-catalysed hydrolysates,
no significant differences could be detected betweenoxidized a-chymotrypsin and the parent enzyme(see Table 1). On analysis of the base-catalysedhydrolysates, however, it appeared that a singlemethionine has been converted into the sulphoxidein oxidized a-chymotrypsin. A small quantity ofthe sulphone is also apparent. The sulphoxidesand sulphone were identified by the positions ofemergence from the long column of the amino acidanalyser. The stereochemical integrity of thesulphoxide in intact oxidized a-chymotrypsin isnot known. Even if the sulphoxide is not racemicin the protein, it is naturally racemized veryrapidly under the conditions of baryta-catalysedhydrolysis, and two peaks of equal area appear onthe analyser trace. The absence of sulphoxide inacid-catalysed hydrolysates of oxidized proteinshas been explained by Ray & Koshland (1962) andby Floyd, Cammaroti & Lavine (1963), who foundthat methionine sulphoxide reverts to the parentcompound under the conditions of acid hydrolysis.
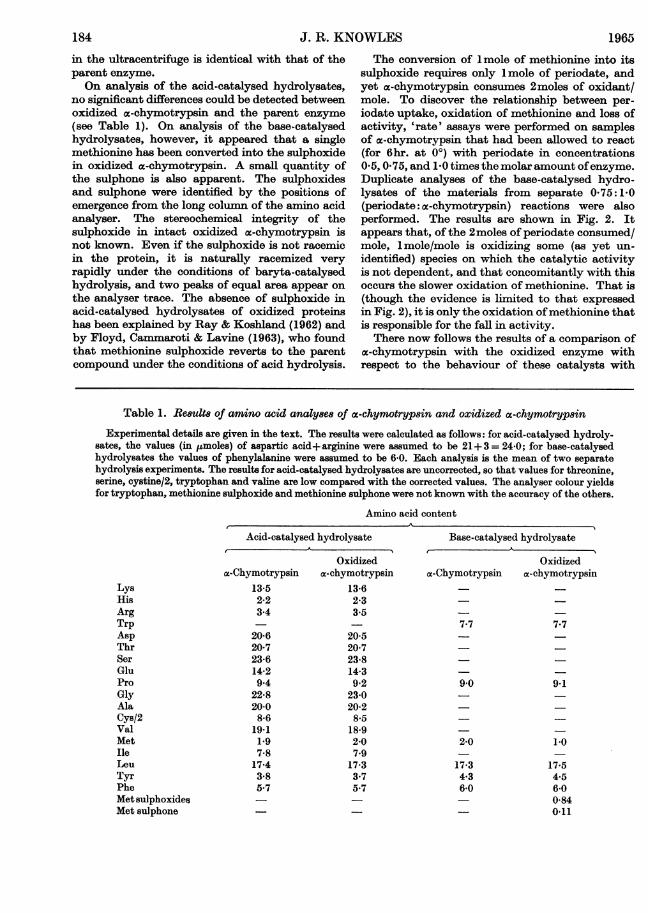
The conversion of 1 mole of methionine into itssulphoxide requires only 1 mole of periodate, andyet a-chymotrypsin consumes 2moles of oxidant/mole. To discover the relationship between per-iodate uptake, oxidation of methionine and loss ofactivity, 'rate' assays were performed on samplesof a-chymotrypsin that had been allowed to react(for 6hr. at 0°) with periodate in concentrations0-5, 0-75, and 1-0 times the molar amount ofenzyme.Duplicate analyses of the base-catalysed hydro-lysates of the materials from separate 0-75: 1-0(periodate: a-chymotrypsin) reactions were alsoperformed. The results are shown in Fig. 2. Itappears that, of the 2moles of periodate consumed/mole, 1 mole/mole is oxidizing some (as yet un-identified) species on which the catalytic activityis not dependent, and that concomitantly with thisoccurs the slower oxidation of methionine. That is(though the evidence is limited to that expressedin Fig. 2), it is only the oxidation ofmethionine thatis responsible for the fall in activity.
There now follows the results of a comparison ofac-chymotrypsin with the oxidized enzyme withrespect to the behaviour of these catalysts with
Table 1. Results of amino acid analy8e8 of oe-chymotryp8in and oxidized oa-chymotryp8in
Experimental details are given in the text. The results were calculated as follows: for acid-catalysed hydroly-sates, the values (in ,umoles) of aspartic acid+ arginine were assumed to be 21+3= 24-0; for base-catalysedhydrolysates the values of phenylalanine were assumed to be 6-0. Each analysis is the mean of two separatehydrolysis experiments. The results for acid-catalysed hydrolysates are uncorrected, so that values for threonine,serine, cystine/2, tryptophan and valine are low compared with the corrected values. The analyser colour yieldsfor tryptophan, methionine sulphoxide and methionine sulphone were not known with the accuracy of the others.
