Acylglycerol kinase promotes tumour growth and metastasis ...

© 2002 Nature Publishing Group664 | SEPTEMBER 2002 | VOLUME 2 www.nature.com/reviews/immunol
R E V I E W S
IL-1 and tumour-necrosis factor (TNF), through an NF-κB-mediated event; by IFN-γ through the Janus-activated kinase (JAK)–signal transducer and activatorof transcription (STAT) pathway; or by activating pro-tein 1 (AP1)-mediated transcription. The transcriptionof chemokine genes is often inhibited by transforminggrowth factor-β (TGF-β) and glucocorticoids1.
As the CXC-chemokines CXCL1 and CXCL8 havebeen associated with tumour growth, metastasis andangiogenesis, I concentrate on these importantchemokines. The biological functions of chemokines aremediated through seven-transmembrane G-protein-coupled receptors. CXCL8 and CXCL6 bind to thechemokine receptors CXCR1 and CXCR2, whereasCXCL1 and other CXC-chemokines that have a Glu-Leu-Arg (ELR) motif at their amino terminus (CXCL2,-3 and-5) bind to and activate CXCR2 only2. There arealso viral receptors and binding proteins for thesechemokines3. Receptor binding initiates a series ofdownstream signals, including calcium mobilizationand the activation of phospholipase-Cβ (PLCβ), phos-phatidylinositol 3-kinase (PI3K), RAS, the RHO familyof GTPases, p21-activated kinase (PAK), extracellular
Chemokines are small, pro-inflammatory peptides;they are often expressed in response to the induction ofexpression of nuclear factor-κB (NF-κB) by cytokinesor other stimuli. Chemokines regulate the transport,activation and, sometimes, proliferation of several celltypes, including myeloid, lymphoid, endothelial andepithelial cells1,2. There are four chemokine subfamilies— CXC, C, CX
3C and CC — based on the positions of
conserved cysteine residues near the amino terminus ofthe proteins1 (TABLE 1). Melanoma growth-stimulatoryactivity (CXCL1) and interleukin-8 (IL-8, CXCL8) aremembers of the CXC-chemokine family, which alsoincludes interferon-γ (IFN-γ)-inducible gene 10(CXCL10), IFN-inducible T-cell α-chemoattractant(CXCL11), monocyte induced by IFN-γ (CXCL9),epithelial-derived neutrophil-activating peptide 78(ENA-78, CXCL5), granulocyte chemotactic protein 2(CXCL6), neutrophil activating peptide 2 (CXCL7), amitogen for B-cell progenitors known as stromal-derived factor (CXCL12) and B-cell-activating factor 1/B-lymphocyte chemoattractant (BCA1/BLC, CXCL13)1.The expression of various members of the chemokinesuperfamily is induced by several cytokines, such as
NF-κB, CHEMOKINE GENETRANSCRIPTION AND TUMOUR GROWTHAnn Richmond
The constitutive expression of angiogenic and tumorigenic chemokines by tumour cellsfacilitates the growth of tumours. The transcription of these angiogenic and tumorigenicchemokine genes is modulated, in part, by the nuclear factor-κB (NF-κB) family of transcriptionfactors. In some tumours, there is constitutive activation of the kinases that modulate theactivity of inhibitor of NF-κB (IκB) kinase (IKK), which leads to the constitutive activation ofmembers of the NF-κB family. This activation of NF-κB is associated with the dysregulation oftranscription of genes that encode cytokines, chemokines, adhesion factors and inhibitors ofapoptosis. In this review, I discuss the factors that lie upstream of the NF-κB cascade that are activated during tumorigenesis and the role of the putative NF-κB enhanceosome inconstitutive chemokine gene transcription during tumorigenesis.
Department of VeteransAffairs and Department ofCancer Biology, VanderbiltUniversity School ofMedicine, Nashville,Tennessee 37232, USA.e-mail: [email protected]:10.1038/nri887

© 2002 Nature Publishing GroupNATURE REVIEWS | IMMUNOLOGY VOLUME 2 | SEPTEMBER 2002 | 665
R E V I E W S
had blockedexpression of KC and vascular endothelialgrowth factor (VEGF), which inhibited tumour growthand angiogenesis12. Similar experiments carried outwith xenografts of human squamous-cell carcinomacell lines into severe combined immunodeficient (SCID)mice gave similar results. In general, the greater the con-stitutive activation of NF-κB, the greater the sensitivityto PS-341-induced apoptosis. Using DIFFERENTIAL DISPLAY
and complementary DNA MICROARRAYS to investigate dif-ferences in gene expression between PAM212 cells thatmetastasize and those that do not, Dong et al.13 foundthat of the genes that are differentially expressed in themetastatic cells, 10 out of 22 are NF-κB dependent intheir expression. CXCL1, certain signalling molecules(c-Met, Syk and Yes-associated protein), proteinsinvolved in cell growth (p21, p27 and cyclin D) and pro-teins involved in apoptosis (cellular inhibitor of apop-tosis 1, Iap1; the phosphoprotein enriched in astrocytesof 15 kDa, Pea15; and Fas ligand, FasL) were among theproteins encoded by these NF-κB-inducible genes. So,genes that are regulated by NF-κB, including chemo-kine genes, modulate squamous-cell carcinoma andmelanoma tumorigenesis.
Regulation of expression of chemokine receptors. Theexpression of chemokine receptors is also modulated byNF-κB. The expression of CXCR2 is regulated throughan NF-κB element, and the expression of this receptor isinduced in response to granulocyte–macrophagecolony-stimulatory factor (GM-CSF), IL-4 and IL-13,whereas lipopolysaccharide (LPS) induces the loss ofexpression of CXCR2 (REFS 14–17). The expression ofCXCR1 and CXCR2 has been detected in melanomatumours, which indicates that melanoma cells might betargets for autocrine stimulation by CXCL8 (REF. 9).However, some melanoma tumour cell lines expressCXCR4, CCR7 and CCR10, but no detectable levels ofother chemokine receptors6. The expression of CXCR3and CXCR4 has been noted in BLM and Sk-Mel37human melanoma cells10. The ligand for CXCR4(CXCL12) is produced by lymph node, liver, lung andbone marrow. Ligands for these receptors6 stimulatesmall GTPases and induce reorganization of the actincytoskeleton, which indicates that these receptors mightbe involved in the migration and, perhaps, metastasis ofthese tumour cells. The activation of NF-κB also occursas a consequence of activation of chemokine receptors.The melanoma cell lines that express moderate levels ofCXCR3 and CXCR4 (BLM and SK-Mel37) have barelydetectable levels of immunoreactive CXCR1 andCXCR2. When B16 melanoma cells were transducedwith a retroviral expression vector for CCR7, enhancedlymph-node metastasis was observed, which could beblocked by antibodies specific for secondary lymphoidtissue chemokine (CCL21), the ligand for CCR7 (REF. 11).Together, these studies indicate that there is differentialexpression of chemokine receptors between melanomacell lines and melanoma tumours. The expression ofthese receptors can often be correlated with the meta-static capacity of the tumour cells and their organ-specific metastasis.
signal-regulated kinases 1 and 2 (ERK1 and ERK2),p38 mitogen-activated protein kinase (p38 MAPK) andNF-κB1,4,5.
Both the CXC-chemokines and their receptors, andthe CC-chemokines and their receptors, are involved intumorigenesis and metastasis6. Expression of CXC-chemokines that have an ELR motif at the amino termi-nus is associated with enhanced tumorigenic capacityand metastasis, whereas expression of ELR-negativeCXC-chemokines, C-chemokines and CC-chemokinesby tumours is often associated with the inhibition oftumour growth or regression3,7. Much research into thedysregulated expression of chemokines during tumourformation has used the melanoma model, in whichCXCL1, CXCL8 and regulated on activation, normal T-cell expressed and secreted (RANTES, CCL5) areexpressed frequently at high levels in metastatic lesions8.This constitutive expression of chemokines occurs dueto transcriptional activation of the encoding genes. Inpart, the endogenous transcription of chemokine genesin melanoma cells is due to altered NF-κB activation.Tumour angiogenesis, growth and metastasis are facili-tated by the NF-κB-modulated transcription ofchemokine genes. In this review, I explore the mecha-nisms by which constitutively activated kinases thatfunction upstream of the NF-κB cascade facilitatechemokine-mediated tumorigenesis. I also examine therole of NF-κB-interacting factors — which are thoughtto form an ENHANCEOSOME-like structure — in the dysreg-ulation of transcription of chemokine genes duringtumour development.
Chemokines and cancerConstitutive secretion of chemokines in melanoma.CXCL1, CXCL8 and CCL5 have been shown to beexpressed constitutively in human melanoma celllines and tumours (see REF. 8 for a full review). The over-expression of these chemokines in melanocytes andmelanoma tumour cells is associated with enhancedtumour-forming capacity and metastatic potential.Antibodies that are specific for these ligands or theirreceptors slow the growth of melanoma tumours inmice8. Human melanoma lesions are reported to expressseveral chemokine receptors6,8–11.
A role for CXCL1 and CXCL8 in tumorigenesis is sup-ported further by data showing that the overexpressionof one of the mouse homologues of CXCL1 — knownas KC (on the basis of the ordinate and abscissa of thecloning position) — enhanced the growth and metasta-sis of KC-deficient PAM212 squamous-cell carcinomacells12. KC-expressing PAM212 cells injected intoBALB/c mice formed tumours that grew more rapidlythan PAM212 parental cells, but this growth advantageassociated with the expression of KC was not observedin BALB/c Cxcr2 −/− mice. When BALB/c mice wereinjected with PAM–Ly2 cells — a metastatic variant ofPAM212 that expresses KC — and the tumour-bearingmice were treated with the NF-κB inhibitor PS-341,which blocks the proteasomal degradation of inhibitorof NF-κB (IκB), the growth of the tumours was inhib-ited by 50%. The mice that were treated with PS-341
ENHANCEOSOME
Gene transcription is achieved bythe assembly of higher-order,three-dimensional transcriptionfactor/enhancer DNAcomplexes, termedenhanceosomes. Enhanceosomesactivate transcription byrecruiting chromatin-modifyingactivities and basal transcriptionfactors to the nearby promoters.
