Nueva Ruta de Síntesis de la 6-Metoxi-5-Metil- α
78
Transcript of Nueva Ruta de Síntesis de la 6-Metoxi-5-Metil- α


UNIVERSIDAD DE LOS ANDES
FACULTAD DE CIENCIAS
DEPARTAMENTO DE QUÍMICA
LABORATORIO DE PRODUCTOS NATURALES
Nueva Ruta de Síntesis de la 6-Metoxi-5-Metil- α-Tetralona y la 6-Metoxi-2,5-Dimetil-α-
Tetralona, Intermediarios para la Obtención de Terpenoides y Esteroides
Trabajo Especial de Grado de Licenciatura en Química
Br. Johan José Pineda Guerrero
Mérida-Venezuela Noviembre, 2006

2
A mi Madre, Hermano, Abuela y Seres Queridos

3
Agradecimientos
• A Dios Todopoderoso por darme las fuerza, compañía, empeño y esperanzas para llegar a esta meta y por regalarme una familia tan solidaria y amorosa como la que tengo. Mama Rosa, Abuela Rosa, Abuelo Rafael, Tía Gladis y mi Hermano Jorge son para mi un ejemplo a seguir. Este es también su triunfo. LOS AMO
• A la memoria de mi abuelo Pedro que donde quiera que se encuentre; para mi abuela Elva, mi papá y mis tíos Oscar, Josefina, Francisco, Belkis espero que reciban éste como un regalo de mi parte.
• Al Dr. Juan Manuel Amaro, por brindarme su sincera y gran amistad llena de ejemplos, conocimientos, consejos y palabras de ánimo que dibujan una sonrisa en el corazón de cualquiera, por tantas horas de trabajo y por la excelente dedicación que me brindó, no tengo palabras para decirle GRACIAS (Papá Noel).
• Al Dr. Ajoy Banerjee por permitirme trabajar en su laboratorio y llevar a cabo mi proyecto; gracias Doctor por confiar en mi trabajo y ofrecerme esa oportunidad. Al Lic. Henry Mora por además de ser un compañero incondicional, me asesoró e impulsó para lograr esta meta. Gracias mi Pana.
• A mis compañeros de Laboratorio del IVIC Liadis, Mariú, Felix, Karla, Manuel Laya, Wiliam Vera, así como mis compañeros de la ULA: Elier, Foción, Freddy, Pablo, Leo, Anita, María Soledad, Kiki, Jennifer, Isaura, Jan, Ciro, José Luis, Leo Barrios, Roger, Vanesa, Camilo, Ada, Carolina, Carlitos, Prof. Irama, Prof. Abad, Prof. Paulino por hacer de esas largas horas de trabajo momentos agradables, con bonitas e inolvidables experiencias compartidas. (Mariú y Felix lo logramos, saludos para mis panas los Orientales).
• A las persona que en Caracas se convirtieron en más que una Familia para mí y me apoyaron de una forma sincera y desinteresada: Geribeht y la familia Cuillo, Dra. Victoria, Sra. Elizabeth, Anlie, Dra. Lourdes, Sra. Candida, Yusely, Dany, Felix, Johanna Cano, Dayana Leal, familia Rodríguez-Guillory y especialmente a Christian. A todos, los llevare siempre en mi corazón.

4
• A mis amigos de toda una vida; aunque sigan distintos caminos continuaremos siéndolo: Johanali, Yenisey, Gerardo y Rebeca. Los quiero.
• A mis amigos que la universidad me regaló; más que amigos son y siempre serán para mi unos hermanos: Mafer, Maru, Moya, Freducho, Guiseppe y Jaldon. De verdad son innumerable la cantidad de experiencias que tengo con cada uno de ustedes; gracias y los quiero mucho.
• A Patty (Yasu), Lenis, Lau (Cuñis), Marbeni, Maregdy, Hugo, Yasmeli, Karen, Kiki, Karina Zabala, Yris por brindarme su amistad en distintos momentos de mi vida. A la Sra. Leo, Sra. Sioly, Sra Isabel, Sra. Aida, Sr. Carlos Castillo, Sra. Alis, Sr. Juan y Sra. Zule por darme el calor de su familia y su hogar.
• A mis primos más queridos Yolimar, Adrianita, Yonar, Julito, Desiree, Yubi, por su solidaridad y a mi familia en general por su constante apoyo.
• A mis Profesores que me brindaron conocimientos durante la carrera, en especial: Xiomara Romero, Luis Rincón, Rafael Almeida, Alí Bahsas, Paulino Delgado.
• Al Instituto Venezolano de Investigaciones Científicas (IVIC), específicamente al Centro de Quimica, en el cual llevé a cabo la realización experimental de este proyecto.
• A la Ilustre Universidad de los Andes y especialmente a la Facultad de Ciencias.

5
Contenido
Página
Resumen……………………………………………… 6
Summary…………………………………………….. 6
Introducción........................................................... 7
Biosíntesis de Terpenoides……………………….... 9
Triterpenos y Esteroides……………………………. 12
Tetralonas……………………………………………. 14
Antecedentes del Problema a Investigar…………. 15
Hipótesis………………………………….………….. 17
Objetivos……………………………….…………….. 17
Resultados y Discusión…………………………….. 18
• Síntesis de [9] y [10]………………………… 19
• Síntesis de [11] y [12]………………………. 32
• Síntesis de [2]……..………………………….. 39
• Síntesis de [3]……..………………………….. 46
• Síntesis de [4]……..………………………….. 53
• Síntesis de [6]……..………………………….. 60
Parte Experimental………………………………….. 68
Conclusiones……………………………………….... 73
Referencias Bibliográficas………………………….. 74

6
Resumen
La síntesis de las tetralonas 6-Metoxi-5-metil-α-tetralona [4] y 6-Metoxi-2,5-dimetil-α-tetralona [6] fue abordada por primera vez en la década de 1940 por Sir. Robinson et al. Ellos demostraron que estas tetralonas son sustratos importantes para la preparación de esteroides y terpenoides. En el presente trabajo de investigación fue posible ensayar una ruta alternativa, frente a la anteriormente propuesta, para la síntesis de las tetralonas [4] y [6] evitando las condiciones drásticas usadas en los procesos anteriores. Los productos intermediarios y finales fueron caracterizados por técnicas espectroscópicas básicas (IR, UV, RMN-1H, RMN-13C y EM); se determinaron los porcentajes de rendimiento parciales y total.
Summary
Synthesis of 6-Metoxy-5-methyl-α-tetralone [4] and 6-Metoxy-2,5-dimethyl-α-tetralone [6] were undertaken by first time at 1940 decade, by Sir. Robinson et al. They demonstrated that these tetralones are important substrates for preparation of steroids and terpenoids. In this research work it was possible to assay an alternative route versus the previously proponed one, for the synthesis of tetralones [4] y [6], avoiding the drastic conditions used in previous processes; intermediate and end products were characterized by basic spectroscopic techniques (IR, UV, 1H-NMR, 13C-NMR and MS); percentages of partial and total yields were determinated.

7
Introducción
Una gran cantidad de productos naturales se aíslan de los extractos de las plantas, cientos de los cuales se han usado como perfumes, saborizantes y medicinas, antes de que la química fuera capaz de estudiar su composición. Las resinas y aceites de muchas plantas son también fuente de productos naturales, y de hecho, los primeros estudios químicos y biológicos que se realizaron en el campo de los productos naturales, utilizaban aceites vegetales como material de partida para el aislamiento de estos productos. En 1818, se encontró que el aceite de trementina tenía una relación C:H de 5:8, y que muchos otros compuestos tenían relaciones C:H semejantes. Todos estos compuestos se caracterizan por tener un origen biosintético común, y a los mismos se les denominó isoprenoides, porque se suponía que el isopreno (2-metil-1,3-butadieno) era su precursor biogenético (Wallach, 1887). Los isoprenoides presentan una gran variedad estructural, la cual se genera de la condensación y fusión repetitiva de dos o más unidades ramificadas de cinco carbonos, basadas en la estructura del isopreno; la unión entre estas hipotéticas unidades de isopreno suele ocurrir entre el extremo ramificado (cabeza) de una de ellas y el extremo lineal de la otra (cola) (Newman, 1972).
Figura 1: Estructura Molecular del Isopreno
Los isoprenoides se subdividen en dos grandes grupos, los esteroides y los terpenoides o terpenos. De manera general, ambos grupos obedecen a lo que se conoce como “Regla del Isopreno”, la cual puede concretarse en el hecho de que la secuencia de los átomos que conforman un isoprenoide responde a varias unidades consecutivas de isopreno (Ruzicka, 1959); sin embargo, esta regla no siempre se cumple estrictamente, por cuanto el número de átomos de carbono, debido a reacciones de degradación, puede no ser múltiplo de cinco; o bien porque suceden reagrupamientos y migraciones que conducen a secuencias anormales de la unidad de cinco átomos de carbonos. Este hecho condujo a Ruzicka et al. (1953) a enunciar la que hoy se denomina “Regla Biogenética del Isopreno”, la cual maneja un concepto mucho más amplio, en el que se incluyen los isoprenoides reagrupados y los nor-isoprenoides o isoprenoides degradados.
H3C
H2C
C C
CH2
H
H2C C
CH3
CH CH2
Cabeza
Cola

8
Centrando nuestra atención en el subgrupo de los terpenoides, es oportuno señalar que los mismos se clasifican, según el número de unidades de cinco carbonos (unidades de isopreno) que contengan, en mono-, sesqui-, di-, tri- y tetraterpenos. Todos ellos se encuentran como sustancias naturales en el Reino Vegetal (Pridham, 1965) y también en el Reino Animal (Prestwich, 1979), integrando una gran variedad de estructuras, tanto acíclicas como cíclicas (Fig. 2).
La mayoría de los terpenos que se han aislado tienen un número par de unidades de isopreno. Los monoterpenos son moléculas con diez átomos de carbono, equivalentes a dos unidades de isoprenos; los diterpenos tienen veinte carbonos (cuatro unidades de isoprenos), los triterpenos tienen treinta carbonos (seis unidades de isoprenos) y los tetraterpenos tienen cuarenta átomos de carbono (ocho unidades de isoprenos). Además, se ha aislado un número significativo de terpenos con tres unidades de isopreno (quince carbonos); estos son los sesquiterpenos. Los sesterterpenos son un grupo más pequeño de terpenos compuestos de cinco unidades de isopreno. Por último se encuentran los hemiterpenos con cinco átomos de carbono (una sola unidad de isopreno).
Figura 2: Clasificación de los Terpenoides o Terpenos
Muchos derivados isoprénicos tienen importancia ecológica porque los mismos juegan un papel importante en los mecanismos de supervivencia de las especies que los producen (Harborne & Tomás-Barberan, 1991; Wink et al., 2003). De igual manera, un número notable de isoprenoides son utilizados en la industria de medicamentos, bien directamente como principios activos o como materiales de partida para la obtención dichos principios activos (Newman et al., 2000).
O
OH
O
OH
O
Cumambrina BMyrcenona
COOH
Ácido (-)-Copálico
CH2OH
HO
Erythrodiol
MONOTERPENOS SESQUITERPENOS DITERPENOS TRITERPENOS

9
Biosíntesis de Terpenoides A partir del conocimiento de las disposiciones y reactividades de las
moléculas precursoras básicas, los químicos han podido predecir algunos esquemas generales de la biosíntesis de terpenoides. Hoy en día se acepta que la biosíntesis de los terpenoides se puede dividir en cuatro etapas generales, que son:
• Etapa 1: Síntesis del isopentenilpirofosfato (IPP), bien sea por la ruta del ácido mevalónico (MVA) o por la ruta del 1-desoxi-D-xilosa-5-fosfato (DOXP).
• Etapa 2: Isomerización del IPP a dimetilalilpirofosfato (DMAPP) y adición repetitiva de IPP y DMAPP.
• Etapa 3: Generación de unidades de prenilpirofosfato.
• Etapa 4: Modificaciones enzimáticas de los esqueletos.
En la primera ruta (Esquema 1), el compuesto clave en la biosíntesis de
terpenos es el ácido mevalónico, el cual se forma a partir de la acetil coenzima A (acetil-CoA), por una serie de reacciones análogas a la condensación de Claisen, a través de las cuales se genera, por reducción, el pirofosfato-dinucleótido de la nicotinamida de adenina (NADPH) (Kuzuyama, 2002). El paso siguiente es la conversión enzimática del ácido mevalónico en pirofosfato de isopentenilo (IPP), por medio de una serie de reacciones de fosforilación y descarboxilación, en las que interviene el pirofosfato de adenosina (Qureshi & Porter, 1981). Posteriormente se produce la isomerización del IPPP a dimetilalil pirofosfato (DMAPP). Ataques sucesivos de los diferentes pirofosfatos formados (IPP y DMAPP), dan origen a varios precursores, a partir de los cuales se forman todos los terpenos (Esquema 2).
Esquema 1: Formación del Isopentenil Pirofosfato (IPP) y del Dimetilalil Pirofosfato (DMAPP), a Partir de la Acetil-CoA, Vía Ácido Mevalónico.
H3C SCoA
O
SCoA
O
H3C
O
OH
OH
-O
O
SCoA
OH
-O
O O
OPP
OH
-O
OOPPH2C
H H
CH3
OPP
CH3
SCoA
O
H
SCoA
O
HB- B-
H+
H2O
NADPH
ATP
H+
IPPDMAPP

10
Esquema 2: Ruta Biosintética de Terpenos (Qureshi & Porter, 1981)
CH3
H3C OPP
CH3
H2CH H
CH3
CH3
CH3
OPP
CH3
OPP
CH3H3C
IPP
GPP
Monoterpenos
OPP
IPP
FPPSesquiterpenos
IPP
OPP
Diterpenos
Sesterterpenos
FPP FPP Triterpenos y Esteroides
GGPP GGPP Tetraterpenos ó Carotenos
PoliterpenosGGPP + nIPP
+
+
+-

11
La segunda ruta biosintética se inicia por reacción del piruvato y el gliceraldehido fosfato para formar el 1-desoxi-D-xilosa-5-fosfato, el cual a través de varios pasos (Esquema 3), conduce a la mezcla en equilibrio de isopentenil pirofosfato (IPP) y dimetilalil pirofosfato (DMAPP) (Dubey et al., 2003).
Esquema 3: Ruta Biosintetica via Piruvato y Gliceraldehído (Dubey et al, 2003)
Piruvato Gliceraldehido-3-P
DOXP sintesis
CO2
1-dioxi-xilosa-5-P
(DOXP)
Fosmidomicina
2-C-metil-D-eritritol-4-P (MEP)
(+CTP) CDP-ME sintesis
4-(CDP)-2-C-metil-D-eritritol (CDP-ME)
(+ATP) CDP-ME quinasa
4-(CDP)-2-C-metil-D-eritritol-2-P (CPD-ME2P)
(-CMP) MECP sintesis
2-C-metil-D-eritritol-2,4-ciclo-PP(MECP)
HMBPP sintesis
1-hidroxi-2-metil-2-(E)-butenil-4-PP (HMBPP)
Isopentenil Difosfato Dimetilalil Difosfat (IPP) (DMAPP)
HMBPP reducido
Tiamina Pyridoxal

12
Triterpenos y Esteroides Este grupo de terpenoides está constituido por numerosos compuestos,
derivados mayoritariamente del óxido de escualeno o en menor número del propio escualeno (Goodwin, 1981). Generalmente suelen estar funcionalizados en C-3, posición en la que predominan fundamentalmente grupos hidroxilos libres o esterificados, o grupos ceto (Xu et al., 2004). Entre los triterpenos y esteroides se encuentran compuestos de gran interés biológico y farmacológico como por ejemplo, los triterpenos antineoplásicos de las series del lupano, oleanano y ursano (Setzer & Setzer, 2003), los ecdyesteroides (Bathori & Pongracz, 2005; Dinan, 2001), las saponinas triterpénicas (Lacaille Dubois & Wagner, 1996), las bufadienolidas (Steyn & van Heerden, 1998), los glicósidos cardiotónicos (Deepak et al., 1996; Hauptman et al., 1999) y los fitoestrógenos (Bolego et al., 2003; Cos et al., 2003).
Los esteroides, tanto los naturales como los sintéticos, tienen una estructura cuyo esqueleto básico es el ciclopentanoperhidrofenantreno (Figura 3). Los mismos se dividen en tres grupos principales, según el número de átomos de carbono que posean:
• La serie de 21 carbonos incluye los corticoides y progestágenos, y su núcleo es el pregnano.
• La serie de 19 carbonos incluye los andrógenos y su núcleo es el androstano. • Los de 18 carbonos son los estrógenos y su núcleo es el estrano.
Figura 3: Esqueleto Fundamental de Compuestos Familias de los Esteroides
El precursor básico para la síntesis de esteroides es el colesterol; este puede
ser producido, en los mamíferos, por todos los órganos con capacidad esteroidogénica, excepto por la placenta, a pesar de que la misma posee la enzimas necesarias para su síntesis. El colesterol es transportado a través del torrente circulatorio por lipoproteínas de baja densidad (LDL), para finalmente ingresar a la célula gracias a la acción de un receptor de membrana. Debido a la importancia fisiológica que tienen los esteroides, en las últimas décadas, ha surgido la necesidad de buscar nuevas rutas sintéticas, para la producción masiva de los mismos.

