
Uptake of -Ketoglutarate by Citrate Transporter CitP Drives Transamination in Lactococcus lactis

55 0 1993 Elacvicr Scicncc Publishera B.V. All rights rcscrwd. 0378-l 119~‘~2’$~)5.00
GENE OhSSl
Sequence of the Kluyveromyces Zact is p -galactosidase: comparison with prokaryotic enzymes and secondary structure analysis ’
(Recombinant DNA; yeast; nucleotide sequence; full-length alignment; mean secondary structure predictions;
fl-glucuronidase)
Olivier Poch ‘, Hervt: L’HBte ‘*, Vincent Dallery b, Franqoise Debeaux b, Reinhard Fleer’ and Regis Sodoyerb
Rcccivcd b! J.-P. Lecocq~W. Szyhalski: 23 Dcccmbcr 1991; Rcvised:Acccptcd: 26 .4pril,27 .4pril 1992: Rcccivcd at puhlishcrs: 2’) .4pril 1992
The UC4 gene encoding the P-galactosidase (BGal) of the yeast, K~uJw~~~~Jw.s lucris, was cloned on a 7.2-kb fragment
by complementation of a lrrcZ-deficient Escherichia co/i strain. The nucleotide sequence of the structural gene, uith 42 bp
and 583 bp of the 5’- and 3’-flanking sequences, respectively, was determined. The deduced amino acid (aa) sequence of
the K. 1rcti.s /JGal predicts a 1025-aa polypeptide with a calculated M,. of 117618 and reveals extended sequence homolo-
gies with all the published prokaryotic jGa1 sequences. This suggests that the eukaryotic /{Gal is closely related, cvolu-
tionarily and structurally, to the prokaryotic BGal’s. In addition, sequence similarities were observed between the highly
conserved N-terminal two-thirds of the flGa1 and the entire length of the p-glucuronidase (BGlu) polypeptides, which suggests
that PGlu is clearly related, structurally and evolutionarily, to the N-terminal two-thirds of the fiGal. The structural anal-
ysis of the BGal alignment, performed by mean secondary structure prediction, revealed that most of the invariant residues
are located in turn or loop structures. The location of the invariant residues is discussed with respect to their accessibility
and their possible involvement in the catalytic process.
INTRODIICTION
The yeast K. 1acti.r is known to produce an inducible
intracellular BGal (EC 3.2.1.32) and can thus use lactose
as carbon and energy source. The mechanisms by which
lactose and galactose induce BGal activity have been ex-
tensively studied (Das et al., 1985; Dickson et al., 1990;
Kuger et al., 1990). Initially, Dickson and Markin (1978)
had shown that a K. k&s DNA fragment can complement
a IaF mutant of E. coli lacking the structural gene for
fiGal. This fragment was then shown to carry the k’. Iactis
LAC4 gene (Sheetz and Dickson, 1981). Subsequent stud-
C~)rvc,c~c”‘~~~,n[,r fry: Dr. 0. Poch, Institut de Biologic Mol&xlairc et Cel-
lulaire de CNRS. 15 rut Dcscartcs. 67084 Strasbourg Ccdex, France.
Tel. (33.88)4l 7040; Fax (U-88)61 0680.
’ This paper is dedicated to the memory of Dr. J.-P. Lecocq.
* Prcscnt address: Transgtne S.A., I1 rut de Molshcim, 67000 Stras-
bourg (France) Tel. (33.88)279100.
Abbreviations: aa, amino acld(s): B.. Brrcillur: /{Gal. [I-galactosidasc;
PGlu, P-glucuronidase: hp, base pair(s); C.. Cbstuic/irm~; K.. Klu~vwron~.v-
c’e~; kb. kilobase or 1000 hp; KI., KlehsirN~r: L.. Ltrcmhucill~r.~: L.4C4.
gene encoding K. ltrcfi.7 /IGal: nt, nucleotide(s); oligo. oligodcoqrihonu-
cleotidc; ORF, open reading frame; PCR, polynerasc cham reaction; S.,
Swpmc’oc’c’ur; SDS. sodium dodcql sulfate; SSC. Il. I5 M NaCl:‘O.Ol5 M
Na;citratc pH 7.6: St.. S/w,nrnn~~r~e.~.

56
ies revealed that regulation of LAG! transcription is com-
plcx and involves the galactose/lactose induction system as
well as the catabolite repression (Riley et al., 1986; Ruzzi
et al., 1986; Salmeron and Johnston, 1986; Dickson et al..
1990; Kuger et al., 1990; Giidecke et al., 1991). The se-
quence of the LAC4 promoter and the deduced N-terminal
part of the protein had been determined (Breunig et al.,
1984; Leonardo et al.. 1987). However, full-length sequence
data for this eukaryotic PGal were missing. Within the last
decade. the complete nt and the deduced aa sequences of
six diffcrcnt prokaryotic BGal have been reported, two from
E. coli (Kalnins ct al., 1983; Stokes et al., 1985) and one
each from KI. pneumoniw (Buvinger and Riley. 1985).