SCID
(Severe combinedimmunodeficiency). Mice withthis defect in their immunesystem do not have B or T cellsand can, therefore, accepttumour cells from anotherspecies without rejection.
DIFFERENTIAL DISPLAY
This is a powerful tool for thecomparison of gene expressionbetween two or more messengerRNA populations.
MICROARRAYS
This technique allows thescreening of messenger RNAextracted from cells against DNAfrom many thousands of genes.The DNA from each gene ispositioned on a solid support ina highly ordered array.

© 2002 Nature Publishing Group666 | SEPTEMBER 2002 | VOLUME 2 www.nature.com/reviews/immunol
R E V I E W S
Table 1 | CXC-, C-, CX3C- and CC-chemokine and receptor families
Systematic name Chromosome Human ligand Mouse ligand Chemokine receptor(s)
CXC-chemokine/receptor family
CXCL1 4q21.1 GROα/MGSAα GRO/MIP2/KC? CXCR2>CXCR1
CXCL2 4q21.1 GROβ/MGSAβ GRO/MIP2/KC? CXCR2
CXCL3 4q21.1 GROγ/MGSAγ GRO/MIP2/KC? CXCR2
CXCL4 4q21.1 PF4 PF4 Unknown
CXCL5 4q21.1 ENA78 GCP2/LIX? CXCR2
CXCL6 4q21.1 GCP2 GCP2/LIX? CXCR1, CXCR2
CXCL7 4q21.1 NAP2 Unknown CXCR2
CXCL8 4q21.1 IL-8 Unknown CXCR1, CXCR2
CXCL9 4q21.1 MIG MIG CXCR3 (CD183)
CXCL10 4q21.1 IP10 IP10/CRG2 CXCR3 (CD183)
CXCL11 4q21.1 ITAC ITAC CXCR3 (CD183)
CXCL12 10q11.21 SDF1α/β SDF1/PBSF CXCR4 (CD184)
CXCL13 4q21.1 BCA1 BLC CXCR5
CXCL14 5q31.1 BRAK/Bolckine BRAK Unknown
(CXCL15) Unknown Unknown Lungkine/WECHE Unknown
CXCL16 17p13 Unknown Unknown CXCR6
C-chemokine/receptor family
XCL1 1q24.2 Lymphotactin/SCM1α/ATAC Lymphotactin XCR1
CX3C-chemokine/receptor family
CX3CL1 16q13 Fractalkine Neurotactin/ABCD3 CX3CR1
CC-chemokine/receptor family
CCL1 17q11.2 I309 TCA3/P500 CCR3
CCL2 17q11.2 MCP1/MCAF/TDCF JE? CCR2
CCL3 17q12 MIP1α/LD78α MIP1α CCR1, CCR5
CCL3L1 17q12 LD78β Unknown CCR1, CCR5
CCL4 17q12 MIP1β MIP1β CCR5 (CD195)
CCL5 17q12 RANTES RANTES CCR1, CCR3, CCR5 (CD195)
(CCL6) Unknown C10/MRP1 Unknown
CCL7 17q11.2 MCP3 MARC? CCR1, CCR2, CCR3
CCL8 17q11.2 MCP2 MCP2? CCR3, CCR5 (CD195)
(CCL9/10) Unknown MRP2/CCF18/MIP1γ CCR1
CCL11 17q11.2 Eotaxin Eotaxin CCR3
(CCL12) Unknown MCP5 CCR3
CCL13 17q11.2 MCP4 Unknown CCR2, CCR3
CCL14 17q12 HCC1 Unknown CCR1, CCR5
CCL15 17q12 HCC/LKN1/MIP1δ Unknown CCR1, CCR3
CCL16 17q12 HCC4/LEC/LCC1 Unknown CCR1, CCR2
CCL17 16q13 TARC TARC/ABCD2 CCR4
CCL18 17q12 DC-CK1/PARC/AMAC1 Unknown Unknown
CCL19 9p13.3 MIP3β/ELC/exodus-3 MIP3β/ELC/exodus-3 CCR7 (CD197)
CCL20 2q36.3 MIP3α/LARC/exodus-1 MIP3α/LARC/exodus-1 CCR6
CCL21 9p13.3 6Ckine/SLC/exodus-2 6Ckine/SLC/exodus-2/TCA4 CCR7 (CD197)
CCL22 16q13 MDC/STCP1 ABCD1 CCR4
CCL23 17q12 MPIF1/CKβ8/CKβ8-1 Unknown CCR1
CCL24 7q11.23 Eotaxin-2/MPIF2 MPIF2 CCR3
CCL25 19p13.3 TECK TECK CCR9
CCL26 7q11.23 Eotaxin-3 Unknown CCR3
CCL27 9p13.3 CTACK/ILC ALP/CTACK/ILC/ESkine CCR10 CCL28 5p12 MEC CCR3/CCR10
Reproduced, with permission, from REF. 123 (© 2001 The Society for Leukocyte Biology).

© 2002 Nature Publishing GroupNATURE REVIEWS | IMMUNOLOGY VOLUME 2 | SEPTEMBER 2002 | 667
R E V I E W S
ANOXIA
Limited oxygen supply.
FOCUS FORMATION
The ability of tumour cells to grow in an anchorage-independent manner byadhering to one another, therebyforming ‘foci’ of tumour cells.
NEMO
(NF-κB essential modulator).This is the regulatorycomponent of the inhibitor ofNF-κB (IκB) kinase (IKK)complex, also known as IKKγ.
which indicates that chemokine autocrine loops mightbe important for perpetuating the constitutive activationof NF-κB in melanoma, but that additional cytokines oralterations in the signal-transduction pathway also contribute to the dysregulation of NF-κB in melanoma23.
CXCL8, anoxia and tumour aggressiveness. Treatment ofhuman malignant melanoma cells with ionizing radia-tion induces the coordinated activation of NF-κB andother regulators of transcription24. The activation ofNF-κB correlates with CXCL8 expression, growth,angiogenesis and metastasis of human melanomacells25,26. The induction of transcription of CXCL8 innecrotic/ANOXIC areas of melanoma tumours is depen-dent on NF-κB and AP1 activity27. Transfection ofmelanoma cells with a dominant-negative inhibitor ofNF-κB reduced tumour growth, metastatic potential,CXCL8 production and growth of blood vessels into thetumour28. These data support an association betweenthe endogenous activation of NF-κB and the enhancedmetastatic potential of malignant melanoma cells, andindicate that targeting NF-κB might have some poten-tial therapeutic effects in clinical trials. In support of thisconcept, the adenoviral-mediated transfer of IκB tomelanoma cells renders them sensitive to TNF-inducedcytotoxicity29. Using a cell-culture system, we haveshown that delivery of the super-repressor form of IκB— which cannot be phosphorylated on Ser32 or Ser36and, therefore, is not targeted for ubiquitylation anddegradation by the proteasome — to melanoma cellsblocks FOCUS FORMATION and targets tumour cells toundergo apoptosis30.
Mechanism of dysregulation of NF-κB. The NF-κB pro-teins constitute a family of proteins that are homologousto the chicken oncogene rel. These proteins are regulatedat several levels — transcription, translation and post-translational processing (BOX 1). Normally, NF-κB (a p65–p50 or p65–p52 heterodimer) is sequestered inthe cytoplasm, owing to the binding of IκB to thenuclear-localization sequence of the p65/RELA NF-κBprotein. Activating signals lead to the phosphorylationand ubiquitylation of IκB and its degradation by the 26Sproteasome, thereby freeing the NF-κB complex to moveto the nucleus and bind specific DNA promotersequences31. Much work has been done to clarify the roleof IκB phosphorylation and ubiquitylation in its targetingto the proteasome32–34. Inducible pathways for the regula-tion of these events involve activation of the IκB kinase(IKK) complex — comprising IKKα, IKKβ and the adap-tor molecule IKKγ (NEMO) — which leads to the phos-phorylation of specific serine residues of IκB (Ser32 andSer36 of IκBα, or Ser19 and Ser23 of IκBβ). In addition,there are non-inducible pathways for the ubiquitylationand degradation of IκB, which result in the targeting ofthis protein for degradation by the proteasome35.
Several members of the mitogen-activated proteinkinase kinase kinase (MAP3K) family, such as NF-κB-inducing kinase (NIK) and MEKK1, affect the activationof NF-κB (FIG. 1). NIK was thought initially to phospho-rylate and activate IKKα preferentially, whereas MEKK1
CXCL8 and its receptors, CXCR1 and CXCR2, areexpressed in ovarian-cancer cells18. CXCR1 is expressedlargely on the luminal side of the epithelial tumour cells,but there is no such polar distribution of CXCR2.CXCL8 and both CXCR1 and CXCR2 are expressed inhuman colon-carcinoma cells, and antibodies specific forCXCR1, CXCR2 or CXCL8 inhibited the invasive capac-ity of colon-carcinoma cells19. Antibodies specific forCXCR1 or CXCR2 also produced a moderate inhibitionof the proliferation of the colon-cancer cells and of theadhesion of colon-carcinoma cells to endothelial cells.
