Pyrolisis and Gasification
704
Commission of the European Communities f \ S > G3 Edited by G.L. FERRERÒ Κ. MANIATIS A. BUEKENS A.V. BRIDGWATER E1S E VI ER APPLI ED SCHNEI
description
Chemical
Transcript of Pyrolisis and Gasification

Commission of the European Communities
f \
S > G3
Edited by G.L. FERRERÒ Κ. MANIATIS A. BUEKENS
A.V. BRIDGWATER
E1S E VI ER A P P L I ED S C H N E I




PYROLYSIS AND GASIFICATION

Proceedings of an international conference held in Luxembourg, 23-25 May 1989.

PYROLYSIS AND GASIFICATION
Edited by
G. L. FERRERÒ Commission of the European Communities, Brussek, Belgium
K. MANIATIS pert to the Commission of the European Communities, Brussels, Belgium
A. BUEKENS Vrije Universiteit Brussel, Belgium
Α. V. BRIDGWATER University of Aston, Birmingham, UK
ELSEVIER APPLIED SCIENCE LONDON and NEW Y O R K | p ^ n i <
N.C./.
CL
/4. 9fø

ELSEVIER SCIENCE PUBLISHERS LTD Crown House, Linton Road, Barking, Essex IG11 8JU, England
Sole Distributor in the USA and Canada ELSEVIER SCIENCE PUBLISHING CO., INC.
655 Avenue of the Americas, New York, NY 10010, USA
WITH 202 TABLES AND 238 ILLUSTRATIONS
© 1989 ECSC, EEC, EAEC, BRUSSELS AND LUXEMBOURG
British Library Cataloguing in Publication Data
Pyrolysis and gasification. 1. Energy sources: Biomass. Thermochemistry I. Ferrerò, G. L. 662'.6
ISBN 1-85166-449-1
Library of Congress CIP data applied for
Publication arrangements by Commission of the European Communities, Directorate-General Telecommunications, Information Industries and Innovation, Scientific and Technical Com
munication Unit, Luxembourg
EUR 12479
Neither the Commission of the European Communities nor any person acting on behalf of the Commission is responsible for the use which might be made of the following information.
No responsibility is assumed by the Publisher for any injury and/or damage to persons or property as a matter of products liability, negligence or otherwise, or from any use or operation of any
methods, products, instructions or ideas contained in the material herein.
Special regulations for readers in the USA This publication has been registered with the Copyright Clearance Center Inc. (CCC), Salem, Massachusetts. Information can be obtained from the CCC about conditions under which photocopies of parts of this publication may be made in the USA. All other copyright questions,
including photocopying outside the USA, should be referred to the publisher.
All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, electronic, mechanical, photocopying, recording, or
otherwise, without the prior written permission of the publisher.
Printed in Northern Ireland by The Universities Press (Belfast) Ltd

PREFACE
Thermochemical processing of renewable resources and solid fuels has become a strong contender to partially replace the energy dependance of the European Community on imported hydrocarbon fuels and the Commission is supporting R & D as well as Demonstration projects In this field. Similarly in the US, Canada and the Developing Countries numerous projects have been carried out on fundamental as well as industrial scale projects on Pyrolysis and Gasification. Such technologies are therefore of increasing importance worldwide, not only because they can provide a source of energy but also because they can be utilized to dispose off various industrial wastes in an environmentally acceptable way.
Though interesting results and experiences have certainly been achieved, several problems still remain and their solution will strongly influence the commercialisation of Pyrolysis and Gasification all over the world.
It was the need to critically evaluate the progress achieved in this field and to draw up recommendations for future work which prompted the Directorate General for Energy of the Commission of the European Communities to organise this International Conference with the assistance of the Directorate General for Information, Marketing and Innovation.
While invited speakers from the Commission, EC countries, US, Canada and the World Bank presented overviews on all aspects of Pyrolysis and Gasification processes such as Feedstock Pretreatment and Characterisation, Gasification and Pyrolysis Technologies, Products Upgrading and Utilization as well as Environmental and Economic aspects, researchers and industrialists from 20 countries presented their results and views in oral as well as poster presentations. The Workshops and Panel Discussions gave the opportunity to all participants to express their opinion so that realistic recommendations for future R & D and Demonstration activities could be drawn up.
About 200 participants representing Administrations, Governmental Institutions, Universities and mainly the Industry attended the Conference. This is a fresh proof of the significance in recent years of Pyrolysis and Gasification technologies.
The Editors express their graditute to all the participants for their contributions as well as to the Chairmen and Rapporteurs who assisted in making this a successful Conference and we are confident that the contents of this proceedings will be a valuable tool and source of information to all those working in the field.
G.L. FERRERÒ Κ. MAN I ATIS Α. BUEKENS A.V.BRIDGWATER


vu CONTENTS
PREFACE
SESSION I
OPENING SESSION AND COUNTRY OVERVIEWS
ACTIVITIES AND RESULTS OF THE COMMISSION DEMONSTRATION PROGRAMME IN THE SECTOR OF GASIFICATION AND PYROLYSIS
G.L. FERRERÒ, General Directorate for Energy, Commission of the European Communities, Brussels, Belgium 3
COMMUNITY R&D STRATEGY IN THE FIELD OF BIOMASS PYROLYSIS AND GASIFICATION
G. GRASSI, Directorate General for Science Research and Development, Commission of the European Communities 7
PROGRESS IN PYROLYSIS AND GASIFICATION OF BIOMASS: AN OVERVIEW OF RESEARCH IN THE UNITED STATES
J. DIEBOLD, D. STEVENS, Solar Energy Research Institute, Golden, Colorado, USA 14
OVERVIEW OF THERMOCHEMICAL CONVERSION OF BIOMASS IN CANADA R.D. HAYES, Bioenergy Research and Development Technology Branch, Energy, Mines and Resources Canada, Ottawa, Canada 28
RAPPORTEUR'S REPORT ON SESSION I OPENING SESSION AND COUNTRY OVERVIEWS
C H . NELS, Federal Office for the Environment, Berlin 40
SESSION II
PRE-TREATMENT AND CHARACTERIZATION
PRETREATMENT AND CHARACTERIZATION OF FEEDSTOCKS C.P. MITCHELL, Forestry Department, Aberdeen University, Aberdeen, UK, A.V. BRIDGWATER, Chemical Engineering Department, Aston University, Birmingham, UK 43
PROCESSING OF URBAN WASTE TO PROVIDE FEEDSTOCK FOR FUEL/ENERGY RECOVERY
J.R. BARTON, Warren Spring Laboratory, Department of Trade and Industry, UK 57
CHARACTERIZATION OF CARBON CONTAINING MATERIALS WITH RESPECT TO PYROLYSIS AND GASIFICATION
H.J. MÜHLEN, W. WANZL, K.H. VAN HEEK, Bergbau-Forschung GmbH, Essen, FRG 72

VIU
KTI ACTIVITIES IN THE FIELD OF BIOMASS PYROLYSIS L. ANTONELLI, Vice President, Director of Alternative Energies Department, Kinetics Technology International SPA, Rome, Italy 85
BIOMASS FUELS AND GASIFICATION J. CARRE, L. LACROSSE, Y. SCHENKEL, Center for Agronomical Researches (CRA), Gembloux, Belgium F. RURIHOSE, Université Catholique de Louvain (UCL), Louvain-la-Neuve, Belgium 93
USAGE OF CARBON BLACK AND ACTIVATED CARBON IN RELATION TO INPUT AND TECHNICAL ASPECTS OF THE PYROLYSIS PROCESS
B. BILITEWSKI, G. HKRDTLE, K. MAREK, Intecus, Associated Engineers for Environmental Protection Technologies, Berlin 98
RAPPORTEUR'S REPORT ON SESSION II PRE-TREATMENT AND CHARACTERIZATION
C. ESNOUF, Cemagref, Antony Cedex, France 106
SESSION III
PYROLYSIS, GASIFICATION AND LIQUEFACTION TECHNOLOGIES
BIOMASS GASIFICATION: PAST EXPERIENCES AND FUTURE PROSPECTS IN DEVELOPING COUNTRIES
M.S. MENDIS, Industry and Energy Department, The World Bank, Washington DC, USA 111
GASIFICATION AND PYROLYSIS OF BIOMASS IN EUROPE A.A.CM. BEENACKERS, Department of Chemical Engineering, University of Groningen, Groningen, The Netherlands A.V. BRIDGWATER, Chemical Engineering Department, Aston University, Birmingham, UK 129
A SURVEY OF BIOMASS LIQUEFACTION PROCESSES R. CAPART, A. ELAMIN, S. AMMAR, M. GELUS, Department of Chemical Engineering, University of Technology, Compiègne, France 158
THE GEORGIA TECH ENTRAINED FLOW PYROLYSIS PROCESS R.J. KOVAC, D.J. O'NEIL, Georgia Institute of Technology, Atlanta, Georgia, USA 169
PILOT PLANT DEMONSTRATION OF USED TIRES VACUUM PYROLYSIS C. ROY, Université Laval, Department of Chemical Engineering, Quebec, Canada J. UNSWORTH, Petro-Tire Inc., Hamilton, Ontario, Canada 180
FLUIDIZED BED PYROLYSIS OF SEWAGE SLUDGE U. BELLMANN, A.B. KUMMER, Y. YING, W. KAMINSKY, Institute for Technical and Macromolecular Chemistry, University of Hamburg, FRG 190

IX
RAPPORTEUR'S REPORT ON SESSION III PYROLYSIS GASIFICATION AND LIQUEFACTION TECHNOLOGIES
A.V. BRIDGWATER, Aston University, Birmingham, UK 195
SESSION IV
PYROLYSIS CASE STUDIES
SUGARS FROM CELLULOSICS BY THE WATERLOO FAST PYROLYSIS PROCESS D.S. SCOTT, J. PISKORZ, D. RADLEIN, S. CZERNIK, Department of Chemical Engineering, University of Waterloo, Waterloo, Ontario, Canada 201
PRODUCTION OF BENZOLES AND ACTIVE CARBON FROM WASTE RUBBER AND PLASTIC MATERIALS BY MEANS OF PYROLYSIS WITH SIMULTANEOUS POST-CRACKING
R. CYPRES, B. BETTENS, Université Libre de Bruxelles (ULB), Brussels, Belgium 209
CHEMICALS FROM ALMOND SHELLS BY PYROLYSIS IN FLUIDIZED BED R. FONT, A. MARCELLA, E. VERDU, J. DEVESA, División de Ingenieria Química, Universidad de Alicante, Spain 230
BIOMASS PYROLYSIS IN MOLTEN SALTS FOR FUEL PRODUCTION J.Κ. MAUND, D.M. EARP, Department of Chemical Engineering and Applied Chemistry, Aston University, Birmingham, UK 238
FLASH PYROLYSIS OF SULCIS COAL L. CONTI, G. SCANO, Dipartimento di Chimica, Università' di Sassari, Sassari, Italy 246
RAPPORTEUR'S REPORT ON SESSION IV PYROLYSIS CASE STUDIES
D.J. O'NEIL, Georgia Institute of Technology, Atlanta, Georgia, USA 250
SESSION V
GASIFICATION CASE STUDIES
PERFORMANCE OF A PILOT SCALE FLUIDIZED BED GASIFIER FUELLED BY RICE HUSKS
HARTINIATI, A. SOEMARDJO, M. YOUVIAL, LSDE-BPP Teknologi, Indonesian Energy Research Laboratory, Puspiptek - Serpong, Indonesia 257
ELECTRICAL ENERGY FROM BIOMASS' F. FONZI, Italenergie S.p.a., Sulmona AQ, Italy 264
FLUIDIZED BED GASIFICATION OF WOOD: PERFORMANCE OF A DEMONSTRATION PLANT
K. MANIATIS, A. BUEKENS, Department of Chemical Engineering and Industrial Chemistry, Free University of Brussels, Belgium A.V. BRIDGWATER, Department of Chemical Engineering, Aston University, Birmingham, UK 274

A NATIONAL PROGRAM ON IMPLEMENTATION OF BIOMASS GASIFICATION PROCESS IN INDONESIA. SCENARIO, PROGRESS AND ECONOMIC EVALUATION
H. SUSANTO, S. REKSOWARDOJO, Department of Chemical Engineering, ITB, Bandung, Indonesia 282
GASIFICATION AND PYROLYSIS OF STRAW - RESEARCH IN DENMARK G. OLSEN, P.H. PEDERSEN, O. HENRIKSEN, E. KOFOED, Laboratory for Energetics, Technical University of Denmark, Lyngby, Denmark 290
AN INVESTIGATION INTO THE GASIFICATION OF LOW QUALITY COAL WITH OXYGEN ENRICHED AIR IN A FIXED BED GASIFIER
A.D. ENGELBRECHT, Division of Energy Technology, CSIR, Pretoria, South Africa 296
PRESSURIZED FLUIDIZED BED GASIFICATION OF PEAT E. KURKELA, P. STAHLBERG, W. MOJTAHEDI, M. NIEMINEN Technical Research Centre of Finland, Laboratory of Fuel Processing Technology, Espoo, Finland 304
RAPPORTEUR'S REPORT ON SESSION V GASIFICATION CASE STUDIES
K. MANIATIS, Free University of Brussels, Belgium A.A.CM. BEENACKERS, Groningen University, The Netherlands 312
SESSION VI
UPGRADING, CLEAN OP AND UTILIZATION OF PRODUCTS
GAS PURIFICATION: A REVIEW OF THE AVAILABLE METHODS OF GAS CLEANING
P. GUIGON, J.F. LARGE, Université de Technologie de Compiègne, France 317
WHAT CAN WE DO WITH PYROLYSIS OILS? E. CHURIN, B. DELMON, Université Catholique de Louvain, Louvain-la-Neuve, Belgium 326
COMPOSITION, PURIFICATION AND USE OF PRODUCER GAS J. VAN DER WEIDE, J.J. SEPPEN, TNO Road-Vehicles Research Institute, Delft, The Netherlands 334
RAPPORTEUR'S REPORT ON SESSION VI UPGRADING, CLEAN UP AND UTILIZATION OF PRODUCTS
J. DIEBOLD, SERI, Colorado, USA 342
SESSION VII
ECONOMIC, ENVIRONMENTAL AND LEGAL ASPECTS
ECONOMIC AND MARKET OPPORTUNITIES FOR BIOMASS DERIVED FUELS A.V. BRIDGWATER, Energy Research Group, Chemical Engineering and Applied Chemistry Department, Aston University, Birmingham, UK 347

XI
ENVIRONMENTAL PROBLEMS IN THE USE OF BIOMASS FUELS GLOBAL AND LOCAL ASPECTS
P. GIRARD, Centre Technique Forestier Tropical, Nogent-sur-Marne, France 372
WASTE MANAGEMENT AND PYROLYSIS: CURRENT SITUATION, TRENDS AND PROSPECTS
C H . NELS, Federal Office for the Environment, Berlin 379
RAPPORTEUR'S REPORT ON SESSION VII ECONOMIC, ENVIRONMENTAL AND LEGAL ASPECTS
R. FABRY, Commission of the European Communities, Brussels, Belgium 387
WORKSHOPS
WORKSHOP 1 - PRETREATMENT AND CHARACTERIZATION Chairman: J. BARTON Rapporteur: J. PISKORZ 391
WORKSHOP 2 - PYROLYSIS A.V. BRIDGWATER, Energy Research Group, Chemical Engineering Department, Aston University, Birmingham, UK C. ROY, Université Laval, Département de génie chimique, Quebec, Canada 394
WORKSHOP 3 - GASIFICATION TECHNOLOGY AND ECONOMICS Y. SOLANTAUSTA, Laboratory of Fuel Processing Technology, Technical Research Centre of Finland A.A.CM. BEENACKERS, Department of Chemical Engineering, University of Groningen, The Netherlands 396
WORKSHOP 4 - PYROLYSIS AND UTILIZATION E. CHURIN, Université Catholique de Louvain, Louvain-la-Neuve, Belgium 399
POSTERS PRESENTED
SECTION I
PRE-TREATMENT, PRODUCTS AND OTHER ASPECTS
FULL-SCALE DEMONSTRATION PROJECTS OF THE EUROPEAN COMMUNITY IN THE FIELD OF PYROLYSIS, GASIFICATION AND CARBONISATION OF BIOMASS AND WASTE
R. FABRY, G.L. FERRERÒ, Directorate General for Energy, Commission of the European Communities, Brussels K. MANIATIS, Free University of Brussels, Belgium 405
THE USE OF WOOD AS FUEL IN MALAYSIA W.K. HOI, Forest Research Institute of Malaysia, Kuala Lumpur, Malaysia A.V. BRIDGWATER, Department of Chemical Engineering, Aston University, Birmingham, UK 411

XU
SELECTED ASPECTS, EXPLANATIONS AND STATEMENTS IN ACCORDANCE AND ANALOGY TO THE BIT GRANULATION-TECHNOLOGY
J.M. DISS, F.W. HOCHHEIM, Directorate General and General Management of the incorporated company B.I.T. SA, Luxembourg 417
THERMOCHEMICAL DENSIFICATION OF BIOMASS - A KINETIC APPROACH TO PROCESS DEVELOPMENT
D.P. KOULLAS, N.S. ABATZOGLOU, E.G. KOUKIOS, Department of Chemical Engineering, National Technical University of Athens, Greece 420
PREPARATION AND USE OF CHARCOAL : WATER SLURRIES C. ESNOUF, S. GAUDEMARD, Cemagref, Antony Cedex, France, G. ANTONINI, O. FRANCOIS, Université de Technologie de Compiegne, Compiegne Cedex, France, C. MEZERETTE, CTFT, Nogent-sur-Marne, France 425
TREATMENT OF PYROLYSIS OIL WITH COMPRESSED CARBON DIOXIDE V. BRANDANI, G. DEL RE, G. DI GIACOMO, D. FLAMMINI University of L'Aquila, Department of Chemistry Chemical Engineering and Materials., L'Aquila, Italy 430
ACTIVATED CARBON FROM EUCALYPTUS KRAFT LIGNIN J.J. RODRIGUEZ, T. CORDERO, J. RODRIGUEZ-MIRASOL, A. SIMON, A. BATALLER, Departments of Chemical Engineering and Mechanical Engineering, University of Malaga, Malaga, Spain 435
ACTIVATED CARBONS FROM CHROMIUM-TANNED LEATHER WASTE M.A. MARTINEZ-SANCHEZ, C. ORGILES-BARCELO, Asociación de Investigación de las Industrias del Calzado y Conexas, Alicante, Spain J.M. MARTIN-MARTINEZ, F. RODRIGUEZ-REINOSO, Departamento de Química Inorgánica e Ingenieria Química. Universidad de Alicante, Alicante, Spain 439
MOTOR FUELS FROM PYROLYTIC LIGNINS J. PISKORZ, P. MAJERSKI, D. RADLEIN, D.S. SCOTT, Department of Chemical Engineering, University of Waterloo, Ontario, Canada 444
ROLE OF CHROMIUM OXIDE IN THE TEXTURE OF CARBONS FROM LEATHER J.M. MARTIN-MARTINEZ, F. RODRIGUEZ-REINOSO, Departamento de Química Inorgánica e Ingenieria Química, Universidad de Alicante, Alicante, Spain M.A. MARTINEZ-SANCHEZ, C. ORGILES-BARCELO, Asociación de Investigación de las Industrias del Calzado y Conexas, Alicante, Spain 452
INFLUENCE OF THE POROSITY OF CARBON IN Fe/Carbon CATALYSTS J.M. MARTIN-MARTINEZ, Departamento de Química Inorgánica e Ingenieria Química, Universidad de Alicante, Alicante, Spain M.A. VANNICE, Department of Chemical Engineering, The Pennsylvania State University, Pennsylvania, USA 457

XIII
DEVELOPMENT AND CONSTRUCTION OF A SAMPLING LINE FOR WOOD PYROLYSIS EMISSIONS
J. LACHENAL, J.M. TOLEDO, Laboratoire National d'Essai, Trappes, France C. MEZERETTE, A.M. VERGNET, Centre Technique Forestier Tropical, Département du CIRAD, Nogent-sur-Marne, France 462
FURNACE FOR BIOFUELS THERMAL UTILIZATION W. BLASIAK, B. ZETHRAEUS, R. COLLIN, Royal Institute of Technology, Department of Heat and Furnace Technology, Stockholm, Sweden W. GAJEWSKI, J. ZAJDEL, Technical University of Czestochowa, Institute of Heat Machinery, Poland 468
INVESTIGATION OF TOXIC COMPONENTS IN PRODUCTS FROM MUNICIPAL WASTE - SEWAGE SLUDGE PYROLYSIS
H. RÖSSLER, U. PRÖSCH, W. KAMINSKY, Institut für Technische und Makromolekulare Chemie, Universität Hamburg, Hamburg, FRG 473
RESEARCH ON TAR CRACKING AND APPLICATION OF TAR G. OLSEN, Laboratory for Energetics, Technical University of Denmark 479
THE FUEL PROPERTIES OF HYDROCARBON LIQUIDS DERIVED FROM PYROLYSIS OF WASTE
P.T. WILLIAMS, D.T. TAYLOR, Department of Fuel and Energy, The University of Leeds, Leeds, UK 486
ENVIRONMENTAL AND PUBLIC HEALTH ASPECTS OF GASIFIER SYSTEMS J. WILLOCX, Consultant, Londerzeel, Belgium A BUEKENS, Professor, Vrije Universiteit Brussel, Brussels, Belgium 492
CHARACTERIZATION OF WOOD CONSTITUENTS BY PYROLYSIS - FIELD IONIZATION MASS SPECTROMETRY
H.R. SCHULTEN, Fachhochschule Fresenius, Department of Trace Analysis, Wiesbaden, FRG 497
CHARACTERIZATION OF WOOD BY PYROLYSIS - FIELD IONIZATION MASS SPECTROMETRY
H.R. SCHULTEN, Fachhochschule Fresenius, Department of Trace Analysis, Wiesbaden, FRG J.M. HALKET, Department of Chemical Pathology, Queen Charlotte's and Chelsea Hospital, London, UK 505
SECTION 2
PYROLYSIS TECHNOLOGY
PYROLYSIS OF HAZARDOUS WASTE OIL U. STEFFENSEN, J. FRANCK, R. RAHNENFÜHRER, W. KAMINSKY Institute for Technical and Macromolecular Chemistry, University of Hamburg, FRG 517

XIV
ENERGAS SEWAGE SLUDGE PYROLYSIS H.F. HINRICHS, H. MULLER, ENERGAS, Gesellschaft zur Energiegewinnung aus Müll und Kohle mbH, FRG 522
COGENERATION PYROLYSIS G. BONINO, Biomass Energies Integrated Systems, Turin, Italy 527
CARBONISATION OF LARGE WOOD PIECES IN A LABORATORY RETORT: PRODUCT ANALYSIS AND ENERGY ASSSESSMENT
N. SHAH, P. GIRARD, Energie Division, CTFT, NogentsurMarne, France R. CAPART, Departement Genie Chimique, UTC, Compiegne, France 530
MILD PYROLYSIS PROCESS IMPROVES STEAM CYCLE EFFICIENCY P. GRAVERSEN, R.M. HUMMELSHØJ, Κ. JENSLEV, COWIconsult, Consulting Engineers and Planners AS, Virum, Denmark 536
FLASH PYROLYSIS OF BIOMASS FOR LIQUID FUELS S.A. BRIDGE, A.V. BRIDGWATER, Energy Research Group, Department of Chemical Engineering and Applied Chemistry, Aston University, Birmingham, UK 541
GASIFICATION OF REFUSE, A PROCESS OF SFW H. HUMMELSIEP, Η. FUNK, SaarbergFernwärme GmbH, Saarbrücken, FRG 547
A CATALYTIC GASIFICATION PROCESS OF BIOMASS J. MUNCK, Dansk Termo Industri/I. Krüger AS, Soborg, Denmark 551
AGRICULTURAL WASTES FOR ELECTRICITY GENERATION C. ESNOUF, M. HEERAH, Cemagref, Antony Cedex, France 554
PYROLYSIS PROCESS FOR RECYCLING FOREST AND AGRICULTURAL WASTES FOR RECUPERATING BIOMASS ENERGY
B. GROUX, R. GUIOL, Ph. POUSAZ, BioAlternative, S.A. Engollon, Switzerland 559
PYROLYSIS OF GREEK LIGNITES A.A. LAPPAS, I.A. VASALOS, Aristotelian University of Thessaloniki, Thessaloniki, Greece 563
DIRECT MASSS SPECTROMETRIC STUDY OF PYROLYSIS BEHAVIOUR OF BIOMASS AND ITS CONSTITUENTS UNDER DIFFERENT IONIZATION CONDITIONS. MS AND MSMS STUDY OF THE PRIMARY PYROLYSIS MECHANISMS
P.L. DESBENE, M. ESSAYEGH, B. DESMAZIERES, C. LANGE, J.J. BASSELIER, Laboratoire de Chimie Organique Structurale, Université" P. et M. Curie, Paris, France 568
A TWIN BED PYROLYSERCOMBUSTOR FLUID BED SYSTEM FOR THERMAL PROCESSING OF URBAN WASTE
H. MASSON, Seghers Engineering, Willebroek, Belgium A. BUEKENS, Κ. MANIATIS, Free University of Brussels, Belgium, J. SCHOETERS, Groep Τ, Leuven, Belgium 574

XV
PMMA PYROLYSIS FUNDAMENTALS AND EXPERIMENTAL INVESTIGATION Α. BUEKENS, F. DE WOLF, Free University of Brussels, Belgium J. SCHOETERS, KIH Groep Τ, Leuven, Belgium 580
PYROLYSIS OF EXHAUSTED OLIVE HUSKS COUPLED WITH TWOSTAGES THERMAL DECOMPOSITION OF AQUEOUS OLIVE OIL MILLS EFFLUENTS
G. DI GIACOMO, G. DEL RE, University of L'Aquila, L'Aquila, Italy, E. BONFITTO, S. IACOBONI, Regione Abruzzo, Avezzano, Italy, Ν. BRUNETTI, E.Ν.E.Α., Centro Ricerche Casaccia, Rome, Italy 586
SECTION 3
GASIFICATION TECHNOLOGY
STUDY OF BIOMASS GASIFICATION UNDER PRESSURE R. CAPART, M. GELUS, N. LESGOURGUES, Z. LI, Department of Chemical Engineering, University of Technology, Compiegne Cedex, France 593
GASIFICATION OF CHARCOAL IN MALAYSIA W.K. HOI, Forest Research Institute of Malaysia, Kuala Lumpur, Malaysia A.V. BRIDGWATER, Department of Chemical Engineering, Aston University, Birmingham, UK 598
UPDRAFT GASIFICATION OF WASTE FUELS P. STÂHLBERG, E. KURKELA, VTT, Laboratory of Fuel Processing Technology, Espoo, Finland H. FILEN, Κ. SALO, Bioneer Oy, Hämeenlinna, Finland 603
PEAT ΑΜΟΝΙΑ PLANT IN OULU SYNTHESIS GAS PRODUCTION FROM PEAT BY FLUIDBED GASIFICATION
K. SIPILÄ, C. WILÈN, E. KURKELA, A. MOILANEN, VTT Laboratory of Fuel Processing Technology, Espoo, Finland J. KOLJONEN, Kemira Oy, Oulu, Finland 608
DEVELOPMENT OF A DOWNDRAFT MOVING BED BIOMASS GASIFIER R. BILBAO, J. LANA, P. GARCIA, J. ARAUZO, Department of Chemical Engineering, University of Zaragoza, Zaragoza, Spain 613
STEAM GASIFICATION OF BIOMASS IN FLUIDIZED BED. EFFECT OF THE TYPE OF FEEDSTOCK
J. CORELLA, J. HERGUIDO, J. GONZALEZSAIZ, Chemical Engineering Department (Faculty of Science), University of Zaragoza, Zaragoza, Spain 618
STEAM GASIFICATION OF BIOMASS IN FLUIDIZED BED WITH A SECONDARY CATALYTIC REACTOR I. RESULTS WITH THE SECONDARY REACTOR EMPTY AND WITH SAND
J. CORELLA, M.P. AZNAR, Ν. CEBRIAN, J.I. IGLESIAS, M.P. MARTINEZ, Chemical Engineering Department, University of Zaragoza, Zaragoza, Spain 624

XVI
STEAM GASIFICATION OF BIOMASS IN FLUIDIZED BED WITH A SECONDARY CATALYTIC BED. - II. TAR CRACKING WITH DOLOMITE(S) IN THE SECONDARY REACTOR
M.P. AZNAR, J. DELGADO, J. CORELLA, J. LAHOZ, Chemical Engineering Department, University of Zaragoza, Zaragoza, Spain 629
FIXED BED GASIFICATION OF LIGNOCELLULOSIC BIOMASS THE CEMAGREF PROCESS
S. GAUDEMARD, J.J. BECKER, Cemagref, Antony Cedex, France 635
STUDY ON MARKED PRODUCTS OF WOOD GASIFICATION MECHANISMS WITH THE AIM OF PRODUCING CLEAN GASES
S. CASTILLO, S. BENNINI, G. GAS, J.P. TRAVERSE, Université Paul Sabatier, Toulouse, France 640
REDUCTION ZONE HEIGHT DETERMINATION IN SOLID WASTE GASIFICATION PROCESS IN A SHAFT FURNACE
J. WANDRASZ, K. WALECZEK, The Silesian Technical University, Poland 646
COMPUTER MODELLING OF FLUIDIZED BED GASIFICATION J.M. DOUBLE, E.L. SMITH, A.V. BRIDGWATER, Energy Research Group, Department of Chemical Engineering and Applied Chemistry, Aston University, Birmingham, UK 651
THREE-PHASE WOOD GASIFIER SYSTEM EASIMODR H. MICHEL-KIM, Efeu GmbH, Research and Development for Energy and Environment, Schwelm, FRG 656
LIST OF PARTICIPANTS 661
INDEX OF AUTHORS 677

SESSION I
OPENING SESSION AND COUNTRY OVERVIEWS


ACTIVITIES AND RESULTS OF THE COMMISSION DEMONSTRATION PROGRAMME IN THE SECTOR OF
GASIFICATION AND PYROLYSIS
G.L. FERRERÒ General Directorate for Energy
Coma I ss Ion of the European Communities 200 rue de la Lol
Β 1049 Brussels Belgium
Summary
The demonstration programme In the "Blomass and energy from waste" sector Is outlined In general, with particular reference to the subject of "Pyrolysls and Gasification".
Comments are made on certain results obtained In the Member States and general trends can be concluded.
The results of finished projects and of measures still In progress Indicate that these technologies, especially pyrolysls, will develop In Mediterranean countries where the conditions of blomass supply lend themselves better to conversion of the blomass In the form of stockable fuels.
The Energy Demonstration Programme Introduced In 1978 by the DirectorateGeneral for Energy of the Commission of the European Communities provides financial support for demonstration projects of an Innovatory nature and Industrial scale in respect of energy saving, renewable energy sources and substitutes for hydrocarbons.
Demonstrations projects are defined as any project which on a real scale amounts to the application of an Innovative technology or a new application of a proven technology and helps to establish the technical and economic feasibility of a process before advancing to commercial exploitât Ion.
These projects form the link between the research and development phase upstream, possibly backed by a pilot project, and the commercial exploitation phase downstream.
Since 1978 an Invitation to submit proposals for demonstration projects has been published each year In the Official Journal of the European Communities, and each year a certain number of projects considered to be of considerable technological and economic Interest are provided with finance by the Commission to cover part of the technical and economic risks Inherent In the project.

From 1978 to 1988 (Table 1) some 1631 projects have been selected from the huge number put forward In response to the annual Invitation to submit proposals Issued by the Commission and financial support totalling 841 Mio ECU (1 007 Mio $) has been awarded. This Community programme, which Is the largest In the world, Is accompanied by national programmes In the Member States of the European Community.
T A B L E
DEMONSTRATION PROJECTS 1978-1988
Total proposals S 176 Total accepted projects 1 631 CEC support 841 Mio ECU - 1 007 Mio $
Under the Βiomass and Energy from Waste Sector of this Programme some 191 projects (Table 2) have been awarded support totalling 87,4 Mio ECU (101,6 Mio $).
T A B L E
BIOMASS AND ENERGY FROM WASTE
Total accepted projects 191
CEC support 87,4 Mio ECU - 101,6 Mio $
Total cost of projects 311 Mio ECU - 361,8 Mio $
Some of these projects will be described to you In the course of the Seminar's technical sessions. The projects which have been selected can be divided Into 11 sub-sectors shown In Table 3. For the most part they fall Into one of the two main catagorles, "biological conversion" and "thermochemlcal conversion" of blomass and waste.

T A B L E
BIOMASS ANO ENERGY FROM WASTE
SUBSECTORS PROJECTS
01. Biomass Harvesting 02. Energy crops 03. Treatment of waste 04. Biogas 05. Refuse Derived Fuel 06. Direct combustion 07. Gasification and Pyrolysls 08. Compost 09. Fuels and chemicals (biol. treat.) 10. Fuels and chemicals (thermo-chem. treat.) 11. Proteins
Total
4 3 18 70 14 40 24 7 1 9 1_
191
Some 90X of biological conversion projects selected, 66 out of 70 projects, concern the production and use of biogas from animal litter, urban waste or effluent from the agro-food Industry. This Is a major sub-sector of the programme.
Some 50X of the 87 thermochemlcal conversion projects selected are concerned with direct combustion, 27% with gasification and carbonization, and 16% with the production and use of refuse-derived fuels (RDF).
In each sub-sector the projects In progress or completed are designed to establish the technical feasibility of new concepts and to evaluate the economic profitability of the projects, and In each of these sub-sectors projects successfully completed have or will give rise to multiplication, with considerable impact In terms of energy and the environment.
Treatment by gasification and pyrolysls of solid urban waste still seems to present certain problems whereas the pyrolysls and gasification of wood seems set for reasonable success.
In the short term It seems unlikely that these technologies are destined for extensive application, and there is clearly a need for demonstration measures before maximum use of their Interesting potential can be achieved.
The results obtained so far In the demonstration programme, although in no great quantity, nonetheless allow certain basic observations to be made on the technologies of gasification and pyrolysls.
The first point to be made is the growing interest In pyrolysls in most southern European countries, with Italy to the fore due to the numerous R&D activities tied up with the Leben-Abruzzo project.

In Germany various gasification of wood and wood waste techniques have had success although marketing of these facilities has not always had equally positive results. In France some experiments Into the pyrolysls of solid urban waste have encountered technical and economic problems whereas wood gasification and torefactlon projects have demonstrated the reliability of these techniques.
In Belgium, finally, certain ongoing experiments have had problems due mainly, as In other countries elsewhere, to the low cost of oil.
For other Community countries too few data are available for any conclusions to be drawn.
The feeling remains, however, that the technologies of gasification and, to a greater degree, pyrolysls are becoming particularly attractive, and the possibilities opened In future by the release of cultivated land for non-food uses may Increase to some extent the use of these processes.
However, there Is still the problem of their profitability.
Studies currently In progress and environmental requirements, which are still difficult to quantify, should provide a better Idea of the economics of these technologies, taking Into account not only the energy value of the products to be treated but also the far more general Impact that the use of blomass for energy purposes can generate.
The demonstration project of the Commission's Directorate-General for Energy will draw to an end In 1989. An Invitation to submit proposals has been published recently in the Official Journal, and projects on the pyrolysls and gasification of biomass and waste in general may still be submitted and accepted. However, regardless of the results of projects still to come. It can already be said that this sector of the demonstration programme has made a solid contribution to the spread of the technologies of gasification and pyrolysls. The projects In process and the results obtained, even though negative In some cases, have shown the way and pinpointed the problems on which greater effort should be concentrated.
We are now starting to reap the benefits of this vast programme, which, complementing measures taken at national level In the individual Member States, has led to technological maturity and process reliability in every respect.
The Directorate-General for Energy is proud to have contributed, and to continue to do so, to solving the common problem, which is not exclusively an energy one, of Increasingly efficient use of blomass and waste In an overall context reflecting nature protection, the use of local resources and the conservation of Jobs.
This conference will be different from many others being held elsewhere on this subject through Its mainly demonstration nature of the results obtained and the guidelines that should emerge for future action In Commission programmes.

COMMUNITY RSD STRATEGY IN THE FIELD OF BIOMASS PYROLYSIS AND GASIFICATION
G. GRASSI Directorate General for Science Research and Development,
Commission of the European Communities
1. INTRODUCTION
The immense stock of chemical energy, represented by vegetal matter (biomass) , constantly produced on the earth will play the role of a strategic and the only renewable industrial energy resource in the long term future. Its exploitation on a large scale will offer supplementary important benefits such as rural development, environmental land and atmosphere improvement, better climatic stability etc.
The biomass potential in the European Community is estimated at around 600 million ton/year (dry matter) and its likely utilisation is as follows:
Energy utilisation Industrial utilisation Chemicals and organic fertilisers
300 million ton/year 80 million ton/year 220 million ton/year
So, the energy sector should get the largest share of the resource. Of course, the development and speed of penetration of the bio-energy sector will depend largely on the future supply cost of biomass (30/60 ECU/ton (dm) actually in Europe); on the cost of the conversion technology; and (mainly) on the cost of imported oil.
Among the several conversion technologies so far developed in the Community (direct combustion, air gasification, advanced gasification, pyrolysis, liquefaction, catalytic conversion, bio-gasification) pyrolysis today appears very promising and best suited to the implementation of large biomass schemes like the LEBEN - Industrial projects. In fact pyrolysis or synthetic-oil can be competitive on a relatively small scale of production. This may also allow for large-scale biomass exploitation, the adoption of modular conversion units with capacity in the range of 2:10 ton/hr, to match in the most convenient way, the more or less productive and/or dispersed resource available.
2. BIOMASS CONVERSION INTO FUELS
The energy content (fixed carbon) of biomass can be exploited by direct combustion or after conversion (and eventual upgrading) into a more valuable fuel by physical, biological or thermochemical processes. My presentation today will be limited to these last conversion methods ; in particular to the state-of-the-art and the future development of pyrolysis and gasification technologies.
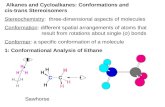
In Table 1, Table 2 and Figure 1 a summary of the main characteristics of thermochemical conversion processes and products is presented (Ref: A. Bridgwater, Aston University - workshop on LEBEN-PROJECTS/ Feb. 1989, Brussels).

TABLE 1. Thermochemical Conversion Technologies and Products
Technology Primary Product Application
Pyrolysis generally gas liquid
Flash pyrolysis
Slow pyrolysis
Liquefaction
Gasification
Combustion
fuel gas oil or liquid fuel substitution solid fuel or slurry fuel solid char
liquid mostly oil or liquid fuel substitution
solid char mostly solid fuel or slurry fuel
liquid oil or liquid fuel substitution
gas fuel gas
heat heating
TABLE 2. Secondary Products and Sources
Secondary Product
Oxygenate Fuels Methanol Fuel alcohol
Hydrocarbo Gasoline
Diesel
Fuel oil
Power Power
Chemicals Ammonia Speciality
η Fuels
chemicals
Source
Gasification Gasification
Pyrolysis Pyrolysis Liquefaction Gasification via
Pyrolysis Pyrolysis Liquefaction Gasification via
Pyrolysis Liquefaction
Pyrolysis Gasification
Gasification Pyrolysis Liquefaction
methanol
methanol
Extraction Extraction
Process
Synthesis Synthesis
Hydrotreating Zeolites
Hydrotreating MTG
Hydrotreating Zeolite + MOGD Hydrotreating
MCGD
Stabilisation Stabilisation
Turbine Engine or turbine
Synthesis and/or Conversion and/or Conversion

• • • / • / , / • • • CONVERSION I
: TECHNOLOGY s
LIQUEFACTION
.V . PRIMARY A ' '/,- PRODUCTS . V • • • / • / • / / • •
;i PROCESSING I TECHNOLOGY
MOONQ
Χ \ \ \ Ύ Ε Ο Ο Ν DAR Y V S \ \ ' .
\ Ν Λ Ν Χ \ -/// PRODUCTS '.-'/Α-';
\ \ \ ν \ \ f / • • • f . \ \ \ \ \ \ t / S / f / .
S Ν Χ Κ Κ \ • • • • • /
\ \ S \ \ S
\ \ \ / • .
V S Ν
/ / S t \ \ \ ' * S S /
Slurry fuel
Fig. 1. Primary and secondary products from thermochemical biomass processing
Primary Products
The primary products can be gas, liquid and/or solid char, depending on the conversion technology employed. Most of the present interest centres on liquid products due to their high energy density and potential for premium liquid fuel substitution.
As far as biomass conversion is concerned, I would like to recall here that:
(a) the basic strategic considerations for the present and future R&D programmes of the Commission suggest the production of two types of liquid "biofuels":
Bio-ethanol from sugar or starch (by advanced technologies and new crops) for the transportation market and which should develop at a significant rate after the year 2000 (presently given low emphasis by the Commission) ;
- Synthetic-oil (pyrolytic-oil) from lignocellulosic material for general thermal application, as a real substitute for the extensive oil import market, which could be conveniently produced (technically and economically) in the medium term (five years).
All other types of biomass fuels can, of course, be regarded as being of interest and deserve consideration, but only as a tactical means to solve local and time limited problems, or due to the particular characteristics of residues (i.e. municipal wastes, manure, sludges, etc.).

10
(b) In the EC the potential displacement by these two types of fuels is very large and it has been estimated at:
2.2 million barrels OE/day for Europe (about 24% of total oil imports).
(c) Biofuels should then be considered an important element in the Community's energy mix, whether viewed from the security perspective (domestic renewable feedstock), socio-economic impact (source of competitive, intensive manpower activity), rural development contribution, market dimensions (not saturable demand), environmental improvement (new uses for agricultural and marginal land, no sulphur, CO2, better climatic conditions etc.) or technological (industrial) competitiveness.
International collaboration could speed up the progress of pyrolytic biomass conversion technologies for the following reasons :
(a) Within the framework of the general EC-Canada Agreement, DG XII has proposed a collaborative programme on "pyrolysis and up-grading of pyrolytic fuels". Furthermore, an international industrial consortium has been constituted for the implementation of the first "fast pyrolysis pilot plant" in Spain, the construction being foreseen at the end of 1989. This technology should be able to convert lignocellulosic biomass into 60% oil + 10% charcoal + gas (energy efficiency > 80%).
(b) DG XII is joining efforts with DG XVII and DG I for a collaboration with Brazil and other countries in the bio-energy sector.
(c) In parallel, transfer of technologies to and from Europe is now under consideration and specific negotiation has already been carried out.
Importance of Regional Biomass Schemes
Biomass is a diluted dispersed resource, therefore there is a need to implement large projects, probably on a regional scale, to appreciate the importance and full value of these benefits.
There is general consensus that large-scale exploitation of biomass by multi-sectorial, innovative and integrated technologies will constitute a real instrument of rural development.
The involvement of regional/national authorities then becomes essential, as a guarantee for large investments as well as their ability to ensure the continuity of supply, the control of the cost of biomass resources in the long term, and to facilitate the market development of this renewable natural resource.
At present, several regions in Europe are considering the possibility of implementing major activities in the sector of biomass and of synthetic oil (pyrolytic fuel) production, as well as other kinds of conversion for energy and industry.
For these types of project, multi-sectoral integration of large market industrial activities with the bio-energy sector makes them more attractive and profitable in terms of economics.
Pyrolysis
Pyrolytic fuel (synthetic oil) has a strategic value because, as a liquid, its handling, storage, transportation and utilisation are similar to that of bunker-oil; its heating power is fair (above 6 000 kcal/kg) and its

11
specific gravity higher (1.2 gr/cm) . As it can be used immediately in the existing utilisation systems, this does not require expensive actions for market promotion and it can be considered as a fuel for general thermal applications (steam and electricity production).
Preliminary research results show the possibility of modern bio-energy technologies reaching promising markets which are:
general thermal applications (heat-steam production); thermal power stations for electricity production ; gas-turbine/steam-turbine electricity plant (combined cycles); conventional refineries.
Through this technology, the penetration and exploitation of biomass for energy production could be accelerated and implemented on a very large scale, by the adoption of modular standardised plants for synthetic liquid fuel production. Furthermore, such a conversion product could also be utilised at a later stage as a raw material for chemicals.
Another activity, inspired by the Commission and already considered of interest by important industrial groups (Mannesmann, VEBA, etc.), is the development of mobile pyrolysis plants, mounted on tracks. These plants are aimed at the production of pyrolytic fuels from biomass harvested by smallholders. This could also allow the creation of service companies, which could contribute to a rapid expansion of this activity.
Concerning the state-of-the-art and the progress of pyrolysis technology, we can confirm that:
This old technology disappeared by and large during the last 40 years. Up to now only a low level of activity in Europe (seven years).
However, the general situation in Europe is now improving solely as a consequence of the RSD programme initiated and managed by DG XII of the Commission. Several experimental activities have been carried out over the past seven years or are now under implementation in several countries (F, I, FRG, B, E, GR) . Large industrial organisations (mainly German, i.e. Mannesmann, Preussag, VEBA, Bayer) are interested.
In some cases European industry has offered a full guarantee of this technology. As a consequence, it was possible to perform on a realistic basis an initial techno-economic evaluation for the bio-energy sector of the LEBEN-Projects (see Figure 2).
It is important to note that advanced pyrolysis technologies also exist outside Europe and in particular in Canada and the USA. These, however, are only at a laboratory stage. European advanced commercial technology can be made available in five to ten years through a continuous R&D effort focussed on conversion efficiency, quality of products and reduction of specific investment.
The forecast on conversion investment costs evolution, is as follows:
at present: 7 US$/barrel OE in 1993: 4 US$/barrel OE around 2000: 2 US$/barrel OE
For comparison, specific investment costs for oil exploration (1987)
North Sea: 8-10 Ş/barrel Middle East: 2 Ş/barrel

5 o $/BARREL OF OIL EQUIVALENT
- $5 (Social and s a l a r i e s )
- $1.8 (Exchange)
I I I
+ $8/bar re l for dssulphur isa t ion to
ECU/t (dm)
F ig . 2.

13
An interesting development concerns pyrolytic oil upgrading, through the adoption of ZSM-5zeolite conversion directly on pyrolysis vapours. A 20% yield of gasoline on wood looks feasible. Assuming a cost for biomass of 35 ECU/t, the gasoline could be produced at a cost of around 175 ECU/t (actual market price: 150 ECU/t), if the phenolic fraction was to be recovered (to obtain phenolic raw material for phenolformaldehyde type of resin) and sold at 335 ECU/t (actual cost 670 ECU/t).
Small capacity decentralised gasoline production plants could result from such activity and would be of great interest especially for remote areas in developing countries.
CONCLUSIONS
Biomass for energy has great potential in the Community (around 300 Mt/year); thus biofuel production should be seriously considered a significant element in the Community's energy mix.
Large-scale exploitation of biomass presents the following benefits :
(1) potential energy contribution: around 10% of primary energy needs; (2) contribution to industrial needs: possibility of covering 100% of the
EC deficit, around the year 2000; (3) social impact: 600 000 new jobs in the Community for the bio-energy
sector alone; (4) large exploitation of biomass could constitute an important instrument
for rural development (5) improvement of environment and quality of life; (6) stimulation of industrial competitiveness in this new sector of
activity;
Multi-sectoral integration of large market activities improves the economic results of a comprehensive massive exploitation of biomass.
Integrated projects on a regional level appear to be the correct dimension for an optimal exploitation of biomass.
As far as the bio-energy by-sector is concerned, the production of synthetic liquid fuels (pyrolysis), as a substitute for oil, for refineries and for general thermal application (heat and electricity production) looks very promising and better suited in the short and medium term in the frame of agro-energy-industrial projects (LEBEN).
By means of integration of markets, progress of technology and development of crops, production of this type of synthetic fuel will, in a relatively short term, become competitive in southern Europe and in five to ten years also in northern Europe.

14
PROGRESS IN PYROLYSIS AND GASIFICATION OF BIOHASS: AN OVERVIEW OF RESEARCH IN THE UNITED STATES
J. DIEBOLD and D. STEVENS Solar Energy Research Institute
Golden, CO 80401
Summary
The United States Department of Energy (U.S. DOE) is conducting research to produce liquid transportation fuels from biomass and municipal waste. Research in the thermochemical conversion area includes production of both methanol and gasoline. Methanol is produced by gasifying the biomass feedstock to produce a medium-energy synthesis gas, which then can be cleaned, conditioned, and catalytically converted to methanol. Gasoline is produced by first generating biocrude oils through high or low pressure pyrolytic processes. Catalytic upgrading of the intermediate, biocrude oil product yields hydrocarbon products, which can be used directly as gasoline or as octane enhancers depending upon the catalytic process employed. A summary of the thermochemical program within the U.S. DOE is presented.
1. INTRODUCTION Until the discovery of large quantities of low cost petroleum and
natural gas in the early 1900's, wood supplied a significant proportion of the energy requirements for heat and power. Combustion of wood in various sized boilers produced steam to heat buildings, power industrial machinery, and even power transportation vehicles such as ships, trains, and farm machinery.
Today, it is estimated that biomass provides about 3 quadrillion BTU's of energy per year in the United States. This corresponds with about 4% of the annual U.S. energy demand. This energy comes primarily from the combustion of wood and other forms of biomass to provide steam and process heat. This contribution is very significant in relation to other energy resources. Nuclear energy generation, for example, provides about 4 to 5 % of the nation's energy supply. With proper resource management and the development of efficient conversion processes, it has been estimated that biomass resources can provide an even greater fraction of this nation's energy supply. (1)
Liquid fuels derived from biomass are expected to contribute significantly to this energy potential. As an abundant, renewable, domestic energy resource, biomass can help the United States reduce its dependence on imported oil. Biomass is the only renewable energy source capable of supplying liquid transportation fuels. Thermochemical conversion processes offer efficient methods for converting biomass to liquid hydrocarbon fuels through a variety of processes. At present, thermochemical research sponsored by the U.S. DOE is focussed on the production of methanol or gasoline. Thermal conversion processes are well suited to the conversion of wood and crop residues, which account

15
for the vast majority of the biomass resources in the United States. These processes can convert all of the organic materials in the feedstocks to liquid fuel products, with water and carbon dioxide as byproducts. In addition, the thermochemical processes and their products are relatively insensitive to variations in the feedstock.
Harvested biomass is typically a solid material having a low energy density, as well as a tendency to biodegrade during storage. With the advent of petroleum fuels, society has become very dependent upon fuels which can be conveniently stored in a form having a very high energy density and which can be transported and metered into a combustor as a fluid. For example, the energy contained in a volume of aromatic gasoline is equivalent to that in 4 volumes of solid softwood or 8 volumes of sawdust. Relative to coal, biomass is extremely easy to liquefy or gasify due to its high reactivity. This characteristic makes biomass a prime candidate for the production of liquids or synthesis gases, which may be upgraded to transportation fuels. The diversity of biomass suggests that fairly small conversion plants would be very desirable, which can be quickly developed and constructed. Thus, the conversion of biomass to liquid transportation fuels would favorably impact local economies by the creation of employment opportunities in rural areas.
Although the extent of the world's petroleum resources have been traditionally underestimated, it is generally agreed that petroleum is a finite resource and the only real question is when, not if, will alternate sources of liquid transportation fuels be needed. However, most of the world is dependent upon oil fields which are remote from the consumer and upon long shipping routes and/or pipelines which will prove to be very difficult to protect. Consequently, the next oil shortage will most likely be caused by political decisions or by accidents, rather than to depletion of the world's petroleum resources. Biomass resources provide a secure fuel source, which is immune to these types of disruptions.
Biomass also provides a method to help ameliorate carbon dioxide emissions to the atmosphere. Recent concerns about the rising levels of carbon dioxide in the atmosphere, implicate the widespread combustion of fossil fuels. Since the use of biomass as a fuel involves the recycling of atmospheric carbon rather than the conversion of fossilized carbon to atmospheric carbon, future methods to reduce carbon dioxide emissions should include the increased use of biomass as a source of fuel. This implies the need for the development of technology related to the conversion of biomass to more conventional forms of fuel.
The U.S. Department of Energy is sponsoring research on the production of fuels through its Biomass and Municipal Waste Technology Division. This research includes the development of energy crops and their conversion to fuels using biological or thermochemical processes. The biological processes typically produce plant oils, methane, or ethanol. The thermochemical processes produce: a) methanol from synthesis gases having a medium energy content; or b) biocrude liquids for subsequent refining to a hydrocarbon gasoline, via catalytic cracking or hydrogénation. The potential impact of this research would be a seven-fold increase in the contribution of biomass to the energy consumed in the United States from the current level of about 3% to a little over 20%. (1) For this large a contribution to the energy supply, successful research must be completed in biomass production, as well as in the conversion processes. Actual deployment of the several thousand biomass

16
refineries would require a major industrial investment, which will not take place until the economics are made more feasible through research.
This paper is an overview of research and development sponsored by the U.S. DOE in the technology area of the pyrolysis and gasification of biomass and municipal-refuse-derived fuel (RDF). This research includes the upgrading of these intermediate liquids an/or gases to methanol or gasoline. The reader is referred to the proceedings of four recent major symposia (1-4) for additional detail in specific areas discussed in this paper.
2. CONVERSION OF BIOMASS TO METHANOL The conversion of biomass to methanol first involves the
gasification of the biomass to a gas which is primarily composed of hydrogen and carbon monoxide, with very little inert gases such as nitrogen or methane, and virtually no sulfur compounds nor tars. Once these synthesis gases have been generated in the proper proportions, the technology to convert them to methanol is commercially available. Therefore, the development of the conversion of syngases to methanol has been left to industry. The gasification of biomass to produce a clean synthesis fuel gas has been studied extensively over the last eight years with the successful development of four process development units (PDU's) involving quite different reactor designs. Two of these used partial combustion with oxygen to directly supply the heat needed for gasification including: a downdraft fixed bed (SERI/Syngas) and a single fluidized bed (IGT). The other two used heat which was transferred indirectly to the gasification zone from separate combustion processes, including: a dual-fluidized bed (Battelle-Columbus) and a single fluidized bed (University of Missouri-Rolla). To better understand pyrolysis and gasification, DOE is continuing research into the mechanisms involved and the catalytic removal of residual tars. DOE is completing limited work on gasification development.
Gasification research Research into the actual mechanisms involved during the gasification of biomass and pelletized RDF is being carried out at SERI and also at the University of Washington. This research will be valuable to the design and operation of fixed-bed or slow-moving-bed gasifiers. In the SERI research, a single cylindrical particle of biomass or pelleted RDF is instrumented and placed in a preheated reactor, which is swept by a flow of a mixture of nitrogen and oxygen. The pellet is heated from all sides. The temperatures of the surface and the center of the particle, the weight, and the energy flux are all measured in real time during the pyrolysis and/or flaming combustion of the single particle. A very thin, visually transparent coating of gold on the glass reactor wall acts as an infrared radiation shield to reduce heat losses to a low level, while allowing the pellet decomposition to be visually observed and photographed. Empirical equations have been derived which predict the time of pyrolysis (or flaming combustion), the heat required for pyrolysis, and the ash content as a function of temperature, initial moisture content, oxygen in the carrier gas, and relative amounts of plastics, metals, and newspaper in RDF pellets. A mathematical model based on scientific principles was shown to predict the temperatures and weight loss of the pellet quite well. (5-7)
In complimentary research at the University of Washington, a single pellet of biomass or RDF is radiantly heated from only one surface to result in a one-dimensional heat flux. The surface and several internal temperatures of the pellet are monitored during the test, and the

17
pyrolysïs products collected. Empirical equations have been derived showing the interactions of the process variables for both softwood pellets of varying density and for RDF pellets. Mathematical models, based on scientific principles, are in progress involving heat transfer considerations and global chemical kinetics. (8,9)
Gasification development U.S. DOE is completing research on one of the promising pyrolysis reactor designs, a fluidized bed. A major technical area, which has been addressed with the past gasification research is how to transfer the necessary heat into the bed that is required to pyrolyze the biomass. In addition to oxygen-blown gasifiers, DOE has examined indirect methods of providing heat. Indirect heating of the fluidized bed through heat-exchange surfaces immersed in the bed has the potential of operational simplicity and possibly a smaller reactor size.
Research into the heat transfer between heat-exchange tubes immersed in a fluidized-bed gasifier at the University of Missouri-Rolla showed that the factor limiting the heat transfer to the bed was the heat transfer between the inside of the tube and the hot combustion gases. Recent research by MTCI, Inc. has been directed toward the improvement of this limiting heat transfer. Rather than merely increasing the hot gas velocities to increase the heat transfer rates (which requires more input of mechanical energy to the blower on the combustor) , MTCI replaced the conventional pressurized combustor with a pulsed combustor. The pulsed combustor concept is over 50 years old, but some of the benefits are only now being realized. The pulsed combustor consists of a combustion chamber fitted with a long exhaust pipe which is sized to produce a resonating, traveling pressure wave. Valves open when the combustor pressure is low to allow a fresh charge of fuel and air to enter without the need for a mechanical blower. In the MTCI design, the mechanical valves are replaced by aerodynamically sized inlet and outlet ports. In the resonance tube, the combustion gases experience very high velocities because the forward and backward velocities of the resonating gases are superimposed upon the net gas velocity. This resonating gas flow resulted in an increase in transfer of heat to a fluidized-bed gasifier by a multiple of between four and five times greater. Data in the literature suggest that an order of magnitude increase in heat transfer over that attained with conventional turbulent flow can be attained through the use of resonating gaseous heat transfer. (10) With the use of calcium carbonate as the fluidized-bed material, relatively high hydrogen and low tar yields were obtained, apparently due to catalytic effects of the calcium. With funding from the Office of Industrial Programs in DOE, this system has also been used to gasify black liquor from the Kraft pulping process in steam. This resulted in gases containing very high levels of hydrogen and carbon dioxide and low levels of tars and carbon monoxide, apparently due to catalytic effects of the sodium present in the black liquor and in the sodium carbonate bed material. (11)
Catalytic reduction of tars in gasification The use of synthesis gases in conventional methanol synthesis systems requires that the levels of pyrolytic tars be very low. Research at the Pacific Northwest Laboratory (PNL) has investigated the use of catalysts to reduce tars. A primary fluidized gasifier was operated, followed by a secondary fluidized bed in which the catalysts were placed. The catalysts appear to function by reacting with the tars to form coke and then to promote the oxidation of that coke to form gases. Inputs to this secondary bed

18
were steam-gasification vapor products and enough air to prevent catalyst deactivation by oxidizing the coke deposits as they formed. The oxidation very specifically gasified the coke on the catalysts to result in greatly increased the gas yields. Even with 0.4 g of air added to the secondary reactor per g of wood feed, the nitrogen composition in the gaseous products did not change significantly from the original value in the dirty gases. The remaining hydrocarbons in the gases were primarily volatile aromatic compounds, which were not be expected to be troublesome for most applications. (12) Using the MB/MS, SERI will soon be initiating a study to screen different catalysts for the reduction of residual pyrolytic tars.
3. CONVERSION OF BIOMASS TO GASOLINE The U.S. DOE is also sponsoring research to convert biomass to
gasoline hydrocarbons. This research is focussed on first converting the biomass to a biocrude oil or vapor and then upgrading the intermediate to gasoline. Gasoline is completely compatible with the existing distribution and vehicle systems; it naturally produces low aldehyde emissions; and it has a greater energy content. With the modern computer-controlled automobile engines coupled with catalytic mufflers, hydrocarbon gasolines would be expected to produce very low levels of pollution in the form of carbon monoxide, aldehydes, and unburned hydrocarbons.
These considerations have led to the development of processes to convert biomass to gasoline. Three process routes have been investigated to make gasoline intermediates from biomass: a) liquefaction in a pressurized solvent to produce a biocrude oil having a low oxygen content; b) liquefaction of biomass at low pressures with slow pyrolysis; and c) fast pyrolysis of biomass at low pressure to form a biocrude oil having a high oxygen content. Two upgrading processes have been investigated: a) catalytic deoxygenation of the biocrude oils with pressurized hydrogen; and b) cracking and deoxygenating the biocrude vapors with zeolite catalysts at atmospheric pressures.
Liquefaction of biomass in pressurized solvents The liquefaction of biomass in pressurized solvents was demonstrated at Albany, OR in the late 1970's. This process was operated at 20.8 MPa pressure, 20 minutes residence time, and with a sodium carbonate catalyst. Since the early 1980's, research in this area was shifted to the University of Arizona and has focussed on improving the solids content of the slurry of biomass solids fed into the high pressure reactor. Higher contents of biomass in the feed, allow a smaller, more economical reactor vessel to be used for a given throughput. Mixtures containing as much as 60% wood flour in product oil have been pumped into pressurized containers, using a modified extruder originally designed to extrude plastics. With this technique, early liquefaction experiments at the University of Arizona were conducted at 375 to 400 C, 5.5 to 21 MPa pressure, 40% wood flour in Albany oil with a residence time of between one and four hours, and both with and without carbon monoxide and sodium bicarbonate catalyst. Recent experimentation was directed toward the recycling of the product oil containing approximately 40% fresh wood flour, along with water and carbon monoxide to result in a carrier oil composed primarily of material made at the University of Arizona. The fluid product distilled from the carrier oil had a heating value of 37 MJ/kg (16,000 BTU/lb), a residual oxygen content of 7 to 10%. The oil yield was close to that theoretically attainable. (13)

19
Liquefaction of biomass at low pressures with slow pyrolvsis The product slates from the slow heating of loose mixtures of RDF materials at atmospheric pressures in a retort swept with helium has been researched at Argonne National Laboratory. Final temperatures of 475 C were used. Sample sizes were varied between 1 and 50 g. The yields of tar and solid residue from newsprint were reported to be 25 to 30% and 21 to 26% respectively at heating rates of 5 C/m. The effect of heating rates between 5 and 30 C/m were found to be negligible. The influence of polyethylene in the feed was seen only in the tars collected after the first of a series of condensers, as deduced through the use of IR spectral analysis. The presence of polyethylene in a kraft paper feed decreased the low viscosity of the tars by a third. (14) The yields of condensates are consistent with those reported for RDF components previously reported by the New York University with similar sample sizes and slow heating rates of between 12 and 136 C/m. (15) It appears that slow heating rates of around 136 C/m and slower in a retort produce lower yields of organic condensates from RDF compared to other processes which use faster heating rates.
Liquefaction of biomass at low pressure with fast pyrolvsis Fast pyrolysis processes can be used for biomass liquefaction at atmospheric pressures, vapor residence times of less than a second, and intermediate temperatures of around 500 C. The fast pyrolysis condensates have about the same oxygen content and energy per unit weight as the feedstock. However, when produced with only the water of pyrolysis present, they are very fluid at room temperature and have a specific gravity of about 1.25. Due its relatively low projected cost, fast pyrolysis is currently thought by the International Energy Agency's Liquefaction Activity to be advantageous depending upon the end use of the product. (16)
Basic research into the pyrolysis of biomass and RDF has been recently studied at the Solar Energy Research Institute (SERI) using the molecular beam mass spectrometer (MB/MS) to study the pyrolysis vapors and gases in real time as they evolve from the pyrolyzing particles. Over 50 different samples of biomass and RDF were pyrolyzed and analyzed by the MB/MS scanning over the mass ranges of 10 to 250. Multivariate analysis of this very extensive data set was used to determine that there were 13 factors that explained over 90% of the variance in the data. Interpretation of the data resulted in the identification of six major chemical compound classes to explain the 13 factors. To determine the effect of process variables, a statistically designed set of experiments was conducted to look at the effects on the six compound classes identified in the feedstock screening tests. Empirical equations were fitted to the data and used to generate parametric plots showing the effect of different variables. In addition, a set of 50 different pyrolysis oils, most of which were part of the IEA set of pyrolysis and gasification oils, were analyzed on the MB/MS. Multivariate analysis was also used to reduce this data set to a similar set of six major chemical compound classes. An empirical equation was fit to the data which predicted the size of the distillable fraction based on the relative amounts of the various compound classes. (17,18)
Applied research in fast pyrolysis at the Georgia Tech Research Institute has used an entrained-flow reactor. In this system, powdered wood (0.30 to 0.42 mm) is entrained in a straight tube by a flow of stoichiometrically combusted flue gases. The heat for pyrolysis is supplied by these carrier gases. If the carrier gases are too hot,

20
significant losses from the first-formed vapors take place to result in higher overall gas yields. Consequently, fairly large amounts of tempered carrier gases at 745 C were used at a carrier-gas-to-biomass weight ratio of about 8 to supply the heat of pyrolysis to maximize the yield of pyrolysis oils. The diameter of the entrained-flow reactor is currently 15 cm and the length is 4.4 m, which results in a residence time of one to two seconds. This residence time is a compromise between the length of time needed to pyrolyze the size of particles fed on a once-through basis and the need to minimize the time which the pyrolysis vapors spend in the reactor. Feeding rates are typically about 15 kg/hr and have resulted in reported yields of 58% organic condensates (moisture free) and 12% char (maf feed) with a total mass closure of 101% (including the large amount of carrier gases). The pyrolyzate is recovered along with the water formed in the combustion used to directly heat the carrier gases, as well as any water formed during pyrolysis, or which was present as moisture in the feed, resulting in condensates containing about half water. (19,20)
A different type of entrained-flow reactor has been developed at SERI specifically for the fast pyrolysis of biomass. In this reactor, the feed is entrained at very high velocities (calculated to be about 400 m/s) in a tube having an inside diameter of 1 cm into a vortex reactor having a diameter of 13 cm and a length of 0.69 m. The feed particles are forced to slide on the hot cylindrical wall in a helical path as they pass through the reactor. The sliding contact of the particles on the wall results in very high heat transfer to the particle so that ablative pyrolysis is thought to take place. Partially pyrolyzed particles exit the reactor tangentially, are mixed with fresh feed, and are recycled back to the carrier gas ejector, where they are re-accelerated by the supersonic carrier gas. The recycle loop decouples the residence times of the solids and the vapors, which allows the vortex reactor to be insensitive to the particle size of the feed. The small amount of char which is formed is also recycled until it is attrited to a fine powder (-50 micrometer). (21) A mathematical model based on first principles suggests that the typical 2-mm thick feed particle will make about 30 passes during a total residence time of 1 to 2 seconds through the reactor before it is completely pyrolyzed. Operation of this small reactor is typically at 13 to 20 kg/hr of dry sawdust (-3 mm) with a carrier-gas-to-biomass weight ratio of 1 to 1.5. Yields on a dry feed basis have been 67% condensates (55% moisture-free organic liquids), 13% char, 14% net pyrolysis gases, and 12% water of pyrolysis for a mass closure of 94% of the feed (a 98% mass closure, if the nitrogen carrier gas is included in the calculations) . (22) This reactor has recently been modified to allow it to pyrolyze RDF, which contains tramp metals and other inert solids. Preliminary operation with RDF indicates that the plastic derived condensates interact with the lignocellulosic derived condensates to form an asphalt appearing material. (23,24) The asphalt nature of these condensates contrasts with the low viscosity reported as a result of slow pyrolysis. (17)
An alternate approach to effect the sliding contact between a hot surface and the biomass to be rapidly pyrolyzed has been under development at the Colorado School of Mines (CSM). The CSM reactor uses two specially grooved disks made of copper, which are stacked one on top of the other. The biomass or RDF is centrally fed between the heated disks. As the bottom disk is rotated at 4 to 80 rpm, the feed particles make their way to the circumference of the disks in a spiral path and are allowed to fall to a cooler zone. The vapors and gases pass between the

21
heated disks and also exit at the circumference of the disks and pass out of the reactor to the condensers. The disks had a diameter of 6.4 cm. Feeding rates were 13 to 210 g/h. Maximum reported liquid yields from sawdust were 54% (including moisture) at disk temperatures of 600 C, a flow of nitrogen purge gas of 0.5 g N2 per g of sawdust, and a feeding rate of 13 g/h. (25,26) If the developmental challenges can be met, the CSM pyrolysis reactor offers a greater decoupling of the purge or carrier gas flow rates than is possible with other fast pyrolysis reactors, e.g. entrained flow, vortex, or fluidized bed reactors.
Characterization of organic condensates formed by thermochemical processes The pyrolysis and gasification processes mentioned above produce condensates which vary in their characteristics depending upon the nature of the process. In particular, time spent at high temperatures increases gas formation, but also changes the nature of the surviving organic condensates. A recent study by PNL of condensates made with short residence times at temperatures from 450 to 900 C revealed that the primary pyrolysis condensates which form at 450-500 C do not contain polycyclic aromatic hydrocarbons (PAH's) and are not carcinogenic to mice. However, as the pyrolysis temperatures are raised, the amount of PAH's increases along with the carcinogenicity to mice. In fact, the tars formed above 800 C appeared to be slightly more carcinogenic to the mice than was the reference material, benzo(a)pyrene. (27)
The nature of the oxygenated oils and tars formed by the various pyrolysis processes is such that they are not equivalent to a petroleum crude oil and they require different refining techniques to be developed. The oils formed in pressurized solvents have tended to have a high phenolic content, whereas, the oils formed by fast pyrolysis have a very high oxygen content. These biocrude oils could be used as boiler or turbine fuels without refining. However, neither of the biocrude oils can distilled into usable gasoline or diesel fractions, rather both of them must be deoxygenated to be converted hydrocarbons and usable transportation fuels. Two such deoxygenation processes have been under development: a) hydrogénation with cobalt-molybdenum catalysts at high pressures, and b) cracking with zeolite catalysts with no added hydrogen at atmospheric pressures.
Upgrading of pyrolysis products by hydrogénation The upgrading of pyrolysis oils, formed at both high and low pressures, through catalytic hydrogénation has been under investigation at PNL. Early work used oils formed at high pressures in the Albany (PERC) process with a sulfided cobalt-molybdenum catalyst at 13.8 MPa hydrogen pressure, 350 to 450 C, and a liquid hourly space velocity (LHSV) of 0.1 volume of oil per volume of catalyst per hour to produce a hydrocarbon product boiling primarily in the gasoline range. (28) More recently, this work has been expanded to oils formed by the fast pyrolysis of biomass at atmospheric pressures made at Georgia Tech, SERI, as well as those from Canada and peat derived oils from Finland. The fast pyrolysis oils must first be stabilized by deoxygenation in the presence of hydrogen at low temperatures to avoid polymerizing the feedstock. After stabilization, the fast-pyrolysis oils are partially deoxygenated and have many properties in common with the oils formed at high pressure. The stabilized pyrolysis oils can then be hydrodeoxygenated at higher temperatures to form a gasoline product similar to that made from the high pressure oils. Originally, this two-step process was conducted separately at a pressure of 13.8 MPa hydrogen, temperatures of 275 and

22
350 C, and liquid hourly space velocities of 0.6 and 0.1 in the first and second reactors, respectively. Yields on a dry wood basis from pyrolysis oils made by Georgia Tech were about 25 wt% of hydrocarbon product containing only 1.3 wt% oxygen. More recently, this two-step process was combined into a single, non-isothermal reactor at 13.8 MPa, which maintained the inlet temperature at around 260 C and the outlet temperature at around 375 C. With a LHSV of 0.1 volume of oil per volume of catalyst per hour, the fast, pyrolysis oil from SERI was converted into a product which contained only 1.3 wt% oxygen, a hydrogen-to-carbon ratio of 1.68, and 73 vol% boiling in the gasoline range (C5 to 225 C). Liquid product yields of about 20 wt% were reported based on the dry wood feed. The cause for the difference in yields has not been addressed. (29) Octane tests of the gasoline products, combined from several preliminary hydrogénation experiments of the Albany oils, indicated a research octane of 77. (29,30) Based on the fairly high hydrogen-to-carbon ratio and the medium octane value, it is highly probable that the octane level could be raised by optimizing the process or by removing some of the hydrogen in a subsequent reforming step to increase the aromatic content, as is commonly done in a typical petroleum refinery, i.e. with "Platforming".
Zeolite upgrading of pyrolysis oil vapors at low pressures An alternate method to the high pressure upgrading of the pyrolysis oil is with the use of zeolite catalysts at atmospheric pressures without added hydrogen. These catalysts were introduced to the petroleum industry about 30 years ago and have found widespread use in the catalytic cracking of heavy petroleum fractions to increase the yield of gasoline. More recently, Mobil has developed zeolite catalysts which have pore sizes small enough to allow production of toluene and similar gasoline compounds. This H-ZSM-5 catalyst is now in commercial use in New Zealand to convert methanol to gasoline. Although the molecular weight of the condensed pyrolysis oils has been found to be typically between 500 and 2000, researchers at SERI determined that the pyrolysis vapors had much lower molecular weights of typically less than 200. The molecular size and shape of most of the identified compounds were small enough to enter the H-ZSM-5 pore. Preliminary experiments with H-ZSM-5 catalyst furnished by Mobil confirmed that a small amount of catalyst was sufficient to change the product slate from the oxygenated pyrolysis vapors to a highly aromatic gasoline product. This process has been studied extensively using the MB/MS which allowed the product slate to be examined in real time as the catalyst aged in a fixed bed of 10 g of H-ZSM-5 catalyst. A process variable study was conducted to identify optimum operating conditions, which were found to be quite different from those used to convert methanol to gasoline. Empirical equations were fit to the data to result in parametric contour plots illustrating the effects of the process variables on the yields. (31,32,33) Concurrently, research has been conducted with a larger fixed-bed reactor having 100 g of catalyst and fed a small slipstream of fresh pyrolysis vapors directly from the vortex reactor at SERI. (34,35) Hydrocarbon product yields, including olefins, have been around 15 wt% of the dry feed, when using steam as the carrier gas and a weight hourly space velocity of between 1 and 4 g wood per g catalyst per hour at 525 C. Recent research has been directed toward recovery of the gaseous olefins as part of the gasoline product. The gasoline produced consists primarily of alkylated benzenes, e.g. toluene, xylenes, ethyl benzene, and isopropyl benzene (eumene). These compounds are present in today's commercial gasolines and their effect is to raise the octane value. (36,37) Similar impure

23
streams (mixed xylenes) in a petroleum refinery are typically worth one and a half times the value of unleaded gasoline. The upgrading of fast-pyrolysis oils with zeolites has also been studied at Georgia Tech Research Institute, but detailed results have not yet been released. (19,20)
Technoeconomic assessments (38) Technoeconomic assessments were recently made by Science Applications, Inc. (SAI) to compare two methods of upgrading fast pyrolysis products to gasoline. Each of the two processes were evaluated at their present stage of development, as well as, at the expected future stage of mature development. In the process being studied at PNL, the condensed fast pyrolysis oils, made by an entrained-flow process similar to that of Georgia Tech, are fed to a high pressure reactor where in the present case the oils are first deoxygenated in the presence of hydrogen at relatively low temperatures to stabilize the oils to prevent polymerization reactions. The stabilized oil is then treated with hydrogen at higher temperatures to complete the hydrodeoxygenation process. Hydrogen is made by steam reforming and shifting of the byproduct gases. This hydrogénation process is relatively complex and capital intensive, but is said to have the potential of very high hydrocarbon yields of about 32% by weight of the dry feed. Liquid hourly space velocities of 1.0 and 0.1 were assumed for the "current" two-step hydrogénation process. A LHSV of 0.5 was assumed for the "future" non-isothermal hydrogénation process in a single reactor.
The other process, which is being studied by SERI, is based on the immediate conversion of the hot organic vapors, formed by fast pyrolysis in the vortex reactor, before they are allowed to cool and condense. In this process at atmospheric pressures, the hot carrier steam, pyrolysis gases, and vapors are passed through a catalytic cracking reactor loaded with H-ZSM-5 catalyst. With a gas residence time of a few seconds, an equivalent LHSV of 2.5 was assumed to give complete conversion of the oxygenated organic pyrolysis vapors to the hydrocarbon products, water, and oxides of carbon. The process stream was then condensed and the olefinic gases adsorbed from the off-gases and recycled. This process is thought to have lower potential gasoline yields of 24% of the dry biomass feed, but is less capital intensive per unit weight of biomass fed than high pressure hydrogénation.
The present technoeconomics of both processes were examined in light of the state-of-the-art as it existed at the time of the beginning of the study: a 15 wt% yield for hydrogénation; and 10% yields for zeolite cracking. The annual cost assumed for the capital was equivalent to a simple amortization over 20 years of the total capital investment at 15% annual interest without any subsidies or tax considerations. The assumed cost of the feedstock was $27.50 per dry tonne. As expected, the research on these processes is only partially completed and established research goals have not yet been reached. For this reason, projected production costs in a plant fed 908 tonnes of dry biomass per day were not economically attractive at $2.64 per gallon via hydrogénation and $2.92 per gallon via catalytic cracking. Incorporation of more recent yields and process information would result in lower predicted production costs for both processes.
If future research and development are successful, the economics improve substantially. The future hydrogénation process is projected to require a capital investment of $74.4 M to result in a cost of $0.96 per gallon of medium octane gasoline. The future zeolite cracking process

24
was projected to require a capital investment of $47.8 M to result in a cost of $1.05 per gallon of high octane gasoline blending stock. These projected production costs are very sensitive to the relative success of research and development to achieve the gasoline yields assumed. Within the accuracy of the study, the projected production costs are equivalent for the two different gasolines.
4. COMMERCIALIZATION OF DOE DEVELOPED TECHNOLOGY There are several private firms which are involved with the
commercialization of biomass gasification in the United States. Most of this work has involved air blown, low-energy gasifiers for fuel purposes. Approximately 20 gasifiers are currently in operation including, for example, a unit which provides boiler fuel for a hospital in Rome, Georgia. Other units are being planned. Technology based on a downdraft gasifier developed by the DOE program at SERI is being used by Syngas, Inc. to develop gasification systems to produce electricity in New York. Another gasifier, based on the vortex reactor is being planned by Pyrotech to operate in California.
There currently are no firms with commercial gasifiers to produce a medium-energy gas, which could be utilized as a synthesis gas. To encourage such a development and to facilitate the commercialization of a process to make methanol from biomass, the U.S. DOE has recently announced that it is looking for an industrial partner to share the cost of the scale-up of a gasifier system to produce a medium-energy gas. This would be at the 15 to 25 tonne per day size. A formal request for proposal is soon to be issued to those U.S. companies indicating an interest to share at least 50% of the cost of the project. The U.S. DOE funding for this project is projected to be about $5M over the life of the development. (42)
5. FUTURE TRENDS IN THERMOCHEMICAL CONVERSION IN THE U.S. The DOE funding available for thermochemical conversion has steadily
declined in recent years. Cutbacks in the research program during the current fiscal year resulted in the cancellation of all thermochemical programs, except those at SERI. The research and development of the zeolite upgrading of the pyrolysis oils to improve yields at lower cost and the pyrolysis of RDF in the vortex reactor to optimize organic vapor yields will continue at SERI, but at reduced levels compared to prior years.
This funding decline is at a time when the world is looking for methods to reduce the emission of carbon dioxide through the curtailment of the use of fossil fuels. Since the use of biomass as a fuel recycles carbon dioxide over the course of a few years, it will not be a net contributor to the "greenhouse" effect. In future years, the funding for alternate sources of methanol and gasoline may be more optimistic.
6. CONCLUSIONS The U.S. DOE has successfully transferred the technology for
gasification to produce low-energy gases and commercialization of this technology is underway. Gasification of biomass to produce medium-energy gases for methanol synthesis is still being researched at a low level of effort and a pilot plant demonstration project is planned, in which the U.S. DOE will share the cost of development with a private firm. Most thermochemical research in the last few years has been directed toward the production of liquid hydrocarbon fuels, i.e. gasoline. Using pyrolysis oils formed during the fast pyrolysis of biomass, two promising

25
processes to make gasoline have been researched concurrently. This work will also examine the use of RDF feedstocks to produce gasoline.
REFERENCES
(1) SCHIEFELBEIN, G., ed. (1988) Thermochemical Conversion Propram Annual Meeting. June 21-22. 1988. Solar Energy Research Institute, Golden, CO 80228, SERI/CP 231-3355, DE 88001187.
(2) BRIDGWATER, A.V., and KUESTER, J.L., eds. (1988) Research in Thermochemical Biomass Conversion. Proceedings of the IEA Conference held in Phoenix, AZ in May 2-6, 1988, Elsevier Applied Science, London and New York.
(3) SOLTES, E.J. and MILNE, T.A., eds. (1988) Pvrolvsis oils from Biomass. Producing. Analyzing, and Upgrading. Proceedings of the symposium held in Denver, CO on April 5-10, 1987, ACS Symposium Series 376, American Chemical Society, Washington, D.C.
(4) KLASS, D., ed. (1989) Proceedings of the symposium on "Energy from Biomass and Solid Wastes XII", held in New Orleans, LA February 13-17, 1989, Institute of Gas Technology (in press).
(5) LEVIE, B.E. (1988) Pvrolvsis of Refuse Derived Fuel Pellets. Ph.D thesis, University of Colorado.
(6) LEVIE, B.E. and DIEBOLD, J.P. (1988) "Pyrolysis of single pellets of refuse derived fuel" in Ref. (1), pp. 259-269.
(7) LEVIE, B.E., DIEBOLD, J.P., and WEST, R. (1988) "Pyrolysis of single pellets of refuse derived fuel" in Ref. (2), pp 312-327.
(8) LAI, W. and KRIEGER-BROCKETT, B.B. (1988) "Single Particle RDF Pyrolysis Properties and Products-Initial Results" in Ref. (1), pp.269-280.
(9) KRIEGER-BROCKETT, B.B. and GLAISTER, D.S. (1988) "Wood Devolatilization - sensitivity to feed properties and process variables" in Ref.(2), pp. 127-142.
(10) DURAI-SWANY, K. , WARREN, D.W., and CHE, S.C. (1988) "Pulse-enhanced biomass gasifier for production of medium-BTU gas" in Ref. (1), pp. 77-86.
(11) DURAI-SWAMY, K. , WARREN, D.W., AGHAMOHAMMADI, B. , and MANSOUR, M.N. (1989) "Pulse-assisted gasification of black liquor and organic wastes for medium-BTU gas" in Ref. (4).
(12) MUDGE, L.K., GERBER, M.A., and WILCOX, W.A. (1988) "Improved gasification by catalytic destruction of tars in biomass-derived gases" in Ref. (1), pp. 87-100.
(13) WHITE, D.H. and WOLF, D. (1988) "Advances in direct biomass liquefaction by the extruder-feeder method" in Ref. (1), pp. 57-66 and Ref. (2), pp. 827-853.
(14) HELDT, J.E. and MALLYA, N. (1988) "Experiments on basic mechanisms of pyrolysis" in Ref. (1), pp. 221-236.
(15) KAISER, E.R. and FRIEDMAN, S.B. (1968) "The pyrolysis of refuse components", Combustion. May, pp. 31-36.
(16) ELLIOTT, D.C, BAKER, E.G., BECKMAN, D., SOLANTAUSTA, Y., TULENHEIMO, V., OSTMAN, Α., GEVERT, S.B., HORNELL, C , and KJELLSTROM, B. (1989) "A technical and economic analysis of direct biomass liquefaction" in Ref. (4).
(17) EVANS, R.J., MILNE, T.A., and FILLEY, J. (1988) "Mass spectrometric studies of municipal solid waste pyrolysis" in Ref. (1), pp.209-219.

26
(18) EVANS, R.J., and MILNE, T.A. (1988) "Mass spectromctric studies of the relationship of pyrolysis oil composition to formation mechanisms and feedstock composition" in Ref. (2), pp. 264-279.
(19) KOVAC, R. (1988) "Production and upgrading of biomass pyrolysis oils" in Ref. (1), pp. 5-20.
(20) KOVAC, R. (1989) "Liquid biofuels production by an entrained pyrolysis process" in Ref. (4).
(21) DIEBOLD, J.P. and SCAHILL, J.W. (1988) "Production of primary oils in a vortex reactor" in Ref. (3), pp. 31-40.
(22) DIEBOLD, J.P. and POWER, A.J. (1988) "Engineering aspects of the vortex reactor to produce primary pyrolysis oil vapors for use in resins and adhesives" in Ref. (2), pp. 609-628.
(23) SCAHILL, J.W. and DIEBOLD, J.P. (1988) "Adaptation of the SERI vortex reactor for RDF pyrolysis" in Ref. (1), pp. 237-246.
(24) DIEBOLD, J.P., EVANS, R.J., and SCAHILL, J.W. (1989) "Fast pyrolysis of RDF to produce fuel oils, char, and a metal-rich byproduct" in Ref. (4).
(25) REED, T.B. (1988) "Principles and operation of a novel "pyrolysis mill" in Ref. (1), pp.247-258.
(26) REED, T.B. (1988) "Contact pyrolysis in a pyrolysis mill" in Ref. (2), pp. 192-202.
(27) ELLIOTT, D.C. (1988) "Relation of reaction time and temperature to chemical composition of pyrolysis oils" in Ref. (3), pp. 55-65.
(28) BAKER, E.G. and ELLIOTT, D.C. (1988) "Catalytic hydrogénation of biomass-derived oils" in Ref. (3), pp. 228-240.
(29) BAKER, E.G. and ELLIOTT, D.C. (1988) "Catalytic upgrading of biomass pyrolysis oils" in Ref. (2), pp. 883-895.
(30) ELLIOTT, D.C. and BAKER, E.G. (1988) "Catalytic hydrotreating processes for upgrading biocrude oils" in Ref. (1), pp. 45-56.
(31) EVANS, R.J. and MILNE, T.A. (1988) "Molecular-beam mass-spectrometric studies of wood vapor and model compounds over an H-ZSM-5 catalyst" in Ref. (3), pp. 311-327.
(32) EVANS, R.J., FILLEY, J., and MILNE, T.A. (1988) "Molecular beam mass spectrometric studies of H-ZSM-5 activity during wood pyrolysis product conversion" in Ref. (1), pp. 33-43.
(33) MILNE, T.A., EVANS, R.J., and FILLEY, J. (1988) "Molecular beam mass spectrometric studies of H-ZSM-5 activity during wood pyrolysis product conversion" in Ref. (3), pp. 910-926.
(34) DIEBOLD, J.P. and SCAHILL, J.W. (1988) "Biomass to gasoline: upgrading pyrolysis vapors to aromatic gasoline with zeolite catalysts at atmospheric pressure" in Ref. (3), pp. 264-276.
(35) DIEBOLD, J.P. and SCAHILL, J.W. (1988) "Conversion of wood to aromatic gasoline with zeolite catalysts", Energy Propress. Vol. 8, No.l, 59-65.
(36) SCAHILL, J.W., DIEBOLD, J.P., and POWER, A.J. (1988) "Engineering aspects of upgrading pyrolysis oil using zeolites" in Ref.(2), pp. 927-940.
(37) DIEBOLD, J.P. and SCAHILL, J.W. (1988) "Zeolite catalysts for producing hydrocarbon fuels from biomass" in Ref. (1), pp. 21-32.
(38) WAN, E.I. and FRASER, M.D. (1988) "Economic assessment of producing liquid transportation fuels from biomass" in Ref. (1) , pp. 111-121.
(39) GRABOSKI, M.S. (1989) Private communication with J.P. Diebold on April 21.
(40) AYRES, W. (1987) "Commercial application of wood derived oil", Enerpv Propress. Vol.7, No. 2, 77-79.

27
(41) AYRES, W. (1989) Private communication with J.P. Diebold on April 21.
(42) ANON,(1989) Commerce Business Daily. Jan. 6.

28
OVERVIEW OF THERMOCHEMICAL CONVERSION OF BIOMASS IN CANADA
R.D. HAYES Bioenergy Research and Development
Technology Branch Energy, Mines and Resources Canada
Ottawa, Canada K1A 0E4
Summa ry
Thermochemical conversion of biomass in Canada has undergone a considerable transformation in approach and expectations over the last ten years. A decade ago, the promise of early commercialization quickly evaporated and was replaced by a more realistic assessment that the potential would likely not materialize except over the longer term. More recently this outlook has been modified slightly in the wake of unanticipated research progress and the surfacing of some special near term market applications.
1. INTRODUCTION AND HISTORY
During the late 1970's and early 80's there was a short-lived perception of panic in energy security accompanied by exaggerated claims of technical readiness of thermochemical conversion applications. Several premature commercial demonstrations in the U.S.A. of pyrolysis and gasification failed technically and financially. Governments and investors became understandably cautious therefore when further approached by technology vendors. American communities that once considered gasification and pyrolysis of municipal solid wastes as environmentally attractive alternatives to relieve pressure on decreased availability of landfill sites for urban refuse, turned their attention to mass incineration and refuse derived fuel (RDF) combustion applications. Though environmentally less attractive in terms of emissions, combustion offered less technical and financial risk than did the more exotic thermochemical conversion options.
Canada was somewhat behind the U.S.A., having felt less apparent impact from the energy supply disruptions of 1973 and 1979. In those days also there was less pressure to find alternatives to landfilling of wastes than there was in many U.S. cities, or, as there is in some Canadian cities today. Canadian thermochemical conversion research, then in its infancy, therefore focussed its effort on the long term. It assumed a lengthy timeframe to mature innovative process developments. In theory, this new breed of technologies would presumably become technically ready for the 1990's or 21 s t century, in a world eager to embrace bioenergy, particularly from wood, as a sustainable and environmentally attractive energy resource.

29
The ultimate vision of some researchers was to refine biomass in a manner analogous to petroleum that would eventually lead to alternative transportation fuels, preferably high quality gasoline, plus a host of high value byproduct specialty chemicals. As the bubble burst on high world oil prices, the sense of urgency to accelerate RSD diminished. Admittedly, severe federal budget reductions for energy RSD also had its predictable effect on the "sense of urgency". Except for one major project, gasification research in Canada all but disappeared by 1984. The pyrolysis/liquefaction research community collectively agreed to back off from upgrading studies, and concentrated instead on perfecting their processes for primary oil production.
Over the past decade, the science has become better understood and technical advances have actually surpassed earlier expectations. Some laboratories have begun to re-activate their upgrading research. Scientists and engineers have broadened the scope of experimental conditions and gained a much deeper understanding of the chemical transformations of complex feedstocks to multiple products.
A new vision of conversion and product opportunities has begun to reshape our view of the timeframe for commercialization. The long term perspective remains paramount, and perhaps even more so since the global call for environmentally sustainable economic development by the Brundtland Commission report, "Our Common Future"l·
In addition to the long-term vision, technical progress in the identification of several high valued specialty chemicale in wood pyrolysis oil may be the market hook (albeit a small market) required to help finance the high costs of staged scale-up from laboratory, process development unit, pilot plant, small commercial demonstration and eventually to full-scale commercial reality.
Several other near term opportunities have also surfaced. Environmental pressures have influenced the trial experimentation of biomass pyrolysis liquefaction technologies on non-woody feedstocks such as used tires, petroleum sludges, sewage sludge, and, once again, municipal solid wastes. Impending environmental regulatory controls on waste incineration and increasing costs for landfilling may create a climate more ideal for fully contained emission-free thermochemical processes.
Researchers, industry and government have all become more creative in building a diversified, yet comprehensive, portfolio of thermochemical conversion opportunities for the commercial world. Though nothing thoroughly new has emerged, the past ten years have seen an evolution from a subject of technical curiosity to one of concerted effort toward cost effective products for the marketplace, some sooner than others.
2. PROSPECTIVE APPLICATIONS OF TECHNOLOGY (PAT)
A broader diversified portfolio, as referred to above, presents a major challenge to the RSD planner. Added to this challenge is the ever-present restraints in government RSD funding. This challenge has necessitated a major overhaul in organizing RSD priorities in the context of assessing a balance in both near and long term commercialization and environmental opportunities and needs.

30
The commercialization process for a new technology as defined within the Energy Diversity Division of Energy Mines & Resources Canada includes everything germane including opportunity identification, the actual research, development, technical evaluation, and demonstration as well as the determination of current and projected economic feasibilities, current and future market, expected timeframes for commercialization, standards development, and many other factors affecting market introduction and penetration. Once these elements have been assessed and analyzed for the various feedstock/conversion' technology pathways, priorities could be established with respect to activities that government can, should, or must, do and those that government should not, or cannot afford, to support.
The first step in the process is to organize the four basic elements of a technology by resource (eg. biomass, wastes, peat etc.), process(es), product(s), and market application. The particular feedstock combined with specific conversion processes and a particular market application of products result in what we define as a Prospective Application of Technology or PAT. The picture can become rather complex especially when considering combinations of thermochemical, biochemical and fractionation technologies that might be otherwise overlooked during conventional analysis. In order to illustrate many of the various combinations pictorially, we have constructed a 'PAT map.'
Figure 1 is a simplified version of a 'PAT map' for thermochemical technologies. One can visualize perhaps one hundred or more probable combinations of resources, processes, products, and market applications. The product possibilities alone include a vast array of oils, gases, chars, carbon black, sugars, fermentation products, high value specialty chemicals, medium value commodity chemicals, olefins, gasoline and gasoline components, diesel fuel, and other materials. Figure 2 is a more general PAT map encompasing most bioenergy technologies.
FIGURE I
Prospective Applications of Technology < Thermochemical)
Thermochemical Processes;
pyrolysis hydrotreating catalytic solvent pressurized partial oxidation oxygen gasification soivoiyss hydrothermolysis product separation etc.
oletins " * diesel /urt
Thermochemie al and/or
biochemical Processes
CMA BTX acetic acid
IzBüfffla.·,!!
" Τ
χ y Ian s
ition
Τ lionln
attractive*/ votatile«
TvTvl Various combinations of thermochemical, biochemical, ex tract lon/fract Jona tion and derlvatlzatlon processes
phenolic denVaoVes synthesis gas gaso/ine components aahe¡áve¡¡lb¡nders lm, mol. m lignns
carbon 'llavoursl aromas black charcoal

31
Figure I I
PROSPECTIVE APPLICATIONS OF TECHNOLOGY
| MATERIALS PREPARATION]
J P R E T R E A T M E N T FRACTIONATION |
THERMOCHEMICAL COMBUSTION GASIFICATION PYROLYSIS CATALYSIS HYDROTHERMOLYSIS SOLVOLVSIS. ETC...
EXTRACTIVES
VARIOUS COMBINATIONS OF BIOCHEMICAL THERMOCHEMICAL. FRACTIONATION, EXTRACTION
AND DERIVATIZATION PROCESSES
| THERMOCHEM-UPGRAOING |
FUEL OIL GAS CMA ACETIC ACID RESINS FIBERS SUGAR ENZYMES OLEFINS BTX METHANE CARBON BLACK GASOLINE ADDITIVES OLIGOMERS
FURFURAL DIESEL FUEL SYNTHESIS GAS HEAT ALCOHOLS ADHESIVES'BINDERS PHENOLIC DERIVATIVES CHARCOAL GASOLINE FLAVOURS/AROMAS LOW MOL WT. LIGNINS
MANY FERMENTATION PRODUCTS ETC.
RESIDENTIAL. COMMERCIAL INSTITUTIONAL AND INDUSTRIAL BLOGS.. PULP AND PAPER , AGRICULTURE & FISHERIES. UTILITIES. FOOD AND BEVERAGE. FOREST PRODUCTS INDUSTRY. NORTHERN COMMUNITIES
TRANSPORTATION. INTERNATIONAL AID AND EXPORT. REFINERIES HEAVY INDUSTRY. BRICK. LIME AND CEMENT ETC.
Our view of the world of thermochemical conversion of biomass is no longer limited to merely the pyrolysis or gasification of wood to heating oil and gas. Thermochemical conversion is not treated in isolation from biochemical, fractionation or derivatization processes. Even considering a single process technology, the variation in process conditions, catalysts, reactants, or feedstocks can impact the range and output of possible product combinations.
The second step will be to select a handful of promising PAT■s, and perform an analysis of expected technical progress, economic feasibility, market penetration, and timeframe for commercialization. In theory, this should provide government (and industry) with a rationale for desirable government (and industry) activities in those PAT's such as research, development, demonstration, detailed engineering, market studies, economic and financial analysis, information dissemination, standards formulation, etc.
Some of the so-called 'thermochemical PAT■s' that are not typically represented at thermochemical conversion of biomass conferences, but which are included or planned for in Energy, Mines and Resources' Bioenergy R&D program planning include the following:
thermochemical treatment of plant oils (oilseed, tall oil) to produce high cetane diesel fuel. biochemical and thermochemical derived lignin that is thermally or thermochemically treated with or without catalysts to produce gasoline and other chemicals.

32
c product derivation of thermochemical and biochemical lignins (eg. resins, adhesives, cement dispersants).
° thermochemical treatment of the steam fractionated cellulose component of biomass.
° direct steam (thermal) production of adhesives with a fibre byproduct suitable for molding, pelletizing, or thermochemical or biochemical processing.
0 calcium magnesium acetate (CMA) production a3 a road de-icer from aqueous effluents (ie. dilute acetic acid) of pyrolysis and biochemical processes.
° investigation of wheat chaff as a thermo- or biochemical conversion feedstock.
° methanation of pyrolysis aqueous effluents. ° separation of short and long fibers of hardwoods where pulp would be
processed from the long fibers and fuels processed biochemically or thermochemically from the lower value short fibers.
° source separated and/or prescreened, or raw, municipal wastes that undergo thermochemical or biochemical conversion with or without pretreatment/fractionation.
3. EMR's BIOENERRY DEVELOPMENT PROGRAM
Table 1 shows the historical trend in Canadian federal government spending on bioenergy RSD including biomass production and conversion. Table 2 is a breakdown of thermochemical R&D expenditures relative to other conversion technology areas.
3.1 BRIEF REVIEW OF NON-THERMOC.HEMICAL R&D
The following is a cursory view of Canadian R&D activities in technical areas other than thermochemical.
The combustion area's main focus of effort is the evaluation and improvement of industrial and residential combusto! performance and emissions. Also included has been support to Environment Canada's National Incinerator Test and Evaluation Program (NITEP) and the development of standards for performance, safety and emissions of residential wood burning appliances.
The biomass handling/preparation area includes research in biomass materials handling, storage, drying, beneficiation, and preparation. This research is seen to provide necessary infrastructural support for all biomass conversion processes, but more importantly, to improve biomass fuel quality with objectives to improve biomass combustion performance and to reduce emissions. Examples of developments include a bin, silo and non-consolidating feeder system that regulates the controlled and non-disrupted feeding of hog fuel and chips to burners, a device to exclude frozen lumps of hog fuel and chips to burners, a continuous moisture sensor to provide improved combustion control of biomass of varying water content, and a low energy grinder to powder wood suitable for suspension-fired boilers.
The biochemical area receives the largest allocation of funds, in part due to increasing industrial leverage provided by cost-shared R&D. Much of the impetus for this area comes from mid-term (1990's) competitive potential to enter the transportation fuel market with biomass derived

33
Table I
25
20
ÏS-
IO-
S/MILUONS
Federal Bioenergy R&D Funding r™™^ , m ^
82
BPsTţjs man
i
83 84 85 86 87 88 89 90
T a b l e I I
1988/89 Bioenergy R&D Expenditure Forecast
35.4%
Combustion Biomass Thermo Bio
Handling/ chemical chemical Preparation
Infor
mation Tech/
Transfer

34
ethanol based blends and oxygenates and the longer term potential of neat ethanol fuel along with value added byproducts. The program is divided into a number of areas including enzymatic and acid hydrolysis of lignocellulosics, steam and extrusion pretreatment and biomass fractionation, novel fermentation engineering, biotechnology of enzymes and microorganisms, anaerobic digestion, characterization and derivatization of lignin, and the development of non-ethanol byproduct streams, among others. The rate of achievements in this area over the past four years rivals that in the thermochemical area. The lure and promise of biotechnology and the recent accelerated progress are likely the major factors in attracting industry participation in an area that was viewed by our program as very long term as of only five or six years ago.
3.2 CANADA'S MAINSTREAM BIOMASS THERMOCHEMICAL CONVERSION RSD
An approach has evolved in Canada whereby, unlike earlier efforts, a whole range of products and reactants are now considered. Products include gases and oils of varying quality, sugar solutions in high yield, chemicals (olefins, phenolics, as well as high value specialty chemicals), gasoline or diesel fuel, and higher value carbon products. Diversification of reactants include whole biomass, fractionated biomass components, peat, and muncipal solid waste, including source-separated wastes such as used tires. Another area of research is the treatment, and especially, the conversion, of waste aqueous effluents from thermochemical conversions into value-added co-products. Left unprocessed, these effluents would otherwise incur a cost for waste treatment.
3.2.1 GASIFICATION
Canadian developments in biomass gasification for the production of low, medium and high energy gases have enjoyed world technical acclaim over the past decade, but there has been a disappointing uptake by Canadian industry. The cautious attitude of our industry appears to have been due largely to premature and unsuccessful attempts in the US to commercialize immature technologies during the latter '70s and early '80s. A possible revival of interest in gasification technology applications appears to be for replacing industrial boilers that use oil and for municipal energy from waste projects. The work to complete ongoing projects will continue, although at a reduced level, and any impetus for an expanded government program will likely wait for stronger industry confidence, interest, and cost share. Application of Canadian technology in developing countries may hold some promise.
Large scale (10 tonnes/hour) gasification technology, developed through the Biosyn project offers a world leading technology in pressurized, air or oxygen fed, fluidized-bed gasification that can be adapted to the production of synthesis gas for methanol production or low energy gas for other applications such as space heating, gas turbines or industrial diesel engine operation.
Another portion of the gasification research funds is directed to the development of a small scale gasifier that is designed to use wet wood and produce a clean gas. The Heuristic gasifier is a two-stage reactor that, in principle, operates similarly to a downdraft design. The first stage is an updraft gasifier where producer gas and condensible vapours (tars)

35
are routed through a hot char bed below. The tars are to be cracked in the second stage. The project now underway is to modify the gasifier, optimize the operating parameters, and enhance cracking in the second stage.
3.2.2 LIOUEFAC.TION/PYROLYSIS
In 1985, research in the direct liquefaction of biomass was expected to wind down because the rapid commercialization that was expected to occur in the early '8 0s did not materialize. The plan was to conclude as much of the work as possible, revert to lower cost basic research, and pursue a strategy of chemicals from liquefaction. Then in 1986 there appeared to be a turnaround in commercial interest. Unprecedented technical achievements started to renew aspirations and industry interest in exploiting selected market niches for pyrolysis products. The following is a brief review of current projects.
McGill University
Over the past several years researchers W.J.M. Douglas and D.G. Cooper at McGill University have been studying an interesting thermochemical approach to wood liquefaction using aqueous hydrogen iodide at fairly mild conditions of pressure and temperature (125°C) . Still far from certain is the exact nature of the liquid products and the techno-economic practicality of hydrogen iodide recovery and recycle. On the positive side, in addition to the low severity conditions of reaction, the process removes about 80% of the oxygen in the wood, and the char yield is low.
University of Toronto
D.G.B. Boocock and co-workers at the University of Toronto have undertaken the investigation of steam pyrolysis or hydrothermolysis of wood. Based on their earlier work, they recently designed and constructed a laboratory scale cascade autoclave which can accommodate up to 100 g of wood chips or 170 g of a single larger piece (3.8 cm reactor I.D., 600ml volume). It is rated at 24.1 MPa (3500 psi.) at 350°C allowing for 7.6 MPa (1100 psi) gas overpressure above the vapour pressure of water at that temperature. Replicated results indicate that oil yield increases with increased chip size. Dry product oil yields are high (up to 50%) with no solids contamination, and the oil is easily separable from the aqueous phase. Coupled with an upgrading process, this technology may someday lend itself well to commercialization.
In addition to their process development work, Professor Boocock's group has contributed greatly to the basic understanding of biomass liquefaction, especially through their scanning electron microscopic studies. The group at the University of Toronto is also performing upgrading studies through the catalytic hydrotreatment of model compounds. Of potential interest too is their discovery in 1984 that a particular clone of hybrid poplar yielded 6% phenol. This discovery raises the prospect of matching processes more closely with specific feedstocks in the interest of optimizing a particular product slate. Efforts are now beginning to genetically engineer trees that are designed for specific process/product applications.

36
Université de Sherbrooke
E. Chornet, R.P. Overend and co-workers at the Université de Sherbrooke have been working on a liquefaction process in pressurized solvent for some years. Their approach involves an overall integration of biomass pretreatment, fractionation, acid processing, thermochemical and biochemical treatment.
Funding for research at Sherbrooke in the areas of peat and wood conversion is provided by a number of sources, including Energy, Mines & Resources. The program there involves a comprehensive variety of fundamental studies, product engineering and technology development at laboratory and pilot plant levels. Examples of approaches under investigation include a thermo-mechanical-chemical treatment to fractionate and liquefy biomass including steam treatment, thermocatalytic conversion of fractionated products such as lignin to monomers, ethylene glycol solvolysis/liquefaction, acid/thermal/shear treatment of biomass and cellulose, and a variety of biochemical investigations in combination with thermochemical techniques. In association with other laboratories, the chemical engineering laboratory of Professor Chornet is especially well equipped for analytical characterization of products derived from their various reaction systems.
University of Waterloo
D.S. Scott, J. Piskorz, D. Radlein and co-workers at the University of Waterloo are well known for their fluidized bed flash pyrolysis development, also known as the WFPP (Waterloo Fast Pyrolysis Process). A number of companies and the European Economic Community have indicated their interest in collaborating with the Waterloo group. The WFPP actually includes five process options as follows :
1. Direct thermal processing at 450-550°C, atmospheric pressure, and about 500 ms vapour residence time. They report high liquid yield (80% including water, based on input wood) that is a suitable fuel for conventional boilers.
2. By varying the process conditions and adopting a mild sulphuric acid pretreatment of wood followed by fluidized bed thermopyrolysis, the WFPP produces a high yield and concentration of anhydro-sugars rather than oil products. Their reproducible yields of sugars from pure cellulose are about 80% of theoretical in a concentrated form. One can easily speculate whether this development could challenge some of the equally exciting biochemical conversion methode of converting lignocellulosics to fermentable sugars.
3. Waterloo's hydrogasification work has been technically highly successful, resulting in 75% conversion of wood carbon to methane via pyrolysis over a nickel-alumina catalyst with hydrogen at about 550°C and 440 ms vapour residence time.
4. Under current investigation is a fourth process option of producing polyolefins from wood in a catalyzed reaction. Apart from the use of catalysts, the process equipment and operating conditions are very similar for all of the above process options.

37
5. The Waterloo group is also performing upgrading research by catalyticly hydrotreating pyrolysis oil fractions, especially lignin, in a continuous pressurized reactor.
University of Western Ontario
Although not currently funded by Energy, Mines & Resources, M. Bergougnou, R. Graham and co-workers have developed an Ultra-Rapid Pyrolysis or Ultrapyrolysis process at the University of Western Ontario. Although there are similarities in this work and the research at the University of Waterloo, there are important differences. Whereas the Waterloo process utilizes fluidized-bed heat transfer. Professor Bergougnou employees a very rapid (30ms) mixing and heat transfer in a vortical contactor or vortactor followed by a plug-flow entrained-bed down flow reactor (50-900ms) and quenching (30ms) with cryogenic nitrogen in a cryovortactor. Dissimilar to the Waterloo process are the process conditions (650-1000°C, 50-900 ms residence time), and the main product at these temperatures is gas rather than liquid. Since pyrolytic fuel gas production has not been of high priority in Canada's bioenergy RSD strategy, the current objective, in collaboration with Ensyn Engineering, (see below) is the production of chemicals and, in particular olefins.
It is interesting to note here that the Universities of Waterloo and Western Ontario conducted an extensive data comparison from each of their reactor systems. Using selected data from both groups at around 500 ms residence time, liquid and gas production data were plotted vs temperature. The temperature ranges were as follows: Waterloo at 400°-750°C, and Western Ontario at 650°-900°C. With combined data for each of the gas yield vs temperature and liquid yield vs temperature respectively there was remarkable agreement of data in overlapping regions. A simple first order kinetic model is able to describe the oil yield over the temperature range of both experiments.
Ensyn Engineering
Ensyn Engineering is a recently formed company whose principal investigator, R. Graham has scaled up the University of Western Ontario Ultrapyrolysis reactor by a factor of 20 to a 5-10 kg/hr capacity RTP (Rapid Thermal Processor). The reactor is designed to accept any carbonaceous feed (solid, liquid, or gas) by injecting it into a turbulent cloud of hot solids. The mixed feed plus solids is carried through a tubular transport reactor to an inerţial separator where vapour products are removed. This project is co-sponsored by the private sector.
Work planned for 1989 involves thermal cracking and catalytic treatment of pyrolysis oils and various lignin preparations including steam exploded biomass derived lignin.
Laval University
Multi-stage vacuum pyrolysis was developed by C. Roy and co-workers, initially at the Université de Sherbrooke and, currently, at the Université Laval. The technology consists of a 40 kg/hr multiple hearth vacuum pyrolysis process development unit located near the Université Laval. There is aleo an industrial cost-shared pilot demonstration of a

38
single-stage process at St-Amable, Quebec. The unit has a capacity of 200 kg/hr and is designed for used tires.
Although the multiple hearth concept suffers from low heat transfer relative to other pyrolysis processes, and, at first glance, is capital cost intensive, it has a number of redeeming features that show commercial promise as follows:
1. A high yield of pyrolysis oil (50% based on wood). 2. The production of co-product carboxylic acids and high value
chemicals. 3. Reactive charcoal at 25% of input wood. 4. The aqueous phase is recovered separately as vapour or liquid as an
integral part of the process leaving a low water content water-free pyrolysis oil ready for upgrading.
5. The multiple hearth performs a product fractionation function that could reduce extraction costs of high value chemicals.
Centralized Analysis
One final work of interest in Canada is a Centralized Analysis project at B.C. Research. In 1984, a trial project was set-up whereby different Canadian bio-oils could be compared in a standard manner. The project embraced a three-pronged approach. Under the guidance of J. Howard and J. McKinley, B.C. Research performed and/or coordinated the centralized analyses of optimized oils produced by each researcher. Individual researchers also did some of their own analyses to obtain immediate experimental feedback.
The second prong of the approach was that all researchers were provided with a standard wood sample, Populus deltoides, by Forintek Canada Corp. The idea was that when each process development became somewhat optimized, the researcher would submit oil from the standard wood sample to the centralized analysis team.
The third prong was a computer communications network link called CoSy, through the University of Guelph, to provide fast communication of analytical data. It was also used to encourage multilateral and bilateral collaboration and problem solving.
The centralized analysis project has entered Phase II. Learning from the successes and pitfalls of the first two and one half years, the scope of this project has changed somewhat. Phase II has two major tasks.
Task 1 is a set of analytical techniques along the same lines as the initial project except the methods that were considered to be less interesting to the entire group are not included. The basic analyses of task 1 include the following:
- Elemental Analysis Water Content Density Carboxylic Acids Gas Liquid Chromatography Carbon-13 NMR Gel Permeation Chromatography

39
Task 2 consists largely of special analytical projects to meet the special needs of individual researchers, Energy, Mines and Resources Bioenergy Development Program or groups of thermochemical conversion researchers that may arise over the next two years.
4. INTERNATIONAL VIEW - IEA
Under the International Energy Agency, eleven countries signed a three year Bioenergy Agreement on Cooperative R&D, effective January 1, 1986 to December 31, 1988. This Agreement has since been extended by another three years. Canada, USA, Sweden and Finland agreed to collaborate on a project entitled Direct Biomass Liquefaction (DBL). A Working Group of engineers and other specialists are preparing a detailed technical-economic assessment (TEA) at commercial size scale-up and operation of the most promising high and low pressure pyrolysis/liquefaction processes. Both primary oil production and upgrading are considered in the TEA. The upgrading work on Canadian atmospheric and vacuum pyrolysis oils has been conducted by D. Elliot at Battelle Pacific Northwest Laboratories. Yields of products in the gasoline boiling range have so far reached 35% of primary oil by hydrotreating. The Working Group is attempting two types of analyses, one based on current state of the art and the second based on projected improvements and developments in the technologies.
ACKNOWLEDGEMENTS
The author wishes to acknowledge the various researchers in North America without whose ideas, open discussion, and dedication to research in thermochemical conversion of biomass, this paper would not have been possible. Each has contributed to this paper, either directly or indirectly. The following list is not complete, but includes the principal investigators in Canadian laboratories and other notable international collaborators. In alphabetical order, many thanks to Narendra Bakhshi, Dave Beckman, Dave Boocock, Jean Bouchard, Maurice Bergougnou, Michel Bertrand, Esteban Chornet, Maurice Charron, Helena Chum, Jim Diebold, Allan Dolenko, Murray Douglas, Guy Drouin, Dick Eager, Doug Elliot, Bob Graham, guy Gravel, Michele Heitz, Ed Hogan, John Howard, Serge Kaliaguine, Björn Kjellstrom, Bill Lowe, Tom Milne, Hugh Menard, Jim McKinley, Ralph Overend, Hooshang Pakdel, Jim Pepper, Jan Piskorz, Desmond Radlein, Tom Reed, Joe Robert, Christian Roy, Tom Tidwell, Don Scott, and their many colleagues, staff, and students.
REFERENCES
This paper contains technical contributions including progress reports from all the Canadian and other workers mentioned here. The reader is referred to these individuals or their published papers as primary references.
1. Brundtland, Gro Harlem. 1987 "Our Common Future". World Commission on Environment and Development.

40
RAPPORTEURS REPORT ON SESSION I OPENING SESSION AND COUNTRY OVERVIEWS
C.H. NELS Federal Office for the Environment, Berlin
The following report covers the first session of the conference. The papers delivered during this session dealt mainly with :
Activities and results of the demonstration program of the Commission in the areas of gasification and pyrolysis as well as European activities planned for the future. International research and development in this area presented by the United States, Canada and Japan.
Because of time constraints follow-up discussion was limited to the EC papers. Topics included details for the forthcoming planned EC research program, how technologies already proven in laboratory and bench scale could be transferred/tested in the field, how many test plants currently exist in the EC and when results will be ready for general use. Special interest was given to problems of developing countries. Issues included how demands of developing countries could be taken into consideration in future RSD programs and whether direct or indirect help could be given by the EC to strengthen their own efforts in this area. Environmentally related issues were also discussed dealing with critical emissions caused by extended agricultural production of biomass, its thermal or chemical conversion and finally the question of its potential for use as a fuel or raw material.
The papers as well as the subsequent discussion led to the following conclusions :
Any future research commitment should have long term orientation. Since funding resources are limited research should be directed toward those types of biomass "crops" which are most widely applicable. Alongside biomass research there should always be an environmental impact assessment. Because of the global nature of the problem there are numerous biomass research programs underway internationally. To avoid duplication and enhance information transfer more conferences such as this one should be organized to coordinate international efforts. Efforts in international cooperation should begin at the earliest stage while planning research programs such as that currently underway in the EC. Such exchanges also provide the missing link to developing countries giving them vital information; the importance of such exchanges should be recognized.

SESSION II
PRE-TREATMENT AND CHARACTERIZATION


43
PRETREATMENT AND CHARACTERIZATION OF FEEDSTOCKS
C Ρ MITCHELL and A V BRIDGWATER Forestry Department Chemical Engineering Department Aberdeen University Aston University Aberdeen AB9 2UD Birmingham B4 7ET
UK UK
SUMMARY Feedstocks generally considered for thermochemical conversion are wood and wood waste, energy crops, agricultural waste and refuse. The main technical criteria for suitability for thermochemical processing is low moisture content, and ash content and characteristics. The main economic criteria are cost which includes production, collection and transport, and quantity which includes availability. Wood will be available from processing residues, unmanaged woodlands, conventional forestry and short rotation forest biomass plantations. Harvesting, processing and transport systems for delivering wood fuel either already exist or are under development. Delivered fuel costs will be between £28 - £36/dt from conventional forestry and in the order of £41/dt from short rotation.
Agricultural wastes, such as cereal straw, can be delivered to the end-user for £22/dryt. Domestic and commercial refuse is widely available and can attract disposal credits of £10 - £20/wet t which has a significant effect on the conversion economics.
1 INTRODUCTION Feedstocks generally considered for thermochemical conversion are wood and wood
waste, energy crops such as short rotation forestry and sweet sorghum, agricultural waste and refuse (1). The main technical criteria for suitability for thermochemical processing are low moisture content, and ash content and characteristics. The main economic criteria are cost which includes production, collection and transport, and quantity which includes availability. There is also the question of competing uses such as pulp and board manufacture, combustion, recycling or material recovery rather than energy recovery.
2 FEEDSTOCKS 2.1 Wood
Wood will be potentially available as a feedstock from residues generated within the wood processing industries, presently unmanaged woodland, conventional forestry and from plantations specifically grown to produce raw material for the biomass industries. Processing Residues
Residues are generated within the forest products industry itself. Processing residues are produced primarily in sawmills; these consist of slabs and blocks of wood, chips and sawdust. However, with the increasing demand for raw material in the pulp and board mills much of these residues go to these industries and are therefore unlikely to be available in large quantities to the energy industry. Unmanaged Woodland
There are several million hectares of woodland within Europe which is currently not managed and which could be brought into production for biomass. In France and Italy alone there are some 5 million hectares of overmature coppice. The maquis systems in the mediterranean region and several other categories of natural vegetation offer good prospects to be brought into management for biomass production (2). In the UK, the potential for production of fuelwood from presently unmanaged woodlands is around 0.15 million wet t/y (3).

44
Conventional Forestry Conventional forestry practice gives several opportunities for harvesting wood for fuel.
Firstly, under current harvesting practice only the merchantable stemwood is removed from the forest. The tops and branches, roots and stumps are left in the forest. The roots and stumps can be harvested but it is a costly operation and there are potentially severe environmental problems which limit their use. However, the tops and branches, so-called forest residues are available for harvest. Secondly, early thinnings can be harvested using whole tree harvesting techniques. A third opportunity lies in adapting harvesting systems to remove both the stemwood and the "energy component" at the same time. These integrated harvesting systems are attracting a great deal of interest world-wide because the overall harvesting system costs are reduced.
Modified conventional forestry management systems involve planting trees closer together initially, taking heavy early thinnings using whole tree harvesting techniques, and using integrated harvesting systems for later thinnings and clearfell.
For the UK alone by the year 2020 the total annual sustainable production of fuelwood from conventional forestry could amount to some 4.8 million wet tonnes. In the longer term modified conventional forestry could provide an additional 8 million wet tonnes per year (4). Short Rotation Forest Biomass Plantations
Woody biomass can be purpose grown for energy in short rotation coppice biomass plantations. These are intensively cultivated plantations using fast growing broadleaved trees which coppice readily and are harvested every 3 to 5 years. The main species used are from the genera Eucalyptus, Populus (Poplars) and Salix (Willows). Growth rates are high giving productivities of around 10 to 15 dry tonnes per hectare and year (dt/ha/y) in commercial operations. Coppice crops need to be grown on good fertile soils. Within Europe the agricultural surpluses problem coupled with the revision of the CAP is leading to land being set aside from agricultural production. Such land is well placed for coppice plantations. Of the 40 million hectares of land coming out of production in the CEC some 5 million hectares has been conjectured to be available for energy plantations (2). In the UK, it has been estimated that if 0.82 million hectares of land currently in agricultural production were to change to short rotation forestry then this would give an annual production of some 2.72 million wet tonnes (4).
2.1.1 Harvesting of Wood Fuel Conventional Forestry
The actual harvesting system chosen for conventional forestry is heavily dependent on the crop, species, size, terrain and the requirements of the end-user. Most current harvesting systems operating in Europe are designed to supply raw material to the pulp, board industries and sawmills. Harvesting systems can be categorised into one and two pass systems. One pass systems are applicable to whole tree operations in either integrated or whole tree comminution, and two pass systems to residue collection and comminution following conventional methods for harvesting the stem wood. The harvesting options available are shown in Figure 1.
Two pass residue harvesting systems are perhaps those that are the most appropriate with short wood harvesting systems. The residues can either be chipped in the stand with mobile machines (terrain chipping) or the residues are collected and extracted to a central point for chipping (landing chipping). Terrain chipping is appropriate to the dryer load bearing mineral soils and landing chipping to the wetter organic soils.
Yields of fuel chips from terrain chipping will reflect the predominance of pine species and are likely to be in the lower end of the range 50-100 wet tonnes per hectare (wt/ha). Production costs for terrain chipping are in the range of £10 - £15/wet L
Yields of fuel chips from landing chipping systems reflect the higher potential of spruce crops and are likely to be in the lower half of a range from 100 - 200 wet tonnes per hectare. Production costs for extraction of residues to landing are estimated to be in the region of £3.00/ wet t and chipping costs in the region of £7.00/wet t
Whole tree comminution systems are appropriate to smaller tree sizes, either in early thinnings or premature clearfell situations where the tree stem size is insufficient to warrant

45
conventional utilization or in clearance or cleaning operations in hardwoods. Chipping can take place either at landing or at the stump (terrain chipping) and the choice of systems will be a factor primarily of the suitability of the terrain and extraction distance.
In landing chipping systems the whole trees are extracted to landing. In the smaller tree sizes appropriate to whole tree chipping, the use of forwarder extraction of whole trees or tree sections, is favoured to reduce extraction costs. Whole tree chipping at landing requires the use of larger chippers with infeeds capable of handling and feeding the whole tree by grapple.
Terrain chipping can be used where the ground conditions permit. Small scale equipment such as small tractor mounted chippers can be used. A self propelled chipper and an infeed at the front fed by a grapple crane can be used in line thinning operations with the chips blown over the chipper into a chip bin at the rear.
Yields of fuel chips from whole tree comminution systems has been found to be in the region of an additional 50 -100% expressed as a percentage of stem wood. Production costs for fuel chips from whole tree comminution systems will vary on the scale of the operation, and as for integrated systems, on the tree size and the terrain. Landing chipping costs will be around £4.80/ wet t and £5.60/ wet t for terrain chipping.
In integrated harvesting systems the whole tree is extracted to landing for processing and product separation. Landing processing, involving delimbing and crosscutting, can be a manual system with the use of chain saws, or be partly or fully mechanised, the availability of equipment and landing size being the factors taken into consideration when deciding the method to be adopted. The residue material on the landing can be chipped in a continuous (hot) system closely following the processing, or, can be allowed to accumulate on the landing for chipping in a separate process (a cold system). The chipping equipment used will be primarily decided by the scale of operation, the use of hand fed tractor mounted chippers will be appropriate to only the smaller operations. Given a sufficient scale of operation, the use of trailer mounted chippers feeding with a grapple crane will give higher outputs, particularly in working a cold system where there are no limiting factors in the processing line. The chipped material can be accumulated on the landing for later loading on to road transport, or, more commonly the chips can be blown directly into containers or lorry trailers for road transport.
Yield of fuel chips from integrated harvesting systems has been found to be in the region of an additional 30% expressed as a percentage of stem wood. Production costs for fuel chips from integrated harvesting operations vary considerably with tree size, terrain and the harvesting system used. On the assumption that integrated systems can produce the traditional assortments as efficiently as comparable shortwood systems, the cost of fuel chips will reflect the "free ride" that they get to landing. The cost of production then amounts to the chipping element which can be as low as £5.10/wet tonne. Short rotation forestry
Current harvesting technology for short rotation crops is limited. The two coppice harvesters currently working are both prototypes; there are no machines commercially available. The principle on which these machines work is for the coppice to be severed by circular saw(s) and the cut coppice rods tied together in bundles before being ejected from the machine. The Loughry coppice harvester produces 300 kg (wet) bundles, the Swedish machine 2.5 wet tonne bundles. The rationale for this approach is to produce bundles which can be left in the field to dry. Coppice willows and poplars are around 55% moisture content (wet basis) at harvest. By stacking the bundles of cut material in the open or with a tarpaulin cover there will be some natural drying. Once stored for 3 months or so the moisture content will drop to around 40% and the bundles are then chipped.
2.1.2 Transport of Wood Fuel The transport system of the raw material to the conversion plant is of utmost
importance. In many respects it is no different from any other wood delivery system to the pulp and board industries which are designed to handle chips. In addition, delivery and reception systems have been developed in Sweden for handling wood fuels to the mill and district heating plants (5).
Converted pulp wagons, containers, tippers and curtainsiders can all be used for transporting wood fuel. Converted pulp wagons show the most potential for use in the present UK situation ie with a small wood fuel market and existing road transport legislation. These

46
allow self loading and unloading, a wide range of available loads and the ability to revert to conventional pulp haulage.
Container systems are used in the UK for several commercial operations. Tippers and curtainsiders showed potential for use in the future when demand for industrial fuelwood will be high allowing full utilisation of independent loaders. It is not feasible to use lorry transport of whole trees and forest residues over long distances without compaction equipment. Transport costs amount to some £0.07/wet tonne/km.
2.1.3 Storage of Wood Fuel Storage of wood fuel will be necessary to bridge the gap between supply and demand
with due allowance made for the time required for drying and pretreatment. The storage can be done in the open, under cover to protect it from rain or an enclosed space such as silos and live bottomed bins which have to be linked to the gasifier feeding system. The type of storage is strongly influenced by scale of operation and climatic factors. The most common type of wood fuel is chipped wood.
If storage is required then it has to be carefully considered. Whole tree chips, due to their high moisture content and foliage content, decompose rapidly (1 - 5% per month) during storage. Such materials also present a fire risk because of heating during decomposition. A further problem associated with the storage of wood fuels in enclosed spaces is the development of microfungi the spores of which represent a serious health hazard (6). From experimental experience, drying during storage will give a moisture change from a delivered 55% to an outgoing 40%. Costs of storage have been calculated to be of the order of £5/wet tonne (7) (Table 1).
2.1.4 Drying of Wood Fuel Flash pyrolysis requires a feedstock at <5% moisture content and gasification <15%
but there is an optimisation to be carried out between moisture content and conversion efficiency. The actual moisture content required for the conversion process will therefore vary somewhat between conversion facilities. Biomass as received will have a moisture content typically in the range 50 - 60% (wet basis).
Passive drying during summer storage can reduce this to perhaps 30%. Active silo drying can reduce the moisture content down to 12% (8). Drying can be accomplished either by very simple means such as near ambient/solar drying or by waste heat flows or by specifically designed dryers operating on solid or hydrocarbon fuels. The selection of the most appropriate system depends on the location, climate, level of sophistication required and scale of operations. Commercial dryers are available in many forms and sizes but the most common are the rotary kilns and shallow fluidized bed dryers.
2.1.5 Costs of Supply of Wood Fuel The costs of supply of wood for fuel have been examined for UK conditions (3, 9).
Short rotation coppice crops biomass can be grown on a four year cutting cycle, harvested, stored, chipped and delivered to the end-user at a cost of £40.83/ dry t (Table 1) for a farmer operated system. In such a system the farmer carries out all the operations to do with growing and tending the crop and uses contractors to carry out the harvesting and chipping operations much as he may use contractors for other specialist farming operations.
Calculation of the costs of harvesting wood for fuel from conventional forestry is not so simple as there are are conventional forest products produced along with the fuelwood assortment. The costs of supply of wood fuel from forestry and the contribution of the elements in the chain are illustrated for a number of options in Figure 1 and for the optimal systems in Table 1. Delivered costs of wood fuel at 40% moisture content range from £28.17/ dry t for integrated harvesting systems in clearfell to £36.33/ dry t for a whole tree chipping operation in an early thinning. Forest residue chips can be delivered for £34.33/ dry L

47
SHORT ROTATION FORESTRY Stumpage ( 3 . 6 0 )
SYSTEM COST
F E L L / B U N D L E
( 6 . 7 0 )
ÏXTRACT
(2 .10)
CHIP ( 2 . 6 0 ] ~*
TRANSPORT ( 4 . 7 0 ) - STORAGE
( 6 . 0 0 ) 2 4 . 5 0
R E S I D U E S Stumpage ( 1 . 0 0 )
COLLECT EXTRACT ( 3 . 3 0 )
CHIP ( 6 . 3 0 )
TRANSPORT ( 4 . 7 0 )
STORAGE ( 6 . 3 0 )
C H I P ( 1 0 . 3 0 )
TRANSPORT ( 4 . 7 0 )
STORAGE ( 6 . 4 0 )
W H O L E TREE C H I P P I N G Stumpage ( 6 . 0 0 )
F E L L / B U N C H (1 .60 )
TERRAIN \
EXTRACT ( 4 . 3 0 )
CHIP ( 6 . 6 0 )
CHIP ( 4 . 8 0 )
TRANSPORT ( 4 . 7 0 )
TRANSPORT ( 4 . 7 0 )
STORAGE ( 6 . 6 0 )
STORAGE ( 6 . 0 0 )
2 1 . 4 0
INTEGRATED ( T H I N N I N G ) Stumpage - residue» (1 .00 )
FELL (1 .20 )
E X T R A C T _ ( 3 . 3 0 )
P R O C E S S ^ ROUNDWOOD
CHIP (6 .10 )
T R A N S P O R 1 _ ( 4 . 7 0 )
STORAGE ( 6 . 3 0 )
INTEGRATED ( C L E A R F E L L ) Stumpage - realduea (1 .00 )
2 0 . 6 0
F E L L (0 .10 )
EXTRACT (1.10)
P R O C E S S ^ ROUNDWOOD
CHIP (6 .10 )
TRANSPORT. ( 4 . 7 0 )
STORAGE ( 4 . 9 0 ) 1 6 . 9 0
Figure 1 Harvesting supply options and costs (£ /w t ) .

48
Table 1
Stumpage* Felling Extraction Comminution Transport Storage
Total £/wet t £/dryt
Total Cost of Wood Fuel,
ŞRF
£/wett
3.50 6.70 2.10 2.50 4.70 5.00
24.50 40.83
* raw material cost at stump
Assumptions: 1 2. 3 4 5
Residues
£/wett
1.00
10.30 4.70 5.30
20.60 34.33
Whole tree chip £/wett
5.00 1.50
5.60 4.70 5.00
21.80 36.33
Moisture content of 55% (wet basis) Transport distance of 70 km one-•way
Integrated clearfell £/wett
1.00 0.10 1.10 5.10 4.70 4.90
16.90 28.17
Covered storage at end user, moisture content reduction to 40% Integrated systems costed on the residue component only. Particle size of chips 5-35 mm.
3 AGRICULTURAL WASTE Considerable quantities of agricultural waste, such as straw, bagasse, corn cobs, and
rice hulls, arise in agriculturally based economies where there may be no ready market for solid fuels. In many cases this material is free and may even have a credit or negative cost of up to £-20/t because of the cost of disposal if it cannot be used. Particular features of agricultural waste are that is is often already collected at one site and has often been subjected to extensive pretreatment such as size reduction and drying. There will also usually be some storage and material handling facilities. In addition such waste often creates a disposal problem with an asociated cost. For all these reasons, agricultural waste can be a very attractive feedstock for conversion and upgrading, since the costs are low or even negative, i.e. attract a disposal credit, and disposal problems can be solved. A summary of a variety of waste arisings in several countries in Europe in given in Table 2.
In the UK, about 7 million t/y of surplus straw arise for which no ready market is available, out of a total arising of about 14 million t/y. Comparable figures pertain to Europe. Prices vary by location from £5 to £50/t, but representative costs have been estimated at about £17/t on-site or £22/t delivered [10].
The wide variety of materials suitable for processing precludes consideration of each individually, and the range is indicated in Table 3.
4 REFUSE 4.1 Resources
Considerable quantities of domestic and commercial refuse are produced from any society which require disposal at a significant cost. Processes that utilise this waste, therefore, can often justify a credit from the disposal authority, as a result of saving the disposal cost. Raw waste attracts the highest credit but requires the most preparation. There is again the advantage of "free" collection as with agricultural wastes. The largest arisings of waste that also attract the greatest disposal credit often occur close to the potential market for the energy products. Estimated arisings for Europe are shown in Table 4.

49
Table 2 Estimated overall national arisings of potential energy from principal agricultural residues, 1979 (11)
(millions of GJ/year unless otherwise stated)
Belgium & Luxemburg Denmark France Italy Holland Ireland UK West Germany
Notes (a) Presented as biogas
not as gross energy, (b) Presented
available. as biogas
Cereal. Livestock Green Plant Total Maize & Rice Wastes Matter
Residues fa) ibi 27.1 27.3 12.3 66.7 98.7 29.2 6.4 134.3
556.6 171.2 59.3 787.1 233.7 81.7 37.3 352.7
16.4 45.6 17.7 79.7 21.9 38.1 3.3 63.3
191.4 99.2 20.7 311.3 327.1 129.7 40.0 496.8
1 472.9 662.0 197.0 2 291.9
available from pig, cattle and poultry by anaerobic animal bedding excluded.
Total in Mtoe
1.52 3.05
17.89 8.02 1.81 1.44 7.08
11.29 52.10
digestion,
availabe by anaerobic digestion: other conversion routes are No data are available for Greece, Spain and Portugal.
Table 3
Alfalfa straw Barley straw Coconut husk Cotton gin stalks Olive pits Rice hulls Tobacco dust
Agricultural Wastes that have been Thermochemically Processed
Almond shells Bark Coconut shells Flax Paper Safflower straw Vegetable fibre
Bagasse Brazil nut husks Coffee grounds Ground nut shells Peach pits Straw Walnut shells
Bamboo Cocoa husks Cotton gin trash Maize cobs Prune pits Sunflower husks
Table 4 Estimated Arisings of Wastes in the EEC in 1982 (12)
Component million tonnes/year
Household waste Agricultural waste Industrial waste Sewage sludge Waste from extractive industries Demolition waste and debris Consumer waste (used vehicles, tyres, etc.)
120 950 160 300 250 170 120
Some thermochemical processes have been devised that accept whole raw waste with minimum pretreatment, such as the now abandoned Purox and Andco-Torrax processes. Current thinking, however, is that some separation of inerts and pre-processing is necessary for reliability and control in handling, feeding and conversion. Pretreatment of refuse to

50
produce a material acceptable for conversion would follow conventional refuse sorting technologies, employing screening, shredding and classification. The products can range from "fluff' (loose, low density material) through "crumb" (ie partially densified material) to pellets. There are many proprietary systems on the market that have been proven for production of refuse derived fuels.
Domestic refuse In the UK, about 18 million t/y of domestic refuse are produced with a heating value of
about 9 GJ/t, and containing about 11 million t/y of combustibles, ie about 300 kg/head/y of raw refuse or 150 kg/head/y daf waste. The average cost of disposal in the UK is claimed to be about £6/t but these are believed to be very conservative with average costs of disposal nearer £12/t, over a range of £5/t to as high as £40/t (13). In Europe, disposal costs are much higher by a factor of up to 5, with typical figures at around £30/t. In some parts of the world, disposal costs can exceed £70/t raw refuse. These figures will have a significant effect on conversion economics if translated into credits. Further details of costs are given below.
Commercial refuse In the UK, arisings of commercial solid waste have been estimated as 8 million t/y (dry
basis) with a heating value of around 16 GJ/t (dry basis) ie about 130 kg/head/y. The material is generally cleaner and drier than domestic refuse with a higher proportion of packaging ie paper, plastics, wood, etc. This is mostly handled by the private sector with a paucity of data on quantity, quality and cost.
Industrial refuse In the UK, combustible waste arisings from the UK industrial sector have been
estimated at 9 million t/y (dry basis) with a heating value of 16 GJ/t (dry basis), i.e. about 150 kg/head/y. This is also mostly handled by the private sector with a paucity of data on quantity, quality and cost.
Other Wastes Sewage sludge is another resource that is being investigated for thermochemical
processing. In the UK, sludge is generated at the rate of about 25 kg dry solids/head/y. A disposal credit of up to £50/dry t is potentially available.
4.2 Refuse Processing Most refuse processing or pretreatment processes are based on refuse separation for
RDF production in which the raw material is shredded to reduce particle size, screened to separate inerts such as dust and glass, demetalled to remove cans, and classified as a refining stage, giving a wet low bulk density (fluff) RDF product, although the sequence of these steps can vary. The fluff can be dried, or pelleted then dried, and an intermediate step in the production of pellets is a partially densified and dried material known as crumb. Each of these operations has a cost associated with it which includes all capital and operating costs. This permits the total cost of production of any RDF product to be estimated as shown below. There will invariably be a residue after recovery of the fuel product which consists of nonfuel materials and inerts, and which will have a much higher bulk density than the raw refuse. This residue would normally be landfilled, and since the bulk density is so much higher, the cost of this operation would be expected to be significantly less than for raw refuse due to the reduced transport and handling costs.
4.3 Costs of Refuse Processing The total cost of the unit operations encountered in refuse processing is summarised in
Table 5. Figure 2 shows a processing flowsheet and mass balance for various levels of separation from which the cost of refuse derived fuels has been estimated in Table 6.
Refuse is normally disposed by landfill or incineration at a cost that reflects the economic, social and environmental constraints. Disposal of residue, as explained above, would be expected to be less than the disposal of raw refuse as costs are usually based on

51
volume rather than weight. It has been assumed that the disposal cost of the residue after processing is half the raw refuse disposal cost.
From the methodology employed in the example in Tables 5 and 6, a general cost model has been derived in Table 7 for the estimation of fuel costs for any raw refuse disposal cost/disposal credit. This has been applied for a range of disposal credits in Table 8 to estimate fuel costs at various levels of disposal cost/credit. This data is used in another paper at this conference to estimate fuel gas and fuel oil costs from gasification and pyrolysis processes (14).
Table 5
Basis Disposal cost
Cost of Refuse Processing Operations
Operations Shredding Demetalling Screening Air classification Disposal of residue Drying - fluff or pellets Pelleting
£10/t raw refuse assumed
£2.75/t raw refuse £2/t raw refuse
£2.75/t raw refuse £1.5/t raw refuse £2.75/t raw refuse
£3/t raw refuse £3/t raw refuse
(self financing)
(50% disposal cost of raw refuse) or £10/t product fuel or £10/t pellets
Cost of Refuse Derived Fuel Products Table 6
A Gross
The gross cost of products are as follows:
Wet "fluff' RDF (25% water)
Dry "fluff' RDF (10% water)
Pelleted RDF (10% water)
Β Nett
£9.75/t raw refuse or £24.40/t product assuming 40% yield of fuel product (with demetalling being self-financing)
£12.75/t raw refuse or £37.50/t product assuming 34% yield of fuel product (with demetalling being self-financing)
£15.75/t raw refuse or £46.30/t dried pellets
A disposal credit may be set against these costs, of, for example, £10/t raw refuse as used above. This gives the following net costs:
Wet "fluff' RDF (25% water) Dry "fluff (10% water) Pelleted RDF (10% water)
-£0.25/t raw refuse or -£0.60/t product £2.75/t raw refuse or £8.10/t product £5.75/t raw refuse or £16.90/t dried pellets.

52
Raw d.a.f. RDF ash water
refuse 0.25 te 0.05 te mme
waste, d.a.f. ash water metal, d.a.f.
0.34 te 0.07 te 0-14 te 0.05 te
Total 1.00te
I Shredding " " " £2.75 Demetalling £2.00 Screening £2.75 Air classification £1.50 Total £9.00
Metals 0.05 te
Waste 0.55 te
Credit £2.00
Disposal£0.275 D
τ Wet "fluff" RDF
d.a.f. RDF 0.25 te ash 0.05 te H20 0.10te Total 0.40 te
JL Drying £3.00
ι H20 0.06 te To atmosphere
Dry "fluff' RDF d.a.f. RDF 0.25 te ash 0.05 te H20 0.04 te Total 0.34 te^
Έ Pelletising £3.00
Σ Pelleted
d.a.f. RDF ash H20 Total
RDF 0.25 te 0.05 te 0.04 te 0.34 te
Note:
Figure 2
Raw refuse disposal cost = £ D / te Processed refuse disposal cost = £ 0.5 / te
Refuse Processing Flowsheet, Mass Balance and Build-up of Cost

53
Table 7 Product Cost as a Function of Disposal Cost Example of build-up of processing costs.
Basis Wet "fluff RDF (see Figure 2)
Assumptions Assume disposal cost = £ D / te. Assume disposal cost of residue after processing is half disposal credit value, as bulk density is much higher thus reducing transport and handling costs, = £ 0.5 D/te
Cost Model Cost of shredding, demetalling, screeening and air classification (Table 5) Cost of waste disposal, 0.55 * 0.5 D Credit from sale of metal Disposal credit
Total cost per tonne of raw refuse Cost per tonne of wet "fluff' RDF Cost per tonne of wet "fluff RDF, d.a.f. basis
Cost of RDF £/te raw refuse
(7.0-0.725D)/0.4 (7.0 - 0.725D) / 0.25
£/te product
£ +9.00
+0.275 D -2.00
-D
7.0 - 0.725 D 17.5- 1.81 D 28.0 - 2.9 D
£/te product daf basis
Wet "fluff RDF Dry "fluff RDF Pelleted RDF
7.0 - 0.725 D 10.0 - 0.725 D 13.0 - 0.725 D
17.5- 1.81 D 29.4-2.13 D 38.2-2.13 D
28.0 - 2.9 D 40.0 - 2.9 D 52.0 - 2.9 D
Table 8 Product Cost for Various Disposal Options Costs derived from cost model in Table 7
Product
Wet "fluff RDF
Dry "fluff RDF
Pelleted RDF
Disposal credit
5 10 15 20
5 10 15 20
5 10 15 20
Note: All costs and credits are
Cost per tonne raw refuse
3.38 -0.25 -3.88 -7.50
6.38 2.75
-0.88 -4.50
9.38 5.75 2.13
-1.50
expressed as £/tonne
Cost per tonne product
8.45 -0.60 -9.63
-18.70
18.75 8.10
-2.55 -13.20
27.55 16.90 6.25
-4.40
Cost per tonne d.a.f. product
13.50 -1.00
-15.50 -30.00
25.50 11.00 -3.50
-18.00
37.50 23.00
8.50 -6.00

54
CHARACTERISTICS
The basic characteristics of biomass are summarised in Tables 9 and 10. Particular features are the fairly high moisture content, low bulk density and wide particle size range.
Table 9 Typical Properties of Biomass
Moisture content (wet basis)
Mean particle size (mm)
Size range 2-150 (mm)
Bulk density (wet kg/m3)
Table 10
Feedstock
Douglas Fir Beech Douglas Fir bark Wood shavings Sawdust Sander dust Charcoal Paper Rice hulls Straw Refuse (UK)
Forest residues
30-60%
5x20
2-75
300
Analyses of
Municipal solid waste (USA)
For comparison Peat Lignite Bituminous coal
Processing residues
20-60%
5x2C
2-100
350
1
Feedstocks
C
52.3 51.6 56.2
80.3 43.4 38.5 48.1 30.7 33.5
58.4 68.4 73.1
Whole trees
40-60%
5x20
2-50
300
SRF
40-60%
5x20
up to 500
350
Ultimate analysis Mass %. drv basis
H
6.3 6.3 5.9
3.1 5.8 5.7 5.9 4.2 4.6
5.4 5.8 5.5
Notes § Ash includes metals, glass etc.
0
40.5 41.5 36.7
11.3 44.3 39.8 40.2 20.5 22.4
26.0 17.8 8.7
Ν
0.1 0.0 0.0
0.2 0.3 0.5 1.0 0.3 0.7
---
S
0.0 0.0 0.0
0.0 0.2 0.0 0.1 0.5 0.4
0.5 0.4 1.2
MŞW (fluff)
15-40%
50
5-1000
100
Ash
0.8 0.6 1.2
3.4 6.0
15.5 4.7
43.8 § 38.4 §
9.7 7.6
11.5
Straw
10-20%
5x200
200
Moisture %,
wet basis
30-60 30-60 25-75 16-40 25-40
2 - 8
16-20 20-35 15-30
6 CONCLUSIONS Wood, municipal solid waste and agricultural residues are all potential feedstocks.
Wood will be available from processing residues, unmanaged woodlands, conventional forestry and short rotation forest biomass plantations. Harvesting, processing and transport systems for delivering wood fuel either already exist or are under development. Delivered fuel costs will be between £28 - 36/ dry t from conventional forestry and in the order of £41/ dry t

55
from short rotation. Agricultural wastes, such as cereal straw, can be delivered to the end-user for £22/dry t. Domestic and commercial refuse is available and can attract disposal credits of up to £20/wet t which has a significant effect on the conversion economics, even after extensive processing to separate the fuel rich fraction. Once at the plant, conventional handling, screening and sorting systems can be utilized for all feedstocks. All aspects of the supply chain are under active research and development which should result in reduced feedstock costs.
7 ACKNOWLEDGEMENTS The work on wood as fuel (CPM) was supported by the UK Department of Energy and
the Biomass for Energy Programme of the CEC. The views expressed are those of this author and not necessarily those of the sponsors.
Much of the data reported in this paper on refuse costs (AVB) were generated for the Energy Technology Support Unit at the UKAEA, Harwell, UK, as an update of earlier work (13). They are intended to be published in due course in a strategic review of thermochemical conversion technologies for biomass and wastes (15). All opinions and statements are the views of this author and in no way reflect any views of the UKAEA or the UK Department of Energy.
8 REFERENCES 1 BRIDGWATER, A V and MITCHELL, C Ρ 1987. Interfacing biomass production and
biomass conversion. In 4th EC Energy from Biomass Conference. Ed Grassi et al. Elsevier Applied Sci ρ 1174 -1178.
2 MITCHELL, C Ρ 1987. European forest energy scene. In Proc 4th EC Energy from Biomass Conference. Ed Grassi et al. Elsevier Applied Sci ρ 54 - 58.
3 HARE, Ρ M; MARTINDALE, L Ρ and MITCHELL, C Ρ 1989. Supply and Use of Wood Fuel in the UK. Energy Technology Support Unit, Harwell. In press.
4 MITCHELL, C Ρ; TRANTER, R B; DOWNING, P; BRANDON, O; PEARCE, M L; BUNCE, R G H; and BARR, C J 1987. Growing wood for energy in Great Britain. ETSUB-1102.
5 DERLER, R 1988. Measurements of tree fuel at eleven district heating plants in Sweden. IEA/BE Report ρ 52.
6 JIRJIS, R 1988. Microfungi problem - health aspects. In Production, storage and utilization of wood fuels. Proc IEA/BE Conference, Uppsala, Sweden Dec 6-7,1988. 2 Vols. Ed B O Danielsson and O Gislerud. Swedish University of Agricultural Sciences, Department of Operational Efficiency. Research Notes No 133/134 1988. ρ 163 - 167.
7 MITCHELL, C Ρ; HUDSON, J B; GARDNER, D Ν A and STORRY 1989. Storage and drying of comminuted wood fuels. In Proc IEA Task VI Workshop "Wood preparation, storage and drying". Lind, Denmark May 16 -18,1989. In press.
8 GUSTAFSSON, G 1988. Forced air drying of chips and chunkwood. In Production, storage and utilization of wood fuels. Proc IEA/BE Conference, Uppsala, Sweden Dec 6 - 7, 1988. 2 Vols. Ed B O Danielsson and O Gislerud. Swedish University of Agricultural Sciences, Department of Operational Efficiency. Research Notes No 133/134 1988. ρ 150 -162.
9 MITCHELL, C Ρ; MARTINDALE, L P; HARE, Ρ M; HUDSON, J B; GARDNER, D Ν A and STORRY 1988. The potential for harvesting fuelwood in the United Kingdom: A systems approach. In Proc IEA Workshop "Economic evaluations of biomass oriented systems for fuel". Ed G Lonner and A Tornqvist Swedish University of Agricultural Sciences, Department of Forest Industry Market Studies, ρ 211 - 217.
10 CLEGG, J M et al., The acquisition and utilisation of straw as fuel, Silsoe College, Report to ETSU, 1985.
11 BUNGAY, H R, "Thermochemical Processes", in Energy: The Biomass Options, (Wiley-Interscience Publication, New York, 1981).
12 BOURDE AU, P H and FERRERÒ, G L, "Introduction and general presentation of the R&D programme on recycling of urban and industrial waste", in Anaerobic digestion and carbohydrate hydrolysis of waste; Eds. Ferrerò, G L, Ferranti, Μ Ρ and Naveau, U, (Elsevier Applied Science, 1987).

56
13 BRIDGWATER, A V; DOUBLE, J M and EARP, D M 1986. Technical and market assessment of biomass gasification in the UK. Report to ETSU 1986.
14 BRIDGWATER, A V, Economic and market opportunities for biomass derived fuels, these proceedings.
15 BRIDGWATER, A V and STRONACH, Ν J, "A Review of Thermochemical Conversion Technologies", Report to ETSU, Department of Energy, 1989.

57
PROCESSING OF URBAN WASTE TO PROVIDE FEEDSTOCK FOR FUEL/ENERGY RECOVERY
J R Barton Warren Spring Laboratory
Department of Trade and Industry
Summary
The UK has over ten years experience in the design, operation and monitoring of Refuse Derived Fuel (RDF) production plants and combustion of the products. Although most plants recover the fuel in pelletised form for use on conventional industrial solid fuel boilers, shredded fuels are also made and used. The paper reviews the technology adopted for fuel recovery and, given the composition and processing characteristics of UK refuse, comments on best practice based on the experience of commercial plants. Brief comment is made on the combustion characteristics of RDF based on laboratory research and practical experience in order to highlight problem areas. The paper concludes that much better guidance can now be given to designers of thermal treatment processes on the role waste pretreatment can play in developing more effective energy from waste systems, whether these involve direct combustion or the application of pyrolysis or gasification.
1. INTRODUCTION Mechanical sorting systems, by separating, concentrating and
altering the physical nature of the waste, permit more effective application of subsequent thermal, biological or other treatment and recovery processes. They also offer the opportunity for improved control over the environmental problems associated with waste treatment and disposal. However, experience of sorting plants for MSW has been mixed and full scale processes have generally not fulfilled expectations. Often equipment capacity and availability has been lower than expected and marketing problems have been experienced with the products due to quality deficiencies, competition from alternatives or the introduction of more stringent regulations governing use. Despite these difficulties, significant progress has been made in terms of achieving reliable plant operation and, more importantly, in understanding the limitations and potential inherent in the approach to permit better matching of feedstock characteristics with process and

58
market requirements. Warren Spring has been closely involved in designing, commissioning and monitoring refuse derived fuel plants in the UK and abroad and this paper summarises the key factors that need to be considered when adopting a pretreatment stage prior to thermal treatment. Obviously sorting waste for fuel recovery results in residues which must be disposed or further treated to recover values. However, whilst effective treatment of these residues is an integral part of the overall concept and influences process selection, it is a subordinate consideration. Fuel and energy recovery is the major weight and volume reducing stage and if this is not effective, extending recovery processes to residues can rarely be justified.
2. SEPARATION AND RECOVERY OF THE FUEL RICH COMPONENTS 2.1 Refuse composition and process selection
Table 1 gives a typical composition for UK refuse along with the ash, moisture and calorific value of the components 'as received', ie as delivered to a processing plant. This input data dictates the mass yield, energy recovery and quality achievable by physical sorting alone. Fig. 1 illustrates the effect of ideal separation (eg by hand sorting) assuming categories are removed in order of increasing fuel value eg starting at point A with raw refuse, removing glass, metals and inert categories first (point Β), then unsorted fines (10 mm) (point C), next putrescibles (point D) and so forth. The effect on mass yield (x axis) energy loss (y axis) and fuel quality at any point can be assessed. Obviously mechanical sorting systems are not so systematic nor are they 100% efficient at removing separate categories but this graph,does provide target values for process designers and a baseline against which to assess plant performance. Theoretical separation efficiences can be calculated at each point using accepted efficiency formulae. The values given on Fig. 1 are calculated using the following formula derived by Douglas'1':
« p £ Separation efficiency = iooC i00f x 100%
where R = % of available energy recovered in concentrate C = weight % of concentrate c = % assay of concentrate (in terms of calorific value) f = % assay of feed (in terms of calorific value)
To obtain values for c and f, an arbitrary maximum calorific value must be selected to represent 100% 'pure' fuel. In this case the average calorific value of the combined plastics categories, ie 28.5 MJ/kg, has been selected. The low values calculated using this formula mainly reflect the unrealistically high target CV for 'pure' fuel which, by definition cannot be reached without rejecting categories such as paper. However, considering the relative changes, these data show that whilst the separation efficiency gains from the removal of non combustibles, fines and putrescibles reach a maximum value and conform to expectations, selective recovery of the plastics alone is equally efficient; this is because the formula used gives equal weight to maximising fuel quality as it does to maximising fuel

59
Table 1. Composition of UK refuse
Paper Plastic film Dense plastics Textiles Misc. combustibles Misc. non-combs Glass Ferrous Non-ferrous Putrescibles Fines -10 mm
TOTAL
As ree'd assay wtZ
33 4 4 4 5 5 8 6 1 20 10
100
Moisture content wtZ
30 25 15 25 25 15 20 15 10 65 40
33
Ash content (as ree'd)
wtZ
8 9 6 8 12 85 90 85 90 8 40
28
Gross calorific value (as ree'd)
MJ/Kg
12 27 30 15 13 ----6 4
9.1
20 40 60 80 100 YIELD OR WEIGHT OF FUEL AS A PERCENTAGE OF INPUT
FIG. 1 EFFECT OF 'IDEAL' SORTING OF REFUSE FOR FUEL RECOVERY

60
recovery. Obviously many other criteria, particularly options for the residues, need to be taken into account when determining an optimal solution but this assessment does highlight that, for refuse, a wide range of fuel yields is compatible with maintaining a high separation efficiency from a strict technical viewpoint.
Even from the fuel viewpoint alone the chemical composition, particularly of elements which cause environmental problems in thermal treatment (eg chlorine, heavy metals), has a significant effect on the costs of converting the 'fuel' to usable energy. Thus to select the optimum fuel recovery and grade for a system and to determine how to achieve it requires more detailed data. Such information has become available from testwork undertaken by WSL and other workers'^)"). For example, size distributions of each category have been measured at 10, 20, 40, 80, 160 and 320 mm and for each sized-category fraction (eg -80+40 mm paper fraction) further analysis gives moisture, ash, calorific value and chemical composition. These data have proved invaluable for optimising plant equipments and process flowlines. At WSL the information is kept on database and manipulative software has been written to permit selection and averaging of sample data and the application of "process function" matrices which represent either real separations achieved by specific process equipments or 'ideal' separations. This work, along with the potential to develop mathematic models of the processes, has been detailed previously^*'; for the purposes of this paper it is sufficient to know that given a specification or requirement based on the thermal treatment step, a rapid assessment of whether this is achievable using sorting technology alone can now be made. 2.2 Practical considerations
Many options can be selected for a process flowline but practical considerations and experience have narrowed these down, particularly during the early treatment steps. Thus whilst the simplified flowline illustrated in Fig. 2 represents a good option for achieving fuel qualities and yields close to the theoretical values given in Fig. 1, variations on the theme, particularly in the latter stages for a more refined fuel with controlled physical characteristics (eg pellets), are entirely acceptable. For discussion purposes, the four stages selected are (i) preliminary liberation and screening, (ii) magnetic separation and coarse shredding, (iii) a refining separation stage and finally (iv) processes to control the physical characteristics of the fuel. The following sections are not intended to be a comprehensive review of the requirements for each stage but to illustrate, with examples, why certain processes are effective, where the major problems are and to indicate where the balance between pretreatment and preparation costs and the costs of the energy raising unit is likely to be in any given circumstances. 2.2.1. Stage 1 liberation and screening
Experience in the UK'^) has clearly identified that screening prior to size reduction has significant benefits in terms of process reliability and fuel quality. It can remove materials which contaminate the fuel categories during shredding as well as problematic items which adversely effect the performance of shredders. The key equipment item is a rotary screen or trommel and, whilst a separate

61
RAW WASTE
wt = 100% cv = 9.1 MJ.Kg"1-· - -
ENERGY = 100%
STAGE 1 LIBERATION AND SCREENING
FINES REMOVAL (^50mm) OVERSIZE REMOVAL (^500mm)
wt = 66% c v = 11.5 M J. K g " 1 * - - I
ENERGY= 83%
STAGE 2
50mm FINES
+500mm OVERSIZE
wt = cv =
ENERGY= wt = cv =
ENERGY=
32% 4M J. Kg"1
14% 2% 13 M J Kg"1
3%
MAGNETIC SEPARATION AND
COARSE SHREDDING
wt = 60% cv = 12.5 MJ.Kg"1-»-
ENERGY = 82%
STAGE 3
-FERROUS METAL w t = 6% cv= 1 MJ.Kg"1
ENERGY= 1%
REFINING (SEPARATIONS) AIR/BALLISTIC CLASSIFIERS
FINES REMOVAL (<10mm)
wt = 48% cv = 13.4 MJ.Kg1 '
ENERGY = 70%
STAGE 4
„HEAVY AND FINE REJECT
wt = 12% cv = 9 MJ.Kg"1
ENERGY= 12%
REFINING (PHYSICAL) SHREDDING
DRYING PELLETING COOLING
wt = 36% cv = 18MJ.Kg-1-*-—I
ENERGY = 70%
MOISTURE LOSS
wt 12%
RDF PELLETS
FIG. 2 STAGES IN PRODUCTION AND REFINING OF REFUSE-DERIVED FUELS

62
liberation stage to open bags and release refuse component (eg at the Byker plant) is advantageous, a trommel can be designed and operated in a manner which ensures sufficient liberation and breakage of friable, non-fuel components (such as glass) to achieve efflicient screening. Effective design criteria for fine screening duties have been developed*·"', the equipment is simple and robust, designs for feedrates up to 40 teh-* for a single unit are proven and scale up for higher feedrates is possible. Running costs are low with specific power consumptions below 1 kWh per tonne of raw waste processed. For UK refuse the aperture size selected for fine screening is normally between 40 and 60 mm, a size which ensures minimal loss of identifiable combustibles categories ( 5%) whilst achieving removal of over 65% of the putrescible category, over 90% of the -10 mm fines and over 85% of the glass. Most metal reports to the topsize, fuel rich fraction, but items such as small batteries and foil from wine bottles tend to report to the fines and this results in reduced heavy metal contamination of the recovered fuel.
One action of a trommel which is detrimental to downstream processes is the tendency to 'create' ragtails; these are sausage like composites of wire, string, textiles and plastics up to several metres in length. Such items, along with the bulky items which 'naturally' occur in refuse (eg matresses, dustbins etc) are difficult to size reduce effectively. Some pulveriser designs, such as vertical shaft, non gridded hammer mills are capable of coping with such materials but the effect on gridded mills can be extremely damaging in terms of throughput and availability. Fortunately a coarse screen section installed in the trommel can reject such items with minimal loss of fuel values and is strongly recommended. Table 2 shows the dramatic improvement achieved at the RZR Herten plant by modifying the trommel to scalp out items above approximately 500 mm in size. Close attention needs to be given to the design of the screen plate to minimise blinding and to ensure easy access to the external surface for cleaning purposes. Experience suggests that downtime for cleaning can be kept to below 10 minutes per 8 hour shift without adversely effecting screening efficiency. Removal of coarse materials also improves the efficiency of subsequent magnetic separation when this is carried out before shredding in Stage 2.
The importance of trommel screening as the first fuel-non fuel separation process is now well established, even without magnetic separation, screening alone proved effective in terms of increasing energy output and boiler efficiency for a conventional mass burn incinerator at Sheffield*·''.
For UK waste, Stage 1 should result in a yield of 60% wt fuel rich material, reporting a calorific value of approximately 11.5 MJ/Kg and containing 83% of the original energy content.

63
Table 2. RZR Plant, Herten - Effect of coarse aperture screening on plant performance
Steady state throughput
Plant availability
Hammer mill specific power consumption
Before coarse aperture scalping
22 teh
72%
-1
25 kWh/tonne
After scalping out oversize at
approx. 500 mm*
27 teh"1
96%
18 kWh/tonne
* weight of oversize removed was less than 2% of the refuse feed.
2.2.2. Stage 2 Magnetic Separation and Coarse Shredding Assuming the fuel rich topsize has been scalped to remove coarse
items, the preferred magnetic extraction system is an overband unit positioned at a head pulley in line with the refuse flow. Recoveries of at least 90% are common but the magnetic product will contain up to 15% contrary materials (eg paper, plastics). Cleaner products are obtained if magnetic separation is delayed until after the shredding operation; however prior metal removal can be considered a protective measure, reducing the risk of explosion damage from volatiles (eg paint thinners, butane) present in cans, aerosols and gas bottles. Irrespective of the sequence, shredders need to be sited in 'explosion proof' housings and preferably fitted with an explosion suppression system. Hammer mill designs are preferred for coarse shredding due to their robust nature and, if scalping has been carried out, a gridded design does assure topsize control for downstream operations is maintained irrespective of hammer wear (hammer condition will still need attention for maintaining desired size distribution and throughputs). Specific power consumptions and flow rates will depend on mill design and feed composition. Values between 15 and 25 kWh/tonne are not untypical. A major pitfall to be avoided is to believe that the size and power of a mill can be significantly reduced by stream splitting. For example, when a trommel was installed before the pulveriser at Byker, the weight throughput to the pulveriser reduced from 30 teh-^ to 20 teh-·'- due to removal of 10 teh-1 of fines. It can be seen from Table 3 that the effect on power usage was minimal as the specific power requirement for the remaining topsize increased from 12 to 17 kWh/tonne. This is not surprising, the volumetric flowrate was largely unaffected and the weight of materials that actually required size reduction, ie the paper, plastics and textiles had not been reduced. Similar experiences at the secondary shredding stage have occurred when little benefit has

64
resulted from prior removal of materials already below the desired shred output size.
From the fuel quality and yield data given on Fig. 2, it can be seen that the bulk of the fuel-non fuel separation has been achieved by the trommelling and magnetic separation stages. Again, without shredding, current testwork on a conventional mass burn unit (at the Edmonton incinerator) is showing encouraging results and the shredded product is similar (but of better quality) to the fuel used at Courtaulds'"' in Grimsby on a large water tube chain grate boiler and the fuel used in the cement kilns at Westbury by Blue Circle'"'. Restricting pre treatment to this stage and using circulating fluid bed combustore is also being given serious consideration in the UK.
For UK waste, Stages 1 and 2 should result in a yield of 60% wt fuel rich material, reporting a calorific value of approximately 12.5 MJ/Kg and containing 82% of the original energy content.
Table 3.
Raw refuse
Screened refuse (-50 mm fines removed)
2.2.3. Stage
Effect of fines removal on hammer mill performance
Throughput Wt. Volume teh m h
30
20
165
155
Size reduction (d,, mm)
From To
80 11
150 24
3. Light-heavy separations and fine
Hammer mill power consumption
Total Specific kWh kWh/te
355
335
screening
11.8
16.8
The separations in Stage 3 are primarily to refine the fuel product. Useful improvements in terms of reducing ash and metal contents can be achieved but residues often report relatively high calorific values. Of equal importance, Stage 3 separations protect more vulnerable downstream equipment such as knife mills and pelletizers by removing heavy or dense items irrespective of composition.
Light-heavy separations for refuse materials have been subject to a number of detailed studies'"''™' and many different designs have been tested at full scale'"'. Products other than fuel from refuse have also been recovered (eg plastics, composts, paper). The separation characteristics of ballistic units are different from air classifiers, for example in certain designs 'bounce' is a more important characteristic than particle density and shape. Such differences can be worth exploiting, eg PVC bottles tend to 'bounce' into the heavy reject for one ballistic design and this can reduce chlorine levels in the fuel. On the other hand fuel quality and fuel flow rates tend to be easier to control in air classifiers (particularly under variable feedrate or surge conditions) and this can

65
bring important benefits in terms of process efficiency of downstream equipment.
Further fines removal is often carried out during this stage to reduce ash content and screening can be incorporated into the light-heavy separation unit or carried out separately. Both trommels and flat bed units have been used, but if a flat bed unit is selected, vibration amplitude should be larger and frequency lower than is normally adopted for screening minerals (eg coal, sand, gravels). To prevent excessive loss of fuel values on aperture size of 10 mm or below is usually selected.
Energy inputs for Stage 3 separations equipments are not particularly high and rarely exceed 10 kWh/tonne for most fuel recovery applications. Careful consideration should be given to the requirements of downstream equipments and the thermal conversion process when designing this section of the plant. Specific problem materials can be rejected and there is scope for more development in the equipments but it must be recognised that in strict energy efficiency terms the separations undertaken are in a regime of diminishing returns.
For UK waste, Stages 1, 2 and 3 should result in a yield of 48% fuel rich material reporting a calorific value of approximately 13.4 MJ/Kg and containing 70% of the original energy value. 2.2.4. Stage 4 Processes to improve the physical properties of the fuel
This stage is concerned entirely with tailoring the fuel to suit the combustion or thermal treatment stage. By further shredding, drying and pelleting, the fuel storage, handling and combustion properties are improved. This stage also lends itself to blending in additives specifically to enhance combustion properties. In terms of capital, maintenance and operating costs per tonne of material processed, the processes in this stage tend to be the most expensive in the plant. Hence, high plant availability is essential and this requires much improved monitoring and control of flowrates, feed composition and equipment. The shredding stage normally requires in excess of 25 kWh/tonne to reduce a coarse shred (say approximately 100 mm) to below 25 mm (mean size approximately 10 mm). The evaporative heat requirement for successful pelleting (reducing moisture levels from approximately 30% to below 12%) is approximately 700 MJ per tonne of fuel and as drying efficiencies are between 50 and 65%, a thermal input of over 1200 MJ per tonne is needed (ie +£4/tonne at natural gas prices or up to 10% of the plants fuel output if an RDF fired combustor is used). For good quality pellet production a specific power consumption of +35 kWh/tonne for the pellet mill alone is typical. Although no physical separation processes are undertaken during Stage 4, moisture removal during drying, pelleting and cooling increases calorific values to around 18 MJ/Kg and the bulk density from approximately 100 Kg m~3 for coarse shred to over 600 Kg m--* for pellets.
There are many views on the best equipment designs and of best process sequence for the production of pellets. There are certainly advantages and disadvantages to all with different plant designers preferring knife mills to hammer mills, cascade dryers to pneumatic

66
dryers, fixed die pelletisers to rolling die machines, single stage pelleting to two stage densification etc. Suffice it to say all systems initially developed failed to meet design objectives, flowrates of half the anticipated levels were not unusual and dedicated effort to modify and improve the process, often by the plant operators rather than individual equipment manufacturers has been needed to achieve satisfactory performance. Today there is no excuse for installing equipments which don't fulfill the process requirements but new plants would be strongly recommended to buy proven systems rather than put together separate units that haven't previously been tested in sequence. Quite simply "the art" in preparing and pelleting waste fuels is as important as the scientific and engineering knowledge of the individual unit processes.
An important consideration in the drying stage is abatement of particulate emissions and odours from the dryer gas. This has caused problems with existing UK plants whether the drying heat has been provided by gas burning or burning RDF. Although satisfactory retrofit solutions have been found (eg increasing chimney heights, increasing efflux velocities, adding odour counteractants and oxydising chemicals to scrub waters, adding additional particulate removal systems etc), this is an area where further investigation is needed to define the problems more closely and develop optimal solutions.
There is undoubtedly scope in Stage 4 processes for significant improvements to be made. The design of pelleting processes has been based mainly on achieving similar handling, storage and feeding to small coal. This has proved expensive but as experience with using waste fuels increases, user requirements will become more specific and this should stimulate development of more cost effective Stage 4 systems.
For UK waste, Stages 1 to 4 should result in a yield of 36% by wt fuel, reporting a calorific value of approximately 18 MJ/Kg and containing 70% of the original energy content. Electrical energy inputs for the full plant are likely to be up to 70 kWh/tonne of refuse input, approximately 200 kWh/tonne of pelletised fuel. 2.1.5. Other processing factors
Before considering fuel characteristics against combustion requirements, brief comment on storage and handling systems is warranted. Within a production plant, short term storage systems have advantages from a design viewpoint in smoothing out flows and matching capacities in different sections of the circuit. However they have frequently resulted in more problems than they have solved. Blockages and uneven discharge characteristics have been experienced with most shred bunker designs, particularly push floor bunkers and bunkers with screw discharge systems. The reasons for failure vary, hopper size and shape is critical (it must diverge), push floor designs generally do not provide sufficient positive motion to be effective if the depth of shred exceeds 1 or 2 metres, the design of screws and discharge outlet must avoid compressing the shred. Recent work at WSL on a multiscrew discharge hopper proved blockages could be avoided by careful design, but even with a fine sized ( 25 mm), relatively dry shred suited for pelleting, fluctuations of 25% about the mean discharge flowrate still occurred. Bunkers with screws that move position within the mass of

67
the shred (eg by traversing the bottom of the hopper or rotating eccentrically within the hopper) largely overcome blockages and a novel design based on a slow moving plate conveyor floor and a rotating rake device to tear off the shred as the mass moved toward the .open discharge side worked successfully at the Rolls Royce plant'11'. Thus progress has been made but problems in this area are still a frequent occurrence at sorting plants and for all new systems or systems to feed shred to a combustion unit, particulary attention needs to be paid to this aspect of plant design.
Handling, storage and feeding of pellets is less of a problem; small amounts, kept in dry and well ventilated conditions, show little sign of degradation after a year or more, but prolonged storage in deep bunkers or piles would not be recommended. There are many examples of localised heating, sweating and fungal growth leading to break up of pellets in bulk storage and potential fire/health hazards. Whilst some of these problems can be attributed to poor control of moisture content during production, changes in climatic conditions (temperature and humidity) also affect pellet stability. Hence storage should be limited to a few months. For undried RDF, a few days is preferable although longer periods can be tolerated if the subsequent handling system is designed to cope with 'lumpy' materials.
The above comments cover a few of the practical difficulties experienced on UK plants. Generally solutions have been found but further progress and experimentation with plant design is still required and to aid this, a few thoughts are given below based on over 10 years of problem solving on RDF plants.
. Reliability is more important than technical efficiency
. Reject problematic materials as soon as possible
. Exploit the natural separation characteristics of the waste first
. Delay energy intensive stages as long as possible
. Ensure the basic processing and separation principles of the equipments are understood
. Volume, not weight throughput tends to be the rate limiting factor
. Once refuse stops moving, it needs a positive kick to start again
. Always remember it is waste until a marketable product is recovered, irrespective of the degree of processing
3. RDF and thermal treatment Although this paper is primarily concerned with production of RDF,
UK experience with combustion of RDF (pellets in particular) is beginning to feed back into the production process and consideration can be given to whether problems are better tackled during feed preparation or by developments in the thermal conversion stage. Table 4 gives basic information on composition and properties of RDF pellets and a typical UK bituminous coal but many of the comments made below are based on more detailed characterisation studies undertaken by WSlA"). The objectives to be realised for an effective energy from waste plant are as follows:
1. Maximum heat release for minimum grate size. 2. Minimising clinker formation.