13
Entre los esteroides de mayor interés fisiológico, destacan las hormonas esteroidales, las cuales se producen en células específicas de los testículos, la corteza adrenal, los ovarios y la placenta (Odel & Parker, 1984; Schneider, 2003). Los testículos serían los encargados de secretar, principalmente, testosterona (andrógenos); la corteza suprarenal produce la aldosterona, el cortisol y la DHEA (dehidroepiandrosterona); los ovarios generan los estrógenos que engloban el estradiol, la 4-androsteno-3,17-diona y la progesterona; y por último estaría la placenta que también secreta estradiol y progesterona, pero además produce otra sustancia, denominada estriol.
Los esteroides anabólicos son derivados sintéticos de la hormona masculina testosterona; estos compuestos promueven el crecimiento del músculo esquelético y aumentan la masa magra corporal. Su síntesis fue desarrollada por Ziegler et al., a partir de 1960 (Blickenstaff et al., 1974), y desde entonces han adquirido una gran importancia farmacológica y comercial, porque los mismos producen efectos similares a la testosterona. Otro grupo de esteroides sintéticos de gran importancia es el de los estrógenos que se utilizan en el control de la fertilidad (Alexander, 1995).
Los esteroides han sido ampliamente utilizados en medicina, sobre todo en pacientes con deficiencias hormonales (Dence, 1980). En las últimas décadas han adquirido una especial relevancia en el tratamiento de enfermedades que conllevan a pérdida progresiva de peso, como por ejemplo, el síndrome de imonudeficiencia adquirida (SIDA). Los esteroides han venido siendo utilizados desde hace más de dos décadas por atletas recreativos con el objetivo de mejorar sus cualidades físicas y estéticas, y también, de manera ilegal, por atletas profesionales para lograr mejores marcas personales y mayor capacidad en la realización de deportes de competencia (Hartgens & Kuipers, 2004.).
En lo que respecta a los triterpenos, cabe destacar que algunos de ellos son los principios activos de muchos adaptógenos y medicinas naturales, utilizadas desde la más remota antigüedad por varias civilizaciones. Ejemplos notables son el de las sapogeninas triterpénicas presentes en el “Ginseng” (Panax gingseng) (Attele et al., 1999; Shibata, 2001) o el de los principios antiinflamatorios de la Calendula officinalis (Akihisa et al., 1996; Della-Logia et al., 1994). Recientemente han adquirido notable importancia los derivados del ácido betulínico, entre los cuales se encuentran sustancias con potente capacidad inhibidora del virus HIV-1 (Evers et al., 1996; Kashiwada et al., 1996).
Las síntesis de varios triterpenos y esteroides ha sido abordada bajo diferentes estrategias. Una de ellas utiliza las tetralonas como potenciales sustratos o como intermediarios para la elaboración de familias completas de estos compuestos. A continuación nos referiremos a las tetralonas.

14
Tetralonas Las tetralonas son cetonas derivadas del naftaleno, cuya nomenclatura viene determinadas por su esqueleto decalínico; en el mismo, las posiciones 1, 4, 5 y 8 son idénticas, así como también las posiciones 2, 3, 6 y 7 (Figura 4).
Figura 4: Esqueleto Fundamental, Representativo de las Tetralonas. Estas cetonas poseen su anillo A completamente insaturado (aromático) y su anillo B completamente saturado; en este último anillo contienen un grupo carbonilo en las posiciones α ó β, siendo éste de significativa importancia, dado que el mismo las capacita como excelentes sustratos para la construcción del esqueleto de moléculas más complejas, mediante reacciones de condensación u otro tipo de reacciones específicas (Jung, 1976). De allí, que existan dos tipos de tetralonas, las 1-tetralonas (α-tetralona) y las 2-tetralonas (β-tetralonas), entre las cuales se encuentran diversas estructuras, dependiendo de los grupos funcionales que existan como sustituyentes en el anillo aromático (Figura 5).
Figura 5: Estructuras de las α-Tetralonas y β-Tetralonas Las tetralonas son intermediarios sintéticos de una considerable cantidad de compuestos con actividad biológica y de agentes quimioterapéuticos, entre los que se encuentran muchos productos naturales (Silveira et al., 2004). En el caso particular de las 1-tetralonas se manejan suficientes referencias, que indican que éstas han sido usadas en la síntesis de terpenoides y esteroides.
12
345
6
78
α
βα
α
α
β
β
β
O
1
R
α O2
R β
α-tetralona β-tetralona

15
Antecedentes del Problema a Investigar
Como ya mencionamos anteriormente, la obtención de tetralonas sustituidas surgió como una alternativa para la síntesis de terpenoides, esteroides y sus derivados. El primer reporte relativo al uso de tetralonas como intermediarios sintéticos se remonta a 1943, cuando Sir Robert Robinson abordó la síntesis de la androstenediona (Martin & Robinson, 1943). A partir de este momento, el uso de las tetralonas, como material de partida en síntesis, se generalizó; prueba de ello es que en la literatura se pueden encontrar muchas referencias de artículos publicados en las décadas del cincuenta y del sesenta, en los cuales, las tetralonas juegan un papel significativo (Doyle et al., 1965; Conforth et al., 1955).
La relevancia de las tetralonas en este aspecto se sigue conservando hasta nuestros días, como lo reflejan varios trabajos de gran impacto en síntesis orgánica. Entre ellos destaca la síntesis de la hormona vegetal ácido abscísico (ABA), la cual fue preparada a partir del 2-metil-1-naftol, retomando la idea planteada en años atrás por Sir R. Robinson (Nyangulu et al., 2006). Otro ejemplo se encuentra en el uso de la 6-metoxi-2,5-dimetil-1-tetralona como producto de partida para la ruta sintética de las clovanonas (cetonas tricíclicas óptimamente activas), utilizando también el método establecido por Sir R. Robinson (Paul & Mukherjee, 2003).
Ante la importancia que tienen como sustratos sintéticos las tetralonas, se consideró interesante explorar la posibilidad de desarrollar un nuevo método para la preparación de la 6-metoxi-5-metil-α-tetralona y la 6-metoxi-2,5-dimetil-α-tetralona. Se escogieron estas dos tetralonas, porque las mismas son la base de partida del método sintético de Sir R. Robinson. De hecho la primera de estas tetralonas fue preparada por Martin & Robinson (1943) mediante la hidrogenación catalítica de un derivado del naftaleno (1-metil-2-naftol) [1] con níquel Raney, en etanol, a 100°C y 100 atm. de presión. Esta reacción condujo a la tetralina [2], la cual fue luego tratada con sulfato de metilo en hidróxido de sodio y agua, para dar el derivado metilado [3]. La oxidación de [3] con trióxido de cromo en ácido acético y agua, condujo a la 6-Metoxi-5-metil-α-tetralona) [4] (Esquema 3). _______ ( ) La 6-Metoxi-5-metil-α-tetralona [4] debería nombrarse más correctamente como 3-Metoxi-4-metil-1,3,5(10)-trien-decalin-9-ona, nombre éste que se utilizará en la descripción de la síntesis planteada en este trabajo.
Esquema 3: Síntesis de Sir R. Robinson de la 6-Metoxi-5-metil-α-tetralona
[1] [2] [3] [4]
H2/Ni Raney
100ºC/100 atm.Etanol
Me2SO4
NaOH/H2O80ºC
CrO3
AcOH/H2O
CH3
HO
CH3
HO
CH3
CH3O
CH3
CH3O
O

16
Unos años más tarde, M. J. Temple Robinson (1957), utilizando como material de partida la tetralina [3], preparó la 6-metoxi-2,5-dimetil-α-tetralona. Para ello, generó por el método de Sir R. Robinson la 6-metoxi-5-metil-α-tetralona [4] a partir de la tetralina [3], y luego, utilizando de manera consecutiva metóxido de sodio y carbonato de metilo en atmósfera de nitrógeno, seguido de yoduro de metilo en metanol, logró funcionalizar la posición α- al carbonilo y obtener el compuesto [5]. Por último, la eliminación del grupo carbometoxi con ácido clorhídrico concentrado en ácido acético/agua, condujo a la 6-metoxi-2,5-dimetil-α-tetralona [6] (Esquema 4).
Esquema 4: Síntesis de M. J. P. Robinson de la 6-Metoxi-2,5-dimetil-α-tetralona En el primer paso de la síntesis de la 6-metoxi-5-metil-α-tetralona [4], se usan
condiciones de hidrogenación enérgicas, bajo las cuales se generan compuestos colaterales no deseados, y si no se controla adecuadamente la alcalinidad del medio se obtiene una mayor proporción de alcohol que de fenol, es decir se hidrogena el anillo aromático no deseado [esto ocurre a pH básicos (Adkins & Krserk, 1948)]. (Esquema 5). Por otro lado, es conocido el peligro que se corre al trabajar en condiciones tan altas de presión de hidrógeno, y generalmente, no se cuenta fácilmente con reactores aptos para presiones altas en laboratorios de investigación.
Esquema 5: Hidrogenación Parcial del β-Naftol (Adkins & Krserk, 1948)
En razón a lo anteriormente expuesto, es evidente que el método de Sir Rober Robinson presenta limitaciones en su primer paso, y el mismo no sería recomendable para desarrollarlo a escala industrial o semiindustrial. De ahí nuestro intento de abordar la síntesis de las tetralonas [4] y [6] (6-metoxi-5-metil-α-tetralona y 6-metoxi-2,5-dimetil-α-tetralona), a través de otra ruta.
OH OH OH
+H2/Ni Raney
90ºC/275 atm.Ph ≤ 7
(86%) (7%)
CH3
CH3O
O
[4]
[3] NaOMe/Me2CO3
reflujo bajo N2
ICH3
MeOH
CH3
CH3O
O
CH3
COOCH3
CH3
CH3O
O
CH3HCl conc.
AcOH/H2O
[5] [6]

17
En conexión con lo anteriormente expuesto se planteó la siguiente hipótesis:
Hipótesis
Es posible ensayar una ruta, por medio de reacciones sencillas distintas a las hasta ahora reportadas, para la síntesis de las tetralonas 6-metoxi-5-metil-α-tetralona y 6-metoxi-2,5-dimetil-α-tetralona, en la cual se reduzcan las condiciones drásticas de los procesos de reacción anteriormente establecidos, elevando así los niveles de obtención de los productos, en términos de manejo y porcentaje de rendimientos.
En concordancia con la anterior hipótesis se formularon los siguientes objetivos:
Objetivos
Objetivo General:
• Realizar la síntesis de las tetralonas: 6-metoxi-5-metil-α-tetralona [4] y 6-metoxi-2,5-dimetil-α-tetralona [6], a través de una ruta alternativa, que reduzca las condiciones drásticas empleadas en los procesos que hasta ahora se han descritos en la literatura, para la obtención de estas tetralonas.
Objetivos Específicos:
• Caracterizar cada uno de los productos intermediarios que se obtengan en las diferentes etapas de la ruta planteada, mediante técnicas espectroscópicas básicas (IR, UV, RMN-1H, RMN-13C y EM).
• Asignar de manera inequívoca todas las señales en los espectros de RMN-1H y de RMN-13C de los productos intermediarios y finales que se obtengan en el proceso sintético.
• Determinar las propiedades físicas de cada uno de los productos que se generen en la síntesis.
• Establecer los porcentajes de rendimiento que se obtengan a los largo de la síntesis.

18
Resultados y Discusión
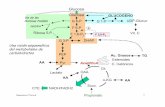
Con la premisa en mente de abordar una síntesis alternativa que permitiera llegar a la 6-metoxi-5-metil-α-tetralona [4] y a la 6-metoxi-2,5-dimetil-α-tetralona [6], a través de procesos viables, eficientes y menos drásticos que los reportados por Martin & Robinson (1943), se planteó el siguiente esquema de reacciones (Esquema 6):
Esquema 6: Ruta Planteada para la Síntesis de la 6-Metoxi-5-metil-α-tetralona
y la 6-Metoxi-2,5-dimetil-α-tetralona.
Al llevar a cabo el proceso sintético según el Esquema 6, se observó que en la transformación de la mezcla [11]/[12] en el ácido carboxílico [13] (paso 3º), la reacción ocurría con bajo rendimiento (< 20%) para generar un único producto. En el EM de este producto se detectó un ión molecular de abundancia 100 % (pico base) a m/z: 162; la magnitud molecular de este ión no correspondía a la del ácido carboxílico esperado [13], pero en cambio concordaba con la del fenol [2]. En razón a ello, se supuso que durante la transformación se generaba el ácido carboxílico [13] y que “in situ” el mismo se transformaba en el fenol [2], mediante un proceso de descarboxilación, favorecido por la estabilidad que confiere la aromatización del anillo A en la transformación [13] [2] . Ante este hecho, el cual simplifica en un paso el esquema sintético, centramos nuestra atención en la optimización de las condiciones de esta reacción para aumentar su rendimiento. De esta manera la ruta se replanteó de acuerdo al Esquema 7.
O
O
O
H3C
Cl
O
CH3
COOCH3
CH3
O
COOCH2
CH3
O
CH3
O
O
O
H3C
+
40% 60%
Mezcla de distribución comercial
+Ac. p-Toluensulfónico
Tolueno (Δ, N2)
+
COOCH2
CH3
O
CH3COOCH3
CH3
O
COOH
CH3
O
H3C
HO
H3C
CH3O
H3C
CH3O
O
DDQdioxano (Δ, N2)
DMSO/t-BuOK
(Δ, N2)
CuO/quinolina
(CH3)3CCN (50º, N2)
(CH3)2SO4/K2CO3
acetona (Δ, N2)
H3C
CH3O
O
CH3CrO3/Ac. Acético
H2O
CH3I/KH
THF (25º, N2)
[7]
[8]
[9] [10]
[11] [12][13]
[2] [4] [6][3]

19
Esquema 7: Ruta Propuesta para la Síntesis de la 6-Metoxi-5- metil-α-tetralona y la 6-Metoxi-2,5-dimetil-α-tetralona.
A. SÍNTESIS DE LA 10-CARBOMETOXI-4-METIL-DECALIN-4-EN-3-ONA [9] Y DE LA
10-CARBOETOXI-4-METIL-DECALIN-4-EN-3-ONA [10]
En un todo de acuerdo con el Esquema 7, el proceso sintético se inició a partir de una mezcla comercial de las ciclohexanonas: 2-carbometoxi-ciclohexanona y 2-carboetoxi-ciclohexanona (2:3) [7] . Al reflujar esta mezcla bajo atmósfera de nitrógeno durante 24 horas con 5-cloro-3-pentanona [8], en tolueno seco, y en presencia de cantidades catalíticas de ácido p-toluensulfónico, se obtuvo con rendimiento del 72% un aceite amarillo, cuya capa fina mostraba la formación de una mezcla de las enonas α,β-insaturadas [9] y [10]. Esta mezcla fue separada de los productos minoritarios de la reacción y de inmediato sometida a estudios espectrales, los cuales, como veremos más adelante, demostraron que se trataba de una mezcla (2:3) de las decalin-4-en-3-onas [9] y [10]. ______________
( ) El empleo de esta mezcla como material de partida, es justificable en razón de que la misma es mucho más barata que los productos puros y porque en el tercer paso se generaría, a partir de la mezcla que se obtendría en esta primera reacción, un único producto, con el cual se continuaría el proceso.
O
O
O
H3C
Cl
O
CH3
COOCH3
CH3
O
COOCH2
CH3
O
CH3
O
O
O
H3C
+
40% 60%
Mezcla de distribución comercial
+Ac. p-Toluensulfónico
Tolueno (Δ, N2)
+
COOCH2
CH3
O
CH3COOCH3
CH3
O
H3C
HO
H3C
CH3O
H3C
CH3O
O
DDQdioxano (Δ, N2)
DMSO/t-BuOK
(Δ, N2)
(CH3)2SO4/K2CO3
acetona (Δ, N2)
H3C
CH3O
O
CH3CrO3/Ac. Acético
H2O
CH3I/KH
THF (25º, N2)
[7]
[8][9] [10]
[11] [12][2]
[4] [6][3]

