Physical Properties of Amino Acids and Prediction of Secondary
33
3/27/02 Physical Properties of Amino Acids and Prediction of Secondary Structure Huan-Xiang Zhou CSIT, Physics, and IMB
Transcript of Physical Properties of Amino Acids and Prediction of Secondary

3/27/02
Physical Properties of AminoAcids and Prediction of Secondary
Structure
Huan-Xiang Zhou
CSIT, Physics, and IMB
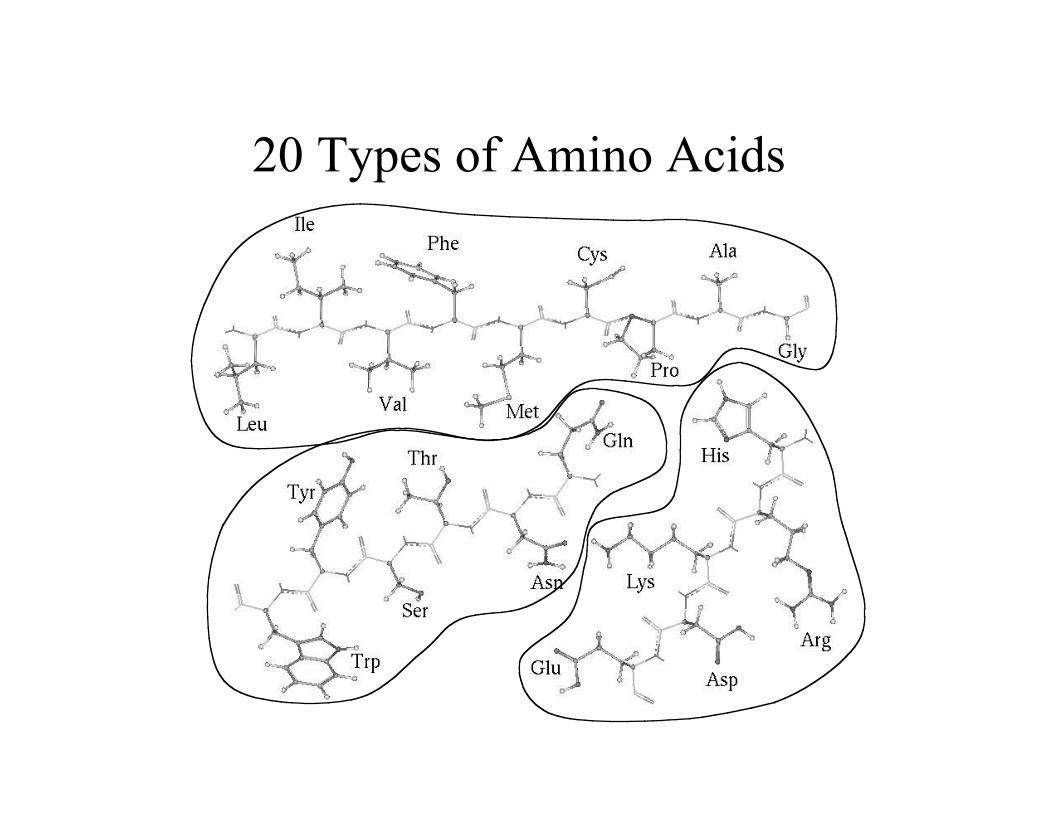
20 Types of Amino Acids

Physical Properties
• Polarity
nonpolar – hydrophobic interactions
polar – hydrogen bonds
charged – salt bridges/charge pair
• Rotational entropy
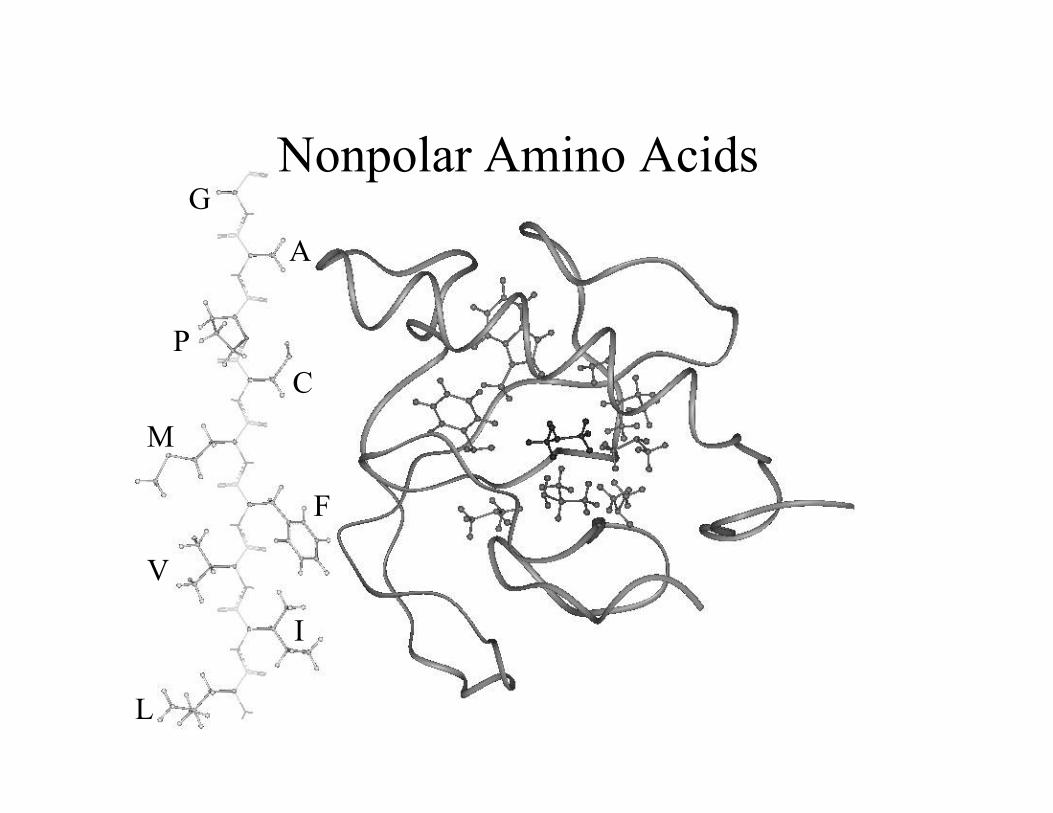
L
V
I
F
M
CP
A
GNonpolar Amino Acids