NF-κκB activation and cancerSeveral tumour systems have been shown to have constit-utive activation of NF-κB. This enhanced NF-κB activityis thought to allow tumour cells to constitutively expressboth angiogenic and angiostatic chemokines, cytokinessuch as VEGF, IL-1 and IL-6, and other factors that arereported to affect melanoma growth and escape fromapoptosis. The expression of CCL5 in melanoma andbreast cancer has been shown to correlate with themetastatic capacity of the tumour20,21. Because the expres-sion of CCL5 is induced by NF-κB22, down-modulationof CCL5 through the inhibition of NF-κB offers promisefor therapeutic intervention.We have observed that oncethe expression of CXCL1 and CXCL8 has been induced,these secreted chemokines feed back on the activation ofNF-κB to keep the cycle going. The endogenous activa-tion of NF-κB in melanoma tumour cells can be inhib-ited by ~50% with an antibody specific for CXCL1,
Box 1 | Members of the REL family and their regulators
Members that activate transcription• v-Rel — an avian retroviral protein
• c-REL — the cellular homologue of v-Rel
• RELA/p65 — the main REL protein
• RELB — a less commonly expressed REL protein
• Dorsal — a Drosophila Rel-like protein
• Dorsal-related immunity factor (Dif) — a Drosophila Rel-like protein
• p50 subunit of nuclear factor-κB (NF-κB) — derived from processing of p105
• p52 subunit of NF-κB — derived from processing of p100
Members that sequester NF-κB in the cytoplasm• Inhibitor of NF-κB, α (IκBα)
• IκBβ
• IκBε — lacks the PEST domain (a sequence that is rich in proline, glutamate, serine and threonine residues)
• IκBγ
• Drosophila Relish
Members that enhance transcription or restrain NF-κB nuclear localization• B-cell leukaemia/lymphoma 3 (BCL3)
Kinases that phosphorylate IκB• IκB kinase-α (IKKα) — phosphorylates IκB and facilitates the processing of p105
• IKKβ — phosphorylates IκBα at Ser32 and Ser36, and IκBβ at Ser19 and Ser23
Adaptor molecules for the IKKs• IKKγ (NF-κB essential modulator, NEMO) — a regulatory factor for IKKα and IKKβ

© 2002 Nature Publishing Group668 | SEPTEMBER 2002 | VOLUME 2 www.nature.com/reviews/immunol
R E V I E W S
The dysregulation of NF-κB in haematopoietictumours, as well as solid tumours, has gradually becomeestablished as a paradigm31 (BOX 2). This alteration in NF-κB activity is often accompanied by altered expres-sion of chemokines, growth factors and cytokines, onco-genes and tumour suppressors30,40–46, adhesion proteinsand proteases47, and inhibitors of apoptosis48. Increasedexpression of p50 or p100 (the precursor of p52) hasbeen reported in biopsies of non-small-cell lung cancerand human breast cancer, as well as head and neck cancercells49–52. During TPA/DMBA (tetradecanoylphorbol 13-acetate/7,12-dimethylbenz(a)anthracene)-mediatedinduction of mouse skin cancer, there is an increase inthe nuclear localization of p50 and B-cell leukaemia/lymphoma 3 (Bcl3) in the developing tumours53. Thetransformation of human thyroid cancer cell lines is anNF-κB-related event54. Nuclear localization of NF-κB isassociated with breast cancer and might be related tothe escape of these tumour cells from apoptosis55.Malignant melanoma cells have elevated constitutiveactivation of IKK compared with normal human epi-dermal melanocytes, which results in the more rapiddegradation of IκB, nuclear localization of p50–p65NF-κB and enhanced transactivating capacity of theNF-κB complex23,56,57. As the activation of NF-κBinduces the transcription of inhibitors of apoptosis, aswell as factors that are associated with tumour angio-genesis, metastasis and growth, we have come to under-stand that NF-κB could be an important potentialtherapeutic target in cancer, particularly if we can iden-tify which of the upstream kinases that affect NF-κBbecome dysregulated during tumorigenesis. Moreover,it has been shown clearly that blocking the activity ofNF-κB targets tumour cells for apoptosis12,58. In cellmodels in which the activation of NF-κB facilitates theescape of tumour cells from apoptosis59,60, the co-administration of an inhibitor of NF-κB, such as theproteasome inhibitor PS-341, would be expected toincrease tumour kill after irradiation14.
NIK-mediated modulation of NF-κB. NIK was firstidentified on the basis of its association with TNF-RECEPTOR-
ASSOCIATED FACTOR 2 (TRAF2)61. NIK associates with, andis reported to activate the phosphorylation of, IKKαand IKKβ. When overexpressed, NIK enhances NF-κBactivity, and the expression of kinase-defective NIKblocks NF-κB activation in response to most inducers.However, NIK-deficient mice still respond to TNF andIL-1 with enhanced NF-κB activity, which indicates thatNIK is not required for the activation of NF-κB by thesecytokines. NIK is involved in the processing of p100 tothe NF-κB p52 subunit62. This p52 subunit eitherhomodimerizes or interacts with p65 to form the NF-κBp65–p52 heterodimer. So, the enhanced NIK activity intumours57 could be associated with the enhanced levelof nuclear p52 that is reported to be present in breastcancers63.NIK is associated also with the phosphorylationand activation of IKKα, which can affect the transactiv-ation potential of p65 (REF. 64). Mice that have a mut-ation in NIK (aly/aly mice) have a loss of signalling inresponse to the chemokines CCL21 and CXCL13 (REF. 64).
was proposed to phosphorylate and activate IKKβ preferentially36,37. However, recent work with Nik−/−
and Mekk1−/− mice indicates that NIK and MEKK1 arenot essential for TNF- and IL-1-mediated induction ofNF-κB, but are essential for the activation of NF-κB by other factors. For example, NIK is required for the lymphotoxin-β (LTβ)-mediated activation of NF-κB38.Once in the nucleus, NF-κB binds DNA; then, additionalproteins, such as CREB-binding protein (CBP)/p300,bind to NF-κB39 and stabilize transactivation throughthe formation of an enhanceosome-like structure.
TNF-RECEPTOR-ASSOCIATED
FACTORS
(TRAFs). A term that originatedwith proteins that were found tobind to the cytoplasmic domainof the tumour-necrosis factor(TNF) receptor in a yeast two-hybrid screen.
p65p50
p65p50
NEMO
p65p50
IκB
TAK1
AKT
PI3K
NIK
TRAF2
ERK1/2
26S proteasome
Nucleus
IKKβ IKKα
PP P
P
P
P
p65p50
IκB
Weak genetranscription
Strong genetranscription
??
TRAF3
LTβ
LTβR
TRAF5
TRAF6
Chemokine
Chemokinereceptor
Ubiquitinligase
PTEN PtdInsP3
Figure 1 | NF-κκB is activated by plasma-membrane receptors that transduce signals tokinases such as PI3K. Phosphatidylinositol 3-kinase (PI3K) can activate AKT, which potentiallyaffects the phosphorylation of both inhibitor of NF-κB (IκB) kinase (IKK; a complex of IKKα, IKKβ andNEMO) and p65 (RELA). In addition, linker proteins, such as tumour-necrosis factor (TNF)-receptor-associated factors (TRAFs), participate in the activation of NF-κB-inducing kinase (NIK), as well astransforming-growth-factor-β-activated kinase 1 (TAK1). NIK-mediated enhancement of NF-κBactivity might be facilitated by its activation of mitogen-activated protein (MAP) kinases, such asextracellular-signal-regulated kinase 1 (ERK1) and ERK2. The activation of IKK leads to enhancedphosphorylation of IκB, followed by its ubiquitylation then degradation by the 26S proteasome. Thisfrees the NF-κB p50 and p65 subunits to be transported to the nucleus. Nuclear NF-κB p50–p65heterodimers that are not phosphorylated on the p65 subunit can only partially activate genetranscription, whereas if p65 is phosphorylated, the enhancement of transcription is greatlymagnified. LTβ, lymphotoxin-β; LTβR, lymphotoxin-β receptor; NEMO, NF-κB essential modulator;PtdInsP3, phosphatidylinositol-3,4,5-trisphosphate; PTEN, phosphatase and tensin homologue.

© 2002 Nature Publishing GroupNATURE REVIEWS | IMMUNOLOGY VOLUME 2 | SEPTEMBER 2002 | 669
R E V I E W S
Ser536 (REFS 69–73). In response to the activation ofPI3K, AKT is recruited to the cell membrane by phos-phatidylinositol-3,4,5-trisphosphate (PtdInsP
3) or
phosphatidylinositol-3,4-bisphosphate, where itbecomes phosphorylated by 3′-phosphoinositide-dependent kinase 1 (PDK1) on Thr308 in the kinasedomain and by PDK2 on Ser473. AKT phosphorylatesseveral substrates at an Arg-Xaa-Arg-Xaa-Xaa-Ser/Thrmotif, some of which mediate escape from apoptosisand contribute to the activation of nuclear NF-κB.Indeed, IKKα has a perfect consensus sequence forAKT-mediated phosphorylation, and Li and Stark havesuggested that AKT-mediated activation of IKKα couldoccur upstream of the phosphorylation of p65 byIKK70. However, work from Delhase et al. does not sup-port this possibility71. Moreover, phosphorylation of thep50 NF-κB subunit by the PI3K–AKT pathway is alsoreported to increase the binding of the NF-κB complexto DNA72.
The lipid phosphatase known as phosphatase andtensin homologue (PTEN) is known to switch off theactivation of AKT by dephosphorylating PtdInsP
3,
which blocks the membrane localization of AKT. PTENis also reported to block NF-κB activation withoutaffecting the IκB degradation pathway72. PTEN is oftendeleted or mutated in melanoma and other types oftumour74,75. The activation of AKT that we observe insome human melanoma cells in vitro is accompanied bythe loss or reduction of expression of PTEN (P. Dhawan,D. Ellis and R. A., unpublished observations). The loss ofPTEN with consequent enhanced activation of AKT hasalso been reported for several other types of tumour,which indicates that this is a frequent step associatedwith transformation.
