
Stimulation of cellular signaling and G protein subunit ... · Stimulation of cellular signaling...
33
Stimulation of cellular signaling and G protein subunit dissociation by G protein βγ subunit binding peptides * Farida Goubaeva‡§§, Mousumi Ghosh‡§§, Sundeep Malik‡, Jay Yang‡, Patricia M. Hinkle‡, Kathy K. Griendling§, Richard R. Neubig†, and Alan V. Smrcka‡** §§These authors contributed equally to this work. *This work was supported by grants from the National Institutes of Health GM60286 and the American Heart Association New York State Affiliate to AVS, NIH DK19974 to PMH, and NIH HL46417 to RRN ‡Department of Pharmacology and Physiology, University of Rochester School of Medicine and Dentistry, 601 Elmwood Avenue, Rochester, NY 14642. §Department of Medicine, Emory University, Atlanta, GA 30322. †Department of Pharmacology, University of Michigan School of Medicine, Ann Arbor, MI 48109. ** To whom correspondence should be addressed Phone: 585-275-0892 Fax: 585-273-2652 e-mail: [email protected] Running title: Peptide activation of G βγ signaling 1 by guest on July 15, 2018 http://www.jbc.org/ Downloaded from
Transcript of Stimulation of cellular signaling and G protein subunit ... · Stimulation of cellular signaling...

Stimulation of cellular signaling and G protein subunit dissociation by G protein βγ subunit binding peptides *
Farida Goubaeva‡§§, Mousumi Ghosh‡§§, Sundeep Malik‡, Jay Yang‡, Patricia M. Hinkle‡, Kathy K. Griendling§, Richard R. Neubig†, and Alan V. Smrcka‡**
§§These authors contributed equally to this work.
*This work was supported by grants from the National Institutes of Health GM60286 and the American Heart Association New York State Affiliate to AVS, NIH DK19974 to PMH, and NIH HL46417 to RRN ‡Department of Pharmacology and Physiology, University of Rochester School of Medicine and Dentistry, 601 Elmwood Avenue, Rochester, NY 14642. §Department of Medicine, Emory University, Atlanta, GA 30322. †Department of Pharmacology, University of Michigan School of Medicine, Ann Arbor, MI 48109. **To whom correspondence should be addressed Phone: 585-275-0892 Fax: 585-273-2652 e-mail: [email protected] Running title: Peptide activation of G βγ signaling
1
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

Summary
We previously developed peptides that bind to G protein βγ subunits and selectively
block interactions between βγ subunits and a subset of effectors in vitro (Scott et al. EMBO J.
20, 767-776). Here, we created cell permeating versions of some of these peptides by N terminal
modification with either myristate or the cell permeation sequence from HIV tat. The
myristoylated βγ binding peptide (mSIRK) applied to primary rat arterial smooth muscle cells
caused rapid activation of ERK1/2 in the absence of an agonist. This activation did not occur if
the peptide lacked a myristate at the N terminus, if the peptide had a single point mutation to
eliminate βγ-subunit binding, or if the cells stably expressed the C terminus of βARK1. An HIV
tat modified peptide (tatSIRK), and a myristoylated version of a second peptide (mSCAR) that
binds to the same site on βγ subunits as mSIRK, also caused ERK activation. mSIRK also
stimulated Jun N-terminal kinase (JNK) phosphorylation, p38 MAP kinase phosphorylation,
phospholipase C activity and caused Ca2+ release from internal stores. When tested with purified
G protein subunits in vitro, SIRK promoted α subunit dissociation from βγ subunits without
stimulating nucleotide exchange. These data suggest a novel mechanism by which selective βγ-
binding peptides can release G protein βγ subunits from heterotrimers to stimulate G protein
pathways in cells.
2
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

Introduction
G protein βγ subunits released from αβγ heterotrimers, in response to G protein coupled
receptor activation, regulate a variety of physiological processes including heart rate, neuronal
excitability and neutrophil chemotaxis. This regulation is mediated by interactions between βγ
subunits and a variety target molecules ranging from inwardly rectifying potassium channels to
soluble enzymes such as phospholipase C (see (1) for a review). βγ subunit-dependent
regulation of the majority of its targets is inhibited by theα-GDP subunit. α-GDP is thought to
sterically occlude a binding site on βγ subunits shared by all its effectors. Activation of βγ
dependent signaling occurs when α-GTP dissociates and uncovers this binding site (2). On the
other hand a number of binding sites for effectors outside the βγ-α subunit interface have been
identified by mutagenesis (3) or peptide crosslinking (4). A model emerging from this analysis
predicts βγ subunit effectors may share a common interaction surface at the βγ-α subunit
interface but individual effectors also have unique interaction surfaces on βγ that could be
targeted pharmacologically.
To probe the nature of common and unique binding sites on βγ subunits we screened
random peptide libraries using purified G protein βγ subunits as the target to identify peptide
sequences required for binding to various surfaces on βγ (5). Four divergent families of peptides
were identified that all bound to a single site on the βγ subunit surface. We hypothesized this
interaction surface represented a protein-protein interaction “hot-spot” with the capacity to
accommodate a diversity of amino acid sequences. Random peptide library screens often target
such protein interaction surfaces (6;7). The peptides inhibited the interaction between βγ
subunits and effector molecules such as phospholipase C (PLC)1 β and PI 3-kinase γ (PI3Kγ).
Interestingly, these peptides did not affect regulation of other effectors such as adenylyl cyclase
type I or voltage gated calcium channels, demonstrating selective interference with particular G
protein βγ subunit-target interactions. This indicates that the proposed “hot spot” is not a
common binding surface for all βγ effectors.
3
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

Most inhibitors of G protein βγ subunit-target interactions, such as the C-terminal domain
of β adrenergic receptor kinase (βARK1ct), are thought to be universal βγ subunit inhibitors,
blocking activation of all downstream target molecules. Thus the βARK1ct has been used
extensively in intact cells as a global inhibitor of βγ subunit signaling (8). The goal of this study
was to introduce peptides derived from the phage display screen into intact cells to investigate
the consequences of selectively interfering with particular G protein βγ subunit-target
interactions in intact cells. To our surprise we found these peptides caused activation of G
protein signaling in the absence of receptor activation. We provide evidence for a mechanism
where these selective peptides bind to βγ subunits and promote dissociation of α subunits but
leave a surface available on βγ subunits for activation of MAP kinase pathways.
Experimental Procedures.
Materials. Peptides were synthesized by Alpha Diagnostics International, purified by HPLC to
greater than 90% purity, and the identity of the peptides was confirmed by mass spectrometry
analysis. The sequences of the peptides were as follows: mSIRK: myristate-
SIRKALNILGYPDYD; SIRK: SIRKALNILGYPDYD; mSIRK(L9A): myristate-
SIRKALNIAGYPDYD; mSCAR; myristate-SCARFFGTPCP-amide, tatSIRK: fluorescein-
GGGYGRKKRRQRRRG-SIRKALNILGYPDYD; tatSIRK(L9A): fluorescein-
GGGYGRKKRRQRRRG-SIRKALNIAGYPDYD. mSCAR and mSIRK were dissolved in
DMSO, all other peptides were dissolved in water. Antibodies to phosphorylated ERK1/2, total
ERK1/2, phosphorylated JNK and phosphorylated p38 were obtained from Cell Signaling
Technology Incorporated. The specific EGF receptor kinase inhibitor AG1487; and src inhibitor,
PP2 were from Calbiochem Corporation. Thapsigargin, EGF and lysophosphatidic acid (LPA)
were from Sigma. Pertussis toxin was from List Biological Laboratories.
4
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

Cell culture. All cell culture reagents were obtained from Invitrogen Corporation. All cells
were grown in DMEM supplemented with 10% Fetal Bovine Serum and 1%
Penicillin/Streptomycin at 37oC with 5% CO2. RASM cells were used between passages 5 and
12. Stable cell lines expressing the βARK1ct were prepared and cultured as described
previously (9).
MAP kinase assays. Rat arterial smooth muscle cells (RASM) or other cells were plated into 35
mm dishes and grown to between 50 and 80% confluency. Serum was removed 16 h before
treatment. Peptides in dimethyl-sulfoxide or dimethylsulfoxide vehicle were diluted 100-400
fold into the media and incubated at 37oC for the indicated duration of time. After treatment, the
cells were transferred to ice, the media quickly aspirated and replaced with 100 µl of 2x SDS
sample buffer. The resulting suspension was sonicated briefly in a bath sonicator and 30 µl was
loaded onto a 12% polyacrylamide gel. After SDS-PAGE the proteins were transferred to
nitrocellulose for 16 h at 25 volts. The transferred proteins were immunoblotted using standard
immunoblotting protocols with 1:1000 dilution of primary antibody (unless otherwise indicated)
and 1:10,000 dilution of anti-rabbit IgG-horseradish peroxidase conjugate. The proteins were
visualized by incubation with Enhanced Chemiluminescence (ECL) reagents (Amersham
Biosciences Inc.) and exposure to film. Visualization of the shift in molecular weight of ERK’s
in the total ERK western blots was somewhat variable. The images for the total ERK western
blots in figures 1A and 1B were expanded to emphasize this difference. In other blots the
purpose of the total ERK blots was to demonstrate equal sample loading in all lanes of the gels
and conditions were not optimized to resolve the masses of the phosphorylated ERK from ERK.
Calcium measurements. For Ca2+ imaging, RASM cells were grown on 25 mm glass coverslips
and loaded with fura2-AM in Hank’s balanced saline solution (HBSS) plus 15 mM Hepes buffer,
pH 7.4. At the start of the experiment, the buffer was switched to Ca2+- and Mg2+-free HBSS,
and Ca2+ imaging was performed as described previously (10). Between 6 and 20 cells were
followed in each experiment, and all experiments were repeated at least 3 times with similar
results. Traces show responses of individual cells.
5
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

