
Targeted Delivery of α-Galactosylceramide to CD8 α ... · Elodie Macho-Fernandez, Luis Javier...
10
of February 3, 2019. This information is current as Based Antitumor Responses - NKT Cell Dendritic Cells Optimizes Type I + α CD8 -Galactosylceramide to α Targeted Delivery of Frisch, François Trottein and Christelle Faveeuw Ghinnagow, Josette Fontaine, Emilie Bialecki, Benoit Elodie Macho-Fernandez, Luis Javier Cruz, Reem http://www.jimmunol.org/content/193/2/961 doi: 10.4049/jimmunol.1303029 2014; 2014; 193:961-969; Prepublished online 9 June J Immunol Material Supplementary 9.DCSupplemental http://www.jimmunol.org/content/suppl/2014/06/07/jimmunol.130302 References http://www.jimmunol.org/content/193/2/961.full#ref-list-1 , 21 of which you can access for free at: cites 55 articles This article average * 4 weeks from acceptance to publication Fast Publication! • Every submission reviewed by practicing scientists No Triage! • from submission to initial decision Rapid Reviews! 30 days* • Submit online. ? The JI Why Subscription http://jimmunol.org/subscription is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/About/Publications/JI/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/alerts Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved. Copyright © 2014 by The American Association of 1451 Rockville Pike, Suite 650, Rockville, MD 20852 The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on February 3, 2019 http://www.jimmunol.org/ Downloaded from by guest on February 3, 2019 http://www.jimmunol.org/ Downloaded from
Transcript of Targeted Delivery of α-Galactosylceramide to CD8 α ... · Elodie Macho-Fernandez, Luis Javier...

of February 3, 2019.This information is current as
Based Antitumor Responses−NKT Cell Dendritic Cells Optimizes Type I +αCD8
-Galactosylceramide toαTargeted Delivery of
Frisch, François Trottein and Christelle FaveeuwGhinnagow, Josette Fontaine, Emilie Bialecki, Benoit Elodie Macho-Fernandez, Luis Javier Cruz, Reem
http://www.jimmunol.org/content/193/2/961doi: 10.4049/jimmunol.13030292014;
2014; 193:961-969; Prepublished online 9 JuneJ Immunol
MaterialSupplementary
9.DCSupplementalhttp://www.jimmunol.org/content/suppl/2014/06/07/jimmunol.130302
Referenceshttp://www.jimmunol.org/content/193/2/961.full#ref-list-1
, 21 of which you can access for free at: cites 55 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2014 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on February 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on February 3, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
Targeted Delivery of a-Galactosylceramide to CD8a+
Dendritic Cells Optimizes Type I NKT Cell–Based AntitumorResponses
Elodie Macho-Fernandez,*,†,‡,x,{ Luis Javier Cruz,‖ Reem Ghinnagow,*,†,‡,x,{
Josette Fontaine,*,†,‡,x,{ Emilie Bialecki,*,†,‡,x,{ Benoit Frisch,# Francois Trottein,*,†,‡,x,{,1 andChristelle Faveeuw*,†,‡,x,{,1
Immunotherapy aiming at enhancing innate and acquired host immunity is a promising approach for cancer treatment. The in-
variant NKT (iNKT) cell ligand a-galactosylceramide (a-GalCer) holds great promise in cancer therapy, although several
concerns limit its use in clinics, including the uncontrolled response it promotes when delivered in a nonvectorized form.
Therefore, development of delivery systems to in vivo target immune cells might be a valuable option to optimize iNKT cell–
based antitumor responses. Using dendritic cell (DC)–depleted mice, DC transfer experiments, and in vivo active cell targeting, we
show that presentation of a-GalCer by DCs not only triggers optimal primary iNKT cell stimulation, but also maintains secondary
iNKT cell activation after challenge. Furthermore, targeted delivery of a-GalCer to CD8a+ DCs, by means of anti-DEC205
decorated nanoparticles, enhances iNKT cell–based transactivation of NK cells, DCs, and gd T cells. We report that codelivery of
a-GalCer and protein Ag to CD8a+ DCs triggers optimal Ag-specific Ab and cytotoxic CD8+ T cell responses. Finally, we show
that targeting nanoparticles containing a-GalCer and Ag to CD8a+ DCs promotes potent antitumor responses, both in prophy-
lactic and in therapeutic settings. Our data may have important implications in tumor immunotherapy and vaccine devel-
opment. The Journal of Immunology, 2014, 193: 961–969.
Immunotherapy aiming at enhancing innate and acquired hostimmunity, including development of tumor-specific cytotoxicCD8+ T cells, is viewed as a promising approach for cancer
treatment (1, 2). Over the last decade, an increasing interest wasgiven to the potent antitumor properties of invariant NKT (iNKT)cells (for reviews, see Refs. 3–7). These cells represent a pop-ulation of unconventional T lymphocytes possessing “innate-like”functions and playing positive roles in tumor surveillance (forreviews, see Refs. 3, 8, 9). Through their TCR, iNKT cells rec-ognize self and exogenous lipid Ags presented by the CD1d
molecule expressed by APCs. In response to the superagonista-galactosylceramide (a-GalCer), iNKT cells rapidly producea wide array of immunostimulatory cytokines, including IFN-g,and upregulate several costimulatory molecules (3, 8). Theseevents contribute to the reciprocal maturation of APCs, for in-stance, the release of IL-12 by dendritic cells (DCs), and to thedownstream activation of NK cells, gd T cells, and B and CD4+
and CD8+ T lymphocytes, with important outcomes on immuneresponses. Thus, a-GalCer is viewed as a potent innate immunestimulator and vaccine adjuvant, and holds great promise to op-timize antitumor responses (3, 8, 10).Studies in the mouse system have highlighted the antitumor
effect of a-GalCer, injected alone in a free form, in a broad rangeof experimental models (11, 12). Attempts to exploit the antitumoreffect of a-GalCer have been conducted in patients with advancedcancers. Systemic administration of free a-GalCer increased in-nate cytokine release, but clinical benefits remained modest (13).A first explanation for these limited results relies in part on thereduced number of iNKT cells in patients with malignant diseases(14). Another issue that limits the use of a-GalCer in clinics is theprofound and long-lasting hyporesponsiveness of iNKT cells afterprimary activation with this molecule (15), a major drawback forpatients who need several immunological stimuli to develop apotent antitumor response. The uncontrolled nature of cells thatuptake free a-GalCer, and thus the uncontrolled response it pro-motes, remains a major issue for the development of a-GalCer inclinics. A possibility to better control iNKT cell functions mightlie on passive or active delivery of a-GalCer to the right APCs,such as DCs (for review, see Ref. 16). Indeed, primary stimulationof iNKT cells by DCs appears to be a key event to limit iNKT cellhyporesponsiveness to further challenges (15, 17, 18), althougha recent report called this into question (19). Presentation ofa-GalCer by DCs also enhances the strength and the quality of
*Institut Pasteur de Lille, Centre d’Infection et d’Immunite de Lille, F-59019 Lille,France; †Universite Lille Nord de France, F-59000 Lille, France; ‡Centre National dela Recherche Scientifique, Unite Mixte de Recherche 8204, F-59021 Lille, France;xINSERM, U1019, F-59019 Lille, France; {Institut Federatif de Recherche 142,F-59019 Lille, France; ‖Department of Endocrinology, Leiden University MedicalCenter, 2333 Leiden, The Netherlands; and #Centre National de la Recherche Sci-entifique, Unite Mixte de Recherche 7199, Universite de Strasbourg, F-67401 IllkirchCedex, France
1F.T. and C.F. contributed equally to this work.
Received for publication November 13, 2013. Accepted for publication May 14,2014.
This work was supported by INSERM, Centre National de la Recherche Scientifique,University of Lille 2, Pasteur Institute of Lille, and Institut National du Cancer(projets libres) under reference R08046EE/RPT08003EEA.
Address correspondence and reprint requests to Dr. Francois Trottein, Center forInfection and Immunity of Lille, INSERM, U1019, Centre National de la RechercheScientifique, Unite Mixte de Recherche 8204, Institut Pasteur de Lille, 1, Rue duProfesseur Calmette, BP 245, 59019 Lille Cedex, France. E-mail address: [email protected]
The online version of this article contains supplemental material.
Abbreviations used in this article: BM-DC, bone marrow–derived dendritic cell; DC,dendritic cell; DT, diphtheria toxin; DTR, diphtheria toxin receptor; a-GalCer, a-gal-actosylceramide; iNKT, invariant NKT; MNC, mononuclear cell; NP, nanoparticle;PLGA, poly(lactic-coglycolic acid).