SY
T
W
N
QPolar Amino Acids

Charged Amino Acids
D
E
K
HR
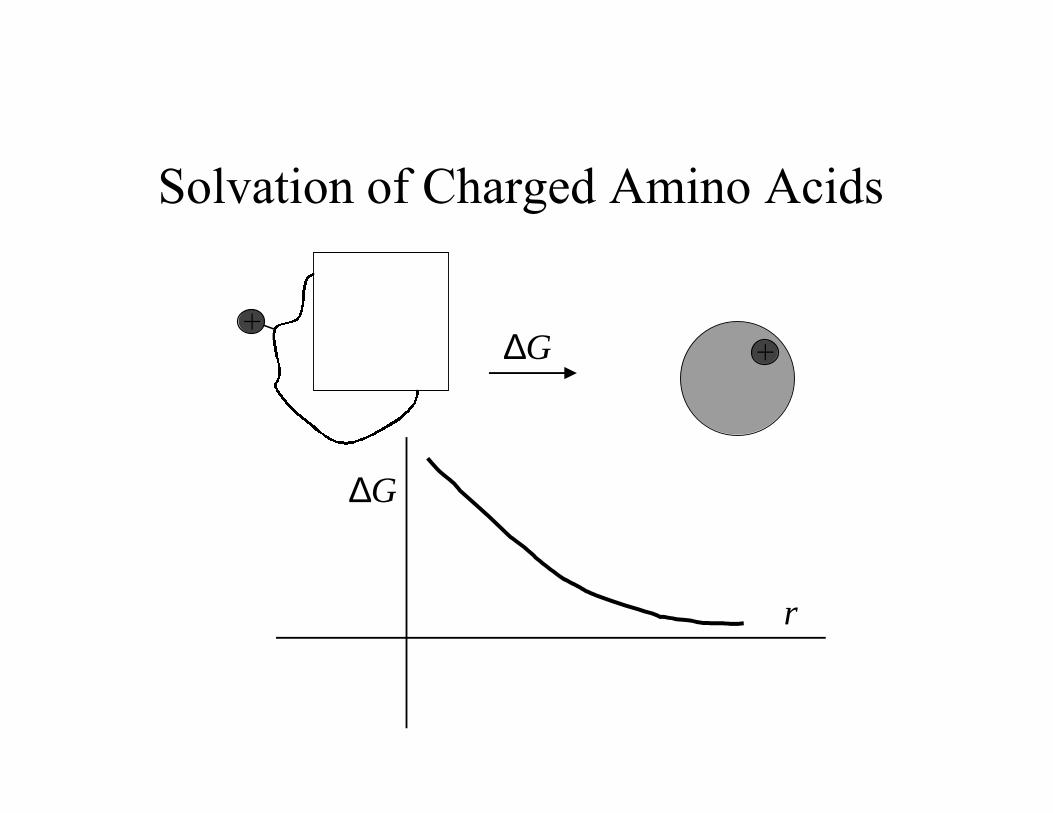
Solvation of Charged Amino Acids
++
∆G
∆G
r
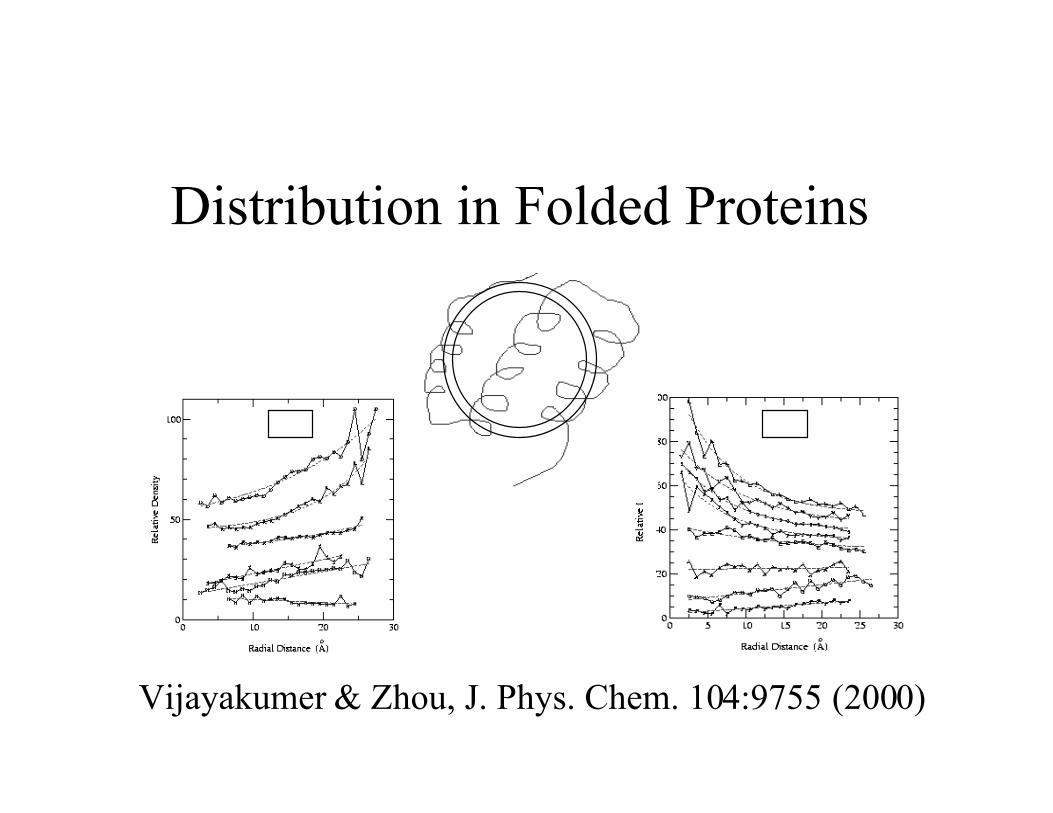
Distribution in Folded Proteins
Vijayakumer & Zhou, J. Phys. Chem. 104:9755 (2000)

