Influence of collision energy on the dynamics of the reaction O(1D) + CH4(X1A1) → OH(X...
7
Influence of collision energy on the dynamics of the reaction O( 1 D) + CH 4 (X 1 A 1 ) ! OH(X 2 P) + CH 3 (X 2 A 00 2 ) R. Sayo´s,* a Jordi Hernando, a Marı´a P. Puyuelo, b Pedro A. Enrı´quez b and Miguel Gonza´lez* a a Departament de Quı´mica Fı´sica i Centre de Recerca en Quı´mica Teo `rica, Universitat de Barcelona, C=Martı´ i Franque `s 1, E-08028 Barcelona, Spain. E-mail: r.sayos@qf.ub.es, miguel@qf.ub.es b Departamento de Quı´mica, Universidad de La Rioja, C=Madre de Dios 51, E-26004 Logron ˜o, Spain Recei v v ed 28th Aug g ust 2001, Accepted 1st Nov v ember 2001 First published as an Adv v ance Article on the web 7th January 2002 We studied the effects of collision energy (E T ) on the dynamics of the title reaction using the quasiclassical trajectory method on an analytical triatomic potential energy surface that we had derived for this system. We compared the dependence of the scalar and two-vector properties of the reaction on E T with experimental data and obtained a quite good agreement. The results can be explained in terms of the coexistence of two microscopic reaction mechanisms: insertion and abstraction. The former mechanism is the most important one, although the contribution of the latter increases with E T . 1. Introduction The reaction of methane with atomic oxygen in the first excited electronic state, 1 Oð 1 DÞþ CH 4 ðX 1 A 1 Þ! OHðX 2 PÞþ CH 3 ðX 2 A 00 2 Þ DH 298 K ¼ 43:1 kcal mol 1 ð1Þ is an important source of stratospheric OH, which affects the chemistry of the Earth’s ozone layer through the HO x cycles. 2–4 The OH + CH 3 reaction channel is the most important one for the O( 1 D) + CH 4 reaction (the quantum yield is equal to 0.75 ± 0.15 at 300 K, ref. 5), whose overall rate constant at room temperature approaches the gas kinetic limit: k ¼ 1.5 10 10 cm 3 molecule 1 s 1 . 5 The dynamics of reaction (1) has been studied experimen- tally. Laser-induced fluorescence (LIF) and chemiluminescence measurements of the internal state distributions of nascent OH have been carried out through the generation of O( 1 D) atoms by photodissociation of either O 3 at 248=266 nm 6–10 [the mean collision energy of the system (hE T i) being around 0.2 eV] or N 2 O at 193 nm 11–13 (hE T i¼ 0.40 eV). Polarized Doppler- resolved LIF spectroscopy 14–18 was used to study the OH product state-resolved kinetic energy and angular distributions at hE T i¼ 0.40 eV and, very recently, global product kinetic energy and angular distributions have been determined at E T ¼ 0.28 eV using molecular beams. 19 The energy distribu- tion of the CH 3 molecules produced in reaction (1) was also measured. 20,21 Reaction (1) has also been studied theoretically. Ab initio calculations at several levels were reported for the lowest potential energy surface (1 1 A PES) of the O( 1 D) + CH 4 sys- tem. 22,23 Using ab initio data and treating the CH 3 group as a pseudoatom, we fitted two versions of a triatomic analytical surface to represent the 1 1 A PES (PES1 23 and PES2 24 ). In addition, quasiclassical trajectory (QCT) calculations were performed on these PESs to examine internal state distribu- tions of nascent OH and the stereodynamics in the reaction conditions explored experimentally (hE T i¼ 0.21 and 0.40 eV). 13,23,24 In this work we perform a thorough dynamic study of reaction (1) using the QCT method and the analytical tri- atomic surface PES2 previously derived by us for the O( 1 D) + CH 4 system. 24 We examine the dependence on the collision energy of the scalar and two-vector properties of reaction (1). This allows us to carry out a detailed analysis of the dynamic behavior for reaction conditions not attained in the experiments, to explore dynamical properties other than those studied in the experimental conditions, and also to compare with the most recent experimental results reported for the O( 1 D) + CH 4 reaction. 19 Finally, the change of the dyna- mical behavior on varying E T has been analyzed in terms of the microscopic reaction mechanism. 2. Computational method The QCT method 25–27 was applied as implemented in the TRIQCT program 28 to study reaction (1), using the analytical triatomic potential energy surface PES2. 24 This analytical sur- face was fitted using ab initio data about the 1 1 A PES through which reaction (1) evolves, ensuring that the assumption of taking the CH 3 group as a pseudoatom of 15 amu (located at its center of mass) did not entail the appearance of a spurious reaction barrier. 24 This PES connects the O( 1 D) + CH 4 reac- tants with the OH + CH 3 products along a minimum energy path (MEP) that does not present an energy barrier above reactants. Also, it allows for either an abstraction microscopic mechanism or an insertion=elimination microscopic mechan- ism that evolves through the CH 3 OH minimum. 24 In a previous study, 24 PES2 quite satisfactorily reproduced the dynamic behavior experimentally established for reaction (1) (final energy of the OH product and other properties such as rate constants, branching ratios, and also translational energy distributions and kk 0 differential cross-sections for OH state-specific products). This was possible because PES2 efficiently describes the insertion=fast elimination mechanism, which is believed to be the most important one for this reaction. 9,12,13,19 Hence, the dynamic properties of the OH product arising from reaction (1) may be reproduced quite 288 Phys. Chem. Chem. Phys., 2002, 4, 288–294 DOI: 10.1039/b107719h This journal is # The Owner Societies 2002 Published on 07 January 2002. Downloaded by UNIVERSITY OF ALABAMA AT BIRMINGHAM on 27/10/2014 11:45:30. View Article Online / Journal Homepage / Table of Contents for this issue
Transcript of Influence of collision energy on the dynamics of the reaction O(1D) + CH4(X1A1) → OH(X...