Copyright� 2014 by The American Association of Immunologists, Inc. 0022-1767/14/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1303029
by guest on February 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

iNKT cell–mediated immune responses. For instance, adminis-tration of a-GalCer–loaded DCs is more potent than a-GalCeralone in both preclinical and clinical tumor studies (18, 20, 21).Finally, simultaneous presentation of a-GalCer and tumor Ag byDCs optimizes CTL-mediated antitumor responses (22, 23). Al-though promising, the transfer of autologous a-GalCer–loadedDCs applied to clinics represents a relatively invasive and costlyprocedure (6). Based on these considerations, in this article, wepropose an underexplored strategy based on controlled and tar-geted delivery of a-GalCer and Ag to DCs to optimally harnessiNKT cell functions and antitumor responses.DCs are heterogeneous and can be classified into different
subtypes according to their phenotype, tissue distribution, andfunctions (24). In the mouse system, DCs in the spleen, an im-portant site of immune responses to blood-borne Ag, are mainlycomposed of CD8a+ DCs, a subset specialized in Ag cross-presentation, and of CD8a2 DCs, a subset more efficient at pre-senting Ag on MHC class II (24). Recent findings indicated thatCD8a+ DCs are particularly potent to activate iNKT cells (25) andto trigger iNKT cell–based innate immune responses (26, 27).These data, and the fact that CD8a+ DCs excel in Ag cross-presentation, prompted us to actively deliver a-GalCer and Agto CD8a+ DCs in vivo. Using Ab-armed poly(lactic-coglycolicacid) (PLGA)–based nanoparticles (NPs), we show for the firsttime, to our knowledge, that targeted delivery of a-GalCer toCD8a+ DCs via the DEC205 endocytic pathway is instrumental toenhance primary activation of iNKT cells and to avoid subsequentiNKT cell hyporesponsiveness. In parallel, compared with con-trols, codelivery of a-GalCer and protein Ag to CD8a+ DCsstrengthens Ag-specific Ab and CTL responses. Finally, wedemonstrate that this strategy is instrumental to amplify antitumorresponses in prophylactic and therapeutic settings. In aggregate,these findings might have important implications in tumor im-munotherapy and vaccine development.
Materials and MethodsMice
Six- to 8-wk-oldmalewild type C57BL/6micewere purchased from Janvier(Le Genest-St-Isle, France) and RAG22/2 3 OTI from Jackson Laboratory(St. Germain sur l’Arbresle, France). The generation of CD1d2/2 andtransgenic CD11c-diphtheria toxin receptor (DTR) mice has been alreadydescribed (28, 29). Animals were handled and housed in accordance withthe guidelines of the Pasteur Institute Animal Care and Use Committee.
Reagents and Abs
a-GalCer was synthesized as previously described (30). Vybrant CFDA SECell Tracer Kit was purchased from Life Technologies (St. Aubin, France).The PKH-26 labeling kit and OVA were purchased from Sigma-Aldrich(St. Quentin-Fallavier, France). Allophycocyanin-conjugated mAbs againstmouse CD5, CD11c, CD86, PE-conjugated anti-NK1.1, anti-TCRd,FITC-conjugated anti-CD8a, anti-TCRb, anti-CD3ε, PerCp-Cy5.5–conjugated anti-CD11b, eFluor450-conjugated anti-Va2 TCR, PE-Cy7–conjugated anti-CD11c, anti-CD8a, anti–PD-1, biotin-conjugated anti-CD1d, Alexa Fluor 700–conjugated streptavidin, and isotype controlswere purchased from BD Pharmingen (Le Pont de Claix, France) orOzyme/Biolegend (Saint-Quentin-en-Yvelines, France). PerCP-eFluor710anti-CD205 was purchased from eBioscience (Paris, France). IFN-g(Alexa Fluor 647 conjugated) and isotype controls were all purchased fromOzyme/Biolegend. PE-conjugated PBS-57 glycolipid-loaded CD1d tetra-mer was from the National Institute of Allergy and Infectious DiseasesTetramer Facility (Emory University, Atlanta, GA). The rat anti-DEC205(CD205) and isotype control (IgG2a) Abs used to arm NPs were fromBIO-X-CELL (West Lebanon, NH).
Preparation and characterization of PLGA-based particles
NPs coated with lipid-PEG and carrying Abs were generated using thebiocompatible polymer PLGA as described previously (31). In brief,endotoxin-free OVA (2.5 mg) and/or a-GalCer (50 mg) were encapsulated
to 100 mg PLGA. Anti-DEC205 Ab and its isotype control were attached tothe lipid-PEG layer as described previously (31). The presence of Abs on theparticle surface was determined by Coomassie dye protein assay. PLGA NPswere characterized by dynamic light scattering and z potential (Table I).
Analysis of NP uptake by DCs and DC-iNKT cocultures
Bone marrow–derived DCs (BM-DCs; 5 3 105 cells/well) or spleenmononuclear cells (MNCs; 1 3 106 cells/well) were exposed with AlexaFluor 647–labeled particles (10 and 100 mg/ml, respectively) during 2 h at37˚C. After extensive washes, BM-DCs (CD11c+ DEC205+/2) and spleenCD8a+ and CD8a2 DCs were analyzed by flow cytometry. BM-DCs (1 3105 cells/well) were cocultured for 24 h with the iNKT cell hybridomaDN32.D3 (1 3 105 cells/well) in the presence of grading doses of free orencapsulated a-GalCer. To study the ex vivo stimulatory capacity ofCD8a+ and CD8a2 DCs, we injected mice i.v. with 2 mg a-GalCer and 2 hlater, DC subsets were sorted using a FACSAria (Becton Dickinson, MD)and cocultured (7 3 104 cells/well) with sorted hepatic NKT cells (CD5+
NK1.1+ cells, .98% pure) or DN32.D3 (1 3 105 cells/well) for 48 and 24h, respectively. Production of IFN-g, IL-4, and IL-2 was measured in theculture supernatants by ELISA.
Role of DCs in iNKT cell responses in vivo
Mice were administrated i.v. with 200 ml PBS containing 5 ng free (or100 ng as a control) or encapsulated a-GalCer. CD11c-DTR mice wereinjected with diphtheria toxin (DT) 24 h before a-GalCer administration.Spleen DCs were sorted (CD11c+ cells), sensitized with 25 ng/mla-GalCer, and injected i.v. to mice. Mice received a second i.v. injectionof free a-GalCer (100 ng/mouse) 1 wk later, to analyze the recall response.Animals were bled and sacrificed 3 h posttreatment. Splenic iNKT cellswere analyzed for intracellular IFN-g expression, and IFN-g and IL-4concentrations in the sera were determined by ELISA.
FACS analysis
Cells were resuspended in the appropriate combination of Abs to allowidentification of DC subsets (anti-CD11c, anti-CD8a, anti-CD11b),iNKT cells (anti-TCRb, PBS-57–loaded CD1d tetramer), NK cells (anti-CD3ε, anti-NK1.1), or gd T lymphocytes (anti-TCRgd, anti-CD3ε). Then,anti–PD-1, -CD86, -CD1d, or isotype controls were added when needed. Tomeasure Cy5–a-GalCer incorporation by splenic DC subsets, we injectedmice i.v. with Cy5-conjugated a-GalCer (20 mg) and 2 h later, incorporationof Cy5 by spleen DC subsets was analyzed by flow cytometry.
Analysis of the CD8+ T cell and Ab responses
Mice received 5 3 106 CFSE-labeled, OVA-specific, CD8+ T cells purifiedfrom RAG22/2 3 OT-I mice. One day later, mice were injected into thefoodpads with NPs containing both a-GalCer (5 ng/mouse) and OVA (250ng/mouse) or with the same quantify of free a-GalCer and OVA. Three dayslater, the proliferation of CFSE-labeled cells in the popliteal lymph nodeswas measured by flow cytometry. Expression of IFN-g by Va2 TCR+CD8a+
cells was determined after in vitro restimulation of popliteal LN cells withthe MHC class I–restricted OVA peptide SIINFEKL (10 mg/ml) for 18 h. Forthe in vivo CTL assay, mice animals were i.v. injected with a mixtureof CFSE-labeled SIINFEKL-primed splenocytes and PKH-26–labeledunprimed splenocytes (23 107 cells/mouse), 6 d after immunization with theNP. Spleens were harvested 2 d later, and the numbers of CFSE- and PKH-26–labeled cells were determined by flow cytometry. The percentage of spe-cific lysis was determined as follows: (1 2 [ratio unprimed/ratio primed]) 3100, where the ratio is equal to number of PHK-26–labeled cells/CFSE-labeled cells. To analyze humoral and memory T cell responses, weinjected mice i.v. twice (at days 0 and 21) with a-GalCer (100 ng/mouse)and OVA (5 mg/mouse) either free or coencapsulated into NPs. Blood weretaken at day 28, and the anti-OVA IgG titers were determined by ELISA.Spleen MNCs were prepared 2 mo after the second immunization andin vitro restimulated with SIINFEKL for 48 h.
Tumor models
Mice were injected with 2.5 3 105 B16F10 melanoma cells expressingOVA 7 d after inoculation of vectorized OVA and a-GalCer. Mice werekilled on day 18, and lung metastases were counted with the aid ofa microscope. Mice were s.c. injected with 2 3 105 OVA-expressingEL4 lymphoma cells (EG7 cells). Tumor size was measured at regularintervals by using a caliper. Tumor size was calculated by using the formula:Tumor size (mm2) = length (mm) 3 width (mm). NP/DEC205(IgG)/OVA/a-GalCer were injected before (prophylactic) or after (therapeutic) inocu-lation of OVA-expressing EG7 cells.
962 OPTIMAL iNKT CELL RESPONSES WITH VECTORIZED a-GalCer
by guest on February 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

Statistics
Results are expressed as the mean 6 SD or SEM. The statistical signifi-cance of differences between experimental groups was calculated by anunpaired two-tailed Student t test (GraphPad Prism 4 software, San Diego,CA). For tumor size, means of “area under the curve” of each individualhave been compared using a Mann–Whitney U test. Results with a p value,0.05 were considered significant.