+∆G_
_+
_+
_+
∂∆G/∂pH = kBT(Qf - Qu)ln10
Charge-Charge Interactions

Charge-Charge Interactions: Unfolded State
+
ru = e2/ε r
Zhou, P. Natl. Acad. Sci. USA 99:3569 (2002)
r is not fixed, but distributed according top(r) = 4πr2(3/2πd2)3/2exp(-3r2/2d2)
average distance d = bl1/2 + s<u> = (6/π)e2/εd
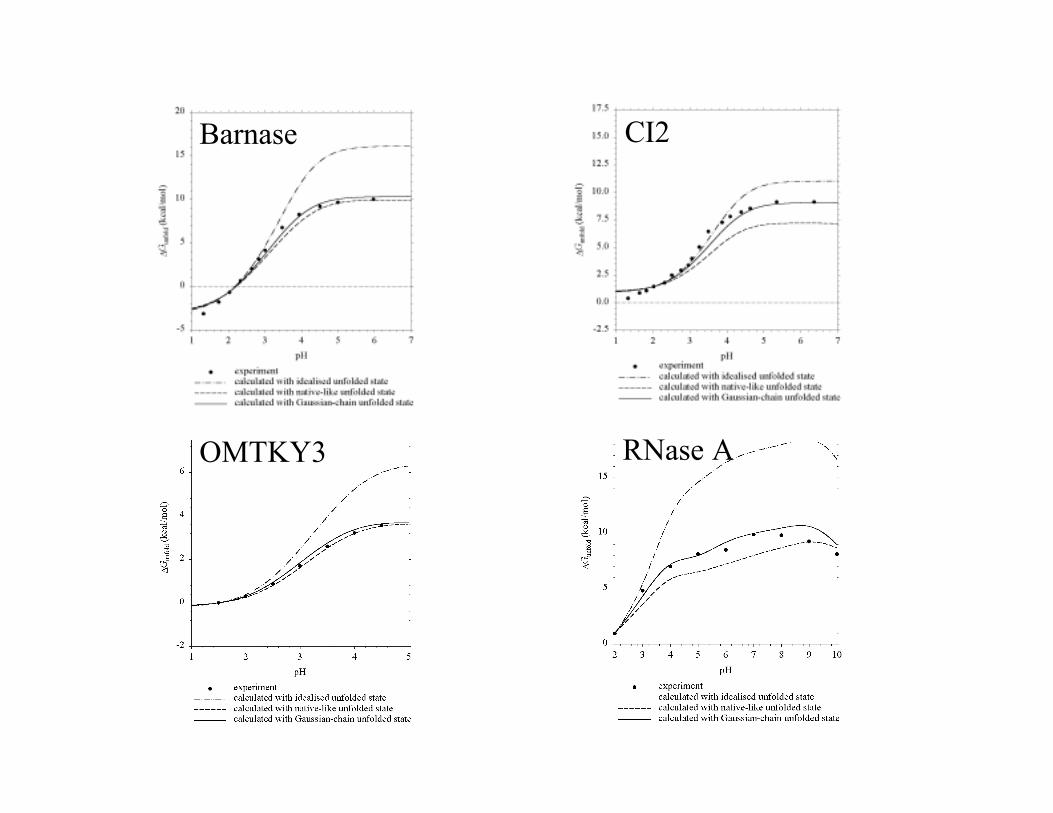
Barnase CI2
OMTKY3 RNase A
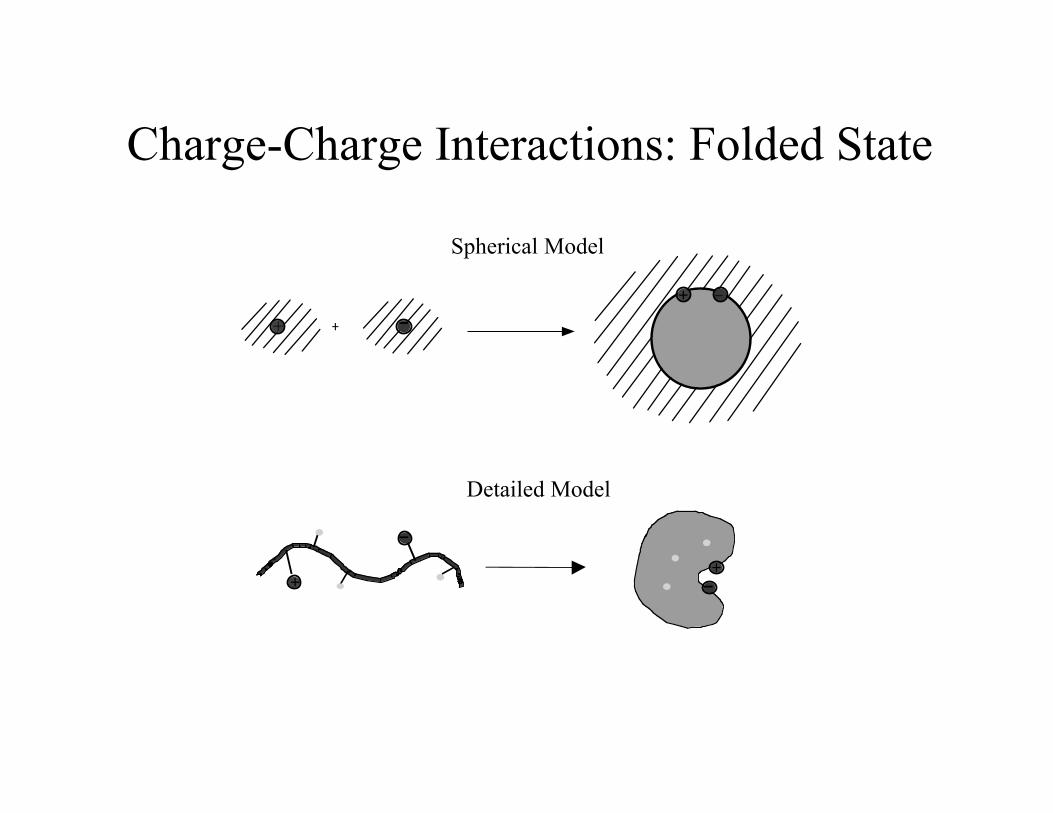