Influence of collision energy on the dynamics of the reaction
O(1D)+CH4(X1A1)!OH(X 2P)+CH3(X
2A002)
R. Sayos,*a Jordi Hernando,a Marıa P. Puyuelo,b Pedro A. Enrıquezb and Miguel Gonzalez*a
a Departament de Quımica Fısica i Centre de Recerca en Quımica Teorica,Universitat de Barcelona, C=Martı i Franques 1, E-08028 Barcelona, Spain.E-mail: [email protected], [email protected]
b Departamento de Quımica, Universidad de La Rioja, C=Madre de Dios 51,E-26004 Logrono, Spain
Receivved 28th Auggust 2001, Accepted 1st Novvember 2001First published as an Advvance Article on the web 7th January 2002
We studied the effects of collision energy (ET) on the dynamics of the title reaction using the quasiclassicaltrajectory method on an analytical triatomic potential energy surface that we had derived for this system. We
compared the dependence of the scalar and two-vector properties of the reaction on ET with experimental dataand obtained a quite good agreement. The results can be explained in terms of the coexistence of twomicroscopic reaction mechanisms: insertion and abstraction. The former mechanism is the most important one,
although the contribution of the latter increases with ET .
1. Introduction
The reaction of methane with atomic oxygen in the first excitedelectronic state,1
Oð1DÞ þ CH4ðX 1A1Þ ! OHðX 2PÞ þ CH3ðX 2A002Þ
DH�298 K ¼ � 43:1 kcal mol�1 ð1Þ
is an important source of stratospheric OH, which affectsthe chemistry of the Earth’s ozone layer through the HOx
cycles.2–4 The OH+CH3 reaction channel is the mostimportant one for the O(1D)+CH4 reaction (the quantumyield is equal to 0.75±0.15 at 300 K, ref. 5), whose overallrate constant at room temperature approaches the gas kineticlimit: k ¼ 1.5 10�10 cm3 molecule�1 s�1.5
The dynamics of reaction (1) has been studied experimen-tally. Laser-induced fluorescence (LIF) and chemiluminescencemeasurements of the internal state distributions of nascent OHhave been carried out through the generation of O(1D) atomsby photodissociation of either O3 at 248=266 nm6–10 [the meancollision energy of the system (hETi) being around 0.2 eV] orN2O at 193 nm11–13 (hETi ¼ 0.40 eV). Polarized Doppler-resolved LIF spectroscopy14–18 was used to study the OHproduct state-resolved kinetic energy and angular distributionsat hETi ¼ 0.40 eV and, very recently, global product kineticenergy and angular distributions have been determined atET ¼ 0.28 eV using molecular beams.19 The energy distribu-tion of the CH3 molecules produced in reaction (1) was alsomeasured.20,21
Reaction (1) has also been studied theoretically. Ab initio
calculations at several levels were reported for the lowestpotential energy surface (1 1A PES) of the O(1D)+CH4 sys-tem.22,23 Using ab initio data and treating the CH3 group as apseudoatom, we fitted two versions of a triatomic analyticalsurface to represent the 1 1A PES (PES123 and PES224). Inaddition, quasiclassical trajectory (QCT) calculations wereperformed on these PESs to examine internal state distribu-tions of nascent OH and the stereodynamics in the reactionconditions explored experimentally (hETi ¼ 0.21 and 0.40eV).13,23,24
In this work we perform a thorough dynamic study ofreaction (1) using the QCT method and the analytical tri-atomic surface PES2 previously derived by us for theO(1D)+CH4 system.24 We examine the dependence on thecollision energy of the scalar and two-vector properties ofreaction (1). This allows us to carry out a detailed analysis ofthe dynamic behavior for reaction conditions not attained inthe experiments, to explore dynamical properties other thanthose studied in the experimental conditions, and also tocompare with the most recent experimental results reported forthe O(1D)+CH4 reaction.
19 Finally, the change of the dyna-mical behavior on varying ET has been analyzed in terms of themicroscopic reaction mechanism.
2. Computational method
The QCT method25–27 was applied as implemented in theTRIQCT program28 to study reaction (1), using the analyticaltriatomic potential energy surface PES2.24 This analytical sur-face was fitted using ab initio data about the 1 1A PES throughwhich reaction (1) evolves, ensuring that the assumption oftaking the CH3 group as a pseudoatom of 15 amu (located at itscenter of mass) did not entail the appearance of a spuriousreaction barrier.24 This PES connects the O(1D)+CH4 reac-tants with the OH+CH3 products along a minimum energypath (MEP) that does not present an energy barrier abovereactants. Also, it allows for either an abstraction microscopicmechanism or an insertion=elimination microscopic mechan-ism that evolves through the CH3OH minimum.24
In a previous study,24 PES2 quite satisfactorily reproducedthe dynamic behavior experimentally established for reaction(1) (final energy of the OH product and other properties suchas rate constants, branching ratios, and also translationalenergy distributions and kk0 differential cross-sections forOH state-specific products). This was possible because PES2efficiently describes the insertion=fast elimination mechanism,which is believed to be the most important one for thisreaction.9,12,13,19 Hence, the dynamic properties of the OHproduct arising from reaction (1) may be reproduced quite
288 Phys. Chem. Chem. Phys., 2002, 4, 288–294 DOI: 10.1039/b107719h
This journal is # The Owner Societies 2002
Publ
ishe
d on
07
Janu
ary
2002
. Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F A
LA
BA
MA
AT
BIR
MIN
GH
AM
on
27/1
0/20
14 1
1:45
:30.
View Article Online / Journal Homepage / Table of Contents for this issue
satisfactorily with the triatomic model considered. This kind ofmodel has also been quite successfully applied in our group tothe study of the O(1D)+N(NO),29 O(3P)+H(CH3)
30,31 andO(1D)+H(OH)32 reactions.The accuracy of the numerical integration of Hamilton’s
differential equations was verified by checking the conserva-tion of total energy and total angular momentum in everytrajectory, and performing back-integrations on some batchesof trajectories. An integration step of 2.5 10�17 s and aninitial distance of 10
+A between the O(1D) and the H–(CH3)
center of mass were selected. This separation ensures that theinteraction energy is negligible compared to the availableenergy of reactants. The rovibrational levels of the OH pro-duct were assigned using the vibrational action variablemethod. As the QCT method does not account for theOH(X 2P) orbital (L ¼ 1) and spin (S ¼ 1=2) electronicangular momenta, we identified the total angular momentumquantum number excluding electronic and nuclear spins, N0,of the OH molecule with the value of the rotational angularmomentum derived from the QCT calculations, j0, plus one.Several collision energies were considered in the QCT study
of reaction (1), ET ¼ 0.28, 0.60, 0.80, 1.0 and 1.4 eV, thuscompleting our analysis at ET ¼ 0.21 and 0.40 eV.24 Theresults obtained at the new values of ET in this study and thosefrom the previous study24 will be shown together. Around200 000–300 000 trajectories were run for each new condition,so the statistical errors estimated for all the calculated dynamicproperties were very small. For each collision energy therovibrational energy levels of the H–(CH3) pseudodiatomicmolecule were sampled according to a 298 K Maxwell–Boltzmann distribution, and the masses of the most abundantisotopes of the O, H and C atoms (16O, 1H and 12C) wereconsidered in the calculations. However, for comparisonwith the experimental results reported in ref. 19, additionalcalculations were carried out using the 18O isotope at theET ¼ 0.28 eV condition. Up to 1000 000 trajectories were runin this case. Very small differences were found when either the16O mass or the 18O mass was employed.