Amino acid content
Acid-catalysed hydrolysate Base-catalysed hydrolysate
LysHisArgTrpAspThrSerGluProGlyAlaCys/2ValMetIleLeuTyrPheMetsulphoxidesMet sulphone
oc-Chymotrypsin13-52-23-4
20-620-723-614-29.4
22-820-08-6
19-11-97-8
17-43-85-7
Oxidizedo-chymotrypsin
13-62-33-5
20-520-723-814-39-2
23-020-28-5
18-92-07-9
17-33.75.7
a-Chymotrypsin
7.7
90
2-0
17-34.36-0
Oxidizedoa-chymotrypsin
7.7
9.1
1-0
17-54.56-00-840-11
184 1965
ROLE OF METHIONINE IN oa-CHYMOTRYPSIN ACTION
120
0 2- 070 *
cec 60k-~50 0 1.
'T 40'00 0-5 1*0 1.5 2-0
Periodate used for oxidation(moles/mole of oc-chymotrypsin)
Fig. 2. Relationship between esterase activity (0) andoxidation of methionine residues (o) of a-chymotrypsinafter treatment with different molar proportions ofperiodate in 0-1M-acetate buffer, pH5-0, at 00 for 6hr.Other details are given in the text.
00)
0cx
0oo-4
0 0-2 0-4 0-6 0-8 1-0
[So] (mm)
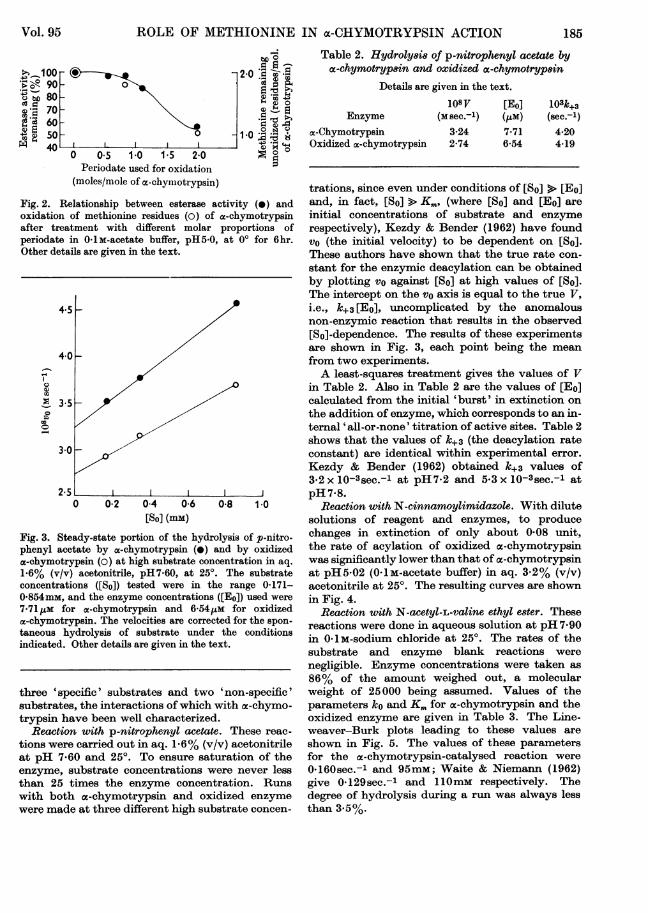
Fig. 3. Steady-state portion of the hydrolysis of p-nitro-phenyl acetate by a-chymotrypsin (e) and by oxidizeda-chymotrypsin (o) at high substrate concentration in aq.1.6% (v/v) acetonitrile, pH7-60, at 250. The substrateconcentrations ([So]) tested were in the range 0-171-0-854mM, and the enzyme concentrations ([Eo]) used were
7-71 zm for a-chymotrypsin and 6-541cM for oxidizedoc-chymotrypsin. The velocities are corrected for the spon-taneous hydrolysis of substrate under the conditionsindicated. Other details are given in the text.
three 'specific' substrates and two 'non-specific'substrates, the interactions ofwhich with a-chymo-trypsin have been well characterized.
Reaction with p-nitrophenyl acetate. These reac-
tions were carried out in aq. 1.6% (v/v) acetonitrileat pH 7-60 and 250. To ensure saturation of theenzyme, substrate concentrations were never lessthan 25 times the enzyme concentration. Runswith both ac-chymotrypsin and oxidized enzyme
were made at three different high substrate concen-
Table 2. Hydroly8i8 of p-nitrophenyl acetate byac-chymotryp8in and oxidized ac-chymotryp8in
Details are given in the text.
Enzymea-ChymotrypsinOxidized a-chymotrypsin
108V(M sec.-l)
3-242-74
[Eo](pM)7*71654
103k43(sec.-I)4-204-19
trations, since even under conditions of [So] > [Eo]and, in fact, [So] > K., (where [So] and [Eo] areinitial concentrations of substrate and enzymerespectively), Kezdy & Bender (1962) have foundvo (the initial velocity) to be dependent on [So].These authors have shown that the true rate con-stant for the enzymic deacylation can be obtainedby plotting vo against [So] at high values of [So].The intercept on the vo axis is equal to the true V,i.e., k+3 [Eo], uncomplicated by the anomalousnon-enzymic reaction that results in the observed[So]-dependence. The results of these experimentsare shown in Fig. 3, each point being the meanfrom two experiments.A least-squares treatment gives the values of V
in Table 2. Also in Table 2 are the values of [Eo]calculated from the initial 'burst' in extinction onthe addition of enzyme, which corresponds to an in-ternal 'all-or-none' titration of active sites. Table 2shows that the values of k+3 (the deacylation rateconstant) are identical within experimental error.Kezdy & Bender (1962) obtained k+3 values of382 x 10-3sec.-1 at pH7-2 and 5-3 x 10-3sec.-l atpH 7-8.
