
John Dewey - Indian ETD Repository @ INFLIBNET: Home
19
Osteopontin Regulates Cyclooxygenase-2 (COX-2) /Prostaglandin E 2 (PGE 2 )-mediated Prostate Cancer Progression and Angiogenesis: Role of Protein Kinase Cα/c-Src/ IκB Kinase α/β dependent Signaling Cascade Scientific principles and laws do not lie on the surface of nature. They are hidden, and must be wrested from nature by an active and elaborate technique of inquiry. John Dewey
Transcript of John Dewey - Indian ETD Repository @ INFLIBNET: Home
Osteopontin Regulates Cyclooxygenase-2 (COX-2) /Prostaglandin E2 (PGE2)-mediated
Prostate Cancer Progression and Angiogenesis: Role of Protein Kinase Cα/c-Src/ IκB Kinase
α/β dependent Signaling Cascade
Scientific principles and laws do not lie on the surface of nature. They are hidden, and must be wrested from nature by an active and elaborate technique of inquiry. John Dewey

47
6. RESULTS
6.1 Osteopontin Regulates Cyclooxygenase-2 (COX-2) /Prostaglandin E2 (PGE2)-mediated Prostate Cancer Progression and Angiogenesis: Role of Protein Kinase Cα-c-Src- IκB Kinase α/β dependent Signaling Cascade
6.1.1 Introduction The prostate cancer is the second most common cause of death in men especially in
western world (2). Cancer progression involves a number of processes which require crosstalk
between several oncogenic molecules, growth factors, hormones, enzymes, proteases and
angiogenic molecules that enable the cancer to spread and evoke angiogenesis (2, 95).
Recent studies have shown that COX-2 functions as one of the important molecule which
controls tumor growth and angiogenesis (95). Three isoforms of COX enzyme (COX-1, COX-2
and COX-3) have been reported (207). Among them, COX-2 is an inducible enzyme. It is
induced in many cell types by mitogens, growth factors, cytokines and tumor promoters and its
increased expression is associated with cancer progression (208-210). Recent reports have
demonstrated that COX-2 promotes tumor cell proliferation, survival, and angiogenesis through
PGE2-mediated pathway (95, 211). PGE2 is the predominant eicosanoid produced by malignant
and benign prostate cells (127). PGE2 regulates tumor development through several mechanisms
including promotion of angiogenesis, inhibition of apoptosis, increased invasiveness/and motility
and modulation of inflammation and immune responses (211-213).
Osteopontin (OPN) plays significant role in determining the oncogenic and metastatic
potential of various cancers and it is recognized as a key marker in the processes of
tumorigenicity and metastasis (6, 7, 30). Although, previous data indicated that, OPN plays
crucial role in chemotaxis and chemoinvasion of PC-3 cells (77) but the molecular mechanism
by which OPN regulates COX-2 dependent prostate cancer progression is still not clear. Recent
data has demonstrated that OPN induces pro-matrix metalloproteinase-2 and -9 (pro-MMP-2 and
-9) activation, urokinase plasminogen activator (uPA) secretion, cell motility, ECM invasion and
tumor growth in melanoma (67, 68).

48
In this study using in vitro and in vivo models, we have shown at least in part, the
molecular mechanism by which OPN regulates PKCα/c-Src/IKK/NF-κB signaling cascades
leading to COX-2-mediated PGE2 production and MMP-2 activation in prostate cancer cells.
Moreover, the data demonstrated that OPN-induced COX-2 regulates tumor cell motility,
angiogenesis and tumorigenesis of prostate cancer through both autocrine and paracrine
pathways. Furthermore, suppression of COX-2 activity by nonsteroidal anti-inflammatory drug
(NSAID), celecoxib or blocking the interaction between PGE2 and its receptor EP2 by using
specific anti-EP2 antibody significantly suppressed OPN-induced in vitro cell motility,
invasiveness and in vivo tumor growth and angiogenesis. The clinical specimen analysis revealed
elevated expression of OPN and COX-2, which correlated with enhanced MMP-2 expression and
angiogenesis in human prostate cancer specimens of higher grades. Consequently, OPN plays
important and essential role in two key aspects of tumor progression: COX-2-mediated PGE2
production and MMP-2 activation by tumor cells leading to tumor progression and COX-
2/PGE2-stimulated angiogenesis. Our findings suggest that blockade of OPN and COX-2 could
be promising therapeutic approaches for the inhibition of prostate tumor growth and
angiogenesis.
6.1.2 Results
6.1.2.1 Regulation of COX-2 Expression in Response to OPN.
To examine the effect of OPN on COX-2 expression, serum starved PC-3 cells were
treated with 0.5μmol/L OPN for 0-24 h. The expression of COX-2 in cell lysate was detected by
Western blot and the data indicated that OPN induced maximum expression of COX-2 at 24 h
(Fig. 9 A). The effects of OPN on COX-2 mRNA levels were also determined by RT-PCR. The
data showed that 0.5 μmol/L OPN induced the maximum expression of COX-2 (Fig. 9 B).
Altogether, our results revealed that OPN enhanced COX-2 expression both at transcriptional
and translational levels.
6.1.2.2 OPN Regulates NIK-IKK-NF-κB-dependent COX-2 Promoter
Activity.