ResultsCD8a+ DCs represent interesting candidates for activea-GalCer delivery
DCs appear to represent the most promising candidates for activea-GalCer delivery to optimize iNKT cell–based immune respon-ses. Because the role of DCs in avoiding (15, 17, 18) or in in-ducing (19) iNKT cell unresponsiveness upon restimulation isstill controversial, we first revisited this issue using two differentapproaches: depletion of endogenous DCs using transgenic CD11c-DTR mice and adoptive transfer experiments.DT treatment depleted splenic DCs (Supplemental Fig. 1A) and,
in agreement with previous studies (17, 27), strongly loweredthe extent of primary iNKT cell activation in response to freea-GalCer, as exemplified by the decreased frequency of IFN-g+
iNKT cells (Fig. 1A) and by the reduced early release of cytokinesin the sera (Supplemental Fig. 1B). As expected, whereas in DC-competent animals, iNKT cells displayed a reduced capacity toproduce IFN-g upon a-GalCer restimulation, the recall responsewas totally blunted when DCs were lacking at the time of primaryiNKT cell stimulation (Fig. 1A). This effect was not due to adefected DC repopulation in the spleen at the time of a-GalCerchallenge (7 d after DT treatment; Supplemental Fig. 1C, leftpanel). Moreover, in non–a-GalCer–experienced animals, thisnewly repopulated DC population was able to promote iNKT cellresponse early after a-GalCer inoculation (Supplemental Fig. 1C,right panel). Together, DCs act as positive regulators of iNKT cellresponse upon repeated a-GalCer challenge.To confirm this finding, we investigated whether primary acti-
vation of iNKT cells by DCs could prevent iNKT cell hypores-ponsiveness. In our experimental conditions, free a-GalCer andin vitro a-GalCer–loaded DCs triggered a similar primary activa-tion of iNKT cells (Fig. 1B). Strikingly, whereas free a-GalCerinduced iNKT cell hyporesponsiveness, iNKT cells from micepreviously inoculated with a-GalCer–loaded DCs maintained theircapacity to reactivate. Of note, free a-GalCer induced enhancedPD-1 expression on iNKT cells, an inhibitory molecule that causesiNKT cell hyporesponsiveness (32, 33), whereas a-GalCer–loadedDCs failed to do so (Fig. 1C). Thus, a-GalCer presentation by DCsleads to efficient primary and secondary iNKT responses.We then investigated the respective role of CD8a2 and CD8a+
DC subsets in the activation of iNKT cells. To address this issue,we inoculated mice with a-GalCer and 2 h later, splenic CD8amice DCs and CD8a+ DCs were purified (Supplemental Fig. 2A)and cultured with iNKT cells. Relative to CD8a2 DCs, CD8a+
DCs promoted a much stronger secretion of IFN-g and IL-4 (Fig.1D). Similarly, CD8a+ DCs triggered an enhanced IL-2 produc-tion by the iNKT cell hybridoma DN32.D3, the activation ofwhich depends solely on CD1d/Ag-mediated TCR triggering(Fig. 1E). Flow cytometry analysis revealed a higher expressionof CD1d on splenic CD8a+ DCs, compared with CD8a2 DCs(Supplemental Fig. 2B). Moreover, using labeled a-GalCer, weobserved that compared with CD8a2 DCs, CD8a+ DCs moreefficiently incorporate a-GalCer in vivo, a finding in line withArora et al. (25) (Supplemental Fig. 2C). Collectively, CD8a+
DCs represent interesting candidates for active a-GalCer delivery.
Targeted delivery of a-GalCer to CD8a+ DCs improvesprimary and secondary iNKT cell responses
The endocytic C-type lectin receptor DEC205 is expressed on thecell surface of spleen and lymph node CD8a+ DCs (34). We tookadvantage of this property to actively target a-GalCer to CD8a+
DCs. To do so, we formulated a-GalCer in PLGA-based NPs dec-orated with Abs recognizing DEC205 (NP/DEC205) (Table I). AsFig. 2A shows, DEC205+ DCs incorporated NP/DEC205, in contrastwith DEC2052 DCs. Moreover, there was no uptake of nontargetedNPs (NP/IgG), used here as a negative control, by DCs. More im-portantly, when incubated with splenocytes, CD8a+ DCs more effi-ciently uptook NP/DEC205 relative to CD8a2 DCs (Fig. 2B). Wethen compared the biological activity of the formulations. DCs in-cubated with grading doses of NP/DEC205/a-GalCer triggereda higher IFN-g production relative to free, nonvectorized, a-GalCerand particularly to NP/IgG/a-GalCer (Fig. 2C). As shown in Fig. 2D,the effect was CD1d dependent. These data collectively show thatencapsulation of a-GalCer in NP/DEC205 selectively targets CD8a+
DCs to efficiently activate iNKT cells in vitro.In vivo, NP/DEC205/a-GalCer promoted a higher iNKT cell
activation, whatever the dose used, relative to free a-GalCer,and particularly to NP/IgG/a-GalCer (Fig. 3A). Of note, DC de-pletion strongly reduced iNKT cell activation after NP/DEC205/a-GalCer administration (Fig. 3B). Relative to free a-GalCer, NP/DEC205/a-GalCer induced a higher transactivation of NK cells,gd T cells, and DCs (Fig. 3C, Supplemental Fig. 3A). We nextinvestigated whether in vivo delivery of a-GalCer to CD8a+ DCscould impact on iNKT cell responsiveness upon a recall response.To address this issue, we injected mice with a-GalCer encapsu-lated in NP/DEC205 (5 ng/mouse) or free a-GalCer (100 ng/mouse), both leading to comparable primary iNKT cell activa-tion (Supplemental Fig. 3B). Strikingly, although free a-GalCerinduced the expected iNKT cell hyporesponsiveness, NP/DEC205/a-GalCer failed to do so (Fig. 3D). Finally, whereas free a-GalCerinduced PD-1 expression on iNKT cells, this was not the caseafter NP/DEC205/a-GalCer administration (Supplemental Fig. 3C).Collectively, in vivo active delivery of a-GalCer to CD8a+ DCsoptimizes primary iNKT cell responses and prevents iNKT cellhyporesponsiveness upon a secondary challenge.
Codelivery of a-GalCer and OVA to CD8a+ DCs optimizesCD8+ T cell and Ab responses
Studies in mice have shown a benefit to target Ags to CD8a+ DCsvia the DEC205 endocytic pathway, particularly to promote Agcross-presentation and to prime CD8+ T cell responses (35, 36).The effects of targeting a-GalCer and Ag in intimate associationwith each other to CD8a+ DCs by means of synthetic deliverysystems have not yet been investigated. To do so, mice recon-stituted with CFSE-labeled OT-I cells were inoculated with NP/DEC205 containing both a-GalCer and the model Ag OVA. NP/DEC205/OVA/a-GalCer induced a higher proliferation of OVA-specific OT-I cells compared with mice inoculated with NP/IgG/OVA/a-GalCer or with soluble a-GalCer plus OVA or OVA alone(Fig. 4A, Supplemental Fig. 4A). Furthermore, upon in vitro peptiderestimulation, the frequency of OVA-specific CD8+ T lymphocytesexpressing IFN-g was higher in mice that received NP/DEC205/OVA/a-GalCer, compared with other animal groups (SupplementalFig. 4B). NP/DEC205/OVA/a-GalCer also elicited a higher CTLresponse, as assessed by the measurement of target cell lysis (Fig.4B). Finally, although the recall response 2 mo after the last im-munization was nearly undetectable in other groups, mice admin-istered with NP/DEC205/OVA/a-GalCer displayed a long-lastingCD8+ T cell memory response (Fig. 4C). Targeting DEC205 can
The Journal of Immunology 963
by guest on February 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
also promote humoral responses (37). Indeed, relative to free OVAplus a-GalCer, NP/DEC205/OVA/a-GalCer promoted a higher titerof OVA-specific IgG (Fig. 4D). Thus, combining OVA and a-GalCerinto the same particle to target CD8a+ DCs via DEC205 is clearly ofbenefit to enhance cellular and humoral immune responses.