Measurements of inositol phosphates (IPs). Cells were plated on 35 mm dishes and labeled by
adding 3-5 µCi of 3H-inositol for 24-48 h in inositol free DMEM. After labeling, the media was
removed and replaced with 1ml of Hepes buffered DMEM containing 10 mM LiCl and
equilibrated for 20 min at 37oC. Ligands or peptides were added in a volume of 50 µl for 30 min
after which the media was aspirated and replaced with 1 ml of ice cold perchloric acid. The
acidified extracts were neutralized by extraction with 1ml of 1:1 octylamine:freon and the
aqueous phase applied to Dowex AG1-X8 columns (BioRad). The columns were washed with
water and 50 mM ammonium formate, followed by elution of the IP containing fraction with 1.2
M Ammonium formate/0.1 M formic acid. The eluted fraction was mixed with scintillation fluid
and analyzed in a liquid scintillation counter.
Preparation of biotinylated βγ subunits. The cDNA for rat β1 subunit was subcloned into a
baculovirus transfer vector for expression of amino terminal fusions of a biotin acceptor peptide
(11). The biotin acceptor peptide is a stretch of 20 amino acids that is the substrate for the
enzyme biotin holoenzyme synthetase (BirA). When a protein fused to the biotin acceptor
peptide is coexpressed with BirA the protein becomes biotinylated in vivo at a specific lysine
residue in the acceptor peptide sequence. The rat β1 subunit was subcloned by PCR with pfu
polymerase (Stratagene) using primers:
5’-ATAAGGCGCGCCAAGTGAACTTGACCAGCTGC3’
5’-CCGGAATTCCGGATCCACCTGCTACTG-3’
and cloned into the baculovirus transfer vector PDW464 in frame with the biotin acceptor
sequence between the AscI and EcoRI restriction sites to yield
MAGGLNDIFEAQKIEWHEDTGGA…β1 sequence, with the lysine residue being the site of
biotinylation. Baculovirus was generated via recombination in bacteria as described in the Bac-
to-Bac system manual (Invitrogen). Biotin-β1γ2 was purified from Sf9 cells using hexahistidine
tagged αi1 following previously published procedures (12). Biotinylation of the purified β
subunit was confirmed by SDS-PAGE followed by western blotting with streptavidin linked
horseradish peroxidase and detection with chemiluminescence. To confirm that the β subunit
was fully biotinylated it was precipitated with streptavidin agarose and greater that 90% of the β
subunit was removed from the supernatant by this procedure.
6
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

Measurement of GTP hydrolysis and GTPγS binding. Steady state GTP hydrolysis was
assayed as [32P]-Pi release from γ[32P]-GTP by standard procedures (13). GTPγS binding was
measured according to (14) except Sf9 cell membranes expressing the M2 muscarinic acetyl
choline receptor were reconstituted with purified recombinant myristoylated αi and β1γ2.
Measurement of α−βγ interactions by flow cytometry. Binding of fluorescein isothiocyanate
(FITC) labeled myristoylated αi1 (F-αi) to biotinylated β1γ2 subunits was measured using a flow
cytometry assay (15;16). Biotinylated βγ subunits (250 pM final concentration) were mixed with
streptavidin beads in HEDNMLG (20 mM Hepes, pH 8.0, 1 mM EDTA, 1mM DTT, 150 mM
NaCl, 0.2 mM free Mg2+, 0.1% C12E10, 10 µM GDP). After 20 min incubation at room
temperature, the beads were washed twice by centrifugation in a microfuge with HEDNMLG
and resuspended in the same buffer at a concentration of 105 beads /mL (250 pM βγ). For α
subunit dissociation experiments, the beads with bound βγ subunits were premixed with 1 nM F-
αi for 10 min prior to addition of competitors. For equilibrium binding measurements 1 nM F-αi
and competitors were added simultaneously. The amount of F-αi bound to beads with
biotinylated βγ was assayed at the times indicated in the figure legends using a Beckman-
Dickson FACscan flow cytometer. Non-specific binding, determined by the simultaneous
addition 1 nM F-αi and 50 nM myristoylated αi, was 10-20% of the total signal and was
subtracted from the data.
7
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

Results To investigate the consequence of selectively inhibiting βγ subunit functions in intact cells,
we created cell permeating peptides by synthesizing one of our phage display selected peptides,
SIRKALNILGYPDYD (SIRK) with myristate (17) at the amino terminus (mSIRK). We tested
the ability of this peptide to affect activation of ERK1 and ERK2 in primary cultures of rat
arterial smooth muscle (RASM) cells. In these cells it has been shown that ERK1 and ERK2 can
be activated by stimulation of G protein-coupled lysophosphatidic acid (LPA) receptors. This
activation is blocked by pertussis toxin and by an adenovirus expressing the βARK1ct,
implicating Gi and βγ subunits in the activation process (18). For these reasons, we predicted
that our cell permeating peptides might block LPA-induced ERK activation. Surprisingly,
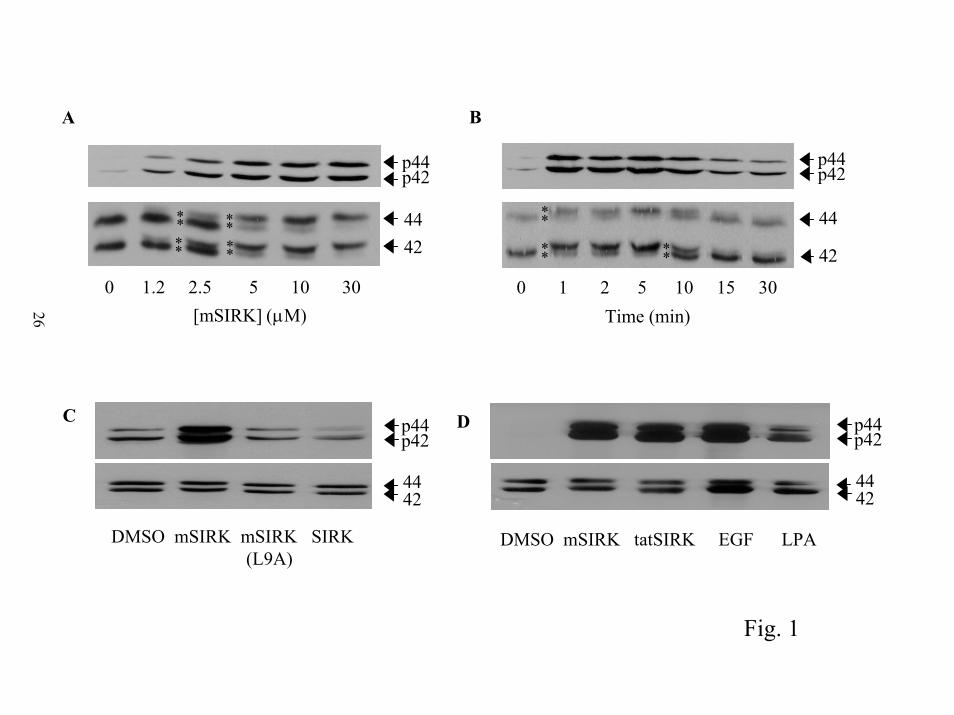
application of mSIRK to RASM cells, in the absence of added LPA, caused a rapid and dose
dependent activation of ERK1/2 as detected by western blotting for the phosphorylated forms of
ERK1/2 (Figure 1 A and B). The concentration of mSIRK required for half maximal activation
of ERK1/2 was 2.5-5 µM, equivalent to the estimated Kd of this peptide for G protein βγ
subunits (5). Full activation was achieved after 1 min of application and diminished over the
course of 30 min. Activation of ERK1/2 is also indicated by the upward shift in molecular
weight of ERK1/2 in a western blot for total ERK1/2. For Figures 1A and 1B the conditions
were optimized to resolve the phosphorylated from the non-phosphorylated ERK1/2 in a total
ERK1/2 western blot (See asterisks in bottom panels, figure 1A and 1B). At full activation, all
of the ERK1/2 was shifted to a higher molecular weight phosphorylated form, indicating all of
the ERK in all of the cells was being activated by this peptide.
To analyze the specificity of this effect we created a second myristoylated version of this
peptide where leucine 9 was changed to alanine (mSIRK(L9A)). We had previously performed
alanine scanning mutagenesis of SIRK and found substitution of A for L9 increased the IC50 of
this peptide for blocking βγ-dependent PLC activation by over 100 fold (from 3 µM to 300 µM)
(5). The results in figure 1C demonstrate mSIRK(L9A) was completely unable to enhance
ERK1/2 phosphorylation. Additionally, a myristoylated-scrambled version (not shown) and a
non-myristoylated version of the peptide (SIRK) (Fig. 1C) were unable to cause ERK1/2
phosphorylation. The necessity for myristoylation of the peptide for cellular activity supports the
8
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