RAS-mediated activation of NF-κB. The activation ofNF-κB can suppress apoptosis induced by oncogenicRAS76. The RAS superfamily is comprised of smallGTPase proteins. One family member, HRAS (namedafter the Harvey-Ras oncogene), is associated withtransformation. Paradoxically, when cells are trans-formed with HRAS, transcriptional activity is mediatedthrough an NF-κB element, although this does notresult in an increase in the nuclear level of p50–p65 (REF. 77). Oncogenic RAS activates NF-κB transcrip-tional activity77. Moreover, inhibitors of PI3K block theendogenous NF-κB activity of malignant melanomacells, although these inhibitors will not block IKK-mediated phosphorylation of IκBα (P. Dhawan, D. Ellisand R.A., unpublished observations). Embryonicfibroblasts from both IKKβ- and IKKα-null mice can-not induce NF-κB transactivation. However, the restora-tion of IKKβ in IKKαβ-null mice will not restorecytokine-stimulated activation of RELA/p65 (REF. 69).Both IKKα and IKKβ are required for the activation ofthe transactivating activity of the RELA/p65 subunit ofNF-κB, presumably because IKKα is required for thephosphorylation and transactivation of p65 (REF. 69). So,one can envision a pathway whereby RAS activatesPI3K, which activates AKT, which activates IKKα-mediated phosphorylation of p65.
The chemotactic response is lost, as is the activation ofNF-κB in response to CCL21-mediated activation ofCCR7 (REF. 65).
Dysregulation of NIK NF-κB activity in melanoma.IKK-associated NIK activity is enhanced in melanomacell lines compared with normal human epidermalmelanocytes (NHEMs)57. A catalytically inactive formof NIK blocks constitutive NF-κB or CXCL1 pro-moter activity in melanoma cells, but not in controlNHEMs. Transient overexpression of wild-type NIKresults in increased phosphorylation of ERK1 andERK2. This phosphorylation is inhibited in a concen-tration-dependent manner by PD98059, an inhibitorof p42/p44 MAPK. Moreover, the NF-κB promoteractivity in melanoma cells decreases with the over-expression of dominant-negative ERK57. Although thesedata support the hypothesis that NIK-mediated ERKactivation contributes to NF-κB activity, they should notbe over-interpreted because transfection/overexpressionmodels were used.
AKT modulates NF-κB transactivation. Once IκB isdegraded by the proteasome, the nuclear-localizationsequence of the p50–p65 or p52–p65 NF-κB complex isexposed and, consequently, this complex can move to thenucleus and bind to the NF-κB element of the promoterof responsive genes. However, simply binding to theDNA is not sufficient. The p65 component needs to bephosphorylated in its transactivation domain to be fullyactive. Zhong et al .66 showed that phosphorylation ofp65 on Ser276 by protein kinase A (PKA) is required forits transactivation functions. In addition, other kinases,including casein kinase II (CKII), phosphorylate p65 atserine residues that differ from those that are phosphory-lated by PKA, and this enhances the transcription activityof NF-κB67. The p65 phosphorylation event might alsobe mediated by IKKβ68 or, possibly, IKKα69. In addition,it has been proposed recently that the serine/threonineprotein kinase AKT, also known as PKB — which wasisolated from the AKR mouse thymoma — might alsofacilitate the phosphorylation of p65 on Ser529 and
Box 2 | Tumours that have elevated NF-κκB
• Melanoma23,26,28,40–41,56
• Mammary carcinoma49,55,63,85
• Non-small-cell lung carcinoma51
• Colorectal carcinoma52,58
• Squamous-cell carcinoma50,53
• Leukaemia47,81,82,114
• Lymphoma47,116
• Thyroid carcinoma54
• Fibrosarcoma117
• Pancreatic cancer118
• Prostate cancer119
• Multiple myeloma113
• Ovarian cancer18

© 2002 Nature Publishing Group670 | SEPTEMBER 2002 | VOLUME 2 www.nature.com/reviews/immunol
R E V I E W S
NF-κB activation have been described, including RAS,RAF and TAX78–83. Protein kinase C (PKC) has also beennoted as a potential modulator of IKK activity, andPKC-mediated activation of IKK is linked to tumorigen-esis84–88. So, when trying to discern why there is dysregu-lation of NF-κB in tumour cells, it is important to lookat several upstream signal-transduction pathways.
Chemokines and NF-κBNF-κB has been shown to be an essential modulator oftranscription of the following chemokines: CXCL1, -2,-3, -5, -8, -9, -10 and -12, and CCL2, -3, -4, -5, -11 and -17. However, the context of the NF-κB element in thepromoter of the 5′ regulatory regions of these genesvaries considerably. This finding probably indicates thatthere are more-subtle mechanisms for the modulationof transcription that involve other co-regulatory factors.The constitutive expression of chemokines in tumourcells has been documented extensively. However, eventhough the expression of many of the chemokines isregulated by NF-κB and tumours have constitutiveactivation of NF-κB, there is variability in the expres-sion of specific chemokines between tumours. As theELR-motif-containing CXC-chemokines CXCL1 andCXCL8 are known for their modulation of tumourangiogenesis and tumour growth, I concentrate hereon the modulation of the transcription of thesechemokines by NF-κB.
Activation of NF-κB enhances chemokine transcription.The constitutive activation of NF-κB facilitates theimmortalization of melanoma and mammary tumourcells, together with their escape from apoptosis89.One consequence of this dysregulated NF-κB activ-ity23,55–58,61–68 is that there is constitutive transcription ofthe NF-κB-dependent chemokines CXCL1 and CXCL8in human tumours90–96. Constitutive expression of thesechemokines can transform immortalized melanocytesand is associated with increased metastasis in colon carci-noma, squamous-cell carcinoma and melanoma19,28,91,97.
Transcriptional regulation of CXCL1 and CXCL8.Although cytokine-mediated activation of NF-κB is themain mechanism for the transcriptional induction ofchemokines, NF-κB does not act alone in this induction.Moreover, the factors that interact with NF-κB to modu-late transcription differ between CXCL1 and CXCL8(FIG. 2). Whereas cooperation between NF-κB and eitherAP1 or the CCAAT/enhancer-binding protein (C/EBP)element is required for the transcriptional modulation ofCXCL8 (REF. 96), for CXCL1, NF-κB cooperates with atranscription factor that binds to GC-rich complexes(SP1), the high-mobility group protein HMGIY (asdetermined by an ELECTROPHORETIC MOBILITY-SHIFT ASSAY andimmediate upstream region (IUR) elements to modu-late the transcription of this chemokine42. SP1 and IURelements are involved in basal and cytokine-inducedtranscription of the CXCL1 gene. The factors that bindto these elements might form an enhanceosome-liketranscriptional response element. The IUR binds bothpositive and negative regulatory factors. The 115-kDa
Other kinases can modulate NF-κB. Although manykinases have been shown to contribute to the activationof NF-κB, it has been difficult to show clearly therequirement for an activating kinase upstream of theIKK complex. NIK is a MAP3K, as are TGF-β-activatedkinase 1 (TAK1), MEKK1 and MEKK3 (see REF. 70 for areview). Deletion of NIK and MEKK1 does not blockTNF- or IL-1-mediated induction of IKK activity, butthe possibility that TAK1 and MEKK3 are potentialmodulators of IL-1- and TNF-mediated induction ofIKK remains open. NIK is required for LTβ-receptor-mediated activation of IKK38. In addition to theMAP3Ks and AKT, several other indirect modulators of
TATA BOX
A DNA motif that binds severalfactors (TATA-binding proteins,TBPs; and TBP-associatedfactors, TAFs) that facilitate theinitiation of transcription.
HDAC1
AP1 CRE NF-κB TATAC/EBP
CBPCBP
IREIRE NF-κB TATA
CBP
SP1 NF-κBIUR TATA
CBP?
CDP
CBP?
–1168
–550
–350
CREB
RBP-Jκ
HDAC1
HDAC1?
NRF
PARP
AP1
p65p50
P
p65p50
P
p65p50
P
HATHAT
HAT
HATHAT
CXCL8
CXCL1
IL-6
C/EBP
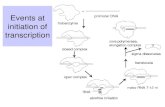
Figure 2 | Similarities between the proposed enhanceosomes for IL-6, CXCL8 andCXCL1. Similar key regulatory elements participate in the modulation of transcription of CXCL8,interleukin-6 (IL-6) and CXCL1. The transcription of CXCL8 is largely modulated through a nuclearfactor-κB (NF-κB) element, which works in concert with adjacent activating protein 1 (AP1) and/orC/EBP elements to modulate the expression of this chemokine. The transcription of CXCL8 isalso modulated by an NF-κB-repressing factor (NRF)120. This negative regulatory element has adual role; in the absence of stimulation, NRF inhibits the transcription of CXCL8, but after an IL-1signal, the NRF is required for the full induction of CXCL8 transcription. An enhanceosome modelof cytokine gene expression, analogous to the CXCL8 paradigm, has been proposed for theregulation of the IL-6 promoter. Both CXCL8 and IL-6 promoters have binding sequences for NF-κB, C/EBP and the TATA BOX121. There is competition between NF-κB and an inhibitory factor,RBP-Jκ for the NF-κB promotor element. Promoter activation relies on the p65 NF-κB subunit,which recruits CREB-binding protein (CBP/p300) to the site122. The binding of CBP/p300, whichhas histone acetylase (HAT) activity, will stabilize transcription from these promoters. In anindependent study, CCAAT displacement protein (CDP) has been shown to interact physicallywith CBP/p300 and is a target for acetylation at specific residues near the homeodomain. So,CDP and CBP seem to have antagonistic roles in the regulation of IL-6 and CXCL8 transcription.The transcriptional repression of CDP is probably associated with its ability to recruit the histone-deacetylase activity of HDAC1. HDAC1 deacetylates the histones and, as a result, the chromatinstructure is closed and unavailable for transcription, with the end result being the consequentsilencing of the chromatin. We propose that similar interactions between NF-κB, CBP and CDPmight be involved in the transcription of CXCL1. c/EBP, CCAAT, enhancer-binding protein; CRE, cyclic AMP response element; IRE, interferon response element; IUR, immediate upstreamregion; PARP, poly-ADP ribose polymerase; RBP-Jκ, recombination signal binding protein-Jκ.

