
Modulation of human estrogen receptor α activity by multivalent estradiol–peptidomimetic...
9
This journal is c The Royal Society of Chemistry 2011 Mol. BioSyst., 2011, 7, 337–345 337 Cite this: Mol. BioSyst., 2011, 7, 337–345 Modulation of human estrogen receptor a activity by multivalent estradiol–peptidomimetic conjugateswz Justin M. Holub, a Michael J. Garabedian b and Kent Kirshenbaum* a Received 6th September 2010, Accepted 24th November 2010 DOI: 10.1039/c0mb00189a Estradiol–peptidomimetic conjugates (EPCs) are linear, sequence-specific peptoid oligomers that site-specifically display multiple copies of 17b-estradiol (E2), a ligand for the human estrogen receptor a (hERa). We evaluate the ability of multivalent EPCs to activate hERa-mediated transcription. EPCs activated the hERa in both a length- and valence-dependent manner, with the highest levels of activation generated by divalent peptoid 6-mers, divalent 18-mers, and trivalent 9-mers. Hexavalent EPCs did not activate hERa, but instead blocked E2-mediated hERa activation. The physicochemical features of EPCs can be precisely tuned, which may allow the generation of a library of chemical tools for modulating specific effects of estrogens. Introduction Multivalent ligands have attracted substantial interest for their potential to modulate molecular interactions of biomedical significance. Multivalent displays enhance the effective local concentration of low affinity ligands to promote strengthened binding to target receptors. Conjugating multiple ligands onto an individual scaffold molecule yields products with strong binding interactions, termed avidities. 1–4 In principle, such chemical constructs can target multimeric protein receptors via multi-site binding contacts. The ability of multivalent ligands to engage cell-surface receptor arrays can enable modulation of a variety of biochemical processes, including chemotaxis, adhesion, and immune responses. The targets of synthetic multivalent ligands include RNA, enzymes, bacterial toxins, and lectins. 5–9 One potential benefit in creating multivalent displays is the ability to tune the physico-chemical features of the scaffold. For example, variation in the spacing between sites of conjugation can be used to influence the density of displayed ligands. Alteration of size, charge, and branching of the scaffold can influence solubility and other pharmacological characteristics, along with cellular uptake. This report describes the characterization of multivalent ligands targeting a steroid hormone receptor. Estrogen is a steroid hormone that mediates its effect primarily thorough the intracellular action of the estrogen receptor. The human estrogen receptor (hER) belongs to a superfamily of ligand- mediated transcription factors. 10 In the classic model for hER-mediated gene transcription, nuclear-localized hER binds its natural ligand 17b-estradiol (E2), undergoes a conformational rearrangement, dimerizes, and binds estrogen response elements (ERE) on DNA, where it participates in the recruitment of additional transcriptional cofactors. 11–13 The hER is known to exist in two isoforms: the widely-studied hERa, and the hERb. 14,15 Compounds that modulate hERa activity are a major target for development of therapeutics that address a range of human diseases, including cancer and osteoporosis. 16–18 In addition to nuclear-localized estrogen receptor, discrete cellular pools of ER at the plasma membrane have been implicated in the rapid ‘‘non-genomic’’ activation of MAPK, contributing to the overall effects of estrogen action. 19,20 Accordingly, several groups have focused on developing macromolecular conjugates that display multiple copies of E2 in an attempt to specifically modulate extranuclear hER. 21–25 These macromolecular constructs can be challenging to synthesize, however, and can exhibit polydispersity in the product composition, uneven loading of bioactive ligand, aggregation, or cleavage of biologically labile linkers. We have previously described the synthesis of multivalent estrogen ligands via conjugation of estradiol groups at multiple sites on a linear peptoid scaffold. 26 Peptoids are oligomers composed of N-substituted glycine units. 27 Step-wise peptoid oligomer synthesis on solid phase support allows the genera- tion of monodisperse, sequence-specific products, and enables precise control over the spacing of reactive groups along the oligomer backbone. 28 Peptoids show substantial potential for the design of bioactive compounds. 29,30 Owing to their ease of synthesis, high proteolytic stability, 31 and favorable cell a Department of Chemistry, New York University, 100 Washington Square East, New York, New York 10003, USA. E-mail: [email protected] b Department of Microbiology, NYU Langone School of Medicine, 550 First Avenue, New York, New York 10016, USA w This article is part of a Molecular BioSystems ‘Emerging Investigators’ issue highlighting the work of outstanding young scientists at the chemical- and systems-biology interfaces. z Electronic supplementary information (ESI) available: Peptoid synthesis protocols, compound characterization and Flu-EPC emission spectra. See DOI: 10.1039/c0mb00189a Molecular BioSystems Dynamic Article Links www.rsc.org/molecularbiosystems PAPER Published on 07 January 2011. Downloaded by Northeastern University on 26/10/2014 21:25:00. View Article Online / Journal Homepage / Table of Contents for this issue
Transcript of Modulation of human estrogen receptor α activity by multivalent estradiol–peptidomimetic...

This journal is c The Royal Society of Chemistry 2011 Mol. BioSyst., 2011, 7, 337–345 337
Cite this: Mol. BioSyst., 2011, 7, 337–345
Modulation of human estrogen receptor a activity by multivalent
estradiol–peptidomimetic conjugateswzJustin M. Holub,
aMichael J. Garabedian
band Kent Kirshenbaum*
a
Received 6th September 2010, Accepted 24th November 2010
DOI: 10.1039/c0mb00189a
Estradiol–peptidomimetic conjugates (EPCs) are linear, sequence-specific peptoid oligomers that
site-specifically display multiple copies of 17b-estradiol (E2), a ligand for the human estrogen
receptor a (hERa). We evaluate the ability of multivalent EPCs to activate hERa-mediated
transcription. EPCs activated the hERa in both a length- and valence-dependent manner, with the
highest levels of activation generated by divalent peptoid 6-mers, divalent 18-mers, and trivalent
9-mers. Hexavalent EPCs did not activate hERa, but instead blocked E2-mediated hERaactivation. The physicochemical features of EPCs can be precisely tuned, which may allow
the generation of a library of chemical tools for modulating specific effects of estrogens.
Introduction
Multivalent ligands have attracted substantial interest for their
potential to modulate molecular interactions of biomedical
significance. Multivalent displays enhance the effective local
concentration of low affinity ligands to promote strengthened
binding to target receptors. Conjugating multiple ligands onto
an individual scaffold molecule yields products with strong
binding interactions, termed avidities.1–4 In principle, such
chemical constructs can target multimeric protein receptors
via multi-site binding contacts. The ability of multivalent
ligands to engage cell-surface receptor arrays can enable
modulation of a variety of biochemical processes, including
chemotaxis, adhesion, and immune responses. The targets of
synthetic multivalent ligands include RNA, enzymes, bacterial
toxins, and lectins.5–9
One potential benefit in creating multivalent displays is the
ability to tune the physico-chemical features of the scaffold.
For example, variation in the spacing between sites of
conjugation can be used to influence the density of displayed
ligands. Alteration of size, charge, and branching of the
scaffold can influence solubility and other pharmacological
characteristics, along with cellular uptake.