To determine the effect of OPN on COX-2 promoter activity, PC-3 cells were transfected
with COX-2 luciferase construct and then treated with OPN in a dose (0-0.5μmol/L) dependent
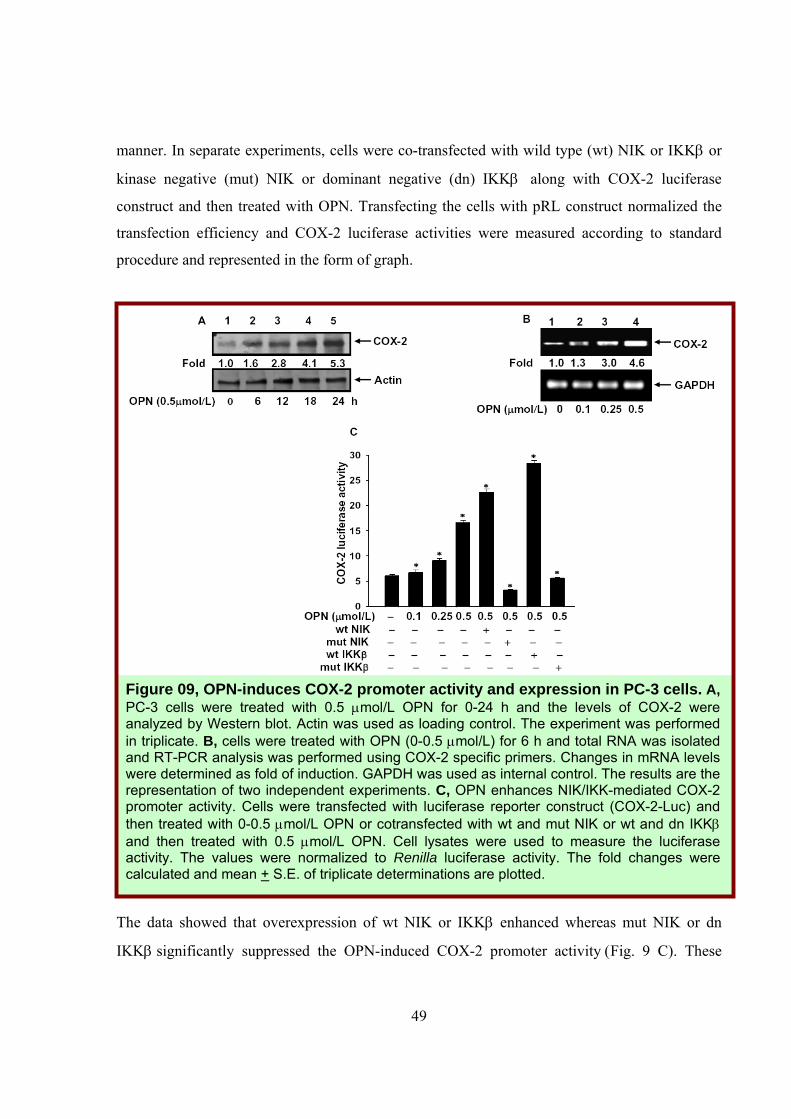
49
manner. In separate experiments, cells were co-transfected with wild type (wt) NIK or IKKβ or
kinase negative (mut) NIK or dominant negative (dn) IKKβ along with COX-2 luciferase
construct and then treated with OPN. Transfecting the cells with pRL construct normalized the
transfection efficiency and COX-2 luciferase activities were measured according to standard
procedure and represented in the form of graph.
The data showed that overexpression of wt NIK or IKKβ enhanced whereas mut NIK or dn
IKKβ significantly suppressed the OPN-induced COX-2 promoter activity (Fig. 9 C). These
Figure 09, OPN-induces COX-2 promoter activity and expression in PC-3 cells. A, PC-3 cells were treated with 0.5 μmol/L OPN for 0-24 h and the levels of COX-2 were analyzed by Western blot. Actin was used as loading control. The experiment was performed in triplicate. B, cells were treated with OPN (0-0.5 μmol/L) for 6 h and total RNA was isolated and RT-PCR analysis was performed using COX-2 specific primers. Changes in mRNA levels were determined as fold of induction. GAPDH was used as internal control. The results are the representation of two independent experiments. C, OPN enhances NIK/IKK-mediated COX-2 promoter activity. Cells were transfected with luciferase reporter construct (COX-2-Luc) and then treated with 0-0.5 μmol/L OPN or cotransfected with wt and mut NIK or wt and dn IKKβ and then treated with 0.5 μmol/L OPN. Cell lysates were used to measure the luciferase activity. The values were normalized to Renilla luciferase activity. The fold changes were calculated and mean + S.E. of triplicate determinations are plotted.

50
results indicated that OPN induces COX-2 promoter activity through NIK/IKK dependent NF-
κB-mediated pathway.
6.1.2.3 OPN Regulates PKCα Activation and its Interaction with c-Src.
Previous data has indicated that PKCα plays crucial role in COX-2 expression (164).
Moreover, other studies have shown the crucial role of PKCα in the survival and growth of
androgen-independent human prostate cancer (PC-3) cells (214-216). OPN induces the c-Src
activation (30). Therefore, to examine the effect of OPN on PKCα activation and to examine the
direction of crosstalk between PKCα and c-Src, PC-3 cells were transfected with dn c-Src,
treated with OPN and cell lysates were immunoprecipitated with anti-PKCα antibody (Fig. 10
A). The immunoprecipitated samples were used for in vitro PKCα kinase assay.
Figure 10, OPN induces PKCα mediated c-Src activation. A&B, PC-3 cells were either transfected with dn-c-Src or pretreated with staurosporine and then treated with OPN. Cell lysates were immunoprecipitated with anti-PKCα or anti-c-Src antibody and the kinase activity of PKCα and c-Src were determined by autophosphorylation experiment and analyzed by autoradiography. C, Diagrammatic representation of OPN induced PKCα dependent c-Src activation.

51
Similarly, in other experiment, PC-3 cells were pretreated with PKCα inhibitor
(Staurosporine) and then treated with OPN and cell lysates were immunoprecipitated with anti-c-
Src antibody. The immunoprecipitated samples were analyzed for c-Src autophosphorylation by
in vitro kinase assay (Fig. 10 B). The levels of total PKCα and c-Src in the same lysates were
detected by Western blot using their specific antibodies as controls. The data revealed that
overexpression of dn c-Src had no effect on OPN-induced PKCα autophosphorylation whereas
staurosporine suppressed OPN-induced c-Src phosphorylation suggesting that OPN regulates
PKCα dependent c-Src activation (Fig. 10 A&B).
6.1.2.4 NIK Plays Differential Role in OPN-induced
IKKα/β Phosphorylation.
To substantiate the role of upstream kinases (PKC, c-Src and NIK) in OPN-induced
IKKα/β phosphorylation, cells were individually transfected with dn c-Src or wt and mut NIK
or pretreated with staurosporine and then treated with OPN. The levels of phospho-serine-
IKKα/β were detected by Western blot and the levels of phospho-tyrosine-IKKα/β were
analyzed by immunoprecipitating the same cell lysates with anti-IKKα/β antibody followed by
immunoblotting with anti-phospho-tyrosine antibody. The levels of IKK α/β were also detected
by Western blot as loading control. These data indicated that both PKC inhibitor and dn c-Src
suppressed OPN-induced serine as well as tyrosine phosphorylation of IKKα/β whereas mut
NIK inhibited only the serine but not tyrosine phosprylation of IKKα/β (Fig.11A &B). These
results suggested that NIK plays differential role in OPN-induced PKCα-c-Src-mediated IKKα/β
activation.
Moreover, to further investigate the role of PKCα, c-Src, NIK, IKK and NF-κB in OPN-
induced COX-2 expression, cells were pretreated with staurosporine or individually transfected
with dn c-Src, wt and mut NIK and IκBα sup. rep., wt and dn IKKα or IKKβ followed by
treatment with OPN. The COX-2 expression was detected by Western blot (Fig. 11C). These
results showed that PKCα and c-Src play an important role in regulating OPN-induced COX-2
expression through NIK-IKK-NF-κB-mediated pathway.