idea that entry of the peptide into the cell is needed to cause ERK activation. To further support
the need for intracellular action, we constructed a peptide where the 11 amino acid cell
permeation sequence from HIV tat (19) was added to the amino terminal end of SIRK (tatSIRK).
Fluorescein was attached to the amino terminus to monitor cellular uptake. TatSIRK was
efficiently taken up into virtually all of cells on the dish as monitored by fluorescence
microscopy (not shown). TatSIRK was able to activate ERK1/2 to an extent comparable to
mSIRK (Fig. 1D) while tatSIRK(L9A) had no effect (data not shown). These cell permeating
peptides activated ERK to a greater extent than LPA and to an extent comparable to EGF in this
assay paradigm (Fig. 1D). Together, these data indicate structural requirements in the peptides
for βγ binding need to be maintained to observe ERK1/2 activation, and that the peptides need to
get inside the cell and are not acting by binding to cell surface receptors. This, in conjunction
with the observation that the concentration requirement for ERK1/2 activation correlates with the
apparent Kd of SIRK for βγ subunits in vitro (5), strongly supports the idea that βγ is the target of
the peptides and this targeting is responsible for ERK1/2 activation.
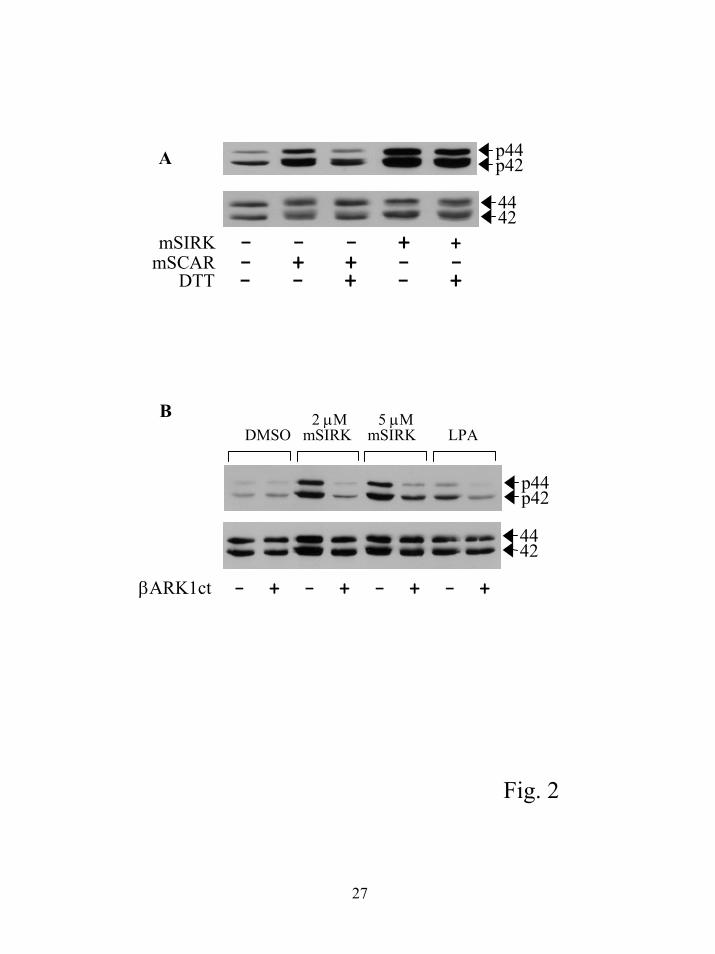
G βγ is the target of the peptides in the intact cell. To further support the idea that βγ subunits
are the target of the peptides in intact cells, we tested a peptide with a completely different amino
acid sequence from SIRK. The peptide, SCARFFGTPCP (SCAR), was also selected in the
phage display screen and we have shown by competition analysis that SCAR peptide binds to βγ
subunits in vitro at the same site as SIRK (5). We predicted a myristoylated version of SCAR
(mSCAR) should have the same effect as mSIRK on ERK1/2 activation in RASM cells. We
have also shown that for SCAR to bind to βγ subunits the cysteine residues must form an
intramolecular disulfide bond. If SCAR is reduced with DTT, it is no longer able to bind to βγ in
a phage ELISA assay. The results in figure 2A show mSCAR activates ERK1/2, pretreatment
with DTT eliminates its ability to activate ERK1/2, while DTT has no effect on the ability of the
linear mSIRK to stimulate ERK1/2. These data emphasize that the structure of the peptide
binding to βγ subunits is important to observe activation of ERK1/2, not just the peptide
sequence.
9
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

To provide further evidence G βγ subunits are the target of mSIRK and related peptides, we
tested the ability of mSIRK to cause ERK1/2 activation in a RASM cell line stably transfected
with the C-terminal region of βARK that sequesters βγ subunits (βARK1ct (Gly495-Leu689))
(8;9). We hypothesized that mSIRK was binding to βγ but leaving a surface available on βγ to
signal to some downstream targets. Based on this hypothesis, we predicted that ERK1/2
activation by mSIRK would be inhibited by the βARK1ct, a general βγ inhibitor that blocks
signaling of βγ to most if not all effectors. The data in figure 2B demonstrate that in cells
expressing βARK1ct, activation of ERK1/2 was almost completely inhibited compared to the
control RASM cells. This indicates that βγ subunit signaling is the target of these peptides.
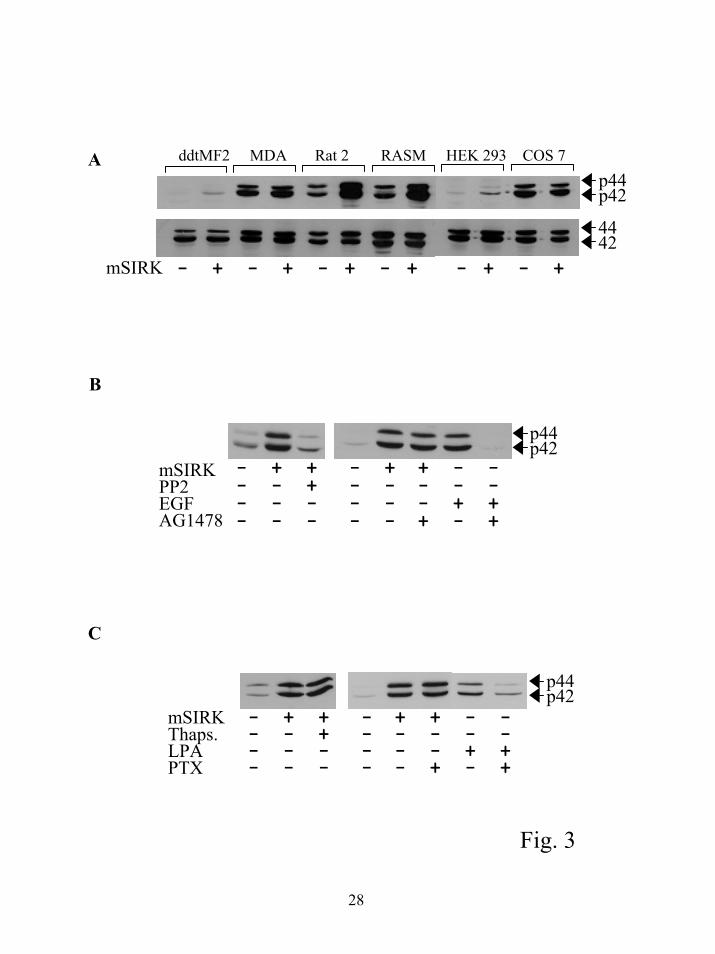
Cell type specificity of ERK activation by mSIRK. All of the experiments described so far
used rat arterial smooth muscle cells. To examine the generality of this response mSIRK was
tested in a variety of cell lines. The results in figure 3A show that the effect of mSIRK is
dependent on the cell type. RASM and Rat2 cells show dramatic activation of ERK1/2 by
mSIRK while other smooth muscle cells, such as ddtMF2 cells show very little, but some
response. These differences could be due to a variety of factors. One possibility is that the
myristoylated peptide is only able to enter certain cell types. We therefore tested the effects of
tatSIRK on HEK293 and Cos7 cells. Similar to mSIRK, tatSIRK had only small effects on
activation of ERK1/2 (not shown). We were able to monitor the uptake of tatSIRK into these
cells with a fluorescein label and confirmed that this peptide did indeed enter the cells. This
indicates that the cell type specific responses are not due to differential uptake of the peptides
and differences in the endogenous MAP kinase signaling systems may be responsible, i.e., in
some cell types βγ dependent MAP kinase activation is stronger than others.
10
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