This report describes the characterization of multivalent
ligands targeting a steroid hormone receptor. Estrogen is a
steroid hormone that mediates its effect primarily thorough
the intracellular action of the estrogen receptor. The human
estrogen receptor (hER) belongs to a superfamily of ligand-
mediated transcription factors.10 In the classic model for
hER-mediated gene transcription, nuclear-localized hER
binds its natural ligand 17b-estradiol (E2), undergoes a
conformational rearrangement, dimerizes, and binds estrogen
response elements (ERE) on DNA, where it participates in the
recruitment of additional transcriptional cofactors.11–13 The
hER is known to exist in two isoforms: the widely-studied
hERa, and the hERb.14,15 Compounds that modulate hERaactivity are a major target for development of therapeutics
that address a range of human diseases, including cancer and
osteoporosis.16–18
In addition to nuclear-localized estrogen receptor, discrete
cellular pools of ER at the plasma membrane have been
implicated in the rapid ‘‘non-genomic’’ activation of MAPK,
contributing to the overall effects of estrogen action.19,20
Accordingly, several groups have focused on developing
macromolecular conjugates that display multiple copies of
E2 in an attempt to specifically modulate extranuclear
hER.21–25 These macromolecular constructs can be challenging
to synthesize, however, and can exhibit polydispersity in the
product composition, uneven loading of bioactive ligand,
aggregation, or cleavage of biologically labile linkers.
We have previously described the synthesis of multivalent
estrogen ligands via conjugation of estradiol groups at multiple
sites on a linear peptoid scaffold.26 Peptoids are oligomers
composed of N-substituted glycine units.27 Step-wise peptoid
oligomer synthesis on solid phase support allows the genera-
tion of monodisperse, sequence-specific products, and enables
precise control over the spacing of reactive groups along
the oligomer backbone.28 Peptoids show substantial potential
for the design of bioactive compounds.29,30 Owing to their ease
of synthesis, high proteolytic stability,31 and favorable cell
aDepartment of Chemistry, New York University,100 Washington Square East, New York, New York 10003, USA.E-mail: [email protected]
bDepartment of Microbiology, NYU Langone School of Medicine,550 First Avenue, New York, New York 10016, USA
w This article is part of aMolecular BioSystems ‘Emerging Investigators’issue highlighting the work of outstanding young scientists at thechemical- and systems-biology interfaces.z Electronic supplementary information (ESI) available: Peptoid synthesisprotocols, compound characterization and Flu-EPC emission spectra. SeeDOI: 10.1039/c0mb00189a
MolecularBioSystems
Dynamic Article Links
www.rsc.org/molecularbiosystems PAPER
Publ
ishe
d on
07
Janu
ary
2011
. Dow
nloa
ded
by N
orth
east
ern
Uni
vers
ity o
n 26
/10/
2014
21:
25:0
0.
View Article Online / Journal Homepage / Table of Contents for this issue

338 Mol. BioSyst., 2011, 7, 337–345 This journal is c The Royal Society of Chemistry 2011
permeability,32 a variety of peptoid oligomers are being
evaluated as molecular tools for chemical biology and other
biomedical applications. These include use as transfection
reagents, modulators of protein activity and protein capture
agents.33–36
We have developed synthetic protocols to expand the
chemical diversity and potential biomedical applications of
peptoid oligomers by conjugating bioactive groups, such as
estradiol, to peptoid scaffolds using the Cu-catalyzed
azide–alkyne [3+2] cycloaddition (CuAAC) ‘‘click’’ reaction.37,38
We determined that multivalent estradiol–peptidomimetic
conjugates (EPCs) are capable of binding the hERain vitro.26 In addition, we successfully demonstrated that
EPC–hERa affinity is enhanced as the valency of ligands
along the oligomer backbone is increased. Here, we investigate
whether EPCs are capable of modulating the activity of the
hERa in vivo. HEK293 cells were chosen as a model because
they lack endogenous hERa, allowing us to express plasmid-
encoded hERa and examine the ability of multivalent EPCs to
modulate hERa-dependent transcription. We used this system
to evaluate libraries of monodisperse estrogen conjugates in
which the length and valency of the ligand presentation is
precisely controlled.
Results and discussion
Synthesis of estradiol–peptidomimetic conjugates
Peptoid oligomers incorporating site-specific azide-functionalized
side chains were synthesized on solid support using a modifi-
cation of the submonomer synthesis protocol established by
Zuckermann et al.28 Following oligomerization, the resin-bound
peptoid oligomers were allowed to react with 17a-ethynyl-estradiol in the presence of a copper catalyst to generate
multivalent EPCs (Fig. 1, ESIz, Scheme S1 and Table S1).
This reaction resulted in a stable triazole linkage formed
between the alkyne-containing estradiol moiety and the
azide-functionalized peptoid side chain. To mitigate steric
hindrance during conjugation and to facilitate receptor
binding, multivalent EPCs 1–4 (Fig. 1a) were designed with
the pendant groups conjugated at every third residue. It
should be noted that the distance between the binding pockets
of dimerized hERa is approximately 24 A (determined from
PDB ID: ERE1). Placement of the E2 moieties at every third
residue along the EPC scaffolds could potentially bridge this
distance when the EPC is in an extended conformation.
Methoxyethyl moieties were included at all other side chain
positions to enhance overall EPC hydrophilicity.
Given that the hERa activates transcription as a homo-
dimer, and that our EPC synthetic protocol enables site-
specific conjugation of bioactive ligands to the peptoid scaffold,
we expanded our multivalent EPC library and generated a
set of divalent EPCs 5–7 (Fig. 1b). To enhance overall
hydrophilicity and facilitate receptor binding, EPCs 5, 6, and
7 were designed and synthesized with the E2 moieties spaced
between the N and C termini of the peptoid oligomer by 5, 8,
and 14 intervening monomer units, respectively. Mass spectra
analysis for all peptoids described herein can be found in the
ESIz, Table S2.
Synthesis of fluorescently labeled EPCs
To evaluate the cell permeability of our EPCs, we synthesized
fluorescently labeled EPCs (Flu-EPCs, Fig. 2) on solid
support using a modification of previously described synthetic
methods39,40 (ESIz, Scheme S2). To mitigate intramolecular
steric clash between the relatively bulky tracer and E2 ligand,
we inserted a six-carbon aliphatic linker between the fluores-
cein and the E2-functionalized peptoid scaffold (Fig. 2).
Estrogenic compounds containing phenol rings are known to
quench fluorescence,41 thus we were aware that the presence of
multiple E2 pendant groups could potentially diminish the
fluorescence emission intensity of our Flu-EPCs. The inserted
aliphatic linker spaces the fluorophore from the E2 ligand and
should mitigate intramolecular quenching. We did observe,
however, that increasing E2 valency diminishes fluorescence
emission intensity of Flu-EPCs (ESIz, Fig. S1).
Toxicity of estradiol–peptidomimetic conjugates
Prior to examining cellular localization, we evaluated the cell
toxicity of the EPCs. We employed a standard MTT cell
Fig. 1 Estradiol–peptidomimetic conjugates for use as estrogen receptor modulators. (a) Compound 1 (n=1); compound 2 (n=2); compound 3
(n = 3); compound 4 (n = 6). (b) Compound 5 (n = 5); compound 6 (n = 8); compound 7 (n = 14).
Publ
ishe
d on
07
Janu
ary
2011
. Dow
nloa
ded
by N
orth
east
ern
Uni
vers
ity o
n 26
/10/
2014
21:
25:0
0.