Charge-Charge Interactions: Folded State
-
++
-
Detailed Model
-+
+ -
+
Spherical Model
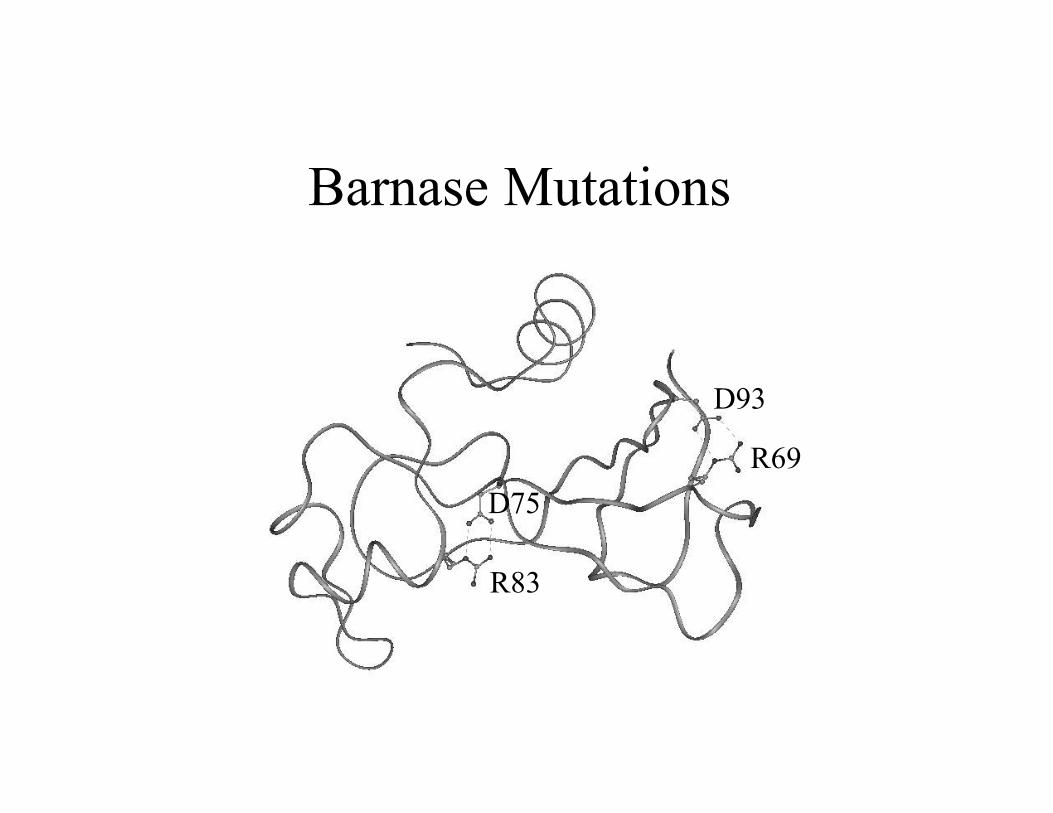
Barnase Mutations
R69
R83
D75
D93
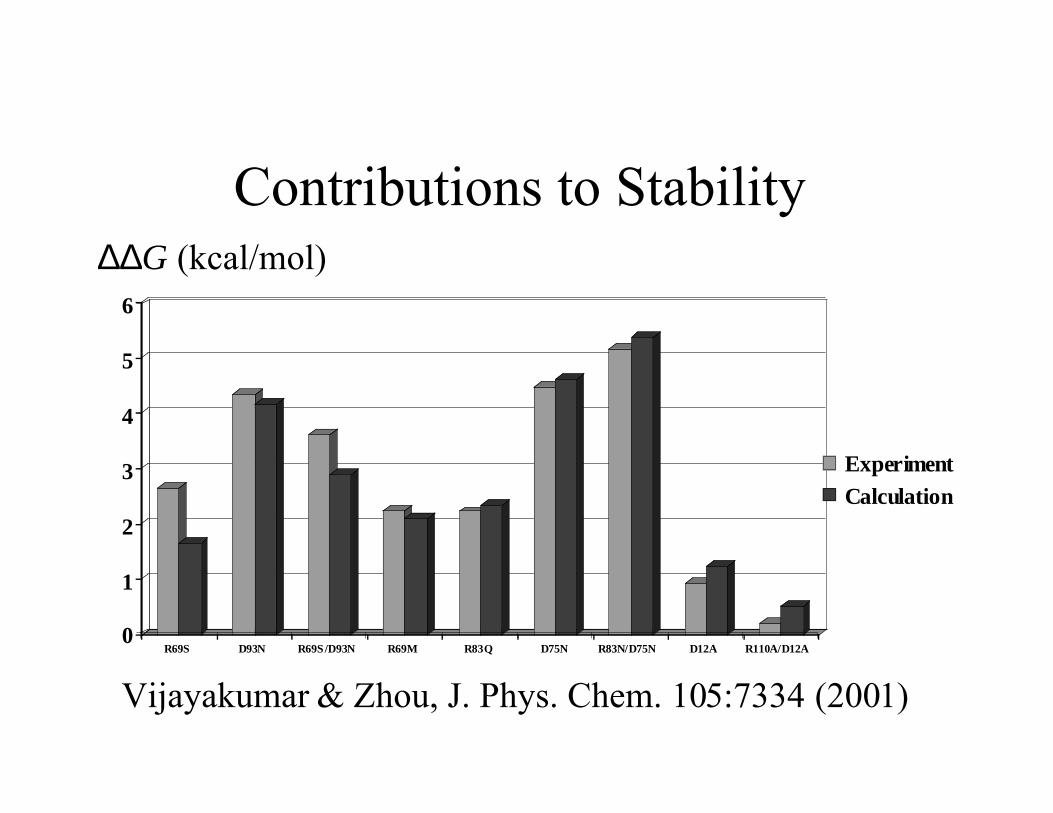
Contributions to Stability∆∆G (kcal/mol)
0
1
2
3
4
5
6
R69S D93N R69S/D93N R69M R83Q D75N R83N/D75N D12A R110A/D12A
ExperimentCalculation
Vijayakumar & Zhou, J. Phys. Chem. 105:7334 (2001)