3. Reaction dynamics
3.1. Scalar properties
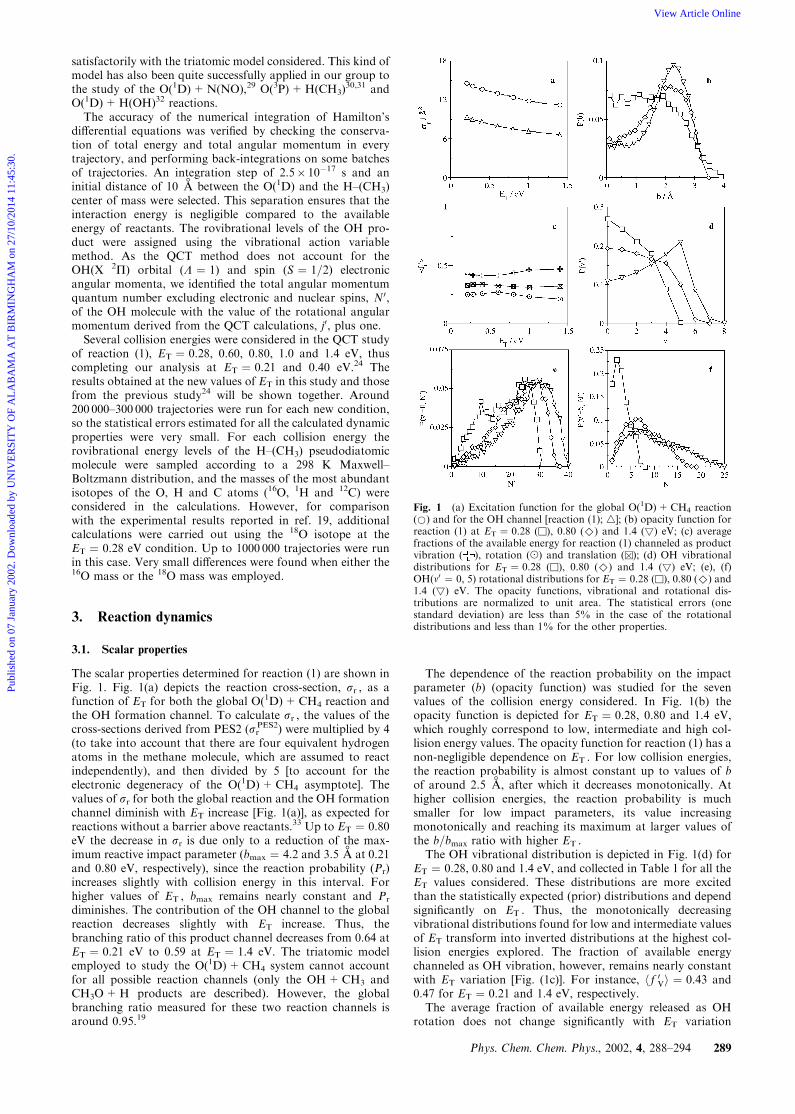
The scalar properties determined for reaction (1) are shown inFig. 1. Fig. 1(a) depicts the reaction cross-section, sr , as afunction of ET for both the global O(1D)+CH4 reaction andthe OH formation channel. To calculate sr , the values of thecross-sections derived from PES2 (sr
PES2) were multiplied by 4(to take into account that there are four equivalent hydrogenatoms in the methane molecule, which are assumed to reactindependently), and then divided by 5 [to account for theelectronic degeneracy of the O(1D)+CH4 asymptote]. Thevalues of sr for both the global reaction and the OH formationchannel diminish with ET increase [Fig. 1(a)], as expected forreactions without a barrier above reactants.33 Up to ET ¼ 0.80eV the decrease in sr is due only to a reduction of the max-imum reactive impact parameter (bmax ¼ 4.2 and 3.5
+A at 0.21
and 0.80 eV, respectively), since the reaction probability (Pr)increases slightly with collision energy in this interval. Forhigher values of ET , bmax remains nearly constant and Pr
diminishes. The contribution of the OH channel to the globalreaction decreases slightly with ET increase. Thus, thebranching ratio of this product channel decreases from 0.64 atET ¼ 0.21 eV to 0.59 at ET ¼ 1.4 eV. The triatomic modelemployed to study the O(1D)+CH4 system cannot accountfor all possible reaction channels (only the OH+CH3 andCH3O+H products are described). However, the globalbranching ratio measured for these two reaction channels isaround 0.95.19
The dependence of the reaction probability on the impactparameter (b) (opacity function) was studied for the sevenvalues of the collision energy considered. In Fig. 1(b) theopacity function is depicted for ET ¼ 0.28, 0.80 and 1.4 eV,which roughly correspond to low, intermediate and high col-lision energy values. The opacity function for reaction (1) has anon-negligible dependence on ET . For low collision energies,the reaction probability is almost constant up to values of bof around 2.5
+A, after which it decreases monotonically. At
higher collision energies, the reaction probability is muchsmaller for low impact parameters, its value increasingmonotonically and reaching its maximum at larger values ofthe b=bmax ratio with higher ET .The OH vibrational distribution is depicted in Fig. 1(d) for
ET ¼ 0.28, 0.80 and 1.4 eV, and collected in Table 1 for all theET values considered. These distributions are more excitedthan the statistically expected (prior) distributions and dependsignificantly on ET . Thus, the monotonically decreasingvibrational distributions found for low and intermediate valuesof ET transform into inverted distributions at the highest col-lision energies explored. The fraction of available energychanneled as OH vibration, however, remains nearly constantwith ET variation [Fig. (1c)]. For instance, h f 0Vi ¼ 0.43 and0.47 for ET ¼ 0.21 and 1.4 eV, respectively.The average fraction of available energy released as OH
rotation does not change significantly with ET variation
Fig. 1 (a) Excitation function for the global O(1D)+CH4 reaction(*) and for the OH channel [reaction (1); 4]; (b) opacity function forreaction (1) at ET ¼ 0.28 (u), 0.80 (^) and 1.4 (
4
) eV; (c) averagefractions of the available energy for reaction (1) channeled as productvibration ( ), rotation (() and translation (u); (d) OH vibrationaldistributions for ET ¼ 0.28 (u), 0.80 (^) and 1.4 (
4
) eV; (e), (f)OH(n0 ¼ 0, 5) rotational distributions for ET ¼ 0.28 (u), 0.80 (^) and1.4 (
4
) eV. The opacity functions, vibrational and rotational dis-tributions are normalized to unit area. The statistical errors (onestandard deviation) are less than 5% in the case of the rotationaldistributions and less than 1% for the other properties.