View Article Online
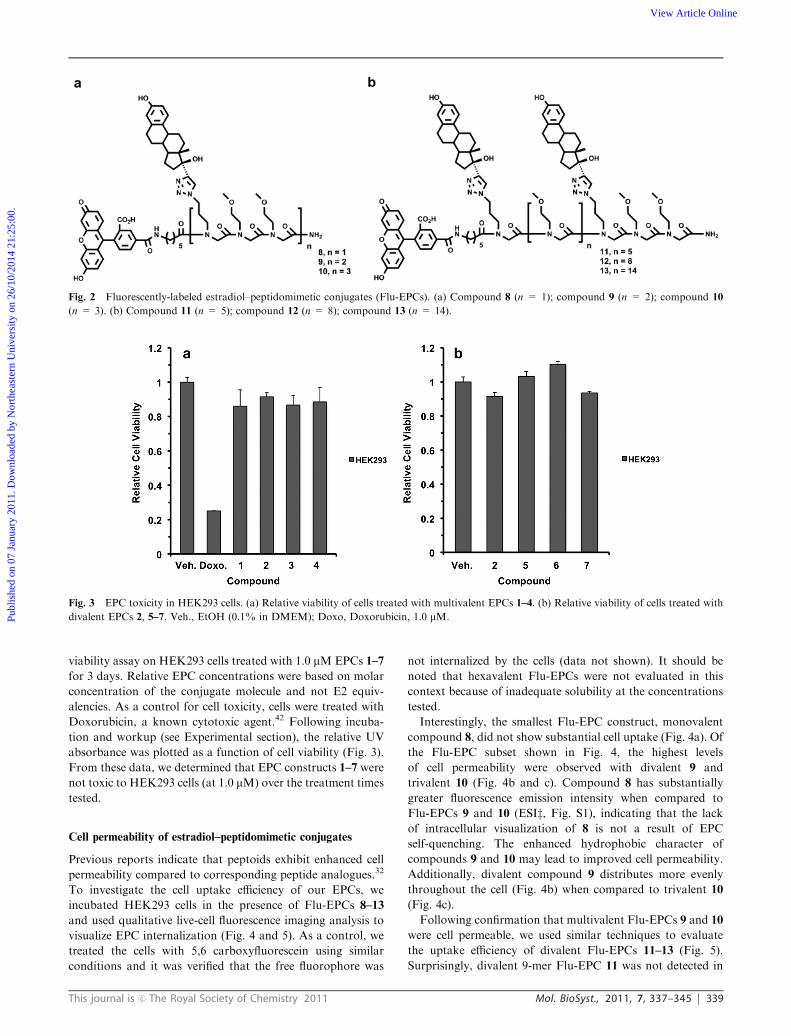
This journal is c The Royal Society of Chemistry 2011 Mol. BioSyst., 2011, 7, 337–345 339
viability assay on HEK293 cells treated with 1.0 mMEPCs 1–7
for 3 days. Relative EPC concentrations were based on molar
concentration of the conjugate molecule and not E2 equiv-
alencies. As a control for cell toxicity, cells were treated with
Doxorubicin, a known cytotoxic agent.42 Following incuba-
tion and workup (see Experimental section), the relative UV
absorbance was plotted as a function of cell viability (Fig. 3).
From these data, we determined that EPC constructs 1–7 were
not toxic to HEK293 cells (at 1.0 mM) over the treatment times
tested.
Cell permeability of estradiol–peptidomimetic conjugates
Previous reports indicate that peptoids exhibit enhanced cell
permeability compared to corresponding peptide analogues.32
To investigate the cell uptake efficiency of our EPCs, we
incubated HEK293 cells in the presence of Flu-EPCs 8–13
and used qualitative live-cell fluorescence imaging analysis to
visualize EPC internalization (Fig. 4 and 5). As a control, we
treated the cells with 5,6 carboxyfluorescein using similar
conditions and it was verified that the free fluorophore was
not internalized by the cells (data not shown). It should be
noted that hexavalent Flu-EPCs were not evaluated in this
context because of inadequate solubility at the concentrations
tested.
Interestingly, the smallest Flu-EPC construct, monovalent
compound 8, did not show substantial cell uptake (Fig. 4a). Of
the Flu-EPC subset shown in Fig. 4, the highest levels
of cell permeability were observed with divalent 9 and
trivalent 10 (Fig. 4b and c). Compound 8 has substantially
greater fluorescence emission intensity when compared to
Flu-EPCs 9 and 10 (ESIz, Fig. S1), indicating that the lack
of intracellular visualization of 8 is not a result of EPC
self-quenching. The enhanced hydrophobic character of
compounds 9 and 10 may lead to improved cell permeability.
Additionally, divalent compound 9 distributes more evenly
throughout the cell (Fig. 4b) when compared to trivalent 10
(Fig. 4c).
Following confirmation that multivalent Flu-EPCs 9 and 10
were cell permeable, we used similar techniques to evaluate
the uptake efficiency of divalent Flu-EPCs 11–13 (Fig. 5).
Surprisingly, divalent 9-mer Flu-EPC 11 was not detected in
Fig. 2 Fluorescently-labeled estradiol–peptidomimetic conjugates (Flu-EPCs). (a) Compound 8 (n = 1); compound 9 (n = 2); compound 10
(n = 3). (b) Compound 11 (n = 5); compound 12 (n = 8); compound 13 (n = 14).
Fig. 3 EPC toxicity in HEK293 cells. (a) Relative viability of cells treated with multivalent EPCs 1–4. (b) Relative viability of cells treated with
divalent EPCs 2, 5–7. Veh., EtOH (0.1% in DMEM); Doxo, Doxorubicin, 1.0 mM.Publ
ishe
d on
07
Janu
ary
2011
. Dow
nloa
ded
by N
orth
east
ern
Uni
vers
ity o
n 26
/10/
2014
21:
25:0
0.
View Article Online
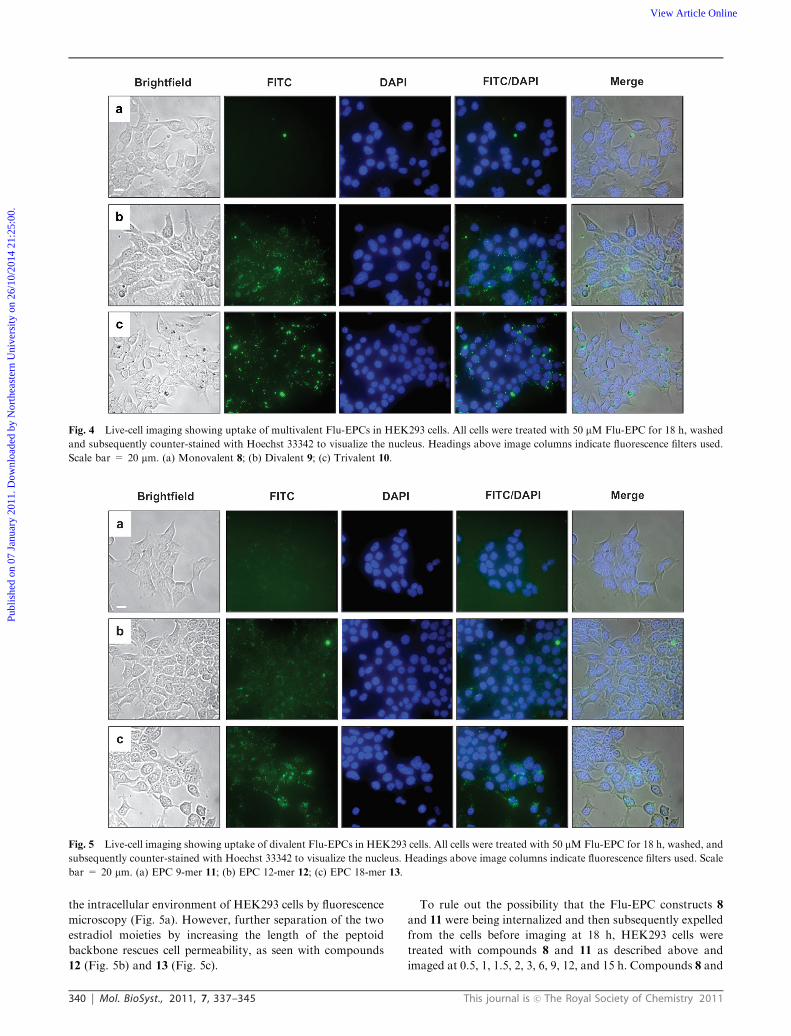
340 Mol. BioSyst., 2011, 7, 337–345 This journal is c The Royal Society of Chemistry 2011
the intracellular environment of HEK293 cells by fluorescence
microscopy (Fig. 5a). However, further separation of the two
estradiol moieties by increasing the length of the peptoid
backbone rescues cell permeability, as seen with compounds
12 (Fig. 5b) and 13 (Fig. 5c).