Activation of ERK1/2 requires src but not EGF receptor transactivation or calcium release
from internal stores. We used a variety of pharmacological inhibitors to examine the pathway
involved in the response to mSIRK. As expected a MEK inhibitor PD98059 was able to
completely block the effect of the peptide (not shown). PP2, a specific src family tyrosine kinase
inhibitor, was able to completely block mSIRK mediated activation of ERK1/2 (Fig. 3B). Src is
thought to be involved in βγ-mediated activation of ERK1/2 by GPCRs, consistent with a
pathway involving βγ subunits (20). The involvement of EGF receptor transactivation was tested
using a specific inhibitor of the EGF receptor, AG1478. This inhibitor completely blocked the
ability of the EGF receptor to activate ERK1/2 but had no effect on ERK1/2 activation by m-
SIRK (Fig. 3B). Thapsigargin treatment, which almost completely emptied the intracellular
calcium stores (data not shown and figure 4B), was unable to block the effect of mSIRK
indicating release of calcium from intracellular stores is not responsible for ERK1/2 activation
(Fig. 3C). Finally, treatment with pertussis toxin did not block the effect of mSIRK (Fig. 3C),
supporting the idea that receptor activation is not involved in the peptide dependent activation of
ERK signaling.
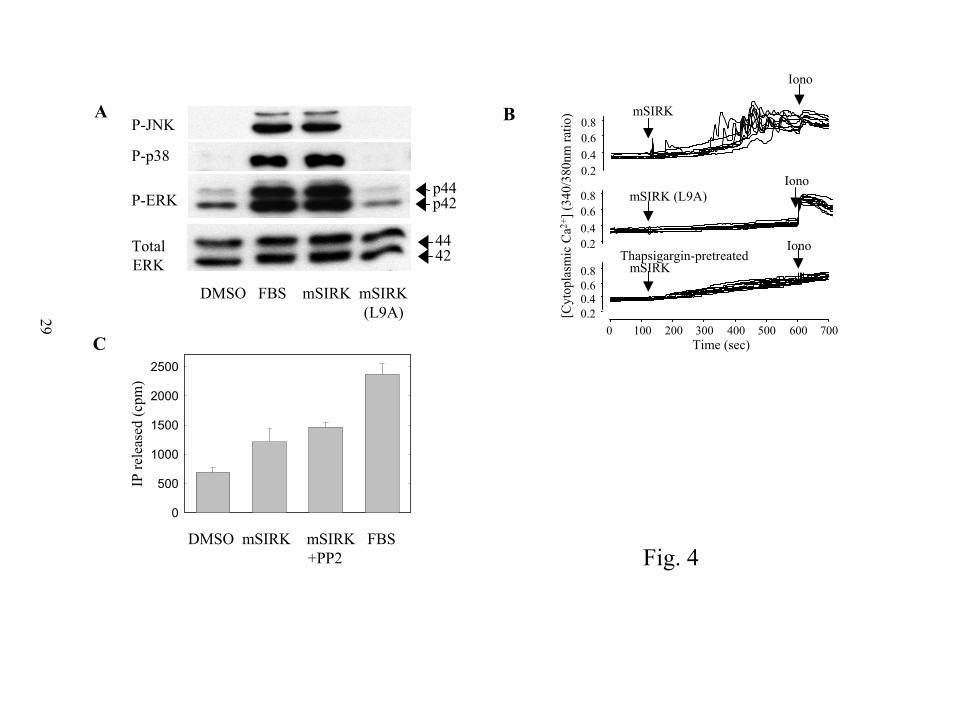
Activation of other GPCR signal transduction pathways by mSIRK. The effects of mSIRK
on other signal transduction pathways downstream of either βγ subunits or α subunits were
examined. First we looked at other members of the MAP kinase pathway. p38 MAP kinase
phosphorylation and Jun Kinase (JNK) phosphorylation are increased by addition of mSIRK
(Figure 4A) but not by the control peptide, mSIRK(L9A).
The effect of mSIRK on intracellular Ca2+ was followed in RASM cells loaded with the
cytoplasmic Ca2+ indicator fura2. At 10 µM, mSIRK caused pronounced oscillations in
cytoplasmic Ca2+ in 22/27 cells, often after a lag of 2 to 5 min (figure 4B, top panel). Ca2+
spiking was not stimulated by addition of buffer, or the control peptide, mSIRK(L9A) (0/34
cells) (figure 4B, middle panel). Ca2+ spikes were completely eliminated by thapsigargin, which
depletes intracellular Ca2+ stores by inhibiting the SERCA ATPase, responsible for maintaining
high Ca2+ concentrations in the endoplasmic reticulum (figure 4B, bottom panel). mSIRK also
stimulated Ca2+ oscillations in HEK293 cells (data not shown). These Ca2+ oscillations resulted
11
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

from the release of intracellular Ca2+, because they were eliminated by thapsigargin and observed
in Ca2+-free media. In HEK293 cells, as the dose of mSIRK was increased from 0.1 to 10 µM,
the fraction of cells responding increased from ~30% to 100%, and the lag time decreased from
3-8 to 0.5-1 min (data not shown). Pertussis toxin treatment did not inhibit the effects of mSIRK
on intracellular Ca2+ (data not shown). To determine whether IP3 production was responsible
for this increase we examined total inositol phosphate (IP) production in cells labeled with 3H-
inositol. mSIRK caused a reproducible increase in intracellular IP release that was unaffected by
treatment with PP2 (Fig 4C). Since PP2 blocks src activation and the subsequent pathway
leading to ERK1/2 activation, the IP3 release is not an indirect consequence of activation of this
pathway.
mSIRK activation of many G protein dependent pathways suggests mSIRK is binding to
βγ subunits and somehow promoting release of βγ subunits, leaving a surface available to signal
to downstream pathways. A potential problem with this hypothesis is that SIRK blocks PLC
stimulation by βγ in vitro (5) and thus should block the βγ surface required to signal to PLC.
The PLC response caused by mSIRK is quite weak compared to G protein coupled receptor
activation of PLC in RASM cells. It takes minutes for significant Ca2+ spiking to be observed
and the cells respond asynchronously in response to mSIRK. The observed increase in total IP
production in 30 min was very small. This is reminiscent of very weak activation by a G protein
coupled receptor and contrasts with the rapid strong activation of ERK in the same cells by these
peptides. One possibility is that the peptide may not completely block PLC activation by free βγ
and this residual ability of the βγ to activate PLC, even in the presence of peptide, is responsible
for the weak IP3 and Ca2+ responses observed. Another possibility is that αq/11 may be released,
that can bind some GTP once released from βγ.
SIRK causes dissociation of α subunits from βγ subunits without stimulating nucleotide
exchange. That all these signaling pathways are being activated in response to a βγ subunit
binding peptide suggests the peptide is a general activator of G protein signaling. One possibility
is that the peptide directly activates the heterotrimer by promoting GTP binding to α subunits.
However, these peptides were selected to bind to G protein βγ subunits and therefore should not
12
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