68
3. Minimising boiler fouling 4. Minimising emissions (acid gases and heavy metals in particular).
Table 4. Properties of RDF pellets and bituminous coal
Gross calorific value MJ/kg
Proximate analysis
Moisture content Zwt Ash content Zwt Volatile matter Zwt Fixed carbon Zwt
Ultimate analysis (DAF)
Coal
27
8 10 26 56
RDF (Byker)
18.5
8 15 67 10
55 7
36 .6 .9 .3
600 16 mm diameter
From Table 4 it can be calculated that almost twice the volumetric throughput of pellets is required to match the same thermal input of coal. This affects fuel handling and feeding equipment more than the combustion appliance because the higher volatile content of RDF and the higher reactivity of RDF char compared to coal greatly reduces the residence time requirements within the combustor for complete burn out. Pellet density, size and shape will affect this but, to date, it has not proved necessary to exploit the ability to change these properties to increase (or reduce) heat release rates.
It is pertinent to note the difference in heat release characteristics of RDF pellets compared to raw waste; the residence time for pellets on a chain grate is approximately 15 minutes compared to approximately 60 minutes for MSW on an incinerator grate. As pellets have approximately 7 times the energy content per unit volume compared to raw waste this suggests a 30 fold advantage over raw waste in terms of grate area required to achieve the same heat release rates. 3.2 Minimising clinker formation
Although clinker formation was a problem experienced on a number of stoker/grate systems during early firing trials^^',
Total carbon H 0 Ν Cl S
Bulk density Size of fuel
1 Heat release
Zwt Zwt Zwt Zwt Zwt Zwt
Kg m" mm 10
84 5 8 1.6 .1 1
900 to 25 mm

69
improvements in processing technology, particularly the use of front end screens, have reduced ash contents in RDF and reduced clinker formation. At Byker, for example, RDF ash contents reduced from approximately 17Z to below 14% on a dry weight basis^) and much of the reduction was due to removal of components with a low fusion temperature such as glass. Most of the remaining ash is inherent in the combustible categories; paper extracted from refuse for example normally assays approximately 10% ash (dry wt basis) and the IDT (initial deformation temperature) is between 1000'C and 1050°C compared with 1150°C for coal. Hence RDF will always report lower ash fusion points than coal but, given efficient processing, clinkering on most commercial systems is not a problem. For systems which still experience problems (eg ram stokers), recent work at Warren Spring has indicated that addition of chemical compounds can increase fusion temperatures and hence offer a potential solution. Obviously for lower temperature thermal processes, eg pyrolysis or gasification, the possibility of clinker formation is much reduced. 3.3. Minimising boiler fouling
Severe fouling of boiler surfaces has been experienced with both raw waste incineration and RDF combustion. The deposits have a high concentration of alkali metals, for example tube entry deposits on a chain grate unit burning pellets assayed over 90% alkali sulphates. The problem is more severe than coal simply because RDF contains ten times more sodium and potassium and these metals are also present in a more easily volatilised form. Alkali salts are added as fillers to all forms of paper and there is no physical separation technique capable of removing these deposit forming chemicals from the feedstock. One processing step that could contribute to some deposition control is the addition of a chemical that reacts with the alkali to form a more stable compound which will not volatilise at the temperature experienced on the fuel bed. Kaolin has been proved effective in retaining alkalis as alumino silicates in the past'^' but is unlikely to offer a complete or cost effective solution. In the longer term, control of deposition will probably lie with the design and operation of the combustion and boiler unit. Boiler fouling has been substantially reduced on a chain grate by a combination of selecting fuel bed depths and grate speeds to reduce the time of the high temperature ash burn out phase (and hence reducing volatilisation), reducing the temperature of the flue gas prior to the first boiler pass (at the cost of reduced boiler efficiency) and use of soot blowers. Again adoption of lower temperature gasification processes as opposed to full combustion would make a significant impact on this problem by retaining the alkalis in the fuel ash residues. 3.4 Minimising emissions
Coal combustion particulates are mainly coarse fly ash entrained through the system and relatively easy to abate using conventional basic units, ie grit arrestors. In RDF combustion a larger proportion of the particulates are sub micron which can only be removed efficiently by more expensive abatement units such as electrostatic precipitators or bag filters. Test work in chain grate stokers has shown RDF particulate emissions are high in metallic chlorides (eg Na, K, Pb, Cd and Zn). As mentioned, the alkali metals are constituents of

70
paper; lead, cadmium and zinc also appear in combustible categories. For combustion processes, improved abatement equipment is the established solution though again lower temperature thermal processes should certainly reduce the emission of these metal chlorides.
For acid gas emissions, SO2 is not a problem compared with coal combustion but HCl emission certainly is. In the UK, combustion of RDF pellets typically gives rise to HCl concentrations of 800 to 1000 mg Nm-3 (9% CC-2). Up to 60% of chlorine in RDF is present in chlorinated plastics (PVC) and this is totally converted to HCl in all forms of thermal treatment.
As discussed previously there is some scope for removing PVC bottles during RDF production but this by itself is unlikely to make a substantial reduction in emission levels. Thus controlling HCl emission from combustion processes requires the appropriate downstream flue gas clean up technology (eg scrubbing). Lower temperature thermal processes may offer more cost effective methods of retaining chlorine within the system, for example gasification with injection of lime is reported' " ' to retain approximately 90% of the fuel's chlorine.
5. Conclusions Whilst burning waste and waste derived fuels for energy recovery
has and continues to be an effective waste treatment option, problems with clinkering, boiler fouling and emissions still occur under full combustion conditions. Whilst some cost effective solutions are available by improved production methods and improved combustion control, other solutions will significantly increase the costs of using RDF on conventional coal fired plant, particularly where stricter emission limits are applied to RDF as compared to coal. Lower temperature processes such as gasification or pyrolysis are looking increasingly attractive as a means of reducing these costs.
In terms of preparing waste for thermal treatment, full scale RDF plants have proved the potential for significantly increasing calorific value, energy density and the provision of fuels with consistent handling, feeding and combustion properties. These are all factors which lead to substantial reductions in the cost of building and running the thermal treatment plant compared to mass burn incineration. The energy loss on converting raw refuse to an RDF can be restricted to 30% by effective plant design and for a range of RDF types there are positive experiences to report on a variety of combustion plant. Equipment requirements and processing costs for the various forms of RDF are established in broad terms and matching fuel characteristics with requirements for the thermal treatment stage is facilitated by the availability of detailed analysis data and improved understanding of the separation processes.
Raw refuse cannot be considered to be the optimum form or composition for thermal treatment and energy recovery. There may be arguments as to the degree of separation and processing warranted for the most effective overall system, but there is no doubt that pretreatment offers excellent opportunities for improving the thermal treatment stage as well as the opportunity to recover additional values from the residues by more appropriate, non thermal processing methods.

71 REFERENCES (1) DOUGLAS, E. Derivation of basic efficiency formula for
concentration operations. Trans. Inst. Mining and Metallurgy 71 (1961-2), pp697-704.
(2) LORBER, K.E. Incineration of RDF and incineration of total waste - comparison of emissions. Paper to EC Seminar, Sorting of household waste and thermal treatment of waste, Luxembourg, Sept. 84. Applied Science, Elsevier 1985 pp208-225.
(3) VAN ROOSMALEN, G.R. et al. Heavy Metal Sources and Contamination Mechanisms in Compost Production. Paper to MER 3 Conference, 18-20 March 1986, Antwerp. Belgium.
(4) BARTON, J.R. et al. The Use of Database and Modelling Techniques in Furthering Developments in Solid Haste Processing in the UK. Paper to 23rd intersociety Energy Conversion Engineering Conference (ECEC), 31 July-5 August 1988. Denver, Colarado, USA.
(5) BARTON, J.R., WHEELER, P.A. The Benefits of Front End Trommelling in Processing Municipal Solid Wastes. Trials at the Byker Plant -June 1987. WSL Report LR661(MR)M, Sept. 1988, 39pp.
(6) WHEELER, P.A. et al. An Empirical Approach to the Design of Trommel Screens for Fine Screening of Domestic Refuse to be published, Resource, Conservation and Recycling (1989).
(7) BARTON, J.R., POLL, A.J. Preparation and Incineration of Screened Refuse: Preliminary Trial, Sheffield, November 1986. WSL Report LR592(MR), March 1987, 49pp.
(8) BARTON, J.R., POLL, A.J., WEBB, M., WHALLEY, L. Waste Sorting and RDF Production in Europe. A report prepared for the Commission of the European Communities R and D programme - Recycling of Urban and Industrial Waste, Barking, England. Elsevier Applied Science, Publishers Ltd, 1985, 209pp.
(9) BARTON, J.R. Development and Application of a Method for Predicting and Assessing Performance of Operating Air Separators in the Processing of Municipal Solid Wastes. Paper to EC Seminar on Sorting of Household Waste and Thermal Treatment of Waste, Luxembourg, 25-27 September 1984, London: Elsevier Applied Science, 1985 ppl09-119.
(10) ROSENBRAND et al. The Separation Performance and Capacity of Zig-Zag Air Classifiers. Paper to EC Seminar, Sorting of Household Waste and Thermal Treatment of Waste, Luxembourg, September 1984. Elsevier Applied Science, 1985, pp208-225.
(11) ENERGY EFFICIENCY OFFICE. Combustion of Shredded Industrial and Commercial Waste in a Circulating Fluid Bed. Project Profile 206, Energy Technology Support Unit, Harwell Laboratory, May 1985.
(12) RAMPLING, T.W.A., HICKEY, T.J. The Laboratory Characterisation of Refuse Derived Fuel. WSL Report LR643(MR), Dec. 1987, 186pp.
(13) BURNLEY, S.J. The ETSU/WSL Refuse Derived Fuel Combustion Trials Programme: A Review of Progress. WSL Report LR595(MR), Sept. 1987, 33pp.
(14) BRINSMEAD, K.H. and REAR, R.W. The Formation of Alkali-Bonded Deposits. Laboratory Studies of the Behaviour of Sodium Chloride during Combustion. BCURA Inf. Circular No. 134, Doc. No. C/4950.
(15) HOS, J.J. et al. Gasification of Organic Solid Wastes in Co-current Moving Bed Reactors. Energy from Biomass and Wastes IV, Florida 1980.

72
CHARACTERIZATION OF CARBON CONTAINING MATERIALS WITH RESPECT TO PYROLYSIS AND GASIFICATION
H.J. Mühlen, W. Wanzl, K.H. van Heek BergbauForschung GmbH, Essen, FR of Germany
Summarv From the broad use of thermochemical conversion of coal and other solid fuels various methods have been established for their characterization with respect to pyrolysis and gasification. The experimental techniques and theoretical methods including those for reactor modeling are reviewed, taking as examples those in use at BergbauForschung. Typical results achieved for coal and lignites are shown, demonstrating how the experimental devices and the theoretical background can be used to characterize other carbonaceous materials like biomass and wastes. The final aim is the design of new or the optimization of existing processes with respect to the feedstock available and the products desired.
1. INTRODUCTION As it is expressed in the introduction to the programme
of this conference, thermochemical processing is most important amongst the available technologies for biomass conversion. Such processes like pyrolysis, gasification and also combustion have been since ever the basis for industrial conversion of coal. Therefore the purpose of this contribution will be to review the main techniques for the characterization of coal with respect to pyrolysis and gasification, thereby showing their importance also for the treatment of other feedstocks. 2. CONVERSION PROCESSES AND RELEVANT FEEDSTOCK PROPERTIES
First of all it has to be noticed that the term "conversion" is used differently in science and technology. As illustrated in fig. 1, taking as an example gasification, it stands in a narrow sense for the heterogeneous reactions of carbon with gasifying agents such as Η,Ο, C02, H2 and 02· In a broader meaning it denotes the gasification reactor, that means, it includes additional processes such as heating up, drying and pyrolysis. In some reactors, in which heat has to be provided for conversion, also combustion may be enclosed. Finally, conversion means the whole process, starting with the storage of the feedstock, its preparation and ending up with valuable products at the one hand and the disposal or utilization of the undesired residues on the other /!/.

73
Feed Propertles
GASIFICATION PROCESS Storage
Grinding
m
GASIFICATION REACTOR Pyrolysls
GASIFICATION REACTIONS C H 2 0
C CO2
C H2
Combustion
Gas Cleaning
Residue Disposal
53 Resultant
Char and Ash
Properties
Fig. 1: Different steps of a thermochemical conversion process taking as an example gas i f icat ion
Table 1; Correlations between coal properties and the ir changes in the s ingle process steps PROCESS STEPS
STORAGE
GRNDNG
PYROLYSS
GASFCATION
COMBUSTION
DISPOSAL
RELEVANT PROPERTES
r o *
r a * — mocera composition — mineral content g r n M t f t y
r a * mocera composition ~ demonta composition
asmen tol composition sirfoce and porosity intrinsic reactivity minora composition
strfoce and porosKy reactivity minora composition
— minera composition seJubïty of ash elements
P0SS8LE EFFECTS
self ¡onrtion ODOQuOn
~ bea hooung release of hydrocarbon
oases aid carbon oxidei
90s ond tor evolution ~ char formation — ÜHHiMjtjtastictty
~ partial consvnption changes η minerals changes in physical structure
bum out partial combustion
eh/tion of traces
»PACT ON PROPERTES
oging reduction of size reduction of swefing
reduction in V i l changes in surface area
shape kitema) surface reactivity elomental composition
~ ash enriched residue reactivity
physical and chemical constitution
Considering characterization of the feedstock i t has to be taken into account that although the properties of the sol i d s influence the reaction in the s ingle process steps , they themselves do undergo changes in them. Under t h i s aspect

74
table 1 tries to summarize the effects of the reactions during storage, grinding, pyrolysis, gasification, combustion and disposal, and the interactions with the relevant properties of the in and outgoing solids. Without going into details it can be stated, that there is a wide number of properties to be considered as relevant to the different process steps. Taking as an example pyrolysis these are rank, maceral composition and elemental composition of the ingoing feedstock. During the process condensable products (tars) gases and char are formed. Also thermoplasticity has to be taken into account for a proper operation of the reactor. The conditions in the pyrolysis process influence shape, internal surface, reactivity and elemental composition of the outgoing char. In a similar way also the other process steps have to be discussed.
To sum up: the solid feedstock must be characterized with respect to every process step, in which generally different properties are relevant. However, the following part will mainly concentrate on the conversion step. 3. EXPERIMENTAL AND THEORETICAL METHODS
Course and product yields during pyrolysis and gasification of carbonaceous solids are mainly influenced by such parameters as final temperature, residence time, rate of heating, gas atmosphere and pressure. Most important is the timetemperature history, which can be described by the rate of heating. As it is shown in table 2 the technical reactor types differ widely by the rates of heating applied. Table 2: Classification of heating rates in coal conversion
slow
medium
fast
flash
Heating Rate K/s
« 1
5 1 0 0
500 100.000
> 105
Heating Time to 1000°C dp = 100 μπι
*· 20 min
10 s U min
10ms 2s
< 1ms
Technical Reactor Types
Coke Oven
Fixed Bed Gasifier
LRCarbonization
Entrained Phase
Fluidized Bed
Plasmareactor
Relativly slow values of some K/s are achieved in cokeovens or fixed bed gasifiers. There the reactions occur in hours. Medium rates of heating are found in carbonizers like the

75
Lurgi/Ruhrgas (LR) type. Much faster heating rates are achieved in entrained phase and fluidized bed reactors, where the reactions occur in less than one second. Plasma reactors applying temperatures up to 3000 Κ lead to very fast heating rates.
In order to characterize the behaviour of the feedstocks in thermochemical processes under these very different heating rate conditions we operate three types of apparatus at BergbauForschung as shown in table 3. In the thermobalance apparatus a sample of about 1 g is heated up with several degrees per minute to a final temperature of 1000 "C As a result from the continuous measurement of the weight loss and gas analysis the formation of products like Ho, CO, CO,, CJJHJJJ, BTX or tar is described, from which the kinetic parameters can be determined as will be mentioned lateron.
For higher heating rates up to 1500 K/s the grid heater technique is used. About 10 mg of the sample are heated on a metallic wire net. Besides the determination of the product formation and yields we use a high speed camera to study the mechanical behaviour of the particles and other visible phenomena like ignition, duration of plasticity phases etc. Very high rates of heating of 10.000 K/s at an adjustable final temperature can be achieved using the Curiepointtechnique. About 5 mg of the solid is housed in a metallic cylinder which is heated up by electrical induction.
Table 3: Laboratory equipment for pyrolysis, hydropyrolysis and gasification; pressure 0.1 30 MPa
apparatus
sample size mg heating rote K/s final temperature °C
results
thermobalance
d t t i f i
"t tilt t Th
1.500 0.05
1.000
kinetics of product formation
E.ko.n.Vo for Hi.COx.CxH».
BIX and tar
wirenettechnique
Th
kinetics of particle swelling from high speed camera studies
Curiepointtechnique
5 10.000
adjustable isothermal experiments
kinetics of product formation
E.ko.n.Vo for Hï.COx.CxH».
BTX and tar
product yield product yield
At its Curietemperature the metallic cylinder loses its ferromagnetic properties and does not take up energy any longer. Thus it stays at this temperature. From this technique we get similar results as in the apparatus mentioned above.

76
As a next step to scale-up we operate process development units (PDU) where the solids can be converted in kg-scale. As given in table 4 these are: fluidized bed reactors operating semi-continuously with a feed of solids between 100 g/hour and 1 kg/hour. Reactions can be performed at a pressure of 1 to 40 bar in steam or air at maximum temperatures up to 900'C. - For pyrolysis, gasification and combustion entrained phase reactors operated as free-fall-reactors have been built up for the same amount of coals. With respect to hydropyrolysis the pressure goes up to 200 bar, other gasifying agents are steam and air. Also inert gas like nitrogen can be used. The maximum temperature is 950°C. Finally, a plasma reactor is used for the conversion of solids in the presence of hydrogen at 1 bar. The temperatures are here extremely high up to 3000-C.
Table 4; Experimental methods applied at Bergbau-Forschung to characterize solid feedstocks for pyrolysis and gasifications processes
apparatus
laboratory : Curie-Point pyrolysis
grid heater
TGA
PDU : fluidized bed
entrained phase - pyrolysis - gssification • combustion
plasma reactor
mode of operation
batch
batch
batch
seml-contlnuous
semi-continuous
semi-continuous
sample size lappr.)
5 mg
10 mg
1000 mg
100g/h - 1kg/h
100g/h - Ikg/h
lOOg/h - Ikg/h
conditions
0.1 - 200 bar (N2,H2I 500 - 950eC ca. 5 > 104 K/s
0.1 - 90 bar (N2,H2,air) 20 - 1000"C ca. 103 K/s 1.0 - 100 bar (N2,H2,H20,airl 20 - 1150eC 1 - 100 K/min
1 - 40 bar IH20,airl 600 - 900eC
1 - 200 bar (H20,N2,iirl up to 950°C
1 bar (H2I up to 3000°C 105 K/s
For the evaluation of the results in laboratory scale isothermal and non-isothermal reaction kinetics are used /2,3/. From the poduct formation as a function of time and temperature kinetic data are established. Also the product yield can be determined for pyrolysis and gasification. These are the input data for reactor models at hand both for fluidized bed or entrained phase reactors. The throughput and degree of conversion calculated can be proven first in kg-scale in the process development units and then be used for the scale-up to industrial reactors.

77
4. RESULTS The following examples for the results on coal characte
rization with respect to thermochemical conversion are mainly taken from thermogravimetric measurements, which in combination with product gas analysis has been proven to be a very powerful method on laboratory scale. As an example for the process development units a result from the plasma pyrolysis reactor is shown lateron also. 4.1 PYROLYSIS
Fig. 2 presents results obtained during the pyrolysis of a hard coal in the thermogravimetric analysis device /4/. As can be seen from the upper diagram on the left the sample was heated at 3 K/min from room temperature to approximately 1000'C within approx. 6 hours. The primary measuring curve relates to the mass left in the reactor. The coke formation per unit time can be derived from this by differentiation. The curves of gaseous product formation are obtained by gaschromatographic analysis performed simultaneously to the TGAmeasurements. The release of higher hydrocarbon gases leads to rather simple curves so that it may be assumed that only one reaction is involved in their formation with possibly 3 reactions at the most in the case of CH4. Obviously several reactions contribute to the generation of Η,Ο, CO and CO,. The formation curve of tar is obtained by subtraction of the gas formation rates from the loss of mass. Obviously tar is formed mainly in one reaction.
moss ioss d G i m g l dllglj
tar
Λ ι CCI *
gaseous HC water
ο ώ ώ 600 m ião irci
200 too sfa ώ rrci
Fia. 2; Product formation during pyrolysis of hard coal (coal: 32 % V.M. daf; 3 K/min; 1.1 MPa, N2)
Fig. 3 shows how the formation of gases during pyrolysis of coal varies with rank, ranging from anthracite to high volatile bituminous coal. In the cases of CH4, C3Hg, CO and N2

78
the gas formation is reduced when rank increases. Thereby the beginning of the curves is shifted torwards higher temperatures indicating that during coalification functional groups with weaker bonds are preferably reduced. H20 and CO2 show a different behaviour: The H20 curves indicate less gas formation with rank. Thereby the comparison leads to the conclusion that all relevant groups had been reduced equally during coalification. The CO2curves show no remarkable differences with rank /3/.
5?
0.4·
O.l·
ft
c m l l
Is >]
1, D
C
L· Ψ à
CHt
V ώ
500 700 WO Temperature I 'C I
Coals : • / .V.M, maf a Heinrich 10.0 b Dickebank 19.5
c Gustav 29.0
d Fürst Leopold 39.5
Fia. 3; Formation of gases during pyrolysis of coal of different rank
These gas formation curves can be fitted by a mathematical model on the basis of nonisothermal reaction kinetics. This is rather easily done if the curves are representing one reaction e.g. in the case of C3H8. A good fit is also possible if one has to assume more than one reaction. In the case shown in fig. 4 the fitting of the measured CH4formation by 3 reactions of second order leads to the lowest least square deviations /5/. In this way kinetic parameters for the different products and for various solid feedstocks have been determined. Table 5 gives as examples values describing the kinetics of pyrolysis of a coal at 1 and 100 bar. For the condensable products tar and benzene and the gases CH4, C2H6 and C2H4 the values for the activation energy E, frequency factor ko and order of the reaction η are given. Without going into details it can be said, that these can be well interpreted by the mechanism of pyrolysis assuming as a first step the thermal breaking of bonds between the aromatic structural elements and in the case of η = 2 a following bimolecular radical reaction as rate controlling. Practical use of these data is made in reactor modelling. Thereby the sound physicochemical basis allows a high security for

79
extrapolation from the laboratory scale into the next higher and, finally into the industrial one.
dV/dt [ cmV(gK| ]
0.3η
3 reactions 1. order S = 0,01
3 reactions 2.order S = 0,003
3X SOO TOO
o measured
— single reaction 1 . . . . i calculated
— total J
Westerholt
CH4
inert gas 10 MPa N2
S: standard deviation
Different kinds of mathematical description of measured CH4 formation curves of a h.v. bit. coal
Table 5; Overview of kinetic parameters measured in the thermobalance at 3 K/min (Coal Zollverein, 32.9 %VM)
Fig.
tar
benzene
CH4
C2H6
C2H4
1) 2) 3)
1) 2) 3)
n=1
n=1 n=2 n=2
n=2 n=2 n=2
n=2
n=2
0.1 E
138
140 142 150
204 158
210
205
N2 pressure MPa
ko
7.6 χ 108
2.1 χ 1011
6.0 χ 107
1.0 χ 107
7.8 χ 1 0 " 2.7 x 10
7
1.5 χ 1013
2.7 χ 1013
10 E
174
136 115 149
214 141 257
224
218
MPa
ko
1.5 χ 1011
2.0 χ 1011
2.0 χ 106
8.0 χ ΙΟ6
7.8 χ 10η
5.3 χ 105
1.8 χ 1011
2.2 χ 1013
7.1 χ 1013
Ε in kJ/mol ; k0 for n = 1 in 1/min ; k0 for n=2 in g/ (cm3 min)

80
4.2 GASIFICATION At first an example shall be shown, how temperature and
pressure influences the rate of the carbonH20reaction. The diagram in fig. 5 is based on experimental results measured in a TGA /6/ taking coal char. As to the influence of temperature an exponential increase of the rate of reaction can be noticed following the Arrheniuslaw. As to the influence of pressure the reaction rate increases in the range between 1 and 10 bar, afterwards the curves come into saturation. This behaviour can be explained by the LangmuirHinshelwoodmechanism, which assumes an adsorption step in the overall reaction /7,8/.
Fig. 6 compares the reactivities of lignites, coals, chars, metallurgical coke and pitch coke against steam at 40 bar depending on temperature. In all cases the rate of reaction is increasing with temperature as has been seen in the previous figure. However, great differences are found for the different solid fuels. The reactivity of the lignite is highest, those of coals and chars have been found in a relatively narrow range, whereby rank is not significant.
0 92
5 90Θ
950 Í
Fia. 5; Rate of CH20gasification depending on pressure and temperature
Finally, coke and pitch coke, that means carbonaceous material which is treated at high temperature significantly exeeding 1200'C, show lowest reactivity. As main factors influencing the scattering of reactivity, chemical structure, physical structure and catalytic influence of the ash constituents have to be taken into account. Especially the coke, treated at high temperatures shows very low reactivity as the active

81
6Û0 650
Fio. 6.; Chemical reactivity against H20 (40 bar) depending on coal rank and pyrolysis conditions
10"2g/gmin
30
20
10
10 bar, 10 Κ /min * Kosovo • Schwarze Pumpe a Rhenish χ Kolubara ° Spanish
Fia.
temperature
7: H,0reactivity of lignites depending on their earthalkaline content
sites, i.e. the rand atoms of the microcristallite structures, their dislocations and the hetero atoms are annealed at high temperature. Responsible for the high reactivities of lignites are the catalytic active constituents of their ashes. Às Fig. 7 shows some correlation exists between the reactivity of the different types of lignites and their contents of earthalcaline compounds. It is known, that these

82
are found finely dispersed in the raw samples as salts of humic acids. This is a precondition for the catalytic activity of these compounds /9/. It should be noticed, that reactivities of other materials such as biomass generally are found to be less than those of lignites. This is understandable as catalytically active compounds are not generally found in these materials. 4.3 PLASMA PYROLYSIS
Plasma pyrolysis for solids has been developed in order to produce acetylene from coal, which is thermodynamically the only stable gas at temperatures exceeding 2500"C. Within a project together with an industrial partner we have built a laboratory scale plasma pyrolyser as shown in fig. 8. Between cathode and anode an electric arc is produced. It rotates in a field of a magnet and produces a hydrogen plasma at temperatures up to 3000"C. Into this coal is injected and pyrolysed. Fig. 9 summarizes some results for different coals. The left handdiagramme shows that the acetylene yield is rising with the specific energy input and goes up to 40 % of the feed coal. Besides acetylene small amounts of ethylene and CO are formed, also tar and char are obtained /10/. Plasma pyrolysis is generally a simple technique to produce directly acetylene from carbonaceous materials. Also it should be taken into account that it could be a tool to destroy organic compounds from e.g. liquid wastes, which are not acceptable for environmental reasons.
cathode
m cool
anode■
A
w,
t
magnetic coil
H 2 0 — = i
V products C2H2
0.1 - 1 bar 2000 - JOOO'C s 10 kg coal/h s Wm„
JH2A
s 30 MW plasma generation
plasma pyrolysis
quench
Fia. 8; Laboratory reactor for the pyrolysis of solid feedstock in a hydrogen plasma

83
acetylene yield In w t - X of feed cod (ar.) 50-1
coal: Cape Breton
40·
30-
20·
10-
Cape Breton^ (Canada)
/ / \S N Luisenthal f f i \ (Saar)
Westerholt (Ruhr)
1 1 1 -
2 4 6 8 4 6 specific energy input in kWi/tcg cool
Fia. 9: Results from laboratory experiments of plasma pyrolysis for different coals
5. CONCLUSIONS With respect to the aims of the Conference to improve
pyrolysis and gasification of biomass in the future the following may be concluded:
From the broad use of thermochemical conversion of coal and other solid fuels experimental and theoretical methods are established for their characterization with respect to their use in pyrolysis and gasification processes. Especially it is possible to determine the course of the formation of different products with temperature and thereby to investigate the influence of the conditions in the reactor i.e. pressure and kind of atmosphere and rate of heating on the amount and composition of the product. Mathematical models are available to improve the design of reactors and processes.
All these methods and results can be used to characterize other carbonaceous material like biomass and wastes for the design of new or the optimization of existing processes, with respect to the feedstock available and the products desired. REFERENCES ( 1) VAN HEEK, K.H.
Char Properties nology, 15 (1987
( 2) JÙNTGEN, H. and nicht-isothermen sehen Forschung Berlin
( 3) JUNTGEN, H. and isothermal coal
and H.-J. MÜHLEN, Effect of Coal and on Gasification. Fuel Processing Tech-), p. 113/133 K.H. VAN HEEK, Reaktionsabläufe unter Bedingungen. Fortschritte der chemi-
13 (1970), p. 601/99, Springer Verlag, K.H. VAN HEEK, An update of German non-pyrolysis work. Fuel Processing Techno-

84
logy, 2 (1979), p. 261/93 ( 4) ARENDT, P. and K.H. VAN HEEK, Comparative Investiga
tions of Coal Pyrolysis under Inert Gas and H, at low and high Heating Rates and Pressures up to 10 MPa. Fuel 60 (1981), p. 779/788
( 5) VAN HEEK, K.H., P. KASSLER and W. WANZL, Übertragung von Laborergebnissen zur Pyrolysekinetik von Kohlen auf in situ-Reaktionen. Erdöl, Erdgas, Kohle 102 (1986), p. 200/205
( 6) MÜHLEN, H.-J. and A. SULIMMA, High temperature, High Pressure Thermogravimetry of Coal Gasification - Apparatus, Data Acquisition and Numerical Evaluation. Ther-mochimica Acta 103 (1986), p. 163/168
( 7) MÜHLEN, H.-J., K.H. VAN HEEK and H. JÜNTGEN, Kinetic Studies of Steam Gasification of Char in the Presence of Hydrogen, Carbon Dioxide and Carbon Monoxide. Fuel 64 (1985), p. 944/949
( 8) VAN HEEK, K.H., H.-J. MÜHLEN and H. JÜNTGEN, Progress in the Kinetics of Coal and Char Gasification. Chem. Eng. Technol. 10 (1987), p. 411/419
( 9) VAN HEEK, K.H. and H.-J. MÜHLEN, Effect of Coal and Char Properties on Gasification. Fuel Processing Technology, 15 (1987), p. 113/133
(10) BAUMANN, H., D. BITTNER, H.G. BEIERS, J. KLEIN and H. JÜNTGEN, Pyrolysis of Coal in Hydrogen and Helium Plasma. Fuel 67 (1988), p. 1120

85
ΚΤΙ ACTIVITIES IN THE FIELD OF BIOMASS PYROLYSIS
L. ANTONELLI Vice President - Director of Alternative Energies Department
Kinetics Technology International SPA, Italy
Summary Solid fuels use is becoming more and more difficult because of the environmental problems connected with their handling and burning. Strong efforts are carried on to develop suitable pretreatments to overcome handling, combustion and pollution problems. Slurrification of fossil coal (coal-water mixture, CWM) is starting to have a significant role in the industrial installations. The production of slurries based on charcoal from biomasses pyrolysis shows specific difficulties and requires sophisticated technologies and know-how. The ^critical factors and the results obtained with our production process are illustrated. The production of charcoal slurries on semi-industrial scale and their physical and rheological behaviour is discussed. Evaluations of energy consumption in slurry production and comparison with pit-coal slurries is also analyzed.
1. GENERAL Figure 1 shows the programs developed by KTI in fuels production
from biomasses pyrolysis. All the aspects of the production of these alternative fuels was investigated including the ecological impact.
Figure 1 : 1. Pyrolysis process development 2. Charcoal and bio-oil utilization as fuels 3. Ecological impact (pyrolytic waters etc.) 4. Economics
In this paper the charcoal use as industrial fuel is presented.
2. CHARCOAL UP-GRADING The use of solid fuels is becoming more and more difficult because
of the environmental problems connected with their handling, sulphur and ashes content.
A system to overcome these drawbacks has been the up-grading of the coal transforming it in a coal-water slurry (CWM). The advantage is to transfer the coal in a liquid fuel eliminating the pollution problems connected with the storage, transport and handling.
The pollution due to SOx and ashes require further treatments. The advantages are significant and a number of large industrial plants have successfully adopted this system of burning coal and many others are considering this possibility.
With the target of the maximum flexibility in the use of the pyrolysis fuels, we started a program to develop the same system of using charcoal.

86
The difficulties of obtaining a char-slurry with acceptable solid content are well known and are mainly due to:
- high porosity - high surface area - high oxygenated groups concentration on the surface area with
consequent hydrophilic characteristics - low bulk density On the contrary, there are interesting positive points due to the
absence of sulphur, to the relatively low ashes content and to the good reactivity.
The program was developed in three phases: 2.1) Phase 1
Laboratory tests have been started using two different categories of char produced in the Raiano pyrolysis plant from wood chips and olive husks. The tests were oriented to investigate the following:
- grinding techniques - granulometry distribution influence on the viscosity and
stability - dispersion and stabilization agent selection and dosing - influence of the oxygen during char milling - influence of the char pretreatment at high temperature. The results of this first phase were: a) The max. solid content of the char based CWM has been as high as
52% by weight, with a slurry viscosity below 1000 cP. b) The bimodal size distribution curve of the pulverized char is
producing an unstable slurry. c) By using a very finely ground char powder with a monomodal size
distribution, the same solid content and the same viscosity, as for the bimodal distribution, was obtained but with the advantage of a very good slurry stability. Another advantage is the simplicity of the process to obtain the monomodal slurry.
d) None of the programmed surface modifications of the char (heating treatment, wetting, ash leaching) resulted successful in increasing the solid content in the char-water slurry.
e) The grinding of char under controlled atmosphere of nitrogen increases the solid content of the char based CWM by 5 to 10J with respect to the grinding in presence of air.
It is worth noting the peculiar behaviour of the char slurries with respect to the granulometrie distribution. Monomodal is better than bimodal which gives unstable mixtures. This is due to the surface physico-chemical characteristics which are not modified by the thermal and chemical treatments tried during the tests.
Figures2 and 3 summarize the results obtained for four typical mixtures prepared from wood chips char and olive husks char.
Figure 4 shows the Theological behaviour of char slurries compared with pit-coal conventional slurries.
Figure 5 shows the Theological behaviour of two typical slurries with micronized char. The sample containing olive husk char has a lower viscosity.
Figure 6 shows the trend of viscosity against the temperature. 2.2) Phase 2
At this stage of the program, the experimental work was transferred from laboratory to pilot plant. The main targets were:
- Reproduction of the best lab results;

Mixture type
87
Mixture no.1 Mixture no.2 (*)
Mixture no.3 (")
Coarse particles/fine p. Char theoretic content in the final mixture (% weight) Dispersing agent (J weight) Char content in the final mixture (% weight)
60/40
44,9 0,7 46,9
60/40
46,7 0,7 48,8
60/40
49,7 0,7
51,9 * grinding in air ** grinding in nitrogen atmosphere
Fig. 2. Solid Content in the Different Aqueous Slurries Obtained from Wood Chips Char
Mixture type Mixture no.4 (»)
Coarse particles (wt) /fine p. (wt) Char theoretic content in the final mixture (% weight) Dispersing agent (% weight) Char content in the final mixture (J weight)
* grinding in nitrogen atmosphere
60/40 46,7 0,7 48,7
Fig. 3. Solid Content in the Olive Husks-Char Water Mixtures
2 . 0
1 .8
1 . 6
1.4
1 .2
1 . 0
0 . 8
0 . 6
0 . 4
0 . 2
n.n
-
-
-
-
\ TOTBWTUTC « 20 ' c
^ \ ^
A r ø U 5 3-
W R T W T" ^ ™m rø απ« β , . „ „.„, Α Ι Β
****** auwr « Μ ΤΗ« κ« w.tr saio
AQUEOUS SLURRT WITH OUR FRUI CLIVE HJSKS (48.TTC U.CF SOLIDI
ι * 2 0 40 60 8 0 100 120 140 160 180 200 220 240 260 280 300
9€AR RATE 1/S
Fig. 4. Charcoal Based Aqueous Slurry and Pit-Coal Water Conventional Slurry

88
- Better understanding of the influence of the granulometry; - Investigation of the influence of the milling equipment when operating at a scale which can be correlated to the industrial dimension;
- Acquisition of data related to the energy consumption for the milling and mixing stages;
- Definition of the best conditions for increasing the solid content above the limits reacted during the lab tests.
A campaign of tests was carried on using a wet milling in single stage. The simplicity of this scheme is very attractive for an industrial application.
The investigation on the bimodal distribution can be done adding a coarse fraction prepared separately. The additive is the same used in lab tests. The milling is done in the presence of air. Characterization of typical samples can be summarized as follows:
Solid content Values higher than those on lab scale are obtained, concentration
around 55-56J can be produced with micronized char. Slurries viscosity
The samples produced in the campaign have a viscosity in the range 600-2000 cP at 30 deg.C, compatible with the pumpability requirements. Furthermore the behaviour is pseudoplastic with significant advantages in industrial application.
Figures 5 and 6 show the Theological behaviour of some samples. Figures 7 and 8 Show the granulometry distribution of the same samples.
Slurries stability A series of slurries produced during the tests have been stored in
drums and the tendency to solid décantation and water separation checked every 2/3 days. With the same frequency all samples have been roughly mixed by hand. After forty days, the Judgement on the most significant sets of samples can be summarized as follows:
E1 very good E3 excellent Ek very good E5 good E12 acceptable In any case, thé analysis of the results shows that all the
products can be used in an industrial plant if midly mixed with a daily frequency as it is normally done in all the installations using CWM. 2.3) Phase 3
Since the max. content of solid obtained using only char was 55-56J, even using micronized powder, another line of investigation was set up trying to produce slurries with char and pit-coal.
The basic concepts at the basis of this approach are: - The pit-coal can be used for CWM production having solid content up to 65-70J.
- The combination of the two coals makes possible to reconsider the utility of the bimodal distribution using the pit-coal only for the coarse function.
- The dilution of the polluting components (sulphur) makes possible

89
2.0
1.8
1.6
1.4
12
1.0
0.8
0.6
04
0.2
0.0
TEM"ERATURE ■ 20 ' c SOLID CONTENT IN BOTH SLURRIES ■ 52Z W.
MONOMODAL (FINE SIZE} CHAR POVCER FROM WOOD CHIPS
ItïICtCOAL (FINE SIZE) CHAR POWDER FROM OLIVE HUSKS
—1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
0 20 40 60 80 100 120 140 160 180 200 220 240 260 280 300
SHEAR RATE 1/S
Fig. 5. WaterCharcoal Mixtures Viscosity
1.6
1.4
1.2
1.0
0.8
0.6
0.4
0.2
SHEAR RATE· 100 m'
20 1 — 40
i — 60 80
1 — 100
TEMPERATURE " c
Fig. 6. Viscosity Related to Temperature for WaterWood Chips Char Mixture

90
the utilization of large quantities of piteoai remaining in the limits imposed by the antipollution regulations.
The new system consists in incorporating a fossil charcoal having an average granulometry of about 45 microns in a micronized pyrolytic coal slurry (dm. 1216 microns). Such procedure will permit to obtain the following advantages:
Higher solid content in comparison with the pyrolitic carbon slurry only.
Lower energy consumption in comparison with the completely wet procedure.
Better fluidity with the same concentration, due to the bimodal distribution.
Lower sulphur and ash content in comparison with the two separate components.
It is also possible to foresee advantages in the plants operations; for example the char slurry production will be done in the pyrolytic plants spread in the territory, the fossil carbon grinding and its incorporation in the micronized char slurry will be centralized close to the power station which uses the fuel.
The operative diagram of such production is the following: PYROLYSIS PLANT Production of micronized char at 50*
I Transport to the power station
POWER STATION Grinding of fossil coal at 45 microns
^ >
High concentration slurry
■
Mixer
■ ' Burner
Experimental work A first campaign of samples production was carried on obtaining
slurries with 60J of char and pitcoal. The ratio between the two coals is 60 and 40? char. The coarse pitcoal is added to the char slurry (50Í). The behaviour is Newtonian, the resulting slurries have a LHV of 4000 Kcal/kg and are usable as fuel in a burner.
A second campaign aiming to increase the solid content to 65J is carried on with a ratio char/pitcoal 40/60; 50/50 and 54/46. Analysis of the produced samples show increasing viscosities. With small adjustments on the additive quantity, which is increased to almost 1J the slurries obtained are good and usable in a burner. The HV is higher than 4500 Kcal/kg.
In conclusion, this second group of samples confirm the possibility of producing mixed slurries with 65Í solid and good viscosity stability and comparable with the pitcoal slurries.
The final experiments tried to increase furtherly the solid content above 65%. The result was that it is still possible to reach 69/70% but the viscosity is high and at the limits of acceptability for industrial applications.

91
3. ENERGY CONSUMPTION The energy consumption registration shows that during all the tests
the milling process was regular without big variation in power absorption. Indications were also obtained for the consumptions expected on industrial mills with capacity 10/100 times higher. Figure 7 summarizes the results obtained on pilot plant: Figure 7 Run Energy consumption
KW Slurry flow
Kg/h Char flow
Kg/h Consumption KWh/t
a b A B
E1 E3 EU E5 E12
2,2 2,1 2,2 1,9
15,3 20,4 18,2 16,7
(35? di E1 + 65? di E2)
7,81 86 170 43 85 10,52 62 120 31 60 10,51 73 125 37 63 10,75 68 106 34 53
58 114 29 57 Legend: pilot mill a = consumptions referred to slurry
b = consumptions referred to char A = consumptions referred to slurry B = consumptions referred to char
The experience matured on pitcoal allows to foresee a reduction of 4060? of the figures shown in A and B when using industrial scale equipment.
A further reduction can be obtained using multisector ball mills for which the above experimental results allow a forecast of 2540 KWh/t of slurry which with a solid concentration of 56? gives a consumption of only 4570 KWh/t of dry charcoal.
1500
1000
500
0
,
E12
* ^ ^ « ^ _ *» * ^
1 ι ι 50 100
150 200 250
E2o
E1,
E3.
Fig. 8. Different Samples Viscosity at 30°C

92
4. CONCLUSIONS The case of the process investigated in this program allows the
production of excellent slurries with a solid content of 65>. Higher concentrations are technically possible but not usable in industrial burning systems. The resulting slurries have roughly 50J char, 50Í pit-coal with a LHV above 4000 Kcal/kg.
Figure 8 shows the Theological behaviour of some typical products. Energetic consumption: taking into condlderation the products with
solid (char/pit-coal 50:50), the energy consumption is 35 KWh/t of slurry or 54 KWh/t of solid including the milling stage. The same value for slurries with only pit-coal is 70 KWh/t of slurry with 70J solid or 114 KWh/t of coal. This calculation are relative to a slurry production of 20 t/h.

93
BIOMASS FUELS AND GASIFICATION
J. CARRE, L. LACROSSE, Y. SCHENKEL Center for Agronomical Researches (CRA)
Unité."Biomasse" 5800 GEMBLOUX, Belgium
F. RURIHOSE Université Catholique de Louvain (UCL)
Unité "TERM" 1348 LOUVAIN-LA-NEUVE, Belgium
Summary
Fixed bed downdraft gasifiers have specific requirements on fuels . This paper considers the different fuels characteristics that are necessary to obtain an optimal gasification : moisture, mineral and volatiles contents, size, bulk density,... The densified products quality is also considered, particularly their cohesion . Specific testing methods have been developped at the CRA . They allow to fix quality limits for these products .
1. INTRODUCTION Many important studies have been carried out on gas producers designs.
The gasified fuel has often been neglected . Indeed, a well-designed gasi-fier will never work with fuels that do not have a minimum quality .
This paper presents the quality limits that fuels must have to be successfully gasified in fined bed downdraft gasifier .
2. FIXED BED DOWNDRAFT GASIFIERS REQUIREMENTS ON FUELS 2.1. Moisture content The fuel moisture content is of prime importance for the success of
the gasification process . The negative influence of water can be noticed at different levels : - the lower heating value (LHV) of lignocellulosic material decreases when
the moisture content increases . Considering that ashfree dry biomass LHV is about 18.8 MJ/kg, the LHV of wet biomass becomes : THV -IflS 100 2 c H (1) L H VH - 1 8 · 8 100 + H 2 · 5 100 + H LHV = wet biomass lower heating value (in MJ/kg) H = moisture content (% dry basis) For instance, ashfree biomass containing 20% moisture content (dry basis) would have a LHV : LHVH = 18.8 y|g - 2.5 - ^ = 15.25 MJ/kg

94
water in excess induces the hearth cooling . The resulting temperature does not allow the cracking of the heavy hydrocarbons coming from the pyrolisis zone of the gasifier . Moreover, the water itself is not cracked and will condense later in the cooling and cleaning system . it must also be said that, thermodynamically, it is always more interesting to gasify as dry as possible fuels . Indeed, the gas LHV and the gasification efficiency quasilinearly decrease from 0 to 50% moisture content (see fig. 1) . The hydrogen and methane gains in the gas do not compensate the carbon monoxide losses (see fig. 2) .
100
90
80
70
M 60
50
40 10 20 30 40 50
MOISTURE CONTENT (%, d.b.)
40
35
30
Q 25
g 20 ω Η
115 10Η
5
0
Fi g . 1 : Gasification efficiency and gas LHV as functions of moisture content
o CO
• CO,
_ Δ _ Δ _ Λ _ Λ _ Δ Λ _ Λ * * 10 20 30 40 50
MOISTURE CONTENT (%, d.b.) Fig.2 : C0,H , CH4 and C02
contents in the gas as functions of moisture content
2.2. Mineral content The fuel mineral content must be as low as possible . It also affects
the LHV . Introducing the mineral content in relation (1), it becomes :
LHV,, 18.8 100 MC 2.5 Η
Ή 100 + Η 100 + Η MC = mineral content (% dry basis)
For instance, biomass containing 20% moisture content and 10% ash content, would have a LHV :
LHV„ = 18.8 Η
100 10 120
20 2.5 y|^ = 13.68 MJ/kg

95
Moreover, the mineral content can make the gasification impossible . Indeed, the temperatures that are reached in the hottest zone of fixed bed downdraft gasifiers are often higher than the biomass ashes melting points. When the mineral content is too high, clinkers will progressively appear in the hearth . They will finally prevent the gas from passing through .
Usually, it is considered that higher than 5% mineral content, it is nearly impossible to avoid clinkers formation . But it is dangerous to generalize : following the mineral composition, melting happens at more or less high temperatures . For instance, the presence of alkali oxides can induce eutectics formation at relatively low temperatures .
For exemple, the mineral content of rice husks is about 20-25% with more than 90% silica (Si0„) . The latter has a melting temperature of about 1700°C . If it was alone, the risks of melting would be very little. Unfortunately, it is principally accompanied by kalium oxide (K.0) but also by Mg, Al, Ca, Fe, Na oxides . It results a much lower melting temperature .
2.3. Volatiles content The biomass volatiles content may not be considered as a limiting
factor . Fixed bed downdraft gasifiers must actually be designed in order to crack the tars and heavy hydrocarbons liberated in the pyrolises zone .
However, it must be noticed that thermodynamically, biomass must not only be as dry as possible (cfr supra) but should also ideally undergo a thermal treatment (torréfaction) .
The gasification efficiency is optimal for dry biomass having lost an equivalent of 20% moisture content (see fig. 3) i.e. biomass with a formula of about C L . 0. , .
0.9 0.4 100
Gasification efficiency and gas LHV evolutions as functions of moisture content or pyrolisis level

96
2.4. Size The optimal size for a fuel that has to be gasified depends on the
gasifier hearth dimensions . It must be such as to allow an optimum air/ fuel contact .
It is generally considered that the length of the fuel must be between 1/10 and 1/5 of the hearth diameter . Out of these values, problems of irregularities in the gasification process can occur : - too long fuels can generate bridges preventing the fuel from flowing down
to the gasifier hearth . The gasification reaction can tend towards combustion by air excess .
- too small fuels cause high pressure drops by forming a kind of airproof cake . For small size fuels (sawdust, agricultural residues,...) densification can be a solution but the densified products must be of prime quality and answer some criteria (see below) . 2.5. Bulk density The fuel bulk density directly influences the gasification velocity
and the residence time of the fuel in the gasifier hearth . If the bulk density is too low, it is difficult to maintain gasification reactions . The air excess also turns gasification into combustion .
2.6. Ultimate analysis and heating value If the only organic material is considered, it may be said that the
different kinds of dry biomasses more of less have the same composition in carbon, hydrogen and oxygen : C = - 50%
H = I 6% 0 = - 44%
It corresponds to a formula of CH. ,, 0. .. and a lower heating value c ίο α U T Λ 1.44 0.66 ö
of 18.8 MJ/kg . The latter mainly varies with the moisture and mineral contents (cfr
supra) . 2.7. Densified products quality Densified products generally answer the above quality criteria .
Indeed, their moisture content is low (<10%) and their density is high . Moreover, it can be possible to adapt their size to the gasifier hearth dimension .
But another quality must absolutely be satisfied : cohesion Researches carried out at the CRA allowed to develop testing methods
in order to determine this parameter . This methods are described by Carré et al. (1984) . The most important are the ones concerning the measurement of : the density after different conditionings the rate of volume change in immersion the friability index after different conditionings the elongation and swelling at 20"C 95% RH Table 1 give the quality limits corresponding to these tests .

97
Density at 20°C 65% RH (kg/m3)
Density at 20°C 95% RH (kg/m3)
Rate of volume change in immersion (% min)
Friability index at 20°C 65% RH
Friability index at 20°C 95% RH
Elongation at 20°C 95% RH (% initial length)
Swelling at 20°C 95% RH (% initial diameter)
800 1200
Υ//Λ 77Δ
50
0-5
100 ΊΖΖΔ 075
y/A \ 03
ΈΖΣΖ. 0 5
10 25
VM 10 25
WZÅ Bad ////// Acceptable Good
Table 1 : technical prescriptions for densified fuels to be gasified
3. CONCLUSIONS When a gasification unit does not work properly, the gas producer,
i.e. the technology is more or less always suspected . Actually, the operators are very often responsible for the problems
that occur . One of the reasons is that they are careless about the fuel characteristics : the gasified biomass is too wet, or bad calibrated, or contains too much inorganics .
They too often forget that downdraft fixed bed gasifiers require very good quality fuels .
It may be said that the respect of the above prescriptions must allow to avoid many important problems and to have a good working of the gasification installations.
REFERENCE CARRE, J., HEBERT, J. and LACROSSE, L. (1984) Critical analysis of the dry process improvement of ligneous materials for energyproducing purposes . Final report CEC (DG VIII)CRA, 245p.

98
USAGE OF CARBON BLACK AND ACTIVATED CARBON IN RELATION TO INPUT AND TECHNICAL ASPECTS
OF THE PYROLYSIS PROCESS
B. Bilitewski, G. Härdtle, K. Marek
intecus associated engineers for environmental protection technologies
Berlin
SUMMARY
A variety of pyrolysie char made of domestic waste, biomass, sewage sludge, plastics, rubber etc., has been examined in regard to its usage as combustible, carbon black and activated carbon (1). Pyrolysis char made of plastics and waste tyres has a high content of carbon and a low content of ash and thus it is very suitable as
carbon black to enrich low and medium rubber qualities a colour pigment in printing ink, colour and plasticsindustry activated carbon for waste water purification
But in spite of the different possibilities to be used pyrolysis char still has not been produced and commercially sold. The examination shows that the raw material cannot be sold without a further treatment, for example the enlargement of the BETsurface.
1. INTRODUCTION
The degasification of waste tyres produces char with a greater or lesser carbon content. It is essential to find a suitable method of reutilizing this product if the pyrolysis process is to be applied economically. Particularly the pyrolysis char obtainded from waste tyre degasification appears to offer a number of interesting possibilities. With a pyrolysis temperature of 700 °C and a retention time of 50 minutes, we obtain a residual char content of more than 40 Ζ by weight, with an upper heating value of approx. 31,400 KJ/kg and a carbon content of approx. 87 Ζ by weight. Cocombustion trials of this char with mineral coal have yielded positive results, although the high sulphur content (2.8 Ζ by weight) constitutes a considerable problem. Orienting tests to determine the suitability of the char for application as activated carbon, as carbon black for vulcanisâtes, as pigment or in the printing ink industry have shown that the pyrolysis char cannot be satisfactorily reutilized without further treatment.

99
2. ACTIVATED CARBON AND ITS APPLICATION
Pyrolysed waste tyre char can be effectively activated with steam in a temperature range of 750 °C 950 °C. Fig. 1 shows the internal surface area of the different sorts of activated carbon as a function of burnoff. By burnoff, we mean the gasified portion of the carbon in the char, expressed as a percentage (1). The symbols used mean: A700, A700PA waste tyres, 700 pyrolysis temperature (°C), t 10 pyrolysed for 10 minutes, t max pyrolysed for 2 hours, where no retention time given, pyrolysed for 50 minutes. Ρ in powder form, AH waste tyre char from the fluidizedbed reaktor at the University of Hamburg, Prof. Dr. V. Kaminsky, K sewage sludge, Sp paper mill waste.
tm^/g] infernal surface area 600
400
200
A70o',f=10 A700V \ /
A550 ¿yv
T V ^ r ·
r\AH
i A700P
A700.t= max Sp700
K 700 ^ ■ ^
20 40 60 80 1001%] burnoff
Fig. 1 Internal surface area of the different sorts of activated carbon as a function of burnoff (Nz isotherm) (1)
It is evident that neither sewage sludge nor paper mill waste are suitable for the production of activated carbon. Other important factors for the possibility to use char as activated carbon are temperatur, reactor type and the retention time of the pyrolysis of waste tyre. Temperatures around 700 °C and retention times around 1015 minutes during the pyrolysis process are advantageous for the production of activated carbon. The fluidized bed reactor (AH) however produces char which is not as suitable as the char in a rotation drum.
Our results show that the theoretical considerations borrowed from coal gasification (2) are applicable to the partial gasification of waste tyre char within the limits of permissible variation. This means that the characteristics of the activated products can be calculated as a function of burnoff, where the cavity structure of the feed char is known. The correctness of the postulate can also be verified qualita
tively by the change in the pore distribution in the adsorption and macro pore structure as a function of burnoff.
Fig. 2 shows the differential radius distribution of the adsorption pore structure for activated carbon from waste tyre char with varying burnoff characteristics.
To begin with, the char has narrow pores which expand to produce larger pores in the course of partial gasification at higher burnoff values. The maximum number of pores is reached at about 200 A. Comparable samples of activated carbon from mineral coal have their maximum number of pores at 100 A and less. For this reason, such commercial activated carbon has a considerably greater internal (BET)

100
surface area (Hydraffin BS 12 - 1200 mz/g) with the same total pore volume as activated carbon from waste.
radius [Ál Fig. 2 Differential pore radius distribution for activated carbon from waste tyres with varying burnoff characteristics
Activated carbon was contacted with the test substance phenol dissolved in water until an equilibrium condition was reached. As phenol frequently occurs in waste water, it constitutes an interesting test substance for waste water treatment. There are two standardized methods for tests using phenol: the DIN standard test method 19 603, which governs the determination of the isotherms using powdered activated carbon in a mechanical shaker with an input concentration of 10 mg/1; and the AWWA standard test method, according to which the phenol adsorption of a layer of granulated activated carbon is determined when subjected to a phenol solution flow.
The adsorption equilibrium was determined by contacting phenol dissolved in water at a fixed input concentration with a predetermined quantity of activated carbon. If the carbon remains in equilibrium with the solution for a sufficiently long period, and no further changes in the adsorption behaviour are detectable, we speak of an equilibrium adsorption. The concentration corresponding to this state is known as the equilibrium concentration. The equilibrium adsorption QQ is defined as the quantity of the test substance in mg that can be adsorbed from the solution per g activated carbon:

101
(CA. Ce) (mg/g) M
Qo ■ equilibrium adsorption (mg/g) V volume of solution (1) M quantity of carbon (g) CA. input concentration (mg/1) Ca equilibrium concentration (mg/1)
As adsórbate a phenol solution with an input concentration of 10 mg/1 and a bath volume of 10 litres was used. The char sample weighed 1 g and was subjected to a constant flow of phenol solution. The concentrations of the solutions were measured continuously with an ultraviolet spectrometer, and the adsorbed quantities calculated in accordance with equation above. Generally speaking, phenol adsorption is increased by a factor of 8 through an activation process (850 °C and 30 minutes). By way of comparison, the commercial activated carbon Hydraffin BS 12 was also tested. The equilibrium adsorption condition is reached between 16 and 20 hours, whereas Hydraffin reaches an equilibrium condition after only 6 hours.
The slope of the equilibrium isotherms, at the same input concentration and with a constant quantity of solution, is determined by altering the weight of the samples of activated carbon and plotted in terms of loglog coordinates (fig. 3). Fig. 3 shows the phenol adsorption capacity of activated carbon from waste tyre with different burnoff characteristics as a function of the residual concentration.
lwt%) phenol adsorption 5.0
2.0 5,0 10 (mg/Il
residual concentration of phenol
Fig. 3 Representation of phenol adsorption as a function of the residual concentration in terms of loglog coordinates

102
twt 5
3
2
1
0,7
0,5
0,3
0,2
%] DCadsorption
BŞ12, : A700,til0
• Ι OW
Sjp* fop
ÍÍA700F
η
Γ ι
»
1
30 50 100 200 300 [mgDC/l]
Fig.4 Dissolved carbon adsorption from waste water as a function of the residual concentration in terms of loglog coordinates
Taking, on the basis of this figure, the phenol adsorption capacity (in 2 by weight) for a residual phenol concentration of 1.0 mg/1 as a criterion for assessing the activated carbon, the high quality of the waste tyre char is confirmed by increasing burnoff. The capacity of the carbon increases with higher burnoff as the almost parallel moved slope became steeper and the adsorption capacity went down.
The reference carbon samples Hydraffin 214 and BS 12 showed very good results. With a residual phenol concentration of 1.0 mg/1, we obtain, a phenol adsorption capacity of 9.8 Ζ for Hydraffin 214 and 4.6 Ζ for Hydraffin BS 12. Because of a steep gradient both of these activated carbon samples can only be applied successfully in a relatively narrow concentration range.
Adsorption tests designed to demonstrate the characteristics of activated carbon cannot be limited to the adsorption of suitable test substances. An orienting study was therefore carried out in which granular and powdery carbon samples were produced from waste tyres and tested together with two commercial activated carbon samples. In a laboratoryscale sewage treatment plant the carbon was added to the activated sludge tank in accordance with the biocarbon process. First of all, the isotherm was plotted on the basis of tests using a mechanical shaker. Fig. 4 shows the adsorption isotherm for waste water which has not been subjected to biological preclarification, when tested in conjunction with the four different carbon samples. The input concentration of the waste water measured CA. 145.3 mg DC/1. The curves obtained are, as far as the gradient is concerned, comparable with the results obtained by Franke et.al. (3) for activated openhearth carbon and waste water treatment carbon.

η [%] reduction 100
103
IJ [%) reduction 100
90
80
70
60
30O[mgO2/l] BSB5input
Fig. 5 Percentage BOD reduction plotted against BOD input with and without carbon
50
... A700,*10 ° 214 ,' y~)
» BS12 \ ·
without carbon y^
A700P
' . °o 2
« Χ
* X ^
Χ
1000 1500 2000 2500 3000 [mg/I] K2&2O7input
Fig. 6 Percentage COD reduction plotted against COD input with and without carbon
On the basis of the gradient, it is possible to describe the behaviour of the carbon in activated carbon filters. Gradients greater than 1 result in separating factors of more than 1, and thus cause an unfavourable equilibrium. In the case of isotherms such as those in fig. 4, complete removal of the substances in a filter is likely to prove difficult owing to the unfavourable equilibrium. However, it is obvious form the curve that the powdery activated carbon A700P from waste tyres demonstrates the most favourable characteristics.
In fig. 5 and 6 the performance of the carbon sample A700P is confirmed. Here the percentage BOD and COD reduction is plotted against the BOD and COD input and compared with the biological BOD and COD reduction. With the help of the activated carbon, the biological BOD and COD reduction can be increased by approx. 1025 Ζ depending on the type of carbon tested.
3. CARBON BLACK AND ITS APPLICATION
The utilization of residues of the pyrolysis of waste tyre as a filler in the rubber industry is very important. Their utilization as industrial black carbon for low technical rubber articles is undeniable, but the most important field of application would be the utilization of residues as enrichment fillers in the production of tyres. We examined the following possibilities at the TUBerlin:
black carbon from the pyrolysis process as halfenrichment black carbon SRF or GPF black carbon from the pyrolysis process as fullenrichment HAF black carbon, as it is for example used for the top surface of tyres
Fig. 7 shows the characteristics of black carbon of the pyrolysis of waste tyres in comparison with normal black carbon as it is used for the vulcanisation of tyres. The mechanicaltechnological results are represented as a function of the temperature. The statement can be made

104
that the property of all used black carbon don't correspond to the standard. The same results have been achieved in Japan by Kobe Steel, Ltd. (4).
But in case of lower temperatures the black carbon quality generally became better.
Another field of application which we examined concerns the black carbon pigment in the production of printing inks. In a company for printing ink in Berlin we examined the possibilities of utilization of black carbon from the pyrolysis process of waste tyres for book, offset, newspaper rotary and heliogravureink. Only one examined black carbon met the requirements for the newspaper rotary print. The particle size distribution, the adsorption capacity of oil, the set behaviour and the swelling time corresponded the usual commercial requirements. The ink proved to be slightly more blue than normal printing ink. The intensity of ink of black carbon usually becomes better the smaller the particle size granulation is and can be improved by a corresponding treatment.
tear strength a b r a s i o n
[ Κ « N/m] [ mm ]
• H A F Q P F
> τ 1 I I ■» » 5 0 0 6 0 0 7 0 0 8 0 0 BOO f C ]
Χ·
S χ
χ
HAF 900 600 700 βΟΟ 900 f C]
[ Χ ]
elongation tensile strength
500 eoo 700 eoo eoo
[ M * N/m] 15,
HAF G P F
I* C]
Fig. 7 Impact of pyrolysis temperatur on the physical property of vulcanisation products in comparison with two commercial(HAF, GPF) carbon blacks.

105
REFERENCES
1. Bilitewski, Β.: Gezielte Herstellung von Adsorptionskoksen aus Abfällen für die Abwasserreinigung, Dissertation, TUBerlin, 1980
2. Juntgen, H.: Gezielte Herstellung von Adsorptionskoksen für die Wasser und Luftreinigung, Habilitationsschrift, Universität Heidelberg, 1966
3. Franke, F. H., Motadi, M., Bohnke, B.: Forschungsbericht des Landes NordrheinWestfalen, Nr. 2678, Westdeutscher Verlag, 23 pp.
4. Kawakami, S. ; Inone.K.; Tanaka.H.: Pyrolysis Process for Scrap Times in American Chronical Society 1980

106
RAPPORTEURS REPORT ON SESSION II PRE-TREATMENT AND CHARACTERIZATION
Catherine Esnouf Cemagref B.P. 121
92164 Antony Cedex France
PRESENTATIONS
Introduction This session was concerned with feedstock variety and its adaptation to
thermochemical processes; two very different examples of charcoal valorization were also presented.
Feedstock Characteristics The diversity of feedstock was emphasised as being the most important
point, as the feedstock determines the process used and the products obtained. This was clearly shown throughout the conference.
Feedstock diversity Not only do different raw materials have to be considered, such as
agricultural waste, wood, refuse etc., but also the different characteristics of those raw materials. Wood, for instance, is affected by the type of forestry it comes from, the parts of the tree that are collected, and the harvesting techniques, giving rise to as many products as there are processing lines.
This gives very different costs: wood from 48 ECU/dry t to 70 ECU/dry t; agricultural waste 25 ECU/dry t; and municipal solid waste from 34 ECU/dry t (wet fluff) to 53 ECU/dry t (dry fluff) and 65 ECU/dry t (pellets:RDF) without considering disposal credits.
Feedstock influence on thermochemical processes A detailed study has been conducted on coal, showing the influence of
chemical and physical structure, as well as mineral matter content. Experimental and theoretical methods have been developed to link feedstock composition to process, and the use of such methods has been proposed for biomass and wastes. The main parameters influencing biomass gasification were presented, for example moisture and mineral content, size and bulk density, volatile content, and their effects on gasification were emphasised.
Pretreatment Feedstocks may need pretreatment to improve the subsequent process, but
it must be remembered that it involves additional cost. With regard to biomass, a critical study on densification for gasification was presented, showing that good and bad methods exist. The minimum cost of pelletisation is US$100/t.

107
Municipal solid wastes (MSWÌ Pretreatment of municipal solid wastes (MSW) should include as few steps
as possible as set by the feedstock and the required output of the plant. In this respect, several critical steps are recommended for any plant recovering energy: liberation and screening; magnetic separation; and coarse shredding - these operations give separation of the fines, oversize putrescibles and ferrous materials. These are steps with high mass and energy efficiency which provide better plant performance and improved energy recovery. Their viability is due to the low cost of implementation.
The manufacture of refuse derived fuels (RDF), however, is often much more questionable: it entails a high cost and complex process and a market for the products has to be found.
Charcoal valorisation Two different strategies for charcoal valorisation were reported:
(i) A large scale biomass project was presented in which charcoal-coal-water slurries have been manufactured for burning in power stations. Compared to coal slurries, the reduced energy needs and absence of sulphur are advantages. The production cost was reported as US$25/t.
(ii) A project using wastes as feedstock for the specific markets of carbon black and activated carbon was also presented. The effect of products specifications on pyrolysis conditions and the importance of the feedstock were reported as major considerations. An acceptable activated carbon was obtained in a three-step process, but the carbon black produced was not of an acceptable standard so that new markets had to be sought for the product.
Strategies of the latter type are more short-term as they provide answers to environmental concerns and could therefore quickly prove profitable. Short-term opportunities for pyrolysis and gasification mean, therefore, that diversification is necessary such as looking for feedstocks that could match market needs through appropriate processes.
DISCUSSION
Feedstock characteristics A discussion took place on moisture content relating to the presentation by
Dr Lacrosse: what is the influence of water? Is it the same on every process? Is it right to suggest zero moisture content as the optimum?
It was agreed that it depended on the process: gasification uses water for the shift reaction and the performance improvement versus cost of removal of the last remaining moisture is disputable. For pyrolysis, with water being produced in the reaction, the less moisture content in the feed the better. On the optimum gasification efficiency presented for partially carbonised (torrified) wood, Dr Lacrosse said that it was the result of a theoretical computation, no experiments having been performed. Mr Bonino stated that this was confirmed by his own experiments.

108
Pretreatment The discussion focussed on the briquetting technology for gasification where
it was asked what technologies had produced "good" and "bad" briquettes. Dr Lacrosse replied it was very difficult to give a general answer, the methods being as important as the equipment used. Generally screw extruders gave the best results, but a small number of tests in a piston extruder in carefully controlled conditions proved to be better on one occasion. It was suggested that a good characterisation method for pellets was the measurement of mechanical strength at high temperature.
Questions were asked on the cost of such pretreatment. Dr Lacrosse replied that it depended greatly on the feedstocks, whether it had to be dried and/or ground before compression. Whichever method was used, a minimum cost of US$100/t was incurred reaching up to US$150-200/t.
Charcoal Characteristics Questions were asked on lignite behaviour and whether the evolution of
reaction rate as a function of temperature and pressure were the same as those presented for coal char? Mr van Heek replied that no systematic study had yet been conducted, but that one has to be very cautious on extrapolations from one feedstock to another. For instance in the case of ash content, carbonate formation in the case of lignite leads to a reaction rate decrease with pressure increase.
With reference to the specific uses of carbon as higher value products, questions were put forward concerning the value of the carbon black. Mr Bilitewski stated that for an acceptable carbon black to be obtained, specific reaction conditions had to be used in tire pyrolysis. The cost of making good carbon black was 200-350 DM/t (1985 figures). For activated carbon, the process is more complex and results in a production cost of 1200-1400 DM/t. Dr Roy, from the University of Laval, indicated that in his process of vacuum pyrolysis, the resulting char from tires had a better homogeneity but that the steel should be eliminated in order to avoid catalytic formation of pyrolytic carbon.
Examples were requested of large scale uses of char-water slurries as well as information on the economics of the process. Mr Antonelli stated that current slurry usage concerned coal slurries; two large scale boilers in the petrochemical industry presently use these slurries in Italy. Huge projects for coal slurry use exist in the USSR, as slurries allowing the transport of coal in pipelines. As for the economics, the last evaluation indicates a total production cost of 30-40 Lit/kg, i.e. US$25/t; with the cost approximately equally distributed into operating cost, capital cost amortization and energy cost. It was emphasised that charcoal use in combination with coal in slurries permits a reduction in sulphur content.
Economics In conclusion, the chairman asked Dr Bridgwater whether he could give a
breakdown of the biomass costs presented into labour, capital, and energy costs, so that this could be extrapolated for developing countries as very high costs in Europe compromise short-term energy uses through pyrolysis and gasification. It was explained that the major costs are labour and capital and therefore extrapolation must be carried out cautiously because if labour is expected to be a lower cost, equipment could cost more or even be unavailable, even without considering maintenance problems.

SESSION III
PYROLYSIS, GASIFICATION AND LIQUEFACTION TECHNOLOGIES


Ill
BIOMASS GASIFICATION: PAST EXPERIENCES AND FUTURE PROSPECTS
IN DEVELOPING COUNTRIES
Matthew S. Mendis Industry and Energy Department
The World Bank
Summary This paper presents an overview of the history, technology and use of small-scale biomass gasification systems in developing countries. The experiences to date have been mostly disappointing due to several reasons including: inappropriate technology and standards for developing countries; use of unacceptable biomass fuels; lack of trained operators and infrastructure support; lack of adequate user incentives; and finally, marginal economics. Recent developments in utilizing ferrocement instead of metal to fabricate biomass gasifiers have led to drastic reductions in the cost of production and significant increases in reactor service life. The resulting economics present a more favorable picture for the future of small-scale gasification systems.
1. HISTORY The basic principles of biomass gasification have been known since
the late 18th century and commercial applications of the principles were first recorded in 1830. By 1850, large parts of London had gas lights and there was an established gas industry manufacturing "producer gas" from coal and biomass fuels. The use of producer gas to run an internal combustion engine was first tried around 1881. By the 1920s, producer gas systems were being used to operate trucks and tractors in Europe. While it was demonstrated that it was possible to operate engines with producer gas, it was not convenient or reliable and producer gas systems for operating mobile or stationary engines did not gain wide acceptability.1
Biomass gasification systems reappeared with a force in Europe, Asia, Latin America and Australia during World War II as a result of the scarcity of petroleum fuels. In Europe alone, almost one million gasifier-powered vehicles helped keep basic transport systems running during the War. In most cases, the gasified biomass fuels were either wood or charcoal. Gasifier systems were generally abandoned with the reemergence of relatively inexpensive liquid fuels after the War.
The "energy crisis" of the 1970s sparked a renewed interest in biomass gasification systems. The technology was perceived as a relatively cheap indigenous alternative for small-scale industrial and utility power generation in those developing countries that suffered from high world market petroleum prices and had sufficient sustainable biomass resources. By the early 1980s over 15 (mainly European and North American) manufacturers were offering small-scale wood and charcoal gasifier power plants (up to approximately 250 kWe). In addition, at least four developing countries (Philippines, Brazil, Indonesia and India) had started gasifier implementation programmes based on locally developed technologies. As a result of developing country interests and the possibilities for export earnings, a large number of biomass gasification systems were

112
installed in several developing countries for testing and demonstration mostly through donor financed projects. In some cases such as Brazil, China, India and Thailand, the technology was even developed and promoted by local entrepreneurs. At present, biomass gasification systems have found only limited commercial applications in several developing countries including Brazil, China, India and Thailand.
2. TECHNOLOGY Chemistry. Biomass gasification is the process of conversion
through thermal decomposition of a solid biomass feed material to combustible gas. Gasification is achieved in the presence of heat and a limited supply (less than stoichiometric) of oxygen resulting in incomplete combustion of the feed material. The result is a combustible gas. When air (as opposed to pure oxygen) is used as the gasifying agent, the product gas mainly consists of carbon monoxide (CO), nitrogen (N2), hydrogen (H2), small amounts of methane (CH4) and other higher hydrocarbon gases. Due to the nitrogen dilution, the product gas has a low energy or calorific value in the range of 3.8 to 5.6 MJ/m3 as compared to natural gas which is the range of 38 MJ/m3. Thus, producer gas from air gasification is suitable for combustion in adjacent internal combustion (IC) engines, boilers or kilns but is too dilute for economic transport in pipelines over long distances.
Equipment. Biomass gasification systems consists of four principal components :
(a) Fuel preparation, handling and feed system; (b) Gasification reactor vessel; (c) Gas cleaning, cooling and mixing system; and (d) Energy conversion system (e.g., IC engine with generator or pump
set or gas burner coupled to a boiler or kiln). Power Gasifiers. Gasification systems that are coupled to IC
engines to produce shaft power are called power gasifiers. When the gas is to be used in an IC engine it is important that it is cleaned of all particulate, tars and moisture and cooled to near ambient conditions to ensure reliable and efficient operation of the engine. As such, power gasifiers require elaborate gas cleaning, cooling and mixing systems and in general have stricter fuel quality and reactor design criteria. An example of a typical power gasifier system is shown in Figure 1.
Heat Gasifiers. Gasification systems that are used to fuel external burners such as those found in boilers, kilns or driers are referred to as heat gasifiers. Heat gasifiers differ from power gasifiers in one important aspect. Heat gasifiers generally don't require elaborate gas cleaning and cooling systems because the producer gas is usually combusted externally in close proximity to the gasifier. Heat gasifiers are also more energy efficient because they tend to utilize the sensible heat and tars in the producer gas. As a result of the less critical gas quality requirements, heat gasifiers, in general, are simpler to design, construct and operate than power gasifiers and are more versatile in the fuels they can utilize. Figure 2 illustrates a typical heat gasifier system.
Reactor Designs. The vessel or reactor used to convert biomass fuels to gaseous fuels are called "gasifiers". These gasifiers are usually characterized by the design of the "fuel bed" and the method in which the biomass fuels are brought into contact with air and heat for the process

113
of gasification. Three principal fuel bed designs can be identified: fixed bed; fluidized bed; and entrained bed. Fluidized bed and entrained bed
biomass
air —*~—
GASIFIER
t
ι '
gas cooler/ cleaner
' 1
engine power
ash dust condensate
Fig. 1 Typical power gasif ier system
biomass
GASIFIER process heat
ash
Fig. 2 Typical heat gasifier system

114
designs, while more robust and versatile in their operation are generally more difficult to design , build and operate, are more expensive and are presently not considered appropriate for small scale (less than 1 MWe) , developing country applications. As such, fluidized and entrained bed gasifiers are not considered in this study.
Fixed Bed Gasifiers. The most common type of gasifier reactors used in developing countries are the fixed bed type because of their relative simplicity of design, low cost of fabrication and ease of operation. Fixed bed gasifiers can be further characterized by the direction of the flow of biomass fuel and gasification air within the reactor. The principal fixed bed gasifier types are:
(a) Down-draft or co-current; (b) Up-draft or counter-current; (c) Cross-draft or cross-current; and (d) Open-core.
Several variations exists within the four principal fixed bed gasifier types. The exact choice of gasifier design is more a function of the type of fuel to be gasified and the end use of the producer gas (for power or heat). For example, power gasifiers fueled with unprocessed biomass tend to be of the down draft design because of the ability of down draft gasifiers to produce low tar gas. Heat gasifiers tend to be more of the up draft or cross draft design. Charcoal fueled gasifiers can be either up draft, down draft or cross draft. Open-core gasifiers are designed for gasification of rice husks. Simplified block diagrams illustrating the main types of biomass gasifiers considered in this study are presented in Figure 3.
Fuel Characteristics. Given the state-of-the-art of commercially available fixed bed gasifier systems and based on data obtained from the UNDP/World Bank Biomass Gasifier Monitoring Program, at present, only wood, charcoal, rice husks and coconut shells are considered suitable biomass fuels for gasification. Biomass fuels such as wood wastes, coconut shells and rice husks can generally be used with minor processing such as drying, sizing and screening. The typical characteristics of these bioraass fuels are presented in Table 1.
In order to ensure reliable and efficient operation, biomass fuels for gasification must meet certain specifications. All gasifier types have fairly strict fuel requirements with respect to size, moisture content and ash content. Inadequate fuel preparation is an important and frequent cause of technical problems associated with gasification and therefore a strict organization and control of fuel preparation procedures is of utmost importance. Table 2.2 presents a generalized overview of the most important fuel requirements for the different types of gasifiers considered in this study.

115 fuel air fuel qas
up-draft
fuel
open-core
Figure 3 Main types of biomass gasifiers

116
Biomass Fuel
Charcoal
Wood
Rice husks
Table 1:
Moisture Content % Wet
2 - 10
20 - 40
3 - 5
TYPICAL CHARACTERISTICS OF BIOMASS FUELS
Ash Content % Dry
2 - 5
0.1 - 1.0
15 - 25
Volatile Matter % Dry
5 - 30
70 - 80
60
Bulk Density kg/m3
200 - 300
600 - 800
100
Average Higher Heating Value MJAg dry basis
30
20
15
Coconut shells 25 0.8 79 20
Table 2: FUEL REQUIREMENTS FOR DIFFERENT GASIFIER TYPES
Gasifier type: Fuel:
Up-draft (Wood)
Down-draft (Wood)
"Open Core" (Rice Husks)
Cross Draft (Charcoal)
Size (mm) 20 -Moisture (%db) < 25 Ash (% db) < 6
100 5 - 100 < 60 < 25
1 - 3 < 12 approx. 20
40 < 7 < 6
80
Producer Gas Utilization Systems IC Engines. One of the most attractive uses of producer gas is its
use in internal combustion engines for the production of shaft power which in turn can be used for generating electricity, pumping water, milling rice, running compressors, motive power, etc. Although producer gas can be combusted in gas turbines, its application in this area has not been adequately demonstrated to warrant serious consideration in this study. Producer gas has been widely used in reciprocating IC Diesel (compression ignition) and Otto (spark ignition) engines. However, several issues regarding the resulting engine performance, efficiency, output, and life need to be closely evaluated to determine the trade-offs of utilizing the relatively low quality producer gas in engines that are essentially designed to operate on high quality liquid fuels.
Spark ignition engines can be run entirely on gas. The maximum power output of an Otto engine on producer gas depends on the gas heating value, the setting of ignition timing and specific engine characteristics and is normally considerably less then the equivalent value on petrol or natural gas. Diesel engines can only be partly operated on producer gas

117
(dual fuel operation) and therefore always consume a certain amount of diesel fuel. The maximum power output of such an engine depends on the gas heating value, the injected diesel fuel amounts and specific engine characteristics.
Direct Combustion Burners. Producer gas can also be directly combusted in external combustion systems such as boilers, kilns, driers, ovens, etc. In most cases little or no modification to the existing equipment is necessary especially if the system is designed to burn a gaseous or atomized liquid fuel. As in the case of IC engines, a close evaluation of system performance, efficiency, output and life as a result of the use of producer gas needs to be accounted for. However, given the fact that direct combustion systems generally involve no internal moving parts, the impacts of producer gas on these systems is usually minimal.
Safety and Environmental Aspects Operation of gasification systems may give rise to the following
types of hazards : (a) Toxic gaseous emissions; (b) Fire and explosion hazards; and (c) Toxic liquid effluent.
However, with care and proper safety precautions, biomass gasification systems can be made to operate without incident. This requires full recognition of the potential dangers associated with the system and operation by only trained personnel.
Toxic Gases. An important constituent of producer gas is carbon monoxide (CO), which is an extremely toxic and dangerous gas because of its tendency to combine with blood haemoglobin and rob the body of normal oxygen intake. Fortunately, normal power gasification systems work under suction, so that even in case a gas leak occurs in the system, no dangerous gas will escape from the equipment during actual operation. If CO emissions do occur, they are only of concern in the immediate vicinity of the gasifier plant as the CO will quickly react with ambient 02 to form
The situation is different during start-up and shut-down of the installation. During start-up the gas is generally vented and it is necessary to ensure that the vented gases cannot be trapped in an enclosed room. As a rule a suitable chimney will provide sufficient safety. During shut-down of the installation a pressure build-up will occur in the gasifier, caused by the remaining hot and pyrolysing fuel. As a result, for a short period (approximately 15 minutes) carbon monoxide containing gases will be released. Therefore gasifier systems should always be installed in well ventilated buildings, or, if possible, installed externally covered only by suitable roofing.
Fire and Explosions. Fire hazards may result from high surface temperature of equipment; risks of sparks during refueling; and flames emerging from gasifier air inlets. In most cases, burn risks can be eliminated by relatively simple safety measures such as insulation of hot parts, installation of double valve fuel feeding system, installation of back-fire valve in gasifier inlet, etc.
Gas explosions may occur in case the hot gas is mixed with a sufficient amount of air to trigger spontaneous combustion. Air leakage into the system will generally not give rise to explosions, but will lead to local zones of gas combustion resulting in higher gas temperatures and lower gas quality. Pyrolytic gases in the bunker section when mixed with

118
air (as may happen during refueling) may form an explosive mixture. It is not unusual for this to burn in a relatively harmless manner, especially when the fuel level in the bunker is low. Risks to the operator can be obviated by burning-off the gases in the bunker through introduction of a piece of burning paper or the like. Also double sluice fuel feeding systems prevent this type of risks. The major explosion risk lies in the lighting of an un-vented cold gasifier. An explosive mixture may still be present in the equipment and be ignited. Therefore cold systems should always be carefully vented before ignition.
Toxic Effluent. A biomass gasification system produces ashes and tar/phenol containing condensates. Ashes do not constitute an environmental hazard and can be disposed of in the normal way. Condensates amounts from down-draft gasifiers are normally small and also tar/phenol contamination is relatively minor. The situation is different with the large quantities of heavily contaminated condensates from up-draft and "open-core" systems. Although no hard data are as yet available on biodegradation of phenolic and tarry constituents from condensates of such systems, it is clear that untreated disposal of such condensates is environmentally unacceptable and can result in contamination of drinking water, fish kills and other related effects. Therefore, this disposal problem needs careful study before introduction of gasifier systems that produce large quantities of condensate are considered.
Positive Impacts. The primary positive impact of biomass gasification systems is the possibility of effectively utilizing a waste biomass material such as wood residues, rice husks, coconut shells, etc., which could otherwise pose disposal problems and create localized fire hazards, insect infestation, vermin breeding or odor during the decay process. Air emissions from IC engines fueled with producer gas are marginally cleaner than emissions with petroleum fuels. The main advantages are the absence of lead and sulphur emissions with producer gas. Additionally, gasification systems, by virtue of utilizing a biomass fuels, do not contribute to the net increase of C02 in the environment as would liquid fossil fuels.
3. CURRENT COMMERCIAL STATUS Commercialized Power Gasifiers. There are a small number of strictly commercial power gasifiers currently operating globally. No accurate inventory is available, but rough estimates would be in the range of one to three thousand. However, the vast majority of these would be small charcoal gasifiers located in Latin America, primarily Brazil. It is estimated that less than one hundred commercial non-charcoal power gasifiers are currently in operation globally. Of the commercial non-charcoal gasifiers, most are either fueled with wood or rice husks.
Down-draft Charcoal Gasifiers. Several hundred commercially operating down-draft charcoal gasifiers are reported in operation throughout the globe. Most are located in Brazil. About a dozen are located in the Philippines as a result of a major government program to promote the technology in the early and mid 1980s. It should be noted, however, that of nearly one thousand down-draft charcoal gasifiers disseminated through a government subsidy program, none are presently in operation. The gasifiers that are in operation were purchased by the private sector without government subsidy and, in many cases, were modified to meet the users needs. The technical performance of most down-draft charcoal gasifiers have been satisfactory, especially where operator

119
training and maintenance support have been provided. However, rising charcoal prices and lower petroleum prices have resulted in the decommissioning of many of these units.
Down-draft Wood Gasifiers. A few commercial down-draft wood gasifiers are reported to be operating in India and Latin America. The units in India are small scale (5 hp - 100 kW) units used primarily for water pumping and isolated power generation. The seemingly most successful commercial down-draft wood gasifiers are located in the Mennonite community of Loma Plata, Paraguay. The gasifiers are part of a power plant used to supply electricity to the Loma Plata community. It is reported that there are three gasifier units each coupled to a 420 kWe gas engine/generator set. The wood gasifiers are used to meet the base load demands while stand-by diesels are used to meet peak loads. The first unit was installed in 1983. After initial technical problems and after extensive modifications of the gas cleaning section, the system is now reported to function to the satisfaction of the user.
At least one gasifier manufacturer (in France) can point to a small number of commercial projects presently operating in developed countries (primarily Europe). Most of these gasifier systems are situated at isolated sawmill sites. However, operational and financial details with respect to these plants are not available.
Up-draft Gasifiers. A few commercial up-draft wood gasifiers are known to be operating primarily in Latin America. The situation was completely different in the 1940s and 1950s when both in Europe and elsewhere a considerable number of systems were functioning on a diversity of fuels including wood residues and agricultural wastes. The last European updraft power gasifier (in Germany) operating on sawdust was closed down recently for environmental reasons (water pollution resulting from disposal of tarry effluent). Up-draft gasifier plants currently in operation perform technically satisfactory on fuels of a fairly wide range of specifications. However, as explained earlier, updraft gasifiers produce large quantities of tar, with potentially serious health and environmental hazards. As such, their commercial applications have not been aggressively pursued.
Cross-draft Gasifiers. A large number of cross-draft charcoal gasifiers have been in commercial operation in Brazil and other Latin American countries. Two such installations were systematically monitored under the UNDP/World Bank BGMP.2 The results indicated that the units experienced considerable operational problems primarily due to inexperienced operators, sensitivity to fuel quality, operation of the gasifier below minimum loads and inconsistent maintenance of the plant and equipment. In cases where these issues were addressed and where plant utilization factors were high, the charcoal cross-draft gasifiers were commercially attractive.
Open-core Gasifiers. A number (at least twenty) commercial open core rice husk gasification plants have been in operation in the Peoples Republic of China (PRC) for decades. Since 1967 identical units were installed at a rice mill in Dogofiri, Mali. The performance of these units were monitored under the UNDP/World Bank BGMP.3 It was concluded that plants operate technically satisfactorily and, at given locations are able to produce electricity at marginally lower financial cost than equivalent diesel engines. As a result of the general success of these units in Mali, it was recently (1986) decided to install an additional unit in Mali.

120
However, it should be borne in mind that the units, as presently designed, produce considerable amounts of potentially harmful tars which are a problem to dispose in an environmentally acceptable manner.
Over the past five years a number of locally designed and manufactured open core gasifiers have been commercially installed at rice mills in Thailand. No performance data, other than that the systems generally work to the satisfaction of the users, are available on the Chinese and Thai rich husk gasifiers.
Commercialized Heat Gasifiers. By far the most successful commercialization of gasification technology has been with heat gasifiers of varying capacities, designs and applications. Most of the heat gasifiers currently on the market have output capacities in the range of 0.1 to 10 MWt. Heat gasifiers operating on fuels of widely differing specifications have been used commercially to produce process heat for use in kilns; ovens; driers; boilers; heaters; etc.
Heat gasifiers have been manufactured and applied in Europe and North America as well as several developing countries of the Pacific, South-East Asia and South America. During the mid-1980s, Brazil had over dozen active heat gasifier manufacturers and over 50 known commercially operating heat gasifier installations.* Heat gasifiers have also been locally manufactured and commercially adopted in Malaysia, the Philippines, Indonesia and India as well as several other Asian and Latin American countries. New Zealand manufactured heat gasifiers are reported to be commercially operating in significant numbers in Papua New Guinea and several other Pacific Island countries. Attempts are now being made to promote similar heat gasifier technology in several African countries.
There appears to be no significant technical problems associated with heat gasifiers. Unlike power gasifiers, the gas produced by heat gasifiers is combusted externally, generally in close proximity if not directly adjacent to the gas outlet of the gasifier. Therefore, heat gasifiers usually do not require elaborate gas cleaning systems, are more efficient because they utilize the sensible heat and condensible tars in the gas and do not produce any toxic effluent. An exception are heat gasifiers applied in situations where the gas has to be transmitted long distances or a clean flame is required as is the case in food drying or baking or ceramic production.
4. RESEARCH. DEVELOPMENT AND DEMONSTRATION PROJECTS Donor Financed Proprams. Over the past decade there have been numerous donor organizations that have financed biomass gasification research, development and demonstration (RD&D) projects in developing countries. It is beyond the scope of this report to outline the extent or results of these efforts. However, a few key points can be summarized. Most donors have concentrated on demonstrating down-draft power gasifier systems for applications in isolated power generation, irrigation, water pumping or rural industries. In a majority of the cases, the gasifiers being demonstrated were designed and manufactured in the donor countries and exported to the developing countries. In many cases, a wide variety of fuels were experimented with, however, positive results were achieved primarily with the well established fuels of charcoal, wood, coconut shells and rice husks. In all cases, the donor financed projects are important

121
to review as their history and experience are usually much better documented than the strictly commercial operations.
The donor projects were, for the most part, designed on the assumption that the technology would work in the developing country context and therefore had the objective of demonstrating economic and social viability while simultaneously promoting the technology for wider use. In reality, most of these demonstration projects failed to achieve normal, reliable operation due to several diverse reasons including those listed below:
(a) Use of biomass residues that were unacceptable for down-draft power gasification systems;
(b) Use of equipment that was previously untested in developing country environments ;
(c) Mismatching of gasifier system capacity to energy demand profiles resulting in systems operating at turn-down ratios below specification or systems being made to respond to rapid and wide load changes ;
(d) Insufficient training of operators and provision of competent technical back-up resulting in initial operating errors and damage to equipment;
(e) Lack of sufficient incentives to motivate operators to accurately perform additional, and usually time consuming, laborious and dirty tasks, associated with gasification systems;
(f) Poor system design including mismatching of imported and local system components, inattention to local operator skills and biases and little or no documentation or operation manuals; and
(g) Finally, disappointing economics due to poor system performance, accounted costs or simply disregard for economic criteria at the outset of project design.
Nationally Financed Programs. In addition to the donor financed gasifier demonstration projects, there have been several nationally initiated efforts over the past decade. The most notable in the developing countries include: The Philippines; Brazil; India; Thailand; and Indonesia. Brief highlights of the key points associated with each program are presented below.
The Philippines. A government backed and United States Agency for International Development (USAID) supported program to commercialize locally designed and manufactured charcoal gasifiers was initiated in the early 1980s. The program was aimed at providing options for rural irrigation water pumping, rural electrification and motive power for jeepneys and bancas (trucks and boats). Nearly one thousand charcoal gasifiers were manufactured under the program. The program, terminated in 1986, was a failure for several reasons including poor design and quality of the gasifier systems, no control on input fuel specifications, inadequate operator training, no manufacturer technical and maintenance back-up, and, perhaps most importantly, disregard for the normal economic incentives necessary to assure success. The Philippine program was subsidized. Gasifiers were essentially given away by the government to local farmer cooperatives without instilling adequate economic incentives to ensure commitment by the farm cooperatives to the overall success of the systems. It is interesting to note that of the few gasifier systems that did succeed in the Philippines, almost all were purchased and operated by the private sector without any subsidy or other form of government assistance.

122
Brazil. Unlike the Philippines, the Brazilian government program to promote gasifiers was essentially passive and relied on demand side incentives rather than on the supply side subsidies. In addition to instituting phased in quotas of petroleum consumption for certain industries and thereby creating a demand for alternative technologies, the Brazilian government also sponsored a few select research efforts to help develop appropriate gasification systems for Brazilian needs. However, no subsidies or direct government assistance were provided for the promotion of gasification systems. The results were dramatic. By 1985 there were between 15 to 20 commercial gasifier manufacturers and well over a thousand power and heat gasifiers that were privately purchased and in operation.5 While gasification activity in Brazil has declined somewhat recently, this has been primarily in reaction to easing of petroleum quotas and prevailing lower petroleum prices.
India. Recently (1987) a government-backed commercial down-draft wood gasification programme was started in India, based on locally developed and manufactured technology and aimed at small-scale irrigation applications. To date a total of 2.5 MWs of capacity, mainly in 5 kW size units (i.e., approximately 500), has been installed. The programme is heavily subsidized. A recent evaluation concludes that approximately 60 % of the units are not in regular operation for technical, organizational and financial reasons. A more detailed evaluation quantifying the above factors is presently underway.
Thailand. Relatively independently from several donor and private sector efforts to promote gasification in Thailand, the Thai Ministry of Interior, through its Department of Public Works (DPW) has, for the past few years, undertaken a program to promote rural electrification using biomass gasification technology. With a budget of approximately 25 million Bhats (one million USŞ) , the DPW has installed 143 - 15 kW charcoal gasifiers throughout Thailand. DPW's objective were to install 4000 units by 1991.6 Like the Philippine government's program, the Thai DPW program has run into several difficulties including technical problems with the design of their gasifier system as well as economic, social and institutional issues that have hampered the program.
Indonesia. The potential for biomass fuelled gasifiers to substitute petroleum consumption has attracted much attention in Indonesia. In 1987, the President of the Republic of Indonesia (ROI) mandated that 10 Indonesian manufactured biomass gasifiers be placed in field operations, to help demonstrate the technical and economic viability of such systems. Over the past decade several donor financed activities in Indonesia have also promoted gasification systems. As a result of these activities, there are at present over 20 power gasifiers and several heat gasifiers installed in Indonesia. The power gasifiers, all for demonstration and testing purposes, are widely spread over the different provinces of Indonesia. Three units are fueled with rice husks while the rest are fired with different types of wood. Most projects aim at demonstration and/or testing of the technology. To date no truly commercial power gasification systems are in operation in Indonesia. Most of the heat gasifiers are situated in Java or Sumatra and operate on coconut shells or other similar types of residues. These units represent the only truly commercial applications of biomass gasification technology in Indonesia to date.
5. ECONOMICS The economics of small scale power gasifiers hinges on the savings
that can be realized by switching from high-cost liquid fuels (i.e., diesel) to low-cost biomass fuels. These fuel cost savings must be measured against the additional capital costs of the gasifier, the increase in operation and maintenance costs, and the reduced reliability of the

123
system. One way to evaluate the tradeoff between capital costs and operating and maintenance costs is to compare the levelized costs of electricity generated by each system. This has been done in a study based on data collected under the UNDP/World Bank Biomass Gasifier Monitoring Program (BGMP).7 The study compared generic wood and charcoal gasifiers to diesel stand-alone systems from 5 kWe to 1 MWe.
The study evaluated commercially available power gasifiers in the following ranges:
o Manual feed charcoal gasifiers from 5 - 200 kWe; o Manual feed wood gasifiers from 5 - 200 kWe; o Automatic feed wood gasifiers from 100 kwe - 1 MWe.
Figure 4 presents some of the principal findings of the study. The analysis indicates that power gasifier economics is most strongly affected by system size and by the relative cost of petroleum and biomass fuels.
Under the baseline economic price of diesel at US$ 0.20/1, charcoal at US$80/t and fuelwood at US$20/t, none of the gasifiers evaluated in the BGMP were economic. However, if economic price of diesel were assumed to rise to US$ 0.40/1, as is the case in many isolated rural areas of developing countries, then the manual feed charcoal gasifiers and wood gasifiers above 30 kW become competitive. With the current low economic prices of petroleum fuels, it is clear that power gasifiers will have a niche only in remote applications where the economic cost of diesel is high (due to transport costs and unreliable supplies) and the cost of biomass fuels is low (due to surplus availability).
6. RECENT BREAKTHROUGH Recent work at the Asian Institute of Technology (AIT) in Bangkok
has resulted in a radical departure from conventional gasifier systems. Due to the high costs of metal, fabrication of a 10 kWe charcoal gasifiers, even when locally manufactured, is in the range of US$500/kW. As such, the economics of gasification are marginal given current petroleum prices. Under the leadership of Dr. Bob Reines, the AIT has developed a 10 kWe "ferrocement" open-core charcoal gasifier with the resultant costs of only $50/kW for the gasifier. The reactor is lined with refractory brick to manage the normally high temperatures encountered in this zone. The ferrocement components are water cooled which provides a relatively uniform temperature gradient over the ferrocement surface minimizing thermal stress. The water also serves as heat transfer fluid for gas cooling. As a result, the ferrocement approach solves many of the material (thermal stress and corrosion) as well as economic constraints associated with conventional metal gasifiers. To date, ferrocement gasifiers have only been demonstrated for small (less than 18 kWe maximum output) charcoal gasifiers.
New Economics The ferrocement gasifier presents the possibility of a ten fold
decrease in the capital costs of a gasifier. A brief comparison of the economics of a diesel generator set, conventional charcoal gasifier system and a ferrocement gasifier system is presented to illustrate the impact of this new approach. Table 3 outlines the basic assumptions used in the analysis. Figure 5 shows the resulting levelized electricity costs (in cents/kWh) of the three systems. At the current economic price of diesel of US$0.25/1 and charcoal at US$40/t, the conventional gasifier is not economic with a diesel system. However, the ferrocement gasifier has levelized electricity about 11% lower than the diesel system. Figures 6 to 8 show a sensitivity analysis on charcoal and diesel prices, installed

124
Costs of Electricity Production Small Stand-Alone Systems
Electricity Costs ($/kWh)
1000 INSTALLED CAPACITY (kW)
— · — Diesel I 40c/l -β- Woodgas/m $2Q/mt
m = manual / a = automatic
POWER SOURCE Diesel θ 20c/l -*- Clmrgns θ $BD7mt Woodgas/a $20/mt
Figure 4: Electricity Costs: Diesel vs. Gasifier Systems
capital costs (ICC) and annual operating hours. In all cases, the $50/kW ferrocement gasifier significantly increase the range in which the technology is economically competitive with diesel systems
7. SUMMARY Based on the above discussion, the current status of biomass
gasification technology can be broadly summarized as follows:
(a) Commercially proven power and heat biomass gasifiers are available especially when the fuels are charcoal, wood, coconut shells and rice husks. Heat gasifiers are more tolerant of other types of biomass fuels. However, only limited experience with "non-conventional" biomass fuels is available for small-scale power gasifiers.
(b) The economics of biomass gasification, at present, are at best marginal. When compared to the counterpart petroleum system, the economics of heat gasifiers are more favorable at present than conventional power gasifiers.
(c) The recent developments in ferrocast charcoal gasifiers could help change the negative picture for biomass power gasifiers. However, further testing especially in field applications are necessary to confirm the viability of this technology.

125
Table 3: BASIC MODEL OF THE ANALYSIS
Diesel Price Charcoal Price ICC Diesel/Gas Ann.Op. Hours PARAMETER
Capacity Ann. Op. Hours Avg. Op. Load Annual Output (
(c/1) (cAg) ($AW) (hrs)
(kW) (hrs) (%)
;kWh/yr) Installed Capital Cost Eng/Gnenset Gasifier
Total ICC Expected Lif Discount Rat
Annual ICC ICC/kWh ICC A « ANNUAL O&M Fuel System Eff. Annual Unit Costs
Labor Monthly Wage Operators Labor Costs Unit Costs
Maintenance Maint. Costs Unit Costs
Total Ann. O&M Tot Ann. O&MA COST/KWH
($) ($) ($) (yr) (%)
($/yr) (cents)
($)
(%) ($/yr) (cAWh)
($/mo)
($/yr) (c/kWh) ($AWh) ($/yO (cAWh) ($/yr)
(c)
25.00 4.00
600.00 3000.00
DIESEL SYSTEM
10.00 3000.00
0.80 24000.00
6000.00
6000.00 6.00 10.00
1377.64 5.74
600.00
20.00 3000.00 12.50
50.00 1.0
600.00 2.50 0.015 360.00 1.50
3960.00 16.50
22.24
LHV (MJ/unit) 36.00 30.00 500.00
CONV. CHAR GASIFIER
10.00 3000.00
0.80 24000.00
7500.00 5000.00
12500.00 4.00 10.00
3943.39 16.43
1250.00
15.00 768.00 3.20
50.00 1.5
900.00 3.75 0.025 600.00 2.50
2268.00 9.45
25.88
Energy Cost (c/MJ) 0.69 0.13 50.00
FEROCEMENT CHAR GASIF
10.00 3000.00
0.80 24000.00
7500.00 500.00
8000.00 4.00 10.00
2523.77 10.52 800.00
15.00 768.00 3.20
50.00 1.5
900.00 3.75 0.025 600.00 2.50
2268.00 9.45
19.97

126
cents/kWh 25.881
19.966
Maint Costa/kWh Labor Costa/kWh Fuel Coata/kWh Cap. Coata/kWh
□¡•«•I Sytten »εοθ/kW Qatifier »60/kW Qasifier
15 2.5 2.5 2.5 3.75 3.75 12.5 3.2 3.2 5.74 16.431 10.516
■>■ Cap. Costs/kWh
EZ] Labor Costs/kWh 2 2 Fuel Costs/kWh
! S i Maint. Costs/kWh
Figure 5 . E l e c t r i c i t y Cost Components
Cost of Electricity vs. Charcoal Price
Coat of Electricity (c/kWh)
3 4 5 6 7 8 Charcoal Price (c/kg)
$eo/kW Diesel · 20 e/l
»200/kW
Diesel · 40 c/l
»600/kw
10
Figure 6. Sensitivity to Charcoal Price

127
COST OF ELECTRICITY vs. ICC of Gasifier System
35 [c /kWh]
100 200 300 400 500 600 700 BOO ICC of Gasifier System ($/kW)
25 c/l Diesel
S40/t Charcoal
40 c/1 diesel
380ft Charcoal
S20/t Charcoal
Figure 7: Sensitivity to Installed Capital Costs
Cost of Electricity vs. Operating Hours
130 Cost of Electricity (c /kWh)
1000 1500 2000 2500 3000 3500 40GG 450G 5000 5500 6000 Annual Operating Hours
^ D i e s e l System — ι »50/feW Gasifier * 1500/kW Gasifier
Figure 8: Sensitivity to Operating Hours

128
1. Reed, T.B., Biomass Gasification: Principles and Technology. Noyes Data Corporation, Park Ridge, NJ, 1981.
2. Furtado, P. et.al., Biomass Gasifier Monitoring Proiect in Brazil. Final Report, prepared by Cetec under World Bank/UNDP contract, January 1987.
3. S tassen, H., et.al., Biomass Gasifier Monitoring Project in Hali. Report prepared by Twente University of Technology, Biomass Technology Group, under World Bank/UNDP contract, 1987.
4. Mendis, M.S., "Biomass Heat Gasifiers: Status and Potential", Internal Working Paper, New and Renewable Energy Unit, The World Bank, Washington, DC, May 17, 1984.
5. Furtado, P. and Antunes, R. , "Commercial Applications of Biomass Gasifiers in Brazil", Presented at the Second International Producer Gas Confernece. Bandung and Jakarta, Indonesia, March 19 -23, 1985.
6. Coovattanachai, N., "Final Report on the Assessment of the Performance and Identification of Technical Problems of DPW's Gasification System", Prince of Songkala University, Haadyai, Thailand, January 28, 1988.
7. Terrado, E., Mendis, M. and Fitzgerald, Κ., "Impacts of Lower Oil Prices on Renewable Energy Technologies", Energy Series Paper No.5. World Bank, Washington, DC, 1988.

129
GASIFICATION AND PYROLYSIS OF BIOMASS IN EUROPE
A A C M Beenackers Department of Chemical Engineering University of Groningen Nijenborogh 16 9747 AG Groningen The Netherlands
A V Bridgwater Chemical Engineering Department
Aston University Aston Triangle
Birmingham B4 7ET UK
ABSTRACT Biomass, as the only renewable source of fixed carbon, has attracted
considerable attention as a renewable energy resource after the oil crises of the last 15 years. Thermochemical processing has attracted considerable attention from the variety of technologies available for converting biomass into more useful and valuable energy products. As pyrolysis, gasification or liquefaction, this technology has been researched and developed for the economic production of fuel products that may be readily integrated into the energy infrastructures of both industrialised and developing countries.
The characteristics of the generic types of thermochemical conversion -gasification, pyrolysis and liquefaction - are described in this paper. The range of products derivable from each system are discussed and related to each technology, and the applications that have been researched to date are reviewed.
Current interest lies in relatively simple conversion technology to produce low Joule gas and basic liquid fuels. Longer term possibilities include gaseous and liquid hydrocarbon fuels, petrochemicals, bulk organics such as methanol and ammonia, and chemical specialities.
1 INTRODUCTION The potential offered by biomass and solid wastes for solving some of the world's
energy problems is widely recognised. The energy in biomass may be realised either by direct use as in combustion, or by upgrading into a more valuable and usable fuel such as fuel gas or fuel oil or higher value products for the chemical industry. This upgrading may be by physical, biological, chemical or thermal methods to give a solid, liquid or gaseous fuel. This paper is concerned with thermochemical conversion, and it reviews the state-of-the-art in gasification, pyrolysis and liquefaction of biomass.
2 THERMOCHEMICAL TECHNOLOGY Except for direct combustion which is outside the scope of this paper, there are three
thermochemical methods of converting biomassrgasification, pyrolysis and liquefaction. Each gives a different range of products and employs different equipment configurations operating in different modes. These are summarised below in Table 1, and the characteristics of the technologies are described in Table 2. The basis of a fuel or chemical production system is that the feedstock is converted to a useful primary energy product in a sequence of operations: pretreatment, conversion, and primary upgrading by simple physical processing such as gas scrubbing. Secondary higher value products may be produced by additional processing as shown overall in Figure 1.

130
Table 1 Thermochemical Conversion Technologies and Products
Technology Pyrolysis generally
Flash Pyrolysis Slow Pyrolysis
Liquefaction
Gasification
Combustion
Primary Product gas liquid solid char liquid mostly solid char mostly
liquid
gas
heat
Application fuel gas
oil or liquid fuel substitution solid fuel or slurry fuel
oil or liquid fuel substitution solid fuel or slurry fuel
oil or liquid fuel substitution
fuel gas
heating
Table 2 Characteristics of Thermochemical Conversion Technologies
Figures quoted are typical, and can vary considerably according to technology and operating conditions
Pvrolvsis Liquefaction Gasification
FEEDSTOCK Feed size Moisture content
PARAMETERS Temperature, °C Pressure, bar Maximum throughput, t/h,
achieved so far
PRODUCTS Gas yield, %wt on dry feed
heating value, MJ/Nm3 Liquid yield, %wt on dry feed
heating value, MJ/kg Solid yield, %wt on dry feed
heating value, MJ/kg
Slow
any low
500-700 0.1-1 5
up to 40 5-10 up to 30 22 30 30
Flash
small very low
500-900 1 0.05
up to 70 10-20 up to 70 22 up to 20 30
small very low
250-350 100-200 0.1
20 2-6 up to 50 27 up to 25 30
mixed-large 50% max
800-1500 up to 30 20
100-250 5-15 up to 5 22 nil (ash) -
Feedstocks generally considered for thermochemical conversion are agricultural waste, wood and wood waste, energy crops, and refuse (MSW). The main technical criteria for feedstocks in terms of their suitability for thermochemical processing are low moisture content and low ash content The main economic criteria are cost and quantity available, both of which have a considerable influence on costs.
3 PRODUCTS 3.1 Introduction
The possible primary products and their processing origins are summarised in Table 3. This primary product can be used directly, or it may be subjected to further chemical processing to give a higher quality fuel or chemical product as shown in Figure 1. The

131
technologies for upgrading and likely viable size ranges are summarised in the relevant sections below.
CONVERSION TECHNOLOGY
/·'/■'·· ' PRIMARY '\,\< SSS PRODUCTS ν Λ S S S S S S S S S -
V A . V WATER ^ . V >
Figure 1 Primary and Secondary Products from Thermochemical Biomass Processing
Table 3 Primary Thermochemical Conversion Products and Technologies
Product
Fuel gas
Liquid
Aqueous
Solid
Heat
Form
LHV MHV
OU
Char-oil slurry Char-water slurry
Waste water
Charcoal
Hot gas
Components
CO, H2, C02, CH4, N2 CO, H2, C02, CH4,
higher hydrocarbons
Water insoluble oxygenates of high boiling point
Charcoal and oil Charcoal, water and
stabiliser
Source
Pyrolysis, Air gasification Pyrolysis, 02 gasification
Pyrolysis, Liquefaction
Pyrolysis Pyrolysis
water soluble } Pyrolysis oxygenates of low boiling j Liquefaction point.eg acetic acid } Gasification
Pyrolysis
Combustion

132
3.2 Primary products The primary products can be gas, liquid, and/or solid char depending on conversion
technology employed. Much of the present interest in thermochemical conversion centres on liquid products due to their high energy density and potential for premium liquid fuel substitution.
The gas is a low to medium heating value fuel gas that may be used as such, or physically and chemically upgraded to higher value products, including liquid products such as gasoline. This is depicted in Figure 1 above.
The liquid, when formed, approximates to biomass in elemental composition, and is composed of a very complex mixture of oxygenated hydrocarbons. The complexity arises from the degradation of lignin, and the broad spectrum of phenolic compounds that result from uncontrolled degradation. The liquid is often referred to as "oil" or "bio-oil", but is more like tar. This also can be upgraded to liquid hydrocarbon fuels. Utilisation of this material is discussed later.
The solid product from pyrolysis processes is char, which has limited application in developed countries for metallurgical and leisure industries. Water is also produced from moisture in the biomass feed and as a reaction product from pyrolysis. An alternative approach to a liquid product lies in grinding the char and slurrying it with water with a stabiliser as in coal-water mixtures. A slurry can also be made from the bio-oil and char The significance of the energy density is shown in Table 4 below.
3.3 Secondary Products These include power, fuels and chemicals and are summarised in Table 4.
Table 4 Secondary Products and Sources
Secondary product
Oxygenate Fuels Methanol Fuel alcohol
Hydrocarbon Fuels Gasoline
Diesel
Fuel oil
Power Power
Chemicals Ammonia Speciality chemicals
Source
Gasification Gasification
Pyrolysis Pyrolysis Liquefaction Gasification via Pyrolysis Pyrolysis Liquefaction Gasification via Pyrolysis Liquefaction
Pyrolysis Gasification
Gasification Pyrolysis Liquefaction
Process
Synthesis Synthesis
Hydrotreating Zeolites
Hydro-treating Methanol MTG
Hydrotreating Zeolite + MOGD
Hydrotreating Methanol MOGD
Stabilisation Stabilisation
Turbine Engine or turbine
Synthesis Extraction and/or Conversion Extraction and/or Conversion

133
4 GASIFICATION Of the conversion technologies discussed in this paper, biomass gasification by now is
the most widely applied technology, except for charcoal production. However, relative to the already significant contribution of biomass combustion to energy generation, the impact of gasification of biomass for energy generation is still fairly limited. The main reason for this is probably that the technology still has not reached full maturity. However, research efforts to develop improved biomass gasification technologies have been impressive during the last 15 years. This is likely tö result in an increasing number of successful commercial biomass gasification applications in the future and justifies a review of recent developments including low joule and medium joule gasification and the role of catalysis in gasification.
4.1 Low Joule Gasification If air is used as the gasifying agent, a so called low joule gas or producer gas is
obtained having a heating value of typically 5MJ/Nm3. Wood based gasifiers for heat applications have operated successfully for many years.
Both updraft moving bed gasifiers and (fast) fluidised bed gasifiers have been installed. European plants have been described by Bierback et al. [1] McKeough et al. [2] and Salo [3]. New American plants were listed annually in the reviews of Klass [4, 5] but are no longer published. A thermochemical biomass conversion database has been established through the IEA Bioenergy Agreement which includes over 300 gasification activities around the world in a total database of 650 thermochemical activities [6].
Recent progress at 150 kg/h dry biomass has been realised by Framatome with high carbon conversion. Carbon to gas efficiencies as high as 96% have been obtained by introducing secondary air above the bed and recycling char to the bed from the first cyclone (of a set of two). In this way the char content in the ash collected from the second cyclone could be as low as 0.7%.
Gasification of wood wastes followed by power generation via a steam cycle has been investigated by the Florida Power Corporation at a scale of 2 MWe in an updraft moving bed gasifier [7]. Some problems were encountered with handling the condensibles which were separated from the wood gas on its way to the boiler by centrifugation. Initial problems to combust the liquids in the burner could be solved. The project confirmed that adapting a gasifier to an existing boiler can be accomplished with only minor effort. The economic breakeven point of the facility was determined at $16 per ton of raw fuel with actual fuel wood prices at the time varying between $13 and $25/ dry tonne.
Figure 2 gives an overview of the capacities of the units constructed in the industrialised world during the last five years and implemented in both the industrialised world and in developing countries. As can be seen, typical capacities in Europe and North America nowadays are in the range of 80 to 300 kW whilst for developing countries these are slightly smaller typically from 40 to 200 kW. Further, the maximum capacity presently is in the range of 1 to 5 MW. Despite the commercial availability of the technology, only limited operating experience is available from the open literature. There is no doubt, however, that significant scope still exists to improve the available technology. To show this in more detail, a diagram of these systems is presented in Figure 3.
Although some counter current moving bed gasifiers have been installed, nearly all modern systems for power generation are of the downdraft moving bed type. This is because of the cleaner gas produced by downdraft gasifiers resulting in a relatively less complicated cleaning process and less environmental problems with respect to waste water. However, downdraft gasifiers show little flexibility with respect to feedstock moisture content (15-20%) and feedstock size. Well defined chips or pellets with a moisture content not exceeding 25% are necessary to produce a gas that has a really low tar content. Most commercial systems operate on wood chips while maize cops and coconut shells can be successfully gasified too.
Loose materials such as straw, rice hulls and MSW are difficult to gasify at a small scale. Pelletisation has been successfully applied but increases costs of the feedstock to $25 per tonne. A potentially very large market exists for small scale rice hull gasification. Initial efforts by Kaupp to gasify it continuously at a scale of 30-60 kWe in a downdraft gasifier were unsuccessful [8]. The main problems are:

134
poor flow due to low density and swelling in the pyrolysis zone poor oxygen distribution due to small particle size sintering arising from poor oxygen distribution lack of a well designed continuous ash removal system.
Figure 2 Typical Distribution of Gasification Based Electrical Power Generation Units
Manufactured in the Industrialized World. I - operating in industrialized world II - operating in developing countries III = I + II
Biomass
Air-Hot, dirty
product gas
* Β
Air (Pilot diesel)
Cool clean gas
D Exhaust
Tar, ash, soot, water
Electricity
Figure 3 Gasifier-Reciprocating Engine Systems
A = Gasifier C = Gas-air mixer Β = Cleaning and cooling section D = Engine-generator set

135
Recently Manurung and Beenackers succeeded in solving all these problems by developing a new type of gasifier optimally designed for rice hulls [9]. The system is shown in Figure 4. Smooth flow of rice hulls is obtained by eliminating the throat of conventional down draft gasifiers. Hot spots, causing ash sintering, is avoided by eliminating the conventional local air inlets. Air is sucked into the gasifier over the whole cross section of the surface of the bed resulting in a uniform combustion zone over the cross section close to the top of the bed. Finally, a scraper slowly rotating over a grate effectively continuously removes the ash from the gasifier.
GASIFIER
ASH FLUSHING VESSEL
WATER SEAL
Figure 4 Small Scale UT/ITB Downdraft Rice Husk Gasifier with Ash Removal System
[9]
Such a device is essential because the rice hulls keep their original shape after gasification. After successful trials both at Twente University, The Netherlands and the Institute of Technology, Bandung, Indonesia, the first field unit of 10 kWe was installed at a rice mill in an Indonesian village in 1986. It is operated by trained but otherwise unskilled people. Now, after more than a year of successful operation the conclusion is that this technology is promising and the Indonesian government has decided to start an implementation programme.
On a larger scale, at 1-10 MWe, gasification of loose waste materials with relatively high ash contents such as straw and rice hulls may be successfully gasified in fluidised beds. For rice hulls van den Aarsen et al. have demonstrated this on a pilot scale [10]. Two methods

136
of gas cleaning are available: dry and wet. Dry dust removal can be carried out, for example, by two cyclones in series followed by an impingement separator, filtration with high temperature resistant fabrics and a cooler-condenser for tar and water removal [11]. The wet method also uses cyclones followed by a wet scrubber, either of conventional or venturi type design [12].
4.2 Low Joule Fuel Gas Utilisation Gasification is a method to transfer the heating value of solid biomass into the heating
value of a combustible gas (and some sensible heat) while in combustion the heating value of the solids is completely transferred into sensible heat. In principle, gasification offers some advantages over combustion. Firstly, a gas has better burning properties relative to a solid. The burning process is easier to control, it needs less excess air, it allows for simpler burner construction, it causes no particle emissions, less air pollution and less fouling of the heat exchange equipment. Further, gases can be burned in internal combustion engines (gas turbines or reciprocating engines) and can be applied easily in combined cycles. Basically, there are three main routes for combustion of the fuel gas produced (see Figure 5): A) external combustion for heating or drying purposes or for electricity
generation with a steam cycle B) combustion in a gas turbine C) internal combustion in either a diesel engine or a spark ignition engine, both
for shaft power and electricity generation.
STEAM CENERATOI!
ELECTRICITY
ELECTRICITY
DUST
COOLING +
TAR CLEANING
GAS TURBINE
DIESEL ENGINE . ELECTRICITY
ELECTRICITY
Figure 5 Low Joule Gas Production and Power Applications
4.2.1 External Combustion of Producer Gas The direct use of the gas in a furnace (route A in Figure 5) is the simplest application
and this generally requires little or no gas treatment except for dust removal. For efficiency reasons it is important to preserve the sensible heat of the product gas which requires close coupling of the gasifier and the furnace. In many cases it is possible to convert an existing natural gas or fuel oil fired facility to biomass firing by adding a gasifier. This requires some

137
repiping and change of burners etc., but not necessarily a derating of the facility. If a completely new installation has to be constructed the situation is somewhat different and clearly direct combustion is a competing alternative.
It should be realised that in many wood combustion installations some gasification occurs, and complete combustion is realised by the introduction of secondary air. Thus, two stage gasification with combustion and direct combustion systems are essentially the same. There remains, however, a difference in controllability, especially under varying load conditions. Generally it can be said that in gasification units combustion of the product fuel gas can be expected to have less impact on the environment than in direct combustion. In retrofitting an existing unit special attention must be paid to the burner. In most cases natural gas or fuel oil will be replaced by a hot dirty gas of low heating value. The characteristics of an ideal burner to cope with such a gas were defined recently as follows [13]: • the burner should maintain stable combustion over a wide range of gas compositions and
gas energy content and should be adjustable without modification to accommodate different gas types
• it should have low pollutant characteristics • the burner should be able to handle dirty gas without clogging or frequent servicing • the burner should be able to operate at low gas pressures • if possible, the burner should be able to simulate the heat release patterns of a natural gas
burner so that retrofit modifications could be attempted. Many types of commercially available burners for low Joule gases have been tested by
the Canadian Gas Research Institute but none were found entirely satisfactory, mainly because of susceptibility to clogging or erosion/corrosion with dirty tar laden gases [13]. Therefore, the Canadian Gas Research Institute has developed a modified two stage burner which, reportedly, meets the characteristics described above. It can handle a gas with a calorific value as low 3 MJ/Nm·' standard cubic meter which is heavily laden with dust and tar as results from counter-current updraft moving bed gasification systems.
4.2 2 Combustion in a Gas Turbine. This is shown as route Β in Figure 5. The favourable properties of gas turbines such as
long running periods between overhaul, low cost of maintenance, potential high inlet temperatures favouring high thermodynamic efficiencies and the possibility of using the exhaust gas in a steam generation cycle (combined cycle), make turbines potentially very attractive for use in combination with a gasifier. Further developments in turbine technology are probably needed such as in prevention of corrosion by alkaline metal vapours, improved tolerance to trace amounts of dust, and in control technology of the gasifier-engine system. For an optimally integrated system the gasifier has to be pressurized [14-16]. For an air blown gasifier the optimum pressure will be in the range of 10-30 bar. Operation under pressure increases the complexity of both the gasifier, the reeding and the ash removal system and can probably only be done economically for relatively large capacities. Efficient filtering, preferably at high temperature, will be required to protect the gas turbine.
4.2.3 Internal Combustion in Engines. A gasifier/engine system is currently the most attractive way of generating shaft power
or electricity from biomass in the power range from a few kilowatts to several megawatts. This option is made attractive by the simple arrangement of a gasifier/engine system with the relatively high overall efficiency in producing shaft power at different loads (with diesel engines up to 30%), and the possibility of immediate use of other fuels. The technology is already relatively old, having started in the 1890's. The wide spread application in the second world war was caused by a shortage of liquid fuels and its moderate revival in the last two decades was stimulated by sharply increased crude oil prices. Re-introduction of this technology for traction will probably remain a curiosity in the industrialised world, unless a dramatic shortage of liquid fuels arises. For stationary applications, however, particularly for electricity generation, the R&D work of the last 15 years has resulted in a commercially successful technology which now is being implemented, especially in the developing countries.
A major problem is that minimum gas quality requirements for engines are still unclear.

138
For dust, values vary from <0.5 to <20 mg/m^ whilst for tar a maximum value of 100 rng/m^ is quoted. There is much uncertainty on the efficiency of the gas cleaning methods. On dust filtration some results have been published recently [17]. Primary cyclones were found to be not effective for particles below 5 micrometer, Barrel filters not effective for diameters in the range 0.3-3 micrometer and bag house filters not efficient for particles between 0.3 and 1.0 micrometer. However, two stage or two sleeve fibre glass filters were found to be effective for any particle size. Tar removal seems to remain the major problem, particularly at the small scale below several MWe where secondary gasification is not economical. Under specific, but poorly understood conditions, particularly with respect to specifications on fuel size, downdraft gasifiers can produce a relatively tar free gas. However, with irregular solids or with no-throat gasifiers as developed for rice hulls, tar removal still requires further additional R&D.
Control devices for optimizing the fuel/air ratio to the engine under varying loads are proven technology, as are the engines [18]. Whether to choose for a diesel engine or a spark ignition engine will vary from application to application. For a discussion on this topic see Kohan [19] for example.
For biomass based power stations of capacities of 10 MWe and higher, direct combustion of biomass is technically proven and an economically feasible technology in some applications [20]. It follows that there is still a need to develop biomass based power generating systems in the capacity range of 1-10 MWe. It is here that gasification in fluidised beds and/or in fast circulating fluidised beds may have potential. Both atmospheric gasification and pressurised gasification may be considered, with the latter option probably in combination with route B of Figure 5 (power generation by gas turbines).
For biomass based power generation in engines Fritz Werner has sold units up to 1 MWe [21] and Ahlstrom Oy in Finland has developed units for combined heat and power in the range of 2-28 MWe + 3-35 MWt [2]. European operating experience both with a counter-current Smaus system and a two-stage Michel Kim (Easimod) gasifier each at a capacity of 500 KWe have been collected by Friedrich Wahl GmbH. [22]. For operating experience in developing countries, reference should be made to the reports of the World bank [23].
4.2.4 Gasification for Ammonia Production A pressurised high temperature Winkler fluidised bed gasifier is operated in Finland for
ammonia production [24]. The feed is 23 ton peat per hour and the pressure is up to 4 bar. The longest uninterrupted operation time so far has been 31 days.
4.3 Medium Joule Gasification Medium Joule gas can be produced if oxygen instead of air is used as the gasifying
agent. Alternatively, steam can also be used as the gasifying agent but then some provision should be made to supply sufficient heat to the gasifier because steam gasification is an endothermic process. Various options are available as indicated in Table 5. The so called Oxygen Donor Gasification process may lead to a Medium Joule Gas using air as the gasifying agent. For a review of the various process principles and technologies, see Beenackers and van Swaaij [25, 26].
Although none of these medium Joule processes are commercial yet, much development work has been done during the past decade, particularly in the European Community under the second E C Biomass Development Programme "Methanol from Wood". An overview of the companies involved in medium joule gasification and of the process types they selected to develop is presented in Table 5 above. Under the E C programme four pilot plants were operated at design capacities ranging from 4.8 to 12 tons dry wood/day. An overview of the results is presented in Table 6; a more comprehensive analysis of the results of this programme has been published elsewhere [27]. The main result of this programme is that atmospheric gasification with oxygen both in a classical fluid bed (Framatome) and in a fast circulating fluid bed (Lurgi) are technically proven.

139
Table 5 Principle Gasification Routes for Medium Joule Gas from Biomass
Gasifying Agent Process Types Processes
Oxygen Downdraft Updraft
Cross flow Huid bed
Circulating fluid bed Entrained bed
Steam With heat carrier (double fluid bed)
With indirect heat supply (fluid bed) With recycle of synthesis gas (fluid bed)
Air With heat and oxygen carrier (double fluid bed)
SERI SFW-Funk
Purox Simplex
Foster Wheeler Framatome*
ΜΓΝΟ IGT
Omnifuel Lurgi* Texaco Baillie
Battelle TNEE Compiegne*
AVSA* C02 Acceptor
ALI 'bN/ltaienergie* ΚΉ
John Brown/Wellman*
* Sponsored by the Commission for the European Communities.
Table 6 Main Characteristics of Pilot Plant Projects for EC Methanol from Wood
Programme
Organ- Press isarion bars
Framatome 1
Lurgi 1
John Brown 1 /Wellman
Italenergie 1 /AGIP
:. Capacity kg dry wood/h
350
200
400
500
Reactor ţy£S
fluid bed+ 2° empty tube gasifier
circulating (fluid) bed
double fluid bed + chemically active solids
Gasifying agent
02&H20 for 1° & 2° gasifier
02&H20
Air
Duration of test run
24 hrs
36 hrs
9.5 hrs
1° fluid bed steam H20 for 1° 100 hrs gasifier heated gasifier, 02 through wall + 2° for 2° gasifier fluid bed
H'carbon Reagent in svngas. use. kg/ vol % kg wood
0.6
6.5
12.5
12.4
02:0.57 H20:0.08
02:0.453 H20:0.02
Air only
N/A

140
Progress on circulating fluidised beds has been realised with both the AVSA process [28] (G4) and the TNEE-Compiegne [29] (G5) dual fluidised bed. Due to significant improvements in the slot designs of the former, the early problems of excessive gas-leakage between the compartments have been largely eliminated.
The EC programme aimed to develop the large scale methanol from biomass technology. With present oil prices below US$20/bbl this technology is not economically feasible. However, application for power generation is already a viable option, depending on site specific conditions. Prospectives for large scale power generation via pressurised gasification of coal using low cost oxygen enrichment techniques such as membrane technology, means that medium joule gas might also be viable from biomass. If so, there is an extra incentive for pressurisation of the gasifier because of the increased flow ratiorproduct gas/gasifying agent, in oxygen gasification relative to air gasification. This conclusion holds for either methanol synthesis or power generation. In the latter technology the use of gas turbines seems to be particularly attractive because of efficiency considerations. Although this route is not commercially available yet, significant development work on pressurised medium joule gasification is in progress; particularly in fluidised beds (see Table 7).
The Framatome project [30] (Gl, Gil) is the only pressurised pilot plant within the EC. Operating results will probably become available from 1990 onwards. In parallel, fundamental research is carried out by Gelus et al, on the kinetics of gasification under pressure [31] (G10). This is considered to be essential research for pressurised units because hardly any information is available on the influence of pressure on pyrolysis and gasification but for the fact that such an influence exists.
Table 7 Pressurised Oxygen-Wood Fluid Bed Gasification Projects [30, 32-35]
Company
Framatome MINO Biosyn/Omnifuel IGT
Pressure bar
5-30 10-30 14 20
CaDacitv kg drv wood/hr
2500 300 1400 400
Moisture wt%
15-40 50 5-45 —
Temperature Ώ
800-1000 700-850 — ~
4.4 Catalysis in Medium Joule Gasification Results obtained outside the E C have shown that a potential exists for catalysis both in
enhancing gasification rates in double fluidised bed processes and in improving the synthesis gas quality, reviewed in [26]. A major conclusion of the Venice Workshop on thermochemical conversion routes in 1985 [36] was that also the E C should take up fundamental research in this area. As a result, the third C E C . R&D subprogramme Energy from Biomass, which started in 1986 included several topics such as the project of TNEE on the improvement of the dual fluidised bed wood fast pyrolysis/gasification process by use of catalysts [37] (G5). The project consists of: • fundamental studies on the catalytic activity of dolomite;
development of a catalyst with favourable characteristics under operating conditions in a double fluidised bed (sintering, erosion, poisoning);
• solids circulation studies on a fluidised bed gasifier - fast circulating combustor system; • pilot unit tests at a capacity of 10-20 kg/hr.
A special feature of the TNEE process is a counter-current gas-solids trickle flow contacting device above the fluid bed. Here pyrolysis gases are heated with the hot solids from the combustor raining down into the bed over a packing. The main challenge of the project is in finding suitable catalysts for product gas composition improvements and/or reduction of the solids circulation flow rate. The main problems here will be catalyst deactivation, particle attrition and catalyst effectivity to produce a hydrocarbon free gas. As far as the project concentrates on alkali carbonates as catalysts still much work remains to be done at the

141
fundamental level on a small scale. A second project is at the University of Zaragoza [38] on catalyst steam gasification,
which includes testing catalysts on activity, selectivity, deactivation rates and attrition risks. Results obtained with a single fluid bed are reported at this conference [39]. Future work will be directed towards a circulating multi-solids fluid bed such as developed by Battelle Columbus [40].
4.5 Hydrogen From Producer Gas Traditionally, the E C "Energy from Biomass" Programmes have also supported the
exploration of speculative new ideas on a laboratory scale to explore long-term potential. An example is the recovery of pure hydrogen from low joule producer gas by a slurry of hydridable metal alloys. Under the previous programme very reactive metal alloy slurries have been developed [41] (G3).
The objective now is to select suitable reactors to apply the technology at a continuous scale, and to evaluate the design rules for scaling-up and to optimise the process conditions. Opportunities are in synthesis gas production from simple single bed air gasification, in providing the hydrogen both for pyrolysis oil upgrading and direct hydro-liquefaction of biomass, and, who knows, in cold fusion if it ever becomes practical.
5 PYROLYSIS 5.1 What is Pyrolysis?
Pyrolysis is thermal degradation either in the complete absence of oxidising agent, or with such a limited supply that gasification does not occur to an appreciable extent which may be described as partial gasification. Relatively low temperatures are employed of 500-800°C, compared to 800 to 1100°C in gasification. Three products are usually produced: gas, liquid and char, the relative proportions of which depend very much on the pyrolysis method and reaction parameters. Fast or flash pyrolysis is used to maximise either gas or liquid products according to the temperature employed.
5.2 Why is Pyrolysis Interesting? Pyrolysis is attractive because solid biomass and wastes which are difficult and costly
to manage, can be readily converted to liquid products. These liquids, as crude bio-oil or slurry of char and water or char, have advantages in transport, storage, combustion, retrofitting and flexibility in production and marketing. The energy density advantages are summarised in Table 8.
Table 8 Energy and Density Characteristics of Biomass and Derived Products
Feed
Straw Woodchips Pyrolysis liquid Char Char-water slurry (50/50) Char-oil slurry (20/80)
The crude pyrolysis liquid is a black fluid which is often referred to as bio-oil, pyrolysis oil, or just "oil". The other main liquid product is a slurry which can be made from water and ground charcoal with chemicals added to stabilise the suspension. Stable and mobile concentrations of up to 60% wt charcoal have been reported. Slurries can also be made from the oil and char.
In pilot plant work to date the gas is usually flared but in a commercial process it would
Bulk density kp/m3
-100 -400
-1200 -300
-1000 -1150
Heatine value drv basis
GJ/t
20 20 25 30 15 23
Enerev densitv GJ/m3
2 8
30 9
15 26

142
be used to drive the process or use it as a fuel gas for fuel drying or power generation [42]. In transport bulk density is important, and some estimated values are given in Table 8
above. Oil and slurry mixtures have a clear advantage over woodchips and straw in transport bulk densities and notably in energy density. For longer distances this difference may be a decisive factor.
Storage and handling may be important because of seasonal variations in production and demand and some storage will always be required [43]. Apart from the bulk density and energy density considerations, it is important to appreciate that crude biomass (such as wood chips and straw) will deteriorate during storage due to biological degradation processes. Char, however, is very stable and will not deteriorate. Another important factor is handling, in which liquids have significant advantages over solids.
Potential disadvantages can arise from the chemical and physical instability of bio-oil, bio-slurries and mixtures, although there are mixed reports on such problems. These are discussed later, but unless the properties of these fluids can be completely controlled, the advantages of liquid fuels cannot be fully realised.
Combustion, retrofitting, market flexibility. Generally liquid (or gaseous) products are easier to handle in the combustion process and this is important in retrofitting existing equipment. Existing oil fired burners cannot be fuelled directly with solid biomass without major reconstruction of the unit, which may not be attractive in uncertain fuel markets. However bio-oils, char-oil slurries and char-water slurries are likely to require only relatively minor modifications of the equipment or even none in some cases [44]. Powdered coal fired furnaces can relatively easily accept charcoal as a partial fuel replacement, as long as the volatile content is compatible with the furnace design.
It is likely that gas turbines can be readily fired with bio-oil and slurry fuels although care is needed with the alkali ash residue in the char content of the slurry [45]. Modified diesel engines may also be modified to accept upgraded char/water slurries or related products, but there is little recent practical experience as yet. In most countries there is a small market for charcoal lumps and briquettes for leisure and industrial applications and small regional markets for firewood, usually as logs. There is reported to be a growing demand for charcoal for specialist steel and non-ferrous metal production [46].
5.3 Pyrolysis Technologies The heat required for pyrolysis can be added indirectly in a variety of ways such as
indirect firing, hot gas, or hot liquid such as metal or molten salt [47](PI2), or directly by partial gasification with limited addition of oxidising agent such as air to give direct heating [48KP5).
Pyrolysis has been practiced for centuries for production of charcoal. This requires relatively slow reaction at low temperatures to maximise solid char yield (P6). More recently, studies into the mechanisms of pyrolysis have suggested ways of substantially changing the proportions of the gas, liquid and solid product. This is achieved by changing the rate of heating, and the final temperature (P7).
High heating rates, of up to a claimed 1000°C/s or even 10 000°C/s, at temperatures below about 650°C and with rapid quenching, causes the liquid intermediate products of pyrolysis to condense before further reaction breaks down higher molecular weight species into gaseous products. These high reaction rates also minimise char formation, and under some conditions no char is apparently formed. At higher maximum temperatures the major product is gas. Pyrolysis at these high heating rates is is known as fast, flash, or ultra pyrolysis according to the heating rate and residence time, although the distinctions are blurred. Other work has attempted to exploit the complex degradation mechanisms by carrying out pyrolysis in unusual environments. The variations are summarised in Table 9.
A wide range of processes based on flash pyrolysis have been researched and developed in these various modes in the last few years either to produce liquid bio-oil in high yield, or to produce chemicals. In a few cases a reactive environment has been included to influence the type of products. Examples of the main technologies and reactor configurations employed are listed in Table 10, which is not intended to be exhaustive but indicative of the variety of processes under development [50].

143
Table 9 Characteristics of Pyrolysis Technologies [49]
Slow pyrolysis Carbonisation Conventional
Fast pyrolysis Fast Flash - liquid Flash - gas Ultra Vacuum
Reactive pyrolysis Hydropyrolysis Methanopyrolysis
Pvrolvsis technology and
Residence Heating time rate
hrs-days very low 5-30 m low
0.5-5 s fairly high <1 s high <1 s high <0.5 s very high 2-30s medium
<10s high
Table 10
Temp. °Cmax
400 600
650 <650 >650 1000 400
<500
Examples of Pyrolysis Technologies [50]
I main product(s) Reactor
gas,
Maior product
solid , liquid & solid
Organisation.
liquid liquid
gas gas
liquid
liquid
Country
Liquids Conventional for liquids Cyclonic for liquids and gases Fast entrained flow for liquids Vacuum for liquid fuels and chemicals Ablative for liquids and chemicals Low temperature for liquids & charcoal Flash fluid bed for liquids
Solids Conventional for charcoal & liquids
Stirred bed Alten (KTI + Italenergie), Italy Cyclone Ensyn Engineering, Canada Entrained upflow Georgia Inst Tech., USA Multiple hearth Laval University, Canada Vortex Solar Energy Research Inst., USA Auger Tübingen University, West Germany Fluid bed Waterloo University, Canada
Downdraft
Gases Molten salt Batch Methanopyrolysis for gases & chemicals Entrained flow Hydropyrolysis for gases & hydrocarbons Autoclave Twin fluid bed fast pyrolysis Twin fluid bed Fluid bed Fluid bed
Bio-Alternative SA, Switzerland
Aston University, UK Brookhaven National Lab., USA
Toronto University, Canada TNEE, France
University of Zaragoza, Spain
Chemicals Vacuum for liquid fuels & chemicals Ablative for liquids & chemicals Molten salt for gases & chemicals Methanopyrolysis for gases & chemicals Hydropyrolysis for gases & hydrocarbons Autoclave
Multiple hearth Laval University, Canada Vortex Solar Energy Research Inst., USA Batch Aston University, UK Entrained flow Brookhaven National Lab., USA
Toronto University, Canada
One of the more innovative processes being developed is based on the principle of ablative pyrolysis in which biomass "liquifies" if it is pressed onto a hot moving surface at below about 650°C [51]. At these temperatures the liquid vapourises and if it is removed sufficiently rapidly from the high temperature zone and quenched, high liquid yields with very

144
low char yields result The most advanced research of this type is being earned out at the Solar Energy Research Institute [52] and a diagram of the equipment is shown in Figure 6. Other processes based on a range of technologies are also well advanced [50] and await a suitable opportunity for larger scale demonstration [53].
Biomass pins
To liquid condensation train and collection
Char cyclone
Screw feeder
400 to 750 °C Steam
Steam ejector
Vortex reactor
Char receiver
Figure 6 Vortex Ablative Pyrolyser at SERI [52]
5.4 Pyrolysis Products
5.4.1 Liquid Product The process of pyrolysis is complex, but a recent theory is that primary vapours are
first produced, the characteristics of which are most influenced by heating rate. These primary vapours then further degrade to secondary tars and gases, the proportions and characteristics of which are a function of temperature and time [54]. Yields of liquids from pyrolysis can thus be influenced by the rate of reaction, with fast or flash pyrolysis at lower temperatures of typically 450-650°C giving the highest liquid yields.
This liquid product may be readily bumed [44] and has been employed for this purpose [55]. There are, however, some precautions which have to be taken in handling, storage and combustion due to the water and high oxygen content (P6). For these reasons, pyrolysis liquids cannot be directly assimilated into a conventional fuel marketing infrastructure and some conversion or upgrading is necessary to give a product that is compatible with conventional fuels. Upgrading technology is not well developed with most attention being paid to either hydrotreating or zeolite decarboxylation to give synthetic gasoline and other hydrocarbons.
Characteristics and Utilisation The liquid product is a highly oxygenated hydrocarbon with an appreciable proportion
of water from both the original moisture and reaction product. Solid char may also be present Raiano). These properties can make it relatively unstable in both chemical and physical terms and have been reported to cause some problems in utilisation and upgrading. It is readily combustible, but care has to be taken in storage, handling and atomisation. Some of the other characteristics are discussed below [56].
Water content is important as it has several effects: it reduces the heating value, affects the pH, reduces the viscosity, influences both chemical and physical stability, reduces potential pollution problems from waste water disposal and could affect subsequent upgrading processes [57]. The interactions are poorly understood. The water is difficult to measure and remove, since evaporation or distillation at normal temperatures of around 100°C can cause significant

145
and potentially deleterious physical and chemical changes in the liquid. Lower temperature drying is not successful due to the nature of the relationship between water and the organic component in which the water seems to be chemically combined, analogous to water of hydration. This phenomenum makes claims of water content and consequently oxygen content of liquids on a dry basis subject to some uncertainty. Water appears to be completely miscible up to 20% by weight of total liquid, but above which an aqueous layer separates. Any water that does separate must be carefully managed and this is discussed further below. Utilisation and consideration of oil on a "wet" basis therefore seems to be more sensible. A much more attractive approach appears to be to not condense the water by maintaining the pyrolysis vapours above the dew point of water i.e. above about 110°C. The principle has been tried by Roy in his vacuum multiple hearth pyrolyser [58] and successfully practiced by Bio-Altemative [59].
Particulate levels may be high from char and ash carry-over. Separation of solids and liquids is poorly understood with reliance placed on primary separation in the vapour phase downstream of the reactor before condensation. Efficient separation inevitably causes some condensation or precipitation and careful design is essential. Solid separation in the liquid phase is not believed to have been studied, but is very likely to be troublesome. However, it is clear that a fairly high level of charcoal can be assimilated in the liquid product, for example Alten reported up to 15% [48] although some lumpiness was evident in the bio-oil. Both char particle size and proportions will influence the liquid product quality. This is why research into char-oil mixtures could prove valuable.
Oxygen content of the pyrolysis liquid is very high, at up to 40% wt. When produced from dry or low moisture content feeds it typically has a heating value a little above that of the biomass feed in the range 20 - 25 MJ/kg, which has caused it to be referred to as "liquid biomass". The oxygen content arises from oxygenated compounds including phenols and polyphenols, which can be recoveered as a valuable chemical fraction [60].
Low pH arises from the organic acid content (e.g. acetic and formic acids), and is therefore corrosive. Mild steel is not suitable for handling or storage. Polypropylene piping has been used to overcome this problem.
Polymerisation or deterioration of the liquid can be caused by temperatures above around 100°C which adversely affect physical properties such as viscosity, phase separation, and deposition of a bitumen-like substance. Heating the liquid to reduce viscosity for pumping or atomisation needs to be considered carefully and thoroughly tested. Exposure to air also causes deterioration, but at a slower rate than temperature increase. Maintenance in a sealed enclosure has been claimed to cause substantial pressure increases, so some minimal venting is necessary to avoid pressure build-up, but minimise exposure to oxygen. Pyrolysis liquid has been stored in this way in a useable form for up to two years without problems (P6). Liquids produced from refuse/MSW appear to be much more unstable [52, 61, 62].
Health hazards associated with pyrolysis liquids are also poorly understood. It has been claimed that these are no worse than coal tar or crude oil [63].
Compatibility with conventional fuels is variously reported as immiscible but compatible (P4). Pyrolysis liquids cannot be expected to be assimilated into a conventional fuel marketing infrastructure without some conversion or upgrading to give a product that is compatible with conventional fuels. One alternative is to feed to crude pyrolysis liquid into a refinery for upgrading in orthodox refinery operations, utilising the hydrogen availability and blending opportunities [64]. The alternative is to create a discrete pyrolysis liquids storage, distribution and utilisation system, that is managed by experts who understand the special problems of this fuel.
Some properties that have been reported are summarised and compared in Table 11.
Stabilisation The crude liquid product can be used directly if in a single phase. If more than about
20% water is present, it can be processed to overcome some of the above problems by emulsification with the water content if this separates (at typically above 20%wt water).. This controls the stability of the liquid to a certain extent, and also enables the contaminated water to be effectively disposed of, but at the expense of a lower heating value product. The cost is relatively high with additives costing about half the value of the product as fuel [46,48].

146
Table 11 Comparison of Pyrolysis Technologies - Typical Data [1]
Conventional Fast Alten * GIT
Temperature, °C 500 480 Products, yield on daf feed
% wt gas 68 % wt liquid (dry) 21 51 % wt water 26 29 % wt char 21 20
Liquid characteristics oxygen (raw product), % wt - 53 oxygen (dry product), % wt 15 42 water, % wt 14.6 17 viscosity, cps@40°C 300 220 pour point, °C 27 -23 density, g/cm^ 1.195 1.26 pH 2 HHV, MJ/kg raw product 26.3 18.3 HHV, MJ/kg dry product - 22.1 Elemental analysis, dry product Cwt % 61.9 52.2 Hwt % 6.0 6.3 Owt % 14.9 41.5 H:C molar ratio 1.16 1.45 0:C molar ratio 0.18 0.60 Char content, % wt 9.2
* (P5).
Upgrading
Flash Waterloo
510
10 66 10 14
-39 18 40
-1.19 2.4
16.3 -
54.7 6.4
38.9 1.40 0.53
Upgrading technology is based either on orthodox hydrogénation technology to produce successively lower oxygen content hydrocarbons, or the evolving zeolite technology to produce hydrocarbon fuels directly.
Hydrotreating is based on technology that is established in the petroleum industry and is in principle readily adaptable to reducing or removing the oxygen content of the bio-oil. Preliminary results indicate that conventional hydrotreating processes may be readily adapted to pyrolysis liquids [65](P8) and [66]. An alternative approach is to send the crude bio-oil to a conventional refinery for upgrading with fossil oil. This has not yet been examined.
Zeolite based synthesis of hydrocarbons has been extensively demonstrated for alcohol feeds. Some experience has been gained on upgrading products of cellulose pyrolysis by decarboxylation [52, 67, 68], but there is concern over the problems of coking which would require a regenerative process. No reliable results are yet available.
Neither technology is yet available commercially, nor have robust mass balance and performance data been produced, although a comprehensive technoeconomic assessment suggests that atmospheric flash pyrolysis gives gasoline costs approximately double that of conventional fuels with the potential to reduce this considerably in the medium term [69]. The zeolite decarboxylation route gives potentially higher quality products due to preservation of the aromatic structures resulting in high levels of benzene, toluene and xylene.
An alternative approach is to reduce the oxygen content to a sufficiently low level that it may be satisfactorily blended with conventional fuels. This might be achieved by less complete hydrogénation, or by simple distillation over activated clay such as bentonite which is claimed to give a stable and storable product in one low cost step. No work on blending requirements is known to have been carried out, but has been considered as a possible route to utilisation [64].

147
5.4.2 Gas product The gaseous product from pyrolysis is usually a MHV fuel gas around 15-22 MJ/Nm3,
or a LHV fuel gas of around 4-8 MJ/Nm3 from partial gasification depending on feed and processing parameters. It has a high level of hydrocarbons, particularly methane, and saturated and unsaturated hydrocarbons from the complex thermal dgradation processes. The heating value is enhanced if the gas is used and kept hot, from the sensible heat, and the relatively high tar content. The gas may be used to drive the pyrolysis process if an indirectly heated process is used, or it can be employed to dry the feed, or generate power.
5-4.3 Solid product When pyrolysis is optimised for charcoal production, yields of up to 30 or 40% wt on
dry feed are obtained. This occurs in slow pyrolysis with reaction times of hours or days. Partial carbonisation gives the higher yields, when the product contains a high level of volatiles, and this is also referred to as torrified wood. At the very high heating rates encountered in fast and flash pyrolysis, very low char yields result, and have been reported as approaching zero under some process conditions. This avoids the marketing and design problems of a multiple product process, since the associated byproduct - gas in the case of liquid production for example - can be used as process heat [46, 53](P5). Char yields can be optimised for production of material for char-water slurries, although an integrated approach is necesary for maximum energy recovery.
5.4.4 Slurries A possible outlet for the char is slurrying with the oil, or with water, or with both oil
and water. Only a limited amount of char can be introduced into oil as unacceptably high viscosities result from a char concentration higher than about 30% wt [46] (P4, PIO). The maximum concentration of char in water that can be handled is about 60% to retain mobility [42]. Costs of the additive are significant at about 1/3 of the slurry preparation cost [46]. Three phase slurries of bio-oil, char and water or waste-water are not feasible.
Coal-water slurries are increasingly used in large boilers and these slurries can be simply and/or partially replaced by char-water slurries. The char/water slurry cannot, however, be an outlet for pyrolytic waste water as unstable sludge formation results. The ash content of the char is an important consideration in developing liquid fuels, and de-ashing processes are being examined [70] (P6).
Although in principle it seems to be attractive to remix all products of pyrolysis process into one single liquid biomass derived fuel, this does not currendy seem possible.
5.4.5 Chemicals Several hundred chemical constituents have been identified to date, and increasing
attention is being paid to recovery of individual compounds or families of chemicals [52, 58, 71] (P7). The potentially much higher value of speciality chemicals compared to fuels could make recovery of even small concentrations viable. An integrated approach to chemicals and fuels production offers interesting possibilities for shorter.term economic implementation.
5.4.6 Water A key feature of the pyrolysis process is that water is produced in significant quantities
of typically between 20 and 40% wt on the feed, depending on feed moisture content. The water phase is highly contaminated with dissolved and suspended organics, with a COD of typically 150 000. This therefore represents a major problem of disposal or utilisation. In the selection of the primary pyrolysis products this waste water must be considered (PIO). If biological treatment is not appropriate or too expensive, part of the heat of combustion of the products will be required for incineration of this heavily contaminated water fraction. The pyrolysis gas should primarily be used for this purpose but this may not be enough in cases where the primary feedstock has a high water content and the gas is required for feed drying.
A potentially more attractive alternative route than incineration is oil condensation above the dew point of water, i.e. about 110-120°C. The water then stays in the vapour phase and can be burned with the product gas [59].

148
5.4.7 Secondary Products The range of secondary products derivable from pyrolytic oil and char is summarised in
Table 12. The secondary products obtainable from the gas are not different from those obtained by gasification (see Section 4).
Table 12 Secondary Products from Pyrolytic Oil and Char
Primarv product
LHVgas
MHVgas
Liquid
Process
-Engine Turbine Conversion
_ Engine Turbine Conversion Conversion Conversion Conversion
Hydrogénation
Zeolites
Secondary product -Power Power Ammonia
_ Power Power Methane Methanol Methanol Hydrocarbons
Intermediate
Hydrocarbons
Process
---Conversion
. ---Refine Conversion Refine
Refine
Refine
Market
Fuel gas Electricity CHP Fertilisers
Fuel gas Electricity CHP SNG Methanol Gasoline Gasoline, diesel, fuel oil
Gasoline, diesel, fuel oil Gasoline, diesel, fuel oil
Size ranee \Jh input
0.1-5 0.1-5 1-10 2-20+
1-5 1-5 1-10 10+ 10+ 10+ 10+
5-20+
5-20+
Char
Notes: + upper size limited by feed supply
(briquetting) Solid fuel 0.2-5
5.5 Pyrolysis Implementation Bio-oil is claimed to be a relatively easy fuel to use, provided the viscosity is not too
high. Preheating to reduce viscosity is not usually favourable due to thermal degradation of the bio-oil. The water content can be considered an advantage both for the combustion process and because it reduces the viscosity of the liquid. Therefore the oil can be considered an outlet or disposal route for some of the pyrolysis water. Phase separation is likely to occur at water concentrations greater than 20% which could only be counteracted by costly emulsifiers. Moreover the water could render the oil more unstable and more corrosive. Methanol has been suggested as a possible additive [42].
An alternative to upgrading bio-oils on a relatively small scale is to introduce them into some sector of mineral oil refining operations [64]. Bio-oil has a relatively large oxygen content however, and oxygen is not easy to remove, being placed in this respect between nitrogen and sulphur contaminants in oil. In a refinery biomass oil would profit from the economics of large scale processing used in mineral oil refining as well as the ready availability of low cost hydrogen. Research is planned in this area. One problem is that the resultant value attributed to the bio-oil by the refinery may be too low to justify implementation.
5.6 Pyrolysis Status in Europe and North America A demonstration plant of 500 kg/h is currently operating in Italy for liquid bio-oil
production with plans for a series of small commercial units there, in Spain and in Greece. A number of demonstration plants for flash pyrolysis for bio-oil production are operating in

149
North America at a scale of up to 25 kg/h with plans for several commercial developments ranging up to 20 t/h, including a commercial installation planned for California based on the SERI ablative pyrolyser [72]. In addition to conventional and well established units for charcoal production, a number of new carbonisation processes have become available for production of slurry fuels from the charcoal and also recovery of the oil as a liquid fuel [73, 74]. These are, however, slow pyrolysis processes that give a low liquid yield. Examples of current activities are listed in Table 13, all of which are orientated to liquid or slurry fuels production.
Table 13 Examples of Pyrolysis Processes
Status Pilot
Demonstration
Commercial
Reactor type Cyclonic reactor Cyclonic reactor Entrained flow Fluid bed Multiple hearth Auger kiln Fixed bed Fluid bed Fluid bed or stirred bed Rotary kiln Fixed bed Horizontal moving bed Cyclonic reactor (planned)
Example, (country') Ensyn (Canada)
SERI (USA) GIT (USA)
Waterloo (Canada) Laval (Canada)
Waste Water Treatment Centre (Canada) Cemagref (France)
Waterloo (Spain) Alten (Italy)
Kiener (West Germany) Bio-Alternative (Switzerland)
Pyrosol (USA) Pyrotech (USA)
5.7 Pyrolysis Costs Of major importance in implementation of pyrolysis technologies is the economics of
production. A preliminary economic analysis is given in Figure 7 to show the effect of scale of operation and feedstock cost on product price [49].
Current typical fuel oil prices are around 125 ECU/t, which is equivalent to 80 ECU/t of pyrolysis oil on an equivalent heating value basis of 25 MJ/kg although it is not yet established what conventional fuel product can be equated to bio-oil. This is shown as the lighter shaded part of Figure 7 which identifies the plant capacities and feed costs that can be justified at this price level. The larger darker shaded area represents an oil price 50% higher, showing the sensitivity of production costs to scale of operation and feed cost. Special credit might be given to bio-oil because of the extremely low sulphur content and possibly low NOx production. Thus pyrolysis oil could be competitive on a relatively small scale of production.
A more comprehensive assessment of bio-oil and bio-slurry fuel costs has recently been completed [AVB 164/165].
6 LIQUEFACTION
6.1 What is Liquefaction? Liquefaction is low temperature, high pressure thermochemical conversion in the liquid
phase, usually with a high hydrogen partial pressure and also a catalyst to enhance the rate of reaction and/or to improve the selectivity of the process. This approach gives a more physically and chemically stable liquid product requiring less upgrading to produce a marketable hydrocarbom product. Catalysts are also employed to provide enhanced hydrogénation and de-oxygenation, as well as some selectivity in product formation.
The attractions of this technology include heat transfer to a liquid phase which is more effective than to a gas phase, the liquid phase reduces the reactor and ancillary equipment volume requirements, and the product is a higher quality liquid than from pyrolysis processes in terms of higher heating value and lower oxygen content.

150
Pyrolysis Liquid Production Cost ν Feed Cost
400-
Product cost, ECU/t
300'
200'
100'
2t/h 3t/h 5t/h
20 Vh 100t/h
40 50 60 Feed cost, ECU/t
Figure 7 Pyrolysis Oil Production Cost in relation to Fuel Oil Prices now &+50%
The high cost of high pressure processing, and unresolved problems of feeding biomass slurries at high pressure, product separation from solvent if used, and use of high pressure hydrogen have all caused significantly less activity in this area of thermochemical conversion. Most work has been on a batch scale of operation (for example [75, 76] in Europe, with only a few examples of continuous processing, including a 25 kg/h plant that was built at Albany in the USA but has now been dismantled; a 1 kg/h plant in the USA [77] and another in the UK [78].
More activity has concentrated on black liquor as a feedstock in recent years due to the adverse economic situation for biomass derived fuels, and the economic and environmental attractions of reducing the waste management problems surrounding black liquor [79].
The potential processing and reaction advantages of this conversion route should not be ignored for longer term possibilities
6.2 Why is Liquefaction Interesting? The particular interest of liquefaction is that a lower oxygen content product is produced
that is more stable and requires less upgrading to a hydrocarbon product. There are also processing advantages with a liquid phase system that requires a lower volume reactor and ancillary equipment, and also lower reaction temperatures that result in lower heat losses and easier materials of construction problems.
A particular disadvantage is the high pressure requirement which is costly and potentially more hazardous. This gives rise to feeding problems associated with slurries. Hydrogen is often added to effect reducing reactions which increases problems and costs, and In addition catalysts are often employed to improve yields of desired products which can lead to more complex catalyst recovery systems or high costs from catalyst losses. A solvent is also necessary as a solid carrier which requires separation and recovery and this also increases process complexity and cost.