To rule out the possibility that the Flu-EPC constructs 8
and 11 were being internalized and then subsequently expelled
from the cells before imaging at 18 h, HEK293 cells were
treated with compounds 8 and 11 as described above and
imaged at 0.5, 1, 1.5, 2, 3, 6, 9, 12, and 15 h. Compounds 8 and
Fig. 4 Live-cell imaging showing uptake of multivalent Flu-EPCs in HEK293 cells. All cells were treated with 50 mM Flu-EPC for 18 h, washed
and subsequently counter-stained with Hoechst 33342 to visualize the nucleus. Headings above image columns indicate fluorescence filters used.
Scale bar = 20 mm. (a) Monovalent 8; (b) Divalent 9; (c) Trivalent 10.
Fig. 5 Live-cell imaging showing uptake of divalent Flu-EPCs in HEK293 cells. All cells were treated with 50 mMFlu-EPC for 18 h, washed, and
subsequently counter-stained with Hoechst 33342 to visualize the nucleus. Headings above image columns indicate fluorescence filters used. Scale
bar = 20 mm. (a) EPC 9-mer 11; (b) EPC 12-mer 12; (c) EPC 18-mer 13.
Publ
ishe
d on
07
Janu
ary
2011
. Dow
nloa
ded
by N
orth
east
ern
Uni
vers
ity o
n 26
/10/
2014
21:
25:0
0.
View Article Online
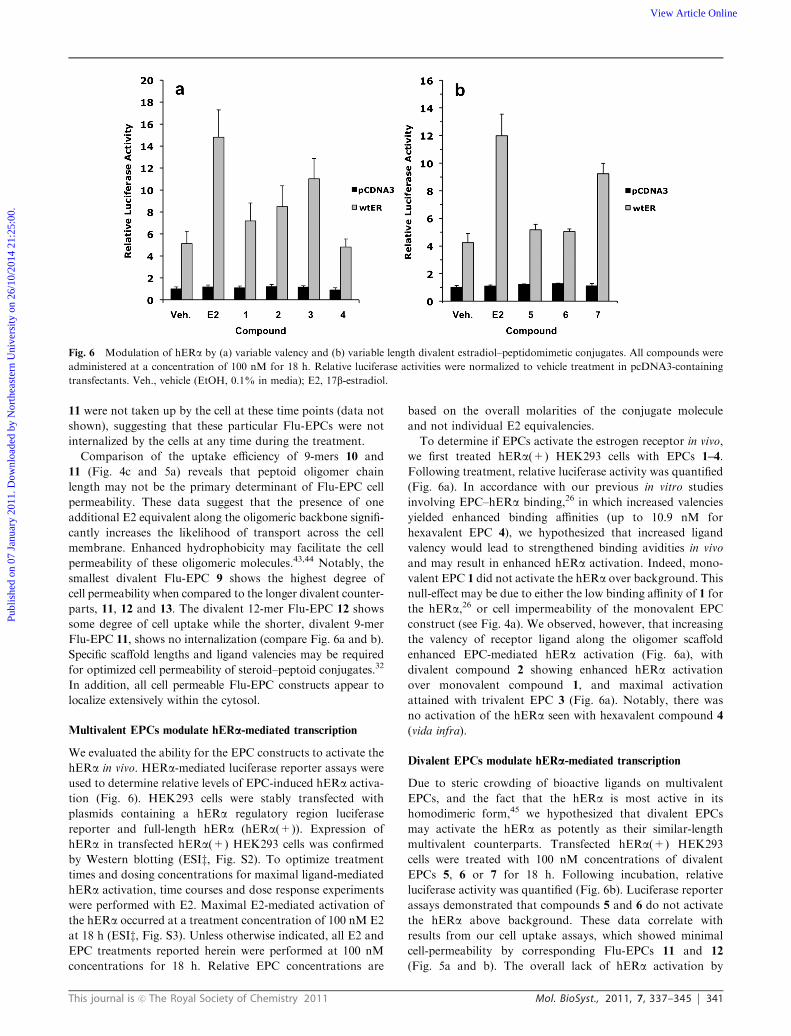
This journal is c The Royal Society of Chemistry 2011 Mol. BioSyst., 2011, 7, 337–345 341
11 were not taken up by the cell at these time points (data not
shown), suggesting that these particular Flu-EPCs were not
internalized by the cells at any time during the treatment.
Comparison of the uptake efficiency of 9-mers 10 and
11 (Fig. 4c and 5a) reveals that peptoid oligomer chain
length may not be the primary determinant of Flu-EPC cell
permeability. These data suggest that the presence of one
additional E2 equivalent along the oligomeric backbone signifi-
cantly increases the likelihood of transport across the cell
membrane. Enhanced hydrophobicity may facilitate the cell
permeability of these oligomeric molecules.43,44 Notably, the
smallest divalent Flu-EPC 9 shows the highest degree of
cell permeability when compared to the longer divalent counter-
parts, 11, 12 and 13. The divalent 12-mer Flu-EPC 12 shows
some degree of cell uptake while the shorter, divalent 9-mer
Flu-EPC 11, shows no internalization (compare Fig. 6a and b).
Specific scaffold lengths and ligand valencies may be required
for optimized cell permeability of steroid–peptoid conjugates.32
In addition, all cell permeable Flu-EPC constructs appear to
localize extensively within the cytosol.
Multivalent EPCs modulate hERa-mediated transcription
We evaluated the ability for the EPC constructs to activate the
hERa in vivo. HERa-mediated luciferase reporter assays were
used to determine relative levels of EPC-induced hERa activa-
tion (Fig. 6). HEK293 cells were stably transfected with
plasmids containing a hERa regulatory region luciferase
reporter and full-length hERa (hERa(+)). Expression of
hERa in transfected hERa(+) HEK293 cells was confirmed
by Western blotting (ESIz, Fig. S2). To optimize treatment
times and dosing concentrations for maximal ligand-mediated
hERa activation, time courses and dose response experiments
were performed with E2. Maximal E2-mediated activation of
the hERa occurred at a treatment concentration of 100 nM E2
at 18 h (ESIz, Fig. S3). Unless otherwise indicated, all E2 and
EPC treatments reported herein were performed at 100 nM
concentrations for 18 h. Relative EPC concentrations are
based on the overall molarities of the conjugate molecule
and not individual E2 equivalencies.
To determine if EPCs activate the estrogen receptor in vivo,
we first treated hERa(+) HEK293 cells with EPCs 1–4.