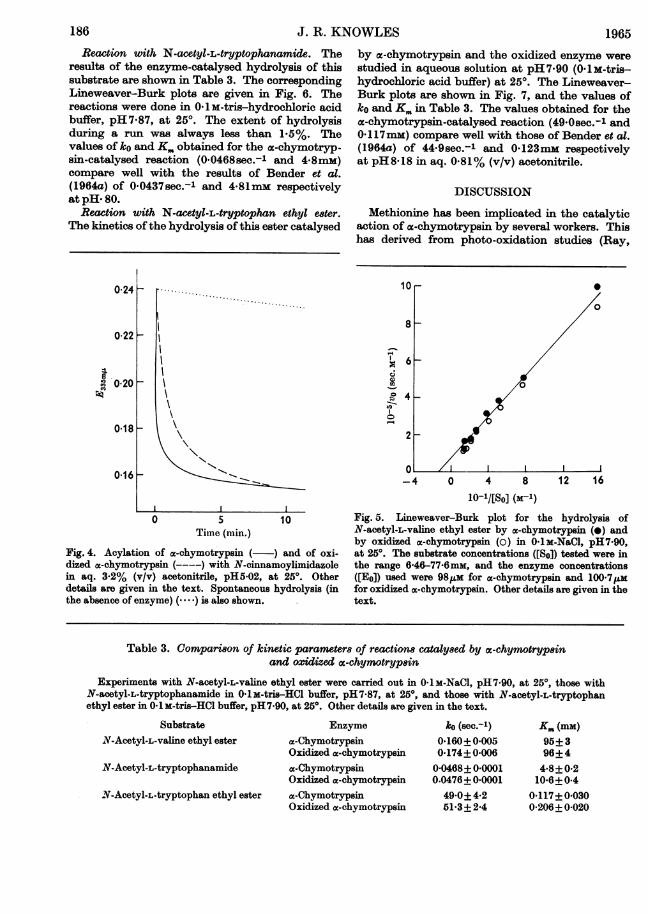
Reaction with N-cinnamoylimidazole. With dilutesolutions of reagent and enzymes, to producechanges in extinction of only about 0-08 unit,the rate of acylation of oxidized a-chymotrypsinwas significantly lower than that of a-chymotrypsinat pH5-02 (OlM-acetate buffer) in aq. 3.2% (v/v)acetonitrile at 250. The resulting curves are shownin Fig. 4.
Reaction with N-acetyl-L-valine ethyl e8ter. Thesereactions were done in aqueous solution at pH 7-90in 0-1 M-sodium chloride at 250. The rates of thesubstrate and enzyme blank reactions werenegligible. Enzyme concentrations were taken as86% of the amount weighed out, a molecularweight of 25 000 being assumed. Values of theparameters ko and Km for a-chymotrypsin and theoxidized enzyme are given in Table 3. The Line-weaver-Burk plots leading to these values areshown in Fig. 5. The values of these parametersfor the a-chymotrypsin-catalysed reaction were0-160sec.-l and 95mM; Waite & Niemann (1962)give 0-129sec.-l and 110mM respectively. Thedegree of hydrolysis during a run was always lessthan 3.5%.
Vol. 95 185
186 ~~~~~J.R. KNOWLES 16
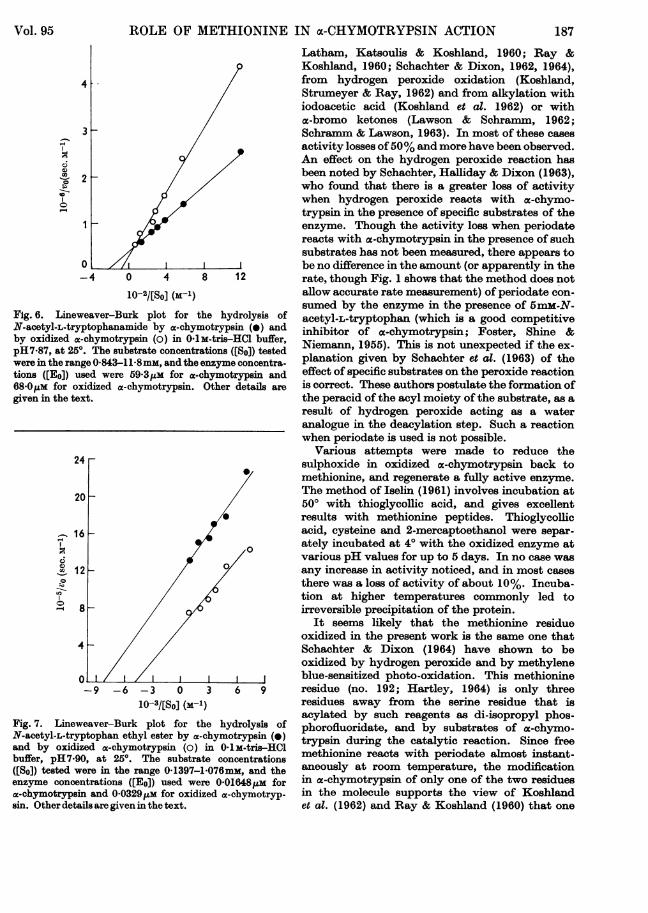
Reaction with N-acetyl-L-tryptophanamide. Theresults of the enzyme-catalysed hydrolysis of thissubstrate are ishown in Table 3. The correspondingLineweaver-Burk plots are given in Fig. 6. Thereactions were done in 0.1IM-tris-hydrochloric acidbuffer, pH7*87, at 250. The extent of hydrolysisduring a run was always less than 1 5%. Thevalues of ko and Km obtained for the z-chymotryp-sin-catalysed reaction (0-0468sec.-l and 4.8mm)compare well with the results of Bender et al.(1964a) of 0-0437sec.-l and 4-81mM respectivelyatpH- 80.