20
Es evidente que este primer paso (Esquema 8), procedió a través de una reacción tipo Michael, favorecida en este caso por un buen grupo saliente (-Cl) para generar las dicetonas [7a] y [7b] , las cuales en medio ácido se encuentran en equilibrio con los respectivos enoles [7c] y [7d]. Estos enoles experimentan luego una condensación intramolecular (Anelación de Robinson) (Gawley, 1976), que implica un ataque nucleofílico de los enolatos de la cadena alquílica sobre los carbonilos cetónicos de las respectiva ciclohexanonas, con la consiguiente generación en cada sustrato de un nuevo anillo, para formar los alcoholes decalínicos [7e] y [7f]; por último, estos alcoholes, en el medio ácido, experimentan una eliminación de agua, transformándose en las enonas [9] y [10].
Esquema 8: Mecanismo de la Anelación de Robinson que Conduce a la Formación de la 10-Carbometoxi-4-metil-decalin-4-en-3-ona [9]
y la 10-Carboetoxi-4-metil-decalin-4-en-3-ona [10]
( ) El mecanismo se inicia mediante la captación de un protón donado por el catalizador (ácido p-toluensulfonico), por el carbonilo de la ciclohexanona y la posterior eliminación de un hidrógeno para formar los respectivos enoles. El hidrógeno que se elimina es lógicamente H-10, el cual es el más acídico por encontrarse situado sobre un carbono α a dos carbonilos.
O
O
O
HH3C
++
Adición de Michael
+
O
COOCH2CH3
OHO
COOCH3
OH+
O
COOCH3
O
H
O
COOCH2CH3
O
H
+ +
Anelación de Robinson
+H +H
+H +H +H
O
COOCH3
O
COOCH2CH3
O
COOCH2CH3
H OH..
..
+H+H
....
O
COOCH3
H OH
O
O
O
H
H3CO
O
H3C
HO..
..HO
H3CO
O
..
..
+H
[7]
[7b][7a]
[8]
[7c] [7d]
[7e] [7f] [9] [10]
12
3
45
6'7
8910
10
11
12 13 13'12'
11'
10'9' 8'
7'
65'
4'
3'
2'
1'
14'
Cl
O

21
Debido a que en la reacción realizada se produce agua, era indispensable que el solvente usado (tolueno) estuviera suficientemente seco, ya que de lo contrario, el agua desplazaría el equilibrio hacia los productos de partida. Por otro lado, la “cuantificación” del agua eliminada permitía el seguimiento de la reacción ya que la misma podía detenerse en el momento en que dejaba de recogerse agua en la trampa de Dean-Stark.
Los productos de la reacción fueron separados por cromatografía en columna de gel de sílice (ver parte experimental), y en este caso se lograron obtener en forma pura ambas enonas [9] y [10], las cuales fueron caracterizadas por sus datos espectrales. Así pues, sus espectros IR (Fig. 9A y 10A; Tablas 9A y 10A), muestran bandas típicas de una cetona α,β-insaturada [νmax : 1.727 cm-1 (C=O) y νmax : 1.618 cm-1 (C=C)] y de un grupo éster [νmax : 1.667 cm-1 (C=O) y νmax : 1.230, 1.195 y 1.172 cm.-1 (C-O-C)] . ____________ ( ) Los valores de νmax indicados corresponden al espectro de la enona [9] (Fig. 9A; Tabla 9A)
Figura 9A: Espectro Infrarrojo en Película de KBr, de la 10-Carbometoxi-4-Metil-Decalin-4-en-3-ona [9]
Tabla 9A: Bandas de Absorción Significativas en el Espectro Infrarrojo (KBr), de la 10-Carbometoxi-4-Metil-Decalin-4-en-3-ona [9]
υmax (cm- 1) 2.935-2.864 1.727 1.667 1.618 1.452 1.230; 1.195 y 1.172
Asignación C-H C=O C=O C=C C-H C-O-C
2.935-2.864C-H
1.727-1.667C=O
1.618C=C
1.452C-H
1.230-1.175C-O-C
O
CH3
O OCH31
2
3
45
6
7
8
9
10
11
12
13

22
Figura 10A: Espectro Infrarrojo en Película de KBr de la 10-Carboetoxi-4-Metil-Decalin-4-en-3-ona [10]
Tabla 10A: Bandas de Absorción Significativas en el Espectro Infrarrojo (KBr), de la 10-Carboetoxi-4-Metil-Decalin-4-en-3-ona [10]
υmax (cm- 1) 2.934-2.862 1.724 1.669 1.618 1.452 1.257; 1.227 y 1.183
Asignación C-H C=O C=O C=C C-H C-O-C
En los espectros de RMN-1H (Fig. 9B y 10B; Tablas 9B y 10B) de ambas
enonas se observa, de manera indiferenciada, el singulete típico de un metilo sobre un doble enlace a δH: 1,77 para H-11 y δH: 1,77 para H-11’ (en [9] y [10]); la principal diferencia entre ambos espectros, como es lógico, reside en las señales propias de cada éster. Así pues, en el espectro de [9] se distingue un singulete propio de un grupo metoxilo a δH: 3,70 [-OCH3; (H-13)], mientras que en el de [10] se aprecia un cuartete a δH: 4,14 [-CH2-O- (H-13’)], el cual se encuentra acoplado (J ≅ 7 Hz) a un triplete que resuena a δH: 1,20 [-CH3 (H-14’)], caracterizando a un grupo etóxido.
En los espectros de RMN-13C (Fig. 9C y 10C; Tablas 9C y 10C) se aprecian diferencias más acusadas, ya que en el de [9] se observan trece (13) picos, mientras que en el de [10] se detectan catorce (14). Esta diferencia se debe a que en el espectro de [9] aparece la señal del carbono metoxílico [δC: 52,3 (C-13); -OCH3], mientras que en el de [10], esta señal es reemplazada por las dos picos de los carbonos del grupo etóxido [δC: 61,1 (C-13’); –O-CH2- y δC: 14,1(C-14’); -CH3].
2.934-2.862C-H
1.724-1.669C=O
1.618C=C 1.452
C-H
1.252-1.183C-O-C 11
O
CH3
O O CH31'
2
3
45
6
7
8
9
10
12
'
'
''
'
'
'
''
'
13'
14'
'

23
Al comparar los espectros de ambas enonas se observan similitudes en los valores de los desplazamientos químicos de las señales de los restantes carbonos, siendo particularmente significativas las correspondientes a los picos asignables a los metilos vinílicos [δC: 10,9 (C-11) y δC: 10,8 (C-11’)], a los carbonos del doble enlace [δC: 131,0 (C-4) y δC: 130,8 (C-4’); δC: 155,7 (C-5) y δC: 155,9 (C-5’)], a los carbonilos cetónicos [δC: 198,3 (C-3) y δC: 198,3 (C-3’)] y a los carbonilos de los ésteres [δC: 174,7 (C-12) y δC: 174,0 (C-12’)].
Por último, en los EM de estas enonas [Fig. 9D y 10D; Tablas 9D y 10D], se observa que los patrones de fragmentación son prácticamente idénticos, pero se aprecia la diferencia en los iones moleculares {m/z: 222 para [9] y m/z: 236 para [10]}. En los Esquemas 9D y 10D se muestran los patrones de fragmentación de estos ésteres.
O
CH3
O oCH3
11
10
1312
7
9
4
2 8
6
5
3
1
131,0
29,5
198,3
26,1
38,7
155,7
174,7
34,2
34,4
22,9
10,9
49,5
52,3
H-1
H-8
H-2
H-11
H-6
H-13
H-7
δ: 1,99 (m)
δ: 1,22 (m)
δ: 2,30 (m)
δ: 3,70 (s)
δ: 1,22 (m)
δ: 2,19 (m)
δ: 1,77 (s)
δ: 2,00 (m)H-9
H-9'δ: 1,98 (m)
O
CH3
O o CH3
11'
10'
13'12'
7'
9'
4'
2' 8'
6'
5'
3'
1'
130,8
29,5
198,3
26,0
38,7
155,9
174,0
34,1
34,4
22,8
10,8
49,4
61,1
H-1'
H-8'
H-2'
H-11'
H-6'
H-13'
H-7'
δ: 2,06 (m)
δ: 1,21 (m)
δ: 2,30 (m)
δ: 4,14 (t)
δ: 1,21 (m)
δ: 2,20 (m)
δ: 1,78 (s)
δ: 1,20 (c)H-14'J(Hz): 7
J(Hz): 7
14'14,1
10-Carbometoxi-4-Metil-Decalin-4-en-3-ona [9]
10-Carboetoxi-4-Metil- Decalin-4-en-3-ona [10]

24
CDCl3
H-13H-11,11’
CDCl3
H-13H-11,11’
Figura 9B: Espectro de RMN-1H [CDCl3, 400 MHz], de la 10-Carbometoxi-4-Metil-Decalin-4-en-3-ona [9]
Tabla 9B: Desplazamientos Químicos (δ) en el Espectro de RMN-1H [(CDCl3), 400 MHz], de la 10-Carbometoxi-4-Metil-Decalin-4-en-3-ona [9]
H H-1 H-2 H-6 H-7 H-8 H-9 H-11 H-13
δ (ppm) ≅ 1,99 ≅ 2,19 ≅ 2,30 ≅ 1,22 ≅ 1,22 ≅ 2,00 1,77 3,70
m m m m m m m s s
J (Hz) - - - - - - - -
O
CH3
O OCH31
2
3
45
6
7
8
9
10
11
12
13

25
Figura 10B: Espectro de RMN-1H [CDCl3, 400 MHz], de la 10-Carboetoxi-4-Metil-Decalin-4-en-3-ona [10]
Tabla 10B: Desplazamientos Químicos (δ) en el Espectro de RMN-1H [(CDCl3), 400 MHz], de la 10-Carboetoxi-4-Metil-Decalin-4-en-3-ona [10]
H H-1’ H-2’ H-6’ H-7’ H-8’ H-9’ H-11’ H-13’ H-14’
δ (ppm) ≅ 2,06 ≅ 2,20 ≅ 2,30 ≅ 1,21 ≅ 1,21 ≅ 1,98 1,78 4,14 1,20
m m m m m m m s c t
J (Hz) - - - - - - - 7 7
H-14’
H-11’
H-13’
11
O
CH3
O O CH31'
2
3
45
6
7
8
9
10
12
'
'
''
'
'
'
''
'
13'
14'
'

26
Figura 9C: Espectro de RMN-13C [CDCl3, 100 MHz], de la 10-Carbometoxi-4-Metil-Decalin-4-en-3-ona [9]
Tabla 9C: Desplazamientos Químicos (δ) en el Espectro de RMN-13C [(CDCl3), 100 MHz], de la 10-Carbometoxi-4-Metil-Decalin-4-en-3-ona [9]
C C-1 C-2 C-3 C-4 C-5 C-6 C-7
δ (ppm) 34,4 34,2 198,3 131,0 155,7 261 29,5
Tipo >CH2 >CH2 >C=O >C= =C< >CH2 >CH2
C C-8 C-9 C-10 C-11 C-12 C-13
δ (ppm) 22,9 38,7 49,5 10,9 174,7 52,3
Tipo >CH2 >CH2 >C< -CH3 -O-C=O O-CH3
Valores δ con
referencia al TMSi
C-3 C-12C-5
C-4
C-13
C-10
C-9
C-1C-2
C-7
C-6
C-8
C-11
C-3 C-12C-5
C-4
C-13
C-10
C-9
C-1C-2
C-7
C-6
C-8
C-11
O
CH3
O OCH31
2
3
45
6
7
8
9
10
11
12
13

27
Figura 10C: Espectro de RMN-13C [CDCl3, 100 MHz], de la 10-Carboetoxi-4-Metil-Decalin-4-en-3-ona [10]
Tabla 10C: Desplazamientos Químicos (δ) en el Espectro de RMN-13C [(CDCl3), 100 MHz], de la 10-Carboetoxi-4-Metil-Decalin-4-en-3-ona [10]
C C-1’ C-2’ C-3’ C-4’ C-5’ C-6’ C-7’
δ (ppm) 34,4 34,1 198,2 130,8 155,6 26,0 29,5
Tipo >CH2 >CH2 >C=O >C= =C< >CH2 >CH2
C C-8’ C-9’ C-10’ C-11’ C-12’ C-13’ C-14’
δ (ppm) 22,8 38,7 49,4 10,8 174,0 61,1 14,1
Tipo >CH2 >CH2 >C< -CH3 -O-C=O -O-CH2 -CH3
C-3’C-12’
C-5’ C-4’
C-13’
C-10’
C-9’
C-1’C-2’
C-7’C-6’
C-8’
C-10’
C-11’
C-3’C-12’
C-5’ C-4’
C-13’
C-10’
C-9’
C-1’C-2’
C-7’C-6’
C-8’
C-14’
C-11’
C-3’C-12’
C-5’ C-4’
C-13’
C-10’
C-9’
C-1’C-2’
C-7’C-6’
C-8’
C-10’
C-11’
C-3’C-12’
C-5’ C-4’
C-13’
C-10’
C-9’
C-1’C-2’
C-7’C-6’
C-8’
C-14’
C-11’
11
O
CH3
O O CH31'
2
3
45
6
7
8
9
10
12
'
'
''
'
'
'
''
'
13'
14'
'

28
Figura 9D: Espectro de Masas (IE, 70 eV.) de la 10-Carbometoxi-4-Metil-Decalin-4-en-3-ona [9]
Tabla 9D: Fragmentos (m/z) más Significativos en el Espectro de Masas (IE, 70 eV.) de la 10-Carbometoxi-4-Metil-Decalin-4-en-3-ona [9]
m/z 223 207 192 163 147 135
Abundancia Relativa (%) 63 10 9 100 15 29
m/z 121 107 91 79 77 67
Abundancia Relativa (%) 55 27 42 23 21 10
O
CH3
O OCH31
2
3
45
6
7
8
9
10
11
12
13

29
Esquema 9D: Interpretación del Patrón de Fragmentación Observado en el Espectro de Masas (IE, 70 eV.) de la 10-Carbometoxi-4-Metil-
Decalin-4-en-3-ona [9]
O O
CH3
O
....O O
CH3
CH3
O
..
+ CH3.
m/z: 207m/z: 222 [M ]+
C14H20O3
..O OCH2
CH3
O
+
+
+
+
CH3
O
CH2OO ..
+
m/z: 207m/z: 193
CH3
O
.++O C O CH3..
+
m/z: 163(Pico base)
O O
CH3
O
..
.+
+ .. CH3
m/z: 207
+
+ O C O
..
..O
CH3 H3C
+ O C..+ ..
m/z: 135
+
H3C
++
..+ CH2
m/z: 121
H3Cm/z: 121
CH3
..+
++
..
CH3
+
m/z: 107 m/z: 107
+
..
+.H
m/z: 221
..O O
CH3
O
CH2
+
[ ]
m/z: 163(Pico base)
.
+ CH2..+
+
.. CH2 +
+
.. CH2 +

30
Figura 10D: Espectro de Masas (IE, 70 eV.) de la 10-Carboetoxi-4-Metil-Decalin-4-en-3-ona [10]
Tabla 10D: Fragmentos (m/z) más Significativos en el Espectro de Masas (IE, 70 eV.) de la 10-Carboetoxi-4-Metil-Decalin-4-en-3-ona [10]
m/z 237 221 207 193 163 147
Abundancia Relativa (%) 51 10 16 10 100 18
m/z 135 121 107 91 73 55
Abundancia Relativa (%) 42 50 25 40 49 9
11
O
CH3
O O CH31'
2
3
45
6
7
8
9
10
12
'
'
''
'
'
'
''
'
13'
14'
'