Following treatment, relative luciferase activity was quantified
(Fig. 6a). In accordance with our previous in vitro studies
involving EPC–hERa binding,26 in which increased valencies
yielded enhanced binding affinities (up to 10.9 nM for
hexavalent EPC 4), we hypothesized that increased ligand
valency would lead to strengthened binding avidities in vivo
and may result in enhanced hERa activation. Indeed, mono-
valent EPC 1 did not activate the hERa over background. This
null-effect may be due to either the low binding affinity of 1 for
the hERa,26 or cell impermeability of the monovalent EPC
construct (see Fig. 4a). We observed, however, that increasing
the valency of receptor ligand along the oligomer scaffold
enhanced EPC-mediated hERa activation (Fig. 6a), with
divalent compound 2 showing enhanced hERa activation
over monovalent compound 1, and maximal activation
attained with trivalent EPC 3 (Fig. 6a). Notably, there was
no activation of the hERa seen with hexavalent compound 4
(vida infra).
Divalent EPCs modulate hERa-mediated transcription
Due to steric crowding of bioactive ligands on multivalent
EPCs, and the fact that the hERa is most active in its
homodimeric form,45 we hypothesized that divalent EPCs
may activate the hERa as potently as their similar-length
multivalent counterparts. Transfected hERa(+) HEK293
cells were treated with 100 nM concentrations of divalent
EPCs 5, 6 or 7 for 18 h. Following incubation, relative
luciferase activity was quantified (Fig. 6b). Luciferase reporter
assays demonstrated that compounds 5 and 6 do not activate
the hERa above background. These data correlate with
results from our cell uptake assays, which showed minimal
cell-permeability by corresponding Flu-EPCs 11 and 12
(Fig. 5a and b). The overall lack of hERa activation by
Fig. 6 Modulation of hERa by (a) variable valency and (b) variable length divalent estradiol–peptidomimetic conjugates. All compounds were
administered at a concentration of 100 nM for 18 h. Relative luciferase activities were normalized to vehicle treatment in pcDNA3-containing
transfectants. Veh., vehicle (EtOH, 0.1% in media); E2, 17b-estradiol.
Publ
ishe
d on
07
Janu
ary
2011
. Dow
nloa
ded
by N
orth
east
ern
Uni
vers
ity o
n 26
/10/
2014
21:
25:0
0.
View Article Online
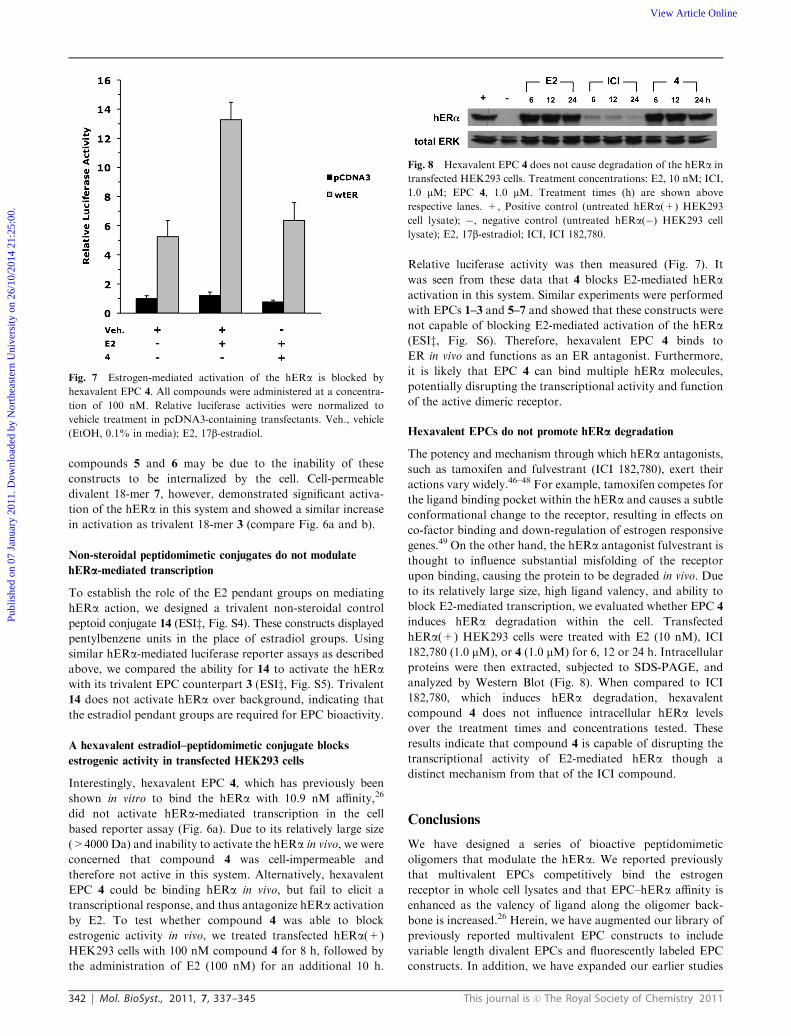
342 Mol. BioSyst., 2011, 7, 337–345 This journal is c The Royal Society of Chemistry 2011
compounds 5 and 6 may be due to the inability of these
constructs to be internalized by the cell. Cell-permeable
divalent 18-mer 7, however, demonstrated significant activa-
tion of the hERa in this system and showed a similar increase
in activation as trivalent 18-mer 3 (compare Fig. 6a and b).
Non-steroidal peptidomimetic conjugates do not modulate
hERa-mediated transcription
To establish the role of the E2 pendant groups on mediating
hERa action, we designed a trivalent non-steroidal control
peptoid conjugate 14 (ESIz, Fig. S4). These constructs displayedpentylbenzene units in the place of estradiol groups. Using
similar hERa-mediated luciferase reporter assays as described
above, we compared the ability for 14 to activate the hERawith its trivalent EPC counterpart 3 (ESIz, Fig. S5). Trivalent14 does not activate hERa over background, indicating that
the estradiol pendant groups are required for EPC bioactivity.
A hexavalent estradiol–peptidomimetic conjugate blocks
estrogenic activity in transfected HEK293 cells
Interestingly, hexavalent EPC 4, which has previously been
shown in vitro to bind the hERa with 10.9 nM affinity,26
did not activate hERa-mediated transcription in the cell
based reporter assay (Fig. 6a). Due to its relatively large size
(>4000 Da) and inability to activate the hERa in vivo, we were
concerned that compound 4 was cell-impermeable and
therefore not active in this system. Alternatively, hexavalent
EPC 4 could be binding hERa in vivo, but fail to elicit a
transcriptional response, and thus antagonize hERa activation
by E2. To test whether compound 4 was able to block
estrogenic activity in vivo, we treated transfected hERa(+)
HEK293 cells with 100 nM compound 4 for 8 h, followed by
the administration of E2 (100 nM) for an additional 10 h.
Relative luciferase activity was then measured (Fig. 7). It
was seen from these data that 4 blocks E2-mediated hERaactivation in this system. Similar experiments were performed
with EPCs 1–3 and 5–7 and showed that these constructs were
not capable of blocking E2-mediated activation of the hERa(ESIz, Fig. S6). Therefore, hexavalent EPC 4 binds to
ER in vivo and functions as an ER antagonist. Furthermore,
it is likely that EPC 4 can bind multiple hERa molecules,
potentially disrupting the transcriptional activity and function
of the active dimeric receptor.