Conformations of Peptides
φφφφ
ψψψψ
χχχχ1
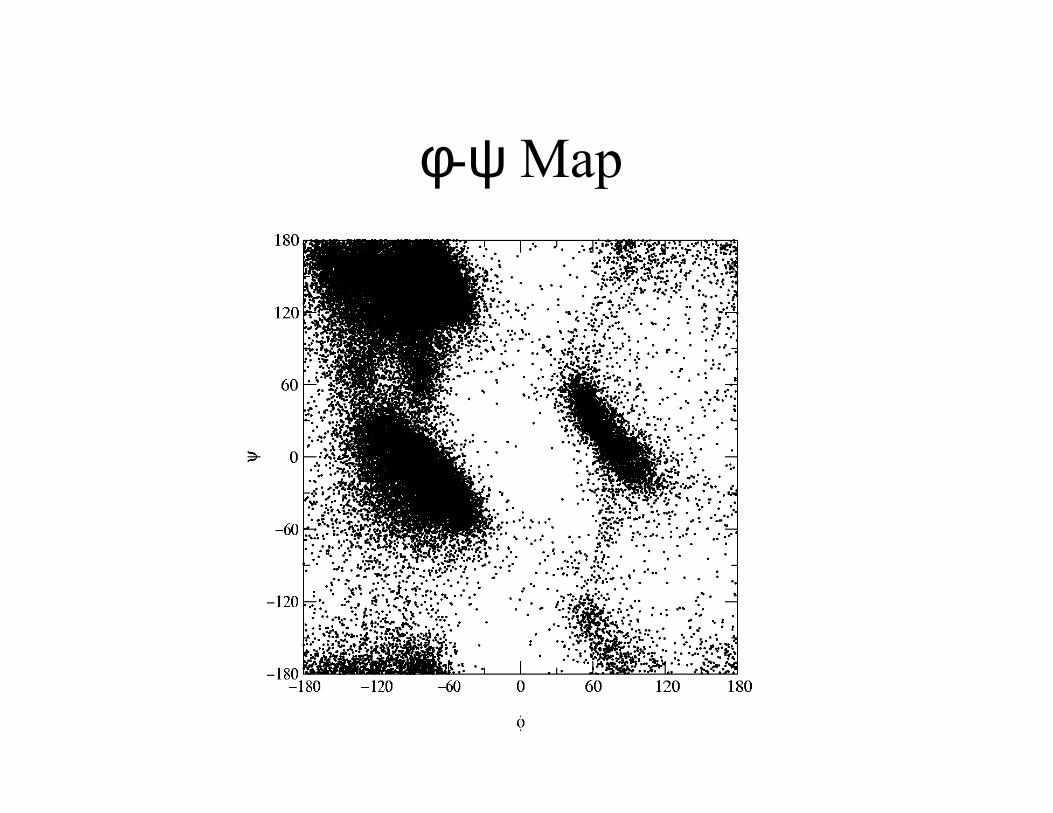
φ-ψ Map

• In the unfolded state, sidechains have morerotational freedom.
• Loss of sidechain entropy depends on type ofamino acids, backbone conformation, andtertiary contacts.
Sidechain Rotational Entropy

Helix-Forming Propensities
• Propensities are manifestedby the occurrencefrequencies of amino acidsin helices and can bemeasured experimentallyby mutations. Order: Ala >Leu > Ile > Val > Ser, Thr> Asp, Asn.