L. hulg~~ricus (Schmidt et al., 1989), C. ac~etohut~licum
(Hancock et al., 199 1) and C. thermosu(furogene.~. EM 1
(Burchhardt and Bahl, 1991). In addition, the nt sequences
encoding the N-terminal regions of the fiGal from St. lir-
ides (Eckhardt et al., 1987) and S. thermophilus (Poolman
et al., 1989) have been published. Previous sequence anal-
ysis of the prokaryotic enzymes revealed the existence of
extensive aa sequence homologies and allowed the delin-
eation of different highly conserved sequence regions
(Schmidt et al.. 1989). The most conserved region exhibits
two invariant residues (Glu3”’ and Tyrso3) shown to be
critical for the catalytic activity of the E. coli enzyme (Ring
et al., 1985: Edwards et al., 1990; Cupples ct al., 1990). In
addition, sequence similarities have been reported between
the fiGa from E. coli and the eukaryotic and prokaryotic
BGlu (Fowler and Zabin, 1978; Nishimura et al., 1986).
The aims of the present study were cloning and sequenc-
ing of the entire LAC4 gene of K. 1ucti.v CBS2359 strain and
comparison of the deduced aa sequence to all the available
/IGal and PGlu aa sequences.
RESULTS AND DISCUSSION
(a) Isolation of the Kluyvevomyces lactis CBS2359 LAC4
gene
In order to verify that the LAC4 gene of the K. kmtis
CBS2359 strain exhibits a restriction pattern similar to that
reported for the K. luctis isogenic strain Y 1140 (Sreekrishna
and Dickson, 1985). a Southern hybridization analysis of
restriction enzyme-digested CBS2359 genomic DNA was
done (Fig. 1). The probe was obtained by PCR amplifica-
tion on CBS2359 genomic DNA using two oligo primers
(5’ primer from nt + 1 to + 33 and 3 ’ primer complemen-
tary to nt + 322 to + 35 1 as derived from the sequence data
available on the Yl 140 strain; Leonardo et al., 1987). A
7.2-kb Xh~l1 fragment strongly hybridized with the PCR
probe (Fig. 1). This result agrees with the restriction map
published by Sreekrishna and Dickson (1985) for the Y 1140
strain and indicates that the CBS2359 LAC4 gene is in-
1 2 3 4
kb
d - 23.1
- 9.4
w 7.2
- 6.5
Fig. I. Southern nnnlysis of the K. /uri,v LACY4 gent. Gcnomic DNA of
CBS2359 strain was isolated by the method of Kaback and Davidson
(1979). DNA \%as digested with PvuII (lane 1). Strll (lane 2). XhoI (lane
3), .YhoI (lane 4) and clcctrophorcacd in a I”,, agarosc gel. Follou ing
Southern blotting onto nitroccllulosc, the filter was probed at 65 C as
described in Sambrook et al. (19X9), with it “P-radiolabelled PCR frag-
mcnt corresponding to the CBS2359 LAC4 ORF extending from positions
+ I to + 3.50 (Leonardo et al.. 1987). The filter wu subscquentl) washed
with 0.2 x SSCI “Cl SDS. Sires (kb) of a Hirzdlll-digested phagc i DNA
arc indicated to the right. The arrow indicatcb the unique 7.2.kb ,YhrrI
fragment containing the LACJ gene.
eluded in a 7.2-kb X&I fragment as shown for the Y 1140
strain (Sreekrishna and Dickson, 1985).