Hexavalent EPCs do not promote hERa degradation
The potency and mechanism through which hERa antagonists,
such as tamoxifen and fulvestrant (ICI 182,780), exert their
actions vary widely.46–48 For example, tamoxifen competes for
the ligand binding pocket within the hERa and causes a subtle
conformational change to the receptor, resulting in effects on
co-factor binding and down-regulation of estrogen responsive
genes.49 On the other hand, the hERa antagonist fulvestrant is
thought to influence substantial misfolding of the receptor
upon binding, causing the protein to be degraded in vivo. Due
to its relatively large size, high ligand valency, and ability to
block E2-mediated transcription, we evaluated whether EPC 4
induces hERa degradation within the cell. Transfected
hERa(+) HEK293 cells were treated with E2 (10 nM), ICI
182,780 (1.0 mM), or 4 (1.0 mM) for 6, 12 or 24 h. Intracellular
proteins were then extracted, subjected to SDS-PAGE, and
analyzed by Western Blot (Fig. 8). When compared to ICI
182,780, which induces hERa degradation, hexavalent
compound 4 does not influence intracellular hERa levels
over the treatment times and concentrations tested. These
results indicate that compound 4 is capable of disrupting the
transcriptional activity of E2-mediated hERa though a
distinct mechanism from that of the ICI compound.
Conclusions
We have designed a series of bioactive peptidomimetic
oligomers that modulate the hERa. We reported previously
that multivalent EPCs competitively bind the estrogen
receptor in whole cell lysates and that EPC–hERa affinity is
enhanced as the valency of ligand along the oligomer back-
bone is increased.26 Herein, we have augmented our library of
previously reported multivalent EPC constructs to include
variable length divalent EPCs and fluorescently labeled EPC
constructs. In addition, we have expanded our earlier studies
Fig. 7 Estrogen-mediated activation of the hERa is blocked by
hexavalent EPC 4. All compounds were administered at a concentra-
tion of 100 nM. Relative luciferase activities were normalized to
vehicle treatment in pcDNA3-containing transfectants. Veh., vehicle
(EtOH, 0.1% in media); E2, 17b-estradiol.
Fig. 8 Hexavalent EPC 4 does not cause degradation of the hERa in
transfected HEK293 cells. Treatment concentrations: E2, 10 nM; ICI,
1.0 mM; EPC 4, 1.0 mM. Treatment times (h) are shown above
respective lanes. +, Positive control (untreated hERa(+) HEK293
cell lysate); �, negative control (untreated hERa(�) HEK293 cell
lysate); E2, 17b-estradiol; ICI, ICI 182,780.
Publ
ishe
d on
07
Janu
ary
2011
. Dow
nloa
ded
by N
orth
east
ern
Uni
vers
ity o
n 26
/10/
2014
21:
25:0
0.
View Article Online

This journal is c The Royal Society of Chemistry 2011 Mol. BioSyst., 2011, 7, 337–345 343
by demonstrating that EPCs are capable of modulating the
hERa in vivo.
Qualitative live-cell imaging experiments indicated that
there is a marked length and valency dependence on cell
permeability of Flu-EPCs, with divalent 6-mer 9, trivalent
9-mer 10, and divalent 18-mer 13 having the highest levels of
internalization. EPCs are intrinsically amphipathic, consisting
of a relatively hydrophilic backbone and large, bulky, hydro-
phobic pendant groups. The low levels of cell permeability that
are seen with Flu-EPCs monovalent 3-mer 8, divalent 9-mer
11, and divalent 12-mer 12 may be due to a number of factors:
the presence of an anionic fluorophore, overall hydrophilicity,
or folded conformation in solution.50 Nevertheless, an obvious
advantage of working with EPCs is their intrinsic length and
valence ‘‘tunability’’, which affords precise control over ligand
spacing and can facilitate physicochemical optimization of
these bioactive ligands for enhanced cell permeability.
In addition, we have shown that our EPC constructs are
capable of modulating the hERa in a living system, with
maximal activity coming from the most cell-permeable
constructs: divalent 6-mer 2, trivalent 9-mer 3, and divalent
18-mer 7. The minimal activation of monovalent 3-mer 1,
divalent 9-mer 5, and divalent 12-mer 6 may be due to their
inability to traverse the cell membrane. The ability for
compounds 2, 3, and 7 to activate nuclear actions of the hERais apparent despite the fact that their fluorescently labeled
counterparts do not seem to penetrate to the nucleus. This
result may be due the presence of the fluorophore perturbing
the ability for these constructs to cross the nuclear envelope.
Surprisingly, hexavalent 18-mer 4, which has the highest
receptor affinity of any EPC tested,26 did not activate the
hERa in vivo. Conversely, divalent 18-mer 7, which is identical
in length but contains substantially fewer E2 equivalents than
EPC 4, shows enhanced activation of the hERa over
background levels. This result indicates that increased local
concentration of receptor ligand and high EPC-receptor
binding affinity does not necessarily translate to enhanced
hERa activation. To the contrary, hexavalent EPC 4 is able
to block estrogenic-mediated activation of the hERa when
co-administered with E2, indicating that the characteristics of
EPC activity are markedly dependent on the valency of the
displayed ligands.
The ability to tune precisely the length and valency of our
EPCs allows for the generation of novel bioactive constructs
that may be optimized for use in a variety of experimental
settings. There is now evidence that pools of estrogen receptor
localize at the cell membrane, in the cytosol, and in the
nucleus, and that each of these receptor pools are capable
of independently activating various ER-mediated pathways
within the cell.20,51–53 The EPC synthetic model outlined
herein affords us the capacity to tailor the physicochemical
properties of these bioactive oligomer constructs. We are
currently exploring the ability for EPC conjugates to activate
specific subsets of estrogen receptors based on subcellular
localization in hERa(+) breast cancer cell lines. Importantly,
the approach outlined herein can be extended to display a
variety of steroid hormones or other ligands,5,26,37 suggesting a
general strategy for modulating protein receptor mediated
signaling pathways.
Experimental
Detailed peptoid synthesis protocols, compound characteriza-
tion and Flu-EPC fluorescence emission spectra are outlined in
the ESI.z
Cell culture
HEK293 cells were maintained at 5% CO2 in a 37 1C
incubator and cultured in Dulbecco’s Modification of Eagles
Medium (DMEM) supplemented with 10% fetal bovine
serum (FBS) and penicillin/streptomycin (PS). Typically, cells
were grown on 10 cm tissue-culture dishes (BD/Falcon) to
approximately 80% confluence before subculture.
EPC toxicity assays
HEK293 cells were plated (5000 cells/well) in triplicate on
treated 96-well plates (BD Falcon) in DMEM supplemented
with 10% FBS and PS, and allowed to attach overnight. The
cells were then switched to media supplemented with either
vehicle (0.1% EtOH), doxorubicin (1.0 mM) or EPCs (1.0 mM).
The final EtOH concentration in the treatment media was
0.1%. The cells were allowed to incubate in a humidified
atmosphere at 37 1C for 3 d. Following incubation, relative
cell viability was quantified using a colorimetric MTT assay
(Promega) performed according to the manufacturer’s
instructions.
Cell uptake of fluorescently labeled EPCs
HEK293 cells were seeded on poly-D-lysine-coated clear-
bottom 96-well plates (Corning) at a density of 5000 cells
per well in DMEM supplemented with 10% FBS and PS.
Following overnight attachment, the cells were switched to
phenol red-free DMEM supplemented with 5% charcoal-
stripped FBS (CS-FBS) and L-Glutamine (L-Gln). The cells
were allowed to starve for 48 h before treatment. Purified
Flu-EPCs (10 mM in EtOH) or 5,6 carboxyfluorescein
(10 mM in EtOH) were dissolved to a final concentration of
50 mM in phenol red-free DMEM supplemented with 5%
CS-FBS and L-Gln. The final EtOH concentration in the
treatment media was 0.5%. The cells were allowed to incubate
in the treatment solution for 18 h at 37 1C in the dark.