52
6.1.2.5 OPN Stimulates PKCα-c-Src-mediated p65 Phosphorylation.
Studies have revealed that the phosphorylation of p65 subunit of NF-κB is essential for the
nuclear translocation and activation of NF-κB (217) and the response elements for NF-κB have
Figure 11, NIK plays differential role in OPN-induced IKK phosphorylation. A, cells were individually transfected with dn c-Src, wt or mut NIK or pretreated with staurosporine and then treated with OPN. Half of the cell lysates were analyzed by Western blot using anti-phospho-S-IKKα/β (Ser-180/181) antibody and the remaining part of cell lysates were immunoprecipitated with IKKα/β antibody and detected by Western blot using anti-phospho-tyrosine antibody. The levels of IKKα/β were also detected by Western blot. All figures are the representation of three independent experiments. B, schematic diagram of OPN-induced PKCα-c-Src depencent phosphorylation of IKKα/β. Role of kinases upstream of NF-κB in OPN-induced COX-2 expression. C, cells were pretreated with staurosporine or individually transfected with dn c-Src, IκBα sup. rep., wt and dn IKKα or IKKβ and wt or mut NIK and then treated with OPN. The COX-2 expression was detected by Western blot. Actin was used as loading control. All figures are the representation of three independent experiments.
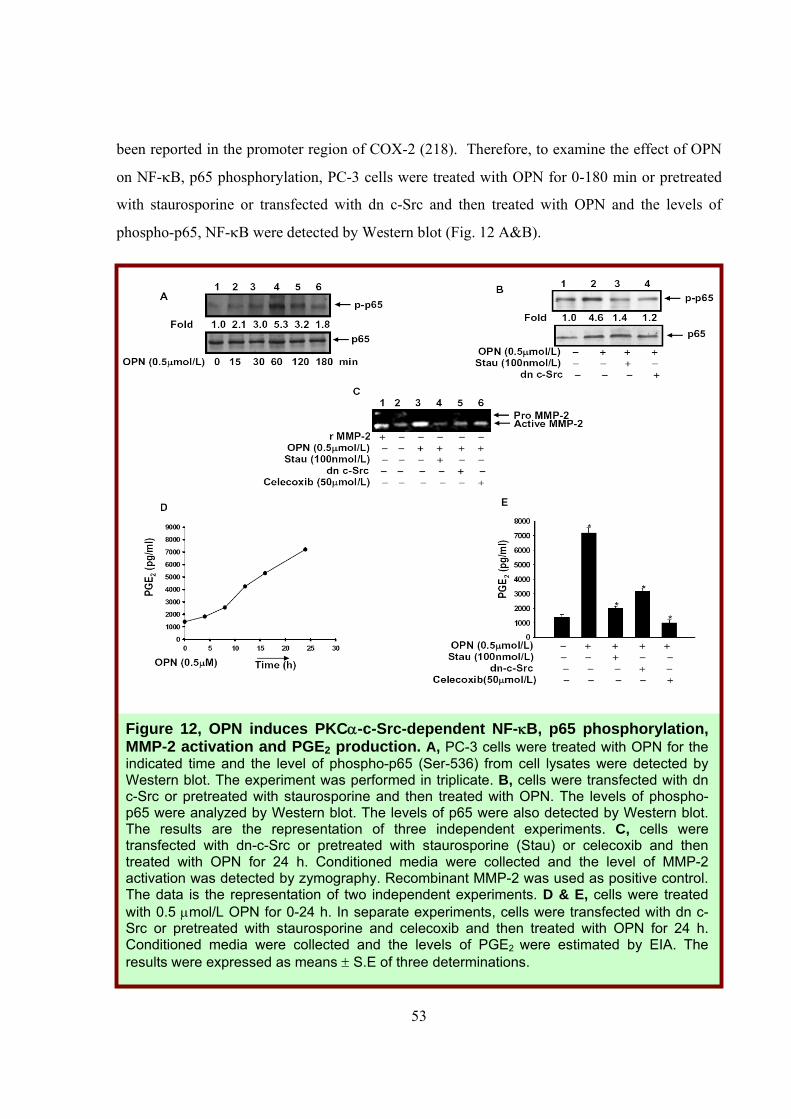
53
been reported in the promoter region of COX-2 (218). Therefore, to examine the effect of OPN
on NF-κB, p65 phosphorylation, PC-3 cells were treated with OPN for 0-180 min or pretreated
with staurosporine or transfected with dn c-Src and then treated with OPN and the levels of
phospho-p65, NF-κB were detected by Western blot (Fig. 12 A&B).
Figure 12, OPN induces PKCα-c-Src-dependent NF-κB, p65 phosphorylation, MMP-2 activation and PGE2 production. A, PC-3 cells were treated with OPN for the indicated time and the level of phospho-p65 (Ser-536) from cell lysates were detected by Western blot. The experiment was performed in triplicate. B, cells were transfected with dn c-Src or pretreated with staurosporine and then treated with OPN. The levels of phospho-p65 were analyzed by Western blot. The levels of p65 were also detected by Western blot. The results are the representation of three independent experiments. C, cells were transfected with dn-c-Src or pretreated with staurosporine (Stau) or celecoxib and then treated with OPN for 24 h. Conditioned media were collected and the level of MMP-2 activation was detected by zymography. Recombinant MMP-2 was used as positive control. The data is the representation of two independent experiments. D & E, cells were treated with 0.5 μmol/L OPN for 0-24 h. In separate experiments, cells were transfected with dn c-Src or pretreated with staurosporine and celecoxib and then treated with OPN for 24 h. Conditioned media were collected and the levels of PGE2 were estimated by EIA. The results were expressed as means ± S.E of three determinations.

54
The data indicated that inhibition of PKCα and c-Src significantly suppressed OPN-induced
phosphorylation of p65, NF-κB. The data showed that OPN stimulates p65 phosphorylation
through PKCα-c-Src dependent pathway, which leads to the NF-κB activation that ultimately
regulates COX-2 expression.
6.1.2.6 PKCα, c-Src and COX-2 Regulate PGE2 Production and MMP-2
Activation in Response to OPN.
Earlier, it has been reported that MMP-2 plays crucial role in prostate cancer cell
metastasis (219). The overexpression of COX-2 leads to upregulation of arachidonic acid
metabolism, which in turn results in increased production of PGE2 and cancer progression (127).
To examine whether PKC, c-Src and COX-2 are involved in OPN-induced MMP-2 activation
and PGE2 production, cells were either pretreated with staurosporine or COX-2 inhibitor
(celecoxib) or transfected with dn c-Src and then treated with OPN. The MMP-2 activation was
analyzed by gelatin zymography (Fig. 12 C). Moreover, to examine the effect of OPN on PGE2
production, PC-3 cells were treated with OPN for 0-24 h and the PGE2 levels were measured in
the conditioned media by using PGE2 EIA kit according to the manufacturer’s instructions. The
results showed that OPN induced PGE2 production in these cells (Fig. 12 D). Moreover, the data
also indicated that inhibitors of PKC and COX-2 and dn c-Src drastically suppressed the OPN-
induced PGE2 production and MMP-2 activation (Fig. 12 C&E). These results demonstrated that
COX-2 plays crucial role in OPN-induced PKC-c-Src-mediated PGE2 production and MMP-2
activation in prostate cancer cells.