NPs incorporating a-GalCer and Ag protect against tumordevelopment
Induction of robust CTL responses is essential for the immuno-therapy against cancer. To investigate the consequences of a-GalCer
and Ag vectorization on the control of tumor development, wevaccinated mice with NP/DEC205/OVA/a-GalCer before inocula-tion of OVA-expressing B16F10 melanoma cells. As Fig. 5A shows,and compared with mice receiving NP/IgG/OVA/a-GalCer, vacci-nated mice were fully protected against the development of lungmetastases. At the dose used, free OVA plus a-GalCer failed topromote antitumor response (data not shown). To confirm thisfinding in another system, we used a model of solid tumor, namely,OVA-expressing T cell lymphoma (EG7). Compared with NP/IgG/OVA/a-GalCer, prior inoculation of NP/DEC205/OVA/a-GalCer
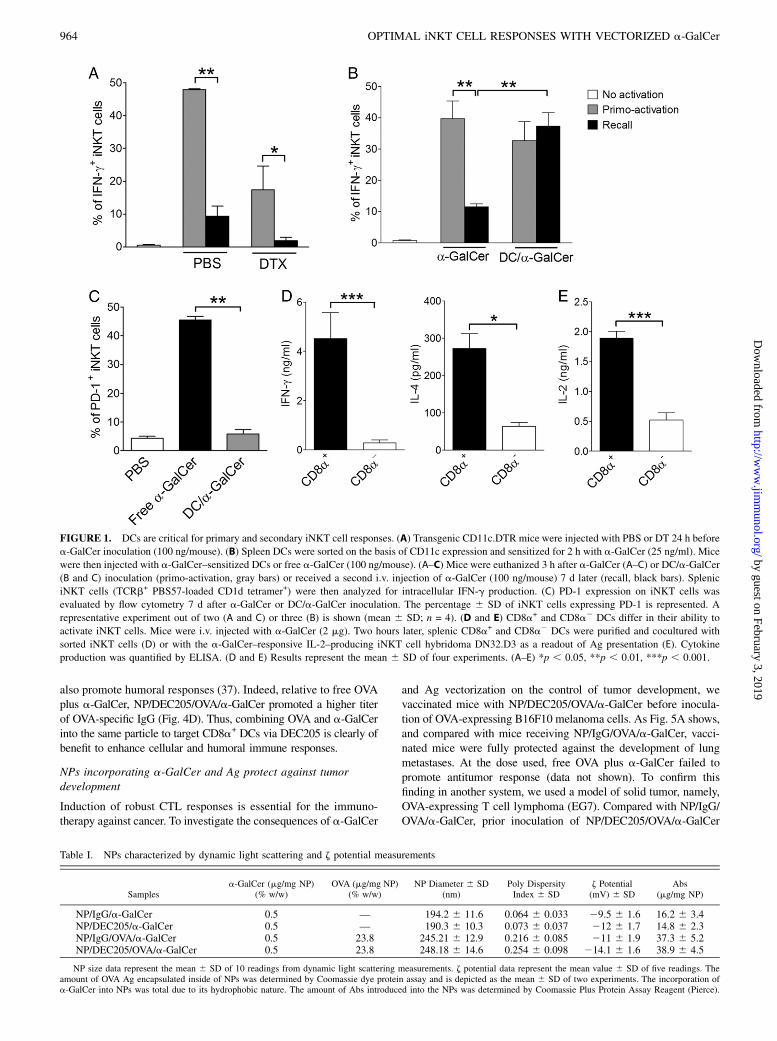
FIGURE 1. DCs are critical for primary and secondary iNKT cell responses. (A) Transgenic CD11c.DTR mice were injected with PBS or DT 24 h before
a-GalCer inoculation (100 ng/mouse). (B) Spleen DCs were sorted on the basis of CD11c expression and sensitized for 2 h with a-GalCer (25 ng/ml). Mice
were then injected with a-GalCer–sensitized DCs or free a-GalCer (100 ng/mouse). (A–C) Mice were euthanized 3 h after a-GalCer (A–C) or DC/a-GalCer
(B and C) inoculation (primo-activation, gray bars) or received a second i.v. injection of a-GalCer (100 ng/mouse) 7 d later (recall, black bars). Splenic
iNKT cells (TCRb+ PBS57-loaded CD1d tetramer+) were then analyzed for intracellular IFN-g production. (C) PD-1 expression on iNKT cells was
evaluated by flow cytometry 7 d after a-GalCer or DC/a-GalCer inoculation. The percentage 6 SD of iNKT cells expressing PD-1 is represented. A
representative experiment out of two (A and C) or three (B) is shown (mean 6 SD; n = 4). (D and E) CD8a+ and CD8a2 DCs differ in their ability to
activate iNKT cells. Mice were i.v. injected with a-GalCer (2 mg). Two hours later, splenic CD8a+ and CD8a2 DCs were purified and cocultured with
sorted iNKT cells (D) or with the a-GalCer–responsive IL-2–producing iNKT cell hybridoma DN32.D3 as a readout of Ag presentation (E). Cytokine
production was quantified by ELISA. (D and E) Results represent the mean 6 SD of four experiments. (A–E) *p , 0.05, **p , 0.01, ***p , 0.001.
Table I. NPs characterized by dynamic light scattering and z potential measurements
Samplesa-GalCer (mg/mg NP)
(% w/w)OVA (mg/mg NP)
(% w/w)NP Diameter 6 SD
(nm)Poly DispersityIndex 6 SD
z Potential(mV) 6 SD
Abs(mg/mg NP)
NP/IgG/a-GalCer 0.5 — 194.2 6 11.6 0.064 6 0.033 29.5 6 1.6 16.2 6 3.4NP/DEC205/a-GalCer 0.5 — 190.3 6 10.3 0.073 6 0.037 212 6 1.7 14.8 6 2.3NP/IgG/OVA/a-GalCer 0.5 23.8 245.21 6 12.9 0.216 6 0.085 211 6 1.9 37.3 6 5.2NP/DEC205/OVA/a-GalCer 0.5 23.8 248.18 6 14.6 0.254 6 0.098 214.1 6 1.6 38.9 6 4.5
NP size data represent the mean 6 SD of 10 readings from dynamic light scattering measurements. z potential data represent the mean value 6 SD of five readings. Theamount of OVA Ag encapsulated inside of NPs was determined by Coomassie dye protein assay and is depicted as the mean 6 SD of two experiments. The incorporation ofa-GalCer into NPs was total due to its hydrophobic nature. The amount of Abs introduced into the NPs was determined by Coomassie Plus Protein Assay Reagent (Pierce).
964 OPTIMAL iNKT CELL RESPONSES WITH VECTORIZED a-GalCer
by guest on February 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
delayed the growth of OVA-expressing EG7 cells (Fig. 5B). Toassess the efficacy of the vaccine in a therapeutic setting, we firstimplanted mice with OVA-expressing EG7 cells and 1 and 5 d later,animals were administered with NP/DEC205/OVA/a-GalCer. Com-pared with controls, tumor growth delay was observed in micevaccinated with NP/DEC205/OVA/a-GalCer (Fig. 5C). Collec-tively, in vivo targeted delivery of a-GalCer and Ag to CD8a+
DCs promotes antitumor responses in both prophylactic and thera-peutic settings.
Discussiona-GalCer is a strong immunostimulatory molecule that holds greatpromise for antitumor therapeutic purposes and vaccine develop-ment (3, 7, 38). However, several concerns limit its use in clinics,including the uncontrolled response it promotes when delivered ina nonvectorized form. Cell-based antitumor therapy (transfer ofDCs pulsed with a-GalCer) to better control iNKT cell functionsis an attractive possibility (6, 7), although this procedure remainscomplex and expensive. Another strategy is to passively or ac-tively deliver a-GalCer to DCs. We and others have shown thata-GalCer vectorized in PLGA-based NPs (30), silica particles(39), liposomes (40), or included into virus-like particles (41)activated iNKT cells but failed to prevent their hyporesponsive-
ness upon restimulation, a likely consequence of uncontrolled celltargeting. Therefore, a controlled delivery of a-GalCer is a requi-site to optimize iNKT cell responses. A recent study showed thatspecific targeting of a-GalCer to CD169+ macrophages leads to anefficient iNKT cell activation in vivo but fails to trigger trans-activation of innate immune cells (42). The consequence on therecall response was not reported in the later study. In this study, weshow that targeted delivery of a-GalCer to CD8a+ DCs, a DCsubset that excels in Ag cross-presentation, is instrumental notonly to enhance primary activation of iNKT cells and to avoidiNKT cell hyporesponsiveness. In parallel, active codelivery ofa-GalCer and Ag to CD8a+ DCs promotes strong CD8+ T cellresponses, with a positive effect on the control of tumor devel-opment.Our data confirmed that DCs are primary initiators of iNKT
cell activation after systemic administration of nonvectorizeda-GalCer, in line with other reports (17, 18, 27, 39, 43, 44). DCsfrom the spleen are heterogeneous, and recent studies suggestedthat DC subsets could differ in their ability to stimulate iNKT cells(26, 45, 46). For example, we have previously reported that amongthe CD8a2 DC subset, CD42 DCs were more efficient at acti-vating iNKT cells, relative to CD4+ DCs (45). Consistent with theenhanced level of cell-surface CD1d and the higher incorporation
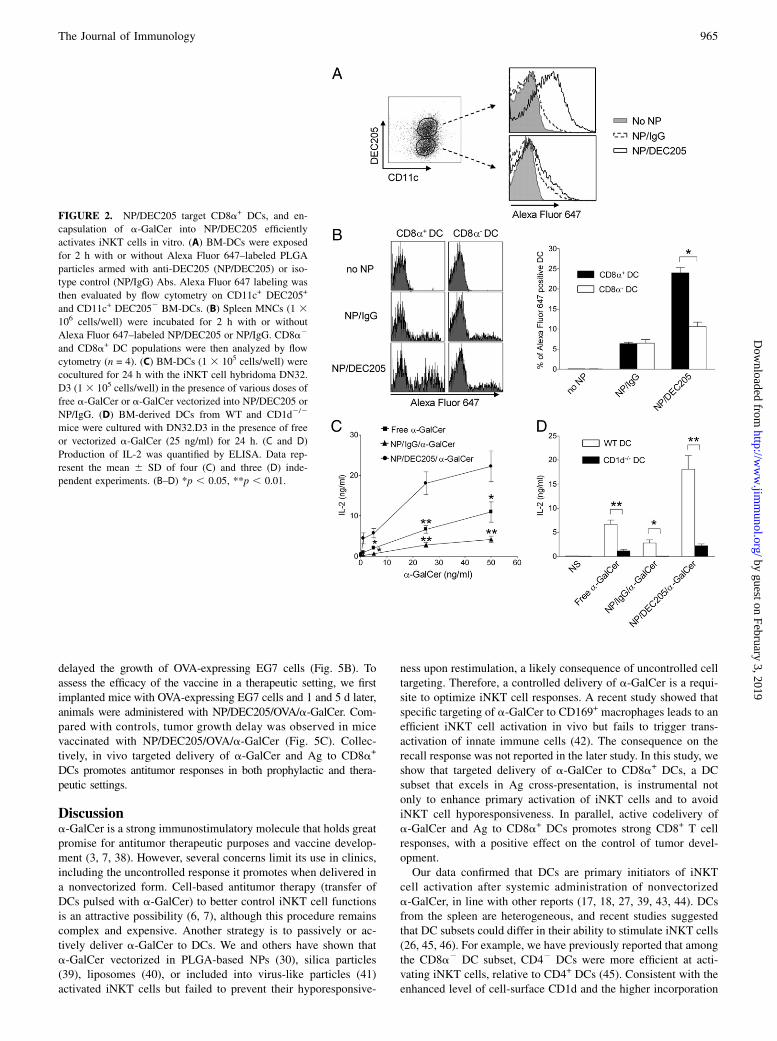
FIGURE 2. NP/DEC205 target CD8a+ DCs, and en-
capsulation of a-GalCer into NP/DEC205 efficiently
activates iNKT cells in vitro. (A) BM-DCs were exposed
for 2 h with or without Alexa Fluor 647–labeled PLGA
particles armed with anti-DEC205 (NP/DEC205) or iso-
type control (NP/IgG) Abs. Alexa Fluor 647 labeling was
then evaluated by flow cytometry on CD11c+ DEC205+
and CD11c+ DEC2052 BM-DCs. (B) Spleen MNCs (1 3106 cells/well) were incubated for 2 h with or without
Alexa Fluor 647–labeled NP/DEC205 or NP/IgG. CD8a2
and CD8a+ DC populations were then analyzed by flow
cytometry (n = 4). (C) BM-DCs (1 3 105 cells/well) were
cocultured for 24 h with the iNKT cell hybridoma DN32.