The genomic DNA from K. hctis CBS2359 was isolated
and digested with X&I. The fragments ranging from 6 to
8 kb were electro-eluted (6-8 X&I fraction). The 6-8 X~LII
fraction was cloned in a pSVL vector (Pharmacia LKB
Biotechnology AB, Uppsala, Sweden) and used to trans-
form E. co/i JMlOl strain devoid of BGal activity. Over
8000 transformants were obtained and screened for fiGal
activity. Plasmid DNA isolated from five blue colonies all
contained a 7.2-kb XbaI insert with identical restriction
maps (data not shown). Fig. 2 shows the 3703-nt sequence
containing the 3078-nt aGal coding sequence capable of
encoding a 1025-aa protein and 42 and 583 nt of the 5’ and
Fig. 2. Complete nt scqucnce of the LAC3 gene. Rankmg region, and Its
dcrivcd aa acqucnce. Putative -35 and IO promoter elemcnta arc in bold
type and the putative polyadcnylation signal is underlined. The noncoding
sequences arc in lower-case letters. The ocgative numerals rcfcr to the
5’-flanking region and positive numerals begin at the A of the start codon
(ATG). To construct the library. the 6-8 .YhuI fraction (see aectwn a) was
isolated from H 0.X”,, agarose gel b> clcctro-elution and cloned into the
.Y/uI site of plasmid pSVL (Pharmacia) by standard methods (Samhrooh
ct al.. 1989). The clone GL A IO which contained the L.4C.d pcnc was uxxl
as the vu-cc for nt sequencing. Scquencmg was done h! the dideorc>

MSCLIPENLRNPKKVHENRL AlGlXXTG
PTRAYYYDQDIFESLNGPWA. CC~QZXI
FALFDAPLDAPDA'KNLDWET TTl?xmT
A K K'W S T ; S V P'S H W'E L Q i D W K' G-m
Y G K'P I Y i! N V Q-Y P 1-P I D i P N P' T-p-
PTVNPTGVYARTF'ELDSKSI' CCCACIGTA?+ATCCCGAAATCGATI
ESFEHRLRFEGVDNCYELYV cLuxuzrT--m
N G Q'Y V G i N KG'S R N'G AE $ D I Q' APLPGC;PCAFLCATGn;GG--n
KYVSEGENLVVVKVFKWSDS A-
TYIEDQDQWWLSGIYRDVSL 200 AClT?U!&---m 600
LKLPKKAHIEDVRVTTTFVD 220 C!TAAAAT---w 660
SQYQDAELSVKVDVQGSSYD' TC-G?.XtXCAGXX~~
HINFTLYEPE'DGS'KVYIiASS' CcrAAAG-m
LLNEENGNTTFSTKEFISFS TT--
TKKNEETAFKINVKAPEHWT A--m-
AENPTLYKYQLDL'IGSDGSV' Gp
IQSIKHHVGFRQVELKDGNI ATPCAATCP
TVNGKDiLFR'GVNkHDHHPR' ACCAkGG
FGRAVPLDFV'VRDLILMKKF TT
NINAVRNSHY'PNHPKVYDLF' A
DKLGFWVIDE'ADL'ETHGVQE GATAAGclmxcrT~
PFNRHTNLEAEYPDTKNKLY c--lKZ?AC
D V N'A H Y i S D N'P E Y-E V A ; L D R' G-m-m-
ASQLVLRDVN-HPS'III!iSLG' TccpAAGp13FITGTcApLpmmn
NEACYGRNHKAMYKLIKQLD A-
PTRLVHYEGDLNA'LSADIFS CCl!ACCAG?+ClTG~---
FMYPTFEIMERWRKNHTDEN' TT
GKFEKPLILCEYGHAMGNGP GGmAGT--m-m
G S L'K E Y 6 E L F'Y K E'K F Y 6 G G F' GGcrcrcT~~Tl-ITAC~~
1WEWANHGIEFED;STADGK A-w-
LHKAYAYGGDFKEEVHDGVF 620 T-ATGClT--TGl,lC 1860
-1
20 60
40 120
60 180
00 240
100 300
M 360
140 420
160 480
100 540
240 720
260 780
280 840
300 900
320 960
340 1020
360 1080
380 1140
400 1200
420 1260
440 1320
460 1380
480 1440
500 1500
520 1560
540 1620
560 1680
5fJo 1740
600 1800
IMDGLCNSEHNPTPGLVEYK A ACPCCGGGCCrr-TATAAG
KVIEPViIKI'AHGSVTITNK A
HDFITTDHLLFIDKDTGKTI 600 C-c!AA!rc 2040
DVPSLKPEESVTIPSDTTYV 700 GAcxrrc~'-A!rGrI 2100
VAVLKDDAGVLKAGHEIAWG GTlGCLiG--TFGCCTGGGGC
Q A E'L P L i V P D'F V T'E T A i K A A' -mm-
720 2160
740 2220
KINDGKRYVSVESSGLHFIL A--m
DKLLGKIESLKVKGKEISSK
760 2280
780 2340
FEGSSITFWRPPTNNDEPRD 800 TGGAGaC~-w.%AC 2400
FKNWKKYNIDLMKQNIHGVS 820 TT 2460
VEKGSNGSLAVVTVNSRISP CCCCCA
VVFYYGFETVQKYTIFANKI 860 GTXT?XSTlTACT-ACXCZA- 2580
N L N'T S M i L T G'E Y Q'P P D 'i P R V' -w--m 2640
GYEFWLGDSYESFEWLGRGP 900 B---C 2700
GESYPDiKES'QRF'GLYiSKD' 920 --p-m 2760
VEEFVYDYPQENGNHTDTHF 940 G--pCCXCTFI 2820
L N I'K F E i A G K'L S I'F Q K i K P F' TTGAACATCAAPLPT 2880
NFKISDEYGVDEAAHACDVK 980 2940
RYGRHYLRLD'HAIHGVGSEA 1000 A-- 3000
CGPAVLDQYRLKAQDFNFEF 1020 TG 3060
57
640 1920
660 1980
840 2520
1025
3120
3180 3240 3300 3360 3420 3480 3540 3600 3661
chain-terminating method (Sang-z et al., 1977) for single-stranded or
double-stranded templates. Large fragments spanning the gcnc region
wcrc subcloned in M 13 and subjcctcd to cxonuclcasc digestion (Cyclone
system; Dale et al., 1985) giving about 50”,, of the sequcncc data. The
missing regions were obtained by ‘gene walking‘ usmg a set of 30 spccitic
primers synthesized on an Applied Biosyatcm 38lA DNA S>nthesi,w
and used without purification. The complete nt scquencc was dctcrmincd
independcntlg on both strands. Premixed scqucncing rcagcnts (Scqucnasc
11 kit) were obtained from IJnited States Biochemical Corporation (Clcvc-
land. OH). Sequencing reactions Mere carried out according to the sup-
plier’s specifications. The ‘SS-labelled nt used for the scqucncing reactions
~vcrc purchased from Amersham International (Bucka., UK). The acccs-
sion No. M84410 in the GenBank database has been assigned to this nt
sequence.