6.1.2.7 COX-2 and PGE2 Regulate OPN-induced PC-3 Cell Motility and
Invasiveness.
Previous studies have indicated that COX-2 plays crucial role in tumor cell invasion and
metastasis (95). Therefore, to study the role of COX-2 and PGE2 in OPN-induced PC-3 cell
migration and to examine the role of upstream kinases (PKCα, c-Src, NIK and IKK) in this
process, the wound migration assay was performed. Cells were either pretreated with
staurosporine, celecoxib or anti-EP2 receptor antibody or transfected with dn c-Src. Wounds
with a constant diameter were made and cells were treated with OPN. The wound photographs
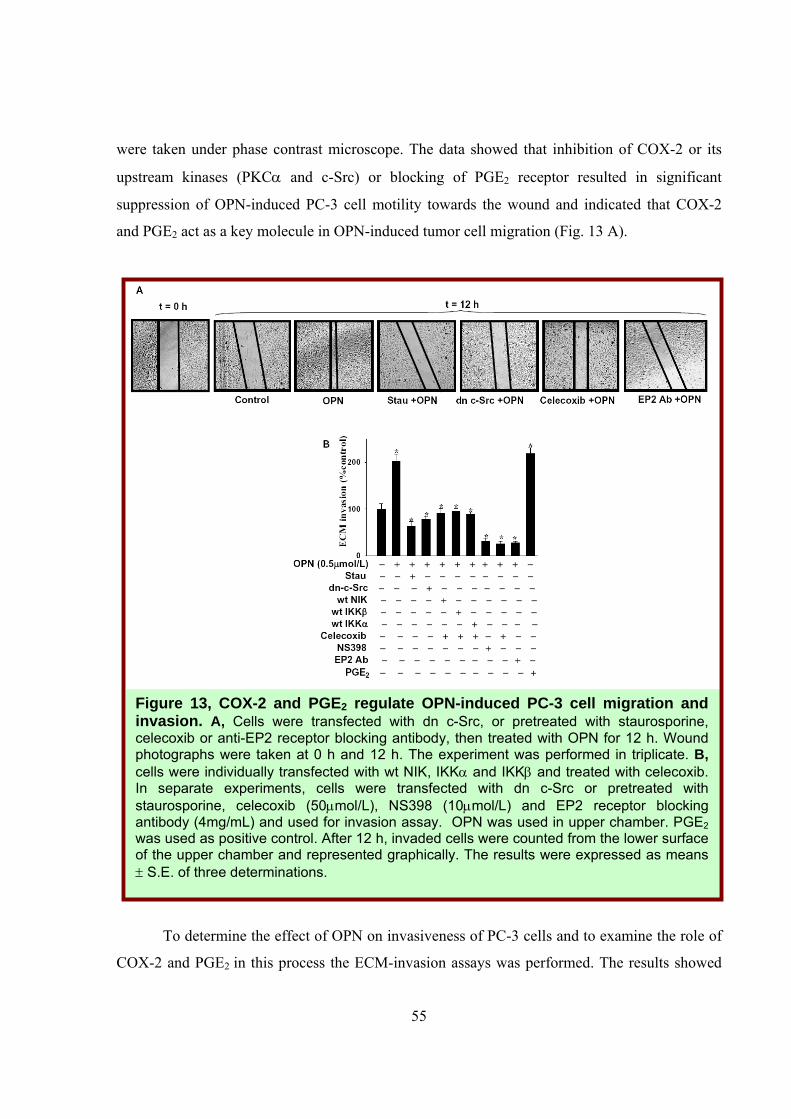
55
were taken under phase contrast microscope. The data showed that inhibition of COX-2 or its
upstream kinases (PKCα and c-Src) or blocking of PGE2 receptor resulted in significant
suppression of OPN-induced PC-3 cell motility towards the wound and indicated that COX-2
and PGE2 act as a key molecule in OPN-induced tumor cell migration (Fig. 13 A).
To determine the effect of OPN on invasiveness of PC-3 cells and to examine the role of
COX-2 and PGE2 in this process the ECM-invasion assays was performed. The results showed
Figure 13, COX-2 and PGE2 regulate OPN-induced PC-3 cell migration and invasion. A, Cells were transfected with dn c-Src, or pretreated with staurosporine, celecoxib or anti-EP2 receptor blocking antibody, then treated with OPN for 12 h. Wound photographs were taken at 0 h and 12 h. The experiment was performed in triplicate. B, cells were individually transfected with wt NIK, IKKα and IKKβ and treated with celecoxib. In separate experiments, cells were transfected with dn c-Src or pretreated with staurosporine, celecoxib (50μmol/L), NS398 (10μmol/L) and EP2 receptor blocking antibody (4mg/mL) and used for invasion assay. OPN was used in upper chamber. PGE2 was used as positive control. After 12 h, invaded cells were counted from the lower surface of the upper chamber and represented graphically. The results were expressed as means ± S.E. of three determinations.
56
that the OPN-induced enhanced tumor cells invasion due to overexpression of wt NIK, IKKα or
IKKβ was suppressed by the celecoxib suggesting the crucial role of COX-2 and PGE2 in OPN-
induced NIK-IKK dependent tumor cell invasion (Fig. 13 B). To further elucidate the specificity
of COX-2 in OPN-induced cell invasion, NS398 (NSAID), a COX-2 specific inhibitor was also
used and the data showed that NS398 inhibited OPN-induced tumor cell invasion. PGE2 was
used as a positive control. Taken together, these results showed the crucial role of COX-2 and
PGE2 in OPN-induced tumor cell motility and invasiveness.