Phys. Chem. Chem. Phys., 2002, 4, 288–294 289
Publ
ishe
d on
07
Janu
ary
2002
. Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F A
LA
BA
MA
AT
BIR
MIN
GH
AM
on
27/1
0/20
14 1
1:45
:30.
View Article Online

[Fig. 1(c)], as occurs in the case of OH vibration. The averagevalues are h fR0i ¼ 0.23 and 0.21 for ET ¼ 0.21 and 1.4 eV,respectively. The average rotational levels for each populatedvibrational level are given in Table 1 for all ET values con-sidered, while Figs. 1(e) and (f ) depict the rotational dis-tributions for low-n0 (n0 ¼ 0) and high-n0 (n0 ¼ 5) levels atET ¼ 0.28, 0.80 and 1.4 eV. For the lowest populated vibra-tional levels (n0 ¼ 0–4) the distributions are more excited thanthe statistical ones (prior distributions) and their populationmaxima appear at high N0 values. The values of the mostpopulated rotational level and of the highest rotational levelreached diminish with the vibrational quantum number, asexpected. On the other hand, though the OH(n0 ¼ 0–4) rota-tional distributions are slightly more excited as ET rises, theydo not vary appreciably with collision energy. This can be
attributed to the large exothermicity of the reaction and thedepth of the minimum through which the reactivity mainlytakes place. The OH rotational distributions for the highestvibrational levels (n0 5 5) are less excited than the ones forn0 4 4, and their population maxima appear at low N0 values.Moreover, they show a somewhat more pronounced depen-dence on ET , the distributions becoming clearly more excitedand displaying a clear bimodal feature as ET increases.The average fraction of available energy channeled as pro-
duct relative translation is also nearly constant with ET
[(Fig. (1c)]. The average value of the product relative transla-tional energy (hET
0i), however, clearly increases as collisionenergy rises, as expected due to the increasing amount ofavailable energy (hET
0i ¼ 0.69 and 1.0 eV for ET ¼ 0.21 and1.4 eV, respectively). Fig. 2(a) depicts the experimental19 and
Table 1 Vibrational populations and average rotational levels of the OH product arising from the O(1D)+CH4!OH+CH3 reactiona
P(n0) and hN0in0
ET=eV n0 ¼ 0 n0 ¼ 1 n0 ¼ 2 n0 ¼ 3 n0 ¼ 4 n0 ¼ 5 n0 ¼ 6 n0 ¼ 7
0.21 0.26 (18.3) 0.25 (16.6) 0.23 (13.8) 0.18 (11.1) 0.08 (7.2)0.28 0.27 (18.8) 0.25 (17.2) 0.21 (14.2) 0.18 (11.5) 0.09 (7.9) 0.003 (3.0)0.40 0.27 (19.9) 0.24 (18.2) 0.20 (15.5) 0.18 (12.7) 0.10 (9.3) 0.01 (5.3)0.60 0.24 (21.5) 0.22 (19.6) 0.20 (17.0) 0.17 (14.3) 0.12 (10.3) 0.05 (6.7) 5 10�4
0.80 0.19 (22.5) 0.19 (20.6) 0.18 (18.2) 0.17 (15.8) 0.16 (12.6) 0.10 (7.8) 0.01 (5.0)1.0 0.15 (23.8) 0.16 (21.4) 0.16 (19.5) 0.16 (17.1) 0.17 (13.7) 0.16 (8.4) 0.03 (6.2) 2 10�4
1.4 0.11 (26.0) 0.12 (23.7) 0.13 (21.5) 0.15 (19.5) 0.18 (15.8) 0.21 (10.1) 0.09 (8.0) 0.01 (7.4)
a For each condition and vibrational level, the first value given corresponds to the vibrational population, while the second one in parentheses is theaverage rotational level. For some of the highest OH populated vibrational levels there was not enough statistics to obtain reliable values of theaverage rotational levels. The average statistical errors (one standard deviation) corresponding to the vibrational populations and the averagerotational levels are less than 1 and 5%, respectively.
Fig. 2 (a), (b) Experimental ( , ref. 19) and QCT (u) product relative translational energy distributions (a) and kk0 angular distributions ex-pressed as solid angle differential cross-sections (b) of reaction (1) at ET ¼ 0.28 eV. All distributions are normalized to unit area. The statisticalerrors of the QCT results (one standard deviation) are less than 1%. (c) Velocity–scattering angle distributions for the 18OH product expressed as a3D map as well as a contour map.
290 Phys. Chem. Chem. Phys., 2002, 4, 288–294
Publ
ishe
d on
07
Janu
ary
2002
. Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F A
LA
BA
MA
AT
BIR
MIN
GH
AM
on
27/1
0/20
14 1
1:45
:30.