Reaction with N-acetyl.L-tryptophasn ethyl e8ter.The kinetics of the hydrolysis of this ester catalysed
by a-chymotrypsin and the oxidized enzyme werestudied in aqueous solution at pH 7'90 (0.1 m-tris-hydrochloric acid buffer) at 250. The Lineweaver-Bark plots are shown in Fig. 7, and the values ofko and K. in Table 3. The values obtained for theac.chymotrypsin-catalysed reaction (49-Osec.-1 and0 117mm) compare well with those of Bender et al.(1964a) of 44-9sec.-l and 0-123mm respectivelyat pH8*18 in aq. 0.81% (v/v) acetonitrile.
DISCUSSION
Methionine has been imnplicated in the catalyticaction of a-chymotrypsin by several workers. Thishas derived from photo-oxidation studies (Ray,
101
8
-4 0 4 8
lO'1/[So] (M'I)0 5
Timne (min.)10
Fig. 4. Acylation of cx-chymotrypsin ( ~) and of oxi-dized a-chymotrypsin (----) with N-cinnamoylimidazolein aq. 3.2% (v/v) acetonitrile, pH5-02, at 25'. Otherdetails are given in the text. Spontaneous hydrolysis (inthe absence of enzyme) (.. is also shown.
Fig. 5. Lineweaver-Burk plot for the hydrolysis ofN-acetyl.L-vallne ethyl ester by ae.chymotrypsin (0) andby oxidized ac.chymotrypsin (0) in 0-lM-NaCl., pH7-90,at 250. The substrate concentrations ([So]) tested were inthe range 6-46-77-6mm, and the enzyme concentrations([Eo]) used were 98ILm for x.chymotrypsin and 100-7,umfor oxidized ac-chymotrypsin. Other details are given in thetext.
Table 3. Comparison of kinetic parameters of reactions catalysed by oz-chymotryp8inand oxidized oc.chymotryp8in
Experiments with N-acetyl.L-valine ethyl ester were carried out in 0-lm-NaCl, pH7-90, at 250, those withN-acetyl.L-tryptophanamide in 0-lx-tris-HOl buffer, pH7-87, at 250, and those with N-acetyl.L-tryptophanethyl ester in 0-lx-tris--HClbuffer, pH7.90, at 250. Other details are given in the text.
SubstrateN-Acetyl-L-valine ethyl ester
N-Acetyl-L-tryptophanamide
N-Acetyl.L-tryptophan ethyl ester
Enzymeoe-ChymotrypsinOxidized a-chymotrypsinoc-ChymotrypsinOxidized oz-chymotrypsinoc-ChymotrypsinOxidized oc-chymotrypsin
0-24 H
0-22 F-
aW.mcli
rr4.0-20k
0-18 I
0-16 H
0)104
0
12 16
ko (sec.'l)0.160+ 0-0050.174+ 0-0060-0468+ 0-00010.0476+ 0-0001
49-0+ 4-251-3+±2-4
Km (mm)95+396+44.8+ 0.210.6+ 0-4
0-117+ 0-0300-206+ 0-020
186 1965
ROLE OF METHIONINE IN oc-CHYMOTRYPSIN ACTION
2
-4 0 4 8 12
10-2/[So] (Mf-1)
Fig. 6. Lineweaver-Burk plot for the hydrolysis ofN-acetyl-L-tryptophanamide by a-chymotrypsin (L) andby oxidized a-chymotrypsin (0) in 0 IM-tris-HCl buffer,pH7.87, at 250. The substrate concentrations ([So]) testedwere in the range 0X843-118 mm, and the enzyme concentra-tions ([Eo]) used were 59 3pm for ac-chymotrypsin and68 0ul.m for oxidized a-chymotrypsin. Other details are
given in the text.