© 2002 Nature Publishing GroupNATURE REVIEWS | IMMUNOLOGY VOLUME 2 | SEPTEMBER 2002 | 671
R E V I E W S
Within the IUR element, there is an overlapping con-sensus sequence (GGGATCGATC) that binds the170–180-kDa CDP protein. Mutations in the IUR ele-ment that map to the putative CDP-binding site inhibitthe binding of CDP to the IUR element and favourincreased transcription from the CXCL1 promoter. CDPcan negatively regulate the expression of CXCL1,whereas PARP can potentiate the transcription of thisgene99–101. CDP has been shown previously to repressactively the transcription of several other genes101–105
poly-ADP ribose polymerase (PARP) binds to theTCGATC sequence of the IUR98, and thereby positivelymodulates CXCL1 transcription, perhaps by displacingthe binding of the negative modulatory factor CCAAT-displacement protein (CDP)99. Inhibitors of the ADP-ribose-polymerase function of PARP reduce CXCL1promoter activity, which indicates that not only DNAbinding, but also the enzymatic activity of PARP, mightbe required for the co-activating function of PARP inCXCL1 transcription98,99.
ELECTROPHORETIC MOBILITY-
SHIFT ASSAY
A technique for detectingDNA–protein complexformation. It involves theincubation of nuclear extractswith a radiolabelledoligonucleotide probe, thenseparating the probe that hasbound to nuclear proteins fromthe free radiolabelled probe bygel electrophoresis, followed byautoradiography.
NEMO
RAS
MEKK3
AKT
PI3KNIK
26S proteasome
Nucleus
IKKβ IKKα
p65p50
P
PP
p65p50
IκB
TRAFs
LTβ
LTβRChemokine
Chemokinereceptor
NF-κB activation
p16
SP1 NF-κBIUR TATA
CBP
PARP
HDAC1
p65p50
PCXCL1 transcription
I II III IV
CDP
HAT
NSAIDs
NEMO-binding peptide
PS-341
Degradation
CBP
PTEN PtdInsP3MEKK1
Figure 3 | Model of potential components involved in the constitutive activation of NF-κκB and enhanced expression ofCXCL1 and CXCL8 in melanoma. We postulate that the constitutive activation of nuclear factor-κB (NF-κB) is modulated byreceptor-mediated signals, such as those mediated through chemokine receptors and the LTβ receptor, which result in thepersistent activation of NF-κB-inducing kinase (NIK), AKT and, potentially, mitogen-activated protein (MAP) kinase kinase kinase 1(MEKK1). This activation of the inhibitor of NF-κB (IκB) kinase (IKK; a complex of IKKα, IKKβ and NEMO) leads to thephosphorylation and degradation of IκB and the phosphorylation of RELA/p65, which can, in turn, lead to constitutive expression of chemokines. We postulate that chemokine expression can be blocked by treatment with non-steroidal anti-inflammatory drugs(NSAIDs), NEMO-binding peptide and PS-341, which would target tumour cells for apoptosis. CBP, CREB-binding protein; CDP, CCAAT displacement protein; HAT, histone acetyltransferase; HDAC1, histone deacetylase 1; IUR, immediate upstreamregion; LTβ, lymphotoxin-β; LTβR, lymphotoxin-β receptor; NEMO, NF-κB essential modulator; PARP, poly-ADP ribose polymerase; PI3K, phosphatidylinositol 3-kinase; PtdInsP3, phosphatidylinositol-3,4,5-trisphosphate; PTEN, phosphatase and tensinhomologue; TRAF, tumour-necrosis-factor-receptor-associated factor.

© 2002 Nature Publishing Group672 | SEPTEMBER 2002 | VOLUME 2 www.nature.com/reviews/immunol
R E V I E W S
into the developing tumour. However, this negativeeffect is, apparently, offset by the enhanced apoptosis ofthe tumour cells due to the inhibition of NF-κB.Indeed, treatment with NON-STEROIDAL ANTI-INFLAMMATORY
DRUGS (NSAIDS) has been shown to inhibit both NF-κBand tumour growth, without detrimental effects on thehost31. So, reagents such as sulindac or other NSAIDs,the proteasome inhibitor PS-341 and the IKK adaptorprotein NEMO-binding peptide are potential therapeu-tic reagents for tumours60,110–112 (FIG. 3). Indeed, initialpreclinical and clinical data with PS-341 indicate that itwill provide a powerful means of killing tumour cells bytargeting them for apoptosis113–115.
ConclusionsIt has become clear that the dysregulation of NF-κB is apotential target for therapeutic intervention for cancers.This is due, in part, to the role of NF-κB in the modula-tion of expression of chemokines and inflammatorycytokines, and also to the role of NF-κB in the regula-tion of expression of inhibitors of apoptosis. An impor-tant point for consideration, however, is that NF-κB alsomodulates the expression of the angiostatic chemokinesthat participate in the recruitment of tumour-infiltratinglymphocytes and NK cells into the developing tumour.So, blocking NF-κB could be a double-edged sword,were it not for the fact that NF-κB does not act alone,but cooperates with several other transcription factors tomodulate gene expression. The angiostatic chemokinesare transcriptionally activated by IFN-γ, in addition toNF-κB. Clearly, characterizing the enhanceosome for theangiostatic chemokines, the chemokines that suppresstumorigenesis, and those that stimulate tumour growthand angiogenesis will allow investigators to developmore-precise ways to block the growth of tumours whilemaintaining full participation of the immune system inthis blocking process.
either by displacing positive transactivating factors orthrough the recruitment of histone DEACETYLASE (HDAC),which would affect the open/closed state of the chro-matin and thereby affect transcription. The action ofCDP might be antagonized by the binding of CBP/p300,which has been shown to bind NF-κB and SP1, and —through its histone-acetylase activity — to stabilize theenhanceosome and enhance transcription25,106,107.
The transcription of CXCL8 is modulated mainlythrough an NF-κB element that works in concert withadjacent AP1 and nuclear factor induced by IL-6 (NF-IL-6) elements. There is some cell-type specificity withregard to the requirement for transactivation througheither the AP1 and NF-κB elements or the NF-IL-6 andNF-κB elements97. The transcription of CXCL8 is alsomodulated by an NF-κB-repressing factor (NRF).
Inhibitors of NF-κκB for tumour therapyIt is now clear that many types of tumour have constit-utive activation of NF-κB (BOX 2). This activation ofNF-κB leads to the endogenous expression of factorsthat are associated with tumorigenesis (VEGF, CXCL1and CXCL8), escape from apoptosis (inhibitor of apop-tosis proteins, IAPs) and metastasis (metallopro-teinases). However, whereas the expression of CXCL1,CXCL8 and CCL2 is associated with increased tumori-genicity and metastasis — CCL1, CXCL1 and CXCL8 inmelanoma and colon adenocarcinoma19,108,109 — thetreatment of various types of tumour with CXCL9,CXCL10, CCL1, CCL3, CCL5, CCL16, CCL19, CCL20,CCL21 and XCL1 is associated with tumour regressionand increased survival, possibly due to the recruitmentof infiltrating leukocytes and dendritic cells (DCs). Asthe expression of many of these chemokines is modu-lated by NF-κB also, the inhibition of NF-κB presentsthe risk of inhibiting the migration of natural killer(NK) cells, tumour-infiltrating lymphocytes and DCs
DEACETYLATION
Acetylation is a post-translational modification ofchromatin components,particularly histones. Histonedeacetylases have been identifiedas components of nuclear co-repressor complexes.
NON-STEROIDAL ANTI-
INFLAMMATORY DRUGS
(NSAIDs). Drugs, such asaspirin, that are used to ablatethe inflammatory response.These drugs can stimulateapoptosis and inhibitangiogenesis, therebysuppressing malignanttransformation and tumourgrowth. They work, in part, bysuppressing NF-κB activation.
1. Rossi, D. & Zlotnik, A. The biology of chemokines and theirreceptors. Annu. Rev. Immunol. 18, 217–242 (2000).
2. Luster, A. D. The role of chemokines in linking innate andadaptive immunity. Curr. Opin. Immunol. 14, 129–135(2002).
3. Homey, B., Muller, A. & Zlotnik, A. Chemokines: agents forthe immunotherapy of cancer. Nature Rev. Immunol.2, 175–184 (2002).
4. Chensue, S. W. Molecular machinations, chemokine signalsin host–pathogen interactions. Clin. Microbiol. Rev. 14,821–835 (2001).
5. Devalaraja, M. N. & Richmond, A. Multiple chemotacticfactors, fine control or redundancy? Trends Pharmacol. Sci.20, 151–156 (1999).
6. Muller, A. et al. Involvement of chemokine receptors inbreast cancer metastasis. Nature 410, 50–56 (2001).This article reviews the role of chemokines asregulators of tumour growth and metastasis, or theinhibition of tumour growth.
7. Strieter, R. M. Chemokines: not just leukocytechemoattractants in the promotion of cancer. NatureImmunol. 2, 285–286 (2001).A review of opposing roles of angiogenic andangiostatic CXC-chemokines in tumour growth.
8. Payne, A. S. & Cornelius, L. A. The role of chemokines inmelanoma tumor growth and metastasis. J. Invest.Dermatol. 118, 915–922 (2002).This article reviews the roles of CXCL1, CXCL8 andCCL5 in melanoma.
9. Luan, J. et al. Mechanism and biological significance ofconstitutive expression of MGSA/GRO chemokines in
malignant melanoma tumor progression. J. Leukocyte Biol.62, 588–597 (1997).
10. Robledo, M. M. et al. Expression of functional chemokinereceptors CXCR3 and CXCR4 on human melanoma cells. J. Biol. Chem. 276, 45098–45105 (2001).
11. Wiley, H. E., Gonzalez, E. B., Maki, W., Wu, M. T., & Hwang,S. T. Expression of CC-chemokine receptor-7 and regionallymph node metastasis of B16 murine melanoma. J. NatlCancer Inst. 93, 1638–1643 (2001).
12. Sunwoo, J. B. et al. Novel proteasome inhibitor PS-341inhibits activation of nuclear factor-κB, cell survival, tumorgrowth, and angiogenesis in squamous-cell carcinoma. Clin. Cancer Res. 7, 1419–1428 (2001).