View Article Online

calculated kinetic energy distributions at ET ¼ 0.28 eV for the18OH+CH3 products. Although the two distributions havesimilar shape and maximum position, the QCT one is widerthan the experimentally reported one, indicating that moretranslational excitation is expected from the calculations(hET
0i ¼ 0.52 and 0.70 eV for the experimental and QCT data,respectively). This can be explained in terms of the triatomicmodel used, which neglects the internal degrees of freedom ofthe methyl fragment. As a result, the available energy cannotbe released as internal energy of the CH3 co-product, resultingin a higher fraction of energy channeled as translation.Nevertheless, the QCT and experimental ET
0 distributions aresimilar. This, along with the fact that the triatomic model givesa quite good description of the internal energy distributions ofthe OH product,24 suggests that little available energy shouldbe expected to be channeled as CH3 internal excitation, asreported experimentally.20,21 However, this is not fullyaccomplished if detailed OH state-specific channels of reaction(1) are considered. Thus, it was measured that for the reactionchannel leading to OH(n0 ¼ 0, N0 ¼ 5, 19),17,18 an importantamount of the available energy was released as product kineticenergy, which was not reproduced by the triatomic model.24
Otherwise, it was proved in the experiments that the produc-tion of vibrationally excited OH molecules [OH(n0 ¼ 4,N0 ¼ 8) channel] was accompanied by a minor translationalexcitation of products,17,18 the triatomic model leading in thiscase to a good agreement with experiment.
3.2. Two-vector properties
The analysis of the total angular momentum (J ) transforma-tion from reactants to products was examined at all the colli-sion energies considered. In all cases, J l! l 0, as expectedfor a heavy–light–heavy (H–L–H) reaction.The angular distributions arising from reaction (1) for the
two-vector correlations between initial (k) and final (k0) rela-tive velocity vectors (kk0), between k and the final rotationalangular momentum j 0 (kj 0), between k0 and j 0 (k0j 0) andbetween l 0 and j 0 (l 0j 0) were also examined. The results areshown as solid angle differential cross-sections (DCSs) in Fig. 3for representative collision energies (ET ¼ 0.28, 0.80 and 1.4
eV), which roughly correspond to low, intermediate and highET values.The kk0 DCSs [Fig. (3a)] show similar features at all collision
energies. They are quite isotropic in the range kk0 ¼ 30�–150�
and they present quite pronounced forward (kk0 ¼ 0�) and lesspronounced backward (kk0 ¼ 180�) peaks. Thus, the kk0 DCSsshow non-negligible forward behavior. This situation some-what resembles that expected for a microscopic mechanismevolving through a collision complex, which favors the scat-tering of products in all possible directions.33 In that case, thelonger-lived the collision complex, the more isotropic the kk0
distribution is expected to be. For the O(1D)+CH4 reactionthe dominant mechanism is an insertion=fast eliminationone9,12,13,19 and the collision complexes through which theprocess evolves are mainly short-lived, which explains thesomewhat forward feature of the calculated kk0 DCSs. Thisforward feature has also been found experimentally, as shownin Fig. 2(b), where the experimental19 and QCT kk0 angulardistributions for the 18OH product at ET ¼ 0.28 eV are com-pared. The agreement between theory and experiment isexcellent in this case. This is confirmed by the calculatedvelocity–scattering angle distribution for the 18OH product[Fig. (2c)]. The maps show the existence of backward and,especially, forward peaks. The experimental center of massproduct flux 3D map depicted in Fig. 7 of ref. 19 for ET ¼0.28 eV has very similar features.Despite the similarity between the kk0 DCSs at different ET
values, some discrepancies arise when increasing the collisionenergy. For low and intermediate values of ET (ET< 1.0 eV)the forward feature of the kk0 angular distributions remainsnearly constant as the collision energy rises. Otherwise, thescattering in the forward hemisphere is accentuated for highervalues of ET . As energy increases the system can evolve fromreactants to products through more repulsive regions of thePES, which would lead to progressively more backward scat-tering. However, in reaction (1) the impulsive effect dominatesand because of this a tendency to more forward scattering isobserved as ET increases. This trend is illustrated by the valuesof the average scattering angle (hykk0i) and the ratio ofOH products scattered in the forward (ykk0 < 90�) and back-ward (ykk0 ¼ 90�) hemispheres ( f/b scattering ratio). ForET ¼ 0.21, 0.80 and 1.4 eV, we obtained hykk0i ¼ 75.3�, 76.4�,68.9� and f=b ¼ 1.7, 1.6, 2.0, respectively.The l 0j 0 DCSs are depicted in Fig. 3(b). They are mainly
symmetric with a minimum at 90� and two maxima at 0� and180�, which means that the l 0 and j 0 vectors have some ten-dency towards parallel alignment. For low and intermediatevalues of ET the l 0j 0 DCSs have a slightly antiparallel orien-tation (yl0j0>90�), which is softened as collision energy increa-ses. For high ET values the l 0j 0 distributions display a preferredparallel orientation (yl0j0<90�). The values of the average l 0j 0
angle (hyl0j0i) and the ratio between parallel and antiparallelarrangements of the two vectors (p=ap scattering ratio) werehyl0j0i ¼ 99.0�, 92.5�, 81.3� and p=ap ¼ 0.70, 0.89, 1.4 forET ¼ 0.21, 0.80 and 1.4 eV. As a result of the angularmomenta transformation, the lj 0 correlation was similar tothat described for l 0j 0.The kj 0 and k0j 0 DCSs are collected in Figs. 3(c) and (d)
respectively, and correspond to symmetric distributions, asthey must be. They are also quite isotropic, though slightlypeaked at 90�, as expected due to the above-mentionedtendency of the system to align the j 0 vector parallel to l and l 0.No significant changes for the kj 0 and k0j 0 DCSs were observedas ET increased.The previously described slight trend of the k and j 0 vectors
to be perpendicular can also be inferred from the analysis of theOH rotational alignment parameter (A
ð2Þ0 � h3 cos2 ykj0 � 1i),
whose limiting values correspond to a fully perpendicular(A
ð2Þ0 ¼ � 1) or parallel (A
ð2Þ0 ¼ +2) kj 0 alignment. Here small
negative values of Að2Þ0 were found for all collision energies
Fig. 3 The (a) kk0, (b) l 0j 0, (c) kj 0 and (d) k0j 0 two-vector angulardistributions of reaction (1) expressed as solid angle differential cross-sections for ET ¼ 0.28 eV (u), 0.80 eV (^) and 1.4 eV (
4
). All dis-tributions are normalized to unit area. The statistical errors (onestandard deviation) are less than 1%.
Phys. Chem. Chem. Phys., 2002, 4, 288–294 291
Publ
ishe
d on
07
Janu
ary
2002
. Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F A
LA
BA
MA
AT
BIR
MIN
GH
AM
on
27/1
0/20
14 1
1:45
:30.