24
,-
C;C)
0
-9 -6 -3 0 310-3/[So] (M-1)
Fig. 7. Lineweaver-Burk plot for the hydrolysis ofN-acetyl-L-tryptophan ethyl ester by a-chymotrypsin (0)and by oxidized a-chymotrypsin (o) in 01M-tris-HClbuffer, pH7.90, at 250. The substrate concentrations([So]) tested were in the range 0-1397-1 076mm, and theenzyme concentrations ([Eo]) used were 0-01648jum fora-chymotrypsin and 0-03291uM for oxidized a-chymotryp-sin. Other details are given in the text.
Latham, Katsoulis & Koshland, 1960; Ray &Koshland, 1960; Schachter & Dixon, 1962, 1964),from hydrogen peroxide oxidation (Koshland,Strumeyer & Ray, 1962) and from alkylation withiodoacetic acid (Koshland et al. 1962) or witha-bromo ketones (Lawson & Schramm, 1962;Schramm & Lawson, 1963). In most of these casesactivity losses of 50% and more have been observed.An effect on the hydrogen peroxide reaction hasbeen noted by Schachter, Halliday & Dixon (1963),who found that there is a greater loss of activitywhen hydrogen peroxide reacts with a-chymo-trypsin in the presence of specific substrates of theenzyme. Though the activity loss when periodatereacts with a-chymotrypsin in the presence of suchsubstrates has not been measured, there appears tobe no difference in the amount (or apparently in therate, though Fig. 1 shows that the method does notallow accurate rate measurement) of periodate con-sumed by the enzyme in the presence of 5mM-N-acetyl-L-tryptophan (which is a good competitiveinhibitor of a-chymotrypsin; Foster, Shine &Niemann, 1955). This is not unexpected if the ex-planation given by Schachter et al. (1963) of theeffect of specific substrates on the peroxide reactionis correct. These authors postulate the formation ofthe peracid of the acyl moiety of the substrate, as aresult of hydrogen peroxide acting as a wateranalogue in the deacylation step. Such a reactionwhen periodate is used is not possible.
Various attempts were made to reduce thesulphoxide in oxidized a-chymotrypsin back tomethionine, and regenerate a fully active enzyme.The method of Iselin (1961) involves incubation at500 with thioglycollic acid, and gives excellentresults with methionine peptides. Thioglycollicacid, cysteine and 2-mercaptoethanol were separ-ately incubated at 40 with the oxidized enzyme atvarious pH values for up to 5 days. In no case wasany increase in activity noticed, and in most casesthere was a loss of activity of about 10%. Incuba-tion at higher temperatures commonly led toirreversible precipitation of the protein.
It seems likely that the methionine residueoxidized in the present work is the same one thatSchachter & Dixon (1964) have shown to beoxidized by hydrogen peroxide and by methyleneblue-sensitized photo-oxidation. This methionineresidue (no. 192; Hartley, 1964) is only threeresidues away from the serine residue that isacylated by such reagents as di-isopropyl phos-phorofluoridate, and by substrates of a-chymo-trypsin during the catalytic reaction. Since freemethionine reacts with periodate almost instant-aneously at room temperature, the modificationin a-chymotrypsin of only one of the two residuesin the molecule supports the view of Koshlandet al. (1962) and Ray & Koshland (1960) that one
Vol. 95 187

J. R. KNOWLES
methionine residue is 'accessible' and the otherone 'buried'.
It is tempting to suggest that the decrease inenzyme activity observed in the present work isdue to a direct effect of oxidation of methionine,rather than a second-order effect on the tertiarystructure of the enzyme as a whole. The kineticand specificity evidence that follows substantiatesthis rather fine distinction.The existence of a minimally modified, partially
active, enzyme makes it feasible to study thedetailed effect of the modification by looking at thenature of the residual activity. Niemann and hisco-workers (see, e.g., Hein & Niemann, 1961, 1962)have usefully obtained a great deal of informationabout the specificity of ao-chymotrypsin, and Benderand his co-workers (see, e.g., Zerner & Bender,1964) have discussed the meaning of the kineticconstants obtained from Michaelis-Menten break-downs ofthe reaction-rate data, and the dependenceof these constants on the rate-determining step ofthe catalytic reaction. Since these data are avail-able, experiments can be devised to discover inwhat way the activity ofa modified oc-chymotrypsinhas been altered, with the intention of defining thefunction of the modified residue, and with the hopeof casting some light on the general questions ofmechanism and specificity.The evidence that the hydrolytic reactions
catalysed by a-chymotrypsin proceed via theformation of an acyl-enzyme intermediate seemsnow to be compelling, both for many non-specificand, more importantly, for specific substrates (forreferences, see Bender & Kezdy, 1964). The presentresults are discussed in terms of the kinetic scheme:
E+S -k- ES k+2 >acyl-E +3- E+P2 (1)k-i
P1
where the formation of a loose 'Michaelis-Menten'complex is succeeded by acylation to an acyl-enzyme, which is deacylated to yield the enzymeand the acid moiety of the substrate.From a Michaelis-Menten breakdown of the reac-