bind to α subunits. Receptors and receptor-mimicking peptides such as mastoparan act at least
in part through binding to α subunits. It is possible, however, that mSIRK may non-specifically
interact with α subunits or may promote nucleotide exchange by an undefined mechanism
through binding to βγ subunits. To determine if the βγ binding peptide directly activates the G
protein promoting GDP release and GTP binding, we tested whether the peptide caused an
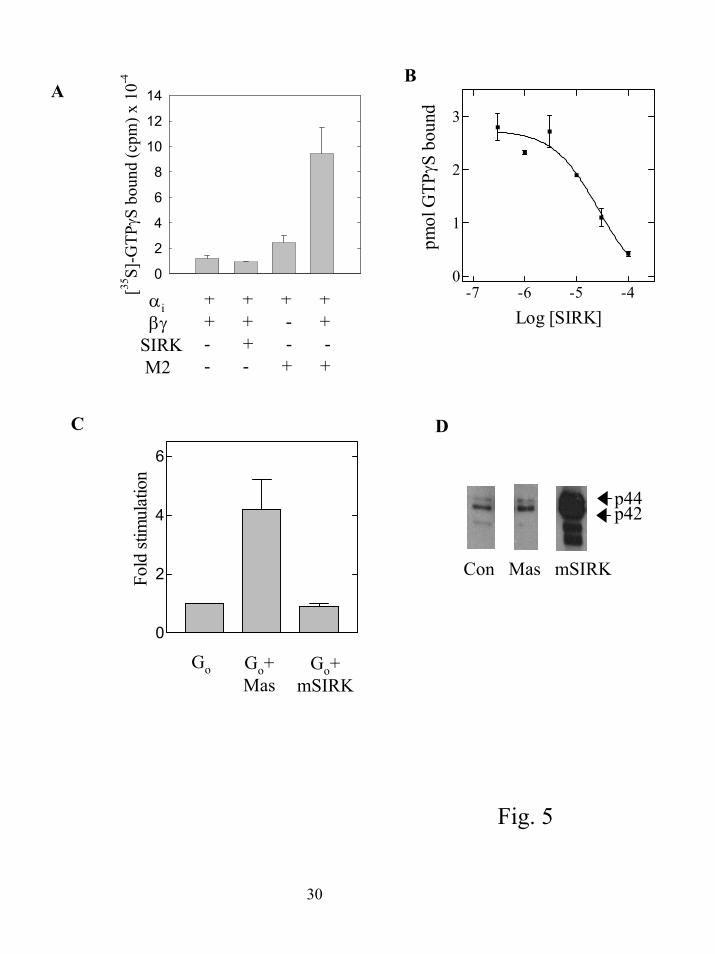
increase in binding of [35S]-GTPγS to purified αi1β1γ2 (21) and found it did not (figure 5A). If
the same subunits were reconstituted with urea-stripped Sf9 cell membranes expressing the M2
muscarinic receptor and carbachol, [35S]-GTPγS binding was stimulated in a βγ subunit-
dependent manner. To determine if SIRK could influence M2 receptor stimulated nucleotide
exchange in this assay, SIRK was tested at various concentrations in the presence of M2 receptor
and carbachol. Here, SIRK inhibited receptor dependent nucleotide exchange in a dose
dependent manner (figure 5B). Relatively high concentrations of SIRK were required to observe
this inhibition (IC50 25 µM) probably because of the relatively high concentration of G protein
subunits used in this assay. We hypothesize the peptide inhibits receptor dependent nucleotide
exchange either by interfering with receptor-G protein interactions or G protein subunit
interactions. Experiments described later in this section suggest the peptide is acting through
disruption of interactions between α and βγ subunits.
Nucleotide exchange can also be assayed by measuring the steady state rate of GTP
hydrolysis on the α subunit. In this assay, the rate of hydrolysis is limited by the rate of
exchange of GDP for GTP. The amphipathic peptide mastoparan stimulated the rate of
hydrolysis of γ[32P]-GTP by Go while mSIRK did not (figure 5C), indicating that mSIRK cannot
stimulate GDP dissociation. Also shown in figure 5D is that mastoparan has virtually no effect
on ERK activation in RASM cells while mSIRK stimulates significant ERK activation.
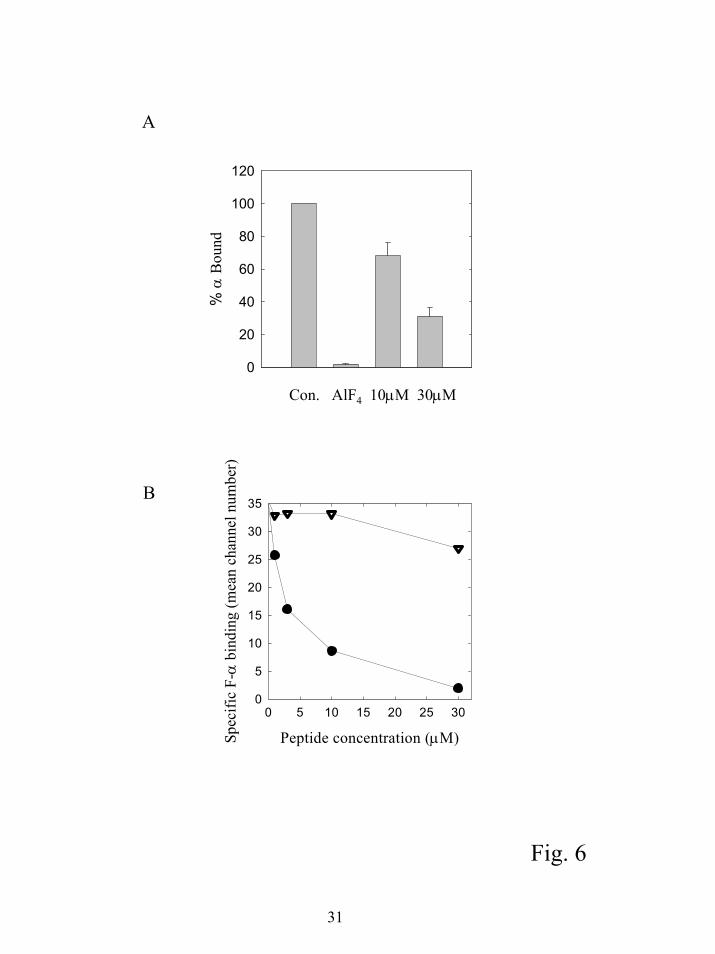
To determine if SIRK could stimulate dissociation of α from βγ in the absence of
nucleotide exchange, we assayed the binding of α subunits to biotinylated βγ subunits bound to
streptavidin agarose beads. G protein α subunits (10 nM) were incubated with 10 nM
biotinylated βγ subunits bound to streptavidin agarose beads and incubated with mSIRK or AlF4-
for 20 minutes. The beads were centrifuged, the supernatant removed and the bound α subunits
13
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

estimated by quantitative western blotting (Figure 6A). At 10 and 30 µM mSIRK the amount of
α subunit bound to βγ subunits was significantly lower than in the absence of added mSIRK.
A drawback of the pulldown assay is the high concentrations of α and βγ required to
detect binding and the relatively crude nature of the quantitation. At 10 nM, each of the subunits
was used at concentrations significantly above the estimated Kd for α-βγ interactions of about 1
nM. A more quantitative assay to measure α-βγ interactions at very low subunit concentrations
has recently been developed that uses flow cytometry to assess the amount of fluorescent α
subunit bound to biotinylated βγ subunits immobilized on beads (15;16). In this assay, where
SIRK and FITC labeled αi (F-αi) are simultaneously mixed with βγ subunits, there was a dose
dependent decrease in the amount of α subunit bound to βγ subunits while SIRK(L9A) had no
effect (Figure 6B). At 10 µM SIRK, F-αi bound to βγ is reduced by 75% compared to only 25%
in the pulldown assay. This probably reflects that the G protein subunit concentration in this
assay is near the Kd for the α-βγ interaction making it easier to observe a decrease in binding.
A possible mechanism for the increase in α-βγ dissociation might involve direct
competition between the peptide and the α subunit for the α subunit-binding site on βγ subunits.
In this model, the peptide would increase net subunit separation by preventing rebinding of α-
GDP subunits after dissociation. This process would be inherently slow because it would depend
on spontaneous dissociation of tightly bound α-GDP from βγ. A second possibility is the
peptide promotes dissociation of α from βγ subunits by a mechanism that does not involve direct
competition. These two possibilities can be distinguished by measuring the α subunit
dissociation rate. If the peptide simply competes for the α subunit-binding site on βγ subunits
then the dissociation rate should be equivalent to the intrinsic α subunit dissociation rate. If the
rate of α subunit dissociation in the presence of peptide is greater than the intrinsic dissociation
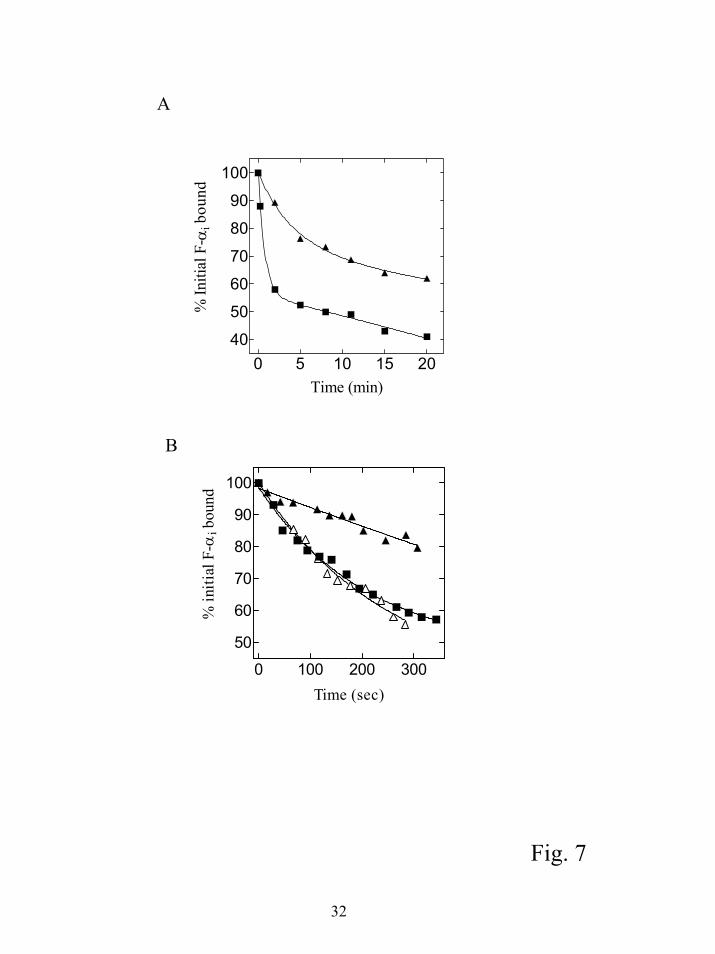
rate, it would suggest the peptide can promote G protein subunit dissociation. We measured the
intrinsic rate of F-αi dissociation from βγ by adding a 50 fold excess of unlabeled myristoylated
αi subunit at time 0 and measuring the amount of F-αi bound to βγ at various times using flow
cytometry (Fig. 7A). The intrinsic off rate was slow, with a koff between 0.05 and 0.08 min-1 (t ½
~ 9-14 min) in four separate experiments. These data are similar to previously published results
14
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