Prs
dic
#i
-f-
ir,rs
asss
- am
fw
mm
65
0 70
0
ME
RW
RK
NW
TiE
NG
KF
E
00
0
00
0
0
0
. . .
I
EF
EO
”ST
AO
GK
L ---
-PLM
NEFG
EYPH
LP
. .
“FY.
. .I.
. . .
KH
DC
i*
A(lo
tlW--
--.G
Y
PK
WS
!Krn
SLP
GE
T
*FR
____
___.
.Qyp
R
LIKY
DEN.
---
G
VPm
GlK
KWlS
LPO
EO
.,....
AF
*~--.
...-.E
YPR
!RKT
FdO
----.G
---
-KYI
EEYL
TNKP
A I”
_...
LE_“
“_._
._..K
”PH
LEK+
.*,---
-.-
----A
ilEEY
LNDN
PK
LED.
“__.
.““.
KYLM
LY
RKLP
D-.
.--G
NK
P~~K
~EVT
GLE
~N
. .
. .
. .
EHCL
------
..RHL
R 1.
. _.
_. . _
_G
EKVL
EKEL
LAM
QEK
LH
L)TL
*GL
AWLD
MYW
RVFD
..RYS
TS
O..~
--GIL
AY
OLO
LATO
FE%
VYKK
YQ
ETIA
GF
SLLE
QYH
LGLD
CIKR
RK
TE
Q”
-SPT
R”
OP*
LNSG
FEta
rfKT
M
DAfP
GI
AVLE
NYHS
VLDQ
KRKE
TN
R....
-SPL
RY
OLQ
LTSQ
FENW
YKM
YO
QAV
SGL
ALLE
NYHL
ILDE
KRKE
TN
*.~“.
-SPL
R”
++
+ +
+ t*
++
BB
Bi?B
~f
iliti~
J+f”
75
0
La04
Eb
QA
LaCZ
La
cL
Gal

3’ flanking regions, respectively. The 3’-noncoding region
contains a consensus polyadcnylation signal (5’-AAT-
AAA) located 280 bp downstream from the stop coding.
(b) Alignment of the fiGal aa sequences
Pairwise sequence comparisons were performed between
the deduced aa sequence of the K. 1wti.s /Gal and those
published for the E’. co/i (Kalnins et al., 1983: Stokes et al.,
1985), KI. ptzeuttwnicre (Buvinger and Riley, 19X5), L. hul-
gcwicu.s (Schmidt et al.. 1989). C. trwtohutylicut~t (Hancock
et al., 1991), C. rhc~rttto.sulfirrogette.s EM I (Burchhardt and
Bahl, 1991) and the N termini of Sr. 1ividcttl.s (Eckhardt
et al.. 1987) and S. thertmphihrs (Poolman et al., 1989). As
shown in Fig. 3 (upper sequence lines), strong homologies
exist over the entire length of the PGal, only falling off
markedly in the C-terminal regions (roughly from aa 760 to
the end in the arbitrary numbering of Fig. 3). In more than
80”,, of the aligned /IGal sequences. there are 385 conser-
vatively maintained na of which 150 are strictly invariant.