tion rates, according to vo = ko[Eo][So]/(Km + [So]),the observed quantities ko and Km are obtained.The mechanism of eq. (1) leads to the relationships:
ko = k+2k+3/(k+2+k+3)and Km = (kl1 + k+2)k+3/k+l(k+2 + k+3)
When acylation is genuinely rate-determining, kobecomes k+2, and Km becomes K' [K,. is here theoperational parameter; K' is defined as the theor-etical quantity (k-1 + k+2)/k+l, which is sometimescalled Km in the American literature]; and if deacyl-ation is much the slower step, ko = k+3, and
Km = K'. k+3/k+2. These facts make simple com-parisons of Km values (as if they were K' values)dangerous if not meaningless, if it is not certainwhat the rate-determining step is. For specificsubstrates, then, the rate-determining step mustbe identified. If a series of acid derivatives ofwidely different base-catalysed hydrolysis rateshave the same ko values, then a common inter-mediate is implied, and deacylation can be assumedto be rate-determining. This approach has beenused by Zerner & Bender (1963) for the N-acetyl-L-tryptophan derivatives. It is clear from theirresults that, for the methyl, ethyl and p-nitrophenylesters, deacylation is rate-limiting, and that, for theamide, acylation is the slow step. This conclusionhas been confirmed by direct observation (at lowpH) of the 'burst' of p-nitrophenol from the N-acetyl-L-tryptophan p-nitrophenyl ester-oc-chymo-trypsin system under conditions when [So] >[Eo] (Kezdy, Clement & Bender, 1964). Thereforefor two of the specific substrates (the tryptophanderivatives) used in the present work the signifi-cance of the observed kinetic constants is known.For the third specific substrate, N-acetyl-L-valineethyl ester, the rate-determining step cannot yetbe defined, though knowledge of the actual k+2/k+3ratio for this ester is not crucial to the presentdiscussion, as shown below.For the reagent N-cinnamoylimidazole, which
can be treated as an essentially irreversible acylat-ing agent of a-chymotrypsin, the initial reactionvelocity will be proportional to k+2/K'. As shownin Fig. 4, no attempt was made in this case toextract quantitative estimates of initial rates fromthe experimental results. For p-nitrophenyl acetateit is known (Hartley & Kilby, 1952, 1954) that theturnover rate of the hydrolysis under conditions of[Eo] < [So] is a measure ofthe rate-limiting deacyla-tion of the acetyl-enzyme (Kezdy & Bender, 1962).
It is now possible to discuss the results obtainedwith these various substrates with a-chymotrypsinand the oxidized enzyme, as far as possible interms of the effect on the individual rate constantsof eqn. 1. The results can be summarized asfollows: (1) the rate of acylation of oxidizeda-chymotrypsin by N-cinnamoylimidazole is sig-nificantly lower than that of a-chymotrypsin;(2) the deacylation rates of acetyl-a-chymotrypsinand acetyl-(oxidized a-chymotrypsin) (k+3) areidentical; (3) the ko values for N-acetyl-L-trypto-phanamide (k+2) are identical, but the Km value(K') for oxidized a-chymotrypsin is about twicethat for oc-chymotrypsin; (4) the ko values forN-acetyl-L-tryptophan ethyl ester (k+3) are identi-cal, but the Km value (K'.k+3/k+2) for oxidizeda-chymotrypsin is about twice that for a-chymo-trypsin; (5) the ko values and Km values for N-acetyl L-valine ethyl ester are the same for oxidized
1965188

ROLE OF METHIONINE IN oc-CHYMOTRYPSIN ACTIONoe-chymotrypsin and a-chymotrypsin; [(6) therate of reaction of L-tyrosine ethyl ester withoxidized a-chymotrypsin is 55% of that witha-chymotrypsin].The experiments with p-nitrophenyl acetate and
with N-acetyl-L-tryptophan ethyl ester eachdemonstrate that oxidation of a-chymotrypsinhas not affected the rate of deacylation either of'non-specific' acetyl-a-chymotrypsin or of 'specific'N-acetyl-L-tryptophanyl-oe-chymotrypsin. More-over, the ko values for N-acetyl-L-tryptophanamideshow that the rate of acylation is also unaffected, atleast for this substrate. The fact that ko forN-acetyl-L-valine ethyl ester is unaltered is furthersupport for the postulate that oxidation of theenzyme affects neither of the catalytic (as distinctfrom the binding) rate constants. Koshland et al.(1962) have reported that a-chymotrypsin oxidizedwith hydrogen peroxide shows no change in ko forN-acetyl-L-tyrosine ethyl ester (for which ko almostcertainly equals k+3), yet Km is raised threefoldcompared with unoxidized enzyme. Moreover,Lawson & Schramm (1962) found that a more bulkymethionine modification [the conversion of amethionine residue into its S-(acetyl oc-aminoiso-butyric acid) sulphonium salt] led to an 11-foldincrease in Km (for N-acetyl-L-tyrosine ethyl ester).For N-acetyl-L-tryphanamide (Km = K') there isan approximately twofold increase in Km onoxidation of the enzyme. Paralleling this, the Km(i.e., K'. k+3/k+2) for N-acetyl-L-tryptophan ethylester with oxidized c-chymotrypsin is about twicethat with a-chymotrypsin. As shown above, k+2and k+3 (for N-acetyl-L-tryptophan derivatives, atleast) are unaltered on oxidation of the protein,so that change in K'. k+3/k+2 must be due to achange in K'. It is natural to extend this to theresults for the hydrolysis of L-tyrosine ethyl ester,and ascribe the fall in rate with oxidized ac-chymo-trypsin (to 55% of that with a-chymotrypsin) to aneffect on K'. However, Dixon and co-workers(reported in a footnote by Schachter & Dixon,1964) find that their preparation of oxidizedoc-chymotrypsin does not show an increase inKm towards N-acetyl-L-tryptophan ethyl ester atpH8'0. This result does not fit into the picture thatemerges from the present work and that of Kosh-land et al. (1962) and Lawson & Schramm (1962).No simple explanation is apparent, though theremay be real differences in the behaviour ofthe hydrogen peroxide- and periodate-modifiedenzymes.Thus far, the differences in activity of a-chymo-