31
Esquema 10D: Interpretación del Patrón de Fragmentación Observado en el Espectro de Masas (IE, 70 eV.) de la 10-Carboetoxi-4-Metil-
Decalin-4-en-3-ona [10]
O OCH2
CH3
O
.....O O CH3
CH3
O
..
+ CH3.
m/z: 221m/z: 236 [M ]+
C14H20O3
..O OCH2
CH3
O
+ CH2..+
+
+
+
+
CH3
O
CH2OO ..
+
m/z: 207m/z: 193
..+
O OCH2
CH3
O
m/z: 179
CH3
O
.+
+O C OCH3
..+
m/z: 163(Pico base)
O O
CH3
O
..
..+
+ .. CH2CH3
+...O O
CH3
O
..
.CH2+
m/z: 207 m/z: 207
+ +
CH3
O
+ O C O.. ....
O C O+ [ ].
m/z: 163 m/z: 163
..
..
..O
CH3H3C
+O C..+ ..
m/z: 135
+
H3C
++
..+ CH2
m/z: 121H3C
m/z: 121 CH3
..+
+ +..
CH3
+
m/z: 107 m/z: 107
+
+ CH2..+
+
.. CH2 ++ CH2..+

32
B. SÍNTESIS DE LA 10-CARBOMETOXI-4-METIL-DECALIN-1,4-DIEN-3-ONA [11] Y DE
LA 10-CARBOETOXI-4-METIL-DECALIN-1,4-DIEN-3-ONA [12] El segundo paso del esquema sintético contemplaba la formación del nuevo doble enlace Δ1,2, α- a la cetona, mediante la deshidrogenación de la mezcla de las 10-carboxi-3-enonas [9] y [10]; de esta manera se generaría, en el anillo A, el sistema dienónico precursor del núcleo aromático. Esta reacción se desarrolló utilizando como reactivo DDQ (2,3-dicloro-5,6-diciano-1,4-benzoquinona) en exceso y como solvente dioxano seco , a reflujo y bajo atmósfera de nitrógeno, según la metodología reportada por Banerjee & Hurtado (1985).
La reacción transcurre a través de un proceso radicalario que puede ser concertado o por etapas (Walker & Hiebert, 1967). En este último caso (Esquema 9), el proceso se iniciaría con la abstracción del hidrógeno α- al carbonilo cetónico, (para generar un radical estabilizado por resonancia), seguida de la abstracción del hidrógeno β- a dicho grupo y la posterior estabilización del biradical generado, mediante la formación del doble enlace Δ1,2
Esquema 9: Mecanismo de la Deshidrogenación con DDQ que Conduce a la Formación de la 10-Carbometoxi-4-metil-decalin-1,4-dien-3-ona [11]
y la 10-Carboetoxi-4-metil-decalin-1,4-dien-3-ona [12] ________
( ) El exceso de DDQ (iniciador de radicales) es fundamental para optimizar la eficacia de esta reacción. Es bien sabido que la presencia de agua en un proceso radicalario altamente energético, puede disminuir el rendimiento del proceso; por ello la reacción tuvo que hacerse en condiciones completamente libres de humedad (solvente seco y atmósfera inerte).
Cl
Cl CN
CN
O
O
Cl
Cl CN
CN
O
O
.
.
. .Cl
Cl CN
CN
O
OH
O
O
H
RO O
O
ROH
O
O
RO
..
O
O
RO
R: CH3
R: CH2 CH3
R: CH2 CH3
R: CH3
[9]
[10]
[11]
[12]
DDQ
6
13
12
11
109 8
754
3
21
13' 14'

33
El seguimiento de la reacción por cromatografía de capa fina fue difícil, dado que los productos de partida y de reacción poseen Rf similares. En consecuencia, la evolución del proceso se monitorió por CG-EM, observándose que en los EM los picos a m/z: 220 (ión molecular de [11]) y m/z: 234 (ión molecular de [12]) alcanzaban su máxima intensidad a las 72 horas. Como producto de reacción se obtuvo un aceite de color marrón claro, en el cual no se detectó la presencia de producto de partida, pero sí la de DDQ como contaminante. Este aceite se purificó mediante percolado a través de una columna de gel de sílice eluida con mezclas hexano-éter (17:3), obteniéndose así, con un rendimiento del 85%, un aceite de color amarillo-naranja libre de DDQ. Los datos espectrales de este aceite indicaron que el mismo era una mezcla (≅ 2:3) de [11] y [12]. Así pues, su espectro IR (Fig. 11/12A; Tabla 11/12A), muestra bandas típicas de una cetona doblemente conjugada [νmax : 1.660 cm-1 (C=O)], de dobles enlaces [νmax : 3.080 y 835 cm-1 (=C-H); νmax : 1.635 cm-1 (C=C)] y de ésteres [νmax : 1.731 cm-1 y 1.731 cm-1 (C=O); νmax : 1.214 y 1.021 cm-1 (C-O-C)] (Bellamy, 1975).
Figura 11/12A: Espectro Infrarrojo en Película de KBr, de la Mezcla de 10-Carbometoxi-4-Metil-Decalin-1,4-dien-3-ona [11] y
10-Carboetoxi-4-Metil-Decalin-1,4-dien-3-ona [12]
Tabla 11/12A: Bandas de Absorción Significativas en el Espectro Infrarrojo (KBr), de la Mezcla de 10-Carbometoxi-4-Metil-Decalin-1,4-dien-3-ona [11] y 10-Carboetoxi-4-Metil-Decalin-1,4-dien-3-ona [12]
υmax (cm- 1) 3.080 2.937-2.862 1.731 1.660 1.635 1.214 y 1.021 835
Asignación =C-H C-H C=O C=O C=C C-O-C =C-H
3.080=C-H
2.937-2.862C-H
1.731-1.660C=O
1.635-1.611C=C
1.214-1.021C-O-C
+
[11] [12]
O
CH3
O OCH31
2
3
45
6
7
8910
11
12
13
O
CH3
O O CH31'
2'
3'
4'
5'
6'7'
8'9'10'
11'
12' 13'
14'

34
Cuando se compara el espectro de RMN-1H de la mezcla de ésteres [11] y [12] (Fig. 11/12B; Tabla 11/12B), con los de los productos de partida ([9] y [10]), se nota que el cambio más significativo es la aparición de un par de dobletes mutuamente acoplados (J ≅ 10 Hz) a δH: 6,62 (H-1) y 6,61 (H-1’) y δH: 6,25 (H-2 y H-2’), asignables a los protones del nuevo doble enlace generado. Por lo demás, este espectro sigue mostrando las señales propias del metilo vinílico sobre C-4 [δH: 1,88 (H-11 y H-11’)] y las de los restos de la parte alcohólica de cada éster [δH: 3,62 (H-13), -OCH3, en [11]; δH: 4,09 (H-13’) –CH2O- y δH: 1,17 (H-14’),-CH3, en [12]]. Las señales de los protones del anillo alifático, con excepción de las de H-6 y H-6’, no sufrieron modificaciones significativas, y continúan observándose como multipletes complejos.
El espectro de RMN-13C (Fig. 11/12C; Tabla 11/12C), también pone en evidencia que la mezcla obtenida en la reacción, corresponde a las dienonas [11] y [12]. Cuando se compara dicho espectro con los de [9] y [10], es particularmente notable el reemplazo de las señales de los carbonos metilénicos (C-1 y C-2), por dos nuevos picos, propios de metinos olefínicos [δC: 154,3 (C-1 y C-1’) y δC: 128,9(C-2 y C-2’)]. Otro cambio notable es el desplazamiento hacia campos más altos de los picos atribuidos a los carbonilos cetónicos [δC: 170,7 (C-3 y C-3’)] y también a los ésteres [δC: 185,9 (C-12 y C-12’)]; en los primeros, este efecto es producido por la conjugación del carbonilo cetónico al nuevo doble enlace y en los segundos por el efecto de protección que produce la nube de electrones π de este nuevo doble enlace α al carbonilo de los ésteres (Kalinowski et al., 1988).
Por último en el EM (Fig. 11/12D; Tabla 11/12D) se aprecian dos iones moleculares de similar intensidad a m/z: 220 y m/z: 234, lo cual demuestra que el producto obtenido es una mezcla de las dienonas [11] y [12]. La interpretación del patrón de fragmentación de este espectro de masas se representa en el Esquema 11/12D.
H-9/9'δ: 1,95 (m)
δ: 1,88 (s)
δ: 6,25 (d)
δ: 1,45 (m)
δ: 2,61 (dd)
δ: 1,45 (m)
δ: 6,17 (d)
H-7/7'
H-6/6'
H-11/11'
H-2/2'
H-8/8'
H-1/1'
53,1
10,3
26,8
154,3
128,9
185,9
147,2
37,4
28,8
170,7
30,0
130,7
1/1'
3/3'
5/5'
6/6'
8/8'2/2'
4/4'
9/9'
7/7'
12/12'
10/10'
O
CH3
O oR
J(Hz): 10
J(Hz): 10
J(Hz): 13
11/11'
R11 = CH3
R12 = CH2-CH3
13
13' 14'
61,7
52,8 14,1
H-13
H-14'
H-13'δ: 1,17 (t)
δ: 4,09 (c)
δ: 3,62 (s)
J(Hz): 7
J(Hz): 7

35
Figura 11/12B: Espectro de RMN-1H [CDCl3, 400 MHz], de la Mezcla de 10-Carbometoxi-4-Metil-Decalin-1,4-dien-3-ona [11] y
10-Carboetoxi-4-Metil-Decalin-1,4-dien-3-ona [12]
Tabla 11/12B: Desplazamientos Químicos (δ) en el Espectro de RMN-1H [(CDCl3), 400 MHz], de la Mezcla de 10-Carbometoxi-4-Metil-Decalin-1,4-
dien-3-ona [11] y 10-Carboetoxi-4-Metil-Decalin-1,4-dien-3-ona [12]
H H-1/1’ H-2/2’ H-6/6’ H-7/7’ H-8/8’ H-9/9’ H-11/11’ H-13 H-13’ H-14’
δ 6,17 6,25 2,61 1,45 1,45 1,95 1,88 3,62 4,09 1,17
m d d dd m m m s s c t
J 10 10 13 - - - - - 7 7
Nota: Los valores δ correspondientes a H-1/1’, H-2/2’, H-6/6’, H-7/7’, H-8/8’, H-9/9’ y H-11/11’ están promediados y corresponden a las señales superpuestas de [11] y [12].
+
[11] [12]
O
CH3
O OCH31
2
3
45
6
7
8910
11
12
13
O
CH3
O O CH31'
2'
3'
4'
5'
6'7'
8'9'10'
11'
12' 13'
14'

36
Figura 11/12C: Espectro de RMN-13C [CDCl3, 100 MHz], de la Mezcla de 10-Carbometoxi-4-Metil-Decalin-1,4-dien-3-ona [11] y
10-Carboetoxi-4-Metil-Decalin-1,4-dien-3-ona [12]
Tabla 11/12C: Desplazamientos Químicos (δ) en el Espectro de RMN-13C [(CDCl3), 100 MHz], de la Mezcla de 10-Carbometoxi-4-Metil-Decalin-1,4-
dien-3-ona [11] y 10-Carboetoxi-4-Metil-Decalin-1,4-dien-3-ona [12]
C C-1/1’ C-2/2’ C-3/3’ C-4/4’ C-5/5’ C-6/6’ C-7/7’ C-8/8’
δ (ppm) 154,3 128,9 170,7 130,7 147,2 22,8 30,0 26,8
Tipo =C-H =C-H >C=O >C= =C< >CH2 >CH2 >CH2
C C-9/9’ C-10/10’ C-11/11’ C-12/12’ C-13 C-13’ C-14’ -
δ (ppm) 37,4 53,1 10,3 185,9 61,7 52,8 14,1 -
Tipo >CH2 >C< -CH3 -O-C=O -O-CH3 -O-CH2 -CH3 -
C -12,12’
C -3,3’C -1,1’
C -5,5’
C -4,4’
C -2,2’
C -13
C-10,10’
C-13’
C -9,9’
C -7,7’C -8,8’
C -6,6’
C -14’
C -11,11’
+
[11] [12]
O
CH3
O OCH31
2
3
45
6
7
8910
11
12
13
O
CH3
O O CH31'
2'
3'
4'
5'
6'7'
8'9'10'
11'
12' 13'
14'

37
Figura 11/12D: Espectro de Masas (IE, 70 eV.) de la Mezcla de 10-Carbometoxi-4-Metil-Decalin-1,4-dien-3-ona [11] y
10-Carboetoxi-4-Metil-Decalin-1,4-dien-3-ona [12]
Tabla 11/12D: Fragmentos (m/z) más Significativos en el Espectro de Masas (IE, 70 eV.) de la Mezcla de 10-Carbometoxi-4-Metil-Decalin-1,4-
dien-3-ona [11] y 10-Carboetoxi-4-Metil-Decalin-1,4-dien-3-ona [12]
m/z 235 221 207 175 163 147
Abundancia Relativa (%) 30 15 25 18 100 75
m/z 134 115 105 91 79 51
Abundancia Relativa (%) 95 33 68 63 58 23
+
[11] [12]
O
CH3
O OCH31
2
3
45
6
7
8910
11
12
13
O
CH3
O O CH31'
2'
3'
4'
5'
6'7'
8'9'10'
11'
12' 13'
14'

38
Esquema 11/12D: Interpretación del Patrón de Fragmentación Observado en el Espectro de Masas (IE, 70 eV.) de la Mezcla de 10-Carbometoxi-
4-Metil-Decalin-1,4-dien-3-ona [11] y 10-Carboetoxi-4-Metil- Decalin-1,4-dien-3-ona [12]
O
CH3
H3C
O
CH3
.+
C12H16O2
+ m/z: 204 [M ]
..
..
O
CH3
O
..CH3+
m/z: 175
..
..OH3C
CH3
+
..
..+
..
..O
CH3
+
+
m/z: 147
m/z: 189
..
..
m/z: 175
OH3C
O
C O.. +
. .. .
.. ..
.. ..
+CH3
m/z: 134
..
..O
H3C
O
H3C
..
...
O....+
m/z: 105
+CH2..
+
. .. .
..
..O
CH3
H3C
O
++ CH3
..
+.
m/z: 176
.
.
+
m/z: 147
+..C O+
OH3C ..
.. .
+
+
..
..O
H3C
CH3
+ C O.. +
m/z: 162
..
m/z: 147
+
+
O
CH3
..
..
CH3. .
CH3
.+
+
m/z: 134m/z: 134
+
.....O
H3C
O....
+
m/z: 119
. . .
.
.
CH3.
+
+
+ CH...+

39
C. SÍNTESIS DEL 4-METIL-1,3,5(10)-TRIEN-DECALIN-3-OL [2] En la ruta inicialmente propuesta (Esquema 1), el tercer paso consistirá en la hidrólisis de los ésteres y para ello se usaría ter-butóxido de potasio (t-BuOK) en dimetil sulfóxido (DMSO), a reflujo y bajo nitrógeno; en este proceso se esperaba obtener el ácido [13] (Esquema 10).
Esquema 10: Mecanismo de la Reacción que Conduciría a la Formación del 10-Carboxi-4-metil-decalin-1,4-dien-3-ona [13]
Sorprendentemente, cuando se llevó a cabo dicha reacción, no se obtuvo el ácido esperado sino el producto generado en la descarboxilación de éste, acompañada de la aromatización reductiva del anillo A; es decir, el fenol [2]. Este hecho resultó altamente positivo ya que no afectaba en nada al proceso sintético global, y adicionalmente, simplificaba el mismo en un paso. Desde el punto de vista mecanístico (Esquema 11), la reacción es perfectamente explicable si se supone que, previamente, el ter-butóxido de potasio (t-BuOK) transforma la mezcla de ésteres en la sal potásica; luego, durante el proceso de extracción del ácido carboxílico, el ácido clorhídrico (al 10%) que se utiliza en dicho proceso, hidroliza la sal potásica generando el correspondiente anión, el cual, en vez de captar un protón, procede a descarboxilarse; esta descarboxilación se produce al captar el oxígeno cetónico un protón del medio acuoso, proceso éste que induce la ruptura del enlace C-10/C-12 con pérdida de CO2, y la consecuente aromatización del anillo A.
Esquema 11: Mecanismo de la Reacción que Conduce a la Formación del 4-Metil-1,3,5(10)-trien-decalin-3-ol [2]
esperadoproducto
[13]
O
O OH
KCl+
H Cl
K+
O
O O-
K+ CCH3
CH3
CH3
-O
O
O O R
14'13'
12
3
45 7
8910
11
12
13
6
[12]
[11] R: CH3
R: CH2 CH3
R: CH2 CH3
R: CH3[11]
[12]
6
13
12
11
109 8
754
3
21
13' 14'
O
O O R
CCH3
CH3
CH3
-OK+
O
O O- K+
OH
H
HO
[2]
+ CO2obtenidosproductos