D3 (13 105 cells/well) in the presence of various doses of
free a-GalCer or a-GalCer vectorized into NP/DEC205 or
NP/IgG. (D) BM-derived DCs from WT and CD1d2/2
mice were cultured with DN32.D3 in the presence of free
or vectorized a-GalCer (25 ng/ml) for 24 h. (C and D)
Production of IL-2 was quantified by ELISA. Data rep-
resent the mean 6 SD of four (C) and three (D) inde-
pendent experiments. (B–D) *p , 0.05, **p , 0.01.
The Journal of Immunology 965
by guest on February 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
rate of a-GalCer, our results are in line with a recent study (25)showing that, relative to CD8a2 DCs, CD8a+ DCs are potenttriggers of iNKT cell activation. The fact that CD8a+ DCs localizein the marginal zone of the spleen and that iNKT cells rapidlymobilize in this zone early after a-GalCer inoculation is in linewith these observations (34, 39, 44, 47).We next sought to investigate the impact of a-GalCer delivery
to CD8a+ DCs on immune responses. We show that NP/DEC205target splenic CD8a+ (DEC205+) DCs and that a-GalCer vectorized
in NP/DEC205 can be loaded onto CD1d to activate iNKT cells.Of importance, NP/DEC205/a-GalCer was ∼2- to 10-fold more ef-fective at activating iNKT cells than free a-GalCer in vivo, a processthat depended on DCs. The enhanced iNKT cell response is probablydue to the more rapid (DEC205-mediated) uptake of a-GalCer byCD8a+ DCs. It is also likely that the DEC205-mediated endocyticpathway facilitates a-GalCer accessibility to the CD1d molecule inendosomes/lysosomes of DCs. Indeed, DEC205 is known to delivercargo specifically in this compartment, which is also well documented
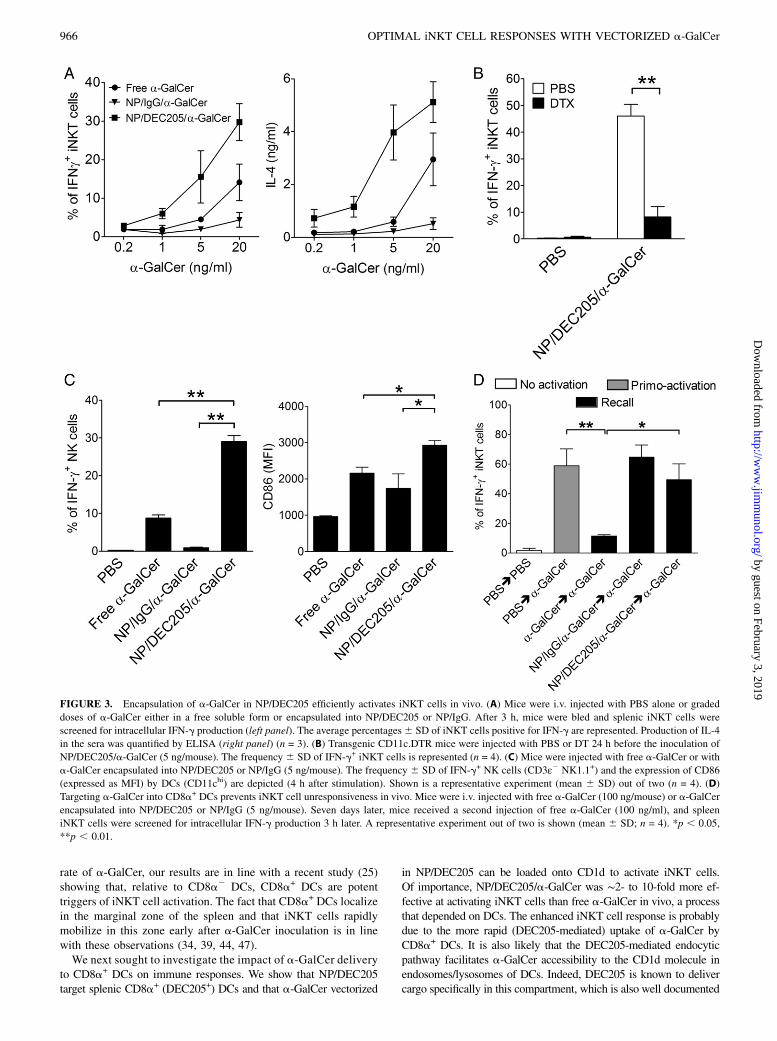
FIGURE 3. Encapsulation of a-GalCer in NP/DEC205 efficiently activates iNKT cells in vivo. (A) Mice were i.v. injected with PBS alone or graded
doses of a-GalCer either in a free soluble form or encapsulated into NP/DEC205 or NP/IgG. After 3 h, mice were bled and splenic iNKT cells were
screened for intracellular IFN-g production (left panel). The average percentages6 SD of iNKT cells positive for IFN-g are represented. Production of IL-4
in the sera was quantified by ELISA (right panel) (n = 3). (B) Transgenic CD11c.DTR mice were injected with PBS or DT 24 h before the inoculation of
NP/DEC205/a-GalCer (5 ng/mouse). The frequency 6 SD of IFN-g+ iNKT cells is represented (n = 4). (C) Mice were injected with free a-GalCer or with
a-GalCer encapsulated into NP/DEC205 or NP/IgG (5 ng/mouse). The frequency 6 SD of IFN-g+ NK cells (CD3ε2 NK1.1+) and the expression of CD86
(expressed as MFI) by DCs (CD11chi) are depicted (4 h after stimulation). Shown is a representative experiment (mean 6 SD) out of two (n = 4). (D)
Targeting a-GalCer into CD8a+ DCs prevents iNKT cell unresponsiveness in vivo. Mice were i.v. injected with free a-GalCer (100 ng/mouse) or a-GalCer
encapsulated into NP/DEC205 or NP/IgG (5 ng/mouse). Seven days later, mice received a second injection of free a-GalCer (100 ng/ml), and spleen
iNKT cells were screened for intracellular IFN-g production 3 h later. A representative experiment out of two is shown (mean 6 SD; n = 4). *p , 0.05,