The gaps introduced in order to maximize the similarities
are of limited lengths, except for two regions (aa 330-351
and 502-524) where large insertions arc mainly due to
K. lacti.~ PGal.
A measure of the pair\visc relationships bct\veen the dif-
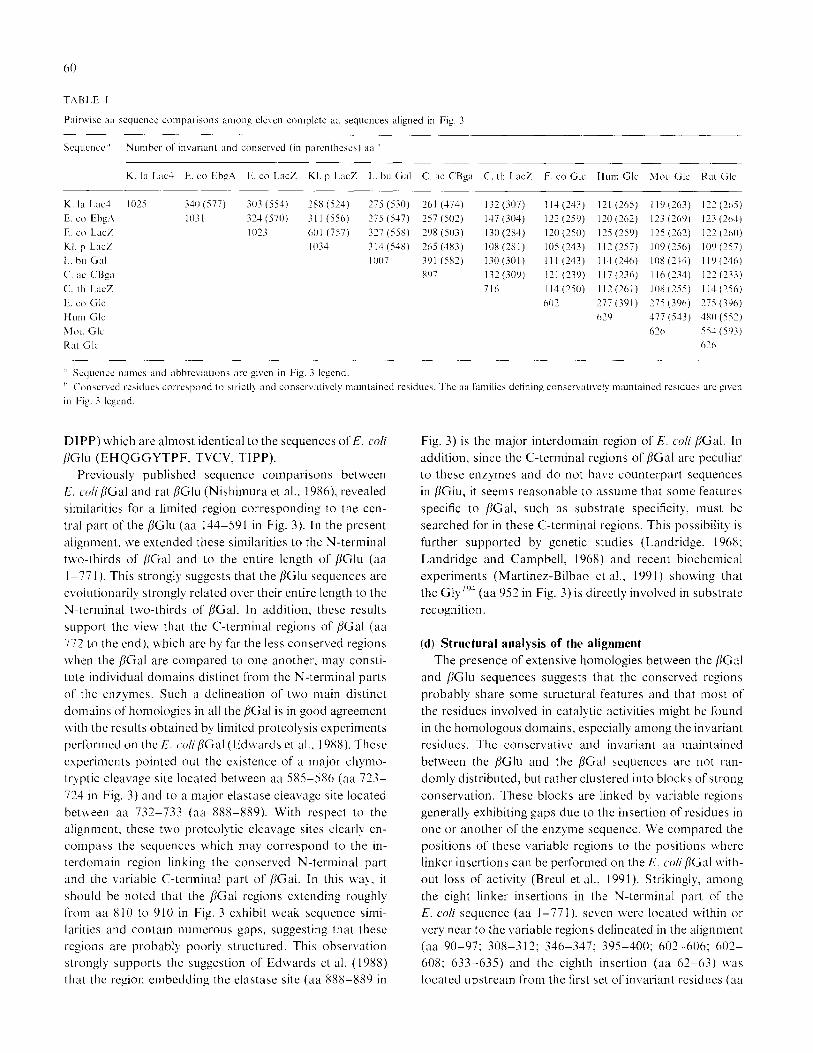
ferent complete /IGal proteins is given in Table I which
shows a compilation of the strictly and conservativelq
maintained aa according to the alignment of Fig. 3. With
respect to the numerous residues conserved bet\veen the
K. Otis cnzymc and the other bGa.1. it is clear that the
eukaryotic protein is closely evolutionarily related to the
prokarqotic ones. In addition, the data shown in Table 1
may suggest that K. /ctcti.v BGal is somcivhat more closclq
related to the E. co/i enzyme encoded by the ehgA gent (577
conservatively maintained aa among which 340 are invari-
ant). The two [IGal encoded by the 1rrc.Z genes from E. co/i
and KI. ptteutmt~iae arc so far the most closely related.
while the /~‘Gal from L. hu/garicu.s and C. ct~rtohu!\Gwttt
may define a third group. The thermostable /IGal from
59
C. thern?o.sulfirroRelle.v EM 1 is of special interest since it
appears to be extensively different from the six other PGal.
Based on the number of strictly maintained rcsiducs, there
is a slightly closer relationship bet\vccn C’. t/7erttto.~ul/lrro-
gettes EM I PGal and the ehgA gene product nhilc the lcast
related enzyme is KI. peutnc~ttirre. Howcvcr, thcsc data ma)
reflect structural analog), rather than evolutionary links
since these relationships are different ~vhen the conserva-
tive replacements are taken into account.
(c) Alignment of the fiGal and BGlu aa sequences
Additional searches for similaritics bctwecn the K. k/i.s
/IGal sequence and the proteins present in the Slviss-Prot
data bank allo\s,ed the detection. at statisticall\ significant
levels, of all reported PGlu. i.c., the E. co/i (JefTerson et al.,
1986). human (Oshima et al.. 1987), mouse (Gallaghci
et al.. 1988) and rat (Nishimura et al.. 1986) enzymes. In
Fig. 3, we propose an optimal alignment of the /iGal and
PGlu sequences in which I94 aa arc conservatively main-
tained of which 71 arc invariant among at least 80”,, of the
eleven aligned complete aa sequences. Analysis of the data
shoed in Table I reveals that none of the /jGal scqucnccs
stems more r&ted to one or another /IGlu scqucncc. In-
deed, the number of strict11 or conservativel) maintained
residues arc somewhat equi\alent. Special attention should
be paid to the C. thrntto.sltl/irro,~etrre.s EM 1 /<Gal which ap-
pears slightly mot-c closely related to the PGal than to the
/IGlu sequences. This ma!’ reflect an intermediate evolu-
tionary position of the thermostable enzyme between the
PGal and PGlu proteins. Such an intcrmediatc evolution-
ary position is further supported b) two observations: first,
the length of the C. IhL~rtrto.slrl/irrft~~~tt~,.~ EM 1 cn~\ mc which
is intcrmcdiate bct\\ecn the /jGal and [IGlu protein Icngths:
second. the presence. in the C’. tl7ertt7O.FItI/ilR)~~~,tte.~ EM I
BGal. of a region (aa 186-237) more similar to the /iGlu,
especially to the E. co/i enzynlc. than to the [IGal sequences.