40
Inicialmente el rendimiento de esta reacción fue muy bajo (< 20%); cuando se indagó sobre este hecho, pudo observarse que la causa que principalmente incidía sobre el rendimiento, dependía de la utilización del HCl que se añadía en exceso en el proceso de extracción con el fin de recuperar el ácido que debía formarse. Como era evidente que dicho ácido no se formaba, la acidificación de la fase acuosa en el proceso de extracción no tenía ya sentido; además, es bien sabido que la extracción de fenoles en medio ácido no resulta eficiente y por ello se pensó que el proceso se optimizaría si se prescindía de utilizar HCl en el mismo.
Es innegable que la formación del fenol [2] se lleva a cabo, ya que la reacción transcurre bajo un control termodinámico, favorecido por la aromatización del anillo A y por la eliminación de una molécula estable (CO2) como grupo saliente, hecho éste que imposibilita la reversibilidad del proceso. No obstante, es evidente que en el transcurso de la reacción es necesaria la presencia de agua para que se pueda formar el fenol [2] (Esquema 11). En consecuencia, se procedió a repetir la reacción bajo dos condiciones experimentales consecutivas: En una primera etapa se realizó la reacción en DMSO seco, a reflujo, manteniéndose estas condiciones durante aproximadamente 40 minutos. Posteriormente se añadió agua a la mezcla de reacción y la misma se mantuvo en agitación durante 30 minutos; transcurrido este tiempo se inició el proceso de extracción con éter etílico, lográndose de esta manera aislar el fenol con un rendimiento del 92 %.
El fenol [2] fue purificado por cromatografía en columna y una vez confirmado su estado de pureza, se abordó su estudio espectral. Su espectro IR (Fig 2A; Tabla 2A), carece de bandas de carbonilos y muestra absorciones típicas de un hidroxilo fenólico [νmax : 3.229 cm-1 (O-H)] y de aromáticos [νmax : 3.010 y 801 cm-1 (=C-H); νmax : 1.587 cm-1
(C=C) y υmax : ≅ 2000 (sobretonos)]. En su espectro de RMN-1H (Fig. 2B; Tabla 2B] se aprecian dos dobletes que conforma el típico sistema AB del par de protones aromáticos “orto”-acoplados de un anillo bencénico 1,2,3,4-tetrasustituido [δH: 6,79 y δH: 6,56; d (J≅ 8 Hz) (H-1 y H-2)]; a campos más altos se distingue un singulete asignable a un metilo unido a un núcleo aromático [δH: 2,10, -CH3 (H-11)], dos tripletes atribuibles a dos metilenos bencílicos [δH: 2,61; t (J ≅ 6 Hz) (H-6) y δH: 2,69; t (J ≅ 7 Hz) (H-9)] y dos multipletes superpuestos propios de dos metilenos cicloalifáticos contiguos [δH: 1,75 (H-7 y H-8)]; la señal del protón del hidroxilo fenólico fue localizada como un singulete a δH: 4,57. Su espectro de RMN-13C (Fig. 2C; Tabla 2C) muestra once (11) picos, seis (6) de los cuales corresponden a carbonos sp2 aromáticos y el resto a alifáticos. Entre estos picos destacan los asignados a los metinos aromáticos [δC: 126,9 (C-1) y δC: 112,3 (C-2)], el correspondiente al carbono que soporta el hidroxilo fenólico [δC: 151,1; =C-O- (C-3)] y el del grupo metilo [δC: 10,9; -CH3 (C-11)]. Por último su EM (Fig. 2D; Tabla 2D] exhibe un pico a m/z: 162 correspondiente a un ión molecular de fórmula C11H14O.

41
Figura 2A: Espectro Infrarrojo en Película de KBr, del 4-Metil-1,3,5(10)-trien-decalin-3-ol [2]
Tabla 2A: Bandas de Absorción Significativas en el Espectro Infrarrojo (KBr), del 4-Metil-1,3,5(10)-trien-decalin-3-ol [2]
υmax (cm- 1) 3.229 3.010 2.923-2.855 ≅2000 1.587 1.224 801
Asignación O-H =C-H C-H sobretonos C=C C-O =C-H
3.229O-H
3.010=C-H
2.923-2.855C-H
1.585C=C
1.224C-O
801=C-H
3.229O-H
3.010=C-H
2.923-2.855C-H
1.585C=C
1.224C-O
801=C-H
CH311
10
7
9
4
2 8
6
5
3
1
121,8
22.8
151,1
27,1
29,9
136,7
112,3
126,9
23,3
10,9
129,5
H-1
H-8H-2
H-11
H-6
H-7
δ: 6,79 (d)
δ: 1,75 (m)
δ: 2,61 (t)
δ: 1,75 (m)
δ: 6,56 (d)
δ: 2,10 (s)
δ: 2,69 (t)H-9
OH
O-Hδ: 4,57 (s)
J(Hz): 8
J(Hz): 8
J(Hz): 6
J(Hz): 7
4-Metil-1,3,5(10)-trien-decalin-3-ol [2]
CH3
HO
[2]
1
2
3
45
6
7
8
910
11

42
Figura 2B: Espectro de RMN-1H [CDCl3, 400 MHz], del 4-Metil-1,3,5(10)-trien-decalin-3-ol [2]
Tabla 2B: Desplazamientos Químicos (δ) en el Espectro de RMN-1H [(CDCl3), 400 MHz], del 4-Metil-1,3,5(10)-trien-decalin-3-ol [2]
H H-1 H-2 H-6 H-7 H-8 H-9 H-11 OH
δ (ppm) 6,79 6,56 2,61 ≅ 1,75 ≅ 1,75 2,69 2,10 4,57
m d d t m m t s s
J (Hz) 8 8 6 - - 7 - -
CDCl3
H-1 H-2
O-H
H-9 H-6
H-11
CH3
HO
[2]
1
2
3
45
6
7
8
910
11

43
Figura 2C: Espectro de RMN-13C [CDCl3, 100 MHz], del 4-Metil-1,3,5(10)-trien-decalin-3-ol [2]
Tabla 2C: Desplazamientos Químicos (δ) en el Espectro de RMN-13C [(CDCl3), 100 MHz], del 4-Metil-1,3,5(10)-trien-decalin-3-ol [2]
C C-1 C-2 C-3 C-4 C-5 C-6
δ (ppm) 126,9 112,3 151,1 121,8 136,7 27,1
Tipo =CH =CH =C-O- =C< =C< >CH2
C C-7 C-8 C-9 C-10 C-11
δ (ppm) 22,8 23,3 29,9 129,5 10,9
Tipo >CH2 >CH2 >CH2 =C< -CH3
Valores δ con
referencia al TMSi
C-3
C-5C-10
C-1
C-4
C-2 C-9
C-6C-8
C-7
C-11
CH3
HO
[2]
1
2
3
45
6
7
8
910
11

44
Figura 2D: Espectro de Masas (IE, 70 eV.) del 4-Metil-1,3,5(10)-trien-decalin-3-ol [2]
Tabla 2D: Fragmentos (m/z) más Significativos en el Espectro de Masas (IE, 70 eV.) del 4-Metil-1,3,5(10)-trien-decalin-3-ol [2].
m/z 162 174 134 115 105
Abundancia Relativa (%) 59 62 100 18 27
m/z 91 77 65 51 39
Abundancia Relativa (%) 62 22 15 18 18
CH3
HO
[2]
1
2
3
45
6
7
8
910
11

45
En el Esquema 2D, representado a continuación, se muestra la interpretación del patrón de fragmentación del fenol [2].
Esquema 2D: Interpretación del Patrón de Fragmentación Observad en el Espectro de Masas (IE, 70 eV.) del 4-Metil-1,3,5(10)-trien-decalin-3-ol [2]
O
CH3
H
.+
OH
+
+
O
+ CH3.
O
C11H14O
+ m/z: 162 [M ] m/z: 147
m/z: 147m/z: 121
+ O C.. ..
+
m/z: 119
+
OH
+
O
H
H
..
..+....O
H
H
+
m/z: 134(Pico base)
m/z: 134(Pico base)
m/z: 134(Pico base)
H
H
+
m/z: 106
+
....O C
+
+C C HH
H
H
m/z: 134(Pico base)
+
..
..
+.
O C.. ..
+

46
D. SÍNTESIS DE LA 3-METOXI-4-METIL-1,3,5(10)-TRIEN-DECALINA [3] Una vez lograda la hidrólisis y aromatización del anillo A mediante la obtención del fenol [2], se procedió a realizar su metoxilación utilizando sulfato de dimetilo [DMS, (CH3)2SO4] como agente metoxilante. Cabe destacar que al realizar al paso anterior, la ejecución de los siguientes pasos experimentales no implican mayor esfuerzo para alcanzar los objetivos propuestos inicialmente.
La metoxilación del fenol se inicia con el ataque nucleofílico del ión “fenóxido”, sobre uno de los grupos metilos del (CH3)2SO4 . Este ataque se da sin la salida formal del protón proveniente del OH- del fenol (es decir sin la formación real del ión fenóxido), ya que la salida de dicho protón es impulsada por el ataque efectivo al (CH3)2SO4, que deja sus electrones sobre el oxígeno adyacente; el papel que cumple el carbonato de sodio es el de servir de iniciador e impulsor de la salida de este protón fenólico, pero en sí, no actúa como una base directa. Por otro lado, es de hacer notar que la salida espontánea de este protón no se facilita, al menos que exista un centro suficientemente electrofílico como el metilo del (CH3)2SO4, el cual al ser atacado por el “fenóxido” genera una especie capaz de soportar una carga negativa estabilizada por resonancia (H3C-O-SO2-O
-). El mecanismo para esta reacción se ilustra en el Esquema 12.
Esquema 12: Mecanismo de la Metoxilación del Fernol [2] que conduce a la 3-Metoxi-4-metil-1,3,5(10)-trien-decalina [3]
__________ ( ) El ataque nucleofílico se podría dar de forma concertada por parte de dos moléculas de fenol a
ambos extremos del DMS. No obstante es importante resaltar que al margen de que se produzca un ataque uni- o bimolecular, cada molécula de DMS metoxilará dos moléculas de fenol.
S
O
O
OOH3C CH3..........
..
..
..
..OH
CH3
OH3C
CH3
+ CH3SO4H
CH3CH3 O O
O
O
S....
..
..
..
..O
H
CH3
..
..O
H
CH3
OH3C
CH3
+ H2SO4

47
Esta reacción resultó sencilla, rápida y efectiva y no originó mayor problema en un todo de acuerdo con lo indicado en la bibliografía (House, 1972). En efecto, en el primer control de la reacción de prueba se evidenció por TLC que el Rf del producto de reacción era mayor que el del producto de partida, condición ésta esperable al desaparecer el carácter polar que le daba el grupo OH al compuesto de partida; por otro lado, en este control se detectó que la mayor parte de [2] se habría transformado en [3] pero aún se observaba una pequeña fracción (estimada < 20%), de compuesto sin reaccionar. Este problema se solucionó cuando se repitió la reacción, aumentando la cantidad de DMS (recién destilado para asegurar que el mismo estuviera libre de contaminantes u otros aditivos). El rendimiento total resultó aproximadamente del 80%; aunque este rendimiento no es menor al reportado para reacciones similares (Fieser & Fieser, 1967), cabe suponer que el mismo no es mejorable debido a la baja probabilidad de choque de dos moléculas de fenol con una sola de DMS.
El metoxiderivado [3], una vez purificado y cuantificado, se caracterizó estructuralmente por técnicas espectroscópicas. Su espectro IR (Fig. 3A; Tabla 3A), muestra bandas típicas de alifáticos [νmax : 2.942-2864 cm-1 (C-H)] y de un núcleo aromático [νmax : 3.026 y 811 cm-1 (=C-H); νmax : 1.588 cm-1 (C=C) y υmax : ≅ 2000 (sobretonos)], pero carece de la absorción característica del grupo OH.
Figura 3A: Espectro Infrarrojo en Película de KBr, de la 3-Metoxi-4-metil-1,3,5(10)-trien-decalina [3]
Tabla 3A: Bandas de Absorción Significativas en el Espectro Infrarrojo (KBr), de la 3-Metoxi-4-metil-1,3,5(10)-trien-decalina [3]
υmax (cm- 1) 3.026 2942-2864 2.000 1.588 811
Asignación =C-H C-H sobretonos C=C =C-H
O
CH3
H3C
1
2
34
56
7
8
910
11
12

48
En su espectro de RMN-1H (Fig. 3B; Tabla 3B) destaca la aparición de un nuevo singulete atribuible al grupo metoxi [δH: 3,84 O-CH3 (H-12)] y la ausencia (en δH: 4,57) del singulete asignado al hidrógeno del grupo OH. Al margen de estos cambios, se continúan observando los dobletes que conforma el sistema AB del par de protones aromáticos “orto”-acoplados de un anillo bencénico 1,2,3,4-tetrasustituido [δH: 6,93 y δH: 6,71; d (J ≅ 8 Hz) (H-1 y H-2)], los dos tripletes atribuibles a los metilenos bencílicos [δH: 2,68; t (J ≅ 6 Hz) (H-6) y δH: 2,76; t (J ≅ 6 Hz) (H-9)], los multipletes superpuestos que generan los dos metilenos cicloalifáticos contiguos [δH: 1,81 (H-7 y H-8)] y el singulete que identifica a un metilo unido a un núcleo aromático [δH: 2,14 -CH3 (H-11)]; como era de esperar los desplazamientos de estas señales no son notables, en comparación con los de sus análogas en el espectro del fenol [2], dado que el cambio estructural entre ambos compuesto no es significativo.
El espectro de RMN-13C (Fig. 3C; Tabla 3C) muestra la presencia de un carbono, el cual se suma a un total de 12 carbonos para esta estructura. Este pico aparece en la zona de los carbonos metoxílicos a δC: 55,4 [O-CH3 (C-12)]. Como es de esperar se continúan observando los seis picos asignables a carbonos aromáticos, entre los cuales destaca el del carbono enlazado al oxígeno [δC: 155,0; =C-O- (C-3)] y los cinco picos típicos de carbonos alifáticos sp3, entre los cuales sólo uno aparece en la fase normal del DEPT-135 [δC: 10,58; -CH3 (C-11)]. Las señales en su totalidad se ven poco desplazadas en relación a sus análogas en el espectro del fenol, excepto la del metino aromático C-2 (δC: 107,8 ), la cual resuena a campos ligeramente más altos debido al efecto dador de electrones que por resonancia ejerce el grupo metoxi unido al anillo aromático.
CH311
10
7
9
4
2 8
6
5
3
1
124,3
22,6
155,0
26,8
29,2
136,3
107,8
126,3
23,1
10,6
129,1
H-1
H-8H-2
H-11
H-6
H-7
δ: 6,93 (d)
δ: 1,81 (m)
δ: 2,68 (t)
δ: 1,81 (m)
δ: 6,71 (d)
δ: 2,14 (s)
δ: 2,76 (t)H-9
OH3C
H-12δ: 3,84 (s)
J(Hz): 8
J(Hz): 8
J(Hz): 6
J(Hz): 6
12
55,4
3-Metoxi-4-metil-1,3,5(10)-trien-
decalina [3]

49
Figura 3B: Espectro de RMN-1H [CDCl3, 400 MHz], de la 3-Metoxi-4-metil-1,3,5(10)-trien-decalina [3]
Tabla 3B: Desplazamientos Químicos (δ) en el Espectro de RMN-1H [(CDCl3), 400 MHz], de la 3-Metoxi-4-metil-1,3,5(10)-trien-decalina [3]
H H-1 H-2 H-6 H-7 H-8 H-9 H-11 H-12
δ (ppm) 6,93 6,71 2,68 1,81 1,81 2,76 2,14 3,84
m d d t m m t s s
J (Hz) 8 8 6 - - 6 - -
H-6H-9
H-2H-1
H-12
H-9 H-6
H-11
O
CH3
H3C
1
2
34
56
7
8
910
11
12

50
Figura 3C: Espectro de RMN-13C [CDCl3, 100 MHz], de la de la 3-Metoxi-4-metil-1,3,5(10)-trien-decalina [3]
Tabla 3C: Desplazamientos Químicos (δ) en el Espectro de RMN-13C [(CDCl3), 100 MHz], de la 3-Metoxi-4-metil-1,3,5(10)-trien-decalina [3]
C C-1 C-2 C-3 C-4 C-5 C-6
δ (ppm) 126,3 107,8 155,0 124,3 136,3 26,8
Tipo =CH =CH =C-O- =C< =C< >CH2
C C-7 C-8 C-9 C-10 C-11 C-12
δ (ppm) 22,6 23,1 29,2 129,1 10,6 55,4
Tipo >CH2 >CH2 >CH2 =C< -CH3 -O-CH3
C-3
C-2C-1
C-5
C-10 C-4C-12
C-9 C-6
C-11
C-7C-8
O
CH3
H3C
1
2
34
56
7
8
910
11
12

51
Su EM (Fig. 3D; Tabla 3D) muestra un pico a m/z: 176 que corresponde a un ión molecular con formula C12H16O. El patrón de fragmentación observado en dicho espectro (Esquema 3D), es congruente con la estructura [3].
Figura 3D: Espectro de Masas (IE, 70 eV.) de la 3-Metoxi-4-metil-1,3,5(10)-trien-decalina [3]
Tabla 3D: Fragmentos (m/z) más Significativos en el Espectro de Masas (IE, 70 eV.) de la 3-Metoxi-4-metil-1,3,5(10)-trien-decalina [3]
m/z 176 161 148 133 117
Abundancia Relativa (%) 100 66 51 22 42
m/z 105 91 79 65 39
Abundancia Relativa (%) 31 21 14 10 12
O
CH3
H3C
1
2
34
56
7
8
910
11
12

52
Esquema 3D: Interpretación del Patrón de Fragmentación Observado en el Espectro de Masas (IE, 70 eV.) de la 3-Metoxi-4-metil-1,3,5(10)-trien-
decalina [3]
O
CH3
H3C
.+
OH3C
+
+ CH3.
C12H16O
+ m/z: 176 [M ]
m/z: 161
..
..
.CH3+
+O
.
.
m/z: 146
.CH3+
+O....
O
CH3
..
..+
..CH3+
m/z: 161
OH3C
+
m/z: 147
+CH2..
+
.CH3 +
m/z: 132
+O.....
. .... +O
+
..CH2 +
m/z: 118
..
..O
CH3
m/z: 147
+
..CH2+
m/z: 146
..
.. .+
O....
+ CH3.
+CH2..
+O....
.m/z: 132m/z: 118
O....
.+
m/z: 105.++
+CH..
+.