13. Dong, G. et al. Molecular profiling of transformed andmetastatic murine squamous carcinoma cells by differentialdisplay and cDNA microarray reveals altered expression ofmultiple genes related to growth, apoptosis, angiogenesisand the NF-κB signal pathway. Cancer Res. 61, 4797–4808(2001).
14. Cusack, J. C. Jr et al. Enhanced chemosensitivity to CPT-11with proteasome inhibitor PS-341, implications for systemicnuclear factor-κB inhibition. Cancer Res. 61, 3535–3540(2001).This article shows that use of the proteasomeinhibitor PS-341 to block the activation of NF-κBresults in the inhibition of tumour growth when it isused in combination with the chemotherapeutic agentCPT-11.
15. Sprenger, H., Lloyd, A. R., Meyer, R. G., Johnston, J. A. &Kelvin, D. J. Genomic structure, characterization, and
identification of the promoter of the human IL-8 receptor Agene. J. Immunol. 153, 2524–2532 (1994).
16. Bonecchi, R. et al. Induction of functional IL-8 receptors byIL-4 and IL-13 in human monocytes. J. Immunol. 164,3862–3869 (2000).
17. Lloyd, A. R. et al. Granulocyte-colony stimulating factor andlipopolysaccharide regulate the expression of interleukin-8receptors on polymorphonuclear leukocytes. J. Biol. Chem.270, 28188–28192 (1995).
18. Ivarsson, K., Ekerydh, A., Fyhr, I. M., Janson, P. O. &Brannstrom, M. Upregulation of interleukin-8 and polarizedepithelial expression of interleukin-8 receptor A in ovariancarcinomas. Acta Obstet. Gynecol. Scand. 79, 777–784(2000).
19. Li, A., Varney, M. L. & Singh, R. K. Expression of interleukin-8and its receptors in human colon carcinoma cells withdifferent metastatic potentials. Clin. Cancer Res. 7,3298–3304 (2001).
20. Azenshtein, E. et al. The CC-chemokine RANTES in breastcarcinoma progression, regulation of expression andpotential mechanisms of promalignant activity. Cancer Res.62, 1093–1102 (2002).
21. Mrowietz, U. et al. The chemokine RANTES is secreted byhuman melanoma cells and is associated with enhancedtumour formation in nude mice. Br. J. Cancer 79,1025–1031 (1999).
22. Genin, P., Algarte, M., Roof, P., Lin, R. & Hiscott, J.Regulation of RANTES chemokine gene expressionrequires cooperativity between NF-κB and IFN-regulatoryfactor transcription. J. Immunol. 164, 5352–5361 (2000).

© 2002 Nature Publishing GroupNATURE REVIEWS | IMMUNOLOGY VOLUME 2 | SEPTEMBER 2002 | 673
R E V I E W S
23. Yang, J. M. & Richmond, A. Constitutive IKK activitycorrelates with NF-κB activation in human melanoma cells.Cancer Res. 61, 4901–4909 (2001).This article shows that melanoma cell lines haveconstitutive IKK activity, and also constitutivephosphorylation of p65, which results in constitutiveNF-κB transcriptional activity and enhancedexpression of chemokines. The article also showsthat the chemokine CXCL1 contributes to theconstitutive activation of IKK through an autocrineloop.
24. Yang, C.-R. et al. Coordinate modulation of Sp1, NF-κB andp53 in confluent human malignant melanoma cells afterionizing radiation. FASEB J. 14, 379–392 (2000).
25. Vanden Berghe, W., De Bosscher, K., Boone, E., Plaisance, S.& Haegeman, G. The nuclear factor-κB engages CBP/p300and histone acetyltransferase activity for transcriptionalactivation of the interleukin-6 gene promoter. J. Biol. Chem.274, 32091–32098 (1999).
26. Huang, S., De Guzman, A., Bucana, C. D. & Fidler, I. J. Levelof interleukin-8 expression by metastatic human melanomacells directly correlates with constitutive NF-κB activity.Cytokines Cell. Mol. Ther. 6, 9–17 (2000).
27. Kunz, M. et al. Anoxia-induced up-regulation of interleukin-8in human malignant melanoma. A potential mechanism forhigh tumor aggressiveness. Am. J. Pathol. 155, 753–763(1999).
28. Huang, S., DeGuzman, A., Bucana, C. D. & Fidler, I. J.Nuclear factor-κB activity correlates with growth,angiogenesis, and metastasis of human melanoma cells innude mice. Clin. Cancer Res. 6, 2573–2581 (2000).
29. Bakker, T. R., Reed, D., Renno, T. & Jongeneel, C. V.Efficient adenoviral transfer of NF-κB inhibitor sensitizesmelanoma to tumor necrosis factor-mediated apoptosis. Int. J. Cancer 80, 320–323 (1999).
30. Wang, D. et al. MGSA/GRO-mediated melanocytetransformation involves induction of Ras expression.Oncogene 19, 4647–4659 (2000).
31. Baldwin, A. S. Control of oncogenesis and cancer-therapyresistance by the transcription factor NF-κB. J. Clin. Invest.107, 241–246 (2001).An excellent review of NF-κB, its role in cancer andthe potential for therapeutic intervention usinginhibitors of NF-κB.
32. Beg, A. A. et al. IκB interacts with the nuclear localizationsequences of the subunits of NF-κB: a mechanism forcytoplasmic retention. Genes Dev. 10, 1899–1913 (1992).
33. Didonato, J., Mercurio, F. & Karin, M. Phosphorylation ofIκB-α precedes but is not sufficient for its dissociation fromNF-κB. Mol. Cell. Biol. 15, 1302–1311 (1995).
34. DiDonato, J. et al. Mapping of the inducible I-κBphosphorylation sites that signal its ubiquitination anddegradation. Mol. Cell. Biol. 16, 1295–1304 (1996).
35. Pando, M. P. & Verma, I. M. Signal-dependent and -independent degradation of free and NF-κB-bound IκB-α.J. Biol. Chem. 275, 21278–21286 (2000).
36. Ling, L., Cao, Z. D. & Goeddel, D. V. NF-κB-inducing kinaseactivates IKK-α by phosphorylation of ser-176. Proc. NatlAcad. Sci. USA 95, 3792–3797 (1998).An introduction to the biological role of NIK as anactivator of IKKα.
37. Nakano, H. et al. Differential regulation of I-κB kinase-α and -βby two upstream kinases, NF-κB-inducing kinase andmitogen-activated protein kinase ERK kinase kinase. Proc.Natl Acad. Sci. USA 95, 3537–3542 (1998).
38. Smith, C. et al. NF-κB-inducing kinase is dispensable foractivation of NF-κB in inflammatory settings but essential forlymphotoxin-β receptor activation of NF-κB in primaryhuman fibroblasts. J. Immunol. 167, 5895–5903 (2001).
39. Perkins, N. D. et al. Regulation of NF-κB by cyclin-dependent kinases associated with the p300 coactivator.Science 275, 523–527 (1997).
40. Shattuck-Brandt, R. L. & Richmond, A. Enhanceddegradation of I-κB-α contributes to endogenous activationof NF-κB in Hs294T melanoma cells. Cancer Res. 57,3032–3039 (1997).
41. Shattuck, R. L., Wood, L. D., Jaffe, G. J. & Richmond, A.MGSA/GRO transcription is differentially regulated in normalretinal pigment epithelial and melanoma cells. Mol. Cell. Biol.14, 791–802 (1994).
42. Wood, L. D., Farmer, A. A. & Richmond, A. HMGI (Y), andSP1 in addition to NF-κB regulate transcription of theMGSA/GROα gene. Nucleic Acids Res. 23, 4210–4219(1995).
43. Wood, L. D. & Richmond, A. Constitutive and cytokine-induced expression of the melanoma growth stimulatoryactivity/GROα gene requires both NF-κB and novelconstitutive factors. J. Biol. Chem. 270, 30619–30626 (1995).
44. Kondo, S., Kono, T., Sauder, D. N. & McKenzie, R. C. IL-8 gene expression and production in human
keratinocytes and their modulation by UVB. J. Invest.Dermatol. 101, 690–694 (1993).
45. Wang, Y. & Becker, D. Antisense targeting of bFGF and FGFreceptor-1 in human melanomas blocks intratumoralangiogenesis and tumor growth. Nature Med. 8, 887–893(1997).
46. Shih, I.-M. & Herlyn, M. Autocrine and paracrine roles forgrowth factors in melanoma. In Vitro 8, 113–124 (1994).
47. Gilmore, T. D., Koedood, M., Piffat, K. A. & White, D. W.Rel/NF-κB/IκB proteins and cancer. Oncogene 13,1367–1378 (1996).
48. Karin, M. & Lin, A. NF-κB at the crossroads of life and death.Nature Immunol. 3, 221–227 (2002).This article provides an up-to-date view of the role ofNF-κB activation in the modulation of factors that areinvolved in apoptosis and cell survival.
49. Dejardin, E. et al. Highly expressed p100/p52 (NF-κB2)sequesters other NF-κB-related proteins in the cytoplasm ofhuman breast cancer cells. Oncogene 11, 1835–1841 (1995).
50. Tamatani, T. et al. Enhanced IκB kinase activity isresponsible for the augmented activity of NF-κB in humanhead and neck carcinoma cells. Cancer Lett. 171, 165–172(2001).
51. Mukhopadhyay, T., Roth, J. A. & Maxwell, S. A. Alteredexpression of the p50 subunit of the NF-κB transcriptionfactor complex in non-small cell lung carcinoma. Oncogene11, 999–1003 (1995).
52. Bours, V., Dejardin, E., Goujon-Letawe, F., Merville, M. P. &Castronov, V. The NF-κB transcription factor and cancer:high expression of NF-κB and IκB-related proteins in tumorcell lines. Biochem. Pharmacol. 47, 145–149 (1994).