This region encompasses stretches of aa scquenccs in the
C. thertttosu~~irro~pctrr.c ERI 1 /jGal (EH KGGYTPF. TVVV,
TABl,t I
Pairwiac aa squcnce cornparsons among elcvcn complete aa sequences aligned in Fig. 3
Scqucnce ’ Number of invarianl and conaer\ed (in parcnthcacs) aa”
K. la Lx-l E. co Ebg4 E. co LacZ Kl. p LacZ I.. bu Gal C. ac CBga C. th Lx% E. co Glc Hum Glc Mou Glc Rx Glc
K. la Lx-! 102 340 (577) 303 (554) 28X (524) 275 (530) 261 (474) 132 (307) I l-1 (243) 121 (265) 119 (1763) 1’7 (265)
E. co kbg4 1031 323 (570) 3 I I (556) 275 (547) 257 (501) 147 (304) 122 (259) 1X(262) 133 (269) 123 (264)
F. co LacZ 10’3 60 1 (757) 337 (558) 29x (503) I30 (384) 110 (250) 125 (259) 125 (162) 112 (260)
KI. p LacZ IO34 314 (54X) 265 (383) 108 (381) 105 (243) I I? (257) 109 (256) 109 (257)
I.. hu Gal I007 391 (582) 130 (301) lll(243) 114(246) lOS(234) II’)
c‘. aI‘ C‘Bga x97 132 (309) 121 (239) I I7 (236) Ii6 (234) 122 (23.1)
(‘. th Lx% 716 II-1 (250) I I? (XI) 10X (355) I14 (256)
I:. co Glc 602 177 (391) 275 (396) 775 (396)
Hum Glc 619 -177 (513) 4x0 (552)
hlou Glc 616 554 (593)
Rx1 Glc 6 2 6
’ Scqucncc names and abbrcvirttions arc given in Fig. 3 Icgcnd.
” Conscrvcd res~ducs corrcapond to strictly and conscrvalivcly mamtained rcsiducs. The aa families defining ux~servallvcly mamtaincd rcsiducb UC‘ ,g\‘en
111 Fig. 3 Iwcnd.
DIPP) which are almost identical to the sequences of E. cnli
/iGlu (EHQGGYTPF, TVCV. TIPP).
Previously published sequence comparisons between
E. co/i~Gal and rat PClu (Nishimura et al.. 19X6), revealed
similarities for a limited region corresponding to the ccn-
tral part of the BGlu (aa 144-591 in Fig. 3). In the present
alignment, \ve extended these similarities to the N-terminal
two-thirds of BGal and to the entire length of /3Glu (aa
l-77 1). This stronglq suggests that the flGlu sequences are
cvolutionarily strongly related over their entire length to the
N-terminal two-thirds of BGal. In addition, these results
support the view that the C-terminal regions of BGal (aa
772 to the end), which arc by far the less conserved regions
\vhcn the flGal are compared to one another. may consti-
tute individual domains distinct from the N-terminal parts
of the enzymes. Such a delineation of two main distinct
domains of homologies in all the /?Gal is in good agreerncnt
M ith the results obtained by limited proteolysis experiments
performed on the 15’. co/ijiGal (Edwards ct al.. 1988). These
cxperimcnts pointed out the existence of a major chymo-
tryptic cleavage site located between aa 585-586 (aa 723-
724 in Fig. 3) and to a major elastase cleavage site located
between aa 732-733 (aa 888-889). With respect to the
alignment, these two proteolytic cleavage sites clearly en-
compass the scquenccs which may correspond to the in-
terdomain region linking the conscrvcd N-terminal part
and the variable C-terminal part of /IGal. In this w’ay, it
should be noted that the PGal regions extending roughly
from aa 810 to 910 in Fig. 3 exhibit weak sequence sim-
laritics and contain numerous gaps, suggesting that these
regions are probably poorly structured. This observation
strongly supports the suggestion of Edwards ct al. (1988) that the region embedding the elastase site (aa 8X8-889 in
Fig. 3) is the major interdomain region of E. co/i BGal. In
addition, since the C-terminal regions of /IGal are peculiar
to these enzymes and do not have counterpart sequences
in /IGlu, it seems reasonable to assume that some features
specific to /JGal. such as substrate specificity, must bc
searched for in these C-terminal regions. This possibility is
further supported by genetic studies (Landridge, 1968:
Landridge and Campbell, 1968) and recent biochemical
experiments (Martinez-Bilbao ct al., 1991) showing that
the Gly’” (aa 952 in Fig. 3) is directly involved in substrate
recognition.