(0.047 min-1) with this assay, and consistent with the low apparent Kd for α-βγ interactions (15).
The rate of α dissociation from βγ was also measured at various times after addition of SIRK.
The initial rate of dissociation was rapid, with the majority of the dissociation occurring within 2
min (t ½ ~1 min or less). The dissociation with peptide was in two phases, an initial rapid phase
followed by a slower dissociation with a rate constant similar to the intrinsic α subunit
dissociation rate. The extent of initial rapid dissociation by peptide was always at least 75% of
the total dissociation that occurred after α subunit alone was added for at least 1h.
To examine the initial dissociation phase in more detail, data were collected in real time
using the continuous data collection mode of the flow cytometer. Over the time course of 300 s,
the peptide treated samples had a significantly higher rate of dissociation compared to the
intrinsic dissociation rate measured with unlabeled αi as the sole competitor (figure 7B). The
peptide dependent dissociation rate ranged from 5 to 14 fold faster than the intrinsic dissociation
rate in 7 separate experiments. The variation resulted primarily from difficulty in getting an
accurate quantitative estimate of the intrinsic koff over the short time course of the measurement.
Overall, these data indicate SIRK can promote rapid dissociation of α from βγ subunits without
promoting nucleotide exchange on the α subunit.
15
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

Discussion Activation of ERK1/2 by βγ inhibitory peptides
There is extensive evidence in the literature suggesting free βγ subunits released from Gi
heterotrimers, upon α subunit activation and dissociation, can initiate activation of multiple
MAP kinase pathways (22;23). We have done extensive characterization of our peptide system
to show the effects we observe are specifically due to interactions of our peptides with βγ
subunits in cells. The concentration of peptide required to cause ERK1/2 activation in cells is
nearly the same as for inhibition of interactions between βγ subunits and downstream targets in
vitro. A single L-A point mutation previously shown to eliminate the ability of the peptide to
inhibit βγ interactions with downstream targets (5), completely eliminates peptide dependent
ERK1/2 activation. A requirement for intracellular action of the peptide is indicated by the fact
that the peptide has no effect unless it is modified by either a myristate or the tat cell permeation
sequence from HIV. That two completely different chemical modifications of the peptide,
whose unifying feature is their ability to permeate cell membranes, strongly indicates their site of
action is not receptors or other sites on the cell surface. A peptide (mSCAR) with no sequence
homology to the SIRK peptide, but binds to the same surface on βγ subunits, also caused
activation of ERK1/2. The fact that activation of ERK1/2 is blocked by the src inhibitor PP2
indicates our βγ “inhibitory peptides” are acting though a pathway similar to that which has been
previously described as being involved in MAP kinase activation by βγ (20). Finally, if the
peptide is tested in RASM cells that express the βARK1ct, the effect on ERK1/2 activation is
greatly attenuated. All of these data indicate the G protein signaling system, and the βγ subunits
specifically, are the targets for binding these peptides in cells, resulting in the activation of signal
transduction that has been presented.
Hot spot binding peptides promote α-βγ dissociation.
We had previously characterized SIRK and other phage display-selected βγ binding
peptides in vitro and demonstrated they could selectively interfere with the activation of some
effectors by βγ subunits. We also postulated they interact with a protein-protein interaction “hot
spot” on the surface of βγ subunits because all the selected peptides, with diverse sequence
16
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

characteristics, bound to the same site on βγ subunits. Here we demonstrate these effector-
selective βγ blocking peptides initiate activation of certain G-βγ dependent signaling pathways in
intact cells in a receptor-independent fashion. We show the peptides can promote dissociation of
α subunits from βγ subunits without inducing nucleotide exchange. In order to explain the
ability of these peptides to activate cellular signaling, they must bind to βγ subunits to decrease
the binding of α to βγ, but leave the surface on βγ subunits required to activate MAP kinase
pathways unoccupied. A defining characteristic of these peptides is they block interactions with
some downstream targets but not others (5). This unique characteristic of these βγ-blocking
peptides makes this proposed mechanism possible. The precise molecular target of βγ subunits in
ERK1/2 activation has not been defined, so we cannot test the effects of these peptides on this
particular interaction directly.
If these peptides promote βγ release, then why is there cell type specificity to ERK
activation as shown in figure 3A? We believe this reflects the different degrees to which free βγ
can activate ERK in different cell types. RASM cells have previously been demonstrated to have
a robust βγ dependent activation of ERK (18). We suspect that βγ is probably released by
peptide treatment in all the cells tested, but they may have different levels of the signaling
molecules necessary for robust βγ stimulated ERK1/2 activation.
It is becoming increasingly clear, particularly through the identification of AGS proteins
(24), there are multiple mechanisms for G protein activation that do not depend on receptor-
dependent nucleotide exchange. The data we present here indicate binding to a previously
postulated “hot spot” on βγ subunits can promote subunit dissociation in vitro and activate G
protein βγ subunit dependent pathways in cells. This suggests a potential mechanism for
regulating G protein βγ subunit dependent signaling by proteins (either effectors or other types of
proteins such as RGS proteins) interacting directly with the peptide binding “hot spot”. A
protein with properties similar to our peptides is a protein designated AGS2 (activator of G
protein signaling 2) that was found in a yeast screen for receptor-independent mechanisms for G
protein activation. AGS2 was shown to bind to βγ subunits, but not α subunits, and could
activate the pheromone response pathway in yeast. AGS2 is identical to Tctex 1, a component of
17
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

the cytoplasmic motor protein dynein. A possible interpretation of the ability to Tctex 1 to cause
separation of α from βγ is that Tctex1 is a βγ subunit effector that can cause separation of α from
βγ but leaves surfaces available on βγ to interact with the yeast signaling machinery.
Still remaining to be resolved is how SIRK peptide promotes subunit dissociation. One
hypothesis is the peptide causes a conformational change in the βγ subunit that promotes subunit
dissociation. Generally it is not thought that the βγ subunits undergo significant conformational
changes because the conformations of βγ crystallized with and without α subunits are very
similar (25-28). On the other hand the structure of βγ crystallized in the presence of phosducin
shows significant conformational alteration (29;30). Interestingly, a peptide derived from
phosducin that apparently binds outside of the α subunit-binding site alters α-βγ subunit
interactions suggesting that binding to βγ can transmit information to the α subunit interface
(31). The peptides we derived from phage display have similarity to a region of phosducin that
binds outside of the α subunit interface. We previously postulated that the site on βγ subunits
where this region of phosducin binds is where our peptide binds. If true, it suggests a truly novel
mechanism for G protein activation involving conformational alteration of βγ.
An alternative hypothesis is since α-βγ interactions involve two parts of α (i.e. switch
regions and N-terminus) (26;27), the slow dissociation of α and βγ may depend on the
requirement for simultaneous breaking of these two contacts. If each contact were dynamic,
mSIRK peptide binding to βγ (for example at the site where βγ contacts the N-terminus) would
leave only one contact, which would generate a complex with a lower stability and lifetime. In
contrast, addition of excess α subunit could only prevent reassociation following full release of α
from βγ due to steric interference between the incoming and outgoing α subunits. Identification
and characterization of the peptide-binding site is critical to determination of this mechanism.
18
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