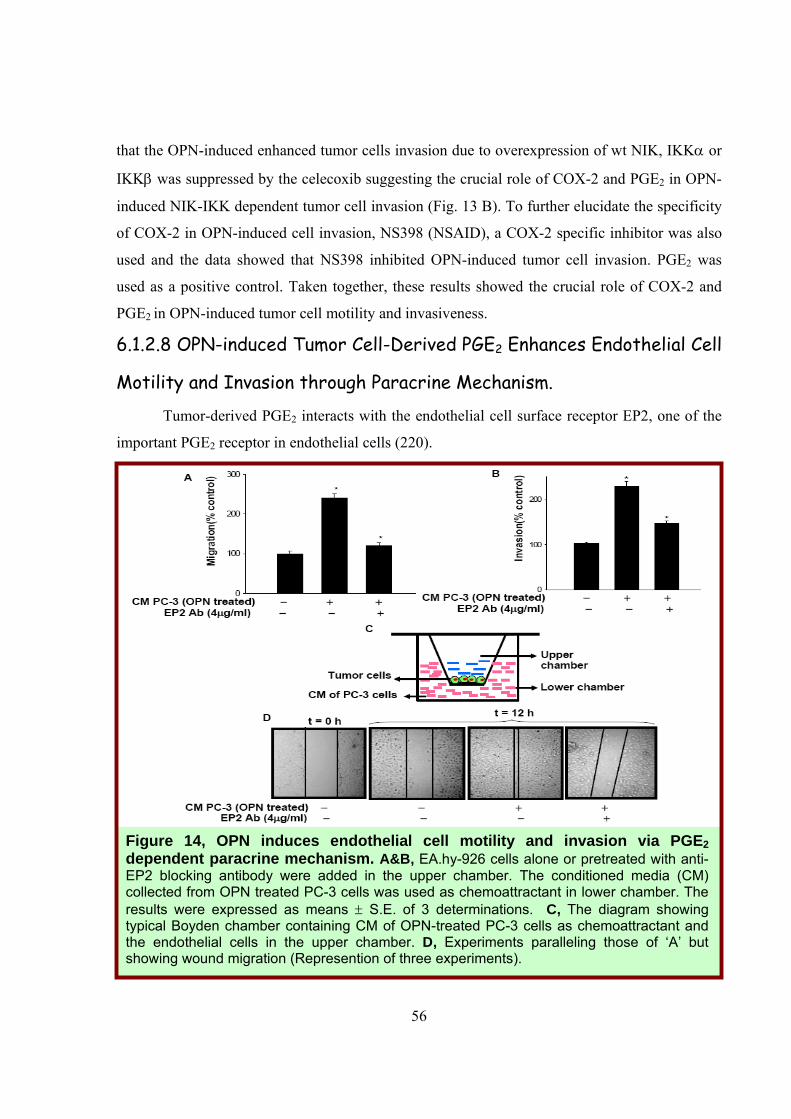
6.1.2.8 OPN-induced Tumor Cell-Derived PGE2 Enhances Endothelial Cell
Motility and Invasion through Paracrine Mechanism.
Tumor-derived PGE2 interacts with the endothelial cell surface receptor EP2, one of the
important PGE2 receptor in endothelial cells (220).
Figure 14, OPN induces endothelial cell motility and invasion via PGE2 dependent paracrine mechanism. A&B, EA.hy-926 cells alone or pretreated with anti-EP2 blocking antibody were added in the upper chamber. The conditioned media (CM) collected from OPN treated PC-3 cells was used as chemoattractant in lower chamber. The results were expressed as means ± S.E. of 3 determinations. C, The diagram showing typical Boyden chamber containing CM of OPN-treated PC-3 cells as chemoattractant and the endothelial cells in the upper chamber. D, Experiments paralleling those of ‘A’ but showing wound migration (Represention of three experiments).
57
Tumor-derived PGE2 interacts with the endothelial cell surface receptor EP2, one of the
important PGE2 receptor in endothelial cells (220). Tumor-derived PGE2 interacts with the
endothelial cell surface receptor EP2, one of the important PGE2 receptor in endothelial cells
(220). To determine whether OPN-induced PC-3 cell derived PGE2 induces endothelial cell
migration and invasion through EP2 receptor-mediated paracrine manner, endothelial cells were
used on the upper side of modified Boyden or Matrigel coated invasion chamber. The
conditioned media collected from OPN-treated PC-3 cells were used as chemoattractant in the
lower chamber (Fig.14 C). In separate experiments, endothelial cells were pretreated with anti-
EP2 receptor blocking antibody and used for migration and invasion assays.
Our data indicated that conditioned media of OPN-treated PC-3 cells significantly enhanced
endothelial cell migration and invasion whereas blocking the EP2 receptor in these cells with its
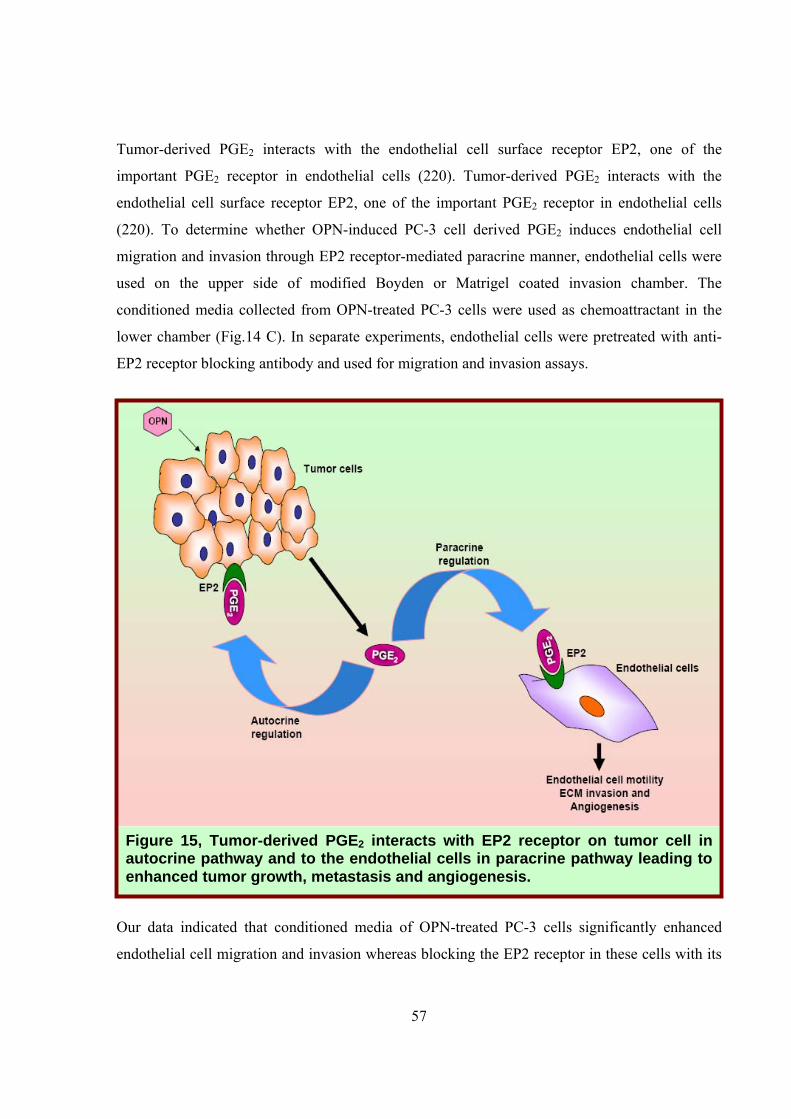
Figure 15, Tumor-derived PGE2 interacts with EP2 receptor on tumor cell in autocrine pathway and to the endothelial cells in paracrine pathway leading to enhanced tumor growth, metastasis and angiogenesis.