53. Budunova, I. V. et al. Increased expression of p50–NF-κBand constitutive activation of NF-κB transcription factorsduring mouse skin carcinogenesis. Oncogene 18,7423–7431 (1999).
54. Visconti, R. et al. Expression of the neoplastic phenotype byhuman thyroid carcinoma cell lines requires NF-κB p65protein expression. Oncogene 15, 1987–1994 (1997).
55. Sovak, M. A. et al. Aberrant nuclear factor-κB/Relexpression and the pathogenesis of breast cancer. J. Clin.Invest. 100, 2952–2960 (1997).
56. Devalaraja, M., Wang, D. Z., Ballard D. W. & Richmond, A.Elevated constitutive IKK activity and IκB-α phosphorylationin Hs294T melanoma cells lead to increased basalMGSA/GROα transcription. Cancer Res. 59, 1372–1377(1999).
57. Dhawan, P. & Richmond, A. The role of endogenous NIKand MEKK1 in the constitutive activation of NF-κB in humanmelanomas. J. Biol. Chem. 277, 7920–7928 (2002).This study shows that, in human melanoma, there isconstitutive activation of NIK, and that blocking NIKblocks the constitutive activation of NF-κB inmelanoma cells. Moreover, this NIK-mediated effecton NF-κB activity requires the activation of MEKK1and ERK1/ERK2.
58. Cusack, J., Liu, R. & Baldwin, A. Inducible chemoresistanceto 7-ethyl-10-[4-(1-piperidino)-1-piperidino]-carbonyloxycamptothe cin (CPT-11) in colorectal cancer cellsand a xenograft model is overcome by inhibition of nuclearfactor-κB activation. Cancer Res. 60, 2323–2330 (2000).
59. Budunova, I. V. et al. Increased expression of p50–NF-κBand constitutive activation of NF-κB transcription factorsduring mouse skin carcinogenesis. Oncogene 19,3003–3012 (2000).
60. Mayo, M. W. & Baldwin, A. S. The transcription factor NF-κB, control of oncogenesis and cancer-therapyresistance. Biochim. Biophys. Acta 1470, M55–M62 (2000).
61. Malinin, N. L., Boldin, M. P., Kovalenko, A. V. & Wallach, D.MAP3K-related kinase involved in NF-κB induction by TNF,CD95 and IL-1. Nature 385, 540–544 (1997).
62. Xiao, G., Harhaj, E. W. & Sun, S. C. NF-κB-inducing kinaseregulates the processing of NF-κB2 p100. Mol. Cell7, 401–409 (2001).
63. Cogswell, P. C., Guttridge, D. C., Funkhouse, W. K. &Baldwin, A. S. Jr. Selective activation of NF-κB subunits inhuman breast cancer: potential roles for NF-κB2/p52 andfor Bcl-3. Oncogene 19, 1123–1131 (2000).
64. Senftleben, U. et al. Activation by IKKα of a second,evolutionary conserved, NF-κB signaling pathway. Science293, 1495–1499 (2001).
65. Fagarasan, S. et al. Alymphophasia (aly)-type nuclearfactor-κB-inducing kinase (NIK) causes defects in secondarylymphoid tissue chemokine receptor signaling and homingof peritoneal cells to the gut-associated lymphatic tissuesystem. J. Exp. Med. 191, 1477–1486 (2000).This article shows that loss of NIK not only affectssignalling through the lymphotoxin-β receptor, butalso alters the response to chemokines, whichindicates that chemokine receptors might alsoactivate NIK.
66. Zhong, H., Voll, R. E. & Ghosh, S. Rearranged NF-κB2 genein the HUT78 T-lymphoma cell line codes for a constitutivelynuclear factor lacking transcriptional repressor functions.Mol. Cell 1, 661–671 (1998).
67. Bird, T. A., Schooley, K., Dower, S. K., Hagen, H.& Virca, G. D.Activation of nuclear transcription factor NF-κB byinterleukin-1 is accompanied by casein-kinase-II-mediatedphosphorylation of the p65 subunit. J. Biol. Chem. 272,32606–32612 (1997).
68. Sakurai, H., Chiba, H., Miyoshi, H., Sugita, T. & Toriumi, W.IκB kinases phosphorylate NF-κB p65 subunit on serine536 in the transactivation domain. J. Biol. Chem. 274,30353–30356 (1999).
69. Sizemore, N., Lerner, N., Dombrowski, N., Sakurai, H. &Stark, G. R. Distinct roles of the IκB kinase α- and β-subunits in liberating nuclear factor-κB (NF-κB) from IκB andin phosphorylating the p65 subunit of NF-κB. J. Biol. Chem.277, 3863–3869 (2002).This article shows the role of AKT in the activation ofIKKα, which potentially phosphorylates the p65subunit of NF-κB to potentiate its transactivatingcapacity.
70. Li, X. & Stark, G. R. NF-κB-dependent signaling pathways.Exp. Hematol. 30, 285–296 (2002).
71. Delhase, M., Li, N. & Karin, M. Kinase regulation ininflammatory response. Nature 406, 367–368 (2000).
72. Koul, D., Yao, Y., Abbruzzese, J. L., Yung, W. K. & Reddy, S. A.Tumor suppressor MMAC/PTEN inhibits cytokine inducedNF-κB activation without interfering with the IκB degradationpathway. J. Biol. Chem. 276, 11402–11408 (2001).
73. Madrid, L. V., Mayo, M. W., Reuther, J. Y. & Baldwin, A. S. Jr.Akt stimulates the transactivation potential of the RelA/p65subunit of NF-κB through utilization of the IκB kinase andactivation of the mitogen-activated protein kinase p38. J. Biol. Chem. 276, 18934–18940 (2001).
74. Celebi, J. T., Shendrik, I., Silvers, D. N. & Peacocke, M.Identification of PTEN mutations in metastatic melanomaspecimens. J. Med. Genet. 37, 653–657 (2000).
75. Zhou, X. P. et al. Epigenetic PTEN silencing in malignantmelanomas without PTEN mutation. Am. J. Pathol. 157,1123–1128 (2000).
76. Mayo, M. W. et al. Requirement of NF-κB activation tosuppress p53-independent apoptosis induced byoncogenic Ras. Science 278, 1812–1815 (1997).
77. Finco, T. S. et al. Oncogenic Ha-Ras-induced signalingactivates NF-κB transcriptional activity, which is required forcellular transformation. J. Biol. Chem. 272, 24113–24116(1997).
78. Norris, J. L. & Baldwin, A. S. Oncogenic Ras enhances NF-κB transcriptional activity through Raf-dependent andRaf-independent mitogen-activated protein kinase signalingpathways. J. Biol. Chem. 274, 13841–13846 (1999).
79. Troppmair, J., Hartkamp, J. & Rapp, U. R. Activation of NF-κB by oncogenic Raf in HEK293 cells occurs throughautocrine recruitment of the stress kinase cascade.Oncogene 17, 685–690 (1998).
80. Yin, M. J. et al. HTLV-1 Tax protein binds to MEKK1 tostimulate IκB kinase activity and NF-κB activation. Cell 93, 875–884 (1998).
81. Chu, Z., DiDonato, J., Hawiger, J. & Ballard, D. The taxoncogene of human T-cell leukemia virus type 1 associateswith and persistently activates IκB kinases containing IKKαand IKKβ. J. Biol. Chem. 273, 15891–15894 (1998).
82. Geleziunas, R. et al. Human T-cell leukemia virus type 1 Taxinduction of NF-κB involves activation of the IκB kinase-α(IKKα) and IKKβ cellular kinases. Mol. Cell. Biol. 18,5157–5165 (1998).
83. Mosialos, G. The role of Rel/NF-κB proteins in viraloncogenesis and the regulation of viral transcription. Semin.Cancer Biol. 8, 121–129 (1997).
84. LaPorta, C. A. & Camolli, R. PKC-dependent modulation ofIκBα–NF-κB pathway in low metastatic B16F1 murinemelanoma cells and in highly metastatic BL6 cells.Anticancer Res. 18, 2591–2597 (1998).
85. Biswas, D. K et al. The nuclear factor-κB (NF-κB): a potentialtherapeutic target for estrogen receptor-negative breastcancers. Proc. Natl Acad. Sci. USA 98, 10386–10391 (2001).
86. Wilson, L., Szabo, C. & Salzman, A. L. Protein kinase-C-dependent activation of NF-κB in enterocytes isindependent of IκB degradation. Gastroenterology 117,106–114 (1999).
87. Han, Y., Meng, T., Murray, N. R., Fields, A. P. & Brasier, A. R.Interleukin-1-induced nuclear factor-κB–IκB-αautoregulatory feedback loop in hepatocytes. A role forprotein kinase Cα in post-transcriptional regulation of IκB-αresynthesis. J. Biol. Chem. 274, 939–947 (1999).
88. Anrather, J., Csizmadia, V., Soares, M. P. & Winkler, H.Regulation of NF-κB RelA phosphorylation and transcriptionalactivity by p21(ras) and protein kinase Cζ in primaryendothelial cells. J. Biol. Chem. 274, 13594–13603 (1999).

© 2002 Nature Publishing Group674 | SEPTEMBER 2002 | VOLUME 2 www.nature.com/reviews/immunol
R E V I E W S
89. Wang, D. & Richmond, A. NF-κB activation by the CXC-chemokine MGSA/GROα involves the MEKK1/p38 MAPkinase pathway. J. Biol. Chem. 276, 3650–3659 (2001).
90. Richmond, A. & Thomas, H. G. Melanoma growthstimulatory activity, a novel growth factor with a tissuedistribution not restricted to melanoma tissue. J. Cell.Biochem. 36, 185–198 (1986).
91. Singh, R. K. et al. Expression of interleukin-8 correlates withthe metastatic potential of human melanoma cells in nudemice. Cancer Res. 54, 3242–3247 (1994).
92. Singh, R. K. et al. Ultraviolet B irradiation promotestumorigenic and metastatic properties in primary cutaneousmelanoma via induction of interleukin-8. Cancer Res. 55,3669–3674 (1995).