**p , 0.01.
966 OPTIMAL iNKT CELL RESPONSES WITH VECTORIZED a-GalCer
by guest on February 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
to load lipid Ags onto CD1d (48). Of interest, and in contrast withspecific targeting of a-GalCer to CD169+ macrophages (42), activedelivery of a-GalCer to CD8a+ DCs also increased the transactivationof NK, gd T lymphocytes, and DCs. Thus, CD8a+ DCs targeting viaDEC205 optimizes the iNKT cell–mediated innate immune responsein agreement with the fact that CD8a+ DCs are a dominant source ofbioactive IL-12 (26, 49).After primary activation, iNKT cells develop a long-lasting
hyporesponsiveness (15). The potential role played by DCs inthis phenomenon is still debated (15, 17–19). Using two com-plementary strategies (DC depletion and DC transfer), our resultsclearly show that presentation of a-GalCer by DCs (primaryiNKT cell activation) does not lead to iNKT cell hyporesponsive-ness after secondary stimulation, in line with previous studies (15,17, 18). Paralleling this, targeting a-GalCer to CD8a+ DCs by
means of NP/DEC205 did not lead to iNKT cell hypores-ponsiveness, confirming the key role played by DCs in main-taining secondary iNKT cell activation. Thus, a-GalCer–loadedCD8a+ DCs not only efficiently trigger TCR signaling iniNKT cells (primo-stimulation), but also maintain secondary ac-tivation after challenge. During primary iNKT cell activation,multiple signals from surface-bound and soluble costimulatoryand/or inhibitory molecules function in concert to stimulate andfine-tune iNKT cell response. Thus, it is likely that, in addition toCD1d, CD8a+ DCs provide additional signals, absent in CD11cnonexpressing cells (e.g., B lymphocytes), to maintain secondaryiNKT cell activation.Codelivering Ag and adjuvants to DCs, including CD8a+ DCs,
has been shown to induce optimal T and B cell–mediated immuneresponses (50–52). Whether in vivo targeted codelivery of a-GalCer
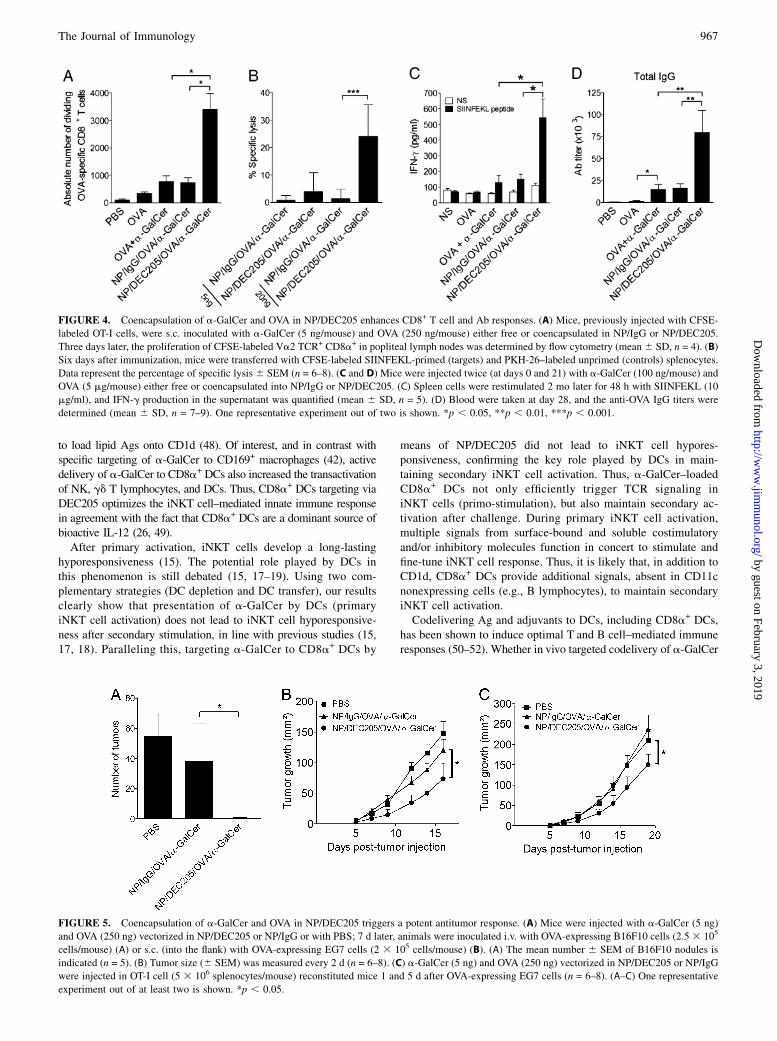
FIGURE 4. Coencapsulation of a-GalCer and OVA in NP/DEC205 enhances CD8+ T cell and Ab responses. (A) Mice, previously injected with CFSE-
labeled OT-I cells, were s.c. inoculated with a-GalCer (5 ng/mouse) and OVA (250 ng/mouse) either free or coencapsulated in NP/IgG or NP/DEC205.
Three days later, the proliferation of CFSE-labeled Va2 TCR+ CD8a+ in popliteal lymph nodes was determined by flow cytometry (mean6 SD, n = 4). (B)
Six days after immunization, mice were transferred with CFSE-labeled SIINFEKL-primed (targets) and PKH-26–labeled unprimed (controls) splenocytes.
Data represent the percentage of specific lysis6 SEM (n = 6–8). (C and D) Mice were injected twice (at days 0 and 21) with a-GalCer (100 ng/mouse) and
OVA (5 mg/mouse) either free or coencapsulated into NP/IgG or NP/DEC205. (C) Spleen cells were restimulated 2 mo later for 48 h with SIINFEKL (10
mg/ml), and IFN-g production in the supernatant was quantified (mean 6 SD, n = 5). (D) Blood were taken at day 28, and the anti-OVA IgG titers were
determined (mean 6 SD, n = 7–9). One representative experiment out of two is shown. *p , 0.05, **p , 0.01, ***p , 0.001.
FIGURE 5. Coencapsulation of a-GalCer and OVA in NP/DEC205 triggers a potent antitumor response. (A) Mice were injected with a-GalCer (5 ng)
and OVA (250 ng) vectorized in NP/DEC205 or NP/IgG or with PBS; 7 d later, animals were inoculated i.v. with OVA-expressing B16F10 cells (2.5 3 105
cells/mouse) (A) or s.c. (into the flank) with OVA-expressing EG7 cells (2 3 105 cells/mouse) (B). (A) The mean number 6 SEM of B16F10 nodules is
indicated (n = 5). (B) Tumor size (6 SEM) was measured every 2 d (n = 6–8). (C) a-GalCer (5 ng) and OVA (250 ng) vectorized in NP/DEC205 or NP/IgG
were injected in OT-I cell (5 3 106 splenocytes/mouse) reconstituted mice 1 and 5 d after OVA-expressing EG7 cells (n = 6–8). (A–C) One representative
experiment out of at least two is shown. *p , 0.05.
The Journal of Immunology 967
by guest on February 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

and Ag to DC subsets impacts on immune and antitumor responseshas not yet been studied. CD8a+ DCs excel in MHC class I cross-presentation, and iNKT cells have been shown to directly licenseCD8a+ DCs for cross-priming, even in the absence of CD4+ T cells(53). In this study, we show that targeted codelivery of a-GalCerand OVA to CD8a+ DCs amplified the primary and memory CD8+
T cell responses compared with the same amount of a-GalCer andOVA given in untargeted form. In the same vein, the OVA-specificAb response was greatly augmented in response to a-GalCer andAg inserted into the same particle and targeted to CD8a+ DCs.Thus, as shown for TLR agonists (52), targeting a-GalCer to CD8a+
DCs improves a-GalCer adjuvanticity. Although this remains to befully demonstrated, we hypothesize that a-GalCer/Ag codelivery toCD8a+ DCs might have superior effects compared with TLR agonist/Ag codelivery. Indeed, TLR agonists exert adjuvant effects by in-ducing direct DC maturation, a process that lowers Ag uptake but iscrucial to efficiently prime naive T cells. In the case of a-GalCer, DCmaturation is indirect and lies on iNKT cell factors produced afterprimo-activation. It is possible that the delayed maturation of DCs inresponse to a-GalCer, relative to TLR agonists, might prolong the Aguptake capacity of DCs, thus leading to amplified immune responses.The fact that iNKT cells can substitute CD4+ Th cells to induce T andB cell responses (53, 54) offers a new avenue for investigating theconsequences of iNKT cell–based adjuvant properties in many set-tings, including in cancer therapy.The ultimate goal of antitumor vaccines is to develop memory
CD8+ CTLs, which are critical mediators of antitumor immunity.Using two different tumor models, our data reveal that a-GalCer/Ag codelivery to CD8a+ DCs triggers antitumor responses, inprophylactic and/or therapeutic settings. Of interest, Shimizu andcolleagues (22, 23) showed that inoculation of irradiated tumorcells previously loaded with a-GalCer promotes antitumor im-munity through Ag cross-presentation and CTL responses. In thissetting, dying tumor cells are selectively taken up by CD8a+ DCs,and the subsequent cross-presentation of a-GalCer to NKT cellsand tumor Ags to CD8+ T cells leads to strong and long-lastingantitumor responses (22). Using a different strategy (DC targetingby means of a synthetic vector), our data are in line with theconcept that having both tumor Ag and a-GalCer on the same cellresulted in more efficient cross-presentation of Ag to CD8+
T cells. These data will prompt us to validate the efficacy ofa-GalCer–containing nanoparticulate vaccines in more physio-logical tumor models (e.g., spontaneous tumors) in combination,or not, with complementary strategies such as depletion of im-munosuppressive regulatory T cells (55).To conclude, we have designed and undertaken an active tar-
geting strategy based on the use of PLGA-based NPs carrying ontheir surface DC-specific Abs to minimize the unwanted effects ofuncontrolled iNKT cell activation, whereas maximizing the abilityof iNKT cells to promote efficient adaptive and antitumor immuneresponses. We show that the in vivo delivery of a-GalCer intoCD8a+ DCs enhances the early activation of iNKT cells but alsoallows these cells to respond to further restimulations. Of impor-tance, the NP-mediated simultaneous codelivery of both a-GalCerand protein Ag in CD8a+ DCs induces optimal CD8+ T cell andantitumor responses in the mouse system. These data may haveimportant implications in the design of novel strategies for cancertherapy and vaccination.