Accounting for the Different Propensities
• Rose (1992) proposed restriction insidechain rotation by helix as a majorfactor.
• This cannot explain lower propensities ofpolar sidechains (Ser, Thr, Asp, and Asn).
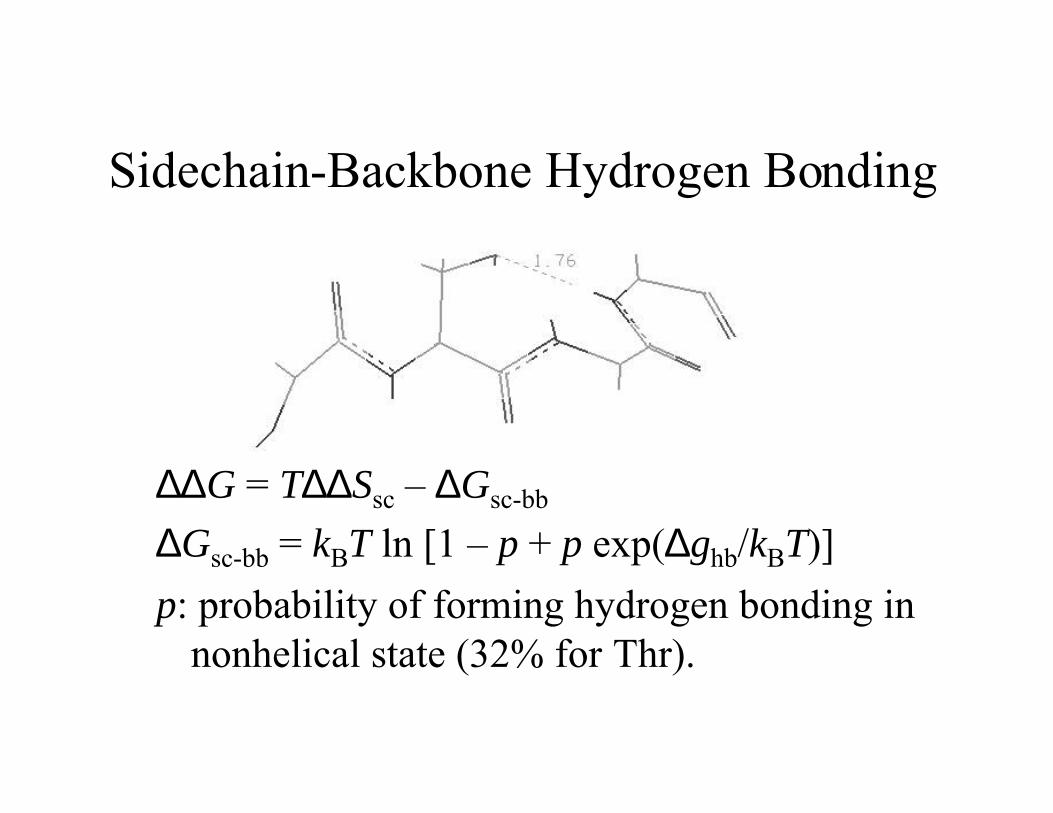
Sidechain-Backbone Hydrogen Bonding
∆∆G = T∆∆Ssc – ∆Gsc-bb
∆Gsc-bb = kBT ln [1 – p + p exp(∆ghb/kBT)]
p: probability of forming hydrogen bonding innonhelical state (32% for Thr).
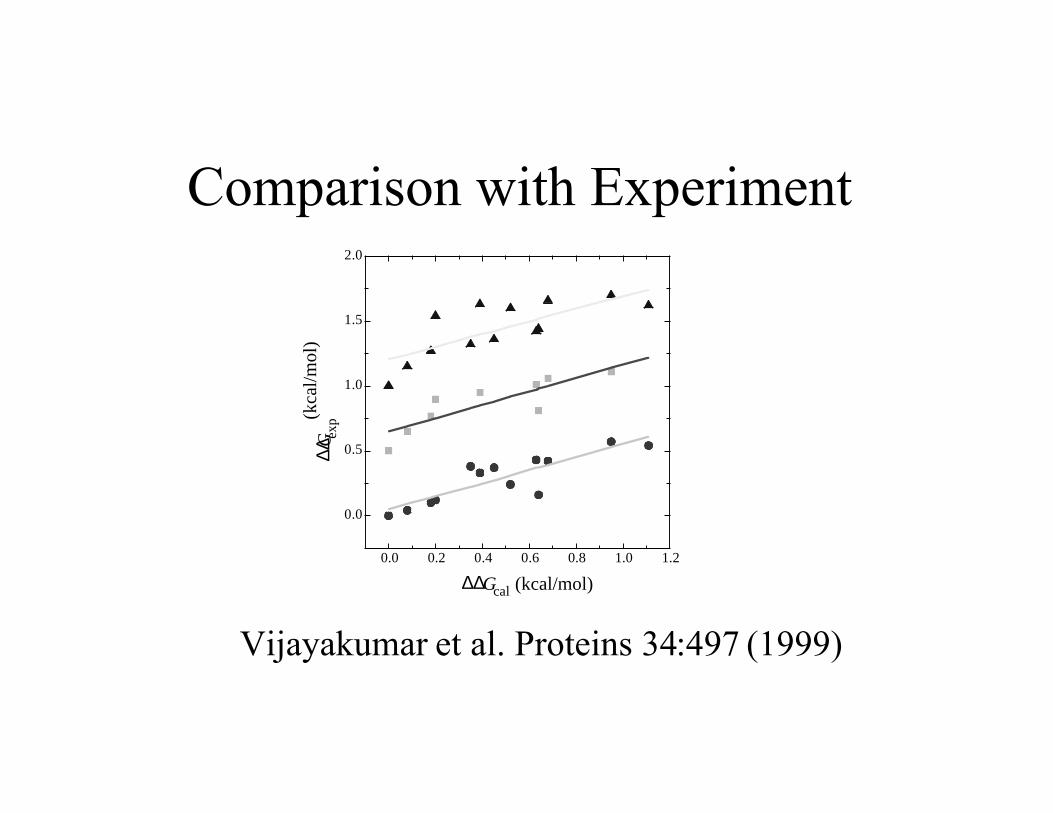
Comparison with Experiment
0.0 0.2 0.4 0.6 0.8 1.0 1.2
0.0
0.5
1.0
1.5
2.0
∆∆G e
xp
(kc
al/m
ol)
∆∆Gcal (kcal/mol)
Vijayakumar et al. Proteins 34:497 (1999)