trypsin and the oxidized enzyme that have beenmentioned are those with 'good' specific substrates,i.e., derivatives of the aromatic amino acidstryptophan and tyrosine. Definitions of 'good-ness' have to be limited to comparative statements,
e.g. that derivatives of aromatic acids generallyappear to have lower K,,, values and higher kovalues than their aliphatic counterparts, andclassification of the 'specificity' of a substrate mustrely on postulates as to the existence of specificinteractions (e.g., at each of the three 'loci' of themodel of Hein & Niemann, 1961, 1962) betweensubstrate and enzyme, reflected in the value ofK, By any criterion of 'goodness' or of specificity,however, valine compounds are less 'good' andless specific substrates of a-chymotrypsin thantryptophan derivatives. It is therefore significantthat the Km values for the hydrolysis of N-acetyl-L-valine ethyl ester by ax-chymotrypsin and theoxidized enzyme are the same. The conclusion thatthis leads to is that oxidation of a-chymotrypsinresults in higher K' values for 'good', aromatic,substrates, but not for less 'demanding' substratessuch as N-acetyl-L-valine ethyl ester. Since thereare strong indications (see Zerner & Bender, 1964)that K'= K, (i.e. k-/lk+i) because k+21 k-1,this is equivalent to saying that oxidation of themethionine residue in a-chymotrypsin affects thebinding of aromatic amino acid derivatives but notof valine derivatives. According to the Hein-Niemann model, one could say that the methionineresidue that has been altered in oxidized a-chymo-trypsin lies in the p2-locus, i.e., the locus in which isbound the aromatic side chains of the 'good'substrates. [The fall in activity was ascribed tooxidation of methionine on the basis of activityassays with L-tyrosine ethyl ester. The differencesin activity with respect to other substrates (e.g.,N-acetyl-L-tryptophan ethyl ester, N-acetyl-L-tryptophanamide) need not be due to oxidation ofmethionine, but could arise from other changes inthe enzyme. This caveat is a necessary one, thoughthe more economical hypothesis is made here.] Thedecrease of the acylation rate observed withN-cinnamoylimidazole may well arise from anincrease in K' (the initial rate being proportionalto k+2/K') due to the effect on the binding of thecinnamoyl side chain in the p2 locus.
Bender, Kezdy & Gunter (1964b) have proposed auseful separation of the specificity concept into'binding' specificity and 'kinetic' specificity. Thesimplest hypothesis is that the same limitednumber of amino acid residues at the active site areresponsible for both types of specificity. Yet thepresent results imply that there are genuinedifferences between these specificities, since bindingefficiency has been altered without affecting therates of the subsequent catalytic steps.In the absence of more data, it is difficult to say
whether the effect of converting a methionineresidue into its sulphoxide is in origin steric, polar,or both. It seems likely, however, that it is largelya steric effect, since, whereas oxidation leads to a
Vol. 95 189

190 J. R. KNOWLES 1965two- to three-fold increase in Km for aromaticsubstrates, sulphonium salt formation (with abulky group; Lawson & Schramm, 1962) leads to an11-fold increase. Thus it is possible that, in thepresent experiments, the isopropyl side chain ofvaline is not large enough to interact with thesulphoxide group in oxidized a-chymotrypsin,whereas the greater binding requirements of thearomatic substrates do result in an unfavourableinteraction. If this thesis is correct, it may berelevant that trypsin, which catalytically is veryclosely similar to cx-chymotrypsin (Bender & Kaiser,1962) but which differs in not being specific foraromatic amino acid derivatives, has no methio-nine residue (actually a glutamine residue) in theposition corresponding to the methionine residueat position no. 192 (Hartley, 1964) in ao-chymo-trypsin.
The author is grateful to Dr Myron L. Bender for copies ofpapers by him and his co-workers before publication, toDr D. B. Hope for the amino acid analyses, and to Dr W. E.van Heyningen for the sedimentation experiment. Thiswork was supported in part by the Department for Scientificand Industrial Research.
REFERENCES
Abrash, H. I., Kurtz, A. N. & Niemann, C. (1960). Biochim.biophys. Acta, 45, 378.
Bender, M. L., Clement, G. E., Kezdy, F. J. & Heck, H. d'A.(1964a). J. Amer. chem. Soc. 86, 3680.
Bender, M. L. & Kaiser, E. T. (1962). J. Amer. chem. Soc.84, 2556.