53
E. SÍNTESIS DE LA 3-METOXI-4-METIL-1,3,5(10)-TRIEN-DECALIN-9-ONA [4]
La síntesis del primero de los derivados funcionalizado en posición α al grupo ceto (3-Metoxi-4-metil-1,3,5(10)-trien-decalin-9-ona [4]) se concibió bajo el concepto de oxidación de un carbono metilénico activado por el hecho de estar conjugado con un sistema aromático; bajo esta premisa, la escogencia adecuada es la de una reacción que implique la generación de un radical libre bencílico, el cual adquiere cierta estabilidad por el efecto resonante que deslocaliza a su electrón desapareado en el seno del núcleo aromático. En consecuencia, se escogió como oxidante el trióxido de cromo (CrO3) en ácido acético/agua (Burnham et al., 1974).
El mecanismo de esta reacción (Esquema 13), se inicia en el momento en que el CrO3 abstrae un hidrógeno a través de la ruptura radicalaria de un enlace C-H en el metileno activo. En este primer paso se reduce el agente oxidante pasando el cromo del estado de valencia 6+ al 5+ (Cr6+ Cr5+). En un segundo paso el CrO3
reducido se une a través de un enlace C-O al carbono metilénico mediante un proceso reductivo en el cual, el estado de valencia del cromo pasa de 5+ a 4+ (Cr5+ Cr4+). A continuación, se produce un ataque por parte del último carbonilo presente en el agente oxidante sobre el segundo hidrógeno metilénico, seguido del consecuente movimiento concertado de electrones, el cual, provoca la salida del grupo oxidante protonado y la generación del carbonilo sobre el carbono metilénico inicial. El acetato en medio acuoso facilita la desprotonación de la especie oxidante, quedando finalmente el cromo con valencia 3+; en este estado de oxidación el cromo forma una especie de coloración verde intensa.
H3CO
CH3
HH
O
O
O
Cr
H3CO
CH3
H
O
O
Cr
OH.
H3CO
CH3
O
OH
O
Cr.. Cr
OH
OH
AcO-
: +H3CO
CH3
H OCr
O
OH
+6+5
+4
+3 +2

54
Esquema13: Mecanismo de Reacción de Oxidación que Conduce a la Formación de la 3-Metoxi-4-metil-1,3,5(10)-trien-decalin-9-ona [4]
El principal problema que se esperaba en esta reacción era la obtención del producto de competencia (la 3-Metoxi-4-metil-1,3,5(10)-trien-decalin-6-ona). La posibilidad de obtener dicho subproducto se debe a que existen dos metilenos activos en la molécula (C-6 y C-9), susceptibles al ataque del grupo oxidante; sin embargo, a la base de los resultados obtenidos, es obvio que la reactividad se vio favorecida hacia el producto esperado (cetona en C-9), por dos razones:
1. El impedimento estérico que ejerce el grupo metilo situado en C-4, en el sentido de impedir el ataque efectivo del grupo oxidante, el cual es bastante voluminoso.
2. Como es conocido, los grupos metoxi son activantes “orto”/”para”, pero este efecto es más notable sobre el sustituyente “para”, haciendo esta posición (C-9), más reactiva al trióxido de cromo en comparación con la posición “meta” (C-6).
Una vez lograda la purificación del producto [4] por medio de cromatografía de capa gruesa, se realizó su estudio espectral y a través del mismo se notó la presencia de una pequeña impureza que puede presumirse sea de la tetralona de competencia; el rendimiento total de esta reacción fue el 75% de la tetralona esperada. Su espectro IR (Fig. 4A; Tabla 4A) muestra de manera destacable una banda característica del carbonilo de una cetona conjugada [νmax : 1.678 cm-1 (C=O)], y además se continúan observando en el mismo absorciones de un sistema aromático [νmax : 3.025 y 824 cm-1 (=C-H); νmax : 1.588 cm-1 (C=C) y υmax ≅ 2000 (sobretonos)] y de otro alifático [νmax : 2.942 cm-1 (C-H)].
El espectro de RMN-1H (Fig. 4B; Tabla 4B) exhibe los dobletes característicos de los hidrógenos aromáticos “orto”-acoplados, observándose un desplazamiento a campo más bajo del doblete asignado al H-1 [δH: 8,00; d (J ≅ 9 Hz)], debido al efecto de desapantallamiento producido por el grupo carbonilo en C-9; por el contrario, la posición del doblete del H-2 no se ve afectada [δH: 6,85; d (J ≅ 9 Hz)]. Otros cambios notables frente al espectro de RMN-1H de [3] (Fig. 3B) son, la desaparición de la señal correspondiente a los H-9 y la posición del triplete atribuido a los hidrógenos (H-8) del metileno α- al carbonilo cetónico [δH: 2,88; t (J ≅ 6 Hz)], el cual, como es de esperar, ante la influencia desapantallante del carbonilo cetónico en C-9, se encuentran a campo más bajo respecto a las señales de los restantes hidrógenos alifáticos [δH: 2,60; t (J ≅ 7 Hz) (H-6) y δH: 2,13; m (H-7)]. Las otras dos señales observadas en el espectro de RMN-1H, corresponden a los singuletes asignados al metilo aromático ubicado en C-4 [δH: 2,16, s, Φ-CH3 (H-12)] y al grupo metoxilo [δH: 3,90, s –O-CH3 (H-12)]; ambos singuletes no varían su posición, dado que el nuevo grupo carbonilo, por su lejanía, no produce efecto alguno sobre los mismos.

55
Figura 4A: Espectro Infrarrojo en Película de KBr, de la 3-Metoxi-4-metil-1,3,5(10)-trien-decalin-9-ona [4]
Tabla 4A: Bandas de Absorción Significativas en el Espectro Infrarrojo (KBr), de la 3-Metoxi-4-metil-1,3,5(10)-trien-decalin-9-ona [4]
υmax (cm- 1) 3.025 2.942 ≅2000 1.678 1.588 824
Asignación C-H (arom.) C-H (alif.) sobretonos C=O C=C =C-H
El espectro de RMN-13C (Fig. 4C; Tabla 4C) muestra como cambios notables,
en relación al espectro de [3] (Fig. 3C], la variación en el desplazamiento del C-9 a campo más bajo [δC: 197,9; C=O] (puesto que éste es ahora un carbono carbonílico), y un desplazamiento similar, más pequeño, del pico asignado al metileno vecino C-8 (δC: 197,9); las restantes señales continúan prácticamente inalteradas.
11
10
7
9
4
2 8
6
5
3
1
123,6
22,7
161,3
26,6
197,9
144,2
108,2
126,2
38,4
11,2
127,0
H-1
H-2
H-11
H-6
H-7
δ: 8,00 (d)
δ: 2,60 (t)
δ: 2,13 (m)
δ: 6,85 (d)
δ: 2,16 (s)
OH3C
H-12δ: 3,90 (s)
J(Hz): 9
J(Hz): 9
J(Hz): 7
12
55,5
CH3
O
J(Hz): 6
H-8δ: 2,88 (t)
3-Metoxi-4-metil-1,3,5(10)-trien-
decalin-9-ona [4]
O
CH3
H3C
O1
2
34
56
7
810
11
12
9
4500,0 4000 3000 2000 1500 1000 450,00,0
10
20
30
40
50
60
70
80
90
100
cm-1
%T
3026,05
2941,63
2863,52
1677,90
1588,10
823,42

56
Figura 4B: Espectro de RMN-1H [CDCl3, 400 MHz], de la 3-Metoxi-4-metil-1,3,5(10)-trien-decalin-9-ona [4]
Tabla 4B: Desplazamientos Químicos (δ) en el Espectro de RMN-1H [(CDCl3), 400 MHz], de la 3-Metoxi-4-metil-1,3,5(10)-trien-decalin-9-ona [4]
H H-1 H-2 H-6 H-7 H-8 H-11 H-12
δ (ppm) 8,00 6,85 2,60 2,13 2,88 2,16 3,90
m d d t m t s s
J (Hz) ≅ 9 ≅ 9 ≅ 7 - ≅ 6 - -
H-8 H-6
CDCl3H-1 H-2
H-12
H-11
O
CH3
H3C
O1
2
34
56
7
810
11
12
9

57
Figura 4C: Espectro de RMN-13C [CDCl3, 100 MHz], de la 3-Metoxi-4-metil-1,3,5(10)-trien-decalin-9-ona [4]
Tabla 4C: Desplazamientos Químicos (δ) en el Espectro de RMN-13C [(CDCl3), 100 MHz], de la 3-Metoxi-4-metil-1,3,5(10)-trien-decalin-9-ona [4]
C C-1 C-2 C-3 C-4 C-5 C-6
δ (ppm) 126,2 108,2 161,3 123,6 144,2 26,9
Tipo =CH =CH =C-O- =C< =C< >CH2
C C-7 C-8 C-9 C-10 C-11 C-12
δ (ppm) 22,7 38,4 197,9 127,0 11,2 55,5
Tipo >CH2 >CH2 >C=O =C< -CH3 -O-CH3
C-9 C-3 C-5
C-10
C-1
C-4
C-2 C-12 C-8C-7
C-11
C-6
O
CH3
H3C
O1
2
34
56
7
810
11
12
9

58
Su EM (Fig. 4D; Tabla 4D) muestra un pico a m/z: 190 que corresponde a un ión molecular de formula C12H12O2. El patrón de fragmentación observado en dicho espectro (Esquema 4D), es explicable para la tetralona [4].
Figura 4D: Espectro de Masas (IE, 70 eV.) de la 3-Metoxi-4-metil-1,3,5(10)-trien-decalin-9-ona [4]
Tabla 4D: Fragmentos (m/z) más Significativos en el Espectro de Masas (IE, 70 eV.) de la 3-Metoxi-4-metil-1,3,5(10)-trien-decalin-9-ona [4]
m/z 190 176 162 134
Abundancia Relativa (%) 100 29 65 83
m/z 119 105 91 65
Abundancia Relativa (%) 30 29 72 30
O
CH3
H3C
O1
2
34
56
7
810
11
12
9

59
Esquema 4D: Interpretación del Patrón de Fragmentación en el Espectro de Masas (IE, 70 eV.) de la 3-Metoxi-4-metil-1,3,5(10)-trien-decalin-9-ona [4]
O
CH3
H3C
O .+
C12H14O2
+ m/z: 190 [M ]
..
..
O
CH3
O
..CH3+
m/z: 175
..
..OH3C
m/z: 147
+
+
..
..+
..
..O
CH3
+
+ C O.. +
m/z: 147
+
m/z: 162
O
CH3
H3C....
+..C O
+.
..
..
m/z: 175
OH3C
O
C O.. +
+
. .. .
.. ..
.. ..
+CH3..
O
H3C
..
..+ + CH.
..+
m/z: 134
m/z: 134
+....O
H3C
O....
+
.
+ CH3..
m/z: 119
O....+
m/z: 105
+ CH2..
+
m/z: 134
.
+
O
H3C
.
+
O
H3C
m/z: 134
.CH3+

60
F. SÍNTESIS DE LA 3-METOXI-4,8-DIMETIL-1,3,5(10)-TRIEN-DECALIN-9-ONA [6] El paso final de la síntesis propuesta, consiste en la introducción de un grupo metilo en el carbono C-8, α- al grupo ceto en C-9. Al realizar esta reacción se completaría la síntesis de la 3-Metoxi-4,8-dimetil-1,3,5(10)-trien-decalin-9-ona [6] (6-Metoxi-5-metil-α-tetralona [6]).
En el proceso sintético reportado por M. J. Temple Robinson et. al. (1957) la obtención de [6] a partir de [4] se llevó a cabo en dos pasos consecutivos: Un primer paso que consistía en la introducción simultánea en C-8 de un carbometoxi y un metilo, utilizando como reactivos NaCH3O/(CH3)2CO3 y CH3I en metanol bajo atmósfera de nitrógeno, y un segundo paso en el que se inducía la eliminación del grupo carboametoxi con ácido clorhídrico concentrado, en agua/ácido acético; con esta estrategia se perseguía evitar la formación del producto de competencia, es decir del derivado dimetilado en C-8 (Esquema 4).
Esquema 4: Síntesis de M. J. P. Robinson de la 6-Metoxi-2,5-dimetil-α-tetralona [6]
En la propuesta planteada en el presente trabajo (Esquema 7, pag. 15) se persigue reemplazar el proceso de dos pasos consecutivos, por una reacción de un solo paso para evitar problemas de rendimiento o pérdida del sustrato. En consecuencia, se optó por una monometilación directa con CH3I en tetrahidrofurano a la temperatura ambiente y bajo atmósfera de nitrógeno. Para evitar o atenuar la formación del derivado dimetilado se incluyó trietilborano como reactivo adicional, en razón de que este reactivo tiene la propiedad de coordinarse con el oxígeno, una vez producida la extracción del hidrogeno α al carbonilo por parte de la base (hidruro) (Esquema 14). Bajo esta óptica se favorecería la formación de una especie “enólica”, que atacaría de manera efectiva al carbono del agente alquilante (CH3I), el cual es susceptible de experimentar el ataque nucleofílico del trietilboranoenolato; al mismo tiempo evitaría, por impedimento estérico ya que el trietilboranoenolato es voluminoso, la posibilidad de un segundo ataque nucleofílico sobre CH3I, atenuando así la formación del subproducto dimetilado. Es evidente pués, que la reacción de metilación será efectiva en la medida en que se asegure la formación del trietilboranoenolato, puesto que es sabido que la reactividad del CH3I es bastante alta.
CH3
CH3O
O
[4]
[3] NaOMe/Me2CO3
reflujo bajo N2
ICH3
MeOH
CH3
CH3O
O
CH3
COOCH3
CH3
CH3O
O
CH3HCl conc.
AcOH/H2O
[5] [6]