(d) Structural analysis of the alignment
The presence of extensive homologies between the BGaI
and /IGlu sequences suggests that the conserved regions
probably share some structural features and that most of
the rcsiducs involved in catalytic activities might bc found
in the homologous domains, especially among the invariant
residues. The conscrvativc and invariant au maintained
between the BGlu and the BGal sequences arc not ran-
domly distributed, but rather clustered into blocks of strong
conservation. These blocks are linked by variable regions
generally exhibiting gaps due to the insertion of residues in
one or another of the enzyme sequence. We compared the
positions of these variable regions to the positions where
linker insertions can be performed on the E. c,o/i/lGal Ivith-
out loss of activity (Breul et al., 1991). Strikingly, among
the tight linker insertions in the N-terminal part of the
E. co/i scquencc (aa l-771). seven were located within or
very near to the variable regions delineated in the alignment
(aa 90-97; 308-312; 346-347; 395-400; 602-606; 602-
608; 633-635) and the eighth insertion (aa 62-63) was
located upstream from the first set of invariant residues (XI

61
63-69). Considering the C-terminal part of the E. co/i en-
zyme, the linker insertions were also mostly located in var-
iable regions (aa 832-835; 957-958; 995-996; 1053-1054
and 1085-1088). This strong correlation observed between
the positions for which linker insertions do not induce loss
of activity and the positions of variable regions constitutes
a very powerful validation of this alignment. Moreover,
these observations allow us to predict with confidence that,
in the structural organisation of PGal and PGlu, the ma-
jority of the delineated variable regions may be involved in
loops allowing the insertion of various residues without
significant perturbation of the overall folding.
It should be pointed out that, among the potential glyc-
osylation sites (NXT/S, where X is any aa except Pro)
conserved in the three eukaryotic /~‘Glu sequences (bold
lettering in shaded boxes in Fig. 3), two are located in
variable regions with large insertions (aa 218-220; 502-
504), the third one being located at the C-terminal end of
the sequences where the sequence homologies fall off mark-
edly. With respect to the two variable regions, the presence
of potential glycosylation sites further reinforces the hypo-
thesis that these regions arc composed of accessible loops.
Consistent with this hypothesis is the presence of numer-
ous strong structure-breaker residues (Pro and Gly; see
Argos, 1987 and references therein) that are observed close
to the two glycosylation sites in the /JGlu sequences.
Among the distinct blocks of conservation, two regions,
from aa 361 to 501 and aa 53 1 to 567, are more conserved.
These regions contain most of the invariant aa (34 out of
72) as well as the longest stretches of conserved peptides
(motifs): GXN(R/K)HE (aa position 428); RXSHYP (aa
position 464); LCDXXG (aa position 477); RDXNHP (aa
position 545) and WSXXNE (aa position 555). Numerous
biochemical experiments pointed to the importance for cat-
alytic activity and Mg’+ binding of the invariant Glu (E)
present in the WSXXNE motif (Herrchen and Leglen,
1984; Badcr et al., 1988; Cupplcs and Miller, 1988; Cup-
pies et al., 1990; Edwards et al., 1990). The critical role of
the region encompassing the RDXNHP and WSXXNE
motifs is supported by the fact that these motifs represent
sequences for which local similarities can be observed bc-
tween BGal encoded by the B. s~euro~herrnophilus ebgA gene
and the PGal sequences (Schmidt et al., 1989).
A second residue, Tyr”” (aa 6 16 in Fig. 3) and the neigh-
bouring Met”” have been shown to be part of the catalytic
site and involved in the catalytic mechanisms of E. coli
PGal (Fowler et al., 1978; Ring et al., 1985; 1988; Ring and
Huber, 1990; Edwards et al., 1990). This residue is thought
to be the proton-donating species needed for catalysis. In-
spcction of the aligned sequences shows that this residue
is located close to the Mg2+ binding motif WSXXNE and
involved in an invariant dipeptide MY (aa 615) specific to
the PGal sequences. It is of special interest to note that
within the BGlu sequences Tyr”” and Tyr”“’ residues. lo-
cated in close proximity to the BGal-specific invariant MY
dipeptide, arc strictly conserved in the four fiGlu sequcnccs.
Therefore, it is tempting to propose that tither of thcsc
tyrosine residues may play, in the BGlu catalytic mecha-
nisms, a role similar to that attributed to the Ty? (aa 616
in Fig. 3) in the E. coli BGal activity.