Bender, M. L. & Kezdy, F. J. (1964). J. Amer. chem. Soc.86, 3704.
Bender, M. L., Kezdy, F. J. & Gunter, C. R. (1964b). J.Amer. chem. Soc. 86, 3714.
Cooper, T. A. & Waters, W. A. (1964). J. chem. Soc. p. 1538.Dube, S. K., Roholt, 0. A. & Pressman, D. (1963). Fed.
Proc. 22, 245.du Vigneaud, V. & Sealock, R. R. (1932). J. biol. Chem. 96,
511.Elmore, D. T., Kingston, A. E. & Shields, D. B. (1963).
J. chem. Soc. p. 2070.Floyd, N. F., Cammaroti, M. S. & Lavine, T. F. (1963).
Arch. Biochem. Biophys8. 102, 343.Foster, R. J., Shine, H. J. & Niemann, C. (1955). J. Amer.
chem. Soc. 77, 2378.Glazer, A. N. & Sanger, F. (1964). Biochem. J. 90, 92.
Hartley, B. S. (1964). Nature, Lond., 201, 1284.Hartley, B. S. & Kilby, B. A. (1952). Biochem. J. 50, 672.Hartley, B. S. & Kilby, B. A. (1954). Biochem. J. 56, 288.Hein, G. E. & Niemann, C. (1961). Proc. nat. Acad. Sci.,
Wash., 47, 1341.Hein, G. E. & Niemann, C. (1962). J. Amer. chem. Soc. 84,
4495.Huang, H. T. & Niemann, C. (1951). J. Amer. chem. Soc.
73, 1541.Iselin, B. (1961). Helv. chim. acta, 44, 61.Jansen, E. F., Curl, A. L. & Balls, A. K. (1951). J. biol.Chem. 189, 671.
Jones, J. B. & Niemann, C. (1963). Biochemistry, 2, 498.Kezdy, F. J. & Bender, M. L. (1962). Biochemi8try, 1, 1097.Kezdy, F. J., Clement, G. E. & Bender, M. L. (1964).
J. Amer. chem. Soc. 86, 3690.Koshland, D. E., Strumeyer, D. H. & Ray, W. J. (1962).Brookhaven Symp. Biol. 15, 101.
Lawson, W. B. & Schramm, H. J. (1962). J. Amer. chem.Soc. 84, 2017.
Margenau, H. & Murphy, G. M. (1956). The MathematicsofPhysics and Chemistry, 2nd ed., chapter 13. Princeton:D. Van Nostrand Co.
Muller, E. & Friedberger, 0. (1902). Ber. dtsch. chem. Ge8.85, 2652.
Ray, W. J. & Koshland, D. E. (1960). Brookhaven Symp.Biol. 13, 135.
Ray, W. J. & Koshland, D. E. (1962). J. biol. Chem. 287,2493.
Ray, W. J., Latham, H. G., Katsoulis, M. & Koshland, D. E.(1960). J. Amer. chem. Soc. 82,4743.
Schachter, H. & Dixon, G. H. (1962). Biochem. biophys.Res. Commun. 9, 132.
Schachter, H. & Dixon, G. H. (1964). J. biol. Chem. 289,813.
Schachter, H., Halliday, K. A. & Dixon, G. H. (1963).J. biol. Chem. 288, Po3134.
Schonbaum, G. R., Zerner, B. & Bender, M. L. (1961).J. biol. Chem. 286, 2930.
Schramm, H. J. & Lawson, W. B. (1963). Hoppe-Seyl. Z.882,97.
Schwert, G. W. & Takenaka, Y. (1955). Biochim. biophys.Acta, 16, 570.
Spies, J. R. (1948). J. Amer. chem. Soc. 70, 3717.Vallee, B. L. (1964). Fed. Proc. 28, 8.Waite, H. R. & Niemann, C. (1962). Biochemistry, 1, 250.Zerner, B. & Bender, M. L. (1963). J. Amer. chem. Soc. 85,
356.Zemer, B. & Bender, M. L. (1964). J. Amer. chem. Soc. 86,
3669.Zerner, B., Bond, R. P. M. & Bender, M. L. (1964). J. Amer.
chem. Soc. 86, 3674.








![Adsorption of α-Chymotrypsin on Plant Biomass Charcoalfile.scirp.org/pdf/JSEMAT_2013101014304054.pdf · combustion under a nitrogen atmosphere [9]. ... The theory accounts for capil-](https://static.fdocument.org/doc/165x107/5b5a6e8d7f8b9ab8578bea95/adsorption-of-chymotrypsin-on-plant-biomass-combustion-under-a-nitrogen-atmosphere.jpg)









![Nano Nickel-Zinc Ferrites Catalysed One-Pot multicomponent ...84-114)V11N11CT.pdf · Microwave assisted organic synthesis (MAOS) [3-4] has emerged as a new “lead” in organic synthesis.](https://static.fdocument.org/doc/165x107/5f33072bf62f7a7bb83b91b2/nano-nickel-zinc-ferrites-catalysed-one-pot-multicomponent-84-114v11n11ctpdf.jpg)