References Reference List
1. Clapham, D. E. and Neer, E. J. (1997) Ann.Rev.Pharmacol.Toxicol. 37, 167-203
2. Hamm, H. E. (1998) J.Biol.Chem. 273, 669-672
3. Panchenko, M. P., Saxena, K., Li, Y., Charnecki, S., Sternweis, P. M., Smith, T. F., Gilman, A. G., Kozasa, T., and Neer, E. J. (1998) J.Biol.Chem. 273, 28298-28304
4. Yoshikawa, D. M., Bresciano, K., Hatwar, M., and Smrcka, A. V. (2001) J.Biol.Chem. 276, 11246-11251
5. Scott, J. K., Huang, S. F., Gangadhar, B. P., Samoriski, G. M., Clapp, P., Gross, R. A., Taussig, R., and Smrcka, A. V. (2001) EMBO J. 20, 767-776
6. Wells, J. A. and de Vos, A. M. (1996) Ann.Rev.Biochem. 65, 609-634
7. Fairbrother, W. J., Christinger, H. W., Cochran, A. G., Fuh, G., Keenan, C. J., Quan, C., Shriver, S. K., Tom, J. Y., Wells, J. A., and Cunningham, B. C. (1998) Biochem. 37, 17754-17764
8. Koch, W. J., Hawes, B. E., Inglese, J., Luttrell, L. M., and Lefkowitz, R. J. (1994) J.Biol.Chem. 269, 6193-7:
9. Ushio-Fukai, M., Alexander, R. W., Akers, M., Lyons, P. R., Lassegue, B., and Griendling, K. K. (1999) Mol Pharmacol 55, 142-149
10. Romoser, V. A., Graves, T. K., Wu, D., Jiang, H., and Hinkle, P. M. (2001) Mol Endocrinol. 15, 125-135
11. Duffy, S., Tsao, K. L., and Waugh, D. S. (1998) Analytical Biochemistry 262, 122-128
12. Kozasa, T. and Gilman, A. G. (1995) J.Biol.Chem. 270, 1734-1741
13. Higashijima, T., Burnier, J., and Ross, E. M. (1990) J.Biol.Chem. 265, 14176-14186
14. Hartman IV, J. L. and Northup, J. K. (1999) J.Biol.Chem. 271, 22591-22597
15. Sarvazyan, N. A., Remmers, A. E., and Neubig, R. R. (1998) J.Biol.Chem. 273, 7934-7940
16. Sarvazyan, N. A., Lindau, M., and Neubig, R. R. (2002) Biochem. 41, 12858-12867
17. Eichholtz, T., de Bont, D. B., de Widt, J., Liskamp, R. M., and Ploegh, H. L. (1993) J.Biol.Chem. 268, 1982-1986
18. Iaccarino, G., Smithwick, L. A., Lefkowitz, R. J., and Koch, W. J. (1999) Proc.Natl.Acad.Sci.USA 96, 3945-3950
19
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

19. Schwarze, S. R., Ho, A., Vocera-Akbani, A., and Dowdy, S. F. (1999) Science 285, 1569-1572
20. Luttrell, L. M., Della, R. G., van Biesen, T., Luttrell, D. K., and Lefkowitz, R. J. (1997) J Biol.Chem. 272, 4637-4644
21. Northup, J. K., Smigel, M. D., and Gilman, A. G. (1982) J.Biol.Chem. 257, 11416-11423
22. Luttrell, L. M., van Biesen, T., Hawes, B. E., Koch, W. J., Krueger, K. M., Touhara, K., and Lefkowitz, R. J. (1997) Adv.Second Messenger Phosphoprotein Res. 31, 263-277
23. Gutkind, J. S. (2001) Sci STKE. 2000, RE1
24. Cismowski, M. J., Takesono, A., Bernard, M. L., Duzic, E., and Lanier, S. M. (2001) Life Sci. 68, 2301-2308
25. Sondek, J., Bohm, A., Lambright, D. G., Hamm, H. E., and Sigler, P. B. (1996) Nature 379, 369-374
26. Lambright, D. G., Sondek, J., Bohm, A., Skiba, N. P., Hamm, H. E., and Sigler, P. B. (1996) Nature 379, 311-319
27. Wall, M. A., Coleman, D. E., Lee, E., Iniguez-Lluhi, J. A., Posner, B. A., Gilman, A. G., and Sprang, S. R. (1995) Cell 83, 1047-1058
28. Sprang, S. R. (1997) Ann.Rev.Biochem. 66, 639-678
29. Loew, A., Ho, Y. K., Blundell, T., and Bax, B. (1998) Structure 6, 1007-1019
30. Gaudet, R., Bohm, A., and Sigler, P. B. (1996) Cell 87, 577-588
31. Bluml, K., Schnepp, W., Schroder, S., Beyermann, M., Macias, M., Oschkinat, H., and Lohse, M. J. (1997) EMBO J. 16, 4908-4915
20
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

1Abbreviations used: PLC, Phospholipase C; PI3K, Phosphoinositide 3-kinase; RGS, regulator of G proteins signaling; RASM, rat arterial smooth muscle; JNK, Jun N-terminal kinase; LPA, lysophosphatidic acid; EGF, epidermal growth factor βARKct, C-terminus of β-adrenergic receptor kinase; DTT, dithiothreitol; C12E10, polyoxyethylene 10-lauryl ether. Acknowledgements. I would like to thank Mamata Hatwar and John Puskas for excellent technical assistance, Dr. Brad Berk’s laboratory for preparation of rat arterial smooth muscle cells and Tabetha Bonacci for kindly providing biotinylated βγ subunits.
21
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

Figure Legends
Figure 1. Cell permeating βγ binding peptides activate ERK1/2 in vascular smooth muscle cells.
A) Concentration dependence. RASM cells were treated with the indicated concentrations of
mSIRK, added in 5 µl of DMSO for 10 min prior to extraction into sample buffer. Asterisks (*)
indicate the presence of doublets of ERK2 (42) and ERK1 (44) that appear with peptide
treatment. B) Time course. RASM cells were incubated with 10 µM mSIRK for the indicated
times. Again (*) indicate the presence of a doublet of ERK2 and ERK1. C) Substitution of L9
with A, or removal of the myristoyl group eliminates the ability of mSIRK to activate ERK1/2.
RASM cells were treated for 5 min with DMSO, 10 µM mSIRK, 10 µM mSIRK with alanine
substituted for leucine at the 9 position, mSIRK(L9A), or 10 µM SIRK with no myristate. D)
HIV tat cell permeation sequence also allows the peptide to activate ERK1/2. Cells were treated
with 5 µM mSIRK, 10 µM tatSIRK, 10 µM LPA and 10 nM EGF or DMSO for 5 min. For all
experiments, samples were all applied to 12 % gels and analyzed by western blotting for either
phosphorylated ERK1/2; p44 and p42 or total ERK1/2 protein; labeled 44 and 42. Each of these
experiments was repeated at least 3 times with similar results.
Figure 2. Activation of ERK1/2 by mSIRK involves a G βγ-dependent signaling pathway. A)
Other peptides that bind to βγ also activate ERK1/2. RASM cells were treated with 5 µM
mSIRK for 5 min or 5 µM mSCAR for 15 min. For the DTT treatment, the peptides in media at
5 µM were treated with 10 mM DTT for 30 min prior to addition to the cells. B) The βARK1ct
inhibits mSIRK mediated activation of ERK1/2. Stable cell lines expressing the βARK1ct or
vector control cells were treated with the indicated concentrations of mSIRK, 10 µM LPA or
DMSO control for 5 min and processed as described. This experiment was repeated 3 times with
similar results.
Figure 3. Cell type specificity and pathway for ERK1/2 activation. A) The magnitude of
activation of ERK1/2 is dependent upon the cell type. The indicated cell types were treated with
DMSO or 10 µM mSIRK for 5 min. All samples were processed and analyzed as described in
“Experimental Procedures” and figure 1. B) The pathway activated by peptide treatment involves
22
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

src but not transactivation of the EGF receptor. Cells were treated with 1 µM AG1478 for 1h or
10 µM PP2 for 30 min prior to addition of 5 µM mSIRK or 10 nM EGF for 5 min. Each of the
treatments was repeated in at least 3 separate experiments. C) Calcium release from
intracellular pools was not responsible for ERK1/2 activation and the activation is not blocked
by pertussis toxin. Cells were treated with 1 µM thapsigargin (30 min) to empty intracellular
calcium pools prior to addition of 10µM mSIRK. For pertussis toxin treatments, cells were
treated for 16 h with 100 ng/ml pertussis toxin followed by treatment with either 5 µM mSIRK
(5 min) or 10 µM LPA (5 min). Measurement of ERK1/2 is as in figure 1.
Figure 4. Multiple signaling pathways are activated by mSIRK treatment. A) p38 MAP kinase
and JNK are activated by mSIRK. RASM cells were treated with 5 µM mSIRK, mSIRK(L9A) or
1% FBS for 5 min and samples were assessed for p38 MAP kinase activation, JNK activation
and ERK activation by western blotting with antibodies specific for phosphorylated p38,
phosphorylated JNK and phosphorylated ERK respectively. Total ERK1/2 is shown as a loading
control. B) mSIRK causes mobilization of intracellular Ca2+. Fura2-loaded RASM cells were
incubated in Ca2+- and Mg2+-free HBSS for Ca2+ imaging. At the times noted by the arrows, 10
µM mSIRK, 10 µM mSIRK(L9A) or vehicle was added. In order to estimate the intracellular
Ca2+pool, 1 µM ionomycin was added at the end of the experiment. In the bottom panel, cells
were exposed to 100 nM thapsigargin during loading and imaging. C) mSIRK causes an
increase in inositol phosphates. Total inositol phosphates produced in RASM cells were
measured as described in “Experimental Procedures”. mSIRK (10 µM), DMSO, or 1% fetal
bovine serum was added for 30 min and PP2 treatment was 10 µM. The data presented are
representative data showing the standard errors of duplicate data points. In 19 experiments the
fold activation by mSIRK ranged from 1.4 to 4 fold with an average of 2.1 fold ± 0.17 SEM.
mSIRK treated samples were significantly different from DMSO treated control (P < 0.0001
using a paired two tailed t-test).
Figure 5. SIRK does not stimulate GDP release from G protein heterotrimers. A. Binding of
[35S]-GTPγS to 200 nM αi in the presence or absence of 400 nM β1γ2 was measured with 1 µM
GTPγS and [35S]-GTPγS (100,000 cpm/assay) and 4 µM GDP for 10 min in the presence or
23
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