58
antibody significantly suppressed the endothelial cell migration and invasion (Fig. 14 A&B). The
OPN-induced migration of endothelial cells was further confirmed by wound assay under the
same experimental conditions (Fig. 14 D). Our results showed that OPN-induced tumor cell-
derived PGE2 enhanced migration and invasion of endothelial cells through EP2 receptor-
mediated paracrine mechanism (Fig. 15).
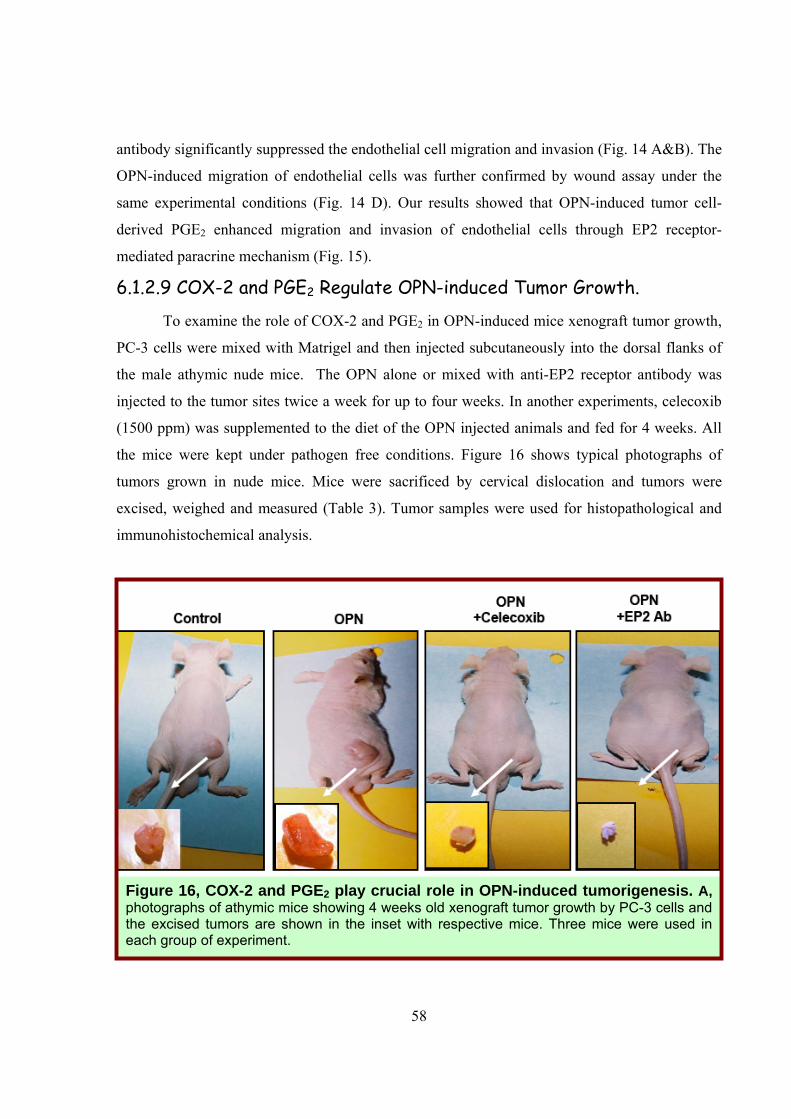
6.1.2.9 COX-2 and PGE2 Regulate OPN-induced Tumor Growth.
To examine the role of COX-2 and PGE2 in OPN-induced mice xenograft tumor growth,
PC-3 cells were mixed with Matrigel and then injected subcutaneously into the dorsal flanks of
the male athymic nude mice. The OPN alone or mixed with anti-EP2 receptor antibody was
injected to the tumor sites twice a week for up to four weeks. In another experiments, celecoxib
(1500 ppm) was supplemented to the diet of the OPN injected animals and fed for 4 weeks. All
the mice were kept under pathogen free conditions. Figure 16 shows typical photographs of
tumors grown in nude mice. Mice were sacrificed by cervical dislocation and tumors were
excised, weighed and measured (Table 3). Tumor samples were used for histopathological and
immunohistochemical analysis.
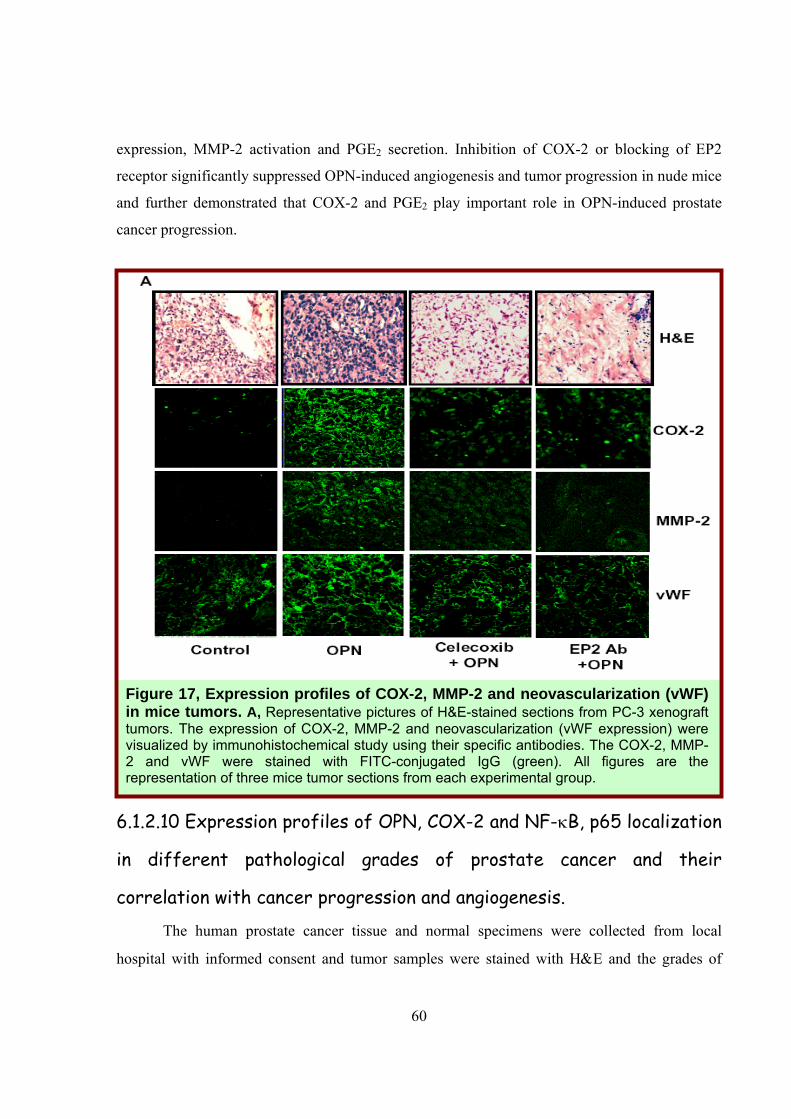
Figure 16, COX-2 and PGE2 play crucial role in OPN-induced tumorigenesis. A, photographs of athymic mice showing 4 weeks old xenograft tumor growth by PC-3 cells and the excised tumors are shown in the inset with respective mice. Three mice were used in each group of experiment.