AcknowledgmentsWe thank Drs. C. Reis e Sousa (London Research Institute, London, U.K.),
L. Van Kaer (Vanderbilt University, Nashville, TN), and A. Bendelac (Uni-
versity of Chicago, Chicago, IL) for the gift of B16F10 melanoma cells
expressing OVA, EG7 cells, CD1d2/2 C57BL/6 mice, and the NKT cell
hybridoma DN32.D3, respectively. We thank the National Institute of
Allergy and Infectious Diseases Tetramer Facility (Emory University,
Atlanta, GA) for their generous support in supplying CD1d tetramers.
DisclosuresThe authors have no financial conflicts of interest.
References1. Andersen, B. M., and J. R. Ohlfest. 2012. Increasing the efficacy of tumor cell
vaccines by enhancing cross priming. Cancer Lett. 325: 155–164.2. Dougan, M., and G. Dranoff. 2009. Immune therapy for cancer. Annu. Rev.
Immunol. 27: 83–117.3. Cerundolo, V., J. D. Silk, S. H. Masri, and M. Salio. 2009. Harnessing invariant
NKT cells in vaccination strategies. Nat. Rev. Immunol. 9: 28–38.4. Dhodapkar, M. V., and J. Richter. 2011. Harnessing natural killer T (NKT) cells
in human myeloma: progress and challenges. Clin. Immunol. 140: 160–166.5. Pilones, K. A., J. Aryankalayil, and S. Demaria. 2012. Invariant NKT cells as novel
targets for immunotherapy in solid tumors. Clin. Dev. Immunol. 2012: 720803.6. Motohashi, S., and T. Nakayama. 2009. Invariant natural killer T cell-based
immunotherapy for cancer. Immunotherapy 1: 73–82.7. Schneiders, F. L., R. J. Scheper, B. M. von Blomberg, A. M.Woltman, H. L. Janssen,
A. J. van den Eertwegh, H. M. Verheul, T. D. de Gruijl, and H. J. van der Vliet. 2011.Clinical experience with a-galactosylceramide (KRN7000) in patients with ad-vanced cancer and chronic hepatitis B/C infection. Clin. Immunol. 140: 130–141.
8. Bendelac, A., P. B. Savage, and L. Teyton. 2007. The biology of NKT cells.Annu. Rev. Immunol. 25: 297–336.
9. Terabe, M., and J. A. Berzofsky. 2007. NKT cells in immunoregulation of tumorimmunity: a new immunoregulatory axis. Trends Immunol. 28: 491–496.
10. Fujii, S., S. Motohashi, K. Shimizu, T. Nakayama, Y. Yoshiga, and M. Taniguchi.2010. Adjuvant activity mediated by iNKT cells. Semin. Immunol. 22: 97–102.
11. Kobayashi, E., K. Motoki, T. Uchida, H. Fukushima, and Y. Koezuka. 1995.KRN7000, a novel immunomodulator, and its antitumor activities. Oncol. Res. 7:529–534.
12. Nakagawa, R., K. Motoki, H. Nakamura, H. Ueno, R. Iijima, A. Yamauchi,S. Tsuyuki, T. Inamoto, and Y. Koezuka. 1998. Antitumor activity of alpha-galactosylceramide, KRN7000, in mice with EL-4 hepatic metastasis and itscytokine production. Oncol. Res. 10: 561–568.
13. Giaccone, G., C. J. Punt, Y. Ando, R. Ruijter, N. Nishi, M. Peters, B. M. vonBlomberg, R. J. Scheper, H. J. van der Vliet, A. J. van den Eertwegh, et al. 2002.A phase I study of the natural killer T-cell ligand alpha-galactosylceramide(KRN7000) in patients with solid tumors. Clin. Cancer Res. 8: 3702–3709.
14. Molling, J. W., W. Kolgen, H. J. van der Vliet, M. F. Boomsma, H. Kruizenga,C. H. Smorenburg, B. G. Molenkamp, J. A. Langendijk, C. R. Leemans,B. M. von Blomberg, et al. 2005. Peripheral blood IFN-gamma-secreting Val-pha24+Vbeta11+ NKT cell numbers are decreased in cancer patients indepen-dent of tumor type or tumor load. Int. J. Cancer 116: 87–93.
15. Parekh, V. V., M. T. Wilson, D. Olivares-Villagomez, A. K. Singh, L. Wu,C. R. Wang, S. Joyce, and L. Van Kaer. 2005. Glycolipid antigen induces long-term natural killer T cell anergy in mice. J. Clin. Invest. 115: 2572–2583.
16. Faveeuw, C., and F. Trottein. 2014. Optimization of natural killer T cell-mediatedimmunotherapy in cancer using cell-based and nanovector vaccines. Cancer Res.74: 1632–1638.
17. Bezbradica, J. S., A. K. Stanic, N. Matsuki, H. Bour-Jordan, J. A. Bluestone,J. W. Thomas, D. Unutmaz, L. Van Kaer, and S. Joyce. 2005. Distinct roles ofdendritic cells and B cells in Va14Ja18 natural T cell activation in vivo. J.Immunol. 174: 4696–4705.
18. Fujii, S., K. Shimizu, M. Kronenberg, and R. M. Steinman. 2002. ProlongedIFN-gamma-producing NKT response induced with alpha-galactosylceramide-loaded DCs. Nat. Immunol. 3: 867–874.
19. Iyoda, T., M. Ushida, Y. Kimura, K. Minamino, A. Hayuka, S. Yokohata,H. Ehara, and K. Inaba. 2010. Invariant NKT cell anergy is induced by a strongTCR-mediated signal plus co-stimulation. Int. Immunol. 22: 905–913.
20. Chang, D. H., K. Osman, J. Connolly, A. Kukreja, J. Krasovsky, M. Pack,A. Hutchinson, M. Geller, N. Liu, R. Annable, et al. 2005. Sustained expansion ofNKT cells and antigen-specific T cells after injection of alpha-galactosyl-ceramideloaded mature dendritic cells in cancer patients. J. Exp. Med. 201: 1503–1517.
21. Nieda, M., M. Okai, A. Tazbirkova, H. Lin, A. Yamaura, K. Ide, R. Abraham,T. Juji, D. J. Macfarlane, and A. J. Nicol. 2004. Therapeutic activation of Val-pha24+Vbeta11+ NKT cells in human subjects results in highly coordinatedsecondary activation of acquired and innate immunity. Blood 103: 383–389.
22. Shimizu, K., M. Asakura, J. Shinga, Y. Sato, S. Kitahara, K. Hoshino, T. Kaisho,S. P. Schoenberger, T. Ezaki, and S. Fujii. 2013. Invariant NKT cells induceplasmacytoid dendritic cell (DC) cross-talk with conventional DCs for efficientmemory CD8+ T cell induction. J. Immunol. 190: 5609–5619.
23. Shimizu, K., Y. Kurosawa, M. Taniguchi, R. M. Steinman, and S. Fujii. 2007.Cross-presentation of glycolipid from tumor cells loaded with alpha-galactosylceramide leads to potent and long-lived T cell mediated immunityvia dendritic cells. J. Exp. Med. 204: 2641–2653.
24. Shortman, K., and W. R. Heath. 2010. The CD8+ dendritic cell subset. Immunol.Rev. 234: 18–31.
25. Arora, P., A. Baena, K. O. Yu, N. K. Saini, S. S. Kharkwal, M. F. Goldberg,S. Kunnath-Velayudhan, L. J. Carreno, M. M. Venkataswamy, J. Kim, et al.2014. A single subset of dendritic cells controls the cytokine bias of natural killerT cell responses to diverse glycolipid antigens. Immunity 40: 105–116.
968 OPTIMAL iNKT CELL RESPONSES WITH VECTORIZED a-GalCer
by guest on February 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

26. Farrand, K. J., N. Dickgreber, P. Stoitzner, F. Ronchese, T. R. Petersen, andI. F. Hermans. 2009. Langerin+ CD8alpha+ dendritic cells are critical for cross-primingand IL-12 production in response to systemic antigens. J. Immunol. 183: 7732–7742.
27. Schmieg, J., G. Yang, R. W. Franck, N. Van Rooijen, and M. Tsuji. 2005.Glycolipid presentation to natural killer T cells differs in an organ-dependentfashion. Proc. Natl. Acad. Sci. USA 102: 1127–1132.
28. Jung, S., D. Unutmaz, P. Wong, G. Sano, K. De los Santos, T. Sparwasser, S. Wu,S. Vuthoori, K. Ko, F. Zavala, et al. 2002. In vivo depletion of CD11c+ dendriticcells abrogates priming of CD8+ T cells by exogenous cell-associated antigens.Immunity 17: 211–220.
29. Mendiratta, S. K., W. D. Martin, S. Hong, A. Boesteanu, S. Joyce, and L. VanKaer. 1997. CD1d1 mutant mice are deficient in natural T cells that promptlyproduce IL-4. Immunity 6: 469–477.