absence of 10 µg of urea stripped membranes expressing the M2 muscarinic acetylcholine
receptor and 100 µM carbachol. Error bars represent standard error from triplicate data points
and data are representative of experiments performed 3 times. B. M2 muscarinic receptor
stimulated binding of 35S-GTPγS binding at various concentrations of SIRK. Assay conditions
were as in A in the presence of urea stripped M2 muscarinic receptor expressing membranes and
carbachol. Results are pmols GTPγS bound minus the basal in the absence of added βγ subunits
of 1.3 pmol. Basal was unchanged by peptide as in A and not shown. Error bars represent
standard error from triplicate data points and data are representative of experiments performed 3
times. C. Steady state GTP hydrolysis was measured with 5 nM recombinant myristoylated Gαo,
10 nM β1γ2, sonicated phospholipid vesicles (PE:PS:PC 5 µM each) 300 nM GTP and [γ32P]-
GTP (30,000 cpm/assay) with 100 µM mastoparan (Mas) or 10 µM mSIRK as indicated. Data
are the combined normalized data from four separate experiments. Control (Go alone) is
significantly different from Go+Mas, P < 0.05, but not significantly different from Go+mSIRK
analyzed with a one way ANOVA and Tukey’s multiple comparison test. D. RASM cells were
treated with vehicle (H2O), 100 µM mastoparan or 10 µM mSIRK for 5 min and assayed for
ERK activation as in figure 1.
Figure 6. SIRK inhibits α-βγ interactions. A) Biotinylated β1γ2 (10 nM) was bound to
streptavidin agarose beads followed by 10 nM myristoylated αi and the indicated concentrations
of mSIRK or AlF4- (30 µM AlCl3, 10 mM NaF, 5 mM MgCl2) in 20 mM Hepes pH 8.0, 1 mM
EDTA, 1 mM DTT, 100 mM NaCl, 10 µM GDP and 0.1% C12E10. After incubation for 30 min
the beads were precipitated and αi associated with the βγ beads was assessed by quantitative
western blotting. Error bars represent standard error from triplicate experiments. B) 1.5 nM
FITC-αi was mixed with 200 pM biotinylated β1γ2 in the presence of the indicated concentrations
of SIRK ( • ) or SIRK(L9A) ( ▼ ) and the amount of F-αi bound was assessed by flow cytometry
as described in Experimental Procedures. Data are representative of experiments performed at
least 3 times each.
Figure 7. SIRK promotes αi dissociation from βγ subunits in the absence of nucleotide
exchange. A) 1 nM F-αi was preincubated with streptavidin beads bound to 250 pM β1γ2 for 10
24
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

25
min. Either 50 nM unlabeled myristoylated αi alone (▲) or 50 nM myristoylated αi plus 25 µM
SIRK ( ■ ) were added to the preformed F-αi-βγ complex. The rate of dissociation of F-αi was
measured using flow cytometry to assess the amount of F-αi bound at various times after
addition of the competitors. For each time point 3000 beads were assessed. Initial binding was
35 (mean channel number) for both curves (After subtraction of a background of 13 measured
for F-αi in the absence of βγ bound to the beads). Curves were best fit with a two-exponent
decay function using GraphPad Prism data analysis software. To test for statistical significance,
the F-αi fluorescence bound after addition of αi alone was compared with αi plus SIRK (25 µM)
at 1 and 2 min using an unpaired two tailed t-test (n=6 for each treatment). The average % initial
F-αi bound 1 min after addition of 100 nM αi as the competitor was 98.25 % ± 1.0 SEM and for
αi plus SIRK was 69.15% ± 0.77 SEM, and were significantly different, P < 0.0001. In a
separate experiment at 2 min after competitor addition the values were 90.0% ±1.77 SEM and
65.71% ±0.82 SEM (means were significantly different P < 0.0001) for αi and αi plus SIRK
respectively. B) Real time measurement of α dissociation. Same as A except after SIRK (25
µM) addition the fluorescence associated with the beads was analyzed using the continuous “list
mode” data acquisition option of the flow cytometer. More than 10,000 data points were
collected over the total time course and randomly selected groups of 200-300 events were
averaged for the indicated times and plotted. ( ) is SIRK added without addition of
myristoylated αi. Curves were best fit with a single-exponent decay function using GraphPad
Prism data analysis software yielding a koff for αi of 0.02 min-1, peptide 0.28 min-1 and peptide +
αi of 0.24 min-1. This experiment was repeated 5 times with qualitatively similar results.
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from
BA
p44p44 p42p42
** **
**
** **** ** 4444
42 42
0 1.2 2.5 5 10 30[mSIRK] (µM)
0 1 2 5 10 15 30Time (min)26
C D p44p44p42p42
4444
DMSO mSIRK mSIRK SIRK (L9A)
42 42
DMSO mSIRK tatSIRK EGF LPA
Fig. 1
by guest on July 15, 2018 http://www.jbc.org/ Downloaded from
p44p42A
mSIRK - - - + + DTT - - + - +mSCAR - + + - -
4442
BDMSO mSIRK mSIRK LPA
2 µM 5 µM
p44p42
4442
βARK1ct - + - + - + - +
Fig. 2
27
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from
A ddtMF2 MDA Rat 2 RASM HEK 293 COS 7
mSIRK - + - + - + - + - + - +
p44p42
4442
B
- + + - + + - -- - + - - - - -- - - - - - + +- - - - - + - +
mSIRKPP2EGFAG1478
p44p42
C
- + + - + + - -- - + - - - - -- - - - - - + +- - - - - + - +
p44p42
mSIRKThaps.LPAPTX
Fig. 3
28
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from
0 100 200 300 400 500 600 700Time (sec)
[Cyt
opla
smic
Ca2+
] (34
0/38
0nm
ratio
)
0.20.40.60.8
0.20.40.60.8
0.20.40.60.8
Thapsigargin-pretreated
mSIRK
Iono
mSIRK (L9A)
mSIRK
Iono
Iono
A BP-JNK
P-p38
p44p42P-ERK
4442Total
ERK
DMSO FBS mSIRK mSIRK(L9A)29
IP re
leas
ed (c
pm)
0
500
1000
1500
2000
2500C
DMSO mSIRK mSIRK FBS+PP2 Fig. 4
by guest on July 15, 2018 http://www.jbc.org/ Downloaded from
-7 -6 -5 -40
1
2
3
Log [SIRK]
pmol
GTP
γ S b
ound
B
αiβγ
SIRKM2
+ + + ++ + - +- + - -- - + +
[35S]
-GTP
γS b
ound
(cpm
) x 1
0-4
0
2
4
6
8
10
12
14A
C D
Go Go+Mas
Go+mSIRK
0
2
4
6
Fold
stim
ulat
ion
Con Mas mSIRK
p44p42
Fig. 5
30
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from
A
% α
Bou
nd
0
20
40
60
80
100
120
Con. AlF4 10µM 30µM
Peptide concentration (µM)
0 5 10 15 20 25 30
Spec
ific
F-α
bin
ding
(mea
n ch
anne
l num
ber)
0
5
10
15
20
25
30
35B
Fig. 6
31
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from
A
0 5 10 15 2040
50
60
70
80
90
100
Time (min)
% In
itial
F- α
i bou
nd
B
0 100 200 30050
60
70
80
90
100
Time (sec)
% in
itial
F-α
i bou
nd
Fig. 7
32
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

Griendling, Richard R. Neubig and Alan V. SmrckaFarida Goubaeva, Mousumi Ghosh, Sundeep Malik, Jay Yang, Patricia M. Hinkle, Kathy K.
subunit binding peptidesγβStimulation of cellular signaling and G protein subunit dissociation by G protein
published online March 20, 2003J. Biol. Chem.
10.1074/jbc.M300052200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from


