59
The histopathological analysis by H&E staining of the tumors is summarized in Table 4
and these data clearly indicated that OPN-induced tumor growth was suppressed by celecoxib
supplementation in diet of the mice or using EP2 receptor blocking antibody. The
immunohistochemical studies showed the enhanced expressions of COX-2 and MMP-2 in mice
tumors injected with OPN (Fig. 17 A). The high expression of vWF (an endothelial cell specific
marker) was also detected (Fig. 17 A).
Taken together our in vivo xenograft study demonstrated that OPN induces prostate tumor
growth and angiogenesis in nude mice, which further correlated with up-regulation of COX-2
Table 4, Characteristics of xenograft tumors generated in mice.
Table 3, Effect of COX-2 inhibition and EP2 blocking on OPN-induced prostate tumor growth in nude mice.
60
expression, MMP-2 activation and PGE2 secretion. Inhibition of COX-2 or blocking of EP2
receptor significantly suppressed OPN-induced angiogenesis and tumor progression in nude mice
and further demonstrated that COX-2 and PGE2 play important role in OPN-induced prostate
cancer progression.
6.1.2.10 Expression profiles of OPN, COX-2 and NF-κB, p65 localization
in different pathological grades of prostate cancer and their
correlation with cancer progression and angiogenesis.
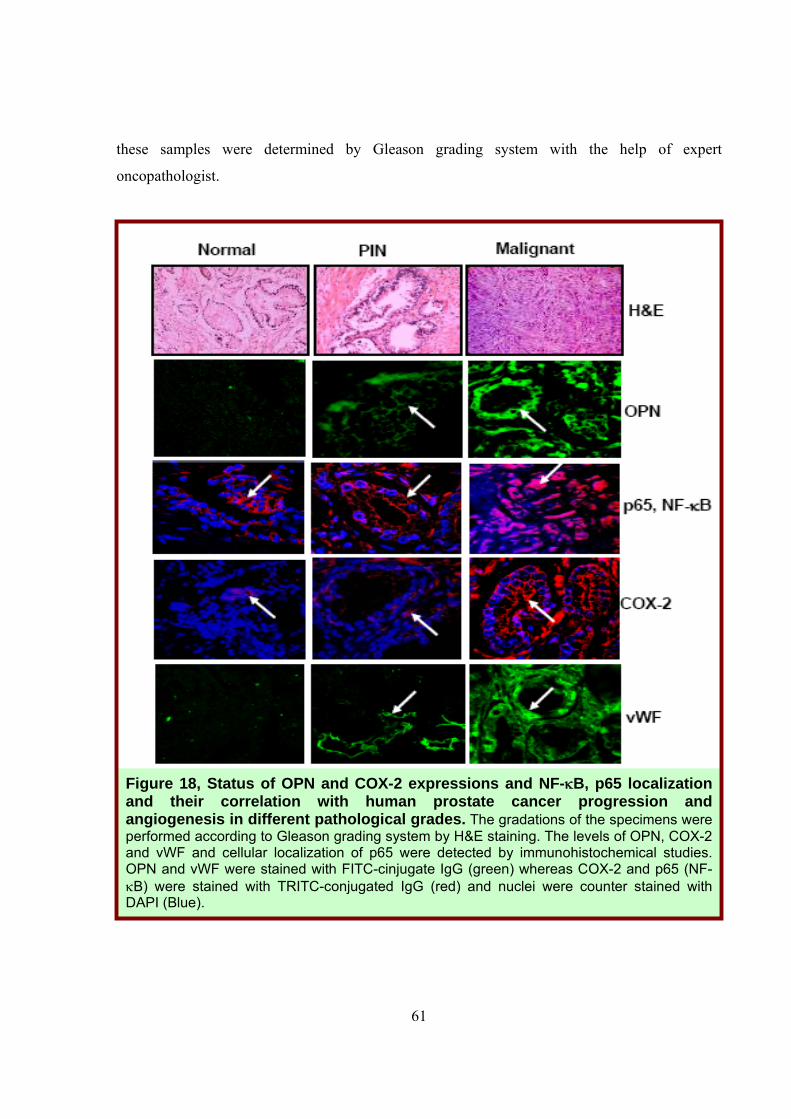
The human prostate cancer tissue and normal specimens were collected from local
hospital with informed consent and tumor samples were stained with H&E and the grades of
Figure 17, Expression profiles of COX-2, MMP-2 and neovascularization (vWF) in mice tumors. A, Representative pictures of H&E-stained sections from PC-3 xenograft tumors. The expression of COX-2, MMP-2 and neovascularization (vWF expression) were visualized by immunohistochemical study using their specific antibodies. The COX-2, MMP-2 and vWF were stained with FITC-conjugated IgG (green). All figures are the representation of three mice tumor sections from each experimental group.
61
these samples were determined by Gleason grading system with the help of expert
oncopathologist.
Figure 18, Status of OPN and COX-2 expressions and NF-κB, p65 localization and their correlation with human prostate cancer progression and angiogenesis in different pathological grades. The gradations of the specimens were performed according to Gleason grading system by H&E staining. The levels of OPN, COX-2 and vWF and cellular localization of p65 were detected by immunohistochemical studies. OPN and vWF were stained with FITC-cinjugate IgG (green) whereas COX-2 and p65 (NF-κB) were stained with TRITC-conjugated IgG (red) and nuclei were counter stained with DAPI (Blue).

62
Expression profiles of OPN, COX-2, vWF and cellular localization of p65 were analyzed
by immunohistochemistry using their specific antibodies (Fig. 18). The higher levels of OPN and
COX-2 were detected in malignant specimens as compared to normal and PIN (prostatic
intraepithelial neoplasia) specimens and that further correlated with the enhanced
neovascularization (vWF expression). Significant nuclear translocation of p65 in the malignant
tumors was also detected. 6.1.3 Discussion
Earlier findings have revealed that the interaction between OPN and prostate cancer cells
may regulate tumor progression and metastatic phenotype of human prostate carcinoma and
further indicated that OPN may be an important factor for prostate cancer progression (76-81).