Following incubation, cells were counter-stained in phenol
red-free DMEM supplemented with 5% CS-FBS and L-Gln
containing Hoechst 33342 (Invitrogen) according to the
manufacturer’s instructions. Live cell imaging was conducted
on a DMIRE-2 fluorescence microscope (Leica) outfitted with a
digital camera (Hamamatsu) using appropriate filters. Images
were processed using FW4000 imaging software (Leica).
EPC-mediated hERa activation
HEK293 cells were seeded in triplicate on poly-D-lysine-coated
24-well plates (Corning) at a density of 15 000 cells per well in
DMEM supplemented with 10% FBS and PS. Following
overnight attachment, the cells were transfected with appropriate
plasmids using the ExGen 500 transfection reagent (Fermentas)
according to the manufacturer’s instructions. Transfectable
plasmids were suspended in phenol red-free DMEM and
administered to cells. Each well received 100 ng ERE-containing
XETL regulatory region-luciferase reporter plasmid,54 3 ng
Publ
ishe
d on
07
Janu
ary
2011
. Dow
nloa
ded
by N
orth
east
ern
Uni
vers
ity o
n 26
/10/
2014
21:
25:0
0.
View Article Online

344 Mol. BioSyst., 2011, 7, 337–345 This journal is c The Royal Society of Chemistry 2011
pcDNA3-hERa or empty vector (pcDNA3), 5 ng pcMV-lacZ,
and 142 ng pBluescript II SK (Stratagene). Following a 5 h
incubation at 37 1C, the transfection solutions were aspirated
from the wells and the cells were allowed to recover overnight
in DMEM supplemented with 10% FBS and PS. The
transfectants were then switched to phenol red-free DMEM
supplemented with 5% CS-FBS and L-Gln and the cells were
allowed to starve for 48 h. E2 or EPCs 1–7 (10 mM in EtOH)
were dissolved to a final concentration of 100 nM in phenol
red-free DMEM supplemented with 5% CS-FBS and L-Gln.
The final EtOH concentration in each treatment solution was
0.1%. Treatment solutions were added and the cells were
allowed to incubate at 37 1C for 18 h. Following incubation,
the cells were washed with PBS and lysed in 1� luciferase cell
culture lysis reagent (Promega) according to the manufacturer’s
instructions. Luciferase activity was quantified in a reaction
mixture containing 10 mL cell lysate and 100 mL 1� luciferase
assay reagent (Promega) using a microplate luminometer
(LMax). Relative luciferase activity values were individually
normalized to b-galactosidase activity and collectively normalized
to vehicle treatment in pcDNA3-containing transfectants. Data
were processed using Microsoft Excel.
hERa degradation assays
Transfected hERa(+) HEK293 cells were grown to approxi-
mately 60% confluence on 6 cm culture dishes (BD Falcon) in
DMEM supplemented with 10% FBS and PS. The cells were
then switched to phenol red-free DMEM supplemented with
5% CS-FBS and L-Gln and the cells were allowed to starve for
48 h before treatment. E2 (10 mM in EtOH) was dissolved to a
final concentration of 10 nM in phenol red-free DMEM
supplemented with 5% CS-FBS and L-Gln. ICI 182,780
or EPC 4 (each 10 mM in EtOH) were dissolved to a final
concentration of 1.0 mM in phenol red-free DMEM
supplemented with 5% CS-FBS and L-Gln. The final EtOH
concentration in each treatment solution was 0.1%. Treatment
solutions containing appropriate concentrations of E2, ICI
182,780, or EPC 4 were added and the cells were allowed to
incubate at 37 1C for 6, 12 or 24 h. Following incubation, the
cells were harvested into ice-cold PBS and pelleted. Whole cells
were lysed in SKL lysis buffer in the presence of 1 mM
Na3VO4 and 1� protease-inhibitor cocktail. Protein concen-
trations were quantified using a standard colorimetric
Bradford assay (Bio-Rad). Samples were loaded onto 10%
acrylamide gels at a concentration of 50 mg protein per well
and subjected to SDS-PAGE. The separated proteins were
then transferred onto Immobilon membranes (Millipore)
and probed with rabbit affinity-purified anti-hERa (HC-20)
(Santa Cruz Biotechnology) or anti-ERK1/2 (Cell Signaling
Technology) primary antibodies. Membranes were then
incubated with anti-rabbit, horseradish peroxidase-conjugated
secondary antibodies (KPL). Signals were detected on X-ray
film (Denville) using an ECL detection kit (GE Health
Sciences) according to the manufacturer’s instructions.
Acknowledgements
The authors gratefully acknowledge the support of the
NSF (CAREER Award #CHE-0645361) and the NIH
(Research Facilities Improvement Grant C060RR-165720).
We thank Jerome Nwachukwu for valuable technical
assistance.
References
1 M. Mammen, S. Choi and G. M. Whitesides, Angew. Chem., Int.Ed., 1998, 37, 2754–2794.
2 D. Schwefel, C. Maierhofer, J. G. Beck, S. Seeberger,K. Diederichs, H. M. Moller, W. Welte and V. Wittmann,J. Am. Chem. Soc., 2010, 132, 8704–8719.
3 L. L. Kiessling, J. E. Gestwicki and L. E. Strong, Angew. Chem.,Int. Ed., 2006, 45, 2348–2368.
4 Y. Lee and N. S. Sampson, Curr. Opin. Struct. Biol., 2006, 16,544–550.
5 M. M. Lee, J. L. Childs-Disney, A. Pushechnikov, J. M. French,K. Sobczak, C. A. Thornton andM. D. Disney, J. Am. Chem. Soc.,2009, 131, 17464–17472.
6 M. Karskela, M. Helkearo, P. Virta and H. Lonnberg, BioconjugateChem., 2010, 21, 748–755.
7 R. J. Vaz, Z. L. Gao, J. Pribish, C. Xin, J. Levell, L. Davis,E. Albert, M. Brollo, A. Ugolini, D. M. Cramer, J. Cairns,K. Sides, L. Feng, J. Kwong, J. S. Kang, S. Rebello, M. Elliot,H. Lim, V. Chellaraj, R. W. Singleton and L. Yi, Bioorg. Med.Chem. Lett., 2004, 14, 6053–6056.
8 S. Liu and K. L. Kiick, Macromolecules, 2008, 41, 764–772.9 Y. L. Zhang, O. A. Li, L. G. Rodriguez and J. C. Gildersleeve,J. Am. Chem. Soc., 2010, 132, 9653–9662.
10 R. M. Evans, Science, 1988, 240, 889–895.11 S. Nilsson, S. Makela, E. Treuter, M. Tujague, J. Thomsen,
G. Andersson, E. Enmark, K. Pettersson, M. Warner andJ. Gustafsson, Physiol. Rev., 2001, 81, 1535–1565.
12 M. J. Tsai and B. W. O’Malley, Annu. Rev. Biochem., 1994, 63,451–486.
13 A. Warnmark, A. Wikstrom, A. P. H. Wright, J. A. Gustafssonand T. Hard, J. Biol. Chem., 2001, 276, 45939–45944.
14 G. G. J. M. Kuiper, E. Enmark, M. Pelto-Huikko, S. Nilsson andJ. A. Gustafsson, Proc. Natl. Acad. Sci. U. S. A., 1996, 93,5925–5930.