Prediction of Solvent Accessibility
• Two-state representation: 0 for buried and 1 forexposed.
• Baseline Method: Those buried >50% of the time in atraining set is predicted to be buried; the rest ispredicted to be exposed. In particular, Leu, Ile, Val,Phe, Trp, and Cys are always predicted to be buried,whereas Asp, Glu, Lys, Arg, His, Asn, Gln, and Proare always predicted to be exposed.

Bayesian Statistical Analysis• Extends the baseline method by considering statistics
of not just one position, but a window of residuescentered at one position.
• Because of low probability for any stretch of residuesin protein sequences, statistically significant resultsfor burial probability of a residue inside a particularstretch of residues cannot be obtained from anytraining set. Assumptions must be made.
• Simplest assumption is probability for a type ofresidue to appear in a site within a segment ofaccessibility states is independent of neighboringpositions.

Linear Regression Analysis• Accessibility state at a position is assumed to be determined
directly by the residue identities at that and neighboringpositions, and the transfer free energies (Gi) and relativemolecular weights (Mi) of the residues occupying thesepositions via
Si = ∑jαj(Si)Rj + ∑j,kβjk(Si)GjGk + ∑j,kγjk(Si)MjMk
The indices j and k run from the beginning to the end of awindow centered at the position i whose accessibility state Siis calculated. The coefficients αj, βjk, and γjk are determinedby minimizing the deviations of calculated accessibility statesfrom actual ones for a training set.
• Rj is an array of 19 zeros and a one representing the particulartype of residue occupying position j.

Multiple Sequence Alignment andSequence Profile
• Proteins are subject to mutations. Residues are likelyreplaced by those with similar properties (divergentevolution). Conversely, a protein structure dictateswhich type of positions are occupied by which typeof residues (convergent evolution).
• When homologous proteins are aligned by sequence,identities of amino acids occupying a given position(sequence profile) hold information about thatposition.
• Multiple-sequence alignment can be readily obtainedPSI-Blast.

MS Information Enhances Accuracy• If a position is always occupied by
residues favoring the buried(exposed) state among a set ofhomologous proteins, that positionis very likely to be buried (exposed).
• ImplementationBaseline Method: ∑lwlpl > 0.5
Bayesian Statistics: Sequence profiles are representedby 28 classes
MLR: Rj replaced by sequence profile
----L--D----
----L--E----
----I--E----
----V--K----

Neural Network Predictor
• Sequence profile is fed as input. Network is trainedon a set of known protein structures.
Shan et al., Proteins 42:23 (2001)
Sequ
ence
Pro
file
s
State
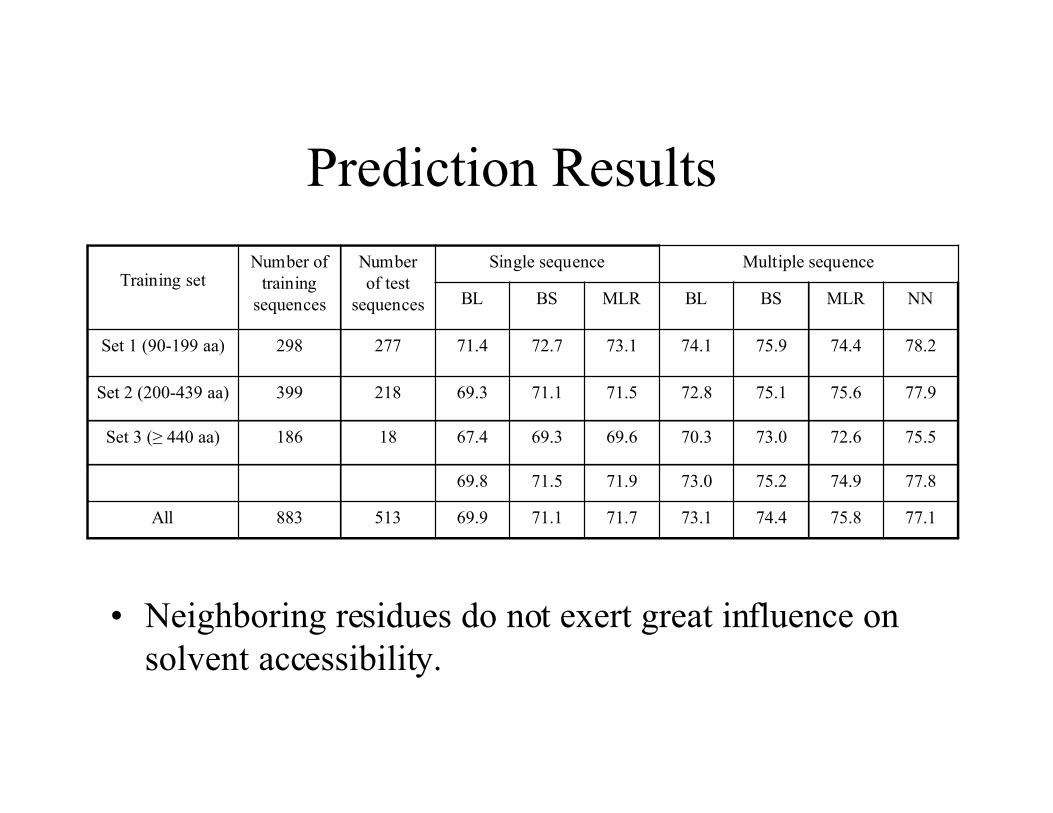
Prediction Results
77.175.874.473.171.771.169.9513883All
77.874.975.273.071.971.569.8
75.572.673.070.369.669.367.418186Set 3 (≥ 440 aa)
77.975.675.172.871.571.169.3218399Set 2 (200-439 aa)
78.274.475.974.173.172.771.4277298Set 1 (90-199 aa)
NNMLRBSBLMLRBSBL
Multiple sequenceSingle sequenceNumberof test
sequences
Number oftraining
sequencesTraining set
• Neighboring residues do not exert great influence onsolvent accessibility.