30. Macho Fernandez, E., J. Chang, J. Fontaine, E. Bialecki, F. Rodriguez,E. Werkmeister, V. Krieger, C. Ehret, B. Heurtault, S. Fournel, et al. 2012.Activation of invariant Natural Killer T lymphocytes in response to the a-gal-actosylceramide analogue KRN7000 encapsulated in PLGA-based nanoparticlesand microparticles. Int. J. Pharm. 423: 45–54.
31. Cruz, L. J., P. J. Tacken, R. Fokkink, B. Joosten, M. C. Stuart, F. Albericio,R. Torensma, and C. G. Figdor. 2010. Targeted PLGA nano- but not micro-particles specifically deliver antigen to human dendritic cells via DC-SIGNin vitro. J. Control. Release 144: 118–126.
32. Chang, W. S., J. Y. Kim, Y. J. Kim, Y. S. Kim, J. M. Lee, M. Azuma, H. Yagita,and C. Y. Kang. 2008. Cutting edge: Programmed death-1/programmed deathligand 1 interaction regulates the induction and maintenance of invariantNKT cell anergy. J. Immunol. 181: 6707–6710.
33. Parekh, V. V., S. Lalani, S. Kim, R. Halder, M. Azuma, H. Yagita, V. Kumar,L. Wu, and L. V. Kaer. 2009. PD-1/PD-L blockade prevents anergy induction andenhances the anti-tumor activities of glycolipid-activated invariant NKT cells. J.Immunol. 182: 2816–2826.
34. Idoyaga, J., N. Suda, K. Suda, C. G. Park, and R. M. Steinman. 2009. Antibodyto Langerin/CD207 localizes large numbers of CD8alpha+ dendritic cells to themarginal zone of mouse spleen. Proc. Natl. Acad. Sci. USA 106: 1524–1529.
35. Bonifaz, L. C., D. P. Bonnyay, A. Charalambous, D. I. Darguste, S. Fujii,H. Soares, M. K. Brimnes, B. Moltedo, T. M. Moran, and R. M. Steinman. 2004.In vivo targeting of antigens to maturing dendritic cells via the DEC-205 re-ceptor improves T cell vaccination. J. Exp. Med. 199: 815–824.
36. Idoyaga, J., A. Lubkin, C. Fiorese, M. H. Lahoud, I. Caminschi, Y. Huang,A. Rodriguez, B. E. Clausen, C. G. Park, C. Trumpfheller, and R. M. Steinman.2011. Comparable T helper 1 (Th1) and CD8 T-cell immunity by targeting HIVgag p24 to CD8 dendritic cells within antibodies to Langerin, DEC205, andClec9A. Proc. Natl. Acad. Sci. USA 108: 2384–2389.
37. Birkholz, K., M. Schwenkert, C. Kellner, S. Gross, G. Fey, B. Schuler-Thurner,G. Schuler, N. Schaft, and J. Dorrie. 2010. Targeting of DEC-205 on humandendritic cells results in efficient MHC class II-restricted antigen presentation.Blood 116: 2277–2285.
38. Berzins, S. P., M. J. Smyth, and A. G. Baxter. 2011. Presumed guilty: naturalkiller T cell defects and human disease. Nat. Rev. Immunol. 11: 131–142.
39. Barral, P., M. D. Sanchez-Nino, N. van Rooijen, V. Cerundolo, and F. D. Batista.2012. The location of splenic NKT cells favours their rapid activation by blood-borne antigen. EMBO J. 31: 2378–2390.
40. Ishii, M., and N. Kojima. 2013. Effective stimulation of invariant naturalkiller T cells by oligomannose-coated liposomes. Int. Immunopharmacol. 15:685–692.
41. McKee, S. J., V. L. Young, F. Clow, C. M. Hayman, M. A. Baird, I. F. Hermans,S. L. Young, and V. K. Ward. 2012. Virus-like particles and a-galactosylcer-
amide form a self-adjuvanting composite particle that elicits anti-tumorresponses. J. Control. Release 159: 338–345.
42. Kawasaki, N., J. L. Vela, C. M. Nycholat, C. Rademacher, A. Khurana, N. vanRooijen, P. R. Crocker, M. Kronenberg, and J. C. Paulson. 2013. Targeted de-livery of lipid antigen to macrophages via the CD169/sialoadhesin endocyticpathway induces robust invariant natural killer T cell activation. Proc. Natl.Acad. Sci. USA 110: 7826–7831.
43. Bai, L., M. G. Constantinides, S. Y. Thomas, R. Reboulet, F. Meng, F. Koentgen,L. Teyton, P. B. Savage, and A. Bendelac. 2012. Distinct APCs explain the cytokinebias of a-galactosylceramide variants in vivo. J. Immunol. 188: 3053–3061.
44. King, I. L., E. Amiel, M. Tighe, K. Mohrs, N. Veerapen, G. Besra, M. Mohrs,and E. A. Leadbetter. 2013. The mechanism of splenic invariant NKT cell ac-tivation dictates localization in vivo. J. Immunol. 191: 572–582.
45. Bialecki, E., E. Macho Fernandez, S. Ivanov, C. Paget, J. Fontaine, F. Rodriguez,L. Lebeau, C. Ehret, B. Frisch, F. Trottein, and C. Faveeuw. 2011. Spleen-resident CD4+ and CD4- CD8a- dendritic cell subsets differ in their ability toprime invariant natural killer T lymphocytes. PLoS ONE 6: e26919.
46. Simkins, H. M., E. Hyde, K. J. Farrand, M. L. Ong, M. A. Degli-Esposti,I. F. Hermans, and F. Ronchese. 2011. Administration of alpha-galactosylceramide impairs the survival of dendritic cell subpopulationsin vivo. J. Leukoc. Biol. 89: 753–762.
47. Edelson, B. T., T. R. Bradstreet, K. Hildner, J. A. Carrero, K. E. Frederick,W. Kc, R. Belizaire, T. Aoshi, R. D. Schreiber, M. J. Miller, et al. 2011. CD8a(+)dendritic cells are an obligate cellular entry point for productive infection byListeria monocytogenes. Immunity 35: 236–248.
48. Barral, D. C., and M. B. Brenner. 2007. CD1 antigen presentation: how it works.Nat. Rev. Immunol. 7: 929–941.
49. Mashayekhi, M., M. M. Sandau, I. R. Dunay, E. M. Frickel, A. Khan,R. S. Goldszmid, A. Sher, H. L. Ploegh, T. L. Murphy, L. D. Sibley, andK. M. Murphy. 2011. CD8a(+) dendritic cells are the critical source ofinterleukin-12 that controls acute infection by Toxoplasma gondii tachyzoites.Immunity 35: 249–259.
50. Bal, S. M., S. Hortensius, Z. Ding, W. Jiskoot, and J. A. Bouwstra. 2011. Co-encapsulation of antigen and Toll-like receptor ligand in cationic liposomesaffects the quality of the immune response in mice after intradermal vaccination.Vaccine 29: 1045–1052.
51. Schlosser, E., M. Mueller, S. Fischer, S. Basta, D. H. Busch, B. Gander, andM. Groettrup. 2008. TLR ligands and antigen need to be coencapsulated into thesame biodegradable microsphere for the generation of potent cytotoxicT lymphocyte responses. Vaccine 26: 1626–1637.
52. Tacken, P. J., I. S. Zeelenberg, L. J. Cruz, M. A. van Hout-Kuijer, G. van deGlind, R. G. Fokkink, A. J. Lambeck, and C. G. Figdor. 2011. Targeted deliveryof TLR ligands to human and mouse dendritic cells strongly enhances adju-vanticity. Blood 118: 6836–6844.
53. Semmling, V., V. Lukacs-Kornek, C. A. Thaiss, T. Quast, K. Hochheiser,U. Panzer, J. Rossjohn, P. Perlmutter, J. Cao, D. I. Godfrey, et al. 2010. Alter-native cross-priming through CCL17-CCR4-mediated attraction of CTLs towardNKT cell-licensed DCs. Nat. Immunol. 11: 313–320.
54. Bai, L., S. Deng, R. Reboulet, R. Mathew, L. Teyton, P. B. Savage, andA. Bendelac. 2013. Natural killer T (NKT)-B-cell interactions promote pro-longed antibody responses and long-term memory to pneumococcal capsularpolysaccharides. Proc. Natl. Acad. Sci. USA 110: 16097–16102.
55. Mattarollo, S. R., K. Steegh, M. Li, H. Duret, S. Foong Ngiow, and M. J. Smyth.2013. Transient Foxp3(+) regulatory T-cell depletion enhances therapeutic an-ticancer vaccination targeting the immune-stimulatory properties of NKT cells.Immunol. Cell Biol. 91: 105–114.
The Journal of Immunology 969
by guest on February 3, 2019http://w
ww
.jimm
unol.org/D
ownloaded from





![pure.au.dkpure.au.dk/portal/files/44301178/1111.2819v1.pdf · arXiv:1111.2819v1 [math.DG] 11 Nov 2011 LIMITS OF BALANCED METRICS ON VECTOR BUNDLES AND POLARISED MANIFOLDS MARIO GARCIA-FERNANDEZ](https://static.fdocument.org/doc/165x107/5fdb920d17c94f6f0002adb7/pureau-arxiv11112819v1-mathdg-11-nov-2011-limits-of-balanced-metrics-on-vector.jpg)