151
6.3 What products are produced? The main product from biomass liquefaction processes is a low oxygen content oil
(around 10-15% wt oxygen on a dry basis), with a heating value of around 35-40 MJ/kg. The byproduct gas may be used to generate hygrogen or used a low heating value fuel gas. Water and a small proportion of char and ash residue are also usually formed. Similar products are derived from black liquor. The oil product may be used directly as a liquid fuel to substitute for fuel oil, or may be upgraded by hydrotreating to hydrocarbon fuels followed by refining for assimilation into the transport fuel market.
6.4 Liquefaction Technologies and Status Most work has concentrated on small scale batch type experimentation (L2), but some
work has also been carried on continuous flow systems (LI). Compared to gasification and pyrolysis, there is comparatively little activity currently due to the high cost of establishing a research facility and low conventional fuel costs. Examples of current activities are listed in Table 14.
Table 14 Examples of Liquefaction Processes
Status Reactor type Example, (country) Bench Autoclave Inst Wood Chemistry (West Germany)
Autoclave VTT (Finland) Autoclave University of Toronto (Canada) Flow reactor T.U.Berlin (West Germany) Fluid bed UMIST/MANOIL (UK) Pressure screw University of Arizona (Canada)
Related to liquefaction is solvolysis in which biomass is dissolved in a solvent at high pressure and elevated temperature, such as [80, 81] and supercritical extraction in which a similar effect is carried out more selectively in a solvent under supercritical conditions, such as [82]. Both technologies are at a relatively early stage of development.
8 CONCLUSIONS 8.1 Gasification
Low joule gasification of biomass for heating applications is a technically proven and often viable technology, provided that the scale of operation is sufficiently large, and that feedstock is available in sufficient quantities at an acceptable cost. • Low Joule gasification of wood for shaft power presently is commercially proven at
capacities up to a few MWe. Smaller units below 0.5 MWe have promising economic potential in developing countries. The economics of retrofitting diesel fuel generator sets with a biomass fueled gasifier can also be attractive in the industrialised world in site specific situations. Significant progress has been made in the field of medium joule gasification. Demonstration projects now have reached the scale of 2-10 tons wood/day operating at pressures up to 30 bar.
• Short term opportunities for medium joule biomass gasification are probably in power generation rather than in methanol production but this remains an interesting long term opportunity.
• The potential for catalysis both in enhancing gasification rates in double fluidised bed processes and in improving synthesis gas quality has been recognised in the E C with fundamental research at bench scale and larger scale applications. There is still much progress to be made [83, 84].
• The most likely short term markets for industrial use of fuel gas from biomass and wastes are where gas quality specifications are undemanding such as in boiler retrofitting and direct firing (in the production of lime, cement, bricks and unglazed

152
pottery). Current and short term economic applications lie in utilising wastes and residues to produce power, and fuel gas where quality requirements are less demanding. There is potential for the production of transport fuels and chemicals in the longer term, particularly in less developed countries and those with few indigenous conventional energy resources.
8.2 Pyrolysis Pyrolysis offers the potential to convert solid biomass into a high energy density
product that is easy and inexpensive to transport and that may be readily utilised in existing installations. The recent EC Energy from Biomass Programme actively examined many aspects of the production, upgrading and utilisation of pyrolytic liquid fuels which will be continued in the successor programme. There are still problems to be resolved, however, if the potential of biomass pyrolysis is to be optimised, including product yield improvement, product quality improvement, new upgrading methods, product testing and utilisation and resolution of environmental problems [84, 85].
8.3 Liquefaction Liquefaction produces a higher quality product in terms of heating value and physiscal
properties, but at the expense of the higher costs of pressurised processing and use of hydrogen. Waste conversion, such as black liquor, is a potentially interesting option.
8.4 General Comments The production of higher value products in combination with fuel production would
enhance the economic viability of the processes. An objective comparison of the various technologies for different applications would be useful.
In general, there are still some technical gaps between biomass production and conversion, and between conversion and application. These will need to be resolved as attention continues to turn to renewable energies.
REFERENCES 1 Bierback, H., Loeffler, J.C., Baguss, I., Knoche, R., The Gasification of Bark and
Sludges in a Lurgi Circulating Fluidized Bed, First Results of the Pilot Plant., in: Proc. 4th E.C. Conference Biomass for Energy and Industry, Elsevier (1987)
2 McKeough, P., Kurkela, E., Sipila, K., Thermochemical Conversion of Peat into Gases & Liquid, Recent developments in Finland, Communication from Technical Research Centre of Finland, 02150 Espo, Finland (1987)
3 Salo, K., :These proceedings 4 Klass, D.L., Resources and Conservation, 11 (1985) 157 5 Klass, D.L. in: D.L. Klass (Ed), Energy from Biomass and Wastes, IX, IGT, Chicago
(1985) 11 6 Bridgwater, A. V., in Bridgwater, A. V and Kuester, J. L. (Eds) Research in
Thermochemical Biomass Conversion, (Elsevier Applied Science 1988) 7 Padilla, A.A., in W.H. Smith (Ed), Biomass Energy Development, Plenum Press,
New York (1985) 377 8 Kaupp, Α., Gasification of Rice Hulls, Theory and Practie, German Appropriate
Technology Exchange, Fed. Rep. of Germany (1983) 9 Manurung, R., Beenackers, A.A.C.M. in W. Palz, J. Coombs and D.O. Hall (Eds),
Energy from Biomass, 3rd EC Conference, Elsevier, London (1985) 900 10 Van den Aarsen, F.G., Beenackers, A.A.C.M., van Swaaij, W.P.M. in: B.
Kjellstrom, H.E.M. Stassen and A.A.C.M. Beenackers (Eds), Producer Gas 1982, The Beyer Inst., Stockholm (1983) 381
11 BECE, Gasification systems, BECE, P.O. Box 498, 7600 AL Almelo, The Netherlands
12 Susanto,H., Institute of Technology, Dept. T.K., Bandung, Indonesia 13 Schaus, O.O., Overall, J.C.K., Lee, G.K. in: D. L. Klass (Ed), Energy from Biomass
and Wastes, IX, IGT, Chicago (1985) 405

153
14 Beenackers, Α.Α.C.M., van Swaaij, W.P.M, in: Α.V. Bridgwater (Ed), Thermocheinical Processing of Biomass, Butterworths, London (1984) 91
15 Kosowski, G.M., Onischak, M., Babu, S.P. in: D. Klass (Ed), Energy from Biomass and Wastes, Vul, IGT, Chicago (1984) 637649
16 Evans, R.J., Knight, R.A., Kosowski, G.M. in: D. Klass (Ed), Energy from Biomass and Wastes, IX, IGT, Chicago (1985) 573
17 Hargrave, R.H., Lundgren, D.Α., Shaw, L.N. in: W.H. Smith (Ed), Biomass Energy Dev., Plenum Press, New York (1985) 389
18 Nolting, E., Leuchs, M. in: W. Palz, J. Coombs and D.O. Hall (Eds), Energy from Biomass, 3rd EC Conference, Elsevier, London (1985) 1141
19 Kohan, S.M. in: D. Klass (Ed), Energy from Biomass and Wastes, IX, IGT, Chicago (1985) 571
20 Beenackers, A.A.C.M., Ismael, S., Power from Biomass via Gasification and Combustion, to be published in Proc. ISES, Hamburg, September 1987
21 Hausen, G.G. in: D. Klass (Ed), Energy from Biomass and Wastes, VIII, IGT, Chicago (1984) 789
22 Friedrich Wahl GmbH, "Bark gasification for combined heat and power production", Final report of C.E.C. Contract EE/287/80 DE,
23 Agence Française pour la Matrise de l'Energie, Biomasse Energie, Le Programme Francais (1987)
24 Sipela, K., C. Wilen, E. Kurkela, A. Moilanen and J. Koljonen: these proceedings. 25 Beenackers, A.A.C.M., van Swaaij, W.P.M., Int. J. Solar Energy 2 (1984) 349 26 Beenackers, A.A.C.M., van Swaaij, W.P.M., Int. J. Solar Energy 2 (1984) 487 27 Beenackers, A.A.C.M., van Swaaij, W.P.M. (Eds), Advanced Gasification, Reidel,
Dordrecht (1986) 1239 28 Platiau D and Ph. Staline in: Euroforum New Energies Congress, Saarbrücken 1988,
Stephens & Associates, Bedford (1980) Vol 3, 660 29 TNEE, in: Euroforum New Energies Congress, Saarbrücken 1988, Stephens &
Associates, Bedford (1980), Vol 3, 653. 30 Lemasle, J.M. in: G. Grassi and H. Zibetta (Eds), Energy from Biomass 1, Elsevier
Applied Science, London (1987) 362 31 Capart R, Gelus, M, Les Courgeus and Li Zhitian, in: Euroforum New Energies
Congress, Saarbrücken 1988, Stephens & Associates, Bedford (1980), Vol 3, 663. 32 Lindman, Ν. in: D L Klass (Ed), Energy from Biomass and Wastes, V, IGT, Chicago
(1981)571 33 Gravel, G, Biosyn, Montreal, personal communication (1982) 34 Schiefelbein, G.F., Sealock Jr., L.J., Ergun, S. in: J.L. Jones and S.B. Radding
(Eds), Thermal Conversion of Solid Wastes and Biomass, A.C.S. Symp. ser. 130 (1980) 13
35 Klass, D.L. in: D.L. Klass (Ed), Energy from Biomass and Wastes, VII, IGT, Chicago (983) 1
36 Bridgwater, A V and Beenackers, A A C M in: W. Palz, J. Coombs and D.O. Hall (Eds), Energy from Biomass, 3rd EC Conference, Elsevier Appl.Sci., London (1985) 247
37 Le Lan, A in: G Grassi and H Zibetta (Eds), Energy from Biomass 1, Elsevier Applied Science, London (1987) 371
38 Corella, J in: G Grassi and H Zibetta, Energy from Biomass 1, Elsevier Appi. Sci, London (1987) 388.
39 Corella, J: These Proceedings 40 Corella, J, Personal communication 41 Knippels J Ρ H M, G F Versteeg and W.P.M. van Swaaij in: Euroforum New Energies
Congress, Saarbrücken 1988, Stephens & Associates, Bedford (1980) Vol 3, 666 42 Antonelli, L, Periodic Reports to the EC 43 Mitchell, C Ρ and Bridgwater, A V, These proceedings 44 Salvi, G and Salvi G, Jnr, Cogis, Final Report to EC, June 1989 45 Zibetta, H, Personal Communication

154
46 Antonelli, L, Personal Communication 47 Maund, J Κ and Earp, D M, in: G. Grassi, D Pirrwitz and H. Zibetta (Eds), Energy
from Biomass 4, Elsevier Appi. Sci., London (1989) 48 Antonelli, L, in: G Grassi and H Zibetta (Eds), Energy from Biomass 2, Elsevier
Applied Science, London (1989) 49 Bridgwater, A V, "Pyrolysis technologies and costs", in: G. Grassi, D Pirrwitz and H.
Zibetta (Eds), Energy from Biomass 4, Elsevier Appi. Sci., London (1989) 50 Bridgwater, A V. and Bridge, S. Α., in: A V Bridgwater (Ed), Pyrolysis Liquids
Upgrading and Utilisation, Elsevier Applied Science, in press 51 Lèdè, J., et al. in: E J Soltes and Τ A Milne (eds) Pyrolysis oils from Biomass, ACS
Symposium Series 376 52 Diebold, J, and Power, A, in: A V Bridgwater and J L Kuester, (Eds) Research in
Thermochemical Biomass Conversion, (Elsevier Applied Science 1988) 53 O'Neil, D, These proceedings 54 Milne, Τ Β, in: Τ Milne and E J Soltes (Eds) Biomass Pyrolysis, ACS Symposium
Series 346, (1988) 55 Bio-Alternative S A, CH-2063 Engollon, Neuchatel, Switzerland, Personal
Communication 56 Bridgwater, A V (Ed), Pyrolysis Liquids Upgrading and Utilisation, Elsevier Applied
Science, in press 57 Bridgwater, A V, in: E Matucci, G Grassi and W Palz, Pyrolysis as a basic technology
for large agroenergy projects, EUR 11382 EEC Luxembourg, (1989) 58 Roy, C, in: A V Bridgwater, and J L Kuester, (Eds) Research in Thermochemical
Biomass Conversion, (Elsevier Applied Science 1988) 59 Bio-Altemative S A, Engollen, Switzerland, Personal Communication 60 Diebold, J, et al., in: A V Bridgwater and J L Kuester, (Eds) Research in
Thermochemical Biomass Conversion, (Elsevier Applied Science 1988) 61 Mallon, G M, Chemical Enginering, July 19 1976, p90 62 Diebold, J, Personal Communication 63 Bridgwater, A V, Rapporteur Report Session 3, these proceedings 64 Rupp, M, in: A V Bridgwater (Ed), Pyrolysis Liquids Upgrading and Utilisation,
Elsevier Applied Science, in press 65 Churin, E, et al. in: A V Bridgwater and J L Kuester, (Eds) Research in
Thermochemical Biomass Conversion, (Elsevier Applied Science 1988) 66 Baker, E G and Elliott, D, et al. in: A V Bridgwater and J L Kuester, (Eds) Research in
Thermochemical Biomass Conversion, (Elsevier Applied Science 1988) 67 Chen, Ν Y and Walsh, D, in: Τ Milne and E J Soltes (Eds) Biomass Pyrolysis, ACS
Symposium Series 346, (1988) 68 O'Neil, D, in: D L Klass (Ed) Energy from Biomass and Wastes XIII, 1989, IGT 69 Elliott, D, et al., in D L Klass (Ed) Energy from Biomass and Wastes ΧΙΠ, 1989, IGT 70 Esnouf, C, et al. in: G. Grassi, D Pirrwitz and H. Zibetta (Eds), Energy from Biomass
4, Elsevier Appi. Sci., London (1989) 71 Stoikos, T, in: G. Grassi, D Pirrwitz and H. Zibetta (Eds), Energy from Biomass 4,
Elsevier Appi. Sci., London (1989) 72 Ayres, W, in Energy from Biomass and Wastes XII, IGT (1988) 73 Bio-Alternative S A, CH-2063 Engollon, Neuchatel, Switzerland 74 Cemagref, Parc de Tourvoie, 92160 Antony, France 75 Meier, D. in: G. Grassi, D Pirrwitz and H. Zibetta (Eds), Energy from Biomass 4,
Elsevier Applied Science, London (1989) 76 Meier zu Kocke, H, and Nelte, A in: G. Grassi, D Pirrwitz and H. Zibetta (Eds),
Energy from Biomass 4, Elsevier Applied Science, London (1989) 77 White, D, and Wolf, D, in: A V Bridgwater, and J L Kuester, (Eds) Research in
Thermochemical Biomass Conversion, (Elsevier Applied Science 1988) 78 Manoil Ltd, c/o G Mortimer, Salford University Business Services Ltd, Business
House, University Road, Salford M5 4PP, UK 79 Solantausta, Y and McKeough, P, in: H Egneus and A Ellegard (Eds) BioEnergy 84
Vol ΠΙ, Elsevier Applied Science, 1985

155
80 Heitz, M, et al. in: A V Bridgwater, and J L Kuester, (Eds) Research in Thermochemical Biomass Conversion, (Elsevier Applied Science 1988)
81 Overend, R P, and Chornet, E, in: A V Bridgwater, and J L Kuester, (Eds) Research in Thermochemical Biomass Conversion, (Elsevier Applied Science 1988)
82 Labrecque, R et al,. I. and E. C. Prod. Res. and Dev. (1984) 23, ppl77 83 Bridgwater, A V, van Swaaij, W Ρ M and Beenackers, A A C M, "Thermochemical
biomass conversion: research development and demonstration requirements", in Biomass for Energy and Industry, 4th EC Conference, edited by G Grassi, Β Delmon, J-F Molle, and Η Zibetta, pp 441-456 (Elsevier Applied Science, 1987).
84 Beenackers, A A C M, Bridgwater A V and van Swaaij, W Ρ M, in: G. Grassi, D Pirrwitz and H. Zibetta (Eds), Energy from Biomass 4, Elsevier Appi. Sci., London (1989)

156
APPENDIX 1 RECENT EC SPONSORED RESEARCH IN EUROPE
References to all these projects may be found in Proceedings of EC Contractors Conferences, including the Biennual ENERGY FROM BIOMASS Conference Proceedings published by Elsevier Applied Science. Projects are identified in the text by the reference numbers below.
Gasification G1 Air gasification of biomass for
fuel-gas production G2 Catalytic steam gasification in fluidized
bed of some biomasses existing in Spain to obtain a methane rich gas
G3 Development of a new method for hydrogen recovery from lean gas mixtures using metal hydride slurries
G4 Further developments of the AVSA biomass gasfication process
G5 Improvement of the dual fluidized bed wood fast pyrolysis gasification process; use of catalyst and optimization of particulate solids circulation techniques
G6 Modelling and application of biomass gasification based power plants
G7 Modelling of biomass conversion processes G8 Production of synthesis gas from biomass
gasification. Final data gathering. G9 R&D of gasification module complete with
filters for rice husk & Biomass residue energy conversion
G10 Study of biomass gasification kinetics under pressure
Gi l Syngas production from wood; the pressurised gasification unit of Clamecy
Pyrolysis Ρ1 Accoustic agglomeration to clean
pyrolysis gas P2 Development of a biomass utilization
system at the power plant in Avezzano P3 Evaluation of potential penetration of
pyrolysis conversion technologies in the mediterranean areas of Italy
P4 Improvement of pyrolysis emulsion/slurry, P5 Improvement of pyrolysis conversion
technology utilizing agricultural and forestry wastes,
P6 New process of suspension pyrolysis and use of charcoal slurry,
P7 Production and utilization of synthetic liquid fuels
P8 Quality improvement of pyrolytic oils from biomass
P9 Study on the production of charcoal slurry based on biomass pyrolysis products
J. M. Lemasle
J. Corella
FRAMATOME, France
W.P.M van Svaaij
H.A.Masson
A. Le Lan
University of Zaragoza Spain
Twente University Netherlands
INIEX, Belgium
T.N.E.E, France
J.Heaton Energy Options Ltd., UK
B. Rhodes A.Bernardini
G. Bonino
M.Gelus
J Carré
CHAM Ltd., UK ALTEN, Italy
B.E.S., Italy
Université de Compiégne France
A.S.C.A.B, France
G. Botti Progettazioni Industriali, Italy
F.Cherubini
F. Uccelli
Consorzio Cooperative della Marsica, Italy
Italy
L Antonelli and F Fonzi, Alten, Italy F Fonzi and L Antonelli Alten, Italy
M Ρ Chassin F Cailliez C Esnouf IA Vasalos
B Delmon L. Leonardini G. Salvi
GRADT, France CIRAD, France;
Cemagref, France CPERI, Greece
UCL, Belgium CRITA, Italy COGIS, Italy

157
PIO Study on biological degradation of the F.Fonzi acidic condensates coming from L.Antonelli pyrolysis of biomass
Pi l Study of optimisation of the use of biomass derived from forest for pyrolysis oil. production
P12 Thermal conversion of biomass JKMaund in molten salt media
ALTEN, Italy
C. Erbaggi Società Cooperativa Agricola Forestale "Collelongo", Italy
Aston University, UK
Liquefaction LI Direct liquefaction of wood and
solid agricultural waste L2 Production of synfuels and chemical
feedstocks by direct hydroliquefaction of ligno-cellulosic biomass,
H Meier zu Köcker Technical University Berlin, West Germany
O Faix Institute of Wood Chemistry West Germany

158
A SURVEY OF BIOHASS LIQUEFACTION PROCESSES
R. CAPART, A. ELAMIN, S. AMMAR, M. GELUS Department of Chemical Engineering University of Technology - B.P. 649
F - 60206 - COMPIEGNE
Summary
From the 1970's, the liquefaction of wood or biomass has been extensively studied in order to provide a liquid product easily pumpable and stockable, very like to gasoline or fuel produced from petroleum Industry. In this paper are exposed different methods to obtain this liquid (oil) at a laboratory scale or from process development units working in a continuous mode. The oil from direct liquefaction or pyrolytic processes are often ameliorated by a subsequent catalytical treatment which makes use of either hydrocraking catalyts (CoMo, NiMo, Pd, etc....) under reductant atmosphere or zeolite - type catalysts (HZSM-5) without reductant gas.
1. INTRODUCTION Thermo-chemical conversion of biomass produces always gases, liquids
and solids. How the products share between these three states depends on the process and temperature is the more important parameter.
Pyrolysis is the breakdown of biomass by heat in the absence of air or oxygène. If pyrolysis is run at slow rates and temperatures less than 500°C, then the process yields a maximum of solid residue, the charcoal. In flash pyrolysis as in fast pyrolysis, heating rates are very high. The solid residue is very small and the production of high quality gases is very important, due to thermal craking of pyrolytic oils.
Liquefaction processes are related to direct high pressure hydrotreatment. The aim is to maximize the liquid yield, with a low oxygen content. It is obvious that liquids, because of their energy density, appears to give the only way to use biomass in transportation. A high yield of liquids could be obtained not only by liquefaction, but also by pyrolysis and upgrading of pyrolytic oils.
There is no clear definition of the different processes of thermochemical conversion. Neither the mass balance, nor the operating conditions could bring a clear understanding of the lot of terms used in the field of thermal biomass valorization. That could be a problem for people in charge of reviewing the state of art of liquefaction.
In liquefaction processes, it is not obvious to evaluate the mass balance and the results depends on the analytical methodology. Oil could be the part which is extracted by a solvent as acetone or benzene or

159
methylene chloride and oil yields depend on the solvent, which is often acetone. In this paper, conversion is evaluated as :
weight of wood - weight of solid residue weight of wood
Water is always produced during thermochemical conversion of wood. So, we can roughly consider that liquefaction leads to form fractions :
- gases - oil - water - solid residue
and comparison of yield could be interesting from an economical point of view.
2. DIRECT LIQUEFACTION OF WOOD - PROCESS DEVELOPMENT UNITS IN CONTINUOUS Appell (1) first studied the liquefaction of lignocellulosic
material at the Pittsburg Energy Research Center (PERC). He obtained a heavy oil by reaction of wood at 350°C in the presence of H2 and/or Co in aqueous Na2C03. This preliminary work encouraged the PERC to. pursue development of studies leading to the conversion of wood into liquid in a single step.
A pilot demonstration unit (PDU) has been constructed in the 1970's at Albany (Oregon) (2). This PDU being able to work following two different processes which have been developped by the PERC and the LBL (Lawrence Berkeley Laboratory).
The PERC process consisted of converting dried wood flour in an anthracene oil, between 300 - 370°C in the presence of Na2C03 as catalyst (2 - 8 wt * based on dry wood), under reductant gas, mixing C0/H2 in the proportion 60/40 and at 200 bar of pressure. Anthracene oil was progressively replaced by recyling the oil produced. The original PDU was submitted to many modifications to improve its working capacity and wood treatment. In the best case it has been working during 572 hours with an average flow rate of 8.75 kg/hour of produced oil. However, the wood concentration in the slurry is dramatically low, no more than 8 * by weight. In addition, serious technical problems due to undissolved solid and increase of oil viscosity prevented the Albany PDU to work after 1981.
In order to increase the wood concentration in the slurry as well as its pumpability, the LBL induced a pretreatment of the wood prior its liquefaction. The pretreatment consisted to a mild hydrolysis in diluted H2S0A at 180°C. After milling, an aqueous suspension containing 30 * of undissolved lignocellulosic material is obtained. The liquefaction reaction is performed from this suspension, the pH of which being increased to 8 by adding Naa C03. Contrarily to the PERC configuration process, the LBL process did not require the recycling of produced oil. The experimental difficulties encountered with the Albany pilot plant conducted the LBL to study the liquefaction through a bench scale laboratory unit (BLU) of much smaller capacity. From litterature data, the LBL process operating either in PDU or in BLU mode provides no advantage with regard to the PERC process. The working time and the oil yield are inferior to those of the PERC process obtained in its best configuration.

160
OPERATING CONDITIONS TYPE OF REACTOR REACTOR TEMPERATURE PRESSURE (bar)
WOOD FEED RATE (Kg/h) SLURRY FEED RATE (Kg/h)
Na2C03/Wood REACTANT GAS (Nm/h) H 2 % IN REACTANT GAS SLURRY VEHICLE
GHV (Kcal/Kg) OIL YIELD
(wt % on Wood Basis) VISCOSITY (CPS)
C H 0
PERC (Run 12) PLUG FLOW
330 210 17,4 245 0,1 14,9 39,2
ANTHRACENE OIL
LBL (Run 7) STIRRED 345 210 7,7 71 0,12 4,5 37,7
WATER
Univ. ARIZONA P.F. EXTRUDER
400 210 3,6 54 0 0 0
WATER
CHARACTERISTICS OF CRUDE OIL
8236,2 53,3
135 ANALYS]
79 8,5 12,5
8025,3 25,1
142
8325 27
103 S (ON DRY BASIS)
79,2 7,8 14,4
80 8,4 10
Table 1 DIRECT LIQUEFACTION OF WOOD CONTINUOUS PROCESS DEVELOPMENT UNITS
The major difficulties encountered with the Albany process plant was the pumpability and the transfer of the slurry througout the installation. To overcome these difficulties, some searchers such as Eager (3) and White (4) used a screw-feeder or an extruder-feeder which allow a certain defibration of the woody Material and a good circulation of the slurry. The experimental device designed by Eager and al consisted of an horizontal tubular reactor with an internal screw (Auger) which vehicles water-wood slurry from a pressurized container through the reaction zone. The liquefaction technique was not different from that of LBL (catalyst Na2C03, reductant gas : CO). The wood/water ratio being about 0.4, Eager obtained good yields in produced oil (37 - 42 %) nevertheless with a certain formation of wood char (1 - 15 * ) . Different types of Auger have been tested. They were all subjected to serious erosion or corrosion problems.
White and Wolf, at the University of Arizona have developped an extrusion technique which permits a good feed and the pressurization of the reactor as well as a mechanical pretreatment. The vertical reactor, of plug flow type, was feeded with steam and CO so that H2 was formed in

161
situ by water gas shift reaction catalysed by Na2C03. Various tests up to 52 hours have been performed, covering a range of pressures from 50 to 210 bar and temperatures from 350 to 430°C. Crude oil containing 6 to 10 % of residual oxygen were obtained and nothing was mention about char formation, probably prevented by steam injection in the reactor.
3. DIRECT LIQUEFACTION OF WOOD WITH REDUCTANT GAS AT LABORATORY SCALE A lot of research works has been devoted to the direct wood
liquefaction in order to prove the feasibility of a process and to define the best experimental conditions, i.e temperature, pressure, choice of carrier solvent or catalyst from numerous repetitive experiments conducted in batch autoclaves generally of small capacity.
Early works due to Appell and the Bureau of Mines were exclusively based on the use of very cheap catalysts such as NaaC03. Boocock (5) (6) has particularly studied the liquefaction of wood in aqueous suspension and discussed the effect of various Ni basis catalysts as Ni Raney, Ni salts or oxydes, at temperatures around 350°C and initial pressures of hydrogen in the range 17 - 100 bar. He obtained relatively low yields of oil containing about 10 wt % of residual oxygen. No noticeable difference was observed between the different Ni - basis catalysts upon the oil yield.
Rogers (7) investigated the catalytic effect of different salts of transition metals in the presence of steam and CO. The nature of chemical bonds on the metal are determinant : the chloride, sulfate, acetate and nitrate are not efficient. Only the oxyde and cyanid have a real catalytic effect.
Soyer (8) liquefied wood sawdust in water suspension by using various Fe basis catalysts under pressure of reductant gas H2 or neutral He at 340°C and 40 - 60 bar. Among all the tested catalysts, finely divided iron powder revealed the more efficient, but its activity is decreasing during the reaction. With a sufficient initial quantity of iron powder (14 wt * based on wood), Soyer obtained a total conversion of wood and observed that the nature of gas (H2 or He) has no significant effect on the yield (about 40 %) and the quality of the oil produced.
From systematic experiments of wood liquefaction in organic solvents (anthracene recycled-oil, pyrolysis oil) under hydrogen pressure, Meier and Faix (9) have tested many current catalyts of hydrocraking. Because of the high wood content in the carrier solvent, total wood conversions were rarely reached and wood char was formed. Meier and Faix concluded that only Pd on activated charcoal support and iron powder meet important requirements of the liquefaction process such as complete recovery of the carrier with simultaneously high yields on the net product oil. The use of iron powder is preconized because of its very low cost. About the influence of pressure, a minimum of 100 bar is necessary to recover the whole carrier oil and to obtain low char formation. Gupta and Weiss (10) made an extensive study of the nickel promoted hydrogénation of celluloslc materials. Using both powdered newspaper and pure cellulose as feedstock, they demonstrated the feasibility of converting celluloslc materials into liquid hydrocarbons by reaction with hydrogen at 425°C and 70 atm in a slurry phase with paraffinic oil and in the presence of 0.2 wt * Ni OH catalyst. Oil yield of 45 % was reported.
Delmon (11) and al. have used mainly tetralin as solvent and the sulfided CoMo as catalyst. They have emphasized the role of CSa to maintain the catalytic activity of CoMo during the reaction.
Araya (12) and al. compared the effect of gas, neutral or reductant

162
on the liquefaction in a typical hydrogen donor solvent : the tetralin. The production of lighter organic liquid is favoured under an atmosphere of hydrogen and with CoMo catalyst, particularly if the catalyst is finely ground and dispersed in the reacting mass.
In the case of liquefaction of pure cellulose, Vasilakos and Austgen (13) have obtained good oil yield for that type of catalyst in conjunction with tetralin as solvent. When using 2-propanol, the best catalyst is the Ni-Raney, giving very good yield in net product oil, about 75 wt % (on the basis of cellulose), with an oxygen content comparable to that of the cellulose derived oil in the tetralin/palladium series (24 - 27 * ) .
4. LIQUEFACTION OR SOLVOLYSIS OF WOOD WITHOUT REDUCTANT GAS In the early 1970 s' the Bureau of Mines (14) conducted some
interesting experiments using formic acid or sodium formate in place of carbon monoxyde. This lowered the operating pressure at 250°C to the range 1000 - 1100 PSIG and still gave good oil yields as shown by the table.
Water ml
100 100 100 50
Table 2
Catalyst Type
HCOaNa 1 5
2.5 2.5
: g. Oil yield Amount HC02H
5 60 1 55
2.5 55 2.5 41
Conversion
99.6 99 99.8 90
: LIQUEFACTION OF WOOD WITHOUT REDUCTANT GAS
The products obtained at these mild conditions were pitches instead of oils and revealed to be instable. The Bureau of Mines studied the effect of recycling wood derived oil. A series of run at 250 to 275°C showed that the product become too viscous to use after only 4 cycles.
Without using catalyst and reactant gas, Yan (15) in 1980 liquefied a variety of wood species and paper in aromatic solvents with high boiling point (340 - 484°C). Various experiments at temperature of 320 - 400°C in batch or semi continuous reactor give yields of oil, gas and water around 60 Ss, 17 % and 23 * based on the original wood. Yu (16) selected organic solvent such as phenol and ethylene glycol and used them with an acid catalyst H 2S0 4 at temperatures lower than in most liquefaction processes i.e 180°C and 250°C. Almost complete conversion of wood into oil was obtained nevertheless with high oxygen content (20 - 30 %).
Continuous liquefaction in mineral oil has been performed by Kaufman and Weiss (17) using a finely divided paper slurry with 20 wt * solid content.
Vanasse, Chornet et Overend (18) have tested two different solvents in a continuous process (known as the UDES-S process) ethylene glycol and creosote oil rich in by-phenol compounds. The reactor of plug flow type was feeded by a wood-solvent slurry in the concentration range 14 - 18 S>. The slurry was obtained after a pretreatment which associates an heating

163
up to 240°C with a mechanical defibrating of the suspension through an homogeneizing valve placed in a recirculation loop. The results are clearly different from a solvent to another. With the creosote oil, the yield in final oil is in the range 61 - 51 % and the wood conversion is almost complete. With the ethylene glycol, the conversion is limited and plateau 65 *. Nevertheless the yield in produced oil between 40 - 55 * is satisfactory. The cellulosic fraction is relatively resistant to the solubilization by ethylene glycol and can be isolated by this way. In both cases, the oxygen content of the produced oil (ranging 21 - 29 %) was much higher than that of the PERC process.
A recent study (19) has been reported about the dissolution of a tropical prototype wood (eucalyptus) in various organic solvents. Simple alcohol (C2 to C Ä), ethylene glycol, water, phenol and phenolic compounds : guaiacol and cresol. The experiments have been conducted in batch autoclaves, heated up to 250°C. Discussed in term of a kinetical parameter, termed severity, the results show that an almost total conversion of wood is obtainable with ethylene glycol and like phenol products, only for a high degree of severity. Selective dissolution of hemicellulose, lignin and cellulose are observed only with the polyol and phenolic solvents.
Recently Boocock (20) has given up the use of Ni-Catalysts with reductant gas and performed the liquefaction of wood samples of different sizes by rapid steam injection at temperature between 335 and 355°C. He obtained good oil yields, around 50 wt % based on the original wood.
Some interests exist to process the biomass with solvents in their supercritical state. As explained by Modell (21) in the case of water (T — 374°C), the hydrogen bonds are weaken in critical conditions and the solvent behaves very much like a polar organic solvent which leads to prevent char formation by keeping intermediates highly solvated and well dispersed. At the same time the presence of hydrogen, a product of reforming can aid in stabilization of the intermediates. In supercritical water, pure cellulose is completely degraded, after Modell. Supercritical methanol has been employed by Grandmaison et al. (22) to perform selective extractions on wood material.
5. THE CATALYTIC HYDROREFINING OF WOOD DERIVED OIL Soltes (23) compared the activity of 20 different catalysts for the
upgrading of the oil produced from wood pine pyrolysis. The catalysts were either metal transition oxydes or noble metals such as Pd, Pt, Rh.
The experiments were conducted with decaline or methylcyclohexane as solvent at 400°C and initial pressure of H2 of 66 atm. The formation of water and hydrocarbons are enhanced by noble metal, however the increasing in temperature leads to coke formation, a decreasing in light products and an increasing in aromatic compounds.
Baker and Elliot (24) have successfully converted both high pressure liquefaction oils and pyrolysis oils to a highly aromatic gasoline range fuel. Their studies were conducted in a one liter continuous flow reactor system. From Baker and Elliot, the production of high quality gasoline boiling range liquid requires hydrodesoxygenation without saturing the aromatic rings in the oil and the sulfided CoMo catalyst is the best choice which satisfy this constraints. As shown by table (3) good quality of oil have been obtained by BAKER particularly in the case of LBL process oil leading to a final product with a 0-content near to zero.

164
PROCESSING CONDITIONS
Source of oil Temperature YIELDS
Total oil 1/1 feed oil Aqueous phase 1/1 feed oil H_ consumption 171 feed oil
Carbon conversion to gas wt %
PRODUCTS INSPECTION Oxygen %
H/C ration CS-225°C Vol. *
LIQUEFACTION PROCESS OIL
LBL (N° 7) 398
0.99
0.20
616
14.1
0.0 (14.4) 1.65(1.18) > 87
PERC (n° 12) 397
0.92
0.20
548
9
0.8 (12.5) 1.5 (1.3)
37
PYROLYSIS PROCESS OIL
GEORG TECH. 353
0.43
0.61
457
17
2.3 (52.6) 1.67 (2.2)
72
LAVAL 258-400
0.42
0.57
711
35.5
0.8(44.2) 1.7(1.77)
87
SERI 259-376
0.37.
0.51.
689.
25.
1.3 (43.) 1.7 (1.7)
73.
Table 3 - HYDROREFINING OF CRUDE OIL (BAKER and ELLIOT) CATALYST CoMo/Ala03 - PRESSURE 140 BAR
( ) Value related to feed oil
Various pyrolysis processes can produce oil at a lower cost than high pressure liquefaction but the oils have a lower quality. The pyrolysis oils have the tendency to form coke quite rapidly and to plug the reactor bed. The Pacific Northwest Laboratory (24) has developped a method to upgrade these oils and produce hydrocarbon fuels similar to those obtained from high pressure liquefaction oils by increasing the reaction temperature from a low value (about 280°C).
GEVERT (29) upgraded PERC-process oil in a batch reactor in the presence of sulfided CoMo finely ground and decahydronaphtalin as solvent. All the distillation products of the upgraded oil i.e lights, heavy gas oil and residue have an oxygen content less than 1 wt % as indicated by table 4.
The pyrolysis oil obtained from wastes of olive industry can be valorized by hydrotreatment. DELMON and CHURIN (30) preconized the same methodology as BAKER and ELLIOT i.e. to avoid the coke formation, the bio-oil must be pretreated at low temperature (< 300°C) before to be heated at the reaction temperature, around 380°C. They reported also a good recovery of the solvent (tetralin) by distillation and the possibility to replace tetralin by the distilled fractions from the produced oil.

165
Boiling range
< 60
50 - 350
> 350
0
0.6
0.7
0.8
C
14
14
9.5
H
85.4
85.2
89.7
H/C
2.0
2.0
1.3
Fraction
Lights
Heavy gas oil 250
Residue
Table 4 : ELEMENTAL COMPOSITIONS OF HYDROGENATED PRODUCTS Wt % GIVEN BY GEVERT (29).
6. THE UPGRADING OF WOOD DERIVED OIL BY ZEOLITE CATALYSTS Another approach to valorize the wood derived oils is to convert
these oils by using an appropriate catalyst in absence of reductant gas. The appropriate catalysts can be classical cracking catalysts such as silica/alumina bust the most used are zeolites known as ΖSM-5 and particularly HZSM-5 in pellet form with silica/alumina as binder. This type of catalyst was already known to have a good efficiency for the upgrading of alcohols, phenolic and many other oxygenated compounds. It has a middle size pore structure and is constituted by cristallites of various oxydes, principally Naa0, Ala03, Si0a, TiOa and eventually ZnO or MnO.
The conversion reaction with zeolite is conducted at atmospheric pressure, under neutral atmosphere, at temperature ranging from 400°C to 550°C. The feeding material can be either a condensed oil or the vapor emitted from a pyrolysis process plant whithout condensation. In the 1980's several laboratories have studied a variety of pyrolysis/liquefaction oil over HZSM-5, the feedstock ranging from high pressure oil (31) (32) to low pressure pyrolytic oils and vapor (33) (34) (35) (36) (37). A review of upgrading possibilities for pyrolytic oils from wood was prepared by Kaliaguine (38).
As for the simple model of oxygenated compounds, the upgrading action of the ZSM catalyst consists of an almost complete removal of the oxygen in the feed material under the form of H20, CO and C03. The upgraded product is particularly rich in C, - C 1 0 gasoline fraction, characterised by boiling points ranging from 70 C to 213°C.
The components of the gasoline fraction are essentially aromatic and are of a great economical interest due to the expected high octane number, between 115 and 135, as reported by Diebold and Scahill. The main compounds identified by mass spectrometry are :
Benzene Indene/Indane Toluene Cresol m, ρ, o Xylene Xylenol m, ρ, Ethylbenzene Naphtalene 1, 2, 4 and 1, 2, 3, 4 Méthyl-Benzene
Milne and al. (39) have established a summary of hydrocarbon yields reported from different works based on the use of HZSM-5. The yield in hydrocarbon C
+2 (in wt * of original wood) is ranging from 12 % to 21 *
while the gasoline fraction C s - C 1 0 is ranging from 7.8 to 15 Ss, depending on the method of pyrolysis which gives more or less char. The

166
ratio of aromatics (benzene, toluene, xylene) to olefin (ethylene, propylene, butene) ranges from 3.8 to 8.3.
However, the zeolite catalysts are rapidly deactived by coke formation which is more important for wood tar or derived oil than for simple model compounds. Different solutions are suitable to reduce the coke formation as the injection of steam in the bed of catalyst or the coprocessing of the wood derived oil with methanol.
Till now, experiments have been performed in microreactor at a laboratory scale. Scahill and Diebold (36) have proposed a technoeconomic study about an industrial process-plant which would include :
- a continuous regeneration of the zeolite in a fluidised bed burner - a separation step of the produced olefins.
7. CONCLUSION Numbers of studies have been carried with different thermochemical
processes, i.e. pyrolysis, direct liquefaction and solvolysis show that, liquid-oil can be obtained from a variety of wood materials. The yield of this oil is in the range of 30 - 70 wt % based on the original solid material. Whatever the type of the process, the oil produced is very oxygenated compared with the petroleum products. The pyrolysis have an oxygen content around 30 - 40 %; solvolysis oil in organic solvent 20 - 30 wt * and the oil obtained with direct liquefaction at high pressure have a less oxygen content about 10 wt %.
Oxygen removal and molecular weight reduction are necessary to produce usable hydrocarbon fuels. This can be actived either by catalytic hydrotreatment on catalytic cracking. Catalytic hydrotreatment if often carried in the presence of catalysts based on transition metals such as CoMo, CoMos, MiMo, NiMos supported on alumine on in the presence of Raney-nickel and noble metals like Pd, Rh under hydrogen pressure around 100 bar and at temperature range of 330 - 400°C. Catalytic cracking could be realized with zeolite supported on silice-alumine at high temperature 450 - 550°C, zeolite such as HZSM-5 could eliminate the need of hydrogen in the upgrading process and can effectively deoxygenate biomass oil to produce hydrocarbons.
We can conclude that, for the feasibility of a liquefaction process for producing hydrocarbon fuels, the research should be oriented to :
- regeneration of the solvent which gives maximum oil yield for continuous liquefaction process ;
- catalysts development for cracking and hydrogénation of the higher molecular weight components.
REFERENCES
(1) APPEL H.R. and al. Conversion of Cellulose Wastes to Oil. US Bureau of Mines - Report of Investigations 8013 (1975).
(2) THIGPEN P.L., BERRY W.L. Liquids fuel from wood by continuous operation of the ALBANY biomass liquefaction facility. Energy from Biomass and Wastes VI. Lake buena vit a Florida, Janv. 1982. IGT.
(3) EAGER R.L., MATHEWS J.F., PEPPER J.M. Can J. of Chem. Eng., Vol. 61, p. 189 - 193.
(4) WHITE D.H., WOLF D. Advances in direct biomass liquefaction by the extruder feeder method, pp. 827 - 842. Research in Thermochemical biomass conversion. Phoenix, Arizona, April 1988. Eds Bridgewater and al Elsevier App. Sci., London
(5) B00C0CK D.G.B and al Can. J. Chem. Eng., 57, pp. 98-101 (1979).

167
(6) BOOCOCK D.G.B, MACKAY D, LEE P. Can J. Chem. Eng., 60, pp. 802 - 808 (1982)
(7) ROGERS D.Ζ., TRAN DINH AN. Examination of alternative catalysts for direct biomass liquefaction, pp. 627 - 645, Proc. of the 14 Biomass thermochem. Conv., Contractors meeting. Arlington, Virginia, June 1982.
(8) BESTUE-LABAZUY C , SOYER N., BRUNEAU C , BRAULT A. Can J. Chem. Eng., 63, pp. 634 - 638 (1985)
(9) MEIER D., FAIX 0. Production and analysis of oil obtained by catalytic hydroliquefaction of wood. pp. 804 - 815. Research in Thermochem. Biomass Conversion Phoenix, Arizona, April 1988. Eds Bridgewater and al. Elsevier App. Sci., London.
(10) GUPTA D.V., KRANICH W.L., WEISS A.H. Ind. Eng. Chem. Process Des. Dev. 15, n° 2, pp. 256 - 260, 1976.
(11) GRANGE P., BURTON Α., DEZUTTER D., CHURIN E., PONCELET G. and DELMON B. Catalytic hydroliquefaction of biomass : Influence of Sulphur on product distribution, pp. 1123 - 1127. 4 E.C. Conference on Biomass for Energy and Industry, ORLEANS, May 1987, Eds Grassi and al. Elsevier, App. S c , London.
(12) ARAYA P.E. and al. Can J. Chem. Eng. G4, pp. 775 - 780 (1986). (13) VASILAKOS Ν.P., AUSTGEN D.M. Ind. Eng. Chem. Process Des. Dev., 24,
pp. 304 311 (1985). (14) APPEL H.R., and al Conversion of cellulosae wastes to oil. US Bureau
of Mines. RI 7560 (15) YAN T.Y. "Liquefaction of wood and paper" Hydrocarbon Technology
Environment, Alternate Energy Sources IV, Vol. 6, TNIGAT VEZIROGLU, Ed. Ann. Arbor S e , pp. 79 - 102 (1980).
(16) YU. SM. Solvolysis liquefaction of wood under mild conditions. Ph D. Thesis. University of California Berkeley (1982).
(17) KAUFMAN JA., WEISS AH "Solid waste conversion : cellulose liquefaction" Worcester Polytechnic Institute, National Technical Information service Report n° PB - 239 509 US Dept of Commerce Washington, DC 203 pp. (1975).
(18) VANASSE C , CHORNET E., OVEREND R.P. Can J. Chem. Eng. 66, pp. 112 - 120 (1988).
(19) HEITZ M., VINCENT D., CHORNET E., OVERED R.P., SASTRE H. Solvent effects on liquefaction : solubilization profiles of a tropical prototype wood : Eucalyptus in the presence of simple alcohols, ethylene glycol, water and phenols. Research in Thermochemical Biomass Conversion. Phoenix, Arizona, April 1988, Eds Bridgewater and al.
(20) BOOCOCK D.G.B and al. Aspects of the steam liquefaction of poplar wood in a gravit fed reactor, pp. 843 - 853. Research in Thermochemical Biomass Conversion, Phoenix, Arizona, April 1988, Elsevier App. Sci. Pub.
(21) MODELL M. Gasification and liquefaction of forest products in supercritical water, pp. 95 - 119. Fundamentals of Thermochemical Biomass Conversion. Estes Park, Colorado, Oct. 1982, Elsevier App. Sci. Pub., Oct. 1982, Eds Overend and al.
(22) GRANDMAISON J.L., AHMED Α., KALIAGUINE S. Solid Residues from supercritical extraction of wood. Characterization of their constituents, pp. 139 - 145. Pyrolysis oil from Biomass, Ed J. Soltes, Ed., ACS Symposium series, 376.
(23) SOLTES EJ. and al Catalyst specificities in high pressure hydroprocessing of pyrolysis and gasification tars. pp. 229 - 239.

168
ACS preprints, Vol. 32, n° 2. Production, Analysis and upgrading of oils from biomass, DENVER, Colorado, April 1987.
(24) BAKER E.G., ELLIOT D.C. Catalytic upgrading of biomass pyrolysis oils. pp. 883 - 895. Research in Therraochemical Biomass Conversion. Phoenix, Arizona, April 1988. Eds Bridgewater and al., Elsevier App. Sci., London.
(25) SCOTT D.S., PISKORZ J. Can J. Chem. Eng. 62, p. 404 (1984). (26) KNIGHT J.Α., GORTON C.W. Oil production via entrained flow pyrolysis
of biomass. Presented at the 20th IECEC, Miami Beach, 1985. SAE
Paper n° 859092 (27) DIEBOLD J. SCAHILL J. Amer. Chem. Soc. Div. Fuel Chem. Prepts. 32
(2) p. 21 (1987). (28) LEMIEUX R. and al. Amer. Chem. Soc. Div. Fuel Chem. Prepts. 32 (2),
p. 12 (1987). (29) GEVERT B.G., OTTERSTEDT J.E. Upgrading of liquefied biomass to
transportation fuels by extrusion. Preprint Energy from biomass and wastes X. Washington DC, April 1986.
(30) CHURIN E., MAGGI R., GRANGE P., DELMONB. Characterization and upgrading of a bio-oil produced by pyrolysis of biomass. pp. 896 - 909. Research in Thermochemical Biomass Conversion. Phoenix, Arizona, April 1988, Eds Bridgewater and al. Elsevier App. Sci., London.
(31) MATHEWS J.F. and al. Can J. Chem. Eng. 63, pp. 686 - 689 (1986) (32) CHANTAL P.D., KALIAGUINE S., GRANDMAISON J.L., MAHAYS. Applied
Catalysts 10, pp. 317 - 332 (1984). (33) DIEBOLD J.P. and al Low pressure upgrading of primary pyrolysis from
biomass and wastes, pp. 801 - 830. Energy from biomass and wastes X. Ed. D.L.K. Klass IGT, Elsevier App. Sci. Pub., London, 1987.
(34) EVANS R.J., MILNE T.A. Molecular beam Mass Spectrometric Studies of wood vapor and Model Compounds over HZSM-5 Catalyst pp. 287 - 296. ACS preprints, Vol. 32, n°2, DENVER, Colorado, April 1987
(35) Renaud J., and al. Conversion of vacuum pyrolytic oils froms populus Deltoides over HZSM-5 , pp. 276 - 286. ACS preprints, Vol. 32, n° 2, DENVER, Colorado, April 1987.
(36) DIEBOLD J.P., SCAHILL J.W. Engineering Aspects of upgrading pyrolysis oil using zeolites. Research in Thermochemical Biomass Conversion. Phoenix, Arizona, April 1987. Eds Bridgewater and al. Elsevier App. Sci. Pub, London.
(37) CHEN Ν.Y. and al. Fluidized Bed Upgrading of wood pyrolysis liquids and related compounds, pp. 264 - 275. ACS Preprints. Vol. 32, n° 2, DENVER, Colorado. April 1987.
(38) KALIAGUINE S. Upgrading pyrolytic oils from wood and other biomass an annotated bibliographic review. Report for the Energy Project Office of the NRCC, March, 1981.
(39) MILNE T.A. and al. Molecular Beam Mass Spectrometries Studies of HZSM-5. Activity during wood pyrolysis product conversion, pp. 910 - 926. Research in Thermochemical Biomass Conversion. Phoenix Arizona, April 1987, Eds Bridgewater and al. Elsevier, App. Sci. Pub., London.

169
THE GEORGIA TECH ENTRAINED FLOW PYROLYSIS PROCESS
R. J. Kovac and D. J. O'Neil Energy and Materials Sciences Laboratory
Georgia Tech Research Institute Georgia Institute of Technology
Atlanta, GA 30332, USA
Summary
The Georgia Tech Research Institute has developed an atmospheric flash pyrolysis process for the conversion of biomass to liquid fuels. The successful demonstration of pyrolysis oil production of 60Z (moisture-and ash-free) has been achieved in a large-scale Process Development Unit (1.4 DRY TONNE/DAY). Nearly fifty steady-state trials have confirmed the technological reliability of the process. The liquid biofuel product is a uniform mixture of oxygenated hydrocarbons and modest levels of stably bound water. Char and non-condensable gases are ancillary products. Gross product thermal efficiency of 94Z and a net thermal process energy efficiency of 73Z are achieved. Oil and char recovery exceeds 70Z (moisture- and ash-free). A conceptual manufacturing process and an economic analysis for oil production for a 200 TPD plant is presented. The plant proves to be profitable even at current fuel costs and investment costs (June, 1989 basis). Rate of return on investment is 39.AZ. The payout period is 2.1 years. Total capital investment is $2.28 million. Manufacturing cost per GJ is $2.70. The process is being prepared for further scale-up from the PDU stage to a commercial prototype.
1. OVERVIEW During the 1960's the Georgia Institute of Technology (Georgia Tech)
began the successful design and development of a patented Moving-Bed (Vertical-Bed) Pyrolysis System for the conversion of biomass, municipal wastes and sludge to high-energy density, transportable fuels (pyrolysis oil and char) and a low-energy process gas which was used on-site for drying of feedstocks (1). In the 1970's the Georgia Tech Moving-Bed Pyrolysis Process was licensed to the Tech-Air Corporation (which became a subsidiary of American Can Company) which, through a sub-license to American Carbons Co., has commercialized the technology. This technology was developed by Georgia Tech and Tech-Air in four large-scale pilot plants and a commercial prototype of fifty tonnes per day (TPD) which was operated continuously at a sawmill, around the clock (24 hours per day) over a period of eighteen months before the demonstration was terminated. That research, development and demonstration program, spanning 1968-1978, confirmed the reliability and high efficiency of pyrolysis technology with biomass and municipal wastes, and the economic viability of vertical-bed pyrolysis plants of 50-250 TPD. That

170
Georgia Tech Moving Bed Pyrolysis Process, in commercial form, remains the standard for commercial biomass pyrolysis technology.
The design for the Georgia Tech Entrained Flow Pyrolysis Process originated in 1978 with an internally-sponsored project for the development of an innovative process for the thermochemical conversion of biomass to synthesis gas via a combined pyrolysis-gasification process. Following a contract award from the U.S. Department of Energy in 1980, and after completion of bench-scale studies and the construction of a preliminary conceptual design and preliminary economic studies, which indicated very high potential for an economically viable, stand-alone process for liquid biofuel production, continuing research and development focused on the design and development of a new pyrolysis process. With the revised objectives, the new Georgia Tech Entrained Flow Pyrolysis Process was focused on the maximal production of the liquid biofuel, pyrolytic oil. A 1.5 TPD Process Development Unit (PDU) was designed and installed in 1983. The purpose of the experimental PDU was to investigate the entrained pyrolysis of wood under steady-state conditions. The experimental parameters which have been studied included feed material characteristics, particle size, pyrolysis temperature and residence time. The data was analyzed in parallel and interactively, to develop mathematical models of the kinetic and transport processes occurring in the pyrolysis process. By this methodology the process model was progressively refined and improved process parameter predictions were developed for maximal oil production. In the later stages of program development, after identification of key parametric parameters, oil yields (dry basis) consistently in excess of 552 and achieving 602 were demonstrated in steady-state operation of the PDU. Modeling indicates that 632 oil yields are achievable in the PDU. The prospect of reaching 702 yields was indicated with process design modification in future prototypes. The program consistently demonstrated mass and energy closures of 100 ± 52 in which all elements of the applicable equations were measured. The validity of engineering data is unlike that of most other pyrolysis projects. It is of significance that the excellent closures have been achieved in a large-scale process demonstration unit. The experimental results have demonstrated a gross product thermal efficiency of 942 and an overall process efficiency of 732. Oil and char yields (dry mass) exceed 702. A detailed summary of the development of the Georgia Tech Entrained Flow Pyrolysis Process has recently been presented (2) . This paper will focus on the conceptual design for a manufacturing process using the proprietary Georgia Tech Process for the production of liquid biofuel and co-products. The results of a detailed economic analysis for a 200-dry TPD plant will be presented.
2. PROCESS DESCRIPTION A manufacturing flow diagram which highlights the key pieces of
equipment and systems for commercial scale operation of the Georgia Tech Entrained Flow Pyrolysis Process is given in Figure 1. There are three principal sections to the manufacturing process: feed preparation, pyrolysis, and product collection. A process flow block diagram is presented in Figure 2. The incoming feed and outgoing product streams are identified in relationship to the key process operations of a 200-dry TPD plant. By reference to both figures, the manufacturing process may be described.
Green wood chips at 502 moisture (wet basis) are brought from a receiving and storage area and conveyed to a wood-burning dryer using a front-end loader. The dryer is a fan-circulatory design fitted with a metering bin and a particle collector. The burner is a co-fuel design and uses wood and product gas. (The exhaust gases from the wood dryer burner will be passed through, and air-cooled in, a heat exchanger and used as a conveying carrier

171
ROTARY SEPARATORS
«AT EXCHANGE* ELOWBl AH) AND PYROLYBB
REACTOR
FEED PREPARATION PYROLYSIS PRODUCT COLLECTION
FIGURE 1 MANUFACTURING FLOV DIAGRAM
gas for the reactor feedstock.) The wood chips are dried to less than 10Z moisture (wet basis) and are transferred to a storage bin which supplies feed to the pyrolysis unit on demand and provides surge capacity. The feed is next comminuted in a hammer mill, fitted with a vibratory screening circuit, where grinding reduces the average particle size of the wood and the moisture content to 6 Ζ (wet basis). The dried feedstock is screened to segregate oversized pieces which are recycled to the hammer mill for regrinding. The screened dry wood feedstock is transported to an enclosed feed bin mounted on a lossinweight feeder. The particulate feed is dropped into a conveying "inert" (nonreacting) gas carrier stream which consists of the combustion products exhausted from the dryerburner. This stream is fed into the pyrolysis reactor mixing zone where the wood and conveying gas are mixed with the entraining hot, inert (nonreacting) combustion gases of a woodfired burner. The wood is rapidly transported upwardly through the vertical pyrolysis reactor and is completely pyrolyzed before exiting the reactor. For a 200 TPD plant the entrained flow reactor will consist of a cluster of five vertical tubes. The stream leaving the pyrolyzer consists of newlyformed char, pyrolysis gases, water vapor (water of reaction, feedstockassociated water, and combustion product water), and conveying/entraining combustion gases. The char is removed by cyclone separation and is cooled before being discharged into a sealed conveyor, whence it is fed via a rotary airlock to a char conveyor. The char is conveyed to a storage bin from which it may be retrieved by gravity flow for subsequent shipment. The char bin incorporates a pressurerelief system. The hot pyrolysis gas and vapor stream passes through a multiple organic spray (pyrolysis oil) quencher system to condense vapors. The pyrolysis oil is collected in a series of patented high
efficiency, Georgia Tech proprietary rotary separators (3) as a relatively narrow molecularweight fraction. This oil consists of a total liquid product in total yield of 70.6Z with moisture content of 15Z (60Z dry oil yield).

172
Recycled gas «id
Wood ractmng
storage
Dried ground wood
X Exhaust gai
Dried ground wood Grinding
and
Recycled go
pstøår st na M
Recycled gas
Oried ground
Dried ground wood
Exhaust 9 »
Pyrotyw
^ J u n t a r
Char
®
Char storage
Dried ground wood
Gas to co-sited users
itary 1©
Oil
®
Oil storage
PRODUCT MASS AND ENERGY YELDS FOR A MIXED HARDWOOD FEEDSTOCK
DRYOLYELO Kg dry ol/ KgrraffMd
0.6
TOTAL CHAR
®
MASS Ko/hr
1056
ENERGY OVrr
28.8
TOTAL OL (15%HO)
® MASS Ko/rr
5837
BBK3Y OVtir
111.4
TOTAL GASEOUS FUEL (DLUTED)
© MASS Kp/lr
15003
ENERGY QJ/rr
13.3
FIGURE 2 GT ENTRAINED FLOW PTROLYSIS SYSTEM; 200 dry TPD
N.B. The water is uniformly dispersed and bound in the pyrolysis oil fraction. Maintenance of operating conditions above the dew point of water prevents 4
3. DESIGN BASIS Mass and energy balances were calculated for a 200-dry TPD in support
of the economic analyses. The product mass and energy yields for a 200-dry TPD plant, based on a mixed hardwood feedstock, is included in Figure 2.

173
The product yields and other design conditions for purposes of performance of the economic analyses are given in Table I.
TABLE I DESIGN BASIS - GT ENTRAINED FLOW PYROLYSIS PLANT 200-dry TPD
Location Southeast USA
FEEDSTOCK - (MIXED HARDWOODS) Average Moisture Content, Wet Basis 50Z Ash Content, Moisture Free 0.77Z Higher Heating Value, Moisture Free 19.5 MJ/kg
PRODUCT YIELDS Pyrolytic Oil, Dry Wt/Wt Dry Wood 0.60 Char, Dry Wt/Wt Dry Wood 0.12 Pyrolysis Gas, Dry Wt/Wt Dry Wood 0.10 Water of Reaction, Wt/Wt Dry Wood 0.18
PRODUCT SPECIFICATIONS Pyrolytic Oil
Specific Gravity 1.10 Higher Heating Value, Moisture Free 22.4 MJ/kg Product Oil Moisture Content, Wet Basis 15Z
Char Higher Heating Value, Moisture Free 29.0 MJ/kg
Pyrolysis Gas (Diluted) Higher Heating Value, Dry Basis 0.9 MJ/kg
COOLING AND COMBUSTION AIR Temperature 25°C Relative Humidity 40Z Pressure 1 bar
PYROLYSIS REACTOR CONDITIONS Temperature 550°C Pressure c.l bar Throughput, kg/hr-m2 2200
4. ECONOMIC ANALYSIS For purposes of the economic analysis, the entrained flow pyrolysis
system was divided into six sections according to the operations performed: wood delivery, wood drying, size reduction and storage, entrained pyrolysis in the reactor, char removal, handling and storage, and oil removal, handling and storage.
4.1 Capital Costs Both purchased and installed capital equipment costs were specified
(Table II) . A number of sources of information were used to arrive at capital equipment costs. For some of the equipment the requirements were discussed with manufacturers and vendors and direct quotes were obtained. The most accurate information was used for critical items, that is, the ones which had a major influence on the total cost. A rotary separator had been recently specified and purchased, giving accuracy to that estimate. The installation cost for each item was determined separately rather than by using a uniform

174
rate of the capital equipment cost to determine installation costs. Installation costs were obtained by estimating material and labor requirements directly for each capital item or by using estimated installation material and labor for various classes and types of equipment. The major reference for the economic analysis was the monograph of Peters and Timmerhaus (4).
TABLE II EQUIPMENT COSTS 200-dry TPD
Purchase Installed Cost ($) Cost (S)
WOOD DELIVERY Front-End Loader Conveyor
DRYING Metering Bin Dryer (with Fan) Dryer Particle Collector Dry Wood Storage Bin Dryer-Burner System
SIZE REDUCTION Hammer Mill (with Screening) Feed Storage Bin
PYROLYSIS Pyrolysis Reactor System Feeder system Gas Blower
CHAR REMOVAL Cyclone Separators Storage Bin
OIL COLLECTION Rotary Separators Quench Spray System Hold-Up Tank Oil Storage Tank
TOTAL
The total capital investment for the 250-day TPD plant (60Z dry oil yield) is given in Table III. No land cost is estimated since (a) the pyrolysis process is based on a rural location where purchase cost is minimal, (b) land may be leased at modest cost, (c) land is provided, and/or (d) the plant only occupies A,460 sq.m. (48,000 sq.ft.) on a 1.0 ha (2.5 acre) site which accommodates a thirty-day supply of wood feedstock.
The total direct plant cost amounts to $1.35 million and the total capital investment (TIC) amounts to $2.3 million for the 200-dry TPD pyrolysis plant. The total purchased cost of equipment was $0.88 million and the installed equipment cost, as noted in Table II, amounted to $1.1 million.
100,000 10,000
1,650 119,800 9,700
34,650 15,500
141,650 105,900
41,400 22,000 26,950
43,600 71,100
30,500 12,000 19,000 73.600
$ 879.000
100,000 11,700
2,250 166,600 13,500 38,100 21,600
198,300 116,500
55,700 27,500 35,050
60,600 78,200
42,450 16,650 22,850 91.250
SI.098.800

175
TABLE III TOTAL CAPITAL INVESTMENT 200-dry TPD
DIRECT COST Installed Equipment Cost Building and Services (5Z of Total
Purchased Equipment Cost) Yard Improvements (4Z of Total
Purchased Equipment Cost) Service Facilities (14Z of Total
Purchased Equipment Cost) Land: 1.0 ha (2.5 acres)
TOTAL DIRECT PLANT COST
INDIRECT COST Engineering and Supervision (15Z of
Total Purchased Equipment Cost) 131,900 Construction Expense (10Z of Total
Direct Plant Cost) 130,100
1,
1,
,098 ,
44,
35,
123,
,800
,000
,200
,100 N.C.
, 3 0 1 , ,100
TOTAL INDIRECT PLANT COST 262,000
TOTAL DIRECT AND INDIRECT PLANT COST 1.563.100
CONTRACTOR'S FEE (5Z of Total Direct Plant Cost) 65,100
CONTINGENCY (8Z of Total Direct and Indirect Cost) 125.000
FIXED CAPITAL INVESTMENT (FCH 1.753.200
WORKING CAPITAL (3 months total labor expenses + 2 months, all other operating expenses + 1 month feedstock supply) 389,200
START-UP COSTS (8Z of FCI) 140.300
TOTAL CAPITAL INVESTMENT (TCI) S2.282.700
4.2 Manufacturing (Product) Costs The total manufacturing costs for a 200-dry TPD plant amounted to $3.25
million as described in Table IV. The operating labor cost is based on four men per shift, three-shift operation, and a labor rate of $8.00/hr.
4.3 Sales Revenue The total annual sales revenue for the products of 200-dry TPD (Table
V) wood pyrolysis plant includes sale of the pyrolytic oil, the char, and the sale of product pyrolysis gas to a co-sited commercial or industrial user. The oil-revenue was based on a selling price indexed on an energy-equivalence basis to a barrel of crude oil at $21.00/barrel, i.e. $0.132 per liter ($0.50/U.S. gallon). This is clearly a conservative value for the pyrolysis oil since petroleum heating fuel derivatives of crude oil, for which it will substitute, will have a higher value than the crude oil itself. No. 2 fuel oil was priced in the market at $0.93/U.S. gallon in 1981 (Wall St. Journal, Oct. 12, 1981). Current inventories, based on earlier cheaper supplies of crude oil, have a depressed current market price. At an expected mid-1990

176
TABLE IV TOTAL MANUFACTURING (PRODUCT) COSTS 60Z OIL YIELD
200 MT/d
RAW MATERIAL ($24.25/DT) 2,1*5,400
OPERATING LABOR 268,800
OPERATING SUPERVISION (15Z of Operating Labor) 40,300
UTILITIES 252,300
MAINTENANCE AND REPAIRS (4Z of Fixed Capital Investment 70,100
OPERATING SUPPLIES (15Z of Maintenance and Repairs 10,500
LABORATORY CHARGES (10Z of Operating Labor) 26,900
DIRECT PRODUCTION COST 2.814.300.
FIXED CHARGES (Depreciation, Taxes, Insurance, Rent; 10Z of Total Product Cost) 325,300
PLANT OVERHEAD COSTS (30Z of Operating Labor, Supervision, Clerical and Maintenance + Repairs) 113,800
TOTAL MANUFACTURING (PRODUCT) COST S3.253.400
TABLE V ANNUAL SALES REVENUE 60Z OIL YIELD 200-dry TPD
OIL PRODUCTION (15Z Moisture) liters per annum 44.6 million gals (US) per annum 11.8 million
CHAR PRODUCTION Tonnes per annum 9,325 Tons (short) per annum 10,260
GAS PRODUCTION GJ per annum 94,750 MMBTU per annum 89,800
OIL REVENUE $3,167,200
CHAR REVENUE 820,500
GAS REVENUE 449.000
TOTAL REVENUE S4.436.700

177
1990 stabilization price of $25.00 per barrel of crude oil, No. 2 fuel oil could range from $0.63 $0.75/U.S. gallon (i.e. $0,166 $0.20 per liter). The char revenue was based on a market price of $80/ton for briquette charcoal (the sales price of the TechAir charcoal in 197678). The gas revenue is based on a value of $5.00/MMBTU ($4.74/GJ) which is significantly less than liquid propane which it would displace ($9.25/MMBTU; $8.77/GJ). The total projected sales revenue is expected to be $4.44 million per annum at current market prices.
4.4 Profitability Analyses Three methods were used for the evaluation of profitability: (a) the
(engineering) rate of return (ROR) on investment expressed on an annual percentage basis, (b) payout (payback) period, and (c) discounted cash flow (DCF) rate of return (ROR) on investment for the book life of the plant (taken as ten years, based on depreciation).
TABLE VI ANNUAL RATE OF RETURN ON INVESTMENT 200dry TPD
Years 15 Years 610 SALES REVENUE $4,436,700 $4,436,700 MANUFACTURING COST 3,253,400 3,253,400 GROSS PROFIT 1,183,300 1,183,300 DEPRECIATION 374,300 4,400 TAXABLE INCOME 809,000 1,178,900 INCOME TAX
Case I Tax at 35Z 283,150 412,615 Case II No tax
NET ANNUAL PROFIT Case I (After tax) $ 525,850 $ 766,285 Case II (No tax) $ 809,000 $1,178,900
Cash Flow* χ 100 Engineering Return on Investment = _ , . , „ ·_ιτ
° ,,. , . Total Capital Investment (Years 15) v
= 39.4 Ζ (After Tax) = 51.8 Ζ (No Tax)
* Cash Flow Net Annual Profit + Depreciation
The engineering rate of return on investment analysis is presented in Table VI. Both a "before income tax" and an "after income tax" case were used.

178
The beforetax case could arise in subsidized situations. The after tax case assumed a net income tax rate of 35Z. The plants were depreciated over ten years with an accelerated rate applicable in years 15 per U.S. Internal Revenue Service directives. The law provides for depreciation of plant equipment over a fiveyear period and, for buildings and services, over a ten year period. The results for both cases demonstrate excellent rates of return on investment: 39.42 (after tax), 51.8Z (without tax). The respective net annual profits were $526,000 and $809,000.
The payout period analysis and the discounted cash flowrate of return analysis (DCFROR) are summarized in Table VII. Both "before tax" and "after tax" cases were used as cited above.
The results of payout period analysis indicate that the capital investment can be "paid back" in 1.6 years in the case of no income taxes, and in only 2.1 years for the "after tax" case. The payout period corresponds to the minimum length of time necessary to recover the original capital investment in the form of cash flow to the project based on total income, less all costs excepting depreciation.
TABLE VII PAYOUT PERIOD AND DISCOUNTED CASH FLOWRATE OF RETURN ON INVESTMENT 200dry TPD
PAYOUT PERIOD DCFROR
Case I (Tax at 352) 2.1 years 30.32
Case II (No tax) 1.6 years AA.9Z
„ .. r> · j ,v, · ,. ^ u > Depreciable FCI Payout Period (No interest charge) = —ττ* r : :—
Avg. Profit Avg. Depreciation per year per year
DCFROR i, where i is calculated from
___ _ Annual Cash Flow to Γ 1 1 . . . 1 "I Project After Taxes [_
1+ί d + i )2 d+i)
5J
Γ Working Salvage"] Γ 1 ^Capital Value J " [d+
1i)
5J
5. SUMMARY The Georgia Tech Entrained Pyrolysis Process is a technicallyproven and
costeffective method for the production of liquid biofuels and related bioenergy products. Based on a detailed economic study for a 200dry TPD wood pyrolysis manufacturing process the Georgia Tech process should be scaledup to a commercial prototype scale and be actively commercialized. A summary of the results of the economic analysis is given in Table VIII.

179
TABLE VIII SUMMARY OF ECONOMIC ANALYSIS 200-dry TPD Plant 60Z Oil Yield
TOTAL CAPITAL INVESTMENT
MANUFACTURING (PRODUCT) COST (ANNUAL)
SALES REVENUE (ANNUAL)
AVERAGE ANNUAL RATE OF RETURN
PAYOUT PERIOD
DISCOUNTED CASH FLOW RATE OF RETURN
MANUFACTURING COST, PER GJ
FIXED CAPITAL INVESTMENT, PER GJ
* Before (or no) tax case.
$2.2 million
$3.25 million
$4.44 million
39.4Z (51.8Z)*
2.1 (1.6)* years
30.3Z (44.9Z)*
$2.56
$1.38
ACKNOWLEDGEMENT
This work was supported by the U.S. Department of Energy through the Biomass Thermochemical Conversion Program, managed by the Battelle-Pacific Northwest Laboratories, under subcontract B-C5863-A-Q.
REFERENCES
(1) BOWEN, M.D. et al. (1978). A Vertical-Bed Pyrolysis System. Chapter 6 in Solid Wastes and Residues: Conversion by Advance Thermal Processes (ed. Jones, J.L. and Radding, S.B.), ACS Symposium 76, Americom Chemical Society, Washington, D.C.
(2) O'NEIL, D.J., KOVAC, R.J. and GORTON, C.W. (13-17 February 1989). Liquid Biofuels Production by an Entrained Flow Pyrolysis Process. Paper presented at Energy from Biomass and Wastes XIII. New Orleans, Institute of Gas Technology.
(3) ELSTON, W.E., KOVAC, R.J. and O'NEIL, D.J. (7-9 October 1986). Proceedings of the Fourth Southern Biomass Energy Research Conference. University of Georgia, Athens, Georgia.
(4) PETERS, M.S. and TIMMERHAUS, K.D. (1980). Plant Design and Economics for Chemical Engineers. Third Edition. McGraw-Hill Book Co., New York.

180
PILOT PLANT DEMONSTRATION OF USED TIRES VACUUM PYROLYSIS
CHRISTIAN ROY Univers i té Laval, Dept. of Chemical Engineer ing, Ste-Foy, Québec, GIK 7P4
and JOHN UNSWORTH
Petro-Tire Inc. , Hamilton, Ontario, L8P 1X1
Summary
Tire recycl ing has become a necess i ty because of the huge piles of t i res which r e p r e s e n t a t h r ea t to the environment . By and large , t he r e is about one worn t i r e produced per year and per person in the developed count r ies . However the used t i res r e p r e s e n t a source of e n e r g y and valuable chemical p roduc te . By vacuum thermal decomposition of r u b b e r , it is possible to recover the initial compounds which cons t i tu te a t i re . We have used a s t e p - t o - s t e p approach, from bench scale batch sys tems, to p rocess development uni t and finally pilot plant uni t to experiment and develop vacuum pyrolys is of used t i r es . Vacuum pyrolys is of r u b b e r yielded 55% oil, 25% carbon black, 9% eteel, 5% fiber and 6% gas . The maximum recovery of oil was performed at 415* C below 2 kPa (abe. p r e s s u r e ) . The specific g rav i ty of th is oil was 17.8"API and i t s g ross heat ing value was 43 000 J / g with a total sulfur content of about 0.8%. It was r ich in benzol and other petrochemical components. The carbon black favorably compared with the s t anda rd g r a d e s and will probably find severa l applications as a semi-reinforcement or reinforcement carbon in r u b b e r p a r t s . I t s main limitation comes from the high level of impurit ies (ash) it contains . The heat of pyro lys is for the react ions is low and has been estimated to be a round 706 k J / k g . The process has been tes ted in a 200 k g / h pilot plant which positively demonstrated the possibili ty to continuously feed unde r a vacuo large chunks of r u b b e r . Work is p r o g re s s ing to optimize the r a t e of heat t r ans fe r in the pilot plant reactor . The process feasibility looks promising, with r e t u r n s on the investment of about 50% after t h r ee yea r s of operat ion.
1. INTRODUCTION Tire recycl ing has become a necess i ty because of the huge piles of t i res
which r e p r e s e n t a significant environmental r i sk . Each year 24 million t i res (220 kt) a re disposed of in Canada and about 250 millions (2.3 Mt) in the U.S. Table I r e p r e s e n t s conserva t ive estimates of used t i res for the EEC and o ther count r ies . While some of these t i res a re recapped or ground up for special u se s , most a re simply dumped in ru ra l farm land or in landfill s igh t s . When buried in landfills they eventual ly float to the surface, and if piled the non biodegradable r u b b e r will cause ser ious harm if ignited by l ightning or vandals .
However, the t i r e s r e p r e s e n t a source of e n e r g y and raw chemicals for the product ion of r u b b e r p a r t s . By thermal decomposition of r u b b e r , it is possible to recover to a cer ta in extent the initial ingred ien t s which cons t i tu te a t i re . There have been numerous a t tempts to in t roduce economically viable techniques of t i re pyrolys is . I t is beyond the scope of this paper to descr ibe the var ious t i re pyrolys is v e n t u r e s worldwide and the many

181
adaptat ions of the technology. A detailed review was publ ished in 1983 by Dodds e t al (3) for the US Department of Energy. To our knowledge t he re a re only a few sc r ap t i re commercial pyro lys is p lan ts in the world, which mainly opera te in Japan as indicated in Table II.
Table I . Tota l Ar is ings of Used T i r e s in Di f fe ren t Countr ies Country Tonnage/Year Reference
EEC USA JAPAN CANADA
1 500 000 2 300 000 579 000 220 000
(1) (2) (1) (2)
Table II. Existing Commercial Scrap Tire Pyrolysis Process or Company
Hyben Recycler
Kobe Steel
Onaharaa Smelting and Refining Kleenair
Capacity
Batch
1 t/h
4 t/h
1 t/h
Location
Britain, Japan
Aioi, Japan
Iwaki, Japan
Centrali a, Wash.
Plants Start-up Date
1977
1978
1981
1986 (discontinued?)
Of all these p lan ts , t h ree opera te on a cont inuous feed mode and one is batch. Typical yields from the Kobe plant a re 31% oil, 29% carbon black, 14.5% gas and 10% steel . The oil is sold to a cement kiln company. The p roduc t s do not overall appear to have met ear l ier expectat ions. The economics of the plant was repor ted to be only marginal (1).
The Kleenair p rocess was developed by Conrad Indus t r i e s back in 1986 for the recycl ing of used r u b b e r in gas , oil and carbon black. The plant capacity was 1000 k g / h , similar to the Kobe plant. In both cases , the feedstock consisted of sh redded t i res . Curren t ly the plant operation has ceased.
The dis t inct feature of the Onahama plant is tha t whole t i r e s a re used as feedstock to the reactor . The major saleable p roduc t s a re a heat ing oil (25-30%), carbon black (35-40%) and steel (10%). The carbon black is used in the adjacent copper smelting plant (4). The economics of the p rocess is good, al though it has been repor ted to be less profitable than bu rn ing the t i res at the plant si te.
Here a re some of the conclusions we have reached after vis i t ing these plants :
• The most successful recycl ing p rocesses a re those which use whole t i res as feedstock. Shredding t i res is expensive. It also r e su l t s in materials which are difficult to handle inside the reactor , eventual ly giving r ise to the so-called "ballmilling" problem.
• The quality of the carbon black p roduc t grea t ly determines i ts end -use and as a consequence, i t s value on the market.
• The plant operat ion should be suppor ted by a h igh-qual i ty control lab which should be operated by low-level technicians . In addit ion, the company should maintain a s t rong R and D act ivi ty for p roduc t improvement and discovery of new markets . None of the p lants than we visited afforded such

182
technical suppor t . • The process is cost sensi t ive to the supply of r u b b e r feedstock. The
process will be more profitable if t h e r e is a t ipping fee or management cost at tached to the recept ion of a t i re .
• Attempts should be made to repl icate the plants elsewhere near la rge piles t h rough licensed or f ranchise agreements , in orde r to sha re the r isk and optimize the investment . Marketing of the pyrolys is technology is r equ i red , s t r e s s ing t he advan tages of the process in terms of the env i ronmental regula t ions . The pyrolys is rou te has been lowprofile compared to the other thermochemical t echniques so far , and it is about time tha t a more agress ive market ing approach be launched in orde r to br ing to l ight the dis t inct advan tages of the process from an environmental point of view.
Our purpose is to meet as many of these goals us ing the vacuum p y r o lysis concept . This paper will explain how th is can be done.
2. VACUUM PYROLYSIS PROCESS Vacuum pyro lys i s is an old concept . I t enables t he product ion of large
quant i t ies of pyrolys is oils from an organic subs tance . When a vacuum pump is at tached to the pyrolys is reac tor , the gas and vapor molecules a re immediately removed from the reaction chamber. This minimizes the extent of secondary react ions such as thermal cracking , repolymerization and r econ densat ion reac t ions , gas phase collision, catalytic cracking and reduct ion and oxidation react ions . If t he vapor phase produc t s a r e proper ly quenched, the yield of organic l iquids such as pyrolys is oils is dramatically increased at the expense of solid r e s idues and gases . Besides, the physicochemical prope r t i e s of the end produc t s a r e specific. For ins tance , t he carbon black produced u n d e r a vacuo is more easily dispersed than t he atmospheric p r e s s u r e pyro ly sis black which is an important charac te r i s t i c in reinforcing r u b b e r par t e .
3. BACKGROUND DATA Laboratory t e s t s were performed in a bench scale reac tor to s tudy the
influence of t empera ture and par t ic le size dur ing pyro lys i s . For these t e s t s approximately 1 kg of feedstock was poured into the reac tor vessel . P r e s s u r e was maintained below 3 kPa dur ing the whole run . Experiment was stopped when the final desi red r u b b e r bed t empera ture was reached. A detailed descr ip t ion of the a p p a r a t u s and procedure used will be found elsewhere (5). Regular t i r e s with no steel belt were used for th i s ser ies of r u n s . The t i res had been previously sh redded to 1/4" 1/2" Tyler sieves (6.3512.70 mm). The chemical composition of the r u b b e r feedstock is given in Table III .
Table III. Chemical composition of the used tires
Elemental Composition Proximate Analysis Gross Calorific Value
81.5% C 65.2% Volatile matter 36800 J/g 7.1% H 28.7% Fixed C 3.4% O 6.1% Ash 0.5% Ν (Moisture 0.5%) 1.4% S (6.1% ash)
The yield of oil, carbon black, pyrolyt ic water and gas produced dur ing pyrolys is at va ry ing final t empera tu res a re summarized in Table IV of the

183
paper. The temperature shown is that of the bed of shredded tires. The maximum oil yield was reached at a plateau temperature of about 415*C as appearing in Table IV. At 415"C the yield of oils was 56.6%.
Table IV. Effect of temperature on product y i e l d during vacuum pyrolys is of used t i r e s (wt. %, t i r e as-received b a s i s ) .
Temperature
250 310 335 363 415 500
CC) Oil (%)
7.2 17.7 27.2 48.3 56.6 56.2
Carbon black and
fibers (X)
91.1 79.4 68.8 45.6 36.6 35.5
Gas (%)
Traces 0.3 1.7 2.9 2.2 4.3
Water (%)
1.7 2.6 2.3 3.2 4.6 4.0
The gas phase was mainly composed of Ha, CO, COj and a few HCs. At 415'C, the gas has a gross calorific value of 36800 J g - 1 with an average molecular weight of 28.3. No H2S could be detected in the gas phase. If any, its concentration must be low since a partial mass balance on the sulfur gave 70.4% for the solid residue while 27.4% of the initial S found its way in the oil, leaving 2.2% for losses and gases.
Two runs were also conducted at identical temperature (420 ± 5'C), one with pieces of rubber sieved between 6.35 and 12.70 mm, and the other run with larger chunks of old rubber of 150 to 400 mm. According to Table V there was no significant difference between the two runs on the product yields:
Table V. Influence used tires
Size (mm)
6.4 - 12.7 150 - 400
of at size in product yields during vacuum pyrolysis of 420'C (wt. %, rubber as-received basis)
Oil (%)
56.6 58.4
Solid residue (%) Gas (%)
36.6 2.2 34.8 2.1
Water (%)
4.6 4.7
Similarly vacuum pyrolysis at 450'C of a steel belt (17.6% fiber + steel) tire sample gave 59.0% oil, 33.8% carbon black, 5.2% gas and 2.0% water on an organic rubber basis which was in line with the results shown in Table V.
4. PROCESS DEVELOPMENT UNIT STUDY These preliminary results were further investigated using a vacuum
pyrolysie Process Development Unit. The system operated on a semi-continuous feed mode using shredded (regular) used tire material sieved to 6.4-12.7 mm mesh size. The reactor was a six hearth furnace, 2 m in height and 0.7 m in diameter. It was externally heated by electric elements surrounding the reactor chamber. The maximum throughput reached with this unit during this study was 13 kg/h . A schematic of the P.D.U. is shown in Figure 1 below and a detailed description of this unit will be found e l se where (5).
At a maximum flowrate and reactor temperature of 513 *C, the oil yield

184
was 54%, 37.8% carbon black and fiber, 4.2% gas and 4.0% water. These results were in straight correlation with the previous bench scale data.
An important engineering parameter to be considered when designing a full scale pyrolysis plant is the quantity of energy required for the pyrolysis reactions. This value has been empirically determined using the P.D.U. described in this paper, and the detailed procedure has been published elsewhere (5).
Figure 1. Schematic of the Vacuum Pyrolysis Process Development
Scrubber 1 Scrubber 2
Chen-tank Flore
leter I l v
Ice P1™? trap
Filters
The quantity of heat required to decompose rubber ( Hr) will depend on the operating conditions used and the extent of reaction which has been reached. Under normal carbonization conditions, the reactions are exothermic. Under vacuum, the reactions were found to be slightly endothermal, reaching 706 kJ/kg. We concluded that the secondary reactions (e.g., thermal and catalytic cracking, repolymerisation, recondensation, oxidation and reduction reactions) which occurred under higher pressure conditions, released heat and hence contributed to the overall exothermicity.
5. PILOT PLANT STUDY The experimental system was a further scale up to the pilot plant stage.
The new system was designed to continuously decompose 200 kg/h of steel belt tires below a pressure of 1.3 kPa. Large chunks of rubber were continuously fed across a column of water which connected a tank on the ground and the top of a horizontal reactor which was elevated 14 m in the air (see Photograph I). The reactor was externally fired with gas and a small portion of the pyrolysis oil. The vapors were sucked at the outlets of the reactor and immediately quenched by two scrubbers set in series. The carbon black was recovered at the bottom of another water head which connected the bottom of the reactor and the ground. A sharp separation of the fiber, steel and carbon black was made in the water phase. The process schematic is illustrated in Figure 2 of the paper.
The objective of the pilot plant phase study was to demonstrate the feasibility of the vacuum pyrolysis process using semi-industrial scale equipment under continuous operation during several hours. The reactor configuration and the reliability of the downstream equipment including the scrubbers, gas cleaning system and pumps were tested. The results obtained so far with this unit are the followings:
• Continous feeding of tires under a vacuo was practicable. • Separation of steel, fiber and carbon black at the reactor outlet is
feasible.

185
• The equipment designed for the condensat ion and recovery of the pyrolys is oils well performed.
• The overall thermal efficiency of the process is high, exceeding 80%. • The optimum t empera ture and p r e s s u r e conditions to produce large
yields of oils have been found. • The t h r ee major produc t s , oil, carbon black and steel , a re saleable and
marketable. • No major problem is expected with the quali ty of the gaseous emis
sions.
Photograph I: Vacuum Pyrolysis Pilot Plant in SaintAmable, Quebec
Figure 2. Schematic of the SaintAmable Pilot Plant
8 f
~1 1 2
ψ Heat for
reactor
1
D L ^ »^ "■» a.
Ώ %
l .Feed conveyor 2.Vacuum reactor 3.Cooling screw 4.Discharge screw 5.Crusher 6.Vibratory screen 7.Magnetic separa to r 8.Heavy oil quencher 9.Light oil quencher
10. Decan ter 11.Vacuum pump 12.Flare stack 13.Carbon black
handling system 14.Steel recovery bin 15.Heavy oil s torage 16.Light oil s torage
The main limitation with th is system was the low ra te of heat t r a n s ferred to the reac tor for each square meter of surface area . Actually the reactor operated at less than half of i ts t h r o u g h p u t capaci ty, due to ineffi

186
cient heat exchange in the reactor chamber. Another improvement on which we are working at the present time is a better system for handling the rubber-material inside the reactor, using large rubber chunks as feedstock.
The pilot plant unit was built and tested during fall 1987 in Saint-Amable near Montreal. Unfortunately the project was momentarily stopped during early 1988 because of the bankruptcy of the licensed company (Petrc— Sun Intern. Inc.) due to facts unrelated to this project. With the financial support of both Université Laval and Energy, Mines and Resources Canada (Bioenergy Development Program), the project wae continued at the P.D.U. level during 1988 up to now. The pilot plant phase will be continued during the second half of 1989 under the auspices of the new licensed company, Petro-Tire Inc., Hamilton, Ontario.
6. OIL AND CARBON BLACK ANALYSIS The two main products obtained from the pyrolyeis of tires are carbon
black and oil. Samples of each of these products obtained in the pilot plant unit were tested in our laboratory and by different customers. Some of the analytical results are reported in this paper.
The oil has a gross heating value of about 43000 J g"1. It has a specific gravity of 0.95 (17.8'API) with a pour point of -6 'C. Its initial boiling point is 112'C with a 50% cut at 376"C (simulated distillation). The residual metal content is < 0.1% for Va, 0.1% for Ni and 0.6% for Na. A typical elemental analysis for thie oil gave 87.3% C, 10.5% H, 0.8% S, 1.2% 0 and 0.2% N. It yielded upon distillation 8% (by vol.) of heavy naphtha, 16% kerosene, 24% light gas oil, 35% cat. feed and 17% bunker. The PONA analysis of the fraction boiling below 204'C which constituted 26.8% of the crude oils gave 24.9% paraffins, 43.3% olefins, 6.6% naphthenes and 25.4% aromatics.
This oil can be used as a heating oil and would be classified as a number 4 ASTM bunker oil. It can alternatively be refined as a crude oil by the petroleum industry and it has also several other potential applications which are under development in our laboratories. As an example, Table VI constitutes a partial list of the major compounds which have been detected in vacuum pyrolysis oils. There is no doubt that some of these compounds have a potential value as petrochemical feedstock.
Table VT. Examples of compounds found in vacuum pyrolysis oils Compound Yield
(wt. %, as-received rubber basis)
Limonene-ld 2.26 Toluene 1.05 Xylene (o-,m-,p-) 0.93 Styrene 0.82 Benzene 0.38 Dimethylcyclopentadiene 0.24 Methylpentene 0.23 Dimethylpentane 0.16 Cyclopentanone 0.15
Table VII summarizes some of the main characteristics of the carbon black produced at 550° C and 1.3 kPa. Pyrolysis carbon black was compared

187
with two reinforcement commercial blacks, N234 and N330, which price on the market varied between 960 and 1040 Can $. As indicated in Table VII, carbon derived from vacuum pyrolysis of used tires has an iodine number greater than the standard blacks. The main disadvantage of the recycled carbon is its high inorganic content (ash). Work is progressing in our laboratory to purify the material in order to increase its potential value on the market. It has numerous usages including reinforcement or semireinforcement for bicycle tires, shoes, auto flaps, footwear, conveyor belts, dock fenders. The carbon black acts primarily to strengthen and increase the impact on abrasion resistance of rubber. Other uses also include pigments for ink and reduction for ore refining.
Table VII. Physicochemical properties of used compared with standard blacks
Physicochemical properties
Iodine index (rag/g) pH Volatile material {%) Ash (%) Composition (%)
C H Ν 0 S
7. PROCESS FEASIBILITY
N234
112.7 4.2 4.2 0.6
94.8 0.84 0.77 2.98 0.68
N330
80.5 7.3 3.3 0.3
96.0 0.66 0.60 1.98 0.74
tirederived carbon black
Obtained at 550
after pyrolysis 'C and 1.3 kPa
144.5 7.9 4.4 14.5
79.2 0.76 0.71 2.10 2.80
Based on the background and engineering data reported in this work, a preliminary feasibility study of the process was performed. The assumptions used are summarized in Table VIII below.
Table VIII. Assumptions used for the process feas ib i l i ty study
Product Yield Price
Oil
Carbon black
Steel Fiber/Kevlar Gas
55%
25%
9% 5% 6%
Regular grade: Improved grade: Regular grade: Improved grade:
90 $/t 2 $/t
Used for makeup
18.0 $/bbl 19.4 $/bbl 100 $/t 485 $/t
heat
Prices are in Can. $. All metric uni ts .

Other assumptions Tipping fee/Lean-up fee Tires per ton Rate of inflation
188
1 $/tire 110 5%/year
No credit given for re-usables or re-caps
Table IX shows that the profitability of a 3 tons per hour or 20 000 tons/year plant with a capital investment of 7 M $ ie attractive if a tipping fee can be collected for the recycling of tires.
Table EC. Prof i tabi l i ty of a 20 000 t / y plant for vacuum pyrolysis of used t i r e s
Sales/Revenues
Oil, regular improved
Carbon black, regular improved
Steel Fiber Tipping fees/Clean-up Re-usables Re-caps
Total Sales/Revenues Cost of sales
Gross margin Adm. and Comm. Expenses Depreciation Financial Expenses Taxes
Profits (loss) of the year Undivided profits (deficit) at the commencement Undivided profits (deficit) at the end
8. CONCLUSIONS
Year 1
733 000
250 000
81 000 1 000
1 100 000 NIL NIL
2 165 000 479 000
1 686 000 136 000 675 000 675 000 NIL
200 000
NIL
200 000
Year 2
1 538 000
525 000
170 000 2 000
2 200 000 NIL NIL
4 436 000 1 160 000
3 276 000 192 000 675 000 675 000 NIL
1 734 000
200 000
1 934 000
Year 3
1 615 000
2 425 000 179 000 2 000
2 200 000 NIL NIL
6 421 000 1 329 000
5 092 000 244 000 675 000 675 000 NIL
3 498 000
1 934 000
5 432 000
A process for the recycling of used tires has been developed from bench scale, to process development and pilot plant units. The project which is still under development at Université Laval under contract with Petro-Tire Inc. has great potential. There is a market for the oil, the carbon black

189
and steel which cons t i tu te the t h r e e main pyro lys i s p roduc t s . The process feasibility would just i fy the cons t ruc t ion of a la rge demonstrat ion p lant (10 000 t / y ) which we foresee in the near fu tu re . This p rocess would simultaneously r e p r e s e n t an e legant solution for the disposal of the t i re piles.
REFERENCES (1) Dufton, P.W. The Value and Use of Scrap Tyres . Rapra Technology
Ltd. England. Report dated 26 November 1987.
(2) The Rubber Association of Canada, Mississauga, Ont. 1989.
(3) Dodds, J., W.F. Domenico and D.R. Evans. Scrap Tires: A Resource and Technology Evaluation of Tire Pyrolysis and Other Selected Alternate Technologies. N.T.I.S. Report # EGG-2241. Presented to the U.S. Dept. of Energy. November 1983.
(4) Kono, H. Onahama Smelting and Refining Co., Ltd. Iwaki City, Fukushima. Pr ivate communication. (1987).
(5) Labrecque, B. Etude du t r ans f e r t de chaleur pa r radiat ion thermique dans un r éac t eu r de pyrolyse sous vide des v ieus pneumatiques . M.Sc.A. Thesis. Universi té de Sherbrooke , Sherbrooke , Qué. 1987. (In French) .
ACKNOWLEDGMENT This work has been suppor ted by the National Research Council of
Canada, the Natural Science and Engineer ing Research Council of Canada, Energy, Mines and Resources Canada, Energie et Ressources Québec, Univer sité Laval and Pet ro-Tire Inc.

190
FLUIDIZED BED PYROLYSIS OF SEWAGE SLUDGE U. BELLMANN, A.B. KUMMER, Y. YING, and W. KAMINSKY Institute for Technical and Macromolecular Chemistry
University of Hamburg, FRG
Summary Digested, thermally conditioned and dried sewage sludge has been pyrolyzed in an indirectly heated fluidized bed reactor at temperatures ranging from 620°C to 750°C. A quantity of up to 40 kg of sewage sludge per hour has been converted into pyrolysis gas, oil, product water and carbonaceous residues in a continuously working pilot plant. The pyrolysis gas has a calorific value of approximately 23 MJ/m3. The main constituents of the pyrolysis gas are hydrogen, methane, ethane, ethene and propene as well as carbon monoxide and carbon dioxide. The oil fraction yielded contains up to 30% aromatic compounds. The heavy metals from the sewage sludge become to a large extent enriched into the solid residue on account of the reducing conditions of the pyrolysis process.
1. INTRODUCTION More elaborate and refined purification methods in the
treatment of communal and industrial crude waste water are leading to a continuous increase of the amount of sewage sludge to be produced. The expected annual amount of sludge is estimated to rise up to 320-430 mio m3 in the EC in 1990 (1). At present, the whereabouts of sewage sludge is distributed as follows (2):
- agriculture + decompostion 32% - sanitary landfill 59% - incineration 9%.
The utilization of sewage sludge as fertilizer seems to be restricted because of the harmful components in the sewage sludge, such a heavy metals, polyaromatic hydrocarbons (PAH) and the polychlorinated biphenyls (PCB).
There are certain requirements to be met prior to the deposition of sewage sludge in landfills with the intention of avoiding any kind of emission. In addition, neither the biological potential nor the chemical energy content are sufficiently exploited. In future, deposition of sewage sludge will be made more difficult resulting from a decreasing deposition ca-

191
pacity on the one hand and increasing cost of deposition on the other hand.
Thermal conversion processes such as combustion or pyro-lysis, offer alternative waste disposal concepts. The combustion process yields a considerable amount of sulphur and nitrogen compounds. Expensive precautions as regards flue gas purification have to be taken for the purpose of running combustion plants on an environmentally tolerable scale. In addition to that, problems arising from the increased elution capability of heavy metals out of the ashes from the oxidizing treatment have to be solved.
The pyrolysis of sewage sludge according to the HAMBURG PROCESS (3) seems to be advantageous as to the minimization of emission due to a closed gas cycle in connection with an indirectly heated fluidized bed and the reduced volume of the de-pyrolyzed sewage sludge. 2. PYROLYSIS RESULTS
Figure 1 shows the flow scheme of the fluidized bed pilot plant by means of which the pyrolysis of sewage sludge has been investigated.
Core of the plant is an indirectly heated fluidized bed, 450 mm in diameter. Right at the start of the pyrolysis, the fluidization material consists of quartz sand with particle sizes between 0.3 and 0.7 mm. In the course of the run the sand is gradually replaced by depyrolized sewage sludge. In order to ensure a constant height of the fluidized bed of approximately 650 mm during continuous input of sewage sludge an overflow vessel has been attached to the reactor.
Next to the reactor, a cyclone separates carbon and solid particles from the product gases. The cyclone is followed by three quench coolers where the pyrolysis oils are subsequently quenched. At the end of the third cooler the temperature goes down to 0-5°C. During the last condensation stage the product gas is freed from aerosols and entrained droplets in an electrostatic precipitator.
The gas is then conveyed into five membrane compressors, connected in parallel, and stored in three gas tanks. One portion of the gas is used to fluidize the sand bed, whereas the rest of the gas may be used as fuel for the radiation heating tubes, thus allowing an autothermal operating mode of the plant.
As feed stock for the pyrolysis experiments all kinds of digested or thermal conditioned and dried sewage sludge from different communitities or industries could be used (4). The water content of the sludge has to be 5-10 weight percent.
The pyrolysis experiments were carried out at temperatures of 620°C, 690°C and 750°C. The sewage sludge was acontinuously and directly fed into the reactor from a silo via two crew conveyors. The conveying capacity was 25-40 kg/h and the whole charge of converted sludge amounted to more than 360 kg.
The product fractions obtained consisted of gas, pyrolysis oil, product water, soot and pyrolysis residue. This solid residue contained the formed pyrolysis coke together with the anorganic fraction of the sludge.

Fig. 1. Flow Scheme of the Fluidized Bed Pilot Plant for Sewage Sludge
cryostat —■
cyclone 1u e n c h c
°°l<=r q u e n ch cooler
s i l o η ■tí
lock Π
ϊ Η , Ο ΐ
3, Η,Ο
compressed air ¡
propane
pjrolysis gas/l
overflow vessel
πι
Γ heat exchanger
= 1 I
quench cooler electrostatic precipitator
r-ςπ
Θ ν
l^ '
gasooeter compressor
d e s t i l l i n g columns
~ = H , 0 :H ,O
steam «_,,_. steam
o i l Ί . , ΐ , j . η — ,
Kighbollinj fraction Xylene Toluene Benzene
fractlor fraction fraction
tìafitì n
flare
&Έ

193
Table 1 shows the composition of the organic pyrolysis products obtained at three different reaction temperatures.
Table I Product Fractions of the Pyrolysis of Digested Sewage sludge (% by Mass of Organic Dry Matter
Temperature ("CI 620 690 750 Sludge mass Çkq) 361.2 126.6. 118.7 Organic dry matter (kg) Product fraction (m%) Pyrolysis gas Oil Product water Soot Pyrolysis coke Loss
185.5 62.9 60.0
2 2 . 7 4 0 . 1 1 2 . 6
3 . 7 2 0 . 0
0 . 9
3 0 . 7 3 4 . 3 1 0 . 8
3 . 1 1 9 . 2
1 . 9
4 0 . 8 2 1 . 1
8 . 0 4 . 9
2 2 . 3 2 . 9
100 100 100
The main contents of the pyrolysis gas are carbon monoxide, carbon dioxide, hydrogen as well as C1-C4 hydrocarbons. Small amounts of carbonoxysulfide and hydrogensulfide are likely to be found. The calorific value of the pyrolysis gas amounts to 23 MJ/m·*, that means up to about 70% of the calorific value of natural gas.
The gaseous portion of the pyrolysis products increases with rising temperature from 620°C to 750°C from 22.7% to 40.8%.
The pyrolysis oil contains hydrocarbons and, in addition to that, some amounts of nitriles, phenols and heterocyclic compounds. With rising reaction temperature the oil yield decreases from 40.1% at 620°C to 21.1% at 750°C. The major portion of the pyrolysis oils is still tar after all, especially at low temperature down to 620°C amounting to 75.5%. This tar consists of substances which cannot be separated by destillation. The increase of pyrolysis temperature leads to a rising percentage of aromatic components due to progressing defunct-ionalization.
Investigations of the pyrolysis residues, composed of the anorganic residue and the pyrolysis coke, show that the heavy metals originated from the sewage sludge are enriched in this fraction. This enrichment holds quantitatively true in the case of all relevant heavy metals except the volatile elements mercury and cadmium which are condenced at the carbon black in the eyeIon.
Elution experiments with water resulted in the finding that heavy metals are evidently stronger incorporated in the matrix of pyrolysis residue than in the incineration ash, produced at 1200°C, or in the sewage sludge itself. Table II shows that chromium, for instance, is more than 250-fold less liable to be eluted from pyrolysis residue than from incineration ash. Moreover, chromium is dissolved from ash essentially in the oxidation state VI as compared to chromium II from pyrolysis residues.

Element ^g/kg): Cadmium Chromium Copper Nickel Lead
8 100 8150 3860 350
14 7750 100 125 55
194
Table II Elutions According to DIN 38.414 T4 Sample Sewage Sludge Incineration Pyrolysis Re-
Ash (1200°Ci sidue (750°C) < 3 30 50 25 50
The investigations have shown that sewage sludge can be pyrolyzed according to the HAMBURG-PROCESS. The gas/oil-ratio is adjustable in a large extent by means of varying the reaction temperature. The use of the aromatic oils for petrochemical application seems to be promising
The reduction of volume and the strong incorporation of the heavy metals in the matrix of the pyrolysis residue cases the problem of sludge decomposition. This shows that the pyrolysis of sewage sludge can be looked upon as environmentally tolerable alternative to the conventional sewage sludge utilization.
This research is sponsored by the BMFT, Federal Republic of Germany. 3. REFERENCES (1) L'HERMITE, P., OTT, H. (1982). Recycling International,
Freitag Verlag Berlin, 342 (2) BLICKWEDEL, P.T., SCHENKEL, W. (1986). Korrespondenz Ab
wasser 33, 680 (3) KAMINSKY, W. (1985).J.Anal. & Appi. Pyrolysis 8, 439 (4) KAMINSKY, W., KUMMER, A.B., BELLMANN, U. (1986). Phosphat
entfernung und Klärschlammnutzung. Hoechst Symposium, Frankfurt

195
RAPPORTEURS REPORT ON SESSION III PYROLYSIS GASIFICATION AND LIQUEFACTION TECHNOLOGIES
A V Bridgwater Energy Research Group
Chemical Engineering and Applied Chemistry Department Aston University Aston Triangle
Birmingham B4 7ET UK
INTRODUCTION
This overview provides what were seen to be the significant points and conclusions from the papers presented in this session, some recommendations and à report on the questions and discussion. It is hoped that the views of the authors are fairly represented, and further details are contained in the individual contributions.
BIOMASS GASIFICATION IN DEVELOPING COUNTRIES Dr M S Mendis
A summary of the World Bank monitoring programme has shown that biomass gasification for heat applications has generally been successful, but for power applications where gas quality is of prime importance, much less success has been realised. Most projects have failed due to unacceptable fuels, untested equipment, mismatch of system to application, poor training, lack of incentives and poor economics. Proven fuels are charcoal, wood, rice husks and coconut shells.
Gasifiers cannot economically compete at present with diesel due to high capital costs. Gasifier capital costs for charcoal are $500/kW, and for wood, $750/kW. Total system costs for power generation for charcoal fuel are $1250/kW, and for wood $1600/kW. A major breakthrough has recently been achieved at AIT in Bangkok, Thailand with a ferrocement gasifier which costs about $50/kW compared to $500 for a fabricated metal gasifier.
The economic attractiveness of biomass gasification systems for fuel and power is very sensitive to fuel prices. The most interesting market is in the 10-300kW size range, operating at more than 60% utilisation. Major inhibitions to development are a lack of trained personnel, poor equipment maintenance, poor incentives for implementation and environmental factors.
Recommendations 1 Continue support for monitoring to obtain better field data. 2 Learn from experiences to date and provide more technical and social
support for any new initiatives.

196
BIOMASS GASIFICATION & PYROLYSIS IN EUROPE A A C M Beenackers and A V Bridgwater
Gasification Low heating value gas production Systems are commercially available, but generally not economic. Applications include process heat, for example drying, and power from engines where considerable experience has been obtained. An interesting system size for Europe is around 1MWe. Turbines offer considerable potential but ideally require pressurised gasification and a particularly clean gas both of which currently present problems.
Medium heating value gas production Considerable experience was gained in the EC sponsored Methanol from Wood pilot programme. The Framatome pressurised fluid bed oxygen gasifier at Clamecy is planned to be restarted which will provide more data. Genedrally the technology is fairly well developed but uneconomic.
Catalytic gasification Good progress has been made in Spain and France on catalytic gasification and in Sweden on secondary processing for gas quality improvement.
Recommendations 1 Obtain more field experience and data from operational gasifiers, 2 Initiate work on gasifier driven turbine power generation, 3 Continue R&D on product gas quality improvement.
Pvrolvsis Pyrolysis has been carried out in diverse processes to produce a wide range of products including liquids, slurries, gases and solids. The technologies are less developed, but there is more potential for production of liquid fuels as either fuel oil substitutes, or by upgrading as synthetic hydrocarbons. More experience is needed at pilot plant scale in Europe for pyrolysis processes and upgrading technologies. Utilisation of the variety of liquid fuels that can be derived from pyrolysis requires assessment and testing.
Recommendations 1 Improve the performance of pyrolysis processes, 2 Build pilot plant scale pyrolysis processes for liquids production at high
efficiency, 3 Continue liquids upgrading, particularly through zeolite synthesis of
hydrocarbons, 4 Provide wider experienceof utilisation of all pyrolysis products.
BIOMASS LIQUEFACTION PROCESSES Prof M Gelus and Dr R Capart
Liquefaction and the related processes of solvolysis, hydrolysis and supercritical extraction are the least developed of the thermochemical conversion technologies. The processes are generally low temperature, high pressure processes which can produce chemicals as well as higher quality oil liquids with a lower oxygen content than pyrolysis oils.

197
Recommendations
As this is the least well developed conversion process, research should be continued into as many variations as possible to more fully evaluate the technical and economic opportunities for both chemicals and fuels production.
THE GEORGIA TECH FLASH PYROLYSIS PROCESS Dr D O'Neill
The entrained flow flash pyrolysis process has been subjected to continued development and 60% weight yield of liquids (dry basis) has now been achieved. A robust computer model has been derived and developed which predicts that yields of more than 70% oil by weight could be achieved in the optimum circumstances.
A 200 t/d commercial plant has been designed and assessed which gives an oil yield of 60% wt at an energy efficiency of 73%. No waste water would be produced by maintaining the vapour temperature above the dew point of water. The 200 t/d plant has a total capital cost of $2.28 million and, with the product oil valued the same as crude oil on a heating value basis, gives attractive profitabilities even in the current economic environment with a payback time of around 2 years and a discounted cash flow rate of return of around 30%.
Development will continue on the piolt plant and opportunities for demonstration will be sought.
VACUUM PYROLYSIS OF SCRAP TYRES Prof C Roy
A novel tyre vacuum pyrolysis proces has been developed based on the application of a biomass conversion technology to alternative feed, and a 250 kg/h pilot plant has been built and operated This provides a more commercially attractive opportunity than a biomass based process in the short term and enables R&D activities to be maintained and provides valuable operating experience. The products are oil, charcoal and carbon black, gas and steel. A 60 t/d (20 000 t/y) plant has an estimated capital cost of CAN$ 7 million, and has been shown to be profitable under current conditions. Development will continue and further opportunities for this technology will be sought.
FLUID BED PYROLYSIS OF SEWAGE SLUDGE Dr W Kaminsky
Fluidised bed pyrolysis of waste materials has been researched since 1970, including earlier work on rubber and plastics which resulted in a demonstration plant of 1 t/h operating in Southern Germany. This has been recently shut down due to poor economics. Recent work on sewage sludge has given oil yields up to 40% by weight at lower temperatures of around 650°C, which reduce as the temperature of pyrolysis increases, while the gas yield increases. Considerable data has been collated but commercial feasibility has not yet been assessed.

198
QUESTIONS and DISCUSSION
QUESTION ANSWER or COMMENT
How does gasification compare with pyrolysis in terms of costs and benefits?
What hazards are involved with pyrolysis oil?
How is water managed in the GTRI process?
What chemicals can be produced from carbonisation plants?
How does atmospheric tire pyrolysis. compare with vacuum pyrolysis?
What are the power costs in tire shredding?
What happens to the sulphur in the products from tire pyrolysis?
How are products from sludge processing denitrified?
What is the cost of the sludge conversion process?
What is the conversion efficiency and economics of sewage sludge pyrolysis?
Gasification gives complete conversion to a single product - gas - which is difficult to transport and can only be readily used locally. Pyrolysis gives multiple products, some which are liquid and can be transported and used remotely. Capital and operating costs are similar, and the only difference, therefore, lies in the product worths.
Pyrolysis oils are no worse than coal tar or crude oil.
Water would be maintained in the vapour phase in a commercial plant. This has been achieved in a fixed bed operation and is planned for the entrained flow process.
Lambiotte operating but with little interest. Extensive recoveryis practised in Brazil.
Vacuum increases the oil yield, and improves the carbon black activity.
As whole tires or 1/4 tires are processed, power costs are very low.
Sulphur is fixed by CaO & ZnO present in the tires
This is not a problem.
A 20 000 t/y dry solids process would cost $2million+ buildings + special costs
The process is not economic without a disposal credit for the sewage sludge.

SESSION IV
FYROLYSIS CASE STUDIES


201
SUGARS FROM CELLULOSICS BY THE WATERLOO FAST PYROLYSIS PROCESS
D.S. Scott, J. Piskorz, D. Radlein and S. Czernik Department of Chemical Engineering, University of Waterloo
Waterloo, Ontario, Canada
Sunarna T-y
It has been found that fast pyrolysis in a fluidized bed of sand at atmospheric pressure can convert cellulose into anhydrosugars and other sugars if certain characteristics are present in the feed. Normally some pretreatment will be required to condition the feed so that high yields of sugars can be obtained. For example, for wood, treatment with 5% sulphuric acid at 100°C will give a product which on pyrolysis will result in a sugar yield corresponding to about an 80% conversion of the cellulose to glucose. Wood pretreated in this way and then pyrolyzed also yields a pentose solution, and an aromatic fraction derived from lignin which has numerous possible uses. The sugar produced by fast pyrolysis would be in the form of a syrup with a concentration of 150 to 200 grams/liter. Results are also given for pyrolysis of wood pretreated by steam explosion, and for some agricultural wastes such as wheat chaff.
1. INTRODUCTION Over the last several years, the Waterloo Fast Pyrolysis Process
(WFPP) has been developed to maximize yields of liquids by the rapid thermal decomposition of lignocellulosic biomass. The process operates at atmospheric pressure and the reaction is carried out in a fluidized bed of sand as a heat transfer medium. Optimal conditions for woody biomass are 450 C to 550 C and about 0.5 seconds gas residence time. The nature of the fluidizing gas has little influence on yields.
Liquid yields from wood at optimal conditions are 70% to 80% of the dry feed, with the organic liquid yields being 60% to 65% of the dry wood fed. A description of the process and the yields obtained with various types of biomass has been published previously by the authors (1)(2). Extensive identification and quantification of many of the individual compounds present in these pyrolytic oils has also been reported (3)(4)(5).
It has been known for many years, as described in the extensive literature on the subject, that slow heating of cellulose under vacuum can lead to significant yields of anhydrosugars, principally levoglucosan. However, pyrolysis of wood, whether slow or rapid, normally gives very low yields of levoglucosan. Instead, lower molecular weight oxygenated compounds are the major products, and, as pointed out by the authors in a previous publication [6], if fast pyrolysis is employed, then hydroxyacet-aldehyde will be one of the principal products. In that publication, a mechanism was also proposed to account for the formation of hydroxyacetal-dehyde, in which it was assumed that levoglucosan was a precursor, and

202
that lower molecular weight products were formed in successive sequential decomposition steps. However, more recent work has led us to believe that levoglucosan and hydroxyacetaldehyde are produced in parallel reactions, a mechanism which has also been proposed recently by Richards (7).
Shafizadeh and Stevenson (8) suggested that the yield of levoglucosan from wood could be increased by pretreatment of the wood to remove hemicelluloses prior to vacuum pyrolysis. Radlein et al. (9) in 1987 reported the results of preliminary experiments which showed that a mild acid hydrolysis of wood followed by fast pyrolysis at atmospheric pressure completely changed the product spectrum, and, in particular, greatly increased the yields of levoglucosan and other sugars. Additional experimental results are given here of investigations of the effects of various pretreatments of wood on the yields of anhydrosugars obtained in the WFPP.
2. EXPERIMENTAL Samples tested included poplar woods, both untreated as well as sub
jected to a variety of pretreatments, and a pretreated pine. The St. Lawrence poplar was prehydrolysed with 0.5% HCl at 165°C for
6 minutes in a plugflow reactor a procedure which led to substantial hemicellulose removal. Wayman poplar was whole wood pretreated with 2% SO2 at 200°C for 45 seconds in a Wenger extruder. Biohol pine was autoclaved with 3% S02 at 110°C for 2 hours. Hemicelluloses were partially removed by this treatment. Stake poplar was steamexploded wood after digestion at 322 psi for 2 minutes.
The acidhydrolysed samples were prepared by boiling the ground wood meal in 5% H2SO4 for 2 or 6 hours as indicated in the tables, except in the case of the Stake wood which was treated with cold (21°C) 5% HOSOA for 21 hours. Following the acid treatment the samples were washed with deionized water till acidfree. "Deionized" wood was prepared by percolating 10 cm g of cold 0.1% HJSOA through a column of clean wood meal at a rate of about 2 cm hr (g wood) , followed by washing with distilled deionized water at a similar rate until the wash was acidfree.
Analysis of acid hydrolysate and of the water extract of the pyrolysis oils was done by HPLC (Aminex ΗΡΧ87Η column at 65°C, eluent 0.07N H3P04, eluent flow rate 0.32 χ 2.25 ml/min).
Except where otherwise noted, pyrolysis of raw or treated wood or cellulose was carried out at standardized conditions near optimal for the Waterloo Fast Pyrolysis Process for all runs, that is at 500°C, 0.46 to 0.50 seconds apparent gas residence time, nitrogen atmosphere, employing the bench scale fluidized bed unit which has been described in detail elsewhere (1).
Ash analysis was carried out by slow controlled combustion of biomass in a muffle furnace at 650°C to prevent loss of volatile inorganic salts. It was estimated that this method gave ash values accurate to 0.01 weight percent.
Cellulose content was determined by gravimetry of the residue remaining after hemicelluloses and lignin were dissolved in a mixture of acetic and nitric acids according to the procedure of Updegraff (10).
A sample of wheat chaff, both untreated and acid pretreated in the same manner as the wood samples, was also pyrolyzed. Pyrolysis conditions and analytical methods were the same as for other samples.
3. RESULTS WOOD The "commercial" feedstocks were pyrolysed without further pretreat
ment with the results shown in Table 1. Results for untreated clean IEA poplar are included for comparison. Pyrolytic lignin is the water

203
insoluble portion of the tar (5). Only the (anhydro) sugars and hydroxy-acetaldehyde from among the water-soluble components are listed. Total liquid yields (water + organics) for the pretreated materials range from -61% (Biohol) to 81% (St. Lawrence) compared to 78% for the un-treated wood. Sugar yields were in all cases relatively low. In the best case (Wayman) 35.5% of the cellulose was converted to sugars.
Some of these materials were then subjected to hydrolysis with HoSO^ as described in the experimental section. This procedure causes extensive hemicellulose removal resulting in an enhanced cellulose content. Ash levels are also reduced. The results of pyrolysis of the hydrolysed substrates are listed in Table 2. In all cases there is a sharp increase to >66% conversion of cellulose to sugars. It should be pointed out that the "char" in these cases is not solely primary but includes a fused material accumulated at the reactor outlet and having the appearance of a carbonized sugar.
An attempt was made to identify the important variables controlling sugar yields. Clean IEA poplar was hydrolysed with f^SO^ in the manner already described and pyrolysed at different temperatures and feed rates, with varying water content and, in one case, with an additive (NHo). The results are summarized in Table 3 which includes a detailed breakdown of the compositions of the gas and the water-soluble organics. In some cases a distinction is made between the primary char and fused materials ("coke"). In all cases primary char yields are nearly zero. Liquid yields are uniformly high and gas yields correspondingly low as would be expected from the pyrolysis of similarly treated substrates (cf. Table 2). In the case of Run S#l, following hydrolysis the lignocellulose was steeped in dilute NHj for one hour, washed till ammonia free and dried. Only in this case is the sugar yield significantly suppressed. Otherwise no clear trends emerge of the effects of hydrolysis time, moisture content, pyrolysis temperature, particle size or feed rate.
We have also made a preliminary investigation of the feasibility of obtaining high sugar yields from wood without preliminary hydrolysis of hemicelluloses. For this purpose "de-ionized" IEA poplar was prepared in the manner already described. In some cases 0.1 wt% of (NH^^SO^ or H2SO4 was added to the de-ionized wood by dissolving in a sufficient amount of water to saturate the wood then drying at 105 C for one hour. The results of these tests are shown in Table 4. Sugar yields are good with a presumably catalytic enhancement by small amounts of (NH^^SO^ or H2SO4. Though less than those from lignocellulose (i.e. acid hydrolysed wood) they are a great deal better than those from untreated wood. To our knowledge they are the highest ever reported from whole wood.
Fermentation tests were carried out by an independent consultant on the liquid products produced by pyrolysis of acid treated poplar in a pilot plant scale reactor (3 kg/hr feed). The anhydrosugars as produced can be fermented only slowly, if at all, by the common ethanol producing yeast strains. After hydrolysis of these anhydrosugars to glucose, however, fermentation proceeded to completion at a normal rate with common bakers yeast, that is, in about 24 hours. If the hydrolyzed sirup of hexoses was treated with activated carbon before fermentation, quantitative conversion to ethanol occurred in 1.5 hours.
4. RESULTS - WHEAT CHAFF Wheat chaff was investigated as a typical agricultural waste avail
able in large quantities which might be a source of fermentable sugars. It is a very different type of biomass from wood, having a lower cellulose content (22.5%) and very substantial amounts of minerals, mainly silica,

204
which are not removed by acid pretreatment. lhe chaff was given two different acid pretreatments, 5% H^SO^ for 20 hours at 20°C, and 5% f^SO^ at 100°C for 2 hours. A summary of these pretreatment results is given in Table 5.
Results of the pyrolysis tests at 500°C for the untreated and the two pretreated wheat chaff samples are given in Table 6. Unlike wood, very substantial amounts of a solid product were collected, which was in a great part probably due to the high ash content of wheat chaff. However, even if calculated on an ash free basis the amount of char product obtained from acid prehydrolyzed wheat chaff was almost twice as high as that from pretreated wood.
The composition of the liquid product obtained from pyrolysis of the untreated wheat chaff was similar to that from wood. Similarly, the influence of acid pretreatment on the change of concentrations of main components of the pyrolytic tar was much the same, that is, a very significant increase was observed in the yield of sugars, mainly levoglucosan, and a drastic drop in the yield of monomer fragmentation products such as hydroxyacetaldehyde and acetol. The pretreatment of wheat chaff with sulphuric acid at room temperature was more efficient in the enhancement of the yield of pyrolytic sugars than was the hot acid pretreatment, in contrast to the behaviour observed for raw poplar wood, but similar to that of steam exploded wood. The conversion of cellulose into anhydro-sugars reached a value of about 71% (glucose equivalent), which is a little less than that for prehydrolyzed wood.
In view of these preliminary and non-optimized results, the processes for the pretreatment and pyrolysis of wheat chaff appear to merit further investigation.
5. DISCUSSION The factors which determine the yield of levoglucosan from cellulose
pyrolysis have been extensively studied. Apart from the hydrodynamic and temperature regime, various intrinsic factors like cellulose morphology and degree of polymerisation have been cited. Definitive evidence has been hard to come by but one of the most clearly established effects is the role of certain types of cations, especially alkaline and alkaline earth, in even trace amounts, in catalysing fragmentation and charring reactions and suppressing levoglucosan yields. Most of the published studies of cellulose pyrolysis relate to slow, low temperature vacuum conditions. The results from de-ionized wood indicate that the deleterious effects of cations persist in the case of fast high temperature pyrolysis of whole wood (cf. Runs A-2 and S#3 in Tables 1 and 4 respectively).
This cation effect appears to be one of the principal reasons why the commercial pretreated wood samples gave only poor to modest yields of sugars. It will be noticed from Table 2 that acid hydrolysis of these samples caused substantial reductions in ash content. (Most of the remaining ash is likely to be inert material like silica.) In fact the data suggest that a mild cold de-ionization of these samples is likely to cause substantial enhancement of sugar yield. The best results in Table 1 are for the SOn treated materials (Wayman poplar and Biohol pine). It is possible that this treatment causes other changes which are conducive to sugar formation. On the other hand the Stake wood gave excellent results after a cold acid wash.
Acid hydrolysis under the conditions used also causes hemicellulose removal. Our data (Table 4) confirm an earlier observation by Shafizadeh and Stevenson [8] that, provided soluble ionic substances are removed, the presence of hemicellulose in wood is not, per se, detrimental to

205
levoglucosan production. (Cations in fact would be expected to associate with carboxylic groups in hemicellulose.) The best results with de-ionized wood are not quite ás good as those with lignocellulose but no definite conclusion can be drawn as the degree of de-ionization has not been optimized.
The other principal question addressed by our data concerns the precise role of acids. Apart from cation and/or hemicellulose removal, the data in Table 4 clearly show what appears to be a positive catalytic effect. Such an effect has been previously postulated by Shafizadeh [8]. (NH4)2S04 decomposes at 235°C to free H 2S0 4 (b.p. 338°C and volatile NH3 gas which probably readily escapes. By this strategy the timing of the release of acid is controlled and the problems of drying samples containing HOSOA are avoided. (When the substrate of Run S#7 was dried some surface charring was clearly visible --a fact which might be contributory to the lower sugar yields.) There remains a great deal of scope for optimizing the chemical nature, the quantity and the manner of incorporating acid into wood or lignocellulose.
One final point concerns the completeness of the characterisation of the water-soluble portion of the organic liquid. We are able to identify by HPLC an average of only 73% for the wood samples in Table 4 compared with 85% to 95% for the lignocelluloses in Table 2. GCMS showed the presence of significant quantities of two unidentified components in the wood tars which were absent from the lignocellulose, suggesting a hemicellulose origin. Preliminary work indicates they are anhydro-xylan substances but positive identification has not yet been established.
The preliminary tests carried out on wheat chaff indicate that the cellulose content of a wide range of biomass materials could be converted to fermentable sugars in good yield by pyrolysis after an appropriate pretreatment. The wheat chaff results, together with other work done in our laboratory (unpublished) show that of the cations naturally present only the alkali and alkaline earth cations appear to have a specific catalytic effect in promoting ring fragmentation rather than depolymerization of the cellulose.
We summarize our principal conclusions as follows (a) It is possible to obtain good yields of anhydro-sugars from wood and
agricultural lignocelluloses by fast high temperature pyrolysis. (b) The highest yields were obtained when the feedstock was pretreated in
a suitable way. Acid hydrolysis or acid washing proved to be very effective leading to over 70% conversion of cellulose to sugars in the most favourable cases.
(c) The acid appears to play at least two critical roles; to remove harmful cations and to catalyse anhydrosugar release.
(d) However, much more fundamental work remains to be done to gain a full understanding of the effects of both pretreatraent and pyrolysis variables on sugar yields.
ACKNOWLEDGEMENTS The authors would like to acknowledge the financial support for a
portion of this work by the Renewable Energy Division of Energy, Mines and Resources Canada, and by the Natural Sciences and Engineering Research Council of Canada.
The valuable assistance of Peter Majerski as operator of our pyrolysis pilot plant is also acknowledged with pleasure.

206
REFERENCES
(1) Scott, D.S. and Piskorz, J., "The Flash Pyrolysis of Poplar Wood", Can. J. Chem. Eng., 60, 666674 (1982).
(2) Scott, D.S., Piskorz, J. and Radlein, D. , "Liquid Products from the Fast Pyrolysis of Wood and Cellulose", Ind. Eng. Chem. Proc. Des Devei., 24, 581588 (1985).
(3) Piskorz, J. and Scott, D.S., Symp. on Production, Analysis and Upgrading of Pyrolysis Oils from Biomass, ACS Meeting, Denver, April 510 (1987).
(4) Radlein, D. , Grinshpun, Α., Piskorz, J. and Scott, D.S., "On the presence of anhydrooligosaccharides in the sirups from the fast pyrolysis of cellulose", J. Anal. Appi. Pyrolysis, 12, 3949 (1987).
(5) Radlein, D. Piskorz, J. and Scott, D.S., "Lignin derived oils from the fast pyrolysis of biomass", J. Anal. Appi. Pyrolysis, 12, 5159 (1987).
(6) Piskorz, J., Radlein, D. and Scott, D.S., "On the mechanism of the rapid pyrolysis of cellulose", J. Anal. Appi. Pyrol., 9, 121 126 (1986).
(7) Richards, G.N., "Glycolaldehyde from pyrolysis of cellulose", J. Anal. Appi. Pyrol., 10, 251256 (1987).
(8) Shafizadeh, F. and Stevenson, T.T., J. Appi. Polymer S c , 27 4577
4585 (1982). (9) Radlein, D., Piskorz, J., Grinshpun, A. and Scott, D.S., "Fast pyrol
ysis of pretreated wood and cellulose", ACS Preprints, Div. of Fuel Chemistry, 32, No. 2, 2936 (1987).
(10) Updegraff, D.M., Anal. Biochem. 32, 420424, 1969
Table 1
Pyrolysis of Raw and PreTreated Wood
Run No.
Feedstock
Cellulose Content, X mf Ash, X mf Temperature,
Yields, wt X Gas Char Water Organics
°C mf
Total Recovery
Levoglucosan Other Sugars Hydroxyac Pyrolytic
etaldehyde Lignin
A2 IEA
Poplar
49.1 0.46 497
10.8 7.7 12.2 65.8
96.5
3.0 6.1 10.0 16.2
1SL
St.Lawrence Poplar
65.4 0.85 488
7.6 10.1 2.9 78.4
99.0
4.5 5.1 9.1 18.8
18 Wayman Poplar
43.1 1.8 500
14.0 11.9 8.5 63.2
97.6
6.7 8.6 7.4 19.3
20 Biohol Pine
54.9 1.0 500
7.5 23.0 10.6 51.0
92.1
10.3 4.7
Trace 21.9
38 Stake Poplar
47.0 0.67 500
13.0 12.4 3.2 58.9
87.3
2.9 1.1 7.5 18.0
X Cellulose Converted to Sugars
18.5 14.7 35.5 29.1 8.5

207
Table 2
Pyrolysis of Some AcidHydrolysed Woods
Run No.
Feedstock
Celluiose Content, X ffl£ Ash. X mf Hydrolysis Time, hours Temperature, C
Yields, wt X mi Gas Char Water Organics
Total Recovery
Levoglucosan Other Sugars Hydroxyacetaldehyde Pyrolytic Lignin
X Cellulose Converted co Sugars
10
IEA Poplar
65.5 0.02 6
480
6.9 S.2 2.2 80.9
98.2
30.0 13.2 0.9 21.0
66.0
14
St.Lawrence Poplar
70.6 0.30 2
480
6.4 10.5 5.2 75.0
97.1
30.1 18.8 0.9 17.9
69.3
46
Stake Poplar
66.5 0.16 2
490
6.6 6.7 3.2 82.1
98.6
25.3 20.2 4.3 18.8
68.4
Table 3
Pyrolysis of IEA LignoCellulose Under Varying Conditions
Run Ho.
Temperature, 'C Feed Rare, g/hr Hydrolysis Tiae, hr Moisture, wt Ζ Particle Size, aa
Yields, wt % mf Gas Char Coke Organics Water
Total Recovery
CO co2 cnu c2+
Oligosaccharides Cellobiosan Glucose Fructose (?) Glyoxal 1,6Anhydroglucofuranose Levoglucosan Hydroxyacecaldehyde Formic Acid Formaldehyde Acetic Acid Acecol
A1
501 46.9 6 16.5 0.25
6.4 0.0 ] 6.7 | 79.6 0.9
93.6
2.79 3.07 0.38 0.10
1.19 5.68 1.89 3.89 0.11 4.50 30.42 0.37 1.42 0.8 0.17 0.06
ST10
480 71.5 6 6.5 0.5
6.9 1 8.2 1 Ι ι 80.9
2.2
98.2
2.13 2.29 0.6
0.69 4.74 1.34 2.21 0.28 •3.90 30.0 0.9
2.36 2.32 0.07
ST11
475 116 2
6.0 0.5
6.4 1 6.3 1 73.6
6.3
92.6
2.5 1.9 0.4
0.89 4.77 1.96 2.12 0.28 3.89
28.7 0.9
1.06 1.03 0.12
S#ll
501 86.9 6 3.5 0.5
4.4 0.0 6.2
77.0 10.0
97.6
2.00 1.96 0.33 0.10
0.50 3.71 Tr 0.50 0.65 1.96
25.56 1.62
) . ,, 1 » · " ...
S « *
485 41.6 2
13.7 0.5
4.3 0.0 4.2 78.1 9.6
96.2
1.96 1.93 0.33 0.11
2.56 6.05 Tr 2.01 2.32 2.40 18.80 3.96 4.96 ... 0.70 ...
Pyrolytic Lignin 19.0 21.0 21.8
*NH3 treated. See text.
23.7 23.9

208
Table 4 Pyrolysis of De-ionized IEA Poplar
Run No. Temperature, "C Feed Rate, g/hr Additive (wt X)* Moisture, wt Ζ Particle Size, mm Yields, wt X mf
Gas Char Coke Organics Water Total Recovery
CO CO 2 CH,, C 2+
Oligosaccharides Cellobiosan Glucose Fructose (?) Glyoxal 1,6-Anhydroglucofuranose Levoglucosan Hydroxyacetaldehyde Acetic Acid
Pyrolytic Lignin X Cellulose Conversion to Sugars
* Ν ■
S#3 530 8.0 0 0 -0.5
14.13 2.5 60.50 18.25 95.38 4.86 8.75 0.38 0.13
4.92 0.94 1.81 2.86 1.12 9.33 3.35 0.67 17.8 37.1
S#4 -530 22.3 Ν (0.1) 3.0 -0.5
7.35 1.66 2.77 66.07 16.20 94.05 3.86 2.90 0.60
18.73 3.36 1.41 -21 53.9
S#5 -530 18.3
Ν (0.1) 28.9 -0.5
6.63 3.93 75.24 5.70 91.50 2.94 3.09 0.47 0.13
1.35 0.81 1.22 2.34 2.24 16.53 2.57 1.15 18.9 45.1
S06 493 23.6
Ν (0.1) 11.4 -0.5
2.61 0.67 4.68 75.93 13.96 97.85 trace 2.37 0.24
1.92 1.12 1.92 2.65 2.87 17.11 1.95 1.34 22.4 50.8
S#7 495 27.1 Η (0.1) 7.0 -0.5
3.22 0.72 5.41 66.10 -17 92.45 0.61 2.27 0.29 0.05
1.68 1.00 1.84 2.26 2.72 15.52 1.65 1.17 23.1 46.4
(NH4)2S04, Η
Properties Pretreatment
None 5X H2S04,
room temp., 20 h 5X H2S04,
b.p. temp., 2 h
Table 5 of Raw and Pretreated Wheat Chaff, mf basis Feed removed Ash content Cellulose content
X 0 15.6
31.6
Average of four tests
X 22.5 28.8
34.2
Table 6 Pyrolysis of Prehydrolyzed Wheat
Run # Temperature, "C Feed
, App. res. time, s Feed rate, g/h Amount fed, g Moisture, w/w X Particle size, mm Yields, w/w X Gas Char Water Organics Total Levoglucosan Other sugars Hydroxyacetaldehyde
57 502
Wheat Chaff acid washed at room temp.
0.5 49.1 36.8 5.7 -0.25
mf maf 8.7 12.2
40.5 16.4 6.0 8.4
42.4 59.6 97.6 96.6 10.5 14.7 6.3 8.8 tr tr
X Cellulose converted 70.8 into sugars (glucose equivalent)
56 500
Wheat Chaff acid washed at 100'C
0.5 83.7 41.8 3.9 -0.25
mf maf 8.4 12.7
44.0 14.9 7.7 11.7
38.4 58.4 98.5 97.7 13.7 20.8 5.5 8.4 tr tr 63.1
X 22.5 26.0*
33.5*
Chaff 66 495
Wheat Chaff untreated
0.5 86.8 36.2 6.1 -0.25
mf maf 11.0 14.1 43.9 27.6 8.6 11.1 37.2 47.9 100.7 100.7 0.9 1.2 0.6 0.8 2.5 3.2
7.3

209
PRODUCTION OF BENZOLES AND ACTIVE CARBON FROM WASTE RUBBER AND PLASTIC MATERIALS
BY MEANS OF PYROLYSIS WITH SIMULTANEOUS POST-CRACKING
PROFESSOR R. CYPRES and DR B. BETTENS Universite Libre de Bruxelles (ULB), Centre de Transposition Semi-Industrielle de la Recherche Appliquée (Centre for the Semi-Industrial Transposition of Applied Research) TSIRA,
Charleroi, and the Association pour l'Incinération, la Collecte et la Destruction des Immondices de la Region de Charleroi (Association for the Incineration, Collection and
Destruction of Waste for the Charleroi Region) ICDI
Summary
The pyrolysis of' rubber has long been the subject of study. The initial aim was to regenerate isoprene, the monomer of natural rubber. Then, after 1967, research was carried out into cracking at temperatures of between 300 and 1 000°C. Temperature was seen to have a marked influence on the kind of products obtained by cracking. At low temperatures, aliphatic hydrocarbons predominate in the liquid phase. At high temperatures, aromatic hydrocarbons, BTX and naphthalenes are formed to the exclusion of all other products.
Pilot installations have been constructed in various countries -by Goodyear and Interco Inc. (37.5 tonnes per day) in the USA, Kobe Steel (20 t/d) and Energy Recovery Research (20 t/d) in Japan, Foster Wheeler in Great Britain, Deutsche Reinfen- und Kunstoffpyrolyse (15 t/d) and Mannesmann Veba Umwelttechnik (48 t/d) in the Federal Republic of Germany, to name but a few.
The process developed by the ULB and the TSIRA Centre is based on two-stage pyrolysis. During the first stage, rubber is depolymerized at a relatively low temperature (around 500°C). During the second stage, the volatile substances thus obtained are post-cracked at temperatures of between 750 and 800°C. This process exploits the fact that aromatization can take place only above 700°C, whereas deploymerization and the formation of mainly C4 olefins produces maximum yields at low temperatures. The aromatization mechanism was studied in great depth by subjecting butane, butene and butadiene to thermal cracking. These are aromatized mainly into benzene, toluene and xylene as a result of Diels Alder cyclization reactions, with two molecules being converted into C4, followed by the dehydrogenation of cyclohexenes into benzene and its derivatives.
Pyrolysis with simultaneous post-cracking maximizes the yield of benzoles. It is also possible, by varying the period of time for which volatiles are retained in the post-cracking zone, to obtain high yields of naphthalenes. This makes for flexibility of operation, as the process can be made to function according to the market prices of benzoles and naphthalenes.
Research was carried out into a full working plant consisting of a pyrolysis furnace with a conveyor belt, a depolymerization zone and a post-cracking zone. The furnace is charged with whole car tyres (lorry tyres are first sheared) and the solid residue is eliminated in the lower section of the furnace.

210
Economic studies showed that it was not profitable to treat more waste tyres than are available within a limited radius of a conurbation. Collection costs become prohibitive if tyres must be collected outside the area covered by municipal collection services. This is why we explored the possibility of setting up units capable of processing approximately 500 t of waste tyres per annum within existing refuse treatment centres. Similar units could be set up in all conurbations. In the case of large or very large cities, where much more waste could be collected, larger installations could obviously be constructed using the same model.
The process is technically feasible and would be highly profitable. The capacity of the installation must correlate with the quantity of tyres available within a radius not exceeding that of an urban centre together with its adjacent centres of population. Treatment of such waste must be combined with that of household waste within existing centres, and suitable arrangements made for its collection.
From an environmental point of view, the dumping of tyres in quarries or elsewhere is not only detrimental to the sites in question, but also encourages the proliferation of rats. The incineration of tyres leads to atmospheric pollution unless expensive devices are installed to clean the fumes. Pyrolysis is the only method which produces aromatic hydrocarbons in the form of benzoles and naphthalenes, together with solid carbon (from the carbon used to reinforce tyres), which can either be converted into active carbon or used for other industrial purposes. The scrap tyre is a unique source of energy in that the only real cost involved is that of collection. The price of benzole and naphthalene fluctuates according to the price of oil.
1. INTRODUCTION AND LITERATURE SURVEY
Like any other organic compound, rubber decomposes when it is heated. Natural rubber is a polymer of 1,3 methyl butadiene. Generally speaking, pyrolysis is governed by the following parameters: temperature, retention time at the reaction temperature, pressure, and type of gaseous atmosphere. There are two stages to pyrolysis: primary cracking and post-cracking. Post-cracking occurs at higher temperatures and enables primary products to be converted into compounds which may have a higher market value.
Work on the pyrolysis of rubber first began in the 1920s. Initial research was carried out with a view to regenerating the monomer of natural rubber, isoprene. Williams (1) used atmospheric distillation to obtain a distillate containing approximately 5% isoprene. The effect of pressure was subsequently explored by Standinger and Fritschi (2) . These two authors showed that distillation at temperatures of between 275 and 350°C and at a reduced pressure of 0.1 mm Hg yields 63.5% of liquid phase, approximately half of which consists of isoprene and its dimer, dipentene. Standinger (3) went on successfully to distil rubber in an inert atmosphere at atmospheric pressure. Using this method, he managed to increase the weight of the liquid phase, which consisted mainly of isoprene dimers such as limonene and other monocyclic terpenes.
The author also reports the presence of cyclohexadiene and methyl-cyclohexadiene in the liquid phase. The rapid decomposition (4) of a crepe

211
of natural rubber at 700°C produced olefins, dienes, cyclohexadiene, as well as aliphatic and aromatic hydrocarbons.
Japanese authors (5) pyrolysed a solution of rubber in heavy oils at temperatures of between 310 and 390°C and at pressures of between 50 and 200 atmospheres. Reaction products consisted mainly of cracked oil, bitumen, carbon and gas.
Since then, research has started up in a number of new areas, in an attempt to discover ways of converting rubber waste into liquid combustibles and fuel. One method is the aromatization of products generated by primary pyrolysis. This has prompted a considerable number of researchers to carry out investigations into the pyrolysis of isoprene and its dimers. Oro, Han and Zlatkis (6), for instance, pyrolysed isoprene at temperatures of between 300 and 1 000°C. The influence of temperature on the composition of the reaction products is very marked. Whilst aliphatic components predominate in the liquid phase at low temperatures, at high temperatures decomposition products are entirely aromatic; they consist principally of naphthalene, methylnaphthalenes, xylenes and trimethylbenzenes, together with toluene. Pyrolysis produces optimum yields of aromatic components at temperatures of between 700 and 800°C.
It was not until 1974 that interest in the solid phase really developed. Most studies concentrated on the recovery of the solid phase with a view to using it in the treatment of waste water once it had been activated. Mention should be made of the work of Tanaka and Gomyo (7), Jo and Yoda (8), Sanga (9), Ishibashi and Noda (10) and Kudo (11).
A number of pyrolysis methods have been, or are being, developed at pilot or industrial scale.
In 1977, Goodyear carried out tests using a rotary drum in which ceramic beads were heated to 650°C and used to pyrolyse shredded tyres. Results showed that the profitability threshold for this type of reactor, which had been developed previously for processing bituminous shale, was of the order of 150 tonnes per day, which was too high.
Two plants came into operation in 1979, one in Japan and the other in the USA. The Japanese plant has a capacity of 20 t per day and is run by Kobe Steel Ltd. It consists of a rotary kiln with scraper blades to remove carbon from the walls. The American plant has a capacity of 37.5 t per day, is situated in Houston, and is run by Intenco Inc.
A third plant with a capacity of 20 t per day was set up in Osaka by the American company Energy Recovery Research Group.
In Great Britain, Foster Wheeler Power Products Ltd uses a process developed jointly by the Warren Spring Laboratory and the National Research Council. Pieces of waste tyre are held in place between two parallel vertical grills and heated by a horizontal stream of hot flue gas from an auxiliary furnace. Some of the gases thus generated are burned in the furnace to produce the hot flue gas.
In the Federal Republic of Germany, Deutsche Reifen- und Kunststoff-pyrolyse GmbH set up a fluidized bed plant with a capacity of 15 t per day in Hamburg in 1982. In this process, which was developed by the University of Hamburg, whole tyres are used as a feedstock for the reactor. These gradually sink into a fluidized bed of sand and carbon black as pyrolysis progresses. The steel casings are collected on a grill and removed.
Mannesmann Veba Umwelttechnik GmbH (Federal Republic of Germany) is currently planning to construct a rotary kiln with a capacity of 48 t per day for pyrolysing rubber, plastics and resins.
Dunlop Holding Ltd has financed a research project at the University of Aston (Great Britain) with a view to using molten carbonate salts as a source of heat for pyrolysis.

212
With the help of subsidies from the US Department of Energy, Rockwell International Corp. has carried out pilot-scale tests into pyrolysis at Canoga Park, California, using molten sodium carbonate salts. Pieces of tyre react with the sodium carbonate at temperatures of between 900 and 1 000°C and produce a combustible gas rich in hydrogen and carbon monoxide when exposed to a reducing atmosphere fuelled by a fixed amount of air.
USBM Firestone pyrolyses scrap tyes in an electric furnace at temperatures of between 500 and 900°C and at a pressure of 150 bars. In spite of the high pressure, products are not qualitatively different from those generated by other processes, nor is the yield.
The Herbold process treats pieces of waste tyres and plastics measuring between 30 and 40 mm at temperatures of between 430 and 450°C. A 24 t/d installation would cost approximately $1 000 000 (1978 prices).
In the Reprox process (Japan), pieces of waste tyre which have been reduced to a size of between 50 and 100 mm are pyrolysed in a fluidized bed. Retention time is less than one minute. Gases from the furnace are burned off after desulphurization, whilst the oils are used as commercial fuel. Research is currently being carried out into the residual carbon which is produced.
In the H. Rubber process, rubber undergoes catalytic hydrogénation after it has been pulverized to a grain size of approximately 0.1 mm and suspended in oil. A desulphurization and hydrogénation catalyst such as cobalt molybdate is used on an aluminium support. Hydrogénation takes place at 450°C and at a pressure of between 35 and 200 bars. Investment for a plant with a capacity of 270 000 t per annum would be of the order of $30 000 000 (1977 prices), and production costs (including hydrogen, labour, energy and maintenance) around $76 per tonne.
In the DSR Firestone process, rubber is heated in aromatic oil to induce depolymerization. The aim is to produce carbon black suspended in the oil, the mixture then being used to make new rubber compounds. Particle size is 35 mesh at a temperature of between 250 and 275°C and after a reaction time of between 12 and 24 hours, unfortunately, the suspension produced by this method is not of a sufficiently high quality to permit its use in the rubber industry.
The H Oil process is also designed to recover high-quality carbon black and fuel oil. Waste is pulverized and then dissolved in oil before being depolymerized. This is done at a temperature of 200°C and a pressure of 3 bars.
Mention should also be made of the low-temperature process (around 300°C) developed at the university of Compiegne.
The Tyrolysis method used in Wolverhampton (GB), is based on thermal decomposition at temperatures which are carefully selected to give the maximum yield of fuel oil. The Wolverhampton plant began operating in 1984. It is designed to treat 50 000 t of waste tyres per year, producing 20 000 t of fuel, 17 500 t of carbon and 7 000 tonnes of steel. It is a self-sustaining process, burning preheated pyrolysis gases. Tyres must be shredded twice in succession before the reactor can be fed with ground feedstock.
Although this list of waste tyre pyrolysis plants is far from exhaustive, it does show that the pyrolysis of waste tyres is widely developed throughout the world. Methods vary according to the technology used.
At low temperatures (below 500°C), fuel oil is produced but not much gas. At high temperatures, light aromatics and lavjer quantities of gas are produced.

213
In our process, advanced depolymerization of rubber takes place at low temperatures. The C3 and C4 olefins thus produced are continuously aromatized into BTX at high temperature.
2. EXPERIMENTAL RESULTS
The results of our laboratory research were published in 1981 (12) and were the subject of a feasibility and development study (13) which was financed by the Walloon region and carried out jointly by the following bodies working together on a temporary basis: the TSIRA Centre, the ICDI, the Centre de Transposition Semi-Industrielle de la Recherche Appliquée (Charleroi), and the Intercommunale de Collecte et de Destruction des Immondices de la Région de Charleroi.
We studied a two-stage process comprising an initial low-temperature pyrolysis phase followed by continuous post-cracking of resultant volatile materials at a higher temperature. This concept is based on a fact revealed by previous studies into the aromatization of short-chain olefins (14, 15, 16), namely that aromatization begins to take place at 750°C as a result of the cyclization of olefins into C2, C3 and C4. Post-cracking at a carefully chosen temperature effectively allowed an additional fraction of the gases corresponding to these ethylene compounds to be aromatized.
2.1 Pyrolysis with Post-cracking
The material to be pyrolysed is heated to a temperature of between 400 and 450°C. The gaseous phase thus produced is entrained by a vector gas stream into a second post-cracking furnace (600 to 800°C). The large isothermal zone in this furnace ensures that the gas remains in the reaction zone for a sufficient length of time to enable aromatization to take place. The composition of mass balances for the different phases is given in Figure 1. The composition of the gaseous phase is given in Figure 2 and that of the liquid phase in Figure 3.
It will be seen that the ethylene compounds disappear at higher temperatures. They are aromatized, producing H and CH in accordance with the following general reactions :
and C4 H8 + C2 H4 - C6H6 + 3 H2
C4 H8 + C3H6 - C6H6 + C H4 + 2H2
In the liquid phase, isoprene quickly disappears above 600°C. At high temperatures, naphthalene and plenanthrene seem to be most common, whereas the concentration of BTX does not vary significantly above the 600°C threshold.
The heating of elastomers induces depolymerization reactions. These depolymerization mechanisms are very complex and are described differently by various authors; they can lead to the formation of dipentene (17), or of numerous dienes of lighter molecular weight. In the case of rubber, butadiene and its methylated derivative probably account for an important fraction of such dienes.
As has been seen, the pyrolysis of rubber leads to the production of ethylene, propene and 1,3 butadiene. These induce aromatization as a result of Diels Alder reactions, which have been amply described in the relevant literature (18, 19, 20). These reactions can be represented as follows:

30
20
6O0
Liquid
700 800 C Post-cracking temperature
Fig. 1. Mass balance for tyre pyrolysis with 450°C hot charge
Moles
%
60
20
600 700 800 C Post-cracking temperature
Fig. 2. Molar percentages of constituents of the gaseous phase obtained by tyre pyrolysis with
45cPC hot charge
Weight
600
% 7 0
5 0
■
■
2 0
1 0
X
\ o
0 \
BTX ■^--^^Styrene
Phenanthrene
/ •^Naphthalei
^ Isoprene
7O0 800 C Post-cracking temperature
Fig. 3. Weight percentage of constituents of the liquid phase obtained by tyre pyrolysis with
450°C hot charge
to l—i

215
2 C 2 H 4 + 1 C 4 H 8 - 1,3 C4H6 + ^
.11—0 1.3 butadiene ethylene cyclohexene
C - r — O 4-methyl-l-cyclohexene
4-vinyl-l-cyclohexene
Dehydrogenation causes cyclic compounds with six carbons to produce aromatic derivatives corresponding to one cycle. As a result of subsequent associative reactions, these may lead to the formation of polycyclic compounds such as naphthalene and phenanthrene.
2.2 Influence of Contact Time
Pyrolysis was carried out three times, with the contact time being varied by changing the rate of flow of vector gas (N ) . Three values were used: 20, 40 and 60 ml per minute.
Michelin X tyres were used in all the tests. The results are given in Tables I, II and III.
TABLE I. Mass balance produced by tyre pyrolysis, as a
Gaseous phase Liquid phase Solid phase
function
Rate
of contact
of flow
20
13.96 34.44 51.60
of
time (%
nitrogen
40
15.12 36.54 48.34
weight)
in ml/min
60
13.42 41.58 45.00

216
TABLE II. Molar % of constituents of the gaseous phase as
H2 C H4 C2 H4 C2H6 C3HR C3H6
a function of the rate
Rate of flow
20
23.28 56.15 14.95 3.21 0.15 2.26
of
of
flow of carrier ga:
nitrogen
40
23.30 53.85 16.78 3.56 -2.52
(ml/min)
60
30.65 47.36 13.56 4.51 0.32 3.60
TABLE ill. Percentage weight of the constituents of the liquid phase as a function of the rate of flow of carrier gas
Rate of flow of nitrogen in ml/min
20 40 60
1.04 2.89 36.41 30.57 16.78 15.66 4.83 5.22 1.35 1.48 4.58 5.83 1.68 2.18 2.47 3.26 9.15 7.97 2.42 2.71 4.06 5.43 4.35 6.13 3.19 2.98 7.68 7.69
An increase in the flow rate N2 appears to improve the yield of liquid phase but somewhat diminish the benzene content. This is because depolymer-ization products are entrained more rapidly at lower temperatures, whereas at high temperatures aromatization is less marked owing to the reduced gas retention time in the hot zone.
2.3 Pyrolysis of Various Tyres and Brands of Tyres
Different brands of tyres were pyrolysed at temperatures of between 450 and 800°C. The results appear in Tables IV to VII.
There do not appear to be any significant differences, although it should be pointed out that the yield of liquid is greater when V 10 tyres are used.
Octene + lights Benzene Toluene m + p-xylenes p-xylene Styrene Indane Indene Naphthalene 2-methylnaphthalene 1-methylnaphthalene Acenaphthene + ... Plenanthrene Heavy fraction
3, 33. 14. 4, 1. 4. 1. 2.
11. 3. 1. 7. 4. 6.
.55
.85
.55
.16
.24
.17
.27
.60
.21
.41
.87
.47 ,58 .08

217
TABLE IV. Mass balances produced by pyrolysing various types of tyre
Michelin Michelin Uniroyal Uniroyal Rad. X Rad. XAS-P Rallye Rallye
Wet Weather
Kleber Kleber V 10 V 10GT
Gaseous phase Liquid phase Solid phase
TABLE V. Molar
16.77 38.46 44.77
percentage
17.55 37.15 45.30
16.05 39.72 44.23
of constituents of
Michelin Michelin
H2 C H4 C2 H4 C2H6 C3 H8 C3H6 C „ H o 4 8
X
31.69 54.88 10.06 2.54 -
0.84 _
XAs
26.50 53.25 12.72 4.38 -3.18 0.18
TABLE VI. Weight percentages of the after pyrolysis
Non-aromatic c6c7 + isoprene Octene Benzene Toluene Ethylbenzene m + p-xylenes o-xylene Styrene (intermediate fraction, propylbenzene, indane, indene, etc) Naphthalene Methyl-naphthalenes (diphenyl, fluorene, etc) Phenanthrene + anthracene Heavy fraction
Michelin X
0.04 0.16 42.07 15.32 2.84 0.62 0.10
t
2.35 12.67
12.97
5.59 1.41
at 450°C-
Michelín XAS-P
0.07 0.27 34.43 15.06 3.95 0.82 0.11
2.42 11.69
14.59
5.38 8.47
Uniroyal
29.01 54.61 11.89 3.51 -0.97 _
19.47 37.02 43.51
16.98 41.34 41.68
the gaseous phase
Uniroyal Kleber Wet Weather V 10
15.75 63.84 14.90 3.96 0.06 1.49 _
constituents of the 800°C
Uniroyal Rallye
0.06 0.25 41.51 17.14 3.52 0.69 0.11
2.98 10.72
9.79
3.92 5.74
Uniroyal
32.91 56.60 7.18 . 2.75 -
0.57 _
liquid pha
Kleber Wet Weather V 10
0.05 0.09 30.42 14.25 3.28 0.66 0.10
2.69 12.96
15.85
5.62 9.52
-0.15 41.89 15.55 2.88 0.65 -
1.87 11.19
10.10
5.04 7.32
16.16 42.17 41.67
Kleber V ÎOGT
39.49 46.98 10.99 2.12 -0.42 _
se
Kleber V ÎOGT
-0.07 33.93 13.04 2.61 0.46 0.09
2.10 14.17
12.29
5.89 12.41

218
TABLE VII. Comparison of various mass balances
Average over Average over Pyrolysis of a 6 pyrolyses 6 pyrolyses mixture of all 6 450-800°C 500-750cC tyres 500-750°C
Gaseous phase 17.16 lb.87 13.82 Liquid phase 39.31 39.02 39.23 Solid phase 43.53 45.11 46.95
Mixtures of tyres give approximately the same results as those obtained for one type of tyre, as can be seen from Table VIII. TABLE VIII. Comparison of the different phases obtained by pyrolysing
a mixture of 6_ tyres with that obtained from an average of 6 pyrolyses of the same tyres
Analysis of the liquid phase as % of weight
C3 H6' C4 H8' C5H10 C5 H8' C5 H6' C6H12 C7H14, Isoprene Cyclohexadiene Octene Benzene Toluene Ethylbenzene m-xylene + p-xylene o-xylene n-propylbenzene Styrene Ol -methylstyrene (unidentified) Indane Indene (unid.) Methylindene (unid.) Naphthalene unid. 2-methylnaphthalene 1-methylnaphthalene Diphenyl (unid.) Acenaphthene (unid.) Dibenzyl (unid.) Fluorene (unid.) Phenanthrene m-phen., pyr., benzanth., H.F.
4.10 1.88 26.15 17.27 6.42 1.86 0.61 0.11 5.97 2.34 2.42 2.47 1.35 5.08 0.21 2.66 2.18 2.67 2.62 2.25 2.91 2.37 4.81
3.59 2.06 27.80 17.81
8.27 1.94
6.51
3.23 4.00
5.52
2.72 5.81
4.71
2.25 3.69
Analysis of the gaseous phase as a % of volume
H2 C H4 C2 H4 C2H6 C3 H8 C3H6
21.05 51.66 13.92 7.48 0.82 5.06
22.37 53.63 12.10 6.66 0.59 4.64

219
3. EXPLOITATION OF RESIDUAL CARBON
Carbon residue accounts for between 35% and 40% of the weight of a pyrolysed tyre. Its minimum value is that of a good smokeless fuel, since all the organic matter has been decomposed thermally and the combustion fumes no longer contain tar.
We studied the adsorption properties of various types of residual carbon obtained from tyre pyrolysis after they had been activated by steam and so converted into active carbon. This is an excellent adsorbent and is well known on the market as a purification medium. Its high price does, however, make it unsuitable for large-scale use as a water purifier.
There is clearly an interesting potential in being able to convert 40% of a particular type of waste, in this case the scrap tyre, into active carbon.
Overall results are given in Figure 4, which shows isotherms of phenol adsorption by carbon, where q represents the amount (in mgs) of phenol adsorbed by a quantity g of carbon.
Activation is by steam heating at approximately 900°C. Activation time is about 30 minutes. Under these conditions, the weight loss of carbon is of the order of 30-40%. The test solution had a phenol concentration of 100 mg/1.
Some types of active carbon obtained from tyre pyrolysis (550, 600, 700°C) have adsorbent qualities which are very similar to those of commercially-produced active carbon. Other types, obtained at pyrolysis temperatures of between 450 and 500°C could also be used provided the higher residual concentration of phenol is acceptable. Carbon produced by pyrolysis could even be used "raw" for the pretreatment of heavily polluted water.
It will be seen from the isotherms that carbon obtained by pyrolysis at 550°C is the best.
As far as abatement rates are concerned (Figure 5) , it will be seen that in the case of high weight concentrations of active carbon (approximately 5 g), carbon obtained at 600°C adsorbs all molecules of phenol (rate = 100%). For other types of carbon, the abatement rate is approximately 95%. At lower concentrations (approximately 1 g) , carbon obtained at 500°C adsorbs the most molecules (90%), followed by carbon obtained at 600°C and 700°C. Rates for carbon obtained at the latter two temperatures are of the order of 75% to 85%.
In order to improve classification of the various types of active carbon obtained, we also studied their adsorption properties vis-a-vis methylene blue and iodine.
The solid phases used in these tests were obtained by steam or carbon dioxide activation. Figures 6 and 7 show the adsorption capacity of these types of carbon vis-à-vis methylene blue (curve a) and iodine (curve b), as a function of activation time.
The specific surfaces of the various types of carbon derived from tyre pyrolysis were measured both before and after activation. The specific surface before activation, of the order of 60 m2/g, is the same whatever the speed of pyrolysis. Steam activation creates specific surfaces of around 390 m2/g for slow pyrolysis and 290 m2/g for rapid pyrolysis.
To sum up, it can be said that this method, in which waste tyres are used as a feedstock, produces oils with a high BTX content and carbon with good adsorbent properties as a result of pyrolysis at a low temperature (450°C) and simultaneous post-cracking at a high temperature (700 to 800°C). Figure 8 shows the mass balance produced by treating one tonne of used tyres.

220
A Commercial Β 550° C 70O° D 600° E 500° F 450° G Non-active
Abatement rate
Residual concentration
Fig. 4. Isotherm of phenol adsorption by carbon
Amount of carbon (in g)
Fig. 5. Rate of phenol adsorption by active carbon
Methyle blue
100.
80 ■·
60 ·-
Iodine
20
.3
.. .2
10 15 20 min
Fig. 6. Steam activation Fig. 7. CO? activation

221
1 t TYRES
P y r o l y s i s
4 5 0 5 0 0 ° C
P o s t c r a c k i n g
75O80O°C
.HEAVY
1 3 2 kg
SOLID
4 6 0 kg
"2 CH4
C2H„ C
2H
6 c
3 M i s e .
3 . 8 O. 35
32 1 8 . 6 2 0 . 1
3 . 1 5
148
_^ LIGHT
BTX 2 6 0 kg
B
T Χ S t y . M i s c .
1 0 5 . 8
6 8 . 6 3 7 . 4 2 4 . 4 2 9 . 8
N a p h t h . 2 1 . 2 P h e n . 1 8 . 0
c a r b o n s t e e l a s h
359 7 0 31
Fig. 8. Mass balance produced from the pyrolysis of 1 t of used tyres
4. STRATEGY FOR THE COLLECTION OF WASTE TYRES
4.1 Availability of Used Tyres
ICDI and TSIRA, working together on a temporary basis, carried out a detailed study into the amounts of used tyres available in Belgium, and the cost involved in collecting these. They considered the Charleroi region for the purposes of the study.
This study, which cannot be described in detail here, concluded that between 45 000 and 50 000 tonnes of scrap tyres are available each year in Belgium. They are the main source of rubber waste. Table IX gives a very approximate breakdown of the different ways in which this waste is used.

222
TABLE IX. Use of waste tyres
Retreading Ground feedstock Export Pyrolysis Artificial reefs Incineration Tipping Regeneration Marginal users
25
20% 0.5% 10% 0% 0%
to 30% + 35%
3% 5%
(difference)
At least 65% of all waste tyres are incinerated or dumped.
Incineration
Incineration exploits the energy content of tyres. They have a calorific value of between 6 000 and 9 000 Kcal/kg, and are thus ideal for this purpose.
Free-air incineration is banned, but has in any case several obvious drawbacks :
it produces large quantities of black, acrid and highly noxious smoke; it produces high concentrations of SO; the considerable calorific value of tyres is wasted.
The main pollutants produced by the combustion of tyres are sulphur oxides, nitrogen, carbon, zinc, very fine "carbon black" dust (carbon black is used to reinforce tyres), and condensed polyaromatic hydrocarbons, which are carcinogenic.
Tyres are incinerated in furnaces specifically designed for the purpose (types include LUCAS, UHDE, CEC-CEA, etc.). Conventional furnaces can also be used if incineration plants are modified by means of scrubbers, electrostatic dedusters, desulphurizers, etc. but appliances such as these are expensive.
Tipping
In many countries, tipping is the most common form of waste tyre disposal.
In Belgium, as in many other European countries, waste disposal sites are becoming increasingly rare. Tyres cannot continue to be disposed of in this way if the environment is to be protected. Because they are not biodegradable, they simply accumulate. Tyre dumps constitute a fire hazard and make the ground spongy and soft, thus creating problems if the site is ever redeveloped. Rats proliferate in dumps which are either poorly covered or left completely uncovered, and this clearly presents a health threat to the general public.
As has been pointed out, tyres have a high calorific value. Tipping does not exploit this potential energy, nor does it exploit the hydrocarbon and carbon content of used tyres.
The cost of tipping varies according to the method used: a simple tipping costs between Bfrs 300 and 400 per tonne, controlled tipping with tyres shredded at the site costs Bfrs 2 500, and controlled tipping with the tyres shredded before they are transported costs Bfrs 4 000.

223
4. 2 Cost of Collection
An evaluation was made both of the cost of collecting tyres in the Charleroi area, and of the scrap tyre potential of the region.
Most used car tyres are located in the main distribution outlets for new tyres. These are, in descending order of importance:
tyre specialists: 57% garages, service stations and dealers: 30% hypermarkets: 13%
Used lorry tyres are mainly found at the following places :
tyre specialists : 60% end users (army, haulage firms, etc.): 35% major distributors: 5-10%
ICDI carried out a detailed investigation into the cost of collecting 1 000 tonnes of used tyres per year in the Charleroi region. Collection is carried out using Berliet Saviem refuse lorries with GDA skips with a capacity of 20.4 m3. Normal manning costs are assumed. When all expenses are taken into consideration, the total cost of collecting one tonne of used tyres is approximately Bfrs 1 550 (1984 prices). This figure assumes that used tyres can be taken away free of charge, and that there are no collection fees.
The remaining tonnage treated at the plant comprises tyres which are delivered to the treatment centre, and for which payment is made. This is the system used for incineration. In 1984, the CBR cement works at Lixhe paid Bfrs 600 for each tonne of tyres delivered to the plant. This figure is much lower than that quoted above for specific collection (Bfrs 1 550 per tonne) . In the case of incineration, it is not possible to increase this price without making it economically unviable to use old tyres as fuel. Higher prices can, however, be paid for pyrolysis, although these must still remain well below the cost of collection if there is to be a real advantage. It has been estimated that, on the basis of the Lixhe experience, 45% of used tyres in the Charleroi region could be obtained for Bfrs 600 per tonne delivered to the plant.
The ICDI study showed that collection price increases rapidly in proportion to distance from the treatment plant, particularly since there are much fewer tyres to be collected near the outer limits of the region concerned.
The conclusions drawn by the ICDI led us to develop a new strategy for the pyrolysis of used tyres. We do not think it reasonable to construct one large plant in Belgium with an annual capacity of between 20 000 and 25 000 tonnes. (This figure corresponds more or less to the quantity of scrap tyres which are not used for other purposes. ) Tyres would have to be collected over long distances and the cost would soon become prohibitive. Moreover, the need to guarantee the supply of tyres to the pyrolysis plant would leave the plant in a weak position vis-à-vis collectors. The price of used tyres would inevitably rise.
If, however, existing centres for the incineration of household refuse were made to include small-scale, "sub-regional" units (i.e. units corresponding to the amounts of used tyre waste produced by any one urban area), this would constitute an extension of existing activities carried out by such bodies. The collection of tyres would be carried out by the same bodies responsible for collecting household waste.

224
5. 2 OOP TONNES PER YEAR PLANT
5.1 Description of the Plant
The tonnage of tyres available in the Charleroi region was used as a basis for designing a 2 000 tonne per annum industrial plant to carry out pyrolysis with simultaneous post-cracking. The plant design is illustrated in Figure 9. The main element is the furnace, the design and dimensions of which were drawn up by EUROFOUR. This consists of a conveyor belt within the horizontal part of the oven, which is heated to between 450-500°C.
The tyres, which are left whole, are fed into a charging chamber, which is kept free of air by circulating nitrogen. It is thus not necessary to cut up the tyres. The estimated saving is between Bf rs 1 000 and 1 500 per tonne.
Depolymerization and primary cracking take place progressively. Volatile material is entrained by the stream of nitrogen into the vertical post-cracking furnace at 700-800°C, where olefins are converted into light aromatics. The post-cracking furnace is heated by radiant tube burners (automatic heat recuperators), which are fitted with fins. These can easily be removed or replaced.
Gases are separated from oils in the condenser. Light oils are drawn towards the top of the fractional condensation column, whilst heavy oils are drawn to the bottom.
To sum up, this installation has the following features:
1. Whole-tyre feedstock, which cuts down on shredding or cutting costs. 2. Depolymerization and primary shredding by heating the tyres to between
450 and 500°C. 3. The aromatization of volatile products produced at low temperatures by
continuous and simultaneous post-cracking.
5.2 Investment and Running Costs
Investment will be of the order of Bfrs 31 million (1984 estimate), divided up as follows :
1. Specific equipment for the plant, comprising the following:
Pyrolysis and post-cracking furnace; Containers for solids; Oil condensers ; Compressors, vibrating separators and other accessories.
22 370 000 2. Installation 800 000 3. Electricity 600 000 4. Miscellaneous (safety, civil engineering,
maintenance , engineering etc.) 6 980 000 TOTAL Bfrs 30 750 000
Running costs including personnel, supply of tyres, electricity and nitrogen, have been estimated at approximately Bfrs 6 000 000 per annum (1984 prices), or Bfrs 3 000 per tonne of tyres for a plant with a capacity of 2 000 tonnes per annum.
The profitability study for this project was carried out by ACKERMANS and VAN HAAREN - N.V. (Antwerp) . This concluded that small units for treating used tyres by means of pyrolysis with simultaneous post-cracking

Tyre circui t — Gas c i rcu i t _
Nitrogen c i rcu i t Water c i rcu i t *.
Extractor ¡ pump
j Light aromatics ι (200 1 barrel)
Production
Heavy oi ls (200 1 barrel)
i»Water reject) jWater
Water entry c i r c u i t point )
Fig. 9. Operating diagram Industrial unit for the pyrolysis of used tyres with simultaneous postcracking. Capacity: 2000 t / a
Carbon con
tainer
Metal con
tainer
Production 780 t /a
Production 140 t / a
to Ln

226
(2 000 tonnes per annum capacity) would not only prove to be a profitable investment, but would also solve a serious environmental problem.
If the hypotheses on which the operation of an industrial unit are based can be shown to be realistic by the pilot plant, it could reasonably be assumed that a considerable number of plants of this type might be marketed both in Belgium and abroad.
6. GENERAL CONCLUSIONS
The following conclusions can be drawn from this study into the pyrolysis of tyres with simultaneous post-cracking:
1. Originality of both the process and the prototype
The process is original because it enables the depolymerization of rubber by thermal cracking and the aromatization of olefins thus produced to take place in one stage and within the same plant. This produces high yields of benzene, toluene and xylene (BTX), which have a higher market value than heating fuel. The prototype as designed is also original, and the plant could be built by companies from the Wallonia region: this is amply demonstrated by the fact that EUROFOORS (Liège), after carrying out a detailed technical study, drew up a cost estimate for a demonstration unit with a capacity of 2 000 tonnes per annum.
2. Tyre pyrolysis: location strategy
The study into the cost of collecting used tyres shows that expenditure increases rapidly in proportion to distance, and that a large number of lorries and containers is needed if they are to be used solely for this purpose. This is why the authors of the study believe that the processing of used tyres must be carried out within existing structures for the collection and treatment of urban waste.
The current collection capacity for the Charleroi region has been estimated at 2 000 tonnes per annum. The tonnage of tyre waste that can be treated by any one pyrolysis plant will depend on the size of the urban centre which supplies it. Treatment capacity has been estimated at between 4 and 5 000 tonnes per year per million inhabitants.
We recommend that the idea of a single, large-capacity plant serving an entire country or region, as has been proposed in the past, be abandoned.
A pyrolysis plant with a capacity of 40 to 50 000 tonnes per annum creates a number of problems. Quite apart from the fact that the distances to be covered within such a large region would drastically increase collection and transport costs, there is also the danger that suppliers might begin to speculate by stockpiling available tyres.
A large plant must be guaranteed regular supplies and would thus be highly vulnerable to any manipulations in this supply. Furthermore, a large plant requires a much greater level of investment and thus constitutes a much higher financial risk for developers.
A large plant also needs the appropriate infrastructure, and since this would have to be created from scratch, investment and running costs would again be pushed up. We thus propose that the treatment of tyres be integrated into existing regional and local centres (this solution could also be adopted for other types of waste). This will effectively constitute an extension of existing activities, and will mean that advantage can be taken of the infrastructure and experience already to hand. It should be emphasized that, under this scheme, the collection of tyres would be carried out by bodies which are already highly experienced in the field of household

227
refuse collection. This does not, however, mean that this type of installation would always have to be limited to small units. Indeed, these could be of a considerable size where large centres of population are concerned. It is clear that large urban centres such as Paris, London, Tokyo, etc., represent a potential of tens of thousands of tonnes per annum.
3. Profitability
The difference between the price of crude and refined products is between 80 and 100% in the case of benzols. For carbon, the difference between the fuel price and the price of either carbon black or active carbon varies by a factor of 1 to 10. Pyrolysis thus has enormous development potential and will have a great impact on job creation within the treatment plants themselves and within industrial undertakings wishing to develop their activities or specialize. Initially, however, it would seem advisable to stick to the collection of raw products separated into fractions which are sufficiently differentiated to enable exploitation to take place. The demand for, and price of, these raw products will help determine which strategy should be adopted in order to optimize the exploitation of these by-products.
A profitability study carried out by Ackermans and Van Haaren S.A. (Antwerp) came to the following conclusions :
"Results of the profitability analysis show that small units for treating used tyres by means of pyrolysis with simultaneous post-cracking and having a capacity of 2 000 tonnes per annum would represent a profitable investment and would also solve a serious environmental problem." "If the hypotheses on which the operation of an industrial unit are based can be shown to be realistic by the pilot plant, it could reasonably be assumed that a considerable number of plants of this type might be marketed both in Belgium and abroad."
The report does, however, draw attention to the sensitivity of benzol and oil prices to fluctuations in the price of oil.
4. Market
Two aspects must be considered here: firstly, the market for pyrolysis products, and secondly, the market for the sale of pyrolysis plants. As far as pyrolysis products are concerned, i.e. BTX and residual carbon, the market is favourable. It has been estimated that the demand for benzene both in Belgium and worldwide will continue to increase steadily. The price of BTX varies according to the price of oil, because light aromatics are obtained by the steam cracking of oil fractions. Marketable quantities of BTX produced by tyre pyrolysis are small in comparison with those produced by the petrochemical industry, and would therefore have little or no effect on prices.
Carbon can fetch prices of up to 10 times its minimum value (based on calorific value) when it is used as a reinforcing medium in the production of tyres, or as active carbon used in water purification (high-quality utilization). However, since there are currently insufficient quantities of this type of carbon to enable industrial tests and demonstrations to be carried out, it is not yet possible to say that it will sell on the market without additional and specific processing being carried out for each individual application.

228
As far as the sale of pyrolysis plants is concerned, it should be possible to build four or five units in Wallonia and an equal number in the north of the country and in Brussels. The export potential is considerable, and the technology could be sold if it were shown that the chosen formula (plants constructed on a decentralized basis and integrated into existing waste treatment centres) were economically viable.
5. Economic and social benefits
One obvious advantage is that this process converts waste into valuable raw materials. In doing so, it produces fuel and combustibles and so results in an overall energy saving. This in turn means that less money must be spent on importing energy from abroad.
Integration of this process into existing waste treatment centres will create new jobs within these centres and within those branches of industry involved in exploiting the by-products obtained.
6. Ecological implications
One of the roles of waste treatment centres is to protect the environment by eliminating waste. If used tyres can be disposed of in a profitable way, numerous ecological benefits can be reaped at no extra cost.
The treatment of used tyres is receiving priority attention from the Commission of the European Communities in the context of environmental protection. This project is thus entirely appropriate in the light of the Commission's current concerns.
7. usefulness of a semi-industrial pilot plant
The process as a whole must be tested at pilot level in order to ensure that all component parts work at optimum efficiency. A pilot plant will also serve to establish yields and will ensure that products are generated in sufficient quantity to be of use to industrial operators.
The pilot plant could provide a number of useful services once the development phase comes to an end: it could be used for training personnel from other treatment centres wishing to acquire a similar unit, or for carrying out research into the pyrolysis of wastes other than tyres (plastic material, wood, etc.). The existence of a training unit for personnel employed in businesses who wanted to purchase such a plant would be extremely useful for promoting the method on a commercial basis .
REFERENCES
(1) WILLIAMS, Philos. Trans. Roy. Soc. London. Ser. A 150, 241 (1860). (2) STANDINGER and FRITSCHI, Helv. Chim. Acta, 5, 758 (1922). (3) STANDINGER and GEIGER, Helv. Chim. Acta, 9, 549 (1946). (4) MIDGLEY and HENNE, J. Amer. Chem. S o c , 26, 1215 (1929). (5) KATO and SOMESHIMA, J. Soc. Chem. Ind. Japan, 38, 596 (1935). (6) ORO, HAN and ZLATKIS, Anal. Chemist., 39, 27 (1967). (7) TANATA and GOMYO, Japan, Kokai 7438, 895 (CI. 14 E 331, 13(9) F2) 11
Apr. 1974, Appli. 7281, 419, 16 Aug. 1972. (8) JO and YODA (Nippon Zeon Co, Ltd). Japan Kokai 74, 102, 595 (Cl. 14 E
331, I, 12(9)F2, 91 C 9 1 ) , 17 Sep. 1974, Appli. 73 14, 588, 05 Feb. 1973.
(9) SONGA, Ger. Offen., 2, 328, 400 (Cl. c 01b) 20 Jan. 1974. (10) ISHIBASHI, NODA and TERADA, Japan Kokai 7545, 799 (Cl. COIB, BOID), 24
Apr. 1975, Appli. 7396, 413, 27 Aug. 1973.

229
(11) KUDO, Japan Kokai 7575, 593 (CI. COIB, BOID), 20 Jan. 1975, Appli. 73 126, 013, 09 Nov. 1973.
(12) R. CYPRES and B. BETTENS, "Traitement pyrolytique des déchets de caoutchouc". Ann. des Mines de Belgique, 10, 873-890, 1981.
(13) Convention Region Wallonne-Association Momentanée ICDI-TSIRA, "Faisabilité de petites unités de pyrolyse de pneus usagés avec postcraquage simultané", 171 pp, 1984.
(14) R. CYPRES and C. BRAEKMAN-DANHEUX, "Processus d'aromatisation dans la formation des goudrons de haute température". Ann. des Mines de Belgique, 11, 1109-1115, 1974.
(15) R. CYPRES, C. BRAEKMAN-DANHEUX, R. DERIE and M. BERKOL, "Aromatisation of a C 4 alkane/alkene/hydrogen mixture obtained by catalytic dehydro-genation of isobutane". J. Anal. Appi. Pyrol., 1, 339-246, 1980.
(16) R. CYPRES, "Aromatic hydrocarbons formation during coal pyrolysis", Fuel Process. Technol., 15, 1-15, 1987.
(17) MADORSKY, "Thermal Degradation of Organic Polymers", p. 219, Inter-science (1969) .
(18) WEIZMANN, BERGMANN, HIGGETT, STEINER and SALZBACHER, Ind. Eng. Chem. 43, 2312-18 (1951).
(19) BREDAEL P., Ann. Mines Belgique, 12, 1045, 1975. (20) BENSON and HAUGEN, J. of Phys. Chem., 71, 6, 1967.
ACKNOWLEDGEMENTS
We should like to thank Miss GILLET and Mr PILATI, engineers at the ICDI and TSIRA respectively, for their contribution in drawing up the feasibility study.
We should also like to thank the Wallonia region and Minister BUSQUIN for the financial support which was granted to TSIRA and ICDI for the purpose of carrying out this feasibility study.