In this study for the first time, we have delineated the molecular mechanism that underlies OPN-
induced COX-2 expression in prostate cancer cells and showed its potential role in regulating in
vitro tumor cell motility and invasion, in vivo tumor growth and angiogenesis.
Moreover, we find that COX-2 plays an important role in OPN-induced PGE2 production
and MMP-2 activation that ultimately regulates tumor progression. Recent studies have revealed
that activation of PKCα is required for the survival and growth of androgen-independent human
prostate cancer cells (215, 216). However, the role of PKCα in OPN-induced downstream
signaling events in prostrate cancer was not clear. In this study, for the first time we have
provided evidences, which indicate that PKCα plays an important role in OPN-induced NF-κB
phosphorylation, which eventually controls COX-2 expression in prostate cancer cells. We
demonstrated that OPN regulates PKCα-c-Src-mediated IKK activation, which results in NF-κB
phosphorylation. Moreover, the results showed that IκBα super repressor significantly
suppressed OPN-induced COX-2 expression suggesting that NF-κB plays important role in
regulation of COX-2 expression and further demonstrated the crucial role of PKCα-c-Src-IKK-
NF-κB signaling cascade in OPN-induced COX-2 expression.
Earlier reports have demonstrated that increased levels of PGE2 production and MMP-2
activation are associated with enhanced expression of COX-2 in many cancers (221-222) and the
COX-2 inhibitors have been reported to suppress PGE2 production and MMP-2 activation in
various types of cancer (121, 223, 224). PGE2 has also been shown to induce MMP-2 activation
in cancer cells (225). Our data showed that inhibition of upstream signaling molecules or COX-2
63
inhibitor (celecoxib) significantly suppressed OPN-induced PGE2 production and MMP-2
activation. Interestingly, our studies showed that NSAIDs like celecoxib and NS398 significantly
suppressed OPN-induced PC-3 cells migration and invasion.
Moreover, our in vivo data demonstrated that mice fed with celecoxib showed significant
reduction in OPN-induced tumor growth suggesting that COX-2 could be an effective
therapeutic target in prostate cancer treatment. Previous reports have shown that tumor cell
derived PGE2 contributes to tumor progression by regulating the cell motility/invasiveness and
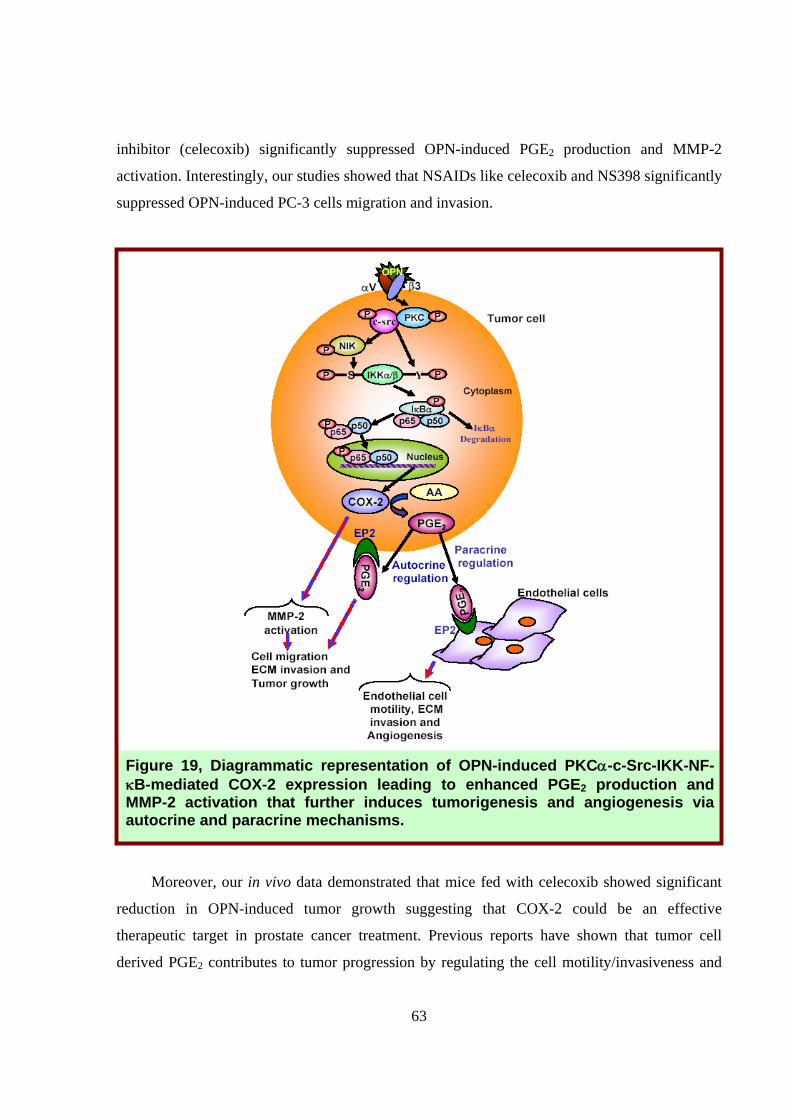
Figure 19, Diagrammatic representation of OPN-induced PKCα-c-Src-IKK-NF-κB-mediated COX-2 expression leading to enhanced PGE2 production and MMP-2 activation that further induces tumorigenesis and angiogenesis via autocrine and paracrine mechanisms.

64
inducing angiogenesis (8-10). Among PGE2 receptors, high expression of EP2 receptor has been
detected in both prostate cancer and endothelial cells (211-213). Moreover, the development of
selective antagonists against the EP2 receptor may have the potential to improve anti-tumor
activity and EP2-receptor antagonists may be more specific than COX-2 inhibitors (226).
In our study we demonstrated that the blocking the EP2 receptor by specific blocking
antibody suppressed OPN-induced prostate cancer (PC-3) and endothelial cell motility and
invasiveness. Furthermore, we also found that anti-EP2 blocking antibody significantly suppressed
OPN-induced mice xenograft tumor progression.
This study showed the in-depth molecular mechanism, at least in part, by which OPN
induces prostate tumor cell migration, invasion and tumor growth and angiogenesis through
induction of COX-2 expression and PGE2 production (Fig. 19). Our results further warrant that
the mechanism demonstrated in the mouse model underlies the human pathology and a clear
understanding of OPN and COX-2 regulation could illuminate cellular changes that accompany
prostate cancer progression and may facilitate the development of novel therapeutic approaches
to suppress OPN-regulated PKCα-IKK-NF-κB-mediated COX-2 expression thereby controlling
tumor progression and angiogenesis.


