View Article Online

considered, thus suggesting a slight tendency for perpendicularkj 0 polarization. For instance, A
ð2Þ0 ¼ � 0.092, � 0.090 and
� 0.12 for ET ¼ 0.21, 0.80 and 1.4 eV, respectively. This weaktendency for perpendicular alignment does not change with theinternal state of the OH product, in contrast with theH+H2O!OH+H2 (ref. 34) and O(3P)+CH4!OH+CH3 (refs. 31 and 35) reactions (abstraction-like pro-cesses), which showed more negative values of A
ð2Þ0 as higher
OH rotational states were considered.We also analysed the OH state-specific two-vector properties
arising from reaction (1). We focused on the ET ¼ 0.40 eVcondition, for which some experimental results have beenreported about the state-specific stereodynamics of theO(1D)+CH4 reaction:14–18 the OH(n0 ¼ 0, N0 ¼ 5) andOH(n0 ¼ 4, N0 ¼ 8) kk0 DCSs. The QCT and experimentalstate-specific kk0 DCSs were previously compared usingPES2.24 Here we investigated other two-vector correlations forthese state-specific reaction channels. We found an l! l 0
transformation, as obtained for the full reaction. The calcu-lated l 0j 0 DCSs are essentially symmetric and exhibit pro-nounced peaks at 0� and 180�. The kj 0 and k0j 0 distributionsare also symmetric (as they must be), showing slight maxima at90�. The A
ð2Þ0 (kj 0) parameter takes small negative values for
the two state-specific reaction channels: � 0.11 [OH(n0 ¼ 0,N0 ¼ 5)] and � 0.075 [OH(n0 ¼ 4, N0 ¼ 8)]. A slight tendencyof the system to arrange the k0 and j 0 vectors perpendicularlyhas also been observed in the experiments for the OH(n0 ¼ 0,N0 ¼ 5) channel.16
4. Microscopic reaction mechanism
To improve our understanding of the dynamics of the titlereaction, the microscopic mechanism was studied. We analysedthe temporal dependence of the interatomic distances andangles, and of the potential energy of the system for thereactive trajectories obtained for a range of values of ET . Theevolution from reactants to products followed two differentreaction pathways: (a) insertion of the oxygen atom into aC–H bond to yield the methanol minimum followed by thedissociation of the resulting collision complex (insertion); and(b) direct abstraction of a hydrogen atom by the oxygen atom(abstraction).To separate the reactive trajectories corresponding to the
two reaction pathways, the minimal value of the potentialenergy reached by each reactive trajectory was analyzed.Abstraction trajectories evolved from reactants to productswith a minimum potential energy above � 7.0 eV (the potentialenergies at the equilibrium geometries of reactants, OH+CH3
products and CH3OH minimum are, respectively, � 4.79,� 6.47 and � 10.54 eV). On the other hand, the insertion tra-jectories take minimal potential energies below � 9.0 eV, andmost of them present values lower than � 10.0 eV.At this point, it might be objected that the energy criterion
chosen to discern between insertion- and abstraction-typereactive trajectories is somewhat misleading, as it implies thatthe abstraction trajectories penetrate the CH3OH well to acertain extent (ca. 0.5 eV). However, the following two findingsshould be considered to clarify this point: (a) a non-negligibleinfluence of the deep CH3OH minimum was found at the ab
initio level (and then reproduced by the analytical PES), evenfor the ‘‘pure ’’ abstraction pathway leading from reactants toproducts (i.e., for a fixed O–H–CH3 collinear arrangement).This was evidenced by the presence of a shallow ‘‘minimum’’(not a true minimum but a consequence of the CH3OH mini-mum) located at the exit channel of the collinear abstractionpathway. Therefore, the minimal potential energy value thatshould be expected for the abstraction reactive trajectories isthat corresponding not to the product asymptote (� 6.47 eV)but to the energy value of the ‘‘minimum’’ (� 6.70 eV for an
O–H–CH3 collinear geometry). (b) Although the energy thresh-old chosen to select the abstraction reactive trajectories was� 7.0 eV, the minimal energy value calculated for nearly all ofthese trajectories was higher than � 6.75 eV, as expected takinginto account the ‘‘minimum’’ in the abstraction pathway.The relative contributions to the global reactivity calculated
for the two microscopic mechanisms at the ET values con-sidered are shown in Table 2. The insertion mechanism is themain reaction pathway, although the contribution of theabstraction mechanism, which is almost negligible atET ¼ 0.21, 0.28 and 0.40 eV, increases with ET and reaches avalue of 0.31 at 1.4 eV [Fig. 4(a)]. This agrees with the
Table 2 Microscopic mechanisms yields for the O(1D)+CH4!OH+CH3 reaction
a
Mechanism
ET=eV Abstraction Insertion
0.21 0 10.28 9 10�4 10.40 0.01 0.990.60 0.04 0.960.80 0.11 0.891.00 0.21 0.791.40 0.31 0.69
a Normalized to unity.
Fig. 4 Global ( ), insertion (4) and abstraction (*) scalar proper-ties of reaction (1): (a) excitation functions; (b) opacity functions forET ¼ 1.0 eV; (c), (d) OH vibrational distributions for ET ¼ 0.60 and1.4 eV, respectively; (e), (f ) OH(n0 ¼ 3, 5) rotational distributions forET ¼ 1.4 eV, respectively. Global opacity functions, and vibrationaland rotational distributions are normalized to unit area. The statisticalerrors (one standard deviation) are less than 5% in the case of therotational distributions and less than 1% for the other properties.
292 Phys. Chem. Chem. Phys., 2002, 4, 288–294
Publ
ishe
d on
07
Janu
ary
2002
. Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F A
LA
BA
MA
AT
BIR
MIN
GH
AM
on
27/1
0/20
14 1
1:45
:30.