230
CHEMICALS FROM ALMOND SHELLS BY PYROLYSIS IN FLUIDIZED BED
R. Font, A. Marcilla, E. Verdú and J. Devesa División de Ingeniería Química. Universidad de Alicante. Spain
Summary
Flash pyrolysis of almond shells at moderate and high temperatures in a fluidized bed reactor and an Analytical Flash Pyrolysis Apparatus (Pyroprobe 100) have been studied, with almond shells nonimpregnated and impregnated with inorganic chemicals. The Analytical Flash Pyrolysis Apparatus was used for screening of catalysts and selecting operation conditions. At moderate temperatures, the yields of the following fractions and chemicals have been obtained: solid, liquids (water, methanol, formaldehyde, acetone, 2propanol, acetic acid, hydroxyacetone, propionic acid, 3methyllbutanol, 2furaldehyde, dry residue at 120 sc, gases ( CO , CO, CH , H ). At high temperatures the gases analyzed have been the following: H , CO, CO , CH , C H , C H , C H , C H , C hydrocarbons. The greatest yields (,Λ.% basis) obtained in the different operating conditions in the fluidized bed reactor have been the following: A) At temperature range 440610 C, pyrolyzing almond shells (without impregnation): 1056 acetic acid; 1.8% hydroxyacetone; 1.0% lhydroxy2butanone; 0.7% 2furaldehyde; 1.0% methanol+formadehyde. B) At temperature range 470610 -C, pyrolyzing almond shells impregnated with CoCl (14.1% g CoCl /total g): 7.0% acetic acid, 7.0% 2furaldehyde, 0.6% methanol + formaldehyde. C) At high temperature (820900
2C) pyrolysis of almond
shells (without impregnation): 30% CO, 16% CO, 0.8% H
5.7 % CH, 2.8% C2H4'
coal formed , 1.0% C Η , 0.7% C H . Recovery ofXoCl from the char as also Been studied. Kinetic expressions for the pyroly
sis of almond shells nonimpregnated and impregnated with also been deduced.
CoCl, have
1. INTRODUCTION Pyrolysis is one of the alternatives to be considered for conversion
of biomass to activated carbon, fuels and chemicals. Pyrolysis processes of biomass at moderate temperatures (380600
aC) yield a pyrolytic oil
which is a mixture of organic chemicals with water, a low BTU gas and charcoal. Flash pyrolysis of lignocellulosic materials leads to an increase in the amount of liquids produced.
High temperature (>700 9C) flash pyrolysis of biomass yields a mixture of gases: carbon oxides, hydrogen and light hydrocarbons. Flash pyrolysis can be achieved discharging the biomass on a hot sand fluidized bed reactor.
The process of biomass decomposition occurs via both primary and secondary reactions. By selecting the operating conditions (temperature, heat transfer and residence time of volatiles), it is possible either to accelerate or to slow down the different reactions involved in the process, according to their apparent activation energy. Using catalysts is another way to change the most favourable reaction paths.
Catalysts may be classified as primary or secondary. Primary catalysts

231
act on the primary reactions (directly from biomass). Secondary catalysts have their effect on the products of the primary reactions (carbon, volatiles).
A research of the pyrolysis of almond shells (an abundant and readily available agricultural byproduct in the area) is being carried out at the Chemical Engineering Division of the Alicante University. The scope of this investigation is to study the chemical products which can be produced at moderate and high temperatures from the almond shells,nonimpregnated and impregnated with chemical compounds.
2. EXPERIMENTAL SECTION Almond shells were washed, dried, crushed and sieved to obtain a uni
form material. The almond shells composition (wt%) is the following: 29% cellulose, 19% hemicellulose, 51% lignin, 0.2% ash (d : 0.2970.500 mm).
Calcinated sand, at 900 2C was used as an inert bed in the fluidized
bed reactor (see references 1, 2, 3). Pyrolysis in the experimental system was run as follows: First, a dried almond shells sample of the selected particle size was placed in the feed hopper. The inert gas flow (from an industrial Ν source) was set and the oven switched on. Once the reactor reached the selected temperature, the feeding valve was opened and the sample fell into the sand bed fluidized reactor.
In order to carry out the experiments at moderate temperature with almond shells impregnated with CoCl , the samples were prepared as follows: Five batches of 60 g of almond shells mixed with catalysts solutions of known concentrations were prepared in a "rotavapor" Buchi, providing agitation and a 60 mmHg vacumm for 2 hours. Afterwards, the samples were dried at 110 9
C for 20 hours. Once the five samples were prepared, they were mixed well.
In addition, an Analytical Flash Pyrolysis Apparatus (Pyroprobe 100) was used for catalyst screening (nominal heating rate 20s
C/ms). Analytical methods and more details of the equipment and procedure
used can be found elsewhere (1, 2, 3).
3. EXPERIMENTAL RESULTS AND CONCLUSIONS A. MODERATE TEMPERATURE. NONIMPREGNATED ALMOND SHELLS
Table I shows the overall yields obtained in the experiments carried out between 365 and 710 aC. The residence time of the vapours inside the fluidized bed is considered lower than 0.5 s, and the residence time of the vapours inside the reactor on the sand bed is around 3 s. The heating rate, according to Scott and Piskorz (4) is about 500 sC/s.
Liquid fractions present a maximum within the 420610 SC temperature
range, decreasing from 610 SC due to their cracking and reforming. The
high yields in liquids obtained (about 65% dry basis) are similar to those obtained by other researchers (5, 6 ,7).
The acetic acid yield is around 10% (dry basis) within the 440610 aC temperature range. This yield is significantly higher than those reported by other authors working with other biomasses, typically around 7% from hardwood and 7.84% from beechwood (5 ,8).
A kinetic study has also been carried out (3). Two schemes of reactions have been considered:
Scheme I (biomass, Β) » a(gases, G)+b(liquids, L)+c(solids, S) 1
w gases, G Scheme II (biomass, Β)^2^liquids, L (g)
^"^ solids, S

232
Assuming first-order reactions, it has been tested that similar expressions can be obtained from both schemes.
On considering the expresions deduced, the correlation of the yields of the total of the gases, the total of the liquids and the solid residue to the schemes suggested is good. The expressions obtained for the kinetic constants in the 400-460 *C temperature range are the following:
k = 7.11.107exp(-32.0/RT) s _ 1
k = 1.49.108exp(-35.1/RT) s"1
k = 14.73exp(-10.8/RT) s - 1 (R in Kcal/molsK)
B. MODERATE TEMPERATURE. ALMOND SHELLS IMPREGNATED WITH CoCl Samples of almond shells impregnated with different catalysts have
been pyrolyzed in an Analytical Flash Pyrolysis Apparatus (AFPA) at 440 aC. From previous results (1), it was concluded that AFPA was a suitable device for experiments to select catalysts and operating conditions. Different catalysts have been tested: KCl, NaCl, BaCl , CaCl , CdCl , MnCl , CoCl , CuCl , NiCl , CrCl and NaOH, NiSO , CoSO . An increase in 2-furalcfe hyde yield can be observed when increasing the acidity of the cation. A re^ markable increase was obtained for the cations of intermediate acidity Cd , Mn and Co . An increase of hydroxyacetone yield with respect to non-catalytic pyrolysis of almond shells was observed with NaOH (basic catalyst).
From the screening carried out, three catalysts (NaOH, MnCl and CoCl ) were selected for study the influence of the catalyst to almond shells ratio, using the AFPA. An increase of some yields has been observed when increasing the amount of catalyst impregnated at 440 2C. With NaOH (3.7 g of NaOH/100 total g of almond shells+NaOH), the yield of hydroxyacetone is 1.3%. With MnCl (14 g MnCl /100 total g) and with CoCl (13.5 g/100 total g), the yields of acetic acid and 2-furaldehyde are about 7-8%.
CoCl was selected for the study of the pyrolysis in the fluidized bed reactor, Because this catalyst allowed a better control of the homogeneity of the impregnation step due to the blue color acquired by the dried almond shells impregnated previously with CoCl .
Table II shows the results obtainea in a series of experiments carried out at 500 2C in a fluidized bed reactor and with different catalyst to almond shells ratios, including the results corresponding to almond shells without catalyst.
A similar variation of the composition of the liquids obtained, compared to that obtained in the test of catalyst selection can be noted. Acetic acid yield was around 7% when using CoCl , as compared with 9.8% when pyro-lyzing without catalyst. On the other hand, 2-furaldehyde yields increased up to 7.5% (at 14.1% of CoCl ) when increasing the catalyst to almond shells ratio, as compared to 0.57% when no CoCl was used. Due to this, the amount of organic liquids analyzed increased up to 16.9% when almond shells were impregnated with 14.1% of CoCl .
A slight increase in the water yield due to the presence of CoCl can also be noticed, although this yield remains almost constant when increasing the catalyst/almond shells ratio. With respect to the rest of the components analyzed, the absence of compounds such as acetaldehyde, hydroxyacetone, isoamyl alcohol and l-hydroxy-2-butanone, and an increase in the yields of methanol+formaldehyde, acetone, 2-propanol and propionic acid, are notable when pyrolyzing with 14.1% of CoCl .
This liquid distribution is typical of the acidic catalysts, since the latter favour the dehydration reaction versus the fission reactions

233
(10). Furthermore, we observed a decrease in tar yields (dry residue at 120
?C ) , which is also typical of this type of catalysts (11, 12). With regard to the gases, no significant variation in the composition
of the gas obatined was observed. Nevertheless, a decrease in the yield of gases as well as an increase in the solid residue yield can be noted.
In order to study the influence of the temperature a series of experiments at 410, 425, 445, 470, 500 and 610 = c was carried out with almond shells impregnated with 14.1% of CoCl . Table III shows the operating conditions and the results obtained in this series of experiments. The following can be observed:
A decrease in solid residue occurs when the temperature is increased, but the drop is not as marked as that observed when pyrolyzing without catalyst (1). This fact may be due to two opposing effects. On one hand, the solid decomposition occurs to a greater extent when the temperature is increased but the presence of CoCl favours the reactions of condensation leading to the formation of carbon flO).
The liquid fraction remains almost constant within the 425610 SC
temperature range, probably due to the fact that the primary reactions are almost completed, and to the small residence time which produces almost no decomposition of the condensable volatiles.
With respect to the liquids analyzed, it can be observed that the yield of acetic acid decreases by 30% at 500 sc when using CoCl , whereas the yield of 2furaldehyde increases up to 1000% within the 425610
SC
temperature range, when using CoCl . Thus, when pyrolyzing almond shells impregnated with CoCl at 500 *C it is possible to obtain a yield of acetic acid around 7.2%. This is similar to values obtained by other researchers working with other biomass without catalyst. Goldstein (8) reported around 7% from hard wood while Beaumont and Schwob (5) obtained around 7.84% from beech hardwood. In addition, a yield of 7.5% of 2furaldehyde, which is very high for a pyrolytic process and similar to that obtained by the conventional methods used for obtaining this compound by acid hydrolysis of lignocellulosic materials can be obtained by using CoCl as catalyst.
Total recovery of the CoCl , from the charcoal formed at 445 sc, is achieved by extraction with a solution of HCl under the following conditions: concentration of HCl lmol/L, extraction time 17 h, ratio g extractant solution/g charcoal 33 and temperature 46 sc.
From the kinetic study carried out in the 410500 ?C temperature range, with almond shells impregnated with CoCl (14.1%), the following expressions for the firstorder reactions of tne scheme II previously presented have been obtained:
k =8.93exp(13.29/RT) s _ 1
k2=0.097exp(4.28/RT) s 1
k3=0.0055 s 1 (R in kcal/molSK)
C. HIGH TEMPERATURE. ALMOND SHELLS NO IMPREGNATED By high temperature pyrolysis around 820900 =C the following yields
(% weight) have been obtained (2): 0.7 1.6%H , 2546% CO , 1528% CO , 58.6% C H , 0.70.8% C H , 2.34.2%C Η , 0.060.09% C H , 0.91.2% CH^, 0.30.5% C ^ 2 , 0.440.60% ^hydrocarbons* 47. 588.0% Tolal gas. 6
Most of the gas produced is formed by secondary reactions corresponding to the cracking of tars in the hot zone of the reactor on the fluidized sand bed (residence time of volatiles around 12 s).
On the other hand, by pyrolysis of the dry residue at 890 sc (obtained from the tar produced in the pyrolysis of almond shells at 610 sc, by distillation up to 120 sc), the following yields in gaseous products are ob

Table I. Constant temperature pyrolysis results
Temperature Solids Liquids
acetic acid 2-furaldehyde water
methanol+formaldehyde acetone 2-propanol hydroxyacetone l-hydroxy-2-butanone 3-methyl-l-butanol propionic acid acetaldehyde dry residue at
Gases CO CO C H4 H2
120 2C
365 69.2 21.1
0.42 0.77 10.6
0.33 0.04 0.01 0.79 0.59 0.67 --7.9 5.3 4.4
0.90 0.04 -
400 54.2 36.3
3.2 0.045 16.6
1.4 0.06* -
0.88 0.40 0.26 0.10 -13.0 8.1 6.0 1.9
0.13 0.09
420 47.0 41.4
3.5 0.46 16.4
1.0 0.14* -
0.92 0.52 0.33 0.13 -18.0 8.3 4.8 2.9
0.36 0.15
425 46.8 41.6
4.0 0.5b 16.7
1.5 0.16* -
0.85 0.50 0.41 0.15 -16.4 8.6 4.9 3.1
0.44 0.15
440 29.4 57.0
10.3 0.91 18.7
0.92 0.12 0.06 2.1 1.1
0.44 0.49 0.06 24.1 13.1 9.0 3.8
0.37 -
440 30.6 52.0
9.0 0.64 16.4
1.3 0.26 0.18 1.4 1.1
0.77 0.28 -20.6 13.3 9.0 3.7
0.47 0.09
460 27.5 56.0
9.0 0.67 17.7
1.1 0.25 0.33 1.5 1.2
0.81 0.33 0.01 22.3 14.2 9.9 3.6
0.61 0.10
495 26.4 58.5
10.0 0.75 16.7
0.56 0.08 0.05 1.9 1.4
0.41 0.10 0.10 27.0 14.4 10.2 3.6
0.61 -
495 26.3 57.2
9.6 0.57 17.7
1.1 0.07 0.06 1.9 1.3
0.90 0.17 0.21 23.0 15.7 10.3 4.7
0.70 -
495 26.4 59.3
9.9 0.40 16.7
1.2 0.06 0.04 1.7 1.2
0.78 0.15 0.24 26.1 14.3 10.0 3.8
0.50 -
610 11.3 65.1
10.4 0.85 16.5
0.70 0.25 _ 1.8 1.3
0.46 0.18 0.10 34.5 22.9 10.6 10.7 1.6 -
710 6.7
43.2
7.7 0.67 14.2
0.58 0.17 0.04 1.4
0.66 0.22 0.26 0.16 18.1 53.2 13.3 34.5 5.4
0.23 T o t a l 95.6 97.3 96.7 95.1 99.6 95.9 97.7 99.2 99.2 100 99.2 103
*acetone+2-propanol

Influence of the CoCl to
Catalyst temperature Solids Liquids
Major compounds acetic acid 2-furaldenyde water
Minor compounds methanol+formaldehyde acetone 2-propanol hydroxyacetone l-hydroxy-2-butanone 3-methyl-1-butanol propionic acid acetaldehyde
dry residue at 120°C 2-furaldehyde yield/added cation Gases
COCO2 CH4
almond
mol
shells
-495 26.4 58.3
9.8 0.57 17.0
0.95 0.07 0.05 1.8 1.3
0.69 0.14 0.18 25.4 -14.8 10.2 4.0
0.60
Table II ratio.
s II Yields (wt%) on
CoCl (3.0%) 500
28.1 54.6
7.4 3.3 23.6
0.50 0.36 0.78 --0.04 0.13 -15.4 143
13.9 9.1 4.0
0.80
moisture
CoCl2
500 37.9 45.9
6.5 5.2 21.1
0.70 0.15 0.37 ---
0.13 -11.7 106
12.7 8.1 4.1
0.54
free almond
(6.4%)
shell
CoCl, C
500 41.3 46.1
7.2 7.5 23.2
1.4 0.19 0.17 ---0.42 -5.7
69.2 12.4 9.3 2.4
0.60
(14.1%)
to
Total 99.5 96.6 96.5 99.8

Table III Influence of the temperature on the pyrolysis of almond shells impregnated with 14.1% of CoCl . Yields
(wt%) on moisture free almond shells. Fluidized bed reactor.
Temperature (aC)
Solids Liquids Major compounds
acetic acid 2-furaldenyde water
Minor compounds methanol+formaidenyde acetone 2-propanol hydroxyacetone l-hydroxy-2-butanone 3-methyl-l-butanol propionic acid acetaldehyde dry residue at 120SC
Gases CO CO
H2
410 57.0 34.6
3.7 6.1 19.4
0.61 --
-----4.8 4.4 4.1
0.23 0.05 0.03
425 46.3 42.8
5.8 7.7 22.7
1.3 0.13 0.07 -----5.1 6.7 5.3 1.1
0.33 0.02
445 45.2 42.3
5.5 6.9 23.2
1.0 0.14* -
--
0.09 --4.2 7.5 5.0 2.0
0.41 0.09
470 44.7 42.1
5.5 6.9 24.2
0.69 0.18 0.08 ---
0.07 -4.5 7.7 4.8 2.3
0.45 0.14
500 41.3 46.1
7.2 7.5 23.2
1.4 0.19 0.17 ---0.42 -5.7 12.4 9.3 2.4
0.60 0.16
610 24.8 42.8
6.4 7.2 21.5
0.34 0.24 0.35 ---0.44 -6.3 26.7 17.5 7.8 0.86 0.55
to σ\
Total 96.0 95.8 95.0 04.5 99.8 94.3
*acetone+2-propanol

237
tained: 0.88% Η , 20.9% CO, 22.8% CO , 6.1% CH , 0.37% C H , 3.0% C H , 0.02% C H , 0.37% C H , 0.18% C Η and 0.1% C hydrocarbons. These yields are expressed in wt% on a dry residue basis.
In order to study the influence of different catalysts on the hydrocarbon yields from high temperature pyrolysis, a set of experiments with almond shells impregnated with different organic chemicals (NaOH, NaCl, KCl, CaCl , BaCl , MnCl , ZnCl , CuCl , NiCl , CoCl , CrCl , NiS04) were carried out using the ATPA '(Pyroprobe 100) at 850 ?C (2). A general decrease can be observed in hydrocarbon yields with respect to the pyrolysis of almond shells without catalysts. On the other hand, no significant change has been observed in the composition of the hydrocarbons.
REFERENCES
(1) Font, R., Marcilla, Α., Verdú, E. and Devesa, J. (1986) FluidizedBed Flash Pyrolysis of Almond Shells. Temperature Influence and Catalyst Screening. Ind. Eng. Chem. Prod. Res. Dev., 25, 491.
(2) Font, R., Marcilla, Α., Devesa, J and Verdú, E. (1988) Gaseous Hydrocarbons from Flash Pyrolysis of Almond Shells. Ind. Eng. Chem. Res. 27, 1143.
(3) Verdú, E. (1988) Thesis Dissertation. Pirólisis de Cascara de Almendra. Universidad de Alicante.
(4) Scott, D.S. and Piskorz, J. (1984)The Continuous Flash Pyrolysis of Bio mass. Can. J. Chem. Eng., 62, 404.
(5) Beaumont, 0. and Schwob, Y. (1984) Influence of Physical and Chemical Pa rameters on Wood Pyrolysis. Ind. Eng. Chem. Process Des. Dev. 23, 6377
(6) Finney, C S . and Garret D.E. (1974) Flash Pyrolysis of Solid Wastes. E nergy Sources, 1, 295.
(7) Sass, A. (1974) Garret's Coal Pyrolysis Process. Chem. Eng. Prog., 70, 72.
(8) Goldstein, I.S. (1981) Organic Chemicals from Biomass. CRC. Boca Raton Fl., Chapter 5.
(9) Thurner, F. and Mann, U. (1981) Kinetics Investigation of Wood Pyrolysis, Ind. Eng. Chem. Process Des. Dev., 20, 482.
(10) Shafizadeh, F. (1975) Industrial Pyrolysis of Cellulosic Materials, Ap plied Polymer Symposium, John Wiley & Sons, 28, 153.
(11) Smicek, S. and Cerny, C.T. (1970) Active Carbon, Elsevier Pubi. Co., Amsterdam.
(12) Gray, M.R., Corcoran W.H. and Gavalas, G.R. (1985) Pyrolysis of a Wood Derived Material. Effects of Moisture and Ash Content, Ind. Eng. Chem. Process Des. Dev., 24, 646.

238
BIOMASS PYROLYSIS IN MOLTEN SALTS FOR FUEL PRODUCTION
J.K.Maund and D.M.Earp Department of Chemical Engineering & Applied Chemistry, Aston University, Birmingham, B4 7ET, United Kingdom.
SUMMARY Molten alkali metal salts are recognised as having considerable potential as high temperature liquid phase media lor the pyrolysis, gasification, combustion and catalytic conversion of organic chemicals. A two litre capacity molten salt system for the pyrolysis of biomass is described. The heating rate of wood, in both air and molten salt, has been studied and compared, and has been found to be between four and ten times faster in molten salt. The pyrolysis of wood in both molten Li/Na/K carbonate and Na/K hydroxide eutectics has been studied and the results of these studies are presented. The hydroxide melt system is favoured as it produces good purity hydrogen. However, the melt system becomes contaminated with carbonate and organic residues and the hydroxide, being consumed, requires regeneration; preliminary studies indicate this to be feasible.
1. INTRODUCTION Molten salt processes are already well established, particularly in the
metallurgical industry, where they are used for thermochemical and heat treatment and for cleaning (1). More recently, molten alkali metal salts, in particular, have been recognised as having considerable potential as high temperature, liquid phase media for the pyrolysis, gasification, combustion, or catalytic conversion of organic materials such as biomass, coal and a variety of waste products. A number of examples, from laboratory scale to demonstration plants, have been developed throughout the world for coal gasification (2)(3)(4); biomass and waste gasification (5) and combustion (6); and plastics pyrolysis (7).
Fused metal salts as reaction media for the thermal conversion of biomass have a number of advantages over conventional gas phase processes:
(i) Chemical stability in the liquid phase at temperatures up to 1000°C; (ii) Appropriate physical properties of high thermal conductivity, low
viscosity, low volatility, and appropriate melting point temperature; (iii)The ability to dissolve biomass into the liquid phase, so that heat and
mass transfer become homogeneous and, therefore, very rapid; (iv)The possibility of homogeneous catalysis or chemical reaction to control
the chemical composition of the products. The Department of Chemical Engineering and Applied Chemistry at Aston
University has been carrying out a 2-year contract with the European Community continuing its research into thermal processing of biomass in molten salts (8)(9). This paper summarises the experimental work performed to date within the contract . Molten salt processing has been studied for some ten years in the Department, and biomass conversion for almost as long, with the thermochemical processing of biomass in molten salts first being studied in 1980 (10).

239
2. EXPERIMENTAL EQUIPMENT The experimental equipment has been described in detail before (11)(12). It
consists of a two litre capacity stirred tank reactor into which the biomass is fed by a sealed laboratory scale screw feeder. Vapour leaving the reactor is cooled by water-cooled heat exchangers which condense the liquid products, which are then collected for analysis. The product gas is burned in a flare. The reactor is heated by a purpose built 3 kW electric resistance furnace, with a three-term electronic temperature controller.
The product gas composition is continuously monitored for H2, CO, CO2 and CH4 using dedicated gas analysers and recorded, at about 10 second intervals, by a micro-computer based data-logger, developed within the Department for use with an open-core downdraft gasifier (13). Reactor pressure is measured using a water manometer, product gas flowrate by rotameter and total volume of gas produced by gas meter; these variables, along with the temperature of the salt bath, are recorded manually.
To prevent the ingress of air into the system and to provide a 'carrier' for the gas analysis system, the rig is operated with a nitrogen purge. This purge is introduced at three points, the stirrer, the feeder and the reactor, the flow of each being metered separately.
3. EXPERIMENTAL WORK The experimental work so far performed has covered three main areas,
particle heating rate tests, pyrolysis in the molten lithium/potassium/sodium carbonate eutectic and pyrolysis in the potassium/sodium hydroxide eutectic.
3.1 Heating Rate Before the experimental rig was finally piped up and closed, a number of
experiments were carried out, firstly to commission the furnace and its temperature controller, and secondly to investigate the heating rate of wooden dowels of different sizes in molten salts at different temperatures and to compare these with rates in air at the same temperatures. These experiments were to enable the relative heat transfer characteristics of the two media to be evaluated.
In order to obtain a reasonable range of temperatures for this work, the sodium/potassium hydroxide eutectic (51 mole % KOH/49 mole % NaOH, melting point 193°C) was used, allowing a temperature range of 250 to 500°C. However, because of the risk of combustion the runs in air were only performed at 250°C. Dowels of 6, 9,12,15 and 22 mm diameter were used, each cut to a length equal to its diameter. The temperature at the centre of the dowel was monitored by drilling a small hole into the centre and fitting it with a chromel/alumel type thermocouple. The temperature history was recorded using the data logging system at frequencies of up to two readings per second. When the dowel temperature had reached the furnace control temperature, it was removed from the furnace.
3.2 Pyrolysis In Molten Alkali Carbonate Eutectic For the initial pyrolysis runs, it was decided to employ the Li/Na/K carbonate
eutectic (43.5 mole % IJ2CO3/31.5 mole % Na2CO3/25.0 mole % K2CO3, melting point 397°C) as this was both non-corrosive and had been used successfully in tyre and plastics pyrolysis at Aston (14)(15). A salt temperature of 500 °C was chosen,as this was the midpoint for the proposed experimental programme range (12). To prevent "overloading" of the melt, a low feed rate was selected. This was initially about 8.4 g/min continuous feeding, although due to operational

240
difficulties (see Section 4), was later reduced to a rate of 4.9 g/min in one minute cycles (1 minute on/1 minute off). The feed material used consisted of in-house manufactured pine 'chips' (without bark) sieved to the size range 0 to 4.75 mm.
3.3 Pyrolysis In Hydroxide Eutectic As a result of difficulties encountered with the carbonate eutectic (see
Section 4), it was decided to employ an alternative salt system. Previous experience at Aston (15) and simple solubility tests indicated that the K/NaOH eutectic, as used in the heating rate experiments (see Section 3.1 above), was a solvent for wood, so it was decided to employ this salt. The bath temperature was set at 300°C. This temperature was chosen as it was about the midpoint of the solubility range for wood, this range of temperatures having been identified in a series of simple solubility tests. A low feed rate of 4.9 g/min was selected in order to prevent "overloading" of the salt, with the feeder being operated both continuously and semi-continuously, as in the carbonate runs. The same feed material as for the carbonate runs was employed.
4. RESULTS AND DISCUSSION
4.1 Heating Rate A summary of the results of the heating rate work is presented in Figure 1.
These show that the particle heating rate in the molten salt was, as anticipated, more rapid than in a gaseous environment. The heating rates measured in molten salt were between four and ten times faster than those measured in air. This is in close agreement with the results of Tada and Yasunishi (16)(17) of Tottori University. It was also observed that in the alkaline melt the cellulose core of the wood was dissolved leaving a honeycomb-like structure of lignin.
4.2 Pyrolysis in Molten Alkali Carbonate Eutectic Each run performed with this system had to be terminated after
approximately 20 minutes due to a 'back-up' of feed in the feed inlet pipe up to the feeder. On each occasion when the rig was stripped down after a run, it was found that the surface of the melt was covered in a layer of char. On top of this char layer there were layers of progressively less pyrolysed feed, until in the feeder tube itself, fresh unreacted feed was found. Initially, it was felt that either the feed rate was too high for the salt system and was therefore 'overloading' the melt or there was insufficient agitation to provide adequate mixing of the salt and feed. However, when these problems persisted at high agitation rates and low feed rates, it was concluded that the molten salt was merely acting as a heat sink causing the biomass to undergo conventional pyrolysis without any solution. This theory was supported by simple solubility tests. Calculations based on previous pyrolysis work at Aston (19) indicated that both the rate of pyrolysis and particle heating rate were rapid at the conditions prevailing in the reactor.
It was decided that the carbonate salt system was unsuitable for the present reactor arrangement when operating in the pyrolysis mode. However, it was concluded that it could be operated as a gasifier, but such a study does not lie within the scope of this project.

241
10°
10u
15 mm Air • 15 mm Salt A 12 mm Salt
20 40 60 80
% Ultimate Temperature 100
Figure 1 Dowel Heating Times in Air and Molten Salt The products formed in the runs using the carbonate melt system were
typical of conventional pyrolysis and, as well as the char consisted of: a thick smoke of fine tar particles. This was difficult to condense and to disentrain from the gas stream. However, some was disengaged from the gas, both in the condensers and the gas rotameter, and proved to be reasonably fluid and readily soluble in acetone. gas, at very low yields, this consisting primarily of CO and C02 (up to 95 %), the balance being CH4 and H2. As some of the feed material added to the reactor was only partially
pyrolysed the mass balances and product yields could not be calculated.
4.4 Pyrolysis in Hydroxide Eutectic A number of successful runs, of up to one hour in duration, have so far been
performed using a hydroxide melt system. In this case the wood fed to the reactor dissolves in the melt to yield gaseous, liquid and solid products.
The gaseous product formed, consists of approximately 98% hydrogen, the balance being mainly methane. The gas evolution was vigorous and resulted in the melt foaming, with a volume increase of approximately 25%. This foaming caused a blockage in the gas outlet line, due to frozen melt, which required minor redesign (11) to prevent its recurrence.
The liquid product formed, consists of 97-99% water, the balance being an organic product, which, due to its low yield and concentration, has not yet been identified. This liquid is also alkaline, probably due to droplets of hydroxide entrained in the product gas which dissolve in the condensate. The gas and liquid products formed are very similar to those found in previous work at Aston on small scale batch pyrolysis and solution of wood in molten hydroxides (10).

242
The work of I rédale and Hatt (10) also showed that the composition of the product gas and the yield of gas were strongly dependent on the melt temperature. This is currently under investigation to determine the optimum operating temperature.
The majority of the carbon in the feed is not liberated as part of the gaseous or liquid products, but is retained in the melt either as an organic residue, which may consist of formate or ethanoate ions (10), or by reaction with the hydroxide to form carbonate. Melt with these 'impurities' is known as 'fouled'. Figure 2 illustrates the effect of the amount of wood pyrolysed on the hydroxide and carbonate levels in the melt. The colour of the melt and of its aqueous solution, also vary with the quantity of wood pyrolysed. Colours vary from that of the fresh melt (which is white when solid and colourless when liquid) with a colourless solution, via a pale yellow, of melt and solution, to a dark brown coloured melt and solution. The light absorption, of the solution is directly proportional to the quantity of wood pyrolysed, with a characteristic absorption at 260 nm. The colouring of the melt is probably due to the organic residue, although the fraction of this residue in the melt has not, as yet, been reliably quantified.
40
SP 'δ
ε o υ
30-
20
3 10
100 200 300
Wood Added (gramme) 400
Figure 2 Hydroxide and Carbonate Content of Melt Against Weight of Wood Pyrolysed
High levels of carbonate and organic residue in the melt lower its effectiveness as a reaction medium. The hydrogen yield drops as more wood is pyrolysed (see Figure 3) and the melt becomes more fouled. There is also evidence to suggest that the wood solution becomes saturated, this being shown by filtration of an aqueous solution of the melt. When small quantities of wood have been pyrolysed (less than 50 g wood/ kg of melt) little or no solid filtrate is produced. However, as the quantity of wood pyrolysed increases, a filtrate of fine char particles is obtained, the number and size of these increasing with the quantity of wood pyrolysed.

243
350
4> ε ε
■e ■3 ¡5 s MD ε
■β >> s
300
250
200 100 200 300
Wood Added (gramme)
400
Figure 3 Hydrogen Yield Against Weight of Wood Pyrolysed This phenomenon indicates that the solution of the wood is being inhibited, so it pyrolyses, with little or no solution, leaving a char residue.
In order to utilise the same melt in a continuous process, it would be necessary to regenerate the melt, for instance, by reaction with steam (20):
CO3" (I) + H20 (g) ^ OH" (I) + C02 (g)
Attempts to carry out this reaction in the molten salt at up to 750°C with a water partial pressure of 0.3 bar showed it to be very slow. There is, however, evidence that it takes place in the presence of live steam at a somewhat higher temperature. This is an area for further investigation.
It has already been shown that the melt may be partially regenerated simply by heating. Fouled melt was heated to a temperature of 400°C and yielded a gas containing hydrogen and methane. This gas production was considered to be due to the breakdown of the organic residue in the melt.
Hatt and Iredale (10) suggested the following overall reaction for wood in the molten hydroxide eutectic:
C6H904 (wood) + 12 OH" + 2 H20 6 C o f * 12.5 H2
However, this is a simplification of the process and the range of products indicates that the actual process is more complicated. A more realistic appraisal of the reactions occurring may be represented by the routes they proposed for the molten salt pyrolysis of cellulose (see Figure 4). Again, however, this is a simplification, as it does not consider the other main constituents of the wood (that is lignin and hemicellulose). The range of products formed could be explained by considering the breakdown of these other components and, in particular.lignin.

244
Cellulose
i S ace hari η ic Acids-
I 1,2 - epoxides 'poxioes 1
κ C H 4 + 3 H 2 < 6C Fl
shift I shift 11 6 CO + 6 H 2 5 CO + C H 4 + 3 H 2 < 6C Fragments
pyrolysisJ
12 H 2 + 6 CO§~ 5 C0|"+ C H 4 + 8 H 2 H 2 / CO§"
Figure 4 Proposed Routes for the Pyrolysis of Cellulose in Molten Sodium/Potassium Hydroxide (10)
5. CONCLUSIONS There is considerable evidence that molten salts are potentially valuable
media for the conversion of biomass into useful fuels and chemicals. The work performed in this project and by other groups has demonstrated the technical feasibility of this type of process.
The salt employed has been demonstrated to have an important influence on the process and hence the products. The salt type determines whether it acts simply as a heat source or as a chemical reagent. Previous work at Aston (21 ) has demonstrated the use of molten salts as liquid phase catalytic reaction media. It should, therefore, be feasible to select a suitable salt and catalyst system to produce specific products.
The sodium carbonate melt acts as a heat source causing the wood to undergo 'normal' pyrolysis, although at higher heating rates than conventional pyrolysis systems which employ a gaseous heating medium. The suitability of this system for gasification and combustion has already been demonstrated, its particular advantage being its ability to absorb contaminants, such as sulphur.
The hydroxide melt system produces hydrogen of high purity and, therefore, has considerable potential to provide hydrogen either as a fuel gas or as a chemical reagent, for instance for the upgrading of pyrolytic oils. A potential problem of this salt system is the fouling of the melt, with carbonate and an organic residue, requiring the melt to be regenerated. However, there is evidence to suggest that this may be feasible by reaction of the fouled melt with steam. Partial regeneration of the melt to recover hydrogen and methane by heat treatment has already been demonstrated. This heat treatment is believed to drive off the organic residue remaining in the melt.

245
REFERENCES (1) Anon., "Guidelines for Safety in Heat Treatment - Part 1 Use of Molten Salt
Baths", The Wolfson Heat Treatment Centre, Aston University B'ham, (1981) (2) Chong V M, "Coal Gasification Process", US Patent 3 770 399, (Nov. 1983) (3) Susie M et al., "Coal Gasification in Molten Salt", Glas Sriska Akademija
Naukai Odelene Prirodus Matematickih 48 (1981) 27. (4) Cover A E & Schreiner W.C., "The Kellogg Molten Salt Process", Energy
Communications 1/2, (1975) 135. (5) Yosim S J & Barclay Κ M, "The Gasification of Wastes Using Molten Salts",
Preprint, ACS Division of Fuel Chemistry 21/1 (1976), 73. (6) Moffat J M & Kohl A L, "Gasification of Wood in a Bath of Molten Alkali
Carbonates", in "5th Canadian Bioenergy R&D Seminar", ed. Hasnain S, Elsevier Applied Science (1984).
(7) Bertolini G E & Fontaine J, "Value Recovery from Plastics Waste by Pyrolysis in Molten Salts", Conservation & Recovery 10/4 (1981) 27.
(8) Maund J K, Bridgwater A V & Smith E L,"Thermal Conversion of Biomass in Molten Salt Media",'Energy from Biomass 1', G Grassi & H Zibetta (eds), Elsevier Applied Science, London (1987)
(9) Maund J K, Bridgwater A V & Smith E L.'Thermal Conversion of Biomass in Molten Salts", '4th EC Conference - Biomass for Energy & Industry', Orléans, France, (May 1987)
(10) Iredale PJ and Hatt BW, "The Pyrolysis and Gasification of Wood in Molten Hydroxide Eutectics", in, "Fundamentals of Thermochemical Biomass Conversion", Overend RP, Milne TA an<-" iVludge LK (eds), Elsevier Applied Science, London (1985).
(11) Earp DM and Maund JK, "Thermal Conversion of Biomass in Molten Salt Media", Third EEC Progress Report - January to September 1988.
(12) Earp DM and Maund JK, "Thermal Conversion of Biomass in Molten Salt Media", paper presented at, "Energy from Biomass - EEC Contractors Meeting", Paestum, Italy (1988)
(13) Earp DM, "The Gasification of Biomass in a Downdraft Reactor", PhD Thesis, Aston University (1988)
(14) Venning NJ, "Fuels from Tyres by Pyrolysis in Molten Salts", PhD Thesis, Aston University, (1981)
(15) Pitt MJ, "Pyrolysis of Plastics in Molten Salts", MPhil Thesis, Aston University, (1979)
(16) Maund JK and Earp DM, "Fuels from Biomass by Conversion in Molten Salts", pp. 542-556 "Research in Thermochemical Biomass Conversion", eds. A V Bridgwater & J L Kuester, Elsevier Applied Science, (1988)
(17) Yasunishi A & Tada Y, 'Wood Pyrolysis in Molten Salt', Kagaku Kogaku Ronbunshu, 11/3, (1985) 346.
(18) Tada Y & Yasunishi A, 'Wood Pyrolysis with Molten Salt as Heating Medium', Kagaku Kogaku Ronbunshu, 13/3, (1987) 376.
(19) Smith EL, Private Communication (20) Maund JK, "Thermal Conversion of Biomass in Molten Salt Media", paper
presented at Institute of Energy Conference: "Gasification - Status and Prospects", Harrogate, UK, (May 1988)
(21) Al-Muslih E F, "Oxidation of Organic Compounds in Molten Salts", PhD. Thesis, Aston University, (1984)

246
FLASH-PYROLYSIS OF SULCIS COAL
L. Conti and G. Scano Dipartimento di Chimica, Universita1 di Sassari
Summary F l a s h - p y r o l y s i s of an i t a l i a n coa l ( S u l c i s c o a l ) has been i n v e s t i g a t e d in f l u i d i z e d bed p y r o l i s e r in n i t r o g e n or methane atmosphere and between 600 and 900 °C. The maximum t a r y i e l d s i s ob ta ined a t 600-620 °C. The p y r o l y s i s o i l can be upgraded a f t e r w a r d s . Experiments in methane atmosphere suppor t a p o s s i b l e r a d i c a l i c mechanism for the r e a c t i o n .
1. INTRODUCTION Continued reduction of crude oil and natural-gas reserves
stimulates the interest of many research groups in coal conversion to gaseous or liquid products, which can represent new raw material for the chemical industry.
In our laboratory, research on the utilization of Sulcis coal as an alternative to direct combustion was started a few years ago1-2
; in this connection, we began to study fluidized bed flash-pyrolysis of this coal.
In the present communication, we report some of the results obtained in this research. 2. RESULTS AND DISCUSSION
Pyrolysis experiments were conducted in a fluidized bed stainless steel pyrolyser able to heat coal powder at 10^ C/sec with short residence time (0.3 sec)3. in this way, the tars undergo only a partial decomposition and can be quickly removed.
Table 1 reports the approximate and ultimate analysis of Sulcis coal : Tab. 1 Analysis of Sulcis coal Proximate analysis Ultimate analysis Moisture (wt%) 7.4 C (wt% maf) 68.2 Ash (wt% mf) 10.9 H (wt% maf) 4.8 Volatile matter (wt% mf) 45.3 Ν (wt% maf) 1.7 Fixed carbon (wt% mf) 43.8 S (wt% maf) 8.7
Fig. 1 reports the yields of total volatile matter, tar, gas and hydrogen sulphide against temperature in nitrogen atmosphere; Fig. 2 reports the tar-component yields (ASTM

247
A Hydrogen sulfide O C1-C3 hydrocarbons Π Tars • Volatile matters
Τ — ι — ι — ι — Γ soo 6βο τ ο β eoe 9oa I D O O
Fig . 1. Trends of product y i e l d s versus pyro ly s i s temperatures
m —
τ — ι — ι — Γ B O O e o o T O O e o o 9 0 0 1 0 0 0
Fig . 2. Yields of l i g h t o i l s and carboids versus temperature

248
270075), relative to the original coal, plotted against temperature.
As can be noted, a maximum tar yield is obtained at 600620 °C; the pyrolysis oil presents a satisfactory H/C ratio and sulphur content and can be upgraded afterwards''.
The characteristics of the tar obtained at 600 °C and the composition of gaseous hydrocarbons are listed in Tables 2 and 3. Tab. Characteristcs of tar Tab. 3 Hydrocarbons C|Cz,
C (wt%) H (wt%) Ν (wt%) S (wt%) Asphalt. Preasphalt. H/C
(wt%) . (wt%)
The trends mechanism : 3
ob
75.2 7.7 1 .0 3.6
41.2 32.3 1 .2
served
CH4 C2H4 C2H6 C3H6 C 4H 8 C 2H 2
(wt%) (wt%) (wt%) (wt%) (wt%) (wt%)
30.5 26.7 9.5
22.8 10.5
traces
in Fig. 2 suggest the following
Coal
primary light oils
sem
carboids ^
ils «««^^ ^^^ second
icoke ^~ ^ residu
ary gas
e
secondary light oils primary gas
chars The coal decomposes essentially with a radical mechanism.
The volatiles formed include unreactive substances as well as reactive ones (carboids). Especially in the intial stages of decomposition, when free radicals are likely to exist, the unreactive volatiles (primary light oils and gases) escape into the atmosphere. Only a part of the reactive volatiles escape, while the most of them polymerize giving a semicoke; or decompose into secondary light oils.
In turn the semicoke reacts to gives gas and a solid residue. The breakdown of the primary light oils, and of the secondary ones perhaps, is noticed at higher pyrolysis temperatures (600750 *C), as well as an increase in carboids. The decrease of carboids in the oil produced at 900 °C can be explained by quick decomposition.
A further check of the validity of such mechanism was provided by some tests carried out in methane atmosphere. In our experimental temperature range, methane cracking gives a high concentration of free radicals5. These react with the radicals deriving from coal pyrolysis, particularly with the carboids that are thus quenched, hindering their decomposition into semicoke and secondary light oils.
Infact, we observed a greater tar production but especially a variation in composition (light oils/carboids ratio) in methane than nitrogen atmosphere, as we can be see

249
in Tab. 4.
Tab. 4 Tar yields at different temperatures (wt % on coal m.a. f. )
Temp .
Tar Light oil Carboid
450
N2 CH4
600
N2 CHz,
750
N2 CH4
13.5 8.7 19.521.0 15.2 20.2 6.8 3.6 5.0 4.1 2.0 1.8 6.7 5.1 14.5 16.9 13.2 18.4
880
N2 CHz,
10.8 11.9 4.6 1.5 6.2 10.4
Light oil/carboid 1.01 0.70 0.3 0.24 0.15 0.10 0.74 0.14
References (1) C. Botteghi, L.Conti and R. Mansani, Boll. Soc. Sarda
Sci. Nat.; 19, 27 (1980) (2) L. Conti, C. Botteghi and R. Rausa, Tecnologie Chimiche,
4, 38 (1981) (3) L. Conti and R. Rausa , Fuel Processing Technology,
17, 107 (1987) (4) S.C. Che, S.A. Quader and E.W. Knell, in "Refining of
Syntetic Crudes". M.L. Gorbaty and B.M. Harney, Editors, Am. Chem. Soc. Washington, D.C. (1979)
(5) D.B. Anthony and J.B. Howard, AICHE Journal; 22, 625 (1976)

250
RAPPORTEURS REPORT ON SESSION IV PYROLYSIS CASE STUDIES
D J O'Neil Georgia Tech Research Institute Georgia Institute of Technology
Atlanta Georgia 30332
USA
Session 4 (Pyrolysis Case Studies) consisted of five papers, with two papers withdrawn. The Session was chaired by Dr G Grassi assisted by Dr A V Bridgwater.
1 . Sugars from Cellulosics by the Waterloo Fast Pyrolysis Process Dr Piskorz Dr Piskorz of the University of Waterloo (Canada) reported the experimental
results achieved for the production of anhydrosugars and other sugars via atmospheric flash pyrolysis in a fluidised bed reactor. Pretreatment of the lignocellulosic feedstocks was a preferred method prior to pyrolysis in order to maximise the conversion of the cellulose fraction to fermentable sugars. Conversion efficiency of cellulose to fermentable sugar of 72% was reported. Sugars were isolated in a water-soluble extract of pyrolyzate with the non-water-soluble fraction producing a "pyrolytic lignin" fraction. Pretreatment with dilute sulphuric acid had yielded hemicellulose derivatives. Success was claimed in fermenting the sugar solution to ethanol but no details were provided on conversion efficiency, ratio, or the need to acclimatise fermenting agents (e.g. baker's yeast).
A question addressed the method of analysis/characterisation which was used to identify laevo-glucosan which had been cited as a key pyrolysis product. It was understood that, while l-glucosan had often been cited as a precursor to formation of cellulose, it had often escaped detection, at least in earlier studies on the structure of cellulose. Dr Piskorz explained that, while GCMS and FTIR were methods of analysis that could be used, he had used standard samples of glucose (and other sugars) and had used HPLC to correlate the elution times of the standards with the pyrolysis components. Some of the sugar standards were also synthesised in his laboratory and were used for HPLC correlation. Additionally, the l-glucosan was isolated, converted to glucose via hydrolysis and the glucose product was fermented to ethanol.
The question was raised of the cost of production of sugars via the flash pyrolysis process in relation to the citation that Brazilian studies had been unsuccessful in producing sugars from lignocellulosics at competitive rates. Dr Piskorz replied that they had estimated the cost of sugar production via the pyrolysis process to be $0.10-$0.15/kg. It was noted that current costs of raffinate sugars were $0.06-$0.08/kg and that his results were not attractive. Dr Piskorz cited the fact that he was reporting experimental results and that his costs were preliminary.
It would appear that the production of fermentable sugars would be at a disadvantage vis-a-vis the well-established prehydrolysis and dilute hydrolysis processes which have been developed in the 1980's which retain a higher yield of fermentable sugars and low levels of toxifying compounds and for which hexose

251
production costs have been reported at $0.07-$0.08/kg. The Waterloo researchers claim only an advantage over enzymatic hydrolysis which has the greater potential for maximal production of fermentable sugars from both the hemicellulose and cellulose fractions assuming further reductions in conversion times. Several questions remain to be answered before a cost-effective biomass conversion process could be economic: cost of acid, disposal of acid solutions, degradation ol hemicellulose and loss of volatile derivatives (furfural-based), fermentation inhibition (furural, hydroxymethyl furfural, phenols, etc), mass market for "pyrolytic lignin" residues etc.
2. Production of Benzols and Active Carbon by Pyrolysls with Simultaneous Post-cracking of Waste Rubber and Plastic Material Dr B Bettens Dr Bettens reported on a "dual pyrolysis" process for the conversion of waste
tires (rubber) and plastics, with emphasis on the former - a whole tire pyrolysis process involving two pyrolytic stages, viz (i) depolymerisation (actually decomposition since a monomer is not the primary product) at a temperature range of 400-500°C, and (ii) a higher temperature cracking process at 700-800°C. Olefin production is characteristic of the first stage and "aromatisation" to BTX (benzene, toluene xylene) production is characteristic of the second stage. Bettens reported mass balance and temperature dependence for the principal product groups, i.e. solid (char), liquids and gases. The liquid phase composition could include 60-70% of BTX compounds (naphthalene and higher fused aromatics could be observed). A simple depolymerisation and aromatisation reaction mechanism was presented. The effects of temperature and process conditions (including flow rate) were examined on depolymerisation and BTX formation.
A feasibility study for a 2000 t/d plant (c.1984) was described and a plant investment of BF 30.4 million was reported (22 million for capital equipment, 0.8 million for assembly, 0.6 million for electricity and 7.0 million for miscellaneous costs). Raw material (waste tire) costs were reported as 1550 BF/ton (if collected -and delivered?) or 600 BF if tires were "turned in" to the plant for payment by individual delivery. Dr Bettens described a manufacturing process for a 2000 t/d plant, critical elements of which were a "feed delivery" system for whole autombile tires (no shredding required), a large first stage direct heating zone (major gaseous volume expansion allowed), and a smaller adjacent cracking reactor zone. Activation of carbon residue was cited as a saleable product.
Considerable questioning arose with regard to the practical operation of the proposed plant. In particular, there was a question on the efficiency and the method of direct heating and the problem of fouling of heating elements. A situation was cited from the experience of one of the audience relative to an acute fouling problem, due to rapid coking of electrical radiant heating elements in a rubber pyrolyzer with loss of heat transfer and failure of the electrical system due to overheating. Dr Bettens recapped the process with emphasis on the conveyor transport system into and through the kiln which was claimed to allow adjustment of the contact (residence) time, facilitating rapid reactions. They do not stress heat transfer as being a problem in the first stage - "not a problem". Dr Bettens noted that a scraping system was included in the design to remove scale (foulants). Pilot tests had not been run which established the optimum "contact" time for the tires in the kiln. A condenser system was described for both the first and second stages. Further work was necessary to balance the take-off (two points) of light fractions and heavy fractions. This was recognised as a problem. No information had been

252
produced to support the claim of activated carbon production. It was speculated that a steam activation process would be used but that a subcontractor would be needed to implement that operation. In summary, basic technical issues remain before a reliable process is established and before reliable process economics can be generated.
3. Low Temperature Conversion of Biomass to Oil Professor Bayer Professor Bayer argues from both an experimental and theorectical
standpoint that, in contrast to conventional .widsom on biomass pyrolysis technology, high carbon-hydrogen organic compounds of low oxygen content need not only be achievable by high-temperature pyrolysis. Evidence was presented that fatty acids and a series of aliphatic hydrocarbons (often associated with olefin analogues) can be produced from biomass which is rich in lipids and proteins. Sewage sludge, because of the high microbial content, was proposed as a productive source of pyrolysis products in which C-C bonds would be retained while heteroatoms (S, Ν, O) may be readily eliminated. Reaction temperatures were in a low range of 280-400°C, with a precaution that temperatures in excess of 380°C should be avoided, normally because of the potential conversion of organic amides (RCONH2) to organic nitrites (or cyanide products) via dehydration.
A principal reaction pathway for formation of hydrocarbons was postulated as involving conversion of glycerol at 280-330°C to fatty acids, followed by conversion of the factty acids to hydrocarbons via decarboxylation at 300-380°C. The limitation of boiling points of the pyrolysis products was such as to indicate their practical use as diesel fuel replacements. Sewage sludge was a source of raw "oil" at yields of 27%. The solid residue was usable as a fuel source for dewatering. Of considerable interest was the observation that not only heteroatoms, but also halogen atoms (CI,F), could be removed under non-oxidative conditions in the presence of inorganic catalysts such as silicates.
One observer noted that the presence of water in the feedstock could modify the catalyst. Bayer speculated on the role of catalysis and noted that over 200 catalysts had been assessed with the goal of maximising the yield of the higher value-added olefins. Lipid-rich plants were desirable and easily manipulated. It was apparent that the aliphatic-to-olefin ratio could be preferentially manipulated to favour olefin production
To the suggestion that the low-temperature pyrolysis may be more significantly applied to destruction of organic components, specifically hazardous waste compounds, Professor Bayer emphasised that it is better, in any event, to maintain the C-C linkage in the organic compounds, making the point that it was very costly to build-up/synthesise higher molecular weight organic compounds, from C-|- compounds, such as methane. He had not addressed the question of the treatment of PCB compounds, only chlorinated dioxins.
When asked to explain how low oxygen content (3-5%) products were realised at low and high temperatures, Professor Bayer postulated that his products (with 85-95% the HHV of petroleum products) were a result of a different chemistry. At higher temperatures he postulated the formation of stable intermediate products, such as ketones, which could only be destroyed under the more extreme conditions. At lower temperatures, the decomposition of carboxylic acids, on the other hand, could be readily achieved via decarboxylation (-C02). Moreover, the formation of methane (CH4), depleted the available hydrogen which would be needed for extensive deoxygenation at higher temperatures.

253
Issues which remain on the practical implementation of the process include process efficiency (dewatering energy requirements?), rate of reaction (20 minutes preferred at small-scale), mass and heat transfer requirements, nature and availability of uniform raw material (process design), cost and efficiency of catalysts, cost of separation processes and refining to extract narrow fuel fractions, etc.
4. Chemicals from Almond Shells by Pyrolvsis in a Fluidised Bed Dr R Fort The University of Alicante paper focussed on the results of pyrolysis in a
laboratory-scale fluidised bed reactor of impregnated almond shells, a biomass waste product. The FBR was equipped with a comprehensive sampling train and, assisted by chemical analysis, product yields and compositional data on oils and gaseous products could be determined. The investigators report oil yields of 46-58% with preferred particle sizes of 0.2-0.3mm.
Several variations on conversion approaches were investigated. Post-pyrolysis of the dried oil residue, arising from pyrolysis at 610°C and
subsequent distillation at 120°C, when conducted at 890°C yielded a "producer gas" with high CO/C02 content (43%), 1% hydrogen, 6% methane and C2 - C4. The balance was not reported. The results correspond to the results of other investigators.
In perhaps the most interesting aspect of the research the almond shells were impregnated with acidic compounds yielding substantial quantities of furfural. Since furfural is produced commercially from oat residues it would be informative to compare yields under comparable conditions with almond shells to determine the potential for economic production of furfural from the locally available biomass resource. Is there a potential for development of an integrated multiple product (furfural, pyrolysis oil) process which could use the oil, the product gases, or char as a process fuel? A remaining technical problem, to be investigated, will be the efficiency of acid impregnation since pore closure has been reported by other investigators (Roy, Canada).
5. Biomass Pyrolysis in Molten Salts for Fuel Production Dr J Κ Maund The application of molten salts as a heating mechanism for the pyrolysis of
wood was reported. A small bench-scale CSTR reactor and condensation train with gas metering was described. The dissolution of wood in the molten salt was considered to have been observed though there was scepticism on the part of one of the audience that wood could be dissolved under non acidic conditions. No firm evidence was presented to support the solubility claim.
The investigator's preference for a thermal regeneration of hydroxide salts due to carbonate formation, rather than by the conventional "wet method", was a judgement based on the avoidance of evaporation costs (high Hv) required by the latter method, principally. The products were reported to be high in hydrogen in the gases (potentially the process has produced more hydrogen than was available in the ligno-cellulosic precursor?), and the condensed phase was mostly water. The latter result was considered to be significant. However, it would appear that the researchers might have overlooked quite serious losses of the organic condensation which, in fact, were observed as aerosols, as smoke and as liquid products in their gas rotameter Their observations of the latter species very pointedly express the serious difficulties in oil collection which face any reliable pyrolysis process. While the problem is solvable, the more significant problem

254
which has not been addressed, and which was raised from the audience, is the effect of build-up of adventitious impurities (e.g. metals in the wood) which can radically and rapidly change the rheology of the molten salt system as well as change the eutectic point or stability of the eutectic mixture. The latter, in turn, will affect the reaction temperature stability and, hence, the process. The investigators reported that their runs were not of such a duration that these problems had been observed.
On a positive note it was suggested to the researcher that he might explore the use of processes for decarboxylation of waste terephthalate compounds (polymer production by-products). Dr Maund reported that they had used the process elsewhere for monomer recovery from waste polymers and were currently investigating the decomposition of glass-reinforced plastic with a goal of recovering the resin decomposition products (possibly monomer derivation) after dissolution of the glass in the molten salt. (NOTE: It appears unlikely that Ε-glass and possibly A-glass would dissolve completely without radically altering the composition, reaction temperature, and rheology of the salt medium).
In summary, a much greater experimental base with the lab-system appears to be required and it would seem that each process application would have to be tailored for each feedstock.

SESSION V
GASIFICATION CASE STUDIES


257
PERFORMANCE OF A PILOT SCALE FLUIDIZED BED GASIFIER FUELED BY RICE HUSK
Hartiniati, A. Soemardjo, and M. Youvial LSDE-BPP Teknologi, Indonesian Energy Research Laboratory
Puspiptek - Serpong, Indonesia.
Sumnary
The performance of a 16 inch fluidized bed rice husks gasifier was determined for the bed temperatures ranging from 1330 -1600°F. The fuel flow rate was varied from 75 -105 kg/hr. No operational problems were observed during 36 hours of continuous operation. The whole study was completed for about 250 hours, and it was found that the gas quality varied between 650 - 1050 Kcal/Kg (4100 - 6300 KJ/Nm3), depending on the bed operating temperature and/or air to fuel ratio. The maximum energy produced was 2920 Kcal-gas/Kg-fuel at the fuel flow rate of 93 Kg/hr and the bed temperature of 1445°F. The lower heating value of the gas in this condition was 975 Kcal/Kg. Fuel to gas energy efficiency was found to be 63-67%.
1. INTRODUCTION. To reduce our great dependence on oil and gas, the biggest commodity
export of Indonesia, since the last decade the government has adopted a policy to conserve its petroleum reserves and prolong the availability of an exportable surplus by promoting the development of alternative energy resources where these can provide an economic substitute for oil. The development for alternative energy resources then raised our interest in biomass gasification.
Gasification is a thermochemical process in which the purpose is to transfer the chemical energy contained in the solid fuel into a gasesous energy carrier, preferably with a minimum " loss " into thermal energy. This process creates many possibilities to generate low, medium or high Btu of gases, depends on the reactor type and the gasifying medium (3).
In Indonesia, agriculture wastes are abundant in many areas and not utilized well. It is estimated that about 29 millions ton wastes per year are produced and as by products, they have a negative value due to the high cost involved in disposing of them. They should be properly handled and several concepts for handling and converting these wastes into a valuable gas fuel have been developed.
To utilize and increase the value of agriculture wastes, the Fluidized Bed Gasification is one of the most promising conversion process because of its ability to use a wide variety of fuels.
Since the gas quality produced by a gasifier depends on the nature of the fuel used and gasifier operating conditions, these conditions need to be optimized in order to be able to provide data for application studies.

258
Gasification of rice husks is comparatively difficult due to the high ash content and the particularly high silica content of the ash.
Major problem of utilizing rice husks is the nature of this feed stock, i.e.the high ash content ( see table I ), the low bulk density (100 Kg/m3) and the poor flow characteristics. Morover, processing of rice husks above 900°C is problematic due to ash sintering( 3 ). Table I presents the characteristic of rice husk used inthe experiment.
2. EXPERIMENTAL A schematic diagram of 16 inch diameter pilot plant fluidized bed
rice husks gasifier is given in figure 1. As shown in this figure, a rotron cyclonair blower supplied fluidizing air to the plenum of the reactor through an LPG start - up burner which was used to preheat the bed before commencement of solids feeding. After the bed temperature reached 950 - 1000°F the starting burner was then shut off and the feeder was simultaneously started. The bed material within the reactor is an aluminum oxide refractory sand having a particle size as shown in table II. The air passed from the plenum through the distributor into the bed to react with the rice husks which have been introduced into the bed via the feeding system.
The rice husks were stored in a surge hopper ι metered through a three-screw feeder, directed through a pressure sealing rotary lock hopper to an injector conveyor, and then injected into the lower part of the bed, just above the distributor. Upon entering the reactor, sufficient amounts of rice husks burn to bring the bed temperature up to the desired set point temperature. During operation the Barber - Colman temperature controller automatically set the gasifier air flow rate. After some initial oscillations in the range of 30°F about the set point, the bed temperature settled down and remain within 8-10°F of the set point. The gas leaving the reactor bed was passed through a double cyclones in which most of particles were separated from the gas stream. Subsequently the gas was burned in a dual fuel burner having an array of LPG/or natural gas and low Btu gas heads. Table II shows the operating variables used in the experiment.
3. MEASUREMENT. SAMPLING AND ANALYSIS. Bed operating temperature and rice husks flow rate were the
variables in these experiments, while the air flow rate and the gas composition were measured. Air flow rates were measured by means of an orifice, the solid fuel flow rate was set at the desired set point by means of switch. The metering feeder was calibrated first before the experiment began. The flow rate of the product gas was derived from the known flow rate of nitrogen entering the gasifier with the air and the nitrogen content of the product gas.
The bed temperature was monitored by a thermocouple penetrating the fluidized bed. The thermocouple temperature was displayed on a Barber -
Colman microprocessor based temperature controller having a resolution of plus or minus 1°F.
The train for sampling product gas is illustrated in Figure 2. A sample of hot gas was extracted from the main gas stream exiting the gasifier. The gas was passed through a hot filter packed with silica wool to separate particulates, then is cooled to an ice condensor to remove condensibles. Subsequently the gas is dried in a dryrite co1umn prior directing to a GLC unit for its composition analysis.

259
Hoppe
Feeder "χ
&â
Gas Duct ITc
rluidi'zed Bed
Injector
/ Cyclones
Ash Orurrs
/
O ner <-
V v Φ Ι
TempXpjTţroller
Exriauşt
älart Up Burner P i ^Ftow Control Valve
>i1ice \d^^er
Figure 1. Schematic Diagram of BPPT gasiti« during test at the Puspiptek Energy Research Laboratory ( Serpong.
Hot Filter (glass wool)
to GLC
\Condensate Flask
Figure 2. Schematic Drawing o1 Gas Sampling System.

260
Table I. Fuel Characteristics
¡ Moisture content ¡ Ash content ¡ Volatile matter ¡ Fixed carbon ¡ Sulphur ¡ Carbon total ! Hydrogen Total ¡ Nitrogen ¡ Oxygen ! Heating value ! [ = kcal/kg ] ¡ Specific gravity
Proximate Analysis ( % weight )
9,96 20,61 54,68 15,02
3220
1,44
Ultimate Analysis! ( %'weight ) ¡
0,02 ! 34,94 ! 5,46 ! 0,11 ! 38,86 !
Table II. Operating Conditions
Fuel Moisture content Operating temperatures Fuel flow rate Air flow rate Bed material
Reactor diameter Static bed height
rice husk 0,14 kg water per kg dry ash free material 1330 - 1600 ° F 75 - 105 kg/hr 100 - 250 kg/hr 0,3 - 0,4 mm = 26 % 0,4 - 0,6 mm = 45 % 0,6 - 0,8 mm = 29 % 16 inch ( 40 cm ) 24 inch ( 60 cm )
Table III. The average gas composition of the product gas (Vol % dry)
! Temp.
! 1330 ¡ 1400 ! 1450 ! 1500 ! 1540 ¡ 1600
H2
3.67 4.74 5.46 5.18 5.04 3.74
CO
8.28 12.23 12.31 10.43 10.14 8.87
C02
13.88 15.04 13.62 14.05 12.18 13.99
N2
68.73 58.84 61.19 62.34 66.54 67.83
CH4
3.63 6.73 5.99 6.17 5.04 3.72
C2H4
1.81 2.42 1.43 1.84 1.06 1.85
LHV ! (KJ/NM3)!
4093 ! 6340 ¡ 5528 ¡ 5585 I 4590 ¡ 4398 i

261
4. RESULTS AND DISCUSSION. The tests were conducted using a 16 inch diameter by 12 feet total
high reactor with bed depth of 24 inches. The parameter study involved varying the feed rate and bed temperature, then monitoring the gas flow rate, and product gas composition. For every given fuel flow rate, the air flow or air fuel ratio increases with rising the bed operating temperature. In these test the fuel flow rates were adjusted between 75 -104 kg/hr. Within the bed temperatures of 1330 - 1600° F, the air fuel ratio was found to be 0.30 - 0.48. Figure 3 shows the correlation between the bed temperature and the air fuel ratio. In all selected conditions, the gasifier was steadily and safely operated for considerable lengths of time.
As one would expect, the encreasing air fuel ratio tends to angment the process yield as shown in figure 3 (Process yield is difined as the ratio of product gas rate to fuel flow rate in dry ash free basis).
Product gas composition. In agreement with previous results done by else, table III
apparently shows a high carbon monoxide at relatively low temperatures. This relates to the theory ( 3 ) that at bed temperature below 800° C (1470° F) the homogeneous water shift reaction seems to be overruled by pyrolysis reaction, and CO is one of the major gas phase components in pyrolysis of biomass. Conversely, the hydrocarbons content in the product gas decline with increasing the bed temperature as a result of thermal cracking of lower hydrocarbons.
The lower heating values of product gas also decrease with increasing the bed temperature. This is due to the fact that the higher the operating temperatures, the more air required per kg daf fuel as more rice husks have to be burned to get a higher operating temperature level. As a result of the decline in gas heating valve, the volume of product gas will increase.
The energy output For every given fuel flow rate, the energy out put of the gasifier
(Kcal-gas/kg-fuel) as a function of operating temperature is shown in Figure 4. Energy output was obtained at 2920 Kcal-gas/Kg-fuel for the daf fuel flow rate of 64.82 Kg/hr,
This maximum value was reached at the operating temperature of 1445° F and air fuel ratio of 34.8%. The lower heating value of the product gas at this condition was 975 Kcal/kg. Gasification efficiency.
Figure 5 shows the effect of operating temperature on the gasification efficiency. The efficiency calculations were made based on the use of lower heating value of the fuel input and the gas output. In other words, the gasification efficiency or cold gas efficiency is the percentage of chemical energy in the fuels that is chemically bound by the product gas. The experimental result shows that the gasification efficiency is 63 - 67%. The total heat losses through the wall, ash, unburned carbon and product gas therefore is 37 - 33%. 5. CONCLUSIONS
The following are the general and specific overall conclusions obtained by this study :

262
100 5,000
A/F Ratio LHV Process Yield
poo TEMPERATURE (F)
0 1,600
Figure 3. LHV,A/F Ratio and Process Yield as a function of temperature
10,000
>-cr
Husks (daf) 52.55 kg/h Husks (daf) 72.28 kg/h Husks (daf) 64.82 kg/h
0 1,325 1,575
TEMPERATURE (F)
Figure 4. Energy in gas as a function of temperature

263
1.300 TEMPERATURE (F) 1,650
Figure 5. Efficiency of fluidi zed bed gasifier
- For every given fuel flow rate, the air fuel ratio increases with rising the bed operating temperature.
- At higher operating temperatures the lower heating value of product gas also decreases due to the more fuel have to be burned to attained that higher temperature level.
- As another result of rising the operating temperature is an increases in process yield.
- The maximum energy output of the gasifier was 2920 Kcal-gas/Kg-daf fuel at the fuel flow rate of 93 Kg/hr and the bed temperature of 1445" F. The air fuel ratio and the lower heating value of the product gas in this condition were 0.35 and 975 Kcal/Kg respectively.
- Fuel to gas energy efficiency of the gasifier was 63-67%.
References :
(1) Adiarso Soemardjo, et al, Unjuk Kerja Gasifier Unggun Terfluidakan skala Pilot dengan Umpan Sekam Padi, LSDE - BPP Teknologi, 1988.
(2) Bing Yan Xu, et al, Design and Operation of a 6.0 inch Fluidized Bed Gasifier for Rice Hulls, University of Missouri - Rolla, Departement of Mechanical and Chemical Engineering, Rolla, Missouri 65401, USA.
(3) F.G. Van Den Aarsen, Fluidized Bed Wood Gasifier, Performance and Modeling, Weerselo, The Netherlands, 1985.
(4) Hartiniati, The potential application of Fluidized Bend Gasifiers as alternative fuel production Facilities in Indonesia, Seminar on Power Generation Technology using Biomass, Jakarta, Indonesia, January 1989
(5) American Rice Ine, Biomass Alternative Fuels Program : Final Report Feasibility Study for Alternative Fuels Production ; Fluidized Bed Gasification of Rice Hulls, ARI, Houston,Texas 77252, USA, March 1982

264
ELECTRICAL ENERGY FROM BIOMASS
FULVIO FONZI Italenergie S.p.a. 67039 Sulmona AQ, Italy
Summary
Electric energy is produced by an electrogenerator, using as fuel a gas of a medium heating value. The gas is produced with a gasifier for biomass. The biomass used is agricultural and forestry waste products. The gasifier is built of two concentric fluidized beds, in which the thermic energy developed by the outer fluidized bed is used to increase the temperature of the internal one until complete gasification of the biomass is obtained. The gas is cleaned, filtered and cooled, and feeds a gas cylinder engine. The electric energy produced is used in the factory.
1. INTRODUCTION
1.1. Aim of the Project
Firstly, a brief review of the reasons for starting the present project.
Biomasses represent a source of renewable energy with low sulphur content, widely available in many areas of the world.
More specifically, the ligneous and cellulosic wastes produced by various agricultural and industrial activities constitute energy sources of particular interest because of their chemico-physical characteristics and because of their low cost.
We refer, in particular, to timber residues, and waste products of the wine making, oil pressing, textile and paper making industries.
In Italy, for example, the production of wastes from the timber and wood-working industries amounts to about 1.5 million tonnes a year. These wastes are partly used for energy production in the individual enterprises ; nevertheless, a large portion remains unused.
Lastly, the coppice is a presently under-used potential biomass resource which can contribute to energy needs.
The Gasification Process
The gasification process of wood passes through successive phases which involve drying, pyrolysis, reduction and oxidation procedures. These phases may be outlined as follows :
1st phase: Drying: Damp wood + heat = dry wood + steam

265
2nd phase: Pyrolysis: Dry wood + heat = charcoal (carbon), CO , CO, CH , organic compound (CnHm, unsaturated and oxygenated compounds)
3rd phase: Gasification: Carbon + Water + heat = CO + H Carbon + CO = 2 CO
The feed reactions may, therefore, be indicated as follows:
C + CO = 2 CO H = + 168 kJ/mole C + H.O = CO + H. H = + 175 kJ/mole 2 2
J O = C0 2 + H2
*2 * C H4
CO + H.,0 = CO, + H, H° = + 2.9 kJ/mole C + 2H_ = CH. H = - 75 kJ/mole
The fourth reaction is not facilitated by operating at low pressure and at high temperature.
The partial oxidation with air of the material to be gasified is the solution commonly used to obtain the necessary process heat. This method makes it possible to use a gasification plant of simple design, but has the disadvantage of producing a gas diluted with the nitrogen contained in the air. Consequently, the gas produced has a low calorific value (900 -1 200 KcalAg) ·
This entails a number of disadvantages : the gasification plant must be directly coupled to the steam generator in order to use the large sensitive heat of the gas produced at high temperature; the steam generator must undergo considerable modification to ensure that high efficiencies are obtained; if the gas is used in internal combustion engines instead of gas oil or petrol, then the gas must be sufficiently purified and cooled.
If it is necessary to produce gas for subsequent use in the synthesis of methane ("syngas") , or if it is required to produce a gas of medium calorific value, this difficulty must be overcome. Numerous technical solutions have been singled out in this respect. For example, the process heat is generated inside the gasification reactor by partial combustion of the biomass with pure oxygen instead of with air (1) , or with oxygen obtained from the thermal decomposition of particular chemical compounds (2) ; the process heat, generated outside the gasification reactor, is supplied to the biomass by an inert material circulating between a combustion chamber and the gasifier (3); lastly, the gasifier is heated from outside.
The process we proposed uses the last of the possibilities listed above. In comparison with gasification with pure oxygen, the process has the advantage of not requiring an oxygen generation plant from the production of syngas.
The process heat is provided by means of an annular chamber placed around the gasification reactor; high temperature flue gases obtained by the combustion of a part of the gas produced with air, pass through this chamber. In order to facilitate heat transfer from the flue gases to the gasifier walls, a granular inert material, which is fluidized by the rising gaseous current, is introduced into the annular chamber. Likewise, the transfer of the heat to the biomass particles is facilitated by a bed of

266
inert particles fluidized by recycling to the gasifier a part of the gas produced together with some superheated steam.
The composition of the gas produced depends, apart from the reaction temperature, on the rate of heating of the wood particles and on the period during which the reaction products remain at a high temperature. The concentration of methane and of oxygenated and unsaturated organic compounds drops if the gasification is made at high temperature and for prolonged residence time of the reaction products. For economic reasons, and in consideration of the behaviour of the materials, the gasification temperature in our process has been fixed at 730°C. In our operational conditions, the heating time of the wood particles is very short (in the order of seconds), and the average time the reaction products remain at a high temperature is much longer (in the order of minutes).
Under these conditions the gas contains an elevated percentage of methane and other low condensing hydrocarbons, which are undesired when the gas has to be used as synthesis gas for methanol production. In this case it is necessary to heat up the gas in a further step. Therefore a second gasification step is foreseen where a partial combustion with oxygen takes place in order to achieve a temperature of 1300°C, which is necessary to crack the hydrocarbons into CO and H . This treatment indeed reduces the advantages of the total independence of oxygen but, nevertheless, the consumption of oxygen is still much less in comparison to the classic oxygen gasification.
Preliminary Design
The preliminary design of the plant is shown in Figure 1. Biomass with a moisture content of 20% is fed into a fluidized bed
with a screw feeder. The gasifier is heated up with heating pipes containing inert fluidizing material. Heat is produced by combustion of a part of the gas produced and the combustion takes place inside the pipes.
Char and ashes are cleaned from the gases leaving the gasifier by two cyclones, and the separated char is fed back into the gasifier. One part of the cleaned gases is used as flushing gas and is recycled to the gasifier by a hot-gas blower. The temperatures in the different sections of the plant are as follows :
fluidized bed of biomass fluidized bed of heating pipes cracking chamber
730°C 930°C 1100°C
The gases leaving the cracking chamber have to be cooled down to 240°C in a first stage. After this, fine solid ash-like particles are separated by a fine mesh filter. In a second cooler the syngas will be cooled down to 450°C.
2. DESCRIPTION OF THE PLANT
2.1 Biomass Charging System
The biomass charging system consists of the following main units :
incoming biomass storage van of a capacity sufficient to ensure two days' work for the gasifier (30 tons of biomass volume 80 mc); biomass transfer and weighting system, consisting of conveyor belts, elevators, granular materials scale, separator of ferrous foreign

o»ygan
Fig . 1. The Vood Gas i f i ca t ion Plan t , December '87
to atmosp.
LOADING SYSTEM; 2. GASIFIER; 3. CYCLONES; 't. CRACKING CHAMBER; 5. QUENCHER; 6. STEAM SUPERHEATER; 7. STEAM GENERATOR; 8. SCRUBBER COOLER; 9. FLARE SYSTEM; 10 EJECTOR; II. COMBUSTION CHAMBER; 12. AIR PREHEATER; 13. ID FAN; ΙΊ. AUXILIARY BOILER
3
OPERATION DATA
DECEMBER '87
FLUIDI ZED BED TEMPERATURE
700 * 750"C
830 ♦ 8B0*C
BIOMASS FLOW RATE
100300 kg /h
SYNGAS FLOW RATE
10O250 Nm/h
GAS COMPOSITION C D DRY BASIS
N 2 : 33 .5 C 2 : 3 . 0 CH 4 :13.3
CO. 24.9 C 0 2 : 2 1 . 9 N 2 . 2 . 4 5
0j:0.05

268
materials, hoppers, etc. in order to guarantee the feeding of the wood chips to the gasifier at a variable rate from 100 to 1 000 kg/h.
2.2 System for Producing Gasifier Heating Fluid
As previously stated, the reaction that occurs in the gasification of the biomass is endothermic; the heat is supplied to the system by means of a fluid which heats the outer walls of the reactor. This fluid consists of flue gases obtained from combustion of a part of the gas produced in the gasifier with excess air.
As will be described in section 2.3, the amount of heat to be supplied to the reactor, under the assumed operating conditions, amounts to 300 000 Kcal/h. Because of the behaviour of the materials, the maximum temperature of the flue gases is fixed at 1 250°C; the temperature difference between the entry and the exit of these flue gases from the reactor is fixed at 400°C. Under these conditions, the rate of flow of the flue gases amounts to 2 500 kg/h. The air fed to the combustion chamber is preheated in a heat exchanger from the flue gases coming from the gasifier, cooled down from 850°C to 340°C. In this experimental plant, these flue gases are no longer used, and are discharged directly into the chimney stack.
It is obvious that in an industrial plant the residual heat would be recovered and used, for instance, for drying the biomass or for generating electric power to serve the plant.
On starting up the plant, when the production of gas from the biomass has not yet begun, or is insufficient, the production of the heating fluid is assured by auxiliary fuel.
A fan, mounted downstream of the combustion chamber and of the preheater, ensures the described flow rate of hot flue gases through the gasifier heating chamber.
Adequate instrumentation prevents the maximum temperature of the flue gases, fixed at 1 250°C, from being exceeded, and acts in such a way as to ensure that the gasification temperature of 730°C is maintained in the reactor.
This is performed by acting on the air and combustible gas flow rate.
2.3 Gasifier
The system consists of a reactor, cyclones for the separation of the solid products from the gas, and a compressor for recycling the fluidizing gas.
The gasification of the biomass, through heating and reaction with steam, occurs in the inner chamber of the reactor. On fixing the hourly flow rate of the biomass (500 kg/h) and the steam biomass ratio (0:4) , the calculated hourly quantity of heat that has to be supplied amounts to about 200 000 Kcal/h. In order to allow for the heat losses through the surface of the equipment, and to ensure an adequate safety margin, the quantity of heat to be provided to the gasifier has been estimated at 300 000 Kcal/h.
To meet the first requirement, the side wall of the gasifying chamber has been constructed of corrugated sheets ; the necessary exchange area is thus obtained with a smaller height of the gasification chamber. Considering an average diameter of 1 200 mm for the gasification chamber, the exchange area of 4 m2 is obtained with a 700 mm high fluidized bed. If the dynamic pressure supplied to the fluid bed process gas by the fan is 800 mm of water, the density of the bed in static conditions must be less than 1.15 kg/dmJ . In practice, allowing for the pressure losses due to the diffuses and to the pipe circuit, the apparent density of the bed must not exceed 0.8-0.9 kg/dm3 .

269
This is why 1-2 mm diameter spheroidal particles of aluminium oxide with a density of 3965 kg/m3 have been chosen as inert material to constitute the fluid bed. Laboratory tests have shown that this material has good mechanical strength and a modest erosion effect on the metal walls.
It is hard to predict the behaviour of the biomass in the fluid bed of alumina particles under the operating conditions. Since the biomass is fed from above, it is very probable that the mixing of the wood chips with the alumina particles would be restricted to the superficial layers. According to this assumption, two overlapping beds would be obtained, one of alumina particles and one of wood chips, mostly heated by the gas flow which in turn has received heat passing through the inert fluidized bed.
Since the volume of the gasification chamber is 1 700 litres, 850 litres of which are taken by the inert material, the biomass (600 kg/h, apparent density 300 kg/m3, equivalent to 2 m3/h) has an available reaction volume of about 850 litres which allows a gasification time longer than the experimental one (20'), determined by the laboratory tests in the same temperature conditions (730°C).
Therefore the wood particles should be pyrolyzed in less than a minute into char and later there will be the gasification of the char because of the action of the CO and of the steam present in the fluidifying gas. The solid particles will be entrained by the gas outside the gasification chamber only when they have attained a very fine grain size, in the order of microns.
The first cyclone is, however, designed to separate from gas all the solid particles with grain size over 500: it is presumed that these particles are still made of char and consequently they are recycled to the gasification chamber.
R = 96
Fig. 2. Double bed gasifier - cross section

270
Two further cyclones separate the finer particles, which are considered to be made of 'ash'; the latter particles are discharged from the plant and collected in an appropriate container. Naturally, these 'ashes' will contain a part of fine-grained char, which has not reacted. The flow rate of the ashes has been estimated at 3.8% of the biomass introduced, i.e. 23 kg/h.
3. MASS AND ENERGY BALANCES
By feeding 600 kg/h of wood chips, containing 20% of moisture by weight, and 240 kg/h of superheated steam to the gasifier, 827 kg/h of gas is obtained, of which 185 kg/h is used to obtain the heating process and 632 kg/h is sent for successive treatments. In addition, 23 kg/h of ashes is produced. According to estimates based on the chemical equilibrium, the gas produced has the following composition :
H2 CO co2 H2° C H4
(% by volume) 11
II
II
II
= 32 = 22 = 15 = 23 = 8
The calorific value of this gas is 2 700 Kcal/kg; it may, therefore, be considered as gas of medium calorific value and, as such, can be used directly for the generation of steam and/or electric power as a substitute for natural gas.
The addition of 98 kg/h of oxygen and the resulting temperature increase of the gas in the cracking chamber permits the almost quantitative completion of the reaction:
CH4 + H20 = CO + 3 H 2
In this way, the reduction of the methane content in the gas produced is accomplished, and 730 kg/h of syngas with the following composition is obtained :
H (% by volume) = 31 CO " = 24 CO " = 14 H20 " = 30 CH " = 1
The process then provides for cooling the gas by direct injection of water, and utilization of its sensitive heat for the production of superheated steam. As a result of this treatment, a large part of the steam present in the gas is condensed, the remaining 543 kg/h of gas having the following composition :
H (% by volume) = 44 CO " = 34 C0 2 " = 16 H20 n = 4 CH " = 2

271
This should, then, meet the specification required for a gas assigned to the synthesis of the methane. This balance shows that from the 1.98 Gcal/h introduced with the biomass into the gasifier, 1.47 Gcal/h is obtained from the system in the form of syngas. This corresponds to an efficiency calculated at 74%.
This balance does not make allowance for the thermal leakages through the surface of the pieces of equipment and for sub-actions and the consumption of energy necessary for working the pumps, the fans, the instruments, etc. However, it should be noted that since we are dealing with an experimental plant the possible energy recoveries have not been optimized; for example, the sensitive heat of the flue gas discharged into the stack at a temperature of 340°C can be used for drying the biomass.
4. DATA GATHERING CAMPAIGN
We carried out some modifications of the plant. The most important was the replacement of the hot gas blower with an ejector. These modifications allowed the final data gathering and test-run to be planned on a sufficiently reliable basis. They also improved the plant safety. Particular care was devoted to :
check of the new equipment installation and the plant status in cold condition; testing of the individual equipment in no-load condition; heating up of the process circuit; running and test of the plant for five days of continuous operation; data collection, elaboration and analysis.
The new equipment was in good condition. The installation of the superheated steam line (2", sch. 40) for ejector feeding has been completed, as well as the new 8" line for recirculation syngas and the flow control butterfly valve.
Some problems occurred during the single machinery tests. We had interlocking problems for the char discharging system. Furthermore, we decided to replace the stored wood chips with new green beechwood because the stored biomass was a mixture of beechwood and sleeper chips partially fermented. The aim was to avoid toxic product formation during the gasification due to the sleeper treatment containing creosote.
After the interventions and substitutions mentioned, the plant was ready. Problems were noted during the heating up of the plant. It is important to point out the following:
the pressure safety valves, located on the steam drum, had to be revised because of tightening problems; so we had to repeat the calibration of the valves ; transformer failure of the motor control centre; it was replaced by another transformer of higher capacity; controller failure of the auxiliary boiler burner; it was replaced after three boiler shutdown; check and calibration of all handling and feeding systems ; particularly the platform scale; the heating up rate was very low to avoid problems caused by ungasified coal deposited inside the gasification circuit. During the previous heating up, it was necessary to clean the plant to avoid burning of these deposits. In this case we had only localized combustion;

272
when the gasification circuit was about 400°C (inner bed temp.), the auxiliary recirculation fan was replaced with an ejector system; during this operation it was necessary to take care of the main equipment to avoid problems related to the several thermal expansions of each system.
When the process temperature into the inner bed was reached, the gasification circuit was recirculated with nitrogen to substitute the hot air used during the heating up. Biomass feeding started only when oxygen content was less than 0.05% (by volume) into the circuit. The feeding system had mainly two problems: clogging and sealing.
In order to assure a good seal of the screw feeder, special attention was devoted to keeping it constantly full of biomass. In this way a good sealing was obtained to prevent the syngas leaking. The analyser showed some problems when the water content was high and, in these conditions, special precautions were required to assure reliable results. However, at the same time many samples were sent to the laboratories for check analysis.
The data collected from the tests were acceptable and satisfied the heat and material balance. These gasification tests prove the innovative concept on which the gasification plant is based; indirect heating by fluidized bed. In addition, the possibility of obtaining a medium Btu gas without using oxygen has been demonstrated. From the data obtained, we calculated a heat transfer coefficient figure of about 200-250 Kcal/h m2 °C.
The certified gas analysis coming from different laboratories confirmed the expected figures.
The main data are :
fluidized bed temperature
biomass flow rate syngas flow rate syngas composition (% vol)
syngas LHV
electrical energy
5. CONCLUSION
: inner bed 700-750°C outer bed 830-880°C
: 600 kg/h : 500 Nm3/h CO H2 co2 C H4 C2H6 C2 H4 N2 °2 3480 ( 14
: 21 : 33 : 24 : 13 : 0 : 2 : 2 : 0
9 5 9 3 30 70 45 05
KcalAg 6 MJAg)
505 kWh/h
We can say we have demonstrated the feasibility of the process on an industrial plant, even though at prototype level. The reactor confirmed the expected high heat transfer performances. The know-how obtained and the figures accumulated in the running period will allow us to design new units, the costs of which we estimate will be lower by about 30%. The test has

273
enhanced the possibility of using this reactor to gasify pellets obtained from organic urban wastes.
The cost of electrical energy production is still very high for the present demonstration plant. The cost for electricity per kWh is not competitive with that of the national grid, at least with this present demonstration plant. However, with a new unit it is possible that a competitive performance can be reached.
In any event, the plant can produce electrical energy at a competitive cost where the electrical energy cost is about 300 Lit/kWh, as is the case in many developing countries (which are very rich in biomass and forest wastes) and where the electricity is produced by big diesel generators.
ELECTRICAL ENERGY FROM BIOMASS
PLANT CAPACITY SUPERHEATED STEAM GAS PRODUCED HEAT TRANSFER COEFFICIENT GAS LHV GAS DENSITY THERMAL ENERGY FLOW
ELECTRICAL ENERGY PRODUCED THERMAL EFFICIENCY
600 kg/h biomass with 20% moisture 240 kg/h 500 kg/h 220-250 KcalAg 3480 Kcal/kg (14.6 MJ/kg) 1.0 kg/Nm3 1*740.000 Kcal/h = = 2.018 kWh/h 505 kWh/h 74%
GAS COMPOSITION: (% vol. H 33.5 CO 21.9 CO 24.9 CH 13.3 C H 0.30 C H 2.70 Ν 2.45 0 0.05
(CALCULATED) (32) (22) (15) ( 8)
ELECTRICAL ENERGY COST: 300 Lit/kWh
Heat transfer coefficient calculation
Η = Q/S Τ / *) (filling coefficient)
1. Considering S.. = 6.03 m2
Ηχ = 188000/6.03/100/ * 0.8 = 249 Kcal/m2h°C = 390 Kcal/m2h°C
2. Considering S 9.45 m2
H2 = 1 8 8 0 0 0 / 9 . 4 5 / 1 0 0 χ 0 . 8 = 159 Kcal /m 2 h"C = 250 Kcal /m 2 h°C

274
FLUIDIZED BED GASIFICATION OF WOOD : PERFORMANCE OF A DEMONSTRATION PI ANT
K. MANIATIS+, A.V. BRIDGWATER*, A. BUEKENS+
+ Department of Chemical Engineering and Industrial Chemistry, Free University of Brussels, Pleinlaan 2,1050 Brussels, Belgium
* Department of Chemical Engineering, Aston University, Birmingham B4, UK
Summary
A Process Development Unit fluidized bed gasifier of 0.8 m and 1.6 m diameter respectively for the bed and freeboard section, was built and tested with the air gasification of chopped wood. The results were correlated with the air factor and were compared with thermodynamic predictions and published data. Reasonably good agreement was found between experimental, published and thermodynamic data. The higher heating value of the gas varied in the range of 4 - 7 MJ/Nm^ under normal operating conditions while the bed temperature varied in the range of 750 -900°C. It was found that the best performance was achieved in the air factor range of 0.20 - 0.4. The gas yield Increased with higher values of air factor but in the region of interest it was about 2.5 kg gas/kg feedstock MAF. It was also concluded that constant feedstock flowrate was a prerequisite for efficient operation, and constant performance.
1. INTRODUCTION Since the energy crisis of the seventies, research on biomass conversion
focused on producing a fuel gas for energy purposes and several R & D programmes on biomass gasification were initiated. Soon It was realised that fluidized bed reactors offer significant advantages over other types of gasifier configurations such as isothermal operation, simple scaling up procedures, high (industrial) capacities a good turn down ratio and multi fuel operation as long as feed size restrictions are respected. Their versatility helped to establish them as the reactor configuration of choise and several R & D projects were initiated in the Universities (eg. 1 - 5) as weel as private companies such as Omnifuel (6) of Canada and Vyncke N.V. of Belgium (7).
In 1980 an R & D programme was initiated between Vyncke N.V. of Harelbeke Belgium - a boiler manufacturer - and the Department of Chemical Engineering and Industrial Chemistry of the Free University of Brussels (V.U.B.). The programme was financed by the Institute of Scientific Research for the Industry and Agriculture and aimed at the commercial production of downdraft and fluidized bed gasifiers. Initially a co-current downdraft moving bed gasifier was designed, built and tested and since 1983, this type of gasifier has been marketed by Vyncke N.V. In 1982, a fluidized bed gasifier process development unit (PDU) was designed by the V.U.B. on the basis of a laboratory scale pilot plant (8) and built at the Vyncker N.V. workshop. The unit was subsequently commissioned and an extensive experimental programme was executed to identify the influence of all

275
major paramaters of the air gasification of wood in a large scale experimental reactor and the effect of scaling up on the performance of the reactor. The purpose of this paper Is to report some results concerning the performance of the fluidized bed gasifier.
2. EXPERIMENTATION It was envisaged that the fluidized bed would be operated under different
conditions and with a variety of feedstocks. In order to provide for every possible experimental condition the reactor was provided with 2 feeding ports (one above the surface of the bed at fluidization conditions and the other 0.1 m above the distributor), 2 ports for overbed preheating burners, several measuring ports for thermocouples and pressure gauges and 2 observation ports. Special consideration was given td the distributor configuration which should allow the passage of heavy and/or bulky material such as stones and conglomerated inerts. It was decided to use a pipe grid distributor which has such capabilities. A layer of refractory bricks forms the inner linning of the shell to give a finished diameter of 0.8 m at the bed section and 1.2 m at the freeboard section. The fluidized bed is shown in Figure 1.
The pilot plant consists of a feeding system (a hopper equipped with a variable speed screw for metering the feedstock, a rotary valve and a conveyor screw), the fluidized bed reactor, air compressor, preheating burner, a flare, a control cabin and a gas sampling system. The latter consists of a cyclone, a cooler (tube and shell), a filter, a gasmeter and a suction pump. Gas samples were taken directly from the gas collector of the flare and were analysed locally by gas chromatography In the control cabin and field laboratory.
The gasification process is depicted in the process flowsheet, Figure 2. The feedstock was delivered by a particle board manufacturer in closed containers. A front end loader transported the feedstock to the conveyor belt for loading the hopper. The feedstock flowrate was calibrated by direct measurement of the weight through an opening below the rotary valve. It entered the reactor above the surface of the bed. A compressor supplied the air required to fluidize the bed, while the air flowrate was measured by a rotameter. The air was fed through the preheating burner to the collectors of the distributor and from there it entered the bed. The feedstock pyrolysed as it fell into the fluidized bed to produce pyrolysis gases and char. The char was gasified by steam and carbon dioxide and partially combusted by oxygen, while the pyrolysis gases participated in secondary reactions. The product gas and fly ash were led to the flare were the gas was burned.
A small fraction of the gas (about 20 % by volume) was removed from the product gas burner and passed through the cyclone to remove the fly ash and through the cooler to condense the steam and tars. The experimental procedure as well as the feedstock properties has been described in detail esewhere (9).
3. RESULTS AND DISCUSSION The results were correlated with the air factor, (defined as the ratio of the
amount of air supplied in the reactor divided by the amount of air required for stoichiometric combustion) and with the bed temperature.
3.1. THE AIR FACTOR The air factor has the strongest influence on the performance of a gasifier
since it strongly influences the bed temperature, the gas quality, the thermal efficiency and in practice all experimental parameters. As the air factor is

276
jt 1200
,600
800 y o o o
u All units in mm
F i g u r e 1: The fluldized bed reactor
Flare
o o o τ
Wood
Compressor
-► To gas analysis
Cooler
Cyclone \ p
Gas
Ash Fluidized bed
Figure 2: Process flowsheet

277
increased after a steady state has been attained, the amount of oxygen supplied in the reactor increases and hence the degree of combustion. Since more heat is liberated the bed temperature Increases (see Figure 3) while the gas quality falls due to higher concentrations of CO2 and H 2 0 . This is illustrated in Figure 4 which also compares the results of the PDU to results obtained at the laboratory scale fluidized bed gasifier at the V.U.B. Due to the higher degree of combustion, the gas yield also increases with higher values of air factor as shown in Figure 5, which also compares data with the bench scale plant at the V.U.B. Figure 6 shows the influence of the air factor on the thermal efficiency (defined as the chemical energy of the gas divided by the chemical energy of the feedstock at moisture, ash free basis).
In general there is good agreement between data obtained from the two plants although the scale up factor was in the order of about 100. Data from bark gasification show consistantly a lower performance but this is due to the very high ash/in erts content (24 wt %) of the bark which resulted in gas of inferior quality. Nevertheless the trend is the same for all parameters and feedstocks. A carefull examination of Figures 4-6 reveals that the best performance was achieved in the air factor range of 0.2 -0.4.
3.2. THE BED TEMPERATURE From Figure 3 it can be seen that the bed temperature was varied in the
range of 680 to 975°C. Operation above 900°C resulted in sand agglomeration, due to the formation of low melting alkali metals euteclics with silica found in the ash of the feedstock and the sand respectively. Loss in fluidization never occured since the agglomerated particles segregated below the distributor due to their larger size. This behaviour was expected but it was decided to perform the experiments at high values of air factor in order to test the operating limitations of the PDU. It was concluded that with a pipe grid distributor it is possible to operate at temperatures above 900°C for short periods of time as long as provisions of sand replenishment are made.
The influence of the bed temperature on the gas composition is shown in Figure 7 for experiments performed in a close range of feedstock flowrate (about 225 kg/h) and of air factor (about 0.3). It is shown that the volume percent concentration of carbon monoxide and hydrogen pass through a maximum at about 820°C, while the concentration of carbon dioxide has a minimum at about the same temperature. Methane has practically a constant concentration of about 5% till 820 °C and drops thereafter. Ethylene and ethane have constant concentrations till about 780 °C and fell thereafter to concentrations below 0.5 and 0.1 vol. % respectively. A similar behaviour is presented by the higher heating value of the
gas, which has an average value of 6 MJ/Nm3 till about 820°C but drops at higher temperatures.
The decrease in the concentrations of carbon monoxide and hydrogen below 800°C can be tentatively explained by the assumption that below this temperature the pyrolysis products (tars and condensable organic vapours) predominate due to the lower temperature and hence the rate of the cracking reactions (which could transform the pyrolysis products to lower hydrocarbons) Is very slow.
Above 830 °C however, more feedstock has to be combusted to maintain the temperature of the bed at that high level and thus the concentrations of the gasification products decrease, while the concentration of carbon dioxide increases.

278
1200
1000
o
ξ 800 4>
i *> 600 ■
400
Thermodynamic prediction
Figure 3: Bed temperature vs the air factor
7
5
41
♦s 2
α> ι Λ ■ to
1. Wood shavings 2. Bark 3. Refuse derived fuel
Experimental data
0 0.1 0.2 0.3 0.4 0.5 0.6 Air factor
0.7 0.8 0.9 1.0
Figure 4: Higher heating value of the gas vs the air factor

279
Air factor
Figure 5: Gas yield vs the air factor
1.0
r 0.6
ΤΟ.8 0.4
Air factor Figure 6: Thermal efficiency vs the air factor
1.0

280
£
700
cr α> ►ι
8
800 Bed temperature
6 «g <
4 SL c o
Figure 7: Gas composition vs the bed temperature
3.3. OPTIMUM PERFORMANCE In the air factor range of 0.2 0.4 the performance shown In Table 1 was
attained repeateally.
Table 1. Optimum performance
Bed temperature Higher heating value Gas yield Thermal efficiency
°C MJ/Nm
3
kg gas/kg feed MAF %
820 6.5 2.5 65
4. CONCLUSIONS The PDU fluldized bed gasifier was scaled up by a factor of about 100 from
a Denen scale plant successfully. Under similar operating conditions the performance of the PDU was better than that of the bench scale plant.

281
REFERENCES
(1) R.S. Burton and R.C. Baillie, Fluid bed pyrolysis of solid wastes materials, Combustion, 13-19 February 1974.
(2) P.T. Raman, W.P. Walawender, V. Shimizu and LT . Fan, Gasification of corn stover in a fluidized bed : effects of superficial gas velocity and feed size fraction, Fuels from biomass and wastes, eds. D.L Klass and G.V. Emert, Ann Arbor Science, 1981.
(3) M. Findely, V. Flanigan and H. Sineath, Phase II, GROW Project, Proc, 13th Biomass Thermochemical contractors meeting, Arlington, Virignia, October 1981
(4) F.G. van der Arsen, A.A.C.M. Beenackers and W.P.M, van Swaaij, Performance of a rice husk fuelled fluidized bed pilot plant gasifier, Proceedings, 1st International Producer Gas Conference, Sri Lanka, November 1982.
(5) J. Schoeters, K. Maniatis and A. Buekens, The fluidized bed gasification of Biomass : Experimental studies of a benchscale reactor, in print with Biomass.
(6) G. Gurnik, K.O. Luke and D.C. Pollock, Application of a fluidized bed gasifier to conversion of forest biomass to an energy source, Report to ENFOR 1980.
(7) K. Maniatis, J. Schoeters and A. Buekens, Fluidized bed gasification of biomass, Reports 1-4 and 8. Reports to Vyncke N.V.
(8) J . Schoeters, K. Maniatis and A. Buekens, Fuel gas from agricultural residues in a fluidized bed reactor, Proc. 2nd World Congress of chemical engineering, Montreal, October 1981.
(9) K. Maniatis, A.V. Bridgwater and A. Buekens, Fluidized bed gasification of wood" Proceedings, Research in Thermochemical Conversion of Biomass, Phoenix, 1988.

282
Α NATIONAL PROGRAM ON IMPLEMENTATION OF BIOMASS GASIFTCATTON PROCESS TN INDONESIA. Scenario. Progress and Economic Evaluation
Herri Susanto and Soehadi Reksowardojo Dept. of Chem. Eng, ITB, Jalan Ganesha 10 Bandung, Indonesia
ABSTRACT Biomass gasification process has received a great
attention in Indonesia, and it shall be a national asset in the near future. Essential steps to be considered in realizing this program are (a) making the existing field test units as a nucleus for the dissemination and cultivation, (b) establishing necessary local institutions for monitoring and technical assistance, (c) building up an infrastructure in the regions concerned in order to provide repair, maintenance as well as production fasilities, (d) creating a funding or credit system for the investment of a gasification unit.
Factors affecting the success of the implementation of biomass gasification process, however, are not only the process reliability, but also a short term profit for the user, motivation of persons involved in the program, supply and preparation of the feedstock, the local management and the financial support during the introduction period.
1. INTRODUCTION Gasifications of various type of biomass including wood,
coconut shell, palmnut shell, corn corb and rice hulls have been tested at ITB. Intensive studies on the gasification of wood and of rice hulls have been done at our laboratory and reported eg. in [1,2,3]. Field tests have also been conducted in several places, in Java Island. Performance of 15 kW wood gasification in Balong, Middle Java, was reported already in [4,5,6]. This unit has now an operating record of about 12000 hours. Experiences in the field operation of rice hulls gasification are presented in [7]. In our experiences, the producer gas is used as an oil substitution upto 80% for diesel engines. The choice of diesel engine instead of gas engine is due to the fact that the diesel engine has been introduced since the sixties, and hence its infrastructure has already been established.
Based on our previous field experiences and on an economic analysis, the implementation of biomass gasification process in Indonesia needs a scenario regarding to the direction and the way of further development, so that the biomass gasification shall become a national asset, and its benefit and usefulness are felt in the social dan economic life of the Indonesian people. A proposal and a progress of the scenario for the implementation of gasification technology are described in this paper. A short discussion on the economic of the gasification process is also presented.

283
2. THE PRESIDENTIAL PROJECT Being aware, that the Indonesian nation will face hard
ships when its oil, gas and coal resources have been pumped out completely, the National Energy Coordination Board has pointed out the need for diversification of energy resources. The biomass gasification process being one of many alternatives, has become a national issue and topic, as at least six Ministries engange in the activities concerning with the biomass gasification. This 'political will' of the government has been responded with research and development conducted by many research institutes, universities and industries. Even foreign parties with various motivations and various ways have taken part in the introduction of the biomass gasification technology. By the end of 1986, the President personally launched a promotion project of the gasification, which in essence constitutes a campaign for the cultivation of biomass gasification process.
In this Presidential project, five wood gasification units with a capacity in the range of 15 - 100 kW and a rice hulls gasification unit with capacity of 40 kW were installed in six provinces (see Appendices A and B). This promotion program is also the last part of our program in research and development of the biomass gasification process within the frame of Project JTA-9A. The objectives of this program are : - promotion and demonstration of biomass gasification as an
alternative energy resource - dissemination of the biomass gasification technology - development of a self supporting energy system in rural areas
- improvement of efficiency and effectiveness of utilization of wood and other agricultural wastes,
- collecting information for further studies, research and development of the next implementation program.
3. SCENARIO and PROGRESS OF THE IMPLEMENTATION A picture of the real conditions of biomass gasification
program in Indonesia can be summarized as follows : a. there are serious political wills of the President and the
government, b. there are activities by many institutions which have
successfully established gasification diesel engine systems in various regions in Indonesia,
c. but there exists limitations on the production, supply, and use of gasification process as an alternative energy resource. The limitations are probably due to various constraints,
such as : a. the process is relatively new, and the field experiences
are only in Java, b. the gasification units must be operated in proper ways,
which requires high dicipline operators, c. there is no infrastucture in the regions for technical
training and assistance, as well as repair, spare parts and after sale services, To overcome the present limitations, it is necessary to
establish a Monitoring and Technical Assistance Team in

284
the region of the Presidential pronotion units for a certain introduction period. The tasks of this team are : a. collecting and evaluation of sociotechnoeconomic field
data, the data could then be used for : determining whether the installed units meet the technical, operational and economical expectation of the local users,
identification of the operating conditions to ensure a successfull implementation of the biomass gasification,
determining a standard gasification technology that is accepted by the users
identifying technological aspects that requires an additional research and development,
defining a scope of application of the biomass gasification in a spesific area,
b. technical and operational support, so that the existing units can be operated properly as a demonstration and promotion one,
c. improvement of the unit, so that it achieves better reliability,
d. transfering the technology from the innovator to the local parties.
The Monitoring and Technical Assistance Team consists of : a. local scientists, technologist and industrialists, b. officials from region institutions and authorities, c. management of local users of the gasification unit, d. central coordination by Project JTA9A, ITB.
The execution of the promotion program is divided into two main phases, settingup the field units and monitoring of the real operating performance at least for one year after the installation. Table I shows the activities and contribution of parties involved in this promotion program. Many institutions have been taking part in this activity for the financial and technical supports, as well as the social and economic aspects, (see also Appendix A).
Appendix Β shows the technical data of the six promotion units. During the second main phases, the daily operation data is recorded by local operator, who have been trained for operation, maintenance and monitoring. The local supervisors will then communicate the data and the technical problems to Project JTA9A/ITB as a central coordinating agent. A technical meeting is planned at the end of the monitoring program.
After about one year installed in sites, we found that the local conditions have influenced very much on the progress of the promotion project. The villagers or the users usually have not yet understood the goal of the project completely. They suppose that this project is as a rural electricity, rather than a self supporting energy with the use of biomass. As a result, they operate the unit with diesel oil, which is furtunately available everywhere, in this time.
The lack of maintenance such as cleaning of the gas piping, periodical ash removal from the ash bin the bottom of the gasifier, and preparation of proper size and moisture content of the feedstock often caused serious technical problem. Since the operation of the gasification unit is

285
cumbersome, the operator often run away from their job, so another training for new operators was necessary. Many other non-technical problems, such as the lack of working capital and mis-management were found to be decisive factors to the the success of the promotion project.
Despite the above problems, Scientific and Industrial Agents have been set-up for the implementation of the biomass gasification process in the future. Institute of Technology in Surabaya and PT BBI, an engine manufacturer will cope the implementation for instance on Java and Small Sunda Islands, including Sumbawa and Timor Islands. The implementation in Kalimantan will be taken by PT Pupuk Kaltim, a fertilizer company. While PT Pupuk Sriwijaya and University of Sriwijaya will carry out the implementation in Sumatra, etc.
The tasks of this Scientific and Industrial Agent are more or less the same as the Monitoring and Technical Assistance Team mentioned previously. But the Scientific and Industrial Agent may also act as a clearing house for credit facilities as the use of gasification unit needs additional investment. Table I : Setting-up the promotion phases/activities central coordinator local parties prem. study and design
-gasification units -sites selection -diesel-genset -capacity
-fuel supply installation -gasification units
-diesel-genset -auxiliary equip's
-electric transmisión -electric distribution -power house
operation -technical ass. -training
-operators -local management
-local supervisor -operating cost for first three months
-self operation in following months
monitoring dissemination and further promotion
-coordination -national program
-operators -local supervisor -government promotion -private interest -local manufacturing
4. ECONOMIC EVALUATION The real economic evaluation of six Presidential promo
tion units could not been evaluated yet, since the units have not been operated in regular and commercial bases. Taking the investment cost of the gasification unit into account, the operation of a gasification unit for the oil substitution in a diesel genset, usually saves only a marginal amount of money. The difference of the diesel oil price and the gasification feedstock is apparently not big enough in this moment to com-

286
pensate the additional investment. Rough economic evaluation showed that this additional investment for the gasification unit should not be more than USD 200 - USD 350 per kW depending on the capacity. Reduction of the investment cost seems possible, if manufacturing of a standardized gasification unit is applied in a commercial production.
The transportation cost of gasification unit must also be taken into consideration in the economic evaluation of the use of gasification process for rural electricity outside Java. As the gasification unit is technically rather simple, manufacturing of gasification units close to the user is considered to be a good solution to reduce partly the additional cost. By a proper arrangement of the transfer of gasification technology from the innovator to the local workshops or industrialists, the implementation of the biomass gasification process would become more feasible. Building up a Scientific and Industrial Agent discussed in the previous section is considered to be a prospective way to realize the local manufacturing of gasification unit in the future.
Our previous field unit in Balong, Middle Java (Appendix C, [7]) has shown, that the life time of a gasification unit upto 8 years could be expected, if the maintenance and operation procedure were carried out properly. But the operation of six promotion units outside Java for about one year have indicated unfavourable conditions with respect to the gasification unit life time. Repair and maintenance cost have also increased due to improper operation of the gasification unit. 5. CONCLUSIONS
The biomass gasification being as one of prospective alternative energy resources has been promoted widely through out the country. This promotion should be followed by a national monitoring program in order to obtain a reliable data for defining the adaptability of the biomass gasification technology. Tranfer of the gasification technology to local scientists and industrialists is necessary in a country like Indonesia for setting up a local network. Acknowledgements
The authors gratefully mention the contribution and cooperation of the Ministry of Forestry, BBI-diesel engine manufacturer, and all parties involved in this promotion program. Literature 1. Ghazali, S, D. Sasongko and S. Hardjosuparto, "Experiences
in using Rubber-tree Wood and Teak Wood as Feedstock for Producer Gas Generator", paper presented in the Second International Producer Gas Conference, Bandung, March 1985.
2. Manurung, R, and A.A.CM. Beenackers, "An Open Core Rice Husk Gasifier for Small Scale Application", paper presented in the Second International Producer Gas Conference, Bandung, March 1985.
3. Sasongko, D, S. Ghazali and S. Hardjosuparto, "Maximum Capacity of A Cocurrent Moving Bed Gasifier", paper presented in ASEAN Workshop on Thermal Conversion of Biomass,

287
Hatyai, Thailand, September 1988. 4. Sudarno H and A. Koopmans, "Balong Demonstration Gasifi
cation Unit, A Field Experience Report", paper presented in the Second International Producer Gas Conference, Bandung, March 1985.
5. Susanto, H, A. Koopman, and Sudarno, H, "Field Experience of 15 and 60 kW Wood Gasification for Rural Electricity", paper presented in ASEAN Conference on Energy from Biomass, Penang, October 1986.
6. Manurung, R, H. Susanto and Sudarno H, "Experiences in the Operation of Rice Husk Gasification for Rural Electricity", paper presented in ASEAN Conference on Energy from Biomass, Penang, October 1986.
7. Susanto, H, "Field Experiences on the Operation of 15 kW Gasification System for Rural Electricity", paper presented in ASEAN Workshop on Thermal Conversion of Biomass, Hatyai, Thailand, September 1988.
8. Groeneveld, M, J, "The Cocurrent Moving Bed Gasifier". PhD thesis, T.H. Twente, 1980.
9. Kjellstrom, B, "Practical Design of Producer Gas Systems", a lecture note in Producer Gas Course, the Second International Producer Gas Conference, Bandung, March 1985.
Appendix A : Location of the Presidential promotion units 1. Irian Jaya Unit
location: Desa (village) Arso III, transmigration site Jayapura
fuel : wood wastes collected from the surrounding local supervisor : Ministry of Transmigration,
Univercity of Pattimura, Ambon end use of energy : rural electricity, at 6 12 p.m.
2. Maluku Unit location : Desa Kaibobo, Seram Island fuel : wood wastes from a nearby playwood industry local supervisor : Ministry of Forestry,
University of Pattimura, Ambon end use of energy : rural electricity, evening
100 houses with 100 W and 90 houses with 60 W cost of electricity Rp 2500 3000/housemonth (vs. to Rp 7000 using pressurized kerosene lamp)
3. Kupang Unit location : Desa Nonbes and Oekabiti, Kecamatan Amaraşi
Kupang, Timor Island fuel : wood (ipilipil) from regular land clearing
for traditional farming local supervisor : Ministry of Forestry, end use of energy : rural electricity, evening
100 houses out of 300 planned 4. Sumbawa Unit
location : Desa Beröra, Kecamatan Lape, Sumbawa Island fuel : rice hulls local supervisor : Centre for Enviromental Study of
University of Mataram, Mataram end use of energy : rural electricity, evening

288
325 houses Samarinda Unit
location : Desa Segihan, Recamatan Sebulu, Tenggarong, East Kalimantan
fuel : wood wastes from wood industry local supervisor : PT. Pupuk Kaltim
(fertilizer company) Institute of Technology in Surabaya
end use of energy : electric power for wood industry rural electricity in the evening
Palembang Unit location : Desa Sungai Buaya, Palembang, South Sumatra fuel :wood wastes from house components industry local supervisor : University of Sriwijaya,
PT PUPUK Sriwijaya (fertilizer comp, end use of energy : electric power for wood industry
Appendix R : Technical Data the Presidential Promotion Units Table II : Summary of technical data Capacity 15 kW 65 kW 100 kW 40 kW Type of gasifier Type of feedstock Capacity of bunker, litre Throat diameter, m Height of red. zone, m Diesel genset Number of cylinder Cylinder volume, litre Compression ratio = 1/17 ; Nomimal power output, kW App. maximum power output
in dual fuel mode, kW Gen. nominal output, kVA (220/380V, 50 Hz, 3 phases)
The wood gasification unit is of a conventional system [8,9]. The oxidation and reduction zones are lined with refractory. The throat is made of stainless steel plate with 12 mm thickness. The rice hulls gasifier is an open core type, that has been developed at ITB and the University of Twente [2], and also in the fields in Majalengka for rural electricity [6] and in Mojokerto for a rice milling.
In the 40, 65 kW and 100 kW units, the gas cooling is accomplished by a forced air-cooled pipe heat exchanger. While in the 15 kW units, the gas is cooled down in a natural draft air-cooled horizontal pipe. The gas filter was filled with a low cost filter medium which may be locally available such as rice hulls and coconut fiber.
In the large units of 65 kW and 100 kW, a blower is used to drive the producer gas during start-up for 5 - 1 0 minutes.
co-current down conventiona
small block of 3 - 6 cm
100 .350 0.11 0.17 0.25 0.30
draft 1 wood 350 0.20 0.30
BBI (Deutz Lisence), air 3 8 2.827 12.763 Rev.per minute 24 117 18 80 25 125
12 19.144
r 176 120 180
open-core rice-hulls 100 no 0.30 cooled 6 5.655 1500 51 40 50

289
In the smaller units, a gas ejector activated by the engine exhaust gas is used to suck the gas during the start-up in place of the blower. The gas/air mixing device is a simple "T" connection.
Appendix C : Balong Field Unit (Middle Java) Balong unit with a capacity of 15 kW electric has been
operated for rural electricity since Februari 1984. Its daily operating hour is about 6 - 8 hours, and now it has a total operating hour of more than 14000 hours. The gasification feedstock is rubber wood waste. The average diesel oil replacement by the producer gas is about 80%. The following table show the operation cost, excluding investment. Table III : Operating and Economic Data (successive 4 months)
month 1 2 3 4 operating hours :
in dual fuel 231 206 146 141 in full oil 0 18 32 0
diesel oil cost, Rp lOOOx 28.6 40.0 93.9 56.7 (147.) (117.) (407) (277) a /
wood cost, Rp lOOOx 14.0 5.0 10.0 4.0 b/ (77) (17.) (47) (27)
wood preparation, Rp lOOOx 51.7 55.3 45.0 29.7 c/ (257) (157) (197) (147)
engine maintenance, Rp lOOOx 17.5 24.7 0.0 15.4 d/ (B7) (77.) (07) (77.)
gasf.unit maintnce, Rp lOOOx 20.0 150 15.0 15.0 e/ (107) (407) (67) (77.)
operators' salary, Rp lOOOx 75.2 97.5 72.5 89.4 f/ (367) (267) (307) (437)
t o t a l , Rp lOOOx 2 0 7 3 7 2 2 3 6 2 1 0 (' 1 O O 7 .)
*/ number in parenthesis is percentage to total op. cost / wood purchase in a respective month °/ transportation and cutting into small block (3x3x8 cm ) / lubricating oil and its filter, air filter etc. ®/ rockwool filter, nut, bolt, packing, welding etc. / one technician and two low skill operators exchange rate : 1 USD = Rp 1650 (May 1989) For comparison, annuity vs annual operating cost : diesel genset (15 years life time) = Rp 1900000,-gasification unit (8 years) = Rp 1610000,-power house and electric distr. (15 years) = Rp 520000,-total operating cost (see the above table) = Rp 3000000,-

290
GASIFICATION AND PYROLYSIS OF STRAW - RESEARCH IN DENMARK G. Olsen, P.H. Pedersen, U. Henriksen and E. Kofoed
Laboratory for Energetics Technical University of Denmark
SUMMARY Straw has been pyrolysed and gasified in batch and con
tinuous reactors. The effect of dolomite as a tar cracking catalyst has been studied and experiments with other catalysts will be performed in the future. The termal cracking of pyrolysis tar and the effect of residence time in the cracking zone has been studied. The heat transmission in straw of various densities formed the basis of dimensioning of a continuous reactor for pyrolysing straw. A continuous reactor for gasification of loose straw has been developed. Superheated steam has been used as the gasifying agent. Future gasification experiments will include C02 and air as gasifying agents.
BACKGROUND In - 1986 the Danish Ministry of Energy decided that
future small scale combined heat and power stations should be based on domestic energy sources like natural gas, wastes and biomass. Additionally the field burning of straw has been banned from 1990, which increases the interest in discovering alternative applications for straw.
For smaller combined heating and power plants, the energy efficiency would improve if an engine was used instead of a steam turbine to deliver the mechanical energy to the generator.
For this reason a research and development program has been started regarding pyrolysis and gasification of biomasses, mainly straw. The research at the Laboratory for Energetics started in February 1988, and has the object of making the Laboratory a center of knowledge in Denmark. The collection of data is obtained by literature studies, through visits to institutions and departments having R&D in gasification of biomass and especially by experimental work. The Laboratory has also participated in the devel-ment of a pyrolysing unit in co-operation with a consulting engineering company. The experiments in this paper are only concerned with the research performed at the Laboratory for Energetics.
The research has been split up into 5 main subjects:

291
1. Know-how. 2. Gas from straw as fuel in small co-generation plants. 3. Gas purification. 4. Tar products used as fuel in internal combustion
engines. 5. Gas from pyrolysis and gasification of straw as fuel in
gas engines.
1. TEST EQUIPMENT AND EXPERIMENTS 1.1 Batch The experimental work started with small scale batch
experiments. Batches of 50 g straw were pyrolysed by external heating in a cylindrical reactor with a heating rate of 10°C per minute. The condensate was collected in a cooling trap (Figure 1).
Gas outlet
Pyrolysis reactor
ß&S Ice bath
Figure 1: Pyrolysis - batch. The same experiments were carried out with a second
reactor, of the same dimensions as reactor no. 1, installed after - the first reactor so that the pyrolysis products were fed immediately through the second reactor. The external heating of the second reactor was independent of the heating of the first reactor (Figure 2).
IS
Pyrolysis reactor
X Gas outlet <
Second reactor
Figure 2: Pyrolysis - batch - second reactor.

292
The tar cracking effect of different materials such as CaCC>3, dolomite and stainless steel chips was studied at various temperatures(1). The CaC03 and the dolomite were precalcinated at 850°C and the experiments were compared to experiments with an empty second reactor at the same temperature. Only dolomite was found to have an effect on the amount of tar produced, as experiments with a second reactor temperature of 900°C showed a colourless, transparent condensate and a clean gas. The dolomite turned black during the experiment, which was explained as deposit of carbon black on the dolomite particles arised from the tar cracking process.
The experiments were repeated with addition of superheated steam as gasifying agent and it was shown that 50g of char was completely gasified in one hour, and that the carbon deposit on the dolomite was gasified.
Studies of heat transfer in straw of various density have been performed in reactors of two different diameters. Both reactors were cylindrical and the temperature in the straw was measured from the center and out to the wall during the heating from 20°C to 900°C. The gas outlet was varied from a central outlet to peripheral outlet.
High density of straw shows a delay in temperature increase and also a delay in reaching the final temperature, but the heating rate is very dependent on the gas outlet position (Figure 3).
Time (minutes)
α Centre 160 (kg/n3) + Centre 240 (kg/m3)
O Periphery 160 (kg/m3) Δ Periphery 240 (kg/m3)
Figure 3: Heat transmission in straw.

293
Recently the batch experimental equipment has been used for pyrolysis data experiments where the gas produced is collected in small cylinders according to the pyrolysing temperature (Figure 4). Each cylinder contains gas produced in 50°C pyrolysis temperature intervals. The gas is analysed and the amount is measured. The condensate is measured volumetrically as a function of pyrolysis temperature. The experiments have not been completed and the results are yet to be published.
Pyrolysis reactor
Second reactor
JU ill ill ã
Gas collect ing cylinders
Figure 4 : Pyrolysis data experiments. 1.2 Continuous
Two continuous reactors have been developed. One is a downdraft gasifier which by means of a piston introduces loose straw into a cylindrical, vertical reactor (Figure 5). The gasifying agent is introduced into the reactor. Ashes are accumulated in the reactor and gas is taken out at the bottom from where it can be fed to a tar cracking catalyst if desired. So far, this reactor has only been used for observations of the continuous gasification process and the equipment still needs to be improved.
The second reactor, which in principle is similar to the one just described, is mainly for pyrolysis of loose straw. The experiments carried out with this equipment are described in the next paragraph.

294
Gas outlet
Gasifying agent
Figure 5: Continuous reactor. 2. TAR One of the main problems in pyrolysing/gasifying straw is
the tar produced by the process. The gas produced by the gasification gas must be suitable for a gas engine. Tar will then be a problem as it is known to cause valves and piston rings to stick when the engine is shut down.
The tar can be cracked thermally or catalytically to gas and carbon black. The thermal cracking of pyrolysis tar has been studied as a function of cracking temperature and residence time in the cracking zone. The pyrolysis products were passed through a chamber of varying size and temperature ranging from 1.5 1 to 10 1 at respectively 600°C to 1100°C. The gas was then cooled in a condenser and led through four gas washing bottles containing acetone (Figure 6).
Second reactor Condenser
Four gas washing bottles in series
Collection of char
Figur« *: Thermal cracking of pyrolyaio tar.

295
The experiments showed a decrease in condensate when the temperature was increased. No significant effect regarding the amount of condensate was observed when the residence time was prolonged. The gas production, though, is slightly increased by increasing residence time and also by increasing temperature. The gas composition is only dependent on the cracking temperature and the heat of combustion of the gas has its maximum at 800-900°C (1).
The catalytic cracking of tar has been successfully performed using precalcinated dolomite. Commercially available catalysts are yet to be studied.
REFERENCES 1. Rensfelt, E., C. Ekström: Fuel gas from municipal waste
in an integrated circulating fluid-bed gasification/ gas-cleaning process. Energy from biomass and wastes XII, New Orleans, Feb. 1988.
2. Olsen, G.: Research in tarcracking and applications of tar. Presented at the international conference: Pyrolysis and Gasification, Luxembourg, May 1989.

296
AN INVESTIGATION INTO THE GASIFICATION OF LOW QUALITY COAL WITH OXYGEN ENRICHED AIR IN A FIXED BED GASIFIER
A D ENGELBRECHT Division of Energy Technology
CSIR, Pretoria, S A
Summary The performance of air and steam blown gas producers deteriorate rapidly when low quality high ash coals are used as feedstock. An investigation was carried out to determine to what extent gas quality and gas output can be improved when the blast air is enriched with oxygen. Extensive testing was done on a small industrial scale gas producer plant (5 GJ per hour output). Using the data obtained, a techno-economic study was done to determine the optimum level of oxygen enrichment to be used for various coal qualities. The investigation showed that enrichment of air with oxygen was economically attractive based on both capital expenditure and running costs.
1. INTRODUCTION In South Africa approximately 1 million tons of coal
is consumed in gas producers annually. In these units the coal is gasified with air and steam to produce a low Btu-gas mainly from bituminous coal and some anthracite. The gas is used in industries where cheap heat is required, such as the brick-making industry for the firing of brick kilns, calcination of calcium carbonate in the paper and board industry and for steam raising in boilers originally designed for oil-firing. In certain heating applications such as the manufacture of refractory materials, furnace temperatures in excess of 1600 c
3a r e required. Due to its
low calorific value (+-6 MJ/NM ) it is difficult to achieve these temperatures with producer gas alone, and it has to be enriched with expensive fuels such as Sasol gas or LPG.
Another disadvantage of standard air and steam-blown gas producers are their low thermal output to diameter ratios. This is due to large amounts of inert nitrogen that passes through the system. This leads to lengthy payback periods on capital.
It has been suggested that oxygen enrichment (see Appendix 1.2 for definition of oxygen enrichment as used in this paper) of the blast air to a gas producer can in the first place produce a cold clean gas to give flame

297
temperatures in excess of 1600 C, and in the second place better thermal output to diameter ratios. Oxygen enrichment has been attempted on some industrial gas producers. These attempts have been mainly unsuccessful due to a lack of understanding of the processes that occur in a gas producer, and the relationship between blast saturation temperature and oxygen enrichment. (For definition of blast saturation temperature see Appendix 1.1). When the blast is enriched with oxygen the oxidation reaction,
C + 02 > CO 405.8 MJ/kg mol occurs at a more rapid rate and more heat is generated in the oxidation zone. To prevent the temperature in the oxidation zone going above the ash fusion temperature of the coal more steam is injected to cool the bed down by the endothermic steam char gasification reaction,
C + Η„0 > Η. + CO + 119.1 MJ/kg mol the other gasification reaction,
C + C02 > 2CO + 160 MJ/kg mol also occurs at a more rapid rate due to more C0„ production by the oxidation reaction. From the above reasoning it seems that the output and calorific value of the gas will increase. It is, however, difficult to reason quantitatively in this way because of the many complex processes occurring simultaneously in the gasifier. A mathematical model taking into account rates of chemical reaction, physical transport processes and the thermodynamic relations can cast some light on the subject. Using this method to predict optimum operating conditions and plant results could be risky because of the lack in available kinetic data as well as heat and mass transfer coefficients. It was, therefore, decided to use a singlestage pilot gas producer to investigate the effect of oxygen enrichment on plant operation and results. It is felt that the results obtained on this singlestage unit will also be applicable to a twostage gas producer. The reason being that the processes of interest occur in the oxidation and gasification zones which are similar for both types of producer.
A prominent oxygen supply company was responsible for the oxygen supply to the plant. The project was carried out with their collaboration. Pure oxygen was added to air to produce oxygen enriched air to the desired level. Pressure swing adsorbtion processes to produce oxygen enriched air

298
are on a steep development curve and could become more economical than addition of pure oxygen. 2. GAS PRODUCER PLANT LAYOUT AND GASIFIER DIMENSIONS
The gas producer plant modified to incorporate oxygen enrichment is given in Fig 1. The gasifier is a 0.83 m internal diameter firebrick lined vessel. The gasifier dimensions are given in Fig 2.
The gasifier is of the fixed bed type with coal being charged at the top and ash being removed at the bottom below the grate. The reactants (air, steam and oxygen) are introduced into the bottom of the gasifier and flow upwards countercurrently to the fixed bed of coal that moves downwards. The gas that is produced is removed from the top of the gasifier. A fixed bed gasifier can be divided into 3 zones. At the bottom of the bed the oxidation reactions occur to provide heat for endothermic reactions in the gasification zones above it. At the top of the bed the volatile matter of the feed coal is driven off by the hot gases moving upwards. This zone is referred to as the devolatilization zone.
The gasifier has pokeholes at the top and bottom so that any large clinker that is formed can be broken up. The pokeholes are also used to determine the height of the coal bed and the position of the fire zone that has to remain fixed. 3. RESULTS
To investigate the effect of oxygen enrichment on gas output and thermal output the total volumetric blast rate was kept constant at each level of enrichment. Only the proportion of 0_, N_ and steam were altered to give the required oxygen enrichment and blast saturation temperature. The above approach was followed since the total volumetric blast rate effects the gas output and thermal output at constant levels of oxygen enrichment and blast saturation temperature.
Before the tests on oxygen enrichment were started, a datum test was done using air (ie. 0% oxygen enrichment). This test was done to measure the improvement that oxygen enrichment has at each level. Results of tests at different levels of oxygen enrichment are given in Table 1. To get a good average result, the duration of each test was 18 hours. Fig 3 and Fig 5 show the increase in calorific value and thermal output at different levels of oxygen enrichment. It can be seen that at 9.9% oxygen enrichment, the calorific value of the gas had increased by 42% and the thermal output by 64%. Fig 4 shows the variation in gas composition at different levels of oxygen enrichment.
4. DISCUSSION From an operational point of view oxygen enrichment

299
has given no problems. This is due to careful operation at correct blast saturation temperature at each level of oxygen enrichment. At the higher levels of oxygen enrichment (6%, 8% and 10%) the blast saturation temperature had to be controlled within +- 1.5 C. In the case of air operation the margin of error was +- 3.0 C. If the BST was allowed to drop too low the maximum temperature in the gas producer rose above the ash fusion temperature of the coal and large clinkers were formed. If the BST rose too high the gas quality decreased and carbon burn-out deteriorated due to a cool fire zone.
The required accuracy in blast saturation temperature can easily be achieved with a fairly cheap control system. It is felt that a lot of problems encountered on industrial gas producers using oxygen enrichment were caused by operation at too high a BST.
From Fig 4 it can be seen that the CO- concentration increased with an increase in oxygen enrichment. Because more oxygen is injected into the gasifier more CO- is formed from the oxidation reaction C + 0_ > CO«.
Because of the slower kinetics of tne C + CO- > 2CO reaction, the rate of CO» conversion is not as rapid as the CO- formation. This results in higher levels of CO-in the product gas at higher levels of oxygen enrichment. This effect makes it unattractive to go to higher levels of oxygen enrichment since the additional oxygen is converted to CO- and not to useful gas.
The amount of steam that can be converted by steam char gasification reaction
C + H-O > CO + H-is limited by the maximum temperature in the reactor and the rate constant of the above reaction. At higher levels of oxygen enrichment most of the additional steam only acts as a dilutant and passes through the system unconverted. REFERENCES (1) CLARK, D., The current status of coal gasification in
South Africa and its prospects for the future. (2) HOWELL, A.N., Gas producer economics. Coal
Gasification Symposium, Wanderers Club, Johannesburg, 9 June 1982.
(3) Gas producers Symposium, Transvaal Coal Owners Association, South Africa, 1974.
(4) PAREKH, D., Handbook of gasifiers and gas treatment systems, DOE/ET/10/59 - T24.

Oi o o
Rotameter Tar extractor Water seals Tar separator overflow to ponds
Fig. 1. Gas producer modified for pressurized operation and oxygen enrichment

301
LEVEL OF ENRICHMENT OF BLAST(X) 0.0 l.B 4.1 6.0 B.O
GAS OUTPUT (NM3/H)
GAS TEMPERATURE (C) 357 .6 33B.1 306 .3 333.2 364.0
7.9 COOL FEEORATE(KB/H) 69.0 72.7 7B.7 B9.2 103.2 llO.O OXYGEN FLOWRATE(NH3/H) 0.0 3.3 7.3 9.6 12.4 14.Β AIR FLOHRATE (NM3/H) 14B.B 143.2 130.7 119.β 110.9 103.6 STEAM FLOWRATECKB/H) 26.3 30.3 33.2 37.7 39.1 42.3 BLAST SAT. TEMP.(C) 33.4 SB.2 60.4 63.7 63.1 66.9
229.7 223.1 23B.9 247.3 269.O CALORIFIC VALUE(MJ/NM3) 3.9 6.0 6.9 7.6 B.l 8.3
(BTU/NCF) 137.6 162.4 IBS.3 203.6 217.6 223.4
HOT RAU BAS EFF.(Ï) B3.1 BO.O B7.7 HOT DETARRED BAS EFF(X) 73.0 73.2 BO.7 COLD CLEAN BAS EFF(X) 69.3 6B.0 76.3
BS.6 BB.l B2.7 BO.4 79.3 74.2 73.Β 73.1 70.3
Steam out
Anular boiler 0.92 m
Coal bed Depth 1.51 m
^y\
I.D. *0.83 m ■
* Gas outlet
Insulation material
Firebrick lining 0.15 m
+ oxygen + a i r '
Pokehole
o Blast pipe 0.15 m
Steam + oxyge
F i g . 2. Gas producer dimensions

302
■a ξ
> Ό V
U U •Η U i*j Iti •Η 4» U O» Ο Ό
ΙΟi 9' β
7
6
5" 4
3
2
I
Fig. 3. Effect of oxygen enrichment on the calorific value of gas
2 3 ι · ι ι ι Ι 5 6 7 β 9 10
Oxygen enrichment (%)
Fig. 4. Effect of oxygen enrichment on gas composition
■Ρ V Ό in
Q
Oxygen enrichment (%)
2,501
2,23 /
Fig. 5. Effect of oxygen enrichment on thermal output at a constant blast rate
« 2,00
1.75
1,50
1,251 2 3 4 5 6 7 8 9 10
Oxygen enrichment (%)

303
APPENDIX 1 1.1 Explanation of the term "blast saturation temperature"
(BST) . Blast saturation temperature refers to the amount of water vapour present in the air blast. The air is saturated with water at the particular temperature (BST) and from the partial pressure of water at this temperature and the total pressure the AIR/HO ratio can be calculated. The blast saturation temperature is a convenient way of measuring the ratio of air flow to steam flow since it requires the measurement of only one patrameter (BST) and of the determination of both flows separately. In the case of oxygen enrichment the blast saturation temperature refers to the ratio of water vapour to enriched air. It can, therefore, be used just as in the standard air blown case to control the 0„/H_0 ratio of the blast. The blast saturation temperature will not give a consistent indication of the 0_/H_0 ratio if the blast is oversaturated or undersaturated. For the blast to be just saturated the air and steam have to meet the following requirement before mixing. For blast saturation temperatures in the range 5070
10 C < Air temperature < 35 C 100 °C < Steam temperature < 125 °C
1.2 Definition of oxygen enrichment The percentage oxygen enrichment used in this paper is the difference between the enriched air concentration and 21. For example:
(AIR FLOW).(0.21) + 0~ FLOW % 0„ enrichment = χ 100 21
AIR FLOW + 0„ FLOW 1.3 Coal composition used for O,. enrichment trials
Proximate analysis ultimate analysis Calorific value(MK/kg) Ash(%) H20(%) Fixed carbon(%) Volatile matter(%)
27.5 15.18 2.70 53.50 28.70
Carbon(%) Hydrogen(%) Nitrogeni %) Sulphur(%) Oxygen(%)
68.2 4.20 1.70 0.80 7.40

304
PRESSURIZED FLUIDIZED BED GASIFICATION OF PEAT
E. KURKELA, P. STÂHLBERG, W. MOJTAHEDI & M. NIEMINEN Technical Research Centre of Finland (VTT) Laboratory of Fuel Processing Technology
SF-02150 Espoo, Finland
Summary
An experimental research project on pressurized buildized-bed gasification of peat and wood waste was initiated at the Technical Research Centre of Finland (VTT) in 1986. This research is related to the development of Gas Turbine Power Plants based on air gasification and hot gas clean-up.The first test runs with the pressurized gasifier were carried out in summer 1988. The gasifier was operated at pressures of up to 10 bar and the maximum thermal input with peat was 400 kW. Air and steam were used as gasification agents. During 175 hours of operation 11 tons of peat was successfully gasified. The effect of operating conditions on gas composition, tar formation and fines elutriation was investigated. Carbon conversion without cyclone dust recycle was 75 - 85 % and the calorific value of the gas (LHV) ranged 4 - 4 . 5 MJ/Nm3. Alkali metals release in fluidized-bed gasification of peat was investigated experimentally and theoretically. Samples were taken from various points and analyzed for the alkali metal content. The results are compared with the theoretically predicted values.
1. INTRODUCTION Power production from coal with Integrated Gasification-Combined
Cycle (IGCC) is becoming an increasingly promising technology for the 1990s. Processes based on oxygen gasification and cold gas cleanup offer an environmentally acceptable alternative for coal utilization. These IGCC processes can already now be considered commercial. Oxygen gasification, however, is not economically competitive in small and medium-size scale power plants. Hot gas cleanup is also an alternative which has potential for increasing the efficiency of power generation significantly IM. Using air gasification and hot gas cleanup seems to be a promising method for simplifying the IGCC process.
A joint research project to study Pressurized Fluidized Bed Combustion (PFBC) and pressurized gasification was initiated in 1988 in Finland in which IVO Oy (the largest utility company in Finland), Helsinki University of Technology and VTT are cooperating. This project is part of the National Combustion Research Program "Liekki", which is financed by the Finnish Ministry of Trade and Industry.
The main emphasis of this research project is on the utilization of peat and wood wastes, which are the only important indigenous solid fuels in Finland. The project was started by designing and constructing the PFBC/G test rig, capable of operating at pressures up to 10 bar. The test rig was commissioned in summer 1987 and it is located at the Laboratory of Fuel Processing Technology of VTT. During 1987 the test rig was used for studying fluidized bed combustion /2, 3/. This paper pre-

305
sents some of the results from the first gasification test period in the summer 1988.
The goals of the first phase of gasification experiments were: a) to study the effect of operating conditions on gasifier performance
(carbon conversion, gas composition), b) to generate basic data on gas impurities, which are released in peat
gasification and might cause problems in gas turbines (alkali metals, particulates) or in high-temperature filters (tars). One of the most important research topics in this project is the be
haviour of alkali metals in gasification. There are very little measured data available on alkali metals release in coal gasification and no data at all on peat or wood waste gasification. Thermodynamic projections /k, 5/ however show that alkali metals volatilization might be a severe problem in IGCC systems applying hot gas cleanup. Vapor phase alkali metals cannot be removed by hot filtration and the conventional standards for gas turbines typically limit the alkali levels to below 0.1 ppm (w) in flue gases entering the turbine.
2. EXPERIMENTAL A schematic diagram of the Pressurized Fluidized Bed Gasification
(PFBG) test facility is shown in Figure 1 and some of the technical data for the rig are presented in Table I. The heart of the facility is the reactor, which is a 5.6 m high pressure vessel of 0.6 m in indiameter. Its inside is refractory-lined to provide a bed diameter of 0.15 m and freeboard diameter of 0.25 m.
Primary gasification air and steam were introduced to the reactor through a multiorifice plate distributor. Secondary air was introduced above the fluidized bed to increase freeboard temperature in order to crack tars and improve carbon conversion. Hot gases leave the freeboard and pass first through two cyclones operating at high temperature (e.g. 800 °C)and then through a third cyclone operating at reduced temperature (e.g. 300 °C). Finally, after the pressure let down, the product gas is burnt in an atmospheric combustion chamber. The elutriated fines captured in the cyclones were collected and weighed. Recirculation of fines was not effected in these experiments .
The test rig included a wide range of measuring devices and sampling systems. The gas analysis included on-line measurement of the major gas components (CO, C0p, Hp, CHjj) as well as sampling systems for chromatographic analysis (Cj-Cc hydrocarbons, H2S). For the determination of tars and NHo a gas slipstream was led through heated lines into gas washing bottles filled with dichlormethane (for tars) or an aqueous solution of 5 % H2S0ij (for NHo). Detailed description of sampling methods for both gas and tars as well as analytical techniques are described elsewhere /6, 7/.
Particle measurements were based on isokinetic sampling. Samples are extracted from the gas duct after the second cyclone. Particle concentrations were determined using an absolute filter, which was placed in an electrically heated casing. The size distribution was measured by coulter counter from collected samples. A detailed description of particle measurements is given in /8/.
For the determination of the concentration of vapour phase alkali metals in product gas a sample was led through an absolute filter into gas washing bottles. The sampling lines and the filter were electrically heated to the same temperature as the gas exiting from the gasifier. The sodium and potassium concentrations in the washing liquid samples were determined by atomic absorption spectroscopy.
The gasification experiments were carried out using crushed peat

306
Gas analysis
Tfb(min)
Secondary air
Primary air
Steam
Particulate t sampling
Rue gas
3rd CYCLONE I
Dust removal
PRESSURE LETDOWN l—l
COMBUSTOR
Waste water
Dust removal
t Bed removal
Figure 1. Schematic diagram of the pressurized fluidized bed gasification test facility.
Table I. Technical data of the Otaniemi PFBG test rig.
Thermal input Fuel Operating pressure Bed temperature Free board temperature Fluidizing velocity Gasification agents Product gas flow rate Bed diameter Freeboard diameter Effective height of the reactor Gas cleanup
400 kW peat, wood, brown coal 4 - 1 0 bar (abs) 750 - 850 °C 750 - 1000 °C 0.6 - 1.5 m/s air and steam 90 - 220 Nm3/h 0.15 m 0.25 m 3.5 m two cyclones, ceramic filter unit (1989-)
pellets as fuel. Peat pellets of 15 - 25 % moisture were crushed in a hammer mill. Two particle size ranges were used in the tests, 0 - 3 mm and 0 - 5 mm. The properties of raw material (feedstock) is shown in Table II. The moisture, the ash, the volatile matter and the sulfur contents of solid samples were determined by routine methods (DIN 51718 - 51820, and ASTM D 4239 respectively). The C, H, and Ν contents were determined with a LECO CHN-600 instrument. The Na and K-contents of

307
Table II. The properties of gasified Particle size range Bulk density Moisture content (wet basis) Volatile matter (dry basis) Dry matter analysis
C H Ν 0 S Ash
Nacontent of ash Kcontent of ash Lower heating value (dry basis)
peat. 0
575 20
67.5
55.5 5.6 1.8
32.4 0.18 4.5 0.67 0.93 20.5
_ ± ± ± ± ± ± ± ± ± ± ± ±
5 mm 25 kg/m
3
5 % w/w 1.5 % w/w
1.5 % w/w 0.2 % w/w 0.4 % w/w 1.5 Í w/w 0.04 % w/w 0.5 % w/w 0.03 % w/w 0.03 % w/w 0.5 MJ/kg
solid samples were analyzed by atomic absorption spectroscopy for ashed samples which were prepared according to DIN 51070.
3. RESULTS AND DISCUSSION A total of 13 tons of peat was gasified during 175 hours of opera
tion. The test runs were conducted at three different pressures and with different air to steam ratios, totaling 13 different operating set points. Some of these results are shown in Table III. The H2O content of the product gas was calculated from hydrogen balance and the fuel gas flow rate from nitrogen balance. Carbon conversion efficiencies were calculated as carbon in products/carbon in gasified peat. Other figures in Table III are average values of the measured data.
The gasification pressure (in the range of 5 10 bar) did not seem to have a significant effect on the carbon conversion. Figure 2 shows a carbon balance for a typical set point. The most important carbon loss was in the form of elutriated fines, which contained 12 20 weightţ of the feed carbon. Adding secondary gasification air to the freeboard and increasing the steam feed improved carbon conversion which was, in the best set points, in the range of 80 85 ί.
Approximately 80 90 wtJ of the elutriated fines were captured by the first cyclone and the dust concentration in the product gas after the second cyclone was 4 8 g/Nm
3 with a particle size range of 0 20 pm.
The net calorific value of the product gas was 3.8 4.5 MJ/Nm3 (wet gas) and the tar content generally ranged 4 6 g/Nm
3, which represents
2 4 % of the energy input of the gasified peat. The major part of tars were light oils like benzene, toluene and naphtalene. Figures 3 and 4 show the effect of freeboard temperature on the naphtalene and phenol concentrations of the product gas.
Set point 3/5B, with an air feed rate of only 1.4 Nm3/kg peat (maf), is in many respects different from the other set points. Since the carbon conversion was only 73 1>, this set point can be considered to correspond to conditions prevalent in partial gasification. The total concentration of tars was high, 10 14 g/Nm3, and the tars were composed of a wide variety of typical pyrolysis products from light oils to heavy tars.
Table IV shows the measured vapour phase alkali metals concentra

308
Table III. Results for different set points.
Gasification pressure, bar (abs) Peat feed rate, g/s Peat moisture content, % w Primary air feed, g/s Secondary air feed, g/s Steam feed, g/s Bed temperature, °C Freeboard temperature (max), °C Temperature before cyclone, °C Gas velocity in freeboard, m/s
Fuel gas output, NnH/h (wet gas) Fuel gas composition, % vol (wet gas)
CO co2 H2 CHn C2Hn C2Hc N2+Ar H20
Tars+oils: Benzene, mg/Nm^ (wet gas) Toluene, mg/ΝπΗ (wet gas) Naphtalene, mg/Nup (wet gas) Tars & oils total, mg/NnP
Carbon conversion efficiency, % w to dry product gas to gas + tars
3/3B 4.97 12.6 17.5 20.2 1.0 1.3 815 806 730
0.48
109
10.6 13.7 13.7 3.3
0.34 0.24 45.3 12.8
3600 510 705
5500
80.7 83.3
3/5B 5.09 15.8 17.2 21.8 1.0 0.0 817 803 700 0.50
117
10.8 12.5 10.1 3.6 0.58 0.31 47.3 14.8
5170 1240 1480 11200
68.7 73.5
4/5 5.01 18.5 14.8 18.7 11.5 2.4 832 862 790 0.73 166
11.9 13.6 13.2 3.6
0.35 0.15 44.8 12.3 3930 130 705 5300
79.3 81.7
4/1 7.07 21.2 16.2 22.5 12.8 5.2 831 868 795
0.65 198
9.6 13.9 12.7 3.4
0.24 0.16 42.7 17.4
3700 65 740
5100
83.6 86.2
4/3B 9.55 21.4 15.5 23.4 12.2 7.5 822 856 800 0.50
207
9.2 13.2 12.1 3.7 0.17 0.19 39.9 21.5 3520 45 670 4800
81.2 83.7
tions of the product gas for three set points. Due to the small number of samples and the generally known problems in the reliability of sampling methods, no final conclusions should be drawn from these figures concerning the effect of operating conditions on the release of alkali metals. However, the total concentration of alkali metals in the product gas seems to be at least an order of magnitude higher than allowable to a gas turbine but not quite as high as predicted by thermodynamic equilibrium calculations.
4. CONCLUSIONS AND FUTURE RESEARCH The calorific value of the product gas from air gasification of peat
was more than 4 MJ/Nm^ and the gas was readily combustible without additional support flame. The carbon conversion of gasification was, however, too low for economic power production. Carbon conversion in fullscale plants will be improved to certain extent by increasing the reactor height and the residence time of the particles. However, conversions >95 % can be achieved only by efficient recycling of all separated particulates. The freeboard temperature should be elevated from 870 °C, which was the maximum temperature reached in these experiments. However, the maximum temperature is limited by the sintering and slagging properties of the peat ash.

309
CONVESWN OF PEAT CARBON TO OFFERENT PRODUCTS:
SET POINT 4 / 5 AR1.eONm3/kgpe«!(m»f) STEAM 0.305 kg/kaP«rt(maf)
PRODUCTQAS 83.7 I
► TARS λ OLS 2.β%
PARTICULATES 1.β%
12%' [ ' ' 0 . 1 %
CYCLONE OUST
Figure 2. Carbon conversion for a typical set point.
780 800 820 840 860 FREEBOARD TEMPERATURE ( C )
2400
2000
1600
1200
800
400
0
+
'·. AR 1.4 Nm3/kgpeat(inaf)
+ \
AR 1.61.8 Nma/kopMtyMf)
X
1 ι 1
+
Fţ_——— ■
■
+
X
V
X
I
r —
<5mm, 45ber
< 3 mm, 5 bar
< 3 mm. 7 bar
<3mm. 9.5 bar
Peel < 3 mm V
x
¿~~ Pert.5mm
— ι 1 1
Figure 3· The effect of freeboard temperature on the naphtalene content of the product gas (dry gas).

310
η E ζ ■*» σ> Ε
111 χ Q.
£ΧΛ(
600
400
200
~
0
+
An 1.4 Nm3/kgpeat(maf)
+
+
■
+ χ
ν
(1 η
< 5 mm. 45 bar
< 3 mm, 5 bar < 3 mm, 7 bar < 3 mm, 9 . 5 bar
+ Am.e1.8Nm3/kep«rt<maf>
+ + χ χ
1 1 1 1 1 — ■ Γ
i —Mt—<f
Jf-\ nil
780 800 820 840 860 FREEBOARD TEMPERATURE ( C )
Figure 4. The effect of freeboard temperature on the phenol concentration in the product gas (dry gas).
Table IV. The measured Na and Κ concentrations in the gasifier product gas (wet gas) and the proportion of peat alkali metals released in the vapour phase.
Set point
4/5 4/1
4/3B
Pressure
bar
5 7
9.55
Tmax
°C
862 868 856
Na % of Na in vapour
ppmw phase
3 .84 .3 4.04 .6 2.1 2.4 2.3 2.7
Κ
ppmw
0.981.1 1.3 0.8
% of Κ in vapour phase
0.760.86 1.1
0.68
Freeboard temperatures of 830 870 °C seem to be high enough to crack the heavy tars to benzene, naphtalene and other light components, which should not be detrimental to hightemperature gas filtration. The results from vapour phase alkali metals sampling seem to support the conclusions drawn from thermodynamic projections, that the concentration of alkali vapours has to be reduced before combusting the product gas in a gas turbine. This can be achieved by cooling the ga3 before filtration or by using special getters like aluminosilicates.
The next phase of the gasification research will be carried out in summer 1989. The experiments will focus on studying the effect of fines recycling on the carbon conversion as well as the applicability of

311
ceramic candle filters to PFB gasification. An online alkali vapour measuring device has been constructed to enable a more accurate measurement of the alkali metal vapours in gasification. This device, based on atomic emission spectroscopy, will be used in the next phase (summer 1989) of the PFB gasification experiments.
ACKNOWLEDGEMENTS The authors wish to acknowledge the assistance of the technical and
analytical staff of the Laboratory. Special thanks are expressed to Mr. Reino Flinkman and Mr. Jarmo Kleemola for operating the test facility.
REFERENCES 1. Solantausta, Y. & Kurkela, E. The production of electricity from
peat by Integrated Gasification CombinedCycle Conversion. IGT Conf. on Biomass and Wastes XII, New Orleans, Feb 15 19, 1988. 12 p.
2. Horvath, Α., Hulkkonen, S. & Jahkola, A. Emissions of a peat burning pressurized fluidizeed bed combustor. Int. FBC Conf., Dec 12
13,.1988, London, UK. 12 p. 3. Hulkkonen, S., Jahkola, A. & Kurkela, E. The Otaniemi PFBC/G
research project. The 10th Int. Conf. on Fluidized Bed Combustion, San Francisco, April 30 May 3, 1989. 8 s.
4. Mojtahedi, W. & Backman, R. Alkali metals volatilisation in pressurized fluidized bed combustion and gasification of peat. 4th Int. Fluidized Combustion Conf., Dec 1213, London, U.K. 16 p.
5. Mojtahedi, W. & Backman, R. Release of alkali metals in pressurised fluidisedbed combustion and gasification of peat. Espoo 1989, Technical Research Centre of Finland, Publications 53. 48 p.
6. Leppälahti, J., Ståhlberg, P., Simell, P. Nitrogen compounds in peat and wood gasification and gas combustion. Conf. on Research in Thermochemical Biomass Conversion, May 2 6 , 1988, Phoenix, Ari
zona, USA. 14 p. 7. Simell, P. Kotimaisten polttoaineiden kaasutuksessa syntyvät terva
maiset epäpuhtaudet (Tarry impurities developed in the gasification of indigenous fuels). Espoo 1988, Technical Research Centre of Finland, Research Reports 631. 53 p. + app. 22 p. (in Finnish).
8. Hulkkonen, S. Particle Measurements of the Otaniemi PFBC/G test rig. Espoo 1988, Technical Research Centre of Finland, Symposium Series 83, p. 251 262.

312
RAPPORTEURS REPORT ON SESSION V GASIFICATION CASE STUDIES
Κ. MANIAŢI S Free university of Brussels, Belgium
A.A.CM. BEENACKERS Groningen University, The Netherlands
1. INTRODUCTION The seven papers presented in this session indicate that extensive research has
been carried out in the field of biomass, wastes and solid fuels gasification in a variety of reactor configurations and at practically every scale of operation varying from the laboratory scale up to full scale demonstration plants.
However, commercialization has been hampered due to the low cost of hydrocarbon fuels and to technical problems concerned with the use of the fuel gas in engines, (see also rapporteur's summary of session 6).
Extensive efforts have been made by several Less Developed Countries and most notably by Indonesia, for the implementation of gasification technologies in rural areas (see also rapporteur's summary of session 3). In Less Developed countries, not only techno-economic problems must be overcome but social barriers as well in order to allow for the successful introduction, operation and maintenance of gasifiers and their supporting systems.
2. DISCUSSION Professor Corella commented that the performance of the ITALENERGIE dual
fluidized bed pyrolyzer/gasifier reactor was not optimal since the operating temperature was too low and the specific throughput was only 100-300 kg/(hr. m 2 while much higher values have been reported by Battelle Columbus at 9000 kg/(hr. m
2)· The low bed height to bed diameter ratio was mentioned as a possible cause. The
chairman of the section, Professor Beenackers, responded that indeed operating problems exist but these were related mainly to the heat transfer through the wall separating the two fluidized beds. Dr. Fonzi added that the main cause for the operating problems was related to the new operating regime of the reactor which was used only as a gasifier to produce a fuel gas and not synthesis gas as originally foreseen.
In response to Dr. Shah's suggestion that local manufacturing of gasifiers in Indonesia and more generally in the Less Developed Countries will accelerate the transfer of technology. Prof. Beenackers reported that indeed through the Dutch Development Cooperation project JTA 9A in Indonesia gasifiers are manufactured locally. Dr. Maniatis added that Darmawan, a private company and Bisma Borna Indra a state owned company, both are involved in manufacturing gasifiers in Indonesia.
Dr. Stiles commented that successful introduction of gasifiers in Less Developed Countries strongly depends on the selection of the right materials for construction. Too often gasifiers are badly corroded after only a few months of operation. This was accepted by the floor but it was also suggested by Dr. Maniatis that better motivation of the operators can improve considerably the lifetime of a gasifier by careful controlling of the particle size of the feedstock by which usually the yield of tars can be reduced significantly.

313
Professor Corolla asked for extra information on the pressurized fluidized bed gasification of peat and more specifically about the type of inert bed material used and on the reduction of tars by catalytic activity of dolomite. Dr. Kurkela responded that although the initial bed material was sand, it was soon replaced by peat ash. Tests with dolomite are planned, however, the emphasis will not be on the reduction of the yield of tars since tars are no problem as the gas is burned hot in a gas turbine, but in improving on the carbon conversion efficiency. Dr Dierven suggested that it would be useful to have continuous in-line measurements of the concentrations of the major gaseous components such as H2, O2 etcetera.
Professor Beenackers wondered why part of the cyclone discharge is not recycled in the Finnish peat gasifier in order to improve on the carbon conversion efficiency. Dr Kurkela commented that the cyclone discharge consists of very fine particles which are difficult to handle due to their low bulk density. On the commercial aspects of turbines fueled by gasifiers Dr. Kurkela commented that such systems can be built soon since there is some interest from private companies on such technology but economics play an important role on the commercial prospects.
Dr. Capart asked whether the effect of heavy metals has been evaluated at all on gasifiers but the Chairman commented that with biomass feedstocks there are no problems with heavy metals in the gasifier.
On the question of Professor Corella concerning experiments with dolimite, Dr. Olsen reported that all their experiments were carried out in a thermal cracking reactor while catalytic cracking with dolomite has been planned for future research. Dr. Stiles reported that with dolimite tars can be converted to gas and transparent liquid while no sintering effect has been observed. His group is working in developing secondary catalytic reactors for the conversion and subsequent elimination of tars. Dr Olsen commented on a question from Dr Shah, that current practice in Denmark for straw disposal involves burning it while some heating plants also use straw as a fuel.
On a question by Dr. Dierven concerning the quality of coal used in the oxygen enriched air updraft gasifier, Mr. Engelbergh replied that the coal had an ash content of 25 wt % and a sulphur content of 1,5 wt %. In response to a question by Dr. Maniatis concerning the economics of using oxygen as a gasifying medium Mr Engelbergh stated that for the experiments in question liquid O2 from a storage tank was used. However, he commented that with advances in pressure swing adsorption techniques it will soon be possible to use of O2 seperation processes on site.
Prof. Beenaeckers enquired about the new aspects of this research in relation to the Lurgi oxygen gasifier. Mr Engelbergh responded that the new aspects were in the operation below the ash fusion temperature of coal and in applying air enriched in oxygen up to 10% instead of 100%. The 10% limit was governed mainly by limitations in steam addition which in used as a temperature moderator. With no further questions from the floor the Chairman acknowledged the valuable contributions of the participants and closed the session.


SESSION VI
UPGRADING, CLEAN OP AND UTILIZATION OF PRODUCTS


317
Gas purification: a review of the available methods of gas cleaning
P. GUIGON and J.F. LARGE Université de Technologie de Compiègne, France
Summary The paper discusses the available methods to be employed for cleaning gas emitted from
Biomass gasifiers. First the extent of treatment, according to utilisation of producer gas is presented then the different gas treatment techniques are reviewed and arrangement of cleaning devices suggested, depending of the use of the gas. Some cost considerations are given.
At the present time many gasification installations depend on the use of gas cleaning systems which are powerful, reliable and economic within a given context and which give all the guarantees required for the environment. The extent and difficulty involved in the cleaning of a gas depends on many factors: the type of installation (fixed bed updraft, downdraft, cross-flow, fluidized bed or circulating bed), the size of the installation, the type of biomass used and its water content, transportation constraints and the eventual use of the gas. At the same time, the solution adopted must take into account the site characteristics (availability of water, electricity supply etc.). Whatever the production system used, the gas will contain varying quantities of dust, tar and water vapour which will give aqueous condensates when cooled. Therefore a series of different gas cleaning systems will be required. In order to prevent the condensation of tars and their mixture with dust to form a semi-solid mixture which could block parts of the apparatus, the first step consists of dust removal at high temperature. This is followed by cooling of the gas by direct or indirect contact to its dew point, causing the formation of liquid tars and aqueous condensates. It is then often necessary to use further techniques in order to trap water and tar mists remaining in the gas. In the particular case where the gas is to be used in chemical synthesis, it is also necessary to remove any catalytic poisons which could affect following processes.
The best method to obtain a clean gas at the exit of the installation is to generate the least amount of tar possible in the first instance. Most manufactureres claim extremely low concentrations and even complete absence of tars (except for fixed bed updraft gasifiers). Practical experience and varied research shows that the removal of tar is a serious problem and that its total elimination is not easy (1).
In figure 1 (2) emission of different gasifiers are given and in particular the tar and solids content of the gas produced. The main method used to obtain low tar contents is to attain very high temperatures either in the gasifier ( in the case of downdraft, DelaCotte type recirculating or high temperature circulating bed) or in a post combustion chamber which may be catalytic. Catalysts have also been used in the gasifier itself to reduce tar emissions and improve the overall energy balance.
1 - TREATMENT (PURIFICATION) ACCORDING TO UTILISATION OF GAS
1.1 Gas for direct combustion When the gas is used to supply a furnace directly, virtually no treatment is necessary. High
temperature dust removal by cyclones is usually sufficient. However, for the heating of brick ovens, the direct drying of seeds, or the heating of greenhouses for example, a clean gas free of tars and dust is required. It is particularly important to control the quantity of polycyclic hydrocarbons when drying seeds (3). These hydrocarbons, contained in the combustion gas, are deposited on the first layer of seeds which then acts as a granular filter.

318
1.2 Gas to supply motors and turbines Gas leaving the gasifier at a temperature of between 300 °C and 800 °C must have both dust
and tar plus part of its water content removed. In diesel and spark ignition engines tars must be reduced to 1050 mg m3. For turbines it is essential to maintain the temperature of the gas above the dew point of condensable hydrocarbons (tars). However the inlet temperature of the gas to the motor must be as low as possible in order to maximize its volumetric efficiency.
1.3 Synthesis gas For gas used in chemical reactions it is not only necessary to remove dust by hot filtration,
but also tar and methane. This operation is either carried out in a secondary catalytic reformer (Mino process) or in a noncatalytic reformer (National Synfuels, Kiener, Creu sotLoire).
Figure 2 (2) gives an overall view of the extent of gas cleaning with respect to the ultimate use of the gas.
2 GAS TREATMENT TECHNIOI JES
2.1 Introduction The methods used for the purification of gas produced by gasifiers have not evolved greatly
since the 1930's. In publication (4) 910 references describing not only the treatment of gas from gasifiers but also that from high temperature furnaces and coke plants are itemized for the period from 1920 1970. Due to interest in combustion and the use of coal gasifiers linked to gas turbines, high temperature gas removal (>700 °C) has been the subject of numerous laboratory experiments resulting in several pilot plants over the last few years. However, opinions are still reserved over which solution to adopt for the industrial scale removal of dust from gas and exhaust fumes at temperatures higher than 400°C (5). Particulate and tar should not be removed in the same cleaning step to avoid any sticky combination of tars and char.
The classification of gas purification techniques is difficult. One approach is to use the collection mechanism (brownian diffusion, inerţial impaction or interception), but, in general, several of these mechanisms play a part in most purification systems. The efficiency of the mechanism depends on the size of the particles or droplets to be trapped. Those which are most difficult to remove are approximately Ιμπι in diameter. The types of equipment are classified as follows: mechanical, hydraulic, electrofilters, porous layers filters and finally various devices combining several of the above. Operating principles and calculating methods will not be given here but can be found in numerous general texts dealing with gas purification, in particular, references (6), (7) and (8). It is intended to concentrate more on the efficiency, implementation and potential of particular devices. In order to facilitate the presentation of devices available for gas treatment, the treatment process is divided into three categories:
1) The trapping at high temperature (higher than the dew point) of most solid material. 2) Condensation and elimination of condensed droplets. 3) Elimination of mists.
2.2 High temperature trapping of particles Purification always begins by the elimination of most of the particles at a temperature above
the dew point of the gas.
2.2.1 Mechanical devices Sedimentation chambers and inertia devices which are only suitable for large particles are
little used in the field with which this report is concerned. Centrifugal devices are, however, very often employed as they allow the separation at high temperature (temperatures at which tars are gaseous) of most dust particles.
Classical cyclones By the use of appropriate refractors little difficulty is encountered in the construction of de
vices capable of operating at 500 600 °C. Using ceramics, cyclones have been fabricated which can operate at 1000 °C. This technique allows dust removal to be accomplished with ease and results in very low dust concentrations. The efficiency with which dust particles are

319
removed by the cyclone depends on the inlet gas velocity and the diameter of the entrance window. Classical cyclones follow certain rules linked to this diameter. Theoretically, both the efficiency of dust removal and the pressure loss increase with increased entry speed and/or reduced diameter. The size of the cyclone and its cost may be correlated to the inlet area.. In order to obtain low exit concentrations (around 300 mg/m3) consisting of small particles (around 5 μτη) it is necessary to use small diameter cyclones which in tum leads to the use of multicyclones. By placing several cyclones (usually 3) in series, it is possible to obtain an output compatible with the tolerances required for gas turbines.
However, the pressure loss and cost of these small cyclones is significant Straightthrough cyclone with reverse flow In order to improve the efficiency of the cyclones by reducing entrainment by bouncing
small particules off the wall, a flow of clean gas is added with a descending circulatory movement . These devices have a very high effeciency (95% for particles > 5 μπι).
Straightthrough cyclone with moving impellers Another solution to giving gases a rotational motion is to use rotating disc devices . The
principle of these devices necessitates careful design, construction and installation. According to reference 9 there are some devices in existance which are capable of functio
ning at flowrates of 8000 nß/h and a maximum temperature of 400 °C. The output dust concentration is of the order of several mg/NirA Devices capable of handling 100,000 Nm^/h are being studied.
2.2.2 Hydraulic dust removal In order to function at high temperature, the cleaning fluid must remain liquid, leading to
the use of organic fluids or salts. This technique results in very high efficiencies. At the present time very little research is being carried out in this field, probably due to problems envisaged concerning corrosion and choice of materials.
2.2.3 Electrostatic filters These filters are mainly used to collect airborn ash in modern pulverizedfuel power plants,
and seem well adapted to the elimination of soot suspended in combustion gases. Much research has been carried out to increase the operating temperature of theses filters. The main difficulty encountered is to find materials which remain insulators at high temperatures. Furthermore, the dust particles must be capable of being attracted. Research appears promising, but the problems of the behaviour of electric insulators, the dimensional stability of ionisation cells and the removal of collected dust remain to be solved.
2.2.4 Porous layer filters Two types can be identified: flexible fibrous or rigid porous devices and granular bed de
vices. Flexible Fibrous or rigid porous layer dust removers A simple and efficient method for the removal of particles from a stream of gas is to pass it
through a fabric or any other porous sheet material. Progress made in the last ten years has led to very low output concentrations (<10 mg/Nm^). The medium serves only as a support, with the trapped particles acting as a filter. The method used to clean the filter is extremely important, as a blocked filter can cause pressure losses to exceed acceptable limits.
Fibres which can be used at high temperatures include metallic, glass, carbon or ceramic. Metallic Fibres
These can be used at temperatures up to 650 CC and have the advantages being capable of being woven and being available in diameters as low as 2 μπι. It is also possible to make sintered filtration media of adjustable permeability. The principle drawback is cost which has precluded their general use except where other fibres are not suitable.
Carbon Fibres These can be used at very high temperatures in reducing atmospheres, but in an oxidising
atmosphere temperature is limited to 350 °C. Furthermore, their mechanical strength is low. Thus, their use for high temperature filtration is limited.