Finally. to get additional insights into the three-
dimensional organisation of these two enzyme types. we
calculated the mean secondary structures over the entire
length of the eleven aligned sequences. WC took into ac-
count the similarly predicted structures (see legend to
Fig. 3) since their concordance over numerous sequences
lend more credence to the predictions (Zvclcbil ct al., 1987).
Eleven b-strands and two a-helices could be deduced from
this analysis. Strikingly, most of the invariant residues be-
longing to the aa known to be frequently involved in cat-
alytic processes (R, H, D and E) (Zvelebil and Sternberg,
1988), are located in positions bordering a b-strand or in
a predicted loop (random coiled) or turn structure. The
location of these invariant residues in turn or loop struc-
tures may reflect their accessibility and their possible in-
volvement in cation and substrate binding or in catalytic
processes. In this way, the two residues Glu’“’ and Ty?
(aa 561 and 616 in Fig. 3) shown to be directly involved in
the catalysis in the E. coli/?Gal are located at the C-terminal
part of the strongly predicted b-strands. Similarly, the long-
est conserved motif NXXRXSHYP (aa position 461)
seems to be part of an extended loop structure. Such a
location suggests that the invariant residues located in the
loop may be accessible for substrate interactions and/or
potential catalytic action.
ACKNOWLEDGEMENTS
We thank Dr. M. Zerbib and Dr. A Mouchaboeuf for
computer assistance. We are pleased to thank Dr. D. Lamy
for helpful discussions.
REFERENCES
Argos, P.: i\ sensitive procedure to compare amino acid scqucnccs. J.
Mol. Biol. 193 (19X7) 3X5-396.
Badcr. D.E., Ring, M. and Huber, R.E.: Sitedmxted mutagenic rcplace-
mcnt of Glu-461 with Gin in P-galactosldase (E. wli): cvidencc that
Glu-461 is important for activity. Biochem. Biophqs. Rcs. Commun.
153 ( 1988) 30 I-306.
Bairoch. A. and Boeckmann. B.: The SWISS-PROT protein sequence
databank. Nucleic Acids Res. 19 (1991) 2237-2219.
Breul. A., Kuchinkc, W., van Wilcken-Bergmann. B. and Miillcr-Hill, B.:
Linker mutagencsia in the IcrcZ gene of Esc~hrrirl~i~/ u>h yields variants
of active P-galactosidase. Eur. J. Biochem 195 (1991) 191-193.
Breunig, K.D.. Dahlcms. U., Das, S. and Hollcnbcrg. C.P.: 2nalysis 01


63
positlvc regulatory gcnc LACY reveals functional homology to. but
scquencc divergence from. the Sacch~ro~~~~ce.s crreaisirre GAL4 gene.
Nucleic 4clds Rca. I4 ( 1986) 7767-778 1. Sambrook. J.. Fritsch, E.F. and Maniatis. T.: Molecular Cloning. 4
Laboratory Manual, 2nd cd. Cold Spring Harbor Laboratory Press,
Cold Spring Harbor, NY, 1989.
Sangcr F.. Nicklen. S. and Coulson, .4.R.: DN.4 sequencing with chain-
terminating inhibitors. Proc. Nutl. .Acad. Sci. USA 74 (1977) 5463-
5467.
Schmidt. B.F.. Adams, R.M.. Rcquadt, C.. Poxver. S. and Mainzer. S.E.:
Expression and nucleotide sequence of the Lacrohrrcillus hulgcvicu.t
fl-galactosidasc gene cloned in the Ewherichitr co/i. J. Bacterial. 17 I (1989) 625-635.
Sheetr, R.M. and Dickson, R.C.: LAC4 is the structural gene for /i-
galactosidasc m K/uyveror~~.~ws k/cti.v. Gcnctics 98 ( I98 I ) 729-745.
Sreekrishna, K. and Dickson, R.C.: Construction of strains of
Strcchmm~~ces cerevititre that grow on lactose. Proc. Natl. Acad. SCI.
us.4 82 (1985) 7909-7913.
Stokes. H.W.. Betts. P.W. and Hall. B.C.: Sequence of the e/7x,4 gene of
E.cchrrichirr dr: comparison with the kwZ gent. Mol. Biol. Evol. 7
(1985) 469-477.
Zvclcbil. M.J. and Stcrnbcrg, M.J.E.: -\nalysia and prediction of the lo-
cation of catalytic residue in enzymes. Protcm Eng. 2 ( 1988) 127-13X.
Zvelcbll. M.J.. Barton. G.J.. Taylor. W.R. and Stcrnberg. M.J.E.: Prc-
diction of protein secondary structure and actwc sites using the align-
ment of homologous sequences. J. Mol. Biol. 195 (1987) 957-962.


