15 M. Byers, G. G. J. M. Kuiper, J. A. Gustafsson and O. K.Park-Sarge, Mol. Endocrinol., 1997, 11, 172–182.
16 Y. Imai, S. Kondoh, A. Kouzmenko and S. Kato,Mol. Endocrinol.,2010, 24, 877–85.
17 F. Holst, P. R. Stahl, C. Ruiz, O. Hellwinkel, Z. Jehan,M. Wendland, A. Lebeau, L. Terracciano, K. Al-Kuraya,F. Janicke, G. Sauter and R. Simon, Nat. Genet., 2007, 39,655–660.
18 S. Sengupta and V. C. Jordan, Adv. Exp. Med. Biol., 2008, 630,206–219.
19 A. Chimento, R. Sirianni, C. Delalande, D. Silandre, C. Bois,S. Ando, M. Maggiolini, S. Carreau and V. Pezzi, Mol. Cell.Endocrinol., 2010, 320, 136–144.
20 K. H. Kim and J. R. Bender,Mol. Cell. Endocrinol., 2009, 308, 3–8.21 H. Y. Kim, J. Sohn, G. T. Wijewickrama, P. Edirisinghe,
T. Gherezghiher, M. Hemachandra, P. Y. Lu, R. E. Chandrasena,M. E. Molloy, D. A. Tonetti and G. R. J. Thatcher, Bioorg.Med. Chem., 2010, 18, 809–821.
22 P. E. Stevis, D. C. Deecher, L. Suhadolnik, L. M. Mallis andD. E. Frail, Endocrinology, 1999, 140, 5455–5458.
23 S. H. Kim and J. A. Katzenellenbogen, Angew. Chem., Int. Ed.,2006, 45, 7243–7248.
24 W. R. Harrington, S. H. Kim, C. C. Funk, Z. Madak-Erdogan,R. Schiff, J. A. Katzenellenbogen and B. S. Katzenellenbogen,Mol. Endocrinol., 2006, 20, 491–502.
25 L. C. Yang, Q. G. Zhang, C. F. Zhou, F. Yang, Y. D. Zhang,R. M. Wang and D. W. Brann, PLoS One, 2010, 5, e9851.
26 J. M. Holub, M. J. Garabedian and K. Kirshenbaum, QSARComb. Sci., 2007, 26, 1175–1180.
27 R. J. Simon, R. S. Kania, R. N. Zuckermann, V. D. Huebner,D. A. Jewell, S. Banville, S. Ng, L. Wang, S. Rosenberg,C. K. Marlowe, D. Spellmeyer, R. Tan, A. D. Frankel,D. V. Santi, F. E. Cohen and P. A. Bartlett, Proc. Natl. Acad.Sci. U. S. A., 1992, 89, 9367–9371.
Publ
ishe
d on
07
Janu
ary
2011
. Dow
nloa
ded
by N
orth
east
ern
Uni
vers
ity o
n 26
/10/
2014
21:
25:0
0.
View Article Online

This journal is c The Royal Society of Chemistry 2011 Mol. BioSyst., 2011, 7, 337–345 345
28 R. N. Zuckermann, J. M. Kerr, S. B. H. Kent and W. H. Moos,J. Am. Chem. Soc., 1992, 114, 10646–10647.
29 S. A. Fowler and H. E. Blackwell, Org. Biomol. Chem., 2009, 7,1508–1524.
30 B. Yoo and K. Kirshenbaum, Curr. Opin. Chem. Biol., 2008, 12,714–721.
31 S. M. Miller, R. J. Simon, S. Ng, R. N. Zuckermann, J. M. Kerrand W. H. Moos, Drug Dev. Res., 1995, 35, 20–32.
32 N. C. Tan, P. Yu, Y. U. Kwon and T. Kodadek, Bioorg. Med.Chem., 2008, 16, 5853–5861.
33 Y. Utku, E. Dehan, O. Ouerfelli, F. Piano, R. N. Zuckermann,M. Pagano and K. Kirshenbaum, Mol. BioSyst., 2006, 2,312–317.
34 J. R. Holder, R. M. Bauzo, Z. Xiang, J. Scott and C. Haskel-Luevano, Bioorg. Med. Chem. Lett., 2003, 13, 4505–4509.
35 M. M. Reddy and T. Kodadek, Proc. Natl. Acad. Sci. U. S. A.,2005, 102, 12672–12677.
36 D. G. Udugamasooriya, S. P. Dineen, R. A. Brekken andT. Kodadek, J. Am. Chem. Soc., 2008, 130, 5744–5752.
37 H. Jang, A. Farfarman, J. M. Holub and K. Kirshenbaum, Org.Lett., 2005, 7, 1951–1954.
38 J. M. Holub, H. Jang and K. Kirshenbaum, Org. Biomol. Chem.,2006, 4, 1497–1502.
39 I. Peretto, R. M. Sanchez-Martin, X. Wang, J. Ellard, S. Mittooand M. Bradley, Chem. Commun., 2003, 2312–2313.
40 T. Schroder, N. Niemeier, S. Afonin, A. S. Ulrich, H. F. Krug andS. Brase, J. Med. Chem., 2008, 51, 376–379.
41 V. Anbazhagan, V. Kandavelu, A. Kathiravan andR. Renganathan, J. Photochem. Photobiol., A, 2008, 193, 204–212.
42 D. A. Gewirtz, Biochem. Pharmacol., 1999, 57, 727–741.43 S. Scarfi, A. Gasparini, G. Damonte and U. Benatti, Biochem.
Biophys. Res. Commun., 1997, 236, 323–326.44 A. Unciti-Broceta, F. Diezmann, C. Y. Ou-Yang, M. A. Fara and
M. Bradley, Bioorg. Med. Chem., 2009, 17, 959–966.45 V. Kumar and P. Chambon, Cell, 1998, 55, 145–156.46 C. Buijs, E. G. E. de Vries, M. J. E. Mourits and P. H. B. Willemse,
Cancer Treat. Rev., 2008, 34, 640–655.47 B. J. Peano, J. S. Crabtree, B. S. Komm, R. C. Winneker and
H. A. Harris, Endocrinology, 2009, 150, 1897–1903.48 J. S. Crabtree, B. J. Peano, X. Zhang, B. S. Komm, R. C. Winneker
and H. A. Harris, Mol. Cell. Endocrinol., 2008, 287, 40–46.49 B. L. Riggs and L. C. Hartmann, N. Engl. J. Med., 2003, 348,
618–629.50 K. Huang, C. W. Wu, T. J. Sanborn, J. A. Patch, K. Kirshenbaum,
R. N. Zuckermann, A. E. Barron and I. Radhakrishnan, J. Am.Chem. Soc., 2006, 128, 1733–1738.
51 M. Razandi, A. Pedram, I. Merchenthaler, G. L. Greene andE. R. Levin, Mol. Endocrinol., 2004, 18, 2854–2865.
52 A. Pedram, M. Razandi, M. Aitkenhead, C. C. W. Hughes andE. R. Levin, J. Biol. Chem., 2002, 277, 50768–50775.
53 A. Pedram, M. Razandi and E. R. Levin, Mol. Endocrinol., 2006,20, 1996–2009.
54 J. R. de Wet, K. V. Wood, M. DeLuca, D. R. Helinski andS. Subramani, Mol. Cell. Biol., 1987, 7, 725–737.
Publ
ishe
d on
07
Janu
ary
2011
. Dow
nloa
ded
by N
orth
east
ern
Uni
vers
ity o
n 26
/10/
2014
21:
25:0
0.
View Article Online


