320
Glass and ceramic fibres Quartz and ceramic fibres are capable of being used at temperatures well above those used
for the inlet gas to a gas turbine. Their main problem lies in the production of woven fabrics and needled felts. Their cost is not prohibitive.
The Acurex corporation has commercialized a filtration system (10) working at 870 °C. Normal glass fibres are limited to use at temperatures below 450 °C.
Granular bed dust removal The idea of a granular bed is not new, but is little used due to its cost, estimated at 125% of
the cost of an equivalent electrostatic precipitator (11). Such filters can be placed in three categories: fixed bed, mobile bed and fluidised bed, and can be considered as efficient but costly. One problem not yet totally resolved is behaviour in the presence of sticky particles, especially during regeneration of the bed. Note that this problem can be avoided by the use of a gasifiable solid so that regeneration of the bed is not necessary.
However, in general these filters cause a high pressure drop and cost more than other filters in terms of maintenance. It is usually, therefore, cheaper to use cyclones, scrubbers and electrostatic filters.
2.2.5 Combined dust removers and particle preconditioning Electrocyclones It has been shown that the most efficient cyclones are those of small diameter. By combi
ning the corona effect and cyclone phenomena, the same efficiency can be obtained for an electrocyclone of 3.7 m diameter as for a normal cyclone of 0.25 m diameter.
Electrically enhanced fabric filter ((8) chapter 7) By charging particles before entry to the filter, a more permeable cake is formed which in
tum allows the use of filtration speeds double those normally adopted. Acoustic agglomeration Particles of around Ιμπι are very difficult to trap and a number of experiments have been
carried out to try and cause them to agglomerate before separation using conventional techniques. Acoustic agglomeration appears promising (6).
2.2.6 Conclusion on hot gas cleaning It would seem that no hot dust removal technique has yet reached the stage of being capable
of reducing dust concentration to levels compatible with gas turbines (5,9). Certain devices accomplish primary dust removal at a reasonable price: multicyclones, straight through cyclones with reverse flow and, in the future, electrocyclones. Granular beds may be interesting if solid regeneration is not required. With new developments in ceramics, both flexible and rigid porous layer filters could become viable in the not too distant future.
2.3 Condensation and elimination of condensate droplets The condensation of water vapour and tars contained in gases can be accomplished by ei
ther direct or indirect heat transfer. Indirect heat transfer is avoided as much as possible due to the possibility of fouling the exchangers. Nearly all fixed gasifiers where a water supply is available use water scrubbers. In certain cases, oü scrubbers may be used.
2.3.1 Scrubbers These devices allow droplets and particles to be collected while cooling the gas. Generally
they are used to remove condensed tar droplets and to eliminate soluable matter. It is then generally necessary to provide for mist elimination. These spray scrubbers exist in numerous forms. In most cases, water or aqueous condensates constitute the scrubbing agent, but occasionally oil, which has an absorption efficiency higher than that of water, may be used. These scrubbers cause problems for the treatment of the liquid effluent as all soluable organic components are recovered in the aqueous phase.
Spray towers. The spray tower is the simplest type of scrubber. It consists of an empty tower in which the
gas flows through a mist of water droplets formed by suitably placed jets. Countercurrent,

321
cocurrent or crossflow configurations may be adopted. These towers are excellent for collecting large particles and for cooling the gas.
Centrifugal spray scrubbers The use of centrifugal force allows the removal efficiency of the spray scrubber to be in
creased by increasing the speed of the droplets in relation to that of the gas. Small baffles give the gas a spiral motion. This increase in efficiency is counterbalanced by a higher pressure drop across the system. Such devices have an efficiency of more than 97% for particles larger than Ιμιτι.
Self induced spray scrubbers In this type of scrubber the gas is introduced at high speed just above or below the surface
of the liquid. The resulting high turbulence give rise to excellent gas/liquid contact. The advantage of these scrubbers is their ability to cope with high solids concentrations.
Impingement plate and packed column scrubbers The gas to be treated passes through a series of liquid layers. These scrubbers are more ef
ficient for the removal of fine particles than evaporative scrubbers, but result in high pressure drops. The design of the columns is analogous to that of plate distillation columns or packed absorbers. The type of packing used can be commercially available or manufactured using a variety of materials.
Disintegrator scrubbers If very small particles must be recovered (smaller than Ιμπι), the scrubber must provide a
very finely dispersed mist. These tiny droplets result in a very high surface area over which contact can occur. In order to obtain this fine mist, the liquid is introduced between the rotor and stator of a special device. The gas must be prepurified (concentrations < 23 g/m3) and have particles greater than 10 μπι eliminated, as these would tend to erode the baffles contained within. Energy requirements are high (around 5.27.0 kJ/m3 of gas treated), but efficiency is excellent with respect to particles below Ιμτη.
Venturi type scrubbers In this type of device the speed of the gas causes atomisation of the liquid. The energy is
supplied by the pressure loss of the gas. The efficiency is improved if conditions are created such that condensation can occur in the throat of the venturi. Venturi scrubbers can be supplied with one or more throats in parallel or in series. Permanent pressure drop is of the order of 0.4 to 1.0 kPa and efficiency can reach 99% for particles smaller than 5μιη.
Free jet washers In this device, atomisation of the water is caused by a jet of compressible fluid and takes
place around the exit nozzle of this fluid. Pollutants are trapped in the turbulent region directly following the jet
2.3.2 Electrofilters Such filters have already been used with success in coking plants to eliminate tars. Various
electrode configurations are possible: a wire placed in the centre of a vertical cyclinder, perforated or corrugated electrodes, or single or double plate electrodes. Efficiency is excellent (greater then 99%). Pressure loss in these devices is low, which suits them to the treatment of large flows. Small electrostatic filters capable of trapping tars emitted from gasifiers are being studied (12).
2.4 Demisters After leaving a cooling and condensation stage, the gas still contains water and tar in the
form of mists which must be trapped in demisters. These usually consist of a porous bed. As the liquid particles agglomerate and flow by gravity there is no need for a cleaning system, but it is of course necessary to provide a drain for the collected liquid. Materials which can be used vary greatly. Woven metal is often employed; fibrous waste such as wood wool may also be used. Large particles are usually trapped by a metallic grid system. These filters require frequent cleaning and replacement and, therefore, regular and careful maintenance. In the case where the gas is to be used in a motor, a simple paper filter which doubles as a security

322
measure is placed at the inlet. If the tar removal system is not cleaned regularly, the filter blocks and stops the motor.
3 ARRANGEMENT OF CLEANING DEVICES. In practice, the devices described can be arranged in very different ways in order to carry
out the cleaning of biomass gas. For each particular type of gasifier, it is necessary to set up a chain giving an efficiency compatible with the final use of the gas. In order to ensure stable operation, the removal of ash and tar is carried out, as much as is possible, in two different steps. If tar removal only is required, wet scrubbers are the current system employed. Table 1 (2) gives the combination of devices necessary for particular applications and different gasifiers.
4 COST OF GAS TREATMENT The cost of the gas treatment is directly linked to its extent. In general, each installation is a
particular case which makes cost prediction difficult. However, in general, the more complicated the installation, the more costly the investment, running and maintenance. Little information on the cleaning of gas is to be found in the literature on gasifiers. Where such information is available, it usually concerns pilot plants, and therefore does not necessarily represent the cost of an industrial scale system. Bridgwater (13) has established a simulation program for the thermal conversion of biomass. The cost of gas cleaning for the simulated installation is determined
The graph shown in figure 3 gives the relative cost of different devices with respect to the performance required.
5 CONCLUSION Existing technology is capable of supplying gas sufficiently clean for uses such as direct
combustion, supply to motors, or for production of synthesis gas. However, in order to be able to dimension gas cleaning systems for various gasifiers, it is absolutely essential to have reliable data on the content, size and composition of the particles. At the present time, wet scrubbers are the only viable technique for removing tars. Further studies concerning their efficiency with respect to the trapping of tars are necessary in order to allow their design.
The cost of an effective treatment remains prohibitive for most applications. For gas turbines, the use of hot dust removal techniques remains to be proven. The methods used to treat water before discharge into the environment have been little studied but the cost of the treatment of water heavily contaminated with organic carbon seems high. For gasifiers installed in developing countries, it is imperative that efficient but cheap treatment systems are found. The most promising method seems to be to reduce tar content by the use of catalysts or postcracking systems.
REFERENCES (1) Rensfelt E. "Practical Achievements in Biomass Gasification " Bioenergie 84
conference Vol.1 ρ 174 ; ed. H.E. Grens et A.Ellogard; Applied Science publisher London (1984).
(2) Brown M.D., E.G.Baker, L.K.Mudge "Environmental design considerations for thermochemical biomass energy" Biomass 11(1986)255270.
(3) Barrett J.R., R.BJacko "Environmental aspects of biomass furnaces used in agriculture air pollution and grain contamination " Bioenergie 84 conference Vol.4 ρ 442 ; ed. H.E. Grens et A.Ellogard; Applied Science publisher London (1984).
(4) Epuration du Gaz de bois pour la combustionSaskatchewan Power CorporationRapport SPC n° 440019791 (énergie de rechange) (1980).
(5) Emerging clean coal technologies ; Noyes data corporation (1986). (6) Strauss W. "Industrial Gas Cleaning " Pergamon (1975). (7) Batel WV'Dust extraction technology" Technicopy Limited (1976). (8) Donovan R.P. "Fabric filtration for combustion sources " Marcel Dekker, Inc
(1985).

323
(9) Cahier de l'utilisation du charbon . CODETEC CERCHAR :" Dépoussiérage sous pression et haute température des fumées du charbon "(1984).
(10) 'Tine particle filtration now possible for 600 °F to 1600 °F gas stream." News release, September 12,1983, Acurex Corporation, 485 Clyde Avenue, Mountain View, California. 94042.
(11) Razgaitis R."An analysis of high temperature particulate collection problem"-Argonne National Laboratory report ANL-77-14 (october 1977).
(12) P.CORTE "Purification des gaz de gazéification" Biomasse actualité ;numero special n°5 avril (1984).
(13) Bridgwater A.V., R.E. Gowers, R.N. Shand, G.J. Walker "Simulation of complete biomass conversion processes"-Bioenergy 84 conference Vol.3 p. 121 ; ed. H.E. Grens et A.Ellogard; Applied Science publisher London (1984).
This work has been done under contract n°86 - Β - 7031 - 11 - 004 - 17
io
Fixed Bed Updraft
Fixed Bed Downdraft
Fixed Bed Cross Flow
Fluid Bed
Entrained Bed
mg / Normal m 3
100 1.000 T "
E S S S S S l
K W W W M
10.000 100.000 T "
ΕΣΣΣΣΣΣΣΣΣ
ESS5S
tVWVv _
Tar Particulates
Figure 1 - Emission of different types of gasifiers (from ref. 2)

324
mg/m'
1000
1 0 0
1 0
Unacceptable
stat· Regulation»
NSPS (Fadaral)
^
^
Accetable
3 m
_ o β «« o c · c β ja ? ** c
c
c t β — o
a Ζ β to
ι
Figure 2 Allowable particulate loadings for various end uses (from ref. 2).
Cost Relative to a Medium
Efficiency Cyclone
ReversedJet "Fabric Filter
0,1
10
'Venturi Scrubber
Electrostatic. Precipitator
L
HighEfficiency
I Cyclone
Mil· MediumEfficiency
I I Cyclone , ' I I I I I |
1 10 Penetration (1 η ) %
100
Figure 3 Relative cost of different apparatus versus efficiency (from ref. 11).

325
Performance standards
Fixed bed updraft *
Fixed bed downdraft
Fluidized bed
M
Entrained bed
Close-coupled boiler
200-1500 mg nr 3
None b
None b
C
2 C b,c
C + WS C + F
ESP M
similar to fluidized bed
Diesel or spark-ignition
engine 10- 50 mg m-
3
WS + F
C b
'e
C + F WS
b
WS + F
C + F C + WS
b
C + ESP M
ι
Gas turbine 1- 80 mg m-
3
WS b
WS + F WS + ESP E S P
b
Cb
2C b
C + F C + ESP
C + WSb
C + F C + ESP
d
f
Syn 1-
gas or SNG 80 mg m-
3
WS b
WS + F WS + ESP E S P
b
Cb
2C b
C + F C + ESP
C + WSb
C + F C + ESP
d
f
C = cyclone: 2C = two cyclones in series: F= fabric filter (baghouse): WS = wet scrubber: ESP = electrostatic precipitator. a Cyclone not effective due to smaller particle size distribution and tar droplets - use wet scrubber to remove tars first if any cleanup is required. b Lower level of contaminants is acceptable - higher level would exceed limits. c Assumes 50% of particulates is char and 90% burns in burner. d ESP is not as effective on particulates with high carbon content and may not be applicable. c Cyclones are effective for this application but wet scrubbers are often used instead because gas cooling is also required. Also cyclone efficiency is affected by large turndown ratio required in some engine applications. f Pressurized operation may restrict the size or applicability of gas cleanup equipment (particularly baghouse and ESPs).
Table 1 - Applicable gas cleaning options (from ref.2)

326
WHAT CAN WF. DO WITH PYROIYSIS OILS?
E. CHURIN and B. DELMON Unité de Catalyse et Chimie des Matériaux Divisés
Université Catholique de Louvain Place Croix du Sud, 1
B-1348 Louvain-la-Neuve (Belgium)
S u m m a r y
The "liquids" obtained by pyrolysis or liquefaction of biomass present the advantage of being easy to handle and have high energy density but because of the high oxygen concentration the volume needed to replace heating oils is around l.S times higher. When dried, the oils are viscous but the water dissolved diminish greatly the viscosity; the nature of the oil affects also the viscosity. If oxygen is removed the composition of the oil is very similar to petroleum-derived fuels. The oxygen removal can be carried out at almost atmospheric pressure or at high pressure. At low pressure, in the presence of a zeolite catalyst oxygen is eliminated as CO, CO2 and principally H2O. Hydrocarbons yields of between 5 and 10 % on a wood basis have been reported. The coprocessing of pyrolysis vapors and methanol allows for the obtention of higher yields. The desoxygentation can also be effected under high hydrogen pressure and the hydrocarbon yield is higher than for atmospheric pressure upgrading.
1- Character is t ics of pvrolvsis ails
Biomass can be converted to "liquids" by pyrolysis, by liquefaction under high pressure or by solvolysis. The "liquids" present the advantage of being easier to handle than biomass and present a much higher energy density. These liquids are intended for use as fuels. In general a fuel consists of one or more compounds containing carbon and hydrogen with or without oxygen, other components (such as Ν and S) have a negative influence on the quality. Biomass "liquids" are well positioned because their Ν and S contents are very low. From an energy point of view, hydrocarbons without oxygen are preferable because it do not add to the energy content of the fuel.
Table 1 shows the composition of biomass pyrolysis oils obtained by different process as well as the composition of petroleum derived fuels. Because of the high oxygen content, the energy density (on a weight basis) of the pyrolysis oils is lower. The commercialization of fuels is generally done on a volume basis and if the energy content is calculated on a volume base, the difference with petroleum fuels diminishes because the density of the pyrolysis oils is higher than 1.
In any case, if a given amount of heat is needed, the volume of pyrolysis oil burned should be between 1.5 and 1.8 times greater than the volume of fuel oil with the consequent influence of the selling price.
The fuel oils with which pyrolysis oils are compared are obtained by mixing different residuums and distillates in order to meet the legal

327
specifications. For a N° 6 fuel oil, distillates account for between 5 and 20% while for a N° 2 fuel oil they account for between 20 and 50%.
Table 1 : Composition of fuel oils and pyrolysis oils.
N°2 fuel oil 33° API
N°6 fuel oil 15.5° API
Waterloo pyrol. oil
Raiano Pyrol. oil
Carbon(%) Hydrogen(%) Oxygen(%) Nitrogen(%) Sulphur (%) H/C ratio Density(kg/1)
87.3 12.6 0.04 -0.22 1.73 -
84.67 11.02 0.38 0.18 3.97 1.56 -
52.6 6.5
40.8 η a η a
1.48 η a
66 7.2
26 0.8
<0.1 1.31 1.2
Due to the increasing demand for transformation fuels such as gasolines, diesels and kerosins, a greater conversion of other fractions to these products has been encouraged resulting in smaller volumes of residual oils. Environmental legislation could place constraints on the use of residual fuel oils due to the presence of sulfur, nitrogen, vanadium, nickel and iron concentrated from crude into residue, the present commercial grades of residual fuel oils have sulfur contents that may vary from 0.7% to 4% and the metal contents can vary between 50 and lOOOppm. In this respect, biomass pyrolysis oils are well placed because they contain no metals at all.
Two additional points should be considered when comparing bio-oils with petroleum products. These are density and viscosity. Table 1 shows the that density of bio-oils is always higher than 1 and this should be taken into account during handling and storage since rain water or water from leaking steam coils rises to the surface of the oil. Oil spillage is another matter that cannot be ignored because with a density higher than 1 kg/1 the oil settles to the bottom of the watercourse and drainage systems. Interceptors should be adapted to cope with such oils.
As for the viscosity of the oil, it depends to a great extent on the nature of the oil and on the water content. The latter depends on the reaction conditions but if water is used for quenching pyrolysis vapours, the oil becomes saturated (and it can disolve a lot of water because of its polar nature)
Table 2 shows dthe influence of the water content and the nature of the oil on viscosity. Figure 1 is a comparison of two oils to petroleum products.
Table 2: Influence of the nature of the oil and the water content on viscosity.
Β D
Water content (%) υ et 60°C (cp)
29 10
10 480
0.15 60
0 450
12 240

328
Maximum viscosity ■ tor typical storage
ƒ Maximum viscosity for pumping and handling
Steam atomization
Mechanical atomization
Fig.l: Viscosities of petroleum products and pyrolysis oils.
Taking these considerations into account, pyrolysis oils can be burned satisfactorily. Minor changes in atomizer design may be needed because some deterioration of the fuel spray has been detected and which is probably due to blockage or incrustation. This problem is apparently related to a decomposition of the oil when heated in the atomizer by the flame or the atomizing steam (11).
For other high value utilizations, the pyrolysis oils must be upgraded and transportation fuels or petrochemical feedstocks can be obtained.
2. Upgrading of the pvrnlvsis nil In view of their properties, pyrolysis oils should be considered as crude
oils which need to be refined and upgraded in order to recover different fractions of a lower viscosity, higher volatility and heat content, presenting a good stability for use as fuels.
The heat value of a fuel can be determined easily using the elemental composition of the material with Dulong formula:
NHV (Kcal/kg)=81xC%+300x(H%-l/8xO%)+25xS%-53.46xH%
When the elemental composition of the pyrolysis oil is compared to the composition of distillates or fuel oils, it can be seen that in order to increase the heat value, oxygen should be removed. The elimination of oxygen results in a diminution of weight and when carbon and hydrogen concentrations are calculated on the new weight basis, composition is similar to that of petroleum cuts. The oxygen removal can be carried out with or without a reducing gas.
2.1. Upgrading in the absence of a reducing gas In the absence of a reducing gas, oxygen is eliminated as H2O, CO or CO2
(to form H2O) and carbon (to form CO and CO2); the yield to hydrocarbons is rather low. Theoretical yields from calculations based on a composition of the

329
pyrolysis oil presented in Table 1 show that if all the oxygen is eliminated as C O 2, the hydrocarbon yield is 43% with an H/C ratio of 2. If all the oxygen is eliminated as CO the hydrocarbon yield is 27% with an H/C ration of 4 while if the oxygen is eliminated as water, the yield is 14.0% with an H/C ratio of 1.4.
A synthetic zeolite catalyst (ZSM-5) has been particularly effective in the conversion of oxygenated compounds, mainly methanol to hydrocarbons (1) (2). In the case of methanol, during the early stages of the reaction, it is rapidly converted to dimethylether and water. The first hydrocarbons produced are mainly olefins which react to give paraffins and aromatics. Typical yields obtained in a fixed bed are hydrocarbons, 43.7%; water, 56.2%; CO and CO2, 0.04%, the rest being coke and oxygenates. Alcohols are good feeds for transformation by this process because of the low tendency to produce coke which deactivates the catalyst. One of the first researchers to study the conversion of pyrolysis vapours over a zeolite catalyst was Frankiewicz. Vapours were produced by the pyrolysis of solid wastes and were contacted with the catalyst in a second reactor (3). Reaction of a model compound, furfuryl alcohol was also studied. Since then, several groups have studied the conversion of pyrolytic vapours of biomass over this catalyst (4)(5)(6).
Using this catalyst, most of the oxygen (between 70 and 80%) is eliminated as water and the rest as CO and CO2. Parallel undesired reactions also take place such as coke formation throught aromatization reactions which contribute to the reduction of hydrocarbon yields due to the consumption of carbon atoms. As disclosed in (6) hydroxyl and methoxy groups tend to reject oxygen in the form of water, aryl ethers reject a nearly equal amount of oxygen as carbon monoxide and water, carbonyl and formate groups reject oxygen as carbon monoxide and carboxyl as carbon dioxide and water.
Since rejection of oxygen as water involves the elimation of hydrogen, Chang et al (7) have introduced the concept of "effective hydrogen index" (EHI) defined as :
EHI-H-20-3N-2S
where H, C, Ο, Ν and S are atoms per unit weight of sample. They show that compounds with EHI<1 cause a rapid deactivation of the catalyst and give a poor product distribution. But they propose also that a lower catalyst deactivation a synergistic yield improvement could be obtained by co-processing the low EHI feed with a sufficient amount of high EHI compound, which could act as some kind of hydrogen donor. Methanol could be used as a high EHI feed.
The yields obtained by Chen et al. (4) when pyrolysis vapour is contacted alone with methanol (1:1 ratio) in a fluid bed with a ZSM-5 catalyst are shown in Table 2.
Table 2: Product distribution from conversion of pyrolysis liquids over ZSM-5 catalyst.
Pyrolysis liquids Pyrolysis liquids + methanol
CO% 002% H20% Cl-C4% C5+ hydrocarb.% Coke%
0.7 10.5 70.7
2.5 6.0 9.6
82.1 2.7
11.2 4

330
The hydrocarbon yield is only around 3% on the basis of the wood processed. The yield is rather low because the percentage of the wood carbon retained in the liquid was only 30% (the retention of carbon in the liquids can be as high as 60%). Researchers from the Université de Laval (8) report yields of between 5 and 10% on a dry wood basis without methanol. The product obtained is a high quality gasoline consituted of mainly alky-substituted benzene .
This process can be accomplished in a fluid bed with constant withdrawal and addition of catalyst. The withdrawn catalyst is regenerated by combustion of the coke and is then put back into the reactor. Another possibility would be a moving bed such as the one used in modern catalytic reforming of naphta with the same regeration process.
If it is proven that a very short contact time is sufficient to obtain the desired conversion, an entrained-bed reactor such as the one used in catalytic cracking of hydrocarbons could be used.
2.2. Upgrading in the presence of reducing gas The upgrading effected in the presence of a reducing gas (hydrogen)
aims at removing oxygen as water and at reducing the molecular weight of the heavy fractions. From the theoretical point of view, and based on the composition of the pyrolysis oil presented in Table 1, it can be calculated that if all the oxygen is rejected as water with externaly supplied hydrogen the hydrocarbon yield could be as much as 58%.
The transformation is conducted under high pressure of hydrogen and in the presence of a catalyst. Typical petroleum hydrotreating catalysts, namely cobalt and molybdenum and nickel and molybdenum in the sulfided form supported on alumina are very active. Though there are some similarities, there are also notable differences between petroleum products and pyrolysis oil components because the presence of the large quantities of oxygen lead to a degradation of the oil during heating. This degradation could be interpreted in terms of free radicals. In fact, upon heating, highly hydroxylated compounds can generate free radicals and the nature of the products obtained depends on the manner in which they are stabilized. The possibilities are:
- Reaction of the highly reactive free radicals with one another to give higher molecular weight materials including coke;
- Free radicals are capable of obstracting hydrogen from neighbouring hydrogen-rich groups or molecules in the mixture.
The latter possibility can be exploited to stabilize the radicals and facilitate the conversion if the concentration of hydrogen-rich compounds which are able to donate hydrogen is high enough.
The effect of the temperature is also very important and we have found, like Elliot (9), that a pretreatment at around 230°C allows for a "stabilization" of the oil and higher yieds are obtained in the second stage of the pretreatment. Without the pretreatment, the polyhydroxylated compounds polymerize and deactivate the catalyst very quickly.
In the test effected in this laboratory to upgrade pyrolysis oils, tetralin was used as hydrogen donor.
Table 3 shows the influence of the tetralin on the nature of the product obtained kwhen pyrolysis oils produced at the Raiano demonstration unit (Italy) are processed in two stages.

331
Table 3: Characteristics of the hydrotreated oils
Without te t ra l in
%HDO 70 %HDN 58 Volatile fraction% 50
Oil:tetralin ratio 1
85 85 95
Table 4 shows the results obtained at the PNL (hydrotreatment in two stages at 274°C and 353°C) by Elliot and Baker on a pyrolysis oil produced at Geòrgie Tech. in an entrained bed. The product obtained in the first stage is used as feed in the second stage.
Table 4: Two-stage upgrading of Geòrgie Tech. Pyrolysis oils
Catalyst Temperature (°C) Pressure (MPa) LHSV (vol.oil/vol.cat.h)
Yields Total oil (L/L feed oil) C5-225°C (L/L feed oil) Hydrogen consumption (L/L feed oil)
Stage 1
C0M0/AI2O3 247 13.5 0.62
0.69 0.07 60
Stage 2
C0M0/AI2O3 353 13.5 0.11
0.62 0.45 576
The total oil yield obtained for the combined two-stage process is 43% and the fraction C5-225°C amounts to 31%.
The results obtained by processing different pyrolysis oils enables Elliot (9) to estimate the following mean yields: 0.5-0.55 1 of oil could be produced per litre of pyrolysis oil processed; the oil could contain 2-3% of oxygen and could have a H/C ratio of 1.5 and a density of 0.9-0.92; about 50-60% vol. of the oil could be in the gasoline boiling range; about 30% is distillable, the rest being a residual oil.
The oxygen content of the oils obtained at different contact times show (Fig. 2) that only an LHSV lower than 0.2 allowed for an oxygen content lower than 2%. This is inconsistent with the facility found to remove oxygen from model compounds (10) and cannot be attributed to the conversion of heavy compounds because petroleum cuts with higher mean molecular weight are processed at higher space velocities. On the contrary, such a low space velocity can be explained by a deactivation. The use of a hydrogen donor allows for higher conversions under the same reaction conditions. The product obtained by hydrotreating pyrolysis oils is highly aromatic and constitutes an excellent BTX or gasoline cut. The cetane index is too low to contribute significantly to a diesel cut.

332
o s
κ O
Pyrolysis Oils
• TR7 O TRI2 ■ Pyrolysis Oils
0.5 0.6 0.7
Fig. 2 : Effect of space velocities on various biomass-derived oils (From (9)).
The product obtained by hydrotreating pyrolysis oils is highly aromatic and constitutes an excellent BTX or gasoline cut. The celane index is too low to contribute significantly to a diesel cut.
Further research needs to be carried out in order to determine the cause of the low reaction rate found when processing pyrolysis oils and to find more active and resistant catalysts. Long duration reaction studies are needed in order to determine the catalyst lifespan. If a severe deactivation is confirmed an ebullating bed reactor such as the one used by Hydrocarbon Research Inc. in the Η-Coal (R) process should be considered for the industrial implementation of the conversion. In this kind of reactors, continuous catalyst replacement restores the activity and yields a product of a constant quality; the catalyst can be continuously regenerated.
R e f e r e n c e s
(1) S.L. Meisel, J.P. Mc Cullogh, CH. Lechthaier and P.B. Weiz, "Gasoline from methanol in one step", Chemtech, Vol. 6, 1976, p. 86.
(2) C D . Chang and A.J. Silvestri, "The conversion of methanol and other oxygenated compounds to hydrocarbons over zeolithe catalysts", J. Calai., Vol. 47, 1977, p. 249.
(3) T.C. Frankiewicz, The conversion of biomass derived pyrolytic vapors to hydrocarbons, p. 123 in Proceedings Specialists' Workshop on Fast Pyrolysis of Biomass, ed. J.P. Diebold, Copper Mountain, Colorado, USA, October, 19-22,1980.
(4) N.Y. Chen, D.E. Walsh and L.R. Kocnig, "Fluidized bed upgrading of wood pyrolysis liquids and related compounds", Am. Chem. Soc. Div. Fuel. Chem., Preprints, Vol. 32, n° 2, p. 264, April 5-10, 1957, Denver, Colorado, USA.
(5) P.D. Chantal, S. Kaliaguine and J.L. Grandmaison, "Reactions of Phenolic compounds over H-ZSM 5", Appi. Cat., 18, 133-145 (1985).

333
(7) CD. Chang, W.H. Lang and A.J. Silvestri, U.S Patent 3.998.898 (1976). (8) M. Renaud, J.L. Grandmaison, Ch. Roy and S. Kaliaguine, "Conversion of
vacuum pyrolytic oils from populus deltoides over H-ZSM 5", Am. Chem. Soc. Div. Fuel. Chem., Preprints, Vol. 32, n° 2, p. 276, April 5-10, 1957, Denver, Colorado, USA.
(9) D. Elliot and E. Baker, "Hydrotreating biomass liquids to produce hydrocarbon fuels", in Energy from Biomass and Wastes-X, Ed. D. Klass, Elsevier Applied Science Publishers, London, 1987, p. 765.
(10) M. Callara , MSc. Thesis, September 1988, University Catholique de Louvain.
(11) Finney CS. and Sottier J.G., AICHE Symposium Series N° 164, Vol. 71, p. 51.

334
COMPOSITION, PURIFICATION AND USE OF PRODUCER GAS
J . VAN DER WEIDE and J . J . SEPPEN TNO Road-Vehic les Research I n s t i t u t e
SUMMARY
The paper discusses the strong variation in the composition of producer gas. This variation is generally caused by insufficient control on the process and quality of the feed stock. The amount of impurities (tar and particles) also tend to increase when the process is not under optimal control. In conventional systems these problems are the cause of a substantial power variation and often too much engine maintenance. The latter can be overcome reasonably by proper purification. Producer gas is generally used in spark ignition engines and diesel engines. In spark ignition engines sometimes petrol is used for start up. Diesel engines are generally started on 100% diesel. The producer gas is used by gradually feeding it into the combustion air and reducing the amount of diesel used simultaneously. These system lay-outs are discussed. The paper concludes that by additional hardware a better process control of the whole system is necessary. Proposed thereby is to use low cost automotive micro-electronic componentry. Good low cost sensors are available for measuring oxygen content, temperatures and pressures. As an output of such an automotive micro-processor the process can be controlled by air injection or grate activation and automatic engine adjustment.
1. INTRODUCTION In the Netherlands the concern for the greenhouse effect is strongly
increasing beside many other environmental concerns. Using biomass materials for energy generation has compared to the conventional fuels the great advantage of no C02-contribution to the atmosphere, because of the closed cycle in which it is used. It will become probable that political pressure with regard to the use of biomass materials for energy generation will arise. Until sofar quite a lot of producer gas projects have failed. Many problems still have to be solved. For the developed world, in particular, an extra problem is that generally rather a lot of manpower attention is necessary to operate a producer gas engine system. This is than an important cost factor. This paper emphasizes on the enumeration of the different problems in gasifier engine systems. Furtheron new technologies to overcome these problems are discussed. The paper ends up by making recommendations for the future research.
2. SUMMARIZATION OF CONSTRAINTS 2.1 PROBLEMS IN GASIFIERS
Generally there is an interaction between problems in the gasifier and the engines. For this reason these problems are summed up in general terms.
The most important problem, with the application of gasifiers, is the fact that the gas quality (calorific value and the amount of dirt) is dependent on the fuel parameters. In particularly in respect to water content, ash content and chop size.

335
Water content of the fuel: Tar formation is the most general problem, due to lower temperatures in the reactor. Tar formation leads to rapid engine failure when it is not filtered out. This means that the possibility to control the gasifier is strongly reduced with increasing water content of the fuel.
Ash content of the fuel: In case the ash content of the fuel exceeds a certain value there is a possibility of slagging in the reactor. Generally this problem is solved (at least in smaller gasifiers) by manual operation of the grate. This needs a lot of attention from the operator.
Chop size: Generally the chop size has to be adapted to the reactor design. Sometimes it is necessary to reduce the chop size. This is labour and mechanical power consuming.
2 . 2 GENERAL PROBLEMS IN ENGINES The most common problem with the application of producergas in combustion engines is the lifetime of the engine. This is quite a general complain although documented cases are known of very good lifetime combined with good gas cleaning.
Engine wear is a result of unwanted components such as dust, tar and corrosive components in the gas. This is strongly dependent on the proper operation of the gas cleaning system.
Dust: Soot, carbon and ash particles are produced in the reactor and carried with the gas. In case the gas cleaning operates insufficient, this material comes into the engine cylinders and creates wear of the cylinder wall and piston rings. Due to the resulting "blow by" these particles can also come into the lubricating oil. These particles, in particular when they are larger than the thickness of the oil film, can also damage the bearings. The information avalaible is insufficient with regard to maximum allowable concentrations and size. Noticed is that the wear is also dependent on the type of engine.
Tar: Tar is formed during the gasifying process. Although some manufacturers of gasifiers claim that no tar is formed it is noticed that tar is always present sometimes as small droplets. Tar as such does not create abrasive wear but can settle down on valves, piston rings, etc. and thereby obstruct the operation. Sometimes tar becomes bitumenous, which can create problems at particularly at starting of a cold engine.
Corrosive components: The corrosive components in the gas (carbon acids, ammonia) do not create problems when the engine is on its working temperature. But during cooling down of the engine condensates can be formed resulting in fast corrosive wear of cylinder and piston rings. Furtheron there is the suspect that small amounts of sodium in the lubricating oil (from the producergas fuel) can create valve corrosion.
A specific problem with the application of producergas in engines is the fluctuating caloric value of the gas. This leads to changing engine power.
The chemical composition of producergas is such that in particularly due to the large carbon monoxide content a relative slow combustion speed occurs. In particularly with fast running engines this results in reduced power and efficiency. Generally power and efficiency start to reduce at engine speeds above 2300 rpm. This means that for obtaining a certain power relative slow running and thus large and expensive engines are necessary.
Good mixing of the gas with the combustion air is necessary to obtain a homogeneous mixture and to avoid wrong fuel distribution over the different cylinders. This can substantially reduce engine power and efficiency.
2.3 PROBLEMS MORE TYPICAL TO OTTO ENGINES The most common problem with the application of producergas in otto engines is the power loss in comparison with the operation on petrol or natural

336
g a s . T h i s i s due t o t h e r e l a t i v e low c a l o r i f i c v a l u e of t h e g a s , r e s u l t i n g g e n e r a l l y i n r e d u c i n g e n g i n e power by some 40-50%. T h i s makes a much more e x p e n s i v e e n g i n e n e c e s s a r y t o r e a l i z e a c e r t a i n power .
Cases a r e known ( i n p a r t i c u l a r l y w i t h one or two c y l i n d e r s ) t h a t o s c i l l a t i o n i n t h e i n l e t m a n i f o l d , a gas s t r e a m o c c u r s , r e s u l t i n g i n power l o s s e s up t o 70%.
I n p a r t i c u l a r l y i n o t t o e n g i n e s due t o ho t p a r t s b a c k f i r e can o c c u r . T h i s can r e s u l t i n damaged e n g i n e p a r t s and i n some c a s e s even t o e x p l o s i o n i n t h e g a s i f i e r .
2 . 4 PROBLEMS MORE TYPICAL TO DIESEL ENGINES D i e s e l e n g i n e s can be o p e r a t e d p a r t l y on p r o d u c e r g a s i n s o c a l l e d d u a l - f u e l o p e r a t i o n . Due t o t h e f a c t t h a t a d i e s e l e n g i n e has r e l a t i v e e x c e s s a i r a v a i l a b l e , t h e power l o s s i s r e d u c e d t o some 10%. I n j e c t i o n over s m a l l q u a n t i t y of d i e s e l o i l (some 10-15% of t h e amount on maximum power) i s a l ways n e c e s s a r y t o i g n i t e t h e g a s / a i r m i x t u r e . E n g i n e s w i t h d i r e c t i n j e c t i o n a r e more f a v o u r a b l e fo r t h i s o p e r a t i o n t h a n e n g i n e s w i t h a p r e - chamber ( p r e - c h a m b e r t e n d s t o c r e a t e p r e - i g n i t o n ) . I t w i l l be c l e a r t h a t a t low e n g i n e power t h e r e l a t i v e amount of d i e s e l o i l r e p l a c e d t e n d s t o become s m a l l e r . T h e r e f o r e t h i s t y p e of o p e r a t i o n i s f a v o u r a b l e f o r e n g i n e s r u n n i n g under c o n s t a n t h i g h l o a d .
Sometimes t h e d i e s e l f ue l i n j e c t o r s i n d u a l - f u e l o p e r a t i o n become o v e r h e a t e d . Due t o t h e f a c t t h a t t h e s m a l l e r amount of d i e s e l o i l g i v e s a s m a l l e r amount of c o o l i n g on t h e i n j e c t o r .
I n d u a l - f u e l o p e r a t i o n somet imes k n o c k i n g o c c u r s . T h i s e n g i n e k n o c k i n g can r e s u l t i n s e v e r e e n g i n e damage . The c a u s e i s p r o b a b l y o v e r f u e l l i n g o r a r e l a t i v e h i g h hyd rogen c o n t e n t i n t h e g a s .
3 . DISCUSSION WITH REGARD TO NEW TECHNOLOGIES The f o r e m e n t i o n e d prob lems w i l l be d i s c u s s e d w i t h an emphas is on t h e
a p p l i c a t i o n of new t e c h n o l g i e s . M i c r o - e l e c t r o n i c s : The s t r o n g e s t e x h a u s t e m i s s i o n r e g u l a t i o n s f o r c a r s
a r e a p p l i e d i n t h e USA and J a p a n . More r e c e n t l y a l s o s t r i n g e n t r e g u l a t i o n s w i l l be a p p l i e d i n E u r o p e . The c a r i n d u s t r y has deve lopped m i c r o - e l e c t r o n i c s y s t e m s t o meet t h e s e r e g u l a t i o n s . The most i m p o r t a n t q u a l i f i c a t i o n s of t h e s e m i c r o - e l e c t r o n i c s y s t e m s a r e : - low cost due to the fact t ha t they are mass produced - very r e l i a b l e . Most of the componentry i s designed for use under the bon
n e t of t h e c a r s . This means t ha t i t i s r e s i s t e n t aga ins t s t rong temperat u r e changes, humidity and v i b r a t i o n . The emission regu la t ions a l so r e qu i re long l i f e t i m e of the componentry
- a v a i l a b l e a re sensors for temperatures , pressures and oxygen content i n gases
- ava i l ab l e a re ac tua to r s to adjus t v a l v e s , e t c . - ava i l ab l e a re improved i g n i t i o n systems with timing control over the
software - ava i l ab l e i s a l a r g e amount of software for many kinds of cont ro l s a l so
under t r a n s i e n t condi t ions - ava i l ab l e i s very users f r i end ly app l i c a t i on equipment. If the proper
software i s a v a i l a b l e , c a l i b r a t i o n s can even be made in the f i e l d - ava i l ab l e i s the software for very simple d i a g n o s t i c s .
This kind of equipment i s a l ready in use for s t a t i o n a r y , na tura l gas engines and veh ic les running on LPG or na tura l ga s . See Figures 1 and 2 .
I t i s qu i t e c l ea r tha t these mic ro-e lec t ron ic systems can be applied on producergas engines . Problems such as varying power, low ef f ic iency a t high engine speeds can be solved to a very l a r g e e x t e n t . With t h i s componentry and software and p a r t l y new to be developed software d i f f e r en t pro-

337
MICROPROCESSOR-CONTROLLED AIR/FUEL RATIO SYSTEM
inputs outputs
O. EitE^-
Lambda sensor
speed
heck engine light ,
steppermoior position
pickup microprocessor control unit
Fig. 1: Schematicei design of a closed loop control based on measurement of the oxygen content in the engine exhaust . . .
Fig. 2: Example of the hardware, at the left the microprocessor, in the middle the oxygen sensor and at the right the carburettor for gaseous fuels with a steppermotor for the final control.
^
M
blems in the gasifier itself can be controlled more accurately with much less attention from an operator. Think e.g. of measurement of the oxygen content in the producergas. That is an important parameter regarding the proper functioning. Furtheron temperatures can be measured in the gasifier in the bed and automatic actions such as some air injection or operation of the grate can be done automatically. By such an accurate control tar and soot formation will be reduced. It will be clear that with such system a very good interaction between the engine and the gasifier becomes possible. This also can be of great advantage for start-up and turn down.
New sensors: When a system is operating under micro-electronic control, new sensors could be added. These new sensors than could influence the process or give a warning. Examples: - sensor for water content in the fuel. Generally a gasifier is designed
for a fuel with a limited amount of water content. If, however, water content would be different and this would be known continuously, the process could probably be influenced to a certain extent. This influencing could be e.g. slowing down the process
- sensor for tar in the gas. Tar is quite dangerous for the engine. If tar is formed, although not expected, a warning would be very useful. Also to

338
a certain extent the process can be automatically controlled to optimise on tar content
- sensor for dust. For dust the same applies as for tar. Such a sensor would give a warning
- sensor for other components in the gas. Also hydrogen sulfide, ammonia and halogen hydrocarbons tend to be wrong for the engine. A warning that such components enter the engine in a large amount, would be very useful. Scrubbing e.g. could be increased to reduce such components
- sensor for lubricating oil condition. A lot of expensive engine damage occurs due to operation on engine oil in a bad condition. This can be due to acids or dust. A sensor which could give global information on this would be very useful
- sensor for filter pollution. Automatic sensing of pollution of filters is quite good possible by measuring the pressure drop over the filter. The micro-electronic system knows engine speed and load and thereby the gas flow. It can compare the measured pressure drop with known normal values and thereby give an alarm in time. This pressure drop can be measured with existing automotive sensors. The incoming signal can also be differentiated against time and thereby give an early warning that the process is not running well.
General remark on sensors: Development of new sensors is generally rather expensive. It is however useful to study what is available on the market for industrial applications. Known are e.g. non-automatic operating equipment for measuring the water content in grains.
Oxygen enrichment: In the Netherlands (and some other countries) development work is done for oxygen enrichment of the air. This is generally for industrial applications. There is a development whereby socalled hollow fiber membrane technic is applied. These are thin hollow fibers, whereby in a limited volume a large surface is created. The material is such that with some overpressure oxygen passes easier through the surface than nitrogen and by that a different oxygen content is obtained. Oxygen enriched air would be very attractive for the gasifier. This is because of the fact that such air makes products more "combustible". Higher temperatures would be created and by that the amount of tar w uld be reduced. Also a fuel with a higher water content could be applied. A system could be such that independence of e.g. water content of the fuel (new sensor) or tar in the gas (new sensor) the amount of oxygen enrichment would be controlled. Of course, mechanical power is needed to drive the compressor for the air.
Engine design: Lifetime: The most important problem in engines is the lifetime. Beside filtering and scrubbing and adaption of the oil (see further), adaption'of engine design is a possibility. However, because of the fact that the amount of engines used is generally small, it means that special manufactured components tend to become expensive. Copper e.g., used in the bearings and oil coolers and nickel in camshafts, tend to be sensitive for in particular sul far corrosion. Application of other materials is quite difficult. It could be manufactured but other disadvantages such as less good heat transfer or sensitivity for abrasive wear would occur. Generally it is concluded that such developments are too expensive, mainly because of small amounts. Quite a different approach to avoid in particularly acids by condensation, is to increase the engine temperature and to keep engine on temperature also when this is stopped. Keeping engine on temperature is already longtime applied in large ship engines. In such system it would also be attractive to blow air through the engine crankcase before stopping. Abrasive wear is of course not reduced by such system. This could be improved by applying a more frequent oil filter change or by applying a larger filter.

339
Power Improvement: Due to the low calorific value of the gas the high power loss generally occurs, which results therein that a more expensive engine has to be applied to obtain a certain power. The following actions can improve engine power: - ignition. Stronger ignition can keep engine longer on good power. Such
electronic ignitions with also more improved timing characteristics and correct spark plugs for this are available on the market
- spark plug engines tend to have a compression ratio which is not optimal for producergas. For producergas engines a compression ratio of about 12.5 is possible and this gives some 10% power increase and some 8% reduction in fuel consumption. Manufacturing of modified pistons is often possible without extreme cost
- oxygen enrichment. When oxygen enrichment would be applied for the gasi-fier, it can also be considered to apply it for the engine. Each volume percent increased oxygen can generate some 5% more power. This enrichment can probably only be done up to some 26 volume percent in air because otherwise probably engine knocking will occur
- more air supply to the engine. Power of the engine will be increased lea-nier with more air for the combustion. For this the following possibilities can be considered: . turbocharging. Turbocharging is widely applied on diesel engines and more recently on spark ignition engines as well. The componentry is available for most of the engines. This method increases engine power easily by some 30%. Until sofar it is nearly not applied with producer-gas, because of insufficient filtering. The speed in the turbo is so high that small particles rapidly create corrosive wear. This means that filtering on in particularly dust has to be nearly total, which makes it neccesary to apply larger and more expensive filters. The application of roots blowers looks to be more attractive for the time being.
. roots blowers. Roots blowers are mechanically driven, so called positive displacement pumps. These blowers are less sensitive to dust and tar. Herewith also the power can be increased by some 30%. However, compression ratio of the engine generally has to be reduced to some 7 tö avoid engine knocking, thus for that other pistons have to be applied as well.
. tuning. The suction of the air for combustion is of a pulsating nature due to the opening and closing of the air intake valve. Due to this waves occur in the intake system. Generally these waves reduce the amount of air sucked in. It is however possible to give the intake system such dimensions that these waves increase the amount of air sucked in. Quite often a power increase of 10-15% is possible. The modification of the engine manifold is such that part of the intake manifold consists of tubes for each cylinder. The method is not sensitive for dust and is as a construction rather simple. The disadvantage of the method is that is generally works in a small engine speed range (which for stationary engines is generally not problematic) and quite specific knowledge is ne-cesary for the designing.
. less air resistance. Sometimes producergas systems are built with a lot of tubing with sharp edges, etc. In such tubing sometimes negative working waves occur. Optimizing on this design is generally quite useful and relative simple.
- micro-electronic control. Maintaining an optimal adjustment will increase the average power output.
Adapted engine oils: Special lubricating oils for engines which run on acid containing gases have been developed. These oils generally have a high

340
Total Base Number (TBN). A high TBN means that the oil has a high acid nu-tralizing capacity. These oils generally nutralize a sul far well, but not always chlorine and fluorine. With an optimal engine jail protection against an hydrogen sulfide concentration of some 2000 mg/m is possible. In that case oil change time has to be reduced by half. Concentrations larger than 5000 mg/m cannot be dealt with by adapted engine oils and reasonable oil change times.
Back-up gas: When gas quality and gas amount changes drasticly and when a rather continuous power is required, the application of a socalled back-up gas can be considered. This could be natural gas or LPG when available. Some engine manufacturters have developed a system whereby can be switched from one gas to another. It is however possible to operate the back-up gas on a continuous base and not just be switching. Such a system operating on a continuous base basicly uses all the producer gas available but takes additional backup gas until the required power is obtained. This system operates by a pressure controller which has some difference to the setting of the producergas pressure. Also the application of micro-electronics simplify the application of such a back-up gas.
4. CONCLUSIONS/RECOMMENDATION FOR FUTURE RESEARCH It is recommended to initiate research and development whereby the
producergas engine is controlled by micro-electronics. The use of low-cost automotive hardware and adapted software is recommended.
It is recommended to initiate research and development whereby the ga-sifier will be controlled with additional micro-electronics. In the first phase low-cost automotive hardware and adapted software shall be applied.
It is recommended to initiate a paper study to investigate the possibility of existing industrial sensors for dust, tar, hydrogen sulfide and halogen hydrocarbons.
It is recommended to initiate a demonstration project whereby the engine power in increased by tuning of the inlet manifold and by the application of a roots blower.
It is recommended to initiate a paper study regarding the cost effec-tiviness on the application of oxygen enrichment of the air via hollow fiber membrane technics.
It is recommended to advise users of producergas engines to use special lubricating oils with a high Total Base Number.
It is recommended to initiate a demonstration in a country where backup gas is available to use this in combination with difficult fuel that generally creates strong fluctuation in the gas quality and gas amount.
If forementioned research has been carried out partly, it is recommended to organize a full-scale demonstration whereby most of the developped new technologies are applied.
REFERENCES
(1) DEKKER, H.J., Pilotinjektie DAF 575, Delft, IW-TNO, 1982. (2) HEYNIS, G. and RAMACKERS, M., Application of Producergas in Otto and
Diesel engines, Delft, IW-TNO, 1983. (3) LYON, D.; HOWLAND, A.H.; LOM, W.L., Controlling exhaust emissions from
a Diesel engine by LPG dual fuelling. Instn. Mech Engrs. C126/71. (4) RELE, R.R.J, ter, Ontwikkeling en toetsing van biogasmotoren op veehou
derijen. Utrecht, Projectenbeheerbureau Energieonderzoek, 1984. (5) WEIDE, J. van der, STASSEN, H.E.M., GEURTS, M.J.G., Diversificatie gas
vormige brandstoffen voor gasmotoren, TNO nr. 7700360698, Delft.

341
(6) WEIDE, J. van der and SEPPEN, J.J. Avanced hardware and combustion technology for gaseous fuels at TNO, Conference Gaseous Fuels for Transportation, Vancouver 1986
(7) Weide, J. van der, et.al. Experiences with CNG and LPG operated heav duty vehicles with emphasis on US HD diesel emission standards, SAE paper 881657.

342
RAPPORTEURS REPORT ON SESSION VI UPGRADING, CLEAN UP AND UTILIZATION OF PRODUCTS
J. DIEBOLD SERI, Colorado, USA
1. GAS PURIFICATION
Techniques exist for the clean up of gasifier gases for engine usage, but they are expensive. They do require periodic filter maintenance or else they fail on a system. It was mentioned that at the Loma Plata plant in Paraguay there are three engines with 40 000 to 60 000 hours of successful operation.
In response to van der Weide suggesting research to couple state-of-the-art computer for engine control to a gasifier, Dr Esnouf commented that such a system, both hardware and software, had been used to control a charcoal gasifier at CEMAGRAF with an engine.
Dr Luengo commented that on-board computer control of engines burning gasahol had not been very successful in Brazil. Dr van der Weide replied that they had to add a fuel analyser before they could successfully control gasahol combustion; a feature which Ford Motor Co will incorporate in a flexible fuel vehicle.
2. PYROLYSIS OIL UPGRADING
Dr Beenackers expressed doubt that a plant as small as 1000 TPD could be economically viable. Dr Luengo commented that for many site specific applications, e.g. remote areas, it is the avoided cost of fuel that will determine the value of a locally produced fuel. Thus, although the cost at the gate of the refinery may be low, the price of fuel delivered to a remote user may be very high. Mr Diebold replied that a US DOE sponsored techno-economic study showed that after successful R&D, both the high pressure hydrogénation and the low pressure zeolite cracking of pyrolysis oils to gasoline are projected to result in fuel at about US Ş1.00 per gallon with a 1000 TPD plant. However, the plant needs to be as simple as possible to achieve this goal. Both Dr Churin and Mr Diebold agreed that more research needs to be completed in the process to convert pyrolysis oil to gasoline.
Dr Meyer commented that pyrolysis oils are rich in phenolics, but lean in actual phenol. Catalysts which dealkylate phenolics to phenol would be very useful to produce a pure chemical (or for use to form methyl aryl ethers, as being researched by Dr Kiokos from Greece).
It was mentioned that the "Mann Oil" process was to be scaled up to 50 TPD. Currently the smaller scale system produces a product which is high in asphaltenes (propane insolubles).
3. RECOMMENDATIONS FOR RESEARCH
The following areas were suggested or were implied from the floor during the discussion, and may well be worth consideration:
A. Gas Clean Up Processes. 1. Lower cost emphasis. 2. Low maintenance emphasis.

343
3. Computer control development. a. Biomass gasifier-engine combination. b. A spin off of automotive engine control.
B. Pyrolysis Oil Upgrading. 1. Think in terms of costs avoided by the user when making economic
conclusions and plant siting. 2. Develop small modular, package plants to avoid the high cost of
field erection. This could help to reduce the typically adverse effects of the "economy of scale", which would be helpful to the biomass industry.
3. More research is needed in the catalytic upgrading of pyrolysis oil a. High pressure hydrogénation. b. Low pressure zeolite cracking
(Both processes 3a and 3b are using catalysts developed for other processes, so catalyst, as well as process development, would seem promising.)
c. Specialty catalysts : phenolics to phenol.


SESSION VII
ECONOMIC, ENVIRONMENTAL AND LEGAL ASPECTS


347
ECONOMIC AND MARKET OPPORTUNITIES FOR BIOMASS DERIVED FUELS
A V Bridgwater Energy Research Group
Chemical Engineering and Applied Chemistry Department Aston University Aston Triangle
Birmingham B4 7ET UK
ABSTRACT
The economic and market opportunities for thermochemical biomass conversion through gasification, pyrolysis or liquefaction are concerned with costs of production and values of products. An analysis of capital and operating costs is presented for fuels that may be produced by either gasification processes (Part 1) or pyrolysis processes (Part 2), from which production costs have been estimated for fuel gas and power. The results suggest that the current lack of interest is not just due to adverse economic viability, since a wide range of attractive opportunities appears to exist for wastes and residues to be utilised in the short term, and energy crops in the longer term. The current low price of conventional energy products contributes to this disinterest, as well as the status of the technology. Lack of long term operation, operating experience and perceived reliability problems all contribute to the relatively poor image of the technology. There seems litde doubt that there will be considerable opportunities for thermochemical conversion of biomass and solid wastes in the future particularly if there is pressure from increasing conventional energy prices and also from environmental concerns over waste management practices. There is need for technology assessment and, if appropriate, support for long term demonstration. This will help ensure that the technologies are sufficiently developed and mature to provide reliable service when the occasion demands.
PART 1 : GASD7ICATION
INTRODUCTION The economics of gasification and pyrolysis is concerned with costs of production,
values of products, and markets. There are a variety of roles in the implementation of a novel idea to a commercial installation: the technology developer, the technology licensee, the equipment manufacturer, the equipment user, and the energy product user. Some of these functions may be combined.
Implementation It is important to distinguish between the different expectations of these different
groups: • The technology developer invests on a relatively speculative basis that he will make a profit
on sales of the equipment and/or the licence in the medium to long term. He will commit resources at relatively high risk, possibly as part of an R&D strategy, or possibly as an opportunistic response to a funding resource.
• The technology licensee acts as an interface or broker between developer and equipment manufacturer. His role can be significant for non-commercial developers of technology such as Universities. The equipment manufacturer will often adopt this role as part of the normal process of commercialisation of new ideas.

348
• The equipment manufacturer may have developed his own technology, or be the technology licensee. He will manufacture and usually market the complete system to the equipment user to produce his own energy product. Alternatively he may set up an operation to use his own process to produce energy which he directly sells into the energy market, in which case he is also the equipment user. He will have conventional industrial profitability targets to meet in terms of overall company profitability, and in terms of venture profitability.
• The equipment user will either buy the equipment for production of an energy product (such as fuel gas or power) for his own use, or to export it These two situations will have different economic criteria to meet If the energy product is generated for internal use, such as to replace a more expensive fossil fuel, the financial target is quite stringent, typically requiring a payback time of less than 2 years as it will have to be funded out of revenue costs. In the case of exporting energy as a utility, the operation would often be viewed as a more conventional investment, and judged against a much longer time horizon according to the orthodox practice of the industrial sector concerned - for example for power production a 20 year equipment lifetime is not unreasonable.
• The energy product user may be the purchaser of the equipment, or he may be the customer of a renewable energy utility. In either case his financial objective is clear: to buy energy as cheaply as possible, and if investment is required, to minimise the payback time, which might be not more than one or two years.
There are two main roles in this development chain - the equipment manufacturer, and the energy product user. Their viewpoints are summarised in Table 1.
Table 1 Requirements and Interactions of Equipment Seller and Energy Product User
Technology
Economics
Plant Manufacturer or Supplier
Gas product quality Alternatives
Performance guarantee needed
Cost of plant to make Economies of scale to make
Profitability
Market Competition Feed availability
Minimum size of plant Product value Market size
Market growth rate Time scale Profitability Uncertainty
Product User or Buyer
Gas product quality Alternatives
Is performance guarantee available? Reliability
Technology status
Capital cost of plant to buy (ie price) Economies of scale in use
Profitability Variable costs of operation
Feed cost Product cost Availability Reliability
Alternatives Feed availability

349
Costs, Incomes and Profitability In all the situations described above, there are a variety of costs to be to be considered:
• Research and development cost, • Capital cost of the gasification or pyrolysis plant, • Variable or operating costs of running the plant, such as feedstock, labour,
utilities, maintenance, overheads, etc., • Production cost of the energy product, • Value of the energy product, which is realised as an income or a saving, • Profitability of the investment required.
Justification for implementation of a new fuel or fuel production system is invariably on the economic grounds of profitability - is the investment justified according to current company financial targets, and/or in comparison with alternative investment opportunities? The techniques usually employed depend on the situation amongst those described above, and can include payback time, return on investment, net present value and discounted cash flow rate of return. Sensitivity analysis is often performed to assess the effect of uncertainty on the economic case. All these aspects are discussed later.
It is important to distinguish between cost and price. It is rare for an investment to be sanctioned without adequate financial justification in terms of profitability. This means that there has to be an adequate margin between cost of production and the price that is charged to justify the investment The expectations for profitability will vary according to sector and role as explained above, and this is also considered in the cost analyses below.
CAPITAL COST Scope
The total plant cost of a gasification or pyrolysis system is the sum of all the equipment costs for the steps required, together with costs of design, installation and commissioning. The complete system starts with delivery of feed material, which will require some handling, possibly storage, and may require pretreatment according to the characteristics of the material and of the gasifier or pyrolyser. Pretreatment is discussed later. After gasification or pyrolysis, the fuel gas will usually require some clean-up, the nature and extent of which depends on the feed characteristics and pretreatment afforded, type of reactor, and application.
If the user is some distance away, a high level of gas clean-up is necessary to avoid deposits in the pipeline, together with a blower or compressor and a pipeline. For power generation, a high degree of clean-up is essential, and there are efficiency advantages from fuel gas cooling. Extensive gas clean-up is usually effected by water scrubbing which gives rise to a waste water problem, the extent of which depends on the feed characteristics and pretreatment afforded, type of gasifier or pyrolyser, and application. The full scope of a biomass gasification system is summarised in Table 2, with comments on system requirements and interactions.
Table 2 Scope and Requirements of Gasification or Pyrolysis Systems
Operation Need and Dependency
Reception of feed Essential - depends on material Storage Essential - depends on material Handung Essential - depends on material Size reduction Optional - depends on material and reactor Screening Optional - depends on material and reactor Drying Optional - depends on material and reactor Gasification or pyrolysis Gas cleaning Not always essential - depends on application Wastewater treatment Optional - depends on application and gas cleaning Gas compression Optional - depends on application and location Pipeline to user Optional - depends on location of application Power generation Optional - depends on application

350
There are, therefore, complex interactions between feed, reactor, and application, which cannot be generalised and defined by a standard system. The more sophisticated systems can cost several times more than a basic system where the feed may be in a form that may be fed directly to a gasifier or pyrolyser, and the application requires minimal gas cleanup. An indication of the relative capital costs of various components of a complete system is summarised in Table 3 - this is only a very approximate guide to illustrate relative magnitudes rather than absolute relationships.
Table 3
Operation
Typical Relative Capital Costs of Gasification or Pyrolysis System Components
Approx. cost relative to
reactor
5 5
10 20 15 40
100 5
20 30 30 20 50 5
30 80
120
Battelle Columbus
(1)
} 54
100
0 28 14
John Brown
(2)
} } 6 }
100
20 32 0
Reception of feed Storage Handling Size reduction Screening Drying Gasification or pyrolysis reactor # Gas cleaning - dry Gas cleaning - wet Heat recovery Wastewater treatment Gas compression for local use Gas compression for pipeline Pipe to user (50 m ) Pipeline to user (1000 m ) Pipeline to user (5000 m ) Power generation (engine)
Notes # This is defined as the reactor system from feed on the ground to hot raw gas from the reactor, and includes feeder, reactor, ash discharge, piping and conveying, and instrumentation.
Analysis of Capital Cost and Relationships Capital cost data broken down by items of equipment or process step is not often
available. As a rule of thumb, the cost of the gasifier/pyrolyser reactor and associated feeder is about 60% of the equipment cost (3). This is supported by analysis of other studies where the gasifier costs between 51 and 63% of the total system equipment cost (4).
There is more data available on the relationship between equipment cost and total installed plant cost of reactor systems. The ratio of total plant cost to equipment cost varies depending on the type of gasifier construction. This can be estimated with a fair degree of confidence by orthodox factor estimating methods and ranges from 2.0 to 4.0 (5)
For packaged and skid-mounted units of typically up to 1 t/h capacity, the gasification or pyrolysis plant is complete except for a concrete area to stand on, and piping up to utilities. A light shed or cover may also be required. Figures quoted by various manufacturers indicate that the total installed cost for such a system would be about 1.2 to 1.4 times the equipment cost (6).
For larger field erected units, over about 1 t/h capacity, much civil and fabrication work is usually required on site, and available data indicates that the total plant cost is about 2.25

351
times the equipment cost. In the paper industry (with similar types of processing equipment), the accepted ratio of total plant cost to equipment cost is about 2.25, confirming this conclusion.
For smaller units which are not skidmounted, less onsite fabrication would be required than for the large units, and the ratio of total plant cost to equipment cost will be smaller than that for large units. A figure of 1.75 has been used for calculating total plant costs from equipment costs where no installed cost data is available.
Cost data In order to provide a consistent basis for analysis and estimation of gasification and
pyrolysis system capital costs, information has been collated from a variety of sources for 48 systems in respect of a basic gasifier/pyrolyser from prepared feed on the ground to a clean, cold fuel gas (3, 7). Included is the feeding system, gasifier or pyrolyser, ash discharge, cyclone, wet scrubber and water recycle. Excluded are all operations prior to the reactor (such as drying, chipping etc), wastewater treatment, gas distribution, and utilisation.
Data are usually available as a single figure representing the equipment cost of a gasifier/pyrolyser system from the feeding equipment to clean gas ie it includes equipment for the feeding, gasification/pyrolysis and gas cleanup steps of the process. In some cases total plant costs are available in addition to or instead of the delivered equipment cost. Where necessary adjustments have been made to exclude or include relevant equipment and/or costs according to the defined scope. All throughputs are based on dry ash free (daf) basis feedstocks.
The capital cost considered is a total plant cost including equipment, installation, design, project management and commissioning, but excluding land costs. Where data have been collected on a delivered equipment cost basis, this has been converted to a total plant cost by multiplying by suitable factors. In all cases it has been adjusted to a 1989 time basis with a process plant cost index, and to a West European location with a location factor. The data is correlated in Figure 1, shown in Figure 2 by type of reactor, and by process in Figure 3. (8)
10 Capital cost,
£1988
10
10'
10:
10
7
6
5
4
:
j
1
I
: f"y
•
•
■ ■ ■■■■ι
J·
*íl^
* ţ >
¿'ν ^r%
High cost level
Low cost level
10"1 10" 10
1
Figure 1 Biomass Gasifier and Pyrolyser Capital Costs and Correlations
10 Capacity, daft/h

352
10ö
Capital cost, £1988
10'
10ö
10°
10*
1
α πα "α
n Π
χ " Χ
1 * *
Χ Χ
χ χ
χ
Q Fixed beds χ Fluid beds
10" 10° 101 102 Capacity, daf t/h
Figure 2 Biomass Gasifier and Pyrolyser Capital Costs by Type of Reactor
10" Capital cost,
£1988 1 0 7 i
106f
10a
10"
u "Β
a i
C t f U
I I I I I l l 4 I I I M I N ) l i l i Ulf |
àî
* Pyrolysers ° Gasifiers
10" 10u
10' 10 Capacity, daf t/h
Figure 3 Biomass Gasifier and Pyrolyser Capital Costs by Type of Process
Capital Cost Relationships The data points generally lie on or close to two parallel lines as shown in Figure 1. The
lower line represents the lower limit of gasifier or pyrolyser capital costs, typically achieved by technically relatively simple gasifier systems and/or systems supplied by small companies, who do not have the resources to provide extensive backup facilities for example for

353
troubleshooting and performance guarantee This probably also represents the target or lowest achievable cost for gasification systems. The upper line represents the cost of high technology systems eg twin fluid bed systems and stirred bed systems and/or systems supplied by major companies who can provide extensive technical support, and performance guarantees. The data is correlated in cost models summarised in Table 4.
In the longer term, learning and development will tend to reduce capital costs towards the lower end and a "target" capital cost has therefore been employed in the economic analysis to represent a short to medium term realistic goal. This target figure has been used for estimation of fuel gas production costs.
Table 4 Capital Cost Correlations for Gasifiers and Pyrolysers (8)
Developed or industrialised country locations:
Lower £i988 = 213 000 (capacity, daft/h)0.72 Systems that are more basic technically, or those marketed by less established companies who are not able to provide the same level of performance guarantees and backup
Highen £i988 = 735 000 (capacity, daf t/h)0.72 High technology systems such as twin fluid bed systems, or systems offered by substantial companies with backup and guarantees.
Average: $1988 = 3 7 0 0 0 ° (capacity, daf t/h)0.72 Average of all data
Target: ^1988 = 2 9 2 0 0 ° (capacity, daf t/h)0.72 This reperesent a reasonable and achievable cost of a system that is developed and attracts multiple sales, and thus benefits from the learning effects of an emerging commercial process. It is the mean of the average and lower capital costs
Developing countries locations:
£l988 = 115 000 (capacity, daf t/h)0.72 Gasifiers that are built in developing countries for local use (not included in Figure.2). This is very location sensitive and therefore approximate.
Power Generation An add-on cost based on a gas engine system is given in the equation below based on
reported data from about 10 sources and discussions with experts. The rule of thumb relationship that 1 t/h daf biomass will generate 1MW power is reasonable at large plant sizes of more than 1MW, but as the plant size decreases the biomass requirement increases due to lower system efficiencies at lower capacities. A relationship between biomass consumption and power generation has been derived and is given in Table 5, together with capital cost correlations for power generation and system costs (9). It should be noted that power costs are

354
conventionally expressed as a cost per installed kW which gives no economy of scale.
Table 5 Capital Cost Correlations for Power Generation (9)
Power generation from clean fuel gas in an engine £l988 = 315 000 (capacity, MW electrical )0.72
Turbine system costs would be higher, and additional gas cleaning plant would be required.
A reasonably efficient biomass to power plant is usually assumed to generate 1 MW for each 1 t/h daf feed. A more realistic relationship is given by the following equation for the number of tonnes of biomass required to generate 1 MWh:
Β = 1.1-O.llogioC where Β = tonnes of biomass to generate 1 MWh and C = capacity of system in MW
Combining these equations gives: ^1988 = 294 000 (capacity, biomass daf t/h)0.75
Total System of Biomass to Power £1988 = 6 8 5 O00 (capacity, MW electrical ) 0 · 7 2
Cost-benefit analysis and trade-offs The design of a complete system has to consider all equipment necessary to handle and
process feed delivered to the plant to a marketable product - a product manufactured to the specifications of the application. As feeds will vary in composition and quality, the process will perform differently according to design and specification, and the quality demands on the product are dependent on the application, there can be a wide range of process configurations. Pretreatment operations such as drying, are expensive in capital and operating cost, and can only be justified by comparing the benefits from a higher quality product or more efficient process, with the higher costs of installing and operating a dryer.
This would be carried out by comparing production costs, or performing an incremental profitability assessment on the marginal capital cost (see later). Generally drying cannot be economically justified for relatively small scale fuel gas production systems, but would be considered for large scale liquid fuel production systems. Similarly feed storage and handling requirements will be assessed according to type of feed material, regularity of arisings, facilities available locally, contractual arrangements for feed delivery and price etc. Feed storage and handling facilities are thus also site and situation specific. There are few known guidelines as to what can be justified, and some suggestions are summarised in Table 6. A discussion of the interactions is given in (10).
VARIABLE OR OPERATING COSTS
The variable or operating costs for a system from prepared feed on the ground to cold clean gas are considered in this section. The variable costs considered here are feedstocks, utilities, maintenance, overheads and labour.

355
Table 6
Operation
Weighbridge Daybin
Screening
Magnet
Bulk storage in open
Bulk covered storage Rechipping
Drying
Densification
Grinding
Silo for holding
Suggested Guidelines for Economic Justification of Peripherals in a Gasification System
Requirements and Limitations
for feed control and management for levelling out feed deliveries before handling, storage or processing. Desirable for larger scale processes for size sensitive reactors; rejects require utilisation. Also important for removal of non-biomass inclusions such as concrete and metal for hazard reduction and equipment protection. Usually specified as protection in early stages of handling and storage operations, as part of screening operation. Some storage is usually required with retrieval facilities. Extent depends on supply chain and reliability, usually not less than one week. for water sensitive materials such as refuse derived fuel pellets. for poorly prepared feedstock and/or for upper size sensitive reactors; rejects require utilisation. usually only justified for large scale operations; and/or where low cost energy is available such as waste heat. can only be justified where biomass is very scarce, and product value is very high. Circumstances are difficult to identify for all feeds except RDF, when improved performance and reliability may outweigh higher cost. is necessary for some highly specific reactors Due to high cost, this can only be justified for production of high value products such as chemicals. to provide intermediate storage in processing chain, typically just before reactor, and particulary is feed material has been extensively pretreated such as drying or grinding
Feedstock Three feedstocks are considered: straw as representative of agricultural waste; refuse or
MSW as domestic, commercial and industrial solid waste; wood as representative of energy crops and forestry products.
Considerable data on arisings in terms of quantity and quality and cost of feedstock have been collated in recent years by the EEC and national bodies, and this section summarises the essential features of available feedstocks in order to relate these to gasification and pyrolysis technology and economics. Costs of feeds relating to UK situations and practice, with some European data, are summarised in Table 7 (8,11). Wastes are likely to attract higher credits in Europe due to higher disposal costs, while energy crops and similar materials are likely to have similar costs. This data, therefore, can be viewed as conservative.
Straw About 7 million t/y of surplus straw arise each year in the UK for which no ready
market is available, out of a total arising of about 14 million t/y (12). Comparable figures pertain to Europe. Prices vary by location from £5 to £50/t, but costs have been estimated at about £17/t or £22/t delivered (12).

356
Table 7 Costs of Feedstock Materials and Effects of Feedstock Characteristics (8)
All costs are in £ per dry ash free tonne All costs are currently achievable. Optimisation would be expected to significantly reduce these figures Straw With bam storage at farm, cost at source Delivered (80 km)
Refuse/MSW Domestic
Wet "fluff RDF
Disposal credit £/raw tonne
5 10 15 20
5 10 15 20
5 10 15 20
£17Λ £22Λ
Cost per daft product £/tonne product
13.50 -1.00
-15.50 -30.00 25.50 11.00 -3.50
-18.00 37.50 23.00
8.50 -6.00
Dry "fluff RDF
Pelleted RDF
Commercial as Domestic
Industrial as Domestic
Wood
Forestry Residues, all grades
Secondary saw mill Residues Wet wastes (eg bark), 50% moisture Dry waste, 25% moisture Energy crops. Conventional forestry: UK, green chips, daf basis Short rotation forestry: UK, green chips, daf basis
Sewage sludge Dry solid basis - 50
Refuse Domestic About 13 million t/y of domestic refuse are produced with a heating value
of about 9 GJ/t , and containing about 8 million t/y of combustibles.(12), ie about 250 kg/head/y. The average cost of disposal in the UK is claimed to be about £6/t but these are believed to be very conservative with average costs of disposal nearer £12/t, over a range of £5/t to as high as £40/t. (2) .In Europe, disposal costs are much higher by a factor of up to 5, with typical figures at around £30/t. This will have a significant effect on conversion economics below if these costs are translated into credits.
Ranee. £/daf t Oto 100
0-30 0-30
5
Mean. £/ daf t 30
20 20
30 45

357
Commercial: Arisings of commercial solid waste have been estimated as 7.4 million t/y (dry basis) with a heating value of around 16 GJ/t ie about 130 kg/head/y. The material is generally cleaner and drier than domestic refuse with a higher proportion of packaging ie paper, plastics, wood, etc.
Industrial: Combustible waste arisings from the UK industrial sector have been estimated at 8.4 million t/y (dry basis) with a heating value of 16 GJ/t, ie about 150 kg/head/y.
Wood and wood waste Wood and wastes are available in a variety of forms:
Forest residues - These have to be removed from existing plantations for silvicultural reasons.
Wood processing residues - These are wastes that arise in the timber production industry, usually adjacent to the forest resource.
Waste from the wood manufacturing industry Fuel wood Energy crops
Typically the forest products industry generate about 50% waste in the production process. A considerable proportion of this is utilised in a variety of industries for combustion and specialist products such as cat litter, horse show ground cover, poultry bedding, etc. There is still a considerable potential resource available.
Other Wastes Sewage sludge is another resource that is being investigated for thermochemical
processing. Sludge is generated at the rate of about 25 kg dry solids/head/y. A disposal credit of up to £50/dry t is potentially available.
Costs The types of feedstock and costs are summarised in Table 8A, with the effect on feed
cost and gasifier performance summarised in Table 8B.
Utilities Utilities include:
• electricity, for driving pumps, blowers and feeding systems, • wash water for product gas scrubbers, and • fuel gas for drying, • fines for drying, and/or separate gasification to produce fuel gas, • steam for steam gasification, or moderation of oxygen gasification. If required, steam
would probably be raised on site within plant battery limits, and probably from a waste heat boiler.
• oxygen for oxygen gasification. If required, oxygen would probably be purchased and charged to feedstock.
• waste water treatment. None of the systems examined require fuel gas, fines, steam or oxygen services.
Noneof the systems allowed for waste water treatment in process or cost terms. It is assumed that all other energy requirements are met in-house without requiring conventional fuels except for start-up.
Power costs of gasifiers and pyrolysers have been collated and analysed for which the mean value is £0.114/GJ (2). However, this does not include any other utilities costs such as water purchase and waste water treatment, where very little data is available. A utilities cost of £0.25/GJ was used for the base case in the production cost calculations which includes a contribution for water purchase and treatment.
Maintenance Yearly maintenance cost is usually estimated as a proportion of the system capital cost,
for which an average value of 2.5% of total plant cost has been found (2).

358
Table 8 Feedstock Characteristics: Effect on Cost and Efficiency
A - Effect on Cost
Material
Straw, on farm Straw, delivered
Refuse, undried Refuse, undried Refuse, undried Refuse, undried
Refuse, dried
Wood, on site Wood, delivered
Moisture Effect on gasifier efficiencv
17% 17%
35% 35% 35% 35%
17%
50% 50%
Β - Effect on Gasifier Efficiency
Moisture %. wet basis
0 10 20 30 40 50
content (%. drv basis')
(0) (11) (25) (43) (67) (100)
78% 78%
71% 71% 71% 71%
78%
62% 62%
Cost, raw basis. £/t
17 22
0 -5
-10 -15
-10
13 17
Eficiencv to cold clean gas. %
82 80 77 73 68 62
Cost, daf basis after processing. £/daf t
20 26
25 15 5
-5
15
26 34
Eficiencv to hot raw gas. %
95 92 89 85 80 74
Overheads Annual overheads cost is usually expressed as a proportion of the capital cost, similar
to maintenance cost. The economic model uses a fraction of the total plant cost to calculate annual overheads which include rates (local tax), insurance and all head office expenses. Payroll overheads have been included in the labour costs.
No relevant published data has been found which can be used to estimate overheads for any one location. The only data available estimated the overheads for a commercial fluid bed system to be 8% of the total plant cost (2). The local rates and insurance for a gasifier or pyrolyser would typically be 4% of the capital cost. Multiplying this figure by 2 to cover head office expenses, gives 8%, which matches the earlier estimate. This figure of 8% of total plant cost was therefore used in the economic analysis.
Labour The cost per shift used for the base case is £25 000/year. This includes the cost of one
operator per shift, plus the costs of supervision and payroll overheads. It has been assumed that the costs of supervision are shared with other plants on a site. Four shifts are required for continuous operation, three shifts per day, plus an extra shift to cover rest days. If operation is restricted to weekdays only, then three shifts will be sufficient. There can be significant variation in labour costs by location and in overheads according to local practice, so each case has to be assessed on its own merits. Labour costs, however, are usually a relatively small part of the total product cost. For less than full time utilisation of the equipment correspondingly less labour is required, for example 4 or 5 shifts may be required for 8000 h/y

359
or 330 d/y operation, and only thre required for 5 or 4.5 d/week operation.
Power Generation It is assumed that no additional raw materials, utilities, or labour are required for power
generation. Maintenance and overheads are allowed for in the relationships to total capital cost.
Plant Availability Plant availability is a measure of the proportion of time that a plant operates compared
to the time that it should be available. Gasification is continuous process that is not conducive to stopping and starting, and it would therefore be usual to expect a gasification system to operate continuously. There are many reasons why this does not happen in practice such as equipment failure, feed problems, planned maintenance requirements, disputes, unfavourable costs, and lack of demand for products. Plant availability can thus be considered to have two components: planned and unplanned shutdown. A fluid bed gasifier, for example, may readily be shut down overnight and restarted from hot within minutes, whereas a fixed bed gasifier can perhaps be turned down significantly, but cannot be turned off, so different systems will have different capabilities.
A full year contains 8760 hours, and it is usual in the process industries to allow about 10% of this time for routine or planned maintenance, leaving typically 8000 hours for operation. Utilisation of equipment for less than 8000 hours per year is thus affecting costs in an adverse way by increasing the capital element of the production cost. Relatively small scale gasifiers, however, will often not need to operate all year round, for example at weekends when the factory may be closed, which would reduce operating hours to around 5000 h/y. For power production, demand may only be for hours of darkness or aound 4000 h/y. These effects can be readily incorporated into economic analyses.
There is also unplanned maintenance that is caused, for example, by equipment breakdown, lack of training, feed blockage, or ash sintering. All are associated with emerging technologies that are not yet fully developed, and which are responsible for many of the unfavourable reports associated with biomass gasification. There are many reasons and always good solutions to these problems, but they do not provide sufficient reassurance to the potential user or purchaser. These effects are more difficult to include in economic assessments due to their unpredictability and lack of data for evalution. It is therefore only possible to account for this aspect of availability by a blanket approximate figure. Subsequent analyses combine all these effects into one overall figure.
One of the largest biomass gasifiers currently operating in the world is achieving an availability of 85% on a scheduled 8000 h/y, from operation for over a year, in Quincy, Florida. It is, therefore, possible to design and operate a gasifier with high reliability and availability, and there is a need to support such iniatives to overcome the uncertainties still associated with this technology.
GASIFICATION FOR FUEL GAS PRODUCTION
Methods of Fuel Gas Production Cost Estimation There are two approaches to the evaluation of a proposed project - calculation of a
product cost with comparison of this cost to the alternatives, or conventional measurement of profitability using criteria such as payback time, return on investment (ROI), net present value (NPV) and discounted cash flow rate of return (DCFRR).
Product cost is conventionally calculated by totalling the feedstock cost and operating costs and adding to them a capital amortisation charge, which covers recovery of the capital and interest payments on the capital employed.. The product cost may then be compared with the product selling price to assess profitability of the initial investment. Product cost can either be an actual cost of production as described above, or a return on the investment can be added to give a notional selling price, which is again compared to the current market price to establish if there is a surplus. This latter approach is less common as two measures are required - setting a target return for the venture, and comparison of calculated product selling price with market place prices.

360
In contrast, conventional profitability measures include all cash flows, both income and expenditure, but do not include charges for interest or capital recovery. This is because they are designed as a method of comparing investment in a project with the investment of a similar sum in a bank for example or similar investment medium. DCFrr and ROI are expressed as a percentage rate of return, which can be compared with the bank interest rate, or more usually with a target rate of return that the company sets for any new investment and which considers cost of capital and risk. NPV is expressed as the value of a project in current money values, at a fixed rate of return which is usually taken as the companies target rate of return. All these measures are usually expressed in real terms. Payback time is the time taken for the capital to be recovered, ignoring interest payments, and is usually expressed in nominal terms.
Information Requirements A considerable amount of information has to be collated or estimated in order to derive
a production cost estimate. This is summarised in Table 9 for a "base case", which is used to estimate production costs of fuel gas and power, and to carry out a sensitivity analysis. The various operating cost estimates have been derived from procedures or estimates set out above and key aspects are amplified below. The effect of small changes in financial data, such as interest rates and escalation rates, can be significant over the long lifetimes anticipated for process plant such as this, (lives of 15 years are commonly quoted), due to the compounding effect of small annual changes. These effects have not been explored here but can be examined through sensitivity analyses. Supplementary data is included in Table 10.
Table 9 Economic and Technical Parameters for Production Cost Estimates
Gasifier technology Air blown fluid bed Gasifier efficiency (see text) 58 to 93% Feedstock Refuse, straw and wood Feedstock heating value 20.00 GJ/t daf Number of shifts (for 8000 h/y operation) 4 Project life 10 y Scheduled operating hours per year 8000 Availablility (Actual operating hours / Scheduled operating hours) 80% Actual operating hours (see Table 10) 6400 Throughput 5 te/h, daf basis Capital cost (including working capital) £928 700 Feedstock cost after processing (see Tables 7 & 10) as stated Utilities cost £0.24/GJ produced Yearly maintenance cost, fraction of capital cost 0.025 Yearly overheads, fraction of capital cost 0.080 Total cost of labour per shift £25 000/y Nominal cost of capital 10 % Inflation rate 5 %
Gasifier technology: An air blown fluid bed is assumed to be employed although the choice will have little effect on costs. Gasifier efficiency: Gasifier efficiencies of 58% to 93% have been used dependant on moisture content of feed and product. Appropriate data has been used in the calculations and cost estimates (8) Feedstock: Three feedstocks are considered - straw, refuse and wood. In each case several variations are included to represent the most likely alternatives. This is one of the major cost sensitivities that is explored in detail below for each energy product Other variationsd are included in the sensitivity analyses later. Feedstock heating value: This is assumed to be 20.00 GJ/t on a dry ash free basis (daf). Number of shifts (for 8000 h/v operationl: This is taken as 4 for continuous operation as it is

361
common practice in the process industries. Lower levels of operation are assumed to require less shifts as summarised in Table 10 below. Hours of planned operation must not be confused with hours of achieved operation - the former requires manning for the hours planned, while the latter is a consequence of unplanned shutdown which reduces output without a concommittant saving in fixed costs suchn as labour.
Table 10
A
Case
Pertinent Data for Estimation of Production Costs and Performance of Sensitivity Analyses
Capital costs (see Table 4)
lt/h 2.5 t/h
Cold clean fuel gas - target cost £291 500 £563 800 (target cost = mean of average and lower cost - see text)
Hot raw fuel gas £247 800 £479 300 (target less 15% for omission of gas cleaning equipment)
Power generation (from Table 5) £606 500 £1 173 100 (1 t/MWh, target cost + generating costs)
Capital cost to cold clean gas (from Table 4) Lower £213 000 £412 000 Average £370 000 £715 700 Higher £735 000 £1 421 700
5t/h
£928 700
£789 500
£1 932 400
£678 600 £1 178 900 £2 341 800
10 t/h
£1 529 800
£1 300 500
£3 183 000
£1 117 800 £1 941 800 £3 857 300
Β Operation, hours per year
Basic h/v No
4 500 5000 6000 7000 8000
of Shifts
2 3 4 4 4
50%
2 250 2 500 3000 3 500 4000
C Refuse feedstock cost for
Product
Wet "fluff' RDF
Dry "fluff' RDF
Pelleted RDF
Disposal credit. £/te
5 10 15 20
5 10 15 20
5 10 15 20
Actual h/v at Availabilities 60%
2 700 3000 3600 4 200 4 800
different cases
Cost raw refuse. £/te
3.38 -0.25 -3.88 -7.50
6.38 2.75
-0.88 -4.50
9.38 5.75 2.13
-1.50
70%
3 150 3 500 4 200 4900 5600
(from Table
Cost product Ms. 8.45
-0.60 -9.63
-18.70
18.75 8.10
-2.55 -13.20
27.55 16.90 6.25
-4.40
80%
3600 4000 4 800 5600 6 400
7)
90%
4 050 4 500 5400 6 300 7 200
:. Cost d.a.f. product. £/te
13.50 -1.00
-15.50 -30.00
25.50 11.00 -3.50
-18.00
37.50 23.00
8.50 -6.00

362
Project life: This taken to be 10 years in common with process plant in the process industries. Scheduled operating hours per year: An 8000 h year is assumed (the availability of 80% gives 6400 h/y). Lower levels of plant utilisation can be considered in a sensitivity analysis. Availablility (Actual operating hours / Scheduled operating hours): 80% is a reasonable and achievable target for well run biomass gasification plants. Actual operating hours (see Table 10): This is the planned operating hours of 8000 h multiplied by the anticipated availability of 80% giving an actual operation of 6400 h/y. Throughput: This is taken as 5 te/h on a daf basis, as being able to exploit the economies of scale at a reasonable and achievable size for short term implementation. This is one of the major cost sensitivities that is explored in detail below for each energy product Capital cost (including working capital): This is taken as the mean of the lower and average capital costs derived in the cost analysis above, and is refered to as the "target" capital cosd. For the 5t/h plant the cost adopted is £928 700, and details of the data employed for the range of plant sizes considered is summarised in Table 10. This approach was taken as there are substantial learning effects to be enjoyed from replication, and the cost figures used will therefore provide a production cost that is representative of short term achievable costs, without adopting an unduly optimistic viewpoint of minimum cost, or an unnecessarily pessimistic view of average or high costs with no further developments possible. Feedstock cost after processing as stated (see Table 7). Utilities cost is taken as £0.24/GJ produced (8). Yearly maintenance cost, is expressed as a fraction of the capital cost at 0.025 which is taken from Table 9.3.5. Yearly overheads is taken as a fraction of the capital cost at 8.0% (2) Total cost of labour per shift of £25 000/y from Table 9 Nominal cost of capital. An average long term view of the cost of money is taken as 10 % in real terms, which is that usually attributed to a low risk investment Sensitivity analyses can be carried out on both on cost of capital and plant life, but previous experience shows that these are not significant Inflation rate is assumed to average 5%/y.
Production Cost Estimates Production cost estimates have ben derived for a range of typical feedstocks for Europe
at different scales of operation. These are summarised in tabular form thus:
Fuel Gas Production Costs by Feed Type - Cold Clean Gas This situation is often considered to the most likely short term opportunity for fuel gas -
low heating value clean and cold fuel gas for retrofitting. The system has been described earlier as an erected plant which processes feed on the ground to a clean fuel gas. All capital costs and direct operating costs are included except: • wastewater treatment - there is wide range of contamination possible depending on
gasification technology and feed, and a wide range of treatment costs depending on extent of contamination, technology, and local requirements,
• a return on the investment - expectations of a commercial return depend on several factors such as the role of the investor, the perceived risk, and short and long term views of the energy market.
The estimated production costs are shown in Table 11.
Fuel Gas Production Costs by Feed Type - Hot Raw Gas Production of hot raw gas instead of cold clean gas as shown in the above analysis,
has been derived by subtracting 15% from the capital cost for loss of water washing facilities, and an increase in gasifier efficiency due to utilisation of the sensible heat in the hot gas which amounts to an increase in gasifier efficiency of about 12% (see Table 11). The results are summarised in Table 12. There is less confidence in the production costs for hot raw gas due to less reliable data on costs and performance of such systems, and definition of realisable sensible heat.

363
Table 11 Fuel Gas Production Costs by Feed Type - Cold Clean Gas
Scope: As in Table 9 The current maximum published interruptible natural gas price is £2.9/GJ. 80% gives a fuel gas price of £2.32/GJ. Costs underlined are those below this current market value of £2.32/GJ
Feedstock Production cost. £/GJ 1 t/h 2.5 t/h 5i/h 10 t/h
STRAW Conversion at 75% efficiency
£22/t delivered (£26/t daf) £17/t on-farm (£20/t daf)
3.72 3.32
REFUSE Wet fluff £-0.60/t product (£-l/t daf) 2.01
Includes disposal credit £10/t raw refuse Conversion at 71% efficiency with moisture at 25%
Dry fluff £8.10/t product (£11/t daf) 2.68 Includes disposal credit £10/t raw refuse Conversion at 76% efficiency with moisture at 10%
WOOD Conversion at 58% efficiency with moisture at 50%
£30/tdaf 5.08 £20/tdaf 4.22
2.93 2.52
1.18
L21
4.07 3.20
2.63
0.86
1.61
3.67 2.81
2.45
0.67
3.44 2.57
Table 12 Fuel Gas Production Costs by Feed Type - Hot Raw Gas
Scope: As in Table 9 Current published interruptible natural gas price is £2.9/GJ. 80% gives a fuel gas price of £2.32/GJ. Costs underlined are those below this current market value of £2.32/GJ
Production cost. £/GJ 1 t/h
STRAW Conversion at 90% efficiency
£22/t delivered (£26/t daf) £17Λ on-farm (£20/t daf)
REFUSE Wet fluff £-0.60/t product (£-l/t daf)
Includes disposal credit £10/t raw refuse Conversion at 87% efficiency with moisture at 25%
Dry fluff £8.10/t product (£11/t daf) Includes disposal credit £10/t raw refuse Conversion at 91% efficiency with moisture at 10%
WOOD Conversion at 73% efficiency with moisture at 50%
£30/tdaf 3.98 £20/tdaf 3.29
2.5 t/h
3.19 2.51
5t/h
3.04 2.72
L52 7o 2.19
2.48 2.08
0.94
1.57
2,17
0,69
1.33
2.90 2.21
10 t/h
2.03
0.54
1Δ2
2.72 2.04

364
Electricity Production Costs Production costs of electricity have been estimated from the capital and operating
cost models above, and are summarised in Table 13. It is assumed that 1 daf t biomass will generate 1 MWh in a state of the art system. This relationship has been widely verified with practical experience. Theoretically, 3.6 GJ = 1 MWh. 11 daf biomass has an energy content of 20 GJ, which is converted at say 70% efficiency to gas, and from there to electricity at say 30% efficiency. This gives an overall conversion of 1 t daf biomass to 1.16 MWh. The 1 t to lMWh (or 1 t/h to 1 MW) relationship has been taken as a conservative estimate.
Table 13 Electricity Production Costs
Scope: As for Table 9 Current bulk supply electricity cost tarif for industry is about 3.0p/kWh (£0.03/kWh), although many smaller users who are more likely to be customers of power from systems considered here will be paying much more. A recently quoted figure is 4.21 p/kWh. A figure of 4.0p/kWh (£0.04/kWh) is therefore used as the cut-off price. Costs underlined are those below this latter figure.
Feedstock lt/h
1MW
Production cost pence/kWh 2.5t/h
2.5 MW 5t/h
5MW lOt/h
10 MW
STRAW Conversion at 75% efficiency
£22/t delivered (£26/t daf) £17/t on-farm (£20/t daf)
6.71 6.11
REFUSE Wet fluff £-0.60/t product (£-l/t daf) 3.99
Includes disposal credit £10/t raw refuse Conversion at 71% efficiency with moisture at 25%
Dry fluff £8.10/t product (£11/t dafi 5.21 Includes disposal credit £10/t raw refuse Conversion at 76% efficiency with moisture at 10%
WOOD Conversion at 58% efficiency with moisture at 50%
£30/tdaf 7.03 £20/tdaf 6.03
5.28 4.68
2.56
3.78
5.59 4.59
4.67
1.95
3.17
4.98 3.98
4.26
1.54
2.77
4.58 3.58
Production Cost Analysis: Cold Clean Gas A detailed analysis of production costs of cold clean fuel gas for a range of cases is
given in Table 14. This shows the significance of feed cost in most situations.

365
Table 14 Production Cost Analysis: Cold Clean Gas
Scope: 5 t/h feed daf; conditions as base case, Table 9.
FREDSTOCK
STRAW £22/t delivered (£26/t daf) £17Λ onfarm (£20/t daf)
REFUSE Wet fluff £l/t daf Dry fluff £ll/t daf
WOOD £30/tdaf £20/tdaf
Total cost £/G.T
2.63 2.22
0.86 1.61
3.67 2.81
Capital cost £/CiJ
0.24 0.24
0.26 0.24
0.31 0.31
Subtotal
2.39 1.98
0.60 1.37
3.36 2.50
Variable cost Feed £/GJ
1.73 1.33
0.07 0.72
2.59 1.72
Labour Utilities O/H&M £/GJ £/GJ g/Ql
0.21 0.21
0.22 0.21
0.27 0.27
0.24 0.24
0.24 0.24
0.24 0.24
0.20 0.20
0.21 0.20
0.26 0.26
PART 2 : PYROLYSIS
PYROLYSIS TECHNOLOGIES AND PRODUCTS Pyrolysis produces gas, liquid and char: the relative proportions of which depend very
much on the pyrolysis method and reaction parameters. Much of the present interest in pyrolysis currently centres on the liquid products due to their high energy density and potential for premium liquid fuel substitution. Several liquid fuels can be produced directly or indirectly oil or biooil and slurries of charcoal with water or oil. In order to carry out the cost analysis it is helpful to briefly review the technology and products (13).
The main products of pyrolysis are: • Liquid. "Oil" or biooil has been produced in yields of up to 70% wt on feed (dry basis)
from flash pyrolysis on laboratory experiments. Commercial operation may not give such high yields and a more conservative figure of 50% wt is used in the calculations below. It has an elemental analysis similar to source biomass and an HHV of 2025 MJ/kg. The product can be used directly as liquid fuel oil or upgraded to a hydrocarbon fuel. Cost analyses for both products are included.
• Solid char can be produced at yields of up to 30% wt on a volatilefree basis (up to 50% with high volatiles) depending on the process. Low char yields accompany high liquid yields. The char has an HHV of 30 MJ/kg and the product can be used as solid fuel directly, or briquetted, or slurried with water or oil to give a liquid fuel. Cost analyses for both slurry fuels are included.
• A fuel gas is also produced: EITHER an MHV fuel gas from indirectly heated processes at yields of up to 80% wt on feed from flash pyrolysis at high temperatures; or up to 20% wt from low temperature flash pyrolysis. It has an HHV of 1520 MJ/Nm3 and the product can be exported or used inplant for process heat, feed drying or power generation. This is not considered further as the process is assumed to be energetically self sufficient by using this byproduct gas. OR an LHV fuel gas is produced from partial gasification processes at yields of 80140% wt on feed. It has an HHV of 48 MJ/Nm3 and again the product can be exported or used inplant for process heat, feed drying or power generation. This is not considered further

366
as the process is assumed to be energetically self sufficient by using this byproduct gas. LIQUID PRODUCTS
Yields of liquids from pyrolysis can be influenced by the rate of reaction, with fast or flash pyrolysis at lower maximum temperatures giving the highest liquid yields, with up to 75% wt being reported. This liquid product may be readily bumed and has been employed for this purpose. Problems have, however, been reported in its use, and special precautions may have to be taken in handling, storage and combustion. For these reasons, pyrolysis liquids cannot be readily assimilated into a conventional fuel marketing infrastructure, although there is adequate evidence that they can substitute for fuel oils in several applications. The discount necessary for these lower quality fuels to be adopted in place of conventional fuels could be up to 20%, but this is insignificant compared to the current range of oil prices in the market place as shown later. Upgrading is necessary to give a product that is compatible with conventional fuels, but this is expensive. The products that are considered in the economic analysis are listed in Table 1 with explanations for selection, and significant parameters and properties.
Table 1 Liquid Products from Flash Pyrolysis
Composition HHV Yield, wt % MJ/kg ondaffeed
Crude pyrolysis liquid (oil or bio-oil) - 22.5 50 Char-water slurry 60% wt char (a) 18.0(b) 50(c) Char-oil slurry 20%wtchar(d) 24.0 60(e)
Notes a this is a probable upper limit for a pumpable slurry b based on a char heating value of 30 MJ/kg c based on 30% wt char yield (volatile free) on daf feed and 60% wt solids in the
slurry d this is a probable upper limit for a pumpable slurry e based on 50% wt oil yield andl0% wt char yield on daf feed and 20% wt solids
in the slurry
COSTS OF PYROLYSIS DERIVED LIQUID FUELS Of major importance in implementation of pyrolysis technologies, as with gasification
technologies, is the economics of production. The costs of production are made up in a very similar way as for gasification (see Part 1), and the relevant parameters are summarised in Table 2. Significant differences do, however, arise in capital cost which are shown in Table 3, and in process yield which is summarised in Table 1.
Production costs for each product have been derived in analogous ways as for gasification and results are summarised in Tables 4 to 6. These are based on directly attributable costs with no allowances for environmental benefits such as low sulphur, or fiscal benefits such replacement/reduction of imports.

367
Table 2 Typical Economic and Financial Parameters for Pyrolysis Base Cases
Pyrolysis technology Entrained flow or fluid bed Pyrolyser efficiency (see text) 58 to 93% Feedstocks Refuse, straw and wood Feedstock heating value 20.00 GJ/t daf Number of shifts (for 8000 h/y operation) 4 Project life 10 y Scheduled operating hours per year 8000 Availablility (Actual operating hours / Scheduled operating hours) 80% Actual operating hours (see Part 1) 6400 Throughput 5 te/h, daf basis Capital cost (including working capital) See Table 3 Feedstock cost after processing (see Part 1) as stated Utilities cost £0.24/GJ produced Yearly maintenance cost, fraction of capital cost 0.025 Yearly overheads, fraction of capital cost 0.080 Total cost of labour per shift £25 000/y Nominal cost of capital 10 % Inflation rate 5 %
Table 3 Capital Costs of Pyrolysis Processes for Bio-oil, Char-water slurry and Char-oil slurry Production
Capital cost of pyrolysis processes 1 t/h 2.5 t/h 5 t/h lOt/h
Basic pyrolysis process Lower (from Part 1) £213 000 £412 000 £678 600 £1117 800 Average (from Part 1) £370 000 £715 700 £1178 900 £1941800 Higher (from Part 1) £735 000 £1 421 700 £2 341 800 £3 857 300 Target capital cost (from Part 1) £291 500 £563 800 £928 700 £1 529 800 (target capital cost = mean of average and lower cost - see Part 1)
Pyrolysis for Bio-oil £437 250 £845 700 £1393 000 £2 294 700 Target plus 50% for feed preparation, feed drying and oil recovery
Pyrolysis for Char-water slurry £320 650 £620 200 £1 021 600 £1 682 800 Target plus 10% for slurry preparation
Pyrolysis for Char-oil slurry £466 400 £902100 £1485 900 £2 447 700 Target plus 60% for feed preparation, feed drying, oil recovery and slurry preparation

368
Table 4 Pyrolysis Liquid (Biooil) Production Costs by Feed Type and Scale
Scope: As in Table 2 Costs include drying and using surplus process gas The current average (scheduled and typical) medium fuel oil price is £2.26/GJ, and cover a range from £1.53 to 3.02/GJ. Costs in bold underlined are those below this current market value. This is probably the nearest product to biooil in terms of ease of handling. The current average (scheduled and typical) light fuel oil price is £2.84/GJ, and cover a range from £2.08 to 3.63/GJ. Costs underlined are those below this current market value The current average (scheduled and typical) heavy fuel oil price is £1.95/GJ, and cover a range from £1.41 to 2.52/GJ.
Production cost. £/GJ
STRAW £22/t delivered (£26/t daf) £17/t onfarm (£20/t daf) REFUSE Wet fluff £0.60/t product (£l/t daf)
Includes disposal credit of £10/t raw refuse WOOD £30/tdaf £20/tdaf
1 t/h
5.34 4.81
2.94
5.70 4.81
2.5 t/h
4.19 3.66
1.79
4.55 3.66
5t/h
3.72 3.19
1.32
4.08 3.19
10 t/h
3.43 2.89
1.03
3.78 2.89
Table 5 CharWater Slurry Production Costs by Feed Type and Scale
Scope: As in Table 2 Costs include drying and using surplus process gas The current average (scheduled and typical) medium fuel oil price is £2.26/GJ, and cover a range from £1.53 to 3.02/GJ. Costs in bold underlined are those below this current market value. This is probably the nearest product to biooil in terms of ease of handling. The current average (scheduled and typical) light fuel oil price is £2.84/GJ, and cover a range from £2.08 to 3.63/GJ. Costs underlined are those below this current market value The current average (scheduled and typical) heavy fuel oil price is £1.95/GJ, and cover a range from £1.41 to 2.52/GJ.
Production cost. £/GJ 1 t/h 2.5 t/h 5 t/h 10 t/h
STRAW £22/t delivered (£26/t daf) £17Λ onfarm (£20/t daf) REFUSE Wet fluff £0.60/t product (£l/t daf)
Includes disposal credit of £10/t raw refuse WOOD £30/tdaf £20/tdaf
6.15 5.48
3.15
4.82 4.15
4.29 3.63
3.98 3.31
LÄ2 L23. fL2&
6.59 5.48
5.26 4.15
4.74 3.63
4.42 3.31

369
Table 6 Char-Oil Slurry Production Costs by Feed Type and Scale
Scope; As in Table 2 Costs include drying and using surplus process gas The current average (scheduled and typical) medium fuel oil price is £2.26/GJ, and cover a range from £1.53 to 3.02/GJ. Costs in bold underlined are those below this current market value. This is probably the nearest product to bio-oil in terms of ease of handling. The current average (scheduled and typical) light fuel oil price is £2.84/GJ, and cover a range from £2.08 to 3.63/GJ. Costs underlined are those below this current market value The current average (scheduled and typical) heavy fuel oil price is £1.95/GJ, and cover a range from £1.41 to 2.52/GJ.
Production cost. £/GJ
STRAW £22/t delivered (£26/t daf) £17/t on-farm (£20/t daf) REFUSE Wet fluff £-0.60Λ product (£- 1/t daf)
Includes disposal credit of £10/t raw refuse WOOD £30/tdaf £20/tdaf
lt/h
3.99 3.60
2.26
4.24 3.60
2.5 t/h
3.14 2.76
1.41
3.40 2.76
5t/h
2.79 2.41
1.06
3.05 2.41
lOt/h
2.57 2.19
0.42
2.83 2.19
DISCUSSION Liquid fuel production from waste with its high disposal credit clearly offers the more
attractive opportunities for commercial implementation, with oil and char-water slurries giving similar production costs. Char-oil slurries offer a significant advantages due to the higher yields of products, but there are more uncertainties in their production and use which require resolution. Both feed cost and product yield are major economic factor in minimising product cost. Both wood and agricultural wastes offer opportunities for oil and char-oil products at higher capacity plants and if the feed cost is sufficiently low. Char-water slurries are less interesting in these situations.
A major uncertainty is the value to be attached to the product It might be compared to a light fuel oil with a scheduled price in the UK as high as £3.63/GJ which opens up wider opportunities for wood, agricultural wastyes such as straw, and energy crops. Alternatively if the product is compared to heavy fuel on the open market, and also requires a discount for successful sales, then only larger capacity high disposal credit waste fed processes have a chance of success. On the market side, therefore, conventional liquid fuel product prices and their movements are a dominant influence on the rate of implementation and degree of success of these technologies.
UPGRADING OIL TO HYDROCARBONS Upgrading technology is not well developed with most attention being paid to either
hydrogénation or zeolite processing to give synthetic gasoline and other hydrocarbons. The third alternative is slurrying with water or oil to give a liquid fuel substitute. This is only at an early stage of development, with few details available on technology complexity, additives or costs.
Some preliminary estimates of production costs of upgraded liquids as hydrocarbons are indicated in Table 7, to give some idea of the performance of the two main technologies for producing gasoline, diesel and fuel oil from biomass by flash pyrolysis and liquefaction. These technologies are at a much less advanced stage of development. The data is taken fom

370
the recently completed IEA DBL (Direct Biomass Liquefaction) study (14), which will be continued as ALPS (Assessment of Liquefaction and Pyrolysis Systems).
Table 7 Liquid Hydrocarbons Production Cost by Flash Pyrolysis and Direct Liquefaction (14)
Scale 1000 daf t/d wood
Product costs it Rash Pvrolvsis
£/GJ
Primary liquid Product cost Primary product Product value *
Final product Product cost Final product Product value *
94.3 5.3 Pyrolysis liquids
48.0 2.1
423.6 9.3 Gasoline, high octane
£225/t
Direct Liquefaction £4 ZIQI
266.1 7.7 Liquefaction liquid
66.0 2.2
512.3 11.2 Gasoline, low octane; + Diesel
£200/t
Note * Current approximate UK figures
CONCLUSIONS The production cost analyses presented above show the short term potential for energy
production from wastes, particularly where there is a substantial credit associated with disposal of the waste, as feedstock contribution is particularly significant. Why, then, has so little been done to gasify these wastes, or any attractively priced biomass resource?
The answers are firstly that considerable attention has been paid to waste conversion throughout the world, and secondly the lack of success is due to many factors, including technology, economics, environment, politics, and social attitudes. A comprehensive assessment of these factors and their interactions cannot be considered here, but some of the salient points are identified. • There arg still technical problems to be resolved in reliability and performance, but these are
not insurmountable and there is evidence in several operating examples in Europe and North America where problems have been satisfactorily resolved.
• The economic attractiveness has been reduced by low conventional energy prices which has removed the pressure to resolve technical problems and develop better systems. This is compounded by the perceived uncertainty of these newer methods of energy production, with a preference by industry and government to invest in low risk ventures in the absence of any significant economic advantage.
• A further factor in the perception of gasification arises from comparisons with conventional fuels i.e. natural gas. This is widely available, totally reliable, and with zero contaminants, none of which applies to biomass or waste derived low or medium heating value fuel gas. There is considered indifference to examination of alternative fuel specifications by many users who take a conservative viewpoint. There is a similar perception of pyrolysis products arising from comparisons with conventional fuels i.e. fuel oils. The lack of understanding of the nature and utilisation constraints of these new fuels is likely to inhibit their use in the short term.
• In a more unstable environment, the incentives to find alternative energy sources will cause more serious attention to be placed on these technologies. The pressures will come from the environmental lobby who expect cleaner and safer waste disposal methods, and the energy lobby who will react to an unstable energy supply situation when it arises.
There seems little doubt that there will be considerable opportunities for thermochemical

371
conversion of biomass and solid wastes in the future particularly if there is pressure from increasing conventional energy prices and also from environmental concerns over waste management practices. There is need for technology assessment and, if appropriate, support for long term demonstration. This will help ensure that the technologies are sufficiently developed and mature to provide reliable service when the occasion demands.
ACKNOWLEDGEMENTS Much of the data reported in this paper were generated for the Energy Technology
Support Unit at the UKAEA, Harwell, UK, as an update of earlier work (4). They are intended to be published in due course in a strategic review of thermochemical conversion technologies for biomass and wastes (8). All opinions and statements are the views of the author and in no way reflect any views of the UKAEA or the UK Department of Energy.
REFERENCES 1 Feldman, H, et al., US DoE Thermochemical Contractors Meetings, Portland 1984,
and Minneapolis 1985 2 Smith, D, John Brown Engineers and Constructors Ltd, Private communication 3 Β H Levelton & Associates Ltd, Enfor project C-258, "A Comparative Assessment of
Forest Biomass Conversion to Energy Forms." Volume V, Data Book of Unit Processes for Primary Conversion by Thermal, Chemical and Biological Methods.
4 Bridgwater, A V, Double, J M, and Earp, D M, "Technical and market assessment of biomass gasification in the UK", report to ETSU, UK Dept Energy, 1986
5 "Revised Guide to Capital Cost Estimation", Institution of Chemical Engineers, UK, 1988
6 Watt Committee: Working Party on Gasification; "The Cost and Competitiveness of Low Calorific Value Fuel Gas from Coal".
7 "Energy from Biomass: A Process Manual for 90 Conversion Routes:. Technical Insights Inc, Fort Lee, New Jersey, USA, October 1983.
8 Bridgwater, A V and Stronach, Ν J, "A Review of Thermochemical Conversion Technologies", Report to ETSU, Department of Energy, 1989
9. Bridgwater, A V and Double, J M, Institute of Energy, May, 1988 10 Bridgwater, A V and Double, J M, 4th EC Conference on Biomass for Industry and
Energy, Orléans, (Elsevier Applied Science, 1987) 11 Mitchell, C Ρ and Bridgwater, A V, These proceedings 12 ETSU personal communications 13 Bridgwater, A V, Beenackers, A A C M and van Swaaij W Ρ M, "Biomass Pyrolysis
and Liquefaction: Status and Opportunities in the European Community", Proceedings of Euroforum New Energies, EEC Conference, Saarbrücken, October 1988
14 Elliott, D C, et al "A Technical and Economie Analysis of Direct Biomass Liquefaction", in Energy from Biomass and Wastes XJU, IGT, February 1989.

372
ENVIRONMENTAL PROBLEMS IN THE USE OF BIOMASS FUELS GLOBAL AND LOCAL ASPECTS
Philippe GIRARD, CENTRE TECHNIQUE FORESTIER TROPICAL DEPARTEMENT DU CENTRE DE COOPERATION INTERNATIONALE EN RECHERCHE AGRONOMIQUE POUR LE DEVELOPPEMENT 45 bis, Avenue de la Belle Gabrielle 94736 NOGENT SUR MARNE CEDEX FRANCE Tel : (1) 48 73 32 95 Telex : CETEFO 211085F
Sumnary In Europe and especially in France, biomass energy use has increased over the last few years. The various issues, concerning biomass energy use seriously dealt with so far are such as : trade balances, market positioning according to sector, fuel supply problems and distribution. On the other hand, related environmental problems are more often ignored. The emission levels depend upon the transformation process utilized viz combustion, gazéification, carbonisation. Industrial units deal reasonably well with the pollution problems. It is not the case for residential heating and small scale charcoal making. In term of quantity, non condensable vapours represents the major tant. Hydrocarbons and pyroligenous oils contain many species of chemical compounds and some of them are known to be carcinogenic. Whereas, the particulates could cause different lung infections. The emissions represent not only the environmental problem but mean also the economic loss due to unburnt combustible. Therefore reducing pollution from unburnt combustibles could also improve the financial gains as a result of efficient performance. Therefore, it is necessary to compare this form of pollution to pollution levels from different fuels.
In Europe, and especially in France, biomass-derived energy production by thermochemical means has increased considerably over the last few years, particularly in the field of residential heating. In France, public bodies have been instrumental in promoting development in this area.
Official commitment has encouraged the development of a new sector of economic activity and has contributed to a proportional reduction in oil imports. Until now, reviews of this sector have always kept to the analysis of trade balances, market positioning according to sector, fuel supply problems and distribution. However, a detailed study of problems related to the use of biomass as fuel reveals a number of environmental aspects which are a cause for grave concern but are more often than not ignored.

373
Woodfuels may be characterized by a generally high and variable moisture content and by high levels of volatile matter (80 % in weight). Levels of emission can only be controlled through the control of heat propagation. Thermal regulation requires great care, which present techniques do not always permit. This results in : - over-estimation of energy yields - difficult and sometimes hampering operating procedure - unacceptable levels of atmospheric pollution.
Awareness of an concern for these problems vary with the different sectors involved in thermochemical biomass transformation. Pollution problems are dealt with reasonably well in the case of high-output industrial units, either directly as such or indirectly through optimization of equipment performance, but in the case of residential heating and small-scale charcoal-making equipment there is considerable room for improvement .
- Combustion : with the recent crisis in oil supplies, many people in industrialized countries have turned to firewood for individual heating needs, primarily for financial reasons. The impact on the environment can be considerable, as certain American studies have down <1> <2> <3>. Pollution in this case is essentially atmospheric (dust and hydrocarbons ). Emission levels can be very high and vary according to the nature of the equipment and fuel used.
- Gasification : the nature of pollutants from gas producers (dust and hydrocarbons) is an obstacle to development in this sector. This is true for heat production in agro-industry where tolerance to certain compounds is very low <4>, as well as for energy production for motors supplying driving power or electricity <5>. In the latter case, the need for reliable and low-cost gas-scrubbing systems is the final obstacle in the development of gas-producing equipment, especially for developing countries where potential demands is very high. At the present stage in technical development, activities in this area are purely exploratory and woodfuel consumption is correspondingly low.
- Charcoal production : this sector has until now been considered of marginal importance, but recent studies by the CTFT, the Laboratoire National d'Essais - LNE and the CITEPA (*) <6> have shown that local atmospheric pollution resulting from charcoal-making activity can be considerable. As a result of these studies, the French Ministry of the Environment has introduced legislation aiming to implement controls on listed units.
We aim here to set out results acquired to date in order to give an overall view of environmental problems resulting from biomass transformation, with particular regard for woodfuels used in residential and small-scale industrial combustion and pyrolyser installations, these sectors being by far the most polluting and the heaviest consumers of wood.
NATURE OF THE PRINCIPAL POLLUTANTS EMITTED THROUGH THERMOCHEMICAL BIO-MASS TRANSFORMATION
Pollutants are generally placed under three different headings : - Particles in suspension in gas emissions. - Hydrocarbons and condensable or pyroligneous organic compounds present in vapour or aerosol form.
(*) CITEPA : Centre Interprofessionnel Technique de la Pollution Atmosphérique .

374
- Noxious gases as 00, NO», SO*. Wood and biomass rarely contain sulphur, which means that fuels derived from them have significant advantages over the conventional fuels.
The mechanisms of the pyrolysis process determine the nature and concentration of pollutants, which vary according to a wide range of parameters. Among these, reaction temperature and the kinetics of thermal transfer are the most significant.
Where industrial combustion units are concerned, pollution is limited thanks to the optimisation of techniques or to the installation of effluent reprocessing systems. The same is true for industrial charcoal production units where emissions are infrequent and very brief when techniques are well mastered, the main polluters are small-scale units using partial-combustion techniques where all smoke is discharged into the air. Production units with clean kilns are uncommon in Europe and almost unknown in developing countries.
The table 1 gives data from different studies in the matter. The figures given are not necessarily directly comparable as the objectives and methods used in the studies were not identical.
These studies show fairly high levels of pollution and wide variation in results, the latter being mostly due to the differing conditions of equipment operation as the table makes clear. For example, the particle emission factor varies from 0.5 to 22 g/kg of wood according to furnace temperature and to the size of wood pieces. Under poor operating conditions, domestic heating appliances become "faulty pyrolysers".
Work by DUSSERRE <15> on wood-fired boilers confirms these results and clearly indicates the prospects that can be opened up by improving equipment.
IMPACT ON THE ENVIRONMENT Where particle emission is concerned, studies carried out in the US
have shown that almost all dust particles can be inhaled. Although they are not a direct cause of illness, these particles can be a contributing factor in the development of lung infections <7>.
There a large numbers of hydrocarbons and condensable organic compounds, over 300 compounds having been identified <7> <10> <11>. Scientists are particularly concerned with the polycyclic hydrocarbons, some of which have been clearly shown to be carcinogenic (see Annexe). A significant proportion of these substances are emitted together with dust particles, thus increasing the risk of propagation and contamination.
Incondensable substances make up the larger part of effluent. The use of wood as fuel or for charcoal production produces large amounts of carbon monoxide whose insidious toxicity has long been proved. The presence of nitrous oxides in woodsmoke is noted in <7>.
According to a CERN study carried out in France <12>, 5 million homes were using wood for fuel in 1984, with wood being the primary fuel in 50 % of cases. Average unit consumption, measured during a field survey and weighted according to local woodland structure, was found to be 16 stacked m3 in a nomai heating season where wood was the primary fuel, and 3 stacked m3 where it was used as an auxiliary fuel. This brings total fuelwood consumption in this sector to 45.5 million stacked m3 (i.e. 17 million tonnes or 6.7 million TPE*).
*TPE : Tonne Petroleum Equivalent.

375
In 1985 wood consumption for French charcoal production amounted to only 400 000 tonnes according to our estimate. When communal wood or bio-mass fuelled heating installations are added to these figures, the national total for biomass-derived fuels is equivalent to 8 million TPE or 4 X of French energy consumption in 1986 <13>.
According to our sources, consumption in some European countries is in all probability higher than this, but it must be noted that we do not at present have any European figures on this.
ECONOMIC IMPACT In addition to environmental problems, high emission levels of un-
burnt effluent have the following consequences : - Corresponding reduction in conversion yields. Energy yields are of less than 50 X for charcoal production by means of partial combustion techniques without heat recovery systems.
- Reduced efficiency and reliability of equipment. Flues in residential heating units require more frequent maintenance and fire hazards are increased with poor performance.
- Wood becomes less competitive in relation to conventional fuels such as coal.
This gives all the more cause for concern for the following reasons : - Wood is superior to other biomass fuels and is the most convenient to use in all the sectors concerned and especially for residential heating purposes. It must also be noted that wood and charcoal meet as much as 90 X of energy needs in some developing countries, where the improvement in energy yields from charcoal production is essential and must be given priority. Any action taken in this area must include measures to reduce pollution.
- Wood has a very good public image, which provides a firm basis for all action taken in view of developing its use for energy production. This is particularly true in the present context of increasing public awareness of environmental and ecological problems.
- Pollution could be significantly reduced if techniques which have now been perfected were applied under appropriate conditions. Manufacturers should be aware of these and provide instructions as to the size of wood pieces to be used, maximum moisture content, loading frequency, etc ...
Besides reducing pollution, lowering emission levels through optimisation, better definition of equipment and effluent recovery during the carbonization process will lead to financial gains as a result of improvements in yields and performance. Prospects are therefore promising in this respect as reducing pollution does not necessarily involve additional expense to the user.
Although pollution resulting from the use of wood for energy production should not be underestimated, woodfuel use must be considered within a wider context.
The table 2 <14> <15> makes a comparison between the different fuels used for heating purposes in units of different sizes :
As for the previous table, care should be taken in comparing these results. These clearly indicate that residential heating is a source of pollution, with coal being the worst offender.
It should also be noted that motorway transport causes the following forms of pollution <16> :

376
For guidance, it should be noted that 25 million cars with a payload under 3.5 Τ are on the road in Prance, travelling 12 000 km per year on average with an average fuel consumption of 9 litres per 100 km for petrol and 7.2 litres per 100 km for diesel oil. (Studies are now under way on a proposal for European norms aiming to reduce pollution).
CONCLUSIONS lhe observations given here and the numerous research programmes now
in progress indicate that there are prospects for significant improvements in equipment performance. Unfortunately, as can be seen from the bibliography, few coordinated programmes have been implemented at European level, in comparison with studies carried out in the US over the last 10 years. It is therefore necessary to : Develope methods and equipment for standard!zed sampling and analysis which are specific to each situation. The methods used by DUSSERRE, for instance, were not applicable to carbonization processes.
Such methods must be recognized and accepted by all laboratories and should be flexible enough to allow for the following :
. application by all laboratories (cost of implementation)
. ease of implementation
. permanent review of cycles allowing sequential sampling of sufficient duration.
Carry out fundamental research on pyrolysis with particular regard for yield improvement and a corresponding reduction in unburnt emission through a better understanding of phenomena and the redefining of apparatus and their implementation.
Carry out a large number of equipment performance tests by working on the parameters affecting thermochemical reactions in the conversion process. The aim is to acquire data needed for further development and finalizing of new techniques. Quantification of effects on the environment must be undertaken at the same time.
Supply training and information to users. Develope new and more efficient techniques and equipment. In the case of charcoal production, this will necessarily require the elaboration of effective smokerecovery systems. The prospects of smokeincinerating systems appear to be promising as these would not only reduce pollution but also supply considerable quantities of heat which can be made use of during the process (drying out of wood before carbonization).
Significant reductions in emission levels and the corresponding improvements in energy yields, together with optimization of equipment performance should make woodfuels more competitive in relation to conventional fuels such as coal or domestic fueloil. Pollution levels generated by residential heating should drop with a switch to such alternative fuels.
A more specific approach to European fuelwood consumption would be of benefit and would certainly clarify the issue at stake.
In this context, an increase in wood and bicmass use for energy production can provide a tenable answer to the problems of sustaining European agriculture, bearing in mind that at the beginning of 1988 it was decided to lay fallow one million hectares of land, and that several thousand hectares more are left fallow each year. If costeffectiveness thresholds can be overcome, progress in genetic engineering can open up prospects for the development of coppice plantations in Europe for energy production purposes.

377
BIBLIOGRAPHY
(1) ROBERT E., MANNING I &. J., COOK W., HAYES T. Preliminary report on a study of the ambient impact of residential wood combustion in Peters-ville, Alabama. Residential solid fuels conference, Portland, Oregon, USA, 1981.
(2) CARLSON J., Residential wood combustion in Missoula, Montana. An overview of its air pollution contributions, health effects, and proposed regulatory solutions. Residential solid fuels conference, Portland, Oregon, USA. 1981.
(3) DECESAR R. The quantitative impact of residential wood combustion and other vegetative burning sources of the air quality in Medförd, Oregon. Residential solid fuels conference, Portland, Oregon, USA, 1981.
(4) S.C. BHATTACHARYA, N. SHAH. Utilization of producer gas for drying agricultural crops. Regional seminar on alternative energy application in agriculture. THAILAND, 1986.
(5) M. STASSEN. Baseline monitoring need. Chevet gazifier station in Se-chelle. UNDP European Bank. Biomass technology group. TWENTE. Juillet 86.
(6) GIRARD P., MEYER C , BOILOT M., FONTELLE. Caractérisation des émissions de carbonisation en four à combustion partielle. Mise au point de systèmes de traitement des fumées de carbonisation. 4ème Conférence européenne. Biomass for energy and industry. 1987.
(7) HUBBLE B.R., STETTER J.R., GEBERT E., HARKNESS J.B.L., FLOTARD R.D. Experimental measurement of missions from residential wood burning stoves. Residential solid fuels conference, Portland, Oregon, USA. 1981.
(8) CITEPA. Monographie. Carbonisation du bois et pollution atmosphérique. 1986.
(9) MEYER C. Détermination des rejets atmosphériques lors de la carbonisation par combustion partielle. Mémoire de fin d'études ENITA-CTFT. 1986.
(10) RUDLING L., AHLING B., LÖFROTH G. Chemical and biological characterization of emissions from combustion of wood and woodchips in small furnaces ans stoves in Sweden. Residential solid fuels conference, Portland, Oregon, USA. 1981.
(11) BEAUMONT 0., Pyrolyse extractive du bois. Ecole des Mines de Paris. 1981.
(12) CERN. Etude de la consommation de bois de chauffage en maisons individuelles (résidences principales). Saison de chauffe. 1984-1985.
(13) P. CORTE., T. DURAND., T. GONARD., B. MOREL DE LA COLOMBE. Thermoche-mical use of biomass in France. Evaluation and trends in AFME strategies. 4ème Conférence europénne. Biomass for energy and industry. 1987.
(14) COOPER. Evironnemental impact of residential wood combustion emission and its application. Air pollution control. Vol. 30. pp. 855 à 861. 1980.
(15) DUSSERE P., Etude des chaudières à bois : contribution à la mesure des émissions des composés imbrûlés, à l'évaluation des rendements énergétiques et à la modélisation de la combustion. INSA. 1986.
(16) DELSEY M., La pollution due aux moyens de transport. INRETS Laboratoire énergie nuisances. Note d'information n' 13. 1979.

378
T a b l e 1 . POLLUTANTS EMISSIONS FROM RESIDENTIAL WOOD COMBUSTION AND CARBONIZATION
Wood
Consumption (kg/h)
Furnace Temperature "C
CO (g/kg wood)
Particles (g/kg wood)
Condensable organic compound (g/kg wood)
P.A.H. (g/kg wood)
Combustion (7)
Residential Heating
Red Oak (large pieces)
0.82 2.58
250 360
138 196
0.8 22.2
3.0 8.4
0.004 0.04
Combustion (7)
Res. Heating
Ditto (small pieces)
3.9 7.7
500 800
92 148
0.5 2.3
0.8 2.1
0.008 0.03
Carbonization (8)(9)
Partial Combustion
Pinus sylvestris
* 400450
80110
5.110.2**
14.525.3
0.100.432
* Cycle l a s t i n g approx. 20 h for a t o t a l load of 600 kg anhydrous wood. ** Measures made in conformity with French standard NF χ 44051 and NF χ 43003. Studies now in progress a t the CTFT and LNE appear t o c a s t doubts on the use of measuring apparatus under condi t ions imposed by c a r
bonizat ion techniques and sugges t the implementation of a more appropria
t e sampling method.
T a b l e 2 . POLLUTANTS EMISSIONS FROM DIFFERENT FUELS USED FOR HEATING PURPOSES
Gaz Fueloil
Coal
Wood (open hearth)
Wood (stoves)
CO g/th
0.036
0.072
6.3 5.4 39.6
NO, g/th
0.144
0.162
0.18
0.45
0.126
SO« g/th
0.001
0.5412.6
0.9 13.6
0.054
Particle g/th
0.036
0.126
1.08
2.34
2.16
B(a)P Mg/th (*)
0.32
3.6 10 000
180 535
(*) Benzo (a) pyrène.
T a b l e 3 . POLLUTANTS EMISSION FROM MOTORWAY TRANSPORT
NO«
Total hydrocarbons..
4star petroKNCV*: 12300 th/t) Diesel g/kg fuel g/th g/kg 200 250 16.26 20.32 8 1 0 10 30 0.81 2.44 10 15 >> 1 5 8
20 50 1.63 4.06 2 3
(NCV : 10900 th/t) g/th
0.73 0.91 0.91 1.37 0.46 0.73 0.18 0.27

379
WASTE MANAGEMENT AND PYROLYSIS: CURRENT SITUATION, TRENDS AND PROSPECTS
C H . NELS Federal Office for the Environment, Berlin
Summary
For the past 15 years researchers from both the scientific world and industry have been attempting to develop new waste treatment processes which might prove more effective than the conventional methods of landfill, composting and incineration in meeting current demands for the exploitation of waste by the recovery of raw materials and energy. One such process is pyrolysis. High hopes were initially vested in this process, so high in fact that the judicious . use of known technology was occasionally prevented. Only a very small number of the waste treatment processes which have proved successful in the Federal Republic of Germany have been, or are yet to be, tested on a large scale. Materials recovery or waste disposal is given main priority depending on the type of waste used as feedstock. Ultimately, the overall criterion by which such processes are judged is their impact on the environment. Although pyrolysis is not expected to revolutionize the treatment of waste in the future, it should, in individual instances, be able to complement existing processes, or offer a practical alternative.
1. INTRODUCTION
In the early seventies, two main methods were used to treat household waste in the Federal Republic of Germany: landfill and incineration. Under the landfill programme, a total of more than 50 000 refuse tips were in the process of being reduced to approximately 500 controlled landfill sites. Waste incineration technology had reached a relatively advanced level, after being given a particularly important boost in 1974 by changes to the technical specifications for air pollution control : these introduced limit values for the emission of noxious gases such as HCL and HF for the first time, and made the use of waste gas scrubbers obligatory in incineration plants.
It was then that the first energy crisis struck. An artificially induced fall in the supply of crude oil, the main source of energy, prompted various countries in the West to step up their search, not just for new reserves of oil and natural gas, but also for alternative organic materials which, after suitable treatment (pyrolysis, degasification, gasification), might yield oil and gas. Consideration was given to anything with a high organic content: household refuse, sewage sludge, special category waste, scrap tyres, the organic component of material from breakers' yards, waste from the plastics industry, etc. Research was carried out into processes for treating this waste, such as the rotary kiln, the vertical reactor and the fluidized bed (direct and indirect heating).

380
Very few of these processes were in fact new. The practice of heating wood to produce charcoal, a clean fuel which gives off very little smoke, was widespread even in the Middle Ages. The degasif ication of coal to produce town gas became popular in the 19th century, though it was rendered largely redundant by the advent of natural gas, and is now confined to the production of coke for the iron industry. The coal and gas industries also produced most of the raw materials for the chemical industry before the development of petrochemistry.
On the basis of past experience, it ought therefore to have been fairly obvious that pyrolysis and gasification would not solve the energy problem in one fell swoop, and that such processes might even lead to new pollution problems. In certain circles, however, euphoria reached such a pitch that experts who were anything but wholly enthusiastic were branded "anti-environmentalist". The words of one inventor summed it all up: "If we pyrolysed all household waste using my process, and then did the same with all plastic waste, scrap tyres, dead wood from the forests and all old asphalt road surfaces, we would have so much oil that we wouldn't have to import any. Plans for new nuclear power stations could be scrapped, and power stations which are already operational could be shut down. So why won't the state promote research into this form of energy?".
That is precisely what the state has done. In the Federal Republic of Germany, approximately DM 400 million was spent on research into waste disposal between 1976 and 1989, of which 10% (DM 40 million) was devoted to pyrolysis and gasification. What exactly has been achieved, and where do we stand today?
2. AIMS OF PYROLYSIS AND GASIFICATION
Pyrolysis and gasification aimed to achieve essentially three different goals. The primary objective as regards the treatment of household and related wastes was that of disposal. The main concern was to reduce the volume of waste and convert it into a residue that could be disposed of as landfill without damage to the environment. The raw materials and energy created in the process were welcome by-products which were sold commercially to reduce running costs.
The pyrolysis and gasification of special category waste with a high calorific value (e.g. plastic wastes, used tyres, organic residues from breakers' yards) was essentially aimed at the recovery of raw materials such as oil, metals and, where possible, coke and carbon black. Unlike processes used for treating household refuse, processes such as these were to be employed only if overall costs could be offset by the sale of the raw materials produced.
Finally, there were pyrolysis processes which involved the careful separation of different types of materials. These types of process, which were generally discontinuous, were used for separating composite organic materials from metal components, or removing organic impurities from metal (e.g. conveyors used in paint shops). This last category will not be discussed in further detail here.
The following four hopes played an important role in generating support for the initial and subsequent development of pyrolysis and gasification processes, particularly as a means of treating household waste:
1. The new processes would make it possible to build easy-to-operate plants for the thermal treatment of waste. These would be cheap to run even when throughput was low (perhaps down to 250 t/d) and so could be used to dispose of waste in the smaller administrative regions.

381
2. These new processes would make it possible to recover not only energy, but also large quantities of valuable materials contained in waste, such as synthesis gas, oil, coke and metals.
3. These new processes would be able to cope with varying quantities of non-homogenous waste, so that all types of waste (including special category waste and sewage sludge) produced within a given collection area could be disposed of together.
4. These new processes, besides achieving all the above, would have minimal impact on the environment.
Since any material that can be incinerated (household waste, plastic waste, used tyres and other organic materials, etc.) can also be either degasified or gasified, supporters of these new processes initially saw themselves as being in direct competition with the waste incineration industry. This occasionally led them to point out in no uncertain terms the inherent weaknesses of the rival process. Incineration, they stressed, produces excessive volumes of waste gas which require scrubbing, as well as a solid residue which has not fully burned out. Furthermore, incineration furnaces often have difficulty coping with wastes having a high calorific value (e.g. scrap tyres) and certain other types of waste such as glass and plastics, the resultant energy cannot be stored, valuable scrap iron separated out of the slag is lost through oxydization, and finally, large quantities of noxious emissions are produced. Sometimes, the disadvantages of the rival process were taken as automatic proof of the advantages of pyrolysis and gasification. This disregarded three points:
1. Waste incineration was an easy target for the critics: the process had been in use for more than 30 years and had thus been subject to rigorous scrutiny. Small wonder, then, that its disadvantages were so well known. By contrast, no pyrolysis or gasification process had yet undergone such a gruelling test.
2. Incineration as a process cannot be dismissed out of hand simply because some plants are badly designed or poorly run. The incineration of waste is itself primarily an environmental protection measure. The fact that it, like any conversion process, has its disadvantages, should and does encourage efforts to improve it. The great potential in this field has not yet been exhausted by any means, as can be seen from the latest developments in waste incineration as a way of reducing harmful substances.
3. The debate tended to become unduly restricted when incineration and pyrolysis/gasification were being considered as alternatives. The new processes also have their advantages and disadvantages, so decisions as to which of these processes to adopt should be made on a case-by-case basis - and other possibilities should also be considered.
Whatever truth there may be in the saying that the better is the enemy of the good, the fact remains that the debate on incineration versus pyrolysis has occasionally produced quite unsatisfactory solutions. Suppose, for example, that all the waste disposal sites in one particular area were full, and that the required reduction in the volume of waste could not be achieved by recycling, then incineration might well be the only solution left. However, it proved impossible to persuade the politicians of this fact: the technology employed in the incineration industry may well be state-of-the-art, but pyrolysis was considered to be better. The upshot was that tips which were already full to capacity continued to be used (perhaps without proper foundation work being carried out and without due attention being given to the problem of water seepage) , or waste was moved to other

382
areas (thereby causing traffic congestion) , or perhaps even exported altogether, which is highly unsatisfactory from a political point of view. The current crisis in waste disposal, much bemoaned by the Minister for the Environment, can be blamed at least partly on the fact that badly needed waste incineration plants were not constructed because people thought they should wait for pyrolysis, which was supposed to be "better".
3. CURRENT STAGE OF DEVELOPMENT
Even more significant than the technical differences between incineration and pyrolysis as alternative means of treating municipal waste is the fact that waste incineration plants have been operating commercially for more than 25 years, and thus have a proven track record. The first waste incineration plant was built in Hamburg at the end of the last century.
The large-scale industrial exploitation of pyrolysis is, by contrast, still very much in its infancy despite approximately 15 years of development. The first full-scale pyrolysis plant for waste disposal, which is situated in Burgau and has a low annual throughput (35 000 t) has been in operation for just two years now (1) .
The processes developed over the past few years have given rise to four different types of household waste treatment plants, all of them based on pyrolysis. The following is a description of the various component parts of pilot and industrial installations (1):
KWU plant (Goldshöfe): scrap reduction; pyrolysis drum heated indirectly via internal off-gas pipe; gas converter (cracking achieved by substoichiometric partial combustion in coke bed at approximately 1 000°C); two-stage scrubber; gas engine with generator; deposition of pyrolysis residues. Alternatively, it has been suggested that the cracked pyrolysis gas be cleaned in a spray dryer producing no waste water, and burned off directly in a waste heat boiler. DBA plant (Burgau): scrap reduction; pyrolysis drum heated indirectly via outside wall; cyclone for partial dust removal; secondary combustion chamber at around 1200°C; boiler with turbine and generator; deposition of pyrolysis residues (further reprocessing of residues in planned). PKA plant (Aalen/Unterkochen): waste sorting plant; reprocessing plant for the "light fraction" (compressed pellets); pyrolysis drum heated indirectly via gas burner; cyclone for partial dust removal; gas convertor (cracking in coke bed); single-stage wet scrubbing; rotary burner; boiler with turbine and generator; burning off of pyrolysis gas together with biogas from a waste composting plant (alternatively, gases can be used in a gas engine); calcination plant for the processing of pyrolysis residues ; ozonation plant for cleaning continuously discharged waste water. KWU low temperature carbonaization plant (Ulm/Wiblingen): waste treatment; pyrolysis drum; sorting of pyrolysis residues; cylinder crushers or swing pipes for pulverizing fine fraction; smelting furnace for pulverized pyrolysis residue and low-temperature carbonization gas (at between 1200° and 1300°C); waste heat boiler with turbine and generator. There are plans to use the smelt granulate as building material or landfill. Two other pyrolysis processes are being developed, mainly for plastic and special category wastes. These are the fluidized bed process (Ebenhausen) and the rotary drum process (Salzgitter) (2). BBC plant (Ebenhausen): special category waste fed in small pieces into a fluidized bed reactor via rotary feeder and worm conveyor; fluidized medium heated to between 500 and 850°C depending on final product desired; removal of dusts and carbon black from pyrolysis gas; cooling of gas and, where

383
possible, distillation of high- and low-boiling pyrolysis products; removal of sand and metal waste from bottom of reactor. Salzgitter pyrolysis (Salzgitter): special category industrial wastes (solid and fluid) fed into rotary tube in the absence of oxygen; degasification of between 650 and 700°C; outward transfer of solid pyrolysis products; separation into pyrolysed coke and metal fractions (where possible); two-stage cooling and scrubbing of pyrolysis gases ; burning off of waste gas in combustion chamber with waste heat boiler; multi-stage treatment of waste water.
The pyrolysis of sewage sludge is not yet sufficiently advanced to enable large-scale industrial plants to be constructed. Further research will thus be carried out into the following two general concepts, each of them involving a pilot plant with a capacity of approximately 0.5 t/h:
medium-temperature pyrolysis (approximately 500°C), with the option of materials recovery and industrial use of products; low-temperature pyrolysis (approximately 300°C) with the option of use of products for heat generation and industrial use of products (e.g. fatty acids) where appropriate.
These plants will generate large quantities of products. Investigations are to be carried out into the exploitability of such products and the possible effects of such plants on the environment, particularly as regards secondary emissions. When the pilot phase comes to an end, both concepts are to be the subject of a detailed scientific and technical evaluation by an independent body.
The above descriptions of initial research in this field show quite clearly that the main priority when the development of pyrolysis first began was the recovery of raw materials such as gas, oil, coke and carbon black. Today, by contrast, the emphasis is on waste disposal. There are three main reasons for this change in direction: firstly, there is a shortage of space for tipping household refuse and waste from breakers' yards; secondly, plastic wastes are not ideally suited to processing, as they yield only a limited amount of new products; and thirdly, the amount of sewage sludge being used in agriculture is decreasing whilst the total volume of sludge produced is on the increase.
It is, of course, better to exploit raw materials and energy produced during the treatment of waste, rather than simply discharging such products into the environment. But there is no prospect of waste being pyrolysed on a regular basis inorder to recover energy and raw materials. Rather, pyrolysis must be regarded as an alternative or complementary way of treating materials which would otherwise have to be disposed of as waste. What role, then, can pyrolysis play in the future?
4. WASTE MANAGEMENT: THE CHALLENGE (3)
The objectives of waste management have been an integral part of the Federal Republic's environmental policy since about 1970. The 1971 programme on the environment contains a report by the waste disposal project group giving a detailed analysis of the situation at the beginning of the decade. This report was critical, but it also pointed to the legal and technical measures which could help solve the problem, as well as the likely cost of such measures. The main requirements were : systematic arrangement for the removal of all waste from households and industrial plants ; the discontinuation of indiscriminate dumping of mixed waste at small tips and the establishment of central plants where waste could be disposed of

384
according to category, or exploited for other purposes. The 1971 environment programme constituted the first large-scale survey of waste management objectives since 1945.
A comprehensive catalogue of objectives was drawn up in a document entitled "The Federal Government's waste management programme '75". Adopted in 1975 and published the following year, this policy programme is as relevant to waste management today as it was when it first appeared.
A 1978 report by the committee of experts on environmental matters gave the first detailed account of the environmental and economic ramifications of waste management problems. The waste management objectives of the various environment policy programmes can be represented in a schematic diagram (cf. Figure 1).
RAW MATERIALS EXTRACTION
WASTE WA
PRODUCTION/CONSUMPTION
MINIMIZATION OF RESIDUES RE-USE
UNAVOIDABLE RESIDUES
INTERMEDIATE PRODUCTION LEVEL
SUITABILITY CHECKS FOR RE-USE
UNAVOIDABLE WASTE
PRIMARY WASTE TREATMENT
SEPARATION OF VALUABLE MATERIAL
WASTE WHICH CANNOT BE SEPARATED
SECONDARY WASTE TREATMENT
CONVERSION INTO MATERIAL WHTrH rhN f^v. FYPT.nTTEn
MATERIAL WHICH CANNOT BE EXPLOITED
TERTIARY WASTE TREATMENT
DUMPING OF RESIDUE
TER/AIR NUISANCE EFFECT 0 F
*
PRIMARY
SECONDARY RECYCLING
RESIDUE
Fig. 1. Waste Management Action Model
This model is linked to the natural ecosystem via input (supply of raw materials) and output (amount of residue). Waste management activities impose a system of internal and external cycles on this flow chart, dividing

385
it up into five separate levels of action. The top two levels correspond to production and consumption, whilst the bottom three represent waste treatment and the various technical processes available.
If a high percentage of targets is met on one level, the amount of materials passed down to the next level will be smaller relative to the quantity of material passed into the recycling chain. The further down this model a type of material moves, the more specialized the technology required. The meeting of waste management targets becomes more elaborate and expensive with every step. Put another way, action taken at production and consumption level is much more effective as a means of conserving resources and protecting the environment than are attempts to treat the waste produced.
The waste management targets for each action level can equally be regarded as the "five principles" of waste management, as follows:
1. The amount of residues and waste of all kinds generated by production, services and consumption should be kept to a minimum, or preferably avoided altogether. This first principle covers activities such as the following: - the development and application of production processes which use a minimum of raw materials and generate little waste;
- the development and production of consumer goods which are more durable;
- the development and production of goods which do not create disposal problems once their service life is exhausted;
- the introduction and further development of repurchase systems. This first law impinges on the freedom of both producers and consumers. It is first and foremost a political principle which requires a change of thinking in a number of areas and the abandonment of a number of convenient consumer habits.
2. Unavoidable waste should be returned to the production or consumption cycle as soon as possible. This is the principle of primary recycling, which requires responsible decisions to be taken, on the basis of careful checks, as to the suitability of waste for the production of secondary raw materials. It also calls for the separate collection of certain types of waste in households and industry. Primary recycling involves collecting certain materials and residues before they become mixed up with other materials to form rubbish.
3. Maximum quantities of usable materials should be obtained from collected waste. This principle is based on the assumption that, generally speaking, waste cannot be avoided. Those responsible for waste disposal must make every effort to remove valuable substances from the waste collected (secondary recycling).
4. Waste which remains after the removal of valuable substances should be further exploited. This fourth principle requires the use of treatment processes (e.g. composting, incineration, pyrolysis) in order to convert waste into usable substances or energy. Activities such as these not only serve to recover valuable products, but help reduce the general volume and thus ease pressure on the ecosystem, and the high degree of mineralization means that only substances which are fairly compatible with the natural background of a particular site need be dumped.
5. Any substances still remaining after having passed through all the stages described above should be carefully deposited in a way that will not harm the environment. This last means of waste disposal should be resorted to only when the particular conditions or

386
characteristics of a certain type of waste do not permit treatment at a higher level.
5. CONCLUSIONS
It is clear from this action programme that pyrolysis and gasification can be used for the following purposes :
as a means of complementing conventional processes for the exploitation of waste (e.g. production of oil from plastic waste); as an alternative to incineration for the purposes of treating waste before dumping (mineralization) .
In the latter case, pyrolysis will gain acceptance only if fractions which are not exploited (pyrolysis residues) are suitable as landfill material. A pyrolysis process in which gases or oils produced after condensation are simply burned off (possibly with pulverized residue containing carbon) is basically no different from incineration: the only real difference is that the individual stages in the process are separate and can thus be better controlled.
Largescale testing of such a process will ensure that realistic comparisons can be drawn with incineration processes, and will also mean that persistent discussion of pyrolysis will not stand in the way of alternative solutions. However, it must be admitted that perhaps one of the greatest virtues of pyrolysis is the fact that the theoretical debate has also made a major contribution towards the further development of incineration.
REFERENCES
(1) SCHOEMPS, H., "Verbrennung oder Pyrolyse?" (Incineration or pyrolysis?) in LWAMaterialen No.2/89. Contains a discussion of the current role of pyrolysis in the treatment of municipal waste.
(2) GABRIEL, H. et al, "Umweltschutztechnik, laufende F+E Vorhaben 1998" (Environmental protection, current R+D projects, 1988), published by the Project group for environmental protection at the German Institute for Aerospace Research and Testing, for the Federal Minister of Research and Technology, p.309 et seq.
(3) JAEGER, Β. "Abfallverwertung in der Bundesrepublik" (Waste exploit
ation in the Federal Republic), published by the Project group for waste management and landfill redevelopment at the Federal Office for the Environment, for the Federal Minister of Research and Technology, p.l et seq. (in press).

387
RAPPORTEURS REPORT ON SESSION VII ECONOMIC, ENVIRONMENTALAND LEGAL ASPECTS
R. FÄBRY Commission of the European Communities, Brussels, Belgium
Session 8, together with Session 1 dealing with a general introduction and with country overviews, was the only session which was not focussed on the technical aspects, but covered other aspects which are also vital for the future of pyrolysis and gasification, i.e. economic, environmental and waste policy aspects.
Dr A.V. BRIDGWATER presented an estimation of the economic viability of gasification and pyrolysis operations which was based on a very detailed and well constructed study.
Firstly, a lot of economic and technical assumptions are needed to define a basic operation configuration, on which the economic calculations are based. Different types of feedstocks and products are envisaged.
The economics are globally more favourable for refuse than for biomass (wooden material, straw) treatment because of the refuse disposal credit.
The economic estimation shows that some opportunities exist (based on the UK situation) for economic operation, even in the short term and at small scale (about 1 t/h).
However, the present low energy prices have tempered the enthusiasm for adopting new technologies which often still need to confirm their long term reliability.
There is a clear need for long-term demonstration operations under the most favourable circumstances in order to prove the reliability and verify the economic viability anticipated by calculation.
Mr P. GIRARD gave a lecture concerning the environmental problems related to the thermal conversion of wood.
Wood is basically a low pollutant fuel and wood conversion has positive effects on the environment : no SO emissions, CO remaining in principle in a closed cycle.
However, a substantial increase in wood consumption for thermal conversion (f.i. 8 Mio toe of wood was consumed in France in 1986, equivalent to 4% of the country's energy consumption) could lead to a non-negligible impact on the environment.
Small scale plants for carbonization and residential heating, whose operational control is not as good as industrial units, may lead to significant pollutant emissions and effluents: small size particulates which increase lung infections, hydrocarbons and pyroligneous oils of which some components are carcinogenic, non-condensable vapours such as CO and NOx.
It is important in the case of the small scale plants to train and inform the users in a better way, while at the same time improving the thermal conversion control and efficiency.
Dr C. NELS presented the evolution of refuse management in West Germany.
In the early seventies, the initial trend was drastically to reduce the number of landfill sites from 5 000 to less than 500. Then came the oil crisis and the subsequent search for alternative energy feedstocks. Refuse pyrolysis was enthusiastically embraced as an alternative technology to municipal waste incineration, whose disadvantages were known from long term operations. It was hoped that refuse pyrolysis would be a simple technology

388
for smaller throughputs at low costs and with low environmental impacts. Important RSD funds were allocated for that purpose and some pyrolysis plants were put into operation. It appeared however that urban waste pyrolysis showed specific disadvantages and still required improvement in order to be competitive.
Anyway, all the technologies are to be considered with their respective advantages and disadvantages on a case by case basis, without leaning too much towards one single technology in particular.

»ORKSHOPS


391
WORKSHOP 1 - PRETREATMENT AND CHARACTERIZATION
Chairman: J. BARTON Rapporteur: J.PISKORZ
Given the limited time available it was agreed that the scope of the discussions should be directed to identifying aspects of pretreatment and characterization where further RSD or coordination activity was required. Based on the conference presentations and experiences of the group members, it was clear that pretreatment requirements for differing feedstocks varied widely and there would be little benefit in detailing the design performance and cost criteria associated with any particular system or equipments. However, differentiation between waste feedstocks and biomass was considered to be an important factor in terms of the approach adopted and scope or degree of pretreatment required.
Generally waste feedstocks attracted a disposal credit, they required and could support (using the disposal credits) a higher degree of feed preparation. Treatment option would be influenced not only by the requirements of the thermal treatment step but also by disposal or recovery option for the residues from pretreatment. In addition, the distribution and fate of toxic or hazardous components in the waste would play a more important role in determining pretreatment requirements.
For biomass, whether specifically 'farmed' or the residues from cropping activities, alternative disposal costs tended to be low and hence the collection and pretreatment costs had to be fully justified by product revenues from the thermal treatment stage. However, biomass was usually less complex in terms of physical and chemical characteristics and thus simpler, less costly feed preparation methods could usually be developed.
In terms of specific processes used in pretreatment, a few general comments were made on the following :
1. Sizing Although good experiences were reported with rotary (trommel) screens, flat bed and disc screens for sizing various feedstocks, the machine selected and screendeck design had to match the feedstock characteristics and process requirements. Trommel and disc screens tended to be more appropriate designs for 'difficult' materials such as refuse.
2. Shredding Although effective size reduction equipments were available for most feedstocks, most group members felt this was an area where improved design could lead to reduced operating costs. For wood, chippers were considered particularly effective in providing suitable feedstocks for pyrolysis/gasification reactors.
3. Drying Whilst problems have been experienced with dust, explosions and poor drying efficiencies, again the group reported that suitable designs were available. Cascade dryers, conveyor 'fluid bed' dryers, flash and pneumatic dryers, had all proved effective in different applications . Attention was drawn to the effect of drying on the

392
physico-chemical proparties of the feedstock. At relatively low temperature (+110°C) changes in crystallinity of cellulose, lignin degradation and volatile losses would affect yield and composition of the products from thermal treatment.
4. Densification The group reported that a range of different designs is available and most have been shown to work despite initial problems. As with shredding equipment the group considered design improvements were achievable for the densification process and it was reported that few machines achieved the reliability and throughputs originally anticipated when processing waste feedstocks. Generally all systems required feed moisture content levels below 15%, preferably below 10%, to achieve a stable, dense product.
It was concluded that whilst many mistakes had been made in selecting pretreatment equipments, there were enough good examples available and reported of the above processes to avoid major problems in the future provided designers and manufacturers took on board past experience. For novel feedstocks prior testing would always be recommended and this should be closely linked with an assessment of the response of the downstream thermal treatment stage.
Whilst the selection of equipments was inevitably feedstock and process specific, a common factor for all systems was the need for effective handling, feeding and storage systems. A catalogue of problems was reported by group members for this aspect with many processes failing due to an inability to maintain and control material flow.
Three main factors were considered to require further study.
1. Development of standard test protocols for assessing the flow and handling characteristics of these feedstocks. Conventional materials handling test methods such as those used in the minerals and chemical industries for hopper design, conveying systems etc. rarely provided satisfactory information for the design and scale-up of equipment for biomass and waste feedstocks. 2. Development of standard test protocols to assess biodegradation of feedstock in storage and its effect on feed composition, handling characteristics and health and safety aspects (e.g. fire, airborne emissions). 3. Development of automatic control systems for feed handling and storage. This was considered a particular problem for the developed nations where labour costs were high. As well as improved system design, development of robust sensing equipment to assess levels, bulk density, volumetric flow etc. were needed.
As a general point regarding the problems experienced in handling these materials, the group agreed that knowledge of the problems and potential solutions generally resided with plant operators rather than equipment suppliers. This knowledge should be gathered together, reviewed and more widely disseminated. Where similar feed materials can be identified, the study should include experience from established industries such as food processing, agriculture and forestry.
In terms of feedstock characterization for thermal treatment the group considered that thermo-chemical characteristics were generally given attention and well reported by researchers in pyrolysis and gasification. However, the physical specification of the feed was frequently overlooked

393
when reporting test work and experience. The size, shape, particle and bulk densities and permeability of feedstocks used affect the thermal process and such information was needed for developing suitable pretreatment options. This was an area where an expert group could develop a checklist of specification requirements, how they should be measured and how they should be reported.
On specific techniques, mention was made that when assessing slagging characteristics, the conventional determination of the deformation and flow temperatures of ash could give misleading results. Initial preparation of the test sample from the feed material using low temperature, plasma techniques, as opposed to using a conventional ashing furnace or ash from the process, could avoid physico-chemical reactions which prevent identification of the presence of low melting temperature components.
Based on the discussions, the priority areas for further study which could be supported by the EC in future programmes were as follows :
1. A consultancy study of full scale handling and storage systems for biomass and waste feedstocks. The study should be directed to obtaining information primarily from the experiences at operating and 'failed' plants rather than equipment suppliers. 2. Development of test protocols to assess the handling and storage properties of biomass and waste feedstocks. 3. Development of automatic storage and feed systems and associated sensing instruments for biomass and waste feedstocks. 4. Development of a common approach to the measurement and reporting of the characteristics of feedstocks for thermal treatment processes, particularly physical properties.

394
WORKSHOP 2 - PYROLYSIS
A V Bridgwater C Roy Energy Research Group Université Laval Chemical Engineering Department Département de génie chimique Aston University Ste-Foy Aston Triangle Quebec Birmingham B4 7ET G1Κ 7P4 UK Canada
INTRODUCTION The chairman opened the session by showing a series of transparencies
which summarised the main characteristics of the pyrolysis technologies available. The advanced pyrolysis technologies are: fast pyrolysis, flash pyrolysis, ultra pyrolysis, vacuum pyrolysis and hydropyrolysis. These were compared with the more conventional carbonisation technologies. As opposed to conventional pyrolysis where the main objective is to produce charcoal, the advanced pyrolysis route leads to a high yield of organic liquids in the first instance.
The general characteristics of pyrolysis oils were described together with the properties of the solid product (charcoal). The oil can be used as a fuel and also contains hundreds of valuable chemicals which can potentially be recovered. There is also a consensus on how slurries of char-oil and char-water can be prepared for power generation. The chairman concluded his brief introduction by showing the different prospective combinations of primary and secondary products from biomass pyrolysis.
SHOULD PYROLYSIS OF MSW BE CONSIDERED? This very simple question kept the workshop participants involved in
animated discussion for a substantial amount of time at the beginning of the workshop. On the one hand there is a growing interest for a better quality of life and a safer environment. On the other hand, there is also a growing concern about the traditional methods of Municipal Solid Waste disposal, i.e. incineration including ash disposal, cost of gaseous emission control etc, and the landfill approach with problems of leachates, land availability, etc. Can pyrolysis significantly contribute to the recycling of MSW? It is reported that the Japanese have already achieved large scale pyrolysis of MSW to gases for power generation. There is no commercial experience at this stage, however, in the world for the conversion of MSW to liquid fuels which are easier to burn than solid fuels in terms of control of emissions.
One problem which was mentioned is the inconsistency of the inlet materials which will also reflect on the varying quality of the char and the pyrolysis oil products. The char is loaded with metal and other inorganic substances. What can be done with it? Should it be burned, gasified or simply dispose of by orthodox methods? Sorting out the inorganic material as a pretreatment step prior to pyrolysis might be a more advisable way to go, although not necessarily economical at the present time. Another problem is what to do with the halogenated hydrocarbon by-products which will possibly find their way into the liquid fuels?
An opinion was expressed that most effort should be spent in primarily mastering feedstocks such as biomass, instead of treating difficult feedstocks such

395
as MSW, in order to give pyrolysis as many chances as possible to emerge as a leading technology. Nevertheless, there appeared to be a consensus that the scientific community should immediately afford time and effort at working on MSW because there is an urgent need for better recycling technologies to get rid of solid wastes.
One additional remark that an attendee expressed is that something should also be done with the enormous surplus of straw which is available in some European countries.
IMPORTANT R&D TOPICS Some of the problems which ought to be addressed by the scientific
community for the further development of pyrolysis technology include the following:
improve heat transfer during pyrolysis, assess the quality of the pyrolysis oil in terms of corrosive properties, chemical stability, mutagenicity and toxicity, and its fuel properties, especially if it is derived from MSW, mention was made about the upgrading of oils. Apart from the deoxygenation of these oils in gasoline, one further possibility is the recovery of valuable fine chemicals such as food aromas and perfumes from these oils, using new separation methods that are more efficient than the simple distillation method which is commercially used elsewhere.
CONCLUSIONS There was a consensus about the fact that finding a market for the pyrolysis
products is vital for the feasibility of the process. However, getting rid of wastes is another goal which is not always compatible with this primary objective of finding a market for the products. As a consequence, it is imperative that the governments contribute to the development of recycling technologies together with the private sector. In fact, there is a growing interest nowadays from large firms for pyrolysis, due to the positive image such an involvement can stir in the public.
Pyrolysis has many applications, both in the lage variety of feedstocks that can be treated and in terms of the many types of products it is possible to obtain during the different processes in development. This development requires time, financial assistance and collaboration between all participants.
This International Congress and the Workshop are examples of collaboration which should be mainyained on a regular basis, perhaps in North America and in Europe.

396
WORKSHOP 3 - GASIFICATION TECHNOLOGY AND ECONOMICS
Y. SOLANTAUSTA Laboratory of Fuel Processing Technology
Technical Research Centre of Finland
A.A.C.M. BEENACKERS Dept. of Chemical Engineering
University of Groningen, Netherlands
INTRODUCTION
A total of about 60 persons participated in the workshop on gasification. The aim of the workshop was to emphasise the aspects of research that should be addressed in future work. An effort was made to define the technologies that should be implemented. The economics of gasification systems were reviewed.
RESEARCH
The following areas of gasification research should be addressed in future CEC biomass programs.
1. Catalytic Gasification
Both gasification rate and product gas composition can be affected by adding catalyst in the gasifier. Gas impurities can be reduced by introducing a second catalytic reactor after the gasifier. Both approaches should be investigated further. Initial results obtained at the University of Zaragossa are encouraging.
2. Gas Cleaning for Turbine Applications
One of the most promising areas, where gasification can be utilized, is power production by combined cycles. However, gas turbines are very sensitive to impurities in the fuel gas (particles, alkali metal vapours). Future gasification combined cycles should be equipped with hot gas cleaning devices to realize the potentially very high efficiency of power production. Research in this area is therefore of great importance.
3. Environmental Effects of Gasification
All the environmental aspects of gasification should be studied. Possible emissions to atmosphere, the nature of the ash, and the quality of possible washwater should be measured, and the effects of these should be evaluated. Gasification combined cycles for coal are assessed as being environmentally superior to conventional power plants. It should be studied whether this is also the case with biomass systems.

397
4. Gasification of Municipal Solid Waste (MSW)
Some participants wanted to promote also research on gasification of MSW, whereas some were reluctant to do this because of the preceding unfavourable experiences. Gasification offers a potentially improved prospect for removal of impurities from the fuel gas compared to mass burning. It is particularly noteworthy to monitor the experience in MSW gasification to be gained from the Sarbrücken plant.
LESS DEVELOPED COUNTRIES (LDC)
Gasification has been applied in numerous cases in LDCs for supplying fuel for engines pumping water etc. Most of the operations have been plagued with considerable technical and financial difficulties. More research is needed to bring about :
reduction of investment cost, improved reliability of the system, improved user friendliness, and gasification of difficult feedstocks.
IMPLEMENTATION
Both low and medium joule gasification should be implemented.
1. Low joule gasification should be implemented in the production of heat, and in power production 1 to 5 MW scale.
2. Medium joule gasification should be implemented in the production of power in combined cycles, in which case the gasification has to be pressurized. It would be of interest if this application could be explored in the Clamecy plant. An additional promising technology is steam gasification in double fluid bed, which enables the production of medium joule gas without purified oxygen.
ECONOMICS
Gasification of biomass has been applied only in a relatively small number of cases in Europe. This is mainly due to the low price of oil and natural gas, which are the main competitors of fuel gas.
Below are presented some economic indicators for different gasification methods. Numbers are only indicative for a superficial comparison, and they are based on Finnish conditions as of 1986. It should be noted that the oil prices were considerably higher than now.
POWER PRODUCTION
Atmospheric gasification, conventional power cycle - competitive in the power range 20-50 MW
Pressurized air gasification, combined cycle - competitive above 150 MW
e Pressurized oxygen gasification, combined cycle
- competitive above 300 MW

398
HEAT PRODUCTION
Fixed bed gasification - competitive above 3 MW
Fluid bed gasification - competitive above 15 MW
CONCLUSIONS
The largest workshop of the conference wishes to make a call not to neglect gasification in either research or implementation. The opportunities of the technology are signficant while considerable expertise has been developed already during the past years.
ACKNOWLEDGEMENTS
The chairman wishes to thank all who participated to the workshop. The input of E. Kurkela and M.S. Mendis is especially acknowledged.

399
WORKSHOP 4 - PYROLYSIS AND UTILIZATION
E. Churin
Unité de Chimie et Catalyse des Matériaux Divisés Place Croix du Sud, 1
134 Louvain-la-Neuve, Belgium
The workshop was organised to discuss the possibilities for the upgrading of biomass-derived oils to transportation fuels or high-value chemicals and other potential uses other than just as a heavy fuel.
A typical characteristic of biomass-derived oils is their very high oxygen content which supposes that they can only be used replace heavy fuel oils.
To obtain high quality fuels or chemicals the oils must be upgradfed in order to reduce the oxygen content, increase volatility and reduce molecular weight.
This transformation can only be obtained in the presence of a catalyst. At present, two major catalytic processes are being investigated to this end. Hydroprocessing of biomass-derived oils is based on the technology applied in the petroleum refining industry. Typical reaction conditions are temperatures of between 250°C and 400°C and pressures of between 7.0 MPa and 20.0 MPa (pressures as high as 35 MP are being tested). Low space velocities are necessary and the best results are obtained between 0.1 and 0.3 volumes of oil per hour per volume of catalyst. During this treatment, hydrodeoxygenation takes place through the elimination of the oxygen constituting the molecules in the form of water, the high hydrogen pressure allows for a hydrocracking of the heavy molecules but, depending on the reaction conditions, side reactions lead to the formation of more or less substantial amounts of gaseous hydrocarbons. High pressure liquefaction oils constitute the best feed for the hydroprocessing because of the lower oxygen contents but pyrolysis oils can be processed provided that a pretreatment is effected at lower temperatures as demonstrated by Elliot at PNL.

400
The catalysts used in this process are CoMo and NiMo supported on alumina, the liftime of which needs to be established. It was stressed that it could be very interesting to conduct a study that would lead to the understanding of the degradation of the oils or hydrotreated oils containing some phenols which could act as antioxydants.
The low pressure upgrading process (almost atmospheric pressure) is based on the ZSM-5 zeolite catalyst. This zeolite exhibited very high yields to gasoline compounds when oxygenated compounds, mainly methanol, were transformed. Pyrolysis vapours from a votex reactor and pyrolysis oils were reacted over this catalyst but pyrolyis vapours were preferred because the pores of the zeolite are narrow and the big molecules produced during the condensation of the vapours would be unable to enter the pores.
It was estimated that about 75% of the primary pyrolytic vapours could enter the pores (as studied by molecular beam mass spectrometry at SERI) which means that the time lapse between pyrolysis vapour production and zeolite contact should be very short. Phenolic compounds exhibit a low reactivity on this catalyst and contribute to coke formation. For this process, research should be oriented towards the modified zeolites in order to diminish coke formation and increase the reactivity of some compounds.
Both processes produce gasoline boiling range compounds constituted of alkyl-substituted benzenes but BTX contents in gasoline is going to be limited by a new legislation because of their toxicity. This will constitute a serious drawback for the commercialisation of the upgraded products as transportation fuel.
A method of upgrading the phenolic fraction of the biomass-derived oi without deoxygenating is the production of aromatic ethers. They can be mixed up to 15% in the gasoline thus improving the octane number.
Concerning the use of the pyrolysis oils without any chemical transformations, the only utilization discussed was the replacement of pure phenol in resin production. The phenolic and neutral fraction extracted after elimination of acidic compounds could replace half of the puie phenol used in phenol-formaldehyde resin. This research is sponsored by the industry and industrial implementation is well advanced.

POSTERS PRESENTED


SECTION 1
PRE-TREATMENT, PRODUCTS AND OTHER ASPECTS


405
FULL-SCALE DEMONSTRATION PROJECTS OF THF, EUROPEAN COMMUNITY IN THE FIELD OF PYROLYSIS, GASIFICATION AND CARBONISATION OF
BIOMASS AND WASTE
By R.Fabry and G.L.Ferrerò Directorate General for Energy
Commision of the European Communities, Brussels and K. Maniatis
Vrije Universiteit Brussel
ABSTRACT
The first part of this article is concerned with a statistical analysis of the proposals for demonstration projects which have been submitted and selected (or not) since 1983 in the framework of the annual calls for offers for the Energy Demonstration Programme of the European Communities in the field of pyrolysis, gasification and carbonization of biomass and waste.
The evolution with time of the number of proposals in function of the technology, the country of origin and the type of waste used is discussed.
The second part reviews the selected projects (terminated, in progress or cancelled) and evaluates the results obtained until now, the problems encountered and the reasons for the cancellation of some of the projects.
1 ■ STATISTICAL ANALYSIS QE THE DEMONSTRATION PROJECT PROPOSALS SUBMITTED AND SELECTED
The Energy Demonstration Programme launched in 1978 by the Directorate General for Energy of the Commission of the European Communities provides financial support for demonstration projects of an innovatory nature at industrial scale in respect to energy saving, renewable energy sources and substitutes for hydrocarbons.
In the period 1978 to 1988, 1631 demonstration projects have been selected from the 5176 proposals submitted; resulting in a total financial support of 841 million ECU, which makes this programme the largest of its type in the whole world. At the beginning of the programme the projects concerning "Biomass and Energy from Waste" were included in the sectors "Energy Savings" and "Solar Energy" of the

406
Programme. However since 1983 due to its increasing importance the sector "Biomass and Energy from waste" became a completely independent one. In this sector 191 demonstration projects have been selected since the initiation of the programme for a total financial support of 87.4 million ECU.
The sector "Biomass and Energy from waste" is divided into 11 subsectors with 1 subsector devoted to pyrolysis, gasification and carbonisation of biomass and waste. Since 1983, 140 proposals for demonstration projects from the annual calls for offers of the Commission have been submitted in the sector "Pyrolysis gasification and carbonization".
Figure 1 presents the evolution in function of time of the number of the proposals submitted and selected since 1983, when the subsector Biomass and Energy form waste was implemented. A distinction has been made between the proposals on carbonisation where the emphasis is on the production of charcoal (or possibly on the production of roasted wood or activated carbon) on one hand, and on the other hand the proposals on pyrolysis and gasification where the aim is the production of pyrolytic oils and/or the production of a fuel gas. It is clear that the number of proposals on pyrolysis/ gasification has been steadily declining until 198 8 when there was however notably a sudden increase in the number of the proposals concerning municipal refuse (4 prop.) and especially various industrial wastes (6 prop.) . It should also be noted that in 1987 the thermal conversion of wastes in general was excluded from the call for offers and the 5 proposals shown in Figure 1 were submitted by error.
It can be noticed that the selection rate of pyrolysis/gasification proposals has been very severe : 15% selected proposals on average (with a zero selection in 1986 and, being normal, in 1987) which is 5% less than the average approval rate for the complete sector "Biomass and Energy from Waste". This very severe selection on pyrolysis/gasification as well as the decline in the number of proposals submitted between 1984 and 1987 is related to the technico-economic difficulties that this branch still has to overcome.
The innovative aspects of the submitted proposals were often related either to the utilization of the fluidized bed technology and/or to the nature of the wastes to be treated, in particular for indutrial wastes.
In the field of carbonization the number of submitted proposals has been inferior but relatively constant. The selection rate of the proposals in this field corresponds to the average selection rate of the entire sector being 20%. The proposals on carbonization which were not approved either had no innovative character or did not offer satisfactory perspectives for economical viability. Generally seen, the innovative aspects of the selected proposals fell in one of

407
the following categories : a ) u t i l i z i n g new types and wider range of dimensions of ag r i cu l tu ra l and forestry res idues; b)improving the thermal e f f i c iency and p r o t e c t i o n of the environment by recycl ing and/or seperate use of the pyrolys is gases; c)new products, such as roasted wood.
The classification of the proposals according to the type of waste utilized is shown in Figure 2. The category of wood waste comprises logs, chips, waste from the wood industry and forestry residues. In the group of industrial waste, wastes of very different origin can be found: textiles, plastics, used oil, tyres, rubber, shredder and various PME wastes . In this group the few proposals (5) concerning commercial wastes, such as carton and packaging waste is also included. As expected, the group of wood waste is the most important in particular for carbonization since it is essential for the production of charcoal and roasted wood.
However, the other types of wastes and more particularly industrial wastes are well represented in the pyrolysis/ gasification proposals. For the industrial wastes, the selection of pyrolysis/gasification proposals has been a little bit more favorable than the average. This is partly often linked either to the fact that materials of high added value are recuperated from specific wastes (such as carbon black from tyres) or to the fact that difficult to be treated wastes (such as wastes with high concentration of plastics, textile waste, shredder waste) can be eliminated with a more accurate process control than by combustion, with a simultaneous fuel gas production. The product gas can either be used in an engine to produce electricity or in a boiler/furnace with little modifications to produce steam. On the other hand, from the 21 submitted proposals on municipal waste only one project has been selected. This reflects the severe techno-economic problems associated with this field.
The classification per country of the submitted proposals is represented in Figure 3. It is clear that 3/4 of the proposals are originating form 3 countries: France, Italy and Federal Republic of Germany. Each of them submitted about 25 % of the proposals. However, for Spain and Portugal, the proposals could only be submitted as from 1986. It should also be mentioned that in certain cases a proposal, not selected one year, has been resubmitted the following years, (such as for straw gasification or municipal waste pyrolysis) . Sometimes also the same basic technology has been used in proposals concerning different wastes and context, (eg. fluidized bed pyrolysis of wood waste). However, the impact of such proposals which can not really be considered as totally distinct, remains-limited.