View Article Online

experimental evidence that suggests that the O(1D)+CH4
reaction mainly evolves through insertion.9,12,13,19
The dynamical properties of the insertion and abstractionmechanisms have been studied. This has allowed us to obtain adeep insight into the two mechanisms and to discern whetherthe abstraction should be considered as a rebound or a strip-ping mechanism.The dependence of the reaction probability on the impact
parameter is different for the insertion and abstraction reactionmechanisms. Thus, trajectories evolving through abstractionare favored for higher values of b than insertion trajectories.The opacity functions corresponding to the insertion andabstraction mechanisms for ET ¼ 1.0 eV are plotted inFig. 4(b). Moreover, the opacity functions associated with theinsertion mechanism shift to higher values of b as ET rises. Thisfinding, along with the increasing contribution of theabstraction mechanism with ET , explains that the globalreactivity for reaction (1) appears for larger values of b ascollision energy rises [Fig. 1(b)].The OH vibrational distributions corresponding to the
insertion and abstraction mechanisms at ET ¼ 0.6 and 1.4 eVare shown in Figs. 4(c) and (d). The results depend on thereaction pathway: abstraction trajectories yield highly invertedvibrational distributions, while insertion trajectories give muchless OH vibrational excitation. This is expected for the twotypes of microscopic mechanisms in the case of an exothermicreaction33. Both the insertion and abstraction OH vibrationaldistributions become rather more excited as ET rises, due to theincrease in available energy. For abstraction, the OH vibra-tional population maximum appears at n0 ¼ 4 for ET ¼ 0.40and 0.60 eV, and at n0 ¼ 5 for ET ¼ 0.80, 1.0 and 1.4 eV.Similarly, the insertion OH vibrational distributions peak atn0 ¼ 0 for ET ¼ 0.21, 0.40, 0.60 and 0.80 eV, at n0 ¼ 1 forET ¼ 1.0 eV, and at n0 ¼ 2 for ET ¼ 1.4 eV. This finding andthe increasing contribution of abstraction as ET increasesexplain the dependence of the global OH vibrational dis-tribution on the collision energy [Fig. 1(d) and Table 1].There are also differences between the insertion and
abstraction mechanisms in the case of the OH rotation. Theinsertion trajectories display excited OH rotational distribu-tions peaking at high N0 values for all populated vibrationallevels. Moreover, they become more excited as ET increases.On the other hand, the shape of the rotational distributionsarising from the abstraction mechanism depends on thevibrational level. For the lowest vibrational levels (n0 4 4)excited distributions similar to the insertion ones wereobtained, while less excited rotational distributions peaking atlow N0 values appeared for the highest vibrational levels(n0 5 5). These trends are shown in Figs. 4(e) and (f ), where theinsertion and abstraction OH(n0 ¼ 3, 5) rotational distribu-tions are depicted for ET ¼ 1.4 eV. For n0 ¼ 3 as the insertionand abstraction trajectories lead to similar OH rotationaldistributions, a unimodal and quite excited global rotationaldistribution is found. The different behavior of the insertionand abstraction reaction modes for n0 ¼ 5, however, results ina clearly bimodal OH full rotational distribution, which peaksat low N0 values since the abstraction mechanism is the mostimportant one producing OH(n0 5 5) for ET ¼ 1.4 eV. Forthese excited OH vibrational levels, the abstraction mechanismyields low excited rotational distributions in all cases, whileinsertion leads to more excited rotational distributions as ET
rises. This finding and the increasing contribution of theabstraction mode as the collision energy increases explains whythe bimodal feature of the OH(n0 5 5) rotational distributionsbecomes pronounced as ET rises [Fig. 1(f)].The two-vector correlations corresponding to the insertion
and abstraction pathways have also been analyzed separately.Fig. 5 shows the kk0 DCSs corresponding to the two differentmechanisms at ET ¼ 0.60 and 1.40 eV. Clearly, abstractionleads to pronounced forward distributions, while insertion
furnishes more symmetric scattering, though with a forwardpredominance too. This together with the finding that theabstraction mechanism was favored for high b values allowedus to classify this reaction mode as stripping-type. On the otherhand, the insertion mechanism furnishes more symmetricscattering, though with a forward predominance too. Thedeviation of the insertion kk0 DCSs from the isotropic situa-tion expected for this mechanism is explained by the shortlifetimes of the collision complexes. The forward feature of theinsertion kk0 DCSs decreases slightly as ET increases, thoughits effect on the global kk0 DCSs is counterbalanced by theincreasing contribution of abstraction. This explains why theforward behavior of the global kk0 DCSs become morenoticeable for higher collision energies [Fig. 3(a)].Similarly, in the case of the l 0j 0 correlation the parallel
feature of the DCSs corresponding to the abstractionmechanism explains the increasing parallel behavior of the fulll 0j 0 DCS with ET [Fig. 3(b)]. For the kj 0 and k0j 0 correlations,minor differences were found between the two mechanisms.Thus, similar values of the rotational alignment parameterwere found for the insertion- and abstraction-like trajectories,which did not show variations of the A
ð2Þ0 value with the final
state of the OH product. As mentioned in section 3.2, thissituation differs from the behavior established for theH+H2O!OH+H2 (ref. 34) and O(3P)+CH4!OH+CH3 (refs. 31 and 35) reactions. However, it must be takeninto account that these two processes evolve throughabstraction mechanisms which are mainly of rebound type (i.e.,they are favored for low b values and yield backward kk0
angular distributions), while the O(1D)+CH4 reaction pro-ceeds via insertion and stripping-type abstraction mechanisms(i.e., it is favored for high b values and yields forward kk0
angular distributions). In the O(3P)+CH4!OH+CH3 (ref.35) reaction those reactive trajectories with non-rebound fea-ture do not display such a strong dependence of A
ð2Þ0 with the
final internal state of the OH product. This tendency maycorrelate with the stronger l 0j 0 parallel alignment found for therebound mechanism than for the non-rebound one, althoughthis is not completely clear.
5. Summary and concluding remarks
A study of the collision energy effects on the dynamics of theO(1D)+CH4!OH+CH3 reaction has been performedusing the quasiclassical trajectory method on an analyticaltriatomic potential energy surface previously derived in ourgroup for this system. The scalar and two-vector propertiesof the reaction have been analyzed in terms of the collisionenergy, considering the following ET values: 0.21, 0.28, 0.40,0.60, 0.80, 1.0 and 1.4 eV. At ET ¼ 0.28 eV the QCT resultsshowed a good agreement with very recent experimental data
Fig. 5 Global ( ), insertion (4) and abstraction (*) kk0 solid angledifferential cross-sections of reaction (1) for ET ¼ 0.60 and 1.4 eV.Global distributions are normalized to unit area. The statistical errors(one standard deviation) are less than 1%.
Phys. Chem. Chem. Phys., 2002, 4, 288–294 293
Publ
ishe
d on
07
Janu
ary
2002
. Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F A
LA
BA
MA
AT
BIR
MIN
GH
AM
on
27/1
0/20
14 1
1:45
:30.