Prediction of Secondary Structure
• Amino acids have different preferences for α-helix(and β-strands). A string of helix-preferring residueswill likely form helix. --AALILA--
• New idea: in a multiple sequencealignment, if position is mostlyoccupied by helix-preferringresidues, that position will likely behelical.
Chou & Fasman, Biochemistry (1974).
----AL----
----AA----
----LL----
----LA----

Neural Network Predictor
• Sequence profile is fed as input. Network is trainedon a set of known protein structures. Consistentlypredicts secondary structure at 75% accuracy.
Shan et al., Proteins 42:23 (2001)
Sequ
ence
Pro
file
State

Prediction of 3-D Structure
• Proteins with similar sequences adopt nearly identicalstructures. Even proteins with very differentsequences (e.g., 10% identity) often adopt similarstructures. Perhaps there is a finite number ofdistinct structure folds.
• New problem: which of the structure folds FITs thesequence best?
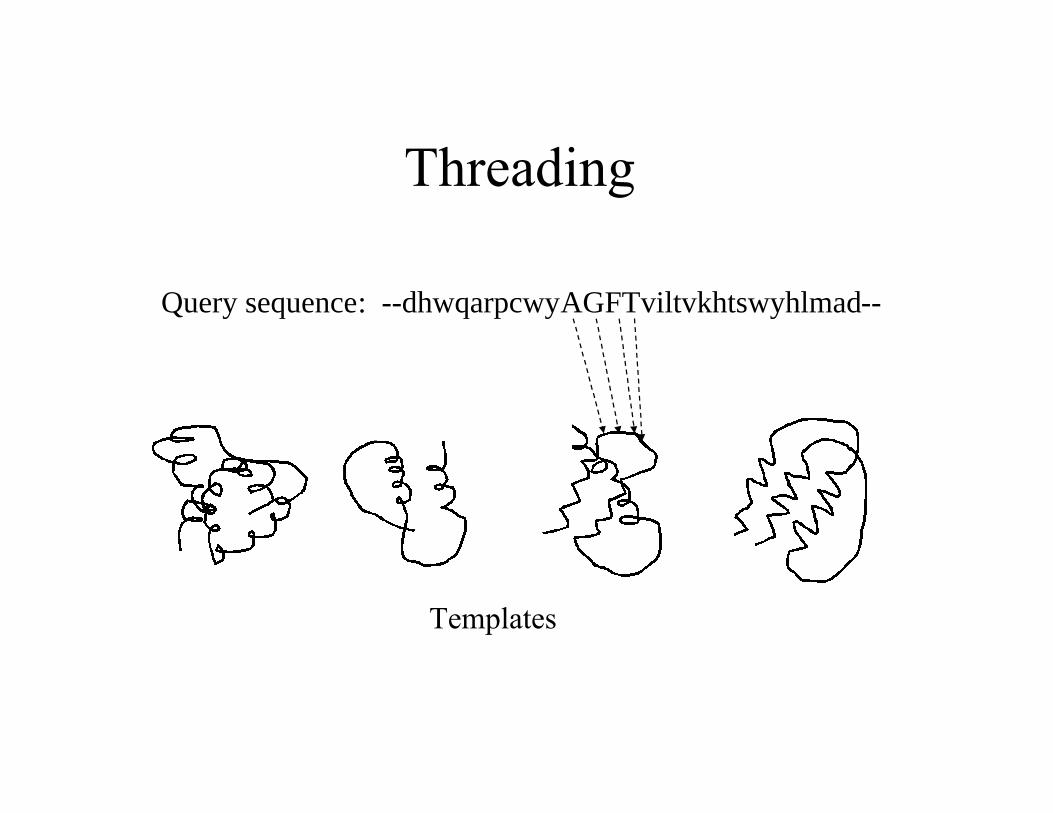
Threading
Query sequence: --dhwqarpcwyAGFTviltvkhtswyhlmad--
Templates

Fitting Function of COBLATHShan et al., Proteins 42:23 (2001)
• When proteins have similar structures, theirsequences do share similarities (e.g., Leu replaced byIle). This similarity can be captured by comparingthe sequence profile of query (from a multiplesequence alignment) with sequences of templates.
• When 3-d structures are superimposable, secondarystructures and solvent accessibilities must also agree.This agreement can be captured by comparingpredicted secondary structure and accessibility ofquery and actual secondary structures andaccessibilities of templates.