408
2. THE SELECTED PROJECTS Before 1983, only 3 projects had been selected; 1 for
each category of : wood, agricultural and industrial wastes. Nevertheless between 1983 and 1988, 21 projects were selected in the subsector pyrolysis, gasification & carbonisation. From the 24 selected proposals, 6 have been completed (the average duration for the realization of a project starting from the selection of the proposal till the complete execution of the programme is about 4 years). 6 projects were cancelled before or shortly after the signature of the contract between the beneficiary and the European Commission and 12 are in progress. The reasons for cancellation of the projects are essentially economic and/or financial: bankrupties, restructuring (3 projects), reduced economic rentability due to increased value of waste (2 projects), financial plus technical problems (1 project). The number of selected projects per country is more or less proportional to the number of the submitted proposals illustrated in Figure 3.
3. RESULTS AND TRENDS
From the 6 completed projects only one can be considered successful. This concerns the production of roasted wood (Project BM/333/84) which can be used as a reducive agent in electro-metallurgy or as fuel for barbecues. The technical reliability of the process has been demonstrated at industrial scale. However, serious technical problems had to be overcome during the optimisation phase with increased investment costs, resulting in lack of rentability of the demonstration plant. This rentability would be secured for further replications.
The other completed projects resulted in different degrees of failures. This was the case for pyrolysis of municipal waste, pyrolysis of used tyres, gasification of flax residue, gasification of bark and carbonisation of different forest and plant residues. The positive results obtained until now in the subsector pyrolysis, gasification and carbonisation of the Energy Demonstration Programme have not been very satisfactory notwithstanding the severity of the selection procedure. This illustrates very well the big difficulties encountered by new technologies or new applications of known technologies in this sub-sector in order to succeed in a breakthrough on the level of economic rentability, while sustaining the technical reliability. Nevertheless, the unsuccessful projects have not been useless since they put emphasis on the technical and economic problems which still have to be overcome. Other projects are still in progress or still have to be implemented which could improve the overall performance. More specifically, in case of success, the demonstration of forest residues pyrolysis in

409
fluidized bed could play an important role in the optimum utilization of the forest resources, especially in the Southern countries of the Community. For more information please contact :
Commission of the European Communities, Directorate General of Energy, 200 rue de la Loi B-1049 Bruxelles
W-wood A-agr¡cultural U-urban 1-lndustriaJ
Figure 1 : Annual distribution of proposals
W A U I
Typ· of watt·
Figure 2: Distribution by type of waste
submitted
selected

410
2.86% 1.43%
22.86%
24.29%
3.57%
5.00%
1.43%
2.86% 2.14%
25.71%
7.86%
Ξ FR 32 ^ IR 2 □H IT 36 ^ NL 11 Π PO 3 ■ UK 4
BE 7 DK 5 FRG 34 GR 4 SP 2
Figure 3: Distribution of submitted proposals by country

411
THE USE OF WOOD AS FUEL IN MALAYSIA
W Κ HOI1 and A V BRIDGWATER
Forest Research Institute of Malaysia Department of Chemical Engineering Kepong Aston University
Selangor Aston Triangle 52100 Kuala Lumpur Birmingham B4 7ET
Malaysia UK
Summary
The efficient use of wood in the solid, liquid and gaseous forms can contribute towards the fuller utilisation of the nation's natural resources. In Malaysia, only wood in its solid form is used as fuel. Fuelwood is still an important energy source for the rural populace and is consumed in substantial quantity for the production of rubber smoked sheets, tobacco curing, brick making, drying of timber as well as for charcoal production. The paper highlights the principal wood-based energy industries in the country where wood is still the predominant source of fuel.
INTRODUCTION Since time immemorial, man has learnt to use fuel for warming up his dwelling and for
cooking his food. In the early days, wood in the solid form provided all the energy needed for both domestic and industrial uses. In the course of development the use of solid wood as a source of energy has undergone considerable change. At present, wood is also used for the production of gaseous and solid fuel.
Perhaps the role of fuelwood as an energy source is closely linked with the process of industrialisation. As society progresses, man becomes less dependent on wood as a source of energy because modern industrial installations require the use of large concentration of fuels which can be stored and easily transported. Wood fuels did not meet these requirements and they were gradually replaced by oil. Furthermore the progress in the development of wood as a source of timber have converted these once wood fuels into an important source of value-added products making them too valuable to be used as fuel. Only wood waste and residues that could not be utilised by current technology are currently used as fuel.
In Malaysia, wood only accounts for about 10% of the total primary commercial energy of the country (Anon, 1986). The total amount of fuelwood in the country in 1986 is estimated to be in the region of 500 000 tonnes. Although the contribution of fuelwood to the overall energy requirement of the country is not very significant, wood is an important and predominant source of energy in many of the rural industries of the country. These industries include: a) Smoking of rubber sheets b) Curing of tobacco leaves c) Brick making d) Drying of timber e) Wood charcoal production
The above industries alone account for almost 90% of the total wood energy consumption of the country. Perhaps one of the main reasons for the popularity of wood as a source of fuel in the rural industries is the abundance in the rural areas and a relatively cheaper fuel when compared with other sources of fuel available in the country. The relative cost of fuel in the country is shown in Table 1 (Hoi, 1987).
AVADLABDLITY OF WOOD FUEL The main supply of sources of fuelwood for the rural industries in the country are
Rubberwood (Hevea brasiliensis) obtained from replanting schemes and wood residues and logging wastes in the form of off-cuts, slabs, trimmings, edgings, tops, loops, branches and Ί Currently FRIM Research fellow at Aston University

412
defective logs from the wood based industries and logging operations.
Availability of rubberwood The economic lifetime of a rubber tree is about 25-30 years. At the end of its economic
life, a typical rubber tree has a height of about 18 m, a diameter of about 70 cm at ground level and an average dry weight of about 3.6 tonnes with composition summarised in Table 2.
Table 1
Enerpv source
Diesel Charcoal
Comparative Cost of Energy in Rural Industries
Calorific value Fuel cost fGJ/tei
44.3 30.5
Wood, wet 12.5 Coal 29.3
ŒCU/GJ1
3.55 1.74 0.70 1.74
Table 2 Composit Trees at Economic
Components
Leaves Leaf-stalks Twigs Small branches Side branches Main branches Main stem Roots TOTAL
ion the Life
of Rubber end of their
»
Drv weipht kg %.
67 1.9 9 0.3
105 2.9 163 4.5 410 11.4
1258 35.1 1032 28.7 547 15.2
3591 100.0
The area of planted rubber plantations in Malaysia is about 1.67 million ha. About 3% of the planted area is replanted every year (Anon, 1987) yielding about 3.6 million tonne of wood. Out of this amount of wood available, it is estimated that only 10% is used as timber and 15% is consumed by the rubber smoking, brick, tobacco and the charcoal industries. The remaining 75% of the rubberwood available is left in the replanting area to be burned later. Hence the amount of rubberwood available for fuel or other further down-stream processing is very substantial (Ambrin et al, 1987).
Availability of wood residues and logging wastes Within the forestry sector, the timber resource of the country generates the necessary
revenue for development while the land provided opportunities for gainful employment and the creation of new wealth. When large tracks of forest have to be cleared within a relatively short period, huge amounts of wood wastes are generated. Studies conducted by the FAO/UNDP (1973) indicated that only an estimated 22% of the gross volume of trees above 61 cm diameter, or less than half of the potential net volume of marketable trees, is removed. A recent study on forest residues in Peninsular Malaysia estimated that the percentage of removal of logs can range from 60% to 65%. From this study, it can be deduced that at least 35% of wood in the form of stumps, roots, branches (of 6" and below) remain in the forest with a composition of 55% stumps and roots, and 45% branches. This will differ from species to species, the size, age and condition of the trees. This method of harvesting yields a wood potential of about 1.2 million t/y with an estimated energy value of about 18 trillion Joules and a market value of about MR$ 50 million annually.
In the saw milling industry, further residues are generated in the form of sawdust, slabs and edgings. Recovery of green timber in the sawmill generally can vary from 35 to 45% depending on the size, quality and sawmilling practice. The approximate breakdown of the residues produced is as follows:
Bark 10% Sawdust 20% Slabs and edgings 70%
The estimated volume of the sawmill waste is estimated to be in the region of 1-1.5 million tonnes. In general the amount of wastes available for energy production is summarised in Table 3.

413
Table 3 Estimated wood-waste available for energy in the country
Activity Type of waste Amount of waste (million t/v)
Rubber replanting scheme Rubberwood billets 2.7 Logging activities Stumps, roots, branches 1.2 Sawmill recovery Bark, sawdust, slabs, edgings 1-1.5
THE USE OF WOOD AS FUEL IN RURAL INDUSTRIES As mentioned earlier, the use of wood as a source of fuel in the rural industries in
Malaysia has been standard practice since the early days of the industries. The devices by which fuelwood is actually burnt are generally based on traditional designs and the use of locally available material. To keep investment low, designs are kept as simple as possible. The main rural industries using wood as fuel are discussed below.
Smoking of rubber sheets Natural rubber is produced from latex tapped from the rubber tree. To produce sheet
rubber, the latex has to be dried by exposing them to flue gas generated from the combustion of wood in the smokehouse. The smokehouse is capable of drying about 25 tonne of rubber sheets per batch operation. The walls of the smokehouse are built of bricks and asbestos and are about 6 m high. The furnace is a simple brick-lined stove located at ground level. Rubberwood is burnt to produce the flue gas for drying the sheets (Hoi and Low, 1986). The hot flue gas will then rise passing through the wet rubber sheets hung horizontally by rattan rods. During the process, moisture is removed from the sheets. The wet gas then leaves the smokehouse through the ventilation vents situated between the roof and the walls. The amount of air passing through the furnace is regulated by simple damper plates at the entrance of the furnace. The total time required for drying in a typical smokehouse can vary from 4-10 days depending on the age of the smokehouse (Enno and Hoi, 1987).
Based on the wood consumption and the rubber production, about 1.5 kg to 2 kg of rubberwood is needed to produce a kilogram of smoked sheets. The fuelwood cost represents about 5% of the value of the final product.
Tobacco curing Fresh tobacco leaves harvested directly from the field has a moisture content of about
70%. In order to produce good quality tobacco, the leaves have to be dried immediately in a tobacco curing barn. The walls of the bam are made of asbestos. The furnace is a simple burner in the form of a chamber located at the base of the bam. Rubberwood is fired through the furnace door when air from combustion is available. The hot flue gas passes through the bam through a series of distribution heat exchanger pipes that eventually lead to the chimney. Heat from the flue pipes is transferred to the air inside the bam which in turn dries the tobacco leaves inside the bam. The process of drying can take between 4 to 6 days. Measurements made by MARDI showed that about 8-10 kg of fuelwood is required to dry 1 kg of the final product (Daham, 1987).
Brick-making The most common method of firing bricks in Malaysia is by the updraft kiln method.
The kiln has no permanent top, but the outer wall, base and fire tunnels are permanently built with bricks. The walls on either side are buttressed and the comers are heavily constructed. Access into the kiln is through a doorway at the end walls. The doorway is filled temporarily with closely laid bricks (without mortar) during the kiln operation.
After the process of preparing the wet bricks, the green bricks are carefully loaded into the kiln. Small spaces are left in between the bricks in order to allow hot gases produced from the fire at the base to rise. Rubberwood is placed in the firing tunnel and is fired, gently at first to allow water to escape from the bricks until the kiln reaches a certain temperature. This is

414
indicated by a red glow at the top of the kiln at night. The sink of the bricks (after shrinkage) against the permanent side walls is an indication of the firing process. Generally the firing period is between 4 to 10 days. A typical amount of fuelwood required to manufacture 1 kg of brick is 0.3-0.4 kg (Hoi, 1987).
Heat for the sawmilling industries The basic use of wood as a fuel in the sawmilling industry for generating steam in
boilers has been a common practice in Malaysia. For most Malaysian wood species, the gross calorific value of oven-dried wood is about 18 MJ/kg. High lignin and resin contents in wood such as for some dense species will result in a higher calorific value. Moisture in the wood will also decrease the calorific value of the wood. In a typical case, air-dried wood with a moisture content of 10% has a gross calorific value of 16 MJ/kg. For freshly sawn wood this value is around 10 MJ/kg (Tan, 1985). Because of the lower heating value of green wood, preliminary drying is always preferable although not always practical. In a typical sawmill the energy requirement per unit of the product is summarised in Table 4.
Table 4 Fuelwood requirement per unit product
Product Equivalent wood bumt/m3 production
Sawmilling 160-340 kg Plywood 400-600 kg
Conversion into charcoal In certain parts of the country, the high cost of transporting wood over long distances
will diminish the relative low cost advantage of wood as fuel. In Malaysia, the economic distance between the source of wood and the demand centres is about 100 km (Hoi et al, 1986). However, the economic value of wood can readily be upgraded if it is converted into charcoal. Charcoal has about twice the calorific value of wood but a fifth of its volume. This enables it to be transported over longer distances for use. Furthermore, because it is a cleaner fuel compared to wood, it will command almost 10 times the price of wood in the major towns.
The production of charcoal in Malaysia has been practiced for centuries. The methods vary from the simple sawdust pit method to the relatively efficient beehive kiln method. The sawdust clamp method is a cottage type industry and is generally located in areas where labour cost is cheap (Hoi, 1983). Although it is simple and inexpensive to build it is a highly labour intensive industry. The quality of the charcoal obtained is not very good and it is sold as domestic charcoal. About 6.5 tonne of wood is required to yield 1 tonne of charcoal.
Bener quality charcoal can be obtained by the beehive kiln method (Hoi, 1986). This method is the most popular and is used to produce domestic mangrove charcoal and industrial charcoal. Beehive kilns are therefore located either in coastal areas with mangrove forest or in rubber growing areas. About 5 tonnes of green wood are required to produce about one tonne of charcoal by this method. The concept of producing charcoal by the transportable metal kiln was introduced into the country about 5 years ago. This method is suitable for small scale on-site production (Hoi and Putri, 1988). A number of sawmills and land development agencies are using these kilns to convert their wood residues into charcoal. About 4.5 tonnes of wood are required to produce 1 tonne of charcoal (Hoi et al, 1986). Currently about 60 000 tonne of charcoal is consumed annually mainly for industrial applications (such as for the reduction of steel, activated carbon and the chemical industries). Only about 20% of the charcoal produced in the country is used for domestic cooking. The amount of fuelwood required per unit production of the three industries is summarised in Table 5.
CONCLUSION The amount of wood wastes available is around 5.4 million t/y, so only a small
percentage is utilised. The rest is left in the field either to be disposed of by burning or by natural deterioration. Hence the amount of unexploited wood resource is very substantial. The total amount of wood wastes used as fuel in the country is summarised in Table 6.

415
Table 5 Fuelwood Requirements
Industry
Rubber smoking Tobacco curing Brick making Charcoal making
Table 6 Estimated Total Wood Consumption
Fuelwood required/unit production Industries
1.5 - 2.0 kg/kg of dry rubber sheet 8.0 - 10.0 kg/kg dry leaves 0.3 - 0.4 kg/kg brick 4.5 - 6.5 kg/kg charcoal
Million t/v
Rubber smoking Tobacco curing Brick making Charcoal production TOTAL
0.5 0.1 0.1 <L3_ 1.0
In the last decade, very little attention has been focussed on the potential of harnessing energy from the forest because of the availability of cheap fossil fuels in the country. However, as the fossil fuels in the country are very limited, it will only be a matter of time before the non renewable resources will be depleted. It has therefore become increasingly urgent for the government to look for alternative resources to satisfy her future energy needs (Salleh and Hoi, 1983). Recently, there has been renewed interest all over the country to reassess the possible contribution of wood to future energy requirements. While the ingredients are already there, means have to be taken by all concerned to exploit the potential fully by intensive research and development efforts. Only then will the long term objective of the government be fully realised.
REFERENCES
1. Anon (1986). Yearbook of statistics 1986. Department of Statistics, Government of Malaysia, Kuala Lumpur, Malaysia.
2. Hoi, W Κ and Baharuddin, Y (1987). Wood as an alternative source of energy. Paper presented at the Asian Physics Symposium, October 1987, Kuala Lumpur, Malaysia.
3. Anon (1987). Lapuran perangkaan tahunan tanaman semula 1987. RISDA, Kuala Lumpur, Malaysia.
4. Ambrin Buang and Mohd. Nazuri Hashim (1987). Current analysis of the rubberwood industry with special emphasis on marketing. Proceedings of the Second Rubberwood Seminar, December 1987, Kuala Lumpur, Malaysia.
5. FAO (1973). A market analysis of Malaysian wood products. Technical Report No.6, FO:DP/MAL72/009 Rome, Italy.
6. Hoi, W Κ and Low, C Κ (1986). The use of fuelwood in the smoking of rubbersheets. Report of consultancy on wood energy use in Malaysia. UNDP, 1986, Rome, Italy.
7. Enno Heijnermanns and Hoi, W Κ (1987). Preliminary report of energy use in tghe natural rubber industry of Malaysia. Forest Research Institute Malaysia, Kuala Lumpur, Malaysia.
8. Daham Mohd. Daud (1987). Pengawetan tembakau dengan menggunakan gas cecair petroleum. Paper presented at National Tobacco Conference, December 1987m, Kuala Lumpur, Malaysia.
9. Hoi, W Κ (1987). Fuelwood trees for rual industries. Paper presented at Confernece of Multi Purpose Tree Species, December 1987, Kuala Lumpur, Malaysia.
10. Tan, Y E (1985). Calorific value of some Malaysian Timber. The Malaysian Forester, 28: 148-153.
11 Hoi, W K, Ali S and Megathewar (1986). Small-scale charcoal from small holders in Malaysia. Paper presented at Rubber Planters' Conference, October 1986. Ipoh, Malaysia.
12 Hoi, W Κ (1983). Country status on charcoal production and technology, Paper presented at experts consultation on charcoal technology, FAO, Bangkok, Thailand.
13 Hoi, W Κ (1986) Charcoal production from beehive kilns. Paper presented at International Union of Forestry Research Organisations Conference, Yugoslavia, 1986.

416
14 Hoi, W Κ and Putri, F (1988). Wood eneigy activities in Malaysia. Paper presented at Regional Workshop on Biotechnology, February 1988, AIT, Bangkok, Thailand.
15 Salley, Μ Ν and Hoi, W Κ (1983). Research in biomass energy - Problems and Prospects. Paper presented at the consultation of wood energy programme in Asia and Pacific region. FAO, 1983, Rome, Italy.

417
SELECTED ASPECTS, EXPLANATIONS AND STATEMENTS IN ACCORDANCE AND ANALOGY TO THE BIT GRANULATION-TECHNOLOGY
J.M. DISS and F.W. HOCHHEIM Directorate General and GENERAL Management
of the incorporated company B.I.T. SA, GR. D. L., L-4807 Rodange, PO Box 9
Summary
Unlike the situation in primary energies, some of the significant advantages of secondary energies (e.g. fuel pellets made from biomass wood waste) are relatively unknown and consequently little utilized. Biomass, especially ligneous materials, is eminently exploitable and the heating capacity expressed in energy flow can be quite remarkable. With an energy conversion efficiency of almost 80% and a favourable cost regime, BIT fuel pellets are an actual and competitive form of secondary energy. It is, however, necessary to consider the supply structure of the biomass wood-waste ('green circuit') and to locate the plant close to forestry resources and woodworking industries. Therefore the biomass wood-waste industry will be increasingly directed to refuse management.
1. INTRODUCTION
The conversion technology for generating fuel pellets from biomass wood-waste, on which BIT granulation technology is based, has been in use for at least ten years mainly in France. It has been used continuously and successfully for this period. The technology has been used for the cost-effective and environmentally benign conversion of forestry and woodworking refuse into wood-waste secondary energy. New applications of this technology can be used to increase energy savings and to transform ligneous materials as well as wood waste to useful fuel.
Consequently the BIT pellet user will benefit from the following advantages :
low investment; good care of boilers ; warm-heaters and generators operating conditions and life are improved; easy operation; automatic feeding of fuel pellets is included; low cost of maintenance and service; high energy efficiency (82%) on heating capacity 4000 kcal/kg to 4200 kcalAg» leads to 80% of specially designed boiler, warm-heater and generator.
Several central granulation plants have been in uninterrupted operation since 1978 without any problems.
There are also many fuel pellet fired heating plants in uninterrupted operation without displaying major problems.

418
2. PRICE COMPARISON BETWEEN FUEL PELLETS AND PRIMARY ENERGIES
A price comparison between secondary and primary energies is quite difficult given the fluctuations in primary energy prices. Unlike primary energy prices, the price of secondary energy generated from wood-waste, wood chips and fuel pellets has not varied greatly during the period from September 1981 and January 1989. The cost of secondary energy has been gradually increasing but at a moderate rate.
The cost of all energies must now be related to the price of fuel oil which is currently relatively cheap. However, up to the first quarter of 1989, although fuel oil does enjoy some advantage, the price of fuel pellets is very competitive.
The study 'Raw Materials from Agriculture and Forestry for Briquettizising and Firing in Heating Plants' was supported by BMFT and performed by TU-Munich.
The conclusion of this study, published in the BINE report No.11, June 1988, states that to be competitive the price of fuel pellets generated from biomass would need to be at least half of the current price.
Therefore those making investment decisions on purely economic grounds would definitely opt for fuel oil under present market conditions. However, if one considers aspects other than pure economics there are several factors which could militate in favour of fuel pellets :
high operating safety without interruptions or breakdowns ; safety of feeding and extraction systems ; safety of residues; non-polluting environmental effects ; high quality and efficiency of conversion technology leads to energy savings; desirability of utilising technologies which generated fuel pellets from biomass derived from wood-waste and refuse.
These later points may become increasingly important with evolving government regulations.
3. INCREASING IMPORTANCE OF REFUSE MANAGEMENT IN SUPPLYING BIOMASS OF LIGNEOUS MATERIALS
An examination of the possibilities for ligneous biomass in various cities and communities has led BIT to conclude that the annual production capacity of generated fuel pellets is approximately 20 000 tonnes.
In our view it is necessary for communal and urban authorities to regulate the further processing and enhancement of biomass from wood-waste. This should be accomplished by collecting and combining wood-waste biomass from forest and waste resources.
This concept of collection and containment would be consistent with the establishment of 'green circuits' within cities and neighbouring communities.
Wood-waste biomass supplied from the 'green circuit' and transformed to fuel pellets represents a real alternative to the compost wood-waste conversion technology.
The importance of this technology is more evident where wood-waste is derived from refuse. Separating and enhancing the wood-waste biomass is preferable to incineration.
There is certainly a strong case for supporting fuel pellet secondary energies as a support for primary energies.

419
However, the economic situation (DM 100-150/m3 of biomass wood-waste in the Federal Republic of Germany) is currently overriding environmental and waste use arguments.
This technology will undoubtedly improve as efforts are concentrated on the limiting problems.
We hope that the presentation 'Selected Aspects, Explanations and Statements in Accordance with the BIT Granulation-Technology' will give encouragement to those proposing this type of technology because environmental and energy saving problems are common throughout the Community.

420
THERMOCHEMICAL DENSIFICATION OF BIOHASS -A KINETIC APPROACH TO PROCESS DEVELOPMENT
D.P. Koullas, N.S. Abatzoglou, and E.G. Koukios Department of Chemical Engineering
National Technical University of Athens, Greece
SUMMARY
Hot densification of conifer sawdust is investigated. The experimental conditions were 0.25-4 h, 100-400 bar and 80-215°C. The effect of temperature, briquetting pressure and time of compression on total process weight loss, volatiles loss, briquette density end briquette moisture content is examined. Moreover, the calorific value of the solid fuel produced is determined and the energy efficiency of the process is calculated. Process yields and product characteristics are mathematically expressed as a function of process input parameters. Under the experimental conditions examined the volumetric energy density of the product is four times than that of original biomass. Furthermore, it was found that the pressure applied during compaction has a major effect on the physical, chemical and energetic aspects of the process.
1. INTRODUCTION Hot densification belongs to the'group of technologies which utilize
biomess as energy source thermochemically (ie. direct burning, pyroly-sis, gasification) (1). The object of briquetting is to produce a solid bio-fuel with higher Volumetric Energy Density {VED), and better transport, handling and combustion properties (2), (3), (4).
During hot densification of lignocellulosic materials, complex competitive physicochemical phenomena are taking place (e.g. development of thermoplasticity, hydrolysis, production of anhydrosugars) (5),(6). These phenomena depend on the pressure, temperature, time, and, in several cases, on other parameters of densification.
Tne object of this work is the understanding of the mechanism of hot densification and the development of mathematical models linking the process yields with the various densification parameters.

421
2 . EXPERIMENTAL The raw materiel of thermochemical densification was conifer saw
dust. The sawdust was densified in a heated mould by a laboratory hydraulic press. The densification temperature, pressure and time varied between 80215°C, 100400 bar and 0.254 h, respectively. The bulk den
sity of both the raw material and the product (briquette) were determined geometrically. Moisture contents were found according to ASTM (D 1037). Total process weight loss, organic volatiles loss and weight loss after weight stabilization at ambient humidity were also determined (7). The volatiles loss were calculated from the weight decrese of the dry matter. The calorific value of the solid fuel produced was analyzed according to ASTM (D 271).
3. RESULTS AND DISCUSSION A part of our experimental data (7) is presented in Table I and II.
As it can be seen in Table I. significant volatiles loss is observed above 130"C. This means that there is a security limit, up to which there is no loss of organic combustible matter. The evaporated water can be partially reabsorbed, since loss after weight stabilization is 34.5* (Table I). Of course, the final briquette moisture after weight stabili
zation is smaller than that of the raw material (Table I), since compression reduces the porosity. This information is useful to one who wants to have high process yields. It should be noted that above 250°C the combustion of the briquette takes place.
Regarding briquetting time, one could claim that around 23 h the values of major densification parameters are stabilized (Table I). The latter remark is valid for all pressures examined. This means that at constant temperature and pressure after certain time there is an optimal utilization of the porosity (ie. the specific surface) for bonding. This is expected, since at prolonged times "secondary reactions" dominate; these reactions are slower than the "primary" ones (8).
Briquettes produced around 170°C and 200 bar have high density (Table I), ie. very good cohesion, if briquette final moisture content is considered as measure of compaction and, therefore, of ita resistance to mechanical stress (9),(10). These conditions can be considered as optimal, since at higher temperatures volatile loss increases. Moreover, higher pressure on the one hand results in increased energy demand and, on the other hand, does not significantly increase briquette density.
The briquette VED is four times higher than that of the raw materiel (Table II). From Table II it can also seen that gross process energy efficiency is rather high.The same is valid for the net energy efficiency (1st Law of Thermodynamics) (7). This encourages the application of this process. These remarks are valid for other raw materials too (e.g. olive kernel) (10).
Regarding mathematical modelling, equations (1) and (5) are derived from the experimental data (6):
yro 0.9620.0379*xt40.0180*XP+0.0911*xT+0.00215*xt**» 0.111*χ,»*χτ0.0687*χτ*χ* +0.0422*xat +0.0166*x
ap0.234*x=T +0.0177*xt*Xp*xT (1)
where,
yTO: * process gross total yield (100total loss, see Table I).

422
X tÄ
Xp·*3
V _ S
t
4
P100
300
T353
(2)
(3)
(4) 488353
yD (lexp(0.00834*t)) + (lexp(0.00002*P))+T°<>o:'s= (5)
where,
yD: % process dry yield
and
t: compression time, h Ρ: pressure, bar Τ: temperature, Κ
For the construction of the above models a nonlinear regression FORTRAN programme based on the LevenbergMarquardt algorithm was used. For all cases, the convergence conditions were satisfied if, on two suc
cessive iterations the residual sum of squares estimates had a relative difference equal to zero. As mentioned earlier the occuring physicochemical phenomena are too complicated. This explains why equations (1) and (5) are different. But, it should be noted that they are not as different as they seem to be, since the exponential term can be analyzed to a polynomial according to Taylor series. This means that equations (1) and (5) are more or less related. Mathematical expressions predicting water loss during densification or the stabilization moisture have not been found, most probably, due to the complexity of the phenomena; from equation (1) one can conclude that temperature linearly or at second power plays a very important role in densification. Interactions such as temperaturepressure or temperaturetime should not be neglected.

423
Table I: Process characteristics and properties of the briquette produced
Tempe
rature
"C — 80 80 80 80 80 130 130 130 130 130 130 170 170 170 170 170 170 215 215 215 215 215
Pressure
Atm
100 100 100 200 200 100 100 100 200 200 200 100 100 100 200 200 200 100 100 100 200 200
Time
h — 1 2 4 2 4 1 2 4 1 2 4 1 2 4 1 2 3 1 2 3 0.25 1
Density
g/cm3
0.29 0.92 0.90 0.89 1.07 1.06 0.83 0.83 0.84 1.11 1.12 1.18 0.98 0.96 1.06 1.19 1.21 — 1.22 1.24 1.24 1.27 —
L Total
%
1.1 2.8 4.0 2.4 2.9 8.0 7.5 7.6 6.8 6.6 6.9 10.5 10.7 10.7 10.4 10.4 9.7 17.1 24.2 24.8 24.4 26.7
o s Vola
tiles * — 0 0 0 0 0 0 0 0 0 0 0 0.6 0.9 0.8 0.6 0.7 7.3 14.3 15.0 14.3 17.0
s after weight stabi 1ization
* — 0.3 3.0 2.8 2.2 2.6 4.4 3.7 3.4 3.4 3.3 3.4 6.6 7.1 7.4 7.0 7.3 6.8 14.8 22.3 22.9 22.4 25.0
Moisture content
% 9.9 9.6 7.1 6.4 6.7 8.5 4.9 4.9 4.9 4.6 4.6 4.6 4.1 3.9 3.5 3.8 3.5 3.3 2.7 2.4 2.5 2.3 2.3
Furthermore, briquette density can be described by the following exponential equation:
ρ a*exp(b*P)
where. (6)
p: density, g/cms a 0.52 0.97 b 0.00060.0007 r2 0.60 0.99
depending on the briquetting conditions

424
Table II: Briquette Volumetric Energy Density (VED). Heat of Combustion (HC) and Net Energy Efficiency (NEE).
Temperature
"C — 130 130 170 170 170 170
Pressure
bar — 200 300 100 100 100 300
Time
h — 4 4 1 2 4 4
VED of Briquette
cal/cm5
1270 5390 5570 4550 4530 4860 5300
HC
cal/g
4430 4565 4565 4640 4720 4580 4570
Efficiency (a) %
103 103 105 107 103 103
NEE (b) %
— 93.5 94.6 91.6 92.9 90.2 —
(a) Efficiency» VED of briquette / VED of raw material. (b) NEE - useful energy / Energy Input (7)
4. KEb'EHENCES
(1) FAST Occasional Paper (1981). Energy from Biomass Technologies: State-of-the-Art Review. No.10.
(2) Reed. T. and Bryant, B. (1978). Densified Biomass: A New Form of Solid Fuel. Report. U.S. Solar Energy Research Institute. Golden. USA.
(3) Koukios, E.G., Mavrokoukoulakis, J.G. and Abatzoglou, N.S. (1982). The Densification of Biomass. Proc. Conf. on Soft Energy Sources 141-150. Thessaloniki. Greece.
(4) Koukios, E.G. and Mavrokoukoulakis, J.G. (1981). Some aspects of biomass densification. Proc. 3rd Symposium on Biotechnology in Energy Production and Conservation. Paper 7. organized by Oak Ridge Lab. USA.
(5) Runkel, R.O.H. and Witt, H. (1953). zur Kenntnis des thermoplastischen Verhaltens von Holz. 3. Mitteilung. Holz als Roh-und Werkstoff 11 457.
(6) Wenzl, E.F.J. (1970). The Chemical Technology of Wood Academic Press 255.
(7) Abatzoglou, N.S. (1982). Densification of Biomass. Dipl. Eng. Thesis. NTU Athens.
(8) Pyle, D.L. and Zaror, C A . (1984). Heat Transfer and Kinetics in the Low Temperature Pyrolysis of Solids. Chemical Engineering Science 39(1) 147-158.
(9) Mobarak, F.. Fahmy. Y. and Augustin. H. (1982). Binderless Lignocel-lulose Composite from Bagasse and Mechanism of Self-Bonding. Holz-forschum 36(3) 131-135.
(10) Koullas. D.P. and Koukios, E.G. (1987). Hot Briquetting of Wheat Straw. Proc FAO/CNRE Workshop on Handling and Processing of Biomass for Energy, (ISES Conference Parallel Session), Hamburg. FRG.

425
Preparation and use of charcoal : vater slurries
C. ESNOUF*, S. GAUDEHARD*, G. ABTOMIHI**, 0. FRANCOIS**, C. HEZERETTE***
* CEXAGREF.BP 121.92164.Antony Cedex.FRANCE. ** université de Technologie de Conpiègne. BP233.60206.Compiegne Cedex.FRANCE. *** CTFT.45b.av.de la Belle Gabrielle.94130. Noqent s/Narne.FRANCE
SumiarY The ai« of the work is to design and develop a whole process of charcoal slurries production for use in siali and medium size boilers and lov speed diesel engines. Two possible flowsheets have been tested : without ash renoval, a binary charcoal/water iiixture contraining 571 charcoal, exhibiting a LCV of 16900 KJ/Kg and a dilatant behaviour has been obtained. Ash renoval, including acid treatment and selective aggloiieration leads to a ternary lixture, containing 471 of charcoal and 12S fuel-oil, of a LCV of 18700 KJ/Kg, exhibiting a pseudo-plastic behaviour and meeting the pollution standards requirements. Preliminary combustion tests shov better burning properties for ternary Bixtures. Economic estiiiate show that slurries would be coiipetitive with a fuel oil at 7.5 ECÜ/GJ. Significant cost reduction can be obtained in the future in formulation optimiza- tion, and pyrolysis liquids valorization. Anyway, tie grants presently needed by farmers for competitivity are less than for other agricultural policies.
1. INTRODUCTION Common agricultural productivity trend shows that european agriculture produce and will
produce more and more surpluses. In year 2000 the area corresponding to surpluses will probably represent 151 of present agricultural area (1). Short rotation forestry is proposed for those areas, but a very large market must be found for such a large wood production. Household and flats heating would be consistent with it, but bioiass is not a fuel easy enough to handle. Charcoal : water slurries considerably improving biomass storage, transportation and handling, might therefore be a right solution. The aim of the work presented here is to design and develop a whole process for such slurries' production and use in small and medium size boilers and in low speed diesel engines. The paper present results on ash removal, slurry formulation, preliminary burning tests and economics.
Slurries must satisfy several objectives, of somewhat contradictory nature : The minimum lov calorific value (LCV) required for the liquid to be usable in the current boilers retrofitted is 16700 KJ/Kg. The slurry must satisfy fluidity specifications : it must be stable (no solids deposit) and its viscosity must not exceed 2 Pa.s (2000 cp) for a shear rate of 100 s . The slurry must respect standards against atmospheric pollution : less than 0.24 g/HJ must be discharged from plants under 3 HW. For small scale boilers, intermittent operation needs easy ignition characteristics for the fuel. Details on the way to obtain those results are given in (2).
2. SLURRIES FORMULATION AND PROPERTIES : Two possible formulations bave been considered and tested, a charcoal/water slurry without
ash removal, and a ternary mix cleaned charcoal/water/fuel. The first steps of the line are the same : the raw material is short rotation coppice (SRC), poplar or eucalyptus, including bark

Γ 0.3 Τ a i r ι ,1.15 Τ volatile»Burned·1*Electricity air i
ο°ο&?ο°ο > Drying ~ ^ 1 ODT wood Pyrolyses ^f Production
ι (15% water Ns» n _„ T , ,
1 ODT wood J' ™ ^ „ < Λ ^0.33 Τ charcoal (502 water content) 0.82 Τ water content)
<i Crushing mill
charcoal 80,
water
NO DEASHING :
1 r ./ addi ti ves
( Γ Q wet ball mill bimodal granulometry : 420 IA
charcoal : water : additives slurry 57 : 42 : 1
ACID AND SELECTIVE AGGLOMERATION CLEANING
ί . rçQ
wet ball mill 813/1
rHCL .water
.ÕO hydrophobic
liquid : fueloil
water + additives
< —
V
Λ purified *■
charcoal + fuel sãfc&gSi
Γ PgSoaJ
Figure 1 charcoal : water : fuel slurry 47 : 40 : 12
bio ultracarbofluid (bioUCF)
acid + water + ashes
N> ON

427
even though it is rich in ash. The wood chips are pyrolyzed in a CemagrefFramatome pyrolyser vith recycling of volatile latter, which enables maximum charcoal yield.
The line is presented in figure 1. Details on paraietric studies for fonulation optimiza tion are given in (2). λ typical slurry obtained fro« a bimodal granulometry(70t of lean diameter 4 μ and 301 of
■ean diaieter 2^)contains 571 charcoal, 421 water, It of Tamol (naphtalene sulfonate) as dispersing agent and 20 to 50ppm of carboxyiethyl cellulose ("Hercules") as stabilizer.
Such a slurry has the specifications required as it has an effective lover calorific value (taking into account the heat a vaporization of vater) of 16900 KJ/Kg and a viscosity of 1900 cp at 100 s . The Theological behaviour of the slurry is dilatancy (apparent viscosity increases with the shearing rate).
When ash removal is included, the production line is sore complex (see figure 1). To achieve satisfying residual ash content, acid and selective agglomeration cleaning lust be coibined (details in (2)).
The hydrophobic liquid used here is domestic fueloil which has the advantage of enhancing the final energy content of the slurry.One may also envisage light fractions of crude oil not currently marketed(presently used mixed with heavy fuels) or light crude oils.
The tvo cleaning steps are complementary,since selective agglomeration preferentially eliminates the silica and the alumina.Neutralization is not necessary since separation of the agglomerates liberate the acid aqueous phase.The agglomerates are just rinsed until a neutral pH is obtained.
A typical ternary ultracarbofluid ("bioDCF") contains 47» of charcoal, 121 of fueloil, 401 vater, 0.8t of a dispersing agent (nonionic vetting type, such as Cemulsol) and 20 ppm of stabilizer. The viscosity is 1900 cp at 100 s"1 and the Theological behaviour is pseudoplastic vhich is a positive effect of tremendous importance. The slurry has a LCV of 18700 KJ/Kg, 751 of which is due to the charcoal, and an ash content of 0.8i.
Preliminary combustion tests have been performed on a 100 KM furnace. Burning properties are much better for a bioDCF than for binary slurries (conversion efficiency of 0.98 as compared to 0.82 at optimum air excess). Flame stability and ignition facility vere satisfactory for bioDCF. Detailed experimentation have still to be carried out.
3. ECOHOHC ESTIMATE : Binary mixtures estimates vere presented in (3). A preliminary estimate is presented here for
bioDCF. The size of production unit is determined by the forestry density around it, to optimize
the transportation/scale economics ratio. A 0.1 density shovs that a 2700 T/day of slurry size of unit is an optimum. For such a capacity, the capital costs are broken dovn as follows (table I).
Capital costs in 10 ECU
»of total
Wood reception
and storage
19
32
Drying
5.7
9
Pyrolysis
9.1
15
Milling and
slurry
18.5
31
Gasbumer and turbo alternator
8
13
Total
60.3
100 Table I

428
The production cost is broken down as follows : the standing charges and staff costs represent 11.71 of total cost ; the operating costs, additive, HCL, and fuel oil, represent 24.91 of total ; the wood transport and storage represent 13.4t of total and the wood itself, 50t of total cost.
As a conclusion, the production cost of bio-DCP is estiiiated at 850F/T (120 ECU/T), that is to say 6,5 ECD/GJ. Considering the present official OPEC price for crude oil (18 $/bbl), bio-DCF is nearly twice the price of doiestic fuel. As coiipared with the binary slurry, the cost is not significantly higher (6.5 and 6.3 ECD/GJ).
The lain iteis taking up that price are the wood, and the fuel and additive. So, finding alternative additives and hydrophobic phases is of treiendous importance for cost reduction.
Considering other possible iiproveients, an increase in short rotation coppice fro· 13 T/ha to 17 T/ha would save a significant 81 for bio-DCF and 111 for binary ikes.
A pyrolysis liquids valorization could also lean a significant cost decrease ; the lost siiple way is to sell their energy content on-site, through steai generation for instance. This would represent a 10< reduction for bio DCF and a 13.51 for binary lixes. Of course, any lore valuable strategy aiongst the eierging technologies for pyrolysis liquids use (upgrading to «otor-fuel for instance) will represent a significant iaproveient.
The coiparaison with other fuels lust be iade in tens of final energy cost, that is to say including the boiler's capital cost and efficiency and the boiler operating tiae per year (the hypothesis here is 4000 h/year). In the case of bio-DCF, the boiler is not equipped with ash reiover and dust recovery in siokes.
The co«parison of bio-DCF with light fuel oil is shown on figure 2 ; it can be seen that the bio-DCF would be coipetitive for a light fuel oil cost of around 7.8 ECD/GJ, that is to say around 40$/bbl crude oil. For binary slurries, the coipetitiveness would be obtained for a price of 6.5 to 7.4 ECO/GJ.
These results can be teiipered in teres of comon agricultural policy in Europe ; table II shows the grant to faners needed for present coipetitiveness with fuel oil and it appears that the proposed energy line is very interesting.
Wheat export production
Ethanol production
binary slurry present advanced
bio-DCF present advanced
3000-4800 7000-8000 5000-12000 2800-4500 1100-2000 3700-4500 2300-3400
Table II Cost of different agricultural policies
required in FF/ha -year grant
In addition, exportation of the technology to developing countries is envisageable since the cost of raw «aterial is lower there (fron 100 to 280 FF/T SRC) and this cost represents 50 to 70S of the final cost price. Low speed diesel engines would represent a significant larket there.

429
bioOCF and light fueloil costs as a function of, oil price, boiler size
U / G J
4
— j — ι — τ ι ι ! ι ι ι ι ! ι ι ι ι ! ι ι ι ι ! ι ι ■ ι j ι ι ι ι ι
_...
_...
_ l
* .,
, , , ,
+ " " " ■ — ' * : .r
:::::
, . , .
¿^^ζΖΖΖΖΪΖ. . A i , =:::::*
+:':"';::::::::£; I
::::::::::""f"" ^
ty..''
* x x -:- ~
x
:::#F?""T
X 4 MW ° 2 . 5 MW
j + 1 MW : · 5 0 0 KW
, , , , ι , , . , i . . . . i . , . .
Light fueloil cost (ECÜ/GJ)
Figure 2
4. CONCLUSION : Two types of slurries satisfying the specification have been obtained. Econoiiics are already
interesting in tens of agricultural policy and of exportation to developing countries, but they could be iiproved by pyrolysis liquid valorization and choice of other conponents in fonulation. The burning properties in boilers and engines bave also to be studied now.
5. REFERENCES : (1) BECKER J.J (1986). Etude des débouchés énergétiques de l'Agriculture en coipléiient des
débouchés agricoles traditionnels. Thèse de Doctorit université Paris Dauphine (2) ANTONINI, G, i al (1988). Fomulation and Theological properties of charcoal liquid fuels
Euroform New Energies, Saarbrücken Vol 3 pp 629 631 N.S Stephens t Associates.
(3) ESNOOF. C , LESANT. V. (1988) New Process of suspension Pyrolysis and use of charcoal slurries id Vol 3 pp.635637.

430
TREATMENT OF PYROLYSIS OIL WITH COMPRESSED CARBON DIOXIDE
V. BRANDANI, G. DEL RE, G. DI GIACOMO and D. FLAMMINI University of L'Aquila, Dept. of Chemistry Chemical Engineering & Materials
1-67040 Monteluco di Roio, L'Aquila, Italy
Summary
This paper deals with the possibility of predicting phase behavior of systems containing compressed carbon dioxide and pyrolytic oils - complex multicomponent mixtures of very different organic compounds-in a wide range of pressure and temperature. A systematic approach based on vapor-liquid equilibrium data of binary and ternary systems containing carbon dioxide is described together with the main characteristics of the equation of state used as theoretical model of the whole system. The results can be utilised to study the technical-economical feasibility of a supercritical solvent extraction process for separation and up- grading of different bio-oils derived from wood pyrolysis and/or liquefation processes.
1. INTRODUCTION Bio-oils derived from wood pyrolysis and/or liquefation processes are
complex mixtures of many organic compounds varying greatly in size, molecular structure and polarity. The recovery from pyrolytic bio-oils of useful products is worthly to be considered especially if it can be regarded as a significative step of an integrated up-grading process. In fact, pyrolytic bio-oils require, in general, to be up-graded since they rarely meet the standards for commercial fuels. On the other hand,depending on the type of wood and on the operating conditions of pyrolysis, bio-oils are usually characterised by an high percentage (from 10 to 60%) of phenol and phenol omologs (1) which are by far the most valuable product of wood pyrolysis. When these compounds are not recovered as chemicals they are usually destroyed during the fuel up-grading process.

431
The use of dense (liquid or supercritical) carbon dioxide as solvent is rapidly increasing in several important areas as enhanced oil recovery (EOR), supercritical fluid chromatografy (SFC) and supercritical fluid extraction (SFE). In comparison to other conventional separation techniques like distillation and liquiliquid extraction, SFE has the advantages of higher efficiency, easy of recovery of the solvent and improved transport properties. Carbon dioxide is also environmental acceptable, inexpensive and readly available. Its critical temperature is 304 Κ and its critical pressure is 73.8 bar. As a consequence, carbon dioxide is completely miscible with many nonpolar liquid hydrocarbons as well as with low molecular weight hydroxil compounds under mild conditions of pressure and temperature (2). Phenol and phenol omologs are not miscible in all propor
tion in dense carbon dioxide, but their solubility is large enough (2,3,4) to allow us to consider dense carbon dioxide as proper solvent for the recovery of phenolic compounds from pyrolytic biooils.
The feasibility of such a process and its optimization is based on the possibility of describing, from a thermodynamic point of view, the phase behavior of the systems carbon dioxidebiooils which is, in general, a multicomponent multiphase mixtures with an high number of very different compounds and one to four cohexisting phases. From a systematic point of view, one is faced with a series of problems which are quite similar to those studied from many years by people involved with heavy petroleum fractions and bitumen characterization:
identification of the mixture's components;
knowledge of the thermodynamic and transport properties of pure components ;
choise of a proper thermodynamic model (Equation of State) capable of describing accurately the complex phase behavior of the system;
collection (from literature and/or from laboratory) of data required for model parametrization;
reduction of the system to an equivalent but less complex system of few pseudocomponents.
In this paper we describe with some details a combined experimental and theoretical approach which can be usefully applied to the thermodynamic characterization and phase behavior prediction of pyrolytic biooils and biooilscarbon dioxide mixture. We also report some preliminary results on the treatment of pyrolysis oil with compressed carbon dioxide.

432
2. CHARACTERIZATION MODEL Classical models for the characterization of complex petroleum
fractions are based on measured properties of pure compounds and/or pseudoCompounds such as boiling point and specific gravity. Recently new correlations have been developed which require, for each petroleum fraction (pseudocomponent), more sophisticated informations: NMR, elemental analysis, and IR spectroscopy. However, for the investigation of the feasibility of our separation process it must be pointed out that much attention has to be paid to the problem of binary and pseudobinary interac
tion parameters. The approach proposed is similar, in principle, to that used by Alessi
et al. (5) and by Yu et al. (6) for heavy ends characterization and super
critical carbon dioxidebitumen phase behavior. It is based upon the attainment from the biooil of five homogeneous fractions. This makes it possible to define and evidence some pseudocomponents for which one determines the average molecular weight. Then, pseudobinary and pseudoternary equilibria data between fractions and carbon dioxide can be experimentally determined at laboratory level: solubility of each fraction (or pure component) and solubility and selectivity of their binary mixture in supercritical carbon dioxide are used as equilibrium data. In addition, when other vaporliquid equilibrium data (bubble and dew points, PTx, PTy, PTxy and PvT) are available from the literature for pure and binary mixture, they can easely inserted in the procedure for determining binary interaction parameters.
As thermodynamic model for the whole system carbon dioxidebiooil we use an equation of state (EOS) which, following Lüdecke and Prausnitz (7), has been obtained by using the equation of MansooriCarnahanStarling (8) for the repulsive term and that of Soave (9) for the attractive term in combination with a density dependent mixing rule proposed by Panagiotopoulos and Reid (10). This equation, which is characterized by three adjustable parameters for each binary or pseudobinary system, has been applied successfully to the prediction of phase behavior of carbon dioxidelemon oil system at pressure from 30 to 110 bar and at temperature from 308 Κ to 323 Κ (11).
The three binary interaction parameters for each binary or pseudobinary system containing carbon dioxide are calculated from the experimental solubility data by minimising the following objective function:
s = Σ.(y y .)? (1) li exp cal ι
where y and y . are experimental and calculated mole fractions in the exp cal
vapor phase and Ν is the number of experimental data. To find the numerical values of the parameters which characterize binary interactions existing

433
between pure and pseudocomponents the following objective function:
Ν Ν S = Σ.(y y )? + Σ.(Y Υ J
2 (2) ii exp cal ι il exp cal ι
is minimised. Here, in addition to yexD and yca^ we have Y e x D and Ycai which are experimental and calculated mole fractions of one solute in vapor phase on carbon dioxide free basis.
3. EXPERIMENTAL APPARATUS The solubility of a single pure or pseudocomponent and of their binary
mixture in supercritical carbon dioxide are obtained by using the single pass flow apparatus. A detailed description of this apparatus and its operation mode have been reported in a previous paper (11). It has been designed to operate up to 1000 bar and 473 Κ and can be used without any modification as a semicontinuous extraction laboratory plant. It is used in combination with an optical cell of few cubic centimeters (Nova Swiss) which allow us to verify the number of cohexisting phase at the same operating conditions of the single pass flow apparatus.
Before starting with the systematic experimental and theoretical study described in the present papar, we used our laboratory extraction plant to extract with supercritical carbon dioxide phenol compounds from a sample of pyrolytic biooil obtained from the fluidised bed reactor of Raiano (Italy). We operated at 313 Κ and 300 bar obtaining an extract which, after depressurization and separation of carbon dioxide, was analysed for phenol and phenol omologs content. We found in appreciable amount phenol, cresols, xilenols and other substitutes.
REFERENCES
(1) CHURIN, E., MAGGI, R., GRANGE, P. and DELMON, B. (1987). Characterization and Upgradind of a Pyrolytic Oil. Pyrolysis as a Basic Technology for Large AgroEnergy Projects, 1516 October, 1987 L'Aquila, Italy

434
(2) DANDGE.D.K., HELLER,J.P. and WILSON,K.V. (1985). Structure Solubility Correlations: Organic Compounds and Dense Carbon Dioxide Binary Systems. Ind. Eng. Chem. Prod. Res. Dev., 24,162166.
(3) VAN LEER,R.A. and PAULAITIS.M.E. (1980). Solubilities of Phenol and Chlorinated Phenols in Supercritical Carbon Dioxide. J. Chem. Eng. Data, 25,257259.
(4) LEE,R.J. and CHAO.K.C. (1988). Extraction of 1Methylnaphtalene and mCresol with Supercritical Carbon Dioxide and Ethane. Fluid Phase Equilibria, 43,329340.
(5) ALESSI.P., CORTESI,Α., FERMEGLIA.M.,FONTANA,M. and KIKIC.I. (1989). Heavy Ends Characterization Method Using Gas Solubility Data. 5th Int. Conf. on Fluid Properties & Phase Equilibria for Chemical Process Design, April 30 May 5, 1989, Banff, Alberta, Canada.
(6) YU.J.M., HUANG,S.H. and RADOSZ.M. (1989). Phase Behavior of Reservoirs Fluids: Supercritical Carbon Dioxide and Cold Lake Bitumen. 5th Int. Conf. on Fluid Properties & Phase Equilibria for Chemical Process Design, April 30 May 5, 1989, Banff, Alberta, Canada.
(7) LÜDECKE, D. and PRAUSNITZ,J.M. (1985). Phase Equilibria of Strongly Nonideal Mixtures from an Equation of State with Density Dependent Mixing Rules. Fluid Phase Equilibria, 22, 119.
(8) MANSOORI.G.A., CARNAHAN.N.F..STARLING,Κ.E. and LELAND.T.W. (1971). Equilibrium Thermodynamic Properties of the Mixtures of Hard Spheres. J. Chem. Phys., 54, 15231525.
(9) SOAVE,G. (1972). Equilibrium Constants from a Modified RedlichKwong Equation of State. Chem. Eng. Sci., 27, 11971203.
(10) PANAGIOTOPOULOS.A.Z. and REÍD,R.C. (1986). Multiphase High Pressure Equilibria in Ternary Aqueous Systems. Fluid Phase Equilibria, 29, 525534.
(11) DI GIACOMO,G., BRANDANI.V., DEL RE,G. and MUCCIANTE.V. (1989). Solubility of Essential Oil Componenents in Compressed Supercritical Carbon Dioxide. 5th Int. Conf. on Fluid Properties & Phase Equilibria for Chemical Process Design, April 30 May 5, 1989, Banff, Alberta, Canada.

435
ACnVATED CARBON FROM EUCALYPTUS KRAFT LIGNIN
J.J. RODRIGUEZ, T. CORDERO, J. RODRÍGUEZMIRASOL, A. SIMON and A. BATAI .1 KR Departments of Chemical Engineering and Mechanical Engineering.
University of Malaga. 29071 Malaga (SPAIN).
Summary
Preparation of activated carbon from acidprecipitated eucalyptus kraft lignin i s studled, following a twostep process consisting in CO2 partial gasification after carbonization in N2 atmosphere. Char yield and composition as well as porosity and surface area development against temperature and solid holding time have been investigated In the carbonization step. CO2 activation has been carried out at 800°C, a bumoff range up to 80% being covered. The porous structure of the resulting products has been studied. Essentially microporous carbons showing an increasingly developed mesoporoslty have been obtained. Apparent surface area values up to 1600 m ^ / g were reached. A 50 k g / d (referred to raw material) pilot plant using a screwconveyor system has been constructed.
1. INTRODUCTION: The development of lignin applications a s a raw material i s being widely
investigated in recent years. One interesting way is pyrorysis and carbonization to obtain chemicals (1,2] and a char which can be used as substrate for the preparation of carbon materials [36].
In this paper, we study the preparation of activated carbons from eucallptus kraft lignin by a twostep procedure consisting In Inert atmosphere carbonization followed by CO2 partial gasification.
2. EXPERIMENTAL: Labscale experiments have been carried out in a stainless steel cylindrical reactor
16 cm length and 8 cm l.d. electrically heated. The system h a s a t ime/temperature programmer which allows to control temperature and heating rate. Gases ( N2 or CO2 ) were continuously passed at a 10 1/h (STP) flow rate. Thermogravimetric experiments were also accomplished using a model Thermoflex Rlgaku thermobalance.
The carbonized products were analized for elemental composition ( C, H, N, S ) using a 240 C PerkinElmer apparatus for C, Η, Ν and a JuliusPeter for S. Ash content was also determined in every sample.
The raw material was eucalyptus kraft lignin supplied by the Empresa Nacional de Celulosas, S.A. from its lignin precipitation pilot plant.The precipitating agent was H2SO4 at a controlled pH and the lignin was centrifuged and dried. The final product has the following typical composition: 53.7% C, 4.5% H, 0.1% N, 1% S, 30.5% O and 10.2% ash content. The relatively high ash content is most probably due to precipitate enmeshment and can be easily reduced by diluted acid washing. As will be emphasized later, this is an Important variable in the carbonization process and thus we have peformed experiments with different ashcontent lignin.
Characterization of the porous structure of the products have been carried out from 77 Κ N2 adsorption isotherms using a Sorptomatic 1800 ( Carlo Erba Strumentazione ).
3. RESULTS: Figure 1 shows the TG and DTG curves obtained for lignin at a 5°C/mln heating
rate. The TG shows an Initial weight loss attributed to moisture followed by a plateau region up to about 200 °C. At this temperature, devolatilizatlon begins and extends up to

436
about 600°C where a constant 55% weightloss level is reached. The DTG exhibits a small maximun due to moisture loss and a wide peak from 200 to 500 °C with a maximun at 370 °C.
CD Ι Ο
0 200 400 600 800 1000 TCC)
Fif. 1. TG and DTG curves obtained for lignin at 5*C/min heating rate.
In labscale carbonization experiments, we observed that when working with lignin samples of low ash content, a plastic phase formation takes place at a relatively low temperature ( 200230°C, depending of the ash content ). Lignin swells and an agglomerate product of pseudo glassy appearance is finally obtained. This would represent a serious technical problem and also could affect negatively to the future porosity development. Then, we investigated the conditions to avoid or reduce this problem. Experimental evidence was obtained in the sense that it becomes less Important as heating rate decreases and ash content Increases. But even at very small heating rates (<5°C/min) an ash content higher than 4% Is necessary to acoid fluid phase formation. Above 8% ash content the problem becomes negligible even at the highest heating rate Investigated (30°C/min).
According to these results, it seams convenient to carry out the carbonization step with a relatively high ash content lignin ( 810% ). Nevertheless, this would lead to a very high ash level in the activated carbon which may be detrimental from a quality point of view. Then, a washing opperatlon has to be Included, the situation of which throughout the whole procces has to be carefully investigated as we have to consider two main aspects: Easy of removal of ashes, which depends on chemical changes occurring throughout carbonization and activation and the potential catalytic effect of some inorganic components mainly in the CO2 gasification step. In the present stage of our research we are working according the following scheme:

437
LIGNIN
( ~ 1 0 % a . c . )
ACTIVATED CARBON
PRECARBONIZATION 350 °C, N,
ACTIVATION 700900 °C, C02
WASHING 2 X oq. H2S04
CARBONIZATION 700900 °C, N,
The results obtained on the carbonization experiments revealed that the most important loss of volatile matter occurs within the 300500 °C range and a nearly constant yield close to 40 % ( referred to organic matter ) is reached beyond 700 °C. The carbon content of the final products becomes very close to 95 % at this temperature. An important feature is the evolution of S content, which goes from 1 % in the initial kraft lignin to less than 0.5 % at 500 °C and 0.25 % at 900 °C.
The apparent surface area of the carbonized products shows an increase up to 800 °C followed by a decrease in the 700900 °C range, which becomes specially significant at 900 °C. Decrease In apparent surface area could be explained as the result of increasing rearrangement of parallel layers of carbon atoms.
Figure 2 shows the N2 adsorptiondesorptlon isotherms corresponding to the 700 and 800°C chars. Both exhibit a shape characteric of microporous solids. This thermal range was adopted as the optlmun for carbonization. At 700°C, the devolatilization process requires no more than one hour and the apparent surface area of the resulting carbon becomes close to 500 m 2 /g , reaching a value of 600 m 2 / g at 800°C.
200 I
al IS)
M E 150 o
«♦« ♦ * _ ■ α
* *α ♦π Π D
D
a 700°C Char o 800
eC Char
100 0,2 0,4 0,6
P/PO 0,8 1,0
Fig. 2 . Adsorptiondesorption isotherms of N2at 77 Κ of chars: open points, adsorption; closed poi nts, desorption.

438
C 0 2 activation of the carbonized samples lead to essentially microporous products, but the micropore distribution is increasingly wider as burn-off increases, according to the shape of the N2 isotherms and the DR and ocs plots. Figure 3 shows the N2 adsorption-desorptlon isotherm of the activation product corresponding to a 55% bum-ofT. An apparent surface area close to 1000 nfi/g was obtained at 27 % burn-off which increased up tol500 m 2 / g at 70% bum-off.
1200
0 4 0 6 P/PO
Fig. 3. Adsorption-desorption isotherm of N2 at 77 Κ of the activation product corresponding to a 55X burn-off: open points, adsorption; closed points, desorption.
Pilot plant studies are at present at the plant start-up stage. The pilot plant has two main sections ( carbonization and activation ), the furnace being in both cases of screw-conveyor type and electrically heated. An intermediate paddle crusher allows to grind the char before CO2 activation. This becomes convenient as same degree of agglomeration is observed in the carbonization step.
ACKNOWLEDGMENT; The authors wish to express their gratitute to the Empresa Nacional de Celulosas,
S.A. for providing the financial support for this research. T. Cordero also acknowledges to the Spanish CICYT-DGICYT for a R+D grant.
REFERENCES
(1) JEGERS, H.E. and .KLEIN, M.T (1985). Ind. Eng. Chem. Process Des. Dev., 24 173 (2) NUNN, T.R et al, (1985). Ind. Eng. Chem. Process Des. Dev. 24 844 (3) OTANI, Cet al, (1984). J.Chim. Phys., 81 887 (4) OTANI, Cet al., (1986). Ceramica, 32. 315 (5) POLIDORO, H. et al., (1986). Ceramica, 32. 181 (6) OTANI, C. et al., (1988). Proc. Carbon'88, p. 648. Newcastle uponTyne.

439
ACTIVATED CARBONS FROM CHROMIUMTANNED LEATHER WASTE.
MA. MARTINEZ-SANCHEZ, C ORGILES-BARCELO, J.M. MARTIN-ΜΑΚΠΝΕΖ* and F. RODRIGUEZ-REINOSO*. INESCOP. Asociación de Investigación de las Industrias del Calzado y Conexas. Elda. Alicante. Spain. 'Departamento de Química Inorgánica e Ingeniería Química. Universidad de Alicante. Alicante. Spain.
SUMMARY The preparation of COa activated carbons from chromiumtanned leather waste has been used as a way to reduce the volume of this residue and to find a practical application. The effect of some preparation variables (temperature, soaking time, heating rate, etc.) on the adsorption characteristics of the carbons has been studied using N2 (77K) and COa (273K) as well as mercury porosimetry. The microporosity does not change much during activation in COa, very likely because of the blocking caused by the Cr203 particles. Since the volume of macropores is very high in all carbons, increasing with burnoff in COa, they could be useful in adsorption from solution processes.
I. INTRODUCTION The tanning and footwear industries produce a large
volume of chromiumtanned leather waste which contains an average 6 wt% of Cr203 , introduced in the leather during the tanning process with chromium (III) salts (1). At present, this waste does not have any practical application and in fact, its elimination is very problematic because of economical and ecological considerations. It is therefore important to find a way of elimination of the waste in which useful materials could be obtained. This paper presents a such new way of elimination by producing activated carbons. This approach has the advantage of considerably reducing the volume of the waste and, additionally, of producing adsorbent carbons with adequate adsorption capacity towards gases and solutes in aqueous solution.
II. EXPERIMENTAL The leather waste used as precursor in this work was
of black boxcalf type with aniline finish. The leather was cut in 2x2x2 mm cubes in order to obtain granular activated carbons although, in some cases, shreded leather was also used. The leather was carbonized in a tubular furnace under a Na flow (80 cm3/min) at different tempera

440
tures (773 and 1123K), heating rates (1.5 and 5K/min) and soak time (2,5 and 10 h). The carbonized material was activated in a flow (80 cm3/min) of C02 at 1098K for 228 h. The nomenclature of the samples includes: precursor, carbonization conditions (2 digits) and time of activation.
The characterization of the porous texture of the carbons was basically carried out by adsorption of N2(77K) and C02(273K) using a conventional gravimetric system fitted with silica spring balances; mercury porosimetry was also used for the larger size pores. III. RESULTS
The chemical analysis of the leather shows that this material may be useful for the preparation of activated carbons since it has a 45 wt% of elemental carbon. The leather has an ash content of about 6 wt%, most of which is Cr203. This high content of chromium in the leather could be considered as an inconvenience for the preparation of activated carbons, but it was shown that the whole amount of chromium remains as Cr203(2) in the final carbons, this being a very stable oxide.
The carbonization process of chromiumtanned leather was studied in a previous report (3) as a function of temperature; the process was almost completed at 1000K, with a yield of 23%, similar to that found for many lignocellulosic materials (4). For this reason, a carbonization (in nitrogen) temperature of 1123K was used in most cases. The carbonized leather was activated in a flow of COa, under different experimental conditions. There follows a short analysis of the role of some of the variables used in the preparation of the activated carbons.
Α. Carbonization heating rate. The N2(77K) adsorption isotherms (Figure 1) corresponding to the carbons prepared using 1.5 and 5K/min carbonization heating rate are characteristic of essentially microporous solids. The analysis of these isotherms (and the corresponding C02 at 273K isotherms) was carried out using the DubininRadushkevich (DR) equation (5). The results obtained are listed in Table 1, where the volume of mesopores larger than 7.5 nm and macropores obtained from mercury porosimetry have also been included. The data show an increase in microporosity for the 5K/min heating rate but almost no change in the volumes
TABLE 1. Some data for carbon
Carbon C8M8 C8R8
H.R. (K/min) 1.5 5
Global Yield (wt%) 17 13
V(cm:
micro N2 C02
0.30 0.25 0.32 0.29
Vg) meso
0.10 0.13
macro
0.84 0.89

441
of meso and macropores (the high value of macropore volume of these carbons should be noted); on the other hand, the global yield is larger for the 1.5K/min heating rate.
Figure 1. N2/77K adsorption isotherms B.- Carbonization time. (Table 2). The global yield
increases with increasing carbonization temperatures but the microporosity remains almost constant from 2 to 10h; there is however an important decrease in meso and macro-porosity for the carbons prepared by 10h carbonization.
TABLE 2. Some data for carbons.
Carbon C8L8 C8M8 C8N8
t(h) 2 5 10
Global Yield (wt%) 14 17 18
N 2
0.30 0.30 0.28
V(cm3/g) micro
CO2
0.25 0.25 0.25
meso
0.11 0.10 0.05
macro
0.80 0.84 0.66
C - Carbonization temperature. (Table 3) . The yield is larger for the higher carbonization temperature. There is almost no change in the microporsity but a decrease at 1123K in mesoporosity takes place.
TABLE 3. Some data for carbons.
Carbon
C5L8 C8L8
T(K)
773 1123
Global Yield (wt%)
11 14
V(cm3/g) micro
N 2 CO2
0.31 0.30
0.24 0.25
meso
0.20 0.11
macro
0.86 0.80

442
D.- Activation time. (Table 4). With increasing activation time, there is a widening of the microporosity. However, for long activation periods there is a decrease in the micropore volume and an increase in the volumes of meso and macropores. This may be a consequence of both the destruction of a proportion of micropores and the progressive accumulation of ash (carbon C8M28 has 55 wt% ash content) in the interior of the carbon particles.
TABLE 4. Some data for carbons.
Carbon C8M2 C8M8 C8M12 C8M28
t(h) 2 8 12 28
Global Yield (wt%) 22 17 15 9
micro N2
0.23 0.30 0.33 0.26
C02
0.23 0.25 0.27 0.19
V(cm3/g) meso
0.04 0.10 0.06 0.22
macro
0.63 0.84 0.79 1.02
IV.- DISCUSSION The micropore volume of the activated carbons prepared
from chromium-tanned leather waste does not change much for the different variables introduced in the carbonization and activation processes, being about 0.30 cm3/g (equivalent surface area: 900 m 2/g). This may be surprising when comparing with other carbon precursors but it may be explained in terms of the high Cr203 content. Cr203 particles may be blocking the entrance of a relatively large proportion of narrow pores; although there are additional data confirming this hypothesis (3), the effect of the Cr203 could be indirectly shown by using both pickled leather (without chromium) and "extracted" leather (after chromium extraction) as precursors for the carbon.
P/Po
Figure 2. N2/77K adsorption isotherms

443
TABLE 5. Some data for carbons.
Carbon Leather Activation Yield (wt% daf)
V micro (cm3/g) Na C02
S8M4 Pickled 58 C8M12 Granular 54 CÎ8M8 Powder 54 D8M12 Extracted 50
0 . 0 8 0 . 3 3 0 . 3 5 0 . 8 5
0 . 0 6 0 . 2 7 0 . 2 8 0 . 5 1
Figure 2 includes the N2(77K) adsorption isotherms for the powder carbons prepared from pickled leather (S8M4), chromium "extracted leather (D8M12) and leather (08M8); for the sake of comparison the isotherm for a granular carbon from leather (C8M12) has been included. In all of these cases the CO2 burn-off for the ash free carbon is around 55%. According to Figure 2 and Table 5, carbon S8M4 has a very low adsorption capacity and this indicates that the tanning process favours the formation of the basic structure needed to form a proper activated carbon. On the other hand, carbon D8M12 exhibits a very high adsorption capacity (about 2500 m2/g in equivalent surface area) and a wide micropore size distribution. Consequently, it is possible to conclude that the Cr203 particles are partially blocking micropores in carbons from chromium-tanned leather; if a portion of these particles is eliminated by previous extraction of the leather, the adsorption capacity is considerably increased.
One important aspect of activated carbons prepared from chromium-tanned leather is the high volume of macro-pores this being important for applications such as adsorption from solutes in aqueous solution, since the macropores would facilitate the access of the solute to the interior of the carbon particles. REFERENCES
J. Soc. Leather Technol. Chem. 69, MARTINEZ-
Conf.
1. LANGERWERF, J.S.A 166 (1985).
2. MARTIN-MARTINEZ, J.M., RODRIGUEZ-REINOSO, SANCHEZ, M.A. and ORGILES-BARCELO, C. Int Pyrolysis and Gasification. Luxembourg. 1989.
3. MARTINEZ-SANCHEZ, M.A., ORGILES-BARCELO, C , MARTIN-MARTINEZ, J.M., RODRIGUEZ-REINOSO, F. and TORREGROSA, R., Proc. Int. Conf. Carbon. Newcastle upon Tyne. p.280. 1988.
4. LINARES-SOLANO, A M. and RODRIGUEZ-REINOSO, Biotechnol. 3_0, 65 (1980).
5. DUBININ, M.M. "Progress in Surface and Membrane Science." Ed. D.A. Cadenhead. Academic Press. New York. 1, 1 (1975).
LOPEZ-GONZALEZ, J.D., MOLINA-SABIO, F., J. Chem. Technol.

444
MOTOR FUELS FROM PYROLYTIC LIGNINS
J. Piskorz, P. Majerski, D. Radlein and D.S. Scott Department of Chemical Engineering, University of Waterloo
Waterloo, Ontario, Canada N2L 3G1
Summary
Pyrolysis of wood by the Waterloo Fast Pyrolysis gives a high yield (70% to 80%) of a homogeneous pyrolysis oil. This oil can be readily separated by extraction into two fractions, one containing mainly water soluble compounds, and one containing components derived from the lignin content of the wood. Such a "pyrolytic lignin", derived from a sawmill waste of mixed coniferous wood and bark, was subjected to catalytic hydrogénation in a bench-scale catalytic flow reactor. The bench scale catalytic reaction apparatus has some novel features, and these are described. The reactor operates as a continuous plug flow unit and is constructed so that a controlled axial temperature gradient can be imposed. Successful hydrogénation runs were carried out in this non-isothermal rector which gave high conversions of up to 78% of carbon fed to a hydrocarbon distillate. Very little coke formation was observed. The product distillate was approximately 60% aliphatic and 40% aromatic in character.
1. INTRODUCTION In an earlier publication (1), it was reported that the conclusion
reached by an International Energy Agency (IEA) study group under the Direct Biomass Liquefaction Project was that the atmospheric pressure flash pyrolysis process was a cost effective and technically feasible method for the production of liquid fuels from wood and peat. However, if hydrocarbon-based transportation fuels were the desired product, further processing of the crude liquid pyrolysis oils was needed to produce marketable gasoline or diesel range products. This IEA report has since become more generally available (2).
In our laboratories one of the most successful atmospheric flash pyrolysis processes for conversion of biomass to liquids has been developed over the past ten years (the Waterloo Fast Pyrolysis Process or WFPP). In our earlier publication with researchers at Battelle, Pacific Northwest Laboratories (1), the pyrolytic liquid obtained from the flash pyrolysis of peat employing WFPP technology was used as a feedstock for the successful catalytic hydrotreating reported then. The present work describes the results obtained using a modified hydrotreating apparatus when a lignin fraction of biomass is used as a feedstock. The lignin used in this work is one of the products which can be obtained from our atmospheric flash pyrolysis process, details of which can be found in earlier publications (3). When wood or other biomass undergoes rapid pyrolysis about 80% to 85% of the lignin present is depolymerized, vapourizes and

445
appears in the liquid product. This material is aromatic in character and of much lower molecular weight than the native lignin. The characteristics of one such lignin have been described in a previous article (4).
The fraction which we have labelled as "pyrolytic lignin" can be recovered from the crude pyrolytic oil, which is a homogeneous single phase liquid, by extraction with water. This extraction removes the water-soluble carbonyl compounds, sugars etc. which are derived from the decomposition of the hemicellulose and cellulose fractions, and leaves a precipitate of material derived largely from lignin. This precipitate is readily removed by centrifuging or filtration, and can be washed and dried if desired to give a light brown powder product. However, for use as a feed for hydrotreating, drying was not necessary. Details of the separation procedure and of the fractions recovered have been given elsewhere (5).
Yields of pyrolytic lignin are normally about 16% to 22% of the feed when wood is pyrolyzed (that is 22% to 28% of the crude pyrolysis oil). Lignin yields will vary with the nature of the biomass used as feedstock. It should be noted also that the aqueous extract from the above lignin recovery step is a source of several valuable acids, aldehydes and ketones derived from the carbohydrate fraction of the biomass (5).
The lignin fractions recovered from processes in which the conversion of cellulose or hemicellulose is the main objective represent a low value by-product at the present time. For example, the conversion of cellulose to glucose for fermentation to fuel alcohol b v most processes leaves a lignin residue of little present value. Similarly, if fast pyrolysis is used to produce oxychemicals or sugars, then a pyrolytic lignin by-product will result. Any conversion of these lignins to higher value-added products than their current use as a low grade fuel would be an economic advantage for any of these biomass conversion processes.
The lignin obtained from fast pyrolysis, or from microbiological processes, should be a good candidate for hydrotreating. It is rich in aromatic structures which are in a less highly condensed form than is normally produced by more severe conversion processes such as conventional pulping. Indeed, it could be anticipated that the difficulty of hydro-treating a lignin fraction might be directly related to the severity of the biomass conversion process used.
2. EXPERIMENTAL A continuous packed bed catalytic reactor was used, modified from
that described by Elliott and Baker (6). A schematic of the apparatus is shown in Figure 1. The reactor itself had a capacity of 96 ml and was only about one-quarter the size of that used by Elliott and Baker. The pyrolytic lignin feed could be readily liquefied by heating, but a controlled minimum temperature was desirable because of the thermal sensitivity of the material. These requirements were met by the use of a unique free-piston feeder. The feed vessel was surrounded by a heating jacket in which thermostatically controlled heating fluid was circulated. The feed vessel contained a free piston which separated the liquefied lignin from water or other appropriate displacement fluid. Feed rate was controlled by a twin head HPLC pump which drew the displacement fluid from a reservoir and which could deliver from 0.25 to 24.0 ml/ min of liquid.
The catalytic reactor had a two zone furnace which allowed an axial non-isothermal temperature gradient to be imposed. Operation was co-current, upflow. Liquid product was collected under pressure in two traps with intermediate cooling. Details of operating conditions are given in the next section.

WATER PUMP
A H2
Figure 1. Continuous hydrogenation-hydrodeoxygenation apparatus.

■6 000
5 000-
3 O 4000 o O 3000
H 2000 o
1000
447
10
nF-i. i- L i u J L *-* ' I 1
h A 6 8 10 TIME (min)
12 14
Figure 2. Total ion Chromatograph of pyrolytic lignin from hog fuel -5. Dimethoxy methyl acetate; 9. Phenol; 10. Trimethyl cyclopentenone; 11,11',11" - substituted methoxy phenols.
o u 2 O
g 2000 b-
^ 9 io
yjL
13 17
14 J IS 16
2 3 TIME (min)
tl
zs β 000 o υ z 6000 O _, 4000 < O 2000
28
iLJJJAiiJUw 7 β TIME (min)
Figure 3. Total ion Chromatograph of light organic product (Run 3) from hydrotreating of pyrolytic lignin -- more volatile fraction
1. Butane 7. Cyclohexane 13. Ethyl benzene 2. Pentane 8. Heptane 14. 2-xylene 3. Ethyl cyclopropane 9. Methyl cyclohexane 15. 4-xylene 4. Hexane 10. Toluene 16. Nonane 5. Methyl cyclopentane 11. Octane 17. Propyl cyclohexane 6. Benzene 12. Ethyl cyclohexane 18. Propyl benzene.

448
The raw material for the tests described here was a sample of hog fuel from the Pacific north-west. Hog fuel is the waste cuttings generated by sawmills, and normally contains a high percentage of softwood bark, in the case of the present sample, about 80%. The sample used consisted of a mixture of waste from two species, Amabilis fir and Western hemlock, which are commonly harvested and processed together. The hog fuel was dried to 9.5% moisture, ground to -1 mm and then pyrolyzed at 525 C and atmospheric pressure in a sand fluidized bed. Apparent vapour residence time was approximately 0.45 seconds. The crude pyrolysis oil was water extracted to give a yield of pyrolytic lignin of 21.2% of the hog fuel fed (both on a moisture free basis). Analysis of this lignin fraction is given in Table 1.
The pyrolytic lignin composition was investigated by means of GC-MS analysis and by C NMR spectroscopy. A typical GC-MS chromatogram is shown in Figure 2, the peaks shown a« tentatively identified in the GC-MS spectra are mainly methoxy and dimethoxy phenols, and substituted methoxy phenols e.g. 2-methoxy-4-(l-propenyl) phenol. The NMR spectra shows some differences in structure of this lignin as compared to those obtained from hardwoods such as poplar (4). The guaiacyl/syringyl ratio is high, consistent with the lignin from the softwood component of the feed. A significant amount of carbonyl groups are also indicated, a feature which is almost completely absent in hardwood lignins. Presumably, these arise from the high extractives content of the bark, and in particular the tannins, resinous and waxy fractions which would contain carboxylic acid or ester linkages.
3. RESULTS The results for two upgrading runs are given in Table 2. In each of
these runs the same catalyst was used without replacement or regeneration and it had already been in operation for over six hours. In both runs, a constant flow of hydrogen was maintained and total reactor pressure was 14 MPa. The catalyst used was sulfided commercial Co-Mo pellet supplied by Katalco Corp., and 65.7 grams were initially charged to the reactor. The reactor was filled with lignin, the desired axial temperature gradient established, and flow started which was recorded as zero time.
The results in Table 2 for the two runs show a high conversion of the feed to a liquid organic fraction, 64% and 61% respectively, representing a carbon conversion of about 78% to liquid hydrocarbons. The oxygen content of this light organic fraction was about 0.5%, and oxygen removal was about 85% as water. Only about 1% of heavy residue remained, which was readily soluble in methanol.
The second test (Run B) was carried out at twice the feed rate but otherwise essentially the same reaction conditions as the previous run. Very similar yields were obtained even though the space velocity was doubled. The short-fall in the overall recovery of products (4% to 9% of feed) appears to be due to the loss of volatile hydrocarbons in the C¿ -Cg fraction when the liquid product was depressurized.
The light organic product had an elemental analysis of C - 88.48%, H - 11.06% and 0 (by difference) - 0.46%. The H/C atomic ratio is 1.50, suggesting an appreciable aromatic content. The results of GC-MS analysis of the light organic fraction are shown in Figures 3 and 4. Material eluted in the first five minutes consisted of normal and cyclic alkanes to CQ, as well as simple aromatics. Small amounts of phenols eluted next and the final group of compounds were largely alkanes, both straight chain and branched, from C-,? t o C?6· Peak numbers shown in Figures 3 and 4 are identified in Figure 3.

449
10000
Z5 8000 o o
6000
_J <I
4 0 O 0
¡Γ 2 0 0 0
32
33 34
τ — 12 13 TIME (min)
35
iL —t
15
10000
3 í ) o •7 0
_l < Ι Ο
8 000
6 000
4 0 0 0
2000
Τ 16
36
A- ι I 17 18 TIME (min)
37 Λ.
19 —I 20
Figure 4. Total ion Chromatograph of light organic product from hydrotreating of pyrolytic lignin less volatile fraction 32. Octadecane; Tridecane 35. Docosane; Eicosane, 7hexyl 33. Eicosane; Hexadecane 36. Docosane; Eicosane, 7hexyl 34. Eicosane, Tetradecane 37. Docosane; Heptadecane, 9octyl
l i
Lu O
3? UJ 2 3 _ l O > u _ l Q2 < CO
6 0
5 0
4 0
3 0
2 0
• 10
0
^ ^ * Jf^*^^
^/^ ^ s *
j r aS"^
^JL - ß*
/ it ^M
/ ~ M
^ _ /
y . Ε Λ
»·—"ι 5 20 45 70 95 120 145 170 195 220 245 270 295 320 345
BOILING POINT C O Figure 5. Simulated distillation curve for light organic product from hydrotreating of pyrolytic lignin (Run 3).

450
The CNMR spectra of the light: organic fraction was also measured. A clean separation of two major groups of hydrocarbons was obtained, that is, aliphatic and aromatic, with almost no indication of any oxygen containing structures. Integration of the aliphatic peaks, most of which were characteristic of nalkanes, indicated that 61.7 ± 0.8% of the carbon was aliphatic. Therefore, the substantial fraction of aromatic carbons, about 38%, suggests that the light organic fraction is more similar to a kerosine than to a diesel fuel. Additional peaks in the NMR spectra indicated the presence of some naphthenic structures also, or of other condensed aromatic or hydroaromatic structures.
A simulated distillation of the light organic fraction was also carried out using the SIMDIS chromatographic method (Sulpeco Inc. Petrocol 3710 column with qualitative calibration mix) according to ASTM method D3710. The resultant cumulative boiling point curve to 345°C (the maximum calibration point) is shown in Figure 5. This analysis was carried out some months after the preparation of the lignin derived hydrocarbon liquid, and it was estimated that 5% to 10% had been lost, representing the most volatile fraction. Figure 5 shows that about 65% or more was below 345°C BP with about 50% boiling within the gasoline range. With fresh product and more careful recovery of volatiles, it is estimated that about 60% of the product would be gasoline range hydrocarbons containing a high percentage of simple aromatic compounds, and cyclic alkanes. The heavier fraction boiling above 225 C would be largely straight chain alkanes and might be suitable for use as a component of diesel fuels.
The successful hydrotreating of this lignin fraction from the pyrolysis of a waste wood and bark mixture suggests that similar lignins from other processes, such as that recovered during fuel alcohol production from lignocellulosics, might also be similarly upgraded with good yields.
4. ACKNOWLEDGEMENTS The authors would like to express their thanks to the Natural
Sciences and Engineering Research Council of Canada for the financial support of this work.
REFERENCES
(1) ELLIOTT, D.C., BAKER, E.G., PISKORZ, J., SCOTT, D.S. and SOLANTAUSTA, Y. (1988). Energy and Fuels, 2, 234235.
(2) ELLIOTT, D.C., BAKER, E.G., OSTMAN, Α., GEVERT, S.B., BECKMAN, D., SOLANTAUSTA, Y. and HORNELL, C. (1989). "A technical and economic analysis of direct biomass liquefaction" Paper 45, IGT Symposium on Energy from Biomass and Wastes XIII, New Orleans, Feb. 1317.
(3) SCOTT, D.S., PISKORZ, J. (1985). Bioenergy 84; Egneus, H.; Ellegard, A. Eds. Elsevier Applied Science, London, Vol. Ill, 1522.
(4) RADLEIN, D., PISKORZ, J., SCOTT, D.S. (1987). J.Anal.Appi. Pyrolysis 12, 5159.
(5) PISKORZ, J., SCOTT, D.S., RADLEIN, D. (1989). ACS Symposium Series "Production, Analysis and Upgrading of Pyrolysis Oils from Biomass" E.J. Soltes, Ed. American Chemical Society, Washington, D.C.
(6) ELLIOTT, D.C, BAKER, E.G. (1987). Energy from Biomass and Wastes X; Klass, D.L. Ed., Institute of Gas Technology, Chicago, 765784.

451
Table 1
Analysis of Pyrolytic Lignin from Hog Fuel
Yield, % of feed, mf
Moisture content, *
Density, gm/ml.
Elemental Analysis, % mf C H o (by diff.)
Moderately Soluble in methanol, pumpable at 60'C
21. 18. 1.
73. 5,
20,
2 ,0 .16
.22
.95
.8
Continuous Fixed Bed Catalytic Upgrading of Lignin from Hog Fuel Moisture 18.OX, C - 73.22, H - 5.95 mf
Run Number
Temperature 'C, outle
Duration, minutes
Amount fed, grams
Yields, grams
Water
Light organic
Total liquid
Heavy residue
Liquid + tar
Cases : CO
co2 CH4 C2H4
C2H6
C3H8 c4+
it/inlet
:s
product
produced
Total recovery
X mf feed as light organica
* C - 88 .48, H -
3
400/240
182
102.1
35.0
53.7*
88.7
1.3
90.0
traces
2.76
2.63
traces
0.80
0.72
0.76
7.67
95.7X
64X 11.06
4
415/230
94
102.6
34.0
50.9
84.9
1.3 86.2
traces
3.08
2.24
traces
0.70
0.63
0.63
7.28
91.IX
61X

452
ROLE OF CHROMIUM OXIDE IN THE TEXTURE OF CARBONS FROM LEATHER
J.M. MARTIN-MARTINEZ, F. RODRIGUEZ-REINOSO, ΜΛ. MARTINEZ-SANCHEZ* and C ORGILES-BARCELO". Departamento de Química Inorgànica e Ingenieria Química. Universidad de Alicante. Alicante. SPAIN. *INESCOP. Asociación de Investigación de las Industrias del Calzado y Conexas. Elda. Alicante. SPAIN.
SUMMARY A series of carbons has been prepared by carbonization in nitrogen followed by activation in carbon dioxide of chromiumtanned leather. The porous texture of carbons has been characterized by adsorption of N2 (77K) and isobutane (261K). There is a progressive increase in porosity upon activation with CO2 up to about 50% burnoff followed by a decrease for larger extents of burnoff. This is due to the progressive increase in the Cr203 content of the carbons. Both Xray diffraction and scanning electron microscopy show a homogeneous distribution of Cr203 particles through the carbons and in addition the mean particle size increases with burnoff. The CraC>3 particles block the entrance of the micropores of the carbon but at the same time they act as skeleton for the structure of the carbons which have appropiate mechanical resistance.
I. INTRODUCTION A previous report (1) has shown that chromiumtanned
leather can be used for the preparation of activated carbons having a large volume of macropores and equivalent surface areas around 1000 m2
/g. The carbons had a very high ash content, most of which were chromium compounds, and it should be admitted that this inorganic matter must somewhat affect the texture and adsorption properties of the carbons. Finding the role of the chromium compounds in these carbons is not an easy task since those compounds could be in the interior of the narrow pores. This paper presents the results found using different experimental approaches, in the study of the effect of the chromium compounds in both the structure and the adsorption properties of carbons prepared from leather. II. EXPERIMENTAL
A series of carbons covering the 770% burnoff range have been prepared from chromiumtanned leather by carbonization in nitrogen at 1173K for 5h followed by activation in carbon dioxide at 1098K for 460h. The nomenclature

453
of the samples includes Ρ followed by the burnoff in C02. The carbons have been characterized by adsorption of N2(77K) and isobutane (261K) using a conventional McBain type gravimetric system. Scanning electron microscopy (Philips 515) and Xray diffraction (Philips PWR 1840) were used in this study. 111. RESULTS AND DISCUSSION
The adsorption of N2(77K) and isobutane (26IK) can be used to show the development of porosity produced in the C02 activation process of the carbonized leather. Figure 1 shows the evolution of micropore volume obtained by application of the DubininRadushkevich (DR) equation (2) to the adsorption data as a function of burnoff. There is an increase in micropore volume from carbon P7 to carbon P58, decreasing thereafter; carbon P7 exhibits a small molecular sieving effect towards isobutane, as expected in a carbon with low burnoff (3). However, if the volume is expressed per gramme of ash free carbon, the micropore volume continuously increases. This different behaviour is not surprising considering the large ash content of the carbons, especially for high C02 burnoff. The char has 21% ash content and it increases (see figure 2) with activation time, especially for burnoff larger than 50%. Consequently, the high ash content for carbons P64 and P70 seems to be responsible for the drop in micropore volume observed in Figure 1. It should be noted that the ash content measured on the carbons is coincident (see Figure 2) with that determined theoretically by assuming that all inorganic matter of the leather remains during the activation process.
0,7
ο,β
E
0.2
, / 7 7 I
~ 1 — 20 8 0 40 BO
Bum—off rø
Figure 1. Variation of micropore volume with burnoff

454
80 70 60 50 4 0 H 30 20 IO 0
A THEORET IC VALUES ■ EXPERIMENTAL VALUES
20 40 60 00 BURNOFF (X)
Figure 2. Evolution of ash content with burnoff In order to understand the evolution of adsorption
capacity of the carbons for large burnoff it is important to know the chemical nature of the ash and its evolution during activation. Xray fluorescency and difraction, together with scanning electron microscopy (SEM) may provide the information required. The SEM pictures for the char obtained at 1173K show prismatic particles uniformly distributed (see Figure 3b); EDAX analysis showed that these particles were basically constituted by chromium. Xray diffraction for the char showed the presence of aCr203 crystals, being the only chromium compound in the carbons.
Figure 3 includes the SEM pictures corresponding to the leather, the char and one activated carbon. Figure 3a shows the fibrous aspect of the leather, with no Cr203 particles on the surface since the leather was treated during tanning with Cr(III) coordination compounds. When this leather is carbonized (Figure 3b) the surface shows the presence of porosity which is developed upon activation (Figure 3c); in both the char and the carbon Cr203 particles are seen in the pictures. The mean Cr203 particle size (calculated from the Xray diffractograms) increases with burnoff as shown in Table 1. Such an increase is more noticeable in the first stages of activation and it takes place to a lesser extent after 50% burnoff, when the increase in ashcontent is more important.
TABLE 1. Some data for carbons
Carbon Char P7 P25 P39 P58 P64 P70
d (nm) (2Θ=65.1) 26.8 26.8 29.9 33.8 34.9 36.1 37.4

455
The SEM micrographs of Figures 3b and 3c show a considerable concentration of Cr203 crystals around and inside the macropores. Since it is not possible to observe Cr203 crystals inside the small pores due to limitations of the technique, the influence of Cr203 on the adsorptive capacity of the carbons may be analysed taking into account the adsorption of N2 (77K) on the ashes obtained from carbon P70 which gives a BET surface area of 7 m2/g , this means that even although the ash content is very high for carbons with large burn-off the contribution to total micropore volume is very small (lower than 0.002 cm3/g). Since the micropore volume for carbon P70 is 0.21 cm3/g and 0.70 cm3/g if the carbon is considered to be ash free this large difference must be due only to blocking of the micropores by Cr203 crystals formed during the heat treatment taking place on activation in C02.
Figure 3. SEM pictures for leather and carbons

456
An additional test would be to extract the Cr203 from the carbons· This is not a simple task because after trying different chemicals and finding that KBr03 was the most effective, only 41% of Cr203 could be removed from the carbons. As a typical example, Figure 4 includes the X-ray diffractograms and Figure 3c and 3d the corresponding SEM pictures for carbon P58 before and after extraction with KBr03 respectively. There is a noticeable reduction in the number of Cr203 particles in the surface of the carbon after treatment and a reduction in crystalinity (the mean particle size decreases from 34.9 nm in P58 to 27.5 nm after partial extraction of Cr203). On the other hand, Figures 3c, 3d and 4 show that there is an important change in the texture, with a decrease in large porosity and some cracks after KBr03 treatment. In addition, the carbon has a much lower mechanical strength after extraction and the adsorption capacity is almost nil. This seems to indicate that although the Cr203 particles are blocking the micropores in the original carbon it also acts as a rigid skeleton maintaining the structure of the carbon.
P58
P58 after Cr„03 extraction
SO 40 2·
Figure 4. X-ray diffractograms for some carbons
REFERENCES
3.
MARTINEZ-SANCHEZ, M.A., ORGILES-BARCELO, C , MARTIN-MARTINEZ, J.M., RODRIGUEZ-REINOSO, F. and TORREGROSA, R., Int. Conf. Carbon. Newcastle upon Tyne. p.280. 1988. DUBININ, M.M. "Progress in Surface and Membrane Science". Ed. D.A. Cadenhead. Academic Press. New York. £, 1 (1985). RODRIGUEZ-REINOSO, F. "Carbon and Coal Gasification". Eds. J.L. Figueiredo and J.A. Moulijn. Martinus Nij-hoff, Dordrecht, p.601 (1986).

457
INFLUENCE OF THE POROSITY OF CARBON IN_Fe/Carbon CATALYSTS
J.M. MARTINMARTINEZ and M.A. vANNICE*
Departamento de Química Inorganica e Ingeniería Química. Universidad de Alicante. ALICANTE. SPAIN. * Department of Chemical Engineering. The Pennsylvania State University. University Park, PA 16802. USA.
Summary. Two activated carbons from olive pits were prepared by carbonization and subsequent activation in COE» for ¿f8 (carbon A) and 82 hours (carbon B> to produce different micro and macroporosi ty. Both carbons were used to support Fe3(C0)ie and both gave welldispersed Fe/C catalysts which retained small Fe crystallites under reaction conditions. Asprepared samples (no initial reduction) were active but a high temperature reduction at 673K increased the TOF for ΑFe whereas the TOF remained the same for BFe. Although catalytic behavior was similar in general, the BFe catalyst produced much less ChL,, which may reflect an effect due to microporosi ty.
1.INTRODUCTION. The suitability of iron catalysts to convert syngas to
hydrocarbons is well known (1). Bulk Fe is employed commercially, but iron supported on carbon <2>, silica (3) and alumina (4) has been frecuently studied. However, it is difficult to obtain very small Fe particles on oxide supports because the precursors do not reduce easily. Thus an inert support with no surface oxygen groups, such as carbon, may be atractive, especially when combined with the use of iron carbonyl clusters such as Fe3(C0)ie and Fe(C0)s (5). On the other hand, Jung et al. (2,6) found that carbons with large surface areas favoured the formation of welldispersed iron. Highly dispersed, unpromoted Fe/C catalysts can be prepared which have high activity and higher that expected olefin/paraffin ratios (6B). This indicated that the microporosity of the carbons might play a role in obtaining highly dispersed Fe (9). However, Kikuchi et al. (10) concluded that the iron particle size is not a strong function of the carbon surface area. Because there are controversial papers on this topic, the major aim of this study was an analysis of the influence of the porosity and the pore size distribution in carbon on the dispersion and kinetic behavior of carbon supported iron catalysts.
S. EXPERIMENTAL. It is well known that the activation of 1ignocellulosic

458
materials in C0 B or steam produces carbons whose porosity can be easily and gradually changed (11,12). Thus, olive pits were carbonized in N e at 1123K for 2 h., then activated in C0 e at 109BK for t*B h. (Carbon A yield = 71'/.) and 82 h. (Carbon Β yield = 36'/.). Both carbons were treated in H e at 1273K for 12 hours to remove sulfur. Carbons were granular with a particle size between 0.5 and 0.S mm. Carbons A and Β were charac
terized by N e adsorption at 77K in a high precision, high vacuum volumetric system. Mercury porosimetry complemented the analysis of the porous texture of the carbons.
Catalysts with 34 wty. iron were prepared using Fe 3(C0) l e cluster dissolved in dry, degassed tetrahydrofurane (THF). The carbonyl was dispersed on the carbons, which had been degassed at IO** kPa for 16 h at 673K, by an incipient wetness impregnation under anaerobic conditions. The samples were then evacuated at 300K to remove excess THF. The catalysts nomen
clature shall be ΑFe and BFe. The catalysts were reduced in H e at 673K for 16 h, then H e desorption (13) and CO chemisorption at 195K and 300K allowed the evaluation of Fe dispersion in both the fresh and used (after use in the syngas reaction) catalysts. It has been proposed that the optimum measure is CO adsorption at 195K to avoid carbonyl formation and CO dis
sociation and the use of an adsorption stoichiometry of CO/Fe» = 1/2 (2, 14) .
Kinetic data were obtained at 0.1 MPa, usually under differential reactions conditions, using a Perkin Elmer Sigma 3 gas Chromatograph. Both asprepared (APno reduction step) and high temperature reduced (HTR 16h at 673K) samples were studied. Each set of kinetic data was obtained after a 20 min. exposure to the reactant gases (Hs/C0 = 3 ) . The mass of catalyst used in each experiment was 0.4 g.
3. RESULTS AND DISCUSSION. Supports. Figure 1 shows the N e adsorption isotherms at
77K for carbons A and B. The application of the DubininRadushkevich (15) and the BET method (16) to the data gave the surface areas <S> and micropore volumes (Ve.) in Table 1, which also includes the C0 S micropore volumes. Mercury porosimetry curves are given in Figure 2. A cross sectional area of 0.162 nm
a was used for N e at 77K and its liquid density was 0.B08 g/ml. Carbon A shows a narrow microporosi ty (Ns and C0 B micropore volumes are quite similar), a poorly developed mesoporosity, and a small macroporosi ty; the distribution of porosity in carbon A is therefore very narrow. However, carbon Β was much more micro and macroporous than carbon A although the mesopore volume remained almost unchanged. Consequently the pore size distribution of carbon Β is well extended.
Chemisorption. Table 2 shows the H e desorption and CO uptakes (values have been corrected for adsorption on the support). In general, hydrogen adsorption gives low surface coverages on very small Fe particles (2,6,14). CO adsorption on Fe at 195K is tipically less than that at 300K because of the different stoichiometry and absence of carbonyl formation (2). Three of the four catalysts conform to this pattern, and the unusually high uptake on the BFe (fresh) sample may be due to CO retention in very small micropores during degassing.

459
Consequently the porosity of these carbons, especially the microporosi ty, does not affect to the iron dispersion significantly.
The used catalysts were also characterized by chemi
sorption; values are given in Table Ξ. Hydrogen desorption remains almost unchanged after the reaction and there is little change in CO chemisorption at either temperature for the ftFe catalyst, so the iron crystallites do not sinter during the syngas reaction. The C0/195K chemisorption is smaller than that of C0/300K for catalyst BFe, and the C0/300K uptake increases after the syngas reaction. The microporosi ty of the carbons seems to prevent the sintering of iron and when the porosity widens, the CO in these very small micropores may be removed more easily by evacuation. Small extents of gasification under reaction conditions could widen these pores. The absence of mesoporosity in the carbons might also play an important role on the distribution of iron, however, this point need a more detailed study (IB).
Kinetic behavior. The CO hydrogénation was carried out on both AP and heattreated iron catalysts. The most significant results are included in Tables 3 and ¿t. The AP catalysts have a lower activity for CO conversion to hydrocarbons than the HTR catalysts, especially for ΑFe. The APAFe catalyst shows a somewhat lower TOF (turnover frecuency values) than the APBFe sample, so the presence of wide porosity in the carbon may favour the initial iron particle size distribution in carbon pores because the TOF is very sensitive to Fe crystal
lite size (2,6). After HTR the ΑFe catalyst becomes much more active and has a larger TOF that the BFe sample. However, the rather high CO conversion levels may generate temperature gradients which have some effect on the activity. Regardless, the porosity of these carbons stabilizes the small Fe particles. Some of these differences in TOF values may due to different concentrations of active sites on the carbon surface, which can stabilize small Fe particles and provide a "dstructure" of Fe, which is zerovalent and superparamagnetic (17,SO).
There is an increase in activation energy for the catalysts after the HTR treatment, but the hydrocarbons chaingrowth parameter (a) at 5*»8K, calculated from the lineal plot of the AndersonShulzFlory equation (19), does not change for the BFe catalyst while that for the ΑFe sample increase somewhat. Select i vi t ies of the catalysts, shown in Table h, have been taken at similar CO conversions. The ΑFe sample is less selective to olefins and produces more methane than the BFe catalyst. Consequently the selectivity to olefin production is more favoured when the microporosi ty widens.
Conelusions. The porosity of the carbon as a support for iron can affect initial dispersion and stabilize small Fe particles. When the porosity is narrow a HTR treatment is beneficial and it increases the activity, and in some cases, it can increase the o lefin/paraffin ratio.Also, a widening of the porosity may lead to iron catalysts that produce less methane.
Acknowledgements. This work was supported by the USSpain Joint Committee for Scientific and Technological Cooperation.

460
REFERENCES. 1. Dry, Μ.E. "The Fischer-Tropsch Synthesis". Catalysis :
Science and Technology. J.R. Anderson and M. Boudart eds. Springer, Berlin. 1, 159 (1981). 2. Jung, H.J., Mulay, L.N., Vannice, M.A., Stanfield, R.M.
and Delgass, W.N., J. Catal. 76, 208 (1982). 3. Ida, T., Tsuiki, H., Ueno, Α., Tohji, Κ., Udagawa, Y., Iwai, Κ. and Sano, Η., J. Catal. 106. 248 (1987). 4. Ruckenstein, E. and Sushumma, I., J. Catal. 97., 1 5. Phillips, J. and Dumesic, J.Α., Appi. Catal. 9, 1 6. Jung, H.J., Walker Jr., P.L. and Vannice, M.A., J.
75. 416 (1982). 7. Jones, V.K., Neubauer,
Phys. Chem. 90., 4832 (1986). 8. Sommen, A.,
277 (1985). Schmitt, J
87 (1972). Kikuchi, E.
Stoop, F. and
L. and Walker Jr., P.L
and Morita, Y
L.R. and Bartholomew,
var der Wiele, Κ., Appi,
(1986). ( 1984) .
Catal. L . Η . , J ■
Catal.
Carbon 9, 791 (1971)
, J. Catal. 57, 27 (1979). Martin-Martinez, J.M., Molina J.D., Carbon 23. Martin-Martinez,
and Garrido, J., J.
and Butt J.
.L.
19 (1985). J.M. , Molina-
Colloid Interf.
lit. 9. 10, 10. 11. Rodriguez-Reinoso, F., Sabio, M. and Lopez-Gonzalez, 12. Rodriguez-Reinoso, F., Sabio, M., Torregrosa, R Sci . 106. 315 (1985). 13. Ameise, J.A., Schwartz, L.H. 95 (1981). 14. Venter, J., Kaminsky, M., Geoffroy, G J. Catal. 103. 450 (1987). 15. Dubinin, M. M. and Radushkevich, L. USSR 55, 331 (1947). 16. Brunauer, S., Emmett, P.H. and Teller Soc. 60, 309 (1938). 17. Chen, A.A., Vannice, M.A. and Phillips, J., J. Phys. Chem 91. 6257 (1987). 18. Martín-Martínez, J.M., and Vannice M.Α., To be published. 19. Snel, R. and Zwart, J., Appi. Catal. 22, 337 (1986). 20. Chen, A.A., Philips, J., Venter, J.J. and Vannice, M.A. J. Catal., In press.
B., J. Catal. 72.
and Vannice M.A.,
Proc. Acad. Sci.
, J. Am. Chem.
ΣνΡ (ml/g)
• A
2
" B y
. Nv _Av \
- \ \
ι ι Λ-~-ι 8 Ρ/ Po A logD
Figure 1 Figure 2

461
TABLE 1 Adsorption Data for the Carbons
Vc.DR (cm3
/g) VMercury poros. (cm3/g ) S
Carbon (mB/g) Na/77K C0B/273K mesopores macropores
159. Ξ 206.9
167.7 Ξ90.2
0.E9 0.29
0.32 0.41
0.47 0.86
0.49 0.55
A 1098 0.39 0.37 0.08 0.23 Β 1420 0.51 0.40 0.10 0.46
TABLE 2 Chemisorption on Fe/C Catalysts (pmDle/gc.t) (after 16h at 673K in H B flowing at 40 cm3
/g)
Fe Catalyst wt% He C0/195K C0/300K CO/Fe D(Fe./Fe„ )"=
ΑFe (fresh) 3.12 15.7 132.1 BFe (fresh) 4.00 27.0 310.2
ΑFe (used) 3.12 17.2 136.4 BFe (used) 4.00 29.8 196.5
<> At 300K. <<"■ From C0/195K, CO/Fe. = 0.5.
TABLE 3 Catalytic Behavior of Fe/C Catalysts (P = 0.1 MPa, H E / C O = 3, Τ = 54BK)
A <nmole/gF„.s) CO TOF CGconv. to HC E
Catalyst HC C/.) C0 B HC CH<* ( s"1. IO3
) ( k J/mole) α
8.37 6.74 2.80 1.3 63 0.46 21.99 19.68 4.38 3.8 65 0.55
60.16 56.73 13.62 9. 9t> 87 0.56 35.98 28.08 6.05 3.8·=· 87 0.57
*■»» Based on C0/300K values on fresh samples. "■* Based on C0/195K values on used samples.
TABLE 4 Select i vi t ies in the CO Hydrogénation Reaction
(Ρ = 0.1 MPa, He/CO =3)
H C
APAFe APBFe
AFe BFe
2.24 7.62
18.86 10.88
Catalyst
APAFe APBFe
AFe BFe
T(K>
548 523
508 538
HC C/.)
2.24 3.86
12.43 9.25
Cl
64 46
67 44
C2
1 1 4
5 10
C2
15 18
12 13
C3
4 e 7 13
C3
2 8
3 3
C4
2 9
3 8
C5
2 8
2 9
(C2+C3)
0.88 0.46
0.80 1 .44

462
DEVELOPMENT AND CONSTRUCTION CF A SAMPLING LINE FOR WOOD PYROLYSIS EMISSIONS
J. LACHENAL and J.M. TOLEDO Laboratoire National d'Essai, 5 rue Enrico Fermi, 78190 Trappes, France
C. MEZERETTE and A.M. VERGNET Centre Technique Forestier Tropical, Departement du C.I.R.A.D.
45bis, Avenue de la Belle-Gabrielle, 94736 Nogent sur Marne Cedex, France
Summary
In France and throughout the world, charcoal-making equipment mainly consists of partial combustion kilns with no provision for smoke recovery. Thus, 70% of the initial mass is discharged into the atmosphere. This situation results in atmospheric pollution and poor energy conversion yields.
At the request of the French Ministry of Environment, the CTFT and the LNE undertook a study to characterize pyrolysis emissions, in order to determine pollution resulting from charcoal-making. Initial experiments showed that standard methods for emission sampling were not appropriate for pyrolysis effluents. Accordingly, the LNE and the CTFT jointly developed a system specifically designed for the isokinetic sampling of a representative fraction of the emission. In this method, gases and condensable compounds (pyroligneous matter and tars) are separated for subsequent chromatographic analysis and their relative proportions are quantified. The flow velocity is monitored continuously in order to regulate the sampling flow.
BACKGROUND
The thermochemical reduction of wood to charcoal using a partial combustion kiln produces a large amount of fumes in the immediate vicinity of the kiln.
At present the French Ministry of the Environment is revising the legislation on charcoal burning (formerly classified as 104 in the nomenclature of classified establishments) and is seeking to harmonize the permitted discharge thresholds for each pollutant (or family of pollutants) in various industrial processes.
There does not appear to be any European legislation as yet in this field.
The equipment for producing charcoal mainly comprises partial combustion kilns which do not recover either the smoke or the other waste products released during the pyrolysis of the wood, which are no longer used since the advent of synthetic chemistry.
EFFECTS OF THE NEW REGULATIONS
The regulations lay down waste product thresholds for dusts and gases (in effluents discharged into the atmosphere) and for liquid effluents. At

463
present it is impossible for all French production units to comply with them.
The only known way of complying with these thresholds would be to install incinerators for burning all the constituents of the smoke. However, this solution has two disadvantages, viz :
its cost, which restricts it to installations above a certain size; the need to know in advance some of the properties of the raw effluents so as to determine the combustion chamber size.
Purification technology specific to this industrial sector must be developed if the great majority of the existing plants in France are to survive.
There have already been some interesting pilot projects, such as the electrofilter with a water film.
However, if we are to develop new equipment and check its effectiveness we must acquire a perfect understanding of the metrological techniques for the pollutants concerned.
JUSTIFICATION FOR THE WORK
In France, the measuring of waste by weight is clearly defined in two standards (AFNOR X43-003 and X44-052) (1, 2). There is a sampling apparatus specific to each of these standards (see Figures 1 and 2). They both work on the principle of a heated filter medium located at the head of a suction line, which is of known capacity and adjustable so as to maintain isokinetic conditions during sampling.
These two items of apparatus were used for a series of on-site measurements made during 1986 to quantify the effluents from charcoal production. This project was the preliminary to a large-scale programme to study the nature of the fumes. Since no precise numerical data were available at the time, the purpose of the project was to obtain information prior to drawing up regulations and designing new processing systems.
The programme was carried out by a number of scientific bodies, the most prominent being :
Centre Technique Forestier Tropical (CTFT) Laboratoire National d'Essais (LNE) Electricité de France (EDF) Centre Interprofessionnel Technique d'Etudes de la Pollution Atmosphérique (CITEPA)
Various papers were published as a result of this project (3, 4, 5). The tests comprised sequences of sampling and analysis during each of
the known phases of a carbonization cycle in a partial combustion kiln. However, difficulties were encountered in obtaining reproducible and completely representative samples for each of the phases under observation. A critical examination of the means and methods used produced the following findings :
With regard to the kiln:
The fume output is of low velocity (2-3 m/s) , and there is a substantial liquid discharge (water or pyroligneous liquid).
As a result, measuring the gas velocity with a Pitot tube and a manometer unit generally produces figures which are too high or completely incorrect, since the orifices for measuring the pressure become clogged.

464
With regard to the sampling apparatus (Figures 1 and 2) :
1' - Probe 2' - Cyclone 3' - Filter 4' - Finned cooler 5' - Electric heating unit
Ι9Γ Condenser Dehydrator Spirometer Manometer Flow regulation valve Flow regulation unit Counter (optional) Thermometer
F i g . 1 . Diagram i l l u s t r a t i n g AFNOR X 43003 method
Diaphragm Regulating | / va lve
j — τ
Fig. 2. Diagram illustrating AFNOR X 44052 method
Since the gas is transferred from the inside of the chimney to the filter medium through a heated tube (a probe) with a nozzle at the open end, it was noted that :
the heating is insufficient to convert the water into vapour before it reaches the filter medium, so that it is filtered very quickly, the sampling period being less than five minutes ;

465
this heating affects the products sampled and radically modifies their proportions, producing cokéfaction and deposition on the inner surface of the probe; the very nature of the sampling equipment prevents recovery of the condensates, except with the first apparatus (see French standard X43-003). However, as mentioned above, the figures for the material recovered are too low and distorted.
With regard to the gas sampling line:
This was installed to process the gases from the kiln before they are analysed by chromatography (CPG) (4). The apparatus consisted of a succession of condensers. It quickly became apparent that the quantity of products to be processed required greater volumes and that the rapid cooling of these gases (heavy tar-like hydrocarbons) quickly clogged the sampling line and modified the measured outputs used for calculating the mass flows.
The findings from this series of preliminary tests are therefore of two types :
we obtained a numerical evaluation of the main waste products discharged into the atmosphere from partial combustion kilns; we learned that it is essential to have sampling methods specific to the effluents given off during wood pyrolysis.
DEFINITION OF THE PROJECT
To meet this need for a reliable metrologicai tool to quantify the gaseous discharges from these carbonizing kilns, close collaboration was set up between the CTFT and the LNE.
A project was drawn up on the basis of the observations made during the preliminary tests to determine the main characteristics of the ideal apparatus, and it is for the two laboratories to test the options as they are selected.
This apparatus must :
sample a known volume of gas for a sufficiently long period to be representative of the emissions from the kiln, which might be from 90 to 120 minutes; separate the condensates from the rest of the gaseous fraction and record their relative proportions in the effluents ; guarantee isokinetic conditions during sampling; make it possible to quantify the constituents of the condensates, tars and other organic pollutants (6) .
In addition to operating this sampling line, it appears essential to measure the velocity of the fumes, the relative humidity of the gases and, if possible, their composition, to determine precisely the isokinetic calibration.
Lastly, the analysis of the condensates must be based on the whole of the sample, without losses due to difficulties in cleaning the equipment.
THE SAMPLING LINE
The preliminary tests quickly revealed the appropriate location for each part of the line :

466
a sampling probe, of small diameter and angled at the end, not heated but insulated; as small a transfer line as possible, also insulated; a condensation unit; a flat filter maintained at a certain temperature, with a porosity compatible with the flow regulator; a device for regulating the intake volume; an airtight pump; a dry gas counter.
The condensation unit must be able to trap all the liquid and particulate phase: water, organic pollutants (acids, methanol, furfurylic compounds, phenolic compounds and tars). Various contact elements such as Raschig rings and glass spheres were not satisfactory (incomplete recovery of the sample) and were abandoned in favour of vessels of the simple washer type followed by sinteredplate vessels. These collected all the tars. An electrostatic filter was also tried, but although this process traps the components satisfactorily, it also alters them by oxidation, and was therefore not adopted.
The flat filter must trap material by filtering the gases after condensation. Its retention capacity must be high. Tests showed that 0.5U was sufficient. Its chemical composition should not affect the products which pass through it. Glass fibre or PTFE are suitable.
If condensation is optimized, this filter becomes the indicator of the efficiency of the upstream processes (in general the fraction recovered is equivalent to 1 0
3 % of the condensate) . It is simply maintained at a temperature too high for residual condensation, which might cause abnormal clogging of the filter.
The purified gases are sampled in sequence downstream of the counter. The pressure and temperature parameters are also recorded to correct the volumes extracted.
The sampling line in its final form is represented diagrammatically in Figure 3.
Temperature sensor Filter / F
1™ »x*«· \ Gas
holder I I ""i" , Simpli no
Gas meter Manometer * 1
Γ"
9 Μ pump
Compressed air
J Chimney flow Trap for condensed particles measurement
Regulation for isokinetic sampling
Fig . 3. Diagram i l l u s t r a t i n g the sampling l i n e for pyro lys i s emissions

467
This was validated in the course of several sessions of measuring the emissions from a Magnientype partial combustion kiln with a capacity of 2 m3 (7) . The task was to improve the calculation of the energy and mass balances by including losses due to waste discharged into the atmosphere. The results obtained made it possible to verify the theoretical balance to within 5%, which confirmed that the line was suitable for sampling and gave reproducible results.
The velocities were measured using a pitot tube linked to a micromanometer. A sequential declogging device was used to monitor these data almost continuously and to deduce from them the kinetics of the flow.
Humidity is measured regularly by an ancillary condensation device. A set of adjustable nozzles at the head of the probe are used to determine the precise calibration for the offtake (isokinetic). This calibration is readjusted constantly during the sampling, which may continue for several hours. It is preferable to carry out a series of samplings in order to quantify the flows more accurately as pyrolysis proceeds.
CONCLUSION
This sampling line for wood pyrolysis emissions operated jointly by the CTFT and the LNE can be used:
to study the kinetics of the occurrence of pollutants as carbonization proceeds ; to determine the chemical properties of the pollutants emitted; to quantify accurately the mass discharges from the pyrolysis plant (during carbonization and roasting) and to deduce from these the emission factors ; to check the efficiency of the waste treatment systems.
So far a large number of measurements have been carried out on site and confirm the suitability of the layout we have adopted.
REFERENCES
(1) Norme Française Homologuée Χ 43003 Methode de détermination ponderale des particules solides entraînées par les gaz de combustion.
(2) Norme Française Homologuée X 44052 Prélèvement de poussière dans une veine gazeuse.
(3) CITEPA monographie: Carbonisation du bois et pollution atmosphérique 1986.
(4) MEYER, C. Determination des rejets atmosphériques lors de la carbonisation partielle. Mémoire de fin d'études. CTFT 1986.
(5) GIRARD, P., MEYER, C , FONTELLE, J. P. and BOILLOT, M. Caractérisation des émissions de carbonisation en four à combustion partielle. Mise au point de systimes de traitement des fumées de carbonisation. 4ème Conference Européenne 1115 May 1987, Orléans.
(6) VILLENEUVE, F. and VERGNET, A.M. Techniques analytiques applicables aux liquides at aux gaz de pyrolyse de la biomasse tropicale. Bois et Foret de Tropiques, cahier scientifique no 9 1988.
(7) SHAH, N., GIRARD, P., MEZERETTE, C. and VERGNET, A.M. Engineering performance evaluation to characterise metal charcoal kiln, for optimisation of wood energy conversion. 5th European Conference: "Biomass for Energy and Industry", 913 October 1989, Lisbon.

468
FURNACE FOR BIOFUKLS THERMAL UTILIZATION W.BLASIAK,Β.ZETHRAEUS,R.COLLIN Royal Institute of Technology
Department of Heat and Furnace Technology S-10044 Stockholm, Sweden
W.GAJEWSKI,J.ZAJDEL Technical University of Czestochowa
Institute of Heat Machinery 42-200 Czestochowa, Poland
Summary The work presents a new type of wood wastes incinerator. Incinerator can be used to utilize different kinds of wood wastes which might be contaminated by pieces of noncombustable wastes like bricks, stones or metallic parts. Low quality non-uniform wood wastes can be burnt in the incinerator without any previous separation, segregation or cleaning process. The incinerator prototype as a typical combustor was investigated using a sectional hot water boiler as a waste heat boiler.
1. INTRODUCTION Two of the applicable utilization methods for wood wastes are combustion or gasification. However, simultaneously the heat from the combustion process must be used in the existing factory heating system. In case of wood wastes and if we can not expect very efficient preprocessing systems (as is normally the case in small factories) the combustion process is difficult to run mainly due to different wastes structure, big water content depending on the season, and due to big contents of non combustible waste like sand, stones, bricks, metallic parts and so on. Taking into account these circumstances and the fact that the waste composition changes within a very wide range even during one day it is obvious that any kind of incierator will have to work in very difficult conditions from the point of view of the combustion process stability.
2. DESIGN OF THE COMBUSTOR The combustor, shown in Fig 1, consists of a vertical cylindrical combustion chamber (1), a conical ash chamber (2), a chamber for initial drying of wastes (3), a flue gas offtake (4) and an external recirculation channel for the hot flue gas (5). Raw wastes are fed gravitationally by means of a standpipe (6) into the initial-drying chamber and finally into the combustion chamber. Air is supplied as primary air, secondary air and complementai air.

469
Primary air is tangentially supplied into the upper part of the combustion chamber. Secondary air is supplied by a suitably shaped channel into the upper part of the drying chamber, being simultaneously the driving force for the swirl in the waste entry. Complementai air can be supplied into the ash chamber in order to burn off the coarse fraction of wastes. To ensure a correct pressure distribution inside the combustor, guaranteeing the possibility of gravitational feeding, it is very important to keep proper velocities and air flow ratios for given geometrical dimensions of the combustor. Especially it is necessary to avoid overpressure near the standpipe inlet. The simple design of the combustor gives possibility to apply a simple gravitational feeding system which is also its main advantage. Wastes supplied through the standpipe (6) undergo a process of segregation i.e, the fines are set in rotational motion and the rest of them falls down. The wastes are dried, degasified and partly burnt up during the time of falling down. The fines are burnt up mainly inside the drying and combustion chamber during their more or less rotational motion, while the coarse wastes are burnt up in a fixed bed on the distributor (11) inside the ash chamber. In the latter case oxygen supply is both by secondary air and by complementai air which infiltrates through the fixed bed. In case of an even more simplified design of the combustor the ash chamber can be eguipped with an ash door (10) which can be used for ash removal and for adjusting the amount of air sucked into the ash chamber. In case of high moisture content of the wastes recirculation of flue gas by means of the secondary air flow is realized. Flue gas is ejected by the jet of secondary air (8) flowing freely through the flue gas channel (4). This kind of design in principle works as a jet pump. The amount of flue gas recirculated in this manner can be adjusted by changing the secondary air nozzle (8) position. To utilize the heat of wastes combustion the combustor was connected to the internal heating system of a small sawmill factory. The new combustor is combined with the existing boiler so as to maintain the same total performance of the whole heating system as before the new combustor installation, using simultaneously much less of the basic fuel.
3. WASTES PROPERTIES Typical properties of the wastes (sawdust, chips of pinewood and sprucewood) utilized in the small industrial scale prototype of the combustor are presented below:
Chemical constitution of the dry substance: C=0.5, H=0.96, 0=0.439, N=0.001;
- Moisture content: W=0.2-0.5; Ash content: A=0.01; Net calorific value: Qw=(7.4-13.4) GJ/kg; - Bulk density gw=(150-180) kg/m3 Total content of volatiles 0.75-0.8.

470
4. PROTOTYPE INSTALLATION OF THE COMBUSTOR IN THE HEATING SYSTEM.
The hot water boiler was used as a waste heat boiler utilizing the heat in the gases coming out of the combustor. The flue gas was led from the exhaust of the combustor via ducting into the boilers chamber. To achieve the neccessary heat output in order to maintain the boiler rating the changes in combustion orgainzation and in the heat transfer processes had to be considered. The hot water boiler was characterized by the following parameters; type of boiler - hand firing sectional hot water boiler, calorific throughput - 0.3 MW, heating surface -32 m2, water pressure - 0.5 MPa, water temperature in the outlet and inlet respectively 95°C and 70°C, outlet flue gas temperature - 200°C, thermal efficiency of the boiler, -0.79, kind of basic fuel - coke (net calorific value - 29 GJ/kg), fuel consumption - 47 kg/h. Calculated total flow of flue gas was 570 nm3/h (excess air factor 1.5). On the basis of the chemical composition of the wastes the amount of air and flue gas for the same value of excess air factor were determined as respectively 4.7 nm3/kg and 5.6 nm3/kg.
MS^h
air I
Fia 1 : Scheme of the combustor

471
Taking into account heat losses due to radiation (sr=0.2) and due to incomplete combustion (s¿n=0.05) the temperatue of the wastes combustion was found using the relation between the flue gs enthalpy (Jfg) and temperature. The calculated values of the adiabatic waste combustion temperatures, t c w are presented in table 1 : Table 1 η (excess air)
J f g [kJ/kg]
tcw [°c]
1.3 8757
1130
1.5
8773
1000
2.0
8812 800
2.5
8857 700
Assuming that the flue gas is cooled to the outlet temperature 200°C and the total thermal throughput of the boiler Qj_, will be kept up as well, the wastes consumption Bw was calculated and presented in table 2, where a is the ratio between flows of flue gas for wastes and coke. Table 2 η
Qb [MW] % [kg/h] V
wf g [nm
3/h]
a2
1.3
0.3
146
727 2.1
1.5
0.3
149
836 2.4
2.0
0.3
158
1140 3.5
2.5
0.3
166
1466 5.1
As shown by a2 in table 2, a retrofit to 100% waste firing would dramatically increase the pressure drops and flue gas volumes. To avoid a change of draft fan in the flue gas system, the boiler was therefore cofired with a coke supply of (1517) kg/h (previously 47 kg/h) and the remainder of the fuel input supplied by wastes. 5. RESULTS OF COMBUSTOR PROTOTYPE RUNNING According to the calculations it is possible to run the combustor together with the hot water boiler only if the combustion in the boiler is run by means of coke (1020 kg/h). In this situation using quite dry wood wastes w=0.25 (experiments were carried out during summer) it is possilbe to achive the rated output of the boiler. If there is no coke combustion in the boiler the temperature is too low for the combustion of fines transported by flue gas from the wood wastes combustor. Running parameters of the combustor during experiments are presented in table 3.

472
Table 3 Amount of wood wastes 96 kg/h Temp, of flue gas in the outlet of the combustor 1000°C Excess air number 1.5 Air flow (total) ( 4.7-5)nm3/kg Flue gas flow from the combustor 5.7 nm3/kg Net calorific value of wood wastes 10.5 GJ/kg Moisture content 25 % Amount of coke (10-20) kg/h Because of the higher flue gas flow some troubles with a slight overpressure inside the boiler were noticed. This was due to the too small capacity of the draft fan which was running under maximal performance. It is necessary to stress that no changes were done in the existing system. Also some fluctuations inside the combustor occurred especially during startup. After some time the heat accumulated in the combustor walls and mass of wastes accumulated inside the ash chamber protected very sufficiently the stability of the combustion process.

473
INVESTIGATIONS OF TOXIC COMPONENTS IN PRODUCTS FROM MUNICIPAL WASTE SEWAGE SLUDGE PYROLYSIS
DIPL. CHEM. H. ROSSLER, DR U. PROSCH, PROF.DR W. KAMINSKY Universität Hamburg
Institut Fur Technische und Makromolekulare Chemie D-2000 Hamburg 13, Bundestrasse 45, Federal Republic of Germany
1. Summary
An increasing amount of municipal waste and sewage sludge has led to new efforts in waste removal management since appropriate recycling technologies are not available on an industrial scale at the moment. The pyrolysis process developed in Hamburg, using a fluidized bed reactor, achieves the conversion of organic waste material into gas, oil and solids. The fluidized bed reactor with a capacity of 20 to 50 kg/h is indirectly heated by four radiation tubes. The pyrolysis gas is partly condensated in packed columns; the remaining part is used for fluidizing the quartz sand bed. The excess gas leaves the plant continuously and is burnt in a torch. The content of toxic components such as PCBs, PCDDs, PCDFs and heavy metals in pyrolysis products determine the suitability of the process and utilization of the pyrolysis products. Our investigations concentrate on the analysis of pyrolysis oils in relation to the highly toxic components such as PCBs, PCDDs and PCDFs, as well as the analysis of solid products relating to heavy metals.
2. PYROLYSIS OF MUNICIPAL WASTE
2.1 Characterization of Educt and Material Balances
The source of the material used is an urban collection of municipal waste in Hamburg-Bergedorf. Table 1 shows the composition of the plastic waste in relation to several classes of plastic; Table 2 elucidates the elementary composition.
TABLE 1. (in we
Polyolefines Polystyrene PVC Other plastics
iight
Inorganic residues Water
Ì' 57 19 13.7 4.8 5.3 0.2
% % % % % %
TABLE 2.
Carbon Hydrogen Chlorine Nitrogen
(in weight
Other elements
Ì' 79. 11 7. 0. 1.
.1
.8
.5
.6
% % % % %
The high content of PVC respectively chlorine is obvious. Therefore we expected the formation of chlorinated organic compounds. We carried out three pyrolysis experiments altogether at temperatures of 690°C, 735°C and 790°C. Table 3 shows the material balances of the experiments performed.

474
TABLE 3. Pyrolysis products (in weight %)
Temperature
Hydrogen Methane Ethene Ethane Propene 1,3-Butadiene Alicycles Other aliphates < C6 Other aliphates > C7 Benzene Toluene Styrene Other Alkylbenzenes Indane Indene, Methylindenes Naphthalene Alkylnaphthalenes Biphenyl, Alkylbiphenyl Anthracen, Phenanthren Other aromatic compounds Tars Carbon black Carbon monoxide Carbon dioxide Water HCl Other components Inorganic ash Products from Dolomit Sedimentation, loss
Sum Mass of pyrolysed waste
680°C
0.195 4.895 5.378 2.96 4.058 0.79 3.271 4.794 6.557 3.274 1.818 5.278 1.046 0.178 0.713 0.381 0.188 0.238 -0.123 17.403 4.159 2.45 5.973 2.5 7.98 2.516 5.27
-5.8 11.414
100 165.6 kg
735°C
0.318 10.563 9.644 3.351 3.648 0.949 2.797 1.886 0.015 7.933 3.244 6.047 2.969 0.221 1.274 1.372 0.672 0.284 0.4 0.82 10.357 5.583 6.589 9.699 4.0 7.98 1.202 5.27
-14.6 5.497
100 135.15 kg
790°C
0.703 17.496 9.782 2.992 1.324 0.442 0.55 0.334 0.006
13.783 5.555 3.105
-4.283 0.144 1.342 3.836 0.899 0.575 0.658 2.24
11.152 6.435 2.377 1.273 2.647 7.98 1.53 5.27
-1.2 1.106
100 130 kg

475 2.2 Analysis of Toxic Compounds
In spite of the high percentage of PVC respectively chlorine, the concentrations determined are below the detection limit, at least within permissible limiting values. Table 4 shows a prospect of PCDD, PCDF and PCB concentrations in high boiling pyrolysis oils (values in pgAg) · In low boiling oils PCDD, PCDF and PCB are not detectable.
TABLE 4. PCDD, PCDF and PCB concentrations in high boiling pyrolysis oils (values in yg/kg) ; (n.d. = not detectable)
Temperature 680°C 735°C 790°C
Sum
Sum
Sum
Sum
Sum
Sum
Sum
Sum PCB
TCDD
PeCDD
HxCDD
HpCDD OCDD TCDF 2,3,7, PeCDF 2,3,4, HxCDF 1,2,3, HpCDF
8
7
6
2,3,7,8, TCDD
1,2,3,7,8
1,2,3,6,7, 1,2,3,7,8, 1,2,3,4,7,
TCDF
,8 PeCDF
,7,8 HxCDF
PeCDD
,8 ,9 ,8
according DIN 51527
HxCDD HxCDD HxCDD
n.d. n.d. n.d. n.d.
n.d. n.d. n.d. 2.1 1.7
16.9 0.38
13.7 0.39 3.2 0.3 5.3
1500
n.d. n.d. n.d. n.d.
n.d. n.d. n.d. n.d. n.d. 4.5 n.d. 1.5 n.d. n.d. n.d. n.d.
<1000
n.d. n.d. n.d. n.d.
n.d. n.d. n.d. n.d. n.d. 14.1 n.d. 4.6 0.2 0.4 n.d. n.d. 162
The distribution of heavy metals in pyrolysis products demonstrates the abundance of heavy metals in solid products. The investigations were carried out by atomic absorption spectroscopy.
TABLE 5. Distribution of environmental heavy metals (% f.s. = % of whole feedstock)
Temperature 680°C mg/kg % f.s.
735 °C mg/kg % f.s.
790CC mg/kg % f.s.
Distribution of lead
Reactor residual Cyclone solids Slurry fraction High boiling oils Water Organic distillates Feedstock : Municipal waste Quartz sand Added substance
547 779 485 10 0.9 0.2
407 0.5 2.7
113.4 20.1 8.5 0.8 0.003 0.05
99.7 0.12 0.18
202 1000 537 23.1 0.2 0.2
407 0.5 2.7
49.8 25.2 15.7 1.4 0.001 0.06
99.64 0.13 0.24
163 805 19 42 0.3 -
407 0.5 -
52.8 18.5 0.3 2.8 0.001 -
99.84 0.16 -

476
TABLE 5. (Contd.)
Temperature
Distribution of cadmium
Reactor residual Cyclone solids Slurry fraction High boiling oils Water Organic distillates Feedstock : Municipal waste Quartz sand Added substance
mg/kg
4.4 117 210 10 0.3 0.05
89 0.02 0.9
3. PYROLYSIS OF SEWAGE SLUDGE
680 °C % f.s.
4.2 13.9 16.8 1.1 0.01 0.05
99.75 0.02 0.23
735°C mg/kg
0.4 49 220 10 0.14 0.15
89 0.02 0.9
% f.s.
0.5 5.6 29.4 2.7 0.002 0.2
99.63 0.03 0.35
790 mg/kg
0.3 54 73.9 29.1 0.08 -89 0.02 -
°C % f.s.
0.4 5.7 4.8 8.8 0.00 -
99.97 0.03 -
We carried out three experiments in pyrolysis of sewage sludge at temperatures of 620°C, 690°C and 750°C in our miniplant. The percentage of chlorinated biphenyls in the feedstock according to Ballschmitter (2) is demonstrated in Table 6.
TABLE 6. Polychlorinated biphenyls
PCB Isomer
in sewage sludge
mg/kg
28 0.07 52 0.04
101 0.06 138 0.08 153 0.09 180 0.04
Table 7 shows the percentage of polychlorinated biphenyls in the high boiling pyrolysis oil of the experiment at 690°C.
TABLE 7. Polychlorinated biphenyls in pyrolysis oil
Isomer (detection limit 0.
Trichlorbiphenyl Tetrachlorbiphenyl Pentachlorbiphenyl Hexachlorbiphenyl Heptachlorbiphenyl Oktachlorbiphenyl Nonachlorbiphenyl
.05 mgAg) mg/kg
0.292 0.331 0.447 0.317 0.097 n.d. n.d.

477
ÍOO"
„ 75.
4J Id u
o
50-
25-
93.5%
Pb
4.4% 0.4%
Charge Residue Soot Oil
Fig. 1. Distribution of lead in sewage sludge pyrolysis Pyrolysis temperature: 690°C

478
Table 8 illustrates the contents of PCDD and PCDF in sewage sludge feedstock and in high boiling oils in the 690°C and 750°C experiments.
TABLE 8. PCDD and PCDF in high boiling pyrolysis oils (values in yg/kg)
Components Feedstock 690°C 750°C
Sum TCDD 2,3,7,8 TCDD
Sum PeCDD 1,2,3,7,8 PeCDD
Sum HxCDD 1,2,3,6,7,8 HxCDD 1,2,3,6,7,8 HxCDD 1,2,3,4,7,8 HxCDD
Sum HpCDD 1,2,3,4,6,7,8 HpCDD OCDD
Sum TCDF 2,3,7,8/2,3,6,8 TCDF
Sum PeCDF 1,2,3,7,8/1,2,3,4,8 PeCDF 2,3,4,7,8 PcDF
Sum HxCDD 1,2,3,4,7,8/1,2,3,4,7,9 1,2,3,6,7,8 HxCDF 2,3,4,6,7,8 HxCDF 1,2,3,7,8,9 HxCDF
Sum HpCDF 1,2,3,4,6,7,8 HpCDF 1,2,3,6,7,8,9 HpCDF OCDF
0.03 n.d.
0.15 n.d.
1.64 n.d. 0.17 0.005
3.2 1.6
15.5
0.122 0.02
0.166 0.012 0.10
0.231 0.01 0.01 0.01 n.d.
0.7 n.d. 0.4 1.0
8.75 0.88
31.50 4.16
23.14 0.54 2.92 2.10
3.4 1.8 0.4
2.58 0.26
2.38 0.14 0.10
0.40 0.04 0.06 n.d. n.d.
n.d. n.d. n.d. n.d.
8.16 0.67
33.40 3.50
13.2 0.46 1.82 1.37
5.5 3.0 2.7
2.72 0.27
2.16 0.24 0.12
0.58 0.06 0.05 n.d. n.d.
n.d. n.d. n.d. n.d.
The distribution of heavy metals is analagous to the distribution of municipal waste pyrolysis. Figure 1 illustrates the distribution of lead.
REFERENCES
(1) U. PROSCH, doctoral thesis, University of Hamburg, 1988. (2) A. KUMMER, doctoral thesis, University of Hamburg, 1989.

479
RESEARCH ON TAR CRACKING AND APPLICATION OF TAR.
G. Olsen Laboratory for Energetics Technical University of Denmark
SUMMARY Straw has been pyrolysed at 600°C in a continous reactor
with s feed rate of 3 kg/h. The tar produced has been fed through an empty reactor of varying size and temperature and finally the gas was led through four washing bottles where the condensate was accumulated. The tar fraction with boiling point between 100°C and 200°C was separated by distillation and the amount, heat of combustion and viscosity were analysed. The tar fraction with boiling point 100°C - 200°C as well as the total amount of condensate is reduced by increased cracking temperature and the residue turns from sticky tar to powderish carbon black. The gas volume increases with increasing temperature and increasing residence time. The gas composition is only dependent on the cracking temperature.
BACKGROUND Pyrolysing biomass produces incinerable gas, char and tar
in various amounts according to the pyrolysing conditions. The char can be gasified and the tar cracked thermally or cataly-tically into gas and carbon .black, leaving ashes as a residue. Instead of converting the biomass energy only into incinerable gas by this procedure, the possibility of producing liquid fuels is of great interest, as these fuels have a wider range of applications and are easier to store and distribute. As a part of the biomass gasification research programme
at the Laboratory for Energetics, the possibility of producing applicable tar by pyrolysing straw has been investigated. The tar has been compared to diesel oil, as it was meant to mainly substitute this fuel in internal combustion engines if possible.

480
EXPERIMENTS The experiments were performed to give information about
the amount and quality of tar according to cracking temperature and residence time. The tar was produced by pyrolysing straw at 600 °C, 1 atm in a continuous reactor with a feeding rate of 3 kg straw/h. The gas and tar were fed through an empty reactor of varying size and temperature and finally washed in acetone in four successive washing bottles. The system was dismantled between each experiment in order to measure the amount of char produced. The secondary reactors had the volumes of 1.5 1 or 10 1 and the temperature of the same reactors was set to 600, 800, 900, 1000 and 1100 °C.
Second reactor
Feec
==H ' '" t
ing of sti
x 1 L — V
*aw
ρ π _ _—
is t u
Piston Pyrolysis reactor
Condenser
Four gas washing bottles in series
Collection of char
Figure 1: Test equipment. From the beginning it was quite clear that untreated tar
was not well suited as fuel for internal combustion engines. For this reason the condensate dissolved in acetone was distilled at 1 atm and the fractions with boiling point from 100200°C were separated and weighed, as was the residue. Initially it has not been necessary to know the composition of the distillates, whereas the heat of combustion and the viscosity of the distillates were more relevant and these analyses were therefore performed. The gas volume as well as the gas composition was
measured for each experiment and the heat of combustion has been calculated for the gas. The part of the experiments in which the tar was produced
and collected lasted approximately 2 hours and in this period no problems concerning the experimental equipment were observed. The condenser installed prior to the washing bottles was cleaned between each experiment. At low temperatures of the secondary reactor, adhesive tar condensed and started to close the condenser. When the

481
temperature was increased to 800°C and 900°C, soot started to build up in the upper part of the condenser, whereas the condenser was clean at the very high temperatures. The losses are found as condensed tar in the char
collector and tar leaving the washing bottles. The water and water soluble tar as well as the possible acetone produced were not measured. The colour of the condensate dissolved in acetone in the
washing bottles, turned from brown to black when the temperature was increased. At 1100°C the condensate consisted of carbon black and a colourless, transparent liquid.
RESULTS The amount of total condensate collected in the washing
bottles is reduced when the temperature of the second reactor is increased as seen in Figure 2. It is remarkable that the total amount of condensate does not exceed 6% of the inserted straw.
Volume of second reactor: 1.5 1
i ι (g/kg straw)
50
ΊΟ
3θ
20
10 -
Total condensate
■H^-f ţ *■ (°C) Temp. 600 70O 8O0 900 1000 1100
Figure 2a: Distillates and residual versus temperature.

60 ■
482
Volume of second reactor: lo 1
(g/kg straw)
Total condensate
» ( c) Temp. O 6O0 700 800 900 1000 1100
Figure 2b: Distillates and residual versus temperature. Accordingly, the gas volume increases when the tempera
ture of the second reactor is increased (Figure 3) . The heat of combustion of the gas has its maximum at 800900°C because of the high content of CH4 and C2H2/C2H4 (Figure 4 and 5).
(1 gas /kg straw)
700
600
500 ■
400
300
200
100
• No second reactor
■k
Volume of second reactor: 10 1
Volume of second reactor: 1.5 1
( C) Temp. O 600 700 800 900 lOOO llOO
Figure 3: Gas VOLUME versus tempersture.

483
Vol .* Volume of second reactor: 1.5 1
30
2 0 -
10
++-T
Vol.%
40
30
20
600 700 800 900 lOOO 1100
Volume of second reactor: 10 1
0 4-4-5
• CO
* CO.
Δ C2H2/C2H4
° C
2H6
( C) Temp. 600 700 800 900 looo lieo
Figure 4: Gas composition.
At 600, 800 and 900°C the reactor temperature and the gas temperature were the same for both the 1.5 1 and the 10 1 second reactor. No difference in gas composition was observed at these experiments. The reactor temperature and the gas temperature were the same for the 10 1 second reactor, 1000 and 1100°C experiments. In the 1.5 1 second reactor, the gas temperature showed a fall of approximately 50°C at the very high temperatures; 1000 and 1100°C. This indicates that the volume of the reator is not big enough for the gas to reach this temperature. The gas composition in the 1.5 1 reactor was different from that of the 10 1

484
reactor. These observations are explained by the difference in temperatures.
(MJ/Nm )
Λ 20 ■ 18 · 16
14 . 12 .
ίο
oí^—r
No second reactor
Volume of second reactor: 10 1
Volume of second reactor: 1.5 1
τ 1 1 ι 1— O 6O0 700 800 900 1000 1100
_ ^ ΓΟ Temp.
Figure 5: Heat of combustion of gas.
The heat of combustion of the distillates is remarkably high for all samples except for the experiment without prolonged residence time (Figure 6 ) . The expected heat of combustion was appr. 22 MJ/kg. This is partially due to the amount of acetone still remaining in the distillates, but as acetone has a heating value of 28.5 MJ/kg, it can not explain the values of 33 37 MJ/kg. The thermal cracking and extended residence time must therefore convert the pyrolysis tar to tar with higher heating value.
(MJ/kg)
40
30
20
IO
Volume of second reactor: 10 1
Volume of second reactor: 1.5 1
No second reactor
+r ι 1 1 Γ O 6O0 700 800 900 ÍOOO 1100
1 ^ ( C) Temp.
Figure 6: Heat of combustion of distillates.

485
The viscosity was measured for five samples of distillates. Sample no. Sample no. Sample no. Sample no. Sample no. The amount
No second reactor. Second reactor 1.5 1, 600°C. Second reactor 10 1, 600°C. Second reactor 1.5 1, 1000°C. Diesel oil. of acetone remaining in the samples after
distillation was measured by gaschromatography.
Sample no. 1 2 3 4 5
μ centistoke 4.890 3.267 2.453 4.056 3.049
g acetone/1 sample 3.5
74.2 116 60.4
Obviously, the acetone is very difficult to separate completely from the tar by distillation. It does not necessarily give any problems with small amounts of acetone in the distillates, but there will be a loss of acetone in the system.
CONCLUSION The tar distillates obtained by this series of experi
ments represents up to 3% of the energy introduced with the straw. The efforts made to collect the tar and to separate the tar fractions by distillation, indicate that producing liquid fuels from straw is not profitable. However the experiments showed that thermal cracking at
1100°C can convert tar to gas and carbon black and because of the latter, the gas is not perfectly clean even at this temperature. Before introducing the gas to a gas engine it has still to be cleaned. The residence time, which was 67 sec. in the experiments
including the 1.5 1 second reactor and approximately 35 sec. in the experiments with the 10 1 volume second reactor, seems only to have influence on the gas volume produced. The reduction in total condensate is to small to measure. The difference in gas composition and total condensate accumulated in the experiments with 1000 and 1100°C 2nd reactor temperature, is due to a difference in gas temperature in these experiments. The 1.5 1 second reactor was not big enough for the gas to reach the reactor temperature, whereas the 10 1 second reactor has a sufficient volume for the gas to reach the applied temperature.

486
THE FUEL PROPERTIES OF HYDROCARBON LIQUIDS
DERIVED FROM PYROLYSIS OF WASTE
PAUL T. WILLIAMS AND DAVID T. TAYLOR Department of Fuel and Energy
The University of Leeds, Leeds, LS2 9JT (UK)
Summary
Pelletised municipal waste (RDF), wood, rubber tyre and crop waste was pyrolysed at 500 C in a 200 cc static batch reactor in the presence of nitrogen. The derived hydrocarbon liquids were condensed in a cold trap, dried and analysed for their properties as fuels in comparison with crude petroleum oil. The properties of the derived oils varied depending on the original waste material. Rubber tyre waste produced a low viscosity oil with a CV of 42.0 MJ/kg, the oils derived from wood, crop and RDF were viscous liquids with CVs of 21.1, 27.3, and 22.1 MJ/kg respectively. Sulphur contents were lower compared to a medium petroleum fuel oil. FTIR was used for functional group characterisation and showed the samples were complex with carboxylic acids, alcohols, alkanes, alkenes, phenols, aromatic and polyaromatic, ketones and aldehydes being identified. Molecular weight ranges of the oils by SEC showed a range similar to petroleum oil and a shift to higher molecular weights was observed on storage indicating polymerisation. Simulated distillation of the lower boiling point range of the oils by pyroprobe gas chromatography showed that similar ranges to petroleum oil were found for the wood and RDF, whilst rubber and crop oil had higher ranges.
INTRODUCTION The main routes investigated for energy recovery from waste have so
far centered on heat recovery from incineration (1,2), gasification (3,4), gas production from landfill sites (5,6) and pelletisation for substitute solid fuel (7,8). However the production of liquid hydrocarbons from waste material and their properties as fuels has received less attention. The production of liquid fuels from waste has advantages over other forms of energy from waste recovery systems, in that since the fuel is liquid it may be stored and transported and hence the fuel product does not have to be used at or near the recycling plant. The derived oils may be upgraded to refined fuel or added to petroleum refinery feedstocks. Also the products may be important for their use as refined chemicals, high concentrations of potentially valuable chemical feedstocks such as benzene, toluene and naphthalene (9) may make the economics of pyrolysis of waste a more attractive proposition.

487
2. MATERIALS AND METHODS The waste material pyrolysed, consisted of pelletised municipal waste
(RDF) from Eastbourne, Sussex UK, the wood waste was pine chips, the crop waste was straw and the rubber tyre waste represented a mixture of heavy and light duty automotive types, all metal core was removed prior to pyrolysis.
The pyrolytic apparatus consisted of a 200 cc stainless steel reactor heated by an electric ring furnace with nitrogen as the carrier gas. The waste was placed in the reactor and heated to 500 C at S C/min and held at 500 C until no significant release of gas was observed. The evolved gases were analysed off line by packed column gas chromatography, the results of which will be reported elsewhere. The hydrocarbon liquids were condensed in a glass liner within an ice trap connected to the reactor. The liquids were separated into an aqueous and oil phase by decanting, then stored in a dessicator over silica gel for several weeks until the samples were dry.
The CV and sulphur content of the raw waste and derived oils were determined by bomb calorimetry. Functional group, compositional analysis was performed using Fourier Transform InfraRed (FTIR) spectroscopy with a Perkin Elmer 1750 system. The molecular weight range of the oils was determined using size exclusion chromatography (SEC), the column used was a Polymer Laboratories PL Gel 10/im, 500Â, 600mm χ 7.5mm with tetrahydrofuran as the mobile phase, the system was calibrated with polystyrene standards. The simulated distillation (10) was carried out using a modified method incorporating pyroprobe gas chromatography (PGC). The derived oils are of high viscosity and contain a high molecular weight fraction and thus are difficult to analyse by gas chromatography. PGC at 500 C causes volatilisation without decomposition of the lower boiling point fraction of the oil, whilst the higher end is retained in the quartz sample tube within the pyroprobe. The system used was a Perkin Elmer 8320 capillary GC coupled to a CDS Pyroprobe 190.
3. RESULTS AND DISCUSSION Table I shows the gross CV and sulphur contents for the waste material
and the dried derived pyrolytic oil. Rubber tyre oil was of low viscosity with a CV of 42.0 MJ/kg, comparable to medium fuel oil and lower sulphur content. Similar CVs for oil from tyre waste have been reported (11,12) but higher reported sulphur levels of between 1.01 and 1.65% (11) and 0.75% (12). Wood waste gave an oil with a CV of 21.1 MJ/kg and <0.1% sulphur, this value compares with literature values of 22.2 MJ/kg and 0.11% sulphur (13) and 28.5 MJAg and 0.1% sulphur (14). The RDF gave a CV of 22.1 MJAg and <0.1% sulphur which compares with municipal solid waste pyrolysis oil with a CV of 26.5 MJ/kg and sulphur 0.10.3% (15) and RDF pyrolysis oil associated with an aqueous phase with a CV of 18.9 MJ/kg and 0.1% sulphur (16). Wood, crop and RDF derived oils were more viscous than the rubber tyre derived oil, the CV were suitable for the direct use of these oils as low grade fuels with the advantage of low sulphur content when compared to a medium fuel oil with a CV of 43.0 MJ/kg and up to 2.5% sulphur.
FTIR absorbance frequency spectra representing functional group, compositional analysis of the waste derived oils compared to crude petroleum oil from the North Sea Brent field are shown in figure 1. The presence of the CΗ stretching vibrations between 2800 and 3000 cm1 and CΗ deformation vibrations between 1350 and 1475 cm1 indicate the presence of alkanes. These appear clearly in the petroleum oil which is known to contain alkanes and also in all the waste derived oils, the absorbance between 2800 and 3000 cm1 is clearly shown although the 1350 to 1475 cm1 is less distinguishable for the wood and RDF oils. The presence of alkanes

488
Petroleum
4000 3000 2000 1600 1200 600 Frequency cm_1
Figure 1. FTIR spectra of pyrolytic oils.
Table I : CV and Sulphur of Waste Material and Derived Pyrolytic Oil.
MATERIAL RAW WASTE DERIVED OIL
Rubber Tyre Crop Wood RDF
CV (MJAg)
39.8 15.5 18.4 21.0
S (%)
1.3 0 . 1 0 . 1 0 . 2
CV (MJAg)
42.0 27.3 21.1 22.1
S (%)
0 . 2 0 . 1
<0.1 <0.1
in wood and rubber tyre waste derived oils has been demonstrated in the literature (9,16,17). Single and polycyclic and substituted aromatic groups are indicated by the absorption peaks between 675 and 900 cm-1 which are present throughout all the samples but to a greater extent in the rubber tyre and crop waste pyrolytic oils. Phenols, and 2 to 4 ring polycyclic aromatic hydrocarbons (PAH) have been identified in wood derived oil (16,17). Benzene, toluene and 2 to 4 ring PAH together with alkyl

489
- Crop Wood
■ Tyre RDF
Petroleum RDF (Fresh)
3000 Mass 0 3000 Mass
Figure 2. Molecular weight histograms of pyrolytic oils.
derivatives have been identified in rubber tyre waste oil (9,18). The presence of O-Η vibrations between 3050 and 3600 cm-1 together with the presence of C=0 stretching vibrations between 1650 and 1850 cm-1 indicates the presence of carboxylic acids. These are most significant in the oils derived from wastes containing a high proportion of cellulose and hemicellulose, i.e. the wood and the RDF which may contain up to 85% paper (16). Carboxylic acids are also indicated, but to a lesser extent in the crop and rubber pyrolytic oils. Carboxylic acids in wood derived oil have been identified in the form of formic, acetic, propionic, and butyric acids (16,17,19). The presence of the C=0 stretching vibrations with absorbance between 1650 and 1850 cm-1 may also indicate the presence of ketones and aldehydes since aldehydes have been identified in pyrolytic oil from wood (16,19) and ketones from RDF (16). The absorbance peak between 1575 and 1650 cm-1 represents C=C stretching vibrations indicative of alkenes and are represented in all the samples. Alkenes have been identified in oil from wood (19) and municipal waste (15). The group of overlapping peaks between 950 and 1325 cm-1 most promonent in the wood and RDF samples most probably represent the presence of primary, secondary and tertiary alcohols and also phenols due to the C-0 stretching and O-Η in-plane deformations of these functional groups. Alcohols and phenols have been identified in wood (16,19) and RDF (16) derived oils.
Figure 2 shows the molecular weight range of the stored pyrolytic oils compared to Brent petroleum oil. Table II shows the highest molecular weight recorded and the number and weight averages, which shows that high

490
500
400
300
200 ί
100
■
»
/ / .
/ /
h //
9
» ê
/ /
/ .
.·' Wooçi.
yS7
,*
Crop^
/ /
/ Tyre / Φ . . · '
. · ■ "
4 A'
r" y/
' Petroleum
^ _ ^ ^ ^ ^ j t 1
y **" ..
**
/ ' ' Λ' ι
Λ /
I?DF
■
Λ ..7
/ /
'Ί å ι i i
0 20 40 60 80 100 % Recovered
Figure 3. Simulated distillation of lower boiling point fraction of pyrolytic oils.
molecular weight fractions are present in all the samples albeit in low concentration. Molecular weight ranges in oil from wood waste of over 2000 (20) and up to 10000 (15) have been recorded. Figure 2 also shows a sample of RDF derived oil analysed after only 4 days compared to the sample stored for several weeks, there is clear evidence of a shift in the histogram to higher molecular weight compounds and Table II shows an increase in both the number and weight average molecular weights, indicating polymerisation of the sample during extended storage. Polymerisation of wood derived oil has been observed after only 3 days storage (20) and cellulose derived oil also exhibits polymerisation on storage (16). The polymerisation of waste derived oils is a disadvantage in that storage for any length of time has a deleterious effect on the oil, increasing the viscosity and consequently the handling problems of these oils.
Figure 3 shows the simulated distillation range of the oils and petroleum oil using pyroprobe GC. The results represent the simulated distillation range only for those compounds that are volatilised below 500 C. In all cases the first distillation fraction was higher than petroleum oil, the middle distillate fraction most closely represented by wood oil and to a lesser extent RDF oil. The highest distillation range was found for crop waste oil and is reflected in the much larger molecular weight

491
Table II : Molecular Weight Data for the Pyrolytic Oil. MW No. Average MW Wt. Average MW Material
Rubber Tyre Crop Wood RDF (Stored) RDF (Fresh)
Max. Re con
3200 3800 2700 3200 1950
335 397 326 314 261
470 584 458 476 340
range and higher number and weight average molecular weights (figure 2 and Table II). Since polymerisation of the samples has taken place during the extended storage period, they will represent a higher boiling point fraction than originally condensed from the reactor. The residue retained in the pyroprobe after volatilisation represents the high molecular weight fraction which is not analysed by this method and thus simple distillation refining of the oils is made more difficult than at first indicated by figure 3. The polymerisation of the samples also leads to difficulties in defining the refinery processing applicable to these oils if they are stored for extended periods. 4.ACKNOWLEDGMENTS
This work was supported by the UK Science and Engineering Research Council under grant number GR/F/06074. We would also like to thank Leeds university personnel, A. Wheeler, J. Taylor, D. Mills and G. Nedjad and Dr. T. Rampling WSL and M. Eastwood E. Sussex C.C. for waste samples.
5. REFERENCES (1) Clark J.A.J. I.Mech.E. Paper No. C08/88 (1988). (2) Dent C. & Krol A.A. MSW conversion to Energy, Harwell HMSO (1987). (3) Finney CS. S Garrett D.E. Energy Sources 1,3 (1974). (4) Preston G.T. Waste Age 7,5 88-91 (1976). (5) Richards K.M. Proc. Energy from Landfill Gas, Solihull OK Oct (1976). (6) Biddle C.A.R. & Naylor E. I.Mech.E. Paper C13/88 (1988). (7) Jackson D.V. & Tron A.R. Int.J.Energy from Wastes 6,1, 31-44 (1985). (8) Porteus A. Refuse Derived Fuel 38-56 App. Sci. Pubs. London (1981). (9) Collin G.in Jones J.L. & Radding S.B. ACS Syp.Sr.130 (1980). (10) ASTM D 2887-84 (1988). (11) Kawakami S. in Jones J.L. fi Radding S.B. ACS Syp.Sr.130 (1980). (12) Suzuki M. & Sato H. Jpn.Kokai Tokkyo JP61/7387 (1986). (13) White E. & Thomson M.in Jones J. fi Radding S. ACS Syp.Sr.130 (1980). (14) Delmon B. et al, in Grassi G. S Zibetta H., Energy from Biomass-1,
Elseveir, London (1987). (15) Pober K.W. s Bauer H. in Anderson L.L. S Tillman D.A. Eds. Fuels from
Waste, Academic Press, New York (1977). (16) Rampling T.W. & Hickey T. WSL LR643(MR) HMSO (1988). (17) Soltis E. fi Lin S.in ACS Div. Fuel Chem. 32, No. 1-2 (1987). (18) Kaminsky W.S Sinn H.in Jones J. & Radding S. ACS Syp.Sr. 130 (1980). (19) Pakdel H. fi Roy C. in ACS Div Fuel Chem. 32, No. 1-2 (1987). (20) Johnson D.K. fi Chum H.L. in ACS Div. Fuel Chem. 32, No. 1-2 (1987).

492
ENVIRONMENTAL AND PUBLIC HEALTH ASPECTS OF GASIFIER SYSTEMS
J. WILLOCX, consultant, Londerzeel (Belgium) A. BUEKENS, professor, Vrije Universiteit Brussel (Belgium)
Summary Gasifier systems usually consist of a reactor with feeding and ash discharge devices,gas cleaning and cooling equipment and gas utilisation installations. Feed preparation is also often accomplished at the same place. During each of the process steps gaseous, liquid or solid effluents may be generated causing damage to public health or the environment on short or longer term if not properly managed. In addition some of these activities may be associated with discomfort by inadmissible noise levels, unpleasant smells or dust emissions. Most attention is however given to the intermediate products of gasification, which are separated as aqueous condensates or are contained in the solid residues. Part of these products are potential water pollutants whereas other may show carcinogenic properties.
1. INTRODUCTION Gasifiers are used for producing a gaseous energy carrier
from solid combustibles in cases where the wanted form of energy may not technically or economically be produced by direct combustion.
The principle of gasification is based on the combustion process in which the air intake is lowered so that the produced gases contain part of the energy of the fuel in the form of combustible gaseous molecules, mainly carbon monoxide and also small portions of lower hydrocarbons. Instead of air as the reacting gas also steam, oxygen or air enriched with oxygen and/or steam may be used.
The unwanted products of gasification, which are steam, higher hydrocarbons and dust are separated in the gasifier system and some of them may cause damage to public health or the environment if not properly managed. Also other adverse effects on the environment may be produced by gasifier systems, including inadmissible noise levels, unpleasant smells or dust emissions.
Gasifier systems usually consist of a reactor with feeding and ash discharge devices, gas cooling and cleaning equipment and a gas utilisation installation. Also fuel preparation may be accomplished at the same place. 2. FUEL PREPARATION AND FEEDING
Gasifier fuels may include coal, peat, wood and wood wastes, forestry and agricultural residues and wastes from

493
agricultural industries. Common operations for preparing fuels such as drying, size reduction, size classification and size enlargement may produce noise and dust. Carbonisation and torréfaction of fuels may in addition give rise to odor problems. Fuel feeding and handling may also be dust generating. The adverse effects of these operations may in general only affect those working at the plant and not the environment, except in cases of large scale operations, which are common at coal gasification plants. 3. GASIFICATION
Reactors for gasification may be classified according to the type of flow of the combustibles as entrained flow, fluidised bed or fixed bed gasifiers. The latter, which are most common, may be subdivided in updraft, downdraft and crossflow gasifiers, indicating the flow direction of the gases in the reactor.
During gasification the bulk of the solid fuel and the reactants are transformed into permanent gases, steam, higher hydrocarbons and dust or particulates, leaving the reactor as a raw gas stream. At certain reactor types part of. the steam and the higher hydrocarbons may leave the reactor as a condensate. At the bottom of the reactor a solid residue is discharged consisting of mineral ash and char.
The higher hydrocarbons, often indicated as tars and oils, are intermediate reaction products, which are formed in varying quantities according to the properties of the fuel, the type of reactor and the operating conditions. Also the characteristics of the tars and oils are influenced by the same variables.
The tar and particulate concentration ranges in the raw gas depend largely on the type of reactor. For fixed bed gasifiers the concentration of particulates is typically below 10 g/Nm whereas for the other types this value is usually exceeded. The ţar loadings are in most reactor types higher than 5 to 10 g/Nm , except for downdraft and crossflow gasifiers, showing values of 10 to 100 times lower than the other types.
The dust and particulate loadings in the raw gas may be specified as fly ash, char particles, soot, carbon black and mineral vapors. Fly ash consists mainly of mineral matter. Char particles represent pyrolysed or partly burned biomass that is fine enough to be entrained in the raw gas stream. Soot or carbon black is very fine pure carbon dust, a product of reverse reactions or cracking of tar liquids, which may adsorb a number of polycyclic aromatic hydrocarbons. Mineral vapors consist of mineral matter boiled off in the oxidation zone.
The solid residue collected at the bottom of the gasifier represents in principle the incombustible fraction of the fuel but also considerable amounts of charcoal may be present. The The disposal of such residues may need special care because of the presence of airborne particles, which may contain polycyclic aromatic hydrocarbons, carcinogenic substances, and because of the fire risks.
The condensate collected from the reactor at certain gasifier types consists mainly of water but may contain considerable amounts of acetic acid and other water soluble organic compounds, such as phenol and furfural. Also small amounts of

494
benzene, napthalene, toluene and xylene may be present besides trace quantities of compounds such as aldehydes, ketones, car-boxylic acids, esters, phenols, polyhydrics, heterocyclic oxygen and nitrogen compounds, polycyclic aromatics and other. Such condensate is environmentally suspect with regard to the risk for water pollution in case of inadequate disposal and they may also produce adverse health effects, including cancer. It may also produce odor problems. 4. GAS CONDITIONING
Conditioning of the gas consists mainly of the separation of tars and particulates from the raw gas and at the same time cooling it in order to make it suitable for a given utilisation. The systems available for gas conditioning may be classified as wet, dry or semi-dry systems.
Wet systems include spray towers, wet packed beds and venturi scrubbers. They usually use water to collect the gaseous, liquid or solid impurities, which implies extra waste water to be disposed of. On the other hand cooling of the gas occurs at the same time as cleaning and fire or explosion risks are minimised or are non-existent.
In dry systems the impurities may be separated as a dry material, which may be disposed of without the environmental care needed for liquid effluents. However dry dust may be hindering or even dangerous when handling and may also give specific disposal problems. Examples of dry systems include cyclones, fabric filters, dry packed beds and electrostatic precipitators.
With semi-dry systems the residue disposal may be less problematic but their use is generally limited to large scale operations, for economic reasons.
The liquid wastes generated during gas cleaning and cooling generally contain less oils and tars than the gasifier condensate discussed in previous chapter. The nature of the organic load may be similar with respect to the number of compounds but the presence of high boiling fractions tends to be favored. Usually this waste contains also dust particles.
Disposal of considerable amounts of contaminated condensate in the environment without adequate measures may cause a risk for water pollution. This may be due to water soluble organics such as phenol, especially if the applied concentrations inhibit or destroy biological life. At lower concentrations some of the potential water pollutants may however be biodegradable in water or soil.
Water pollution may also be caused by compounds such as benz(a)anthracene or benzo(a)pyrene, which show carcinogenic properties. Despite of their insolubility they may be dispersed in water adsorbed on particles and be dangerous for human health after bioaccumulation in the food chain.
In this respect the disposal of dust may need also special attention because char particles may adsorb non-polar organic compounds up to 20 to 40 % of their weight whereas this may be higher for soot and carbon black. Such particles may be dispersed via the atmosphere and contaminate air and food substances.
In order to control such problems several solutions have

495
been proposed. An attractive solution solving two problems at the same time, the disposal of char and of condensate, appears to be the adsorption of the liquid wastes on the ash/char residues, which may be combusted after drying. The condensates may also be disposed of by evaporation of the water and recycling the dry residue to the reactor. Another solution could consist of mixing the fuel with a combination of Ni/La and Κ CO., catalyst which reduces the production of higher hydrocarbons in favor of smaller molecules, especially hydrogen. 5. GAS UTILISATION
For driving internal combustion engines the raw gas needs generally to be cleaned down to 1050 mg/Nm of solid particles and 1001000 mg/Nm of tars and oils. With respect to the pollutants in the exhaust gases from producer gas engines it may in general be stated that the problems will be smaller than with gasoline engines because of more complete combustion and a lower adiabatic combustion temperature. However it has been reported that at low load producer gas engines generate higher carbon monoxide and hydrocarbon emissions than when diesel fuel is used. Other measurements show that in engines fueled with producer gas the hydrocarbon emissions are generally lower than for gasoline, as may be expected, but that the nitrous oxide emissions are roughly the same. The issue may be complicated by the changing gas quality and the manual adjustment of the gasair mixing ratio which is often employed with producer gas engines. The noise level may also be a problem with engine operation.
If the gases are burnt for heating purposes a relatively high tar and particulate content may be allowed since they will in general be consumed in the burner by combustion. The major environmental problems with burning the gases may result from NO and SO in the exhaust gases. However the concern with sulphur is of a smaller extent with gasification than with direct combustion of the solid fuel since it is easier to separate sulphur from fuel gases than from exhaust gases. The sulphur problem may be especially reduced if fuels with low sulphur content, such as biomass, are used. 6. CONCLUSION
This paper atten.pts to highlight the major environmental and public health aspects of gasifier systems. The range of aspects includes minor issues, such as noise, dust and odor problems, and major issues, such as risks for water, air and soil pollution, including cancer risks. The extent to which these adverse effects may be produced depends on the type of reactor, its capacity and operating conditions, the type of feedstock, gas conditioning and gas utilisation. LITERATURE
Brown, M. D., Baker, E. G., Mudge, L. Κ., Environmental design considerations for thermochemical biomass energy, Pacific Northwest Laboratory, Richland, Washington, 1986.
Buekens, Α., Schoeters, J., Thermal methods in waste disposal, Part I : Pyrolysis and gasification, Commission of the European Communities, Brussels, 1984.

496
Evans, R., Knight, R., Omnischak, M., Babu, S., Development of biomass gasification to produce substitute fuels, Proceedings of the 1985 Biomass Thermochemical Conversion Contractors Meeting, Minneapolis, 1986.
Knoef, H., Stassen, H., Hovestad, A., Visser, R., Environmental aspects of condensates from downdraft biomass gasifiers, University of Twente, Enschede, The Netherlands, 1987.
N., Producer gas manual, Producer gas course, Bandung, Indonesia, 11 18 March 1985.
Ν., UNDP/World Bank guidelines for field monitoring of small scale biomass gasifiers, The World Bank, Energy Department, Washington D.C., 1984.

497
CHARACTERIZATION OF WOOD CONSTITUENTS
BY PYROLYSIS - FIELD IONIZATION MASS SPECTROMETRY*
H.-R. SCHULTEN Fachhochschule Fresenius, Department of Trace Analysis, Dambachtal 20
D-6200 Wiesbaden Federal Republic of Germany
Summary
Direct pyrolysis-field ionization mass spectrometry (Py-FIMS) is applied to standard samples of wood constituents such as glucose, celiobiose, cristalline and amorphous cellulose, birch hemicellulose, and birch lignin. The thermal behaviour and chemical characterization of these polar low molecular weight plant constituents as well as complex biomaterials by thermal degradation coupled with soft ionization mass spectrometry is shown. This high-vaccum (in-source) technique allows molecular weight distribution profiles of thermally released material to be rapidly obtained at both low and high mass resolution. The main topic is to demonstrate that the thermally released chemical species are directly related to the original structure of the starting material and artifact formation during pyrolysis plays only a minor role under the selected experimental conditions. Thus, for qualitative and quantitative investigations of biomass, in particular dried, milled wood, the method can be utilized for basic research on wood composition and quality control of biomass conversion processes.
1. INTRODUCTION The global search for alternative energy sources based on biomass and
current problems of tree damage have created an urgent demand for analytical methods directly applicable to plant materials. The first requirement of such methods is that they should provide an immediate "fingerprint" of the whole sample with minimal preparation so that the sample may be identified by eye, library searching or more sophisticated pattern recognition methods. The second requirement is that it should be possible to identify relevant chemical species · present as biomarkers and perhaps responsible for the "fingerprint" differences and variations found.
The importance of biomass as a renewable starting material has steadily increased and in particular its pyrolysis and gasification processes play an important role in the search for alternative energy resources. Around the year 2,000 biomass may already provide approximately 696 of the total primary energy demand in the countries of the European Community (2). As biomass conversion increases in importance so too will the applied analytical methods. The responsibility assigned to such methods in predicting and steering the successful outcome of large-scale chemical engineering operations could well become enormous. The situation is complicated by the intrinsic variability of the naturally produced feedstocks together with quality control of the desired products such as charcoal, gases, liquid fuels. Much interest therefore attaches to the development of rapid, reliable and comprehensive techniques which may be suitable for application to both feedstocks and products initially in pilot studies and later in automated factory environments (3).
Characterization of plant materials by Py-FIMS, Part VI. For Part V see ref.(l).