93. Schadendorf, D. et al. IL-8 produced by human malignantmelanoma cells in vitro is an essential autocrine growthfactor. J. Immunol. 151, 2267–2675 (1993).
94. Schadendorf, D. et al. Metastatic potential of humanmelanoma cells in nude mice — characterization ofphenotype, cytokine secretion and tumor-associatedantigens. Br. J. Cancer 74, 194–199 (1996).
95. Loukinova, E. et al. Growth-regulated oncogene-αexpression by murine squamous cell carcinoma promotestumor growth, metastasis, leukocyte infiltration andangiogenesis by a host CXC-receptor-2-dependentmechanism. Oncogene 19, 3477–3486 (2000).
96. Matsusaka, T. et al. Transcription factors NF-IL-6 and NF-κBsynergistically activate transcription of the inflammatorycytokines, interleukin-6 and interleukin-8. Proc. Natl Acad.Sci. USA 90, 10193–10197 (1993).
97. Balentien, E. et al. Effects of MGSA/GROα on melanocytetransformation. Oncogene 6, 1115–1124 (1991).
98. Nirodi, C. S. & Richmond, A. Role of poly (ADP-ribose)polymerase (PARP) in the transcriptional regulation of themelanoma growth stimulatory activity (CXCL1) gene. J. Biol.Chem. 276, 9366–9374 (2001).This article shows that PARP participates with NF-κBin the modulation of transcription of CXCL1.
99. Nirodi, C. S. et al. The 170-kDa CCAAT displacementprotein (CDP/Cut) selectively binds the IUR cis-element inthe CXCL1 promoter. The role of CDP in the negativeregulation of CXCL1 gene expression. J. Biol. Chem. 276,9366–9374 (2001).This study further defines the CXCL1 enhanceosomeand shows that binding of the transcriptionalrepressor CDP to an element adjacent to the NF-κB-binding site represses the transcription of CXCL1.
100. Hassa, P. O. & Hottiger, M. O. A role of poly (ADP-ribose)polymerase in NF-κB transcriptional activation. J. Biol.Chem. 380, 953–959 (1999).
101. Ludlow, C., Choy, R. & Blochlinger, K. Functional analysis ofDrosophila and mammalian Cut proteins in flies. Dev. Biol.178, 149–159 (1996).
102. Coqueret, O., Berube, G. & Nepveu, A. The mammalian Cuthomeodomain protein functions as a cell-cycle-dependenttranscriptional repressor which downmodulatesp21WAF1/CIP1/SDI1 in S phase. EMBO J. 17, 4680–4694(1998).
103. Mailly, F. G. et al. The human Cut homeodomain protein canrepress gene expression by two distinct mechanisms: activerepression and competition for binding-site occupancy. Mol. Cell. Biol. 16, 5346–5357 (1996).
104. Li, S. et al. Transcriptional repression of the cystic fibrosistransmembrane conductance regulator gene, mediated byCCAAT displacement protein/Cut homolog, is associatedwith histone deacetylation. J. Biol. Chem. 274, 7803–7815(1999)
105. Li, S., Aufiero, B., Schiltz, R. L. & Walsh, M. J. Regulation ofthe homeodomain CCAAT displacement/Cut proteinfunction by histone acetyltransferases p300/CREB-bindingprotein (CBP)-associated factor and CBP. Proc. Natl Acad.Sci. USA 12, 1627–1631 (2000).
106. Xiao, H., Hasegawa, T. & Isobe, K. p300 collaborates withSp1 and Sp3 in p21(Waf1/Cip1) promoter activationinduced by histone deacetylase inhibitor. J. Biol. Chem. 275,1371–1376 (2000).
107. Kundu, T. K. et al. Activator-dependent transcription fromchromatin in vitro involving targeted histone acetylation byp300. Mol. Cell. Biol. 6, 551–561 (2000).This article shows the role of p300 in acetylation ofhistones and the stabilization of the transcriptionalmachinery.
108. Bottazzi, B., Walter, S., Govoni, D., Colotta, F. & Mantovani, A.Monocyte chemotactic cytokine gene transfer modulatesmacrophage infiltration, growth and susceptibility to IL-2therapy of murine melanoma. J. Immunol. 148, 1280–1285(1992).
109. Nakashima, E. et al. Human MCAF gene transfer enhancesthe metastatic capacity of a mouse cachecticadenocarcinoma cell line in vivo. Pharm. Res. 12,1598–1604 (1995).
110. Berman, K. S. et al. Sulindac enhances tumor necrosisfactor-α-mediated apoptosis of lung cancer cell lines byinhibition of nuclear factor-κB. Clin. Cancer Res. 8, 354–360(2002).
111. May, M. J. et al. Selective inhibition of NF-κB activation by apeptide that blocks the interaction of NEMO with the IκBkinase complex. Science 289, 1550–1554 (2000).
112. Yamamoto, Y. & Gaynor, R. B. Therapeutic potential ofinhibition of the NF-κB pathway in the treatment ofinflammation and cancer. J. Clin. Invest. 107, 135–142(2001).This article emphasizes the available data thatindicate that, by developing methods of disrupting theNF-κB pathway, new advances can be made in thetherapeutic intervention of acute and chronicinflammatory conditions, as well as malignancies.
113. Hideshima, T. et al. NF-κB as a therapeutic target in multiplemyeloma. J. Biol. Chem. 277, 16639–16647 (2002).
114. Tan, C. & Waldmann, T. A. Proteasome inhibitor PS-341, apotential therapeutic agent for adult T-cell leukemia. CancerRes. 62, 1083–1086 (2002).
115. Adams, J. Proteasome inhibition, a novel approach tocancer therapy. Trends Mol. Med. 8, 49–54 (2002).This article reviews the current status of the use ofproteasome inhibitors to block tumour growth.
116. Zhang, J., Chang, C. C., Lombardi, L., Dalla-Favera, R.Rearranged NF-κB2 gene in the HUT78 T-lymphoma cellline codes for a constitutively nuclear factor lackingtranscriptional repressor functions. Oncogene 9,1931–1937 (1994).
117. Higgins, K. A. et al. Antisense inhibition of the p65 subunit ofNF-κB blocks tumorigenicity and causes tumor regression.Proc. Natl Acad. Sci. USA 90, 9901–9905 (1993).
118. Shah, S. A. et al. 26S proteasome inhibition inducesapoptosis and limits growth of human pancreatic cancer. J. Cell. Biochem. 82, 110–122 (2001).
119. Mukhopadhyay, A., Bueso-Ramos, C., Chatterjee, D.,Pantazis, P. & Aggarwal, B. B. Curcumin downregulates cell-survival mechanisms in human prostate cancer cell lines.Oncogene 20, 7597–7609 (2001).
120. Nourbakhsh, M. et al. The NF-κB repressing factor isinvolved in basal repression and interleukin (IL)-1-inducedactivation of IL-8 transcription by binding to a conserved NF-κB-flanking sequence element. J. Biol. Chem. 276,4501–4508 (2001).This article models the components of theenhanceosome for CXCL8 and shows that atranscriptional repressor binds to an element flankingthe NF-κB element. So, the models for thetranscription of CXCL8, CXCL1 and IL-6 are similar.
121. Stein, B., Cogswell, P. S. & Baldwin, A. S. Functional andphysical associations between NF-κB and C/EBP familymembers: a Rel domain–bZIP interaction. Mol. Cell Biol. 13, 3964–3974 (1993).
122. Li, S., Aufiero, B., Schiltz, R. L. & Walsh, M. J. Regulation ofthe homeodomain CCAAT displacement/Cut proteinfunction by histone acetyltransferases p300/CREB-bindingprotein (CBP)-associated factor and CBP. Proc. Natl Acad.Sci. USA 97, 7166–7171 (2000).
123. International Union of Immunological Societies/World HealthOrganization Subcommittee on Chemokine Nomenclature.Chemokine/chemokine receptor nomenclature. J. Leukocyte Biol. 70, 465–466 (2001).
AcknowledgementsThe work described in this review was facilitated by funding fromthe Department of Veterans Affairs (Senior Career Scientist Awardand Merit Award) and the National Cancer Institute. The figureswere contributed by C. S. Nirodi and P. Dhawan. I am alsoendebted to M. Boothby (Vanderbilt University School of Medicine)for critical review of this manuscript.
Online links
DATABASESThe following terms in this article are linked online to:Cancer.gov: http://www.cancer.gov/breast cancer | colon carcinoma | head and neck cancer |leukaemia | lung cancer | lymphoma | melanoma | ovarian cancer |pancreatic cancer | prostate cancer | thyroid cancerFlyBase: http://flybase.bio.indiana.edu/Dif | Dorsal | RelishLocusLink: http://www.ncbi.nlm.nih.gov/LocusLink/AKT | AP1 | Bcl3 | CBP | CCL3 | CCL5 | CCL11 | CCL19 | CCL20 |CCL21 | CCR7 | CCR10 | CDP | C/EBP | CKII | c-Met | CXCL1 |CXCL2 | CXCL3 | CXCL5 | CXCL6 | CXCL7 | CXCL9 | CXCL10 |CXCL11 | CXCL12 | CXCL13 | CXCR1 | CXCR2 | CXCR3 | CXCR4 |cyclin D | ERK1 | ERK2 | FasL | GM-CSF | HDAC | HDAC1 | HMGIY |HRAS | IκB | Iap1 | IFN-γ | IKKα | IKKβ | IKKγ | KC | IL-1 | IL-4 | IL-6 |IL-8 | IL-13 | LTβ | MEKK1 | MEKK3 | NF-κB | NIK | p21 | p27 | p38 MAPK | p100 | p105 | PAK | PARP | PDK1 | Pea15 | PI3K |PKA | PLCβ | PTEN | RELA | RELB | SP1 | Syk | TAK1 | TGF-β |TNF | TRAF2 | VEGF | XCL1 | Yes-associated proteinMedscape DrugInfo: http://www.medscape.com/druginfosulindacAccess to this interactive links box is free online.