61
Esquema 14: Mecanismo de Reacción de la Metilación que Conduce a la Formación de la 3-Metoxi-4,8-Dimetil-1,3,5(10)-trien-decalin-9-ona [6]
Para solventar el problema del producto de competencia se tomaron otras medidas adicionales, tales como: A) realizar la reacción con cantidades equimolares exactas, es decir en la proporción 1:1 de CH3I con respecto al sustrato [4]; B) mantener en condiciones absolutas de sequedad el medio de reacción, puesto que es sabido que el hidruro de sodio es altamente reactivo y ante la presencia de humedad en el medio, inhibiría la formación del hidruro o limitaría su eficacia como base frente al sustrato.
Al llevar a cabo la reacción se pudo constatar por medio de TLC, la presencia de tres manchas: Una correspondiente al productos de partida, otra que se presume es el producto de competencia (mancha muy tenue) y una última que presentaba mayor intensidad y la cual resultó ser el producto deseado; el Rf de esta mancha era intermedio entre la del compuesto de partida y la del producto de competencia. La separación de los componentes de esta mezcla se logró por cromatografía preparativa sobre placas de gel de sílice.
El producto mayoritario se identificó a la base de sus datos espectrales. Así pues, su espectro IR (Fig. 6A; Tabla 6A), no sufrió modificaciones significativas respecto al espectro del sustrato de partida [4] (Fig. 4A) y en el mismo se continúan observando absorciones de sistemas alifáticos [νmax : 2.934 cm-1 (C-H)] y aromáticos [νmax : 816 cm-1 (=C-H); νmax : 1.588 cm-1 (C=C) y υmax : ≅ 2000 (sobretonos)] y la banda del carbonilo cetónico [νmax : 1.666 cm-1 (C=O)].
OH3C
CH3
OH
H
H-
.. ..
B
OH3C
CH3
O
H
B-
.. ..
H3C I+-
OH3C
CH3
O
CH3

62
Figura 6A: Espectro Infrarrojo en Película de KBr, de la 3-Metoxi-4,8-Dimetil-1,3,5(10)-trien-decalin-9-ona [6]
Tabla 6A: Bandas de Absorción Significativas en el Espectro Infrarrojo (KBr), de la 3-Metoxi-4,8-Dimetil-1,3,5(10)-trien-decalin-9-ona [6]
υmax (cm- 1) 2.934 2.000 1.666 1.588 816
Asignación C-H (alif.) sobretonos C=O C=C =C-H
Los cambios más resaltante en el espectro de RMN-1H (Fig. 6B; Tabla 6B),
respecto al espectro del producto [4] de partida (Fig. 4B) son: A) la aparición de un nuevo doblete a campos más altos [δH: 1,23; d, -CH3 (J≅ 7 Hz) (H-13)], asignable al nuevo metilo presente en la estructura [6]; B) la complejidad del multiplete atribuible a H-8 (δH: 2,51; m), debido al acoplamiento adicional de este protón con los hidrógenos del nuevo metilo (H-13), y C) la transformación, en un multiplete (δH: 2,88), del triplete generado por los hidrógenos bencílicos (H-6), hecho éste que ocurre a tenor de la variación que experimentan las constantes de acoplamientos J6,7, como consecuencia de la pérdida de equivalencia de los hidrógenos H-7 ante la presencia del nuevo metilo. Al margen de estos cambios, el espectro continúa mostrando los dos singuletes asignables al grupo metoxi [δH: 3,86 O-CH3 (H-12)] y al metilo sobre el núcleo aromático [δH: 2,13, -CH3 (H-11)] y los dobletes de los dos hidrógenos aromáticos “orto”-acoplados [δH: 7,96 y δH: 6,81; d (J≅ 9 Hz) (H-1 y H-2)].
4500,0 4000 3000 2000 1500 1000 450,00,0
10
20
30
40
50
60
70
80
90
100
cm-1
2933,30
2870,07
2841,76
1666,00 1585,66
1095,69
816,55
O
CH3
H3C
O
CH31
2
34
56
7
810
11
12
9
13

63
El espectro de RMN-13C (Fig. 6C; Tabla 6C) confirma la formación del derivado [6], ya que el mismo presenta un total de 13 picos, cuyos desplazamientos químicos y orientación en el DEPT-135, concuerdan con el número, tipo de hibridación y grado de sustitución, esperables para los carbonos que conformarían el esqueleto de una metoxi-α-tetralona dimetilada. El metilo situado sobre el carbono vecinal al grupo ceto resuena a δC: 15,3 (C-13), mientras que el metilo ubicado en el núcleo aromático lo hace a δC: 11,0 (C-11); en la región de los carbonos aromáticos se aprecian seis picos, dos de ellos correspondientes a metinos [δC: 125,9 (C-1) y δC: 108,3 (C-2)] y el resto a carbonos cuaternarios, entre los cuales destaca uno unido al oxígeno [δC: 161,1 (C-3)] que forma parte del grupo metoxilo [δC: 55,5; -OCH3, (C-12)]. En la zona de los carbonos alifáticos se observan dos picos correspondientes a metilenos [δC: 26,2 (C-6) y δC: 30,7 (C-7)] y uno asignable al metino α al grupo carbonilo [δC: 41,5 (C-8)]. Por último, la señal que resuena a más bajo campo corresponde, como es lógico, al carbonilo cetónico [δC: 200,2; -C=O (C-9)].
Su espectro de masas (Fig. 6D; Tabla 6D) muestra un pico a m/z: 204 que se
corresponde con el peso del ión molecular con formula C13H16O2. La interpretación del patrón de fragmentación del espectro de masas de este producto se representa en el Esquema 6D.
3-Metoxi-4,8-Dimetil-1,3,5(10)-trien-decalin-
9-ona [6]
10
7
9
4
28
6
5
3
1
123,5
30,7
161,1
26,2
200,2
143,8
108,3
125,9
41,5
11,0
127,1
H-1
H-2
H-11
H-7
δ: 7,96 (d)
δ: 2,00 (m)
δ: 6,81 (d)
δ: 2,13 (s)
OH3C
H-12δ: 3,86 (s)
J(Hz): 9
J(Hz): 9
12
55,5
CH3
CH3
Oδ: 1,23 (d)H-13
J(Hz): 7
δ: 2,51 (m)H-8
δ: 2,88 (m)H-6
15,3 13
11

64
Figura 6B: Espectro de RMN-1H [CDCl3, 400 MHz], de la 3-Metoxi-4,8-Dimetil-1,3,5(10)-trien-decalin-9-ona [6]
Tabla 6B: Desplazamientos Químicos (δ) en el Espectro de RMN-1H [(CDCl3), 400 MHz], de la 3-Metoxi-4,8-Dimetil-1,3,5(10)-trien-
decalin-9-ona [6]
H H-1 H-2 H-6 H-7 H-8 H-11 H-12 H-13
δ (ppm) 7,96 6,81 2,88 2,00 2,51 2,13 3,86 1,23
m d d m m m s s d
J (Hz) 9 9 - - - - - 7
H2O (Ste.)
H-13
H-11
H-12
H-1 H-2
CDCl3
O
CH3
H3C
O
CH31
2
34
56
7
810
11
12
9
13

65
Figura 6C: Espectro de RMN-13C [CDCl3, 100 MHz], de la 3-Metoxi-4,8-Dimetil-1,3,5(10)-trien-decalin-9-ona [6]
Tabla 6C: Desplazamientos Químicos (δ) en el Espectro de RMN-13C [(CDCl3), 100 MHz], de la 3-Metoxi-4,8-Dimetil-1,3,5(10)-trien-
decalin-9-ona [6]
C C-1 C-2 C-3 C-4 C-5 C-6 C-7
δ (ppm) 125,9 108,3 161,1 123,5 143,8 26,2 30,7
Tipo =CH =CH =C-O- =C< =C< >CH2 >CH2
C C-8 C-9 C-10 C-11 C-12 C-13
δ (ppm) 41,5 200,2 127,1 11,0 55,5 15,3
Tipo >CH >C=O =C< -CH3 -O-CH3 -CH3
Valores δ con
referencia al TMSi
C-9C-3
C-5
C-10
C-1
C-4
C-2 C-12 C-8 C-13
C-11
C-7 C-6
O
CH3
H3C
O
CH31
2
34
56
7
810
11
12
9
13

66
Figura 6D: Espectro de Masas (IE, 70 eV.) de la 3-Metoxi-4,8-Dimetil-1,3,5(10)-trien-decalin-9-ona [6]
Tabla 6D: Fragmentos (m/z) más Significativos en el Espectro de Masas (IE, 70 eV.) de la 3-Metoxi-4,8-Dimetil-1,3,5(10)-trien-decalin-9-ona [6]
m/z 204 189 175 162 147
Abundancia Relativa (%) 100 51 37 53 12
m/z 134 119 105 91 77
Abundancia Relativa (%) 85 26 27 51 25
O
CH3
H3C
O
CH31
2
34
56
7
810
11
12
9
13

67
Esquema 6D: Interpretación del Patrón de Fragmentación en el Espectro de Masas (IE, 70 eV.) de la 3-Metoxi-4,8-Dimetil-1,3,5(10)-trien-decalin-9-ona [6]
O
CH3
H3C
O
CH3
.+
C12H16O2
+ m/z: 204 [M ]
..
..
O
CH3
O
..CH3+
m/z: 175
..
..OH3C
CH3
+
..
..+
..
..O
CH3
+
+
m/z: 147
m/z: 189
..
..
m/z: 175
OH3C
O
C O.. +
. .. .
.. ..
.. ..
+CH3
m/z: 134
..
..O
H3C
O
H3C
..
...
O....+
m/z: 105
+CH2..
+
. .. .
..
..O
CH3
H3C
O
++ CH3
..
+.
m/z: 176
.
.
+
m/z: 147
+..C O+
OH3C ..
.. .
+
+
..
..O
H3C
CH3
+ C O.. +
m/z: 162
..
m/z: 147
+
+
O
CH3
..
..
CH3. .
CH3
.+
+
m/z: 134m/z: 134
+
.....O
H3C
O....
+
m/z: 119
. . .
.
.
CH3.
+
+
+ CH...+

68
Parte Experimental
1. TÉCNICAS GENERALES APLICADAS.
1.1. Cromatografía en Capa Fina y Capa Gruesa:
Para la cromatografía de capa fina analítica se utilizaron placas de gel de sílice con indicador fluorescente, sobre soporte de vidrio [Merck, HF 254 (0,25 mm)]. Las cromatoplacas se desarrollaron en los sistemas de eluyentes adecuados y se observaron directamente bajo luz UV. En algunos casos el revelado se hizo exponiendo las placas a vapores de yodo. Para la cromatografía de capa gruesa o preparativa se utilizaron placas similares pero con un espesor de 0,50 mm.
1.2. Cromatografía en columna:
Para la cromatografía en columna al vacío se usó como adsorbente gel de sílice [Merck 60 (63-200 μm, 70-230 mesh)]. Las columnas se desarrollaron siguiendo las técnicas descritas en la literatura (Coll & Bowden, 1986; Willis, 1991).
1.3. Punto de Fusión:
Los puntos de fusión fueron determinados empleando un aparato de plancha de calentamiento para el rango 20-300 oC (± 1 oC), [Fisher-Johns].
1.4. Espectros de Infrarrojo:
Se realizaron en un espectrofotómetro infrarrojo-FT Perkin Elmer, modelo FT-1725X, en película o en pastillas de KBr.
1.5. Espectros de RMN- 1H y de RMN-13C:
Los espectros de RMN- 1H y de RMN-13C se realizaron en un equipo de , a 400 y 100 MHz, respectivamente.
1.6. Espectros de Masas:
Los espectros de Masas se realizaron en un espectrómetro Hewlett Packard, modelo 5930A, a un potencial de 70 eV.
1.7. Materiales y Reactivos para la Realización de las Reacciones
Para las reacciones se utilizaron planchas de calentamiento con agitación, dotadas de placa refractaria de 18 x 14 cm, [“Corning”, mod. PC-420], y mantas de calentamiento [“Glas-Col”] de diferentes capacidades. Se dispuso de material de vidrio de alta calidad, con uniones esmeriladas, [“Corning”, “Quickfit” ó “Ace Glass”]. Los gases utilizados (nitrógeno y/ó argón) fueron de pureza industrial. Se emplearon reactivos y solventes de pureza analítica, principalmente de las casas comerciales “Aldrich”, “J. T. Baker”, “Merck” ó “Riedel-de Haën”. Cuando se necesitaron solventes secos, estos se secaron por los métodos convencionales descritos en la literatura (Vogel, 1974).

69
Espectrofótometro de infrarrojo con Transformada de Furier
Resonancia Magnética Nuclear
Bruker-Avance DRX 400 Fusiometro
Rotavapor Cromatografo de Gases
Acoplado a Masas
Cromatografía de Columna
Cromatografía de Capa Fina Cromatografía de Capa Gruesa
Equipo de destilación Fraccionada

70
2. SÍNTESIS DE LA 10-CARBOMETOXI-4-METIL-DECALIN-4-EN-3-ONA [9] Y DE LA
10-CARBOETOXI-4-METIL-DECALIN-4-EN-3-ONA [10] En un balón provisto de una trampa Dean-Stark y un refrigerante con tubo terminal de cloruro de calcio (Fig. I) se colocó una mezcla de 2-carbometoxi- y 2-carboetoxi-ciclohexanona [4,35g (25,64 mmol) y 5,51g (35,30 mmol)], disuelta en tolueno seco, 1-cloro-3-pentanona [11,21g (93,05 mmol)] y ácido p-toluensulfónico en cantidades catalíticas [0,35 (1,89 mmol)]. La mezcla de reacción se reflujó por un periodo de 24h. Luego de reposar y alcanzar la temperatura ambiente ésta fue vertida en una solución saturada de bicarbonato de sodio y luego extraída con éter; la fase orgánica se lavó con solución de ácido clorhídrico al 3%, luego con agua y por último con solución saturada de cloruro de sodio; a continuación se secó sobre MgSO4 anhidro.
La fase orgánica seca fue filtrada y luego concentrada a sequedad en un rotavapor. El producto fue purificado por cromatografía de columna sobre silica gel eluyendo con una mezcla hexano-éter (8:2); el producto puro resultó ser un aceite amarillo. Rendimiento: 75 % .
3. SÍNTESIS DE LA 10-CARBOMETOXI-4-METIL-DECALIN-1,4-DIEN-3-ONA [11] Y DE
LA 10-CARBOETOXI-4-METIL-DECALIN-1,4-DIEN-3-ONA [12] A una solución de la mezcla de los carboxilatos [9] [0,48g (2,16mmol)] y [10] [0,72g (3,05 mmol)] en 100 mL de dioxano recientemente destilado, se le adicionó DDQ (2,3-dicloro-5,6-diciano-1,4-benzoquinona) [2,31g (10,16 mmol)]. La mezcla se reflujó bajo atmósfera de N2 por 72h. Transcurrido este tiempo se dejó reposar hasta alcanzar la temperatura ambiente, y luego se eliminó el solvente por medio de un rotavapor. De esta manera se obtuvo un aceite arcilloso de color marrón claro (producto crudo), el cual fue purificado por medio de un percolado sobre columna de gel de sílice eluida con una mezcla hexano-éter (85:15). El producto puro fue un aceite amarillo-naranja. Rendimiento 90%.
Fig I. Equipo del tipo Dean-Stark

71
4. SÍNTESIS DEL 4-METIL-1,3,5(10)-TRIEN-DECALIN-3-OL [2] En un balón de tres bocas provisto de un refrigerante y una trampa de gases en la parte superior se agregó ter-butóxido de potasio [3,7g (33mmol)] recién preparado, sobre una solución con las dienonas [11] [0,18g (0,81mmol)] y [12] [0,27g (1,15mmol)] en 35mL de DMSO (dimetil sulfoxido) previamente destilado sobre hidruro de sodio y luego sobre tamiz molecular, bajo atmósfera a inerte. La solución resultante se reflujó por un periodo de 40 minutos, se dejó enfriar hasta alcanzar la temperatura ambiente y se agregó agua destilada hasta cambiar el color de la solución. La fase acuosa fue extraída con éter y secada con MgSO4 anhidro y luego de filtrada. Al evaporar a sequedad el solvente se obtuvo un aceite negro como producto crudo, el posteriormente se purifico por cromatografía en columna sobre gel de sílice, eluída con una mezcla hexano-éter 14:1. El producto puro resultó ser un sólido blanco (P. F. = 113-114 ºC) (Reportado: 113,5-114.5 ºC; Martin & Robinson, 1943). Rendimiento 92%. 5. SÍNTESIS DE LA 3-METOXI-4-METIL-1,3,5(10)-TRIEN-DECALINA [3]
En un balón provisto de tres bocas, tratado y purgado bajo atmósfera inerte, se colocó el fenol [3] [0,3g (1,85mmol)] disuelto en 10mL de acetona previamente destilada y secada sobre K2CO3. Seguidamente se añadió DMS (sulfato de dimetilo) [3,13mL (32,98mmol)] y K2CO3 [0,30g (3,15mmol)]; por ultimó se adicionaron 9mL más de acetona y la mezcla se dejó en reflujo por un tiempo de 5 h. Al finalizar la reacción, la mezcla se dejó en reposo y luego se centrifugó. El crudo obtenido después de decantar el solvente fue lavado con acetona unas tres veces más, y la mezcla nuevamente centrifugada y decantada. Finalmente se realizó la eliminación del solvente por medio de un rotavapor obteniéndose un sólido contaminado con un aceite amarillento. Para purificar este crudo se pasó el mismo por una columna cromatográfica de gel de sílice, la cual se eluyó con hexano “redestilado”. De esta manera se obtuvo un sólido blanco (P. F. = 48-52 ºC) (Reportado: 51,5-52 ºC; Martin & Robinson, 1943). Rendimiento: 88%. 5. SÍNTESIS DE LA 3-METOXI-4-METIL-1,3,5(10)-TRIEN-DECALIN-9-ONA [4]
A una solución del metoxidecalina [3] [0,095g (0,54mmol)] en ácido acético [1,4 mL (24,5mmol)] se le añadió, gota a gota durante un período de 30 minutos, 2,24 mL de una solución de ácido crómico en mezclas agua/ácido acético (10% v/v). La mezcla de reacción se mantuvo en agitación a una temperatura inferior a la ambiental (entre 17-21º C) y la evolución de la reacción fue controlada por TLC, observándose que la misma se completó al cabo de dos horas.