498
Conventional procedures for the analysis of such complex biomaterials normally involve series of laborious steps including separations, chemical tests and spectroscopic or other instrumental techniques. This process can be especially time-consuming when only the differences between samples are of interest and the chemical species responsible for the difference is completely unknown. Even where fractions have been isolated containing the species of interest, the analytical techniques may lack sensitivity or resolution.
An analytical scheme is introduced, based upon pyrolysis (Py) field ionization (FI) mass spectrometry (MS) and pyrolysis gas chromatography/mass spectrometry (Py-GC/MS) for the rapid profiling of plant material without sample preparation. As demonstrated earlier (Ί), the field ionization method provides strong molecular ion signals for most organic molecules over a wide polarity range with minimal fragmentation. Thus, the technique is well suited to the analysis of complex mixtures with no prior component separation. In comparison with other soft ionization techniques the FI method is relatively background-free, making it a powerful tool for the recognition of small differences between sample spectra. The sum of all spectra obtained during Py-FIMS gives a highly differentiated spectrum of the total sample. Such a procedure has been applied to the analysis of polysaccharides (5), chitin (6), plant mucilages (7), tobacco (8,9), humic substances and soils (10-12) and biomass (13,14).
The present paper reports the application of the improved technique (15) of temperature-programmed Py-FIMS to samples of plant materials such as wood constituents.
2. MATERIALS AND METHODS Wood constituents
The D-glucose (anhydrous dextrose, moisture 0.5%) and cellobiose (from cellulose, moisture 0.10%) were research grade and purchased from Pfanstiehl Laboratories Inc., Waukegan, Illinois, U.S.A. The specimen of amorphous (Sigmacel Type 100) and crystalline (FMC Avicel, PH-102, 90 ium) cellulose were obtained from the Institute of Wood Research, University of Montana, Missoula, U.S.A. The milled birch wood lignin ands birch hemicellulose were gifts of the Federal Research Centre for Forestry and Forest Products, Hamburg, FRG. Pyrolysis-field ionization mass spectroscopy
For direct temperature-programmed Py-FIMS, approximately 100 .ug of dried, ground raw material were placed into the quartz crucible of the modified direct introduction system ( 12 ) of a Finnigan MAT 731 double-focussing mass spectrometer. The sample was heated linearly from 50 C to 750 C within 10 min. In general, the mass spectra were recorded electrically by repetitive magnetic scans in the range m/z 100 to m/z 1200 and acquired by the Finnigan SS200 data system. For accurate mass measurements, some spectra were recorded photographically at high mass resolution (20,000; 10% valley definition).
3. RESULTS AND DISCUSSION The results obtained by application of temperature-programmed Py-FIMS
to samples of glucose and cellobiose (as simple building blocks of wood), crystalline cellulose, amorphous cellulose, birch hemicellulose and birch lignin are given.
Firstly, it was considered crucial to obtain Py-FIMS spectra of single components, e.g. monomer and dimer hexose units of known structure, in order to correlate the structure with the spectral peaks and the thermal processes involved. Such a step is essential for tackling the much more complex and unknown structures, e.g. plant materials such as wood, to be subsequently investigated.

499
Pyrolysis-field ionization mass spectrometry aí Glucose and cellobiose, the building blocks of cellulose
The time-integrated Py-FI mass spectra of D-glucose (mol. wt. 180) and cellobiose (mol. wt. 3^2) are shown in Figures la and b, respectively. These mass spectra are formed by summing the recorded spectra using the routine heating procedure for pyrolysis of macromolecular samples (linear temperature rise from 50 to 750 °C in approx. 10 min). As the soft ionization method employed yields mainly molecular ions with little fragmentation, the spectra provide a direct
6Θ-
29
68 59
iXm
133149 119
182
Ά
163
191216 | l < f l | t l t l | Μ Ι > | Ί Ί
se ise m/z
3 5 8
6e
s i s.
20·
31
Lu
144
96 05 ¡98
204 252 28B j306 ι^·'>ι>1 >I HI 11 ■ ι.,,.■, ι ,
15Β 2ΘΘ 258 380 358 400 458 588
Fig. 1 a) Pyrolysis FI mass spectrum of Dglucose; b) Pyrolysis FI mass spectrum of cellobiose.

500
molecular weight distribution profile of all components of the produced pyrolysate within the mass range covered. Both spectra are characterized by weak molecular ions at m/z 181 ((M+H)+ ion of glucose) and m/z 3Ψ2 ( (M+* ion of cellobiose). In Fig. la, the main peaks within the molecular weight range of glucose are m/z 163 (ΜΗ,Ο+Η)*, m/z 1** (M2 χ H20)+, m/z 133 (ΜΗ,ΟCHO)"1", m/z 116 (MCHjOoK The large lower mass peaks are producea by further water eliminations and direct bond cleavages. Notable is the relatively small m/z 126 (M3xH20)+ peak in Fig. la which is the base peak in the cellobiose spectrum (Fig. lb). Fragment peaks of the dimer (2M) are seen in the glucose spectrum at m/z 306 (2Μ2χΗ2θ)+ and m/z 293 (2MH20CH,0)+. As expected, the same fragments at m/z 306 and m/z 293 also appear in the cellobiose spectrum (Fig. lb ). In this spectrum, however, additional signals due to 2, 3 and Ψ water eliminations at m/z 288, 270 and 252, respectively, as typical products of thermal processes are observed, b) Crystalline cellulose
The summed FI mass spectrum and total ion current (TIC) profile obtained on PyFIMS of the crystalline cellulose sample is shown in Fig. 2. This type of
1.0 ,
6 0 ' 4 3Ί
114
m
io c Ρ ç a :£ S O O
1 " &
0.5
50 ~Ί 1
100 200 Temperature I*C1
I 300
-//-700
t f3 2 8 Í T
3?
6 3 4 8 3 6
2 0 8 3Θ0 ' tifili» ^ h i 40Θ
4 7 0 5 1 0
5 0 0
m/z
Fig. 2 Pyrolysis FI mass spectrum and thermogram (plot of the total ion current (TIC) versus temperature) of crystalline cellulose,
profile will be referred to as a thermogram and the ion current is seen to start around 230°C, maximize at 305°C and end at 335°C heating. When the FI mass spectra in this temperature range are summed, a timeintegrated FI mass spectrum is obtained which summarizes the m/z (mainly molecular ion) contents of the thermogram. The spectral pattern obtained is highly differentiated and free of background revealing the advantages of this ionization technique. The major pyrolysis products have peaks at m/z 162, 1Ψ4 (base peak), 126, etc. Differences observed between this spectrum and that of amorphous cellulose are described below. The creation of such a timeintegrated survey spectrum also has the advantage of telling the analyst which ions are the most significant thereby allowing him to plot individual m/z values in the thermogram. The time (temperature) of occurrence of these ion currents is important in deciding whether a peak is probably a released volatile (low mass, low temperature, short time) or a

501
low mass pyrolysis product (high temperature , long time). This is illustrated by the total and selected ion current plots of pine wood flour in the following paper (17).
c) Amorphous cellulose The thermogram and timeintegrated survey spectrum of amorphous
cellulose are shown for comparison in Figure 3. The thermogram is somewhat ro·
100
o 60u c α ■σ c
5 40
O Φ
1 2 6
BS 98
et
m W f
t» * 1 x ?. 05,
S = £ Φ O S3
50 100 200 Temperature [°C]
JUifc 43 0
<»·η·Ί~>· . · | ) . < . | | 400 500 see
m / z
Fig. 3 Pyrolysis FI mass spectrum and thermogram of amorphous cellulose experimental conditions as Fig. 2.
broader than that in the crystalline case (Fig. 2) as the start of the intense TIC is around 195°, maximum at 295° and end at 335°C. The typical hexose signal at m/z 162 is less significant the intensity ratio m/z 162 / 144 (base peak) averages 16% in the crystalline case (5 determinations) compared to 5% in the amorphous case. This observation should be helpful in an estimation of naturally occuring cellulose as reported, for instance, in soils (12). Moreover, m/z 126 is more abundant in the amorphous case (74% compared with 51%) and, in addition, the amorphous spectrum has a significant peak with m/z 114 (approx. 30% of base) which falls below 15% of base peak in the crystalline case. The signal at m/z 114 is of particular importance in the following discussion of polyoses in birch wood, d) Birch Hemicellulose
Softwood xylans are characterized by arabinofuranose linked by oC(l«—3)glycosidic bonds to the xylan backbone (16). The survey spectrum shown in Figure 4 and is dominated by the base peak at m/z 114 and large ions in the lower mass range e.g. at m/z 96, 86, 74 and 60. In addition to low abundant ions for monomeric pyrolysis products of lignin, the small signals at m/z 210, 332 and 418 are due to lignin contamination (see e) below). The pentose molecular ion (arabinose ) at m/z 150 and the product of direct water eliminations at m/z 132 are clearly discerned but of relatively low abundance. Thus, it appears difficult to identify hemicellulose on the basis of nominal mass peaks by PyFIMS alone.

502
60
4 0 74
M J L
9 6
«
1 3 8 164 140·—
UjJUulv illiy|,rl|H,,|l^i,,Di| ι, |.»τί·, I ι J 3 0 0
Γ" 350
" ■ Ι Ι Ι 4Θ0 45Θ 5 0 8 108 158 200 250
η / ζ
Fig. <t Pyrolysis FI mass spectrum of birch wood hemicellulose.
100
418
2.0
8.3
«34 5
« 5 9 6 Θ.0
^(^Mtf^JJ^^IfUlnHlMi | 100 280 308 408 500 680 780 880 908
n/Z ►
Fig. 5 Pyrolysis FI mass spectrum of birch wood lignin.

503
e) Birch Lignin The survey spectrum of birch lignin is shown in Figure 5 and is
characterized by intense ions having signals of the nominal mass 180, 210 (base peak), 332 and 418. Investigations by PyGC/MS and accurate mass measurements using PyFIMS allowed the assignments of these lignin pyrolysis products. Coniferyl alcohol (180.0786; C . Q H ^ O J ) , sinapyl alcohol (210.0892; CjjH^O^) and syringaresinol (418.1628; Cj^HjgO«) were identified (18). In addition a lignin dimer of yet unknown structure Γ332.Ι26Ό; C , O H 2 Q 0 6 ) was found. A further significant spectral characteristic is the absence of any significant peaks between m/z 54 and m/z 150. In the higher mass range above m/z 418, however, numerous FI signals up to m/z 878 are registered. In general, the sensitivity of PyFIMS for thermally stable lignin subunits is high, as can be derived, for instance, from the weak but distinct FI signals at m/z 210, 332 and m/z 418 in the previous spectrum of hemicellulose (Fig. 4). The method apparently has great potential for the detection of lignin traces in preparations of wood and wood constituents.
4. CONCLUSIONS The results obtained from both two model sugars and of the three major
macromolecular components cellulose, hemicellulose and lignin clearly show that a direct, characteristic relationship exists between the products of thermal degradation monitored by PyFIMS and the original chemical structures of even such complex biological materials as wood.
Raw feedstocks, intermediate fractions and finished products can be directly compared and differences investigated where considered necessary. The potential importance of the combined techniques in production is increased by the suitability of the spectral data for direct incorporation into computerized process monitoring and control systems. The method as described has consequences for both research and production aspects of biomass conversion and the search for alternative energy sources.
REFERENCES
(1) Simmleit, N. and H.R. Schulten (1989). Characterization of Plant Materials by PyrolysisField Ionization Mass Spectrometry V: Pattern Recognition of Spruce Trees An Integrated, Analytical Approach to Forest Damage. Environ. Sci. Technol., in press.
(2) Solar Energy R & D Programme (1985). Results and Conclusions 19791984, Commission of the European Communities, EUR 10249 EN, Directorate General (ΧΠ) for Research and Development, Brussels, Belgium.
(3) Soltes, E. J. and T. J. Elder (1981). Pyrolysis. In: Organic Chemicals from Biomass, I. S. Goldstein (Ed.), CRC Press, Boca Raton, U.S.A., pp. 64101.
(4) Schulten, H.R., H. D. Beckey, H. L. C. Meuzelaar and A. 3. H. Boerboom (1973). High Resolution Field Ionization Mass Spectrometry of Bacterial Pyrolysis Products. Anal. Chem. 45, 191195.
(5) Schulten, H.R. and W. Görtz ŢT978). CuriePoint Pyrolysis and Field Ionization Mass Spectrometry of Polysaccharides. Anal. Chem. 50, 428433.
(6) Kaaden, A. van der, Boon, J. J., de Leeuw, 3. W., de Lange, F., Wijnand Schuyl, P. J., Schulten, H.R. and U. Bahr (1984). Comparison of Analytical Pyrolysis Techniques in the Characterization of Chitin, 56, 21602164.

504
(7) Schulten, H.-R., U. Bahr, H. Wagner and Hermann, H. (1982). Pyrolysis-Field Ionization Mass Spectrometry of Pharmaceutical Plant Mucilages. Biomed. Mass Spectrom. 9, 115-118.
(8) Schulten, H.-R. (1986). Pyrolysis-Field Ionization Mass Spectrometry - A New Method for Direct, Rapid Characterization of Tobacco. Beitr. Tabakforsch. Int. 13, 219-227.
(9) Simmleit, N. and H.-R. Schulten (1986). Differentiation of Commercial Tobacco Blends by Pyrolysis-Field Ionization Mass Spectrometry and Pattern Recognition. Fresenius Ζ. Anal. Chem. 32fr, 9-12.
(10) Haider, Κ. and H.-R. Schulten (1985). Pyrolysis-Field Ionization Mass Spectrometry of Lignins, Soil Humic Compounds and Whole Soil. 3. Anal. Appi. Pyrolysis 8, 317-331.
(11) Schulten, H.-R.~(l987). Pyrolysis and Soft Ionization Mass Spectrometry of Aquatic/Terrestrial Humic Substances and Soils. J. Anal. Appi. Pyrolysis _12, 1*9-186.
(12) Post, B., R. Hempfling, H. Klamberg and H.-R. Schulten (1988). Zur Charakterisierung von Huminstoffen. Fresenius Z. Anal. Chem. 331, 273-281.
(13) Schulten, H.-R. (198fr). Relevance of Analytical Pyrolysis Studies to Biomass Conversion. 3. Anal. Appi. Pyrolysis 6, 251-272.
(lfr) Schulten, H.-R. (1987). Workshop on Pyrolysis as a Basic Technology for Large Agro-Energy Projects, Commission of the European Communities, L'Aquila, Italy, Oct. 15-16.
(15) Schulten, H.-R., N. Simmleit and R. Müller (1987). High-Temperature, High-Sensitivity Pyrolysis-Field Ionization Mass Spectrometry. Anal. Chem. 59, 2903-2908.
(16) Fengel, D. and G. Wegener 1983. Wood : Chemistry, Ultrastructure, Reactions, W. de Gruyter, Berlin.
(17) Schulten, H.-R. and 3. M. Halket (1989). Characterization of Wood by Pyrolysis-Field ionization Mass Spectrometry, Proc. Int. Conf. Pyrolysis and Gasification, Luxembourg, May 23-25, and literature cited.
Acknowledgements This work was financially supported by the Deutsche
Forschungsgemeinschaft, the Bundesministerium für Forschung und Technologie, Bonn-Bad Godesberg, and the Umweltbundesamt, Berlin.

505
CHARACTERIZATION OF WOOD
BY PYROLYSIS - FIELD IONIZATION MASS SPECTROMETRY*
. H.-R. SCHULTEN1 and 3. M. HALKET 2
Fachhochschule Fresenius, Department of Trace Analysis, Dambachtal 20 _ D-6200 Wiesbaden Federal Republic of Germany
Department of Chemical Pathology, Queen Charlotte's and Chelsea Hospital, Goldhawk Road, London W6 OXG United Kingdom
Summary
Direct pyrolysis-field ionization mass spectrometry (Py-FIMS) is used for the investigations of birch and pine woods. The thermal behaviour and global chemical characterization of these complex biomaterials by in-source thermal degradation coupled with soft ionization mass spectrometry is demonstrated. Comparison between the spectrum of a synthetic blend (cellulose, birch hemicellulose and birch lignin) and an authentic birch wood sample clearly shows that artefact formation in temperature-programmed Py-FIMS is negligible. Curie-point pyrolysis gas chromatography/mass spectrometry (Py-GC/MS) is further employed to investigate the identities of selected pyrolysis products. The combined methodology should find use in fundamental research in the thermal analysis of plant materials as well as in applications to environmental problems such as the present tree damage and/or in monitoring feedstocks and products in biomass conversion.
1. INTRODUCTION In a concomitant publication (1) the state of the art of pyrolysis-field
ionization mass spectrometry (Py-FIMS) for biomass investigations is described and an analytical scheme is introduced, based upon this method and pyrolysis-gas chromatography/mass spectrometry (Py-GC/MS) for the rapid profiling of biomass and identification of plant constituents. Recent work has demonstrated the usefulness of direct Py-FIMS in combination with Py-GC/MS in characterizing tobacco (2), food (3), spruce needles (<0 and soils (5,6). The reproducibility of the Py-FI mass spectra of plant materials (7,8) and plant litter in forest soils (9) has been shown by the application of chemometric methods.
The present paper reports the application of the improved Py-FIMS method (7) to birch and pine woods. In particular, the complementary techniques of capillary column Curie-point Py-GC/FIMS and Curie-point Py-GC/EIMS for further characterization of Py products are described.
2. MATERIALS and METHODS Wood samples
The birch wood as well as the pine wood were gifts of the Federal Research Centre for Forestry and Forest Products, Hamburg, FRG. *
Characterization of plant materials by Py-FIMS, Part VII. For Part VI see ref. (1).

506
Pyrolysisfield ionization mass spectroscopy For direct, temperatureprogrammed pyrolysisFIMS, approximately 100
/Ug of dried, ground wood were placed into the quartz crucible of the modified direct introduction system of a Finnigan MAT 731 doublefocussing mass spectrometer. The experimental parameters recently have been given in detail (1).
Curiepoint pyrolysisgas chromatography/mass spectrometry The flash pyrolysis experiments were performed using a Fischer 0310
Curiepoint pyrolyzer (Fischer, Meckenheim, FRG). The total heating time (THT) was 9.9 s and the final temperature 500 °C. The only modification made to the commercially available apparatus was the inclusion of a leak as previously described (10) to enable rapid pressure release after pyrolysis and sample injection. Approximately 1 mg of biomass sample is placed into the sample holder and flushed with helium prior to pushing through the GC inlet septum and activation of pyrolysis. After pyrolysis, the sample holder is removed from the GC inlet system.
Gas chromatography A standard split/splitless injector (Labormechanik Gerstel, Mülheim/Ruhr,
FRG) was employed together with a Varian 3700 gas Chromatograph. The injector was maintained at 250 °C and operated in the splitless mode or at a split of 10 ml/min. The helium carrier gas pressure was 10 Pa.
In PyGC/EIMS experiments, a 30 m χ 0.32 mm i.d. fused silica column was employed (chemically bonded DB1, 0.25 ium film thickness, J&W Scientific, Rancho Cordova, CA, USA). In PyGC/FIMS experiments, a 26 m χ 0.32 mm i.d. fused silica column was used (CpSil5CB, film thickness 1.3 ium. Chrompack, Middelburg, The Netherlands). The columns were maintained at 50 C for 2 min after pyrolysis and then programmed at 20° C/min to 250° C. Connection to the mass spectrometer ion source was via a deactivated 0.1 mm i.d. fused silica line.
Mass spectrometry For flash PyGC/MS, a Finnigan MAT 212 mass spectrometer fitted with a
combined EI/FI/FD ion source was employed under the following conditions: Field ionization: accelerating voltage and emitter potential, 3kV; counter electrode, 8kV; multiplier, 2.2kV; scanning speed, 1.1 s/mass decade; mass range, m/z 501,000. Electron ionization: accelerating voltage, 3kV; ionizing energy, 70eV; multiplier, 2.2kV; scanning speed, 1.Is/mass decade; mass range, m/z 33500.
3. RESULTS and DISCUSSION
The results obtained by application of temperatureprogrammed PyFIMS to birch and pine woods are given. The complementary use of flash PyGC/MS using both EI and FI as ionization modes is illustrated by application to the volatiles produced from wood. Further characterization of the peaks can be carried out by high resolution mass measurement (4).
Pyrolysisfield ionization mass spectrometry ã) Birch wood
The timeintegrated survey spectrum of authentic birch wood is reproduced in Figure la together with the original output of the corresponding thermogram (ordinate : intensity; abscissa : pyrolysis time; number of recorded single spectra; temperature). The spectral features are swamped by peaks due to birch wood lignin at m/z 180, 210 and ^18 (see ref. 1). Of particular interest are the FI signals in the mass range between m/z 250 and 500 as some of these pyrolysis products have been identified as oligomeric lignin building blocks (11). Other peaks of interest in the highmass range are observed at m/z 595, 689, 855

507
6 0
2 1 Θ
1 1 4
il 211 3 3 2
(ίΜ,^μΜΜί,^Ι^ι,ι,,ΐΜ,^ν,.,Λ,,,ί '■
B79 S
iøe 2ββ 3ββ 48β see 6ββ 7ββ see 9ββ m/2 ►
6 0
UJ^Jl
126
154
*ln 332 358
^ l | hh fJ4«if ^ f*t*p rii r<*' ti1! f ■■■ 108 2ΘΘ 38Θ 400 500 608 788 800 988
m/z
Fig. 1 a) Pyrolysis FI mass spectrum of birch wood; b) Pyrolysis FI mass spectrum of a synthetic blend : 42% crystalline cellulose, 35% birch wood hemicellulose, and 21% birch wood lignin.
and 879 which are probably due to lipid fragments. The polyose content is practically indicated by m/z 114 only. The series of ions having m/z 60, 84, 85, 96, 98, 114, 126, 144 and 162 are, at least mainly due to the hexose constituents in wood such as hexose sugars, hemicellulose and cellulose.

508
For analytical pyrolysis of plant materials with respect to biomass conversion (12) and environmental problems e.g. forest decline (13,14), it is of fundamental interest to understand the relationship between observed pyrolysate and the chemical structure of the starting material, wood in this case. Therefore, a synthetic birch wood was prepared by mixing the three major constituents of birch wood,i.e. birch hemicellulose, cellulose and birch lignin. For comparison, Fig. lb shows the survey spectrum of a synthetic blend consisting of 35% birch hemicellulose, k2% cellulose and 21% birch lignin. The high degree of similarity is obvious.
In order to investigate the similarity further, a difference spectrum was obtained by subtracting the synthetic blend spectrum (Fig. lb) from that of authentic birch wood (Fig. la). The result is given in Fig. 2. The differences in the
400Θ0
20000 1 »♦*♦
20000
40000
2ΘΘ
416 3961434
18Θ
596 6 44 689 „ I , , i ι ! r., il » V " '* 1 ' " ' ' " ' 'Ί ft":' " ·'! ' '' ' ' ' ! , ,MV;>., , . .Mr 4 ¿ 0 3 ¿ 0 · 6 ¿ 0 7 β 0 β 0 β
879 Θ55|
ι ι \
2 1 0
Fig. 2 Difference spectrum obtained by subtracting the synthetic blend spectrum (Fig. lb) from that of genuine birch wood (Fig. la).
two spectra are now clearly revealed. Highmass peaks above the xaxis e.g. m/z 596, 64Ψ, 689, 855 or 879 are those which are either absent in the blend or present to a much lesser extent. These significant groups are probably due to aliphatic/paraffinic lipid components which are, of course, not present in the blend. Peaks below the xaxis are those present to a lesser extent in the authentic wood spectrum e.g. m/z 180 and 210. The sensitivity of PyFIMS for these aromatic lignin constituents, coniferyl and sinapyl alcohol, is very high as can also be derived from Fig. 5. Thus, these constituents are overemphasized in the summed spectrum of the synthetic mixture. However, it has to be considered that these subunits may also be overrepresented in the lignin preparation due to the isolation procedure. When the subtraction from the authentic wood spectrum is performed by calculating the corresponding sensitivities of PyFIMS for cellulose, hemicellulose and lignin, only minor intensities of signals below the xaxis are observed.
As an important result, the statement can be made that the method gives a qualitative estimate of wood and wood constituents mixtures and that the formation of artefacts (i.e. pyrolysis products which are not directly related to chemical substructures in the macro molecules) is negligible.

509
b) Pine Wood The survey FI mass spectrum obtained from pine wood is reproduced in
Fig. 3. The survey spectral pattern is highly differentiated and free of background 31
1003
80
60
40
2 0
29
18
KÅ
8 5 114 i+\ 98 i
ML AM *w
15Θ
iti 4VJL
2 2 6
200 2 3 0
2™ 2 8 4 3 0 θ 328
t iX,MÌ 330
¿ l ø ø j
I 80
£ 60i
4 0
2 ØH 3 7 4 Ι 4
?β
ί « « » 4 7 4 S 0 2 ' ι 374 l i Γ " s
?:
øtllHl'<'i'if ll>|i(.i|.ÍlÍvl|Í,|,4„^il|ltÍ|ilJUk„|ÍlÍl ΙΐΙ.μιμ^,,ιίι,ι,νι 59Θ 612 626
500 55β 6ΘΘ 650
Fig. 3 Pyrolysis FI mass spectrum of pine wood.
indicating once again the advantages of this ionization technique. The major products start at the highmass end with m/z 682; a series at 598, 612 and 626 with CH_homologues; and continues with a series at m/z 226, 270, 272, 284, 286, 300, 302and 328 which is partly due to lignin dimers. This has been confirmed by accurate mass measurements giving, for instance, 270.0895 (C . n H ] 4 0 A ) , 272.1048 (C 1 ( .H , 6 Oj , 284.1047 (C, 7 H, ,O a ) , 286.1200 ( Ο , , Η , . Ο Λ 300ϊ099Γ ( C 1 7 H . , 0 5 ) , 302ñl5S (CjyHjoO,.) ancT 328.1344 (C^HjgO^f (15). In this reference fignin monomers have afso'Deen reported from spruce wood and litter at m/z 150, 152, 178 and 180. In addition, two main groups of components are seen in the ranges m/z 300396 and m/z 420550. The large peaks at m/z 18, 31, 43, 60, 84, 96, 98, 114 (partly), 126, 144 and the weak 162 are typical of hexose sugars, mainly cellulose (12).
Figure 4a shows the thermogram for the total ion intensity (Til, plot of the intensity of all ions in the recorded mass range versus temperature) which yields a maximum at 320°C. In order to illustrate the usefulness of the time (and temperature)resolution in deciding whether a peak is a released volatile or higher temperature pyrolysis product, the FI signals for water (18.0106; HO), methoxystilbenol (226.0994; C^H.^O) and tocopherol (430.3811; C2^^-^ were chosen. The first m/z 18 is clearly released volatile water whereas the later m/z 18 (300 to 400°C and around 450°C) indicate water eliminations which originate from higher temperature Py processes. Also, the temperature frame for the release of the volatile wood constituent methoxystilbenol (m/z 226) is completely resolved from the thermal production of tocopherol (m/z 430).
In addition, complete thermal fractions of pyrolyzates can be displayed which give full PyFI mass spectra as illustrated for the temperature range 50 to 250 C in Fig. 4b. As illustrated for soil samples (6), it can be assumed that this third dimension of the pyrolysis mass spectra plus high mass resolution is an

510
500 Temperature [*C]
226
tee-.
B0-
eø-
2 0 - 18
1 1 4
; 9 6 β n
JUJ. 1 0 8 É. Ji.H'l'·
1 8 0
Jk-Λτ Lt
3 1 6
328
Jl l
600 700
T V U +
3 9 6 4 1 4
m/z
Fig. Ψ a) Thermogram of pine wood showing the TIC trace together with the single traces for m/z 18, 226 and 430 illustrating the time-resolution; b) Pyrolysis FIMS survey spectrum of pine wood obtained by summing the mass spectra from 50 to 250 C in Fig. 4a.
essential step for a better understanding of pyrolysis pathways and the identity and thermal behaviour of individual chemical species. Curie-point Py-GC/FIMS
The field ionization mass spectra contained within a whole Py-GC/FIMS run may be summed in a similar way to give a time-integrated survey spectrum.

511
100η
80
60: c α σ c
Q <
Φ
α:
40·
20
60 Ι
58
|61 7 ' [ ^ . ^ i j k tø)l|
74
164
150 I
97 84 ι 112
I
138
m IU miJUL
160 ι
α)
182
2\° æ o ?7η . . . ι Ι ι 1—270
50 100 m/z
150 200 250 300 350
12 ι
10
8
ο χ
4UuA*iJ^A>JyJLJlLA
b)
I B I . A ^ ^ I U . — _ ^
0 — I H 11 1111 11 11 U I I 11 | I l I 11 111111 11 I ì I A I |*H+1 I 11 I 11 [ I 11111 I 111 I |
100 150 200 250 300 350 400 450 500
m/z 180 m/z 178 m/z 164
Scan Number
Fig. 5 a) The timeintegrated survey spectrum of pine wood after Curiepoint pyrolysis GC/FIMS (m/z 50350); b) Curiepoint pyrolysis GC/FIMS of a pine wood sample. Total ion current (m/z 45 450) and single ion chromatograms (m/z 164, 178 and 180); and c) Total ion current chromatogram obtained after Curiepoint pyrolysis GC/EIMS of pine wood (see top next page).

512
C)
οΓ ¿ 0 0 o χ
3 0 0
« 2 0 0
3. 100
α o
I ' I ' I ' I ' I ' I I J ' I I I ' I ' I ' I ' I ' ι 2:00 4:00 6:00 8:00 11:00 12:00 U:00 16:00 Time [min]
ι 1111 1111111111111111111111111111111 1111111111111 1111 I I I 50 100 150 200 250 300 350 400 ¿50 500 Scan Number
The survey spectrum obtained in the pine wood analysis is reproduced in Fig. 5a and shows strong peaks at m/z 12Ψ, 126, 138, 150, 152, 16*l·, 166, 178, 180 and 182. This spectrum summarizes the whole molecular weight contents of the GC/MS run. It is also highly differentiated and may be compared with the low molecular weight region of the direct PyFIMS thermogram (Fig. kb). Indeed, a relatively small group of peaks (e.g. coniferyl alcohol) is evident having a similar profile superimposed on the weak cellulose peak at m/z 180. The thick film capillary column used in this case has a higher sample capacity allowing more information to be obtained from the less sensitive FI technique but discriminates strongly against higher molecular weight or polar components.
Such a timeintegrated PyGC/FIMS spectrum can act as a rough bridge between the direct PyFIMS masses and the fingerprintlike mass spectra obtained after Curiepoint PyGC/EIMS (see below). As in the thermogram case, the survey spectrum tells the analyst which are the most significant masses so that selected ion chromatograms may be directly plotted. Normally, the choice of ions to include in such mass chromatograms can be a very tedious process, involving examination of the spectra corresponding to many GC peaks. An example of such a mass chromatogram formed from the PyGC/FIMS data for the pine sample is shown in Fig. 4b for the m/z values 180, 178 and 16f, together with the TIC chromatogram.
Curiepoint PyGC/EIMS The same sample subjected to PyGC/EIMS gave the TIC profile shown in
Fig. 5c containing a wealth of EI spectra suitable for identification purposes. Mass chromatograms may then be constructed in the same way as in the PyGC/FIMS case, above, and the spectra may be compared by their GC retention indices.
Further investigation of structures : The peaks present in the direct Py survey spectrum may be further
investigated either by high resolution mass measurement (volatile and involatile components) and by PyGC/MS using both FI and EI modes (volatile components). Thus, a powerful strategy emerges for the rapid characterization or identification of survey spectrum components (Schulten et al., 1989b). This may be of particular importance where the survey spectral peaks are found to differ significantly

513
between, for example, 'good' and 'bad' batches of feedstock for a particular application.
Using such a strategy, the following survey spectrum peaks (at least in part) were tentatively identified (volatiles) : m/z 58, acetone; m/z 60, acetic acid; m/z 112, 2-hydroxy-3-methyl-2-cyclopentene-1-one; m/z 124, 2-methoxyphenol and 1,2-nonadiene; m/z 138, phenol-2-methoxy-4-methyl; m/z 150, acetophenone, 2-hydroxy-4-methyl; m/z 164, phenol, 2-methoxy-4-(2-propenyl)- and phenol, 2-methoxy-4-(l-propenyl)- and phenol, 2-methoxy-5-(l-propenyl); m/z 166, acetophenone, 4-hydroxy-3-methoxy and phenol, 2-methoxy-4-propyl; m/z 180, 2-propanone, l-(4-hydroxy-3-methoxy) and phenol, 4-(3-hydroxy-l-propenyl); m/z 182, benzeneacetic acid, 4-hydroxy-3-methoxy-.
The major molecular ions found in the Py-GC/FIMS case are m/z 150, one prominent GC peak, m/z 164, three prominent GC peaks, m/z 180, five prominent GC peaks (plus three others, probably fragment ions owing to somewhat longer retention times).
These results show that the Py-FIMS method is readily applicable to wood and wood components to yield highly differentiated and interpretable mass spectra. Also illustrated is the potential of accurate mass measurement and Curie-point Py GC/MS for further investigation of single components. The additional possibility of cross-correlation between the GC FI survey spectrum containing all the molecular weight information of the GC run and the FI chromatogram containing the time dimension and therefore retention index data, suitable as a further important identification parameter. Thus, it is possible to rapidly obtain a more comprehensive picture of the whole sample contents from occluded volatiles up to the m aero molecular structure.
The GC/MS part of this work will be further improved by ready availability of modern high capacity capillary columns allowing higher mass and more polar Py products to be separated and identified. Cryogenic trapping followed by derivatization for gas chromatography should present no significant difficulties in those cases where considered desirable. In the present work, emphasis has been placed on illustration of rapid profiling so that a fast GC temperature program has been chosen. For research work involving a thorough attempted identification of components, a much slower temperature program or longer GC column may be used thereby improving the separation and information content of the chromatogram.
The potential also exists for creation of libraries of Py-FIMS spectra as well as application of pattern recognition techniques (13,14,16) to aid further investigation both from a fundamental research point of view and for application in commercial biomass conversion. One of the most important aspects of this treatment is the demonstrated power of chemometrics using the integrated approach allowing incorporation of external parameters such as chemical, physical, technical properties and spectroscopic data e.g. IR, NMR etc.
4. CONCLUSIONS
The results obtained from both a real wood samples and a synthetic blend of the three major macromolecular components cellulose, hemicellulose and lignin clearly show that a direct, characteristic relationship exists between the products of thermal degradation monitored by Py-FIMS and the original chemical structures of even such complex biological materials as wood. Artefact formation may occur during pyrolysis but obviously plays only a minor role and can be excluded for the major observed signals.
Thus, it can be expected that technical processes of biomass conversion can be simulated qualitatively and quantitatively in the ion-source microreactor of the mass spectrometer. If sufficient data sets are available, modeling and prediction of biomass properties should be accessible by chemometric evaluations.

514
REFERENCES
(1) Schulten, H.R. (1989). Characterization of Wood Constituents by PyrolysisField Ionization Mass Spectrometry, Proc. Int. Conf. Pyrolysis and Gasification, Luxembourg, May 2325, and literature cited.
(2) Halket, 3. M. and Schulten, H.R. (1985). Rapid Characterization of Tobacco by Pyrolysis Field Ionization Combined PyrolysisMass Spectrometry and PyrolysisGas Chromatography/Mass Spectrometry Studies. 3. Anal. Appi. Pyrolysis 8, 547560.
(3) Halket, 3. M. and H.R. Schulten (1988). Fast Profiling of Food by Analytical Pyrolysis. Z. Lebensm. Unters. Forsch. 185, 112.
(4) Schulten, H.R, Ν. Simmleit, and R. Müller (1989)7 Characterization of Plant Materials by PyrolysisField Ionization Mass Spectroscopy: High Resolution Mass Spectrography, TimeResolved High Resolution Mass Spectrometry and CuriePoint PyrolysisGas Chromatography/Mass Spectrometry of Spruce Needles. Anal Chem. 61, 221227.
(5) Hempfling, R. and Schulten, H.R. (1988). Characterization and Dynamics of Organic Compounds in Forest Humus Studied by PyrolysisGas Chromatography/Electron Impact Mass Spectrometry and Pyrolysis(High Resolution) Field Ionization Mass Spectrometry. 3. Anal. Appi. Pyrolysis _13, 319325.
(6) Hempfling, R., W. Zech and H.R. Schulten (1988). Chemical Composition of the Organic Matter in Forest Soils Π. Moder Profile. Soil. Sei. 146, 262276.
(7) Schulten, H.R., Ν. Simmleit and R. Müller (1987). HighTemperature, HighSensitivity PyrolysisField Ionization Mass Spectrometry. Anal. Chem. 59, 29032908.
(8) Simmleit, N. and H.R. Schulten (1989). Analytical Pyrolysis and Environmental Research. 3. Anal. Appi. Pyrolysis 15, 328.
(9) Schulten, H.R., R. Hempfling, W. Zech (1988). Discriminating Horizons in a Moder Profile by Field Ionization Mass Spectrometry and Pattern Recognition. Geoderma 41, 211222.
(10) Halket, 3. M. and H.R. Schulten (1986). Simple Pyrolysis Chamber Modification for Capillary Column CuriePoint Pyrolysis Gas Chromatography/Mass Spectrometry of Complex Biomaterials. J. High Resolut. Chromatogr. Chromatogr. Commun. 9, 596597.
(11) Kögel, I., R. Hempfling, W. Zech, P. G. Hatcher and H.R. Schulten (1988). Chemical Composition of the Organic Matter in Forest Soils I. Organic Chemical Composition of Forest Litter. Soil Sci. 146, 124136.
(12) Schulten, H.R. 1984. Relevance of Analytical Pyrolysis Studies to Biomass Conversion. J. Anal. Appi. Pyrolysis 6, 251272.
(13) Simmleit, N. and H.R. Schulten (1989). Characterization of Biomaterials by InSource PyrolysisMass Spectroscopy IV : Differentiation of Spruce Needles by Integrated Field Ionization Mass Spectra and Principal Component Analysis. Biomed. Environ. Mass Spectrom. ^8, xxxxxx.
(14) Simmleit, N. and H.R. Schulten (1989). Characterization of Plant Materials by PyrolysisField Ionization Mass Spectrometry V: Pattern Recognition of Spruce Trees An Integrated, Analytical Approach to Forest Damage. Environ. Sci. Technol., in press.
(15) Hempfling, R. (1988). Charakterisierung verschiedener Waldhumusformen und ihrer Dynamik durch analytische Pyrolyseverfahren. Bayreuther Bodenkundl. Ber. 6, 1126.
(16) Windig, W., H. L. C. Meuzelaar, F. Shafizadeh, and R. G. Kelsey 1984. Biochemical Analysis of Wood and Wood Products by PyrolysisMass Spectrometry and Multivariate Analysis. 3. Anal. Appi. Pyrolysis 6, 233250.

SECTION 2
PÏROLYSIS TECHNOLOGY


517
PYROLYSIS OF HAZARDOUS WASTE OIL
U. STEFFENSEN, J. FRANCK, R. RAHNENFÜHRER, and W. KAMINSKY Institute for Technical and Macromolecular Chemistry
University of Hamburg, FRG
Summary The recycling of waste oils which are contaminated with organo-chlorine compounds is a problem of growing economical and ecological importance. The combination of the Recyclon-procees with fluidized bed pyrolysis offers suitable method to dispose high loaded oils. In contrast to other technologies the enormous advantages of the described process are the low emissions, the low PCB-concentration and high quality of the yield products and the minimal amounts of pyrolysis residue.
1. INTRODUCTION More or less viscid liquid waste are used which are
composed of mineral oils, synthetic oils, oil containing residues or water oil mixtures (1). The elemination of waste oils in the Federal Republic of Germany was reorganized in 1986. Now it is part of waste ordinance according to which the originator of the waste has to pay for its elimination.
Because of rising amounts of used lubricants with different compositions (of polychlorinated compounds) in the industrialized world, its recycling gets economical and ecological importance. Especially the possibility of the formation of highly toxic dioxines and furanes from chlorine containing compounds by combustion or any other thermal utilization attracts attention.
Many waste oils contain considerable concentrations of chlorinated hydrocarbons because of the contamination of used motor oils with used transformer oils.
The conventional utilization of waste oils gets impossible because of the emissions of ordinary reprocessing plants. The Recyclon process (2) offers a solution for dehalogenation of waste oils by the reaction of a fine sodium dispersion with chlorinated hydrocarbons to sodium chloride. After distillation, a distillation residue of about 20% of the feedstock is obtained. The combustion or deposition of this viscous, salty residue is very difficult. The conversion to chemical raw materials succeeds by fluidized bed pyrolysis.

518
FLUIDIZED BED PYROLYSIS The core of the plant is an electrically heated reactor.
Its fluidized bed has a diameter of 80 mm and a height of 280 mm. The bed consists of quartzsand with particle sizes of 0,3 to 0,5 mm. A piston pump injects the preheated distillation residue through a water cooled, ceramic isolated injector into the fluidized bed, where the pyrolysis takes place (3). The ceramic isolation inhibits an untimely decomposition of the feedstock surely.
The inert fluidization gas conveys the gaseous or evaporated reaction products out of the reactor through the following cyclone to precipitate dust and fine grained coke and sand. The porous pyrolysis residue adsorbs the melted sodium chloride. Three cooler and an electrostatic precipitator condense the pyrolysis oil. A membrane compressor conveys the obtained pyrolysis gas as fluidization gas back into the fluidized bed.Excessive gas is registrated by a gasmeter and combusted in a torch.
The composition of the pyrolyeis products depends on the temperature, the residence time, the feed, and the composition of the fluidization gas. The most important parameter is the temperature. It was variated between 590°C and 750°C.
As product fractions a pyrolysis gas, a pyrolysis oil, carbon black, and pyrolysis residue, which contains coke and the inorganic components of the distillation residue, are obtained.
ι Pyrolysis oí distillation residues - mass balances
^
Syo'C 6so"C 700'C 75°"C

519
The best temperature to pyrolyse distillation residues is 700°C. The yield of pyrolysis gas increases with increasing temperature, while highboiling tars in the oil decreases.
The formation of the pyrolysis gas depends on the pyrolysis temperature. Important components are methane, ethene and propene.
Tab. I: Important Components of the Pyrolysis Gas in Dependence on the Temperature (% by weight)
Temperature 590*C 650'C 700°C 750°C
Hydrogen CO/CO Methane Ethene Ethane Propene Propane n/iso Butene EC5-Hydrocarbons Other Hydrocarbons
TOTAL
Gas density (kg/m3)
2,00
7,83
13,75
17.76
9,83 17.91
2,94 16,80 5,20
5,98
1 0 0
1,10
1,23
5,16
24,49
22,58
12,06
15.95 1,82
8,75 3,71 4,25
1 0 0
1,05
1,29
2,31 30,10
32,56 8,22
12,43
0,74 3,44 2,83
6,08
1 0 0
0,97
1.37 2,71
40,59 31,34 7,55 7 , 1 1 0,43 1,51 1,67
5.72
1 0 0
0,89
The yield of gaseous products, especially of methane and ethane increases with the temperature. Fropene and ethane show a maximum while the yield of compounds like propane and the C4-and C5~hydrocarbons decreases with increasing temperature. Traces of H2S are also obtained if the feedstock contains sulphur components. COS can't be detected.

520
Tab. II Important Components of the Pyrolyais Oil in Dependence of the Temperature
Temperature
Aliphatic Compounds C , Hydrocarbons C Hydrocarbons
7 C„ Hydrocarbons C Hydrocarbons
9 C Hydrocarbons C Hydrocarbons C Hydrocarbons Cyclopcntaclicnc
Aromatic Compounds Benzene Cyclohexadiene Toluene Ethylbcnzcnc m/p-Xylcne Styrcnc/o-Xylcnc Mcthylstyrenc Indcnc Mcthylindcnc Naphthalene 2-Mcthylnaphthalcne i-Methylnaphthalene Biphenylene Accnaphthcnc Acenaphthylcne Fluorene Phenanthrene Anthracene Other Hydrocarbons Tar C/H ratio of tar
59o"C
0,19
0 , 9
0,24
0,81
1,02
0,76
0,89
n.d.
0,42
0,03
0,25
0,05
0,10
0,13 n.d. 0,14
0,15
0,15
0,01
0,01
0,01
0,01
0,01
n.d. n.d. n.d.
8,13
85.59
I / I , 7 4
6so*C
0,75 1,87
0,193 0,86
0,49 0,86
0,43 0 , 2
2,44 0,38 3,20
0,56
0,71
0,76
0,74
0,44 0,87
0,38
0,22
0,17
0,07
0,12
0,11
0,11
0,09
0,05
16,4
64,79 1/1,67
70o"C
1,99 1,28
o,97
0,39 +
0,49 0,50
0,30
12,93
1,29
8,88
1,01
1.93 2,92
2,04
1,26
i ,99
1,44
0,74
0.59
0,15 0,30
0,34
0,31 0,25
0,15
30,5 25,06
1/1,01
750-C
0,05
0,08
0,09
0,66
0,13
0,38
0,57 0,76
16,34
o,37 10,74
1,27
2,42
6,19
3.49
4,35
2,75
4,58
1,45 1,26
0,24
0,35
0,45 0,25
0,19 0,05
14,6
25,96
1/0,94

521
The pyrolysis oils consists of many different compounds. The yield of the oils increases with temperature and shows a maximum at 700°C.
The most important compounds of the oils are benzene, toluene, xylene, Βtyrene, and naphthalene. The share of these components raises with the temperature. Opposite to this the share of the higher boiling tars decreases.
The C/H ratio of the tars grow less with higher temperature.
Although the waste oil had been contaminated with 600 ppm'β of FCBs. After the pyrolysis no PCBs, PCOOs, and PCDFs could be detected.
REFERENCES (1) Gesetz über die Vermeidung und Entsorgung von Abfällen
(AbfG) (BGBl.) (waste law of FRG) I p. 1410 (2) Patent in FRG no. DE 2813200 C2, 1986 (3) Patent in FRG no. Ρ 3728871.7, 1988

522
E N E R G A S S E W A G E S L U D G E P Y R O L Y S I S
H.-F. Hinrichs, H. Müller ENERGAS, Gesellschaft zur Energiegewinnung
aus Müll und Kohle mbH
Summary Due to the extension of the sewage sludge system in the Federal Republik of Germany the annual amount of sewage sludge is increasing constantly. Considering the restrictions on waste disposal, such as composting and agricultural utilization, thermal processes are becoming more and more significant. Beside the mineralization and sterilization of sewage sludge in refuse incineration plants or flui di zed bed reactors the ENERGAS pyrolysis process (ENERGAS Klärschlamm-Pyrolyse = EKPy) is an interesting alternative for the treatment of sewage sludge. The simple, compact construction of the indirectly fired pyrolysis reactor including its measuring and regulation system is suitable for the thermal treatment of sewage sludge. Additionally it is possible to link noxious substances such as heavy metals, HCl, etc. by additives.
The products obtained are gas and coke. The gas is combusted in order to provide the energy for the degassification process and to produce hot-water, steam and electricity with the surplus energy. Due to its residue content of carbon the coke can be sold as a fuel or used as activated carbon.
The process is especially suitable for catchment areas with a low population density (100.000 EGW = calculated refuse production per inhabitant) where the sewage sludge transport to bigger, thermal disposal plants (refuse incineration plants etc.) is economically unfavourable.
1. General - Sort, Amount and remain of the sewage sludge Sewage sludge is a result of mechanical, biological and chemical treatment of municipal sewage. It is a concentrate of solid substances contained in dissolved or suspended form in untreated waste water. Its condition is not only determined by the components of the sewage given to the purification plant, but is essentially influenced by the purification process as well as the sludge treatment applied in the purification plant.
In 1973 43 mill, tons of raw sludge (solid substances content 4,6 %) or 2 mill, tons of dry matter and in 1986 50 mill, tons of raw sludge or 2,3 mill, tons of dry matter were produced.
Shown in statistics of the Umweltbundesamt for the years 1986/1987 the sludge is disposed off as follows:
- Depositing 46,6 % - Agricultural utilization 24,9 % - Combustion 13,7 % - Composting 1,3 % - Others 13,5 %

523
However, depositing and agricultural utilization of sewage sludge is getting increasingly difficult. This is due to generally less depositing space available resp. the protests against such deposits. Moreover, sewage sludge regulations on heavy metal content impose restrictions on the usage of sewage sludge. 2. Current methods of thermal treatment of sewage sludge The most suitable way of sanitation and decrease in volume is the combustion. Concerning the combustion the fluidized bed incineration is especially approved for dried sewage sludge. By this process sewage containing 25 - 30 % of dry matter can be combusted i. e. mineralized and sterilized without special problems. The emission of noxious material, especially of acid components, can be decreased by additon of limestone to the fluidized bed incineration and by a succeeding flue gas cleaning.
Another way of combusting sewage sludge is to dry it as much as possible and to blow it into the furnace of a refuse incineration plant operating by means of grate firing. This process is also well approved. On the other hand, it is possible to add adequately dried sewage sludge to the refuse and to combust them together.
Although there are technical possibilities for the thermal treatment of sewage sludge in a fluidized bed incineration as well as in a joint combustion in refuse incineration plants, alternative thermal ways of treatment are of high interest. That can be explained by the fact that in the Federal Republik of Germany not even 50 refuse incineration plants are in existence, but more than 8.800 purification plants. This means that now as well as in the future there are not enough thermal plants for the treatment of sludge available.
Therefore, a thermal treatment process is required that can be applied for small and medium sized purification plants at reasonable costs. An alternative to the combustion of sewage sludge is the pyrolysis. Its processing engineering principles are known and need not be explained in detail. The pyrolysis reacts at the temperature range of 300 - 600 °C and does not require as high a temperature as the combustion. 3. Pyrolysis of sewage sludge - EKPy System Basically the pyrolysis can be conducted under 2 different objectives:
- Pyrolysis for sanitation and decrease in volume with recourse recovery at the same time
- Pyrolysis for sanitation, decrease in volume and energetic utilization of the products obtained
Disadvantages of the pyrolysis are, however: - The gets of the obtained products are far from covering the operating costs.
- Experience with the pyrolysis has shown that the aromatics obtained are too highly loaded to be sold.
In a way, at present only the energetic utilization of the sewage sludge is favourable. Its objective is to split the sewage sludge into a usable gas and a residue that can possibly be used as a fuel.

524
The conduction of the sewage sludge pyrolysis EKPy System proceeds as follows:
Punctureproof or dry sewage sludge is given into the inlet device of is conveyed into the reactor by means
stages:
the stationary reactor. From there it of a screw.
The reactor is subdivided into - Drainage up to complete drying - Heating up to 300 °C - Zone of pyrolysis of 300 - 500 °C
The stationary reactor is heated wiht e. g. thermal oil. At the inside transport equipment is provided. It is designed not only to convey sewage sludge, but also to crush newly formed, bigger parts. This is achieved by other stationary devices next to the conveyor, where bigger parts are de-mi ni shed. Furthermore the reactor contains a temperature probe controlling all the individual processes.
4. Results of the sewage sludge pyrolysis by EKPy The loss in weight depending on temperature is shown in figure 1. The first graph shows the loss in weight dependence on temperature at air addition (combustion conditions). The second graph shows the loss in weight dependence on temperature in a nitrogen atmosphere ( pyrolysis conditions).
The decomposition, i. e. a noticeable output of substances, starts at approx. = 190 °C. At higher temperatures there is still some output, though it may be the beginning decomposition of anorganic substances.
200 400 600 800 1000 Temperature (Degree Celsius)
1200
Figure 1 : Loss in weight (%) depending on temperature Graph 1 : Loss in weight at air addition Graph 2 : Loss in weight in a nitrogen
atmosphere

525
According to the graph, at a temperature of 520 °C the weight in loss is 58 weight% related to the raw material. Raw material is the sewage sludge when it is delivered to the purification plant, i. e. the organic substance plus anorganic parts. The standardized value, the actually determining value is 75,3 weight%. Therefore the residue of carbon containing material of the sewage sludge input is 24,7 weight%.
Figure 2 shows the standardized loss in weight. From this graph the loss in weight depending on temperature can be determined.
100 _.
90
— 80 1 70 σ fe 60 o 2 50 LO
¿ 40 30 20 10 0
ω
— · " I
χ / /
remperature °C 200 250 300 350 400 450 500 550 600 βςη
ι Loss in weignt
weight% 6 16 31 50 62 70 75 77 70 70 t; * ** >
200 400 600 800 Temperature (Degree Celsius]
1000
Figure 2 Loss in weight (%) standardized at 77 (%) org. sustances sewage sludge pyrolysis < 650°C
The essential pyrolysis rections happen at the temperature range of 200 and 520 550 °C. At a higher temperature range turnover is neglible. The use of higher temperatures is only suggestive to expedite the pyrolyis reaction.
Additionally it is of high significance that at low temperature ranges of 250 350 °C only 50 % of sewage sludge input can be decomposed and therefore considerable amounts of anorganic substances remain in the residue.
Advisably, the pyrolysis of sewage sludge is conducted up to a final temperature of 520/550 °C. It is also possible to fix the final temperature at 480 500 °C and to prolong the time of treatment accordingly.

526
A product obtained at a temperature of 620 °C was checked on carbon, hydrogen and nitrogen. The temperature of 620 °C was chosen because it could be expected to obtain a completely degassed product. The following data was received:
Element Carbon Hydrogen nitrogen
Unit weight-% II
II
Actual value 34,22 0,92 2,38
Standardized value 77,77 2,09 5,41

527
COGENERATION PYROLYSIS
G. BONINO Biomass Energies Integrated Systems
25 Strada della Viola, 10133 Turin, Italy
Summary
Exhaust gases from a wood or charcoal gasifier generating set can provide the appropriate temperature and heat carrier to sustain a pyrolysis process for charcoal production, as shown by tests which have been conducted with satisfactory results. A commercial application of the system is envisaged when operating gasifier generating sets from 40-50 Kwel installed power upwards. The required time is approximately four hours with a maximum product output of about half a ton, within the 50-80 Kwel power range. Thus it is possible to double the output to approximately one ton of charcoal in the course of an eight hour working day. This process adds new economical flexibility to gasifier generating set utilization, with a new and advantageous cost/benefit ratio. The pyrolysers are of simple construction and the operational requirements are also simple. Unloading is made easy by previously wrapping the wood during the loading phase by means of metal nets. Cogeneration pyrolysis appears to be most appropriate for agricultural residues which are carbonized without wastes, an operation unlikely to be performed with traditional kilns. The integration of systems, namely electricity and charcoal production, increases overall efficiency and can add biomass gasification for engine feeding with a new competitive profile also in European conditions.
1. INTRODUCTION
No author on charcoal making seems to be aware that production of commercial charcoal is also possible in an economically efficient and quick way by using the sensible heat of engine exhaust gases out of a gasifier generating set. Therefore, the scope of this paper is to provide more information on this process which the writer has called 'Cogeneration Pyrolysis' - meaning here the carbonization of biomass first started and then sustained by means of the heat recovered (cogeneration) from the engine thermodynamic cycle. As this process is strictly linked with producer gas for electrical power production, it proposes mainly new flexibilities in gasifier generating set operation, rather than aiming at competing with or replacing other long established charcoal technologies.
In contrast to a traditional diesel fed generating set, the exhaust gases of a gasifier generating set can directly transfer their sensible heat to wood and/or agricultural residues, thus inducing the pyrolysis process for their conversion into commercial charcoal.
Kilns are of simple construction, requiring an exhaust gas inlet at the bottom centre and an outlet at the top leading to a chimney. No air has to be allowed inside the kilns. Since they operate as engine mufflers, the standard engine exhaust muffler becomes redundant and can be removed. Build-up of counterpressure which reduces engine efficency is avoided by the draught action provided by the chimney. The carbonization time span required

528
is reduced to between four and four and a half hours. Several stages of charcoal formation are observed during this period. Although the engine exhaust fumes have a temperature of about 380°C at the beginning, the final emission temperature at the chimney of the kilns remains constant at about 100°C. The wood absorbs heat as it is dried, giving off its moisture as water vapour. Subsequently, the temperature rises and eventually reaches the point where exothermic decomposition of the wood starts. The temperature stabilizes at approximately 500°C.
Kilns of several cubic metres in size can cool off overnight so that the charcoal can be removed on the following day. In order to make this operation easy, rapid and relatively clean, nets are previously laid in the kilns to wrap round the wood contained therein and form packages one over the other. Thus the charcoal can be unloaded simply by hooking up the nets.
2.
This new integration of systems, namely electricity and charcoal, should be investigated further as it can lead to additional applications. One initial result is the opportunity for small charcoal gasifier generating sets to produce the amount of fuel required for their own operation, possibly from locally available agricultural residues, e.g. yellow corn stokes. Charcoal in excess of the gasifier requirements can be used for grill, or in developing countries, for cooking fuel. It is clear that if the gasifier is working on wood or torrefied wood, the total quantity of charcoal produced is then available.
A relevant industrial application of cogeneration pyrolysis lies in the medium to large size sawmills, especially those located in developing countries. Sawmill wood wastes, excluding sawdust, account for 30 to 35% or more of the original timber weight. The 300 Kwel size gas engine generating set, on a three sets layout, can reach the one MW installed power often required. Charcoal conversion of wood wastes by means of cogeneration pyrolysis can cheaply overcome the difficulty of their conversion into useful electrical energy, due to the high humidity content and size differences. Once converted to charcoal, the original large sizes can be reduced to producer gas requirements with little or no effort.
At this level of undertaking, the recovery of the energy contained in the pyroligneous gases and vapours also becomes economically interesting and feasible, thus abating the pollution caused by pyrolysis emissions in the air. In fact, out of the total biomass energy content, 50% remains in the charcoal and 50% is carried away with gases and vapours. From one thousand kilograms of dry wood the following approximate heat quantities, in megajoules (MJ), are available: charcoal 9500 MJ 50%, gas 1500 MJ 8%, condensibles 8000 MJ 42%.
The pyrolysis gas, when cleared of condensibles, has a composition physically resembling that of producer gas inasmuch as the combustible gases - carbon monoxide, methane and hydrogen - have approximately the same volume in both instances. Likewise, the calorific values which average in both cases 1100 to 1200 Kcal per cubic metre. In fact, the non reacting gas is approximately 50% of the total volume in both cases - nitrogen for producer gas and carbon dioxide for pyrolysis gas.
Two main total energy utilization scenarios can be envisaged:
burning the vapours and gases in order to dry the commercial wood produced by the sawmill ; separating gas from condensibles to use it in a dual fuel diesel engine, the condensates to be burnt in the drying furnace.

529
Fig. 1. Wood charcoal from cogeneration pyrolysis
Fig. 2. Agricultural residues are easily converted to charcoal

530
CARBONISATION OF LARGE WOOD PIECES IN A LABORATORY RETORT PRODUCT ANALYSIS AND ENERGY ASSESSMENT
N. SHAH &. P. GIRARD R. CAPART ENERGIE Division Département Génie Chimique CTFT (Département of CIRAD) U.T.C. 45 Bis, Avenue de la Belle Gabrielle BP 233 94736 NOGENT SUR MARNE (FRANCE) 60206 COMPIEGNE (FRANCE)
Summary Pyrolysis of big wood blocks (65 mm* 35 mm* 35 mm) of Oak and plaquettes (250 mm* 40 mm* 25 mm) of Assao was studied in a laboratory retort to investigate : 1. Supplementary Specific Energy required to carbonise 1 kg of dry wood (termed hereinafter as SSE). An attempt is also made to estimate thermicity of pyrolysis reactions and the value is verified later by isothermal calorimetrie analysis. 2. Indicators characterising the retort carbonisation such as :
a) weight & net energy efficiencies, b) effluents produced per kg. of dry wood carbonised (condensi-bles and non-condensibles), c) effluents produced as a function of carbonisation cycle time, d) SSE consumption.
The pyrolysis retort set-up is classical (with possibilities of recuperation of all effluents generated) and uses electrical resistors to provide heat for carbonising wood. Electrical energy utilized during a complete cycle of carbonisation was measured by a w-hr meter to estimate the SSE. Other technical carbonisation process parameters (i.e. initial wood moisture, final temperatures, residence time at final temperatures and heating rates employed) were varied and chosen to approach field carbonisation at industrial scale. This paper reports the typical results achieved with the laboratory set-up used. The results are compared with the industrial retort-kiln measures.
Amongst all the methods of thermo-chemical conversion of wood into upgraded fuels, pyrolysis/carbonisation is most well-known, commonly practised presently and is poised to play even grater role if the conversion efficiencies are further improved and when the oil based fossil-fuels become more expensive and scarce. As an evidence of trade importance France consumed in 1985 about 115,000 tons of charcoal. The traditional methods of charcoal making (without emissions recovery) are known to be less than 50 % energy efficient and could also cause environmental nuisance if charcoal emissions are unutilised or untreated (1).
According to (2) it was discovered that the energy demand of a charcoal process is closely related to the retort capacity, if the operation takes pace batchwise. However, the yields of charcoal and byproducts are very little affected by alteration of the retort capacity.

531
The objective of this research work was to characterise the batch retort pyrolysis process.
MATERIALS AND METHODS
The details about the wood used in this study are given in Table 1. Schematics of the laboratory retort with condensate recuperator and measurement instrumentation used is shown in Fig. 1. Two wood heating rates such as 1.5 'C/min (Tests 1, 2, 5, 6, 7 4 8) and 4,5 'C/min (Tests 3 & 4) were used. The final temperatures reached i.e. 400-600 "C corresponds to the field charcoal making practise and the typical heating rates employed for field carbonisation are mostly between 1-2 *C/min. The emissions of carbonisation were passed through the condensor maintained at 0 "C and additional security condensor traps assured the complete condensation before the emissions pass through a dry filter, gas meter and finally were collected in a rubber balloon.
The condensates recuperated at different temperatures were measured for its weight and were later analysed in the laboratory to know its composition according to the procedure given in (3). The non-condensable fraction was sampled during the cycle at different temperatures and was analysed to know the correct composition by gas chromatography. The calorific values were calculated separately for condensables and non-condensi-bles and were added to give total potential energy of the effluents.
The charcoal was analysed for its humidity, volatile contents and ash to calculate PC (fixed carbon) content (proximate analysis method NF B55101). The calorific values of wood and charcoal were determined by bomb calorimeter. The charcoal friability was measured using a classical method of trommel adapted from coal testing procedures. The friability of charcoal is a commercially important parameter and here it gives a relative measure of physical strength of charcoal which could indicate its resistance to handling losses.
Measurement of SSE of carbonisation and estimation of heat of pyrolysis : the effective energy used to carbonise 1 kg of dry wood was found as below :
Em = ( E(w + r) - E(r) )/Mb where, Em : energy consumption measured giving SSE in MjAg dry wood E(w+r) : energy required to heat wood charge and retort E(r) : energy required to heat only retort without wood Mb : weight of dry wood The following equation calculates Ec : t^f Ec =CMw.Cpw(100 -Ti) + Mw.Lv + Mb.Cpb(270 - Ti) +l7J Mr.CptdT]] /Mb Simplification : Tf( Mr.CptdT = (Mb + Mc)/2).Cpt (Tf - 270) Where, zjo
: weight of moisture in the wood Cpt : specific heat of water, dry wood and residual weight
"C (residual charge is considered as torrefied wood) latent heat of vaporisation of water at 100 *C weight of charcoal produced initial and final temperature respectively residual weight between 270 "C and Tf that changes with temperature and is close to (Mb + Mc)/2
and Em values were compared. Since the measured values turned always inferior to those calculated we are lead to formulate a
Mw Cpw Cpb,
at about 270 Lv Mc Ti, Mr The
out to
Tf
Ec a be a

532
hypothesis that the differential heat comes from reaction heat which is evidently exothermic. Thus an estimation of pyrolysis heat value (Hr) was done as below :
Hr = Ec - Em To verify the results obtained from the retort isothermal calorime
trie analysis was done using Calvet type calorimeter (SETARĂM HT 1 000).
RESULTS AND DISCUSSION Table 1 summarises the principle characteristics of eight carbonisa
tions. Influence of heating rate on carbonisation process : It was observed that rapid heating (4.5 'C/min) provokes gas forma
tion and reduces charcoal yield. Higher heating rate also produced a charcoal of nonacceptable friability. Hence from the limited experimentation in this study (only two heating rates employed) it could be said that the heating rate needs to be optimised to minimize process duration and maximize charcoal yields.
Carbonisation effluents : From the analysis of carbonisation effluents, one could estimate the
product distribution at a given temperature of wood charge. Following was generally observed : Until wood reaches about 150 "C the components other than water are
not found in the effluents whereas acetic acid and furfural starts volatalising when wood reaches about 180200 'C.
The bulk of pyrolysis reactions take place between 270400 "C during which the maximum effluents are produced.
After 540 'C the effluents would contain mostly noncondensible gases and practically no fraction is condensed at 0 *C.
Other technical carbonisation parameters. Weight efficiency : The usual fashion of calculating weight efficiency (charcoal wt./wood
wt.) does not take into account the quality of charcoal and hence in order to have a fair comparison of weight efficiencies of charcoal produced of different qualities, an efficiency indicator (E80) is calculated as below : E80 = Wt. eff (db)* FC (x)/80
where, E80 : Efficiency indicator calculated considering charcoal
having FC of 80 X Wt. eff. (db) : Weight of charcoal/weight of dry wood used FC (χ) : X FC of charcoal obtained in each test. The E80 for this retort lies between 3340 X for carbonisation
between 400600 *C. The higher values corresponded to slow heating rate (1. 5 'C/min). Thus while employing rapid heating rate one may gain time at the cost of reduced charcoal yields. The tradeoffs could be determined for a given technology while product qualities are matched with the market demands.
Initial moisture content : The initial moisture content of wood directly affects two important
parameters of the process viz. cycle duration i SSE. The energy required for drying of initial moisture of wood represents a large fraction of SSE requirement of carbonisation. As seen in Table 1 to carbonise about same quantity of dry wood at initial moisture content of 47 X (wb) required about 1.5 times longer time than the wood at 0 X moisture to obtain nearly same quality charcoal (PC). About 35 X reduction in SSE was obtained when initial moisture content dropped from 47 X to 30 X (wb).

533
Fixed carbon (FC) content : FC is an important indicator of quality of charcoal and is used as a criterion to classify charcoal for trade. As seen from results in Table 1 higher the temperature of conversion higher is the FC to a certain limit. The residence time at the final temperature also has a similar effect on FC.
Friability index : Higher the index value lesser is the charcoal resistance to physical shocks. The values of friability index illustrated in Table 1 indicate that higher the heating rate lower is the resistive quality.
Energy assessment of the process : As seen from the results in the Table 1 the charcoal making in the laboratory retort has energy conversion efficiency between 53-57 % when heating rate employed is 1.5 "C/min. The energy conversion efficiency could drop down to 44-49 % if heating rate of 4.5 "C/min is used. The values of potential energy content of effluents are calculated per kg of dry wood carbonised and are represented in Table 2. This experience of studing the effluents from a retort kiln leads into following general conclusions :
a) GHV (gross heating value) of noncondensable gases varies according to the cycle time and the weighted average value could be taken as 13 MJ/Nm3.
b) About 65-70 % of condensibles is water and the rest has GHV of about 25 MJ/Nm3.
c) The condensible combustibles represent about 50-60 % of the total GHV of the effluents.
Table 3 shows the SSE of carbonisation. Fluegge (4) had reported the SSE values of 1420 kJ/kg dry wood for a batch retort (Reichert) and put forward the advantage gained while using continuous retort system (Sific retort) that demanded only 71 kJ/kg dry wood (enhanced heat exchange rate in continuous retort where wood charge is moving). The average value of reaction heat found in this study is about 435 kJ/kg dry wood which has the same order of magnitude as reviewed by (5). It is widely understood that the pyrolysis of wood till a certain temperature (about 270-280 *C) is endothermic whereafter the reactions turn to be exothermic and if the pyrolysis is continued further till say 400-500 "C the global balance of reaction heat could be slightly exo/endo-thermic. The isothermal calorimetrie analysis on about 60 mg of wood samples in Calvet type calorimeter gave reaction heat value of about 150 kJ/kg (endo).
It was interesting to note that 70 % reduction in SSE could be achieved as a result of emissions recovery for providing carbonisation heat. This explains the recent interest shown by carbonisation industries in valorisation of effluents of carbonisation for optimal wood energy conversion. REFERENCES (1) GIRARD Ph., Atmospheric pollution from charcoal manufacture. State of
production in use, IUFRO 5. Division Conf. Sao Paulo, (May 1988). (2) WALTER E., Handbook of charcoal making, EEC. Solar Energy R & D . Se
ries E, vol. 7, (1985). (3) VERGNET A.M. and VILLENEUVE F., Techniques for the analysis of pyroly
sis gases and liquids from tropical biomass. Bois et Forêts des Trpi-ques. N' 205, III quarter,( 1984), CTFT.
(4) FLUEGGE F., Chemische Technologie des Holzes (56,57), Munich FRG (1954).
(5) WAI-CHUN, R. CAHN, M. KELBON & B. KRIEGER, Modelling and experimental verification of physical and chemical processes during pyrolysis of a large biomass particle, Fuel, vol. 64. (Nov. 1985) pp. 1505-13.

534
3 |—
2
t
1 I. Retort 2. Heating program renul ator 3. Temperature recorder 4.6.7.9 Refrigerant 5. Condensate collector 10. Dry filter II. Security vase 12. Gas meter 13. Gas collector balloon
Fig. 1 : SCHEMATICS OF THE CARBONISATION RETORT & EFFLUENTS RECUPERATION SYSTEMS
Table 1 : GLOBAL ELEMENTS CHARACTERISING CARBONISATION IN LAB RETORT
test ■' tnp of doriti« cubo
(kr) Cc)
1) 14.0
2)9.0
3)5.5
4)5.5
511.0
6)7.3
7)5.5
114.0
550SM
550600
550600
550600
400475
400475
440490
390410
rende nei t IM it TI (kr)
2.0
2,0
2,5
2,5
4,1
1,5
0,6
0
HOD haidit; Dry i t l ib (d)
47
0
31
29
10
10
0
0
1,5
1,56
1,47
1,51
2,65
2,65
2,96
2,49
en
19,8
19,9
19,2
19,2
20,3
20,3
20,3
20,3
CUtCOil Telatile· FC
IA m 7,9
6,4
4,9
5,4
16,2
11.4
16,0
24,4
90,9
92,6
94,1
93,2
11,9
79,9
80,3
73,2
ar V/U
34,3
35,2
31,5
34,7
32,8
32,5
32,7
31,0
Iriibilit» I
17,1
16,3
31,5
34,7
32,8
32,5
32,7
31,0
wiøt XA
35,0
35,5
21,6
21,0
34,0
35,3
35,1
37,8
m i e t i l a CH bern
t I
39,8 56,6
41,1 63,1
33,6 44,1
32,1 41.5
34,1 53,1
35,3 55,3
35,2 56,1
34,6 57,7
irrUHTS/U drr nod I n end Cndeu Tot tol U 0120 tali h3
0,16 434 0,70
0,17 414 0,(1
0,19 426 1.72
0,19 399 0,(9
0,11 450 0,(7
0,12 409 0,63
0,13 433 0,67
0,10 371 0,56

Table 2
535
Potential Energy of Effluents (per kg of dry wood carbonised)
Test N' 1 2 3 4 5 6 7 8
Non cond. gases MJ 2,08 2,21 2,47 2,47 1,43 1,56 1,69 1,30
Condensibles MJ
2,28 2,17 2,24 2,10 2,36 2,15 2,27 1,94
Total MJ 4,36 4,38 4,71 4,57 3,79 3,71 4,96 3,24
MJ/Nm3
6,22 6,44 6,54 6,62 5,66 5,89 7,4 5,78
Table 3 : SSE required & heat of reaction (per kg dry wood)
Test N' 1 2 3 4 5 6 7 8
SSE (kJ) meas (ra 1 370 0 (*) 900 850 400 430 170
0 (*)
calc (c) 2 610 210
1 460 1 360 530 530 430 300
Exothermicity (c) - (m) (kJ)
1 240 210 560 510 130 470 260 300
(*) negligible Table 4 : Comparative SSE requirement data
Retort system 1. Lab retort
(well insulated) capacity 9, 7 lit
2. M.S.T. single retort capacity 3 m3
3. A.C.C. two retorts coupled capacity 5,6 m3 each
Source of suppl. heat
electricity
wastewood
i)wastewood . without emissions recovery . with emissions recovery ii) propane
Wood X humidity
0-47
14 - 23
11,7 - 47
11.7 11,7
SSE cons. Mj/kg dry wood
(*) 0 - 1,37
4,8 - 8,5
3,16 - 7,2
0,88 1,3 - 1,54
(*) negligible

536
MILD PYROLYSIS PROCESS IMPROVES STEAM CYCLE EFFICIENCY
P. Graversen, R.M. Hummelshøj, and Κ. Jenslev COWIconsult, Consulting Engineers and Planners AS
45 Teknikerbyen, DK-2830 Virum, Denmark Telephone 45 42 85 73 11, telex 37 280 cowl dk.
Summary
This paper describes a new design for combining a traditional steam cycle power plant fueled by municipal refuse with a pyrolysis unit for biomass e.g. straw.
The electricity gain from the traditional plant is limited by a superheating temperature of 380-430°C. Above this temperature chlorides in the fluegas mainly from PVC in the refuse will result in high-temperature corrosion.
In this new concept the much less corrosive pyrolysis gas is used in a separate super-heater in order to increase the steam temperature above this limit. Approx. 80% of the energy added to the steam in the super-heater can be converted to electricity. The degassed biomass is utilized in the furnace/boi1er increasing the steamflow and hereby also increasing the electricity production further.
The pyrolysis unit is designed as a simple duct which is heated externally to 600°C by fluegasses and through which the compressed biomass is fed using a hydraulic piston machine.
The overall efficiency and electricity production from the biomass resource, in this case straw, can be compared with the best gasifier-engine based system but without the well-known operational problems associated hereto, e.g. tar-cracking, condensate removal, clogging and slag sintering.
1. Background During participation and work in an adviser group for the Danish
Ministry of Energy, 15 gasification plants, mainly in industrial scale in various countries, have been visited. The aim of the work was to registrate state-of-the-art and to point out R&D areas of interest for Denmark.
The biggest unexploited biomass resource for gasification and pyrolysis purpose in Denmark is straw and municipal organic waste, but energywise utilization of this resource is mainly seen as incineration plants for production of district heating.
Straw is, however, abundantly available in Denmark and the northern agricultural zones in general, and abroad there is hardly no previous experience abroad with pyrolysis and gasification of straw. Therefore the R&D of processes where this resource can be converted to electricity are given a high priority in Denmark.
In the following, one of the new promising concepts is described.

537
2. Limits of traditional processes Through the previously mentioned study of the state-of-art for many
pyrolysis and gasification methods already tested, the basic knowledge gained was to a large extend, how not to do. Therefore, our approach is to develop a very simple process that works and short-cuts the well-known limits and problem areas of traditional concepts.
The limits for the traditional steamcycle concepts are:
* High-temperature corrosion, slag sintering, and erosion in the super-heater due to high content of chlorides - mainly from PVC in the refuse - and fly-ash in the fluegas.
* Low power/heat ratio due to low superheating temperature of max· 380°C-430°C in order to reduce the speed of the above mentioned corrosion.
Corrosion
Surface temperature 500 700 °C 100 300
Fig. 1 Corrosion speed by fluegas from combustion of municipal refuse.
Experience shows that straw is not readily accepted by any of the traditional gasification and pyrolysis processes due to associated technical and economic problems.
The limits for earlier straw gasification concepts are:
* Power and maintenance demanding pretreatment i.e. chopping of straw
* Clogging of silo and gasifier * Slag sintering and ash softening above 800°C * Difficult gas cleaning in combination with engine/gen-set
(tar/condensate formation and dust removal)
3. Principle of the new concept The pyrolysis unit is combined with a traditional steam cycle
power plant fueled by municipal refuse, see dotted line on fig. 2. Non-chopped straw is fed through 1-3 parallel pyrolysis ducts
depending on size - using a hydraulic piston machine. The straw is compressed to a density above 160 kg/m^.

538
The first section of the pyrolysis is perforated and acts as a convective dryer zone using fluegas circulation.
The second section is heated externally with fluegas from a separate super-heater, see fig· 2, in order to pyrolyse the dried straw at approx. 600°C, i.e. below the ash softening temperature.
The pyrolysis process is initiated using another heat source. The hot gas generated incl. the vapours of the tar fraction are
representing more than 50% of the energy content of the energy content in the initial straw, and are utilized in the separate super-heater without letting the temperature of the pyrolysis gas fall below the tar dew point at any place.
Approx. 80% of the energy added to the steam in the super-heater can be converted in the turbine to mechanical energy/electricity.
As the gas has a low content of chlorides and particles, the additional superheating will not create corrosion problems, as would be the case when increasing the superheating temperature in a traditional concept where the superheating is performed by the fluegasses directly from the incinerator plant.
The degassed 'straw coke' is also utilized to increase the electricity production as it is fed into the furnace/boiler which creates the heat necessary to evaporate the steam. The steam is evaporated at a temperature below the limit for high-temperature corrosion. Hereby this straw based top-up fuel raises the calorific value of the municipal refuse basically used, which results in an increased steamflow/electricity production.
The overall efficiency will be above 80% for the straw utilization itself, and with a power/heat ratio of approx. 0.7 comparable with the best gasifier genset based systems but without the well-known problems associated hereto.
The extra capital costs of a complete pyrolysis plant for a 4 t/h waste to energy plant based on municipal refuse incl. the necessary modifications are below 0.7 mill ECU, which amount shall be compared with an increase in the electricity output of 26-58%.
The advantages of the new concept can be summarized in the following headlines:
* Improvement of the electricity gain by 26-58% * Essential reduction of high-temperature corrosion * No risk for ash softening and slag sintering * No demand for gas cleaning or tar cracking * No condensate disposal demand * Low N0 X combustion at approx. 1200°C * Low demand for moisture control of biomass due to built-in
dryer section with heat recovery * Easy handling (no pretreatment, stirring devices,
adding/regeneration of catalysts or need for modification of a gas-engine)
* Due to the simple pyrolysis unit design can presumably be modified to handle other biomass fuels, e.g. wood chips, shells, park and garden waste or a mixture of these and straw
* Low marginal cost

539
ι I
380167 bar
1 Seperate super-heater
Municipal refuse
>
Ruegas to filter/scrubber S
^
4851 66 bar
Pvrolvsls gas
Fvrolvsis unit
■e-
/T\ I District \ K J\ heating/ > \ry IhBatsink /
Fig. 2 Simplified process diagram
Municipal refuse 100%
4t/h 11.6 MJ/s 10440kJ/kg
Furnace Steam 3 8 0 t
Waste
Turbine
Mech. Dower Generator + gear
Condenser
Power/hea
Electricity 17.7% }
n \
56% Heat
/
it ratio = 0.31
2.0 MWe
\ >6.5 MJ/s
Fig. 3 Sankey diagram for a traditional waste to energy plant
760kg/h Straw 26%
f Ruegas
Pyrolysis unit
Pyrolysis gas
η [co ke
Municipal waste 100%
4t/h 11.6MJ/S 10440kJ/Xg
superheater
Furnace
Steam 485°C
Waste i
^ \ ^
Turbine Mech. power Bedridtv
Generator + gear
Condenser
Power/h
28% \
i \
70% Heat
/
eat ratio = 0.4
3.2SMW
VlMJ/s
Fig. 4 Sankey diagram for a waste to energy plant improved with the pyrolysis unit

540
4. Present Stage and Perspective - Our concept has in 1987 been awarded the first prize in a Danish
competition for utilization of non-imported fuels for decentralized co-generation power plants, funded by the Danish power company ELKRAFT.
- Laboratory tests with the pyrolysis unit carried out by the TECHNICAL UNIVERSITY OF DENMARK during last year have shown very positive results.
- Product development and engineering are carried out in collaboration with the Danish manufacturer VØLUND.
- The straw handling and hydraulic piston feeder machines are already developed to commercial scale in the MW size.
- Patent rights have been claimed for the principle and involved machinery.
- A pyrolysis unit for 120 kW is scheduled for test during autumn of 1989.
If the test in pilot scale also confirms the present optimistic views, then the path is open to full size demonstration in the MW size. It is expected that the concept has wide perspectives, especially in countries where end-disposal of waste involves a fee, which, together with sold electricity and in many cases district heating, will serve as a source of income for this type of plant.
REFERENCES
(1) COWIconsult, Vølund Energy, The Technical University of Denmark, Laboratory for Energetics (1989), Development of a Pyrolysis Unit, a) Main Report b) Report of Experimental Work c) Appendices (Danish Language).
(2) Janzen, P., The Danish Corrosion Centre (1988), Proceedings of a conference on corrosion problems associated to biomass fueled decentralized power plants (Danish Language).
(3) Ministry of Energy, Energy Research Programme, Steering Committee for Biomass, Denmark (1987). Report on biomass to energy utilization via thermochemical conversion methods (Danish Language)

541
FLASH PYROLYSIS OF BIOMASS FOR LIQUID FUELS
S A Bridge and A V Bridgwater Energy Research Group
Department of Chemical Engineering and Applied Chemistry Aston University Aston Triangle
Birmingham B4 7ET UK
ABSTRACT
Fast or flash pyrolysis has been shown to give very high yields of liquid "bio-oil" on laboratory scale processes of up to 70%. The principle of flash pyrolysis for liquids production is that very high heating rates are combined with relatively low temperatures and with rapid quenching of liquid intermediate pyrolysis products. Several techniques have been employed including fluid beds, entrained flow reactors, cyclone and vortex reactors; and the principles employed include gas-solid heat transfer as in entrained flow reactors and solid-solid heat transfer as in ablative pyrolysis. This paper reviews the reaction mechanisms, pathways and kinetics of pyrolysis by reference to the flash pyrolysis technologies employed to date. Conceptual designs for liquid producing pyrolysis processes are developed from specifications laid down for process performance by evaluating known theories of reaction pathways and mechanisms, and by examining the resultant technical requirements.
INTRODUCTION Pyrolysis produces a solid char, liquid products (tar and pyroligneous acid) and a fuel
gas, the proportions of which are dependent on the process conditions and the nature of the feedstock. Current interest is focussed on liquid fuels which include the crude pyrolysis liquid or "bio-oil", upgraded products such as gasoline and diesel, char-water slurries and char-oil slurries. The first of these, bio-oil, is of particular interest as it is a liquid that can readily substitute for conventional hydrocarbon fuel oils, and it has been produced on laboratory scale processes at yields of up to 70% using fast or flash pyrolysis technology.
This paper prescribes a specification for an ideal pyrolysis process. By analysis of reaction mechanisms, reaction pathways, models of pyrolysis processes, and evaluation of technical requirements, a conceptual process is outlined and compared to the initial specification.
SPECIFICATION The specifications of an ideal pyrolysis process are set out in Table 1. While clearly
these are not all possible and some are contradictory, this does identify the main features that a successful and competitive process has to address. After consideration of the principles of pyrolysis, these are re-examined to form the basis of a potentially successful plant.
Pyrolysis liquids approximate to the original biomass in elemental composition by the following idealised relationship:
C6H9O4 —> C6H9O4 In practice the following reactions can be considered to occur in flash pyrolysis:
2C6H9O4 —> 2C6H7O3 + 2H2O 4C6H9O4 —> 16CO + 8CH4 + 2H2
2CO + 2H2O —> 2C02 + 2H2
6C6H9O4 —> 2C6H7O3 + 14CO + 8CH4 + 4H2 + 2C02

542
Table 1 Ideal Pyrolysis Process Specification for Liquids Production
PROCESS Minimum feedstock pretreatment and preparation Maximum feedstock versatility and insensitivity to feedstock characteristics Minimum heat input to reactor Minimum heat losses Minimum reaction temperature Good separation of oil and water phases Minimum number of product separation steps Minimum product stabilisation and/or upgrading Scaleability
PRODUCTS QU Char Gas Water Maximum oil yield Minimum char yield Minimum gas energy Minimum yield Maximum oil HHV Minimum volatiles As clean as possible Minimum COD Minimum char content Neutral pH Minimum oxygen content Minimum water content Neutral pH Easy upgradability Low viscosity Single phase High stability
REACTION PATHWAYS AND MECHANISMS Pyrolytic reactions are very complex and consist of both concurrent and consecutive
reactions which results in a variety of products. The general overall mechanism for fast pyrolysis is dehydration, depolymerisation followed by melting and vaporisation to form vapours and gases with minimum char formation. However different pathways may be taken depending on the process conditions.
The most recent hypothesis on reaction pathways of fast pyrolysis has been proposed by Evans and Milne (1) based on molecular-beam, mass-spectrometric sampling (MBMS). They postulated that the primary vapours formed in the rapid pyrolysis of biomass under high pressure (but in the absence of catalysts or reactive environments) appear to have the same composition as the vapours obtained directly from low-pressure pyrolysis. Under high pressure conditions, the direct formation of a liquid product mixture is due to the pyrolysis of wood that has become "plastic" at the temperatures and pressures involved. This has been confirmed by Diebold (2) and Ledè et al. (3) who have shown that under direct-contact fast pyrolysis conditions, wood exhibits many properties of a "molten plastic state". At low pressures they found that the charcoal formed, though retaining structural features of the wood, shrinks, which could indicate a plastic state where pyrolysis products pass directly into a liquid state before devolatilisation. The direct formation of gaseous species from primary pyrolysis reactions, known as prompt gases, are primarily CO2, H2O and CO. These are largely associated with the char forming reactions.
The sequential transformation of the primary products in the vapour phase is shown in Figure 1 as passing through three stages. In the first stage at temperatures between 500-600°C, slight cracking reactions occur before substantial conversion occurs to permanent gases. The higher molecular weight lignin products are cracked to lighter aromatics and oxygenates. The second stage occurs from about 600-700°C with the formation of secondary products characterised by CO, light olefins and aromatics. Aromatics are also formed from the primary products of carbohydrate degradation at this stage. The third stage above 700°C, gives tertiary products from thermal degradation which include CO, CO2, H2, CH4, low molecular weight

543
saturated and unsaturated hydrocarbons and some polynuclear aromatics. These are generally formed only in high temperature conversion processes such as gasification and combustion, and in low yield.
lePyrolytlt severity ι Blomais
Solid P h a i ·
Liquid P h i s ·
Gat Phas ·
Primary procassas ISecondary processes; <| |«Ter t ls ry processes s»|¡ts~
Figure 1 Reaction Pathways in Biomass Flash Pyrolysis (1)
MODELLING Modelling implies mathematical descriptions designed to help in the analysis and
understanding of complex physical or chemical processes. The development of a mathematical model can be theoretical using physicochemical principles, empirical based on experimental data, statistical or any combination of the three. The need to develop models for pyrolysis may be attributed to the following reasons:
• to provide a step for establishing better reactor design techniques, • to develop a diagnostic tool for evaluating the importance of the various system
parameters such as particle size, heat of pyrolysis (reaction) and thermal properties of the feedstock and products,
• to identify system characteristics or conditions useful to researchers, • to predict devolatilisation behaviour as a function of reactor conditions like weight
loss, volatile release rate, product distribution and composition and temperature profiles,
• to be applied when experiments are too expensive to be conducted or are not possible. A knowledge of the system kinetics is necessary in order to satisfactorily design or
simulate the process equipment. Several models have been proposed and a few are outlined in the section below.
Kinetic models of pyrolysis The pyrolysis process has been shown to be very complex and is a dynamic process in
which reaction quenching is a critical part of the process for obtaining liquid products. The pathway generally quoted for the overall pyrolysis process is
biomass —> char + volátiles and the rate of the above reaction is described in the form of a first order Arrhenius type rate law:
dp/dt = kexpE/RT(ppf) where ρ represents the local density, pf the final density, k the preexponential (frequency)

544
factor, E, the activation energy and R, the universal gas constant. If the sample is heated at a constant rate M, then:
dp/dt = -k /Mexp-E/RT(p-pf ) The kinetic parameters, E and k, are usually derived from weight loss data obtained by
either dynamic or static thermogravimetric analysis (TGA). Owing to the different experimental conditions used to carry out these studies, considerable variations exist in the published values of kinetic constants. This makes it very difficult to generate general pyrolysis models due to the specificity of essential data to specific experimental conditions and feedstocks. These models cannot be used to predict product yields. Other methods used for predicting weight loss have modelled the activation energy as a function of the extent of reaction (4), used constant kinetic parameters but different orders of reaction (5), and used different kinetic parameters for different parts of the reaction (6).
The modelling of biomass pyrolysis in terms of individual products has also been proposed (7). However kinetic parameters cannot be predicted a priori and must be estimated from experimental data, a problem that increases as the number of reactions postulated increases. It has been suggested that this model should be viewed as a method for correlating data and comparing results from different biomass materials.
Rate laws based on a competitive mechanism have also been proposed to account for the variations in product yields with temperature and heating rate. Bradbury, Shafizadeh et al. (8) and Diebold (9) have used this technique to investigate the kinetics of cellulose pyrolysis.
Recently kinetic models for the secondary decomposition of primary pyrolysis tars have been proposed (10,11,12). Testing of these models showed that predicted liquid yields agreed with achieved yields within ± 10% for the temperature range 500-700°C with residence times of up to 1 second (13,14).
Different approaches have been used to develop kinetic models. However, most have been developed for predicting the weight loss of biomass rather than the product yield and distribution. One research group found that even a change of 1 kcal/mol in the activation energy of a competitive, multi-step reaction scheme caused changes in the product yield and distribution (15). Hence research in this area of pyrolysis requires further investigation.
For a complete model of pyrolysis the kinetic equation must be coupled with equations describing transport phenomena.
HEAT TRANSFER Several reactor configurations have been employed for flash pyrolysis such as fluid
beds, entrained flow reactors and cyclone reactors, and each configuration heats the feed in a different way. In fluid beds the feed is heated indirectly by convective and radiative heat transfer from hot solids such as sand, in entrained flow reactors heat transfer is mainly by convection from hot gases, and in cyclones heat transfer is by intimate contact of the particles with the hot wall surfaces. In the latter case, solid-liquid-solid heat transfer occurs which is significantly different from the gas phase heat transfer processes in fluid bed and entrained flow reactors, and this is known as ablative pyrolysis. Higher heat transfer coefficients are achieved using this technique resulting in potentially higher reactor specific capacities and smaller reactor volumes as there is no need for a heat transfer gas. Also this very high rate of heat transfer in ablative pyrolysis results in the almost exclusive production of liquids and gases which makes this process attractive for the conversion of biomass to liquid fuels.
Heating rate Heating rate and final temperature both play important roles in the product distribution
and yields from pyrolysis. At low heating rates of less than l°C/s and at relatively low temperatures of less than 500°C, char, gas and liquid products are produced in approximately equal amounts. Flash and fast pyrolysis, including ablative pyrolysis, are claimed to involve extremely high heating rates of up to lxl06°C/s and very short residence times of less than 1 second in order to maximise either liquid or gas production while minimising char formation. If the desired product is liquids, moderate temperatures of between 400°C to not more than 650°C and rapid quenching have to be employed so that pyrolysis vapours are condensed before secondary reactions occur. Higher yields of gases are obtained at temperatures greater

545
than 650°C. The main focus of pyrolysis research in Europe currently, is the production of liquids either for direct use or for upgrading to give high quality hydrocarbon fuels such as gasoline.
CONCEPTUAL DESIGN OF PYROLYSER FOR LIQUID FUELS Theory
From the specifications outlined in Table 1 and the comparisons of the flash pyrolysis technologies mentioned above, ablative pyrolysis appears to be the most attractive route to employ for the production of liquid fuels. This is due to the high heat transfer rates, low reactor volume, better temperature control, and potentially higher liquid yields resulting from these advantages.
Fundamental research on ablative pyrolysis has been carried out by Ledè et al. (3). A stainless steel disc of 7.5 cm diameter, spinning at a constant and controlled speed, was heated to different temperatures by four gas burners underneath. Wood dowels were pressed vertically onto the hot surface by weights on the upper part of the rod. From their experiments, the following relationships were obtained: (i) V = a.p
where V is ablation rate, m/s (speed of consumption of wood dowel) ρ is pressure, Pa a is constant, m slPa1
(Ü) h = 0.017 ρ where h is heat transfer coefficient, W m2K4
ρ is pressure, Pa (iii) Td = 739 Κ (466°C)
where Td is temperature of decomposition Ledè believed that decomposition occurs and is completed when the temperature of
wood reaches 466°C independent of the reaction conditions. This behaviour of wood was likened to the phase change phenomenon of fusion.
Practice Based on this work, a conceptual design study produced the specification outlined in
Table 2. The pressure between the particle and the reactor wall needs to be as high as possible for optimum results, but there will be mechanical considerations in meeting this objective. Other aspects that require resolution in the experimental development is the masking effect of multiple particles on the interaction of particle and wall and volatilisation of liquid products. The vaporised liquids require rapid removal and quenching to avoid secondary reactions. Partial vacuum operation would avoid the need for flushing gas.
Table 2 Specification of an Ablative Pyrolyser for Liquids Production
Biomass throughput, d.a.f. kg/h Temperature, °C Pressure (particle on reactor) Heat input, kJ/kg Effective area for ablation, m^ Estimated actual area, m^ Minimum relative velocity, m/s
Derived from Specified Literature Ledè Estimated Ledè Estimated Ledè
Experimental 5
600 As high as possible
2000 3 χ IO"3
9 χ IO"3
1.2
Commercial 1700
600 As high as possible
2000 1 3
1.2
CONCLUSIONS As a result of the above work, the main conclusions are :
• Ablative pyrolysis is the most promising route to employ for the production of liquid fuels based on the ideal pyrolysis process specifications and characteristics outlined.
• Further research is required in the area of the of pyrolysis kinetics.

546
REFERENCES "1 . Evans R J and Milne Τ A, "Characterization of the Pyrolysis of Biomass 1:
Fundamentals", Energy and Fuels, American Chemical Society Journal, 1, No 2, 1987. 2. Diebold J Ρ and Power A J, "Engineering Aspects of the Vortex Pyrolysis Reactor to
Produce Primary Pyrolysis Oil Vapours for Use in Resins and Adhesives", in Research in Thermochemical Biomass Conversion, Phoenix, Arizona, USA, April 1988, Bridgwater A V and Kuester J L (eds), ρ 609, (Elsevier Applied Science Publishers, London and New York, 1988).
3. Ledè J, Panagopoulos J, Li Η Ζ and Villermaux J, "Fast Pyrolysis of Wood: Direct Measurement and Study of Ablation Rate", Fuel 64, Nov. 1985, ρ 1514.
4. Tran D Q and Rai C, "A Kinetic Model for Pyrolysis of Douglas Fir Bark", Fuel 57, 1978, ρ 293.
5. Madorsky S L, Hart V E and Straus S J, Journal of Research of the Natural Bureau of Standards, 56, (6), 1956, ρ 343-354.
6. Tinney E R, "Tenth Symposium (International) on Combustion". The Combustion Institute : Pittsburgh, 1956, ρ 925.
7. Nunn Τ R, Howard J B, Longwell J Ρ and Peters W A, "Studies of the Rapid Pyrolysis of Sweet Gum Hardwood", in Fundamentals of Thermochemical Biomass Conversion, Estes Park, Colorado, Oct 1982, Overend R P, Milne Τ A and Mudge L K (eds), Elsevier Applied Science Publishers, New York, 1985, ρ 293-314.
8. Bradbury A G W, Sakai Y and Shafizadeh F, Journal of Applied Polymer Science 23, 1979, ρ 3271-3280.
9. Diebold J Ρ and Scahill J W, "Ablative Entrained-Flow Fast Pyrolysis of Biomass" in Proceedings of the 16th Biomass Thermochemical Conversion Contractors' Meeting, Portland, Oregon, 1984, ρ 319.
10. Diebold J, "The Cracking Kinetics of Depolymerized Biomass Vapors in a Continuous Tubular Reactor", MASc Thesis, Colorado School of Mines, Golden, CO, USA, 1985.
11. Liden A G, "A Kinetic and Heat Transfer Modelling Study of Wood Pyrolysis in a Fluidized Bed", MASc Thesis, University of Waterloo, Canada, 1985.
12. Gorton C W and Knight J A, "Oil From Biomass by Entrained-Flow Pyrolysis", Biotech, and Bioeng. Symp., No 14, 1984, ρ 14-20.
13. Scott D S, Piskorz J, Bergougnou M A, Graham R and Overend R P, "The Role of Temperature in the Fast Pyrolysis of Cellulose and Wood", Ind. Eng. Chem. Res., 27, 1988, ρ 8-15.
14. Vasalos I, Stoikos T, Samolada M, Achladas C and Papamargaritis C, "Production and Utilization of Synthetic Liquid Fuels", 'EEC Contractors' Meeting - Energy from Biomass', Paestum, Italy, May 1988.
15. Krieger-Brockett B and Glaister D S, "Wood Devolatilization- Sensitivity to Feed Properties and Process Variables" in Research in Thermochemical Biomass Conversion, Phoenix, Arizona, USA, April 1988, Bridgwater A V and Kuester J L (eds), Elsevier Applied Science Publishers, London and New York, 1988, ρ 127.

547
GASIFICATION OF REFUSE, A PROCESS OF SFW
Dr. H. HUMMELSIEP and Dr. H. FUNK Saarberg-Fernwarme GmbH, Sulzbachstrasse 26, D 6600 Saarbrücken, FRG
Summary
Since 1974 Saarberg-Fernwarme GmbH has been engaged in the technology of gasifying municipal and industrial waste. The first pilot plant was started up in 1975 at Neunkirchen/Saar. This unit was designed to process solid waste at a rate of 100 kg per hour, and was intended to prove the feasibility of this system.
The knowledge gained from this operation was used to establish criteria for the design and construction of a large demonstration plant for the treatment of industrial and municipal.solid waste at a capacity of one ton per hour. This project was supported by the Ministry of Research and Development at Bonn. Having proved the feasibility of the system, this operation has since been terminated after about 15 000 operating hours.
At the present time, plans have been formulated for the construction of a commercial waste gasification plant to treat 120 tons per day of refuse. It is contemplated to start operation of this plant within the framework of a research and engineering project. Once a successful operation has been established, this plant will continue to serve as a solid waste utilization plant to process refuse as delivered.
1. PROCESS DESCRIPTION
The process is carried out in three phases :
- preparation of solid waste; - gasification and gas purification; - water purification.
Solid Waste Preparation
The refuse is transferred from a pit on a conveyor, which carries the material to a screen, where fines are discharged. The larger pieces are dumped into a shredder and the chunks are reduced in size to smaller than 4 inches by 4 inches. Then the Fe-scrap is discharged magnetically and the sorted material is charged to a vibrating screen, and classifier. Thus the material is separated into three fractions. They are: the light fraction, the heavy fraction, and the fines. The fines are composed mainly of inerts and are discharged to a landfill. The light fraction, containing mostly paper and plastics and the like, is shredded once more and compacted to a kind of nodule. These nodules together with the heavy fraction are charged to the reactor for gasification.

548
Gas Generation and Purification
The gas generating reactor is designed as a vertical cylinder at an OD of about 3 metres and a height of 5 metres. The upper section is lined with refractory and the lower one in the area of the reaction and ash holding zone is furnished with a water wall. The reactor is fed from the top where the charge passes through sliding gates preventing any escape of gas. The material is then exposed to the following phases of treatment:
- drying phase ; - degassing phase; - gasification and reduction phase; - combustion phase.
The bottom of the reactor is sealed with water by standing in a tray where the ash is retained. While turning and stirring the bed of ash a scraper disposes the ash into the tray. This churning action stirs the ash and promotes a balanced combustion of the remaining carbon. The required oxidizing fluid is injected into the reactor in the form of air through a cone-shaped grate. Depending on the desired consistency of the gas to be generated, one can inject pure oxygen or oxygen enriched air. A small part of the material is burned in the lower reactor section in the presence of oxygen. The gas free of oxygen is heated to a temperature level of 950-1000°C. This gas passes upwards through the reactor and the reducing zone where carbon and hydrogen compounds are formed to compose the so-called producer gas, after the gasification and pyrolysis phase has been completed.
The product gas is discharged overhead from the reactor and passes through a hot multi-cyclone, designed for high temperature resistance, and to retain fine particles and dust.
The scrubbing of gaseous, water soluble harmful components is carried out with recycled water and lye added. After this 'cooling down phase' a purified gas is available of medium heating value of about 7000 kj/m3 , provided oxygen enriched air is used as the oxidizing means. The measurements of emissions after combustion indicate that no harmful components are detected.
It is possible to separate components such as hydrogen, carbon monoxide and methane, in a further processing step, if so required. Thus a synthesis gas for the production of methanol can be prepared by employing regenerators.
Water Purification
Solid waste generally contains moisture at a rate of 20% to 30%. After its condensation this water has to be separated from tars and oils in a decanter. The oils and tars in turn are recylced to the reactor, unless they are to be utilized otherwise, while the water flows to the first basin as part of a treatment system. There the water is purified in three steps:
- chemical treatment; - biological purification; - filtration through activated carbon.
The chemical treatment results in separation of iron, zinc, lead, and other heavy metals in the form of hydroxides.
Then by adding phosphates in a reactivation unit the water is subjected to biological treatment.

Legend 1) Screening 2) Shredding 3) Magnetic separator 4) Mechanical separator 5) Dryer 6) Shredder 7) Compactor 8) Reactor 9) Dust separator 10) H2O washer 11) Alkali washer 12) Clean gas compressor 13) Boiler 14) Separator 15) Cooler 16) Cooling tower 17) Two-stage water treatment 18) Steam generator 19) Air separator 20) Oxidant heater
A i r ^ | 0
Τ 19
Air
20
Larger fraction,
f =
1 Medium fractior
18
Raw gas
V _ Ό
Fine dust Tar/water mixture
Freshwater 4*. ^5
Tar and o i l Waste water
17
Tar and o i l Out fa l l .
—Fresh lye
Circulating water
Π1 4
. (tfaste lye
Flow Chart - Gasification of refuse by the SFW-Funk process

550
The final step of purification is accomplished in an activated carbon filter.
Some of the data obtained from the results of these test runs are beyond the limits of the required standards of water purification. However, by optimizing the operating condition and the incorporation of an activated carbon filter the required standards of purification can easily be met.
2. POTENTIAL OF APPLICATIONS
Solid waste gasification is closing a gap in various methods of thermal decomposition where annual capacities in the range of 30 000 to 100 000 tons per year of refuse are available for utilization. Therefore it ought to be of special interst for counties or medium sized communities. The operating costs for utilizing solid waste compare with an incinerator handling a capacity of about 200 000 tons per year of garbage.
The application of this process is not limited to the treatment of household waste and similar industrial waste, since this gas generator is capable of consuming a wide range of refuse. Waste from the woodworking industry, agricultural waste, leather and textiles, to name only a few, can be utilized while recovering energy and operating without causing pollution.
The purified gas basically offers the same possibilities of applications and usage as natural or city gas. However, it should not be piped into an existing grid of natural gas because of the high content of CO, but it is useful as a fuel for direct heating in industrial projects, for instance drying kilns or boilers, and water heating facilities (for process heat or district heating).
Pipelines cost less to construct than piping for district heating, hence product gas can be brought economically to the point of consumption to be converted to heat and/or electric energy at district heating stations with gas engines, gas turbines, or absorption heat pumps.

551
A CATALYTIC GASIFICATION PROCESS OF BIOMASS
J. Munck Dansk Termo Industri/I. Krüger AS
Summarv
The paper presents a technical concept for a biomass gasification plant with an output of 1,2 MW supplying gas directly to the gas engine delivering electrical power to the public grid as well as heat to district heating. The plant will be situated in Højreby Municipal in Denmark.
1. INTRODUCTION The gasification process is characterized by coupling of the
following well-known processes: 1. Pyrolysis of biomass
) CO Cat > C 02
Biomass + heat l o o: 6 0 0» c P» ) CH4 J H ) H^O ) ashes/remaining carbon 2. Watergas production
Carbon + H O + heat ) CO
P- ) CO 800-1000° ) H.
Methanization „ C0 + C°2 + » 2 - 3 5 0 ^ ^ CH4 + H20 + heat
By using a suitable catalyst in the first process the following benefits have been achieved: a) production of a gas with a neglicible content of tar b) production of gas with a high concentration of CH, compared with a
conventional technique c) the process takes place at a temperature level lower than for a
conventional process By the second process the remaining carbon from process No. 1 is
converted to a gas by adding of water. In the third process the gas mixture from process No. 1 and No. 2
is converted to methan by using suitable catalyst. The methanization process is an exothermic process and the formed
heat can be utilized in the pyrolytic process.

552
2. RESULTS The key process in the concept is the pyrolytic process. For
nearly one year a pilot plant has been in operation with a capacity of approx. 50 kW measured as gas production.
By using straw as a biomass at the pilot plant the following key figures have been measured: Production of gas: Åpprox. 1 Nm'/kg dry straw Heat value: Âpprox. 12,5 MJ/Nm1 Composition: Approx. CO 12% vol.
CO 30% CHT 16% H 37% Ν, 1% C2
C7 4 %
Åshes: Approx. 0,2 kg/kg dry straw Carbon content in ashes: 50%
An energy calculation for the plant (process No. 1) has given the following results: Straw feeding: 100% Gas production: 77% Remaining carbon: 18% Process heat: 5%
For the process No. 2 (water gas production) only a few tests have been made, but the results have shown that the carbon is very active and consequently more or less can easily be converted to water gas. A more theoretic study has shown that the water gas process can eventually take place in the pyrolytic reactor.
This concept will be tested within the next 6 months. The conversion to methan (process No. 3) is regarded more or less
as a fully developed process which can be supplied from engineering firms specializing in catalytic processes. Therefore, this process will be added when the two other processes have been fully developed.
3. THE DESIGN As the pyrolytic reaction is the key process, the pyrolytic
reactor will be described in more details. The pyrolytic reactor (the pilot plant) has a diameter of 60 cm
and a total height of 100 cm. The reactor is indirectly heated by a heat exchanger built in the
reactor. In the top of the reactor a distributor is situated spreading the
straw all over the area of the reactor. The catalytic layer has a height of approx. 40 cm and is placed in
the bottom of the reactor on a screen. The catalytic pellets have a size of 515 mm. The working
principle is as follows: The disintegrated straw is feeded into the reactor by means of a
screw and is spread over the catalyst by the distributor. In the top of the catalytic layer the temperature is approx. 100°C
and at the bottom approx. 600°C. The straw is forced through the catalyst by a vibration technique,
and during the passage of the catalyst the temperature of the straw is increased from 100°C to 600°C.

553
The mixture of gas and ashes is separated in cyclons and the content of water is condensed and drained.
The total process is shown below:
Catalytic Pyrolysis and Gasification of Biomass
Electric energy
Aah
Î Electric generator and Gasengine .
'GaatorhM
Filter
Water drain
CooUnç. water for dlalrlcl hating
ting of UM reactor»
1 1 Ash
Gas
Straw
1 Disintegrator and Feeding silo
Catalytic pyrolysis reactor
' '
Water gas reactor
Catalytic methai lization
O (
Aah and carbon Drain water
Water drain
Cyclone
Cyclone
1
— t
It is planned that the total concept of the plant should be finished within 1989 so that the civil works can be carried out in 1990 and the production of heat of electricity could begin at the end of 1990.
4. APPLICATIONS The concept can also be used in connection with other biomasses
than straw and should therefore have a high market potential not only in the industrialized countries but also in the developing countries.
A few tests have been made on gasification of rice shells, bagasse, seed wastes, chopped wood and sludge from wastewater treatment plants.

554
AGRICULTURAL WASTES FOR ELECTRICITY GENERATION
C. ESNOOF and N. BEERAI
Centre National du lacbinisie agricole, du Génie Rural des Eaux et des Forêts BP 121 92164 ANTONY Cedex France
Sumary
The ail of the project is power production in siali units in rural areas of developing countries. The selected process is the gasification of charcoal obtained froi agricultural wastes, straw, cotton stalks, grasses. The paper presents the results obtained on a pyrolyser pilot of 100 Kg/h of straw. Straw bales are carbonized in fixed bed in continuous operation. Long t e n stabilized operation has been obtained, with 331 charcoal yield and various volatile content. The charcoal obtained is further aggloierated in a revolving pan and resulting balls are gasified in a 15 Kg/h crossdraft gasifier. Experimental results show a 0.77 energy efficiency and a LCV of gas produced of 42004600 KJ/Ni3
. Preliiinary economics show that the profitability of the line greatly depends on the operating tiie of gasifier unit.
1. INTRODUCTION The ai« of the project is power production with raw «ateríais other than wood (often rare) or
fossil fuels (often «ported, rare and expensive). It concerns siali ans lediui sized units (550 KH) in rural areas of developing countries.
The selected process is the gasification of vegetal charcoal obtained froi agricultural wastes, such as wheat and rice straw, cotton stalks and grasses.
A new concept of pyrolyser was developed and preliiinary results presented in (1). Nore ampíete results are given here, including gasification tests. A preliiinary econoiic estilate is also presented.
2. NATERIEl AND NETHOD
The processing line is presented in figure 1.
The pyrolyser is a continuous fixed bed, processing bioiass in the fori of lediui density rectangular bales (100120 Kg/i3
). It has been previsiouly described (1) The resulting charcoal is crushed to I n , lixed with a binder and then aggloierated in siali bales (20 to 30 η diaieter) in a revolving pan.
The pan used is a laboratory unit producing 50 Kg/h, but this device is comercial in sizes up to 1 ton/h. The balls characteristics are presented in table I.
Table I Charcoal balls characteristics
Coiposition : dry laterial 4Sstarch 25iash 71t charcoal
Lower calorific value (LCV) : 26 200 KJ/Kg density : 325 Kg/i3
volatile latter content : 201 (at 960"C)

« Starch
BKVOLVIKO PMI
Balls
280 to 300 Kg/n3
100 Kg/h
Agricultural Wastes
Pulverulent Charcoal
100 Kg/h
5 χ
Gasifier
Wheat and r i c e Stra·
Cotton s ta lks
300 Kg/h
100 to 120 Kg/m3
STRAW PTOOLYSER : CONTINUOUS FIXED BED WITH
' VOLATILES RECYCLING AND BURNING
COOKING
Charcoal crossdraft gasifier electricity generator
Kffrtn o y 100 KW
•Ό
! O O π> (Λ VI 3
(Ω _ j
3 Π>
π t£3 C
3
Ol

556
The resulting balls have then been gasified in a cross draft gasifier of 15 Kg/h inputffigure 2). This gasifier has been experimented on charcoal in long tiie run (2).
Variable quantities of stea« have been added to the air, so that the gasification temperature would be lowered, in order to avoid clinker formation. The steam/charcoal ratio selected is 0.47 so that a temperature of HOO'C and a very limited fusion of ashes are obtained.
3. RESULTS
Continuous stabilized operations of 7 hours have been regularly obtained on the pyrolyser. The instabilities
previously observed (1) were due to irregular straw feeding (on account of compression at the tight chamber entrance). λ specific device now controls the progression of straw inside the chamber and pilots the feeding device. The pyrolysis temperature is around 600'C with a hot gas attack at 750800'C and the stability is obtained after
3 hours of operation (not counting the first tvo hours of preheating).
Charcoal yield is around 331 on dry straw and 281 on an ash free basis. The charcoal aspect is the same as feeded biomass, straw or cotton stalks. The volatile content of charcoal depends on temperature and residence time, as can be seen in table II.
The energy efficiency of the pyrolyser is 0.55 (LCV charcoal/LCV straw) or 0.87 ( LCV charcoal +
volatiles/LCV straw) if the volatile matter is valorized elsewhere. Figure 2
Table II Volatile content of charcoal
sample
residence time
temperature fc)
volatile content
1
12
450
20.2
2
15
425
16.6
3
17
420
13.2
4
17
460
12.8
5
17
525
11.6
6
20
640
7.8
7
22
480
12.7
8
23
560
10.4
9
27
530
8.1
similar results were obtained with cotton stalks, only the pressure drop of gases in the bale being much lower.
charcoal balls gasification with steam led to a gas containing 22251 CO, 1114* H2, so a lower calorific value of 42004600 KJ/lfir. It represents an energy efficiency (LCV gas/LCV charcoal) of 0.84 without water vaporisation and of 0.77, the latter taken into account.
This means a general efficiency of the process of (LCV gas/LCV biomass) 0.4 (without valorization of volatile matter in excess).
The gas must be cleaned (filtration) before the engine as it contains around 0.16 g/m3 of
dust and 0.058 and solidity.
g/mJ of tar. The balls behaved perfectly during gasification, keeping their form

557
O P E R A T I N G TIME E L E C T R I C I T Y GENERATOR O P E R A T I N G
T I M E : 2 5 0 0 H/YEAR
2 3 4 FUEL-OIL PRICE IN FF/I
O P E R A T I N G T I M E
Α Λ Λ
4 0 0 0 H / Y E A R
Figure 3.
2 3 4 F U E L - O I L P R I C E I N F F / I
8 1 0 1 2 1 4 1 6 1 8 2 0 F U E L - O I L PRICE IN ECU/GJ 45 66 89 1 1 1 CRUOE OIL PRICE J/bbl (WITHOUT T R A N S P O R T )
22,5 33 44.5 56 CRUDE OIL PRICE S/bbl H O O K T R A N S P O R T C O S T )

558
4. PRELIMINARY ECOHOMICS : Tbe coiplete line, carbonisation to electricity production, is estimated. The pyrolyser and
revolving pan are 100 Kg/h charcoal capacity and each electricity unit is 20 KW. (1520 Kg/h charcoal). They are locally lanufactured except for the recycling fan of pyrolyser. The capital costs break down as follows (table III) :
Tbe charcoal Manufacture capital costs break down as follows (table III).
revolving pan
30
pickup baler 5T/h
50
pyrolyser construction
160
recycling fan
2 χ 15 KW
140
Total
380 Table III Capital costs in 103 FF
For the electricity generator, the additional cost as coipared with a fueloil engine is 65 to 85 103 FF.
For the gasifier unit, two operating tiie are studied : 2500 h/year for irrigation of 2 crops per year, and 4000 h for a village or craftsian electricity production. For the pyrolyser, 1000 h/year corresponds to a pluvial rice crop giving 0.8 to 1.3 ton of straw per ha and year, that is an area of 200350 ha ; 2000 h/year for the saie crop on an area of 400700 ha and 4000 h for an irrigated rice crop giving 8 to 13 ton straw/ha and year, on an area of 100 to 140 ha.
Maintenance costs are increased as coipared with european standards, up to 10Î of capital cost per year.
At first the straw is given no price, except for harvest salaries. Given a fueloil price, a laxiiui price for charcoal balls purchase is coiputed, allowing
profitability for the electricity unit. This price is coipared with the liniiui selling price for pyrolyser profitability.
The results are presented on figure 3. One can coipare the selling price of charcoal to those observed in Abidjan (Côte d'Ivoire),
lFF/Kg i 30t ; the prices of balls are equivalent for 2000 and 4000 h operating tiie. Given the fact that the oil price in year 2000 are expected to be froi 20 to 35 $/bbl, it can
be seen that the profitability, essentially a function of electricity unit operating tiie, is probable for 4000 h/year, or for 2000 h/year if the pyrolyser works 4000 h/year, i.e on irrigation crops ; in both cases, the area lust be far froi oil refineries (100t transportation cost).
Hot that those figures dont take into account the tax part in the fueloil present selling price, often leading to a 4 or 5 F/liter price.
5. CONCLDSIOH The carbonisation of straw or cotton stalks bales is technically feasible in a fixed bed
continuous reactor. But the overall process seeis quite coiplex to iipleient so that profitability of the line is only obtained when the electricity generator set is operated for a longtime, irrigation of two crops per year or electricity production for craftsmen.
6. REFEREHCES : (1) ESHODF. C al (1987) Mobile straw pyrolyser. Bioiass for Energy and Industry pp
10631067 Elsevier applied Science. (2) PLANCON. M, GOOPILLOH. J.F, ESHOOF. C (1988) Automated operation of lean gas engines
Research in thenocheiical Bioiass Conversion pp 10491056 Elsevier applied Science.

559
PYR0LY5I5 PROCESS FOR RECYCLING FOREST AND AGRICULTURAL WASTES FOR RECUPERATING BIOMASS ENERGY
B. GROUX, R. GUIOL and Ph. POUSAZ BIO-ALTERNATIVE S.A., Case postale, CH-2063 ENGOLLON
BIO-ALTERNATIVE ITALIA S.R.L., Via Caffaro 23, 1-16124 GENOVA BIO-ALTERNATIVE S.A. ESPAGNE, Calle Felix Bois 18, E-28036 MADRID
ABSTRACT
There is a need for converting biomass wastes into charcoal, oil, and gas which can be utilized in industrial or domestic equipment. Low temperature pyrolysis, which favours coal and oil production (respectively 50?ί and 20% of the energy contained in the biomass), offers particularly promising means of conversion since charcoal and oil products are storable and easily transportable. Bio-Alternative S.A. developed an industrial process in order to emphasize on charcoal production and optimization in qualitative oil extraction with respect of the environment. This technology can be used for different types of biomasses: eucalyptus wood, oak wood, ash wood, fir wood, acacia wood, genista wood, sorghum waste, grapes residues, pine cone, coffee residue, cocoa residue, coconut shell, rice husk, marc of olives, corn residue, straw. Results are given for an industrial plant treating 15'000 tons of wood wastes per year.
1. INTRODUCTION The convertion of organic wastes from broad origin (forest and
agricultural wastes) into a range of products that can be used as fuels, is a good challenge. Pyrolysis is attractive because solid biomass wastes which are difficult to burn in existing equipments can be converted into products (charcoal, oil) with advantages in transport, storage and combustion.
In this field of recuperating energy from biomass, environmental requirements are the most important task for the design of pyrolysis plants.
A technology has been developed by Bio-Alternative S.A. in order to produce charcoal, bio-oil and bio-gas from biomass wastes.
2. TECHNICAL DEVELOPMENT In order to study the processing of any biomass and to optimize
the quality and yield of the products obtained by the pyrolysis process, Bio-Alternative S.A. operates a pilot unit allowing the continuous treatment of 50-100 kg of raw material per hour.
In the fixed bed, the biomass is continuously in motion towards the oxydation zone where the partial combustion of the pyrolysis gases occures in order to maintain the carbonization process (see figure 1). Bio-Alternative S.A. carried out an extensive experimental program on the pilot plant for evaluation of a wide variety of biomasses (see following table).

560
Table 1 : Pyrolysis pilot plant; some relevant examples
BIOHASS
Eucalyptus Acacia GenlBta Beech Olive stones Rice husk Coconut shell Cocoa waste Coffee waste Bagasse of sorghun Haize Pine cone
INPUT
CAPACITY kg/hr
85,0 54,7 60,5 67,5 51,9 «2,0 93,4 65,0 70,0
«5,0 66,5 »0,0
CHARCOAL Vield kg/hr
23,Β 12,5 14,6 17,β 14,5 18, 3
24,3
22,8
24,5
14,4
14,9
27,0
HHV Energy balance kcal/kg S
7200 51
6600 34
7550 44
7275 47
7400 48 4500 48
7900 48
6000 52
6000 52
6600 51
5550 44
7900 54
yield kg/hr
7,7 6,2 4,4 9,2 6,2 ',3
16,8
2,6 5,6
4,5 1,4
12,6
0 U Τ
BIO HHV
kcal/kg
5530
5900
3B20
5350
5000 5520
4850
6300
6300
6200
7800
6100
Ρ U I
OIL Energy balance
13 15
7 18 14 10 20 6 13
15 6 19
BIOGAS
Yield kg/hr
«6,5
92,1
107,4
40,5 51,9 29,4
56,1
B5,3
70,0
40,5
52,3
7B,8
HHV Energy balance kcal/kg 5
1040 14 1064 41
B80 37
1370 20
1055 25 1024 18
1095 15
560 IB
BOO 20
560 13
720 25
4D0 Β
Beside the production of good quality charcoal, the leading points in the BioAlternative technology are 1)optimization of the biooil extraction and 2) meeting the highest antipollution requirements.
1) The selection of proper pyrolytic conditions and effective extraction system allows the production of biooil with good stability and quality, representing a high energetic density. Typically the biooil obtained has 20% maximum water content (determined by azeotropic distillation) and does not give décantation upon standing. The density is around 1,2 and the viscosity between 50cp and 150cp depending on the biomass. Energetic density is around 6500 kcal/dm3.
2) Regarding the environmental requirements, it must be assumed that everything that leaves the plant must be charcoal, biooil and biogas. Most important is the absence of waste water contaminated with toxic constituents. In the process, this problem is worked out by safely burning all gaseous effluents without condensation except of the components recovered in the biooil.
3. INDUSTRIAL UNIT PLANT Our investigations on the pilot plant resulted in the construction
of an industrial pyrolysis plant which is located in Spain (Olivenza). This plant is processing on wood wastes which are collected by a lorry equiped with a grinding machine. Capacity of the plant is 2150 kg per hour of wood chips ( 2 x 2 x 5 cm) with about 20?ó moisture. The operation is running continuously and is controlled by 3 teams of 2 men each.
3.1 Products Results of the pyrolysis process are presented in the following table: Table 2 : Pyrolysis industrial plant; results
Material Capacity Energetic balance
INPUT Oak wood 2150 kg/hr (20?ó moisture) ZXS3ZZ2ESX 100 % ¿ X V W S A X X
OUTPUT Charcoal Biooil Biogas
570 kg/hr 300 kg/hr
1755 kg/hr
m i n i , i n >>?tT*
Ι Ι 1 1 1 V, I ■
48 19 20

561
3.2 Analysis Analysis of the products are the followings Charcoal
Bio-oil
high calorific value ashes volatiles humidity fixed carbon sulfur content
high calorific value water content sulfur content
Bio-qas high calorific value : non-condensable content
7250 kcal/kg 3 %
15 % 2 %
80 % 0,1 %
5350 kcal/kg 15 % 0,3 %
1000 kcal/kg : 15 % CO; 13
6 ro 02: 3/0 % C02; 48 CH4.
% N2; 8 % H2;
3.3 Use of the products The pyrolysis plant of Olivenza is associated to a bricketting unit which production is destined to the barbecue market (total capacity : 2500 kg briquettes per hour).
Presently, the charcoal production is completely supplied to barbecue market but investigations are planned on activation process and utilization as charcoal slurry.
The bio-oil is distributed as industrial fuel and the bio-gas is used on site in the briquetting unit.
Our investigations are in progress to use them for electricity production.
4. CONCLUSION The experimental programme of Bio-Alternative S.A. on the pilot
plant permitted the realization of a "second generation" pyrolysis industrial plant.
Our present R&D programmes are the followings : - Use of charcoal as slurry for substitution to mineral liquid fuels. - Use of bio-oil in turbin and diesel systems (electricity production) - Use of bio-gas in a motor and boiler for electric and thermic pro
duction.

562
Figure 1 : Uio-Alternative S.A. pyrolysis process flow sheet
Ù-&
RAW MATERIAL INLET
OLL OUTLET
CHARCOAL OUTLET
Figure 2: Pyrolysis industrial plant in Olivenza (Spain)

563
PYROLYSIS OF GREEK LIGNITES A.A. Lappas, and I.A. Vasalos
Aristotelian University of Thessaloniki Thessaloniki, Greece
SUMMARY Two different types of Greek lignites were evaluated using the fast pyrolysis process in a fluidized bed reactor. The innovation of our experimental system is that the reactor effluent is connected on line with an FID and a TC detector for the continuous monitoring of total hydrocarbons and total gases respectively. The total yield distribution of the various products was obtained as a function of pyrolysis temperature and lignite particle size. Two kinetic models for coal decomposition were also tested for the determination of the appropriate kinetic parameters. From this work it can be concluded that: i. The produced gases from the pyrolysis of Greek lignites are dominated by oxygen compounds CO«, CO. A number of hydrocarbons are also produced (C,-Cg). ii. The experimental results are interpreted reasonably well with the competing reaction model.
1. INTRODUCTION Pyrolysis, one of the most important coal conversion process, has
special importance for the Greek economy because of the big reserves of lignite in two regions (Ptolemais and Megalopolis). It is possible the big reserves of peat in the region of Philippus will be used in the future to solve the energy problems of Greece.
Pyrolysis involves heating of coal at elevated temperatures in the absence of air and it is a chain of decomposition reactions where the linkages between aromatic clusters are broken and volatile decomposition products are produced(l)
The objective of this investigation was the evaluation of Greek lignites with respect to the distribution of gas and liquid products, and the development of a kinetic model suitable for the decomposition of Greek lignites. 2. EXPERIMENTAL 2.1 Unit description. A schematic diagram of the fluid bed reactor is shown in Fig. 1. The heart of the unit is a stainless steel reactor vessel. A close tube section at the top of the reactor is used to introduce lignite sample to the fluid bed.
The unit is also equipped with a gas feed system which includes the following gas components: N2, Air, H-0. In this work pyrolysis was studied using N- as fluidizaiion gas.
The reactor effluent passes through a stainless steel coil placed inside a cold liquid bath where the condensation of the liquids is

564
taking place. The rest of the pyrolysis products are collected in a gas collection system (in a gas cylinder by liquid displacement).
The key to the successful testing of lignite with this sytem is the withdrawal of two small sidestreams at a location downstream of the reactor and the continuous monitoring of the total hydrocarbon evolution profile with a Flame Ionization Detector (FID) and the total gas evolution profile with a Thermal Conductivity Detector (TCD). The two detectors are connected with a personal computer for the treatment of data. From profiles like these, we can find the kinetic of evolution of total HC and total gases during flash pyrolysis. 2.2 Unit Operation. A fixed amount of heat carrier (silica sand) is introduced in the stainless steel reactor. A sample of dry lignite (2 gr) is introduced in a easily friable glass container when the bed reach the desired temperature. The glass is dropped inside the fluidized bed and thereby the sample is intimately mixed with the inert heat carrier. 2.3 Gas and Liquid Analysis. The gases from the collection system were withdrawn and analyzed in a system of gas chromatographs: an FID (column porapak QS) and a TCD (column carbospheremolecular sieve). The former was used to detect hydrocarbons and the latter CO, CO, H, N, Oj.
The liquid products were collected from the stainless steef coil (which is placed in the cold bath) with a mixture of methyldichloride and methanol (1:1 v/v). The solution was removed by evaporation approximately at 47°C followed by a vacuum treatment using a rotary evaporator. 2.4 Sample Preparation. TABLE I: Elemental analysis Greek lignite samples from the Ptolemais and Megalopolis locations were selected for this study.The preparation of the samples includes: crushing of raw material production of fraction with desired size homogegenization of samples, elemental analysis(tabl .1) and proximate analysis.
3. TOTAL PRODUCT YIELD CALCULATION The flash pyrolysis of lignite produces tar, char and a range of
low molecular weight gases in various proportions and amounts depending on the pyrolysis conditions (temperature, pressure, heating rate, ambient gas, lignite type, particle size and experimental system). 3.1 Tar yield. The liquid yield increases as temperature decreases for the two lignites (Fig. 2). The reduction of the liquid yields with the temperature can be attributed to the secondary reactions that take place in high temperature. 3.2 Gas yield. The produced gases were dominated by oxygen compounds which mainly consist of CO. The final CO» yield increases with temperature with high rate at low temperature.In contrast at high temperature it tends assymptotically to a value of about 20% (of dry lignite) for the Ptolemais lignite and 35% for Megalopolis lignite (Fig. 2)· The CO final yield is influenced by the temperature in a different way than CO yield (Fig. 2). CO» is produced by carboxylic acids decomposition, a relatively low activation energy process (1). Activation
Ptolemais 1 % wt (MF)
C 52.35 H 3.52 Ν 1.89 S 1.10 0(diff.) 18.74 Ash 22.20 Moisture total 100.00
ignite % wt
20.67 1.39 0.74 0.43 7.46 8.80 60.50 100.00
Megalopolis % wt (MF)
29.48 1.83 1.51 3.40 19.96 43.82
100.00
lignite %wt
16.00 0.99 0.83 1.86
10.94 23.88 45.50 100.00

565
energies for the reactions of CO production are rather high and they necessitate elevated temperature (1).
Regarding the yield of light hydrocarbons it should be noted that each product exhibits different trend and behavior. The CpH. yield increases monotonically with temperature. At elevated temperature this increase in yield is extremely rapid (Fig. 3). Except of the hydrocarbons included in Figure 3, many hydrocarbons (C3C6) have been produced and detected from pyrolysis. TABLE II: Megalopolis lignite kinetic parameter [K (sec" ), E(KJ/Kmole)] Κ = 1.8xl06exp(93453/RT) (from Chang et al.) α
Kb = 0.448exp(27727.8/RT) Kc = 96.65exp(38406.7/RT) Kob HC = 01024exp(40253.4/RT)
Koc HC = l546exp(26038.93/RT) G = 1.322+5.47xl0"4(T500)+7.5655xl0"5(T500)2
A0 = 6.8465.89xl0"3(T500)9.251xl0"6(T500)2 [T: °C] A (% wt. dry lignite) 4. KINETIC MODELING
The kinetic model for our case must concern the production of total gas and total HC from pyrolysis as we have the production profiles from TCD and FID respectively.
It must be mentioned here that the non ideality of apparatus affects the TCD and FID signals. The non ideality of the system was revealed using tracer responce analysis tests (CH. was used as a tracer) and obtaining the RTD curve. Many different kinetic models have been proposed to explain the reaction scheme for the decomposition of coal during the pyrolysis. THe nCSTR in series model describes well our system (5). The system effect on TCD and FID signals was taken into account using the convolution integral (5).
The effect of heat and mass resistances on the kinetic model was negligible because for the experiments Megalopolis lignite with small particle size (60 µm) was used (3, 4).
Among the literature models we verified that the two competing rection model described in (2) interprets better our experimental results (Fig. 4, 5). This model refers to a single coal particle and for the total gas and total HC production takes the form:
The preexponential factors Κ Κ. and the activation energies
Aj — — > TAR — — > GAS + CHAR of each kinetic constant of model and the parameters A
Lignite G (2) are presented at Κ Table II. It was considered
A2 — — > GAS + CHAR here that A , G are not constants bat functions of temperature (6).

566
Kh u r ^ t o t a ! HC + CHAR b,HC^
A, ——> TARCCCTKU < 1 ^ ^ ^ G A S , + 1 . CHAR
t o t a l HC + CHAR
GASi + CHAR
5. CONCLUSIONS The investigation of Greek lignites under the process of pyrolysis,
using a fluid bed reactor resulted in the conclusion that Greek lignites produce high gas yield while the yield of liquids were relatively low. The produced gases were dominated by oxygen compounds which mainly consist of CO« especially at low temperature. At higher temperature the yield of CO was also high. Other gases produced in significant amounts were CH., C^H., Η„, CgHg, C,H„, CH,. Another result is that important differences were not observed between the behaviour of the two type of Greek lignites (Ptolemais and Megalopolis) and that lignite particle size has not important influence on the total products yield.
Except from the evaluation of Greek lignites our experimental system has the ability for kinetic modeling studies. The non ideality of the experimental system has been found using tracer response tests.The model of nCSTR in series simulates perfectly the system. It was found that the two competing reaction model interprets reasonably well the experimental data. 6. FUTURE PLANS
A system of traps has already constructed at the pyrolysis reactor exit. With this system we can trap various samples from the produced gases during the pyrolysis as a function of time and temperature. These samples are analyzed in gas analyzers and so the time and temperature distribution of each pyrolysis product can be obtained. These profiles are very important because they can lead to the evolution kinetic of each discrete pyrolysis product. ACKNOWLEDGMENTS
This work was supported by the Greek General Secretariat for Science and Technology and the European Community Contract EN3B/B3/102/EL. REFERENCES (1) Bautista, J.R., Ind. Eng. Chem. Fundam., 1986, 25, pp. 536544 (2) Chang, P.W., Swamy, Κ.D., Knell, E.W., Coal Process. Tech., 6, 20
(1980) (3) Agarwal, P.K., Genetti, W.E., Lee, Y.Y., Fuel., 1984, vol. 63 (4) Gavalas, G.R., Wilks, K.A., AIChE J., vol. 26, 1980 (5) Wen, C.Y., Fan, L.T., "Models for Flow Systems and Chemical
Reactors", Marcel Dekker Inc., New York 1987 (6) Gavalas, G.R., "Coal pyrolysis", Coal Science and Technology 4,
Elsevier, Amsterdam 1982

567
Fig.l Schematic diagram of f l u id bed pyrolysis reactor
N 2 O — H 2 ) ^ ( ^ — «
'5°° * ·
ώ0 ÄUiÖUdr" ^ Fig 2 Effect of temperature on
pyrolysis yields from Megalopolis l ign i te : o, C02; ■, CO; „. ™
R
Fig. 3 Effect of temperature on pyrolysis yields from Megalopolis l ign i te : o, CH^; ■, CjH^; 0 , H2
0 io io èo èo 100 izo 140 ileo tao zoo 220 240
Fig.4 Simulation of TCD pyrolysis response at 700°C:, experimental TCD response; □, two competing reaction model
Ao 100 120 140 180 ISO 200 220 240
Fig. 5 Simulation of FID pyrolysis response at 700°C:, experimental FID response; o, two competing reaction model

568
DIRECT MASS SPECTROMETRIC STUDY OF PYROLYSIS BEHAVIOR OF BIOMASS AND ITS CONSTITUENTS UNDER DIFFERENT IONIZATION CONDITIONS. MS AND MS-MS STUDY OF THE PRIMARY PYROLYSIS
MECHANISMS.
P-L.DESBENE, M.ESSAYEGH, B.DESMAZIERES, C.LANGE
andJ-J.BASSELIER
Laboratoire de Chimie Organique Structurale,
Université P.et M. Curie, 4, Place Jussieu 75230 Paris Cedex 05 (France)
Summary
Pyrolysis of biomass (wood) and its constituents (cellulose and lignin) has been studied directly into the ion source. The different ionization techniques used (NH3, N2, NH3 + N2) for the MS analysis of primary pyrolysis products led to similar pyrograms. The total ion current (T.I.C.) from biomass presented two peaks partially separated which can be attributed to two separate steps. The comparison of cellulosic T.I.C, lignin T.I.C. and fragmentograms corresponding to several ions that are representative of these components indicated that the first biomass pyrolysis products stemmed from lignin decomposition while the last one stemmed from cellulose decomposition.
Introduction
Pyrolysis of biomass, which leads to gas, liquid and char, is a very complex reaction which is difficult to study. The chemical complexity of biomass and the number of parameters influencing pyrolysis (temperature, residence time, thermal flux...) make difficult the "mastering" of this reaction.
However, advances in pyrolysis industrial application will take place only if chemical transformations encountered during the process are understood. Some studies have been published, but the problem of mechanisms and kinetics remains unsolved. These studies can be divided in two classes :
- those using thermal analysis (thermogravimetry (1-6) or GC/MS coupling in association with thermal analyses (7) - those using direct analysis of pyrolysis products by mass spectrometry (8).
This last approach is seducing because it allows to escape partly the inconvénients of thermal analysis.

569
The approach by thermogravimetiy, which uses the lost of mass to characterize conversion rates does not reckon with the nature of reaction products and the factors of their formation.
The GC/MS coupling in association with thermal analysis allows a detailed analysis of products but it is long, complex and gives a low precision. The complex problems of vapors collection and of the extreme reactivity of some species are difficult to solve.
Direct pyrolysis into the ion source of a mass spectrometer combines the advantages of the two methods mentioned above and escape their limitations. "Molecular evolution" is followed directly during the pyrolysis and the resulting vapors are analyzed on line.
Considering the work of Milne et al. (8), we have studied direct pyrolysis into the ion source of a mass spectrometer, using different operating conditions. We analyzed various wood species (eucalyptus, hornbeam, beech, chesnut-tree and birch) and their constituents (lignin and cellulose). We report here the results of this preliminary study.
Results
The samples, either woods of various species or their constituents (lignin and cellulose) were pyrolyzed directly into the ion source by means of a L.I.D. rod, with the same conditions (fast temperature rise : 9°C/s, between 40°C and 800°C, then isothermal at 800°C).
We choose chemical ionization (ionization gas : NH3 or ND3), electronic impact giving too many fragments. Examples of pyrograms and averaged mass spectra recorded during pyrolysis of extracted celluloses and extracted lignins are reported respectively in figures 1 and 2. In order to allow comparison we report the behavior of two woods under the same conditions, characterized by their pyrograms, fragmentograms and averaged mass spectra (figure 3).
Discussion and conclusion
This series of experiments shows that : a) in the case of extracted celluloses
- The shape of pyrograms is the same regardless of the wood species (figure 1). - Averaged mass spectra are also identical.
b) in the case of extracted lignins - The pyrograms are characterized by several broad signals of variable intensity, contrary to extracted celluloses. - These pyrograms are different as a function of wood species (figure 2). Moreover, extracted lignins begin to undergo decomposition during the temperature rise, that is to say , at a lower temperature than in the case of extracted celluloses. - At last, extracted lignins mass spectra averaged on pyrolysis duration are the same regardless of the spectra analyzed.

570
Β 1 0 0 %
20 %
100%.
25%
8 0 0 * 0 800 C
180
I Ifli
132 " U 41 6 2
■ I ) JÌ1I,,.Ì,I,|L
ι x16 222 240 264 282 306
32,4
1 1 |Ul...jifL .. n, .lbj.,|,ll 8 0 100 120 U P 160 180 2 0 0 2 2 0 240 260 280 3 0 0 320
342
384
330 350 370 390 410 430 450 470 4 9 0 510 530 550 570
F r g u r e 1 : P y r o g r a m s ( T . I . C . ) and a v e r a g e d mass s p e c t r u m of e x t r a c t e d c e l l u l o s e s F r o m d i f f e r e n t w o o d s p e c i e s :
A P y r o g r a m of e x t r a c t e d b i r c h c e l l u l o s e . B P y r o g r a m of e x t r a c t e d e u c a l y p t u s ce l l u l ose . C Mass s p e c t r u m r e s u l t i n g f r o m p y r o l y s i s of e x t r a c
t e d b i r c h c e l l u l o s e

571
Β 100% 100°¿
15%
800 C eoo C
209 211222 235 , Q , 301 315 "
J ' V 249 271 285 . , k^L la.1u.^Jh..T lL., i J . l | . ....É^.Ji
80 100 120 140 160 160 200 220 240 260 280 300 320
t x16 ι ¡401 42β
387M*°5*1»
) 541 9 | |
SWHÉ^llllÉiliÉÉtlllillHjt l l t l r t l Mii* iliiiy.lin ιίίΐι yi Kl I Ιΐ|ΐΐΙΙι»Ι» ι il ι
330 350 370 390 410 430 450 470 490 510 530 550 570
Figure 2: Py r og rams (T.I.C. ) and averaged mass spectrum of e x t r a c t e d l i gn ins f r o m d i f f e r e n t wood spec ies:
A P y r o g r a m o f ex t rac ted b i rch l i gn in Β Pyrograrn of ext rac ted beech l ignrn. C Mass spect rum resu l t ing f r o m pyro lys is of e x t r a c
ted b i r ch I i gn in .

Β
M r 193
Μ : 1 8 0
Τ. I.C.
8 0 0 C 8 0 0 C
180
115 134 162
■ ■■ *t H .·Ιΐ!· 222
240
1x16 I I
282
80 120 160 200 240 2 80
342 1
384
330 370 410 450 490 530
324
~320
570
'132
115
Ô0 ■ * ,
J '
'193
150 ,163 174
J 13
< I l i ; . ij. Μ ιιJ.yLjL^...i
!x16 209
120 160
■ 252 244 264 282
, 2Ί1 228 Li. ι I2701 î ,0·
5
L,. Mucuri.. |mdll|MlHi ^iilLnil..., 200 240
¿uu 280 320
333
330 370 410 450 490 1 r 530 570
570
F i g u r e 3 : P y r o l y s i s oF d i f f e r e n t w o o d s p e c i e s :
A and Β
Ç, D and E
P y r o g r a m s a n d f r a g m e n t o g r a m s of r e s p e c t i v e l y b i r c h a n d b e e c h w o o d . : Mass spec t ra c o r r e s p o n d i n g respect ive ly
to d e c o m p o s i t i o n w a v e s of cel lu lose f r o m a b i r c h w o o d and oF l i g n i n s f r o m a b i r c h w o o d a n d a b e e c h w o o d ·

573
c) For every species of wood Pyrograms have at least two broad signals. However, the pyrolytic behavior varies for each species. Beech and chestnut woods undergo decomposition during the temperature rise, while hornbeam and beech woods pyrolysis begins only at 800°C. Fragmentograms (R.I.C.) of ions 193 and 180, respectively specific of lignin and characteristic of cellulose do not vary as a function of the species pyrolyzed. It appears that the first wave of the various woods pyrograms is characteristic of the thermal decomposition of lignin, while the second wave can be attributed to the thermal decomposition of cellulose. At last, mass spectra corresponding to the decomposition of cellulose in the raw wood are identical to those of extracted cellulose. Then, cellulose undergoes the same decomposition after or without extraction from the wood. However, mass spectra of the thermal decomposition of lignin in the raw material are different from those obtained with extracted lignin. The thermal decomposition mass spectra of lignin are different for each species and it seems they can be used as an identification print.
These preliminary results are very encouraging, and we are presently pursuing the study by MSMS spectrometry.
REFERENCES
[1] W.K.TANG and W.E.NEIL, J.Polymer.Sci, & 65,1964
[2] Κ.ΑΚΓΓΑ and M.KASE, J.Polymer.Sci., 5, 833,1967.
[3] A.F.ROBERTS, Combust.Flam., 14,261,1970.
[4] D.FASENEAM, CanJ.Chem., 49_ 632,1971.
[5] K.MIN, Combust.Flam., 3JÌ, 285,1977.
[61 C.VOVELLE, H.MELLOTTEE and J.L.DELFAU, Prepr.Pap.Am.Chem.Soc, Div.Fuel Chem., 28_ 291,1983.
[η M.KOSK, I.SURINA, I.LAPCIK, I.RUCKA and V.REISER, Chem.Zvesti., 2Z 843,1983.
[8] a) R J.EVANS and A.T.MILNE, J.Anal.Appl.Pyrol, 2,57,1985.
b) A.T.MILNE and M.N.SOLTYS, ibid., 5, 93,1983, ibid., 5, 111, 1983.
c) RJ.EVANS, A.T.MILNE and M.N.SOLTYS, ibid., & 273,1984.

574
A TWIN BED PYROLYZERCOMBUSTOR FLUID BED SYSTFM FOR THFRMAI PROCESSINO OF URBAN WASTE
H. MASSON*, A. BUEKENS+, K. MANIATIS +, J. SCHOETERS0
* Seghers Engineering + Free University of Brussels
0 Groep Τ, Leuven
This paper describes the concept, the development and the operation of an interconnected fluid bed system able to generate, autothermally, a medium BTU gas from urban refuse.
In one fluidized cell, the raw waste is pyrolyzed by cooled, recycled pyrolysis gas at about 700° C, leaving char as a residue.
The bed material, mainly sand, ash and remaining char, is sent through non mechanical valves to the fluidized bed combustion chamber where the char is burned at about 850° C in an air flux.
The flue gases pass through a gasair heat exchanger and downstream cleaning equipment before being vented by the stack.
The hot bed material flows back from the combustion to the pyrolysis chamber, again through non mechanical valves. This flux of hot solid provides the heat supply needed to support the heating and the pyrolysis of the feedstock.
Experimental results on solid circulation fluxes and on pyrolysis and combustion kinetics are reported.
1. INTRODUCTION in 1988 the Körber prize was awarded to the team of Professors, Sinn and
Kaminsky for the demonstration of the thermochemical treatment of municipal refuse in an environmentally acceptable way. Prof. Buekens and Dr. Dragala were appointed as mentors to the project, with the task to advise respectively on the technical and economic aspects and on the cleanup of the wastewater generated.
The Laureates envisaged a reactor configuration based on the fluidized bed technology and the combination of gasification and combustion processes as the most promising way in tackling this accute problem. The main reason was that during relatively low temperature gasification process (about 700°C) most of the heavy metals are not vapourised and the final volume of flue gases is significantly decreased. The char remaining after the gasification proceses can be combusted to provide the heat necessary to sustain the gasification process so that the overall system can operate autothermally. There are several possible configurations for suppyling the heat from the combustion reactor to the gasification reactor but the most reliable method seems to be the utilization of a solid heat carrier as was successfully demonstrated in catalytic cracking processes using fluidized bed reactors. This paper presents a conceptual design of one particules reactor configurations to be evaluated in this project.

575
2. PROCESS DESCRIPTION
2.1 .Solid Circulation principles
The reactor configuration is based on the AVSA process comprising a combustor and gasifier fluidized bed reactors. In the combustor solid, liquid or gaseous products are combusted by air, at about 900°C. The heat generated by the combustion process is stored as sensible heat in the inert sand bed of the fluidized bed. This bed is used as heat carrier and sand to the pyrolysis reactor which operates at about 700°C. In the gasifier, the sensible heat of the sand is used to provide the heat requirements of the endothermic pyrolysis process. From the pyrolysis unit the sand carrier is circulated back to the combustor reactor where its temperature is raised again to 900°C. The principle of solids circulation is illustrated in Figure 1.
The expansion of a fluidized bed increases with gas velocity. On the other hand, the pressure drop balances the weight of solid per unit section.
In a communicating vessels fluidized bed system, in which the two sections are fluidized at different velocities, the height of the two beds is different (a and b). If an overflow is allowed, the two levels equilibrate Inducing a solid circulation (c). As shown on figure (2) several units may be placed in series. It is then possible to organize a solid ciurculation loop, without any gas mixing. This circulation principle has been applied in the present pyrolyser-combustor system.
2.2. The AVSA pvrolvser-combustor principle The reactor consits of four cells arranged as shown in figure 3. Two of the cells
operate as combustore and the other two as pyrolysers. The pyrolysis and combustion sections are connected through immerged orifices in such a way that a double communicating vessel system is realised. In each part, the two cells are fluidised at different velocities, to promote solid overflow from one cell to another.
The solid carrier flows over the partition from cell 1 to 2 and from cell 3 to cell 4. It flows through an immerged orifice from cell 2 to cell 3 and from cell 4to cell 1. Smooth operation of the system requires a great and regular transport of heat amongst the various cells but the heat supply to the gasifier is much more critical. The AVSA configuration can achieve this in the following ways : 1 ) through the sensible heat of the circulating solid, 2) by conduction through the partition wall between combustor and gasifier, 3) eventually by heat exchange pipes located in part 1 and 3, as shown on figure (3).
3. SOLID CIRCULATION TESTS ON A COLD MODEL
3.1. Instrumentation Gas flows are determined by means of using Pitot tubes situated in each of the
ducts suppling air to the four cells, of the system. Pressure drops through the distributor and the fluid beds are measured with water manometers. The pressure drop at the immerged orifices is also measured. Solid circulation is studied by injecting a pulse of hot solid (200 g, 150 degrees Celsius) at the top of cell 4. The thermal wave is detected in the several parts of the unit by small thermistors. The solid dispersion mechanism is much more rapid than non stationary conductive heat transfer from particle to particle.

576
The conduction process thus only affects the thermistor responses as a drift of the basis line.
3.3. Effect of operating parameters
3.3.1 .Solid transfer mass flow The specific mass flow is defined as :
„ (mass of solid In the system) (cycling time) (communicating holes area)
and fluctuates between 200 and 1000 kg/s m2 and presents a maximum for Ho/H -0.92. A predictive correlation has been derived from a sensitivity analysis performed on th parameters by stepwise regression, as shown on figure 4.
3.3.2. Pressure drop through the communication orifices In figure 5, a correlation is presented between the pressure drop at the
communication orifice and the solid mass flow rate. A square root law describes the results quite well. This fact allows to measure,
after calibration, the solid transfer rate by a conventional differential pressure sensor.
3.3.3 Pvrolvsis-combustlon experiments Pyrolysis experiments have been performed with wood pellets at 700°C. The gas
stream used was a mixture of steam and nitrogen and results of the experiment have been reported elsewhere (1). It was found that pyrolysis was completed after about 100s while the elemental analysis of the char revealed that about 95% of it was fixed carbon. Tests to combust the char residue in a fluidized bed were successful and showed that the burnout time was in the order of 1000 s (1).
4. Conclusions 1. A new combustor/pyrolyser system has been investigated. It is characterised
by compactness, simplicity and reliability. 2. Cold tests model have shown that mass transfer rates as high as 1000 kg/sm2
are possible between the combustor and the pyrolyser. The main operating parameters have been identified and optimised. It has been shown that the solid transfer rate is measurable by a simple differential pressure measurement, which is very attractive from a viewpoint of regulation and control.
3. Pyrolysis and char combustion studies have been successfully performed.
5. References 1. H.A. Masson, A twin fluid bed pyrolyser combustor system, Research in
Thermochemical Biomass Conversion, ed. A.V. Bridgwater, J.L. Kuester, Elsevier Applied Science, London, 1988.

577
h static
Δρ
Height of the layer
I
I 1 i
f Ü / U
mf reduced gas velocity
U/U mf
I 'U/Umf>l ' U / V ' 2 '»/"mfΊ '"/"mf'2 «"«Wi 'u/u
mf>2
F i g . 1. P r i n c i p l e s of s o l i d c i r c u l a t i o n
ΓΎΊ 'S.
*%%^
ΓΎ1!
"ν w m... m "Ί u 2 ,ûl_ Ú2J
, ui
il!
C^ 1 2 3 4 p
o φ Fig. 2. Avoiding gas mixing in communicating fluidized bed systems

FUEL FEED
O
steam steam aj.r (recycled gas)
air steam (recycled gas)
steam (recycled gas)
F i g . 3 . Layout of t h e AVSA p r o c e s s
H o . 6 9
H m = 0 .2573 I O
3 ( ( ¡ A ) "1·
3 4) ( ( 4 ) '
1 7) ( s in (
H ) π ) R e f ·9 6
°p α
ρ .42
s = h χ 1
S = H X L
Df : hydraulic diameter of the communication window = 4 s/ perimeter d = solid particles mean diameter P ■>£
x Ufa
Ref : Reynolds number relative to window
1.25 10
.75
.50
.25
.50 .75 1.00 1.25 10 Measured flux (kg/s nr)
F i g . 4 . S o l i d f l u x c o r r e l a t i o n
00

579
DP TRANSFER (mmH20)
^ h - .025 m 1 = .054 m □ h = .037 m 1 = .054 m O h = .050 m 1 - .054 m
Ufa/Umf incr.: 2, 2.5, 3 L = partition wall width = H = partition wall height -Ho = static bed height = .45 dp - 300 μ
125 .50 m
O h = X h = + h =
m m
Umf =
.075
.075
.075
.22
m 1 m 1 m 1
1 = h =
m/s
= .054 m = .035 m = .027 m
window width window height Ho/H = .9
Fig . 5. Circu la t ing system - mass flow versus pressure drop

580
PMMA PYROLYSTS FUNDAMENTALS AND EXPERIMENTAT, INVESTIGATION
A. Buekens, F. De Wolf and J. Schoeters
Vrije Universiteit Brussel Department CHIS
Pleinlaan 2 B-1050 BRUSSELS
BELGIUM
INTRODUCTION
This paper describes experiments performed at the Free University of Brussels (VUB), Department of Chemical Engineering and Industrial Chemistry (CHIS). These experiments were performed in order to improve the yield of MMA monomer produced during PMMA waste pyrolysis. The results of this study were used to design a fluidized bed monomer recovery system for the Shanghai Resource Recovery & Utilization Co (Peoples Republic of China).
PYROLYSIS OF POLYMERS
Most polymers decompose according to one or more of the following schemes : 1. Depolymeristation (also called inverse polymerization) into monomer.
Examples are polymethylmethacrylate and PTFE. 2. Random cracking of the main chain into fragments of uneven length.
Polyethylene and polypropylene are important examples of this category. 3. Elimination of reactive substituents or side groups, with formation of a cracking
product on one hand, a charring polymer chain on the other. Examples are PVCandPVAc.
(CH2CHCl)n » (CHCH) + H a
(CH2CH)n » (CHCH)n + CH3CCOH I OOCŒ3
Table 1 gives the yield of monomer during vacuum pyrolysis [1]. The yields reported here are only indicative. Under different pyrolysis conditions (temperature, pressure, residence time) other results will be obtained.
KIH Groep Τ, Vuurkruisenlaan 4,3000 Leuven

581
Polyethylene for example will produce substantial amounts of monomer at higher temperatures. Also polystyrene is capable of much higher yields under appropriate experimental conditions [2].
Polymethylmethacrylate Polytetrafluoroethylene Poly (methylstyrene) Poly (methylstyrene) Polystyrene Polyacrylonitrile Polypropylene Polyethylene
>98% 99 95 70 45 5 2 0
Table 1 : Monomer yield for different polymers [1]
THE MECHANISM OF PMMA PYROLYSIS
PMMA decomposes according to two mechanisms :
1. At low temperatures (220°C) scission at the chain ends occurs; 2. At higher temperatures (270°C) a random homolytic scission takes place.
The low temperature reaction is believed to start at the unsaturated end groups of the PMMA molecule. During free radical polymerization about 50 % of the polylmer chains contain unsaturated end groups with the following structure :
CH3 CH2
I II CH2 C CH2 C
I ß «I C02CH3 OO^O^
The double bond strengthens the bond in αposition and weakens the bond in βposition, so that the molecule decomposes in the latter bond to yield the following radicals:
CH3 CH2
I II CH2 C · "CH2 - C
I I Οθ2α*3 CO^CHj
The first radical then depropagates according to a free radical mechanism [3].

582
A higher temperatures the distinction between stronger and weaker bonds becomes less pronounced so that a random homolytic scission occurs :
CH3 CH3 CH3 CH3
I I I I CH2 - C - CH2 - C - -> CH2 ' C" + OCH2 " C ~
I I I I CO2CH3 C02CH3 C02CH3 CO^CHj
Both radicals react further and produce monomer by unzipping of the main polymer chain.
TECHNICAL UNITS
PMMA is used under two forms : extrusion grade and cast material. The latter has an extremely high molecular weight, so that thermal reprocessing is no longer feasible. Instead, the polymer may be reconverted to monomer by pyrolysis. Because of the relative ease with which PMMA depolymerizes, some dedicated small scale pyrolysis units have been in use.
The techniques which have been used are :
1. Pvrolysis in a bath of molten lead [4]
The PMMA waste is fed batchwise or semi-batchwise in a vessel filled with molten lead at a temperature of 400 to 500°C. The monomer vapours are then condensed and the monomer is recovered after a final purification step (distillation). The units are quite simple to construct and to operate. The main disadvantages are the difficult temperature control resulting in a reduced monomer recovery and the local overheating of the vessel walls which cause carbon depositions, because of fouling the unit has to be cleaned at regular intervals (6 to 8 hours) which is quite cumbersome.
2. Pyrolysis in an extruder [5]
In this process a modified polymer extruder is used. The shredded PMMA waste is molten at 250°C and passes through an reaction zonde at 500 to 600°C. It is claimed that high yields (95%) are possible without excessive carbon depositions.
3. Pvrolvsis in a heated cauldrons
This system has been used by Shanghai Resource Recovery & Utilization Co. PMMA waste is batch fed in a cauldron, placed in a coal-fired brick-lined furnace. After preheating, volatiles are driven off gradually. The remaining carbonized residue is allowed to burn out in situ, at the end of the production cycle, which takes 8 hours (one shift).

583
FLUIDIZED BED PYROLYSIS EXPERIMENTS [6]
i . Experimental The test unit consists of :
a preheating system for the fluidizing gas (steam); a fluidized bed reactor with an internal diameter of 15 cm in the bed zone and of 30 cm in the freeboard zone. The reactor is made of Inconel 600 alloy and is electrically heated; a screw feeder; a cyclone separator; a condensor (shell and tube heat exchanger).
The unit is fluidized with steam.
The reaction products are : a liquid product containing the MMA monomer. This liquid is separated in an organic phase and the condensed water (from the fluidizing steam), in which some MMA is dissolved.
PMHA f e e d
Fluidizing gas
•ngA
Fly-Ash
Organic phase
a ter phase Condensate
Figure 1 : Fluidized bed unit
2. Analysis
The permanent gases (H2, CH4, CO, CO2, N2) were analysed with a gas Chromatograph equipped with two columns : a molecular sieve 13X and a Chromosorb 106 column. The condensate is also analyzed by gas chromatography on a OV-101 column.
Results
The results are summarized in Figures 2 and 3.
Figure 2 gives the MMA content and the yield of the organic phase that was produced as a function of operating temperature. Figure 3 shows the effect of temperature on the total MMA yield (in the organic phase + dissolved in the water phase).

584
100
■ 1 • 2
300 400 SOO Temperatur· (*C)
600
Figure 2 : MMA in organic phase and yield of organic phase 1 = % MMA in organic phase 2 = MMA in organic phase as % of feed
Both figures indicate a decrease in both monomer yield and purity of the liquid phase with temperature. The minimal temperature of operation is set by the reaction rate. At temperatures below about 380°C the rate of pyrolysis is too low and PMMA accumulates in the fluidized bed, when fed at a rate of 1 kg/hour. The total yields recorded were high : up to 98 % at the lowest temperature.
100
90
80
>\ B
■
B \
^ u \
300 400 500 Temperature (*C)
600
Figure 3 : Total MMA yield as % of feed
Conclusions
On the basis of experimental work conducted in a fluidized bed bench scale plant the operating conditions for the fluidized bed pyrolysis of PMMA were optimized. This has allowed to design a commercial scale (300 kg/h) reactor for use by Shanghai Resource Recovery and Utilization Company.

585
LITERATURE
1. Cameron, G., "Patterns and problems in the pyrolysis behaviour of synthetic addition polymers" NBS Special publication 357, "The mechanemisms of pyrolysis, oxidation, and burning of organic materials", Proceedings of the 4th Materials Research Symposium, Oct.2629, 1970, Gaithersburg, Md. (Issued June 1972).
2. Schoeters, J. and Buekens, Α., "Pyrolysis of plastics in a steam fluidized bed", International Recycling Congress, Berlin 1979.
3. MacCallum, J., "The mechanism of initiation of random degradation", Die makromolekulare Chemie, 99,1966.
4. DomingoSegui, E., Cabañero Alarcon, Β., French patent 1,079,107 (25.11.54), "Procédé et dispositif de régénération des monomères à partir de polyméthylacrylate, et, en particulier, de polyéthacrylate de méthyle".
5. Tokushige et al., Japan Steel Works Ltd., U.S. Patent 3,959,357 (May 25,1975), "Method for continuously thermally decomposing synthetic macromolecule materials".
6. De Wolf, F., "The pyrolysis of PMMA", (in Dutch), Engineering Thesis, Free University of Brussels, 1988.

586
PYROLYSIS OF EXHAUSTED OLIVE HUSKS COUPLED WITH TWOSTAGES THERMAL DECOMPOSITION OF AQUEOUS OLIVE OIL MILLS EFFLUENTS
G. DI GIACOMO, G. DEL RE University of L'Aquila, L'Aquila, Italy
E. BONFITTO, S. IACOBONI Regione Abruzzo, Avezzano, Italy
Ν. BRUNETTI E.Ν.E.Α., Centro Ricerche Casaccia, Roma, Italy
Summary
A twostage thermal decomposition process for the purification of aqueous olive oil mills effluents has been studied. In the first stage the vegetation waters concentrates are pyrolyzed along with the corresponding amount of olive stone obtained from the exausted olivehusks. The salts separate into the wood charcoal bed while the organic compounds live the pyrolytic reactor as a gaseous stream together with other decomposition products of olivestones. This stream can be used directly or after a partial condensation process as fuel for the energy requirements of the whole process. Wood charcoal is also obtained as an interesting by product of this waste minimization process. Mass and energy balances are reported together with a simplified flow sheet of the whole process.
1. INTRODUCTION The purification of aqueous olive oil mills effluents, which are u
sually referred as Vegetation Waters ( V.W. ), is a serious problem for major virgin olive oil producing countries like Italy, Spain and Greece. For example in Italy about 3,200,000 tons of olive per year are processed in 11,000 olive oil mills, producing 600,000 tons of oil and 1,600,000 mes of V.W.. These waste waters are brownish and evil smelling and are very polluting, being characterized by a COD ranging from 30 to 200 g/1 and a BOD5 from 30 to 100 g/1.
Disposal of V.W. by soil irrigation is no more allowed by italian law since march 1989. Various methods have been proposed for the disposal of V.W., but until now only incineration of V.W. concentrates is a really efficient treatment (1,2). However the salts contained in V.W. (1.5?2% by weigth) at the temperatures involved in incineration process induce fouling of the equipments compromising the smooth running of the process. We propose a process for thermal decomposition of V.W. which allows to overcome both the problem of energy demand and fouling caused by melting of salts. The process is based on a two stage thermal decomposition of V.W.: in the first stage concentrated V.W. are mixed with olivestones obtained from exhausted olivehusks, dried and pyrolyzed; in the second stage both liquid and gaseous pyrolysis products are burnt. During the pyrolysis the salts separate on the wood charcoal bed while the organic compounds originally contained in the V.W. are extracted as gaseous products. The characteristics of charcoal produced by pyrolysis are not si

587
gnificantly affected by salts coming from V.W.. Usually a liquid organic phase is separated from the gaseous stream
by means of controlled condensation and used as fuel for the concentration of V.V.. The remaining part of the gaseous stream is used to fulfill the energy requirements of the dryer situated just before the pyrolytic reactor.
Wood charcoal is obtained as a by-product of the proposed process and although its amount is about 30% of the olive-stones fed to the plant, its commercial value is at least comparable or even higher than the commercial value of the corresponding olive-stones.
2. MATERIAL AND ENERGY BALANCES The main characteristics of V.V. coming from batch (traditional) as
well as from continuous centrifugation olive oil mills are reported in table 1 (3,4).
Fig. 1 shows a block diagram of the proposed purification process. The energy and mass balances are made on the basis of a 2000 Kg/hr pyrolytic reactor. The ratio at which V.W. and olive stones are fed to the plant is the same as that one coming from olive oil mills.
V.W. coming from olive oil mills are first concentrated up to 50% of water by a conventional two or three stage evaporator. Energy required for concentration is supplied by pyrolytic oil. Steam coming from concentration unit can be usually discharged into municipal sewers after condensation, as it has a sufficently low BOD.
Concentrated V.W. are fed to a mixing unit together with olive stones (about 8% humidity). The solid like stream leaving the mixing unit is fed to a dryer in order to lower humidity from about 24% to 6%. The energy requirement of the dryer is fulfilled by pyrolytic gas.
The solid stream leaving the dryer is fed to the pyrolytic reactor, from which solid charcoal and a gaseous stream are continuously withdrawn. The gas stream is condensed in part under controlled temperature to give a liquid organic phase (pyrolytic oil); the uncondensed portion of gaseous stream is used to fulfill the energy requirements of the dryer.
TABLE I. Main average characteristics of V.W.
Extr.
BATCH CENTR:
TABLE
YELD HEAT (
Syst
[F.
II.
DF CO
oil [g/kg]
2 6.5
dr: at
Led residue 110 C[g/kg]
110 80
Experimental results of (basis 100 kg, 8% humid:
CHARCOAL
29 MB. 7400
[kg] [kca: 1/kg]
salt
olive-stones ity, heat of
Oil
12 5000
J
[kg] [kca!
cont.
20 6.4
[g/kg] COD
: pyrolysis test comb. 4300 kcal/kg)
1/kg]
GAS
80 1500
[g/1]
146 86
[NmJ] [kcal/Nm3]

588
2U át Jt
<$> T l
T I
3 &r
<·>
&J
^k o
C = CO Ν CENTR ATO*
M = MIXER
S = SEPARATO«
P=RVROLYTIC REACTOR
^Γ7 «Γ
«k
T I = CO WC. KM, TANK
D = D R Y E R
T 2 = PYROtVTIC OIL TANK
Fig. 1 Simplified flow sheet of the plant.
A pyrolytic reactor of vertical moving bed type is particularly suitable for the proposed process.
Yeld of charcoal, gas and oil from pyrolytic reactor are based on a test of pyrolyzation of olive stones performed on a pilot plant by BioAlternative (NeuchatelSuisse); the results of the test are reported in table II.
In the mass balance of the pyrolytic reactor it has been assumed that all the organic matter in V.W. (about 10% in V.W. coming from batch process and about 8% for V.W. coming from continuous centrifugation process) leaves the reactor with the gas stream. Then it is condensed to give pyrolytic oil.
In table III are reported the mass balances of the process both for V.W. coming from batch and continuous centrifugation olive oil mills: the only difference is in the concentration unit.
In table IV are reported the energy balance of the process. It has been assumed that the concentration of V.W. requires 420 kcal/kg of vaporized water and that the dryer requires 900 kcal/kg of evaporated water (taking into account the combustion efficiency of pyrolytic gas) .
As can be seen from data reported in table IV, energy available from pyrolytic oil and gas is larger than the energy demand of the whole process.

589
TABLE III. Mass balance.
STREAM 1 2 3 4 5 6 7 8
9 11 12 13
V.W. cone.V.W. steam cone.V.W. oliveston. 4+5 steam dried olive stones+V.W. charcoal gas pyrol. oil pyrol. oil
BATCH V.W.
FLOW RATE[kg/hr] 4300 940
3360 940 1530 2470 470 2000
510 1224[Nm
3/hr]
603 280
%H20 89 50 100 50 8 24 100 6
~"
CENTRIFUGATION
FLOW RATE[kg/hr] 5530 940
4590 940
1530 2470 470 2000
510 1224[Nm
3/hr]
603 474
V.W.
%H20 92 50 100 50 8 24 100 8
~*
TABLE IV. Energy balance.
ENERGY DEMAND [kcal/hr]
V.W. CONCENTRATOR DRYER TOTAL
AVAILABLE ENERGY
OIL 3.0*10b
GAS 1.8*106
TOTAL 4.8*106
BATCH V.W 1.75*10
6
0.60*106
2.35*106
[kcal/hr]
CENTRIFUGATION V.W. 2.37*10
6
0.60*106
2.97*106
3. CONCLUSION The proposed twostage thermal decomposition process for the purifi
cation of aqueous olive oil mills effluents allows to overcome both the problem of energy demand and fouling caused by melting of salts. Charcoal of commercial value is produced as a byproduct thus allowing a decrease of the operating costs.
REFERENCES
(1) BACCIONI, L.,(1981). Riciclo delle acque e loro incenerimento: una soluzione per la depurazione delle acque nei frantoi. La Rivista Italiana delle Sostanze Grasse, LVIII 3437.
(2) LANZANI, Α., BONDIOLI, P., FEDELI, E., PONZETTI Α., PIERALISI, G., (1988). Un processo per lo smaltimento integrale delle acque di vegetazione con contemporanea valorizzazione delle sanse nella lavorazione delle olive. La Rivista Italiana delle Sostanze Grasse, LXV

590
117-124. (3) DI GIOVACCHINO, L., (1985). Sulle caratteristiche delle acque di
vegetazione delle olive-Nota I, La Rivista Italiana delle Sostanze Grasse, LXII 411-417.
(4) DI GIOVACCHINO, L., (1985). Sulle caratteristiche delle acque di vegetazione delle olive-Nota II, La Rivista Italiana delle Sostanze Grasse, LXV 481-487.