View Article Online

on the product translational energy distribution, 18OH kk0
differential cross-section (DCS) and 18OH velocity–scatteringangle distribution. Two different microscopic reaction mecha-nisms have been found to contribute to the reactivity: aninsertion mechanism and an abstraction mechanism, the for-mer being the more important one though the latter increasesits contribution as ET rises. The features of the dependence ofthe global dynamical behavior of the system on the collisionenergy can be explained in terms of the different dynamicalproperties observed for each mechanism.
Acknowledgements
This study was supported by the ‘‘Direccion General deEnsenanza Superior ’’ of the Spanish Ministry of Educationand Culture through the DGES projects refs. PB98-1209-C02-01 and -02. Financial support from the ‘‘Generalitat de Cat-alunya ’’ (Autonomous Government of Catalonia) ref.2000SGR 00016 is also acknowledged. J.H. thanks the CIRITfrom the ‘‘Generalitat de Catalunya ’’ for a predoctoralresearch grant. The authors are also grateful to the ‘‘Centre deSupercomputacio i Comunicacions de Catalunya (C4-CES-CA=CEPBA) ’’ for computer time.
References
1 M. W. Chase, Jr., C. A. Davies, J. R. Downey, Jr., D. J. Frurip,R. A. McDonald and A. N. Syverud, J. Phys. Chem. Ref. Data,1985, 14, Suppl. 1.
2 J. R. Wiesenfeld, Acc. Chem. Res., 1982, 15, 110.3 P. Warneck, Chemistry of the Natural Atmosphere, Academic
Press, San Diego, 1988.4 E. B. Burnett and C. R. Burnett, J. Atmos. Chem., 1995, 21, 13.5 R. Atkinson, D. L. Baulch, R. A. Cox, R. F. Hampson, Jr., J. A.
Kerr, M. J. Rossi and J. Troe, J. Phys. Chem. Ref. Data, 1999, 28,206 and references therein.
6 A. C. Luntz, J. Chem. Phys., 1980, 73, 1143.7 P. M. Aker, J. J. A. O ’Brien and J. J. Sloan, J. Chem. Phys., 1986,
84, 745.8 S. G. Cheskis, A. A. Iogansen, P. V. Kulakov, I. Yu. Razuvaev,
O. M. Sarkisov and A. A. Titov, Chem. Phys. L ett., 1989, 155, 37.9 C. R. Park and J. R. Wiesenfeld, J. Chem. Phys., 1991, 95, 8166.
10 Y. Rudich, Y. Hurwitz, G. J. Frost, V. Vaida and R. Naaman, J.Chem. Phys., 1993, 99, 4500.
11 M. Gonzalez, J. Hernando, R. Sayos, M. P. Puyuelo, P. A.Enrıquez, J. Guallar and I. Banos, Faraday Discuss., 1997, 108,453.
12 S. Wada and K. Obi, J. Phys. Chem. A, 1998, 102, 3481.13 M. Gonzalez, M. P. Puyuelo, J. Hernando, R. Sayos, P. A.
Enrıquez, J. Guallar and I. Banos, J. Phys. Chem. A, 2000, 104,521.
14 M. Brouard, S. P. Duxon and J. P. Simons, Isr. J. Chem., 1994, 34,67.
15 M. Brouard, J. P. Simons, in T he Chemical Dynamics and Kineticsof Small Radicals, Part II, ed. K. Liu and A. Wagner, WorldScientific, Singapore, 1995, p. 795.
16 M. Brouard, H. M. Lambert, J. Short and J. P. Simons, J. Phys.Chem., 1995, 99, 13 571.
17 M. Brouard, H. M. Lambert, C. L. Russell, J. Short and J. P.Simons, Faraday Discuss., 1995, 102, 179.
18 J. P. Simons, J. Chem. Soc., Faraday T rans., 1997, 93, 4095.19 J. J. Lin, J. Shu, Y. T. Lee and X. Yang, J. Chem. Phys., 2000, 113,
5287.20 T. Suzuki and E. Hirota, J. Chem. Phys., 1993, 98, 2387.21 R. Schott, J. Schlutter, M. Olzmann and K. Kleinermanns, J.
Chem. Phys., 1995, 102, 8371.22 H. Arai, S. Kato and S. Koda, J. Phys. Chem., 1994, 98, 12.23 M. Gonzalez, J. Hernando, I. Banos and R. Sayos, J. Chem. Phys.,
1999, 111, 8913.24 M. Gonzalez, J. Hernando, M. P. Puyuelo and R. Sayos, J. Chem.
Phys., 2000, 113, 6748.25 R. N. Porter and L. M. Raff, in Dynamics of Molecular Collisions,
Part B, ed. W. H. Miller, Plenum, New York, 1976, p. 1.26 D. G. Truhlar and J. T. Muckerman, Atom–molecule Collision
T heory: A Guide for the Experimentalist, ed. R. B. Bernstein,Plenum, New York, 1979, p. 505.
27 H. R. Mayne, Int. Rev. Phys. Chem., 1991, 10, 107.28 R. Sayos and M. Gonzalez, TRIQCT (unpublished program).29 M. Gonzalez, D. Troya, M. P. Puyuelo, R. Sayos and P. A.
Enrıquez, Chem. Phys. L ett., 1999, 300, 603.30 M. Gonzalez, J. Hernando, J. Millan and R. Sayos, J. Chem.
Phys., 1999, 110, 7326.31 R. Sayos, J. Hernando, M. P. Puyuelo, P. A. Enrıquez and M.
Gonzalez, Chem. Phys. L ett., 2001, 341, 608.32 R. Sayos, C. Oliva and M. Gonzalez, J. Chem. Phys., 2000, 113,
6736.33 R. D. Levine and R. B. Bernstein, Molecular Reaction Dynamics,
Oxford University Press, New York, 1987.34 D. Troya, G. Lendvay, M. Gonzalez and G. C. Schatz, Chem.
Phys. L ett., 2001, 343, 420.35 M. Gonzalez, J. Hernando and R. Sayos, Unpublished.
294 Phys. Chem. Chem. Phys., 2002, 4, 288–294
Publ
ishe
d on
07
Janu
ary
2002
. Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F A
LA
BA
MA
AT
BIR
MIN
GH
AM
on
27/1
0/20
14 1
1:45
:30.
View Article Online