72
Transcurrido este tiempo, la mezcla de reacción fue vertida en agua destilada y extraída exhaustivamente con éter. La fase orgánica fue lavada con una solución saturada de bicarbonato de sodio y luego con agua; posteriormente fue secada sobre MgSO4 anhidro y una vez filtrada fue concentrado a sequedad en un rotavapor. El residuo obtenido en la concentración fue purificado por cromatografía preparativa en placas de gel de sílice, eluídas con una mezcla hexano-éter 19:1. De esta manera se obtuvo un sólido blanco que se recristalizó en hexano fijando su punto de fusión en 112-112,5 ºC (Reportado: 112-113 ºC; Martin & Robinson, 1943). Rendimiento. 76%.
6. SÍNTESIS DE LA 3-METOXI-4, 8-DIMETIL-1,3,5(10)-TRIEN-DECALIN-9-ONA [6]
En un balón de dos bocas, provisto de una trampa de gases y un agitador magnético, se colocó, bajo atmósfera inerte, hidruro de sodio disuelto en 3 mL de THF seco. Posteriormente se agregó trietilborano [0,034 mL (0,31mmol)] y finalmente se adicionó la cetona [4] [0,043g (0,24mmol)] disuelta en 3 mL de THF seco. La mezcla se dejó en agitación por una hora a temperatura ambiente bajo flujo de argón; transcurrido este tiempo se adicionó a la mezcla, gota a gota, yoduro de metilo [0,017 mL (0,27mmol)] y se dejó en agitación por treinta minutos más. Luego se adicionó lentamente HCl al 10% y de inmediato se extrajo con éter. La fase etérea fue lavada con solución saturada de cloruro de sodio y con agua destilada, se secó sobre MgSO4 anhidro, se filtró y finalmente se eliminó el solvente a presión reducida. De esta manera se obtuvo un aceite amarillo claro con presencia de un sólido blanco, cuyo TLC evidenció la presencia de dos producto de reacción y restos del producto de partida. Esta mezcla fue separada y el producto mayoritario purificado por cromatografía en placas preparativas de gel de sílice eluidas en hexano. El producto puro resultó ser un sólido blanco de punto de fusión 112º-113 ºC (Reportado 112-113 ºC; J. T. Robinson 1957). Rendimiento: 71%.

73
Conclusiones
1. Se logró ensayar de manera efectiva una nueva ruta sintética para la obtención de la 3-Metoxi-4-metil-1,3,5(10)-trien-decalin-9-ona (6-Metoxi-5-Metil-α-Tetralona [4]) y de la 3-Metoxi-4,8-dimetil-1,3,5(10)-trien-decalin-9-ona (6-Metoxi-2,5-Dimetil-α-Tetralona [6]), las cuales son excelentes intermediarios para la obtención de esteroides y tepenoides.
2. En el desarrollo de este proceso sintético,
• Se demostró que la descarboxilación, en medio básico, de un éster angular α,β insaturado, es una reacción adecuada para generar un sistema aromático. En esta reacción la formación del anillo aromático se ve favorecida por factores termodinámicos y por la salida de una molécula estable como el CO2.
• Se constató que es posible la obtención del intermediario fenólico [2], evitando una deshidrogenación parcial y poco práctica.
• Se pudieron establecer los porcentajes de rendimiento, tanto de los intermediarios sintéticos como la de los productos de síntesis, así como el porcentaje total de la síntesis a partir del producto de partida, siendo éste: 30%.
• Se lograron caracterizar cada una de los intermediarios sintéticos, así como los productos de síntesis [4] y [6], a través de técnicas espectroscópicas básicas: IR, RMN-1H, RMN-13C y EM. De igual manera se asignaron, de manera inequívoca, las señales en los espectros de RMN-1H y RMN-13C de cada uno de estos compuestos.

74
Referencias Bibliográficas
1. ADKINS H. and KRSEK G., 1948. Comparison of Nickel Catalysts in the Hydrogenation of β-Naphthol. J. Am. Chem. Soc., 70, 412.
2. AKIHISA T., YASUKAWA K., OINUMA H., KASAHARA Y., YAMANOUCHI S., TAKIDO
M., KUMAKI K. and TAMURA T., 1996. Triterpene Alcohols from the Flowers of Compositae and their Anti-Inflammatory Effects. Phytochemistry, 43, 1255-60.
3. ALEXANDER N. J., 1995. Future Contraceptives. Sci. Am., 273, 136-41.
4. ATTELE A. S., WU J. A. and YUAN C. S., 1999. Ginseng Pharmacology: Multiple Constituents and Multiple Actions. Biochem. Pharmacol., 58, 1685-93.
5. BANERJEE A. and HURTADO H., 1985. Acid Catalized Novel Rearrangement of Bicyclic Dienones. Tetrahedron, 41, 3029-32.
6. BATHORI M. and PONGRACZ Z., 2005 Phytoecdysteroids. From Isolation to their Effects on Humans. Curr. Med. Chem., 12, 153-72.
7. BELLAMY L. M., 1975. “The Infrared Spectra of Complex Molecules, Vol. 1 and 2.” Chapman and Hall. London.
8. BLICKENSTAFF R. T., GHOSH A. C. and WOLF G. C., 1974. “Total Synthesis of Steroids”. Academic Press. New York.
9. BOLEGO C., POLI A., CIGNARELLA A. and PAOLETTI R., 2003. Phytoestrogens: Pharmacological and Therapeutic Perspectives. Curr. Drug Targets, 4, 77-87.
10. BURNHAM J. W., DUNCAN W. P., EISENBRAUM E. J., KEEN G. W. and HAMMING M. C., 1974. Effects of Alkyl Substituents in the Chromic Acid Oxidation of Tetralins. J. Org. Chem., 39, 1416-1420.
11. COLL J. C. and BOWDEN B. F., 1986. The Application of Vacuum Liquid Chromatography to the Separation of Terpenes Mixtures. J. Nat. Prod., 49, 934-936.
12. CONFORTH J. W., KAUDEN O., PIRE J. E. and ROBINSON R. (Sir.), 1955. Experiment on the Synthesis of Substances Related to the Steroids, Part LIII. Stereospecific Synthesis of a Tricyclic Ketone. J. Chem. Soc., 3348-61.
13. COS P., DE BRUYNE T., APERS S., VANDEN BERGHE D., PIETERS L. and VLIETINCK
A. J., 2003. Phytoestrogens: Recent Developments. Planta Medica, 69, 589-99.

75
14. DEEPAK D., SRIVASTAVA S., KHARE N. K. and KHARE A., 1996. Cardiac Glycosides. En, W. HERZ, G. W. KIRBY, R. E. MOORE, W. STEGLICH and C. H. TAMM (Eds.) “Progress in the Chemistry of Organic Natural Products”, 69, 71-155.
15. DELLA LOGGIA R., TUBARO A., SOSA S., BECKER H., SAAR S. and ISAAC O., 1994. The Role of Triterpenoids in the Topical Anti-Inflammatory Activity of Calendula officinalis Flowers. Planta Medica, 60, 516-20.
16. DENCE J. B., 1980. “Steroids and Peptides: Selected Chemical Aspects for Biology, Biochemistry, and Medicine”. John Wiley. New York.
17. DINAN L., 2001 Phytoecdysteroids: Biological Aspects. Phytochemistry, 57, 325-39.
18. DOYLE P., MACLEAN I. R., MURRAY R. D. H., PARKER W. and RAPHAEL R. A., 1965. Bridged Ring Systems. Part VI. The Total Synthesis of (±)-Clovene. J. Chem. Soc., 1344-51.
19. DUBEY V. S., BHALLA R. and LUTHRA R., 2003. An Overview of the Non-Mevalonate Pathway for Terpenoid Biosynthesis in Plants. J. Biosci., 28, 637-46.
20. EVERS M., POUJADE C., SOLER F., RIBEILL Y., JAMES C., LELIEVRE Y., GUEGUEN J. C., REISDORF D., MORIZE I., PAUWELS R., DE CLERCQ E., HENIN Y., BOUSSEAU
A., MAYAUX J-F., LE PECQ J-B. and DEREU N., 1996. Betulinic Acid Derivatives: A New Class of Human Immunodeficiency Virus Type 1 Specific Inhibitors with a New Mode of Action. J. Med. Chem., 39, 1056-58.
21. FIESER L. F. and FIESER M., 1967. “Reagents for Organic Synthesis”, pag. 295-96. Jonh Wiley & Sons. New York., y referencias ahí citadas.
22. GAWLEY R. E., 1976. The Robinson Annelation and Related Reactions. Synthesis, 777-94
23. GOODWIN T. W., 1981. Biosynthesis of Plant Sterols and Other Triteerpenoids. En, J. W. PORTER and S. L. SPURGEON (Eds.) “Biosynthesis of Isoprenoid Compounds”, pag. 443-80. John Wiley. New York.
24. HARBORNE J. B. and TOMAS-BARBERAN F. A. (Eds.), 1991. “Ecological Chemistry and Biochemistry of Plant Terpenoids ”. Oxford University Press. Oxford (UK).
25. HARTGENS F. and KUIPERS H., 2004. Effects of Androgenic-Anabolic Steroids in Athletes. Sports Med., 34, 513-54.
26. HAUPTMAN P. J., GARG R. and KELLY R. A., 1999. Cardiac Glycosides in the Next Millennium. Prog. Cardiovasc. Dis. 41, 247-54.

76
27. HOUSE H. O., 1972. “Modern Synthetic Reactions, 2nd Ed.”, W. A. Benjamin Inc. London.
28. JUNG M. E., 1976. A Review of Annulation. Tetrahedron, 32, 3-31.
29. KALINOWSKI H. O., BERGER S. and BRAUN S., 1988. “Carbon-13 NMR Spectroscopy.” John Wiley & Sons. Chichester.
30. KASHIWADA Y., HASHIMOTO F., COSENTINO L. M., CHEN C. H., GARRETT P. E. and LEE K-H., 1996. Betulinic Acid and Dihydrobetulinic Acid Derivatives as Potent Anti-HIV Agents. J. Med. Chem., 39, 1016-17.
31. KUZUYAMA T., 2002. Mevalonate and Nonmevalonate Pathways for the Biosynthesis of Isoprene Units. Biosci. Biotechnol. Biochem., 66, 1619-27.
32. LACAILLE DUBOIS M. A. and WAGNER H., 1996. A Review of the Biological and Pharmacological Activities of Saponins. Phytomedicine, 2, 363-86.
33. MARTIN R. H. and ROBINSON R. (Sir.), 1943. Experiment on the Synthesis of Substances Related to the Steroids, Part XLI. Androstenedione. Part I. J. Chem. Soc., 491-97.
34. NEWMAN A. A. (Ed.), 1972. “Chemistry of Terpene and Terpenoids”. Academic Press. New York.
35. NEWMAN D. J., CRAGG G. M. and KENNETH M. S., 2000. The Influence of Natural Products upon Drug Discovery. Nat. Prod. Rep., 17, 215-34.
36. NYANGULU J. M., NELSON K. M., ROSE P. A., YUANZHU G., LOEWEN M., LOUGHEED B., WILSON QUAIL J., CUTLER A. J. and ABRAMS S. R., 2006. Synthesis and Biological Activity of Tetralone Abscisic Acid Analogues. Org. Biom. Chem., 4, 1400 -12.
37. ODELL W. D. and PARKER L. N., 1984. Control of Adrenal Androgen Production. Endocr. Res., 10, 617-30.
38. PAUL T. and MUKHERJEE D., 2003. Stereocontrolled Total Synthesis of (±)-Clovan-3-one and (±)-epi-Clovan-3-one and a Facile Total Synthesis of (±)-Pseudoclovene A. Tetrahedron Lettes, 44, 4985-88.
39. PRESTWICH G. D., 1979. Chemical Defense by Termite Soldiers. J. Chem. Ecol., 14, 175-91.
40. PRIDHAM J. B., 1965. “Terpenoids in Plants”. Academic Press. London
41. QURESHI N. and PORTER J. W., 1981. Conversion of Acetyl-Coenzyme A to Isopentenyl Pyrophosphate, En, J. W. PORTER and S. L. SPURGEON (Eds.) “Biosynthesis of Isoprenoid Compounds”, pag. 47-94. John Wiley. New York.

77
42. RUZICKA L., 1959. History of the Isoprene Rule. Proc. Chem. Soc., 341-60.
43. RUZICKA L., ESCHENMOSER A. and HEUSER H., 1953. The Isoprene Rule and the Biogenesis of Terpenic Compounds. Experientia, 9, 357-67.
44. SCHNEIDER H. P., 2003. Androgens and Antiandrogens. Ann. N. Y. Acad. Sci., 997, 292-306.
45. SETZER W. N. and SETZER M. C., 2003. Plant-Derived Triterpenoids as Potential Antineoplastic Agents. Mini Rev. Med. Chem., 3, 540-56.
46. SHIBATA S., 2001. Chemistry and Cancer Preventing Activities of Ginseng Saponins and Some Related Triterpenoid Compounds. J. Korean Med. Sci., 16 Suppl, S28-S37.
47. SILVEIRA C. S., BRAGA A. L., KAUFMAN T. S. and LENARDA E. J., 2004. Synthetic Approaches to 2-Tetralones. Tetrahedron, 60, 8295-328.
48. STEYN P. S. and VAN HEERDEN F. R., 1998. Bufadienolides of Plant and Animal Origin. Nat. Prod. Rep., 15, 397-413.
49. TEMPLE ROBINSON M. J., 1957. Steroids and Related Compounds I. Synthesis of 1:2:3:4:9:10:11”β”:12”α”-Octahydro-7-methoxy-2”α”:8:11”β”-trimethyl-3-oxophennthrene. Tetrahedron, 1, 49-66.
50. VOGEL A. I., 1974. “Practical Organic Chemistry”. 3st. Edition. Longman. London.
51. WALKER D. and HIEBERT J. D., 1967. 2,3-Dichloro-5,6-dicyanobenzoquinone and Its Reactions. Chem Rev., 67, 153-95.
52. WALLACH O., 1887. Zur Kenntnis der Terpene und ätherische öle, vierte Abhandlung. Liebigs Ann. Chem., 238, 78-88.
53. WILLIS W. B., 1991. “Laboratory Experiments in Liquid Chromatography”. CRC Press. Boca Ratón. USA.
54. WINK M., 2003. Evolution of Secondary Metabolites from an Ecological and Molecular Phylogenetic Perspective. Phytochemistry, 64, 3-19
55. XU R., FAZIO G. C. and MATSUDA S. P. T., 2004. On the Origins of Ttriterpenoid Skeletal Diversity. Phytochemistry, 65, 261-91.