SECTION 3
GASIFICATION TECHNOLOGY


593
STUDY OF BIOMASS GASIFICATION UNDER PRESSURE
CAPART, M. GELUS, M. LESGOURGUES, Z. LI Departement of Chemical Engineering University of Technology - B.P. 649
F - 60206 - COHPIEGNE Cedex
Abstract
In this work are presented results concerning the effect of pressure on the gasification of wood-char by C02 and steam, and on the catalytic power of K 2C0 3 for gasification. The experimental set-up is a pressurized thermobalance which enables experiments under isothermal conditions up to 25 bar and 1000°C. The mathematical model presented here involves a kinetic law of LANGHUIR-HINSHELWOOD type and takes into account the intra-particular diffusion of the reactant gas.
1. INTRODUCTION Biomass gasification under atmospheric pressure in order to produce
syngas as a raw material for ammonia or methanol synthesis is today feasible. However, gasification under pressure appears to be highly profitable because the energy cost of the methanol production is lower and the processing of pressurized gas (rich in CO, H 2 and CH^) requires smaller equipments and is therefore less expensive.
Whatever the method of working of a gasifier (fixed bed or fluidized bed), the biomass is volatized in the same various stages. These are mainly pyrolysis of virgin wood, and combustion and gasification of the wood charcoal, by-product of the pyrolysis.
The chemical gasification step is the slowest and undoutbly it limits the rate of the whole process.
The main reactants of gasification are carbon dioxide and steam. The reactions can be written as :
C + C0 2 -» 2 CO C + H20 -> CO + H2
To obtain information and values about gasification reactions, thermogravimetric analysis is undoutly the most effective method. The thermobalance set in out laboratory allows such an investigation into the kinetics of gasification with a pure gas (C02, H20) or with mixed gas, in isothermal conditions up to 1000°C and under pressure up to 25 bar.
2. KINETICS AND MECANISM OF GASIFICATION REACTIONS The (C + C02) reaction is slower than the reaction (C + H 20). It can
be twice to ten time slower according to the temperature value and the nature of the carbon. Gadsby (1), Blackwood (2) and Turkdogan (3) have studied this reaction for the case of coconut charcoal, carbonaceous vegetable material. It is usually agreed that the decomposition of CO is

594
a two step process which creates an instable oxide C(0) formed from a free active site of carbon : C«
Cf + C0 2 C(0) + CO C(0) CO + η Cf
The second step is irreversible and decomposition of the oxide C(0) leads to the formation of several active sites of carbon Cf.
This mechanism is consistent with a rate expression of LangmuirHinshelwood type such as that proposed by Blackwood and Ingeme (4).
k, PCO, + k= PCO* "CO,
1 + k, PCO, + k, PCO (mole g/g/mn)
In this equation, the term k4 PCO of the denominator characterises the inhibiting effect due to carbon monoxide. Gadsby (1), Ergun (5) and Turkdogan (3) have established more simple relation without the term k^ PC02
2. Beenackers and Van Smaaij (6) have considered the kinetics using a
power law rich reveals the complexity of the reaction process : V = k PCO, 0
·0 3
The mecanism involved in the reaction C + H20 is more complex. Some instable chemisorbed products appear during the reaction and for most authors, the kinetics is in accord with a LangmuirHinshelwood type rate expression as far as the water gas shift reaction is not at equilibrium. Blackwood and Ingeme (4) have found a rate expression whose simplified form is the following :
k, PH,0 + k= PH„02 H.O
1 + k. + k_ PH„
3. EXPERIMENTAL SETUP The reactor of the thermobalance
is a refractory steel tube of 5 cm internal diameter, heated by a tubular oven (4 kw). A steam generator (10 kw) and a superheater (3 kw) are connected to the reactor.
The thermobalance is designed to in isothermal conditions. The charcoal sample is contained 5 mm mesh basket which is quickly (5 seconds) into the zone of the reactor, by an
operate wood or in a 0 lowered heated electrical winch itself hung from the balance plate.
The weighing system is a modified SARTORIUS 1264 balance set in the pressurized housing, the electronics remaining outside. The reactor is swept by the contained gas (C02, steam) and the balance housing by, a slight counterflow of N 2 in order to protect the weighing systems.

595
4. CHARCOAL SAMPLES PREPARATION Wood cubes (oak) are pyrolysed slowly In an inert atmosphere (N )
from ambient temperature to 700°C for six hours and maintained at 700 C for a further one hour. The samples of charcoal are then cut into smaller cubes (1,5 χ 1,5 χ 1,5 cm) of which the mass is about 1.4 g and the average density 0.4 ± 0.02 g/cm3. It is of a great importance to heat the wood pieces slowly in order to avoid bursting them and to provide charcoal samples of homogeneous density.
5. PROCEDURE AND EXPERIMENTAL RESULTS The velocity of the gas in the reactor is fixed at 5 cm/s whatever
the conditions of the experiments. After the stabilisation of pressure, temperature and gas flowrate, the sample container was lowered in the heated zone of the reactor and the massloss of the charcoal was recorded.
Figure (2) represents the relation of the massloss with the time. During a short period, a rapid decreasing in the mass was observed, due to the loss of volatile products, following this the curve of mass loss is approximately linear up to a rate of conversion of about 60 70 %. The rate of gasification can be defined by the slope of the linear portion, represented by the following relation :
1 dm dX m0 : initial mass of sample V — — — — X : rate of conversion
m„ dt dt c
40 60 80 100
TIME (minutes)
Fig.2: CHARCOAL GASIFICATION : MASS LOSS VS TIME (T = 900°C, m0 1.5 g, u 3 cm s"1)
Fig. 3 : CHARCOAL GASIFICATION BY C02 : Influence of pressure on the gasification rate (m ^ 1,5 g u ■ 5 cm/s).
The gasification rate increases with temperature and pressure. As shown by figure (3) the rate increases at first rapidly with pressure up to 6 7 bar and them much more slowly.
Experiments with steam have been more difficult to perform due to condensation problems. Similar results as with C02 have been obtained at 900°C with higher rates of gasification (see figure 4).

596
dm m. dt
5.10
4.10
ΐ.10
2.ΙΟ
Ι 0"
Fig. 4
(mn 1)
900°C
15 20 PRESSURE
CHARCOAL GASIFICATION BY HjO : Influence of pressure on the gasification rate (m » l . S g « = S cm/s).
Φ 5.10
2
4.IO"'
LIO"*
2.10' '
io»
0
■ " " )
C . c o 7 2 CO ICJCOJ « I
l i n n t i T y i f i l
20 PRESSURE
Fig. S : CHARCOAL GASIFICATION : Influence of presaure on the catalytic effecc.
Gasification can be catalysed by alkali metal salts, among which potassium carbonate (K2C03) is known as the more efficient. The rate of gasification is approximately (see figure 5) four times higher when the charcoal is impregnated with about 5 * in mass of Κ CO .
6. MATHEMATICAL MODEL AND DETERMINATION OF KINETIC PARAMETERS In order to determine the kinetic constants of the reaction certain
basic assumptions must be clearly defined : the wood char particle is isotropic, of spherical shape with an
equivalent radius of 0.93 cm ; the reaction is complete and no chemical equilibrium is reached ; the external mass or heat transfer is neglected ; no gradient of temperature and pressure exists in the particle ; the pseudo steady state is appropriate for calculating the concentration of the gaseous reactant within the particle.
With these assumptions, the mass balance equation relative to the reactant gas (CO_ or H_0) can be written as follows :
d*p dx
a
with 1 + ρ
dP ,
( — )a
dx 2
+ — χ
dp — + V dx
RTLa
De P„ (1 + P)
ρ : volumic fraction of the reactant gas χ : radial coordinate L(m) : radius of particle P„(Pa) : total pressure R (J mole~1
K"1) : ideal gas contanst
De (ma s_1) : mass diffusivity of the gas
χ — 1 , ρ — ρ V (mole m 3 s1) : rate of reaction
T (K) : temperature of gasification The mass balance equation was numerically solved by the Runge Kutta
method and the profile of the reactant gas concentration optimized by a "shooting method" (criterion P(L) — Ps).
The mass diffusivity of the reactant gas De is uncertain ; however, from the GROENEVELD'S investigations on the diffusivity of C02 throughout slices of wood char (7), its value was fixed at
dp — 0 dx

597
0.15 IO* ma s 1
, which seems a suitable value with respect to the temperature and the high porosity of the woodchar.
In order to fit the experimental results of the rate of gasification with C02 in the pressure range 1 2 5 bar and the temperature range 800 950°C. The relation due to Blackwood is appropriate :
k, PC02 k2 PC0a
1 + k3 PC02 + kA PCO
Each constant kt obeys to the Arrhénius law : E<
exp — and a good fit is obtained by taking the following RT
values of kj° and Et
k 1 0 131 ( s 1 atm"1) k
a ° 7.7 10" a ( s 1 a tm ' 2 ) k 3 ° 2 (a tm 1 ) k . ° 1.26 1 0 a (a tm 1 )
E t 30000 cal mole 1
E2 20000 cal mole 1
E 3 0 cal mole 1
E„ 45500 cal mole 1
Influence of pressure Influence of temperature
'1 0 . χ χ
Fig. 6 : Profiles of intreparticular gas concentration
The aspects of the concentration profile of the reactant gas leads to the following remarks : the gradient of reactant gas concentration decreases when the total pressure increases and that gradient increases with temperature. Indeed the rate of reaction is augmented by temperature so that the rate control by internal diffusion is favored.
REFERENCES
(1) GADSBY J. and al. Proc. Roy. Soc. London (1948), p. 35776. (2) BLACKWOOD J.D., Coke and Gas, (1960) 22, p. 190194. (3) TURKD0GAN E.T., VINTERS J.V., Carbon (1970) 8, p. 3953. (4) BLACKWOOD J.D., INGEME A.J., Australian J.
p. 194209. (5) ERGUN S.J., Phys. Chem. (1956) 60, p. 480485. (6) VAN DEN AARSEN E.G., BEENACKERS A.A., VAN SWAAIJ
Thermochem. Biomass. Conv., Estes Park, Colorado (7) GR0ENEVELD M.J., The cocurrent moving bed gasifier
Twente University (1980).
Chem. (1960) 13,
Fund, of 1822 Oct. (1982).
Ph. D Thesis,

598
GASIFICATION OF CHARCOAL IN MALAYSIA
W Κ HOI ! and A V BRIDGWATER Forest Research Institute of Malaysia Department of Chemical Engineering
Kepong Aston University Selangor Aston Triangle
52100 Kuala Lumpur Birmingham B4 7ET Malaysia UK
Summary
The paper highlights the practical experience and potential of using residue charcoal for small-scale power generation in rural areas. Trials conducted showed that with very slight modifications diesel and petrol engines can be adapted to work satisfactorily on producer-gas. The conversion efficiencies of diesel (13 kW) and petrol (45 kW) engines working on producer gas was found to be 22% and 17% respectively. With its simple operation and maintenance, the down-draft gasifier was found to be suitable for providing a reliable and independent source of electricity to the rural areas in Malaysia.
INTRODUCTION
Solid biomass such as wood and charcoal can be converted into useful gaseous fuel by a process known as gasification. During the Second World War, producer gas from gasification was used extensively in Northern Europe and many countries in the Asian and Pacific region as fuel for driving buses, tractors, cars and other mechanical equipment (1). Since wood was abundantly available at that time, wood charcoal was the main feedstock. After the War, the availability of cheap oil led to a complete halt to the use of producer gas. However, the sudden increases in the price of fossil fuels in the 1970s injected new interest in this old technology. This has led to the development of new gasifiers that are safe, efficient and economical to use (2). Today, gasifiers are not only designed to use charcoal as feedstock but can also be adapted to utilise forestry and agricultural residues with relatively high efficiencies compared to pre-war gasifiers.
In Malaysia, producer gas technology has a good potential in the generation of mechanical and electrical power, especially for some cottage industries in rural areas which rely heavily on liquid fossil fuel. Although it is still possible for rural population to make use of conventional prime movers, it is expensive to depend entirely on liquid fossil fuel even when subsidised. As a result, these industries are facing the problem of high operating cost because of high fuel price.
Research and Development in charcoal gasification was initiated by the Forest Research Institute, Malaysia in 1986. This work was partially supported financially by The Federal Republic of Germany through the Malaysian-German Forestry Research Project (3). In this project emphasis was placed on the development of producer gas systems that are easy to operate, be maintained, and fabricated using locally available materials.
The main aim of this paper is to assess the performance of a 7 kW downdraft charcoal gasifier which has been installed at the Institute for trials.
THE GASIFIER
The charcoal gasifier installed at FRIM consists of four main components (Figures 1 and 2). They are: a) A reactor made of stainless steel to generate the gases. b) A cooler to condense the tar and condensibles in the gas. c) A cloth filter to remove soot and ash. d) A diesel and petrol generating set
1 Currently FRIM Research Fellow at Aston University

599
The reactor has the following specifications:
Height of reactor Diameter at nozzle level Primary air supply Height of reduction zone Insulation of reduction zone Gas cleaning Gas cooling Volume of reactor Capacity
85 cm 20 cm 3 nozzles 24.5 cm Refractory brick lining Cyclone Semi-circular air cooled tubes 0.09 m3 2.5 kg/h charcoal
The reactor is a downdraft system, cylindrical in shape and lined with a thick refractory brick lining. On the top of the reactor is a fuel feeding lid through which charcoal can be fed. When fully charged the reactor contains approximately 7 kg of charcoal and the gasifier can then run for about 4 hours. However, the reactor chamber can be extended upwards if there is a need to put in more charcoal in order to reduce the recharging rate. Air for the oxidation zone is supplied through a valve located at the side of the reactor. In the reduction zone, an inspection door is provided for inspection and starting the gasifier. An ash door is located at the bottom of the reactor for periodic cleaning of the gasifier and removal of the ash. A shaker arm which can be rotated helps to loosen the burning charcoal at the reduction zone in order to prevent bridging.
Fuel hopper lid
Blower
Inspection gate
Shaker arm Gas test valve
Air filter
Mixer To engine
Gas cooler Gas cleaner Safety stop
Figure 1 Gasifier Layout
The gas passes through the grate and then passes upwards in an annular space around the reduction zone and leaves the gasifier near the top via the gas outlet pipe. The gas is then passed through a simple cyclone which removes a large proportion of the dust carried with the gas stream. The temperature of the gas at this point is about 120°C. The gas is then passed through a simple pipe cooler which cools the gas by natural convection. Moisture and wood tar condense along the sides of the pipe and can be drained off at the bottom of the cooler. The gas temperature after passing through the cooler is found to be between 27 - 30°C.
To further remove the remainder of the dust, the gas is passed through a simple cloth filter which is installed between the cooler and the generating set. The gas temperature at this point is about 25°C.

600
Figure 2 Gasifier
The diesel engine using a dual fuel system has the following specifications: Type : Two cylinder, 4 stroke direct injection Volume : 2 litre, bore 87.5 mm, stroke 110 mm Compression ratio : 17.5 :1 Power : 13 kW at 2000 rpm Governor : mechanical Cooling : water cooled Starter : hand cranking
The petrol engine which is operated fully on gas has the following specifications: Model Cylinder volume Compression ratio Maximum power Cooling system Starter
Toyota model 4K 1.29 litre, bore 75 mm, stroke 75 mm 9:1 45 kW at 5600 rpm water cooled electric starter
Both the engines are coupled to a simple generator with the following specifications: KVA Rpm AC volt AC amps Frequency
7.5 1500 415 10.5 50 Hz

601
OPERATION OF THE GASIFIER
Fuel preparation During all the trials, the gasifier was fed exclusively with rubberwood charcoal
produced by the transportable metal kiln (4). The charcoal has a fixed carbon of 80-85%, ash content of 3-5%, volatile content of 10-15% and moisture content of 7-10% (5).
In order to prevent bridging just above the nozzles the charcoal was broken into small pieces. The ideal size distribution of the gasifier fuel was found to be between 0.5-10 cm. As charcoal is very hydroscopic, it must be stored in a closed shed to minimise water absorption.
Starting procedure Before starting, the reactor is filled with about 2 kg of charcoal. A blower with suction
operation is fitted to the flare pipe of the gasifier. The charcoal is ignited with the help of burning material at the inspection gate. The fuel lid at this time is left open and the charcoal is left to ignite freely. As soon as the charcoal in front of the nozzle is glowing well, the reactor is filled with charcoal. The lid and the nozzles are then shut tighdy and the blower is then transferred to the primary air inlet valve of the gasifier with pressure operation.
The gas produced can be tested at the flare located on top of the gas cleaner. The quality of the gas is determined by igniting it. A certain amount of practical experience is needed in order to assess the quality of the gas. A good gas is normally indicated by an almost transparent flame which bums steadily without blowing out. A bright yellow flame indicates that the gas is contaminated with tar and vapour and is unsuitable for the engine. Sparks in the flame mean that carbon particles are present in the gas and this indicates that the filter system is not working very well.
As soon as the gas burns consistently with a transparent flame, the engine can then be started and the blower removed. In order for the engine to work uniformly the volume of the gas provided by the blower must not be less than 30 m3/h.
RESULTS AND DISCUSSIONS
The producer gas has the following typical analysis: H2 : 10.2% vol; CO : 27.6% vol; CO2 : 4.0% vol; CH4 : 1.0% vol; N2 : 55.0% vol; O2 0.2% vol. The analysis does not vary by more than ± 10% under any operating conditions. Taking the heating value of the gas to be about 4.5 MJ/m3, it has been found that the thermal efficiency of the gasifier is about 70%. This means that conversion of the chemical energy in the charcoal into gas energy is 70%. The loss of combustible material (such as charcoal fines) in the ash accounts for about 10% of the energy supplied by the charcoal. The remaining energy is lost as sensible heat in the gas and from the reactor as heat to the surrounding. The results of gasification trials using rubberwood charcoal as feedstock can be summarised as below:
Power output Fuel consumption Start-up time Gasifier refuelliing interval Fuel replacement Thermal efficiency of engine
In general there have been no serious problems in converting diesel and petrol engines to work on producer gas. Diesel engines seem to be more suitable when there are load fluctuations. Petrol engines are, however, more suitable when a constant load is needed. It must be remembered that engines working on gas are extremely dependent on the moisture content of the feedstock. Wet charcoal with a moisture content of about 18% causes the output power of the petrol engine to be between 5-10% lower and the diesel replacement of the diesel engine to be reduced to 70%.
Charcoal with sizes larger than 3 cm has been found to be unsuitable as serious
Diesel engine 5.2 kW 1.5 kgft 15-20 mins 4h 75% 21.6% overall
Petrol engine 4.0 kW 2.0kg/h 20-30 mins 3h 100% 12% on gas

602
bridging occurs in the region just above the air injection nozzles resulting in poor gas production. When this occurs the fuel lid has to be opened (although it is extremely dangerous) and a stick has to be inserted into the fuel bed in order to relieve the blockage.
The gasifier can be started easily with a battery and good gas production can be attained within 10-15 minutes. If the moisture content and sizes of the feedstock used are correct the gasifier system can be left to run for hours without much supervision. Refueling can be carried out with the engine running but idling. The charcoal bed in the reducing zone, however, has to be changed after every 20 hours of operation because the ash and fines will block the flow of the gas and the output power will be significantly reduced. This process normally takes about 1 hour.
The cloth filter has to be changed after every 20 hours of operation. The cloth filter can be reused after cleaning by shaking. The process of changing the filter normally takes about 1 hour. In order to maintain a constant output power, the cylinder head, valve port and valve seating has to be cleaned after every 100 hours of operation. The time needed for this operation is normally 1 hour. Because of high temperatures in the reactor chamber, the asbestos gaskets in the reduction zone, fuel lid and the ash door have to be replaced after 300 hours. Failure to replace these gaskets can adversely affect the performance of the gasifier due to leakage of air.
It has been found that the paint on the walls of the reactor will start to peel badly after 300 hours of operation. For long term protection of the gasifier it is necessary to repaint the walls of the reactor and cyclone with heat resistant paint after every 300 hours of operation.
CONCLUSION
Studies on the gasification of rubberwood charcoal and the utilisation of producer gas in internal combustion engines has shown that it is possible to develop small inexpensive down-draft gasifier systems for rural applications by using locally available material. The main constraint in the utilisation of producer gas in engines is the cleaning of the gas to that it is free of impurities. Not much work has been carried out to develop a simple but efficient gas cleaning for the gasifier.
The assessment of the results has shown that low power output systems fuelled by charcoal have considerable potential for a number of rural applications including water pumping, drying of food and shaft power production. Apart from being reliable, these systems are easy to operate and maintain. The main problems in the utilisation of this system lie in the inconvenience in the start up and the maintenance of the filter system by the operator.
REFERENCES (1) Anon, "Producer gas: another fuel for motor transport:", National Academy Press,
United States, 112pp, 1983. (2) Anon, "Producer gas technology", Papers presented at International Producer Gas
Conference, Bandung, Indonesia. The Beijer Institute, Sweden, 1985. (3) Graf, U, "Charcoal gasification at the Forest Research Institute Malaysia. "Report of
short-term expert", Deutsche Gesellschaft fur Technische Zusammenarbeit, West Germany, 1986.
(4) Hoi W K, Low, C Κ, and Wong, W C, "The production of charcoal by the improved transportable metal kiln.", Paper presented at International Conference on Rural Technology, Kuala Lumpur, 1985.
(5) Hoi, W K, "The production of rubberwood charcoal by the transportable metal kiln", Paper presented at Rubber Research Institute Colloquium on Research and Development in Rubberwood, Kuala Lumpur, 1985.

603
UPDRAFT GASIFICATION OF WASTE FUELS
P. STÅHLBERG», E. KURKELA», H. FILEN»« & Κ. SALO«» «Technical Research Centre of Finland (VTT)
Laboratory of Fuel Processing Technology SF02150 Espoo, Finland
««Bioneer Oy SF13101 Hämeenlinna, Finland
Summary
The aim of the study was to extend use alternatives for gasification technology by studying the suitability of updraft gasification for gasifying waste fuels. The waste fuels studied were municipal waste, forest residues, straw and car scrapyard waste. The present version of the gasifier is well suited for forest residues, pieceshaped municipal waste and car scrapyard waste. Faultless operation of the gasifier with crushed and chopped municipal waste and straw requires the use of a fuel mixture of larger piece size, for example, sod peat or wood chips, or changes in the construction of the gasifier. The flue gas emissions are fairly low, although the plant is not equipped with flue gas purification. A scrubber used in the experiments reduced the particulate contents of the flue gas by 70 % to 50 mg/m2n and the heavy metal contents in solids by 30 99 %. The scrubber had no significant effect on gaseous emissions. As the specific emissions of certain components of unpurified flue gas are much lower than those from grate and fluidized bed combustion, the flue gases can be purified further at lower investment costs. The competitiveness of the gasification plant seems to be good in the size class of 5 15 MW and municipal waste as the fuel. Two Bioneer gasification plants for forest residues with 4 and 6 MW gasification outputs have been in operation in Sweden since 1986. The first gasification plant designed especially for municipal waste will be put into operation during spring 1989 in Italy.
1. INTRODUCTION The use of updraft fixedbed gasification for producing district
heat was developed to a commercial level in the early 1980s in Finland. There were a total of nine Bioneer gasification plants in operation in 1988 in Sweden and in Finland. These plants produce district and process heat and are fueled with sod peat, wood chips and wood residues. Bioneer gasifiers are manufactured for the output class of 1 15 MW.
In the Bioneer gasifier the fuel is gasified with air and steam in a reactor of shaft furnace type. The process was primarily developed for the gasification of sod peat and other pieceshaped biofuels. The flowchart of the Bioneer gasification district heating plant is shown in Figure 1.
The temperature of the combustion zone in the lower part of the gasifier can be controlled by the amount of steam fed along with the

604
β 10 14
1. Fuel bin 2. Fuel feed conveyor 3. Fuel feeder 4. Gas generator 5. Ash removers β. Ash conveyor
7. Ash ballette 8. Drop separator 9. Humidifier
10. Gasification air fan 11. Plate heat exchanger 12. Gas pipe
13. Gas burner 14. Combustion air fan 15. Gas boiler 16. Economizer 17. Rue gas fan 18. Stack
Figure 1. Flowchart of the Bioneer district heating plant.
gasifying air. In this way, ash melting, occurring even at low temperatures, and problems due to it in the operation of the grate and in the ash removal can be prevented. The steam reacts with hot carbon in the gasification zone and forms carbonmonoxide and hydrogen.
The proportion of tar condensed in updraft gasification is as high in the product gas that the gas cannot be used as such for example for the fuel of gas engines or conveyed long distances in pipelines. In the existing Bioneer plants the gas is burnt close to the gasifier.
The competitiveness of the Bioneer boiler plant compared to conventional solid fuel boilers is based on the following factors:
high reliability, automatic operation, high degree of utilization high efficiency even at partload low specific emissions, small need of flue gas purification possibility to employ different feedstocks in the same plant. In the last few years, the research and product development work on
updraft gasification, done in cooperation with the Laboratory of Fuel Processing Technology of VTT and Bioneer Oy, has focused on the gasification of different waste fuels. Tests have been performed mainly at a test station of 1 MW gasification output, where the gas is burnt in a multiple tubular boiler. Experiments have also been carried out with municipal waste at a district heating plant of 6 MW.
2. TEST STATION AND TESTS The test station constructed by VTT and Bioneer Oy for joint re
search and product development was completed in 1985. The nominal gasification output of the station is 1 MW (Figure 2). The test station is equal to the commercial district heating plants, but its instrumentation is more extensive than at the commercial plants and it is also equipped with sampling units and data collectors required in research and product development.
The waste fuels used in the gasification tests were municipal waste, forest residues, straw, rubber and plastics waste from car scrap

605
Figure 2. Test gasification plant, gasification output 1 MW.
yards, and mixtures of these, sod peat and wood chips. Properties of the waste fuels are presented in Table I.
The gasification tests with different waste fuels took 3 1 0 days. In the determinations, attention was paid especially to adjustments of the plant, to tar contents of the product gases and, for municipal waste, to flue gas emissions, in addition to fuel and process measurements.
Table I. Test fuels. Fuel
MUNICIPAL WASTE waste briquettes
crushed waste I crushed waste II chopped waste FOREST RESIDUES STRAW barley straw wheat straw CAR SCRAPYARD WASTE
waste I waste II
Code
A Β C D E
F G
H I
Sod
0
0 0
0 0
10 2
size
mm
35 Ν Ν 20 30
100 70
60 20
Moisture
? 8 29 46 42 52
30 16
16 19
Ash content
%
11.8 16.8 13.6 13.5 4.2
5.7 4.1
19.4 30.9
»very unhomogeneous, largest metal pieces and glass removed

606
3. RESULTS ftHD DISCUSSION The composition of the dry product gas (main components) and the net
calorific value calculated on the basis of these are presented in Table II. In addition, the product gas contains light hydrocarbons and tar, which increase the net calorific value by 0.5 4 MJ/m3n [m3n = m3 (0 °C, 1.013 bar)]. Typical compositions and heat values of the product gas of wood chips and peat gasification are also presented as reference values.
The dry product gas of wood chips, forest residues and sod peat contains 50 100 g/m3n tar. The tar content of municipal waste ranges 10 20 g/m3n, of straw 1 0 4 0 g/m3n and of car scrapyard waste 80 200 g/m3n.
The present construction of the Bioneertype gasifier is well suited for the gasification of forest residues, municipal waste briquettes and car scrapyard wastes. When chopped and crushed municipal waste and straw were gasified, occasional fuel arching occurred at times. Arching re
sults in variations in the quality of the product gas and in the gasification output. The operation of the gasifier is improved if a fuel mixture in piece form is used.
Emissions developed in the combustion of gas produced from municipal waste are presented in Table III. The experiments with waste types A and Β were carried out at the test station of 1 MW and those with wastes C and D at the district heating plant of 6 MW. Flue gas purification
Table II. Composition and net calorific value of dry product gas.
Fuel and code
MSW1 A MSW B/wood chips 2 MSW B/wood chips 3 MSW C/sod peat 4 MSW C/sod peat 5 MSW D Forest residue chips E Straw F Straw G Scrapyard waste H Scrapyard waste I Sod peat Sod peat Sod peat Sod peat Small wood chips Large wood chips
Moist, cont.
% 8
29/42 29/45 116/36 46/36 42
52 29 16 16 19 25 33 42 52 43 41
CO
% 17.8 17.9 20.6 18.4 18.7 14.7
23.1 18.9 14.9 22.5 18.9 24.5 24.2 21.6 16.1 29.0 30.2
"2
% 14.6 14.8 18.5 13.8 16.3 14.8
14.9 17.6 11.3 19.0 14.3 19.0 18.8 16.8 16.7 15.4 10.8
CH4
% 2.6 1.9 2.0 1.5 1.7 2.0
2.3 2.3 2.9 1.4 1.6 3.0 3.0 2.4 2.0 1.6 2.5
C02
t 11.0 13.0 12.0 12.7 12.4 12.5
9.9 13.8 14.8 9.0 10.6 10.2 10.3 12.5 14.3 6.8 7.1
Heat value Hu
MJ/m3n 4.7 4.5 5.3 4.4 4.7 4.2
5.3 5.1 4.1 5.4 4.5 6.2 6.2 5.4 4.5 5.9 5.9
1 MSW = municipal solid waste. Dry matter contents: 2 wood chips 45 %, 3 r wood chips 60 %, 4 sod peat 25 %, 5 sod peat 42 %

607
equipment was not used at the test station. At the district heating plant, the flue gases were purified with a wet scrubber.
The contents of heavy metals presented in Table III were determined only for the solid matter, except that of mercury. The mercury contents also include steam phase. The PAH contents include 7 compounds, chlor benzenes 11 compounds and chlor phenols 23 compounds. 15 compounds were determined for chlorinated dibenzofuranes and 12 compounds for chlor
inated dibenzopdioxines. One of the greatest advantages of updraft gasification is a low
particulate content of unpurified flue gas, when further purification is more simple and cheaper than in most combustion processes.
The emission values presented in Table II cannot be considered as typical emissions from a plant based on the gasificationcombustion process. The values are primarily test results for dimensioning the essential flue gas purifiers (e.g. wet scrubber and fiber filter) and for evaluating purification costs.
Table III. Contents of emission components in flue gases in the gasification tests with municipal waste. The values are reduced to 10 t CO2 content.
Particulates mg/m3n Heavy metals: Hg mg/m3n Cd mg/m3n Pb mg/m3n Cr mg/m3n Zn mg/m3n PAH pg/m3n PCB pg/m3n Chlorobenzenes pg/m3n Chlorophenols pg/m3n Furanes ng/m3n Dioxins ng/m3n N0X mg/m3n SO2 mg/m3n
Type of municipal waste A
350 0.09 0.42 14 37 12.3 0.2 7.6 6.2 0.04
240 405
B/wood chips* 330 0.03 0.06 5.0 40 11.6 1.0 8.2 6.5 <0.04
390 600
C/sod peat** 42
0.02 0.02 1.7 0.01 3.3 5.0 5.6 2.5 8.1 <1.0 <1.0
D
60
0.05 4.0 0.01 7.8 8.3 3.6 2.2 8.4 <1.0 <1.0
η - 0 UC, 1.013 bar < = smaller than analytical accuracy Dry matter content: * wood chips 45 Í, ** sod peat 25 %
4 CONCLUSIONS The tests indicated that the updraft gasifier is well suited for the
gasification of pieceshaped waste fuels. When using chaff and fuels with a high fines content, the use of a support fuel is required either in the gasifier or in the combustion of gas to guarantee the faultless operation of the gasification plant. The flow of the fuel can also be improved by equipment technical measures. As the product gas contains an abundance of condensing compounds, the gas should be burnt in the im
mediae vicinity of the gasifier. The need for flue gas purification is lower in gasification than in direct combustion.

608
PEAT AMMONIA PLANT IN OULU - SYNTHESIS GAS PRODUCTION FROM PEAT BY FLUID-BED GASIFICATION
K. SIPILÄ«, C. WILÈN», E. KURKELA·, A. MOILANEN« & J. KOLJONEN«· »Technical Research Centre of Finland (VTT)
Laboratory of Fuel Processing Technology SF-02150 Espoo, Finland
»»Kemira Oy SF-90101 Oulu, Finland
Summary
A peat ammonia factory was put into operation at the Oulu Works of Kemira Oy in summer 1988. The factory uses about 1.2 million MWh sod peat for producing 80 000 t/a ammonia for fertilizer production. The total investment in replacing the old heavy-oil based plant with a peat-based one was FIM 225 million, and it concerned peat handling, drying and gasification and gas cleanup. The plant is the only plant producing synthesis gas from peat in the world. Similar technology can also be used for biomass. Sawdust has been gasified mixed with peat. Sod peat is crushed and dried in a pressurized steam dryer, and the dry peat is then gasified in a pressurized HTW gasifier with oxygen and steam. The trial runs of the plant were started in May 1988, and ammonia production in August 1988. The longest uninterrupted operation time has so far been 31 days.
In the research work carried out in co-operation between Kemira Oy and the Technical Research Centre of Finland, the main topics have been crushing and drying of raw material, feeding into a pressurized reactor, safety, reactivity and gas impurities in gasification, ash behaviour, and waste waters. Research results are reviewed in this paper.Further research will focus on peat handling and on gas impurities. A new project has been initiated on IGCC applications, where hot gas cleanup and biomass utilization in pressurized reactors are of major interest. The work is a part of a national fuel processing research programme JALO.
1. RAW MATERIAL HANDLING The feed rate of sod peat (moisture content 40 %) into the plant is
42 t/h. The sods are crushed and fed into a pneumatic back-pressure steam dryer, and the moisture content of the product is 15 % after the dryer. The dryer is operating at 4 bar pressure. The heat obtained from the dryer is used for district heating. The condensate of the dryer is led among the waste waters of the factory and combustible components to the power plant. The flow-sheet of the plant is shown in Figure 1.
One research topic of significance in the peat ammonia project has been peat properties, which are essential in pretreatment and feeding devices. These include flow-technical characteristics of peat and bio-mass, feed into pressurized reactors, as well as self-ignition and dust explosion characteristics. Attention has been paid especially to the

609
Sod 41 t/K moUtun 40%
Outotmhol tcnibb* ISOOkmolfo
t siuda» Dual ttpåntion
Figure 1. Flow-sheet of the peat ammonia plant of Kemira Oy.
significance of pressure up to 10 bar, and to the behaviour of dried peat in the lockhoppers and feeding lines of the gasifier. The peat leaves the lines at 1 - 4 bar pressure and at 95 °C temperature.
The attritive effect of the dryer is shown in Figure 2. The particle size distribution of sod peat crush is shown after the crusher and in the outflow from the dryer. Dried lignite crush, which is used as the raw material of the Berrenrath plant in the F.R.G., is presented as reference.
The properties of various peat grades were determined by an annular ring shear cell (Walker). The flow curve and function (FF) can be determined by carrying out the measurements at several different consolidations /1, 21. When the internal friction and effective internal friction, volume weight and unconfined yield strength is known, the minimum dimensions of the discharge opening of the bin can be calculated on that basis that the gravitational flow of the bulk solids is able to break the arch formed in the discharge opening. The results of the shear tests are presented in Table I.
2. GASIFICATION PROCESS The peat is fed with the nominal flow of 25 t dm/h into the HTW
gasifier. The gasifier has operated as expected. The large amount of fines among the peat and the high naphtalene content of the gas have caused malfunctions. A new parallel crusher is being installed at the plant to improve the particle size distribution of the peat. Benzene scrubbing has been intensified to remove naphtalene. The naphtalene content might also be reduced by using limestone in the fluidized bed. The typical operation values for the gasifier are as follows: bed temperature 700 °C, free-board temperature up to 900 °C; gas composition: carbon monoxide 35, hydrogen 32, methane 6 - 8 , and carbon dioxide 22 vol?. The peat feed is 23 t/h, oxygen demand 290 Nm3/t peat (maf) and steam 160 kg/t peat; synthesis gas output 925 Nm3/t peat (CO + H2). The

610
* . ζ o κυ < oc u. κ X o iii 5 LU > I
3 s => υ
100.0 79.4 63.1 60.1 39.8 31 .β 25.1 20.0 16.8 12.8 10.0 7.9 β.3 S.0 4.0 3.2 2.6
ι.082 0.106 0.21 0.6 1 1.88 3.15 4.78 9.52
PARTICLE SIZE, mm
LIGNITE o PEAT AFTER CRUSHER x PEAT AFTER DRYER
Figure 2. Attrition of sod peat crush in the MoDo steam dryer.
Table I. Results of shear tests (Annular Shear Cell (Walker)).
Material
Sod peat crush Crush 1 (dried) Crush 2 (dried) Crush 3 Crush 4 Crush 5 (dried) Crush 6 (dried) Crush 7 (dried) Crush 8
Milled peat 1 Milled peat 2
Pellet crush
Lignite
Particle size
d|ç value
0.63 0.17 0.16 0.4 < 0.4 0.37 < 0.37 0.5 < 3 < 3
0.5 <0.27
Moisture content wtJ
12.1 11.9 30.9 27.9 14.0 14.8 14.5 21.4 11.0 36.9
21.7
16.7
NFF
10.8 17.4 9.4 5.8 14.2 12.4 9.4 8.7 5.9 3.2 7.5 24.7
Minimum opening
cm
29.9 14.9 22.1 14.0 18.1 46.4 25.2 29.1 10.4 39.8
4 31.6
vol. wt kg/nP
355 512 470 470 512 292 311 348 200 300 550 653

611
carbon conversion is 88 % and the ash is fed into the steam boiler for total carbon utilization.
Research has focused on the reactivity of peat and the behaviour of ash in gasification. The reaction rate of different sod peat chars has been slightly lower than that of lignite. The attrition of peat grains in the bed can be significant /3/. The reactivity of char is reduced by densification.
The deposit formation of ash is in accordance with the theory of sintering by viscous flow in peat gasification. According to this theory, glassy particles are sticked to each other as a result of viscous flow. The siliceous ash molten in the oxygen flame forms glass. The peat ash is typically siliceous and hence, deposit formation with this mechanism can be expected /4/. A laboratory method based on the measurement of compression strength was developed for testing the sintering tendency of ash /5, 6/ and viscosities were also measured for different peat ashes. The compression strength of ash pellets as a function of the B/A index is shown in Figure 3, which indicates a good correlation at 800 °C in these tests. This index describes the ratio of alkaline and acid oxides in the ash. The viscosity of ash as a function of temperature, measured at the Institute of Gas Technology, is presented in Figure 4. The viscosity measured at 1 000 °C also correlates with the B/A index well /4/.
3. OUTLOOK FOR THE FUTURE The plant is operating parallel with an existing oilfired gasifier,
and hence, the full capacity can be achieved either by peat or by heavy oil. In summer 1989, accurate measurements will be carried out for gas impurities and waste waters. A new object of research will be the application of experience obtained from this plant for IGCC applications. Preliminary plans have been made to lead a byflow for hot gas cleanup experiments, in order to study by longterm tests the durability of ceramic and metal filters in use for biofuels. On the basis of the present experience it may be concluded that it is possible to produce ammonia and chemicals from peat.
É (3 Ζ UI
et
0.75
Figure 3 . B/AINDEX
Compression s t reng th as a function of the B/A index.

612
Wc"
8# »g
1400
TEMPERATURE [°C] ■ KMNEVA + JOUTENNEVA O ILOMANTSI
Figure 4. Ash viscosi ty as a function of temperature.
Δ VIIDANSUO
REFERENCES
1. Rautalin, A. & Wilén, C. Peat handling and safety at combustion and gasification plants. 10th Int. Conf. on Fluidized Bed Combustion, San Francisco, CA, April 30 May 3, 1989.
2. Thun, R. & Rautalin, A. Technical flow problems and safety risks connected with peat handling. Bulk Solids Handling 6(1986)5, p. 845 851.
3. Sipilä, Κ. Reactivity of biomass chars in fluidbed steam gasification. Proc. Research in Thermochemical Biomass Conversion, Phoenix, Arizona, 1 6 May, 1988. 11 p. + app. 2 p.
4. Moilanen, A. Ash behaviour in gasification. Proc. Symp. on LowGrade Fuels, Helsinki, Finland, June 12 16, 1989. Espoo 1989, VTT Symp. Series. In press.
5. Hupa, M., Skrifvars, B.J. & Moilanen, A. sintering tendency by a laboratory method Foundation Conf. on Mineral Matter and Ash in Coal, Santa Barbara, Ca, Feb 21 26, 1988.
6. Moilanen, Α., Hupa, M. & Skrifvars, B.J. The effect of temperature, chemical composition and gas atmosphere on sintering of peat fly ash. Int. Conf. on Coal Science, Tokyo, Japan, Oct. 1989.
7. Koljonen, J. Bulk chemicals ammonia. Proc. Int. Conf. Biomass for Energy & Chemicals in Europe, Industry & Agriculture, London, Nov 26, 1987.
8. KolJonen, J. Synthesis gas and fuel gas from peat for industrial and residential applications. Proc. 17th World Gas Conf., Washington, DC, June 5 9 , 1988.
9. Fagernäs, L. & Wilén, C. Steam drying process for peat and their organic condensates. Proc. 8th Int. Peat Congr., Leningrad, Aug 14 20, 1988. Moscow 1988, Int. Peat Society. Vol. II, p. 261 271.
Measuring the ash Proc. Engineering

613
DEVELOPMENT OF A DOWNDRAFT MOVING BED BIOMASS GASIFIER
R. Bilbao, J.Lana, P.Garcia, J. Arauzo Department of Chemical Engineering. University of Zaragoza
50009 Zaragoza. SPAIN.
Summarv Thermochemical processing of agricultural (corncob, cereal straw, ...) and forest residues (branches, chips, thinning residues, ...) is an interesting alternative for small scale energy production. In particular, air gasification in a downdrat moving bed gasifier yields a LHV gas with a low tar content, which is useful for electricity and/or heat production in isolated communities farms and small industries (ceramics, dryers, wood processing, ...). Two gasifiers of the type described above have been developed in order to process agricultural and forest residues which are produced in important amounts in the regions of Aragon and Cast i l la-León (Spain). The first one is capable of processing 50 kg. of biomass per hour and the second 200 kg. of biomass per hour. Both gasifiers have a similar structure although there are substancial differences between them, These differences were introduced in the design of the second gasifier in view of the experience gained with the first one, and of the modelling studies carried out. Both installations are described and some results obtained in them are presented.
1. DESCRIPTION OF EXPERIMENTAL SYSTEMS The 50 kg/h plant consists in the following elements: Gasifier,
Ash removal system, Blower, Rotameter, Biomass feeding system, Cyclone, Heat exchanger, Venturi meter, Gas sample point, One way valve, Torch. A scheme of the gasifier is shown in Figure 1, where the dimensions have been indicated.
The building materials for the gasifier must withstand the high temperatures developed in the oxidation zone (about 1200°C), as well as the corrosion caused by an enviroment where tars, H2 and CH4 are present. The attrition caused by the solid which moves downwards must also be taken into account. The gasifier wall consist of two

614
concentrici 10 mm. thick brick layers. The inner one is built with a refractory material of a high alumina content (82-84% A l 2 0 3 , 12-14% S i0 2 , 1 % Fe203 .) , whereas the outermost layer is built with a thermally insulating material in order to avoid heat losses. The external wall is covered with a 5 mm. thick F-lll carbon steel layer, which also serves as a support for the whole ensemble.
Under the gasifier there is a wedged compartment built on refractory concrete, on whose sloped walls the ashes slide to a screw conveyor, at the bottom of the cavity. This conveyor transports the ashes to a hopper where they are stored. The compartment also contains the exit pipe for the gases leaving the gasifier.
The biomass feeding system is connected directly to the gasifier, in order to avoid leaks. It consists of a 700 I. hopper in which the biomass required for a given run is loaded at one time. The feed is then introduced in the gasifier by means of an electrically driven ram. The whole ensemble is made of 3 mm. thick carbon steel plate.
Several thermocouples are located at different heights in the gasifier, as shown in Figure 1. Two of them Fe-Konstantan are in the drying-pyrolysis zone, and there are also thermocouples (Ni-Cr-Ni) just beneath the throat in the combustion zone and also under the ash-removal grid. A portable temperature probe was also available to pinpoint temperatures in different parts of the installation.
The 200 kg/h plant is basically similar to the plant of 50 kg/h, the main differences being due to the higher scale employed.
Moving upwards from the bottom, the gasifier of this plant can be described as follows: It consists of 1000 mm. i.d., 1200 mm. length cylinder which narrows, reaching a diameter of 500 mm., 250 mm. above the end of the straight section. A symmetric widening follows, reaching again 1000 mm. i.d. in a distance of 250 mm. This is continued by a second cylinder of constant, 1000 mm. i.d. and 350 mm. lenght. A scheme with the dimensions of gasifier is shown in Figure 2.
A ceramic material with a density of 350 kg/m3 was used to make the body of the gasifier. This material was chosen in view of its good insulating properties. However, its resistance against erosion was not so good. Thus, it was internally covered with 1/8" thickness of an erosion-resistant material with a density of 3450 kg/m3. Between both layers of materials an annulus of 100 mm. thickness was filled with ceramic wool, made of the same nature as the ceramic material used for the outer layer. Finally as in the 50 kg/h. Plant, the external wall of the gasifier is surrounded by a F-lll carbon steel shell.
The ashes are removed using an eccentric stainless steel rotating grid. Its capacity for ash removal has been determined in previous calibration measurements.

615
A new feeding system has been designed, in order to ensure plant autonomy, independently of the size of the feeding hopper (unlike the 50 kg/h. plant). This consists in a compartment closed in both ends by two gate valves, which ensure air tightness. Biomass is transported to the top of the system from the floorlevel, usign a belt conveyor. The air feeding system, is similar to that described previously, although, given the higher diameter of the throat in this gasifier the air distribution had to be improved somewhat.
2. EXPERIMENTAL The experimental program which is being developed studies the
influence of the following process variables: a)Air flow rate to the system. This variable controls the oxigen
input to the gasifier for the partial combustion of the pyrolysis products. By this means the energy requirements of the process are sat isf ied.
b)Residence time of biomass in the gasifier. In the 50 kg/h. plant, this is inversely proportional to the percentage of reaction time in which the grid vibrator is working. In the 200 kg./hr plant, the residence time is inversely proportional to the rotating velocity of the grid.
During the experiments gas samples are taken at regular intervals from the exit pipe. These samples are collected for GC analysis. At the end of each experiment the amount of biomass consumed is determined. The weight of ashes and fines is also determined.
The results of previous test in the 50 Kg/h. Plant were used to select the following ranges of operating conditions for experiments with wood chips: Air flow rate: 40-70 Nm3/h., % Vibration:15-50 (which corresponds to approximately 50-80 min. of residence time in the gasifier), Air to biomass ratio: 1.1-2 Nm3/kg. Table I shows the operating conditions for two series of runs with wood chips in the 50 kg/h. plant and 29 % vibration were used as reference in these experiments. Some of the results obtained in the 50 kg/h. plant are shown in Table II. These correspond to the two series of experiments mentioned above. Table III shows some results obtained whith wood chips in the 200 kg/h. plant.
The average composition (volume percent) of the gas obtained falls into the following ranges: N2 : 45-60 %, CH4: 0.25-2.5%, H2: 10-22%, CO: 13-25%, C02 : 8-19%, C2H2+C2H4: traces.
ACKNOWLEDGMENTS The authors express their gratitude to C.A.I.C.Y.T. for providing
financial support for the building of the 50 kg/h plant and to Consejería de Economía y Hacienda de la Junta de Castilla-León, I.D.A.E.

616
and C.I.E.M.A.T. for providing financial support for the building of the 200 kg/h plant.
Table I. Operating conditions in experiments with wood ch Run
1 2 3 4 5
Air flow (Nm3/h.)
50 50 50 50 50
% Vibration
50 38 29 22 16
Biomass (kg./h.)
46.1 35.8 44.7 28.1 25.4
Residence Time(min)
53 69 55 88 97
6 7 8 9
40 50 60 70
29 29 29 29
31.7 44.7 39.3 44.9
79 55 62 55
Table II. Resu Run
1 2 3 4 5
ts obtained in two series of Gas flow rate
(Nm3/h.) 73.2 72.1 79.1 71.7 79.3
LHV gas (kcal./ Nm3)
939.7 1011.1 1006.3
925.7 1061.2
6 7 8 9
66.0 79.1 95.4
119.1
1292.1 1006.3 1147.4 1187.7
runs with wood chips Gas Yield
(Nm3gas/kg. biomass) 1.59 2.02 1.77 2.64 3.12
2.08 1.77 2.42 2.44
Table Results obtained with wood chips in 200 kg/h plant Air flow (Nm3/h.)
250. 250. 300.
Biomass (kg/h.)
197.5 223.6 182.0
Gas flow (Nm3/h.)
223.6 364.8 375.4
LHV gas (kcal/Nm3)
1143.7 1051.7 1000.3

617
co CO co
ω co
en o
ο ο
CVJ
13

618
STEAM GASIFICATION OF BIOMASS IN FLUIDIZED BED. EFFECT OF THE TYPE OF FEEDSTOCK
J.CORELLA, J. HERGUIDO and J. GONZALEZ-SAIZ Chemical Engineering Department (Faculty of Science). University of Zaragoza.
50009 Zaragoza. Spain.
Summary Steam gasification of four different biomass (pine sawdust, pine wood chips, cereal straw and thistles (Cynara Cardunculus ) from energetic crops) has been carried out in a 15 cm. i.d. continuous fluidized bed gasifier. The gas, tar and char yields, the exit gas composition and the conversion of carbon have been determined at temperatures between 650 and 780°C (923-1053 K) for each type of biomass. At the same gasification conditions and due to the different structure and size of the particles, important differences are observed in the product distribution depending on the gasification temperature and on the biomass used. The differences in H2, CO and C02 in the gas product are important at low gasification temperatures. These differences decrease on the temperature increasing up to 780°C at which a gas composition similar for all types of biomass is obtained.
1. INTRODUCTION The type of biomass is an important factor to be considered in the gasification of
biomass in fluidized beds. A same gasifier should process the different types of biomass produced in the surroundings at the different seasons. Therefore, we considered necessary to know how the product distribution changes and what problems can appear when the biomass to be processed is changed.
Scott and Piskorz, Nunn et al., Prasad and Kuester, etc... have studied the pyrolysis and gasification in fluidized beds of maple, poplar bark, bagasse, wheat straw, etc... Regarding this work it is important the previous one of Walawender and Fan of the Kansas State University who studied the steam gasification in a 5 cm i.d. fluidized bed of cotton wood (branches) (1), of pure cellulose (2), and of dried feedlot manure (3). Their work is interesting and useful; however, in our region the most abundant types of biomass are different from the ones they use. As well, we believed that there was still work to be done in this specific field. Therefore, we have studied the steam gasification of different types of biomass. This study has been carried out in a 15 cm. i.d. fluidized bed gasifier at temperatures between 650 and 780°C.
2. EQUIPMENT The installation used in this work is shown in Figure 1. The gasifier is a 15 cm. i.d.
fluidized bed of 1.2 m. total height. The gas distributor plate was made of special nozzles. Its good operation has really been of key importance in the good progress of this research. The bed is silica sand (from beach), 32 cm. height at u = umf and t=0. The biomass feeding point is at the bottom of the bed, at 5 cm. above the gas distributor, and it is also fundamental for obtaining high throughputs and results similar to those of large gasifiers (4). The biomass is continuously fed to the gasifier at a prefixed constant flow rate. In principle, this operation is not easy because of the characterestics (form, size,

619
easy pyrolysis,...) of the biomass and the high temperature of the gasifier, but this problem was solved in our installation. The gasifier was externally heated by an electric oven. The plant has two lines in parallel for introducing the gasifying agent (steam) or other gases (air, N2...). The gases from the gasifier pass through a cyclone for separating the dragged fines (ash, char, sand), by a water-cooled heat eschanger in which the steam and the tars are condensed, and by a demixter consisiting of five glass-bulbs arranged in parallel and with a glasswool filling. After the continuous measurement of the gas flow, gas samples are taken and analyzed by gas chromatography.
CYCLONE
Figure 1.- Scheme of the experimental installation.
3. OPERATING CONDITIONS The main operation parameters are given in Table I. The bed temperature was varied
from one run to another, maintaining constant the other parameters. The effect of the H20/biomass ratio and of the space-time (Ho/uo) for the gas were previously studied (5,6).
4. FEEDSTOCKS Four different types of biomass have been processed. Pine sawdust, small pine wood
chips with a small quantity of bark, cereal (wheat) straw (one of the most abundant and available residues in Spain) and, finally, thistles from energetic crops have been processed. In Table II the main physicochemical characteristics of these biomass are shown. Concerning their fluidization, Aznar et al. (7,8) determined the umf, ucf, terminal velocities, bed and particle densities and the segregation In the biomass-sand and char-sand mixtures, for the biomass used here and with sand (and other solids) of different sizes. In Table II it can be observed how the four types of biomass have a similar composition (dry-ash-free). According to Prasad and Kuester (9) the hydrogen/oxigen ratio (H/O) in the biomass is a important parameter for the composition of the gas produced.
5. RESULTS AND DISCUSSION Product yield
The variation of the gas yield with the temperature of the gasifier is shown in Figure 2. An important increase in the gas yield with the temperature is observed. However, important differences among some biomasses and others are observed, these differences

620
being more acute at high temperatures. Thus, for example, at 780°C (1050 K) the gas yield is of 1.2 Kg. of gas/Kg. of sawdust, whilst with thistles it is only of 0.63. This increase of the gas production with the temperature may be due to the greater production of gas in the initial pyrolysis (more rapid at higher temperatures) and to the endothermal reactions of gasification of the char: on increasing the temperature the tar cracking and steam reforming reactions are favoured with the resulting decrease of the same and the gas yield increase.
TABLE Gaslfler Operating Conditions
Reactor temperature range 650780°C (9231053 K) Fluidizing gas mixture 90% H ¿O 10% N2 Superficial gas velocity at inlet, (u 1 0.25 m/s Bed height at U Oand 4 , ' ^ f (Ho) 0.32 m Total pressure in the gasifier 1.1 atm Steam/biomass ratio 0.80 Second or fluidizing solid Silica sand from beach
( 8 kg) dp = 300 + 200 μπι Umf » 6 cm/s Ucf = 11 cm/s
650°C 700° C 750°C 780°C
Flow rates: Biomass (kg/hr) 3.94 Steam (kg/hr) 3.39
Nitrogen (Nl/s) 0.131 Ng purge (Nl/s) 0.008
3.71 .22 0.123
0.008
3.56 3.06
0.120 0.008
3.46 2.98 0.115
0.008
TABLE II Main characteristics of feedstocks
Sawdust Straw Wood Chips
Particle size dp~ 500 μπι L > 5 mm. 1 0 x 5 X 2 mm
Inmediato analysis (% wt basis) moisture 8.5 7.8 11.1 ash 1.2 14.1 2.1 volatiles 77.4 61.6 74.4 fixed carbon 12.9 16.5 12.4
Elemental analysis (% daf basis) carbon 42.5 43.7 41.8 hydrogen 6.3 6.1 5.3
nitrogen 0.2 0.4 0.2 H/O molar ratio 2.02 2.73 1.68
Low Heating Value (Kcal/Kg daf) 4500 3790 4450
§: Thistle is constitued of fibers and pith in different proportions; it is very difficult to give a medium particle size.
Thistle
10.0 13.6 60.7 15.7
40.8 5.5 0.3
2.19
3860

621
M 1 . ¡
υ
o.i
0.1
0.'
o.:
* Sawdust t Straw * Wood Chips * Thistle
600 650 700 750 6
TEMPERATURE fC)
Figure 2.Gas yield vs temperature
E Ζ
9 UJ
> 3
1G)
1
•t· Straw . Wood Chips
■* Thistle ♦ Sawdust
600 800 650 700 750 TEMPERATURE (°C)
Figure 3.Gas yield (Nm3/Kg daf) vs temp.
For comparing which is the production of gas by Kg of net biomass, the volume of gases produced per Kg of biomass dryasffree has been calculated. This gas yield at different temperatures is shown in Figure 3. In this Figure two groups of well differentiated biomasses can be observed now: on the one hand sawdust and straw and on the other chips and thistles. It is observed how the sawdust and the straw give a gas yield quite a lot higher than that of the chips and thistles. The difference is high and it can be attributed to the different sizes (and shapes) of the particles of each biomass used. Thus, for the sawdust particles (spherical and of a very small size) the effectiveness factor (h) is near the unit. In the case of the chips their surface/volume ratio is much less and their char is gasified more slowly (h<1). Something similar happens with the thistles. In the case of the triturated straw its particles have a laminar shape, with a very small thickness. Then, the preferential direction of the gasification reaction is the transversal. Therefore, their characteristic dimension respect to the gasification (its thickness) is very small with a behaviour similar to that of the sawdust particles (h = 1). The night gas yield obtained with the straw can also be explained by a possible catalytic effect of its ash: it is abundant in the straw and it has a high content in K20 which favours the charsteam reaction.
Char and Tar production The char yield, referred to biomass daf, is presented in Figure 4. The sawdust and
the straw are those biomasses which produce less char. This is in agreement with the greater gas yield they give.
50
40
30
20
10
600 650 700 750 800
TEMPERATURE (X) Figure 4. Char yield vs. temperature
S «
+ Straw • Wood Chips H Thistle » Sawdust
600 650 700 750 800 TEMPERATURE (°C)
Figure 5. Tar yield vs. temperature

622
Besides the already known decrease of the tar yield with the temperature.in Figure 5 it is observed how this value is very similar for the different biomasses. The smaller production of tar occurs with the sawdust, coinciding with its greater yield to gases, coming from the cracking and reforming of the primary tars.
Gas composition The [H2] [C02]/[CO] [H20] ratio of the exit gas for each biomass and the curve
corresponding to the thermodinamic equilibrium constant are shown in Figure 6. These curves are similars to those presented by
Figure 6
Influence of gasifier temperature on the equilibrium of the water gas shift reaction theoretical and experimental.
600 650 700 750
TEMPERATURE (°C)
800
Maniatis and Buekens (10). The direct reaction of the watergas shift reaction has a kinetic constant greater than the inverse reaction at temperatures below 810°C (1083 K). At this temperature both constants (direct and inverse) are equal: Κ » k1/k2 = 1 (Figure 6), Prasad and Kuester (9), Walawender et al. (2). Besides, in our reactor the partial pressure of the steam (in the denominator of the above ratio) is much greater than that of the other components (1,5 to 3 times greater than that of the H2, which is the component which is found in greater proportion in the dry gas). Then, on increasing the temperature, the experimental ratio comes near the theoretical one, Figure 6. In simple words, a progressive increase of the concentrations of H2 and C02, and a reduction of that of CO is produced when the gasification temperature is increased. More details about the gas composition will be given in the extended version of this work (11).
ACKNOWLEDGMENTS The present work has been carried out within the Shared Project between the CAICYT
(Madrid) and the CEE (DG XII), Contracts ns AG3/84 and EN 3B0103E. The authors
acknowledge the F.P.I, grant awarded to J. Herguido by the Spanish Ministry of Education and Science.

623
REFERENCES (1) Singh, S.K., Walawender, W.P., Fan, LT. , Wood and Fiber Science. 18 (2), 1986,
327344. (2) Walawender, W.P., Hoveland, D.A., Fan.L.T., Ind. Eng. Chem. Process Des. Dev.. 2á
1985, 813817. (3) Raman, K.P., Walawender, W.P., Fan, LT. , Ind. Eno. Chem. Process Des. Dev.. l à ,
1980, 623629. (4) Corella, J., Herguido, J., Alday, F.J., 'Pyrolysis and steam gasification of biomass
in fluidized bed. Influence of the type and location of the biomass feeding point on the product distribution'. In 'Research in Termochemical Biomass Conversion', Bridgwater, A.V. and Kuester, J.L. (eds), Elsevier Appi. Sci. Pu. London, 1988, 384398.
(5) Corella.J., Adánez.J., GonzálezSáiz.J., Herguido.J., 'Steam gasification of biomas i fluidized bed reactor", in 'Biomass for Energy and Industry. 4th E.C.Conference'. Grassi, Delmon, Molle and Zibetta (eds). Elsevier Appi. Sci. Pu.,1987, 11071111.
(6) Agorreta, E., Aznar, M.P., Corella, J., 'La gasificación de serrín de pino con vapor de agua en un pequeño de lecho en continuo; Estudio de las variables del proceso'. 11 Jornadas Biomasa. Soria (Spain), 1987, pp. 4.131/4.135.
(7) Aznar, M.P., GráciaGorrfa, F.A., Corella, J., An. Quim. (Madrid^. 8_4_,1988a, 379
385. (8) Aznar, M.P., GráciaGorria, F.A., Corella, J., An. Quim. (Madrid), 8_4_,1988b, 386
394. (9) Prasad, B.V.R.K., Kuester, J .L , Ind. Eng. Chem. Res.. 27 , 1988, 304310.
(10) Maniatis, Κ., Buekens, Α., EPE, Vol XVII, na 34, 1982, 3539.
(11) Corella, J., Herguido, J., GonzálezSáiz, J., to be published.

624
STEAM GASIFICATION OF BIOMASS IN FLUIDIZED BED WITH A SECONDARY CATALITIC REACTOR. ■ I. RESULTS WITH THE SECONDARY REACTOR
EMPTY AND WITH SAND.
J. CORELLA, M. P. AZNAR, Ν. CEBRIAN, J.I. IGLESIAS, M.P. MARTINEZ
Chemical Engineering Department. University of Zaragoza. 50009 Zaragoza. Spain.
ajumara We are going to improve the gas composition from a fluidized bed gasifier by using catalysts
placed in a secondary bed in series with the gasifier. Catalysts for tar cracking, methanation and steam reforming will be used. To know their activity preliminary results in the same installation without catalyst are needed. Then, in this part I preliminary runs have been carried out with the secondary reactor empty and filled with silica sand. A bench scale facility based In a 6 cm. id. fluidiced bed gasifier, a porous metallic filter and a 4 cm. id. secondary vessel have been used. Working at 750°C in the gasifier, steam/biomass ratio of 0.9-1.1, and 6-9 g. biomass/min. the tar and CH4 contents in the exit gas are about 100 g/Nm
3 and 6-9% vol. respectively. The product distribution and gas composition are given for several operating conditions
1. INTRODUCTION It is well known that the gasification of biomass in fluidized beds still has technical problems or, at
least, that it could be improved. One of these problems is the quality of the gas produced. When air is used to gasify, not much tar is produced but the gas only serves to be burnt and, sometimes, with dificufty. When steam is used as gasifiing medium, the exit gas is composed of H2, CO, CO2, CH4, some C2 and C3, tars and elutriated char and ashes. This gas could be used as syntesis gas (CO+H2,H2+N2), as H2 rich gas, or to be burnt as medium heating value gas. In the first case the 5-
10% vol CH4 content should be eliminated. In the later case the CH4 content should be increased. And the tars should always be entirely eliminated for many and well know reasons.
There appears to be a clear necesity of reforming or modifiing the gas composition. Together with other possible technical solutions (O2 Introduction, use of very high temperatures, etc..) the use of catalysts could be a good or possible solution.
Thus, catalysts could be used for, at least, tar cracking, methanation or steam reforming of the CH4. It is curious to observe how these processes or solutions have been widely used and studied in the petroleum and coal refineries and industries but not in the thermochemical processing of biomass. So, this and forthcoming papers will be devoted to the experimental study of the use of catalysts in the steam gasificaton of biomass in fluidized beds.
As an starting point one had to face to, at least, three questions: 1st) Which catalysts to use? 2nd) Where to place them? 3rd) Which is the most commedable scale to investigate these processes?
The catalysts we have used have been of three very different types: tar cracking, methanation and steam reforming of CH4. The results with the last two will be shown in the next EC Conference in Lisbon (September 1989). Here we will only show the preliminary results (part I) and the results with some tar cracking catalysts (part II).
About the important question of where to place the catalyst, we think we already have defined ideas but it is also necesary to say that each author seems to have his own opinions. At least three different sites or emplacements can be envisaged for the catalyst: 1) impregnated in the biomass, 2) in the same gasifier, mixed or instead of the silica sand, and 3) in a secondary reactor in series with the gasifier. In our oppinion the methanation and the steam reforming catalysts have to be placed in a secondary reactor for, at least, three other reasons: 1st) these commercial catalysts are made to be used in fixed bed, not in fluidized beds (like the gasifier); 2nd) the temperature of these two

625
processes can be different from that of the gasifier; 3rd) the gasifier atmosphere can deactivate the catalyst. About the cracking catalysts, these catalyst could be used in the same gasifier (1) or in a secondary reactor. In part II of this work only its use in a secondary reactor will be studied and shown. Two stage processes are know for coal gasification (2) but these processes (and even studies) for gasification of biomass are very scarce.
We think that the scale of the research (i.e. the size of the gasifier and of the catalytic reactor) is an important and previous decision that can influence not only further important results like the product distribution and gas composition (see, for instance ref. 3) but also future decisions about the technical and economical viability of the process. Between very small gasifiers (i.e. 2-3 cm i.d.) and pilot plant gasifiers (larger than 15 cm i.d.), for the reasons exposed in (4) we have chosen here an intermediate or bench scale installation. This installation has a continuous feeding of biomass and a continuous reforming of real exit gas from the gasifier of biomass.
To compare results, to clearly see the effect of the catalyst and to differentiate it from the effects (thermal cracking, for instance) of the installation, previous runs have been made: i) without the secondary catalytic reactor; ii) with this reactor being empty and iii) with this reactor filled with inert silica sand. Only these results will be shown in this part I.
2. INSTALLATION A scheme of the installation can be seen in figure 1. The gasifier is a 6 cm i.d. fluidized bed, 30
cm height, externally heated by a 4 Kwatt furnace, followed by a disengaging zone 20 cm i.d., and 14 cm high. This upper zone was always thermally isolated to diminish tar condensation. The gas distributor was a plate with 9 bubble cups of 8mm diameter with 10 holes each one of 0.8mm diameter. At not very hight superficial velocities this plate gives small bubbles (good gas-solid contact). Before the gas distributor plate there was an elicoidal pipe to preheat the fluidizing and gasiting gas. This fluidizing gas was always steam.
Gas sample
Gas to vent
Secondary bed
Figure 1. Scheme of the experimental installation

626
The continuous feeding system of the biomass has had special importance. Althought several improvements were made some runs had to be stopped by the plugging of this system. Main aspects of this feeding systems are: the downpipe or standpipe has to be externally refrigerated (with several quenching air flows). This downpipe has a 3 cm. diameter to dinimish its plugging. The screw feeder has 3.5 cm i.d. The usual biomass flow rate was 6-10 g/min. (0.36-0.6 kg/hr). As biomass, pine (pinus pinaster) sawdust was always used (C:40.8%wt, H: 6.3%wt, 0:52.9%). Its size was always -630+250 mm, its humidity about 10-14% wt and its LHV 4400 kcal/kg daf.
After the gasifier there was a porous metallic filter (10 mm nominal pore size and 50 cm length). Then, there was the catalytic bed of 4 cm i.d. and 15 cm height. At its bottom there was a bubble cup (2 cm o.d.) as gas distributor. At the top there was another bigger zone of 8 cm i.d. and 20 cm. high.
At the exit of this catalytic reactor there is an heat exchanger, a tar and water receiver, an effective demixter (with glass wool), a continuous flowmeter, a gas sampling device and the exit gas pipe. About every 10 minutes a gas sample was taken which was analyzed by conventional gas cromatography. Sometimes, in the same run, several samples of the condensed liquid were taken. This liquid was analyzed by several methods: liquid cromatography, total organic carbon, tar extraction with solvents, etc... All these analysis allowed us to know the evolution and deactivation of the catalyst when it was used.
It is necessary to cite the complexity of this installation, the difficulty of carrying out the runs in good contitions (maintaining constant the flowrates, temperatures, pressures, etc. ) , the high number of plugs and conections which made escapes of gas highly probable, the difficulty in closing the mass balaces, etc... The % of 'valid runs' was increasing with time, improvements, experience,...
In the gasifier there was always silica sand of -200 +125 mm (umf = 5 cm/s; Ucf = 9-2 cm/s), with Ho = 30 cm(at t=0 and u=umf). The superficial gas velocity at the gasifier inlet was 15,18 or 21 cm/s. This variable is very important since it determines the segregation or not of the char in the upper part of the bed, the gas velocity at the exit, the gas velocity at the inlet of the secondary reactor and the space-time or mean residence time in this secondary reactor.
Char and tar were collected in different quantities (and of different types!) in the different parts of this installation. Unless stated, char and tar yields will be refered to all the installation and for the whole of the run.
3. RESULTS WITH THE SECONDARY REACTOR EMPTY To stablish what results are obtained without catalyst, runs were made with the secondary
reactor empty and in the following conditions:
gasification temperature 750°C height of sand in the gasifier 30cm temperature in the filter chamber 515-520°C steam flow rate 5-7 g/min. biomass flow rate 6.2-6.6 g daf/min.
The product (gas, tar, char) distribution and the gas composition in different runs with different temperatures in the secondary reactor (empty) are shown in figures 2 and 3. To compare with further results with catalyst, two data are important and should be remenbered: the tar and the CH4 contents in the gas. The tar content is between 76-120 g/Nm3 equivalent to tar yields of 5.9-8.1 Kg/Kg daf. The CH4 content in the gas is between 5 and 9 % vol.
Compariing these results with those previously obtained in the same rig but without fitter and secondary vessel, less tar and more gas yield are now obtained. We think this is due to the existence of thermal cracking in the filter and in the secondary vessel. Also, there is now more H2 and CO2 and less CO. This can be easily attibuted to the existence now of more volume at high temperatures for the gas phase shift reaction (CO+H2O = CO2+H2).

627
=· 60. 0 > 50
Τ 40. 0. O 30υ (0 20
° 10
0.
α π
•
•
■ Λ
— Γ — · —
□
■
ι—' 300 400 500 600 700 800 900
TEMPERATURE (°C) Figure 2. Effect of the temperature of the
secondary reactor (empty) on the product dis t r ibut ion (■ gas; ° char; · tar)
300 400 500 600 700 TEMPERATURE (°C)
800 900
Figure 3. Effect of the temperature of the secondary reactor (empty) on the gas composition at the exit of the installation <H, CD, CO. CH4)
4. RESULTS WITH SILICA SAND IN THE SECONDARY REACTOR With silica sand of 297+200mm (umf = 6.0 cm/s; ucf= 110 cm/s) in the secondary reactor the
effects of the bed height and of the temperature of this bed have been studied. The effect of the bed height is shown in figures 4 and 5 in which the gas yield and the gas composition at the exit of the installation are shown. The temperature of the secondary reactor was 745 ± 10°C (gasifier temperature = 750°C, other conditions: the same as above). The gas yield does not change appreciably with the use of sand or whit the height of the sand in the bed. Only the H2 content increases a little and the CO decreases (attributed to the shift reaction in the bed)
5 10 HEIGHT (cm)
Figure 4. Gas yield vs. height of the secondary bed with sand
70
vo
l.)
οι
σι
o
0
ι .
ι
t 40 o. S 30 O U 2 0
< 10 . O 0 .
C
.♦
■
*
El
»
5 HEIGHT
•
α
ι ' 10 (cm)
»—
Β
Ι ' 15
Figure 5. Gas composition vs. height of the secondary bed with sand (297 + 200 Mm). T^ secondary bed 745 ± 10°C

628
The effect of the temperature of the secondary bed with silica sand (being the filter temperature = 550°C), for two sands and two different bed heights, is shown in figures 6 and 7. Comparing with the results obtained in the empty reactor there is not a sensible influence of the sand, indicating this solid is inert. The product distribution and gas composition will be used in further parts of this work as the basis to see the effect of several catalyst placed in the secondary reactor.
I a.' 2 8 <
IV
6 0
5 0
4 0
3 0
ÜU
1 0
o
*
El
»
•
H
•
Β
•
« 300
Figure 6.
400 500 600 700 800
TEMPERATURE (°C)
Effec t of the temperature of the secondary bed with sand of 297 + 200 um, 10 cm high
3 0 0 4 0 0 500 6 0 0 700 8 0 0 900
TEMPERATURE (°C)
Figure 7. Gas composition at the e x i t of the I n s t a l l a t i o n v s . temperature of the secondary reactor with sand of 630 + 297 μιη, 14 cm high (u ( g a s i f i e r ) = 15 cm/s u (secondary bed) = 57 cm/s)
5. REFERENCES
(1) Corella.J., Herguido, J., GonzálezSáiz, J., Alday, F.J., RodriguezTrujillo.J.L, in Research in Thermochemical Biomass Conversion A.V. Bidgwater and J.L. Kuester (eds.); Elsevier Appi. Sci., London (1988),754765.
(2) Juneja, M.N.; Mazundar, A; Biswas, D.K.; Rao, S.K. Fuel Sci. Techn. £(2), 1987, 4560.
(3) Corella.J., Herguido, J., Alday.F.J. in Research in Thermochemical Biomass Conversion A.V. Bidgwater and J.L. Kuester (eds.); Elsevier Appi. Sa'., London (1988), 384398.
(4) Corella.J., Aznar, M.P.,Cebn'án,N., Brana, P. in Adas XI Iberoamerican Symposium on Catalysis Guanajuato, Méjico, May 1988. IMPMéxico D.F. (ed), pp 10191027.

629
STEAM GASIFICATION OF BIOMASS IN FLUIDIZED BED WITH A SECONDARY CATALITIC BED. - II. TAR CRACKING WITH DOLOMITES IN
THE SECONDARY REACTOR
M. P. AZNAR, J. DELGADO, J. CORELLA, J. LAHOZ
Chemical Engineering Department . University of Zaragoza. 50009 Zaragoza. Spain.
Summary
The use of dolomites for the cracking of tars in gases from steam gasification of biomass in fluidized beds has been studied. The installation was based in a bench scale facility with a 6 cm i.d. fluidized bed gasifier operating at 750 °C, a porous metalic filter at 530°C, and a 4 cm i.d. secondary bed filled with dolomite and operating between 400 and 950°C, mainly between 800 and 900 °C. Dolomites from Spain, Sweden and United Kingdom have been used. Above 800-850°C tar conversions higher than 95% and tar concentrations in the gas less than 200 mg/Nm3 have been obtained. The deactivation of the dolomite along the time-on-stream has been studied and the life of the dolomite is given.
1. INTRODUCTION In part I we indicated how the product distribution and the gas composition from
biomass gasifiers or pyrolyzers can and have to be improved. In particular, tar and CH4 contents in the gas obtained are far from the optimal ones. This part II will be only devoted to the tar elimination in the exit gas by catalytic cracking . The problems that tar gives are well known, and this tar has to be eliminated, or at least diminished to a great extent. This tar could be eliminated by using very high temperatures or introducing oxigen in the gasifing gas.The first solution is not easy when fluidized beds are used as gasifiers and the use of oxigen Introduces the necesity of an auxiliary O2 production plant. So, here we are going to continue the study of another solution for tar elimination. This solution is based in the use of cracking catalysts.
In the petroleum refineries very difficult feedstocks and heavy oil fractions (end b.p. >600°C) are easily cracked in riser reactors operating between 500 and 620°C (at the bottom) and contact times with the catalyst of 2-4 seconds. These commercial catalysts (fesh or 'in equilibrium') could also be used in blomass gasification/pyrolysis for tar cracking but its very low mean diameter (about 70 μηι) makes them easily elutriables (terminal velocity, ut=20 cm/s). So they can be used only in riser gasifiers or circulating systems (l)Therefore, here we are going to study another type of cracking catalysts: dolomites.
At least, in recent years in four countries (Sweden, France, Spain and USA) studies have been made about tar cracking with dolomites. Important and previous works have already been made. Among others, there are the studies in Sweden of the Royal Institute of Technology (2) and of Studsvik Energy (3), in France of Magne, Deglise and colls. (4), in USA of Battelle-Pacific NorthWest (i.e. 5), of Longwell, Peters and colls. (6) and of Wen and Cain (8) and in Spain our previous work on this subject (9). To make here a comparative study of these works is imposible by the obliged length of this communication. So we can only summarize the previous findings in two main ideas:

630
Calcined dolomites are very active for cracking of tars coming from biomass or coal gasification or pyrolysis, and there is still a lot of work to be done to optimize the use of dolomites in these processes.
The specific fields in which there is not much knowledge are: the deactivation rate of the dolomite, its fluiddynamic properties and the effect of the origin or provenance of the dolomite in its activity. This work is a step more in the study of these factors.
Associated with the cracking of oils, hidrocarbons or tars there is always coke formation on the catalyst surface that in oil cracking deactivates the catalyst in a few seconds. So, in tar cracking we are going to find this deactivation of the dolomite by coking.
The catalyst (dolomite) can be placed in several places. In a previous work (10) we placed it in the same gasifier. In this work only its use in a secondary reactor in series with the gasifier will be studied.
The realtionship between the particle size of the dolomite and the superficial gas velocity (u) in the cracking reactor is very important. Remember that the particles with a terminal velocity (or chocking velocity in risers) less than u will be carried out of the bed. Thus, for a fixed superficial gas velocity at the gasifier inlet, and given the diameters of the gasifier (or pyroliser) and of the cracking reactor, the dolimite has to have a given minimun particle diameter to be fluidized and not carried out from the bed.
2. INSTALLATION AND OPERATING CONDITIONS The bench scale installation was shown in part I. It basically consists of constituted
by a 6 cm i.d. fluid bed gasifier continuously fed with biomass from the top, a porous metallic filter and a 4 cm i.d. secondary catalytic reactor filled in this work with dolomite.
The main operating variables were:
fluidizing or gasifiing gas pressure superficial gas velocity at
gasifier ¡nlet(u0l) biomass
gasifier bed
steam/biomass ratio temperature of gasification temperature of the filter temperature of the
secondary bed of dolomite height (at t=0) of the
bed of dolomite spacetime (τ) for the bed
of dolomite, defined as (H0/U02)
spacetime (τ') for the bed of dolomite, defined as kg dolomite/kg biomass daf fed/hr
steam atmosferic
15 cm/s pine (pine pinaster) sawdust, 630+250μπι
silica sand, 200 +125μπι, 30 cm high
0.800.95 750800°C 520550°C
400950°C, mainly 800900°C
10cm
0.17 s
0.210.26 hr
Gas samples were collected each about about every 10 minutes and analysed by gas cromatography. The condensed liquid (mainly water) at the cracking reactor exit was

631
taken about evry 10 minutes. It was analysed by liquid cromatography and its TOC (Total Organic Carbon) content determined. The TOCvalue was converted to the equivalente amount of tar on the assumption that the molecular weight of tar was equal to that of phenol, like Sjöström et al. did it (2)
3. DOLOMITES USED We have used three different dolomites from three different countries: 1. Spanish, from 'Dolomitas del Norte' of Cantabria (North of Spain). Its size was
innitially 1000+600 \im but given that u02 = 5070 cm/s and that there was some carry over it was increased to 2.0+1.0 mm.
2. Swedish Sala Dolomite kindly given by the R.I.T. of Stockholm. It was of the same origen as that used in ref. 2.
3. English, the 'stone' used in the John Brown/Wellman gasifier. It had a size of
2.5+0.6mm and it was really a calcite. Their detailed characterization is more than length of this paper. We ca only
say that they were calcinated at 900°C first in a furnace and afterwards in the same cracking vessel previous to its use and that this variable, for instance, had some importance.
4. SOME RESULTS The gas yield obtained with the Spanish dolomite at different temperatures of the
dolomite bed is shown in figure 1. Above 700°C a gas yield increase is observed due to the tar cracking as will be shown bellow.
's Ό
*:
Figure 1. Gas yield at different temperatures of the bed with dolomite.
> < (3
100
80
6 0
40
?ο· ■ I I I
s-υκ>·
ι1—ι—■—ι—■— 400 600 800 1000
TEMP. OF THE SECONDARY BED (°C)
The most impresive and spectacular effect of the dolomite is in the conversion or cracking of tars. In figure 2 the tar conversion with the Spanish dolomite at different temperatures is shown. Above 800850°C tar conversions (cracking) higher than 95% are obtained. The tar concentration in the exit gas at different bed temperatures is shown in fig. 3.

632
100
600 700 800 900 TEMP. OF THE SECONDARY BED (°C)
Figure 2. - Tar conversion (average for 1 hr) vs. temperature of the bed of dolomite.
600 700 800 900 TEMP. OF THE SECONDARY BED (°C)
Figure 3.- Tar concentration in the exit gas (average for the 1 hr length run).
There is a very clear diminution of the tar content in the exit gas when dolomite is used at temperatures above 800°C aprox. But these results are the average for each run of 1 hr. length and τ of 0.17 seconds or τ' of 0.21-0.26 hr, like in ref.(2). Let us see what happens in the first minutes of reaction.
In figure 4 the tar conversion in the secondary bed and at two temperatures (of the cracking bed), 780 and 840°C, is shown at different times-on-stream of the (Spanish) dolomite. The tar concentration in the exit gas from the dolomitic bed at different times-on-stream is shown in figure 5. Several clear and important effects can be seen in these figures .For instance: the deactivation of the dolomite affects the tar conversion or the tar content in the gas. The deactivation occurs in several minutes. The temperature is an important factor. With the space-time used, at 840°C a tar conversion of 99% and a tar content in the gas of less than 200 mg/Nm
3 can be achieved, but this only in the first 40 minutes (of using the dolomite).
20 40 60 80 100 TIME ON STREAM (min.)
Figure 4.- Tar conversion in the bed of dolomite vs. time on stream.
m E
LU O ζ o o cr < - ι -
Ο 20 40 60 80 TIME ON STREAM (min.)
Figure 5.- Tar concentration in the exit gas from the bed of dolomite vs time on stream.
The deactivation by coking of the dolomite also influences the gas composition which varies with time-on-stream of the dolomite, as can be seen in figure 7. At t>60 minutes, the gas composition is the same as those obtained when sand was used in the secondary bed.

633
Figure 6. Gas compos, at the exit of the bed of dolomite.
780 °C 840 °C
ζ O t co O
8 (Λ
S τ ■ 1 ■—*π ■ Γ 20 30 40 50
TIME ON STREAM (min.) 70
Intimated asociated with 840°C curve in figures 5 and 6 are the cromatograms (liquid cromatography) shown in figure 7. They correspond to the condensed water at the exit of the bed of the Spanish dolomite (at 840°C). In the first three samples the dolomite was still active and there are few components and in small quiantity in the condensed water. After about 40 minutes (see also figs 5 and 6) the bed begins to be deactivated and the cromatogram now shows more componentes and in more quantity which corresponds to a lower tar conversion in the bed of dolomite.
REFERENCES
(1) Corella.J.; Herguido, J.; RodrfguezTrujillo, J.L.; Paper to be presented in the 5th EC Conference on Biomass for Energy and Industry. Lisbon, September 1989
(2) Sjöström, K.; Taralas, G.; Liinanki, L. in Thermochemical Processing of Biomass Α.V. Bridgwater and J.L. Kuester L(eds), Elsevier Appi. Sci., London, 1988, 974986.
(3) Aldén, Η.; Espenäs, B.G.; Rensfelt, E. in Thermochemical Processing of Biomass. A.V. Bidgwater and J.L. Kuester (eds.); Elsevier Appi. Sci., London 1988, 9 8 7 1 0 0 1 .
(4) Donnot, Α.; Reningovolo, J.; Magne, P.; Deglise, X., J. Anal. Appi. Pyrolysis. 1985, 8_, 401414.
(5) Elliot.D.C; Baker, E.G., Biomass, 1986, â, 195203. (6) Yeboah, Y.D.; Longwell, J.P.; Howard, J.B.; Peters, W.A. Ind. Eng. Chem.
Process Des. Dev. 1980, l i , 646653. (7) Eilig, D.L.; Lal.C.L.; Mead, D.W.; Longwell.J.P.; Peters, W.A. Ind. Eng. Chem.
Process Des. Dev. 1985, 24, 10801087. (8) Wen, W.Y.; Cain, E. Ind. Eng. Chem. Process Des. Dev, 1984, 23, 623637. (9) Corella, J. ; Herguido, J. ; GonzálezSáiz, J. ; Alday, F.J.; RodrlguezTrujillo,
J .L , in Thermochemical Processing of Biomass A.V. Bidgwater and J.L. Kuester (eds.); Elsevier Appi. Sci., London 1988, 754766.

ON W -Ρ-
Figure 7.- Liquid cromatograms of the condensed water collected at the exit of the secondary bed of dolomite, at different intervals of time-on-stream.

635
Fixed bed gasification of lignocellulosic biomass the CEMAGREF process
S.GAUDEMARD and JJ.BECKER Centre d'Etudes du Hachinisiie Agricole du Génie Rural des Eaux et des Forêts
Parc de Tourvoie ANTONY 92 FRANCE Summary
The use of biomass as an energy carrier could solve some of the main energy problems especially in developing countries,and gasification of lignocellulosic material in order to produce lean gas suitable to fuel an engine seems to be one of the most promising techniques. Nevertheless,among t h e different technologies available (fixed bed,updraft o r downdraft,fluidized bed...) none appears able to face properly the major problem related to the presence of tars in the gas produced.CEMAGREF developped a new technology which enables the elinination of the tars produced during pyrolysis i n an internal burner in order to obtain a clean gas.An automation of the wood feeding system and the internal running of the gasifier was carried out to ensure a good gas quality even in non stationary operating o f t h e system.Tests results obtained o n a pilot unit developed in CENAGREF showed l o w t a r content a n d good gasification efficiency in the range o f 5 0 t o 2 0 0 kg/H o f d r y wood chips. Therefore a generating set including a POYAUD g a s engine of 120 kW was implemented at the end of t h e line,and t h e power is consmed i n a flexible resistance.Long r u n tests will be conducted in order to prove the industrial feasibility of the process.
l.INTRODUCTION Among the various energy from biomass lines CEMAGREF identified the gasification technique as
one of the most promising especially in Developing Countries. The aim of the research described here is indeed to produce a tar free lean gas,from high or
medium grain size lignocellulosic biomass. Moreover,the thermal engine fueled with lean gas must be as flexible and reliable as with an oil derived fuel.It is therefore necessary to automate the part of the line upstream from the engine (biomass feeding,control of the inner operating parameters of the gasifier according to the power level required by the engine).
2.0PERATING PRINCIPLE OF THE CENAGREF GASIFIER The pyrolysis of bioiass produces charcoal and volatile matters,both products supplying
approxiiately half the energy available.Although the gasification of charcoal (action of C02 and H20) is widespread,the required elimination of the heavy components of volatile Batters is no easy task and very few analytical data are available.Nevertheless,we noticed that this problem could be solved by treating them at high temperature and by using the catalytic effect of charcoal.
The horizontal positioning of the gasifier and the use of a high teiperature recycling fan (300 to 5O0*C)in tbe CENAGREF process enable to solve the above-ientioned probiere: - horizontal position for a better distribution of coibustion gases through the bioiass bed. - high teiperature recycling in order to boost heat transfers as soon as the load enters the
reactor (thermal shock at 400 C instead of 80*C for the DELACOTTE gasifier)and to control the temperature range in the combustion chamber from 1000' to llOO'C even in case of complete gasification of the charcoal.
The Bachine obtained is shown on diagram l.It is a fixed bed process and therefore the feed Bust be large grain sized bioiass (woodchips,coconutshells,...)

636
3.TECHNICAL DESOJIPTIOH OF THE PILOT QUIT The pilot unit at ÀHT0NY described here is smal («annua vood iiput of about 200 kg/h) and
easy to operate ; the aim of this operation was threefold : - defining the autoiiated and control systems required for an industrial unit - determining all the elements necessary for an accurate dimensioning of the unit for a given pover.
- carrying out long-run tests proving the industrial feasibility of the process This unit includes the folioving equipment (diagrai 2) :
a) an agricultural moving floor trailer driven by an electric ïotor and containing the vood chips used as raw ïaterial for the gasifier, b) a belt conveyor c) a recycling gasifier,CEMAGREF process, d) a vet filtration unit e) a Lov BTU gas burner,CEKAGKEF process f) an air heating systei operating on fuel oil in order to obtain air températures around 400'c. On the industrial scale,a lean gas/air exchanger is to be used instead of the systei operating on fuel oil g)a 120 kv generating set including a six cylinder gas engine and an alternator yielding the produced current in a flexible resistance.
The analysis of the lean gas produced ,Boreover the composition into non condensable elements by gas chromatography, includes the content of condensable elements (vater and tars) and particles with a device designed for the purpose.
4.PRINCIPLE OF AUTOMATION OF THE LIME The objective of this operation is to obtain an automatic setting of the value of the process
parameters in order to provide the power level required by the engine. The automation of the vood feeding system is a standard operation vhich is not vorth
describing here. On the opposite,the automation of the gasifier by it self has to be considered in detail. The system includes four control parameters :
a) three gas flovs (air,lean gas and recycling gas). b) the vood input.
The following constraints must be taken into consideration in order to ensure an adequate running of the system : a) the lean gas outflow released by the gasifier must contain the amount of energy required by the engine b) the pressure in the gasifier must remain close to that of atmosphere (in fact,a little depression is set to avoid the exhaust of pyrolysis gases through the vood feeding system). c) wood residence time in the reactor must be long enough to ensure a complete pyrolysis of the wood when leaving the pyrolysis grate and a complete gasification before falling into the ash box. d) the temperature in the combustion chamber must be high enough to enable a complete cracking of the tars. e) the temperature of the recycling gas must be high enough to avoid tar condensation in the recycling circuit.
Whatever the output of the gasifier the geometry of the vood is maintained by controlling its height at the end of the gasification grate.
Thus the automation carried out includes tvo control levels: -firstly:a subsequent change in the pover level required by the engine leads immediatly to a resetting of the three control parameters(air flov,recycling flov,piston speed (according to the values provided by the model and confirmed by experiments. - secondly:the regulation around the setted values during a steady state is obtained by measuring and treating continuously the following parameters : - pressure level inside the gasifier controlling the air flov. - height of the charcoal bed controlling the speed of the biomass piston. - combustion and recycling temperatures controlling the recycling flov.

637
5.EXPERIMENTAL RESOUS A first set of experiuents aimed at obtaining a steady-state operating of the line at
different lean gas outflow levels.Table 3 gives some results obtained during twenty hours of autonatic operating.As long as the engine was not functioned at the end of the line,the variation in gasifier loading was obtained by changing Manually the position of the exhaust gas valve.This operation was not perfectly representative of the actual control by an engine as it did not take into account a possible change in the heating value of the lean gas occuring during the transient phases.A quick switch of the lean gas production fro« 600 Ni3/h (200 kg/h of dried wood chips) to 300 Hi3/h and vice versa could be realized while Maintaining the adequate temperature levels and the geometry of the biomass bed,the change in operating paraneters being done autonatically.Moreover no unexpected production of tars was collected in the gas analysis system.
6.FUTURE DEVELOPMENTS The good results obtained so far encourage us to carry on with the project.Two aspects will be
worth developping in the future. It would also be interesting to use the experiiiental data obtained on the prototype at ANTONY
and co«pleientary analyti caldata («ainly obtained with a big sized thermobalance developed at CEMAGREF) to develop a «odel for the optinal diiiensioning of the gasifier for a given power level,depending on the biomass (especially its grain size) to be processed.
Long-run tests(around 1000 hours)should highlight the feasiblity of the line on an industrial scale.But,these tests are significant only when the lean gas produced fuels the engine which is the weak point of such a power line.
Moreover,it could be checked if the response time of the gasifier to a variation of the load has the saee îagnitude as the response tiae of the engine,in order to obtain the sate flexibility as with diesel oil.
Therefore a 120 kW POYAUD spark engine is implemented at the end of the line.The engine runs an alternator and the power is consuied in a flexible resistance.
7.ECONOMIC ANALYSIS With the relative coeplexity of the CEMAGREF process and the cost of the autonation,the
comercial size of the gasifier will be in the range of 500 to 5000 kg/h of dry wood. An economic calculation related to an unit of 1000 kg/h of dry wood was carried out and the results are shown in diagram 4. Two raw laterial scenarios were considered : -wood as a by product in an agroindustrial complex(palm or hevea plantations...)at a cost of 15$/ODT(collecting cost only), -wood produced in energy plantations at a cost of 35$/0DT (Eucalyptus in Brasil or Congo...)
It caie out that even if wood has to be produced in energy plantations,it lakes sense to iipleient the gasification technology in areas of Developing Countries far from the coast (because of the high transportation cost by road or railway of the diesel oil).
8.REFERENCES (1) BECKER,J.J,Fixed bed gasification of lignocellulosic bioiass the CEMAGREF process
Conference "Research in Termochemical Bioiass Conversion",Hay 1988,Phoenix,USA (2)BECKER,J.J and GAUDEMARD ,S.,Développèrent d'une ligne de production d'électricité à partir
de biomasse.Société française des thermiciens,journée d'études "Les loteurs à biogaz", Noveiber 1988,Paris,FRANCE

638
air_ J I
DIAGRAM 1 CEMAGREF Fixed bpd Gasifipr
wood
I J , recycling gas
(from recycling fan)
gas exhaust recycling gas (towards recycling fan) pyrolysis area
Q(bed of wood)
gasification area (bed of charcoal only)
DIAGRAM 2 Schematic of the pilot unit in ANTONY
Wood feeding system
Engin«

639
TABLE 3 EXPERIMENTAL RESULTS
Feed rate (kg/h)
Gas flow rate (N»3/h dry gas)
Moisture content of the feed ({)
Heating value of dry gas (HJ/Hr3)
Hater content (KG/Ni3 dry gas)
Tars (g/Hi3 dry gas) (1)
Particulates (g/Hi3 dry gas)(l)
Energy balance (\) (2)
210
200/600
15
4.9
0.076
0.025
5
78
Dry gas ««position (1) H2
00
C02
H2
CE4
02/Ar
45/52
18/21
12/13
16/20
1
1.3
(1) Before filtration (2) LHV of lean gas over LLHV of wood
(one assîmes that gasification air is heated by means of an exchanger operating with lean gas outflow)
DIAGRAM 4
C o m p e t i t i v i t y of Low BTU gas
l o c a t i o n : a f r i k a n coast 2 on equipment France b a s i s )
l e an gas
1 ^ oil price 3 0 «/bbl
$/MBtu
» location : continental Afrika (x2 on diesel oil ; χ 1,6 on equipment France
. basis) ψ y' diesel oil
"* oil price î/bbl
basis : gasification unit of 1 ODT/h biomass (cost A00 000 $ France basis),
running 60O0h/year, amortized in 10 years at 8 X

640
STUDY ON MARKED PRODUCTS OF WOOD GASIFICATION MECHANISMS WITH THE AIM OF PRODUCING CLEAN GASES
S. CASTILLO, S. BENNINI, G. GAS*, J.P. TRAVERSE Laboratoire de Recherche sur l'Energie,
*U.A. CNRS No 241, Centre de Physiologie Vegetale, Université Paul Sabatier, 31400 Toulouse, France
Summary
This work concerns research into the thermochemical upgrading of lignocellulose materials. It describes a method which was developed for analysing the rupture mechanisms of the macromolecules of wood under the effect of intense thermal flow. A knowledge of these mechanisms is necessary for understanding the process of gasification and determining the respective proportions of the three phases produced (solid, liquid and gaseous) and in particular for preventing the formation of tars during gasification of wood.
This method uses * 4C for the specific marking of the lignin fraction of lignocelluloses. A study of the distribution of the marked atoms in the different phases obtained after pyrolysis - in particular the gaseous phase - gives information on the mechanisms of rupture under the effect of thermal shock.
1. PRESENTATION OF THE METHOD AND ASSOCIATED TECHNIQUES
1.1 Preparation of Marked Lignocelluloses
Lignocelluloses are made by the association of polysaccharides (cellulose, hemicelluloses) and lignin. We used the fact that it was possible to mark the lignin fraction of the wood specifically by exposing poplar branches to ^C-marked metabolites which are precursors of the phenyl-propane groups constituting lignin. When one of the four precursors represented is incorporated (Table I) we obtain lignocelluloses which are marked only on the lignin fraction.
When a solution containing the marked precursor has been absorbed the poplar branches are transferred to a vessel containing distilled water, in a ventilated room at a temperature of 22°C with 5W/m2 lighting so that the precursor can metabolize.
The branches are then stripped of their leaves and bark, fixed in liquid nitrogen and lyophylized. They are then comminuted and screened (250 ym). The lignocelluloses are extracted successively by aqueous and organic solvents (1) . The residue is rinsed in absolute alcohol and dried under vacuum. The specific radioactivity of these lignocelluloses (Table II) is obtained by measuring the radioactivity of the ^4C02 obtained by combustion of a known quantity of powder in the oxidizer. The l^COo is absorbed in a suitable solution and its radioactivity measured by liquid scintillation.

641
TABLE I . L i g n i n p r e c u r s o r s and monomers
Monomer precursors of l i g n i n Monomers of l i g n i n
ÇH=CHCH 2 OH
Ç H J Î Ç H C O O H *ÇH=*CHCH2OH * N H ,
Phenylalanine 14 OH
Cinnamic acid
H=CHCOOH
( cyc le C)
ÇH = CHCH2OH
n i ^ ^ Î O C H 3
OH
CH=CHCOOH
Ì] OCH,
OH
ÇH = CHCH2OH
OCH,
*CH=CHCOOH
il ÇH = CHCH2OH
(f
Cinnamic acid , 14 . (α C)
1 Τ 3 OH
: coumaryl a lcoho l
R. = Η, R = OCH : c o n i f e r y l a lcoho l ) main poplar
R, = R, = OCH : s i n a p y l a lcoho l ) monomers
14. TABLE II. Specific radioactivity of the various C lignocelluloses
Precursor Specific
Lignocellulose Radioactivity (Bq/mg)
14 Phenylalanine (U C)
14 Cinnamic acid (cycle C)
Ferulic acid (014CH )
14 Cinnamic acid (alpha C)
642 000
1 128 000
618 000
1 526 000

642
1.2 Thermal Treatment Conditions
The samples undergoing pyrolysis are made up of a mass of ligno-
celluloses weighing approximately 0.2 g. The sample is moulded in a press at 140 kg/cm2 to produce a disk about 12 mm in diameter and approximately 1 mm thick. The disk is placed in an image furnace powered by a Xenon lamp, the rays of which are concentrated by a system of mirrors. The sample is pyrolysed under a quartz bell with nitrogen flushing which removes the gases generated by pyrolysis from the bell. We decided on 4 Kw for the power of the lamp and 0.55 1/min for the nitrogen flow. These conditions give complete pyrolysis of the sample in less than one minute and a volume of gas suitable for the analysis technique can be recovered.
2. ANALYSIS OF PRODUCTS OBTAINED
After pyrolysis, a solid residue is obtained consisting exclusively of carbon, a liquid tar phase deposited on the base, and a gaseous phase which is recovered - for analysis - in a spherical container.
2.1 The Gaseous Phase
The pyrolysis gas (approximately 0.5 L.N/g of material) is analyzed by gas chromatography. H2, CH4 and CO are separated in a molecular sieve column (60/80 mesh; vector gas: argon); CO2, C2H
4' C'j/ftţ, C2H2 are separated in a column of Porapak Ν (80/100; vector gas: helium). The quantitative analysis of the mixture is deduced by comparing the chromatogram obtained with the chromatogram of a standard mixture containing the various constituents of the pyrolysis gas in known proportions.
The radioactivity of the six carbonaceous gases which can be marked is determined by coupling a proportional counter to the Chromatograph in the gaseous phase. After separation on the Chromatograph columns the gases pass in turn into the counter where they are completely oxidized at 600°C on the copper oxide, so that the radioactivity of the resultant carbonic gas can be measured.
2.2 The Solid Phase
The solid residue is weighed and its radioactivity determined by measuring the radioactivity of the carbonic gas produced by complete combustion.
2.3 The Liquid Phase
It is difficult to recover the tars completely because they are dispersed on the plate and in the gas-collecting circuit. To present our results, the radioactivity of the liquid phase was estimated by taking the difference between the initial radioactivity of the sample and the radioactivity of the gaseous and solid phases.
3. RESULTS AND DISCUSSION
Table III gives the distribution of carbon atoms in the different pyrolysis products. It is deduced from the initial carbon content of the lignocelluloses used (46.7%) and from chromatographic analysis for the gaseous phase and weighing the pyrolysis residue for the solid phase. The carbon content of the liquid phases is determined by taking the difference.

643
TABLE III. Distribution of carbon in the different phases resulting from the pyrolysis (as a percentage of the number of moles of initial carbon) and marked carbons in the same phases (as a percentage of the initial radioactivity)■
Products of
Gas phase
Solid phase
Liquid phase
pyrolysis
C%
R%
C%
R%
C%
R%
a
33
6
25
25
42
50
b
36
15
40
40
24
37
LIGNOCELLULOSES
c
32
34
34
34
34
45
d
33
31
44
44
23
21
The gaseous phase contains approximately one third of the carbon atoms of the starting material. Analysis of the radioactivity of the gaseous and solid phases shows significant differences depending on how the lignocelluloses are marked. With lignocelluloses synthesized from uniformly marked phenylalanine (a) , the radioactivity of the whole gaseous phase represents less than 6% of the initial radioactivity, while the solid residue and the tars represent 44% and 50% respectively. This shows the low contribution of the lignin to the formation of pyrolysis gas, as already noted for the lignins extracted (2, 3). In the cases where a single carbon atom is marked (c, d) , the total radioactivity of the gas represents a significant fraction of the initial radioactivity (34% and 41% respectively) : in this case the atoms marked have been for the most part incorporated into the gaseous phase.
The results of a more precise study of the radioactivity of the various constituents of the gaseous phase are given in Table IV. For each gas the expression of the results is based on a comparison of the following ratios :
C %
and
Number of moles of carbon in the gas
Number of moles of carbon in the gaseous phase
Radioactivity of gas i R% =
Total radioactivity in the gaseous phase
The most abundant constituents are carbon monoxide and hydrogen. The latter represents approximately 45% of the volume of the gas obtained but is not mentioned in this study, which is concerned with determining the origin of the carbonaceous gases from the macromolecules marked with 1 4C.

644
TABLE IV. Distribution of carbon (in brackets ) and marked carbon in the various constituents of the gaseous phase for a given test. The average value of the carbon content of the gases produced by the different lignocelluloses is also given in the Table.
Average carbon content Radioactivity of gases
LIGNOCELLULOSES b c
CO
co2 C H4 C2H6 C2 H4 C2 H2
(59)
(21)
(11)
(0.8)
(5.2)
(3)
78.6
8.5
6.6
0
3.4
2
(61)
(17)
(12)
(1)
(6)
(3)
76
10.8
6.3
0
4
2.7
(57)
(22)
(11)
(0.5)
(5)
(4)
41
5.7
43.5
3.7
3.3
2.8
(59)
(21)
(12)
(0.5)
(5)
(4)
67
22.2
6.3
0
4.5
0
(59)
(24)
(9)
(1)
(5)
(2)
The marked atoms are present in all the gases (the small quantities of CjHg detected preclude a significant measurement). This fact is observed even in the two cases where only one type of atom is marked (c and d) , showing that flash pyrolysis causes ruptures of links which cause the formation and subsequent recombination of marked atomic entities. In all these cases, the radioactivity measured for a gas is not proportional to its carbon content (see in particular CO and CH^ for the lignocelluloses a,b,d and CO and CO2 for lignocelluloses a,b,c). These results show that under the conditions of flash pyrolysis lignin degrades mainly into CO and to a lesser degree into CH4 and CO2, and hydrocarbons into C2.
In cases where the lignin is marked on the extracyclic methoxyl group (c), the radioactivity of CH4 is equivalent to that of CO. The result shows that the OCH 3 group forms CH. and CO in equal quantities. It confirms the theory that the origin of the methane produced by the degradation of the lignins is the methoxyl group (4).
With the C lignin lignocelluloses, of which the precursor is cinnamic acid marked on the carbon of the lateral chain, the high radioactivity of the CO2 suggests a large proportion of the x; in the formation of the CO_.
CONCLUSIONS 14. The marking with C means that we can study specific sites in the
vegetable macromolecules. Marked lignocelluloses, extracted in almost their natural state, are very representative of the wood used for gasification in industrial processes.
The thermal processing, carried out in a concentrated radiation furnace, involves the application of an anisotropic thermal flow to a small sample in the form of a disk of solid material studied in isolation. The general thermal conditions reasonably approximate those in the industrial gasification process, even though the experiment is conducted on a much smaller scale.

645
We have shown the predominant role of lignin in the formation of tars. The contribution of this constituent of wood to the formation of pyrolysis gas has also been partly elucidated: the lignin forms mainly carbon monoxide and methane, most of the latter coming from the extra-cyclic methoxy groups (5).
All the results constitute a theoretical basis for the monitoring and direction of gasification reactions with a view to generating clean gas free of tar.
ACKNOWLEDGEMENT
This work was carried out with financial support from the Agence Française pour la Maîtrise de l'Energie and the Conseil Regional de Midi-Pyrenees.
REFERENCES
(1) G. ALIBERT and A.M. BOUDET, Physiol. Veg., 1979, 17, 67. (2) S. CAUBET, P. CORTE, C. FAHIM and J.P. TRAVERSE, Solar Energy, 1982,
29(6), 565. (3) J. DOAT and X. DEGLISE, Bois et Forets des Tropiques, 1982, 198, 59. (4) E. AVNI, R.W. COUGHLIN, P.R. SOLOMON and HSIANG HUI KING, Fuel, 1985,
64, 1495. (5) S. CASTILLO, S. CENNINI, G. GAS and J.P. TRAVERSE, Fuel, 1989, 68,
174.

646
REDUCTION ZONE HEIGHT DETERMINATION IN SOLID WASTE GASIFICATION PROCESS IN A SHAFT FURNACE
J. WANDRASZ and K. WALECZEK The Chair of Thermal Apparatus and Waste Disposal
Mechanical and Power Engineering Faculty The Silesian Technical University, Poland
Summary
The paper deals with computing of the reduction zone height in a shaft furnace used for gasification of waste wood by means of experimental research on the kinetics of reduction reactions (charcoal reacting with CO2 and H2O) . The problem has been narrowed to the analysis of the process carried on the average temperature where ash is drained from the combustion zone in a solid state. The research results show that in a temperature range of 700-1100 °C the reactions go in a kinetic-diffusive area and the charcoal particle diameter has no effect on the rate constant. Coefficients in Arrhenius equation are evaluated. Two alternative mathematical models of the reduction zone have been worked out: one for air-flue gas blast and another one for air-steam blast. It has been confirmed that at the analysed temperatures in the combustion zone reduction reactions cease before a state of equilibrium is reached in the system.
1. INTRODUCTION
Gasification processes can be divided according to the temperature in the combustion zone into mean temperature processes (at temperatures 1000-1200°C) and high temperature ones (1400-1650°C). In the first case the solid residue is ashes, in the second one granulated product of melted slag. In practice all large units for waste gasification work in the high temperature range. Mean temperature processes are applied in the smaller units in an industrial plant where constructional simplicity, readiness to start and stop the process is required. Processes with a combustion temperature up to 1100°C do not require a water jacket, mobile rotary grid and water bowl, which facilitates the design of the furnace.
Furnaces with the double receipt of gas, where part of the hot comparatively pure combustible gas is carried away from above the reduction zone, seem to be better. Designing such a system requires parameters of the reduction zone to be defined including its height. In the mean temperature process it can be easily done by determining parameters of reduction reaction kinetics if the reaction rate constant depends on the temperature of the process only. High temperature processes are not discussed in this paper.
2. DESCRIPTION OF REDUCTION ZONE IN A SHAFT FURNACE IN WOOD WASTES GASIFICATION PROCESS
In the reduction zone in gas generators there are five main reactions defining the process :

647
C + CO = 2 CO
C + 2 H20 = C02 + 2H2
C + H20 = CO + H 2
(5)
(6)
(7)
CO + H20 = C0 2 + H2
C + 2H2 = CH4
(8)
(9)
Reaction of methane formation (9) is important mainly in pressure processes. When pressure equals atmospheric pressure the reaction is so slow that it can be omitted in the description of the process. Reactions (5), (6) , (7) and (8) are dependable and two of them are sufficient for the description of the process. Reaction (8) in contact with coal is carried on by means of a solid phase and thus has, like reactions (5), (6), (7), a heterogeneous character. During research on the kinetics of the reduction process in gas generators it is simplified and reactions of coal with CO2 and H2O are of the first order. In the kinetics equation the difference of gas content to equilibrium content has to be taken into consideration. The rate of reaction of a given component refers to the mass of a solid material (coal) assuming that it is proportional to the material surface.
Therefore:
-(1/m) (dnoo /dr) = koo. (Coo - Coo.R) kmolAg s (10)
-(1/m) (dnH20/dr) = kH 0 (CH 0 - CH 0 R) kmolAg s (11)
Reaction rate constant k depends on temperature according to Arrhenius equation :
k = A e-E/(MR)T ( m 3 / k g s (12)
After some tests two reactions describing the whole process (e.g. reactions (5), (6) or (7), (8)) should be chosen. The combustion zone influences the reduction zone. To lower its temperature to 1000-1200°C, steam or flue gases should be added to air blast. Apart from carbon dioxide and steam, the gases leaving the combustion zone consist also of some amounts of carbon monoxide and hydrogen oxide which are difficult to define. The content and temperature of the gas leaving the combustion zone give some details necessary for reduction zone analysis.
3. SOME RESEARCH ON THE REDUCTION REACTION KINETICS
The research has been carried out mainly on charcoal being a gasification product of oak wood which belongs to a group of waste materials with high volatile content. A flow power reactor has been used, where CO2, H2O, mixture CO2-N2, H2O-N2, C02-H20-N2 were passed through a sample of charcoal, within the temperature range of 700-1100°C. After having analysed the gas content downstream before and after the sample, constant reaction speeds have been defined as koo2 and kH20 in different temperatures, and then the value of coefficients A and E have been computed in equation (12).
It seems to be interesting that experiments carried on for different charcoal granulation proved there is no influence of particles granulation on values kcc>2 and kH2Û. Introductory measurements proved that the flow rate as well as content of entering gas influence the value of ko02 and kH2Û. Therefore the research has been carried on when flow rate and content of entering gas are similar to those occurring in industrial gas generators. An analysis of the results showed that CO as well as CO, are formed as a result of coal reaction. For a model description of a simultaneous reaction of

648
2,5 H,
<,S HOP __
\AU¿.
"LHK_
1000 10 BO »00 t,, 'C 1100
^~—~~ M J £ l _
Hop
—M>t_
100
MO kmol
SO
W
60
50
2.Ï H, m
1,5
i
0,5 Hg_—
Hx^
Mhfa -
0,003 0,004 0.003 ή«, <""°' 0,007 ' m
1 a 2,5 H,
0,5
' o.õõl õÕõí ο,οοί n t , fr¡"g' õ[Õoy
ifOO MV«
H »
6» 100 f , JUL. HO ao fOÕ S, ±í_ m»
MWH,
MJ .moi
80
10
SO
,50
2,5
H, m
<ι5
0
\
. Hgp
MA/rf
Λ . . * · <*
Fig. 1. Dependence of reduction zone height Hop and H90 and calorific value of the gained gas MWd on temperature in the combustion
zone ts, blast stream ns, solid phase density ρ and heat losses Qs (for Ot = °°)
a : airflue gas blast, b airsteam blast

649
charcoal with H2O and CO2, reaction systems (5) , (6) are the most applicable. The following results have been gained:
Reaction A, m3 A g s, E, J/kmol
C + CO = 2 CO 1007600 184650000
C + C0 2 = 2 CO 25230655 212650000
C + 2 H O = CO + 2H 2946990 188278000
The values of constant reaction speed koo obtained in the research seem to be lower in the case of charcoal reaction with CO2 than in simultaneous reaction of charcoal and CO and H O . It is caused by reaction (7) omitted in the model description.
4. MATHEMATICAL MODEL OF REDUCTION ZONE AND RESULTS OBTAINED
A mathematical model of the reduction zone for gasification of charcoal wastes has been designed according to the method of elementary energy balances dividing reduction zone into differential elements. This model has been designed in two versions :
for airflue gas blast, assuming that there is no H O in the blast, for airsteam blast. Computation of reduction reaction kinetics has been done using the
experimentally obtained values given above. The most problematic is the description of heat exchange between the gas and solid phases in the reduction zone. Therefore, computations have been repeated several times with variable volumetric coefficient of heat penetration a, W/m
3 Κ in the range 0100000. The influence of such parameters as temperature in the combustion zone, blast stream, density of solid phase and heat losses up to the height of the reduction zone, calorific value and temperature of the gas obtained have been tested. It has been established that at analysed temperatures in the combustion zone below 1200"C, reactions in the reduction zone cease long before a state of equilibrium is reached in the system. In higher parts of the reduction zone temperature is lowered mainly because of heat losses. It has been established that there is an optimal height of the reduction zone, Hop, above which total physical and chemical enthalpy of the gas is lowered. This height is in practice the top limit border of the reduction zone. To define the phenomena better the height of 90% CO2 conversion (or CO2 and H20 in the airsteam blast) has been calculated. In Figure 1 this height has been denoted as H90.
The following proportions of the components in the gases leaving the combustion zone have been assumed: CO2/CO = 1, H2O/H2 = 2. It has been shown that any change of the assumed values influences the calorific values of the reduction gas but not the height of the reduction zone. A similar conclusion concerns changeability of volume heat penetration coefficient Ct between the solid and gas phases (Figure 2). It has been computed that calorific value of the reduction gas MWd depends mainly on temperature in the combustion zone ts, less on apparent density of charcoal ρ and heat losses Qs. There is no dependence on blast stream viz ñs. Optimal height of the reduction zone Hop and height Hso depend on heat losses Qs and blast stream viz ns. The influence of other parameters is negligible (Figure 1).
Definitely higher calorific value of reduction gas has been obtained in airsteam blast than in airflue gas blast.

650
Figure 3 shows an example of temperature distribution and concen
tration of the gas components in the reduction zone for airflue gas blast.
iOODOO
Fig. 2. Dependence of reduction zone height Hop and calorific value of the gained gas MWd on volume heat penetration coefficient α in
reduction zone in airflue gas blast
0,3 f J 0,25
0,2
0,1S
0,1
D,0S
Hjo
^t
[Ό]
fc j
]«&__
H»
1Z0O
t, «c 1100
1000
900
BOO
roo
Ο,Β 1,5 H, m 2 600
Fig. 3. Distribution of gas components concentration and temperature of solid phase in the reduction zone in airflue gas blast
(a = 20000 W/m3K, ρ = 100 kg/m3
, ts = 1100°C, CO2 content in the blast 7.8%), R state of equilibrium

651
COMPUTER MODELLING OF FLUIDISED BED GASIFICATION
J M DOUBLE, E L SMITH and A V BRIDGWATER Energy Research Group
Department of Chemical Engineering and Applied Chemistry Aston University, Birmingham, B4 7ET, England
Summary The objective of this study has been to provide a greater understanding of the biomass gasification process through the development and use of computer models. A new theoretical equilibrium model of gasification is described, based on the operating condition called the adiabatic carbon boundary. This represents an ideal gasifier working at maximum efficiency -the point where the carbon in the feedstock is completely gasified. It is used as a "target" against which to compare the results of real gasifiers. A second model has been developed which uses a stagewise approach in order to model fluid bed gasification, and the results have indicated that pyrolysis and the reactions of pyrolysis products play an important part in fluid bed gasification. Results from both models are presented.
1. INTRODUCTION The work described in this paper was undertaken in order to obtain a better
understanding of the chemical processes which occur in a biomass gasifier. The first stage of the work was to construct an idealised model of gasification against which the performance of real gasifiers can be measured. Some results of this work have been presented elsewhere [1]. The second stage was to construct a more sophisticated model of fluid bed gasification which uses a stagewise approach. Both models described here are relatively simple, and both are designed to predict the steady-state performance of a gasifier in terms of the compositions and flowrates of its input and output streams. A representation of this is shown in Figure 1. The models are not kinetic models, and they cannot be used to size gasifiers or predict the spatial variation of reaction conditions within the reactor.
Biomass
Gasifying agent
Temperature Flo wr ate Composition: C. H, O, Ash, H20
Temperature Flo wr ate Composition: 02 , N2, H20
Gasifier
Temperature | Pressure |
Temperature \ Flowrate Composition: H2, CO, C02, CH4.C2S, N2. H20.
/ Temperature \ Flowrate Composition: Ash, Char .
Product gas
Solid residues
| Heat losses |
Figure 1 Variables in the Models of Gasification

652
2. EQUILIBRIUM MODELLING The basis of any model of gasification is the material and energy balances
over the gasifier, but these balances are not sufficient on their own to calculate the values of the unknown variables. The relationships of chemical equilibrium have frequently been used as well [eg 2,3]. In addition to the mass and energy balances and equilibrium relationships, other assumptions are needed, and the number of assumptions used determines the number of degrees of freedom in the system. Some of the variables in the system will be determined by circumstances, for example the feedstock composition, flowrate and temperature. Others will be chosen by the designer, typically the gasifying agent temperature and composition and the pressure of the gasifier. If it is assumed that the reactor is well mixed, as might be the case for a fluid bed gasifier, the outlet temperatures will be equal to the reaction temperature. This leaves only one additional variable to be specified, which can be either the gasifying agent flowrate or the reaction temperature.
There are two cases of chemical equilibrium, the first when there is excess carbon present in the system and char appears in the solid residue, and the second where all the carbon is gasified in an excess of gasifying agent. There is a third special case at the point between these two cases where the carbon is completely gasified without an excess of gasifying agent. This point is known as the carbon boundary. Previous work has enabled models of gasification to be constructed for the cases either side of the carbon boundary, but in the current work it was decided to concentrate on the carbon boundary itself, as explained below. In the case of a carbon boundary equilibrium model with the assumptions and design variables as described above both the temperature and gasifying agent flowrate are fixed by the material and energy balances. Thus, for a given feedstock, gasifying agent composition and operating pressure the carbon boundary condition is achieved at a unique set of operating conditions.
In an ideal gasifier, the carbon boundary condition will give the maximum chemical energy in the product gas. In the case where there is excess carbon in the system, the char residue will contain chemical energy, allowing less chemical energy in the gas phase. However, excess gasifying agent would reduce the amount of chemical energy in the system by boosting the temperature of the system and converting energy to sensible heat of the product gases. Thus, the equilibrium carbon boundary model represents an ideal gasifier working at optimum operating conditions for a particular feedstock and gasifying agent, and can be used as a theoretical optimum against which the performance of real gasifiers can be compared.
The results of a sensitivity analysis performed using the carbon boundary model have already been published [1]. Figure 2 is an example of the output obtained: it shows the effect of biomass moisture content on performance for an oxygen gasifier. The sensitivity analysis predicted that the parameters which have the most effect on gasification were biomass moisture content and gasifying agent composition.
The reaction temperatures predicted by the model are lower than are achieved in real gasifiers. The equilibrium model takes no account of reaction rates and in real gasifiers higher temperatures must be used in order to achieve reasonable gasification rates. If an equilibrium model is to be used for predicting the performance of a real gasifier rather than providing an idealised target, a fixed temperature model should be used operating with an excess of gasifying agent over the carbon boundary case, with the equilibrium temperature being based on actual values.

653
o E
o α. E 8 to ro O
Figure 2
Γ1073
Key » H2 CO * C02 -*- CH4 M H20 ·»■ Temperature
20 40 60 80 Biomass moisture, % dry basis
Typical Results of the Carbon Boundary Equilibrium Model
Whilst equilibrium models are useful in presenting an idealised picture of gasification, they do not always accurately predict the results from real gasifiers. A comparison between the carbon boundary equilibrium model and real gasifier results has already been published [1]. One reason for the differences between real and ideal gasifiers is that some of the volatile and gaseous products of pyrolysis may pass through the reactor without attaining equilibrium. A more accurate model may be produced by adopting a stagewise approach with separate submodels of pyrolysis and gasification.
3. STAGEWISE MODELLING OF FLUID BED GASIFICATION A stagewise model is one in which a process is subdivided into steps or
stages. In the case of gasification, the steps are drying, pyrolysis, tar cracking, and char gasification. In a fixed bed gasifier, the stages would take place in different parts of the bed, but in a well mixed fluid bed reactor each reaction stage would take place throughout the bed. A stagewise model of fixed bed gasification can include both material and energy balances for each stage, whereas in a fluid bed only an overall energy balance can be used in conjunction with the material balances for each stage. A generalised stagewise model of a fluid bed reactor is shown as Figure 3.
It is easy to model the char gasification step using equilibrium methods, but pyrolysis is a very complex process which is sensitive to reaction conditions and cannot yet be modelled theoretically. An empirical model of pyrolysis must therefore be used, based on data obtained under the same reaction conditions as occur in a fluid bed gasifier (although an inert atmosphere must be used to avoid gasification). Such pyrolysis data is rare. There are also little or no quantitative data available on tar cracking within fluid bed gasifiers.
For the model described here it was decided to reduce the complexity of the stagewise model by considering only two stages: a first stage incorporating the drying, pyrolysis and tar cracking, and a second stage of gasification, modelled using an equilibrium method assuming an excess of gasifying agent. Volatile and gaseous material from the first stage might either pass directly to the outlet stream or might be involved in the gasification stage. A number of possibilities were tested, and the results which gave the closest fit to real data were obtained

654
when the hydrocarbons and tars from pyrolysis passed to the product gas, whilst all of the other pyrolysis products passed to the char gasification stage. This is a model of the case where there is good mixing within the reactor, but reaction rates prevent the decomposition of the hydrocarbon products of pyrolysis. A conceptual diagram of this model is presented in Figure 4.
Product gas
Biomass £{ Drying π/Pyrolysisp
VGasifier';
Figure 3 A Generalised Stagewlse Model of a Fluid Bed Gasifier
= ^ _ Product gas >
Drying, pyrolysis and tar cracking
Gasifier
Figure 4 The Simplified Stagewlse Model of Fluid Bed Gasification
The model has only been tested over a limited range of reaction conditions, because of the lack of pyrolysis data. The pyrolysis data used are from Belleville and Capart [4], and were obtained under reaction conditions similar to those found in a fluid bed gasifier.
Table I shows a comparison with published results from two fluid bed gasifiers. The results were obtained using the same operating conditions in the model as were reported with the actual gasifier results. No experimental parameters have been used to fit the results of the model to gasifier results. There is good agreement with the results of the Lurgi Circulating Fluid Bed gasifier, but poor agreement with the results of the Mino gasifier. This is probably because the Lurgi CFB has good mixing within the reactor, as assumed in the the computer model, whereas in the Mino gasifier there may be poor mixing. There is also the possibility that the pyrolysis data used in the model are not applicable to the feedstock used in the Mino gasifier.

655
Table I Comparison of Model and Real Gasifier Results
Model Lurgi CFB Mino
Gas yield Nm
3/kg feed
1.37 1.36
Dry gas composition, H2 CO C 0 2 CH 4 33.0 35.0 24.7 6.0 33.5 33.6 26.7 4.9 18.7 35.5 30.3 10.0
mol % HCs 1.3 1.3 1.8
Ref.
[5] [6] [7]
Note : Compositions adjusted to a nitrogen-free basis. Gas yield is expressed as Nm3 dry gas per
kg dry, ash free feedstock. Feedstock is wood, gasifying agent is oxygen/steam and reaction temperature is 780°C.
The stagewise model does not provide the information required for sizing a gasification reactor. Models for calculating the char gasification rate are well established [eg 8]: this is the rate determining step in gasification [9].
4. CONCLUSIONS The equilibrium carbon boundary model of gasification represents an
idealised gasifier and may be used as a target against which to compare the results of practical gasifiers. Equilibrium models do not predict accurately the results of fluid bed gasifiers because of the effects of pyrolysis, but stagewise models which use equilibrium for char gasification calculations and empirical pyrolysis models can predict the results of well-mixed fluid bed gasifiers.
5. ACKNOWLEDGEMENT The authors are grateful to the Science and Engineering Research Council
for their support of J M Double.
REFERENCES 1 Double J M, Bridgwater A V, "Sensitivity of Theoretical Gasifier Performance to System
Parameters", in Palz W, Coombs J, Hall DO (eds), "Energy from Biomass: 3rd EC Conference", Elsevier Applied Science, 1985.
2 Gumz W, "Gas Producers and Blast Furnaces: Theory and Methods of Calculation", J Wiley and Sons Inc, New York, 1950.
3 Desrosiers R, Thermodynamics of Gas-Char Reactions", in Reed Τ Β (ed), "A Survey of Biomass Gasification", Solar Energy Research Institute, Colorado, USA, 1979.
4 Belleville P, Capart R, "A Model for Predicting Outlet Gas Concentrations from a Wood Gasifier", in Bridgwater A V (ed), Thermochemical Processing of Biomass", Butterworths, London, 1984.
5 Double J M, "The Design, Evaluation and Costing of Biomass Gasifiers", PhD thesis, Aston University, Birmingham, England, 1988.
6 Mehrling P, Reimert R, "Synthetic Fuel from Wood via Gasification in the Circulating Fluid Bed", in Beenackers AACM (ed), "Advanced Gasification, Methanol Production from Wood - Results of the EEC Pilot Plant Programme", D Reidel Publishing Company, Dordrecht, 1986.
7 Hall D O, Overend R P (eds), "Biomass: Regenerate Energy", Wiley, New York, 1987, p248. 8 Buekens A G, Schoeters J G, "Modelling of Biomass Gasification", in Overend R Ρ et al (eds),
"Fundamentals of Thermochemical Biomass Conversion" Elsevier Applied Science, Barking England, 1985.
9 Smith E L, Shand R N, "Design and Evaluation of Biomass Gasification Systems", in Grassi G et al (eds), "Biomass for Energy and Industry, 4th EC conference", Elsevier Applied Science, 1987.

656
THREE-PHASE WOOD GASIFIER SYSTEM EASIMODR
H. MICHEL-KIM Efeu GmbH
Research and Development for Energy and Environment D-5830 Schwelm
1. THREE-PHASE WOOD GASIFIER WITH LOW POLLUTION VALUES EASIMODK SYSTEM MICHEL-KIM
Many of the attempts to develop wood gasifiers to a stage where they could be put on the market, however, failed due to the fact that most one-phase processes could not prevent the formation of tar and phenol or waste products. The technical advantage of the Michel Kim/EFEU process is based on a systematic breaking down of the various process phases of gasification and an optimization of the individual stages providing for all necessary interventions in the process parameters. Traditional gasification processes were primarily one phase only, whereby the internal gasification process with its complex transitions in the areas of material transport and heat was left largely to coincidence. Tar and phenol formations occurred continually, along with bridge and channel formations as well as slag formation. Users of traditional gasifiers often had to put up with large and unwieldy apparatus, effluent pollution and unpleasant odors, as well as high maintenance costs.
2. ADVANTAGES OF THE EFEU PROCESS The EFEU process of coordinated, multiphase gasification offers the
following advantages: Overcoming of previous technical difficulties by a thorough development of the gasification technology; avoidance of feared tar and phenol wastes; A wide variety of fuels, also with high moisture levels, can be converted into gas in a manner friendly to the environment; Gasification efficiency and load flexibility are considerably better than in previous gasification systems as well as in all known methods of firing.
3. SPECIAL FEATURES OF THE EASIMODR GAS GENERATOR The working principal of the Michel-Kim EASIMODR-gas generating
system has become known during recent years, and, most recently, in a project actually tested in Schwelm with a gas output of 2500 kW. The following drawings, fig. 1 - 3, help to illustrate the characteristics of the latest stage of development. Figure 1) illustrates the dosing silo, the bottom loaded primary reactor with level control, the intermediate gasification with flue charcoal circulation, activated charcoal remover and coke reactor with integrated air heater.

657
The dosing silo has a reliable air buffer delivery system. Fuel is fed continually from the silo into the bottom of the primary reactor, whereby the rate of fuel feeding is controlled automatically depending on the various process parameters. A portion of the fuel is fed into a circumjacent primary chamber and ignited with preheated air. The combustion in the primary chamber stabilizes gasification in the primary reactor and allows the removal of stones at this stage. During the intermediate gasification, gas conversion takes place at approx. 900° C in a rotating Venturi-burner; whereby gas and flue charcoal are circulated. The conversion temperature is regulated with a secondary air supply. Tar and phenols are hereby practically fully converted. Depending on the direction of rotation of the Venturi-burner during the intermediate gasification, flue charcoal (wood charcoal) from the primary reactor can be either refed to the Venturi-burner or extracted via an integrated worm gear. Depending on how long the charcoal remains in the intermediate gasification (a regulable timespan) a high level of activation can be achieved so that the use of wood charcoal as activated charcoal is possible. The coke reactor also prevents tar and phenols from escaping from the primary reactor and stabilizes the heating value of the generator gas at a high level. Coke cosumption lies at around 1 % of the primary fuel quantity. (The coke reactor is equipped with a stepped grate into which the air heater is integrated. The primary air is pre-warmed up to approx. 520° C, the generator gas cooled down to approx. 330° C.)
V¡' ,< ,',','.< ,t ,i ,< ,·
Dosing-silo 1. Dosing-silo 2. Air bag 3. Screw conveyor
Underfed primary-reactor
4. Vertical conveyor 5. Agitator 6. Guide blades 7. Rotating grate 8. Venturi-burner 9. Worm gear
for charcoal extraction
Coke-reactor
10. Air-heater 11. Step grate
Figure 1)

658
Figure 2) illustrates the pre-drying and excess heat utilization. The drier has an outer jacket of gill plate and a rotating cone made from perforated plate which carries a progressive spiral conveyer. The fuel is in a ring channel around the spiral cone. The rotation of the cone is such that the fuel is loosened and carried upwards. Extraction of the fuel takes place at the bottom. The drying air flows radially from the outside to the inside of the drier in the bottom area, then upwards through the interior of the rotating cone and finally in a radial direction from inside to outside through the fuel bed. The drying air is pre-warmed up to over 100° C by using the hot generator gas and hot water in the gas scrubber.
Pre-drying and excess heat utilization
1 Rotating cone 2 Gill plate 3 Progressive spiral conveyor 4 Perforated plate
5 Gas/Air heat exchanger 6 Water/Air heat exchanger 7 Water/Air heat exchanger

659
Figure 3) illustrates the 4-stage gas scrubber (2 jet scrubbers and 2 packing scrubbers) with subsequent cooling tower, gas blower and burn off. During the first three scrubbing stages the level of condensate is regulated and, in contraflow to the gas, enriched with ammonium. The removal of the ammonium-concentrate takes place dependent on the level of concentration in the first scrubbing stage. Condensation of the water contained in the cooled gas takes place principally in the fourth scrubber. The condensate has only traces of ammonium at this stage.
Figure 3)
4-stage Gas scrubber
1 Jet scrubber 2 Jet scrubber 3 Packing scrubber 4 Packing scrubber
5 Cooling tower 6 Gas blower 7 Gas torch

660
4. STATE OF DEVELOPMENT AND MARKET INTRODUCTION The EASIMODK technology is currently being introduced to the market.
The European Licence rights were bought by Schwelm Anlagen + Apparate GmbH, Schwelm, who put the first industrial gas generator into operation with a gas output of about 2500 kW in march 1989. Following the first test weeks, it can be assumed that the initial problems have been solved and the market introduction phase can begin. Schwelm Anlagen + Apparate GmbH will subject the existing 2500 kW gas generator to a long term test and use this test period to make final adjustments before beginning small series production. It is plannend to introduce 3 standard sizes to the market for the generation of 500 kW (el), 1000 kW (el) and 2000 kW (el). A large number of inquiries for the technology have been registered up to date. The first EASIMODR gasgenerator for the generation-size of 1000 kW (el) will be delivered in 1990.

LIST OF PARTICIPANTS


663
ANTONELLI, L. Vice President KINETICS TECHNOLOGY INTERNATIONAL S.P.A. Via Monte Carmelo 5 I - 00166 ROME
BEENACKERS, A.A.CM. Prof. Chemical Engineering GRONINGEN UNIVERSITY Mijenborgh 16 NL - 9747 AG GRONINGEN
ARAUZO, J. Professor UNIVERSITY OF ZARAGOZA Facultad de Ciencias Ciudad Universitaria E - 5009 ZARAGOZA
BELLMANN, U. UNIVERSITÄT HAMBURG Institut für Technische und Makromolekulare Chemie Bundesstraße 45 D - 2000 HAMBURG 13
AZNAR, M.-P. Ass. Prof. Chem. Eng. Dept. Chem. Eng. Facultad de Ciencias UNIVERSITY OF ZARAGOZA E - 50009 ZARAGOZA
BETTENS, B. Chef de Travaux U.L.B. 50, Avenue Fr. Roosevelt B - 1050 BRUXELLES
BABU, S.P. Director, Process Research INSTITUTE OF GAS TECHNOLOGY 3424 South State Street USA - CHICAGO, IL. 60616
BILBAO, R. Professor Dpt. de Ingenieria Quimica Facultad de Ciencias UNIVERSIDAD DE ZARAGOZA E - 50009 ZARAGOZA
BALDELLI, C. Engineer ITABIA Via Archimede, 161 I - 00127 ROMA
BILITEWSKI, B. INTECUS Krefelderstraße 20 B - 1000 BERLIN 21
BARTON, J.R. Civil Servant WARREN SPRING LABORATORY (WSL) Gunners Wood Road Stevenage UK - HERTS SGI 2BX
BLASIAK, W. Assistant Professor THE ROYAL INSTITUTE OF TECHNOLOGY Department of Heat & Furnance Technology S - 10044 STOCKHOLM
BAYER, E. Professor Institut für Organische Chemie UNIVERSITÄT TUBINGEN Auf der Morgenstelle 18 D - 7400 TUBINGEN 1
BONFITTO, E. Technical Officer - Engineer REGIONE ABRUZZO - E.R.S.A. Piazza Torlonia 91 I - 67051 AVEZZANO (AQ)

664
BONINO, G. Engineer BIOMASS ENERGIES 25, Strada della Viola I 10133 TURIN
CAMP, J.P. S.E.M. PAG VAPEUR 1, rue du Four F 38190 CROUES
BONSANG, R. Ingenieur Civil INIEX 200, rue des Chêra Β 4000 LIÈGE
CAPART, R. Enseignant, chercheur UNIVERSITY OF TECHNOLOGY OF COMPIËGNE 10, Square Sutterlin F 60200 COMPIÊGNE
BOUGARD, J. Director BOUGARD ENGINEERING S.A. 33, Chaussée de Mons Β 1400 NIVELLES
CARLSSON, K.B. Project manager Dept. FUM FLÄKT IP S 35187 VÄXJÖ
BRENNDÖRFER, M. Dipl.Ing. KURATORIUM FUR TECHNIK UD BAUWESEN IN DER LANDWIRTSCHAFT e.V. (KTBL) Bartningstraße 49 D 6100 DARMSTADT
CARRE. J. Administrateur ASCAB 27, rue Louis Vicat F 75015 PARIS
BRIDGE, S.A. Research Student ASTON UNIVERSITY Dpt. Chemical Engineering Aston Triangle UK BIRMINGHAM B4 7ET
BRIDGWATER, Α. Reader in Chemical Engineering UNIVERSITY OF ASTON Department of Chemical Engineering Aston Triangle UK BIRMINGHAM B4 7ET
CARVANI, L. Researcher ENIRICERCHE 26, Via Maritano I 20097 S. DONATO MILANESE
(MILANO)
CASTILLO, S. Teaching and Research Laboratoire de Recherche sur l'Energie UNIVERSITÉ PAUL SABATIER F 31062 TOULOUSE Cedex
BRUNETTI, Ν. Project leader in thermochemical biomass conversion ENEA Dpt. Fare CRECASACCIA C.P. 2400 I 00100 ROMA
CHURIN, E. Researcher UNIVERSITE CATHOLIQUE DE LOUVAIN Unité CATA 1, Place Croix du Sud Β 1348 LOUVAINLANEUVE
BUEKENS, A. Professor VRIJE UNIVERSITEIT BRUSSEL Pleinlaan 2 Β 1050 BRUSSELS
CORELLA, J. Prof. Chem. Eng. Dept. Chem. Eng. Facultad de Ciencias UNIVERSITY OF ZARAGOZA E 50009 ZARAGOZA

665
CORTE', P. COMMISSION OF THE EUROPEAN COMMUNITIES Directorate General for Energy 200, rue de la Loi Β 1049 BRUSSELS
DELMON, Β. Professeur UNIVERSITÉ CATHOLIQUE DE LOUVAIN Unité CATA 1, Place Croix du Sud Β 1348 LOUVAINLANEUVE
CRANSVELD, J. TPR (TECHNOLOGIES POUR DES PRODUITS RESIDUAIRES) 190 Β Quai du Halage Β 4131 AWIRS
CYPRES, R. ULB Chimie génerale et Carbochimie CP 165 Β 1050 Bruxelles
DABORN, G.R. Department of Trade & Industry WARREN SPRING LABORATORY, DTI Gunnels Wood Road UK STEVENAGE HERTFORDSHIRE SGI 2BX
DARIF, M. Thesard LABORATOIRE DE CHIMIE ORGANIQUE Faculté des Sciences Ile du Sauley F 57000 METZ
DEARMAN, C R . VECUS ENERGY SYSTEMS Ltd Longmace House Mill Lane Arundel UK WEST SUSSEX BN18 9AH
DE BOER, F. HEINEKEN BREWERIES P.O. Box 510 NL 2380 BD ZOETERWOUDE
DEHAYE, J. AQUITAINERGIE 14, rue FrançoisdeSourdis F 33077 BORDEAUX Cedex
DEJAK, C. ENEA Viale Regina Margherita 125 I 00198 ROMA
DELRE, G. Professor UNIVERSITY OF L'AQUILA Dpt. Chimica Ing. Chim. e Materiali I 67040 MONTELUCO DI ROIO
(L'AQUILA)
DENIS, E. Research & Development Engineer CHI (Cockerill Mechanical Industries Seraing) et C.R.l.G. (Centre de Recherches de l'Institut Gramme) 25, rue de la Sablonnière Β 4900 ANGLEUR
DERBYSHIRE, F. Research and Development Director SUTCLIFFE SPEAKMAN CARBONS LTD Guest Street Leigh UK LANCASHIRE WN7 2HE
DESMAZIERES, Β. Research Engineer UNIVERSITY PARIS VI Laboratoire de Chimie Organique Structurale Bâtiment F 4 Place Jussien F 75005 PARIS
DEVAL, M. Administrateur de Société S.H. DEIRA 21, Square Larouse Β 1060 BRUXELLES
DE WILDE, J.P. Research Assistant DELFT UNIVERSITY OF TECHNOLOGY Faculty of Aerospace Engineering P.O. Box 5058 NL 2600 GB DELFT

666
DHEE, Α. Gérant CADET INTERNATIONAL 43, rue Paton F 59800 LILLE
EBBAGGI, C. TECHNICONSULT 595/C, Via Cassia I 00189 ROMA
DIEBOLD, J. Engineer SOLAR ENERGY RESEARCH INSTITUTE Cole Blud USA Co 80401 GOLDEN
EKSTRÖM, G.G.V. Process Engineer STUDSVIK ENERGY (A division of STUDSVIK AB) Dpt. Applied Technology Studsvik Energy S 61182 NYKÖPING
DIRVEN, P. Energy Program Manager SCK (BELGIAN NUCLEAR RESEARCH CENTRE) Boeretang 200 B 2400 MOL
DOEHRER, K. Forstdirektor VERSUCHS UND LEHRBETRIEB FORSTTECHNIK/ HOLZHOF WALDECK/ HESS. FORSTAMT Postfach 1108 D 3549 DIEMELSTADT
ELAMIN, A. Etudiant UNIVERSITY OF COMPIËGNE Appt. 239 3, Square Blaise Pascal F 60200 COMPIËGNE
ENGELBRECHT, A.D. Research Engineer DIVISION ENERGY TECHNOLOGY, CSIR P.O. Box 217 SOUTH AFRICA PRETORIA 0001
DOS SANTOS, A.M. Associate Professor Dept. of Heat and Furnace Technology THE ROYAL INSTITUTE OF TECHNOLOGY S 100 44 STOCKHOLM
ESNOUF, C. Research Engineer C.E.M.A.G.R.E.F. Bte 121 F 92164 ANTONY Cedex
DOUBLE, J. Chemical Engineer UNIVERSITY OF ASTON Department of Chemical Engineering Aston Triangle UK BIRMINGHAM B4 7ET
FABRY, R. Expert COMMISSION OF THE EUROPEAN COMMUNITIES Directorate General for Energy 200, rue de la Loi B 1049 BRUSSELS
DUPRÉ, F. Geschäftsführer C.DUPRÉ GmbH & Co KG FranzKirrmeierStraße 17 D 6720 SPEYER
FARGIONE, P. Engineer UNIVERSITY OF ROME 244, Corso Vittorio Emanuele I 00186 ROME
II
EARP, D.M. Research Assistant Dpt. of Chemical Engineering & Applied Chemistry ASTON UNIVERSITY Aston Triangle UK BIRMINGHAM B4 7ET
FENZL, W. Projektmanager VOESTALPINE INDUSTRIEANLAGENBAU GES.m.b.H. Turmstraße 44 A 4031 LINZ

667
FERRERÒ, G.L. COMMISSION OF THE EUROPEAN COMMUNITIES Directorate General for Energy 200, rue de la Loi Β 1049 BRUSSELS
FONT, R. Professor of Chemical Engineering UNIVERSITY OF ALICANTE R. Font. División de Ingeniería Química Facultad de Ciencias Apartado 99 E ALICANTE
FONZI, F. Gen. Manager ITALENERGIE SPA 39, Via Repubblica I 67039 SULMONA (AQ)
FROMENT, L. R&D Chemical Engineer Dept. Environment & Chemical Procese Studies S.C.Κ. / C.E.N. 200 Boeretang Β 2400 MOL
GALLASTEGUI, S. BEAZ S.A. Alameda Recalde, 18 E 48009 BILBAO
GAMBINO, G. Membro Commissione Amministratrice FIORENTINAMBIENTE AZIENDA SERVIZI NETTEZZA URBANA 52, Via Baccio da Montelupo I 50142 FIRENZE
FRANCAVILLA, G. AERIMPIANTI S.P.A. 21, Via Bergamo I 20135 MILANO
FRANCK, J. UNIVERSITÄT HAMBURG Institut für Technische und Makromolekulare Chemie Bundesstraße 45 D 2000 HAMBURG 13
FRANCOIS, 0. Ingénieur UNIVERSITÉ DE TECHNOLOGIE DE COMPIËGNE BP 649 F 60206 COMPIEGNE Cedex
FRISCHKORN, C. Chemist KFA JÜLICH GmbH Kernforschungsanlage E D 5170 JÜLICH
GARCIA, P. Investigator Dpt. de Ingenieria Química Facultad de Ciencias UNIVERSIDAD DE ZARAGOZA E 50009 ZARAGOZA
GAUTIER, E. Technical Director S.E.S. SOFT ENERGY SYSTEMS S.P.A. 64, Via Marino Ghetaldi I 00143 ROMA
GAVR0, S. Bachelor of Mechanical Engineering ENERGOINVESTWO Institute for Thermal Technique and Nuclear Technique Tvornicka br. 3 YU 71000 SARAJEVO
GIL, I. BEAZ S.A. Alameda Recalde, 18 E 48009 BILBAO
FRITZSCHE, A. Diplom Ingenieur FLEUS UNIVERSITÄT BREMEN Kutsteinerstraße NWl D 2800 BREMEN 33
GIRARD, P. Ingénieur de Recherche CENTRE TECHNIQUE FORESTIER TROPICAL 45 bis, Avenue de la Belle Gabrielle F 94736 N0GENY/MARNE

668
GRABIK, V. Deputy Director UNITED NATIONS INSDUSTRIAL DEVELOPMENT ORGANIZATION (UNIDON) Loewenstraße 1 CH 8001 ZURICH
HAYES, R.D. Chief Bioenergy R&D ENERGY MINES & RESOURCES CANADA 460 O'Connor Street CANADA OTTAWA K15 5H3
GRASSI, G. COMMISSION OF THE EUROPEAN COMMUNITIES Directorate General for Science, Research and Development 200, rue de la Loi Β 1049 BRUSSELS
HECKA, C. VEBA OEL AG Alexandervon HumboldtStraße Postfach 20 10 45 D 4650 GELSENKIRCHEN
GRGUREVIC, S. M. A. Econ. Research Associate INSTITUTE FOR INDUSTRIAL ECONOMICS Marsala Tita 16/11 P.O. Box 317 YU 11000 BEOGRAD
HEERAH, M. Ph.D. Student CEMAGREF Parc de Tourvoie BP 121 F 92164 ANTONY Cedex
GRIMM, H.P. W.I.P. MÜNCHEN Sylvensteinstraße 2 D 8000 MÜNCHEN 90
GUIGON, P. Professeur UNIVERSITE DE COMPIEGNE U.T.C. Dpt. de Genie Chimique BP 649 F 60206 COMPIEGNE
GUIOL, R. Technical Managerr BIOALTERNATIVE S.A. CH 2063 ENGOLLONNEUCHÂTEL
GULYURTLU, I. Principal Research Engineer LABORATORIO NACIONAL DE ENGENHARIA E TECNOLOGIA INDUSTRIAL Departamento de Energias Convenctonaie Edificio J. Azinhaga Dos Lameiros Ρ 1699 LISBOA Codex
HEESINK, A.B.M. Dpt. of Combustion Technology TNO Division of Technology for Society (MTTNO) P.O. Box 342 NL 7300 AH APELDOORN
HENIUS, U.M. Engineer THE TECNOLOGICAL INSTITUTE Gregersenevej DK 2630 TÅSTRUP
HERBOLD, 0. HERBOLDENGINEERING GmbH SUdetenstraûe 2 D 6920 SINSHEIM
HINGER, K.JIngenieur WERNER & PFLEIDERER GmbH Theodorstraße 10 D 7000 STUTTGART 30
HAANAPPEL, V.A.C. Research Engineer BIG BIOMASS TECHNOLOGY GROUP B.V. P.O. Box 217 NL 7500 AE ENSCHEDE
HOCHHEIM , F.W. General Manager BIOMATERIAL INTERNATIONAL TECHNOLOGY S.A. Grossheide 323 D 4050 MOENCHENGLADBACH 1

669
HUMMELSHOEJ, R.M. Project Engineer COWIconsulC, Consulting Engineers and Planners A.S. 45, Teknikerbyen DK - 2830 VIRUM
KITTELMANN, J.-F. Engineer VØLUND FORSKNINGSCENTER Centervej 2 DK - 6000 KOLDING
HUMMELSIEP, H. Abteilungsleiter SAARBERG-FERNWÄRME GmbH Sulzbachstraße 26 D - 6600 SAARBRÜCKEN
KNIGHT, J.A. Consultant KNIGHT CONSULTANTS, Inc. 2117 Kodiak Dr. NE USA - ATLANTA GA 30345
IACOBONI, S. Head Department FACHHOCHSCHULE PRESENIUS Department of Trace Analysis Dambachtal 20 D - 6200 WIESBADEN
KOULLAS, D. Chemical Engineer, Researcher NTU A NATIONAL TECHNICAL UNIVERSITY OF ATHENS Department of Chemical Engineeerlng, Div. IV 42, Patision street GR - 10682 ATHENS
JACCARD, L. Administrateur ENS0F0R S.A. CH - 6985 CURIO
JACKSON, D. Consultant to DG XII El 11 Orchard Crescent STEVENAGE UK - HERTS SGI 3EN
JØRGENSEN, L. Engineer DANSK TERMO INDUSTRI A/S Induetrivaenget 27 DK - 3400 HILLERØD
KAMBALE, K. Ingenieur Forestier Centre d'adaptation des techniques de l'energie bois - "CATEB" MINISTÈRE DE L'ENVIRONNEMENT BP 11596 ZAIRE - KIN I
KAMINSKY, W. Prof. Dr. Institut fUr Technische u. Makromolekulare Chemie UNIVERSITÄT HAMBURG Bundesstraße 45, 2 D - HAMBURG 13
KROMSBEIN, G. Process Engineer UHDE GmbH Friedrich-Uhde-Straße 15P.O. Box 105062 D - 4600 DORTMUND 1
KRISTENSEN, 0. Engineer VØLUND FORSKNINGSCENTER Centervej 2 DK - 6000 KOLDING
KURKELA, E. Research Scientist TECHNICAL RESEARCH CENTRE OF FINLAND Biologinkuja 5 SF - 02150 ESPOO
KUTUBUDDIN, M. Chemist Institut flir Organische Chemie UNIVERSITÄT TUBINGEN Auf der Morgenstelle 18 D - 7400 TUBINGEN 1
LACROSSE, L. Gasification Coordinator CENTER FOR AGRONOMICAL RESEARCHES 23, Avenue Maréchal Juin B - 5800 GEMBLOUX

670
LAPPAS, Α. Graduate Studene CHEMICAL PROCESS ENGINEERING RESEARCH INSTITUTE PO Box 19517 GR 54006 UNIVERSITY CITY
THESSALONIKI
MARTINMARTINEZ, J.M. Ph.D. Chemistry INESCOP Instituto Espafiol del Calzado Polig. Ind. campo Alto PO Box 253 E 03600 ELDA (Alicante)
LAURIDSEN, T.L. Manager Dpt. R & D AALBORG BOILERS A/S P.O. Box 209 DK 9100 AALBORG
LUENGO, C A . Physicist UNIVERSITY OF CAMPINAS Applied Physics Dept. CXP 6165 BRASIL 13081 CAMPINAS
MARTINEZSANCHEZ, M.A. Ph.D. Chemistry INESCOP Instituto Espafiol del Calzado Polig. Ind. campo Alto PO Box 253 E 03600 ELDA (Alicante)
MARURI, J. BABCOCK WILCOX ESPAÑOLA S.A. 6, Centre International Rogier Bte 442 Β 1210 BRUXELLES
LYNGE JAKOBSEN, J.H. M.Sc. CARL BRO AS Granskoven 8 DK 2600 GLOSTRUP
MALY, V. Engineer KFA PBE Postfach 1913 D 5170 JÜLICH
MANIATIS, Κ. Expert to the Commission of European Communities V.U.B.; CHIS, Pleinlaan 2 Β 1050 BRUSSELS
MARITANO, M. Manager CASTAGNETII S.P.A. (FIAT Group) Via Acqui 86 I 10090 CASCINE VICARIVOLI
(Torino)
MARSCHEIDER, F. Dipi. Chem. UNIVERSITÄT HAMBURG Institut für Technische und Makromolekulare Chemie Bundesstraße 45 D 2000 HAMBURG 13
MASS0N, H. R & D Manager N.V. SEGHERS ENGINEERING Molenweg 107 Β 2660 WILLEBROEK
MAUND, J.K. Lecturer Dpt. of Chemical Engineering & Applied Chemistry ASTON UNIVERSITY Aston Triangle UK BIRMINGHAM B4 7ET
MEIER, D. Research Scientist INSTITUTE FOR WOOD CHEMISTRY Leuschnerstraße 91 D 2050 HAMBURG 80
MELAN, C. Directeur adjoint PAUL WURTH S.A. Β.P. 2233 L 1022 LUXEMBOURG
MENDIS, M.S. Economist Engineer THE WORLD BANK 1818 Η Street NW USA WASHINGTON DC 20433

671
MEZERETTE, C. Ingénieur Chimiste CT.F.T. / C.I.R.A.D. 45 bis, Avenue de la belle Gabrielle F 94736 NOGENTSURMARNE
NOVAK, M. Manager ENERGAS GottliebDunkelStraße D 1000 BERLIN 42
21
MICHELKIM, H. GeschäfsfUhrer EFEU Loher Straße 1 D 5830 SCHWELM
MININNI, G. Senior Scientist CNR Instituto di Ricerca Sulle Acque 1, Via Reno I 00198 ROMA
MOLINA, L. Engineer R&D AERIMPIANTI S.P.A. 21, Via Bergamo I 20135 MILANO
0ASMAA, A. Research Scientist TECHNICAL RESEARCH CENTRE OF FINLAND Laboratory of Fuel Processing Technology Biologinkuja 35 SF 02150 ESP00
OLSEN, G. Chemical Engineer LABORATORY OF ENERGETICS, TECHNICAL UNIVERSITY OF DENMARK DTH Building 403 DK 2800 LYNGBY
0'NEIL, D.J. Research Director GEORGIA TECH RESEARCH INSTITUTE Georgia Institute of Technology USA ATLANTA GA 30332
MÖLLER MADSEN, S. M.Sc. Engineering I KRUGER A/S Gladsaxevej 363 DK 2860 SOEBORG
MUNCK, J. Chemical Engineer I. KRUGER AS Gladsaxevej 363 DK 2860 SØBORG
NELS, C. Beamter UMWELTBUNDESAMT Postfach D 1000 BERLIN 33
NICOLAY, D. COMMISSION OF THE EUROPEAN COMMUNITIES DirectorateGeneral Telecommunications, Information, Industries and Innovation L 2920 LUXEMBOURG
PARK, D. Research Scientist KOREA ADVANCED INSTITUTE OF SCIENCE & TECHNOLOGY (KAIST) P.O. Box 131 Cheongryang KOREA SEOUL
PASSANO, E. AERIMPIANTI S.P.A. 21, Via Bergamo I 20135 MILANO
PEREIRA, F.J. Professor UNIVERSIDADE DE AVEIRO Departamento de Ambiente Ρ 3800 AVEIRO
PETERSEN, Β. Engineer, Managing Director INNOSYS Heimdalsgade 14 DK 2200 COPENHAGEN

672
PETERSEN, E. Manager of research cenere VØLUND FORSKNINGSCENTER Centervej 2 DK 6000 KOLDING
PISKORZ, J. Research Engineer UNIVERSITY OF WATERLOO DepC. of chemical Engineering WATERLOO CANADA ONTARIO N2L3GI
POHLMANN, K. Dipl. Chem. UNIVERSITÄT HAMBURG Institue fUr Technische und Makromolekulare Chemie Bundesstraße 45 D 2000 HAMBURG 13
POOS, Α. COMMISSION OF THE EUROPEAN COMMUNITIES DirectorateGeneral Personnel Administration L 2920 LUXEMBOURG
POUSAZ.P. Ingénieur en chimie BIOALTERNATIVE S.A. CH 2063 ENGOLLONNEUCHATEL
and
RENSFELT, E. Area Manager STUDSVIK ENERGY S 61182 NYKÖPING
RICK, F. Dipl.Ing. TUV RHEINLAND E.V. Postfach 10 17 50 D 5000 KLON
RITAPAL, K. Editor ERD0L & KOHLE ERDGAS PETROCHEMIE/ HYDROCARBON TECHNOLOGY Postfach 100 252 D 7022 LEINFELDENECHTD.
RODRIGUEZ JIMENEZ, J.J. Professor Dpto. Ingenieria Química Facultad de Ciencias UNIVERSIDAD DE MALAGA Campus Universitario de Teatinoe, S/N E 29071 MALAGA
RODRIGUEZ MIRASOL, J. Research Felo« Dpto. Ingenieria Quimica Facultad de Ciencias UNIVERSIDAD DE MALAGA Campus Universitario de Teatinoe, S/N E 29071 MALAGA
RAHNENFUHRER, R. UNIVERSITÄT HAMBURG Institut fUr Technische und Makromolekulare Chemie Bundesstraße 45 D 2000 HAMBURG 13
ROSSI, C. ENEL THERMAL AND NUCLEAR RESEARCH CENTRE CR.T.N. Via A. Pisano I 56100 PISA
RAMPLING, T.W.A. Scientist WARREN SPRING LABORATORY, DTI Gunnel8 Wood Road UK STEVENAGE HERTFORDSHIRE SGI 2BX
RÖSSLER, H. UNIVERSITÄT HAMBURG Institut für Technische und Makromolekulare Chemie Bundesstraße 45 D 2000 HAMBURG 13
RAYMOND, G.A. Ingénieur PYROPNEU 31, rue Sompré B 4120 IVOZRAMET
ROY, C. Associate Professor Dpt. of Chemical Engineering UNIVERSITÉ LAVAL CANADA SAINTEFOY (Québec) GIK 7P4

673
SALO, Κ. R&D Manager A. AHLSTROM OSAKEYHTIO BIONEER OY PL 537 SF 13111 HXMEENLINNA
SOLANTAUSTA, Y. Research Scientist TECHNICAL RESEARCH CENTRE OF FINLAND Biologinkuja 5 SF 02150 ESPOO
SCHÄDEL, S. UNIVERSITÄT HAMBURG Institut für Technische und Makromolekulare Chemie Bundesstraße 45 D 2000 HAMBURG 13
SORACE, G. Managing Director FIORENTINAMBIENTE AZIENDA SERVIZI NETTEZZA URBANA 52, Via Baccio Da Montelupo I 50142 FIRENZE
SCHIEBER, A. FRA GmbH Postfach 1645 D 7080 AALEN
SPINOSA, L. Senior Scientist CNR Instituto di Ricerca Sulle Acque 5, via F. de Biasio I 70123 BARI
SCHNEIDER, T. CivilEngineer ÖKOINSTITUT e.V. Bliro Darmstadt PrinzChristiansWeg 7 D 6100 DARMSTADT
SCHULTEN, H.R. Head Department FACHHOCHSCHULE PRESENIUS Department of Trace Analysis Dambachtal 20 D 6200 WIESBADEN
STÂHLBERG, P. Research Engineer TECHNICAL RESEARCH CENTRE OF FINLAND Laboratory of Fuel Processing Technology Biologinkuja 35 SF 02150 ESP00
STASSEN, H.E.M. Director BTG (BIOMASS TECHNOLOGY GROUP B.V.) P.O. Box 217 NL 7500 AE ENSCHEDE
SHAH, N. Research Engineer C.T.F.T. / C.I.R.A.D. 45 bis, Avenue de la belle Gabrielle F 94736 NOGENTSURMARNE
SIB0N0, A. BIO ALTERNATIVE ITALIA Sri 1, Via Caffaro I 16124 GENOVA
SIPILÄ, Κ. Director of Laboratory TECHNICAL RESEARCH CENTRE OF FINLAND Laboratory of Fuel Processing Technology Biologinkuja 35 SF 02150 ESPOO
STEFFENSEN, U. UNIVERSITÄT HAMBURG Institut für Technische und Makromolekulare Chemie Bundesstraße 45 D 2000 HAMBURG 13
STENHOLM, M. DKTEKNIK Gladsaxe Møllevej 15 DK 2860 SØBORG
STEPPAT, H. Dipl.Ing. Kufsteinerstraße 18 D 8205 KIEFERSFELDEN

674
STILES, H.N. Consulting Engineer BTG BIOMASS TECHNOLOGY GROUP B.V. P.O. Box 217 NL 7500 AE ENSCHEDE
VAN DER WEIDE, J. Research Manager TNO DELFT PO Box 237 NL 2600 AE DELFT
STOIKOS, T. Researcher CHEMICAL PROCESS ENGINEERING RESEARCH INSTITUTE PO Box 19517 GR 54006 UNIVERSITY CITY
THESSALONIKI
VAN HEEK, K.H. Professor BERGBAUFORSCHUNG GmbH FranzFischerWeg 61 D 4300 ESSEN 13
STREHLER, Α. Professeur TECHN. UNIVERSITY MUNICH Weihenscephan Bayer Landesanstalt flir Landtechnik Am Staudengarten 3 D 805 FREISING
STRIPP, O.F. HERBOLDENGINEERING GmbH Südetenstraße 2 D 6920 SINSHEIM
VELCICH, G. Development Engineer DANEC0 DANIELI ECOLOGIA S.p.A Via Linussio Z.I.U. I 33100 UDINE
WANDRASZ, J. Professor POLITECHNIKA SLASKA Wyckiat Mechaniczny 111 Konarskiego 18 POLEN 44100 GLIWICE
SWISTER, M. Etudiant LABORATOIRE DE CHIMIE ORGANIQUE Faculté des Sciences Ile du Sauley F 57 045 METZ
WANZL, W. Dr.rer.nat BERGBAUFORSCHUNG GmbH FranzFischerWeg 61 D 4300 ESSEN 13
SWITTEN, S. Secrétaire Administration MINISTÈRE DE LA RÉGION WALLONNE (IGE) Avenue Prince de Liege, 7 Β 5100 NAMUR
WICHERT, J. Dipl. OK FLEUS UNIVERSITÄT BREMEN Kufsteinerstraße NWI D 2800 BREMEN 33
TARALAS, G. Msc. THE ROYAL INSTITUTE OF TECHNOLOGY Department of Chemical Technology S 10044 STOCKHOLM
WILLIAMS, P. Lecturer Department of Fuel and Energy UNIVERSITY OF LEEDS UK LEEDS LS2 9JT
THESSÊN, G. MSc (ME) FLÄKT AB Box 81001 S 10481 STOCKHOLM
WINTHER, E.B. B.Sc.Μ.E. ELKRAFT POWER COMPANY Ltd. 5, Lautruphfit DK 2750 BALLERUP

675
WOLF, Β. Rechtsanwalt PKA Robert Bosch Straße 82 D 7080 AALEN
WOLPERT, V.M. WOLPERT & JONES (Studies) Ltd. Hunters, Holly Hill Colemans Hatch UK EAST SUSSEX TN7 4EP
YING, Y. UNIVERSITÄT HAMBURG Institut fUr Technische und Makromolekulare Chemie Bundesstraße 45 D 2000 HAMBURG 13
ZANZI, R. MSc. THE ROYAL INSTITUTE 0F TECHNOLOGY Department of Chemical Technology S 10044 STOCKHOLM
ZEPPI, C. Research Engineer ENEL / CRTH 120, Via A. Pisano I 56100 PISA


677
INDEX OF AUTHORS
ABATZOGLOU, N.S., 420 AMMAR, S., 158 ANTONELLI, L. , 85 ANTONINI, G., 425 ARAUZO, J., 613 AZNAR, M.P., 624, 629
BARTON, J.R., 57, 391 BASSELIER, J.J., 568 BATALLER, Α., 435 BECKER, J.J., 635 BEENACKERS, A.A.CM., 129, 312, 396 BELLMANN, U., 190 BENNINI, S., 640 BETTENS, B., 209 BILBAO, R., 613 BILITEWSKI, Β., 98 BLASIAK, W., 468 BONFITTO, E., 586 BONINO, G., 527 BRANDANI, V., 430 BRIDGE, S.A., 541 BRIDGWATER, A.V., 43, 129, 195, 274,
347, 394, 411, 541, 598, 651 BRUNETTI, Ν., 586 BUEKENS, Α., 274, 492, 574, 580
CAPART, R., 158, 530, 593 CARRE, J., 93 CASTILLO, S., 640 CEBRIAN, Ν., 624 CHURIN, E., 326, 399 COLLIN, R., 468 CONTI, L., 246 CORDERO, Τ., 435 CORELLA, J., 618, 624, 629 CYPRES, R., 209 CZERNIK, S., 201
FABRY, R., 387, 405 FERRERÒ, G.L., 3, 405 FILEN, H., 603 FLAMMINI, D., 430 FONT, R., 230 FONZI, F., 264 FRANCK, J., 517 FRANCOIS, O., 425 FUNK, H., 547
GAJEWSKI, W., 468 GARCIA, P., 613 GAS, G., 640 GAUDEMARD, S., 425, 635 GELUS, M., 158, 593 GIRARD, P., 372, 530 GONZALEZSAIZ, J., 618 GRASSI, G., 7 GRAVERSEN, P., 536 GROUX, B., 559 GUIGON, P., 317 GUIOL, R., 559
HALKET, J.M., 505 HÄRDTLE, G., 98 HARTINIATI, 257 HAYES, R.D., 28 HEERAH, M., 554 HENRIKSEN, U., 290 HERGUIDO, J., 618 HINRICHS, H.F., 522 HOCHHEIM, F.W., 417 HOI, W.K., 411, 598 HUMMELSHeíJ, R.M., 536 HUMMELSIEP, H., 547
IACOBONI, S., 586 IGLESIAS, J.I., 624
DELGADO, J., 629 DELMON, B., 326 DEL RE, G., 430, 586 DESBENE, P.L., 568 DESMAZIERES, Β., 568 DEVESA, J., 230 DE WOLF, F., 580 DIEBOLD, J., 14, 342 DI GIACOMO, G., 430, 586 DISS, J.M., 417 DOUBLE, J.M., 651
EARP, D.M., 238 ELAMIN, Α., 158 ENGELBRECHT, A.D., 296 ESNOUF, C , 106, 425, 554 ESSAYEGH, M., 568
JENSLEV, Κ., 536
KAMINSKY, W., 190, 473, 517 KOFOED, E., 290 KOLJONEN, J., 608 KOUKIOS, E.G., 420 KOULLAS, D.P., 420 KOVAC, R.J., 169 KUMMER, A.B., 190 KURKELA, E., 304, 603, 608
LACHENAL, J., 462 LACROSSE, L., 93 LAHOZ, J., 629 LANA, J., 613 LANGE, C , 568

678
LAPPAS, A.A., 563 LARGE, J.F., 317 LESGOURGUES, Ν., 593 LI, Ζ., 593
MAJERSKI, P., 444 MANIATIS, Κ., 274, 312, 405, 574 MARCILLA, Α., 230 MAREK, Κ., 98 MARTINEZ, M.P., 624 MARTINEZSANCHEZ, Μ.A., 439, 452 MARTINMARTINEZ, J.M., 439, 452, 457 MASSON, H., 574 MAUND, J.K., 238 MENDIS, M.S., Ill MEZERETTE, C , 425, 462 MICHELKIM, H., 656 MITCHELL, C.P., 43 MOILANEN, A., 608 MOJTAHEDI, W., 304 MÜHLEN, H.J., 72 MÜLLER, H., 522 MUNCK, J., 551
SALO, Κ., 603 SCANO, G., 246 SCHENKEL, Y., 93 SCHOETERS, J., 574, 580 SCHULTEN, H.R., 497, 505 SCOTT, D.S., 201, 444 SEPPEN, J.J., 334 SHAH, N., 530 SIMON^ A., 435 SIPILÁ, K., 608 SMITH, E.L., 651 SOEMARDJO, Α., 257 SOLANTAUSTA, Y., 396 STÂHLBERG, P., 304, 603 STEFFENSEN, U., 517 STEVENS, D., 14 SUSANTO, H., 282
TAYLOR, D.T., 486 TOLEDO, J.M., 462 TRAVERSE, J.P., 640
UNSWORTH, J., 180
NELS, C.H., 40, 379 NIEMINEN, M., 304
OLSEN, G., 290, 479 O'NEIL, D.J., 169, 250 ORGILESBARCELO, C , 439, 452
VAN DER WEIDE, J., 334 VAN HEEK, K.H., 72 VANNICE, Μ.Α., 457 VASALOS, I.A., 563 VERDU, E., 230 VERGNET, A.M., 462
PEDERSEN, P.H., 290 PISKORZ, J., 201, 391, 444 POUSAZ, Ph., 559 PRÖSCH, U., 473
RADLEIN, D., 201, 444 RAHNENFUHRER, R., 517 REKSOWARDOJO, S., 282 RODRIGUEZ, J.J., 435 RODRÍGUEZMIRASOL, J., 435 RODRIGUEZREINOSO, F., 439, 452 RÖSSLER, H., 473 ROY, C , 180, 394 RURIHOSE, F., 93
WALECZEK, K., 646 WANDRASZ, J., 646 WANZL, W., 72 WILÊN, C , 608 WILLIAMS, P.T., 486 WILLOCX, J., 492
YING, Υ., 190 YOUVIAL, M., 257
ZAJDEL, J., 468 ZETHRAEUS, B., 468






Thermochemical processing of renewable resources and solid fuels has become a strong contender partially to replace the energy dependence of the European Community on imported hydrocarbon fuels; the Commission is supporting R&D as well as demonstration projects in this field. Similarly in the USA, Canada and the developing countries numerous projects have been carried out on fundamental, as well as industrial scale, projects on pyrolysis and gasification. Such technologies are therefore of increasing importance worldwide, not only because they can be utilized to dispose of various industrial wastes ¡ri an environmentally acceptable way.
Though interesting results and experiences have certainly been achieved, several problems still remain and their solution will strongly influence the commercialisation of pyrolysis and gasification all over the world.
It was the need to evaluate critically the progress achieved in this field and to draw up recommendations for future work which prompted the Directorate-General for Energy of the Commission of the European Communities to organise this international conference with the assistance of the Directorate-General for Telecommunications Information Industries and Innovation.
While invited speakers from the Commission, EC countries, the USA, Canada and the World Bank presented overviews on all aspects of pyrolysis and gasification processes such as feedstock, pretreatment and characterisation, gasification and pyrolysis technologies, products upgrading and utilization as well as environmental and economic aspects, researchers and industrialists from 20 countries presented their results and views in oral as well as poster presentations. The workshops and panel discussions gave the opportunity to all participants to express their opinion so that realistic recommendations for future R&D and demonstration activities could be drawn up.
About 200 participants representing administration, governmental institutions, universities and industry attended the conference. This is fresh proof of the significance in recent years of pyrolysis and gasification technologies.
ISBN 185166 4491